カタログ表
|
第1部:
|
ページ
|
|
|
第1項。
|
業務.業務
|
5
|
|
第1 A項。
|
リスク要因
|
32 |
|
項目1 B。
|
米国証券取引委員会のコメントは未解決だ
|
62 |
|
第二項です。
|
属性
|
62 |
|
第三項です。
|
法律訴訟
|
63 |
|
第四項です。
|
炭鉱安全情報開示
|
63 |
|
第二部です。
|
||
|
五番目です。
|
登録者普通株市場、関連株主事項及び発行者による株式証券の購入
|
63 |
|
第六項です。
|
保留されている
|
64 |
|
第七項。
|
経営陣の財務状況と経営成果の検討と分析
|
64 |
|
第七A項。
|
市場リスクの定量的·定性的開示について
|
73 |
|
第八項です。
|
財務諸表と補足データ
|
73 |
|
第九項です。
|
会計と財務情報開示の変更と相違
|
73 |
|
第9条。
|
制御とプログラム
|
73
|
|
プロジェクト9 B。
|
その他の情報
|
74 |
|
プロジェクト9 Cです。
|
検査妨害に関する外国司法管区の開示
|
|
|
第三部
|
||
|
第10項。
|
役員·幹部と会社の管理
|
74 |
|
第十一項。
|
役員報酬
|
74 |
|
第十二項。
|
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項
|
74 |
|
十三項。
|
特定の関係や関連取引、取締役の独立性
|
74
|
|
14項です。
|
チーフ会計士費用とサービス
|
75 |
|
第4部
|
||
|
第十五項。
|
展示品と財務諸表の付表
|
75 |
|
第十六項。
|
表格10-Kの概要
|
77 |
|
サイン
|
79 |
2
カタログ表
前向きに陳述する
本年度報告(以下、“年次報告”と略す)は、リスクおよび不確実性に関連する前向き表現と、これらの前向き表現が実現されていないか、または誤りであることが証明された場合、このような前向き表現に明示または示唆された結果とは大きく異なる結果をもたらす可能性がある。私たちは1995年の“個人証券訴訟改革法”と他の連邦証券法の安全港条項に基づいてこのような前向きな声明を出した。本年度報告では歴史的事実に関する陳述を除き,他のすべての陳述は前向き陳述である。場合によっては、“目標”、“予想”、“信じる”、“できる”、“考慮”、“継続”、“可能”、“設計”、“推定”、“予想”、“未来”、“計画”、“可能”、“可能”、“計画”、“潜在”、“予測”、“項目のような前向きな陳述を単語
によって識別することができる。“追求”、“求める”、“すべき”、“戦略”、“目標”、“br}”、“将”またはこれらの語の否定または他の類似用語。これらの前向きな陳述は、以下の態様に関する陳述を含むが、これらに限定されない
| • |
臨床研究の開始とタイミングの面で、著者らは各種の候補薬物に対する期待マイルストーンを満たすことができる
|
| • |
米国食品医薬品局(FDA)の現在または任意の他の将来の候補製品に対する規制を含む連邦、州、および非米国規制要件
|
| • |
FDAに規制文書を提出する時間と私たちの能力、ならびにFDAまたは他の規制機関の私たちの候補製品に対する承認または他の行動を獲得して維持する能力;
|
| • |
私たちの競争相手の活動は、競合製品の発表時間、定価、割引の決定を含む
|
| • |
私たちの臨床試験の安全性と有効性の結果、および私たちの候補製品を承認するための他の必要なテストは、臨床試験の進展、潜在的な規制承認、または私たちの任意の候補製品のさらなる開発を保証するためにデータを提供するかどうかを保証する
|
| • |
私たちは候補製品を開発、取得し、推進する能力、十分な数の患者の参加を募集し、臨床研究を成功させる能力、および現在予想されている時間範囲内で、または規制機関がそのような候補製品を承認することを申請し、獲得する能力を獲得する
|
| • |
私たちは私たちの候補製品と任意の他の未来の候補製品のための重要な協力とサプライヤー関係を構築することができます
|
| • |
私たちは私たちの販売とマーケティング能力を発展させたり、第三者と合意して、私たちの任意の候補製品を販売したりすることができます
|
| • |
私たちは計画された研究と開発を行うために追加資金を得ることができます
|
| • |
買収された業務の統合は、統合プロセスがキー従業員の流失を招く可能性があること、私たちが行っている業務中断、または場合によっては、標準、制御プログラム、または政策の不一致が、買収予想利益を達成する能力に悪影響を及ぼす可能性があることを含む多くのリスクに関連している。
|
| • |
私たちが許可を得ることができる任意の技術や製品の開発と商業化に成功しました
|
| • |
製造困難による予期しない遅延は、我々のAAVプロジェクトの直接製造能力の発展、および任意の供給制限または法規環境の変化を含む;私たちが現在または未来に業務を展開している非米国司法管轄区で成功して運営する能力は、適用される法規要件と法律を遵守することを含む
|
| • |
私たちの候補製品を保護するために特許を取得して実行することに関連する不確実性と、予測できない第三者侵害クレームを防御することに成功した能力
|
| • |
私たちのビジネスと私たちが経営している市場の予想傾向と挑戦
|
| • |
自然災害と人為災害は、新冠肺炎などの流行病とその他の不可抗力を含み、私たちの運営、私たちのパートナーと医療業界の他の参加者に影響を与える可能性があり、そして私たちの臨床研究、臨床前研究活動と薬品供給に悪影響を及ぼす可能性がある
|
| • |
インフレ上昇と資本市場の中断、ウクライナの現在の衝突、経済制裁と経済減速、または衰退など、世界経済と政治発展が私たちの業務に与える影響は、私たちの研究開発努力と私たちの普通株の価値と私たちの資本市場に入る能力を損なう可能性がある
|
| • |
私たちの資本需要の見積もりは
|
| • |
私たちは追加融資を受け、必要に応じて資本を調達し、運営に資金を提供したり、ビジネス機会を求めたりすることができる。
|
私たちはあなたに上記のリストに本年度報告書で行われたすべての展望的な陳述が含まれていないかもしれないということを想起させます。
本年度報告中の任意の前向き陳述は、既知と未知のリスク、不確定性と他のbr要素に関連する未来の事件または私たちの未来の財務表現に対する私たちの現在の見方を反映しており、これらの要素は私たちの実際の結果、業績または業績を招く可能性があり、これらの前向きな陳述が明示的あるいは暗示する任意の未来の結果、業績あるいは成果とは大きく異なる。実際の
結果が現在の予想と大きく異なることをもたらす可能性のある要素は、第1の部分1 A項に列挙された要素を含む。危険要素と本年度報告書の他の部分。このような不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。法的要求がない限り、私たちは未来に新しい情報があっても、これらの前向きな陳述を任意の理由で更新または修正する義務がない。
本年度報告はまた、これらの市場の推定規模およびいくつかの疾病の発病率と流行率に関するデータを含む、私たちの業界、業務およびいくつかの疾病市場の推定、予測およびその他の情報を含む。見積り,予測,予測,市場研究や類似手法に基づく情報自体が不確実性や実際のイベントの影響を受けるか,あるいは
の場合はその情報に反映されるイベントや状況と大きく異なる可能性がある.他に明確な説明がない限り、私たちは、報告、研究調査、研究、および市場研究会社および他の第三者によって準備された類似データ、業界、医療および一般出版物、政府データ、および同様のソースから、当業界、企業、市場、および他のデータを取得する。別の説明がない限り、本年度報告で言及された“私たち”、“私たち”、“私たち”または私たちの“会社”および同様の用語は、ロケット製薬会社を指す。
3
カタログ表
私たちの業務に関する重大なリスクの概要
私たちの業務は多くのリスクと不確実性の影響を受けており、あなたは私たちの業務を評価する際にこれを認識しなければならない。これらのリスクと不確定要素は含まれているが、これらに限定されない
| • |
新冠肺炎を招くSARS-CoV-2疫病はすでに著者らの臨床前と臨床研究を含む著者らの業務に不利な影響を与える可能性がある。
|
| • |
もし私たちが販売およびマーケティング能力を確立できない場合、あるいは第三者と合意して私たちの任意の候補製品を販売し、販売することができなければ、これらの候補製品が承認された場合、私たちはそれを商業化することに成功できないかもしれない。
|
| • |
もし私たちが計画した研究と開発を行うために追加の資金を得ることができなければ、私たちは私たちの製品開発計画や商業開発を延期、減少、またはキャンセルさせることを余儀なくされるかもしれない。
|
| • |
私たちは製品販売から何の収入も得られなかったし、永遠に利益を上げないかもしれない。
|
| • |
私たちは臨床試験の開始、登録または完成に重大な遅延に遭遇する可能性があり、あるいは安全性と有効性が関連する監督管理機関を満足させることができない可能性があり、これは私たちが現在と未来の候補製品を適時に商業化することを阻止するかもしれない(もしあれば)。
|
| • |
もし私たちが計画通りに臨床試験を行うのに十分な数の患者を募集することが困難であれば、計画中の臨床試験を延期、制限或いは中止する必要があるかもしれず、いずれの状況の発生も私たちの業務、財務状況、運営結果と将来性を損なう。
|
| • |
著者らが行っている臨床研究の初歩的あるいは中期的な結果は、これらの研究が完成した後に得られた結果を代表しないかもしれない。そのほか、早期臨床研究の成功は後の研究結果を代表しないかもしれない。
|
| • |
必要な前臨床研究と臨床試験を成功させても、いつあるいは規制部門の承認を得て、候補製品を商業化するかどうかを予測することはできませんし、承認の範囲は私たちが求めているものよりも狭いかもしれません。
|
| • |
私たちの任意の候補製品は決してアメリカ、EU、あるいは他の司法管轄区で承認されないかもしれません。これは私たちが市場の潜在力を十分に発揮する能力を制限するだろう。
|
| • |
私たちが製品候補の規制承認を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けるだろう。
|
| • |
私たちの候補製品は不良と予見できない副作用を引き起こす可能性があり、あるいは公衆に安全ではないと思われる可能性があり、これは臨床試験や規制承認への進出を延期または阻止し、商業潜在力を制限したり、重大な負の結果を招く可能性がある。
|
| • |
私たちは生産問題に直面し、私たちの開発や商業化計画の遅延を招き、私たちの製品の供給を制限したり、他の方法で私たちの業務を損害したりする可能性があります。
|
| • |
私たちの製造面での経験は限られていて、私たちが製品を成功的に製造できるという保証はない。
|
| • |
私たちの製造施設は重大な政府法規や承認の制約を受けており、私たちが法規を遵守したり、承認を維持しなければ、コストが高く、私たちの業務に悪影響を及ぼす可能性があります。
|
| • |
私たちの候補製品の開発と商業化に成功した能力は、生成された薬物や関連治療費用の精算資金の利用可能性に大きく依存するだろう。
|
| • |
承認されても、私たちは私たちの候補製品を商業化することに成功できないかもしれない。
|
| • |
私たちが他の候補製品チャンネルを拡大する努力は成功しないかもしれない。
|
| • |
著者らは主に研究開発活動、臨床テストと商業化の成功かどうかはまだ確定していない。
|
| • |
第三者に依存して我々の薬品製品の製造,研究および臨床前と臨床試験の一部または全部を行うことが予想されるが,これらの第三者の表現は満足できない可能性がある。
|
| • |
キーパーソンのサービスを失ったり、キーパーソンを引き付けることができない場合、私たちの業務は影響を受ける可能性があります。
|
| • |
私たちは私たちの組織を拡張する必要があるかもしれないし、このような成長を管理する時に困難に直面する可能性があり、これは私たちの運営を混乱させるかもしれない。
|
| • |
私たちの従業員、主要な調査者、コンサルタント、ビジネスパートナーは、規制基準と要求を守らないこと、私たちの業務を損なう可能性のあるインサイダー取引を含む不当な行為やその他の不適切な活動に従事する可能性があります。
|
| • |
米国以外のビジネス関係、特にEUを考慮すると、国際業務に関連する様々なリスクが私たちの業務を損なう可能性があります。
|
| • |
私たちが製品や関連技術の十分な特許保護を獲得して維持できなければ、製品を商業化することに成功する能力が損なわれる可能性がある。
|
| • |
もし私たちが許可協定に違反すれば、私たちの候補製品の商業化努力に実質的な悪影響を及ぼすかもしれない。
|
| • |
私たちは私たちの特許と他の知的財産権の発明権または所有権のクレームに疑問を受けるかもしれない。
|
| • |
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの業務と競争地位は損なわれるかもしれない。
|
| • |
もし私たちが私たちの候補製品に関連する知的財産権を獲得したり保護したりできなければ、私たちは私たちの市場で効果的に競争することができないかもしれない。
|
| • |
私たちはRenovacorの買収のような潜在的な買収や業務合併の期待的な利益を達成できないかもしれない。
|
| • |
我々の最大株主RTW Investments,LPは株主承認に提出された事項に大きな影響を与える可能性がある.
|
4
カタログ表
上述したリスク要因要約は、以下および本年度報告に列挙された他の情報(当社の連結財務諸表および関連説明を含む)、および米国証券取引委員会に提出された他の文書の完全なリスク要因と共に読まれなければならない。このようなリスクや不確実性が実際に発生すれば、私たちの業務、見通し、財務状況、運営結果は重大で不利な影響を受ける可能性がある。以上の概要または本年度報告の他の部分的に完全に説明されたリスクは、私たちが直面している唯一のリスクではない。私たちは現在知らないか、あるいは現在重要ではないと考えている他のリスクや不確実性は、私たちの業務、将来性、財務状況、運営結果にも大きな悪影響を及ぼす可能性があります。
第1部
| 第1項。 |
業務.業務
|
概要
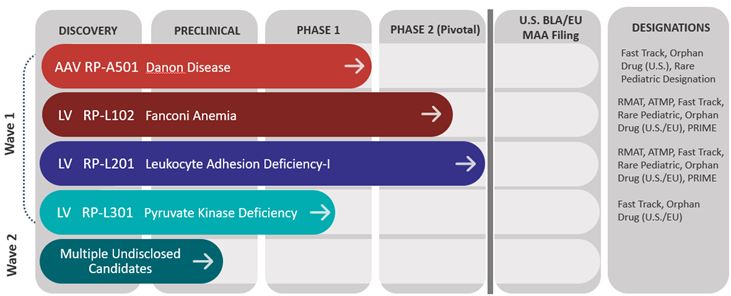
著者らは臨床段階、多プラットフォームの生物技術会社であり、第一種、唯一と同類の中で最も良い遺伝子療法の開発に専念し、直接の標的作用機序と明確な臨床終点を有し、希と壊滅性疾患に応用している。3つの臨床段階の体外レンチウイルスベクター(“LV”)計画があります。これらの方案はファンコニ貧血(“FA”)を含み、これは1種の骨髄中の遺伝欠陥であり、血細胞の産生を減少或いは欠陥血細胞の産生を促進する;白血球粘着欠陥-I(“LAD-I”)、1種の免疫系機能障害を招く遺伝性疾患である;及びピルビン酸キナーゼ欠乏症(“PKD”)は1種の稀な赤血球常染色体劣性遺伝性疾患であり、慢性非球形溶血性貧血を招く。このうち,第2段階FA計画と第2段階LAD−I計画はいずれも米国で登録されている可能性がある。ヨーロッパ(“EU”)ですあまり一般的でないFAサブタイプCおよびGに対する遺伝子治療計画の追加作業が行われている。米国でもDanon病の臨床生体段階腺関連ウイルス(“AAV”)計画があり,Danon病は多臓器リソソーム関連疾患であり,心不全により早期死をきたす。ダノン計画は現在進行中の第一段階試験にある。また,不整脈源性心筋症(“PKP 2−ACM”)に対するAAVベクター計画もあり,PKP 2−ACMは遺伝性心疾患であり,心筋質量の進行性低下,右室の重篤な拡張,発育不良,心筋線維脂肪代替および不整脈や突然死の高い傾向を特徴としている。Renovacor,Inc.(“Renovacor”)を買収したため, われわれは現在,AAV 9による組換え遺伝子療法によりBAG 3拡張型心筋症(DCM)の進展を緩和または阻止することが可能であり,DCMは最もよく見られる心筋症形式であり,心壁が徐々に薄くなり,心腔が拡大し,ポンプ血ができないことが特徴である。印税許可協定によると、私たちはこれらすべての候補製品の世界的な商業化と開発権を持っている。
2021年12月からロケット賛助を行わないことが決定したRP−L 401の臨床評価は,学術革新者に返還された。遺伝子治療は、このような疾患を有する患者に有益である可能性があると信じているが、納得できる臨床データおよびこれらの深刻な児童および若年疾患の治療進展に基づいて、我々は、RP−A 601、RP−A 501、RP−L 102、RP−L 201、RP−L 301およびBAG 3−DCMの進行に利用可能なリソースを集中させることを選択する。
遺伝子治療の概要
遺伝子はデオキシリボ核酸(“DNA”)配列から構成され、デオキシリボ核酸はタンパク質に暗号を提供し、これらのタンパク質はすべての生きている生物の中で広範な生理機能を実行する。遺伝子は代々受け継がれているが,この過程で遺伝子変化,いわゆる変異が起こる可能性がある。これらの変化は,タンパク質の産生に乏しい,あるいは機能低下や異常な変化を引き起こすタンパク質を産生する可能性があり,逆に疾患を引き起こす可能性がある。
遺伝子治療は1種の治療方法であり、単離された遺伝子配列或いはDNA断片を患者に応用し、最もよく見られる目的は遺伝子突然変異による遺伝病を治療することである。現在、多くの遺伝性疾患の既存の治療方法は大蛋白或いは酵素の応用に重点を置いており、通常は疾病の症状のみを対象としている。遺伝子治療は遺伝子配列の機能コピーを直接患者の細胞に伝達することによって、欠損或いは機能失調の遺伝子の発病影響を解決し、それによって簡単に症状を解決するのではなく、遺伝病を治愈する潜在力を提供することを目的としている。
我々は改良された非病原性ウイルスを用いて我々の遺伝子治療法を開発している。ウイルスは細胞を透過し,細胞内で遺伝物質を輸送することが得意であるため,輸送手段として特に適している。我々のウイルス伝達ツールを作成する際には,ウイルス(原因)遺伝子が除去され,代わりに欠失や変異遺伝子の機能形態があり,これが患者の遺伝性疾患の原因となる。欠失或いは突然変異遺伝子の機能形式は治療用遺伝子、或いは“遺伝子組換え”と呼ばれる。遺伝子組換えを体内に移植する過程を“遺伝子組換え”と呼ぶ。遺伝子組換えウイルス遺伝子を用いてウイルスを修正すると,修正されたウイルスは“ウイルスベクター”と呼ばれる.ウイルスベクターは遺伝子組換えを標的組織或いは器官(例えば患者の骨髄内の細胞)に導入する。LVとAAVの2種類のウイルスベクターが開発されていますLVおよびAAVに基づくわれわれの計画は,患者に持続的(長期)の著明な治療利益を提供する可能性があると信じている。
遺伝子療法は2つの療法のうちの1つであることができる(1)離体する(インビトロ)、この場合、患者の細胞が抽出され、ベクターがbr制御された安全な実験室環境内のこれらの細胞に送達され、その後、修正された細胞が患者に再注入されるか、または(2)体内にあるこの場合、ベクターは患者に直接注入され、br}静脈内注射(“IV”)または標的部位の特定の組織に直接注入され、トランスジェニックを標的細胞に搬送することが目的である。
5
カタログ表
科学の進歩、臨床の進歩及び遺伝子療法に対するより広範な監督管理は、遺伝子療法製品の発展を推進するために希望に満ちた環境を創造したと信じている。これらの製品は細胞機能の回復と臨床結果の改善を目的としており、多くの場合早期死亡の予防を含む。FDAは近年いくつかの遺伝子療法を承認し、遺伝子療法製品に前方調節の経路が存在することを表明した。
基本用語
LVおよびAAV遺伝子治療を検討する際に使用されるいくつかのキータームおよび最適値範囲のサムネイルインデックスを以下に示す。
|
用語.用語
|
定義する
|
最適範囲
|
|
LV療法(血液病)
|
||
|
CD 34+細胞
|
造血幹細胞(ほとんどのCD 34+細胞は真の幹細胞ではないが、これは依然として臨床的に最も有用な方法である)
|
一般的に>100万CD 34+細胞/kgの潜在的な疾患に依存する。
|
|
ベクトルコピー数(VCN)[製品]
|
各入力幹細胞の平均遺伝子コピー数(DNA分析によって決定される;これは正確な値ではなく平均値である)
|
いくつかのLV臨床研究では、0.5から2が目標となっている(一般に最大値は5.0と考えられる)
|
|
ベクトルコピー数(VCN)
[体内にある後処理する]
|
末梢血液または骨髄細胞あたりの平均遺伝子コピー数(DNA分析によって決定される;これは正確な値ではなく平均値である)
|
潜在的な障害物にかかっていますが、多くの障害物は是正できるかもしれません体内にあるVCNs
|
|
AAV療法
|
||
|
ベクトルコピー数(VCN)
[体内にある後処理する]
|
関心のある器官における1細胞あたりの平均遺伝子コピー数(DNA分析によって決定される;これは正確な値ではなく平均値である)
|
潜在的な無秩序にかかっていますが生体生体VCNs
|
パイプの概要
次のグラフはロケット計画と候補製品の現在の開発段階を示している
心血管プロジェクト
達農病
ダ能病(“DD”)は多臓器リソソーム関連疾患であり、心不全による早期死亡を招くことができる。DDはオートファジーであるリソソーム関連膜蛋白2(“LAMP−2”)をコードする遺伝子変異によるものである。この変異は自食空胞の蓄積を招き,主に心筋や骨格筋にある。男性患者は心臓移植を必要とすることが多く,通常10代あるいは20代で進行性心不全で死亡する。深刻な心筋症以外に、DDに関連する表現は骨格筋無力と知的障害を含むことができる。DD治療の特効薬は現在のところなく,通常うっ血性心不全(“CHF”)の治療に用いられる薬剤も終末期CHFに進展することは変化しないと考えられる。終末期うっ血性心不全患者は心臓移植を受ける可能性があり,現在少数の患者のみが心臓移植を受けることができ,これは重篤な短期や長期合併症に関与しており,長期的には治癒できない。RP−A 501臨床試験中です体内にあるDDの治療は米国とEUで15,000から30,000人の患者が推定されている。
6
カタログ表
DDはX連鎖の優性,単遺伝子まれな遺伝性疾患であり,進行性心筋症を特徴としており,心臓移植が利用可能な場合でも男性ではほぼ一般的に致命的である。DDは主に生命早期の男性に影響を与え,その特徴は欠乏であるLAMP 2 B心臓や他の組織に発現していますDDの前臨床モデルは、AAVを介した心臓形質導入による心臓再建を証明しているLAMP 2 B心機能の発現と改善。
我々は現在,DDに対する腺関連ウイルスベクター計画,すなわちRP−A 501を持っている。著者らはRP-A 501段階1臨床試験で7名の患者を治療し、この試験は成人/老年青少年と児童男性DD患者を組み入れた。これは、15歳以上の成人/老年青少年患者の低用量(6.7 e 13ゲノムコピー(GC)/kg)を評価する第1のキュー(n=3)、15歳以上の成人/老年青少年患者のより高い用量(1.1 e 14 GC/kg)を評価する第2のキュー(n=2)、および低用量レベル(6.7 e 13 GC/kg;n=2)の小児科キューを含む。先に開示されたように、高用量キュー(1.1 e 14 GC/kg用量)の治療を受けた患者は進行性心不全を発症し、治療後5ヶ月で心臓移植を受けた。この患者の病態は,他の4名の低用量および高用量治療を受けた成人/老年青少年患者よりも深刻であったことは,治療前の心エコー図上のベースライン左心室(LV)駆出率(35%)の低下と有意に上昇したLV充満圧から証明された。患者の臨床経過はDD進展の特徴がある。患者は移植後良好であった。
低用量群で観察された予備治療効果、および高用量群(血栓性微小血管疾患(TMA))で観察された補体媒介安全性の問題から、FDAとのプロトコルでは、低用量(6.7 e 13 GC/kg)に焦点を当て、本試験では、1.1 e 14 GC/kg以上の用量は使用しなくなる。追加のセキュリティ対策が実施され、更新された試験
スキームに反映されている。これらの措置は、末期心不全患者の排除と、補体活性化を防止することに重点を置いた一過性B細胞およびT細胞媒介阻害を含む改善された免疫調節レジメンを含み、同時に、より低いステロイド用量およびより早いステロイド削減を可能にし、すべての免疫抑制治療はRP−A 501使用後2~3カ月で停止する。
第一段階の臨床研究において、著者らは患者の利益の将来性を評価するために、各種の治療効果評価を行っている。これらの評価には以下のようなものがある
| • |
ニューヨーク心臓協会(“NYHA”)の機能分類は最もよく使われる心不全分類システムである。NYHA II級は患者が軽微な体力活動制限を示し、休憩時に快適であるが、普通の体力活動は疲労、動悸及び/或いは呼吸困難を招く。クラスIとは,患者が体力活動の制限を示さず,通常の体力活動が過度の疲労,動悸および/または呼吸困難をもたらさないことである。レベルIIIおよびレベルIVは、より深刻またはより深刻な心不全と考えられている。
|
| • |
脳性ナトリウム利尿ペプチド(“BNP”)は血液を基礎とする評価指標であり、心不全の重要なマーカーであり、CHFと心筋症に対して予後意義がある。BNP上昇は心不全悪化と心血管疾患の予後不良と密接に関連している。
|
| • |
高感度トロポニンI(HsTnI)は血液を基礎とする評価指標であり、心臓損傷の重要なマーカーでもあり、DD患者の中(例えばBNP)はよく上昇し、すでに末期疾病患者の中で明らかに上昇することが証明された。
|
| • |
心臓厚の心エコー測定で最も注目すべきは左室質量(“LVM”)と最大左室壁厚(“MLVWT”)であり,心臓肥厚の程度を示している。
|
| • |
カンザスシティ心血管調査アンケート(“KCCQ”)は有効であり、患者報告の結果評価であり、患者の心不全症状、疾病が身体と社会機能に与える影響、及び心不全が全体の健康状況と生活の質に与える影響を評価する。評価点数は0(健康状態が非常に悪い)から100(健康状態良好)まで様々であった。KCCQは+/−5点に分類される変化
は有意と考えられ,結果と相関していることが証明されている。
|
| • |
ヘマトキシリン−エオジン(“H&E”)組織学と電子顕微鏡により心内膜心筋生検を組織学的に検査し,自己貪食空胞の存在と筋原線維構造の破壊を含むDD関連組織障害の証拠を検出し,いずれもDD関連心筋障害の特徴である。
|
| • |
心内膜心筋生検標本におけるLAMP 2 B遺伝子の発現を免疫組織化学とWesternブロットにより測定し,RP−A 501治療後のDD心筋組織にLAMP 2 B蛋白が存在することを確認した。
|
2022年5月25日にこれは…。アメリカ遺伝子と細胞治療学会(“ASGCT”)年会で、著者らは低用量6.7 e 13 GC/kg治療を受けた3人の患者と高用量1.1 e 14 GC/kg治療を受けた2人の患者の安全性と治療効果の更新、および低用量6.7 e 13 GC/kg治療を受けた2人の小児科患者(児童列年齢8-14歳)からの初歩的な安全性情報を含む、我々の第1段階研究のデータを開示した。
7
カタログ表
治療後12−24カ月で高用量および低用量成人/高齢若年者群(年齢15歳)において,RP−A 501は良好な耐性を維持し続けたことを報告した。前述したように,低用量(6.3 e 13 GC/kg)の成人/老年青少年列には薬物製品に関連するSAEは認められなかった。最もよく見られる不良事件は主に軽微であり、臨床症状と関係なく、治療後のトランスアミナーゼの上昇と関係がある。すべての3つの低用量成人/老年青少年行列患者において、トランスアミナーゼとクレアチンキナーゼの上昇が観察され、治療後の最初の1~2ケ月以内にベースラインレベルまで回復した。その中の3名の患者の中で2名の患者の血小板は一過性と可逆性の低下が出現した。これらの変化はコルチコステロイドや他の免疫抑制療法に対する反応である。低用量群の2人の患者と高用量群の1人の患者は,ステロイド減量治療により軽快したコルチコステロイド治療に関連する骨格筋症を経験した。2020年に開示されるように、高用量キュー内の1人の患者(同じ患者が後に上述した心臓移植を受けた)は、医薬製品に関連する深刻な有害事象に分類される非持続性免疫関連事象を経験する。このTMA事象(その後突発的に新たに分類された意外な重篤な副作用)は,免疫を介した補体活性化による可能性が高いと考えられている, 可逆性血小板減少症と急性腎損傷を招き、Eulizumabと一時血液透析が必要である。この患者は絶対投与量が最も高く、次いで高ベースライン体重と比較的に高い用量レベル(1.1 e 14 GC/kg)である。支持治療により,TMAは完全に消失し,患者は3週間以内に正常な腎機能を回復した。患者の心臓移植進展はこの事件とは無関係であり,治療時の晩期DDによるものである。成人/老年青少年患者では,治療後の最初の2−4カ月に追加のSAEは認められず,治療に関連する有害事象はすべて可逆的であり,持続的な腎臓,肝臓,または他の後遺症はなかった。2名の小児科患者の初歩的な安全性評価により、成人/老年青少年行列と比べ、血小板は正常であり、補体活性化は減少し、補体に関連する不良イベントはなかった。この2名の小児科患者は治療後10日目からコルチコステロイドホルモンを徐々に減少させ,RP−A 501治療後数週間でDD関連のベースライン骨格筋症は有意に悪化しなかった。トランスアミナーゼや肝臓炎症パラメータ(血液中のγ−グルタミルトランスフェラーゼ,ビリルビン,凝固パラメータを含む)は治療後数週間で有意に増加しなかったことに注意されたい。
すべての成年/老年青少年(年齢15歳)患者は、免疫調節方案のコンプライアンスを観察し、そしてベースライン時に(>40%)左心室駆出率を保留し、分子、心エコーと機能パラメータに疾病の改善を示した。これらの患者は免疫組織化学により心臓LAMP 2 Bの持続発現が確認され,組織学的評価は自食空胞レベルの低下を示した。心エコー図はこれらの患者の心壁の厚さが安定或いは減少し、駆出率の改善或いは安定を示した。成人/老年青少年行列中の患者はBNPとNYHA分級,6分間歩行試験と報告された体力活動の増加において持続的な改善あるいは安定を示した。
2022年9月、著者らはアメリカ心不全学会(HFSA)会議で行われているRP-A 501第一段階試験の中期データを公表し、児童列の最新の安全性と初期治療効果パラメータ、及び低用量と高用量成人/老年青少年行列(15歳及び以上の患者;n=5)の長期治療効果パラメータ(データ締め切りは2022年9月27日)を含む。このデータは2022年11月に75%のこれは…。アメリカ心臓協会(“AHA”)年次総会。これらのプレゼンテーションの間、私たちはキューにわたるインクリメンタル安全な更新を提供します。以前と同じ要約すると、RP-A 501は6.7 e 13 GC/kg用量レベルで全体的な耐性は良好であり、成人/老年青少年と児童低用量列には薬品に関連する意外と深刻な不良事象或いは深刻な不良事象は観察されなかった。すべて観察された2種類の用量の副作用はすべて可逆的であり、成人/老年青少年グループは治療後2-3年間フォローアップし、児童グループは6-11ケ月フォローアップし、持続的な後遺症は観察されなかった。いかなる早期トランスアミナーゼとクレアチンキナーゼの上昇はベースライン或いは低下に回復し、任意の一過性加重のDD関連骨格ミオパチーはコルチコステロイド治療を中止した後に解決された。2022年9月にHFSAと2022年11月にAHAで提出した最新の安全データは再び証明され、RP-A 501は低用量で全体的な耐性が良好で、小児科と成人/老年青少年列における安全性は制御可能である。
小児科コホートでは,RP−A 501術後6カ月と9カ月のフォローアップで,両患者のNYHA分類(II級からI級)が改善した。成人/老年青少年コホートでは,免疫調節を密接にモニタリングした3名(低用量2名と高用量1名)でNYHA
級(II級からI級)の改善が認められ,密接なモニタリングを受けていない成人患者1名にNYHA級の安定が認められた。6カ月と9カ月のフォローアップでは,両小児科患者とも心不全のキーマーカーであるBNPの著明な改善(減少)が認められ,これらの評価のレベルは基準値の50%を下回った。br}両小児科患者では6カ月と9カ月のフォローアップでhsTnIの改善(減少)が認められ,これらの評価のレベルは基準値の20%未満であった。成人/老年青少年列では、3人の低用量患者と1人の高用量患者のhsTnIの低下が観察され、この4人の患者は少なくとも1回の評価でベースラインレベルの50%を超え、24-36ケ月のフォローアップによって持続的に低下した。少なくとも1つの評価において、3人の低用量患者および1人の高用量患者のBNPは、基準値よりも少なくとも25%低下した。2人の成人/年上青少年患者のうち, 最近の評価では、BNPレベルはベースラインよりやや高かったが、この2人の患者のベースラインBNPレベルは正常範囲内であるか、またはやや上昇した。密接なモニタリング免疫調節を受けた成人/老年青少年コホート患者
(2つの低用量および1つの高用量)患者において、左心室(LV)後壁の厚さが改善され(治療前と比較して約15%~25%減少)、4人の患者において左室重量の減少が認められ、免疫調節を密接に監視していない低用量キュー中のbr}患者を含む。深刻な進行性心室壁肥厚は達能症肥大型心筋症の特徴であり、男性患者の早期死亡の主要な原因でもある。免疫組織化学は心臓LAMP 2 B遺伝子の持続発現を示し、標準H&Eと電子顕微鏡下の心臓組織構造と空胞の品質改善を示し、2種類の用量レベルの成人/老年青少年列中の5名の患者と児童行列中の2名の患者のうち4名の患者を観察した。24ケ月のフォローアップを通じて、免疫組織化学方法を通じて厳密なモニタリングを受けた免疫調節方案のすべての3名の成人/老年青少年患者の心臓LAMP 2 B遺伝子の持続発現を観察した。重要なのは,遺伝的修復(例えば心筋ベクターコピー数(“VCNs”)やLAMP 2蛋白発現証明)は,心筋面積全体に対する自食空胞の相対面積の減少に伴う, ベースラインと比較して、4人の成人/老年青少年プラトゥーン患者において、この比率は少なくとも20%の低下が認められた(うち、3人の患者の低下幅は少なくとも50%であった)。治療後6ケ月にこのパラメータを評価できる1名の小児科コホート患者においても、空胞面積の有意な減少(>50%ベースライン)が認められた。NYHA分類で認められた改善を除いて,KCCQにより報告された生活の質(QOL)の改善は,免疫調節を密接にモニタリングした成人/老年青少年患者3名および小児科コホート患者2名で注目された;ベースラインのKCCQスコアは最初の小児科患者で50点,最近の9カ月評価で93点,ベースラインのKCCQスコアは2位の小児科患者で52点,予備の3カ月評価で81点であった。
8
カタログ表
2022年12月22日、RP−A 501に関するFDAとの第1段階終了会議の最新状況を発表した。会期中にFDAとともに陽性の第一段階データセットを検討し,進行中の研究遺伝子療法の臨床開発のための研究設計と終点を提案した。FDAとの検討後,6.7 e 13 GC/kgの投与量を継続する予定であり,単腕開放ラベル試験
設計と強力な自然病歴比較器を用いる予定であるが,これはFDAが達農病における無作為対照試験の挑戦を認めているためである。FDAはまた,機能や生活の質評価支援のバイオマーカーによる複合終点を患者が利益を得る尺度とすることを考慮したいと述べている。研究の適切な外部制御や加速承認の適切なゴールを支援することを含め,FDAと我々が提案したキー実験の設計について対話を継続することを期待している.著者らは現在FDAと実験設計を検討しており、この設計は著者らの内部cGMP AAV施設で生産された薬物製品brを使用して治療した小児科患者を2名評価することができ、適度な規模の全世界の肝心な研究の初期構成部分とする。
2023年1月9日、41年間に実施されたRP-A 501第1段階研究の追加的な積極的な効果更新を公表したST年に一度のモルガン大通医療会議。提出されたデータには,数か月の追加追跡が含まれており,重要なバイオマーカー,心エコー,機能測定のさらなる改善が示されている。次の表は,これらの更新の要約を提供する著者らはまた追加の自然病歴比較データを提供し、これらのデータはI期患者の病気経過が重要なバイオマーカー(BNP)と機能指標(NYHA分類)において未治療患者の病気経過と明らかに異なることを示している。また,成人2名の治療後2−3年でもRP−A 501耐性は良好であった/年齢の高い青少年高用量および低用量プラトゥーン、ならびに8~13カ月の小児科キュー。小児科列では,著明な即時あるいは遅延毒性,著明な骨格筋症あるいは末期トランスアミナーゼの上昇は認められなかった。
1期RP−A 501研究において観察された重要なバイオマーカー、エコー結果、および機能対策の改善または安定
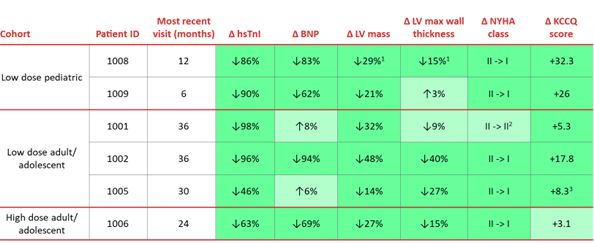
深い緑色=改善;浅い緑色=最小変化(安定)
PH 1試験で末期心不全を有しEFを伴うPT 1007
は含まれていない
1患者1008個の心エコーパラメータはM 9訪問であった(M 12未定)。
2患者1002は、M 30訪問NYHAレベル(M 36未定)として記述される。
3患者1005 KCCQスコアは、M 24アクセス(M 30保留)として記述されている。
これらの臨床更新に加えて、私たちの内部製造活動の最新の情報を提供します。我々は、力価および満粒子および空粒子の両方において第1段階材料よりも優れた2ロットのcGMP RP−A 501の製造に成功した。我々の内部で製造された製品品質の向上は、より低い総ウイルス粒子の全量注入を可能にし、RP−A 501の安全性
を潜在的にさらに最適化することができると信じている。さらに、FDAの同意を得て、商業化および我々の比較可能な方法および効力分析によってHEK−293細胞ベースのプロセスを継続することに同意した。
現在行われている達農病の第一段階試験の結果は、任意の心臓疾患に対する最も全面的な研究遺伝子治療データセットの一つを代表している。Rp−A 501の耐性は一般に良好であり,9カ月に及ぶフォローアップを得た小児科患者でも,36カ月に及ぶフォローアップを得た成人/老年青少年4名においても,RP−A 501は持続的な治療活性と達農病の改善を有することが証明されている。すべての密接なモニタリングを受けた免疫調節方案の成人/老年青少年と児童患者は組織、実験室と画像に基づくバイオマーカー及びNYHA分類(II級からI級まで)とKCCQ採点においてすべて改善があり、6~36ケ月のフォローアップを行った。
9
カタログ表
所期の一里塚
2023年2月7日、RP−A 501がFDAのRMAT認証を取得したことを発表した。我々は,FDAと達成できる疾患を治療するRP−A 501との高度な連携の持続的な対話に非常に鼓舞し,FDAとの持続的な対話と合意に基づいて,2023年第2四半期にグローバル研究の初期部分を開始する予定である。
不整脈心筋症(PKP 2−ACM)
不整脈を引き起こす心筋症(ACM)は1種の遺伝性心臓疾患であり、不整脈と突然死の高い傾向、心筋質量の進行性低下、深刻な右心室拡張、発育不良と心筋繊維脂肪の代替を特徴とする。心筋症は右室遊離壁によく見られるため、不整脈源性右室発育不良/心筋症(ARVD/ARVC)と呼ばれる。しかし,左主型と二室型も観察されたため,最近の用語ACMの使用を招いている。PKP 2遺伝子突然変異は家族性ACMの最もよく見られる遺伝発病要素である。PKP 2は親和性素-2蛋白をコードし、それは橋粒の1つの構成部分であり、橋粒は細胞間接着に参与する細胞間複合体である。PKP 2は心筋細胞間カルシウムシグナルの転写制御にも参与する。PKP 2変異を有する患者は通常ヘテロ接合体であり,PKP 2の心筋における発現の減少を示した。平均発症年齢は35歳であり,心室性不整脈,構造性心室性異常,心臓性突然死(SCD)を発生するリスクは非常に高い。
現在、ACMに対して非常に有効であることが証明されていない特定の利用可能な医学療法はまだ証明されておらず、現在の治療方案は標準的な心室性不整脈と心筋症のガイドラインに従い、その中に生活様式の変化(即ち運動制限)に関連し、β受容体遮断薬、抗不整脈薬物と利尿剤などの薬物治療を含む。これらの治療法の使用は不整脈負担や心筋症の重症度によって推進される。これらの療法は疾患の進行を変えることはなく,通常は対症療法および/または緩和支援のみを提供する。確定診断後、かなりの割合の患者は植入型心臓除細動器(ICD)を受け、一次或いは二次的に心室性不整脈とSCDの予防に用いられた。注目すべきは,ICDは治癒できず,画期的な生命を脅かす不整脈は死亡リスクが持続する可能性があり,ICDは終末期心不全への進展を阻止できないことである。ICD解雇は命を救ったが,身体的にも感情的にも創傷事件であった。末期心不全に進展した患者は心臓移植が考えられ,心臓移植は基礎疾患を治癒したが,それ自体も著明な発症率や死亡率に関与している。そのため,この群には高度に満足されていない医療ニーズが存在する。PKP 2−ACMの米国とEUにおける罹患率は5万人と推定されている。
我々は現在,PKP 2 aを発現する組換えAAVr.74ベクターであるPKP 2−ACMに対する腺関連ウイルスベクタープログラム,すなわちRP−A 601を有している。PKP 2-ACMは通常PKP 2遺伝子ヘテロ接合性発病突然変異によって引き起こされ、心筋中のPKP 2発現の低下を招く。病気経過の早期に一回の遺伝子治療を行い、疾病の根本的な原因(PKP 2欠乏)を解決し、早期の電気再構築を緩和することができ、そしてACMと関連する生命に危害を及ぼす不整脈とSCDのリスクを低下させ、それによって潜在的に不可逆的な心臓構造変化の発展を阻害する。シナプス発作の予防、生命に危害を及ぼす不整脈、SCD、ICDショック及びそれによる焦慮、不快感と入院治療は生活の質と生存効果を極めて大きく高めることが期待できる。また、この方法は患者に多種の不整脈や心不全薬物を一生服用する必要がなく、これらの薬物はPKP 2-ACM非特異性かつ自身の副作用と関連し、患者に運動制限がない状況で生活する機会を与え、不整脈、動悸、ICDショックと末期心不全への進展に対する懸念を減少させることができる。
スポンサーによる非臨床研究では,RP−A 601はPKP 2駆動を変更したACMの自然病歴の有効性を証明している。薬物治療を研究したPKP 2 CKO動物の生存時間は測定された最長時点(5ケ月)まで延長し、心臓拡張と心筋繊維脂肪代替/繊維化が減少し、左心機能が保護され、不整脈表現型は緩和された。未治療のPKP 2 CKOマウスの平均生存期間は約1ケ月であった。
所期の一里塚
われわれはすでにPKP 2−ACMに代表される動物モデルにおいてRP−A 601の臨床前概念検証を実現し,薬理学とGLP毒理学研究を完了し,GMP薬物製品を生産し,適切な効力分析を開発してI期研究を支援した。私たちは2023年第2四半期にINDを提出する予定です。
BAG 3拡張型心筋症
拡張型心筋症(“DCM”)は最もよく見られる心筋症であり,心壁が徐々に薄くなり,心腔が拡大し,血液をポンプできないことが特徴である。拡張型心筋症患者の20−50%に拡張型心筋症の家族性関連が認められ,40%と高い家族性患者が識別可能な遺伝的原因を有していた。BAG 3遺伝子変異(bcl-2に関連するathanogene 3)は家族性DCMで観察されるよりよく見られる発病遺伝変異の一つであり、これらの変異は高度な透過性を有し、約80%の人がBAG 3発病遺伝変異を有する 遺伝子は40歳以上でDCM
が発生する。BAG 3蛋白は多種の細胞機能と関係があり、心筋収縮、蛋白質品質制御(補助パートナーとして)、心筋細胞構造支持と抗アポトーシスを含む。BAG 3関連性拡張型心筋症(BAG 3-DCM)は発病が早く、進展が速く、病死率が高い。BAG 3の流行は-米国の関連DCMの数は30,000人にのぼる。
10
カタログ表
現在,BAG 3を有する拡張型心筋症患者 変異患者は、アンギオテンシン変換酵素阻害剤、アンギオテンシン受容体遮断薬、ネプリシン阻害剤、β-アドレナリン受容体アンタゴニストまたはβ-受容体遮断薬、アルドステロン拮抗薬および/または利尿剤、およびいくつかの生活様式の変化を含む心不全治療基準に従って治療されるが、疾患の根本的な原因を解決することはできない。特定のパラメータに適合する患者はまた、埋め込み型除細動器、心臓再同期装置、または両方の組み合わせを受けることができる。BAG 3関連拡張型心筋症の潜在的機序に対する直接治療法はなく,BAG 3関連拡張型心筋症と診断された患者は,BAG 3変異のない拡張型心筋症患者よりも終末期心不全や死亡に急速に進展しているようである。例えば、約19%のBAG 3 DCM患者は、確定診断後12ヶ月に機械的心臓支持、心臓移植または心不全関連死を必要とし、この割合は、分期に類似した非BAG 3 DCM患者のほぼ2倍である。
2022年12月、我々はRenovacorの買収を完了し、RocketにRenovacorの最先端の計画を提供し、これはAAV 9に基づく組換え遺伝子療法であり、心筋細胞中のBAG 3蛋白のレベルを増強し、BAG 3 DCMの進展を緩和または阻止するために、機能的に整ったBAG 3遺伝子
を提供することを目的とした。AAV 9−BAG 3の概念はBAG 3ノックアウトマウスモデルの研究で初歩的に検証されており,注射後4週間と6週間の時点で治療したBAG 3ノックアウトマウスの駆出スコアは未治療のノックアウトマウスより改善し,駆血スコアは歩行試験対照群に相当することが示されている。
所期の一里塚
私たちはこの計画の最適な発展経路を評価し、2024年上半期にBAG 3-DCMのINDを提出する予定だ。
血液学の授業
ファンコニ貧血補充A群(FANCA)
FAは1種の稀な生命を脅かすDNA修復障害であり、通常単一FA遺伝子の突然変異によって引き起こされる。60%から70%の症例がFanconi−A(“FANCA”)遺伝子変異によるものと推定されており,
がわれわれの計画の重点である。FAは骨髄不全、発育異常、骨髄性白血病とその他の悪性腫瘍を招き、通常生命の早期と数十年に発生する。骨髄再生不良性貧血はもういかなる或いは極めて少ない赤血球、白血球と血小板を産生しなくなり、感染と出血を招く骨髄であり、FAの早期発病率と死亡率の最もよく見られる原因であり、中位の発病年齢は10歳前である。白血病は次の最もよく見られる死亡原因であり、最終的に約20%の患者の晩年に発生する。固形臓器悪性腫瘍、例えば頭頚部癌も発生する可能性があり、生命の頭の20年から30年の間に発病率は比較的に低いにもかかわらず。
同種(ドナーを介した)造血幹細胞移植(“HSCT”)は現在FAを治療する最もよく使われる治療方法であり、この治療法の改善はFAの血液学的是正を更に頻繁にさせるが、HSCTは依然として急性と長期リスクと関係があり、移植関連死亡、移植片対宿主病(GVHD)、異遺伝子移植の時々致命的な副作用を含み、その特徴は消化管潰瘍、肝臓毒性と皮疹、及び後続癌リスクの増加である。我々はFAの遺伝子治療計画において,患者自身の幹細胞を用いて生命の早期に最も毒性の低い血液矯正を行うことを目指している。広く適用されている自己遺伝子療法の発展は,これらの患者に革命的な影響を与えると信じている。
著者らの各血液学プロジェクトは第三世代自己不活化レンチウイルスベクターを用いて患者のHSCsの欠陥を是正し、HSCsは骨髄で発見された患者の一生に血球を産生できる細胞である。造血幹細胞遺伝子コードの欠陥は深刻で、生命を脅かす可能性のある貧血を招く可能性があり、即ち患者の血液は十分な正常機能の赤血球が不足して酸素を全身に運ぶことができる。幹細胞欠陥はまた深刻な、生命を脅かす可能性のある白血球減少を招き、感染感受性を招き、及び血液凝固を招く血小板減少を招く可能性があり、これは深刻で、生命を脅かす可能性のある出血事件を招く可能性がある。FA患者の遺伝欠陥は骨髄中の血球内遺伝子と染色体の正常な修復を阻害し、これはよく急性骨髄性白血病(AML)の発展を招き、これは血液癌の一種、及び骨髄不全と先天性欠陥である。FA患者の平均寿命は30歳から40歳と推定される。米国とEUにおけるFAの罹患率は合計約4,000名の患者と推定されている。非条件患者に対する治療効果を考慮して、現在アメリカとEUの毎年の潜在市場機会は合計400から500名の患者であると考えられている。
11
カタログ表
我々は現在,FAを標的とした体外LVプログラム,すなわちRP−L 102を持っている。RP-L 102は私たちがリードしているレンチウイルスベクターベースのプログラムであり、私たちはスペインマドリードの有力な研究機関Centro de Investigacy Energéticas,
Mediobientales y Tecnol≡gicas(“CIEMAT”)から許可を得た。Rp-L 102は現在、著者らの第二段階登録臨床試験で研究されており、これらの臨床試験はスタンフォード大学医学院(“Stanford”)、ロンドンのミネソタ大学オーモンデストリート病院(“GOSH”)、スペインの乳児ニノ-イエス病院(“HNJ”)の最終的かつ根治医学センターでFA患者を治療している。この試験は米国とEUから10名の患者を募集した。他の2名の患者はスタンフォード大学の米国第一段階研究で治療を受け,計12名の患者がRocket協賛の臨床試験でRP−L 102治療を受けた。患者は新鮮な細胞を用いたRP-L 102静脈注入と、改善された幹細胞濃縮過程、形質導入増強剤および商業レベルの担体と最終薬物製品を結合した“B過程”を受けた。
治療後少なくとも1年の時点で,DNA損傷剤であるマイトマイシン−Cに対する骨髄幹細胞の耐性が,我々が行っている第2段階研究の主な終点である。FDAおよびEMAとのプロトコルによれば、骨髄修復が10%以上のマイトマイシン-C耐性閾値を超えるインプラントは、上場申請の承認をサポートすることができる。
2021年12月には63カ国で研究開発アメリカ血液学会(“ASH”)年次総会。ASHに掲示された1/2期試験の初歩的な結果は11名の小児科患者から来て、ますます多くの証拠を示して、8名の少なくとも12ケ月のフォローアップを行った患者の中で、6名の患者はMMCを移植し、6名の患者を含む骨髄前駆細胞のMMCに対する抵抗率は16%-63%から様々である(FA患者の骨髄細胞はMMCを含むDNA損傷剤に高度に敏感である;このようなDNA損傷に対する敏感性はFA関連骨髄不全と悪性病変感受性の原因の一つと考えられている。骨髄造血細胞がMMCに耐性を産生する以外、少なくとも12ケ月のフォローアップを行った7名の患者の中で、6名の患者の末梢VCNレベルは持続的に上昇した。1人の患者は治療を受けた後約9ケ月にB型インフルエンザ感染が出現し、そして進行性血液学的不全を伴い、同種造血幹細胞移植が必要であり、治療は成功した;残りの患者は輸血を必要としなかった。RP−L 102は高度に良好な耐性を示し,すべての被験者は細胞毒性調節なしに治療を受け,異型増殖の徴候もなかった。これまで,RP−L 102に関連する唯一の重篤な副作用は,1人の患者の2段階一過性輸液関連反応であった。
2022年5月、ASGCTの第25回会議でRP-L 102のバックラインデータを示したこれは…。忘年会です。2022年4月4日までに評価可能な9名の患者のうち,5名の患者の骨髄コロニー形成細胞のMMCに対する耐性が増加し,12~18カ月で21%から42%,18~21カ月で51%から94%に増加した。主要な終点はすでに実現されており、これは、統計的および臨床的意義がベースラインよりも少なくとも10%高いMMC抵抗を得るために、少なくとも5人の患者が2つ以上の時点でMMC抵抗、およびそれに伴う遺伝的是正および臨床的安定の証拠を得る必要がある試験スキームに基づく。6例目の患者の末梢VCNは徐々に増加した遺伝校正の証拠を示した。他の3名の患者は治療後12カ月以内に症状が出現した。1人の患者は治療後に進行性骨髄不全が出現し、先に開示されたように成功した同種移植を受けた。RP−L 102の耐性は良好で,異型増生,クローン優性あるいは発癌統合の徴候はなく,先に報告したように,1名は2段階一過性輸液関連反応を経験し,この反応は消失した。
2022年10月にヨーロッパ細胞と遺伝子治療学会29でRP−L 102のデータを発表しましたこれは…。ASGCT 2022会議で提出された臨床活動結果を含む年次会議。また、我々のRP-L 102によるFA治療の第2段階試験において、他の3人の患者のうち少なくとも1人の患者のフォローアップ時間は12ヶ月未満であり、彼らの初歩的な移植証拠(例えば、骨髄マイトマイシンC耐性と血液と骨髄中のVCN)は5人の患者に見られるレベルに相当し、この5人の患者はより長期的な移植と表現型是正証拠を持っていることを開示した。この試験で最初にインプラントの証拠があった5名のうち,RP−L 102治療を受けて約22カ月後にT細胞リンパ芽球性リンパ腫に進展したことも明らかになった。リンパ腫の手術生検では,血液と骨髄に伴うVcnがそれぞれ0.003.26と0.42の場合,無視できる遺伝子マーカー(Vcn 0.42)を示した。これらの発見は,腫瘍には遺伝子マーカーがほとんどないため,リンパ腫はLVを介した挿入によるものではないことが最終的に示唆された(非常に低いが検出可能なVCNは腫瘍標本中の血球の結果である可能性が高い)。FAは癌感受性症候群であり、癌は10歳以下の患者に発生する可能性があります。重要なのは、リンパ腫の誘導化学療法に明らかな合併症がなく、現在完全に緩和されている状態です。遺伝子矯正の造血細胞の存在は,化学療法に対する患者の全体的な耐性に寄与する可能性がある。
2022年12月に64カ月のRP−L 102陽性臨床データを公表したこれは…。12ケ月のフォローアップの中で、10名の評価可能な患者のうち少なくとも6名は表現型是正を獲得し、骨髄由来のコロニー形成細胞のマイトマイシンCに対する耐性が増加し、それに伴う遺伝是正と血液学的安定を示した。7名の患者の末梢血液と骨髄VCNにより、MMC抵抗の最新発展と36ケ月のフォローアップの可能な血液学的安定性指標に伴い、すでに次第に増加した遺伝校正の証拠を示した。主要な終点はすでに達成し、1つの試験方案に基づいて、この試験方案の中で、統計と臨床意義は少なくとも5人の患者が2つ以上の時点でベースラインより少なくとも10%高いMMC抵抗の増加を獲得し、遺伝的是正と臨床安定を伴う証拠を得る必要がある。RP−L 102の安全性は非常に良好であり,この治療は何の細胞毒性調節もなく,耐性は良好である。RP−L 102に関連する骨髄異常増生,クローン性優性あるいは挿入変異は認められなかった。
12
カタログ表
所期の一里塚
我々の重要なFA第2段階研究で定義された主要な終点の実現に基づいて,我々はすでにバイオ製品ライセンス申請(BLA)に関するFDAの対話を開始し,FAを治療するRP−L 102のための計画を提出し,2023年第4四半期にこのような申請を提出する予定である。
白血球粘着欠陥-I(LAD-I)
LAD-Iは稀な常染色体劣性白血球粘着と遷移疾患であり、β-2インテグリン成分CD 18をコードするITGB 2遺伝子突然変異によって引き起こされる。CD 18欠損brは好中球(抗感染白血球の1つの亜群)が血管から組織に入る能力を損傷させ、これらの細胞は感染に対抗するためにこれらの細胞を必要とする。多くのまれな疾患の場合と同様に,発症率の正確な推定を確認することは困難であるが,これまでに数百例が報告されている。LAD−I患者の多くは重篤な疾患と考えられている。同種造血幹細胞移植を受けていない患者では,重篤なLAD−Iが再発し,生命を脅かす感染や大量の乳児死亡率に注目すべきである。異遺伝子HCSTがない場合,重篤なLAD−Iの死亡率は2歳前に60%から75%と報告されている。
私たちは今1軒例えばLAD-I、RP-L 201のための生体プログラム。RP−L 201はCIEMATから許可を得た臨床プログラムである。我々はすでにカリフォルニア大学ロサンゼルス校と協力し,米国LAD−I計画の臨床開発をリードしている。カリフォルニア大学ロサンゼルス校及びそのELIとEdythe BRoad再生医学と幹細胞研究センターはLAD-I登録臨床試験の主要なアメリカ臨床研究センターであり、HNJとGOSHはそれぞれスペインとロンドンで主要な臨床サイトを担当している。この研究はLAD−I遺伝子療法の臨床開発を支援するためにカリフォルニア再生医学研究所(CIRM)660万ドルのCLIN 2助成金を獲得した。
これまで,RP−L 201の開放ラベル,単腕,1/2期登録臨床試験は9名の重症LAD−I患者を治療し,RP−L 201の安全性と耐性を評価した。1人目の患者は2019年第3四半期にカリフォルニア大学ロサンゼルス校でRP-L 201治療を受けた。この研究の第1段階と第2段階の登録は現在完了しており,9名の患者は米国とヨーロッパの3つの研究センターでRP−L 201治療を受けている。
2021年12月、著者らは第63回ASH年次総会で陽性臨床データを提出した。ASH口頭報告には9名の重症LAD−I患者のうち8名の予備データが含まれており,CD 18発現が2%未満と定義されており,2021年11月8日にRP−L 201治療を受けており,データ締め切りは2021年11月8日である。8人の患者は少なくとも3ヶ月のフォローアップデータを有し、そのうち4人の患者のフォローアップ時間は12ヶ月以上である。すべてのRP-L 201の注入耐性は良好であり、薬品と関連する深刻な副作用報告は見られなかった。評価可能な8名のすべての患者に初歩的な治療効果の証拠が観察された。すべての8名の患者は好中球CD 18発現が4-10%を超える閾値を示し、成人期の生存と関連し、そして深刻なLAD-I表現型逆転と一致し、その中の6名の患者は少なくとも6ケ月のフォローアップを受けた。末梢血VCNレベルはずっと安定しており、各ゲノム0.54-2.94コピー範囲内である。RP-L 201術後にLAD-I関連性感染は1例もなく、入院治療が必要であった。2022年1月に発表された他の更新は、9人の患者が3カ月でCD 18発現が61%に達し、9人の患者のうち9人の患者がRP-L 102術後3~24カ月の時点で26%~87%のCD 18発現を示し、各患者が3カ月後にCD 18発現レベルが安定していることが初歩的に観察された。
2022年5月、ASGCT第25回年次総会で最新データを提出した。報告には,全9名の治療を受けた患者の輸液後3~24カ月のフォローアップ時の治療効果と安全性中期データ,および全体生存データが含まれており,2022年3月9日までの輸液後少なくとも12カ月フォローアップした7名の患者の生存データを含む。3カ月から9歳までのすべての患者はCD 18の持続的な回復を示し、10%以上の好中球HILに発現した(範囲:20%-87%、中央値:56%)。Kaplan−Meierの推定によると,1年後,コホートに異遺伝子造血幹細胞移植がない全体生存率は100%であった。データ遮断日まで、9名の患者はすべて生存し、臨床状況は安定していた。治療前と比較して,すべての患者の全原因入院率と重篤な感染発生率は統計的に有意に低下した。持続的な表現型矯正に伴い,LAD−I関連の皮疹の消退と創部修復能の回復の証拠が示されている。RP−L 201に関連する副作用がないすべての患者では,RP−L 201の耐性は非常に良好であった。他の研究プログラムに関連する有害事象は,ブチルチオダン調節を含め,以前に開示されており,これらの薬剤やプログラムの耐性プロファイルと一致している。
2022年12月私たちは64ヶ月のこれは…。ASH年次総会です。報告brは、以前に開示されたすべての患者がRP-L 201注射後3~24ヶ月間フォローアップした主要なデータと、7人の患者の注射後12ヶ月以上の全体生存データとを含む。Kaplan Meier推定では,注入後12カ月の総生存率は100%であり,全9名のLAD−I患者は3~24カ月の利用可能なフォローアップで,すべての入院,感染,炎症関連入院と入院期間の延長は統計的に有意に減少した。データはLAD−I関連皮疹の消退と創傷修復能の回復の証拠を示した。これまでRP−L 201に関連する重篤な副作用がなかったすべての患者において,RP−L 201の安全性は非常に有利であった。
13
カタログ表
所期の一里塚
第二段階重要LAD-I試験の積極的な治療効果と安全性データに基づいて、WEは、深刻なLAD−Iを治療するためのRP−L 201のBLA申請計画をFDAと検討し始めており、2023年第2四半期にこのような申請を提出する予定である。
ピルビン酸キナーゼ欠乏症
赤血球PKDは稀な常染色体劣性遺伝病であり、赤血球解糖経路の1つの構成部分をコードするピルビン酸キナーゼL/R(“PKLR”)遺伝子突然変異によって引き起こされる。PKDの特徴は慢性非球形溶血性貧血であり,これは赤血球が正常球形を呈しず分解され,細胞に酸素を輸送する能力が低下する疾患であり,貧血の重症度は軽度(無症状)から重篤であり,児童の死亡や頻繁,生涯輸血が必要となる可能性がある。児童群はPKD患者の中で最もよく見られ、最も深刻な亜群であり、PKDは通常脾腫大(脾異常増大)、黄疸と慢性鉄負荷を招き、これは慢性溶血と疾病治療に応用した赤血球注入の結果である可能性がある。貧血の重症度の変異性は,PKLR遺伝子に影響を与える可能性のある大量の異なる変異による部分と考えられている。アメリカとEUの白人人口の発病率は100万人当たり3.2から51例と推定されている。業界推定では,FDAの承認された分子標的療法が乏しいにもかかわらず,米国とEUでは少なくとも2500例の症例が診断されている。市場研究により、遺伝子療法をより広範な人群に応用することは市場機会を毎年約250名の患者から500名の患者に増加させることができる。
私たちは今1軒例えば生体LVに基づくPKDのためのプログラム、RP-L 301。RP−L 301はCIEMATから許可を得た臨床段階計画である。2019年10月、世界第1段階研究を開始したRP-L 301のINDが承認された。その計画はアメリカとEMA孤児薬物疾患の称号を与えられた。
この世界第一段階開放ラベル、単一腕臨床試験はアメリカとヨーロッパで4~5名の成人と児童PKD患者を募集する予定である。この実験は、子供(8~17歳)および成人群におけるRP−L 301を評価するために、2つのキューからなる。この試験はRP-L 301の安全性、耐性と初歩的な活性を評価することを目的とし、初歩的な安全性評価は成人行列で行い、その後児童患者に対して評価を行う。スタンフォード大学はアメリカ成人と小児科患者のトップになり、HNJはヨーロッパ小児科のトップになり、ディアス病院はヨーロッパ成人患者のトップになる。2020年7月,RP−L 301臨床試験で1位の患者を治療した。
2021年12月私たちは63ヶ月の研究開発ASH年次総会です。ASHポスターbrは,重篤な貧血と大量輸血需要を有する成人患者2名の初歩的なデータを紹介し,2021年11月3日締め切りに治療を受けた。これらの患者のすべては肝臓鉄過負荷を含む広範なPKD関連疾患合併症がある。両患者ともヘモグロビンレベルは有意に改善し,それぞれベースラインの7.4と7.0 g/dLから12カ月後の13.3 g/dLと14.8 g/dLに向上し,これは重篤な(Hb
)からの改善を表している

2022年5月に第25回会議で最新データを発表しましたこれは…。ASGCT年次会議。報告には,2022年4月13日現在の重篤または輸血依存性貧血を有する成人患者2名のデータが含まれている。注入18ケ月後、2名の患者は持続的な遺伝子組換え発現があり、ヘモグロビン正常化、溶血改善、移植後に赤血球注入要求がなく、生活の質を改善したことは逸話報告であり、正式な生活の質評価記録でもある。注入18カ月後、RP−L 301の耐性曲線は良好であり、RP−L−301に関連する深刻な有害事象は発生しなかった。両患者とも治療/調整後に一過性トランスアミナーゼの上昇が出現し,肝障害の臨床症状は出現せず,その後症状は消失し,臨床後遺症はなかった。小児科キュー
は現在募集中である。
2022年12月私たちは64ヶ月のこれは…。ASH年次総会です。報告には,重篤な貧血を有する成人患者2名からの積極的な更新データが含まれている。輸血24ケ月後、2名の患者はすべて強力と持続的な治療効果があり、正常化したヘモグロビン(ベースラインレベルは7.0-7.5 g/dL範囲内)、改善した溶血パラメータ、赤血球注入と独立及び生活の質を改善することを表現し、この逸話報告もあり、正式な生活の質評価記録もある。2人の成人患者は注入後24ケ月以内にRP-L 301と関連する深刻な副作用が出現しなかった。2名の成人患者のRP-L 301術後12ケ月の末梢血と骨髄中の挿入部位分析により、高度ポリクローナルのパターンは、突然変異を挿入する証拠がないことを示した。
所期の一里塚
第一段階研究でPKD成人と小児科列の登録を完了した。第2段階のキー試験は2023年第4四半期に開始される予定だ。
14
カタログ表
CGMP製
私たちはニュージャージー州クランベリーにある103,720平方フィートの新しい製造工場はすでに規模を拡大し、計画中の達農病の第二段階の重要な研究のためにAAV薬物製品を生産した。この施設は実験室
の研究開発と品質空間も持っている。我々はFDAと我々の内部施設でAAV cGMP生産を開始する化学,製造と制御要求および達農病第二段階キー試験の効力分析計画について了承した。2022年に私たちの製造と商業能力をさらに強化するために、私たちはMayo Pujolsを私たちの首席技術官に任命し、Mayo Pujolsは業界で最も経験の豊富な細胞と遺伝子治療技術運営と製造リーダーの一人である。
戦略.戦略
潜在的治癒による一流遺伝子療法の開発と商業化により,壊滅的で十分な治療が得られていないまれな小児科疾患を有する患者に希望と緩和をもたらすことが求められている。このような目標を達成するために、私たちは全面的に統合されたバイオテクノロジー会社に発展するつもりだ。最近と中期には、満足されていない大量の需要を有する破壊的疾患に対して、独自の内部分析および製造能力を開発し、現在計画されている計画の登録試験を継続して開始する当社一流の候補製品を開発する予定である。中長期的には,われわれの残りの臨床プロジェクトにBLASを提出し,われわれの遺伝子治療プラットフォームを構築し,われわれのチャネルを拡大し,われわれの遺伝子治療技術と互換性があると考えられるより多くの適応を狙う予定である。また、その間、私たちが現在計画している計画は、FDAが提供する加速的な審査の優先審査証明書を取得する資格があると信じている。著者らは細胞と遺伝子治療、稀な疾病の薬物開発と製品審査の面で専門知識を持つ指導者と研究チームを結成した。
私たちの競争優位性は私たちが疾病の選択方法に基づいていることであり、これは厳格な過程であり、目標疾患を識別する明確な基準があると信じている。この資産開発方法は私たちを遺伝子治療会社として頭角を現し、先発優位を提供してくれる可能性があると信じています。
知的財産権
我々は、内部開発でも第三者から許可を得ても、特許権を求め、維持し、擁護することを含む、我々の業務発展に重要なビジネス的意義を有するノウハウ、発明、改善を保護し、強化するために努力している。私たちはまた、私たちのノウハウプラットフォームおよびノウハウに関連するビジネス秘密、持続的な技術革新、許可内の機会に依存して、遺伝子治療分野における私たちの独自の地位を強化し、維持することが、私たちのビジネス発展に重要であるかもしれません。また,孤立した薬物指定,データ独占,市場独占,特許期限延長(あれば)による規制保護にも依存する予定である。
私たちのビジネス成功は、私たちのビジネスに関連する重要なビジネス技術、発明およびノウハウの特許および他の独自保護を取得および維持する能力があるかどうか、私たちの特許を保護および実行する能力があるかどうか、私たちのビジネス秘密を秘密にし、第三者が効果的に強制的に実行可能な特許および独自の権利を侵害することなく運営される可能性がある。第三者がその将来の製品を製造、使用、販売、提供、または輸入することを阻止する能力は、これらの活動をカバーする有効かつ強制的に実行可能な特許または商業機密の下で当社が所有する権利の程度に依存する可能性がある。許可されたおよび会社が所有する知的財産権については、私たちの任意の未解決特許出願または将来提出された任意の特許出願が特許を得ることを保証することはできません。私たちはまた、私たちの商業製品を保護し、これらの製品を製造する方法において、私たちの既存のいかなる特許または将来私たちに付与される可能性のあるいかなる特許も保証することはできません。
我々は,大量の特許や特許出願を開発·許可し,遺伝子治療製品の開発や商業化に関する多くの技術的ノウハウやビジネス秘密を持っている。特許および非特許知的財産権を含む我々の独自の知的財産権は、通常、遺伝子発現ベクターおよびそれを用いた遺伝子治療の方法である。2023年2月22日現在、私たちの特許組み合わせは、私たちの候補製品および関連技術に関連する自社およびライセンス特許シリーズを含み、以下では、より包括的な議論を行う。
ファンコニ貧血
我々のFanconi貧血特許の組み合わせは、オーストラリアおよび日本における付与された特許と、Fanconi貧血相補群遺伝子を含む多核酸カセットおよび発現ベクター組成物と、そのようなベクターを用いて哺乳動物細胞においてFanconi貧血を治療する遺伝子治療を提供する方法とを含む米国、ヨーロッパ、日本、中国および他の国で出願されている特許を含む。これらのアプリケーションは、CIEMAT、CIBER、Fundacion Instituto de Investigation Sanitaria Fundacion Jimenez Diaz(“FISFJD”)およびFundacion Para Investigacion Biomedica del Hospital Del Ninoによって独占的に許可されている。私たちは、このシリーズの任意の特許が発行され、適切なメンテナンス、更新、年金、または他の政府費用が支払われた場合、2037年に満期になり、いかなる特許期限の調整や延長も行われないと予想する。
15
カタログ表
ピルビン酸キナーゼ欠乏症
我々のPKD特許の組み合わせは、ヨーロッパ、中国、香港、日本、メキシコ、韓国および米国の許可された特許、ならびに米国、EU、日本、中国および他の国および地域の係属中の特許出願(Br)が、ピルビン酸キナーゼ遺伝子を含む多核酸カセットおよび発現ベクター組成物を含み、そのようなベクターを使用して哺乳動物細胞においてピルビン酸キナーゼ欠乏症を治療する遺伝子治療を提供する方法を含む。これらの
アプリケーションはCIEMAT,CIBER,FIISFJDによって独占的に許可されている.私たちは、この製品の組み合わせのいずれかの特許が発行され、適切なメンテナンス、更新、年金、または他の政府費用が支払われた場合、2037-2038年に満了し、特許期限の調整または延長は行われないと予想する。
達農病
私たちのダ農病特許の組み合わせは、固有の知的財産権と、米国、ヨーロッパ、日本、中国、および他の国/地域の特許出願を含むカリフォルニア大学サンディエゴ校によって許可された特許シリーズを含み、これらの特許は、達農病の治療を必要とする。この製品の組み合わせのいずれかの特許が発行され、適切なメンテナンス、更新、年金、または他の政府費用が支払われた場合、いかなる特許期限の調整または延長も行わずに、
は2037年に満了すると予想される。我々は米国特許を有し,EU,日本,中国などの特許を出願しており,達成能br病に対する遺伝子治療ベクターであると主張しており,この米国特許は2020年に発行されている。適切なメンテナンス、更新、年金、または他の政府費用が支払われた場合、これらの特許出願によって生成された任意の特許は、特許期間の調整または延長を必要とせずに2039年に満了すると予想される。私たちはまた、達能病を治療する方法に対する追加特許出願を提出した。適切な保守、更新、年金、または他の政府費用が支払われた場合、これらの特許出願によって生成された任意の特許は、いかなる特許期間の調整または延長を行うことなく、2040−2041年に満了すると予想される。
白血球粘着欠陥(LAD-I)
私たちの特許の組み合わせは、我々のLAD-I計画に関連する可能性がある同種造血幹細胞移植の形質導入に関する米国、EU、日本、中国、および他の国の係属中の特許出願を含む。私たち
は、これらの特許出願によって生成された任意の特許が発行され、適切なメンテナンス、更新、年金または他の政府費用が支払われた場合、2039年に満期になり、いかなる特許期限の調整や延長も行われないと予想する。
私たちの目標は、私たちの遺伝子治療製品の候補および製造プロセスを保護するために、その特許と特許出願の組み合わせを拡大し続けることである。私たちはまた、私たちが所有または独占的に許可された特許と特許出願との組み合わせを再許可する機会を時々評価することができ、私たちは時々このような許可を締結するかもしれない。個別特許の期限は,特許を取得した国·地域の法的期限に依存する。私たちが出願を提出したほとんどの国では,特許期間は非仮出願を提出した日から20年である。米国では、特許期限は、特許期限
を調整することによって延長することができ、これは、特許権者が米国特許商標局が特許を付与する際の行政遅延による補償であるか、または前に提出された特許によって特許が最終的に放棄された場合、特許期間を短縮することができる。
FDAによって承認された薬物をカバーする特許期限は、FDA規制審査中に失われた特許期間を補償するために、米国特許の特許期限を回復することを可能にする特許期間延長資格に適合する可能性もある。ハッジ-ワックスマン法は特許期間を特許満了後最大5年間延長することを許可している。特許期間の延長の長さは,薬物が規制審査を受ける時間の長さと関係がある。1つの特許の残り期間を延長することは、製品承認日から合計14年間を超えることができず、承認された薬物に適用される特許を延長することしかできない。また,1つの特許が1回しか延期できないため,1つの特許が複数の製品に適用される場合には,1つの製品に基づいて延期するしかない.欧州や他の外国司法管轄区域にも同様の規定があり、承認薬をカバーする特許の有効期限を延長する。可能な場合には,臨床試験の時間の長さやBLA提出に係る他の要因から,我々の候補製品とその使用方法をカバーする特許出願のために特許期間を延長する予定である。
場合によっては、私たちは商業秘密に依存して私たちの技術を保護するかもしれない。しかし、商業秘密は保護するのが難しいかもしれない。私たちは、従業員、コンサルタント、科学コンサルタント、第三者と秘密保持協定を締結することで、私たちのノウハウとプロセスを保護することを求めています。我々はまた,その場所の物理的セキュリティとその情報技術システムの物理的および電子的セキュリティを維持することで,我々のデータやビジネス秘密の完全性と機密性を保護しようとしている.私たちはこれらの個人、組織、そしてシステムに自信がありますが、合意やセキュリティ措置は違反される可能性があり、私たちはどんな違反に対応するのに十分な救済措置がないかもしれません。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。私たちのコンサルタントまたは協力者が、私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連するまたはそれによって生じるノウハウおよび発明の権利について議論される可能性がある。
16
カタログ表
材料契約
CIEMATとのライセンス契約
2016年3月、CIEMAT、CIBER、FIISFJD(総称して“CIEMAT”と呼ぶ)とライセンス契約を締結し、PKDの治療分野でのみヒトPKLR遺伝子を含むレンチウイルスベクターに関連するいくつかの特許、ノウハウ、その他の知的財産権を付与するグローバル独占権利を付与した。協定条項によれば、(A)ライセンス知的財産権がカバーする1つまたは複数の製品またはプロセスの規制承認を開発および取得し、そのような製品またはプロセスを商業市場に導入し、それを合理的に公衆に提供する義務があり、(B)規制承認後少なくとも2年以内に少なくとも1つの許可された知的財産権にカバーされた製品またはプロセスを少なくとも1つの国で開発または商業化し、(C)ライセンス知的財産権を十分に使用する。道徳的で合法的な方法ですライセンスの交換としては,CIEMATへの前払い,任意のライセンス知的財産権に係る製品やプロセスに基づく純売上高の使用料支払い,開発と規制マイルストーン(Br)支払いおよび再許可収入支払いが義務付けられている。CIEMATと協力して、特許の起訴と維持を担当し、費用は私たちが負担します。また、CIEMATと協力して特許を侵害および/または挑戦から実施および保護する第一の責任を負っています。許可協定が発効してから5年以内に, 私たちはCIEMATが市場価値で獲得した許可知的財産権のいかなる改善も優先的に許可する権利がある。私たちは、非商業用途のために、CIEMAT許可(無料)に、私たちが作成した許可知的財産権を任意に改善する義務があります。
ライセンス権の対価格として,研究開発(R&D)コストとしてCIEMATに予備許可料10万ユーロ(約30万ドル)を支払った。指定された開発と規制のマイルストーンを達成した後、CIEMATに合計140万ユーロ(約150万ドル)のマイルストーン支払いを義務づける。PKD
ライセンスがカバーする任意の商業化製品については、純売上高の中桁パーセントまでの印税を支払う義務がありますが、特定の調整を行う必要があり、私たちまたは私たちの許可者または付属会社が支払います。我々が従属ライセンス者と再ライセンス契約を締結した場合,我々は,その従属ライセンシーから受け取った任意の対価格の一部を特定の場合に支払う義務がある.
私たちは90日前にCIEMATに通知して、いつでも本合意を終了することができます。ライセンスは、その国/地域において許可された製品またはプロセスをカバーするライセンス権利が存在する限り、または各国/地域がライセンス権利のために取得すべき任意の追加の法的保護が終了するまで、本プロトコルで定義される各国/地域の有効期間内で有効である。
2016年7月、我々は、FA-A遺伝子を含むレンチウイルスベクターに関連する特定の特許、ノウハウ、データ、および他の知的財産権を世界的に独占的に所有する許可協定をCIEMATと締結し、これらのレンチウイルスベクターは、FAタイプA遺伝子治療のためのVSV-Gパッケージを分割レンチウイルスベクターに統合したヒト治療用途にのみ適用可能である。本ライセンスは,あらかじめCIEMATの同意を得た場合にのみ再許可が可能であり,無理に差し押さえられてはならない.合意条項によれば、(A)ライセンス知的財産権がカバーする1つまたは複数の製品またはプロセスの規制承認を開発し、取得し、そのような製品またはプロセスを商業市場に導入し、その後、それを合理的に公衆に提供する義務があり、(B)規制承認後少なくとも2年以内に少なくとも1つの許可された知的財産権にカバーされた製品またはプロセスを少なくとも1つの国/地域で開発または商業化し、(C)許可知的財産権を適切、道徳的かつ合法的な方法で使用することができる。ライセンスの交換としては、CIEMATに前金、任意のライセンス知的財産権に関連する製品またはプロセスに基づく純売上高に基づく印税支払い、規制および融資マイルストーン支払い、およびライセンス収入支払いが義務付けられています。CIEMATと協力して、特許の起訴と維持を担当しています。費用は私たちが負担します。私たちはまた、CIEMATと協力して、特許を侵害および/または挑戦から許可し、保護する第一の責任を負っています。許可協定が発効してから5年以内に, 私たちはCIEMATが市場価値で獲得した許可知的財産権のいかなる改善も優先的に許可する権利がある。私たちは、CIEMATライセンスbrへの私たちが作成したライセンス知的財産権の任意の改善を非商業用途のために(無料)義務があります。
ライセンス権の対価格としてCIEMATに10万ユーロ(約10万ドル)の初期許可料を支払い,この費用を研究開発コストとして支出した。指定された開発と規制のマイルストーンを達成した後、CIEMATに合計500万ユーロ(約600万ドル)のマイルストーン支払いを義務づける。ライセンスがカバーする任意の商業化製品については、純売上高1桁の中央値パーセント印税を支払う義務がありますが、特定の調整を行う必要があり、私たちまたは私たちの許可先または付属会社が支払う必要があります。もし私たちが従属ライセンス者と再ライセンス契約を締結した場合、特定の場合にその従属ライセンシーから受信した任意の代価の一部を支払う義務があります。
私たちは90日前にCIEMATに通知して、いつでも本合意を終了することができます。ライセンスは、その国/地域において許可された製品またはプロセスをカバーするライセンス権利が存在する限り、または各国/地域がライセンス権利のために取得すべき任意の追加の法的保護が終了するまで、本プロトコルで定義される各国/地域の有効期間内で有効である。
LAD-IとCIEMATおよびUCLBのライセンスプロトコル
我々は2017年11月にCIEMATおよびUCL Business PLC(“UCLB”)(総称して“許可側”)とライセンス契約を締結し、LAD-I領域でのみヒトLAD-I遺伝子を含むレンチウイルスベクターに関連するいくつかの特許、ノウハウ、その他の知的財産権を世界的に治療する独占的権利を付与した。合意条項によれば、(A)ライセンス知的財産権がカバーする1つまたは複数の製品またはプロセスの規制承認を開発し、取得し、そのような製品またはプロセスを商業市場に導入し、それを合理的に公衆に提供することが義務であり、(B)少なくとも規制承認後の少なくとも2年以内に、少なくとも1つのライセンス知的財産権がカバーする製品またはプロセスを少なくとも1つの国/地域で開発または商業化する。そして(C)許可された知的財産権を適切、道徳的、合法的な方法で使用する。ライセンスの交換としては,任意のライセンス知的財産権に係る製品やプロセスに基づく純売上高,開発と規制マイルストーン支払い,再ライセンス収入支払いの中央値から1桁までの印税支払いを許可者側に支払う義務がある。私たちは許可者たちと協力して、ライセンス特許の起訴と維持を担当し、費用は私たちが負担します。私たちはまた、ライセンス側と協力して、ライセンス特許を侵害および/または挑戦から実行および保護する第一の責任を負っている。ライセンス契約の発効日から5年以内
, 私たちは許可者が市場価値で獲得した許可知的財産権のいかなる改善も優先的に許可する権利がある。我々は,我々が作成した許可知的財産権を許可側に許可(無料)し,非商業用途のために任意の改善を行う義務がある.
17
カタログ表
ライセンス権の対価格として,研究開発コストとして許可側に予備許可料10万ユーロ(約40万ドル)を支払った。指定された開発と規制マイルストーンを実現した後,合計140万ユーロ(約150万ドル)にのぼるbrをライセンス側に支払う義務がある。LAD-Iライセンスがカバーする任意の商業化製品については、純売上高1桁の中央値パーセント印税を支払う義務がありますが、特定の調整を行う必要があり、私たちまたは私たちの許可者または付属会社が支払います。もし私たちが従属ライセンス者と再ライセンス契約を締結した場合、特定の場合にその従属ライセンシーから受信した任意の代価の一部を支払う義務があります。
私たちは90日前に許可者に通知して、いつでもこの合意を終了することができる。ライセンスは、許可された製品またはプロセスをカバーする許可権が存在する限り、または各国/地域が許可権として取得すべき任意の追加の法的保護が終了するまで、本(Br)プロトコルで定義された各国/地域の有効期間内で有効である。
UCSDと締結したDanon病許可協定
2017年2月、カリフォルニア大学サンディエゴキャンパス(“UCSD”)に代表されるカリフォルニア大学取締役会とライセンス契約を締結し、この合意に基づき、UCSDはリソソーム貯蔵疾患(達農病を含む)の治療のための独占的で再許可可能な世界的許可を授与した。ライセンスの交換としては、前金、いくつかの臨床および商業マイルストーン支払い、印税支払い(ライセンス知的財産権の範囲内の有効な声明に基づいて対象となる製品の純売上高)、維持費、および再許可収入支払いを支払う義務があります。ロケットは5万ドルの前払い許可料を支払った。我々は,達農病治療の特定開発と規制マイルストーンの実現後,UCSDに合計150万ドルまでの記念碑的支払いを義務付けている。減少マイルストーン支払い計画は
他の適応を実現する同じマイルストーンに適用される。協定がカバーする任意の商業化製品については、純売上高に1桁パーセントの印税を支払う義務があるが、特定の調整が必要である。もし我々が再ライセンス者と再ライセンス契約を締結した場合,特定の場合,その再ライセンシーから受け取った任意の対価格の一部を支払う義務がある.本プロトコルによりUCSDから許可を得た知的財産権を用いて製品を開発するには,何らかの職務調査マイルストーンも遵守しなければならない.
UCSDとのライセンス契約期間はライセンス特許によって満期となり,その中のいくつかの特許はまだ未定出願段階にある。
Regenxbio,Inc.ライセンス
2018年11月19日、我々はRegenxBioInc.(“RGNX”)とライセンス契約を締結し、この協定に基づいて、RGNXのNAV AAV-9キャリアに関連するすべての米国特許および特許出願の独占許可を得て、ヒトのDanon病を治療するために使用した体内にあるAAV−9を用いて遺伝子治療を行い、任意の既知のLAMP 2トランスジェニック異性体およびLAMP 2トランスジェニック異性体のすべての可能な組み合わせ(
“場”)を提供し、この場で他の2つのNAV AAVベクターにすべての米国特許および特許出願の独占的選択権(“選択権”)を付与する(各特許は“許可特許”、総称して“許可特許”と呼ばれる)。
ライセンス契約に基づいて私たちに与えられた権利を考慮して、2018年の総合運営報告書に研究開発コスト支出として700万ドルの前金をRGNXに支払いました。もし私たちが追加キャリアを購入する選択権を行使すれば、各追加キャリアは200万ドルの費用を支払うことになります。ライセンス契約は,ライセンス使用料期限内に,ライセンス特許を含む製品(“ライセンス製品”)の純売上高に応じて,RGNXに1桁までのライセンス使用料を支払うことを規定している.成功すれば、米国とEUが指定された臨床開発と規制のマイルストーンに達した後、RGNXに各許可製品あたり1300万ドルの記念碑的支払いを要求される。また、ライセンス製品に関連しているか、または他の方法でライセンス製品に関連する優先審査券から受け取ったRGNXの20%の支払い費用を支払います。第三者の追加許可が必要な場合、これらの印税義務は具体的に減少する可能性があります。また,再許可者から受け取ったすべての非特許権使用料再許可収入(ある場合)の一部をRGNXに支払わなければならない.RGNX協定によると,2019年に1人目の達成患者の調剤時に100万ドルの許可料を支払った。2022年12月31日と2021年12月31日までの年間で、追加のマイルストーンや支払いは実現されていない。
市場に計画を提供する
2022年2月28日、私たちはCowenと市場発売計画について販売契約を締結し、この計画によると、Cowenを私たちの販売代理として株式を提供し、売却することを随時全権的に決定することができる。販売契約に基づいて提供および販売される株式は、S-3テーブルにおける当社の棚登録宣言(文書番号333-253756)に基づいて提供および販売される。我々は2022年2月28日に米国証券取引委員会に株式発売に関する目論見書補充書類を提出し、販売契約に基づいて、売却株式総収益の3.0%に相当する現金手数料をコーエンに支払うことに同意した。私たちはまた、コーエンに慣用的な賠償と貢献権を提供し、コーエンの販売協定に関連するいくつかの費用を清算することに同意した2022年12月31日まで私たちはすでに市場発売計画に基づいて330万株の普通株を売却し、総収益は4800万ドル、140万ドルを引いた手数料の純収益は4660万ドルだった。
18
カタログ表
競争
生物技術と製薬業界は、遺伝子治療領域を含み、その特徴は技術が迅速に進歩し、競争が激しく、そして特許製品と新療法を高度に重視していることである。私たちの経験と科学知識はそれに競争優位を提供していると信じていますが、私たちはより規模が大きく、資金が豊富な製薬とバイオテクノロジー会社、新しい市場参入者と新しい技術、学術機関、政府機関、民間および公共研究機関からの競争を含む多くの異なる源からの潜在的な競争に直面しています。これらの機関は将来、パイプラインではまだ構想されていない適応を治療するために製品を開発するかもしれません。我々が開発と商業化に成功した任意の候補製品は、骨髄移植および将来出現する可能性のある新しい療法のような既存の治療法と競合するであろう。私たちの候補製品が承認されれば、その成功に影響を与える重要な競争要素は、治療効果、安全性、利便性、価格、薬物経済価値、耐性、および政府当局と他の第三者支払人が提供する保険と十分な補償である可能性があると考えられる。また,臨床適応に対する単一治療療法を開発し,死亡率や高発症率の問題を解決する予定であり,慢性や重複治療が必要となる可能性のある代替競争療法の潜在競争相手とは異なる。
他の早い段階にある会社も、大手や成熟会社との協力手配で競争する可能性がある。製薬とバイオテクノロジー業界の合併と買収は、遺伝子療法を開発する少数の会社により多くの資源を集中させる可能性がある。これらの会社はまた合格した科学と管理人員を募集と維持し、臨床試験場所と臨床試験患者登録を確立し、著者らの計画を補充し、或いは必要な技術を提供する方面で私たちと競争している。
新薬と治療方式の市場進出、先進技術の出現に伴い、激しい競争と日々の競争に直面することが予想される。もし私たちの潜在的な競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの潜在的な競争相手
はまた、私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができるかもしれない。
製造業
著者らの遺伝子治療プラットフォームは2つの主要な構成部分がある:LVベクターとAAVベクターの産生及び標的細胞形質導入過程、それによって薬物製品を産生する。私たちは2022年にニュージャージー州クランベリーにある工場でGMPの生産を開始した。私たちは私たちのAAVプロジェクトと第三者メーカーのために私たち自身の直接製造能力を補充する予定です。私たちのLV計画に対して、私たちは現在第三者メーカーに依存して私たちの臨床試験のためにbrプラスミド、ベクター、細胞バンクと最終薬物製品を生産している。適用されるプライマリサービスと供給契約に基づいて、仕入先と注文に基づいてこのような生産を管理します。私たちはこの製造業者たちと長期的な合意を締結した。可能性があれば、私たちはリスクを低減するために余分な源と複数の源から材料を調達するだろう。もし私たちの既存の任意の第三者サプライヤーがどんな理由でも提供できなければ、私たちは遅延に遭遇する可能性があるにもかかわらず、代替サプライヤーの能力を得ることができる潜在的な代替サプライヤーの数があると信じています。もし私たちの候補製品が登録されていれば、私たちは現在商業消耗品を生産する契約関係もありません。将来の我々の候補製品の商業的生産については,活性br薬物(薬物物質)成分と最終薬物製品(薬物製品)を直接生産·アウトソーシングして契約製造組織に提供し,これらの製品が適用規制機関の承認を得て上場許可を登録すれば,様々な選択を行う予定である。
直接製造または契約製造施設でコスト効果のある方法で生産できる候補薬剤の開発を継続したい。もし私たちの生産候補製品が依存しているサプライヤーや製造業者が欠陥のある製品を提供してくれたり、そのような製品が後にリコールされたり、あるいは私たちが直接生産した自分の製品を直接製造することでこのような問題に遭遇した場合、私たちはbrの遅延と追加コストに遭遇する可能性があり、すべてが重大である可能性がある。
政府の監督管理
FDA法規と上場承認
アメリカでは、FDAは連邦食品、薬物と化粧品法案(“FDCA”)に基づいて薬品を監督し、公衆衛生サービス法(Public Health Service Act)、法律と他の連邦、州と地方法規に基づいて公布された法規に基づいて生物製品を監督する。製品開発過程,承認過程又は承認後のいずれかの場合,出願人が適用される米国の法規の要求を遵守しなければ,行政又は司法制裁及び候補製品の承認を受けない可能性がある。他の事項に加えて、これらの制裁は、FDAが臨床一時停止試験を実施することを含むことができ、FDAは、未解決の申請または関連サプリメントの承認を拒否し、承認の撤回、タイトルまたは警告状のない、製品のリコール、製品の差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、返還、返還、民事罰または刑事起訴を含むことができる。政府機関のこのような行動はまた私たちがこのような行動に対応するために多くの資源を使う必要があるかもしれない。どんな機関や司法法執行行動も私たちに実質的な悪影響を及ぼすかもしれない。
19
カタログ表
FDAと州と地方司法管轄区及び国外の類似監督管理機構は薬品の臨床開発、承認、製造、流通とマーケティングに対して実質的な要求を提出した。これらの機関と他の連邦、州と地方実体監督管理研究開発活動及び私たちの製品のテスト、製造、品質管理、安全、有効性、ラベル、包装、貯蔵、流通、記録保存、承認、承認後の監視、広告、販売促進、サンプリングと輸出入。Rocketの薬物は遺伝子治療候補製品に適したBLA承認プログラムを通過しなければ,生物製品として米国で合法的に発売されることができる。
FDA内部では,FDAの生物製品評価·研究センター(“CBER”)が遺伝子治療製品を規制し,これらのタイプの製品の開発に関する指導文書を発表している。FDAはまた,遺伝子治療製品に関する一般的な指導文書,その臨床前評価,遅延不良事象の遺伝子治療研究に関与している被験者の観察,効力試験および遺伝子治療INDにおける化学,製造と制御情報を発表した。
FDAが米国でバイオ医薬品を発売する前に必要なプログラムには、一般に以下のような態様が含まれる
| • |
“良好な実験室規範”或いは他の適用法規による非臨床実験室テスト、動物研究と調合研究を完成した
|
| • |
FDAが30日以内に反対しない限り、臨床試験の開始を可能にするINDを提出した
|
| • |
FDA法規と良好な臨床実践(GCP)に基づいて行われ、GCPは国際道徳と科学的品質基準であり、試験参加者の権利、安全と福祉が保護され、データの完全性を維持することを目的とした、その予期される用途に使用される薬剤または生物の安全性と有効性を決定するために、十分かつ制御された良好な人体臨床試験を行う
|
| • |
BLAを作成し食品医薬品局に提出しました
|
| • |
BLAの使用料を審査するためにFDAを提出します
|
| • |
適切な場合、または適用される場合、製品は、FDA諮問委員会によって審査される
|
| • |
GCP要件に適合する状況を評価するために、現在の良好な製造規範(CGMP)要件に適合するかどうかを評価するために、生産製品またはそのコンポーネントの製造施設および臨床試験場所の承認前検査を満足的に完了させ、適用されれば、FDAの現在の良好な組織規範(CGTP)要件、およびGCP要件に適合する状況を評価するために選択された臨床試験地点を含む;
|
| • |
BLAに対するFDAの承認は、生物が発売または販売される前に発生しなければならない。
|
臨床前研究
臨床前研究は製造した薬物物質或いは活性薬物成分と調合薬物或いは薬物製品の純度と安定性の実験室評価、及び体外と動物研究を含み、人体における薬物の初歩的な試験の安全性と活性を評価し、そして治療使用の原理を確立する。臨床前研究の進行はGLP法規を含む連邦法規と要求の制約を受けている。臨床前試験の結果は,生産情報,分析データ,任意の利用可能な臨床データや文献や臨床研究計画などとともに,INDの一部としてFDAに提出される。
会社は通常、生殖不良事件と発ガン性の動物テストのような長期的な臨床前テストを完成しなければならず、また薬物化学と物理特性に関する追加情報を開発し、(“cGMP”)要求に基づいて商業大量生産薬物の過程を最終的に決定しなければならない。製造技術は一貫して高品質の候補薬物ロットを生産できる必要があり、また、メーカーは最終薬物製品の特性、強度、品質と純度をテストする方法を開発しなければならない。また,適切な包装を選択·試験し,候補薬物が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
INDと臨床試験
臨床試験はGCP要求に符合する合格した研究者の監督の下で人類被験者に研究製品を服用することに関連する。臨床試験は書面による研究案に基づいて行い,その中で研究の目標,安全性モニタリングのためのパラメータ,評価すべき有効性基準を詳細に説明した。最初の臨床試験を開始する前に、臨床前試験の結果および製品化学、製造および制御に関する情報および提案のような他の情報が含まれた初期INDをFDAに提出しなければならない。INDはFDAが受領してから30日以内に自動的に発効し、FDAが30日以内に薬物製品或いは臨床試験の進行に対して関心或いは問題を提出し、臨床保留を実施しなければならない。IND発効期間中は,いつでも臨床見合わせが可能である。この場合,INDスポンサーは臨床試験開始または再開前にFDAの未解決の問題を解決しなければならない。したがって,INDの提出によりFDA
が臨床試験の開始や継続を許可することもない可能性がある。
20
カタログ表
米国で臨床試験を開始する前にFDAにINDを提出する以外に、いくつかの組換えまたは核酸分子の合成に関連するヒト臨床試験は、米国国家衛生研究院(NIH)、組換えまたは核酸分子の合成に関連する研究ガイドラインまたはNIHガイドラインに記載されているように、機関生物安全委員会(IBC)の監督を受けなければならない米国国立衛生研究院のガイドラインによると、組換えおよび合成された核酸は、(1)核酸分子を連結することによって構成される生細胞中で複製可能な分子(すなわち、組換え核酸)と、(2)化学的または他の方法で修飾されているが、自然に産生される核酸分子(すなわち、合成核酸)と塩基対を行うことができる分子を含む化学的または他の方法で合成または増幅された核酸分子と定義される。または(Iii)(I)または(Ii)に記載されたものを複製することによって生成される分子具体的には,米国国立衛生研究院のガイドラインによると,ヒト遺伝子転移試験の監督にはIBCによる評価と評価が含まれており,IBCは地方機関委員会であり,この機構の組換えや合成核酸分子を用いた研究の審査·監督を担当している。IBCは研究の安全性を評価し、公衆衛生または環境に対する任意の潜在的リスクを決定し、このような審査は臨床試験開始前のいくつかの遅延をもたらす可能性がある。NIHガイドラインは強制的ではないが,関連研究がNIH組換えや合成核酸分子研究助成を受けた機関で行われているか,あるいはその助成によって行われていない限り,多くの会社や他のNIHガイドラインに拘束されていない機関は自発的にこれらのガイドラインに従っている。
米国国外で臨床試験を行うスポンサーはFDAの認可を得ることができるが,INDによる臨床試験を希望している。外国の臨床試験がINDで行われていなければ,臨床試験がGCPに従って行われている限り,スポンサーはBLAやINDを支援するために臨床試験のデータをFDAに提出することができ,FDAは機関が必要と考えている現場検査で研究のデータを検証することができる。
製品開発期間中に行われたすべての後続臨床試験は,既存のINDに単独で提出しなければならない。また,臨床試験を行う地点ごとの独立機関審査委員会(“IRB”)は,臨床試験がその地点で開始される前に審査·承認を行わなければならない。また,各被験者からインフォームドコンセントを得なければならない。FDAまたはIRB、
またはスポンサーを含む監督機関は、参加者が受け入れられない健康リスクに直面していることを発見すること、または臨床試験がFDAの要求に従って行われていないことを含む、様々な理由で臨床試験を一時停止または終了することができる。さらに、いくつかの臨床試験は、データ安全監視委員会または委員会と呼ばれる臨床試験スポンサーによって組織された独立した合格専門家グループによって監督される。このグループは,試験のいくつかのデータへのアクセスに基づいて,指定されたチェックポイントで試験が可能かどうかを許可し,被験者に受け入れられない安全リスクやbr}の他の原因があると判定された場合(治療効果を示していなければ),臨床試験の中止を推奨する可能性がある。
BLA承認のためのヒト臨床試験は、通常、いくつかの段階が重複または合併する可能性があるが、3つの段階のプロセスに関連する。第一段階は最初の臨床評価であり、投与と安全性と耐容量のテスト、及び稀な疾患のようないくつかの適応を含み、人類に有効な初歩的な証拠とする。第2段階は、特定の適応における薬剤の有効性を評価し、最適な用量および用量間隔を決定し、より大きな患者群において可能な副作用およびリスクを決定するための研究を含む。1つの製品が安全であることが発見され、第2段階で初歩的な効果が決定された後、第3段階の臨床試験で評価される。第三段階試験は、治療された疾患に関連する研究薬物の全体的利益リスク指数を評価するための拡大多地点有効性および安全性試験を含む。臨床前とヒト臨床試験の結果は,商業販売開始の承認のためにBLAとしてFDAに提出される。
すべての臨床試験はFDA法規、GCP要求及びその規程に従って行わなければならず、データが信頼できると思われ、監督管理目的を達成する。臨床試験結果を詳細に説明する進捗報告は少なくとも年に1回FDAに提出しなければならず,重篤な有害事象が発生すればより頻繁である。第1段階、第2段階、および第3段階の臨床試験は、いかなる指定された時間でも成功しない可能性があり、
または全く成功できない。政府の規制は、候補製品や新薬の販売をかなり長い間延期または阻止し、私たちの活動に費用の高い手続きを適用するかもしれない。
臨床試験情報の開示
FDAが監督する製品(薬品を含む)の臨床試験スポンサーはいくつかの臨床試験情報を登録し、開示しなければならない。製品、患者集団、調査段階、研究場所と研究者、および臨床試験の他の方面に関する情報は、その後、登録の一部として公開される。スポンサーも完成後にその臨床試験の結果を開示する義務がある。これらの試験の結果を開示することは,研究中の新製品や新適応が承認された後に延期することができ,最長2年遅らせることができる。競合他社はこれらの公開情報を用いて
開発計画の進展に関する情報を取得する可能性がある.
“BLA”承認プロセス
米国での上場承認を得るためには,FDAに上場申請を提出しなければならず,この申請はFDAが満足するデータを提供し,提案された適応に対する研究薬の安全性と有効性を証明しなければならない。この出願は、否定または曖昧な結果および積極的な発見、ならびに製品の化学、製造、制御および提案されたラベルなどに関する詳細な情報を含む、関連する非臨床または臨床前研究および臨床試験から得られたすべての関連データを含む。データは、使用製品の安全性および有効性を試験することを目的とした会社のスポンサーからの臨床試験から来ることができ、研究者によって開始されたGCP要求に適合した研究を含む多くの代替源から来ることもできる。
21
カタログ表
新薬開発期間中、スポンサーはいつかFDAと会う機会がある。これらの要件は、INDを提出する前、第1段階または第2段階の終了時
およびBLAの提出前にある可能性がある。他の時間に会議を開催することを要求することができます。これらの会議は,スポンサーにこれまで収集してきたデータに関する情報を共有させ,FDAにアドバイスを提供させ,スポンサーとFDAに次の段階の開発について合意させる機会を提供することができる。
製品開発,非臨床研究と臨床試験の結果,および製造プロセス,薬物化学に対する分析テスト,アドバイスのラベルやその他に関する情報の説明をBLAの一部としてFDAに提出し,この製品の期待適応の発売承認を要請した。FDAは、それらが届出を受ける前に十分に完全であり、実質的な審査
を行うことができることを確実にするために、すべての提出されたBLASを審査する。それはBLAの記録を受け入れるのではなく、追加的な情報を提供することを要求するかもしれない。この場合,BLAおよび付加情報を再提出しなければならない.再提出された申請はFDAが届出を受ける前にも審査
を受けなければならない。FDAは、BLAを受信した日から60日の間、実質的な審査を可能にするために出願が十分に完全であるかどうかに関するFDAの敷居に基づいて、出願を受け入れるか否かを決定するために予備審査を行う。FDAは、提案された製品がその予期される用途に対して安全、有効または有効であるかどうか、および許容可能な純度プロファイルを有するかどうか、および製品の識別、安全、強度、品質、効力、および純度を保証および維持するために、cGMPに従って製造されたかどうかを決定するためにBLAを審査する。FDAはBLAの具体的な業績目標の検討に同意した。具体的には、FDAは、改正された“処方薬使用者費用法”(PDUFA)によって達成された目標および政策に基づいて、FDAが元のBLAの予備審査を完了し、出願人に応答するのに10ヶ月の時間がある, 優先審査のために指定された元のBLAの提出日から6ヶ月間。FDAは、いくつかの遅延された情報を考慮するために、または提出中に提供された情報を明確にすることを目的とした情報を考慮するために、審査プロセスをさらに3ヶ月延長することができる。FDAがBLAの実質的な審査を完了した後、それは、BLAが現在の形態で承認されないことを伝達するために、薬剤が承認されることをスポンサーに通知するか、またはBLAが現在の形態で承認されないことを伝達するために、または申請が承認される前に受信されなければならない追加の臨床、非臨床または生産データを通知するが、申請の最終承認またはそのような承認のいずれかの時間(あれば)を示唆しない。または、これらの欠陥がBLAの再提出時にFDAによって満足的に解決されている場合、FDAは承認書を発行する可能性がある。FDAは、含まれる情報タイプに依存して、そのような再提出を2~6ヶ月以内に検討することを約束した。FDAは、新薬または医薬製品の申請を諮問委員会に提出することができ、一般に、申請を承認すべきかどうかを審査、評価、提案するための臨床医および他の専門家を含むグループであり、どのような条件下で承認されるべきであるかを審査する。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。
BLAを承認する前に、FDAは通常、この製品を生産する施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していると判断しなければ、製品が要求された仕様内で一貫して生産されることを保証するのに十分であることを決定しない限り、この製品を承認しないだろう。さらに、BLAを承認する前に、FDAは、GCPに適合することを確実にするために、1つまたは複数の臨床サイトを検査する可能性がある。遺伝子治療製品については,メーカーがcGTPSに適合していなければ,FDAもこの製品を承認しない。これらは、ヒト細胞、組織、および細胞および組織に基づく製品またはHCT/Pを製造するための方法および使用のための施設および制御を管理するFDAの法規であり、これらのHCT/Pは、ヒトレシピエント内に移植、移植、注入または転移するためのヒト細胞または組織である。CGTP要求の主な目的は,細胞や組織に基づく製品の製造方式の確保であり,感染症の導入,伝播,伝播を防止することを目的としている。FDAの規定では,組織機関がFDAにそのHct/Pを並列登録し,適用した場合に適切なスクリーニングとテストによりドナーの評価を行うことも求められている。FDAがアプリケーション、製造プロセス、または製造施設が受け入れられないと判断した場合、それは、通常、不足点を列挙し、追加のテストまたは情報の提供を要求することが多い。これは申請に対する追加的な検討を大幅に延期するかもしれない。FDAが臨床サイトを発見した場合GCPによる臨床試験が行われていない, FDAは臨床現場で発生したデータをBLAが提供する主要な奏効率分析から除外することを決定する可能性がある。さらに、任意の要求された追加情報が提出されているにもかかわらず、FDA
は、最終的に、その申請が承認の規制基準を満たしていないことを決定する可能性がある。
FDAは要求するかもしれないし、会社は製品が承認された後に追加の臨床試験を行うかもしれない。これらのいわゆる4期試験や承認後試験は薬物承認を継続する条件となる可能性がある。4期試験の結果,候補製品の有効性が確認でき,重要なセキュリティ情報を提供することができた.また,FDAはスポンサーに上場後の試験を要求し,同機関が決定したセキュリティ問題を具体的に解決する権利がある。以下の“マーケティング後要件”を参照してください。
FDAはまた、薬物の利益がそのリスクよりも大きいことを保証するために、リスク評価と緩和戦略(“REMS”)を制定することをメーカーに要求する権利がある。スポンサーはまた、REMSをBLAとして提出することを自発的に提案することができる。“作業重点法”の審査の一部として決定された再生可能エネルギー管理システムの構築が必要である。法定基準によれば、REMSの要素は、“尊敬する医師書”、薬品使用ガイドライン、より詳細な的確な教育計画を含むことができ、場合によっては、安全な使用を確保する要素(“ETASU”)と呼ばれる配布および使用制限も含まれる。ETASUは、処方または調剤のための特殊なトレーニングまたは認証、場合によっては調剤のみ、特殊な監視、および患者登録簿の使用を含むことができるが、これらに限定されない。これらの要素はBLA承認の一部として交渉されており,場合によっては承認日が延期される可能性がある.採用されると、REMは定期的な
評価および修正を受ける。
22
カタログ表
承認申請において決定されたいくつかの条件を変更するには、適応、ラベル、生産プロセス、または施設の変更を含み、変更を実施するためには、新しいBLAまたはBLAサプリメントを提出し、FDAの承認を得る必要がある。新適応に対するBLAサプリメントは通常オリジナル申請と類似した臨床データが必要であり,FDAがBLAサプリメントを審査する際に使用するプログラムや行動はBLASを審査する場合と同様である。
候補製品が監督部門の承認を得ても、承認は特定の疾病状態、患者数と投与量に限定される可能性があり、あるいは使用に対する重大な制限を含む可能性があり、形式はbr警告、予防或いは禁忌症、或いは煩雑なリスク管理計画、分配或いは使用制限、或いは発売後の試験要求である。また、監督部門の許可を得た後であっても、後に製品に以前未知の問題があることが発見された場合、安全ラベルの実施やREMSの実施、発売後の研究或いは臨床試験を要求し、甚だしきに至ってはその製品を市場から完全に撤退させることを含む製品の制限を招く可能性がある。私たちの製品に対する規制機関の承認が遅れたり、承認されたりできなかったり、承認されたりしましたが、用途は明らかに限られており、私たちの業務を損なうことになります。また、将来のアメリカや外国政府の行動にどのような不利な政府法規が生じる可能性があるのか予測できない。
“ハッジ·ワックスマン修正案”
1984年の“医薬品価格競争および特許期限回復法”、すなわち“ハッジ·ワックスマン改正案”によれば、臨床開発およびFDA規制審査中に失われた製品の米国特許期間の一部は、新製品またはその用途をカバーする特許の5年間にわたる特許寿命を返上することによって回復することができる。この期間は、通常、INDの発効日(特許発行後)とBLA提出日との間の時間の半分であり、BLA提出日と出願が承認されたとの間の時間に加えて、発起人が全力で行動すべきであることが前提である。しかしながら、特許期限の回復は、特許の残り期間を製品承認日から合計14年間延長することはできず、承認された薬物に適用される特許のみを延長することができ、特許が満了する前に延長を出願しなければならない。米国特許商標局はFDAと協議し,任意の特許期間の延長または回復の出願を審査·承認する。
市場排他性
2010年3月23日に署名された法律となった“平価医療法案”(略称ACA)は、FDAによって承認された参考生物製品と類似または交換可能であることが証明された生物製品のための短い承認経路を開いた副題“2009年生物製品価格競争と革新法案”(BPCIAと略称する)を含む。生物類似性は分析研究、動物研究と1つ以上の臨床試験を通じて証明することができ、それは生物製品と参考製品が安全性、純度と効力の面で臨床的に意義のある差異がないことを要求する。互換性は、製品が参照製品と生物学的類似性を有することを必要とし、この製品は、参照製品と同じ臨床結果を生成することが予想され、複数回投与された製品の場合、生物製剤および参照生物製剤は、安全リスクを増加させることなく、または参照生物製剤の独占使用に関連する治療効果がリスクを低下させることなく、以前の投与後に交換することができることを証明しなければならない。しかし、生物製品のより大きく、よく複雑な構造に関連する複雑性、及びこのような製品の製造技術は、FDAが制定している実施に対して重大な障害を構成している。
参考生物製品は製品が初めて許可を得た日から4(4)年と12(12)年の専門期間がある。FDAは、参照製品が初めて許可された日から4年まで、基準生物製品に基づく生物学的類似または交換可能製品の申請を受け入れず、FDAは、参照製品が初めて許可された日から12(12)年まで、基準生物製品に基づく生物類似または交換可能製品の申請を承認しないであろう。“最初の許可”とは、一般に、米国で特定の製品が承認された初期日を意味する。最初に許可された日は生物製品の許可日を含まず(かつ新しい専門期間は適用されない)、許可が生物製品を補充するためのものである場合、または生物製品の同一発起人または製造業者のための後続の出願のために、新しい適応、投与経路、投与スケジュール、剤形、剤形の変更をもたらす(生物製品構造の修正を含まない)。投与システム、投与装置または強度、または生物製品の構造を、安全性、純度または効力の変化を引き起こさない修飾を行う。したがって、新しい
製品が以前に承認された製品の構造の修正を含むかどうかを決定し、それによって、新製品の許可がそれ自体の独占期間をトリガする最初の許可であるかどうかを評価するために、安全性、純度、または効力を変化させることを決定しなければならない。承認された場合,その後の出願であるかどうか, 生物製品の“初許可”はスポンサーが提出したデータに基づいて逐一決定されるため,排他性が保証されている。
さらに、孤児医薬品法によれば、生物製品がまれな疾患または疾患の治療に使用されることが意図されている場合(通常、米国では200,000人未満に影響を与えることを意味するか、または米国で疾患または疾患を治療するための生物製品の開発および生産がコストが製品の販売から回収されることを合理的に期待できない場合)、FDAは生物製品を“孤児薬”として指定することができる。BLAを提出する前に,孤立製品の指定を要求しなければならない.FDAが孤児製品の指定を承認した後、FDAは、治療剤の識別情報およびその潜在的な孤児の使用を開示する。孤立製品を指定することは、監督審査と承認過程においていかなる利点も伝達することはなく、監督管理承認プロセスの持続時間を短縮することもない。孤児状態を有する製品が、このような指定された疾患または状況に対するFDAの最初の承認を得た場合、この製品は、孤児製品の排他性を得る権利があり、これは、FDAが7年以内に他の出願を承認することができず、限られた場合を除いて、同じ適応で同じ薬剤または生物製品を販売することを意味する。Brは、例えば、孤児に対して排他的な製品に対する臨床的優位性を示すか、または排他性を有する方が、疾患患者または指定された薬物が対象とする場合の需要を満たすために十分な数の薬物が利用可能であることを確保できない場合である。しかし,競争相手は孤立製品と排他的な同じ適応を持つ異なる製品の承認を得るか,あるいは同じ製品の承認を得ることができるが,
孤立製品は排他的な異なる適応を持つ.EUでは、孤児薬品の地位は似たような利点を持っているが、同じではない。
23
カタログ表
小児科専門権は、米国の別のタイプの非特許マーケティング専門権であり、付与された場合、任意の既存の規制専用権(非特許専有権を含む)の条項に追加の6ヶ月間の市場保護を追加することを規定する。BLAスポンサーから提出された小児科データがこのようなデータに対するFDAの書面要求に公平に応答すれば,この6カ月の排他性を与えることができる。
まれな小児科疾患指定と優先審査証明書
FDCAにより、FDAは“稀な小児科疾患”の定義に符合する薬物と生物製品の開発を奨励した。“深刻または生命に危険を及ぼす疾患として定義され、深刻または生命を脅かす表現は、主に出生から18歳までの個人に影響を与え、米国では200,000人未満、米国では200,000人を超える影響を与え、brは、このような疾患または疾患を治療する薬剤または生物製品を米国で開発および製造するコストが、米国でそのような薬剤または生物製品を販売することから得られることが合理的に予想されていない。稀な小児科疾患候補製品のスポンサーは、稀な小児科疾患薬物または生物製品が承認された日後に、優先審査クーポン(PRV)と呼ばれる後続のヒト薬剤または生物製品申請を得るための優先審査のために使用することができるクーポンを得る資格がある可能性がある。スポンサーは、そのBLAを提出する前に、FDAに稀な小児科疾患の指定を要求することができる。まれな小児科疾患指定は、スポンサーがそのBLAが承認された後にPRVを受信することを保証しない。さらに、稀な小児科疾患指定要求を提出しないスポンサーを選択することは、そのマーケティング申請が承認された後にPRVを受信することができ、前提として、彼らが元のマーケティング申請においてこのような証明書を要求し、すべての資格基準を満たすことを前提とする。PRVを受信した場合、それは何度も販売または譲渡される可能性がある。国会はPRV計画を2024年9月30日まで延長し、2026年9月30日までPRVを承認する可能性がある。
開発と審査計画を加速する
FDAはいくつかの方法でBLASの検討を加速させることを許可された。Fast Track計画によると,候補生物製品のスポンサーは,INDの届出と同時にあるいはその後,特定の適応の製品をFast Track製品として指定することをFDAに要求することができる。もし生物製品が深刻或いは生命に危害を及ぼす疾病を治療することを目的とし、そしてこのような疾病が満たされていない医療需要を満たす潜在力を示すならば、迅速なチャンネル指定を受ける資格がある。高速チャネルは,候補製品と研究中の特定の適応に適した組合せを指定する.他の利点に加えて,FDAとより大きな
インタラクションが可能であれば,FDAは申請完了前にFast-Track BLAの各部の審査を開始する可能性があり,この過程をスクロール審査と呼ぶ.
迅速チャネル計画を含むFDAに提出されたマーケティングのための任意の製品は、再生薬(br}高度治療(RMAT)指定、優先審査、および承認の加速など、FDAが開発および審査を加速するために意図された他のタイプの計画の資格に適合する可能性がある。RMAT認証を受ける資格があるためには、候補品は再生医学療法でなければならない それは細胞療法、治療用組織工学製品、ヒト細胞と組織製品、またはこのような療法または製品を使用する任意の組み合わせ製品と定義されているが、“公衆衛生サービス法”第361条と連邦法規第21編1271部分によってのみ規制されているものは除外されている;深刻または生命に危険な疾患または状況を治療、修正、逆転または治癒することを目的としている;初歩的な臨床証拠は、この製品がこのような疾病或いは状況が満たされていない医療需要を満たす可能性があることを示している。遺伝子治療製品はRMATが指定した再生医学療法の定義に適合する可能性があるRMAT
認証を取得した候補製品のBLAは、長期的な臨床的利益を合理的に予測する可能性のあるエージェントまたは中間端末を使用することによって、または大量のbr}サイトから取得されたデータに依存して、優先審査または加速承認を得る資格がある可能性がある。RMATを指定する利点はまた、承認の加速をサポートするための任意の潜在的代替品または中間終点を検討するために、FDAとの早期相互作用を含む。加速的承認を得、承認後に制約を要求されるRMAT認証を有する候補製品は、電子健康記録のような臨床研究、患者登録、または他の真の証拠源からの臨床証拠の提出、より大きな検証データセットの収集、または承認前にそのような治療を受けたすべての患者の承認後に監視することによって、これらの要件を満たすことができる。
Fast TrackまたはRMATで指定された製品を取得する候補製品を含みます処理が深刻であれば 承認されれば,既存療法と比較して重篤な疾患の治療,診断または予防の安全性または有効性において有意に向上するであろう。FDAの目標は、標準審査の10ヶ月間ではなく、優先審査申請の審査を6ヶ月以内に完了させることである。
さらに、生物学的製品が生命に深刻なまたは危険な疾患または状況を治療し、代替終点への影響が臨床利益を合理的に予測する可能性があることを証明し、または生存または不可逆的な発病率または死亡率または他の臨床的利益ではなく、臨床終点への影響に基づいて、病状の重症度、希少性および流行率、および代替治療を利用可能または不足していることを考慮すると、加速承認を得る資格がある。承認の一つの条件として,FDAは承認を加速させる候補薬物やバイオ製品のスポンサーに十分かつ良好に制御された発売後の臨床試験を要求する可能性がある。また,FDAは現在,その機関が別途通知しない限り,あらかじめ宣伝材料を承認しなければならないことを要求している発売承認後120日以内に
を発表または出版する予定です.
24
カタログ表
製品が1つまたは複数の計画の条件に適合していても、FDAは、製品がもはや資格条件を満たしていないと後で決定する可能性があり、またはFDAが審査または承認する期間が短縮されない可能性がある迅速な指定、優先審査、承認の加速は承認基準を変更することはありませんが、開発や承認過程を加速させる可能性があります。
発売後要求
新製品が承認された後、製薬会社および承認された製品は、監視および記録保存活動を含むFDAの規制を継続し、適用される規制機関に製品の副作用を報告し、規制機関に最新の安全および治療効果情報を提供し、製品のサンプリングおよび流通要求、および宣伝および広告要求を遵守することを含み、その中には、他に加えて、消費者向けの広告基準、薬物または患者集団における薬物承認のラベルに記載されていない薬物の普及のための制限が含まれている。またはタグ外使用、業界賛助の科学的および教育活動の制限、およびインターネットに関連する販促活動への要求。医師はその独立した専門医学判断に基づいて、合法的な薬品をラベル外用途のために処方することができるが、メーカーは通常このようなラベル外用途を販売或いは普及させることができない。製品またはそのラベルの修正または強化、または生産場所の変更は、通常、FDAおよび他の規制機関の承認を得る必要があり、彼らは承認される可能性があり、承認されない可能性があり、または長い審査過程を含む可能性がある。
処方薬の広告は連邦、州、そして外国の規制によって制限されている。米国では、FDAは消費者向けの広告を含む処方薬販売促進を規制している。処方薬販売促進材料は初めて使用時にFDAに提出しなければならない。どの処方薬製品と薬品サンプルの流通も“米国薬品サプライチェーン安全法”と“処方薬販売法”を遵守しなければならず、この2つの法律はいずれもFDCAの一部である。
米国では,製品が承認されると,その生産はFDAの全面的かつ持続的な規制を受ける。FDAの規定は,製品は承認された特定の施設で生産され,cGMPに適合しなければならないことを要求している。CGMPは他の事項のほかに,品質管理と品質保証およびそれに応じた記録やファイルメンテナンスが要求され,cGMPから外れた状況を調査·是正する義務がある.薬品メーカーと他の生産と流通許可薬品に参与する実体はFDAとある州機関にその機関を登録し、FDAとある州機関の定期的な抜き打ち検査を受けて、cGMPと他の法律を遵守する状況を理解しなければならない。また、処方薬製品サプライチェーン内の製造業者および他の参加者は、製品追跡および速度要求を遵守し、偽、移転、窃盗、故意に偽の製品、または米国での流通に適していない製品をFDAに通報しなければならない。そのため、製造業者は、cGMPコンプライアンスを維持するために、生産および品質管理に時間、お金、エネルギーをかけ続けなければならない。これらの法規はまた製造と品質保証活動に対して一定の組織、プログラムと文書要求を提出した。契約メーカー、実験室または包装業者を使用するBLA所有者は、合格した会社を選択し、監督することを担当し、場合によっては、これらの会社の合格サプライヤーも担当する。これらの会社とそのサプライヤーは随時FDAの検査を受け、違反条件を発見します, CGMPに準拠しないことを含み、法執行行動がそのような製品の運営を中断させる可能性があり、または製品のリコール、市場からの製品の撤回、または輸入の拒否を含む製品、製造業者、または承認されたBLA所有者の制限をもたらす可能性がある。また,承認されたBLAのメーカーおよび/または
保有者は年間製品費と開設費を支払う必要がある.これらの費用は通常毎年増加します。
FDAはまた、承認された製品の影響を監視するために、またはREMSによって承認に条件を適用して、その製品の流通または
使用を制限するための第4段階試験とも呼ばれる上場後試験を要求する可能性がある。製品に以前未知の問題が存在することが発見されたか、または適用されたFDA要件を遵守できなかったことは、負の結果をもたらす可能性があり、負の宣伝、司法または行政法執行、FDAが命名されていないbrまたは警告状、広告の強制訂正または医師とのコミュニケーション、承認撤回、民事または刑事罰などを含む。新たに発見または開発された安全性または有効性データは、新たな警告や禁忌症の追加を含む製品承認のラベルを変更する必要がある場合があり、他のリスク管理措置を実施する必要があるかもしれない。また、新しい立法による要求、またはFDAの政策が変更される可能性がある新しい政府要求を確立することが可能であり、規制部門が私たちが開発している製品を承認することを延期または阻止する可能性がある。
保証と精算を請け負う
私たちが監督部門の許可を得て商業販売のための任意の製品の販売は、政府医療計画行政当局、管理型医療機関、個人医療保険会社、その他の実体を含む第三者支払者の精算状況にある程度依存する。その病態を治療するために処方薬を服用している患者とその処方医は,通常,その処方薬に関する全費用を第三者支払者に依存して精算する。保険を提供しない限り、患者は私たちの製品を使用することができなくて、私たちの製品の大きなコストを支払うのに十分な費用を精算します。したがって、私たちの製品が承認されると、保険を提供し、私たちの製品の大部分のコストを支払うのに十分でなければ、市場から受け入れられない可能性があります。
25
カタログ表
第三者支払い者が薬品に保険を提供するかどうかを決定するプロセスは、一般に、薬品価格を設定すること、または保険が承認された後に支払者が薬品に支払うべき送達率を決定するプロセスとは分離される。第三者支払者は、承認リスト(処方とも呼ばれる)上の特定の医薬品製品に保証範囲を制限するか、または他の方法で健康br技術評価を行うことができる。いずれの場合も、支払者カバールールは、FDAによって承認されたいくつかの薬剤を特定の適応から除外する可能性がある。第三者支払者が私たちの候補製品に保険を提供しないと決定すれば、承認されると、医師の私たちの製品に対する使用率が減少する可能性があります。また、第三者支払者が薬品に保険を提供することを決定することは、適切な販売率を承認することを意味するものではない。製品開発投資の適切なリターンを達成するために十分な価格レベルを維持できるように、十分な第三者精算が得られない可能性がある。また、薬品の保証範囲と精算範囲は支払人によって異なる。第三者支払者は、ある特定の薬品又はサービスを保証することを決定し、他の支払者も当該医療製品又はサービスに保証を提供するか、又は適切な販売率で保証を提供することを確保することができない。したがって、保証範囲の決定過程は、各支払人にそれぞれ私たちの製品を使用する科学的かつ臨床的支援を提供する必要があり、これは時間のかかる過程になる。
医療費の抑制はすでに連邦、州と外国政府の優先事項となっており、薬品価格はずっとこの努力の重点である。第三者支払者は,安全性と有効性を疑問視するほか,薬品や医療サービスの価格,医療の必要性を検査し,薬品や医療サービスのコスト効果を審査することが多くなってきている。もしこれらの第三者支払者が私たちの製品が他の利用可能な療法と比較して費用効果があると思わない場合、FDAの承認後、彼らは私たちの製品をカバーしないかもしれないし、もし彼らがそう思う場合、支払いレベルは私たちの販売製品を利益にするのに十分ではないかもしれない。
2009年の“米国回復·再投資法案”は、同一疾患に対する異なる治療法の有効性を比較するための資金を連邦政府に提供した。この研究の計画は2012年に衛生·公衆サービス部、医療保健研究·品質局、国家衛生研究院によって発表され、研究状況や関連支出の報告が定期的に国会に提出される。有効性研究の結果を比較した結果、公共や個人支払人に保険政策を強制的に要求するためではないが、研究が我々の候補製品の販売にどのような影響を与えるかは不明である。もしそのような製品やそれが治療しようとしている場合が研究のテーマだ。比較有効性研究により競争相手の製品が優位であることが示されれば、承認されると、候補製品の販売に悪影響を及ぼす可能性がある。第三者支払者が、私たちの製品が他の利用可能な療法と比較して費用対効果があると思わない場合、彼らは承認された後に私たちの製品をその計画の下の福祉としてカバーしないかもしれない、または、もし彼らがそう思う場合、支払いレベルは利益に基づいて私たちの製品を販売するのに十分ではないかもしれない。
また、一部の外国の国では、薬品の提案価格は必ず承認されなければならず、合法的に発売されることができる。各国の薬品定価に対する要求は大きく異なる。例えば、EUは、その国の医療保険システムが精算を提供する医療製品の範囲を制限し、使用する医療製品の価格を制御するために、その加盟国に様々な選択を提供している。加盟国は医薬製品の具体的な価格を承認するか、あるいは医薬製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることができる。薬品の価格制御や精算制限を実施することが保証されないいかなる国/地域でも、私たちのどの製品に対しても割引の精算と定価手配が許可されます。歴史的に見ると、EUで発売された製品は米国の価格構造に従わず、通常価格を著しく下げる傾向がある。
逆控除及び虚偽申告法その他の規制事項
米国では、FDAに加えて、医薬品や医療機器の研究、製造、流通、販売、普及は、司法省、医療保険、医療補助サービスセンター、米国衛生·公衆サービス部の他の部門(例えば、監察長事務室)、薬品監督管理局、消費財安全委員会など、様々な連邦、州、地方当局の監督·法執行を受ける可能性がある。連邦貿易委員会、職業安全·健康管理局、環境保護局、州総検事長、その他の州と地方政府機関。私たちの現在および未来の業務活動は、販売、マーケティング、および科学/教育援助計画を含み、適用される医療規制法を遵守しなければならず、その中には、改正された“連邦反減税法令”、“連邦虚偽請求法”、改正された“健康保険携帯および責任法案”(HIPAA)に基づいて公布されたプライバシーおよび安全法規、医師支払い透明法、およびbr}のような州法が含まれる可能性がある。定価と返却点計画は改正後の1990年の“総合予算調節法”と改正された1992年の“退役軍人医療法案”中の医療補助薬品返却計画の要求に適合しなければならず、この法案は退役軍人管理局と他の連邦機関及びある安全網提供者(340 Bカバー実体と呼ばれる)に特殊な価格設定を要求する。もし製品が総務庁連邦供給スケジュールの許可ユーザーに提供されたら
, 他の法律と要求が適用される。このようなすべての活動はまた連邦と州消費者保護と不正競争法によって制限される可能性がある。
医薬製品の流通は他の要求と法規の制約を受け、広範な記録保存、許可、保存と安全要求を含み、br}の無許可による薬品の販売を防止することを目的としている。
26
カタログ表
連邦反リベート法規は、処方薬製造業者(またはそれを代表する当事者)を含む任意の個人または実体を規定し、知られている場合、直接または間接的に現金または実物の形態で要求、受け入れ、提供、提供または支払いを行うことができ、購入、レンタル、注文または手配または購入の推奨、レンタルまたは推薦購入、レンタルまたは任意の商品、施設、物品またはサービスの注文を含む任意の報酬を不正であると規定する。すべてまたは一部は、連邦医療保険または医療補助のような連邦医療計画に基づいている。“報酬”という言葉は価値のあるものを含むと広く解釈されている。連邦反リベート法規は、一方では、薬品メーカーと処方者、購入者および処方マネージャーの間の手配に適用されると解釈される。いくつかの法定例外と規制避難港保護のいくつかの一般的な活動は起訴されないが、例外と避難港の範囲は狭い。処方、購入、または推奨される報酬を誘導することを目的としていると告発される可能性がある慣行に関連し、例外または安全港の資格に適合していない場合、審査される可能性がある。特定の適用された法定例外或いは安全港を監督するすべての要求を満たすことができず、連邦反リベート法規が規定する違法行為を構成しない。代わりに、そのすべての事実と状況の累積審査に基づいて、この手配の合法性を逐案的に評価する。また,“患者保護と平価医療法案”は連邦反リベート法規下の意図基準を改正した, “衛生保健、教育と和解法案”(総称して“ACA”と呼ぶ)の改正後、より厳格な標準を採用し、個人或いは実体が実際に法規或いは法規違反の具体的な意図を理解する必要がなく、違反行為を実施することができる。また、“連邦虚偽申告法”によると、“連邦反リベート条例”違反による物品やサービスのクレームが虚偽または詐欺的クレームを構成している。この法律に違反した人は最高5年の禁錮刑、刑事罰金、行政民事罰金を科され、連邦医療計画から除外されることができる。しかも、多くの州は連邦反リベート規制のような法律を採択した。その中のいくつかの州禁止は紹介患者が任意の保険会社によって精算された医療サービスを受けることに適用され、MedicareとMedicaidのような連邦医療計画だけではない。これらの連邦および州反リベート法の広範性、およびこの分野の他の法律または法規の変化の可能性から、私たちの将来の業務活動は、私たちの販売およびマーケティング実践および/または私たちの将来の医師や医学界との関係を含めて、反リベート法の挑戦を受ける可能性があり、これは私たちを傷つけるかもしれない。
民事虚偽請求法案を含む連邦虚偽請求および虚偽陳述法は、任意の個人またはエンティティが知られている場合に提出することを禁止するか、またはそれを引き起こす(MedicareおよびMedicaidを含む)連邦計画(MedicareおよびMedicaidを含む)に虚偽または詐欺性を支払うプロジェクトまたはサービス(薬品を含む)請求を提出することを禁止する。支払者に直接クレームを提出することはありませんが、これらの法律によれば、メーカー
が顧客に不正確な請求書やコード情報を提供したり、ラベルの外で製品を普及させることによって“原因”とみなされて虚偽または詐欺的なクレームを提出しているとみなされている場合、製造業者は責任を負う可能性があります。また、私たちの製品の卸売業者や推定小売価格を報告するなど、医療補助フィードバック情報を計算するための価格や他の連邦、州、第三者精算に影響を与える情報、および私たちの製品の販売やbrマーケティングは、この法律の審査を受けています。例えば、連邦民事虚偽請求法案によると、製薬会社はその薬品のラベル外普及に責任があることが発見された。民事虚偽請求法案違反の処罰には,政府が実際に受けた被害の3倍を含み,個々の虚偽クレームに対する強制的な民事罰が含まれており,連邦医療保健計画への参加から除外される可能性があり,連邦虚偽クレーム法案は民事法規であるにもかかわらず,虚偽クレーム法案違反を招く行為には様々な連邦刑事法規が関与している可能性がある。もし政府が私たちがこれらの虚偽申告法に違反していると非難したり、私たちがこれらの虚偽申告法に違反したと判定したら、私たちは巨額の罰金を科され、株価が下落する可能性があります。また、, 個人は連邦民事虚偽申告法に基づいて訴訟を起こす能力があり、ある州はすでに連邦虚偽申告法にならって法律を制定している。
さらに、HIPAAは、個人第三者支払者を含む任意の医療福祉計画を知りながら故意に実行または実行しようとする追加の連邦刑事法規を制定し、重大な事実を偽造、隠蔽または隠蔽すること、または医療福祉、プロジェクトまたはサービスの交付または支払いについて任意の重大な虚偽、架空、または詐欺的な陳述を行うことを含む追加の連邦刑事法規を制定する。
メーカーが州政府に価格設定とマーケティング情報を報告することを要求する州法律も増えている。その中の多くの法律はこのような法律を遵守するために必要な条件に対して曖昧だ。例えば、連邦政府価格報告法は、複雑な価格指標を正確かつタイムリーな方法で計算し、政府プロジェクトに報告することを要求する。さらに、以下に述べるように、“医師支払い陽光法案”の下で同様の連邦は、あるメーカーに、過去1年間に医師および教育病院に支払われたいくつかのお金を追跡し、連邦政府に報告することを要求し、医師(医師、歯医者、検眼師、足科医および脊椎マッサージ師を含むと定義される)およびそれらの直系親族が所有するいくつかの所有権および投資権益を要求する。これらの法律は私たちに行政とコンプライアンスの負担をもたらし、それによって私たちの販売、マーケティング、その他の販売促進活動に影響を与えるかもしれません。また、これらの法律とその実施は明確性が不足していることから、私たちの通報行動は関連州と連邦当局の処罰条項によって制限される可能性がある。これらの報告義務は,2022年1月1日から,医師アシスタントや看護師従業員などのある非医師提供者への価値移転を含むように拡大された。
また、私たちは連邦政府と私たちが業務を展開している州のデータプライバシーと安全規制によって制限されるかもしれない。“経済と臨床衛生情報技術法案”によって改正されたHIPAA及びそのそれぞれの実施条例は、2013年1月25日に発表された最終総合規則を含み、特定のタイプの個人と組織の個人が健康情報のプライバシー、安全と伝送を識別できることに対して具体的な要求を提出した。また、ある州の法律は、場合によっては健康情報のプライバシーやセキュリティを管理しており、その多くの法律は互いに異なり、HIPAAとは大きく異なり、同じ効果が生じず、コンプライアンス作業を複雑化させる可能性がある。
27
カタログ表
規制要求が私たちを可能な法律や規制行動に直面させることができなかった。具体的な状況によると、適用される法規要件を満たすことができないことは、重大な刑事、民事および/または行政処罰、損害賠償、罰金、返還、MedicareおよびMedicaid、禁止、リコールまたは差し押さえ、生産の完全または部分的な一時停止、製品の承認の拒否または撤回、政府契約、契約損害、名声損害、行政負担、利益減少および将来収益、ならびに私たちの業務の削減または再編を含む供給契約の締結を許可することを拒否する可能性があります。いずれも私たちの業務運営能力と運営結果に悪影響を及ぼす可能性があります。
我々は,法律や計画要件の遵守を促進するための包括的なコンプライアンス計画を開発し,連邦医療計画や他の政府医療計画に基づいて精算可能な製品を商業化するために,これらの要求の主体となる可能性がある。
法律の変化または既存の法律の解釈は、(I)私たちの製造スケジュールを変更すること、(Ii)製品br}ラベルを追加または修正すること、(Iii)私たちの製品をリコールまたは停止すること、または(Iv)追加の記録保存要件を要求することなど、私たちの将来の業務に影響を与える可能性がある。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。
医療立法改革
米国や一部の外国司法管轄地域では,医療システムの立法や規制が多く変化しており,これは我々の製品販売収益力に影響を与える可能性がある。特に、2010年に“患者保護と平価医療法案”が公布され、この法案は2010年の“医療保健と教育和解法案”によって改正され、あるいは総称して“ACA”と呼ばれ、その中で他を除いて、生物製品をより低コストの生物模倣薬の潜在的な競争を受けさせる;吸入、注入、点滴、移植或いは注射の薬物に対して、メーカーが医療補助薬物リベート計画の下で不足しているリベートを計算する新しい方法を解決した;大多数のメーカーが医療補助薬物リベート計画の下で不足している最低医療補助リベートを高めた。医療補助薬品還付計画を医療補助管理保健組織に参加する個人処方の使用に拡張し、メーカーにあるブランドの処方薬に新しい年会費と税金を支払うことを要求する。新しいMedicare Part D保証不足割引計画を作成し、メーカーは保証間隔期間内に条件を満たす受益者に適用ブランド薬品交渉価格の50%(2018年両党予算法に従って70%に向上)の販売時点割引を提供することに同意しなければならず、メーカーのbr}外来薬品としてMedicare Part Dの条件を組み入れた;そして連邦政府の比較有効性研究の計画を増加するために激励を提供した。
また、諮問委員会法案が制定されて以来、他の立法改正も提案され、採択された
| • |
2011年8月、オバマ総裁は赤字削減合同特別委員会の創設を含む“2011年予算制御法案”に署名し、2013年から2021年までに少なくとも1.2兆ドルの赤字削減を国会に提案した。赤字削減合同特別委員会は的確な赤字削減を実現しておらず、立法がいくつかの政府プロジェクトに自動的に削減された。このbrには、各財政年度にサプライヤーに支払われる連邦医療保険の総減少幅が含まれており、最大2%に達し、その後の立法改正により、国会が追加的な行動を取らない限り、2030年まで有効になる。これらの削減は2013年4月に施行され,その後法規制が立法改正されたため,国会がさらに行動しない限り,これらの削減は2030年まで有効である。
|
| • |
2017年4月13日、CMSは、各州が個人や小団体市場の保険会社に基準を設定する上でより大きな柔軟性を得ることを可能にする最終ルールを発表し、このような市場で販売される計画に求められる基本的な健康福祉
を緩和する可能性がある。
|
| • |
2018年5月30日、“裁判権法案”が法律に署名された。他の事項以外に、この法律はある患者に連邦フレームワークを提供し、いくつかの第一段階の臨床試験が完成し、FDAの許可を得た研究用新薬製品を獲得するために調査を行っている。場合によっては、条件に適合する患者は、臨床試験を登録することなく、FDAの許可を得ることもなく、FDA Expanded Access計画に従って治療を求めることができる。“試用権法案”によると,製薬業者はその薬品を条件に該当する患者に提供する義務はない。
|
| • |
CMSは2019年5月23日、Advantage計画が2020年1月1日からB薬剤の一部に階段療法を使用することを許可する最終ルールを発表した。
|
28
カタログ表
アメリカでは、特殊薬品の価格設定に関する立法と法執行の関心が増加している。具体的には,米国議会は最近いくつかの調査を行い,薬品定価の透明性を向上させ,連邦医療保険下の処方薬のコストを低減し,定価とメーカー患者計画との関係,brおよび政府計画を改革する薬品精算方法を検討するための連邦と州立法を提案した。連邦レベルでは、総裁·バイデンは2021年7月9日に行政命令に署名し、政府の政策、すなわち(I)処方薬と生物製品の価格を下げるために立法改革を支持することを確認し、連邦医療保険の薬品価格の交渉を許可し、インフレ上限を設定し、低コストの模造薬と生物模倣薬の開発と市場参入を支持する;および(Ii)公共医療保険オプションの制定を支持する。その他の事項に加えて、行政命令は、処方薬の価格設定が高すぎ、国内の薬品サプライチェーンを強化し、連邦政府が薬品のために支払う価格を下げ、業界価格詐欺を解決するための行動を説明する報告書をHHSに提供するよう指示し、FDAに2003年の“連邦医療保険、改善と現代化法案”およびFDA実施条例に基づいて第804条の輸入計画を制定することを提案した州とインディアン部族との協力を指示した。FDAは2020年9月24日にこのような実施条例を発表し、2020年11月30日に発効し、各州のカナダ薬品輸入計画の制定と提出に指導を提供した。2020年9月25日, CMSは、各州がこの規則に基づいて輸入した薬品は“社会保障法”第1927条に基づいて連邦税金還付を受ける資格がなく、メーカー
は“最適価格”や平均メーカー価格の目的でこれらの薬品を報告しないと声明した。これらの薬物は対象とされていない外来薬であるため,CMSはさらに,これらのbr薬の全国平均薬品調達コストを公表しないことを示している。実施すれば、カナダからの薬品輸入は私たちの任意の候補製品の価格に実質的かつ不利な影響を与えるかもしれない。また、2020年11月30日、HHSは薬品メーカーがD部分でスポンサーの値下げを計画している安全港保護
を取り消し、法律が値下げを要求しない限り、直接或いは薬局福祉マネージャーを通過する法規を発表した。この規則はまた、販売時点での値下げを反映するための新しい安全港を作成し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための安全港を作成する。裁判所の命令により,上記の安全港の除去と増加は延期され,最近の立法はこの規則の実施を2032年1月1日に一時停止した。その中のいくつかの措置および他の提案された措置は、追加の立法許可によって発効する必要があるかもしれないが、バイデン政府とバイデン政府はこれらの措置を撤回または他の方法で変更する可能性があるが、バイデン政府と国会は薬品コストを制御するための新しい立法措置を求め続けると表明している。
最近、総裁·バイデンは2022年8月16日に“インフレ低減法案”に署名し、(I)政府はFDA承認日から9年(小分子薬)や13年(生物製品)を超える一部の高コスト連邦医療保険D部分(2026年から)と連邦医療保険B部分薬(2028年から)の価格設定や交渉を行うことができると規定している。(Ii)メーカーは2022年に連邦医療保険D部分の薬品と2023年の連邦医療保険B部分の薬品の価格増加がインフレより速い時に連邦医療保険B部分とD部分の薬品にリベートを支払い、(Iii)現在の保険不足条項の代わりに連邦医療保険D部分を再設計し、2025年から連邦医療保険受益者の自己負担上限に2,000ドルの上限を確立し、メーカーが10%のコストを担当し、最高上限は2,000ドルであり、上限に達した後は20%である。アイルランド共和軍の実施は規制当局が間もなく取る行動によって行われると予想され、その結果はまだ確定していない。
州レベルでは、立法機関は、価格または患者の精算制限、割引、いくつかの製品参入の制限、およびマーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法し、実施しており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。
私たちは、すでに採用され、将来取られる可能性のある医療改革措置が、より厳しい保険基準をもたらす可能性があり、私たちが承認された任意の製品の価格に追加的な下振れ圧力をもたらし、将来の収入を深刻に損なう可能性があると予想している。連邦医療保険や他の政府計画精算のいずれの減少も、個人第三者支払者の支払いの同様の減少を招く可能性がある。
外国、連邦と州の各レベルはすでに立法と監督管理提案を引き続き提出することが可能であり、医療保健の獲得性を拡大し、医療保健コストを制御或いは低減することを目的としている。コスト抑制措置や他の医療改革を実施することは、私たちの収入の創出、利益の実現、あるいは私たちの製品の商業化を阻止するかもしれない。このような改革は、私たちが開発に成功し、規制承認を得る可能性のある候補製品の予想収入に悪影響を及ぼす可能性があり、私たちの全体的な財務状況と候補製品を開発する能力に影響を及ぼす可能性がある。
EUの薬品審査と承認
臨床試験許可
EUでは,臨床試験の認可申請者は,臨床試験を行うEU加盟国の国家主管当局の承認を事前に得なければならない。また,出願人は,関連独立倫理委員会が関連加盟国の法律に基づいて賛成意見を発表した後にのみ,特定の研究地点で臨床試験を開始することができる。2014年4月,EUは新たな臨床試験条例(EU)第536/2014号を採択し,2022年1月31日に臨床試験指令2001/20/ECに代わった。それはEUの臨床試験の承認制度を徹底的に改革する。具体的には、すべてのEU加盟国に直接適用されるこの新しい立法(各EU加盟国で国家立法を実施する必要がないことを意味する)は、EU臨床試験の承認を簡略化し、簡略化することを目的としている。例えば,新たな臨床試験条例は,単一入口点による申請手続きの簡略化(試験を行う加盟国の各国の主管機関や倫理委員会に単独で提出するのではなく)を規定し,臨床試験申請評価の最終期限を厳格に規定している。臨床試験条例はまたEU加盟国のより効率的な共同評価と許可申請を可能にする, 臨床試験情報システムを介して。臨床試験認可請求が新たな臨床試験条例施行後1年以内(すなわち2023年1月31日)に提出された場合、新たな臨床試験条例の一時的な条項は、スポンサーが以前の臨床試験指令と臨床試験条例の要求との間で選択することを可能にする。2023年1月31日から、すべての申請は“臨床試験条例”に基づいて提出する必要がある。スポンサーが“臨床試験指令”に基づいて臨床試験を提出することを選択した場合、要求に応じて、この臨床試験は、新しい“臨床試験条例”の施行後3年まで、指示および各EU加盟国の関連執行法規の管轄を継続する。“臨床試験条例”の施行後に臨床試験が3年以上持続すれば、“臨床試験条例”はこの臨床試験への適用を開始する。臨床試験条例の実施には、私たちの臨床試験が適用される法律に適合することを確実にするために、追加のステップと手順をとる必要があるかもしれない。
29
カタログ表
マーケティング許可
EUでは、医薬製品はマーケティングの許可を得た後にのみ商業化されることができる。2つのタイプのマーケティング許可がある:(1)集中許可は、欧州委員会が人用薬品委員会(“CHMP”)(欧州薬品管理局の一機関)の意見に基づいて集中手続きによって発行され、ヨーロッパ経済区全体またはヨーロッパ経済地域(EU加盟国にノルウェー、アイスランド、リヒテンシュタインを加えて構成されている)で有効である。(2)国家マーケティング許可は、(EC)1234/2008号に規定された手続きを含む欧州経済地域全体ではなく、EU加盟国主管当局によって発行され、最初の申請(“分散手続き”)またはその後、(EC)1234/2008号に規定されたプログラムに従って初期国家マーケティング許可(“相互承認手順”)が発行された後、いくつかの加盟国で統一的に販売許可が発行される。
特定のタイプの製品、例えば、バイオテクノロジー医薬製品、孤児医薬製品、高度治療薬(すなわち、遺伝子治療、身体細胞治療および組織工学薬)、ならびにHIV/エイズ、癌、神経変性疾患、糖尿病、自己免疫疾患および他の免疫機能障害およびウイルス疾患を治療するための新しい活性物質を含む医薬製品については、集中プログラムを実行しなければならない。EUで許可されていない新しい活性物質を含む製品、または重大な治療、科学的または技術的革新を構成する製品、または公衆健康に有利な製品については、集中手順が任意である。遺伝子治療製品はEU先進治療薬物製品(ATMPと略称する)の一種である。ATMP上場許可申請に対する科学評価は主に専門的な科学委員会によって行われ、この委員会は高級治療委員会(CAT)と呼ばれる。CATは、マーケティング許可申請の主題であるATMPの品質、安全性、および有効性に関する意見草案を用意し、CHMPに最終承認を送信する。そして、CHMP提案はすべての欧州経済圏加盟国に対して拘束力がある決定を採択した欧州委員会に送信された。ATMPのマーケティング許可出願を評価する最長期限は、有効な出願を受信した日から210日であり、拷問禁止条約および/またはCHMPの質問に回答したときに、出願人が補足情報または書面または口頭解釈を提供する時間を含まない。停止タイミングは、評価を申請する時間範囲を210日以上に延長する可能性がある。CHMPが積極的な意見を出したところで, EMAは、EMAの提案を受けて67日以内にマーケティング許可を付与する最終決定を下すEU委員会に意見および支援文書を提供する。特殊な場合、公衆衛生の観点、特に治療革新の観点から見れば、1種の医薬製品は重大な利益を持っており、CHMPは加速評価を承認することができる。CHMPがそのような要求を受け入れる場合、210日間の評価期限は150日(クロック停止を含まない)に減少するが、CHMPが申請が加速評価にもはや適していないと判断した場合、中央プログラムの標準時限に回復する可能性がある。遺伝子治療薬製品の開発と評価は関連EUガイドラインを背景に考慮しなければならず、EMAは遺伝子治療薬物製品の開発とマーケティング許可に関する新しいガイドラインを発表し、これらの新しいbrガイドラインを遵守することを要求する可能性がある。
国家マーケティング許可は集中手続きの強制範囲に属さない製品を対象としている。製品がEU加盟国で販売されることが許可されている場合、このマーケティング許可は、相互認識手順によって別の加盟国で認められることができる。その製品が申請時にどの加盟国でも国家マーケティング許可を得ていない場合は、分散手続きによって各加盟国で同時に承認を得ることができる。分権手続きに従って、権限を求める各加盟国の主管当局に同じ書類を提出し、申請者はそのうちの1つを参考加盟国(“RMS”)として選択する。RMSが製品の許可を提案し、他の会員国が反対意見を提出しなかった場合、製品は、許可を求めるすべての加盟国で国家マーケティング許可を得るであろう。(EC)1234/2008号で規定されている手続きは、加盟国の関係当局間で評価に相違がある場合を含めて、すべての関連加盟国で統一されている。
上記の手順により,MAAを承認する前に,EU環境管理機関またはEU加盟国主管当局は,製品の品質,安全性,有効性に関する科学的基準に基づいて,製品のリスク−利益バランスを評価する。
イギリス(イギリスおよび北アイルランドを含む)がEUを離れたので、イギリスはもはや集中マーケティング許可のカバーを受けないだろう(北アイルランド議定書によれば、北アイルランドは集中マーケティング許可を認め続けるだろう)。現在の集中マーケティング許可を有するすべての医薬製品は、2021年1月1日にイギリスのマーケティング許可に自動的に変換される。2021年1月1日からの2年間、イギリスの医薬品および保健製品規制機関(MHRA)は、新しいイギリスのマーケティング許可をより迅速に承認するために、欧州委員会が集中手続きで新しいマーケティング許可を承認する決定に依存する可能性がある。しかし、まだ個別的な申請が必要になるだろう。
30
カタログ表
規制排他性
EUでは、発売が許可された革新製品(すなわち参考製品)は、8年間のデータ独占期間と追加2年の市場独占期間を得る資格がある可能性がある。brデータ独占期は、模倣薬または生物類似申請者が初めてEUの許可を得た日から8年以内に、EUが模倣薬または生物類似薬の発売許可を申請する際に参照製品ファイルに含まれる臨床前と臨床試験データに依存する。市場排他期は成功した模倣薬或いは生物類似申請者がその製品をEUで商業化することを禁止し、参考製品が初めて許可を得た10年後までである。10年前の8年間に、マーケティング許可保持者が1つまたは複数の新しい治療適応の許可を得た場合、10年の市場独占期間は最長11年に延長することができ、これらの新しい治療適応は、許可前の科学的評価中に既存の治療法と比較して有意な臨床的利益をもたらすと考えられる。しかしながら、1つの革新的な医薬製品が所定のデータ独占期間を取得しても、別の会社がマーケティング許可アプリケーションに基づいてマーケティング許可を取得した場合、同社は、完全に独立した薬物試験、臨床前試験、および臨床試験データパケットを有する製品の別のバージョンをマーケティングすることができる。
孤児指定と排他性
EUでは、孤児医薬品を指定する基準は、原則として米国の基準に類似している。(EC)141/2000号条例第3条によると、以下の基準を満たす場合、医薬品を孤児に指定することができる:(I)生命または慢性衰弱にかかわる疾患の診断、予防または治療に用いることができる。(2)または(A)申請時に、この状況は、EUの10,000人当たり5人以下に影響を与え、または(B)この製品に孤児身分によるメリットがなければ、EUで十分な見返りが生じず、その開発に必要な投資が合理的であることを証明する。(Iii)EU市場で販売されているそのような疾患の診断、予防または治療を許可していない好ましいbr方法、またはそのような方法が存在する場合、製品は、(EC)
847/2000号で定義されている疾患の影響を受ける人に大きな利益を得るであろう。孤児医薬製品は費用を下げたり費用を免除したりするなどの経済奨励を受ける資格があり、マーケティング許可を得た後、承認された治療的適応の10年間の市場独占経営権を得る権利がある。上場許可を申請する前に、孤児指定申請を提出しなければならない。孤児指定が承認された場合、出願人はマーケティング許可申請の費用減免を受けるが、マーケティング許可を提出したときにその指定が待っている場合は、そうではない。孤児指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の継続時間を短縮することもない。
5年目の終了時に、製品が孤児として指定された基準に適合しなくなったと判定された場合、例えば、製品の利益が十分に高く、市場排他性を維持するのが合理的であることを証明するのに十分でない場合、10年間の市場排他性は6年に減少することができる。そうでなければ、孤児薬品のマーケティング排他性を撤回することは、非常に特殊な場合にのみ、例えば:
| • |
第2の出願人は、その製品が許可製品と類似しているが、より安全で、より効果的であり、または臨床的に優れていることを証明することができる
|
| • |
ライセンス製品の販売許可保持者は、第二次孤児医薬製品申請に同意する;または
|
| • |
許可された製品の販売授権者は十分な孤児薬品を供給することができない。
|
上記の連合規則は一般的にヨーロッパ経済地域に適用される。
素数ラベル
2016年3月,EMAは適応候補の開発を促進するイニシアティブを開始したが,これらの適応はまれであり,現在のところ治療法はほとんどない。優先薬品(Prime)計画
は満たされていない医療需要領域の薬物開発を奨励し、そして重大な革新を代表する製品に対して加速評価を提供することを目的としており、その中で上場許可申請は集中プログラム
を通じて行われる。条件に適合する製品は、医療需要を満たしていない条件、すなわちEUに満足できる診断、予防または治療方法がないこと、または、ある場合、新薬は主要な治療優位性をもたらす)でなければならず、それらは早期臨床データに基づいて医療需要を満たしていない患者に利益をもたらす潜在力を示さなければならない。中小企業の製品は大企業よりも早くPrime計画に参加する資格があるかもしれません。Primeの称号を持つ候補製品のスポンサーは多くのbrのメリットを獲得し、しかし限らず、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素について頻繁に討論し、
及びファイルを提出した後にマーケティング許可申請評価を加速する。EMAのCHMPまたは高度治療委員会の専任連絡先および調査委員は、EMA委員会レベルの製品のより多くの理解を促進するために、Primeプログラムの早期に任命されることが重要である。会議を開始してこれらの関係を開始し、EMAを含む1つの多学科専門家チームは、全体発展と監督戦略に関する指導を提供する。研究開発過程で薬物が資格基準に適合しなくなった場合, Prime計画の下での支援は撤回されるかもしれない。
31
カタログ表
イギリスの離脱とイギリスの規制枠組み
2016年6月、英国の有権者はEU離脱(通称離脱)に賛成票を投じ、英国は2020年1月31日にEUから正式に離脱した。期間中EU薬品法は英国に適用され続け,この移行期間は2020年12月31日に満了する過渡期がある。しかし、EUとイギリスは、2021年1月1日から暫定的に適用され、2021年5月1日から正式に適用される貿易·協力協定、すなわちTCAを達成した。TCAには薬品に関する具体的な条項が含まれており,その中には相互承認GMP,医薬製品の製造施設の検査や発表されたGMPファイルが含まれているが,イギリスとEUの薬品法規を大規模に相互承認することはないと予想される。現在、イギリスは“2012年人類薬品条例”(改正)(北アイルランド議定書によると、EU規制枠組みは引き続き北アイルランドに適用される)を採択し、医薬製品のマーケティング、普及、販売に関するEUの立法を実施している。そのため,イギリスの規制制度は現在のEU法規と大きく一致しているが,イギリスの規制制度はEUから独立しており,TCAはイギリスとEUの薬品立法を相互に認めることを規定していないため,これらの制度は将来的に異なる可能性がある。例えば、イギリスはすでに“2004年ヒト用薬物(臨床試験)条例”(改正)により、現在廃止された“臨床試験指令2001/20/EC”を国家法律として定着させた。イギリスの臨床試験に対する監督管理がすでに発効した新しい臨床試験規制規定をどの程度反映するかは不明である, しかし、イギリスの薬品監督機関の薬品と保健製品の監督管理機関(“MHRA”)はすでにイギリスの臨床試験立法の改善と強化を目的とした一連の提案について相談を展開した。この協議は2022年3月14日まで続いた。
人力資本
2022年12月31日現在、私たちは240人のフルタイム従業員を持っており、そのうち230人はアメリカにあり、5人はスペインに、2人はスイス、イギリス、スウェーデン、ポルトガルでは各1人です。私たちはまたアルバイトを利用して私たちの業務需要に柔軟性を提供する。私たちの職員たちは労働組合代表もなく、集団交渉協定のカバー範囲もない。
私たちの人的資本目標には、私たちの既存と新入社員、コンサルタント、コンサルタントを識別、吸引、採用、維持、激励、発展と統合することが含まれている。私たちの株式と現金インセンティブ計画の主な目的は、株式と現金に基づく報酬奨励を付与することで、これらの従業員を激励してできる限りのことをし、私たちの目標を実現することで、株主価値と会社の成功を増加させることです。
多様化、公平、包容的な文化を発展させることは、私たちの成長戦略を実現するために必要なトップレベルの人材を引き続き誘致し、維持するために重要だと考えている。そのため、私たちは、私たちの従業員が毎日最高の職場を提供できるように、多様で包括的で安全な労働環境を作成し、維持することに投資しています。すべての従業員はロケット行動とロケット行動基準を維持する責任があり、これは私たちの政策と実践の基礎を構成している。
企業情報
私たちは1999年にイノーク製薬会社というデラウェア州に設立しました2018年1月、イノクはRocket PharmPharmticals,Ltdと合併し、Rocket PharmPharmticalsと改名し、Inc.我々の主な実行オフィスはニュージャージー州クランベリーシダブルック通り9号に位置し、郵便番号:08512、電話番号は(6096598001)。私たちのインターネットアドレスはwww.rocketpharma.comです。我々は,重要な非公開情報を開示する手段として我々のサイトを用い,FD法規下での開示義務を遵守している.我々は、これらの材料を米国証券取引委員会(“米国証券取引委員会”)に電子的に提出または提供した後、合理的で実行可能な範囲内で、私たちの10-K年間報告、10-Q四半期報告、8-K現在の報告、および取引法第13(A)または15(D)節に提出または提供されたこれらの報告の任意の改正をできるだけ早く無料で提供する。私たちのアメリカ証券取引委員会報告書は私たちのサイトの投資家の一部を通じてアクセスすることができます。米国証券取引委員会には、www.sec.govで提出された文書に関する報告書、依頼書、情報声明、その他の情報が含まれているウェブサイトがあります。私たちのウェブサイト上の情報は、参照によって本報告書に組み込まれていないか、または私たちが米国証券取引委員会に提出または提供している任意の他の報告書に組み込まれていない。私たちの普通株はナスダック世界市場に発売され、コードは“RCKT”です
| 第1 A項。 |
リスク要因
|
私たちがいる産業には多くの危険と不確実性が含まれている。これらのリスクに関する以下の情報と、本年度報告書の他の場所に出現する他の情報と、私たちの財務諸表や関連説明をよく考慮しなければなりません。以下のいずれのリスクが発生しても、私たちの業務、財務状況、br}運営結果、将来の成長見通しに重大な悪影響を及ぼす可能性があります。以下に説明するリスクおよび不確実性は、時間の経過とともに変化する可能性があるが、他のリスクおよび不確実性は、現在重大ではないと考えられているリスクおよび不確実性を含めて、私たちの業務を損なう可能性がある。この場合、私たちの普通株の市場価格は下落するかもしれない。
新冠肺炎の大流行関連リスク
SARS-CoV-2の爆発は、新冠肺炎を招き、あるいは未来の他の類似した大流行は著者らの臨床前と臨床研究を含む著者らの業務に不利な影響を与える可能性がある。
大流行や流行のような公衆衛生危機は私たちの業務に悪影響を及ぼすかもしれない。新冠肺炎とその変種伝播に対する私たちの初歩的な反応の一部として、私たちのほとんどの会社の従業員は遠隔アルバイトに転換し、少数の従業員の役割は私たちがニュージャージー州クランベリーにある工場現場でフルタイムで働く必要がある。
32
カタログ表
持続的な新冠肺炎の発生或いは類似の流行病のため、私たちはすでに未来に私たちの業務、臨床前研究、臨床試験の中断に深刻な影響を与える可能性があることを経験する可能性がある
| • |
患者を臨床試験に参加させるのは遅延したり困難です
|
| • |
臨床サイト起動の遅延または困難は、臨床サイト調査員と臨床サイトスタッフを募集する上での困難を含む
|
| • |
患者のフォローアップ、病院を私たちの臨床試験場所とする能力、臨床試験を支援する病院スタッフの移転など、医療資源を臨床試験の実施から移行させる
|
| • |
臨床試験現場データ監視のような臨床試験活動を中断する理由は、連邦または州政府、雇用主および他の人によって課せられたまたは提案された旅行制限、または臨床試験対象のアクセスおよび研究手順(特に不要と考えられる可能性のある任意のプログラム)の中断であり、これは、対象データおよび臨床研究終点の完全性に影響を与える可能性がある
|
| • |
私たちの内部で作られた建設が遅れたり困難に直面したりします
|
| • |
製造槽または材料の安全を確保する上で遅延または困難が発生する
|
| • |
我々の学術パートナーや契約研究機関の施設では,自ら実験室作業を行う臨床前研究遅延や困難が必要である
|
| • |
FDAおよび/または同様の外国規制機関の動作中断または遅延は、承認スケジュールに影響を与える可能性がある。
|
新冠肺炎がどの程度私たちの業務、臨床前研究と臨床試験に影響を与える可能性があり、これは引き続き未来の発展に依存し、これらの発展は高度な不確定性を有し、自信を持って予測できない、例えばウイルス新変種の識別、疫病発生の持続時間、旅行制限と疫病コントロールの行動、例えばアメリカと他の国/地域の社会的距離と隔離或いは封鎖、業務閉鎖或いは業務中断、及びアメリカと他の国が疾病の制御と治療のために取った行動の有効性。また、新冠肺炎の伝播による景気後退、不況、あるいは他の持続的な不利な市場事件。
私たちの財務状況と資金需要に関連するリスク
私たちの財務状況に関連するリスク
私たちは運営赤字の歴史があり、私たちは利益を達成したり維持することができないかもしれない。私たちは予測可能な未来に、私たちが引き続き損失を受けると予想する。もし私たちが計画した研究と開発を行うために追加資金を得ることができなければ、私たちは私たちの製品開発計画や商業開発を延期、減少、またはキャンセルさせられるかもしれません。
私たちは初期段階にある遺伝子治療会社で、運営歴史が限られており、あなたの投資意思決定の根拠とすることができます。遺伝子治療製品開発は投機性の強い仕事であり、大きなリスクに関連している。今まで、私たちの業務は主に私たちの会社、業務計画、資金の調達、製品と技術権利の買収と開発、私たちの研究開発と製造能力の強化、及び私たちの候補製品のために臨床前と臨床研究開発活動を展開することに限られている。私たちは製品販売から何の収入も得たことがない。私たちのどの候補製品も規制部門の承認を得ておらず、これまで株式売却の収益を通じて私たちの運営に資金を提供してきた。
設立以来、私たちは純損失を被った。2022年12月31日、2021年、2020年12月31日までの年度の純損失はそれぞれ2.219億ドル、1.691億ドル、1.397億ドルだった。2022年12月31日までの累計赤字は7.138億ドル。我々のほとんどの運営損失は,我々の研究開発計画に関するコスト,我々の製造能力の建設,および我々の運営に関する一般コストと行政(G&A)コストによるものである.私たちは今後数年と予測可能な未来に、研究開発、臨床試験、コンプライアンス活動、内部と外部製造活動を継続する予定であり、もし私たちの任意の候補製品が承認され、販売とマーケティング活動があれば、予想されるG&A費用に加えて、予測可能な未来に重大な損失をもたらす可能性があるため、私たちは引き続き重大な費用と運営損失を発生させるだろう。
私たちの限られた経営の歴史は、私たちの業務のこれまでの成功度を評価することを難しくし、私たちの未来の生存能力を評価することも難しいかもしれません。
今まで、私たちの運営は主に私たちの会社、業務計画、資金調達、私たちの技術の獲得、管理と拡張、潜在的な候補製品の決定、私たちの候補製品の研究、臨床前研究と臨床試験、私たちの研究開発と製造能力の強化、及び許可手配とbr}協力の確立に集中している。私たちはまだ候補製品の臨床試験を完了していません。市場の承認を得たり、商業規模の製品を生産したり、商業化に成功するために必要な販売やマーケティング活動を行っています。したがって、私たちの将来の成功や生存能力のどの予測も、私たちがより長い運営履歴を持っている場合ほど正確ではないかもしれません。私たちは現在薬物発見と臨床段階の会社で、後の時点で、私たちはbrを商業段階の会社に移行する必要があります。私たちは私たちがこの過渡期で成功するということを保証できない。
将来的には、私たちは販売およびマーケティング能力を確立することができない場合、または第三者と合意して私たちの任意の候補製品を販売およびマーケティングすることができず、これらの候補製品が承認された場合、私たちはそれを商業化することに成功できないかもしれない。
33
カタログ表
私たちの販売やマーケティングインフラは限られていて、医薬製品の販売、マーケティングあるいは流通の面で会社の経験がありません。私たちが販売とマーケティングの役割を保留している任意の承認された候補者が商業的に成功するためには、私たちの販売とマーケティング組織を発展させ、あるいはこれらの機能を第三者にアウトソーシングしなければならない。将来的には、私たちのいくつかの候補製品が承認された後、私たちのパートナーと一緒にこれらの製品の販売活動に販売または参加するために、集中的な販売、マーケティング、および商業支援インフラを構築し続けることを選択するかもしれない。
私たち自身のビジネス能力の確立と第三者とのこれらのサービス提供の合意はリスクに関連している。例えば、販売員の募集と訓練やbr精算専門員は高価で時間がかかり、いかなる製品の発表も延期される可能性がある。マーケティングおよび他の商業化能力を確立する候補製品の商業発表が何らかの理由で遅延または発生していない場合、これらの商業化費用を早期または不必要に発生させる。これは費用がかかるかもしれないし、もし私たちが私たちの商業化者を維持したり再配置できなければ、私たちの投資は損失するだろう。
もし私たちが第三者と合意して、販売、マーケティング、商業支援、流通サービスを実行すれば、私たちの製品収入またはこれらの製品収入が私たちにもたらす収益性は、私たちが自分たちが開発した任意の薬をマーケティングし、販売する場合よりも低くなるかもしれない。さらに、私たちは私たちの候補製品を商業化する計画を第三者と達成することに成功できないかもしれないし、私たちに有利な条項の下でそうすることができないかもしれない。私たちはこれらの第三者に対してほとんどコントロール権がないかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの薬品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが商業化能力を確立することに成功できなければ、私たち自身も第三者と協力しても、私たちの候補製品を商業化することに成功しないだろう。
税法の変化は私たちの業務と財政状況に悪影響を及ぼすかもしれない。
私たちが運営している司法管轄区域で、私たちは絶えず変化して複雑な税法の制約を受けている。アメリカ連邦、州、地方、非アメリカ所得税に関する規則は絶えず立法過程に参与する人員及びアメリカ国税局、アメリカ財務省と非アメリカ税務当局の審査を受けている。税法の変更(これらの変更は追跡力を有する可能性がある)は、私たちまたは私たちの普通株の保有者に悪影響を及ぼす可能性がある。近年、このような変化は多く発生しており、未来も変化し続けるかもしれない。税法の将来の変化は、私たちの業務、キャッシュフロー、財務状況または経営業績に実質的な悪影響を及ぼす可能性があります。私たちは投資家に税法の潜在的な変化が私たちの普通株に投資する影響について彼らの法律と税務顧問に相談することを促します。
私たちは純営業損失と研究開発控除を使用して、未来の課税収入の金額と能力はある制限と
不確定性の影響を受ける可能性があります。
2017年12月31日以降に開始された課税年度で発生した連邦純営業損失は一般にこれまでの納税年度に繰り越すことはできず,2017年12月31日以降に開始された課税年度で発生するこのような連邦純営業損失は満期にならないが,いずれの課税年度においてもこのような純営業損失の控除は当該年度の課税所得額の80%以内に制限され,当該年度に課税所得額が確定した場合には,純営業損失控除自体は考慮されない。しかし、コロナウイルス援助、救済、経済安全法案は、2017年12月31日以降から2021年1月1日までの課税年度におけるこのような連邦純営業損失の80%の使用制限を廃止し、2017年12月31日以降から2021年1月1日までの納税年度で発生する連邦純営業損失は、損失が発生した納税年度までの5つの納税年度の毎年に遡ることが許可されている。この法律変化は連邦純営業損失の繰越を一時的に許可し、発行者に実質的な利益をもたらすことはないと予想される上述したように、私たちは過去3つの納税年度に純損失が発生し、予測可能な未来に損失が続くと予想されています。したがって、私たちは私たちの純営業損失や税収控除を利用するために必要なアメリカ連邦や州課税所得額が発生するかどうかはわかりません。
一般的に、米国国税法第382条及び383条の規定によると、ある会社が所有権変更が発生した場合、その利用は、変更前の純営業損失又は純営業損失、税収控除又は控除(連邦研究開発税収控除を含む)を利用して将来の課税収入又は税収を相殺する能力が制限される。これらの目的のため、所有権変更は、通常、指定テスト期間中に、会社の株式の少なくとも5%を有する1つまたは複数の株主または株主団体の総持株率が、その最低持株率より50ポイント以上増加する場合に発生する。私たちの株式所有権の将来の変化は、その多くが私たちの制御範囲内ではなく、アメリカ国税法第382条と383条による所有権の変化を招き、純運営損失と信用を利用する能力を制限する可能性があります。州法によると、私たちの純営業損失や信用も損なわれる可能性があります。したがって,このような純運営損失やクレジットをすべて使用する前に所有権変更を行うと,その純運営損失やクレジットの大部分を利用できない可能性がある。
資本需要に関連するリスク
私たちは受け入れ可能な条件で提供できないかもしれないし、根本的にできないかもしれない追加資金を集める必要があるかもしれない。必要に応じて必要な資金を得ることができない場合、いくつかの許可活動、製品開発作業、または他の操作を延期、制限、または終了させることができる可能性がある。
34
カタログ表
私たちは未来に大量の資金が必要であり、私たちの遺伝子治療プラットフォームを拡大し、私たちの現在の候補製品と他の未来の候補製品の臨床前と臨床開発を推進し、これらの候補製品を商業化する可能性がある。私たちは私たちの臨床前と臨床活動によって私たちの支出レベルが増加すると予想する。さらに、私たちが現在または未来の任意の候補製品が市場の承認を得た場合、私たちは製品販売、医療事務、マーケティング、製造、および流通に関連する巨額の費用が発生すると予想される。また、上場企業の運営に関連する追加コストが予想され、特に新興成長型企業やより小さな報告会社の資格を備えておらず、大型加速申告会社としていくつかの追加開示やコンプライアンス要件の制約を受けています。したがって、私たちは私たちの持続的な運営に関連した多くの追加資金を得る必要があるだろう。もし私たちが必要な時や受け入れ可能な条件で資金を調達できない場合、私たちは私たちの製品開発努力または他のbr操作を延期、制限、または終了させることを余儀なくされるかもしれない。さらに、私たちが株式または転換可能な債務証券をさらに発行することで追加資金を調達すれば、私たちの既存の株主は重大な希釈を受ける可能性があり、私たちが発行した任意の新しい株式証券は普通株式保有者よりも高い権利、優遇、特権を持っている可能性がある。私たちが未来に獲得したどんな債務融資も、私たちの資金調達活動や他の財務や運営事項に関する制限的な契約に関連する可能性があり、これは私たちが追加的な資本を獲得し、ビジネス機会を求めることをより難しくするかもしれない, 潜在的な買収も含めて。また、最近の資本市場の変動、金利上昇、証券市場価格の一般的な下落は、私たちに有利な条項で新資本を獲得する能力に影響を与える可能性があり、これは私たちの流動性を損なう可能性があり、私たちの業務の成長を制限し、インフラを買収または改善する能力を制限し、市場での競争能力を制限することができる。
設立以来、私たちの業務は大量の現金を消費した。2022年12月31日現在、私たちの現金、現金等価物、投資は3兆997億ドルです。私たちの将来の資本需要は多くの要素に依存します
| • |
私たちの臨床前研究と臨床試験の登録時間、開始時間、完了時間と結果
|
| • |
アメリカ、ヨーロッパ、その他の管轄区域で私たちの候補製品の規制承認のコストを求めています
|
| • |
LVおよびAAV遺伝子治療候補製品を製造し、臨床前および臨床需要をサポートする
|
| • |
私たちの現在の候補製品の臨床前研究と任意の後続臨床試験の結果
|
| • |
私たちの候補製品の研究と開発及び臨床前研究と臨床試験を行う範囲、進捗、結果とコスト
|
| • |
私たちの候補製品に対する規制審査のコスト、時間、結果
|
| • |
将来の活動のコストは、製品販売、医療事務、マーケティング、製造、流通を含み、私たちが発売許可を得た任意の製品候補製品について;
|
| • |
もし承認されれば、臨床試験と商業発売のコストを支援するために臨床レベルと商業レベルの製品を生産する
|
| • |
組織や財団の寄付金を含む追加の非希釈資金を得ることができます
|
| • |
その候補製品が発売許可を得た場合、その製品の商業販売によって得られる収入
|
| • |
特許出願を準備し、提出し、起訴し、私たちの知的財産権を維持し、実行し、知的財産権に関するクレームを弁護するコスト;
|
| • |
私たちが有利な条件で協力を確立し維持する能力があれば
|
| • |
私たちは私たちが買収する可能性のある相補的な業務や技術の能力を統合することに成功しました
|
| • |
他の候補品や技術の程度を得ることができます
|
| • |
現在または将来の候補製品(ある場合)の時間、収入および販売金額、または私たちの特許使用料に関連するマイルストーン支払い。
|
その中の多くの要素は私たちがコントロールできることではない。潜在的な候補製品を決定し、臨床前テストと臨床試験を行うことは時間がかかり、高価で不確定な過程であり、完成するのに数年かかり、しかも著者らは監督と市場の許可を得て、製品販売を実現するために必要なデータ或いは結果を永遠に生成できないかもしれない。しかも、私たちの候補製品が承認されれば、商業的成功を得られないかもしれない。したがって、私たちは追加的な資金調達に依存して私たちの業務目標を達成し続ける必要があるだろう。
私たちは製品販売から何の収入も得られなかったし、永遠に利益を上げないかもしれない。
私たちが収入を創出し、利益を達成する能力は、私たち単独または戦略協力パートナーと製品開発を成功させ、私たちの候補製品を商業化するために必要な規制、定価、精算承認を得る能力にかかっています。予測可能な未来に、私たちは製品販売から収入を得られないことが予想されます。もしあれば。私たちが製品販売から将来の収入を得る能力は、私たちの以下の成功に大きくかかっている
| • |
私たちの候補製品の研究、臨床前、臨床開発を完成させる
|
| • |
臨床研究に成功した候補製品のために規制とマーケティングの承認を求めています
|
| • |
私たちのキャリアおよび候補製品のための持続可能で商業的な、反復可能かつ譲渡可能な製造プロセスを開発すること
|
| • |
(数量および品質上)十分な(数量および品質上の)製品およびサービスを提供して、臨床前および臨床開発および私たちの候補製品に対する市場の需要を支援することができる第三者との供給および製造関係を確立し、維持することができる
|
| • |
私たちが規制およびマーケティングの承認を得た候補製品を発表し、商業化する方法は、パートナーと協力するか、または独立して起動される場合、販売チーム、マーケティング、および流通インフラを構築することによって行われる
|
35
カタログ表
| • |
私たちの製品のために有利な市場保護を獲得し、維持すること、例えば、EUにおいて市場排他性孤児の称号を獲得(保持)することは、逆に第三者の活動および私たちが影響を与えない他の要因に依存する可能性がある
|
| • |
私たちの製品候補者のために個人と政府支払者から十分な価格設定と補償を受けた
|
| • |
市場に候補品を受け入れて遺伝子療法を実行可能な治療選択としました
|
| • |
競争的な技術や市場の発展に対応しています
|
| • |
新しい候補遺伝子治療製品を識別し検証します
|
| • |
私たちが参加する可能性のある任意の協力、許可、または他の手配で有利な条件を交渉する;
|
| • |
特許、商業秘密、およびノウハウを含む、私たちの知的財産権の組み合わせを維持し、保護し、拡大します。
|
開発された1つ以上の候補製品が商業販売に承認されても、承認された候補製品の商業化に関連する巨額のコストが発生することが予想される。FDA、EMAあるいは他の国内外の監督管理機関が現在予想されている基礎の上で臨床とその他の研究を行うことを要求すれば、私たちの費用は予想を超えるレベルに増加する可能性がある。承認された製品の販売から収入を得ても、利益が得られない可能性があり、運営を継続するために追加資金が必要かもしれません。
臨床開発と製品規制に関するリスク
私たちの候補製品の臨床開発に関するリスク
私たちは臨床試験の開始、登録或いは完成に重大な遅延に遭遇する可能性があり、あるいは安全性と有効性を証明できない可能性があり、適用された監督管理機関を満足させることができ、これは私たちが現在と未来の候補製品を適時に商業化することを阻止するかもしれない(もしあれば)。
規制部門の許可を得て私たちの現在と未来の候補製品を販売する前に、私たちの候補製品の安全性と有効性を証明するために、広範な臨床試験を行わなければならない。臨床試験は高価で時間もかかり、結果も確定していない。
私たちの臨床試験での経験は限られている。我々はロケットスポンサーのFA,LAD−I,PKD,Danon病の臨床試験を開始したが,これまで何の臨床試験も完了していない。いずれの臨床試験が計画通りに行われるか,あるいは計画的に完了することは保証されない(あれば)。臨床試験は、試験のどの段階でも様々な原因で遅延または停止する可能性がある
| • |
患者は予想された速度で研究に参加することができませんでした
|
| • |
私たちの候補製品は効果がありません
|
| • |
治療中に予期せぬ副作用または他の安全問題が出現した患者;
|
| • |
政府の規制や行政行為の変化
|
| • |
要求された臨床実践に従って研究されていない
|
| • |
FDA、EMA或いはその他の監督機関による臨床研究操作或いは研究場所の検査は、臨床一時停止を招く
|
| • |
財力が足りない
|
| • |
私たちが行って計画中の臨床試験では、薬物製品の供給が不足し、患者を治療できない
|
| • |
国内外の臨床場所の政治的動揺や自然災害
|
| • |
FDAを含む米国政府は閉鎖しています
|
| • |
大流行や流行病などの公衆衛生危機。
|
また、もし私たちが外国で私たちの候補製品の規制承認を得ることを求めるなら、私たちが外国で臨床研究を開始、登録、完成することに成功する能力は、外国で業務を展開するために独自の多くのリスクの影響を受ける
| • |
CROや医師との関係を構築したり管理したりすることは困難です
|
| • |
臨床試験を行う異なる基準;
|
| • |
一部の国では、LVおよびAAV遺伝子治療プログラムを検討するのに十分な規制専門知識を有する既存のグループが不足している
|
| • |
このような臨床試験のために合格した地元のパートナーや協力者を見つけることはできません
|
| • |
各種の外国法律、医療標準と監督管理要求の潜在的負担を遵守し、薬品と生物技術製品と治療に対する監督管理を含む。
|
もし私たちが計画通りに臨床試験を行うのに十分な数の患者を募集することが困難であれば、計画中の臨床試験を延期、制限あるいは中止する必要があるかもしれないが、いずれの状況の発生も私たちの業務、財務状況、運営結果、将来性を損なう。
36
カタログ表
患者が著者らの候補製品の臨床試験に参加する資格を確定し、参加させることは著者らの成功に重要である。臨床試験を迅速に完了するために、十分な患者、または必要なまたは必要な特徴を有する患者を識別、募集、募集することができない可能性がある。患者登録と試験完了は多種の要素の影響を受ける
| • |
調査中の病気の重症度は
|
| • |
学習プログラムの設計
|
| • |
患者集団の規模は
|
| • |
研究に関する資格基準
|
| • |
類似または競合療法において観察された副作用の結果を含む、研究された候補製品の知覚的リスクおよび利点
|
| • |
潜在患者に臨床研究場所の近似性と可用性を提供する
|
| • |
競争療法と臨床研究の有用性
|
| • |
臨床研究への参加を促進するために努力しています
|
| • |
医師の患者への転職方法と
|
| • |
治療期間と治療後に患者を十分にモニタリングする能力がある。
|
特に,われわれの現在の候補製品のいずれもまれな遺伝性疾患であり,臨床研究に利用可能な患者池は限られていることを評価する予定である。患者を識別し診断する過程は高価であることが証明されるかもしれない。場合によっては、潜在的な患者は米国以外に位置する可能性があり、政府政策の変化を含む移民に関する問題は、登録過程に追加の遅延をもたらす可能性がある。最後に,我々LV計画の治療過程は,患者から細胞を取得し,必要な時間線内で細胞を形質導入施設に搬送することを要求しており,この過程に許容できない輸送遅延
をもたらす可能性がある。
私たちの現在の候補製品はまだどんな臨床研究も完成していない。われわれが行っている臨床研究における初歩的,中期あるいは主要な結果
は,これらの研究が完了した結果を代表しない可能性がある。また、早期臨床研究の成功は今後の研究結果を代表しない可能性がある。
我々が後援しているFA,LAD−I,PKD,DDの臨床試験を開始したが,これまで何の臨床試験も完了していない。私たちはいかなる臨床試験が計画通りに行われるか、あるいは予定通りに完成することを保証できない。以前あるいは行われている研究と臨床試験の研究設計と結果は必ずしも未来の研究或いは臨床試験結果を予測するとは限らず、初歩的或いは中期結果は研究或いは試験完了後に継続或いは確認されない可能性がある。私たちの臨床研究中の被験体または私たちが行っているまたは未来の臨床研究のいずれの未来の被験体についても、積極的なデータは継続または出現しない可能性があり、私たちの候補製品に関連する進行中または将来の研究において重複または観察されない可能性がある。また,我々の候補製品も臨床開発の後期段階で必要な安全性と有効性を示すことができない可能性があり,初歩的な臨床研究に成功しているにもかかわらず。著者らはこれらの研究のいずれかが最終的に成功することを保証することはできず、臨床前或いは早期臨床研究が著者らの候補製品の更なる臨床進歩或いは監督管理承認を支持することを保証することもできない。
私たちは時々私たちの臨床前研究および臨床試験の中期または主要なデータを公開する可能性があり、これらのデータは当時利用可能なデータの初歩的な分析に基づいて、特定の研究または試験に関連するデータをより全面的に審査した後、結果と関連するbr}結果と結論が変化する可能性がある。データ分析の一部として,仮説,推定,計算,結論も行い,
我々はすべてのデータを網羅的かつ詳細に評価する機会がないか,または機会がないかもしれない.したがって、我々の報告の裏線または予備結果は、同じ研究の将来の結果と異なる可能性があり、または他のデータが受信されて十分に評価されると、異なる結論または
考慮要因がこれらの結果を合格させる可能性がある。バックラインデータはまた監査と検証手続きを受ける必要があり、これにより、最終データが私たちが以前に発表した予備データと実質的に異なる可能性がある。したがって、最終データが利用可能になる前に、バックラインデータは慎重に表示されなければならない。私たちが完成する可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、あるいは私たちの臨床試験中の患者が他の治療を継続するにつれて、1つまたは複数の臨床結果が実質的に変化する可能性があるリスクの影響を受ける可能性がある。初期または中期データと最終データとの間の不利な違いは、私たちのビジネスの将来性を深刻に損なう可能性があります。
さらに、規制機関を含む他の人は、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品、およびわが社の全体的な商業可能性に影響を与える可能性がある。さらに、公開開示された特定の研究または臨床試験に関する情報を選択することは、一般的に広範な情報に基づいており、あなたまたは他の人は、私たちが決定した重要な情報または他の適切な情報が私たちの開示に含まれることに同意しない可能性がある。
臨床前と臨床活動から得られたデータは異なる解釈を受ける可能性があり、これは監督部門の承認を延期、制限或いは阻止する可能性がある。また、多くの要素、製品開発期間中の監督管理政策の変化を含むため、監督管理の遅延や拒否に遭遇する可能性がある.
私たちの候補製品は不良と予見できない副作用を引き起こす可能性があり、あるいは公衆に安全ではないと思われる可能性があり、これは臨床試験や規制承認への進出を延期または阻止し、商業潜在力を制限したり、重大な負の結果を招く可能性がある。
遺伝子療法は依然として比較的新しい疾患治療法であり、私たちの候補製品は副作用を生じる可能性がある。
37
カタログ表
遺伝子治療製品の治療に出現する可能性のある副作用には,投与直後の免疫反応が含まれており,治療の有効性や持続性を大きく制限する可能性がある。もし私たちの潜在的な候補製品をテストする時にいくつかの副作用が観察された場合、私たちは候補製品のさらなる臨床開発を停止または延期することを決定または延期することができるかもしれない。FDAあるいは他の規制機関
は候補製品の臨床開発を停止または延期することを要求する可能性があり、原因は観察された新薬関連安全事件と関係がない。例えば,Danon病の治療に用いられたRP−A 501の第1段階臨床試験は2021年5月にFDAにより臨床的に放置され,これまで免疫を介したと考えられる補体活性化による血栓性微小血管病変が発生してきた。研究案と他の支援文書を修正し,患者選択と安全管理ガイドライン
を改訂した。臨床は2021年8月に解除された。
候補製品によって引き起こされる副作用に加えて、所与の候補製品に関連する管理プロセスまたは関連プロセスも、不良副作用をもたらす可能性がある。このような有害事象が発生した場合、私たちの臨床試験は一時停止または終了する可能性がある。場合によっては、FDA、欧州委員会、EMA、または他の規制機関は、任意またはすべての目標適応を承認する私たちの候補製品のさらなる開発を停止または拒否するように命令することができる。さらに、私たちが私たちの任意の候補製品の進行中または将来の臨床試験を開始または遅延、一時停止または終了しないことを選択または要求された場合、これらの候補製品の商業的見通しが損なわれる可能性があり、これらの候補製品から製品収入を得る能力が延期またはキャンセルされる可能性がある。
さらに、規制当局が候補製品を承認した後(例えば、長期追跡研究において)私たちの候補製品による副作用が発見された場合、いくつかの潜在的な重大な負の結果をもたらす可能性がある
| • |
規制部門は、製品候補製品の承認を一時停止または撤回することができる
|
| • |
規制部門はラベルに警告を追加することを要求するかもしれない
|
| • |
候補製品の投与方法を変更したり追加の臨床試験を行うことが要求されるかもしれません
|
| • |
私たちの名声は損なわれるかもしれない。
|
これらの状況のいずれも、他の候補製品を開発する能力を損なう可能性があり、私たちの業務、財務状況、および将来性を深刻に損なう可能性があります。
政府の規制に関連するリスク
我々の候補遺伝子治療製品は新技術に基づいているため、候補製品の開発及びその後監督部門の許可を得る時間とコストを予測することは困難である。現在、少数の遺伝子と細胞治療製品だけがアメリカとEUで承認されている。
これまで、著者らは研究開発努力を遺伝子治療プラットフォームに集中し、著者らの未来の成功は実行可能な遺伝子治療製品の候補製品の成功開発にかかっている。
FDA、EMAとその他の監督管理機関の臨床研究要求及びこれらの監督管理機関が候補製品の安全性と有効性を確定するための標準は潜在製品のタイプ、複雑性、意外性及び期待用途と市場によって大きく異なる。我々のような新製品候補製品の規制承認過程は、他のより有名またはより広く研究されている医薬品や他の製品候補製品の規制承認過程よりも高価である可能性があり、時間もかかる可能性がある。現在、ノワ製薬のKymriahとZolgensma(AveXisによって開発された)、Kite PharmaのYescarta、グラクソ·スミスクラインのStrimvelisおよびSpark TreeuticsのLuxturnaを含む少数の遺伝子および細胞治療製品だけが米国またはEUでマーケティング許可を得ている。したがって、私たちの製品がアメリカ、EU、または他の司法管轄区域で規制承認を得るのにどのくらい時間がかかるか、またはいくらかかるかを決定することは難しい。EMAの承認はFDAが何かを承認する必要があるかもしれないということを見せてくれないかもしれない。潜在的な製品を市場に出すために必要な監督管理の承認を遅延したり、獲得するのに必要な意外なコストを得ることができないことは、私たちが十分な製品収入を生成する能力を低下させ、私たちの業務、財務状況、運営結果、将来性に重大な損害を与える可能性がある。
遺伝子治療製品に対する規制要求はすでに変化し、未来に変化し続ける可能性がある。例えば、FDAの生物学的製剤評価および研究センター(“CBER”)は、私たちの開発コストを増加させ、規制の立場および解釈の変化を招き、候補遺伝子治療製品の承認および商業化を延期または阻止すること、またはbr}が重大な承認後制限または制限をもたらす可能性がある追加の非臨床研究または臨床試験の実行を要求する可能性がある。また,FDAは遺伝子や細胞治療製品を評価する方法の開発を続けている。例えば、この機関は遺伝子治療製品の開発、審査、承認の各方面に関連し、遺伝子治療製品に関連する臨床と製造問題を含む一連の草案と最終指導文書を発表した。2020年1月,FDA
はヒト遺伝子療法を服用している患者の長期追跡研究を提案し,遺伝子療法不良や予測不可能な結果のリスクが増加するため,
遅延の有害事象として表現される可能性がある最終ガイドラインを発表した。最終ガイドラインは、統合ベクター(例えばレンチウイルスベクター)の遺伝子療法治療を受けた患者は治療後15年以内に長期安全性と有効性フォローアップを受け、AAVベクターを統合した遺伝子療法治療を受けた患者は治療後5年間に及ぶ長期安全性と有効性フォローアップを行うことを提案した。私たちは、そのような指導またはFDAが発表する可能性のある他の指導が、私たちの遺伝子治療候補または任意の適用可能な規制開発および審査プロセスの持続時間または費用に悪影響を及ぼすかどうかを決定することができない。
38
カタログ表
米国で臨床試験を開始する前にFDAにINDを提出するほか,組換えあるいは核酸分子の合成に関連するあるヒト臨床試験は機関生物安全委員会(IBCs)の監督を受けなければならないことはNIHガイドライン“組換えあるいは核酸分子の合成に関連する研究ガイドライン”に掲載されている。米国国立衛生研究院のガイドラインによると、組換えおよび合成された核酸は、(1)核酸分子を連結することによって構成され、生細胞中で複製可能な分子(すなわち、組換え核酸)と、(2)化学的または他の方法で修飾されているが、自然に産生される核酸分子(すなわち、合成核酸)と塩基対を行うことができる分子を含む化学的または他の方法で合成または増幅された核酸分子と定義される。または(Iii)(I)または(Ii)に記載されたものを複製することによって生成される分子。具体的には,米国国立衛生研究院のガイドラインによると,ヒト遺伝子転移試験の監督にはIBCによる評価と評価が含まれており,IBCは地方機関委員会であり,この機構の組換えや合成核酸分子を用いた研究の審査·監督を担当している。IBCは研究の安全性を評価し、公衆衛生または環境に対する任意の潜在的リスクを決定し、このような審査は臨床試験開始前のいくつかの遅延をもたらす可能性がある。NIHガイドラインは強制的ではないにもかかわらず、関連研究がNIH組換えまたは合成核酸分子研究助成を受けた機関で行われているか、またはその助成によって行われていない限り、多くの会社および他のNIHガイドラインに拘束されていない機関は、自発的にこれらのガイドラインに従っている。
さらに、EMAの先進療法委員会(“CAT”)および他の規制審査委員会および諮問グループおよびその公布された任意の新しいガイドラインは、規制審査過程を延長する可能性があり、追加的な研究を要求し、私たちの開発コストを増加させ、規制の立場や解釈の変化を招き、候補製品の承認および商業化を延期または阻止し、あるいは重大な承認後の制限または制限を招くことができる。私たちが私たちの候補製品を推進する時、私たちはこれらの規制や諮問グループと協議し、適用されるガイドラインを遵守することを要求されるだろう。もし私たちがこれをできなかったら、私たちは私たちのいくつかの候補製品の開発を延期または停止することを要求されるかもしれない。このような追加的な手続きは私たちが予想していたより長く検討と承認過程を招くかもしれない。潜在的な製品を市場に出すために必要な監督管理の承認を遅延したり、獲得する際に意外なコストが発生したりすることは、私たちの製品収入を創出する能力を低下させる可能性があり、私たちの業務、財務状況、運営結果、将来性は深刻な損害を受けるだろう。
私たちがいくつかの候補製品の孤立した指定を得ても、私たちは候補製品の潜在的な市場排他性(承認されれば)を含むこのような指定の利点を実現できないかもしれない。
米国および他の主要市場を含むいくつかの司法管轄区の監督機関は、比較的小さい患者集団に影響を与える疾患または疾患を治療することを目的とした薬物をbr孤立薬物として指定する可能性がある。まれな疾患または疾患の治療のために使用される場合、FDAは、候補製品を孤立薬として指定することができ、一般に、米国での患者数が200,000人未満であるか、または米国での患者数が200,000人を超えると定義され、薬剤開発コストが米国の販売から回収されることが合理的に予想できない。EUでは、欧州委員会は、生命や慢性衰弱にかかわる疾患の診断、予防または治療のための製品の開発を促進するために、欧州医薬品局孤児薬物製品委員会の提案に基づいて、孤児薬物の称号を付与し、(I)EUでは、このような疾患の影響は10,000人に5人以下である。または(2)奨励措置がない場合、欧州連合における薬剤の販売は、薬剤または生物製品の開発に必要な投資が合理的であることを証明するのに十分ではない。この2つの場合、孤児として指定された出願人は、EUによって許可された疾患の診断、予防、または治療に関連する好ましい方法がないこと、または、そのような方法が存在する場合、疾患に使用可能な製品と比較して有意な利益を有する必要があることを証明しなければならない。
FDAおよび欧州委員会の孤児薬物指定、ファンコニ貧血の治療のためのRP-L 102、白血球接着欠陥-1の治療のためのRP-L 201、ピルビン酸キナーゼ欠乏症の治療のためのRP-L 301、Danon病の治療のためのRP-A 501、および乳児の悪性骨化症を治療するためのRP-401を取得した。これまで、私たちは他の候補製品に孤児薬物の称号(または外国同等製品)を得ることを要求していませんでしたが、私たちが将来申請しても、FDAや外国規制機関が私たちのどの候補製品もそのような称号を得ることを承認する保証はありません。さらに、私たちの任意の候補製品を孤立製品として指定することは、どの規制機関がその候補製品の規制審査または最終承認を加速させるかを意味するわけではなく、いかなる規制機関も、私たちの候補製品が独占的に発売承認される前に、私たちの候補製品と同じ適応を持つ他社の候補製品に孤立薬物指定の能力を付与することを制限しない。
一般に、孤児薬物の称号を有する候補製品が、その称号を有する適応の最初の発売許可を得た場合、製品は、許可された孤児医薬製品に含まれる1つまたは複数の類似活性物質を含む医薬製品として定義されたFDAまたは外国規制機関が、同じ薬物を構成する製品(または欧州医薬品局における“類似医薬製品”)を承認する権利を有する別のマーケティング申請を排除する。また,同じ治療適応に用いられる)は,この市場独占期に同じ適応を治療しているが,限られた場合は除外した。もし他のスポンサーが私たちの前にこのような承認を得た場合(私たちの孤児薬物名にかかわらず)、あるいは私たちが私たちの製品のために市場許可を得る前にEUで“類似の医薬製品”を承認した場合、私たちbrは適用される専門期間内に私たちの製品の市場承認を得ることを禁止されるだろう。適用期間は米国では7年、EUでは10年。申請者がその製品情報に追加の小児科研究結果を含む報酬および報酬を享受している場合、欧州連合の専門期間はさらに2年間延長することができる。一方、1つの製品が孤児薬物指定基準に適合しなくなり、このような指定がスポンサーによって撤回または満了された場合、EUの専門期間は6年に短縮されることができる, 製品が十分な利益を持っている場合を含めて、市場排他性はこれ以上合理的ではない。任意の規制機関が、指定された要求に重大な欠陥があると判断した場合、または製造業者が、このようなまれな疾患または疾患を有する患者の需要を満たすのに十分な数の製品を保証できない場合、孤立薬物の排他性を解除することができる。
39
カタログ表
候補製品の孤立薬物排他性を獲得しても,この排他性は候補製品を競合から効果的に保護できない可能性があり,異なる薬物は米国で同じbr条件に対して承認されるからである。孤児薬物が承認された後であっても、FDAが別の薬剤が同じ薬剤ではないと結論した場合、またはより安全で、より有効であることが証明されたので、または患者のケアに大きな貢献をしたので、FDAはその後、別の薬剤が同じ疾患を治療することを承認する可能性がある。FDAは孤児指定と孤児薬物排他性に関連する法規や政策をさらに再評価するかもしれない。FDAがいつ、いつ、あるいは未来に孤立薬物の法規や政策をどのように変化させるか、またどのような変化が私たちの業務に影響を与える可能性があるかどうかは分からない。FDAがその孤児の薬物法規や政策に変化する可能性があることによって、私たちの業務は悪影響を受ける可能性がある。
欧州連合では、以下の場合、同じ孤児適応の類似医薬製品にマーケティング許可を与えることができる
| • |
第2の出願人は、その薬剤が許可された孤児医薬品と類似しているが、より安全で、より効率的で、または他の態様で臨床的利点を有することをその出願で証明することができる
|
| • |
元孤児薬品発売授権書所持者は孤児薬品の再申請に同意した
|
| • |
元孤児薬品発売授権書を持っている人は、十分な量の孤児薬品を供給することができない。
|
FDAが指定したFast Track或いは再生医学高度療法(RMAT)或いはEMAが指定した優先薬物或いはPrimeは、私たちに現在或いは未来の任意の候補製品を与えても、開発或いは監督審査或いは承認過程を加速することができない可能性があり、現在の候補製品と任意の未来の候補製品が発売許可を得る可能性を増加させることはない。
候補製品が深刻または生命に危険な疾患を治療するために使用され、その製品が満たされていない医療需要を解決する潜在力を有する場合、スポンサーは、特定の適応についてFDA Fast Track指定を申請することができる。Danon病のためのRP−A 501、FAのためのRP−L 102、LAD−IのためのRP−L 201、およびPKDのためのRP−L 301の高速チャネル名を受信した。私たちは将来の候補製品のための迅速なチャネル認証を求めるかもしれないが、FDAがこの地位を私たちが提案した任意の候補製品に与える保証はない。
会社はその候補製品に対するRMAT認証を要求することができ、製品が以下の基準を満たす場合、FDAはこのような認証を付与することができる:(I)それは細胞療法、治療用組織工学製品、ヒト細胞と組織製品、またはそのような治療法または製品を使用する任意の組み合わせ製品であるが、限られた例外がある;(Ii)それは治療、修正、逆転または治癒を目的としている;および(Iii)初歩的な臨床証拠は、このような疾患または状態が満たされていない医療需要を満たす可能性があることを示している。Danon病の治療のためのRP−A 501、FAのためのRP−L 102、およびLAD−IのためのRP−L 201のRMATが受信された。RMAT
の指定は、FDAとより頻繁に会議を開いて候補製品の開発計画を検討すること、および潜在的なスクロール審査および優先審査の資格を含む潜在的な利点を提供する。RMAT認証を取得した製品も加速承認を得る資格があり、その根拠は、長期的な臨床的利益を予測することが可能な代替物または中間終点、または試験を他の場所に拡大することを含む大量の場所から取得されたデータに依存する。
良質な指定はEMAが提供する計画であり,満たされていない医療ニーズに対する薬物開発の支援を強化することを目的としている。Prime認証を取得する資格があるためには,候補製品は早期br臨床証拠が必要であり,この療法が既存の治療法よりも良い治療優位性を提供し,あるいは治療選択のない患者に利益を与える可能性があることを証明した。FAのためのRP−L 102およびLAD−IのためのRP−L 201の良質な名前を受信した。Primeの利点は、マーケティング許可申請の前に継続的な支援を提供し、知識の蓄積を支援する調査委員を任命すること、重要な開発マイルストーンで早期の対話および科学的提案を行うこと、および申請過程がより早い時期に製品の資格鑑定を加速的に検討する可能性があることを含む。
FDAは広範な裁量権を持ち,Fast TrackやRMAT認証が付与されているかどうか,EMAは広範な自主権を持ち,Prime認証が付与されているかどうかから,特定の製品
候補製品がこのような認証を受ける資格があると考えても,FDAやEMAがその製品を付与することを決定する保証はない.Fast Track,RMATあるいはPrime資格を取得しても,従来の開発,審査,承認スケジュールと比較して,より速い開発プロセス,審査や承認を経験することができず,Fast Track,RMATまたはPrime資格を得ることは製品承認の基準を変更することはないかもしれない.また,FDAはFast TrackやRMATの指定を取り消す可能性があり,EMAがわれわれの臨床開発計画からのデータがこの指定を支持しなくなったと考えた場合,Prime指定を取り消す可能性がある。
FDAの加速承認やEMAの条件付き承認は,より速い開発過程や規制審査をもたらさない可能性があり,我々のbr候補製品が上場承認される可能性も増加しない。もし私たちがこの過程で成功しなければ、私たちは承認または条件付き承認の加速を求める候補製品の開発や商業化が延期されるか、放棄されるか、明らかに高価になる可能性がある.
40
カタログ表
私たちはFDAの加速承認とEMAの条件付き承認経路を使用して私たちの候補製品の承認を求めることができる。試験設計を用いて承認の加速を支援することができるが,このような製品
候補製品は,より速い開発や規制審査スケジュールによって制限されない可能性がある.
1つの製品が深刻または生命に危険な疾患を治療する場合、通常、既存の治療法よりも意義のある利点を提供し、代替終点に対して合理的に臨床的利益を予測する可能性のある効果を示す場合、製品はFDAの加速承認を得る資格がある可能性がある。承認を加速させる条件として,FDAは十分かつ良好な制御を行う上場後臨床試験を含む特定の義務を所定のスケジュール内で強制的に実行する可能性がある。このような検証的な実験は職務調査の方法で行われなければならない。さらに、FDAは、機関から別の通知がない限り、承認を加速させた製品の販売促進材料を予め承認しなければならないことを要求しており、製品の商業発表時間に悪影響を及ぼす可能性がある。FDAまたはEMAが承認の加速または条件付き承認の候補製品を求めるのではなく、マーケティング申請を提出する前に1つまたは複数の完全な3期臨床試験を完了することを要求する場合、そのような候補製品の開発および商業化スケジュールは延期されるであろう。たとえ私たちが加速的な承認やbrの条件付き承認を得たとしても、私たちは最終的に規制機関の完全な承認を得られないかもしれない。市販後の臨床試験により生成された他のデータは,加速承認を得た候補製品の利益−リスクバランスが積極的であること,あるいはさらなる義務達成の負担が高すぎる可能性があることを確認できない可能性がある。また、2023年12月29日に公布された“2023年総合支出法”, 承認プロセスの改訂を加速し、FDAに追加的な権限を与えて上場後の研究要求を実行し、承認を加速した保有者が承認後の臨床研究要求を遵守できなかった場合には、より迅速に承認を撤回することを含む。
EUでは、条件付きマーケティングライセンスは年間更新プログラムを遵守し、マーケティングライセンス所有者がライセンス遵守の具体的な義務を遵守する場合を評価しなければならない。条件を満たしていない場合、EMAは、既存の義務のスケジュールを延長すること、そのような義務の範囲を変更すること、または新たな義務を追加することを決定することができ、これは、追加の財源および時間を必要とする可能性がある。私たちは、このような変更または追加の義務を遵守できない可能性があり、マーケティング許可を撤回する必要があるかもしれない。EMAは、このような措置が実際にはあまり適用されないにもかかわらず、条件付きマーケティング許可を更新しないことを決定することもできる。欧州連合の条件付認可薬品の精算決定の分析により、積極的な保健技術提案を達成するスケジュールにいくつかの遅延があることを表明した。条件付き承認を求める任意の候補製品がこのような状況になれば、このような製品の商業化の時期と成功を遅らせる可能性がある。最後に、条件付き許可を満たす条件または他の態様から得られた新しいデータが示された場合、我々の製品の利点は、もはやそのリスクを超えない場合、EMAは、条件付きマーケティング許可を一時停止または撤回するような規制行動をとることができる。
Danon病に対するRP−A 501,FAに対するRP−L 102,LAD−Iに対するRP−L 201のまれな小児科疾患名を受信した。しかし,これらの候補製品のマーケティング申請が承認された場合,まれな小児科疾患優先審査券の資格基準を満たしていない可能性がある。
Danon病に対するRP−A 501,FAに対するRP−L 102,LAD−Iに対するRP−L 201のまれな小児科疾患名を受信した。生物製品を1種の稀な小児科疾患の製品に指定することは、この生物製品のBLAが申請が許可された時に稀な小児科疾患の優先審査証明書の資格基準に符合することを保証することはできない。連邦食品、薬物、および化粧品法案(“FDCA”)によると、私たちが稀な小児科疾患の称号を獲得した候補製品について、私たちは私たちの原始BLAで稀な小児科疾患の優先審査証明書を申請する必要がある。FDAは、このような候補製品のBLAが承認された場合、優先審査証明書の資格基準を満たしていないと判断することができる。
FDAが2024年9月30日以降に生物製品まれな小児科疾患優先審査券を付与する権限は現在、2024年9月30日または以前に稀な小児科疾患指定を獲得した生物製品に限られており、FDAは2026年9月30日までの稀な小児科疾患優先審査券しか授与できない。しかし、FDAが稀な小児科疾患の優先審査証明書を付与する権限は議会によってさらに延長される可能性がある。
必要な前臨床研究と臨床試験を成功させても、いつあるいは規制部門の承認を得て、候補製品を商業化するかどうかを予測することはできませんし、承認の範囲は私たちが求めている範囲よりも狭いかもしれません。
適切な規制機関が候補製品を審査して承認するまで、私たちは候補製品を商業化することができない。私たちはまだbr司法管轄区域の規制機関の許可を得ていません。私たちの任意の候補製品を販売することができます。著者らの候補製品が臨床試験中にその安全性と有効性の終点に達しても、監督部門は適時に審査プロセスを完成できず、
の完全な返信状を発行し、或いは最終的に監督部門の許可を得ることができない可能性がある。さらに、FDA諮問委員会が使用を承認したり制限したりすることを提案しない場合、私たちは遅延や拒否に遭遇する可能性がある。また,製品開発,臨床試験,審査過程において,将来の立法や行政行動における追加政府法規や規制機関政策の変化により遅延や拒否が生じる可能性がある。監督管理機関は審査過程中にかなりの自由裁量権を持っており、いかなる申請を受け入れることを拒否することができ、著者らのデータが承認を得るのに十分ではないことを決定することができ、追加の臨床前、臨床或いは他の
研究を行う必要がある。そのほか、臨床前と臨床試験から得られたデータの異なる解釈は候補製品の上場承認を延期、制限或いは阻止する可能性がある。
41
カタログ表
規制当局はまた、要求よりも限られた適応を有する候補製品を承認することができ、または彼らは、狭い適応、警告、または他のラベル変更の形態で重大な制限を適用する可能性がある。これらの監督管理機関は予防措置や禁忌症を要求するか、あるいは高価な発売後の臨床試験の表現に基づいて承認することを要求する可能性がある。監督管理機関はリスク評価と緩和策略或いはREMS或いは同等の要求の形で製品の流通、処方或いは調剤に制限と条件を加えることができる。さらに、規制当局は、私たちの候補製品の商業化に必要または必要なラベル宣言を承認しないかもしれない。上記のいずれの状況も、私たちの候補製品の商業見通しに重大な損害を与え、私たちの業務、財務状況、運営結果、将来性に重大な損害を与える可能性があります。
私たちが製品候補の規制承認を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けるだろう。
私たちが管轄区域で規制の承認を受けても、私たちは持続的な規制義務と持続的な規制審査の制約を受けるだろう。候補製品が承認されると、適用される規制機関は、候補製品の指定用途またはマーケティングに重大な制限を加えることができ、またはコストの高い可能性のある承認後の研究、発売後の監視または患者または薬物制限に持続的な要求を加えることができる。また,承認されたBLAの保持者は,不良イベントおよび製品がBLAにおける規格に適合していないいかなる故障も監視·報告する義務がある.承認されたBLAの所有者はまた、承認された製品、製品ラベル、または製造プロセスをいくつかの変更することができるように、新しい申請または追加の出願を提出し、FDAの承認を得なければならない。FDAガイドラインはあるタイプの遺伝子治療を受けた患者に15年間にわたる追跡観察を受け、潜在的な不良事件を理解することを提案した。他の適用可能な連邦および州法律に加えて、広告および販売促進材料はFDAの規定に適合し、FDAの審査を受けなければならない。
また、製品メーカーおよびその施設は、使用料を納付し、GMPに適合することを確保し、現在の良好なティッシュ規範を遵守し、“BLA”で行われている約束を遵守するために、FDAおよび他の規制機関の持続的な審査および定期検査を受ける必要がある。いくつかの商業処方薬生物製品の場合、製造業者およびサプライチェーンに関連する他の当事者はまた、流通チェーン要件を満たし、製品を追跡および追跡するための電子的、相互運用可能なシステムを確立し、偽、移転、盗難、および意図的に偽の製品、または米国での流通に適していない他の製品をFDAに通報しなければならない。もし、私たちまたは規制機関が、ある製品に以前に未知の問題、例えば意外な深刻性または頻度の不良事象が存在することを発見した場合、または製品の製造施設に問題がある場合、監督管理機関は、リコールを要求するか、または市場から製品を撤回するか、または生産を一時停止することを含む、製品または製造施設に制限を加えることができる。
もし私たちが私たちのすべての候補製品を承認した後に適用される規制要求を遵守できなければ、監督管理機関は様々な行動を取るかもしれません
| • |
私たちは法律に違反していると警告状を出しました
|
| • |
禁止令を求めたり、民事または刑事罰や罰金を科したり
|
| • |
現在行われている臨床研究を中断します
|
| • |
私たちが提出したBLAまたは補足BLAのような未解決のマーケティング申請の承認を拒否する;
|
| • |
製品を差し押さえる
|
| • |
政府契約を含めて私たちが供給契約を締結することを許可することを拒否する。
|
政府は違法の疑いのあるいかなる調査にも対応するために多くの時間と資源を必要とする可能性があり、否定的な宣伝が生じる可能性がある。上記のいずれかのイベントまたは処罰が発生すると、候補製品を商業化し、収入を創出する能力を抑制し、私たちの業務、財務状況、運営結果、および将来性を損なう可能性があります。
また、FDAの政策や同様の外国規制機関の政策は変化する可能性があり、我々の候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布される可能性がある。米国や海外の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちがbrの既存の要求の変化に適応できない場合、あるいは新しい要求や政策を採用することができない場合、あるいは私たちが法規遵守を維持できない場合、私たちは私たちが得る可能性のあるいかなるマーケティング承認も失う可能性があり、私たちは利益を達成したり維持することができないかもしれません。これは私たちの業務、財務状況、運営結果、将来性に重大な損害を与えます。
私たちのどの候補製品もアメリカやEUでFDAやEMAの承認を得られないかもしれません。たとえ私たちがそうしても、私たちはいかなる他の司法管轄区域でも承認されたり、私たちの任意の候補製品を商業化したりすることはありません。これは私たちが市場の潜在力を十分に発揮する能力を制限するだろう。
最終的に任意の特定の管轄区域で私たちの任意の候補製品を販売するためには、各管轄区域に基づいて、多くの異なる安全性と有効性法規要件を確立し、遵守しなければならない。米国FDAやEU EMAの承認を得ても、他の国や管轄区域規制機関の承認を得ることは確保できない。また、ある国で行われている臨床前研究や臨床試験は、他の国の規制機関に受け入れられない可能性があり、一国の規制承認は他のどの国の規制承認も保証されていない。承認の流れは国/地域によって異なり、追加の製品テストと検証、追加の行政審査期間に及ぶ可能性があります。外国の監督管理機関の承認を求めることは私たちに困難とコストをもたらす可能性があり、追加の臨床前研究或いは臨床試験
が必要であり、これは高価で時間がかかるかもしれない。各国の規制要求には大きな違いがある可能性があり、私たちの製品がこれらの国で発売されることを延期または阻止する可能性がある。外国の監督管理審査プロセスに関連するリスクはFDAとEMA承認プロセスと類似している。私たちは国際市場を含めてどの候補製品も司法管轄区域で販売されることを許可されていないし、私たちはこのような承認を得ようとしていない。もし私たちが国際市場の規制要求を遵守できなかったり、必要な承認を獲得して維持できなかったり、あるいは国際市場の規制承認が延期された場合、私たちの目標市場は減少し、私たちの製品の全面的な市場潜在力を実現する能力は実現できないだろう。
42
カタログ表
承認されれば、私たちの候補製品は生物模倣薬からの競争に直面する可能性があり、これらの模倣薬は短い規制経路で承認される。
“患者保護および平価医療法案”は、2010年の“医療·教育調整法案”によって改正され、または総称してACAと呼ばれ、“2009年生物製品価格競争および革新法案”、またはBPCIAと呼ばれるサブタイトルを含み、FDA許可の参照生物製品と類似しているか、または交換可能な生物製品のための短い承認経路を作成する。BPCIAによると、バイオ類似製品の出願は、参照製品がFDA許可を初めて得た4年後にFDAに提出することができる。また,生物類似製品の承認は,参考製品が初めて許可を得た日から12年後にFDAによって発効する可能性がある。この12年間の独占期間内に、FDAが競合製品のBLAを承認した場合、別の会社は、スポンサー自身の臨床前データと、他の会社の製品の安全性、純度、および有効性を証明するために、十分かつ制御された良好な臨床試験からのデータとを含む参照製品の競合バージョンを販売する可能性がある。この法律は複雑であり、FDAはまだ説明して施行している。したがって、その最終的な影響、実施、そして意味には不確実性がある。
我々のBLAによりバイオ製品として承認された候補製品はいずれも12年の専門期間を得る資格があるべきであると考えられる。しかしながら、国会の行動または他の理由により、このような独占性が短縮される可能性があり、またはFDAは、我々の研究薬を競合製品の参考製品とみなさないことは、予想よりも早く模倣薬の競争のための機会を創出する可能性がある。BPCIAの他の面では,そのいくつかがBPCIAの排他的条項に影響を与える可能性があり,最近の訴訟のテーマでもある.さらに、許可が得られると、生物学的類似体は、我々のいずれかの参照製品の代わりに非生物学的製品と類似した伝統的な模倣薬でどの程度置換されるかは、まだ発展中の多くの市場および規制要因に依存するであろう。
もし競争相手が私たちの製品を参照する生物模倣薬の市場許可を得ることができれば、私たちの製品はこのような生物模倣薬の競争を受ける可能性があり、それに伴い競争圧力と結果がある。
医療立法改革措置は私たちの業務と運営結果に実質的な悪影響を及ぼす可能性がある。
米国および多くの外国司法管轄区域は、医療システムに影響を与える立法および規制改革を公布または提案しており、これらの改革は、私たちの候補製品や将来の候補製品の上場承認を阻止または延期し、承認後の活動を制限または規制し、マーケティングの承認を得た任意の製品を収益的に販売する能力に影響を与える可能性がある。規制、規制の変更、または既存の規制の解釈は、(I)私たちの製造スケジュールの変更、(Ii)製品ラベルの追加または修正、(Iii)私たちの製品のリコールまたは停止、または(Iv)
追加の記録保存要件のような、私たちの将来の業務に影響を与える可能性があります。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。タイトルを見て“企業−政府規制−医療立法改革−”.
また、米国や海外の政府や第三者支払者が医療コストの制限や低減に力を入れていることは、このような組織が新承認製品の保証範囲や精算レベルを制限する可能性があるため、候補商品に保険を提供したり、十分な支払いを提供できない可能性がある。アメリカの専門薬品価格設定の実践における立法と法執行の興味はますます大きくなっている。具体的には、米国議会は最近数回の調査を行い、薬品定価の透明性を高め、連邦医療保険制度下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査し、政府計画の薬品精算方法を改革するための連邦と州立法を提出し、公布した。私たちは将来的に追加のアメリカ連邦医療改革措置
を取ることが予想され、どの措置もアメリカ連邦政府が医療薬品とサービスのために支払う金額を制限する可能性があり、これは私たちの候補薬物に対する需要の減少または追加の価格設定圧力を招く可能性がある。
アメリカの個別州もますます積極的に立法と実施を通じて、価格或いは患者の精算制限、割引、ある薬品への参入の制限、マーケティングコストの開示と透明性措置を含む薬品と生物製品の定価を制御するための法規を実施し、他の国からの輸入と大量購入を奨励することを目的としている。法律で規定されている価格
第三者支払者の支払い金額の制御またはその他の制限は、私たちの業務、財務状況、運営結果、見通しを損なう可能性があります。また,地域医療当局や個別病院では,どの薬品やサプライヤーがその処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これは私たちの薬品の最終需要を減少させ、あるいは私たちの薬品の価格設定に圧力を与える可能性があり、これは私たちの業務、財務状況、運営結果と将来性に負の影響を与えるかもしれない。
イギリスのEU離脱やイギリスの離脱は、規制や法律の複雑さを増加させる可能性があり、これは、欧州での業務をより難しくし、私たちの製品候補製品がヨーロッパおよび/またはイギリスの規制承認を得ることを確保する上で追加的な挑戦をもたらすかもしれない。
43
カタログ表
著者らは現在イギリスに臨床試験基地があり、イギリスに契約実験室があり、私たちの全世界の臨床試験のためにテストを行い、イギリスとヨーロッパ全体に他の協力者と潜在的な協力者がいる。EU条約50条によると、イギリスは2020年1月31日にEU加盟国として停止した。英国とEUは貿易協定を結び、2021年1月1日から臨時適用され、2021年5月1日から正式に適用される。協定条項によると、EUとイギリスは薬品に対して単独の規制制度を持っており、いくつかの条項は相互承認基準、例えばGMPに関する基準を規定しているにもかかわらず。例えば、イギリスはEU範囲内の医薬製品マーケティング許可を得る集中手順にはもはや適用されないが、イギリスは単独の医薬製品許可プロセスを必要とし、許可はイギリスまたは大ブリテン(イングランド、スコットランド、ウェールズ)のみをカバーすることになる。現在、イギリスは“2012年人類薬品条例”(改正)(北アイルランド議定書によると、EU規制枠組みは北アイルランドに引き続き適用される)を採択し、医薬製品のマーケティング、普及、販売に関するEUの立法を実施している。そのため,現在イギリスの規制制度はEUの規制制度とほぼ一致しているが,イギリスの規制制度はEUから独立しており,TCAは英国とEUを相互に認める製薬立法を規定していないため,これらの制度は将来的に異なる可能性がある。
規制枠組み中断の累積影響は、EUおよび/またはイギリスにおける製品のマーケティング許可および商業化の開発サイクルを大幅に増加させる可能性がある。もっと規制の複雑さがあるかもしれませんが、これは私たちの臨床試験と規制承認の時間を乱すかもしれません。また、国と国際法律法規の変化と法的不確実性は、私たちの臨床と規制戦略に困難をもたらす可能性がある。
また、イギリスの離脱により、他の欧州諸国はEUに滞在し続けるかどうかについて国民投票を求める可能性がある。これらの可能性と他の我々が予見できない可能性を考慮すると、同様の前例がないことを考慮すると、イギリスのEU離脱がどのような財務的、規制、法的影響をもたらすか、そしてこのような脱退が私たちにどのように影響を与えるか、そして私たちの業務が悪影響を受ける可能性の全面的な程度は不明である。
適用される法律や法規を守らないことに関するリスク
もし私たちがどんな製品を商業化することに成功すれば、私たちと顧客と第三者支払者との関係は、適用される反リベート、詐欺、乱用、および他の医療保健法律法規の制約を受けることになり、これは、私たちを刑事制裁、民事処罰、政府医療計画から除外され、契約損害、名声損害、利益、および将来の収入減少に直面させる可能性がある。
医療提供者、医師、そして第三者支払者は、私たちが監督管理の許可を得た任意の製品の推薦と処方において主な役割を果たすだろう。私たちの第三者支払者、医療提供者、医師との手配は、私たちが規制部門の許可を得た任意の製品をどのように研究、マーケティング、販売、流通するかを含む、広範に適用される詐欺と乱用、および他の医療保健法律法規に直面する可能性があります。タイトルを見て“企業−政府規制−反リベートと虚偽クレーム法律その他規制事項.”
これらの法律の範囲も執行も不確定であり、現在の医療改革環境では急速に変化する可能性があり、特に適用例や法規が不足している場合には、連邦と州執行機関は最近、医療会社と医療提供者との相互作用の審査を強化し、医療業界のいくつかの調査、起訴、有罪判決、和解を招いている。私たちが第三者との業務配置や私たちの業務が全体的に適用される医療法律や法規に適合していることを確保するために努力しており、多くのコストがかかります。政府当局は、私たちの業務実践が現在または未来に適合していない可能性があり、詐欺および乱用または他の医療保健法律および法規の適用に関する現行または将来の法律、法規、機関指導または判例法に適合していない可能性があると結論するかもしれない。もし私たちの運営がこれらの法律または他の私たちに適用される可能性のある任意の政府法規に違反していることが発見された場合、私たちは重大な行政、民事と刑事罰、損害、罰金、返還、連邦および州医療計画から除外され、個人監禁、名誉損害、および私たちの運営の削減または再編を受けるかもしれない。これらの法律に準拠していないという疑惑を解決するために、br社の誠実な合意または他の合意の制約を受けていれば、いずれも私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性がある追加の報告義務および監督。このような操作を防御するにはコストが高く、時間がかかる可能性があり、大量の財力と人的資源が必要かもしれない。だから…, たとえ私たちが私たちに提起される可能性のあるどんな訴訟も防ぐことに成功したとしても、私たちの業務は損害を受ける可能性がある。もし私たちがそれと業務を展開する任意の医師または他の医療提供者または実体が適用法に適合していないことが発見された場合、彼らは政府が援助する医療計画から除外することを含む重大な刑事、民事または行政制裁を受ける可能性がある。
44
カタログ表
もし私たちが適用されるアメリカと外国のプライバシーやデータ保護法律法規を遵守できなければ、私たちは私たちの業務、運営、財務業績に悪影響を及ぼす責任を負うかもしれません。
私たちはまた、私たちが収集した臨床試験および米国および海外の私たちの他の業務に関連する患者および医療提供者の情報のような個人データの収集、使用、開示、安全、送信および保存を管理する監督管理指導を含む、外国の法律および法規の制約または影響を受ける可能性がある。例えば、EUの一般的なデータ保護条例(GDPR)は、すべての個人データの処理に厳しい要求を導入し、いくつかのカテゴリの敏感な情報に、健康に関する情報を含むより厳しい要求を加えている。GDPRは、私たちがどのように彼らに関する情報を収集、使用、開示、保持、および利用するかを制御するために、ファイル要件を課し、個人に特定の権利を付与することを含むコンプライアンス負担を増加させる。GDPRは違反報告要件、より強力な規制執行、2000万ユーロ以上の世界年収4%までの罰金を規定している。GDPRは、様々な要求をどのように遵守するかを決定する上で一定の柔軟性を提供しているが、持続的なコンプライアンスを確保するために、多くの努力および費用を投入し続けている。さらに、GDPRでの要求は、定期的に変更されるか、またはEU諸国の法律によって修正される可能性があり、コンプライアンスコストが現在の要求よりも大幅に高い場合、私たちの業務運営に影響を与える可能性があります。
また、GDPRはイギリスの離脱後にイギリスに適用されなくなったにもかかわらず、イギリスの2018年の“EU(離脱)法案”はイギリスのGDPRをイギリスの法律に組み入れた。英国GDPRと2018年の英国データ保護法は,EUのデータ保護制度から独立した英国のデータ保護制度を規定しているが,EUのデータ保護制度と一致している。イギリスのGDPRを守らないことは、1750万GBまたは世界的な収入の4%の罰金につながる可能性がある。英国はEU GDPR下の第3の国とされているにもかかわらず、欧州委員会(EC)は現在、英国がEU GDPRで十分な保護を提供していることを認める決定を発表しているため、EUからの個人データの英国への移行は制限されていない。EU GDPRと同様に,イギリスGDPRは個人データをイギリス以外の国に移しており,これらの国はイギリスが十分な保護を提供しているとはみなされていない。イギリス政府は、イギリスからヨーロッパ経済圏への個人データ転送が依然として自由な流れを維持していることを確認した。
個人データをヨーロッパ経済区やイギリス以外の地域に移行できるようにするためには,ヨーロッパとイギリスのデータ保護法に従って十分な保障措置を実施しなければならない。2021年6月4日、EUは、EUデータ保護命令に従って以前に採用されていた標準契約条項の代わりに、EU/欧州経済領域(または他の方法でGDPRによって制約されている)のコントローラまたはプロセッサからEU/欧州経済領域(GDPR制約を受けない)以外のコントローラまたはプロセッサにデータを送信するための新しい形態のbr標準契約条項を発表した。イギリスはEUの新標準契約条項の制約を受けないが、イギリスに対する移転メカニズム草案バージョンが発表されており、決定されると、イギリスからの移転が許可される。EUやイギリスのGDPRで制限されたデータ転送を行う際には,これらの新たな保障措置の実施が要求され,多大な努力とコストが必要となる。
米国では、多くのプライバシー法が私たちの活動に適用される可能性があり、州と連邦の2レベルの一連の法執行機関は、一般消費者保護法に基づいて会社のプライバシーやデータセキュリティ問題を審査することができる。州と連邦の二級もまた新しい法律が考慮されたり施行されている。例えば、“カリフォルニアプライバシー権·実行法”(2023年1月1日施行)によって改正された“2018年カリフォルニア消費者プライバシー法”(2020年1月1日施行)は、カリフォルニア州住民情報を処理する会社(“消費者プライバシー法”の定義に基づいて)にそのデータ収集、使用、開示方法を具体的に開示し、消費者に敏感な個人情報の使用を制限することを含む個人データプライバシー権を消費者に提供する。保証業務に新たなbr運営要求を適用し、データ保持制限を適用し、データ漏洩に個人訴訟権利を提供し、法定損害賠償枠組みを作成し、CCPAの実施と実行の権限を付与された新しい州機関カリフォルニアプライバシー保護機関を作成する。CCPAの臨床試験データに対する免除は限られているにもかかわらず、CCPAと他の類似した法律は私たちの将来の業務活動
に影響を与える可能性があり、私たちの収入増加、私たちがどれだけの消費者データを処理し、このような法律をどのように解読するかに依存する。また、4つの州がプライバシー法を公布しており、これは私たちの潜在的な責任を増加させ、私たちの将来の業務に悪影響を及ぼす可能性がある。特に,バージニア州消費者データ保護法(VCDPA)が1月1日に施行された, 2023年;コロラド州プライバシー法案(CPA)とコネチカット州データプライバシー法案(CTDPA)が2023年7月1日に発効し、ユタ州消費者プライバシー法案(UCPA)が2023年12月31日に発効する。これらの法規はCCPAと類似した概念を多く含むが、それらは範囲、応用、および法執行においてもいくつかの重要な違いが存在し、これらの違いは、規制された企業が個人の敏感なデータを収集し、処理し、データ保護評価を行い、付属会社に個人データを送信し、消費者の権利要求に応答する方法に影響を与える。
他の州も似たような立法を考慮しており、連邦レベルでも広範な立法措置が導入されている。このような提案立法が通過すれば、複雑性、要求変化、制限、および潜在的な法的リスクを増加させる可能性があり、コンプライアンス計画、影響戦略、および以前の有用なデータの利用可能性についてより多くのリソースを投入する必要があり、コンプライアンスコストの増加および/またはビジネス実践および政策の変化をもたらす可能性がある。国家別州包括的プライバシー法の存在は、私たちのコンプライアンス義務をより複雑で高価にし、私たちが法執行行動を受けたり、他の方法でコンプライアンスによって責任を負う可能性を増加させる可能性がある。
さらに、“健康情報技術促進経済および臨床健康法案”によって改正されたHIPAAによって公布された条例およびその実施条例、または総称されたHIPAAに基づいて、カバーエンティティと呼ばれる特定の医療保健提供者、健康計画および医療情報交換所およびそれらの商業パートナーにプライバシー、安全、および違反通知義務が適用され、これらのサービスは、そのようなカバーエンティティまたはそのカバー下請け業者のための個別に識別可能な健康情報を作成、受信、維持、または送信することに関する。HIPAAはプライバシーと安全基準を制定し、個人が健康情報と保護された健康情報(PHI)の使用と
漏れを識別できることを制限し、そしてPHIのプライバシーを保護し、電子PHIの機密性、完全性と可用性を確保するために行政、物理と技術保障措置を実施することを要求する。大多数の医療保健提供者は、私たちがそれから患者の健康情報を取得する研究機関を含み、HIPAAが公布したプライバシーと安全法規の制約を受け、“経済と臨床衛生情報技術法案”によって改正された。我々は現在HIPAAでの保証実体や業務パートナーであるとは考えていないため,HIPAAの要求や
の罰を直接受けることはない.しかし、誰でもHIPAAの刑事条項に基づいて、直接、または協力および教唆または共謀の原則に従って起訴されることができる。だから事実や状況によります, 私たちが知っている限りでは、HIPAAがカバーするヘルスケア提供者または研究機関から個人識別可能な健康情報を受信し、そのヘルスケア提供者または研究機関が、個人識別可能な健康情報の開示に関するHIPAAの要求を満たしていない場合、重大な刑事罰に直面する可能性がある。
45
カタログ表
プライバシーとデータ保護の世界的な立法と規制構造は引き続き発展しており、予見可能な未来には、実施基準と法執行実践はまだ確定していない可能性がある。このような変化は私たちの業務に不確実性をもたらし、私たちが責任を負うか、あるいは私たちに追加的なコストをもたらすかもしれない。このような法律、法規、そして基準を遵守するコストは高く、未来には増加し続けるかもしれない。
プライバシー法の解釈と適用は私たちの接近と一致しないかもしれない。もし私たちが連邦、州、または外国の法律または自律基準を遵守できなかったか、または遵守できなかった場合、否定的な宣伝、管理時間と精力の移転、および政府の実体または他の人が私たちに訴訟を提起する可能性がある。多くの司法管轄区域で、法執行行動と規定を守らない結果が上昇している。私たちが他の国や司法管轄区域に拡張し続けるにつれて、私たちは追加の法律と法規の制約を受けるかもしれません。これらの法律と法規は私たちが業務を展開する方法に影響を与えるかもしれません。
法執行活動と潜在的な契約責任に関連するリスクに加えて、私たちの連邦と州レベルで変化する法律と法規を遵守するための持続的な努力は費用がかかる可能性があり、私たちの政策、手続き、システムを絶えず修正する必要がある。さらに、私たちの第三者協力者、サービスプロバイダ、請負業者、またはコンサルタントが、データプライバシーまたはセキュリティに関連する適用される法律、法規、または契約義務を遵守できない場合、政府エンティティまたは他の人が私たちに訴訟を提起する可能性があります。
個人情報および/または他の機密情報の収集、処理、使用および開示に関するプライバシーポリシーおよび他の文書を発行することも可能である。私たちは適用された法規、私たちが発表した政策、他の文書を遵守しようと努力しているにもかかわらず、私たちは遵守できないかもしれないし、守られていないとみなされるかもしれない。私たちが努力したにもかかわらず、私たちの従業員やサプライヤーが私たちが発表した政策や文書に従わなかったら、コンプライアンスを達成することができなかったかもしれません。もしこれらの失敗が詐欺的、不公平、あるいは私たちの実際のやり方の歪曲であることが発見されれば、私たちは潜在的な国際、地方、州、そして連邦行動に直面するかもしれない。私たちがプライバシー権を侵害したり、データ保護法や適用されたプライバシー通知を遵守できなかったりすると、私たちが責任を負わなくても、弁護の費用が高く時間がかかる可能性があり、brの負の宣伝を招き、私たちの業務を損なう可能性があります。これらの法律や個人情報の全面的な保護に関する消費者集団訴訟の脅威にも直面している。私たちがこれらの法律に違反していると判断されなくても、政府のこれらの問題の調査には通常、大量の資源がかかり、否定的な宣伝が必要であり、これは私たちの名声と私たちの業務、財務状況、運営結果、または将来性を損なう可能性がある。
私たちは環境、健康、安全法律法規に制約されており、私たちは環境コンプライアンスや救済活動に関する責任と巨額の費用を負担するかもしれない。
私たちの業務は、私たちの開発、テスト、製造活動を含めて、多くの環境、健康と安全法律法規の制約を受けています。その他の事項以外に、これらの法律と法規はまた、危険物質と生物材料の制御使用、処理、放出と処置及び登録、例えば化学溶媒、人体細胞、発ガン化合物、変異原化合物、生殖、実験室プログラム及び血液伝播病原体に接触する毒作用を有する化合物を管理する。もし私たちがこのような法律法規を守らなければ、私たちは罰金や他の制裁を受けるかもしれない。
似たような活動をしている他社と同様に,危険や生体材料の放出や接触に関する責任を含む,我々の活動に固有の環境責任リスクに直面している。環境、健康、そして安全の法律法規はもっと厳しくなっている。私たちは将来の環境コンプライアンスや救済活動に多くの費用を発生させる必要があるかもしれませんが、この場合、私たちの第三者メーカーの生産努力や私たちの開発努力は中断または遅延する可能性があります。
私たちの候補製品の製造、商業化、開発に関するリスク
私たちの候補製品を作ることに関連するリスク
遺伝子治療用製品は斬新で複雑で製造が困難である。私たちは生産問題に直面し、私たちの開発や商業化計画の遅延を招き、私たちの製品の供給を制限したり、他の方法で私たちの業務を損害したりする可能性があります。
私たちは現在第三者と開発、製造、テスト協定を締結して、私たちの候補製品を生産しています。いくつかの要素は生産中断を招く可能性があり、設備故障、施設汚染、原材料不足或いは汚染、自然災害、流行病などの公共衛生危機、公共事業サービスの中断、人為的エラー或いはサプライヤーの運営中断を含む。
46
カタログ表
私たちの候補製品は小分子薬物よりも複雑な加工工程が必要だ。私たちの契約メーカーが私たちの製品を生産するための施設
候補製品はFDAが承認前検査に基づいて検査しなければなりません。これらの検査はFDAにマーケティング申請を提出した後に行われます。私たちはコントロールせず、私たちの契約メーカーが私たちの候補製品を生産する時にcGMPに完全に依存するだろう。もし私たちの契約製造業者が私たちの規格やFDAまたは他の機関の厳格な規制要件に適合した材料を生産することに成功しなければ、彼らは規制検査および/またはその製造施設の規制適合性を維持することができないだろう。しかも、私たちの契約製造業者が十分な品質管理、品質保証、合格者の能力を維持することを制御することはできません。FDAや同様の外国規制機関が、これらの施設に欠陥があることを発見した場合、またはこれらの施設を承認して私たちの候補製品を生産しない場合、またはそれが欠陥を発見したり、将来的にそのような承認を撤回したりすれば、代替製造施設を探す必要があるかもしれません。これは、私たちが規制機関の承認を得たり、私たちの候補製品を販売する能力に深刻な影響を与えます。承認されれば、代替製造施設を探す必要があるかもしれません。
私たちの製造プロセスや私たちと契約している施設でのどんな問題も、潜在的なパートナー(大きな製薬会社や学術研究機関を含む)の魅力の低いパートナーになる可能性があり、魅力的な開発計画を得ることを制限することができます。第三者製造プロセスや施設における問題も、臨床試験をタイムリーに完成させたり、わが製品に対する市場の需要を満たす能力を制限したりする可能性があります。さらに、私たちと第三者との製造プロトコルが任意の理由で終了した場合、限られた数の製造業者が適切な代替者とすることができ、製造を代替者に移行するためにかなりの時間を必要とする可能性がある。場合によっては、私たちの製品または候補製品を製造するために必要な技術的スキルは、元の製造業者固有または独自である可能性があり、困難に遭遇する可能性があり、またはそのようなスキルを予備または代替サプライヤーに譲渡することを禁止する契約制限が存在する可能性があり、またはそのようなスキルを全く譲渡できない可能性があります。製造プロセスの変更や新たな製造施設の移転や設置には,臨床や商業製造活動を継続するために接続研究が必要となる可能性がある。臨床用品の比較可能性の証明には成功しない可能性があり,追加の臨床試験が必要かもしれない。さらに、このような転換は、特に新しい工場が必要な法規要件を遵守する必要があるため、またはその工場で生産された任意の製品を販売する前にbr承認が必要である可能性があるため、高価で時間がかかる可能性がある。
私たちはcGMPを生産するために私たちの製造施設を準備している。我々の製造面での経験は限られており,我々が製品を製造できる保証はない.
私たちは以前から第三者に依存して私たちの候補製品のための供給品を製造してきた。我々はすでにニュージャージー州クランベリーに位置する新製造工場の拡張を完了し,最近2ロットのAAV cGMPの生産を完了した。
私たちの一部の従業員は以前他の会社で働いていましたが、バイオ製薬製品の製造経験はありますが、会社としては、私たちの以前の製造経験は非常に限られています。また,我々が製造施設を運営するには政府の承認が必要となり,承認を得るのに時間がかかる可能性があり,このような承認を得る保証はない。医薬品メーカーとして、私たち
はまた、生産過程、品質管理と保証、記録保存に関するcGMP要求に適合していることを証明し、維持することが要求される。さらに、製造業務の確立と維持には、他の資源、特に私たちのいくつかの上級管理職の時間と注意力、および潜在的な巨額の資本支出を再分配する必要があるかもしれません。私たちの製造能力開発のどんな失敗や遅延も私たちの候補製品の開発や商業化に悪影響を及ぼす可能性があります。
私たちの製造施設は重大な政府法規や承認の制約を受けており、私たちが法規を遵守したり、承認を維持しなければ、コストが高く、私たちの業務に悪影響を及ぼす可能性があります。
私たちは規制、法規、そしてガイドラインに規定されたcGMP要件を守らなければならない。私たちは品質管理と品質保証を実現する上で困難に直面する可能性があり、合格者不足の問題に直面する可能性がある。私たちは適用された規制要件に適合することを確実にするために、FDAと他の管轄区域の類似機関の検査を受けた。CGMPまたは他の規制要件を遵守できなかった場合、または私たちの施設または第三者の施設または運営が法規要件を遵守できなかったか、または任意の規制機関の検査を通過できなかったことによる、私たちの候補製品の製造、充填、包装または保存中に発生した遅延、中断、または他の問題は、私たちの臨床試験の医薬製品の供給の著しい遅延、または臨床試験の終了または一時停止を含む、私たちの候補製品を開発および商業化する能力を著しく弱める可能性がある。私たちの候補製品のマーケティング申請の提出または承認を遅延させたり阻止したりします。重大な違反はまた、罰金、禁止、民事処罰、規制機関が私たちの候補製品に上場承認、遅延、一時停止または承認撤回、許可証の取り消し、製品の差し押さえまたはリコール、運営制限、および刑事起訴を授与できなかったことを含む制裁を実施する可能性があり、いずれも私たちの名声を損なう可能性がある。もし私たちがコンプライアンスを維持できなければ、私たちは私たちの候補製品の販売を許可されないかもしれないし、および/または製品のリコール、差し押さえ、禁止、br、または刑事起訴を受ける可能性がある。
私たちの候補製品の商業化に関するリスクは
私たちが私たちの候補製品の開発に成功し、それを商業化できるかどうかは、発生した薬物や関連治療費用の精算状況に大きく依存する。
47
カタログ表
米国や他の国の市場では,患者は通常第三者支払者によって治療に関連する費用の全部または一部を精算する。政府と個人支払者の獲得性と精算範囲は大多数の患者が高価な治療費用を負担できる鍵である。私たちの候補製品の販売は、政府当局や他の第三者支払者(例えば、個人健康保険会社や健康維持組織)が私たちの候補製品の費用をどの程度支払い、支払うかに大きく依存し、保証できません。私たちはまだ私たちの候補製品に政府または第三者支払人の補償を受け始めていません。保険や精算が得られない場合、あるいは限られたレベルに限られていれば、私たちの製品を商業化することに成功できないかもしれません。保険を提供しても、承認された精算金額は、十分な投資リターンを達成するのに十分な定価を確立したり維持したりするのに十分ではないかもしれません。支払者が製品に保険を提供するかどうかを決定するプロセスは、支払者が製品のために支払う支払率を設定するプロセスと分離することができる。支払者は、承認されたリストまたは処方内の特定の製品に保証範囲を制限することができ、特定の適応のすべてのFDAによって承認された製品を含まない可能性がある。支払側は私たちの候補製品を保証しないことを決定し、承認されると、医師の私たちの製品に対する使用を減少させ、私たちの販売、運営結果、財務状況に実質的な悪影響を与える可能性があります。タイトルを見て“企業-政府規制-保険範囲と補償.”
新たに承認された製品の保険カバーと精算に関する不確実性が大きい。アメリカでは、新薬の保証と精算に関する主な決定は通常医療保険と医療補助サービスセンター(CMS)によって行われ、CMSはアメリカ衛生と公衆サービス部(HHS)内の一つの機関であり、CMSは新薬がMedicareの下で保険と精算を受けるかどうか、及びどの程度精算するかを決定する。個人支払者はCMSに大きく従う傾向がある。CMSが我々のような根本的な新製品の精算にどのような決定を下すかを予測することは困難である。これらの新製品には既定のやり方や前例がないからである。支払人が精算を決定する際に考慮する要素は、製品があるかどうかに基づいている
| • |
健康計画の下で保障された福祉
|
| • |
安全で効果的で医学的に必要です
|
| • |
特定の患者に適しています
|
| • |
費用対効果があります
|
| • |
実験的でも調査的でもない。
|
近年,米国では医療制度を変更する多くの提案がなされている。これらの改革提案には、特定の医療費の支払いを制限または禁止したり、薬品の価格を政府のコントロール下に置く措置が含まれている。また、多くの外国、特にEU諸国では、処方薬の定価は政府によってコントロールされている。もし私たちの製品がbr政府の法規によって制限されたり、支払いが禁止されたり、あるいは私たちの製品の価格が政府によってコントロールされていれば、私たちは収入を作ったり、利益を達成したり、私たちの製品を商業化することができないかもしれません。
また、第三者決済者は新薬のカバー範囲と精算レベルを制限することが増えている。彼らはまた、FDAが承認された発売された製品以外の医療適応のための任意の
の承認製品のカバー範囲の提供を拒否する、厳格な事前許可要件を実施することができる。したがって,第三者決済者が患者に新たに承認された薬物を使用するかどうか,患者にどの程度の費用を支払うかについては,大きな不確実性がある。政府の医療保健計画や個人支払者が要求する強制的な割引やリベート、および将来的に現在の薬品から米国より低い価格で販売されている国/地域からの薬品の輸入制限を緩和する法律により、薬品の純価格を下げることができる。ますます多くの第三者支払人は製薬会社に価格に基づいて所定の割引を提供し、医療製品の料金に挑戦することを要求している。私たちは私たちが商業化されたどの候補製品も精算できることを確実にすることはできない。もし精算できるなら、精算のレベルもそうだ。また、多くの製薬業者
は平均販売価格やASPと最適価格などのいくつかの価格報告指標を計算し、政府に報告しなければならない。場合によっては、これらの指標が正確かつタイムリーに提出されていない場合には、処罰が適用される可能性がある。また,これらの薬品価格
は,政府医療計画が要求する強制割引やリベートにより低下することができる。私たちの候補製品に関するサービス(例えば、患者に私たちのbr製品を管理する)のための精算の仕方とレベルを提供することは、私たちの候補製品の商業化に成功するためにも重要である。
また、一部の外国の国では、薬品の提案価格は必ず承認されなければならず、合法的に発売されることができる。各国の薬品定価に対する要求は大きく異なる。例えば、欧州連合は、その国の健康保険制度が補償を提供する医療製品の範囲を制限し、人間が使用する医療製品の価格を制御するために、その加盟国に様々な選択を提供している。精算または定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較する。加盟国は医薬製品の具体的な価格を承認するか、あるいは医薬製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることができる。Brが薬品に対して価格制御や精算制限を実行することを保証できないいかなる国/地域も、私たちの任意の候補製品に優遇された精算と定価手配を提供することを許可する。歴史的に見ると、EUで発売された製品はアメリカの価格構造に従わず、通常価格ははるかに低くなることが多い。場合によっては、ある会員国の補償価格は、他の加盟国の価格設定レベルに影響を与える可能性があり、これは、他の市場での値下げまたは侵食を防止するために、特定の市場で製品を販売しないことをもたらす可能性がある。
48
カタログ表
私たちは激しい競争と迅速な技術変革に直面しており、私たちの競争相手は私たちよりも先進的で効果的な治療法を開発するかもしれませんが、これは私たちの財務状況と候補製品を商業化することに成功した能力に悪影響を及ぼすかもしれません。
我々は深刻な遺伝性とまれな疾患の遺伝子治療に従事しており,競争が激しく,変化が迅速な領域である。競合他社が我々が準備中のものと同じ適応を扱っている遺伝子治療があることは知られていないが,米国や国際的には大手多国籍製薬会社,バイオテクノロジー会社,大学,その他の研究機関を含む競争相手がいる可能性がある。
私たちの潜在的な競争相手は、より多くの研究開発者、製造能力、および経験豊富なマーケティングおよび製造組織のようなより多くの財務、技術、および他の資源を持っている可能性がある。これらの競争相手は、独占的な基礎の上で開発、買収に成功するかもしれないし、私たちが開発する可能性のある任意の候補製品よりも効率的でコストの低い製品、あるいは私たちよりも早く特許保護、監督管理承認、製品商業化、市場浸透を実現することができるかもしれない。また、私たちの競争相手が開発した技術は私たちの潜在的な候補製品を経済的あるいは時代遅れにする可能性があり、私たちは競争相手の製品に対して私たちの候補製品をマーケティングすることに成功できないかもしれません。
さらに、我々の特許権が満期または成功的に挑戦された場合、競争相手製品に関連する特許の有効性および/または範囲に関するより多くの訴訟に直面する可能性がある。競争相手の製品供給は、私たちが開発および商業化する可能性のある任意の製品の需要と価格を制限する可能性があり、それによって、私たちの業務、財務状況、運営結果、および将来性に損害を与える可能性があります。
私たちのすべての候補製品の商業成功は医者、患者、第三者支払人と医学界の他の人の市場に対する受け入れの程度に依存するだろう。
遺伝子治療の倫理、社会、法律、その他の面への懸念は、追加の法規制や私たちの製品の禁止を招く可能性がある。米国FDA、EU EMAと他の国際規制機関の必要な承認を得ても、私たちの候補製品の商業成功はある程度医者、患者と医療保険支払者が遺伝子治療製品、特に私たちの候補製品が医療利益、コスト効果と安全性の受け入れ程度を持っているかどうかに依存する。私たちが商業化したどの製品も医師、患者、医療支払者、医学界の他の人に受け入れられないかもしれない。もし私たちが商業化したどの製品も医師、患者、医療支払者、医療界の他の人の十分な受容度を得ていなければ、著しい製品収入が生じず、利益を上げることができないかもしれない。遺伝子治療製品および候補製品に対する市場の受容度(商業化販売が許可された場合)は、いくつかの要因に依存する
| • |
臨床前研究と臨床試験により証明されたこれらの候補製品の有効性と安全性
|
| • |
代替療法に対する候補製品の潜在的かつ知覚可能な利点;
|
| • |
代替治療に対する私たちの治療のコストは
|
| • |
FDAまたはEMAによって承認された候補製品の臨床適応;
|
| • |
患者の遺伝子治療に対する認識と意思;
|
| • |
医者は新しい治療法を提案しました
|
| • |
医師は私たちの製品候補者の管理に関する専門訓練を受けたいと思っています
|
| • |
ターゲット患者集団が新しい治療法を試みる意欲は
|
| • |
副作用の流行率や重症度は
|
| • |
製品承認ラベルに含まれる任意の制限または警告を含むFDA、EMAまたは他の規制機関の製品ラベルまたは製品挿入要件;
|
| • |
相手が便利で管理しやすい
|
| • |
有力なマーケティングと流通支援
|
| • |
製品が市場に投入されるタイミングを競う
|
| • |
私たちの製品や競合製品や治療法の宣伝;
|
| • |
十分な第三者支払者は範囲と精算をカバーする。
|
潜在的な製品が臨床前研究と臨床試験において良好な治療効果と安全性を示しても、市場のこの製品に対する受容度はその製品が承認され、市場に投入された後に完全に確定することができ、承認後に不利な長期追跡データを獲得すれば、その製品は時間の経過とともに変化する可能性がある。もし私たちのすべての候補製品が市場の承認を得られなければ、私たちの業務、財務状況、運営結果、将来性に深刻な損害を与える可能性があります。
わが国のパイプライン開発と研究開発活動に関するリスク
私たちは私たちの追加候補製品開発チャンネルの努力を拡大することに成功しないかもしれない。
我々のビジネスモデルは,まれな遺伝疾患における我々の専門知識を応用することを中心に,重点のある選択基準を構築することで候補遺伝子治療製品の組合せを開発·推進し,br開発により商業化に入っている。我々のこれまでの努力で生じた候補製品パイプラインを除いて,新たな候補製品を認識·開発し続けることができない可能性がある。私たちが私たちのルートを拡大することに成功しても、私たちが確定した任意の潜在的な候補製品は臨床開発に適していないかもしれない。もし私たちが候補製品の識別、開発、商業化に成功できなければ、私たちは将来的にbr製品の収入を得ることができなくなり、これは私たちの財務状況と運営結果に大きな損害を与える可能性がある。
49
カタログ表
著者らは主に研究開発活動、臨床テストと商業化の成功かどうかはまだ確定していない。
私たちの主な関心は私たちの研究開発活動と私たちの候補製品の臨床テストと商業化であり、私たちは不確定だがかなり長い期間、主にこれらの活動に従事し続けることが予想される。2022年12月31日までの1年間、研究開発は私たちの最も重要な運営支出だ。研究開発活動は,臨床研究の進行を含め,その性質に基づいて,ある目標を達成するのに要する時間と関連するコストの最終声明を排除した。そのため,実際の研究開発コストは予算金額を大きく超える可能性があり,想定される時間枠は大幅に延長される可能性がある。コスト超過、意外な監督管理遅延或いは要求、意外な不良副作用或いは治療効果不足は著者らの研究開発を阻害或いは大幅に緩和し、著者らの業務は最終的に影響を受ける可能性がある。
第三者に関わるリスク
著者らは第三者に依存して臨床前研究と臨床試験のいくつかの方面を行い、そして私たちのために他の任務を実行する。もしこれらの第三者
が契約の職責を成功的に履行できず、期待期間内に監督管理要求を達成または遵守できない場合、私たちは監督部門の私たちの候補製品と業務の承認を得ることができないか、あるいは商業化できない可能性があり、brの財務状況と運営結果は深刻な損害を受ける可能性がある。
私たちはCRO、医療機関、契約実験室を含む第三者に依存し、計画してきました。私たちが行っている臨床前と臨床プロジェクトのいくつかの側面のために使用されています。それにもかかわらず、私たちはすべての臨床試験と臨床前研究が適用された方案、法律、法規、科学的基準に従って行われることを保証する責任があります。私たちのこれらの第三者への依存は私たちの規制責任を免除しません。私たちと私たちのサプライヤーは、GMP、良好な臨床実践(GCP)、および良好な実験室規範(GLP)の現在の要求を遵守するように要求されており、これらの要件は、FDA、EMAまたは同様の外国当局が臨床開発中に私たちの候補薬物に対して実行する法律および法規のセットである。
監督管理機関は定期的に臨床前研究と臨床試験スポンサー、主要な研究者、臨床前研究と臨床試験場及びその他の請負業者を検査することによって、これらの規定を実行する。もし私たちまたは私たちの任意のサプライヤーが適用された法規を遵守できなかった場合、私たちの臨床前研究および臨床試験で生成されたデータは信頼できないと考えられる可能性があり、FDA、EMAまたは同様の外国当局は、私たちのマーケティング申請を承認する前に追加の臨床前研究および臨床試験を行うことを要求するかもしれない。特定の規制機関が検査を行った後、この監督機関は私たちのいかなる臨床試験がGCP規定に適合しているかどうかを確認することを保証することはできません。また,われわれの臨床試験はGMP規定に適合した製品を用いて行わなければならない。もし私たちがこれらの規定を守らなければ、私たちは臨床試験を繰り返す必要があるかもしれません。これは開発と規制承認過程を延期します。
もし私たちがこれらの第三者、医療機関、臨床研究者、あるいは契約実験室との任意の関係が終了すれば、私たちは商業的に合理的な条項でCROの代替と合意することができない、あるいは全く手配を達成できないかもしれない。また,われわれのCROはわれわれの従業員ではなく,このようなCROとの合意に基づいて我々に提供されている救済措置を除いて,われわれが行っている臨床前および臨床計画に十分な時間と資源を投入しているかどうかを制御することはできない。
もし私たちのCROがその契約の義務または義務を成功裏に履行できなかった場合、または予想された期限内に完了した場合、もし彼らが交換する必要がある場合、または彼らが得たデータの品質または正確性が私たちの方案、法規の要求、または他の理由を遵守できなかった場合、私たちの臨床試験は延長、延期、または終了される可能性があり、私たちは規制機関の私たちの候補製品の承認を得ることができないか、または商業化に成功する可能性がある。CROはまた予想よりも高いコストを発生させる可能性がある。したがって、私たちの業務、財務状況、運営結果、私たちの候補製品の商業見通しは重大な悪影響を受ける可能性があり、私たちのコストは増加する可能性があり、私たちの収益能力は遅れる可能性があります。
追加のCRO、医療機関、臨床研究者、または契約実験室は追加コストを交換または増加させ、管理時間および重点を必要とする。また,新しいCROが以前のCROの置換を開始すると,自然な
過渡期がある.したがって,遅延が生じる可能性があり,期待される臨床開発スケジュールを満たす能力に実質的な影響を与える可能性がある。私たちはCROとの関係
を慎重に処理しているにもかかわらず、私たちは未来に類似した挑戦や遅延に遭遇しない保証はなく、これらの遅延や挑戦が私たちの業務、財務状況、または運営結果に実質的な悪影響を与えないことを保証することはできない。
我々は第三者に依存して我々の薬品製品の製造,研究および臨床前と臨床テストの一部あるいは全部を行うことが予想されるが,これらの第三者
の表現は満足できない可能性がある。
著者らは独立に遺伝子治療生産、製品製造、研究及び臨床前と臨床テストのすべての方面を行うことを望まない。その中のいくつかのプロジェクトについて、私たちは現在、第三者に依存し続けると予想している。場合によっては、これらの第三者は、いくつかのビジネス環境で使用される同じ品質管理プロトコルに適用されない可能性がある学術、研究、または同様の機関である。
50
カタログ表
これらの第三者の研究開発活動への依存は,これらの活動の制御を減少させるが,必要なすべての法規や研究
合意を遵守することを確保する責任は免除されない.もしこれらの第三者が法規の要求または私たちが規定した研究計画と合意に従ってその契約の職責を成功裏に履行し、期待された期限内に私たちの研究を完成または行うことができない場合、私たちは将来の製品提出と私たちの候補製品の承認を支援するために必要な臨床前および臨床研究の完成を完成または遅延させることができないだろう。
一般的に、これらの第三者は通知を受けた後に私たちとの雇用関係を勝手に終わらせることができる。もし私たちが代替計画を達成する必要があれば、私たちの製品開発活動を延期するかもしれない。
私たちはすべてのLV候補製品を含む第三者メーカーに依存して、私たちのいくつかの候補製品のための供給品を製造したいです。
第三者製造商会に依存してリスクをもたらし、もし私たちが自分ですべての候補製品を製造すれば、私たちはこれらのリスクの影響を受けません
| • |
ビジネス上合理的な条件で第三者と製造協定を交渉することはできない
|
| • |
製造活動のいくつかの態様では、第三者製造業者が使用されるため、制御が減少する
|
| • |
このような活動は私たちの研究計画や合意に従ったリスクがありません
|
| • |
第3者との製造契約を終了または更新しない方法で、私たちに代価または損害を与える方法で、
|
| • |
私たちの業務または運営とは無関係な条件による第三者製造業者またはサプライヤーの運営中断は、製造業者またはサプライヤーの倒産を含む。
|
これらの事件のいずれも、臨床研究の遅延や規制部門の承認を得られなかったり、将来の製品を商業化することに成功した能力に影響を与える可能性がある。いくつかの事件は、禁止、リコール、差し押さえ、または生産の完全な一時停止、または部分的な一時停止を含むFDAの行動の基礎となる可能性がある。
私たちは私たちのいくつかの候補製品を開発し続けるか、私たちの候補製品を商業化に成功させるための戦略的パートナーを見つけることができないかもしれない。
候補製品を開発するのに必要な資金コストが相対的に高い、br製造制限、または他の理由から、私たちのいくつかの候補製品を開発および/または商業化するために、戦略的パートナーシップを確立することを求めることができる。私たちの研究開発パイプラインが不足している可能性があり、私たちの候補製品が協力努力の開発段階に早すぎると考えられるかもしれない、または第三者が私たちの候補製品がbrの効果や市場機会に必要な潜在力を示していないと思うかもしれないから、私たちの候補製品のためのこのような戦略的パートナーシップまたは他の代替計画を構築することに成功できないかもしれない。また、既存の合意によれば、潜在的な協力者と将来の合意を締結することができないように制限される可能性がある。
もし私たちが適時に、受け入れ可能な条項で、または適切な許可者やパートナーと合意できない場合、私たちは候補製品の開発を削減し、私たちのbr開発計画を減少または延期し、私たちの潜在的な商業化を延期し、いかなる販売またはマーケティング活動の範囲を縮小し、または私たちの支出を増加させ、自費で開発または商業化活動を行わなければならないかもしれない。もし私たちが独立して開発または商業化活動を支援することを選択した場合、私たちは追加の専門知識と追加の資本を得る必要があるかもしれないが、これらは受け入れ可能な条項や根本的に得られないかもしれない。もし私たちが協力計画を達成できず、そして
必要な開発と商業化活動を展開するのに十分な資金や専門知識がなければ、私たちは私たちの候補製品をさらに開発することができないかもしれません。私たちの業務、財務状況、運営結果、およびbr}の見通しは実質的に損なわれる可能性があります。
資金不足や世界的な健康問題によるFDAや他の政府機関の中断は、重要な指導部や他の人員の採用、保留、配置の能力を阻害する可能性があり、あるいは新製品や修正された製品のタイムリーまたは開発、承認、商業化を他の方法で阻止することは、私たちの業務にマイナスの影響を与える可能性がある。
FDAが新製品を審査および承認する能力は、政府予算および資金レベル、法律、法規および政策の変化、FDAのキーパーソンの雇用および保留、ユーザ費用の支払いを受ける能力、およびFDAが通常の機能を履行する能力に影響を与える可能性のある他の事件を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また,研究開発活動を援助する他の政府機関への政府の援助は政治過程の影響を受けており,この過程は本質的に不安定で予測不可能である。FDAおよび他の機関の中断はまた、生物製品または承認された生物製品の修正が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼす。例えば、過去数年間、2018年12月22日から35日間
を含めて、米国政府は何度も閉店しており、FDAのようないくつかの規制機関は、FDAのキー従業員を休暇させ、キー活動を停止しなければならない。
51
カタログ表
また、2021年5月26日まで、アメリカ食品薬品監督管理局は、それは引き続きそのユーザー費用業績目標に基づいて、新冠肺炎大流行期間の医療製品申請を適時に審査することを確保すると指摘した;しかし、アメリカ食品薬品監督管理局は現在の速度を維持できない可能性があり、承認期限は延長する可能性があり、新冠肺炎の大流行の新しい変種或いは未来の任意の大流行による承認前検査或いは臨床場所検査を必要とする状況を含む。FDAが承認を得るために検査を行う必要があると判断し、旅行制限のために審査期間内に検査を完了することができず、FDAが遠隔相互作用評価が不十分であると判断した場合、機関は、検査が完了するまで、完全な返信状を発行するか、または申請に対する行動を延期することができる。新冠肺炎突発公共衛生事件の期間中、アメリカ食品薬品監督管理局はその申請に対する規定検査を完成できないため、多くの会社は完全な返信状を受け取ることを発表した。米国以外の規制機関は、新冠肺炎の大流行や将来の任意の大流行に対応するために、類似した制限や他の政策措置をとる可能性があり、監督活動に遅延が生じる可能性がある。FDAが現在の性能レベルを維持し続けることができない場合、私たちの候補製品および私たちが求める可能性のある任意の承認を遅延させ、挫折する可能性があり、これは私たちの業務に悪影響を及ぼすかもしれません。
私たちの知的財産権に関するリスクは
候補製品の開発と商業化された知的財産権は、他の側が私たちに付与されたライセンスの条項と条件によって制限されています。
私たちは、当社の製造プロセスおよび私たちの遺伝子治療製品候補製品に関連する技術を含む、当社の技術および製品の開発に非常に重要または必要な第三者からの特定の特許権およびノウハウの許可に深刻に依存しています。これらの許可および他の許可は、すべての関連する使用分野および将来、私たちのプラットフォームを許可したり、当社の技術および製品を開発または商業化したいすべての地域でそのような知的財産権および技術を使用する独占的な権利を提供しないかもしれません。したがって、私たちは競争相手が私たちのライセンスに含まれていない地域で競合製品を開発し、商業化することを阻止できないかもしれない。
我々の許可または開発計画に必要とされる可能性のある他の第三者技術の許可は、将来的には得られない可能性があり、または商業的に合理的な条項または全部で取得できない可能性があり、これは、私たちの業務および財務状況に重大な損害を与える可能性がある。
場合によっては、私たちは、第三者から許可を得る技術を含む、特許出願の準備、提出および起訴を制御する権利がないか、または特許を維持または強制的に実行する権利がないかもしれない。私たちの許可者がそのような特許を保持できなかった場合、またはこれらの特許または特許出願の権利を失う場合、私たちが許可した権利は減少またはキャンセルされる可能性があり、私たちがそのような許可権利主体に属する任意の製品を開発および商業化する権利は影響を受ける可能性がある。上記に加えて、第三者から付与された特許権に関するリスクは、将来所有可能な特許権にも適用される。
また、私たちのいくつかの特許権と技術の研究はアメリカ政府によって資金援助されている。したがって、政府はこれらの特許権および技術に対していくつかの権利を持っているか、または権利を持っている可能性がある。政府資金を利用して新しい技術を開発する場合、政府は通常、政府が非商業目的のために発明を使用することを許可する非独占的許可を含む、生成された任意の特許のいくつかの権利を得る。これらの権利は、政府が第三者に私たちの機密情報を開示し、第三者が私たちが許可した技術を使用または許可する先行権を行使することを可能にするかもしれない。政府が行動する必要があると判断した場合、政府が援助する技術の実用化を実現できないので、健康や安全需要を緩和し、連邦法規の要求を満たしたり、米国工業を優先したりするために行動しなければならないので、政府はその進行権を行使することができる。さらに、このような発明に対する私たちの権利は、米国でそのような発明を含む製品を製造するいくつかの要件によって制約される可能性がある。br}政府のそのような権利のいかなる行使も、私たちの競争地位、業務、財務状態、運営結果、および見通しを損なう可能性がある。
もし私たちが製品や関連技術の特許保護を獲得して維持できない場合、あるいは取得した特許保護範囲が十分でなければ、私たちの競争相手は私たちと似ているまたは同じ製品や技術を開発して商業化する可能性があり、私たちが製品を商業化することに成功する能力は損なわれる可能性がある。
私たちの成功は、私たちがアメリカや他の国で私たちの候補製品や製造技術の特許保護を獲得し、維持する能力に大きく依存している。私たちの許可者はすでにアメリカと海外に私たちの多くの新技術や候補製品に関連する特許出願を提出することによって、私たちの特許地位を保護しようとしているかもしれません。これらの技術と製品は私たちの業務に非常に重要です。
特許訴訟プロセスは高価で、時間がかかり、複雑であり、私たちは合理的なbrコストで、または適時に提出、起訴、維持、強制執行、またはすべての必要または理想的な特許出願を許可することができないかもしれない。さらに、遺伝子治療分野のいくつかの特許は、本来、私たちのいくつかの候補製品に特許保護を提供する可能性があるが、私たちの製品の商業化発表前に満了する可能性があり、このような特許満了リスクは、FDAの10年間の生物製品規制排他性を求めて獲得することによって部分的に解決することができ、FDAは今後特許満了が存在しない可能性がある場合に保護を提供することになる。場合によっては、遺伝子治療分野のある学術研究者の仕事が公共の分野に入っており、このような仕事に関連するいくつかの発明のために特許保護を受ける能力は排除されていると考えられる。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果の特許可能性を特定できない可能性もある.
52
カタログ表
私たちはいくつかのエンティティと知的財産権許可協定を締結しており、各エンティティは私たちの業務に重要であり、将来的にもより多くのライセンス契約を締結することが予想される。私たちの特許の組み合わせには、これらのライセンス協定に基づいて許可された複数の特許と特許出願が含まれており、これらのプロトコルは、将来のライセンス協定が、私たちに様々な勤勉、開発、商業化スケジュール、マイルストーン義務、支払い、およびその他の義務を課すことが予想される。私たちまたは私たちのライセンシーがこれらの合意の下で私たちの義務を履行できなかった場合、または私たちが破産状態にある場合、許可者は、許可がカバーする製品を販売する能力を含む許可提供のいくつかの権利を失う可能性があります。
バイオテクノロジーや製薬会社の特許地位は通常高度に不確定であり,複雑な法律や事実問題に関連しており,近年多くの訴訟のテーマとなっている。したがって,我々の特許権の発行,範囲,有効性,実行可能性,商業的価値は高い不確実性を持っている.未解決および将来の特許出願は、我々の候補技術または製品を保護する特許を発行すること、または他社が競合技術および候補製品を商業化することを効果的に阻止する特許をもたらすことができないかもしれない。米国や他の国/地域特許法または特許法解釈の変化は,我々の特許権の価値を低下させたり,我々の特許保護範囲を縮小したりする可能性がある。
我々の知的財産権は現在の開発計画を継続できると信じているが,いくつかの会社や学術機関は遺伝子治療の代替方法を求めており,これらの方法や方法をめぐって知的財産権を構築している。例えば,バステッド研究所はレンチウイルス遺伝子療法のベクター成分に関連する特許ファミリーを制御している。この特許は核の現地化を改善する要素に関するものだ。これらの特許は2019年から満期になり、2023年に完全に満期になりますが、私たちの製品が2023年第4四半期までに発売される場合、ライセンスを取得する必要があるかもしれません。また,我々は我々の技術や候補製品に関連する可能性のあるすべての
第三者知的財産権を知らないかもしれない.科学文献で発見された発表は、実際の発見よりも遅れがちであり、米国および他の司法管轄区域の特許出願は、通常、提出されてから18ヶ月後に発行されるか、または場合によっては全く発行されない。したがって、私たちは、任意の所有または任意の許可された特許または係属中の特許出願において最初に特許請求を提出した発明であるか、またはそのような発明のために特許保護を出願した最初の会社であることを決定することはできない。
私たちが許可したまたは将来所有可能な特許出願が実際に特許の形態で発表されても、それらの発表形態は、私たちに意味のある保護、競争相手または他の第三者が私たちと競争することを阻止し、または他の方法で私たちにいかなる競争優位性を提供してくれないだろう。我々の競争相手または他の第三者は、1984年の“薬品価格競争および特許期限回復法”(Hatch-Waxman修正案)下の安全港を利用して研究および臨床試験を行うことができ、類似または代替技術または製品を非侵害的に開発することによって、我々の特許権を回避することができる。
特許の発行は、その発明性、範囲、有効性または実行可能性に対して決定的ではなく、我々の特許権は、米国および海外の裁判所または特許庁で挑戦される可能性がある。このような
挑戦は、排他的な喪失または特許主張の縮小、無効、または実行不能をもたらす可能性があり、これは、他人が類似または同じ技術および製品を使用することを阻止するか、またはそれを商業化する能力を制限するか、またはIS技術および候補製品の特許保護期間を制限する可能性がある。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に期限切れになる可能性がある。したがって、私たちの知的財産権は他の人たちが私たちと似ているか同じ製品を商業化することを排除するのに十分な権利を提供していないかもしれない。
もし私たちが許可協定に違反すれば、私たちの候補製品の商業化努力に実質的な悪影響を及ぼすかもしれない。
私たちが私たちの候補製品や第三者技術の使用、開発、商業化権利に関する任意の知的財産権ライセンス協定に違反すれば、私たちの業務に非常に重要な許可権を失う可能性があります。知的財産権許可は複雑な法律、商業、そして科学的な問題に関する私たちの業務に重要だ。ライセンス契約によると、私たちと私たちのライセンシーとの間に知的財産権紛争が発生する可能性があります
| • |
ライセンス契約に基づいて付与される権利範囲;
|
| • |
私たちの技術とプロセスが、ライセンス契約に拘束されていないライセンス側の知的財産権をどの程度侵害しているかどうか
|
| • |
私たちは協力開発関係の下で特許と他の知的財産権を第三者に再許可する権利;
|
| • |
我々のIS候補製品の開発や商業化に関するライセンス技術の使用に関する職務義務と、どのような活動がこれらの職務義務を満たしているか
|
| • |
私たちの許可者、私たちと私たちのパートナーが知的財産権を共同で創造または使用することによって生成された発明およびノウハウの所有権;
|
| • |
発明者およびその権利を我々に譲渡できるライセンシーにどの程度異議を唱えることができるか。
|
もし私たちの許可範囲内の知的財産権紛争が許容可能な条項で現在の許可手配を維持する能力を妨害したり、損害を与えたりすれば、影響を受けた候補製品の開発に成功し、商業化することができないかもしれない。また、知的財産権の所有権を許可する上で議論が生じた場合、特許権を追求または実行する能力が損なわれる可能性がある。もし私たちまたは私たちの許可者がこの知的財産権を十分に保護できなければ、私たちの製品商業化能力は影響を受ける可能性がある。
53
カタログ表
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの業務と競争地位は損なわれるかもしれない。
特許提供の保護に加えて、私たちは、非特許の商業秘密保護、非特許の技術的ノウハウ、および持続的な技術革新に依存して、私たちの競争的地位を発展させ、維持しています。私たちは、請負業者、協力者、従業員、およびコンサルタントと秘密保護協定を締結することによって、私たちのノウハウやプロセスを保護することを求めています。しかしながら、一般に秘密協定および他の契約制限が存在するにもかかわらず、これらの合意の当事者が許可されていない場合に、私たちの技術的ノウハウまたは他の商業秘密を開示または使用することを阻止することはできないかもしれない。許可されていない使用と漏れを監視することは困難であり、私たちは私たちのノウハウを保護するために私たちが取った手順が有効かどうか分からない。これらの合意当事者である任意の請負業者、協力者、従業員、およびコンサルタントがこれらの合意のいずれかの条項に違反または違反する場合、私たちは、任意のこのような違反または違反に対して十分な救済措置をとることができない可能性がある。したがって、私たちは私たちの商業機密を失うかもしれない。特許訴訟のように、高価で時間がかかり、結果的に予測できないように、第三者に我々の商業秘密を不正に取得して使用することを強制した。しかも、アメリカ以外の裁判所は商業秘密をあまり望まないか、保護したくないことがある。
そうでなければ、私たちの商業秘密は私たちの競争相手に知られたり独立したりするかもしれない。競争相手は、私たちの開発作業から得られた競争優位性の一部または全部をコピーし、故意に私たちの知的財産権を侵害し、私たちが保護された技術をめぐって設計したり、彼ら自身の私たちの知的財産権に属さない競争力のある技術を開発しようとするかもしれない。もし私たちの任意の商業秘密が競争相手によって合法的に取得または独立して開発された場合、私たちは彼らまたは彼らが情報を伝達する人がその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちのビジネス秘密が十分に保護されていない場合、または競争相手に対する優位性を提供するのに十分でなければ、私たちの競争地位は悪影響を受ける可能性があり、私たちの業務も影響を受ける可能性がある。また,我々のビジネス秘密を保護するための措置が不十分であると考えられれば,我々のビジネス秘密を盗用する第三者に対する十分な追跡権
がない可能性がある.
私たちは、私たちの開発プロセスにおける遺伝子治療製品コンポーネントおよびプロセスに必要な権利を買収および許可によって取得または維持することができないかもしれない。
現在、私たちは知的財産権を持っており、第三者からの許可と私たちが持っている特許を通じて、私たちの候補遺伝子治療製品を開発しています。私たちの計画は他の候補製品に関連する可能性があり、これらの製品は第三者が保有する独占権を使用する必要があるかもしれないので、私たちの業務の成長は、私たちがこれらの独占権を取得、許可、または使用する能力にある程度依存するかもしれない。さらに、私たちの候補製品は、効率的かつ効率的に動作するために特定の配合物を必要とする場合があり、これらの権利は他の人によって保持されている可能性がある。私たちは、私たちが決定した第三者から、任意の成分、使用方法、プロセス、または他の第三者が可能かもしれない知的財産権を得ることができないかもしれない。第三者知的財産権の許可·買収は競争分野であり、一部のより成熟した企業も魅力的と考えられる第三者知的財産権許可または買収戦略を求めている。これらの老舗会社は私たちより競争優位を持っているかもしれません。それらの規模、現金資源及びより良い臨床開発と商業化能力のためです。
例えば,米国や外国の学術機関と協力し,これらの機関との書面合意に基づいて,われわれの臨床前研究や開発を加速する場合がある。一般に、これらのbr機関は、その機関が協働から得た任意の技術的権利の許可を交渉するためのオプションを提供してくれる。知的財産権のこのような最初の交渉権にかかわらず、私たちは指定された時間範囲内で、またはその許容可能な条項の下でライセンスを交渉することができないかもしれない。もし私たちがそれができなければ、その機関は知的財産権を他の側に提供する可能性があり、これは私たちが私たちのbr計画を継続することを阻止するかもしれない。
しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた、投資から適切なリターンを得ることを可能にする条項の下で第三者知的財産権を許可または取得することができないかもしれません。もし私たちが必要な第三者知的財産権を得ることに成功しなければ、私たちの業務、財務状況、成長の見通しは影響を受ける可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関が提出した様々なプログラム、書類提出、費用支払い、および他の要求
を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
定期維持費、継続費、年会費、および特許および/または出願に関連する様々な他の政府費用は、特許および/または出願の有効期間内に、米国特許事務室および米国以外の様々な政府特許代理機関に段階的に支払われる。私たちの知る限り、私たちと私たちのライセンシーは、私たちと彼らがこれらの費用を支払うことをシステム的に注意して、私たちと私たちの許可者は外部会社を雇って、私たちと彼らのそれぞれの外部弁護士に依存して、これらの費用を非アメリカ特許代理機関に支払うように注意しています。米国特許商標局および様々な非政府特許機関は、特許出願中にいくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。私たちと私たちのライセンシーは、信頼の良い法律事務所や他の専門家を招いて、私たちと彼らが遵守するのを助けて、多くの場合、滞納金を支払うことによって、あるいは適用規則に基づいて他の方法で不注意を正すことができます。しかしながら、場合によっては、規定を遵守しないことは、特許または特許出願の放棄または失効をもたらす可能性があり、それにより、関連する管轄区域の特許権の一部または全部が失われる可能性がある。この場合、私たちの競争相手が市場に参入する可能性があり、このような状況は私たちの業務に実質的な悪影響を及ぼすだろう。
54
カタログ表
私たちの候補製品をカバーする発行された特許が法廷で疑問視されれば、無効または実行不可能と認定される可能性がある。
私たちまたは私たちの許可パートナーのうちの1つが第三者に対して法的訴訟を提起して、私たちの候補製品のうちの1つをカバーする特許を強制的に執行する場合、被告は、私たちの候補製品をカバーする特許を無効および/または強制的に実行できないと反訴することができる。米国の特許訴訟では,被告が無効および/または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、特許資格に適合する主題、新規性の欠如、明らかな、または有効化されていないことを含む、複数の法定要件のいずれかを満たすことができなかったと言われている可能性がある。実行不可能な断言の理由は,その特許の起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った声明をしたりしたためかもしれない.第三者も米国や海外の行政機関に類似したクレームを出すことができ、訴訟範囲外でも同様である。このようなメカニズムには、再審、贈与後審査、外国司法管轄区の同等の訴訟手続(例えば、反対訴訟手続)が含まれる。このような訴訟は、私たちまたは私たちの許可brパートナーの特許が撤回または修正され、私たちの候補製品をカバーしないようにする可能性がある。法的に無効と実行不可能と断言された後の結果は予測できない。例えば、有効性の問題については、私たちは無効な以前の技術がないとは確信できないが、私たちと特許審査員は起訴中にこれを知らない。我々が米国特許商標局(“USPTO”)に開示した従来技術は、我々のいくつかの特許主張の範囲または有効性に影響を与える可能性がある。被告が無効および/または強制執行できない法律で勝訴した場合, 私たちは私たちの候補製品に対する少なくとも一部、さらにすべての特許保護を失うだろう。このような特許保護の喪失は私たちの業務に実質的な悪影響を及ぼすだろう。
米国特許法または他の国または管轄区域特許法の変化は、特許の全体的な価値を低下させ、製品を保護する能力を弱める可能性がある。
他のバイオテクノロジー会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存する。バイオテクノロジー業界で特許を取得および実施することは、技術的な複雑さと、法律的な複雑さとも関連するため、バイオテクノロジー特許の取得および実施は高価で時間がかかり、本質的に不確実である。議会は私たちに不利な特許改革法案を採択するかもしれない。
近年、米国最高裁判所はいくつかの特許事件に対して裁決を下し、場合によっては入手可能な特許保護範囲を縮小し、場合によっては特許所有者の権利を弱める。米国議会、米国裁判所、USPTO、および他の国の関連立法機関の将来の行動によると、特許を管理する法律·法規は予測不可能な方法で変化する可能性があり、それにより、私たちが新しい特許を獲得したり、私たちの既存の特許と私たちが将来獲得する可能性のある特許を強制的に執行する能力を弱める可能性がある。
同様に、他の国/地域または司法管轄区域の特許法律および法規の変化またはそれらを実行する政府機関の変化または関連政府機関が特許法律または法規を実行する方法の変化は、私たちが新しい特許を取得したり、私たちが許可または将来獲得可能な特許を実行する能力を弱める可能性がある。例えば、欧州特許法の複雑さと不確実性もここ数年増加している。欧州では、新しい単一特許制度が2023年末までに導入される可能性があり、このような制度を導入する前に付与された特許を含む欧州の特許に大きな影響を与えるだろう。単一特許制度の下で,欧州の出願はすぐに特許付与後に単一特許裁判所(UPC)によって管轄される単一特許となる権利がある。UPCは新しい裁判所制度であるため、br裁判所は前例がなく、いかなる訴訟の不確実性を増加させた。UPC実施前に付与された特許は,UPCの管轄から脱退することを選択し,UPC国の国家特許として保持することができる。UPC管内に残っている特許は,UPCによる単一撤回挑戦を受けやすい可能性があり,成功すれば,UPC署名国のすべての国/地域で特許を無効にする可能性がある。私たちはどんな潜在的な変化の長期的な影響を確実に予測することができない。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
世界のすべての国/地域で候補製品特許を申請、起訴、保護する費用は目を引くほど高く、私たちの米国以外のいくつかの国での知的財産権は米国ほど広くないかもしれない。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の司法管轄区で私たちの発明を使用して製造された製品を販売したり、輸入したりすることを阻止することができないかもしれない。競争相手のbrは、私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っている地域に輸出することもできるが、法執行力はアメリカ
に及ばない。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護、特にバイオテクノロジー製品に関する保護の強制執行を支持しておらず、これは、私たちの特許の侵害や私たちの独占権を侵害する方法で競争製品を販売することを阻止することを困難にするかもしれない。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは、巨額のコストを招き、私たちの努力と注意を私たちの業務の他の側面から移転させる可能性があり、私たちの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者からのクレームを引き起こす可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償または他の救済措置は商業的な意味がないかもしれない。したがって、私たちが世界各地で知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著な商業的優位性を得るのに十分ではないかもしれない。
55
カタログ表
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許の寿命は限られている。米国では、すべての維持費がタイムリーに支払われる場合、特許の自然失効時間は、通常、米国で最初の非臨時出願日から20年である。様々な延期がある可能性があるが、特許の寿命およびその提供される保護は限られている。我々の候補製品をカバーする特許を取得しても,特許有効期限が満了すると,模倣薬や生体模倣薬を含むbr競合製品からの競争に直面する可能性がある。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
知的財産権はすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは私たちの競争優位性を維持させることができないかもしれない。例えば:
| • |
他の人は私たちと似た候補製品を作ることができるかもしれないが、これらの製品は私たちが持っている特許請求の範囲内ではない
|
| • |
私たち、または私たちのライセンスパートナーまたは現在または未来の協力者は、私たちが許可または将来所有する可能性のある発表された特許または係属中の特許出願によってカバーされる発明を最初にした人ではないかもしれない
|
| • |
私たち、または私たちの許可パートナー、または現在または未来の協力者は、私たちまたは彼らのいくつかの発明に関する特許出願を最初に提出することではないかもしれない
|
| • |
他の人は、私たちが持っているまたは許可されていない知的財産権を侵害することなく、類似または代替技術を開発したり、私たちの任意の技術を複製したりすることができる
|
| • |
私たちの競争相手は、特許権のない国で研究や開発活動を行い、これらの活動から学んだ情報を利用して競争力のある製品を開発し、私たちの主要な商業市場で販売するかもしれません
|
| • |
私たちのいかなる特許、私たちの任意の係属中の特許出願(発行された場合)、または私たちの許可者の特許出願は、私たちの候補製品を保護するのに十分な範囲を有する権利要件を含むことを保証することはできません
|
| • |
私たちに発行されたいかなる特許や私たちの許可者が私たちの商業的に実行可能な候補製品に独占市場の基礎を提供するか、あるいは私たちにどんな競争優位性を提供するかを保証することはできません
|
| • |
私たちは私たちの商業活動や候補製品が他人の特許を侵害しないという保証はない
|
| • |
私たちが持っている関連特許やライセンスが満期になる前に、承認されれば、私たちの候補製品を大規模に商業化することに成功することを保証することはできない
|
| • |
私たちは他の特許を申請できる独自技術を開発しないかもしれない
|
| • |
他人の特許や知的財産権は私たちの業務を損なうかもしれません;
|
| • |
いくつかの商業秘密またはノウハウを保護するために、私たちは特許を出願しないことを選択することができ、第三者はその後、これらの知的財産権をカバーする特許を提出する可能性がある。
|
これらの事件が発生すると、それらは私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちは私たちの特許と他の知的財産権の発明権または所有権のクレームに疑問を受けるかもしれない。
私たちはまた、元従業員、協力者、または他の第三者が私たちの特許または他の知的財産権に対して所有権権益を持っているというクレームを受ける可能性がある。将来、私たちは、例えば、私たちの候補製品の開発に参加したコンサルタントや他の人の義務紛争に遭遇するかもしれません。これらと他の挑戦在庫や所有権のクレームに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償を支払うことに加えて、貴重な知的財産権、例えば貴重な知的財産権の独占所有権や使用権を失う可能性がある。この結果
は我々の業務に実質的な悪影響を与える可能性がある.私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
知的財産権侵害に対する第三者のクレームは、私たちの開発と商業化努力を阻害または延期する可能性がある。
私たちの商業的成功は第三者の特許と固有の権利の侵害を避けることにある程度依存する。アメリカ国内外に大量の訴訟があり、生物技術と製薬業界の特許とその他の知的財産権に関連し、特許侵害訴訟、妨害、異議、一方的再審、許可後審査及び各方面間米国特許商標局又は米国特許商標局及び対応する外国特許庁において手続を審査する。我々が開発候補を求める分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する.バイオテクノロジーや製薬業界の拡張とより多くの特許の発行に伴い,我々の候補製品が第三者特許権侵害請求を受けるリスクが増加する可能性がある。
56
カタログ表
第三者は私たちが許可されていない状況で彼らのノウハウを使用していると主張するかもしれない。我々の候補製品の使用または製造に関連する第三者特許または特許出願が存在する可能性があり、これらの特許請求項は、材料、製剤、製造方法、または治療方法である。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、我々の候補製品が発行されたbr}特許を侵害する可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。管轄権のある裁判所が、任意の候補製品の製造プロセス、製造中に形成された任意の分子または任意の最終製品自体をカバーするために任意の第三者特許を保有している場合、そのような特許の所有者は、適用される特許に基づいてライセンスを取得し、またはその特許が満了するまで、その候補製品
を商業化する能力を阻止することができるかもしれない。同様に、管轄権のある裁判所が、共同療法を含む私たちの処方、製造プロセス、または使用方法の様々な態様をカバーするために、任意の第三者特許を保有している場合、そのような特許の所有者は、brライセンスまたはその特許の満了を取得しない限り、適用可能な候補製品を開発し、それを商業化する能力を阻止することができるかもしれない。いずれの場合も、そのような許可は商業的に合理的な条項や全く存在しないかもしれない。
我々にクレームをつけた当事者は、禁止または他の公平な救済を受ける可能性があり、これは、私たちの1つまたは複数の候補製品のさらなる開発と商業化を効果的に阻止することができる。これらのクレームを弁護するには、その是非にかかわらず、巨額の訴訟費用がかかり、私たちの業務中の従業員資源を大量に分流させる。もし私たちの権利侵害クレームが成功した場合、私たちは3倍の損害賠償と故意に侵害した弁護士費の支払い、印税の支払い、私たちの侵害製品の再設計、または第三者から1つ以上の許可を得ることができないかもしれない、または大量の時間とお金の支出を必要とするかもしれない巨額の損害賠償を支払う必要があるかもしれない。
人員や会社の拡張に関するリスク
私たちの関係者のリスクは
キーパーソンのサービスを失ったり、キーパーソンを引き付けることができない場合、私たちの業務は影響を受ける可能性があります。
私たちは私たちの最高経営責任者Gaurav Shah医学博士、私たちの首席医療官兼臨床開発担当Jonathan Schwartz医学博士、私たちの総裁兼最高経営責任者Kinnari Patel、製薬博士、工商管理修士、そして私たちの財務副社長、財務担当、最高会計官兼臨時財務責任者John Militelloを含む、私たちの上級管理職の努力に強く依存しています。これらの人員や私たちの他の高級管理者を失ったサービスは、研究、開発、マーケティング、または製品の商業化目標の実現を遅延または阻害する可能性がある。私たちとキーパーソンの採用計画は“勝手”だ。私たちはどんな重要な従業員にも“キーパーソン”保険を提供しないし、このような保険を購入するつもりもない。また、私たちの業務は専門的な科学的性質であるため、私たちは私たちがbrに合格した科学技術者と顧問を誘致し、維持する能力に強く依存している。大手製薬と化学工業会社、専門バイオテクノロジー会社及び大学と他の研究機関の間では、私たちの業務分野の合格人材に対する競争は非常に激しく、私たちはこれらの人員の誘致と維持に成功できないかもしれない。
私たちの従業員、主要な調査者、コンサルタントと商業パートナーは監督管理の基準と要求を守らないこと、およびインサイダー取引を含む不当な行為或いはその他の不当な活動に従事する可能性がある。
私たちは従業員、コンサルタント、そして商業パートナー詐欺や他の不適切な行為の危険に直面している。これらの当事者の不正行為は、FDAおよび非米国規制機関の規定を故意に遵守しないこと、FDAおよび非米国規制機関に正確な情報を提供すること、米国および国外の医療詐欺および乱用法律法規を遵守し、財務情報またはデータを正確に報告すること、または許可されていない活動を私たちに開示することを含む可能性がある。特に、医療業界の販売、マーケティング、業務配置は、詐欺、不正行為、リベート、自己取引、その他の乱用行為を防止するための広範な法律法規によって制約されている。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。このような不正行為はまた、臨床研究中に得られた情報を不当に使用する可能性があり、これは規制制裁を招き、私たちの名声に深刻な損害を与える可能性があり、あるいは規制機関が私たちの候補製品を承認しない可能性がある。私たちはすべての従業員に適用される商業道徳と行動基準を持っているが、常に従業員または第三者の不正行為を識別し、阻止できるわけではなく、私たちがこのような行為を発見し、防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御できないかもしれないし、これらの法律や法規を遵守できないことによる政府の調査または他の行動や訴訟から私たちを保護することができないかもしれない。もし私たちにこのような訴訟を提起して、私たちは自分のために弁護したり、自分の権利を維持することに成功できなかったら、これらの行動は私たちの業務に大きな影響を与えるかもしれません, 巨額の罰金や他の制裁が含まれている。
私たちは、私たちの従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示したか、または私たちの従業員がその前の雇用主が主張した商業機密を誤って使用または開示したという疑惑の影響を受けるかもしれない。
57
カタログ表
私たちが雇った人は以前、私たちの競争相手や潜在的な競争相手を含む大学や他のバイオテクノロジーや製薬会社に雇われていた。私たちは、私たちの従業員、コンサルタント、および独立請負業者が、私たちのために働いているときに他人の独自情報またはノウハウを使用しないことを保証するために努力しているが、私たちまたは私たちの従業員、コンサルタントまたは独立請負業者は、商業秘密または他の独自情報を含む、当社の従業員の任意の元雇用主または他の第三者の知的財産権を不注意または他の方法で使用または漏洩する可能性がある。訴訟を通じてこのような
クレームに対抗する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償の支払いに加えて、貴重な知的財産権や人員を失う可能性があり、これは私たちの業務に悪影響を及ぼす可能性がある。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
私たちの拡張と成長計画に関連するリスク
私たちは私たちの組織を拡張する必要があるかもしれないし、このような成長を管理する時に困難に直面する可能性があり、これは私たちの運営を混乱させるかもしれない。
私たちの業務活動の拡大に伴い、私たちは私たちの全従業員基盤を拡大し、より多くのコンサルタントや請負業者を雇うことができるかもしれない。私たちの経営陣は、不比例な注意を日常活動から移し、これらの成長活動を管理するために多くの時間を投入する必要があるかもしれない。私たちは私たちの業務の拡張を効果的に管理できないかもしれません。これは私たちのインフラが弱く、運営が挫折し、ビジネスの機会を失うこと、従業員の流失、余剰従業員の生産性の低下を招く可能性があります。私たちの予想成長は大量の資本支出を必要とする可能性があり、より多くの候補製品を開発するなど、財務資源を他のプロジェクトから分流する可能性がある。もし私たちの経営陣が私たちの成長を効果的に管理できなければ、私たちの支出は予想よりも増加するかもしれません。私たちは収入を創出および/または増加する能力が低下する可能性があり、私たちは私たちの業務戦略を実施できないかもしれません。
私たちはRenovacorの買収のような潜在的な買収や業務合併の期待的な利益を達成できないかもしれない。
買収や事業合併の成功、例えばRenovacorの買収は、他の事項に加えて、私たちが開発と運営協同効果を実現することを可能にする方法で私たちの業務を統合する能力に依存します。統合過程は、キー従業員の流失を招く可能性がある;私たちが行っている業務中断、または標準、制御プログラム、または政策の不一致は、予想収益を達成する能力に悪影響を及ぼす可能性がある。両業務間の統合努力はまた、我々の中核業務や他の我々の株主に有利になる可能性のある機会から経営陣の注意をそらすことになる。買収を実現できないすべてまたは任意の期待収益、および統合過程で遭遇したいかなる遅延も、私たちの業務や運営結果に悪影響を及ぼす可能性があり、買収完了後の私たちの普通株の価値に影響を与える可能性がある。もし私たちがこれらの目標を達成できなければ、買収の予想収益は完全に達成できないかもしれないし、完全に実現できないかもしれないし、予想よりも長い時間やコストが必要かもしれない。特に、買収や業務合併は短期的または長期的に私たちの株式価値を増加させない可能性がある。さらに、Renovacorの買収や事業組合の買収など、我々の普通株の市場価格に影響を与える可能性があり、これは私たちの株主に大きな損失をもたらす可能性がある。
さらに、将来の潜在的なビジネス、技術、製品買収については、
| • |
現在の株主の持株比率を希釈する株式証券を発行する
|
| • |
多くの借金を招き、これは私たちの運営に圧力を与えるかもしれない
|
| • |
多くの実際または負債を負担しています
|
| • |
私たちの開発計画の優先順位を再決定し、いくつかの候補製品の開発と商業化を停止したり、
|
| • |
他の会社と合併したり、他の会社と業務合併を行ったりすると、我々の株主は、ある株主が望ましくないと考えられる条項で他の会社の現金または株を得ることができる。
|
将来の買収、再編、業務合併を評価して考慮する予定ですが、現在のところ、買収、再編、業務合併に関する合意や了解はありません。
将来的に第三者と戦略同盟や合弁企業を結成することは、私たちの業務を乱し、私たちの財務状況と経営業績を損なう可能性があります。
私たちは第三者と戦略同盟を結成したり、合弁企業を設立したりするかもしれませんが、これは私たちの既存の業務を補完または拡大すると思います。私たちは戦略連合や買収によって生まれた任意の新製品を開発、製造、マーケティングする時に多くの困難に直面する可能性があります。これらの新製品は私たちが期待した利益を達成したり、私たちの業務を強化したりすることを延期したり阻止したりすることができます。このような戦略連合や合弁企業のいずれかの後に、予想される相乗効果を達成して、取引が合理的であることを証明することは保証できません。私たちが戦略連合や合弁企業で直面しているリスクは
| • |
管理時間と重点を私たちの業務を運営することから統合課題に対応することに移します
|
| • |
研究開発を協調させることです
|
| • |
どんな製品買収や戦略的位置づけによっても戦略パートナーとの関係が変化した
|
| • |
従業員の統合に関する文化的課題
|
| • |
任意の合弁企業で制御、プログラム、および政策を実施または改善する必要性
|
| • |
知的財産権侵害請求、違法、商業紛争、税務責任、その他の既知の責任を含む、任意の戦略連合または合弁企業の前の活動における任意の組合会社の責任
|
58
カタログ表
| • |
予期しなかったログアウトや費用;
|
| • |
従業員、顧客、前株主、または他の第三者のクレームを含む訴訟または他のクレーム
|
過去または未来の戦略連合で遭遇したこれらのリスクや他の問題を解決できなかったことは、これらの取引の予想される収益を達成できず、予期しない責任を生じ、全体的に業務を損なう可能性がある。将来の戦略連合または合弁企業はまた、債務、または負債、償却費用、または増加した運営費用をもたらす可能性があり、いずれも私たちの財務状況または経営業績を損なう可能性があります。
アメリカ以外のビジネス関係、特にEUでは、国際業務に関連する様々なリスクが私たちの業務を損なう可能性があります。
私たちはアメリカ以外で様々なビジネス関係に従事しており、私たちはアメリカ国外で私たちの候補製品を商業化するかもしれません。多くの他の国/地域では、他の人が私たちのアメリカの法律や法規(“海外腐敗防止法”を含む)に適用される禁止されたビジネスに従事することが一般的です。これらの法律や政策を遵守するために設計された政策やプログラムを実施する可能性があるにもかかわらず,我々の従業員,請負者,エージェントがこれらの法律や政策を遵守することを保証することはできない.もし私たちが国際拡張と運営の挑戦に成功しなければ、私たちの業務と運営結果は損なわれる可能性がある。
米国以外の候補製品をある程度商業化する可能性があり、国際運営に関連する様々なリスクの影響を受けることが予想される
| • |
国外では薬品と生物製品の審査に対する監督管理要求が異なる
|
| • |
知的財産権の保護を減らすことです
|
| • |
関税、貿易障壁、規制要求の意外な変化
|
| • |
インフレ、特に外国経済と市場の政治的不安定を含む経済的疲弊
|
| • |
外国に居住または旅行する従業員は、税収、雇用、移民、労働法の規定を遵守する
|
| • |
外国為替変動は、営業費用の増加と収入の減少、他の国での業務展開に付随する他の義務を招く可能性がある
|
| • |
労働騒乱がアメリカよりも一般的な国では労働力の不確実性
|
| • |
海外の原材料供給や製造能力に影響を与える事件による不足
|
| • |
地政学的行動(戦争およびテロを含む)または自然災害(地震、台風、洪水および火災を含む)、公衆衛生危機(例えば、流行病および流行病)または経済的または政治的不安定による業務中断;
|
| • |
GDPRおよび英国GDPRを含む、個人データの収集、使用、開示、保持、セキュリティ、および移行に関する外国の法律、法規、標準、および規制ガイドラインを遵守する;
|
| • |
アメリカ以外の管轄区域で私たちの契約を実行することはもっと難しいです。
|
これらのリスクおよび関連リスクは、私たちの業務、財務状況、運営結果、および見通しに実質的な損害を与える可能性がある。
もし私たちが私たちの協力者や戦略的パートナーと衝突すれば、これらの当事者たちは私たちに不利な方法で行動し、私たちのbr戦略を実施する能力を制限するかもしれない。
もし私たちの企業や学術パートナーや戦略パートナーが私たちと衝突した場合、相手は私たちに不利な方法で行動し、私たちの戦略を実施する能力を制限するかもしれません。私たちのいくつかの学術協力者と戦略パートナーは、私たちが協力している分野ごとに複数の製品開発を行っています。しかし、我々の協力者や戦略パートナーは、関連分野の製品を単独で開発することができ、他のパートナーと協力してこれらの協力テーマの製品または潜在的な製品と競争力のある製品を開発することもできる。協力者または戦略パートナーによって開発された競合製品であっても、協力者または戦略パートナーによって権利を有する競合製品であっても、私たちの協力者またはパートナーが候補製品の支援を撤回することをもたらす可能性がある。
私たちのいくつかの協力者や戦略的パートナーたちはまた私たちの未来の競争相手になるかもしれない。私たちの協力者や戦略パートナーは競争製品を開発し、私たちがその競争相手との協力を阻止し、適時に規制の承認を得ることができなかった、私たちとの合意を早期に終了したり、私たちの候補製品を開発し商業化するのに十分な資源を投入することができなかったかもしれない。これらの開発のいずれも私たちの製品開発を損なう可能性があります。
私たちの普通株式所有権に関連するリスク
将来的に私たちの普通株の公開市場での販売は私たちの普通株の市場価格を大幅に低下させる可能性があります。たとえ私たちの業務が良好であっても。
公開市場で私たちの大量の普通株を売ることはいつでも起こる可能性がある。これらの売却、あるいはそのような売却が起こりうるという見方や見方は、私たちの普通株の市場価格
を低下させる可能性がある。1933年証券法(“証券法”)の下で第144条及び701条の規則で許可された範囲内、又は証券法に基づいて登録され、当社の非関連会社が保有している範囲内で、我々が発行した普通株式は、いつでも公開市場で自由に販売することができる。さらに、私たちの従業員、役員、役員、および関連株主は、時々私たちの普通株を売却することを規定する規則
10 b 5-1計画に加入しているか、または加入する可能性があります。ルール10 b 5-1計画によれば、仲介人は、従業員、上級管理者、または関連株主
のさらなる指示を必要とすることなく、従業員、取締役または上級管理者が計画に入る際に確立されたパラメータに従って取引を実行する。規則10 b 5−1計画は、場合によっては修正または終了される可能性がある。当社の従業員、役員、取締役、および関連株主は、重大な非公開情報を把握することなく、ルール10 b 5-1計画以外の追加株式を購入または売却することもできます。さらに、将来的には、融資、買収、訴訟和解、従業員手配、または他の態様のために、普通株または他の普通株に変換可能な株式または債務証券を増発する可能性がある。どのような発行でも、私たちの既存株主の株式が大幅に希釈され、私たちの株価が下落する可能性があります。
59
カタログ表
もし証券アナリストが私たちの業務に関する研究や報告を発表しなければ、あるいは彼らが私たちの株に対するマイナス評価を発表したら、私たちの株価は下落するかもしれません。
私たちの普通株の取引市場は、業界や金融アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存している。私たちはアナリストの報告を得ていますが、私たちの業務を追跡している一人以上のアナリストが私たちの株に対する評価を引き下げたら、私たちの株価は下落するかもしれません。もしこれらのアナリストのうちの1人以上が私たちの株を追跡したり、私たちに関する報告書を定期的に発表できなかったら、私たちの株の市場での可視性を失い、ひいては私たちの株価を下落させる可能性がある。
私たちの普通株の価格は大きく変動する可能性があり、これは私たちの株主に大きな損失をもたらすかもしれない。
私たちの株価は変動するかもしれません。総じて,株式市場,特にバイオ製薬会社の市場は,極端な変動を経験しており,この変動は特定の会社の経営業績とは無関係であることが多い。このような変動のため、私たちの株主は彼らが普通株を購入したかそれ以上の価格で彼らの普通株を売ることができないかもしれない。私たちの普通株の市場価格は多くの要素の影響を受けるかもしれません
| • |
候補製品や競争相手の臨床試験結果
|
| • |
競争力のある製品や技術の成功
|
| • |
協力の開始または終了
|
| • |
アメリカや他の国の法規や法律の発展
|
| • |
特許出願、発行された特許または他の固有の権利に関連する発展または紛争;
|
| • |
キーパーソンの採用や退職
|
| • |
私たちの候補製品や臨床開発計画に関連する費用レベルは
|
| • |
私たちは他の候補製品の結果を発見、開発、取得、または許可するために努力している
|
| • |
財務結果、発展スケジュール、または証券アナリストの提案に関する推定の実際または予想変化;
|
| • |
一般的な遺伝子治療に関する否定的な宣伝や私たちの製品候補
|
| • |
私たちの財務業績や私たちと似ていると思われる会社の財務業績の違い
|
| • |
医療支払い制度の構造を変え
|
| • |
マクロ経済状況と19型新冠肺炎の大流行の経済影響、インフレと金利上昇及び世界的な衝突、ロシア-ウクライナ戦争を含む
|
| • |
製薬とバイオテクノロジー産業の市場状況;
|
| • |
一般的な経済、産業、そして市場状況。
|
もし私たちの四半期の経営業績が投資家や証券アナリストの予想を下回れば、私たちの普通株の価格は大幅に低下する可能性がある。また、私たちの経営業績のどの四半期の変動も、私たちの株価を大幅に変動させる可能性があります。我々の財務業績を四半期比較することは必ずしも意味があるとは限らず,我々の将来の業績の指標とすべきではないと考えられる。
従来、ある会社の証券市場価格が変動した後、同社に対して証券集団訴訟が提起されるのが一般的であった。私たちにこのような訴訟を提起すれば、私たちがこのようなクレームを弁護するために巨額の費用を発生させ、経営陣の注意と資源を移転させる可能性があり、これは私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性がある。
RTW Investments,LP,我々最大の株主は,株主に承認されたすべての事項に大きな影響を与える能力がある可能性がある.
RTW Investments,LP(“RTW”)の合計実益は我々普通株流通株の約25.2%を持っている.投票権の集中によりRTWは我々の株主承認に提出されたすべての事項および我々の管理や事務に大きな影響を与える権利がある.例えば、RTWは、取締役の選挙および私たちのすべてまたは実質的にすべての資産の任意の合併、合併、または売却の承認に著しく影響を与えることができる。さらに、これは、私たちの株主の一つとして、これがあなたの最良の利益に合致すると思うかもしれませんので、私たちの株式に対する能動的な買収提案や要約を阻止または阻止することができます。
私たちは予測可能な未来に私たちの株に何の現金配当も支払わないと予想されるので、資本増加(あれば)は株主が収益を得る唯一の源になるだろう。
60
カタログ表
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、すべての未来の収益を維持するつもりで、もしあれば、私たちの業務の成長と発展に資金を提供します。しかも、未来の債務協定のいかなる条項も私たちが配当金を支払うことを阻止するかもしれない。したがって、私たち普通株の資本増価(あれば)は株主が予測可能な未来に唯一の収益源になるだろう。
私たちの業務に関わる他のリスクは
もし私たちが財務報告書に対して適切かつ有効な内部統制を維持できなければ、私たちが正確かつ適時に財務諸表を作成する能力は損害を受ける可能性があり、これは私たちの経営業績、投資家の私たちに対する見方を損害し、それによって私たちの普通株の価値を損なう可能性がある。
上場企業として、財務報告書の内部統制を維持し、このような内部統制におけるいかなる重大な弱点も報告することが求められている。Sarbanes-Oxley Actの404節(“404節”)によると、私たちは財務報告書の内部統制に対する経営陣の有効性に関する報告書を提出しなければならない。第404条によれば、私たちの独立公認会計士事務所は、財務報告書の内部統制に対する私たちの有効性を証明することを要求されるであろう。このような認証レポートの準備やレポート要求遵守のコストは,我々の費用を増加させ,大量の管理時間を要する可能性がある.追加的なコンプライアンスコストのため、投資家は私たちの普通株の吸引力が低下していることを発見するかもしれない。したがって、一部の投資家が私たちの普通株の吸引力が低下していることを発見すれば、私たちの普通株はそれほど活発ではない取引市場が出現する可能性があり、私たちの株価はもっと変動するかもしれない。
経営陣と我々の独立公認会計士事務所が財務報告内部統制を評価するために達成しなければならない基準のルールは複雑であり、大量の文書、テスト、可能な救済措置が必要である。私たちと私たちの独立公認会計士事務所の財務報告の内部統制の評価については、情報技術を含め、追加の財務·管理制御、報告システムおよびプログラムを実施し、追加の会計·財務担当者を招聘するシステムをアップグレードする必要があるかもしれません。
必要な新しい制御措置や改善された制御措置を実施できなかったり、実行中に遭遇した困難は、私たちの報告義務を履行できない可能性があります。さらに、私たちまたは私たちの独立公認会計士事務所が第404条に基づいて行った任意のテストは、財務報告の内部統制における私たちの欠陥を明らかにすることができ、これらの欠陥は、重大な欠陥とみなされるか、または私たちの財務諸表を前向きにまたは遡及的に変更する必要がある場合があり、またはさらなる関心または改善が必要な他の分野を決定することができる。悪い内部統制はまた、投資家が私たちが報告した財務情報に自信を失う可能性があり、これは私たちの普通株の取引価格にマイナス影響を与える可能性がある。内部統制の欠陥はまた私たちが未来に財政的業績を再説明することにつながるかもしれない。私たちは、株主または他の第三者訴訟、および米国証券取引委員会、ニューヨーク証券取引所、ナスダック、または他の規制機関の調査を受ける可能性があり、これは追加の財務および管理リソースを必要とし、罰金、停止、損害賠償の支払い、または他のbr救済措置をもたらす可能性がある。さらに、第404条の監査人認証条項を遵守する上でのいかなる遅延も、短い転売登録の資格の取り消し、米国証券取引委員会の行動、および私たちの普通株が一時停止または退市されることを含む様々な行政制裁を受ける可能性があり、これは、私たちの普通株の取引価格を低下させ、私たちの業務を損なう可能性があります。
わが社の定款書類やデラウェア州法律の条項は、私たちを買収することをより困難にする可能性があり、これは私たちの株主に有利になるかもしれませんし、私たちの株主が現在の経営陣を交換または更迭しようとするのを阻止するかもしれません。
会社登録証明書および定款の条項は、株主が有利と思われる可能性のある会社の合併、買収、またはその他の支配権変更を阻止、延期、または阻止する可能性があり、あなたの株から割増取引を得る可能性があります。これらの条項はまた、投資家が将来私たちの普通株株に支払うことを望むかもしれない価格を制限し、私たちの普通株の市場価格を下げる可能性がある。また、私たちの取締役会は責任を持って私たちの管理チームのメンバーを命じているため、これらの規定は株主が私たちの取締役会のメンバーを交換しにくくし、株主が現在のbr経営陣を交代または罷免しようとしていることを挫折または阻止する可能性があります。他にもこれらの条項には
| • |
取締役会だけが取締役数を決定することを許可した
|
| • |
私たちが再説明した会社証明書および再記載された定款のいくつかの条項を修正するために絶対多数の票が必要である
|
| • |
株主の書面同意による行動を禁止することは、すべての株主の行動が私たちの株主会議で行われなければならないことを要求する
|
| • |
指名が取締役会に入るか、株主が年次株主総会で行動できる事項の事前通知要求を作成する。
|
また、我々は、“デラウェア州一般会社法”第203条の規定によって管轄されており、この条項は、規定された方法で承認されない限り、私たちが発行した議決権株の15%以上を保有する者が、取引日後3年以内に私たちと合併または合併することを禁止する。私たちの憲章文書やデラウェア州法律のどの条項も、場合によっては私たちの普通株の市場価格を下げることができる。
我々の内部計算機システム,あるいは我々の第三者協力者や他の請負者のシステムは,障害やセキュリティホールに遭遇する可能性があり,我々の開発計画が実質的に中断される可能性がある.
61
カタログ表
私たちの内部コンピュータシステム、ならびに私たちの現在および未来の任意の協力者および他のコンサルタントおよび請負業者のコンピュータシステムは、コンピュータウイルス、許可されていないアクセス、ネットワーク攻撃、データ漏洩、自然災害、テロ、戦争、ならびに電気通信および電気故障の破壊を受けやすい。私たちはこれまで、このような重大なシステム障害、事故、攻撃、またはセキュリティホールを経験していませんが、このような事件が発生して私たちの運営が中断された場合、私たちの開発計画や業務運営に大きな中断を招く可能性があります。私たちのビジネス機密や他の独自の情報の損失によるものであっても、他の同様の中断によるものでもあります。例えば、完成したまたは未来の臨床試験における臨床試験データの損失は、私たちの監督管理の承認作業を遅延させ、データを回復または複製するコストを著しく増加させる可能性がある。もし任意の中断またはセキュリティホールが私たちのデータやアプリケーションを紛失したり、破損させたり、機密または独自の情報を適切に開示しない場合、私たちは責任を負う可能性があり、私たちの競争地位が損なわれる可能性があり、私たちの候補製品のさらなる開発と商業化が遅れる可能性がある。
ソーシャルメディアプラットフォームの使用はますます多くなり、新たなリスクと挑戦をもたらしている。
ソーシャルメディアは私たちの臨床開発計画や私たちの候補製品を交流するためにますます使用されており、治療のための疾患が開発されている。私たちの候補製品が承認された後、私たちは商業化作業で適切なソーシャルメディア
を利用するつもりです。バイオ製薬業界のソーシャルメディア実践は発展し続けており,このような使用に関する法規は常に明確ではない。この変化
は我々の業務に適用される法規に適合しない不確実性とリスクをもたらしている。例えば、患者は、ソーシャルメディアチャネルを使用していわゆる有害事象を報告する可能性がある。このような開示が発生した場合、私たちは適用される有害事象報告義務を監視し、遵守することができないかもしれないし、あるいはソーシャルメディアによって生じる政治的および市場的圧力の下で、私たちの業務または公衆の合法的な利益を守ることができないかもしれません。これは、私たちが私たちの調査製品が発表する可能性のある発言を制限したためです。さらに、任意のSNS上で敏感な情報または否定的または不正確な投稿またはコメントを開示しないリスク、または私たちの任意の従業員がSNS上で投稿した投稿が不適切な販促とみなされる可能性があるリスクもある。もしこのような事件が発生したり、私たちが適用された法規を遵守できなかった場合、私たちは責任を負い、br規制行動に直面したり、私たちの業務に他の損害を与える可能性があります。
不利な国や世界的な経済状況や政治的発展は、私たちの業務、財務状況、または経営結果に悪影響を及ぼす可能性がある。
私たちの経営結果は国内または世界経済と金融市場の全体的な状況の悪影響を受ける可能性があります。例えば、政府声明、行動または政策、政治的動揺、世界金融危機は、資本と信用市場の極端な変動と混乱を招く可能性がある。深刻または長期的な経済低迷、政治的動揺、またはそれ以上の世界金融危機は、新冠肺炎の大流行と現在の露烏戦争による危機を含み、我々の製品に対する需要が弱まる(承認されれば)、あるいは必要なときに許容可能な条件で追加資本を調達する能力(すべてであれば)を含む様々なリスクを私たちの業務にもたらす可能性がある。経済が疲弊したり下落したりすることは、私たちのサプライヤーに圧力を与え、供給中断を招く可能性もある。上記のいずれも私たちの業務を損なう可能性があり、現在の経済環境、さらなる政治発展、金融市場状況が私たちの業務に悪影響を及ぼす可能性のあるすべての方法を予見することはできません。
| 項目1 B。 |
米国証券取引委員会のコメントは未解決だ
|
ない。
| 第二項です。 |
属性
|
本社、研究開発、GMP製造施設、ストレージ施設
Rocketの会社の本社はニュージャージー州クランベリーにあり、オフィス、プロセス開発、研究開発実験室、および50,000平方フィートのAAV現在の良好な製造仕様(CGMP)製造を含む103,720平方フィートのレンタル施設であり、私たちのパイプをサポートしています。この施設内のより小さい面積は、2018年8月に最初にレンタルされ、賃貸契約は、建物全体(改訂された、すなわち“ニュージャージー州賃貸協定”)を含むために2019年6月に改訂された。ニュージャージー州賃貸プロトコルの初期期限は2034年に終了し、5年連続の2つの継続期間を選択することができる。また、私たちはニューヨークのエンパイアステートビルで空間をレンタルしました。2024年7月に満期になった賃貸によると、エンパイアステートビルは約6600平方フィートのオフィス空間からなります。ロケットはニュージャージー州デイトン市で別の4666平方フィートの貯蔵施設を借りた。
マサチューセッツ州レキシントンにある工場
私たちは現在マサチューセッツ州レキシントンで約15,000平方フィートのオフィススペースをレンタルしています。賃貸契約は2023年2月28日に満期、つまりレンタル期間の終了になります。この空間はINote kの前
本部であり,レンタル期間の残り時間内に転貸する.
62
カタログ表
ニュージャージー州ホプウェルにある工場は
Renovacorの買収の一部として、我々は、2033年3月に満了する約15,463平方フィートの賃貸契約をニュージャージー州ホプウェルで締結したと仮定する。レンタル期間の残り時間で
場所全体を転用するように努力します。
マサチューセッツ州ケンブリッジ市にある工場
Renovacorの買収の一部として、2024年4月に満了するマサチューセッツ州ケンブリッジ市の約5945平方フィートのオフィスビルの分譲協定を担当した。私たちは賃貸期間の残り時間を通じてこの不動産を転用するために努力するつもりだ。
| 第三項です。 |
法律訴訟
|
私たちは時々他の様々な法的手続きやクレームの影響を受けるかもしれません。これらの訴訟とクレームは私たちの正常な業務活動の過程で発生します。訴訟やクレームの結果は確実に予測できないが、私たちは他のクレームや訴訟の当事者であるとは信じておらず、これらのクレームまたは訴訟の結果が私たちに不利であると判定された場合、その結果が個別または全体的に私たちの業務に重大な悪影響を与えることを予想する理由がある。結果にかかわらず,弁護や和解コスト,管理資源分流などにより,訴訟は我々に悪影響を与える可能性がある。
| 第四項です。 |
炭鉱安全情報開示
|
適用されません。
第II部
|
五番目です。
|
登録者普通株市場、関連株主事項、発行者購入株式証券市場
情報
|
私たちの普通株はナスダック世界市場とナスダックバイオテクノロジー指数(ナスダック:NBI)で取引されている。2023年2月22日、我々の普通株のナスダック世界市場における最後の報告価格は1株19.20ドルであった。
株式表現グラフ
以下の図は2017年1月1日から2022年12月31日までの著者らの普通株の株主累積総リターンと同期(A)ナスダック生物技術指数
と(B)ナスダック総合指数の累積総リターンを比較した。このグラフは、私たちの普通株ナスダックバイオテクノロジー指数とナスダック総合指数の2017年1月1日の投資を100ドルと仮定し、配当金が再投資を行うと仮定している。
次の図に示す比較は履歴データに基づく.次の図に示す株価表現は必ずしも代表的ではなく,我々の普通株の未来の潜在表現を予測するためでもないことに注意されたい.
63
カタログ表
株主.株主
2023年2月22日現在、登録されている株主は45人で、その株は仲介人が代名人や街頭名義で保有している株主は含まれていない。 我々はこれまで
がInote k PharmPharmticals Corporation(“INote k”)との逆統合を完了し,2018年1月4日に完了した.2018年1月4日までの株式業績情報は、我々の前身会社Inote kの逆合併前の株価を代表して、逆合併後に完了した4:1逆分割により調整しました。
配当政策
私たちは私たちの株のどんな現金配当金も発表したり支払ったりしたことがない。私たちは現在、すべての利用可能な資金と任意の将来の収益(もしあれば)を維持し、私たちの業務の発展と拡張に資金を提供するつもりで、私たちは予測可能な未来にいかなる現金配当金も支払わないと予想しています。将来現金配当金を派遣するかどうかは、当社の取締役会が自ら決定する。投資家たちは現金配当金を受け取る期待を持って私たちの普通株を購入してはいけない。
最近売られている未登録証券
ない。
発行人が株式証券を購入する
2022年12月31日までの年間で、私たちの普通株は買い戻しされていません。
| 第六項です。 |
保留されている
|
| 第七項。 |
経営陣の財務状況と経営成果の検討と分析
|
以下、当社の財務状況及び経営結果の検討及び分析は、本年度報告に他の部分に含まれる総合財務諸表、関連付記及びその他の財務情報とともに読まなければならない。本討論は展望性陳述を含み、私たちの計画、目標、期待と意図のようなリスクと不確定要素に関連する。私たちの実際の結果はこのような前向きな陳述で議論された結果と大きく違うかもしれない。このような差異をもたらすか、または促進する可能性のある要因には、以下の決定された要因および本年度報告の他の部分が“リスク要因”で議論される要因が含まれるが、これらに限定されない。
別の説明がない限り、言及された“合併会社”、“ロケット”、“会社”、“私たち”は、いずれもロケット製薬会社(前身はイノク製薬会社)およびその子会社を意味する。用語“Rocket Ltd”とは、Rocket PharmPharmticals,Inc.の完全子会社ローマと合併する前の私有するRocket PharmPharmticals株式会社を意味する。用語“Inote k”
は、逆合併前のInote k製薬会社およびその子会社を意味する。会計目的については、米国公認会計原則によると、逆合併は“逆買収”とされ、Rocket Ltdは会計上の買収側とされている。そのため、別の説明や文意が別に指摘されている以外、本年度報告に含まれる歴史財務情報はすべてロケット有限会社の逆合併前の財務情報である。
経営陣の財務状況と経営結果の議論と分析(“MD&A”)は、ロケット財務諸表の読者に管理の観点からの記述を提供することを目的としている。私たちの経営陣の討論と分析は、当社の合併財務諸表と本10-K年度報告書の他の部分に添付されている補足として、読者が私たちの運営結果と財務状況を理解するのを助け、私たちの合併財務諸表と付記を結合して読むことを目的としています。
序言:序言
著者らは臨床段階、多プラットフォームの生物技術会社であり、第一種、唯一と同類の中で最も良い遺伝子療法の開発に専念し、直接の標的作用機序と明確な臨床終点を有し、希と壊滅性疾患に応用している。3つの臨床段階の体外レンチウイルスベクター(“LV”)計画があります。これらの方案はファンコニ貧血(“FA”)を含み、これは1種の骨髄中の遺伝欠陥であり、血細胞の産生を減少或いは欠陥血細胞の産生を促進する;白血球粘着欠陥-I(“LAD-I”)、1種の免疫系機能障害を招く遺伝性疾患である;及びピルビン酸キナーゼ欠乏症(“PKD”)は1種の稀な赤血球常染色体劣性遺伝性疾患であり、慢性非球形溶血性貧血を招く。このうち、第2段階FA計画および第1段階/第2段階LAD−I計画の両方が米国に登録されている可能性がある。ヨーロッパ(“EU”)ですあまり一般的でないFAサブタイプCおよびGに対する遺伝子治療計画の追加作業が行われている。米国でもDanon病の臨床生体段階腺関連ウイルス(“AAV”)計画があり,Danon病は多臓器リソソーム関連疾患であり,心不全による早期死亡をきたす。ダノン計画は現在進行中の第一段階試験にある。また,不整脈源性心筋症(“PKP 2−ACM”)に対するAAVベクター計画もあり,PKP 2−ACMは遺伝性心疾患であり,心筋質量の進行性低下,右室の重篤な拡張,発育不良,心筋線維脂肪代替および不整脈や突然死の高い傾向を特徴としている。私たちがRenovacorを買収したため, われわれは現在,AAV 9による組換え遺伝子療法によりBAG 3拡張型心筋症(DCM)の進展を緩和または阻止することが可能であり,DCMは最もよく見られる心筋症形式であり,心壁が徐々に薄くなり,心腔が拡大し,ポンプ血ができないことが特徴である。印税許可協定によると、私たちはこれらのすべての候補製品に対して世界の商業化と開発権を持っている。
64
カタログ表
2021年12月からロケット賛助を行わないことが決定したRP−L 401の臨床評価は,学術革新者に返還された。遺伝子治療はこのような疾患を有する患者に有益である可能性があると信じているが、我々は、納得できる臨床データおよびこれらの児童および若年性深刻な障害の治療進展の潜在力に基づいて、RP−A 601、RP−A 501、RP−L 102、RP−L 201およびRP−L 301の進展に利用可能なリソースを集中させることを選択している。
最新の発展動向
市場に計画を提供する
2022年2月28日、Cowen and Company LLC(“Cowen”)と市場発売計画の販売協定を締結し、この計画により、時々私たちの販売代理であるCowenを通じて株を提供し、販売することができます。販売契約に基づいて提供および販売される株式は、S-3テーブルにおける当社の棚登録宣言(文書番号333-253756)に基づいて提供および販売される。販売協定に基づき、2022年2月28日に米国証券取引委員会に株式要約及び売却に関する目論見補足文書を提出した。吾らは、販売契約に基づいて株式を売却して得られた毛収入の3.0%の現金手数料をCowenに支払い、Cowenに慣用的な補償及び出資権を提供し、Cowenの販売協定に関する若干の支出を返済することに同意した。通り抜ける
December 31, 2022,市場発行計画によると、私たちはすでに330万株の普通株を売却し、総収益は4800万ドル、140万ドルを引いた手数料の純収益は4660万ドルである。
Renovacorを買収する
2022年9月19日、当社はデラウェア州のある会社Renovacorと合併協定と計画(“合併合意”)を締結し、これにより当社はRenovacor(“Renovacor
買収事項”)を買収した。Renovacorの買収は12月1日に完了した2022年です合併協議の条項と条件に基づいて、Renovacor買収の発効日直前に発行された1株当たり額面0.0001ドルのRenovacor普通株はすでにログアウトし、合併協定に記載されている交換式に基づいて決定された0.1763株(“交換比率”)当社の普通株完納金及び免税普通株1株当たり0.01ドルの権利に変換した。♪the the the2022年12月31日までの年間で、3,391,976株のRenovacor買収に関する普通株が発行され、約320万ドルの買収関連コストが発生した。
公開発行する
2022年10月6日、782万株の普通株の公開発行を完了し、公開発行価格は1株当たり14.75ドル。今回の公募株によるRocketの総収益は約1.153億ドルで、720万ドルの発売コスト、手数料、法律、その他の費用を差し引いた純収益は1.081億ドルだった。
財務概要
設立以来、著者らはほとんどの資源を会社の組織と人員の配備、業務計画、資金調達、候補製品の買収或いは発見と関連知的財産権の保護、候補製品の発見、研究開発活動及び潜在的な商業化を計画する。私たちは販売を許可された製品もなく、製品販売から何の収入も得ていない。設立から2022年12月31日まで、株式と転換可能な債券融資により投資家から約8億356億ドルの現金純収益を調達し、経営活動に資金を提供した。
収入.収入
今まで、私たちは製品販売を含むいかなる収入も得ていません。近い将来、製品販売から何の収入も生じないと予想しています。候補製品の開発に成功し、規制部門の承認を得たり、第三者と許可を得たりすれば、将来的に製品販売から収入を得ることができるかもしれない。
研究と開発費
私たちの研究開発計画費用には主に私たちの候補製品を開発するための外部コストが含まれています。これらの費用には
| • |
技術開発、臨床前、臨床活動を含む研究開発活動を展開する研究機関との合意に基づいて発生する費用
|
| • |
契約製造業者への費用および内部製造プロセスで使用される製造投入コストを含む、プロセス開発、臨床前および臨床材料生産に関連する費用
|
| • |
プロセス開発と規制活動を支援するコンサルタント;
|
| • |
私たちの候補製品の組み合わせの権利の開発と商業化の許可に関連する費用。
|
65
カタログ表
我々は,計画活動と一致した契約支払いスケジュール,すでに作業が発生した伝票,および第三者が発生したコスト
に対応するマイルストーンから外部開発コストを確認する.将来的に受信された研究開発活動のための貨物またはサービスの払い戻し不可能な前払いは前払い費用として記録される。
著者らは候補製品の直接研究開発費用を計画通りに追跡し、主に外部コスト、例えば著者らの臨床前研究、技術開発、製造と臨床開発活動に関連する研究協力と第三者製造プロトコル
を含む。私たちが計画通りに行った直接研究開発費にはライセンス契約による費用も含まれています。我々の人員,非計画,および未分配計画支出には,我々内部の研究開発組織が行っている活動に関するコストが含まれており,通常複数の計画に利益を与える.これらのコストは候補製品ごとに個別に分配されたものではなく、主に:
| • |
私たちの研究開発活動に従事している科学者の給料と人員に関連する費用は、福祉、出張、株式報酬を含む
|
| • |
施設賃貸料と修理費、減価償却費などの施設やその他の費用
|
| • |
内部研究開発活動のための実験室用品と設備。
|
著者らの直接研究開発費用は主に外部コスト、例えば著者らの臨床研究に関連する研究者、コンサルタント、実験室とCROに支払う費用、及び臨床研究材料の取得と製造に関連するコストを含む。私たちは特定の計画に直接関連した賃金と福祉費用を分配する。私たちは、関係者に関連する自由支配可能ボーナスまたは株式ベースの報酬コスト、私たちの一般的な発見プラットフォームの改善に関連するコスト、減価償却、または複数の開発されているプロジェクトにまたがる他の間接コストを割り当てないので、これらのコストは、個別に他の研究開発費に分類される。
次の表に,2022年12月31日,2021年12月31日と2020年12月31日までの年度内に,計画および我々の候補製品費のタイプと性質追跡の研究開発費を示す。
|
12月31日までの年度
|
||||||||||||
|
2022
|
2021
|
2020
|
||||||||||
|
直接費用:
|
||||||||||||
|
達農病(AAV)RP-A 501
|
$
|
28,524
|
$
|
15,804
|
$
|
18,459
|
||||||
|
血小板親和性素−2による不整脈心筋症(AAV)RP−A 601
|
11,724
|
1,071
|
122
|
|||||||||
|
白血球接着欠乏症(LV)RP−L 201
|
20,617
|
24,222
|
5,531
|
|||||||||
|
ファンコニ貧血(LV)RP-L 102
|
23,917
|
15,453
|
15,015
|
|||||||||
|
ピルビン酸キナーゼ欠損症(LV)RP−L 301
|
2,744
|
4,206
|
4,990
|
|||||||||
|
乳幼児悪性骨化症(LV)RP−L 401 (1)
|
271
|
2,236
|
2,057
|
|||||||||
|
他の候補製品
|
3,580
|
3,504
|
1,199
|
|||||||||
|
直接費用総額
|
91,377
|
66,496
|
47,373
|
|||||||||
|
未分配費用
|
||||||||||||
|
従業員報酬
|
$
|
32,274
|
$
|
20,780
|
$
|
14,137
|
||||||
|
発行権証に関する非現金研究開発費
|
-
|
12,781
|
26,562
|
|||||||||
|
株に基づく報酬費用
|
12,465
|
11,954
|
7,121
|
|||||||||
|
減価償却および償却費用
|
4,037
|
5,130
|
2,770
|
|||||||||
|
研究室と関連費用
|
17,405
|
3,359
|
4,240
|
|||||||||
|
専門費
|
3,601
|
1,797
|
1,443
|
|||||||||
|
その他の費用
|
4,411
|
3,179
|
1,792
|
|||||||||
|
他の研究·開発費総額
|
74,193
|
58,980
|
58,065
|
|||||||||
|
研究と開発費用総額
|
$
|
165,570
|
$
|
125,476
|
$
|
105,438
|
||||||
(1)2021年12月から、ロケットスポンサーのRP-L 401の臨床評価を行わないことが決定され、学術革新者に戻る予定である。研究完了費用
は2022年に発生した。
私たちは現在または未来の候補製品の臨床研究を完了する持続時間とコストを決定することができず、私たちがいつ、いつ、あるいはどの程度商業化と販売のいずれの規制部門の許可を得た候補製品から収入を得るかどうかを決定することもできない。私たちは私たちのどんな候補製品も規制部門の承認を得ることに決して成功しないかもしれない。候補製品の臨床研究と開発の持続時間、コスト、および時間は、様々な要素に依存するであろう
| • |
私たちが行っている臨床研究や他の研究開発活動の範囲、進捗、費用
|
| • |
将来の臨床研究の結果
|
| • |
臨床研究の生存率の不確実性
|
66
カタログ表
| • |
規制の承認基準を変更し
|
| • |
すべての規制承認の時間と受け入れ。
|
予測可能な未来には,我々の計画が後期開発段階に入り,より多くの臨床試験が行われるにつれて,製造への投資を含めて開発候補製品に関する研究開発活動に投資し続け,研究開発費が増加することが予想される。必要な臨床研究を行い、監督部門の許可を得る過程は高価で時間がかかり、候補製品の開発成功も非常に不確定である。そのため,研究開発プロジェクトの継続時間や完成コストを決定することもできず,いつ,どの程度候補製品の商業化や販売から収入を得るかを決定することもできない.
私たちの将来の研究開発費用は私たちの候補製品の臨床成功と、これらの候補製品の商業潜在力の持続的な評価に依存するだろう。また,どの候補製品が将来の協力の影響を受ける可能性があるか,いつこのような手配が確保されるか(あれば),そのような手配が我々の開発計画や資本要求にどの程度影響するかは予測できない。
予測可能な未来には,候補製品の開発完了を求めるにつれて,我々の研究開発費が増加することが予想される.
私たちの候補製品の成功と商業化は大きな不確実性を持っている。これは、製品開発と商業化に関連する多くのリスクと不確実性が原因であり、以下のような不確実性を含む
| • |
私たちの臨床試験や他の研究開発活動の範囲、進捗、結果、コスト
|
| • |
代替療法と比較して、私たちの候補製品の治療効果と潜在的利点は、任意の標準的な看護を含む
|
| • |
私たちの候補製品の市場受容度は
|
| • |
特許請求の範囲および他の知的財産権の取得、維持、擁護、および実行;
|
| • |
重大で変化し続ける政府の規制;
|
| • |
すべての上場承認の時間、領収書、そして条項。
|
我々が開発可能な候補製品の開発に対して,これらの変数のいずれかの結果の変化は,我々の候補製品の開発に関するコストと時間の大きな変化を意味する可能性がある.例えば、FDAまたは他の規制機関が、現在開発可能な任意の候補製品の臨床開発を完了するために行われている試験または他の試験ではなく、臨床試験または他の試験を要求する場合、または任意の臨床試験の登録に重大な遅延がある場合、候補製品の臨床開発を達成するために多くの追加の財源と時間を要する可能性がある。
一般と行政費用
一般と行政費用は主に従業員の給料と関連福祉コストを含み、従業員の商業、行政、運営、財務、法律、業務発展と人的資源機能における従業員の株式給与と出張費用を含む。さらに、他の重大な一般および行政費用には、法律、相談、投資家および公共関係、監査および税務サービスの専門費用、および一般および行政活動で使用される施設のレンタル料およびメンテナンス、保険およびその他の用品の他の費用が含まれる。予想される将来、従業員数の増加により、一般的かつ管理費が増加し、候補製品の持続的な発展と商業運営への発展を支援することが予想される。私たちはまた、ますます複雑になっている上場企業として運営を続けるにつれて、より多くの会計、監査、法律、規制、コンプライアンス、取締役と役員の保険コスト、投資家と広報費用を発生させ続けると予想しています。
利子支出
2022年の利息支出は、ニュージャージー州クランベリー工場に対する私たちの融資リース義務と関連がある。2021年8月2日に普通株に変換された2021年転換手形(定義は後述)、2021年4月に普通株に償還され変換された2022年転換手形(定義は後述)、およびニュージャージー州クランベリー施設に対する融資リース義務に関する2021年と2020年の利息支出。
利子収入
利息収入は投資と現金等価物から稼いだ利息と関係がある。
67
カタログ表
経営成果
2022年と2021年12月31日終了年度比較
次の表は、2022年12月31日と2021年12月31日までの年間経営実績をまとめたものです
|
|
12月31日までの年度
|
|||||||||||
|
2022
|
2021
|
変わる
|
||||||||||
|
(単位:千)
|
||||||||||||
|
運営費用:
|
||||||||||||
|
研究開発
|
$
|
165,570
|
$
|
125,476
|
$
|
40,094
|
||||||
|
一般と行政
|
58,773
|
41,772
|
17,001
|
|||||||||
|
総運営費
|
224,343
|
167,248
|
57,095
|
|||||||||
|
運営損失
|
(224,343
|
)
|
(167,248
|
)
|
(57,095
|
)
|
||||||
|
インセンティブを開発する
|
500
|
1,000
|
(500
|
)
|
||||||||
|
利子支出
|
(1,862
|
)
|
(2,977
|
)
|
1,115
|
|||||||
|
利息とその他の収入,純額
|
3,889
|
3,068
|
821
|
|||||||||
|
投資割増純額
|
(47
|
)
|
(2,912
|
)
|
2,865
|
|||||||
|
その他の収入を合計して純額
|
2,480
|
(1,821
|
)
|
4,301
|
||||||||
|
純損失
|
$
|
(221,863
|
)
|
$
|
(169,069
|
)
|
$
|
(52,794
|
)
|
|||
研究と開発費
2021年12月31日までの年度と比較して、2022年12月31日までの年間研究開発支出は4010万ドル増加し、1兆566億ドルに達した。研究開発費の増加は主に製造と開発コストが2630万ドル増加し、実験室用品が660万ドル増加し、研究開発者が1150万ドル増加し、直接材料が360万ドル増加したことと、コンサルティングと専門費用が270万ドル増加したためである。注目された増加は、2021年12月31日までの年度内に普通株株を購入する引受権証に関する費用が減少したため、許可料1290万ドルの減少によって相殺された。
一般と行政費用
2021年12月31日までの年度と比較して、2022年12月31日までの年間G&A支出は1700万ドル増加し、5880万ドルに達した。G&A費用の増加は主に商業準備費用の増加によるものであるそれは商業戦略、医療事務、市場開発、そして価格分析を含む490万ドルのうち、報酬と福祉は440万ドルで、G&A従業員数と320万ドルの買収関連費用が増加したためだ。
その他の収入,純額
2022年12月31日までの1年間で、他の純収入純額は2021年12月31日までの年間より430万ドル増加し、250万ドルに達した。この変化は、主に2022年4月に全額償還された2022年転換手形と2021年8月に転換された2021年転換手形に関する利息支出が110万ドル減少し、利息やその他の収入純額が80万ドル増加し、投資償却額が290万ドル増加したためだ。
2021年12月31日までと2020年12月31日までの年度比較
次の表は、2021年12月31日現在と2020年12月31日までの年度の経営成果をまとめたものである
|
12月31日までの年度
|
||||||||||||
|
2021
|
2020
|
変わる
|
||||||||||
|
(単位:千)
|
||||||||||||
|
運営費用:
|
||||||||||||
|
研究開発
|
$
|
125,476
|
$
|
105,438
|
$
|
20,038
|
||||||
|
一般と行政
|
41,772
|
28,865
|
12,907
|
|||||||||
|
総運営費
|
167,248
|
134,303
|
32,945
|
|||||||||
|
運営損失
|
(167,248
|
)
|
(134,303
|
)
|
(32,945
|
)
|
||||||
|
インセンティブを開発する
|
1,000
|
-
|
1,000
|
|||||||||
|
利子支出
|
(2,977
|
)
|
(6,967
|
)
|
3,990
|
|||||||
|
利息とその他の収入,純額
|
3,068
|
2,150
|
918
|
|||||||||
|
投資割増純額
|
(2,912
|
)
|
(580
|
)
|
(2,332
|
)
|
||||||
|
その他の費用の合計
|
(1,821
|
)
|
(5,397
|
)
|
3,576
|
|||||||
|
純損失
|
$
|
(169,069
|
)
|
$
|
(139,700
|
)
|
$
|
(29,369
|
)
|
|||
68
カタログ表
研究と開発費
2020年12月31日までの年度と比較して、2021年12月31日までの年間研究開発支出は2000万ドル増加し、1兆255億ドルに達した。増加の主な原因は、製造と開発コストが1,480万ドル増加し、研究開発者の増加による給与と福祉が660万ドル増加し、非現金株式報酬支出が480万ドル増加し、実験室用品と事務費用が290万ドル増加し、減価償却と償却が190万ドル増加したが、2020年12月と2021年8月と2021年12月に株式証明書を発行したことによる新しい研究協定非現金支出は1380万ドル減少したことである。
一般と行政費用
2020年12月31日までの年度と比較して、2021年12月31日までの年間G&A支出は1290万ドル増加し、4180万ドルに達した。M&A費用の増加は、主に非現金株の報酬支出が580万ドル増加し、給与·福祉が180万ドル増加し、給与·福祉が180万ドル増加し、事務·行政コストが250万ドル増加し、保険コストがbr増加し、商業準備費が220万ドル増加したが、12月31日までの年間記録された債務転換費用の減少によって相殺された。2021年に転換可能な手形が2020年2月に再融資されるため、2020年には200万ドルが発生する。2021年12月31日までの年度は債務転換費用を記録していない。
その他の費用、純額
2021年12月31日までの1年間、他の費用の純額は180万ドルだったが、2020年12月31日までの年間純額は540万ドルだった。この変化は,主に2021年4月に全額償還された2022年転換手形と2021年8月に転換された2021年変換可能手形に関する利息支出の減少と,我々の投資に関する付加価値収入が230万ドル減少したためであり,2021年12月31日現在の年間金利が2020年12月31日までの年度を下回っているためである。
流動性と資本資源
設立以来、私たちは何の収入も発生せず、損失が発生した。同社の運営は、候補薬物開発の不確実性、技術的不確実性、特許および独自の権利に関する不確実性、商業製造経験のない、マーケティングまたは販売能力または経験、キーパーソンへの依存、政府規定の遵守、および追加融資を必要とするいくつかのリスクおよび不確実性の影響を受ける。現在開発中の候補薬物は大量の追加研究開発が必要であり、広範な臨床前と臨床テスト及び監督部門の許可を含め、それから商業化することができる。このような努力は多くの追加資本、十分な人員インフラ、そして広範囲なコンプライアンス報告能力を必要とする。
私たちの候補薬は開発と臨床段階にある。私たちの研究開発が成功することは保証されず、私たちの知的財産権は十分に保護され、brによって開発されたいかなる製品も必要な政府の承認を得ることができ、あるいはいかなる承認された製品も商業可能性があるだろう。私たちの製品開発が成功しても、いつ(もしあれば)製品販売から相当な収入を得ることができるかどうかはわかりません。私たちの経営環境は技術の急速な変化と製薬やバイオテクノロジー会社からの激しい競争です。
我々の総合財務諸表は正常業務過程における業務連続性、資産現金化と負債返済状況に基づいて作成された。発足以来、ロケットの毎年の運営は純損失とマイナスキャッシュフローになっている。ロケットは2022年、2021年、2020年12月31日までの会計年度にそれぞれ2億219億ドル、1億691億ドル、1億397億ドルの純損失を出した。2022年12月31日現在,我々の運営キャッシュフローは負であり,累計赤字は7.138億ドルである。2022年12月31日現在、私たちは3億997億ドルの現金、現金等価物、投資を持っている。私たちはこのような資源
が2024年までに私たちの運営費用と資本支出要求を満たすのに十分になると予想している。私たちは主に株式を売却することで私たちの運営に資金を提供する。
CIRMは2019年4月30日、LVベースのRP−L 201遺伝子療法の臨床開発を支援するために、CLIN 2に応じて660万ドルまでの資金を同社に奨励した。この寄付金の収益は、カリフォルニア大学ロサンゼルス校(UCLA)美泰児童病院米国臨床サイトに登録された1/2期患者の臨床試験費用および医薬製品の生産を支援するために使用され、この研究は、カリフォルニア大学ロサンゼルス校(UCLA)微生物学、免疫学および分子遺伝学、小児科(血液学/腫瘍学)、分子および医学薬理学教授、ELIおよびEDY再生医学および幹細胞研究センターのドナルド·コーエンによって指導される。2019年には、会社はCIRMから最初の2つの合計120万ドルの贈与を得て、研究開発費の相殺とした。2020年には,会社はより多くのCIRMマイルストーンを達成し,追加110万ドル
マイルストーンを獲得し,2020年の研究開発費の減少と記す。同社は2021年1月と4月にそれぞれ110万ドルと100万ドルの追加マイルストーン支払いを受けた。2022年3月にはCIRMの次のマイルストーンに達し、2022年4月5日に90万ドルのマイルストーン支払いを受けた。2022年12月31日現在、これ以上のマイルストーンは実現されていない。
69
カタログ表
長期的に見れば、私たちの将来の生存能力は、経営活動から現金を得る能力や、追加資本を調達して私たちの運営に資金を提供する能力にかかっている。もし私たちが株式証券を発行することでより多くの資金を調達すれば、私たちの株主は希釈されるだろう。私たちが将来参加する任意の債務融資は、私たちの留置権または追加のbr債務の発生を制限すること、配当金の支払い、私たちの普通株の買い戻し、特定の投資、および特定の合併、合併、または資産売却取引に従事する能力を含む、私たちの業務を制限する追加契約を適用するかもしれない。私たちが調達した任意の債務融資または追加株式には、私たちまたは私たちの株主に不利な条項が含まれているかもしれない。私たちが必要な時に資金を調達できなかったことは、私たちの財務状況や業務戦略を実行する能力に悪影響を及ぼす可能性がある。
市場に計画を提供する
2022年2月28日、Cowen and Company LLC(“Cowen”)と市場発売計画の販売協定を締結し、この計画により、時々私たちの販売代理であるCowenを通じて株を提供し、販売することができます。販売契約に基づいて提供および販売される株式は、S-3テーブルにおける当社の棚登録宣言(文書番号333-253756)に基づいて提供および販売される。我々は2022年2月28日に米国証券取引委員会に株式発売に関する目論見書補充書類を提出し、販売契約に基づいて株式売却所得の3.0%に相当する現金手数料をコーエンに支払うことに同意した。私たちはまた、Cowenに慣用的な賠償と貢献権を提供し、Cowenの販売協定に関連するいくつかの費用を返済することに同意する。通り抜ける December 31,
2022,市場発行計画によると、私たちはすでに330万株の普通株を売却し、総収益は4800万ドル、140万ドルを引いた手数料の純収益は4660万ドルである。
契約義務
通常の業務過程で、私たちは未来に支払う義務があるように契約と約束を締結します。当社の所得税及びリース手配に関する責任に関する資料は、当社合併財務諸表の“付記13.所得税”及び“付記14.負担及び又は有事項”に記載されており、“第8項.財務諸表及び補足データ”に掲載されている
私たちは私たちの財務状況や経営結果に重大または合理的に重大な影響を及ぼす可能性のある表外手配は何もありません。
キャッシュフロー
次の表は私たちの毎期のキャッシュフローをまとめています
|
12月31日までの年度
|
||||||||||||
|
2022
|
2021
|
2020
|
||||||||||
|
経営活動のための現金純額
|
$
|
(178,142
|
)
|
$
|
(121,163
|
)
|
$
|
(74,640
|
)
|
|||
|
投資活動が提供する現金純額
|
(69,326
|
)
|
18,853
|
(96,591
|
)
|
|||||||
|
融資活動が提供する現金純額
|
155,288
|
37,681
|
282,989
|
|||||||||
|
現金、現金等価物、および制限的現金純増加
|
$
|
(92,180
|
)
|
$
|
(64,629
|
)
|
$
|
111,758
|
||||
経営活動に使われている現金純額
2022年12月31日までの1年間、経営活動は1億781億ドルの現金を使用したが、これは主に私たちの純損失2.219億ドルと私たちの運営資産と負債の純変化610万ドルが原因であるが、3760万ドルの非現金費用純額によって部分的に相殺され、3100万ドルの株式給与支出と630万ドルの減価償却と償却費用が含まれている。2022年12月31日までの年度、我々の営業資産と負債の変化には、970万ドルの売掛金と売掛金の増加、360万ドルの前払い費用やその他の資産の増加が含まれている。
2021年12月31日までの年間で、経営活動は1.212億ドルの現金を使用しており、主な原因は、私たちの純損失1.691億ドルと、私たちの運営資産と負債の純変化340万ドルですが、5130万ドルの非現金費用純額によって部分的に相殺されており、株式承認証の発行に関連する費用1280万ドル、株式ベースの給与支出2920万ドル、投資プレミアムの償却290万ドル、減価償却と償却費用540万ドルが含まれています。2021年12月31日までの年度、我々の営業資産と負債の変化には、480万ドルの売掛金と売掛金の減少、130万ドルの前払い費用およびその他の資産の減少が含まれている。
2020年12月31日までの年間で、経営活動は7,460万ドルの現金を使用しており、主な原因は、私たちの純損失1.397億ドルと営業資産と負債の純変化990万ドルだったが、5510万ドルの非現金費用純額によって部分的に相殺され、株式承認証の発行に関連する費用2660万ドル、株式ベースの給与支出1860万ドル、転換手形の割引額280万ドルの増加、減価償却·償却費用110万ドルが含まれている。2020年12月31日までの年度の運営資産および負債変動には,融資リース負債が50万ドル増加し,売掛金および売掛金が1,100万ドル増加すること,前払い費用およびその他の資産が150万ドル増加することが含まれる。
70
カタログ表
投資活動が提供する現金純額
2022年12月31日現在の投資活動のための現金純額は6,930万ドルであり,投資満期日からの収益2.729億ドルおよびRenovacorの買収収益br}4,270万ドルを含むが,購入投資3.763億ドル,購入物件および設備840万ドルおよび購入使用権資産30万ドルに相殺されている。
2021年12月31日までの年間で、投資活動が提供する現金純額は1,890万ドルで、2.724億ドルの投資満期日収益を含み、2.459億ドルの投資の購入と760万ドルの不動産·設備の購入によって相殺された。
二零年十二月三十一日現在の投資活動のための現金純額は9,660万ドルであり、投資満期日に得られた1.418億ドルを含むが、購入投資2.093億ドル、購入物件及び設備2,060万ドル及び使用権資産取得のために支払われた850万ドルが相殺される。
融資活動が提供する現金純額
2022年12月31日までの1年間に、融資活動が提供する現金純額は1.553億ドルで、主に2022年10月の公開株式に関する1.081億ドルの収益と、市場での株式発行計画に関する普通株発行4660万ドルを含む。
2021年12月31日までの年間で、融資活動が提供する現金純額は3770万ドルで、2021年8月の私募に関する普通株の発行による1130万ドルと、株式オプション行使による普通株の発行による2640万ドルが含まれている。
2020年12月31日までの年間で、融資活動が提供する現金純額は2億83億ドルで、主に普通株の発行で得られた2兆808億ドルを含む。2020年12月14日、引受業者が1株56.00ドルの公開発行価格で696,428株の普通株を承認する選択権を含む5,339,286株の普通株の公開発行を完了した。ロケットが2020年12月の公募株から得た純収益は約2兆808億ドル。
重要な会計政策と試算
私たちの経営陣の財務状況と経営結果の討論と分析は私たちの連結財務諸表に基づいています。これらの報告書は
は米国公認の会計原則(“米国公認会計原則”)に適合している。これらの総合財務諸表を作成する際には、財務諸表の日付の報告済み資産および負債額、または有資産および負債の開示、および報告期間内に発生した報告済み費用に影響を与える推定および仮定を行う必要がある。私たちの見積もりは、私たちの歴史的経験と、当時の状況で合理的だと思う様々な他の要素に基づいており、これらの要素の結果は、資産や負債の帳簿価値を判断する基礎を構成しており、これらの資産や負債は他の出所からはあまり明らかではないように見える。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。環境、事実、そして経験の変化に伴い、私たちは定期的に私たちの推定を検討する。予算における重大な改訂の影響は見積もり変更の日から財務諸表に反映されることが予想される。
我々の重要会計政策の説明については、本報告の他の部分の総合財務諸表付記の“付記3-重要会計政策概要”を参照されたい。私たちは最も重要な会計政策は、私たちの計算すべき研究と開発費用、株式ベースの報酬、商業権、無形資産に関する政策だと考えている。
商誉
企業合併は買収法の下で計上される。買収の総コストは,買収日
それぞれの推定公正価値に応じて関連識別可能な純資産に分配される。買収された資産と負担する負債の公正価値を決定するには管理職の判断が必要であり、通常は将来の現金流入と流出、割引率、資産寿命、市場倍数などの項目に関する仮定を含む重大な見積もりと仮説の使用に関連する。買収した資産と負担した負債はその推定公正価値に基づいて入金される。買収価格は買収純資産の見積もり公正価値の一部を超えて営業権に計上されている。12月31日から、毎年営業権の減価テストが行われ、またはより頻繁にイベントまたは環境変化が資産が減値可能であることが示された場合にテストが行われる。このようなイベントまたは状況の例は、法律またはビジネス環境の重大な不利な変化、不利な規制行動、または意外な競争を含むが、これらに限定されない。当社には分部と報告単位がありますので、総合レベルでの減値商誉
を審査します。営業権をテストする時、会社はまず営業権を持つ報告単位を評価する定性的要素を選択することができる。定性評価は報告単位の公正価値或いは帳簿価値に影響する関連イベントと状況の全体状況を評価することを含む。これらの事件と状況はマクロ経済状況、業界と競争環境状況、全体財務表現、報告単位の特定事件と市場考慮要素を含む。当社は報告先の最近の推定値も考慮している, 最新の公正価値推定と帳簿との間の差額の大きさ、ならびに正面および不利なイベントおよび状況、ならびに決定された各イベントおよび状況が報告単位の公正価値とその帳簿価値との比較に及ぼす影響の程度を含む。定性的評価で報告単位の公正価値が帳簿価値を超える可能性が高いと結論すれば,その報告単位のさらなるテストは行われない。
71
カタログ表
当社はその営業権に対して定性評価を行い、報告単位の公正価値が報告単位の帳簿価値を超える可能性が高いことを確定した。そのため、当社は2022年まで、2021年および2020年12月31日までに営業権減額はないことを決定しました。
無形資産
進行中の研究開発(“IPR&D”)プロジェクトに関する無形資産は、関連研究開発事業が完了または放棄されるまで無期限とされている。開発が完了すれば、通常、規制機関の許可を得て製品を販売する際に、関連資産は有限寿命とみなされ、その後、その時点での
のそれぞれの推定使用寿命に基づいて償却される。公正価値が低下すると判断された知的財産権研究開発無形資産は下方調整され、合併
経営報告書における研究開発費で費用が確認される。これらの知的財産権研究開発無形資産は少なくとも毎年1回テストし、あるいは事件が発生した時、研究開発活動の進展、資産予想発展変化、監督管理環境と未来の商業市場変化などを含む指標に基づいて、潜在的な減値が存在する可能性があることを表明した。
研究と開発費用を計算すべきである
連結財務諸表作成過程の一部として、我々が計上すべき研究·開発費用を見積もる必要がある。この流れは、未完了契約および調達注文を審査し、私たちの担当者とコミュニケーションして、私たちを代表して実行されたサービスを決定することと、実際のコストを他の方法で通知していない場合に、実行されたサービスレベルおよびサービスによって生じる関連コストを推定することとを含む。私たちのほとんどのサービスプロバイダは予定のスケジュールに従って、あるいは契約マイルストーンに達した時に私たちに借金の領収書を発行しますが、一部のサービスプロバイダは前払い金を必要とします。我々は、当時知られていた事実と状況に基づいて、連結財務諸表において、貸借対照表毎の日付までの課税費用を推定する。予想される研究および開発費用の例には、以下の項目に支払われる費用が含まれる
| • |
私たちを代表して研究開発サービスを実行することに関連するCRO
|
| • |
臨床試験に関連する研究場所や他の提供者
|
| • |
非臨床開発活動に関連するサプライヤー;
|
| • |
臨床用品の製品製造·開発·流通に関するサプライヤー。
|
我々は,我々を代表して非臨床研究や臨床試験を行って管理している複数のCROと締結した契約に基づいて,我々が受け取ったサービスと費用を見積もり,臨床試験に関する費用を決定した。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われる費用は、提供されたサービスレベル
を超え、それによって、臨床費用の前払いをもたらす可能性がある。その中のいくつかの契約下の支払いは患者の成功登録と臨床試験マイルストーンの完成などの要素に依存する。サービス料金を計算する際には,サービスを提供する時間帯,患者登録人数,アクティブなサイト数,および期間ごとにかかる作業レベルを見積もる.サービス実行の実際の時間または作業レベルが私たちの推定と異なる場合、私たちはそれに応じて計算すべきまたは前払いを調整します。実際に発生した金額と実質的に差がないと予想されるにもかかわらず,実行されたサービスの実態や時間に対する実行サービスの状態や時間の理解が異なる可能性があり,任意の特定の時期に我々が報告する費用が高すぎるか低すぎる可能性がある.
株に基づく報酬
私たちは通常、株式オプションと制限株式単位の形で、従業員と非従業員に株式ベースの報酬を支給する。我々は、FASB ASCテーマ
718、報酬-株式報酬(“ASC 718”)に基づいて、株式ベースの報酬報酬を計算した。ASC 718は、株式オプションおよび制限株式単位の付与、および既存の株式オプションの修正を含む株式ベースのすべての支払いを要求し、その公正価値に基づいて総合経営報告書および全面損失で確認しなければならない。私たちは付与日付与持分ツールの公正価値に基づいて、獲得した従業員と非従業員サービスの補償費用を測定した。従業員および非従業員が報酬と引き換えにサービスを提供することを要求されている間、このコストは直線的に確認されている。日オプションを付与する公正価値は,期待変動率や期待期限などの重要な仮定に基づいて,Black-Scholesオプション定価モデルを用いて計算される.これらの仮定は主に会社株の取引価格、歴史データ、同業者会社データ及び未来の傾向と要素の判断に基づいている。
我々の運営報告書において株式に基づく報酬費用を分類する方式は,受賞者の賃金コストやサービスを分類する方式や受賞者のサービスを分類する方式と同様である.同社は付与された賠償の少なくとも一部の補償費用を確認した。没収は発生状況に応じて計算されます。
72
カタログ表
最近の会計公告
最近、当社または総合財務諸表に重大な影響を与えることが予想される会計声明は何もありません。
| 第七A項。 |
市場リスクの定量的·定性的開示について
|
適用されない
| 第八項です。 |
財務諸表と補足データ
|
本プロジェクト8に要求される財務諸表は、本年度報告項目15に記載されている。
| 第九項です。 |
会計と財務情報開示の変更と相違
|
ない。
| 第9条。 |
制御とプログラム
|
情報開示制御とプログラムの評価
本年度報告で述べた期間が終了した時点で、我々の経営陣は、我々の主要幹部と我々の主要財務·会計官の参加の下で、我々の開示制御及び手続の有効性を評価した。我々の2022年12月31日までの開示制御及び手続の評価によると、我々の最高経営責任者及び臨時最高経営責任者及び会計官は、この日までに、我々の開示制御及び手続は合理的な保証レベルで有効であると結論した。取引法第13 a−1 i)及び第15 I 5(E)条に定義されている“開示制御及び手続”という言葉は、会社が取引法に基づいて提出又は提出された報告において開示を要求する情報が米国証券取引委員会規則及び表で指定された期間内に記録、処理、集計及び報告されることを確実にするための会社の制御及びその他の手続を意味する。開示制御およびプログラムは、取引法に基づいて提出または提出された報告書で開示を要求する情報が蓄積され、必要な開示に関する決定をタイムリーに行うために、我々の最高経営者および一時的な財務および会計官を含む我々の経営陣に伝達されることを保証することを目的としているが、制御およびプログラムに限定されない。管理層
は,どのような制御やプログラムも,どんなに設計や操作が良好であっても,その目標を実現するために合理的な保証を提供することしかできないことを認識しており,我々の管理層は,可能な制御とプログラムの費用対効果関係を評価する際にその判断を適用しなければならない.
経営陣財務報告内部統制年次報告書
私たちの経営陣は財務報告書の十分な内部統制を確立して維持する責任がある。財務報告の内部統制は、取引法に基づいて公布された規則13(A)-15(F)および15(D)-15(F)
において、我々の主要行政者および主要財務官が設計またはその監督の下で我々の取締役会、管理層および他の人員によって実施されるプログラムとして定義され、公認された会計原則に基づいて財務報告の信頼性および外部目的の財務諸表の作成に合理的な保証を提供し、以下の政策および手順を含む
| • |
私たちの資産を合理的、詳細、正確かつ公平に反映した取引および処置の記録を保存することと関連がある
|
| • |
公認された会計原則に従って財務諸表を作成するために必要に応じて取引を記録することを確保するための合理的な保証を提供し、私たちの収入と支出は私たちの経営陣と取締役の許可のみに基づいて行われる
|
| • |
財務諸表に重大な影響を及ぼす可能性のある不正取得、使用、または処分について、財務諸表に重大な影響を与える可能性のある資産の防止またはタイムリーな発見について合理的な保証を提供します。
|
経営陣(主要幹部·財務担当者を含む)の監督·参加の下、テレデビル委員会後援組織委員会(COSO)が発表した“内部統制-総合枠組み(2013)”で確立された財務報告の有効な内部統制基準に基づいて、2022年12月31日現在の財務報告内部統制を評価した。私たちの経営陣の財務報告の内部統制の有効性の評価には、内部統制の設計と運用有効性のテストと評価が含まれています。我々の経営陣の考えでは、COSO 2013の枠組みで確立された基準に基づいて、2022年12月31日まで、財務報告に対して有効な内部統制を維持しています。
2022年12月31日現在、財務報告の内部統制に対する我々の有効性は、独立公認会計士事務所EisnerAmper LLPによって監査されており、その報告
は本報告に含まれている。
73
カタログ表
内部統制の内在的限界
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。したがって、有効と判定されたシステムであっても、財務諸表の作成や列報の面で合理的な保証を提供することしかできない。将来の間にどの有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは
が政策やプログラムを遵守する程度が悪化する可能性がある.
財務報告の内部統制の変化
本報告に記載されている間、私たちは財務報告の内部統制に重大な影響を与えていないか、または合理的に私たちの財務報告の内部統制に大きな影響を与える可能性のある変化を生じていない。
| プロジェクト9 B。 |
その他の情報
|
ない。
| プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示
|
適用されません。
第3部-その他の情報
| 第10項。 |
役員·幹部と会社の管理
|
このプロジェクトに関する資料は,2023年株主総会の依頼書(“依頼書”)に“役員選挙著名人”,“br}”当社行政者に関する資料“,”取締役会及び会社管治に関する資料“のタイトルを以下に掲載し,ここに組み込んで参考にする。依頼書は,本年度報告に含まれる会計年度終了後120日以内に米国証券取引委員会
に提出される。
| 第十一項。 |
役員報酬
|
この項目に関する情報は、依頼書“役員報酬”および“役員報酬”のタイトルの下で述べられ、“株式報酬計画情報”は参照によって本明細書に組み込まれる。依頼書は、本年度報告書に含まれる会計年度終了後120日以内に米国証券取引委員会に提出される。
| 第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項
|
このプロジェクトに関する情報は、委託書“いくつかの利益所有者および管理層の保証所有権”および“持分補償計画情報”のタイトルの下に記載され、ここに組み込まれて参考となる。依頼書は、本年度報告書に含まれる会計年度終了後120日以内に米国証券取引委員会に提出される。
| 十三項。 |
特定の関係や関係者の取引、取締役の独立性
|
この項目に関する情報は、依頼書“関係者との取引”および“取締役会および会社のガバナンスに関する情報”のタイトルで述べられ、引用によって本明細書に組み込まれる。依頼書は、本年度報告書に含まれる会計年度終了後120日以内に米国証券取引委員会に提出される。
| 14項です。 |
チーフ会計士費用とサービス
|
独立会計士事務所はEisnerAmper LLP、ニューヨーク、ニューヨーク、PCAOB監査員IDです.
この項目に関する情報は、委託書“独立公認会計士事務所の任命を承認する”というタイトルの下に記載され、引用により本明細書に組み込まれる。依頼書は、本年度報告書に含まれる会計年度終了後120日以内に米国証券取引委員会に提出される。
74
カタログ表
第4部
| 第十五項。 |
展示品、財務諸表、付表
|
(A)本年度報告のアーカイブの一部として、以下のファイルを保存します
(1)財務諸表:
|
独立公認会計士事務所報告
|
F-2
|
|
2022年と2021年12月31日までの連結貸借対照表
|
F-5
|
|
2022年·2021年·2020年12月31日までの総合業務報告書
|
F-6
|
|
2022年,2021年,2020年12月31日までの総合全面損失表
|
F-7
|
|
2022年まで、2021年、2020年12月31日まで年度株主権益変動表
|
F-8
|
|
2022年、2021年、2020年12月31日までの統合現金フロー表
|
F-9
|
|
連結財務諸表付記
|
F-10
|
(2)財務諸表付表:
これらは、適用されない、不要、または必要な資料が財務諸表またはその付記に列挙されているので、すべての財務諸表の添付表は省略される。
75
カタログ表
(三)展示品:
|
展示品
|
展示品索引
|
|
番号をつける
|
展示品説明
|
|
2.1
|
イノク製薬会社、ロケット製薬株式会社とローマ合併子会社との間の合併再編協定と計画は、2017年9月12日(会社の現在の報告書8-K表の添付ファイル2.1として提出され、2017年9月13日に米国証券取引委員会に提出され、引用により本明細書に組み込まれる)
|
|
2.2
|
ロケット製薬会社、Renovacor社、ゼブラフィッシュ合併子会社とゼブラフィッシュ合併子会社の間で2022年9月19日に調印された合併協定と計画(会社の現在の8-Kレポートの添付ファイル2.1として提出され、2022年9月20日に米国証券取引委員会に提出され、引用により本明細書に組み込まれる)
|
|
3.1
|
ロケット製薬会社の7回目の再登録証明書の改訂は、2015年2月23日から発効した(会社10-K年次報告書の添付ファイルとして3.1を会社に提出し、2015年3月31日に米国証券取引委員会に提出し、引用により本明細書に組み込む)
|
|
3.2
|
7回目の改訂後の登録者登録証明書(逆株式分割)は、2018年1月4日から施行される(会社現在報告8-K表の添付ファイル3.2として提出され、2018年1月5日に米国証券取引委員会に提出され、参照により本明細書に組み込まれる)
|
|
3.3
|
2018年1月4日に施行された7つ目の修正された登録者登録証明書(改称)(会社の現在の報告書8-K表の添付ファイルであるbr}3.2アーカイブは、2018年1月5日に米国証券取引委員会に提出され、参照により本明細書に組み込まれる)
|
|
3.4
|
2018年6月25日に施行された第7回改正登録者登録証明書修正案(会社現在報告8-K表の添付ファイル3.1として提出され、2019年6月25日に米国証券取引委員会に提出され、参照により本明細書に組み込まれる)
|
|
3.5
|
改訂·改訂されたロケット製薬会社規約は、2018年3月29日から施行される(会社登録説明書の添付ファイル3.4として提出され、2018年1月11日に米国証券取引委員会に提出され、引用により本明細書に組み込まれる)
|
|
4.1
|
ロケット製薬会社普通株証明書表(会社現在報告8-K表の添付ファイル4.1として提出し、2018年1月5日に米国証券取引委員会に提出し、引用により本明細書に組み込む)
|
|
4.2
|
証券説明(会社年間報告10−K表の添付ファイル4.8として提出され、2021年3月1日に米国証券取引委員会に提出され、引用により本明細書に組み込まれる)
|
|
10.1#
|
2004年株式オプションおよびインセンティブ計画(会社レジストリS-1(333-199859)の添付ファイル10.1として提出され、2014年11月5日に米国証券取引委員会に提出され、参照により本明細書に組み込まれた
|
|
10.2#
|
ロケット製薬会社第2次改正と2014年株式オプション·インセンティブ計画の見直し(会社の現在の報告書の添付ファイル10.1としてbr社に提出された現在の報告Form 8-Kは,2018年6月25日に米国証券取引委員会に提出され,引用により本明細書に組み込まれる)
|
|
10.3#
|
株式オプションインセンティブ協議表(従業員)(2018年8月14日に米国証券取引委員会に届出し、会社四半期報告書10-Q表添付ファイル10.2として提出し、引用により本明細書に組み込む)
|
|
10.4#
|
無保留株式オプションプロトコル表(従業員)(会社四半期報告10-Q表の添付ファイル10.3アーカイブとして、2018年8月14日に米国証券取引委員会
に提出され、参照により本明細書に組み込まれる)
|
|
10.5#
|
非限定株式オプション協定表(非従業員取締役)(会社四半期報告書10-Q表添付ファイル10.4として提出し、2018年8月14日に米国証券取引委員会に提出し、参考として本明細書に組み込む)
|
|
10.6#
|
非限定的株式オプション協定表(コンサルタント)(会社四半期報告書10-Q表の添付ファイル10.5として提出され、2018年8月14日に米国証券取引委員会
に提出され、参照により本明細書に組み込まれる)
|
|
10.6.1#
|
限定株式報酬プロトコル表(会社年次報告書10-K表の添付ファイル10.6.1アーカイブとして、2021年3月1日に米国証券取引委員会に提出され、参照により本明細書に組み込まれる)
|
|
10.7#
|
ロケット製薬有限会社2015年株式オプション計画(会社年間報告10-K表の添付ファイル10.3として米国証券取引委員会に提出し、2018年3月7日に提出し、引用により本明細書に組み込む)
|
|
10.8#
|
登録者とDavid P.Southwellとの間で2014年7月28日に署名された書簡協定(会社登録声明としての添付ファイル10.3は表S-1(333-199859)で提出され、2014年11月5日に米国証券取引委員会に提出され、改訂され、引用により本明細書に組み込まれた)
|
|
10.9#
|
イノクとDavid·ソスウェル間の招待状に対する修正案は、2017年9月1日に施行された(会社の現在報告されている8-K表の添付ファイル10.2アーカイブとして、2017年9月1日に米国証券取引委員会に提出され、引用により本明細書に組み込まれる)
|
|
10.10#
|
登録者とRaj Prabhakarの間の招待状は、2017年9月29日。
|
|
10.11#
|
招待状は,日付は2021年4月29日であり,登録者とイザベル·カモナが提供している。
|
|
10.12**
|
改訂·再署名された賃貸契約は、2019年6月26日に、Rocket PharmPharmticals,Inc.とCedar Brook 12 Corporation Center,L.P.(会社Form 10-Q四半期報告書の添付ファイル10.1として提出され、2018年8月8日に米国証券取引委員会に提出され、参照により本明細書に組み込まれる)
|
|
10.13#
|
ロケット製薬会社は2014年従業員株購入計画を改訂して再策定した(会社年次報告8−K表の添付ファイル10.10として提出し、2018年3月7日に米国証券取引委員会に提出し、引用により本明細書に組み込む)
|
|
10.14#
|
賠償協議表は、登録者及びその取締役によって締結される(2018年1月5日に米国証券取引委員会に提出された会社の現在の報告である8−K表の添付ファイル10.1,
,参照により本明細書に組み込む)
|
|
10.15#
|
賠償協議表は、登録者及びその上級管理者によって締結される(会社の現在の報告である8−K表の添付ファイル10.2が提出され、2018年1月5日に米国証券取引委員会に提出され、引用により本明細書に組み込まれる)
|
|
10.16#
|
登録者がその一部の上級管理者と締結したサービス契約及び制御権変更協定のフォーマット(会社年次報告10−K表の添付ファイル10.16アーカイブとして、2021年3月1日に米国証券取引委員会に提出され、参照により本明細書に組み込まれる)
|
|
10.17
|
ロケット製薬会社とESRTエンパイアステートビル有限責任会社との間のリース契約は、2018年6月6日(会社10-Q表四半期報告書の添付ファイル10.6アーカイブとして、2018年8月14日に米国証券取引委員会に提出され、参照により本明細書に組み込まれます)
|
76
カタログ表
|
10.18†
|
ライセンス契約は、Rocket PharmPharmticals,Ltd.とRegenxbio Inc.によって署名され、日付は2018年11月19日である(会社が2019年3月8日に米国証券取引委員会に提出した10-K表年次報告書の添付ファイル10.26として提出され、参照により本明細書に組み込まれる)
|
|
10.19
|
登録者と海王星コンサルティング会社の間で普通株を購入する引受権証は、日付は2020年12月21日。(会社年間報告書10-K表の添付ファイル10.23として提出され、2021年3月1日に米国証券取引委員会に提出され、参照により本明細書に組み込まれる)
|
|
10.20
|
系列優先株を購入する引受証は、2013年6月28日、イノク製薬会社とスカイライン技術金融会社の間で保有されている(会社年次報告10-K表の証拠として10.24提出され、2021年3月1日に米国証券取引委員会に提出され、引用により本明細書に組み込まれる)
|
|
10.21
|
イノク製薬会社と砦信用有限責任会社との間の日付が2013年6月28日のシリーズ優先株の引受証を購入する(添付ファイルとして10.25からbr}会社は2021年3月1日に米国証券取引委員会に提出されたForm 10−K年度報告書を引用して本明細書に組み込む)
|
|
10.22
|
登録者と海王星コンサルティング会社の間で普通株を購入する引受権証は、2021年12月17日となる。(最初の指示)
|
|
10.23
|
登録者と海王星コンサルティング会社の間で普通株を購入する引受権証は、2021年12月17日となる。(第2の指示)
|
|
10.24
|
証券購入協定は,期日は2021年8月27日であり,Rocket PharmPharmticals,Inc.と買い手である各人が締結され,この契約の買い手リストは添付されている買い手リストとして添付されている(会社の現在の報告8-K表の添付ファイル10.1として提出され,2021年8月30日に米国証券取引委員会に提出され,引用により本明細書に組み込まれる)
|
|
10.25
|
登録権利協定は、日付が2021年8月27日であり、Rocket PharmPharmticals,Inc.と投資家である各人とが締結され、その付表Aである発明者
付表(会社の現在の報告である8−K表の添付ファイル10.2が提出され、2021年8月30日に米国証券取引委員会に提出され、参照により本明細書に組み込まれる)に記載されている各人の間で締結される
|
|
10.26
|
当社とコーエン有限責任会社との間の販売協定は、2022年2月28日(当社現在報告8-K表の添付ファイル10.1として提出され、2022年3月1日に米国証券取引委員会
に提出され、引用により本明細書に組み込まれる)
|
|
21.1
|
子会社リスト(会社年間報告10-K表添付ファイル21.1として提出し、2021年3月1日に米国証券取引委員会に提出し、引用により本明細書に組み込む)
|
|
23.1*
|
EisnerAmper LLPの同意
|
|
24.1*
|
授権書(署名ページに含まれる)
|
|
31.1*
|
2002年サバンズ-オキシリー法第302条に基づいて可決された1934年“証券取引法”第13 a-14条又は第15 d-14(A)条に基づいて発行された最高経営責任者証明書
|
|
31.2*
|
2002年サバンズ·オキシリー法第302条に基づいて可決された1934年“証券取引法”第13 a−14条(A)条又は第15 d−14(A)条に基づく首席財務官の証明
|
|
32.1*
|
2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条による最高経営責任者と最高財務責任者の認証
|
|
101.INS
|
XBRLインスタンスドキュメントを連結する.
|
|
101.書院
|
インラインXBRL分類拡張アーキテクチャ文書.
|
|
101.カール
|
XBRL分類拡張計算文書を連結する.
|
|
101.def
|
XBRLソート拡張を連結してLinkbase文書を定義する.
|
|
101.介護会
|
XBRL分類拡張ラベルLinkbase文書を連結する.
|
|
101.Pre
|
インラインXBRL分類拡張プレゼンテーションリンク文書.
|
|
104
|
表紙相互データファイル(図101に含まれるイントラネットXBRL形式)
|
|
*
|
本局に提出します。
|
|
#
|
契約または補償計画を管理すること。
|
|
†
|
本展示品のいくつかの部分に対して、秘密保護待遇が与えられた。漏れた部分は米国証券取引委員会に個別に提出された。
|
|
**
|
本展示品のいくつかの部分は排除されています。それらは実質的ではありませんので、公開開示すれば会社に競争損害を与える可能性があります。
|
| † |
本10-K表年次報告添付ファイル32.1の認証は、米国証券取引委員会に届出されたものとみなされるものではなく、引用によってRocket製薬会社が1933年証券法(改訂本)または1934年証券取引法(改訂本)によって提出されたいかなる文書にも、本10−K表年次報告日の前または後に提出されたものであっても、このような文書に含まれる任意の一般的な合併言語にかかわらず、適用されてはならない。
|
|
第十六項。
|
表格10-Kの概要
|
適用されません。
77
カタログ表
サイン
1934年の証券取引法第13節または15(D)節の要求によると、Rocket PharmPharmticals,Inc.(登録者)は、2023年2月28日にニュージャージー州クランベリー市で正式に署名され、正式に許可されたbr人によって代表されて本報告書に署名した。
|
ロケット製薬会社です。
|
||
|
差出人:
|
メリーランド州ゴラフ·シャア
|
|
|
ガウラフ·シャア医学博士
|
||
|
社長と最高経営責任者
|
||
授権依頼書
個人署名は、以下に示すように、各個人がここで構成され、Gaurav Shah,MDおよびJohn Militelloを任命し、彼ら皆が完全な代替および再代替の権力を有し、他の人がいない場合には、彼または彼女の真および合法的な事実代理人および代理人として、彼または彼女の名義、場所およびエージェントとして行動し、各人の名義および以下に説明するすべての人を代表して行動し、本年度報告の任意およびすべての修正を表格10-Kの形で提出し、本年度報告を提出する。すべての証拠品およびこれに関連する他の文書と共に、米国証券取引委員会は、上述した事実弁護士および代理人、ならびに各行為および事柄のすべての権力および許可を行い、実行することができ、これらのすべての事実弁護士および代理人またはそれらのいずれかまたは彼らまたはそれらの代替者が合法的にbrを行うことができるか、またはそれによって
を行うことができることを承認および確認する。
本報告は、1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
|
名前.名前
|
タイトル
|
日取り
|
||
|
メリーランド州ゴラフ·シャア
|
取締役CEO兼最高経営責任者
|
2023年2月28日
|
||
|
ガウラフ·シャア医学博士
|
(首席行政主任)
|
|||
|
ジョン·C·ミリトロ
|
財務副総裁、上級財務総監兼財務主管
|
2023年2月28日
|
||
|
ジョン·C·ミリトロ
|
(臨時首席財務官、校長
会計主任)
|
|||
|
/s/カス·ボス
|
|
|
||
|
カス·ボイス
|
役員.取締役 | 2023年2月28日 | ||
|
/s/ペドロ·グラナディロ
|
|
|||
|
ペドロ·グラナディロ
|
役員.取締役 | 2023年2月28日 | ||
|
メリーランド州ゴタン市マークル
|
|
|||
|
メリーランド州ゴタン市マークル
|
役員.取締役 | 2023年2月28日 | ||
|
デヴィッド·P·ソスウェル
|
|
|
||
|
デヴィッド·P·ソースウェル
|
役員.取締役 | 2023年2月28日 |
||
|
ロデリック·ワン医学博士
|
|
|
||
|
ロデリック·ワン医学博士
|
役員.取締役 | 2023年2月28日 |
||
|
/s/ナヴィン·ヤラマンチ、医学
|
|
|||
|
ナヴィン·ヤラマンチ医学博士
|
役員.取締役 |
2023年2月28日 |
||
|
/s/エリザベス·ビヨーク
|
|
|
||
|
エリザベス·ビヨーク
|
役員.取締役 | 2023年2月28日 |
||
|
/s/Fady Malik
|
|
|||
|
ファディ·マリク
|
役員.取締役 |
2023年2月28日 |
79
カタログ表
ロケット製薬会社です。
連結財務諸表索引
カタログ
|
独立公認会計士事務所報告
|
F-2
|
|
2022年と2021年12月31日までの連結貸借対照表
|
F-4
|
|
2022年·2021年·2020年12月31日までの総合業務報告書
|
F-5
|
|
2022年,2021年,2020年12月31日までの総合全面損失表
|
F-6
|
|
2022年まで、2021年、2020年12月31日まで年度株主権益変動表
|
F-7
|
|
2022年、2021年、2020年12月31日までの統合現金フロー表
|
F-8
|
|
連結財務諸表付記
|
F-9
|
F-1
カタログ表
独立公認会計士事務所報告
当社の取締役会と株主へ
ロケット製薬会社です。
財務諸表のいくつかの見方
ロケット製薬会社とその子会社(“当社”)2022年12月31日と2021年12月31日までの総合貸借対照表,および2022年12月31日までの3年間の各年度に関する総合経営報告書,全面赤字,株主権益変動とキャッシュフロー,および関連するbr付記(総称して“財務諸表”と呼ぶ)を監査した。財務諸表は,すべての重要な面で当社の2022年12月31日と2021年12月31日までの財務状況,および2022年12月31日までの3年間の各年度の経営業績とキャッシュフローを公平に反映しており,米国公認の会計原則に適合していると考えられる。
また、米国上場企業会計監督委員会(PCAOB)の基準に従って、2022年12月31日までの財務報告内部統制を監査した内部制御-統合フレームワーク (2013トレデビル委員会が主催する組織委員会(“COSO”)が発表し、2023年2月28日の報告書は保留なしの意見を表明した。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちはPCAOBに登録されている公共会計士事務所であり、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と条例に基づいて、私たちは会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかの合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。私たちの監査には、経営陣が使用する会計原則の評価と重大な推定、財務諸表の全体的なレポートの評価も含まれています。私たちは私たちの監査が私たちの意見に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、今期の財務諸表監査において発生した事項であり、(1)財務諸表に対して重大な意義を有する勘定又は開示に関するものであり、(2)特に挑戦性、主観性又は複雑性を有する判断に関するものである。重要監査事項の伝達は、財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、次の重要な監査事項を伝達することによって、重要な監査事項又はそれに関連する勘定又は開示について個別の意見を提供することもない。
研究と開発費用はプロジェクトに応じて計算される
財務諸表付記3に開示されているように、当社は、既存のbr契約の計算すべき研究開発費用、当社に提供されているサービスを評価および決定し、領収書が発行されていないか、または実際のbrコストを他の方法で通知する際に提供されるサービスのレベルおよびサービスによって生じる関連コストを推定し、達成された契約マイルストーンを評価する。同社は,複数の契約研究組織(CRO),臨床試験に関する研究地点と契約製造組織(CMO)との契約により受けたサービスとかかる努力から,行っている臨床試験のコストを推定している。2022年12月31日現在、売掛金と売掛金の研究開発費は約1910万ドル。
第三者CRO,CMO,調査サイトに関する計算すべき項目の見積りは非常に複雑であるため,研究開発活動に関する計算すべき項目を重要な監査事項として決定した.当社のこれらの計算すべき項目の見積もりの複雑さは、主に進捗を決定することと、これらのスケジュールによって生じる直接的かつ間接的なコストであり、その中でコストとマイルストーンの領収書は、これまで提供されてきたサービスの時間と一致しない可能性がある。そのため、監査人は、プログラムを実行し、研究開発費課税項目に関する監査証拠を評価するために判断する必要がある。
この問題を解決することは、財務諸表に対する私たちの全体的な意見を形成するための実行手順と監査証拠の評価に関するものである。我々は、設計を評価し、開発すべきプロジェクトの推定を決定する制御に対する会社の操作有効性をテストし、管理層の推定のための投入および推定に使用されるデータの完全性と正確性の制御を含むことを理解し、評価した。我々の監査プログラムには、契約、領収書、支払いのサンプルをチェックし、2022年12月31日までに会社が第三者研究開発サプライヤーサンプルに対する借金を確認し、会社の進捗推定を契約、作業説明書、第三者仕入先が確認したデータ、br}領収書とそれによって発生した計算すべき項目の支払いと比較することも含まれている。
/s/EisnerAmper LLP
2016年以来、当社の監査役を務めてきました。
2023年2月28日
F-2
カタログ表
独立公認会計士事務所報告
取締役会と株主へ
ロケット製薬会社
財務報告の内部統制については
Rocket PharmPharmticals,Inc.およびその子会社(“当社”)の2022年12月31日までの財務報告内部統制を監査しました内部制御--統合フレームワーク(2013)テレデビル委員会(“COSO”)協賛組織委員会(“COSO”)により発表されました。2022年12月31日現在、当社はすべての重要な面で財務報告に対する有効な内部統制を維持しており、その根拠は内部制御--統合フレームワーク (2013)
はCOSOから発行されます。
著者らもすでにアメリカ上場会社会計監督委員会(“PCAOB”)の基準に従って、ロケット製薬会社と付属会社に対して2022年12月31日までの総合貸借対照表及び2022年12月31日までの3年間の各年度の関連総合経営報告書、全面赤字、株主権益変動及び現金流量を監査し、関連付記及び著者らが2023年2月28日に提出した報告はすべて保留意見がない。
意見の基礎
当社経営陣は、効果的な財務報告内部統制を維持し、付随する経営陣財務報告内部統制年次報告に含まれる財務報告内部統制の有効性の評価を担当する。私たちの責任は私たちの監査に基づいて、会社が財務報告書の内部統制について意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所であり、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と条例に基づいて、私たちは会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを合理的に決定するために、監査
を計画し、実行することを要求する。財務報告の内部統制の監査には、財務報告の内部統制を理解すること、重大な弱点があるリスクを評価すること、評価されたリスクテストに基づいて内部統制の設計と運用有効性を評価することが含まれる。私たちの検討はまた、私たちがこのような状況で必要だと思う他の手続きを実行することを含む。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
実体の財務報告に対する内部統制は、財務報告の信頼性に合理的な保証を提供することを目的とした過程であり、公認された会計原則に基づいて外部目的の財務諸表を作成する。実体の財務報告に対する内部統制は、(1)合理的で詳細かつ正確かつ公平に実体資産の取引および処分を反映する記録を保存することに関連する政策および手順、(2)公認された会計原則に従って財務諸表を作成するために取引が必要な記録を保証するために合理的な保証を提供し、実体の収入と支出は実体管理層と取締役の許可のみに基づいて行われる、という政策と手続きを含む。(3)財務諸表に重大な影響を与える可能性のある不正取得、使用、または処分実体資産の防止またはタイムリーな発見について合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的に任意の有効性評価を行う予測は,条件の変化により制御不足のリスクが生じたり,政策やプログラムの遵守度が悪化したりする可能性がある.
/s/
EisnerAmper LLP
2023年2月28日
F-3
カタログ表
ロケット製薬会社です。
合併貸借対照表
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
十二月三十一日
2022
|
十二月三十一日
2021
|
|||||||
|
資産
|
||||||||
|
流動資産:
|
||||||||
|
現金と現金等価物
|
$
|
|
$
|
|
||||
|
投資する
|
|
|
||||||
|
前払い費用と他の流動資産
|
|
|
||||||
|
流動資産総額
|
|
|
||||||
|
財産と設備、純額
|
|
|
||||||
|
商誉
|
|
|
||||||
|
無形資産
|
||||||||
|
制限現金
|
|
|
||||||
|
預金.預金
|
|
|
||||||
|
投資する
|
||||||||
|
経営的リース使用権資産
|
|
|
||||||
|
融資リース使用権資産
|
|
|
||||||
|
総資産
|
$
|
|
$
|
|
||||
|
負債と株主権益
|
||||||||
|
流動負債:
|
||||||||
|
売掛金と売掛金
|
$
|
|
$
|
|
||||
|
賃貸負債を経営し、流動
|
|
|
||||||
|
融資リース負債流動
|
|
|
||||||
|
流動負債総額
|
|
|
||||||
|
非流動経営賃貸負債
|
|
|
||||||
|
融資リース負債、非流動
|
|
|
||||||
|
その他負債
|
|
|
||||||
|
総負債
|
|
|
||||||
|
引受金及び又は事項(付記12)
|
|
|
||||||
|
株主権益:
|
||||||||
|
優先株、$
|
||||||||
|
Aシリーズ転換可能優先株;
|
|
|
||||||
|
Bシリーズ転換可能優先株;
|
|
|
||||||
|
普通株、$
|
|
|
||||||
|
国庫株は、原価で計算する
|
( |
) | ||||||
|
追加実収資本
|
|
|
||||||
|
その他の総合損失を累計する
|
(
|
)
|
(
|
)
|
||||
|
赤字を累計する
|
(
|
)
|
(
|
)
|
||||
|
株主権益総額
|
|
|
||||||
|
総負債と株主権益
|
$
|
|
$
|
|
||||
付記はこれらの連結財務諸表の構成要素である。
F-4
カタログ表
ロケット製薬会社です。
連結業務報告書
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
|
12月31日までの年度
|
||||||||||||
|
2022
|
2021
|
2020
|
||||||||||
|
収入.収入
|
$
|
|
$
|
|
$
|
|
||||||
|
運営費用:
|
||||||||||||
|
研究開発
|
|
|
|
|||||||||
|
一般と行政
|
|
|
|
|||||||||
|
総運営費
|
|
|
|
|||||||||
|
運営損失
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||
|
インセンティブを開発する
|
|
|
|
|||||||||
|
利子支出
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||
|
利息とその他の収入,純額
|
|
|
|
|||||||||
|
投資割増純額
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||
|
その他の収入を合計して純額
|
( |
) | ( |
) | ||||||||
|
純損失
|
$
|
(
|
)
|
$
|
(
|
)
|
$
|
(
|
)
|
|||
|
普通株主1株当たり純損失−基本損失と希薄損失−
|
$
|
(
|
)
|
$
|
(
|
)
|
$
|
(
|
)
|
|||
|
加重平均発行済み普通株式-基本普通株式と希釈普通株
|
|
|
|
|||||||||
付記はこれらの連結財務諸表の構成要素である。
F-5
カタログ表
ロケット製薬会社です。
合併全面損失表
(単位:千)
|
12月31日までの年度
|
||||||||||||
|
2022
|
2021
|
2020
|
||||||||||
|
純損失
|
$
|
(
|
)
|
$
|
(
|
)
|
$
|
(
|
)
|
|||
|
その他総合損失
|
||||||||||||
|
投資は純損失を実現していない
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||
|
全面損失総額
|
$
|
(
|
)
|
$
|
(
|
)
|
$
|
(
|
)
|
|||
付記はこれらの連結財務諸表の構成要素である。
F-6
カタログ表
ロケット製薬会社です。
合併株主権益変動表
(単位は千で、シェアは含まれていない)
|
普通株
|
財務局 |
その他支払い済み
|
積算
他にも
全面的に
|
積算 |
株主合計
|
|||||||||||||||||||||||
|
株
|
金額
|
在庫品
|
資本
|
収入/(赤字)
|
赤字.赤字
|
権益
|
||||||||||||||||||||||
|
2019年12月31日の残高
|
|
$
|
|
$
|
(
|
)
|
$
|
|
$
|
|
$
|
(
|
)
|
$
|
|
|||||||||||||
|
普通株発行は発行コストを差し引く
|
|
|
|
|
|
|
|
|||||||||||||||||||||
|
株式オプションの行使により普通株を発行する
|
|
|
|
|
|
|
|
|||||||||||||||||||||
|
株式承認証の行使により普通株を発行する
|
- | - | - | - | - | - | ||||||||||||||||||||||
|
手形転換により普通株式を発行する
|
||||||||||||||||||||||||||||
|
株式承認証を発行する
|
- | - | - | - | - | |||||||||||||||||||||||
|
株の買い戻し
|
( |
) | ( |
) | ( |
) | ||||||||||||||||||||||
|
在庫株を売却する
|
|
|
|
(
|
)
|
|
|
|
||||||||||||||||||||
|
株式オプションの行使に応じて在庫株を発行する
|
( |
) | ( |
) | ||||||||||||||||||||||||
|
有価証券は総合損失を実現していない
|
-
|
|
|
|
(
|
)
|
|
(
|
)
|
|||||||||||||||||||
|
株に基づく報酬
|
-
|
|
|
|
|
|
|
|||||||||||||||||||||
|
純損失
|
-
|
|
|
|
|
(
|
)
|
(
|
)
|
|||||||||||||||||||
|
12月31日までの残高2020
|
|
|
|
|
(
|
)
|
(
|
)
|
|
|||||||||||||||||||
|
株式オプションの行使により普通株を発行する
|
||||||||||||||||||||||||||||
|
手形転換により普通株式を発行する
|
||||||||||||||||||||||||||||
|
普通株発行は発行コストを差し引く
|
|
|
|
|
|
|
|
|||||||||||||||||||||
|
株式承認証を発行する
|
-
|
-
|
-
|
|
-
|
-
|
|
|||||||||||||||||||||
|
有価証券は総合損失を実現していない
|
-
|
|
|
|
(
|
)
|
|
(
|
)
|
|||||||||||||||||||
|
株に基づく報酬
|
-
|
|
|
|
|
|
||||||||||||||||||||||
|
純損失
|
-
|
|
|
|
|
(
|
)
|
(
|
)
|
|||||||||||||||||||
|
12月31日までの残高2021
|
|
|
|
|
(
|
)
|
(
|
)
|
|
|||||||||||||||||||
|
普通株発行は発行コストを差し引く
|
||||||||||||||||||||||||||||
|
株式オプションの行使により普通株を発行する
|
||||||||||||||||||||||||||||
|
帰属制限株式単位に応じて普通株を発行する
|
||||||||||||||||||||||||||||
|
買収に関連する普通株を発行する
|
||||||||||||||||||||||||||||
|
利回り制限株式単位決済に関する普通株を発行する
|
||||||||||||||||||||||||||||
|
市場発行計画に基づいて普通株を発行し,発行コストを差し引く
|
||||||||||||||||||||||||||||
| 在庫株買い戻し |
( |
) | ( |
) | ||||||||||||||||||||||||
|
有価証券は総合損失を実現していない
|
- | ( |
) | ( |
) | |||||||||||||||||||||||
|
株に基づく報酬
|
- | |||||||||||||||||||||||||||
|
純損失
|
- | ( |
) | ( |
) | |||||||||||||||||||||||
|
12月31日までの残高2022
|
$ | $ | ( |
) | $ | $ | ( |
) | $ | ( |
) | $ | ||||||||||||||||
付記はこれらの連結財務諸表の構成要素である。
F-7
カタログ表
ロケット製薬会社です。
統合現金フロー表
(単位:千)
|
12月31日までの年度
|
||||||||||||
|
2022
|
2021
|
2020
|
||||||||||
|
経営活動:
|
||||||||||||
|
純損失
|
$
|
(
|
)
|
$
|
(
|
)
|
$
|
(
|
)
|
|||
|
純損失と経営活動で使用される現金純額の調整:
|
||||||||||||
|
転換手形割引の増加
|
|
|
|
|||||||||
|
財産と設備の減価償却と償却
|
|
|
|
|||||||||
|
使用権資産の償却
|
||||||||||||
|
財産と設備の減記,純額
|
|
|
|
|||||||||
|
株に基づく報酬
|
|
|
|
|||||||||
|
投資割増純額
|
|
|
|
|||||||||
|
株式引受証の発行に係る支出
|
||||||||||||
|
経営性資産と負債の変動、買収を差し引く:
|
||||||||||||
|
前払い費用と他の資産
|
(
|
)
|
|
(
|
)
|
|||||||
|
売掛金と売掛金
|
|
(
|
)
|
|
||||||||
|
リース負債を経営する
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||
|
融資リース負債
|
|
|
|
|||||||||
|
その他負債
|
(
|
)
|
(
|
)
|
|
|||||||
|
経営活動のための現金純額
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||
|
投資活動:
|
||||||||||||
|
購入投資
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||
|
投資満期で得られた収益
|
|
|
|
|||||||||
|
買収業務の現金収益は,支払われた現金を差し引く
|
||||||||||||
|
使用権資産取得のための金
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||
|
財産と設備を購入する
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||
|
投資活動が提供する現金純額
|
(
|
)
|
|
(
|
)
|
|||||||
|
融資活動:
|
||||||||||||
|
株式オプションの行使により普通株を発行する
|
|
|
|
|||||||||
|
普通株発行は発行コストを差し引く
|
||||||||||||
|
在庫株を売却して得られた収益は,現金を支払った純額を差し引く
|
|
|
|
|||||||||
|
オプション行使時の源泉徴収税金の支払い
|
|
|
(
|
)
|
||||||||
|
在庫株買い戻し
|
( |
) | ( |
) | ||||||||
|
転換可能手形の貸手への再融資コスト
|
|
|
(
|
)
|
||||||||
|
市場発売計画に基づいて普通株を発行し、発行コスト
を差し引く
|
||||||||||||
|
融資活動が提供する現金純額
|
|
|
|
|||||||||
|
現金、現金等価物、および限定的な現金純変化
|
(
|
)
|
(
|
)
|
|
|||||||
|
期初現金、現金等価物、および限定現金
|
|
|
|
|||||||||
|
期末現金、現金等価物、および制限現金
|
$
|
|
$
|
|
$
|
|
||||||
|
非現金融資と投資活動の追加開示:
|
||||||||||||
|
課税財産と設備購入·期末残高
|
$
|
|
$
|
|
$
|
|
||||||
|
投資が未実現損失
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||
| 転換可能手形を普通株式に変換する |
||||||||||||
|
買収に関連する普通株を発行する
|
||||||||||||
|
融資リース使用権、資産、賃貸負債
|
|
|
|
|||||||||
|
金融使用権資産からみた建設工事の再分類
|
|
|
|
|||||||||
|
キャッシュフロー情報の追加:
|
||||||||||||
|
利子を支払う現金
|
$
|
|
$
|
|
$
|
|
||||||
付記はこれらの連結財務諸表の構成要素である。
F-8
カタログ表
ロケット製薬会社です。
連結財務諸表付記
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない)
| 1. |
業務の性質と列報根拠
|
Rocket PharmPharmticals,Inc.(“Rocket”或いは“会社”)は臨床段階、多プラットフォームの生物技術会社であり、第一種、唯一と一流の遺伝子療法の開発に専念し、直接の標的作用機序と明確な臨床終点を有し、稀と壊滅的な疾病に適用される。その会社は所有している三つ 臨床段階の体外レンチウイルスベクター(LV)計画。これらの計画はファンコニ貧血(FA)を含み、これは1種の骨髄中の遺伝欠陥であり、血球の産生を減少或いは欠陥血細胞の産生を促進する;白血球粘着欠陥-I(LAD-I)、1種の免疫系機能障害を招く遺伝性疾患;ピルビン酸キナーゼ欠乏症(“PKD”)、1種の稀な赤血球常染色体劣性遺伝性疾患であり、慢性非球形溶血性貧血を招く。このうち,第2段階FAプランと第1/2段階LAD-Iプランはいずれも米国に登録されている可能性がある.ヨーロッパ(“EU”)ですあまり見られないFA CやGサブタイプに対する遺伝子治療計画の他の作業が行われている。米国では、Danon病の臨床段階に対する腺関連ウイルス(“AAV”)計画もあり、Danon病は多臓器リソソーム関連疾患であり、心不全により早期死亡する。達能計画は現在進行中の第一段階試験中である。また,同社は不整脈源性心筋症(“PKP 2−ACM”)に対するAAVベクター計画を打ち出しており,PKP 2−ACMは遺伝性心疾患であり,心筋質量の進行性低下,右室の重篤な拡張,発育不良,心筋線維脂肪代替および不整脈や突然死の高い傾向を特徴としている。社がRenovacor Inc.(“Renovacor”)(付注17−“Renovacor
買収”参照)を買収したため,会社はAAV 9による組換え遺伝子療法を用いてBAG 3拡張型心筋症(“DCM”)の進展を緩和または阻止することができる, これは最もよく見られる心筋症であり,心壁が徐々に薄くなり,心腔が拡大し,血液をポンプできないのが特徴である。印税許可協定によると、同社はこれらすべての候補製品に対して世界の商業化と開発権を持っている。
2021年12月からロケット賛助を行わないことが決定したRP−L 401の臨床評価は,学術革新者に返還された。同社は、遺伝子療法がこのような疾患を有する患者に有益である可能性があると信じているが、RP−A 601、RP−A 501、RP−L 102、RP−L 201、RP−L 301、およびBAG 3−DCMの発展を促進することに既存のリソースを集中させることを選択している。
| 2. |
リスクと流動性
|
当社は設立以来何の収入も生じておらず、損失を出しています。同社の運営は、候補薬物開発の不確実性、技術的不確実性、特許および独自の権利の不確実性、商業製造経験のない、マーケティングまたは販売能力または経験、キーパーソンへの依存、政府法規の遵守、および追加融資の必要性を含むいくつかのリスクおよび不確実性の影響を受ける。現在開発中の候補薬物は商業化する前に大量の追加の研究と開発が必要であり、広範な臨床前と臨床テストと監督管理承認を含む。このような仕事は多くの追加資本、十分な人員インフラ、そして広範囲なコンプライアンス報告能力を必要とする。
同社の候補製品は開発と臨床段階にある。会社の研究と開発が成功することは保証されず、会社の知的財産権が十分に保護される保証はなく、開発されたいかなる製品も必要な政府の承認を得ることが保証されず、いかなる承認された製品も商業可能性があることを保証することはできない。同社の製品開発事業が成功しても、その会社がいつ(あれば)製品販売から相当な収入を得るかは定かではない。同社は技術の急速な変化や製薬やバイオテクノロジー会社からの激しい競争環境で運営されている。
当社の総合財務諸表は、業務の連続性、資産現金化、正常業務過程における負債返済状況に基づいて作成されています。同社の運営キャッシュフローはマイナスで、累計損失は#ドル713.8 2022年12月31日まで。2022年12月31日現在、同社は399.7 百万の現金、現金同等物、そして短期と長期投資。当社はこのような資源が当社の2024年までの運営支出および資本支出需要を支払うのに十分であると予想している。
当社は2022年2月28日にCowen and Company,
LLC(“Cowen”)と1つの市場発売計画について販売協定(“販売協定”)を締結し、この計画により、当社は時々適宜普通株式を発行·売却することができ、額面は$である0.01 1株当たりの総発行価格は最高$に達する200
はコーエンによりその販売代理となる百万株(“株”)である.同社は2022年12月31日までに販売された3.3 百万株普通株、純収益は$46.6 2,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000株の市価発売計画(付記9-“株主権益”参照)
2022年10月6日、同社は後続発売(“発売”)を完了し、同発売に応じて販売した7,820,000 普通株式、純収益は$108.1
百万ユーロ(付記9--“株主権益”参照)。
長期的には、当社の将来の生存能力は、経営活動から現金やbrを発生させて追加資本を調達してその運営に資金を提供する能力に依存する。同社が必要なときに資金を調達できなかったことは、その財務状況やその業務戦略を実施する能力にマイナス影響を与える可能性がある。
F-9
カタログ表
| 3. |
重要会計政策の概要
|
合併原則
総合財務諸表は、米国公認会計原則(“米国公認会計原則”)に基づいて当社及びその付属会社を合併する勘定である。合併で、すべての会社間口座がキャンセルされた。
予算の使用
米国公認会計原則に基づいて総合財務諸表を作成する際、管理層は財務諸表の日付の資産及び負債額、或いは有資産及び負債の開示、及び報告期間内の支出金額に影響するため、推定及び仮定を行わなければならない。これらの総合財務諸表に反映される重大な推定および仮定br}は、営業権減価、無形資産、研究開発費用の計算、株式取引の推定値、および株式ベースの報酬を含むが、これらに限定されない。
推定と仮説の変化は報告の結果に反映され,その間に既知である.実際の結果はこれらの推定とは異なる可能性がある。
現金·現金等価物 制限された現金
現金、現金等価物、および限定的な現金には、銀行預金、預金、および金融機関の通貨市場口座が含まれています。
現金等価物は、その短期的な性質のため、公正な価値に近いコストで計算されており、当社はこれらの現金等価物に重大な信用リスクがないと考えています。当社は購入日から三ヶ月以下のすべての高流動性投資を現金等価物と見なしています。会社の現金と現金等価物口座は連邦保険の限度額を超える場合があります。当社はこのような勘定で何の損失も出ていません。
制限された現金には、当社がリース発行した信用状について担保銀行が発行した預金(より多くの開示は付記br 14“承諾およびまたは事項”を参照)と、当社のクレジットカードを担保して発行した銀行が発行した信用状を担保する預金が含まれています。現金、現金等価物、および限定的な現金は、
|
2022年12月31日
|
2021年12月31日
|
|||||||
|
現金と現金等価物
|
$
|
|
$
|
|
||||
|
制限現金
|
|
|
||||||
|
$
|
|
$
|
|
|||||
政府補助金
研究開発費はカリフォルニア再生医学研究所(“CIRM”)の精算後の純額を差し引いたものであり,会社がCIRMの条件を遵守して精算を受ける可能性が高い場合には,精算が関連コストにマッチするまでの間にこれらの精算を確認する。会社は2022年12月31日、2021年12月31日、2020年12月31日までの年間で相殺した0 , $0.1 100万ドルと$3.6 CIRMは、それぞれ研究開発(“R&D”)費用(より多くの開示は付記16“CIRM
贈与”)に資金を贈与します。
信用リスクと表外リスクの集中度
会社を信用リスクに直面させる金融商品には、主に現金と現金等価物、販売可能な証券が含まれる。当社は良質な金融機関と現金および現金等価物残高を保持しているため,当社は同社などの基金の信用リスクが最も低いと信じている。同社の有価証券には、米国債、商業手形、社債、国債、機関債券が含まれる。会社の投資政策は、会社が任意の投資タイプに投資できる金額を制限し、会社が保有するすべての投資が少なくともAA+/Aa 1格付けであることを要求し、信用リスクを低下させる。
投資する
投資には米国債商業手形会社への投資が含まれています政府は機関債券を持っています経営者は、買収時にこれらの証券の適切な分類を決定し、各貸借対照表の日付にそのような分類の適切性を評価する。当社は、財務会計基準委員会(FASB)会計基準編纂(ASC)320に基づいて、その投資を販売可能に分類した投資--債務と持分証券それは.投資は公正価値によって入金され、損益計上株主権益累積その他の全面収益(損失)の構成部分を実現せず、総合全面損失表に全面損失総額の構成部分を計上し、実現までに。実現した収益と損失は、特定の確認に基づいて投資収益に計上される。いくつありますか違います。 0.2 百万人 $0.1 百万人そして $0.1 それぞれ100万ドルです
F-10
カタログ表
無形資産
進行中の研究開発(“IPR&D”)プロジェクトに関連する無形資産は、関連研究開発事業が完了または放棄されるまで無期限とされている。
であり、開発が完了した場合(一般に規制部門の許可を得て製品を販売する場合)、関連資産は有限寿命とみなされ、その後、その時点での推定使用寿命に基づいて償却される。価値が低下すると判断された知的財産権研究開発無形資産は下方に調整され、合併経営報告書の研究開発費用で費用が確認される。これらの知的財産権研究開発無形資産は少なくとも年に1回のテストを行うか、あるいは潜在的な減値を示す可能性のあるトリガイベントが発生した場合にテストを行い、これらの指標は研究開発活動の進展、予想資産発展の変化、監督管理環境と未来の商業市場の変化を含む.
商誉
業務
組合せは買収方式で計算する.買収の総コストは,買収日それぞれの推定公正価値に応じて関連する識別可能な純資産に分配される。買収された資産と負担する負債の公正価値を決定するには管理職の判断が必要であり、通常は将来の現金流入と流出、割引率、資産寿命、市場倍数などの項目に関する仮定を含む重大な見積もりと仮説の使用に関連する。買収した資産と負担した負債はその推定公正価値に基づいて入金される。買収価格は買収純資産が公正価値を推定した部分を超えて営業権に計上されている。12月31日から、毎年営業権の減価テストが行われ、またはより頻繁にイベントまたは環境変化が資産が減値可能であることが示された場合にテストが行われる。このようなイベントまたは状況の例は、法律またはビジネス環境の重大な不利な変化、不利な規制行動、または予想外の競争を含むが、これらに限定されないその会社は所有している1つは 区分和1つは その報告書は、したがって、総合的な水準での減価商業権を検討する。営業権をテストする時、会社は
を選択して、まず営業権を持つ報告単位の定性的要素を評価することができる。定性的評価は、報告単位の公正価値または帳簿価値に影響を与えるすべての関連イベントと状況を評価することを含む。これらの事件と状況はマクロ経済状況、業界と競争環境状況、全体的な財務業績、報告単位の特定の事件と市場考慮要素を含む。当社は、報告単位の最近の推定値も考慮し、最新の公正価値推定と帳簿額面との差額、正面および不利なイベントおよび状況の大きさ、および識別された各イベントおよび
状況が報告単位の公正価値と帳簿価値の比較に与える影響の程度を含む。定性的評価で報告単位の公正価値が帳簿価値を超える可能性が高いと結論すれば,その報告単位のさらなるテストは行われない
当社はその営業権に対して定性評価を行い、報告単位の公正価値が報告単位の帳簿価値を超える可能性が高いことを確定した。そのため、同社は存在を確定しました違います。
財産と設備
財産と設備はコストから減価償却累計を引いて申告する.減価償却費用は資産の耐用年数内に直線法で確認します。推定使用寿命は至れり尽くせり15年 それは.当社は、これらの資産が当社の将来の代替用途を有することが決定されたので、実験室設備、機械および設備、家具および固定装置、およびニュージャージー州クランベリーに位置する施設に関連する賃貸改善を資本に計上する。修理と維持資産の支出は発生時に費用を計上する。廃棄又は販売時には、資産を処分するコスト及び関連減価償却は勘定から差し引かれ、それによって生じるいかなる収益又は損失も運営損失に計上される。 エンティティソフトウェアライセンスを含む内部使用ソフトウェアおよびクラウドコンピューティング手配の開発または購入に関連するコストは、資本に計上される。償却は資産の予想寿命内で直線的に計算される、すなわち
6年 それは.資本化されたソフトウェアは、合併貸借対照表中の財産及び設備に計上される。
長期資産減価準備
事件や環境変化が資産の帳簿金額を回収できない可能性があることを示すたびに、当社は長期資産の減値を審査する。当社はASC 360-10-15“長期資産減価または処分”に基づいて長期資産減価分析を行っている。ASC 360-10-15は、他の資産および負債のキャッシュフローと実質的に無関係であることを識別可能な最低レベルで資産および負債をグループ化し、割引されていない将来のキャッシュフローの合計に基づいて資産グループを評価することを要求する。割引キャッシュフローが資産を表示していない帳票金額
が回収可能であれば、減価費用は割引キャッシュフロー分析または評価に基づいて、資産グループ別の帳票金額がその公正価値を超えた金額で計量される。1種類あります違います。
F-11
カタログ表
公正価値計量
当社は、報告書の公正価値を決定する際に使用される投入を評価するために、公正価値報告書のすべての資産および負債の情報を開示しなければならない。FASB ASC 820、 公正価値計量と開示利用可能な投入の階層構造を構築する。
観察できる投入は、企業とは独立したソースから得られた市場データに基づいて市場参加者が資産または負債を定価する際に使用される投入である。観察できない投入は、市場参加者が資産や負債の価格設定のために使用されるという会社の仮定を反映し、その時点で入手可能な最適な情報に基づいて制定される。公正価値レベルは投資報告の公正価値を確定する際に使用する推定値投入にのみ適用され、投資信用品質の評価基準ではない。公正価値レベルの3つの階層は以下のとおりである
第1レベル-当社が計量日に取得する能力のある同じ資産または負債の未調整オファーに対するアクティブ市場の推定値
に基づいて。
レベル2-非アクティブまたはすべての重要な投入が直接または間接的に観察されることができる市場における同様の資産または負債の見積もりに基づいて推定される。
第3レベル−会社自身の仮説を反映した推定値が必要であるが,これらの仮定は公正価値計測に重要であり,
は観察できない。
推定値は市場で観察または観察できないモデルや投入に基づくため、公正価値の決定にはより多くの判断が必要である。そのため、当社が価値を公平に決定する際に下した判断程度は、第3級のツールに分類されることが最大である。公正価値レベル内の金融商品レベルは、公正価値計量に重大な意義がある任意の投入中の最低レベルに基づいている。当社の金融商品の公正価値は、現金及び現金等価物、制限性現金、預金、支払すべき帳簿金及び売掛金を含み、大部分のこのようなツールは短期的な性質であるため、その公正価値はそのそれぞれの帳簿価値と一致する。
株式承認証
ASCテーマ480に基づいて、株式承認証を権益ツール、負債または派生負債として会計処理し、負債と持分を区別する (“ASC 480”)および/またはASC主題815、派生ツールおよびヘッジ (“ASC 815”),
は引受権証プロトコルの具体的な条項に依存する.負債分類株式証は、行使、終了、再分類、または他の方法で決済されるまで、各報告期間ごとにその推定公正価値で入金される。負債分類権証の推定公正価値変動を当社総合経営報告書の権証負債公正価値変動に計上する。株式分類権証は発行時に追加実収資本を計上し、再計量する必要はない。
研究と開発費
研究開発コストは、賃金と従業員コスト、ライセンスコスト、製造·開発コスト、臨床試験費用、監督·科学相談費、および契約研究と株式ベースの給与支出を含み、ASCテーマ730に基づいて、研究·開発による会計処理を行う。同社には現在、商業生物製薬製品は何もなく、数年以内には何の製品もないと予想されている。そのため,研究開発コストは発生時に費用を計上する。会社のいくつかの研究開発コストは将来の収益をもたらす可能性があるが、すべての研究開発支出を支出する政策は、企業が候補製品の商業化に成功した歴史がなく、これを将来の利益期間の数のいかなる推定としてもしていないという事実に基づいている。
外貨取引
2022年12月31日、2021年、2020年12月31日までの年間で、いくつかの取引はユーロとポンドで取引される。2022年、2021年、2020年12月31日までの年間では、外貨取引の損益は顕著ではない。
在庫株
当社は原価計上で在庫株を計上しています。
F-12
カタログ表
株に基づく報酬
会社は、通常、株式オプションおよび限定株式単位の形で、従業員および非従業員に株式ベースの奨励金を支給する。当社
は、FASB ASCテーマ718、報酬-株式報酬(“ASC 718”)に基づいて、株式ベースの報酬を会計処理する。ASC 718は、株式オプションおよび制限株式単位の付与、および既存の株式オプションの修正を含む株式ベースのすべての支払いを要求し、その公正価値に基づいて総合経営報告書および全面損失で確認しなければならない。当社は付与日に付与された持分ツールの公正価値に基づいて、受け取った従業員と非従業員サービスの補償費用を計量する。このコストは、報酬と交換するために従業員および非従業員にサービスを提供することを要求している間、直線的に確認される。日オプションを付与する公正価値は,期待変動率や期待期限などの重要な仮定に基づいて,Black-Scholesオプション定価モデルを用いて計算される.会社のこれらの仮定に対する見積もり
は主に会社株の取引価格、履歴データ、同業者会社データ及び未来の傾向と要素の判断に基づいている。
当社がその総合経営報告書において株式に基づく報酬費用を分類する方式は、受賞者の賃金コストやサービスを分類する方式と同じか、受賞者のサービス支払いを分類する方式と同じである。同社は付与された賠償の少なくとも一部の補償費用を確認した。没収は発生時に
に計上する.
ニューヨーク州生命科学研究と開発税控除
ニューヨーク州は、バイオテクノロジーに集中する新興技術会社の投資家および所有者が、ニューヨーク州で発生したいくつかの支出(適用可能な研究開発関連支出を含む)についてニューヨーク納税申告書の税収免除を申請することを可能にする。資格と金額がニューヨーク証券取引所の承認を得た場合、この控除は研究開発インセンティブとして確認されます。brは、2022年、2021年、2020年12月31日までの年間で、会社が記録した研究開発インセンティブ収入は0.5 百万、$1.0 百万ドルと$0 ニューヨーク生命科学研究と開発税収控除と関係がある。
所得税
当社は貸借対照法で所得税を計算します。当社は、将来の繰延税金資産と負債
が、既存の資産と負債の帳簿金額とそれぞれの課税ベースとの差、および営業損失と税額控除の課税結果によるものであることを確認した。当社は、当社がその等の一時的な差額を回収または決済することが予想される年度の課税所得額に適用される予定の策定税率を用いて繰延税金資産と負債を計測します。当社は、公布日を含む期間内に、税率変動が繰延税金資産及び負債に及ぼす影響を経営実績において確認している。当社はさらに繰延税金資産の一部または全部を現金化しない可能性があり、当社は必要に応じて繰延税金資産の計量を減少させるために推定値を設定します。
当社の繰延税金資産は主に純営業損失、繰越及びその他の貸借対照表の差額に関連しています。ASC 740“所得税”によると、当社は2022年および2021年12月31日にこのような繰延税金資産に関連する将来的な利益を実現する可能性が低いため、繰延税項目純資産を完全に相殺するために全額推定値を計上する準備をしている。
当社は財務諸表で確認された所得税の不確実性を計算し、二歩法で確認すべき税収割引額を決定する方法を採用した。まず、税務機関が外部審査後にこのような状況を維持する可能性を決定するために、税収状況を評価しなければならない。税務状況がより継続する可能性があると考えられる場合、財務諸表で確認すべき利益金額を決定するために税務状況を評価する。確認可能な受益額は,最終和解時に実現される可能性が50%を超える最大額である。所得税の準備には、それによって生じる税収準備金または未確認の税収割引の影響が適切であると考えられ、関連する純利息および罰金が含まれる。
1株当たり純損失
当社はFASB ASC 260に従って1株当たり純損失を計算した “1株当たり収益”.普通株株主が1株当たり基本純損失を占めるべき計算方法は、普通株株主が純損失を当期発行普通株の加重平均で割るべきである。普通株株主が償却純損失を占めるべきであることは、普通株株主が純損失を占めるべきであることを調整し、希釈性証券の潜在的影響に基づいて未分配収益を再分配することによって計算される。普通株株主が希釈1株当たり純損失を占めるべき計算方法は:普通株株主は希釈純損失を当期発行普通株で割った加重平均を占めるべきであり、その中に潜在的な希釈性普通株を含む。この計算については,既発行オプションは潜在的な希釈性普通株とみなされている。
細分化市場報告
運営部門は企業の構成要素として定義され、その独立した離散情報は、首席運営意思決定者または意思決定グループが資源をどのように割り当てるかを決定し、業績を評価する際に評価することができる。会社は以下の点でその運営·管理業務を確認します1つは 運営部門です。
総合損失
総合損失とは、企業が一定期間内に非所有者源の取引及びその他の事件と状況によって発生した権益変動であり、純損失と投資未実現損益の変動を含む。
最近の会計公告
最近、当社または総合財務諸表に大きな影響を与えることが予想される会計声明には影響していません。
F-13
カタログ表
| 4. |
金融商品の公正価値
|
公正な価値に応じて恒常的に計量するプロジェクトは会社の投資です。以下の表は、当社が公正価値体系内で公正価値を段階的に日常的に計量する財務ツールを示している
|
公正価値計量
December 31, 2022 Using:
|
||||||||||||||||
|
レベル1
|
レベル2
|
レベル3
|
合計する
|
|||||||||||||
| 資産: | ||||||||||||||||
|
現金等価物:
|
||||||||||||||||
|
貨幣市場共同基金
|
$ | $ | $ | $ | ||||||||||||
|
商業手形
|
||||||||||||||||
|
アメリカ国庫券
|
||||||||||||||||
|
社債
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|||||||||||||
|
投資:
|
||||||||||||||||
|
商業手形
|
||||||||||||||||
|
アメリカ国債
|
|
|
|
|
||||||||||||
|
社債
|
|
|
|
|
||||||||||||
|
機構債券
|
|
|
|
|
||||||||||||
|
総資産
|
$
|
|
$
|
|
$
|
|
$
|
|
||||||||
| 負債: |
||||||||||||||||
|
株式証法的責任
|
$ | $ | $ | $ | ||||||||||||
|
総負債
|
$ | $ | $ | $ | ||||||||||||
|
公正価値計量
December 31, 2021 Using:
|
||||||||||||||||
|
レベル1
|
レベル2
|
レベル3
|
合計する
|
|||||||||||||
|
資産:
|
||||||||||||||||
|
現金等価物:
|
||||||||||||||||
|
貨幣市場共同基金
|
$
|
|
$
|
|
$
|
|
$
|
|
||||||||
|
|
|
|
|
|||||||||||||
|
投資:
|
||||||||||||||||
|
アメリカ国債
|
|
|
|
|
||||||||||||
|
社債
|
|
|
|
|
||||||||||||
|
市政債券
|
|
|
|
|
||||||||||||
|
機構債券
|
|
|
|
|
||||||||||||
|
|
|
|
|
|||||||||||||
|
総資産
|
$
|
|
$
|
|
$
|
|
$
|
|
||||||||
当社はその通貨市場共同基金と米国国庫券を公正価値レベルの下の1級資産に分類しており、これらの資産は活発な市場の見積もりで評価されており、何の推定調整も行われていないからである。同社は、その商業手形と社債、市政債券、機関債券を2級資産に分類し、これらの資産は活発な市場で取引されておらず、第三者価格設定サービスを通じて類似資産の見積もりに基づいて推定されている
F-14
カタログ表
株式証法的責任
当社の株式証負債は総合貸借対照表において他の負債の一部として記録されており、公正価値の経常使用観察不可能な投入(第3級)に従って計量され、帳簿状況は以下の通りである
公平な価値に応じて恒常的に計量される第3級負債変動
|
|
捜査命令
|
|||
|
|
負債.負債
|
|||
|
残高、2022年1月1日
|
$
|
|
||
|
Renovacorを買収する
|
|
|||
|
バランス、2022年12月31日
|
$
|
|
||
責任公平価値を決定する際に用いる仮説−分類株式証−
当社は報告期間ごとにBlack-Scholesモデルを用いて私募株式権証(付記11-“株式承認証”参照)を推定し、総合経営報告書の中で公正価値変動を確認した。株式証負債の推定公正価値は第三級投入を用いて確定された。オプション定価モデルに固有の仮定は、期待株価変動、期待寿命、無リスク金利、配当率と関係がある。私募株式証の期待残存寿命を考慮して、当社は同業グループの歴史変動率から普通株の予想変動率を推定した。無リスク金利は、推定日に基づく米財務省のゼロ金利収益率曲線であり、この期限は私募株式証の期待残存期限と類似している。私募株式証の期待寿命は,その残存契約期間と同じであると仮定した。配当率は歴史金利に基づいており、会社は歴史金利を維持すると予想しています.
私募株式証の公正価値は以下の仮定に基づいて推定される
|
|
2022年12月1日
|
|||
|
株価.株価
|
$
|
|
||
|
行権価格
|
$
|
|
||
|
予想変動率
|
|
%
|
||
|
無リスク金利
|
|
%
|
||
|
期待配当収益率
|
|
|||
|
予想寿命(年)
|
|
|||
|
1部当たり株式証明書の公正価値
|
$
|
|
||
公正価値の2022年12月1日から2022年12月31日までの変化は重要ではない。
| 5. |
財産と設備、純額
|
その会社の財産と設備は:
|
2022年12月31日
|
2021年12月31日
|
|||||||
|
実験室装置
|
$
|
|
$
|
|
||||
|
機械と設備
|
|
|
||||||
| コンピュータ装置 |
||||||||
|
家具と固定装置
|
|
|
||||||
|
賃借権改善
|
|
|
||||||
|
内部使用ソフト
|
|
|
||||||
|
|
|
|||||||
|
減算:減価償却累計と償却
|
(
|
)
|
(
|
)
|
||||
|
$
|
|
$
|
|
|||||
2022年12月31日現在、2021年12月31日と2020年12月31日までの年次償却·償却は3.9 百万、$3.2 百万ドルとドル1.1 それぞれ100万ドルです
F-15
カタログ表
|
6.
|
無形資産と商業権
|
当社の無限生体無形資産には,Renovacorを買収して得られた知的財産権研究開発資産とMICEコロニーモデルがある。
2022年12月31日現在の無形資産の概要は以下の通り
|
|
総帳簿価値
|
累計償却する
|
無形資産、純額
|
|||||||||
|
現在行われている研究と開発
|
$
|
|
$
|
-
|
$
|
|
||||||
|
マウス集団モデル
|
|
-
|
|
|||||||||
|
無形資産総額
|
$
|
|
$
|
-
|
$
|
|
||||||
無形資産の帳簿総生産の増加はRenovacorの買収によるものである(付記17-“Renovacor買収”参照)。
営業権の帳簿価値は1ドルに増加した39.2 2022年12月31日現在、100万ドルであるのに対し、30.8 2021年12月31日現在、Renovacor買収による100万ユーロ(付記
17-“Renovacor買収”参照)。
| 7. |
売掛金と売掛金
|
2022年12月31日と2021年12月31日までの会社の売掛金と売掛金は以下の通り
|
十二月三十一日
2022
|
十二月三十一日
2021
|
|||||||
|
研究開発
|
$
|
|
$
|
|
||||
|
従業員報酬
|
|
|
||||||
|
財産と設備
|
|
|
||||||
|
専門費
|
|
|
||||||
|
買収関連費用
|
|
|
||||||
|
支払うべき政府補助金
|
|
|
||||||
|
他にも
|
|
|
||||||
|
$
|
|
$
|
|
|||||
| 8. |
転換可能な手形
|
2021年の変換可能チケット
2018年1月4日,Inote k PharmPharmticals Corporation(“Inote k”)との逆合併について,当社は未償還の変換可能チケットによりInote kの債務を負担し,オリジナル元金総額は$である52.0 100万元です(“2021年転換可能手形”)。
2022年変換可能手形
当社は2020年2月20日と2020年6月5日に、それぞれ2021年に転換可能な手形のある所有者と単独の非公開交渉交換協定(“交換協定”)を締結した。交換協定によると、当社は2020年2月20日に、2021年の交換可能手形の元金総額約3,940万ドルを(A)
約$に交換します39.4 元金総額は百万ドルである6.25 %変換可能優先チケットの有効期限が切れます(“2022年変換可能チケット”)。また、取引所協定によると、当社は2020年6月12日にドルを両替しました7.5 2021年変換可能手形元金総額
(A)$7.5 新発行債券元金総額6.25 2022年満期の変換可能優先チケットの割合。
2021年4月26日、会社は残りの約$
を償還した38.4 2022年の交換手形元金金額百万ドル会社の株式取引価格として30 初期変換価格の%を超える20 1取引日以内に30 --連続取引期間。2022年に変換可能なチケット残り元本の所持者
は、交換プロトコルの条項に従ってこのようなチケットを約10%に変換する1.3 百万株会社の普通株と現金は、断片的な株式の代わりになる。ASC 470によると-債務2022年の交換可能手形には有益な転換特徴と2022年の交換可能手形の帳簿金額が含まれていないため、2022年の交換可能手形の決済は転換に計上される。2022年12月31日までに違います。 2022年に転換可能な手形は返済されていない
2021年の換算チケット割引の増加額は$0 , $0.3 百万ドルと$1.3 それぞれ2022年12月31日,2021年12月31日,2020年12月31日である。2022年の換算チケット割引の増加額は$0 , $0.5 百万ドルとドル1.5 2022年、2022年、2021年、2020年12月31日までの年度の利息支出は、総合経営報告書に利息支出を計上する。
F-16
カタログ表
| 9. |
株主権益
|
普通株
その会社は現在最も多くの発行を許可されている120,000,000 $の株0.01 額面普通株。発行されたすべての普通株は1株/株の額面で投票する権利がある1 投票を基礎とする。
Br 2014年株式オプションとインセンティブ計画の2回目の改訂と見直し
2018年3月、ロケット取締役会は、会社株主が2018年6月25日に開催された年次総会で承認した第2回改訂·再改訂の2014年株式オプション·インセンティブ計画(以下、“改訂後2014年計画”と称する)を承認した。
在庫株
2022年度に会社が記録した在庫株はbr}$である0.05 百万差し引かれた株式については,RSUに帰属する賃金税の支払いが義務付けられている。あったことがある 違います。 2021年12月31日現在の国庫株。
私募する
当社は2021年8月27日に当社最大株主RTW Investments,LP(“買い手”)の付属ファンドと証券購入プロトコル(“購入プロトコル”)
を締結し,これにより,当社は私募方式(“私募”)で買い手に証券を発行·売却する812,516 会社普通株,買い取り価格は$32.48 1株当たり純収益の合計は約$である26.4 見積もりのbrを差し引いて提供費用を支払うと、百万ドルになります。私募は2021年8月31日に終了します。
市場に計画を提供する
当社は2022年2月28日に、コーエンと市場発売計画について販売合意を締結し、この計画により、当社は時々適宜コーエンを介してその販売代理として株式を発売·売却することができる。販売契約により発売及び売却予定の株式(あれば)は、当社が表S-3を採用した棚上げ登録声明に基づいて発売及び販売を行う。当社は2022年2月28日に米国証券取引委員会に目論見書補充書類を提出し、販売契約に基づいて株式の要約及び売却に関係する内容となっている。会社はコーエンに現金手数料を支払います3.0 販売契約に基づいて株式を売却して得られた総収益の%は、Cowenに慣用的な賠償および出資権を提供している。その会社はCowenに販売協定に関するいくつかの費用を返済したThrough
December 31, 2022その会社はすでに販売しています3.3 市場で計画された100万株を発行し,総収益は
$である48.0 百万ドル、手数料を引きます1.4 100万ドルの純収益46.6 百万ドルです。
公募する
2022年10月6日,会社は公開発行を完了した7,820,000 普通株の公開発行価格は $14.75 1株あたり
。Rocketが公募株から得た総収益は約$115.3 百万ドル純額は$7.2 発売コスト、手数料、法律、その他の費用のうち百万ドルは、純収益$の発売に使用されます108.1 百万ドルです。
| 10. |
株に基づく奨励
|
株式オプション推定値
ブラック·スコアーズ定価モデルにおいて、従業員、非従業員、および取締役に付与される株式brオプションの公正価値を決定するための会社の加重平均は、以下のように仮定される
|
十二月三十一日までの年度
|
||||||||||||
|
2022
|
2021
|
2020
|
||||||||||
|
無リスク金利
|
|
%
|
|
%
|
|
%
|
||||||
|
予想期限(年単位)
|
|
|
|
|||||||||
|
予想変動率
|
|
%
|
|
%
|
|
%
|
||||||
|
期待配当収益率
|
|
%
|
|
%
|
|
%
|
||||||
|
行権価格
|
$
|
|
$
|
|
$
|
|
||||||
|
普通株主公正価値
|
$
|
|
$
|
|
$
|
|
||||||
F-17
カタログ表
改定後の2014年計画に基づき、2022年12月31日と2021年12月31日までの年度の株式オプション活動をまとめた
|
量
株
|
重みをつける
平均値
トレーニングをする
値段
|
重みをつける
平均値
契約書
期限(年)
|
骨材
固有の
価値がある
|
|||||||||||||
|
12月31日までの未返済債務2020
|
|
$
|
|
|
$
|
|
||||||||||
|
授与する
|
|
|
|
|||||||||||||
|
鍛えられた
|
(
|
)
|
|
- |
|
|||||||||||
|
キャンセルします
|
(
|
)
|
|
|||||||||||||
|
12月31日までの未返済債務2021
|
|
$
|
|
|
$
|
|
||||||||||
| Renovacor賞の変換 | ||||||||||||||||
|
授与する
|
|
|
|
|||||||||||||
|
鍛えられた
|
(
|
)
|
|
|
||||||||||||
|
キャンセルします
|
(
|
)
|
|
|||||||||||||
|
12月31日までの未返済債務2022
|
|
$
|
|
|
$
|
|
||||||||||
|
12月31日までに付与され行使可能なオプション2022
|
|
$
|
|
|
$
|
|
||||||||||
|
12月31日までの未帰属オプション2022
|
|
$
|
|
|
$ |
|||||||||||
二零二年、二零二一年及び二零二年十二月三十一日までに、授出された株式購入権は、授出日の加重平均一株当たり公正価値を$とする9.88 , $31.07 , そして$15.10 ,それぞれ分析を行った。
二零二二年、二零二一年及び二零年十二月三十一日までの年間における帰属オプションの総公平価値は34.9 百万、$22.6 百万ドルとドル15.6 それぞれ100万ドルです
制限株単位
(“RSU”)
次の表は、2022年12月31日と2021年12月31日までの年次RSU活動をまとめたものである
|
量
株
|
重みをつける
平均値
授与日
公正価値
|
|||||||
|
12月31日までは帰属していない2020
|
|
$
|
|
|||||
|
授与する
|
|
|
||||||
|
既得
|
||||||||
|
没収される
|
||||||||
|
12月31日までは帰属していない2021
|
|
$ | ||||||
| Renovacor賞の変換 | ||||||||
|
授与する
|
||||||||
|
既得
|
( |
) | ||||||
|
没収される
|
( |
) | ||||||
| 2022年12月31日現在帰属していません | $ | |||||||
内在的なRSUの価値2022年12月31日,2021年,2020年12月31日までの年間帰属額は0.8
million, $0.4 百万ドルと$0.5 それぞれ100万ドルです
2022年12月31日までに13.1 100万ドルの未確認賠償コストRSUに関連する許可していません。この金額は加重平均期間中に確認される予定です2.5 何年もです。
株に基づく報酬
報酬タイプ別に確認された株価報酬は以下の通りです
| 十二月三十一日までの年度 |
||||||||||||
| 2022 |
2021 |
2020 |
||||||||||
|
株式オプション
|
$
|
|
$
|
|
$
|
|
||||||
|
制限株式単位
|
|
|
|
|||||||||
|
取り分で計算した総報酬費用
|
$
|
|
$
|
|
$
|
|
||||||
F-18
カタログ表
合併
経営と総合損失表におけるカテゴリ別株式による補償費用は以下のとおりである
|
十二月三十一日までの年度
|
||||||||||||
| 2022 |
2021 |
2020 |
||||||||||
|
研究開発
|
$
|
|
$
|
|
$
|
|
||||||
|
一般と行政
|
|
|
|
|||||||||
|
取り分で計算した総報酬費用
|
$
|
|
$
|
|
$
|
|
||||||
2022年12月31日現在、同社の総資産は45.6 未確認株による報酬支出は、加重平均期間中に確認される予定です1.3
years.
|
11.
|
株式承認証
|
2022年12月31日まで、ロケット株式証の概要は以下の通り
|
行権価格
|
卓越した
|
付与/負担日
|
期日まで
|
||||
|
|
|
|
|
||||
|
|
|
|
|
||||
|
|
|
|
|
||||
|
|
|
|
|
||||
|
|
|
|
|
||||
| 適用されない | |||||||
|
合計する
|
|
||||||
次の表は、2022年12月31日、2021年12月31日、2020年12月31日までの年間ロケット普通株式購入株式証の変化の概要です
|
|
量
株式引受株式
卓越した和
Exercisable |
行権価格
1株当たり
|
||||||
|
12月31日までの残高は2020
|
|
|||||||
|
2021年8月に承認
|
|
$ | ||||||
|
2021年12月に承認されました
|
||||||||
|
鍛えられた
|
|
|
|
|||||
|
12月31日までの残高は2021
|
|
|||||||
|
Renovacor持分証明書の責任を負う-
|
|
$ |
||||||
|
Renovacor持分証-持分を承認すると仮定する
|
|
|||||||
|
Renovacor持分証-持分を承認すると仮定する
|
||||||||
| 授与する | ||||||||
|
鍛えられた
|
|
|
|
|||||
|
12月31日までの残高は2022
|
|
|||||||
当社は二零二一年十二月三十一日及び二零二年十二月三十一日までに関連側に株式証明書を発行し、非現金研究開発費$を発生させます12.8 百万ドルとドル26.6 (付記18--“関連先取引”参照)違います。
は2022年12月31日までの年度内に,関連側に株式承認証を発行した。
F-19
カタログ表
2021年ロケット権証の公正価値はブラック-スコアーズ公正価値定価モデルを用いて計算され、以下のように計算される
|
現在までの年度
十二月三十一日
2021
|
||||
|
無リスク金利
|
|
%
|
||
|
予想期限(年単位)
|
|
|||
|
予想変動率
|
|
%
|
||
|
期待配当収益率
|
|
%
|
||
|
行権価格
|
$
|
|
||
|
普通株主公正価値
|
$
|
|
||
Renovacorが株式証明書を公開したと仮定する
Renovacorの買収と同時に(“Note 17-Renovacor買収”参照),Rocketは優れたオリジナル製品を仮定している8,622,644
Renovacorが2020年4月に初公開発売に関連して発行した買収前公開株式証(“株式公開株式証”)。すべての公共持分証は最初に所有者に購入させる権利がありますRenovacor普通株で、最初の行権価格は#ドルです11.50 1株1株につき,調整することができる.公開株式証を行使する際には、断片的な株式は発行されない。したがって、公共持分証は以下の倍数で行使されなければならない二つ 以下の事項について出された公共の手令1つは 会社の普通株のシェア。株式公開承認証は満期になります5年 SPAC合併終了日後またはそれ以前に償還または清算時に。買収の結果,これら従来のRenovacor公共株式承認証はRocket引受権証に変換され,購入権がある760,086 ロケット普通株で、使用価格は1ドルです65.23 一株ずつです
会社は一部公開株式証ではなくすべてを償還することができ,償還価格は$とする0.01 公共の権限によって演習中のいつでも少なくとも30 もし会社の普通株の最後の販売価格が$以上の場合にのみ、数日前の書面償還通知が必要です90.75 1株当たりで計算する10 1取引日以内に30 ·当社が株式承認証所有者に償還通知を発行する日前の第3営業日までの取引日期間;かつ償還時および償還期間全体にのみ、当該等の株式公開承認に関連する普通株式について有効な登録声明がある30 −上記取引期間は、その後、償還の日まで毎日継続される。
これまで、上記の条件の中のいくつかの条件はまだ公募株式証の償還を満たしていない。もし当社が公共株式証明書の償還を要求した場合、管理層はすべての公共株式承認証の行使を希望するすべての所有者に、株式承認契約に記載されている“キャッシュレス基礎”に従って公共持分証を行使することを要求する権利がある。引受権証を行使する際に普通株式を発行することができる使用価格および株式数は、配当金または資本再編、再編、合併または合併を含む場合によって調整することができる。また、いずれの場合も、当社は現金純額決済による株式承認証の公開を要求されません
2022年12月31日まで、同社は公共株式証明書が株式分類のすべての基準に符合すると認定した。したがって,合併完了時に公開株式証は
追加実収資本$の構成要素として記録される3.4 百万ドルです。
Renovacor私募株式証明書を仮定して
買収前にRenovacorは突出していました3,500,000 私募取引により、Renovacorと初公開(“IPO”)と同時に発行された引受権証(“私募株式証”)が公開された。各個人持分証はすべて行使して購入することができる1つは Renovacorの普通株、行使価格は$11.50 それは.私募株式証明書は公開株式証と同様であり、私募株式証明書(I)は、所持者に応じて現金で選択することができる(当該等株式証を行使する際に発行可能な普通株式に関する登録声明
は無効)又は無現金で行使することができる点であり、(Ii)いずれの場合も、当該等株式証が初期購入者又はその譲渡者によって所有されることが許可されている限り、当社は償還できない。もし私募株式証明書が非初期購入者またはその譲渡者が所有することが許可された場合、私募株式証は自社で償還することができ、このような所有者は公開持分証と同じ基準で行使することができる。Chardan資本市場で購入した私募株式証明書の使用権は超えてはならない5年 Renovacor IPO発効日から、FINRAルール5110(F)(2)(G)(I)によれば、Chardan Capital Marketsまたはその任意の関係者実益がこれらの私募株式権証を持っていればよい。
買収の結果,個人株式承認証は購入権のある引受権証に変換される617,050
ロケット普通株、行使価格$65.23 一株ずつです。
私募株式承認証はASC 815-40-15が想定する方式で会社の普通株にリンクするのではなく、このツールの保有者は株式に固定オプション定価の投入br株を固定交換するものではないからである。当社はその総合貸借対照表において私募株式証を派生負債に分類している。当社は報告期間終了時に株式証の公正価値を計量し、当社の今期の経営業績において公正価値が前期間より変動していることを確認した。権証負債の公正価値計量討論は付記4を参照されたい。
F-20
カタログ表
Renovacorが事前に持分証を出資したと仮定する
SPAC合併協定に署名すると同時に、Renovacorはいくつかの投資家(“PIPE投資家”)と引受契約(“引受協定”)を締結し、Chardan Healthcare、Old Renovacorのいくつかの株主及びいくつかの他の機関及び認可投資家を含み、これにより、SPAC完了日及びSPAC業務合併が完了すると同時に、PIPE投資家は共同で購入する2,284,776 株式会社の普通株、価格は$10.00 1株当たり,及びあらかじめ出資した引受権証を発行して,その所有者に購入権を持たせる715,224
会社普通株(“予融資権証”)は,初期買付価格は$である9.99 1株当たりの収益は,総収益は約$である30.0 100万ドルですあらかじめ出資した引受権証は直ちに行使でき、行使価格は$となる0.01 そして無期限に行使することができ、事前計画権証の所有者が当該予資権証を行使することを禁止する金額が、当該保有者の私たち普通株に対する実益所有権を超えることを前提としている9.99 %は、この制限を最大で向上させることができます19.99 %はいつでもbr所有者が選択します。
買収の結果、これらの予め出資した権証は権利購入の権利証に変換される126,093
ロケット普通株、行使価格$0.06 一株ずつです。これらの株式承認証は2023年1月に行使される。
2022年12月31日まで、br社は予融資権証が株式分類のすべての基準に符合すると認定した。そのため、合併が完了した場合、公開株式証は追加実収資本#ドルの構成要素として記録される2.3 百万ドルです。
| 12. |
1株当たり純損失
|
普通株主は1株当たりの基本と償却純損失を以下のように計算する
|
12月31日までの年度
|
||||||||||||
|
2022
|
2021
|
2020
|
||||||||||
|
分子:
|
||||||||||||
|
普通株主は純損失を占めなければならない
|
$
|
(
|
)
|
$
|
(
|
)
|
$
|
(
|
)
|
|||
|
分母:
|
||||||||||||
|
加重平均発行済み普通株式-基本普通株式と希釈普通株
|
|
|
|
|||||||||
|
普通株主1株当たり純損失−基本損失と希薄損失−
|
$
|
(
|
)
|
$
|
(
|
)
|
$
|
(
|
)
|
|||
当社は、期末毎の発行済み金額に基づいて以下の潜在的な普通株を記載しており、これらの株式を計上することにより逆償却効果が生じるため、前記期間中に普通株株主が1株当たりの純損失を占めるべき計算範囲には含まれていない
|
12月31日までの12ヶ月間
|
||||||||||||
|
2022
|
2021
|
2020
|
||||||||||
|
2021年交換手形変換後発行可能株式
|
|
|
|
|||||||||
|
2022年交換手形転換後発行可能株式
|
|
|
|
|||||||||
|
普通株式行使可能な引受権証
|
|
|
|
|||||||||
| 普通株に変換可能な制限株式単位 |
||||||||||||
|
普通株購入の選択権
|
|
|
|
|||||||||
|
|
|
|
||||||||||
| 13. |
所得税
|
F-21
カタログ表
法定連邦所得税率で計算される所得税利益と財務諸表に反映される所得税の入金状況は以下のとおりである
|
12月31日までの年度
|
||||||||||||
|
2022
|
2021
|
2020
|
||||||||||
|
法定税率で徴収されるアメリカ連邦税
|
|
%
|
|
%
|
|
%
|
||||||
| 外貨利回り |
( |
%) | ( |
%) | % | |||||||
|
国の税収分配の変化
|
(
|
%)
|
|
%
|
|
%
|
||||||
|
株の報酬
|
|
%
|
|
%
|
|
%
|
||||||
|
譲渡定価調整
|
|
%
|
(
|
%)
|
|
%
|
||||||
|
推定免税額
|
|
%
|
|
%
|
(
|
%)
|
||||||
| 連邦NOL整備 | ( |
%) | % | % | ||||||||
|
税金控除
|
(
|
%)
|
|
%
|
|
%
|
||||||
|
他にも
|
(
|
%)
|
(
|
%)
|
(
|
%)
|
||||||
|
実際の税率
|
|
%
|
|
%
|
|
%
|
||||||
制定された会社税率を採用した後、会社の繰延所得税資産と負債の重要な構成部分は以下の通りである
|
12月31日まで
|
||||||||||||
|
2022
|
2021
|
2020
|
||||||||||
|
繰延所得税資産(負債)
|
||||||||||||
|
研究開発単位
|
$
|
|
$
|
|
$
|
|
||||||
|
純営業損失(“NOL”)と信用繰越
|
|
|
|
|||||||||
|
資本化研究開発コスト
|
|
|
|
|||||||||
|
株に基づく報酬
|
|
|
|
|||||||||
|
債務割引
|
|
|
(
|
)
|
||||||||
|
株式承認証
|
|
|
|
|||||||||
| 無形資産 | ( |
) | ||||||||||
|
他にも
|
(
|
)
|
(
|
)
|
|
|||||||
|
推定免税額
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||
|
繰延所得税純資産(負債)
|
$
|
(
|
)
|
$
|
|
$
|
|
|||||
2022年12月31日現在、同社の連邦純営業損失(NOL)は約$に転換している142.5 百万ドルのうち約$は3.4 100万ドルは以下の時間で満期になります国のNOLとの繰越契約$76.6 百万ドルのうち約$は4.8 100万ドルは#年で満期になりますそれは.減税と雇用法案によると、2017年12月31日以降に開始された納税年度に発生する連邦NOLの有無制限の繰越期間は、毎年使用が許可されているNOL金額が課税収入の80%に制限されている。同社は連邦研究開発信用限度額を持っています21.0
は#年から満期になります2038年12月31日 .
ASC 740に要求されるように所得税そのほか、Rocket管理層はすでにその繰延税金項目資産の現有のプラスと負の影響に対する証拠を評価し、主にNOL繰り越し及び資本化研究開発コストを含む。Rocketは設立日から税務損失が発生したため、経営陣はRocketが連邦と州繰延税項純資産の収益を確認しない可能性が高いと認定したため、2022年、2022年、2021年、2020年12月31日までの繰延税項純資産について、全額推定値を確立した。Rocketは繰延税金資産でいくつかの繰延税金負債を相殺し、これらの資産は同時期に相殺相殺が生じると予想される。2022年12月31日と2021年12月31日までの年間で,推定手当は#ドル減少した2.1 百万ドルとドル7.2 それぞれ100万ドルです繰延税金資産の現金化は未来の課税所得額の発生にかかっている。
米国国税法第382条によると、ある会社が“所有権変更”を経験した場合、同社は変更前の純資産繰越や他の変更前税収属性を用いて変更後の収入を相殺する能力が制限される可能性がある。当社は、当社が第382条で定義された“損失会社”となって以来、所有権変更
が発生したか、または複数回の所有権変更が発生したか否かを評価する研究を完了している。同社は2005年、2007年、2015年、2018年に何度も所有権変更を経験した。所有権変更にはbrがあり,我々の所有権変更前のNOL繰り越しが年間制限され続けることは,所有権変更後の期間にそれらを使用して課税収入を相殺する能力を大きく制限することになる.一般的に、毎年の使用限度額は所有権変更時に私たちの株式の総価値に指定された免税金利を乗じたものに等しい。所有権変更のため,同社の限度額は約$である1.7 私たちは合併前のNOLと開発信用の能力を利用して年間100万ドルに制限しています。この制限により約$が91.2 百万ドル127.1 百万合併前に連邦NOLが満期になり、未使用は累積制限金額として超過します20年間 繰越期間は$35.8 百万ドルです。さらに、$4.9 何百万人もの連邦研究開発信用が満期になり、利用されないだろう。そのため、同社は連邦NOLと連邦研究開発控除に関する繰延税金資産
を合計$減少させた4.9 100万ドルは、推定免税額のそれに応じて相殺される。Renovacorの買収と同時に(付記17-“Renovacor買収”参照)、RocketはRenovacorの連邦NOL$を買収した44.5 百万ドルです。382節の研究は行われていないが,Renovacor NOLと関係があるため,Renovacor NOLの制約は不明である.
F-22
カタログ表
私たちの納税義務を計算する際には、連邦税収およびRocket運営または業務が存在する複数の州の適用における複雑な税金法律法規の不確実性を処理する必要がある。ASC 740は、技術的な是非に応じて、任意の関連する控訴または訴訟手続きの解決策を含む審査によって、不確定な税金状態を維持する可能性がより高い場合、不確定な税金状態からの税金優遇を確認することができると規定している。
米国会計基準第740条によれば、当社は、不確定な税務状況を負債として記録し、評価以前に入手できなかった新たな情報により当社の判断が変化した場合にこれらの負債を調整する。いくつかの不確実性の複雑さのため、最終解決策がもたらす可能性のある支払いは、現在確認されていない税金優遇負債の推定とは大きく異なる。これらの差は、新しい情報が得られる間に所得税費用の増加または減少として反映されるであろう。2022年、2021年、2020年12月31日までに、会社は違います。
当社は、添付されている合併経営報告書において、所得税支出項目が確認されていない税収割引に関する利息と罰金を確認します。2022年2021年2020年12月31日までに違います。
| 14. |
引受金とその他の事項
|
♪the the the会社は最初から一つの手配がレンタルかどうかを確認しています。経営リースおよび融資リースは、会社の総合貸借対照表において、リース、現在の賃貸負債、長期賃貸負債の使用権資産として列報されている。当社のある賃貸契約には契約更新選択権が含まれていますが、当社は、当社がレンタル開始時またはトリガイベント発生時に更新を合理的に決定しない限り、契約更新期間の使用権資産または賃貸負債を確認しません。当社の借款は暗黙的な金利を提供していないため、当社は賃貸支払いの現在値を計算する際に逓増借款金利を推定し、当社の類似期限債務に対する担保借入金利の推定
を用いた。同社はバイオテクノロジー業界が会社の長期借入コストを比較できることに基づき,その逓増借入金利を利用している。Br社は各レンタル構成部分及び関連する非レンタル構成部分を1つの合併賃貸構成部分として計算することを選択したため、すべての契約対価格は合併レンタル構成部分に割り当てられた。同社のいくつかのレンタルプロトコル
には、レンタルバージョンアップ条項(インデックスベースのアップグレードを含む)が含まれている。運営賃貸については、当社は賃貸手配の固定構成要素に基づいて、直線原則で最低賃貸料支出を確認した。Br社はレンタル開始日からのレンタル期間内にこの費用を償却します。可変リース構成部分は,本質的に固定されておらず,指数や料率に関係のない金額を表し,発生した
であることが確認された.
融資リース
同社はニュージャージー州クランベリーに工場の賃貸契約を持っています103,720 オフィス、工芸開発、研究開発実験室、50,000 平方フィートは、会社のパイプラインをサポートするために、AAV現行の良好な製造仕様(“cGMP”)製造施設に特化している。この施設内の比較的小さい面積
は最初に2018年8月にレンタルされ、2019年6月に賃貸契約が改訂され、建物全体が含まれている(このテナントは改訂された、すなわち“ニュージャージー州賃貸協定”)。ニュージャージー州の賃貸契約には十五年 期限は2019年9月1日から、更新がお選びいただけます二つ
連続5年制 条項を更新する。
当社は賃貸開始日を2020年3月15日と決定し、当時所有者が所有していたすべての内装工事がほぼ完成し、当社はその賃貸改造を貸借対照表に計上し、設備をこの空間に移すことを開始した。ニュージャージー賃貸契約が発効した場合、当社が確認した使用権資産総額は$です47.7 百万ドル、相応の賃貸負債は#ドルです20.2 百万ドルです。その会社はドルを再分類します26.5 100万ドルの建設コストが進行中であり1.1 レンタル開始日2020年3月15日使用権資産の一部として前払い賃貸料。
2022年12月31日と2021年12月31日までの年間で、会社は追加の$を再分類しました0.3 百万ドルとドル0.1 進行中の建築コストはそれぞれ100万ドルで、2022年12月31日までの総再定級を32.4 百万の建設中の費用は使用権資産の一部として使用される。
ニュージャージー州賃貸契約のレンタル料は#ドルと推定される1.2 毎年百万ポンド、月の分割払いで、レンタル空間の性質によって、年によって基本的なレンタル料が増加します3 %です。借約の総承諾額から約#ドルと見積もられています29.3 百万ドル以上十五年 レンタル期間。同社は現金br保証金$を支払った0.3
2022年12月31日と2021年12月31日まで、会社の運営と融資リースの制限された現金残高総額は#ドル0.8
F-23
カタログ表
賃貸借契約を経営する
2018年6月7日、当社は締結しました3年制 ニューヨークエンパイアステートビルオフィススペースリース協定(“ESB賃貸契約”)。ESBリース契約について、会社
は取り消すことのできない予備信用状(“帝国信用状”)を設立し、金額は#ドルである0.9 百万ドルです。当社は2021年3月26日に“ESB賃貸契約第1号改正案”(“ESB賃貸改正案”)を締結し、賃貸契約期間を延長するJune 30, 2024 ,
は将来の賃貸料支払いを減少させ,帝国LOCを$に減少させる0.8 百万ドルです。帝国保証金は当社の賃貸借保証金であり、大家は当該借款の受益者であり、賃貸借期間は満了する2024年8月29日 それは.当社は、ESB賃貸契約の改訂入金としてESB賃貸改訂を行い、リース負債を再計測し、経営リース使用権資産を調整します$1.1 2021年12月31日までおよび2021年12月31日までの年度。
会社はドルの預金を持っています0.8 百万ドルとドル0.9 1つの銀行を帝国LOCの担保とした100万ユーロ
は,それぞれ2022年12月31日と2021年12月31日の連結貸借対照表において制限された現金の一部に分類されている.
2018年1月4日、イノクとの逆合併について、当社はマサチューセッツ州レキシントンにあるイノクの前本社の運営リースを担当し、レンタル期間を延長しました2023年2月28日 それは.2018年7月、会社はマサチューセッツ州レキシントン空間部分を転用する協定に調印し、2018年9月にマサチューセッツ州レキシントン空間残り部分を転用する協定に調印した。分譲契約により受け取った賃貸料収入の合計は
$である0.4
当社は2022年12月1日にRenovacorの買収(付記17-“Renovacor買収”参照)を完了し、ニュージャージー州ホプウェルとマサチューセッツ州カンブリッジにある施設を経営する空間を負担し、残りのレンタル条項は約10.25
and 1.3 それぞれ数年です。したがって,会社が確認した使用権資産総額は#ドルとなる1.2 100万ドル相当の賃貸負債総額は$1.0 百万ドルです。さらに、2022年12月31日現在、ニュージャージー州ホプウェルに位置する施設の一部のレンタル開始日はまだ来ていない。したがって、将来の賃貸支払いは約#ドルだ4.3 100,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000ドルは、以下の将来経営賃貸支払表には含まれていません。会社は残りの賃貸条項でこの2つの不動産を転貸するつもりです。
家賃費用は$1.2
million, $1.1 百万ドルと$0.9 2022年12月31日まで、2021年12月31日、2020年12月31日までの年度はそれぞれ100万ユーロ。
|
レンタル料
|
2022年12月31日
|
|||
|
リースコストを経営する
|
$
|
|
||
|
融資リースコスト
|
||||
|
使用権資産の償却
|
|
|||
|
賃貸責任の利子
|
|
|||
|
総賃貸コスト
|
$
|
|
||
F-24
カタログ表
次の表は、2022年12月31日までの会社賃貸負債の未割引キャッシュフローに基づく満期日をまとめ、会社貸借対照表で確認された経営賃貸負債と照合した
|
賃貸負債満期日を経営する
|
2022年12月31日
|
|||
|
2023
|
|
|||
|
2024
|
|
|||
|
2025
|
|
|||
| 2026 | ||||
| 2027 | ||||
| その後… | ||||
|
賃貸支払総額
|
$
|
|
||
|
差し引く:利息
|
(
|
)
|
||
|
リース負債総額を経営する
|
$
|
|
||
|
融資リース負債が満期になる
|
2022年12月31日
|
|||
|
2023
|
|
|||
|
2024
|
|
|||
|
2025
|
|
|||
|
2026
|
|
|||
| 2027 | ||||
|
その後…
|
|
|||
|
賃貸支払総額
|
$
|
|
||
|
差し引く:利息
|
(
|
)
|
||
|
融資リース負債総額
|
$
|
|
||
|
2022年12月31日
|
||||
|
経営的使用権資産
|
$
|
|
||
|
流動賃貸負債を経営する
|
|
|||
|
経営的非流動賃貸負債
|
|
|||
|
リース負債総額を経営する
|
$
|
|
||
|
融資使用権資産
|
$
|
|
||
|
流動賃貸負債への融資
|
|
|||
|
融資非流動賃貸負債
|
|
|||
|
融資リース負債総額
|
$
|
|
||
F-25
カタログ表
|
その他の情報
|
||||
|
賃貸負債に含まれる金額を計量するために支払う現金:
|
||||
|
レンタル経営からの経営キャッシュフロー
|
$
|
|
||
|
融資リースキャッシュフロー
|
$
|
|
||
|
加重平均残余賃貸期間--レンタルを経営します
|
|
|||
|
加重平均残余賃貸期間-融資リース
|
|
|||
|
加重平均割引率-レンタル経営
|
|
%
|
||
|
加重平均割引率-融資リース
|
|
%
|
||
訴訟を起こす
会社はその正常な業務活動に現れる様々な法的手続きやクレームの影響を時々受ける可能性がある。訴訟及び請求の結果は確実に予測できないが、当社は任意の他の請求又は訴訟の一方であるとは信じておらず、当該等の請求又は訴訟の結果が当社に不利であると判断されれば、その個別又は全体がその業務に重大な悪影響を及ぼすことが合理的に予想される。結果にかかわらず、弁護と和解コスト、管理資源の移転、その他の要因により、訴訟は会社に悪影響を及ぼす可能性がある。
弁済手配
会社の定款によると、デラウェア州の法律で許可されている場合、会社は取締役、高級管理者、会社の従業員又は代理人又はそのような職務を担当している者のいずれかに対して賠償義務を負う。同社が将来支払いを要求される可能性のある最大の潜在的な支払い金額は無制限だ。同社はその通貨リスクを減らす保険を持ち、将来支払う任意の金額の一部を回収できるようにしている。したがって、当社はこれらの賠償承諾の推定公正価値はわずかであると考えている。
通常業務全体において、会社は、会社が業務を運営するために必要な商品やサービスを提供するサプライヤーと合意を締結した。場合によっては、サプライヤー協定は、サプライヤーの製品および/またはサービスの使用によるサプライヤーのいくつかの損害の賠償を会社に要求する条項を含む。当社は保険
を持っており、これらの賠償により生じる可能性のある任意の将来の金額の一部を回収することができます。したがって、当社はこれらの賠償承諾の推定公正価値はわずかであると考えている。
| 15. |
知的財産権に関する合意
|
同社はその子会社を介して宇宙船を7人LLCには様々な許可、研究、協力計画がある。これらの取引の主な結果は
が臨床前段階にあり、安全性或いは実行可能性テストを経ていない知的財産権を獲得したことである。すべての場合、同社は有形資産、プロセス、プロトコル、またはオペレーティングシステムを買収していない。当社は,研究や開発活動のための無形資産を他者から購入するコストに基づいて,将来的には他の用途がないうえで,買収日までに買収された知的財産権を支出する。
CIEMATとのライセンス契約
2016年3月、当社はCIEMAT、CIBER、FIISFJD(総称して“CIEMAT”と呼ぶ)とライセンス契約を締結し、Rocket WorldwideがPKD分野でヒトPKLR遺伝子を含むレンチウイルスベクターに関連するある特許、ノウハウ、その他の知的財産権を治療する独占権利を付与した。合意条項によれば、当社は、(A)ライセンス知的財産権によってカバーされる1つまたは複数の製品またはプロセスの規制承認を開発および取得し、そのような製品またはプロセスを商業市場に導入し、その後、それを合理的に公衆に提供する義務があり、(B)許可された知的財産権によってカバーされる少なくとも1つの製品またはプロセスを少なくとも1つの国/地域で開発または商業化する規制部門の承認を得た後は継続的に知的財産権を使用する;(C)許可された知的財産権を適切、道徳的、合法的な方法で使用する。ライセンスの交換として、Rocketは、CIEMATに前金、任意のライセンス知的財産権に関連する製品またはプロセスに基づく純売上に基づく印税支払い、開発および規制マイルストーン支払い、および再許可収入支払いを義務化する。同社はCIEMATと協力し、特許の起訴と維持を担当し、費用は会社が負担する。ロケットはまた、CIEMATと協力して、br侵害および/またはライセンス特許の強制執行および保護に挑戦する第一の責任を負う。ライセンス契約が発効した日から5年以内に、当社はCIEMATが市価で獲得したライセンス知的財産権の任意の改善を優先的に許可する権利がある。ロケットはCIEMAT許可(無料)に創造された許可知的財産権を非商業用途に任意に改善する義務がある。
F-26
カタログ表
許可権の対価格として,ロケットはCIEMATに予備許可料を支払い,金額はユーロである0.03 百万ドル0.03 100万ドル)を研究開発(“R&D”)コストとして支出しています同社には合計ユーロまでのマイルストーン支払いが義務付けられている1.4 百万
(約ドル1.5 100万ドル)は、具体的な開発と規制のマイルストーンを実現する際にCIEMATを付与する。PKDライセンスがカバーする任意の商業化製品については、Rocketは、会社またはその許可可能な人または付属会社の特定の調整に応じて、純売上高に応じて中央桁のパーセントに低い印税を支払う義務がある。ロケットと分許可者とが分許可協定を締結した場合、特定の場合、ロケットは、その分被許可者から受信した任意の対価の一部を支払う義務がある。
ロケットはいつでも本プロトコルを終了することができます。方法はCIEMATに提供します90 数日前にお知らせします。ライセンスは、各国/地域が許可権として取得すべき任意の追加の法的保護が終了するまで、本プロトコルで定義されている各国/地域の有効期間内に有効であり、または各国/地域が許可権として取得されるべき任意の追加の法的保護が終了するまで、許可されている製品またはプロセスをカバーする許可権が存在する限り、有効である。
2016年7月、RocketはCIEMATとライセンス契約を締結し、FA-Aタイプの遺伝子治療のためのVSV-Gパッケージを分割レンチウイルスベクターに統合したヒト治療用途に限定されるいくつかの特許、ノウハウ、データ、および他の
知的財産権を世界的に独占的に所有する許可協定を締結した。本ライセンスは,あらかじめCIEMATの同意を得た場合にのみ再許可することができ,無理に差し押さえられてはならない.合意条項によれば、Rocketは、商業的に合理的な努力(A)ライセンス知的財産権によってカバーされる1つまたは複数の製品またはプロセスの規制承認を開発し、取得し、そのような製品またはプロセスを商業市場に導入し、その後、それを合理的に公衆に提供する義務がある(B)許可された知的財産権によってカバーされる少なくとも1つの製品またはプロセス
を少なくとも1つの国/地域で開発または商業化する規制部門
の承認後数年間連続して使用され、(C)許可された知的財産権が適切、道徳的、合法的な方法で使用される。ライセンスの交換として、会社はCIEMATに前金、製品純売上高または任意の許可知的財産権に関連するプロセスに基づく印税支払い、規制および融資マイルストーン支払い、および再許可収入支払いを義務化している。会社はCIEMATと協力して、特許の起訴と維持を担当し、費用は私たちが負担します。ロケットはまた、CIEMATと協力して、特許侵害および/または挑戦から許可特許を実行し、保護する第一の責任を負う。ライセンス契約が発効した日から5年以内に、当社はCIEMATが市場価値で獲得したライセンス知的財産権のいかなる改善も優先的に許可する権利があります。ロケットは、CIEMAT許可(無料)に、私たちが作成した許可知的財産権の任意のbrを非商業用途に改善する義務があります。
許可権の対価格として,ロケットはCIEMATに予備許可料を支払い,金額はユーロである0.1 百万ドル0.1 百万)は,研究開発コストとして
を支出する.同社には合計ユーロまでのマイルストーン支払いが義務付けられている5.0 百万ドル6.0 100万ドル)は、具体的な開発と規制のマイルストーンを実現する際にCIEMATを付与する。ライセンスがカバーする任意の商業化製品
については、Rocketまたはその許可先または付属会社の純売上高に中央桁から1桁のパーセントの印税を支払う義務があるが、特定の調整が必要である。当社がbr次ライセンサーと再ライセンス契約を締結した場合、当社はその次のライセンシーから受け取った任意の対価の一部を特定の場合に支払う義務があります。
ロケットはいつでも本プロトコルを終了することができます。方法はCIEMATに提供します90 数日前にお知らせします。ライセンスは、各国/地域が許可権として取得すべき任意の追加の法的保護が終了するまで、本プロトコルで定義されている各国/地域の有効期間内に有効であり、または各国/地域が許可権として取得されるべき任意の追加の法的保護が終了するまで、許可されている製品またはプロセスをカバーする許可権が存在する限り、有効である。
LAD-IとCIEMATおよびUCLBのライセンスプロトコル
当社は二零一七年十一月にCIEMAT及びUCL Business PLC(“UCLB”)(総称して“ライセンシー”)とライセンス契約を締結し、LAD−I治療分野のみにヒトLAD−I遺伝子を含むレンチウイルスベクターに関するいくつかの特許、ノウハウ及びその他の知的財産権を世界に独占的に所有している。合意条項によれば、Rocketは、(A)ライセンス知的財産権によってカバーされる1つまたは複数の製品またはプロセスの規制承認を開発および取得し、そのような製品またはプロセスを商業市場に導入し、その後、それを合理的に公衆に提供すること、(B)少なくとも1つの国/地域で少なくとも開発または商業化ライセンス知的財産権によってカバーされる少なくとも1つの製品またはプロセスを使用することを商業的に合理的に使用する義務があるまた(C)許可された知的財産権を適切かつ道徳的かつ合法的に使用する。
は、許可の交換として、会社は、任意の許可に係る知的財産権の製品またはプロセスに基づく純売上の1桁中央値の使用料支払い、br}開発と規制マイルストーン支払い、およびライセンス収入支払いを許可者側に支払う義務がある。ロケットは許可側と協力し,ライセンス特許の起訴と維持を担当し,費用はそれが負担する。当社はまた、ライセンス側と協力して、ライセンス特許を侵害および/または挑戦から実行および保護する第1の責任を負っている。Rocketは、ライセンス契約が発効した日から5年間、ライセンス者が市場価値に応じて獲得したライセンス知的財産権のいかなる改善も優先する権利がある。会社は,それが創造したライセンス知的財産権の任意の改善許可(無料)をライセンス者に非商業用途に使用する義務がある。
許可権の対価格として,ロケットは許可側に初期許可料を支払い,金額はユーロとした0.03 百万ドル0.04 ミリオン),
を研究開発コストとして支出する.その会社には総額ユーロまでの支払いが義務付けられている1.4 百万ドル1.5 百万ドル)は、指定された開発と規制のマイルストーンを実現する際に許可者に交付される。LAD-Iライセンスがカバーする任意の商業化製品
については、Rocketは、会社またはその許可可能な人または関連会社の具体的な調整に応じて、純売上高に1桁の印税を支払う義務がある。当社が再ライセンス者と再ライセンス契約を締結した場合、特定の場合、会社は、当該再ライセンシーから受信した任意の対価格の一部を支払う義務がある。
F-27
カタログ表
ロケットはいつでも許可者に以下のように本プロトコルを終了することができる90 数日前にお知らせします。本ライセンスは、その国/地域において許可された製品またはプロセスをカバーする許可権が存在する限り、または各国/地域が許可権のために取得すべき任意の追加の法的保護が終了するまで、本プロトコルで定義される各国/地域の有効期間内で有効である。
ライセンス カリフォルニア大学サンディエゴ校とダ農病で合意しました
2017年2月、会社はカリフォルニア大学サンディエゴキャンパス(“UCSD”)を代表とするカリフォルニア大学取締役会と許可協定を締結し、この協定に基づき、UCSDはリソソーム貯蔵疾患(達農病を含む)のある知的財産権を治療するための世界的に独占的で再許可可能な許可を与えてくれた。ライセンスの交換として、br社は、プリペイド、いくつかの臨床および商業マイルストーン支払い、使用料支払い(許可知的財産権の範囲内の有効なクレームによってカバーされる製品の純売上高)、維持費、およびbr}の二次許容収入支払いを支払う義務がある。前払い許可証料$0.05 2020年、同社の研究開発費は100万ユーロ。会社には合計$までのマイルストーン支払いが義務付けられている1.5 達農病治療の具体的な開発と監督管理マイルストーンが実現した後、カリフォルニア大学サンディエゴ校に100万ドルを寄付した。マイルストーンの支払いスケジュールを減らすことは、他の適応を達成するための同じマイルストーンに適用される。合意がカバーする任意の商業化製品については,純売上高に低い1桁パーセントの印税を支払うことが義務付けられているが,特定の調整が必要である。再ライセンス者と再ライセンス契約を締結した場合、特定の場合、再ライセンシーから受信した任意の対価格の一部を支払う義務がある。会社はUCSDを用いて本プロトコルにより許可された知的財産権を用いて製品を開発する際には,何らかの職務調査マイルストーンを遵守しなければならない.UCSDとのライセンス契約
の期限はライセンス特許満期であり,その中のいくつかの特許はまだ未定出願段階にある
Regenxbio,Inc.ライセンス
2018年11月19日、会社はRegenxBioInc.(“RGNX”)とライセンス契約を締結し、これにより、会社はRGNXのNAV AAV-9キャリアに関連するすべての米国特許と特許出願の独占許可を取得し、以下の方法で達農病の治療に使用する
in vivoAAV−9を用いて遺伝子治療を行い、任意の既知のLAMP 2トランスジェニック異性体およびLAMP 2トランスジェニック異性体のすべての可能な組み合わせ(“フィールド”)を提供し、(“選択権”)のすべての米国特許および特許出願の独占的許可を付与する二つ この分野の他のNAV AAVキャリア(いずれも“ライセンス特許”であり、総称して“ライセンス特許”と呼ばれる)。
ライセンス契約の条項によると、会社は商業的に合理的な努力を用いて、ライセンス特許を含む製品(“ライセンス製品”)を開発、商業化、マーケティング、普及、販売する義務がある。以下の規定に従ってライセンス契約を早期に終了しない限り、RGNXのライセンスは、ライセンス特許の最後の有効な権利要件の最後の有効な権利要件を、国/地域、ライセンス製品、およびライセンス製品の方法で終了する10年
は、この国/地域で初めてライセンス製品を販売した後です。ライセンス契約は、会社が規定された通知期限内に重大な違約行為を是正していない場合、会社がRGNXまたはそのいくつかのライセンシーに挑戦し始め、ライセンス特許の無効または強制実行ができないことを宣言するか、または会社が倒産または資金が相殺されない場合、RGNXは合意を終了することができる。会社は以下の時間に合意全体を終了するか、または1つまたは複数の許可キャリアを終了することができる6か月 “通知。当社のオプションは満期になりました4年 ライセンス契約の日付から始めます。
ライセンス契約により当社に付与された権利の対価格として,当社はRGNXに$を前払いした7.0 百万ドルです。ライセンス契約では,ライセンス使用料期限内に,ライセンス製品の純売上高に応じてRGNXに支払うライセンス使用料
が1桁から10代まで低いことが規定されている.成功すれば、同社はRGNXに$までの記念碑的支払いを要求されるだろう13.0 米国とEUが指定されたbr臨床開発と規制マイルストーンに達した時、各許可製品は100,000ポンドを獲得した。さらに会社はRGNXを支払わなければなりません20 %ライセンス製品に関連する、または他の方法でライセンス製品に関連する優先審査証明書から受信された支払い費用。第三者からの追加許可が必要であれば,これらの印税義務は
指定の削減を受ける.当社はまた,再許可者から受け取ったすべての非特許権使用料再許可収入(ある場合)の一部をRGNXに支払わなければならない。その会社は$を支払った1.0 RGNX協定によると、2019年に1位に達した患者の調剤後に100万元の許可料を支払う。いくつありますか違います。
| 16. |
CIRM贈与金
|
LAD-I CIRMからの贈与
2019年4月30日、CIRMは同社にガンダムを授与6.6 CLIN 2による贈与奨励により,LVによるRP−L 201遺伝子療法の臨床開発を支援した。この寄付金の収益は、カリフォルニア大学ロサンゼルス校(“UCLA”)美泰児童病院米国臨床サイトに登録された1/2期患者の臨床試験費用と薬物生産に援助するために使用され、同社はカリフォルニア大学ロサンゼルス校(UCLA)微生物学、免疫学と分子遺伝学、小児科(血液/腫瘍学)、分子と医学薬理学の首席研究員Donald Kohn医学博士がリードし、UCLA再生医学と幹細胞研究のELIとEdythe BRoadセンターのメンバーでもある。2019年、会社は最初の二つ CIRMが提供した贈与総額は#ドル1.2 研究開発費の相殺として百万ドル。2020年にはより多くのCIRMマイルストーンを達成し追加ドルを獲得しました1.1 百万マイルストーンは2020年の研究開発費の減少を記録している。会社は追加のマイルストーン支払い#ドルを受け取った1.1 100万ドルと$1.0 2021年1月と4月はそれぞれ100万人。2022年12月31日現在、会社は次の
マイルストーンに達していないため、売掛金は記録されていません。
F-28
カタログ表
国際海事機関CIRMは寄付金を提供します
2020年11月12日、CIRMは同社の最高可達を奨励する3.7 CLIN 2により付与された100万ドルは,LVベースの遺伝子療法RP−L 401の臨床開発を支援し,IMOの治療に用いられた。同社は$を受け取りました1.0 2021年1月4日の支出によると,CIRM IMO賞に関する費用は100万ドルであり,削減された研究開発費$を記録している0.9
2020年12月31日までの年間は百万ドルです。会社が記録した研究開発費は#ドル減少しました0.1 2021年12月31日までの年間
は百万ドルである。2021年12月からロケット賛助を行わないRP−L 401の臨床評価を決定し,学術革新者に返還する予定である
|
17.
|
Renovacorを買収する
|
当社は2022年9月19日にデラウェア州のRenovacor社と合併協議及び計画(“合併合意”)を締結し、これにより、当社は2022年12月1日にRenovacor(“Renovacor買収事項”)を買収する。二零二二年十二月一日、合併協議の条項により、(I)合併第I回合併(“第一次合併”)が当社(“第一次合併”)及び(Ii)合併第II回合併後に存続する会社として合併し、合併第II回合併は第二次合併後も存続する。合併協定の条項と条件により,Renovacor買収完了時に,1回目の合併発効時刻直前に発行されたRenovaor普通株1株が解約され,受領権に変換される0.1763 当社の普通株は、合併協議に記載されている交換
式に基づいて決定され、この式は、取引時のRenovacorの現金純資産レベルに基づいて調整することができる。2022年12月1日の開市前に,Renovacor株はニューヨーク証券取引所での取引を停止し,買収完了後,Renovacorの発行済み普通株を普通株に変換した3,391,976 ロケット普通株。
今回の買収の総対価は$である72.3 $を含む百万ドル62.4 発行された普通株式は百万ドル、$2.7 合併前のサービス期間中に占めるべき株式報酬部分は100万ドルである7.2 株式承認証として百万ドルです。対価は(I)購入日の推定公正価値に基づいて計算される3,391,976 Renovacor普通株のために発行された発行済み普通株,(2)買収予定の従業員株式オプションの推定公正価値367,852 当社普通株、(三)28,798 従業員タイミングにRSU発行の普通株,および(Iv)株式取得承認証を付与する1,503,229 普通株式(“付記11”参照)。
Renovacorを買収する総対価は$72.3 百万ドルには次のようなものがあります
|
|
株
|
価値がある
|
合計する
|
|||||||||
|
株の掛け値
|
|
$ |
|
$
|
|
|||||||
|
現金で値段を合わせる(1)
|
|
|||||||||||
|
株式オプション
|
|
|
||||||||||
|
時間はRSUに帰属する
|
|
|
||||||||||
|
株式承認証を仮定する(2)
|
|
|
||||||||||
|
総掛け値
|
|
$
|
|
|||||||||
|
(1)
|
|
|
(2)
|
|
今回の買収はすでに買収会計方法に従って業務合併として入金され、この方法は買収した資産と負担した負債を買収日にその公正価値で確認することを要求し、買収した知的財産権研究開発資産の公正価値を無期限資産に分類し、成功するまで或いは
が関連研究と開発仕事を放棄するまでに分類する。
F-29
カタログ表
予備買付価格配分は、以下に概説する予備公允価値に基づいて、買収日に買収された資産と負担する負債に以下の金額を分配する
|
運営資本(1)
|
$
|
(
|
)
|
|
|
現金と現金等価物
|
|
|||
|
財産と設備
|
|
|||
|
経営的リース使用権資産
|
|
|||
|
他の非流動資産
|
|
|||
|
知的財産権研究開発
|
|
|||
|
その他無形資産
|
|
|||
|
リース負債を経営する
|
(
|
)
|
||
| 繰延税金負債 |
( |
) | ||
|
取得した純資産
|
|
|||
|
商誉
|
|
|||
|
購入注意事項
|
$
|
|
|
(1)
|
|
買収された知的財産権開発に割り当てられた公正価値は、BAG 3-DCMに対するRenovacorの最先端のAAVベース遺伝子治療によって産生される予想税後キャッシュフローの現在値に基づく。予想される税引後キャッシュフローの現在値は、商業的に実行可能な製品に開発完了した税引後コストを推定し、将来の収入および生産の持続費用を推定し、それによって生成された純現金流量を現在値に割引することによって決定される。使用するコストと収入予測は,異なる開発段階の評価確率に基づいて減少する.主要市場が規制を受けたり開発を停止したりする前に、買収された知的財産権の研究開発は無期限無形資産とみなされる。
購入価格が、識別可能な資産および負担された負債に割り当てられた公正価値を超える部分は、営業権金額#ドルである8.3 買収から生まれた百万ドル。買収の一部の記録としての営業権は主に私たちのポートフォリオと研究能力の拡大、繰延税金と
集合の労働力に起因する。2022年12月31日現在、買収による営業権は非流動資産として当社の総合貸借対照表に記録されており、償却はありませんが、毎年減値審査を行う必要があります。会社
が$を生み出した3.2 2022年12月31日までの年間で、買収に関する一般的·行政コストは100万ドル。
| 18. |
関係者取引
|
2018年4月、会社は取締役会のメンバーと業務発展コンサルティングサービスについて協定を締結した。契約により支払われるサービス料は$28 四半期ごとに当社は14日間 “通知同社が発生した費用は#ドルです0 2022年12月31日までの年間とドル0.1
2020年10月、当社は当社の役員の配偶者と情報技術相談サービスについて相談合意を締結しました。契約に基づいて提供されるサービスの交換として,同社は10,000 1つ以上の限定株式単位に帰属する3年制 ピリオド。
2020年12月21日、当社は関連先と相談協定を締結しました。コンサルティング契約に基づき、関連方向当社はいくつかの業務発展と資産確認コンサルティングサービスを提供しています。相談契約の期限は3年 終了することができます60日 “どちらか一方から通知があります。契約に基づいて提供される業務開発サービスの交換として,当社は以下の権利を行使できる引受権証を発行した603,386 普通株株。コンサルティング契約によると、関連側は、自社の参入許可のために、新資産を決定した後に普通株を行使可能な追加株式権証を取得する権利がある。同社が記録した非現金研究開発費は#ドル26.6 2020年12月31日までの年度は、株式証明書の発行に関連している。
2021年8月9日、br社は行使可能な引受権証を発行した301,291 普通株を同一関連側に譲渡して業務発展と資産識別コンサルティングサービスに利用する(“2021年8月株式承認証”)。同社が記録した非現金研究開発費は#ドル7.6 2021年12月31日までの年間で、2021年8月の権証の発行に関する百万ユーロ。2021年12月17日,当社は行使可能な引受権証を発行した153,155
and 153,155 普通株はそれぞれ同一関連側に業務発展と資産識別のためのコンサルティングサービス(“2021年12月株式承認証”)を付与する。同社が記録した非現金研究開発費は#ドル5.2 2021年12月31日までの年間で、2021年12月の権証の発行に関する百万ユーロ。非現金研究開発費総額は$12.8 2021年12月31日までのbr年度では,2021年8月と2021年12月の権証の発行に関する百万ドルである。
カタログ表
二零二一年八月二十七日、当社は当社の最大株主(“買い手”)RTW
Investments,LPの付属基金と証券購入プロトコル(“購入契約”)を締結し、これにより、当社は私募で買い手(“私募”)を売却·発行することに同意した812,516 会社普通株,買い取り価格は$32.48
1株当たり純収益合計は約$である26.4 当社が支払うべき推定発売費用
を差し引く前に、当社に1,000,000ドルを支払います。私募は2021年8月31日に終了します。また、購入契約を実行するとともに、当社は買い手と登録権契約を締結し、これにより、会社は買い手の要求に応じて、買い手が要求した後に合理的に実行可能な場合に保有する普通株式をできるだけ早く転売することを含むS-3表の登録説明書を証券取引委員会に提出する60日 このようなニーズです
2021年9月、会社は取締役会メンバーとパイプ開発、新資産評価、会社戦略についてコンサルティング契約を締結した。現金支払いの代わりに協議書の下で1年制 期限内に、当社は取締役会メンバーに購入選択権を付与します20,000 同社の株公正価値$の普通株0.4 百万ドルです。
| 19. |
401(K)貯蓄計画
|
1986年の国内収入法第401(K)条によると、会社には固定納付貯蓄計画(以下、計画と略す)がある。この計画は、最低年齢およびサービス要件に適合するほぼすべての従業員をカバーし、参加者が税前に基づいて年間報酬の一部を延期することを可能にする。本計画に対する会社の貢献は会社の取締役会が適宜決定することができます。その会社は4 計画に対する従業員の支払いの割合は、いくつかのbrによって制限される。2022年12月31日、2021年12月31日、2020年12月31日までの年度までの会社の貢献は0.7 百万、$0.6 百万ドルと$0.3 それぞれ百万,
である.
F-25