アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号、市外局番を含む:(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してください。はい、そうです☒
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示す。はい、そうです☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきた
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ |
|
☐ |
|
ファイルマネージャを加速する |
|
☐ |
|
☒ |
|
規模の小さい報告会社 |
|
||
|
|
|
|
新興成長型会社 |
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する。 ☐
これらのエラーのより真ん中に登録者の任意のエンタルピーCER幹部が相関回復期間内に§240.10 D−1(B)に基づいて受信したインセンティブベースの補償に基づいて回復分析を行う必要があるかどうかを再選択マークで示す。 ☐
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、そうです☐ No
登録者の非関連会社が保有する投票権と無投票権普通株の総時価は#ドルである
2023年2月27日現在登録者が発行した普通株式数かつては…
引用で編入された書類
2023年株主年次総会の登録者最終委託書の一部に提出する登録者は,登録者が2022年12月31日までの財政年度後120日以内に証券取引委員会に提出した書類を,本年度報告の第III部分のForm 10−Kに引用して組み込む予定である。
カタログ表
|
|
ページ |
前向きに陳述する |
1 |
|
リスク要因の概要 |
3 |
|
|
|
|
第1部 |
|
|
第1項。 |
業務.業務 |
5 |
第1 A項。 |
リスク要因 |
74 |
項目1 B。 |
未解決従業員意見 |
140 |
第二項です。 |
属性 |
140 |
第三項です。 |
法律訴訟 |
140 |
第四項です。 |
炭鉱安全情報開示 |
140 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
141 |
第六項です。 |
[保留されている] |
143 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
144 |
第七A項。 |
市場リスクの定量的·定性的開示について |
157 |
第八項です。 |
財務諸表と補足データ |
158 |
第九項です。 |
会計と財務情報開示の変更と相違 |
158 |
第9条。 |
制御とプログラム |
158 |
プロジェクト9 B。 |
その他の情報 |
159 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
159 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
160 |
第十一項。 |
役員報酬 |
160 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
160 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
160 |
14項です。 |
最高料金とサービス |
160 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
161 |
第十六項。 |
表格10-Kの概要 |
163 |
i
前向きに陳述する
このForm 10-K年度報告書は、重大なリスクと不確実性に関する前向きな陳述を含む。歴史事実に関する陳述以外に、本10-K表年次報告に含まれるすべての陳述は、私たちの戦略、未来運営、未来財務状況、未来収入、予想コスト、見通し、計画と管理目標に関する陳述を含み、すべて前向きな陳述である。“予想”、“信じる”、“考慮する”、“継続する”、“可能”、“推定”、“予想”、“予定”、“可能”、“可能”、“計画”、“潜在”、“予測”、“プロジェクト”、“すべき”、“目標”、“到着する”、“会する”、またはこれらの言葉の否定または他の同様の表現は、前向き陳述を識別することを目的としている。すべての前向きな陳述がこのような識別語を含むわけではないにもかかわらず。
本年度報告におけるForm 10−Kに関する前向きな陳述には、以下についての記述が含まれている
私たちは私たちの展望声明で開示された計画、意図、または予想を実際に達成できないかもしれません。あなたは私たちの展望的声明に過度に依存してはいけません。実際の結果または事件は、私たちが前向きな陳述で開示した計画、意図、および予想とは大きく異なるかもしれない。私たちは
1
本10−K表年次報告に含まれる警告要因,特に“リスク要因”の部分には,実際の結果やイベントが我々が行った前向き陳述とは大きく異なる可能性が考えられる重要な要素が含まれている。私たちの前向きな陳述は、私たちが行う可能性がある任意の未来の買収、合併、処置、協力、合弁、または投資の潜在的な影響を反映していない。
あなたはこのForm 10-K年次報告書と私たちがForm 10-K年次報告書で引用した文書を読むべきであり、これらの文書は証拠としてアメリカ証券取引委員会に提出された他の文書に完全に提出されており、私たちの未来の実際の結果は私たちが予想していたものと大きく異なるかもしれないことを理解しなければならない。本年度報告に含まれるForm 10−Kに含まれる前向き陳述は、本年度報告におけるForm 10−Kにおいて行われ、法的要件が適用されない限り、新たな情報、未来のイベント、または他の理由でいかなる前向き陳述を更新する義務も負わない。
本年度報告書の用語“私たち”、“私たちの会社”、“私たちの業務”は、文意が別に言及されているか、または別の指示がある場合を除いて、Verve Treateutics社およびその合併子会社を意味する。
2
リスク要因の概要
私たちの業務は多くのリスクに直面しており、投資決定を下す前に、あなたはこのようなリスクを認識しなければならない。以下では,我々の業務が直面していると考えられる主なリスクと,本報告に含まれる本年度報告第I部分表10−Kにより網羅的に記述されているリスク項目“リスク要因”その他の情報をまとめた。以下に説明するリスクと不確実性は私たちが直面している唯一の危険と不確実性ではない。私たちは現在知らないか、あるいは現在あまり重要ではないと考えている他のリスクや不確実性も私たちの業務運営を損なう可能性があります。
以下のいずれかのリスクが発生した場合、私たちの業務、財務状況および経営結果、および将来の成長見通しは重大な悪影響を受ける可能性があり、本報告で作成された前向き陳述の実際の結果は、このような展望的陳述で予想される結果とは大きく異なる可能性がある
3
4
第1部
プロジェクト1.ビジネス
概要
著者らは臨床段階の遺伝子薬物会社であり、心血管疾患を治療する新しい方法を開拓し、治療を慢性治療から単一治療コースの遺伝子編集薬物に転換した。過去50年間に治療方面で進展を得たにもかかわらず、心血管疾患は依然として全世界の主要な死亡原因である。現在の慢性看護モデルは脆弱である−厳格な患者コンプライアンス,広範な医療インフラ,定期的な医療サービスが必要であり,多くの患者が適切な看護を受けられないようにしている。私たちの目標は新しい単一治療コースの治療法を提供することでCVDの慢性ケアモデルを覆すことです体内にある遺伝子編集治療の重点はこのような高度な流行と生命を脅かす疾患の根本的な原因を解決することである。著者らの最初の2つの項目はそれぞれPCSK 9とAngptl 3に対して、この2つの遺伝子は低密度リポ蛋白質コレステロール(LDL-C)などの脂質低下の目標として広く検証されている。著者らは、これらの遺伝子を編集することは、粥状動脈硬化性心血管疾患(最もよく見られる心血管疾患形態)に罹患しているか、あるいは直面する患者の一生において、低密度リポ蛋白-Cを有効かつ持続的に低下させることができると信じている。
私たちの方法は21年間の多くの突破を利用してST世紀の生物医学--ヒト遺伝分析、遺伝子編集、メッセンジャーリボ核酸またはメッセンジャーリボ核酸--治療および脂質ナノ粒子(またはLNP)送達に基づく療法--主に肝臓で発現する遺伝子を標的とし、心血管疾患を引き起こすタンパク質の産生を妨害する。私たちは単行コースのパイプを進めています体内にある遺伝子編集プログラムは、各プログラムは自然抗病性突然変異をシミュレーションし、特定の遺伝子を閉鎖し、血中脂質を低下させ、それによってASCVDのリスクを低下させるように設計されている。著者らは最初にこれらの計画を開発して家族性高コレステロール血症(FH)患者を治療する予定であり、FHは遺伝病であり、一生深刻に上昇する血液低密度リポ蛋白-Cを招き、それによって早発性ASCVDのリスクを増加させる。もし私たちの計画がFHで成功すれば、私たちはそれらもより広範なASCVD患者に潜在的な治療を提供できると信じている。最終的に,これらの治療法はASCVDハイリスク群の管理のために開発される可能性があり,予防策として,あるワクチンが感染症に対する長期保護を提供する方式と類似していると考えられる。
長期にわたる低密度リポ蛋白の接触は粥状動脈硬化プラークの形成を招き、それによってASCVD患者の動脈硬化を招く。累積低密度リポ蛋白質コレステロール暴露の低下とASCVDリスクの低下との関係は医学的に最もよく知っている関係の一つである。研究により、確定診断されたASCVD患者の中で、5年以内に低密度リポ蛋白-Cを39 mg/dL下げることは更なる事件のリスクを21%下げることができ、一生中の類似程度の低密度リポ蛋白-C差異は初めてASCVD事件が発生するリスクを88%下げることができる。このことは,低密度リポ蛋白を大幅に低下させるだけでなく,患者の一生の間この低下を保つことであることを示唆している。ASCVDの治療と予防の基盤は,低密度リポ蛋白質を早期,可能な限り長時間積極的に低下させることでなければならないと考えられる。
現在の看護標準は1種の慢性看護モードであり、その治療方法の持続性と終身性及び固有の依存性の問題のため、常に総低密度リポ蛋白-C暴露を十分に制御できない。そのため、大部分の確定診断されたASCVD患者の低密度リポ蛋白-Cレベルはアメリカ心臓協会(AHA)とアメリカ心臓病学会(ACC)が提案した目標より高く、これは彼らにASCVD事件の再発リスク、及び侵襲性医療プログラム甚だしきに至っては死亡の可能性に直面させた。また、低密度リポ蛋白質コレステロール上昇による損害の無音性質を考慮して、多くのASCVDリスクのある患者は持続治療の治療メリットと前述の治療の巨大なリスクを正確に認識しておらず、毎日の服薬、生活様式の変更と他の慢性方法による重い生涯薬物負担に重点を置いている。蓄積低密度リポ蛋白-C暴露を有効かつ持続的に制御する単一治療コース遺伝子編集治療は、ASCVD患者あるいはASCVDリスクのある患者の慢性看護モードを根本的に混乱させ、患者、提供者、医療保健システムの重大な負担を軽減することができると信じている。
私たちの主要な候補製品VERVE-101は、肝臓中のPCSK 9遺伝子を永久的に閉鎖することを目的としている。PCSK 9は高度に有効な標的であり、それは低密度リポ蛋白受容体(LDLR)を調節することによって血液低密度リポ蛋白-Cを制御する上で重要な役割を果たしている。血液中PCSK 9蛋白の減少は,肝臓が血液から低密度リポ蛋白−Cを除去する能力を向上させた。VERVE−101はLNPを介した送達を用いて肝臓を標的化し,塩基編集技術を用いてPCSK 9遺伝子の特定の位置で単一塩基変化を行い,PCSK 9タンパク質の産生を撹乱する。
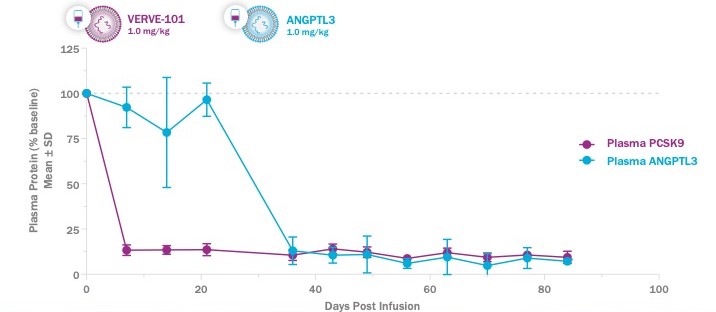
1つは体内にある非ヒト霊長類動物におけるVERVE−101前駆体製剤の概念検証研究では,低密度リポ蛋白レベルの有意な低下が観察され,その後長い間不変であった
5
治療します。この研究では,PCSK 9に対する塩基エディタを単回静注したところ,治療後2週間で血液PCSK 9蛋白が平均89%低下するとともに,血液低密度リポ蛋白レベルの平均59%の低下が認められた。この低密度リポ蛋白−Cの減少幅は治療後2年間で平均71%を維持していた。
NHPにおけるVERVE−101を用いた臨床前研究では,15日目に肝臓生検組織におけるPCSK 9標的遺伝子位置に1.5 mg/kgを単回投与したところ,70%の平均編集が認められた。この研究では,治療後2週間で血液PCSK 9蛋白が平均79%低下するとともに,血液低密度リポ蛋白レベルの平均62%の低下が認められた。治療1年後に評価を行ったところ,これらの低下は持続し,血液PCSK 9蛋白は平均89%,血液低密度リポ蛋白レベルは68%低下した。
また,われわれのNHPに対する前臨床研究では,VERVE−101は単回投与後耐性が良好であり,肝機能テストはわずかに上昇し,2週間で消失した。VERVE−101で処理した初代ヒト肝細胞では,PCSK 9標的の標的編集が観察されたが,約3,000個の識別された潜在的非標的のいずれにも有意な編集は認められなかった。
われわれの臨床前データに基づき,VERVE−101が最初にヘテロ家族性高コレステロール血症(HeFH)の治療に応用されている。われわれはHeFHを超えてVERVE−101の臨床開発を徐々に拡大し,確定したASCVD患者の治療に用いる予定であり,これらの患者は経口治療において低密度リポ蛋白−Cの目標を達成しておらず,世界数億の潜在患者を代表している。最終的に,VERVE−101はASCVDハイリスク群に対して有用である可能性が考えられ,一般群の予防策として有用であると考えられる。
CHART-1試験は約40名のASCVDと診断されたHeFH成人患者を募集し、VERVE-101投与の安全性と耐性を評価し、薬物動態と血液PCSK 9蛋白と低密度リポ蛋白-Cの低下に対して追加的な分析を行うことを目的とした。試験は、(A)単回漸増用量部分を含み、その後、(B)より多くの参加者が選択された潜在的治療用量を得る単回用量キューを拡大し、(C)選択可能な第2の用量キューで、A部分の低用量キューにおいて、条件に適合する参加者が、選択された潜在的治療用量で第2の治療を受けることを選択することができる、3つの部分を含む。ニュージーランドやイギリスの規制当局との相互作用の中で、各国/地域の資格、設計、行動の様々な修正を考慮して、特定の国/地域に対する合意を策定した。
われわれはすでにニュージーランドとイギリスでVERVE−101の臨床試験申請(CTA)の承認を得ており,2022年7月にわれわれの心臓1号臨床試験で1人目の患者がVERVE−101投与量を受けていると発表した。2022年11月、我々は、全世界1 b期開放ラベル臨床試験である心臓1号臨床試験用量増加部分の第1用量列中のVERVE-101用量を完了したことを発表した。ニュージーランドとイギリスでは学生募集が行われている。著者らは2023年下半期に心臓1号臨床試験用量増加部分の初歩的な安全性と薬効学データを報告する予定である。
2022年10月、私たちはアメリカ食品医薬品局(FDA)に私たちの試験新薬IND申請を提出し、HeFH患者のVERVE-101臨床試験評価を要求し、その後FDAは私たちのIND申請が保留されたことを通知した。2022年12月、我々はFDAから臨床的座礁信頼を受け取り、(I)ヒトと非ヒト細胞との間の潜在的差異、(Ii)生殖系編集のリスク、および(Iii)非肝細胞タイプの非標的分析を含む、棚上げを解決するために必要な情報を概説した。ニュージーランドとイギリスで行われている心臓1号臨床試験の臨床データはFDAに提出されたIND申請パッケージには含まれていない。臨床保留状では,FDAは試験の臨床データの提供を要求している。さらに、FDAは、追加の避妊措置を取り入れ、参加者用量間の交互間隔の長さを増加させるために、米国の試験計画を修正することを要求している。私たちは私たちの反応をできるだけ早くFDAに提出するつもりだ。
我々はAngptl 3の開発候補薬物Verve−201に対して肝臓中のAngptl 3遺伝子を永久的に閉鎖することを目的としている。アンギオテンシン変換酵素3はコレステロールとトリグリセリド代謝の重要な調節因子である。Angptl 3蛋白の産生を妨害することは,PCSK 9とは異なる機序により低密度リポ蛋白−Cやトリグリセリドレベルの低下を引き起こす可能性が考えられる。われわれは,米国では約1300人,難治性高コレステロール血症の治療に関与しているホモ接合体家族性高コレステロール血症(HoFH)の治療に用いる計画を開発する予定であり,ASCVD患者では低密度リポ蛋白−C目標に達していないヒトとPCSK 9阻害剤の経口治療と定義されている。最終的に,VERVE−201はASCVDハイリスク群において一般群においても予防策として有用であると考えられる。我々はVERVE−201臨床開発開始の規制申請を支援するための前臨床研究を行っており,2024年に1 b期臨床試験を開始する予定である。
6
VERVE−201では,Angptl 3遺伝子に対する塩基エディタを内部開発したGalNAc−LNPを用いて肝臓に送る予定である。HoFH患者では,LDLRの欠乏により,標準LNPを持つ塩基編集を肝臓に送ることは挑戦的であり,LDLRはLNP取り込みを介した経路が知られている。我々はすでにGalNAcリガンドを有する独自のLNPsを開発し,肝臓中のデシアル酸糖蛋白受容体(ASR)に結合し,LDLRを迂回し,HoFH患者の肝臓を摂取できるようにすることを目的としている。
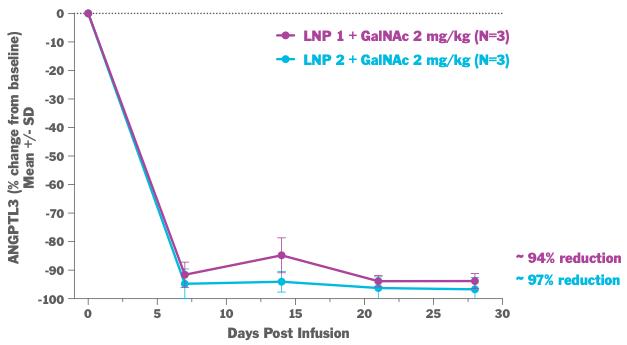
VERVE−201前駆体製剤の臨床前研究では,我々独自のGalNAc−LNPsの2種類の異なる製剤を単一処理し,Angptl 3を標的とした塩基エディタを提供した。血液Angptl 3蛋白は約94%(n=3)と97%(n=3)の低下が認められ,低密度リポ蛋白(LDL−C)は100 mg/dL近く低下し,ベースラインより約35%低下した。これらの研究は,野生型NHP中のLDLR遺伝子を編集し,標準LNPに封入されたCas 9と二重誘導RNA戦略を用いてNHP肝臓におけるLDLR発現を除去することにより作成した内部開発のHoFH NHPモデルで行った。このモデルでは,LDLR遺伝子上で70%近くの全肝DNA編集を実現し,肝臓ではLDLR蛋白が約94%減少し,血液中の低密度リポ蛋白−Cが6倍に増加した。
NHPにおけるAngptl 3塩基編集の概念検証研究(n=4)では,血液Angptl 3蛋白がベースラインより約96%低下することが観察され,2年間のフォローアップを行った。また,治療をアラニンアミノトランスフェラーゼやビリルビンレベルで測定したところ,肝臓毒性マーカーへの長期的な影響は認められなかった。
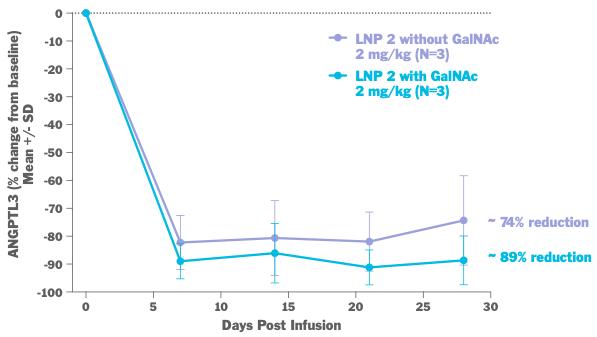
我々はまた、正常肝臓を有する野生型HHPにおけるGalNAc-LNPの使用またはGalNAcを使用しない標準LNPの送達効率を評価する臨床前研究において、Angptl 3標的塩基エディタを送達するための当社独自のGalNAc-LNP方法の潜在的広範な実用性を評価した。これらの研究では,GalNAc−LNPにより伝達されたAngptl 3標的塩基エディタで処理した野生型NHPのAngptl 3蛋白が約89%減少し,標準LNPで処理した野生型NHPのAngptl 3蛋白が約74%減少することが観察された。
我々は,新たかつ最適化されたGalNAc標的リガンドの開発能力,最適なアンカー,最適なLNP成分の組成および割合,および標的リガンドとの付加およびLNP形成の最適プロセスに投資し,確立している。著者らはGalNAcがこの2種類の形式のFH患者に1つの伝達プラットフォームを提供し、他の肝臓をガイドとする伝達を優勢とする応用に適用できると信じている。またデータを生成し,そこでGalNAc−LNPがPCSK 9に対する塩基エディタを効率的に提供できることが観察された。この研究では,野生型NHPにGalNAc−PCSK 9 LNPs標的PCSK 9の塩基エディタを用いたところ,血液中PCSK 9蛋白が約87%減少することが観察された。このデータはGalNAc−LNP送達が他の適応の肝臓編集において広範な実用価値を有する可能性を示唆しており,GalNAc−LNP送達のPCSK 9塩基編集の臨床前開発を推進していると考えられる。
著者らは卓越した会社を構築することに集中し、遺伝子編集薬物を開発して心血管疾患患者を治療し、心血管疾患は世界で死亡を招く主要な原因である。我々のチームの専門知識と能力を利用して,我々のチャネルをPCSK 9とAngptl 3に拡張し,我々の単授業遺伝子編集手法を他に適用する予定である体内にある肝臓遺伝子編集治療、例えば著者らはリポ蛋白質(A)或いはLp(A)の計画に対して、疾病の根本的な原因に対する単一治療コースの遺伝子編集薬物を開発する。
私たちのチーム
我々は2018年に設立され、Sekar Kathiresan、M.D.,Kiran Musunuru,M.D.,Ph.,公衆衛生修士,J.Keith Joung,M.D.,Ph.D.,Burt Adelman,M.D.,Issi Rozen,MBAとBarry Ticho,M.D.,Ph.D.を含む世界的に有名な心血管遺伝学研究者、遺伝子編集先駆者、成熟したビジネスリーダーから構成されている。私たちの設立以来、私たちは才能あふれる個人チームによって駆動される組織と文化を構築し、彼らは私たちの名前の背後の意義-活力を体現している。精神と情熱--彼らには共通の目標があり、それは心血管疾患患者や心血管疾患のリスクのある患者の看護を変えることだ。
著者らの指導チームのメンバーは人類遺伝学、遺伝子編集、心血管疾患看護及び薬物開発と商業化の面で豊富な集団経験を持っている。私たちの最高経営責任者であるKathiresan博士は心臓病予防の専門家であり、彼はCVDに対する抵抗力を引き起こす遺伝子突然変異を引き起こす上で突破的な発見を得た。アンドリュー·アーシュ、J.D.,私たちの首席運営官兼総法律顧問総裁は、フィ然を達成したバイオテクノロジー幹部であり、20年以上の運営と法律管理経験を持っている。アンドリュー·ベリンガー、医学博士、私たちの首席科学官と首席医療官は、心臓病の専門家であり、薬物輸送、薬物開発と転化医学の面で公認された専門知識を持っている。私たちの最高財務官Allison Dorvalは、財務、会計、財務報告、投資家関係について20年以上の指導経験を持っています。私たちの首席行政官Joan Nickersonは人的資源の面で25年以上の経験を持っている。
7
私たちは心臓病学、人類遺伝学、転化医学、交付技術、商業と金融分野の有力な専門家を含む科学諮問委員会があり、医学博士ユージン·ブロンウォルド、医学博士Daniel·J·レッド、医学博士アンドリュー·ジル、医学博士アントニー·フィリパキス、博士、キラン·ムスルール、医学博士、医学博士、公衆衛生修士、医学博士ペニー·M·シートンを含む。ブリグム女性病院心血管内科専門家、ハーバード医学院西洋医学研究所教授のブロンワルド博士が私たちの科学顧問委員会の議長を務めている。心臓病学の中で最も多く引用された著者とされ、米国国家科学院院士に選ばれた初めての心臓病専門家でもある。
我々は,塩基編集とCRISPRヌクレアーゼ,および複数のLNPを含む様々な遺伝子編集要素をカバーする認可技術と知的財産権を有しており,これらのLNPはBEAM治療会社やBEAM,ブロド研究所,BROAD,Editas Medicine,Inc.,ハーバード大学総裁および研究員,ハーバード大学,マサチューセッツ州総病院,Acuitas治療会社,Acuitas,ノワ製薬またはノワ製薬会社の許可を得ている。
心血管ケアを変える
過去50年間に治療面で進展を得たにもかかわらず、心血管疾患は依然として全世界流行病である。現在の慢性看護モデルは脆弱である−厳格な患者コンプライアンス,広範な医療インフラ,定期的な医療サービスが必要であり,多くの患者が適切な看護を受けられないようにしている。世界保健機関のデータによると、心血管疾患は依然として全世界の主要な死亡原因であり、ほぼ3分の1の死亡は心血管疾患によるものである。それも期待寿命の減少を招く要因であり、米国で最も高価な健康状態の一つでもある。アメリカ疾病コントロール·予防センター(CDC)のデータによると、心血管疾患は毎年アメリカの医療システムに与えるコストと生産力損失は3500億ドルを超える。我々の目標は,新たな治療法を提供することにより,心血管疾患の慢性看護パターンを撹乱し,このような高度な流行と生命に危険な疾患の根本的な原因の解決に専念することである。
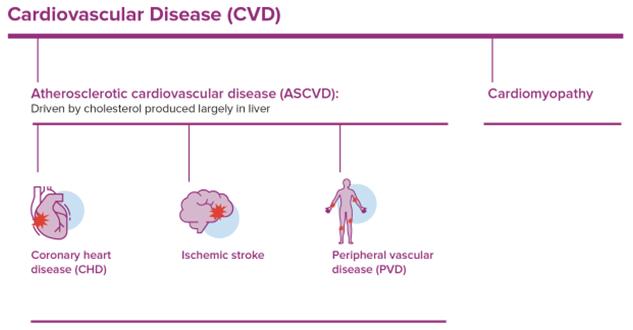
心血管疾患は総称して心臓や血管疾患と呼ばれ,ASCVDなどと診断され,次の図に示す。ASCVDにおいて、コレステロールはCVDの大きなサブセットであり、それは粥状動脈硬化プラークの発展を招き、粥状動脈硬化は血管壁中のコレステロール、細胞と細胞破片の混合物であり、動脈硬化を招く。
低密度リポ蛋白(LDL)、トリグリセリドに富むリポ蛋白質(TRL)或いはリポ蛋白質(A)のいずれも血コレステロールを持っており、生涯高累積暴露血コレステロールはASCVDの根本的な原因である。下図は、コレステロールおよびトリグリセリドを含み、表面にアポリポタンパク質Bまたはアポリポタンパク質Bを含む、これらの肝臓産生リポ蛋白質が血液中に分泌されるおよびその典型的な組成を示す。この3種類のリポ蛋白質のそれぞれはASCVDの1つの独立した危険経路を代表し、著者らはこれらの経路中の複数の経路が運ぶ血中脂質を同時に低下させることはASCVDの治療に追加のメリットを提供するべきであると考えている。
8

現在、低密度リポ蛋白-Cを下げる治療方法は持続、生涯治療を採用し、このような慢性看護モードの局限性のため、多くのASCVD患者の低密度リポ蛋白-Cに対する累積暴露は依然として十分に制御されていない。ASCVD患者の最もよく見られる治療方法は毎日スタチン類薬物を服用することであり、同時に治療方式を変更することを提案する。いくつかの非スタチン類の毎日錠剤があり、エゼブラブ、胆汁酸分離剤とフェナバドール酸を含み、ASCVD患者が推薦された低密度リポ蛋白-C目標を達成するのを助けるために、単独で或いは順番にスタチン類薬物治療に添加することができる。2つのFDAによって承認されたモノクロナル抗体、またはモノクロマブ、volocumabおよびalirocumabと呼ばれ、それらはPCSK 9タンパク質を標的化し結合し、通常月に2回注射される。そのほか、InlisiranはPCSK 9に対する小干渉RNA、或いはsiRNAと呼ばれ、毎年2回皮下注射し、すでにFDAとヨーロッパ薬品管理局(European Medicines Administration、EMAと略称する)の許可を得た。これらの承認された治療方法があるにもかかわらず、長期的に有効にASCVD患者或いはASCVDハイリスク患者の低密度リポ蛋白レベルを制御することは依然として重要な満足されていない需要である。
累積低密度リポ蛋白質コレステロール暴露の低下とASCVDリスクの低下との関係は医学的に最もよく知っている関係の一つである。ヒト遺伝学研究により、遺伝性疾患FHを有するヒトは生涯血液低密度リポ蛋白-Cが深刻に上昇し、これは早発性ASCVDリスクの増加を招く可能性がある。逆に,生来の耐性突然変異は肝臓に発現するコレステロール上昇遺伝子,例えばPCSK 9を閉じ,彼らの一生の低密度リポ蛋白−Cレベルは低く,ASCVDに罹患することは少ない。これらの知見は,患者の一生における低密度リポ蛋白曝露を減少させるために早期積極治療の重要性を指摘している。確定診断されたASCVD患者の場合、以前心臓病を有する患者のように、AHA/ACCが発表した臨床治療ガイドラインは、血液低密度リポ蛋白-Cを70 mg/dL未満に低下させる目標を提案し、ヨーロッパ心臓病学会(ESC)は血液低密度リポ蛋白-Cを55 mg/dL未満に低下させる目標を提案する。血液低密度リポ蛋白が十分な時間を維持すれば、最初のASCVD事件のリスクは心臓発作を含み、著しく低下することができる。研究により、確定診断されたASCVD患者の中で、5年以内に低密度リポ蛋白-Cを39 mg/dL低下させ、更なる事件のリスクを21%低下させることができ、一生中の類似程度の低密度リポ蛋白-C差異は初めてASCVD事件が発生するリスクを88%まで下げることができる。
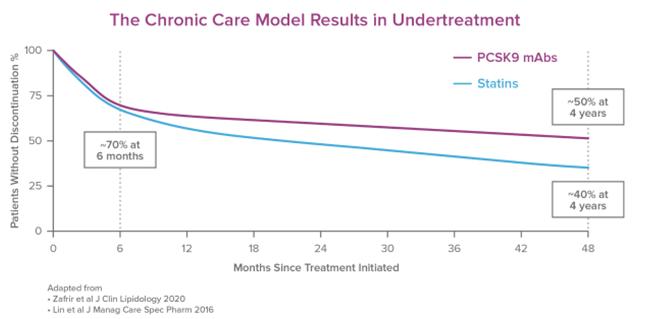
スタチン類と非スタチン類薬物が使用可能であるにもかかわらず、多くのASCVD患者において、低密度リポ蛋白-Cの累積暴露はよく十分に制御されていない。そのため、大部分の確定診断されたASCVD患者の低密度リポ蛋白レベルは臨床治療ガイドラインより高い。アメリカ全国外来の心血管看護登録において、ASCVD臨床事件を経験した260万人の患者の中で、53%の患者はいかなるコレステロール降下治療を受けておらず、72%の患者は依然としてAHA/ACCが提案した低密度リポ蛋白レベルより高い。また,心臓発作後1年間に約6000名の患者に対して行った臨床試験データによると,無料で薬物を提供している約3000名の患者のうち,39%のみがスタチンの治療を完全に遵守していると報告している。
ASCVD患者またはASCVDリスクのある患者の大きな割合は、毎日の服薬または頻繁な注射に関連する重い生涯薬物負担のため、開始しないか、または治療を継続することを選択する。低密度リポ蛋白質コレステロール上昇による損害の無音性質を考慮して、多くのASCVDリスクのある患者は持続治療の治療メリット及び前述の治療の巨大なリスクを正確に認識せず、長期治療の重い、一生の薬物負担に重点を置いている。以前のスタチン類薬物と注射可能なモノクロナル抗体PCSK 9阻害剤の大量の研究により、治療を中止することはよくあることが示唆された。次の図は
9
その中の2つの研究により、50%以下の患者は4年以内にまだPCSK 9阻害剤モノクロナル酸或いはスタチン類薬物の治療を受けていることを表明した。
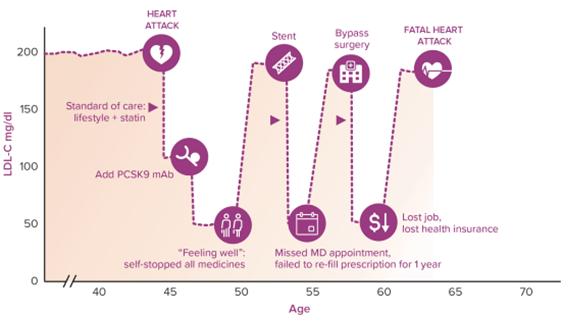
完全に治療を堅持しないことは患者の一生中の血液低密度リポ蛋白レベルの著しい変動を招く可能性がある。下のイラストは仮想的なFH患者の旅を描いており、彼は44歳時に心臓病発作後に標準看護治療を開始し、当時患者はASCVDと診断され、依存性差と医療ルート不足による数年間低密度リポ蛋白質コレステロールを完全にコントロールしなかった潜在的な結果を描いている。不完全な低密度リポ蛋白制御は臨床ASCVD事件の再発を招くことができ、そして冠状動脈内ステント移植と冠状動脈バイパス手術などの侵襲性医療プログラムが必要であり、そして致命的である可能性がある。これらの繰り返し発生するイベントやプログラムは,患者,治療提供者,医療システム全体に大きな負担を与え,医療サービスのコストや使用を増加させている。
ASCVDを治療する1コースの遺伝子編集療法の利点は
ASCVD患者の単一治療コース遺伝子編集治療は慢性看護モードの多くの挑戦を解決し、このような高度に流行した疾患の治療のための新しい例を作る可能性があると信じている
10
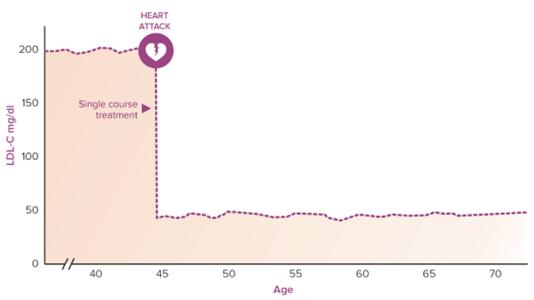
命を危険にさらす病気です患者の一生における累積低密度リポ蛋白-C暴露を有効かつ持続的に制御することによって、著者らの遺伝子編集薬物はASCVD患者或いはASCVDリスクのある患者の慢性看護モードを根本的に撹乱し、患者、提供者と医療システムの重大な負担を軽減できると信じている。次のイラストは同じ仮想的なFH患者の旅を描いています この場合,WHOは心疾患発作後に単クールの遺伝子編集治療を受けたため,ASCVD事件の再発を回避した。
ASCVD治療法を変更する目標を達成するために、著者らは一連の単一治療コース遺伝子編集治療法を開発しており、21の技術を利用した多くの突破を実現しているST世紀生物医学--ヒト遺伝分析、遺伝子編集、メッセンジャーリボ核酸に基づく治療とLNP介在性伝達。私たちは私たちの方法が次の潜在的な利点から利益を得ると信じている
11
私たちの戦略
私たちは次のような重要な要素を含む戦略を実行している:
12
私たちの方法は
著者らは全身カスタマイズの方法を採用しており、単一治療コースの遺伝子編集薬物を開発し、心血管疾患患者に対する治療を変化させることを目的としている。我々の遺伝子エディタは,肝臓で検証された遺伝子に対して,広くヒト遺伝学やヒト薬理学データに支持されており,心血管疾患に関与していることが知られている。我々の初期プログラムでは,VERVE−101とVERVE−201を含む塩基編集を用いて,DNA二本鎖切断を招くことなく,ゲノムの単塩基レベルで正確かつ効率的な編集が可能な次世代遺伝子編集方法である。我々の遺伝子エディタには,遺伝子や塩基エディタをコードするmRNAをカプセル化するLNPsと,肝臓に発現する興味のある遺伝子に対するgRNAがある.著者らは、著者らの方法の以下の重要な要素は、全世界範囲で数百万の心血管疾患患者に遺伝子編集治療を提供する目標を実現するのを助けると信じている。
編集者選択
我々は,遺伝子療法やRNA療法を含む他の遺伝医学的方法と比較して,遺伝子改変型において持続的で有効かつ多機能的な潜在力を提供していると信じているため,核技術としてCVDに対する単クール遺伝子編集療法を開発している。我々は塩基編集やCRISPRヌクレアーゼを含むライセンスにより様々な遺伝子編集技術を獲得している。私たちはまた新しい遺伝子編集技術を発見することを求めている。著者らは、異なる遺伝子編集技術を異なるCVD単一治療コース治療に柔軟に応用し、任意の所与の治療応用のために最適な潜在的選択を決定できると信じている。
13
CRISPR-CAS編集
CRISPR-Casはヌクレアーゼに基づく遺伝子編集形式であり、gRNA配列とDNA配列の間のマッチングを評価することによって、ヒト細胞において高度に特異的なゲノムDNA配列を標的とすることができる。GRNAはCas蛋白がDNA配列の相補部分を認識することを可能にする。RNA-DNAペアリングが発生すると,Cas酵素は二本鎖DNA切断を招き,細胞の自然DNA修復機構はゲノムを改変あるいは修復する。修復が故障した場合,目的遺伝子,いわゆるノックアウトを破壊する可能性がある。CRISPR−Casは妨害により標的遺伝子を効率的にノックアウトまたはサイレンシングした。しかし、標準CRISPR-Cas遺伝子編集の潜在的限界は遺伝結果の予測可能性の欠如と二本鎖DNA切断に関連する潜在毒性を含む。
基礎編集者
塩基編集は、DNAの二本鎖切断を引き起こすことなく、ゲノムの単塩基レベルで正確かつ効率的な編集を行うことができる次世代遺伝子編集方法である。CRISPR-Cas遺伝子編集方法がゲノムの“はさみ”に類似しているとすれば、塩基編集は“鉛筆”に類似し、遺伝子中の1文字を消去および書き換えする。
我々とビムの許可プロトコルにより、2種類の異なるタイプの塩基編集-アデニン塩基編集(ABES)とシトシン塩基編集(CBE)を用いることができ、各編集は1つの修飾したCas 9蛋白がgRNAに結合し、標的ゲノム配列の能力を保持し、同時に二本鎖DNA切断を回避した。塩基編集はデアミナーゼの種類で区別され,デアミナーゼは化学修飾を行う塩基編集酵素であり,Cas 9と融合している。アデニンやA,塩基またはシトシンまたはC塩基上のアミン基をデアミナーゼで予測可能な化学修飾を行ったものを脱アミノ基と呼び,次の図に示す。
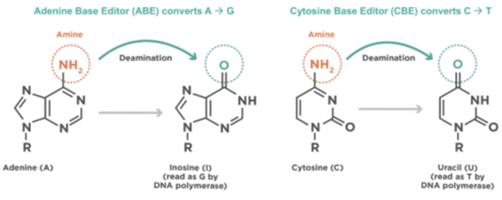
VERVE−101とVERVE−201では,ABEを用いてAのアミン基をイノシンやI塩基に変換し,DNAポリメラーゼはグアニンやG塩基と読み,最終的にAからGへのスペル変化をもたらす。初期修飾が起こると,中間DNAは編集後の鎖と未編集の鎖からなり,前者は標的にIを含み,後者はチミンやT塩基を持つ。I:T塩基対は一致せず、細胞は通常、編集が失われる可能性がある中でそれの修復を試みる。編集を保留するために、私たちの基礎編集は未編集DNA一本鎖を切断し、二本鎖切断を作成するのではなく、刻印と呼ばれる。しかしながら、未編集の鎖上のノッチの存在は、未編集の鎖ではなく、新たに編集された鎖を修復テンプレートとして使用するように細胞を誘導することによって、編集の効率を向上させ、I:C塩基対を生成する。DNA修復または複製時に、IはGと読み、G:C塩基対を生成し、A:T塩基対からG:C塩基対への永久変換を完了する。PCSK 9あるいはAngptl 3遺伝子内の特定の位置のこの一塩基対の変化は遺伝子を変化させ、機能的なPCSK 9或いはAngptl 3蛋白を産生できず、循環血中脂質レベルの上昇を維持する作用を乱す。
目標選択
著者らは肝臓-心血管軸の有効な遺伝子に集中し、これらの遺伝子は主に肝臓で発現し、蛋白質の生産を乱し、或いは有益な突然変異を導入することは心血管疾患の潜在的な原因を有効に治療する可能性がある。私たちが計画した目標を考慮すると、私たちは以下の基準を評価する
14
非目標編集者の評価
遺伝子編集はゲノム中の特定の位置で正確に変更することができるが、人気のない位置で変更することも可能であり、いわゆる非標的編集である。塩基編集は,一塩基レベルでの編集の精度と効率,二本鎖DNA切断を起こさずに編集する能力を考慮すると,CRISPR−Casヌクレアーゼ編集よりも固有に低い非標的編集リスクを有する。
非ターゲット編集の方法を最大限に削減することは、複数の直交分析を使用して、我々の編集者と非ターゲット編集を行う可能性を全面的に評価することを含む。これらの方法は、DNA配列決定によって単一ヌクレオチド分解能で編集されたインビトロ方法、例えばOne-SEQを検出することを含み、その配列は、標的上の遺伝子座上の配列と類似している計算設計された合成DNAライブラリーを使用するか、またはDigenome-配列番号であり、細胞から抽出されたDNAは、編集遺伝子座の無偏見評価を提供する。この二つの方法は候補サイト指名に相補的で厳格な作業プロセスを提供する。また,これらの指名候補部位を評価し,ゲノムおよび転写群における構造変異および非ガイド効果を評価する高感度ハイブリッド捕捉試験の解析方法を開発した。我々は,多様な革新技術を用いて非目標編集における内部専門知識を評価することで,この分野をリードし,将来のプロジェクトを迅速に推進することができると信じている.
脂質ナノ粒子投与方式の選択
遺伝子編集治療にはmRNAとgRNA分子を細胞内で標的細胞型に伝達する必要がある−われわれの例では,肝臓中の肝細胞であるわれわれのすべてのプログラムは非ウイルス法LNPsを用いて伝達されている。LNPsは承認された製品でも他者と他の薬剤による臨床試験でもよく確認されており,全身投与後優先的に肝臓に蓄積されている。われわれが非ウイルスLNP送達を選択したのは,既存のウイルス送達方法よりもLNPの方が潜在的に高い安全性を有し,またLNPの肝臓への天然配向により,LNPを用いて効率的な肝臓編集が可能であるためである。
LNPsによって非ウイルス的にLNPsを肝臓に輸送することは、潜在的な利点を有する
これまで、我々のLNP発見プラットフォームはすでに著者らが設計、合成した新しい特許イオン化可能脂質を産生し、それらが遺伝子編集ペイロードをマウス肝臓に輸送する潜在力を評価した。私たちはNHPで評価するために、これらの処方をさらに最適化して拡大している。LNPsに添加すると,RNAペイロードをより効率的に肝臓に輸送することができる新たな標的リガンドも開発された。
ターゲットごとに、外部パートナーまたは私たちの内部LNP発見プラットフォームからの最適なLNP配信オプションを評価します。われわれの先行プロジェクトVERVE−101では,AcuitasからLNP技術の許可を得ており,Acuitasは成熟した会社であり,臨床使用のLNPの協力と開発に良好な記録を有している。我々のAcuitasとの協力には,VERVE−101のためのAcuitas LNPを選択する前に様々なLNP製剤およびRNAペイロードを評価するためのいくつかのNHP研究が含まれている。Verve−201については,すでにノワール社からLNP技術の許可を得ており,内部開発したGalNAc−LNP技術を用いて納入する予定である。
15
われわれの内部LNP発見プラットフォームを将来の治療計画の重要な交付技術源とした。私たちは以下の点に集中して私たちの内部LNP発見プラットフォームを最適化しています
私たちの内部LNP発見プラットフォームは私たちの現在と未来の計画の候補製品を改善すると信じています。
我々は,新たかつ最適化されたGalNAc標的リガンドの開発能力,最適なアンカー,最適なLNP成分の組成および割合,および標的リガンドとの付加およびLNP形成の最適プロセスに投資し,確立している。著者らはGalNAcがこの2種類の形式のFH患者に1つの伝達プラットフォームを提供し、他の肝臓をガイドとする伝達を優勢とする応用に適用できると信じている。
単独治療コース
われわれはわれわれの単一治療コース遺伝子編集治療を設計しており,静脈輸液により単回用量レジメンとして管理しており,NHPの臨床前研究で生じたデータの支持を得ている。しかし,LNPsを使用する利点の1つは,分回投与の潜在力である。私たちの遺伝子エディタの場合、私たちは、安全性および有効性を向上させるために、短時間で限られた数の分回用量を行うことを含む単一の短期治療コースを患者に使用することを選択することができる。単一治療コースで十分な治療効果が得られない可能性のある患者では,われわれの方法は再投与の選択を可能にする可能性がある。PATSIRANはトランスサイレチンアミロイドーシス(ATTR)患者の治療に用いられる承認されたLNP被覆siRNAであり,長期使用であり,安全性や有効性の問題はない。これはウイルスベクターと異なり,ウイルスベクターは繰り返し注射時に安全性と有効性の挑戦に直面している。
単一治療コースの遺伝子編集治療の価値はその期待効果の安全性、有効性と持続性に依存する。VERVE−101の単一治療コース治療は,ASCVD患者またはASCVDリスクのある患者の一生において低密度リポ蛋白−Cを持続的に低下させることができると信じられている。われわれの遺伝子編集治療は肝細胞のDNAを恒久的に変化させることを目的としている。VERVE−101を用いて,肝細胞におけるABEタンパク質の瞬時発現は,PCSK 9遺伝子の永久編集をもたらすように設計されている。肝細胞は主にPCSK 9が編集した肝細胞分裂を携帯することで反転するため,編集による効果は持続すると信じられている。
これは遺伝子療法とは対照的であり,遺伝子療法の治療効果は持続性の欠如により挑戦されている。遺伝子治療は通常、ウイルス伝達或いはウイルス発現mRNAを通じて外因性mRNAを発現するように設計されている。治療効果の持続性は、ゲノムに組み込まれていないウイルスベクターのメッセンジャーリボ核酸発現喪失によって制限される可能性がある。これは、ゲノム中の予測不可能な位置でのウイルス統合に依存するか、これが安全課題をもたらすか、または反復用量に依存する可能性があり、これはウイルスの伝達に独自の挑戦を有する。
著者らは、単一治療コースの遺伝子編集治療は持続性と変革性の結果を提供し、心血管疾患患者に持続的な健康利益をもたらすことができると信じている。
拡張可能な製造
著者らの遺伝子編集治療をmRNAとgRNAを包むLNPsとして設計することにより、著者らは拡張可能かつ費用効果のある製造技術の潜在力から利益を得、それによって数百万の心血管疾患患者を治療する機会があると予想される。
我々の候補製品は、SARS-CoV-2刺突タンパク質を含む長メッセンジャーリボ核酸のLNPであるLNP被覆siRNA、例えばPatisiran、およびLNP被覆メッセンジャーリボ核酸ベースの新冠肺炎ワクチンの2つの検証および承認された薬剤クラスと類似している。多くの組織はメッセンジャーリボ核酸生産、ニトロプルナトリウム生産、充填剤に関連するすべてのコンポーネントとプロセスのサプライチェーンを強化するための重大かつ持続的な投資を行っており、特に世界が大量の新冠肺炎ワクチンの生産に努力していることを考慮している。今後数年間、私たちは最終的にLNP被覆mRNA生産の世界生産能力の増加から利益を得ると信じている。
私たちは現在GMPサプライヤーと協力して、私たちの臨床試験ロットのために私たちの候補薬物のすべての成分を生産している。これらはプラスミドDNAの調製、インビトロ転写反応によるメッセンジャーリボ核酸、gRNAの産生を含む
16
固相合成,脂質合成,LNP配合と充填整理により合成した。このような供給者たちと密接に協力して、私たちは臨床規模でロットを実行することに成功した。
MRNA生産とLNP配合における内部プロセス開発能力の確立にも投資しており,将来のコア能力の一つとなると信じている。このような内部過程開発能力の目標はプラスミドDNA、mRNAとLNP生産ロットを拡大し、品質、一致性と安定性を高め、コストを下げることである。また、生物活性および効力分析を含む分析方法の開発に投資しており、さらなる製品開発、バッチ比較性評価、追加の製造業成長に重要である。
私たちの遺伝子編集プログラムは
私たちは単行コースのパイプを進めています体内にある遺伝子編集プログラムは肝臓中の心血管疾患に関連する遺伝子を持続的に閉鎖することを目的としている。我々の遺伝子エディタは,LNPsと肝臓に発現する関心遺伝子に対するgRNAを含み,LNPsは遺伝子エディタをコードするmRNAをカプセル化している。著者らの研究の重点は血中脂質制御に関連する遺伝子、及び心血管疾患内外の肝臓を介した他の標的である。我々は我々の先行計画を策定しており,最初は様々な形式のFH患者の治療に用いられており,FHは遺伝性疾患であり,生涯深刻に上昇する血液低密度リポ蛋白−Cを招き,早発性ASCVDのリスクを増加させる。FH患者は主にLDLR遺伝子突然変異であり、肝細胞が循環から低密度リポ蛋白を除去する能力を影響する。FHは臨床的には,よりよく見られるハイブリッド型,HeFH,およびよりまれな純型と呼ばれ,HoFHと呼ばれる2つの形式で表現される。
次の図は我々の計画パイプラインをまとめたものである. 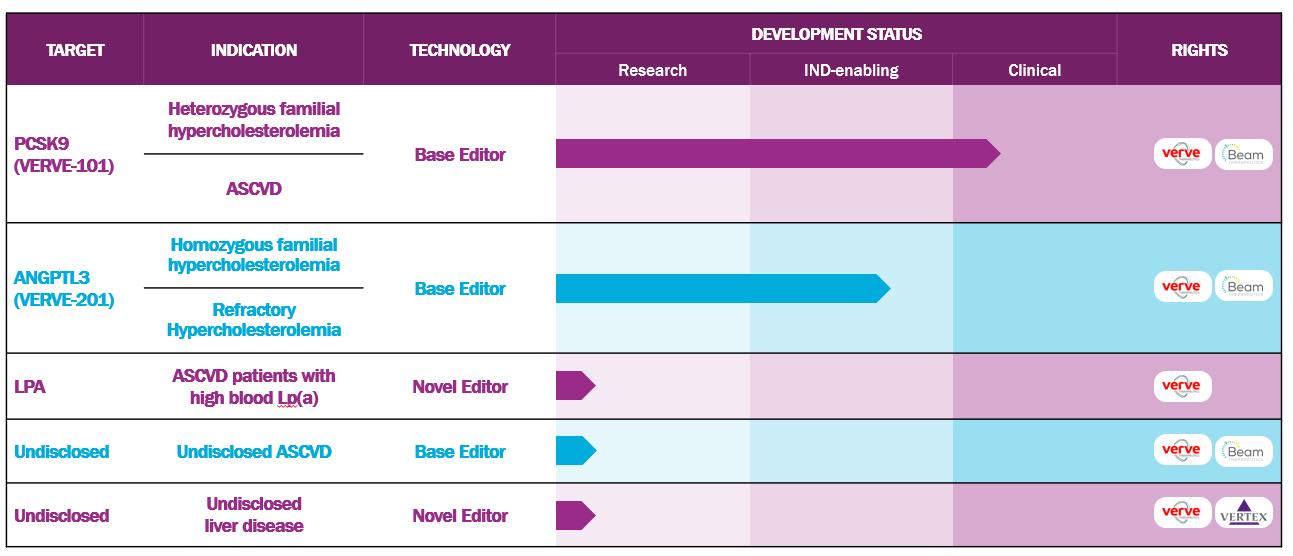
われわれの最先端の候補製品であるVERVE−101はPCSK 9遺伝子に対して現在心臓−1研究を行っており,ニュージーランドやイギリスのHeFHハイリスク患者におけるVERVE−101の安全性と耐性を評価する1 b期の臨床試験である。遺伝子定義のHeFHは米国では約130万人,EUやイギリスでは210万人,世界では約3100万人に影響している。著者らは2023年下半期に心臓1号臨床試験用量増加部分の初歩的な安全性と薬効学データを報告する予定である。
われわれはVERVE−101を戦略的に開発しており,最初にHeFH患者に用いられ,これらの患者の満足されていない需要が最も高く,収益−リスクプロファイルの方が有利である可能性が認識された。われわれはVERVE−101の漸進的臨床開発計画を用いて,まずハイリスク群の有効性と安全性を評価し,成功すればより広範なASCVD患者群に拡大する予定であり,これらの患者は経口治療の低密度リポ蛋白−C目標ではなく,最終的に一般群のASCVDリスク患者に拡張する。
我々はAngptl 3遺伝子に対する開発候補である類似した漸進的手法を用いてVERVE-201を開発する予定である.われわれは,米国で約1300人に影響を与え,経口治療の低密度リポ蛋白−C目標やPCSK 9阻害剤,あるいは難治性高コレステロール血症に達していないHoFHの治療に開発する予定である。最終的に,VERVE−201はASCVDのハイリスク群が一般群においても予防策として有用であると信じている。私たちは臨床前研究を行っています
17
VERVE−201臨床開発開始の規制申請を支援し,2024年に1 b期臨床試験の研究を開始する予定である.
我々は,ASCVDリスクに関する異なる経路に対して広範な遺伝子エディタパイプラインを開発する予定である.また,我々の遺伝子編集手法は,高度に満たされていない医療ニーズや肝臓に発現する検証された遺伝子標的を有する他の適応により広く応用できると信じている。重要なのは体内にある遺伝子編集治療において、著者らは疾病の根本的な原因を解決するために、単一治療コースの遺伝子編集薬物を開発する予定である。
家族性高コレステロール血症:著者らの単一治療コース遺伝子編集治療の第一の目標
FHは遺伝性疾患であり,患者は一生重篤な低密度リポ蛋白−Cが上昇し,早発性ASCVDのリスクが増加する可能性がある。FHは常染色体優性遺伝病であり、よくLDLR遺伝子突然変異によって引き起こされる。FHを有する個体は1つの変異対立遺伝子を持つ可能性があるため,疾患のヘテロ接合体であり,HeFH,あるいは2つの変異の対立遺伝子と呼ばれるため,疾患のホモ接合体であり,HoFHと呼ばれる。HoFHは通常HeFHよりも深刻である.
未治療のHeFH男性と女性の低密度リポ蛋白−Cレベルは通常200~400 mg/dLであり,それぞれ50歳と60歳までにASCVDに進展した。遺伝子定義のHeFHの罹患率は約250人に1人がHeFHを有しており,米国の約130万人に相当すると推定されている。HoFHを有する男性と女性の低密度リポ蛋白レベルは500 mg/dLより高く,通常20歳までにASCVDに進展し,関与しなければ30歳までに死亡する。遺伝子定義のHoFHの罹患率は約25万人に1人がホジキンリンパ腫を患っており,米国の約1300人に相当すると推定されている。
FHは多種の要素によって臨床診断を行うことができ、血中低密度リポ蛋白濃度、体格検査、個人或いは家族高コレステロール血症歴及びASCVDの早期発病を含む。伸筋腱黄色腫は,典型的なアキレス腱,膝蓋骨下,手伸筋腱であり,極めて高い低密度リポ蛋白質コレステロールレベルを伴い,FH特有と考えられている。しかし、FHは通常沈黙しており、若い時に心臓病が発作するまで、その時ASCVD家族歴と上昇した低密度リポ蛋白レベルは通常唯一の発見である。FH表現型の分析において、FH表現型は通常低密度リポ蛋白質コレステロールレベルが190 mg/dLより大きいことを意味し、6つの展望性コホート研究から、30年間フォローアップし、FH表現型は30年のASCVDリスクの5倍の増加と関係がある。FH表現型患者では,ASCVDの進展速度は男性で10~20歳,女性で20~30歳であった。HoFH患者では,通常幼少期に粥状動脈硬化が出現し,最初に大動脈根部に発生し,大動脈弁上狭窄をきたし,冠動脈に伸長する。低密度リポ蛋白−Cレベルを有効に低下させなければ,HOFH患者はASCVDで早期に死亡する。FH患者の粥状動脈硬化の重症度は血低密度リポ蛋白レベルの上昇程度と持続時間に比例する。
FHの診断は臨床的特徴に基づいて行うことができるが、遺伝子検出は心臓リスクおよび診断に対する追加的な洞察を提供する可能性がある。最近26,000人以上の個体のデータ分析により、任意の所与の低密度リポ蛋白質コレステロールレベルにおいて、FH突然変異はASCVDリスクと同じ低密度リポ蛋白レベルを有するが明らかな発病突然変異がないASCVDリスクより明らかに高いことを発見した。この分析では、低密度リポ蛋白レベルが190 mg/dL以上で病原性FH変異がない個体がASCVDに罹患するリスクは、低密度リポ蛋白レベル130 mg/dL以下の参照群の6倍である。しかし、低密度リポ蛋白レベルが190 mg/dL以上と病原性FH突然変異の個体がASCVDに罹患するリスクは参照群の22倍であり、これはFH患者の生涯低密度リポ蛋白の上昇の粥状動脈硬化程度が晩年に獲得した低密度リポ蛋白の上昇よりもっと大きいことを反映しているかもしれない。
飲食と生活様式の変化はFH患者の低密度リポ蛋白を下げるために重要であるが、通常は推薦された低密度リポ蛋白レベルを達成するために多種の薬物治療が必要である。FH患者の推薦低密度リポ蛋白質コレステロールレベルは非FH合併ASCVD患者のレベルと類似している。FH患者の治療はよくASCVD或いはASCVDのリスクがある患者より早く始まり、ASCVDの早期発症のリスクが増加することを考慮すると、FH患者は通常より積極的な多薬治療過程に従う。FH患者が使用する薬剤は、非FH患者が使用する薬剤と類似しているが、FH患者の慢性ケアは、早期介入およびより多くの薬剤のために、通常より重い。また,多くの患者,特にHoFH患者では,低密度リポ蛋白−Cレベルは十分に制御されておらず,臨床治療ガイドラインの推奨目標には達していない。
VERVE-101:PCSK 9計画
私たちの主な候補製品VERVE-101はシングルコースとして設計されています体内にあるPCSK 9遺伝子に対する遺伝子編集治療。著者らはまずHeFH患者のためにVERVE-101を開発する予定であり、成功すれば、より広範なASCVDが確立され、かつ低密度リポ蛋白の経口治療目標に達していない患者のために開発範囲を拡大する。
18
HeFH患者では,LDLR遺伝子の遺伝子変異が発現を低下させた 低密度リポ蛋白受容体の発現は,肝細胞が血液から低密度リポ蛋白を除去する能力を制限し,血液中の極めて高い低密度リポ蛋白−Cレベルを招く。時間の経過に伴い、高密度リポ蛋白質コレステロールは動脈に蓄積し、粥状動脈硬化プラークの形成を招き、血液流動が減少或いは閉塞し、最終的に心臓病発作或いは脳卒中を招く。PCSK 9遺伝子の不活化によりPCSK 9蛋白レベルが低下し,LDLRの発現が増加し,低密度リポ蛋白−Cレベルが低下し,ASCVDのリスクが低下すると考えられる。他のPCSK 9阻害剤を評価する臨床試験では,潜在的な変異にかかわらず,標的PCSK 9がHeFH患者に作用する可能性が示唆された。
Verve−101は、ABEとgRNAをコードするmRNAを封入したLNPからなり、以下の図に示すようになる。4種類の脂質成分はRNAとともに組み立てられ,直径約60ナノメートルの緻密で安定したLNPを形成した。

Verve−101は,約1~2時間以内に患者に静脈内投与し,その後肝臓に蓄積するように設計されている。VERVE−101を服用する前に,抗ヒスタミン薬とステロイドからなる先行投与レジメンが与えられる。肝臓に入るとVERVE−101は肝細胞に取り込まれ,細胞質に脱出し,そこで塩基編集蛋白が瞬時に発現される。そして,gRNAは塩基編集タンパク質に結合し,この複合体は核に担持され,gRNAの20ヌクレオチド間隔配列で指定された遺伝子標的を位置決めする。AbeはDNAに結合し,目的位置でAからGへの単一綴り変更を行い,PCSK 9遺伝子を閉じる。ABE mRNA構造はコドン最適化されており,mRNAを介した免疫反応を低下させるための化学修飾の可能性が含まれている。GRNA配列はいくつかの化学修飾が増強しなければならない体内にあるエンドヌクレアーゼとエキソヌクレアーゼの安定性を向上させる。
PCSK 9を目標とする
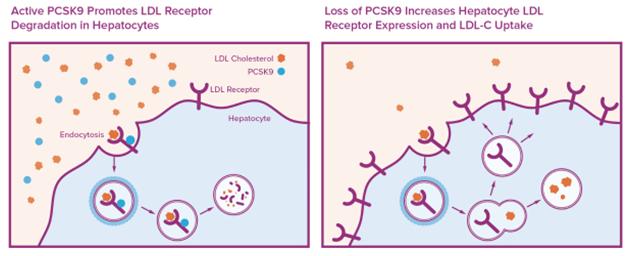
PCSK 9遺伝子は低密度リポ蛋白受容体遺伝子の制御を通じて、血低密度リポ蛋白-Cの調節に重要な役割を果たしている。PCSK 9の正常機能を左下図に示す。PCSK 9遺伝子は肝臓で血液中に放出されるタンパク質を産生する。低密度リポ蛋白受容体は肝細胞表面に存在し,低密度リポ蛋白に結合し,低密度リポ蛋白を循環から除去する。低密度リポ蛋白受容体に結合した低密度リポ蛋白は肝細胞に摂取され,低密度リポ蛋白粒子が分解される。そして,低密度リポ蛋白受容体は細胞表面に循環され,低密度リポ蛋白の摂取過程が繰り返される。血液中のPCSK 9蛋白はLDLRの循環過程を中断する。具体的には,血液中のPCSK 9蛋白がLDLRに結合し,LDLRを破壊する。このようにする過程で,PCSK 9は肝細胞表面の低密度リポ蛋白受容体の数を減少させ,肝臓が血液中の低密度リポ蛋白を除去する能力を低下させた。右の数字は損失を描いています
19
PCSK 9遺伝子機能が低下し、PCSK 9蛋白の減少を招き、それによって低密度リポ蛋白受容体の発現と低密度リポ蛋白の摂取を増加させる。
中で述べたとおりニューイングランド医学雑誌ある研究により、PCSK 9遺伝子が自然にLoF変異を起こした成人の低密度リポ蛋白-Cレベルは、変異のない成人より38 mg/dL低く、変異を有する成人のASCVDのリスクは88%低下することが分かった。人類遺伝学研究により、PCSK 9遺伝子の1つ或いは2つのコピーに自然に発生した機能喪失突然変異を携帯することは深刻な不良健康結果と関係がないことを表明した。
ヒト遺伝学研究に加え,ヒト薬理学研究ではPCSK 9の標的としての有効性が確認された。PCSK 9抑制による心血管結果への影響は2つの大型、ランダム、二重盲検、プラセボ対照の研究によって確定され、この2つの研究はすでに許可されたPCSK 9蛋白と結合し、その活性を遮断するモノクロナル抗体に対して行われ、この2つの試験はそれぞれFOURIER試験とオデッセイ結果試験である。FOURIER試験により、FOURIER開放ラベル拡張研究において、背景スタチン類薬物治療以外に、2.2年の中央値時間内に、EUROCURABMを用いてASCVD患者を治療した主要な心血管イベントは15%減少し、持続的な安全性と増加した心血管イベントの減少メリットは他の5.0年のフォローアップで蓄積したことを証明した。オデッセイの結果により、確定診断されたASCVD患者の中で、2.8年の中位時間内に、スタチン類薬物治療の基礎の上にalirocumab治療を加えることは、主要な心血管イベントを追加的に15%減少させることができる。臨床試験では,これらのモノクロナル抗体を用いた治療はプラセボ治療と比較して低密度リポ蛋白−Cが平均約60%低下した。注目すべきは,両試験では,注射部位の反応を除いて,プラセボや薬物治療を受けた患者の全体的な有害事象発生率は類似しており,新たな糖尿病,血糖コントロールの悪化や神経認知不良事象の増加は認められなかったことである。
InlisiranはさらにPCSK 9の目標を検証し,Inlisiranは2020年にEMAの承認を得,2021年12月にFDAの承認を得た。Orion−9試験では,InlisiranがHeFH患者を治療する重要な3期試験では,510日後のInlisiran治療群のPCSK 9レベルの百分率変化がベースラインより60.7%減少し,510日後の低密度リポ蛋白−Cがベースラインより39.7%低下した。
著者らは、ヒト遺伝学研究とPCSK 9阻害剤のヒト薬理学研究は大量の証拠を提供し、標的PCSK 9は低密度リポ蛋白-Cを低下させ、ASCVDリスクを低下させる潜在的安全かつ有効な方法であることを表明した。
臨床前研究
著者らは大量の候補gRNAライブラリーの広範なスクリーニング、多種のLNP配合の評価とABE mRNA構造の最適化を通じて、VERVE-101を発見した。VERVE-101のマウス代替品、VERVE-101の前駆体製剤をテストし、ABE-PCSK 9前駆体製剤、およびVERVE-101自体と呼んでいます体外培養そして体内にある複数の動物モデルにまたがっていますこれらの研究では,以下の点が観察された
20
ABE−PCSK 9マウス代替物の体内検証
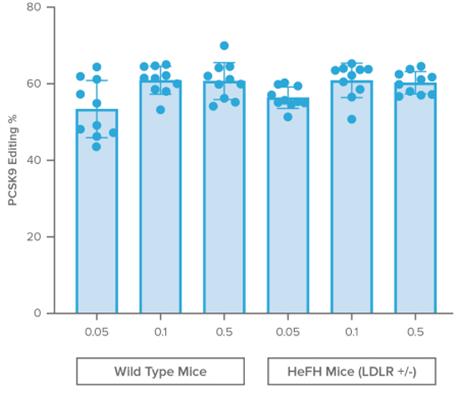
われわれVERVE−101の最初のターゲット患者群はHeFH患者であり,機能的低密度リポ蛋白受容体レベルの低下を生じ,血液中の低密度リポ蛋白−Cレベルを上昇させた。ハイブリッドLDLRノックアウトマウスを用いてHeFHの疾患状態をシミュレーションした。このモデルに用いるために,同じPCSK 9部位を標的とした相同遺伝子を含むVERVE−101のマウスエージェントバージョン,およびVERVE−101と同じ2成分−ABE mRNAとLNPを開発した。以下の図に示すように,野生型とハイブリッド型LDLRノックアウトマウスに0.05,0.1および0.5 mg/kgのマウス代替物VERVE−101を一度に投与すると,肝臓でのPCSK 9編集量が類似して強力であることが観察された。
21
ABE−PCSK 9前駆体配合のNHP検証
次に,この方法をNHPモデルに適用し,ABE−PCSK 9前駆体製剤を用いて臨床前概念検証を構築した。この研究では,健康なNHPを単回投与した。次の図において、各処理されたNHPは1つの紫色のストリップで表され、各処理された車両制御は1つの青色のストリップで表される。われわれのABE−PCSK 9前駆体製剤単一治療後,投与2週間後に肝生検で採取した肝臓組織全体におけるPCSK 9の平均67%の編集を1枚目の図に示す。これは血液PCSK 9蛋白の平均89%の減少に伴い,血液低密度リポ蛋白濃度は平均59%減少し,次の2つのグラフに示す。
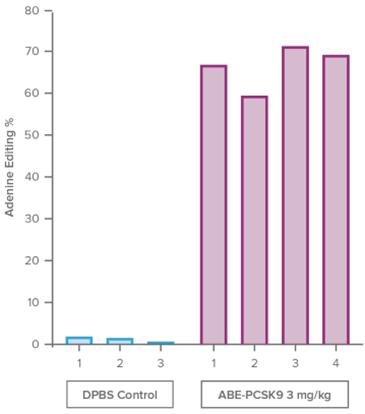
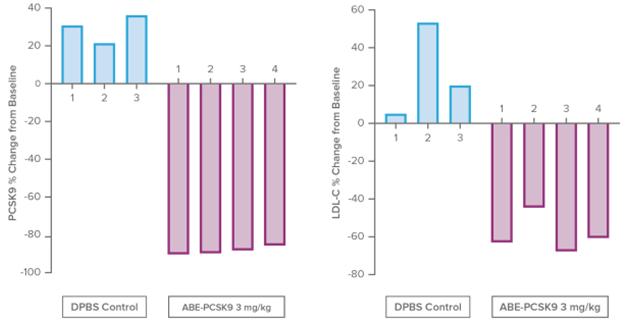
22
重要なことは,この臨床前研究では,血液PCSK 9蛋白と血液低密度リポ蛋白レベルの低下が持続することが観察された。以下の図に示すように,ABE−PCSK 9単回静注2年後,NHPの血液PCSK 9蛋白は平均90%,血低密度リポ蛋白は平均71%の低下が認められた。
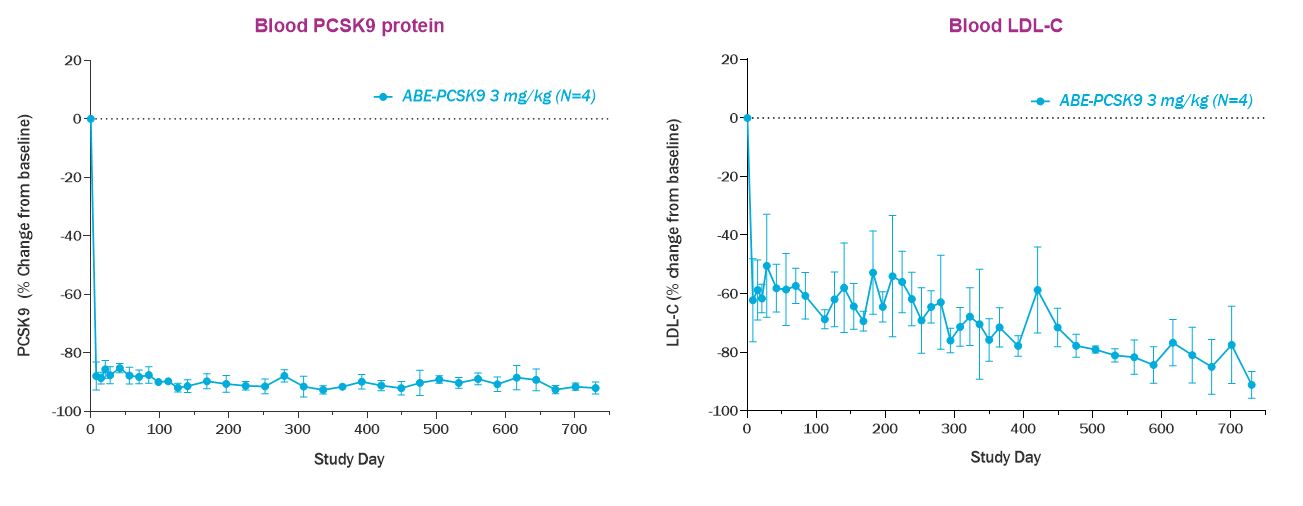
成熟肝細胞の肝臓での回転は平均200~300日ごとに発生すると推定されている。新しい肝細胞の由来はまだ確定していないが、証拠により、成熟肝細胞は動態平衡の肝臓回転とその後の肝損傷期間に新しい肝細胞の産生を担当することを表明した。不可能なのは,肝に再生能の強い肝細胞の一部が存在する可能性があることである。いずれの場合もABE−PCSK 9前駆体製剤を用いた臨床前研究で示された耐久性データは,再生を担う肝細胞がPCSK 9遺伝子部位で編集されていることを示している。また,持続性炎症や肝障害を認めた証拠はなく,肝細胞のより速い回転や免疫を介した編集肝細胞郭清が示唆されている可能性がある。
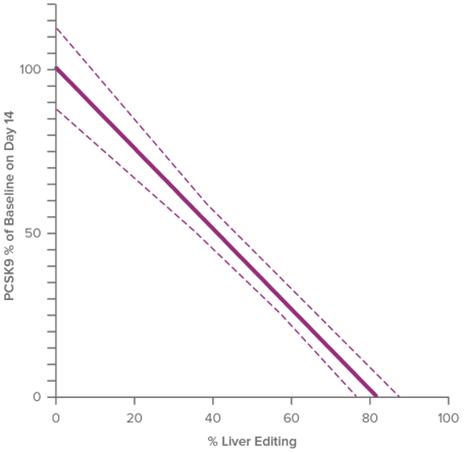
肝臓編集の薬効学とそれによる血液PCSK 9蛋白レベルへの影響を探索した 多くのNHP反復研究。肝細胞におけるPCSK 9遺伝子の編集と血液中のPCSK 9蛋白レベルとの線形関係が確認された。次の図は,各NHPからの大量のデータ点を表す信頼区間付き最適フィッティング線を示している。NHPではPCSK 9蛋白の60%以上の減少を実現しており,肝臓全体の編集率は約50%から55%である。ヒトでは,肝臓全体の編集とPCSK 9減少との関係は類似しているはずであると考えられる。
23
VERVE-101臨床前治療効果データ
ABE−PCSK 9前駆体製剤の臨床前研究によりVERVE−101が開発された。
VERVE−101の短期臨床前研究
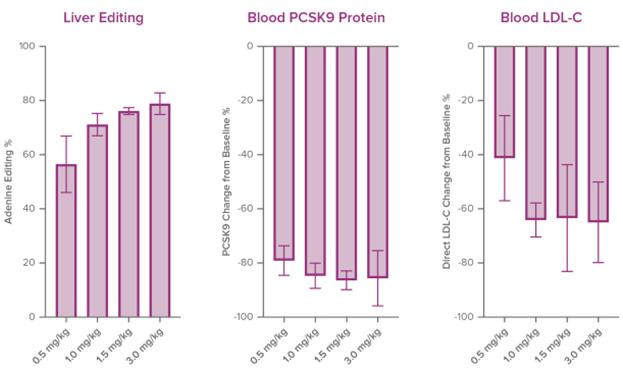
次の図のVERVE−101の臨床前用量反応研究では,NHPでは,VERVE−101,各用量レベルで3つのNAMPを4つの用量レベルで投与した。次の図では、各ストリップは、0.5 mg/kgから3.0 mg/kgまでの異なる用量グループを表す。投与量1 mg/kgの場合,肝臓全体の編集レベルは約71%に認められ,次の図に示すように,多くの肝細胞の編集を代表していると考えられる。編集レベルが血液PCSK 9蛋白と血液低密度リポ蛋白に変換される用量依存の減少も観察された。PCSK 9蛋白は1 mg/kg用量で約85%の減少が認められたが,低密度リポ蛋白(LDL−C)は約64%の低下が認められた。
24
NHPにVERVE−101を投与すると編集が速く,大部分の編集が投与後1~2日以内に発生することが観察された。検討では,NHPS(n=2)群に同じ1 mg/kg投与し,1日目,2日目,7日目,14日目,28日目に連続して剖検を行った。24時間以内の効率的な編集が観察され,後続時点の付加編集が最も少なく,次の図に示すようになった.
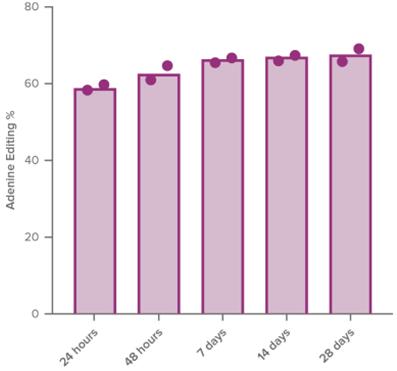
血液PCSK 9蛋白と低密度リポ蛋白−Cへの影響は服薬後2週間以内にピークに達した。LNPの主成分であるイオン化性脂質は,生分解性で2週間以内に血液から除去されるように設計されており,服薬後2週間で肝臓からピーク濃度の10%以下がほぼ除去されることが観察された。投与後1週間以内に、肝臓ABE遺伝子発現レベルは97%低下した。
25
VERVE−101の長期臨床前研究
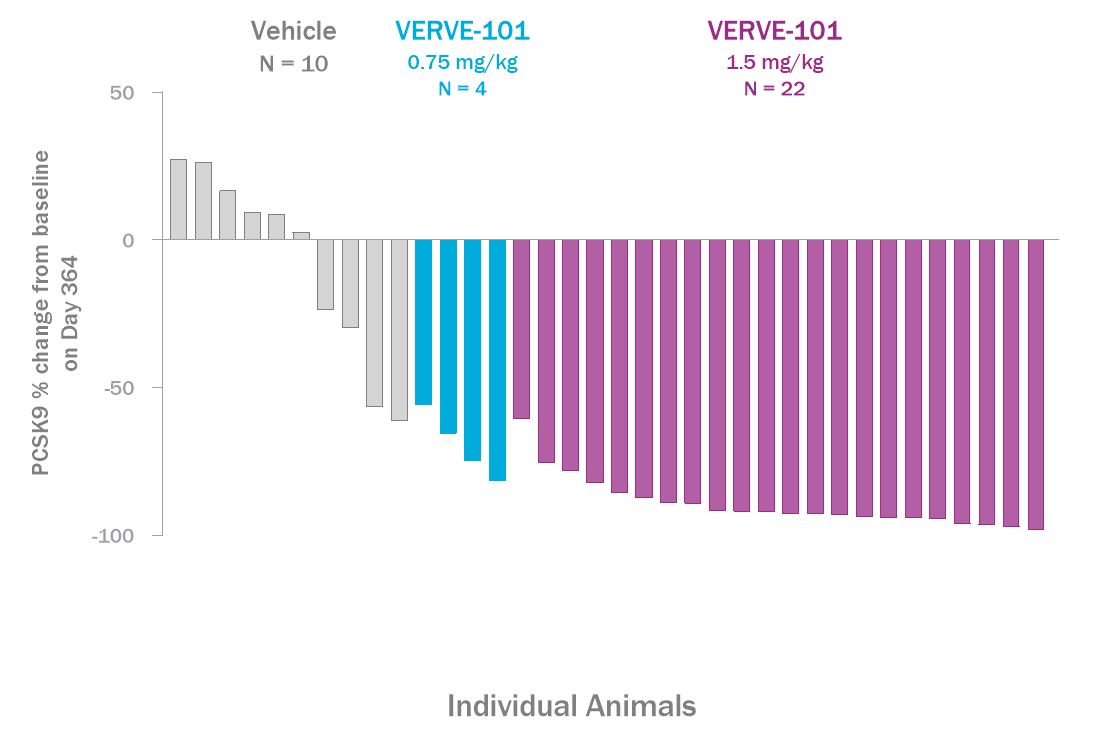
36個のNHPに対する長期NHP研究では,VERVE−101(n=22)と0.75 mg/kg(n=4)のVERVE−101を1.5 mg/kg投与し,対照群(n=10)と比較した。この研究は肝臓全体の編集、血液PCSK 9蛋白レベルと血液低密度リポ蛋白レベルを測定することを目的としている。この研究は,我々が臨床生産現場で生産する予定の最終VERVE−101薬物製品を利用している。私たちは投与量のNHPでこの研究を3年以上継続する予定だ。
1.5 mg/kg(n=22)投与量の場合,15日目にPCSK 9標的部位の平均70%の全肝編集が認められ,以下の図に示すように,多くの肝細胞の編集を表していると考えられる。
1.5 mg/kg用量でPCSK 9蛋白は治療後2週間で約79%,低密度リポ蛋白(LDL−C)は約62%の低下が認められ,治療後1年でそれぞれ89%,68%に改善した。0.75 mg/kg用量レベル(n=4)では,PCSK 9蛋白は治療後2週間で約54%,低密度リポ蛋白(LDL−C)は約38%の低下が認められ,治療後1年でそれぞれ69%,50%に改善した。
マウス肝部分切除モデル
著者らは一部の肝切除マウスモデルにおいて耐久性挑戦研究を行い、肝細胞回転後の編集レベルが不変であるかどうかを評価した。この研究では,代用VERVE−101を服用したマウスで11日後に肝臓の3分の2を切除する部分肝切除術や偽手術を行った。部分肝切除を受けたマウスでは,残りの肝臓は約9日以内に再生して肝臓重量を回復させた。そして治療後22日か95日に検死を行いました治療後22日と95日に再生肝葉中PCSK 9の持続編集を認めた。PCSK 9蛋白レベルは治療後22日と95日の持続的な低下も認められた。これらのデータは,肝臓の再生に伴い,VERVE−101による編集レベルが強く持続することが予想されるという我々の信念を支持している。
VERVE−101生物分布データ
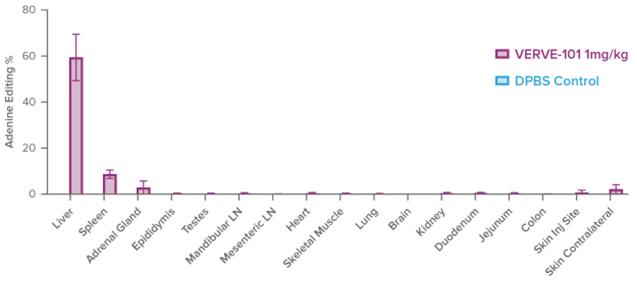
VERVE−101をLNPベースの方法で肝臓に輸送している。VERVE−101のNHPへの単回投与1 mg/kg後の生物分布の分析では,大多数の編集は肝臓で発生し,用量依存関係を呈し,脾と副腎の編集比率は以下の図のように低かった。検査した他の組織によると,編集量は約2%未満であった.
26
NHPSにおけるVERVE−101の耐性
NHP研究では,VERVE−101の全体耐性は良好であった。VERVE−101の治療を1 mg/kg以下の対照群と比較したところ,アラニンアミノトランスフェラーゼ(ALT)は服薬後1~2日で一過性に上昇し,軽度急性肝障害と一致し,投与後2~3日でピークに達し,1 mg/kg投与後の平均値は約300 U/Lであった。アラニンアミノトランスフェラーゼ(ALT)は肝障害の常用血液マーカーである。投与1週間でALTの平均値は正常範囲内であり,回復を示したものを以下の図に示す。これらの知見は,承認された静脈内投与LNP製品に対する非臨床研究の所見と一致している。
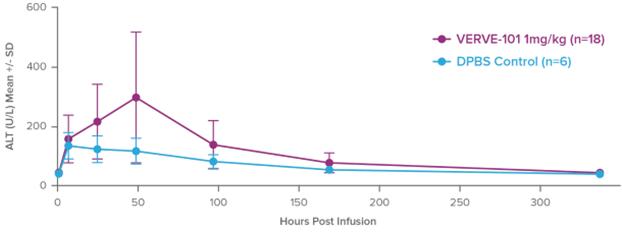
肝酵素結果は標準的な臨床実験室テストを通じてモニタリングすることができ、その性質は常に一時的かつ温和であり、そして1週間から2週間以内に完全に正常化する。これらの発現はウイルスベクター伝達法よりも有利であり,後者は予測不可能な急性肝障害をきたす可能性が考えられる。
VERVE−101の長期肝臓安全性を評価するために,ABE−PCSK 9前駆体製剤の長期持続性研究において肝酵素をモニタリングした。肝臓に高レベルのPCSK 9編集を経験したNHPの肝臓に持続的な炎症を認めた証拠は認められなかった。対照的に、ウイルスベクターに対する自己免疫反応により、ウイルスベクター伝達は亜急性と慢性肝障害を引き起こす可能性がある。
われわれが行っているVERVE−101の長期臨床前研究では,肝機能テストへの長期影響は認められず,ALTレベルの軽微で一過性の上昇のみが認められ,上記の研究と類似していた。この研究では,全身性PCSK 9が血糖恒常性に影響するかどうかも評価した
27
力を抑える。VERVE−101投与後1年間は血糖恒常性への影響は認められなかった。
6個性成熟男性HHPに対する1.5 mg/kg用量のVERVE−101の研究では、治療後11週間にPCSK 9部位で種系編集を行った証拠は認められず、精子発生の完全な周期を超えていた。
VERVE−101代用品を436匹服用した雌マウス子孫の研究では,子孫の遺伝子分類では,PCSK 9遺伝子編集は子孫に遺伝していないことが示された。
LNPsは免疫系を刺激することが知られているため,NHPにVERVE−101を単回投与した後のよく見られるサイトカインのセットも評価した。1 mg/kg以下の用量で、IP-10またはMCP-1のようないくつかのサイトカインは、対照群動物と比較して軽微な瞬間活性化を有することが観察された。この活性化は投与後24時間以内に明らかであり,1週間後の次の観察点では完全に消失した。腫瘍壊死因子を含む他のサイトカインはa対照動物よりも大きな変化は示さなかった。
VERVE−101を単回投与したNHPの補体活性化も評価した。1 mg/kg以下の用量では,ごく少量の活性化のみが対照群動物よりも高いことが認められた。この最小の活性化は投与後約2時間で検出されたが,24時間後には消失した。
NHPにおける前臨床非標的編集
ヒトゲノムは非標的編集を評価する関連ゲノムであるが,NHPにおける非標的編集の評価は初代肝細胞の非標的解析能を支持すると信じている体外培養投与時の肝臓の非標的編集を予測するには体内にある.
私たちが目標から外れている位置を識別する方法はバイオインフォマティクスと体外培養ABE-Digenome-SeqとOne-Seqという最先端技術を含む生化学技術。One-Seqは全面的で敏感なものです体外培養方法は、編集が起こり得る潜在的シーケンスをスクリーニングおよび識別するために使用される。One−Seqを用いて,NHPゲノム中で我々の標的配列に最もマッチする25,000個の配列を評価した。編集が発生する可能性のある45個の潜在サイトを優先順位付けし、PCSK 9ターゲットサイトが第一選択サイトとして決定された。
次に,VERVE−101で処理した初代NHP肝細胞におけるこれらの部位の編集を次世代DNAシークエンシングを用いて評価した。以下の図に示すように、PCSK 9ターゲットサイトの編集を除いて、C 5として指定されたサイトを除いて、評価された44個の潜在的な非ターゲットサイト(紫色のドットによって描かれた)内の任意の非ターゲット編集は観察されない。C 5部位はヒトゲノムに存在しない。
次に,NHPをVERVE−101で処理し,NHP肝臓試料を採取し,初代NHP肝細胞で評価した同じ部位を配列決定した。NHP肝臓試料では,C 5部位にのみ非標的編集が認められた。これらのデータは目標から外れた場所を正確に予測することができるという私たちの信念を支持しています体内にある初代肝細胞に基づく非標的解析体外培養.
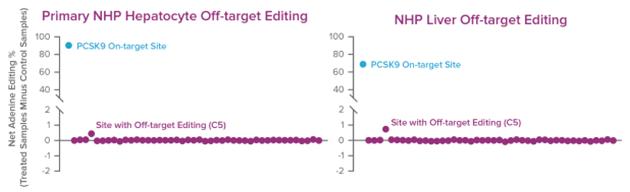
初代ヒト肝細胞の非標的解析
細胞内の非標的解析を接続する方法を確立しました体内にある編集にあたっては,VERVE−101ヒトゲノムの評価に移った。2種類の直交技術であるOne−Seq法とABE−Digenome−Seq−を用いて3,000以上の潜在的な部位を優先的に選択し,高感度なハイブリッド捕捉試験を用いて初代ヒト肝細胞の編集を評価した。次の図に示すように何も観察されませんでした
28
未処理細胞と比較して、約3,000個の潜在的な非標的部位(黒丸)のいずれにおいても有意な純編集があり、PCSK 9標的部位にのみ標的編集(紫点)が観察された。
2022年4月、複数のドナーからの初代ヒト肝細胞においてVERVE-101が意外または非標的DNA編集をもたらす可能性を評価する包括的な臨床前評価データを公表した。我々は,FDAの最近のガイドラインに一致する様々な方法を用いて,ターゲットサイトと最大の実験またはバイオインフォマティクス類似性を有する3000以上のサイトを識別する.次に,VERVE−101の服用がこれらの位置の非標的編集を引き起こすかどうかをシークエンシング解析を用いて決定した。VERVE−101で治療したところ,統計的有意な非標的編集は認められなかった。非標的細胞(脾細胞,副腎細胞と造血幹細胞)と他の細胞環境(小児肝細胞とヒト肝細胞系)における非標的編集の可能性を評価し,未処理対照において統計学的に有意な2つの編集位置を決定した。この2つの非標的編集の発生用量は、私たちが予想していた目標上の編集が飽和した用量よりも大きい。これらの評価から,VERVE−101の非標的ゲノム修飾リスクは低く,関連する臨床副作用が生じることが予想される。
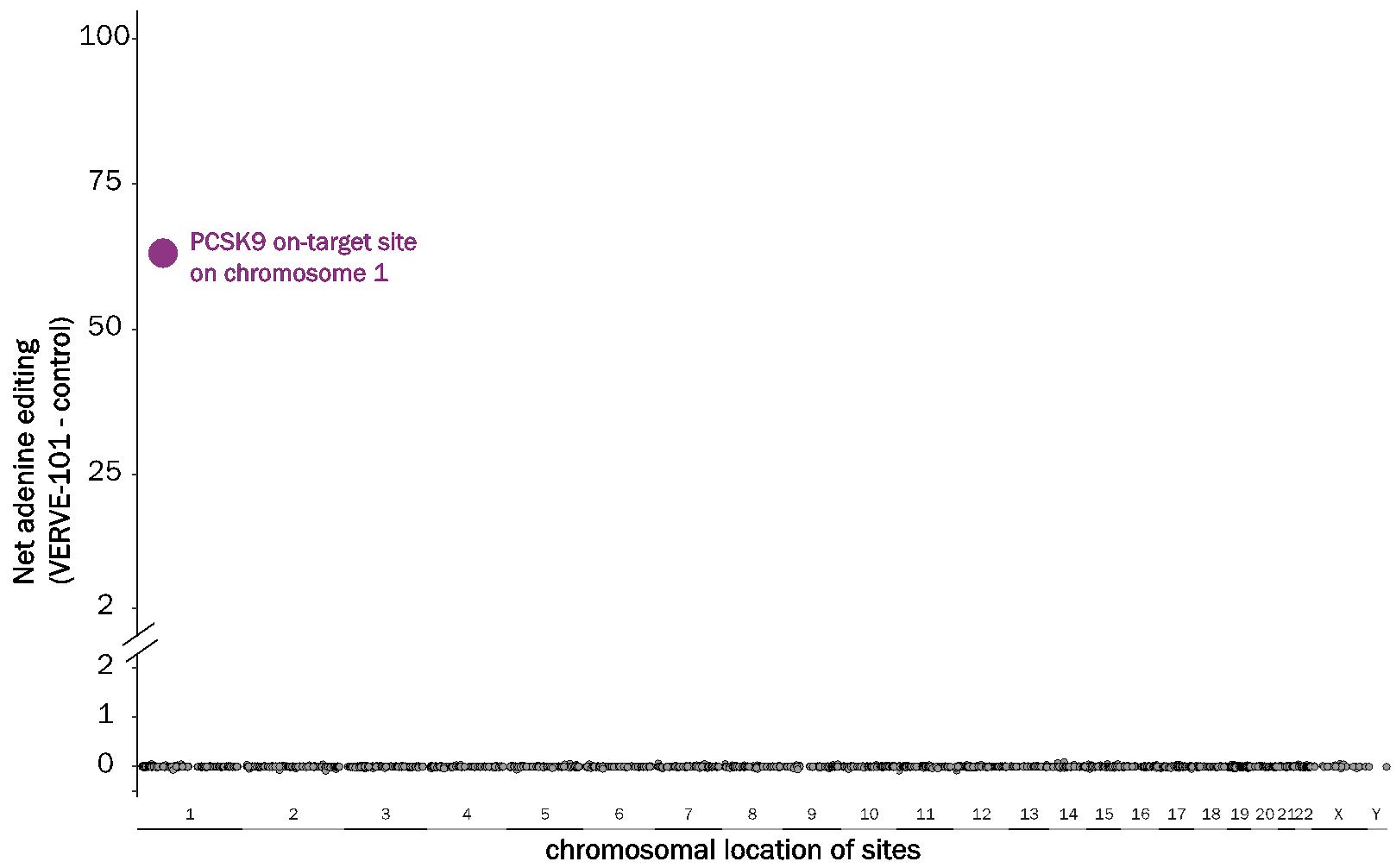
以上の分析に加えて,塩基編集によるRNAの編集とDNAの転座の2つの理論的リスクを評価した。初代ヒト肝細胞では,対照よりも高いRNA編集やDNAシフトは認められなかった。
多用量VERVE−101の臨床前研究
我々は単一治療コースの遺伝子編集薬としてVERVE−101を開発している。しかし、ASCVD患者の治療の複雑さを考慮すると、いくつかの患者は任意の単一薬物治療後、追加の脂質低下から利益を得る可能性があると考えられる。著者らは4つのNHPの中で90日間のVERVE-101臨床前研究を行い、患者の再投与の可能性を探索した。この研究では,VERVE−101前駆体を1,30,60日目に0.5 mg/kg服用した。われわれは14,46,75日目に肝臓生検によりPCSK 9の編集を測定し,90日目に肝臓剖検によりPCSK 9の編集を測定した。以下の図に示すように,研究過程では,PCSK 9の編集量の増加,14日目の平均29%,46日目の36%,75日目の53%,90日目の59%の増加が観察された.これらのデータは,低用量のPCSK 9塩基エディタを繰り返すことで高レベルの肝臓編集が可能であることを示していると考えられる。私たちは用量後の肝臓損傷の証拠は何も認められなかった。
29

これらのデータは,LNPsの遺伝子編集薬物送達方法としての潜在的な鍵となる利点の1つを強調していると考えられる。
また,我々が生成したデータは,我々の独自GalNAc−LNPがPCSK 9に対する塩基エディタを効率的に提供することができ,野生型NHPにおけるPCSK 9の約87%の減少を実現していることを示している。われわれはGalNAc−LNP交付のPCSK 9塩基エディタが臨床前開発段階に入ることを進めており,このデータはGalNAc−LNP送達が他の適応の肝臓編集において広範な実用価値がある可能性を示唆していると信じている。
心臓1号臨床試験
HEART-1臨床試験は約40名のASCVDと診断されたHeFH成人患者を募集し、VERVE-101投与の安全性と耐性を評価し、薬物動態と血液PCSK 9蛋白と低密度リポ蛋白-Cの低下に対して追加的な分析を行うことを目的とした。試験は、(A)単回漸増用量部分を含み、その後、(B)より多くの参加者が選択された潜在的治療用量を得る単回用量キューを拡大し、(C)選択可能な第2の用量キューで、A部分の低用量キューにおいて、条件に適合する参加者が、選択された潜在的治療用量で第2の治療を受けることを選択することができる、3つの部分を含む。ニュージーランドやイギリスの規制当局との相互作用の中で、各国/地域の資格、設計、行動の様々な修正を考慮して、特定の国/地域に対する合意を策定した。
われわれはすでにニュージーランドとイギリスでVERVE−101のCTA許可を得ており,2022年7月,われわれの心臓1号臨床試験では,最初の患者がVERVE−101投与量を受けていることを発表した。心臓1号臨床試験用量増加部分の第1用量列中のVERVE−101の用量を完了した。ニュージーランドとイギリスでは学生募集が行われている。著者らは2023年下半期に心臓1号臨床試験用量増加部分のすべての用量列の初歩的な安全性と薬効学データを報告する予定である。
Verve-101は最近、革新許可と参入経路(ILAP)によってイギリス薬品と保健品監督局(MHRA)が発行したHeFH治療の革新パスポートを獲得した。革新パスポート指定はILAPの切り口であり、この計画は発売時間を加速し、そして患者がイギリスで生命を脅かす或いは深刻な虚弱な疾病或いは重大な患者或いは公共衛生需要のある薬物を治療することを便利にすることを目的としている。
われわれは2022年10月にIND申請をFDAに提出し,HeFH患者のVERVE−101の臨床試験評価を求めたが,その後FDAからIND申請が放置されていることが通知された。2022年12月、我々はFDAから臨床的座礁信頼を受け取り、(I)ヒトと非ヒト細胞との間の潜在的差異、(Ii)生殖系編集のリスク、および(Iii)非肝細胞タイプの非標的分析を含む、棚上げを解決するために必要な情報を概説した。ニュージーランドとイギリスで行われている心臓1号臨床試験の臨床データはFDAに提出されたIND申請パッケージには含まれていない。臨床保留状では,FDAは試験の臨床データの提供を要求している。また、
30
FDAは、追加の避妊措置を取り入れ、参加者用量間の交互間隔の長さを増加させるために、米国の試験計画を修正することを要求している。私たちは私たちの反応をできるだけ早くFDAに提出するつもりだ。
Verve-201:Angptl 3プログラム
我々はAngptl 3の開発候補薬物Verve−201に対して肝臓中のAngptl 3遺伝子を永久的に閉鎖することを目的としている。アンギオテンシン変換酵素3はコレステロールとトリグリセリド代謝の重要な調節因子である。我々は,HoFHと難治性高コレステロール血症の治療に用いられる計画を開発し,HoFHは米国で約1300人に影響し,難治性高コレステロール血症はASCVD患者と定義され,経口治療では低密度リポ蛋白−C目標に達しておらず,PCSK 9阻害剤を使用している。最終的に,VERVE−201はASCVDハイリスク群において一般群においても予防策として有用であると考えられる。
我々はVERVE−201臨床開発開始の規制申請を支援するための前臨床研究を行っており,2024年に1 b期臨床試験を開始する予定である。VERVE−201では内部で開発されたGalNAc−LNP技術を用いて,Angptl 3遺伝子に対する塩基エディタを肝臓に提供する予定である。我々はすでにGalNAcリガンドを有する独自のLNPsを開発し,肝臓中のASRに結合し,LDLRを迂回し,HoFH患者の肝臓を摂取できるようにした。
目標としたAngptl 3
Angptl 3遺伝子は最近すでに深刻な高脂血症治療の新しい標的になり、将来性が高い。Angptl 3蛋白はほとんど肝臓でのみ産生され,血液中に放出される。自然に産生される低コレステロール、低トリグリセリド、および低循環脂肪酸のマウスの遺伝的研究によって、コレステロールおよびトリグリセリド代謝の調節剤として初めて決定された。Angptl 3蛋白の主要な機能はリポ蛋白リパーゼを抑制することであり、リポ蛋白リパーゼは心臓、骨格筋と脂肪血管表面の酵素であり、循環中のトリグリセリドの分解と除去を担当する。Angptl 3蛋白もLDLR発現に依存しない機序により低密度リポ蛋白−Cを調節することが証明されており,PCSK 9が低密度リポ蛋白−Cを調節する機序とは逆である。
著者らの創始者が行ったヒト遺伝学研究により、Angptl 3遺伝子が自然に発生する機能喪失突然変異はトリグリセリド、低密度リポ蛋白-Cと高密度リポ蛋白コレステロールレベルが極めて低いことを確定した。その後の研究により、天然アンギオテンシン変換酵素3機能が乏しい患者に明らかな不良健康結果が観察されなかったことが確認された。また,一致した対照群家族メンバーと比較してAngptl 3遺伝子機能が完全に乏しい個体は,一致した対照群家族と比較して,冠動脈コンピュータ断層撮影(CT)で評価された冠動脈粥状硬化プラークはなかった。単一Angptl 3変異コピーを持つ個体に対する2つの独立集団遺伝学研究により、Angptl 3機能の部分喪失はASCVDに対して保護作用があり、Angptl 3変異を何も持たない個体と比較して、リスクはそれぞれ34%と41%低下した。つまり,これらの研究はAngptl 3が高脂血症とASCVDリスクを低下させる潜在的治療標的として強力な証拠を提供している。
Angptl 3に対する多くの治療法が開発または臨床評価されており,Angptl 3を標的としてさらなる検証を提供している。EvinacumabはAngptl 3に対するモノクロナル抗体であり,HoFHとHeFH患者の低密度リポ蛋白−Cとトリグリセリドの低下に有効であることが証明されている。HoFH患者に対するevinacumabの3期試験では,24週間後にプラセボと比較して低密度リポ蛋白−Cが49%,トリグリセリドが50%低下した。これらのデータに基づいて、2021年にFDAはHoFH患者の治療のためにevinacumabを承認した。
エビナマブは低密度リポ蛋白質コレステロールを低下させる作用とPCSK 9を抑制する作用を加えている。HeFHによる難治性高コレステロール血症患者の晩期臨床試験では,多くの症例でPCSK 9阻害剤にevinacumabを添加し,プラセボと比較して56%の低密度リポ蛋白−Cをさらに低下させた。また,Angptl 3に対する他の研究薬は,矢印製薬会社Angptl 3(ARO−ANG 3)と礼来社に対するsiRNA計画の2つの異なる高トリグリセリド血症または心血管疾患患者で評価されている。
31
臨床前研究
著者らの早期臨床前研究では、すべての形態のFH患者、および多種のエディタとgRNAオプションを治療できることを期待するために、多種のLNP製剤を評価した。これまでに生成された前臨床データでは,以下の点が観察された
LNPsの発見と検証
開発候補薬剤としてVERVE−201を指名する前に,臨床前安全性と有効性を最適化するために厳しいプロセスを用いた。Angptl 3を標的とした塩基エディタの前駆体製剤と,我々独自のGalNAc−LNPsの複数の前駆体製剤を評価してこのエディタを交付した多くの研究を行った。
HoFH患者の中で、LNPを介した肝臓輸送はHeFH患者より挑戦的である。これは,LDLR遺伝子の欠陥がHoFHの病理生理を駆動することが多いのに対し,LNPsの摂取は主にLDLRに依存する経路を介していると考えられるためである。LDLRを迂回する方法の一つは,LNPsに標的リガンドを添加し,このリガンドがLDLR以外の受容体を介して作用することである。
我々はLNPsに統合できる独自のGalNAc標的リガンドをスクリーニング·開発した。GalNAcリガンドは肝臓中のASRと結合し、siRNAsの肝臓への輸送を増強するために使用されている。ASCPRは肝臓に高度に発現し,約15分で迅速に回転し,LDLRを介した肝臓への取り込みとは独立した高い能力を有している。
著者らは低密度リポ蛋白受容体が完全に不足しているマウスで臨床前研究を行い、著者らの独自のGalNAc標的LNPsの治療効果を評価した。以下の図に示すように,LNPにGalNAcリガンドを添加するとLDLR−/−マウス肝臓の編集が増加した。GalNAc標的LNPsは,野生型,LDLR+/−マウスおよびLDLR−/−マウスにおいて類似した見かけ効力を有することが観察された。
32
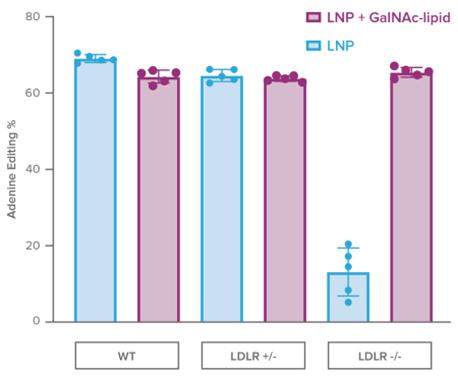
我々は,新たかつ最適化されたGalNAc標的リガンドの開発能力,最適なアンカー,最適なLNP成分の組成および割合,および標的リガンドとの付加およびLNP形成の最適プロセスに投資し,確立している。著者らはGalNAcがこの2種類の形式のFH患者に1つの伝達プラットフォームを提供し、他の肝臓をガイドとする伝達を優勢とする応用に適用できると信じている。
HoFHのNHPモデル
NHP中のHoFHのモデルを構築するために、野生型NHP中のLDLR遺伝子を編集し、標準LNPsに包まれたCas 9と二重誘導RNA戦略を用いてLDLRの肝臓における発現を除去し、それによって70%近くの全肝DNAをLDLR遺伝子に編集し、肝臓LDLR蛋白の約94%の減少を招き、血液低密度リポ蛋白-Cを6倍増加させた。
国内で開発されたGalNAc−LNPsを用いたHoFHのNHPモデルの検証
この新規なHoFH NHPモデルを用いて,我々独自のGalNAc−LNPsの2種類の異なる処方を用いて臨床前研究を行い,Angptl 3を標的とした塩基エディタを提供した。この研究では,標準LNPを用いた基礎エディタの交付はHoFHのNHPモデルの肝臓で有効なAngptl 3編集を実現していないことが観察された。以下の図に示すように,Angptl 3を標的とした塩基エディタをGalNAc−LNPとともに処理したNHPでは,血液Angptl 3蛋白がそれぞれ約94%(n=3)と97%(n=3),低密度リポ蛋白(LDL−C)が100 mg/dL近く低下し,ベースラインより約35%の低下が認められた。
33
GalNAc−LNPの正常肝臓への送達
Angptl 3標的塩基エディタを渡すための我々独自のGalNAc−LNP法の潜在的広範な実用性も評価し,ある臨床前研究では,正常肝臓を有する野生型HHPにGalNAc−LNPとGalNAcを使用しない標準LNPを用いたAngptl 3塩基エディタの送達効率を評価した。これらの研究では,我々のGalNAc−LNPにより伝達されたAngptl 3標的塩基エディタで処理した野生型NHPのAngptl 3蛋白が約89%減少し,標準LNPで処理した野生型NHPのAngptl 3蛋白が約74%減少することが観察された。GalNAc−LNP送達がLDLRの適応に有用であることが示唆されたと考えられる。
34
我々は34個の野生型NHPの大型検証用量-反応研究を完了した。本研究では,Angptl 3塩基を1.5 mg/kg(n=6)と3.0 mg/kg(n=16)のGalNAc−LNPを用いて編集し,対照群(n=12)と比較した。血液循環の96%の減少が観察されました Angptl 3蛋白3.0 mg/kg投与群。
ABE−Angptl 3前駆体配合のNHP検証
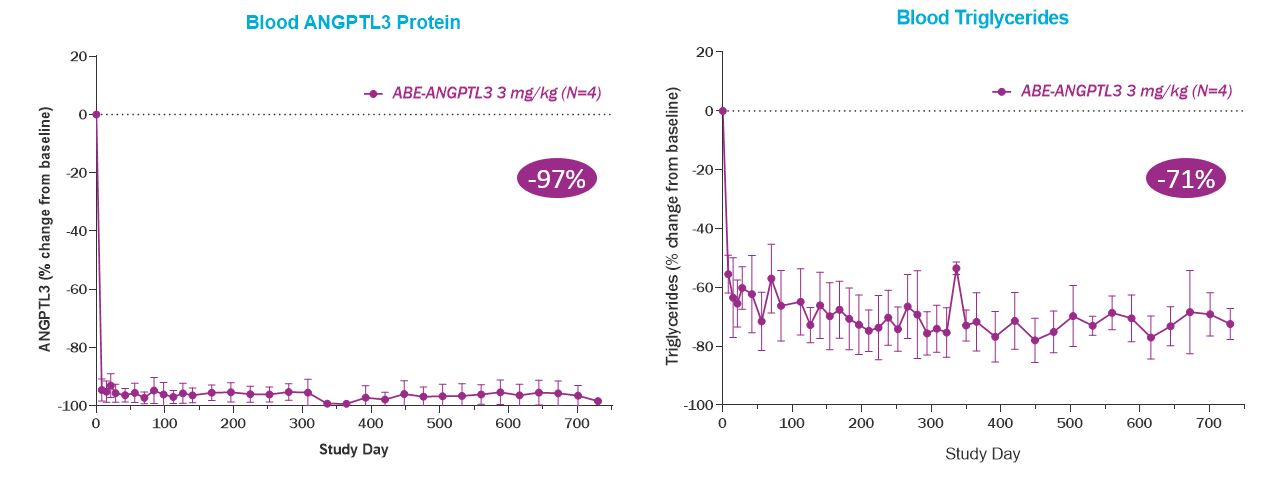
ABE−Angptl 3前駆体製剤を用いて臨床前概念検証研究を行った。この研究では,健康なNHPを単回投与した。次の図において、各処理されたNHPは1つの紫色のストリップで表され、各処理された車両制御は1つの青色のストリップで表される。われわれのABE−Angptl 3前駆体製剤単一治療後,投与2週間後に肝生検で採取した肝組織全体でAngptl 3の平均60%の編集が認められた。それに伴い血中アンギオテンシン変換酵素3蛋白は平均95%,血中トリグリセリド濃度は平均64%低下した。
重要なことは,この臨床前研究では,血液Angptl 3蛋白と血中トリグリセリドレベルの低下が持続することが観察された。以下の図に示すように,ABE−Angptl 3単回静注2年後にNHPsの血液Angptl 3蛋白とトリグリセリドの平均低下幅はそれぞれ97%と71%に認められた。
35
ABE−Angptl 3前駆体製剤の臨床前研究および我々独自のGalNAc−LNPs前駆体製剤がVERVE−201の開発につながった。
Verve-201 Next Steps
開発候補薬剤としてVERVE−201を指名する前に,臨床前の安全性と有効性を最適化するための厳しいプロセスを用いた。最適な構成を選択し、初代ヒト肝細胞においてVERVE-201を評価し、有効で標的編集されたAngptl 3遺伝子を示し、検出可能な脱標的および高被覆率全ゲノム光学プロファイル評価の構造変異がなかった。我々はVERVE−201臨床開発開始の規制申請を支援するための前臨床研究を行っており,2024年に1 b期臨床試験を開始する予定である。
逐次投与する
低密度リポ蛋白レベルが非常に高い患者や高い密度リポ蛋白レベルと高トリグリセリドレベルを同時に有する高脂血症患者は,PCSK 9やAngptl 3のような2つの脂質経路に対する遺伝子編集薬の恩恵を受ける可能性が考えられる。私たちは4つのNHPの中で90日間の臨床前研究を行い、私たちの基礎編集者の連続投与の可能性を評価した。この研究では,1日目に1.0 mg/kgのVERVE−101前駆体,その後30日目に1.0 mg/kgのAngptl 3塩基エディタを投与した。以下の図に示すように,逐次投与後のPCSK 9とAngptl 3の血漿蛋白レベルの有意な低下が認められた。15日目に肝生検にてPCSK 9編集を測定したところ,平均71%の編集が認められた。45日目に肝生検によりAngptl 3の編集を測定したところ,平均52%の編集が認められた。90日目に肝臓剖検を行い,平均69%のPCSK 9編集と63%のAngptl 3編集を認めた。また検討期間中に血漿PCSK 9とAngptl 3蛋白レベルをモニタリングし,1回目の投与後に血漿PCSK 9蛋白の90%以上の低下,2回目の投与後に血漿Angptl 3蛋白の90%以上の低下が認められ,研究終了時に類似の低下が認められた。これらのデータは,連続用量のPCSK 9塩基エディタ,次いでAngptl 3塩基エディタであり,2つのキー脂質経路を制御する2つの遺伝子を編集できる可能性を示している。
36
LP(A)計画
私たちのLP(A)計画は設計に集中しています体内にあるゲノム編集薬は,正確なDNA変化により持続的に肝臓中のLPA遺伝子を不活化する。我々は,最初にASCVDと循環中のLp(A)濃度の高い患者のための計画を開発する予定である。
Lp(A)は低密度リポ蛋白に類似した粒子であり、そのアポリポタンパク質Bはアポリポタンパク質(A)に共有結合し、アポリポタンパク質(A)は肝臓で産生され、血液中を循環する。LPA遺伝子標的は疫学、人類遺伝学と薬理学研究によって確定され、これらの研究はLp(A)がASCVDリスクの重要な原因と変更可能な駆動要素であることを実証した。この増加のリスクはLp(A)濃度が非常に高い個体の中で最も顕著であった(例えば,150 nmol/L)。ASCVD患者の20%のLp(A)濃度がこの閾値を超えていると推定されている。Lp(A)濃度はほぼ完全に遺伝ライフスタイル療法によって決定されており,現在承認されている脂質低下療法の影響は小さいか影響していない。
ヒト遺伝学と薬理学研究はLp(A)を減少させる薬物の潜在的な治療効果と安全性を実証した。循環Lp(A)の増加を招くDNA変異はASCVDとある心臓弁膜疾患(例えば大動脈弁狭窄)のリスクの最も強い遺伝性駆動要素の一つである。対照的に、LPA遺伝子の1つまたは2つのコピーで自然に発生する機能喪失突然変異はこれらの疾病の保護と関係があり、深刻な健康結果は検出されなかった。
これらのヒト遺伝学的研究に加えて,最近の肝臓LPA発現に対する研究的治療のヒト薬理学的研究は,循環中のLp(A)濃度を80%以上低下させるのに有効である。これらの薬剤が高Lp(A)患者のASCVD再発リスクを低下させる可能性があるアンチセンスオリゴヌクレオチドpelecarsenとsiRNA olpasiranの心血管結果試験で試験を行っている。
私たちはこれらの以前の研究は体内にあるASCVDを治療するためのゲノム編集薬物-単一治療コースの薬物が同時に高いリスクと高い需要を満たしていない患者集団におけるLp(A)の潜在的効用を低下させるために大量の証拠を提供した。我々のLP(A)計画は早期研究段階にある.
未来のチャンス
私たちは他の人を特定するために投資しています体内にある肝臓遺伝子編集治療、そして単一治療コースの遺伝子編集薬物を開発し、疾病の根本的な原因を解決する予定である。目標生物がヒト遺伝学的に検証されるプロジェクトに専念し続ける予定であり,多くの場合,他の方式を用いた臨床開発プロジェクトにより検証を行う予定である。
37
製造業
私たちは現在製造施設を所有したり運営したりしていない。著者らは現在第三者契約製造組織(CMO)とサプライヤーに肝心な出発原料、薬物物質(gRNA、mRNA)と私たちの薬物製品を提供することに依存している。私たちは第三者CMOを使用して私たちのIND研究を支援し、私たちの臨床試験と商業活動を支援する予定です。私たちが製造規模を拡大するにつれて、私たちは引き続き私たちのCMOネットワークを拡大して強化するつもりだ。私たちの候補製品製造に必要なすべての材料は複数のソースと、私たちの候補プロジェクトコンポーネントを組み立てることができる複数のCMOがあると信じています。
新規かつ最適化されたGalNAc標的リガンド、脂質アンカー、LNP成分の最適な組成および比率の開発、標的リガンドによる付加およびLNP形成の最適なプロセスを含むメッセンジャーリボ核酸生産およびLNP製剤の内部製造能力の確立に投資し続ける。生物活性と効力分析を含む分析方法の開発にも投資しており、さらなる製品開発、バッチ比較性評価、追加の製造業成長に重要である。
製造は手続きと文書要求を強制的に執行する幅広い規制によって制限されている。これらの規定は記録保存、製造過程と制御、人員、品質管理と品質保証を管理する。私たちのCMOはこのような規定を遵守し、定期的な監視と正式な監査によって評価されなければならない。私たちの第三者製造業者は、現在の良好な製造仕様またはcGMP要件および他の適用される法律法規に基づいて、私たちが開発した任意の候補製品を生産しなければなりません。
私たちは豊富な技術、製造、分析、品質経験を持つ人員を持って、私たちが契約した製造とテスト活動を監督します。
競争
生物技術と生物製薬業界、特にCVD領域は、技術進歩が迅速で、競争が激しく、知的財産権保護が有力であるという特徴がある。私たちが開発と商業化に成功したどの候補製品も、既存の治療法や将来出現する可能性のある新しい療法と競争しなければならない。私たちは心血管疾患、遺伝子編集と製造方面の技術、開発経験と科学知識が私たちに競争優勢を提供すると信じているが、私たちは主要な製薬、専門製薬と生物技術会社、学術機関、政府機関及び公共と個人研究機関を含む多くの異なる源からの潜在的な競争に直面している。
私たちと比較して、私たちが競争しているか、あるいは将来競争する可能性のある多くの会社は、研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング承認製品を獲得する上で、より多くの財務資源と専門知識を持っている。製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。
もし承認されれば、心血管疾患治療のために開発したすべての候補製品の成功を影響する肝心な競争要素は治療効果、安全性、利便性、価格、模造薬の競争レベル及び政府と他の第三者支払い者が精算できるかどうかである可能性がある。
もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。また、多くの場合、私たちの競争能力は、後発薬の使用を奨励することを求める保険会社または他の第三者支払者の影響を受ける可能性がある。もし私たちの候補製品が発売承認されたら、私たちはそれらの価格が競争相手の模造薬より著しく高いと予想します。
いくつかの承認された製品は低密度リポ蛋白質の低下或いは心血管リスクの低減に応用でき、例えばスタチン類薬物、エゼミブ、フェニルペドド酸、ロミチート、ミパモソンとエイコサペンタエン酸などである。いくつかの承認された製品はPCSK 9蛋白を標的とし、低密度リポ蛋白とASCVDリスクを低下させる機序としている。Evocumabこれは
38
安進が発売したRepathaというモノクロナル抗体はHeFH、HoFHおよびASCVD患者の治療にFDAによって許可された。AlirocumabはサイノフィとRegeneron製薬会社によって販売されたモノクロナル抗体であり、名称はPRALUENTであり、すでにアメリカ食品と薬物管理局によってASCVD患者の治療とHeFHを含む原発性高脂血症患者の治療に許可されている。承認されたモノクロナル抗体治療は細胞外でPCSK 9蛋白を抑制することにより作用する。Inclisiranは小干渉RNAであり、ノファ社はこれをLeqvioと命名し、アメリカでは低密度リポ蛋白-Cをさらに低下させる必要がある臨床ASCVD或いはHeFH患者の治療に許可され、ヨーロッパではHeFH或いは混合性脂質異常を含む高コレステロール血症患者の治療に許可されている。Inclisiranは肝細胞内PCSK 9の合成を抑制することで機能し,細胞外蛋白抑制とは異なる。臨床発展の異なる段階で、3種類の経口小分子製品候補薬物はPCSK 9蛋白を標的とし、低密度リポ蛋白の低下とASCVDリスクを低下させる機序として知られている。これらの薬物は、最近完成した2 b期成人高コレステロール血症患者試験で研究され、2023年第1四半期に結果を公表する予定であるメルク社のMK-0616を含み、Esperion Treeutics社が許可したSerometrix LLCの経口小分子薬であり、2022年にINDを提出する計画、2024年末または2025年初め、およびAZD 0780、アスリコンがDogma治療会社から買収され、第1段階の臨床試験で評価されている。
臨床前開発にはPCSK 9遺伝子に対するもう2つの遺伝子エディタがあることが知られている。Precision Biosciences,Inc.,またはPrecisionは,以下の場合NHPにおけるPCSK 9とLDL-Cレベルの長期安定低下を示す臨床前データを発表した体内にあるその遺伝子編集プラットフォームを利用してPCSK 9遺伝子に対して遺伝子編集を行った。2021年9月、PrecisionはiECUREと提携し、iECUREは2022年にPrecisionのPCSK 9配向ヌクレアーゼ候補製品をFH治療の第1段階臨床試験に進める予定である。2023年1月、Precisionは、iECUREとの協力による計画の実施を中止することを決定し、薬剤が将来的に臨床試験に入るかどうかについて、より多くの指導を提供することを計画していることを発表した。また、2022年にCRISPR治療会社またはCRISPRはCTX 330を発表し、これがその研究段階である体内にあるPCSK 9に対する遺伝子エディタ。
EvinacumabはRegeneronによって販売されているAngptl 3蛋白に対するモノクロナル抗体であり,HoFH患者の治療にFDAによって承認されており,また難治性高コレステロール血症,ASCVDあるいはHeFH患者および重篤な高トリグリセリド血症患者の第二段階研究で評価されている。臨床開発にはAngptl 3を標的とした候補製品がいくつかあり,これを低密度リポ蛋白−CとASCVDリスクを低下させる機序としてARO−ANG 3,Angptl 3に対するsiRNA,矢印製薬会社がHoFH患者と混合性脂質異常患者の第2段階臨床試験を行っていることが知られている。2022年、矢印社は2023年下半期にHoFH患者とHeFH患者においてARO-ANG 3の重要な3期研究を開始する計画を発表した。また、礼来社はAngptl 3タンパク質に対するsiRNAを評価しており、混合性脂質異常を有する成人に対する第2段階研究である。2022年、CRISPRはAngptl 3に対する遺伝子編集プログラムCTX 310を発表し、INDイネーブル研究を行っており、2023年に患者投与を開始する予定である。
Lp(A)を減少させるためのいくつかの研究薬が現在開発されている。その中には,2019年にノワ社がIonis PharmPharmticalsから許可を得たアンチセンスオリゴヌクレオチドPelecarsenが含まれており,Lp(A)上昇と心血管疾患患者の3期LP(A)Horizon心血管結果研究で評価されており,2025年にはTOPLINE結果が予想される。OlpasiranはLp(A)に対する研究siRNA薬であり,安進社が矢印製薬会社から許可を得ており,最近ASCVD患者や高Lp(A)濃度の患者のLp(A)濃度を低下させることが証明されている。Opasiranが現有のASCVDと高Lp(A)患者の心血管イベントを減少する潜在力はOcean(A)研究の中で評価を行い、この研究は2022年にスタートし、2026年に研究を完成する予定である。また,SLN 360はSilence Treeutics plcが開発している研究性siRNA薬であり,リポ蛋白質(A)濃度の高さとASCVD事件高リスク患者に対する第2段階研究で評価されており,CRISPRは2022年にCTX 320を発表しており,その研究段階である体内にあるLp(A)に対する遺伝子エディタ.
知的財産権
私たちは、私たちの候補製品の構成、それらの使用方法、関連技術、および私たちの業務に重要な他の発明をカバーするために、特許保護を求めて維持することを含む、私たちの業務に重要と考えられるノウハウを保護するために努力しています。特許保護に加えて、私たちは、私たちの業務において特許保護から保護されているか、または私たちの技術のいくつかの態様を含む、特許保護に適していないと思う態様を商業秘密に依存しています。
私たちのビジネス成功は、私たちが商業的重要性を有する技術、発明、および私たちの業務に関連する技術、発明および技術の特許および他の独自の保護を獲得し、維持する能力があるかどうかにある程度依存する
39
私たちの知的財産権、特に私たちの特許権を実行し、私たちの商業秘密の機密性を保護し、他人の有効かつ実行可能な知的財産権を侵害することなく運営する。
私たちのようなバイオテクノロジーと製薬会社の特許状況は通常不確実であり、複雑な法律、科学、および事実の問題に関連しているかもしれない。さらに、特許出願において要求されるカバー範囲は、特許発行前に大幅に縮小することができ、その範囲は、特許発行後に再解釈または挑戦することができる。したがって、私たちは私たちのすべての候補製品が強制的に実行可能な特許によって保護されるか、または引き続き保護されることを保証できない。私たちが現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるかどうか、または任意の発行された特許の権利主張が競争相手の影響を受けないように十分な特許保護を提供するかどうかを予測することはできない。私たちが持っているどんな特許も第三者によって挑戦され、回避され、無効に発表される可能性がある。
2022年12月31日現在、私たちの特許権は、PCSK 9およびAngptl 3標的に対する私たちの遺伝子編集プログラム、ならびに私たちのRNA配信および他のプラットフォーム技術を含む、私たちのプログラムおよび技術の様々な側面をカバーしています。発行または未解決の米国または外国特許は、可能な特許期間の調整または延長を考慮することなく、2041年から2043年までの異なる日に満了する予定であり、すべての適切な保守、更新、年金、および他の政府費用が支払われると仮定する。我々の特許組合せのいくつかの部分に関するさらに詳細な情報は以下のとおりである.
PCSK 9計画
我々のVERVE-101計画については、2022年12月31日現在、我々の特許権は、PCSK 9遺伝子を標的とする誘導RNA配列、アデニン塩基編集プログラムをコードするmRNAおよびそれらの組成物、これらの組成物を治療する方法、PCSK 9遺伝子を標的とする誘導RNA配列、アデニン塩基エディタをコードするmRNAおよびその組成物を含む15件以上のVERVE-101計画の様々な側面をカバーしている体内にある遺伝子編集、処方、投与プラン、併用療法。
Angptl 3プログラム
我々のVERVE−201計画については、2022年12月31日現在、我々の特許は、係属中の米国特許出願、係属中の国際PCT出願、およびわれわれが保有または制御している15件以上の外国出願対応出願を含み、Angptl 3遺伝子を標的とする誘導RNA配列、アデニン塩基編集をコードするmRNAおよびその組成物、適応治療用組成物、適応治療方法を含むVERVE−201計画の様々な側面をカバーしている体内にある遺伝子編集、処方、投与プラン、併用療法。
許可と協力協定
私たちは多くのライセンス契約の当事者であり、これらの合意に基づいて、私たちは第三者から特許、特許出願、その他の知的財産権の許可を取得します。許可された知的財産権部分は,CRISPRに関連する物質成分とその基礎編集のための状況をカバーしている。このような許可証は私たちに様々な勤勉さと財政的支払い義務を課している。私たちは未来にこのような種類の許可協定を締結し続けると予想する。
BEAM Treeuticsとの協力とライセンス契約
2019年4月に、吾らはビム或いは原始ピムプロトコルと協力及び許可プロトコルを締結し、ビムのいくつかの塩基編集技術及び遺伝子編集及び交付技術に基づいて、吾らは独占的、世界的、再許可可能な許可を得て、ビムを使用したCRISPR関連蛋白12 b或いはCas 12 b技術の基礎編集製品及びヌクレアーゼ製品を開発、製造、使用、販売、販売及び輸入し、それぞれの場合、4つの冠状動脈疾患リスクの増加に関連する遺伝子標的(PCSK 9とAngptl 3遺伝子を含む)又は許可製品を獲得した。元のピム協定調印後、協定によって私たちに与えられた権利の一部の代価として、私たちはビムに276,075株の私たちの普通株を発行した。
2022年7月、吾はピムと改訂及び再署名された協力及び許可協定、又は改訂されたピム協定を締結した後、元のピム協定を改訂及び再記述することに等しい。改訂されたビムプロトコルによると、ビムはビムのいくつかの塩基編集技術に基づいて、第3の肝臓媒介心血管疾患標的に対する製品の開発と商業化のために、独占的、世界的範囲内の再許可可能な製品を付与し、原始ビムプロトコルに従って許可されたPCSK 9およびAngptl 3遺伝子標的を開発し、商業化した。我々は許可された遺伝子標的に対する製品の開発と商業化を担当しており,いずれの場合もビムの選択加入権利に制約されている。以下に述べる以外に、改訂されたBEAMプロトコルに従ってライセンス製品の開発を完全に担当しています。
40
Angptl 3およびPCSK 9遺伝子標的については、認可製品の第1段階臨床試験において最終患者にこのような遺伝子標的を投与した後、ビムは、世界のこのような許可製品開発費用の33%を分担し、米国で商業化されたこのような許可製品の利益および費用を50/50の割合で共同商業化し、共有する権利がある。第3の遺伝子標的については、認可された製品の第1段階の臨床試験において最終患者にこのような追加の遺伝子標的を注入した後、ビムは、許可された製品開発の世界的費用の35%を共有し、許可された製品を商業化し、世界で商業化された利益および費用の35%を共有することを選択する権利がある。
ビームが特定のライセンス製品の選択加入権利を行使した場合、提携製品と呼び、協力製品の開発および商業化コストの特定の割合を支払う義務があり、その提携製品の任意の販売から指定されたパーセントの利益を得る権利がある。各協働製品について、私たちとピムは、この協力製品が米国で販売されることが予想される前に後続の共同普及協定を締結し、この合意に基づいて、私たちとピムは、その協力製品を普及させるために必要な50%の普及作業をそれぞれ提供する。米国以外の連携製品については,合計560万ドルの臨床·規制マイルストーンと,合計750万ドルの販売ベースのマイルストーンの支払いが義務付けられている。
私たちは、ピムが(I)その選択加入権利を行使していないか、または(Ii)ピムがその選択加入権利を行使している場合、私たちまたはピムは、割り当てられた開発および商業化コストを支払わず、その許可製品の商業化に参加する任意の許可製品を非協働製品と呼ぶことを選択する。このような非協力製品については,世界的に合計1130万ドルの臨床·規制マイルストーンと,合計1500万ドルの販売ベースのマイルストーンを支払う義務がある。
提携製品が米国国外または非協力製品で世界的に販売されている場合、純売上高の中央値パーセントまでの料率でBeamに等級版税を支払うことが要求されるが、指定された減額で支払わなければならない。このような使用料の支払いは、(I)当該製品をカバーする特許権に基づいて当該国/地域の最後の有効な特許請求が満了したとき、(Ii)当該製品の国/地域に関連する規制排他期間、又は(Iii)当該製品が当該国/地域で初めて商業販売されてから10年以内に、以下に準ずる国/地域及び製品/製品の終了時に終了する。
我々の知的財産権の下で,我々のGalNAc−LNP交付技術を含めて,我々が開発した臨床前計画に関する独占的,世界的に再許可可能で全額支払いの許可をビムに付与した。ビムは我々が制御するノウハウと特許の非独占的許可を持ち,共同連携技術に権益を持ち,ビムが合意した研究·開発計画に基づいて活動することを許可している。
吾ら及びヒラメ表はいずれも吾等の許可を得る権利を再許可する権利があるが,何らかの制限を受けなければならず,再許可協定は改訂されたヒラメ表合意及び任意の適用可能な許可協定の条項に適合しなければならない。
改訂されたBEAMプロトコルは、特定の基礎編集製品の開発および商業化のための当社のGalNAc-LNP配信技術の非独占的、世界的、再許可可能なライセンスを取得するために、各選択権、特定の規制および商業販売マイルストーン、およびGalNAc-LNP配信技術を使用した基礎編集製品の純売上について、BEAMが私たちに支払うべき費用について、BEAMに個々の目標に基づく選択権を付与する。
改訂されたピム協定によると、ビム社は自費でそれぞれの特許権の起訴を制御している。私たちは権利がありますが、義務はありません。ビーム適用の許可内で合意が許可された範囲内で、費用申請、起訴、改訂されたピム協定下の特定製品特許権を自己負担し、独占権利出願、起訴、起訴及びメンテナンス技術項目の下の特許権、並びに改訂されたピム協定に従ってビムの他の特許権に付与することができます。
一方が合意義務を履行するためにビムとVerveが共同開発した知的財産権については,プロトコルにより,その知的財産権はその性質に応じて連携技術とみなされ,ビムとVerveが共同所有しており,我々とベムは誰が共同所有する特許権の申請,起訴,維持を善意に基づいて決定する.
41
改訂されたBEAMプロトコルの期限は、任意のライセンス製品の任意のライセンス使用料期限の最終期限まで継続される。我々は、90日間の終了通知をビムに送信することにより、任意のライセンス製品の改訂後のピム合意を終了する権利があるが、ビムがその選択加入権利を行使しないことを選択したこと、またはその選択加入権利を行使する期限が満了したことを前提として、いかなる協力製品の合意も終了することはできない。ピムは私たちに90日間の終了通知を出すことで、いくつかの製品に関する改訂されたピム協定を終了する権利がある。改訂されたBEAMプロトコルは、(I)他方に重大な違約が存在し、指定された治癒期間内に違約を訂正できなかった場合、または(Ii)他方が破産または清算された場合に、いずれかの一方によって終了することができる。Beamは改訂されたBeamプロトコルを終了することができ、吾らは、修正されたBeamプロトコルに従ってBeamに付与された許可を直ちに終了することができ、相手が改訂されたBeamプロトコルに従って許可を付与する任意の特許権の実行可能性、有効性、または範囲に疑問を提起することを前提とすることができる。
Acuitas協定
PCSK 9遺伝子標的の許可プロトコル
2020年10月には,我々のVERVE−101候補製品のコンポーネントとしてAcuitasとの開発とオプションプロトコルやAcuitas開発プロトコルにより最適化されたLNPを選択した。この選択については,LNP技術の使用に関する選択権を行使し,AcuitasまたはAcuitasライセンス契約と非排他的グローバルライセンスを締結し,許可されたLNP技術の下で複数のレベルで再許可を行う権利があり,研究,開発,開発,製造,製造,保持,使用,販売,輸入および輸出入,その他の方法でLNP技術を使用したライセンス製品を商業化および開発する権利があり,これらの製品はPCSK 9遺伝子標的とすべてのヒト治療または予防用途に使用されている。Acuitasライセンス協定によると、私たちはライセンス製品の開発と商業化に勤勉に努力する義務がある。
AcuitasはLNP技術に関連する特許を提訴·維持する権利を保持し,費用はAcuitasが負担する。もしAcuitasがLNP技術に関連する特許を提出、起訴、または維持しないことを選択した場合、それは私たちに権利があるが、Acuitasにこのような特許の提出、起訴、または維持を要求する義務はなく、費用は私たちが負担し、私たちのこのような特許の許可は自動的に撤回不可能、永久、全額支払い、および印税免除になるが、その後、このような特許はその国の許可技術の一部ではないだろう。
我々とAcuitasは,AcuitasライセンスプロトコルとAcuitas開発プロトコルのさらなる規定に基づき,共同所有特許について共同特許起訴と保守協定を締結する。
我々はAcuitasに200万ドルの前払い許可料(これまでに支払われた目標予約費を差し引く)を支払い,ある開発ベースのマイルストーンに達するまで年間80万ドルのライセンス維持費の支払いを要求された。その許可技術が私たちに譲渡されたCMOに関する従業員と合理的な外部費用を四半期ごとにAcuitasに精算する義務があります。
私たちはまたAcuitasに合計980万ドルの臨床と規制マイルストーン費用、950万ドルの販売マイルストーン費用を支払う義務がある。私たちは私たち、私たちの付属会社、あるいは私たちの許可された販売許可製品の年間純売上に基づいて、より低い一桁の割合で印税を支払うことを要求されます。LNP技術に関連する技術に基づいて第三者から許可を得ると,このような特許権使用料が減少する可能性がある.任意のこのような使用料の支払いは、(I)当該国/地域でライセンス製品をカバーするライセンス技術の有効クレームが最終的に満了する日、(Ii)当該国/地域の法規の排出期間が満了した日、および(Iii)当該国/地域で初めて商業販売された日から10年以内に、国/地域およびライセンス製品/ライセンス製品に基づいて支払われなければならない。
Acuitasライセンス契約は、このようなライセンス製品の当該国/地域での最終満了印税期限に基づいて、ライセンス製品および国/地域で終了する。事前にAcuitasに書面で通知しておけば、私たちは理由なくAcuitas許可協定を終了することができる。以下の場合、いずれの場合も、Acuitas許可プロトコルを終了することができる:(I)他方に重大な違約が存在し、指定された救済期間内にその違約を是正できなかった場合、または(Ii)他方が破産または債務を相殺しない場合には直ちに通知を出す。Acuitasに書面で通知した後、私たちは合意を終了するのではなく、Acuitasの未治癒の重大な違約によって合意を終了するのではなく、適用されたマイルストーンと特許使用料を特定の割合に減らすことを選択することができる。
ノワール許可協定
42
2021年10月、私たちは、脂質技術の非独占的な許可を得るために、ノファ社とライセンス契約を締結し、VERVE-201を含むいくつかの候補製品の研究および開発に使用している。協定により付与されたライセンスと権利の対価格として、2021年12月31日までの年間に80万ドルを一度に支払い、払い戻しはできません。ライセンス契約は,特許脂質技術を用いた製品に合計1,000万ドルの臨床·規制マイルストーン費用,および3,500万ドルの販売ベースマイルストーン費用を支払うことを求めている。
2022年6月には、他の3種類のライセンス製品を非独占ライセンスの範囲に入れる協定を修正しました。追加の許可製品を考慮して、私たちはノバ会社に一度に280万ドルを支払うことを要求されて、払い戻しできません。
遠大学院とハーバード学院総裁や研究員と署名したCAS 9許可協定
2019年3月には、遠大とハーバードと特定特許権についてライセンス契約を締結し、2019年12月には、このライセンス契約または改訂されたCas 9ライセンス協定の修正案に署名しました。Cas 9ライセンス契約に基づいて付与される許可は、(I)ハーバード大学またはハーバードCas 9-I特許権によって独占的に所有される特定の特許および特許出願、マサチューセッツ工科大学またはマサチューセッツ工科大学およびブロダーが共同所有するいくつかの特許および特許出願、ロックフェラー大学またはロックフェラー大学およびブロダーが共同所有する特定の特許および特許出願、およびマサチューセッツ工科大学、ブロダーおよびハーバードが共同所有する特定の特許および特許出願の権利を含み、Cas 9ライセンス協定に従って許可された特許および特許出願をハーバード/ブロダーCas 9-I特許権と呼び、(Ii)MITブロダーが共同所有する特定の特許および特許出願を含む。ハーバード大学及びアイオワ大学研究基金会、又はアイオワ州大学は、Cas 9ライセンス契約により付与された特許及び特許出願を、ハーバード/ブロスCas 9−II特許権、及びハーバード/ブロスCas 9−I特許権、ハーバード/ブロスCas 9特許権と呼ぶ。
2017年2月、ブロイドとロックフェラーは、ロックフェラーがその独占的かつ独占的な代理として、ハーバード/ブロイドCas 9-I特許権におけるロックフェラーの権利を付与する機関間合意に達した。
2014年12月、2016年8月に改訂されたマサチューセッツ工科大学、アイオワ州、ブロイドは、ハーバード/ブロスCas 9-II特許権における権利を許可するために、アイオワ州ライセンスブロダーを彼らの独占的かつ独占的代理として締結した共同発明管理協定を締結した。
Cas 9ライセンス契約下のライセンス
Cas 9許可協定によると、遠大とハーバードは私たちのハーバード/遠大Cas 9特許権のグローバル、印税と再許可許可を授与し、私たちが人類疾患の予防と治療のために使用、製造、使用、使用、販売、輸出入、PCSK 9、Angptl 3と2つの追加目標に対する製品を許可することを可能にし、しかしいくつかの制限と保留された権利の制限を受ける。ハーバード/遠大Cas 9−I特許権とあるハーバード/遠大Cas 9−II特許権またはCAS 9−II A組特許権については,ライセンスはEditas Medicine,Inc.またはEditasと共同で独占されている。ある他のハーバード/遠大Cas 9-II特許権またはCas 9-II Bグループ特許権については、ライセンスは非排他的である。このライセンスは、ブロード、マサチューセッツ工科大学、ハーバード大学が制定した包括的な革新戦略に従っている。
遠大とハーバード大学はまた、ハーバード/遠大Cas 9特許権の非独占的、世界的に、印税のある、再許可可能な許可を授与し、内部研究目的に使用した;Editasが遠大とハーバードと締結した独占許可協定分野以外のヒト疾患の予防または治療のための製品の研究、開発、商業化のための製品;および目標、製造、製造、使用、使用、販売、輸出入はCas 9許可製品ではないがCas 9を支持する製品を提供した。
ブロイドおよびハーバードは、Cas 9許可協定に従って私たちに付与された許可は、ハーバード/ブロイドCas 9特許権に保持されている米国政府の権利によって制限され、ブロイド、ハーバード、マサチューセッツ工科大学、ロックフェラーおよびアイオワ州代表自身および他の学術、政府および非営利エンティティが研究、教育または教育目的のためにハーバード/ブロイドCas 9特許権を実践する権利を受ける。また,Cas 9ライセンス契約に基づいて我々に付与されたハーバード/遠大Cas 9−I特許権のいくつかの権利は,ハワード·ヒューズ医学研究所が研究目的のための非独占的許可に制約されている。私たちの共同独占許可権はまた、遠大、ハーバード、マサチューセッツ工科大学、ロックフェラーおよびアイオワ州に保持されている権利によって制限されており、それらは、それぞれおよび任意の第三者(非営利性および営利団体を含む)が、研究、開発、製造、製造、使用、要約、輸入、または他の方法でハーバード/遠大Cas 9特許権および許可製品を研究製品または研究ツールとして利用する権利を有している、または研究目的のために使用されている。
43
私たちは私たちが許可された権利を再許可する権利があるが、再許可協定がCas 9許可協定の条項に適合しなければならないことを前提としている。いかなる再許可協定も,ブロイドとハーバードの書面で同意せずに再許可を譲渡する権利を含むことはできない。さらに、任意の再許可協定には、Cas 9許可協定に規定されている同じ条項に従って、ハーバード、ブロダー、マサチューセッツ工科大学、ロックフェラー、アイオワ州、ハワード·ヒューズ医学院への再許可受給者の賠償を要求する条項、および特定の目的のために、ブロイド、ハーバード、マサチューセッツ工科大学、ロックフェラー、アイオワ州、ハワード·ヒューズ医学院が再許可協定の第三者受益者であることを宣言する条項が含まれなければならない。
私たちは、商業的に合理的な努力を使用するか、または私たちの関連会社または再ライセンシーのうちの少なくとも1人に商業的に合理的な努力を使用させること、(I)許可分野でCas 9許可製品を研究開発すること、(Ii)許可分野のそのような製品を商業市場に導入すること、および(Iii)そのような製品が市場に入った後に、許可分野でそのような製品をマーケティングし、そのような製品を合理的に公衆に提供することを義務化する。さらに、私たち自身または私たちの任意の関連会社または分割ライセンシーを通じて、特定の期間内に特定の開発マイルストーンを達成する義務があります。もし私たちが発展マイルストーンを実現できなければ、遠大とハーバードはCas 9許可協定を終了する権利があるが、私たちは特定の手続きに従ってこのマイルストーンを延長または修正する権利がある。もしブロードとハーバードが開発マイルストーンに達しずにCas 9許可協定の終了を選択した場合、ハーバードとブロッドが合理的に受け入れた証拠を提供し、私たちがその目標に対する開発マイルストーンの職務調査義務に違反していないことを証明し、私たちまたは私たちのある関連会社または分割許可者が(A)その目標に対する許可分野のCas 9許可製品を研究·開発しており、(B)商業的に合理的な努力を使用して、その目標に対する許可分野のCas 9許可製品を商業市場に導入する(適用される場合)、その停止権は特定の目標にのみ適用されない。(C)Cas 9許可製品を市場に投入した後,商業的に合理的な努力を用いて許可分野でその目標に対するCas 9許可製品を販売し,そのCas 9許可製品を合理的に公衆に提供し(適用すれば),その後,残りの期限内に継続する, または上記(A)~(C)項によれば、我々の少なくとも1つの関連会社または分割ライセンシーに、その目標のためのCas 9ライセンス製品の開発および商業化を継続させる。
Cas 9許可協定によると、遠大とハーバードはまた権利を保持し、その包摂的な革新戦略を通じて、特定の場合、心血管疾患領域以外の特定の遺伝子に対する製品の開発と商業化を望む第三者(指定実体を除く)により多くの許可を付与し、そうでなければ、これらの製品は遠大とハーバードから得た共同独占許可の範囲に属する。もし第三者がハーバード/遠大Cas 9-I特許権に基づいて製品の開発と商業化申請許可を申請すれば、Cas 9許可協定に基づいて、遠大とハーバードは共同で私たちの独占許可を授与し、遠大とハーバードはこの要求を私たちに通知する可能性があり、私たちはこれをCas 9第三者が提案した製品要求と呼ぶ。Cas 9サードパーティが提案する製品要求には、提案の目標またはカテゴリを含む第三者の誠実なアドバイスが添付されなければならない。遠大はCAS 9の第三者提案製品要求を承認することができない:(I)もし私たちが私たちの任意の関連会社または再許可者を直接または間接的に通過すれば、CAS 9第三者に対して製品要求またはCAS 9被許可側製品の同じ遺伝子目標の製品を研究、開発または商業化しており、このような持続的な努力が遠大に満足していることを証明することができ、または(Ii)もし私たちが直接または間接的に私たちの任意の関連会社または再許可業者を通じてそうすることを望むならば、私たちは遠大に興味を持って研究することができる。Cas 9被許可側製品の開発と商業化は、ビジネス的に合理的な研究、開発、商業化計画を持っており、その計画に基づいた合理的なビジネス努力を開始し、継続している。また、もし私たちが直接または間接的に私たちのいかなる付属会社または分割許可者を通じても研究していないならば, Cas 9被許可側の製品を開発または商業化しているが,再許可を付与したいと考え,第三者の名称を開示する義務があり,第三者と再許可協定を締結する可能性がある。もし私たちが私たちのいかなる付属会社や分譲許可者を直接または間接的に通過しなければ、Cas 9の被許可者製品を研究、開発、商業化しなければ、遠大でハーバードを満足させる計画を制定し、実施することができなかったり、第三者と分許可合意を達成することができなかったりする。遠大かつハーバードは、特定の第三者標的または特定のカテゴリに対する私たちの権利を終了する権利があり、(A)特定の第三者標的について当社が独占的または共同独占的に許可する特許権に基づいて、または(B)当特定のカテゴリで独占的または共同独占的に許可されている特許権に基づいて、無料で第三者に許可を付与する権利があり、このような許可が心血管疾患分野のための製品を商業化する権利を付与しないことを前提としている。
44
支払条件
Cas 9ライセンス契約に基づき,遠大とハーバードに10万ドルの前払い許可料を支払い,遠大とハーバードに合計138,037株の普通株を発行した。遠大とハーバードも反償却権利を持っており、これにより、著者ら(I)は優先株融資を完成した後、すでに遠大とハーバードに計309,278株の普通株を発行し、及び(Ii)は初めての公開募集を完成した後、すでに遠大とハーバードに878,098株の普通株を追加発行した。
毎年のライセンス維持費も支払わなければなりません。例年によっては低いものから中五桁まで様々です。この年度のライセンス維持費の一部は、維持費の支払いと同じ年に許可または有効化された製品に対して不足している印税を相殺することができる。
ブロードとハーバードの合計は、(I)米国、EU、および日本において、米国で一定数未満の患者のヒト疾患の臨床および規制マイルストーン支払いを予防または治療するための権利があり、各特許製品は最大570万ドルに達する;(Ii)米国、EUおよび日本では、各特許製品の臨床および規制マイルストーン支払いは、合計1740万ドルに達し、米国で少なくとも一定数の患者を悩ませるヒト疾患の予防または治療に使用される。もし私たちがCas 9許可協定中に統制権変更が発生すれば、これらの臨床と規制マイルストーン支払いのいくつかは一定の割合を増加させるだろう。各ライセンス製品にいくつかの販売ベースのマイルストーンが発生した場合、ブロドとハーバードに合計5400万ドルの追加金を支払う義務があります。
もし私たちの平均時価が指定されたハードルを超えて、9桁の高いドルの金額から100億ドルに上昇したり、時価が成功して支払いに成功したり、これらの閾値を超えたことを犠牲にして私たちの会社を売却したり、会社が成功した支払いを販売して、時価成功支払いと一緒に、成功支払いと呼ばれ、私たちは遠大とハーバードに等級別成功支払いを支払う義務もあります。時価成功支払いは吾らが現金、普通株または普通株の形で支払うことを目的としており、この目的では、吾らの普通株が当時ナスダック証券市場に上場していた場合、その株式の推定値は、吾等の普通株が支払日直前の取引日にナスダック証券市場で発表された終値となる。当社の支配権が変更されたり当社が売却されたりした場合、私等は事件発生後指定期間内に関連会社の販売成功金を現金で支払う必要があります。Success支払いは累積的であり,任意のトリガ日の平均時価によって,複数のSuccess支払いが満期になって支払う可能性がある.私たちが支払うことができる最高成功支払総額は3130万ドルです。いくつかの成功した支払いは、許可された製品が経過した場合、または臨床試験で評価された場合にのみ支払うことができる。私たちがこのような成功した支払いを満たすために普通株を発行する限り、私たちはこのような株を登録し、ハーバードで転売するために、アメリカ証券取引委員会に登録声明を提出する義務がある。
2021年9月、関連測定日までに、私たちの平均時価は3つの指定されたハードルを超えました。Cas 9許可協定によると、合計約630万ドルの支払い成功を支払い、2021年11月に現金で決済します。
ブロイドとハーバードの合計は、ライセンス製品の純売上高から中一桁パーセントの印税を得る権利があり、私たち、私たちの付属会社、または私たちの分被許可者が製造したライセンスサポートの他の製品の純売上高からより低い一桁パーセント印税を得る権利があります。印税率は許可または有効化された製品の純売上高の合計に依存する。第三者がライセンス製品をカバーする特許権を持っているので、ライセンス製品の純売上高で第三者に印税を支払うことを法律で要求された場合、私たちは、同時期にブロドとハーバードに支払われるべき印税を相殺するために、第三者に支払う金額の特定の割合をクレジットすることができる。目標ごとに、Editasが開始した計画がハーバード/遠大Cas特許権がカバーする技術を使用し、そのうちの1つの目標に対して、特定の目標のマイルストーンおよび印税支払いを一定のパーセント減少させるべきである。我々の印税支払い義務は、以下の遅い時間で製品および国/地域で終了する:(I)ハーバード/遠大Cas 9特許権の有効な権利は、各国/地域の組成、製造または使用の最後の終了日、または(Ii)許可または有効な製品の初商業販売の日の10周年を主張する。もし私たちがハーバード/遠大Cas 9特許権のいずれかの再許可を第三者に再許可すれば、遠大とハーバードは共同で10%から20%の再許可収入を得る権利があり、著者らはある臨床マイルストーンに達した後、この割合は比較的に高い一桁に下げるべきである。
45
条文を検察·強制執行する
ブロドとハーバードはそれぞれの特許権に対する起訴統制権を保持している。米国特許商標局のいかなる妨害手続き、欧州特許庁のいかなる反対手続、または任意の他の手続に関連する費用を含む、ハーバード/ブロイドCas 9特許権の起訴および維持に関連するいくつかの費用をブロッドおよびハーバードに返済する義務がある各方面間あるいは私たちは特許保護のこれらや他の管轄区域の他の許可された手続きを求めている。遠大とハーバードはハーバード/遠大特許権の範囲内でいかなる申請或いは特許を維持しなければならず、私たちが遠大とハーバードに起訴に関連する費用を返済する義務を履行すれば、このようにすることは善意の基礎があり、しかもこのようにすることは遠大或いはハーバードの特許起訴戦略に符合する。もし私たちがどのハーバード/遠大Cas 9特許権を起訴する費用を支払うことを停止すれば、私たちに付与されたハーバード/遠大Cas 9特許権に関するいかなる許可も終了する。
ある条件を満たせば、私たちは第一の権利がありますが、義務はありません。私たちの許可製品に対してハーバード/遠大Cas 9-I特許権を強制的に執行し、例えば遠大とハーバードに証拠を提供し、第三者に対する訴訟の善意の基礎を証明し、Editasと協調します。私たちは私たちが始めたいかなる訴訟の費用も一人で負担して、ブロイドとハーバード(およびマサチューセッツ工科大学、ロックフェラー、アイオワ州、適用されれば)の事前書面同意がなければ、私たちは和解を達成することができません。このような訴訟で戻ってきたどんなお金も私たち、ブロドとハーバードの間で共有されるだろう。
終了条項
事前に終了しない限り、Cas 9許可プロトコルの有効期限は、ハーバード/Cas 9特許権の最後の有効主張が満了した時点で満了する。しかし、私たちの特許使用料とマイルストーン支払い義務は、上述したように、満期または終了後に引き続き存在する可能性がある。私たちは4ヶ月前にブロダーとハーバードに書面で通知した後、勝手に合意を終わらせる権利がある。私たちまたは遠大およびハーバードは、反対側の治癒されていない実質的な違約の特定の通知期間内に合意を終了することができ、この通知期間は違約の性質によって異なる。もし私たちまたは私たちの関連会社または再許可者が私たちの能力の範囲内でいかなるハーバード/遠大特許権の実行可能性、有効性または範囲に疑問を提起したり、第三者がそうすることを協力したり、あるいは私たちが破産したり、借金をしない場合、遠大とハーバードは直ちにCAS 9許可協定を終了することができる。ブロイドとハーバードの単独行動はCas 9許可協定を終わらせる権利がない。しかし,遠大とハーバードでは,両機関が共同でCas 9許可協定を終了するような同じ事件が発生した場合には,それぞれの特許権に関する我々の許可を終了する可能性がある.
Vertexとの連携と許可合意
2022年7月18日、我々はVertexと戦略連携と許可協定、あるいはVertex連携協定と呼ばれ、4年間の独占的なグローバル研究協力を行い、開発に重点を置いている体内にある遺伝子編集は単一肝疾患を治療する未開示目標になることが期待される。また,2022年7月18日にVertexと株式購入プロトコルを締結し,この合意に基づき,私募でVertexに普通株を売却·発行することに同意した。
Vertex協力協定によると、新しい薬物の発見、研究、いくつかの臨床前開発を担当します体内にある遺伝子編集開発は人々が興味を持つ目標である.我々の研究活動は(I)特定の遺伝子編集システムの認識と設計に集中する体内にある目標に対する配信システム,および(Ii)候補開発を評価·最適化し,Vertex連携プロトコルで規定されている標準を実現する.Vertexは合意された予算に従って私たちの研究費用を精算するだろう。研究期間の初期期間は4年であり,頂点会社はたかだか1年延長することができる.
Vertexは独自に我々の研究で生成された任意の候補製品の後続開発,製造,商業化を担当する.私たちは2022年7月20日にVertexの2500万ドルの前払いを受けた。私たちは(I)開発基準を適用した候補製品あたり最高2200万ドルの成功支払い(最高6600万ドル)と(Ii)の合計3億4千万ドルの開発と商業マイルストーン支払いを得る資格がある。純売上高別の一桁特許権使用料を得る資格もありますが、指定された減額で計算しなければなりません。このような使用料の支払いは、(I)製品をカバーする特許権に基づいて、その国/地域の最後の有効な特許請求に基づいて満了した場合、(Ii)当該製品が国/地域に関連する法規に従って排他的期間、または(Iii)当該製品が当該国/地域で初めて商業販売された10年後に発生する場合に終了する。
Vertex連携プロトコルに従って開発された第1の候補製品の第1の段階の臨床試験の第1の患者用量の前に、私たちはまた、利益共有スケジュールに参加することを選択する権利がある
46
私たちはVertexと協力から生成されたすべての候補製品のコストと純利益を共有する。マイルストーンや特許権使用料ではなく、私たちの選択権を行使すれば、特定の割合の開発·商業化コストを支払う義務があり、任意の協力の下で進められる候補製品の販売には、指定されたパーセントの利益を得る権利があります。我々が選択権を行使する際には,40%までの利益/コストシェア(Vertexは少なくとも60%保持)を選択することができる.私たちの選択加入権利を行使するためには、私たちが選択した利益/コストパーセントと、Verveが権利加入を選択する際に、最先端の候補製品に含まれるVerve許可技術に応じて、2,500~7,000万ドルの様々な費用を支払う必要があります。すべての利益共有スキームの下で、Vertexは、協力によって生成される任意の候補製品の世界的な開発および商業化を制御する。
Vertex協力協定には、このような取引の慣行陳述と保証、契約、および賠償義務が含まれている。我々とVertexはそれぞれ通知を出した後に他方の実質的な違約や破産のプロトコルを終了する権利があり,適用すればプロトコルを終了する権利がある.便宜上,Vertexは90日の通知後にVertex連携プロトコルのすべてを終了することも可能である.
Vertex連携プロトコルの実行については,吾らもVertexと株式購入プロトコルを締結し,私募でVertexに1,519,756株自社普通株を売却·発行し,1株当たり23.03ドルであり,2022年7月15日までの5日出来高加重平均株価に相当し,総購入価格は3,500万ドルであった。今回の私募は2022年7月20日に完了した。
政府の監督管理
その他の事項以外に、アメリカ連邦、州と地方各級及び欧州連合を含む他の国と司法管轄区の政府当局は生物製品を含む薬品の研究、開発、テスト、製造、定価、精算、販売、品質管理、承認、包装、貯蔵、記録保存、ラベル、広告、販売促進、流通、マーケティング、承認後の監視と報告及び輸出入などの方面に対して広範な監督管理を行った。米国や他の国や司法管轄区で上場承認を得る過程、その後適用される法規や法規、その他の規制機関の遵守には、多大な時間と財力が必要である。
アメリカの生物製品の許可と規制
米国では,我々が開発可能な任意の候補製品は,生物製品または生物製品として“公衆衛生サービス法”(Public Health Service Act,PHSAと略す)および“連邦食品,薬物および化粧品法”(Federal Food,Drug and Cosmetic Act,FDCAと略す)およびその実施条例およびガイドラインの規制を受けるであろう。製品開発過程中の任意の場合、臨床前試験、臨床試験、承認過程または承認後過程を含み、適用される米国の要求を遵守できない場合、スポンサーは研究、監督審査および承認および/または行政または司法制裁を行う上で遅延を受ける可能性がある。
FDAはアメリカで発売されるために、治療適応の候補製品を承認しなければならない。 このような製品の臨床開発計画の発起·管理を担当する会社,機関,あるいは組織をスポンサーと呼ぶ。米国での新生物製品の販売と流通の承認を求めるスポンサーは、以下の各ステップを満足的に達成しなければならない
47
臨床前研究と研究性新薬の応用
人体で任意の生物候補製品をテストする前に、候補製品は臨床前テストを経なければならない。臨床前試験は製品の化学、調合と安定性の実験室評価、及び動物実験において治療効果と毒性潜在力を評価する研究を含む。これらの研究は一般にINDを支援する研究と呼ばれる.臨床前試験の進行および試験に用いる化合物の配合は、GLP規制および基準、米国農務省の動物福祉法(適用される場合)を含む連邦法規および要求に適合しなければならない。臨床前試験の結果および生産情報と分析データはIND申請の一部としてFDAに提出された。
INDはFDCAの免除であり、未承認候補製品が州間商業で臨床研究のために輸送されることを許可し、FDAにこのような研究製品をヒトに使用することを許可することを要求する。このような許可は、承認されていない任意の新薬出願またはNDAの候補製品を州間輸送および管理する前に取得されなければならない。INDの申請を支援するためには、スポンサーは各臨床試験にプログラムを提出しなければならず、任意の後続のプログラム修正はINDの一部としてFDAに提出されなければならない。INDはFDAが受領した30日後に自動的に発効し、それ以前にFDAが提案された臨床試験の製品または懸念または問題を提起しなければ、人体研究対象が不合理な健康リスクに直面することを懸念することを含む。この場合、INDスポンサーおよびFDAは、臨床試験が開始または再開される前に、FDAの任意の未解決の問題を解決しなければならない。
IND下での臨床試験開始後,FDAもこの試験を臨床放置あるいは一部の臨床放置を実施することができる。臨床保留はFDAがスポンサーに発表した命令であり,提案された臨床研究の延期や進行中の研究の一時停止が要求されている。一部の臨床保留はIND要求の一部の臨床仕事を遅延或いは一時停止することである。例えば、一部の臨床的保留は、特定のプロトコルまたはプロトコルの一部が継続できないことを宣言する可能性があり、プロトコルの他の部分または他のプロトコルはそうすることができる。臨床保留或いは一部の臨床保留を実施した後30日を超えない後、FDAはスポンサーに棚上げ根拠に関する書面解釈を提供する。臨床保留あるいは一部の臨床保留を発表した後、FDAがスポンサー調査が継続可能であることを通知した後にのみ、臨床調査を回復することができる。FDAは、スポンサーによって提供された情報に基づいて、調査が継続または再開可能であることを決定し、これらの情報は、上述した欠陥を修正するか、または他の方法でFDAを満足させるであろう。臨床研究被験者に安全問題の製造問題をもたらす可能性があるため,臨床休止を実施することがある。
スポンサーは選択可能であるが,必要ではなく,IND下で海外臨床研究を行っている。ある国外の臨床研究がIND下で行われる時、FDAが放棄しない限り、すべてのIND要求を満たさなければならない。外国の臨床研究がINDの下で行われていない場合、スポンサーは、この研究をINDまたは上場承認申請の支持として使用するために、この研究がFDAのいくつかの法規要件に適合することを確実にしなければならない。具体的には,独立倫理委員会の審査·承認を受けること,被験者のインフォームドコンセントを求めて受けることなど,GCPに従って行わなければならない。GCPは臨床研究の倫理とデータ完全性基準を含むことが要求される。FDAの規定は、非IND外国臨床研究に参加したヒト被験者の保護、及び結果データの質と完全性を確保することを目的としている。
48
また,米国国立衛生研究院(NIH)組換えDNA研究資金を受けた機関で行われた遺伝薬物臨床試験は,NIH科学政策オフィス内の新型特殊技術·研究諮問委員会(NExTRAC)という委員会の審査を受ける可能性もある。2019年現在、この審査チームの規約は、標準監督機関が評価できず、異常リスクを構成する臨床試験に公衆審査の重点を置くようになっている。ある遺伝的薬物プロトコルに対して,NExTRACがこのプロトコルを包括的な公共審査を行う必要があると決定した場合,FDAはINDの審査や承認を延期する可能性がある。
臨床試験結果を報告する
PHSAによれば、処方薬および生物学的製品を含むいくつかのFDA規制製品の臨床試験のスポンサーは、NIHによって維持されている共通レジストリ(Clinicaltrials.gov)に登録され、特定の臨床試験情報を開示しなければならない。特に,臨床試験登録の一部として,臨床試験の製品,患者群,調査段階,研究場所,調査者,その他に関する情報が公開されている。スポンサーも試験完了後に臨床試験結果を開示する義務があるが,場合によっては結果の開示が試験完了日後2年に延期される可能性がある。NIHの臨床試験登録と報告要求に関する最終規則は2017年に発効し,NIHとFDAは最近,政府が規定に適合しない臨床試験スポンサーに対してこれらの要求を実行し始めることを希望していることを示している。
具体的には,PHSAは米国衛生·公衆サービス部(HHS)部長に臨床試験情報の要求に応じて提出できなかったため,責任者に規定を遵守しない通知を行う権限を付与した。しかし、責任者は、規定を遵守しないことを是正し、必要な情報を提出する30日の時間がある。Clinicaltrials.govに臨床試験情報を提出しないこともFDCAが禁止した行為であり,違反行為は1日10,000ドルまでの民事罰金を受け続ける可能性がある。民事罰金に加えて、違反は禁止および/または刑事起訴や連邦支出資格の取り消しなど、他の規制行動を招く可能性がある。HHSの発表最終実施規定の長期遅延により,FDAは従来これらの報告要求を実行していなかったが,これらの規定は現在発表されており,FDAは2021年4月以降,規定に適合しないいくつかの通知を発表している。
治療のための薬を研究する機会を広げる
使用を拡大することは、“同情的使用”と呼ばれることがあり、臨床試験以外に研究製品を使用し、比較可能または満足できる代替治療案がない場合には、重篤または直ちに生命を脅かす疾患または条件を有する患者を治療する。参入拡大に関する規則や条例は,研究療法の恩恵を受ける可能性のある患者が研究製品を獲得する機会を改善することを目的としている。FDAの法規は、個別患者(緊急時および非緊急時に治療された単一患者IND申請)、中規模の患者集団、および治療レジメンまたは治療INDに従って研究製品の使用を申請したより大きな集団のために、会社または治療医がINDの研究製品を治療目的で使用することを可能にする。
患者または患者のグループを治療するために研究製品を使用するIND申請を拡大することを考慮すると、スポンサーおよび治療医または調査者は、患者が深刻または直ちに生命を脅かす疾患または状態を有し、疾患または状態を診断、監視または治療するための類似または満足できる代替療法がなく、潜在的な患者利益が治療の潜在的リスクが合理的であり、治療すべき背景または条件の下で不合理ではないことを証明するすべての基準が適用される場合に適切であるかどうかを決定するであろう。要求された治療に対して、研究薬物の使用を拡大することは、製品の発売承認を支持する可能性のある臨床研究の起動、進行または完成、または他の方法で製品の潜在的開発を損害する可能性がある。
スポンサーはその薬品をより多くの人に提供する義務はないが、2016年に採択された“21世紀治療法”の要求に基づいて、スポンサーが獲得拡大の要請にどのように対応するかに関する政策があれば、この政策を公開しなければならない。スポンサーは、2期または3期試験の開始が早いとき、または研究薬または生物学的薬剤が突破的療法、迅速チャネル製品または再生医学高度療法として指定された後15日以内にこのような政策を開示しなければならない。
また、2018年5月30日には、“裁判権法案”が法律に署名された。他の事項に加えて、この法律は、ある患者に連邦フレームワークを提供し、彼らが第1段階の臨床試験を完了し、FDAの承認を得るために調査を行っているいくつかの研究製品を使用することを可能にする。場合によっては条件を満たした患者は
49
臨床試験への参加やFDA拡大参入計画によりFDA許可を得ることなく治療を求めることができる。“試用権法案”によると,メーカーはその研究製品を条件に適合した患者に提供する義務はない。
血中乳酸を支持するヒト臨床試験
臨床試験はGCP要求に基づいて、合格した首席研究者の監督の下で、研究製品候補を健康ボランティア或いは疾病或いは状況を有する患者に治療することに関連している。臨床試験は,試験目標を詳細に説明し,基準の組み入れと排除,安全性をモニタリングするためのパラメータおよび評価する有効性基準のプロトコルの下で行った。INDの一部として,各臨床試験の案と任意の後続の案修正案をFDAに提出しなければならない。
米国国外での臨床試験のスポンサーが望ましいが,必要なくFDAの認可を得,INDによる臨床試験を行っている。IND下で外国臨床試験を行う場合,放棄しない限り,FDAのすべてのIND要求を満たさなければならない。外国の臨床試験がINDの下で行われない場合、スポンサーは試験をIND或いは上場承認申請の支持として使用するために、試験がFDAのある規制要求に適合することを保証しなければならない。具体的には、FDAは、独立した倫理委員会によって審査·承認され、参加者のインフォームドコンセントを得ることを含むGCPに従って行わなければならないことを要求している。GCPは臨床試験の倫理とデータ完全性基準を含むことが要求される。FDAの規定は非IND外国臨床試験に参加した人体被験者の保護、及び結果データの品質と完全性の確保を助けることを目的としている。これらは,非IND外国試験の進め方の確保にも寄与しており,米国の臨床試験に必要な方式と同等である。
また,各臨床試験は,臨床試験を行う各機関のIRBが集中的または単独で審査·承認されなければならない。委員会が考慮する事項は,臨床試験設計,患者インフォームドコンセント,倫理的要因,被験者の安全,および機関が担う可能性のある責任である。IRBの運営はFDAの規定に適合しなければならない。FDA、IRB或いは臨床試験スポンサーは随時各種の原因で臨床試験を一時停止或いは中止することができ、臨床試験がFDAの要求に従って行われていないことを発見すること、或いは参加者が受け入れられない健康リスクに直面していることを含む。臨床検査はまた広範なGCP規則とインフォームドコンセントの要求を満たさなければならない。
また、いくつかの臨床試験は臨床試験スポンサーによって組織された独立した合格専門家グループによって監督され、このグループはデータ安全監視委員会、あるいはDSMBと呼ばれる。このグループは,計画的に実験を継続し,実験を変更して行ったり,実験の何らかの利用可能なデータに基づいて指定されたチェックポイントで実験を停止したりすることが提案されており,これらのデータはdsmbのみで得られる.
臨床試験は通常3つの連続段階に分けて行われるが、これらの段階は重複或いは合併する可能性がある。承認された後に追加的な研究が必要かもしれない。
1つの臨床試験は複数の段階の要素を結合する可能性があるが、FDAは通常、候補製品の発売承認を支持するために複数の第3段階試験を必要とする。ある会社が臨床試験を特定の段階に指定することは、プログラムおよびデータが完成するまで決定できないので、この研究がこの段階に対するFDAの要求を満たすのに十分であることを必ずしも示しているわけではない
50
FDAに提出され、FDAによって検討される。さらに、上述したように、キー試験は、候補製品の安全性および有効性に対するFDAの評価要件を満たすと考えられ、規制承認を支援するために、他のキー試験または非キー試験と共に使用することができる。一般的に、重要な試験は3期試験であるが、設計が臨床利益の良好な制御と信頼できる評価を提供すれば、特に医療需要を満たしていない領域では、それらは2期試験である可能性がある。
場合によっては、FDAは製品のBLAを承認する可能性があるが、承認後に製品の安全性および有効性をさらに評価するためにスポンサーに追加の臨床試験を行うことが要求される。この承認後の試験は通常4期臨床試験と呼ばれる。これらの研究は、予期される治療適応患者の治療から追加の経験を得るために使用され、加速承認条例によって承認された生物学的製品の場合に臨床的利益を証明するために使用される。FDAが製品を承認し、ある会社が承認を必要としない臨床試験を行っている場合、会社は、これらの臨床試験のデータを使用して、任意の4期の臨床試験のすべてまたは一部の要件を満たすか、または製品ラベルの変更を要求することができる。4期臨床試験を行った職務調査を行わなければ,製品の承認撤回につながる可能性がある。
2022年12月、食品·薬物総合改革法案(FDORA)の成立に伴い、国会は、各新生物製品の第3段階臨床試験または任意の他の“重要な研究”のための多様な行動計画の制定と提出をスポンサーに要求した。これらの計画はより多くの異なる患者群がFDA監督製品の後期臨床試験に参加することを奨励することを目的としている。具体的には,行動計画には,スポンサーの募集目標,これらの目標の基本原理,スポンサーがこれらの目標をどのように実現しようとしているかの解釈が含まれなければならない。このような要求に加えて、立法はFDAに多様な行動計画に関する新しいガイドラインを発表するように指示した。
臨床開発計画中のFDAとの相互作用
INDが承認され臨床試験を開始した後,スポンサーはFDAとの相互作用を継続する。臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出しなければならず,深刻な有害事象が発生すればより頻繁に提出される。さらに、IND安全報告は、深刻かつ予期しない疑わしい副作用、他の研究または動物またはインビトロ試験からの結果、製品に曝露された人体に重大なリスクがあることを示し、レジメンまたは研究者マニュアルに記載されている場合と比較して、任意の臨床的に深刻な疑わしい副作用の発生が増加することを示すFDAに提出されなければならない。第1段階、第2段階、および第3段階の臨床試験は、任意の指定された時間内に成功しないか、または全く成功しない可能性がある。マーケティング申請をサポートするために臨床データが提出されるとき、FDAは、通常、GCPおよび提出された臨床データの完全性を保証するために、1つまたは複数の臨床サイトを検査する。
また,スポンサーは臨床開発計画のあるときにFDAと会う機会がある。具体的には、スポンサーは、INDまたはINDを提出する前に会議を申請し、2期臨床試験またはEP 2会議の終了時に、NDAまたはBLAを提出する前、またはNDAの前またはBLAの前にFDAと面会することができる。他の時間に会議を開催することを要求することもできる。スポンサーとFDAの間には4つの種類の会議がある。Aクラス会議は,もともと停滞していた製品開発計画を継続したり,重要なセキュリティ問題を解決したりするために必要な会議である.クラスB会議には,IND申請前会議とNDA/BLA前会議,EOP 2会議のようなB段階終了会議がある.クラスC会議とは,AクラスまたはBクラス会議以外のいずれかの製品開発やレビューに関する会議である.最後に、D類会議は一連の狭い問題(2つの重点議題を超えないことに限定されるべき)に重点を置き、3つの学科あるいは分部を超える投入を要求すべきではない。
これらの会議はスポンサーにこれまで収集してきたデータの情報をFDAと共有する機会を提供し,FDAに次の段階の開発に関する提案を提供した。例えば,EOP 2会議では,スポンサーはその第2段階の臨床結果を検討し,新製品の承認を支援すると考えられる重要な第3段階臨床試験計画を提出することができる。この会議は自ら行うことができ,電話会議/ビデオ会議や書面回答で行うことができ,議事録はスポンサーからFDAへの質問とFDAからの回答のみを反映している.FDAは、議事録および諮問書簡で伝達された応答は、スポンサーに対する提案および/または提案のみを構成するため、スポンサーは、そのような提案および/または提案の制約を受けないと述べている。しかし,実践的には,スポンサーがFDAの提案に沿って臨床計画を設計していないことは,その計画を大きな失敗リスクに直面させる可能性がある。
51
小児科研究
2003年の“小児科研究公平法”またはPREAによれば、BLAまたはそのサプリメントは、すべての関連する小児科亜集団において主張される適応の安全性および有効性を評価するのに十分なデータを含み、安全で有効な各小児科亜群に対する製品の用量および投与をサポートしなければならない。スポンサーは、第2段階会議終了後60日以内に、またはスポンサーとFDAとの間で合意した場合に、初歩的な小児科研究計画を提出しなければならない。スポンサーはまたデータを評価する前に小児科研究計画を提出しなければならない。これらの計画は、研究目標および設計、任意の延期または免除要求、および法規要件の他の情報を含む、提案された1つまたは複数の小児科研究の大綱、またはスポンサー計画による研究を含まなければならない。スポンサー、FDA、FDAの内部審査委員会はその後、提出された情報を審査し、協議し、最終計画について合意しなければならない。FDAやスポンサーはいつでも計画の修正を要求することができる。
深刻な或いは生命に危害を及ぼす疾病或いは状況を治療することを目的とした研究製品に対して、FDAはスポンサーの要求に応じて会議を開催し、初期小児科研究計画の準備或いは討論を討論し、小児科評価を延期或いは放棄しなければならない。また,FDAは開発過程の早期に会議を開催し,小児科研究計画をスポンサーと検討し,FDAは重篤あるいは生命に危険な疾患よりも遅くない第1段階会議が終了する前と,FDAが研究計画を受け取ってから90日後にスポンサーと面会しなければならない。
FDAは、成人のために製品が使用されるか、または小児科データ要件を完全にまたは部分的に免除するまで、スポンサーの要求に能動的にまたは対応することができ、承認は、小児科データの一部または全部の提出を延期することができる。延期は、小児科試験が完了する前に、製品または候補治療薬が成人で使用を許可する準備ができていることを発見するか、または小児科試験が開始される前に追加の安全性または有効性データを収集する必要があることを含むいくつかの理由があるかもしれない。現在、法律は、FDAが、PREA要求を提出できなかった小児科評価、延期または延期を求めることができなかったか、または必要な小児科処方の承認を要求できなかったスポンサーにPREA不適合書簡を送信することを要求する。それはさらにFDAにPREAコンプライアンスとスポンサーの反応を公開することを要求する。
規制が別途要求されない限り、小児科データ要件は、孤児として指定された製品には適用されないが、FDAは最近、PREAにおいてこの法定免除を乱用すると考えられる行為を制限する措置を講じているにもかかわらず、まれな小児科亜群に追加的な孤児薬物指定を付与することを意図していないことを宣言し、一般的な疾患である。FDAはまた、児童人口中の疾病罹患率が比較的に低いため、PREA要求を免除する疾病リストを保留した。
遺伝子治療製品に関する特別規定とガイドライン
遺伝子治療製品に適用されるプログラムや基準は,我々が開発可能な任意の候補製品に適用されると予想される。FDAは、遺伝子治療製品を、治療のために遺伝子発現の修正または操作または生細胞の生物学的特性の変化を求める製品として定義する。この製品は細胞を修飾するために使うことができる体内にある患者に投与する前に体外で細胞に転移するかもしれません
FDA内部では、生物製品の評価と研究センター(CBER)が遺伝子治療製品の監督管理を担当している。CBER内部では,組織と高度療法室が遺伝子療法と関連製品の審査を統合し,FDAは細胞,組織,遺伝子療法諮問委員会を設置し,その審査についてCBERにアドバイスを提供している。NIHは,NExTRACを含め,遺伝子治療問題や他の新興バイオテクノロジーに関する問題についてFDAに提案している。FDAとNIHは遺伝子治療案の開発と提出に関するガイドラインを発表している。
FDAは2020年1月に発表された最終指導文書、遺伝子治療INDの化学、製造と制御情報、遺伝子治療製品を用いた後の長期フォローアップ、稀な疾患の遺伝子治療と網膜疾患の遺伝子治療、及び2022年10月に発表されたヒト神経変性疾患遺伝子治療の最終指導文書を含む各種の遺伝子治療に関する指導文書を発表した。FDAは、これらは以前に発表された他の指導文書と法的拘束力がないことを示しているが、これらの文書を遵守することは、任意の遺伝子治療製品候補承認を得るための必要条件である可能性がある。ガイドラインは、FDAが上述した各開発段階で考慮する他の要因を提供し、適切な遺伝子療法の臨床前評価、INDアプリケーションに含まれるべき化学、製造、および制御情報、INDまたはBLAアプリケーションをサポートする製品の効力を測定するための適切な設計テスト、およびそのような影響の潜在的リスクに基づいて、研究遺伝子治療を受けた参加者の潜在的遅延副作用を観察するための措置に関する。特にAAVキャリアのためにFDAは
52
通常、スポンサーは参加者の潜在的な遺伝子治療関連不良事件を5年間監視し続けることを提案している。他のタイプの遺伝子治療或いは遺伝子編集製品はもっと長いフォローアップ時間を必要とする可能性があり、最長15年に達する可能性がある。
CGMPの要件に合致する
臨床試験と同時に、会社は通常追加の臨床前研究を完成しなければならず、候補生物製品の物理的特徴に関する追加情報を開発し、cGMP要求に基づいて最終的に商業大量生産候補製品の技術を決定しなければならない。BLAを承認する前に、FDAは通常、製品を生産する1つまたは複数の施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に完全に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分であると判断しない限り、申請を承認しないであろう。PHSAは,外来製剤の導入や生物製品の使用による他の有害事象のリスク低減を支援するために,属性が正確に定義できない製品の製造制御の重要性を強調している。製造過程は一貫して高品質の候補製品ロットを生産することができなければならず、他の要求以外に、スポンサーは最終生物製品の特性、強度、品質、効力と純度をテストする方法を開発しなければならない。また,適切な包装を選択·試験し,候補生物製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
製造業者や他の製品の製造と流通に参加する人たちもまたFDAと特定の州機関に彼らの工場を登録しなければならない。国内でも海外の製造企業でも,最初に生産過程に参加する際には,FDAに登録して追加的な情報を提供しなければならない。登録されていない工場で製造または輸入されたどの製品も、外国でも国内でも、FDCAの下に誤ったブランドが貼られているとみなされている。機関はcGMPや他の法律の遵守を確保するために、政府当局の定期的な抜き打ち検査を受ける可能性がある。2022年12月に公布された“大流行病予防法”は、1種の薬物または生物が米国に輸入または提供される前に、米国国外の別の機関でさらなる製造、製造、繁殖、複合または加工を行っても、外国の薬品製造機関は登録と発売要求を守らなければならないことを明らかにした。検査は“リスクに基づくスケジュール”に従わなければならないが、これはいくつかの機関がより頻繁に検査されることを招く可能性がある。メーカーはまた、その工場に関する電子または実物記録の提供を要求しなければならないかもしれない。遅延、拒否、制限、またはFDAの検査拒否は、製品が偽とみなされる可能性があります.
製造業を管理する規制要件
FDAの規定では,薬品は特定の承認施設で生産され,cGMPに適合しなければならないことが求められている。CGMP条例には、人員、建物および施設、設備、アセンブリおよび薬品容器および閉鎖的な制御、生産およびプロセス制御、包装およびラベル制御、保有および分配、実験室制御、記録および報告、ならびに返品または回収された製品に関する要件が含まれる。承認された医薬品の生産および流通に参加するメーカーおよび他のエンティティは、FDAおよびいくつかの州機関にその工場を登録し、cGMPおよび他の要求を遵守することを確実にするために、FDAの定期的な抜き打ち検査を受けなければならない。検査は“リスクに基づくスケジュール”に従わなければならないが、これはいくつかの機関がより頻繁に検査されることを招く可能性がある。メーカーはまた、その工場に関する電子または実物記録の提供を要求しなければならないかもしれない。FDAの検査の延期、拒否、制限、または拒否は、製品が偽とみなされる可能性がある。承認された製品の製造プロセス,仕様や容器閉鎖システムの変更は厳しく規制されており,通常はFDAの承認を得て実施する必要がある。FDAの法規はまた、cGMPとの任意の偏差を調査および是正し、NDAスポンサーおよび承認製品の生産に参加する任意の第三者製造業者に報告および文書要求を提出することを要求する。
BLAの受け入れと審査
必要な臨床試験,臨床前研究および臨床試験の結果,および製品の化学,製造,制御,安全更新,特許情報,乱用情報および提案されたラベルに関する情報が,出願の一部としてFDAに提出され,候補製品を1つまたは複数の適応に押し出すことの承認を要求すると仮定する。データは、製品使用の安全性および有効性を試験するために、または研究者によって開始された研究を含む多くの代替源からの臨床試験からのものである可能性がある。上場承認を支持するために、提出されたデータは品質と数量で薬物製品の安全性と有効性、および生物製品の安全性、効力と純度を決定し、FDAを満足させるのに十分でなければならない。書類の提出と審査にかかる費用
53
PDUFAでの出願金額は大きく(例えば、2023年度の申請料は約330万ドル)、承認されたNDAのスポンサーは年間計画費を支払う必要があり、2023年度の合格処方薬1個あたりの年会費は394,000ドルを超える。これらの費用は、一般に年に1回調整され、孤児薬指定を有する製品は、申請料を免除することができ、場合によっては、公衆の健康を保護するために必要な免除が必要である場合があり、費用は、革新に重大な障害を構成するか、またはスポンサーは、審査のためにその最初の人間治療申請を提出する小規模企業である。
FDAは、すべての出願を受信してから60日以内にすべての出願を予備審査し、実質的な審査のためにスポンサー申請が十分に完全であるかどうかをその時点で通知しなければならない。関連部分では、FDAの法規は、FDAがすべての関連情報およびデータを受信する前に、申請は提出されたとみなされてはならないと規定している。FDAが申請がこの基準を満たしていないと判断した場合、それはスポンサーに提出拒否またはRTF決定を発行する。一般に、技術移転フレームワークの根拠は、明らかに情報または必要な情報の部分を見落としているような行政上の不完全さであり、安全性、純度および有効性の評価を見落としたり、説明を適切に使用するために必要なキーデータ、情報または分析を提供するなど、科学的な不完全さ、または情報の内容、紹介または組織が不十分であり、実質的かつ有意義な審査を行うことができない。FDAは申請を受け入れるのではなく、より多くの情報を提供することを要求するかもしれない。この場合、申請は追加情報と共に再提出されなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。
出願が受け入れられた後、FDAは出願の深い実質的な審査を開始した。FDAは、提案された製品がその予期される用途に対して安全に有効であるかどうか、許容可能な純度プロファイルを有するかどうか、および製品がcGMPに従って製造されているかどうかを決定するために出願を審査する。FDAがPDUFAで合意した目標と政策によると、FDAは10ヶ月間、新しい分子実体としての標準出願の予備審査を完了し、“優先審査”を有する出願については6ヶ月の期間がある。FDAは、新しい情報を考慮するために、またはスポンサーが明確な提供を提供する場合に、FDAが最初の提出後に発見した突出した欠陥を解決するために、審査プロセスをさらに3ヶ月延長することができる。これらの審査目標にもかかわらず,FDAによる申請の審査がPDUFA目標日を超えることは珍しくない。
審査申請の過程で、FDAは通常、スポンサーに情報要求を提出し、回答の最終期限を設定する。FDAはまた,新製品の製造施設の承認前検査を行い,製造プロセスや施設がcGMPに適合しているかどうかを確認する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、この製品を承認しないであろう。
FDAはまた、IND申請およびGCP要件に適合することを保証し、FDAに提出された臨床データの完全性を保証するために、スポンサーおよび1つまたは複数の臨床試験場所を検査することができる。FDORAの通過に伴い,国会はFDAに提出された臨床や非臨床研究の準備,進行や分析に係る施設,研究記録を持ったり研究過程に参加したりする他の人の検査を明確に許可し,FDAが検査を行う権限を明らかにした。その従業員と第三者請負者がcGMPやGCPを遵守することを確保するために、スポンサーは訓練、記録保存、生産、品質管理に多くの時間、お金、エネルギーを費やす可能性がある。
さらに、FDAは、安全性または有効性の問題を提起する新製品候補出願を含む申請を、審査、評価、および提案を行って、申請を承認すべきかどうか、およびどのような条件で承認すべきかを決定するために諮問委員会に提出することができる。通常,諮問委員会は臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、最終承認決定を下す際にこれらの提案を考慮する。臨床試験のデータは常に決定的ではなく,FDAやその諮問委員会は異なる方法でデータを解釈する可能性があり,スポンサーが同じデータを解釈する可能性がある。FDAは臨床試験データを再分析する可能性もあり,FDAやスポンサーの審査過程で広く議論される可能性がある。
FDAはまた、製品の利益がそのリスクよりも大きいことを保証し、製品の安全な使用を保証するために、REMSを提出する必要があると判断した場合、REMSの提出を要求することができる。REMSは、制限された分配方法、患者登録、または他のリスク最小化ツールのような薬物ガイドライン、医師コミュニケーション計画、評価計画、および/または安全使用を保証する要素を含むことができる。アメリカ食品医薬品局は
54
REMSに対する要求,および具体的なREMS条項は,具体的な状況に応じて決定される.FDAがREMSが必要であると考えている場合、申請されたスポンサーは提案されたREMSを提出しなければならず、FDAはREMSのない出願を承認しないであろう。
BLASの決定について
FDA審査申請は,他の事項に加えて,製品が安全であるかどうか,およびその期待用途に有効であるかどうかを決定し,後者の決定は大量の証拠に基づいている。米国食品薬品監督管理局の規定によると、“実質的な証拠”という言葉は、“科学的訓練と経験を経た専門家が臨床調査を含み、関連製品の有効性の証拠を評価するための十分かつ良好な調査を行い、その上で、これらの専門家は、そのラベルまたは提案のラベルに規定、推薦または提案の使用条件下でその主張または表示の効果を有すると公平かつ責任を持って結論を出すことができる”と定義されている
FDAのこのエビデンス基準の解釈は,新製品の有効性を確認するためには,少なくとも2回の十分かつ良好な制御の臨床調査が必要である。しかしながら、場合によっては、FDAは、いくつかの特徴および追加情報を有する単一の実験がこの基準を満たす可能性があることを示している。このやり方は1998年に国会で認められました立法は関連部分で規定されています[林業局]関連する科学的決定に基づいて、良好な臨床調査からのデータおよび確認性証拠(調査の前または後に得られる)が有効性を決定するのに十分である場合、FDAは、そのようなデータおよび証拠を実質的な証拠と見なすことができる。法のこの改正は,FDAが十分かつ良好に制御された臨床調査を発見する可能性があることを認識しており,対照試験外の支持性データを含めて有効性を確立するのに十分である。2019年12月、FDAはガイドライン草案を発表し、有効性の実質的な証拠を確立するために必要な研究をさらに説明した。それはまだ最終的にこの指針を決定していない。
申請とすべての関連情報を評価した後,諮問委員会のアドバイス(あれば)や製造施設や臨床試験地点の検査報告を含めて,FDAはCRLや承認書を発行する。この結論を達成するために、FDAは、この薬剤が有効であることを決定しなければならず、その期待利益は、患者に対する潜在的リスクよりも大きい。この“利益−リスク”評価は、NDAまたはBLAにおいて製品の安全性および有効性に関する大量の証拠によって提供される。この評価はまた他の要素の影響を受け、潜在疾病の深刻性及び現有の治療法がどの程度患者の医療需要を満たしているか;発売前の臨床試験証拠はどのようにこの製品の発売後の環境における実際の使用状況の不確定性を推定するか;及びリスク管理ツールが特定のリスクを管理する必要があるかどうかを含む。この評価に関連して,FDA審査チームはすべての個別審査と他の文書を1つの“行動パッケージ”にまとめ,FDA審査の記録となっている。FDA審査チームはその後、FDAの高官が決定を下す提案を発表した。
CRLは,申請の審査周期が完了したことを示しており,申請は現在の形で承認されない.CRLは、通常、提出中の不足点を列挙し、FDAが申請を再検討するために、大量の追加のテストまたは情報を必要とする可能性がある。CRLは、追加の臨床または他のデータ、追加の重要な第3段階臨床試験、および/または臨床試験、臨床前研究または生産に関連する他の重要で時間のかかる要件を必要とする可能性がある。CRLが発行された場合、スポンサーはFDAが決定した欠陥に1年間応答することができ、FDAは申請が撤回されたと考えるか、スポンサーに追加的な6ヶ月の延期を与えて対応することができる。FDAは、含まれる情報のタイプに応じて、発行されたCRLに応答するために、そのような再提出を2ヶ月または6ヶ月以内に検討することを約束している。しかしながら、この補足情報を提出しても、FDAは最終的に、その申請が承認された規制基準を満たしていないと決定する可能性がある。FDAは,CRLが最終的な機関行動ではなく,その決定を司法審査を受けるようにする立場である.
一方,その製品の商業マーケティングを承認し,特定の適応に関する具体的な処方情報を提供する。すなわち、承認は、FDA承認のラベルに記載されている使用条件(例えば、患者数および適応)に限定される。さらに、解決すべき特定のリスクに応じて、FDAは、製品ラベルに禁忌症、警告または予防措置を含むことを要求することができ、承認後に製品の安全性をさらに評価するための第4段階の臨床試験を含む承認後試験を行うこと、および/または製品商業化後に製品を監視するための試験および監視計画を実施すること、または販売および使用制限またはREMS下の他のリスク管理メカニズムを含む他の条件を適用することは、製品の潜在的な市場および収益性に大きな影響を与える可能性がある。FDAは発売後の試験或いはモニタリング計画の結果に基づいて、製品の更なるマーケティングを阻止或いは制限することができる。承認後、新たな適応の追加、製造変更、および追加のラベル宣言など、承認された製品のいくつかのタイプの変更は、さらなるテスト要件およびFDAの審査および承認を受けることになる。
55
2021年4月に法律となった“革新確保法案”に署名することにより,FDAは新薬と生物製品を承認してから30日以内に行動案を公表し,新薬と生物製品を承認する決定について概説しなければならない。これまでCRLは利用可能なファイルを公開していない.
審査計画を速める
FDAはいくつかの方法でBLASの検討を加速させることを許可された。高速チャネル計画によれば、候補製品のスポンサーは、IND提出と同時にまたは後に、FDAに特定の適応の製品を高速チャネル製品として指定することを要求することができる。もし候補製品が深刻な或いは生命に危害を及ぼす疾患を治療することを目的とし、このような疾病が満たされていない医療需要を満たす潜在力を示す場合、迅速なチャンネル認証を受ける資格がある。高速チャネルは,候補製品と研究中の特定の適応に適した組合せを指定する.他の利点に加えて、FDAとより大きな相互作用が可能であれば、FDAは、申請が完了する前にFast Track申請の部分の審査を開始する可能性があり、この過程をローリング審査と呼ぶ。
迅速チャネル計画に従って、画期的な治療指定、優先審査、加速承認、または再生医学高度治療指定のようなFDAの開発および審査を加速することを目的とした他のタイプの計画に参加する資格がある可能性があるFDA上場候補製品を提出する任意のもの。
FDORAの2022年12月の採択に伴い、国会は薬品や生物製品の承認を加速させるためのいくつかの条項を改正した。具体的には、新しい立法許可FDAは、スポンサーが加速承認を得る前に検証的臨床試験を行うことを要求し、加速された承認された製品のスポンサーが6ヶ月ごとに承認後の研究の進展報告をFDAに提出することを要求し、検証試験が製品の臨床的利益を検証できなかった後、加速手順を使用してNDAまたはBLAの加速承認を撤回することを要求する。また,FDORAは,承認加速後にこのような研究を要求しないことを決定した場合には,そのサイト上で“なぜ承認後研究に適していないのか,あるいは不要な理由”を公表することを求めている。
これらの加速計画はいずれも承認基準を変更することはないが,候補製品の開発や承認過程の加速に役立つ可能性がある.
56
承認後法規
製品発売の規制承認または既存製品の新しい適応が得られた場合、スポンサーは、すべての通常の承認後の規制要件、およびFDAが承認プロセスの一部として適用される任意の承認後要求を遵守することを要求されるであろう。スポンサーは、いくつかの不良反応や生産問題をFDAに報告し、最新の安全性および有効性情報を提供し、広告や販売促進ラベル要求に関する要求を遵守することを要求される。製造業者およびそのいくつかの下請け業者は、FDAおよびいくつかの州機関に彼らの工場を登録し、製造業者にいくつかのプログラムおよび文書要件を適用するcGMP法規を含む現行の法規要件に適合しているかどうかを知るために、FDAおよびいくつかの州機関の定期的な抜き打ち検査を受けなければならない。したがって、スポンサーおよびその第三者メーカーは、cGMP法規や他の法規要件の遵守を維持するために、生産および品質管理の分野で時間、お金、エネルギーをかけ続けなければならない。
製品はまた正式なロット発表が必要である可能性があり、これはメーカーが製品が発表される前に、製品の各ロットに対していくつかのテストを行わなければならないことを意味する。製品が正式なバッチ発行を必要とする場合、製造業者は、各バッチのサンプルをFDAに提出し、バッチの生産履歴要約および製造業者がバッチに対して行ったすべての試験結果を示す放出スキームを提示しなければならない。さらに、FDAは、いくつかの製品のバッチに対していくつかの検証的試験を行い、その後、これらのロットを流通させるかもしれない。最後に、FDAは薬品の安全性、純度、効力と有効性に関する実験室研究を行う。
承認された場合、規制要求や基準の遵守が維持されていない場合、または製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または規制要件を遵守できなかったことを含む、以前に未知の問題が存在することが発見され、新しいセキュリティ情報を追加するために承認されたラベルの改訂を招く可能性がある;新しい安全リスクを評価するために発売後研究または臨床試験を実施すること、またはREMS計画に従って流通または他の制限を実施することが可能である。他の他の潜在的な結果には
薬品は承認された適応と承認されたラベルの規定でしか宣伝できない。医療保健提供者は彼らの専門的な判断において、ラベル外用途と呼ばれる薬物ラベルに記載されていない用途のための製品を開発する可能性があるが、薬品メーカーが許可されていない製品の用途を誘致、奨励、または普及することを禁止する。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。
FDAは市場に投入された処方薬製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。他にも、この規定には、消費者向け直接広告、未承認用途に関する通信、業界スポンサーに関する科学的および教育活動、ならびにインターネットおよびソーシャルメディアに関連する販促活動の基準および規定が含まれる。薬物が承認されるまで,薬物の安全性や有効性の宣伝は禁止されている。承認された後、医薬品は一般にFDAの承認されていない使用に使用されてはならず、これはこの製品の処方情報に反映される。FDAは2021年9月、薬物または生物の予期される用途を決定する際に考慮される証拠のタイプを記載する最終規定を公表した。
57
非常に具体的で狭い条件下では、製造業者が、科学または医学定期刊行物情報の配信のようなラベル外情報に関する非販売促進、非誤解的伝播に従事することを可能にすることができる。また,2022年12月の承認前情報交換法の成立に伴い,承認されていない製品のスポンサーは,製品承認後に患者の接触を加速させるために,開発中の製品に関する何らかの情報を支払者に能動的に伝達することができる。これまで,このような通信はFDAの指導の下で許可されていたが,新たな立法はスポンサーの保護を明確にしており,これらのスポンサーは未承認の製品用途を含む開発中の製品に関するいくつかの情報を支払者に伝えてきた。
ある会社が非ラベル用途の普及を発見した場合、それはFDA、司法省、または衛生·公衆サービス部監察長事務室、および州当局の不利な公共関係および行政および司法法執行の影響を受ける可能性がある。これは、民事および刑事罰金、および会社の薬品の宣伝または流通を実質的に制限する方法の合意を含む一連の重大な商業的影響を及ぼす可能性のある一連の処罰を企業に受ける可能性がある。連邦政府は、不当な販売促進の疑いのある会社に対して巨額の民事と刑事罰金を科し、企業に同意法令または永久禁止令を締結し、これらの法令または永久禁止に基づいて、特定の販売促進行為を変更または制限することを要求する。
最後に、製品に適応、ラベル、または製造プロセスまたは施設の変化を含む修正がある場合、スポンサーは、新しいBLAまたはBLAサプリメントの承認を得るために提出および提出を要求される可能性があり、これは、スポンサーに追加のデータの開発を要求するか、または追加の臨床前研究および臨床試験を行うことを要求する可能性がある。FDAが新しい適応を承認することを確保する過程は、元の適応を承認する過程と類似しており、新適応における製品の安全性および有効性を証明するために、十分かつ制御された臨床試験データを提出する必要がある。このような実験を行っても,FDAはタイムリーに使用するためにラベル適応のいかなる拡張も承認しないか,あるいは全く承認しない可能性がある。継続的な年間使用料要求もあり,現在は何らかの承認された薬物の計画費用として評価されている。
孤児薬の指定と排他性
米国の孤児薬物指定は,スポンサーにまれな疾患や疾患の治療のための製品の開発を奨励することを目的としている。米国では、法律は、まれな疾患または疾患を、米国で20万人未満または米国で20万人を超える影響を及ぼす疾患と定義しており、米国での製品の販売からその疾患または疾患に対する生物製剤の開発および提供のコストを回収することができる合理的な期待はない。
FDAが承認すれば、孤児薬物指定は会社が製品発売許可日から7年以内に税収控除と市場排他性を得る資格を持つことになる。孤児製品に指定された出願は、当該製品の発売を承認する申請を提出する前のいつでも提出することができる。規制規定により提出された受け入れ可能な機密要求によると、1つの製品がFDA孤児製品開発事務所の孤児薬物指定を受けた場合、その製品は孤児となる。そして、その製品はどんな他の製品のように審査と承認手続きを通過しなければならない。
スポンサーは、以前承認されていなかった製品を孤児薬として指定したり、すでに発売されている製品のために新たな孤児適応を申請することを要求することができる。さらに、1つの製品が他の態様で承認された孤児薬物と同じ製品である場合、製品が信頼できる仮定を提示することができる場合、すなわち、その製品が第1の薬剤よりも臨床的に優れている可能性がある場合、製品の発起人は、同じ稀な疾患または疾患の後続製品に対する孤児薬物名を求めることができ、取得することができる。複数のスポンサーは、同じ製品のために同じまれな疾患または疾患の孤児薬物指定を得ることができるが、孤児薬物指定を求める各スポンサーは、完全な指定申請を提出しなければならない。
孤児として指定された製品が、このような指定された疾患または条件を有するFDAの最初の承認を受けた場合、またはまれな疾患または条件下での特定の適応または使用として指定された場合、製品は、通常、孤児薬物排他性を得る。孤立薬物排他性とは、いくつかの限られた場合を除いて、FDAが同じ適応に対する別のスポンサーの同一の製品に対するマーケティング申請を7年以内に承認しない可能性があることを意味する。孤児薬として指定された製品が最終的に発売承認された場合,その適応範囲は孤児薬物申請で指定された範囲よりも広く,排他性を得る資格がない可能性がある。
専門期間はFDAが上場申請を承認した日から,この製品が指定した適応にのみ適用される。FDAは、同じ製品の第2の申請を異なる使用のために承認することができ、または製品の臨床的に良いバージョンのための第2の申請を承認することができる
58
同じ用途です。場合によっては、孤児薬物排他性は、孤児薬物排他性を有する会社が市場需要を満たすことができない場合、または同じ場合に同じ薬物を使用する後続製品が、より良い治療効果または安全性に基づいて臨床的に承認された製品よりも優れていることが証明された場合、または患者ケアに大きく貢献することを含む、別の製品の承認を阻止しない。孤児薬物法案は孤児薬物の排他性を認めることをFDAに明確に要求していたが,その臨床的優位性にかかわらず,早い時期の裁判所の意見であった。トランプ総裁が2020年12月27日に署名した総合的な立法によると、製品の臨床的優位性を示す要求は、2017年のFDA再認可法案が公布される前に孤児薬物指定を受けたが、FDAの承認や許可を得ていない薬物や生物製品に適用される。
2021年9月,第11巡回控訴裁判所は,排他的範囲を決定するために,法規中の“同一疾患または状況”という言葉は指定された“まれな疾患または状況”を指すと判断し,FDAはそれを“適応または使用”と解釈することはできない。したがって,裁判所は孤児薬物は“適応や使用”ではなく,指定された疾患や状況全体に排他的に適用されると結論した。この決定を覆すための立法提案があったが、それらはまだ法律になっていない。FDAは2023年1月23日、裁判所の命令範囲を超える事項において、FDAは、孤児薬の独占性と孤児薬の使用または適応のために許可された使用または適応とを束ねて、その既存の法規を適用し続けると発表した。
小児科排他性
小児科排他性は米国の別のタイプの非特許マーケティング排他性であり、承認されれば、非特許排他性と孤児排他性を含む任意の既存の規制排他性の期限をさらに6ヶ月延長する。BLAスポンサーから提出された小児科データがこのようなデータに対するFDAの書面要求に公平に応答すれば,この6カ月の排他性を与えることができる。これらのデータは,この製品が研究されている小児科群で有効であることを証明する必要はなく,逆に臨床試験がFDAの要求に公平に応答していると考えられれば,追加的な保護が得られる。要求された小児科研究報告が法定期限内にFDAに提出され、FDAの受け入れを受ける場合、製品をカバーする任意の法定または規制された非特許専門期間は6ヶ月延長される。
生物製品を管理する規制の排他性
ある生物製品が食品·薬物管理局の承認の下で発売が許可された場合、その製品はあるタイプの市場とデータ排他性を有する権利がある可能性があり、FDAが特定の時間帯に競争製品を許可することを禁止する。2010年3月、米国は2010年の“医療·教育和解法案”(または総称して“PPACA”)によって改正された“患者保護·平価医療法案”を公布し、その中には“2009年生物製品価格競争と革新法案”、略称“BPCIA”という副題が含まれている。BPCIAはPHSAを改訂し、FDAが許可した参考生物製品の生物と類似或いは交換可能な生物製品のために短い承認経路を作成した。FDAはこれまでにいくつかの生物模倣薬を承認しており、第1の交換可能な生物類似製品は2021年7月30日に承認され、第2の以前に生物類似製品として承認された第1の交換可能な生物類似製品は2021年10月に交換可能に指定されている。FDAはまた、2020年11月に発表された指導草案を含むPHSAに従って生物模倣薬および交換可能な生物模倣薬を審査および許可する方法を概説し、交換可能な生体模倣薬製造業者に追加の透明性を提供することを目的としたガイドライン草案を発表した。
BPCIAによれば、製造業者は、以前に承認された生物製品“生物類似”の製品の生産を要求する申請を提出することができ、この製品は“参考製品”と呼ばれる。FDAに生物類似製品を承認させるためには、参考製品と提案された生物類似製品が安全性、純度と効力の面で臨床的に意義のある差がないことを発見しなければならない。生物類似スポンサーは、分析研究、動物研究、および1つまたは複数の臨床研究のデータに基づいて、その製品が参考製品の生物と類似していることを証明して、1つまたは複数の適切な使用条件下での安全性、純度、および効力を証明することができる。また、スポンサーは、ラベル、投与経路、用量および強度における生物類似製品と参照製品の使用条件が同じ作用機序を有することを証明しなければならず、生産施設は製品の安全、純度および効力を確保するための基準に適合しなければならない。
FDAが生物類似製品と参照製品の交換を許可するためには、FDAはこの製品が参照製品と生物的に類似していることを発見するだけでなく、この製品が参照製品と同じ臨床結果を生成することができることを発見しなければならず、参照生物製品の独占使用に関連する安全リスクを増加させることなく、または治療効果のリスクを低減することなく2つの製品を交換することができる。FDAの承認後、基準製品の衛生保健提供者の介入を開くことなく、交換可能な生物類似体で参照製品を代替することができる。交換可能な生物類似製品が承認された後
59
第1の交換可能な生物類似製品が初めて商業販売されてから1年後、FDAは任意の第2の生物類似製品の交換地位を付与することができる。2022年12月、FDAは、これらの製品がそのような製品が基準製品と交換可能であると承認された初日に承認される限り、複数の第1の交換可能な生物類似生物製品を承認することができるFDORAによって明らかにされた。
参考生物製品は製品が初めて許可を得た日から12年以内に独占経営権が付与され、FDAは参考製品が初めて許可を得た日から4年後にこの基準製品に基づく生物類似または交換可能製品の申請を受ける。しかしながら、1つの製品が独占特許を取得する資格がある参考製品と考えられていても、FDAが製品の完全なBLAを承認する場合、製品は、スポンサー自身の臨床前データと、その製品の安全性、純度、および効力を証明するために十分かつ制御された臨床試験データとを含み、別の会社は、製品の競合バージョンを販売することができる。最近,政府は12年の参考製品専門期間の短縮を提案したが,これまで公布されていない。同時に、BPCIAが成立して以来、多くの州では、生物学的類似製品に関連する薬局慣行を解決するために、法律または法律が改正されている。
特許期限の回復と延長
米国では,ハッジ·ワックスマン法によれば,新生物製品,その使用方法または製造方法を有する特許は,製品開発およびFDA規制審査中に失われた特許期間の最大5年間の延長を可能にする限られた特許期間延長を得る資格がある可能性が主張されている。出願延期された特許が承認されたと仮定すると、製品をカバーする特許の回復期は、通常、IND出願の発効日とBLA提出日との間の時間の半分であり、BLA提出日と最終承認日との間の時間を加える。特許期限回復は特許の残存期間を延長するために使用できず,製品が米国で承認された日から合計14年を超える。承認された製品に適用される特許は1つのみ延期する資格があり,延期出願は延期された特許が満了する前に提出されなければならない。複数の製品をカバーする特許は、そのうちの1つの承認に関連して延期することしかできない。米国特許商標局は,FDAと協議した後,任意の特許期限延長の出願を審査·承認する。
連邦と州のデータプライバシーとセキュリティ法
我々が実験や将来業務を行う可能性のある米国や他の国/地域では、複数のプライバシーやデータセキュリティ法律がビジネス活動に影響を与える可能性があります。このような法律は変化しており、私たちの義務と未来の規制リスクを増加させるかもしれない。一般的に、医療業界では、1996年の連邦健康保険携帯性および責任法(HIPAA)に基づいて、HHSは、いくつかの医療提供者、健康計画、およびヘルスケア交換所を含む、保護された健康情報(PHI)のプライバシーおよび安全を保護するための法規を発行している。HIPAAはまた,医療取引で使用されるデータ内容,コードとフォーマットの標準化,医療計画と提供者の識別子の標準化を規範化している。HIPAAはまた、保護された健康情報を取得した保証エンティティの商業パートナーに対して、保証エンティティまたは代表的な保証エンティティにサービスを提供する際に何らかの義務を課す。HIPAAは場合によっては私たちに適用される可能性があり、私たちのビジネスパートナーにも適用される可能性があり、その方法は私たちと彼らの関係に影響を与える可能性があります。私たちの臨床試験はHIPAAの共通の規則によって規制され、この規則は特定のプライバシー関連条項も含む。連邦プライバシー法規のほかに、多くの州の法律が健康情報のセキュリティと安全性を管理しており、これらの法律は私たちの業務に適用される可能性がある。州総検察長は、HIPAA違反行為に対して連邦民事·刑事処罰を行う可能性があるほか、連邦裁判所に民事訴訟を提起し、損害賠償や禁令を求めてHIPAAを執行し、連邦民事訴訟に関連する弁護士費と費用を求める権利がある。また、, 州総検察長(および個人原告)は、HIPAAのプライバシーや安全ルール違反の疑いで禁止令と損害賠償を求める民事訴訟を起こしている。州総検事長はまた州のプライバシーと安全法を執行する権利がある。未来にはまたプライバシーと安全に関する新しい法律と規制が採択されるかもしれない。
州レベルでは、カリフォルニア州で米国初のGDPRのような法律と呼ばれる法律が公布された。カリフォルニア消費者プライバシー法案、またはCCPAと呼ばれ、消費者のための新しいプライバシー権(法律で広く定義されているように)を創出し、消費者または家庭の個人データを処理するエンティティにより多くのプライバシーおよびセキュリティ義務を課す。CCPAは2020年1月1日に施行され、カバーする会社がカリフォルニアの消費者に新たな開示情報を提供することを要求し、これらの消費者に特定の個人情報の販売から撤退することを選択し、データ漏洩に対して新たな訴訟理由をとることを可能にする。また、2023年1月1日から、カリフォルニアプライバシー権法案(CPRA)は、ある敏感な個人情報に対する消費者の権利を拡大することを含むCCPAを大きく改正する。CPRAはまた、CCPAの実施と実行の権限を付与される新しい国家機関を作成した
60
シーピーRAと。CCPAやCPRAは我々の業務活動に影響を与える可能性があり,これをどのように解釈するかに依存し,我々の業務はネットワークの脅威を受けやすいだけでなく,個人データや個人が識別できる健康情報に関する変化する規制環境の影響を受けることを示している。このような規定は私たちのいくつかの商業活動に適用されるかもしれない。また,バージニア州やコロラド州を含む他の州では州プライバシー法が成立しており,他州は近い将来類似した法律を考慮する可能性がある。
これらの法律の汎用性、およびこれらの法律によって提供される法定例外および規制安全港の範囲が狭いため、私たちの現在または未来のいくつかの業務活動は、いくつかの臨床研究、販売およびマーケティング実践、および特定のプロジェクトおよびサービスを私たちの顧客に提供することを含み、1つまたは複数のこのようなプライバシーおよびデータセキュリティ法律の挑戦を受ける可能性がある。ますます高いコンプライアンス環境、および複数の司法管轄区域の異なるプライバシーコンプライアンスおよび/または報告要件を遵守するために強力かつ安全なシステムを確立し、維持する必要があり、医療会社がそのうちの1つまたは複数の要件を完全に遵守できない可能性を増加させる可能性がある。私たちの運営が、我々のプライバシーまたはデータセキュリティに適用される上記の任意の法律または法規に違反していることが発見された場合、私たちは、潜在的な重大な刑事、民事および行政処罰、損害賠償、罰金、契約損害、名声損害、利益減少および将来の収益減少、追加の報告要件および/または規制を含む罰を受ける可能性があり、これらの法律違反に関する告発を解決するために、法令または同様の合意に同意する制約を受けた場合、いずれも私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性があります。ある程度、私たちが開発する可能性のある任意の候補製品は、承認されると、海外で販売され、私たちは似たような外国法の制約を受けるかもしれない。
FDAは診断を承認しました
FDAは2014年8月に最終指導意見を発表し,治療製品や体外随伴診断承認への適用要求を明らかにした。ガイドラインによれば、新薬のための、キット診断装置およびその対応する治療装置は、製品ラベルにおいて示される治療のためのFDAの承認または承認を同時に得るべきである。キット診断装置の承認または許可は、装置が十分に評価され、ターゲット集団において十分な性能特徴を有することを保証するであろう。FDAは2016年7月、製品の共同開発に関する問題について、薬物治療や体外随伴診断装置のスポンサーを支援するためのガイドライン草案を発表した。
2014年のガイドラインはまた、候補生物製品の臨床試験において治療決定を行うためのキット診断装置は、装置が承認または承認された予期される用途に使用されない限り、研究装置とみなされるであろうと説明している。患者選択のような重要な治療決定を行うために使用される場合、FDAの調査デバイス免除またはIDE法規に従って、診断デバイスは、一般に重大なリスクデバイスとみなされる。したがって、診断デバイスのスポンサーはIDEの規定を遵守することを要求されます。ガイドラインによると、診断装置と製品がそれぞれの承認を支援するために一緒に研究しなければならない場合、IDE法規とIND法規の要求を同時に満たすことができれば、この2つの製品は同じ調査研究で研究することができる。ガイドラインは、研究計画やテーマの詳細に応じて、スポンサーが単独でINDを提出したり、INDとIDEを同時に提出したりすることができる。
2020年4月、FDAは、適切な多様な薬物または生物腫瘍学製品の指定用途をサポートするために、マーカーセット診断装置の開発とマーカーの考慮事項を記述する追加のガイドラインを発表した。本ガイドラインは,診断に伴うラベルに関する既存の政策に基づいている。FDAはその2014年のガイドラインで、診断に伴う診断が特定のカテゴリの治療製品と共に使用するのに適していると結論するのに十分な証拠がある場合、診断に伴う予期される用途または使用適応は、特定の製品ではなく、特定の治療製品群と命名すべきであることを示している。2020年ガイドラインは、2014年ガイドライン中の政策声明を拡張し、セット診断開発者は、彼らのテストを開発できるかどうかを決定する際に、一連の要因を考慮するか、または(単一の治療製品を列挙するのではなく、特定のグループのための腫瘍学治療製品のようなより広いラベル宣言をサポートするために、承認されたセット診断のラベルを補充修正することによってもよい)ことを提案する。
FDCAにより体外診断,随伴診断を含め,医療機器として規制されている。アメリカでは、医療機器の設計と開発、臨床前と臨床試験、発売前の承認或いは承認、登録と上場、製造、ラベル、貯蔵、広告と販売促進、販売と流通、輸出と輸入及び発売後の監督などの方面はFDCA及びその実施条例と他の連邦と州法規と条例の管轄を受けている。適用免除が適用されない限り、診断テストは、商業流通の前に、前の市場承認またはFDAの承認を通知する必要がある。
61
FDAは以前、治療製品候補を承認しながら発売前に承認或いはPMAを得るために、候補製品に反応する患者を選択するために体外随伴診断を要求してきた。臨床および臨床前データの収集、およびFDAおよびFDAへの審査の提出を含むPMAプロセスは、数年またはそれ以上を要するかもしれない。これには厳格な上場前審査が含まれており、その間、スポンサーは設備の安全性と有効性の合理的な保証、設備設計、製造、ラベルなどに関する設備とその部品の情報を準備してFDAに提供しなければならない。PMA申請は申請料を払わなければなりません。2023連邦財政年度の標準費用は441,547ドル、小企業費用は110,387ドル。
EUの薬品審査の法規と手続き
アメリカ国外で任意の製品をマーケティングするために、会社はまた他の国と司法管轄区域の品質、安全性と有効性及び製品に対する臨床試験、マーケティング許可、商業販売と流通などの方面の多くの異なる監督管理要求を守らなければならない。製品がFDAの承認を得ているか否かにかかわらず、スポンサーは外国の監督管理機関のような必要な承認を得なければならず、その後、これらの国あるいは司法管轄区でその製品の臨床試験やマーケティングを開始することができる。具体的には,EUの医薬製品承認の流れは米国とほぼ同じである。製品の安全性と有効性の各提案の適応を決定するために、臨床前研究と十分かつ良好に制御された臨床試験を満足できるように完成させる必要がある。また、関係主管当局にMAAを提出し、これらの主管当局がマーケティング許可を与え、その後、製品を欧州連合で販売·販売することができるように要求されている。
臨床試験許可
2014年4月,EUは新たな臨床試験条例(EU)第536/2014号を採択した。この法規は2022年1月31日に施行され、これまで欧州委員会は2020年に独立監査により臨床試験情報システムのすべての機能を確認した。臨床試験法規はEUの臨床試験の承認を簡略化と簡素化することを目的としている。この条例の主な特徴は、単一入口点による申請手続きの簡略化、即ち“EUポータルサイト”である;申請のために準備と提出する単一文書、及び簡略化された臨床試験スポンサー報告プログラム;及び統一的な臨床試験申請評価プログラムは、2つに分けられる。第1部は、臨床試験許可申請が提出されたすべてのEU加盟国(関係加盟国)の主管当局によって評価される。二番目の部分は関連する各会員国によって個別的に評価される。臨床試験申請の評価には厳しい期限が設定されている。評価手続きにおける道徳管理委員会の役割は、欧州連合加盟国に関する国家法律によって引き続き管轄される。しかし、全体的な関連スケジュールは臨床試験条例によって定義されるだろう.
米国と同様に,ある臨床試験を行う締約国はEudraCTサイト上でEUの臨床試験情報を公表しなければならない:https://eudract.ema.uropa.eu。
EUのトップクラスの呼称
2016年3月,EMAは適応候補の開発を促進するイニシアティブを開始したが,これらの適応はまれであり,現在のところ治療法はほとんどない。優先薬物計画、又はPrimeは、満たされていない医療需要分野の薬物開発を奨励し、中央プログラムの下で審査された重大な革新を代表する製品に対して加速評価を提供することを目的としている。中小企業の製品は大企業よりも早くPrime計画に参加する資格があるかもしれません。Primeの称号を持つ候補製品のスポンサーは多くのメリットを得ることができ、しかし限らず、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素を頻繁に討論し、及びファイルを提出した後にマーケティング許可申請の評価を加速する。重要なのは、ヒト医薬製品委員会(CHMP)または高級治療委員会の専任EMA連絡先と調査委員がPrime計画の早期に任命され、EMA委員会レベルで製品のより多くの理解を促進することである。会議を開始してこれらの関係を開始し、EMAを含む1つの多学科専門家チームは、全体発展と監督戦略に関する指導を提供する。
マーケティング許可
EU規制制度の下で製品のマーケティング許可を得るためには,スポンサーはMAAを提出しなければならないか,EMAが管理する中央手続きに基づいているか,EU加盟国主管当局が管理している手続きのうちの1つ(分散プログラム,国)に基づいている
62
プロセス,または相互承認プロセス).マーケティング許可は連合に設立されたスポンサーだけに付与されることができる。(EC)第1901/2006号条例は、EUのマーケティング許可を得る前に、EMAによって承認された小児科人口のすべてのサブセットをカバーする小児科調査計画(PIP)に含まれるすべての措置を、EMAがPIPに含まれる1つまたは複数の措置について特定の製品の免除、カテゴリ免除、または延期を承認しない限り、EMAによって承認されなければならないと規定している。
中央手続きは欧州委員会によってすべての欧州連合加盟国に有効な単一マーケティング許可を付与することを規定している。(EC)第726/2004号条例によれば、特定の製品、特定のバイオテクノロジーによって生産された医薬品、孤児医薬品に指定された製品、高度な治療製品、および癌の治療のための特定の疾患を治療するための新しい活性物質を含む製品については、集中的な手順が実行されなければならない。他の疾患の治療のための新しい活性物質を含む製品、および高度な革新性または集中処理を有する患者の利益に有利な製品については、集中処理がオプションである可能性がある。メーカーはEMAにその製品の品質,安全性,有効性を証明しなければならず,EMAはMAAに意見を提供しなければならない。欧州委員会はEMAによって提供された意見に基づいてマーケティング許可を承認または拒否する。
具体的には、遺伝子治療薬の販売許可のような生存可能なヒト組織または細胞を含む製品は、欧州議会および欧州理事会の第2001/83/EC号命令と組み合わされた高度治療薬に関する1394/2007/EC号条例によって管轄されており、この命令は、一般に欧州共同体の医療製品規則と呼ばれている。第1394/2007/EC条例は遺伝子治療薬物製品、体細胞治療薬物製品と組織工学製品の許可、監督管理と薬物警戒について具体的な規則を規定した。高級治療薬物メーカーは必ずEMAにその製品の品質、安全性と有効性を証明し、EMAは発売許可の申請に対して意見を提供しなければならない。欧州委員会はEMAによって提供された意見に基づいてマーケティング許可を承認または拒否する。
集中プログラムの下で,EMAに設置されたCHMPが製品の初歩的な評価を担当する。欧州連合の中央手続きによると、重大な影響評価を評価する最長期限は210日であり、スポンサー側が“気候変動管理計画”問題に回答する際に補足資料や書面または口頭で説明する時間は含まれていない。特殊な場合、公衆衛生の観点、特に治療革新の観点から見ると、1種の医薬製品が重大な意義を持っている場合、CHMPは加速評価を承認することができる。CHMPがそのような要求を受けた場合、210日間の制限時間は150日に減少するが、CHMPが加速評価にもはや適していないと判断した場合、集中手順の標準時限に回復する可能性がある。
国家認可手続き
他の2つの可能な方法はいくつかのEU加盟国の医薬製品を許可することができ、これらの経路は集中プログラムの範囲に属さない研究用医薬製品に使用することができる
上記の手順によれば、販売許可を付与する前に、欧州経済区EU加盟国または欧州経済区主管当局または欧州経済区主管当局は、製品の品質、安全性および有効性に関する科学的基準に基づいて、製品のリスク·利益バランスを評価する。
条件付き承認
63
特定の場合、EU立法(第14条-a条例(EC)第726/2004号(条例(EU)2019/5及び条例(EC)第507/2006号改正))により、スポンサーが全面販売許可を申請するために必要な全面臨床データを取得する前に条件付き販売許可を得ることができる。以下の場合、候補製品(孤児薬品として指定された薬剤を含む)に条件付き承認を与えることができる:(1)候補製品は、深刻な衰弱または生命に危険な疾患の治療、予防または医学診断のために使用される、(2)候補製品は、患者が満たされていない医療需要を満たすことを目的としている、(3)全面的な臨床データを提出する前に、即時発売に関するメリットが依然として補充データが必要であるという事実に固有のリスクを超えることを前提として、上場許可を承認することができる。(4)候補製品のリスク−収益バランスが積極的であり,(5)スポンサーが必要な包括的な臨床試験データを提供できる可能性が高い。条件付き販売許可には,現在行われているあるいは新たな研究の完了や薬物警戒データの収集義務など,販売許可保持者が履行しなければならない具体的な義務が含まれていることができる。条件付きマーケティング許可の有効期間は1年であり、リスク-収益バランスが正のままであれば、毎年更新することができる, 追加または修正された条件または特定の義務の必要性を評価した後である。上記の集中プログラムのスケジュールは、条件付きマーケティング許可申請の審査にもCHMPに適用される。
遺伝子治療の専門的なプログラム
欧州連合における遺伝子治療製品の販売許可は、欧州議会および欧州理事会の第2001/83/EC号命令と組み合わされた高度治療薬製品に関する1394/2007/EC号条例によって管轄されており、この命令は、一般に欧州共同体医療製品規則と呼ばれている。第1394/2007/EC号条例は、遺伝子治療薬物製品の許可、監督、および薬物警戒に関する具体的な規則を含む。先進治療薬物製品のメーカーはEMAにその製品の品質,安全性,有効性を証明しなければならず,EMAはMAAに関する意見を提供しなければならない。欧州委員会はEMAによって提供された意見に基づいてマーケティング許可を承認または拒否する。
小児科研究
EUのマーケティング許可を得る前に、スポンサーは、EMAが特定の製品の免除、カテゴリ免除、またはPIPに含まれる1つまたは複数の措置を承認しない限り、EMA承認に適合する小児科集団のすべてのサブセットをカバーするPIPに含まれるすべての措置を証明しなければならない。(EC)第1901/2006号条例、いわゆる“小児科条例”は、すべての販売許可手続のそれぞれの要求を規定する。会社が認可された薬物のために新たな適応,薬物形式あるいは投与経路を増加させようとした場合にも,この要求は適用される。EMAの小児科委員会、またはPDCOは、ある薬物の開発を延期することを承認する可能性があり、会社が成人に対する有効性および安全性を証明するのに十分な情報があるまで、小児薬の開発を延期することを可能にする。小児薬の開発を必要としない場合、または適切でない場合、PDCOは、高齢者人口のみに影響を与える疾患のような免除を与えることもできる。MAAを提出するか、または既存のマーケティング許可を修正する前に、EMAは、会社が各関連PIPに列挙された合意された研究および措置を実際に遵守していると判断する。
EUの規制データ保護
EUでは、改正された(EC)第726/2004号条例および改正された2001/83/EC命令に基づいて、完全な独立データパケットによって承認された新しい化学物質に基づいて、8年間のデータ独占性と追加2年間の市場独占性を得る資格がある。データ排他性により、EUの監督当局は8年以内にイノベーターのデータを参考にして汎用(略語)アプリケーションを評価することができない。追加の2年間の市場専門期間内に、後発薬の発売許可申請を提出することができ、革新者のデータを参考にすることができるが、市場専門権が満期になるまで、いかなる後発薬も発売できない。この10年の最初の8年間に、マーケティング許可所有者が1つまたは複数の新しい治療適応の許可を得た場合、10年全体の期間は最大11年に延長され、許可前の科学的評価では、これらの適応は既存の療法と比較して有意な臨床的利益をもたらすことができると考えられる。ある化合物が新しい化学物質であると考えられていても、革新者が所定のデータ独占期間を取得し、別の会社がMAAベースのマーケティング許可を取得した場合、同社は薬物試験、臨床前試験、および臨床試験の完全な独立したデータパケットを有しており、同社は製品の別のバージョンを販売することができる。
64
EUおよびその他の管轄区域の特許期間の延長
EUはまた,補完保護証明書(SPC)による特許期間の延長を規定している。SPC獲得のルールと要求は米国と類似している。最高特許委員会は、特許の有効期限を予定期限の後5年まで延長することができ、1種の薬物に最大15年の市場排他性を提供することができる。場合によっては、小児科専門権が取得された場合、これらの期間は、さらに6ヶ月延長することができ、これは以下で詳細に説明される。SPCはEU全体で入手できるにもかかわらず、スポンサーは各国に基づいて申請しなければならない。EU以外のいくつかの他の外国司法管轄区域にも同様の特許期間延長権が存在する。
授権期間と継続期間
原則として、上場許可の有効期間は5年であり、5年後にEMAまたはライセンス加盟国の主管当局によるリスク-収益バランスの再評価に基づいて更新することができる。そのため、販売許可保持者は、販売許可が失効する少なくとも6ヶ月前に、販売許可が付与されてから導入されたすべての変化を含む、品質、安全、および有効性に関する文書の統合バージョンをEMAまたは主管当局に提供しなければならない。一旦更新されると、上場許可の有効期限は無期限であり、欧州委員会または主管当局が薬物警戒に関する正当な理由に基づいて、追加の5年間の継続を決定しない限り。いかなる認可の後も、認可が失効してから3年以内に欧州連合市場に薬品が投入されていない場合(集中手続きの場合)、または認可加盟国の市場に投与されていない場合、その許可は無効となる。
上場認可後の規制要件
承認を得た後、上場許可の保有者は医薬製品の製造、マーケティング、普及と販売に適用される一連の要求を守らなければならない。これらの措置には,欧州連合の厳格な薬物警戒や安全報告規則の遵守が含まれており,これらの規則により,認可後の研究や追加的なモニタリング義務を実施することができる。また、許可製品の製造は、欧州薬品管理局のGMP要求とEUの他の規制機関の類似した要求を厳格に遵守しなければならず、これらの要求は、薬品の製造、加工、包装に使用される方法、施設、制御措置を規定し、その安全性と身分を確保する。最後に、EUは改訂された2001/83 EC号指令に基づいて、許可製品のマーケティングと普及、業界賛助の継続医学教育と薬品処方者及び/或いは公衆向けの広告を含み、厳格な管理を行った。
孤児薬の指定と排他性
条例(EC)第141/2000号及び条例(EC)第847/2000号によると、製品は、そのスポンサーが診断、予防又は治療を目的としていることを前提として、欧州委員会によって孤児薬として指定されることができ、(1)申請時にEUの影響が万分の5を超えない生命又は長期衰弱した疾患、又は(2)欧州連合において生命、深刻な虚弱又は深刻かつ慢性的な疾患を危険にさらすことができ、インセンティブ措置がなければ、欧州連合における販売が十分なリターンを生じる可能性がなく、必要な投資が合理的であることを証明することができる。上記2つの場合のいずれについても、スポンサーは、欧州連合が関連疾患の診断、予防または治療に満足できる方法を許可していないこと、または、そのような方法が存在する場合、薬剤が疾患の影響を受ける人に大きな利益をもたらすことを証明しなければならない。
孤児薬物指定は、費用低減、規制援助、EU中央マーケティング許可の申請の可能性を含む一連のメリットを提供する。孤児薬の発売許可は10年間の市場排他期をもたらすだろう。この市場排他期間内に、欧州薬品管理局、欧州委員会あるいは加盟国は申請を受け入れることができず、“類似医薬製品”の販売許可を承認することもできない。類似医薬製品“の定義は、承認された孤児医薬製品に含まれる1つ以上の類似活性物質を含む医薬製品であり、同じ治療適応のためのものである。しかしながら、5年目の終了時に、製品が指定された孤児薬の基準をもはや満たしていないと判定された場合、適応の治療を許可する市場専門期間は、例えば、製品の利益が十分に高く、市場専門性が合理的であることを証明するのに十分ではないので、6年に短縮することができる。
65
小児科排他性
スポンサーがすべてのEU加盟国でマーケティング許可を取得した場合、または欧州委員会が集中手続きで付与されたマーケティング許可を取得し、小児科集団に対する研究結果が製品情報に含まれている場合、否定的であっても、補充保護証明書(SPC)の期限を延長することによって追加6ヶ月の合格特許保護期間を得る資格がある。
セット診断設備の承認
EUでは,診断などに伴う医療機器は,従来適用されていたEU医療機器指令(理事会指令93/42/EEC)の代わりに,EU医療機器条例(EU)2017/745)添付ファイルIで詳細に説明されている一般安全と性能要件(SPR),あるいはMDRに適合しなければならない。SPR遵守とセット医療機器への適用の付加的な要求は,医療機器にヨーロッパ基準に適合したマークを貼り付けることができる前提条件であり,このマークがなければ,医療機器は販売や販売できない。SPRに適合することを証明するためには,メーカーは医療機器のタイプとその分類によって異なる合格評価プログラムを受けなければならない。MDRは、EU全体にわたって統一的、透明、予測可能かつ持続可能な医療機器規制枠組みを構築することを目的としている。
また,欧州連合の規制当局は2022年5月に施行された新たな体外診断条例(EU)2017/746)を採択した。新法規は“体外診断指令”(IVDD)98/79/ECに取って代わった。その体外診断医療機器の合格評価を通知機関に申請したいメーカーは,要求を満たし,新たなより厳しい法規を遵守するために,2022年5月までにその技術文書を更新しなければならない。新しい規定には次のようなものがあります
イギリスの離脱とイギリスの規制枠組み
イギリスは2020年1月31日にEUから離脱した。EUと英国は、2021年1月1日に臨時発効し、2021年5月1日に発効する貿易·協力協定における新たなパートナーシップについて合意した。この協定は主に自由貿易に注目し,医療製品を含む商品貿易に関税や割当を徴収しないことを確保している。その後、EUと連合王国は2つの独立した市場を形成し、2つの異なる規制と法制度によって管理される。そのため、この協定は貨物貿易障壁を最大限に減らすことを図るとともに、連合王国が単一市場の一部ではないため、国境検査は避けられないことを認めている。2021年1月1日より,MHRAはイギリスの薬品や医療機器の監督を担当し始め,国内法によりイギリスにはイングランド,スコットランド,ウェールズが含まれているが,北アイルランドは北アイルランド議定書に基づくEUのルールの制約を受け続けている。MHRAは“2012年ヒト薬品条例”(SI 2012/1916)(改正された)、あるいはHMRに依存し、薬品管理の基礎となる。HMRはすでに医薬製品を管理するEUの法律文書主体を国内法に組み入れており、これらの法律文書は連合王国がEUから離脱する前から存在している。
イギリスの薬品監督管理の枠組みの中で薬品の品質、安全性と有効性、臨床試験、マーケティング許可、商業販売と流通をカバーする大部分はEU指令と法規に由来するため、イギリスの離脱はイギリスの候補製品の開発、製造、輸入、承認と商業化における私たちの規制制度に実質的な影響を与える可能性がある。例えば、イギリスは、EMAからEU範囲内のマーケティング許可を取得する集中的なプログラムによって保護されなくなり、個々のマーケティング許可が必要となるであろう
66
イギリスです。2023年12月31日までに、MHRAは、集中手続きによって新しいマーケティング許可を承認する欧州委員会の決定に依存することができる。
また、連合王国がEUから離脱したにもかかわらず、2018年のEU(離脱)法案第3条により後に改正され、連合王国法の一部となり続けているため、いわゆる“連合王国GDPR”(すなわちEU一般データ保護条例)を実施することにより、GDPRは、連合王国に機関を設立して行われる処理業務に実質的に同等の形態で適用され、連合王国で個人に商品やサービスを提供し、および/または連合王国での行為を監視することに関する任意の処理を継続している。
しかし,GDPRによりヨーロッパ経済圏からイギリスへのデータ移行が合法であるかどうかは不明である。貿易·協力協定は、2021年1月1日から、英国が個人データの処理と移転についてEU加盟国とみなし、4ヶ月間移行期間を規定する。これはあと2ヶ月延長されるかもしれない。その後、英国はGDPR下の“第三国”となり(欧州経済圏から英国へのデータ移転には標準契約条項のような“移行メカニズム”が必要となる)、欧州委員会が英国への個人データの移転について適切な決定をしない限り。欧州委員会は英国に関する十分な決定草案を公表しているが、これらの決定はさらに審査が必要であり、いつこのような決定を通過するかどうかは観察される必要がある。英国政府は、すべてのEUと欧州経済圏加盟国がデータ保護の面で十分であり、イギリスからEUと欧州経済区に流れるデータが影響を受けないようにすることを決定した。しかし、GDPRおよび適用されるEU加盟国およびイギリスプライバシー法によると、これらの法律を遵守するための任意の措置により、責任、費用、コスト、および他の運営損失を招く可能性があります。また,全体的には,データ保護法の適用,解釈,施行における連合王国と欧州経済区の違いは現在ますます大きくなる。
一般資料保障規程
欧州経済地域内の機関活動および/または欧州経済地域に商品またはサービスを提供し、および/または健康データを含む欧州経済地域内の個人行動を監視する背景で、個人データを収集、使用、開示、移転または他の処理する行為は、2018年5月25日に施行されたGDPRによって管轄される。上述したように、いわゆる“連合王国GDPR”を実施することにより、GDPRは、連合王国、イギリス機関、イギリスに重点を置いた加工業務に実質的に同じ形で適用され続ける--したがって、本節でGDPRに言及する場合には、文脈が他に要求されない限り、連合王国のGDPRをも指す。
GDPRの範囲は広く、個人データを処理する会社に対して多くの重大で複雑な要求を提出しており、例えば、個人データを処理する法的基礎の確立を要求すること、個人データの定義を拡大すること(臨床試験関連の場合に通常処理される“仮名”またはキーコードデータを含む);場合によっては管理者と処理者がデータ保護官を任命する義務があること、データ主体に対する透明性義務を増加させること、個人データを保持することを制限すること;データ主体のためにより多くの権利を尊重する義務を導入すること;データ主体の同意を正式に決定することのより高い基準;特定の技術および組織保障措置を実施して個人データの安全と機密性を保護する義務を確立する;第三者プロセッサまたは共同制御者と協力する際に特定の具体的な契約条項に同意し、いくつかの措置をとる義務を規定する;規制当局および影響を受けた個人に特定の重大な個人データ違反行為を通報する義務があることを規定し、場合によっては連合王国および/または欧州連合の代表者を任命しなければならないと規定する。特に,“特殊種別個人データ”(例えば健康や遺伝子情報に関する個人データ)の処理は,我々の臨床試験における業務に関連しており,GDPRでのコンプライアンス負担が増加し,関連規制機関が積極的に興味を持つ話題である。また、, GDPRは、欧州経済圏加盟国は、例えば、臨床試験または他の態様で処理可能な健康データのような特殊なカテゴリの個人データの処理に関する具体的な要件を導入することができると規定している。連合王国では,2018年のイギリスデータ保護法はこの点でイギリスGDPRを補完している。より広く言えば、欧州データ保護当局は、GDPRおよび国家法律を異なる方法で解釈し、追加の要件を適用する可能性があり、これは、欧州経済領域および/または連合王国で個人データを処理するか、または欧州経済領域および/または連合王国から個人データを処理する複雑さをもたらす。実行およびコンプライアンスに関するガイドラインは、しばしば更新され、または他の方法で修正される。この事実は、欧州経済地域および/またはイギリスが個人データ処理に適した法的により大きな相違があることをもたらす可能性があり、これは私たちのコストと全体的なコンプライアンスリスクを増加させる可能性がある。このような特定の国に対する規制はまた、ヨーロッパ経済地域および/または連合王国で関連する個人データを処理する能力を制限する可能性がある
67
運営は最終的に私たちの業務に悪影響を与え、私たちの業務と財務状況を損なう。
GDPRはまた,移転した個人データを保護する具体的な保障措置が実施されていない限り,米国への移行を含めて個人データをヨーロッパ以外の国に移転するための厳しいルールを実施している。欧州連合裁判所の最近の裁決とその後の監督指導によると、以前提供されたいくつかの保障措置は失効しており、場合によっては代替保障措置に依存することは複雑または不可能である可能性がある。もし私たちがヨーロッパ経済区とイギリスからの個人資料の移転のために有効な解決策を実施することができなければ、例えば個人の明確な同意を得てその個人資料をアメリカや他の国に移転することを含めて、私たちはより多くの規制行動、巨額の罰金、ヨーロッパ経済区とイギリスからの個人データの移転禁止に直面する。欧州経済地域およびイギリスから個人データを輸出できないことも、ヨーロッパ経済区およびイギリス以外での私たちの活動を制限する可能性がある;欧州経済地域およびイギリス以外のパートナーおよび他のサービスプロバイダ、請負業者および他の会社との私たちの協力能力を制限すること、および/または巨額の費用でヨーロッパ経済区および/またはイギリスでの私たちの処理能力を向上させること、または他の方法で地理的位置や関連システムおよび運営の分離を変更することを要求することは、いずれも、私たちの業務または財務業績に悪影響を及ぼす可能性がある。さらに、欧州経済地域やイギリス以外の他の国は、同様の国境を越えたデータ伝送制限やローカルデータ常駐を要求する法律の制定を検討しており、サービスや運営業務を提供するコストや複雑さを増加させる可能性がある。
GDPRはまた、より強力な規制法を規定し、規制機関が、金額が大きい者を基準として、2000万ユーロまたは前会計年度の世界年収の4%に達する可能性のある罰金を含む、以前の欧州データ保護法よりも高い処罰を行うことを可能にしている。行政罰金に加えて、潜在的かつGDPR違反の疑いがある行為については、監督当局は、広範な監査および検査権、および不正行為者が個人データのすべてまたは一部を処理することを一時的または永久的に禁止することを命令する権限を含む広範な他の潜在的な法執行権力を持っている。GDPRはまた、データ主体と消費者協会に個人訴訟権利を与え、監督当局に苦情を提出し、司法救済を求め、GDPR違反による損害について賠償を受けることができる。GDPRを守ることは厳格で時間のかかる過程となり、ビジネスコストを増加させたり、会社にそのビジネスやり方を変えて完全な遵守を確保したりする可能性がある。
また、2022年10月、総裁·バイデンはEU-米国プライバシーの盾に代わるEU-米国データプライバシーの枠組みを実施する行政命令に署名した。欧州委員会は2022年12月にEU-米国のデータプライバシーの枠組みを十分に決定するプロセスを開始した。この枠組みがいつ最終的に決定されるか、法廷で挑戦されるかどうかはまだ分からない。この問題をめぐる不確実性はEUでの私たちの業務運営にさらに影響を及ぼすかもしれない。
保証範囲·定価·精算
FDAや他の政府機関の規制承認が求められる可能性のある任意の候補製品のカバー範囲や精算状態には、重大な不確実性がある。米国や他の国の市場では,病状により処方治療を受けた患者や処方サービスを提供する提供者は,通常,第三者支払者に依存して関連する医療費の全部または一部を精算する。患者は、保険を提供し、そのような候補製品を支払うのに十分なコストの大部分を清算しない限り、私たちが開発する可能性のある任意の候補製品を使用することはあまりできない。私たちが開発可能な任意の候補製品が承認されても、これらの候補製品の販売は、MedicareおよびMedicaid、商業健康保険会社および管理型医療保健組織などの米国の政府医療計画を含む第三者支払者にある程度依存し、これらの候補製品に保険を提供し、十分な精算レベルを確立する。支払者が製品に保険を提供するか否かを決定するプロセスは、保険が承認された後に支払者が製品に支払う価格または販売率を設定するプロセスと分離することができる。第三者支払者は、徴収された価格に挑戦し、医療の必要性を審査し、医療製品およびサービスの費用対効果を審査し、コストを管理するために制御を実施することが増えている。第三者支払者は、承認リスト上の特定の製品に保証範囲を制限することができ、特定の適応のすべての承認製品を含まない可能性があるレシピ表とも呼ばれる。
販売が許可される可能性のある任意の製品の保険および精算を確保するために、会社は、製品の医療必要性および費用効果を証明し、fdaまたは他の同様のマーケティングに必要な費用を証明するために、高価な薬物経済学的研究を行う必要があるかもしれない
68
承認する。それにもかかわらず、候補製品は医学的に必要または費用効果があると考えられないかもしれない。第三者支払者は、私たちが開発する可能性のある任意の候補製品を保証しないことを決定し、承認されると、医師のこのような候補製品の使用を減少させ、私たちの販売、運営結果、財務状況に重大な悪影響を与える可能性がある。また、支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。また、1つの支払者が1つの製品に保険を提供する決意は、他の支払者も当該製品に保険·精算を提供することが保証されておらず、支払者によって保険や精算レベルが大きく異なる可能性がある。第三者精算や保険は、製品開発への投資の適切なリターンを実現するために、十分な価格レベルを維持することができないかもしれません。また,いずれの随伴診断テストも単独の保証と精算が必要であり,それに伴う薬品や生物製品の保証や精算は含まれていない。医薬品や生物製品に適用される保険や精算に適用される類似の挑戦は、任意の付随的な診断に適用される。
医療コストの抑制も連邦、州と外国政府の優先事項となっており、薬品価格はずっとこの努力の重点である。各国政府はコスト制御計画の実施に大きな興味を示し、価格制御、精算制限と代替後発薬の要求を含む。価格制御及びコスト制御措置を講じ、既存の制御及び措置を有する司法管区においてより限定的な政策をとることにより、任意の承認された製品の販売から得られる収入をさらに制限することができる。保証政策と第三者精算料率は随時変化する可能性がある。1つの会社またはその協力者がマーケティング承認を得た1つまたは複数の製品が有利な保証·精算状態を獲得しても、将来的にはあまり有利ではない保証政策および精算料率を実施することが可能である。
もし私たちが将来アメリカで私たちが開発する可能性のある任意の候補製品を販売することを許可されたら、私たちは連邦医療計画(例えばMedicaid)の保険を得るために、政府医療計画に基づいて、または特定の政府および個人購入者に割引またはリベートを提供する必要があるかもしれない。このような計画に参加することは私たちが特定の薬品価格を追跡して報告する必要があるかもしれない。もし私たちがそのような価格を正確に報告しなければ、私たちは罰金と他の処罰を受けるかもしれない。
アメリカ以外では、私たちが開発可能な任意の候補製品に十分なカバー範囲と支払いを提供することを確保することは挑戦に直面するだろう。多くの国で、処方薬の価格設定は政府によって規制されている。政府当局との価格交渉は、製品の監督マーケティング承認を受ける範囲をはるかに超えている可能性があり、私たちが開発する可能性のある任意の候補製品の費用対効果を他の利用可能な治療法と比較するために臨床試験を要求することができるかもしれない。このような臨床試験を行うことはコストが高く、私たちの商業化努力の遅延を招く可能性がある。
連合では、様々な国の価格設定と補償プログラムの差が大きい。一部の国では、補償価格を合意した後にのみ、製品を販売することができると規定されている。いくつかの国は、精算または価格設定の承認を得るために、特定の候補製品の費用対効果を現在利用可能な治療法(いわゆる衛生技術評価)と比較することを追加的な研究の完了を要求するかもしれない。例えば、欧州連合は、その国の健康保険制度が補償を提供する製品範囲を制限し、人が使用する医療製品の価格を制御するために、その加盟国に様々な選択を提供している。EU加盟国は製品の具体的な価格を承認することができ、製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることもできる。他の会員国は会社が自分の製品価格を固定することを許可しているが、処方数を監視し、処方を制限するために医師に指導意見を発表した。最近、EUの多くの国が薬品割引要求を高め、各国が医療支出を管理しようとしていることに伴い、これらの努力は継続される可能性があり、特にEUの多くの国が深刻な財政危機と債務危機を経験した場合である。全体的には,医療コスト,特に処方薬の下り圧力が大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。政治、経済、規制の発展は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉は継続される可能性がある。EU加盟国が使用する参考価格と平行貿易(低価格と高価な加盟国間の裁定), さらに値引きできます。薬品に対して価格制御や精算制限を実施することが保証されていない国は、これらの国で承認されれば、私たちのいかなる製品に対しても有利な精算と定価手配を許可する。
医療保健法律法規
医療保健提供者と第三者支払人は推薦と処方が発売承認された薬品の面で主要な役割を果たしている。提供者コンサルタント第三者支払者や
69
顧客は、広く適用される詐欺および乱用、リベート、虚偽申告法、患者プライバシー法、および業務および/または財務的手配を制限する可能性のある他の医療法律および法規の制約を受ける。
適用される連邦および州衛生保健法律法規の制限は、個人および実体がインフォームドコンセントおよび意図的な場合に、個人の転転または購入を誘導または奨励するために、直接または間接的に現金または実物の形態で報酬を請求、提供、支払い、受け入れまたは提供することを禁止し、任意の商品またはサービスを注文または推薦することを含み、これらの商品またはサービスは、連邦医療保険および医療補助などの連邦保健計画に従って全部または部分的に支払うことができる。個人または実体が連邦政府に故意に連邦政府に虚偽、架空または詐欺的な支払いクレームを提出するか、または連邦政府への支払い義務を回避、減少または隠蔽するために、個人または実体が連邦政府に虚偽、架空または詐欺的な支払い請求を行うことを禁止する“民事虚偽申告法”および民事罰金法を含む連邦民事および刑事虚偽申告法。HIPAAは、他の事項に加えて、個人第三者支払人、および重大な事実を偽造、隠蔽、または隠蔽し、または医療福祉、プロジェクトまたはサービスの交付または支払いについて任意の重大な虚偽、架空または詐欺的な陳述を行うことを含む、任意の医療福祉計画を知りながら故意に実行または実行しようとするいかなる人も禁止する追加の連邦刑法を制定した。“海外腐敗防止法”、または“海外腐敗防止法”と呼ばれ、会社およびその中間者が業務を獲得または保留するために、または他の方法で優遇待遇を求めるために、米国ではない役人に不当なお金を支払うこと、提供または承諾することを禁止し、特定の薬品、機器メーカーに要求する連邦“医師支払い陽光法案”を禁止する, 生物製品や医療用品は毎年HHS内の医療保険·医療補助サービスセンター(CMS)にこの実体の医師(医師,歯科医,視光師,足科医と脊椎マッサージ師を含むと定義される)や教育病院への支払いやその他の価値移転に関する情報,および医師およびその直系親族が持つ所有権と投資権益を報告し,2022年から適用を要求するメーカーに前年に医師アシスタント,勤務看護師,臨床看護師専門家,登録看護師麻酔科医アシスタント,登録看護師,麻酔科医アシスタント,登録看護師アシスタント,麻酔科医アシスタント,看護師助産師を登録しています
また、いくつかの州の法律は、製薬会社が製薬業界の自発的コンプライアンスガイドラインと連邦政府が公布した関連コンプライアンスガイドラインを遵守することを要求し、また、メーカーに医師および他の医療保健提供者への支払いまたはマーケティング支出に関する情報を報告することを要求している。また、いくつかの州と地方の法律はこの司法管轄区に薬品販売代表を登録することを要求している。州法や外国法は健康情報のプライバシーやセキュリティを管理する場合もあり、多くの法律は互いに大きく異なり、HIPAAに先を越されず、コンプライアンス作業を複雑化させることが多い。
医療改革
アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。過去数年間、連邦と州政府は薬品と生物製薬製品の定価制限、薬品と他の医療製品のカバー範囲と精算、政府制御及びアメリカ医療保健システムの他の改革に関する多くの提案を提出した。
2010年3月、米国議会は、政府医療計画下の薬品のカバー範囲の変更と支払い方法を含むPPACAを公布した。PACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月、“2011年予算制御法案”(Budget Control Act Of 2011)などの法案は国会のための支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これらの変化には、各年度に提供者に支払われる連邦医療保険総額が2%に達し、2013年4月に発効し、2031年まで続くことが含まれている。コロナウイルス援助,救済,経済安全法案,あるいはCARE法案,その後の立法により,これらの連邦医療保険の自動減額は2022年6月に一時停止され,その後丸2%の削減が残っている。2012年の“米国納税者救済法”は,いくつかの医療サービス提供者に支払う連邦医療保険を減少させ,政府が提供者に多額の金を取り戻す訴訟時効を3年から5年に延長した。これらの法律は、連邦医療保険および他の医療資金のさらなる減少をもたらし、他の方法で、規制によって承認される可能性のある任意の候補製品の価格、またはそのような任意の候補製品の処方または使用頻度に影響を与える可能性がある。
70
PACAが公布されて以来、この法律の条項を廃止し、代替するために、多くの法的挑戦や国会行動が続くだろう。例えば、税法は“個人強制令”を廃止した。この条項は、ほとんどのアメリカ人が最低レベルの医療保険を購入することを要求する条項が2019年に施行される。また、2018年12月14日、テキサス州北区の米国地域裁判所裁判官は、PPACAの個人権限部分はPACAの基本的かつ不可分な特徴であるため、この許可は税法の一部として廃止され、PPACAの残りの条項も無効であると判断した。米国最高裁は2020年11月10日にこの事件を審理し、原告がPPACAの合憲性に挑戦する資格がないことが分かったため、2021年6月17日にこの訴訟を却下した。PACAをめぐる訴訟と立法は継続される可能性があり、結果は予測不可能で不確実だ。
トランプ政権はまた、PPACAの実施を破壊または延期するための行政行動をとっており、PPACAに基づいて権力および責任を有する連邦機関がPPACAの実施を放棄、延期、免除、または延期することを指示する任意の条項を含み、これらの条項は、各州、個人、医療保健提供者、医療保険会社または医薬品または医療機器製造業者に財政的または規制的負担をもたらす。しかし、2021年1月28日、バイデン総裁はこれらの命令を撤回し、連邦機関に米国人の医療保険取得を制限するルールや他の政策を再考するよう指示し、この獲得を保護·強化するための行動を検討するように指示した。この命令によると、連邦機関は、新冠肺炎に関連する合併症を含む以前の疾患を有する人の保護の政策を弱めること、連邦医療補助計画および“全米政治行動計画”に基づくデモおよび免除は、仕事要件を含むカバー範囲または破壊計画を減少させる可能性のある政策、健康保険市場または他の医療保険市場を破壊する政策、連邦医療補助計画および汎米政治行動計画への参加の難しさを増加させる政策、および扶養者に対する負担能力を含む保険または経済援助の負担能力を低下させる政策を含む、再検討を指示されるであろう。
薬品価格
アメリカでも処方薬の価格はずっと話題になっています。最近、米国議会は数回の調査を行い、大統領行政命令を発表し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険と医療補助下の薬品価格を下げることを目的とした州と連邦立法を提案し、公布した。2020年には、総裁·トランプ氏が処方薬の価格を下げるためのいくつかの行政命令を発表し、これらの命令のいくつかが条例に盛り込まれている。これらの規定には、価格最恵国モデルを実施し、連邦医療保険B部分のある医師が管理する薬品に対する支払いを他の経済先進国が支払う最低価格とリンクさせ、2021年1月1日から発効する臨時最終規則が含まれている。しかし、この規則は全国的な初期禁止によって制限され、2021年12月29日、CMSはそれを廃止するための最終規則を発表した。CMSは、この規則の発表に伴い、それは価値をMedicare B部分の薬品支払いのすべての選択に組み入れ、そして受益者が根拠に基づく看護を獲得する機会を改善することを探索する。
また、2020年10月、HHSとFDAは、各州と他のエンティティが804条の輸入計画、すなわちSIPを制定し、いくつかの処方薬をカナダから米国に輸入することを許可する最終規則を発表した。最終規則は現在行われている訴訟のテーマであるが、少なくとも6つの州(バーモント州、コロラド州、フロリダ州、メイン州、ニューメキシコ州、ニューハンプシャー州)がFDAの審査と承認のためにSIPsを開発することを目的としてカナダからの薬物の輸入を許可している。また、2020年11月20日、HHSは薬品メーカーが連邦医療保険Dの一部の下でスポンサーの値下げを計画する避風港の保護を廃止し、法律が値下げを要求しない限り、直接或いは薬局福祉マネージャーを通過する法規を決定した。この規定はまた、販売所での値下げを反映するための新しい避難港を創出し、薬局福祉マネージャーとメーカーとの間のいくつかの固定費用手配のための新しい避難港を創出した インフレ削減法案によると、同法案の実施は2032年1月1日に延期された。
2021年7月9日、総裁·バイ登は14063号行政命令に署名し、その中で薬品価格などの問題に重点を置いた。この命令はHHSに45日以内に1つの計画を制定し、処方薬の過度な定価を打撃し、国内の薬品サプライチェーンを強化し、連邦政府がこのような薬品に支払う価格を下げ、繰り返し出現した価格詐欺問題を解決するよう指示した。2021年9月9日、HHSは薬品価格を下げる計画を発表した。この計画の主な特徴は、メーカーとの薬品価格交渉を支持することによって、すべての消費者と医療保健システム全体がより負担し、より公平な薬品価格を提供することである;サプライチェーンの強化を支持し、生物模倣薬と後発薬の市場変化を促進することによって、処方薬業界全体の競争を改善と促進すること;及び科学革新を促進することである
71
公共および個人研究を支援し、市場インセンティブを確保することにより、価値と入手可能な新しい療法の発見を促進し、より良い医療保健と健康改善を促進する。
最近、2022年8月16日、総裁·バイデンは2022年インフレ削減法案、あるいはアイルランド共和軍と署名した。新しい立法は連邦医療保険D部分に影響を与え、D部分は計画であり、連邦医療保険A部分または連邦医療保険B部分に加入する個人は毎月外来処方薬保険料を支払うことを選択することができる。他の事項を除いて、アイルランド共和軍はある薬品のメーカーに連邦医療保険との価格交渉(2026年から)を要求し、価格は交渉できるが上限があり、連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施し、インフレを超える価格上昇を処罰し(2023年に初めて満了)、D部分のカバーギャップ割引計画の代わりに新しい割引計画を用いる(2025年から)。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。
具体的には、価格交渉において、国会はMedicareが、競合する模倣薬または生体模倣薬を有さず、Medicare B部分およびD部分によって精算されるいくつかの高価な単一由来薬剤および生物製品のための低い価格を交渉することができる。CMSは、2026年からMedicare D部分によって支払われる10種類の高コスト薬剤の価格を交渉することができ、その後、2027年の15種類のD部分薬剤、2028年の15種類のB部分またはD部分薬剤、および2029年以降の20種類のB部分またはD部分薬剤の価格を交渉することができる。この規定は、少なくとも9年間承認された医薬品および許可13年の生物製品に適用されるが、単一のまれな疾患または疾患のための許可された医薬および生物製品には適用されない。また、この立法は、製薬業者が提供する価格が法律で規定された交渉で達成された“最高公平価格”以下でない場合、または値上げ幅がインフレを超える場合、製薬業者は民事罰金と潜在的な消費税を受けることになると規定している。この立法はまた、メーカーに連邦医療保険D部分の価格上昇幅がインフレを超えた薬品にリベートを支払うことを要求している。新法律では,2024年の連邦医療保険の自己払い薬品コスト上限は年間4000ドルと規定されており,その後2025年から年間2000ドルが上限となっている。
州レベルでは、各州はますます積極的に、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法と実施し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。例えば、いくつかの州は、健康担体、薬局福祉マネージャー、卸売流通業者を含む医薬品製造業者と薬品サプライチェーン中の他のエンティティに要求し、薬品定価に関する情報を開示する。また、地域医療機関や個別病院は、どの薬品やサプライヤーがその処方薬や他の医療保健計画に組み込まれるかを決定するために入札プログラムを使用することが増えている。これらの措置が承認されると、私たちの製品に対する最終的な需要を下げたり、私たちの製品の価格設定に圧力をかけるかもしれません。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
従業員と人的資本
2022年12月31日現在、204人の常勤従業員がおり、そのうち58人が医学博士、薬学博士、または博士号を持っている。これらの常勤従業員のうち,164人が研究·開発活動に従事し,40人が一般·行政活動に従事している。私たちの従業員の中の一人も労働組合代表でもなく、集団交渉協定のカバー範囲もない。私たちは私たちが従業員と仲がいいと思う。
著者らは発見、臨床前研究と臨床開発及び遺伝子編集技術と遺伝子薬物の製造と交付において異なる専門家チームを吸引した。私たちのグループはいくつかの核心的な価値観に基づいて、これらの価値観は私たちの日常活動を推進し、私たちの長期的なビジョンを刺激します
私たちの人的資本資源目標には、適用、識別、採用、維持、インセンティブ、そして私たちの既存の、より多くの従業員を統合することが含まれています。私たちは、私たちの採用、昇進、発展実践を含む、組織のすべての面で多様性、公平性、包摂性を実現するために努力しています。毎年私たちは
72
従業員人口統計情報を審査して、会社のすべての機能とレベルでの多様な努力を評価します。著者らは各従業員に対して年間業績と発展評価を行い、個人の優勢と発展機会、職業発展目標と業績目標を討論した。私たちはまた、従業員の尊敬度と満足度を評価するために定期的に従業員を調査している。私たちの株式インセンティブ計画の主な目的は、株に基づく報酬奨励を付与することによって、選定された従業員と役員を吸引、維持、激励することである。私たちは従業員を重視し、短期と長期給与、401(K)貢献、健康、福祉、生活の質の福祉、有給時間など、私たちが提供するすべての報酬を定期的に基準にしています私たちの業界の同業者と競争して、私たちが競争力を維持し、潜在的な新入社員への魅力を確保する。
私たちの会社情報は
我々は2018年3月9日にデラウェア州法律に基づいて登録が成立し、名称はEndcadia,Inc.です。2019年1月15日、Verve Treateutics,Inc.と改称しました。
私たちの主な実行オフィスはマサチューセッツ州ボストンブルックリン通り201601 Suit 601、ボストン、郵便番号:02215、電話番号は(6176030070)です。私たちのサイトはhttp://www.vervetx.comです。当社サイトに含まれているか、または本サイトで取得可能な情報は、本年度報告の一部を構成していません。私たちは本年度報告書に私たちのサイトアドレスを含めて、非アクティブなテキストとしてのみ参考にします。
利用可能な情報
私たちのサイトはhttp://www.vervetx.comです。我々のForm 10-K年次報告、Form 10-Q四半期報告、Form 8-K現在の報告は、証拠物、依頼書、および情報宣言、および取引法第13(A)および15(D)条に基づいて提出または提供される報告書の修正案を含み、米国証券取引委員会または米国証券取引委員会にこれらの材料を電子的に提供した後、合理的で実行可能な範囲内でできるだけ早く当サイトの“投資家”部分を介して無料で提供される。当サイト上の情報は、本報告書に明示的に組み込まれない限り、本年度報告または他の任意の証券届出文書の一部ではありません。また、米国証券取引委員会に提出された文書は、米国証券取引委員会のインタラクティブデータ電子申請システムを介して閲覧することができる。私たちが任意の証券届出文書で下したすべての陳述は、すべての前向きな陳述または情報を含み、その陳述を含む文書の日付で行われ、法律が私たちにそうすることを要求されない限り、私たちはこれらの陳述または文書を更新するいかなる義務も負わない。
73
第1 A項。リスク要因です
以下に述べるリスクと不確実性のため、当社の将来の経営業績は、本10-K表年次報告に記載されている結果とは大きく異なる可能性がある。私たちの業務を評価する時、あなたは以下のリスクに関する情報を慎重に考慮しなければならない。もし実際に以下のいかなるリスクが発生すれば、私たちの業務、財務状況、経営業績、未来の成長見通しは重大な不利な影響を受ける可能性がある。この場合、私たちの普通株の市場価格は下落するかもしれない。しかも、私たちは私たちの仮定と期待が正しいことが証明されることを投資家に保証することはできない。重要な要素は私たちの実際の結果が展望性陳述で表明したり暗示したりする結果と大きく異なる可能性がある。これらのリスク要因に制限されたいくつかの前向き陳述の議論については,本年度報告10−K表の3ページを参照されたい。このような差異をもたらすか、または促進する可能性のある要因は、以下に説明する要因を含む。
私たちの財務状況と追加資本需要に関連するリスク
設立以来、私たちは大きな損失を受けて、どんな製品も販売を許可されなかった。私たちは予測可能な未来に損失が出て、永遠に達成されたり利益を維持したりしないかもしれないと予想している。
設立以来、著者らはほとんどの財務資源と努力を研究と開発に投入し、臨床前研究と臨床試験を含み、重大な運営損失を発生した。2022年、2021年、2020年12月31日までの年度の純損失はそれぞれ1.574億ドル、1.203億ドル、4570万ドルだった。2022年12月31日現在、私たちの累計赤字は3.442億ドルです。私たちは製品販売から何の収入も得ていません。私たちは主に私たちの優先株と普通株を私募し、公募で普通株を売却し、2022年7月にVertex製薬会社またはVertex社の戦略協力と許可協定またはVertex協定に関連する支払いを受けることで、私たちの業務に資金を提供します。
予測可能な未来に、私たちは引き続き巨額の運営費用と純損失が発生すると予想される。私どもの運営費と純損失は四半期と年度の間に大きく変動する可能性があります。私たちの費用は大幅に増加すると予想されています
74
さらに、以下の場合、私たちの支出はさらに増加するだろう
1つ以上の候補製品のマーケティング承認を得て商業化に成功しても、より多くの候補製品を開発·マーケティングし、および/または任意の上場製品の承認適応を拡大するために、多くの追加の研究開発や他の支出が生じることが予想される。私たちは予測できない費用、困難、合併症、遅延、および私たちの業務に悪影響を及ぼす可能性のある他の未知の要素に直面するかもしれない。私たちの未来の純損失の規模は私たちの未来の支出の成長率と私たちが収入を作る能力にある程度依存するだろう。
私たちは製品販売から収入を得たことがなく、永遠に達成されたり利益を維持したりしないかもしれない。
私たちは最近私たちの最初の候補製品の臨床開発を始めたばかりで、まだ何年もかかると予想されていますが、もしあれば、商業化された候補製品を準備することができます。利益を実現し、維持するためには、必要な規制承認を成功させ、最終的に大量の収入を生じる1つまたは複数の製品を商業化しなければならない。この成功を達成する能力は、一連の挑戦的な活動において有効であることを要求するであろう
私たちはこのような活動の初期段階にいるだけで、私たちがこれらの活動で成功するという保証はありません。たとえ私たちが成功しても、利益を達成するために十分な収入が生まれないかもしれません。私たちはまだ候補製品の臨床試験を終えていない。医薬品開発に関連する多くのリスクと不確実性のため、費用を増加させる時間や金額、あるいは私たちがいつ収入を生み出すことができるか、または利益を達成できるかどうかを正確に予測することはできない。
たとえ承認された製品の販売から収入を得ることができても、利益を得ることができない可能性があり、運営を継続するために追加の資金が必要かもしれない。私たちの収入は、私たちが規制の承認を受けた地域の市場規模、製品の受け入れ可能な価格、保険と補償を受ける能力、そして私たちがその地域の商業権を持っているかどうかにある程度依存するだろう。私たちの潜在的な患者数が私たちが推定しているほど多くなければ、規制部門が承認した適応は私たちが予想していたより狭い、あるいは治療者たちは競争、医師の選択、または治療ガイドラインによって縮小され、承認されても、このような製品の販売から大量の収入を得ることができないかもしれない。
75
私たちは多くの追加資金が必要になるだろう。もし私たちが必要な時に資金を集めることができなければ、私たちは私たちの製品開発計画や商業化努力を延期、減少、またはキャンセルさせることを余儀なくされるかもしれない。
特に我々が行っているVERVE−101の1 b期臨床試験,VERVE−201の臨床前研究の完了,継続研究,開発と臨床前試験,より多くの臨床試験を開始し,VERVE−101,VERVE−201,我々が開発する可能性のある他の候補製品の発売承認を求める場合には,我々が行っている活動および計画中の活動に多くの資金が投入される予定である。われわれが行っている計画中の活動に関する費用は大幅に増加し,特に臨床前活動および計画中の臨床試験を進めている場合には大幅に増加することが予想される。また、いずれかの候補製品が市場承認されれば、製品の製造、販売、マーケティング、流通に関連した巨額の商業化費用が発生すると予想される。また、上場企業の運営に関連した追加コストが引き続き発生すると予想される。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。私たちは現在信用計画もなく、約束された資金源もない。もし私たちが必要な時や受け入れ可能な条件下で資金を調達したり、十分な資金を得ることができない場合、私たちは私たちの研究開発計画や任意の将来の商業化努力を延期、制限、減少または終了させ、あるいは私たちが自分で開発とマーケティングをより望んでいた候補製品を開発する権利を与えることを余儀なくされるかもしれない。
私たちの将来の資本需要は多くの要素に依存します
潜在的な候補製品を決定し、臨床前テストと臨床試験を行うことは時間がかかり、高価かつ不確定な過程であり、完成するのに数年かかり、しかも私たちは永遠に市場の許可を得て製品販売を実現するために必要なデータ或いは結果を生成できないかもしれない。また、候補製品の決定と開発に成功して承認されても、私たちは商業的に成功しないかもしれない。私たちの商業収入は、もしあれば、私たちの運営を維持するのに十分ではないかもしれない。したがって、私たちは追加的な資金調達に依存して私たちの業務目標を達成し続ける必要があるだろう。
2022年12月31日現在、私たちは約5.548億ドルの現金、現金等価物、有価証券を持っている。私たちの既存の現金、現金等価物、有価証券は、2025年下半期の運営費用と資本支出需要に資金を提供できると信じています。しかし私たちは
76
このような誤った仮説であることが証明される可能性のある見積りや,我々の運営計画は,現在我々が知らない多くの要因によって変化する可能性がある.したがって、私たちは現在予想されているよりも早く私たちの資本資源を枯渇させ、計画よりも早く追加資金を求めることを余儀なくされるかもしれない。
いかなる追加的な拠出努力も、私たちの管理職の彼らの日常活動への関心を移す可能性があり、これは私たちが任意の候補製品を開発し、商業化する能力に悪影響を及ぼすかもしれない。私たちは許容可能な条件で追加資金を提供するかどうか、あるいは根本的にできないかどうかを確認することができない。例えば、経済やその他の要因は最近、世界金融市場に大きな妨害を与えており、このような状況が続く可能性があり、私たちの資本獲得能力を低下させる可能性があり、これは将来的に私たちの流動性に悪影響を及ぼす可能性がある。私たちには追加資本や外部資金の約束源がありません。もし私たちが十分な追加資本を調達できない場合、あるいは私たちが受け入れられる条項で追加資本を調達できない場合、私たちは候補製品や他の研究開発計画の開発や商業化を大幅に延期、削減、または停止しなければならないかもしれません。私たちは、他の場合ではなく、私たちが開発する可能性のある候補製品のためのパートナーを探すことを要求されるかもしれないし、他の方法よりも不利な条項でパートナーを探したり、不利な条項で私たちが開発する可能性のある候補製品の権利を放棄したり、許可したりすることができます。そうでなければ、私たちは自分で開発または商業化された市場を求めることができるかもしれません。
上記のどの事件も、私たちの業務、将来性、財務状況と経営結果を深刻に損害し、私たちの普通株価格の下落を招く可能性があります。
追加資本の調達は私たちの株主に希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません。
これまで、製品販売から相当な収入を得ることができれば、株式発行、債務融資、協力、戦略連合とマーケティング、流通、または許可手配の組み合わせで現金需要を満たす予定です。私たちは資本や外部資金の供給源を約束していない。私たちが株式または転換可能な債務証券を売却することで追加資本を調達する場合、私たちの株主利益は希釈され、これらの証券の条項は清算または他の特典を含む可能性があり、普通株主としての私たちの権利に悪影響を及ぼす。任意の債務融資および優先株融資に関連する可能性のあるプロトコルは、追加債務の生成、おそらく私たちの資産の売却、資本支出の実施、配当金の発表、または将来の債務を確保するために、私たちの特定の行動をとる能力を制限または制限する契約を含む。
もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時や私たちが受け入れた条件下で株式や債務融資または他の手配を通じて追加資金を調達できない場合、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少または終了することを要求され、あるいは私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を与えることができます。
私たちの限られた経営の歴史は、株主が私たちの業務のこれまでの成功度を評価することを困難にし、私たちの将来の生存能力を評価することも困難になるかもしれない。
私たちは2018年に運営を開始し、臨床期会社です。これまで,我々の業務は組織と我々を備えた会社,業務計画,資金調達,我々の技術の開発,潜在的な候補製品の決定,知的財産権の保護,臨床前研究と早期臨床試験に限られてきた。著者らは2022年7月に著者らの最初の臨床試験、すなわちVERVE-101の1 b期臨床試験を開始した。我々の他の研究プロジェクトは,VERVE−201を含め,まだ研究や臨床前開発段階にあり,失敗のリスクが高い。私たちは、いかなる臨床試験を完成させ、市場の承認を得て、臨床開発または商業規模の製品を生産し、あるいは第三者代表が私たちにそうすることを手配したり、成功した製品の商業化に必要な販売とマーケティング活動を行うことができることを証明していません。一部は経験が乏しいため、著者らは著者らが行っている臨床前研究と臨床試験が時間通りに完成するかどうか、あるいは計画中の臨床前研究と臨床試験が時間通りに開始或いは完成するかどうかを確定することができない。したがって、もし私たちがより長い運営歴史や開発成功と商業化遺伝子編集製品の歴史を持っていれば、株主は私たちの将来の成功や生存能力に対するいかなる予測もそれほど正確ではないかもしれない。
私たちの限られた運営の歴史、特に急速に発展する遺伝薬物分野を考慮すると、私たちの技術や業界を評価し、私たちの将来の表現を予測することが困難になるかもしれません。運営会社として、私たちの限られた歴史は、私たちの未来の成功や生存能力のどの評価も重大な不確実性の影響を受けるようにしている。私たちは
77
急速な発展分野で初期段階にある会社がよく遭遇するリスクと困難に直面するだろう。もし私たちがこのような危険にうまく対応できなければ、私たちの業務は影響を受けるだろう。
さらに、業務の増加に伴い、私たちは予見できない費用、制限、困難、複雑な状況、遅延、および他の既知および未知の要素に遭遇する可能性がある。私たちはある時点で研究開発に集中している会社からビジネス活動を支援できる会社に転換する必要があります。そのような移行で、私たちは成功しないかもしれない。
私たちは私たちの純営業損失と研究開発税収控除を使用して、未来の課税収入或いは税金を相殺する能力はいくつかの制限を受けるかもしれません。
私たちは累積損失の歴史があり、予測可能な未来に私たちは引き続き重大な損失が発生すると予想しています。そのため、私たちは私たちの純運営損失、NOL、研究開発税収の繰越を利用するために必要な課税収入が発生するかどうかを知りません。2022年12月31日現在、私たちは1.736億ドルの連邦NOL繰り越しと1.57億ドルの州NOL繰り越しがある。
一般的に、改正された1986年の国税法第382条及び383条、又はこの法典及び州法の該当条項によると、会社は“所有権変更”を経験し、ある株主が3年以内にその持分所有権の変化が50ポイントを超える(価値で計算される)と定義され、その利用変更前のNOL及び研究開発税収は繰越を免除して、変更後の課税所得額又は税金を相殺する能力が制限される。私たちはまだこのような所有権変更が発生したかどうかを評価するための研究を行っていない。私たちは過去にこのような所有権の変化を経験したことがあり、未来は私たちの株式所有権の後続の変化(これは私たちがコントロールできるものではないかもしれない)のためにこのような所有権の変化を経験するかもしれない。したがって,課税所得純額を稼ぐと,変動前のNOLや研究開発税収を用いてこのような課税収入を相殺する能力が制限される可能性がある。州法によると、私たちのNOLや信用もまた損なわれる可能性がある。
もう一つのリスクは、NOLの使用停止や他の予測不可能な理由のような法規の変化により、私たちの既存のNOLが満期になるか、または他の方法で将来の所得税債務を相殺できない可能性があるということだ。以下に述べるように、税法またはその実施または解釈の変化は、米国連邦税率およびNOL繰り越し管理規則の変化を含む、我々の業務および財務状況に悪影響を及ぼす可能性があり、減税および雇用法案、またはコロナウイルス援助、救済および経済安全法案またはCARE法案によって改正された税法は、米国連邦税率およびNOL繰り越し管理規則の変化を含み、将来的にNOLを用いて課税収入を相殺する能力に著しく影響を与える可能性がある。このような理由で、私たちが利益を達成しても、私たちは私たちのNOLと他の税金属性の大きな部分を使うことができないかもしれない。
発見と開発に関するリスク
私たちの開発は非常に早い段階にあり、私たちはまだ候補製品の臨床試験を完成していません。したがって、私たちは私たちがどんな候補製品を商業化すれば、何年もかかると予想する。もし私たちが臨床試験を通じて私たちの現在または未来の候補製品を推進できなければ、マーケティングの承認を得ることができず、最終的に私たちの候補製品を商業化したり、その過程で重大な遅延に遭遇した場合、私たちの業務は実質的に損害を受けるだろう。
我々の開発は非常に早期の段階にあり,これまでわれわれの努力は主に研究努力と臨床前開発に集中してきた。私たちは2022年7月に私たちの最初の臨床試験、すなわちVERVE-101の1 b段階の臨床試験を開始しましたが、私たちはまだ候補製品の臨床試験を完成していません。私たちが製品収入を作る能力は、私たちの候補製品の成功開発、マーケティング承認、最終商業化に大きく依存しています。これは決して起こらないかもしれません。私たちはまだ製品販売から収入を得ていません。私たちは永遠に適切な製品を開発したり商業化することができないかもしれません。
アメリカで臨床試験を開始することはFDAのINDに対する受け入れに依存し、FDAと他の監督機関との討論に基づいて最終的に試験設計を確定する。
FDAまたは他の規制機関は、追加の臨床前研究を完成させることを要求するか、またはそれぞれの国が臨床試験を開始する前に他の要求を満たすことを要求することができ、これは、私たちの臨床試験を私たちの計画のスケジュール外に遅延させるかもしれない。例えば,2022年11月,FDAは米国でVERVE−101臨床試験評価を行うIND申請を休止し,本年度報告Form 10−Kの日までINDは放置されている。私たちがこれらの規制機関の指導を受けて組み込まれた後であっても、FDAまたは他の規制機関は、VERVE-101に関する要求を含む臨床試験を開始する要求を満たしているかどうか、または彼らへの変更を含むかどうかに同意しないかもしれません
78
著者らの試験設計或いは選択された臨床終点は、追加の臨床前研究或いは臨床試験を完成し、私たちの臨床試験の登録を延期する必要があるかもしれない、あるいは現在期待されているよりも厳しい承認条件を適用する。他の国では、EU諸国を含めて、同様の手続きやリスクが臨床試験申請に適用されている。
私たちが開発する可能性のある任意の候補製品の商業化には、臨床前および臨床開発、FDAおよびEMAを含む複数の司法管轄区域の規制およびマーケティング承認、製造供給、能力および専門知識、商業組織、および重大なマーケティング努力が必要となる。VERVE−101、VERVE−201、および我々が決定し開発する可能性のある他の任意の候補製品の成功は、以下の要因を含む多くの要因に依存する
もし私たちがこれらの要素のうちの1つまたは複数でタイムリーにまたは根本的に成功できなければ、私たちは私たちが開発する可能性のある任意の候補製品を迅速にまたは根本的に商業化することに成功したり、私たちの業務に実質的な損害を与えるかもしれない。もし私たちが臨床開発を通じて私たちの候補製品を推進できなければ、規制部門の承認を得ることができず、最終的に私たちの候補製品を商業化したり、その過程で重大な遅延に遭遇したら、私たちの業務は実質的に損害を受けるだろう。
遺伝子編集は、塩基編集を含む、新しい技術であり、安全かつ有効なヒト治療用途として臨床的に検証されていない。我々が講じている新しい療法の発見と開発方法は検証されておらず,適切な製品が生じない可能性がある。
私たちは遺伝子編集技術を利用して薬物を開発することに集中していますが、これは新しい技術であり、大きく実証されていません。我々が許可を得てVERVE−101やVERVE−201とともに使用している基本編集技術は,完成した臨床試験では評価されておらず,第三者が我々の基本編集や同様の技術を用いて行われている臨床試験も知られていない。遺伝子編集技術に基づく候補製品の開発の実行可能性を支持する科学的証拠は初歩的であり、限られている。我々の候補製品の開発成功には,遺伝子エディタを目的細胞に安全に輸送し,このような候補製品の効率と特異性を最適化し,そのような候補製品の治療選択性を確保する必要がある。塩基編集技術や他の遺伝子編集技術が遺伝子薬物の発展を招くことは保証されず,任意あるいはすべての問題の解決に成功する保証はない。
79
著者らの未来の成功は遺伝子編集技術、伝達技術方法とこの技術の治療応用の成功開発に高度に依存している。我々は我々の最初の計画を変更または放棄することを決定するかもしれないが,新たなデータがあるため,遺伝子編集療法の開発で経験を得た。私たちの技術が満足できる製品を生成することは確認できません。私たちの初期適応または私たちが追求している任意の他の適応において、これらの製品は安全で効果的、拡張可能、または利益があります。他の遺伝子編集技術会社の臨床開発の不利な発展は、私たちの努力や投資家と監督機関に私たちの候補製品の見方に悪影響を与える可能性がある。
同様に、もう1つの発見されていない新しい遺伝子編集技術は、第三者によって開発されている可能性があり、塩基編集技術を用いて追求されている遺伝子標的が塩基編集よりも魅力的であることが決定される可能性がある。
我々はまた,Vertexとの連携の一部として,関心のある目標に対する特定の遺伝子編集システムや輸送システムの識別と設計を求めることを含む新たな遺伝子編集開発候補の開発を求めている.私たちは未来のプログラムのための新しい遺伝子編集技術を開発することを求めるかもしれない。我々はこれまでに新たな遺伝子編集技術を開発したこともなく,第三者から許可を得る遺伝子編集技術もない.私たちは私たちが目標や他の任意の目標のために新しい遺伝子編集システムを開発することに成功するとは確信できない。
また,他の遺伝子編集技術を開発するために必要ないかなる権利を得ることができるかどうかは確認できない.私たちは現在、基礎編集技術の分野でコンサルティングやコンサルティングサービスを提供してくれたすべての創始者が、彼らが提供してくれたサービスについて発明義務を譲渡しているにもかかわらず、これらの発明義務の譲渡は制限されており、他の分野での彼らの仕事にも適用されず、それぞれの学術·研究機関によって作られた知的財産権にも適用されていない。これらの創始者がこれらの機関に割り当てられた知的財産権を得るためには、商業的に合理的な条項で得られないか、あるいは全く得られない可能性があるライセンス契約をこれらの機関と締結する必要がある。これらの要素のいずれも、私たちのビジネス機会を減少または除去し、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性があります。
遺伝子編集分野の開発活動は現在,ある知的財産権の所有権や使用に関するいくつかのリスクに直面しており,これらのリスクは米国では特許干渉プログラムの影響を受け,ヨーロッパでは反対プログラムの影響を受けている。私たちおよび私たちのライセンシーに適用される可能性のある知的財産権のリスクに関するより多くの情報については、より多くの情報を知るために“-当社の知的財産権に関連するリスク”というタイトルの節を参照してください。
また,新たな治療法や新たな治療法の採用に関する公衆の見方や関連メディアの報告,遺伝子編集に特に関連する倫理的懸念は,被験者の臨床試験への参加意欲に悪影響を及ぼす可能性があり,あるいは任意の治療法が承認された場合,医師や患者はこれらの新たかつ個性的な治療を受ける可能性がある。医師、医療保健提供者と第三者支払人が新製品、新技術と新しい治療方法を採用する速度はしばしば遅く、特に追加の前期コストと訓練を必要とする可能性のある製品、技術と治療方法である。医師はこれらの新規かつ潜在的な個人化療法を用いた研修を受けたくない可能性があり,特定の療法が複雑すぎたりリスクがある可能性があり,適切な訓練なしには採用できず,施行しないことを選択する可能性がある。また、健康状態、遺伝子特徴或いはその他の原因により、ある患者は治療に適していない可能性がある。さらに、連邦や州機関、国会委員会、外国政府の公衆に対する否定的な見方、道徳的関心、または財務的考慮に対する反応は、新しい立法、法規、または医療基準を招く可能性があり、任意の候補製品の開発または商業化、規制承認を得たり、維持したり、他の方法で利益を達成する能力を制限する可能性がある。新しい政府要求を制定し、規制部門が私たちが開発している候補製品を承認することを延期または阻止するかもしれない。立法が変わるかどうか,条例,政策やガイドラインが変わるかどうか,機関や裁判所の解釈が変わるかどうか,あるいはこれらの変化の影響(あれば)が何になるかは予測できない。これらの要素や他の要素に基づいて, 衛生保健提供者や支払人は決定する可能性があり、これらの新しい療法のメリットはそれらのコストを超えないか、または超えることはない。
遺伝子編集分野は比較的新しい分野であり、急速に発展している。我々は塩基編集技術を用いた遺伝子編集に重点を置いているが,他の遺伝子編集技術が塩基編集よりも著しく優れており,我々の業務に実質的な被害を与える可能性がある.
我々はこれまで塩基編集を用いた遺伝子編集技術に取り組んできた.他社もこれまで亜鉛フィンガーヌクレアーゼを用いた遺伝子編集技術の研究·開発を行ってきた
80
工学的メガヌクレアーゼと転写活性化物様エフェクターヌクレアーゼであるが、これまでに候補製品の1種が発売許可されていない。塩基編集技術が遺伝子薬物の開発を招くことは肯定できない、あるいは他の遺伝子編集技術は薬物開発により良いあるいは魅力的とは考えられない。例えば,マサチューセッツ工科大学の張峰とブロイド,およびコロンビア大学のサミュエル·スターンバーグはそれぞれトランスポゾンの使用,すなわち“ジャンプ遺伝子”を発見したと発表した。トランスポゾンは、ゲノムの異なる位置に自身を挿入することができ、DNA中に二本鎖切断を引き起こすことなく、特定のDNA配列を特定の位置に運ぶようにプログラムすることができる。BEAMは一次編集技術を使用し、この技術はCRISPR蛋白を利用してDNA中の突然変異位置を定位し、標的DNAの一本鎖を切断する。誘導RNAは、CRISPRタンパク質が誘導RNAに相補的なDNA配列を認識することを可能にし、逆転写のためのプライマーおよび置換鋳型を担持する。逆転写酵素はノッチ部位のテンプレート配列を複製し,編集を実装する.
他社はIntellia Treateutics,Intellia Treateutics Inc.,NTLA−2001の1 b期試験の臨床データを報告しており,CRISPR/Cas 9に基づく遺伝子編集製品候補案であり,遺伝性トランスサイレチンアミロイドーシス合併多発性神経障害の治療や甲状腺貫通ホルモン(ATTR)アミロイドーシス合併心筋症の治療に用いられている。同様に,他に発見されていない新しい遺伝子編集技術は塩基編集よりも魅力的である可能性がある.また,他の遺伝子編集技術を開発または使用する権利を得ることができるかどうかは確認できない.これらの要素のいずれも、私たちのビジネス機会を減少または除去することができ、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちは潜在的な候補製品を探して開発する努力で成功しないかもしれない。もしこのような努力が成功しなければ、私たちは決して商業舞台会社にならないかもしれないし、何の収入も生まれないかもしれない。
私たちの業務の成功は主に遺伝子編集技術を用いて候補製品を識別、開発、商業化する能力にかかっている。私たちは最近ニュージーランドとイギリスで初めてのVERVE-101臨床試験を開始しました様々な理由から,われわれの研究計画は臨床開発の潜在的候補製品を決定できない可能性がある。私たちの研究方法は他の潜在的な候補製品の識別に成功できない可能性があり、私たちの潜在的な候補製品は臨床前に有害な副作用があることが証明される可能性がある体外培養実験または動物モデル研究では、それらはこのような実験または研究において有望な治療効果信号を示さない可能性があり、またはそれらは他の特徴を有する可能性があり、候補製品が生産に適用されない、販売できない、または発売承認を得ることができない可能性がある。
新冠肺炎疫病は現在或いは未来の臨床前研究と臨床試験を開始し、完成する能力に影響し、監督管理活動を混乱させ、或いは著者らの業務と運営に他の不利な影響を与える可能性がある。また、この大流行は世界各地の経済に悪影響を与え、私たちの業務、運営、将来性に悪影響を及ぼす可能性がある。
新冠肺炎の疫病はすでに多くの国の政府に措置を取り続け、隔離、旅行制限、国境審査とその他の措置を強化することで疫病の伝播を遅らせる可能性がある。大流行と政府の対応措置も企業と商業に直接と間接的な重大な影響を与え、労働力不足が発生したため、サプライチェーンの中断、施設と生産の一時停止、医療サービスや用品などのある商品やサービスに対する需要が急増し、旅行などの他の商品やサービスに対する需要が低下した。
大流行の将来の進展とそれが私たちの業務や業務に与える影響は不確実だ。私たちと私たちの契約製造組織(CMO)と契約研究組織(CRO)は、研究規模の生産といくつかの臨床前研究を実行する能力の低下を経験しており、私たちはすでに私たちの臨床前研究と臨床試験を開始し、完成する能力に影響を与える中断、および私たちの研究開発活動に重要なプロジェクト調達の中断に直面している可能性がある:
81
私たちと私たちのCROおよびCMOはまた、臨床前研究の遅延、生産中断、必要な機関審査委員会またはIRB、機関生物安全委員会またはIBCまたは他の必要な現場承認を得る能力、および臨床試験場所の他の遅延を含む、私たちおよび将来のINDイネーブル研究および臨床試験に関連する中断に直面する可能性がある。
新冠肺炎の疫病に対応することはまた、監督管理と知的財産権事務の資源配分を変更する可能性があり、それによって、私たちの監督管理の承認と私たちの知的財産権を保護する能力に悪影響を与える可能性があり、例えばアメリカ食品·薬物管理局や他の監督管理機関の運営中断や遅延を招き、これは審査と承認スケジュールに影響を与える可能性がある。新冠肺炎の疫病のため、私たちは食品と薬物管理局との協力が遅延した。また、対面インタラクションを制限するための措置により、規制会議や承認の障害や遅延に直面する可能性があります。私たちは新冠肺炎疫病が私たちの業務に与える全体的な影響を確定することができません。上述の原因で、それは私たちの業務、財務状況、運営結果と将来性に不利な影響を与える可能性があります。
臨床薬物開発は長くて高価な過程に関連し、結果は不確定である。もし私たちが最終的に規制部門の私たちの候補製品の承認を得ることができなければ、私たちの業務は深刻な損害を受けるだろう。
私たちはすべての候補製品の失敗の危険が高い。私たちの候補製品がいつ、人体で有効または安全を証明するか、あるいは発売許可を得るかどうかを予測することはできません。FDA、EMA、または他の同様の外国規制機関から承認を得るのに要する時間は予測できないが、通常は臨床試験開始後数年後に必要であり、監督機関の重大な裁量権を含む多くの要素に依存する。監督部門の許可を得て任意の候補製品を販売する前に、私たちは臨床前開発を完成し、それから広範な臨床試験を行い、私たちの候補製品の人体における安全性と有効性を証明しなければならない。われわれは最近ニュージーランドとイギリスでVERVE−101の臨床試験を開始したが,いずれの臨床試験も完了していない。臨床試験は私たちの候補製品が人類にとって安全であり、指定用途に有効であることを証明できないかもしれない。たとえ私たちが開発可能な任意の候補製品の初期臨床試験が成功したとしても、私たちが開発する可能性のあるこれらの候補製品は、臨床開発の後期段階で期待される安全性と有効性を示すことができないかもしれないが、臨床前研究と予備臨床試験に成功したにもかかわらず。臨床試験中の薬物や生物製品の失敗率が高かった。製薬と生物技術業界のいくつかの会社は後期臨床試験で重大な挫折を経験し、早期臨床試験においても奮い立つ結果を得た。また、臨床試験が成功しても、開発期間中の上場承認政策の変化、付加法規の変化或いは公布, 各提出された製品申請について、法規または指導意見または規制審査の変化は、申請の承認または拒否の遅延をもたらす可能性がある。
候補製品の臨床試験を開始する前に、私たちは米国と海外で提出される予定のINDおよび他の規制文書を支持するために、広範な臨床前試験と研究を完成させなければならない。私たちは私たちの臨床前テストと研究の適時な完成或いは結果を確定することができず、また私たちの臨床前テストと研究の結果が最終的に私たちの現在或いは未来の候補製品の更なる開発を支持するかどうか、あるいは監督機関が私たちの提案した臨床計画を受け入れるかどうかを予測することができない。したがって、私たちは、私たちが予想している時間内に米国のINDまたは同様の外国の申請を提出して臨床開発を開始することができない可能性があり、これらの申請の提出は、規制機関が臨床試験の開始を許可することを引き起こさない可能性がある。
例えばFDAは2022年11月にIND申請を臨床試験評価に提出しました
VERVE-101はアメリカで一時停止状態にある。著者らは2022年12月にFDAの臨床座礁信頼を受け取り、その中で、以下に関連する追加の臨床前データを含む臨床棚上げを解決するために必要な情報を概説した:(I)ヒトと非ヒト細胞間の潜在的差異、(Ii)生殖系編集のリスク、および(Iii)非肝細胞タイプの非標的分析。FDAは,進行中の心臓1号臨床試験の臨床データの提供も求めている。さらに、FDAは、追加の避妊措置を取り入れ、参加者用量間の交互間隔の長さを増加させるために、米国の試験計画を修正することを要求している。
米国が試験を開始する前に、私たちはIND申請の棚上げ問題の解決を要求されるだろう。一時停止が速やかに解除されるかどうか,あるいは全くできないかどうかは確認できず,米国でVERVE−101の臨床試験を開始できない可能性がある。一時停止により,米国でVERVE−101臨床試験を開始する能力のいかなる遅延も,あるいはVERVE−101の臨床試験を開始することができず,我々のVERVE−101臨床開発計画を遅らせる可能性があり,追加の臨床前または臨床開発コストが必要となる可能性があり,最終的にFDAの承認を得たVERVE−101の能力を低下させる可能性がある。VERVE−101のいずれの臨床試験も遅延完了が可能である
82
私たちのコストを増加させ、私たちの候補製品の開発と承認の流れを遅くし、製品の販売を開始し、収入を創出する能力を遅延または危険にさらす可能性があります。
また,候補製品は臨床前安全性研究を継続する必要があり,これらの研究はわれわれの臨床試験と同時に行われる可能性がある。これらの安全性研究の結果は,将来の臨床試験の開始や登録を遅らせる可能性があり,臨床試験を継続する能力に影響する可能性がある。
臨床試験は費用が高く,設計と実施が困難であり,完成まで数年かかる可能性があり,結果はまだ確定していない。私たちのどの臨床試験も計画通りに行われるか、計画通りに完成されるか、あるいは全くできないことは保証されない。1つまたは複数の臨床試験の失敗は、研究設計の欠陥、用量選択問題、プラセボ効果、患者登録基準、および良好な安全性または有効性特徴を証明できなかったことを含むが、これらに限定されない、試験の任意の段階で発生する可能性がある。
臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、しかし依然としてその製品のマーケティング許可を得られなかった。さらに、私たちの任意の候補製品が任意の臨床試験において安全性および有効性を証明できない場合、私たちの他の候補製品の感想に負の影響を与える可能性があり、および/またはFDA、EMA、または他の規制機関が、私たちの任意の候補製品を承認する前に追加の試験を要求する可能性がある。
私たちの現在と未来の候補製品は、以下の理由を含む、多くの理由で規制部門の承認を得ることができないかもしれない
この長い承認過程および臨床試験結果の予測不可能性は、規制部門の承認を得ることができず、私たちが開発したすべての候補製品を市場に出すことができず、私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性がある。
FDA、EMA、および他の同様の外国規制機関は、承認過程においてかなりの自由裁量権を有し、私たちが開発した任意の候補製品がいつ、または規制承認を受けるかどうかを決定する。私たちが行っているまたは将来の候補製品臨床試験から収集されたデータが有望であると信じていても、これらのデータは、FDA、EMA、または任意の他の類似した外国規制機関の承認をサポートするのに十分ではないかもしれない。
私たちが承認されても、規制機関は、私たちが要求しているものよりも少ないか制限されているか、高価な発売後の臨床試験の表現によって承認されるかもしれない、またはその候補製品の商業化に必要または必要なラベル宣言を含まないラベルを承認することができる任意の候補製品を承認することができる。また、アメリカ以外では、規制機関は私たちが私たちの製品に徴収しようとしている価格を承認しないかもしれない。上記のいずれの場合も、私たちの候補製品のビジネス見通しに実質的な損害を与える可能性があります。
83
新冠肺炎の大流行に対応するため、アメリカ食品薬品監督管理局は2020年3月18日に指導意見を発表し、その後2020年7月2日、2021年1月27日と2021年8月30日に指導意見を更新し、大流行期間中に臨床試験を行う問題を解決した。指導意見は疫病の影響を受ける臨床試験のスポンサーにいくつかの考慮事項を提出し、臨床研究報告(或いは単独の文書として)に管理研究のための緊急措置、及び新冠肺炎が研究に与えるいかなる妨害を含むことを要求することを含む;唯一の被験者識別子と研究場所に従って新冠肺炎関連研究妨害の影響を受けるすべての研究参加者リストをリストし、そして個人参加状況がどのように変化したかを説明する。実施された緊急対策(例えば、参加者が研究製品および/または研究を停止し、重要な安全性および/または有効性データを収集するための代替プログラム)と、研究報告のための安全性および有効性結果への影響の分析および対応する議論とを含む。このガイドラインの最新更新では,FDAは過去1年間に受けた臨床従事者からの問題を解決し,これらの事業者は大流行環境での操作に適応している。他にも、これらの問題は、いつ試験を一時停止、継続または開始するか、およびINDスキームの修正および遠隔現場監視アクセスをどのように提出するかに集中している。大流行期間中に臨床研究を指導するガイドラインが継続的に有効であることは保証されない,あるいは有効であっても,これらのリスクや挑戦への対応に寄与する保証はない。2023年1月30日、バイデン政府は、2023年5月11日に新冠肺炎に関する公衆衛生緊急声明を終了すると発表した。2023年1月31日, アメリカ食品薬品監督管理局は、それはすぐに連邦登録通知を発表し、公衆衛生突発事件の終了がどのようにこの機関の新冠肺炎関連ガイドラインに影響するかを記述し、臨床試験ガイドラインと更新を含む。
したがって,十分な数の患者を臨床試験に参加することができないことは重大な遅延を招くか,あるいは1つ以上の臨床試験を完全に放棄する必要があるかもしれない。私たちの臨床試験の登録遅延は、私たちの候補製品の開発コストを増加させ、候補製品の開発と承認過程を減速または停止させ、製品の販売開始と収入創出に必要なマーケティング承認を求める能力を危険にさらす可能性があり、これはわが社の価値を低下させ、必要に応じて追加融資を得る能力を制限する。
そのため、新冠肺炎の疫病は引き続き世界経済と金融市場に重大な影響を与える可能性があり、これは私たちの業務と運営に不利な影響を与える可能性があり、公募株を通じてより多くの資金を調達する能力に影響を与え、私たちの株価と取引の変動性に影響を与える。私たちは新冠肺炎疫病が私たちの業務に与える全体的な影響を確定することができなくて、それは私たちの業務、財務状況、運営結果と将来性に不利な影響を与える可能性がある。
臨床前研究と早期臨床試験の結果は未来の結果或いはその後の臨床前研究と臨床試験の成功を予測できないかもしれない。
私たちは最近始まって臨床試験を始めましたそのため、私たちのプロジェクトの潜在力に対する信念は研究と臨床前研究に基づいている。しかし、臨床前研究の結果は後の臨床前研究或いは臨床試験の結果を予測できない可能性があり、いかなる早期臨床試験の結果も後の臨床試験の結果を予測できない可能性がある。そのほか、臨床試験の初歩的な成功はこれらの試験が完成した後に得られた結果を代表しないかもしれない。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、しかし依然としてその製品のマーケティング許可を得られなかった。我々の候補品については非ヒト霊長類動物において数回の臨床前研究が行われているが,これらの研究で観察された結果が我々の候補品がヒトでの臨床試験で類似した結果に変換されるかどうかは決定できない。私たちが行っているあるいは未来の臨床試験は最終的には成功しないかもしれないし、私たちが開発する可能性のある任意の候補製品のさらなる臨床開発を支持しないかもしれない。臨床試験を通過した候補製品は高い失敗率を示した。製薬と生物技術業界のいくつかの会社は臨床開発において重大な挫折を経験し、早期の研究においても鼓舞的な結果を得た。私たちの臨床開発におけるどのような挫折も、私たちの業務と運営結果に実質的な損害を与える可能性がある。
84
私たちは私たちの候補製品の開発と商業化を完成したり、最終的に完成できなかったりする過程で意外なコストや遅延が生じるかもしれません。
臨床試験中や臨床試験の結果では、私たちは多くの予見できない事件に遭遇する可能性があり、これらの事件は、私たちの上場承認を延期したり、私たちの候補製品を商業化することを阻止したりする可能性があります
臨床試験が我々、そのような試験を行う機関のIRBsまたはその倫理委員会、そのような試験のデータ審査委員会またはデータ安全監視委員会またはFDA、EMAまたは他の外国規制機関によって一時停止または終了された場合、遅延に遭遇する可能性がある。このような機構は多種の要素のために臨床試験を一時停止或いは中止する可能性があり、これらの要素は法規の要求或いは著者らの臨床規程に従って臨床試験、FDA、EMA或いは他の外国の監督機関による臨床試験操作或いは試験場所の検査を行うことができなかったことを含み、臨床一時停止、予見できない安全問題或いは不良副作用を招き、著者らの候補製品が属する製品種別に関連する問題を含む。
もし私たちの候補製品に対して、現在予想されている以上の追加の臨床試験または他の試験を行うことを要求された場合、もし私たちが候補製品の臨床試験または他の試験を成功させることができなければ、これらの試験または試験の結果が陽性でないか、または軽微な陽性である場合、または安全問題がある場合、私たちは:
85
もし著者らが臨床前研究或いは臨床試験或いは上場承認を得る上で遅延に遭遇すれば、著者らの開発コストも増加する。われわれのいかなる臨床前研究や臨床試験が計画通りに開始されるかどうか,再構成が必要かどうか,あるいは予定通りに完成するかどうか,あるいは全く知られていない。私たちはまた、より多くの患者または腕を増加させることを含む、1つまたは複数の臨床試験の設計または計画を変更することを決定することができ、これは、コストおよび費用の増加および/または遅延をもたらす可能性がある。重大な臨床前研究または臨床試験遅延は、候補製品を商業化する独占的な権利を有する私たちの任意の期限を短縮するか、または私たちの競争相手が私たちの前に製品を市場に出すことを可能にし、候補製品を商業化することに成功する能力を弱化させ、私たちの業務と運営結果を損なう可能性がある。
臨床前薬物の開発は不確実である。私たちの臨床前計画の一部またはすべてが遅延する可能性があり、または決して臨床試験に入らない可能性があり、これは、市場の承認をタイムリーに得るか、またはこれらの候補製品を商業化する能力に悪影響を与え、これは私たちの業務に悪影響を及ぼすであろう。
FDAの承認を得て新しい生物製品を販売するためには,製品の純度(または製品品質)および人体に対する安全性と効力または効果の証明を証明しなければならない。これらの要求を満たすためには,十分かつ良好な制御の臨床試験が必要である。候補製品の臨床試験を開始する前に、米国のINDを支持するために、広範な臨床前試験と研究を完成させなければならない。私たちは私たちの臨床前テストと研究の適時な完成或いは結果を確定することができず、FDAが私たちが提案した臨床計画を受け入れるかどうか、あるいは私たちの臨床前テストと研究の結果が最終的にこれらの候補製品の更なる開発を支持するかどうかを予測することもできない。したがって,我々が期待しているスケジュール上でINDや同様の臨床前計画申請を提出できることを保証することはできず,INDや同様の申請の提出がFDAや他の規制機関が臨床試験の開始を許可することを保証することもできない。例えば,FDAは2022年11月,米国におけるINDの評価VERVE−101の臨床試験の申請を一時停止した。
臨床試験を行う前は長く、時間がかかり、高価な過程である。時間長は、候補製品のタイプ、複雑性、新規性、および予期される用途によって大きく異なる可能性があり、各候補製品は、通常、数年またはそれ以上の時間とすることができる。私たち自身が臨床前テストや研究を行った候補製品に関する遅延は、追加の運営費用を発生させる可能性があります。また,我々は潜在的なパートナーによるいくつかの候補製品の臨床前テストや研究に関連する遅延の影響を受ける可能性があり,これを制御することはできない。候補製品の臨床前研究および臨床試験の開始および完成速度は、例えば、多くの要因によって遅延する可能性がある
また、他の候補製品の臨床試験を確実に開始しても、私たちの開発は成功しない可能性があり、私たちまたは第三者が私たちを代表して行った臨床試験は、製品の純度(または品質)および私たちの任意の候補製品または私たちの技術を使用した候補製品のために必要なマーケティング承認を得るために必要な安全性および有効性または有効性証明を証明できない可能性がある。われわれが前臨床研究や予備臨床試験から積極的な結果を得ても,将来の試験で同様の成功を収めることはできないかもしれない。
もし私たちが臨床試験の患者登録過程で遅延や困難に遭遇した場合、必要な規制承認を受けることは延期または阻止される可能性がある。
患者に著者らの候補製品に参加する資格を有する臨床試験を確定し、参加させることは著者らの成功に重要である。2022年には,特定の国の合意に基づいてニュージーランドとイギリスでVERVE−101の心臓1号臨床試験を開始し,国ごとに資格を異なる修正を行った。臨床試験の成功とタイムリーな完成には,試験が終了するまで十分な数の患者を募集する必要がある。もしそうであれば、候補製品のために他の臨床試験を開始したり、継続することができないかもしれません
86
これらの試験に参加するのに十分な数の条件を満たす患者を見つけることができず,FDAや米国以外の同様の規制機関の要求である。アテローム性動脈硬化性心血管疾患(ASCVD)の患者数が多いため、もし著者らがASCVDと診断された患者を治療するためのVERVE-101或いはVERVE-201の臨床開発を拡大すれば、監督機関のこの適応の許可を得るために臨床試験を行うために必要な患者の数は非常に高い可能性があり、私たちは十分な数の患者を募集できない可能性があるため、著者らはすでに確定診断されたASCVD患者を治療するためのVERVE-101臨床試験を起動或いは完成できない可能性がある。ホモ接合体家族性高コレステロール血症(HoFH)の患者数が少ないため,患者募集が困難である可能性があり,VERVE−201によるHoFH治療の臨床試験を開始あるいは完了できない可能性がある。
患者の入選は様々な他の要素の影響を受けている
他の製薬やバイオテクノロジー社は,新冠肺炎の流行により,彼らが行っている臨床試験の登録が遅れており,このような遅延に遭遇する可能性があると報告している。著者らは十分な数の患者を発見し、著者らの臨床試験に参加することができず、重大な遅延を招き、1つ以上の臨床試験を完全に放棄し、必要な監督管理許可を得ることを延期或いは阻止する必要があるかもしれない。私たちの臨床試験の登録遅延は私たちの候補製品の開発コストを増加させる可能性があり、これはわが社の価値を低下させ、追加融資を受ける能力を制限する。
将来の臨床試験のために十分な数の患者を募集することができても,われわれの臨床試験で患者を維持することは困難である可能性がある。最終的にプラセボ治療を受けた多くの患者は、試験を受けた候補製品を受けていないことを認識するかもしれず、彼らは試験を継続するのではなく、代替療法を求めるために、私たちの臨床試験から撤退することを決定するかもしれない。もし私たちが十分な数の患者を募集したり維持したりすることが困難な場合、私たちは臨床試験を延期、制限、あるいは中止する必要があるかもしれません。その中のいずれも私たちの業務、財務状況、運営結果、将来性を損なうことになります。
もし私たちが開発する可能性のある任意の候補製品または私たちがこれらの候補製品の配送パターンを管理する場合、深刻な不良イベント、不良副作用または意外な特性をもたらし、このようなイベント、副作用または特性は、規制部門の候補製品の承認を遅延または阻止し、商業潜在力を制限し、または任意の潜在的なマーケティング承認後に重大な負の結果を招く可能性がある。
われわれは最近VERVE−101の心臓1号臨床試験を開始した。また,遺伝子編集技術を用いた臨床試験の数は限られており,完成していない臨床試験は,我々がVERVE−101で用いている遺伝子編集技術のような塩基編集技術に関する。また、現在、遺伝子編集製品候補製品は監督部門の許可を得て人間に使用されていない。私たちは私たちが開発する可能性のある任意の候補製品がいつ、あるいは人間にとって安全であることが証明されるか予測できない。遺伝子編集技術が副作用を起こさない保証はありません
87
患者DNAの不適切な編集はリンパ腫、白血病或いは他の癌或いは他の機能異常の細胞を招く可能性がある。
任意の候補遺伝子編集製品の1つの重大なリスクは、深刻な有害事象、不良副作用、または予期しない特徴をもたらす可能性がある“非目標”編集が発生する可能性があることである。著者らは著者らが行っている或いは未来のいかなる臨床研究においても非標的編集が発生しないことを確定することはできず、臨床前研究で観察されなかった副作用はこのような副作用が人類の臨床研究で発生しないことを保証することはできない。DNA編集の潜在的永久性或いは遺伝物質の候補製品を携帯するための他の成分のため、遺伝子編集者への曝露後の遅延或いは遅延は不良事象を呈する潜在的なリスクも存在する。また,遺伝子編集は永久的に変化するため,副作用を認めた後でも治療を取り消すことはできない。
私たちはLNPsを使って私たちの遺伝子エディタを肝臓に送っています最近,LNPsはファイザーやファイザー社が開発した新冠肺炎ワクチンや,BioNTech SEやModerna社が開発したワクチンを含むメッセンジャーリボ核酸をヒト内で伝達するために使用されており,LNPsは臨床試験で治療用のメッセンジャーリボ核酸を伝達するために用いられている。LNPsは肝臓損傷および/または全身炎症反応を引き起こす可能性があり,いずれも致命的である可能性がある。我々のLNPsの最適化を継続することを目標としているが,我々のLNPsが悪影響を与えない保証はない.我々のLNPは、肝臓損傷、免疫反応、輸液反応、補体反応、オプソニン反応、抗体反応、IgA、IgM、IgEまたはIgGまたはそれらの何らかの組み合わせ、またはLNPに関連するいくつかの脂質またはポリエチレングリコール成分のポリエチレングリコールまたはポリエチレングリコールに対する反応のうちの1つまたは複数をもたらす可能性がある。著者らは薬物のいくつかの方面はmRNA或いは脂質からの免疫反応、及び肝臓経路内の副作用或いはmRNA或いはLNPの分解を引き起こす可能性があり、その中のいずれも著者らが行っている1つ以上の臨床試験における重大な有害事象を引き起こす可能性がある。他のLNPsでもいくつかのタイプの悪影響が観察された。このような有害事象の根本的な原因には不確実性が存在する可能性があり,進行中や将来の臨床試験における副作用を正確に予測することは困難であり,われわれの計画の深刻な遅延を招くであろう。
我々がVERVE−201で使用する予定のGalNAc−LNPsは,遺伝子エディタを肝臓に運ぶ新しい送達機構であり,ヒトでは研究されていない。
もし私たちが開発した任意の候補製品が深刻な有害事象、副作用、または予期しない特徴に関連している場合、私たちはその開発を放棄する必要があるかもしれないし、深刻な有害事象、副作用、または他の特徴があまり一般的ではない、あまり深刻ではない、またはリスク効果の観点からより受け入れやすい特定の用途または人々に制限する必要があり、これらはいずれも私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす。
将来私たちが上記のいかなる有害事象が私たちの候補製品以外の要素によって引き起こされているかを証明できなければ、FDA、EMA、または他の規制機関は、私たちが任意のまたはすべての目標適応に対して開発できる任意の候補製品の開発を停止または拒否するように命令することができる。もし発売後の後続研究で深刻な安全問題が発見された場合、彼らはマーケティング許可を取り消すこともできる。将来のすべての深刻な有害事象が製品に関連していないことを証明できても,このような事件は患者募集や入選患者の試験完了能力に影響を与える可能性がある。さらに、私たちが私たちが開発する可能性のある任意の候補製品の臨床試験を延期、一時停止、または終了することを選択または要求された場合、その候補製品の商業的将来性が損なわれる可能性があり、これらの候補製品から製品収入を生成する能力は延期またはキャンセルされる可能性がある。これらの状況のいずれも、候補製品を識別し開発する能力を損なう可能性があり、私たちの業務、財務状況、運営結果、および将来性を深刻に損なう可能性があります。
公衆の遺伝子薬物に対する否定的な見方、特に遺伝子編集と塩基編集は、監督管理部門が著者らの潜在製品の承認及び/或いは需要に負の影響を与える可能性がある。
私たちの計画にはヒトゲノムの編集が含まれています著者らの候補製品の臨床と商業成功は遺伝子編集療法を用いたヒト疾患の予防或いは治療に対する公衆の理解と受け入れにある程度依存する。公衆の態度は遺伝子編集の不安全、不道徳あるいは不道徳な説の影響を受ける可能性があるため、私たちの候補製品は公衆或いは医学界の受け入れを得ることができない可能性がある。公衆の悪い態度は私たちが臨床試験を募集する能力に悪影響を及ぼすかもしれない。さらに、私たちの成功は、医師が処方した処方および彼らの患者が受けることを望む治療に依存し、これらの治療は、彼らがよく知っている既存の治療方法を代替または補充するために、私たちが開発可能な候補製品を使用することに関連し、より多くの臨床データを得ることができる。
また,倫理的懸念により,遺伝子編集技術は公衆の議論とより厳しい規制審査を受けている。
88
より厳格な政府法規または否定的な世論は、私たちの業務や財務状況に負の影響を与え、候補製品の開発および商業化、または私たちが開発する可能性のある任意の候補製品の需要を遅延または損害する可能性がある。我々の前臨床研究または臨床試験または我々の許可者、パートナーまたは競争相手、または遺伝子編集技術を使用する学術研究者の有害事象は、最終的には、我々が識別し開発する可能性のある候補製品に起因することができなくても、それによって生じる宣伝は、政府規制の増加、不利な公衆認識をもたらす可能性があり、私たちは、識別および開発された潜在的候補製品テストまたは承認の潜在的規制遅延、承認された候補製品に対するより厳しいラベル要求、およびそのような任意の候補製品に対する需要の減少を識別することができる。第三者や政府が遺伝子編集技術を利用して米国の国家安全を脅かす生物製剤や製品を開発することも,我々に同様の負の影響を与える可能性がある。
私たちが時々発表したり公表したりする臨床試験の中期と初歩的な結果は、より多くの参加者のデータの獲得によって変化し、監査と検証プログラムの制限を受ける可能性があり、これは最終データの重大な変化を招く可能性がある。
私たちは時々私たちの臨床試験の中期または初歩的な結果を発表したり報告したりするかもしれない。我々が達成可能な臨床試験の中期結果は、参加者登録の継続およびより多くの参加者データの取得に伴い、1つまたは複数の臨床結果が実質的に変化する可能性があるというリスクに直面する可能性がある。データ分析の一部として、私たちはまた仮定、推定、計算、および結論を下すだろうが、私たちはすべてのデータを完全に評価する機会がないか、または完全に評価する機会がないかもしれない。初期、中期、またはトップラインデータもまだ監査および確認手続きを受ける必要があり、これは、最終データが私たちが以前に公表した予備データまたは中間データと大きく異なる可能性がある。したがって、最終データが利用可能になる前に、予備、中期、または主要データを慎重に見なければならない。初期または中期データと最終データとの間の不利な差は実質的である可能性があり、私たちの名声および業務の将来性を深刻に損なう可能性があり、私たちの普通株の取引価格の大幅な変動を招く可能性がある。
遺伝子薬は複雑で製造が難しい。私たちは規制機関の要求を満たす遅延や生産の問題に直面するかもしれません。これらの問題は、私たちの開発計画の遅延、私たちが開発する可能性のある候補製品の供給を制限したり、他の方法で私たちの業務を損なうことになります。
私たちが開発可能などの候補製品も、ほとんどの化学薬に必要な加工工程よりも複雑な加工工程を必要とするかもしれない。また,化学薬とは異なり,生物の物理的·化学的性質は,我々が開発しようとしている候補製品のように,通常完全には表現できない。したがって、候補完成品の分析は、候補製品が予期される方法で動作することを確実にするのに十分ではない可能性がある。製造過程の問題は、正常過程との微小な偏差であっても、製品欠陥或いは製造失敗を招く可能性があり、それによって大量故障、製品リコール、製品責任クレーム或いは在庫不足、或いは私たちの潜在的なIND申告の進展を遅延させる可能性がある。もし私たちが候補製品の開発に成功したら、私たちは問題に直面する可能性があり、十分な数量と品質の臨床レベルの材料を得ることができません。これらの材料はFDA、EMAあるいは他の同様の適用外国標準や規範に適合し、一致して受け入れ可能な生産生産量とコストを持っています。さらに、私たちが開発可能な候補製品は、製造過程に追加の複雑さを導入するLNPsのような複雑な送達モデルを必要とするだろう。
さらに、FDA、EMA、および他の規制機関は、任意のバッチで承認された製品のサンプルを提出し、適用試験結果を示すプロトコルをいつでも提出することを要求するかもしれません。場合によっては、FDA、EMA、または他の規制機関は、機関が発行を許可する前に大量の製品を配布しないように要求するかもしれません。製造過程中の微小な偏差は、品質属性や安定性に影響を与える偏差を含み、製品に許容できない変化を招き、大量故障や製品リコールを招く可能性がある。ロット失敗や製品リコールは臨床試験や製品発表を延期させる可能性があります。そうでなければ、私たちの業務、財務状況、運営結果、将来性を損なうかもしれません。
私たちはまた、私たちの製造プロセスを管理するために必要な経験豊富な科学、品質管理と製造人員を採用と維持することに問題がある可能性があり、これは私たちの生産遅延或いは適用法規の要求を維持することを維持することを招く可能性がある。
生物製品生産の性質を考慮して、生産過程において汚染リスクが存在する。どの汚染も私たちが計画通りに候補製品を生産する能力を深刻に損なう可能性があり、私たちの運営結果を損害し、名声の損害をもたらす可能性がある。私たちは製造過程で必要ないくつかの原材料が生物源から来ると予想している。このような原材料は入手が難しく、汚染されたりリコールされる可能性がある。私たちが開発する可能性のある任意の候補製品の製造に生体由来物質を使用する材料不足、汚染、リコール、または制限は
89
商業製造または臨床材料の生産に悪影響または中断をもたらし、これは私たちの開発スケジュールおよび私たちの業務、財務状況、運営結果、将来性に実質的な損害を与える可能性がある。
私たちの製造プロセスや私たちと契約した施設のいずれの問題も、潜在的なパートナー(大きな製薬会社や学術研究機関を含む)の魅力の低いパートナーになる可能性があり、これは、より多くの魅力的な開発プロジェクトを獲得することを制限する可能性があります。第三者製造プロセスまたは施設における問題は、私たちが行っているまたは計画されている任意の臨床試験に十分な臨床材料を提供する能力を確保し、私たちが開発および商業化する任意の候補製品の市場需要を満たすことを制限する可能性もある。
もし私たちの候補製品が発売承認された場合、私たちまたは他の人は後にその薬の効果が以前に考えられていたほど効果的ではないことを発見したり、以前に発見されなかった副作用を招いたりして、私たちがその薬を販売する能力が影響を受ける可能性がある。
我々の候補製品の臨床試験は,入念に定義された臨床試験に同意した患者のサブセットで行われた。したがって,われわれの臨床試験では,候補製品の有意なプラス効果が実際のプラス効果(あれば)よりも大きいか,あるいは不良副作用を認識できない可能性がある。もし私たちの1つ以上の候補製品が監督部門の承認を得た場合、私たちまたは他の人はその後、それらの効果が以前に考えられていたほど効果的ではないことを発見し、あるいは不良な副作用をもたらし、多くの潜在的な重大な負の結果を招く可能性がある
これらの事件のいずれも、特定の候補製品に対する市場の受容度を達成または維持することを阻止することができ、承認されれば、私たちの業務、財務状況、および運営結果を深刻に損なう可能性がある。
私たちは、より利益的または成功可能性の高い候補製品または適応を利用することなく、特定の候補製品または適応を追求するために限られたリソースを費やす可能性がある。
私たちの財務と管理資源が限られているので、私たちは他の候補製品を探す機会を放棄したり、延期したり、その後、より大きなビジネス潜在力を持つことが証明された他の兆候の機会を放棄したり延期したりするかもしれません。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。私たちの現在と未来の研究開発計画および特定の適応候補製品への支出はいかなる商業的に実行可能な製品も発生しないかもしれない。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の印税手配によって候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利だ。資源の配分や利用戦略に成功しなければ、我々の業務、財務状況、運営結果に悪影響を及ぼす。
90
われわれは臨床試験を行っており,米国以外の地点でより多くの臨床試験を行う予定である。FDAはこれらの地点で行われた試験のデータを受け入れない可能性があり,米国以外で試験を行うことで余分な遅延や費用に直面する可能性がある。
私たちは、ニュージーランドとイギリスの試験地点で行われているVERVE−101の1 b段階試験を含む、米国以外の1つまたは複数の試験地点で1つまたは複数の追加の臨床試験を行うことを計画している。FDAは米国国外で行われた臨床試験のデータを受け入れることができるが,これらのデータの受け入れはFDAが適用した条件に依存する。例えば、臨床試験は入念に設計と進行し、合格した研究者が道徳原則に従って行わなければならない。必要であれば,FDAは現場検査により試験データを検証できる必要がある。試験群はまたアメリカ人口を十分に代表しなければならず、データはFDAが臨床的意義があると考えられる方式でアメリカ人口とアメリカの医療実践に適用しなければならない。また,これらの臨床試験は適用される現地法に制限されているが,FDAがこれらのデータを受け入れるかどうかは,試験がすべての適用された米国の法律や法規にも適合しているかどうかに依存する。FDAが米国国外で行った試験データを受け入れる保証はない。FDAが米国国外で行ったいかなる試験のデータも受け入れなければ、追加の試験が必要になる可能性が高く、これは高価で時間がかかり、適用候補製品の開発を延期または永久に停止する可能性がある。
また,米国以外での臨床試験はわれわれに重大な悪影響を及ぼす可能性がある。国際臨床試験に固有のリスクは
私たちの第三者への依存に関するリスク
我々は依存し,第三者に依存して我々の製品製造,研究および臨床前と臨床試験の一部または全部に依存し続けることが予想され,これらの第三者の表現は満足できない可能性がある。
私たちは私たちの製品製造、研究、そして臨床前と臨床テストのすべての方面を独立して行うことを望まない。我々は現在依存しており,臨床前または臨床開発で試験された任意の候補製品を製造するCMOと,我々の動物試験や研究を行うCROを含む第三者に依存し続けていると予想される。これらの第三者のいずれかは、いつでも私たちとの協力を終了することができ、またはサプライチェーン不足に直面する可能性があり、または私たちの計画された開発活動を支援するために、私たちの前臨床試験のための動物のような必要な資源を得ることができない可能性がある。もし私たちの開発計画を修正したり、代替手配を達成する必要があれば、これは私たちの製品開発活動を延期するかもしれません。これらの第三者の研究開発活動への依存は、これらの活動の制御を減少させるが、必要なすべての法規や研究案を遵守することを確保する責任は免除されない。例えば,我々が独自に開発·商業化した候補製品については,INDを有効にするすべての研究や臨床試験が研究計画や合意に従って行われることを確保していきたい。
我々が開発可能な任意の候補製品のための臨床試験を設計しようとしているが,CROは一部またはすべての臨床試験を行う。したがって、私たちの開発計画の多くの重要な側面は、それらの実施とタイミングを含めて、私たちの直接制御範囲内ではないだろう。我々は第三者に依存して進行中と将来の臨床前研究や臨床試験を行い,完全に我々自身に依存している従業員に比べて,前臨床研究や臨床試験により開発されたデータ管理の直接制御も減少している。外部の当事者とのコミュニケーションも挑戦的である可能性があり、ミスや協調活動の困難を招く可能性がある。外部の当事者は
91
これらの要素は第三者が著者らの臨床前研究と臨床試験を行う意志或いは能力に重大な不利な影響を与える可能性があり、そして著者らのコントロール範囲を超える意外なコスト増加に直面させる可能性がある。CROや他の第三者が満足できる方法で臨床前研究および進行中および将来の臨床試験を行わなかった場合、彼らの私たちの義務に違反したり、規制要求を遵守できなかったりする場合、私たちが開発する可能性のある任意の候補製品の開発、規制承認、商業化が遅れる可能性があり、私たちは規制部門の承認を得られず、私たちの候補製品を商業化することができないかもしれません。あるいは私たちの開発計画は実質的で不可逆的な損害を受ける可能性があります。もし私たちのCROおよび他の第三者が収集した臨床前および臨床データに依存できなければ、私たちが行った任意の臨床前研究または臨床試験の規模を繰り返し、延長または増加させる必要があるかもしれないが、これは商業化を著しく延期し、より大きな支出を必要とするかもしれない。
第三者が法規の要求または私たちが宣言した研究計画および方案に従ってその契約義務を成功裏に履行し、予想された期限内に私たちの研究を完了または実行しなかった場合、私たちは、将来のIND提出をサポートし、私たちが開発する可能性のある任意の候補製品を支援するために必要な臨床前研究および臨床試験を完了または遅延させることができないであろう。
バイオ製品の製造は複雑で、様々な原因で製品損失を招きやすい。我々は第三者と契約を結び,臨床前および臨床試験のための候補製品を生産し,商業化のために継続していく予定である。このような第三者への依存は、許容可能なコストまたは品質で十分な数の候補製品または製品またはそのような数を得ることができないリスクを増加させ、これは、私たちの開発または商業化努力を延期、阻止、または損害する可能性がある。
私たちは所有したり経営したりしないし、現在は製造施設を設立する計画もない。私たちは依存し、引き続き第三者生産VERVE-101と、臨床前および臨床試験のための私たちの他の候補製品、ならびに私たちの任意の候補製品が市場承認を得た場合の商業製造に依存することが予想される。私たちはまたこれらの第三者に依存して包装、ラベル付け、殺菌、貯蔵、配送、その他の生産物流を行う。このような第三者への依存は、許容可能なコストまたは品質で十分な数の候補製品または製品またはそのような数を得ることができないリスクを増加させ、これは、私たちの開発または商業化努力を延期、阻止、または損害する可能性がある。私たちは第三者製造業者とどんな合意にも到達できないかもしれないし、受け入れられる条項でそうすることもできないかもしれない。たとえ第三者製造業者と合意できても、第三者メーカーに依存することは、追加的なリスクをもたらす
生産能力制限または原料または原料薬市場の遅延または中断により、私たちまたは私たちの第三者メーカーは、私たちの候補製品を生産するために必要な原材料または活性医薬成分または原料薬の不足に遭遇する可能性があり、これらの原料または原料薬の数は、私たちの臨床試験に必要な数を必要とするか、または、私たちの候補製品が承認された場合、競争相手や他社がそのような原材料または原料薬を購入することによる不足を含む、商業化または需要の増加に十分な数を使用することができる。もし私たちまたは私たちの第三者メーカーが十分な数の候補製品を生産するために必要な原材料や原料薬を得ることができなければ、私たちの業務に大きな悪影響を及ぼす可能性があります。
商業販売または末期臨床試験のための完成治療製品のために許可された成分は、cGMPに従って生産されなければならない。私たちの第三者メーカーは、私たちのどの候補製品の製造と販売を始めることができる前に、規制機関の検査と承認を受け、その後も不定期な検査を受けなければならない。第三者メーカーは、cGMP法規や米国以外の類似した法規要件を遵守できない可能性がある。私たちまたは私たちの第三者製造業者が適用された法規に従わないことは、FDA Form 483の観察通知、警告状、または臨床所持、罰金、禁止、民事処罰、遅延、許可の一時停止または撤回、許可証の取り消し、候補製品または製品の差し押さえまたはリコール、運営制限および刑事起訴などの規制行動をもたらす可能性があり、いずれも私たちの製品供給に重大な悪影響を及ぼす可能性があります。
92
VERVE−101のようなバイオ製品を製造することは、複雑で、特に大量生産されている。生物製品は一貫的に生産され、明確な定義に符合する製造技術でなければならない。したがって,製造過程を検証·制御し,再現性を確保する必要がある。生物製品の生産は汚染、設備故障或いは設備の設置或いは操作が不適切、サプライヤー或いはオペレータの誤り、生産量の不一致、製品特性の変化及び製品技術の大規模化が困難であるため、製品損失を招きやすい。私たちはまだ私たちの潜在的な商業化候補製品のために製造プロセスを拡大していない。正常製造プロセスとの微小な偏差でも生産量の低下、製品欠陥、その他の供給中断を招く可能性がある。私たちの候補製品または私たちの候補製品を製造する製造施設で微生物、ウイルス、または他の汚染が発見された場合、汚染を調査および修復するために長い間、そのような製造施設を閉鎖する必要がある可能性があり、これは私たちの運営結果を損なう可能性があり、潜在的な名声被害をもたらす可能性がある。私たちの候補製品と私たちが開発する可能性のあるどの製品も他の候補製品や製品と製造施設を競争する可能性があります。したがって、私たちはこのような施設を優先的に使用することができないかもしれないし、全く使用できないかもしれない。CGMP法規の下で運営されているメーカーの数は限られており、私たちのために製品を製造する能力があるかもしれない。
私たちの既存または未来のメーカーのどんな業績失敗も、臨床開発やマーケティング承認を延期する可能性がある。我々は現在,大量の薬物物質に余分な供給や源を提供する予定はなく,第三者メーカーと長期的な商業供給について何の合意も達成していない。もし私たちの未来の契約メーカーが約束通りに契約を履行できなければ、私たちはこれらのメーカーの交換を要求されるかもしれない。いくつかの潜在的な代替メーカーが私たちの候補製品を生産できると信じていますが、私たちはそのような代替製品を決定して同定する際に追加のコストや遅延が生じたり、代替メーカーと合意できない可能性があります。
私たちの現在と未来の他人が私たちの候補製品や製品を生産することへの依存は、私たちの将来の利益率と、マーケティングの承認をタイムリーかつ競争力的に獲得する製品の商業化能力に悪影響を及ぼす可能性があると予想されています。
もし私たちの候補製品のいかなる第三者メーカーも、私たちの候補製品の生産規模を拡大することができず、および/またはその製造製品の良率を向上させることができなければ、私たちが候補製品を製造するコストが増加する可能性があり、商業化が遅れる可能性がある。
臨床試験の需要を満たすのに十分な数を生産するために、承認されれば、私たちが開発する可能性のある任意の現在または将来の候補製品は、その後商業化され、私たちの第三者メーカーは、製品の品質を維持しながら生産量を増加させ、製造プロセスを最適化することを要求されるであろう。より大規模な生産への移行は難しいことが証明されるかもしれない。また、もし私たちの第三者メーカーが私たちの候補製品の生産量を向上させるために彼らの製造プロセスを最適化することができない場合、あるいは彼らが製品の品質を維持しながら私たちの候補製品の生産量を増加させることができなければ、私たちが行っているあるいは未来の臨床試験や市場需要を満たすことができない可能性があり、これは私たちの利益を創出する能力を低下させ、私たちの業務と運営結果に実質的な悪影響を与える可能性がある。
我々はすでに第三者とプロジェクトや候補製品の研究、開発、製造、商業化について協力し、より多くの協力を行うことが可能である。もしこのような協力が成功しなければ、私たちの業務は不利な影響を受けるかもしれない。
私たちの戦略の一部として、私たちは協力して、私たちの1つまたは複数の計画または候補製品について第三者とより多くの協力を求めるつもりです。例えば、2019年4月には、BEAMの特定の塩基編集、遺伝子編集、配信技術を独自に許可するBEAMプロトコルを締結し、2022年7月に改訂および再記述を行った。2020年10月には、VERVE-101で使用されているLNP配信技術の許可をAcuitasから取得し、2021年10月には、VERVE-201で使用されている特定の脂質技術の許可を得たノワール協定を締結し、2022年7月にVertex協定を締結し、4年間のグローバル協力研究を行い、開発に専念した体内にある遺伝子編集は単一肝疾患を治療する未開示目標になることが期待される。私たちの他の協力パートナーには、大中型製薬会社、地域的、全国的な製薬会社、バイオテクノロジー会社が含まれています。BEAMプロトコル、および任意の第三者と締結する可能性のある任意の他の手配によれば、私たちは、協力者が私たちの候補製品の開発または商業化に取り組んでいるリソースの数および時間に対して限られた制御を持っています。私たちがこれらの製品から収入を作る能力は
93
計画は私たちの協力者がこのような計画で彼らに割り当てられた機能を成功的に履行する能力にかかっているかもしれない。
私たちが参加した協力は成功しないかもしれないし、どんな成功もこのような協力者たちの努力と活動に大きく依存するだろう。協力は以下のようなリスクを含む多くのリスクをもたらす
94
協調プロトコルは、候補製品の開発や商業化を最も効果的な方法で招くことができないか、または全くそうではない可能性がある。現在または未来のいずれの協力も製品の開発に成功して商業化されていない場合、または私たちの協力者が私たちとの合意を終了した場合、私たちは未来の研究資金や協力の下でマイルストーンや特許権使用料の支払いを受けないかもしれない。もし私たちがこれらの合意に基づいて期待した資金を受け取っていなければ、私たちの候補製品の開発は延期されるかもしれません。私たちは私たちの候補製品を開発するために追加の資源が必要かもしれません。本“リスク要因”の節で述べた製品開発,規制承認,商業化に関するすべてのリスクは,我々の協力者の活動にも適用される.
協力協定は、非日常的な費用や他の費用を発生させ、私たちの短期的かつ長期的な支出を増加させ、既存の株主を希釈した証券を発行したり、私たちの管理と業務を混乱させることを要求するかもしれません。たとえば,ピムプロトコルを実行する際には,ビムに276,075株の普通株を発行し,Vertexプロトコルの実行に関連してVertexとの私募を完了し,このプロトコルによりVertexに1,519,756株の普通株を発行した.また,Cas 9許可プロトコルに基づき,ブロードとハーバードに138,037株の普通株を発行した。ブロイドとハーバードは逆希釈権利も持っており、この権利により、私たちは優先株融資を完了した後、ブロードとハーバードに309,278株の普通株を追加発行した。Cas 9許可協定によると、IPO終了時に遠大とハーバードに878,098株の普通株を追加発行した。もし私たちの平均時価が指定されたハードルを超えて、9桁に達するドルの金額から100億ドルに上昇したり、これらのハードルを超えたと引き換えに私たちの会社を売却したりすれば、私たちもハーバード大学と遠大グループに成功費用を支払う義務がある。当社の支配権が変更されたり、当社が売却されたりした場合、当社は事件発生後の指定期間内に任意の関連する成功した支払いを現金で支払わなければなりません。そうでなければ、成功した支払いは、私たちの選択に応じて現金または私たちの普通株の株または現金と私たちの普通株の株の組み合わせで支払うことができる。2021年9月、私たちはハーバードとブロイドに通知しました。関連する測定日まで、私たちの平均時価は3つの指定されたハードルを超えて、Cas 9許可協定に基づいて、合計約630万ドルの成功支払いを支払います, 私たちは2021年11月に現金で決済します。
適切な協力者を探す上で、私たちは激しい競争に直面する可能性があり、交渉過程は時間がかかって複雑だ。我々が最終的な協調合意を達成する能力は,協力者の資源や専門知識の評価,協調の条項や条件の提案,提案した協力者のいくつかの要因の評価に依存する.もし私たちまたは私たちの協力者が開発する可能性のある任意の候補製品の権利を許可すれば、私たちがこれらの取引を私たちの既存の運営と会社文化と組み合わせることに成功できなければ、私たちはこのような取引の利点を達成できないかもしれない。
さらに、私たちの契約義務に対する制約の下で、私たちの協力者が業務合併に参加する場合、その協力者は、私たちの許可された任意の候補製品の開発または商業化を淡水化または終了する可能性がある。もし私たちの協力者が私たちとの合意を終了したら、私たちは新しい協力者を引き付けるのがもっと難しいことを発見するかもしれないし、ビジネスや金融界での私たちの見方は不利な影響を受けるかもしれない。
もし私たちがビジネス上合理的な条件下で協力を構築したり維持したりできなければ、私たちの開発と商業化計画を変えなければならないかもしれません。私たちの業務は悪影響を受ける可能性があります。
私たちは適切な協力者を誘致する上で激しい競争に直面しており、いくつかのより成熟した会社も私たちが魅力的だと思う第三者知的財産権をライセンスしたり獲得したりする戦略を求めているかもしれない。これらの老舗会社はその規模、財務資源及び更に強い臨床開発と商業化能力のため、私たちより競争優位を持っているかもしれない。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちが協力について最終的な合意に到達するかどうかは、他に加えて、パートナーの資源と専門長の評価、提案された協力の条項と条件、提案されたパートナーのいくつかの要因の評価に依存する。これらの要因は、臨床試験の設計または結果、FDA、EMAまたは他の規制機関によって承認される可能性、候補製品の潜在的市場、そのような候補製品の製造および患者への送達のコストおよび複雑さ、競合製品の潜在性、挑戦の利点、任意の既存の協力協定の条項、および一般的な業界および市場条件を考慮せずにこのような所有権に挑戦する場合、このような不確実性が存在する可能性がある。協力者は,適応のような他の候補製品や技術について連携する機会も可能であり,我々の候補製品にとって,このような連携が我々との連携よりも魅力的であるかどうかを評価せざるを得ないであろう.
95
既存または将来のライセンス契約によれば、私たちも制限される可能性があり、潜在的な協力者と特定の条項の合意を締結することはできません。
協力は複雑で時間のかかる交渉、記録、そして実行だ。また、大手製薬とバイオテクノロジー会社との統合は将来の潜在的協力者の数を減少させた。
私たちはタイムリーに、受け入れ可能な条項で、あるいはもっと多くの協力について交渉することができないかもしれない。それができなければ、私たちが協力を求めている候補製品の開発を減らし、その開発計画や私たちの1つ以上の他の開発計画を減らしたり、潜在的な商業化を延期したり、販売やマーケティング活動の範囲を縮小したり、私たちの支出を増やして自費で開発や商業化活動を行わなければならないかもしれません。もし私たちが自分で援助して開発や商業化活動に従事することを選択すれば、私たちはより多くの専門知識と追加的な資本を得る必要があるかもしれないが、これらは私たちが受け入れられない条件や根本的には得られないかもしれない。私たちが協力できず、必要な開発や商業化活動を展開するのに十分な資金や専門知識がなければ、私たちの候補製品をさらに開発したり、市場に出すことができないかもしれません。
私たちの候補製品で使用されるいくつかの部品と材料は単一源のサプライヤーに依存している。
私たちの候補製品で使用されるいくつかの部品と材料は単一源のサプライヤーに依存している。私たちは、これらのサプライヤーやサービスプロバイダが経営を継続し、私たちの需要を満たすのに十分な能力や供給を持っていることを保証することができず、私たちの競争相手や他の私たちと協力し続けることに関心のある会社に買収されないことを保証することはできません。私たちは原材料、部品、肝心な工程と完成品の単一ソースサプライヤーを使用して、供給中断、価格上昇あるいは納品遅延を含むいくつかのリスクに直面させます。一般的に、代替部品の代替供給源は比較的少ない。これらのサプライヤーは未来の臨床試験或いは商業販売に対する私たちの需要を満たすことができないか、或いは満足したくないかもしれない。これらの部品、材料、およびプロセスのための追加または代替サプライヤーを確立するには多くの時間を要する可能性があり、規制要件に適合した代替サプライヤーを確立することは困難である可能性がある。任意の単一ソース供給者またはサービスプロバイダの任意の供給中断は、供給遅延または中断を引き起こす可能性があり、これは、私たちの業務、財務状態、運営結果、および将来性を損なうことになる。
もし私たちがサプライヤーを交換しなければならない場合、私たちが開発する可能性のある任意の候補製品の製造と納品は長い間中断する可能性があり、これは私たちの業務に悪影響を及ぼす可能性があります。必要であれば、追加的または代替的な供給者を確立することはすぐに完了しないかもしれない。代替サプライヤーを見つけることができれば、代替サプライヤーは合格を必要とし、追加の規制機関の承認が必要になる可能性があり、これはさらなる遅延を招く可能性がある。製品で使用される単一ソースコンポーネントおよび材料の十分な在庫を維持することを求めているが、コンポーネントまたは材料供給の中断または遅延、または代替ソースからコンポーネントまたは材料を許容可能な価格で得ることができないことは、候補製品の需要を満たす能力を弱める可能性がある。
私たちの知的財産権に関するリスクは
もし私たちまたは私たちのライセンシーが、私たちの遺伝子編集技術および候補製品をカバーする特許権を獲得、維持、擁護、実行できない場合、または取得した特許保護範囲が十分でなければ、私たちの競争相手は、私たちと似ているまたは同じ技術および製品を開発し、商業化する可能性があり、私たちの技術および候補製品の開発と商業化に成功する能力は不利な影響を受ける可能性がある。
私たちの成功は、私たちが単独で他人と共同で所有する可能性のある知的財産権の保護を獲得、維持、擁護、強化する能力があるかどうか、または私たちが開発した独自技術および候補製品について米国および他の国/地域の他の人から許可、特に特許を得ることができるかどうかに大きく依存する。私たちの遺伝子編集技術と候補製品を保護することは難しくて高価で、私たちはそれらの保護を確保できないかもしれない。私たちは、これらの活動をカバーする有効かつ実行可能な特許または商業秘密の下で当社が所有する権利の程度に応じて、許可されていない第三者が、我々が開発する可能性のある候補製品または運営上同様の製品を製造、使用、販売、提供、販売、輸入、または他の方法で商業化する能力を阻止する。
私たちはアメリカと海外で私たちの業務の重要な候補品に関連する特許出願を提出し、許可を通じて私たちとの
96
技術と候補製品です。私たちがどんな独自技術または候補製品の特許保護を獲得または維持できない場合、私たちの業務、財務状況、運営結果、および見通しは深刻な損害を受ける可能性があります。特許保護を含む保護を得ることができなかったのは、特定の法律および事実のためである可能性があり、私たちの候補製品が米国または任意の所与の国/地域で保護される可能性を排除する可能性がある。例えば、不十分、欠陥、または誤りのある特許訴訟は、私たちの製品を十分にカバーする特許権の減少、損失、または利用できない可能性があります。我々の候補製品をカバーすることを目的とした特許開示および宣言は、1つの国/地域では十分または許容されるが、他の国/地域では不十分または許可されない可能性がある。米国における特許出願の要求は、米国以外の国または地域での特許出願の提出をサポートするのに十分ではない可能性がある。
特許訴訟過程は高価で、時間がかかり、複雑であり、私たちは合理的なコストまたはタイムリーな提出、起訴、維持、弁護、またはすべての必要または望ましい特許出願を許可することができないかもしれない。さらに、有効かつ強制的に実行可能な特許を取得して保持する能力は、我々の発明と従来技術との差が、我々の発明が従来技術よりも特許を出願することを可能にするか否かに依存する。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.さらに、場合によっては、私たちは、第三者から許可された技術を含む特許出願の準備、提出、起訴、または特許を維持、実行、擁護する権利がない。したがって、これらの許可内の特許および出願は、私たちの業務の最良の利益に適合する方法で準備、提出、起訴、維持、弁護、実行されてはならない。
製薬とバイオテクノロジー会社の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連しており、近年多くの訴訟のテーマとなってきた。特に遺伝子編集分野は、広範な特許活動と訴訟のテーマとなってきた。また、米国以外の特許保護範囲は不確定であり、外国法律は米国法律のように私たちの権利を保護しない可能性があり、その逆も同様である。例えば,欧州特許法は米国法よりも人体治療法に対する特許性制限が多い。また、2023年第2四半期よりも早く、欧州の出願は、単一特許裁判所またはUPCによって管轄される特許付与後に単一特許となる選択権をすぐに有することになる。これは欧州特許実践の大きな変化になるだろう。UPCは新しい裁判所制度であるため、裁判所には前例がなく、いかなる訴訟の不確実性も増加している。
所有およびライセンス内の特許権については、我々および我々のライセンシーが現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるか否か、または任意の発行された特許の権利主張が競争相手から十分な保護を提供するかどうかを予測することはできない。しかも、私たちは私たちの候補製品に関連する可能性のあるすべての第三者知的財産権を知らないかもしれない。
さらに、科学文献に発表された発見は、実際の発見よりも遅れがちであり、米国および他の管轄区域の特許出願は、通常、出願18ヶ月後に発表されるか、または場合によっては全く発表されない。したがって、私たちまたは私たちのライセンシーは、私たちまたは私たちのライセンスが、私たちが現在または将来可能な特許および特許出願に要求された最初の発明であるか、または私たちまたは私たちのライセンシーが最初にそのような発明のために特許を出願して保護されたものであることを確実に知ることはできない。したがって、私たちが所有していると許可されていない特許権の発行、範囲、有効性、実行可能性、商業的価値は高い不確実性を持っている。さらに、私たちが持っているおよび許可されていない保留および将来の特許出願は、私たちの技術および候補製品の全部または一部を保護するために、または他社が競争技術および製品を商業化することを効果的に阻止するために、特許の発行を招くことができないかもしれない。米国や他の国の特許法または特許法解釈の変化は、私たちの特許の価値を低下させ、私たちの特許権を獲得、保護、維持、擁護、実行する能力を低下させ、私たちの特許保護の範囲を縮小し、より広く言えば、私たちの特許権の価値や範囲に影響を与えたり縮小したりする可能性がある。
さらに、私たちまたは私たちの許可者は、既存技術を米国特許商標局またはUSPTOの第三者に提出したり、反対、派生、撤回、再審査に参加したりする必要があるかもしれません各方面間私たちの特許権または他人の特許権の介入手続きを審査、許可後に審査または挑戦します。このような提出、訴訟、または訴訟における不利な裁決は、私たちの特許権の範囲を縮小したり、無効にしたりして、第三者が私たちの技術または候補製品を商業化し、私たちに支払うことなく、直接競争することを可能にしたり、第三者特許権を侵害することなく薬物を製造または商業化することができなくなる可能性があります。もし私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、結果にかかわらず、会社が私たちと協力し、現在または将来の候補製品を認可、開発、または商業化することを阻止することができる。
97
また、特許出願において要求されるカバー範囲は、特許発行前に大幅に縮小することができ、その範囲は特許発行後に再解釈することができる。私たちが所有して許可された特許出願が特許の形で発表されても、それらの発表形態は私たちに何の意味のある保護も提供してくれず、競争相手が私たちと競争することを阻止したり、他の方法で私たちにどんな競争優位性を提供してくれたりしない。特許の発行は、その発明性、範囲、有効性、または実行可能性に対して決定的ではなく、私たちが持っている特許および許可されていない特許は、米国および海外の裁判所または特許庁で挑戦される可能性がある。このような挑戦は、独占的または経営の自由を失うこと、または特許主張の全部または部分的な縮小、無効、または実行不能をもたらす可能性があり、それにより、他人が類似または同じ技術および製品を使用することを阻止するか、またはそれを商業化する能力を制限するか、または我々の技術および候補製品の特許保護期間を制限することができる。最終的な結果が私たちに有利であっても、このような訴訟は巨額のコストを招く可能性があり、私たちの経営陣や従業員に時間がかかる必要がある。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。さらに、私たちの競争相手は、同様の技術または代替技術または製品を非侵害的に開発することによって、私たちが所有または許可している特許を迂回することができるかもしれない。したがって、私たちが所有し、許可された特許の組み合わせは、他社が私たちの任意の候補技術や製品と似ているか、または同じ技術および製品を商業化することを排除するために、十分な権利を提供してくれないかもしれない。
私たちの遺伝子編集技術と候補製品の権利は、他の人が私たちに付与するライセンスの条項と条件によってある程度制限されています。
私たちは第三者から許可された知的財産権に依存しており、私たちの許可者たちはいつも私たちの最善の利益に従っているわけではないかもしれない。もし私たちが知的財産権の許可下での私たちの義務を履行できなかった場合、許可が終了された場合、またはこれらの許可に関する紛争が発生した場合、私たちは私たちの業務に重要な重要な権利を失うかもしれない。
我々は、我々の遺伝子編集技術および候補製品の開発に非常に重要または必要である第三者からの特定の特許権およびノウハウに許可し、依存している。例えば,我々はBEAMプロトコル,Cas 9ライセンスプロトコル,Acuitasプロトコル,ノワールプロトコルおよび他のライセンスプロトコルの締約国であり,これらのプロトコルにより,遺伝子編集技術,LNP技術および候補製品のキー特許および特許出願を取得した。これらの許可協定は、様々な職務調査、マイルストーン支払い、特許権使用料、保険、その他の義務を私たちに課している。もし私たちがこれらの義務を履行しなければ、私たちの許可者は私たちの許可を終了する権利があるかもしれません。この場合、私たちはこれらの合意によって許可された知的財産権によって保護された私たちの遺伝子編集技術や製品候補製品を開発またはマーケティングすることができません。
これらのライセンスおよび他のライセンスは、すべての関連する使用分野および将来、私たちの遺伝子編集技術および候補製品のすべての地域でこのような知的財産権および技術を使用することを望んでいる独占的な権利を提供しないかもしれません。私たちに付与されたいくつかの許可および取得された特許は、許可者またはいくつかの第三者が所有するいくつかの以前に存在する権利に明示的に制限される。したがって、競争相手がある地域や分野で競合製品を開発し、商業化することを阻止することはできないかもしれない。もし私たちがこれらの排除された分野の権利が私たちの候補製品を商業化したり、私たちの競争優位性を維持するために必要だと判断すれば、私たちは私たちの候補製品を開発、製造、またはマーケティングを継続するために第三者から許可を得る必要があるかもしれない。私たちは独占的に基づいて、商業的に合理的な条項やそのような許可を得ることができないかもしれません。これは、候補製品を商業化することを阻止したり、私たちの競争相手や他の人が私たちの業務に重要な技術を得る機会を得ることができるかもしれません。
また,Cas 9ライセンスプロトコルによると,ある特定の場合,ハーバードと遠大は,このようなライセンスプロトコルの標的に属する特許を我々が許可されている分野と同じ分野の第三者に付与することができる.このように、第三者はCas 9許可プロトコルのすべての権利を持っている可能性があり、これは私たちの競争地位に影響を与え、第三者が私たちの潜在的な未来の候補製品や技術のような製品を商業化することができるかもしれない。この場合、第三者に付与されるいかなる権利も、ハーバードおよびブロダーに付与された特許および特許出願の独占的権利の範囲を縮小するだろう。
私たちは、特許および特許出願の準備、提出、起訴、維持、実行、および弁護に関して完全な制御権を有しておらず、これらの特許および特許出願は、我々が許可または第三者から取得した技術をカバーしている。私たちのライセンシーが侵害者に対して特許を実行したり、有効性の挑戦または実行可能な主張を弁護する力は、私たち自身が実行するよりも強くないかもしれないし、私たちの最適な利益に合わないかもしれない。私たちはこのような特許と特許出願の準備、提出、起訴、維持、強制執行、そして弁護の方法と
98
私たちの業務の最大の利益のために。もし私たちの許可者がこれらの特許を起訴、維持、強制執行および保護できなかった場合、またはこれらの特許または特許出願の権利を失った場合、私たちが許可した権利は減少またはキャンセルされる可能性があり、私たちが開発する可能性のある任意の製品候補製品の開発および商業化の権利は不利な影響を受ける可能性があり、私たちは競争相手の競合製品の製造、使用、販売を阻止できないかもしれない。
私たちのライセンス者は第三者コンサルタントや協力者または第三者からの資金に依存する可能性がありますので、私たちのライセンス者は私たちが許可を得た特許の唯一および独占所有者ではありません。他の第三者が私たちの許可内の特許を所有している場合、このような許可を付与するために共通の所有者の同意が必要な司法管轄区域内では、私たちの許可は無効になる可能性があり、これらの共通の所有者は、このような特許許可を私たちの競争相手に与える可能性があり、私たちの競争相手は競争製品および技術を販売することができる。さらに、ライセンス内特許および特許出願に対する私たちの権利は、そのような認可内特許と特許出願の共同所有者との間の機関間または他の運営プロトコルにある程度依存する。1つまたは複数の共通の所有者がそのような機関間または運営プロトコルに違反している場合、私たちは、このようなライセンス内の特許および特許出願の権利に悪影響を受ける可能性がある。これらの事件のいずれも、私たちの競争地位、業務、財務状況、運営結果、および見通しに重大な悪影響を及ぼす可能性があります。
さらに、私たちのいくつかのライセンス特許と特許出願に含まれる発明は、アメリカ政府の資金を用いて行われている。私たちは、私たちが許可した特許および特許出願に関連する義務である、このような資金によって生成された適用義務、例えばタイムリーな報告を遵守することを保証するために、我々の許可者に依存する。もし私たちの許可者がその義務を履行できなかった場合、関連特許の権利喪失または強制執行を招く可能性がある。例えば、米国政府は、このような許可内の特許に対して、米国政府が発明を使用することを許可するか、または米国に代わって米国に発明を使用させる非排他的許可を含むいくつかの権利を有することができる。もしアメリカ政府がこのような権利を行使することを決定すれば、私たちをその請負業者として雇う必要はない。米国政府の権利はまた、援助された発明および技術を第三者に開示し、米国政府の資金開発を使用する許可技術を第三者が使用または使用することを許可する先行権を行使することを可能にするかもしれない。米国政府が、我々または我々の許可者が米国政府が援助している技術の実用化を実現できなかったため、健康や安全需要を緩和し、連邦法規の要求を満たしたり、米国工業を優先したりするための行動が必要であると考えている場合、米国政府もそのデモ権利を行使することができる。さらに、このような許可された米国政府によって援助された発明の権利は、米国でそのような発明を含む候補製品を製造するために、いくつかの要件によって制限される可能性がある。上記のいずれも、私たちの業務、財務状況、経営結果、見通しに重大な損害を与える可能性があります。
もし私たちの第三者許可者が認定した場合、私たちは努力したにもかかわらず、私たちは許可協定または許可協定の下のいくつかの義務に深刻に違反し、適用可能な許可プロトコルを終了するか、または場合によっては適用許可プロトコルの下の1つまたは複数の許可を終了することを選択する可能性があり、この終了は、この許可プロトコルがカバー可能な候補製品および技術を開発および商業化することができなくなるであろう。第三者入網許可が終了した場合、あるいは第三者入網許可下の基礎特許が予想される排他性を提供できなかった場合、競争相手は規制部門の承認を求め、私たちと同じ製品を市場に出す権利がある。これらの事件のいずれも、私たちの競争地位、業務、財務状況、運営結果、および見通しに重大な悪影響を及ぼす可能性があります。
私たちが所有し許可している特許および特許出願および他の知的財産権は、優先権または発明権紛争、妨害、および同様のプログラムの影響を受ける可能性がある。もし私たちまたは私たちのライセンシーがこれらの訴訟で失敗した場合、私たちは第三者から許可を得ることを要求されるかもしれません。これらの許可は商業的に合理的な条項では得られないかもしれません。あるいは私たちが開発する可能性のある1つ以上の候補製品の開発、製造、商業化を停止することは、私たちの業務に大きな悪影響を及ぼす可能性があります。
私たちが選択権を持っているいくつかのアメリカ特許と1つのアメリカ特許出願はブロドとマサチューセッツ工科大学が共同で所有しており、場合によってはブロダー、マサチューセッツ工科大学、ハーバードが共同所有しており、私たちは総称してボストン許可側と呼ばれ、米国干渉番号106,048がカリフォルニア大学、ウィーン大学、Emmanuelle Charpentierと共同で所有する米国特許出願に参加しており、私たちは総称してCVCと呼ばれている。2018年9月10日、連邦巡回控訴裁判所(CAFC)は、事実介入がないと判断した米国特許商標局(PTAB)の特許裁判と控訴委員会を確認した。干渉は、異なる当事者によって提出された特許請求の範囲の標的の発明優先権を決定することを目的とする、米国特許商標局内のプログラムである。
99
2019年6月24日、PTABは、CVCが共同所有する14の米国特許出願と13の米国特許と1つの米国特許出願(ボストンライセンス者が共同所有する)との間に第2の干渉が存在することを発表した(米国干渉番号106,115)。宣言された介入では,CVCは一次当事者,ボストン許可側は上級当事者に指定されている。2022年2月28日,PTABは,ボストン許可側が干渉の第1項においてCVC:真核細胞に作用する単一RNA CRISPR−Cas 9システムに優先することを裁定した。したがって、CVCがこの干渉に関連する特許出願は特許を出願できないと考えられる。2022年9月,CVCはPTABの決定をCAFCに上訴し,控訴は行われている。
2020年12月20日、PTABは、ToolGen,Inc.が所有する1つの米国特許出願とボストンライセンス者が共同所有する14件の米国特許と2つの米国特許出願との間に干渉が存在すると発表した(米国干渉番号106,126)。宣言の介入では,ボストン許可側は初級側,ToolGen,Inc.は高級側に指定されている.
PTABは現在ToolGenとSigma−Aldrichに対する後続介入手続きを一時停止しており,CVCとボストン許可者の間で2回目の介入の結果についてCAFCが裁決を下すのを待っている。
2021年6月21日、PTABは、Sigma-Aldrich Co.,LLCが所有する米国特許出願とボストンライセンス者が共同所有する14件の米国特許と2つの米国特許出願との間に干渉があることを発表した(米国干渉番号106,133)。発表された介入では,ボストン許可側が初級側,Sigma−Aldrich Co.,LLCが高級側に指定された。
PTABは現在ToolGenとSigma−Aldrichに対する後続介入手続きを一時停止しており,CVCとボストン許可者の間で2回目の介入の結果についてCAFCが裁決を下すのを待っている。
干渉を宣言した結果,PTAB前に米国特許商標局で対抗プログラムが起動され,このプログラムは優先度,具体的には,どちらがまず保護が要求されるテーマを最初に発明したかを最終的に決定するためであることを宣言した.干渉は通常2段階に分けられる.第1段階は運動段階または予備運動段階と呼ばれ、第2段階は優先段階と呼ばれる。第1段階では、各当事者は、一方の当事者が既存技術、書面記述、および許可された権利要件に基づく特許性に関連する問題を含むが、これらに限定されない問題を提起することができる。一方の当事者は,より早い優先権利益を求めたり,まず干渉が適切であるかどうかを宣言したりすることも可能である.干渉の第2段階で優先権を決定するか、または誰が一般的に保護を必要とする発明を最初に発明したかを決定する。各段階が実際にどのくらいかかるかを正確に予測することはできないが,各段階はPTABによって決定されるまで約1年以上かかる可能性がある。予備動議段階で提出された動議は、第2優先段階に達しないように、干渉手続きから除外される可能性がある。
現在の控訴またはこれらの未解決の米国介入手続きがボストン許可者に有利な方法で解決されることは保証されない。第2の干渉における控訴がCVCに有利である場合、または106,126または106,133干渉がそれぞれToolGen,Inc.またはSigma-Aldrich Co.,LLCに有利な判断を行う場合、またはボストン許可側の特許および特許出願が縮小され、無効または実行できない場合、選択された特許および特許出願を許可する能力を失い、我々が候補製品をカバーする関連第三者特許の許可を得ることができない場合、候補製品を許可する能力を失うことになり、候補製品を商業化する能力は悪影響を受ける可能性がある。私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。私たちが許可を得ることができても、それは非排他的である可能性があり、それによって、私たちの競争相手や他の第三者が私たちに許可された同じ技術にアクセスできるようにすることができ、これは私たちが大量の許可と印税を支払う必要があるかもしれない。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができなければ、私たちの遺伝子編集技術や候補製品を商業化できないかもしれません。あるいはこのような商業化努力は著しく遅れる可能性があり、逆に私たちの業務を深刻に損なう可能性があります。
私たちまたは私たちの許可者はまた、元従業員、協力者または他の第三者が、発明者または共同発明者として、私たちが所有している特許出願内に特許または特許出願または他の知的財産権において権益を有するクレームの制約を受けることができるかもしれない。このような第三者共通所有者のこのような特許出願における権利の独占的許可を得ることができない場合、そのような共通所有者の権利は、または将来的には、我々の競争相手を含む他の第三者に譲渡されることができるかもしれない。さらに、私たちは、そのような特許出願から発行された任意の特許を第三者に強制的に実行するために、このような共同所有者の協力が必要である可能性があり、そのような協力は私たちに提供されないかもしれない。
もし私たちまたは私たちのライセンシーが、私たちまたは彼らが直面している任意の妨害プログラムまたは他の優先権、有効性(任意の特許異議を含む)または発明権紛争に失敗した場合、私たちは貴重な知能を失うかもしれない
100
私たちが所有している、許可されている、または任意の特許を失っているか、またはそのような特許主張が縮小され、無効にされているか、または実行不可能とみなされているか、または我々が所有しているまたは許可内の特許の独占所有権または独占使用権を失ったことに起因する財産権である。このような紛争が特許権の喪失を招いた場合、そのような妨害手続または他の優先権または在庫紛争に参加する当事者を含む第三者から許可を取得し、維持することを要求される可能性がある。このような許可は商業的に合理的な条項では得られないかもしれないし,まったく存在しない,あるいは非排他的である可能性がある.もし私たちがそのようなライセンスを取得して維持できなければ、私たちが開発する可能性のある1つまたは複数の候補製品の開発、製造、商業化を停止する必要があるかもしれない。排他性の喪失や私たちの特許主張の縮小は、他の人が類似または同じ候補技術および製品を使用または商業化することを阻止する能力を制限するかもしれない。我々または我々のライセンシーが訴訟、他の同様の優先権紛争または在庫または所有権紛争に介入して勝訴しても、巨額のコストを招き、管理職および他の従業員の注意を分散させる可能性がある。上記のいずれも、当社の業務、財務状況、経営結果、または将来性に重大な悪影響を及ぼす可能性があります。
第三者との知的財産権ライセンス協定における私たちの義務を履行できなかった場合、あるいはライセンス側との業務関係が妨害された場合、私たちは私たちの業務に重要な知的財産権を失う可能性があります。
私たちは合意の当事者であり、私たちは第三者と他の計画を達成するかもしれないし、勤勉さ、開発と商業化スケジュール、マイルストーン支払い、特許権使用料、保険、その他の義務を私たちに強要するかもしれない。我々は既存の合意を有しており、これらの合意に基づいて、候補製品又は関連技術の製品純売上高のために合意がカバーする範囲内の印税を支払う義務がある。もし私たちが現在または未来の合意の下でこのような義務を履行できなかった場合、私たちの取引相手は、これらの合意を終了する権利があるか、または私たちに特定の権利を与えることを要求する権利があるかもしれない。このような状況は、任意のこのようなプロトコルに従って開発された任意の候補製品の価値に実質的な悪影響を及ぼす可能性がある。これらの合意を終了したり、これらの合意の下での私たちの権利を減少またはキャンセルすることは、私たちがあまり有利ではない条項で新しい合意または回復された合意を交渉しなければならないことを招き、または重要な知的財産権または技術に対する私たちの権利を含むこれらの合意の権利を失うことになり、これは私たちの業務、財務状況、運営結果、および見通しに大きな悪影響を及ぼすだろう。私たちはまだこれらの合意に関連したすべてのリスクに直面しているが、私たちは第三者がこれらの技術にもアクセスすることを防ぐことはできない。しかも、私たちの免許は私たちの未来のビジネス機会に制限を加えるかもしれない。
ライセンス契約によると、知的財産権に関する論争が発生する可能性があります
また,我々が現在第三者から知的財産権や技術許可を得ているプロトコルは複雑であり,このようなプロトコルのいくつかは様々な解釈の影響を受ける可能性がある.可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。また、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で現在の許可スケジュールの能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた技術や候補製品の開発および商業化に成功できない可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに大きな悪影響を及ぼす可能性がある。
私たちの現在または未来の許可者は、第三者コンサルタントまたは協力者または第三者からの資金に依存する可能性があり、したがって、私たちの許可者は、私たちが許可している知的財産権または知的財産権の唯一および独占所有者ではありません。他の第三者が知的財産権または私たちが許可した知的財産権の所有権を持っている場合、彼らはそのような知的財産権または知的財産権を私たちの競争相手に許可することができ、私たちの競争相手はそれと競争する製品および技術を販売することができる。これはあるかもしれません
101
私たちの競争地位、業務、財務状況、経営結果、将来性に重大な悪影響を及ぼす。
私たちが最善を尽くしたにもかかわらず、私たちの許可側は、許可協定に深刻に違反している可能性があるので、許可協定を終了し、これらの許可協定がカバーする候補製品や技術を開発し、商業化することができないかもしれません。これらの許可が終了した場合、または基礎知的財産権が予想される排他性を提供できなかった場合、競争相手は規制部門の承認を求め、私たちと同じ製品や技術を市場に出す権利があるだろう。これは私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすかもしれない。
もし私たちが商業的に合理的な条項で第三者からライセンスを取得できない場合、あるいはこのような合意の下で私たちの義務を履行できなければ、私たちの業務は損害を受ける可能性があります。
我々は現在知的財産権の権利を有しており,第三者の許可により候補製品を決定·開発し,キー技術を許可する権利の一部を通過する候補製品パイプラインの拡大を図る予定である.私たちは過去にハーバード、遠大、ビム、Acuitas、ノワールを含む第三者許可者から技術許可を得ることに成功したが、私たちは許容可能な条項または根本的に受け入れられない条項で第三者から任意の候補製品や技術の許可または権利を得ることができることを株主に保証することはできない。
様々な第三者が競争の激しい技術分野で業務を展開し、将来的に特許として発行される特許または特許出願が発行されている可能性があり、候補製品を商業化する能力を阻害または排除する可能性がある。我々の候補品に関連する可能性のある任意の第三者特許については,米国法第35編271(E)(1)節に規定されている“安全港”または研究免除にある程度依存し,FDAの医薬品承認を求めることに関連する特許侵害活動を免除している。しかし,米国特許法はこの条項に基づいて我々の臨床候補製品にこのような“安全港”を提供しているにもかかわらず,INDやBLAを提出すると免除は無効になる。臨床試験の不確実性を考慮すると、それらの完了時間を決定することはできず、1つまたは複数の関連する第三者特許が発効したときに、1つの候補製品にBLAを提出することができる。
したがって,第三者の特許やノウハウを用いて我々の製品を商業化する必要がある可能性があり,この場合,これらの第三者から許可を得ることが求められる.もし私たちがこのような技術を許可できない場合、あるいは私たちが不利な条項でそのような技術を許可することを余儀なくされた場合、私たちの業務は実質的な損害を受ける可能性がある。もし私たちが必要な許可を得ることができない場合、私たちは影響を受けた候補製品を開発したり商業化することができなくなり、これは私たちの業務に実質的な損害を与える可能性があり、このような知的財産権を持つ第三者は、私たちの販売禁止または私たち側が印税および/または他の形態の賠償を支払う義務を求めることができる。我々が許可を得ることができても,非排他的である可能性があり,我々の競争相手が我々に許可された同じ技術にアクセスできるようにする.
また,遺伝子編集分野には広範な特許出願活動が存在し,製薬会社,バイオテクノロジー会社,学術機関が遺伝子編集技術分野で我々と競合しているか,あるいは我々と競合することが予想され,我々の業務に関連する可能性のある特許出願を提出し,第三者特許出願がある可能性があり,発表されれば,第三者が我々の特許権を迂回することを可能にする可能性がある.私たちの分野では大量の特許が発行され、特許出願が提出されているので、これらおよび他の第三者は、私たちの候補製品、技術、または方法を含む特許権を持っていると主張するかもしれない。私たちの候補製品をマーケティングするためには、このような第三者知的財産権所有者から許可を得ることが必要または慎重であることが分かるかもしれません。しかしながら、私たちは、このような許可を得ることができないかもしれないし、他の方法で第三者から取得することができないかもしれないが、私たちが開発する可能性のある候補製品および遺伝子編集技術に必要な任意の成分、使用方法、プロセス、または他の知的財産権を考えることができるかもしれない。我々はまた、我々が開発可能な候補製品と共に使用するために、我々が評価または将来評価可能ないくつかの送達および遺伝子編集組成物および方法を含む、第三者からいくつかの遺伝子編集技術のライセンスを取得する必要があるかもしれない。さらに、私たちのいくつかの自己特許出願および許可内特許および特許出願は、第三者と共同所有すると判断される可能性がある。第三者と共同所有するいかなる特許についても、私たちはこのような共通の所有者のこのような特許の利益に許可を提供する必要があるかもしれない。もし私たちがこのような第三者共同所有者のこのような特許または特許出願における権利の独占的許可を得ることができない場合、これらの共通所有者は、私たちの競争相手を含む他の第三者にその権利を許可することができるかもしれない, 私たちの競争相手はそれと競争する製品と技術を販売することができる。さらに、私たちは、第三者に対してそのような特許を強制的に実行するために、私たちの特許のどのような共通所有者の協力も必要とするかもしれませんが、そのような協力は私たちに提供されないかもしれません。
102
また,学術機関と協力し,これらの機関と合意した書面により,われわれの臨床前研究や開発を加速する可能性がある。場合によっては、これらの機関は、協力によって生成された機関の技術的な任意の権利について交渉することができるオプションを提供してくれる。私たちがそのようなオプションを持っていても、私たちは指定された時間範囲内で、あるいは私たちが受け入れられる条項の下で機関からライセンスを交渉することができないかもしれない。もし私たちがそれができなければ、その機関は他の人たちに知的財産権を提供するかもしれないし、私たちの計画を実行し続けることを阻止するかもしれない。
また、第三者知的財産権のライセンスまたは買収は競争の激しい分野であり、一部の成熟した企業も魅力的または必要と考えられる第三者知的財産権ライセンスまたは買収戦略をとっている。これらの老舗会社はその規模、資本資源及び更に強い臨床開発と商業化能力のため、私たちより競争優位を持っているかもしれない。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちが投資から適切な見返りを得ることを可能にする条項に従って許可したり、第三者知的財産権を取得することができないかもしれない。もし私たちが必要な第三者知的財産権を得ることに成功したり、私たちの既存の知的財産権を維持することができなければ、関連計画や候補製品の開発を放棄しなければならないかもしれません。これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
もし私たちが必要な第三者知的財産権を得ることができない場合、あるいは私たちの既存の知的財産権を維持することができない場合、私たちは私たちの技術、候補製品、またはそれらを製造する方法を再設計するために多くの時間と資源を必要とするかもしれません、または代替技術を開発または許可することは、技術的にも商業的にも不可能かもしれません。もし私たちがそれができなければ、影響を受けた技術や候補製品を開発または商業化することができないかもしれません。これは、私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性があります。
さらに、許可プロトコルの下での義務を履行できない場合、私たちの取引相手は、これらのプロトコルを終了する権利がある可能性があり、この場合、私たちは、開発、製造、またはマーケティングを行うことができない場合があり、または、これらのプロトコルによってカバーされる任意の製品の開発、製造、またはマーケティングを停止させることを余儀なくされる可能性があり、またはそのようなプロトコルの下での他の処罰に直面する可能性がある。このような状況は、任意のこのようなプロトコルに従って開発された候補製品の価値に実質的な悪影響を及ぼす可能性がある。これらのプロトコルを終了するか、またはこれらのプロトコルの下での私たちの権利を減少または廃止するか、または私たちの業務の利益に適合した場合に、私たちのこのような合意の下での私たちの権利を自由に譲渡または再許可することを制限することは、重要な知的財産権または技術に対する私たちの権利、またはそのようなプロトコルに依存する1つまたは複数の候補製品のさらなる開発または商業化を阻害または遅延または禁止することを含む、これらの合意の下での私たちの権利を失ってしまう可能性がある。
ゲノム編集技術(塩基編集を含む)をめぐる知的財産権構造は高度に動的であり、第三者は法的訴訟を提起する可能性があり、私たちが彼らの知的財産権を侵害、流用または他の方法で侵害することを告発する可能性があり、その結果は不確定であり、私たちの製品の発見と開発を阻止、延期、または妨害する可能性がある。
私たちのビジネス成功は、私たちと私たちの協力者が、私たちの候補製品を研究、開発、製造、マーケティング、販売し、第三者の知的財産権を侵害、流用、または他の方法で侵害することなく、私たちの独自技術を使用する能力にかかっています。ゲノム編集分野,特に塩基編集技術分野はまだ初期段階であり,現在のところこのような製品候補は発売されていない。私たちと私たちの競争相手を含むいくつかの会社がこの分野で緊張した研究·開発を行っているため、知的財産権の構造が変化しており、今後数年間も不確定である可能性がある。バイオテクノロジーと製薬業界の特徴は、特許と他の知的財産権に関連する広範かつ複雑な訴訟と、干渉、派生、派生を含む特許に挑戦する行政訴訟である各方面間米国特許商標局の再審、認可後の再審及び再審手続又は外国司法管轄区の異議及びその他の同様の手続。将来的には知的財産権に関する重大な訴訟や、私たちが所有しているものや許可されていない他の第三者、知的財産権、独自の権利に関する訴訟があるかもしれない。私たちは、私たちの遺伝子編集プラットフォーム技術と私たちが開発する可能性のある任意の候補製品に関連する知的財産権の対抗性訴訟や訴訟の影響を受ける可能性があり、将来的には、妨害手続き、許可後の審査、各方面間米国特許商標局の審査及び派生手続、並びに外国司法管区の同様の手続
103
ヨーロッパ特許庁です。我々が我々の候補製品を開発している分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在し,それらは既存特許または将来付与される可能性のある特許に基づいて,その是非曲直にかかわらず侵害請求を行う可能性がある.
バイオテクノロジーや製薬業界の拡張や特許の発行に伴い,我々の遺伝子編集技術や候補製品は他人の特許権侵害のクレームを引き起こすリスクが増加する可能性がある。さらに、私たちを含む業界参加者は、どの特許が様々なタイプの療法、製品、またはそれらの使用または製造方法をカバーするかを常に明確にしているわけではない。私たちはいくつかの第三者特許出願が発表されれば、私たちの遺伝子編集技術および候補製品をカバーすると解釈されるかもしれないことを知っている。我々が現在知らない第三者特許、すなわち、私たちの候補製品の使用または製造に関連する技術、製造方法、または治療方法の請求項もあるかもしれない。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、我々の候補製品が発行された特許を侵害する可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。
私たちの候補製品と手続きが侵害される可能性のある関連する第三者特許またはアプリケーションを識別できなかったのかもしれません。特許出願は、発行するのに数年かかる可能性があるため、提出後18ヶ月以上の機密保持が必要となる可能性があり、発行前に修正することができるので、待っている出願は、私たちが開発する可能性のある任意の候補製品または私たちの技術の製造、使用、販売または輸入によって侵害される可能性があり、これらの特許を知らない可能性があるかもしれない。さらに、2000年11月29日までに提出された出願およびその日以降に提出されたいくつかの出願は、米国国外では提出されず、特許が発行される前には秘密にされている可能性がある。さらに、特許検索は、特許間の用語の違い、データベースの不完全さ、および特許請求の範囲の意味の評価が困難であるため、私たちを含む業界参加者が、私たちが開発する可能性のある任意の候補製品および私たちの技術に関連する可能性のあるすべての第三者特許権を決定することは困難である。私たちは、関連する特許または特許出願を識別することができないか、または潜在的利益の係属中の特許出願を識別することができないかもしれないが、そのような特許出願が我々の技術に関連するクレームを提出する可能性を誤って予測している。さらに、私たちは第三者特許が無効で、強制的に実行できない、または私たちの活動によって侵害されていないという結論を誤って導出するかもしれない。さらに、いくつかの制限の下で、公表された係属中の特許出願は、我々の技術、私たちが開発する可能性のある任意の候補製品、または私たちが開発する可能性のある任意の候補製品の使用をカバーするために、後で修正することができる。
第三者は、既存の特許または将来付与される可能性のある特許に基づいて、その是非曲直にかかわらず、侵害請求を私たちに提起するかもしれない。第三者が彼らの特許権を強制的または他の方法で主張するために私たちと訴訟を行うことを選択する可能性があるリスクがある。このような主張に法的根拠がないと考えても、管轄権のある裁判所は、これらの第三者特許が有効で、強制的に実行可能であり、侵害されていると判断することができ、これは、私たちの候補製品または主張する第三者特許をカバーする任意の他の候補製品または技術を商業化する能力に悪影響を及ぼす可能性がある。連邦裁判所でこのような米国特許の有効性に挑戦することに成功するためには,有効性推定を克服する必要がある。この負担が重いため,このような米国特許主張の無効について明確で納得できる証拠を提出することが求められているため,管轄権のある裁判所がこのような米国特許の主張の無効を宣言する保証はない.
我々が候補製品を開発している分野には,第三者米国や外国から発行された特許や係属中の特許出願が多く存在する.我々の候補製品はCRISPRに基づく遺伝子編集技術を使用しており,特許出願が非常に活発な分野である.CRISPRおよびCAに関連する大量の特許出願は、我々の遺伝子編集技術および候補製品およびそれらの使用または製造をカバーすることができる関連特許および係属中の出願の範囲を全面的に評価することを困難にする。我々の遺伝子編集技術および候補製品の使用または製造に関連する材料、製剤、製造方法、または治療方法に対して権利を有する競合他社によって所有または制御される特許を含む第三者特許または特許出願が存在する可能性がある。
第三者の有効かつ強制的な知的財産権の侵害、流用、または他の方法で侵害が発見された場合、私たちは、当社の候補製品および技術の開発、製造、およびマーケティングを継続するために、第三者からライセンスを取得することを要求される可能性があります。しかし、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。私たちが許可を得ることができても、それは非排他的である可能性があり、私たちの競争相手や他の第三者が私たちに許可された同じ技術にアクセスできるようにするためには、大量の許可と印税を支払う必要があるかもしれない。私たちは裁判所の命令、開発、製造、商業化侵害技術または候補製品の開発を中止することを強要されるかもしれない。さらに、私たちが顧客に賠償するために特許や他の知的財産権を故意に侵害していることが発見された場合、私たちは3倍の損害賠償と弁護士費を含む金銭損害賠償責任を負われる可能性があります
104
協力者でもあります権利侵害の発見は、私たちの候補製品の製造と商業化を阻止したり、いくつかの業務運営を停止させたりする可能性があり、これは私たちの業務を損なう可能性があります。第三者の機密情報や商業秘密を盗用したと主張することは、私たちの業務、財務状況、運営結果、将来性に類似した負の影響を与える可能性があります。
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許の寿命は限られている。個別特許の条項は、特許が付与された国の特許法律条項に依存する。米国を含む大多数の国では、すべての維持費が適時に支払われれば、特許の自然失効時間は、通常、適用国における最初の非臨時出願日から20年である。しかしながら、特許によって提供される実際の保護は、国によって異なり、特許のタイプ、そのカバー範囲、規制に関連する延長の利用可能性、特定の国の法的救済の利用可能性、および特許の有効性および実行可能性を含む多くの要因に依存する。様々な延期があるかもしれないが、特許の有効期限とその提供される保護は限られている。私たちの候補製品が特許を取得しても、特許有効期限が切れると、生物模倣薬を含む競争製品からの競争に直面するかもしれません。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
私たちの候補製品は生物模倣薬からの競争に直面する可能性があり、これらの模倣薬は短い規制経路で承認された。
患者保護および平価医療法案は、2010年の医療保健および教育調整法案、または2009年の生物製品価格競争および革新法案と呼ばれる副タイトルを含むPPAAと総称され、またはBPCIAは、FDAによって承認された参考生物製品と類似したまたは交換可能な生物製品のための短い承認経路を作成する。BPCIAによると、生物類似製品の出願は、参照製品が初めてFDA承認を得た4年後にFDAに提出されなければならない。また,FDAの生物類似製品の承認は,参考製品が初めて承認された日から12年後に発効する可能性がある。この12年間の独占期間内に、FDAが競合製品の完全なBLAを承認した場合、スポンサー自身の臨床前データおよび十分かつ制御された良好な臨床試験データが含まれており、別の会社の製品の安全性、純度、および有効性を証明するために、別の会社は依然として参照製品の競合バージョンを販売する可能性がある。この法律は複雑であり、FDAはまだ説明して施行している。したがって、その最終的な影響、実施、そして意味には不確実性がある。
BLAによりバイオ製品として承認されたいずれの我々の候補製品も12年の専門期間を得る資格があるべきであると考えられる。しかしながら、国会の行動または他の理由により、このような排他性は短縮される可能性があり、またはFDAは、私たちの候補製品を競合製品の参考製品とみなさないかもしれず、これは、予想よりも早く生物学的類似競争のための機会を創出する可能性がある。BPCIAの他の面では,そのいくつかがBPCIAの排他的条項に影響を与える可能性があり,最近の訴訟のテーマでもある.さらに、承認されると、生物類似体が私たちのいずれかの参考製品をどの程度置換するかは、非生物製品の伝統的な模造薬代替と類似しており、これはまだ発展中のいくつかの市場および規制要因に依存するかどうかは不明である。
もし私たちが“ハッジ·ワックスマン法案”に基づいて米国や外国で類似した法律によって特許期間を延長しなければ、私たちが開発する可能性のある任意の候補製品のマーケティング排他的期間を延長することができ、私たちの業務は実質的に損害を受ける可能性がある。
米国では、FDA承認された薬物をカバーする特許期限は、FDA規制審査中に失われた特許期限の補償として、限られた特許期限延長を得る資格がある可能性がある。1984年の“薬品価格競争と特許期限回復法”は、ハッジ·ワックスマン法案とも呼ばれ、特許期限が特許満了後最大5年間延長されることを許可している。特許期間延長の長さは,薬物が臨床開発と規制審査にある時間の長さと関係がある。特許延期は、特許の残り期間を製品承認日から合計14年間延長してはならず、承認された薬物に適用され、カバーされた特許のみを延長することができる。ヨーロッパにも同様の規定があります保護証明書を補充するなど
105
承認された薬物をカバーする特許の期限を延長する他の非米国司法管轄区域と。将来的には,我々の候補製品がFDAによって承認されれば,これらの候補品をカバーする特許出願特許期間の延長が予想されるが,適用当局がこのような延長を承認すべきかどうか,そのような延長の長さを承認すべきかどうかの評価に同意する保証はない.例えば、試験段階または規制審査中に職務調査を行うことができなかったため、適用された最終期限内に出願を提出できなかったため、関連特許の満了前に出願を提出することができなかったか、または適用要件を満たすことができなかったため、米国または他のどの国/地域でも特許期間延長の許可を得ることができない可能性がある。また、政府当局が提供する延期期限およびそのような延期期間のいずれかの特許保護範囲は、私たちが要求しているものよりも短い可能性がある。もし私たちがいかなる特許期間の延長を得ることができない場合、あるいはどのような延長の期限が私たちが要求したものよりも短い場合、私たちの競争相手は私たちの特許権が満期になった後に競争製品の承認を受ける可能性があり、私たちの業務、財務状況、運営結果、見通しは大きな損害を受ける可能性があります。
私たちは“ハッジ·ワックスマン法案”に基づいて、私たちの任意の候補製品をカバーするアメリカ特許の特許期間延長を獲得しない可能性があります。たとえその特許が特許期間を延長する資格があるところを決定することができても、あるいはそのような延長を受けた場合、その期間は私たちが求めているよりも短いかもしれません。
米国や他の管轄区域特許法の変化は特許の全体的な価値を低下させ、私たちの遺伝子編集プラットフォーム技術と候補製品を保護する能力を弱める可能性がある。
他のバイオテクノロジーや製薬会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存している。生物製薬業界で特許を獲得と実施することは技術上の複雑性と法律上の複雑性にも関連するため、コストが高く、時間がかかり、しかも内在的な不確定性を持っている。
“Leahy-Smith America発明法”や“Leahy-Smith Act”のような特許改革立法を含む米国特許法または特許法解釈の変化は、私たちの所有および認可内の特許出願をめぐる起訴および私たちの所有および認可内の認可された特許をめぐる不確実性およびコストを増加させる可能性がある。“ライシー·スミス法案”は米国特許法を多くの重大な改正を行った。これらの変化には、特許出願起訴方式に影響を与える条項、既存技術を再定義し、競争相手に特許の有効性を疑問視するためのより効果的かつ費用対効果的な経路を提供し、特許起訴中に米国特許商標局が以前の技術を第3の方向に提出することを可能にすることと、付与後審査を含む米国特許商標局が管理する付与後プログラムにおいて特許有効性を攻撃する追加プログラムとが含まれる各方面間審査と派生手続き。USPTO手続きの証拠基準は、米国連邦裁判所が特許請求の無効を宣言するために必要な証拠基準よりも低いため、第三者は、USPTO手続において、USPTOが権利請求を無効にするのに十分な証拠を提供する可能性があり、同じ証拠が最初に地域裁判所訴訟で提出された場合には、権利請求を無効にするのに十分ではない。したがって,第三者は米国特許商標局の手続きを用いて我々の特許主張の無効を宣言しようとする可能性があり,第三者が地域裁判所訴訟で最初に被告として疑問を提起すれば,我々の特許主張は無効ではない.
特許性の他の要件が満たされていると仮定すると,2013年3月までに,米国では,まず発明により保護された発明を発明した者が特許を取得する権利があり,米国以外では,最初に特許出願を提出した者が特許を取得する権利がある。2013年3月以降、“ライシー·スミス法案”(Leahy-Smith Act)によれば、米国は先に出願を提出する制度に移行し、この制度の下で、特許性の他の法定要件が満たされたと仮定すると、最初に特許出願を提出した発明者は、第三者が最初にその発明を発明した者であるか否かにかかわらず、発明の特許を取得する権利がある。したがって、Leahy-Smith法案およびその実施は、私たちの特許出願をめぐる起訴および私たちが発行された特許の実行または保護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務、財務状況、運営結果、および見通しに大きな悪影響を及ぼす可能性がある。
また,生物製品や薬品の開発や商業化における会社の特許地位は特に不確定である。米国最高裁判所の過去の裁決は、場合によっては特許保護範囲を縮小し、場合によっては特許所有者の権利を弱体化させる。この一連の事件は特許取得後の有効性と実行可能性の面で不確実性をもたらしている。米国議会、連邦裁判所、および米国特許商標局の将来の行動によれば、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、私たちの特許権および私たちの将来の特許権の保護、擁護、執行能力に実質的な悪影響を及ぼす可能性がある。例えば、この場合、アソークです。分子病理学はMyriad Genetics,Inc.を訴える米国最高裁判所は,あるDNA分子の権利主張は特許を申請できないと判断した。最近ではアンはセノフィを訴えたまた,連邦巡回裁判所は,関数的言語を用いた宣言は,広範な宣言の起用要求を満たすことに高い障害となる可能性があると考えている
106
関数式言語です。私たちは裁判所、アメリカ議会、またはUSPTOのこの決定と未来の判決が私たちの特許価値にどのように影響するか予測できない。他の管轄区域特許法の任意の類似した不利な変化は、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を及ぼす可能性もあります。
私たちの候補製品をカバーする発行された特許が法廷で疑問視されれば、無効または実行不可能と認定される可能性がある。私たちは法廷で私たちの商業秘密を保護できないかもしれない。
もし私たちまたは私たちの許可パートナーのうちの1つが第三者に対して法的訴訟を提起して、私たちの候補製品のうちの1つをカバーする特許を強制的に執行する場合、被告は私たちの候補製品をカバーする特許が無効または強制執行できないと反訴することができる。米国の特許訴訟では,被告が無効または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、明らかな書面、または実施できないことを含む、いくつかの法定要件のいずれかを満たしていないと言われているからかもしれない。さらに、場合によっては、特許有効性挑戦は、非法定明顕二重特許に基づいている可能性があり、成功すれば、明顕型二重特許に対する権利請求の無効が発見される可能性があり、または明顕二重特許の裁決を排除するための終了免責声明が提出された場合、米国特許商標局によって付与された特許期限調整を含む特許期間の損失をもたらす可能性がある。実行不可能な主張の理由は,特許起訴に関連する人が起訴期間中に米国特許商標局に特許実行可能性に関する情報を隠蔽したり,誤った声明をしたりしたためかもしれない.第三者も米国や海外の行政機関に類似したクレームを出すことができ、訴訟範囲外でも同様である。これらの仕組みには再審査、贈与後の審査、各方面間外国の管轄区域での検討と同等の手続き。このような訴訟は、私たちの特許がこれ以上私たちの候補製品をカバーしないように、私たちの特許を撤回、キャンセル、または修正する可能性がある。法的に無効と実行不可能と断言された後の結果は予測できない。例えば、有効性の問題については、私たちは、特許審査員および私たちまたは私たちの許可パートナーが起訴中に知らない無効な以前の技術を決定することができない。もし被告が無効または強制不可能な法的主張に勝った場合、私たちは私たちの1つまたは複数の候補製品の少なくとも一部またはすべての特許保護を失うかもしれない。このような特許保護の喪失は私たちの業務に実質的な悪影響を及ぼすかもしれない。
特許提供の保護に加えて、我々は、特許を出願できない独自技術、特許を実施しにくいプロセス、および私たちの候補製品発見および開発中に特許がカバーされていないノウハウ、情報または技術に関する任意の他の要素を保護するために、商業秘密保護および秘密保護プロトコルに依存して保護する。しかし、商業秘密を保護することは難しいかもしれないが、アメリカ国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、あるいは保護したくない。私たちは、従業員、コンサルタント、科学コンサルタント、請負業者と秘密保持協定を締結することで、私たちのノウハウやプロセスを保護することを求めています。私たちは私たちが可能であるか、または私たちの商業秘密またはノウハウとプロセスに接触したすべての当事者とこのような合意に到達したことを保証することができない。また、私たちは私たちのビルの実体安全と、私たちの情報科学技術システムの実体と電子安全を維持して、私たちの資料と商業秘密の完全性とセキュリティを維持することに力を入れています。私たちはこのような個人、組織、そしてシステムに自信がありますが、合意や安全措置は違反される可能性があり、私たちはどんな違反にも対応する十分な救済措置がないかもしれません。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。
知的財産権訴訟や他の知的財産権に関する法律手続きは、私たちに大量の資源を費やし、私たちの人員のその正常な職責に対する関心を分散させる可能性がある。
解決策が私たちに有利であっても、知的財産権クレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させ、私たちの技術や管理者の正常な責任を分散させる可能性があります。また、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や訴訟手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源を持っているので、私たちよりも効率的にこのような訴訟や訴訟の費用を負担するかもしれません。そして、彼らのより成熟して発展した知的財産権の組み合わせのために、彼らはこのような訴訟においても優勢である可能性があります。知的財産権訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争の能力を損なう可能性がある。
107
特許保護の獲得と維持は、政府特許機関が提出した様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要件に適合しない場合、私たちの特許保護は減少またはキャンセルされる可能性がある。
私たちが所有し許可されている特許および特許出願の有効期間内には、定期的なメンテナンス、更新および年金費用、ならびに発行および処理された特許出願の任意の様々な他の政府費用を、いくつかの段階に分けて、または毎年米国特許商標局および外国特許代理機関に支払わなければならない。米国特許商標局および各種外国政府特許機関は、特許出願過程において、いくつかのプログラム、文書、費用支払い、およびその他の同様の規定を遵守することを要求する。場合によっては、私たちは、私たちの許可パートナーに依存して、関連する特許エージェントにこれらの費用を支払うか、またはその手続きおよび文書規則を遵守するかもしれません。私たちの特許については、私たちは外部会社と外部弁護士に依存して納期を注意し、私たちが彼らにそうするように指示した後に費用を支払う。多くの場合、適用規則に従って滞納金を支払うか、または他の方法で不注意を是正することによって失効を是正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効され、関連法ドメインの特許権の一部または全部の喪失をもたらす可能性がある。特許または特許出願が放棄または失効される可能性のある規定を遵守しないイベントは、規定された期限内に公式行動に応答することができなかったこと、費用を支払わなかったこと、および適切に合法化されず、正式な文書を提出することを含む。この場合、潜在的な競争相手は、同様または同じ製品または技術で市場に参入することができる可能性がある。もし私たちまたは私たちの許可者が私たちの候補製品をカバーする特許および特許出願を維持できなかった場合、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を与えます。
私たちは限られた外国の知的財産権を持っており、世界各地で私たちの知的財産権と独自の権利を保護できないかもしれない。
私たちのアメリカ以外の知的財産権は限られている。世界のすべての国で候補製品の申請、起訴、特許を守る費用は目を引くほど高く、外国の法律はアメリカの法律のように私たちの権利を保護できないかもしれない。また、一部の国の法律は知的財産権の保護程度は米国の連邦や州法律に及ばず、名目上このような保護があっても、このような知的財産権に対する司法や政府の法執行が不足している可能性がある。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、私たちに特許保護や許可証を持っているが、法執行力がアメリカの地域に比べて他の侵害製品を輸出することもできる。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護、特にバイオテクノロジー製品に関する保護の実行に賛成せず、私たちの特許を侵害したり、私たちの知的財産権および独自の権利に違反する競争製品を販売することを阻止することを困難にする可能性がある。また、ある司法管轄区域は新しい治療方法を構成する発明に対して同程度或いは完全な保護を与えていない。
外国の管轄区域で私たちの知的財産権と独自の権利を強制的に執行する訴訟手続きは巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移すことは、私たちの特許を無効または狭義に解釈されるリスクに直面させる可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者が私たちにクレームを提起する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちの知的財産権および独自の権利を世界各地で強制的に実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネス的優位性を得るのに十分ではないかもしれません。
多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちまたは私たちの任意の許可者が第三者に私たちの業務に関連する任意の特許の許可を付与することを余儀なくされた場合、私たちの競争的地位が損なわれる可能性があり、私たちの業務、財務状況、運営結果、および見通しは不利な影響を受ける可能性があります。
また、アメリカと外国の地政学的行動は不確実性を増加させる可能性がある
そして私たちの特許出願または現在または未来の特許出願の起訴または維持に関連する費用
108
ライセンス者および私たちが発行した特許または現在または未来の特許の維持、強制執行または保護
許可証発行人。例えばアメリカと外国政府はロシアのウクライナ侵攻に関する行動です
ロシアでの特許出願の提出、起訴、そして維持を制限または阻止する可能性がある。政府の行動はまたロシアで発行された特許を維持することを阻止するかもしれない。これらの行動は、私たちが許可した特許または特許出願が放棄または失効され、ロシアにおける特許権の一部または全部を喪失させる可能性がある。このような事件が発生すれば、私たちの業務に実質的な悪影響を及ぼす可能性がある。また、ロシア政府は2022年3月に、ロシアの会社や個人がロシアで友好的ではないと考えている米国や他の国で、同意または補償なしに、米国市民権または国籍を有し、米国および他のロシアが友好的でないと考えられる商業または利益活動の主要地点に登録されている特許権者が所有する発明を利用することを許可する法令を採択した。したがって、私たちは、第三者がロシアで私たちの発明を実践したり、私たちの発明を使用して製造された製品をロシア国内で販売したり輸入したりすることを防ぐことができないだろう。したがって、私たちの競争地位は損なわれる可能性があり、私たちの業務、財務状況、経営業績、見通しは不利な影響を受ける可能性があります。
私たちは、私たちの従業員、コンサルタント、または請負業者が第三者の機密情報を誤って使用または開示したと主張する第三者のクレームを受けるかもしれないし、私たちは彼らの現在または前任雇用主のいわゆる商業機密を誤って使用または開示したり、私たちが彼らの知的財産権を流用したと主張したり、私たちが私たち自身の知的財産権を所有していると主張したりするかもしれない。
私たちの多くの従業員、コンサルタント、および請負業者は、以前、私たちの競争相手または潜在的な競争相手を含む大学または他の製薬またはバイオテクノロジー会社に雇われていた。私たちは、私たちの従業員、コンサルタント、および請負業者が、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちは、商業秘密または他の固有情報を含む任意のそのような個人の現職または前任雇用主の知的財産権を使用または開示していると告発される可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。
また、私たちの政策は、知的財産権開発に参加する可能性のある私たちの従業員、コンサルタント、請負業者が合意に署名し、このような知的財産権を私たちに譲渡することを要求しているにもかかわらず、実際に私たちが自分の知的財産権を開発しているすべての側とこのような合意を実行することができないかもしれません。私たちと彼らの知的財産権譲渡協定は自動的に実行されないかもしれないし、違反される可能性があります。私たちは第三者にクレームを出させたり、私たちが私たちの知的財産権の所有権だと思うことを確認するために、私たちが提起したクレームを弁護することを余儀なくされるかもしれません。このようなクレームは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
もし私たちがこのような任意のクレームを起訴または弁護できなければ、金銭損害賠償の支払いに加えて、貴重な知的財産権や人員を失う可能性があり、これは私たちの競争業務の地位と将来性に実質的な悪影響を及ぼす可能性がある。このような知的財産権は第三者に付与することができ、私たちは、私たちの技術または製品を商業化するために、第三者から許可を得る必要があるかもしれません。この許可は、商業的に合理的な条項では得られないかもしれません、または全く存在しないかもしれません。このようなクレームの起訴や抗弁に成功しても、訴訟は巨額のコストを招き、経営陣や従業員の注意を分散させる可能性がある。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
私たちのいくつかの候補技術および製品のための特許を申請することに加えて、私たちは、私たちの競争地位を維持するために、ビジネス秘密および秘密協定に依存して、私たちの非特許ノウハウ、技術、および他の固有情報を保護します。私たちは、私たちの従業員、会社協力者、外部科学協力者、CRO、CMO、コンサルタント、コンサルタント、および他の第三者のような、これらの秘密にアクセスする権利のある当事者と秘密協定を締結することによって、私たちのビジネス秘密および他のノウハウを保護することを求めています。私たちはまた、私たちの従業員やコンサルタントと秘密および発明または特許譲渡協定を締結していますが、私たちが私たちの商業秘密またはノウハウに接触している可能性があるか、または接触したことがあるすべての当事者とこのような合意を締結した保証はありません。このような努力にもかかわらず、どちらも合意に違反し、私たちのビジネス秘密を含む私たちの固有の情報を漏洩する可能性があり、私たちは十分な救済措置を得ることができないかもしれない。商業秘密の漏洩や流用を検出し、当事者に商業秘密の不法開示或いは流用を要求することは困難であり、高価で時間がかかり、結果は予測できない。
契約措置に加えて、物理的および技術的セキュリティ対策のような他の適切な予防措置を通じて、私たちの独自の情報の機密性を保護しようとしています。しかしビジネスの秘密と
109
独自技術は保護するのが難しいかもしれない。例えば、従業員またはアクセス許可権限を有する第三者が商業秘密を盗用する場合、これらの措置は、私たちの独自の情報に十分な保護を提供しない可能性がある。私たちの安全措置は、従業員やコンサルタントが私たちのビジネス秘密を盗用して競争相手に提供することを阻止できないかもしれませんが、私たちはこのような不正行為に対するいかなる救済措置も、私たちの利益を十分に保護するための十分な救済措置を提供できないかもしれません。また、商業秘密は、私たちが法的追跡権を得ることを阻止する方法で他の人によって独立して開発された可能性がある。私たちのビジネス秘密のような私たちの任意の機密または独自の情報が漏洩または流用されている場合、またはこれらの情報のいずれかが競争相手によって独立して開発されている場合、私たちの競争地位は損なわれる可能性がある。
しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。もし私たちの任意の商業秘密が競争相手または他の第三者によって合法的に取得または独立して開発された場合、私たちは彼らまたは彼らが情報を伝達する人がその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちの任意の商業秘密が競争相手または他の第三者に漏洩された場合、または競争相手または他の第三者によって独立して開発された場合、私たちの競争地位は実質的かつ不利な損害を受けるだろう。
もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は悪影響を受けるかもしれません。
任意の登録商標または商号は、汎用商標として疑問、回避、または発表される可能性があり、または他の商標が侵害されたと認定される可能性がある。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれません。私たちは私たちが関心のある市場の潜在的なパートナーや顧客の中で知名度を確立するために必要です。時々、競争相手は私たちと似たような商品名や商標を採用して、ブランド表示を確立する能力を阻害し、市場の混乱を招く可能性があります。さらに、他の登録商標または商標の所有者は、我々の登録または未登録商標または商号の変異体を含む商号または商標侵害クレームを提出することができる。長期的には、私たちの商標や商号に基づいて名称を確立することができなければ、効果的に競争できない可能性があり、私たちの業務は悪影響を受ける可能性があります。商標、商業秘密、ドメイン名、著作権または他の知的財産権に関連する専有権を実行または保護する努力は無効である可能性があり、大量のコストおよび資源移転を招き、私たちの財務状況または運営結果に悪影響を及ぼす可能性があります。
知的財産権はすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。例えば:
110
これらの事件が発生すると、それらは私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
商業化に関連するリスク
私たちが現在または未来の任意の候補製品が市場承認を得ても、医師、患者、第三者支払人、および医学界の他の人が商業成功に必要な市場受容度を達成できない可能性があり、そのような候補製品の市場機会は、承認されれば、私たちが予想しているよりも小さいかもしれない。
もし私たちが現在または未来の任意の候補製品が市場の承認を得たら、それはまだ医者、患者、第三者支払人、および医学界の他の人の十分な市場受容度を得ることができないかもしれない。例えば、現在の心血管疾患の治療法、例えばスタチン類薬物、エゼミブ、フェニルパドール酸、ロミチート、ミパモソンとエイコサペンタエン酸エチルは、医学界で広く認められており、医師はこれらの治療法に依存し続ける可能性がある。
我々が開発したVERVE−101,VERVE−201,あるいは任意の他の候補製品が臨床試験においてその安全性と有効性の終点に達しても,臨床試験の成功が商業製品としての成功を確保することは確認できない。例えば,2022年9月,アスリコンとIonis製薬会社は月に1回皮下注射したアンチセンスオリゴヌクレオチドPCSK 9阻害剤を高コレステロール血症治療の第三段階臨床開発に用いないことを決定し,これまで行われてきた2 b期臨床試験はその主要な終点に達し,28週間後に統計的に有意な低密度リポ蛋白質コレステロール(LDL−C)の62.3%低下を実現した
プラセボは、結果がアスリカン目標製品の概要基準を満たしていない上で、広範な第三段階開発計画に投資した。
教育医療界や第三者支払者が我々の候補製品のメリットを知るには大量の資源が必要であり,成功しない可能性がある。もし私たちが現在または未来の候補製品が実現できなければ
111
受容度が十分に高ければ、著しい製品収入が生じないかもしれませんし、利益が出ないかもしれません。もし私たちの現在または未来の候補製品が商業販売のために許可された場合、市場の受け入れ度は多くの要素に依存する
現在または未来の候補製品の潜在的な市場機会の評価は、業界出版物、研究、調査、および第三者による研究から得られた業界と市場データ、およびこれらのデータ、研究、調査と研究の分析に基づいている。業界出版物および第三者研究、調査および研究は、一般に、それらの情報は、そのような情報の正確性または完全性を保証しないにもかかわらず、信頼できるソースから得られることを示している。私たちはこれらの業界の出版物と第三者研究、調査と研究が信頼できると信じているが、私たちはこれらのデータを独立して確認していない。候補製品の潜在的な市場機会の推定は著者らの業界知識、業界出版物と第三者研究、調査と研究に基づく重要な仮定を含み、これらの仮説は小サンプルに基づく可能性があり、市場機会を正確に反映できなかった。私たちは私たちの内部仮定が合理的だと信じているが、これらの仮定を証明する独立したメッセージ源はない。もし私たちの任意の仮定や推定、あるいはこれらの出版物、研究、調査、または研究が不正確であることが証明された場合、私たちの任意の候補製品の実際の市場は私たちが予想していたより小さいかもしれないので、私たちの製品販売収入は制限されるかもしれません。私たちは利益を達成したり維持したりすることがもっと難しいかもしれません。
私たちは激しい競争に直面しており、これは他の人たちが私たちの前に製品を発見し、開発したり、商業化したり、あるいは私たちよりも成功するかもしれない。
新薬や生物製品の開発と商業化競争が激しい。これらの製品に対する看護基準が確立されているため、心血管疾患の新製品を治療する上で特に競争力がある。私たちの現在の候補製品は競争に直面しており、将来的に開発または商業化を求める可能性のある任意の候補製品も、世界各地の主要な製薬会社、専門製薬会社、バイオテクノロジー会社からの競争に直面するだろう。現在,多くの大手製薬やバイオテクノロジー会社が製品をマーケティング·販売しているか,あるいは我々が開発している製品候補の多くの疾患適応を治療するための製品が開発されている。競争力のある製品や療法のいくつかは私たちの方法と似た科学的な方法に基づいており、もういくつかは完全に異なる方法に基づいている。潜在的な競争相手はまた学術機構、政府機関とその他の公共と個人研究組織を含み、これらの組織は研究を展開し、特許保護を求め、研究、開発、製造と商業化のための協力手配を確立する。
いくつかの承認された製品は低密度リポ蛋白質の低下或いは心血管リスクの低減に応用でき、例えばスタチン類薬物、エゼミブ、フェニルペドド酸、ロミチート、ミパモソンとエイコサペンタエン酸などである。いくつかの承認された製品はPCSK 9蛋白を標的とし、低密度リポ蛋白とASCVDリスクを低下させる機序としている。Evocumabこれは単一抗体やmAbで市場名はRepathaです® 安進により開発され,米国食品医薬品局によってヘテロ家族性高コレステロール血症患者,HOFH患者の治療として承認された
112
ASCVDです。AlirocumabこれはPRALUENTという市販の単一抗体です®セノフェイとRegeneron製薬会社あるいはRegeneronはASCVD患者の治療とHeFHを含む原発性高脂血症患者の治療にRegeneronを許可した。承認されたモノクロナル抗体治療は細胞外でPCSK 9蛋白を抑制することにより作用する。Inclisiranは小さな干渉RNAやsiRNAで市場名はLeqvioです®ノワール社により承認され,米国では低密度リポ蛋白−Cをさらに低下させる必要がある臨床ASCVDやHeFH患者の治療に用いられ,ヨーロッパではHeFHや混合性脂質異常を含む高コレステロール血症患者の治療に用いられている。Inclisiranは肝細胞内PCSK 9の合成を抑制することで機能し,細胞外蛋白抑制とは異なる。臨床発展の異なる段階で、3種類の経口小分子製品候補薬物はPCSK 9蛋白を標的とし、低密度リポ蛋白の低下とASCVDリスクを低下させる機序として知られている。これらの薬物は、最近完成した2 b期成人高コレステロール血症患者試験で研究され、2023年第1四半期に結果を公表する予定であるメルク社のMK-0616を含み、Esperion Treeutics社が許可したSerometrix LLCの経口小分子薬であり、2022年にINDを提出する計画、2024年末または2025年初め、およびAZD 0780、アスリコンがDogma治療会社から買収され、第1段階の臨床試験で評価されている。
臨床前開発にはPCSK 9遺伝子に対するもう2つの遺伝子エディタがあることが知られている。Precision Biosciences,Inc.,またはPrecisionは,臨床前データを発表し,以下の場合,非ヒト霊長類動物のPCSK 9とLDL−Cレベルが長期にわたって安定して低下することを示している体内にあるその遺伝子編集プラットフォームを利用してPCSK 9遺伝子に対して遺伝子編集を行った。2021年9月、PrecisionはiECUREと提携し、iECUREは2022年にPrecisionのPCSK 9配向ヌクレアーゼ候補製品をFH治療の第1段階臨床試験に進める予定である。2023年1月、Precisionは、iECUREとの協力による計画の実施を中止することを決定し、薬剤が将来的に臨床試験に入るかどうかについて、より多くの指導を提供することを計画していることを発表した。また、2022年にCRISPR治療会社またはCRISPRはCTX 330を発表し、これがその研究段階である体内にあるPCSK 9に対する遺伝子エディタ。
EvinacumabはRegeneronによって販売されているAngptl 3蛋白に対するモノクロナル抗体であり,HoFH患者の治療にFDAによって承認されており,また難治性高コレステロール血症,ASCVDあるいはHeFH患者および重篤な高トリグリセリド血症患者の第二段階研究で評価されている。臨床開発にはAngptl 3を標的とした候補製品がいくつかあり,これを低密度リポ蛋白−CとASCVDリスクを低下させる機序としてARO−ANG 3,Angptl 3に対するsiRNA,矢印製薬会社がHoFH患者と混合性脂質異常患者の第2段階臨床試験を行っていることが知られている。2022年、矢印社は2023年下半期にHoFH患者とHeFH患者においてARO-ANG 3の重要な3期研究を開始する計画を発表した。また、礼来社はAngptl 3タンパク質に対するsiRNAを評価しており、混合性脂質異常を有する成人に対する第2段階研究である。2022年、CRISPRはAngptl 3に対する遺伝子編集プログラムCTX 310を発表し、INDイネーブル研究を行っており、2023年に患者投与を開始する予定である。
リポ蛋白質(A)やLp(A)を低下させるためのいくつかの研究薬が開発されている。その中には,2019年にノワ社がIonis PharmPharmticalsから許可を得たアンチセンスオリゴヌクレオチドPelecarsenが含まれており,Lp(A)上昇と心血管疾患患者の3期LP(A)Horizon心血管結果研究で評価されており,2025年にはTOPLINE結果が予想される。OlpasiranはLPAに対する研究siRNA薬物であり,安進社が矢印製薬会社から許可を得ており,最近ASCVD患者と高Lp(A)濃度患者のLp(A)濃度を低下させることが証明されている。Olpasiranは現有のASCVDと高Lp(A)患者の心血管イベントを減少する潜在力はOcean(A)研究の中で評価を行い、この研究は2022年にスタートし、2026年に研究を完成する予定である。また,SLN 360はSilence Treeutics plcが開発している研究性siRNA薬であり,リポ蛋白質(A)濃度の高さとASCVD事件高リスク患者に対する第2段階研究で評価されており,CRISPRは2022年にCTX 320を発表しており,その研究段階である体内にある遺伝子エディタ標的LPA.
もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。また、多くの場合、私たちの競争能力は、後発薬の使用を奨励することを求める保険会社または他の第三者支払者の影響を受ける可能性がある。もし私たちの候補製品が発売承認されたら、私たちはそれらの価格が競争相手の模造薬より著しく高いと予想します。
私たちと比較して、私たちが競争しているか、あるいは将来競争する可能性のある多くの会社は、研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング承認製品を獲得する上で、より多くの財務資源と専門知識を持っている。
113
製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。
販売、マーケティング、流通能力を確立することができない場合、または第三者と販売、マーケティング、および流通協定を締結することができなければ、現在および将来の候補製品が承認された場合、商業化に成功できない可能性がある。
私たちは販売やマーケティングインフラもなく、製品商業化会社としての経験もありません。私たちが発売許可を得た任意の製品を商業的に成功させるためには、販売、マーケティング、流通組織を構築する必要があり、私たち自身であるか、第三者との協力やその他の手配を通じて行う必要がある。
将来、私たちのいくつかの候補製品が承認されれば、アメリカでマーケティングを行う販売·マーケティングインフラを構築したいと思います。私たち自身の販売、マーケティング、そして流通能力を確立することはリスクに関するものだ。例えば、販売チームの採用と訓練は高価で時間がかかり、どんな製品の発表も延期される可能性がある。販売チームを募集し、マーケティング能力を確立する候補製品の商業発表が何らかの理由で延期または発生していない場合、これらの商業化費用を早期または不必要に発生させる。これらの努力は費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり、再配置できなければ、私たちの投資は損失します。
私たち自身が製品を商業化するのを阻害するかもしれません
もし私たちが自分の販売、マーケティング、流通能力を確立することができなければ、私たちは第三者と合意してこれらのサービスを提供し、私たちの製品収入と私たちの収益力(もしあれば)は私たち自身が開発したいかなる製品もマーケティング、販売、流通を下回るかもしれません。さらに、私たちは第三者との販売、マーケティング、流通に成功できないかもしれません。あるいは私たちが受け入れられる条項でそうすることができないかもしれません。私たちはこのような第三者に対して支配権がほとんどないかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが販売、マーケティング、流通能力を成功的に確立できなければ、私たち自身も第三者と協力しても、私たちの候補製品を商業化することに成功しないだろう。
私たちは現在依存しており、私たちの候補製品を生産するためにCMOに依存し続ける予定だ。もし私たちが予想された計画を達成できない場合、あるいはこれらの組織が私たちの供給要求を満たすことができなければ、私たちの候補製品の開発および/または商業化は延期されるかもしれない。
私たちは現在依存していて、引き続き第三者が私たちの候補製品を生産する臨床用品と私たちの製品の商業用品に依存して、関連する監督管理機関が発売を許可した場合、包装、滅菌、貯蔵、流通、その他の生産物流に依存すると予想されています。もし私たちが予想していた条項やスケジュールに従ってこのような計画を達成できなければ、私たちの候補製品の開発および/または商業化は延期されるかもしれない。もしこれらの第三者が成功しなければ
114
もし私たちが契約義務を履行し、期待期限を満たしたり、法規の要求に基づいて私たちの候補製品を製造する時、もし私たちとこれらの当事者との間に食い違いがある場合、あるいはこれらの当事者がマーケティング許可を得た任意の候補製品の商業化を支援する能力を拡張できない場合、私たちは私たちの供給要求を満たすために十分な候補製品の生産を満たすことができないか、または遅延する可能性があります。これらの施設はまた、流行病(持続的な新冠肺炎の大流行、テロ、戦争または他の武力衝突を含む)、地政学的緊張(例えば、ロシアとウクライナとの間の持続的な衝突)、自然災害(例えば、洪水または火災)、またはそのような施設が規制検査後の汚染または規制懸念のような製造問題に直面する可能性があるという悲劇的な事件の影響を受ける可能性がある。この場合、適切な代替第三者施設を見つけ、契約関係を確立する必要があるかもしれませんが、これは既製または許容可能な条項ではないかもしれません。これは、追加の要求されたFDA承認の結果を含む追加の遅延および増加した費用をもたらし、私たちの業務に実質的な悪影響を及ぼす可能性があります。
私たちの第三者メーカーはFDAの検査と承認を受けて、その後、私たちの任意の候補製品の製造と販売を開始し、その後、時々FDAの検査を受けることができます。もし私たちの第三者製造業者がこのような検査を通過し、他の方法でFDAの私たちの候補製品に対する承認案を円満に完成させなかった場合、FDA Form 483の観察通知、警告状、または禁止または運営許可証の取り消しなどの規制行動を引き起こす可能性がある。
我々の競争相手や他社がこのような原料または原料薬を購入することによる不足を含む生産能力制限または原料または原料薬市場の遅延または中断により、我々または我々の第三者メーカーはまた、我々の候補製品を生産するために必要な原材料または原料薬の不足に遭遇する可能性があり、これらの原料または原料薬の数は、私たちの臨床試験に必要な数を必要とし、または、候補製品が承認された場合、商業化または需要の増加のために十分な数を使用することができる。もし私たちまたは私たちの第三者メーカーが十分な数の候補製品を生産するために必要な原材料や原料薬を得ることができなければ、私たちの業務に大きな悪影響を及ぼす可能性があります。
任意の候補製品を商業化することができても、これらの製品は、不利な価格設定法規、第三者保険、清算やり方、医療改革措置の制約を受ける可能性があり、これは私たちの業務を損なう可能性があります。
新薬製品の発売審査、定価、カバー範囲と精算を管理する規定は国によって異なる。現在と未来の立法は承認要求を大幅に変更する可能性があり、これは追加のコストに関連し、承認の遅延を招く可能性がある。一部の国は薬品の販売価格が承認されてから発売されることを要求している。多くの国で、定価審査期間はマーケティングまたは製品許可を承認した後から始まる。一部の外国市場では、処方薬の定価は初歩的な承認を得た後も、政府の持続的なコントロールを受けている。したがって、特定の国/地域での製品のマーケティング承認を得ることができるかもしれませんが、その後、価格法規の制約を受けて、これらの法規は私たちの製品の商業発表を延期し、長い時間遅延し、その国/地域で製品を販売することによって生じる収入に悪影響を与える可能性があります。不利な価格設定制限は、私たちの候補製品が市場承認を得ても、1つ以上の候補製品への投資を回収する能力を阻害する可能性がある。
私たちが任意の候補製品を商業化することに成功した能力はまた、政府衛生行政当局、個人健康保険会社、および他の組織がこれらの製品および関連治療に提供する保険範囲と十分な補償にある程度依存するだろう。政府当局や他の第三者支払人、例えば個人健康保険会社や健康維持組織は、どのような薬剤を支払うかを決定し、精算レベルを確立する。第三者支払者の製品使用状況の決定を含む第三者支払人の保証範囲と精算は多くの要素に依存する可能性がある
米国では、第三者支払者の間に統一された製品カバーや精算政策はない。したがって、政府や他の第三者支払人から製品の保証範囲と補償承認を得ることは時間がかかり、費用がかかるプロセスであり、各支払人に提供する必要があるかもしれない
115
科学的、臨床的、費用効果的なデータを支持し、支払者ごとの方法で私たちの製品を使用するが、保険と十分な精算を受ける保証はない。承認されれば、政府医療計画(例えば、MedicareおよびMedicaid)、個人健康保険会社および他の第三者支払者が提供する保険および精算範囲の利用可能性および十分性は、大多数の患者が私たちの候補製品を負担することができるために重要である。もし政府当局、個人健康保険会社、他の組織の承認を得たら、私たちの候補製品のために受け入れ可能な保証と補償レベルを実現する能力があるかどうかは、私たちの候補製品を商業化することに成功する能力に影響を与えるだろう。第三者決済者が特定の製品に保険を提供することによる精算支払率が十分でない可能性があるか,あるいは患者の自己払い費用が必要となる可能性があると仮定すると,患者は受け入れられないほど高いと考えている。
アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。政府当局と第三者支払者は,特定の薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。ますます多くの第三者支払人は製薬会社に価格に基づいて所定の割引を提供し、医療製品の定価に挑戦することを要求している。私たちが商業化しているどの製品も保険や精算を受けることができないかもしれません。あっても、精算レベルは満足できないかもしれません。精算は私たちが市場で承認された任意の候補製品の需要や価格に影響を及ぼすかもしれない。私たちの製品のために十分な精算を得て維持することは難しいかもしれない。私たちは、カバー範囲と精算または他の治療法に対する精算レベルを証明するために、高価な薬物経済学的研究を要求されるかもしれない。カバー範囲や十分な精算がない場合、あるいは精算が限られたレベルに限られている場合、マーケティングの承認を得た任意の候補製品を商業化することに成功しない可能性があります。
新たに承認された薬物については,保険獲得や精算に重大な遅延がある可能性があり,保険範囲はFDAや米国以外の同様の規制機関がこの薬剤を承認する目的よりも限られている可能性がある。また、保険や精算を受ける資格があるということは、すべての場合に薬物の費用を支払うことを意味するのではなく、研究、開発、製造、販売、流通費用を含む私たちのコストを支払うことを意味するわけではない。もし適用されれば、新薬の一時精算レベルも私たちのコストを支払うのに十分ではないかもしれないし、永久的にならないかもしれない。販売率は薬物の使用や臨床環境によって異なる可能性があり,すでに低コスト薬物のために設定された精算レベルに基づいている可能性があり,他のサービスの既存支払いにも組み込まれている可能性がある。医薬品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、および将来、米国価格よりも低い価格で販売される可能性のある国から医薬品を輸入することを制限する法律の緩和によって低下する可能性がある。第三者支払者が自分の精算政策を設定する際には,通常連邦医療保険カバー政策と支払制限に依存する。私たちは政府の資金援助と個人支払人から私たちが開発してくれた任意の承認された製品から保険と十分な販売率を迅速に得ることができません。これは私たちの経営業績、製品の商業化に必要な資金を調達する能力、そして私たちの全体的な財務状況に実質的な悪影響を及ぼすかもしれません。
私たちの候補製品がアメリカや他の国/地域で販売されることが承認されても、私たちの候補製品が医学的に合理的であることは保証されず、特定の適応や第三者支払者にとって必要であり、第三者支払者が提供する保険範囲と十分な精算レベルを保証することもできず、あるいは第三者支払者の精算政策が私たちの候補製品を販売する収益力に悪影響を与えないことは保証されない。
私たちの将来の成長は海外市場に進出する能力にある程度依存しており、そこでは追加の規制負担や他のリスクや不確実性の影響を受け、これらのリスクや不確実性が現実になれば、私たちの業務を損なう可能性があります。
私たちの将来の収益性はアメリカ以外の市場で私たちの候補製品を商業化する能力にある程度依存するだろう。もし私たちの候補製品を海外市場で商業化すれば、私たちは追加のリスクと不確定要素に直面します
116
これらの不確実性のいずれかに関連するリスクが現実的になれば、我々の業務に実質的な悪影響を及ぼす可能性がある。
私たちの臨床試験と製品責任訴訟は私たちの資源を移転し、私たちが重大な責任を負い、私たちが開発する可能性のある任意の製品の商業化を制限する可能性があります。
私たちは人体臨床試験で私たちの候補製品をテストすることに関連する内在的な臨床試験と製品責任暴露のリスクに直面し、私たちが開発する可能性のある任意の製品を商業化販売すれば、私たちはより大きなリスクに直面する。現在、商業販売のための製品は承認されていませんが、臨床試験で使用されている候補製品、将来使用される候補製品、将来の承認された製品の販売は、責任クレームに直面する可能性があります。これらのクレームは、その製品を使用する患者、ヘルスケア提供者、製薬会社、またはそのような製品を販売する他の人によって提起される可能性がある。もし私たちが私たちの候補製品や製品に被害を与えるクレームを自己弁護することに成功できなければ、私たちは大きな責任を招くだろう。是非曲直や最終結果にかかわらず、賠償責任は
私たちは現在どんな臨床試験責任保険も受けていません。私たちは私たちの臨床試験を拡大したり、私たちの候補製品を商業化し始めた時に保険を受ける必要があるかもしれない。保険範囲はますます高くなっています。私たちは合理的な費用や出現可能な責任を支払うのに十分な金額で保険を受けることができないかもしれない。成功した臨床試験または製品責任クレームまたは一連のクレームが、保険されていない負債または保険を超える負債によって私たちに提起された場合、私たちの資産はそのようなクレームを支払うのに十分ではない可能性があり、私たちの業務運営は損害を受ける可能性がある。
規制承認およびその他の法律コンプライアンス事項に関するリスク
遺伝子編集は新奇であり、私たちが開発する可能性のある任意の候補製品を管理する規制構造は不確定であり、変化する可能性がある。したがって、私たちが開発する可能性のある任意の候補製品について、もし私たちが本当に規制承認を受けた場合、私たちは規制承認を受ける時間とコストを予測することができない。
我々が開発した任意の新しい候補遺伝子編集製品の規制要求は完全に明確ではなく,変化する可能性がある。より広範な遺伝子薬物領域では、FDAとEMAマーケティングの許可を得た遺伝子治療製品の数が限られていることが知られている。より成熟した遺伝子療法や細胞療法のカテゴリーに適した製品であっても,規制機関は
117
景観はまだ発展中である.遺伝子治療製品と細胞治療製品の監督管理要求は常に変化し、将来も引き続き変化する可能性がある。また、既存の遺伝子治療製品と細胞治療製品の監督を担当する人員の間には大量の重複があり、時には調和していない。例えば,米国では,FDAはその生物製剤評価·研究センター(CBER)内に遺伝子治療や関連製品の審査を統合するための組織と高度な治療オフィスを設置し,細胞,組織,遺伝子治療諮問委員会を設置し,その審査についてCBERにアドバイスを提供している。遺伝子治療臨床試験もIBCの審査と監督を受ける可能性があり、IBCは地方機関委員会であり、臨床試験に参与する機関で行われている基礎と臨床研究を審査と監督する。FDAは個別遺伝子治療プログラムを継続できるかどうかを決定しているにもかかわらず,他の審査機関の審査過程や決定は臨床試験の開始を阻害または延期する可能性があり,たとえFDAがこの試験を審査して起動を許可したとしても。
同じ状況は連合にも適用される。欧州連合では,遺伝子治療薬製品の開発と評価は関連EUガイドラインを背景に考慮しなければならない。EMAは遺伝子治療薬製品の開発とマーケティング許可に関する新しいガイドラインを発表し、これらの新しいガイドラインを遵守することを要求する可能性がある。また,高度治療薬製品については,ヒト用医薬品委員会(CHMP)の審査に加え,マーケティング申請許可はEMAの高度治療委員会(CAT)の審査を受ける。したがって,遺伝子治療製品や細胞治療製品に適用されるプログラムや基準は,我々が開発可能な任意の候補製品に適用可能であるが,現在のところ確定していない。
遺伝子治療製品、細胞治療製品、または塩基編集または他の遺伝子編集技術を適用して開発された製品の発売後の経験または臨床試験における不利な発展は、FDA、EMAおよび他の規制機関が、我々が開発する可能性のある任意の候補製品の要求を修正または承認すること、または塩基編集技術を使用する製品の使用を制限することをもたらす可能性があり、いずれも、私たちの業務に実質的な損害を与える可能性がある。そのほか、FDA、EMAと他の監督機関の臨床試験要求及びこれらの監督機関が候補製品の安全性と有効性を確定するための標準は、潜在製品のタイプ、複雑性、意外性及び期待用途と市場によって大きく異なる。我々が開発する可能性のある候補新製品の規制承認プロセスは、他のより有名またはより広く研究されている医薬品または他の製品候補製品と比較して、より高価である可能性があり、時間がかかる可能性がある。既存又は将来の条例又は立法を管理する管理機関は、基本編集技術を利用して製品の生産及び販売をタイムリー又は技術的又は商業的に実行可能な条件下で許可してはならない。さらに、規制行動または個人訴訟は、私たちの研究計画や最終製品の商業化に費用、遅延、または他の障害をもたらす可能性がある。
上述した規制審査委員会および諮問グループおよびその発表された新しいガイドラインは、規制審査プロセスを延長する可能性があり、追加的な研究または試験を行い、私たちの開発コストを増加させ、規制の立場や解釈の変化を招き、これらの候補治療案の承認および商業化を延期または阻止するか、または承認後の重大な制限または制限を招くことができる。我々の研究計画や将来の候補製品の開発を進める際には、これらの規制·諮問チームと協議し、適用されるガイドラインを遵守することが求められる。もし私たちがこれをできなかったら、私たちは私たちが決定して開発した任意の候補製品の開発を延期または停止することを要求されるかもしれない。
遺伝子治療や遺伝子編集を含む遺伝子薬物分野の候補製品を開発しているため,この分野では臨床経験がほとんどないため,FDA,EMAあるいは他の規制機関は,臨床的に意義のある結果を提供するために我々の臨床試験の終点を考慮していない可能性があり,これらの結果の分析が困難である可能性があり,リスクを増加させている。
規制審査過程において、FDA、EMAまたは他の監督管理機関が私たちが開発する可能性のある任意の候補製品の臨床治療効果と安全性を決定できるように、成功基準と終点を決定する必要がある。遺伝子編集手法を用いない臨床経験のない疾患の治療のための候補製品の決定と開発が求められているため,FDA,EMAあるいは他の規制機関は,臨床的に意義のある結果(患者への確実なメリットを反映)を提供するために提案した臨床試験の終点を考慮しない可能性があり,リスクを増加させる。また,それによる臨床データや結果の解析は困難である可能性がある。FDAがわれわれの成功基準が十分な検証と臨床意義を得ていることを確実に発見しても,われわれはあらかじめ指定された終点に到達できず,ある程度の統計学的意義を達成できない可能性がある。また,あらかじめ指定された基準を達成しても,予測不可能な結果や,非主要端点の結果や他の関連データと一致しない可能性がある.FDAも製品のメリットとリスクをトレードオフし、FDAは安全性の観点から治療効果の結果を見るかもしれません
118
規制部門の承認を支持する。連合と他の国の他の規制機関はこのようなサイトとデータについて似たような論評をするかもしれない。私たちが開発する可能性のあるどの候補製品も新しい技術に基づいており、開発とその後の規制承認の時間とコストを予測することは困難である。現在のところ遺伝子編集治療製品はアメリカやヨーロッパで承認されていない。
たとえ著者らが必要な臨床前研究と臨床試験を完成しても、上場審査過程は高価で、時間と不確定であり、著者らが開発したいかなる候補製品の商業化審査を獲得することを阻止するかもしれない。もし私たちが必要な規制承認を得ることができない場合、あるいは規制承認を得る上で遅延が生じた場合、私たちが開発した候補製品を商業化することができない、あるいは商業化を遅らせることになり、私たちの収入を創出する能力は深刻な損害を受けるだろう。
我々が開発した任意の候補製品およびその開発および商業化に関連する活動は、設計、テスト、製造、安全、効果、記録保存、ラベル、貯蔵、承認、広告、販売促進、販売、流通を含み、FDAと米国の他の規制機関、および他の国の類似機関によって全面的に規制されている。候補製品のマーケティング承認が得られなければ、指定された司法管轄区域で候補製品を商業化することはできないだろう。私たちはまだどのような管轄区域の規制機関からもどんな候補製品のマーケティングの承認を得ていない。会社としては、マーケティング承認を得るために必要な申請の提出と支援には経験がなく、その過程で第三者CROに依存して助けてくれると予想されています。監督管理の承認を得るためには、各治療適応の生物候補製品の安全性、純度と効力を決定するために、生産情報を含む広範な臨床前と臨床データと支持情報を各監督機関に提出する必要がある。監督管理の承認を得るためには、製品の製造過程に関する情報を関連監督機関に提出し、関連監督機関が製造施設を検査する必要がある。私たちが開発した任意の候補製品は効果がないかもしれません。中程度の効果しかないかもしれません。あるいは不良または予期しない副作用、毒性、または他の特徴があることが証明される可能性があり、上場承認を受けることを阻止したり、商業使用を阻止したり制限したりする可能性があります。
アメリカと国外では、発売承認を得る過程はすべて高価であり、もっと多くの臨床試験が必要であれば、数年を要するかもしれないが、本当に承認された場合、しかも関連する候補製品のタイプ、複雑性と新規性、および治療すべき特定の疾病或いは状況を含む様々な要素によって大きく異なる可能性がある。大量に開発されている製品のうち、一部のみがFDAや外国規制機関の承認手続きに成功し、商業化されている。たとえ私たちが開発する可能性のある候補製品が臨床試験で安全かつ有効であることが証明されても、規制機関は適時に審査過程を完成できないかもしれないし、監督部門の承認を得られないかもしれない。FDA諮問委員会または他の規制機関が承認または制限を提案しない場合、追加の遅延を招く可能性がある。開発期間中の市場承認政策の変化、追加法規又は法規の変化、又は各提出製品申請に対する規制審査の変化は、出願の承認又は拒絶の遅延を招く可能性がある。FDAと他国の類似機関は承認過程において大きな自由裁量権を有しており、いかなる申請も拒否することができ、私たちのデータが承認を得るのに十分ではなく、追加の臨床前、臨床、あるいは他の研究を行う必要があると決定することもできる。そのほか、臨床前と臨床試験から得られたデータの異なる解釈は候補製品の上場承認を延期、制限或いは阻止する可能性がある。私たちが最終的に得たどのマーケティング承認も限られているかもしれないし、制限されたり、承認された後の約束は、承認された製品が商業的に実行できないようにすることができる。
もし私たちが承認を得る上で遅延に遭遇した場合、あるいは私たちが開発した任意の候補製品が承認されなければ、これらの候補製品のビジネス見通しが損なわれる可能性があり、私たちが収入を作る能力は深刻な損害を受けるだろう。
米国で私たちの候補製品のマーケティング承認や商業化を獲得し、維持することは、他の司法管轄区で私たちの候補製品のマーケティング承認を得ることに成功するという意味ではありません。外国の管轄区域でマーケティングの許可を得ることができなければ、私たちが開発したどの候補製品もこれらの管轄区で販売できなくなり、逆に収入を創出する能力を著しく弱めることになる。
マーケティングと販売のために、EUや他の多くの外国司法管轄区で開発される可能性のある任意の候補製品を販売するためには、私たちまたは私たちの協力者は単独のマーケティング承認を得て、多くの異なる現地法規の要求を遵守しなければならない。承認手続きは国·地域によって異なり、関連する可能性がある
119
追加的なテスト。承認を得るのに要する時間は、FDA承認を得る時間とは大きく異なる可能性がある。米国以外の規制承認手続きには、通常、FDA承認の取得に関するすべてのリスクが含まれる。また、米国以外の多くの国では、製品がその国での販売を許可される前に、精算承認を受けなければならないことが求められている。私たちやこれらの第三者は(あれば)米国以外の規制機関の承認をタイムリーに得ることができないかもしれない。FDAの承認は、他国又は管轄区域の規制機関の承認を確保するものではなく、米国以外の1つの規制機関の承認も、他の国又は司法管区の規制機関又はFDAの承認を確保することができない。私たちはマーケティング承認を申請できないかもしれませんし、どの司法管轄区でも私たちの候補製品を商業化するために必要な承認を得ることができないかもしれません。これは私たちの収益能力を著しく弱化させます。
2016年6月23日、イギリスの有権者はEU離脱、いわゆる離脱に賛成票を投じた。長引く交渉を経て、イギリスは2020年1月31日にEUを離脱し、EUルールと法規は2021年1月1日からイギリスに適用されなくなった。医薬品および保健製品規制機関、またはMHRAは、現在イギリス(北アイルランドを除く)の医薬品マーケティング許可の唯一の決定者である。MHRAは“2012年ヒト薬品条例”(SI 2012/1916)(改正された)、あるいはHMRに依存し、薬品管理の基礎となる。HMRは連合王国がEUから離脱する前にあらかじめ存在していた医薬製品に関するEUの法律主体を連合王国の国内法に組み入れている。
イギリスの薬品に対する監督管理の枠組みは薬品の品質、安全と治療効果、臨床試験、マーケティング許可、商業販売と流通をカバーしているため、EUからの指令と法規、イギリスの離脱の結果と未来にイギリス製品と候補製品の承認に適用される監督管理制度の影響はまだ不明である。イギリスの離脱やその他の理由により、いかなるマーケティング承認を得ることができないかに関するいかなる遅延も、イギリスで私たちの候補製品のために規制承認を求める努力を制限または延期させる可能性があり、これは私たちの業務に重大で実質的な損害を与える可能性がある。イギリスの離脱により、計画中のEMAマーケティング許可申請に加えて、イギリスでのマーケティング承認を得るために、MHRAに単独の申請を提出する必要があると予想されています。
私たちは私たちの候補製品を商業化する際に、追加のリスクに直面すると予想しています
関税、貿易障壁、規制要件を含む米国以外の市場での承認
インフレ、特に外国経済と市場の政治的不安定を含む経済的疲弊
外国に住んでいるあるいは旅行する従業員は税金、就職、移民、労働法を遵守する;外国人
為替レートの変動、これは運営費用の増加と収入の減少、その他を招く可能性があります
別の国で商売をすることの付帯義務;労働力不足国における労働力の不確実性
騒ぎはアメリカよりもっと一般的だ。
私たちは私たちの候補製品のためにいくつかの指定を求めるかもしれません。アメリカの迅速なチャネル、突破的な治療、再生医学の高度な治療と優先審査指定、イギリスの革新的な許可と訪問経路の指定、そしてEUのPrime指定を含むかもしれませんが、私たちはこのような指定を受けないかもしれません。私たちが受け取っても、このような指定はより速い発展や規制審査や承認過程をもたらすことはありません。
候補製品が深刻または生命に危険な疾患の治療に使用され、候補製品がこのような疾患が満たされていない医療需要を解決する潜在力を示す場合、スポンサーはFDAに迅速チャネル指定を申請することができる。Fast Track製品については,スポンサーがFDAとより多くのインタラクションを行う可能性があり,FDAは申請完了前にFast Track製品申請部分の審査を開始する可能性がある。FDAがスポンサーから提出された臨床データを初歩的に評価した後にFast Track製品が有効である可能性があると判断すれば,スクロール審査が可能である。
さらに、出願人は、その製品を突破的療法として指定することを求めることができ、これは、1つまたは複数の他の薬剤と単独で、または1つまたは複数の他の薬剤と組み合わせて、重篤または生命に危険な疾患または状態を治療することを意図した薬剤であり、初期臨床証拠は、1つまたは複数の臨床的重要終点において、臨床開発早期に観察される重大な治療効果のような既存の療法よりも実質的な改善を示す可能性があることを示す。画期的な治療法として指定された薬物や生物製品に対して,FDAと試験スポンサーとの相互作用とコミュニケーションは,臨床開発の最も有効な方法の決定を助けることができ,同時に無効なコントロールレジメン中の患者数を最小限に抑えることができる。
120
さらに、製品が深刻または生命に危険な疾患または状態を治療、修正、逆転または治癒することを意図し、予備臨床証拠が、このような疾患または状態が満たされていない医療需要を解決する潜在力を有することを示す場合、製品は再生医学高度療法またはRMAT認証を受ける資格がある。RMAT指定の利点は、開発および審査、潜在的優先審査資格、および代替または中間終点に基づく加速承認を含むFDAとの早期相互作用を含む画期的な治療指定と同様である。
さらに、FDAが候補製品が治療において大きな進展を遂げたと決定した場合、または適切な治療方法が存在しない治療方法を提供する場合、FDAは、候補製品を優先的に検討するように指定する可能性がある。顕著な改善は,疾患治療の有効性の増加,治療を制限する産物反応の除去あるいは大幅な減少,記録されている患者のコンプライアンスの向上が重篤な結果の改善をもたらす可能性があること,新たな亜群における安全性と有効性の証拠であることが示唆された。優先審査指定は、FDA審査申請の目標が6ヶ月であり、標準的な10ヶ月の審査期間ではないことを意味する。
私たちは私たちの候補製品のためにこれらと他の称号を求めるかもしれない。FDAは候補製品などの称号を付与するか否かについて広範な裁量権を有しているため,特定の候補製品がこのような称号や地位を得る資格があると考えても,FDAは付与しないことを決定する可能性がある。また、従来のFDAプログラムと比較して、高速チャネル、突破的療法或いはRMAT指定は必ずしもより速い規制審査過程を意味するわけではなく、必ずしもいかなる承認面の優位性をもたらすとも限らない。したがって、私たちは私たちの候補製品のためにこれらの指定を求めて獲得するかもしれませんが、従来のFDAプログラムに比べて、より速い開発過程、審査、または承認を経験しないかもしれません。また,FDAがわれわれの臨床開発計画のデータがこれらの指定を支持しなくなったと考えた場合,これらの指定を撤回する可能性がある。
連合で、私たちは未来に私たちのいくつかの候補製品のための良質な称号を求めるかもしれない。PRIMEはEMAの役割を強化し、科学と監督管理支持を強化し、開発を最適化し、未満足の医療需要を解決する潜在力を有する重大な公衆衛生利益の新薬の評価を加速することを目的とした自発的計画である。この計画の重点は,EUに満足できる治療法がない疾患に対する薬物,あるいはそのような方法が存在しても,既存の治療法よりも大きな治療優位性を提供することである。Primeは開発中で欧州連合で許可を得ていない薬品に限られており,出願人は集中手順で予備上場許可申請を申請しようとしている。Primeとして受け入れられるためには、候補製品はその主要な公共健康利益と治療革新方面の資格基準に適合しなければならず、この基準は声明を実証できる情報に基づいている。Prime指定の利点は、マーケティング許可申請の前に継続的な支援を提供し、知識の蓄積を支援するCHMP調査委員を任命することと、重要な開発マイルストーンで早期対話および科学的提案を行うことと、製品の加速的な審査を行う可能性があることとを含み、これは、申請中に承認の程度に関する意見をより早く発表するために、審査時間を減少させることを意味する。Primeはまた申請者が同時にEMA科学提案と衛生技術評価提案を要請し、適時な市場進出を促進することを奨励した。いずれの候補製品の良質な認証を得ても,従来のEMAプログラムと比較して,この認証は実質的に速い開発過程,審査または承認をもたらさない可能性がある。さらに進む, 良質な称号を得ることは、EMAがマーケティング許可を付与する可能性を保証したり増加させたりしない。
私たちはまた、150日間の評価、審査手順のスクロール、および革新的な許可および取得経路、またはILAPのような患者に利益をもたらす新薬を優先的に得るために、イギリスの退欧後のMHRAのいくつかの手順に従うかもしれない。ILAPは発売時間を加速し、患者が新しい化学実体、生物薬物、新しい適応と用途を再調整する薬物を含む薬物の獲得を便利にすることを目的としている。我々は2023年2月14日にMHRAから革新的なパスポートを取得し,これがILAP進出の起点である。ILAPから受益した製品開発者は臨床試験設計方面の提案を獲得し、監督管理の許可と衛生技術評価に最適なデータ生成を提供することを確保する。
我々が開発する可能性のある任意の候補製品の孤立薬物排他性を得ることができない可能性があり,たとえそうしても,この排他性はFDAやEMAが他の競合製品を承認することを阻止しない可能性がある。
孤児医薬品法によれば、1つの製品がまれな疾患または疾患を治療するための医薬または生物学的製剤である場合、FDAはその製品を孤児薬として指定することができる。EU EMAは孤児製品の承認に対しても似たような規制制度を持っている。一般的に、孤児薬物指定を有する候補製品がその後、このような指定された適応を有する最初の発売許可を得た場合、その製品は市場排他期を有する権利があり、これはFDAまたはEMAが別の発売を許可することを阻止するであろう
121
この時間帯に同一治療適応のために同一製品を申請する。適用期間はアメリカでは7年、EUでは10年だ。1つの製品が指定された孤児薬の基準をもはや満たしていない場合、特にその製品の利益が十分に高く、市場排他性がもはや合理的でない場合、EUの排他的期限は6年に短縮されることができる。
FDAが私たちの製品が孤児薬物の独占経営権を獲得することを承認するために、この機関は、米国の年間患者数が20万人未満の疾患または疾患の治療に指定されていることを発見しなければならない。FDAは孤児薬物の排他的な条件や疾患がこの基準を満たしていないと結論するかもしれない。私たちが製品の孤児薬物排他性を獲得しても、この排他性は、異なる製品が同じ条件で承認されることができるので、その製品を競争から効果的に保護することができない可能性がある。特に,遺伝子療法の背景には,孤児薬物の排他的な目的の下で,何が“同一薬物”を構成するかという概念が変化しており,FDAは最近,遺伝子組換えやベクターのわずかな違いだけで両遺伝薬物製品が異なる薬物であるとは考えられないという最終指導意見を発表した。さらに、孤児薬が承認された後であっても、FDAが、より安全で、より効果的であることが証明されているので、後の製品が臨床的に優れていると結論した場合、FDAはその後、同じ疾患に対して同じ製品を承認することができる。FDAまたはEMAが、指定された要求に重大な欠陥があると判断した場合、または製造業者が稀な疾患または疾患を有する患者の需要を満たすのに十分な数の製品を保証できない場合、孤児薬の排他性を失う可能性もある。
2017年、国会はFDA 2017年再認可法案、FDARAと略称した。その他の事項以外に、FDARAはFDAが以前に存在した監督管理解釈を編纂し、薬品スポンサーに1種の孤児薬物の臨床優位性を証明することを要求し、この薬物は他の方面で以前許可された同じ稀な疾病を治療する薬物と同じであって、孤児薬物の排他性を獲得することができる。トランプ総裁が2020年12月27日に署名した総合的な立法によると、製品の臨床的優位性を示す要求は、2017年FDARA公布前に孤児薬物指定を受けたが、FDAの許可や許可を得ていない薬物や生物製品に適用される。
FDAと議会は“孤児薬物法案”とその法規と政策をさらに再評価するかもしれない。第11巡回控訴裁判所の2021年9月の判決が特にそうである可能性があることを考慮すると、この判決は、排他的範囲を決定するために、用語“同じ疾患または状態”は、指定された“まれな疾患または状態”を意味し、FDAは、それを“適応または使用”を意味すると解釈することはできないと考えている。したがって,裁判所は孤児薬物は“適応や使用”ではなく,指定された疾患や状況全体に排他的に適用されると結論した。この決定を覆すための立法提案があったが、それらはまだ法律になっていない。FDAは2023年1月23日、裁判所の命令範囲を超える事項において、FDAは、孤児薬の独占性と孤児薬の使用または適応のために許可された使用または適応とを束ねて、その既存の法規を適用し続けると発表した。FDAや国会がその孤児の薬物法規や政策を変更する可能性があることによって、私たちの業務は悪影響を受ける可能性がある。
遺伝子編集に対する否定的な世論、及び遺伝子編集と遺伝子研究の強化に対する監督審査は、国民が私たちの未来の製品候補者の見方に不利な影響を与える可能性がある。
私たちの潜在的な治療製品は遺伝物質を患者細胞に導入することを含む。著者らの潜在製品の臨床と商業成功はある程度公衆の遺伝子編集と遺伝子監督管理による人類疾病の予防或いは治療に対する受け入れ程度に依存する。公衆の態度はこのような言い方の影響を受ける可能性があり、即ち遺伝子編集と遺伝子規制は安全ではなく、不道徳或いは不道徳であるため、私たちの製品は公衆或いは医学界の受け入れを得ることができないかもしれない。公衆の悪い態度は私たちが臨床試験を募集する能力に悪影響を及ぼすかもしれない。さらに、私たちの成功は、医師が処方した処方および彼らの患者が受けることを望む治療に依存し、これらの治療は、彼らがよく知っている既存の治療方法を代替または補充するために、私たちが開発可能な候補製品を使用することに関連し、より多くの臨床データを得ることができる。
より厳格な政府法規または負の世論は、私たちの業務や財務状況に負の影響を与え、候補製品の開発および商業化を遅延または損害するか、または承認されると、任意の製品の需要を損なう可能性がある。例えば,2003年には,宿主細胞のDNAに結合して宿主細胞のDNAを変化させる早期バージョンのマウスガンマレトロウイルスを用いた試験が,報告された白血病症例を含めていくつかのよく知られている有害事象を招いた。私たちの臨床試験における有害事象は、最終的に私たちの候補製品によるものでなくても、それによって生じる宣伝は、政府の規制増加、不利な公衆認識、潜在的な規制遅延、私たちの候補製品のテストまたは承認、承認された候補製品に対するより厳しいラベル要求、およびそのような候補製品に対する需要の減少をもたらす可能性がある。癌のリスクは遺伝子編集の問題です
122
私たちが計画したり未来のどんな臨床試験でもこのようなことが起こらないことを確実にする。このような有害事象が発生した場合、私たちの候補製品の商業化や臨床試験のさらなる推進は停止または延期される可能性があり、これは私たちの業務と運営に負の影響を与えるだろう。
私たちまたは私たちが所有する可能性のある任意のパートナーが私たちが開発した任意の候補製品のためにマーケティング承認を得ても、承認条項と私たちの製品の持続的な規制には大量の資源支出が必要であり、私たちまたは彼らが私たちの製品を製造してマーケティングする方法を制限するかもしれません。これは私たちの収益能力を深刻に弱めるかもしれません。
私たちが発売許可を得た任意の候補製品、及びその製品の製造プロセス、承認後の臨床データ、ラベル、広告と販売促進活動は、FDAと他の監督管理機関の持続的な要求と審査を受ける。これらの要求は、安全および他の上場後の情報および報告の提出、登録および上場要求、品質管理、品質保証および対応する記録およびファイル維持に関連するcGMP要求、および医師へのサンプルの配布と記録の保存に関する要求を含む。たとえば,承認されたBLAの所有者は,不良イベントおよび製品がBLAの規格に適合していない任意の障害を監視および報告する義務がある.FDAは通常、遺伝子薬物治療を受けた患者に15年間にわたる潜在的有害事象の追跡観察を受けることを提案している。所持者
承認されたBLAはまた、いくつかの変更を行うために、新しいまたは追加の出願を提出し、FDAの承認を得なければならない
承認された製品、製品ラベル、または製造プロセス。候補製品の上場が承認されても、承認は、製品が発売される可能性のある指定用途の制限または承認条件によって制限される可能性があり、または製品の安全性または有効性を監視するために、高価な上場後のテストおよび監視の要求を含む可能性がある。
したがって、私たちまたは私たちが所有する可能性のある任意の協力者が私たちが開発した1つまたは複数の候補製品のマーケティング承認を得たと仮定すると、私たちとそのような協力者と私たちと彼らの契約製造業者は、製造、生産、製品監視、品質管理を含むすべてのコンプライアンス分野で時間、お金、エネルギーをかけ続けます。もし私たちとそのような協力者が承認後の規制要求を遵守できなければ、私たちとそのような協力者は規制機関によって私たちの製品のマーケティング承認を撤回される可能性があり、私たちまたはそのような協力者が未来の製品をマーケティングする能力が制限される可能性があり、これは私たちが利益を達成したり維持したりする能力に悪影響を及ぼすかもしれない。また、承認後の法規を遵守するコストは、私たちの業務、経営業績、財務状況、見通しにマイナス影響を与える可能性があります。
私たちが発売許可を得たどの候補製品も市場から制限されたり撤退したりする可能性があります。もし私たちが規制要求を守っていない場合、あるいは私たちの製品が予期せぬ問題に遭遇した場合、その中のどの製品も承認された場合、私たちは重大な処罰を受けるかもしれません。
FDAおよび他の規制機関は、承認されたラベルの規定に基づいて販売されることを確実にするために、医薬品の承認後の販売および販売促進を密接に規制する。医師が彼らの専門医療判断において製品のラベルに記載されていない用途、いわゆるラベル外用途を開く可能性があるにもかかわらず、FDAおよび他の規制機関は、ラベル外使用に関するメーカーのコミュニケーションに厳格な制限を加え、承認されたラベルと一致しない方法で私たちの製品を販売すれば、FDAおよび他の州法執行機関(司法省または司法省を含む)のラベル外マーケティング法執行行動の影響を受ける可能性がある。“虚偽請求法”を含む“連邦食品、製品および化粧品法”および処方製品販売促進および広告に関する他の法規に違反し、連邦および州医療詐欺および法律乱用および州消費者保護法違反を調査または告発する可能性もある。2021年9月、FDAは、薬物または生物の予期される用途を決定する際にFDAが考慮すべき証拠タイプを記載する最終規定を公表した。
さらに、私たちの候補製品、製造業者、または製造プロセスには、以前に未知の問題が存在したり、法規の要求を遵守できなかったりして、様々な結果が生じる可能性があることが分かった
123
政府は違法の疑いのあるいかなる調査にも対応するために多くの時間と資源を必要とする可能性があり、否定的な宣伝が生じる可能性がある。上記のいずれかのイベントまたは処罰が発生すると、私たちが開発した任意の候補製品を商業化する能力を抑制し、当社の業務、財務状況、運営結果、および将来性に悪影響を及ぼす可能性があります。
米国食品医薬品局、米国証券取引委員会および他の政府機関の資金不足は、政府の閉店やこれらの機関の運営の他の中断を含め、重要な指導部や他の人員の能力を雇用·保留し、新製品やサービスのタイムリーな開発や商業化を阻止するか、あるいはこれらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止することを阻害する可能性があり、これは私たちの業務に負の影響を与える可能性がある。
FDAの新製品の審査と承認能力は、政府予算と資金レベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、及び法定、監督と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。FDAや他の機関の中断も、新製品候補製品が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。また,政府は米国証券取引委員会(Securities and Exchange Commission,米国証券取引委員会と略す)や我々の業務が依存する可能性のある他の政府機関に資金を提供し,研究開発活動に資金を提供する機関を含め,政治過程の影響を受けるが,政治過程自体が不安定で予測不可能である.
FDAや他の機関の中断も、新製品候補製品が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、ここ数年間、米国政府は何度か閉鎖されており、食品·医薬品局や米国証券取引委員会などのいくつかの規制機関は、肝心な従業員を休暇にし、キー活動を停止させなければならない。政府が長期的に停止すれば、FDAが私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。また、将来的に政府の閉店は、適切な資本化と事業継続のために、私たちが公開市場に進出し、必要な資本を得る能力に影響を与える可能性がある。
また、新冠肺炎疫病に対応するため、一部の会社は2021年に完全な返信を受けたことを発表し、原因は食品と薬物管理局がそのアプリケーションの必要な検査を完成できないからである。オミック変異による一定期間の起動ミスや一時停止を経験した後、FDAは2022年2月に国内検査を再開し、2022年4月から海外検査を優先すると表明した。しかし、FDAは現在の速度を継続できない可能性があり、審査期限は延長される可能性があり、承認前検査や臨床部位の検査や、持続的な新冠肺炎の流行や旅行制限により、FDAは審査期間中にこのような要求された検査を完了することができない。新冠肺炎疫病に対して、アメリカ以外の監督管理機関は類似の制限或いは他の政策措置をとる可能性があり、監督活動の遅延に遭遇する可能性がある。政府が長期的に停止したり、他の中断が発生したりすれば、FDAが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。今後の停止や他の中断は、米国証券取引委員会のような他の政府機関にも影響を与える可能性があり、これは、私たちの公開届出文書の審査(必要があれば)を延期し、私たちが公開市場に入る能力によって、私たちの業務に影響を与える可能性もあります。
124
顧客、医療提供者、専門家、第三者支払者などとのいかなる関係も、適用されるリベート、詐欺、乱用、および他の医療法律法規の制約を受けることになり、刑事制裁、民事処罰、契約損害、名声損害、罰金、返還、政府医療計画から除外され、私たちの業務を削減または制限し、利益と将来の収益の減少を含む罰に直面する可能性があります。
医療提供者、医師、第三者支払者は、市場の承認を得ることができる任意の製品の推薦と処方において主な役割を果たすだろう。私たちが医療保健提供者、第三者支払者、顧客と達成したどんな計画も、広く適用される詐欺と乱用、他の医療法律法規の影響を受けるだろう。法律と法規は、私たちが市場の承認を得た任意の製品の業務または財務配置と関係を制限することを制限するかもしれません。これらの措置には
125
私たちが第三者と達成したどんな業務計画も、私たちの業務が全体的に適用される医療法律や法規に適合していることを確保するために努力しており、多くのコストがかかります。政府当局は、私たちの業務実践が現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関連する現行または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちの運営がこれらの法律または任意の他の私たちの政府法規に適用される可能性があることが発見された場合、私たちは重大な民事、刑事と行政処罰、損害賠償、罰金、個人監禁、追加の報告要件および監督に直面する可能性があり、これらの法律を遵守しない、製品を政府援助の医療計画(例えばMedicareおよびMedicaid)から除外すること、返還、契約損害、名声損害、および私たちの業務の削減または再編を解決するために、会社の誠実な合意または同様の合意の制約を受ける可能性がある。このような行動を防御するには費用がかかり、時間がかかる可能性があり、大量の財政と人的資源が必要かもしれない。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。さらに、私たちがそれと業務を展開することを期待している任意の医師または他の医療提供者またはエンティティが、適用された法律に適合していないことが発見された場合、彼らは、政府の援助された医療計画から除外されることを含む刑事、民事または行政制裁を受ける可能性がある。
現在と未来の法律は、私たちと任意の協力者が市場の承認を得て、私たちの候補製品を商業化する難しさとコストを増加させ、私たちまたは彼らが獲得する可能性のある価格に影響を与えるかもしれない。
米国や一部の外国司法管轄地域では、医療システムの立法や法規の変更および提案された変更が複数あり、これらの変更は、私たちの候補製品の上場承認を阻止または遅延させ、承認後の活動を制限または規範化し、私たちまたは任意の協力者が利益的に販売または商業化し、私たちまたは彼らがマーケティング許可を得た任意の候補製品の能力に影響を与える可能性がある。私たちは、現在の法律、および将来取られる可能性のある他の医療改革措置は、より厳しいカバー基準をもたらす可能性があり、私たちまたは任意の協力者が受ける可能性のある任意の承認された製品の価格に追加の下振れ圧力をもたらす可能性があると予想している。
2010年3月、総裁·オバマ氏はPACAに署名し、法律にした。また、PACAが公布されて以来、他の立法改正も提案され、採択された。2011年8月、“2011年予算制御法案”(Budget Control Act Of 2011)などの法案は国会のための支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これらの変化には、各年度に提供者に支払われる連邦医療保険総額が2%に達し、2013年4月に発効し、2031年まで続くことが含まれている。CARE法案とその後の立法により,これらの連邦医療保険の自動減額は2022年6月末に一時停止されたが,その後2022年7月1日に2%の全面削減が再開された。2012年の“米国納税者救済法”は,いくつかの医療サービス提供者に支払う連邦医療保険を減少させ,政府が提供者に多額の金を取り戻す訴訟時効を3年から5年に延長した。これらの新しい法律は、連邦医療保険および他の医療資金のさらなる減少をもたらし、他の方法で、規制によって承認される可能性のある任意の候補製品の価格、またはそのような任意の候補製品の処方または使用頻度に影響を与える可能性がある。
PACAが公布されて以来、この法律の条項を廃止し、代替するために、多くの法的挑戦や国会行動が続くだろう。例えば、税法は“個人強制令”を廃止した。この条項は、ほとんどのアメリカ人が最低レベルの医療保険を購入することを要求する条項が2019年に施行される。また、2018年12月14日、テキサス州北区の米国地域裁判所裁判官は、PPACAの個人権限部分はPACAの基本的かつ不可分な特徴であるため、この許可は税法の一部として廃止され、PPACAの残りの条項も無効であると判断した。しかし,2021年6月17日,米国最高裁はこの事件を却下し,PPACAを維持した。PACAをめぐる訴訟と立法は継続される可能性があり、結果は予測不可能で不確実だ。
トランプ前大統領政府はまた、PPAAの実施を破壊または延期する行政行動をとり、PPACAに基づいて権力と責任を持つ連邦機関にPPACAの実施を放棄、延期、承認免除、または延期するよう指示する任意の条項を含み、これらの条項は、各州、個人、医療保健提供者、医療保険会社または医薬品または医療機器メーカーに財政的または規制的負担をもたらす。しかし2021年1月28日に総裁·バイデンはこれらの命令を撤回し
126
新たな行政命令を発表し,連邦機関に米国人のヘルスケア取得を制限するルールや他の政策を再検討し,このような医療を得る機会を保護·強化するための行動を考えるよう指示した。この行政命令によると、連邦機関は、新冠肺炎に関連する合併症を含む以前の疾患を有する人の保護の政策を弱めること、連邦医療補助および“全米政治行動計画”によるデモおよび免除は、仕事要件を含むカバー範囲または破壊計画を減少させる可能性のある政策、健康保険市場または他の医療保険市場を破壊する政策、連邦医療補助計画および汎米政治行動計画への参加の難しさを増加させる政策、および扶養者への負担能力を含む保険または経済援助の負担能力を低減する政策を含む再検討を指示されるであろう。この行政命令はまた、衛生と公衆サービス部に健康保険市場のために特殊な保険期間を作成し、新冠肺炎の大流行に対応するように指示した。
我々は,これらの医療改革措置,および将来とりうる他の医療改革措置は,連邦医療保険や他の医療資金のさらなる減少,より厳しいカバー基準,新たな支払い方法,および任意の承認された製品のために得られる価格および/または医師が市場に進出する可能性のある任意の承認された製品を管理することによって得られる清算レベルの追加的な下り圧力をもたらす可能性があると予想している。精算レベルの低下は私たちが受け取った価格や私たちの製品の処方や管理頻度に悪影響を及ぼす可能性があります。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。したがって、これらの改革が発効すれば、市場で承認された候補製品の期待収入の開発に成功する可能性があり、我々の全体的な財務状況および候補製品の開発または商業化の能力に影響を与える可能性がある。政府はまた新冠肺炎の流行に対応するためにもっと多くの行動をとる可能性がある。
処方薬の米国や外国の司法管轄区域における価格はかなりの立法や行政行動の影響を受けており、許可を得ると、我々の製品の価格に影響を与える可能性がある。
アメリカでも処方薬の価格はずっと話題になっています。アメリカ議会は最近数回の調査を行い、州と連邦立法を提案し、公布し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険と医療補助下の薬品コストを下げることを目的とした。2020年には、総裁·トランプ氏が処方薬のコスト削減を目的としたいくつかの行政命令を発表し、これらの命令のいくつかが条例に盛り込まれている。これらの規定には、価格最恵国モデルが実施され、連邦医療保険B部分のある医師が管理する薬品に対する支払いを他の経済先進国が支払う最低価格とリンクさせ、2021年1月1日から発効する臨時最終規則が含まれている。しかし、この規定は全国的な予備禁止令によって制限され、2021年12月29日、医療保険·医療補助サービスセンター(CMS)はそれを廃止するための最終規則を発表した。CMSは、この規則の発表に伴い、それは価値をMedicare B部分の薬品支払いのすべての選択に組み入れ、そして受益者が根拠に基づく看護を獲得する機会を改善することを探索する。
また、2020年10月、HHSとFDAは、各州と他のエンティティが804条の輸入計画、すなわちSIPを制定し、いくつかの処方薬をカナダから米国に輸入することを許可する最終規則を発表した。最終規則は現在行われている訴訟のテーマであるが、少なくとも6つの州(バーモント州、コロラド州、フロリダ州、メイン州、ニューメキシコ州、ニューハンプシャー州)がFDAの審査と承認のためにSIPsを開発することを目的としてカナダからの薬物の輸入を許可している。また、2020年11月20日、HHSは薬品メーカーのD部分下でスポンサーの値下げを計画する安全港保護を廃止し、法律が値下げを要求しない限り、直接或いは薬局福祉マネージャーを通過する法規を決定した。この規定はまた、販売時点での値下げを反映するための新しい安全港を創出し、薬局福祉マネージャーと製造業者との間のいくつかの固定費用スケジュールのための新しい安全港を創出し、この計画の実施は、インフレ低減法案によって2032年1月1日に延期された。
最近、2022年8月16日、総裁·バイデンは2022年インフレ削減法案、あるいはアイルランド共和軍と署名した。新しい立法は連邦医療保険D部分に影響を与え、D部分は計画であり、連邦医療保険A部分または連邦医療保険B部分に加入する個人は毎月外来処方薬保険料を支払うことを選択することができる。その他の事項を除いて、アイルランド共和軍はある薬品のメーカーが連邦医療保険と価格交渉を要求し(2026年から)、価格は交渉できるが、上限がある;連邦医療保険B部分と連邦医療保険D部分に基づいてリベートを実施して、インフレを超えた価格上昇を処罰する(初めて2023年に満期になる);そして新しいカバーギャップ割引計画でD部分の代わりにノッチ割引計画をカバーする
127
割引計画(2025年から)。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。
具体的には、価格交渉において、国会はMedicareが、競合する模倣薬または生体模倣薬を有さず、Medicare B部分およびD部分によって精算されるいくつかの高価な単一由来薬剤および生物製品のための低い価格を交渉することができる。CMSは、2026年からMedicare D部分によって支払われる10種類の高コスト薬剤の価格を交渉することができ、その後、2027年の15種類のD部分薬剤、2028年の15種類のB部分またはD部分薬剤、および2029年以降の20種類のB部分またはD部分薬剤の価格を交渉することができる。この規定は、少なくとも9年間承認された医薬品および許可13年の生物製品に適用されるが、単一のまれな疾患または疾患のための許可された医薬および生物製品には適用されない。それにもかかわらず、CMSは価格交渉でこれらの製品の最高価格を決定する可能性があるため、もし私たちの製品がMedicare価格交渉の対象であれば、私たちは完全に政府行動のリスクに直面するだろう。また、存在する可能性のあるリスクを考慮すると、金利協定のこれらの条項は、もし私たちの薬物製品が市場に発売されて9年後に価格を制定しなければ、医薬品の期待リターンや私たちの製品の特許のすべての価値を保護することができないというリスクをさらに増加させる可能性がある。
また、この立法は、製薬業者が提供する価格が法律で規定された交渉で達成された“最高公平価格”以下でない場合、または値上げ幅がインフレを超える場合、製薬業者は民事罰金と潜在的な消費税を受けることになると規定している。この立法はまた、メーカーに連邦医療保険D部分の価格上昇幅がインフレを超えた薬品にリベートを支払うことを要求している。新法律では,2024年の連邦医療保険の自己払い薬品コスト上限は年間4000ドルと規定されており,その後2025年から年間2000ドルが上限となっている。
州レベルでは、各州はますます積極的に、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法と実施し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。さらに、医療機関および個別病院は、どの薬品およびサプライヤーが彼らの処方薬および他の医療計画に含まれるかを決定するために入札プログラムを使用するようになってきている。これらの措置が承認されると、私たちの製品に対する最終的な需要を下げたり、私たちの製品の価格設定に圧力をかけるかもしれません。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
EUでは、承認されれば、同様の政治、経済、規制発展が候補製品を利益的に商業化する能力に影響を与える可能性がある。米国やEU以外の市場では,精算や医療保健支払い制度は国によって異なり,多くの国で特定製品や療法に価格上限が設定されている。一部の国、特に欧州連合国では、処方薬の価格設定は政府によって統制されている。これらの国では、製品の発売許可を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。一部の国で精算或いは定価の承認を得るためには、私たちの候補製品のコスト効果を他の利用可能な治療法と比較する臨床試験を行う必要があるかもしれない。もし私たちの製品が精算を受けられない場合、あるいは範囲や金額が制限されている場合、あるいは定価が満足できないレベルに設定されている場合、私たちの業務は損害を受ける可能性があり、実質的かもしれません。
グローバルなプライバシーやデータセキュリティ要件を遵守することは、追加的なコストと責任をもたらしたり、グローバルでデータを収集して処理する能力を抑制したりする可能性があり、これらの要求を守らないことは、私たちを巨額の罰金と処罰に直面させる可能性があり、これは、私たちの業務、財務状況、または運営結果に大きな悪影響を及ぼす可能性があります。
私たちは、個人情報の収集、送信、記憶、および使用に適したデータプライバシーおよび法律法規の保護の制約を受けており、その中には、米国、EU、英国の包括的な規制制度を含む、個人情報のプライバシー、セキュリティ、および伝送に何らかの要求を加えることが含まれている。プライバシーとデータ保護の立法と規制構造は世界各地の司法管轄区域で発展し続けており、プライバシーやデータ保護問題にますます注目されており、これは私たちの業務に影響を与える可能性がある。このような法律及び法規を遵守できなければ、罰金、影響を受けた者が損害賠償を要求し、当社の名声及び名誉損失を損害することを含む当社に対して法執行行動をとる可能性があり、上記のいずれの事項も当社の業務、財務状況、運営結果又は将来性に重大な悪影響を及ぼす可能性がある。
128
アメリカには多くの連邦と州の法律法規が個人情報のプライバシーと安全に関連している。特に、HIPAAが公布した条例に基づいてプライバシーおよびセキュリティ基準が規定され、個別に識別可能な健康情報または保護された健康情報の使用および開示が制限され、保護された健康情報のプライバシーを保護し、保護された電子健康情報の機密性、完全性および可用性を確保するための行政、物質および技術保障措置の実施が要求される。保護された健康情報が適用されたプライバシー基準や我々の契約義務に従って処理されているかどうかを決定することは複雑である可能性があり,変化する解釈の影響を受ける可能性がある.このような義務は私たちの現在または未来の業務活動の一部または全部に適用されるかもしれない。
もし私たちが保護された健康情報のプライバシーと安全を適切に保護できなければ、私たちは私たちの業務パートナーとのいくつかの契約に違反していることが発見されるかもしれない。また、適用されるHIPAAプライバシーやセキュリティ基準を含めて適用されるプライバシー法に従わなければ、民事や刑事罰に直面する可能性があります。HHS法執行活動は財務的責任と名声の損害を招く可能性があり、このような法執行活動に対する応答は大量の内部資源を消費する可能性がある。また、州総検察長は民事訴訟を起こし、禁止令や損害賠償を求め、州住民のプライバシーを脅かす侵害行為に応える権利がある。私たちはこのような規定が私たちの業務にどのように解釈、実行されるかを決定することができない。法執行活動や潜在的な契約責任に関連するリスクに加えて、私たちは連邦や州レベルで変化する法律と法規を遵守するために努力している可能性が高く、私たちの政策、手続き、システムを絶えず修正する必要があります。
2018年、カリフォルニア州は“カリフォルニア消費者プライバシー法”(California Consumer Privacy Act)を採択し、CCPAと略称し、2020年1月1日に施行され、カリフォルニア住民の個人情報を処理する企業に多くの要求を提出した。CCPAの多くの要件は、収集された彼らに関する情報およびそのような情報の使用および共有方法に関する通知をデータ主体に提供することを企業に要求することと、データ主体にそのような個人情報へのアクセスを要求する権利と、場合によってはそのような個人情報の削除を要求することとを含む、“一般データ保護条例”(GDPR)における要件と同様である。CCPAはまた,カリフォルニア住民にその個人情報を販売しないことを選択する権利を与えている。CCPAにはその要求に違反した会社に対する重大な処罰が含まれている。2020年11月、カリフォルニア州有権者は、2023年1月1日に施行されるカリフォルニアプライバシー権法案(CPRA)を採択し、CCPAを大幅に拡大し、カリフォルニア州住民に個人情報の使用、保持、共有を要求することが合理的に必要であり、収集または処理の目的に応じて、敏感な個人情報の追加保護を提供し、住民への保留情報の通知に関する内容をより多く開示することを含むGDPRのような追加条項を盛り込んだ。CPRAはまた新しい法執行機関であるカリフォルニアプライバシー保護局を設立した-その唯一の責任はCPRAを実行することであり、これはコンプライアンスリスクをさらに増加させるだろう。CPRAの規定は私たちのいくつかの商業活動に適用されるかもしれない。また,バージニア州,コロラド州,ユタ州,コネチカット州を含む他の州では州プライバシー法が成立している。バージニア州のプライバシー法も2023年1月1日に施行されました, 他の三つの統計の法律は今年遅く施行されるだろう。他の州は今後このような法律を考慮するだろうし、国会も連邦プライバシー法が採択されるかどうかを議論してきた。これらの法律は、私たちの研究対象の決定、ビジネスパートナーとの関係、そして最終的に私たちの製品のマーケティングと流通を含む、私たちのビジネス活動に影響を与えるかもしれません。
米国の法律と同様に、ヨーロッパや他の国にも重要なプライバシーやデータセキュリティ法が適用されている。欧州経済地域(EEA)に位置する個人の個人データ(個人健康データを含む)の収集、使用、開示、転送または他の処理、およびEEAで行われる個人データの処理については、2018年5月に発効し、当業界で運営する会社に個人データの処理や国境を越えたこのようなデータの処理に義務を負うことを要求するGDPRによって規制される。GDPRは重い問責義務を規定し、データ管理者と処理者にそのデータ処理と政策の記録を保存することを要求した。私たちまたは私たちのパートナーまたはサービスプロバイダのプライバシーまたはデータセキュリティ対策がGDPR要件を遵守できない場合、私たちは、個人データを使用する方法および/または2000万ユーロまたは前期の世界の年商4%までの罰金、および影響を受けた個人の賠償要求、負の宣伝、名声被害、および潜在的な商業および名誉損失を変更することを要求する訴訟、監督管理調査、法執行通知に直面する可能性があります。
GDPRは、個人データをEUから欧州委員会に国境を越えて移行させ、十分なデータ保護立法を提供していないと考えている国(例えば米国)に制限を加えている。会社が個人データをEUから他国に移す能力について、人々は懸念してきた。2020年7月、欧州連合裁判所(略称CJEU)は欧州連合-米国プライバシー盾(EU-U.S.Privacy Shield)の無効を宣言し、欧州経済圏の個人データ移転を合法化するためのメカニズムの一つである
129
アメリカへ行きます。CJEUの決定は、もう一つのデータ伝送手段である標準契約条項である欧州経済圏から米国への個人データの長期的な可能性が疑問視される。EU−米国プライバシー保護法による自己認証は行われていないが,CJEUのこの決定は,EEAから米国へのデータ転送をより厳密に審査し,データプライバシー法規を遵守するコストや,サプライヤーやビジネスパートナーと適切なプライバシー·セキュリティ協定を交渉するコストを増加させる可能性がある。
また、2022年10月、総裁·バイデンはEU-米国プライバシーの盾に代わるEU-米国データプライバシーの枠組みを実施する行政命令に署名した。欧州委員会は2022年12月にEU-米国データプライバシーの枠組みの十分性決定を採択するためにこのプロセスを開始した。この枠組みがいつ最終的に決定されるか、法廷で挑戦されるかどうかはまだ分からない。この問題をめぐる不確実性はEUでの私たちの業務運営にさらに影響を及ぼすかもしれない。
連合王国のEU離脱後、連合王国の“2018年データ保護法”は、連合王国で行われた個人データ処理に適用され、GDPRが定めた義務と平行する義務が含まれている。データ転送については,連合王国と欧州連合ともに単独の“十分性”によって決定され,2つの司法管轄区域間のデータ転送がそれぞれイギリスの“データ保護法”とGDPRに適合することが決定された.このような十分な決定のどんな変化や更新も私たちの業務に影響を及ぼす可能性がある。
GDPRに加えて,世界ではプライバシーやデータセキュリティ法が制定されている国が増えている.多くの法律はGDPRに倣って手本としているが、他の法律は異なるまたは互いに衝突する条項を含む。これらの法律は、コンプライアンスコスト、契約に関連するコスト、および潜在的な法執行行動を増加させることによって、私たちの臨床試験および商業製品の販売および流通を含む業務活動を展開する能力に影響を与えるだろう。
最近のデータプライバシー規制の変化の影響を扱い続けているが,新法規の発効と継続的な法的挑戦に伴い,データプライバシーは国内レベルでも国際的にも変化しており,変化するデータ保護ルールを遵守する努力は成功しない可能性がある.このような法律の解釈と適用は私たちの接近と一致しない可能性がある。私たちはこの変化する状況を理解して順応するために多くの資源を投入しなければならない。データ保護に関する法律を守らないことは、欧州経済区や他の地方データ保護当局が法執行行動をとるリスクに直面し、法律違反が発見されれば、重大な処罰を受ける可能性がある。同様に,米国連邦や州の個人情報のプライバシーやセキュリティに関する法律を守らなければ,このような法律の罰を受ける可能性がある.このようなデータ保護およびプライバシー法を遵守しない行為は、政府に罰金や命令を科す可能性があり、私たちのやり方、クレームまたは他の責任、規制調査および法執行行動、訴訟、および巨額の救済費用を変更することを要求し、いずれも私たちの業務に悪影響を及ぼす可能性がある。私たちがこれらの法律に違反していると判断されなくても、政府のこれらの問題の調査には通常、大量の資源がかかり、否定的な宣伝が必要であり、これは私たちの名声と私たちの業務、財務状況、運営結果、または将来性を損なう可能性がある。
私たちの従業員、主要な調査者、コンサルタント、ビジネスパートナーは、規制基準と要求を遵守しないこと、およびインサイダー取引を含む不正行為または他の不正活動に従事する可能性があります。
私たちは従業員、サプライヤー、コンサルタント、パートナーの詐欺や他の不正行為のリスクに直面しており、私たちの臨床試験では、私たちの主要な研究者やCROも詐欺や他の不正行為のリスクに直面している。これらの当事者の不正行為は、FDA法規またはEUおよび他の司法管轄区に適用される法規を故意に遵守しないこと、FDA、欧州委員会、および他の規制機関に正確な情報を提供すること、米国および海外の医療詐欺および法律法規を乱用し、財務情報またはデータを正確に報告すること、または不正な活動を開示することを含む可能性がある。特に、医療業界の販売、マーケティング、商業配置は、詐欺、不正行為、リベート、自己取引、その他の乱用行為を防止するための広範な法律法規によって制約されている。これらの法律法規は、広範な定価、割引、マーケティングと販売促進、販売手数料、顧客激励計画などの業務手配を制限または禁止している。このような不正行為はまた、臨床試験過程中あるいはFDA或いは他の監督機関との相互作用過程で得られた情報の不適切な使用に関連する可能性があり、これは規制制裁を招き、著者らの名声に深刻な損害を与える可能性がある。私たちはすべての従業員に適用される行動基準を通過しましたが、常に従業員の不適切な行為を識別し、阻止できるわけではありません。私たちがこのような行為を発見し、防止するための予防措置は、未知または未管理のリスクや損失を効果的にコントロールできないかもしれません。あるいは政府の調査や他の行動から私たちを保護することができません
130
このような法律や法規を守らないことによる訴訟。もし私たちにこのような行動を取って、私たちが自分の権利を弁護したり、維持することに成功しなかったら、これらの行動は、巨額の罰金や他の制裁を加えることを含む、私たちの業務、財務状況、運営結果、将来性に大きな影響を及ぼすかもしれない。
私たちが将来所有する可能性のある任意の国際業務を管理する法律法規は、米国以外でいくつかの候補製品を開発、製造、販売することを阻止し、コストの高いコンプライアンス計画の開発と実施を要求するかもしれない。
私たちが事業を展開しているアメリカ以外のすべての司法管轄区域では、私たちは多くの法律と法規の制約を受けている。国際ビジネス慣行コンプライアンス計画の作成、実施、維持のコストが高く、特に第三者に依存する必要がある場合には、このような計画の実行は困難である。
“反海外腐敗法”(FCPA)は、個人または企業が業務を獲得または保持するのを助けるために、任意の米国人または企業が、任意の外国人官僚、政党または候補者に直接的または間接的に支払い、提供、許可支払い、または任意の価値のあるものを提供することを禁止する。“海外腐敗防止法”はまた、米国に上場する証券会社にある会計条項を遵守することを要求し、これらの条項は、会社(国際子会社を含む)のすべての取引の帳簿と記録を正確かつ公平に反映し、国際業務のために適切な内部会計制御システムを設計し、維持することを要求する。“海外腐敗防止法”の反賄賂条項は主に米司法省によって執行されている。米国証券取引委員会は“反海外腐敗法”における帳簿と記録条項の執行に関与している。
“海外腐敗防止法”や我々の業務に適用される可能性のある他の反腐敗法律を遵守することは高価で困難であり、特に腐敗が公認問題である国である。また、製薬業では、多くの国で病院が政府によって運営されているため、医師や他の病院従業員が外国人官僚とされているため、“海外腐敗防止法”や他の反腐敗法の遵守が特別な挑戦をもたらしている。臨床試験やその他の仕事に関連して病院に支払われた何らかの金は、政府関係者に支払われた不正金と考えられ、“海外腐敗防止法”の法執行行動につながった。
私たちはまた、適用される輸出規制法律、国と個人への経済制裁、税関要求を含む、私たちの国際業務を管理する他の法律と法規に制限されている。さらに、様々な法律、法規、および行政命令は、米国国外での使用および伝播を制限するか、または国家セキュリティ目的のために秘密にされた情報、および特定の製品およびこれらの製品に関連する技術データを特定の非米国国民と共有することも制限される。私たちのアメリカ以外の拡張は、これらの法律を遵守するためにより多くの資源を投入することを要求し続けています。これらの法律は、アメリカ以外の場所で特定の薬剤や候補薬の開発、製造、販売を阻止するかもしれません。これは、私たちの成長潜在力を制限し、私たちの開発コストを増加させるかもしれません。
私たちが“海外腐敗防止法”や他の適用される反腐敗、輸出、制裁、税関法律を遵守することを完全に効果的に確保することは保証されない。国際ビジネス慣行を管理する法律を遵守しない場合、政府契約の資格停止または取り消しを含む重大な処罰を受ける可能性がある。これらの法律に違反し、“海外腐敗防止法”を含め、重大な民事·刑事罰を招く可能性がある。“反海外腐敗法”による訴訟だけで、未解決のクレームが解決されるまで、米国政府とのビジネスを一時停止する権利を招く可能性がある。海外腐敗防止法違反の有罪判決は、政府請負業者の資格を長期的に廃止する可能性がある。私たちは国際ビジネス慣行法に基づいて私たちのいかなる義務も履行できなかったため、政府契約や関係の終了を招き、私たちの運営にマイナス影響を与え、私たちの名声と政府契約を得る能力を損なうだろう。米国証券取引委員会は、発行者が海外腐敗防止法の会計規定に違反したため、発行者の米国取引所での証券取引を一時停止または禁止する可能性もある。
もし私たちまたは私たちが現在または将来雇用している任意の第三者製造業者が環境、健康、安全の法律法規を遵守できなかった場合、私たちは罰金や罰金を受けたり、私たちの業務に重大な悪影響を及ぼす可能性のあるコストや責任を生じるかもしれない。
私たちが今私たちと雇用している第三者製造業者、そして私たちが将来雇用する可能性のあるいかなる第三者メーカーも、研究室手続きおよび危険材料と廃棄物の処理、使用、貯蔵、処理、処理を管理する法律法規を含む多くの環境、健康、安全な法律法規の制約を受けるだろう。私たちの行動は化学物質と生物学的材料を含む危険で燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物製品を発生させるだろう。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。汚染や傷害のリスクを取り除くことはできません
131
これらの材料から。もし私たちが危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に責任を負い、いかなる責任も私たちの資源の範囲を超える可能性がある。私たちはまた民事や刑事罰金と処罰に関連した巨額の費用を発生させるかもしれない。
危険材料の使用により従業員が負傷したコストや支出を支払うために一般責任保険と労働者補償保険を維持しているが、この保険は潜在的な責任に対応するには不十分である可能性がある。私たちは私たちが生物、危険または放射性物質を貯蔵したり処分したりすることによって、私たちが提起した環境責任や有毒侵害に対して保険を維持することはできません。
また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究、開発、または商業化努力を損なうかもしれない。このような法律法規を遵守しないことはまた巨額の罰金、処罰、または他の制裁につながる可能性がある。
さらに、私たちの現在および将来の任意の第三者契約製造業者の運営について、彼らが適用される環境、健康および安全法律法規に準拠していない場合、または私たちの製品に関連する廃棄物を適切に処理することができない場合、私たちは、それによって生じるいかなる損害、名声損害、または私たちの候補製品または製品の製造と供給中断に責任を負わなければならないかもしれない。さらに、私たちの第三者契約製造業者が環境、健康、安全法律法規を守らないことで禁止または他の制裁を受けた場合、私たちのサプライチェーンは悪影響を受ける可能性があります。
従業員の事務と管理成長に関するリスク
私たちの未来の成功は私たちが肝心な幹部を維持する能力、及び合格した人材を吸引、維持、激励する能力にかかっている。
私たちは最高経営責任者Sekar Kathiresan医学博士、最高運営官兼総法律顧問のアンドリュー·アシュJ.D.,最高財務官Allison Dorval、首席科学官兼首席医療官Andrew Bellinger医学博士と私たちの管理、科学と臨床チームの他の主要なメンバーの研究開発、臨床、財務、運営とその他の業務専門長に高度に依存している。私たちは私たちの幹部と雇用協定を締結したが、彼らの誰もが私たちとの雇用関係をいつでも終わらせることができる。私たちは私たちの役員や他の職員たちに“キーパーソン”保険を提供しない。合格した科学、臨床、製造、会計、法律及び販売とマーケティング人員を募集と維持することも著者らの成功の鍵となる。
幹部や他の重要な従業員を失ったサービスは、私たちの研究開発と商業化目標の実現を阻害し、業務戦略を成功させる能力を深刻に損なう可能性がある。また、幹部やキーパーソンを交換することは困難かもしれませんし、私たちの業界では開発に成功し、マーケティング承認や製品商業化に必要なスキルや経験を持っている個人数が限られているので、時間がかかるかもしれません。この限られた人材バンクから募集する競争は非常に激しく、多くの製薬と生物技術会社の間の類似人員に対する競争を考慮して、私たちは受け入れ可能な条件でこれらの肝心な人員を採用、訓練、維持或いは激励することができないかもしれない。私たちはまた、大学や研究機関から科学や臨床人を募集する競争に直面している。また、私たちは、科学と臨床コンサルタントを含むコンサルタントとコンサルタントに依存して、私たちの研究開発と商業化戦略の制定を助けてくれます。私たちのコンサルタントやコンサルタントは、私たち以外の雇用主に雇われる可能性があり、他のエンティティと締結された相談または相談契約に基づいて約束することができ、これは、私たちが彼らを得る機会を制限するかもしれない。私たちの成功はまた内部統制と私たちの財務報告書の正確性と即時性を実施して維持することにかかっている。もし私たちが引き続き高い素質の人材を誘致し、維持することができなければ、私たちが成長戦略を推進する能力は制限されるだろう。
私たちは私たちの開発と規制能力を拡大し、販売、マーケティング、流通能力を実施する可能性があると予想されているので、私たちは私たちの成長を管理することが困難になるかもしれません。これは私たちの運営を混乱させるかもしれません。
私たちの発展に伴い、私たちは私たちの従業員の数と業務範囲が大幅に増加することを予想して、特に薬物開発、臨床、監督管理事務、製造と品質管理の分野で、もし私たちの任意の候補製品がマーケティングの承認、販売、マーケティング、流通を獲得すれば。私たちが予想している将来の成長を管理するためには、私たちの管理、運営、財務制度を継続して実施し、改善し、私たちの施設を拡大し、より多くの従業員を募集し、訓練し続けなければならない
132
合格者。私たちの財務資源が限られていることと、私たちの管理チームがこのような成長を期待している会社を管理する上での経験が限られているため、私たちの業務の拡張を効果的に管理したり、より多くの合格者を募集したりすることができないかもしれません。私たちの業務の拡張は巨大なコストを招き、私たちの管理と業務発展資源を移転する可能性があります。成長を管理できないどんな状況も、私たちの業務計画の実行を延期したり、私たちの運営を妨害したりする可能性がある。
将来の買収や戦略連合は私たちの業務を混乱させ、私たちの財務状況と運営結果を損なうかもしれない。
私たちは、より多くの業務、技術、または資産を買収し、第三者と戦略同盟を結んだり、合弁企業を作ったりする可能性があり、これらは私たちの既存の業務を補完または拡大すると信じています。将来性のある市場や技術のビジネスを買収すれば、既存の運営や会社文化と組み合わせることに成功できなければ、これらの事業を買収するメリットは実現できないかもしれません。私たちは、戦略連合や買収によって生じた任意の新製品や候補製品を開発、製造、マーケティングする際に、多くの困難に直面する可能性があり、予想される利益を達成したり、私たちの業務を強化したりすることを延期または阻止する可能性があります。このような買収の後、取引が合理的であることを証明するために、予想される相乗効果を達成することを株主に保証することはできません。買収に直面しているリスクは
これらのリスクや過去や将来の買収や戦略同盟で遭遇した他の問題を解決することができず、これらの取引の予想される利点を実現できず、予期しない債務を発生させ、全体的に業務を損なう可能性がある。もう一つのリスクは、将来の買収は、債務、または負債、償却費用、または増資運営費用をもたらし、これらのいずれも、私たちの財務状況や運営結果を損なう可能性があるということだ。
我々の内部情報技術システム、または私たちの協力者、サプライヤーまたは他の請負業者またはコンサルタントのシステムは、故障やセキュリティホール、データ損失、および他の中断を受ける可能性があり、これは、私たちの製品開発計画の深刻な中断を招き、私たちの業務に関連する敏感な情報を危険にさらしたり、私たちの重要な情報へのアクセスを阻止したり、契約や法的義務をトリガしたりして、私たちに責任、名声の損害、または他の方法で私たちの業務および財務業績に悪影響を及ぼす可能性があります。
私たちは情報技術システム、インフラ、データに依存して私たちの業務を運営している。通常のビジネスプロセスでは、知的財産権、独自のビジネス情報、および個人情報を含むが、これらに限定されない機密情報を収集、格納、および送信します。重要なのは、このような機密情報の利用可能性、セキュリティ、セキュリティ、プライバシー性、および完全性を維持するために、私たち、私たちのサプライヤー、協力者、または他の請負業者、またはコンサルタントが、このような機密情報の利用可能性、セキュリティ、セキュリティ、プライバシー性、および完全性を維持するために、安全な方法でそうしなければならないということである。
セキュリティ対策が実施されているにもかかわらず、私たちの内部情報技術システムおよび任意の協力者、サプライヤー、請負業者、またはコンサルタントのシステムは、コンピュータウイルス、コンピュータハッカー、悪意のあるコード、従業員エラー、窃盗または乱用、サービス拒否攻撃、複雑な民族国家および民族国家によって支持される行為者、許可されていないアクセス、自然災害、テロ、戦争、または他の要因の破壊または中断を受けやすい
133
武力衝突、電気通信、そして電力故障、または他の妥協。ロシアとウクライナ間の持続的な衝突と、それによる米国と欧州各国政府による制裁、およびそれらが将来取る任意の追加制裁または他の行動により、サイバーセキュリティ攻撃は全体的に増加する可能性がある。
ネットワーク攻撃の頻度,複雑さ,強度が増加しており,発見されにくくなってきている.ネットワーク攻撃は、サービスの信頼性に影響を与え、情報の機密性、完全性、および利用可能性を脅かすために、有害なマルウェアの配備、恐喝ソフトウェア、サービス攻撃の拒否、ファイルの不正アクセスまたは削除、社会工学、および他の手段を含む可能性がある。ネットワーク攻撃はまた、支払いまたは情報が予期されない受信者に送信されることをもたらすために、ネットワーク釣りまたは電子メール詐欺を含む可能性がある。私たちはすべての種類の安全脅威を予見できないかもしれないし、これらすべての安全脅威に対して効果的な予防措置を取ることができないかもしれない。サイバー犯罪者が使用する技術は常に変化し,起動前には認識されない可能性があり,外部サービスプロバイダ,組織犯罪分岐機関,テロ組織や敵対する外国政府や機関などの外部団体を含む様々なソースから来ている可能性がある.私たちは私たちが今まで取ってきた措置と私たちが未来に取る可能性のある行動が未来の違反を防ぐのに十分だという保証はない。
我々が重大なシステム障害、事故、ネットワーク攻撃、またはセキュリティホールを経験する程度では、それは、我々の商業機密または他の独自または機密情報の損失によるものであっても、他の中断によるものであっても、我々の開発計画および業務運営の重大な中断をもたらす可能性がある。例えば、進行中または将来の臨床試験における臨床試験データの損失は、我々の規制承認作業を遅延させる可能性があり、データを回復または複製するコストを著しく増加させる可能性がある。適切な技術およびネットワークセキュリティインフラを確立し、維持するために必要なリソースを割り当てて効率的に管理しなければ、取引エラー、サプライチェーンまたは製造中断、処理効率の低下、データ損失または知的財産権、または他の固有情報の損失または破損を含む深刻な業務中断を受ける可能性があります。
任意の中断またはセキュリティホールが、私たちまたは私たちのサプライヤー、協力者または他の請負業者またはコンサルタントのデータやアプリケーションを紛失したり破損したり、あるいは機密または独自の情報を適切に開示しない場合、私たちは訴訟の暴露、処罰、罰金を含む責任を招く可能性があり、私たちは規制行動や調査の対象になる可能性があり、私たちの競争地位と名声が損なわれる可能性があり、私たちの候補製品のさらなる開発と商業化は延期される可能性がある。このような事件のせいで、私たちは私たちの契約義務に違反するかもしれない。さらに、不正アクセス、使用または個人情報(私たちの顧客または従業員に関する個人情報を含む)をもたらすこのような事件は、私たちの名声を損なう可能性があり、連邦および/または州の違反通知法と外国と同等の法律を遵守させ、是正措置を強制するように強要され、そうでなければ、個人情報のプライバシーおよび安全を保護する法律および法規に基づいて責任を負うことになり、重大な法律および経済的リスクおよび名声の損害を招く可能性がある。以上のいずれも、当社の業務、財務状況、運営結果、または将来性に重大な悪影響を及ぼす可能性があります。
上記の事件の財務リスクは、私たちが維持している任意の保険によって保険を行うことができないか、または完全にカバーできない可能性があり、私たちの業務、財務状況、運営結果、または将来性に重大な悪影響を及ぼす可能性があります。また、私たちの既存の保険範囲が受け入れ可能な条項で提供され続けるか、あるいは私たちの保険会社が将来のクレームを拒否しないことを保証することはできません。私たちの契約における責任制限が強制的または十分であること、または上記の事件による責任または損害から私たちを保護することは保証されない。
私たちの普通株式所有権と上場企業としての私たちの地位に関するリスク
我々の役員、取締役及びその付属会社は、彼らが共同行動を選択すれば、株主に承認されたすべての事項に重大な影響を与える能力がある。
2023年1月31日現在、私たちの役員と取締役とその関連会社が所有する株式は、私たちの普通株の約21.0%を占めています。したがって、これらの株主が共同行動を選択すれば、彼らは私たちの株主に提出されたすべての事項と、私たちの管理と事務に効果的に影響を与えることができるだろう。例えば、この人たちが一緒に行動することを選択した場合、取締役の選挙および私たちのすべてまたはほとんどの資産の合併、合併、または売却の承認に大きな影響を与える可能性がある。
このような所有権制御の集中可能性:
134
わが社の定款書類やデラウェア州法律の条項は、わが社の買収をより困難にする可能性があり、これは私たちの株主に有利になる可能性があり、私たちの株主が現在の取締役や経営陣のメンバーを交換または罷免しようとすることを阻止するかもしれません。
私たちが再記述した会社登録証明書および私たちの改正および再記載された定款の条項は、株主が有利と思われる可能性のある会社の合併、買収、または他の支配権変更を阻止、延期、または阻止する可能性があり、私たちの株主がその株式から割増取引を得る可能性があることを含む。これらの条項はまた、投資家が将来私たちの普通株に支払いたいかもしれない価格を制限し、それによって私たちの普通株の市場価格を下げる可能性がある。また、我々の取締役会が責任を持って我々の管理チームのメンバーに命じているため、これらの規定は、株主が取締役会のメンバーを交換する難しさを増やすことで、現在の経営陣の任意の試みを交換または罷免することを阻害または阻止する可能性がある。他にもこれらの条項には
また、私たちはデラウェア州に登録して設立されたので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、規定された方法で合併または合併が承認されない限り、私たちが発行した議決権のある株を15%以上保有している人が取引日後3年以内に私たちと合併または合併することを禁止しています。
私たちの普通株の活発な取引市場は引き続き発展したり持続しないかもしれない。
私たちの普通株は2021年6月17日にナスダック世界ベスト市場で取引を開始した。私たちの普通株の取引の歴史が限られていることから、私たちの株の活発な取引市場は発展し続けたり持続できないかもしれない。もし私たちの普通株の活発な市場が引き続き発展したり持続したりしなければ、私たちの株主は株式市場価格を下げることなく彼らの株を売ることが難しいかもしれません。
証券アナリストが研究報告書を発表したり停止したりしなければ、私たちの業務の誤解性、不正確または不利な研究報告を発表したり、彼らが私たちの株に対する否定的な評価を発表したりすると、私たちの株式の価格や取引量が低下する可能性がある。
私たちの普通株の取引市場は、業界や金融アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存している。既存のアナリストが私たちを追跡し続ける保証はなく、新しいアナリストが私たちを追跡し始めるという保証もない。どんな報告書もアナリストが有利な報告書を提供するという保証はない。私たちはアナリストの報告を得ていますが、1人以上の私たちの業務を追跡しているアナリストが私たちの株の評価を下方修正したり、私たちの業務の不正確または不利な研究報告を発表したり、私たちの競争相手に関するより有利な相対的な提案を提供したりすれば、私たちの株価は下落する可能性があります。もしこのようなアナリストの一人以上が私たちの株を追跡しなければ、私たちは市場での私たちの株の可視性を失うかもしれません。これは逆に私たちの株価と取引量を低下させる可能性があります。
135
私たちの普通株の価格はずっと不安定で、大幅に変動する可能性があり、これは私たちの株主に大きな損失をもたらす可能性があります。
私たちの株価はずっと変動していて、変動し続ける可能性が高い。株式市場、特に小さいバイオ製薬会社の市場は極端な変動を経験しており、この変動は往々にしてある会社の経営業績とは無関係である。このような変動により、私たちの株主は彼らが株式を購入したかそれ以上の価格で彼らの普通株を売ることができないかもしれない。私たちの普通株の市場価格は多くの要素の影響を受けるかもしれません
過去には、ある会社の証券市場価格が変動した後、同社に対して証券集団訴訟が提起されることが多かった。私たちは一方としてのいかなる訴訟も、正当な理由の有無にかかわらず、不利な判決を招く可能性がある。私たちはまた不利な条件で訴訟を解決することに決定するかもしれない。このような負の結果は、巨額の損害賠償または罰金の支払い、私たちの名声を損なうこと、または私たちの製品またはビジネス実践に悪影響を及ぼす可能性があります。このような訴訟は、このようなクレームを弁護し、経営陣の注意と資源を移転させるために、他の巨額の費用を発生させる可能性もある。また、公聴会、動議、または他の一時的な手続きや事態の発展結果に対する負の公開公告は、私たちの普通株の市場価格に負の影響を与える可能性がある。
我々は現金,現金等価物,有価証券の使用に広範な自由裁量権を有しており,それらを有効に使用できない可能性がある.
私たちの経営陣は、私たちの現金、現金等価物、有価証券を運用する上で広範な裁量権を持っており、私たちの経営業績を改善したり、私たちの普通株価値を向上させることなく、これらの資金を使用することができます。私たちの経営陣がこれらの資金を有効に使用できなかった場合、財務損失を招く可能性があり、これは私たちの業務に実質的な悪影響を与え、私たちの普通株価格の下落を招き、候補製品の開発を延期する可能性があります。それらが使用される前に、私たちは収入や切り下げを生じない方法でこのような資金に投資するかもしれない。
136
私たちの総流通株の大部分は近い将来に市場に売る資格があります。これは私たちの業務が良好であっても、私たちの普通株の市場価格を大幅に低下させる可能性があります。
公開市場で私たちの普通株の大量株を売ったり、市場で大量の株を持っていると思っている人が株を売却しようとしたりすると、私たちの普通株の市場価格が下がる可能性があります。私たちが初めて公募するまで私たちの株主だった人は私たちの普通株の大量の株式を持ち続けました。もしこの人たちが公開市場で私たちの普通株を大量に売却したり、意図的に私たちの普通株を売却したりすると、私たちの普通株の取引価格が下がるかもしれません。
また、私たちの一部の役員、役員、および私たちの取締役に関連する株主は、時々私たちの普通株を売却することを規定している規則10 b 5-1に加入しているか、または加入する可能性があります。規則10 b 5-1計画によれば、ブローカーは、役員、取締役、または関連株主のさらなる指示を必要とせずに、計画に入る際に確立されたパラメータに従って取引を実行する。規則10 b 5−1計画は、場合によっては修正または終了される可能性がある。我々の役員、取締役、および我々取締役に関連する株主は、重大·非公開情報を把握することなく、ルール10 b 5-1計画以外の追加株式を購入または売却することができる。
また、特定の条件に適合する場合には、相当数の普通株を保有する保有者は、彼らの株式に関する登録声明を提出することを要求する権利があり、または彼らの株式を、自分または他の株主のために提出する可能性のある登録声明に含める権利がある。私たちはまたS-8表の登録声明を提出して、私たちが株式補償計画に従って発行できるすべての普通株を登録しました。S-8表に基づいて登録された株式は、発行時に公開市場で自由に販売することができるが、関連会社、帰属手配、行使オプションに適用される数量に制限される。
我々は“新興成長型会社”であり、新興成長型会社に適用される開示要求を低減し、我々の普通株の投資家への吸引力を低下させる可能性がある。
私たちは“新興成長型会社”やEGCで、2012年のJumpStart Our Business Startups ActやJOBS Actの定義に基づいています。私たちは2026年末までEGCになるかもしれませんが、それまでのいずれかの6月30日に、非関連会社が保有する私たちの普通株の時価が7億ドルを超えている場合、または任意の会計年度の年間毛収入が10.7億ドルを超えた場合、適用年度の12月31日からEGCになることを停止します。もし私たちが3年以内に10億ドルを超える転換不能債券を発行すれば、私たちはまたEGCではないだろう。私たちがまだEGCである限り、私たちは他のEGCではない上場企業に適用されるいくつかの開示要求の免除に依存することを許可され、意図されている。これらの免除には
私たちがこのようないくつかまたはすべての免除に依存すれば、投資家が私たちの普通株の吸引力の低下を発見するかどうかは予測できない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場がある可能性があり、私たちの株価はもっと変動するかもしれない。
また、雇用法案は、EGCが、これらの基準が民間企業に適用されるまで、上場企業に適用される新しい会計基準または改正された会計基準を遵守することを延長する移行期間を利用することを可能にする。私たちは、“選択脱退”延長の移行期間を選択したが、これは、基準が発表または改正された場合、それが公共または民間会社に対して異なる適用日を有する場合、私たちは、民間会社が新しい基準または改正基準を採用する際に、私たち(I)が“選択脱退”延長の移行期間を撤回できなくなるまで、または(Ii)EGCの資格に適合しなくなるまで、このようにしていくことを意味する。
137
上場企業として、我々はコストを増加させ続けており、我々の経営陣は、新たなコンプライアンスやコーポレートガバナンスを実施するために多くの時間を投入し続けることが求められている。
上場企業として、私たちはすでに発生しています。特に私たちがEGCでなくなった後、私たちは引き続き大量の法律、会計、その他の費用を発生させます。これらの費用は私たちが以前個人会社として発生しなかったことです。2002年サバンズ-オクスリ法案、ドッド-フランクウォール街改革と消費者保護法、ナスダック世界精選市場の上場要求及びその他の適用された証券規則と法規は上場企業に対して各種の要求を提出し、有効な開示、財務制御と会社管理やり方を確立と維持することを含む。私たちの経営陣と他の人たちはこのようなコンプライアンス計画を実施するために多くの時間を投入し続ける必要があるだろう。また、これらの規則および法規は、特に上場企業の内部統制および財務報告要件を満たすために追加の財務および会計従業員を雇用する場合には、いくつかの活動を私たちが個人会社の場合に比べてより時間とコストを高くするであろう。例えば、これらの規制は、私たちが取締役や上級管理職責任保険を獲得することをより困難かつ高価にする可能性があり、逆に、合格した取締役会メンバーを引き付け、維持することが困難になる可能性があると予想されます。
私たちはこのような規則と規定を評価しており、私たちが発生する可能性のある追加コスト金額やそのようなコストの時間を予測または推定することができない。これらの規則や条例は往々にして異なる解釈を持ち,多くの場合特殊性に欠けるため,規制機関や理事機関が新たな指導意見を提供するにつれて,実践における適用は時間とともに変化する可能性がある。これは遵守事項に関する持続的な不確実性と、開示と統治慣行を絶えず修正するために必要なより高いコストをもたらす可能性がある。
第404条によれば、本年度報告書を提出したForm 10−Kから、財務報告書の内部統制に関する報告書を我々の経営陣が提出しなければならない。しかし、私たちはまだEGCですが、私たちは私たちの独立公認会計士事務所が発行した財務報告書の内部統制に関する認証報告書を含むことを要求されません。規定された時間内に第404条を遵守するために、私たちは費用が高く挑戦的な財務報告書に対する私たちの内部統制を記録して評価する過程を行っている。この点では、より多くの財務·会計担当者を雇用することを含む内部資源を提供し続ける必要があり、外部相談者を招聘することが可能であり、財務報告内部統制の十分性を評価·記録することにより、財務報告内部制御プログラムを適宜改善し、制御措置が文書に規定された方法で動作しているか否かをテスト検証することにより、継続的な報告を実施し、財務報告内部制御プログラムを改善する必要がある。私たちは努力したにもかかわらず、私たちは規定された時間枠内で結論を出すことができないかもしれない、すなわち私たちは財務報告書の内部統制に有効であり、404条の要求に適合する可能性がある。財務報告書の内部統制に1つまたは複数の重大な弱点があることが発見されれば、財務諸表の信頼性への自信を失った金融市場の不良反応を招く可能性がある。
私たちは予測可能な未来に私たちの株に何の現金配当も支払わないと予想されるので、資本付加価値(あれば)が私たちの株主の唯一の収益源になるだろう。
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、私たちの未来のすべての収益を維持するつもりで、もしあれば、私たちの業務の成長と発展に資金を提供します。したがって、私たち普通株の資本増価(あれば)は私たちの株主が予測可能な未来に唯一の収益源になるだろう。
我々が再記述した会社登録証明書は、デラウェア州衡平裁判所とアメリカ合衆国連邦地域裁判所が私たちの株主のために開始する可能性のあるいくつかのタイプの訴訟および訴訟の唯一および独占フォーラムを指定し、これは、私たちまたは私たちの役員、上級管理者、および従業員との紛争において有利な司法フォーラムを得る私たちの株主の能力を制限するかもしれない。
私たちが書面で代替裁判所を選択することに同意しない限り、デラウェア州衡平裁判所(または、デラウェア州裁判所に管轄権がない場合、デラウェア州連邦地方裁判所)は、デラウェア州成文法または一般法の下で以下のタイプの訴訟または手続きの唯一かつ独占的な裁判所となることが規定されている
138
これらの裁判所条項の選択は、取引法に規定されている義務または責任を執行するための訴訟には適用されない。また、証券法第22条は、連邦裁判所と州裁判所は、このようなすべての“証券法”訴訟に対して同時に管轄権を持っていると規定している。したがって、州裁判所と連邦裁判所はこのようなクレームを受理する管轄権を持っている。複数の司法管区でクレームを提訴せざるを得ないことや、異なる裁判所が不一致や逆の裁決を下す脅威などの考慮要因を回避するために、我々が書面で代替裁判所を選択することに書面で同意しない限り、アメリカ合衆国連邦地域裁判所は、証券法によって生じた任意のクレームを解決する唯一かつ独占的な裁判所であるべきである。デラウェア州裁判所は、このような選択の裁判所条項が事実上有効であることを決定しているが、株主は依然として専属裁判所条項が指定された場所以外の場所でクレームを出すことを求めることができる。このような状況では,我々が再記述した会社登録証明書の独占フォーラム条項の有効性と実行可能性を強く主張する予定である.これは、他の法ドメインでこのような訴訟を解決することに関連する多くの追加費用を必要とする可能性があり、これらの規定がこれらの他の法ドメインの裁判所によって実行されることを保証することはできない。
これらの排他的フォーラム条項は、私たちの株主が司法フォーラムで、私たちまたは私たちの役員、上級管理者、または従業員との紛争に有利だと思うクレームを提出する能力を制限する可能性があり、これは、私たちと私たちの役員、上級管理者、および従業員に対するこのような訴訟を阻止するかもしれない。もし裁判所が私たちが再記載した会社登録証明書に含まれる任意の独占裁判所条項が訴訟で適用されないか、実行できないことを発見した場合、私たちは他の管轄区域でこのような訴訟の解決に関連するさらなる重大な追加費用を発生する可能性があり、これらはすべて私たちの業務、財務状況、および運営結果に重大な悪影響を及ぼす可能性がある。
一般リスク因子
私たちの開示統制と手続きはすべてのミスや詐欺を阻止したり検出できないかもしれない。
私たちは取引法の特定の報告書の要求事項を守らなければならない。我々の開示制御および手続きは、取引所法案に基づいて提出または提出された報告書において、開示を要求する情報が蓄積され、米国証券取引委員会規則および表で指定された期間内に管理層、記録、処理、まとめおよび報告に伝達されることを合理的に確保することを目的としている。任意の開示制御およびプログラムまたは内部制御およびプログラムは、発想および動作がどのように完全であっても、絶対的な保証ではなく、合理的な保証を提供することしかできず、制御システムの目標が達成されることを確保することしかできないと信じている。これらの固有の限界は,意思決定過程における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという現実を含む.さらに、ある人の個人的な行動、2人以上の結託、または無許可超越制御は、制御を回避することができる。したがって、我々の制御システムの固有の制限により、エラーや詐欺によるエラー陳述や開示不足が発見されることなく発生する可能性がある。
税法またはその実施または解釈の変化は、私たちの業務および財務状況に悪影響を及ぼす可能性がある。
税法の変化は私たちの業務や財務状況に悪影響を及ぼすかもしれない。2017年12月22日、米国政府は“税法”を公布し、同法の重大な改革を行った。CARE法案などの改正税法には、会社税率を35%の最高限界税率から21%に下げる統一税率を含め、2017年12月31日以降に開始される納税年度に生じるNOLの控除額を今年度の課税収入の80%に制限する会社税への大きな変化が含まれている。また、2022年から税法は現在の研究開発支出を差し引く選択を廃止し、通常企業に5年以内に資本化と償却を要求している。
CARE法案に加えて、国会の新冠肺炎流行への対応の一部として、2020年と2021年に税収条項を含む経済救済立法が公布され、いくつかの新しい税収条項を導入したインフレ低減法案やアイルランド共和軍が2022年8月に法律に署名した。IRAは、特に、上場企業のある株の買い戻しに1%の消費税を課しており、これは、通常、上場企業(またはその特定の関連会社)が金銭または他の財産(会社自体の株式を除く)と引き換えに上場企業の株式を買収するいかなる取引にも適用されるが、遵守しなければならない
139
最小の例外。したがって、消費税は非伝統的な株の買い戻しのいくつかの取引に適用される可能性がある。税法、アイルランド共和軍、そしてこのような追加立法によって提供される規制指導は継続されており、このような指導は最終的に私たちの業務と財務状況に及ぼすそれらの影響を増加または減少させる可能性がある。また,各州が税法,アイルランド共和軍,その他の税法をどの程度遵守するかは不明である。私たちは、普通株式の潜在的投資家に、最近公布された任意の税法または提案された法律の変化と、私たちの普通株を投資または保有する潜在的な税金結果について、彼らの法律および税務顧問に相談することを促す。
不利なグローバル経済状況は、私たちの業務、財務状況、株価、経営業績に悪影響を及ぼす可能性があります。
私たちの経営業績は世界経済と世界金融市場の全体的な状況および経済安定性の不確実性の悪影響を受ける可能性があります。世界経済·金融市場はまた、ロシアとウクライナの間の紛争、テロ、または他の地政学的事件を含む軍事衝突の現在または予想される悪影響を受ける可能性がある。米国や他の国がこのような紛争に対応するために実施している制裁には、ロシアに関する制裁が含まれており、金融市場や世界経済に悪影響を及ぼす可能性もあり、影響を受けた国や他の国の経済対策は市場や経済の不安定を悪化させる可能性がある。信用と金融市場のさらなる悪化と経済状況への自信が起こらない保証はない。深刻または長期的な経済低迷は、私たちが開発する可能性のある任意の候補製品に対する需要の減弱、必要に応じて許容可能な条件で追加資本を調達する能力など、私たちの業務に様々なリスクをもたらすかもしれない。経済が疲弊したり下落したりすることは、私たちのサプライヤーに圧力を与え、供給中断を招く可能性もある。株式市場と信用市場が悪化すれば、任意の必要な債務や株式融資をより困難にし、コストをより高く、希釈度をより高くする可能性がある。有利な条件で任意の必要な融資を得ることができなければ、成長戦略を実現する能力を弱める可能性があり、財務業績や株価を損なう可能性があり、臨床開発計画の延期または放棄を要求する可能性がある。さらに、私たちの現在または未来のサービス供給者、製造業者、または他の協力者は、このような困難な経済期に生き残ることができないかもしれない, これは私たちが時間通りで予算通りに運営目標を達成する能力に直接影響を及ぼすかもしれない。私たちは現在の経済気候と金融市場の状況が私たちの業務に悪影響を及ぼす可能性のあるすべての方法を予測することができない。
項目1 B。未解決の従業員のコメント。
ない。
項目2.財産
私たちは現在マサチューセッツ州ボストンで105,182平方フィートのオフィスと実験室空間を借りて、レンタル契約は12月に期限が切れます 2032年には、あと5年延長することができる。
2021年10月、マサチューセッツ州ケンブリッジ市に11,931平方フィートのオフィスと実験室スペースを追加したビーム治療会社と転貸協定を締結しました。分譲は2021年12月に始まり、2022年12月に満了する。
私たちの施設は私たちの現在の需要を満たすのに十分で、必要な時に適切な追加空間を提供すると信じている。
項目3.法的訴訟
私たちは現在どんな実質的な法的手続きの当事者でもない。
時々、私たちは通常の業務過程で発生した訴訟や他の法的手続きに巻き込まれるかもしれない。結果にかかわらず、弁護と和解コスト、管理資源の移転、その他の要素により、訴訟は私たちの業務、財務状況、運営結果、将来性に悪影響を及ぼす可能性がある。
第4項鉱山安全情報開示
適用されません。
140
第II部
第五項登録者普通株式市場、関連株主事項及び発行者が株式証券を購入する。
市場情報
私たちの普通株は2021年6月16日からナスダック世界精選市場で公開取引され、取引コードはVERVである。その前に、私たちの普通株は市場を公開しなかった。
所持者
2023年2月27日現在、私たちの普通株は約17人の登録保有者がいます。この数字には,その株式が街中の被指名者が所有する実益所有者は含まれていない.
配当をする
設立以来、私たちはどんな現金配当金も発表したり支払ったりしたことがない。私たちは将来の収益(あれば)を残し、私たちの業務の運営と拡張に資金を提供し、予測可能な未来に普通株式保有者に現金配当金を支払うことはないつもりです。
株式表現グラフ
以下の株式表現グラフは、2021年6月17日(我々の普通株がナスダック全世界精選市場で取引された日)から2022年12月31日までの、我々の普通株ナスダック総合指数とナスダックバイオテクノロジー指数の株主累積リターン総額の比較を示している。グラフは2021年6月17日の初期投資を100ドル、寄り付き価格を1株19.00ドルとし、すべての配当金が再投資を行ったと仮定しているが、私たちの普通株はまだ配当を発表していない。グラフ中の比較は米国証券取引委員会が必要としているものであり,我々の普通株の将来可能性を予測または指示するための表現ではない.
141
取引法第18節の場合、本第5項の業績グラフは、“募集材料”または米国証券取引委員会に“保存されている”とみなされないか、またはこの節の下の責任を他の方法で負うものとみなされず、参照によってそのような文書に明示的に組み込まれない限り、Verve Treateutics,Inc.が証券法または取引法に従って提出された任意の文書とみなされてはならない。
最近売られている未登録証券
本Form 10−K年次報告がカバーする期間内に、当社は未登録の株式証券を発行していないが、当社の現在のForm 8−K年報に開示されている取引は除く。
証券登録所得収益の使用
2021年6月21日、米国証券取引委員会が2021年6月16日に発効を発表したS-1表登録書(第333-256608号文書)と、証券法第462(B)条に基づいて提出され、米国証券取引委員会が2021年6月16日に発効を宣言したS-1表登録書(第333-257158号文書)に基づいて普通株式初公募を完了した。
引受割引と私たちが支払うべき2,510万ドルの発売費用を差し引いて、私たちが得た純発行収益は2.816億ドルです。2022年12月31日現在、初公募株の純収益は使用されていません。私たちは今回発行された純収益を通貨市場基金と短期投資に投資した。我々が初公募株を用いて得られた資金純額の計画に実質的な変化はないことは、ルール424(B)に基づいて米国証券取引委員会に提出された日が2021年6月16日である最終入札説明書に記載されている。
発行者または関連購入者が株式証券を購入する
2022年12月31日までの年次内に、吾らまたは任意の関連バイヤーまたは吾を代表する任意の代表または関連バイヤーを代表して行動する者は、当社の普通株のいかなる株式も購入していない。
142
第六項です。
[保留します。]
143
プロジェクト7.経営陣の議論財務状況や経営成果の分析もあります
以下、我々の財務状況及び経営結果の検討及び分析は、我々の連結財務諸表及び本年度報告(Form 10-K又は年次報告)の末尾に関する注釈と共に読まなければならない。本議論および分析に含まれるまたは本年度報告の他の部分に記載されている情報は、リスクおよび不確実性要因に関する前向きな陳述を含む、我々の業務および関連融資の計画および戦略に関する情報を含む。本年度報告の“リスク要因”の一部に列挙された要素を含む多くの要素の影響により、我々の実際の結果は、以下の議論と分析に含まれる前向き陳述に記述または示唆された結果とは大きく異なる可能性がある。
概要
著者らは臨床段階の遺伝子薬物会社であり、心血管疾患を治療する新しい方法を開拓し、治療を慢性治療から単一治療コースの遺伝子編集薬物に転換した。過去50年間に治療方面で進展を得たにもかかわらず、心血管疾患は依然として全世界の主要な死亡原因である。現在の慢性看護モデルは脆弱である−厳格な患者コンプライアンス,広範な医療インフラ,定期的な医療サービスが必要であり,多くの患者が適切な看護を受けられないようにしている。私たちの目標は新しい単一治療コースの治療法を提供することでCVDの慢性ケアモデルを覆すことです体内にある遺伝子編集治療の重点はこのような高度な流行と生命を脅かす疾患の根本的な原因を解決することである。著者らの最初の2つの項目はそれぞれPCSK 9とAngptl 3に対して、この2つの遺伝子は低密度リポ蛋白質コレステロール(LDL-C)などの脂質低下の目標として広く検証されている。著者らは、これらの遺伝子を編集することは、粥状動脈硬化性心血管疾患(ASCVD)(最もよく見られる心血管疾患形態)を有する患者の一生において、低密度リポ蛋白-Cを有効かつ持続的に低下させることができると信じている。
我々の方法は21世紀の生物医学の多くの突破-ヒト遺伝子分析、遺伝子編集、メッセンジャーRNA或いはメッセンジャーRNA、mRNAに基づく療法と脂質ナノ粒子(LNP)送達-主に肝臓で発現する遺伝子を標的とし、心血管疾患を引き起こすタンパク質の産生を撹乱する。私たちは単行コースのパイプを進めています体内にある遺伝子編集プログラムは、各プログラムは自然抗病性突然変異をシミュレーションし、特定の遺伝子を閉鎖し、血中脂質を低下させ、それによってASCVDのリスクを低下させるように設計されている。著者らは最初にこれらの計画を開発して家族性高コレステロール血症(FH)患者を治療する予定であり、FHは遺伝病であり、一生深刻に上昇する血液低密度リポ蛋白-Cを招き、それによって早発性ASCVDのリスクを増加させる。もし私たちの計画がFHで成功すれば、私たちはそれらもより広範なASCVD患者に潜在的な治療を提供できると信じている。最終的に,これらの治療法はASCVDハイリスク群の管理のために開発される可能性があり,予防策として,あるワクチンが感染症に対する長期保護を提供する方式と類似していると考えられる。
私たちは2018年3月に登録が成立し、その後間もなく運営を開始した。私たちの設立以来、私たちはほとんどの資源を私たちの遺伝子編集とLNP技術を構築し、私たちのプロジェクトの組み合わせの発展を推進し、私たちの知的財産権を確立し、保護し、研究と開発活動を行い、私たちの会社、業務計画、資金を組織し、配備し、これらの業務に一般的かつ行政的な支援を提供してきた。これまで、私たちは主に私たちの優先株を売却し、私たちの初公募株(IPO)、私たちの後続公募株で私たちの普通株を売ること、私たちの市場やATM(ATM)株式発行計画、そしてVertex製薬会社(Vertex)との戦略的パートナー関係を通じて、私たちの運営に資金を提供してきました。
2022年12月31日現在,非公開配給の優先株と普通株および公募した普通株を売却することで合計8.616億ドルの毛収入を集めた。
私たちは臨床段階の会社です。これまで、私たちは製品販売から何の収入も得ておらず、予測可能な未来にも製品販売から収入を得ることはない。設立以来、私たちは重大な運営損失を受けた。2022年、2021年、2020年12月31日までの年間純損失はそれぞれ1.574億ドル、1.203億ドル、4570万ドルだった。2022年12月31日現在、私たちの累計赤字は3.442億ドルです。
2022年、2021年、2020年12月31日までの年間の総運営費はそれぞれ1億676億ドル、8710万ドル、4060万ドルです。われわれが行っている心臓1号臨床試験におけるVERVE−101の推進に伴い,われわれの計画組合せに関する開発活動は巨額の費用と増加した運営損失を招き続けることが予想され,他の臨床前開発を継続している
144
製品候補;これらの候補製品の臨床開発への推進;基礎編集と新しい遺伝子編集技術、交付技術、製造能力のさらなる開発;Angptl 3に対する私たちの開発候補製品VERVE-201を含む他の候補製品の発見と開発を求める;法執行を維持、拡大し、私たちの知的財産権の組み合わせを保護する;研究開発と臨床者を招聘する;最終的に販売、マーケティング、流通インフラを構築し、私たちが市場の承認を得る可能性のある任意の製品を商業化する;商業製造源と安全供給チェーン能力を確立し、規制承認を得る可能性のある任意の候補製品の商業数量を提供する。私たちの研究、製品開発、将来の商業化努力、上場企業としての運営を支援するために、運営、法律、コンプライアンス、財務と管理情報システムと人員を増やします。
したがって、私たちは私たちの持続的な運営を支援し、私たちの戦略を実施するために多くの追加資金を必要とするだろう。製品販売から相当な収入を得ることができるまで、もしあれば、株式発行、債務融資、および他の資本源の組み合わせによって、他社との協力や許可手配、または他の戦略取引を含む可能性がある運営融資を提供する予定です。もし私たちが必要な時や受け入れ可能な条件下で資金を調達したり、十分な資金を得ることができない場合、私たちは私たちの研究および開発計画や将来の商業化努力を延期、制限、減少または終了させることを余儀なくされるかもしれないし、開発とマーケティングは、私たちが自分で開発とマーケティングをより望んでいた候補製品の権利を付与するかもしれない
製品開発に関連する多くのリスクや不確実性のため、費用を増加させる時間や金額を予測することもできず、いつまたは利益を得ることができるかどうかを予測することもできない。たとえ私たちが製品販売から収入を得ることができても、私たちは利益を上げることができないかもしれない。もし私たちが利益を上げることができない場合、または持続的に利益を上げることができない場合、私たちは計画通りに運営を継続できず、私たちの運営を減少または終了させることができないかもしれない。
2022年12月31日現在、私たちは5.5448億ドルの現金、現金等価物、有価証券を持っている。私たちの既存の現金、現金等価物、有価証券は、2025年下半期の運営費用と資本支出需要に資金を提供できると信じています。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは予想よりも早く利用可能な資本資源を枯渇させるかもしれない。この時点で私たちの運営に資金を提供するためには、追加の資本を調達する必要があり、これは保証されないだろう。“流動性と資本資源”を見てください
新冠肺炎がわれわれの業務に与える影響
2020年3月、新冠肺炎は世界保健機関によって全世界大流行と発表され、今まで、新冠肺炎の大流行は引き続き世界各地で重大な公衆衛生と経済挑戦を構成している。新冠肺炎疫病は直接或いは間接的に著者らの業務、運営結果と財務状況の時間長と全面程度に影響する可能性があり、未来の発展に依存し、これらの事態の発展は高度な不確定性があり、変化が発生する可能性があり、予測が困難である。我々は、我々の契約製造組織、またはCMO、ならびに我々の契約研究組織、またはCROは、研究規模の生産およびいくつかの臨床前研究を実行する能力の一時的な減少を経験している。これらの行動はそれ以降正常化しているが,我々は我々のCMOやCROとともに,新冠肺炎の大流行がこれらの行動に及ぼす影響を密接にモニタリングしている。
著者らはまた、新冠肺炎疫病が著者らの従業員とその他の業務運営に与える持続的な影響を引き続き密接に注目する予定である。従業員に安全な作業環境を提供するために、我々は、オフィスおよび実験室施設内の従業員を作業活動に現場参加を必要とする従業員に制限し、オフィスおよび実験室施設の消毒リズムを増加させ、すべての対面会議の代わりに仮想インタラクションを使用し、オフィスおよび実験室施設内の従業員に個人保護装置を提供することを含む様々な社交距離措置をオフィスおよび実験室施設で実施した。最近、より多くの従業員が限られた身分で私たちのオフィスと研究室に戻ってきた。私たちは新冠肺炎の流行の影響と影響と私たちの対応を監視し続けており、政府当局が要求したり提案したりする可能性がある、あるいは私たちの従業員や他の業務パートナーの最適な利益に合致すると考えられる行動を継続する予定です。
許可と協力協定
様々な許可や協力協定に基づいて、将来的に支払い可能な意義の大きなマイルストーンと成功した支払いを義務化し、これらの合意がカバーする任意の候補製品の販売に印税を支払い、これらの合意は最終的に規制承認と商業化を実現する。これらの合意に関する情報は、本年度報告表格10-K第I部分第1項の“営業許可証と連携協定”を参照されたい。
145
私たちの運営結果の構成要素は
収入.収入
2022年12月31日までの1年間に190万ドルの協力収入を確認した。私たちは開発に成功し、規制部門から1つ以上の候補製品の承認を得ない限り、近い将来、販売製品から何の収入も得られないと予想している。
運営費
研究開発費
研究·開発費用には、研究·開発活動を行うことによるコストが含まれている
私たちは発生した費用に応じて研究と開発費用を支払う。私たちが将来受け取る研究開発活動のための商品やサービスのために支払われた払い戻し不可能な前払いは前払い費用として記録されています。前払い額は福祉の消費に伴って支出される。
開発の初期段階では,我々の研究開発コストは一般に概念検証研究に用いられており,これらの研究は必ずしも特定の目標に割り当てられるとは限らないため,我々の費用を1つずつ計画的に追跡するようになっていない.
研究開発活動は私たちのビジネスモデルの核心だ。予測可能な未来には,我々の計画や候補製品を臨床開発に進め,より多くの候補製品を開発し,我々の製造能力を建設し,我々の遺伝子編集とLNP技術を発展させることに伴い,我々の研究·開発費は増加し続けると予想される。また,我々の発見研究作業や関係者コストが増加することも予想されるため,株式報酬に関するコストを含めて過去レベル以上に増加することが予想される.さらに、マイルストーンや特許使用料の第三者への支払いに関連する追加費用が生じる可能性があり、将来の候補製品の権利を得るために、これらの第三者と許可、買収、オプション協定を締結する可能性があります。
現在、私たちは私たちの任意の候補製品或いは計画の臨床前と臨床開発を完成し、監督部門の承認を得るために必要な努力の性質、時間とコストを合理的に推定或いは知ることができない。私たちの候補製品が開発に成功するかどうかは大きな不確実性を持っている。これは製品開発に関連する多くのリスクと不確実性のためです
146
私たちの現在または未来の任意の候補製品について、これらの変数のいずれかの変化は、候補製品開発に関連するコスト、タイミング、および生存能力を著しく変化させる可能性がある。私たちは規制部門が私たちが開発する可能性のあるすべての候補製品の承認を得ることに決して成功しないかもしれない。
一般と行政費用
一般と行政費用は主に人事関連費用を含み、幹部、知的財産権、業務発展と行政機能者の給料、福祉、株式ベースの給与を含む。一般及び行政費用には、知的財産権及び会社事務に関する法律費用、会計、監査、税務及びコンサルティングサービスの専門費用、保険料、出張費、施設に関する直接及び分配費用、その他の運営コストも含まれる。
私たちは今後、より多くの研究開発活動を支援するために、私たちの一般的かつ行政的費用が増加すると予想している。また、ナスダックと証券取引委員会または米国証券取引委員会が要求する会計、監査、法律、規制および税務関連サービスの維持に関連するコスト、取締役および幹部の保険コスト、投資家および広報コストなど、上場企業に関連するコストが増加し続けることが予想される。
その他の収入(費用)
反希釈権利負債の公正価値変動
逆希釈権は、優先株融資や他の株式融資を完了した後にハーバードとブロダーに追加普通株を発行する義務を表しており、私たちの初公募株終了時に十分に満たされている。協議開始時には,権利を逆希釈した負債を公正価値で入金し,コストを研究開発費とし,報告期間ごとに再計量し,ツールが返済されていない場合には他の収入(費用)に変化を記録する。
成功支払負債の公正価値変動
もし私たちの平均時価が指定されたハードルを超えて、9桁に達するドルの金額から100億ドルに上昇したり、これらのハードルを超えたと引き換えに私たちの会社を売却したりすれば、私たちもハーバード大学と遠大グループに成功費用を支払う義務がある。当社の支配権が変更されたり、当社が売却されたりした場合、当社は事件発生後の指定期間内に任意の関連する成功した支払いを現金で支払わなければなりません。そうでなければ、Success支払いは私たちの選択に基づいて現金または私たちの普通株の株、または現金と私たちの普通株の株の組み合わせで決済することができます。私たちが支払う可能性のある残りの潜在的なSuccess支払い総額は2500万ドルです。協議開始時には、成功支払負債は公正価値で入金され、コストは研究及び発展費用と記入され、各報告期間内に他の収入(支出)で未清算ツールに記入された費用は再計量される。
147
私たちの推定によると、成功した支払い負債の公正価値と、私たちが経営報告書に記録した公正価値はそれに応じて変化し、異なる時期に大幅に変動する可能性があります。
利息とその他の収入,純額
利息と他の収入は主に私たちの有価証券から稼いだ利息と私たちの核心業務とは関係のない他の雑収入と支出を含む。
所得税
2022年12月31日現在、連邦純運営損失(NOL)は1兆639億ドル、州NOLは1.48億ドルに転換した。連邦NOL繰り越しに無期限があり、将来の課税収入の80%を相殺することができるが、州NOL繰り越しは2042年前の異なる期日に満期になる。最終的な現金化の不確実性のため、私たちは私たちの繰延税項純資産に全額推定準備金を計上した。
行動の結果
2022年12月31日までと2021年12月31日までの年次比較
次の表は、2022年12月31日と2021年12月31日までの年間経営実績をまとめたものです
|
|
現在までの年度 |
|
|
|
|
||||||
(単位:千) |
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
協力収入 |
|
$ |
1,941 |
|
|
$ |
— |
|
|
$ |
1,941 |
|
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
130,095 |
|
|
|
68,202 |
|
|
|
61,893 |
|
一般と行政 |
|
|
37,533 |
|
|
|
18,865 |
|
|
|
18,668 |
|
総運営費 |
|
|
167,628 |
|
|
|
87,067 |
|
|
|
80,561 |
|
その他(費用)収入: |
|
|
|
|
|
|
|
|
|
|||
反希釈権利負債の公正価値変動 |
|
|
— |
|
|
|
(25,574 |
) |
|
|
25,574 |
|
成功支払負債の公正価値変動 |
|
|
1,486 |
|
|
|
(7,815 |
) |
|
|
9,301 |
|
利息とその他の収入,純額 |
|
|
6,867 |
|
|
|
142 |
|
|
|
6,725 |
|
その他の収入を合計して純額 |
|
|
8,353 |
|
|
|
(33,247 |
) |
|
|
41,600 |
|
所得税準備前の損失を差し引く |
|
|
(157,334 |
) |
|
|
(120,314 |
) |
|
|
(37,020 |
) |
所得税支給 |
|
|
(53 |
) |
|
|
— |
|
|
|
(53 |
) |
純損失 |
|
$ |
(157,387 |
) |
|
$ |
(120,314 |
) |
|
$ |
(37,073 |
) |
148
協力収入
2022年12月31日までの1年間の連携収入は190万ドルであり,すべて頂点プロトコルによる研究サービスに関係している.2021年12月31日までの1年間、私たちは何の収入も記録しなかった。
研究開発費
次の表は、2022年12月31日と2021年12月31日までの年間研究開発費をまとめています
|
|
現在までの年度 |
|
|
|
|
||||||
(単位:千) |
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
従業員関連の費用 |
|
$ |
46,439 |
|
|
$ |
19,859 |
|
|
$ |
26,580 |
|
外部コンサルタント(第三者CROを含む)による臨床前研究に関する外部費用 |
|
|
26,952 |
|
|
|
17,681 |
|
|
|
9,271 |
|
製造活動に関連する原材料コストと外部費用、第三者CMOを含む |
|
|
20,584 |
|
|
|
16,628 |
|
|
|
3,956 |
|
実験室用品 |
|
|
11,883 |
|
|
|
4,630 |
|
|
|
7,253 |
|
施設に関するコスト(減価償却を含む) |
|
|
10,486 |
|
|
|
3,583 |
|
|
|
6,903 |
|
許可証とマイルストーン支払い |
|
|
4,515 |
|
|
|
2,031 |
|
|
|
2,484 |
|
臨床試験コスト |
|
|
3,037 |
|
|
|
— |
|
|
|
3,037 |
|
他の研究と開発コスト |
|
|
6,199 |
|
|
|
3,790 |
|
|
|
2,409 |
|
研究開発費総額 |
|
$ |
130,095 |
|
|
$ |
68,202 |
|
|
$ |
61,893 |
|
2022年12月31日までの年間研究開発費は1兆301億ドルだったが、2021年12月31日までの年間は6820万ドルだった。6190万ドルの成長は、主に、私たちのパイプラインの推進を支援するために、私たちの研究開発組織の成長に起因しています
私たちは、予測可能な未来に、私たちの計画と候補製品を臨床開発に進めるとともに、より多くの候補製品を開発し続け、私たちの技術と製造能力に投資し、私たちの研究と開発費用は引き続き増加すると予想しています。
149
一般と行政費用
2022年12月31日までの年間の一般·行政費は3750万ドルであるのに対し、2021年12月31日までの年間は1890万ドルである。1870万ドル増加しました主な理由は以下の通りです
私たちは今後、より多くの研究開発活動を支援するために、私たちの一般的かつ行政的費用が増加すると予想している。
その他の収入(費用)
反希釈権利負債の公正価値変動
逆希釈権利負債の公正価値が2,560万ドル増加したのは、主に2021年12月31日までの1年間に、878,098株の我々の普通株を発行することにより、この負債を返済したためである。和解金額は3,250万ドルであり、和解時に発行された株式の公正価値の増加により、反償却権利負債の公正価値は2,560万ドルに調整された。反償却権利責任は2019年と2020年に部分的に清算され、2021年6月に当社の初公募が完了した時にすべて返済されたため、2022年12月31日までの年度内にさらなる調整はなかった。
成功支払負債の公正価値変動
成功した負債の公正価値が930万ドル増加したのは、主に私たちの普通株の公正価値が減少し、2022年12月31日までの1年間に他の費用を150万ドルの公正価値調整したためである。2021年12月31日までの1年間に、ある成功した支払い義務が触発され、ハーバードとブロダーに借りられた金額は合計630万ドルに達した。これらの金額は2021年11月に現金で決済される。2022年12月31日までの年間で、支払い成功のトリガや支払いはありません。残りの成功的な支払い債務は各報告期間の終了時に再評価されるだろう。
利息とその他の収入,純額
2022年12月31日までの1年間で、利息やその他の収入(支出)が670万ドル増加したのは、主に我々のより高い有価証券残高の金利上昇によるものである。
2021年12月31日までと2020年12月31日までの年度比較
2021年12月31日までの年度経営実績と2020年12月31日までの年度比較に関する検討は、本10−K年度報告に漏れているが、米国証券取引委員会が2022年3月14日に米国証券取引委員会に提出した2021年12月31日現在の10−K年度報告書の“第7項.経営層の財務状況及び経営成果の検討及び分析”で見つかり、この検討内容は参考にして米国証券取引委員会サイトwww.sec.govに組み込まれている。
流動資金と資本資源
流動資金と資金源
2018年の設立以来、私たちは重大な運営赤字が発生した。予見可能な未来には,われわれのプロジェクトの臨床前と臨床発展に伴い,巨額の費用と運営損失を招くことが予想される。今まで、私たちは主に株式発行を通じて私たちの業務に資金を提供してきた。2022年12月31日現在、私たちは私募により優先株と普通株を売却し、IPO、後続公開発行、ATM株式発行計画で普通株を売却し、合計8.616億ドルの毛収入を調達した。2022年12月31日現在、私たちは5.5448億ドルの現金、現金等価物、有価証券を持っている。
150
2021年6月、私たちは初公募株を完成し、2,105,368株が引受業者が引受権を全面的に行使することによって売却された普通株を含む16,141,157株の普通株を発行し、公開発行価格は1株19.00ドルである。引受割引と私たちが支払うべき発売費用を差し引いた後、IPOから2兆816億ドルの純収益を得た。2021年6月、私たちは反希釈権利責任の最終解決策として、ハーバードとブロダー社に878,098株の普通株を発行した。
2022年7月20日、VertexがVertexプロトコルに従って支払った2500万ドルの前金を受け取りました。また,2022年7月20日にVertexに1株23.03ドルで1,519,756株の普通株を売却·発行し,総購入価格は3,500万ドルであった。
2022年7月25日、私たちは9,583,334株の普通株を発行·売却した。その中には1,250,000株の普通株が含まれており、これは引受業者が追加普通株を購入する選択権を十分に行使することによって販売され、公開発行価格は1株27ドルであり、引受割引と私たちが支払うべき約1,590万ドルの発売費用を差し引いた純収益総額は約2.429億ドルである。
2022年7月、私たちはJefferiesと販売協定を締結し、この合意に基づいて、私たちは時々現行の市場価格で私たちの普通株の株を提供して売る権利があります。私たちは、Jefferiesが販売契約に従って販売した任意の株の総販売収益の3.0%の最高3.0%の手数料をJefferiesに支払うことに同意する。販売契約下のいかなる販売も、2022年9月23日に発効し、総発行価格は最大2億ドルに達する当社のS-3表の登録声明(第333-267578号文書)に基づいて行われます。2022年12月31日までの年間で、販売契約に基づいて1,280,168株の普通株を売却し、支払うべき手数料と発売費用を差し引いた純収益総額は4,290万ドルです。
キャッシュフロー
次の表は私たちの各時期の現金源と用途をまとめています
|
|
現在までの年度 |
|
|||||
(単位:千) |
|
2022 |
|
|
2021 |
|
||
経営活動用の現金 |
|
$ |
(122,332 |
) |
|
$ |
(77,880 |
) |
投資活動用の現金 |
|
|
(155,955 |
) |
|
|
(239,098 |
) |
融資活動で提供された現金 |
|
|
328,956 |
|
|
|
377,089 |
|
現金、現金等価物、および限定的な現金の純増加 |
|
$ |
50,669 |
|
|
$ |
60,111 |
|
経営活動
2022年12月31日までの年間で、経営活動で使用されている現金純額は1.223億ドルで、主に私たちの純損失1.574億ドルと、支払い成功負債の公正価値の変化に関する非現金プロジェクトの150万ドルの減少と、投資プレミアム償却に関する100万ドルを含む。これらの額は、株ベースの2250万ドルの給与、280万ドルの減価償却費用、390万ドルの非現金賃貸費用、および私たちの経営資産と負債の純増加840万ドルによって相殺される。
2021年12月31日までの1年間で、経営活動に用いられた現金純額は7790万ドルで、主に1.203億ドルの純損失と290万ドルの運営資産と負債の純減少を含む。これらの額は、逆希釈権利および成功支払負債の公正価値変化3340万ドル、減価償却支出150万ドル、株式ベースの補償710万ドル、非現金賃貸支出180万ドル、および有価証券割増150万ドルによって相殺される。
投資活動
2022年12月31日までの1年間、投資活動のための現金純額は1.56億ドルで、4.794億ドルの有価証券の購入と約1330万ドルの財産と設備の購入を含み、主に実験室設備と関係があり、これらの金額は3.367億ドルの有価証券の満期日によって部分的に相殺された。
2021年12月31日までの1年間、投資活動のための現金純額は2.391億ドルで、3.715億ドルの有価証券の購入と440万ドルの財産と設備の購入を含み、主に実験室設備に関連しており、これらの金額は1.368億ドルの有価証券満期日によって部分的に相殺されている。
151
融資活動
2022年12月31日までに、融資活動が提供する現金純額は3.29億ドルで、主に当社の普通株を売却して得られた純額2.865億ドル、Vertex協定に関連する株式購入プロトコルに基づいてVertexに1,519,756株を発行した純額4000万ドル、株式購入権を行使して得られた金220万ドルおよび従業員が株式を購入して株式を発行する予定で得られた金110万ドルを含むが、支払いによって発売された80万ドルの部分が相殺される。
2021年12月31日までの1年間に、融資活動が提供する現金純額は3.771億ドルで、Bシリーズ優先株発行の純収益9380万ドル、初公募株で我々の普通株を売却した純収益2.852億ドル、株式オプションを行使した収益100万ドルと、我々従業員株購入計画により株約70万ドルを発行し、360万ドルのIPO費用の一部が相殺された。
資金需要
私たちがプロジェクトの組み合わせを推進し続けるにつれて、私たちの運営費用と将来の資金需要は大幅に増加すると予想される。
具体的には、以下のような状況に伴い、私たちの支出が増加するだろう
2022年12月31日現在、私たちは5.5448億ドルの現金、現金等価物、有価証券を持っている。私たちは私たちの既存の現金、現金等価物、そして有価証券が私たちが運営費用に資金を提供できるようにすると信じている
152
そして資本支出は2025年下半期まで要求される。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは予想よりも早く利用可能な資本資源を枯渇させるかもしれない。
潜在的な候補製品を確定し、臨床前テストと臨床試験を行うことは時間がかかり、高価かつ不確定な過程であり、完成するのに数年かかり、しかも私たちは永遠に市場の許可を得て製品販売を実現するために必要なデータ或いは結果を生成できないかもしれない。しかも、私たちの候補製品が承認されれば、商業的成功を得られないかもしれない。私たちの商業収入は、もしあれば、数年以内に商業使用できないと予想される製品の販売から来ます。したがって、私たちは私たちの業務目標を達成するために多くの追加資金を得る必要があるだろう。
私たちが現在計画している業務に資金を提供する能力の予想は、リスクと不確実性の影響を受ける推定に基づいている。経営陣には現在未知の多くの要因があるため、私たちの運営計画は変化する可能性があり、現在の運営計画が予想される時間枠で実現される保証はなく、計画よりも早く追加資金を求める必要があるかもしれません。
受け入れ可能な条件で、私たちは十分な追加資金を得ることができないかもしれないし、全くないかもしれない。私たちは約束された外部資金源を持っていない。市場変動はまた私たちが必要な時に資本を得る能力に悪影響を及ぼすかもしれない。株式または転換可能な債務証券の売却によって調達された追加資本は、清算または他の特典を含む可能性がある。債務融資および優先株融資に関与する可能性のあるプロトコルは、追加債務の生成、我々の資産の売却、資本支出を行うことができるか、または配当を発表することができるかもしれないなど、私たちが具体的な行動をとる能力を制限または制限する契約を含み、株式承認証を発行する必要があるかもしれない。
もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に、あるいは私たちが受け入れ可能な条件下で株式または債務融資または他の手配を通じて追加資金を調達できない場合、私たちは、私たちの製品開発または将来の商業化努力を延期、制限、減少または終了することを要求されるか、または私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を付与することができるかもしれない。
契約義務
私たちはキャンセルできない経営レンタル方式である資産をレンタルして、レンタル期間は2032年までです。賃貸契約は主に事務空間と実験室空間に関するものだ。2022年12月31日現在、私たちがこれらのオフィスと実験室で借りた将来の最低約束総額は、関連する公共地域維持費や不動産税を含まない1兆192億ドルです。これらの賃貸契約の詳細については、付記7を参照されたい賃貸借証書私たちの連結財務諸表まで。
著者らは正常な業務過程中にCRO、CMOとその他の第三者と臨床試験、臨床前研究と製造サービスについて契約を締結した。このような契約には最低購入約束は含まれておらず、私たちは事前に書面で契約をキャンセルすることを通知することができる。キャンセル時に支払うべき金額には、キャンセル日までに提供されるサービスの支払いまたは発生した費用のみが含まれており、当サービス提供者のキャンセル不可義務が含まれています。
私たちはまた許可協定を締結し、このような合意に基づいて、私たちはいくつかのお金を支払う義務があるかもしれない。例えば、2021年11月、私たちはハーバード/遠大許可協定に基づいて遠大とハーバードに約630万ドルの成功裏の支払いを支払った。もし追加の成功した支払いをトリガすれば、ハーバード/遠大許可協定によると、私たちは遠大とハーバードに2500万ドルの追加費用を支払う義務があります。私たちの知的財産権許可協定には、協定によって許可された知的財産権を使用した製品の開発に依存し、開発または規制承認マイルストーンの実現、および商業および成功支払いマイルストーンに依存する潜在的なマイルストーン支払いが含まれています。このような支払い義務は将来発生する事件に依存しており,このような潜在的義務の時間や可能性は不明である。当社のライセンス契約およびこのようなプロトコルの下で将来支払われる可能性のある金額のより多くの情報については、“ビジネスライセンスおよび提携プロトコル”および付記8を参照されたい許可協定私たちの連結財務諸表まで。
新興成長型会社の地位
新興成長型会社、またはEGCとして、2012年にJumpStart Our Business Startups ActまたはJOBS Actによると、これらの基準が民間会社に適用されるまで、いくつかの会計基準の採用が延期される可能性がある。雇用法案に規定されている他の免除及び低減の報告要件は、最初の公募株式の登録説明書に2年間の監査済み財務諸表のみを列記することを含み、これは免除である
153
2002年の“サバンズ-オキシリー法案”第404(B)節の財務報告の内部統制に関する監査役報告の要求を受けず、上場企業会計監督委員会が採用する可能性のある強制監査会社のローテーションに関するいかなる要求の免除も受けず、当社の役員報酬スケジュールをそれほど広く開示しない。
また、雇用法案は、EGCは、新たなまたは改正された会計基準を遵守するために延長された過渡期を利用することができると規定している。この規定は、これらの基準が民間会社に適用されるまで、企業会計基準委員会が特定の会計基準の採用を延期することを可能にする。私たちは、私たち(I)がもはや新興成長型企業または(Ii)が“雇用法案”に規定されている延長移行期間から撤退することを明確かつ撤回できないまで、この延長された移行期間を使用して、新しい会計基準または改正会計基準を遵守することを選択することを選択した。したがって、我々の連結財務諸表は、上場企業の発効日までに新たなまたは改訂された会計声明を遵守している会社と比較できない可能性がある。
2026年12月31日までの会計年度が終了するまで、EGCに分類される可能性がありますが、それまでのいずれかの6月30日に、非関連会社が保有する私たちの普通株の時価が7億ドルを超えている場合、または任意の年度の年間毛収入が10.7億ドルを超えた場合、適用年度の12月31日から新興成長型会社ではなくなります。もし私たちが3年以内に10億ドルを超える転換不能債券を発行すれば、私たちはまたEGCではないだろう。
重要な会計政策と重大な判断
経営陣は私たちの財務状況と経営結果の討論と分析は私たちの連結財務諸表に基づいていて、私たちはアメリカ公認会計原則に従って作成しました。これらの財務諸表および関連開示を作成するためには、私たちが連結財務諸表中の資産、負債および費用の報告金額、または資産および負債の開示に影響を与える推定、判断、および仮説を作成することが要求される。我々は,歴史的経験,既知の傾向や事件,および当時の状況で合理的と考えられる様々な他の要因に基づいて推定し,これらの要因の結果は資産や負債の帳簿価値の判断の基礎を構成しているが,これらの資産や負債の帳簿価値は他の源から容易に見られるものではない.私たちは持続的な基礎の上で私たちの推定と仮定を評価する。異なる仮定や条件の下で、私たちの実際の結果はこれらの推定とは異なる可能性がある。
私たちの重要会計政策は私たちの総合財務諸表の付記2、重要会計政策の概要により詳細に記述されていますが、以下の会計政策は私たちが総合財務諸表を作成する際に使用する判断と推定に最も重要であると考えられます
収入確認
会計基準編纂またはASCトピック606の範囲での協力協定を締結しました取引先と契約した収入またはASC 606は、プロトコルに従って、我々のいくつかの候補製品に研究開発サービスを許可し、実行する。これらの取り決めの条項は、一般に、払い戻し不可能な前払い費用、研究開発コストの精算、開発、規制、および商業マイルストーン支払い、および製品の純売上を許可する印税のうちの1つまたは複数の費用を支払うことを含む。
ASC 606によれば、エンティティは、そのクライアントが約束された商品またはサービスの制御権を取得したときに収入を確認し、その金額は、エンティティがこれらの商品またはサービスと交換するために予期される対価格を反映する。ASC 606の範囲内の手配によって確認されるべき適切な収入金額を決定するために、(I)契約において約束された貨物またはサービスを決定するステップと、(Ii)契約において異なるかどうかを含む約束された貨物またはサービスが履行義務であるかどうかを決定するステップと、(Iii)可変対価格の制限を含む取引価格の測定と、(Iv)取引価格を履行義務に割り当てるステップと、(V)各履行義務を履行するときに収入を確認するステップと、の5つのステップを実行する。エンティティが顧客に譲渡された商品またはサービスと交換するために獲得する権利のある対価格を受け取る可能性がある場合にのみ、5段階モデルを契約に適用する。
154
私たちの手配で約束された商品またはサービスは、通常、私たちの知的財産権と研究開発サービスの許可権を含む。契約中の他の項目にオプションを提供し、顧客がこのようなオプションを行使することを選択した場合、これらのプロジェクトは、オプションが顧客に実質的な権利を提供しない限り、別個の契約として入金される。私たちは、顧客の物質的権利オプション、または無料または割引で追加の商品またはサービスを得るオプションを評価します。顧客選択権が実質的な権利を表すと決定された場合、実質的な権利は、手配の開始時に別個の履行義務として確認される。履行義務とは、(I)顧客が単独でまたは他のいつでも利用可能な資源と共に貨物またはサービスから利益を得ることができるとき、および(Ii)約束された貨物またはサービスが契約内の他の約束とは別に識別できる場合、異なるとみなされる、契約において一意の貨物またはサービスを顧客に譲渡することを約束する貨物またはサービスを意味する。約束された製品やサービスがユニークであるかどうかを評価する際には、基礎知的財産権の発展段階、顧客が自ら知的財産権を開発する能力、必要な専門知識がいつでも利用可能かどうか、製品やサービスと契約中の他の製品やサービスが不可分であるか、相互に依存しているかを考慮する。
私たちは譲渡契約で約束された貨物やサービスの予想受取額に基づいて取引価格を推定します。対価は固定対価または変動対価を含むことができる。可変価格を含む各手配の開始時に、潜在的な支払いの数と支払いを受ける可能性を評価します。我々は、最も可能な金額法または予想金額法を用いて予想受信金額を推定し、どの方法に基づいて予想受信金額を最も予測することができるか。取引価格に含まれる可変対価金額は制限される可能性があり,確認された累積収入金額が将来的に大きく逆転しない可能性が高い場合にのみ,取引価格に含まれる.
私たちの契約には通常、開発と規制マイルストーン支払いが含まれています。これらの支払いは最も可能な金額法によって評価され、重大な収入逆転が発生する可能性が高い場合、制限されます。規制承認のような我々または被許可者の制御範囲内でのマイルストーン支払いは、これらの承認を受ける前に実現可能であるとは考えられない。各報告期間が終了した時点で,吾らはこのような発展マイルストーンを達成する可能性や任意の関連制限を再評価し,必要に応じて全体の取引価格の見積りを調整する。いずれの調整も累積追跡に基づいて記録されており,これは調整期間中の協調収入に影響を与える.私たちはこれまで、私たちのどのような協力計画の実現に伴う開発、規制、またはビジネスマイルストーン収入の実現に関するいかなる考慮事項も確認していません。
販売ベースの特許使用料を含む手配については、販売レベルに基づくマイルストーン支払いを含み、許可が特許使用料に関連する主要項目とみなされる場合には、(I)関連販売が発生した場合、または(Ii)特許使用料の一部または全部が割り当てられた履行義務が履行された(または部分的に履行されている)ときに収入を確認する。これまで、私たちの任意の協力計画による販売ベースの印税収入に関する考慮事項は何も確認されていません。
私たちは契約義務ごとの推定独立販売価格に基づいて取引価格を分配します。契約で決定された契約義務ごとの独立販売価格を決定するために判断する必要がある仮説を立てなければならない。私たちは、他の比較可能な取引、取引交渉で考慮される価格設定、および推定コストを含む可能性があるサービス義務の独立販売価格を決定するためにキー仮説を使用する。また,材料権利の独立販売価格を決定する際には,比較可能取引,臨床試験成功確率,オプション行使可能性の推定を利用した。可変対価格は、契約のうちの1つまたは複数の履行義務に具体的に割り当てられ、可変対価格の条項が義務の履行状況に関連している場合、割り当てられた金額は、各義務を履行するために期待される金額と一致する。
関連貨物またはサービスの制御権が移転した場合には、履行義務毎に割り当てられた対価格が収入として確認される。ライセンスと他の承諾からなる履行義務については,合併履行義務が時間の経過とともに履行されているか,ある時点で履行されているかを決定する判断を用いて,合併履行義務の性質を評価し,時間の経過とともに進行を測定する適切な方法を決定する。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。
前金と費用は、これらの手配の義務を履行するまで、受領または満了時に繰延収入と表記されています。私たちの価格権が無条件の時、金額は売掛金として記録されます。
155
研究と開発費用を計算すべきである
連結財務諸表作成過程の一部として、何らかの計算すべき研究·開発費用を見積もる必要がある。このプロセスは、請求書を受信していない場合、または他の方法で実際のコストを通知する場合に、提供されるサービスのレベルおよびそのサービスのために生成される関連コストを推定することに関する。私たちは、私たちが当時知っていた事実と状況に基づいて、私たちの連結財務諸表において、各貸借対照表の日付までの課税費用を推定します。我々は定期的にサービスプロバイダと推定の正確性を確認し,必要に応じて調整する.計算すべき研究および開発費用の推定例には、以下の者に支払われる費用に関する費用が含まれる
サービス及び供給材料を提供する複数のCRO及びCMOとの見積及び契約に基づいて、受信したサービス及び費用の努力を推定し、契約研究及び製造に関する費用を記録する。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。サービス料を計算する際に、サービスを提供する時間帯と各時間の努力度を見積もります。サービスパフォーマンスの実際の時間や努力の程度が見積もり値と異なる場合は、対応する項目を調整します。実際に発生した金額と実質的に異なることはないと予想されるが、提供されたサービスの実際の状態および時間に対する提供サービスの状態および時間の理解が異なる可能性があり、報告された金額が任意の特定の時期に高すぎたり、過小になったりする可能性がある。これまで,我々が計算した研究や開発費の先行推定には何の大きな調整もなかった.私たちのほとんどのサービスプロバイダは、所定のスケジュールに従って、または契約マイルストーンに達したときに、提供されたサービスの借金の請求書を支払ってくれますが、一部のサービスプロバイダは前払いを必要としています。場合によっては、私たちのサプライヤーに支払われるお金は、提供されたサービスレベルを超え、事前支払い費用につながる可能性があります。私たちはこれらの費用を前払い費用として私たちの合併貸借対照表に記録します。
株に基づく報酬
我々は、従業員、取締役、コンサルタントまたは創業者に付与された株式オプションおよび他の株式ベースの報酬の公正価値に基づいて、彼らに付与された株式オプションおよび他の株式ベースの報酬を測定し、必要なサービス期間(通常は対応する報酬の帰属期間)内の株式ベースの報酬支出を確認する。私たちは罰金が発生しなかった時に確認します。
株式ベースの報酬報酬は、サービスまたは業績の帰属条件に基づいて制限される。費用確認の直線方法をすべてのサービス帰属に基づく報酬に適用し、付与日に基づいて株式に基づく業績奨励補償を確認し、サービス中に加速帰因法を用いて、業績条件が実現可能な場合に業績奨励の株式補償を確認する。
私たちはブラック-スコアーズオプション定価モデルを使用して、付与日に各株式オプション付与の公正価値を推定し、このモデルは、私たちの普通株式の公正価値、私たちの普通株式変動性の仮定、私たちの株式オプションの期待期限、私たちの株式オプションの期待期限に近い期間のリスク費用金利、および私たちの期待配当収益率を使用している。私たちの普通株の公正価値は制限株式奨励の公正価値を決定するために使用される。
私たちの普通株の授与日の終値を用いて、付与日制限株式単位の奨励の公正価値を推定します。
私たちの普通株は2021年6月に初めて公募される前に市場を公開しなかった。したがって、私たちが初めて公募する前に、私たちの普通株式の推定公正価値は、私たちの取締役会が各オプション付与日に決定したものであり、経営陣の意見は、私たちが最新に利用可能な第三者普通株推定値と、他の客観的かつ主観的要因の評価を考慮して、これらの要素は関連しており、最近の推定日から付与日まで変化した可能性があると考えられている。私たちが初めて株式を公募した後、私たちの普通株の公正価値は私たちの普通株の見積市場価格によって決定されました。
156
公正価値計量
成功支払責任
私たちが最初の10-Q四半期報告書を提出して以来、私たちの平均時価が指定されたハードルを超えて、9桁に達するドルの金額から100億ドルに上昇したり、これらのハードルを超えて私たちの会社を売却したりすれば、私たちはハーバードと遠大に成功金を支払わなければならない。当社の支配権の変更や当社が売却された場合、当社は事件発生後の指定期間内に現金で支払う必要があります。そうでなければ、私たちは現金または普通株、または現金と普通株の組み合わせでお金を支払うことを選択することができる。成功した支払いはASC 815項目に記載されている派生ツールおよびヘッジそして、最初に公正価値によって記録し、それに応じて研究開発費を計上した。公正価値のいずれの後続変動も、財務諸表の他の収入(費用)で確認される。支払い義務の達成または満期の早い者まで、価値変動を公正に許可する負債を調整し続ける。成功した支払いの推定公正価値を決定するために、イベント発生確率、イベント発生時間、および私たちの普通株式価値を含むいくつかのキー変数に基づいて負債価値をモデル化するモンテカルロシミュレーションモデルを使用した。
最近発表された会計声明
付記2を参照して、本年度報告書10-K表末の総合財務諸表付記には、“重要会計政策概要--最近発表された会計公告”が添付されている。
第七A項。数量と品質開示市場リスクについて。
金利リスク
私たちは金利の変化と関連した市場リスクに直面している。2022年12月31日現在、米国政府支援証券および国債に主に投資される標準小切手口座および通貨市場口座基金を含む1億154億ドルの現金および現金等価物を持っている。また、2022年12月31日現在、米国債や機関証券を含む4億394億ドルの有価証券を保有している。金利収入は一般的な金利レベルの変動に非常に敏感であるが、私たちの現金等価物の満期日が短いことと、私たちの有価証券のリスクが低いため、金利が直ちに10%変化することは、私たちの現金等価物と有価証券の公平な市場価値に大きな影響を与えない。
外貨両替リスク
私たちは現在、外貨為替レートの変化に関する重大な市場リスクに直面していません。しかし、私たちは確かにアメリカ以外のサプライヤーと契約を締結しており、これらのサプライヤーは外貨為替レートの変動の影響を受ける可能性があります。私たちは将来アメリカ以外のサプライヤーと追加の契約を締結するかもしれません。これは私たちの外貨両替リスクを増加させるかもしれません。
インフレ率
インフレは一般的に私たちの労働コストと目標開発コストを増加させることで私たちに影響を及ぼす。2022年12月31日までの1年間、インフレは我々の業務、財務状況、あるいは運営結果に実質的な影響を与えないと考えられる。
157
項目8.財務諸表と補足データ
第8項の要求に基づいて作成された財務諸表は、本年度報告第15項の表格10−Kから引用され、F−1ページから列報される。
項目9.変更と異議会計と財務開示専門。
ない。
第9条。制御とプログラム.
開示制御とプログラムの有効性に関する結論
我々は、米国証券取引委員会に提出された定期的かつ現在の報告で開示すべき情報が、米国証券取引委員会規則および表で指定された期間内に記録、処理、集約および報告されることを保証し、必要に応じて必要な開示に関する決定をタイムリーに行うために、必要に応じて私たちの管理層に伝達されることを保証するために、開示制御および手続きを維持する。開示制御およびプログラムを設計および評価する際、管理層は、任意の制御およびプログラムは、設計および動作がどんなに良好であっても、予想される制御目標を達成するために絶対的な保証ではなく合理的な保証を提供することしかできないことを認識している。合理的な保証レベルを達成するためには、管理層は必然的にその判断を用いて可能な制御とプログラムのコスト-利益関係を評価する必要がある。さらに、任意の制御システムの設計も、将来のイベント可能性のいくつかの仮定に部分的に基づいており、任意の設計がすべての潜在的な将来の条件でその目標を成功的に達成する保証はなく、時間の経過とともに、制御が条件の変化によって不十分になる可能性があり、またはポリシーまたはプログラムを遵守する程度が悪化する可能性がある。費用対効果を有する制御システムの固有の制限により、エラーまたは詐欺によるエラー陳述が発生し、発見されない可能性がある。
我々の経営陣は、最高経営責任者及び最高財務官の参加の下、本年度報告書に記載されている期間が終了した時点で、取引所法第13 a−15(E)及び15 d−15(E)規則に基づいて定義された開示制御及び手続の有効性を評価した。同等の評価によると、我々の主要行政総裁及び主要財務官は、当該日までに、我々の開示制御及び手続が合理的な保証レベルで有効であると結論した。
経営陣財務報告内部統制年次報告書
我々の経営陣は、財務報告書の十分な内部統制の確立と維持を担当している(取引法第13 a-15(F)及び15 d-15(F)条に記載されている)。我々の財務報告の内部統制は、我々の主要執行者及び主要財務官の監督の下で設計されたプログラムであり、我々の財務報告の信頼性の合理的な保証を提供し、公認された会計原則に基づいて外部目的の財務諸表を作成する。経営陣は2022年12月31日までの財務報告書の内部統制を評価した。経営陣は、テレデビル委員会後援組織委員会が発表した“内部統制--総合枠組み”(2013年枠組み)で決定された基準に基づいて評価を行った。この評価に基づき、我々の経営陣は、財務報告書の内部統制が2022年12月31日から有効であると結論した。
公認会計士事務所認証報告
雇用法案による“新興成長型会社”の免除により、本年度報告にはわが公認会計士事務所の認証報告は含まれていません
財務報告の内部統制の変化
ルール13 a-15(F)および15 dで定義されているように、財務報告の内部統制に変化はありません‑15(F)は、2022年12月31日までの3ヶ月以内に、財務報告の内部統制に重大な影響を及ぼすか、または合理的に可能性がある。
158
プロジェクト9 B。他の情報。
ない。
プロジェクト9 Cです。情報開示報告検査を阻止する外国司法管轄区域を分類する。
適用されません。
159
第三部
プロジェクト10.役員、執行幹事a会社を管理しています
本第10項で要求される情報は、米国証券取引委員会または米国証券取引委員会に提出される2023年株主年次総会に関する最終委託書に、“第1号提案−第2種取締役の選出”、“コーポレート·ガバナンス-商業行為及び道徳基準”、“コーポレート·ガバナンス-取締役会委員会”、“コーポレートガバナンス-報酬委員会のインターロック及び内部人参加”、“延滞第16条(A)条報告書”(適用される場合)と題する章に含まれる。
私たちは、私たちの最高経営責任者、最高財務官、最高会計官、財務総監、または同様の機能を実行する者を含む、私たちのすべての役員、高級管理者、および従業員に適用される書面による商業行為および道徳的規則を採択した。規則の最新コピーは、Investors.vervetx.comのウェブサイトの投資家部分で取得できます。私たちは米国証券取引委員会規則に基づいて開示されなければならない私たちのコードの任意の修正または免除を私たちのウェブサイトで開示するつもりだ。
プロジェクト11.役員報酬.
第11項の要件の情報は、S-K規則402(V)項に従って開示された情報に加えて、必要であれば、参照によって本明細書に組み込まれる、2023年年次総会に関する取締役に提出された最終委託書の“役員および米国証券取引委員会報酬”と題する部分に含まれる。
プロジェクト12.特定の保証所有権実益所有者と経営陣と関連株主について。
本第12項に要求される情報は、米国証券取引委員会2023年株主総会について米国証券取引委員会に提出された最終委託書に“役員および米国証券取引委員会報酬-株式報酬計画情報”および“特定の利益所有者および管理層の保証所有権”と題する部分に含まれ、引用によって本明細書に組み込まれる。
プロジェクト13.特定の関係や関連取引や役員が独立しています
本第13条に要求される情報は、2023年年次総会に関する取締役に提出された最終委託書の“関係者との取引”及び“コーポレート·ガバナンス−米国証券取引委員会独立性”と題する部分に含まれ、引用により本明細書に組み込まれる。
プロジェクト14.主な課金サービスしています
本第14項に要求される情報は、米国証券取引委員会に提出された2023年株主総会に関する最終委託書の“第2号提案である独立公認会計士事務所の承認”と題する部分に含まれ、引用により本明細書に組み込まれる。
160
第4部
プロジェクト15.物証、財務諸表スケジュールです。
1. 財務諸表。
Verve Treateutics,Inc.の財務諸表および独立公認会計士事務所安永会計士事務所(PCAOB ID:00)の財務諸表
2. 財務諸表明細書。
財務諸表の添付表は、必須でないか、適用されないか、またはこれらの情報が連結財務諸表またはその付記に含まれているので省略されている。
3. 陳列品
展示品 番号をつける |
|
説明する |
3.1 |
|
再記載された登録者登録証明書は、2021年6月21日から発効する(添付ファイル3.1を参照して登録者の現在の報告書に組み込まれているタブ8-K、ファイル番号001-40489、2021年6月21日に提出) |
3.2 |
|
2回目の改訂及び再改訂された登録者規約は、2023年2月14日から施行される(添付ファイル3.1を参照して登録者2023年2月17日に提出されたタブ8−K、書類番号001−40489の現在の報告書に組み込まれる。) |
4.1 |
|
普通株式株式を証明する株式サンプル証明書(2021年5月28日に提出されたS-1表登録宣言書類第333-256608号の添付ファイル4.1を参照して組み込む) |
4.2 |
|
第二次改正及び再署名された投資家権利協定は、期日が2021年1月14日であり、登録者及びその他の当事者によって署名される(2021年5月28日に提出された登録者登録声明表S-1の333-256608書類添付ファイル10.1を参照して編入) |
4.3 |
|
取引法第12条に基づいて登録された証券説明(登録者が2022年3月14日に提出した10−K表年次報告書の添付ファイル4.3参照) |
10.1# |
|
2018年株式インセンティブ計画(2021年5月28日に提出されたS-1表登録声明ファイル333-256608号の添付ファイル10.2を参照して編入) |
10.2# |
|
2018年株式インセンティブ計画下の株式オプション契約表(2021年5月28日に提出されたS-1表登録声明の添付ファイル10.3を参照して登録者登録声明、文書番号333-256608) |
10.3# |
|
2021年株式インセンティブ計画(2021年6月14日に提出されたS-1表第333-256608号登録声明登録者修正案第1号添付ファイル10.4参照) |
10.4# |
|
2021年株式インセンティブ計画下の株式オプション契約表(添付ファイル10.4を参照して登録者が2022年3月14日に提出した10-K表年次報告書に編入) |
10.5#* |
|
“2021年株式インセンティブ計画”制限株式単位プロトコルフォーマット |
10.6# |
|
2021年従業員株式購入計画の改訂と再作成(2021年6月14日に提出された登録者による登録声明の第1号修正案の添付ファイル10.7を参照してS-1表に組み込まれ、書類番号333-256608) |
10.7* |
|
非従業員役員報酬計画の概要 |
10.8 |
|
改正及び再署名された協力及び許可協定は、期日が2022年7月5日であり、登録者とビム治療会社との間の協力及び許可協定(2022年11月7日に提出された登録者四半期報告書10-Q表の添付ファイル10.2を参照して編入) |
10.9 |
|
2020年10月6日までの“開発·選択協定”の改正·再署名、由和 登録者とAcuitas Treateutics,Inc.(登録者登録宣言の添付ファイル10.10を参照してS−1表に組み込まれ、文書番号333−256608、2021年5月28日に提出) |
10.10 |
|
非排他的許可協定は、日付が2020年10月14日であり、登録者及びAcuitas Treateutics,Inc.(登録者の添付ファイル10.11を参照することにより編入される 表S-1登録声明、アーカイブ番号333-256608、2021年5月28日提出) |
161
10.11 |
|
CAS 9許可協定は、日付が2019年3月15日であり、登録者、ハーバード大学、ブロイド研究所の総裁と研究員によって署名され、修正された(登録者登録声明を引用することによりS-1表アーカイブ添付ファイル10.12に編入される No. 333-256608, filed May 28, 2021) |
10.12 |
|
登録者とVertex PharmPharmticals Inc.との間の戦略的協力および許可協定は、2022年7月18日(登録者を引用して2022年11月7日に提出されたForm 10-Q四半期報告書の添付ファイル10.3に編入される) |
10.13 |
|
賃貸契約は,期日は2021年8月19日であり,登録者とARE-MA地域第87号の間で締結される テナント有限責任会社(登録者が2021年11月10日に提出した10-Q表四半期報告添付ファイル10.1登録成立参照) |
10.14 |
|
“賃貸借契約第一修正案”は、期日が2022年1月4日であり、登録者及びAre−MA地域第87号テナント有限責任会社(2022年3月14日に提出された登録者年次報告表格10−Kの添付ファイル10.14を参照して編入) |
10.15 |
|
第二次リース改正案は,期日は2022年6月17日であり,登録者とAre−MA Region No,87 Tenant,LLC(2022年8月9日に提出された登録者四半期報告10-Q表の添付ファイル10.2を引用して合併した) |
10.16# |
|
登録者とその執行者との間の賠償協定フォーマット、及び 取締役(2021年5月28日に提出されたS-1表、書類番号333-256608、登録者登録声明の添付ファイル10.17を引用して統合) |
10.17# |
|
登録者とSekar Kathiresan,M.D.との間の雇用契約は,2021年6月11日(2021年6月14日に提出された表S−1登録声明の第1号改正案添付ファイル10.18を参照して登録者修正案,333−256608号文書に編入) |
10.18# |
|
登録者とアンドリュー·アッシュ,J.D.が2021年6月11日に締結した雇用契約(2021年6月14日に提出された登録者S-1表登録声明修正案第1号添付ファイル10.19を参照して編入、文書番号333-256608) |
10.19# |
|
登録者とAndrew Bellinger,M.D.,Ph.D.が2021年6月11日に締結した雇用契約(2021年6月14日に提出された登録者S-1表第1号修正案の添付ファイル10.20を参照してS-1表に組み込まれ、文書番号333-256608) |
10.20# |
|
登録者とアリソン·ドバルとの雇用契約は,2021年11月26日(添付ファイル10.19を参照して登録者が2022年3月14日に提出した10−K表年次報告書に組み込まれている) |
10.21 |
|
公開市場販売協定SM登録者とジェフリー有限責任会社との間で署名され、日付は2022年7月1日(登録者を引用して2022年7月1日に提出されたS-3表登録声明の添付ファイル1.2合併、書類番号333-265996) |
10.22* |
|
登録者とVertex PharmPharmticals Inc.との間の株式購入契約は,2022年7月18日である |
21.1 |
|
登録者の子会社(引用21.1により登録者に合併する 表S-1登録声明、アーカイブ番号333-256608、2021年5月28日提出) |
23.1* |
|
独立公認会計士事務所安永法律事務所同意 |
31.1* |
|
2002年サバンズ-オキシリー法第302節で可決された1934年“証券取引法”第13 a-14(A)及び15 d-14(A)条に従って発行された最高経営責任者証明書 |
31.2* |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
32.1** |
|
2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条に規定されている最高経営責任者証明書。 |
32.2** |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による首席財務官の認証。 |
101.INS |
|
連結されたXBRLインスタンス文書−インスタンス文書は、XBRLタグがイントラネットXBRL文書に埋め込まれているので、相互作用データファイルには表示されない。 |
101.書院 |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
101.カール |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
101.def |
|
インラインXBRL分類拡張Linkbase文書を定義する |
101.介護会 |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
101.Pre |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
104 |
|
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
162
*アーカイブをお送りします。
**関数で提供されます。
S-K条例第601(B)(10)(Iv)項によれば、本展示品の部分は省略されている。
#は、管理契約または補償計画を示します。
項目16.表格10-Kの概要
ない。
163
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
|
|
VERVE治療会社 |
|
|
|
|
|
日付:2023年3月2日 |
|
差出人: |
/s/Sekar Kathiresan |
|
|
|
セカール·カイザー·レイサン |
|
|
|
最高経営責任者 首席執行幹事 |
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
/s/Sekar Kathiresan |
|
最高経営責任者 |
|
March 2, 2023 |
セカール·カイザー·レイサン |
|
(首席行政主任) |
|
|
|
|
|
|
|
/s/アリソン·ドバル |
|
首席財務官 |
|
March 2, 2023 |
アリソン·ドバル |
|
(首席財務官と首席会計官) |
|
|
|
|
|
|
|
/s/Burt Adelman |
|
役員.取締役 |
|
March 2, 2023 |
バート·アデルマン |
|
|
|
|
|
|
|
|
|
/s/ロングコート |
|
役員.取締役 |
|
March 2, 2023 |
ロンネルのコート |
|
|
|
|
|
|
|
|
|
/s/Alexander Cumbo |
|
役員.取締役 |
|
March 2, 2023 |
アレクサンダー·カンポ |
|
|
|
|
|
|
|
|
|
/s/マイケル·マクレーン |
|
役員.取締役 |
|
March 2, 2023 |
マイケル·マクレーン |
|
|
|
|
|
|
|
|
|
/s/ヒラ·ミハイル |
|
役員.取締役 |
|
March 2, 2023 |
ヒラ·ミハイル |
|
|
|
|
|
|
|
|
|
/s/Krishna Yeshwant |
|
役員.取締役 |
|
March 2, 2023 |
Krishna Yeshwant |
|
|
|
|
164
連結財務諸表索引
|
ページ |
|
|
監査済み財務諸表 |
|
|
|
独立公認会計士事務所レポート(PCAOB ID番号0042) |
F-2 |
|
|
合併貸借対照表 |
F-3 |
|
|
合併経営報告書と全面赤字 |
F-4 |
|
|
転換可能優先株と株主権益連結報告書(損失) |
F-5 |
|
|
統合現金フロー表 |
F-7 |
|
|
連結財務諸表付記 |
F-8 |
F-1
Indepの報告公認会計士事務所
Verve治療会社の株主と取締役会に。
財務諸表のいくつかの見方
Verve Treateutics,Inc.(当社)2022年12月31日までと2021年12月31日までの連結貸借対照表,2022年12月31日までの3年間の年間関連総合経営表と全面赤字,転換可能優先株と株主権益(赤字)と現金流量および関連付記(総称して“総合財務諸表”と呼ぶ)を監査した。総合財務諸表は、すべての重要な面で、2022年12月31日、2022年12月31日、2021年12月31日までの会社の財務状況、および2022年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
ASU番号2016-02を採用
連結財務諸表付記2で述べたように、会計基準更新(ASU)第2016−02号を採用したため、当社は2021年にリース会計方法を変更した賃貸借証書(主題842)、および関連する修正案。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性に対する意見を表明するためではありません。したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/s/
2020年以来、当社の監査役を務めてきました。
March 2, 2023
F-2
VERVE治療会社
合併貸借対照表
|
|
十二月三十一日 |
|
|||||
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない) |
|
2022 |
|
|
2021 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
有価証券 |
|
|
|
|
|
|
||
協同して応収する |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
その他長期資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用を計算する |
|
|
|
|
|
|
||
賃貸負債、当期分 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
長期賃貸負債 |
|
|
|
|
|
|
||
成功支払責任 |
|
|
|
|
|
|
||
収入を繰延し、流動ではない |
|
|
|
|
|
|
||
その他長期負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
優先株、$ |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
F-3
VERVE治療会社
合併経営報告書と全面赤字
|
|
十二月三十一日までの年度 |
|
|||||||||
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない) |
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
協力収入 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
|
|
|
|
|
|
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
総運営費 |
|
|
|
|
|
|
|
|
|
|||
運営損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他の収入(支出): |
|
|
|
|
|
|
|
|
|
|||
優先株部分負債の公正価値変動 |
|
|
|
|
|
|
|
|
|
|||
反希釈権利負債の公正価値変動 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
成功支払負債の公正価値変動 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
利息とその他の収入,純額 |
|
|
|
|
|
|
|
|
|
|||
その他の収入を合計して純額 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
所得税準備前の損失を差し引く |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
所得税支給 |
|
|
( |
) |
|
|
|
|
|
|
||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
普通株株主は普通株1株当たり純損失、基本損失と希釈損失を占めなければならない |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
加重平均普通株、普通株株主は1株当たり純損失、基本損失と希釈損失に用いる |
|
|
|
|
|
|
|
|
|
|||
総合的な損失: |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
その他の全面的な損失: |
|
|
|
|
|
|
|
|
|
|||
有価証券は赤字を実現していない |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
総合損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
付記はこれらの連結財務諸表の構成要素である。
F-4
VERVE治療会社
転換可能な優先株合併報告書株と株主権益
F-5
|
|
交換可能優先 |
|
|
|
普通株 |
|
|
その他の内容 |
|
|
積算 |
|
|
|
|
|
合計する |
|
||||||||||||||
(共有を除いて千単位で |
|
株 |
|
|
金額 |
|
|
|
株 |
|
|
金額 |
|
|
支払い済み |
|
|
全面的に |
|
|
積算 |
|
|
株主権益 |
|
||||||||
2019年12月31日の残高 |
|
|
|
$ |
|
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
||||||
Aシリーズ転換優先株を発行して#ドルのバッチ権利債務を返済する |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
A-2シリーズ転換可能優先株を発行し、発行コストを差し引く$ |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
許可機関に普通株を増発する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
制限された普通株の帰属 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
証券売却可能な未実現損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
株式オプションの行使 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2020年12月31日残高 |
|
|
|
$ |
|
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
||||||
Bシリーズ転換優先株を発行し、発行コストを差し引く$ |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
||
初公開終了時に優先株を普通株に変換することができます |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
初公募株から普通株を発行し、発行コストを差し引く$ |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
許可機関に普通株式を発行する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
制限された普通株の帰属 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
0 |
|
|
|
— |
|
|
|
— |
|
|
株式オプションの行使 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
ESPPにより普通株を購入する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
証券売却可能な未実現損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2021年12月31日の残高 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
株式オプションの行使 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株式単位の帰属を制限する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
ESPPにより普通株を購入する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
頂点合意に関する普通株式を発行する |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
後続公開時に普通株を発行し、発行コストを差し引いて#ドル |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
市場から普通株を発行し,発行コストを差し引くと#ドルになる |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
証券売却可能な未実現損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
株に基づく報酬 |
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
2022年12月31日の残高 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
付記はこれらの連結財務諸表の構成要素である。
F-6
VERVE治療会社
C統合現金フロー表
|
|
現在までの年度 |
|
|||||||||
(単位:千) |
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
経営活動のキャッシュフロー: |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営活動で使用される現金純額の調整: |
|
|
|
|
|
|
|
|
|
|||
減価償却 |
|
|
|
|
|
|
|
|
|
|||
非現金レンタル費用 |
|
|
|
|
|
|
|
|
|
|||
有価証券割増の純償却 |
|
|
( |
) |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
|
|
|
|||
優先株部分負債の公正価値変動 |
|
|
|
|
|
|
|
|
( |
) |
||
反希薄化権利の公正価値変動 |
|
|
|
|
|
|
|
|
|
|||
成功支払負債の公正価値変動 |
|
|
( |
) |
|
|
|
|
|
|
||
経営性資産と負債変動状況: |
|
|
|
|
|
|
|
|
|
|||
協同して応収する |
|
|
( |
) |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
売掛金 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
費用とその他の負債を計算すべきである |
|
|
|
|
|
|
|
|
|
|||
成功支払責任 |
|
|
|
|
|
( |
) |
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
|
|
|
|||
リース負債を経営する |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
繰延賃料負債 |
|
|
|
|
|
|
|
|
|
|||
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
投資活動によるキャッシュフロー: |
|
|
|
|
|
|
|
|
|
|||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券の満期日 |
|
|
|
|
|
|
|
|
|
|||
投資活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
融資活動によるキャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
優先株発行で得られた金の純額 |
|
|
|
|
|
|
|
|
|
|||
普通株を発行して得られる収益は,発行コストを差し引く |
|
|
|
|
|
|
|
|
|
|||
頂点合意に関する普通株式を発行して得られる収益 |
|
|
|
|
|
— |
|
|
|
— |
|
|
持分発行費用を支払う |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
株式オプションを行使して得られる収益 |
|
|
|
|
|
|
|
|
|
|||
従業員による株購入計画による株購入の収益 |
|
|
|
|
|
|
|
|
|
|||
融資活動が提供する現金純額 |
|
|
|
|
|
|
|
|
|
|||
現金、現金等価物、および制限現金の増加 |
|
|
|
|
|
|
|
|
|
|||
現金、現金等価物、制限現金--期初 |
|
|
|
|
|
|
|
|
|
|||
現金、現金等価物、制限された現金--期末 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
非現金投資と融資活動を追加開示します |
|
|
|
|
|
|
|
|
|
|||
売掛金及び売掛金に含まれる財産及び設備の増加 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
分権債務弁済 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
初公開終了時に優先株を普通株に変換することができます |
|
$ |
|
|
$ |
|
|
$ |
|
|||
普通株を発行することで派生債務を解決する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
新しい経営リース負債と引き換えに使用権資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
付記はこれらの連結財務諸表の構成要素である。
F-7
VERVE治療会社
連結財務諸表付記
1. 業務の性質と列報根拠
組織する
Verve治療会社(“会社”あるいは“Verve”)は臨床段階の遺伝子薬物会社であり、心血管疾患を治療する新しい方法を開拓し、治療を慢性治療から単一治療コースの遺伝子編集薬物に転換する。同社は2018年3月9日に設立され、Endcadia,Inc.というデラウェア州の会社で、その後間もなく運営を開始した。2019年1月、会社は会社登録証明書を修正し、Verve Treateutics,Inc.と改称しました。会社の主な事務所はマサチューセッツ州ボストンにあります。
流動資金と資本資源
設立以来、会社は主に研究開発と資金調達に取り組んできた。同社は生物技術業界の早期会社によく見られるリスクと不確定要素の影響を受け、これらのリスクと不確定要素は候補製品の成功研究、開発と製造に関連する技術リスク、競争相手の新技術革新の開発、肝心な人員への依存、ノウハウの保護、政府法規の遵守及び追加資本を獲得して運営を援助する能力を含むがこれらに限定されない。現在と未来のプロジェクトは広範な臨床前と臨床テスト及び商業化前の監督管理承認を含む大量の研究と開発努力が必要となる。このような努力は多くの追加資本、十分な人員とインフラ、そして広範囲なコンプライアンス報告能力を必要とする。同社の開発努力が成功しても、同社がいつ(あれば)製品販売から相当な収入を得るかは定かではない。
付随する総合財務諸表は持続経営を基礎として作成され、正常業務過程における資産の現金化及び負債の返済及び負担を考慮している。当社は現金、現金等価物及び有価証券を期待しています$
陳述の基礎
添付されている総合財務諸表は、米国公認会計原則(“公認会計原則”)に基づいて作成される。本付記内の適用指針に対するいかなる言及も財務会計基準委員会(“FASB”)の会計基準編纂(“ASC”)及び“会計基準更新”(“ASU”)に掲載されている権威公認会計原則を指す。
2. S重要会計政策の概要
合併原則
予算の使用
♪the the the公認会計基準に従って財務諸表を作成することは、報告期間及び報告期間内に報告された資産、負債及び費用額、又は有資産及び負債の開示に影響を与えるために、管理層に推定と仮定を要求する。当社は過去の経験に基づいて推定及び仮説を立て,その根拠と考えている
F-8
現金と現金等価物
制限現金
限定現金とは、会社がその会社の施設をレンタルすることに関連する保証金として発行された信用状のための担保である
|
|
十二月三十一日 |
|
|||||
(単位:千) |
|
2022 |
|
|
2021 |
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
制限現金 |
|
|
|
|
|
|
||
現金総額、現金等価物、および限定現金 |
|
$ |
|
|
$ |
|
||
有価証券
有価証券の公正価値が償却コストよりも低く、有価証券の帳簿価値が合理的な時間内に回収できないという証拠がある限り、会社は有価証券の非一時的減値を審査する。会社に信用損失が発生した場合、有価証券を意図的に売却したり、会社が償却コストベースで回収する前に有価証券を売却する必要がある可能性があれば、投資の非一時的減値は総合経営報告書で確認される。今回の評価で考慮した証拠には,減値原因,当社の投資政策の遵守状況,減値の重症度と持続時間および期末後の価値変化がある。同社はその証券を非一時的減値評価を行い,証券時価低下の要因は現在の経済と市場状況であると考えている。当社がこの証券の売却を要求される可能性はあまりなく、当社は償却コストベースで回収する前にその証券を売却するつもりはありません。この分析によると,これらの有価証券は一時的に減価されたとは考えられない2022年12月31日と2021年12月31日.
信用リスクが集中する
会社を深刻な集中信用リスクに直面させる可能性のある金融商品は主に現金、現金等価物、有価証券、制限された現金を含む。当社は定期的に金融機関で政府の保険限度額を超える預金を維持することができます。経営陣は、当社の預金は経営陣が高い信用要素を持つと考えている金融機関に保管されているため、当社には重大な信用リスクはないと信じているが、当社は当該等の預金によって何の損失も受けていない。
♪the the the同社は通常、余分な資本を通貨市場基金、米国国庫券、機関証券に投資しており、これらはすべて最小の信用と市場リスクの影響を受けている。ポートフォリオは
F-9
一致する同社の投資政策は許可された投資を定義し、信用品質基準を規定し、いかなる単一発行者の信用開放も制限している。
繰延発売コスト
株式発行が完了する前に、同社は株式発行に直接関連する逓増法律、専門会計、その他の第三者費用を他の非流動資産として資本化する。発売完了後、これらのコストは株主権益(損失)に計上され、発売による追加実収資本の減少となる。
金融商品の公正価値
ASCテーマ820、公正価値計量(“ASC 820”)は、公正価値計量のためのツールのための公正価値レベルを確立し、市場データに基づく仮説(観察可能な投入)と会社自身の仮説(観察不可能な投入)とを区別する。観察できる投入は,市場参加者が当社以外のソースから得られた市場データに基づいて資産や負債を定価する際に使用する投入である。観察できない投入は、市場参加者が資産や負債の価格設定のために使用されるという会社の仮定を反映し、その時点で入手可能な最適な情報に基づいて制定される。ASC 820は、公正価値を、計量日市場参加者間の秩序ある取引において資産を売却することによって受信された価格または負債を移動させることによって支払われた価格として決定する。公正価値計量において市場参加者の仮説を考慮する基礎として、ASC 820は以下の項目を区別した三級価値階層構造を構築した
第1レベル-同じ資産または負債の活発な市場見積もり。
第2レベル-第1レベル以外の直接または間接的に観察可能な投入、例えば見積市場価格、金利、収益率曲線。
第3級-資産または負債の観察不可能な投入(すなわち、市場活動の支援が少ないか、またはない)。第3レベルの投入には、管理層自身が資産または負債のために価格を設定する際に市場参加者自身が使用する仮説(リスクの仮定を含む)が含まれる。
推定値が市場で観察または観察できないモデルまたは投入に基づく場合、公正価値の決定にはより多くの判断が必要である。そのため、当社が価値を公平に決定する際に下した判断程度は、第3級のツールに分類されることが最大である。公正価値レベル内の金融商品レベルは、公正価値計量に重大な意義がある任意の投入中の最低レベルに基づいている。
財産と設備、純額
財産と設備はコストから減価償却累計を引いて申告する
資産種別 |
|
使用寿命を見込む |
コンピュータ装置及びソフトウェア |
|
|
事務家具 |
|
|
実験室装置 |
|
|
賃借権改善 |
|
長期資産減価準備
♪the the the事件や環境変化が長期資産の額面が回収できない可能性があることを示した場合、当社はその長期資産の減値を評価する。保有·使用する資産の回収可能性は,資産の帳簿価値と資産予想による将来の未割引現金流量を比較することで測定した。このような資産が減値とみなされる場合、減値は
F-10
独立した金融商品と派生商品
当社は、貸借対照表に負債記録として、公正価値に応じて個別に計算する金融商品を確認した。
優先株部分負債会社は、その後の成約に応じて、Aシリーズ優先株(“Aシリーズ優先株”)の追加株式を発行する義務と、会社投資家がAシリーズ優先株を購入する権利が独立した金融商品であることを決定した。独立優先株部分負債(“部分負債”)は最初に公正価値で入金され,公正価値変動による損益は経営報告書中の他の収益(費用)と全面損失で確認された。分割払い負債は、各報告期間および債務行使または満了時に再計量される。2020年12月31日現在、Aシリーズ優先株はすべて閉鎖され、優先株部分の債務はすべて返済されている。より多くの議論については、付記9、優先株部分負債を参照してください。
(I)ハーバード学院(“ハーバード”)総裁及び研究員及び遠大研究院有限会社(“遠大”)との許可協定(“ハーバード/遠大許可協定”)及び(Ii)遠大許可協定(“遠大許可協定”)(付記8、許可協定参照)に基づいて、当社は以下の金融商品を発行する。
権利を逆に薄める·逆希釈権利とは、同社の初公募株を含む追加融資を完了した後、ハーバードおよびブロダー社に普通株を増発する義務である。これらの逆希釈権利はASC 815に規定されている派生ツールおよびヘッジ("ASC 815"),そして、初歩的に公正価値に従って入金し、相応して研究開発費用を計上した。負債は報告期間ごとに再計量され,公正価値変動は経営報告書中の他の収入(費用)で確認され,そのツール未返済期間に全面損失が確認された発行された総額は
収入確認
同社は、ASC主題606“顧客との契約収入”(“ASC 606”)の範囲に属する協力協定を締結し、同協定に基づいて、会社のいくつかの候補製品に権利を付与し、研究開発サービスを提供する。これらの取り決めの条項は、一般に、払い戻し不可能な前払い費用、研究開発コストの精算、開発、規制、および商業マイルストーン支払い、および製品の純売上を許可する印税のうちの1つまたは複数の費用を支払うことを含む。
ASC 606によれば、エンティティは、そのクライアントが約束された商品またはサービスの制御権を取得したときに収入を確認し、その金額は、エンティティがこれらの商品またはサービスと交換するために予期される対価格を反映する。ASC 606の範囲内で確認すべき適切な収入額が決定されたことを決定するために、会社は、(I)契約において約束された貨物またはサービスを決定するステップと、(Ii)契約において異なるかどうかを含む約束された貨物またはサービスが履行義務であるかどうかを決定するステップと、(Iii)可変対価格の制限を含む取引価格の測定と、(Iv)取引価格を履行義務に割り当てるステップと、(V)会社が各履行義務を履行するときに収入を確認するステップと、の5つのステップを実行する。♪the the the
F-11
エンティティが顧客に譲渡された商品またはサービスと交換するために獲得する権利のある対価格を受け取る可能性がある場合にのみ、会社は5ステップモードを契約に適用する。
会社の手配で約束された貨物またはサービスには、通常、会社の知的財産権と研究開発サービスの許可権が含まれる。当社は、契約中の他の項目にオプションを提供し、顧客がこのようなオプションを行使することを選択した場合、これらの項目は、オプションが顧客に実質的な権利を提供しない限り、別個の契約として入金される。同社は、顧客の物質的権利選択を評価するか、または追加商品またはサービスの選択を無料または割引して得る。顧客選択権が実質的な権利を表すと決定された場合、実質的な権利は、手配の開始時に別個の履行義務として確認される。履行義務とは、(I)顧客が単独でまたは他のいつでも利用可能な資源と共に貨物またはサービスから利益を得ることができるとき、および(Ii)約束された貨物またはサービスが契約内の他の約束とは別に識別できる場合、異なるとみなされる、契約において一意の貨物またはサービスを顧客に譲渡することを約束する貨物またはサービスを意味する。承諾された商品やサービスが独特であるかどうかを評価する際に、当社は、基礎知的財産権の発展段階、顧客が自ら知的財産権を開発する能力または必要な専門知識が既製品であるかどうか、およびそのような商品またはサービスが契約中の他の商品またはサービスと不可分であるかどうか、または関連する要素を考慮する。
当社は譲渡契約で約束された貨物またはサービスの予想受取額に基づいて取引価格を推定します。対価は固定対価または変動対価を含むことができる。可変価格を含む各手配の開始時に、会社は潜在的な支払いの数と支払いを受ける可能性を評価する。当社は最大可能金額法または予想金額法を採用しており、どの方法で予想受信金額を最も予測できるかによって予想受信金額を推定しています。取引価格に含まれる可変対価金額は制限される可能性があり,確認された累積収入金額が将来的に大きく逆転しない可能性が高い場合にのみ,取引価格に含まれる.
同社の契約には通常、開発と規制マイルストーン支払いが含まれており、これらの支払いは最も可能な金額法に基づいて評価され、重大な収入逆転が発生する可能性が高い場合には制限される。規制承認のような会社統制または被許可者統制の範囲内でのマイルストーン支払いは、承認を受ける前に実現可能とは考えられない。報告期間が終了するごとに、当社はこのような発展マイルストーンを達成する可能性や任意の制限を再評価し、必要に応じて全体の取引価格の見積もりを調整します。いずれの調整も累積追跡に基づいて記録されており,これは調整期間中の協調収入に影響を与える.これまで、会社は開発、監督、または商業マイルストーン収入の実現に関連するいかなる代価も確認しておらず、これらの収入は会社のいかなる協力手配から来ている。
販売ベースの特許使用料を含む手配については、販売レベルに基づくマイルストーン支払いを含み、許可が特許権使用料に関連する主要項目とみなされる場合、当社は、(I)関連販売が発生した場合、または(Ii)特許権使用料の一部または全部が割り当てられた履行義務が履行された(または部分的に履行されている)ときに収入を確認する。当社はこれまで、当社のいかなる協力手配による販売による特許権使用料収入に関する対価格も確認していません。
当社は契約義務ごとの推定独立販売価格に基づいて取引価格を分配します。会社は仮説を立てなければならず,契約で決定された契約義務ごとの独立販売価格を決定する必要がある。同社は、他の比較可能な取引、取引交渉で考慮される価格、および推定コストを含む可能性があるサービス義務の独立販売価格を決定するためにキー仮説を使用する。また,材料権利の独立販売価格を決定する際には,同社は比較可能取引,臨床試験成功確率,オプション行使可能性の推定を利用している。可変対価格の条項が義務の履行状況に関連している場合、可変対価格は契約のうちの1つまたは複数の履行義務に具体的に割り当てられ、割り当てられた金額は、会社が各履行義務を履行するために得られると予想される金額と一致する。
関連貨物またはサービスの制御権が移転した場合には、履行義務毎に割り当てられた対価格が収入として確認される。ライセンスや他の承諾からなる履行義務については、会社は、合併履行義務が時間の経過とともに履行されるか、ある時点で履行されるかを決定するために、合併履行義務の性質を判断する判断を利用して、時間の経過とともに適切な義務を履行する
F-12
進展を測る方法。当社は報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整します。
前払いおよび費用は、会社がこれらの手配の義務を履行するまで、受領または満了時に繰延収入と表記する。当社には権利がある考えは無条件です。
研究開発コスト
株に基づく報酬
その会社の株式ベースの報酬計画は、特定の持分奨励を付与することを許可する。補助金は役員を含む従業員と非従業員に付与される。
当社はASCテーマ718に基づいて株式ベースの報酬を計算している報酬--株式報酬(‘’ASC 718‘’)。ASC 718が従業員、非従業員、および取締役に支払うことを要求するすべての株式ベースの支払いは、総合経営レポートにおいて費用および付与日公正価値に基づく完全な損失として確認されなければならない。当社はブラック·スコアーズオプション定価モデル(‘ブラック·スコルス“)を用いて従業員と非従業員の株式オプションの公正価値を付与することを想定している。会社普通株の公正価値は制限株式単位奨励の公正価値を決定するために使用される。会社は会社普通株を用いて付与日の終値で会社限定株式単位が付与日の公正価値を奨励すると推定する。
その会社の株式ベースの報酬報酬はサービスまたは業績に基づく帰属条件に依存する。サービスを基礎とする帰属条件の従業員、取締役及び非従業員が獲得した奨励に関する補償支出は、付与日に応じて関連サービス期間(一般に帰属期限)の公正価値を直線基準で確認する。報酬業績帰属条件の従業員に関する報酬支出は、業績マイルストーンを実現する可能性があると考えられる暗黙的なサービス期間内に確認される。当社は、各報告期間内にどれだけの報酬が帰属可能とみなされるかを判断に基づいて決定します。
株式に基づく報酬の公正価値推定には、経営陣にオプションの推定寿命や会社普通株の変動性などの推定と判断が求められる。このような判決は確認される補償費用の額に直接影響を及ぼす。
賃貸借証書
2021年9月30日までの四半期内に、会社は改正されたトレーサビリティ法を採用し、ASCテーマ842“レンタル”(以下、“ASC 842”と呼ぶ)を初歩的に採用し、発効日である2021年1月1日を初申請の日とした。したがって、従来の期間は、ASC 840における以前の指導によって提案されている。ASC 842を採用したため、当社は(1)経営リース負債#ドルを記録した
F-13
手配開始時に、当社は、特定の事実および状況、識別された資産(例えば、存在)の存在、および識別された資産使用に対する当社の制御(例えば、適用される)に基づいて、そのスケジュールがレンタルであるかどうか、またはレンタルを含むかどうかを決定する。1つの手配がレンタルまたはレンタルを含むと決定された場合、レンタルは、リースの経済的特徴に基づいて、レンタル開始日(賃貸資産が当社が使用可能であると定義される日)において経営的または融資的賃貸として評価される。レンタル負債は将来の賃貸支払いの現在価値で計量し、レンタル開始日の割引率で割引します。レンタル契約に隠されている金利は一般的に確定しにくい。そこで、当社は、類似経済環境下で、賃貸支払いに相当する金額を担保方式で類似期間内に借り入れた金利である逓増借入金利を採用している。当社は、該当する賃貸使用権(“ROU”)資産を確認し、初期計量をリース負債額とし、レンタル開始前またはレンタル開始時に支払われる任意の初期リースコストまたはリース支払いに応じて調整し、任意のリースインセンティブから減算する。
同社の賃貸契約には経営賃貸契約のみが含まれています。経営リースは貸借対照表でROUリース資産、リース負債流動、リース負債非流動であることが確認された。固定賃貸料はレンタル残高の計算に計上されるが、いくつかの運営および伝達コストに支払われるいくつかの変動コストは含まれていない。レンタル料金は予想期間内に直線的に確認します。
所得税
総合損失
総合損失には,純損失および株主との取引や経済事件以外の取引や経済事件による株主権益の他の変化がある。2022年、2021年及び2020年12月31日まで、当社唯一のその他総合損失とは有価証券の未実現損失のことです。
1株当たり純損失
当社は2段階法で1株当たり純損失を計算しており、当社は参加証券の定義に合った株式を発行しているため。2級法は、発表または累積された配当金および未分配収益の参加権に基づいて、各種類の普通株および参株証券の1株当たり純損失を決定する。二段階法は、この期間中に得られる収入が、その期間のすべての収入が割り当てられているかのように、普通株主がそれぞれ配当を得る権利に基づいて、普通株式と参加証券との間で分配されることを要求する。
普通株株主が1株当たり基本純損失を占めるべき計算方法は、普通株株主が純損失を当期発行普通株の加重平均で割るべきである。普通株主が償却純損失を占めるべきであることは、普通株株主の純損失を調整して、希釈証券の潜在的な影響に基づいて未分配収益を再分配することによって計算される。普通株株主が希釈1株当たり純損失を占めるべき計算方法は、普通株株主は希釈純損失を当期発行普通株で割った加重平均を占めるべきであり、その中に普通株等価物の希釈影響を仮定する潜在希釈性普通株を含む。
当社の転換可能優先株は、契約に応じて当該等の株式の保有者を配当に参加させる権利がありますが、契約要求に応じて当該等の株式の保有者が自社の損失を分担することはありません。したがって,会社が純損失を報告している間,このような損失はこのような参加証券に割り当てられない。会社が普通株株主が純損失を占めるべき期間を報告する時、普通株株主は希釈1株当たり純損失と普通株株主が1株当たりほぼ純損失を占めるべきであり、希釈性普通株は以下の場合は既発行とみなされないからである
F-14
市場と地理情報を細分化する
後続事件
最近発表された会計声明
2012年のJumpStart Our Business Startups Actは、このような基準が非上場企業に適用されるまで、延長された過渡期間を利用して、上場企業に適用される新しい会計基準や改正された会計基準を遵守することを新興成長型会社(“EGC”)に許可する。EGCとして、同社はある新しい会計基準を利用した移行期間の延長を選択した。
2016年6月、FASBはASU第2016-13号を発表した金融商品--信用損失(主題326):金融商品の信用損失の計測。この基準は、現在使用されている発生した損失モデルではなく、期待損失モデルを使用して信用損失を報告することを要求し、信用リスクに関連する追加開示を確立する。未実現損失のある売却可能債務証券については、本基準は、投資の償却コストを削減するのではなく、準備を計上することを要求する。新基準は2023年1月1日から当社に対して施行される。会社のポートフォリオの構成、現在の市場状況と歴史信用損失活動に基づいて、会社はこの基準を採用することは合併財務諸表と関連開示に重大な影響を与えないと予想している。
3.有価証券
証券タイプ別の有価証券には、以下のものが含まれる
|
|
2022年12月31日 |
|
|||||||||||||
(単位:千) |
|
償却する |
|
|
毛収入 |
|
|
毛収入 |
|
|
公平である |
|
||||
アメリカの国庫券と手形 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
アメリカ機関証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
合計する |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
|
|
2021年12月31日 |
|
|||||||||||||
(単位:千) |
|
償却する |
|
|
毛収入 |
|
|
毛収入 |
|
|
公平である |
|
||||
アメリカの国庫券と手形 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
アメリカ機関証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
合計する |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
F-15
非一時的の2022年12月31日または2021年12月31日に被害を受けた。当社の有価証券はいずれも連続した未実現損失は出ていません
4. P不動産と設備、網
財産と設備、純額は、以下を含む
|
|
十二月三十一日 |
|
|||||
(単位:千) |
|
2022 |
|
|
2021 |
|
||
実験室装置 |
|
|
|
|
$ |
|
||
賃借権改善 |
|
|
|
|
|
|
||
家具と固定装置 |
|
|
|
|
|
|
||
コンピュータ装置 |
|
|
|
|
|
|
||
総資産と設備 |
|
|
|
|
|
|
||
減価償却累計を差し引く |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
|
|
十二月三十一日までの年度 |
|
|||||||||
(単位:千) |
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
減価償却費用 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
5. f金融商品の空気価値
ハーバード/遠大許可協定と遠大許可協定によると、当社は公正な価値によって恒常的に計量する金融商品は通貨市場基金、有価証券と派生負債(成功支払負債)を含む。2021年12月31日までの年度内に、反ダンピング権利責任を全面的に清算する。
|
|
2022年12月31日まで |
|
|||||||||||||
(単位:千) |
|
公平である |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
||||
資産 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカの国庫券と手形 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ機関証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
総資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
負債.負債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
成功支払責任 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
総負債 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
|
|
2021年12月31日まで |
|
|||||||||||||
(単位:千) |
|
公平である |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
||||
資産 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカの国庫券と手形 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ機関証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
総資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
負債.負債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
成功支払責任 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
総負債 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
F-16
現金等価物-現金等価物$
有価証券は当社は公正価値に応じて有価証券を恒常的に計量し、これらのツールを公正価値レベルの第二級に分類している。有価証券は、価格設定がアクティブ市場のオファーとは異なり、報告日に直接または間接的に観察されることができるので、公正価値レベルの第2レベルに分類され、公正価値は、モデルまたは他の推定方法を使用することによって決定される。
権利を逆に淡水化する-この義務は追加のものを発行しています
成功支払責任-会社の平均時価が指定された一定期間内に指定されたハードルを超え、9桁までのドル金額からドルに上昇した場合、会社はハーバードと遠大等級への支払いに成功する義務がある
成功支払負債は公正価値に従って列報され、その公正価値計量部分が市場で観察されていない重大な投入に基づいているため、第3級とみなされる。同社は、イベント発生確率、イベント発生時間、および会社普通株の価値を含むいくつかのキー変数に基づいて負債価値をモデル化するモンテカルロシミュレーションモデルを使用する。
当社は公正な価値に応じて負債を再計量し,増加した$
2021年9月、複数回の支払い成功がトリガされ、ハーバードとブロイドの借金の金額は合計$となった
会社の優先株の売却、初公募株、または会社がある評価ハードルを実現することに関する成功した支払負債の主な投入を評価するための2022年12月31日、2021年12月31日、2020年12月31日の状況は以下の通り
|
|
はい。 |
|
|
はい。 |
|
|
はい。 |
|
|||
普通株公正価値(1株当たり) |
|
$ |
|
|
$ |
|
|
$ |
|
|||
株式変動性 |
|
|
% |
|
|
% |
|
|
% |
|||
トリガイベントの累積確率 |
|
適用されない |
|
|
適用されない |
|
|
|
% |
|||
予想期限(年単位) |
|
適用されない |
|
|
適用されない |
|
|
|
|
|||
2020年12月31日、当社の普通株の公正価値は、経営陣が独立第三者評価専門家の協力を得てAICPA推定ガイドラインと一致する方法で決定された。株式変動率の計算は,期待期限の仮定に合致した上場企業株の履歴変動率に類似した既存情報を用いて推定される.また、同社は負債を計算する際に、将来の事件発生の時間と可能性を取り入れている。会社は一つ申請した
2021年2月、当社は遠大に書面通知を出し、遠大許可協定を中止することを選択し、この協定は2021年6月に発効した。
F-17
第三級投入に基づく金融商品の公正価値変動の照合状況は以下の通りである
(単位:千) |
|
逆希釈する |
|
|
成功 |
|
|
合計する |
|
|||
2020年12月31日残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
第1次優先株を発行する |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
普通株発行 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
価値変動を公平に承諾する |
|
|
|
|
|
|
|
|
|
|||
2021年12月31日の残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
価値変動を公平に承諾する |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
2022年12月31日の残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
6. 費用を計算する
計算すべき費用には以下が含まれている
|
|
十二月三十一日 |
|
|||||
(単位:千) |
|
2022 |
|
|
2021 |
|
||
従業員補償と関連福祉 |
|
$ |
|
|
$ |
|
||
外部研究開発費を計算しなければならない |
|
|
|
|
|
|
||
専門費 |
|
|
|
|
|
|
||
許可証とマイルストーン支払い |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
||
7. 賃貸借証書
同社の経営リース活動には、マサチューセッツ州ボストンのオフィスと実験室スペースのキャンセル不可施設レンタルが含まれています。
同社はまた、ASC 842の範囲内の埋め込みリースを含む複数の契約研究および契約製造サービス契約を第三者と締結している。埋め込まれた借約は、契約期間が12ヶ月以下であるため、短期借約とみなされる。したがって、賃貸負債や純資産収益率は記録されていない。
経営リース費用の構成は以下のとおりである
(単位:千) |
|
2022年12月31日 |
|
2021年12月31日 |
|
||
リースコストを経営する |
|
$ |
|
$ |
|
||
可変リースコスト |
|
|
|
|
|
||
短期賃貸コスト |
|
|
|
|
|
||
合計する |
|
$ |
|
$ |
|
||
経営リースに関する補足キャッシュフロー情報は以下のとおりである
(単位:千) |
|
2022年12月31日 |
|
2021年12月31日 |
|
||
賃貸負債に含まれる金額を計量するために支払う現金: |
|
|
|
|
|
||
経営リースに関する経営キャッシュフロー |
|
$ |
|
$ |
|
||
2022年12月31日現在、会社の経営リースは加重平均増量借入金金利計測を採用していますのです
2021年8月19日、当社はデラウェア州有限責任会社(“オーナー”)Are-MA Region No 87 Tenant,LLCとリース契約を締結し、これにより、当社は約をレンタルした
F-18
この物件は2022年8月に初めて当社に提供された。リース開始時、会社が記録した経営リース純資産は#ドル
当社が物件の基本賃貸料を支払う責任は2023年1月に始まった(“賃借日”)。基本賃貸料は最初は$
ボストンのレンタル会社のレンタル期間は
ボストン賃貸契約の締結と保証金として、会社は大家に約#ドルの信用状を提供した
2022年7月、当社はその先の賃貸契約に基づいて家主に通知し、定期賃貸契約が満期になる前にその賃貸契約を終了する予定です。2022年12月31日現在、前回のレンタルにはROUとレンタル責任が残っていません。
201年10月に当社は
借款を取り消すことができない将来の最低承諾額2022年12月31日の状況は以下の通り
12月31日までの年度 |
|
金額 |
|
|
|
|
(単位:千) |
|
|
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
その後… |
|
|
|
|
賃貸支払総額 |
|
$ |
|
|
差し引く:利息 |
|
|
( |
) |
リース負債現在価値を経営する |
|
$ |
|
|
8.許可協定
ハーバード/遠大許可協定と遠大許可協定
2019年3月、当社は同時にいくつかの基礎編集技術についてハーバード/遠大ライセンス協定及び遠大ライセンス協定(“ライセンス合意”)を締結し、これにより、当社は指定特許権下の独占、世界範囲内、再ライセンス可能なライセンス使用料許可、及びいくつかの特許権下の非独占、グローバル範囲内、再ライセンス使用可能なライセンス使用料許可を取得し、ライセンス製品を研究及び開発する。当社は、開発計画に基づいて、商業的に合理的な努力で許可製品を開発し、規制許可を得た任意の許可製品を商業市場に投入し、このような製品を市場に投入した後に監督管理許可を得た許可製品を販売し、監督管理許可を得た許可製品を合理的に公衆に提供することに同意する。協定の有効期限は最後の有効請求が満期になるまで続くだろう。当社はいずれのライセンスも終了することができる
F-19
協定は4ヶ月前に事前に終了しない限り、ハーバード大学とブロード大学に書面で通知された。当社は2021年2月に遠大に書面通知を出し、遠大許可協定を終了する意向を示し、この協定は2021年6月に発効した。
同社はハーバード/遠大許可協定と遠大許可協定によって付与された権利の一部として$を支払った
権利を逆に薄める−ハーバード大学およびブロダー社に発行された普通株の初期株式は、逆希釈条項によって制限される。会社が確定した優先株融資総水準を実現することに関する希薄化権利は2019年に部分的に満たされ、2020年に完全に満たされ、和解金額は合計#ドルとなる
支払いに成功する-公平付記5のさらなる説明によると、会社はライセンス契約に従って成功した支払いを支払わなければならない 金融商品の価値。2021年9月、いくつかの成功的な支払いが触発され、ハーバードとブロダーに借りられた金額は合計#ドルです
賠償協定
通常の業務過程において、会社は、ある事項について、供給者、レンタル者、契約研究機関、業務パートナー、および他の当事者に、このような合意または第三者による知的財産権侵害クレームに違反することによる損失を含むが、これらに限定されない異なる範囲および条項の賠償を提供することができる。また、当社は取締役会メンバー及び行政者と合意を締結し、取締役又は行政者としての身分又はサービスとして生じる可能性のあるいくつかの法的責任について補償を行うことを当社に要求している。これらの賠償協定によると、会社が将来支払う必要がある可能性のある最大潜在金額は多くの場合無制限である。同社はこのような賠償によりいかなる物質コストも発生しておらず、賠償要求があることも知られていない。
その他の支払い-当社は、下位5桁から下位6桁までのライセンスメンテナンス費を毎年支払うことに同意しています 各ライセンス契約に基づいて特定のカレンダー年。当社はハーバードと遠大のライセンス特許に関するいくつかの特許訴訟と維持費の支払いを担当しています。実現した範囲では,会社は総額$までの支払いが義務付けられている
BEAMライセンスプロトコル
2019年4月、当社はビム治療会社(“ビム”)と協力と許可合意に達した。ビムプロトコルによれば、会社は、ビムのCRISPR関連タンパク質12 bまたはCas 12 b技術を使用した塩基編集製品およびヌクレアーゼ製品の開発、製造、使用、販売、および輸入のための世界的範囲での独占的再許可を取得し、それぞれの場合、PCSK 9およびAngptl 3遺伝子を含む、冠動脈疾患リスクの増加に関連する4つの初期遺伝子標的のいずれかについて。また、同社は、そのいくつかの交付技術に基づいて、候補製品および製品を開発、製造、販売、輸入するために、世界的に独占的かつ再許可可能なライセンスをビムに付与したが、Verveにライセンスされた基礎編集製品は除外した。
双方は合意した研究および/または開発計画に基づいていくつかの活動を展開することができる。初期遺伝子標的のために最終患者に投与された許可製品の1 b期臨床試験の後TSビムは共有に参加する権利があります
F-20
製品アメリカの50/50に基づいていますビームが初期遺伝子標的の特定の許可製品に対して選択権を行使する場合、これを協力製品と呼び、協力製品の開発および商業化コストの特定の割合を支払う義務があり、協力製品の任意の販売から指定されたパーセントの利益を得る権利がある。BEAMプロトコルの期限は、どの製品のどの印税期限の最終期限が切れるまで続いています。当社は、ビムに90日間の終了通知を発行し、任意の特許製品のピム合意を終了する権利があるが、ビムがその選択加入権利を行使しないことを選択したこと、またはその選択加入権利を行使する期限が満了したことを前提として、いかなる提携製品の合意も終了することはできない。
当社は、研究計画、いかなる開発計画の下で活動を行うことによるすべてのコストと支出、およびビムがまだ加入していない特許製品開発のいかなるコストと支出を選択していないかを担当しています。
BEAMがBEAMプロトコルによって許可権を付与した代償の一部として、会社は発行することで
双方はまた、双方のそれぞれの交付技術によって付与された許可証をさらに考慮するために、それぞれが開発に基づくマイルストーン支払いを他方に支払い、最高で#ドルに達することを約束した
当社は2022年7月5日に、ビームと改訂及び再署名された協力及び許可協定(“改訂されたピム協定”)を締結した。
改訂されたビム協定によると、ビム社は、ビム社のいくつかの基礎編集技術の下で、別の肝臓媒介心血管疾患標的の製品を開発し、商業化するために、独占的、世界的に再許可可能なライセンスを同社に付与した。同社は追加遺伝子標的に対する製品の開発と商業化を担当しているが、ビムの選択権に適合する必要がある。ライセンス製品の1 b期臨床試験で最終患者にこの追加の遺伝子標的を注射した後、ビムは共有に参加することを選択する権利がある
交換として,会社は会社の知的財産権に基づいて,会社のGalNAc−LNP交付技術を含めて,会社が開発した臨床前計画に関する独占的,世界的に再許可可能な全額支払い許可証をビムに付与した。
改訂されたピム協定はまた、会社のGalNAc-LNP交付技術に関連する知的財産権を明確にした;目標ごとに、ビームが会社GalNAc-LNP交付技術を獲得する非独占的、世界的、再許可可能な許可証の選択権を付与し、いくつかの基礎編集製品を開発し、商業化するために、すなわち、各選択権、ある規制と商業販売マイルストーン、およびGalNAc-LNP配信技術を使用した基礎編集製品の純販売の低桁の特許権使用料を行使する時、ビームは会社に費用を支払うべきである。当社のBEAMプロトコルの下で未開示遺伝子に関する権利と経済的義務を終了し、当社とBEAMがこれらの遺伝子標的のための製品を独立して開発および商業化することを可能にし、BEAMプロトコルに従って双方が開発した他の技術開発および商業化基礎編集製品を交付および商業化するために申請する他のライセンスを締結する。
納入技術製品の販売があれば、双方は毎回の納入による世界的な年間純売上高に応じて他方に中下位桁の使用料を支払う
F-21
支払元の技術製品;ただし,このような印税は連携製品やライセンス製品の純売上には適用されない.同社は、2022年12月31日現在、改訂されたBEAM協定下のいかなるマイルストーンや特許権使用料の支払いも受けられないと結論した。
Acuitas協定
開発と選択プロトコル
2019年12月、当社はAcuitas Treateutics,Inc.(以下Acuitas)と開発·協力協定を締結した2020年10月、CH協定は改正され、再記述された。同社はAcuitasが当該計画活動に関連するサービスを四半期ごとに補償することに同意し、補償の根拠は約束した全従業員数であり、従業員1人当たりの年率で計算し、合理的な外部コストの精算を含む。同社が確認した研究·開発費は約$
許可協定
2020年10月、同社はAcuitasに返却不可能な前払い許可料#ドルを支払った
実現した範囲では,会社には総額#ドルの費用を支払う義務がある
ノワール許可協定
2021年10月、同社は、VERVE-201を含むいくつかの候補製品を研究開発する際に使用される脂質技術の非独占的許可を得るために、ノワーズ製薬会社(“Novartis”)とライセンス契約を締結した。本契約により付与されたライセンスと権利の対価格として、当社はE使い捨て、払い戻しできない前金#ドル
2022年6月、同社は協定を修正し、他の3種類の許可製品を非独占許可の範囲に入れた。追加の許可製品を考慮して、同社は製造を要求されたE使い捨て、払い戻し不可能な前金$
F-22
9.協力と許可協定
頂点合意
プロトコルの概要
当社は2022年7月、Vertex PharmPharmticals Inc.(“Vertex”)と戦略協力および許可プロトコル(“Vertexプロトコル”)を締結し、4年間のグローバル独占研究協力を行い、開発に専念した体内にある遺伝子編集は単一肝疾患を治療する未開示目標になることが期待される。また、会社はVertexと株式購入協定(“株式購入協定”)を締結し、同協定に基づき、会社は株式を売却した
Vertexプロトコルによると、会社は新型薬物の発見、研究、ある臨床前開発を担当している体内にある遺伝子編集開発は人々が興味を持つ目標である.同社の研究活動は(I)特定の遺伝子編集システムの識別と設計に集中している体内にある目標に対する交付システム;(Ii)頂点プロトコルに規定された基準を実現するために開発候補案を評価し最適化する.Vertexは,双方が合意した研究計画と予算(“研究計画”)に従って会社の研究費を精算する義務がある。研究期間の初期期間は4年であり,頂点から最大1年間の追加期限(“研究期間”)を延長することができる.研究計画は共同研究委員会(“JRC”)が監督し、具体的な内容はVertexプロトコルを参照。研究計画に対するいかなる実質的な修正も司法審査委員会の共同同意を得る必要がある。
研究期間中頂点はいくつかの遺伝子編集システムと体内にあるターゲットをターゲットとした配信システムは,証明書エージェントとなる.ライセンスエージェントを指定すると,Vertexは使用許可エージェントのライセンスを取得し,ライセンスエージェントはJRC同意のいくつかの開発候補基準を実現するために研究計画に基づいた開発を継続する.研究条項によると、Vertexは、ライセンスエージェントによって生成された任意の候補製品の後続開発、製造、および商業化を個別に担当する。
会社はVertexから$の前金を受け取った
Vertexプロトコルに従って開発された第1の候補製品の第1の1 b期臨床試験の第1の患者用量の前に、会社はまた、利益共有スケジュールに参加することを選択する権利があり、このスケジュールによれば、Vertexおよび会社は、協力して生成されたすべての候補製品のコストおよび純利益を共有する。企業が加入を選択する権利を行使する場合、マイルストーンや特許権使用料ではなく、特定の割合の開発·商業化コストを支払う義務があり、協力して推進される任意の候補製品の任意の販売から指定されたパーセントの利益を得る権利があるだろう。会社が選択権を行使する際には、利益/コストシェアを最大で選択することができる
Vertex協定はこのような取引の慣例的な陳述と保証、契約、そして賠償義務を含む。当社とVertexはそれぞれ通知を出した後にプロトコルを終了する権利があり,他方が実質的に違約したり債務を返済できない場合はプロトコルを終了する権利があり,適用すればプロトコルを終了する権利がある.便宜上,Vertexも90日前に通知し,Vertexプロトコルの全内容を終了することができる.
同社は、Vertexプロトコルで約束された商品およびサービスをASC 606に従って評価した。最初に,“頂点協定”には,(1)研究サービス義務,研究計画(“研究計画”)に関連して提供される研究開発サービスが含まれる
F-23
研究サービス“)および(Ii)は、保有エージェントの選択権を利用するために割引価格でライセンスを取得することに関連する3つの保有エージェント物質権利。
その会社は$を確定した
当社の結論は,研究サービス債務の費用返済に関する可変対価格はその債務に完全に分配され,費用返済は具体的には研究計画の下で行われているサービスに及ぶためである。研究サービスの返済額は市場レートで計算されていると考えられていることから,受信予定のこの債務の推定額を説明した。そのため、会社は固定対価格#ドルを割り当てた
義務を果たす |
|
金額 |
|
|
|
|
(単位:千) |
|
|
サービス義務を研究する |
|
$ |
|
|
最初に許可を得た代理材料権利 |
|
|
|
|
第二世代所有代理店物権 |
|
|
|
|
第三者が代理店の物権を持つ |
|
|
|
|
合計する |
|
$ |
|
|
|
|
|
|
|
研究サービス債務に割り当てられた額は、投入された研究総費用計量をサービス期間中に比例して使用して確認し、各報告期間終了時に行われ再計量された割合を推定する。カードエージェントに割り当てられた重大な権利の金額は繰延収入として記録され,オプション行使ごとに確認を開始し,オプションを行使していない場合は検討期間満了時に全額確認する.
2022年12月31日までの年度内、会社は$を確認しました
Vertexプロトコルによると,会社の協力プロジェクトに関するコストには,内部研究と外部研究コストがあり,主に賃金と福祉,臨床前研究がある。これらのコストは,会社の2022年12月31日年度までの総合経営報告書における研究と開発費用に計上されている.
10. 優先株と普通株
会社(The Company)発行されて販売される
F-24
2021年6月、会社はその会社登録証明書を修正して再発行し、許可する
2021年6月に、当社は初の公募を完了し、これにより当社は発行及び販売します
初公募終了時には、会社優先株のすべての流通株が自動的に
2022年7月にVertexプロトコルを実行し、当社はVertexとも株式購入プロトコルを締結し、売却および発行した
2022年7月,会社は普通株の後続公開発行を完了し,これにより,会社は普通株を発行して売却した
当社は2022年7月にJefferies LLC(“Jefferies”)と公開市場販売協定(“販売協定”)を締結し、これにより、当社は時々現行市価で自社普通株を発売·販売する権利がある。同社はジェフリーにガンダムを支払うことに同意しました
Cの所有者普通株は普通株ごとに一票を投じる権利がある。
11.株ベースの報酬
取締役会が2018年8月に採択した“2018年株式インセンティブ計画”(“2018年計画”と略す)は、会社員、上級管理者、取締役、コンサルタント、外部コンサルタントに、条件に応じたインセンティブ株式オプション、非法定株式オプション、株式付加価値権、制限株式および制限株式単位を付与することを規定している当社の普通株を発行または購入します。2018年計画に基づき承認発行された普通株式最高株式数は
2021年6月、会社取締役会が可決し、会社の株主の承認を得た“2021年株式インセンティブ計画”(以下、“2021年計画”と略す)が2021年6月16日に正式に発効した。2021年計画では、会社員、取締役、コンサルタントおよび外部コンサルタントに合格および非適格株式オプション、株式付加価値権、制限および非制限株式および株式単位、業績奨励およびその他の株式ベースの奨励を付与することを規定します。2021年計画によると発行保留の株式は2031年1月1日までに年ごとに増加する。2022年12月31日までその会社は予約しました
総合経営表と総合損失表に計上されている株式補償費用は以下の通り
|
|
現在までの年度 |
|
|||||||||
(単位:千) |
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
株式に基づく報酬総支出 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
F-25
株式オプション
ブラック·スコアーズが株式オプションを付与する際に使用した仮定は以下のとおりである
|
|
現在までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
予想変動率 |
|
|
% |
|
|
% |
|
|
% |
|||
加重平均無リスク金利 |
|
|
% |
|
|
% |
|
|
% |
|||
期待配当収益率 |
|
|
— |
|
|
|
|
|
|
— |
|
|
予想期限(年単位) |
|
|
|
|
|
|
|
|
|
|||
次の表は,この年度までの株式オプション活動の概要を提供する2022年12月31日:
|
|
量 |
|
|
重みをつける |
|
|
重みをつける |
|
|
骨材 |
|
||||
2021年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
授与する |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
没収される |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日に行使できます |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日以降に帰属する予定です(1) |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日までの年度内に、すでに株式購入権の加重平均授与日の公正価値を授授したはい$です
2022年12月31日まで一ドルあります
限定株及び限定株式単位
2018年、当社は発行しました
2022年12月31日までの年度内会社が承認します
未帰属制限株式単位の状況と変化の概要2022年12月31日の状況は以下の通り
|
|
株 |
|
|
|
重み付けの- |
|
||
2021年12月31日現在の未帰属制限株式単位 |
|
|
|
|
$ |
|
|
||
承認された制限株式単位 |
|
|
|
|
$ |
|
|
||
帰属制限株式単位 |
|
|
( |
) |
|
$ |
|
|
|
没収された制限株式単位 |
|
|
( |
) |
|
$ |
|
|
|
2022年12月31日現在の未帰属制限株式単位 |
|
|
|
|
$ |
|
|
||
F-26
2022年12月31日一ドルあります
2021年に改正·再策定された従業員株購入計画
2021年6月、取締役会は改正·再記述された2021年従業員株式購入計画を採択し、会社株主は2021年6月16日に発効する計画を承認した。ESPPにより留保発行された株式は2031年1月1日まで年ごとに増加する。2022年12月31日まで,
12.普通株主の1株当たり純損失
以下の表は、会社の普通株主が1株当たりの基本純損失と償却純損失を占める計算方法をまとめた
|
|
十二月三十一日までの年度 |
|
|||||||||
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない) |
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
分子: |
|
|
|
|
|
|
|
|
|
|||
普通株主は純損失を占めなければならない |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
分母: |
|
|
|
|
|
|
|
|
|
|||
基本普通株と希釈普通株の加重平均 |
|
|
|
|
|
|
|
|
|
|||
普通株株主は普通株1株当たり純損失、基本損失と希釈損失を占めなければならない |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
当社の潜在的希薄化証券は、転換可能な優先株、未帰属制限株、未帰属制限株式単位、および普通株オプションを含み、その影響は逆薄となるため、1株当たりの純損失の計算から除外されている。したがって、普通株株主が基本純損失を占めるべきであることと希釈後の1株当たり純損失を計算するための発行済み普通株加重平均は同じである
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
転換可能優先株 |
|
|
|
|
|
|
|
|
|
|||
無帰属制限株 |
|
|
|
|
|
|
|
|
|
|||
未帰属限定株式単位 |
|
|
|
|
|
|
|
|
|
|||
普通株購入の未償還オプション |
|
|
|
|
|
|
|
|
|
|||
合計する |
|
|
|
|
|
|
|
|
|
|||
同社はハーバード大学とブロード大学とのライセンス契約の一部としてSuccess支払いを支払わなければならない。会社は会社の普通株式を発行することでこれらのお金を支払うことを選択することができる。2022年12月31日までの総金額は
13. 所得税
同社の所得税前損失は国内業務の損失のみを含み,合計$
所得税費用(福祉)は以下のようにまとめられる
F-27
|
|
十二月三十一日までの年度 |
|
|||||||||
(単位:千) |
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
現在: |
|
|
|
|
|
|
|
|
|
|||
連邦制 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
状態.状態 |
|
|
|
|
|
|
|
|
|
|||
外国.外国 |
|
|
|
|
|
|
|
|
|
|||
総当期に準備する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
延期: |
|
|
|
|
|
|
|
|
|
|||
連邦制 |
|
|
|
|
|
|
|
|
|
|||
状態.状態 |
|
|
|
|
|
|
|
|
|
|||
外国.外国 |
|
|
|
|
|
|
|
|
|
|||
繰延準備金総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
連邦法定所得税率を用いて計算された所得税費用と会社の有効所得税率との入金は以下のとおりである
|
|
十二月三十一日までの年度 |
|
|||||||||
(単位:千) |
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
連邦法定金利 |
|
|
% |
|
|
% |
|
|
% |
|||
評価免除額を変更する |
|
|
( |
)% |
|
|
( |
)% |
|
|
( |
)% |
株に基づく報酬 |
|
|
( |
)% |
|
|
% |
|
|
( |
)% |
|
役員報酬 |
|
|
( |
)% |
|
|
( |
)% |
|
|
% |
|
永久品 |
|
|
( |
)% |
|
|
( |
)% |
|
|
% |
|
連邦福祉を差し引いた州所得税 |
|
|
% |
|
|
% |
|
|
% |
|||
研究開発税収控除 |
|
|
% |
|
|
% |
|
|
% |
|||
他にも |
|
|
% |
|
|
% |
|
|
% |
|||
有効所得税率 |
|
|
% |
|
|
% |
|
|
% |
|||
F-28
繰延税項は、財務諸表の資産と負債ベースと所得税との一時的な差異で確認されている。2022年12月31日と2021年12月31日まで、会社の繰延税金資産と負債の重要な構成要素が含まれている
|
|
十二月三十一日 |
|
|||||
(単位:千) |
|
2022 |
|
|
2021 |
|
||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失が繰り越す |
|
$ |
|
|
$ |
|
||
資本化コスト--純償却額 |
|
|
|
|
|
|
||
研究開発税収控除 |
|
|
|
|
|
|
||
資本化研究コスト |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
リース責任 |
|
|
|
|
|
|
||
費用を計算する |
|
|
|
|
|
|
||
繰延税金資産総額 |
|
|
|
|
|
|
||
繰延税金負債: |
|
|
|
|
|
|
||
財産と設備 |
|
|
( |
) |
|
|
( |
) |
使用権資産 |
|
|
( |
) |
|
|
( |
) |
繰延税金負債総額 |
|
|
( |
) |
|
|
( |
) |
繰延税項目の総資産,純額 |
|
|
|
|
|
|
||
減算:推定免税額 |
|
|
( |
) |
|
|
( |
) |
繰延税金資産は,推定準備後の純額を差し引く |
|
$ |
|
|
$ |
|
||
会社は設立以来毎年純営業損失を出しています。管理層はすでに当社の繰延税項目の純資産の現金化に影響を与えるプラスと負の証拠を評価し、このなどの資産は主に営業損失の純額、税務項目の相殺及び税務目的の資本化のコストから構成されている。経営陣は、当社の米国における累積純損失の歴史と予想される将来の税務損失を考慮しており、当社は繰延税項純資産の収益を確認しない可能性が高いと考えています。そこで,会社は2022年12月31日と2021年12月31日に全額推定手当を記録した。見積もり免税額が増加した$
将来の税収優遇の実現は、会社が純営業損失の繰越期間内に課税収入を発生させる能力を含む多くの要素に依存する。会社が国税法382に基づいて所有権変更が発生した場合、会社がこれらの連邦や州の純営業損失および開発信用繰越の能力を利用することは将来的に制限される可能性がある
2022年12月31日まで同社は約$を持っています
2022年12月31日と2021年12月31日まで会社が所有しています
F-29
14.関連する当事者取引
ビムの幹部は2022年8月まで会社の取締役会のメンバーだった。
2020年10月に当社はビムと材料交換協定を締結し,ビームは当社にいくつかのメッセンジャーリボ核酸,gRNAおよびタンパク質を提供することに同意したが,当社は提供する材料ごとの協定価格でいくつかの遺伝子リボ核酸をビムに提供することに同意した。2022年12月31日及び2021年12月31日まで、当社は確認しました
当社は2021年12月、ピムとマサチューセッツ州ケンブリッジ市の実験室とオフィススペースの転貸契約を締結し、2022年12月31日に転貸を終了した。このテナント契約のレンタル料総額は$
遠大グループの幹部は同社の取締役会のメンバーだ。取締役会のメンバーが辞任し、2021年5月から発効する。2019年3月、当社は同時にいくつかの基礎編集技術についてハーバード/広汎許可協定および広範許可協定を締結し、これにより、当社は指定特許権下の独占、世界範囲内、再許可可能、特許権使用料を徴収できる許可、およびいくつかの特許権下の非独占、グローバル、再許可可能、特許権使用料許可を取得し、許可製品を研究および開発する。許可協定によると、他の対価格は逆希釈権利と成功的な支払いを含む。付記8、ライセンス契約を参照してください。
15.従業員福祉計画
会社は国税法第401(K)節(“401(K)計画”)に基づいて、基本的にすべての従業員を対象とした固定納付計画を確立した。従業員は雇われた初日から401(K)計画に参加する資格がある
16. S後続事件
当社はすべての後続イベントを評価し、重大な確認されたまたは確認されていない後続イベントが開示される必要がないことを確認した。
F-30