
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
本財政年度末まで
あるいは…。
手数料書類番号
(登録者の正確な氏名はその定款に記載)
|
|
|||
(法団として設立された国又はその他の司法管区) |
|
(委員会ファイル番号) |
|
アメリカ国税局の雇用主は |
(主な執行機関の住所、郵便番号を含む)
(
(登録者の電話番号、市外局番を含む)
適用されない
(前の名前または前の住所は、前回の報告から変更された場合)
取引法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
取引記号 |
登録された各取引所の名称 |
(ナスダック世界選りすぐり市場) |
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してくださいはい、そうです
登録者が当該法第13条又は第15条(D)に従って報告書を提出する必要がないか否かを、再選択マークで示すはい、そうです
登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告を提出したかどうか、および(2)このような提出要求を過去90日以内に遵守してきたかどうかを、再選択マークで示す
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
|
|
|
ファイルマネージャを加速する |
|
|
|
|
|
|
|
|||
非加速ファイルサーバ |
|
|
|
規模の小さい報告会社 |
|
|
|
|
|
|
|
|
|
新興成長型会社 |
|
|
|
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オキシリー法案”(“米国連邦法典”第15編7262(B)条)第404(B)条に基づいて、その監査報告書を作成または発表する公認会計士事務所の財務報告内部統制の有効性を評価した
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する¨
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す¨
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)はい、そうです
2022年6月30日まで(登録者が最近完成した第2財期の最終営業日),登録者の非関連会社が保有する普通株の総時価は約$である
2023年2月21日現在、登録者の発行済み普通株式数は
Alector、Inc.
表格10-Kの年報
カタログ
|
|
|
ページ |
第1部 |
|
|
|
第1項。 |
業務.業務 |
|
3 |
第1 A項。 |
リスク要因 |
|
45 |
項目1 B。 |
未解決従業員意見 |
|
97 |
第二項です。 |
属性 |
|
97 |
第三項です。 |
法律訴訟 |
|
97 |
第四項です。 |
炭鉱安全情報開示 |
|
97 |
|
|
|
|
第II部 |
|
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
|
98 |
第六項です。 |
[保留されている] |
|
99 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
|
99 |
第七A項。 |
市場リスクの定量的·定性的開示について |
|
107 |
第八項です。 |
財務諸表と補足データ |
|
108 |
第九項です。 |
会計と財務情報開示の変更と相違 |
|
130 |
第9条。 |
制御とプログラム |
|
130 |
プロジェクト9 B。 |
その他の情報 |
|
131 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
|
131 |
|
|
|
|
第三部 |
|
|
|
第10項。 |
役員·幹部と会社の管理 |
|
132 |
第十一項。 |
役員報酬 |
|
132 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
|
132 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
|
132 |
14項です。 |
最高料金とサービス |
|
132 |
|
|
|
|
第4部 |
|
|
|
第十五項。 |
展示·財務諸表明細書 |
|
133 |
第十六項。 |
表格10-Kの概要 |
|
133 |
|
サイン |
|
136 |
|
|
|
|
前向き陳述に関する特別説明
このForm 10-K年間報告書は前向きな陳述を含んでいる。本年度報告に含まれる歴史的事実に関する陳述を除いて、本年度報告に含まれるすべての陳述は、私たちの未来の運営結果と財務状況、業務戦略、候補製品、計画中の臨床前研究と臨床試験、臨床試験の結果、研究開発コスト、監督許可、成功のタイミングと可能性、及び未来運営の管理計画と目標に関する陳述を含み、すべて前向きな陳述である。これらの表現は、既知および未知のリスク、不確実性および他の重要な要素に関連しており、これらの要素は、場合によっては私たちが制御できず、私たちの実際の結果、業績または業績が展望性表現に明示または暗示されている任意の未来の結果、業績、または業績と大きく異なることをもたらす可能性がある。
場合によっては、前向き陳述は、“可能”、“会議”、“はず”、“予想”、“予想”、“計画”、“予想”、“可能”、“意図”、“目標”、“プロジェクト”、“考慮”、“信じる”、“推定”、“予測”、“潜在的”または“継続”またはこれらの用語の否定または他の同様の表現によって識別することができる。本報告書に含まれる前向きな陳述は、以下の態様に関する陳述を含むが、これらに限定されない
1
これらの展望的な陳述は主に、私たちの現在の私たちの業務、私たちが経営している業界、および私たちの業務、財務状況、経営結果、および見通しの財務傾向の期待と予測に基づいており、これらの展望的な陳述は未来の業績や発展の保証ではない。これらの前向き陳述は,本報告発表日までの状況のみを代表しており,“リスク要因”の節や本報告の他の部分で述べたいくつかのリスク,不確実性,仮説の影響を受ける可能性がある。展望性陳述は、リスクおよび不確実性の影響を固有に受けているので、いくつかのリスクおよび不確実性は予測または定量化できないので、未来のイベントの予測として、これらの前向き陳述に依存してはならない。著者らの展望性陳述に反映された事件と状況は実現できない或いは発生できない可能性があり、実際の結果は展望性陳述中の予測結果と大きく異なる可能性がある。法的要件が適用されない限り、私たちは、任意の新しい情報、未来のイベント、または他の理由でも、本明細書に含まれる任意の前向きな陳述を公開または修正するつもりはありません。
また、“私たちが信じている”という声明と類似した声明は、関連テーマに対する私たちの信念と意見を反映している。これらの陳述は,本報告日までに我々が把握した情報に基づいており,これらの情報がこのような陳述の合理的な基礎を構成していると考えられるが,このような情報は限られているか不完全である可能性があり,我々の陳述は,我々が入手可能なすべての関連情報を詳細に調査または検討していることを示していると解釈されてはならない.このような陳述は本質的に不確実であり、あなたにこのような陳述に過度に依存しないように想起させる。
2
第1部
プロジェクト1.BU無邪気ですね。
概要
我々の使命は,神経変性や他の疾患を治癒するために,免疫系の能力を増強する療法を開発することである。
著者らは臨床段階の生物製薬会社であり、免疫神経学の先駆者であり、これは神経変性を治療する新しい治療法である。免疫神経学的目標は免疫機能障害を多様な病理の根本的な原因とすることであり,これらの病理は退行性脳疾患の駆動因子である。我々は,脳の健康な免疫機能を回復させることで,これらの病理変化に同時に対抗する治療法を開発している。我々の研究と薬物発見プラットフォームは我々の科学的方法を支持し,人類遺伝学的検証を経た広範な候補製品の組合せを進めることができ,より短い開発時間で技術成功の可能性を高めると信じている。そこで私たちは100個以上の免疫系標的を決定しました3つの候補製品、latozinemab(AL 001とも呼ばれる)、AL 002、およびAL 101は臨床開発中であり、私たちは私たちの臨床前および研究パイプラインの開発を続けている。私たちは最近、PROGROGINとTREM 2計画に開発資源を集中させ、私たちの現金滑走路を2025年まで延長する優先順位審査を完了しました。私たちは私たちの既存の資源を利用して、私たちのパートナーであるグラクソ·スミスクライン(グラクソ·スミスクライン)とエバーヴィバイオテクノロジー株式会社(AbbVie)の子会社グラクソ·スミスクライン英国有限会社と協力して、私たちの臨床候補製品と研究パイプラインを推進している。
2021年7月1日、私たちは、latozinemabおよびAL 101を含む、グラクソ·スミスクラインと協力して、latozinemabおよびAL 101を含む、GSKプロトコル(GSKプロトコル)を締結し、世界的に協力して前粒子タンパク質のモノクロナル抗体を開発および商業化した。GSK協定の条項によると、私たちは7億ドルの前金を受け取り、そのうち5億ドルは2021年第3四半期に受け取り、2億ドルは2022年第1四半期に受信した。グラクソ·スミスクラインとともに,前頭部痴呆(FTD),アルツハイマー病(AD),パーキンソン病,筋萎縮性側索硬化症(ALS)の候補治療薬が求められている。
Latozinemabは原顆粒(PGRN)を調節し、これは脳中の免疫活動の重要な調節因子であり、多種の神経退行性疾患と遺伝的な関係がある。LatozinemabはFTDの治療に開発されており、これは深刻で進展の速い神経変性疾患であり、アメリカでは5万から6万人、EUでは約11万人に影響する。
Latozinemabは現在全世界の重要な3期試験を行っており、FERRONT-3は、原顆粒遺伝子突然変異(FTD-1)によりFTDリスクがある或いは症状のある成人の潜在的治療に用いられているGRN)である。私たちは2023年に規制部門と接触する準備ができている。会議の目的は,この分野で新たに出現した知識に基づく統計分析計画を検討することであり,これらの計画は,より少ない患者および/またはより短い治療コースで重要なFRONT−3臨床試験を達成できる可能性がある。同社は2025年初めにINFRONT−3試験のデータを読み出し、規制機関からのフィードバックを待つことを目標としている。以前の臨床研究では,latozinemabは原粒子レベルを正常範囲まで向上させることに成功し,バイオマーカーや臨床活動の早期シグナルを奨励した。著者らの1 a期、1 b期と2期の臨床試験において、Latozinemabは健康ボランティアとFTD患者における耐性が良好であった。
2021年には,進行中のオープンタグ第2段階臨床試験FRONT−2がFTD患者で使用されているラトジンモノクロナル抗体のデータを公表したGRNそれは.FORONT−2の設計目的は,治療用量を長期投与したlatozinemabの安全性と耐性を決定し,疾患および臨床結果のバイオマーカーを測定することである。ラトジンモノクロナル抗体の治療耐性は良好であり、治療期間中、血漿と脳脊髄液(CSF)中のPGRNは健康ボランティア年齢が対照群と一致するレベルまで回復した。ベースラインと年齢が一致する対照群と比較して、疾患に関連するリソソーム、炎症とアストログリア細胞増加症のバイオマーカーは12ケ月以内に正常になり、血漿と脳脊髄液神経糸軽鎖(NFL)の平均レベルは12ケ月以内に安定を維持した。容量磁気共鳴画像により、遺伝FTDイニシアティブ(GENFI 2)患者登録中に一致した歴史対照キュー参加者と比べ、latozinemab治療を受けた患者の脳室拡大と脳萎縮の速度が減速することを示した。CDRとNACC FTLD−SBスケールを用いた臨床結果評価では,GENFI 2マッチの対照行列と比較してラトジンモノクロナル抗体治療が48%の臨床進展を遅らせることが分かった。
2022年3月にFTD患者にラトジンモノクロナル抗体を用いたFRONT−2試験の追加データを公表したC 9 ORF 72遺伝子変異(FTD-C 9 ORF 72)は,FTD患者の結果をもとに−GRNそれは.CDR+NACC FTLD−SBスケールを用いた臨床結果評価では,FTD−の治療が認められたC 9 ORF 72病気の患者
3
ALLFTD連盟のマッチング対照行列と比較して、Latozinemabは疾病の経年化進展が約54%遅れる傾向を招いた。また、Latozinemabの治療耐性は良好であり、脳脊髄液(CSF)と血漿中の原顆粒蛋白はそれぞれ生理レベルより2~3倍増加し、神経変性バイオマーカーGFAPが減少した。FTDに関するより多くのデータ更新を提供する予定ですC 9 ORF 722023年下半期の行列。
2021年9月、C 9 orf 72変異ALS患者に対するLatozinemabの第2段階研究を開始した。しかし,筋萎縮性側索硬化症の将来性を考慮して,登録者数を増加させるためにこの研究を閉鎖した。我々とグラクソ·スミスクラインは現在,筋萎縮性側索硬化症におけるlatozinemabの開発計画を評価している。
AL 101は、アルツハイマー病およびパーキンソン病を含むより大きな適応の治療のためのlatozinemabと類似したプロト顆粒タンパク質レベルを向上させることを目的とした、我々PGRN製品の組み合わせの第2の候補である。PGRN発現レベルを適度に低下させる突然変異はアルツハイマー病とパーキンソン病のリスク増加と関係がある。動物モデルでは,PGRNレベルの増加がこれらの疾患に対して保護作用を有することが証明されている。2021年、著者らは著者らの第1段階臨床試験の中期データを公表し、単剤AL 101静脈或いは皮下投与の安全性、耐性、薬物動態、薬効学及びバイオアベイラビリティを測定した。AL 101は末梢と脳中の原顆粒レベルを上昇させ、1ケ月間持続した。AL 101はすべての用量で耐性が良好であった。静脈内および皮下注射の多用量AL 101を試験するために、追加列の登録を完了した。
2022年11月、第1段階研究からの多用量キューのデータを公表した。著者らはAL 101耐性が良好であり、用量依存方式で血漿と脳脊髄液中のPGRNレベルを増加させることを証明した。AL 101の薬物動態学と薬効学特徴はADとPDなどの更に大きな適応の未来の発展を支持する。著者らとグラクソ·スミスクラインは2023年にAL 101計画を推進し、アジア薬物動態学(PK)移行研究を開始し、その後AD早期に全世界第二段階の臨床試験を行った。
著者らのAL 002候補製品は髄様細胞に発現する受容体2(TREM 2)を誘発し、TREM 2シグナル伝達の機能を増加させ、ミクログリア細胞の活性化を増強する。我々は現在AbbVieと協力してアルツハイマー病治療のためのAL 002を開発している。
著者らの第一段階臨床試験において、AL 002は健康ボランティアの中枢神経系において耐性、標的性と機序検証を示した。2021年1月、私たちはInvoke-2を開始しました。これはAL 002の無作為対照第2段階臨床試験であり、私たちの最初の目標は約265人の早期アルツハイマー病を有する参加者を募集することです。我々は現在,研究中に中断した参加者の代わりにAPOE e 4/e 4参加者を含む64名までの参加者を増加させる予定である.2023年1月、最初の患者は私たちのInvoke-2段階2臨床試験の長期延長(LTE)に参加した。2023年2月、私たちはAbbVieとAbbVieプロトコルを修正しました。これは、LTE研究における最初の患者の用量のために1780万ドルの記念碑的支払いを受けることになります。
われわれが行っているADのInvoke−2段階2臨床試験でアミロイド関連像異常(ARIA)を認めた。ARIAのMRI表現は血管源性水腫或いは鉄含有ヘモフラビン沈着を示した。アルツハイマー病のある治療薬、即ち抗β-アミロイド抗体の使用は、アルツハイマー病患者のARIA発生率が増加することを示している。われわれのInvoke−2段階2臨床試験では,ARIA症例の多くは無症状と非重篤であった。しかし,先に報告したように,少数のARIAに関連する重篤な副作用はAPOE e 4/e 4遺伝子型の患者でほぼ完全に発生している。ARIAに関するリスクを低減するために,われわれのInvoke−2段階2臨床試験では,APOE e 4/e 4参加者の用量と登録を自発的に中止した。これらの変化の後,少量のARIAに関連する重篤な有害事象はAPOE e 4対立遺伝子非ホモ接合体の患者で発生した。われわれは早期のMRIモニタリングを継続し,最近発表されたARIAモニタリングと管理ガイドラインと一致している。私たちは独立データ監視委員会(IDMC)の指導の下でこの研究を行い、この委員会は非盲目的データの審査と実験提案を許可された。
AL 044膜貫通に対する4ドメインサブファミリーA(MS 4 A)は、アルツハイマー病の主要なリスク部位であるMS 4 A遺伝子ファミリーメンバーは脳中の髄系細胞に選択的に発現し、ミクログリア細胞機能の制御に関連し、ミクログリア細胞の活性と関係がある可能性がある膜貫通受容体タンパク質をコードする。2022年9月、アルツハイマー病および潜在的孤児の兆候の候補薬剤を開発することを目的として、AL 044の最初のヒト段階試験を開始した。初期PKと耐性データに基づいて,実験を終了することにした.我々はINDに使用可能な候補薬剤を開発するための予備MS 4 A計画を積極的に推進しており、その用量と耐性は改善される可能性がある。
4
脳の神経免疫系はヒトの先天性免疫系の一部であり,我々の免疫神経学における先駆的な仕事に基づいて,われわれのいくつかの治療計画の腫瘍学への潜在的応用が確認されている。天然免疫生物学に集中した製品は,現在適応免疫系に対する免疫腫瘍学的薬物の治療効果を補完·拡大できると信じている。
AL 009は私たちの天然免疫腫瘍候補製品の一つです。AL 009は1種の多シグナル抑制物であり、免疫抑制を駆動する重要な糖鎖チェックポイント経路を遮断することによって、先天性と獲得性免疫系の腫瘍に対する反応を増強することを目的としている。著者らは引き続きAL 009上で臨床前研究と技術活動を行った。私たちはAL 009のグローバル中継権を持っている。
AL 008は私たちのもう一つの天然免疫腫瘍候補製品です。AL 008は1種の抗体であり、CD 47 Sirp-α(Sirpα)経路を抑制することを目的とし、これは腫瘍が免疫系から逃避するために選択した有効な免疫チェックポイント経路である。AL 008はSIRP-αと結合し、免疫抑制と免疫刺激を促進する二重作用機序を有する。我々は2020年にInnoventBiologics(Innovent.)とライセンス契約を締結し,この合意によると,Innoventは中国でAL 008を開発·商業化し,Alectorは世界の他の地域での開発権と商業化権利を保持する.InnoventはAL 008の開発を継続することはなく,AL 008の開発と商業化のためのInnoventtの権利を新たに獲得する予定である.Innoentは中国規制部門にINDを提出し,AlectorはInnovent.から関連材料と情報を取得している。Alectorプログラムは、IND申請のデータおよび文書を評価して、米国におけるINDの提出を潜在的にサポートする。
我々が臨床開発により我々の計画を推進し,“業務−我々の戦略”という節で概説した戦略的方法を実行する努力の一部として,Alectorは時々他の生物製薬会社とパートナーシップを構築する可能性がある。私たちはこれまで、私たちが準備中のいくつかのプロジェクトのために3つの許可、共同商業化、または共同開発協定に署名してきた。
免疫系は神経変性の中枢である
細胞老化或いは肝心な免疫細胞の遺伝子突然変異を調節するため、脳中の健康な免疫機能が失われ、多種の神経退行性疾患の発生と発展の基礎である。ゲノム分析により、神経退化を招きやすい遺伝子突然変異と免疫系機能障害の間に強い相関性があることを表明した。これらの遺伝子変異により脳の免疫機能が悪化し、重要な活動ができなくなります
免疫系が脳内でこれらのすべての重要な機能を実行する能力を回復することは神経変性変化を解決するために重要であると考えられ、過去に単一の退行性病変に集中した方法が疾病進展に与える影響は限られているか、あるいは影響していないからである。
正常な生物老化の一部として,あるいは神経変性に関与し脳免疫細胞の老化加速に関与する有害遺伝子変異により,脳免疫系の機能特徴が徐々に悪化していく。神経変性変化における遺伝子突然変異の作用の理解に基づいて、著者らは著者らの候補製品を設計し、神経変性変化に関連する突然変異遺伝子に対して、脳免疫細胞の退化を緩和或いは逆転し、治療効果を実現することを目標とした。脳の健康な免疫機能を回復することで、神経変性を引き起こす多くの独立した病理に同時に対抗できると信じている。
5
私たちの戦略
我々の目標は,神経変性疾患に対する免疫系を用いた治療法の開発である。この目標を達成するために策定された業務戦略の主な原則は、
私たちの方法は
神経変性における先天性免疫系とミクログリア細胞の役割
過去10年間の重要な証拠により、アルツハイマー病、パーキンソン病、FTDとALSなどの神経退行性疾患は、大脳免疫系機能失調と関係があることを表明した。より広範なヒト免疫系特有の二重適応性と先天性成分と異なり、脳の免疫系は主にミクログリア細胞と呼ばれる先天性免疫細胞から構成されている。これらの脳内に存在するマクロファージは、脳内のすべての細胞の10%~15%を占め、脳の健康と維持の多くの面を担当する。ミクログリア細胞は脳中の重要な先天性免疫細胞として、感染と損傷に反応し、細胞破片と病理蛋白を除去し、神経細胞と脳支持細胞を培養し、神経細胞間の連結の数量と機能を制御する。ミクログリア細胞はずっと著者らが注目している焦点であり、新しい科学進歩は脳中のこれらの肝心な先天性免疫細胞がどのように潜在的な神経変性疾患の治療或いは予防の肝心な焦点を代表するかを理解する可能性がある。
6
図1.我々の臨床段階の神経変性疾患計画
重要な科学データは私たちの仮説を支持しています
私たちの考えでは、脳の免疫細胞が正常と疾病状態でどのようにその構造と機能に影響するかを理解することは、多くの神経疾患を理解するキーポイントである。ヒト遺伝的証拠は脳と先天性免疫系との相互作用の重要性を支持している。例えば、遺伝連鎖研究、候補遺伝子解析、全ゲノム関連研究(Gwas)および全ゲノムまたは全エクソン群配列決定によって決定されるアルツハイマー病の最高リスク遺伝子の多くは、脳の免疫機能を調節する。その中の多くの危険遺伝子は主にミクログリア細胞に発現し、そしてこれらの細胞の機能を制御する。
ミクログリア細胞はすでに脳全体の維持、健康と機能の重要な細胞であることが証明され、脳免疫防御の最初の防御線である。これらの生来の免疫細胞は“ミクログリア感覚体”を持ち、絶えず脳細胞を観察し、病理或いは機能障害の微細な兆候を識別し、反応することができる。ミクログリア細胞は、脳中の有毒な誤った折り畳まれたタンパク質、細胞断片、損傷または不必要な神経細胞、機能失調または老化のシナプスおよび感染性病原体を除去する。また、ミクログリア細胞は新しいニューロンとシナプスの生成を支持し、ニューロン回路を再構築する。ミクログリア細胞はまたアストログリア細胞と少突起グリア細胞の生存と機能を制御し、この2種類の細胞は大脳新陳代謝と血液供給を制御する主要な脳支持細胞であり、損傷後に老化或いは損傷した神経繊維を補充する。そのほか、ミクログリア細胞はすでに血液脳関門の透過性を調節でき、それを末梢免疫細胞に接触させ、感染或いは損傷を防ぐことを助けることが証明された。ミクログリア細胞はまたその形態、機能と数量を変化させ、絶えず変化する脳環境に応答することができる。
正常と疾病脳内ミクログリア細胞の単細胞レベルでの遺伝子転写分析により、ミクログリア細胞は多種の亜型が存在し、それらは脳中の特定の疾病病理に反応する可能性がある。私たちの候補製品はミクログリア細胞の生存、増殖、遊走と機能を制御するミクログリア細胞チェックポイントタンパク質を標的にすることによって、ミクログリア細胞サブタイプを募集することを目的としている。これにより、所定の変性脳疾患に対抗するために、必要に応じてミクログリア細胞の活動を調節することができる。
7
ヒト遺伝学、免疫学と神経科学領域の研究結果により、正常な老化或いは遺伝子突然変異により、ミクログリア細胞の有益な機能退化により、神経細胞の接続機能障害、神経細胞の大量死亡と神経変性を招く。
ミクログリア細胞の力を利用した治療法は神経変性疾患の結果を改善する可能性があることに焦点を当て,単独治療でも抗アミロイドβ蛋白標的治療との併用も信じている。抗アミロイドβ抗体は誤って折り畳まれた凝集体を標識し,ミクログリア細胞を募集して除去した。我々の療法は,我々の原粒子系の療法を含めて,ミクログリア細胞が誤って折り畳まれたタンパク質を除去する能力を増強し,これらのタンパク質を標識する抗アミロイドβ抗体に結合することが予想される。
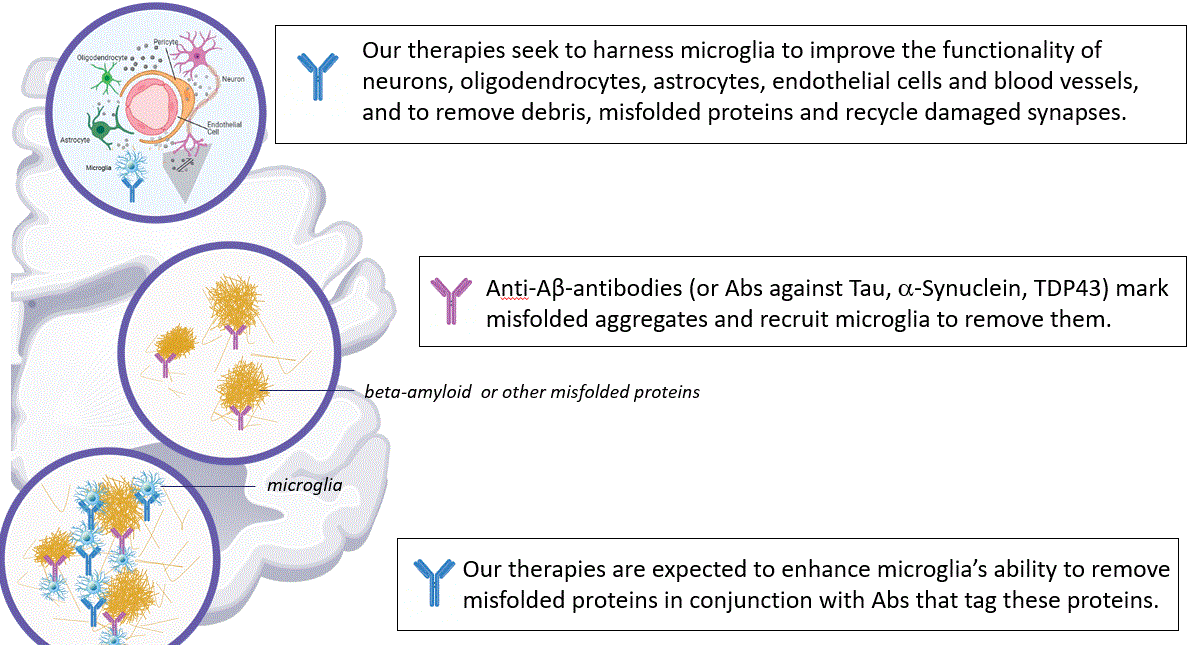
図2.われわれのチェックポイント療法は独立して作用すると予想される
私たちの研究と発見プラットフォームは
著者らの研究と薬物発見プラットフォームはヒト遺伝子データセット、生物情報学と画像化方面の先進的なツール及び神経変性疾患と免疫学方面の知見を利用した:(1)多種の神経変性疾患の発展過程において重要な役割を果たす免疫システム標的を決定し、これらの標的に対する抗体療法を迅速に開発する;(2)バイオマーカーと関連する独自の分析及び臨床前モデルを用いてこれらの標的を尋ね、その活性の優先順位を決定する;(3)候補臨床試験製品をテストし、治療に最も反応する可能性のある遺伝子定義の患者集団でテストを行う。これらのプラットフォーム能力は、神経変性候補薬物の開発に関連する概念と技術的課題を解決するためのツールを提供してくれると信じている。
著者らは独自の免疫神経学バイオインフォマティクスアルゴリズムと方法に基づいて、疾患と健康個体からの大型遺伝子データセット、脳に基づく遺伝子発現プロファイルとプロテオミクス、およびヒト病理学を分析した。これらの特許機能は神経退化を引き起こす免疫機能異常に関連する処理しやすい標的、薬効バイオマーカーと患者集団を迅速に識別することができる。具体的には、私たちのプラットフォームの仕事の優先順位は
8
著者らは遺伝子発現スペクトル、プロテオミクス、脳画像と疾病病理データ及び著者ら自身の臨床前と臨床データを用いて、著者らの独自の免疫神経学アルゴリズムと方法を絶えず完備した。我々の薬物発見プラットフォーム能力を用いて、ヒト遺伝学、疾患バイオマーカー、および反応の敏感な患者集団検証を経た標的を決定し、神経変性歴史薬物開発と比較して、私たちはより有効なスケジュール上でより大きな技術成功確率に定位すると信じている。
我々のルート計画は
図3.下表は著者らの臨床計画、臨床前と研究計画、BBB技術と知的財産権の組み合わせを重点的に紹介した。
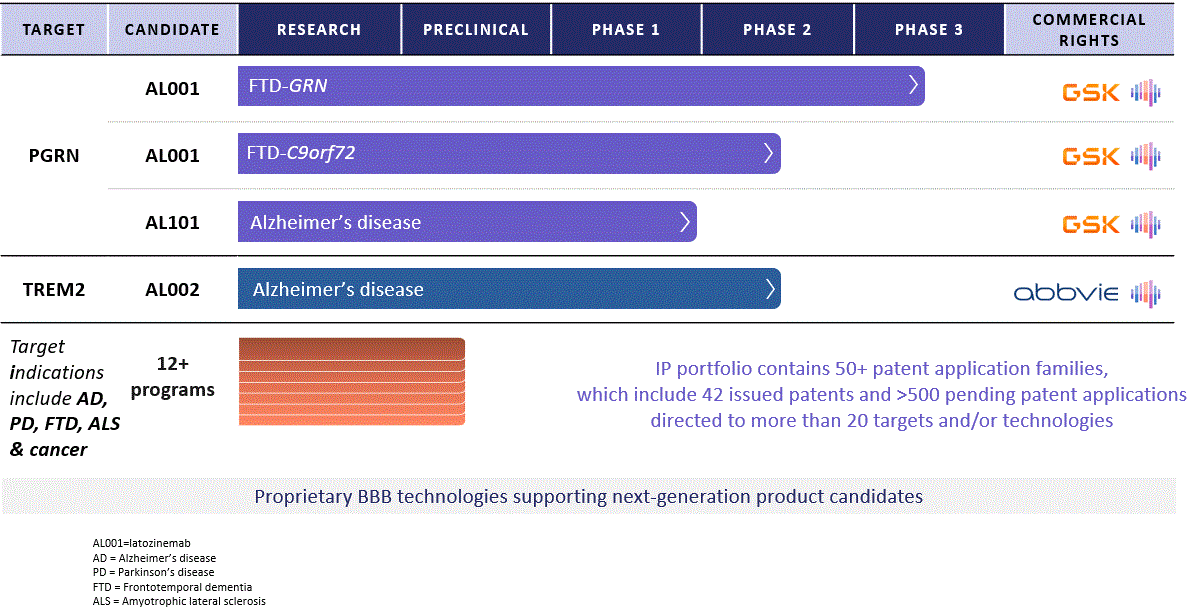
私たちの臨床レジメンはLatozinemab、AL 101、そしてAL 002を含む。また、著者らはアルツハイマー病、パーキンソン病と腫瘍学などの適応を探すために、いくつかの臨床前と研究計画を引き続き推進している。
9
私たちの当初の粒子計画は
著者らの最初の開発計画はPGRNのレベルを調節することに集中し、PGRNは大脳中小グリア細胞機能の重要な調節因子であり、FTDとその他の神経退行性疾患と密接な遺伝関係がある。個体は2コピーのPGRN遺伝子を持ち,それらが共に作用し,全身に健康レベルのPGRNを産生する。PGRN遺伝子の2つのコピーの突然変異は神経細胞蝋様リポ褐素沈着症と呼ばれる神経変性疾患を招き、児童痴呆、視力喪失、てんかんと死亡を典型的な症状とする。PGRN遺伝子の単コピー突然変異はPGRNレベルの50%-70%低下を招き、それによって75歳時のFTDの発生を招き、露出率は約90%である。そのほか、大規模なヒト遺伝学研究により、PGRN遺伝子の制御性突然変異はPGRNレベルを少し低下させ、それによってアルツハイマー病とパーキンソン病のリスクを増加させ、PGRNもこれらの疾病の重要なリスク遺伝子になることを表明した。
健康なPGRNレベルは多くの細胞過程と関係があり、正常なミクログリア細胞活動、神経細胞生存とリソソーム機能を含む。PGRNは機能障害のミクログリア細胞を介して細胞毒性サイトカインと補体因子を放出することが不足し、脳内ミクログリア細胞-ニューロンの動態バランスを破壊し、神経変性を促進する。また,これらのミクログリア細胞はアストログリア細胞を活性化し,ニューロンを損なう。そのため、PGRNの欠乏は神経細胞とミクログリア細胞の健康と機能損傷を招き、是正しなければ、迅速な神経退化を招く。
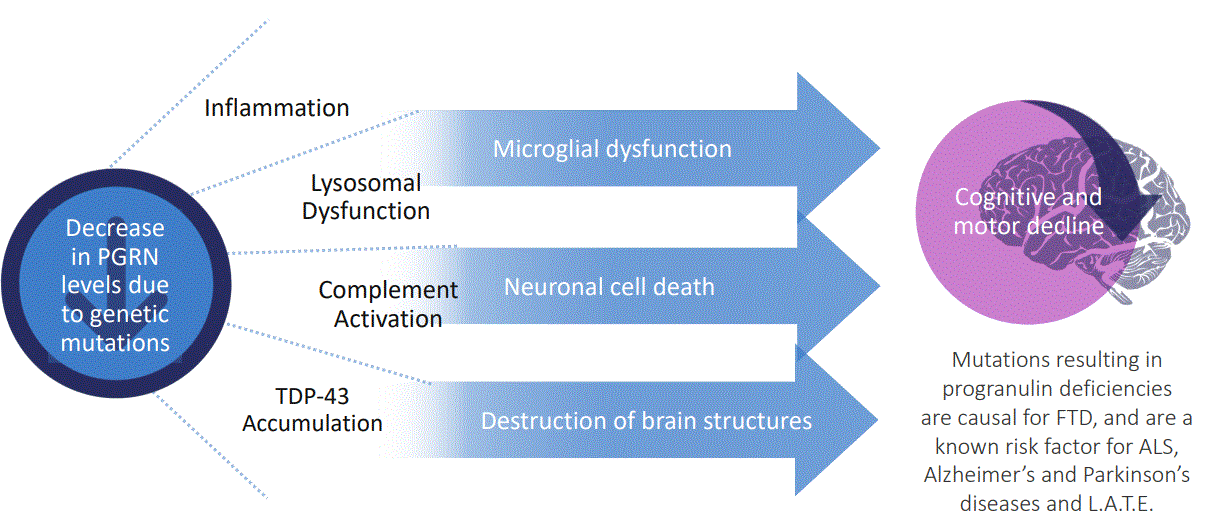
図4.PGRN欠乏はミクログリア細胞と神経細胞の間の動態バランスを破壊し、そして老化過程中に神経退化を促進する。
SORT 1は体内のPGRNレベルを制御する
ヒトとマウスの遺伝学研究により、神経栄養因子PGRN分解受容体Sortilin(SORT 1)は血漿と脳中のPGRNレベルの主要な負調節因子であることを発見した。SORT 1は細胞表面と細胞内小胞体ゴルジ体上の選別受容体である。SORT 1は血漿と脳中の細胞外PGRNと結合し、そしてそれを細胞内に輸送し、リソソームから分解し、細胞外PGRNレベルを低下させる。マウスモデルではSORT 1欠乏はPGRN血漿と脳中レベルを2倍から3倍に増加させるが,ヒトではSORT 1発現がやや低下した変異体はPGRNレベルを増加させる。
また,マウスではSORT 1の遺伝的欠失はPGRNの遺伝的欠失に悪影響を及ぼすことはなく,SORT 1が不足している場合にはPGRNは期待される機能を発揮し続けている。これらの研究や他の研究は,薬物理学的薬物によるSORT 1遮断は安全かつ有効な方法であり,脳における機能性PGRNのレベルを向上させる可能性が示唆された。
著者らはすでに2種類のSORT 1に対する異なる候補製品、latozinemabとAL 101を開発し、患者の脳中のPGRNレベルを向上させ、神経変性疾患における低レベルのPGRNによる損害を相殺することを目的とした。私たちの最初の候補品latozinemabは、PGRN遺伝子機能のコピーを欠く患者(FTD-のような遺伝的形態のFTDを含む孤児疾患の治療を目的としているGRN)である。著者らの第二の候補PGRN製品AL 101はアルツハイマー病とパーキンソン病、およびFTDなどの広範に流行する神経変性疾患の治療を目的としている。グラクソ·スミスクラインと協力しました
10
私たちのPGRN候補製品を開発し商業化します。グラクソ·スミスクラインとの連携については、“グラクソ·スミスクラインとのビジネス戦略連盟”というタイトルの章を参照されたい
LatozinemabとAL 101は米国食品医薬品局(FDA)がFTDを治療するための孤児薬物と、FTD-GRN患者を治療するためのFast Track指定を獲得した。一般に、孤児の薬物名を有する製品が、その後、そのような名称を有する疾患の特定の活性成分に対するFDAの最初の承認を得る場合、この製品は、孤児の薬物排他性を得る権利がある。これは、FDAが7年以内に他のNDAまたはBLA申請を承認しない可能性があり、同じ適応の下で同じ薬物または生物学的製剤を販売している可能性があり、限られた場合、例えば孤児の独占的地位を有する製品に対する臨床的利点を示さない限り、FDAが孤児薬物の指定を撤回した場合、またはFDAが孤児独自の薬物の保持者が指定された薬物を有する疾患または状態を有する患者の需要を満たすのに十分な数の孤児製品を確保していないことを意味する。FDAはFTDを治療するLatozinemabおよびAL 101の孤児薬を承認したが、FDAは依然としてFTDの治療のために異なる有効成分を有する他の薬物を承認することができる。また、孤児薬物の独占性は、孤児の独占期間が満了する前に、FDAが同一の薬物製品の別の異なる適応のマーケティング申請を承認することを阻止しない。孤児薬物の指定は、監督審査と承認過程においていかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。迅速チャネル指定は,重篤な疾患の治療と満たされていない医療ニーズを満たす療法の開発と審査の加速を促進することを目的としている。高速チャネル指定を有する計画は、FDAとの早期および頻繁なコミュニケーション、潜在的な優先審査、およびマーケティング申請のスクロール提出から利益を得ることができる。
ラトジンモノクロナル抗体によるFTD治療の臨床研究
私たちの最初の候補品latozinemabはヒト組換えモノマブであり、FTD患者の脳内のPGRNレベルを増加させることができるGRN病人です。静脈末梢注入により、LatozinemabはPGRNのSORT 1分解機序を遮断することと脳内機能性PGRNの循環半減期を増加させることによって作用する。われわれは最初に前顆粒蛋白遺伝子変異による症候性FTDの治療に用いられるLatozinemabを開発している。
FTDの概要
FTDは進展が迅速で、深刻な退行性の脳疾患であり、現在まだ承認されていない治療方法である。FTDは認知症であり,確定診断時に65歳以下の人に最もよく見られる。FTD患者は強制行為、制約不足、無関心、焦慮及び言語と行為問題を含む一連の個性に関連する症状を表現した。FTDの疾患進展速度はアルツハイマー病より速い。FTD患者の平均期待寿命は症状開始後7年から10年であった。FTD症状は潜在性発症を有し,臨床症状は通常45歳から65歳の間に出現し,平均年齢は58歳である。したがって,FTDは遅発性アルツハイマー病と比較して早発性認知症と考えられ,60歳以下の早発性認知症ではアルツハイマー病よりもよく見られる。
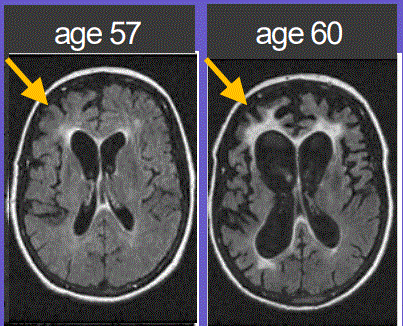
図5.FTD患者の前頭葉と側頭葉萎縮のMRI表現。
人々はFTDについてあまり知られておらず、しかも珍しいと考えられているが、過去10年間に、科学界はすでにFTD生物学に関する知識及び疾病流行に対する認識を獲得した。FTDは米国では約5万人から6万人,EUでは約11万人に影響している。ここにあります
11
多種の遺伝可能な形式のFTD;これまでに、研究者は70個以上のFTDを引き起こすPGRNの遺伝的機能欠損突然変異を確定した。FTD-GRN患者はすべてのFTD患者の5%から10%を占めた。
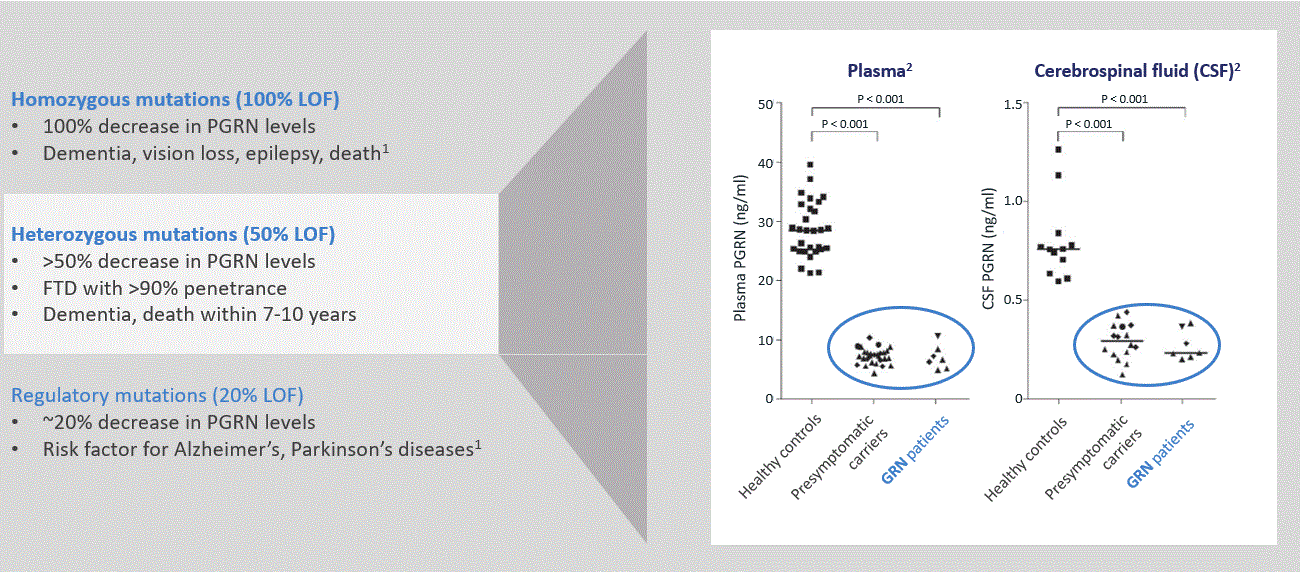
図6.単一PGRNコピーの突然変異は、PGRNレベルの50%以上の低下をもたらし、FTDが発生する可能性が90%を超える(1)Rhinn H,Tatton N,McCeh S,Kurnellas M,Rosenthal A.prograinを神経変性疾患の治療標的とする。薬理学的発展傾向。2022年8月;43(8):641-652。DOI:10.1016/j.tips.2021.11.015。35039149。(2)Dement Geriatr Cogn Disord Extra 2016;6:330-340
FTDでは-GRN患者は,latozinemabによるSORT 1の抑制は潜在的な機序であり,PGRNの50%を超える減少を補償することができる。LatozinemabはSORT 1とPGRNの結合と分解能力を低下させ、その循環半減期を延長することによってPGRNレベルの増加を招くことを目的としている。私たちは様々な動物モデル、健康ボランティア、FTDでPGRNプログラム抗体をテストしましたGRN静脈投与後,患者の脳PGRNレベルは有意に上昇し,持続時間は長かった。
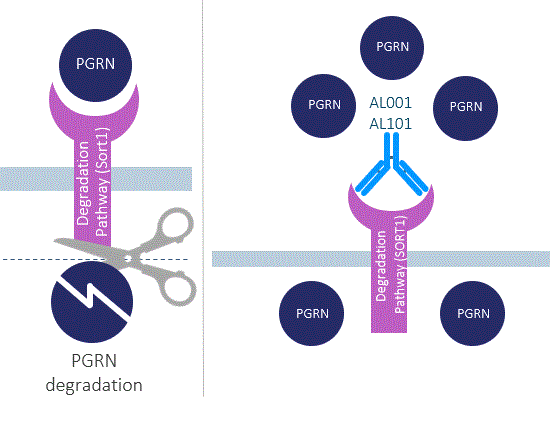
図7.我々のPGRNプログラムの動作機構.LatozinemabはSORT 1と結合し、PGRNの分解を防止し、その循環半減期を著しく増加させる。類似した作用機序はAL 101にも適用される。
12
これまでのPGRN候補製品開発計画と臨床試験結果は
Latozinemabは現在、FTDリスクと症状を有する参加者を含む世界的に重要な段階の3期試験を行っているGRNFORENT−3と命名した。この無作為、二重盲検、プラセボ対照の試験は現在、180名ものFTD-を募集する予定ですGRN変異キャリアはアメリカ、カナダ、ヨーロッパ、オーストラリアの約50の臨床地点に広がっています症状およびリスクのある参加者はランダムに2つのグループに分けられ、4週間ごとにラトジンモノクロナル抗体またはプラセボ静注を受ける。参加者はまた、オープンタグ拡張(OLE)研究における治療を継続することを選択することができる。2022年6月、FORONT-3試験の最初の参加者は、FORONT-3試験において96週間の参加者がLatozinemabを服用した長期的な安全性、耐性、および有効性を評価し、OLEに参加し続けることを目的としたオプションのOLEに参加した。著者らの重要な3期試験の主要な終点はCDR+NACC FTLD-SB評価を使用することによって、臨床低下に対するLatozinemabの影響を測定することであり、この評価は試験参加者の行為、言語、記憶、判断と機能活動における臨床損害を評価する。また、著者らの第三段階試験は二次臨床終点、複数のバイオマーカーと安全性を評価する。私たちは2023年に規制部門と接触する準備ができている。会議の目的は,この分野で新たに出現した知識に基づく統計分析計画を検討することであり,これらの計画は,より少ない患者および/またはより短い治療コースで重要なFRONT−3臨床試験を達成できる可能性がある。同社は2025年初めにINFRONT−3試験のデータを読み出し、規制機関からのフィードバックを待つことを目標としている。
2021年には、進行中のオープンタグ2期FRONT−2臨床試験におけるLatozinemabデータを公表した。FORONT−2の設計目的は,治療用量を長期投与したlatozinemabの安全性と耐性を決定することであり,疾患および臨床結果のバイオマーカーも測定した。12個もの症候性FTDの結果-GRN開放ラベル研究では,12カ月を超える治療を受けた結果,ラトジンモノクロナル抗体耐性は良好であった。治療期間中、ラトジンモノクロナル抗体は血漿および脳脊髄液中のPGRNを年齢に適合した健康ボランティア対照群のレベルに回復させた。
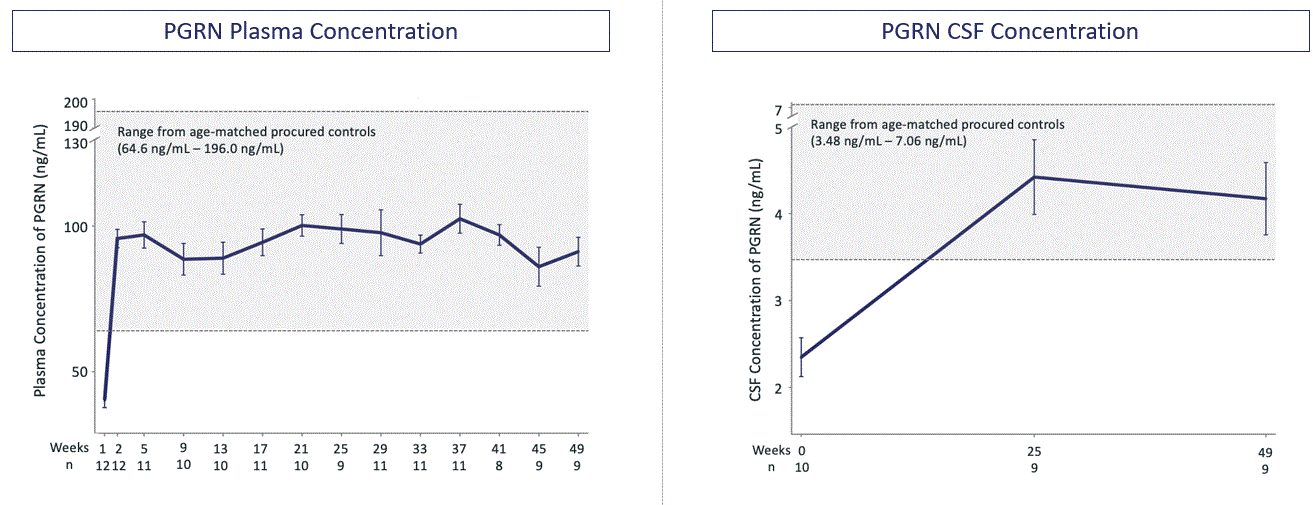
図8:Latozinemabは血漿および脳脊髄液中のPGRNを年齢に適合した健康ボランティア対照群のレベルに回復させた
血漿と脳脊髄液中のPGRNレベルを評価するほか、リソソーム(例えばCTSD、LAMP 1)、補体(C 1 QB)、アストログリア細胞増殖症(GFAP)およびNFLを含む疾患に関連する蛋白を評価した。われわれが2021年に公表した第2段階試験結果では,ベースラインと年齢が一致した対照群と比較して,12カ月の治療では,リソソーム機能,補体活性化,アストログリア細胞増殖症,ニューロン健康の多くの疾患に関連するバイオマーカーが正常化あるいは安定していた。
13
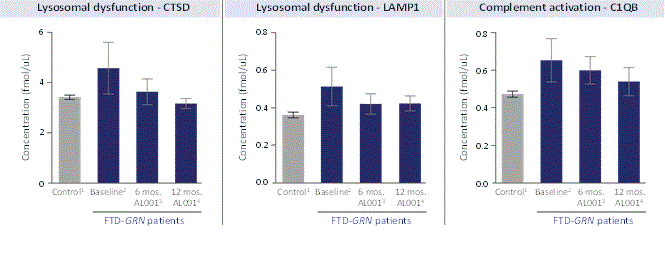
図9.Latozinemab治療は脳脊髄液症候性FTD患者のリソソームと補体バイオマーカーを正常化するGRN患者は我々の第二段階試験に参加した。(1)制御グループは、N=44個の年齢一致の取得対照サンプル、(2)ベースラインN=11個のFTD-GRN参加者、(3)6ヶ月でLatozinemab N=9個のFTD-GRN参加者を使用し、(4)12ヶ月間にLatozinemab N=10個のFTD-GRN参加者を使用する
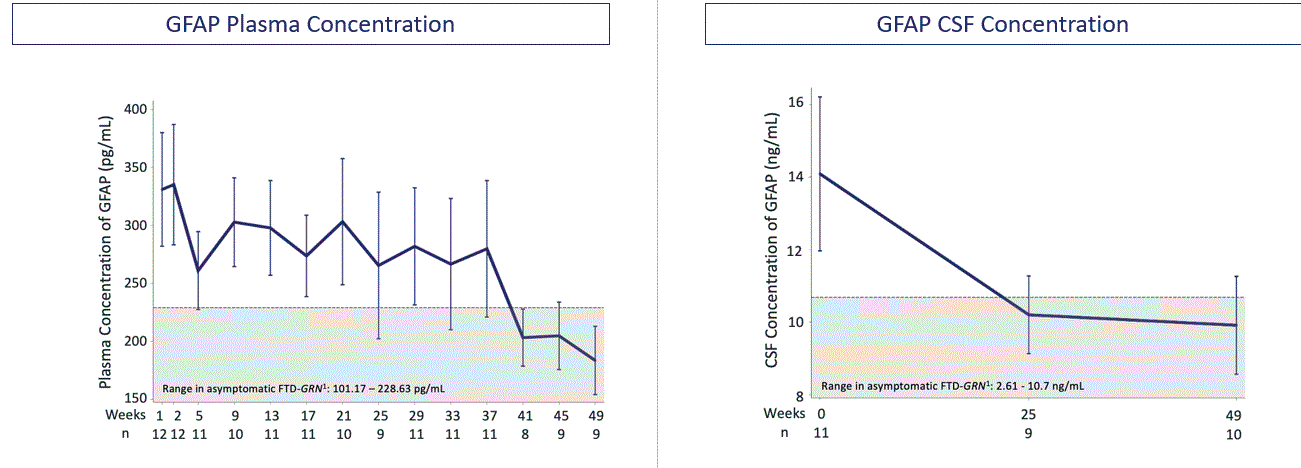
図10.Latozinemab治療は無症状FTD患者の血漿および脳脊髄液中のグリア線維酸性タンパク質(GFAP)レベルを正常レベルまで低下させたGRN参加者は我々の第2段階試験に参加し,アストログリア細胞増加症の減少を示した。
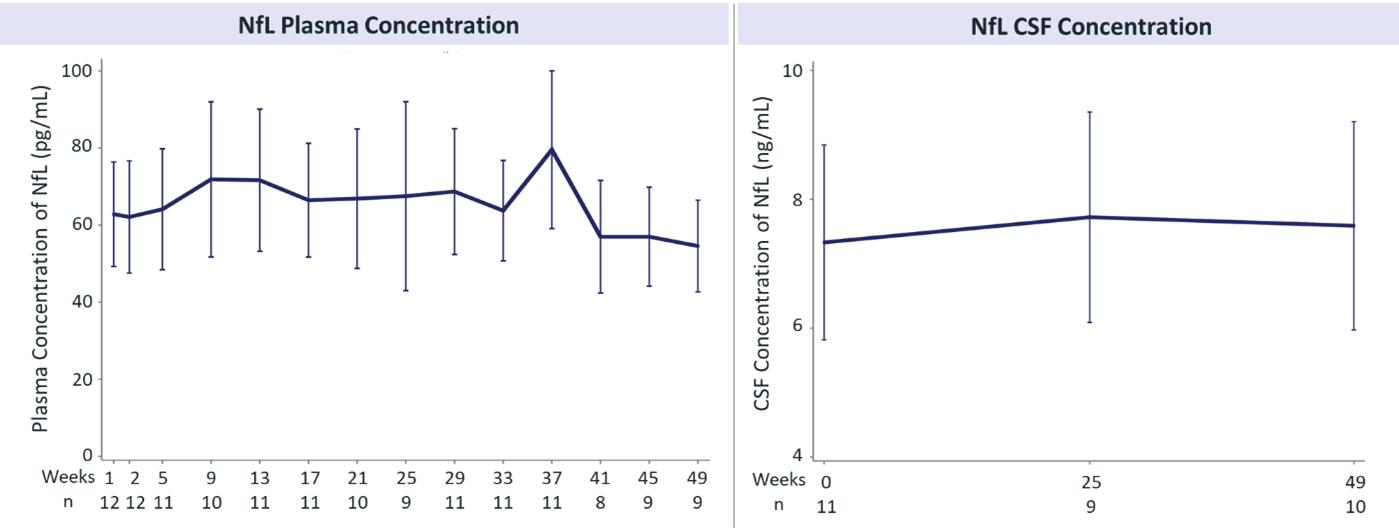
図11.ラトジンモノクロナル治療の症候性FTD患者では,血漿と脳脊髄液中のNFLレベルは12カ月以内に安定していたGRN参加者登録は私たちの第二段階実験に参加した。
オープンタグINFRONT−2試験で観察された臨床結果の背景を提供するために,一致した対照チーム列は10個のFTD−2であったGRNGENFI 2連合からの参加者は,傾向スコアマッチング技術を用いて作成した.GENFI 2患者10人はCDRNACCFTLD SBによるベースラインとさらに
14
年齢,NFLレベルとベースラインの臨床診断によるマッチングは,いずれも盲目的に行われており,縦方向の結果は得られていない。
容量磁気共鳴イメージングを用いて,ラトジン単抗体治療を用いたFTD−FTDの萎縮率の10%以上の低下が認められたGRN全脳と前頭葉皮質の患者群を測定したところ,マッチング対照のFTD−GENFI 2行列と比較して脳室拡大の割合が約50%低下したGRN.
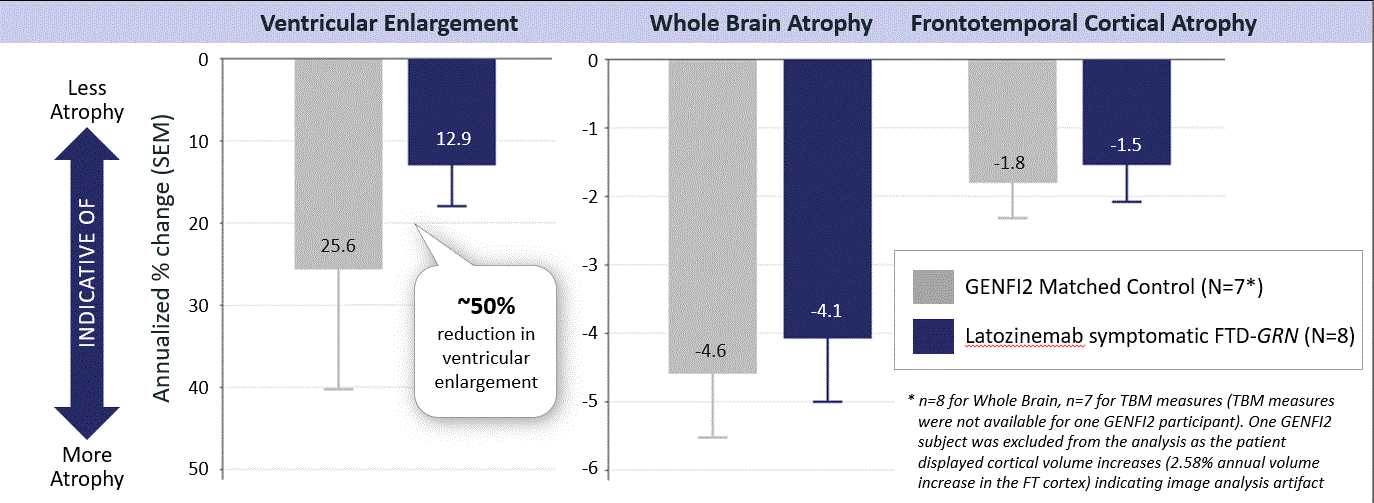
図12.vMRIデータは,ラトジン単抗体治療のFTD患者では,脳室拡大と脳萎縮の速度が低下していることを示しているGRN患者は我々の第二段階試験に参加した。
われわれの第二段階試験では,CDRとNACC FTLD−SBスケールを用いて臨床結果を評価した。CDRPlus NACC FTLD-SBはFTD患者のために開発した臨床痴呆評価量表と国家アルツハイマー病協調中心額側頭葉変性総和評価表である。12カ月後,ラトジンモノクロナル治療は12名の患者の疾患進展速度を48%低下させると推定されている。
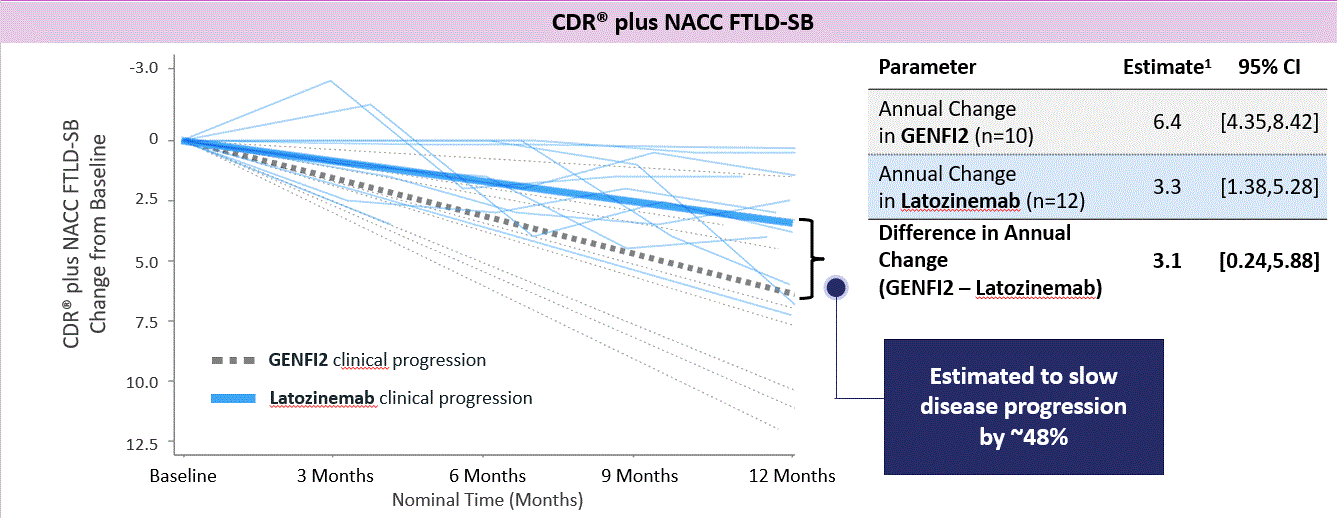
図13.ラトジンモノクロナル治療はFTDの臨床進展が遅いことを示したGRNわれわれの2期試験に参加した患者は,適合したGENFI 2対照群と比較したベースラインを含む繰返し測定のランダム係数モデル&すべての利用可能なベースライン後測定は,最長12カ月に達した。
先の臨床試験ではLatozinemab耐性が良好であり,その作用機序が証明された。健常ボランティアで行った1 a期試験(n=50)では,ラトジンモノクロナル耐性は良好であった。我々の実験段階1 b部分(n=14)では,FTD-GRNあらかじめ指定されたフォローアップ時点で、混合プラセボと比較して、患者の血漿と脳脊髄液中のPGRNレベルはベースラインに対して統計学的に有意に上昇した。またこれらの研究の結果は
15
第一段階の研究により、ラトジンモノクロナル抗体の全体的な耐性は良好であり、試験中に薬物と関連する深刻な不良事件或いは投与量制限不良事件は報告されなかった。

図14.Latozinemabは有症状と無症状FTD患者のPGRNレベルを回復できるGRN患者は健康ボランティアの正常範囲に回復した。
我々のPGRNは潜在的な他のアプリケーションを計画しています
Latozinemabを用いて任意の他の神経変性疾患とより広範なFTD患者集団を治療するために、著者らは特定の患者集団に適した承認を得るために追加の臨床研究を要求される。私たちは追加のFTD患者の遺伝子セット(FTD-C 9 ORF 72)我々のラトジン単抗開放ラベル第二段階臨床試験では、将来的により多くの適応に拡大する可能性がある。
2022年3月、C 9 orf 72遺伝子変異(FTD−FTD)を有するFTD患者においてLatozinemabを用いたFRONT−2試験の追加データを公表したC 9 ORF 72)は,FTD患者の結果をもとに−GRNそれは.CDR+NACC FTLD−SBスケールを用いた臨床結果評価では,FTD−の治療が認められたC 9 ORF 72Latozinemabを服用した患者はALLFTD連盟からの一致対照行列と比較して、疾患の年間化進展が約54%遅延した。

図15.ラトジンモノクロナル治療はFTDの臨床進展が遅いことを示したC 9 ORF 72著者らの第二段階試験に参加した患者は一致したALLFTD対照群と比較した。ALLFTD-ベースライン後の時点で、約12ヶ月。
16
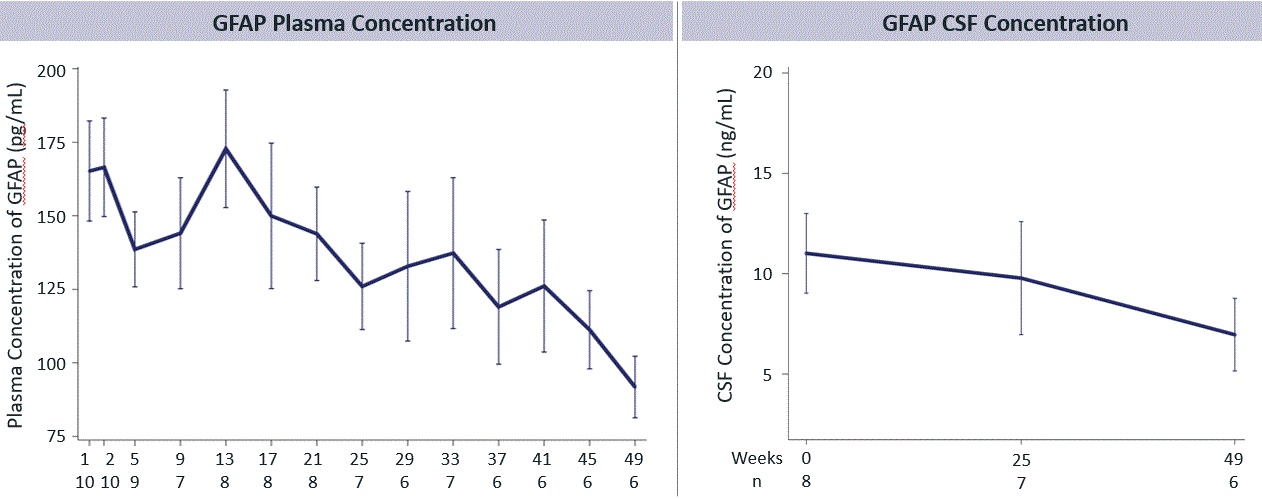
図16.Latozinemab治療はFTD患者の血漿と脳脊髄液中のGFAPレベルを正常レベルまで低下させるC 9 ORF 72参加者登録は私たちの第二段階実験に参加した。
前顆粒蛋白質レベルの低下と9番染色体開放リーディングボックス72(C 9 ORF 72)遺伝子は、TAR DNA結合タンパク質43(TDP-43)の異常蓄積に関連する。TDP-43の脳細胞における過度な凝集は神経細胞死を引き起こすと考えられ、FTD-43-を含む多種の神経変性疾患と関係があるC 9 ORF 72筋萎縮性側索硬化症
2021年、著者らはALS患者におけるラトジンモノクロナル抗体の安全性、耐性、薬物動態と薬効学を評価する第2段階の臨床試験を開始したC 9 ORF 72突然変異です。多種の急性と慢性神経変性モデルを用いた臨床前研究において、文献により、原顆粒レベルの増加はTDP-43の病理変化を逆転でき、そしてそれに対して保護作用があることが示唆された。筋萎縮性側索硬化症の将来性を考慮して,登録者数を増やすためにこの研究を閉鎖した。我々とグラクソ·スミスクラインは現在,筋萎縮性側索硬化症におけるlatozinemabの開発計画を評価している。
筋萎縮性側索硬化の概要
筋萎縮性側索硬化症は壊滅的、致命的、進行性神経変性疾患である。筋萎縮性側索硬化症では,脳や脊髄中の運動ニューロンが死亡し,虚弱,筋萎縮,麻痺をきたし,認知障害をきたすことが多く,呼吸不全をきたす。米国では毎年5,000人以上が筋萎縮性側索硬化症と診断されており,米国では31,000人の患者がこの疾患を有していると推定されている。複数の遺伝子内の変異はC 9 ORF 72遺伝子は,この疾患の原因と考えられている。この変異はTDP−43の細胞への蓄積を招き,ニューロン死を招くことができ,ALS症例の95%がTDP−43の病理に関与していると推定される。家族性筋萎縮性側索硬化症の約40~50%と散発性筋萎縮性側索硬化症の10%までの症例はC 9 ORF 72突然変異です。現在承認されている筋萎縮性側索硬化症の治療薬物は適度な生存利益しかもたらさず、新しい治療方案が切実に必要である。
AL 101アルツハイマー病とパーキンソン病の治療
我々は,ヒト組換えモノクロナル抗体であり,SORT 1に対してもlatozinemabに類似したプロ顆粒蛋白レベルの向上を目的としたPGRN計画における第2の候補製品AL 101を開発している。アルツハイマー病やパーキンソン病などの大型慢性神経変性疾患の治療のためにAL 101を開発している。
PGRN発現レベルを適度に低下させる多型変異はアルツハイマー病やパーキンソン病のリスクを増加させることが証明されており、動物モデルにおいて、PGRNレベルを増加させることはこれらの疾患に対して保護作用があることが証明されている。
2022年、著者らは著者らの第1段階臨床試験の中期データを公表し、単剤AL 101静脈或いは皮下投与の安全性、耐性、薬物動態、薬効学とバイオアベイラビリティを測定した。AL 101は末梢と脳中の原顆粒レベルを上昇させ、1ケ月間持続した。AL 101はすべての用量で耐性が良好であった。静脈内および皮下注射の多用量AL 101を試験するために、追加列の登録を完了した。AL 101耐性は良好であり、血漿と脳脊髄液中のPGRNレベルは用量依存性に上昇した。2回の複数回の投与で
17
(MD)キュー、27名の健康ボランティアがAL 101 30 mg/kgの静注を受け、4週間ごと(Q 4 W)、計4回[n=11]またはAL 101 300 mg、2週間ごとに皮下注射(Q 2 W)、計7剤[n=13]それは.3人のボランティアはMD IVプラセボを受けた。MD IV(Q 4 W)とSC(Q 2 W)投与後,AL 101は全体的な耐性が良好であった。以前に公表された単回用量後のデータと一致して、AL 101は複数回の静注およびSC用量後の脳脊髄液中で測定することができる。投与後の血漿と脳脊髄液中のPGRNレベルは上昇し、AL 101 30 mg/kg MD IV群の上昇幅はAL 101 300 mg MD SC群より高かった。30 mg/kgのAL 101を複数回静脈注射することは、血漿中のPGRNレベルをベースラインの約160%~200%(2.6~3倍)、および脳脊髄液中の約80%(1.8倍)のベースラインレベルに増加させ、維持することができる。単回と複数回の静脈注射後のAL 101のPKとPDスペクトルはADとPDなどの慢性神経退行性疾患の未来の発展を支持する。

図17:我々の第1段階試験では,AL 101治療は健常ボランティアのPGRNレベルを増加させた。
アルツハイマー病の概要
アルツハイマー病は慢性神経変性疾患であり,通常65歳以上の人では発症が遅く,時間の経過とともに悪化する。これは認知症の最もよく見られる原因であり,全例の60%から70%を占めている。アルツハイマー病の最もよく見られる早期症状は最近起こったことを覚えにくいことだ。疾病の発展に伴い、症状は言語問題、方向性障害、情緒変動、動力喪失、自己ケアと行為問題を含む可能性がある。人の病状が低下すると、彼らは往々にして家庭と社会から離れるだろう。徐々に身体機能が失われ、死に至る。進行速度が異なる可能性があるが、確定診断後の典型的な期待寿命は8年から10年である。
アルツハイマー病罹患率の推定はそれぞれ異なるが,アルツハイマー協会は2022年,65歳以上の米国人のうち650万人を超える人がアルツハイマー病を患っていると推定しており,2060年にはこの数は2倍近く増加すると予想されている。アルツハイマー病はアメリカの六番目の死因であり、65歳以上の人の第五の死因でもある。
アルツハイマー病は患者の認知と日常機能を弱める以外に、医療保健システムに重大な負担をもたらした。アルツハイマー協会のデータによると、2022年の米国アルツハイマー病と他のタイプの認知症患者の総介護コストは3210億ドルと推定され、その3分の2近くは連邦医療保険システムが負担している。2050年までに,米国でアルツハイマー病や他の認知症患者に提供される医療,長期ケア,ホスピスの総支出は1.1兆ドル以上に増加すると予想される。
パーキンソン病概要
パーキンソン病は長期の中枢神経系退行性疾患であり、主に運動系に影響する。疾患早期に最も顕著な症状は震え,硬直,行動遅延,歩行困難であった。認知や行動問題も起こりうる。認知症は病気の末期によく見られるようになった。抑うつと焦慮もよく見られ、3分の1以上のパーキンソン病患者は抑うつと焦慮が発生する。他の症状には感覚、睡眠、情緒問題が含まれる。パーキンソン病
18
通常60歳以上の人たちの中で起きています確定診断後の平均期待寿命は症状出現後3年から10年の間であった。
パーキンソン病に対する疾患修飾療法は現在のところなく,患者の選択は症状を改善する治療に限られている。最初の治療は通常抗パーキンソン病薬であるレボドパであり,レボドパがそれほど有効でなくなるとドパミン作動薬が使用される。疾病の発展と神経細胞の消失に伴い、これらの薬物の効果はますます悪くなり、同時に不随意捻転を標識とする合併症を産生する。
パーキンソン財団のデータによると、世界で1000万人以上がパーキンソン病を患っている。毎年約9万人の米国人がパーキンソン病と診断され,2030年には120万人の米国人がパーキンソン病に罹患すると推定されている。パーキンソン財団のデータによると、米国だけでは、パーキンソン病の直接と間接コストは、治療、社会保険支払いと収入損失を含めて、毎年520億ドル近くと推定され、2037年までに、アメリカの毎年のコストは約800億ドルと推定されている。
我々のTREM 2計画は
TREM 2は膜貫通受容体蛋白の一種であり、一部の天然免疫細胞に発現し、脳中のミクログリア細胞に選択的に発現する。ミクログリア細胞上のTREF 2は細胞の損傷部位への遊走を促進し,細胞生存率を向上させ,貪食機能を増加させ,細胞増殖を促進すると考えられている。ホモ接合TREM 2突然変異或いは2つの染色体コピー突然変異を有する稀な個体は40歳前に神経退行性変化に発展する可能性があり、確定診断後の平均寿命は10年である。研究により、TREM 2の2つのコピーの1つの遺伝子変異はアルツハイマー病のリスクを3倍に増加させることを発見した。単コピーTREF 2の変異はアルツハイマー病のリスクを著しく増加させるだけでなく、TREM 2変異を持たない人と比較して、TREM 2変異を有するアルツハイマー病患者の症状出現時間は3年早く、脳体積損失率は増加した。証拠はまた、機能突然変異の獲得はTREF 2発現の増加を招き、それによってアルツハイマー病に保護性表現型を提供することを表明した。
2013年にTREM 2はアルツハイマー病と強い遺伝的関連があることが発見され、これは大規模なゲノム解析を使用して稀な遺伝子変異を識別し、遅発性アルツハイマー病のリスク増加に関連した最初の例の一つである。
TREM 2は通常脳に発見されるアポリポタンパク質E(ApoE)などの膜脂質やリポ蛋白質に結合する。ApoE遺伝子の多型もアルツハイマー病のリスクを著しく増加させると考えられ,アルツハイマー病の単一最高リスク因子である。
アルツハイマー病治療のAL 002
著者らの候補製品AL 002はヒト化し、TREM 2を活性化するモノクロナル抗体であり、静脈と末梢輸液による送達を目的としている。AL 002はTREF 2受容体を調節するミクログリア細胞調節因子である AbbVieと協力してアルツハイマー病治療のための薬剤を開発している。AbbVieとの連携に関するより多くの情報は、“AbbVieとのビジネス-戦略連合”というタイトルの部分を参照してください
19
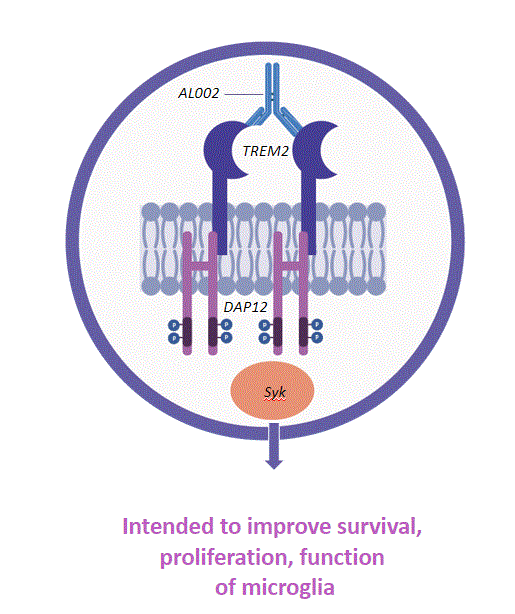
図18.我々のTREF 2が候補製品AL 002を活性化する作用機序。
アルツハイマー病を治療する方法はまだありません。FDAはすでに2種類の疾病の潜在病理に対する治療方法を許可し、アミロイドβ蛋白プラークが減少し、そして潜在的な臨床メリットがあることを証明した。他の承認された治療カテゴリは、アセチルコリンエステラーゼ阻害剤およびグルタミン酸作動性調節剤を含む対症療法を含む。これらの薬物はニューロン通信の保存を助けることを目的としているが、一時的な利点のみを提供し、神経細胞の死亡を遅らせることや阻止することはない。また,抗うつ薬や抗精神病薬はラベル外に処方されることが多く,騒動,攻撃的行動,精神病,抑うつを有する患者の重篤なアルツハイマー病症状の治療に用いられる。
最近開発されているアルツハイマー病治療の候補薬には、合成を遮断し、脳内の誤ったフォールディングを増強するアミロイドβタンパク質またはtauタンパク質の除去または脱重合、慢性炎症の逆転、血管機能障害の修復、代謝障害、および神経毒性の増強に焦点を当てた薬剤がある。これらの候補薬剤のほとんどは,複数のアルツハイマー病病態の1つに対して設計されており,これまでこれらの候補薬剤の多くは有意な利点を示さなかった。
脳中のアミロイドβプラークとtau蛋白はこの疾患の物理的病理を代表し、脳中のニューロン結合の喪失とニューロンの死亡を引き起こすと考えられているが、最近の科学データはより複雑な図を描いている。より有効な治療はミクログリア不全に関連する問題を含む多種の病理問題を解決する必要がある可能性が考えられる。
私たちのTREF 2臨床計画は
2021年1月、私たちはアルツハイマー病早期患者で私たちの第2段階試験を開始した。このランダム、二重盲検、プラセボ対照、用量範囲の大きさのマルチセンター第二段階試験の目的は、APOE e 4/e 4参加者を含む研究中の中断の代わりに、世界最大90地点で約265人の早期ADを有する参加者を募集することであり、私たちは現在、APOE e 4/e 4参加者を含む研究中の中断の代わりに64人の参加者を増加させることを計画している。著者らの第二段階試験の主要な終点は臨床認知症分級総和(CDR-SB)を用いて疾病の進展を測定する。この試験はまた、複数の液体とイメージングバイオマーカーを測定し、いくつかの二次臨床、薬物動態学と薬効学的終点を評価し、肝心な第三段階研究を支持するデータを生成する安全性を評価する。2023年1月、Invoke-2研究の第1の参加者は、私たちの第2段階臨床試験のLTEに薬物を登録して服用した。私たちは2023年第3四半期にInvoke-2の試験登録を完了する予定で、2024年第4四半期にトップラインデータがある予定です。
20
われわれが行っているADのInvoke−2段階2臨床試験でARIAが観察された。ARIAのMRI表現は血管源性水腫或いは鉄含有ヘモフラビン沈着を示した。アルツハイマー病のある治療薬、即ち抗β-アミロイド抗体の使用は、アルツハイマー病患者のARIA発生率が増加することを示している。われわれのInvoke−2段階2臨床試験では,ARIA症例の多くは無症状と非重篤であった。しかし,先に報告したように,少数のARIAに関連する重篤な副作用はAPOE e 4/e 4遺伝子型の患者でほぼ完全に発生している。ARIAに関するリスクを低減するために,われわれのInvoke−2段階2臨床試験では,APOE e 4/e 4参加者の用量と登録を自発的に中止した。これらの変化の後,少量のARIAに関連する重篤な有害事象はAPOE e 4対立遺伝子非ホモ接合体の患者で発生した。われわれは早期のMRIモニタリングを継続し,最近発表されたARIAモニタリングと管理ガイドラインと一致している。我々はIDMCの指導の下でこの研究を行い,IDMCは非盲目的データの審査と実験提案を許可された。
2019年,AL 002を有する健常ボランティアで1 a段階臨床試験(n=56)を完了した。われわれの第1段階試験では,AL 002は単回漸増用量部分耐性が良好であった。そのほか、治療過程中に脳脊髄液中の可溶性TREM 2(STRM 2)とミクログリア機能下流バイオマーカーの用量依存性と統計学的意義上の著しい変化を観察し、これは健康ボランティアの中に標的の参与があり、また機序の検証があることを表明した。われわれの第1段階健常ボランティア試験で観察された耐性,および鼓舞的なバイオマーカーデータに基づいて,アルツハイマー病患者でAL 002を用いて試験の1 b段階を開始した。しかし,これまで臨床前研究および健康ボランティアで収集してきたデータに基づき,我々のパートナーであるAbbVieと一致し,新冠肺炎の大流行の影響を受けた1 b段階試験の登録を終了し,第2段階試験の開始に移行した。著者らの第一段階の臨床試験において、AL 002は健康ボランティアとアルツハイマー病患者の中枢神経系において耐性、標的性と機序証明を示した。

図19.我々のAL 002期臨床試験では、健常ボランティアにおいて、ミクログリア活性化バイオマーカーであるsTREM 2の用量依存性低下およびCSF−1 Rの増加が観察された脳脊髄液サンプルは5つの最高用量の列から採取した。表示されたデータは、34人の健常ボランティアの脳脊髄液サンプルの分析から(*はpを表す)
TREM 2の臨床前データは
AL 002はミクログリア細胞表面のTREF 2に結合し、脾臓関連チロシンキナーゼ(Syk)のリン酸化によってミクログリア細胞の活性を最適化することを目的としている。著名な学術協力者と共に、AL 002 Sは機能的にAL 002と類似しているがマウスTREM 2と交差反応する抗体であり、アルツハイマー病に関連する遺伝子発現特徴を正常化し、アルツハイマー病マウスモデルにおいて病理を減少させることができることを証明した。また、AL 002 cは、マウスFc領域を持つAL 002であり、ヒトTREM 2の正常或いは遺伝リスク変異体を発現する深刻なアミロイドマウスモデルにおいて、ミクログリア細胞増殖を誘導でき、アミロイドプラークが緻密であり、そして損傷ニューロンに関連する栄養不良軸索を減少できることが証明された。
21
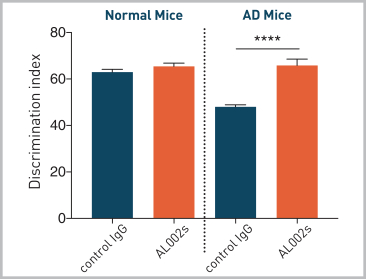
図20.AL 002はアルツハイマー病マウスモデルの認知欠陥を統計学的に有意に改善した(*はpを表す
MS 4 A計画は
MS 4 A遅発性アルツハイマー病の最も顕著な遺伝リスク遺伝子の一つである。リスクバリエーションがありますMS 4 A遺伝子座はアルツハイマー病罹患率の増加と発病年齢の低下と関係があるMS 4 A遺伝子ファミリーメンバーは脳中のミクログリア細胞に選択的に発現し、ミクログリア細胞機能の制御に関連し、ミクログリア細胞の活性と関係がある可能性がある膜貫通受容体タンパク質をコードする。当社のAL 044候補製品は相殺することを目的としていますMS 4 A遺伝子ファミリーが遺伝子のリスク変異体を機能的に変換しますMS 4 A遺伝子ファミリーの保護的変異。2022年9月、アルツハイマー病および潜在的孤児の兆候の候補薬剤を開発することを目的として、AL 044の最初のヒト段階試験を開始した。初期PKと耐性データに基づいて,実験を終了することにした.我々はINDに使用可能な候補薬剤を開発するための予備MS 4 A計画を積極的に推進しており、その用量と耐性は改善される可能性がある。
Alectorの新興天然免疫腫瘍学的パイプライン
免疫腫瘍学
私たちの発見プラットフォームを免疫腫瘍学のような他の適応にも拡張しました天然免疫生物学に集中した製品は,現在適応免疫系に対する免疫腫瘍学的薬物の治療効果を補完·拡大すると信じている。ミクログリア細胞の遺伝子発現と機能は末梢或いは非脳の先天性免疫系の細胞と類似している。これらの末梢天然免疫細胞、例えばマクロファージ、単球、NK細胞などは、癌、炎症と自己免疫性疾患を含む多種の慢性疾患において重要な役割を果たしている可能性がある。我々は,SIRP蛋白ファミリーやSiglec蛋白ファミリーに対する計画を含め,末梢疾患,特に癌を治療するための,我々の先天免疫系に関する専門知識を用いて,他の先天免疫チェックポイントに重点を置いた計画を開発している。
22
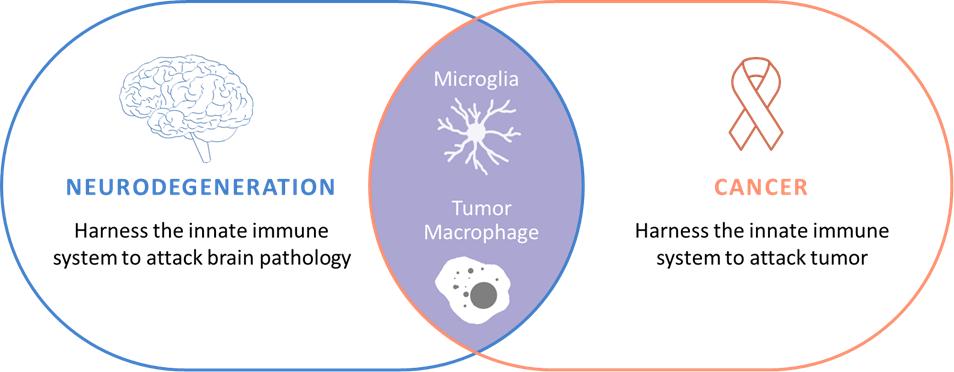
図21である.神経変性と癌は先天性免疫系に集まっている。腫瘍関連マクロファージとミクログリア細胞の間には多くの機能重複が存在すると考えられる。
AL 009、私たちのマルチシンボル計画
AL 009は私たちの天然免疫腫瘍候補製品の一つです。AL 009は1種の多シグナル抑制物であり、免疫抑制を駆動する重要な糖鎖チェックポイント経路を遮断することによって、先天性と獲得性免疫系の腫瘍に対する反応を増強することを目的としている。AL 009は1種の標的シアル酸トラップであり、先天性免疫細胞上のSiglec阻害シグナルを優先的に遮断し、腫瘍学的適応において治療メリットを提供することを目的としている。著者らは引き続きAL 009上で臨床前研究と技術活動を行った。私たちはAL 009のグローバル中継権を持っている。
AL 008、我々のSIRP-Alphaプログラム
AL 008はSIRP-αを抑制するモノクロナル抗体である。AL 008の二重作用機序はマクロファージ上の受容体の内化と分解を誘導することによって免疫抑制を解除し(“私のシグナルを食べないで”を閉鎖する)と同時にFcガンマ受容体と結合して免疫刺激経路を促進し、それによってCD 47-SIRP-α経路を非競争的に拮抗し、それによって抗腫瘍免疫を推進する。主に髄系細胞に発現するSIRP-α受容体を標的とすることにより、AL 008はいくつかのCD 47標的薬で観察された抗原沈下と標的副作用を回避できる可能性があると信じている。さらに、AL 008は、SIRP−Gammaのような他のSIRPファミリーメンバーと交差反応することなく、SIRP−αに選択的に結合するように設計されている。この特異性は、AL 008がCD 47−SIRP−α経路を活性化しながら、CD 47−SIRP−ガンマ経路を活性化するT細胞を保持しながら抑制性CD 47−SIRP−α経路を拮抗させることを可能にする。我々は2020年にInnoventとライセンス契約を締結し,この協定によると,Innoventは中国でAL 008を開発·商業化し,Alectorは世界の他の地域での開発権と商業化権利を保持する。InnoventはAL 008の開発を継続することはなく,AL 008の開発と商業化のためのInnoventtの権利を新たに獲得する予定である.Innoentは中国規制部門にINDを提出し,AlectorはInnovent.から関連材料と情報を取得している。Alectorプログラムは、IND申請のデータおよび文書を評価して、米国におけるINDの提出を潜在的にサポートする。
総合療法
われわれの療法も相互作用したり,病的タンパク質を除去するための他の実験薬とともに機能している可能性がある。アミロイドβ蛋白、tauフィラメント或いは誤って折り畳まれたα-シヌクレインに対する抗体などの治療法は病理性蛋白質を標識することを目的とし、そしてミクログリア細胞を募集して薬物/病理性蛋白質複合体を処理する。老化したミクログリア細胞がこの機能を有効に実行することはあまり不可能であり,われわれの免疫神経学的療法はこの欠陥を改善することができる。われわれは臨床前モデルにおける各種の組合せ策略を探索し、将来的に臨床前モデルの結果に基づいてこの策略を臨床に応用することを考える予定である。
23
グラクソ·スミスクラインとの戦略同盟
概要
2021年7月、私たちはグラクソ·スミスクラインと、latozinemabおよびAL 101を含む、グラクソ·スミスクラインと世界的に協力して、latozinemabおよびAL 101を含む前の顆粒タンパク質のモノクロナル抗体を開発および商業化する協力および許可協定に署名した。グラクソ·スミスクライン協定は2021年8月17日に施行された。
GSK協定の条項によると、私たちは7億ドルの前金を受け取り、そのうち5億ドルは2021年8月に受け取り、2億ドルは2022年1月に受け取った。さらに、私たちは15億ドルまでの追加臨床開発、規制、商業発表に関連したマイルストーン支払いを得る資格があるかもしれない。アメリカでは、各当事者はLatozinemabとAL 101の商業化された利益と損失を二等分する。アメリカ以外では、私たちは2桁の等級版税を得る資格があります。
双方はlatozinemabとAL 101を共同開発し,グラクソ·スミスクラインはアルツハイマー病,パーキンソン病と他の孤児適応の第三段階臨床試験を行う。開発コストはグラクソ·スミスクラインが60%,40%を分担するが,開発計画下の初期二期臨床試験の開発コストを独自に負担し,双方が製造開発コストを平均的に分担する。
アメリカでは、双方はLatozinemabとAL 101の商業化を共同で担当し、私たちは孤児適応の商業化をリードし、グラクソ·スミスクラインはアルツハイマー病、パーキンソン病と他の孤児適応の商業化をリードする。米国以外では,グラクソ·スミスクラインは独自にすべての適応のLatozinemabとAL 101の商業化を担当する。私たちは製品ごとに開発コストと米国で商業化された損益を分担しないことを選択することができる。この場合、製品の開発や商業化は行われなくなり、会社は米国での純売上高に応じて利益や損失を共有するのではなく、等級別特許権使用料を得ることになる。グラクソ·スミスクラインはいつでも180日前に契約終了を通知することができるが、会社は受け取った部分支払いを返済する必要はない。
統治する。協力は共同指導委員会(JSC)によって管理される。司法員叙用委員会は、特定のプロジェクトや活動を監視するための追加のグループ委員会を設置することができる。“GSK協定”に規定されている制限に適合する場合、適用される管理委員会は協議一致方式で決定することができず、各当事者が問題を各当事者が指定した高級管理者に報告することによって問題を解決することができない場合、この問題は紛争解決案に代わるものにグレードアップするが、各当事者が保留している最終決定権の制限を受ける。
排他的である。GSKプロトコルの有効期間内には,AlectorおよびGSKは排他的な規定を遵守し,GSKプロトコル以外の何らかの活動がGSKプロトコルでの目標を指すことを禁止する必要がある.
知的財産権。“グラクソ·スミスクライン協定”に基づいて設立された知的財産権の所有権は一般に在庫状況に基づいて決定される。一般的に、私たちは、私たちが単独で開発または双方が共同開発した特許を含む、米国内でのライセンス特許の起訴と維持を制御する権利があり、グラクソ·スミスクラインは、アメリカ国外でこのような特許の起訴と維持を制御する優先権を持っている。グラクソ·スミスクラインは特定の第三者製品がこのような特許を侵害したことを起訴する優先権を持っている。双方は,どちらがGSK合意の標的となるいずれかの計画に基づいて開発された製品が第三者の知的財産権を侵害するクレームに対する抗弁を制御すべきであることを共同で合意すべきであり,双方がこのような合意に達していない場合には,このようなクレームを提出する側が優先的に抗弁する権利がある.
任期と解約期間。グラクソ·スミスクライン協定期間内のいつでも、便宜上、グラクソ·スミスクラインは、指定された通知期間後に、グラクソ·スミスクライン協定全体を随時終了することができる。さらに、GSKまたは私たちは、他方がGSKプロトコルに深刻に違反したためにGSKプロトコルを終了することができ、このプロトコルは、指定された期間にわたって治癒されていない。
エバーヴィとの戦略同盟
概要
2017年10月、私たちはエバービーと共同開発とオプション協定に署名した。AbbVieとのグローバル戦略協力の主な目標は、アルツハイマー症や他の神経変性疾患を治療する療法を共同開発し、商業化することである。
24
AbbVieプロトコルによると,我々は我々のTREM 2とSiglec 3プロジェクトにAbbVieのグローバル開発と商業化の独占的選択権を付与した.AbbVie協定の条項は2.05億ドルの初期前払いと2000万ドルの株式売却を含む。著者らは第一段階と第二段階の研究の設計と実行を担当し、アルツハイマー病の臨床試験を実行する上で著者らの重要な内部専門知識を利用した。2022年6月、AbbVieはSiglec 3計画の終了通知を提供した。2023年2月、私たちはAbbVieとAbbVieプロトコルを修正し、これは、研究中の最初の患者の用量を長期延長するための1780万ドルの記念碑的支払いを受けることになるだろう。もしAbbVieがTREM 2計画の選択権を行使すれば、私たちは2.5億ドルを得るだろう。また、AbbVie協定の条項によると、規制部門の最大3種類の適応の承認と関連がある2.25億ドルまでの追加マイルストーン支払いを受ける資格がある。AbbVieがTREM 2計画に対して選択権を行使した後、開発コストや利益を共有しないか、製品販売を得る等級別印税に変更することを選択する可能性があります。AbbVieがTREM 2計画の選択権を行使した後、AbbVieはその全世界の臨床試験の専門知識と商業化ネットワークを利用して、ある開発活動と全世界の商業化を担当する。このパートナーシップにより,両組織の優位性を有効に利用し,期待した結果を最適に実現することを目標としている.
オプションを行使する。AbbVieは選択権の期限が満了するまで、TREM 2計画に対する選択権を随時行使することができる。オプション期間は,AbbVieが第2段階臨床試験を完了した後にパケットを受信した一定時間後に終了し,TREF 2計画の研究や開発活動に関するいくつかの情報が含まれている。AbbVieが候補製品のオプション期間内にそのオプションを行使できなかった場合、TREF 2計画のすべての権利を保留する。もしAbbVieがそのTREM 2計画の選択権を行使すれば、AbbVieは世界の開発と商業化活動をリードするだろう。AbbVieがある候補製品を選択すると、AbbVieは商業的に合理的な努力をし、世界規模で相応の製品を開発し、それを商業化しなければならない。
統治する。協力は共同指導委員会(JSC)によって管理される。司法員叙用委員会は、特定のプロジェクトや活動を監視するための追加のグループ委員会を設置することができる。“AbbVie協定”に規定されている制限に適合する場合、適用される統治委員会は、協議一致方式で決定することができず、各当事者が問題を各当事者に指定された上級執行幹事に報告することによって問題を解決することができない場合、この問題は紛争解決方法に代わるものに格上げされるが、各当事者が保持している最終決定権を遵守しなければならない。
排他的である。AbbVieプロトコルの期間内に、私たちとAbbVieは排他的な要求を遵守し、AbbVieプロトコル以外のAbbVieプロトコルの下で目標とするいくつかの活動を禁止しなければならない。
知的財産権。“AbbVie協定”によって設定された知的財産権の所有権は一般に在庫状況に基づいて決定される。一般的に、すべての当事者は自分の特許を優先的に起訴して維持する権利を持っている。AbbVieがこのような特許に関連する計画に対して選択権を行使する前に、私たちは通常連合特許を起訴して維持する権利があり、AbbVieはこのような選択権を行使した後に共同特許を起訴し、維持する権利がある。AbbVieはAbbVieプロトコルによって開発された共同所有特許とAbbVieプロトコルによって許可された私たちの特許の任意の侵害に対して訴訟を提起する権利がある。しかも、AbbVieは自分の特許を起訴する権利がある。AbbVieはTREM 2計画に基づいて開発された製品が第三者の知的財産権を侵害した疑いに対して抗弁する権利があり、この製品はAbbVie協定のテーマである。
任期と解約期間。AbbVieプロトコルの間のいつでも、研究、開発、および臨床試験中に含まれ、AbbVieはAbbVieプロトコルを完全に終了することができ、または便宜上、AbbVieプロトコル下のTREF 2計画を終了することができる。この場合、TREM 2計画に関連するすべての権利は私たちのものだ。さらに、AbbVieまたは私たちは、他の当事者がAbbVieプロトコルに実質的に違反したためにAbbVieプロトコルを終了することができ、そのプロトコルは指定された期間にわたって治癒されていない。
Adimab協力協定
概要-2014年Adimab連携協定(2014 Adimabプロトコル)
2014年、私たちは2014年のAdimab協力協定(2014 Adimab協定)を締結した。2014年のAdimab協定によると、私たちは資金を提供する必要があり、私たちとAdimab LLC(Adimab)はビジネス上の合理的な努力を使用していくつかの研究を行い、発見と最適化のために使用しなければならない
25
私たちが選んだ目標。私たちはLatozinemabとAL 101候補製品でAdimabが発見した抗体を開発しており、私たちはAdimabによって最適化された抗体を私たちのAL 002製品候補に開発している。
統治する。Adimabとの私たちの協力は、双方の少なくとも2人の代表で構成された研究委員会によって管理されている。研究委員会は研究計画の中で優先順位を決定し,新たな提案研究計画,その他の活動を準備·決定した。もし研究委員会が協議一致の方法で決定を下すことができず、各当事者が各方面が指定した高級行政員に問題をエスカレートさせることで問題を解決できない場合、どちらも仲裁を求めることができる。
排他的である。2014年Adimab協定により、それぞれが協力範囲外の他方が提供する情報又は材料を使用する能力が制限される。
知的財産権。研究によって生成された知的財産権の所有権は、一般に、特定のカテゴリの知的財産権が一方または他方に専門的に譲渡されているにもかかわらず、発明または創造に適用される知的財産権の当事者の所有に属する。例えば、研究中に発明されたAdimabのバックグラウンドプラットフォーム技術の改善に関連する特許権は、Adimabに譲渡される。我々が以下の選択権を行使する前に,我々とAdimabは,双方が研究に関する権利と義務を履行することを可能にするために,それぞれ我々の持つ知的財産権の非排他的許可を相手に付与し,Adimabが保持している自己のライブラリの使用と許可の権利を除いて,研究に関連する権利や義務を履行する以外のいかなる目的でも,その所有可能かもしれない研究を実践するための特許を作成しないことに同意した.一般的に、すべての当事者は、自分の特許を起訴、維持、擁護、強制執行する義務があるが、私たちが研究で生成したいくつかの特許を起訴、実践、許可する能力は、いくつかの契約によって制限されている。私たちがこのような特許に対して以下に述べる選択権を行使すると、これらの制限は撤廃される。
オプションを行使する。2014年のAdimabプロトコルは、Adimabが発見または最適化した特定の数の抗体に関連する特定の権利を得ることができる独占的選択権を付与し、これらの抗体は私たちが選択した目標に向けている。選択権は、特許権に拡張され、そのような抗体の配列を具体的にカバーし、研究、開発、製造、製造、使用、販売、販売、提供、輸入、および輸出のために、そのような抗体に基づく製品を研究、開発、製造、製造、販売、提供、輸入、および輸出する権利がある。これらのライセンスは排他的であり,Adimabの背景やプラットフォーム技術,およびAdimabが保持している継続使用および自分のライブラリを許可する権利に加えて,これらのライセンスは非排他的である.私たちはAdimabのこのような背景やプラットフォーム技術を含まない、私たちが申請したAbbVieと協力するプログラムに関連する重要な特許が、私たちが独占的に所有している発明を要求することをAdimabに書面で確認した。私たちは2014年のAdimab協定に基づくすべてのオプションが満期になったか、行使中か、または複数の目標と数百種類の抗体(AbbVieと協力する目標計画を含む)について行使された。我々が目標に対して選択権を行使する際には,ビジネス上の合理的な努力に投入し,その目標に対する選択権を利用して製品を商業化する義務がある。これらのオプション実践から得られた譲渡及び許可の特許権は、上記“商業−知的財産権”と題する部分により詳細に記載されている
財務条項です。私たちは2014年のAdimab協定に記載された条項と制限に基づいて、Adimabと私たちの協力に関する研究に資金を提供する。また,抗体を用いた各項目に潜在的な記念碑的支払いを支払い,このような抗体を含む製品の商業販売に中央値以下の印税を支払った。しかしながら、もし私たちが発明の権利を付与する取引または第三者との協力によって作られた製品を販売する場合、私たちは、そのような販売の印税ではなく、それによって生じる収入の一部を支払うことを選択することができる。
任期と解約期間。私たちは、3ヶ月前にAdimabに書面で通知した場合、2014年のAdimabプロトコルを完全に終了するか、または特定の目標に対する製品または抗体を終了することができる。また、いずれも2014年のAdimabプロトコルを完全に終了することができ、またはいくつかの制限の場合、違約側に90日の通知を出しても是正されていない重大な違約行為については、特定の選択権を終了することができる。2014年のAdimab協定が満了する前に終了した場合、私たちはAdimabに対していくつかの持続的な支払い義務を負うか、または協力成果に関するいくつかの制限を遵守することが要求されるだろう。2014年の“アディマブ協定”は、協力して作られた製品を製品と国別に初商業販売した12周年で満期になる。私たちとAdimabが相互に付与したライセンスは継続されないだろうが、いくつかの制限を受けている。
26
概要-2019 Adimab連携プロトコル(2019 Adimabプロトコル)
2019年、私たちは別のAdimab協力協定(2019 Adimab合意)に合意した。2019年のAdimabプロトコルによると、私たちは資金援助が必要であり、私たちとAdimabは、私たちが選択した目標に対する抗体を発見し、最適化するために、商業的に合理的な努力を使用していくつかの研究を行わなければならない。私たちはまだ2019年のAdimab協定の下でどんな研究プロジェクトも確定していない。
統治する。Adimabとの私たちの協力は、双方の少なくとも2人の代表で構成された研究委員会によって管理されている。研究委員会は2019年のAdimab協定に基づいて研究面のコミュニケーションを促進し、限られた権力を持って研究計画を修正し、その方式は締約国に必要な資源に実質的な影響を与えない。もし研究委員会が協議一致の方法で決定を下すことができなければ、何の決定もしないだろう。
排他的である。2019年Adimabプロトコルにより、それぞれが協力範囲外の他方が提供する情報又は材料を使用する能力が制限される。
知的財産権。研究によって生成された知的財産権の所有権は、一般に、特定のカテゴリの知的財産権が一方または他方に専門的に譲渡されているにもかかわらず、発明または創造に適用される知的財産権の当事者の所有に属する。Adimabのバックグラウンドプラットフォーム技術に関連するいくつかの知的財産権は、研究中に発明されたこれらの技術の任意の改善を含み、Adimabに譲渡される。協力標的抗体をカバーする特許は私たちが所有している;しかし、私たちが以下の選択権を行使する前に、2019年のADIMAB協定に基づいて規定されている研究義務を履行する以外は、いかなる目的でもこのような特許を行使してはならない。以下に述べるオプション期間が満了した後,抗体に対して選択権を行使しないことを選択した場合,このような特許の所有権はAdimabに譲渡される.我々が以下の選択権を行使する前に,我々とAdimabはそれぞれ我々が持つ関連知的財産権の非独占的許可を相手に付与し,それぞれの側が研究に関する権利と義務を履行することを可能にする.一般的に、すべての当事者は、自分の特許を起訴、維持、擁護、強制執行する義務があるが、私たちが研究で生成したいくつかの特許を起訴、実践、許可する能力は、いくつかの契約によって制限されている。私たちがこのような特許に対して以下に述べる選択権を行使すると、これらの制限は撤廃される。
オプションを行使する。2019年Adimabプロトコルは、Adimabが発見または最適化した特定の数の抗体に関連する特定の権利を得ることができる独自の選択権を付与し、これらの抗体は私たちが選択した目標に向けている。この選択権は、適用可能なオプションの抗体の所有権に延長され、Adimabが所有または開発したいくつかの技術に従って世界的に、印税、再許可可能な非独占的許可を得る権利を取得し、研究、開発、製造、製造、使用、販売、販売、提供、輸入、および輸出のために、すべてのヒト治療、予防および診断用途のために使用される。我々が目標に対して選択権を行使する際には,ビジネス上の合理的な努力に投入し,その目標に対する選択権を利用して製品を商業化する義務がある。
財務条項です。私たちは、2019年のAdimab協定に記載されている条項と制限に基づいて、Adimabと私たちの協力に関する研究に資金を提供します。私たちはまたいくつかの開発費用を担当して、もし私たちがオプションを行使すれば、オプション費用を支払う義務があります。我々はまた,抗体を用いた各製品に対して潜在的な記念碑的支払いを行っているが,任意の所与の目標に対する総支払いには制限があり,このような抗体を含む製品の商業販売には1桁以下の印税を支払う。
任期と解約期間。私たちは、60日前にAdimabに書面で通知した後、2019年のAdimabプロトコルを完全に終了するか、または特定の目標に対する製品または抗体を終了することができます。また、違反者に90日の通知を出しても是正されていない重大な違反については、いずれも2019年のAdimab合意を完全に終了することができる。2019年にAdimabプロトコルが満了する前に終了した場合、私たちは協力成果の使用を禁止されるだろう。2019年Adimabプロトコルは、製品および国ごとに満了し、そのような製品が同国で初めて商業販売されて12周年と、その製品をカバーする最後の特許がその国で満期になる遅い者を基準とするか、または、2019年のAdimabプロトコルによって製品が選択されていない場合は、最後のオプションが満了した時点で満了する。期限が切れた後、Adimabが選択権を行使した製品に関する私たちに付与された許可は、非独占的で著作権免除に基づいて存在するだろう。
27
概要-2021年Adimab協調プロトコル(2021年Adimabプロトコル)
2021年、私たちは別のAdimab協力協定(2021年Adimab合意)に到達した。2021年ADIMAbプロトコルによると,我々は我々が選択した標的の抗体工学研究計画に資金を提供することが求められている。2021年のAdimabプロトコルはまた、Adimabによって発見または最適化され、私たちが選択した目標のための特定の数のエンジニアリングシーケンスを取得するための独占的選択権を付与される。私たちは各方面が行った研究計画のテーマである目標を選択した。我々はこの研究計画に関連するいかなる工程系列の選択権も行使しておらず,他の研究計画に追加的な目標を指名していない.2021年のAdimabプロトコルは、行われた研究計画の選択期間が終了した時点で満了します。
製造業
我々はcGMP規定に従って臨床試験のための候補製品を生産しなければならない。CGMP条例には、人員、建物および施設、設備、アセンブリおよび薬品容器および閉鎖的な制御、生産およびプロセス制御、包装およびラベル制御、保有および分配、実験室制御、記録および報告、ならびに返品または回収された製品に関する要件が含まれる。私たちの候補製品の製造施設はcGMP要求とFDA或いは類似の外国監督管理機関の満足を満たさなければ、任意の製品を臨床試験に使用することができる。我々の第三者メーカーは,FDAや他の外国当局によるそれぞれの工場のcGMP一般適合性の定期検査も受ける。これらの検査には、適用される法規に適合するかどうかを評価するために、私たちの製品をテストして製造するためのプログラムや操作が含まれている可能性があります。
私たちは現在、臨床試験と商業化のための候補製品を生産するためのインフラや内部能力を持っていない。我々は依存し,第三者cGMPメーカーや我々のパートナーに依存して人体臨床試験用製品を生産し,FDAや他の外国機関のこのような製品の規定に適合することが予想される。著者らはCDMOの製造と供給に依存し、著者らの臨床前と臨床材料を供給し、著者らの候補製品の臨床前と臨床開発に用いられる。私たちの広範な製造戦略の一部として、私たちの候補製品の製造を加速し、製造リスクを最小限に抑えるために、私たちは現在いくつかのCDMOと私たちの候補薬物或いは製品を生産する関係を確立した。
私たちは長期供給協定を持っていません。私たちは製造サービス協定を開発することによって必要な薬物製品を購入します。私たちは引き続き第三者メーカーや私たちのパートナーに依存して、マーケティングの承認を得た任意の候補製品に商業的な供給を提供したい。私たちは豊富な技術、製造、分析、品質、規制(cGMPを含む)とプロジェクト管理経験を持っていて、私たちの第三者メーカーを監督し、規制の目的を達成するために製造と品質データと情報を管理します。
法律および規制要件を遵守しない場合、製造業者は、警告状、製品の差し押さえまたはリコール、禁止、製造業務に重大な制限または一時停止を加える同意法令、および民事および刑事罰を含む可能性のある法律または規制行動に直面する。契約メーカーは生産生産量、品質管理と品質保証面の困難に直面し、合格者が不足している。このような行動または事件のいずれかは私たちの製品の供給に実質的な影響を及ぼす可能性がある。
商業化計画
私たちは現在承認された薬を何も持っておらず、私たちは短期的にも何の承認もないと予想している。そのため、私たちは販売、マーケティング、あるいは商業製品を流通する能力もなく、一社として薬品をマーケティングした経験もありません。私たちの候補製品が商業化されることが承認された時、私たちはこれらの製品のためにアメリカ、ヨーロッパ、アジア、およびいくつかの他の重要な市場で商業化インフラを開発するつもりです。私たちはまた、エバービーやグラクソ·スミスクラインのようなパートナー関係に依存して、販売とマーケティング、商業流通などの商業的なインフラを提供することができます。
28
知的財産権
私たちの成功は、私たちの候補製品、技術、およびノウハウのために独自の保護を獲得し、維持し、他人の固有の権利を侵害することなく運営し、他の人が私たちの固有の権利を侵害することを防止する能力があるかどうかにある程度依存する。私たちの戦略は、米国および米国以外の司法管轄地域で、当社のノウハウ、発明、改善および候補製品に関連する特許保護を求め、獲得することを求めることであり、これらの特許技術、発明、改善、および製品候補は、私たちの業務の発展と実施に重要である。私たちの特許の組み合わせは、私たちの候補製品および関連コンポーネント、それらの使用方法および製造プロセス、私たちの独自の試薬および分析、ならびに私たちのビジネスに重要なビジネス的意義を有する任意の他の発明をカバーすることを目的としています。私たちはまた、当社のノウハウ、プラットフォーム、および候補製品に関連する機密情報およびノウハウを保護するために、商標およびビジネス秘密に依存しています。私たちは私たちの候補技術と製品に関する多くの技術的ノウハウと商業秘密を持っていると信じている。
2022年12月31日現在、私たちの特許組み合わせは、42個の発行された特許および500件以上の係属中の特許出願を含む50以上のシリーズを含み、20個以上の異なる目標および/または技術に関連しており、これらの特許は、私たちが独占的に所有するか、または独占的に許可する権利がある。私たちの候補製品については、一般に、候補製品と標的タンパク質との結合エピトープに基づく物質組成、候補製品の機能的特徴、候補製品の劣化配列、および/または候補製品の特定の配列をカバーする多段階の特許保護が求められる。物質の構成範囲に加えて,候補製品の製造方法,核酸,配合,使用方法についてクレームをつけるのが一般的である。患者に対する選択基準、バイオマーカー、疾患サブセット、薬効学的および臨床的終点、ならびに用量レジメンの請求項をさらに含む、請求項1~4のいずれか一項に記載の使用方法。以下に述べるように、私たちはより多くの特許出願を通じて、私たちの候補製品および技術の特許保護を強化する予定です。
PGRN計画
私たちは、私たちのPGRN計画のための6つの特許シリーズ、Latozinemab、およびAL 101を持っています。これには、私たちのPGRN計画候補製品の構成および用途をカバーする7つの発行された米国特許が含まれています。最初の2つの特許家族は2036年に満了する予定であり、第3特許家族は2039年に満了する予定であり、第4特許家族は2040年に満了する予定であり、第5特許家族は2041年に満了する予定であり、第6特許家族は2042年に満了する予定であり、すべての場合、いかなる特許期限調整およびいかなる特許期限延長も含まれていない。
TREM 2計画
私たちはTREM 2計画に対する7つの特許シリーズを持っており、その中には発行された2つの米国特許が含まれており、私たちのTREM 2計画候補製品の組成と用途をカバーしている。第1特許家族は2035年に満了する予定であり、第2特許家族は2036年に満了する予定であり、第3特許家族は2038年に満了する予定であり、第4特許家族は2040年に満了する予定であり、第5および第6特許家族は2041年に満了する予定であり、第7特許家族は、必要な非臨時特許出願が直ちに提出され、米国仮特許出願の他のすべての適用要件を満たし、2043年に満了する予定であり、すべての場合、いかなる特許期限調整および任意の特許期限の延長も含まれていない。
MS 4 A計画
2つの特許シリーズを持っていますMS 4 A私たちの潜在力の構成と用途をカバーする計画MS 4 A製品候補を計画する。第1の特許家族は2039年に満了する予定であり、第2の特許家族は2040年に満了する予定である。
個別特許の期限は,特許を付与した国の特許法的期限に依存する。米国を含む多くの国では,特許期間は一般に適用国の非臨時特許出願の最初に出願された日から20年である。米国では、場合によっては、特許の期限を特許期限調整によって延長することができ、これは、特許権者が米国特許商標局の審査および特許付与時の行政遅延による損失を補償することができ、または1つの特許が共通所有特許または共通発明者と命名された特許によって最終的に放棄され、満期日が早い場合には、特許期限が短縮される可能性がある。1984年の“医薬品価格競争および特許期限回復法”(Hatch-Waxman Act)は、特許発効中の薬物が規制審査を受ける時間の長さの一部として、米国特許の満了後に特許期間を最大5年間延長することを可能にした。1つの特許
29
特許の延長の残り期間は、製品が承認された日から合計14年間を超えることはできず、各規制審査期間は1つの特許しか延長できず、承認された薬物、その使用方法、または製造方法に関する権利要件を延長することしかできない。
EUおよび他のいくつかの外国司法管轄区域にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。将来,我々の候補製品がFDAや外国規制機関の承認を得た場合,各薬剤の臨床試験時間や他の要因に応じて,これらの製品をカバーする発行された特許の特許期間の延長を申請する予定である。以上言及した満期日は,我々が入手可能な潜在的特許期間の延長や他の市場排他性とは無関係である。
場合によっては、私たちはまた私たちの技術を保護するために商業秘密に依存する。しかし、商業秘密は保護するのが難しいかもしれない。私たちは、従業員、コンサルタント、科学コンサルタント、請負業者と秘密保持協定を締結することで、私たちのノウハウやプロセスを保護することを求めています。また、私たちは私たちのビルの実体安全と、私たちの情報科学技術システムの実体と電子安全を維持して、私たちの資料と商業秘密の完全性とセキュリティを維持することに力を入れています。
競争
生物技術と製薬業界、神経変性疾患領域を含む生物技術と製薬業界は、その特徴は技術が迅速に進歩し、競争が激しく、知的財産権を重視することである。私たちは大型と専門製薬と生物技術会社、学術研究機関、政府機関及び公共と個人研究機関を含む多くの異なる源からの激しい競争に直面している。いくつかの製薬と生物技術会社は現在神経変性疾患の適応を治療するための製品を開発しており、著者らはこれらの適応について研究を行い、FTD、アルツハイマー病、パーキンソン病とALSを含み、その中には大量の資金を持つ大企業、例えば生物遺伝会社、礼来会社、メルク会社と羅氏ホールディングス会社を含む。その中のいくつかの会社は、私たちの適応と同じまたは類似した候補製品を探しており、場合によっては、同じ目標に基づいて、または類似した行動メカニズムによって行動する。私たちのすべての候補製品の成功に影響を与える重要な競争要素は、有効性、安全性、管理方法、コスト、発売時間、販売促進活動レベルと知的財産権保護を含むと信じている。
我々の候補製品は,神経変性疾患の症状治療のための承認された療法や,神経変性疾患の進展を阻止または緩和するための臨床研究で承認されている療法と競合することが予想され,これらの療法は複数の会社や機関によって開発されている。
政府の監督管理
アメリカ連邦、州と地方各級及びその他の国の政府当局はその他の以外に、薬品と生物製品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、普及、広告、流通、承認後のモニタリングと報告、マーケティングと輸出入などの方面に対して監督管理を行う。一般的に、新薬や生物製剤が発売される前に、その品質、安全性、有効性を証明するデータを大量に取得し、各規制機関特有のフォーマットに組織し、審査を提出し、監督機関の承認を得なければならない。
アメリカの薬物開発
アメリカでは、FDAは“食品、薬物と化粧品法”(FDCA)に基づいて薬品を監督し、FDCAと公衆衛生サービス法(PHSA)に基づいて生物製品を監督する。医薬品と生物製品はまた他の連邦、州、そして地方法規によって制限されている。規制の承認を得て、その後、適切な連邦、州、地方、外国の法規と条例を遵守する過程には、多くの時間と財政資源が必要だ。製品開発過程,承認過程又は上場後のいずれかの場合,出願人が適用される米国の要求を遵守しなければ,行政又は司法制裁を受ける可能性がある。他の行動に加えて、これらの制裁は、FDAが未解決の申請の承認の拒否、承認の撤回、臨床棚上げ、無見出しまたは警告状、製品のリコールまたは市場撤回、製品の差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、原状回復、返還、返還、および民事または刑事罰を含むことができる。どんな機関や司法法執行行動も私たちに実質的な悪影響を及ぼすかもしれない。
30
任意の将来の候補製品は、生物製品許可証申請(BLA)または新薬申請(NDA)プログラムによってFDAの承認を得なければならず、その後、米国で合法的に発売されることができる。このプロセスは、一般に以下のことを含む
NDAまたはBLAを支持するために必要なデータは、2つの異なる発展段階で生成される:臨床前および臨床。臨床前と臨床テストと承認過程は大量の時間、精力と財力を必要とし、私たちは未来のいかなる候補製品のいかなる承認も適時に或いは根本的に承認されないかどうかを確定することができない。
臨床前開発段階は通常薬物化学、製剤と安定性の実験室評価、及び後続の臨床試験を支持する動物毒性評価研究を含む。スポンサーは,臨床前研究の結果を,生産情報,分析データ,任意の利用可能な臨床データや文献,提案された臨床案とともにINDの一部としてFDAに提出しなければならない。INDはFDAがヒトに研究製品の使用を許可する要求であり,ヒト臨床試験が開始される前に発効しなければならない。
臨床前研究には製品化学と処方の実験室評価があります体外培養動物研究と、有害事象の可能性を評価し、場合によっては治療使用の理由を確立する。臨床前研究の進行はGLPの安全/毒理学研究に関する法規を含む連邦法規と要求の制約を受けている。INDスポンサーは,臨床前試験の結果を生産情報,分析データ,任意の利用可能な臨床データや文献,臨床研究計画などとともにFDAに提出し,INDの一部としなければならない。いくつかの長期的な臨床前試験、例えば生殖不良事象や発ガン性の動物試験は、IND提出後も継続する可能性がある。INDは、FDAが1つまたは複数の提案された臨床試験に対して懸念または問題を提起しない限り、FDAが受信してから30日後に自動的に有効になり、試験を保留する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。したがって,INDの提出はFDAが臨床試験の開始を許可しない可能性がある。
31
神経変性疾患を治療するための治療法を承認する規制要求が変化している。例えば、2つの新しい薬物aducanumabとlecanemabは最近FDAの加速承認を得ており、これは代替終点、すなわち脳におけるアミロイドβプラークの減少に基づいている。FDAの加速承認経路によると、患者の臨床利益を合理的に予測する可能性のある代替終点は、後続の検証性研究に依存する加速承認の基礎となる可能性がある。対照的に、FDAがADのもう一つの製品Donanemabの承認を加速することを拒否した理由は、少なくとも12ヶ月の薬物曝露患者数が不足しているからである。FDAは最近、24週間の第2段階研究に基づく治療効果データに基づいて、フェニルブタン酸ナトリウム/タウリンジオールのALS治療の加速承認を承認した。承認に賛成票を投じたFDA諮問委員会は、第2段階研究で薬剤の生存データを取得し、別のAD第2段階研究で薬剤のバイオマーカーデータを取得した。また,最近発表されたFTD−GRNを含む遺伝的形態に関するFTDの疾患進展モデルは,試料量のより正確な推定やバイオマーカーを代替終点として使用して試料量を減少させるためのこのようなFTDに基づく臨床試験の設計に役立つ可能性がある。私たちは、これらの開発の私たちの臨床プロジェクトへの適用性を評価し、その評価に基づいて、規制機関と接触し、私たちの候補製品の承認経路を検討するかもしれない。
臨床試験
臨床開発段階は、合格した研究者の監督の下で、GCP要求に基づいて健康ボランティアまたは患者に研究製品を提供することであり、通常は試験スポンサーに雇用されない、または試験スポンサーの制御下にある医師であり、すべての研究対象に任意の臨床試験への参加についてインフォームドコンセントを提供することを含む。臨床試験は,臨床試験の目標,用量プログラム,被験者の選択と排除基準,および被験者の安全性をモニタリングし,治療効果を評価するためのパラメータを詳細に説明した場合に行われる。INDの一部として、すべての議定書とその後の議定書のいかなる修正もFDAに提出されなければならない。また,各臨床試験は,臨床試験を行う各機関の内部審査委員会によって審査·承認されなければならず,臨床試験に参加する個人が直面するリスクが最小限に減少することを保証し,期待される利益については合理的である。IRBはまた、各臨床試験対象またはその法律代表に提供されなければならないインフォームドコンセントを承認し、完成まで臨床試験を監視しなければならない。行っている臨床試験や完成した臨床試験結果を公的登録機関に報告することも求められている。
米国国外で臨床試験を行うスポンサーはFDAの認可を得ることができるが,INDによる臨床試験を希望している。海外の臨床試験がINDに基づいて行われていなければ,スポンサーはNDAやBLAを支援するために臨床試験のデータをFDAに提出することができる。試験がGCP要求に基づいて行われ,FDAが必要と考えた場合に現場検査でデータを検証できれば,FDAはINDではない工夫と良好な外国臨床試験を受ける。
米国の臨床試験は通常3つの連続段階で行われ,第1段階,第2段階,第3段階と呼ばれ,重なる可能性がある。
32
承認後試験は,4期臨床試験と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの試験は,治療適応が予想される患者の治療から追加的な経験を得るために用いられている。場合によっては,FDAはNDAまたはBLAを承認する条件として4期臨床試験を強制的に実行することができる。
その他の情報に加えて,臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出されなければならず,書面のIND安全報告はFDAや調査者に提出されなければならず,深刻かつ意外な疑わしい有害事象を発見するためには,他の研究からの結果は薬物や生物に曝露されたヒトに重大なリスクがあり,動物や生物からの発見であることが示唆された体外培養テストにより、人類ボランティアに対して重大なリスクがあり、及び方案或いは研究者マニュアルに記載されたテストと比べ、深刻な不良反応の発生率は臨床で任意の重要な増加があることを表明した。
第1段階、第2段階、および第3段階の臨床試験は、もしあれば、任意の指定された時間で成功しない可能性がある。FDA或いはスポンサーはいつでも様々な理由で臨床試験を一時停止或いは中止することができ、研究対象或いは患者が受け入れられない健康リスクに直面していることを発見することを含む。同様に、臨床試験がIRBの要求に従って行われない場合、または薬物または生物が患者に予期せぬ深刻な傷害を受けることに関連している場合、IRBは、その機関の臨床試験の承認を一時停止または終了することができる。さらに、いくつかの臨床試験は、データ安全監視委員会または委員会と呼ばれる臨床試験スポンサーによって組織された独立した合格専門家グループによって監督される。このグループは,実験のあるデータへのアクセスにより,許可試験が指定されたチェックポイントで行えるかどうかを決定する.臨床試験と同時に,会社は通常追加の動物研究を完成させ,薬物や生物の化学的および物理的特性に関する追加情報を開発し,cGMP要求に基づいて商業量産製品のプロセスを最終的に決定しなければならない。製造過程は一貫して高品質の製品ロットを生産できる必要があり、その中で、会社は最終製品の特性、強度、品質、純度をテストする方法を開発しなければならない。また,適切な包装を選択·テストし,我々の候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
新冠肺炎の大流行の結果により、アメリカ食品薬品監督管理局は各種の生物製薬メーカーと臨床試験スポンサーに適した新冠肺炎関連指導文書を発表した。例えば、FDAは、2020年3月に、疫病の影響を受けた臨床試験スポンサーのいくつかの考慮要因を説明し、臨床試験を管理するための緊急措置、新冠肺炎の大流行による臨床試験中断、実施された緊急措置(例えば、参加者が製品および/または研究を停止し、重要な安全性および/または有効性データを収集するための代替プログラムを含む)が報告された臨床試験の安全性および有効性結果への影響などを含む疫病期間中の臨床試験に関する指導意見を発表する。2020年と2021年に、FDAは以前の指導意見の更新、良好な製造規範、薬品製造と生物研究モニタリング施設に対する遠隔相互作用評価、及び薬品製造とサプライチェーン検査などを含む一連の業界指導文書を発表した。これらと将来のガイドラインや規制要件は、将来の立法を含めて、新たな政策や手順の制定と実施、私たちの臨床試験の重大な調整、あるいはコンプライアンスに要する時間や資源を増加させることが要求される可能性があり、これは私たちの臨床開発計画やスケジュールに影響を与える可能性があります。新冠肺炎突発公共衛生事件が著者らの業務(非臨床研究と臨床試験を含む)に対する影響程度は未来の発展に依存し、これらの発展は高度な不確定性があり、自信に満ちて予測できない。
NDA/BLAレビュープロセス
臨床試験が完了した後、研究製品が提案された1つまたは複数の指示用途に対して安全に有効であるかどうかを評価するために、データを分析する。臨床前研究および臨床試験の結果は、その後、NDAまたはBLAの一部としてFDAに提出され、製品品質および他の関連データを確保するために、提案されたラベル、化学および製造情報が提示される。簡単に言うと、NDAまたはBLAは、薬物または生物学的製剤を販売するための1つまたは複数の適応を指定する承認要求であり、薬物の安全性および有効性または生物学的製剤の安全性、純度および効力の証拠を含まなければならない。応用は臨床前研究と臨床試験の陰性とファジィ結果、及び陽性結果を含む可能性がある。データは、製品使用の安全性および有効性を試験するために、または研究者によって開始された研究を含む多くの代替源からの臨床試験からのものである可能性がある。上場承認を支援するために提出されたデータは
33
品質と数量は、研究製品の安全性と有効性を確定し、FDAを満足させる。薬物や生物製剤が米国で発売される前に,NDAやBLAに対するFDAの承認を得なければならない。
改正された“処方薬使用料法案”(PDUFA)によると,NDAまたはBLAごとに使用料が付加されなければならない。FDAは毎年PDUFAユーザ料金を調整する。FDAが2022年10月1日に発効し,2023年9月30日まで続く2023年の処方薬使用料スケジュールによると,臨床データを必要とするアプリケーション(例えばNDAやBLA)の使用料は約320万ドルである。PDUFAは,市販されているヒト薬物や生物製品ごとに年間計画費(2022年は369,413ドル)を徴収し,処方薬や生物製品を生産するための施設に年間建造費を徴収している。場合によっては、小企業が初めて出願した出願料を免除することを含む、費用を免除または減免することができる。さらに,孤児薬として指定された製品については,この製品が孤児適応も含まれていない限り,NDAまたはBLAに対して使用料を評価しない。
FDAは、提出されたすべてのNDAおよびBLAを受け入れる前にそれらを検討し、NDAまたはBLAの提出を受け入れるのではなく、追加の情報の提供を要求する可能性がある。FDAは、受信後60日以内にNDAまたはBLA提出申請を受け入れるか否かを決定しなければならない。提出された申請が受け入れられると、FDAはNDAまたはBLAの深い審査を開始する。FDAがPDUFAで合意した目標および政策によれば、FDAは、新しい分子実体NDAまたは元のBLAの予備審査を完了し、出願人に応答し、優先審査のために指定された新しい分子実体NDAまたは元のBLAの提出日から6ヶ月間、FDAが10ヶ月の時間を有する。FDAは、そのPDUFA規格および優先NDAまたはBLAの目標日を常に満たすわけではなく、審査プロセスは、FDAがより多くの情報を提供または明確にすることを要求することによって延長されることが多い。
NDAまたはBLAを承認する前に、FDAは、それらがcGMP要件に適合しているかどうかを決定するために、新製品の製造施設を承認前に検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、この製品を承認しないであろう。FDAはまた、GCP要求に適合することを確保するために、臨床試験のデータを監査することが可能である。さらに、FDAは、新薬または医薬製品の出願を諮問委員会に提出することができ、一般に、申請が承認されるべきかどうか、およびどのような条件下で(ある場合)かを審査、評価、および提案するために、臨床医および他の専門家を含むグループである。FDAは諮問委員会の提案に制限されていないが、承認決定を下す際にこれらの提案を考慮する。FDAは臨床試験データを再分析する可能性があり,FDAや出願人の審査過程で広く議論される可能性がある。FDAがNDAまたはBLAを評価した後、それは承認書または完全な返信を発行する。この薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。完全な返信は、申請の審査期間が終了し、現在の申請が承認されないことを示している。完全な応答文は、一般に、FDAによって決定されたNDAまたはBLA内のすべての特定の欠陥を記述する。完全な返信は、追加の臨床データ、追加の重要な段階3期の臨床試験、および/または臨床試験に関連する他の重要で時間のかかる要件を必要とする可能性がある, 臨床前研究や生産。完全な返信が発行された場合、出願人は、手紙で決定されたすべての不足点を解決するために、または出願を撤回するために、秘密保持プロトコルまたはBLAを再提出することができる。このようなデータや情報を提出しても,FDAはNDAやBLAが承認基準を満たしていないと決定する可能性がある.臨床試験から得られたデータはつねに決定的ではなく,FDAのデータ解釈は我々の同じデータに対する解釈とは異なる可能性がある。
孤児薬
孤児医薬品法によれば、FDAは、米国では20万人未満または米国で20万人を超える影響を与える疾患または疾患であることが一般的であり、米国ではこのような疾患または疾患を治療する製品を開発および提供するコストが製品の販売から回収されるという合理的な期待がない、まれな疾患または疾患の治療のための医薬または生物製品に孤児の称号を付与することができる。
NDAやBLAを提出する前に,指定孤児薬を申請しなければならない。FDAが孤児薬の指定を承認した後、治療剤のアイデンティティ及びその潜在的孤児の使用
34
アメリカ食品医薬品局です。指定孤児薬は、規制審査と承認過程でいかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。
孤児薬物名を有する製品が、その後、そのような名称を有する疾患の特定の活性成分に対するFDAの最初の承認を得た場合、この製品は、孤児薬物排他性を得る権利がある。これは、FDAが7年以内に他のNDAまたはBLA申請を承認しない可能性があり、同じ適応の下で同じ薬物または生物学的製剤を販売している可能性があり、限られた場合、例えば孤児の独占的地位を有する製品に対する臨床的利点を示さない限り、FDAが孤児薬物の指定を撤回した場合、またはFDAが孤児独自の薬物の保持者が指定された薬物を有する疾患または状態を有する患者の需要を満たすのに十分な数の孤児製品を確保していないことを意味する。FDAはFTDを治療するLatozinemabおよびAL 101の孤児薬を承認したが、FDAは依然としてFTDの治療のために異なる有効成分を有する他の薬物を承認することができる。また、孤児薬物の独占性は、孤児の独占期間が満了する前に、FDAが同一の薬物製品の別の異なる適応のマーケティング申請を承認することを阻止しない。もし競争相手が我々の前にFDAで定義された同じ製品の承認を得た場合、すなわち私たちが承認を求めている同じ適応、またはある候補製品が競争相手の製品範囲に含まれていると決定された場合、孤立薬物独占も7年以内に私たちの製品の承認を阻止する可能性がある。孤児薬に指定された我々の製品がある適応の発売が承認され,その適応が孤児薬物の称号を与えられていない場合,この製品はこの非孤児適応において孤児薬物排他性を持たないであろう。EUの孤児薬の地位は似たような要求と利益を持っているが、同じではない。
2021年の訴訟では、ある裁判所は、孤児薬物排他性は、指定された疾患または条件全体のすべての用途または適応には適用されず、条件を満たす疾患内の承認用途または適応にのみ適用されるFDAの長期的な立場に同意しない。この控訴裁判所の判決は孤児薬物の排他的な応用に不確実性をもたらした。FDAは2023年1月、この機関が適用された裁判所の判断を遵守しているにもかかわらず、“連邦紀要報”に通知を発表した。FDAは、孤児薬物の排他的な範囲を、薬物が承認された用途または適応とバンドルし続けることを意図しており、これは、他のスポンサーが、未承認の同一の孤児が疾患または状態を指定した場合に、薬物の新しい用途または適応の承認を得ることを可能にする。将来の訴訟、立法、機関決定と行政行動がどのように孤児薬物の専有範囲に影響するかはまだ不明である。
開発と審査計画を加速する
FDAは特定の基準に適合する新薬と生物製品の審査過程を加速または促進するための迅速なチャネル計画を持っている。具体的には、新薬および生物製品が深刻または生命に危険な疾患を治療することを目的とし、臨床前または臨床データが、このような疾患が満たされていない医療需要を解決する可能性があることを示す場合、高速車線指定を受ける資格がある。高速チャネル指定は製品にも,検討中の特定の適応にも適用可能である。スポンサーは、NDAまたはBLAの承認を得る前の任意の時間に、FDAに製品を高速チャネル状態として指定することを要求することができるが、NDA前またはBLA前の会議よりも遅くないことが好ましい。
迅速チャネル計画に従って、優先審査および承認の加速など、FDAの開発および審査を加速するための他のタイプの計画に参加する資格がある可能性があるFDA上場製品を提出する任意のもの。重篤または生命に危険な疾患を治療する製品は、優先審査を受ける資格があり、承認されれば、既存の療法と比較して、安全性および有効性の面で有意な改善を提供するであろう。
製品が重篤または生命に危険な疾患を治療し、通常、既存の療法よりも有意な利点を提供する場合、加速承認を得る資格もある。さらに、臨床的利益を合理的に予測する可能性のある代替終点への影響、または不可逆的発症率または死亡率(IMM)よりも早く測定することができる臨床終点への影響を証明しなければならず、後者はIMMまたは他の臨床的利益の影響を合理的に予測する可能性がある。承認の一つの条件として,FDAは承認を加速させた薬物や生物のスポンサーに十分かつ良好に制御された上場後臨床試験を要求する可能性がある。FDAが有効であることが証明された薬物や生物が流通や使用が制限された場合にのみ安全に使用できると結論すれば,その製品の安全使用を確保するために必要と考えられる市販後の制限が要求される可能性がある。
さらに、1つの医薬または生物学的製剤が、1つまたは複数の他の薬剤または生物学的製品と共に、深刻または生命に危険な疾患の治療のために単独でまたは1つまたは複数の他の薬剤と共に使用されることが意図されている場合、予備的な臨床的証拠は、製品が1つまたは複数の臨床的に重要な終点で現在承認されている療法よりも実質的に改善されている可能性があることを示している場合、医薬または生物学的製剤は、突破的療法として指定される資格がある可能性がある。突破によるメリット
35
治療指定には,迅速チャネル指定と同様の利点と,有効な薬物開発計画を確保するためのFDAの密な指導が含まれている。迅速チャネル指定、優先審査、加速承認、および画期的な治療指定は、承認の基準を変更することはありませんが、開発または承認プロセスを加速させる可能性があります。
生物類似または交換可能な生物製品の簡明な許可経路
2010年に法律となった患者保護および平価医療法案(ACA)に署名したのは、FDA許可の参照生物製品と高度に類似していることが証明された生物製品のための短い承認経路を作成するBPCIAを含む。BPCIAは重複テストを最大限に減少させ,開発コストを低減し,患者が負担できる治療を得る機会を増加させることを試みている。FDAが別途決定しない限り、生物学的類似製品の許可申請は、以下の内容に基づいて生物学的類似性を証明する情報を含まなければならない
生物類似性は生物製品と参照製品が高度に類似していることであり、臨床では活性成分の微小な差異がないにもかかわらず、製品の安全性、純度と効力について言えば、生物製品と参照製品の間に臨床上意義のある差異がない。さらに、法律は、基準製品と生物類似製品との間の“互換性”を規定しており、それにより、処方参照製品の医療提供者の介入を必要とすることなく、生物類似製品を参照製品の代わりに使用することができる。より高い互換性基準は、示すために十分な情報によって証明されなければならない
生物類似体が米国で発売される前に,FDAの承認が必要である。しかし,生物製品の膨大で複雑な構造やこのような製品を製造するプロセスに関する複雑さは,FDAがこのような制定中の法律を実施することに大きな障害となっている。例えば、FDAは、特許生物製品の生物と類似していることを証明するために必要な科学的証拠の種類および数--実験室、臨床前および/または臨床--に対して裁量権を有する。
FDAはスポンサーが提供するすべての証拠を考慮して生体類似性の証明を支援する予定であり,スポンサーがその生体類似性を開発する際に漸進的な方法を用いることを提案している
36
製品です。したがって、生物類似製品の応用は、参考製品の潜在的な安全性と有効性を決定するための全臨床前と臨床試験を繰り返す必要がないかもしれない。しかしながら、有効成分が同一であるか、または有効成分中の任意の不純物または差異が生物学的類似製品の安全性、純度または効力に影響を与えないことを証明するのに十分な情報がない場合、FDAは、生物学的類似製品の承認の申請を拒否することができる。また、BLASと同様に、生物類似製品の出願は、生物製品の安全性、純度および効力を確保および維持するための施設で生産されない限り、承認されないであろう。
生物学的類似出願の提出は、FDAが十分ではないと考えられる出願を受け入れることを拒否する可能性があるので、FDAが申請の届出および審査を受けることを保証しない。他の理由を除いて、2012年の“生物類似使用者費用法案”に基づいて評価された任意の適用可能な使用料が支払われていない場合、FDAは、生物類似申請またはサプリメントを不完全とみなす。また,FDAは申請を受け入れることができるが,スポンサーが生体類似性を証明していないことを理由に承認を拒否した場合,スポンサーはさらなる分析,臨床前あるいは臨床研究を行い,新たな生物製品としてBLAを提出して許可を得ることができる。
FDAが生物類似製品の商業流通のために最終的に承認する時間は、ブランド製品の製造業者が1つ以上の法定排出期間を有する権利があるかどうかを含む様々な要因に依存し、その間、FDAはブランド製品生物に類似した任意の製品の承認を禁止される。FDAは参考製品が初めて許可を得た日から12年以内に生物類似申請を承認することができない。また,生物類似製品スポンサーは,参考製品が初めて許可を得た日から4年以内に申請を提出してはならない。他の法律規定によると、参考製品も排他性を有する権利がある。例えば、稀な疾患または疾患のための参照製品(孤児薬物)を指定することは、7年間の排他性を有する権利がある可能性があり、この場合、生物学的類似性法規に規定されている12年の期限が終了するか、または7年の孤児薬物排出期間が終了するまで、参照製品の生物に類似した製品は承認されず、両者のうち、より遅く発生するものを基準とする。場合によっては、規制排他期間は、特許満了日または後に生物類似性出願が承認されることを阻止するために、特許の有効期限を超える可能性がある。また、場合によっては、FDAがメーカーにその製品の児童への影響の研究、すなわちいわゆる小児科延長を要求すれば、FDAは参考製品の専門期間をさらに6ケ月延長することができる。
任意の使用条件下でブランド製品と交換可能であると決定された第1の生物学的製品も、排他的期間を有する権利があり、その間、FDAは、別の製品が任意の使用条件下で基準製品と交換可能であることを決定することができない。この特許期間は、最初の交換可能製品の最初の商業マーケティングから1年後、最初の交換可能製品出願を提出した出願人の特許侵害問題が解決されてから18ヶ月後、訴訟におけるすべての特許に関する裁判所の最終裁決または却下訴訟(損害の有無にかかわらず)に基づいて、最初の交換可能製品を承認してから42ヶ月後、最初の交換可能製品出願を提出した出願人に対する特許侵害訴訟が行われている場合、または最初の交換可能製品出願を提出した出願人が起訴されていない場合、最初の交換可能製品を承認するための18ヶ月間に延長される。
承認後に要求する
新製品が承認された後、製造業者および承認された製品は、監視および記録保存要件、不良体験を報告する要求、および宣伝および広告要件を遵守することを含むFDAによって持続的に規制されるであろう。ここには、未承認用途または患者集団のための薬剤の普及、いわゆる“ラベル外使用”の制限、および業界支援のための科学的および教育活動の制限が含まれる。医師はラベル外の用途のために合法的な薬品を処方するかもしれないが、製造業者はこのような用途を販売または普及させない可能性がある。処方薬宣伝材料は初回使用時にFDAに提出されなければならない。さらに、適応、ラベルまたは製造プロセスまたは施設の変化を含む医薬または生物学的に何らかの修正がある場合、出願人は、新しいNDA/BLAまたはNDA/BLAサプリメントの承認を得るために提出および提出を要求される可能性があり、これは、追加のデータまたは臨床前研究および臨床試験を開発する必要があるかもしれない。
FDAはまた、製品の安全な使用を保証するために、REMSの要件を含む承認時に他の条件を追加することができる。REMSは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全な使用を確保する要素を含むことができる。どの項目でも
37
これらの承認またはマーケティングの制限は、製品の商業普及、流通、処方、または配布を制限する可能性があります。製品承認は、規制基準を満たしていないことや、初期マーケティング後に問題が発生したことで撤回される可能性があります。
規制要件や基準が守られていない場合、あるいは製品発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または規制要件を遵守できなかったことを含む、以前に未知の問題が存在することが発見され、新しいセキュリティ情報を追加するために承認されたラベルの改訂につながる可能性がある;新しい安全リスクを評価するために発売後研究または臨床研究を実施するか、または流通制限またはREMS計画下の他の制限を実施することができる。他の他の潜在的な結果には
FDAは市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。薬品や生物製品は承認の適応と承認されたラベルの規定に基づいてしか普及できない。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。
他のアメリカの規制事項
FDA以外に、製品承認後の製造、販売、販売促進とその他の活動もアメリカの多くの監督管理機関によって監督され、医療保険と医療補助サービスセンター、衛生と公衆サービス部の他の部門、司法省、薬品監督管理局、消費財安全委員会、連邦貿易委員会、職業安全と健康管理局、環境保護局及び州と地方政府を含む。
例えば、アメリカでは、販売、マーケティング、科学と教育プロジェクトも州と連邦の詐欺や乱用法律を守らなければならない。これらの法律には、処方薬製造業者(またはそれを代表する側)を含む任意の人が、インフォームドコンセントおよび意図的な場合には、連邦医療保険または医療補助などの連邦医療計画に基づいて支払われる可能性がある代替薬の購入、推薦、注文または処方を含む任意の報酬を請求、受け入れ、提供、または支払いすることが規定されている連邦反リベート法規が含まれている。この法律に違反した人は最高5年の禁錮刑、刑事罰金、行政民事罰金、連邦医療計画から除外されることができる。また、“反リベート法”では、政府は、“虚偽申告法”の規定により、連邦“反リベート条例”違反による物品又はサービスのクレームが虚偽又は詐欺的クレームを構成することを含むと主張することができる。
定価と返却計画は,米国の1990年の“総合予算調節法”の医療補助帰点要求およびACAの最近の要求に適合しなければならない。総務省連邦供給スケジュールの許可されたユーザに製品を提供する場合は、他の法律および要求が適用される。製品はアメリカの“毒物防止包装法”に適用される児童保護包装要求に適合しなければならない。製造、販売、販売促進、その他の活動はまた、連邦と州消費者保護および不正競争法によって制限される可能性がある。
生物と医薬製品の流通は広範な記録保存、許可、貯蔵と安全要求を含む追加の要求と条例を遵守し、許可されていない医薬製品の販売を防止しなければならない。
38
これらの法律または規制要件のいずれかを守らない場合、会社は可能な法律または規制行動に直面するだろう。状況に応じて、適用される規制要件に適合しないことは、刑事起訴、罰金またはその他の処罰、禁止、リコールの要求、製品の差し押さえ、生産の完全または部分的な一時停止、製品の承認の拒否または撤回、または政府契約を含む会社の供給契約の締結の許可を拒否する可能性がある。これらの法律に違反して私たちにとった行動は、たとえ私たちが弁護に成功しても、巨額の法的費用を招き、私たちの経営陣の業務運営への注意をそらす可能性があります。私たちが販売している未来の製品の販売を禁止または制限または撤回することは、不利な方法で私たちの業務に大きな影響を与えるかもしれません。
規制、法規、または既存の規制の解釈の変化は、私たちの製造スケジュールの変更、製品ラベルの追加または修正、私たちの製品のリコールまたは生産停止、または追加の記録保存要件のような、私たちの将来の業務に影響を与える可能性があります。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。
アメリカの特許期限回復と市場排他性
FDAが将来の候補製品を承認する時間、期限、詳細によると、私たちのいくつかの米国特許は、ハッジ·ワックスマン法に基づいて限られた特許期間の延長を受ける資格がある可能性がある。ハッジ·ワックスマン法は、製品開発およびFDA規制審査中に失われた特許期間の補償として、最長5年間の特許期間の回復を可能にする。しかしながら、特許期限の回復は、特許の残り期間を製品承認日から合計14年間延長することはできない。特許期間回復期は、一般にINDの発効日とNDAまたはBLAの提出日との間の時間の半分であり、NDAまたはBLAの提出日とその出願の承認との間の時間の半分を加えるが、出願人が職務調査を行っていない間に審査期限が短縮される。承認された薬物に適用される特許は1つのみ延期する資格があり,延期出願は特許が満期になる前に提出されなければならない。米国特許商標局(USPTO)はFDAと協議し,任意の特許期間の延長または回復の出願を審査·承認する。将来的には、臨床試験の期待長および関連秘密協定またはBLAの提出に関連する他の要因に依存する現在の満期日後の特許寿命を延長するために、現在所有または許可されている特許の特許期間を回復することを申請することができる。
FDCAにおける市場排他性条項はまた、いくつかの申請の提出や承認を延期する可能性がある。FDCAは新しい化学実体秘密協定の承認を得た最初の申請者に5年間の米国内の非特許マーケティング排他性を提供した。FDAが以前に同じ活性部分を含む他の新薬を承認していなければ,薬物は新しい化学実体であり,活性部分は薬物物質の作用を担う分子やイオンである。排他期間内に、FDAは、出願人が承認のために必要なすべてのデータを合法的に参照する権利を有していない場合、FDAは、薬剤の別のバージョンのために別の会社に提出された簡略化された新薬出願(ANDA)または505(B)(2)NDAを受け入れない可能性がある。しかしながら、出願が特許無効または非侵害の証明を含む場合、4年後に提出することができる。FDAが、出願人が行っているまたは後援する新しい臨床研究(バイオアベイラビリティ研究を除く)が承認申請に不可欠であると考えている場合、FDCAはまた、NDA、505(B)(2)NDAまたは既存のNDAの補充のために、既存の薬剤の新しい適応、用量または強度のような3年間の市場排他性を提供する。この3年間の排他性には,新たな臨床研究に関する使用条件のみが含まれており,FDAが原始活性物質を含む薬物のANDAを承認することは禁止されていない。5年と3年の排他性は完全な秘密協定の提出や承認を延期したり承認したりしないだろう。しかしながら、完全なセキュリティプロトコルを提出する出願人は、安全かつ有効であることを証明するために、必要なすべての臨床前研究および十分かつ良好に制御された臨床試験を参照する権利を行うか、または得ることを要求されるであろう。
参照生物製品は、製品が初めて許可を得た日から12年間のデータ独占権を付与され、FDAは、参照製品が初めて許可された日から4年まで、参照生物製品に基づく生物類似または交換可能製品の申請を受け入れないであろう。“初許可”とは、一般に、米国で特定の製品が許可された初期日を意味する。最初に許可を得た日は、生物製品が許可された日を含まず(および新しい特定期間は適用されない)、許可が生物製品を補充するためのものである場合、または生物製品の同じ発起人または製造業者(おそらく人、利害関係者または他の関連エンティティ)のために変更を申請し(バイオ製品構造の修正を含まない)、それにより、新しい適応、投与経路、投与スケジュール、剤形、送達システム、送達デバイスまたは強度または強度をもたらす
39
生物製品の構造の変化は、安全性、純度あるいは効力の変化を招くことはない。したがって、新製品が以前の許可製品の構造の修正を含むかどうかを決定し、それにより、安全性、純度、または効力の変化をもたらし、新製品の許可がそれ自身の排他期間をトリガする最初の許可であるかどうかを評価する必要がある。その後の出願は、承認された場合には、生物製品としての“第一次許可”の排他性が保証されるか否かは、具体的な状況やスポンサーが提出したデータに依存する。
EU薬物開発
アメリカと類似して、EUの臨床前と臨床研究の各段階は重要な監督管理によって制御されている。EU臨床試験指令2001/20/ECはEU臨床試験監督管理枠組みを調整し、EU臨床試験の制御と許可のために共通規則を制定したが、EU加盟国は異なる方法でこの指令の条項を交換と応用した。これは会員国制度の大きな変化を招いた。現在の制度では、臨床試験を開始する前に、各EU諸国で承認されなければならず、これらの国では、試験は2つの異なる機関によって行われる:国家主管機関(NCA)と1つ以上の道徳委員会(ECs)。現行制度によると,臨床試験期間中に調査された薬物に対して発生するすべての疑わしい意外重篤な副作用は,これらの反応が発生した加盟国の国家薬品管理局と欧州薬品監督管理局に報告しなければならない。
EU 536/2014号臨床試験法規は“臨床試験指令”の代わりに、そして2022年1月31日に発効し、現在の臨床試験許可規則を簡略化し、臨床試験許可を調整と簡略化し、不良事件報告プログラムを簡略化し、臨床試験の監督を改善し、そしてその透明性を増加することを目的としている。
EUの薬品審査と承認
欧州連合27加盟国(ノルウェーを含む、クロアチアを含まない)、アイスランド、リヒテンシュタインからなる欧州経済地域では、医薬製品はマーケティング許可(MA)を得た後にのみ商業化できる。二つの種類のマーケティング許可があります。
40
上記の手順により,MAを付与する前に,欧州環境管理局または欧州経済区加盟国主管当局は,製品の品質,安全性,有効性に関する科学的基準に基づいて,製品のリスク−利益バランスを評価する。
保証と精算を請け負う
私たちの製品の販売は、政府医療計画、商業保険、信託医療機関など、私たちの製品がどの程度第三者支払者にカバーされるかにある程度依存します。米国では,薬品や生物製品の保険や精算に統一された政策はない。したがって、私たちのどの製品の保険範囲や精算金額に関する決定は支払者ごとに決定されます。そのため、保証範囲の決定過程は通常時間がかかり、高価な過程であり、各支払人にそれぞれ私たちの製品を使用する科学的かつ臨床的な支持を提供する必要があり、保証と十分な補償を得ることができない。
アメリカ政府、州立法機関と外国政府はコスト制御計画の実施に大きな興味を示し、政府が支払う医療コストの増加を制限し、価格制御、精算制限とブランド処方薬の代わりに模造薬を要求することを含む。例えば,ACAに含まれる条項は,医療補助計画で精算された薬品のリベートを増やすこと,医療補助リベートを医療補助管理保健計画に拡大すること,ある連邦医療保険Dの一部の受益者を強制的に割引すること,製薬会社の連邦ヘルスケア計画における販売シェアに応じて年会費を徴収することで薬品の収益性を低下させる可能性がある。一般的な制御と措置を採用し、既存の制御と措置の司法管轄区で制限的な政策を強化することに加え、薬品への支払いを制限する可能性がある。
医療補助薬品還付計画は製薬業者が衛生と公衆サービス部部長と締結し、全国的な税金還付協定を発効することを要求し、各州がメーカーが医療補助患者に提供する外来薬物の連邦マッチング資金を獲得する条件とする。ACAは医療補助薬品還付計画に対していくつかの変更を行い、製薬業者の税金還付責任を増加させ、大多数のブランド処方薬の最低基本医療補助税金還付をメーカーの平均価格(AMP)の15.1%からAMPの23.1%に高め、そしてブランド製品の固体経口剤形の“シリーズ延長”(即ち徐放製剤のような新しい調合)の新しい税金還付計算を増加し、そしてAMPの法定定義を修正することによってその還付責任に影響を与える可能性がある。ACAはまた,製薬業者に医療補助管理の医療使用に税金還付の支払いを要求することにより,医療補助薬物福祉を受ける資格のある潜在人口を拡大することにより,薬品還付の影響を受ける医療補助使用範囲を拡大した。医療保険や医療補助サービスセンターも医療補助税還付責任を米国領に拡大することを提案している。
2003年に“連邦医療保険処方薬、改善と現代化法案”(MMA)は連邦医療保険D部分計画を創立し、連邦医療保険受益者に自発的な処方薬福祉を提供した。D部によると、連邦医療保険受益者は、個人実体が提供する処方薬計画に参加することができ、これらの計画は外来処方薬の保険を提供する。連邦医療保険A部やB部と異なり,D部のカバー範囲は標準化されていない。すべての連邦医療保険薬物計画は少なくとも連邦医療保険が設定した標準保険レベルを提供しなければならないが、D部分の処方薬計画発起人はすべての保険を受けたD部分の薬物に費用を支払う必要はなく、各薬物計画は自分の薬物処方を開発し、それがどの薬物および被覆のレベルまたはレベルをカバーするかを決定することができる。しかしながら、D部分処方薬処方は、必ずしも各カテゴリまたはカテゴリのすべての薬剤を含むとは限らないにもかかわらず、各治療カテゴリおよびカバーされたD部分薬剤カテゴリの薬剤を含む必要がある。D部分の処方薬計画に使用されるどの処方も薬局と治療委員会が開発·審査しなければならない。政府が処方薬の費用の一部を支払うことは、私たちが発売許可を得た製品に対する需要を増加させる可能性がある。しかし、D部分の処方薬計画がカバーしている私たちの製品のいかなる交渉価格も私たちが獲得する可能性のある価格を下回るかもしれません。また,MMAは連邦医療保険受益者の薬品福祉にのみ適用されるが,個人支払者は自分の支払率を設定する際に連邦医療保険カバー政策や支払制限に従うことが多い。MMAによる任意の支払い減少は、非政府支払者支払いの同様の減少をもたらす可能性がある。
MedicaidまたはMedicare Part B計画に従って連邦補償を獲得するか、または米国政府機関に直接販売する薬品の場合、製造業者は割引を340 B薬品定価計画に参加する資格のあるエンティティに拡大しなければならない。製品を与えるのに必要な340 B割引は,メーカーから報告されたAMPと医療補助返金金額から計算される。
41
アメリカは特殊薬品定価やり方に対する立法と法執行への興味はますます大きくなっており、アメリカ議会の調査と提案された連邦と州立法を含み、これらの立法は薬品定価の透明性を高め、連邦医療保険下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査し、政府計画の薬品精算方法を改革することを目的としている。連邦レベルでは、トランプ政権の指導の下で、HHSとCMSは様々なルールを発表し、これらのルールは、D部分の薬品メーカーの計画スポンサーの値下げ、薬局福祉マネージャーとメーカー間の費用スケジュール、医療補助薬品バックオフ計画下のメーカー価格報告要求に影響を与えることが予想され、薬局福祉マネージャーアキュムレータ計画といくつかの価値に基づく調達スケジュールに関連する最適な価格報告に影響を与えるメーカーが後援する患者援助計画の規定を含む。HHSに対する複数の訴訟は、これらの新しい規則の様々な側面に挑戦してきた。バイデン政権とHHSは実施を延期したり、トランプ時代のいくつかの政策を廃止する規定を公表したりした。
2021年1月1日に施行される“2021年米国救援計画法案”によると、メーカーが州医療補助計画に支払う医療補助薬品還付計画還付の法定上限が撤廃される。この上限を廃止することは、販売承認製品よりも多くのリベートの支払いを製薬業者に要求する可能性があり、これは私たちの業務に実質的な影響を与える可能性がある。また、国会では、可決されれば、連邦医療保険がカバーする処方薬の価格に大きな影響を与える可能性があり、薬品価格の上昇を制限し、連邦医療保険があるカバー薬品の定価について交渉することを許可する立法が検討されている。コスト抑制措置や他の医療改革を実施することは、収入を創出し、利益を達成すること、または候補製品を商業化することを阻止するかもしれない(承認されれば)。
さらに、多くの州は、バイオ製薬メーカーに独自の価格設定情報を開示すること、または国家機関が購入した薬品に対して最高価格上限を設定することを要求するなど、間接的または直接に薬品価格を規範化するための立法を提出または公布した。例えば、多くの州が州薬品価格透明性と報告法を検討しているか、最近公布されており、これは私たちのコンプライアンス負担を大幅に増加させ、規制部門の任意の候補製品に対する承認を得て商業化を開始した後、このような州法律に基づいてより大きな責任を負うことができるかもしれない。これらの措置および立法は、私たちが規制承認を受ける可能性のある任意の候補製品の価格に影響を与えるかもしれないし、承認された場合、そのような候補製品の需要に影響を与える可能性がある。
上述したように、政府や第三者支払者が十分な保険や補償を提供できない場合、規制部門の承認を得て商業販売を行う任意の製品の適正性が影響を受ける可能性がある。米国のコスト抑制措置への重視が増しており、薬品価格の圧力を増加させ続けることが予想される。保証政策と第三者精算料率は随時変化する可能性がある。規制部門の承認を得た1つまたは複数の製品が有利な引受·精算状態を獲得しても、将来的にはあまり有利ではない引受政策や精算料率が実施される可能性がある。
また、大多数の外国の国では、薬品の提案価格は許可されなければならず、合法的に発売されることができる。各国の薬品定価と精算に対する要求は大きく異なる。例えば、欧州連合は、その国の健康保険制度が補償を提供する医療製品の範囲を制限し、人が使用する医療製品の価格を制御するために、その加盟国に様々な選択を提供している。加盟国は医薬製品の具体的な価格を承認することができ、医薬製品を市場に投入する会社の収益力に対して直接或いは間接的に制御制度をとることもできる。薬品に対して価格制御や精算制限を実行する国が私たちのいかなる製品にも有利な精算と定価手配を許可することは保証されません。歴史的に見ると、EUで発売された製品はアメリカの価格構造に従わず、通常価格ははるかに低くなることが多い。
42
科学顧問委員会
著者らは高い素質の科学顧問委員会を創立し、顧問から構成され、彼らは共に神経変性疾患、ゲノム学、蛋白質工学、薬物開発と薬物発見及び転化医学方面の深い専門知識を持っている。著者らの科学者はこれらのコンサルタントと協力して、新しい疾病標的を確定し、バイオマーカー戦略を開発し、発見と開発を加速した。
名前.名前 |
付属実体 |
|
|
アダム·ボクサー医学博士 |
カリフォルニア大学サンフランシスコ校神経科 |
アーロン·ギトラー博士です |
スタンフォード大学遺伝学科 |
スティーヴン·ハウザー医学博士 |
カリフォルニア大学サンフランシスコ校神経科 |
マイケル·ヘネカ医学博士 |
ルクセンブルク大学ルクセンブルクシステム生物医学センター |
マーティン·キャンプマン博士です |
カリフォルニア大学サンフランシスコ校ウィルキンス科学研究所生化学と生物物理学部 |
ルイス·ラニール博士 |
カリフォルニア大学サンフランシスコ校微生物学と免疫学学部 |
羅立群博士。 |
アメリカ国立科学アカデミーとスタンフォード大学生物学部は |
リチャード·シェラー博士です |
アメリカ国立科学院と国家医学研究所は |
トーマス·クリスティアン·スデホフ医学博士 |
スタンフォード大学の分子と細胞生理学や神経外科は |
ロバート·ヴァサ博士 |
西北大学神経病学科 |
ベリスラフ·ズロコビッチ医学博士 |
南カリフォルニア大学生理学と神経科学科 |
従業員と人的資本
2022年12月31日現在、私たちは273人のフルタイム従業員を持っており、そのうち78%以上が研究開発活動に従事している。私たちの職員たちは労働組合代表者もなく、集団交渉協定のカバー範囲もない。
私たちの人的資本目標には、私たちの既存と新しい従業員、コンサルタント、コンサルタントを識別、採用、維持、激励、統合が含まれている。私たちの株式と現金インセンティブ計画の主な目的は、株式と現金に基づく報酬奨励を付与することで、これらの従業員を激励してできる限りのことをし、私たちの目標を実現することで、株主価値と会社の成功を増加させることです。過去数年間、米国の雇用市場は競争の激しい求人や給与環境の影響を受けてきたが、これは逆に一部の従業員の流失を招いている。
新冠肺炎の疫病とその後の変種に対応するため、著者らは健康と安全仕事の重点を従業員とその家族の保護に重点を置いた。我々は,疾患コントロール·予防センターの指導と一致し,適用される州や地方法規に適合していると考えられる従業員と我々のコミュニティの利益に最も適した変化を実施した。これには、私たちの一部の従業員が在宅で働くようにするとともに、重要な現場作業を継続する従業員のための追加的な安全措置を実施することが含まれる。
企業情報
私たちは2013年5月にデラウェア州に有限責任会社として設立され、Alector LLCと呼ばれ、2017年10月に再編が完了し、デラウェア州の会社となり、Alector,Inc.となりました。私たちの主な実行オフィスはOyster Point Boulevard 131 Oyster Point Boulevard,Suit 600,Suite 600,California 94080にあります。私たちの電話番号は415-231-5660です。私たちのサイトの住所はwww.alector.comです。当社のウェブサイトに含まれている、または当社のウェブサイトを介してアクセス可能な情報は、本10-Kフォーム年次報告書または米国証券取引委員会に提出された他のいかなる文書にも引用されません。
1934年に改正された証券取引法(取引法)によると、私たちは、米国証券取引委員会に提出されたいくつかの報告書と、これらの報告書の修正を、私たちのウェブサイト上または私たちのサイトを通じて提供します。
43
これらの報告には,Form 10−Kに関する我々の年次報告,Form 10−Qに関する我々の四半期報告,および我々の現在のForm 8−Kに関する報告,取引法第13(A)または15(D)節に提出または提出された報告書の修正が含まれている。私たちが電子的に情報をアーカイブしたり、アメリカ証券取引委員会に提供したりした後、私たちは合理的で実行可能な範囲でできるだけ早く私たちのサイトでこれらの情報を無料で提供します。
米国や他の国/地域における商標としてAlector,Alectorロゴ,その他のマークを用いた。本報告書は、私たちの商標およびサービスマーク、ならびに他のエンティティに属する商標およびサービスマークへの参照を含む。便宜上、本報告で言及されている商標および商品名は、ロゴ、イラスト、および他の視覚的表示を含む、存在しない可能性がある®または商標記号であるが、このような参照は、適用法に基づいて、これらの商標および商品名に対する私たちの権利または適用許可者の権利を最大限に主張しないことを意味するわけではない。私たちは、任意の他のエンティティとの関係を示唆するために、または任意の他のエンティティによって裏書されたり、後援されたりするために、他のエンティティの商号、商標またはサービスマークを使用または提示するつもりはない。
情報開示のルート
投資家および他の人は、私たちの投資家関係サイト(http://investors.alector.com)、アメリカ証券取引委員会の記録ファイル、ネットワーク放送、ニュース原稿、私たちのサイトで提供されている会社のプラットフォームおよび電話会議を使用して、投資家に重要な商業および金融情報を発表する可能性があることに注意しなければならない。私たちはこれらのメディアを使って、私たちのウェブサイトを含めて、私たちのメンバーや公衆と私たちの会社、私たちの製品、その他の問題について交流します。私たちが提供する情報は重要な情報とみなされるかもしれない。したがって、私たちは私たちの投資家やわが社に興味を持っている他の人が私たちがサイトで提供している情報を見ることを奨励します。
44
第1 A項。RISK因子です。
私たちの普通株に投資することは高い危険と関連がある。私たちの業務を評価する際には、以下に説明するリスク、および当社の財務諸表および関連説明、および“経営陣の財務状況および運営結果の議論および分析”と題する部分、ならびに私たちの他の公開申告文書中の他の情報を含む本Form 10-K年次報告書の他の情報を慎重に考慮しなければなりません。次のいずれの事件や事態が発生しても、私たちの業務、財務状況、経営結果、成長の見通しを損なう可能性があります。この場合、私たちの普通株の市場価格は下落する可能性があり、あなたは投資の全部または一部を失うかもしれない。私たちは今知らないか、あるいは私たちは今どうでもいいと思っている他のリスクと不確定要素もまた私たちの業務運営と私たちの普通株の市場価格を損なう可能性があります。
リスク要因の概要
私たちの業務は多くのリスクと不確実性の影響を受けていますが、以下に述べるように、わが社に投資する前にこれらのリスクと不確実性を考慮すべきです。わが社に投資するリスクの主な要素と不確実性は、
45
私たちの業務、財務状況、資本要求に関連するリスク
私たちは薬物開発の異なる段階にあり、運営歴史が限られており、商業販売が許可されていない製品は、現在の業務を評価し、将来の成功と生存能力を予測することを困難にする可能性がある。
著者らは臨床段階の生物製薬会社であり、運営歴史は限られており、主にFTD、アルツハイマー病、パーキンソン病と筋萎縮性側索硬化症、及び先天性免疫生物学に集中した腫瘍学的治療を含む神経変性疾患を治療する薬物の開発に集中している。私たちは2013年5月に運営を開始した。これまで、私たちは主に株式証券の売却とエバービーやグラクソ·スミスクラインとの協力で受け取った前金を手配することで、私たちの業務に資金を提供してきました。私たちは商業販売を許可された製品もなく、製品販売から何の収入も得ていない。薬物開発は高度に不確実な仕事であり、大きなリスクに関連している。候補製品latozinemabの第2段階と第3段階臨床試験を行っており,候補製品AL 002の第2段階臨床試験を行っており,AL 101の第1段階臨床試験を完了している。初期PKと耐性データに基づき,AL 044候補製品の第1段階臨床試験を終了することにした。また,AbbVieとAlectorが連携してこの計画に基づいて開発した資産AL 003の後続手順を審査し,AL 003をさらに開発する必要はないと結論した後,AbbVieはCD 33連携計画を終了することにした.これまで重要な臨床試験を終えておらず、候補製品のマーケティング承認を得ておらず、ビジネス規模の製品を製造していないし、第三者代表も手配しておらず、成功した製品の商業化に必要な販売やマーケティング活動も行っていない。会社として、私たちの限られた経営歴史は、私たちの未来の成功と生存能力のどの評価も重大な不確実性の影響を受けるようにした。
私たちは臨床段階の生物製薬会社が急速に発展している分野でよく遭遇するリスクと困難に直面するが、私たちはまだこれらのリスクと困難を克服する能力を示していない。もし私たちがこのような危険と困難に成功的に対応できなければ、私たちの業務は影響を受けるだろう。
私たちが設立して以来、私たちは毎年大きな純損失が発生し、予測可能な未来に純損失が続くと予想されています。
設立以来、私たちはほとんどすべての報告期間に純損失を出した。2022年12月31日、2021年12月31日、2020年12月31日までの純損失はそれぞれ1.333億ドル、3630万ドル、1.902億ドルだった。2022年12月31日までの累計赤字は5兆797億ドル。
著者らは研究開発活動に大量の財政資源を投入し、著者らの臨床前と臨床候補製品を含む。私たちは数年以内に製品販売から収入が出ないと予想しています。もしあれば。私たちが現在エバービーとグラクソ·スミスクラインとの協力計画から得ている収入は可変で、金額は限られている。我々とエバービーやグラクソ·スミスクラインとの協力については,完全な義務履行の進捗を測ることで協調収入を確認し,これは計画コストに基づいて測定した。私たちの未来の純損失額は私たちの未来の支出と収入水準に部分的に依存するだろう。また,我々の純損失は四半期ごとに大きく変動する可能性があるため,我々の運営結果を経時的に比較することは我々の将来の業績の良い指示ではない可能性がある。
46
2021年7月1日、私たちは、latozinemabおよびAL 101を含む、グラクソ·スミスクラインと世界的に協力して、latozinemabおよびAL 101を含む前粒子タンパク質のモノクロナル抗体を開発および商業化することで合意した。GSK協定の条項によると、私たちは7億ドルの前金を受け取り、そのうち5億ドルは2021年8月に受け取り、2億ドルは2022年1月に受け取った。さらに、LatozinemabとAL 101の臨床開発、規制、商業発売に関連するマイルストーン支払いを取得する資格があり、最高15億ドルを追加することができます。
私たちの候補製品を開発するのは高価で、私たちの早期研究プロジェクトに資金を提供し、臨床前と臨床開発を通じて私たちの計画を進めていくために、引き続き多くの資金がかかると予想される。私たちが候補製品の開発に成功し、規制部門の承認を得たり、任意の候補製品を発売して商業化したりしても、大量の追加資金が必要だ。
私たちは予測可能な未来に、巨額の費用とますます高い運営損失が発生すると予想している。私たちの費用は大幅に増加すると予想されています
私たちの以前の損失と予想された未来の損失はすでに私たちの株主権益と運営資本に悪影響を与え続けるだろう。いずれの特定の四半期においても、私たちの経営業績は証券アナリストや投資家の予想を下回る可能性があり、これは私たちの株価を下落させる可能性がある。
薬物開発は高度に不確実な仕事であり、大きなリスクに関連している。
私たちは商業販売を許可された製品がありません。私たちの候補製品の販売から収入を得るためには、これらの製品の規模は利益を達成するのに十分な規模であり、単独でも第三者と協力しても成功しなければならない
47
締約国は開発、監督管理の承認、製造とマーケティング療法の面で重大な商業成功を得た。私たちが収入を創出し利益を達成する能力は多くの要素に依存しています
これまで、私たちの二つの候補製品の臨床開発は終了した。AbbVieとAlectorが連携してAL 003の後続ステップを審査した後,AbbVieはCD 33連携計画を終了することを決定し,AL 003はその計画に基づいて開発された資産であり,さらなるAL 003を開発する必要はないと結論した.また,初期PKと耐性データから,われわれのAL 044候補製品の第1段階臨床試験を終了することにした。薬物開発に関連する多くのリスクや不確実性のため、私たちが支出した時間や金額を予測することはできず、私たちがいつどんな意味のある収入を生み出すことができるか、あるいは利益を達成したり維持したりすることも予測できない。さらに、FDAまたは外国規制機関が現在予想されている研究以外の研究を要求している場合、または私たちまたは私たちの現在または未来の協力者の任意の臨床試験または私たちの任意の候補製品の開発に遅延が生じた場合、私たちの費用は現在の予想を超えるまで増加する可能性がある。私たちの1つ以上の候補製品が商業販売のために承認されても、任意の承認された候補製品の発売および商業化および持続的なコンプライアンス努力に関連する巨額のコストが生じることが予想される。
私たちは、私たちの候補製品の開発と商業化を達成するために大量の追加資金を得る必要があり、必要な場合に受け入れ可能な条件で必要な資本を得ることができない場合、あるいは必要な資金を全く得ることができない場合、私たちの商業化努力、製品開発、または他の運営を延期、制限、減少、または終了させることができるかもしれない。
設立以来、私たちの業務は大量の現金を必要としてきましたが、予測可能な未来に、私たちの支出は大幅に増加すると予想されています。これまで、私たちは主に株式証券の売却とAbbVieの協力手配に関する前金を受け取ることで、私たちの業務に資金を提供してきました
48
グラクソ·スミスクラインです我々の候補製品を開発し、FTD、アルツハイマー病、筋萎縮性側索硬化症とパーキンソン病及び腫瘍学を含む神経変性疾患を治療する臨床試験を行い、大量の資金が必要となる。私たちはまた、私たちの候補製品をさらに開発するための大量の資金が必要であり、これらの候補製品のいずれかが承認されれば、承認された任意の製品を商業化するための大量の資金が必要になるだろう。
2022年12月31日現在、私たちは7億129億ドルの現金、現金等価物、有価証券を持っている。私たちの現在の運営計画によると、私たちは私たちの既存の現金、現金等価物、有価証券が2025年までの運営を計画するのに十分な資金を提供すると信じている。既存の現金、現金等価物、および有価証券がどのくらいの間、私たちの運営に資金を提供するために使用できるかの推定は、不正確であることが証明される可能性があるという仮定に基づいており、私たちは現在予想されているよりも早く利用可能な資本資源を使用するかもしれない。さらに、インフレ率が上昇した時期を含めて変化する環境は、私たちの支出増加の速度が私たちの現在の予想よりも大きく速くなる可能性があり、そして私たちがコントロールできない状況のため、私たちは現在予想されているよりも多くのお金が必要かもしれない。もし私たちが現在の予想よりも速い速度で拡張することを選択すれば、私たちは予想よりも早く追加資金を調達する必要があるかもしれない。
世界市場は最近、インフレの激化、サプライチェーンと新冠肺炎の流行の他の経済影響、ロシアのウクライナ侵攻を含む地政学的事件などによるマクロ経済衰退が原因で変動と不安定を経験している。また、バイオテクノロジー会社の公開市場と株価は過去数年間で著しい下落を経験した。これらの要因により、予測可能な未来には、公開市場で資金を調達する能力が深刻な影響を受ける可能性がある。私たちが必要な時、私たちは私たちが受け入れられるか、あるいは全くないという条件で追加的な資本を得ることができないかもしれない。もし私たちが十分な資本をタイムリーに得ることができなければ、私たちは私たちの研究開発計画や任意の候補製品の商業化を大幅に延期、削減、停止することを要求されるかもしれません(承認されれば)、あるいは私たちの業務を継続または拡大することができない、あるいは他の方法で私たちのビジネスチャンスを利用することができません。これは、私たちの業務、財務状況、運営結果、成長の見通しに大きな影響を与え、私たちの普通株価格を下落させる可能性があります。
私たちが株式または転換可能な債務証券を売却することで追加資本を調達する場合、私たち株主の所有権権益は希釈され、これらの証券の条項は清算または他の特典を含む可能性があり、普通株主としての権利に悪影響を及ぼす可能性がある。債務融資が可能であれば、金利を含む不利な条項である可能性があり、追加債務を招く、資本支出を行う、または配当を宣言するなど、特定の行動をとる能力を制限または制限する契約を含む可能性がある。もし私たちが製薬パートナーとの協力、戦略連合、または許可手配を通じてより多くの資金を調達すれば、私たちは私たちの技術、将来の収入源、研究プロジェクト、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項で許可を与えなければならないかもしれない。
私たちの候補製品開発には大量の資源が必要であるため、私たちが資金を得る能力に応じて、いくつかの候補製品を優先的に開発しなければならない。さらに、私たちは、成功した候補製品を生成しない計画に限られたリソースを使用するか、またはより利益または成功の可能性が高い可能性の高い候補製品または適応を利用することができないかもしれない。
私たちは100個以上の免疫系標的を決定した。我々の3つの候補製品Latozinemab,AL 002,AL 101は臨床開発を行っており,我々は研究パイプラインの開発を継続している。私たちの候補製品の一つAL 003の開発は2022年6月に終了しました。結論的に、このようなプロジェクトと候補製品を開発するには大量の資本投資が必要だ。私たちの計画と候補製品の開発には大量の資源が必要であるため、私たちの計画と候補製品を特定の疾病と疾病経路に集中させ、どの候補製品を追求し、推進するか、各候補製品に割り当てられた資源量を決定しなければならない。我々の薬物開発戦略は,我々の候補製品の臨床試験を行い,概念検証データを迅速に生成できる適応を最も多くの証拠があると考えられる上で規制承認を求めることである。そして,臨床試験に拡張し,主要な適応の遺伝や機序と重なることにより,他の神経変性適応の規制承認を求める予定である。
しかし,我々の候補製品が1つの適応で規制されていても,他の適応で承認される保証はなく,このような承認を求めるために多くの資源がかかる可能性がある.また,神経変性以外の適応を求めることに資源を集中させる可能性があり,どの発見項目に重点を置くかを決定する際に用いる遺伝や機序原理と同様である。私たちは分配研究、開発、協力、管理、
49
特定の候補製品または治療分野のための財政資源は、いかなる実行可能な商業製品の開発にもつながらず、より良い機会からリソースを移転することが可能である。たとえば,InnoventはAL 008の開発を続けることはなく,AL 008の開発と商業化のためのInnoventtを付与する権利を新たに獲得する予定である.Innoentは中国規制部門にINDを提出し,AlectorはInnovent.から関連材料と情報を取得している。Alector計画は、IND申請中のデータおよび文書を評価して、INDの米国での提出を潜在的に支援することを計画している。同様に、いくつかのプロジェクトについて延期、終了、または第三者と協力する潜在的な決定は、その後、次善であることが証明される可能性があり、予想される貴重な機会を逃す可能性がある。もし私たちが私たちの任意の計画や候補製品の実行可能性や市場潜在力に対して誤った判断をしたり、生物製薬業界の傾向を誤読したりすれば、特に神経変性疾患に対して、このような事件は私たちの業務、財務状況、および運営結果に実質的な悪影響を与える可能性がある。したがって、私たちは、実行可能な商業製品または利益の市場機会を利用することができず、他の候補製品または他の疾患および疾患経路との機会を追求する機会を放棄または延期することが要求される可能性があり、これらの疾患および疾患経路は、後に、私たちが選択したよりも大きな商業的潜在力を有することが証明されるか、または協力、許可によってこれらの製品候補製品に貴重な権利を放棄する可能性がある, または他の特許権使用料配置は、この場合、独占的開発および商業化権利を保持するために追加資源に投資することが有利である。著者らは遺伝子スクリーニングとバイオマーカーの使用に依存して患者のリスク特徴を方向性干与と一致させ、最終的に著者らはセット診断方法の開発と使用が必要である可能性があり、これは製品開発コストとスケジュールに影響を与える可能性があり、具体的には特定の診断方法と任意の需要に依存してその使用に必要な適用法規の要求を満たす。
私たちの候補製品の発見、開発、商業化に関するリスク
生物製薬製品の開発自体に危険がある。私たちの業務は私たちの候補製品の成功開発に大きく依存しており、これらの候補製品は臨床前と臨床開発の異なる段階にある。私たちは私たちの候補製品がマーケティングと承認を含めて規制されることを保証することはできません。これはそれらが商業化できる前に必要です。
私たちが現在計画している多くの候補製品は臨床開発段階にある。私たちはこれまで、知的財産権を決定、取得し、私たちの計画や候補製品を開発し、これらの業務に一般的かつ行政的な支援を提供するために、多くの努力と財政資源を投入してきた。私たちの将来の成功は、私たちが開発に成功し、規制機関の承認を得て、私たちの候補製品を商業化することに成功する能力にかかっており、私たちは多くの理由でこれができないかもしれません
50
上記のいずれかの事件が発生した場合、私たちは1つ以上の計画のための開発作業を放棄することを余儀なくされる可能性があり、これは私たちの業務に実質的な悪影響を与え、運営を停止させる可能性があります。たとえば,AL 003の第1段階実験を完了し,我々と連携計画を検討した後,AbbVieはAL 003を開発しているCD 33連携計画を終了することを決定した.
私たちは私たちの現在の候補製品をさらに開発することに成功しないかもしれない。FDAや同様の外国規制機関の規制承認を得るまで、私たちは私たちの候補製品のマーケティングや普及は許可されておらず、私たちはいかなる候補製品の規制承認も得られないかもしれない。私たちの多くの候補製品は初期開発段階にあり、私たちが製品販売から任意の収入を得る前に、大量の追加の臨床開発、臨床前、臨床と製造活動の管理、監督管理の承認、十分な製造供給、商業組織と重大なマーケティング努力が必要である。
私たちは臨床開発計画を完成させたことがない。候補製品latozinemabの第2段階と第3段階臨床試験を行っており,候補製品AL 002の第2段階臨床開発を行っており,AL 101の第1段階臨床試験を完了している。しかも、私たちは私たちのどの候補製品も臨床試験で成功すると確信できない。私たちが臨床試験に入った任意の候補製品について、私たちはその完成前にこのような試験または臨床計画を終了するかもしれない。
もし私たちのすべての候補製品が臨床試験に成功すれば、私たちは通常、規制部門の承認を求め、アメリカ、EU、その他の可能なビジネス機会があると思う国で私たちの候補製品を販売する予定です。私たちは申請を始めたり、作成したり、提出したことがありません。規制部門がどんな候補製品の販売を許可するかを求めています。これらの候補製品が臨床試験に成功しても、私たちの生存能力に悪影響を及ぼすかもしれないが、規制部門の承認を得ず、どんな候補製品も販売することができるかもしれない。米国以外の国で規制承認を得るためには、これらの国/地域の私たちの候補製品の安全性、有効性、製造と制御、臨床試験、商業販売、定価、流通に関する多くの異なる規制要件を守らなければならない。私たちはまた、私たちの1つまたは複数の候補製品の規制承認申請を支援し、承認を求めるために、私たちの協力者またはパートナーに必要な活動に依存する可能性がある。私たちは私たちの協力者やパートナーが私たちが望む時間範囲でこのような活動を展開すると確信できない。たとえ私たち(または私たちの協力者やパートナー)が司法管轄区域で成功的に承認されたとしても、私たちは他のどんな管轄区でも承認されることを確実にすることはできない。もし私たちの候補製品が複数の管轄区域で承認されなければ、私たちの業務、財務状況、運営結果、成長の見通しはマイナス影響を受ける可能性があります。
規制部門の承認を得ても、神経変性疾患の治療にも他の疾患の治療にも、どのような候補製品も商業化に成功し、市場に広く受け入れられているか、または他の商業的に利用可能な代替製品よりも有効であることは保証されない。
生物製薬製品開発への投資は重大なリスクに関連し、即ちどの候補製品も十分な有効性或いは許容可能な安全性を証明できず、監督管理部門の承認を得ることができず、商業的に実行可能ではない。私たちが私たちの候補製品を開発過程に進めることに成功することができる保証はありません。あるいは、承認されれば、私たちの任意の候補製品は商業化に成功します。
私たちの研究と薬物発見プラットフォームから候補製品パイプラインを作成したり、商業的に成功した製品を開発する努力は成功しないかもしれません。私たちの研究や薬物発見プラットフォームからより多くの候補製品の識別と開発に成功できなければ、私たちのビジネス機会は制限されるかもしれない。
私たちの戦略の一つはより多くの候補製品の臨床開発を決定して追求することだ。神経変性疾患および他の疾患を治療するための追加候補製品の決定、開発、監督管理の承認と商業化は大量の追加資金を必要とし、薬物開発固有の失敗リスクが出現しやすい。私たちは、他の候補製品の識別または獲得に成功し、開発中にこれらの他の候補製品のいずれかを推進し、そのような候補製品の商業化に成功するか、または承認された場合、識別、取得、開発、または(承認された場合)他の候補製品を商業化するのに十分な資源を集約することができる保証はない。もし私たちが
51
もし私たちが他の候補製品を識別、獲得、開発、商業化することに成功できなければ、私たちのビジネス機会は制限されるかもしれない。
私たちは私たちが承認した候補製品の追加的または適応拡大の承認を得ることに成功できないかもしれない。
我々の薬物開発戦略は,我々の候補製品の臨床試験を行い,概念検証データを迅速に生成できる適応を最も多くの証拠があると考えられる上で規制承認を求めることである。そして,臨床試験に拡張し,主要な適応の遺伝や機序と重なることにより,他の神経変性適応の規制承認を求める予定である。我々の候補製品のためにより多くの適応の臨床試験を行うには大量の技術、財政と人的資本資源が必要であり、しかも薬物開発固有の失敗リスクが出現しやすい。私たちは私たちが予備適応の承認を得ても、規制部門の私たちの候補製品に対する追加適応の承認に成功することを保証することはできない。
私たちは商業販売のための製品は何も承認されておらず、製品販売から何の収入も得られていない。
我々は神経変性疾患の治療に大部分の研究と開発努力を集中させ、薬物開発において限られた成功を収めた領域である。また,我々の候補製品は新しい方法と新技術に基づいており,疾患や状況の兆候である新たなバイオマーカーを識別·開発しなければならず,我々の候補製品の疾患進展への影響を測定することができ,製品候補開発とその後の規制承認の時間とコストを予測することは困難である。
私たちはかなりの部分の研究と開発努力を神経変性疾患の解決に集中している。全体的に、生物製薬会社の神経変性疾患領域における努力は薬物開発における成功は限られている。現在、FTD、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症とその他の神経退行性疾患患者の治療選択は限られている。私たちの未来の成功は神経変性疾患を治療する私たちの候補製品の成功開発に強く依存している。候補製品を開発し、承認された後、神経変性疾患の治療に使用される製品を商業化し、目標組織において疾病修正活性と有効量を獲得し、FDAと他の監督管理機関の監督許可を得ることを含む多くの挑戦に直面させ、これらの監督機関は限られた一組の前例だけに依存することができる。
著者らは神経変性疾患を治療する方法はミクログリア細胞と他の髄系免疫細胞に富む標的を識別し、選択することを目的とし、これらの細胞は神経変性疾患の遺伝と関連している。脳内の有効量を達成し、期待される標的を達成するために、十分な量と効力で血液脳関門を横断することができる候補製品を識別し開発し、疾患や状態の兆候を正確に識別し、適切な患者集団を選択し、目標参加、経路参加、および疾患進行に対する候補製品の影響を測定することができるバイオマーカーおよびバイオマーカー分析を識別し開発しなければならない。この戦略は成功的であることが証明されないかもしれない。私たちは私たちの方法が安全で効果的、拡張可能、または利益になる満足できる治療製品を生成すると確信できない。
われわれは臨床試験において重大な遅延に遭遇する可能性があり,あるいは予想される時間内に臨床試験を行うことや完了できない可能性があり,まったくなければ。
臨床テストは高価で時間がかかり、不確実性も存在する。もしあれば、どんな臨床試験も計画通りに行われるか、予定通りに完成することは保証できません。INDや臨床試験申請(CTA)の提出はFDAやEMA(適用すれば)が臨床試験のタイムリーな開始を可能にすることを決定することはできない(あれば)。また,これらの試験が開始されても,このような臨床試験を一時停止または終了する可能性があるという問題がある。1つまたは複数の臨床試験の失敗は試験の任意の段階で起こる可能性があり、私たちの将来の臨床試験は成功しないかもしれない。成功またはタイムリーな臨床試験の開始または完了を妨げる可能性のあるイベントは、
52
臨床試験の開始や完了に成功できない場合は、私たちの追加コストをもたらしたり、収入を創出する能力を弱める可能性があります。さらに、私たちが私たちの候補製品を製造したり、調合を変更したりすれば、私たちは修正された候補製品を以前のバージョンに関連付けるために追加的な研究を行うことを要求されるかもしれません。臨床試験遅延はまた、私たちの製品が特許保護を持っている任意の期限を短縮することができ、私たちの競争相手が私たちの前に製品を市場に出すことを可能にすることができ、これは候補製品を商業化することに成功する能力を弱める可能性があり、私たちの業務と運営結果を損なう可能性がある。
53
例えば、私たちはアルツハイマー病患者の治療にAbbVieを使用してAL 003を開発してきましたが、2022年6月30日にAbbVieは私たちに書面通知を提供し、CD 33協力計画を中止することを正式に決定しました,AL 003はこの枠組みの下で開発された.また,InnoventはAL 008の開発を継続することはなく,AL 008の開発と商業化のためのInnoventtの権利を新たに獲得する予定である.Innoentは中国規制部門にINDを提出し,AlectorはInnovent.から関連材料と情報を取得している。Alectorは,INDアプリケーションのデータと文書を評価し,米国でのIND提出を潜在的に支援することを計画している.また,初期PKと耐性データからAL 044候補製品の第1段階臨床試験を終了することにした。
臨床試験が、私たち、データ安全監視委員会またはFDA、EMA、または任意の他の規制機関によって一時停止または終了された場合、またはそのような試験を行った機関のIRBsが、その臨床研究者およびその審査を受けた場所の参加を一時停止または終了する場合、遅延に遭遇する可能性もある。このような主管機関は多種の要素のために臨床試験を一時停止或いは中止する可能性があり、これらの要素は監督管理要求或いは著者らの臨床規程に従って臨床試験、FDA、EMA或いはその他の監督機関の臨床試験操作或いは試験地点の検査を行うことができず、臨床一時停止、予見できない安全問題或いは副作用の強制実施を招き、候補製品の使用によるメリット、政府法規或いは行政措置の変化、十分な資金不足により臨床試験、新冠肺炎の大流行及び後続変異の影響を証明できないことを含む。
私たちは将来、候補製品を事前に臨床試験に入れ、完成する前にこのような試験を終了するかもしれません。これは私たちの業務に悪影響を及ぼすかもしれません。
私たちの候補製品のすべての臨床試験の完成を遅延させることは私たちのコストを増加させ、私たちの候補製品の開発と承認過程を遅くし、そして遅延して、あるいは私たちの製品販売と収入を創造する能力を脅かす可能性があります。さらに、臨床試験の開始または完了遅延をもたらす多くの要因は、最終的には、私たちの候補製品が規制部門の承認を得ることを拒否される可能性もある。
われわれは臨床試験で患者を募集することが困難である可能性があるため,われわれの臨床開発活動は延期されたり,他の悪影響を受けたりする可能性がある。
臨床試験方案に基づいて適時に臨床試験を完成し、他の事項以外に、試験が終了するまで十分な数の患者を募集する能力があるかどうかに依存する。私たちは引き続き私たちの第3段階前-3と第2段階Invoke-2実験を登録する措置を取った。例えば,より多くの臨床試験サイトを開設し,募集を拡大し,INFRONT−3試験を募集している。しかし,様々な理由から,臨床試験で患者を募集する際に困難に遭遇する可能性がある
54
新冠肺炎の大流行と後続変種の影響により、著者らの臨床試験で患者を募集することと、著者らが行っている臨床試験で患者を維持することはすでに遅延或いは制限を受け続ける可能性がある。さらに、任意の新しい冠肺炎変異体を含むより多くの新冠肺炎懸念または制限が出現した場合、患者は、旅行の追加的な制限、連邦または州政府によって課せられたまたは提案された新しい物理的距離、または疾患流行中に患者が臨床試験サイトにアクセスすることを望まないため、臨床試験サイトにアクセスできないか、または訪問することができない可能性がある。新冠肺炎の大流行の影響は軽減しているが、新冠肺炎の大流行と後続変種による影響は引き続き著者らの臨床試験による予想の読み取りを延期或いは阻止する可能性がある。
私たちの臨床試験は深刻な有害事象、毒性、または他の副作用を示す可能性があり、私たちの候補製品の安全性と有効性を証明する実質的な証拠がない可能性があり、これは規制承認と商業化の範囲を阻止、延期、または制限するだろう。
私たちの任意の候補製品の商業販売が規制される前に、私たちは長い、複雑で高価な臨床前研究と臨床試験を通じて、私たちの候補製品が各目標適応において安全かつ有効であることを証明しなければならない。バイオ医薬品として規制されている候補製品については,安全で純粋で有効であり,その目標適応に利用できることを証明する必要がある。各候補製品は、その目標患者集団およびその目標用途において十分なリスクおよび収益状況を証明しなければならない。
臨床試験費用は高価であり,完成まで数年かかる可能性があり,その結果自体も確定していない。臨床試験では,いつでも失敗する可能性がある。著者らの候補製品の臨床前研究結果は早期或いは後期臨床試験の結果を予測できない可能性があり、著者らの候補製品の早期臨床試験結果は後期臨床試験の結果を予測できないかもしれない。健常ボランティアまたはグループの患者または疾患適応において行われた臨床試験結果は、別のグループの患者または疾患適応で得られた結果を予測することができない可能性がある。いくつかの場合、多くの要素のため、同じ候補製品の異なる臨床試験間の安全性或いは有効性結果は有意差が存在する可能性があり、方案に規定されている試験プログラムの変化、患者群の大きさとタイプの差異、投与方案の変化と投与方案の遵守、及び臨床試験参加者中の他の臨床試験方案要素と中退率を含む。開放ラベルや長期拡張研究も臨床計画の時間とコストを大幅に延長する可能性がある。臨床前研究と初歩的な臨床試験で進展を得たが、臨床試験後期段階の候補製品は必要な安全性と有効性を示すことができないかもしれない。生物製薬業界のいくつかの会社は高級臨床試験で重大な挫折を受け、早期の試験で良好な結果を得たが、治療効果の不足或いは受け入れられない安全性の問題が存在する。神経変性疾患の中で特にそうであり、歴史的に見ると、これらの疾病の失敗率は多くの他の疾病領域より高い。臨床試験を開始した候補製品の多くは規制機関の商業化承認を得たことがない。
例えば,われわれが行っているアルツハイマー病に対するInvoke−2段階2臨床試験では,ARIA症例が観察された。ARIAのMRI表現は血管源性水腫或いは鉄含有ヘモフラビン沈着を示した。アルツハイマー病のある治療薬、即ち抗β-アミロイド抗体の使用は、アルツハイマー病患者のARIA発生率が増加することを示している。われわれのInvoke−2段階2臨床試験では,ARIA症例の多くは無症状と非重篤であった。しかし,先に報告したように,少数のARIAに関連する重篤な副作用はAPOE e 4/e 4遺伝子型の患者でほぼ完全に発生している。ARIAに関するリスクを低減するために,われわれのInvoke−2段階2臨床試験では,APOE e 4/e 4参加者の用量と登録を自発的に中止した。これらの変化の後,少量のARIAに関連する重篤な有害事象はAPOE e 4対立遺伝子非ホモ接合体の患者で発生した。われわれは早期のMRIモニタリングを継続し,最近発表されたARIAモニタリングと管理ガイドラインと一致している。我々はIDMCの指導の下でこの研究を行い,IDMCは非盲目的データの審査と実験提案を許可された。
著者らは設計臨床試験の経験が限られており、臨床試験を設計と実行して上場承認を支持できないかもしれない。私たちの現在の臨床試験あるいは他の未来の臨床試験が成功するかどうかは確認できない。さらに、我々の目標適応のいずれの臨床試験で観察される任意のセキュリティ問題も、これらの適応および他の適応において規制部門の承認を得る見通しを制限する可能性があり、これは、私たちの業務、財務状況、および運営結果に実質的な悪影響を及ぼす可能性がある。
55
また,このような臨床試験が成功しても,FDAや外国規制機関が我々のように結果を解読する保証はなく,承認のための候補製品を提出する前に,より多くの試験が必要となる可能性がある。もし試験結果がFDAや外国の監督機関を満足させず、マーケティング申請を支援することができなければ、私たちは費用がかかるかもしれません
私たちが獲得できないかもしれない大量の資源は、私たちの候補製品が承認される可能性があることを支援するために追加的な実験を行うために使用されるかもしれない。たとえ私たちの候補製品が規制部門の承認を得ても、このような承認の条項は私たちの候補製品の範囲と用途を制限する可能性があり、これはその商業的潜在力を制限する可能性がある。
我々の業務と財務業績は新冠肺炎の疫病及びアメリカと世界の他の地域での後続変異の影響を受ける可能性がある。
新冠肺炎の大流行は、最近世界各地に蔓延した変種疫病を含み、全世界の商業活動に不利な影響を与えた。新冠肺炎疫病の影響は弱まっているにもかかわらず、このような事件はすでに金融市場の大幅な変動と不安定を招き、インフレ率の上昇を含む。新冠肺炎の疫病と政府の対応措置は全世界のサプライチェーンを混乱させ、多くの業界に不利な影響を与えた。大流行はインフレ率が引き続き急速に上昇し、世界経済のさらなる減速を招くなど、経済と市場状況に実質的な悪影響を及ぼす可能性がある。著者らは引き続き新冠肺炎の大流行の影響を密接に注目している。新冠肺炎疫病がどの程度著者らの運営或いは財務業績に影響するかはまだ確定していない。
新冠肺炎疫病と政府が取った対応措置も商業に直接と間接的な重大な影響を与えており、例えば労働力不足、サプライチェーン問題と中断、施設と生産の一時停止、インフレ率の上昇、及び医療サービスと用品のようないくつかの商品とサービスに対する需要が急増している。新冠肺炎疫病が著者らの業務と財務業績に与える影響程度はまだ確定していないが、持続的かつ長引く公衆衛生危機、例えば新冠肺炎疫病は、著者らの業務、財務状況と運営業績に実質的な不利な影響を与える可能性がある。もし世界の新冠肺炎の大流行と後に出現する可能性のあるいかなる新しい変種によっても、私たちは中断、特にサプライチェーンの中断を経験する可能性があり、これは私たちの業務と臨床試験に深刻な影響を与える可能性がある
56
私たちは、対象を新冠肺炎ウイルスから保護するのを助けるために、追加の臨床試験政策とプログラムを制定、実施、維持する必要があるかもしれないが、これは、私たちが期待している臨床研究と規制承認スケジュールを延期し、私たちの臨床研究コストを増加させる可能性がある。例えば、FDAは、2020年3月に、大流行期間中の臨床試験に関するガイドラインを発表し、その後更新され、大流行の影響を受ける臨床試験スポンサーが考慮すべきいくつかの事項が記載されている。2020年と2021年、アメリカ食品薬品監督管理局は薬品と生物製品製造業従業員の新冠肺炎感染、薬品製造と生物研究モニタリング施設の遠隔相互作用評価及び製造、サプライチェーン及び薬品と生物製品検査などの良好な製造実践注意事項への対応に関する指導意見を発表した。新冠肺炎変異体の伝播を考慮して、アメリカ食品と薬物管理局、アメリカ食品と薬物管理局及び類似の外国監督機関は他の指導と政策を発表する可能性があり、これらの指導と政策は私たちの業務と臨床開発スケジュールに実質的な影響を与える可能性がある。既存の政策と法規のさらなる変化は、私たちのコンプライアンスコストを増加させたり、私たちの臨床計画を延期したりするかもしれない。もし私たちの臨床研究が新冠肺炎関連要素によって遅延したり、私たちの臨床研究データが影響を受けた場合、私たちは追加の資源をかけてより多くの研究を行う必要があるかもしれないし、より多くの参加者を募集する必要があり、これは私たちの業務運営に不利な影響を与え、監督部門の承認を遅らせる可能性がある。
新冠肺炎疫病は依然として発展し続けており、著者らの業務、臨床前研究と臨床試験に影響を与える程度は未来の事態の発展に依存する可能性があり、これらの事態の発展は非常に高い不確定性を持っており、例えば疫病の持続時間、旅行制限、アメリカと他の国の社会疎遠、企業閉鎖或いは商業中断、アメリカと他の国がこの疾病をコントロールと治療するための行動の有効性、及び新しい変種が出現する可能性がある。もし新冠肺炎ウイルス及びその変種が未来に引き続き発展し、任意の特定の国或いは地区で巻き返していれば、私たちも上述のいずれかの妨害を受ける可能性がある。
新冠肺炎及びその後続変異体の伝播は、より多くの遠隔従業員を雇用すること、混合作業モードで運営することを含む業務慣行を修正した。さらに、政府当局や法規の要求や、私たちの従業員、顧客、パートナー、サプライヤーの利益に最も適合していると思う行動に行動するかもしれません。私たちの多くの職員たちは遠隔作業を続けている。私たちが重要な機能を実行する能力は損なわれるかもしれない。私たちは遠隔従業員の出張に関連した増加費用を発生するかもしれません。私たちはこの状況を監視し続けるつもりです。このような費用は私たちの運営結果に悪影響を及ぼす可能性があります。
また、私たちと業務往来のある第三者は、私たちの協力者、契約組織、第三者メーカー、サプライヤー、臨床試験場所、監督機関と他の私たちと業務往来のある第三者を含み、同様に新冠肺炎疫病と後続変異の影響に基づいて運営を調整し、それぞれの能力を評価した。これらの第三者が閉鎖や業務中断に遭遇した場合、現在計画されている方法およびスケジュールに従って業務を展開する能力は実質的かつ負の影響を受ける可能性がある。
例えば、私たちのいくつかの臨床試験サイトは臨床試験アクセス遅延を経験しており、しばらくの間、私たちが行っていた試験の参加者の一部は、時間通りに所定の用量または評価を受けていないことに気づいた。これらの事件は著者らの臨床試験データの完全性、信頼性或いはロバスト性に負の影響を与える可能性がある。我々と我々CROはFDAが発表した指導意見に基づいてこのような試験の操作を何らかの調整を行い,患者のモニタリングと安全を確保し,大流行中の試験完全性のリスクを最小限に抑え,将来的には新冠肺炎の灰再発によりさらなる調整が求められる可能性がある。
持続的な新冠肺炎の大流行あるいは他の疾病の爆発が私たちの運営と財務業績に不利な影響を与える場合、本“リスク要素”部分に記載されている多くの他のリスクを増加させる効果もある可能性がある。
技術や科学が急速に変化する環境では、私たちは激しい競争に直面しており、私たちの競争相手は、私たちの前に規制部門の承認を得たり、私たちよりも安全で、より先進的で、より効果的な療法を開発する可能性があり、これは、私たちが開発する可能性のある任意の候補製品の能力に悪影響を与え、最終的には私たちの財務状況を損なう可能性がある。
新薬製品の開発と商業化競争は激しい。そのほか、神経変性疾患領域の特徴は競争が激しく、日々激しくなり、知的財産権を強く強調することである。私たちは将来開発や商業化を求める任意の候補製品の面で主要な製薬会社、専門製薬会社、
57
世界中のバイオテクノロジー会社です潜在的な競争相手はまた学術機構、政府機関とその他の公共と個人研究組織を含み、これらの組織は研究を展開し、特許保護を求め、研究、開発、製造と商業化のための協力手配を確立する。
現在、多くの大型製薬と生物技術会社はFTD、アルツハイマー病、パーキンソン病とALS、及び癌の治療を含む神経変性疾患を治療する製品を開発している。多くの既存あるいは潜在的な競争相手は、単独またはその戦略パートナーであっても、研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング許可を得た製品の面で、私たちよりも多くの財務資源と専門知識を持っている。例えば、FDAは2023年1月、衛材会社(Eisai Co.,Eisai)と生物遺伝会社(Biogen)によって開発されたアルジマー病の治療のための抗アミロイドβ原線維抗体であるlecanemabを承認した。衛材はまた日本とヨーロッパでマーケティング許可申請を提出した。また、Biogenは2022年7月、スーパーオキシドジスムターゼ1(SOD 1)の変異に関連するALSを治療する薬剤であるFDAがtofersenのNDAを受けたことを発表した。2022年9月、Amylyx製薬会社はFDAが成人ALSの治療にRELYVRIO(フェニルブタン酸ナトリウムとタウリン酸ジオール)を承認することを許可したと発表した。他の競争相手は、同じ目標に適合したり、類似した行動メカニズムを介した候補製品を追いかけている。
さらに、製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場所と臨床試験の患者登録を確立する方面で私たちと競争し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。さらに、現在承認されている製品は、神経変性疾患の治療への適応が発見される可能性があり、これは、このような製品を我々の任意の候補製品と比較して顕著な規制と市場タイミング優勢を有する可能性がある。私たちの競争相手は私たちよりも早くFDA、EMA、または他の規制機関のその製品の承認を得ることができ、私たちの候補製品に対する兆候によってFDAから孤立した薬物独占経営権を獲得する可能性があり、これにより、私たちの競争相手は私たちが市場に入る前に強力な市場地位を確立することができるかもしれない。また、私たちの競争相手が開発した製品や技術は、私たちの潜在的な候補製品を経済的あるいは時代遅れにする可能性があり、私たちは私たちが開発する可能性のある任意の競争相手の候補製品をマーケティングすることができないかもしれません。
さらに、私たちは、私たちの競争相手の製品に関連する特許の範囲、所有権、有効性、および/または実行可能な訴訟または他の訴訟に直面する可能性があり、私たちの競争相手は、私たちの製品の侵害、流用、または他の方法で彼らの知的財産権を侵害すると主張するかもしれない。私たちの競争相手の製品供給は、私たちが開発し商業化する可能性のある任意の製品の需要と私たちが受け取ることができる価格を制限するかもしれません。
私たちの候補製品は複雑に作られており、生産中に困難に直面する可能性がある。もし私たちまたは私たちの任意の第三者製造業者がそのような困難に遭遇したり、厳格に実行された規制基準に達しなかったりすれば、私たちは臨床試験や患者に候補製品や製品を提供する能力が(承認された場合)延期または停止される可能性があり、または商業的に実行可能なコスト構造を維持できないかもしれない。
私たちの候補製品を製造する過程は複雑で、高価で、監督管理が厳しく、そして多重リスクの影響を受ける。また,候補製品の開発が臨床前研究から後期臨床試験の承認と商業化に伴い,製造方法など開発計画の様々な面で,プロセスや結果の最適化に努める過程で変更されることが一般的である。これらの変化はこれらの予想される目標を達成できない可能性があるが、これらの変化のいずれも私たちの候補製品の表現を異なり、計画中の臨床試験あるいは他の未来の臨床試験の結果に影響を与える可能性がある。
私たちの候補製品の臨床試験、あるいは商業製品を提供するためには、承認されれば、これらの製品を大量に生産する必要があります。私たちのCDMOは、私たちの任意の候補製品の製造能力をタイムリーにまたは費用効果的に向上させることに成功できないかもしれない、または全くできないかもしれない。また,拡張活動中に品質の問題が生じる可能性がある.もし私たちのCDMOが拡張できなければ
58
私たちの候補製品を十分な品質と数量で生産すれば、その候補製品の開発、テスト、および臨床試験は延期または不可能になる可能性があり、いかなる最終製品の規制承認や商業発表が延期または獲得できない可能性があり、これは私たちの業務を深刻に損なう可能性がある。もし私たちが未来に内部製造能力を構築することを決定すれば、同じリスクは私たちの内部製造施設にも適用されるだろう。また、内部製造能力の建設は、複雑なプロジェクトを計画、設計、実行することができず、タイムリーかつ費用効果のある方法で製造施設を構築することができないため、重大なリスクをもたらす。
さらに、私たちが開発する可能性のあるどの製品の製造過程もFDA、EMA、および外国規制機関の承認手続きと持続的な監督を受けており、現在の良好な製造規範(CGMP)を継続的に遵守することを含む、すべての適用可能なFDA、EMA、および外国規制機関の要求に適合するメーカーと契約を締結する必要がある。FDAが医薬品および生物製品生産従業員における新冠肺炎感染に対応する良好な製造規範に関するFDAの最近の指導意見、および将来の指導および法規を含む、FDA、EMAまたは他の規制機関が許容可能な規格に適合した製品を確実に生産できない場合、私たちは、このような製品を商業化するために必要な承認を得ることができないかもしれない。私たちの任意の候補製品が規制機関の承認を得ても、私たちまたは私たちのCDMOがFDA、EMA、または他の規制機関が許容できる規格で承認された製品を生産することができ、それによって製品が発売される可能性のある要求を満たすのに十分な数の製品を生産することができ、あるいは将来の潜在的な需要を満たすことができる保証はない。これらの挑戦のいずれも臨床試験の完成を遅らせる可能性があり、移行臨床試験或いは1つ以上の臨床試験を繰り返し、臨床試験コストを増加させ、私たちの候補製品の承認を延期し、商業化努力を損害し、私たちの商品コストを増加させ、そして私たちの業務、財務状況、運営結果と成長の将来性に不利な影響を与える必要がある。
将来的には、我々が開発する可能性のある任意の候補製品を販売およびマーケティングするために、販売およびマーケティング能力を確立することができない場合、これらの候補製品が承認された場合、商業化に成功できないかもしれない。
私たちは医薬製品を販売したり、マーケティングしたり、流通した経験もありません。販売およびマーケティング責任を保留する任意の承認された製品をビジネスに成功させるためには、販売およびマーケティング組織を構築するか、またはこれらの機能を第三者にアウトソーシングしなければならない。将来、私たちのいくつかの候補製品が承認されれば、集中的な販売、マーケティング、およびビジネス支援インフラを構築して、私たちのいくつかの候補製品を販売したり、私たちの協力者と一緒にビジネス活動に参加することを選択するかもしれません。
私たち自身のビジネス能力の確立と第三者とのこれらのサービス提供の合意はリスクに関連している。例えば、販売員の募集と訓練や専門員の精算は高価で時間がかかり、どんな製品の発表も延期される可能性がある。販売チームを募集し、マーケティングや他の商業化能力を確立する候補製品の商業発表が何らかの理由で延期または発生していない場合には、これらの商業化費用を早期または不必要に発生させる。これは費用がかかるかもしれないし、もし私たちが私たちの商業化者を維持したり再配置できなければ、私たちの投資は損失するだろう。
私たち自身が承認された製品を商業化するのを阻害するかもしれません
59
もし私たちが第三者と合意して、販売、マーケティング、商業支援、流通サービスを行えば、私たちの製品収入または製品収入の収益力は、私たちが自分たちが開発した任意の製品をマーケティングし、販売することを下回るかもしれません。また、私たちは第三者と私たちの候補製品を商業化する計画を成功させることができないかもしれないし、私たちに有利な条項でそうすることができないかもしれない。私たちはこれらの第三者に対して支配権がほとんどないかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが商業化能力を確立することに成功しなければ、私たち自身も第三者と協力しても、私たちの候補製品が承認されれば、私たちはそれを商業化することに成功しないだろう。
我々が開発したすべての候補製品が市場承認を得ても,医師,患者,医療支払者,医学界の他の人がビジネス成功に必要な市場受容度を達成できない可能性がある。
私たちのすべての製品の商業成功はそれが医者、患者、第三者支払人、医学界の他の人に受け入れられる程度に依存するだろう。たとえ私たちが開発可能なすべての候補製品が市場の承認を得ても、それらは医師、患者、医療保険支払人、医学界の他の人の十分な市場受容度を得ることができないかもしれない。もし私たちが開発したすべての候補製品が商業販売に使用されることが許可された場合、市場のその製品に対する受け入れ度は複数の要素に依存する
もし私たちが開発したすべての候補製品が十分な受容度に達していなければ、私たちは著しい製品収入が生じないかもしれません。私たちは利益を上げることができないかもしれません。
60
私たちが商業化しているどの製品も、不利な価格設定法規、第三者精算やり方、または医療改革措置の制約を受ける可能性があり、これは私たちの業務を損なうだろう。
新薬の発売承認、定価と精算を管理する規定は国によって異なる。米国では、最近公布された、または将来発表される可能性のある立法は、承認要求を著しく変更する可能性があり、これは追加のコストに関連し、承認の遅延を招く可能性がある。一部の国は薬品の販売価格が承認されてから発売されることを要求している。多くの国で、定価審査期間はマーケティングまたは製品許可を承認した後から始まる。一部の外国市場では、処方薬の定価は初歩的な承認を得た後も、政府の持続的なコントロールを受けている。したがって、特定の国/地域での製品のマーケティング承認を得ることができるかもしれませんが、その後、価格法規の制約を受けて、これらの法規は私たちの製品の商業発表を延期し、長い時間遅延し、その国/地域で製品を販売することによって生じる収入に悪影響を与える可能性があります。不利な価格設定制限は、私たちが開発可能な任意の候補製品が市場承認を得ても、1つ以上の候補製品への投資を回収する能力を阻害する可能性がある。
私たちが開発する可能性のあるいかなる製品を商業化することに成功する能力があるかどうかは、政府衛生行政部門、個人健康保険会社、その他の組織によるこれらの製品と関連治療の清算程度にもある程度依存するだろう。政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、どのような薬剤を支払うかを決定し、精算レベルを確立する。アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。政府当局と第三者支払者は,特定の薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。政府当局は現在、ある患者群に対して強制的な割引を実施しており、例えば医療保険、医療補助と退役軍人事務部病院であり、随時このような割引の増加を求める可能性がある。もし承認されれば、未来の規制は私たちの製品の価格に否定的な影響を及ぼすかもしれない。ますます多くの第三者支払人は製薬会社に価格に基づいて所定の割引を提供し、医療製品の定価に挑戦することを要求している。私たちは私たちが商業化されたどの候補製品も精算できることを確実にすることはできない。もし精算できるなら、精算のレベルもそうだ。精算は私たちが市場で承認された任意の候補製品の需要や価格に影響を及ぼすかもしれない。精算を得るためには,医師は標準看護薬よりも患者が我々の製品を使用することが,価格の低い標準看護薬の後発薬を含むより良い治療結果を有することを証明する必要があるかもしれない。精算ができない場合や数量限定で精算します, 私たちは市場で承認された候補製品を商業化することに成功できないかもしれない。米国では、第三者支払者の間に統一された製品保証と精算政策がなく、製品の保証と精算レベルは支払人によって異なる。したがって、保証範囲の決定プロセスは、通常、時間がかかり、高価なプロセスであり、保証範囲と十分な補償が一致するか、または最初に得られることを保証することなく、各支払人にそれぞれ私たちの製品を使用する科学的および臨床的支援を提供する必要があるかもしれない。
新たに承認された薬物の精算には大きな遅延がある可能性があり,カバー範囲はFDA,EMAあるいは他の類似した外国規制機関がこの薬物を承認する目的よりも限られている可能性がある。さらに、精算を受ける資格があるということは、いかなる薬物もすべての場合に支払われることを意味するわけではなく、または支払いの費用率は、研究、開発、製造、販売、流通を含む私たちのコストをカバーする。もし適用されれば、新薬の一時精算レベルも私たちのコストを支払うのに十分ではないかもしれないし、永久的にならないかもしれない。販売率は薬物の使用や臨床環境によって異なる可能性があり,すでに低コスト薬物のために設定された精算レベルに基づいている可能性があり,他のサービスの既存支払いにも組み込まれている可能性がある。医薬品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、および将来、米国価格よりも低い価格で販売される可能性のある国から医薬品を輸入することを制限する法律の緩和によって低下する可能性がある。第三者支払者が自分の精算政策を設定する際には,通常連邦医療保険カバー政策と支払制限に依存する。私たちは政府援助や個人支払者から、私たちが開発する可能性のある任意の承認された製品の保証範囲と利益の支払率を迅速に得ることができません。これは、私たちの経営業績、候補製品を商業化するために必要な資金を調達する能力、および私たちの全体的な財務状況に実質的な悪影響を及ぼすかもしれません。
現在および将来のCMSは、私たちのアルツハイマー病を治療する候補薬物を含む、私たちの候補製品を含む薬物カテゴリの制限を含み、承認されれば、候補製品を商業化し、収入を創出し、利益を達成する能力に実質的な悪影響を及ぼす可能性がある。将来のCMSカバー決定と政策がどのように我々の業務に影響を与えるかはまだ不明である。
61
私たちは承認された候補製品が予想よりも早く生物学的に似た競争に直面する可能性があることを求めるつもりだ。
競争相手の前に規制部門の承認を得て候補製品を商業化することに成功しても、私たちの候補製品は生物類似製品からの競争に直面する可能性がある。米国では,我々の候補製品はバイオ製品としてFDAによって規制されており,BLA経路に基づいてこれらの候補製品の承認を求める予定である。2009年の“生物製品価格競争と革新法”(BPCIA)は、生物類似および交換可能な生物製品を承認するために簡略化された道を開いた。簡略化された規制経路は、生物類似体と既存ブランド製品との類似性に基づいて、それを“交換可能”な生物類似体として指定することを含む、FDAのための生物類似生物製品を審査および承認する法的権威を確立する。BPCIAによると、バイオ類似製品の申請は、最初のブランド製品がBLAによって承認されて12年後にのみFDAの承認を得ることができる。この法律は複雑であり、FDAはまだ説明して施行している。したがって、その最終的な影響、実施、そして意味には不確実性がある。FDAがいつこれらのBPCIAを実施するためのプロセスを完全に採用する可能性があるかは不明であるが、どのようなプロセスも、我々の候補製品の将来のビジネス見通しに重大な悪影響を及ぼす可能性がある。
我々のBLAによりバイオ製品として承認された候補製品はいずれも12年の専門期間を得る資格があるべきであると考えられる。しかしながら、国会の行動や他の理由により、この排他性は短縮される可能性があり、またはFDAは、私たちの候補製品を競合製品の参考製品とみなさず、予想よりも早く競争機会を創出する可能性がある。さらに、承認されると、生物類似製品が我々のいずれかの参考製品をどの程度置換するかは、従来の非生物学的製品の非生物模倣薬代替に類似しており、これはまだ発展中のいくつかの市場および規制要因に依存するかどうかは不明である。また,競争相手は生物類似の承認手順を放棄し,自分の臨床前研究と臨床試験を完了した後に完全なBLAを提出することを決定することができる。この場合、BPCIAによっては、その製品が承認された直後に競合他社がその製品を販売することを阻止する資格がある可能性のある排他性はない。
ヨーロッパでは、欧州委員会は、過去数年間に発表された生物類似承認に関する一般的かつ特定の製品カテゴリに関するガイドラインに基づいて、いくつかの生物類似製品のマーケティング許可を承認した。ヨーロッパでは、競争相手は革新生物製品の承認を支持するデータを参考にすることができるが、革新製品が承認されてから10年後に市場に出すことができる。この10年前の8年前に、マーケティング許可者が承認され、1つまたは複数の新しい治療適応が承認され、既存の療法と比較して有意な臨床的利益をもたらす場合、この10年間の市場排出期間は11年に延長される。また、会社は他の国で生物類似製品を開発する可能性があり、承認されれば、これらの製品は私たちの製品と競争する可能性がある。
もし競争相手が私たちの候補製品を参考にする生物模倣薬のマーケティング許可を得ることができれば、もし承認されれば、このような製品はこのような生物模倣薬の競争を受ける可能性があり、それに伴う競争圧力と潜在的な不利な結果である。これらの競争力のある製品は、私たちの候補製品が承認される可能性のあるすべての指標で直ちに私たちと競争するかもしれない。
私たちの法的手続きやクレームに関連したり、どのようなものも高価で時間のかかる弁護である可能性があり、結果がどうであれ、私たちの名声を損なうかもしれない。
私たちは将来、知的財産権、データプライバシー、製品責任、雇用、集団訴訟または派生製品、告発者および他の訴訟クレーム、ならびに政府および他の規制調査および訴訟手続きを含む法的訴訟および正常な業務過程で生じるクレームの影響を受ける可能性がある。このようなことは時間をかけて、経営陣の注意力や資源を分散させ、巨額の費用や責任を発生させたり、私たちの業務やり方を変えることを要求したりする可能性があります。また、訴訟費用やこの費用の時間を期間ごとに見積もることは困難であり、変化する可能性があり、我々の財務状況や運営結果に悪影響を及ぼす可能性がある。訴訟の潜在的なリスク、費用、不確実性のため、私たちは正当なクレームや抗弁があっても、時々和解合意に同意することで紛争を解決するかもしれない。上記のいずれも、我々の業務、財務状況、および経営結果に悪影響を及ぼす可能性がある。
62
私たちに製品責任訴訟を提起すれば、私たちは多くの責任を負い、候補製品の商業化を制限することを要求されるかもしれません。
私たちの候補製品は臨床テストを行うため、私たちは固有の製品責任のリスクに直面して、私たちはいかなる製品を商業化する時、私たちはもっと大きなリスクに直面します。例えば、私たちの候補製品が臨床試験、製造、マーケティング、または販売中に傷害をもたらしたり、不適切なことが発見されたりした場合、私たちは起訴されるかもしれない。このような製品責任クレームは、製造欠陥、設計欠陥、製品固有の危険、不注意、厳格な責任、または保証違反の告発を含む可能性があります。州消費者保護法によると、クレームも主張することができる。もし私たちが製品責任クレームで自分を弁護することに成功できなければ、私たちは大きな責任を招くかもしれないし、あるいは私たちの候補製品のテストと商業化を制限されることが要求されるかもしれない。成功的な防御であっても、多くの財政的で管理的な資源が必要だ。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を得ることができず、これは、私たち単独またはパートナーと開発した製品の商業化を阻止または阻害する可能性がある。私たちの保険証書には様々な例外があるかもしれません。製品責任クレームの影響を受けるかもしれませんが、私たちは保険範囲を持っていません。私たちは私たちの保険範囲の制限を超えたり、私たちの保険カバー範囲内でない裁判所の裁決または和解合意で達成された任意の金額を支払う必要があるかもしれません。私たちはこれらの金額を支払うために十分な資本を持っていないか、または得ることができません。私たちが未来の会社のパートナーと合意しても、私たちは損害賠償を受ける権利があります。何かクレームがあれば、この賠償は利用できないか十分かもしれません。
規制承認およびその他の法律コンプライアンス事項に関するリスク
FDA、EMAと類似の外国監督管理機関の監督管理審査過程は長く、時間がかかり、しかも本質的に予測不可能である。もし私たちが最終的に規制部門の候補製品の承認を得ることができなければ、私たちは製品収入を生むことができなくなり、私たちの業務は実質的に損害を受けるだろう。
FDA、EMA、および類似の外国規制機関の承認を得るのに要する時間は予測不可能であり、通常、臨床試験開始後数年後に必要であり、関連する候補製品のタイプ、複雑性、および新規性を含む多くの要素に依存する。さらに、承認政策、法規、または承認を得るために必要な臨床データのタイプおよび数量は、候補製品の臨床開発過程で変化する可能性があり、司法管轄区域によって異なる可能性があり、これは、承認の遅延または承認申請の不承認の決定を招く可能性がある。監督管理機関は審査過程中にかなりの自由裁量権を持っており、いかなる申請を受け入れることを拒否することができ、著者らのデータが承認を得るのに十分ではなく、追加の臨床前、臨床或いはその他の研究を行う必要があることを決定することもできる。私たちはまだどの候補製品にも提出したり、規制部門の承認を得たりしていません。私たちの既存の製品候補製品や私たちが将来開発を求める可能性のあるどの製品候補製品も、規制部門の承認を得られないかもしれません。
また、私たちの候補製品の開発および/または規制承認は、私たちがコントロールできない理由で延期される可能性があります。例えばアメリカ連邦政府は未来に停滞しているかもしれません
63
または予算の自動減額は、FDAの予算、従業員、および運営を大幅に減少させる可能性があり、これは、応答時間が遅く、審査期間がより長くなる可能性があり、候補製品開発を推進したり、規制部門の候補製品の承認を得る能力に影響を与える可能性がある。FDAおよび他の規制機関が、規制承認前に私たちの規制申請や会議および/または指導要請を審査し、規制承認前に製造施設を検査する上で何らかの遅延や資源が限られている場合、新冠肺炎の大流行の影響やその他の理由で、臨床研究および/または規制承認を求める予想スケジュールに重大な遅延が生じる可能性があり、これは私たちの業務に悪影響を及ぼす可能性がある。
私たちの候補製品の申請は、多くの理由で、初期または後続の指示で規制部門の承認を得ることができないかもしれませんが、以下の理由に限定されません
この長い承認過程と、臨床試験結果の予測不可能性は、規制部門の承認を得ることができず、私たちのすべての候補製品を市場に出すことができず、私たちの業務、運営結果、成長の将来性を深刻に損なう可能性がある。
著者らの候補製品は不良副作用或いはその他の特性を招く可能性があり、その臨床開発を阻止し、その監督管理の承認を阻止し、その商業潜在力を制限し、或いは深刻な負の結果を招く可能性がある。
私たちの候補製品によって引き起こされる有害事象や他の副作用は、私たちまたは規制機関の臨床試験の中断、延期、または停止を招く可能性があり、より厳しいラベルやFDA、EMA、または他の同様の外国規制機関の規制承認遅延または拒否を引き起こす可能性がある。
薬物に関連する副作用は、患者の募集、入選患者が研究を完成する能力、および/または潜在的な製品責任クレームを招く可能性がある。私たちのいくつかの開発と商業化協定によると、私たちは製品責任保険の維持を要求された。私たちは責任による損失から私たちを守るために、合理的な費用や十分な金額で保険範囲を維持することができないかもしれない。成功した製品責任クレームあるいは一連の私たちに対するクレームは私たちの運営、業務と
64
名声。また、是非曲直または最終結果にかかわらず、製品責任クレームは、私たちの商業的名声の損傷、臨床試験参加者の撤退、関連訴訟によるコスト、管理層の私たちの主要業務に対する注意分散、監督管理機関の調査、患者や他のクレーム者が巨額のお金の奨励を得ることができ、私たちの候補製品を商業化することができず、商業販売のために承認された場合、私たちの候補製品に対する需要は減少する可能性がある。
われわれが行っているADのInvoke−2段階2臨床試験でARIAが観察された。ARIAのMRI表現は血管源性水腫或いは鉄含有ヘモフラビン沈着を示した。アルツハイマー病のある治療薬、即ち抗β-アミロイド抗体の使用は、アルツハイマー病患者のARIA発生率が増加することを示している。われわれのInvoke−2段階2臨床試験では,ARIA症例の多くは無症状と非重篤であった。しかし,先に報告したように,少数のARIAに関連する重篤な副作用はAPOE e 4/e 4遺伝子型の患者でほぼ完全に発生している。ARIAに関するリスクを低減するために,われわれのInvoke−2段階2臨床試験では,APOE e 4/e 4参加者の用量と登録を自発的に中止した。これらの変化の後,少量のARIAに関連する重篤な有害事象はAPOE e 4対立遺伝子非ホモ接合体の患者で発生した。われわれは早期のMRIモニタリングを継続し,最近発表されたARIAモニタリングと管理ガイドラインと一致している。我々はIDMCの指導の下でこの研究を行い,IDMCは非盲目的データの審査と実験提案を許可された。
さらに、もし私たちの1つまたは複数の候補製品が発売承認され、私たちまたは他の人が後にこのような製品によって引き起こされる不良副作用または有害事象を発見した場合、多くの潜在的な重大な負の結果をもたらす可能性があるが、これらに限定されない
これらの事件のいずれも、特定の候補製品に対する市場の受け入れ度を達成または維持することを阻止することができ、承認されれば、私たちの業務、財務状況、運営結果、および成長の見通しを深刻に損なう可能性がある。
私たちは現在、将来アメリカ国外で私たちの候補製品の臨床試験を継続している可能性があり、FDA、EMA、適用された外国の規制機関はこのような試験のデータを受け入れないかもしれない。
私たちは現在、ヨーロッパ、ラテンアメリカ、アジア、またはオーストラリアを含む米国以外の地域で1つ以上の臨床試験を行い、将来的に選択し続ける可能性がある。FDA、EMAあるいは適用される外国監督管理機関がアメリカ或いは他の司法管轄区域以外で行われた臨床試験を受ける研究データはいくつかの条件によって制限される可能性がある。外国の臨床試験のデータがアメリカでの発売承認の基礎としようとする場合、FDAは通常、(I)データがアメリカの人口とアメリカの医療実践に適用されない限り、外国のデータのみに基づいて申請を許可しない;および(Ii)試験は公認能力を有する臨床研究者によって行われ、CGCP規定に符合する。そのほか、十分な患者群と統計能力を含むFDAの臨床試験要求を満たさなければならない。多くの外国の規制機関もまた似たような承認要求を持っている。また、このような外国裁判は、裁判を行う外国司法管轄区域に適用される現地法によって管轄される。FDA、EMA、または任意の適用可能な外国規制機関が、米国または適用司法管轄区域以外で行われた試験からのデータを受け入れることは保証されない
65
実験設計や実験多様性に関する要求が含まれている.FDA、EMA、または任意の適用可能な外国規制機関がこのようなデータを受け入れない場合、追加の試験が必要になり、これは高価で時間がかかり、私たちの業務計画の様々な側面を遅延させ、適用司法管轄地域で私たちの候補製品が商業化承認や許可を得られない可能性があります。著者らは患者のリスク特徴を的確な干与と一致させるために遺伝子スクリーニングとバイオマーカーへの依存を行い、最終的にはそれを使用するために追加の監督管理要求を満たす必要がある可能性がある。
一つの管轄区域で私たちの候補製品に対する規制承認を獲得し、維持することは、私たちが他の管轄区域で私たちの候補製品の規制承認を得ることに成功するという意味ではない。
1つの管轄区域で私たちの候補製品に対する監督管理承認を獲得し、維持することは、他の任意の管轄区で規制承認を得ることができるか、または維持できる保証はありませんが、1つの管轄区で規制承認を得ることができなかったり、遅延したりすることは、他の管轄区の監督管理承認の流れに悪影響を及ぼす可能性があります。例えば、FDAまたはEMAが候補製品の発売を承認したとしても、外国司法管轄区の比較可能な規制機関は、候補製品のこれらの国での製造、マーケティング、および普及を承認しなければならない。承認手続きは司法管轄区域によって異なり、1つの管轄区で行われる臨床試験は他の管轄区の監督機関に受け入れられない可能性があるため、米国とは異なる要求と行政審査期限に関連する可能性がある。米国以外の多くの管轄区では、候補製品は先に精算許可を得てから、その管轄区で販売を許可することができる。場合によっては、私たちが私たちの製品のために受け取る価格もまた規制部門の承認を受ける必要がある。
外国の監督管理の承認を得て、外国の規制要求を遵守することは、私たちに重大な遅延、困難、コストをもたらす可能性があり、私たちの製品がある国/地域で発売されることを延期または阻止する可能性がある。もし私たちまたは私たちと協力している任意のパートナーが国際市場の規制要求を遵守できなかったり、適用されたマーケティング承認を得られなかったりすれば、私たちの目標市場は減少し、候補製品の市場潜在力を十分に発揮する能力は損なわれるだろう。
私たちが規制機関の候補製品の承認を得ても、私たちの製品は広範な発売後の要求と規制審査を受けるだろう。
もし私たちの任意の候補製品が承認されたら、それらは製造、ラベル、包装、貯蔵、広告、販売促進、サンプリング、記録保存、上場後の研究、および安全性、有効性および他の上場後の情報の提出に関する持続的な法規要求を受け、アメリカの連邦と州要求、比較可能な外国監督機関の要求を含む。
メーカーとメーカーの工場は、品質管理および製造プロセスがcGMP法規に適合することを確保することを含む、FDA、EMAおよび同様の外国規制機関によって適用される広範な要求を遵守することが要求されている。したがって、私たちと私たちの契約製造業者は、cGMPの遵守状況および任意のNDA、BLAまたはマーケティング許可申請(MAA)で行われた約束の遵守状況を評価するために、持続的な審査および検査を受けるだろう。したがって、私たちと他の私たちと協力している人たちは、製造、生産、品質管理を含むすべての規制コンプライアンスの分野で時間、お金、エネルギーをかけ続けなければならない。
私たちの候補製品のために得られた任意の規制承認は、マーケティングおよび普及のために製品が使用される可能性のある承認された用途を示す制限または承認条件(リスク評価および緩和戦略を実施するための任意の要求を含む)、またはコストが高い可能性のある上場後テスト要件を含む。私たちはFDA、EMA、そして似たような外国の規制機関にいくつかの副作用と生産問題を報告することを要求されるだろう。薬物安全問題を解決するいかなる新しい立法も、製品開発や商業化の遅延を招き、あるいはコンプライアンスを確保するコストを増加させる可能性がある。FDA及び司法省を含む他の機関は、製品の承認後のマーケティング及び販売促進活動を密接に監督し、承認のみの適応を確保し、承認されたラベルの規定に基づいて製品の製造、販売及び流通を行う。私たちは私たちの製品の広告と販売促進に関する要求を守らなければならない。処方薬に関する販売促進情報は様々な法律や法規によって制限されており,製品が承認されたラベル上の情報と一致しなければならない。したがって、私たちは私たちの製品を未承認の適応や用途に使用しないかもしれない。承認された秘密保持協定、BLAまたはMAAの所有者は新しいものを提出しなければならない
66
申請を追加し、承認された製品、製品ラベル、または製造プロセスのいくつかの変更の承認を受ける。著者らはまた、一般或いは特定の患者亜群におけるわが製品の安全性と有効性を検証するために、発売後の臨床試験を要求されることができる。もし最初の上場承認が承認ルートを加速することによって得られた場合、私たちは成功した発売後の臨床試験を行い、私たちの製品の臨床治療効果を確認する必要があるかもしれない。成功しなかった上場後の研究やこのような研究が完成できなかったことは、上場承認の撤回につながる可能性がある。
規制当局が、予期せぬ深刻度または頻度の不良事象など、製品に以前に未知の問題があることを発見した場合、または製品の製造施設に問題がある場合、または製品の販売促進、マーケティング、またはラベルに同意しない場合、規制機関は、製品の市場からの撤退を要求することを含む、製品または私たちに制限を加えることができる。もし私たちが適用される規制要求を遵守できなければ、規制機関や法執行当局は他の措置を取るかもしれない
政府は違法の疑いのあるいかなる調査にも対応するために多くの時間と資源を必要とする可能性があり、否定的な宣伝が生じる可能性がある。現行の法規の要求を守らないいかなる行為も、私たちの製品の商業化と創造能力に重大な悪影響を及ぼす可能性がある。規制制裁を実施したり、規制承認を撤回したりすれば、わが社の価値と私たちの経営業績は悪影響を受けるだろう。
私たちはFDAからFTDを治療するラトジンモノクロナル抗体およびAL 101の孤児薬物指定を取得し、私たちの他のいくつかの候補製品のために孤児薬物指定を求める可能性があるが、市場排他性を含むこのような指定や孤児薬物状態の維持に関連する利点を得ることができない可能性があり、これは私たちの収入を減少させる可能性がある(あれば)。
孤児医薬品法によれば、FDAは、米国では患者数が20万人未満、または米国では患者数が20万人を超える疾患または疾患であると定義されているまれな疾患または疾患を治療するための薬剤または生物を孤児として指定することができ、合理的な期待がなければ、米国で薬剤または生物学的薬剤を開発および提供するコストは、薬剤または生物学的薬剤または生物の米国販売から回収される。NDAやBLAを提出する前に,指定孤児薬を申請しなければならない。米国では,臨床試験費用に贈与資金を提供する機会,税収割引,ユーザ費用減免など,孤児薬を指定して一方が財政的インセンティブを得る権利がある。FDAが孤児薬物指定を承認した後、FDAは、この薬剤の模倣薬識別情報およびその潜在的孤児用途を開示する。孤児薬物の指定は、監督審査と承認過程においていかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。FTDを治療するラトジンモノクロナル抗体やAL 101の孤児薬物指定はFDAから得られているが,孤児薬物状態に関する利点は得られない可能性がある。さらに、私たちは将来的に私たちの他の候補製品のために孤児薬物名を求める予定ですが、他の候補製品のために孤児薬物名を得ることができないかもしれません。
孤児薬物名を有する製品が、その後、そのような名称を有する疾患の特定の活性成分に対するFDAの最初の承認を得た場合、この製品は、孤児薬物排他性を得る権利がある。これは、FDAが7年以内に他のNDAまたはBLA申請を承認しない可能性があり、同じ適応の下で同じ薬物または生物学的製剤を販売している可能性があり、限られた場合、例えば孤児の独占的地位を有する製品に対する臨床的利点を示さない限り、FDAが孤児薬物の指定を撤回した場合、またはFDAが孤児独自の薬物の保持者が指定された薬物を有する疾患または状態を有する患者の需要を満たすのに十分な数の孤児製品を確保していないことを意味する。アメリカの食品医薬品局が
67
ラトジンモノクロナル抗体とAL 101はFTDを治療する孤児薬物として承認されたが、FDAは依然として他の異なる有効成分を含む薬物をFTDの治療に許可することができる。また、孤児薬物の独占性は、孤児の独占期間が満了する前に、FDAが同一の薬物製品の別の異なる適応のマーケティング申請を承認することを阻止しない。
上述したように、2021年の訴訟では、ある裁判所は、孤児薬物排他性は、指定された疾患または条件全体のすべての用途または適応には適用されず、条件を満たす疾患内の承認用途または適応にのみ適用されるFDAの長期的な立場に同意しない。この控訴裁判所の判決は孤児薬物の排他的な応用に不確実性をもたらした。FDAは2023年1月、適用された裁判所の判断を遵守しているが、FDAは“連邦登録簿”に通知を発表した。FDAは、孤児薬物の排他的な範囲を、薬物が承認された用途または適応とバンドルし続けることを意図しており、これは、他のスポンサーが、未承認の同一の孤児が疾患または状態を指定した場合に、薬物の新しい用途または適応の承認を得ることを可能にする。将来の訴訟、立法、機関決定と行政行動がどのように孤児薬物の専有範囲に影響するかはまだ不明である。
Latozinemabに対するFast Track指定と,顆粒蛋白遺伝子特定の遺伝子変異を有するFTD患者の治療のためのAL 101はFDAから得られているが,Fast Track指定に関連する利点を得ることができない可能性がある。
迅速チャネル指定は,重篤な疾患の治療と満たされていない医療ニーズを満たす療法の開発と審査の加速を促進することを目的としている。高速チャネル指定を有するプロジェクトは、FDAとの早期および頻繁なコミュニケーション、潜在的な優先審査、および規制審査のためにローリング申請を提出する能力から利益を得ることができる。高速チャネル指定は製品にも,検討中の特定の適応にも適用可能である。
2019年12月、FDAはlatozinemabのFast Track称号を承認し、2020年1月、FDAはAL 101のFast Track称号を許可し、各薬物は顆粒蛋白遺伝子の特定の遺伝子突然変異を持つFTD患者の治療に用いられた。われわれの臨床開発計画がFast Track指定基準を満たし続けることができなければ,あるいはわれわれの臨床試験が意外な有害事象や臨床供給問題で一時停止または終了したり,臨床放置されたりすると,Fast Track計画に関連するすべての利点を実現できない可能性がある。また,高速チャネルを指定すると承認基準は変更されない.高速チャネル指定だけではFDA優先審査プログラムに適合する資格は保証されない。
医療コストを低減するための医療立法措置は,我々の業務や運営結果に実質的な悪影響を及ぼす可能性がある。
第三者決済者は,国内でも海外でも,政府のものでも商業的でも,ますます複雑な方法を開発して医療コストを抑えている。米国や一部の外国司法管轄地域では、医療システムに多くの立法や規制の変化が発生しており、私たちの販売製品の収益性に影響を与える可能性がある。
特に,2010年に患者保護·平価医療法案(ACA)が公布され,その中で他を除いて,生物製品を低コストの生体模倣薬の潜在的競争を受けさせ,吸入,注入,点滴,インプラントあるいは注射の薬物計算メーカーが医療補助薬物還付計画の下で不足している税金還付を解決し,医療補助薬物還付計画の下で大多数のメーカーが不足している最低医療補助税還付を増加させ,医療補助薬物還付計画を医療補助管理の医療機関に登録されている個人の処方に拡大し,メーカーがあるブランドの処方薬に新たな年費と税を徴収する方法を解決した。連邦政府の比較有効性研究のプロジェクトを増加させるためのインセンティブを提供した。2021年6月、米最高裁はテキサス州と他の挑戦者がACAの法的地位に挑戦していないと判断し、この事件を却下したが、ACAの合憲性を具体的に裁くことはなかった。したがって,ACAは現在の形で依然として有効である.最高裁のこの裁決、将来の訴訟あるいはバイデン政府が公布した医療措置が私たちの業務、財務状況と運営結果にどのように影響するかはまだ不明である。いかなる新しい立法や医療法規の遵守にも多大な時間とコストがかかり、我々の業務に実質的な悪影響を及ぼす可能性がある。
2021年1月1日に施行される“2021年米国救援計画法案”によると、メーカーが州医療補助計画に支払う医療補助薬品還付計画還付の法定上限が撤廃される。この上限を廃止して製薬業者に販売承認の支払いを要求する可能性があります
68
製品、これは私たちの業務に実質的な影響を及ぼすかもしれない。2022年8月、国会は“2022年インフレ低減法案”を可決し、その中には製薬業と連邦医療保険受益者に重大な影響を与える処方薬条項を含み、連邦政府がある高価な単一源連邦医療保険薬物の最高公平価格について交渉することを許可し、薬品価格交渉要求を守らないメーカーに罰と消費税を加え、すべての連邦医療保険B部分とD部分の薬物がインフレリベートを獲得することを要求し、もしその薬品価格の増加がインフレより速い場合、限られた例外、及び連邦医療保険D部分を再設計して受益者の自己払い処方薬コストを下げるなどの変化を含む。コスト抑制措置や他の医療改革を実施することは、収入を創出し、利益を達成すること、または候補製品を商業化することを阻止するかもしれない(承認されれば)。
多くの州では、バイオ製薬メーカーが独自の価格設定情報を公開すること、あるいは国家機関が購入した薬品に対して最高価格上限を設定することを要求するなど、間接的または直接に薬品価格を規範化するための立法が提出または公布されている。例えば、多くの州が州薬品価格透明性と報告法を検討しているか、最近公布されており、これは私たちのコンプライアンス負担を大幅に増加させ、規制部門の任意の候補製品に対する承認を得て商業化を開始した後、このような州法律に基づいてより大きな責任を負うことができるかもしれない。このような措置や立法は、私たちが獲得または要求する可能性のある任意の候補製品の価格に影響を与える可能性があり、私たちは規制部門の承認を得ることができるかもしれない。
さらに、CMSは2022年4月に、アルドゥカヌモノクロナル抗体およびFDAによって承認された任意の将来のアミロイドに対するモノクロナル抗体をカバーする国家政策を発表し、アルツハイマー病の治療への適応を指摘した。CMSは2023年1月にlecanemabの承認についてこの政策を再確認した。2つの部分からなる国家保険決定計画(NCD)によれば、連邦医療保険は、アルツハイマー病の治療のためのアミロイド(またはプラーク)に対するモノクロナル抗体をカバーし、以下の証拠開発保険項目で指定されたカバー基準に従って提供される場合、これらの抗体はFDAの伝統的な承認を得るであろう。CMSはまた、通常の臨床実践または登録によってデータを収集するような、CMSによって承認された研究に参加する医療保険患者により良い機会およびカバー範囲を提供するであろう。また,FDAが臨床的利益を示すか加速的な承認を得ているかが決定されていない薬物では,連邦医療保険はFDAや米国国立衛生研究院が承認した臨床試験で保険を提供する。現在および将来のCMSが私たちの候補製品を含む薬物カテゴリに対するカバー制限は、候補製品を商業化し、収入を発生させ、利益を達成する能力に実質的な悪影響を及ぼす可能性がある。将来のCMSカバー決定や政策が我々の業務にどのように影響するかは不明である.
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入の制限、マーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。多くの州では、州薬品価格透明性と報告法が検討されているか、最近公布されており、これは私たちのコンプライアンス負担を大幅に増加させ、規制部門の承認を得て商業化を開始した後、このような州法律に基づいてより大きな責任を負わせる可能性がある。
米国連邦や州医療立法の将来の行方を予測することはできません。これらの立法は、医療の獲得性を拡大し、医療コストをコントロールまたは低減することを目的としています。これらや法律や規制枠組みの任意のさらなる変化は、私たちの収入を減らしたり、コストを増加させたりして、私たちの業務、財務状況、運営結果に重大で不利な影響を与える可能性もあります。
政府、保険会社、医療組織および医療サービスを管理する他の支払者は、医療コストの制御または低減、および/または価格制御の実施に悪影響を及ぼす可能性がある
69
これらの医療改革措置や将来とりうる他の措置は,医療保険や他の医療資金のさらなる減少,より厳しいカバー基準,より低い精算,および新たな支払い方法につながる可能性が予想される。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。これは私たちが受け取ったすべての承認された製品の価格を下げるかもしれない。バイデン政府と各州は、価格または患者の精算制限、割引、特定の製品への参入およびマーケティングコスト開示の制限、および透明性措置を含む薬品と生物製品の価格設定を制御するために、さらなる立法と法規を通過する可能性がある。連邦医療保険または他の政府援助計画の支払いを拒否または減少させるいかなる精算も、同様の私的支払者の支払いを拒否または減少させる可能性があり、これは、十分な収入を生成することができ、利益を達成することができるか、または候補製品を商業化することができることを阻止することができるかもしれない(承認されれば)。また、米国議会のFDA承認過程に対するより厳格な審査は上場承認を著しく延期または阻止する可能性があり、より厳格な製品ラベルと上場後のテストとその他の要求の影響を受ける可能性がある。
当社の従業員、独立請負業者、コンサルタント、ビジネスパートナー、およびサプライヤーは、法規基準および要件を遵守しないことを含む、不適切な行為または他の不適切な活動に従事する可能性があります。
私たちは、従業員、独立請負業者、コンサルタント、ビジネスパートナー、サプライヤーの詐欺、不正行為、または他の不正活動のリスクに直面しています。これらの当事者の不正行為には、故意、無謀、不注意な行為が含まれている可能性がある
もし私たちがFDAの任意の候補製品の承認を得て、アメリカでこれらの製品を商業化し始めたら、私たちのこのような法律の下での潜在的なリスクは著しく増加し、私たちはこのような法律を遵守することに関連するコストも増加する可能性がある。特に、医療業界の研究、販売、マーケティング、教育、その他のビジネス配置は、詐欺、リベート、自己取引、および他の乱用行為を防止するための広範な法的制約を受けている。これらの法律および法規は、幅広い価格設定、割引、教育、マーケティングおよび販売促進、販売および手数料、特定の顧客インセンティブ計画、および他のビジネススケジュールを制限または禁止することができます。私たちはビジネス行為と道徳的規範を通過していますが、従業員や第三者の不正行為を常に識別して阻止できるわけではありません。このような活動を発見し、防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御できない可能性があり、またはそのような法律を遵守できないことによる政府の調査や他の行動や訴訟から保護されている可能性があります。もし私たちにこのような行動を取って、私たちが自分の権利を弁護したり、維持することに成功しなかったら、これらの行動は巨額の罰金や他の制裁を加えることを含めて、私たちの業務に大きな影響を与えるかもしれない。
もし私たちが医療保険法を守らなければ、私たちは巨額の処罰に直面する可能性があり、私たちの業務、運営、財務状況は不利な影響を受けるかもしれない。
もし私たちの候補製品がFDAの承認を得て、米国でこれらの製品の商業化を開始すれば、私たちの運営は様々な連邦や州詐欺や法律乱用の制約を受けるだろう。私たちの運営に影響を及ぼす可能性のある法律には
70
71
これらの法律の範囲は広いため、法定例外状況と利用可能な避難港の範囲は狭く、私たちはこれらの法律を遵守しようと努力しているにもかかわらず、私たちのいくつかの商業活動は1つ以上のこのような法律の挑戦を受けるかもしれない。私たちの業務計画が適用される医療保険法に適合することを確保する努力は巨額のコストに及ぶ可能性がある。政府および法執行当局は、私たちの業務実践が、詐欺および乱用または他の医療保健法律および法規を適用する現在または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちにこのような訴訟を提起した場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの訴訟は、民事、刑事および行政処罰、損害賠償、返還、罰金、Medicare、Medicaidおよび他の連邦ヘルスケア計画への参加から除外される可能性があり、契約損害、名声損害、利益および将来の収益の減少、および私たちの業務削減を含む、私たちの業務運営能力および運営結果に悪影響を及ぼす可能性があります。また,我々の任意の候補製品の米国国外での承認と商業化は,上記の医療保健法や他の外国法の外国等価物の制約を受ける可能性もある。
もし私たちまたは私たちが雇用した任意の契約製造業者やサプライヤーが環境、健康、安全の法律法規を遵守できなかった場合、私たちは罰金や罰金を受けたり、私たちの業務に重大な悪影響を及ぼす可能性のあるコストが発生する可能性があります。
私たちと私たちが雇用しているどんな契約製造業者も、多くの連邦、州、地方の環境、健康と安全の法律、法規、許可要件、それらの管理実験室手続き;危険と規制された材料と廃棄物の発生、処理、使用、貯蔵、処理と処理、地面、空気と水中への危険物質の排出と排出、そして従業員の健康と安全を守らなければならない。私たちの行動は化学物質、生物学的、そして放射性物質を含む危険で燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物を発生させるだろう。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。私たちはこのような材料が汚染や傷害をもたらす危険を除去することができない。もし私たちが危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に責任を負い、いかなる責任も私たちの資源の範囲を超える可能性がある。いくつかの環境法により、私たちは現在または過去の施設および第三者施設の任意の汚染に関する費用に責任を負わなければならないかもしれない。私たちはまた民事や刑事罰金と処罰に関連した巨額の費用を発生させるかもしれない。
適用される環境法律と法規を遵守することはコストが高い可能性があり、現在または未来の環境法律と法規は私たちの研究、製品開発、製造努力を損なう可能性がある。しかも、私たちはこのような材料や廃棄物が意外なダメージや汚染をもたらすリスクを完全に除去することはできない。危険材料の使用による従業員の負傷により生じる可能性のあるコストや支出を支払うために労働者補償保険を維持しているが、潜在的な責任を支払うのに十分ではない可能性がある。私たちは特定の生物或いは危険廃棄物保険を受けていません。私たちの財産、意外と一般責任保険は生物或いは危険廃棄物の暴露或いは汚染による損害と罰金は明確に含まれていません。したがって、汚染や傷害が発生した場合、私たちは損害賠償責任を負うことを要求されるか、または私たちの資源を超えた罰金に処せられる可能性があり、私たちの臨床試験または監督管理の承認が一時停止される可能性があり、これは私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちの業務は安全、プライバシー、データ保護に関する複雑で変化するアメリカと外国の法律法規によって制約されています。これらの法律と法規は変化と不確定な解釈が発生する可能性があり、クレーム、私たちの業務やり方や罰金を変える可能性があります。そうでなければ、私たちの業務を損なう可能性があります。
様々な国、国、および国際法律および法規は、セキュリティおよびネットワークセキュリティ要件、ならびに個人データの収集、使用、保持、保護、開示、転送、および他の処理に適用される。これらのセキュリティとデータ保護およびプライバシーに関連する法律·法規が進化しており、規制や公共審査が強化され、法執行と制裁レベルがエスカレートしている可能性がある。私たちはこのような法律を遵守するために努力し続けており、私たちは私たちのコンプライアンス努力を満たすために多くの追加資源を投入する必要があると予想される。新しい立法は似たような問題に新しい義務と要求を加えるかもしれない
72
このような法律の解釈と適用は司法管轄区域によって一致しないかもしれないし、私たちの現在の政策や慣行と一致しないかもしれない。私たちは、実際に、またはセキュリティ、プライバシーおよびデータ保護に関連する適用された法律および法規を十分に遵守できていないと考えられているか、または私たちが処理または維持しているシステム、個人データおよび他のデータを保護することができず、規制罰金、調査および法執行行動、処罰および他の責任、影響を受けた個人が損害賠償を要求すること、および私たちの名声が損なわれている可能性があり、これらは、私たちの業務、財務状況、運営結果、および将来性に大きな影響を与える可能性があります。
FDAや他の政府機関の資金不足は、重要な指導部や他の人員を採用して保持する能力を阻害し、新製品やサービスのタイムリーな開発や商業化を阻止したり、これらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止したりすることができ、これは私たちの業務にマイナスの影響を与える可能性がある。
FDAが新製品を審査·承認する能力は、政府予算と資金レベル、肝心な人員の雇用と維持及びユーザー費用の支払いを受ける能力、及び法律、法規と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また、私たちの行動に依存する可能性のある他の政府機関への政府の援助は、研究開発活動に資金を提供する機関を含め、政治プロセスの影響を受け、このプロセスは本質的に不安定で予測不可能である。FDAや他の機関の中断も、新薬が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。過去、米国政府は予算停止を経験し、FDAなどのある規制機関はFDAと他の政府の重要な従業員を休暇させ、重要な活動を停止せざるを得なかった。政府が長期的に停止すれば、FDAが私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。また、当社の上場企業としての運営では、将来的に政府の閉鎖が公開市場に参入し、必要な資本を得る能力に影響を与える可能性があり、適切な資本化と運営を継続することができます。
私たちの業務活動は“反海外腐敗法”(FCPA)、類似の反賄賂と反腐敗法律、その他の法規によって制約される可能性がある。
私たちの業務活動は、イギリスの“反賄賂法”を含む“海外腐敗防止法”および私たちの国の反賄賂または反腐敗法律、法規または規則のような制約を受ける可能性がある。“海外腐敗防止法”は、一般に、公式行動に影響を与えるか、または他の方法で業務を獲得または保留するために、非米国政府関係者への提供、承諾、他人に直接または間接的に価値のあるものを与えることを禁止する。“海外腐敗防止法”はまた、上場企業に会社の取引を正確かつ公平に反映した帳簿や記録を作成·保存し、適切な内部会計制御制度を制定·維持することを求めている。私たちの業務は厳しく規制されているため、非米国政府関係者を含む公職者との大きな相互作用に関連している。また、多くの他の国では、私たちの臨床試験や薬品を処方した医療提供者は彼らの政府に雇われており、薬品の購入者は政府の実体であり、そのため、私たちはこれらの調査者、処方者、購入者との取引は“海外腐敗防止法”によって規制されている。
最近,米国証券取引委員会と司法省はバイオテクノロジーや製薬会社に対する“反海外腐敗法”の法執行活動を増加させた。私たちのすべての従業員、代理店、請負業者、または協力者、または私たちの付属会社の従業員がすべての適用された法律と法規を遵守するかどうか、特にこれらの法律の高度な複雑さを考慮することはできません。これらの法律と法規に違反することは、私たち、私たちの役人、または私たちの従業員への罰金、刑事制裁、私たちの施設の閉鎖、輸出許可証の取得、制裁を受けた国での業務活動の停止、コンプライアンス計画の実施、そして私たちの業務の禁止につながる可能性があります。このような違反は、私たちが1つ以上の国または地域で私たちの製品を提供することを禁止し、私たちの名声、私たちのブランド、私たちの国際拡張努力、私たちの従業員を引き付け、維持する能力、ならびに私たちの業務、将来性、経営業績、および財務状況を深刻に損なう可能性があることを含む可能性があります。
73
私たちの第三者への依存に関するリスクは
私たちは、第三者との協力によって、私たちが開発可能ないくつかの候補製品を研究·開発し、商業化したい。もしそのような協力が成功しなければ、私たちはこのような候補製品の市場潜在力を達成できないかもしれない。
私たちは現在、AbVie、GSK、およびAdimabとの手配を含む、第三者パートナーを使用して、私たちが開発する可能性のある候補製品の研究、開発、商業化を継続していく予定です。
私たちの他の協力計画の協力者は、大中型製薬会社、地域的、全国的な製薬会社、バイオテクノロジー会社、学術機関を含む可能性がある。任意の第三者とのこのような配置は、一般に、私たちの協力者が開発または潜在的な商業化に取り組んでいることを制御し、彼らと開発された任意の候補製品の資源の数および時間を求めることができる共有または限られた制御を提供してくれる。私たちが商業実体とのこれらの計画から収入を得る能力は、私たちの協力者がこれらの手配の中で彼らに割り当てられた機能を成功的に履行する能力に依存するだろう。私たちは私たちの現在の協力や私たちが入るかもしれないどんな協力の成功も予測できない。
私たちの研究計画や私たちが開発する可能性のある任意の候補製品の協力は私たちに次のようなリスクをもたらすだろう
74
たとえば,AbbVieは我々と一緒にCD 33連携計画を検討した後,AL 003を開発しているCD 33連携計画を終了することにした.また,イノベーターは中国でAL 008の第1回人体研究を行うことはなく,イノベーターはAL 008の開発を継続することはなく,革新者に付与された権利を新たに獲得し,AL 008の開発と商業化に利用する予定である。Innoentは中国規制部門にINDを提出し,AlectorはInnovent.から関連材料と情報を取得している。Alectorプログラムは、IND申請のデータおよび文書を評価して、米国におけるINDの提出を潜在的にサポートする。
適切な協力を求めることで、私たちは激しい競争に直面するかもしれない。例えば、バイオテクノロジーと製薬会社との間の業務合併は潜在的な協力者の数を減少させる。また、交渉過程は時間も複雑であり、私たちはタイムリーで、受け入れ可能な条件で協力を交渉することができず、交渉することさえできないかもしれない。それができなければ、私たちが協力を求めている候補製品の開発を減らし、その開発計画や私たちの1つ以上の他の開発計画を減らしたり、潜在的な商業化を延期したり、販売やマーケティング活動の範囲を縮小したり、私たちの支出を増やして自費で開発や商業化活動を行わなければならないかもしれません。もし私たちが私たちの支出を増やし、私たち自身の開発や商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれないし、私たちはこれらの資本を得ることができないかもしれない
75
受け入れ可能な条件で、あるいは全く受け入れられない。もし私たちが十分な資金がなければ、私たちは候補製品をさらに開発したり、それらを市場に出して製品収入を生成することができないかもしれない。
私たちまたは私たちの協力者が合意によって付与された権利を行使しないことを選択した場合、または私たちまたは私たちの協力者が候補製品を既存の運営および会社文化に統合することに成功しなかった場合、私たちは協力のメリットを実現できないかもしれません。さらに、もし私たちと私たちの任意の協力者との合意が終了すれば、私たちの協力者が私たちに権限を与えた技術と知的財産権へのアクセスが制限されたり、完全に終了したりする可能性があり、これは、私たちが協力者の技術や知的財産権を利用して私たちの候補製品を開発し続けることを延期するか、あるいはこれらの候補製品の開発を完全に停止することを要求するかもしれない。私たちはまた、適切な代替協力者を見つけたり、新しい協力者を誘致することが難しいことを発見するかもしれませんし、私たちの開発計画が延期されるかもしれませんし、商業·金融界での私たちのイメージが悪影響を受ける可能性があります。本“リスク要因”の節で述べた製品開発、規制承認、商業化に関連する多くのリスクは、私たちの協力者の活動にも適用され、私たちの協力者にどんな負の影響も私たちに悪影響を及ぼす可能性があります。
我々は第三者に依存して著者らの臨床試験および著者らの研究と臨床前テストのいくつかの方面を行うことが予想されるが、これらの第三者の表現は締め切り前にこのような試験、研究或いはテストを完成できないことを含む満足できないかもしれない。
著者らは現在、CRO、臨床データ管理組織、医療機関と臨床研究者などの第三者に依存し、引き続き著者らの研究、臨床前テストと臨床試験のある方面に依存することを予想している。これらの第三者のいずれも、私たちとの契約関係を終了したり、その契約義務を履行できない可能性があります。もし私たちが代替計画を達成する必要があれば、これは私たちの製品開発活動を延期するだろう。
これらの第三者研究開発活動への依存は,これらの活動に対する我々の制御を減少させたが,我々の責任を軽減していない.例えば、私たちは私たちのすべての臨床試験が試験の全体的な研究計画と方案に従って行われることを確実にする責任がある。さらに、FDAは、データおよび報告の結果が信頼性、反復可能かつ正確であることを保証し、試験参加者の権利、完全性、および機密性を保護するために、cGCPの実施、記録、および臨床試験結果の報告の規定を遵守することを要求する。現在行われている臨床試験を登録し,完了した臨床試験の結果を一定時間範囲で政府後援のデータベースに発表することも求められている。もし私たちの契約の第三者が私たちの臨床試験を不当に使用した患者の募集過程で得られた情報、CGCPがプライバシー法の下での規定に適合していないか、または適用されない規定に関連する活動に従事すれば、私たちはまた追加の責任に直面する可能性があり、これは規制制裁を招き、私たちの名声と業務運営に深刻な損害を与える可能性がある。そうしないと罰金、否定的な宣伝、そして民事と刑事制裁につながるかもしれない。
これらの第三者が規制要件や私たちが規定した規程に従って契約責任を成功的に履行し、期待された期限内に私たちの臨床試験を完了または行うことができない場合、私たちは私たちが開発する可能性のある任意の候補製品の発売承認を得ることができないか、または延期することができなくなり、私たちの薬物の商業化に成功する努力を遅らせることができないだろう。
私たちはまた他の第三者に依存して私たちの臨床試験のための薬品の貯蔵と配布を望んでいる。私たちの流通業者のどんな業績ミスも、どんな薬品供給の出荷も含めて、私たちが開発する可能性のある任意の候補製品の臨床開発やマーケティング承認、あるいは私たちの薬品の商業化を遅延させ、追加の損失をもたらし、私たちの潜在的な製品収入を奪う可能性があります。
私たちは第三者と契約を結び、私たちの研究計画、臨床前研究、臨床試験のために材料を製造し、そして私たちが開発可能な任意の候補製品を商業化する。また、それぞれのプロトコルにより、グラクソ·スミスクラインとエバービーは一定の製品製造権を持っている。このような第三者への依存は、私たちがそのような材料、候補製品、または私たちが開発および商業化する可能性のある任意の薬剤を十分な量、または許容可能なコストでそのような供給を得ることができず、私たちの開発または商業化努力を延期、阻止または損害する可能性があるというリスクを増加させる可能性がある。
私たちには何の生産施設もありません。著者らは現在CDMOに依存して、臨床前研究と臨床試験のための材料を製造し、臨床前研究、臨床試験、著者らが開発する可能性のある任意の候補製品の商業供給に引き続きこのようにする予定である。私たちは現在いくつかのCDMOと関係を築き、私たちのすべての候補製品の製造とグラクソ·スミスクラインとの関係を構築しました,そして
76
AbbVie、LatozinemabおよびAL 101のための,AL 002と。私たちはCDMOとさらなる合意を作ることができないかもしれないし、受け入れられる条項でそうすることもできないかもしれない。たとえ第三者製造業者と合意できても、CDMOに依存することは、追加のリスクをもたらす
第三者メーカーは、米国以外のcGMP法規や同様の規制要件を遵守できない可能性がある。私たちの失敗、または私たちのCDMOまたはパートナーが適用された法規を遵守できなかった場合、臨床的に私たちの試験を一時停止させ、罰金、禁止、民事処罰、遅延、承認の一時停止または撤回、候補製品または薬物の押収またはリコール、運営制限、刑事起訴を含む制裁を加える可能性があり、これらはいずれも私たちの薬品供給に重大な悪影響を与え、私たちの業務、財務状況、運営結果、将来性を損なう可能性があります。
私たちが開発する可能性のあるどんな薬物も他の候補製品や製品と生産施設を競争する可能性がある。CGMP法規の下で運営されているメーカーの数は限られており、私たちのために製品を製造する能力があるかもしれない。
私たちの既存または将来の第三者製造業者の任意の性能障害は、臨床開発またはマーケティング承認を延期する可能性があります。もし私たちの既存の契約製造業者のいずれかが約束通りに契約を履行できない場合、私たちは製造業者の交換を要求される可能性があり、そのような代替製品を決定して同定する際に追加コストと遅延が生じる可能性があります。しかも、契約製造業者との生産能力の確保と保留は大きなコストを招く可能性がある。
私たちの現在と期待されている未来は、私たちが開発する可能性のある任意の候補製品や薬物を他人に生産することに依存しており、私たちの将来の利益率と、私たちが適時かつ競争力のある上場承認された薬物を商業化する能力に悪影響を及ぼすかもしれない。
私たちおよび私たちが依存しているCDMOパートナーは、第三者サプライヤーに依存して、私たちの製造過程で使用される重要な原材料を提供してくれますが、これらの第三者サプライヤーの流失や彼らは私たちに十分な原材料を提供することができません。新冠肺炎疫病の影響でも他の原因でも、私たちの業務を損なう可能性があります。
私たちと私たちが依存しているCDMOパートナーは、第三者サプライヤーに依存して私たちの候補製品を生産するために必要な原材料を提供し、引き続き第三者メーカーに依存して上場承認された任意の候補製品の商業供給を提供する予定です。これらの第三者サプライヤーへの依存及び十分な原材料供給を獲得する上で直面している挑戦はいくつかのリスクに関連し、新冠肺炎疫病の影響によるサプライチェーン問題、定価、このような材料の獲得可能性、このような材料の品質及び納品スケジュールの制御が限られている。小さな会社として、私たちの交渉手段は限られており、私たちは私たちの規模の大きい競争相手よりも低い優先順位を得ることができるかもしれない。私たちは長期供給協定を持っていません。私たちは開発製造サービス契約や購入注文に基づいて必要な薬物製品を購入します。私たちはサプライヤーが必要な数量のこれらの原材料を提供し続けるか、あるいは予想される仕様と品質要求を満たすかどうかを確認することができません。新冠肺炎の疫病および後続変異による供給中断を含む、限られたまたは独占的な供給源の供給中断は、新しい供給源(もしあれば)が見つかり、資格認証を得るまで、候補製品を生産する能力を実質的に損なう可能性がある。この場合、私たちは合理的な時間内にまたは商業的に合理的な条件下で十分な代替供給ルートを見つけることができないかもしれない。サプライヤー側のいかなる不振も、臨床試験の制限や規制承認に必要な供給の制限を含む候補製品の開発と潜在的な商業化を遅らせる可能性があり、これは私たちの業務に重大な悪影響を与える。
77
私たちの知的財産権に関するリスクは
もし私たちが開発したすべての候補製品のために特許保護を得ることができなければ、私たちの競争相手は私たちと類似または同じ製品を開発し、商業化する可能性があり、私たちが開発した任意の候補製品を商業化することに成功する能力は不利な影響を受ける可能性がある。
私たちの成功は、私たちがアメリカや他の国で私たちの特許候補製品と私たちが開発する可能性のある他の技術の特許保護を獲得し、維持する能力があるかどうかに大きくかかっている。私たちは、私たちのコア計画や候補製品、および私たちの業務に重要な他の技術に関する特許出願を米国と海外に提出することで、私たちの独自の地位を保護することを求めています。著者らの候補製品が臨床開発段階に入り、進展を得ることに伴い、著者らは引き続きこれらの候補製品のある方面に対して知的財産権保護を行う。例えば、私たちは、私たちの技術およびコア製品候補について特許出願を提出することを意図しているか、または特許出願を提出しようとしているが、そのような特許出願がライセンス特許として発行される保証はない。さらに、我々が我々の技術および製品候補のいくつかの態様についてのみ仮特許出願を提出した場合、これらの仮特許出願のいずれも、適用される仮特許出願が提出された日から12ヶ月以内に非仮特許出願が提出されるまで、発行された特許となる資格がない。この時間内に非一時的特許出願を提出できなかったいかなる行為も、関連する一時的特許出願に開示された発明を特許保護する能力を失う可能性がある。さらに、場合によっては、コアプログラムおよび候補製品、ならびに私たちのビジネスに重要な他の技術に関連する他の組み合わせの発表された声明を得ることができず、そのようなコアプログラム、候補製品の使用方法、および/または製造方法を保護するための宣言を含む特許出願を提出する必要がある場合がある, 他の技術もありますこのような特許出願がライセンス特許として発行されることは保証されず、たとえ彼らが確かに特許出願を発行したとしても、これらの特許主張は、我々の競争相手のような第三者を阻止するのに十分ではない可能性がある。私たちのコア計画および候補製品に関連する特許保護を取得または維持できなかった場合、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性があります。
もし私たちの任意の特許出願または私たちの協力者の特許出願がどの司法管轄区でも特許として発行されていない場合、私たちは効果的に競争することができないかもしれない。
米国および他の国の特許法またはその解釈の変化は、私たちの発明、取得、維持、および私たちの知的財産権を保護する能力を弱める可能性があり、より広く言えば、私たちの知的財産権の価値に影響を与えたり、私たちの候補製品に関する私たちまたは私たちの協力者の特許の範囲を縮小したりする可能性がある。私たちと私たちの協力者が私たちの候補製品に関連する知的財産権について、私たちと私たちの協力者が現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるかどうか、または発行された特許の権利主張が競争相手や他の第三者の影響を受けないように十分な保護を提供するかどうかを予測することはできません。
特許訴訟プロセスは高価で、時間がかかり、複雑であり、私たちまたは私たちの協力者は、必要または理想的なすべての特許出願を合理的なコストまたはタイムリーに提出、起訴、維持、強制執行または許可することができない可能性がある。我々の研究開発成果における出願可能な特許の側面をタイムリーに決定できず,特許保護を得ることができない可能性もある.私たちは、私たちの従業員、会社の協力者、外部科学協力者、CRO、CDMO、コンサルタント、コンサルタント、および他の第三者などと、私たちの研究開発成果の機密または特許を申請可能な側面にアクセスする権利がある当事者と秘密および秘密保護協定を締結したが、いずれも協定に違反し、特許出願を提出する前にこのような成果を開示し、それによって、私たちの特許保護を求める能力を危険にさらす可能性がある。さらに、有効かつ強制的に実行可能な特許を取得して保持する能力は、我々の発明と従来技術との差が、我々の発明が従来技術よりも特許を出願することを可能にするか否かに依存する。また、科学文献で発表された発見は実際の発見よりも遅れがちであり、米国および他の司法管轄区の特許出願は通常、出願18ヶ月後に公表され、時には全く公表されない場合もある。したがって、私たちまたは私たちの協力者がこのような発明のために特許保護を申請した最初の人であることを確認することはできない。
78
もし私たちが獲得した任意の特許保護の範囲が十分に広くない場合、あるいは私たちがいかなる特許保護を失った場合、競争相手が類似または同じ技術および候補製品を商業化する能力が悪影響を受けることを阻止する。
バイオテクノロジーと製薬会社の特許地位は通常高度に不確定であり、複雑な法律と事実問題に関連しており、近年多くの訴訟のテーマとなってきた。したがって,我々の特許権の発行,範囲,有効性,実行可能性,商業的価値は高い不確実性を持っている.私たちまたは私たちの協力者の未解決および将来の特許出願は、私たちの候補製品または他の技術を保護する特許の発行、または他社が競争技術および候補製品を商業化することを効果的に阻止する特許をもたらすことができないかもしれない。
また,特許出願に要求されるカバー範囲は特許発行前に大幅に縮小することができ,その範囲は特許発行後に再解釈することができる.私たちまたは私たちの協力者が現在または将来許可または所有している特許出願が特許として発行されても、それらは、私たちに任意の意味のある保護を提供してくれる、競争相手または他の第三者が私たちと競争することを阻止する、または他の方法で私たちにどんな競争優位性を提供してくれるかの形で発表されないだろう。私たちまたは私たちの協力者が権利を持つどの特許も、第三者によって挑戦され、範囲が縮小され、回避され、または無効に宣言される可能性がある。したがって、候補製品または他の技術が効果的かつ実行可能な特許によって保護されるかどうか、または保護され続けるかどうかは分からない。私たちの競争相手または他の第三者は、類似または代替技術または製品を非侵害的に開発することによって、私たちの特許を回避することができ、これは、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を及ぼす可能性があります。
特許の発行は、その発明性、範囲、有効性、または実行可能性に対して決定的ではなく、我々の特許は、米国および海外の裁判所または特許庁で挑戦される可能性がある。私たちまたは私たちの協力者は、第三者によって、米国特許商標局(USPTO)または外国特許庁に既存技術を予め提出しておくか、または反対、派生、撤回、再審査、許可後、当事者との間の審査、発明権紛争、妨害訴訟、または私たちまたは私たちの協力者特許権に挑戦する他の類似訴訟に巻き込まれる可能性がある。このような提出、訴訟、または訴訟における不利な裁決は、そのような特許権の範囲を縮小したり、無効にしたり、実行できないようにしたりして、第三者が私たちの候補製品または他の技術を商業化し、私たちに支払うことなく、直接私たちと競争することを可能にする可能性がある。さらに、私たちまたは私たちのいずれかの協力者は、例えば、外国特許庁の異議において、第三者が、私たちまたは私たちの協力者の特許および特許出願の特許可能性の特徴に疑問を提起するように、許可された挑戦手続に参加しなければならない可能性がある。このような挑戦は、特許権の喪失、排他性の喪失、または特許主張の縮小、無効または実行不能をもたらす可能性があり、これは、他人が類似または同じ技術および製品を使用することを阻止するか、またはそれを商業化する能力を制限するか、または候補製品および他の技術の特許保護期間を制限する可能性がある。最終的な結果が私たちに有利であっても、このような手続きは大量のコストを招く可能性があり、私たちの科学者と経営陣に多くの時間がかかる必要がある。もし私たちまたは私たちの協力者がこのような訴訟や他の発明権紛争で敗訴すれば, 私たちは第三者から許可証を取得して維持する必要があるかもしれない。このような許可は商業的に合理的な条項では得られないかもしれないし,まったく存在しない,あるいは非排他的である可能性がある.もし私たちがそのようなライセンスを取得して維持できなければ、私たちが開発する可能性のある1つまたは複数の候補製品の開発、製造、商業化を停止する必要があるかもしれない。排他性を失ったり、私たちが所有して許可したりする特許主張を縮小することは、他の人が類似または同じ技術および製品を使用または商業化することを阻止する能力を制限するかもしれない。
さらに、新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちの知的財産権は、他の人が私たちと似ているか同じ製品を商業化する権利を十分な時間で排除することを提供してくれないかもしれない。
私たちのいくつかの特許と特許出願は第三者と共同で所有することができる。さらに、協力者または将来の許可者は、私たちと直接関係のない他の第三者と彼らの特許および特許出願を共同で所有する可能性がある。特定の特許および特許出願に対する私たちの権利は、これらの特許と特許出願の共通所有者との間の機関間または他の運営協定にある程度依存する可能性があり、彼らは私たちの許可協定の一方ではない。もし私たちの協力者または未来のライセンシーが、任意の第三者共通所有者がそのような特許または特許出願の権利の下で独占的許可を付与していない場合、または私たちが他の方法でそのような独占的権利を得ることができない場合、これらの共通所有者は、その権利を私たちの競争相手を含む他の第三者に許可することができ、私たちの競争相手は、私たちの知的財産権が含まれていない範囲で競合製品および技術を販売することができる。さらに私たちは
79
そのような特許は、第三者にそのような特許を強制的に実行するために、いかなる特許も共同所有者間の協力であり、そのような協力は私たちに提供されない可能性がある。上記のいずれも、我々の競争地位、業務、財務状況、経営結果、および見通しに重大な悪影響を及ぼす可能性がある。
私たちの開発と商業化権利は、知的財産権に関する条項と条件を含む、他人との合意の条項と条件にある程度制限されている。
私たちは第三者の特定の特許権およびノウハウに依存しており、これらの特許権およびノウハウは、私たちの候補製品の開発に非常に重要または必要であり、私たちの候補製品の開発および商業化は、第三者とのいくつかの協力協定の条項および条件によって制約されている。例えば、2014年、私たちはAdimabとAdimab協力協定を締結した。2014年のAdimab協力協定によると、私たちはAdimabが私たちのlatozinemabとAL 101候補製品で発見された抗体を開発しており、私たちは私たちのAL 002製品候補のためにAdimabによって最適化された抗体を開発している。2019年8月、私たちはAdimabと新しい協力協定を締結し、未来のプロジェクトのための抗体を開発した。2021年には、指定された数のエンジニアリングシーケンスの独占的選択権を付与するAdimabと別の協力協定を締結しました.Adimabによって発見または最適化され、私たちが選択した目標に向けられている。また、2017年10月、私たちはAbbVieと共同でアルツハイマー病や他の神経変性疾患を治療する薬剤を開発し、商業化することで合意した。2021年7月、我々は、前顆粒タンパク質のモノクロナル抗体LatozinemabおよびAL 101を世界的に協力して開発および商業化するグラクソ·スミスクライン協定に合意した。
私たちのAdimab、AbbVie、GSKとの合意、および私たちが将来達成した他の合意は、すべての関連する使用分野および私たちが将来私たちの技術および製品を開発または商業化することを望む可能性のあるすべての地域で、協力者によって保持されている特定の知的財産権および技術を使用することを提供しないかもしれません。したがって、このような協力者が保持している技術を使用した競争製品を競争相手や他の第三者が開発し、そのような製品が私たちの知的財産権の範囲内にない限り、それを商業化することはできないかもしれない。
さらに、このような合意のいずれかの条項によれば、私たちは、準備、提出、起訴、および維持を制御する権利がないかもしれませんし、私たちの開発候補製品に関連したり、影響を与えたりする特定の特許および特許出願の強制執行および弁護を制御する権利がないかもしれません。さらに、グラクソ·スミスクライン協定は、いくつかの特許および特許出願の準備、提出、起訴、維持、実行、および弁護におけるグラクソ·スミスクラインに特定の権利を与える。
私たちは特許と特許出願の準備、提出、起訴、維持、執行、または弁護が私たちの業務の最適な利益に合った方法で準備、提出、起訴、維持、強制執行、そして弁護されるかどうかを決定することができない。もし私たちの協力者がこれらの特許を起訴、維持、強制執行および保護できなかった場合、またはこれらの特許または特許出願の権利を失った場合、私たちのこれらの特許に対する権利は減少またはキャンセルされる可能性があり、私たちはそのような権利に制約された候補製品を開発し、商業化する権利は悪影響を受ける可能性があり、私たちは競争相手の競合製品の製造、使用、販売を阻止する能力を低下させるかもしれない。さらに、私たちが協力者または協力者から許可された特許出願への起訴を制御する権利があっても、私たちは特許起訴の日までに発生した協力者の行動や不作為の悪影響や損害を受ける可能性がある。
さらに、私たちまたは私たちの協力者の特許は、1つ以上の第三者の権利によって維持される可能性がある。例えば、我々は神経栄養因子PGRN分解受容体SORT 1に対する新しい治療性抗体の生産と特徴づけの研究を支援するために、国家衛生研究所から授与された賞を受賞した。したがって、アメリカ政府はそれによって生成された知的財産権に対していくつかの権利を持っているかもしれない。米国政府の援助の下で新しい技術を開発する場合、米国政府は、米国政府が発明を使用することを許可するか、または他の人にその発明を使用させることを代表させる非独占的許可を含む、生成された任意の特許のいくつかの権利を得ることができる。米国政府の権利はまた、援助された発明および技術を第三者に開示し、米国政府の資金開発を使用する技術を使用または許可する第三者の先行権を行使することを可能にする可能性がある。米国政府が政府援助の技術の実用化を実現できなかったために行動する必要があると考えている場合,あるいは健康や安全需要を緩和するために行動する必要があるため,連邦法規の要求を満たすため,あるいは米国工業を優先するためにデモの権利を行使することができる。さらに、このような発明の権利は、米国のいくつかの施設において、そのような発明を具現化する製品を製造するといういくつかの要件によって制約される可能性がある
80
この要求を放棄しなければ、どんな状況にも適用されない。このような権利に対する米国政府または任意の第三者の行使は、私たちの競争地位、業務、財務状況、経営結果、および将来性に実質的な悪影響を及ぼす可能性がある。
もし私たちが合意の義務を履行できなかった場合、すなわち私たちは私たちの協力者または未来の許可者に可能な知的財産権を選択したり、私たちの協力者や未来の許可者との業務関係が妨害されたりすれば、私たちは私たちの業務に非常に重要な知的財産権を失うかもしれない。
私たちは、私たちの研究を進めたり、私たちが開発する可能性のある候補製品の商業化を可能にするために、いくつかの知的財産権を選択するために、他の人から追加の知的財産権を得る必要があるかもしれないという合意を締結しました。私たちはもしあれば、合理的な費用や合理的な条件で追加的な知的財産権を得ることができないかもしれない。この場合、私たちは、私たちの技術、候補製品、またはそれらを製造する方法を再設計するために、または代替技術を開発または許可するために、多くの時間および資源を必要とすることができ、これらすべては、技術的または商業的に不可能である可能性がある。もし私たちがそれができなければ、影響を受けた候補製品を開発したり商業化することができないかもしれません。これは、私たちの業務、財務状況、運営結果、将来性を深刻に損なう可能性があります。私たちは、第三者特許が存在しないことを保証することはできません。これらの特許は、私たちの現在の技術、製造方法、候補製品、または未来の方法または製品に対して強制的に実行される可能性があり、それによって、私たちの製造または将来の販売を禁止すること、または私たちの将来の販売について、第三者に印税および/または他の形態の賠償を支払う義務があるかもしれません。
しかも、私たちは協力者とのすべての合意が、私たちの未来の合意が様々な経済、開発、勤勉、商業化、そして他の義務を私たちに強要すると予想している。私たちのいくつかの協力協定はまた、開発スケジュールを遵守したり、商業的に合理的な努力をして、許可された製品を開発し、商業化することを要求しています。私たちは努力しているにもかかわらず、私たちの協力者は、私たちがこのような合意規定の義務に深刻に違反していると結論するかもしれないので、合意項目の下での損害賠償を中止したり、求めたりして、これらの合意がカバーする製品や技術を開発し、商業化する能力を取り消したり制限したりする可能性がある。これらの合意を終了することにより、いくつかの特許または他の知的財産権の権利が失われた場合、または基礎特許が予期される排他性を提供できなかった場合、競争相手または他の第三者は、規制部門の承認を自由に求め、私たちと類似しているまたは同じ製品を販売することができ、私たちのいくつかの候補製品の開発および商業化を停止することを要求されるかもしれない。上記のいずれも、我々の競争地位、業務、財務状況、経営結果、成長見通しに重大な悪影響を及ぼす可能性がある。
また、協力協定によると、知的財産権に関する紛争が発生する可能性がある
さらに、私たちは現在、知的財産権または技術が可能かもしれないプロトコルを第三者から選択する権利が複雑であり、そのようなプロトコルのいくつかは様々な解釈の影響を受ける可能性がある。可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および成長の見通しに大きな悪影響を及ぼす可能性があります。さらに、もし私たちが選択したり許可したりする知的財産権紛争が、商業的に許容可能な条項で現在手配されている能力を維持したり弱体化したりすれば、私たちは開発と商業化に成功できないかもしれない
81
影響を受けた候補製品は、これは私たちの業務、財務状況、運営結果、および成長見通しに実質的な悪影響を及ぼす可能性がある。
私たちは世界各地で私たちの知的財産権と固有の権利を保護できないかもしれない。
世界のすべての国で私たちの候補製品や他の技術出願、起訴、特許保護に対する費用は目を引くほど高く、外国の法律はアメリカの法律のように私たちの権利を保護できないかもしれない。
したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちの特許保護があるが法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護、特にバイオテクノロジー製品に関する保護の実行に賛成せず、私たちの特許を侵害したり、私たちの知的財産権および独自の権利に違反する競争製品を販売することを阻止することを困難にする可能性がある。外国の管轄区域で私たちの知的財産権と独自の権利を強制的に執行する訴訟手続きは巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移すことは、私たちの特許を無効または狭義に解釈されるリスクに直面させる可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者が私たちに反訴する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちの知的財産権および独自の権利を世界各地で強制的に実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネス的優位性を得るのに十分ではないかもしれません。
多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置が限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たち、私たちの協力者、または私たちの未来の任意のライセンシーが、私たちの業務に関連する任意の特許の許可を第三者に付与することを余儀なくされた場合、私たちの競争的地位が損なわれる可能性があり、私たちの業務、財務状況、運営結果、および見通しは不利な影響を受ける可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
私たちが所有または許可している特許および出願の有効期間内に、定期維持費、継続費、年会費、および様々な他の政府特許および出願費用は、米国特許商標局および米国以外の様々な政府特許機関によって支払われる。場合によっては、私たちは、私たちの協力者または許可パートナーに依存して、これらの費用を米国および非米国の特許エージェントに支払う。米国特許商標局および様々な非米国政府機関は、特許出願過程において、いくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。私たちはまた、私たちの許可された知的財産権に関するこれらの要求を守るために、私たちの協力者に依存しているかもしれない。場合によっては、不注意は、滞納金を支払うことによって、または規則を適用する他の方法によって救済することができる。しかしながら、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効される可能性があり、それにより、関連法ドメインの特許権の一部または全部が失われる可能性がある。この場合、潜在的な競争相手は、同様または同じ製品または技術で市場に参入する可能性があり、これは、私たちの業務、財務状況、運営結果、および成長見通しに実質的な悪影響を及ぼす可能性がある。
ロシアに対する制裁を含むロシアとウクライナの間の持続的な衝突は、ロシア、ウクライナ、ユーラシア特許庁の特許出願の提出、起訴申請、発行された特許の維持を妨害する可能性がある。例えば、紛争と制裁は、申請料、延長費、および年金の支払いを妨害する可能性がある。紛争および制裁はまた、ロシア、ウクライナ、およびユーラシア特許庁によって発行された特許の実行または保護を妨害する可能性がある。紛争や関連制裁の可能性は
82
したがって、私たちの特許出願を起訴すること、および将来発行される任意の特許を実行または保護することをめぐる不確実性およびコストが増加し、これらのすべては、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
米国特許法の変化は特許の全体的な価値を低下させ、製品を保護する能力を弱める可能性がある。
米国特許法または特許法解釈の変化は、特許出願をめぐる起訴および発行された特許の実行または弁護の不確実性およびコストを増加させる可能性がある。例えば、“ライシー·スミス米国発明法”(“米国発明法”)によれば、米国で最初に特許出願を提出した発明者は、他方が保護を要求する発明を最初に発明したか否かにかかわらず、1つの発明の特許を得る権利がある。したがって、2013年3月以降、我々が以前に米国特許商標局に特許出願を提出した第三者は、当該第三者が発明を行う前に本発明を作成したとしても、我々の発明をカバーする特許を付与することができる。この可能性は私たちが発明から特許出願までの時間を認識することを要求する。米国およびほとんどの他の国の特許出願は、提出後または発表前の期間は秘密であるため、私たちは私たちの候補品または他の技術に関連する特許出願を最初に提出した会社であることを確認することはできない。
米国発明法によれば、米国特許商標局のいくつかのプログラムは、特許出願の起訴方式に影響を与える可能性があり、特許訴訟に影響を与える可能性もある。これらの措置には、第三者が特許訴訟中に米国特許商標局に以前の技術を提出することを可能にすることと、米国特許商標局によって管理されたライセンス後のプログラム(ライセンス後審査を含む)が特許有効性を攻撃することを可能にする追加のプログラムとが含まれる各方面間審査と派生手続き。USPTO訴訟における証拠基準は、米国連邦裁判所が特許主張の無効を宣言するために必要な証拠基準を下回っているため、第三者は、USPTO訴訟において、USPTOが権利要求を無効にするのに十分な証拠を提供する可能性があり、同じ証拠が地域裁判所訴訟で提出された場合には、権利請求を無効にするのに十分ではない。したがって,第三者は米国特許商標局の手続きを用いて我々の特許主張の無効を宣言しようと試みる可能性があり,第三者が地域裁判所訴訟で被告として疑問を提起すれば,我々の特許主張は無効にされない.したがって、米国発明法およびその実施は、私たちの特許出願をめぐる起訴および私たちが発行した特許の実行または保護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
また、薬品研究開発と商業化における企業の特許地位は特に不確定である。米国最高裁判所と米国連邦巡回控訴裁判所の裁決は、場合によっては入手可能な特許保護範囲を縮小し、場合によっては特許所有者の権利を弱める。この一連の事件は特許取得後の有効性と実行可能性の面で不確実性をもたらしている。米国議会、連邦裁判所、および米国特許商標局の将来の行動によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、私たちの既存の特許の組み合わせおよび私たちの将来の知的財産権の保護と実行能力に重大な悪影響を及ぼす可能性がある。
法廷または米国または海外の行政機関で疑問が提起された場合、私たちの候補製品および他の技術に関連する発行された特許は、無効または実行不可能と認定される可能性がある。
もし私たちが第三者に対して法的訴訟を起こして、私たちの候補製品または他の技術をカバーする特許を強制的に執行する場合、被告はその特許が無効または強制執行できないと反訴することができる。米国の特許訴訟では,被告が無効または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、明らかな書面記述、または実施を含む、いくつかの法定要件のいずれかを満たすことができなかったと言われている可能性がある。主張を実行できない理由は,特許起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った声明をしたりしたためかもしれない.第三者は,訴訟の範囲外であっても,米国や海外の行政機関に我々の特許の有効性や実行可能性を疑問視するクレームを出すことができる.このようなメカニズムには、再審査、付与後再審、当事者間の再審、派生手続き、外国法ドメインにおける同等の手続き(例えば、反対手続き)が含まれる。このような訴訟は、私たちの候補製品または他の技術をカバーしないように、私たちの特許が撤回、キャンセル、または修正される可能性がある。法的に無効と実行不可能と断言された後の結果は予測できない。例えば、有効性の問題については、私たちは無効な以前の技術がないことを確認することはできないが、私たちまたは私たちの許可パートナーと特許審査員は起訴中にこれを知らない。3番目なら
83
もし一方が法的に無効または強制執行できないと主張した場合、私たちは私たちの候補製品または他の技術の少なくとも一部またはすべての特許保護を失うだろう。このような特許保護の喪失は、私たちの業務、財務状況、運営結果、および成長の見通しに実質的な悪影響を及ぼすだろう。
もし私たちが開発する可能性のある候補製品のために特許期間の延長とデータ独占権を得なければ、私たちの業務は実質的に損害を受ける可能性がある。
FDAが私たちが開発する可能性のある任意の候補製品の上場承認の時間、期限、詳細によると、私たちの1つ以上の米国特許は、“ハッジ·ワックスマン法案”によって限られた特許期間を延長する資格がある可能性がある。ハッジ·ワックスマン法は、FDA規制審査中に失われた特許期間の補償として、特許期間を最大5年間延長することを許可している。1つの特許期間の延長は、製品が承認された日から計14年の期間を超えてはならず、1つの特許を延長することしかできず、承認された薬物、その使用方法又はその製造方法に関する請求項を延長することしかできない。一部の外国国·地域では、例えばヨーロッパでは、補完保護証明書に基づいて、規制審査中に失われた特許期間の補償として同様の延長を得ることもできる。しかし、私たちは、テスト段階または規制審査中に職務調査を行わなかったこと、適用された最終期限内に出願を提出できなかったこと、関連する特許が満了する前に出願を提出できなかったこと、または適用要件を満たしていなかったことなどの理由で、米国および/または他の国および地域で延期許可を得ることができない可能性がある。しかも、特許保護の適用期間や範囲は私たちが要求しているものよりも短いかもしれない。もし私たちが特許期間の延長を得ることができない場合、あるいはこのような延長の期限が私たちが要求したものよりも短い場合、私たちの競争相手は私たちの特許が満期になった後に競争製品の承認を受ける可能性があり、私たちの業務、財務状況、運営結果、成長の見通しは実質的に損なわれる可能性がある。
私たちは私たちの特許と他の知的財産権発明権のクレームに疑問を受けるかもしれない。
私たちは、発明者または共同発明者として、元従業員、協力者、または他の第三者から、私たちの特許、商業秘密、または他の知的財産権の利益について告発されるかもしれない。例えば、私たちは、従業員、コンサルタント、または私たちの候補製品または他の技術の開発に参加する他の技術者の義務衝突によって在庫紛争が生じる可能性があります。訴訟は、これらおよび他の私たちに挑戦する特許、商業秘密、または他の知的財産権の在庫または所有権のクレームに対抗するために必要かもしれない。このようなクレームの弁護に失敗した場合、金銭損害賠償の支払いに加えて、私たちの製品候補製品や他の技術に重要な知的財産権の独占所有権や使用権など、貴重な知的財産権を失う可能性があります。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。上記のいずれも、我々の業務、財務状況、経営結果、成長見通しに重大な悪影響を及ぼす可能性がある。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
私たちの候補製品および他の技術のための特許出願に加えて、私たちは、ビジネス秘密および秘密保護協定によって、私たちの非特許ノウハウ、技術、および他の固有情報を保護し、私たちの競争的地位を維持します。私たちはビジネス秘密と技術的ノウハウが私たちの知的財産権の主要な源の一つだと思う。ビジネス秘密と技術的ノウハウを保護することは難しいかもしれない。私たちは時間の経過とともに、私たちの商業秘密と技術ノウハウが独立して開発、記述方法の定期刊行物文章を発表し、人員が学術職から業界科学職に移って業界内に伝播することを望んでいる。
私たちは、私たちの従業員、会社および学術協力者、外部科学協力者、CRO、CDMO、コンサルタント、コンサルタント、および他の第三者など、これらの秘密にアクセスする権利のある当事者と秘密協定を締結することによって、これらの商業秘密および他のノウハウの保護を求めています。私たちはまた、私たちの従業員やコンサルタントと秘密および発明または特許譲渡協定を締結し、私たちの従業員は、前の雇用主の独自の情報や技術を私たちまたは彼らの仕事に持ってこないように訓練し、前の従業員が退職した時に守秘義務を注意する。私たちは私たちが可能であるか、または私たちの商業秘密またはノウハウとプロセスに接触したすべての当事者とこのような合意に到達したことを保証することができない。私たちは努力したにもかかわらず、どちらも合意に違反し、私たちのビジネス秘密を含む私たちの固有の情報を漏洩する可能性があり、私たちはできないかもしれない
84
このような違反のための十分な救済措置が得られた。しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。もし私たちの任意の商業秘密が競争相手または他の第三者によって合法的に取得または独立して開発された場合、私たちは彼らがその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちの任意の商業秘密が競争相手または他の第三者に漏洩された場合、または競争相手または他の第三者によって独立して開発された場合、私たちの競争地位は実質的かつ不利な損害を受けるだろう。
私たちは買収や他の方法で私たちの候補製品や他の技術の必要な権利を得ることができないかもしれない。
神経変性治療分野で我々と競合する製薬会社、バイオテクノロジー会社、学術機関の多くは特許を有している可能性があり、我々の業務に関連する可能性のある特許出願を提出し、提出している可能性がある。これらの第三者特許の侵害を回避するためには、これらの第三者知的財産権所有者からこのような特許の許可を得ることが必要または慎重であることが発見される可能性がある。私たちはまた、未来の候補製品と一緒に使用するために、第三者からのいくつかの技術的ライセンスが必要かもしれない。また、私たちが第三者と共同で所有している任意の特許については、これらの共同所有者の当該特許に対する利益を満たすために許可を得ることを望んでいる可能性がある。しかし、私たちは、このような許可を得ることができないか、または他の方法で第三者から取得することができないかもしれません。私たちの将来の候補製品に必要な成分、使用方法、プロセス、または他の知的財産権です。第三者知的財産権の許可または取得は競争分野であり、より多くの老舗企業は、魅力的または必要と考えられる第三者知的財産権許可または取得戦略をとる可能性がある。これらの老舗会社はその規模、資本資源及び更に強い臨床開発と商業化能力のため、私たちより競争優位を持っているかもしれない。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちの投資を適切なリターンを得るための条項で許可したり、第三者の知的財産権を取得することができないかもしれないし、全くできないかもしれない。もし私たちが必要な第三者知的財産権を獲得したり、私たちの既存の知的財産権を維持することができなければ、関連プログラムや候補製品の開発を放棄しなければならないかもしれない, これは私たちの業務、財務状況、運営結果、および成長見通しに実質的な悪影響を及ぼすかもしれない。
私たちは、私たちの従業員、コンサルタント、またはコンサルタントが、彼らの現在または前任者のいわゆる商業機密を誤って使用または開示したり、私たち自身の知的財産権を持っていると主張したりするという疑惑を受けるかもしれない。
私たちの多くの従業員、コンサルタント、そしてコンサルタントは現在あるいは以前、私たちの競争相手と潜在的な競争相手を含む大学や他のバイオテクノロジーや製薬会社に雇われています。私たちは、私たちの従業員、コンサルタント、およびコンサルタントが、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちは、商業秘密または他の固有情報を含む、私たちまたはこれらの個人が、商業秘密または他の固有情報を含む任意のそのような個人の現職または前任雇用者の知的財産を使用または開示しているという疑惑を受ける可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権や人員を失う可能性がある。このようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。
また、私たちの政策は、知的財産権の概念や開発に参加する可能性のある私たちの従業員と請負業者が合意に署名し、このような知的財産権を私たちに譲渡することを要求しているにもかかわらず、実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を実行することができないかもしれない。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームをつけさせられたり、私たちが私たちの知的財産権の所有権だと思うことを決定するために、私たちが提起する可能性のあるクレームを弁護したりすることができます。このようなクレームは、私たちの業務、財務状況、運営結果、および成長見通しに実質的な悪影響を及ぼす可能性があります。
私たちまたは私たちの協力者の知的財産権侵害、流用、または他の侵害に対する第三者クレームは、私たちの候補製品および他の技術の開発および商業化を阻止または延期する可能性があります。
神経変性疾患の治療方法領域の競争が激しく、活力に満ちていることを発見した。我々と我々の競争相手を含む複数の会社がこの分野の重点研究と開発を行っているため、知的財産権の構造が変化しており、将来はまだ不確定である可能性がある。また、
85
我々の候補製品で使用されている技術はまだ開発中であり,類似技術を用いた製品は市場に進出していない.したがって、将来的には知的財産権に関する重大な訴訟と、私たちおよび他の第三者、知的財産権、独自の権利に関する訴訟があるかもしれない。
私たちのビジネスの成功は、私たちと私たちのパートナーが私たちが開発した任意の候補製品を開発、製造、マーケティング、販売する能力と、第三者の特許および他の知的財産権を侵害、流用、または他の方法で侵害することなく、私たちの独自技術を使用する能力にある程度依存します。バイオテクノロジーや製薬業界では、特許や他の知的財産権に関する多くの複雑な訴訟があり、私たちは将来、その是非にかかわらず、このような訴訟の影響を受けたり、そのような訴訟の脅威にさらされたりする可能性がある。さらに、米国特許商標局による付与後、派生および再審手続き、または外国司法管轄区で行われた異議および他の同様の訴訟を含む、第三者特許に挑戦するためにコストの高い行政訴訟を提起する可能性がある。
我々が我々の候補製品を開発している分野には,大量の米国や外国から発行された特許と,第三者が所有する未解決特許出願が存在する.バイオテクノロジーや製薬業界の拡張や特許の発行に伴い,我々の候補製品や他の技術は他人の特許権侵害のクレームを引き起こすリスクが増加する可能性がある。私たちの候補製品と、私たちが開発している、開発されている、または将来開発される可能性のある他の技術が、第三者が持っている既存または未来の特許を侵害しないという保証はありません。私たちは、すでに発行された特許、および第三者、例えば、私たちが候補製品分野を開発している競争相手、および私たちの候補製品または他の技術の成分、配合、製造方法、または使用または処理方法をカバーするクレームを含む、現在または将来の候補製品または他の技術によって侵害される可能性があると主張する可能性がある。私たちは第三者が持っている特許を知っていますが、これらの特許は私たちの候補製品や他の技術とは関係なく、私たちの候補製品や他の技術によって侵害される可能性もあると思います。さらに、特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、我々の候補製品または他の技術が発行された特許を侵害する可能性がある。
第三者は特許を持っているか、または将来的に特許を取得し、私たちの候補製品または他の技術がこれらの特許を製造、使用または販売していると主張する可能性がある。もし第三者がいたら-一方は私たちが彼らの特許を侵害したと主張したり、私たちが彼らの独自技術を不正に使用したと主張して、私たちに訴訟を提起して、私たちはこのようなクレームに法的根拠がないと思っても、管轄権のある裁判所はそのような特許が有効で強制的に実行可能であり、私たちの候補製品や他の技術によって侵害される可能性があると判断するかもしれない。この場合、これらの特許の所有者は、適用された特許に従って許可されない限り、またはこれらの特許が満期になるまで、または最終的に無効または実行不可能と判定されない限り、私たちが適用可能な候補製品または技術を製造または商業化することを阻止することができるかもしれない。そのような許可は商業的に合理的な条項や根本的に存在しないかもしれない。私たちが許可を得ることができても、許可は私たちに許可料や印税、あるいは両方を支払うことを要求する可能性が高く、私たちに与えられた権利は非排他的である可能性があり、これは私たちの競争相手が同じ知的財産権を得ることをもたらす可能性がある。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができなければ、私たちの候補製品や他の技術を商業化できないかもしれない、あるいはこのような商業化努力が著しく遅延する可能性があり、逆に私たちの業務を深刻に損なう可能性がある。
侵害クレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、私たちの業務の経営陣や他の従業員資源を大量に移転し、私たちの名声に影響を与える可能性がある。私たちに対する侵害クレームが成功した場合、私たちは、私たちの権利侵害候補製品または他の技術のさらなる開発または商業化を禁止される可能性がある。さらに、故意に侵害された3倍の損害賠償および弁護士費を含む大量の損害賠償を支払わなければならない可能性があり、第三者から1つ以上のライセンスを取得し、印税を支払うこと、および/または私たちの侵害製品候補製品または技術を再設計することは不可能であるかもしれないし、多くの時間とお金の支出が必要である可能性がある。この場合、私たちの候補製品や他の技術をさらに開発して商業化することはできません。これは私たちの業務を深刻に損なう可能性があります。
第三者の告発に対抗するために訴訟に参加することは、私たちが彼らの特許や他の知的財産権を侵害したり、他の方法で侵害したりすることに非常に高価であり、特に私たちのような規模の会社には時間がかかる。私たちのいくつかの競争相手は私たちよりも訴訟や行政訴訟の費用を効率的に負担するかもしれない。なぜなら私たちはより多くの財政資源を持っているからだ。特許訴訟と他の訴訟もまた多くの管理時間を取るかもしれない。起動と起動の過程で生じる不確実性
86
引き続き私たちに特許訴訟や他の訴訟を提起することは私たちの市場での競争能力を弱めるかもしれない。上記のいずれの状況の発生も、我々の業務、財務状況、経営業績、成長見通しに重大な悪影響を及ぼす可能性がある。
私たちは私たちの特許と他の知的財産権を保護したり強制したりする訴訟に巻き込まれたり、特許侵害疑惑を弁護したりする可能性があり、これは高価で時間がかかり、成功しないかもしれない。
競争相手は私たちの特許を侵害したり、パートナーの特許を許可したりする可能性があり、あるいは私たちは侵害クレームに対する抗弁を要求されるかもしれない。さらに、私たちの特許や私たちの許可パートナーの特許も在庫や有効性紛争に巻き込まれる可能性があります。このような告発に反撃したり抗弁したりすることは高価で時間がかかるかもしれない。侵害訴訟では,裁判所は我々の利害関係のある特許を無効または強制執行できないと判断することができ,他方の我々の特許技術の使用は“米国法典”第35編271(E)(1)節に規定する特許侵害の安全港に属するか,または我々の特許が関連技術をカバーしていないことを理由に他方の論争のある技術の使用を阻止することを拒否することができる.どんな訴訟手続きの不利な結果も、私たちの1つ以上の特許を無効または狭義に解釈されるリスクに直面させる可能性がある。さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。
解決策が私たちに有利であっても、知的財産権クレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させ、私たちの人員の正常な責任を分散させる可能性があります。また、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。私たちはそのような訴訟や訴訟手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源とより成熟して発展した知的財産権の組み合わせを持っているので、このような訴訟や法的手続きの費用を私たちよりも効率的に負担するかもしれない。特許訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争能力に重大な悪影響を及ぼす可能性がある。
もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は悪影響を受けるかもしれません。
我々の登録または未登録商標または商号は、挑戦、侵害、回避、または汎用商標として宣言されるか、または他の商標が侵害されたと認定される可能性がある。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれません。私たちは私たちが関心のある市場の潜在的なパートナーや顧客の中で知名度を確立するために必要です。時々、競争相手や他の第三者は、私たちと同様の商号や商標を採用して、ブランド表示を確立する能力を阻害し、市場の混乱を招く可能性があります。さらに、他の登録商標または商標の所有者は、我々の登録または未登録商標または商号の変異体を含む商号または商標侵害クレームを提出することができる。長期的には、私たちの商標や商号に基づいて名称を確立することができなければ、効果的に競争できない可能性があり、私たちの業務は悪影響を受ける可能性があります。商標、商業秘密、ドメイン名、著作権、または他の知的財産権に関連する専有権を実行または保護する努力は無効である可能性があり、大量のコストおよび資源移転を招き、私たちの業務、財務状況、運営結果、および成長の見通しに悪影響を及ぼす可能性があります。
知的財産権はすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。例えば:
87
これらの事件のいずれかが発生した場合、私たちの業務、財務状況、運営結果、および成長見通しに実質的な悪影響を及ぼす可能性がある。
私たちの運営に関わるリスクは
私たちは私たちのキーパーソンに強く依存しています。もし私たちが高素質の人員を誘致、激励、維持することに成功できなければ、特に競争の激しい採用と給与環境では、私たちの業務戦略を成功的に実施することができないかもしれません。
競争の激しい生物技術と製薬業界で競争する能力は私たちが高い素質管理、科学と医療人員を誘致、激励し、維持する能力にかかっている。私たちは最高経営責任者アノン·ローゼンタール博士を含む私たちの指導部に強く依存している。幹部、他の重要な従業員、他の科学·医学コンサルタントが提供するサービスを失うこと、およびこのような損失の場合に適切な代替者を見つけることができず、あるいは高級管理者を誘致して空席を埋めることができず、特に競争の激しい採用や賃金環境では、候補製品の開発遅延を招き、私たちの業務を損なう可能性がある。
私たちはカリフォルニア州サンフランシスコ南部の工場で業務を展開しています。この地域は多くの他の生物製薬会社と多くの学術·研究機関の本部所在地です。技術人材に対する競争は非常に激しく、離職率が高い可能性があり、特に最近の競争が激しい求人や賃金環境では、受け入れ可能な条件で高素質の人員を採用し、引き留める能力を制限する可能性がある。私たちは、私たちの地域以外から人材を募集する必要があるかもしれないと予想している。
価値のある従業員をわが社に引き付けるために、賃金と現金インセンティブに加えて、限定的な株式、株式オプション付与、および時間とともに付与された株式奨励を提供していきます。時間が経つにつれて、これらの株式が従業員に付与される価値は、私たちの株価変動の大きな影響を受ける可能性があり、これらの変動は私たちの制御範囲を超えており、いつでも他社が提出したより利益のある見積もりを相殺するのに十分ではないかもしれない。私たちは私たちの重要な従業員と雇用協定を持っていますが、これらの雇用協定は自由に雇用できることを規定しています。これは、私たちのどの従業員も通知するかどうかにかかわらず、いつでも私たちの仕事を離れることができることを意味します。私たちが受け入れ可能な条件で素質の高い従業員を吸引して激励することができない場合、あるいは根本的にできなければ、私たちの業務や運営業績に影響を与える可能性があります。
私たちは私たちの組織の規模と能力を効果的に管理する必要がある。
2022年12月31日までに、273人のフルタイム従業員がいます。私たちの発展計画と戦略の発展に伴い、私たちは管理、運営、財務、そして他の人員を増やす必要があるだろう。私たちは必要になります
88
経営陣のメンバーの重大な責任を通じて、私たちの組織の規模と能力、未来のどんな成長も効果的に管理しています
私たちの将来の財務業績と私たちは私たちの候補製品を開発し続け、承認された後にそれを商業化する能力は、私たちが将来どのような成長を効果的に管理する能力にも部分的に依存するだろう。これらの成長活動を管理するためには、私たちの経営陣はまた、日常活動から過剰な注意を移さなければならないかもしれない。
現在、予測可能な未来に、私たちはいくつかの独立した組織、コンサルタント、およびコンサルタントに大きく依存していくつかのサービスを提供するだろう。これらの独立した組織、コンサルタント、コンサルタントのサービスが必要な時に適時に提供され続けることは保証されず、合格した代替者を見つけることも保証されない。さらに、私たちのアウトソーシング活動を効率的に管理できない場合、あるいはコンサルタントが提供するサービスの品質や正確性が何らかの理由で影響を受ける場合、私たちの臨床試験は延長、延期、または終了される可能性があり、規制部門の候補製品の承認や他の方法で私たちの業務を進めることができないかもしれません。私たちは、既存のコンサルタントを経済的に合理的な条件で管理したり、他の適任な外部請負業者やコンサルタントを見つけることができるという保証はありません。
もし私たちが新入社員を雇用し、私たちのコンサルタントや請負業者チームを拡大することで、私たちの組織を効果的に拡大することができなければ、私たちは私たちの候補製品をさらに開発するために必要な任務を実行することができないかもしれません。私たちの臨床試験は延長、延期、または終了される可能性があり、規制部門の私たちの候補製品の承認を得ることができないかもしれませんので、私たちは私たちの研究、開発、商業化目標を達成できないかもしれません。
私たちは戦略的協力を行っており、将来的に買収、協力、または戦略的協力を行う可能性があり、これは私たちの資本要求を増加させ、私たちの株主を希釈し、債務または負債を発生させ、他のリスクに直面させる可能性がある。
私たちは過去にAbbVie、Innoent、GSKとの戦略協力のような戦略的協力を行ったことがあり、将来私たちは相補的な製品、知的財産権、技術、または業務の許可または獲得を含む様々な買収、協力、戦略協力を行うことができるかもしれない。買収、協力、または戦略的パートナーシップは、多くのリスクをもたらす可能性がある
89
また、このような取引を行うと、希釈証券を発行し、債務を負担または発生させ、巨額の使い捨て費用を発生させ、重大な将来の償却費用につながる可能性のある無形資産を買収することができる。
我々の内部コンピュータシステム、または我々の第三者研究機関の協力者、CROまたは他の請負業者またはコンサルタントによって使用されるシステムは、障害または他の障害、ネットワーク攻撃、または情報セキュリティホールを受ける可能性があり、これらは、そのようなシステムおよびデータの機密性、完全性および可用性を危険にさらし、私たちの開発計画および業務運営が深刻に中断され、機密、財務または独自の情報のリスク開示をもたらし、私たちの名声に影響を与える可能性がある。
セキュリティ対策が実施されているにもかかわらず、我々の内部コンピュータシステムおよび将来のCROおよび他の請負者およびコンサルタントのコンピュータシステムは、コンピュータウイルスおよび不正アクセスによって容易に破壊される可能性がある。ネットワーク脅威構造の変化に伴い、特に私たちのある従業員は新冠肺炎の大流行或いはその後に遠隔或いは混合仕事に従事し、これらの攻撃の頻度、複雑性と強度はすべて増加しており、ますます検出が困難になっている。このような攻撃は、恐喝ソフトウェアまたは他のサービス拒否を含むキーレコーダまたは他の有害および致命的なマルウェアの使用を含むことができ、悪意のあるウェブサイト、社会工学、および/または他の手段を使用して配備することができる。障害、ネットワーク攻撃、あるいは他の情報セキュリティホールが発生し、我々の運営中断を招く場合、私たちの知的財産権や財務情報を含む機密情報が流用され、我々の開発計画や業務運営に実質的な中断をもたらす可能性があります。例えば、完了した、行われている、または将来の臨床試験における臨床試験データの損失は、私たちの規制承認作業の遅延を招き、データを回復または複製するコストを著しく増加させる可能性がある。同様に,我々は第三者研究機関協力者とCROに依存して我々の候補製品を研究·開発し,他の第三者に依存して我々の候補製品を製造し臨床試験を行うが,彼らのコンピュータシステムに関連する類似イベントも我々の業務に大きな悪影響を与える可能性がある.任意の中断またはセキュリティホールは、私たちのデータまたはシステムの損失または破損をもたらし、または私たちの関係者に関連するデータを含む機密、財務、または独自の情報を不適切に開示します, 私たちは機密、財務、または独自の情報の責任またはリスク開示を招く可能性があり、私たちの候補製品のさらなる開発と商業化は延期される可能性がある。当社および当社のビジネス相手側が、システムまたはデータを正常に検出、防止または完全に回復し、当社の業務および運営に悪影響を及ぼす可能性があり、および/またはキーまたは敏感なデータ損失をもたらす可能性のあるすべての障害、サービス中断、攻撃、またはシステム脆弱性を回避することは保証されません。これは、財務、法律、業務、または名声をもたらす可能性があります。
世界的な疫病の結果を含む業務中断は、私たちの将来の収入や財務状況を深刻に損なう可能性があり、私たちのコストと支出を増加させる可能性がある。
私たちの業務、および私たちの協力者、CRO、CDMO、サプライヤー、および他の請負業者およびコンサルタントの業務は、病気の伝播、地震、電力不足、電気通信障害、水不足、洪水、ハリケーン、台風、火災、極端な気象条件、医療流行病、政治的不安など、大流行事件および他の私たちがコントロールできない事件の影響を受ける可能性があり、私たちはこれらの事件のために保険をかけていないか、または完全にまたは部分的に保険をかけていません。また,我々は我々の第三者研究機関協力者に依存して我々の候補製品を研究·開発し,政府の閉鎖や資金撤回の影響を受ける可能性がある.このような業務中断の発生は、私たちの運営と財務状況を深刻に損害し、私たちのコストと支出を増加させる可能性がある。私たちは私たちの候補製品を生産して加工するために第三者メーカーに依存する。もしこれらのサプライヤーの運営が人為的あるいは自然災害、全世界流行病、あるいは他の業務中断の影響を受けた場合、候補製品の臨床供給を獲得する能力は中断される可能性がある。
90
私たちの業務の大部分は、私たちの会社の本社を含めて、カリフォルニア州サンフランシスコ南部の施設にあります。火災、自然災害、世界的な流行病、停電、通信障害、許可されていない進入、地震、または他の事件により、会社、開発または研究施設の破損または長時間の中断は、候補製品の一部またはすべての開発を停止または延期させる可能性があります。これらの施設に対して財産損失と業務中断保険を維持していますが、この場合、私たちの保険はすべての損失をカバーできない可能性があり、私たちの業務はこのような遅延と中断によって深刻な損害を受ける可能性があります。
私たちの業務は国際業務に関連する経済、政治、規制、その他のリスクの影響を受けている。
私たちの業務は国際業務の展開に関連するリスクの影響を受けています。例えば私たちのCDMOや臨床試験場所はアメリカ以外にありますしたがって、私たちの将来の業績は様々な要素の影響を受けるかもしれない
私たちが計画している国際業務に関連するこれらのリスクや他のリスクは、私たちの収益業務を実現する能力に大きな悪影響を及ぼす可能性があります。
私たちの純営業損失の繰越と他の税務属性を使用する能力は限られているかもしれません。
2022年12月31日現在、我々の連邦と州の純営業損失(NOL)の繰越はそれぞれ約4030万ドルと2.035億ドルだった。連邦NOL繰越無期限では、控除額は課税収入の80%を超えてはならない。米国国税法第382条と383条によると、所有権が3年以内に累計変動して50%を超えた場合、会社の純営業損失と研究開発信用繰越の年間使用が制限される可能性がある。私たちが初めて公開した結果として
91
2019年2月の初公募株と2020年1月の後続公募株、および我々の設立以来発生した他の取引は、このような所有権変更を経験しているかもしれません。私たちはまた、私たちの株式所有権がその後変化し、その中のいくつかは私たちがコントロールできないので、未来に所有権の変化を経験するかもしれない。そのため,変動前純営業損失繰越やその他の変動前税項属性を利用して変動後の課税収入や税項を相殺する能力が制限される可能性がある。また、2020年にコロナウイルス援助、救済、経済安全法案(CARE法案)が改正された通常の2017年減税·雇用法案(TCJA)と呼ばれる立法では、2017年12月31日以降に開始される納税年度のNOLは、2022年12月31日以降の課税年度では、現在の課税所得額の80%を超えないことが毎年相殺されることが規定されている。私たちのNOLはまた州法によって制限されるかもしれない。
税法またはその実施または解釈の変化は、私たちの業務および財務状況に悪影響を及ぼす可能性がある。
連邦、州と地方司法管轄区と私たちが業務を展開しているいくつかの外国司法管轄区によると、私たちはアメリカで所得税と非所得税を納めているかもしれません。これらの管轄区域の税務法律、法規、行政慣行は、事前に通知されたり、事前に通知されない場合に大きな変化が生じる可能性がある。例えば、2022年1月1日、TCJAの条項が発効し、この条項は、発生年に国内研究開発コストを差し引くオプションを廃止し、納税者に5年以内に国内コストのこのようなコストを償却し、外国コストのために15年以内にこのようなコストを償却することを要求している。その会社は現在潜在的な影響を評価している。また、米国は最近、帳簿収入に15%の最低税率を徴収し、株式買い戻しに1%の消費税を徴収する“インフレ低減法案”を公布した。税法(インフレ低減法案の規定を含む)、法規または裁決の変化、既存の法律法規解釈の変化または会計原則の変化は、私たちの財務状況、キャッシュフローおよび経営結果にマイナスまたは実質的な影響を与える可能性がある。
一般リスク因子
私たちの普通株の市場価格は引き続き変動する可能性があり、これは投資家に大きな損失をもたらす可能性がある。
私たちの普通株はナスダック世界ベスト市場に上場していますが、私たちの株式市場はある程度の取引が活発です。私たちの普通株の取引価格はずっと高度に変動し続ける可能性があり、様々な要素の広範な変動を受ける可能性があり、その中のいくつかの要素は私たちがコントロールできない。私たちは私たちの普通株の取引価格を予測できない。今後の1つまたは複数の時期に、私たちの運営結果や私たちの製品ラインの進展が公開市場アナリストや投資家の予想に合わない可能性があるため、これらや他の要因により、私たちの普通株価格が低下する可能性がある。普通株市場の価格変動や下落を引き起こす可能性があるいくつかの要因は
92
近年、株式市場全体、特に製薬やバイオテクノロジー会社の市場は、重大な価格や出来高変動を経験しており、これらの変動は、その株式がこれらの価格や出来高変動を経験した会社の経営業績の変化に関係なく、あるいは比例しないことが多い。インフレへの懸念のような広範な市場や業界要素は、私たちの実際の経営業績にかかわらず、私たちの普通株の市場価格に深刻な影響を与える可能性がある。ある会社の証券市場価格にこのような変動が生じた後、同社に対して証券集団訴訟が提起されることが多い。私たちの株価の潜在的な変動性のため、私たちは未来に証券訴訟の目標になるかもしれない。証券訴訟は巨額のコストを招き、経営陣の注意と資源を私たちの業務から移す可能性がある。
もし証券アナリストが私たちの業務に関する研究や報告を発表しなければ、あるいは彼らが私たちの株に対するマイナス評価を発表したら、私たちの株価は下落するかもしれません。
私たちの普通株の取引市場は、業界や金融アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存している。もし私たちの業務を担当する一人以上のアナリストが私たちの株の追跡を停止したり、彼らの私たちの株の評価を下げたり、あるいは私たちの経営業績に対する彼らの推定を達成できなければ、私たちの株価は下落するかもしれません。もしこのようなアナリストの一人以上が私たちの株を追跡しなければ、私たちは市場での私たちの株の可視性を失うかもしれないし、これは逆に私たちの株価を下落させるかもしれない。
公開市場で私たちの普通株を大量に販売するか、あるいはこのような売却が発生する可能性があると考えられ、たとえ私たちの業務が良好であっても、私たちの普通株の市場価格が大幅に低下する可能性がある。
公開市場で私たちの大量の普通株を売ることはいつでも起こる可能性がある。もし私たちの株主が公開市場で私たちの普通株を売却したり、市場が私たちの株主が大量の普通株を売却しようとしていると思ったら、私たちの普通株の市場価格は大幅に低下する可能性があります。
条件によると、私たち普通株のいくつかの株主は、彼らの株式に関する登録声明を提出することを要求する権利があり、または彼らの株を私たち自身または他の株主のために提出する可能性のある登録声明に含める権利がある。証券法によるこれらの株の登録は、これらの株を公開市場で自由に取引できるようになるが、私たちの関連会社は第144条の制限を受けている。これらの株主のどの証券売却も私たちの普通株の市場価格に実質的な悪影響を及ぼす可能性がある。
追加資本の調達は、私たちの既存の株主を希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません。
私たちは公共およびプライベート·エクイティの発行、債務融資、戦略的パートナーシップと連合、および許可手配の組み合わせによって追加資本を求めるかもしれない。私たちや間接的に株主が負担します
93
このような証券の発行とサービスコスト。なぜなら私たちが未来のどの発行でも債券や株を発行する決定は市場状況と私たち以外の要素にかかっているからだ 制御は、私たちは未来に発行された金額、時間、または性質を予測したり、推定することができない。私たちが株式または債務証券を売却することによって追加資本を調達する場合、あなたの所有権資本は希釈され、条項は清算または株主としての権利に悪影響を及ぼす他の特典を含む可能性があります。債務の発生は、固定支払義務の増加を招き、例えば、追加債務を発生させる能力の制限、知的財産権を得ることができるかもしれない能力の制限、および業務を展開する能力に悪影響を及ぼす可能性のある他の運営制限など、限定的な契約に関連する可能性がある。我々は、表S-3の形式で米国証券取引委員会に総合的な棚登録声明を提出する予定であり、この声明が発効した後、4億ドルまでの普通株式、他の株式証券および/または債務証券の発行を許可する。さらに、私たちが第三者と達成した任意の未来の協力は短期的に資金を提供する可能性があるが、私たちの未来の潜在的なキャッシュフローと収入を制限するだろう。もし私たちが戦略的パートナー関係と連合と第三者との許可手配を通じてより多くの資金を調達すれば、私たちは私たちの技術や候補製品に対する貴重な権利を放棄したり、私たちに不利な条項で許可を与えなければならないかもしれない。
私たちの主要株主と経営陣は私たちのかなりの割合の株を持っており、株主の承認が待たれる事項に大きな影響を与えることができるだろう。
2023年2月21日現在、我々の役員、役員、5%を超える流通株保有者と、それぞれの関連会社実益は、私たちが発行した普通株の50.2%を持っています。したがって、これらの株主が一緒に行動すれば、取締役選挙や重大な会社取引の承認を含む株主の承認を必要とするすべての事項に大きな影響を与える可能性がある。このような所有権集中はわが社の支配権の変更を遅延または阻止する可能性があり、私たちの他の株主はこれが彼らの最適な利益だと思うかもしれない。これは逆に私たちの株価に実質的な悪影響を及ぼす可能性があり、私たちの株主が取締役会や経営陣を交代または罷免しようとすることを阻止する可能性がある。
上場企業としては、著しい追加コストを発生させ続けており、我々の経営陣は、新たなコンプライアンスやコーポレートガバナンスを実施するために多くの時間を投入する必要があるだろう。
上場企業として、私たちはすでに大量の法律、会計、その他の費用を発生させ続けており、これらの費用は私たちが民間会社として発生していない。サバンズ-オクスリ法案、ドッド-フランクウォール街改革法案と消費者保護法、ナスダックの上場要求、その他の適用される証券規則と法規は、有効な開示、財務制御、会社管理の確立と維持を含む上場企業に対して様々な要求を提出した。私たちはすでに招聘して、私たちが上場企業になることに関連する追加会計、財務、その他の人員を引き続き採用する予定で、私たちは上場企業の要求を守るために努力して、私たちの経営陣と他の人員はすでにこれらの要求を遵守するために多くの時間を投入し続けます。このような要求は増加し、私たちの法律と財務コンプライアンスコストを増加させ続け、いくつかの活動をより時間的で高価にするだろう。例えば、上場企業としての私たちの規制に適用されることで、取締役や上級管理職責任保険を維持することがより難しくなり、高価になる可能性があり、合格した取締役会のメンバーを引き付けることが難しくなる可能性があります。私たちは現在、私たちが発生する可能性のある追加コスト金額やそのようなコストの時間を予測または推定することができないこれらの規則と規則を評価している。これらの規則と条例はよく異なる解釈を受けます。多くの場合、特殊性が不足しているので、, 規制や理事機関が新たな指導を提供するにつれて、それらの実践への応用は時間の経過とともに変化する可能性がある。これは遵守事項に関する持続的な不確実性と、開示と統治慣行を絶えず修正するために必要なより高いコストをもたらす可能性がある。
もし私たちが効果的な内部統制を維持できなければ、私たちの業務、財務状況、そして経営結果は不利な影響を受けるかもしれない。
上場企業としては、経営陣に財務報告の内部統制の有効性を年間評価することを要求する“取引法”に規定されている報告書その他の義務を遵守しなければならない。サバンズ-オキシリー法404(B)条はまた、私たちの独立監査人にこの管理評価を証明して報告することを要求する。
94
管理管理層は、財務報告の内部統制に達成しなければならない基準を評価するルールは複雑であり、規則下の詳細な基準を満たすためには、大量の文書、テスト、および可能な救済措置が必要である。テスト過程で、私たちの経営陣は重大な弱点や欠陥を発見する可能性があり、これらの欠陥や欠陥は適時に修復できない可能性があり、2002年の“サバンズ-オックススリー法案”で規定された期限内に完成できない可能性がある。これらの報告とその他の義務は、会計資源を含む我々の管理、行政と業務資源に対して、極めて大きな要求を提出した。もし私たちが第404条の要求を遵守できない場合、あるいは私たちまたは私たちの独立公認会計士事務所が財務報告の内部統制に対する私たちの有効性を証明できない場合、投資家は私たちの財務報告の正確性と完全性に自信を失う可能性があり、私たちの株式の市場価格は下落する可能性があり、私たちはナスダック、米国証券取引委員会、または他の規制機関の制裁または調査を受ける可能性があり、これには追加の財務および管理資源が必要になるだろう。
私たちの業務はインフレ率上昇の影響を受けている。
アメリカは最近過去最高のインフレ水準を経験した。米労働統計局のデータによると、2022年12月31日までの12カ月間、米国消費者価格指数の年間上昇幅は約6.5%だった。インフレ率が引き続き上昇すれば、従業員の給与と研究開発費のような私たちの支出に影響を及ぼすだろう。研究開発費は私たちの運営費用の大きな部分を占めている。私たちが候補製品を市場に投入している間、このような増加した費用は簡単に回収できないかもしれない。また、米国は深刻な労働力不足を経験しており、これは逆に非常に競争力のある賃金環境を創出し、会社の運営コストを増加させる可能性がある。インフレにより金利が上昇し、市場に他の悪影響を及ぼす場合、それは私たちの総合的な財務状況や経営業績に悪影響を及ぼす可能性がある。
私たちは予測可能な未来に何の配当もないと予想している。投資家たちは決して彼らの投資から見返りを得ることができないかもしれない。
あなたは私たちの普通株への投資に依存して配当収入を提供してはいけない。私たちは予測可能な未来に、私たちは普通株式保有者に何の配当も支払わないと予想している。代わりに、私たちは私たちの既存の業務を維持して拡大するためにどんな収益も維持する予定だ。さらに、任意の将来の信用スケジュールには、私たちの普通株が発表または支払い可能な配当金の金額を禁止または制限する条項が含まれている可能性がある。そのため、投資家は、投資リターンを実現する唯一の方法として、価格上昇後に普通株を売却することに依存しなければならない。したがって、現金配当金を求める投資家は私たちの普通株を購入してはいけない。
デラウェア州の法律と私たちが改正して再説明した会社の登録証明書と定款の条項は、わが社の支配権の変更や私たちの経営陣の変更を阻止、延期、阻止し、それによって私たちの普通株の取引価格を下げる可能性があります。
私たちが改正して再記述した会社の登録証明書および定款の条項は、株主が有利と思うかもしれない合併、買収、または他の支配権の変化を阻止、延期、または阻止する可能性があり、私たちの普通株を保有することによって割増取引を得ることができる可能性があります。このような規定はまた私たちの株主が私たちの経営陣を交代または更迭しようとしていることを阻止したり挫折させたりする可能性がある。したがって、このような規定は私たちの普通株の価格に悪影響を及ぼすかもしれない。他の点では私たちの憲章文書:
95
また、デラウェア州一般会社法(DGCL)第203条は、取引日後3年以内にデラウェア州上場企業が利害関係のある株主(通常、その関連会社と所有しているか、過去3年以内に議決権株を有する者)との商業合併を禁止しており、当該商業合併が所定の方法で承認されない限り、商業合併を行うことを禁止している。
当社の会社登録証明書の改訂と再記載、改訂と再記載の会社定款やデラウェア州法律の遅延または制御権の変更を防止する条項は、私たちの株主が彼らが保有する私たちの株式の株式から割増の機会を得ることを制限する可能性があり、一部の投資家が私たちの普通株に支払う価格に影響を与える可能性もあります。
私たちの改正と再記述の定款は、デラウェア州衡平裁判所は、私たちと株主との間のほとんどの紛争の独占法廷であり、これは、私たちの株主と私たちまたは私たちの役員、役員、または従業員との紛争が有利な司法フォーラムを得る能力を制限するかもしれない。
我々の改正及び重述の別例規定は、デラウェア州衡平裁判所(又は、衡平裁判所に管轄権がない場合、デラウェア州の他の州裁判所又はデラウェア州地域の連邦地方裁判所)は、以下の事件の専属裁判所(当該裁判所の管轄権の管轄を受けない不可欠な方が存在すると認定されている(かつ必要不可欠な一方が当該裁決を下してから10日以内に当該裁判所の属人管轄権に同意しない)のいずれかのクレームを除く)、当該裁判所以外の裁判所又は裁判所の排他的管轄権に属する、又は当該裁判所が当該裁判所に対して標的管轄権を有さない事件を除く
この規定は、取引法で規定されている義務や責任を執行するための訴訟にも適用されず、米国連邦裁判所が排他的管轄権を有する他のクレームにも適用されない。
私たちが改正して再記述した付例は、米国連邦地域裁判所が証券法に基づいて提出された任意の訴因を解決するための独占的なフォーラムになるとさらに規定している。これらの排他的フォーラム条項は、司法フォーラムにおいて、私たちまたは私たちの役員、役員、または他の従業員との紛争に有利であると考える株主のクレームを提出する能力を制限する可能性があり、これは、私たちと私たちの役員、役員、および他の従業員に対する訴訟を阻止するかもしれない。任意の個人またはエンティティが、私たちの任意の証券の任意の権益を購入または他の方法で取得することは、これらの規定に了承され、同意されたとみなされるべきである。裁判所がこれらの規定を強制執行するかどうかには不確実性があり,他の定款文書では選択のような裁判所条項の実行可能性が法的手続きで疑問視されている。裁判所はこれらのタイプの条項が適用されないか、または実行できないことを発見するかもしれません。もし裁判所が私たちの改正と再述の定款における排他的な裁判所条項が訴訟で適用されないか、または実行できないことを発見すれば、私たちは
96
他の管轄区域での紛争解決に関連した追加コストが生じる可能性があり、これは私たちの業務を深刻に損なう可能性がある。
項目1 B。未解決従業員コメント。
ない。
プロジェクト2.ニュースオペラです。
私たちの会社の本社は現在カリフォルニア州サンフランシスコ南部にあり、そこで約105,000平方フィートのオフィスと実験室空間を借りました。レンタル契約の期限は2029年5月に満了し、レンタル期間をさらに10年延長する権利がある。賃貸契約はまた、ビル内の利用可能なオフィススペースに拡張できる優先購入権を提供してくれます。私たちは2021年11月に約7132平方フィートの会社本社を転貸し、レンタル期間は2022年12月に満期になりました。また、2022年5月に約13,150平方フィートの会社本社を分譲し、レンタル期間は2023年11月に満期になります。2023年2月に約9,275平方フィートの会社本社を転貸し、レンタル期間は2025年1月に満期になります。カリフォルニア州のニューアークで約18,700平方フィートの追加オフィスと実験室空間を借りました。私たちはこのような施設が私たちの最近の必要性を満たすのに十分だと信じている。必要があれば、私たちは後日商業的に合理的な条項で、適切な追加または別の用地を提供すると信じています。
項目3.法律法律手続き。
時々、私たちは訴訟や他の法的手続きに巻き込まれるかもしれない。私たちは現在、私たちの経営陣が私たちの業務に重大な悪影響を及ぼす可能性があると考えている訴訟や法的手続きに参加していません。結果にかかわらず、訴訟や他の法的手続きは、法的費用および和解費用、管理資源の移転などの要因によって私たちに悪影響を及ぼす可能性がある。
プロジェクト4.地雷安全安全に開示する。
適用されません。
97
第II部
項目5.登録者普通株·関連株の市場保有者は重要であり,発行者は株式証券を購入する.
普通株式市場情報
我々の普通株はナスダック全世界の精選市場で公開取引され、コードは“ALEC”である
記録保持者
2023年2月21日現在、私たちの普通株には約7人の登録株主がいます。実際の株主数はこの記録保持者の人数を超えており、受益者である株主を含むが、その株式は仲介人や他の被命名者が街頭名義で保有している。
配当政策
設立以来、私たちはどんな現金配当金も発表したり支払ったりしたことがない。私たちは将来の収益(あれば)を残し、私たちの業務の運営と拡張に資金を提供し、予測可能な未来には何の現金配当金も支払わないつもりだ。将来的に現金配当金を派遣することは、私たちの取締役会が様々な要素を考慮して適宜決定し、これらの要素は私たちの財務状況、経営業績、現在と予想される現金需要、当時の既存債務ツールの要求と契約制限、そして私たちの取締役会が関連すると思う他の要素を含む。
株式表現グラフ
取引法第18節の場合、本グラフは“資料募集”ではなく、米国証券取引委員会“保存”ともみなされず、同節に規定された他の責任の制約も受けず、参照によってAlectorに組み込まれたものと見なすべきではなく、Inc.1933年証券法(“証券法”)に基づいて提出されたいかなる文書であっても、当該文書が本文書の日付の前または後になされたものであっても、どのような文書において使用される一般的な合併言語であってもかまわない。
次の図はナスダック総合指数とナスダック生物技術指数に対する私たちの普通株の累積株主リターンの累積総リターンを比較した。2019年2月7日(私たちの普通株取引の初日)に私たちの普通株と各指数に100ドルの投資が行われたと仮定し、2022年12月31日までその相対的な表現を追跡した。米国証券取引委員会が適用する規則によると、すべての価値はすべての配当金に全額再投資すると仮定しているが、これまで私たちの普通株は配当を発表していない。以下のグラフに示す株主リターンは歴史的結果に基づいており、必ずしも未来の業績を代表するとは限らず、将来の株主リターンについては何の予測も認めません。
98
発行者および関連購入者が株式証券を購入する
ない。
第六項です。 [保留されている].
項目7.経営陣の以下の問題の議論と分析財務状況と経営実績。
以下の財務状況と経営結果の検討と分析、および当社の連結財務諸表と本年度報告書の他の場所に関する付記(Form 10-K)を読むべきです。本議論は、“前向きな陳述に関する特別な説明”の節に記載された説明を含む、リスクおよび不確実性に関連する前向きな陳述を含む。我々の実際の結果と選定活動の時間は以下の議論と大きく異なる可能性がある.このような差異をもたらすか、または促進する可能性のある要因には、以下の決定された要因および本報告の他の部分“リスク要因”の部分に列挙された要因が含まれるが、これらに限定されない。
概要
著者らは臨床段階の生物製薬会社であり、免疫神経学の先駆者であり、これは神経変性を治療する新しい治療法である。免疫神経学的目標は免疫機能障害を多様な病理の根本的な原因とすることであり,これらの病理は退行性脳疾患の駆動因子である。我々は,脳の健康な免疫機能を回復させることで,これらの病理変化に同時に対抗する治療法を開発している。我々の研究と薬物発見プラットフォームは我々の科学的方法を支持し,人類遺伝学的検証を経た広範な候補製品の組合せを進めることができ,より短い開発時間で技術成功の可能性を高めると信じている。そこで私たちは100個以上の免疫系標的を決定しました3つの候補製品、latozinemab(AL 001とも呼ばれる)、AL 002、およびAL 101は臨床開発中であり、AL 009およびAL 008を含む我々の研究パイプラインの開発を継続している。私たちは最近、PROGROGINとTREM 2計画に開発資源を集中させ、私たちの現金滑走路を2025年まで延長する優先順位審査を完了しました。私たちは私たちの既存の資源を利用して、私たちのパートナーであるGSKとAbbVieと協力して、私たちの臨床候補製品と研究パイプラインを推進している。
私たちの運営資金は、主にエバービーとグラクソ·スミスクラインとの協力と、初公募株(IPO)と後続発行完了後に転換可能な優先株と普通株の発行と販売に由来している。私たちは2019年2月にIPOを完了し、引受割引と手数料および発行費用を差し引いた純収益は1.682億ドルでした。私たちは1つ完成しました
99
引受割引と手数料および発行費用を差し引いた後,2020年1月に後続発行を行い,純収益は2億245億ドルであった。
今まで、私たちは何の製品も販売を許可されていませんし、製品販売から何の収入も得ていません。また、候補製品の1つの開発を成功させ、市場承認を得ることができるまで、製品販売から収入を得ることはないと予想される。私たちは引き続き私たちの候補製品を開発し、予測可能な未来に運営に資金を提供するために追加の資金を必要とするだろう。成立以来、私たちは毎年純損失を出しており、予測可能な未来にも純損失が続くことが予想される。私たちが製品収入を作る能力は私たちの1つ以上の候補製品の成功と最終商業化にかかっているだろう。2022年、2021年、2020年12月31日までの年度の純損失はそれぞれ1.333億ドル、3630万ドル、1.902億ドルだった。2022年12月31日までの累計赤字は5兆797億ドル。我々のほとんどの純損失は,我々の研究や開発計画に関するコストと我々の運営に関する一般的かつ行政的コストによるものである.私たちは持続的な活動に関連する費用が大幅に増加すると予想しています
経営成果の構成部分
収入.収入
私たちはまだ製品販売から何の収入も得ておらず、近い将来もそうしない予定です。これまで,我々の収入は主にAbbVieプロトコルやGSKプロトコルに関連しており,候補製品の許可や共同開発に用いられてきた.サービスの提供に伴い、時間が経つにつれて、私たちはAbbVieの前払い収入を確認した。グラクソ·スミスクラインが開発許可証を取得した時点と、時間の経過とともに研究開発サービスのための前金の収入を確認した。研究·開発サービスの収入は計画コストとして確認され,これまでに発生した実コストと業績義務履行に要する総期待コストの比を測ることで決定された。
私たちAbbVie協定の条項によると、AbbVieから前金を得ることに加えて、開発と規制マイルストーン支払い、AL 002概念検証後に開発を継続する選択加入支払い、およびこのような計画候補製品の商業化後に利益共有または印税によって支払われる他の未来支払いを得る権利がある可能性がある。
私たちのGSK協定の条項によると、私たちは7億ドルの前金を受け取りました。そのうち5億ドルは2021年8月に受け取り、2億ドルは2022年1月に受け取りました。さらに、LatozinemabとAL 101の臨床開発、規制、商業発売に関連するマイルストーン支払いを取得する資格があり、最高15億ドルを追加することができます。AlectorとGSKはLatozinemabとAL 101を共同開発している。
米国では,AlectorとGSKはLatozinemabとAL 101の商業化の利益と損失を二等分する。私たちは製品ごとに開発コストと米国で商業化された損益を分担しないことを選択することができる。この場合、製品の開発や商業化は行われなくなり、利益の一部ではなく、米国での純売上高の印税を得ることになる。米国以外では,グラクソ·スミスクラインはすべての適応を担当するLatozinemabとAL 101の商業化を担当し,2桁の階層印税を得る資格がある。
私たちは今後数年間私たちの収入が主にエバービーとグラクソ·スミスクラインの合意から来ると予想している。2022年12月31日現在、AbbVieとGSKプロトコルに関する繰延収入残高は4.916億ドルである。繰延収入は,AL 002の概念検証とラトジンモノクロナル抗体とAL 101の特定適応の初期第二段階臨床試験により,計画の研究と開発期間中に確認される予定である。
100
研究と開発費
研究開発費は私たちの運営費用の大きな部分を占めている。私たちは研究と開発費用を発生した費用として記録した。研究開発費には、私たちの候補製品の発見と開発によるコストが含まれています
私たちは研究開発費が発生している間にすべての研究開発費を支出する。いくつかの開発活動のコストは,我々のプロバイダ,パートナー,および第三者サービスプロバイダが提供してくれた情報やデータを用いて特定のタスクを達成する進捗を評価することによって確認される.将来の間に受信される研究および開発活動のための貨物またはサービスの払戻不可能な前払いは延期され、資本化される。資本化された金額はその後、関連貨物の交付とサービス提供時に費用を計上する。
具体的な計画費用には,我々の最先端の候補製品の開発に関する費用が含まれている:Latozinemabは重要な第3段階臨床試験を行っており,FINRONT−3は第2段階臨床試験を行っており,AL 002は第2段階臨床試験を行っており,AL 101は第1段階臨床試験を完了している。将来の候補製品の発見や開発に関する費用と,臨床前研究から第一段階の臨床試験に入る予定の項目に関する単独追跡費用もある。これらの費用は,主に賃金と福祉,株による給与,減価償却を含む施設費用,実験室消耗品に用いられる。
我々がパートナーとコストを分担する場合,たとえば我々のGSKプロトコルでは,研究開発費には我々パートナーの費用分担精算や支払いが含まれている可能性がある.
現在、私たちは、私たちの任意の候補製品の開発を完了し、規制部門の承認を得るために必要な努力の性質、時間、見積もりコストを合理的に推定または知ることができない。予測可能な未来には,我々の候補製品の開発に関する研究·開発活動に投資し続けるとともに,我々の候補製品が開発の後期段階に入るにつれて,我々がより大規模な臨床試験を開始するにつれて,規制機関に臨床試験に成功した候補製品の承認を求めるにつれて,研究開発費が大幅に増加し,より多くの人員の募集に伴う研究開発作業を支援する費用が生じることが予想される。必要な臨床研究を行い、監督部門の許可を得る過程は高価で時間がかかり、著者らの候補製品の開発成功も非常に不確定である。
一般と行政費用
一般および行政費用は、主に株式ベースの報酬を含む行政、法律、財務と会計、情報技術、人的資源、その他の行政機能者の人事関連費用を含む。一般及び行政費用には、知的財産権及び会社事務に関する法律費用、会計、監査、相談及び税務サービスのために支払われる専門費用、保険料、並びに研究及び開発費用に含まれていない施設コストも含まれる。
101
その他の収入、純額
その他の収入には、純額には、我々の現金等価物と有価証券が稼いだ利息、および期間内に発生した外国為替取引収益と損失が含まれる。
経営成果
2022年と2021年12月31日終了年度比較
|
|
現在までの年度 |
|
|
ドル |
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
協力収入 |
|
$ |
133,617 |
|
|
$ |
207,085 |
|
|
$ |
(73,468 |
) |
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
210,418 |
|
|
|
189,407 |
|
|
|
21,011 |
|
一般と行政 |
|
|
61,033 |
|
|
|
55,038 |
|
|
|
5,995 |
|
総運営費 |
|
|
271,451 |
|
|
|
244,445 |
|
|
|
27,006 |
|
運営損失 |
|
|
(137,834 |
) |
|
|
(37,360 |
) |
|
|
(100,474 |
) |
その他の収入、純額 |
|
|
7,778 |
|
|
|
1,031 |
|
|
|
6,747 |
|
所得税前損失 |
|
|
(130,056 |
) |
|
|
(36,329 |
) |
|
|
(93,727 |
) |
所得税費用 |
|
|
3,254 |
|
|
|
— |
|
|
|
3,254 |
|
純損失 |
|
$ |
(133,310 |
) |
|
$ |
(36,329 |
) |
|
$ |
(96,981 |
) |
収入.収入
2022年12月31日までの事業年度は1兆336億ドルであったが、2021年12月31日現在の事業年度は2.071億ドルであった。7,340万ドルの減少は主にLatozinemab FTDで確認された1.734億ドルの協調収入によるものである-GRN2021年にGSKプロトコルの一部として提供されるライセンス。これはAbbVieプロトコルでの協調収入の純増加5580万ドルによって相殺され,これはAL 003計画の終了により義務履行の推定コストが変化したが,AL 002計画が期待する総コストの増加分がこの増加を相殺したためである.この低下は,グラクソ·スミスクラインが最初の第2段階臨床試験で確認した4420万ドルの収入増加によっても相殺された。
研究と開発費
2022年12月31日までの1年間の研究開発費は2.104億ドルだったが、2021年12月31日までの1年間の研究開発費は1兆894億ドルだった。2100万ドルの増加は、従業員数の増加と従業員への株式贈与により、株式報酬を含む1750万ドルの支出が増加したためである。また,AL 002の臨床試験のさらなる進展に伴い,われわれのAL 002は890万ドル増加し,施設やその他の未分配研究開発費は590万ドル増加し,追加の賃貸料支出,従業員数,その他の間接コストが増加した。これは主にグラクソ·スミスクラインとの費用分担協定によるlatozinemabの960万ドルの減少によって相殺される。
102
|
|
現在までの年度 |
|
|
ドル |
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
直接研究開発費 |
|
|
|
|
|
|
|
|
|
|||
Latozinemab |
|
$ |
22,118 |
|
|
$ |
31,689 |
|
|
$ |
(9,571 |
) |
AL 101 |
|
|
7,751 |
|
|
|
6,728 |
|
|
|
1,023 |
|
AL 002 |
|
|
34,805 |
|
|
|
25,941 |
|
|
|
8,864 |
|
AL 044 |
|
|
7,546 |
|
|
|
9,935 |
|
|
|
(2,389 |
) |
他の初期プロジェクトは |
|
|
37,385 |
|
|
|
37,740 |
|
|
|
(355 |
) |
間接研究開発費 |
|
|
|
|
|
|
|
|
|
|||
関係者(株ベースの |
|
|
76,063 |
|
|
|
58,519 |
|
|
|
17,544 |
|
施設や他の未分配の研究と |
|
|
24,750 |
|
|
|
18,855 |
|
|
|
5,895 |
|
研究開発費総額 |
|
$ |
210,418 |
|
|
$ |
189,407 |
|
|
$ |
21,011 |
|
一般と行政費用
2022年12月31日までの年間の一般·行政費は6100万ドルであるのに対し、2021年12月31日までの年間は5500万ドルである。600万ドルの増加は、従業員数の増加と従業員への株式贈与による従業員関連支出の490万ドルの増加であり、株式報酬を含む。私たちはまた110万ドルを増加させ、主に情報技術と関連して、私たちの成長を支持する。
その他の収入、純額
2022年12月31日までの1年間の他の収入純額は780万ドルだったが、2021年12月31日までの年間は100万ドルだった。670万ドルの増加は、私たちの有価証券の投資収益が前年より高いためです。
所得税費用
所得税支出は2022年12月31日までの年間330万ドルであるが、2021年12月31日までの年間はゼロである。TCJAは2022年1月1日から、米国国税法174条に基づく即時控除ではなく、研究開発費の資本化と償却を納税者に要求した。330万ドル増加した主な理由は、GSK協定から受け取った現金に対するこの新しい法律の影響だ。
流動性と資本資源
会社設立から2022年12月31日まで、私たちの運営資金は主にエバービーとグラクソ·スミスクラインとの協力と、初公募株と後続発行が完了した後、転換可能な優先株と普通株の発行と販売に由来している。
2022年12月31日現在、私たちは7億129億ドルの現金、現金等価物、有価証券を持っている。2022年12月31日までの累計赤字は5兆797億ドル。
将来の資金需要
私たちの現金の主な用途は私たちの運営に資金を提供することで、主に私たちの計画に関する研究と開発支出、次いで一般と行政支出です。私たちが行っている活動に関連する費用は引き続き増加し、特に私たちが引き続き私たちの候補製品と発見計画を推進することに伴い、私たちは引き続き増加すると予想される。また、上場企業の運営に関する追加コストも発生すると予想されています。
私たちの現在の運営計画によると、私たちは既存の現金、現金等価物、有価証券が2025年までの運営と資本支出需要に資金を提供できると信じている。私たちはすでに
103
このような誤った仮説であることが証明される可能性のある推定に基づいて、私たちは現在予想されているよりも早く私たちが利用できる資本資源を利用することができる。私たちはまた追加的な資金調達を日和見的に求めることができる。私たちは、株式または債務融資、許可協定、他社との協力協定または他の手配、資産売却、または他の融資源を公開することで資金を調達することを求めることができる。私たちは未来に私たちの研究開発活動と持続的な運営のために多くの追加資金を得る必要があると予想する。もし私たちが必要な時や有利な条件下で資金を調達できなければ、私たちは私たちの研究開発計画や将来の商業化努力を延期、減少、または廃止することを余儀なくされるだろう。
私たちの将来の資本需要は多くの要素に依存します
我々の任意の候補製品の開発に関連するこれらまたは他の変数のいずれの結果も変化し、候補製品の開発に関連するコストおよび時間を著しく変化させることができる。また、私たちの運営計画は将来的に変化する可能性があり、運営需要とそのような運営計画に関連する資本要求を満たすために追加の資金が必要になるかもしれません。
キャッシュフロー
次の表は、示す期間のキャッシュフロー(千単位)をまとめた
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
経営活動提供の現金 |
|
$ |
(20,329 |
) |
|
$ |
298,551 |
|
|
$ |
(166,734 |
) |
投資活動用の現金 |
|
|
(159,014 |
) |
|
|
(49,663 |
) |
|
|
(105,051 |
) |
融資活動で提供された現金 |
|
|
4,514 |
|
|
|
30,295 |
|
|
|
232,113 |
|
経営活動
2022年12月31日までの1年間、経営活動に用いられた現金は2,030万ドル。これは純損失1.333億ドルが繰延収入2億ドルから6,640万ドル増加して相殺されたためである
104
前金で受け取った収入は少ない。しかも、私たちは株式報酬のために4610万ドルの非現金費用を持っている。
2021年12月31日までの1年間に、経営活動が提供した現金は2兆986億ドル。これは純損失3630万ドルが、2021年第3四半期にGSKから受け取った5億ドルの前払いによる繰延収入2兆929億ドルの増加で相殺されたためだ。また、私たちは4080万ドルの株式ベースの給与と630万ドルの減価償却と償却費用の非現金費用がある。
2020年12月31日までの年間で、経営活動のための現金は1兆667億ドル。これは主にAbbVie協定に関連する収入が確認されたため、純損失1.902億ドルと繰延収入2110万ドルの減少によるものである。株式給与の非現金費用は3050万ドル、減価償却と償却費用は590万ドルで、この数字を部分的に相殺した。我々の負債と課税臨床用品コストも1,220万ドル増加し,経営活動に使用されている現金を相殺している。
投資活動
2022年12月31日までの1年間で,投資活動のための現金は1.59億ドルであり,主に4.02億ドルの有価証券満期日に関係し,5.569億ドルの有価証券購入によって相殺された。
2021年12月31日までの年間で,投資活動に用いられる現金は4970万ドルであり,主に2.863億ドルの有価証券満期日に関係しており,3.344億ドルの有価証券購入と1070万ドルの有価証券販売によって相殺されている。
2020年12月31日までの年間で,投資活動のための現金1.051億ドルは主に5.068億ドルの有価証券の購入に用いられ,4.068億ドルの有価証券満期収益が相殺された。さらに、私たちは現金で500万ドルの財産と設備を購入した。
融資活動
2022年12月31日までの1年間に、融資活動が提供する現金450万ドルは、主に普通株を購入するオプションの行使から来ている。
2021年12月31日までの1年間に、融資活動が提供した現金3030万ドルは、主に普通株を購入するオプションの行使から来ている。
2020年12月31日までの1年間に、融資活動が提供した現金2.321億ドルは、主に後続の公開発行完了後に9,602,500株の普通株を発行する純収益から来ている。また、普通株を購入するオプションの行使から630万ドルの現金を獲得しました
重要な会計政策と試算
経営陣の財務状況と経営結果の検討と分析は、米国公認会計原則(GAAP)に従って作成された我々の総合財務諸表に基づいている。これらの財務諸表を作成する際には、財務諸表日に報告された資産及び負債の報告金額及び又は有資産及び負債の開示、並びに報告期間内に報告された収入及び発生費用に影響を与える推定及び仮定を行う必要がある。我々の見積もりは,我々の歴史的経験と,当時の状況では合理的な様々な他の要因に基づいており,これらの要因の結果は資産や負債の帳簿価値を判断する基礎を構成しているが,これらの資産や負債の帳簿価値は他の源からは明らかではないように見える。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性があり、そのような違いは、いずれも実質的である可能性がある。以下で議論する会計政策は、これらの政策がより重要な分野に関連しているため、経営陣の判断と見積もりに関連しているため、我々の歴史や将来の業績を知るために重要であると考えられる。
収入確認
約束された商品またはサービスの制御権が顧客に転送されると、収入を確認し、金額はこれらの商品またはサービスが期待して受け取った対価格を反映している。私たちが手配された義務を履行する際に確認すべき適切な収入額を決定する時、私たちは履行します
105
以下の手順:(1)顧客との契約の決定,(2)契約中の履行義務の決定,(3)取引価格の決定,(4)契約に取引価格を割り当てる履行義務,および(5)エンティティが契約履行義務を履行する際に収入を確認する.複数の履行義務が存在することが確定した場合,プロトコル開始時に相対独立販売価格(SSP)に基づいて決定されたすべての履行義務に取引価格を割り当てる.外部ソースの証拠がある場合、外部ソースの証拠は、各交付物の相対SSPを推定するために使用される。外部ソースの証拠がなければ、配信可能製品のSSPの最適な推定値を使用します。
約束された製品やサービスの制御権がクライアントに転送された場合,ある時点で連携収入を確認する.我々は投入測定指標を用いて業績義務の完全履行の進展状況を測定することにより、経時的な協力収入を確認した。研究開発期間の収入を確認するために,これまでに発生した実コストと業績義務履行に要する総期待コストの比較を測定した。収入はプロジェクトコスト発生時に確認します。我々は,報告期間ごとに履行義務を履行する期待コスト推定数を再評価し,任意の重大な変化を調整した。臨床試験費用は高価であり,完成まで数年かかる可能性があり,結果自体も確定していない。時間の経過に伴い、著者らの予測コストは方案中に規定された臨床試験プログラムの変化、製造コスト推定の変化或いは監督機関の著者らの臨床試験設計或いは操作に対するフィードバックによって変化する可能性がある。私たちは異なる時期の履行義務を満たすために全体的な予想コストを調整した。2022年12月31日までの1年間に,AbbVieプロトコルでの協調収入が純増加を記録したのは,AL 003計画終了による義務履行の推定コストが変化したが,AL 002計画予想総コストの増加分がこの増加を相殺したためである.
研究と開発費用を計算すべきである
推定された臨床前研究と臨床試験費用の課税費用を記録した。見積数は,研究機関,臨床研究に関するCRO,臨床研究に関する研究地点,臨床前開発活動に関するサプライヤー,および臨床試験材料の生産に関する代理工組織が締結した契約に基づいて提供されるサービスである。また,関連プロトコルに基づき,患者の登録や活動レベルに応じて,臨床試験に関する費用を計算すべきである。著者らは合理的に可能な範囲内で患者の登録レベルと関連活動を監視し、各報告期間の計算すべき残高を決定する際に判断と推定を行った。もし私たちが提供されたサービスのレベルやこれらのサービスのコストを過小評価したり過大評価したりすれば、私たちの実際の支出は私たちの推定とは異なるかもしれない。これまで、著者らは臨床前研究と臨床試験の計算すべき費用の見積もりに大きな変化はなかった。
株に基づく報酬
株式の報酬に基づいて付与された日に計算され、報酬の推定公正価値に基づいて、従業員に必要なサービス期間(通常は帰属期間)の費用が確認される。我々は,Black-Scholesオプション定価モデルを用いて,普通株を購入するオプション付与日の公正価値と,それによる株式ベースの補償を推定した.
ブラック·スコアーズオプション価格設定モデルは、株式報酬の公正価値を決定するために、高い主観的仮定を使用することを必要とする。これらの仮説には
所期期限·予想期間は、株式ベースの報酬が突出すると予想される期間を表す。期待期間は、株式報酬の平均帰属期限と契約満期日との間の中点を使用する簡略化された方法を用いて導出される。
予想変動率-我々の株式変動性に関する情報は限られています。2019年2月7日まで、私たちの普通株はどの公開市場でも活発な取引がないからです。予想変動率は、業界内の同種の上場企業の歴史株式変動率から算出された。これらの会社は、株式ベースの奨励金の予想期間に相当する期間内に私たちの業務に匹敵すると考えられている。2020年に私たちは予想変動率を決定する際に私たち自身の歴史的変動率を考慮し始めた。
無リスク金利·無リスク金利は、測定日に発効した米国債収益率曲線に基づいており、満期日は予想期限に実質的に等しい。
106
配当を期待する予想配当率はゼロである。なぜなら、私たちは歴史的に支払われていないし、予測可能な未来に私たちの普通配当金を支払うことも望んでいないからだ。
RSUに関連する株式ベースの補償は、私たちの普通株の授与日における公正価値に基づいており、これは私たちの普通株の授与日の終値に等しい。私たちは奨励許可期間内の費用を確認する。オプションとRSUの費用は直線的に確認される.
私たちはまたある幹部に市場条件を持つRSUを付与した。モンテカルロシミュレーションモデルを用いて市場条件を持つRSUの公正価値を推定した。モデルに用いられる仮説と推定には,授権日の株価,無リスク金利,配当収益率,期待株価変動率,市場条件に達した推定期限が含まれる.この費用は市場状況の達成を考慮することなく、参加者の持続的な雇用確認に基づいている。加速帰因法を用いて市場条件を持つRSUに関する費用を確認した。
私たちはすべての補償が発生した没収について説明する。
第七A項。数量化と高質市場リスクの開示について。
金利リスク
私たちは正常な業務過程で市場リスクに直面している。このような危険は主に金利敏感性を含む。私たちの投資活動の主な目標は資本を保存し、私たちの運営に資金を提供することだ。私たちはまた大きなリスクを負うことなく投資から最大の収益を得ることを求めている。我々の目標を実現するために,我々の政策に基づいて,様々な信用品質が高く,期限が一般的な短い証券に対してポートフォリオを行った.
2022年12月31日現在、我々は7.129億ドルの現金、現金等価物、および有価証券を持っており、その中には主に銀行預金、通貨市場基金、短期政府有価証券が含まれている。このような利息を稼ぐ道具はある程度の金利リスクを持っているが、歴史的に利息収入の変動は私たちにとってそれほど大きくない。我々の現金等価物と有価証券の満期日は一般的に短く、有価証券のリスク状況が低いため、金利が直ちに上昇または低下することで、100ベーシスポイント低下することで、2022年12月31日までの公正価値の変化は約260万ドルになる。
外貨リスク
私たちの費用は普通ドルで計算されます。しかし、私たちは、ユーロを含む外貨支払いのために、研究開発サービスサプライヤーと限られた数の契約を締結した。私たちは外貨建ての契約で外貨取引収益や損失の影響を受けています。今まで、外貨取引損益は私たちの財務諸表にとって重要ではなく、私たちも正式な外貨ヘッジ計画はありませんでした。現在の為替レートの上昇または10%の低下は私たちの財務業績に実質的な影響を与えないだろう。
107
プロジェクト8.財務諸表Sと補足データ。
ALECTOR,Inc.
財務諸表索引
|
|
ページ |
監査された連結財務諸表 |
|
|
独立公認会計士事務所レポート(PCAOB ID: |
|
108 |
合併貸借対照表 |
|
110 |
合併経営報告書と全面赤字 |
|
111 |
株主権益合併報告書 |
|
112 |
統合現金フロー表 |
|
113 |
連結財務諸表付記 |
|
114 |
“インディペンデント”公認会計士事務所
Alector、Inc.の株主と取締役会へ。
財務諸表のいくつかの見方
Alector,Inc.(当社)2022年12月31日現在と2021年12月31日までの連結貸借対照表,2022年12月31日までの3年度の関連総合経営表と全面赤字,株主権益とキャッシュフローおよび関連付記(総称して“総合財務諸表”と呼ぶ)を監査した。総合財務諸表は、すべての重要な面で、2022年12月31日、2022年12月31日、2021年12月31日までの会社の財務状況、および2022年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
また、米国上場企業会計監督委員会(PCAOB)の基準に基づき、テレデビル委員会協賛組織委員会が発表した“内部統制-総合枠組み(2013枠組み)”で確立された基準に基づいて、2022年12月31日までの財務報告内部統制を監査し、2023年2月28日に発表した報告書に対して保留意見を発表した。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、監査委員会に伝達または要求が監査委員会に伝達された当期財務諸表監査によって生じる事項である:(1)財務諸表に対して重大な意義を有する勘定または開示に関するものであり、(2)特に挑戦的で主観的または複雑な判断に関するものである。重要な監査事項のコミュニケーションは何の方法でも私たちを変えないだろう
108
吾らは総合財務諸表について全体的な意見を提供していないが、吾等は下記の重要な監査事項を透過的に伝達することなく、重要な監査事項又はそれに関連する勘定又は開示について独立した意見を提供している。
収入確認
|
|
関係事項の記述
|
同社の協業収入は2022年12月31日までの年度で1兆336億ドル。付記2で述べたように、協調収入は、投入評価指標を用いて業績義務の完全履行の進捗を測定することにより確認される。研究開発期間中の協力収入を確認するために、同社はこれまでに発生した実際のコストと業績義務履行に要する総期待コストの比を測定した。収入は計画コスト発生時に確認. 監査協力収入は、企業が業績義務を履行する総予想コストを決定することに関する高度な判断を評価する推定に関連するため、挑戦的である。 |
私たちが監査でどのようにこの問題を解決したのか |
我々は,設計を評価し,関連する研究開発計画予算更新の制御を含む管理層の関連する研究開発計画予算更新の制御を含む,企業が業績義務を履行するための予想総コストを決定するための識別されたリスクを解決した内部制御の操作有効性をテストしたことが分かった. 協力収入をテストするために、私たちの監査プログラムは会社の業績義務履行の見積もりコストを理解し、管理層の関連研究開発計画予算の更新を評価することを含む。また,開発計画に記録された費用サンプルをテストし,関連する研究開発計画に対する経営陣の予算推定の歴史的正確性を評価し,開発計画を直接監督する会社員に聞いた。 |
/s/
2017年以来、当社の監査役を務めてきました。
2023年2月28日
109
Aレクター, INC.
強固な基礎スプレー銃のシーツ
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
有価証券 |
|
|
|
|
|
|
||
協力パートナー売掛金 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
その他の資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
臨床供給コストを計算しなければならない |
|
|
|
|
|
|
||
負債を計算すべきである |
|
|
|
|
|
|
||
繰延収入,当期分 |
|
|
|
|
|
|
||
賃貸負債を経営し、今期の部分 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
収入、長期分を繰延する |
|
|
|
|
|
|
||
賃貸負債長期部分を経営しています |
|
|
|
|
|
|
||
その他長期負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
株主権益: |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
110
Aレクター, INC.
業務所合併報告書損失と全面的損失
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
$ |
|
|
$ |
|
|
$ |
|
||||
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
|
|
|
|
|
|
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
総運営費 |
|
|
|
|
|
|
|
|
|
|||
運営損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他の収入、純額 |
|
|
|
|
|
|
|
|
|
|||
所得税前損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
所得税費用 |
|
|
|
|
|
— |
|
|
|
— |
|
|
純損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券の未実現収益 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
総合損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
1株当たり基本と希釈して純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
1株当たり純損失を計算するための基本株式と償却株式 |
|
|
|
|
|
|
|
|
|
|||
付記はこれらの連結財務諸表の構成要素である。
111
ALECTOR,Inc.
合併報告書株主権益
(単位:千、共有データを除く)
|
|
普通株 |
|
|
その他の内容 |
|
|
その他の総合収益を累計する |
|
|
積算 |
|
|
合計する |
|
|||||||||
|
|
株 |
|
|
金額 |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
残高-2019年12月31日 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||
普通株の発行 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
株式オプションの行使 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
以下の条項により普通株を購入する |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
無制限普通株 |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
有価証券の未実現収益 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
残高-2020年12月31日 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||||
株式オプションの行使 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株式単位の帰属を制限する |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
以下の条項により普通株を購入する |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
無制限普通株 |
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
有価証券は赤字を実現していない |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
残高-2021年12月31日 |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
|
||||
株式オプションの行使 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株式単位の帰属を制限する |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
以下の条項により普通株を購入する |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
無制限普通株 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
株に基づく報酬 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
有価証券は赤字を実現していない |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
残高-2022年12月31日 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
付記はこれらの連結財務諸表の構成要素である。
112
Aレクター, INC.
合併状態キャッシュフロープロジェクト
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
経営活動のキャッシュフロー: |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営に使用する現金純額を調整する |
|
|
|
|
|
|
|
|
|
|||
減価償却および償却 |
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬 |
|
|
|
|
|
|
|
|
|
|||
有価証券の割増と割引が増加する |
|
|
( |
) |
|
|
|
|
|
|
||
使用権資産の償却 |
|
|
|
|
|
|
|
|
|
|||
使用資産減価損失 |
|
|
|
|
|
|
|
|
|
|||
財産·設備処分損失純額 |
|
|
|
|
|
|
|
|
|
|||
経営性資産と負債変動状況: |
|
|
|
|
|
|
|
|
|
|||
協力パートナー売掛金 |
|
|
|
|
|
( |
) |
|
|
|
||
前払い費用と他の流動資産 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
その他の資産 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
売掛金 |
|
|
( |
) |
|
|
|
|
|
|
||
負債と臨床供給費を計算すべきである |
|
|
( |
) |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
|
|
( |
) |
||
賃貸負債 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他長期負債 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
経営活動提供の現金純額 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
投資活動によるキャッシュフロー: |
|
|
|
|
|
|
|
|
|
|||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券を売る |
|
|
|
|
|
|
|
|
|
|||
有価証券の満期日 |
|
|
|
|
|
|
|
|
|
|||
投資活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
資金調達活動のキャッシュフロー: |
|
|
|
|
|
|
|
|
|
|||
普通株を公開発行して得られる収益は,発行コストを差し引く |
|
|
|
|
|
|
|
|
|
|||
普通株式オプションを購入して得た収益を行使する |
|
|
|
|
|
|
|
|
|
|||
従業員が株を購入して株式を発行する予定で得られた金 |
|
|
|
|
|
|
|
|
|
|||
融資活動が提供する現金純額 |
|
|
|
|
|
|
|
|
|
|||
現金、現金等価物、および制限的現金純増加(減少) |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
期初現金、現金等価物、および限定現金 |
|
|
|
|
|
|
|
|
|
|||
期末現金、現金等価物、および制限現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
非現金投資と融資活動: |
|
|
|
|
|
|
|
|
|
|||
購入した財産と設備はすでに口座に入っている |
|
$ |
|
|
$ |
|
|
$ |
|
|||
付記はこれらの連結財務諸表の構成要素である。
113
Alector,Inc.(AlectorまたはThe Company)はカリフォルニア州サンフランシスコ南部に本社を置くデラウェア州の会社です。Alectorは臨床段階の生物製薬会社であり,免疫神経学の先駆者であり,神経変性を治療する新しい治療法である。
後続サービス
2020年1月30日,会社は発行と販売により後続発行を完了した
陳述の基礎
予算の使用
公認会計基準に従って連結財務諸表を作成することは、連結財務諸表の日付の資産及び負債額、又は有資産及び負債の開示、並びに報告期間内の収入及び費用の報告金額に影響を与えるために、管理層に推定及び仮定を要求する。同社は、収入確認、製造すべき項目、臨床計算項目、資産および負債の公正価値、所得税不確実性、株式に基づく報酬および関連仮定に関する推定を含む歴史的経験およびその他の要素に基づいてその推定を評価し、事実および状況が必要な場合にこれらの推定および仮定を調整する。実際の結果はこれらの推定とは大きく異なるかもしれません.
信用リスクが集中する
会社を深刻な集中信用リスクに直面させる可能性のある金融商品は主に現金、現金等価物、短期有価証券を含む。現金および現金等価物は、金融機関の小切手口座および現金等価物口座に格納される。そのような預金は時々連邦保険の限度額を超えるかもしれない。
現金、現金等価物、制限された現金
当社は購入の日に原始期限が三ヶ月以下のすべての高流動性投資は現金と現金等価物であると考えています。現金等価物は、通貨市場基金に投資された金額を含み、公正価値に応じて列報される。
2022年12月31日までの限定現金は、2018年6月に締結されたリースのために設立された信用状に関する。
114
次の表は、統合貸借対照表内に報告されている現金、現金等価物、および制限現金の入金を提供し、これらの現金合計は、統合キャッシュフロー表に示される同じ金額の合計である
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
現金と現金等価物 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
制限現金 |
|
|
|
|
|
|
|
|
|
|||
現金総額、現金等価物、制限された現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
有価証券
すべての有価証券は“販売可能”に分類され、市場オファーに基づいて公正価値に基づいて勘定される。同社は販売可能なポートフォリオが現在の業務に利用できると考えている。したがって、会社は特定の投資を短期有価証券に分類することができ、たとえ声明の満期日が現在の貸借対照表の日付よりも1年以上遅れている可能性がある。売却可能な債務証券については、未実現収益(いかなる関連税収影響も控除)は収益に含まれず、他の包括的収益に計上され、実現前に株主権益の単独構成要素として報告される。同社は、売却可能な債務証券を四半期ごとに評価し、未実現損失があるかどうかは信用に関連する要因によるものであるかどうかを決定する。減値が信用に関連しているかどうかを決定する際に、考慮される要素は、投資の公正価値がそのコストベースより低い程度、公表された信用格付けの低下、金利の変化、および証券に関連する任意の他の不利な要素を含む。信用に関する減値が存在することが確定した場合、当社は割引キャッシュフローモデルに基づいて信用損失を計測する。債務証券を売却できる信用関連減値は信用損失準備として確認され、会社の総合経営報告書中の他の収入に対して相応の調整を行った。信用とは関係のない未実現損失は、いかなる関連する税務影響を差し引いて、実現前に他の全面収益を計上する。いくつありますか
証券を売るコストは特定の識別方法に基づいている。証券の償却コストは割増の償却と満期割引の増加に応じて調整された。我々の投資政策によると、管理層は通貨市場基金、米国債、社債、商業手形に投資する。同社の現金、現金等価物、有価証券預金には何の損失も出ていない。
金融商品の公正価値
同社の金融商品には、現金および現金等価物、有価証券、パートナーの売掛金、流動および非流動前払い費用、売掛金および売掛金が含まれる。納期が相対的に短いため、当社の金融商品は公正価値に近い。
当社は推定技術を採用し,観察可能な投入を最大限に利用し,観察できない投入をできるだけ少なくしている。当社は、市場参加者が元本または最も有利な市場が資産または負債の定価である場合に採用されている仮定に基づいて、その金融商品の公正価値を決定する。公正価値計測における市場参加者の仮定を考慮した場合、以下の公正価値レベルは、観察可能な投入と観察不可能な投入とを区別し、この2つの投入は、以下のレベルの1つに分類される
レベル1−投入は、計量日と同じ資産または負債のアクティブな市場で調整されていないオファーである
レベル2−投入とは、アクティブ市場における同様の資産または負債の観察可能、調整されていないオファー、非アクティブ市場上で同じまたは同様の資産または負債の調整されていないオファー、または資産または負債に関する観察可能な市場データによって観察または確認可能な他の投入を意味する
115
財産と設備
賃貸借証書
当社は賃貸借開始時に賃貸借契約であるか否か又は賃貸借契約を含むか否かを決定します。リースは貸借対照表で使用権資産と賃貸負債であることが確認された。賃貸負債及びそれに応じた使用権資産は、予想リース期間内の賃貸支払いの現在値に基づいて入金される。当社は、似たような経済環境下で賃貸支払いに相当する金額を担保方式で類似期限内に借入することによる金利を適切な逓増借款金利を採用している。支払いされた初期直接費用または受信された報酬、および任意の前払いまたは計算されたレンタル料などの項目については、使用権資産を何らかの調整する必要がある場合がある。経営リースの賃貸料支出はリース期間内に直線法で確認し,経営報告書と総合損失の経営費用を計上する。可変レンタル支払いにはレンタル運営費用が含まれています。
当社は12ヶ月以下の経営リース(短期賃貸)の貸借対照表を含まず、長期賃貸のリース組成物と非賃貸組成物とを分離していないことを確認している。
長期資産減価準備
事件や環境変化がある資産の帳簿価値が回収できない可能性があることを示すたびに、当社は長期資産の減値を審査する。回収能力は,帳簿金額と資産予想による将来の未割引純現金流量を比較することで測定した。割引されていない将来のキャッシュフロー総額が当該資産の帳票価値よりも少ない場合には、その資産の帳票価値がその公正価値を超えた金額について減値損失を確認する。2022年12月31日までの年度2021年には
収入確認
約束された商品又はサービスの制御権が顧客に移転する場合、会社は収入を確認し、金額はこれらの商品又はサービスが期待して受信した対価格を反映する。会社が手配された義務を履行する際に確認すべき適切な収入額を決定する場合、会社は、(I)顧客との契約を決定するステップ、(Ii)契約中の履行義務を決定するステップ、(Iii)取引価格を決定するステップ、(Iv)取引価格を契約に割り当てる履行義務、および(V)エンティティが契約義務を履行する際に収入を確認するステップを実行する。複数の履行義務が存在することが確定した場合,プロトコル開始時に相対独立販売価格(SSP)に基づいて決定されたすべての履行義務に取引価格を割り当てる.外部ソースの証拠がある場合、外部ソースの証拠は、各交付物の相対SSPを推定するために使用される。外部ソースの証拠がなければ、配信可能製品のSSPの最適な推定値を使用します。
約束された商品やサービスの制御権が顧客に転送された場合,会社はある時点で連携収入を確認する.時間の経過に伴い、会社は投入測定指標を用いて業績義務の完全履行の進捗状況を測定し、協力収入を確認した。研究開発期間の収入を確認するために、同社はこれまでに発生した実際のコストと業績義務履行に要する総予想コストの比を測定した。収入はプロジェクトコスト発生時に確認します。会社は報告期間ごとに期待コスト推定数を再評価し、契約履行義務を履行する。
116
研究開発コスト
研究·開発コストは発生時に費用を計上し,主に新製品開発を含む。研究および開発コストには、賃金および福祉、顧問費、プロセス開発コスト、株式ベースの給与および実験室用品、および会社を代表する特定の研究および開発活動を行う第三者に支払われる費用が含まれる。また、研究開発コストには、プロジェクト、実験室用品、第三者研究開発活動によって生成された賃金単コストが含まれる協力プロトコルによる精算可能コストが含まれる。
同社が行っている研究·開発活動の大部分は第三者サービスプロバイダが行っている。同社は推定した臨床前研究と臨床試験費用の計上費用を記録している。見積数は,研究機関,臨床研究に関するCRO,臨床研究に関する研究地点,臨床前開発活動に関するサプライヤー,および臨床試験材料の生産に関する代理工組織が締結した契約に基づいて提供されるサービスである。また,同社は関連協定に基づき,患者登録や活動のレベルに応じて,臨床試験に関する費用を計算しなければならない。同社は合理的に可能な範囲内で患者の登録レベルと関連活動を監視し、各報告期間の計算すべき残高を決定する際に判断と推定を行う。会社が提供されるサービスレベルまたはこれらのサービスのコストを過小評価または過大評価した場合、実際の費用は推定とは異なる可能性がある。これまで,同社の臨床前研究や臨床試験にかかる費用の見積もりには大きな変化はなかった。
株に基づく報酬
株式に基づく報酬は、授与日に奨励の公正価値に基づいて測定される。普通株購入オプションの公正価値はブラック·スコアーズオプション定価モデルを用いて測定した。制限株式単位(RSU)に関する株式ベースの補償は、付与日会社普通株の公正価値に基づいており、この価値は、付与日会社普通株の終値に等しい。会社は奨励授権期間内の費用を確認します。サービス条件のみで付与されたオプションとRSUの費用は直線的に確認される.
2021年、会社はある幹部に市場条件を持つRSUを授与した。モンテカルロシミュレーションモデルを用いて市場条件を持つRSUの公正価値を推定した。モデルに用いられる仮説と推定には,授権日の株価,無リスク金利,配当収益率,期待株価変動率,市場条件に達した推定期限が含まれる.この費用は市場状況の達成を考慮することなく、参加者の持続的な雇用確認に基づいている。加速帰因法を用いて市場条件を持つRSUに関する費用を確認した。
当社はすべての奨励で発生した没収行為を計算します。
総合損失
総合損失には、純損失と株主権益のいくつかの変化が含まれており、これらの損失は、株主との取引や経済事件ではなく、取引および経済事件の結果である。同社の他の全面赤字の唯一の要素は、有価証券の未実現純収益(赤字)である。
所得税
当社は貸借対照法に基づいて所得税を計算し、この方法は次の事件の予想される将来の税務結果について繰延税金資産と負債を確認することを要求します 財務諸表に含まれていますこの方法によれば、繰延税金資産および負債は、財務諸表と資産および負債の税ベースとの差に基づいて決定され、公布された税率および法律を用いて計量され、これらの税率および法律は、差の予想が逆転したときに発効する。税率変動が繰延税金資産や負債に及ぼす影響は、公布日を含む期間の収入で確認されている。
繰延税金資産は、当社がこれらの資産がより現金化する可能性があると考えている範囲で確認します。繰延税金資産の一部または全部が現金化できない可能性が高い場合には、推定準備を提供する。会社の歴史上の経営業績と
117
過去の期間に記録された累計純損失を除いて、繰延税項目の純資産はすでに推定値から完全に相殺された。
同社は二歩法を用いて不確定な税務状況を記録した。まず、当社は税務倉庫位の技術的価値に基づいて、当該等の税務倉位を維持する可能性が高いかどうかを決定します。次に、確認の可能性が高い税務頭寸に該当する税務について、当社は最終的に関連税務機関と和解して実現可能な最大税収割引額が50%を超えることを確認しました。
会社は添付されている営業報告書の中で、利息支出項目と他の支出項目と税収割引に関する未確認利息と罰金をそれぞれ確認した。これまで、未確認の税収割引について利息や罰金を徴収していません。
従業員401(K)計画
会社は“アメリカ国税法”(以下、“規則”と略す)401(K)条に基づいて、Alectorのほとんどのアメリカ人従業員をカバーした合格した払込型貯蓄計画を制定した。401(K)計画は、セクション401(K)の規定に従って繰延納税の退職給付を提供することを意図している。資格に該当する従業員は最大で守ることができます
細分化市場
その会社は細分化市場で運営されている。会社の運営意思決定者兼最高経営責任者が総合管理会社の運営を担当し、資源を分配することを目的としている。
118
以下の表は、同社が公正価値システム内で階層的に公正価値の経常的に計量する金融資産をまとめたものである
|
|
2022年12月31日 |
|
|||||||||||||||
|
|
公正価値 |
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
公平な市場 |
|
||||
|
|
(単位:千) |
|
|||||||||||||||
貨幣市場基金 |
|
レベル1 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
アメリカ政府国債 |
|
レベル1 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
商業手形 |
|
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
社債 |
|
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
現金等価物総額と販売可能性 |
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
|
|
2021年12月31日 |
|
|||||||||||||||
|
|
公正価値 |
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
公平な市場 |
|
||||
|
|
(単位:千) |
|
|||||||||||||||
貨幣市場基金 |
|
レベル1 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
アメリカ政府国債 |
|
レベル1 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
現金等価物総額と販売可能性 |
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
同社の二級証券は第三者定価源を用いて評価を行っている。定価サービスは業界標準評価モデルを使用し、すべての重要な投入が観察される。同社は、現在の業務に資金を提供するために利用可能な有価証券を流動資産に分類する。2022年12月31日まで残りの契約満期日は#ドルです
4. 協力協定
グラクソ·スミスクライン
2021年7月1日、会社はグラクソ·スミスクライン(グラクソ·スミスクライン)の子会社グラクソ·スミスクライン英国有限公司と協力·許可協定を締結し、協定によると、会社はグラクソ·スミスクラインと協力してラトジン·モノクロナル抗体(AL 001とも呼ばれる)とAL 101(グラクソ·スミスクライン協定)を含むプロト粒子タンパク質のモノクロナル抗体を世界で開発·商業化した。グラクソ·スミスクライン協定は#年に発効した
グラクソ·スミスクライン協定によると、同社は$を受け取りました
119
商業化するラトジンモノクロナル抗体とAL 101。米国以外では、同社は2桁の等級別印税を受ける資格がある。
同社とグラクソ·スミスクラインはlatozinemabとAL 101を共同開発し、グラクソ·スミスクラインはアルツハイマー病、パーキンソン病、その他の非孤児適応の第3段階臨床試験を行う。同社とグラクソ·スミスクラインは開発コストを分担します
同社の結論は、GSKプロトコルはASC 808協力計画の範囲に属し、双方は活動の積極的な参加者であり、重大なリスクとリターンに直面しているため、これはAL 001とAL 101の商業化成功に依存する。いくつかの要素は、ASC 606、顧客との契約収入で計算される必要があり、取引相手が異なる会計単位である商品またはサービスの顧客である場合。
当社は、ASC 606項の明確な履行義務には、(I)AL 001 FTDのライセンスとノウハウがあると認定しているGRN現在、第3段階の臨床開発、および(Ii)ライセンス権およびノウハウを含む第2段階またはそれ以上の開発段階の製品に関する研究および開発活動にある。
初期および2022年12月31日までの取引価格には、前金を含む固定対価格が含まれています
各履行債務はそれぞれの独立した価値が取引価格に割り当てられる。各履行義務の推定SSPはAL 001とAL 101の商業化が期待される割引キャッシュフロー及び会社が初期第二段階の臨床試験で発生する推定研究と開発コストに基づいて決定された。各業績義務のSSP推定は管理層の仮定を反映し、その中に予測された収入、開発スケジュール、割引率及び技術と監督管理成功の可能性が含まれている可能性がある。FTDのライセンス-GRNその際、当社はグラクソ·スミスクラインがライセンス授与時にナンバープレートから恩恵を受けることができると認定したため、履行に関する義務はある時点で履行されている。第2段階またはより早い開発段階にある候補製品の場合、会社は、会社が提供する対応する開発サービスを承諾していない場合、GSKは、これらのライセンスの開発がより早い段階にあるので、ライセンスから利益を得ることができないと判断する。別の約束がない限り,同社は初期第2段階臨床試験が終了するまで研究·開発活動を行う。研究や開発活動の進行に伴い,収入は時間の経過とともに確認される。同社はこれまでに発生した実際のコストと履行義務を履行する期待総コストとを比較して進捗状況を測定する。
第3段階臨床開発製品の研究および開発活動はASC 808の範囲内に決定された。双方は製品開発、製造と商業化の積極的な参加者であり、製品商業の成功に依存する重大なリスクとリターンに直面する。当社とグラクソ·スミスクラインは、双方の研究開発過程におけるコスト分担に基づいて、プロジェクトごとの損益分担に参加しています。ASC 808は、識別および測定ガイドを提供しない。そこで,プロトコルのコスト分担条項の性質から,当社はASC 730(研究と開発)クラス比を適切にすることにした.同社は,これらのサービスに関する金をGSKに支払うか返済するかは,それぞれ研究と開発費用の増加または減少に計上すると結論している。
2022年12月31日までの年間GSKプロトコル下の連携収入 and 2021, was $
120
合意による共同開発活動に関するコストは,総合経営報告書の研究開発費に含まれており,グラクソ·スミスクラインのコストのどの返済もこのような費用の減少に反映されている。2022年12月31日までの年度2021年には研究開発費の削減を確認しました
AbbVie
同社は2017年10月にAbbVieバイオテクノロジー株式会社(AbbVie)と合意し,臨床前開発における2つの計画目標に対する抗体(AbbVie合意)を共同開発した。AbbVie協定の条項によると、AbbVieは#ドルを稼いだ
初期および2022年12月31日までの取引価格には、前金を含む固定対価格が含まれています
2022年12月31日までの年度のAbbVie協定での連携収入, 2021, and 2020 was $
革新者
当社は2020年3月にInnoventBiologics(Innovent.)と協定を締結し,中国のAL 008(Innoventプロトコル)をライセンス,開発,商業化した。AL 008は同社のCD 47-SIRP-Alpha経路に対する新型抗体であり、CD 47-SIRP-Alpha経路は腫瘍が天然免疫システムから逃れるために選択した有効な生存経路である。革新協定の条項によると、革新は同社に最高$を支払う
121
完全に2022年12月31日から制限される。2022年12月31日まで,
財産と設備、純額
財産と設備、純額は以下の各項目からなる
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
コンピュータ装置 |
|
$ |
|
|
$ |
|
||
家具と固定装置 |
|
|
|
|
|
|
||
実験室装置 |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
財産と設備、毛額 |
|
|
|
|
|
|
||
減価償却累計と償却を差し引く |
|
|
( |
) |
|
|
( |
) |
財産と設備の合計 |
|
$ |
|
|
$ |
|
||
負債を計算すべきである
計算すべき負債には以下の内容が含まれる
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
研究と開発コストを計算すべきである |
|
$ |
|
|
$ |
|
||
従業員の報酬を計算する |
|
|
|
|
|
|
||
専門サービスに応じる |
|
|
|
|
|
|
||
課税財産と設備 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
負債総額を計算すべきである |
|
$ |
|
|
$ |
|
||
2018年6月、当社はレンタル契約に調印し、レンタル契約を締結しました
2019年5月、当社はテナント契約を締結しました
2021年11月に、当社はテナント契約を締結しました
122
7,132 2平方フィートの空間を分けて
レンタル料金の構成は以下のとおりである
|
|
|
|
|
十二月三十一日 |
|
|
|
|
|||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
リースコストを経営する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
可変リースコスト |
|
|
|
|
|
|
|
|
|
|||
転貸収入と可変賃貸コストの精算 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
合計する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
2022年12月31日まで,運営リースの重み付き平均残存テナント期間は
以下は当社の経営賃貸契約に基づいて借りた賃貸金である2022年12月31日:
|
|
(単位:千) |
|
|
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
その後… |
|
|
|
|
未割引賃貸支払総額 |
|
|
|
|
減算:現在値調整 |
|
|
( |
) |
リース総負債 |
|
$ |
|
|
会社が確認した株式ベースの報酬は以下の通り
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
123
公正価値の決定
同社が普通株を購入したすべてのオプションの付与日における推定公正価値は、以下の仮定に基づいてブラック·スコアーズオプション定価モデルを用いて計算された
|
|
十二月三十一日までの年度 |
||||
|
|
2022 |
|
2021 |
|
2020 |
予想期限(年単位) |
|
|
|
|||
予想変動率 |
|
|
|
|||
無リスク金利 |
|
|
|
|||
配当率 |
|
|
|
|||
各株式購入の公正価値は、当社が以下に議論する方法と仮定を使用して決定される。これらの投入のすべては主観的であり、一般的に管理職が重要な判断と見積もりを下す必要がある。
予想期限-予想期間は株式ベースの奨励が突出すると予想される期限を表す。期待期間は、株式報酬の平均帰属期限と契約満期日との間の中点を使用する簡略化された方法を用いて導出される。
予想変動率-株式は2019年2月7日まで公開市場では何も活発に取引されていないため、当社の株式オプション変動性に関する情報は限られている。予想変動率は同業比上場企業の歴史的株式変動性に基づいて得られた。これらの会社は、株式ベースの奨励金の予想期間に相当する期間内に会社の業務に匹敵すると考えられている。2020年、当社は予想変動率を決定する際に自身の歴史変動率を考慮し始めた。
無リスク金利-無リスク金利は、ゼロ金利米国国庫券発行日に発効した米国国庫券収益率曲線をもとに、その満期日は予想期限にほぼ等しい。
期待配当率-予想配当金はゼロであり、同社は配当金を支払っていないため、予測可能な将来にその株式オプションに関連する普通株のいかなる配当も支払うことは期待されていない。
2019年株式インセンティブ計画と2022年インセンティブ計画
2019年2月6日、会社は“2019年株式インセンティブ計画(2019計画)”を採択し、取締役会は会社従業員、取締役、コンサルタントに奨励的株式オプション、非法定株式オプション、制限株、制限株式単位、株式付加価値権、業績単位、業績株を発行することができる。当社の2017年株式オプションおよび付与計画(2017計画)は終了しましたが、この計画に基づいて付与された株式は引き続き2017年計画に管轄されます。発行のために保留されているが、2017年計画に基づいて付与された奨励発行または2017年計画により付与された奨励制限を受けていない株式は、2019年計画の利用可能株式に追加される。2017年に奨励される予定の株式は、会社が買い戻したり没収したりした株式も2019年の計画に基づいて発行するために保留されます。取締役会又は取締役会により任命された委員会は、誰にオプション又は株式、株式数、期限、行使価格を付与するかを決定する権利がある。もし個人が持っている株式代表が
124
2022年1月1日、会社は“2022年インセンティブ計画”(インセンティブ計画)を採択して保留した
オプション活動は以下のとおりである
|
|
量 |
|
|
重みをつける |
|
|
重みをつける |
|
|
骨材 |
|
||||
|
|
|
|
|
|
|
|
(単位:年) |
|
|
(単位:千) |
|
||||
2020年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
没収される |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2021年12月31日現在の未返済債務 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
没収される |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2022年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日から行使可能 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
12月31日に帰属する予定だ |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
総内的価値は指標の株式オプションの行使価格と会社普通株の現金株式オプションの公正価値との差額である。行使されたオプションの内的価値の合計は#ドルである
制限された株式活動
125
|
|
量 |
|
|
重みをつける |
|
||
2020年12月31日現在の非帰属限定株式奨励及び限定株式単位 |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
|
|
||
既得 |
|
|
( |
) |
|
|
|
|
没収される |
|
|
( |
) |
|
|
|
|
2021年12月31日現在の未帰属制限株式単位 |
|
|
|
|
|
|
||
授与する |
|
|
|
|
|
|
||
既得 |
|
|
( |
) |
|
|
|
|
没収される |
|
|
( |
) |
|
|
|
|
未帰属制限株式単位 |
|
|
|
|
$ |
|
||
2022年12月31日まで従業員に発行された非帰属制限普通株に関する未確認株式報酬総額は#ドルである
2019年従業員株購入計画
2019年従業員株購入計画(2019 ESPP)は、条件を満たした会社員が割引価格で普通株を購入できるようにする。各サービス期間は
2022年、2021年、2020年12月31日終了年度の連邦と州所得税の計上概要は以下の通り(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
現在: |
|
|
|
|
|
|
|
|
|
|||
連邦制 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
状態.状態 |
|
|
|
|
|
|
|
|
|
|||
所得税支給 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
126
連邦法定税率と会社の実際の税率との入金は以下のとおりである2022年12月31日、2021年12月31日、2020年12月31日(単位:千):
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
連邦法定税率で税収割引を受ける |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
州所得税 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
税金控除 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
不確定税収状況 |
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬 |
|
|
|
|
|
( |
) |
|
|
|
||
評価免除額を変更する |
|
|
|
|
|
|
|
|
|
|||
他にも |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
所得税支給 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
会社の繰延税金資産と負債の重要な構成要素の一時的な違いをもたらす税金の影響は(千計):
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失 |
|
$ |
|
|
$ |
|
||
ボーナスを計算する |
|
|
|
|
|
|
||
税金控除 |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
||
リース責任 |
|
|
|
|
|
|
||
第174節研究開発資本化 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
繰延税項目総資産 |
|
|
|
|
|
|
||
推定免税額を差し引く |
|
|
( |
) |
|
|
( |
) |
繰延税金資産総額 |
|
$ |
|
|
$ |
|
||
繰延税金負債: |
|
|
|
|
|
|
||
減価償却および償却 |
|
$ |
( |
) |
|
$ |
( |
) |
使用権資産 |
|
|
( |
) |
|
|
( |
) |
繰延税金負債総額 |
|
|
( |
) |
|
|
( |
) |
税金資産を繰延し,純額 |
|
$ |
|
|
$ |
|
||
繰延税金資産の現金化を評価する際には、経営陣は繰延資産の一部または全部が現金化される可能性が高いかどうかを考慮する。繰延税金資産の最終的な現金化は、これらの一時的な差額が控除可能な期間に生じる将来の課税所得額になることに依存する。繰延税金資産の推定値を準備する必要があるかどうかを評価するには、近年の累積損失と将来の課税収入を含む既存のすべてのプラスと負の証拠を判断し、分析する必要があり、繰延税金資産の全部または一部が現金化できないかどうかを決定する必要がある。2022年12月31日まで当社は繰延税項目の純資産が完全に現金化される可能性が低いと信じているため、当社は全額推定値を利用して繰延税項目の純資産を相殺している。繰延税金資産の評価免除額は#ドル増加した
2022年12月31日まで同社の連邦と州の純営業損失(NOL)は約$に転換した
127
一般に、所有権変更制限により、NOL繰越及び積分の使用は年次制限を受ける可能性があり、その中で第382節はNOL繰越及び所有権変更後のある固有損失の制限を規定し、第383節は規則のある超過積分等の特殊な制限、及び類似の国家規定を規定する。そのため,同社がNOL繰り越しを利用する能力は,このような“所有権変更”によって制限される可能性がある。繰越は年間制限される可能性があり、推定支出を考慮する前に繰延税金総資産が減少する可能性がある。さらに、繰越の一部は将来の収益を減らすために使用される前に満了する可能性がある。当社は現在所有権のいかなる変化も第382条に規定する重大な制限を招くことを知らない。
下表は,同社が税収割引に関する活動を確認していない年度までの活動をまとめたものである2022年12月31日、2021年12月31日、2020年12月31日(単位:千):
2020年12月31日の残高 |
|
$ |
|
|
年内の税務頭寸に関する減幅 |
|
|
( |
) |
年内の税務倉庫位の増加 |
|
|
|
|
2021年12月31日現在の残高 |
|
|
|
|
年内の税務倉庫位の増加 |
|
|
|
|
2022年12月31日現在の残高 |
|
$ |
|
2022年12月31日までの不確定税収の未確認税収割引が確認されれば、税収割引は税収純資産を増加させるため、有効税率に影響を与えない。現在、この部分の資産は全額推定手当で相殺されている。同社の政策は、未確認の税収優遇に関する利息と罰金(あれば)を総合経営報告書の税収支出に計上することだ。当社は2022年12月31日までに年度中にいかなる利息や罰金も発生せず、利息や罰金の税務頭寸もありません。当社には何の税収もなく、税収割引総額が2022年12月31日以降の12ヶ月以内に大きな変化が生じる可能性があることが確認されていない。
当社は、税務機関が技術的利点に基づいて審査を行った後、不確定な税務状況が持続可能である可能性が高いと考えた場合にのみ、当該不確定税務状況の税務利益を確認する。2022年12月31日までに年度末そして2021年、当社は不確定な税収状況を$と記録しました
2014年、同社はAdimab、LLC(Adimab)と協力し、同社はAdimabがそのLatozinemabおよびAL 101計画で発見した抗体を開発し、AdimabがそのAL 002計画で最適化した抗体を開発している(2014 Adimab合意)。2014年のAdimabプロトコルはまた、同社がAdimabによってそのAL 003計画において最適化された抗体を開発することを規定している。しかし,AbbVieとAlectorが連携してこの計画に基づいて開発した資産AL 003の次のステップを審査し,AL 003をさらに開発する必要はないと結論した後,AbbVieはCD 33連携計画を終了することにした.2019年8月、同社はAdimabと追加抗体を開発する協力協定(2019 Adimab協定)に署名した。2021年12月、同社はAdimabと別の抗体工学研究プロジェクト協力協定(2021年Adimab協定)に署名した。2021年のAdimabプロトコルは,このプロトコルによる研究項目の選択期間が終了した時点で満了する.この2022年5月までのAdimab元最高経営責任者はAlectorの共同創業者と取締役会長だった。2022年12月31日までの年度, 2021年と2020年にAlectorが起こりました
128
$
以下に発行された潜在的希薄化株式は、その逆希薄化作用により、列報期間中の1株当たり償却純損失の計算には計上されていない:
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
未来帰属制限株 |
|
|
|
|
|
|
|
|
|
|||
普通株購入オプション |
|
|
|
|
|
|
|
|
|
|||
2019年ESPPが約束した株式によると |
|
|
|
|
|
|
|
|
|
|||
合計する |
|
|
|
|
|
|
|
|
|
|||
2023年2月、会社とAbbVieはAbbVie協定を改訂し、これにより会社は#ドルを受け取ることになります
129
項目9.Accouとの変更と分岐会計と財務情報開示の専門。
ない。
第9条。制御するプログラムがあります
情報開示制御とプログラムの有効性に関する結論
2022年12月31日現在、経営陣は、我々の最高経営責任者及び財務·会計官の参加の下、取引法第13 a-15(E)及び15 d-15(E)条に定義されている開示制御及びプログラムの設計及び動作の有効性を評価している。我々の開示制御および手続きは、取引所法案に基づいて提出または提出された報告書において開示を要求する情報が、米国証券取引委員会規則および表で指定された期間内に記録、処理、集約および報告され、これらの情報が、必要な開示に関する決定をタイムリーに行うために、最高経営者および最高財務会計官を含む私たちの管理層に蓄積されて伝達されることを目的としている。どのような制御やプログラムも,設計や操作がどんなに良好であっても,予想される制御目標を実現するために合理的な保証を提供することしかできないが,管理層は,可能な制御とプログラムのコスト-利得関係を評価する際にその判断を運用しなければならない.この評価に基づき、我々の最高経営責任者および最高財務会計官は、2022年12月31日までに、我々の開示制御およびプログラムの設計·運営が合理的な保証レベルで有効であると結論した。
財務報告の内部統制の変化
2022年12月31日までの四半期内に、財務報告の内部統制には何の変化もなく、これらの変化は、私たちの財務報告の内部統制に大きな影響を与えたり、合理的になったりする可能性がある。
財務報告の内部統制に関する経営陣の報告
当社の経営陣は、当社の財務報告の信頼性及び公認会計原則に基づいて対外総合財務諸表を作成する合理的な保証を提供するために、適切な財務報告の内部統制(例えば、取引法第13 a-15(F)条で定義されている)を確立及び維持する責任がある。最高経営責任者と最高財務責任者の監督·参加の下、我々の経営陣は、トレデビル委員会後援組織委員会が“内部統制-総合枠組み”(2013)で提案した基準に基づいて、2022年12月31日までの財務報告書の内部統制に対する有効性を評価した。この評価に基づき、経営陣は、財務報告書に対する内部統制は2022年12月31日から有効であると結論した。
本稿で述べたように、2022年12月31日現在、財務報告の内部統制の有効性は、独立公認会計士事務所安永会計士事務所によって監査されている。
独立公認会計士事務所報告
Alector、Inc.の株主と取締役会へ。
財務報告の内部統制については
我々は、テレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み”(2013フレームワーク)(COSO規格)で作成された基準に基づき、Alector,Inc.2022年12月31日までの財務報告内部統制を監査した。Alector,Inc.(当社)はCOSO規格に基づき、すべての重要な点で2022年12月31日までの財務報告に対して有効な内部統制を維持していると考えられる。
130
アメリカ上場企業会計監督委員会の基準に基づいて監査を行いました) 当社の2022年12月31日まで、2022年12月31日及び2021年12月31日までの総合貸借対照表、2022年12月31日までの3年度の関連総合経営及び全面赤字、株主権益及びキャッシュフロー表及び関連付記、及び当社の2023年2月28日までの報告について保留のない意見を表明した。
意見の基礎
当社経営陣は、効果的な財務報告内部統制を維持し、添付されている“経営陣財務報告内部統制報告”に掲載されている財務報告内部統制の有効性を評価する責任がある。私たちの責任は私たちの監査に基づいて、会社の財務報告書の内部統制に意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。
我々の監査には,財務報告の内部統制を理解すること,重大な弱点があるリスクを評価すること,評価されたリスクテストに基づいて内部統制の設計·運用有効性を評価すること,および状況下で必要と考えられる他のプロセスを実行することが含まれる。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分に関する記録の保存、(2)公認された会計原則に従って財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、会社の収入および支出は会社の経営陣と取締役の許可のみに基づいて行われる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用、または処分会社の資産を防止またはタイムリーに発見する合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
/S/安永法律事務所
カリフォルニア州サンマテオ
2023年2月28日
プロジェクト9 B。他にも情報です。
ない。
プロジェクト9 Cです検査を妨害する外国司法管轄区域を開示する。
適用されません。
131
第三部
プロジェクト10.役員·役員職ICERSと会社管理。
本プロジェクトに要求される情報は、2022年12月31日120日以内に米国証券取引委員会に提出された2023年株主総会に関する付表14 Aの最終委託書(以下、“依頼書”と略す)に含まれ、参照により本明細書に組み込まれる。
我々の取締役会は、すべての上級管理者、役員、および従業員に適用されるビジネス行動および道徳基準を採択しました。この基準は、私たちのサイトにあります(Https://investors.alector.com/コーポレート·ガバナンス/ガバナンス-概要)の下の“管理ファイル”。私たちは、上記で指定されたウェブサイトのアドレスおよび位置にこれらの情報を掲示することで、Form 8-K第5.05項のビジネス行為および道徳基準条項の改正または免除に関する開示要件を満たす予定です。
プロジェクト11.行政官イー補償します。
本プロジェクトが要求する情報は、2022年12月31日から120日以内に米国証券取引委員会に提出された依頼書に含まれ、参照によって本明細書に組み込まれる。
プロジェクト12.特定の受益者の保証所有権従業員と経営陣と関連した株主問題。
本プロジェクトが要求する情報は、2022年12月31日から120日以内に米国証券取引委員会に提出された依頼書に含まれ、参照によって本明細書に組み込まれる。
本プロジェクトが要求する情報は、2022年12月31日から120日以内に米国証券取引委員会に提出された依頼書に含まれ、参照によって本明細書に組み込まれる。
第14項.元金口座TING料金とサービスです。
本プロジェクトが要求する情報は、2022年12月31日から120日以内に米国証券取引委員会に提出された依頼書に含まれ、参照によって本明細書に組み込まれる。
132
第4部
プロジェクト15.展示品、資金エーアールアイチェックリストです。
Alector,Inc.の連結財務諸表は,本報告の一部として,項目8.財務諸表と補足データの下で表格10−Kの形で提出される。
これらは必要ではなく、適用されないわけでもないので、他のすべての付表は省略され、または要求された情報は、連結財務諸表または付記に含まれる。
添付ファイルインデックスに記載されたファイルは、参照によって本報告に組み込まれるか、または本報告と共に保存され、各場合は本明細書に示すようになる(S−K法規601項目に従って番号)。
項目16.表10-Kの概要
ない。
展示品索引
|
|
|
|
|
|
|
|
|
引用で編入する |
|
|||
番号をつける |
展示品名 |
表 |
書類番号. |
展示品 |
保存する 日取り |
保存済み ここから声明する |
3.1 |
登録者登録証明書の改訂と再予約。 |
8-K |
001-38792 |
3.1 |
2/11/2019 |
|
3.2 |
登録者の付例を改訂して再編成する。 |
8-K |
001-38792 |
3.1 |
10/6/2020 |
|
4.1 |
登録者とその特定の株主との間の登録権協定が改正され、再署名され、日付は2018年4月26日である。 |
S-1 |
333-229152 |
4.1 |
1/7/2019 |
|
4.2 |
登録者普通株式証明書サンプル |
S-1 |
333-229152 |
4.2 |
1/7/2019 |
|
4.3 |
登録者の証券記述。 |
10-K |
001-38792 |
4.3 |
2/25/2021 |
|
10.1+ |
登録者とその各役員と行政者との間の賠償協定フォーマット。 |
S-1 |
333-229152 |
10.1 |
1/7/2019 |
|
10.2+ |
改訂された2017年株式オプションと付与計画とその合意フォーマット |
S-1 |
333-229152 |
10.2 |
1/7/2019 |
|
10.3+ |
2019年株式インセンティブ計画とその合意の形態。 |
S-1 |
333-229152 |
10.3 |
1/7/2019 |
|
10.4+ |
2019年従業員株購入計画 |
S-1 |
333-229152 |
10.4 |
1/7/2019 |
|
10.5+ |
改正された2022年インセンティブ株式インセンティブ計画とそのプロトコルフォーマット |
8-K |
333-229152 |
10.1 |
1//3/2022 |
|
10.6+ |
登録者とアノン·ローゼンタール博士との間の確認的招待状。 |
S-1/A |
333-229152 |
10.5 |
1/29/2019 |
|
133
10.7+ |
招待状は2021年11月30日にAlector,LLCとSaraswati(Sara)Kenkare-Mitra博士が執筆した。 |
10-K |
001-38792 |
10.18 |
2/24/2022 |
|
10-8+ |
Alector,LLCとMarc Grasso,M.D.は2022年2月4日に発行された招聘状である。 |
10-K |
001-38792 |
10.19 |
2/24/2022 |
|
10.9+ |
2022年1月31日Alector、LLCとGary Romano、M.D.,Ph.D. |
8-K |
001-38792 |
10.1 |
03/29/2022 |
|
10.10+ |
役員は報酬計画を奨励する。 |
S-1 |
333-229152 |
10.10 |
1/7/2019 |
|
10.11+ |
役員報酬政策外。 |
|
|
|
|
X |
10.12+ |
登録者とその特定の執行者との間の統制権変更及び離職契約のフォーマット。 |
S-1 |
333-229152 |
10.12 |
1/7/2019 |
|
10.13 |
登録者とHCP Oyster Point III,LLCとの間のリースは,2018年6月27日である。 |
S-1 |
333-229152 |
10.14 |
1/7/2019 |
|
10.14# |
3回目の改訂と再署名は、登録者とAdimabとの間の協力協定であり、日付は2016年9月19日であり、改訂された。 |
S-1 |
333-229152 |
10.15 |
1/7/2019 |
|
10.15# |
登録者とAbbVie Biotech,Ltd.の共同開発とオプション協定は,2017年10月16日である。 |
S-1 |
333-229152 |
10.16 |
1/7/2019 |
|
10.16# |
登録者とAdimab,LLC間の2019年連携協定は,2019年8月16日である. |
10-Q |
001-38792 |
10.17 |
11/12/2019 |
|
10.17# |
協力と許可協定は,2021年7月1日にグラクソ·スミスクライン英国有限公司とAlector,Inc.によって署名された。 |
10-Q |
001-38792 |
10.19 |
8/3/2021 |
|
10.18# |
登録者とAdimab,LLCとの間で2019年の協力協定の第1改正案が,2022年8月16日に発効した。 |
|
|
|
|
X |
21.1 |
登録者子会社リスト。 |
10-K |
001-38792 |
21.1 |
2/24/2022 |
|
23.1 |
独立公認会計士事務所が同意します。 |
|
|
|
|
X |
24.1 |
授権書(本年報10-K表の署名ページに掲載)。 |
|
|
|
|
X |
31.1 |
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
|
|
|
|
X |
31.2 |
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
|
|
|
X |
134
32.1* |
2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条に規定されている最高経営責任者証明書。 |
|
|
|
|
X |
32.2* |
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による首席財務官の認証。 |
|
|
|
|
X |
101.INS |
XBRLインスタンスドキュメントを連結する |
|
|
|
|
X |
101.書院 |
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
|
X |
101.カール |
インラインXBRL分類拡張計算リンクライブラリ文書 |
|
|
|
|
X |
101.def |
インライン分類拡張Linkbase文書の定義 |
|
|
|
|
X |
101.介護会 |
XBRL分類拡張ラベルLinkbase文書を連結する |
|
|
|
|
X |
101.Pre |
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
|
|
|
|
X |
104 |
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
|
|
|
|
|
+管理契約または補償計画を示します。
#米国証券取引委員会の規則によると、この展覧会の一部(スター番号で示す)は省略されています。
*本10-K表年次報告に添付されている証拠物32.1および32.2に添付されている証明は、米国証券取引委員会に提出されたものとはみなされず、参照によって登録者が1933年の“証券法”(改正)または1934年の“証券取引法”(改正)に従って提出された任意の文書に組み込まれてはならない。このような文書に含まれる任意の一般的な登録言語にかかわらず、本10-K年次報告日の前または後に行われてはならない。
135
サイン
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者がその代表を代表して本報告書に署名することを正式に手配した.
|
|
ALECTOR,Inc. |
|
|
|
|
|
日付:2023年2月28日 |
|
差出人: |
/s/アノン·ローゼンタール |
|
|
|
アーノン·ローゼンタール博士。 |
|
|
|
共同創業者兼最高経営責任者 |
授権依頼書
本人は、以下の署名の者は、その真の合法的な事実代理人および代理人であり、十分な代替権および代替権(その役員および/または会社の高級社員の身分を含む)を有することを宣言する。本年度報告の任意又はすべての修正案に表格10-Kで署名し、それをすべての証拠物及び他のすべての関連文書と共に証券取引委員会に提出し、上記実際の受権者及び代理人、並びに彼ら一人一人に完全な権力及び権限を付与し、本人が可能又は自ら行うことができるすべての意図及び目的に応じて、事務所内及び周囲で必要及び必要なすべてのことを行い、ここで上記事実上の所有者及び代理人又はその任意の代理人又はその本人を承認及び確認し、または彼女の1人以上の代替者は、本条例によってなされたことを合法的に行うことができるか、または手配することができる。
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
サイン |
タイトル |
日取り |
|
|
|
/s/アノン·ローゼンタール
アーノン·ローゼンタール博士。 |
共同創業者兼最高経営責任者兼取締役 |
2023年2月28日 |
|
|
|
/s/Marc Grasso
マーク·グラッソ医学博士 |
首席財務官 (首席財務会計官) |
2023年2月28日 |
|
|
|
/s/ティルマン·グロス
ティルマン·グロス博士 |
取締役会議長 |
2023年2月28日 |
|
|
|
/s/エリザベス·ガロファロ エリザベス·ガロファロ医学博士 |
役員.取締役 |
2023年2月28日 |
|
|
|
/s/ポーラ·ハモンド
ポーラ·ハモンド博士です |
役員.取締役 |
2023年2月28日 |
|
|
|
ルイ·J·ラヴィネ,Jr.
ルイス·J·アヴリル |
役員.取締役 |
2023年2月28日 |
|
|
|
/s/Terry McGuire
テリー·マグワイア |
役員.取締役 |
2023年2月28日 |
|
|
|
136
/s/リチャード·シェラー
リチャード·シェラー博士です |
役員.取締役 |
2023年2月28日 |
|
|
|
/s/David·ウィーナ
デヴィッド·ウェナ |
役員.取締役 |
2023年2月28日 |
/s/クリスチャン·ヤフィー
クリスチャン·ヤフィー医学博士 |
役員.取締役 |
2023年2月28日 |
137