swtx-202212310001773427誤り2022会計年度Http://Fasb.org/us-GAAP/2022#ライセンスとサービスメンバーHttp://Fasb.org/us-GAAP/2022#ライセンスとサービスメンバーHttp://Fasb.org/us-GAAP/2022#ライセンスとサービスメンバーP 3 YP 3 Y00017734272022-01-012022-12-3100017734272022-06-30ISO 4217:ドル00017734272023-02-22Xbrli:共有00017734272022-12-3100017734272021-12-31ISO 4217:ドルXbrli:共有00017734272021-01-012021-12-3100017734272020-01-012020-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバー2019-12-310001773427米国-公認会計基準:財務省株式公開金メンバー2019-12-310001773427US-GAAP:AdditionalPaidInCapitalMembers2019-12-310001773427アメリカ公認会計原則:他の総合収入メンバーを累計2019-12-310001773427アメリカ-公認会計基準:前払いメンバーを保留2019-12-3100017734272019-12-310001773427US-GAAP:AdditionalPaidInCapitalMembers2020-01-012020-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバー2020-01-012020-12-310001773427アメリカ公認会計原則:他の総合収入メンバーを累計2020-01-012020-12-310001773427アメリカ-公認会計基準:前払いメンバーを保留2020-01-012020-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバー2020-12-310001773427米国-公認会計基準:財務省株式公開金メンバー2020-12-310001773427US-GAAP:AdditionalPaidInCapitalMembers2020-12-310001773427アメリカ公認会計原則:他の総合収入メンバーを累計2020-12-310001773427アメリカ-公認会計基準:前払いメンバーを保留2020-12-3100017734272020-12-310001773427US-GAAP:AdditionalPaidInCapitalMembers2021-01-012021-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバー2021-01-012021-12-310001773427アメリカ公認会計原則:他の総合収入メンバーを累計2021-01-012021-12-310001773427アメリカ-公認会計基準:前払いメンバーを保留2021-01-012021-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバー2021-12-310001773427米国-公認会計基準:財務省株式公開金メンバー2021-12-310001773427US-GAAP:AdditionalPaidInCapitalMembers2021-12-310001773427アメリカ公認会計原則:他の総合収入メンバーを累計2021-12-310001773427アメリカ-公認会計基準:前払いメンバーを保留2021-12-310001773427US-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバーSWTX:GSKMember2022-01-012022-12-310001773427SWTX:GSKMemberUS-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001773427SWTX:GSKMember2022-01-012022-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバーUS-GAAP:PrivatePlacementMembers2022-01-012022-12-310001773427US-GAAP:PrivatePlacementMembersUS-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001773427US-GAAP:PrivatePlacementMembers2022-01-012022-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバーSWTX:MarketOfferingMember2022-01-012022-12-310001773427SWTX:MarketOfferingMemberUS-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001773427SWTX:MarketOfferingMember2022-01-012022-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバー2022-01-012022-12-310001773427米国-公認会計基準:財務省株式公開金メンバー2022-01-012022-12-310001773427アメリカ公認会計原則:他の総合収入メンバーを累計2022-01-012022-12-310001773427アメリカ-公認会計基準:前払いメンバーを保留2022-01-012022-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバー2022-12-310001773427米国-公認会計基準:財務省株式公開金メンバー2022-12-310001773427US-GAAP:AdditionalPaidInCapitalMembers2022-12-310001773427アメリカ公認会計原則:他の総合収入メンバーを累計2022-12-310001773427アメリカ-公認会計基準:前払いメンバーを保留2022-12-310001773427SWTX:GSKMember2022-01-012022-12-310001773427SWTX:GSKMember2021-01-012021-12-310001773427SWTX:GSKMember2020-01-012020-12-310001773427US-GAAP:PrivatePlacementMembers2021-01-012021-12-310001773427US-GAAP:PrivatePlacementMembers2020-01-012020-12-310001773427SWTX:MarketOfferingMember2021-01-012021-12-310001773427SWTX:MarketOfferingMember2020-01-012020-12-31SWTX:試用版0001773427US-GAAP:PrivatePlacementMembers2022-09-072022-09-070001773427US-GAAP:PrivatePlacementMembers2022-09-070001773427SWTX:GSKMemberUS-GAAP:PrivatePlacementMembers2022-09-062022-09-060001773427SWTX:GSKMemberUS-GAAP:PrivatePlacementMembers2022-09-06Xbrli:純0001773427SWTX:CowenAndCompanyLLCMメンバーSWTX:AtTheMarketOfferingMember2021-02-252021-02-250001773427SWTX:CowenAndCompanyLLCMメンバーSWTX:AtTheMarketOfferingMember2022-01-012022-12-310001773427US-GAAP:PrivatePlacementMembers2020-10-132020-10-130001773427アメリカ公認会計基準:超過割当オプションメンバー2020-10-132020-10-130001773427アメリカ公認会計基準:超過割当オプションメンバー2020-10-13SWTX:セグメント0001773427SRT:最小メンバ数2022-01-012022-12-310001773427SRT:最大メンバ数2022-01-012022-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバー米国-GAAP:ShortTermInvestmentsメンバー2022-12-310001773427米国-公認会計基準:外国政府債務証券メンバー米国-GAAP:ShortTermInvestmentsメンバー2022-12-310001773427アメリカ-公認会計基準:会社債務証券メンバー米国-GAAP:ShortTermInvestmentsメンバー2022-12-310001773427米国-GAAP:ShortTermInvestmentsメンバー米国-GAAP:ビジネス紙のメンバー2022-12-310001773427アメリカ-公認会計基準:会社債務証券メンバーSWTX:LongTermInvestmentsメンバー2022-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバー米国-GAAP:ShortTermInvestmentsメンバー2021-12-310001773427アメリカ-公認会計基準:会社債務証券メンバー米国-GAAP:ShortTermInvestmentsメンバー2021-12-310001773427米国-GAAP:ShortTermInvestmentsメンバー米国-GAAP:ビジネス紙のメンバー2021-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバーSWTX:LongTermInvestmentsメンバー2021-12-310001773427アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバー2022-12-310001773427アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバー2022-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:アメリカ政府債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427アメリカ-GAAP:公正価値は再帰的メンバーを測定する米国-公認会計基準:外国政府債務証券メンバー2022-12-310001773427アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバー米国-公認会計基準:外国政府債務証券メンバー2022-12-310001773427アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する米国-公認会計基準:外国政府債務証券メンバー2022-12-310001773427アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する米国-公認会計基準:外国政府債務証券メンバー2022-12-310001773427SWTX:上場可能証券のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001773427SWTX:上場可能証券のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001773427SWTX:上場可能証券のメンバーアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001773427アメリカ-公認会計基準:公正価値投入レベル3メンバーSWTX:上場可能証券のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2022-12-310001773427米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバー2022-12-310001773427米国-GAAP:ビジネス紙のメンバーアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427米国-GAAP:ビジネス紙のメンバーアメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバーSWTX:MarketableSecuritiesNon Currentメンバ2022-12-310001773427アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:会社債務証券メンバーSWTX:MarketableSecuritiesNon Currentメンバ2022-12-310001773427アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバーSWTX:MarketableSecuritiesNon Currentメンバ2022-12-310001773427アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバーSWTX:MarketableSecuritiesNon Currentメンバ2022-12-310001773427アメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバー2022-12-310001773427アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001773427アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバー2021-12-310001773427アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバーSWTX:上場可能証券のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバーSWTX:上場可能証券のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバー2021-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバーSWTX:上場可能証券のメンバーアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:アメリカ政府債務証券メンバーSWTX:上場可能証券のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2021-12-310001773427アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:会社債務証券メンバー2021-12-310001773427アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2021-12-310001773427アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:会社債務証券メンバー2021-12-310001773427米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバー2021-12-310001773427米国-GAAP:ビジネス紙のメンバーアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427米国-GAAP:ビジネス紙のメンバーアメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するSWTX:MarketableSecuritiesNon Currentメンバ2021-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバーSWTX:MarketableSecuritiesNon Currentメンバ2021-12-310001773427アメリカ-公認会計基準:アメリカ政府債務証券メンバーアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するSWTX:MarketableSecuritiesNon Currentメンバ2021-12-310001773427アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:アメリカ政府債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するSWTX:MarketableSecuritiesNon Currentメンバ2021-12-310001773427アメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル1メンバー2021-12-310001773427アメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001773427アメリカ-公認会計基準:リース改善メンバー2022-12-310001773427アメリカ-公認会計基準:リース改善メンバー2021-12-310001773427アメリカ-公認会計基準:リース改善メンバーSRT:最大メンバ数2022-01-012022-12-310001773427US-GAAP:ComputerEquipmentMembers2022-12-310001773427US-GAAP:ComputerEquipmentMembers2021-12-310001773427US-GAAP:ComputerEquipmentMembersSRT:最小メンバ数2022-01-012022-12-310001773427US-GAAP:ComputerEquipmentMembersSRT:最大メンバ数2022-01-012022-12-310001773427アメリカ-GAAP:家具と固定機器のメンバー2022-12-310001773427アメリカ-GAAP:家具と固定機器のメンバー2021-12-310001773427アメリカ-GAAP:家具と固定機器のメンバー2022-01-012022-12-310001773427米国-GAAP:ソフトウェアとソフトウェア開発コストメンバー2022-12-310001773427米国-GAAP:ソフトウェアとソフトウェア開発コストメンバー2021-12-310001773427SRT:最小メンバ数米国-GAAP:ソフトウェアとソフトウェア開発コストメンバー2022-01-012022-12-310001773427SRT:最大メンバ数米国-GAAP:ソフトウェアとソフトウェア開発コストメンバー2022-01-012022-12-310001773427アメリカ-アメリカ公認会計基準:建設中のメンバー2022-12-310001773427アメリカ-アメリカ公認会計基準:建設中のメンバー2021-12-310001773427SWTX:StamfordConnecticutMember2018-10-012018-10-310001773427SWTX:StamfordConnecticutMember2018-10-310001773427SWTX:StamfordConnecticutMemberSWTX:レンタル運営リース更新オプションのメンバー2022-01-31SWTX:オプション0001773427スイッチ:レンタル運営リース更新オプション2つのメンバーSWTX:StamfordConnecticutMember2022-01-310001773427SWTX:StamfordConnecticutMember2022-01-310001773427SWTX:ダラムノースカロライナ州のメンバー2018-08-310001773427SRT:最小メンバ数SWTX:ダラムノースカロライナ州のメンバー2018-08-012018-08-310001773427SWTX:ダラムノースカロライナ州のメンバーSRT:最大メンバ数2018-08-012018-08-310001773427米国-公認会計基準:研究·開発費メンバー2022-01-012022-12-310001773427米国-公認会計基準:研究·開発費メンバー2021-01-012021-12-310001773427米国-公認会計基準:研究·開発費メンバー2020-01-012020-12-310001773427アメリカ-公認会計基準:一般と行政費用メンバー2022-01-012022-12-310001773427アメリカ-公認会計基準:一般と行政費用メンバー2021-01-012021-12-310001773427アメリカ-公認会計基準:一般と行政費用メンバー2020-01-012020-12-310001773427SWTX:持分インセンティブ計画2019年2020-01-012020-12-310001773427SWTX:持分インセンティブ計画2019年2022-12-310001773427SWTX:持分インセンティブ計画2019年SRT:最小メンバ数Swtf:ShareBasedPaymentArrangementEmployeeAndNonEmployeMember2022-01-012022-12-310001773427SWTX:持分インセンティブ計画2019年SRT:最大メンバ数Swtf:ShareBasedPaymentArrangementEmployeeAndNonEmployeMember2022-01-012022-12-310001773427SWTX:従業員株式購入計画2019メンバー2019-08-300001773427SWTX:従業員株式購入計画2019メンバー2022-12-3100017734272019-01-012019-12-310001773427米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001773427米国-公認会計基準:従業員株式オプションメンバーSRT:最小メンバ数2022-01-012022-12-310001773427米国-公認会計基準:従業員株式オプションメンバーSRT:最大メンバ数2022-01-012022-12-310001773427SRT:CEO実行官メンバ米国-公認会計基準:従業員株式オプションメンバーSWTX:CeoPerformanceAwardMember2019-06-012019-06-30SWTX:分割払い0001773427SRT:CEO実行官メンバ米国-公認会計基準:従業員株式オプションメンバーSWTX:CeoPerformanceAwardMember2019-06-300001773427米国-公認会計基準:従業員株式オプションメンバーSWTX:CeoPerformanceAwardMember2022-12-310001773427米国-公認会計基準:従業員株式オプションメンバーSWTX:CeoPerformanceAwardMember2022-01-012022-12-310001773427米国-公認会計基準:制限された株式メンバー2019-12-310001773427米国-公認会計基準:制限された株式メンバー2020-01-012020-12-310001773427米国-公認会計基準:制限された株式メンバー2020-12-310001773427米国-公認会計基準:制限された株式メンバー2021-01-012021-12-310001773427米国-公認会計基準:制限された株式メンバー2021-12-310001773427米国-公認会計基準:制限された株式メンバー2022-01-012022-12-310001773427米国-公認会計基準:制限された株式メンバー2022-12-310001773427米国-GAAP:制限株式単位RSUメンバー2021-12-310001773427米国-GAAP:制限株式単位RSUメンバー2022-01-012022-12-310001773427米国-GAAP:制限株式単位RSUメンバー2022-12-310001773427SWTX:PfizerMembersUS-GAAP:ライセンスプロトコル用語メンバ2017-08-012017-10-310001773427スイッチ:JuniorSeriesAConvertiblePferredUnitsMemberSWTX:PfizerMembersUS-GAAP:ライセンスプロトコル用語メンバ2017-10-310001773427スイッチ:JuniorSeriesAConvertiblePferredUnitsMemberSWTX:PfizerMembersUS-GAAP:ライセンスプロトコル用語メンバ2017-08-012017-10-310001773427SWTX:PfizerMembersUS-GAAP:ライセンスプロトコル用語メンバSWTX:NeroacestatMembers2017-08-012017-10-310001773427SWTX:MirDametinibメンバーSWTX:PfizerMembersUS-GAAP:ライセンスプロトコル用語メンバ2017-08-012017-10-310001773427SWTX:BeigeneLtd.メンバーSwtf:ClinicalCollaborationAgreementメンバ2022-01-012022-12-310001773427SWTX:BeigeneLtd.メンバーSwtf:ClinicalCollaborationAgreementメンバ2021-01-012021-12-310001773427SWTX:BeigeneLtd.メンバーSwtf:ClinicalCollaborationAgreementメンバ2020-01-012020-12-310001773427SWTX:KatholiekeUniversity-LeuvenMembersスイッチ:資産購入とライセンス契約メンバーの排除2021-05-012021-05-310001773427SWTX:KatholiekeUniversity-LeuvenMembersスイッチ:資産購入とライセンス契約メンバーの排除2021-05-310001773427SWTX:GSKMemberUS-GAAP:PrivatePlacementMembers2022-09-012022-09-300001773427SWTX:GSKMemberUS-GAAP:PrivatePlacementMembers2022-09-300001773427SWTX:Jazz PharmPharmticals Ireland Limitedメンバースイッチ:資産購入とライセンス契約メンバーの排除2020-10-012020-10-310001773427SWTX:Jazz PharmPharmticals Ireland LimitedメンバーSRT:最大メンバ数スイッチ:資産購入とライセンス契約メンバーの排除2020-10-310001773427SWTX:MapkureMember米国-GAAP:シリーズAPReferredStockMembers2019-06-012019-06-300001773427SWTX:MapkureMember米国-GAAP:シリーズAPReferredStockMembers2019-06-300001773427米国-GAAP:シリーズAPReferredStockMembers2020-06-012020-06-300001773427SWTX:MapkureMember米国-GAAP:シリーズAPReferredStockMembers2020-06-300001773427SWTX:MapkureMemberアメリカ-アメリカ公認会計基準:シリーズBPferredStockMember2022-06-012022-06-300001773427アメリカ公認会計基準:副次的事件メンバーSWTX:MapkureMemberアメリカ-アメリカ公認会計基準:シリーズBPferredStockMember2023-01-012023-01-310001773427アメリカ公認会計基準:副次的事件メンバーSWTX:MapkureMemberアメリカ-アメリカ公認会計基準:シリーズBPferredStockMember2023-01-310001773427SWTX:MapkureMember米国-GAAP:シリーズAPReferredStockMembers2022-12-310001773427SWTX:MapkureMember2022-01-012022-12-310001773427SWTX:MapkureMember2022-12-310001773427アメリカ-公認会計基準:州と地方法律法規のメンバー2022-12-310001773427SWTX:都市のメンバー2022-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバー2022-01-012022-12-310001773427アメリカ-アメリカ公認会計基準:普通株式メンバー2021-01-012021-12-310001773427米国-GAAP:制限株式単位RSUメンバー2022-01-012022-12-310001773427米国-GAAP:制限株式単位RSUメンバー2021-01-012021-12-310001773427米国-公認会計基準:制限された株式メンバー2022-01-012022-12-310001773427米国-公認会計基準:制限された株式メンバー2021-01-012021-12-31
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表10-K
| | | | | |
| ☒ | 1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで十二月三十一日, 2022
あるいは…。
| | | | | |
| ☐ | 1934年証券取引法第13条又は15条に基づいて提出された移行報告 |
依頼書類番号: 001-39044
スプリングウォークス治療会社は
(登録者の正確な氏名はその定款に記載)
| | | | | | | | | | | |
| デラウェア州 | 83-4066827 |
| (法団又は組織の他の司法管区として設立された国) | (国際税務局雇用主身分証明書番号) |
| |
| ワシントン通り百号 | スタンフォード | CT | 06902 |
| (主にオフィスアドレスを実行) | (郵便番号) |
登録者の電話番号は市外局番を含んでいます(203) 883-9490
同法第12条(B)に基づいて登録された証券:
| | | | | | | | |
| クラスごとのタイトル | 取引コード | 各取引所の名称 それに登録されている |
| 普通株、1株当たり0.0001ドル | SWTX | ナスダック世界ベスト市場 |
同法第12条(G)により登録された証券:なし
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。
はい、そうですx違います¨
登録者がこの法第13節または第15節(D)節に基づいて報告を提出する必要がないかどうかを再選択マークで示す。
はい、そうです¨ 違います。x
登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告を提出したかどうか、および(2)このような提出要求を過去90日以内に遵守してきたかどうかを、再選択マークで示すはい、そうですx違います¨
登録者が電子的に提出されたかどうかをチェックマークで示す;過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間内)に、S−T規則405条(本章232.405節)に従って提出を要求した各対話データファイルはい、そうですx違います¨
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
| | | | | | | | | | | | | | | | | | | | | | | |
| 大型加速ファイルサーバ | ☒ | ファイルマネージャを加速する | ☐ | 非加速ファイルサーバ | ☐ | 規模の小さい報告会社 | ☐ |
| | | | | | 新興成長型会社 | ☐ |
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる☒
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意の幹部が関連回復中に§240.10 D−1(B)に従って受信されたインセンティブベースの報酬に基づいて回復分析を行う必要があるかどうかを再選択マークで示す。◻
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。はい、違いますx
2022年6月30日(登録者が最近完成した第2財期の最終営業日)の終値によると、登録者の非関連会社が保有する登録者普通株の総時価は$である906,368,657.
2023年2月22日現在、登録者普通株の流通株数は62,498,784.
法団に成立した文書を引用する
登録者の年次株主総会に関する最終委託書は,登録者が2022年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出され,本明細書に記載された範囲内で参照により第3部に組み込まれる.
SpringWorks治療会社
表格10-Kの年報
カタログ表
| | | | | | | | |
| | ページ |
第1部 | | |
第1項。 | 業務.業務 | 4 |
第1 A項。 | リスク要因 | 46 |
項目1 B。 | 未解決従業員意見 | 99 |
第二項です。 | 属性 | 99 |
第三項です。 | 法律訴訟 | 99 |
第四項です。 | 炭鉱安全情報開示 | 99 |
| | |
第II部 | | |
五番目です。 | 登録者普通株市場、関連株主事項及び発行者による株式証券の購入 | 100 |
第六項です。 | [保留されている] | 101 |
第七項。 | 経営陣の財務状況と経営成果の検討と分析 | 101 |
第七A項。 | 市場リスクの定量的·定性的開示について | 114 |
第八項です。 | 財務諸表と補足データ | 115 |
第九項です。 | 会計と財務情報開示の変更と相違 | 141 |
第9条。 | 制御とプログラム | 141 |
プロジェクト9 B。 | その他の情報 | 144 |
プロジェクト9 Cです。 | 検査妨害に関する外国司法管区の開示 | 144 |
| | |
第三部 | | |
第10項。 | 役員·幹部と会社の管理 | 145 |
第十一項。 | 役員報酬 | 145 |
第十二項。 | 特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 | 145 |
十三項。 | 特定の関係や関連取引、取締役の独立性 | 145 |
14項です。 | 最高料金とサービス | 145 |
| | |
第4部 | | |
第十五項。 | 展示品と財務諸表の付表 | 146 |
第十六項。 | 表格10-Kの概要 | 146 |
前向き陳述に関する特別説明
本年度報告は,リスクと不確実性に関する前向き陳述をForm 10−Kまたは年次報告の形で含む。我々は1995年の個人証券訴訟改革法と他の連邦証券法における安全港条項に基づいてこのような前向きな声明を行った。本年度報告では歴史的事実に関する陳述を除き,他のすべての陳述は前向き陳述である。場合によっては、これらの前向き記述は、“可能”、“将”、“すべき”、“予想”、“意図”、“計画”、“予想”、“信じ”、“推定”、“予測”、“潜在”、“継続”、またはこれらの用語または他の同様の用語のような否定語を使用することによって識別することができる。これらの前向きな陳述は、以下の態様に関する陳述を含むが、これらに限定されない
•私たちの製品開発活動と臨床試験の成功、コストと時間は、私たちが行っているミダミニ2 b期の臨床試験の時間スケジュール及び任意の他の臨床試験と関連する準備の開始と完成時間、私たちの臨床試験の結果が利用できる予想時間、ニガレスタット3期の臨床試験の登録性質、及びミダミニ2 b期の臨床試験の潜在的な登録性質を含む
•私たちの臨床研究のバックラインまたは中期データは、そのような研究の最終的またはより詳細な結果、または進行中または未来の研究の他の結果を予測することができないかもしれない
•私たちの候補製品の潜在的な属性と利点は
•私たちは単独でまたは他の会社と協力して承認された候補製品を商業化する予定です
•私たちは私たちの候補製品のさらなる開発を完成させるために必要な資金を含めて、私たちの運営のために資金を得ることができ、承認されれば商業化される
•既存の現金、現金等価物、および有価証券の期限は、私たちの運営費用および資本支出要件を支払うのに十分であると予想される
•私たちのビジネス発展努力の潜在力は、私たちのポートフォリオの潜在的価値を最大限に発揮する
•私たちは他の候補製品を識別、許可、または取得する能力
•私たちの第三者パートナーは、開発中の共同療法を含む、私たちの候補製品に関する研究·開発活動の能力と意思を継続している
•私たちは、私たちの候補製品に対する規制承認と、承認された候補製品ラベル上の任意の関連制限、制限、または警告を得ることができ、維持することができる
•私たちが計画している規制提出と相互作用の時間は、2024年上半期に提出される予定のミダメチニブの新薬申請またはNDA、米国食品医薬品局またはFDAが決定した時間および結果を含み、FDAが2023年2月に分配された処方薬ユーザ費用法案(PDUFA)優先審査を受けて獲得したナイル西司彼のNDA申請に関する決定、目標行動日は2023年8月27日、他の規制機関、臨床試験場所の調査審査委員会、および出版物審査機関の決定を含む
•1つまたは複数のこれらの指定が得られる可能性のある私たちの任意の他の候補製品について、孤児薬、高速チャネル指定、および突破的治療指定の潜在的利点を指定する;
•我々は現在、硬線維腫、NF 1-PNおよび他の腫瘍学およびまれな疾患適応の治療法の開発にマーケティングまたは参加している会社と競争することができる
•私たちは、製品候補製品の知的財産権保護または市場排他性を獲得し、維持する能力、およびそのような保護の持続時間を期待している
•私たちは、臨床前研究、臨床試験、および(承認されれば)商業用途のための候補製品の製造に成功し、既存の契約製造組織(CMO)が候補製品の臨床供給および商業規模生産を支援する能力、および将来的に薬物物質および完成品を製造するためにより多くのCMOを求める能力を選択することができる能力と潜在力がある
•私たちの候補製品の市場規模と成長潜在力、そして私たちが単独でまたは他の会社と協力してこれらの市場にサービスする能力
•承認されれば、私たちの候補製品の市場受容率と程度
•アメリカやアメリカや他の国の規制動向
•私たちは第三者サプライヤーと製造業者と契約を締結する能力と彼らが契約を十分に履行する能力
•発売されたか発売される可能性のある競争製品の成功
•現在行われている新冠肺炎の大流行に関連するリスクは、私たちの業務、運営、臨床前研究、臨床試験、サプライチェーン、戦略、目標と予想スケジュールに悪影響を及ぼす可能性がある
•私たちは重要な科学、医学、商業、管理人材の能力を誘致し、維持している
•費用、将来の収入、資本需要、追加融資需要の推定
•私たちの財務的表現は
•私たちの競争相手や私たちの産業に関連した発展と予測。
本年度報告中の任意の展望性陳述は未来の事件と未来の財務表現に対する著者らの現在の見方を反映し、既知と未知のリスク、不確定要素とその他の要素に関連し、私たちの実際の結果、業績或いは成果はこれらの展望性陳述と明示或いは暗示する任意の未来の結果、業績或いは成果とは大きく異なるかもしれない。実際の結果が現在の予想と大きく異なる可能性がある要因には、他に加えて、本年度報告第1 A項のリスク要因および他の部分に記載されている要因がある。このような不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。法的要求がない限り、私たちは未来に新しい情報があっても、これらの前向きな陳述を任意の理由で更新または修正する義務がない。
私たちは時々、これらの市場の潜在規模および特定の疾患の推定発病率および流行率の推定を含む、私たちの業界、一般的な商業環境、および特定の疾患に関する市場の推定、予測、および他の情報を提供するかもしれない。推定、予測、予測、市場研究或いは類似方法に基づく情報自体は不確定性の影響を受け、実際のイベント、状況或いは数字は、実際の疾病罹患率と市場規模を含み、提供した情報とは大きく異なる可能性がある。他に明確な説明がない限り、市場研究会社および他の第三者、業界、医療および一般出版物、政府データおよび同様のソースが用意された報告、研究調査、研究、および類似データから当業界情報、商業情報、市場データ、流行情報、および他のデータを取得し、いずれの場合も、信頼できると考えられるソースからこれらの情報を取得し、場合によっては私たち自身の仮説および分析を適用し、これらの仮説および分析は将来的に不正確であることが証明される可能性がある。
第1部
プロジェクト1.ビジネス
概要
著者らは臨床段階の生物製薬会社であり、精確な医学方法を用いて、壊滅的な稀な疾病と癌を受けた十分なサービスを受けていない患者群のために生活を変える薬物を獲得、開発、商業化している。著者らは差別化された小分子標的腫瘍学候補製品の組み合わせがあり、そして2つの稀な腫瘍タイプに対する末期臨床試験、1つの登録と1つの潜在登録、及び他のいくつかの高度に流行している遺伝子に対して癌を定義する計画を進めている。著者らは研究、転化科学と臨床開発における戦略方法と卓越した運営により、私たちの2つのリード候補製品を迅速に後期臨床試験に推進することができ、その中の1つの候補製品が2023年2月に承認されると同時に、業界リーダーと複数の共有価値のパートナー関係を構築し、私たちの製品組み合わせを拡大することができるようにした。その上で、著者らは引き続き差別化された、全面的に統合された生物製薬会社を構築し、患者とその疾病を理解することに集中し、変革性の標的薬物を開発と提供する。
我々の最先端の候補製品であるniroacestatは、研究されている経口小分子ガンマ分泌酵素阻害剤やGSIであり、硬線維腫を治療する単一療法として開発されている。硬線維腫はまれで、常に人を衰弱させ、破壊させる軟組織腫瘍であり、米国食品·薬物管理局(FDA)が許可した治療法はまだない。2022年12月、成人硬線維腫の治療のためのネオノセットのNDAをFDAに提出した。2023年2月27日、我々はFDAがNDAの届出を受け、優先審査を与えることを発表した。FDAは処方薬使用料法案(PDUFA)の目標行動日を2023年8月27日としている。また,FDAは,現在諮問委員会会議を開催してこの申請を検討するつもりはないと述べている。提出されたNDAはFDAのリアルタイム腫瘍学審査(RTOR)計画に基づいて審査されており,3期DEFI試験の積極的なデータの支持を得ており,靭帯様腫を有する成人患者に対するグローバル,無作為,二重盲検,プラセボ対照試験である。2022年5月,DEFI試験の陽性背線試験結果を発表し,2022年9月の欧州医学腫瘍学会大会(ESMO)でDEFI試験のより多くのデータを公表した。DIFI試験は無進展生存期間(PFS)の改善の主要な終点に達し、プラセボと比較してニロシ他群は統計学的に有意に改善し、疾患進展のリスクは71%(危険比(HR)=0.29(95%CI:0.15,0.55);pを低下させたことを示した
米国で発売される可能性のある成人硬線維腫治療のナイルキセチンを支持するためのビジネス準備を積極的に行っている。また、2024年にEUの欧州医薬品局にマーケティング許可申請またはMAAを提出する予定だ
我々はまた、ニューノキシートによる卵巣顆粒細胞腫、または卵巣顆粒細胞腫、卵巣癌のサブタイプの治療を評価している。2022年9月には,進行中の第2段階試験において,1人目の患者が薬物治療を受けており,ナイロキシットが再発性卵巣GCT患者の単一療法として評価されていることを発表した。
我々の第2の候補は,研究中の経口小分子MEK阻害剤であるミダメチニブであり,現在開発中であり,神経線維腫症1型関連叢状神経線維腫,あるいはNF 1−PNの治療に用いられており,まれな末梢神経鞘腫瘍であり,重篤な疼痛や破壊を引き起こす。ミダミニーは,このような患者に必要な長期治療を可能にするために,他のMEK阻害剤と比較して一流のプロファイルを提供する可能性があると信じている。FDAはNF 1−PNのミダミニー孤児薬物指定および高速チャネル指定を許可しており,欧州委員会はNF 1のミダミチニブ孤児薬物指定を承認している。2021年11月、NF 1−PNを有する小児および成人患者のための登録可能な2 b期臨床試験であるReNeu試験の全面募集を発表し、2023年下半期にReNeu試験の背線データを報告する予定である。
悪性血液病において、著者らは新薬とB細胞成熟抗原(BCMA)の併用による多発性骨髄腫治療の新しい方案を評価している。私たちはいくつかの異なるBCMA指導の治療と組み合わせたナイルシタンを評価するために、業界パートナーといくつかの臨床協力協定を締結した。私たちの業界協力のほかにフレッド·ハッチンソン癌研究センターや
賛助研究協定の一部として,Dana−Farber癌研究所はナイルキャストがBCMA指導治療を強化する能力をさらに探索する
遺伝子定義の転移性固形腫瘍において、著者らの現在の臨床段階の努力はマイトジェン活性化プロテインキナーゼ或いはMAPK経路に集中している。単一療法や併用療法におけるMAPK異常を含む固形腫瘍に対するミダミニーの治療作用を評価している。我々は百済神州や百済神州と協力して,ミダミニと百済神州のリフィニヌスの併用によるRAS変異や他のMAPK遺伝子変異の癌の治療を探索している。さらに,MapKure,LLCまたはMapKure(私たちと百済神州が共有する実体)を介してユニークな遺伝子で定義されたBRAF変異腫瘍のセットにBGB 3245を使用することを探索している。2022年6月、我々はミダミニとリフェラファニーの併用によるMAPK活性化変異を有する末期固形腫瘍患者の1/2段階試験を評価する予備臨床データと、BRAFまたはRAS変異を有する末期固形腫瘍患者のBGB-3245治療の第1段階試験を評価する予備臨床データを公表し、各レジメンがMAPK活性化変異を有する様々な固形腫瘍タイプに管理可能な安全性と臨床活性の証拠を提供した。2023年上半期の医学会議で行われている1/2期試験におけるミダミニとリフェラファニーの併用に関するより多くのデータと,行っている1期試験における追加BGB−3245単一療法データを公表する予定である。2023年2月、最初の患者は1/2 a期開放ラベル、用量増加と拡大試験を受け、ミダミニーとBGB-3245の併用によるMAPK突然変異を有する末期固形腫瘍患者の治療効果を評価した。SpringWorksが支持する学術後援の研究では,ミダミニーは小児や若年者の低レベルグリオーマや他の種々のMAPK活性化変異を有する末期固形腫瘍の治療に評価されている
また,我々はKatholieke University LeuvenやKU Leuvenやフランダースバイオテクノロジー研究所から許可を得たTEA領域やTEAD阻害剤計画,Dana−Farber癌研究所から許可を得た表皮増殖因子受容体小分子阻害剤の組み合わせなど,強力な生物学的基盤と検証された作用機序を有する資産を利用して我々のポートフォリオを構築していく予定である。2022年第4四半期に、私たちはTEAD阻害剤開発候補SW-682を指名し、2023年にSW-682に研究性新薬申請を提出する予定である。私たちは私たちの薬物発見能力と私たちの開発計画のための転換医学活動を支援するために、私たちの研究開発インフラに投資し続けている
我々は,共有価値パートナーシップを継続して使用し,われわれの治療法の患者サービスの潜在力を最大限に発揮する予定である。著者らはリードする臨床前、臨床、医療と商業能力の面で投資を行い、そして革新的な協力パートナーシップの構築に集中し、各方面の調整激励措置に参与し、業務成果を最適化することを求めた。私たちは、この方法は引き続き革新者との協力関係を拡大し、私たちの既存と未来のポートフォリオの潜在力を最大限に発揮し、拡張可能かつ持続可能な業務の構築を支持し、効率的に候補製品を推進し、それを商業化することに集中し、これらの候補製品は腫瘍患者の生活を変える潜在力があると信じている。
私たちの戦略
サービス不足の患者群に対する変革性薬物の買収,開発,商業化により,差別化された世界的なバイオ製薬会社の構築を継続することを目標としている。われわれの目標は,まれな疾患と腫瘍学分野を標的とした業界の先頭になることである。
私たちの戦略の主な内容は
•現在開発されているまれな腫瘍学的適応では,われわれの主要候補製品niroacestatとmirdametinibの市場承認を効果的に推進している私たちのリードする薬物開発能力は、私たちの候補製品の発売承認を引き続き効果的に推進することができ、可能な状況で加速された規制経路を利用することができると信じている。2017年8月の設立以来、私たちは2つの候補製品の上場承認を推進する上で急速な進展を遂げてきた。著者らの最初の候補製品niRogacestatはFDAから硬繊維腫を治療する孤児薬物の称号、快速通路の称号と突破的な治療称号を授与され、そして欧州委員会から軟組織肉腫を治療する孤児薬物の称号を授与された。2022年5月,我々は3期DEFI試験の陽性結果を発表し,2022年9月のヨーロッパ医学腫瘍学会大会でDEFI試験のより多くのデータを公表した。2022年12月、成人硬線維腫の治療のためのネオノセットのNDAをFDAに提出した。FDAは2023年2月にNDA申請を受け,優先審査を承認し,PDUFAに割り当てられた目標行動日は2023年8月27日であった。我々の第2の候補製品であるミダミチニブは、FDAおよび欧州委員会の孤児薬物指定を取得し、NF 1−PNの治療のためのFDAの高速チャネル指定を取得した。ミダミチニブは現在潜在的な登録ReNeu試験の評価を受けている;2021年6月、2 b期ReNeu試験に参加した最初の20名の成人患者の中期臨床データは児童腫瘍基金NF会議で公表された。2021年11月、ReNeu試験の全面募集を発表し、2023年下半期にTOPLINE結果を報告する予定です。
•新規ノキシットを多発性骨髄腫BCMA標的治療の礎として確立し,高度に流行している転移性固形腫瘍のバイオマーカーで定義された亜群の治療にわれわれの正確な腫瘍学的方法を応用した著者らはすでにいくつかの臨床協力協定を締結し、ナイルキセチンとBCMA標的治療の多種の方法の併用治療を評価し、抗体-薬物結合体、CAR T細胞療法、二重特異性抗体およびモノクロナル抗体を含み、多発性骨髄腫患者の治療に用いられる。ニロシド併用BCMA標的治療は,BCMA標的治療単独と比較して多発性骨髄腫患者の臨床結果を改善する可能性があると信じられている。著者らは引き続き精確な医学方法を用いて、遺伝子定義癌患者に対する潜在的な治療方法を決定し、開発した。われわれの臨床段階での取り組みは,MAPK異常を携帯する固形腫瘍に対する単一療法と併用療法の開発に集中している
•戦略的パートナーシップを通じて私たちの製品グループの潜在力を最大限に発揮し、私たちの価値志向の方法を展開して新薬を識別、買収、開発し、私たちの製品の組み合わせをさらに拡大しますわれわれはすでに戦略的パートナーシップを構築し,革新的な併用療法を開発し,癌駆動の基本的な機序に対する新たな知見を利用している。我々の戦略は,選定された協力プロジェクトのために開発コストと下流経済を分担することで,各方面のインセンティブを調整することである。この戦略を実行することにより,生物製薬業界全体で開発されている有望な治療法が得られると信じており,これらの治療法には科学的かつ臨床的理由が既存の候補製品と組み合わされている。私たちはいくつかの業界や学術パートナーと協力することを発表し、将来的により多くの戦略的パートナーシップを実行しようとしている。
•もし私たちの候補製品が承認されれば、単独でも他社と協力しても、重点と有効な方法を使用して新薬をサービス不足の患者集団にもたらすために商業化される。もし私たちの候補製品が承認されれば、私たちは単独でまたは他の会社と協力して、アメリカと選定された国際マーケティングと私たちの候補製品を商業化するつもりです。私たちは、私たちのビジネス準備活動を完了しており、私たちの医療組織や商業インフラの建設を含め、アメリカで初めての商業打ち上げを行う予定です。著者らは引き続き集中と効率的な方法を採用してこれらの努力を行い、関連製品の機会に適した的確な方式で市場参入、販売とマーケティング能力を初歩的に構築した。この方法は、私たちが製品候補製品を開発している患者と医師に効果的に接触させ、私たちの製品の組み合わせのビジネス潜在力を最大限に発揮することができると信じています。
•患者提唱団体,キーオピニオンリーダー,研究機関,ヘルスケア提供者からなる密に統合されたネットワークを育成し続け,患者とその家族の生活を変える治療法の開発に情報を提供する我々のポートフォリオを効率的かつ影響力のある方法で発展させるためには,重要な利害関係者ネットワークを育成しなければならないと考えられる.これらの利害関係者からの経験と知見を統合し,硬線維腫研究財団,小児腫瘍基金,有力学術医や研究者を含め,壊滅的にまれな疾患や癌を有する患者とその家族の生活を変化させる方法の開発を指導し続けている。
•多元化、包摂性、専門的で卓越した文化を維持することに対する我々の確固とした約束を通じて、最も優秀な人材を誘致、維持、支持している患者を中心とした使命と組織力で支援する文化の構築に取り組んでおり,この文化は経験や視点の多様性によって駆動されている
私たちの候補製品
私たちの販売ルートは次のグラフにまとめられています。
___________________________
*孤児薬物、快速通路、突破的治療の称号を獲得した。
メダカは孤児薬と快速通路の称号を得た。
(1)私たちと百済神州が共同で持つ実体MapKureが開発された。
本報告では、ここで“潜在登録試験”を指す場合、候補製品の有効性および安全性を評価する臨床試験を指し、適用可能な規制機関への候補製品のマーケティング申請の提出を潜在的に支援する。このような試験は2/3期あるいは3期臨床試験あるいは肝心な試験と呼ばれることもある。
ナイルキャストは
概要
著者らの最先端の候補製品Niroacestatは1種の研究中の経口、選択的GSIであり、著者らは靱帯様腫瘍、卵巣顆粒細胞腫と多発性骨髄腫を含むいくつかの腫瘍学的適応を治療するための薬物を開発しており、現在治療方法があるにもかかわらず、これらの疾患は依然として重大な満足されていない医療需要が存在する。ガンマ分泌酵素は1種の酵素複合体であり、アミロイド前駆体蛋白、あるいはAPP、Notch、HER 4、E-cadherin、N-cadherin、BCMAとCD 44を含む多くの膜貫通蛋白を分解することができる。これらの膜貫通タンパク質はガンマ分泌酵素によって切断され,様々なシグナル事象を招き,これらのシグナル事象はこれらのタンパク質の細胞質ドメインが解離した結果である。ガンマ分泌酵素のいくつかの基質は多種の疾病と関係があり、アルツハイマー病のAPPと癌のBCMAとNotchを含み、ガンマ分泌酵素を治療標的とする理論的基礎を形成した
2018年6月、FDAは、硬線維腫を治療するためのナイルキャストの孤児薬物の称号を承認し、2018年11月に進行性、切除不可能、再発または難治性硬繊維様腫瘍または深層線維腫症の成人患者の治療のためのナイルカル司快速チャネルの称号を承認した。2019年8月、FDAはネオノカスタット突破療法を許可し、進行性、切除不可能、再発或いは難治性硬繊維様腫瘍或いは深部線維腫症の成人患者の治療に指定した。また、2019年9月、欧州委員会は軟組織肉腫を治療するナイルキャストの孤児薬名を承認した。
第一段階と第二段階の臨床試験が硬繊維腫患者における鼓舞的な結果に基づいて、2019年5月、著者らはDEFI試験を開始することを発表し、これは登録された第三段階の全世界無作為二重盲検プラセボ対照臨床試験であり、そして2020年7月に全面的な募集試験を発表した。2022年5月、著者らはDEFI試験の陽性TOPLINE結果を発表し、2022年9月のヨーロッパ医学腫瘍学大会でDEFI試験のより多くのデータを公表し、これらの試験の中で、ナイルキャストはプラセボと比較して、すべての主要と副次的な治療効果と生活の質の終点において統計的に顕著かつ臨床的に意義のある改善を示した
2022年12月、成人硬線維腫の治療のためのネオノセットのNDAをFDAに提出した。2023年2月27日、我々はFDAがNDAの届出を受け、優先審査を与えることを発表した。FDAはPDUFAの目標行動日を2023年8月27日としている。また,FDAは,現在諮問委員会会議を開催してこの申請を検討するつもりはないと述べている
卵巣癌のサブタイプであるニューノキシートによる卵巣顆粒細胞腫の治療効果も評価されている。2022年9月、第1の患者が第2段階試験で用量を受け、再発性卵巣顆粒細胞腫患者の単一療法としてニトロクロロキセチンを評価することを発表した。
われわれの単一療法開発努力に加え,BCMA標的治療と組み合わせた薬剤として多発性骨髄腫を治療する薬剤を評価している。GSISは多発性骨髄腫細胞上膜結合のBCMAの切断を減少させ,BCMA標的治療の活性を高めることが証明されている。我々は,いくつかの業界パートナーとの臨床協力により,BCMA目標モデルでこのような新たな併用治療法を評価している。
ナイルキャストは硬線維腫を治療した
疾患背景
硬繊維腫は、侵襲性線維腫症或いは硬繊維様型線維腫症とも呼ばれ、稀な非転移性軟組織腫瘍であり、児童と成人に発生することができる。これらの腫瘍の特徴は,成長パターンが関節,筋肉,内臓を含む周囲の健康組織を侵すことができることである。腫瘍の大きさ、成長方式と部位の侵襲性によって、硬繊維腫は深刻な疾病、例えば疼痛、毀容、内出血と運動範囲の衰弱障害を招くことができる。2つのデンマーク肉腫センターであるオーフス大学病院とコペンハーゲン大学病院で治療した硬線維腫患者179名の疫学研究により,硬線維腫と診断された後,患者の医療資源利用率は約3年で有意に向上し,一部の原因は入院と外来受診回数の増加および在院日数の増加であり,この疾患による発症率の重要性が示唆された。
硬線維腫は通常15歳から60歳の間に発生し、よりよく見られるのは生命の3年目と40年目に診断され、女性の罹患率は2~3倍高い。アメリカでは、毎年1,000から1,650人の新たに確定診断された硬線維腫患者がいると推定されています。硬線維腫症例の多くは自発的に発生しており、以下のような変異の一つに関係していますCTNNB 1遺伝子、β-カテニンをコードする。普通の人群と比べ、家族性腺腫性ポリープ症の家族歴或いはFAP家族歴を有する患者の硬線維腫の発生リスクは約850倍高い。靭帯様腫瘍を有するFAP患者の腫瘍は通常装甲運搬車この遺伝子はβ-カテニン分解に関与するタンパク質をコードする。いつですか装甲運搬車あるいは…CTNNB 1もし突然変異、組織創傷が存在すれば、手術、妊娠或いは軟組織損傷を含み、靱帯様腫瘍の形成を招くことができる。
硬線維腫の臨床経過は患者群によって異なる。任意の所与の患者において、靭帯様腫瘍は、急速成長と安定期間との間で交互にすることができ、長年のフォローアップ期間において、約5%~10%の患者が自発的消退を認めた。靭帯様腫は形態と放射線学的表現に大きな差がある可能性があるが,通常は通常診断である。硬繊維腫は通常健康診断或いは各種画像技術、例えば超音波、コンピュータ断層撮影或いはCT、磁気共鳴画像或いはMRIの時に初めて発見される。組織学的には,硬線維様腫は異なるコラーゲン沈着を示し,境界不明瞭であった。確定診断は免疫組織化学染色によるβ-カテニンの核定位に依存する。診断は遺伝子変異をスクリーニングすることでも確認できますCTNNB 1そして装甲運搬車遺伝子です。
靭帯様腫瘍は病態が強いにもかかわらず,死亡率への影響は通常限られている。このような全体寿命への限られた影響と現在の悪い治療選択により,かなり大きな靭帯様腫患者流行池があると考えられる。現在の靱帯様腫瘍を治療する方法は成功率が低いことが多い。77%と高い手術を受けた患者が再発することは,患者の年齢,腫瘍位置,腫瘍の大きさに依存する。また,400人以上の靭帯腫患者を管理する医師からのフィードバックの市場調査研究によると,約50%が特定の系治療(例えば化学療法やチロシンキナーゼ阻害剤)や局所介入(例えば手術)を受けた患者は満足できる治療結果を持たず,彼らの靭帯腫の後続治療が必要であると考えられる。多くの市場研究に基づいて、私たちは90%以上の患者が最終的に積極的な干与を受けると信じており、私たちは今後10年の任意の年に、約5,500~7,000人の硬繊維腫患者がアメリカで積極的に治療を受けると推測している。
靱帯様腫の治療現状
FDAが承認した靭帯様腫の治療法は現在のところない。歴史的には靭帯様腫は手術切除により治療されてきたが,後硬膜腫の存在を示す証拠が多くなってきたため,あまり歓迎されなくなった
外科腫瘍の再発率は77%に達し、これは疾病の負担を増加させる可能性があり、追加の干与が必要である。再発率が高いほか,手術自体にも合併症のリスクがあり,非常に病態である可能性があり,まれに切断が必要である。手術の潜在的な発病率、およびその利益の大きさと持続性の不確定性を考慮すると、医師は現在、歴史的に手術対象である可能性のある患者に対して積極的なモニタリング方法をとる。手術はその局限性があるが、硬繊維腫に顕著な発病率或いは死亡率のリスクが存在する時、依然として手術を使用することができ、例えば頭と頚部に発生する腫瘍である。放射線治療は単独あるいは手術との併用も可能であるが,報告されている二次性腫瘍の発生リスクを考慮すると,通常第一選択ではない。
これらの局所治療に加えて,系統療法はラベルにも用いられており,異なる程度の活性と耐性を有している。これらの療法は、リポソームアドリアマイシンおよびビンクリスチン/メトトレキサートなどの化学療法薬、非ステロイド性抗炎症薬、抗ホルモン療法およびチロシンキナーゼ阻害剤、例えば、ソラフェニブ、イマチニブおよびパゾパニを含む。これらの薬物の中で、ソラフィニだけが繊維様腫瘍患者のランダム、二重盲検、プラセボ対照臨床試験で研究を行った;この3期の臨床試験は研究者が発起したものであり、生物製薬業界のスポンサーがない。ソラフェニはプラセボと比較してPFSにおいて統計的に有意に改善したにもかかわらず,耐性は大きな問題であり,治療を受けた患者の20%が有害事象で中止され,また22%の治療を受けた患者が同意を撤回した。27カ月の中位フォローアップ期間中,ソラフィニ治療を受けた患者の61%が治療を中止した。全体的には,既存の非ラベル系療法は硬線維腫の治療にはあまり適しておらず,この群では許容可能な安全性と活動度のバランスを示していないと考えられる。したがって,靭帯様腫の治療には重大な満たされていない医療ニーズがあると考えられる。
作用機序
ナイルキャストは経口的、有効、選択的、可逆的、非競争的なガンマ分泌酵素小分子阻害剤であり、Notchを含む多くの重要な機能を有する膜貫通タンパク質を溶解することができる完全なプロテアーゼ複合体である。Notchに対するガンマ分泌酵素の切断はNotch細胞内ドメインやNICDの放出をもたらし,NICDは核を行き来し,下流標的遺伝子の転写を活性化する。Notchシグナルは細胞増殖の調節器であり、その失調は多くの形態の癌と関係がある。靭帯様腫瘍細胞系では,ナイルキセチンがNICDの放出を著しく減少させ,Notchシグナル経路の下流活性を低下させ,腫瘍細胞の遊走,浸潤,成長を減少させることが観察された。
ニロシドによる硬線維腫治療の臨床研究進展
3期DEFI試験開始前に,両項目の成人靭帯腫患者を募集した臨床試験でニロゼスタットの臨床活性が認められた。その中の第一項はファイザー社が固形腫瘍患者の中で行った一期用量増加臨床試験であり、その中の一組の患者は靱帯様腫瘍と診断された。評価可能な靭帯様腫瘍患者7名のうち,5名の患者はRECIST v 1.0により部分緩解(PR)を獲得し,客観緩解率(OOR)は71%,疾患コントロール率は100%であった。この第一段階臨床試験で治療を受けた硬線維腫患者におけるニトロクロロキセチンの活性を考慮して,NCIは進行中の硬線維腫患者に対してニロシドが1日2回150 mg,あるいはBIDを評価し,われわれのDEFI試験で用いたのと同じ用量で評価した第2段階臨床試験を行った。NCI協賛の第2段階試験では,研究に参加した硬線維腫患者17名中5名がRECIST V 1.1でPRを確認した結果,ORRは29%で進展性疾患はなかった。ナイルキャストは2つの臨床試験で耐性が良好であり、2022年にアメリカ臨床腫瘍学会(ASCO)年会で報告された治療期間の中央値は4年を超えた。これらの硬線維腫患者に対する1期と2期臨床試験では,ナイル西特は良好な臨床活性と安全性を示し,3期DEFI試験の開始を支持している
2020年8月,ミネソタ大学とDana−Farber癌研究所の研究者がデータを発表し,われわれの拡大参入計画の下で,小児4名と若年成人硬線維腫患者のニロシド治療の臨床的メリットが観察された。この4例の患者の臨床活動の中で、1例は完全に緩和し、2例は部分的に緩和し、1例は病状が安定し、3級或いは4級の不良事件の報告がなかった。著者らはこの4人の児童と若い成人患者のデータを鼓舞し、この第二段階の臨床試験は若い靭帯腫患者におけるニロシドの潜在的利益を更に評価することを信じている。2020年9月、児童腫瘍学グループ(COG)と協力して、進行性、手術切除できない硬繊維腫を有する児童と青少年に対するニロシットの治療効果を評価するための2期臨床試験を開始した。2022年第4四半期に、この試験は計算すべき目標を達成した。
硬線維腫の3期DEFI試験
第一段階と第二段階の臨床試験で観察された硬線維腫患者の臨床利益程度、及び著者らとFDAの討論に基づいて、著者らは2019年5月にDEFI試験を開始し、そして2020年7月に全面的な組み入れを発表した
DEFI試験は登録された第三段階全世界二重盲検無作為プラセボ対照臨床試験であり、北米とヨーロッパの37の臨床地点で行われる。
DIFI試験はプラセボと比較して、進行性硬線維腫患者に対するニロシドの有効性、安全性と耐性を評価することを目的とした。組織学的に進展性疾患を有する硬線維腫患者142名(RECIST v 1.1測定により,過去12カ月以内の腫瘍成長と定義)を募集し,幼稚で手術不可能な硬線維腫を治療するか,難治性または再発のある疾患(1一連の治療後)である。
実験は、二重盲検段階とオプションの開放ラベル拡張またはOLE段階の2つの段階を含む。患者は1日150 mgのナイルカルスタットまたはプラセボの治療を1:1の割合でランダムに28日間受けた。患者は臨床または放射線学的疾患の進展が出現するまで、ニロシットまたはプラセボ治療を受け、受け入れられない毒性を経験し、または同意を撤回した。試験中に疾患進展を確定診断した条件に適合した患者は選択可能なOLE段階に入り、150 mgのニロキセチン治療を受け、1日2回治療を受けることができる。
主な終点は無進行生存,またはPFSであり,ランダム化から任意の原因の進行や死亡の評価日までの時間と定義される.進展はRECIST V 1.1を用いて放射線学検査を行う或いは臨床盲法、独立、中心放射線学或いは臨床回顧によって確定する。副次的および探索的終点は、安全性と耐性測定、客観応答率またはOOR、反応持続時間、磁気共鳴画像評価の腫瘍体積変化および患者報告の結果を含み、疼痛、症状負担、身体/役割機能および全体生活の質の測定を含む。
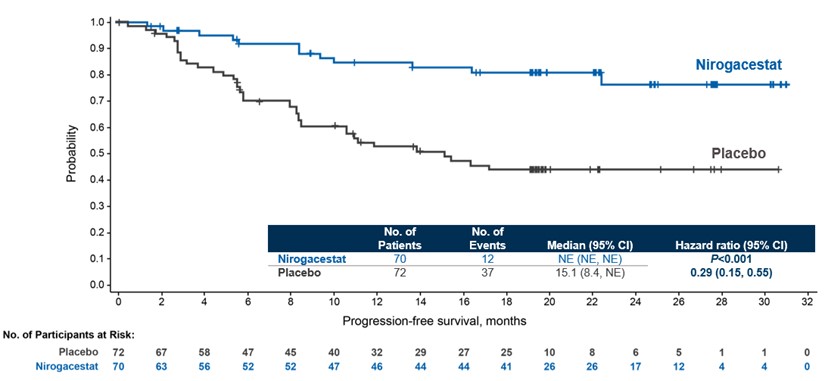
2022年5月にDEFI試験の陽性結果を発表し,2022年9月の欧州医学腫瘍学会大会でDEFI試験のより多くのデータを公表した
DIFI試験は無進展生存期間(PFS)の改善の主要な終点に達し、プラセボと比較してニロシ他群は統計学的に有意に改善し、疾患進展のリスクは71%(危険比(HR)=0.29(95%CI:0.15,0.55);pを低下させたことを示した
以下の患者報告の結果の中で、予め指定された二次治療効果の終点は客観的に有効であることが確認され(完全に有効或いは部分的に有効な参加者の割合と定義される)、そして第10周期の患者報告の結果の中で比較的にベースラインの変化が発生し、或いは利点:簡明疼痛アンケート表(BPI-SF)の平均最悪疼痛強度;Gounder/硬繊維様腫瘍研究基金会硬繊維様症状/影響尺度(GONDESS)硬繊維様腫瘍症候状尺度(DTSS)総症状採点;女神硬繊維様腫瘍影響尺度(DTIS)身体機能領域採点;及びヨーロッパ癌研究と治療組織生活品質アンケート-CORE 30[EORTC QLQ-C 30]全世界の健康状況/生活の質、身体機能と役割機能量表の得点。
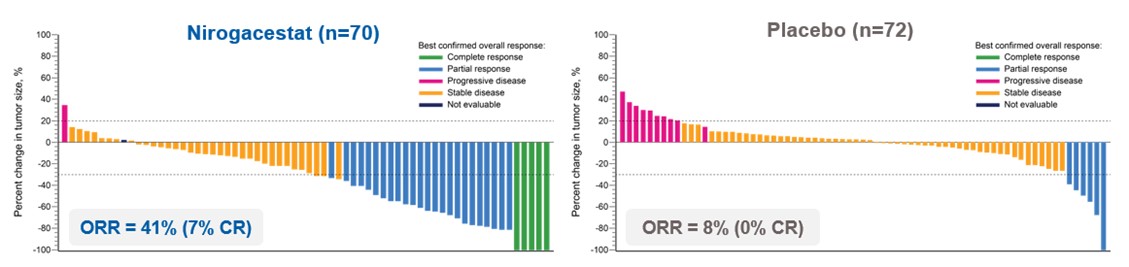
確認された客観的有効率(完全緩和+部分緩和)ニロシタン群41%,プラセボ群8%(p
ナイルキャストはPROSにおいて顕著な統計学的意義と臨床意義の改善を示した。具体的には、ナイルキャストは痛みを著しく軽減した(p
NIROGACEATはDEFI試験で管理可能な安全性を示し,TEAEの95%が1級または2級と報告されている。以下の表にすべてのTEAE(および対応する3級TEAE)を示し,報告は両群とも25%を超えている。プラセボ群と比較してニロセタ治療を受けた参加者の中で最もよく見られるTEAEは下痢,嘔気,疲労であった。ナイルカートリッジ服用群では42%の患者がTEAES服用により用量減少が必要であったが,プラセボ群では0%であり,ナイルカートリッジ服用群では20%の患者がTEAEs服用により治療を中止したが,プラセボ群では1%であった。研究者が報告した閉経、閉経、閉経と卵巣不全イベントは卵巣機能障害と定義され、75%(27/36)の生育潜在力のある女性はニロシター治療を受ける。これらのイベントは74%(20/27)の影響を受けた参加者で解決され、その中の64%(9/14)の参加者はまだナイルシター治療を受けており、100%(11/11)の参加者は任意の理由で治療を中止した。
| | | | | | | | | | | | | | |
| 安全人口、n(%) | ナイルキャスト(n=69) | プラセボ(n=72) |
| 薬物曝露持続時間,中央値(範囲),月の研究 | 20.6 (0.3, 33.6) | 11.4 (0.2, 32.5) |
| 用量強度、中央値(範囲)、mg/日 | 288.3 (169, 300) | 300.0 (239, 300) |
| どんな学年でも | ≥レベル3 | どんな学年でも | ≥レベル3 |
どんなTEAEでも | 69 (100) | 39 (57) | 69 (96) | 12 (16) |
報告されたどの学年のTEAEも両腕とも25%の患者が |
| 腹をくだす | 58 (84) | 11 (16) | 25 (35) | 1 (1) |
| 吐き気がする | 37 (54) | 1 (1) | 28 (39) | 0 |
| 疲れている | 35 (51) | 2 (3) | 26 (36) | 0 |
| 低リン血症 | 29 (42) | 2 (3) | 5 (7) | 0 |
| 皮疹、斑丘疹 | 22 (32) | 4 (6) | 4 (6) | 0 |
| 頭が痛い | 20 (29) | 0 | 11 (15) | 0 |
| 口内炎 | 20 (29) | 3 (4) | 3 (4) | 0 |
TEAEにより死亡しました | 0 | 1 (1)a |
TEAEによって減少しているかどうか | 29 (42) | 0 |
TEAEによる休職 | 14 (20)b | 1 (1)b |
______________________________
A)敗血症で死亡した。
B)1患者の薬物使用停止をもたらすTEAEは胃腸疾患(n=5)を含む[4%]卵巣機能不全(n=4)[3%])、アラニントランスアミナーゼ上昇(n=3[2%])、アスパラギン酸アミノトランスフェラーゼが上昇した(n=2[1%])と代謝/栄養障害(n=2[1%]).
ナイルキャストは卵巣顆粒細胞腫を治療した
卵巣顆粒細胞腫は性索間質腫瘍の中で最もよく見られる一種であり、すべての卵巣癌の約5%を占める。これらの腫瘍は2つのサブタイプに分けられます成人GCTと青少年GCTです患者に発生しています
年齢は30歳以下。GCTの約95%が成人GCTであると診断された。両亜型とも成長遅延が特徴であり,多くの成人GCTは早期に診断されている。GCTの症状は腹痛と異常膣出血を含み、一部の病例は月経過多、月経不順或いは出産適齢女性の閉経が出現する可能性がある。FDAが承認したGCT治療法は現在のところない。手術は主要な治療選択であり、組織学的診断、適切な分期と疾病の完全な除去を実現することを目的としている。疾患早期と出産適齢の患者では,片側卵管卵巣切除に腹膜分期を加えることが可能である。閉経後の女性と疾病の比較的に末期の女性に対して、典型的な外科治療方法は経腹全子宮切除、両側卵管卵巣切除、腹膜生検、すべての見られる疾患を切除することである。白金をベースとした化学療法および/または放射線治療は、メリットが大きくないにもかかわらず、末期または切除できない疾患に使用されている。しかし,成人GCT患者は再発の可能性が高く,約40%であり,通常確定診断の10年前に発生する。したがって,再発した疾患に対しては,追加的な治療選択が必要である。アメリカでは、GCT患者の年間発病率は約1500から2000名の患者である。
多種の細胞シグナル経路、例えば転化成長因子-β、Notch、PI 3 K/AKTなどは成人マクロファージ癌の発生発展に参与している。GCTの約97%はC 124 W突然変異によって駆動されていますFOXL 2FOXL 2蛋白をコードする遺伝子,FOXL 2は顆粒細胞の発育と機能に必要な転写因子である。臨床前のデータによるとNotchシステムは生存経路としてFOXL 2突然変異顆粒腫細胞株は、顆粒腫細胞の増殖を誘導し、アポトーシスを減少させる。ガンマ分泌酵素阻害剤はNotchシグナル経路の阻害剤であることが証明されている。臨床前、ガンマ分泌酵素阻害剤はNotch経路を抑制することは細胞の増殖と生存率を低下させることができるFOXL 2−変異顆粒腫細胞株。この理論的基礎に基づき,われわれはナイルキャストのGCT患者としての治療の潜在力を臨床的に評価している。2022年9月,1人目の患者が第2段階試験で用量を受け,単一療法治療として再発した成人GCT患者のナイルキセチンを評価したと発表した。
ナイルキャストとBCMA標的薬の併用
BCMAは多発性骨髄腫(MM)細胞に広く発現する細胞表面蛋白であり、BCMA標的治療薬物の臨床活性はすでにこの適応において実証された。GSISはMM細胞におけるBCMAの発現を増加させることができる。この結合機構の活性は複数の臨床前モデルで観察されており,これらのモデルはBCMA指導の治療を用いてGSIと結合し,最初の臨床データである。この組み合わせは,BCMAのみで指導された療法と比較して,応答率を向上させ,臨床的利益時間を延長したり,より低用量で投与することで副作用を減少させ,MM患者に有意義な臨床的メリットを提供する可能性があると信じている。
我々は,いくつかの業界パートナーとの臨床協力により,このような新たな併用治療法を評価している。2019年6月、著者らはグラクソ·スミスクラインと臨床協力を行い、再発或いは難治性多発性骨髄腫(RRMM)患者におけるニロシットと彼らのBCMA標的ADC Belamafの連合応用を探索した。Belamafを含めたBCMA指導の治療は,ニロシドのようなGSIを添加することで臨床活性を増強することができると考えられる。2020年6月,この組み合わせの適応性を評価する1 b期臨床試験の第1名患者の投与量を発表した。この試験の初歩的な臨床データは2022年に発表される予定だ。2021年10月、私たちは、第1の段階キューで観察された鼓舞的な予備データに基づいて、ベラマフおよびニューノキシットの0.95 mg/kg用量を3週間毎に評価する第1の同時用量レベルに基づいて拡大された第2の段階キューを開始することを発表した。著者らはまた、2つの新しいサブ研究を追加し、ベラマフとナイル西特、ポマドアミンとデキサメタゾンの併用及びレナドアミン+デキサメタゾンの併用によるRRMM患者の治療を探索することを発表した。グラクソ·スミスクラインは試験の進行と費用を担当し、試験は各方面が平等に代表する共同開発委員会が管理する。
2022年9月、著者らはグラクソ·スミスクラインと拡大した全世界の非独占的許可と協力協定を締結し、ナイルスタチンとbelantamab mafodotin(Belamaf)を多発性骨髄腫患者に併用した。拡大されたプロトコルは、早期の治療シリーズにおいてベラマフとナイルキャストの組み合わせを開発し、新たな診断のための多発性骨髄腫患者を含む商業化の潜在力を継続することを含む。私たちはNIOROCASTATのすべての商業権を維持し続けている。また,今後のベラマフ臨床試験にナイル西塔を供給し続け,グラクソ·スミスクラインとベラマフとの併用の承認が求められている市場での商業化を図る。グラクソ·スミスクラインは併用療法研究に関するすべての開発費に資金を提供しているが,ニロシット供給に関する費用や知的財産権に関する何らかの費用は除外している。共同療法の一部として、私たちはナイルキャストを商業化する費用を担当している
我々はいくつかの業界パートナーと他の単独の非排他的臨床協力を達成しており、各パートナーは1つまたは複数のBCMA標的療法を開発している。各パートナーは、RRMMにおけるニロシットとそのそれぞれのBCMA薬剤との組み合わせを評価するために、臨床試験の進行および費用を担当する。
著者らの臨床協力以外に、2020年9月、著者らはFred Hutchと賛助研究協定を締結し、Fred Hutchの研究者によって開発された各種の臨床前と患者由来の多発性骨髄腫モデルの中で、ナイルキャストはBCMAを調節し、BCMA標的治療の能力を増強する;2021年8月、著者らはDana-Farberと賛助研究協力を締結し、各種の臨床前多発性骨髄腫モデルにおいてBCMA標的治療のNagacestatを更に研究する。
疾患背景−多発性骨髄腫
MMは形質細胞腫瘍であり、発病率と死亡率はすべて非常に高く、アメリカで二番目によく見られる血液系悪性腫瘍であり、すべての血液系腫瘍の約10%を占める。NCIモニタリング、疫学と最終結果計画は、2016年にアメリカには約13万人のMM患者がいると推定されている。その中の約3万人が再発したり、現在利用可能な治療方法に無効であり、これは患者群がほとんど治療選択がないため、満足されていない医療需要が大きいことを意味する。2022年に米国では約13,000人が多発性骨髄腫で死亡すると推定されている。
MMの特徴は悪性形質細胞が骨髄中に拡張と異常に凝集し、正常な骨髄機能を撹乱し、時間の経過とともに貧血、高カルシウム血症、血小板減少、骨痛、疲労と体重減少を招くことである。疾病の発展に伴い、それは周囲の骨髄を破壊し、そして腎不全、感染感受性の増加、骨格退化と神経系疾患を招く可能性がある。
多発性骨髄腫の治療現状
多発性骨髄腫の治療は過去10年間に著しい進歩があり、これは疾病過程に対する更に深い理解と順序或いは多薬併用の方法によるものである。新たに診断された多発性骨髄腫患者は、幹細胞移植を受けるか、複数の治療薬の単独または併用治療を受けて、彼らの疾患の進行を制御しようとしている。これらの薬剤には、Bortezomibなどのプロテアソーム阻害剤、免疫調節薬、例えばレナドアミン、daratumabなどのモノクロナル抗体、Panobinostatなどのヒストン脱アセチル化酵素阻害剤、馬フランジなどのアルキル化剤、抗炎症薬、例えばデキサメタゾン、およびアドリアマイシンなどの化学療法薬が含まれる。現在これらの選択があるにもかかわらず、臨床反応と利益の持続性はしばしば短い。現時点では治癒と考えられる治療法がないため,最初の治療で生き残ったほとんどの患者は最終的に利用可能な治療法に抵抗力や難治性があると考えられ,彼らの疾患は進行している。これらの大量の前処理を経た患者がこのような末期状態に達した場合,彼らは通常臨床試験に導かれ,実験試薬で治療を行う。これらの患者の病状末期により,その疾患の難治性や従来の治療が彼らの健康に与える損失は,治療に対する反応が通常悪い。
BCMAは形質母細胞と分化した形質細胞表面でのみ発現の制限があるため、BCMAガイドの薬物はすでにRRMM患者を治療する潜在的な有望な方法になり、過去数年間に、いくつかのこのような薬物はすでに監督部門の許可を得た。臨床前と臨床開発では,少なくとも20個のBCMAに対する異なる計画が知られており,これらの計画はADDC,モノクロナル抗体,二重特異性抗体,自己CAR T細胞,同種異体CAR T細胞を含む様々な治療パターンを代表している。
GSIとBCMAガイドの薬物を組み合わせてRRMMを治療する他の人の努力も知られている。百時美施貴宝社の子会社Celgeneは現在、BCMA指導のADC CC-99712と2017年12月に礼来社から許可を得たGSI Crenigacestatとの組み合わせを評価している;この組み合わせは現在第1段階の臨床試験にある。CelgeneはCrenigacestatとその承認された自己BCMA指導のCAR T細胞療法ABECMA(ide−cel)との併用による1/2期臨床試験も評価している
タバコ曲司彼とBCMA標的治療の併用
BCMAを標的とした治療法は将来のMM治療モデルにおいて重要な役割を果たすと信じており,我々の各パートナーはBCMAを標的とした治療法を検討している中で独自の優位性を有している。ADCおよび二重特異性抗体は、従来の輸液プログラムおよび標準的な薬物製造、貯蔵および投与プロセスを含むいくつかの魅力的な特徴を有する。さらに,全治療過程において,ADCおよび二重特異性抗体の用量は容易に変化することができる。同種異体CAR T細胞療法の利点は,“既製”の細胞療法製品を一度に注入することにより,深い臨床的メリットを生じる可能性があることである。所与のBCMA標的治療方法に対する医師および患者の選好は、患者が看護を受ける治療環境、所与の患者の臨床的特徴、および治療の有効性および耐性に依存する可能性がある。
われわれの臨床経験とその耐性特徴から、臨床前MMモデルに基づくBCMA標的治療に基づいて、BCMA標的治療と結合し、NIOROCASTATは活発な用量であると考えられ、NIOROCASTATは注目された差別化GSIであり、BCMA標的治療と組み合わせて使用できる可能性があると信じている。NIOROCASTATはBCMA標的治療と比較して、MM細胞表面BCMAの高レベル発現を維持し、脱落した可溶性BCMA細胞外領域(ECD)による末梢抗原沈着を減少させることにより、臨床結果を改善する潜在力があると信じている。特に,この組み合わせは応答率を向上させ,臨床的利益持続時間を延長したり,BCMA標的治療を低用量でニロシドと併用することで副作用を減少させる可能性が考えられる。
組合せ作用機序
ある研究により、ガンマ分泌酵素は細胞膜に結合したBCMAを直接溶解でき、BCMA ECDが細胞表面から放出されることが明らかになった。γ分泌酵素を抑制することにより,膜結合したBCMAが保持され,標的密度を増加させるとともに,可溶性BCMA ECDのレベルを低下させることが可能であり,餌受容体である可能性がある。多発性骨髄腫の複数の臨床前モデルでナイルカル司がBCMA指導治療活性を増強する能力が観察され,2019年12月に61回であったST著者らの協力者グラクソ·スミスクラインとbelantamab mafodotin(Belamaf)が共同でアメリカ血液学会年会で発表した初歩的な臨床データ、及び著者らの協力者精密生物科学会社が2021年12月にアメリカ血液学会年会で提出した初歩的な臨床データ、及び著者らの協力者グラクソ·スミスクラインが2022年ASCO年会で提出した初歩的な臨床データである。
BCMAを発現するMM細胞に対するBCMA指向抗体-薬物結合体の活性は、(1)その細胞毒性ペイロードを標的に伝達すること、(2)抗体依存性細胞毒性、(3)リガンド結合を遮断することによりBCMA受容体のシグナル伝達を阻害すること、および(4)免疫原性細胞死の4つの可能な機序に起因する。同種異体CAR T細胞治療のBCMAを発現するMM細胞に対する活性は、T細胞を直接介したBCMAを発現するMM細胞の殺傷によって駆動される。BCMAxCD 3二重特異性抗体の活性はT細胞の募集と活性化によって駆動され、BCMAを発現するMM細胞を殺傷する。BCMAに対するモノクロナル抗体の活性はBCMAを介した生存促進と増殖シグナルを遮断し、抗体依存の細胞貪食作用を媒介し、抗体依存の細胞毒性を増強したためである。
下図はガンマ分泌酵素を介した膜結合BCMA切断を減少させ,癌細胞上の標的密度(BCMA)の増加と誘導受容体(可溶性BCMA ECD)レベルの低下をもたらすGSI(BCMA配向ADCとの結合を示す)の効果を示した。
ミダメチニブ
概要
ミダミニーは研究中の経口小分子MEK 1とMEK 2阻害剤である。MEK蛋白はMAPK経路において重要な地位を占め、MAPK経路は細胞成長と生存を調節する重要なシグナルネットワークであり、多種の腫瘍学と稀な疾病適応において核心的な役割を果たしている。
著者らは最初にミダミニーを単一療法としてNF 1-PN患者を治療することを研究しており、これは稀な疾患であり、その特徴はMAPK経路突然変異による末梢神経鞘腫の成長を招き、深刻な疼痛、容破壊と発病率を招く。2018年10月と2019年7月、FDAと欧州委員会はそれぞれNF 1を治療するミダミニー孤児薬物称号を授与し、2019年5月、FDAはNF 1-PNを治療するミダミニー快速チャネル称号を授与した。
神経線維腫症臨床試験連盟は2期臨床試験を行い,19名のNF 1−PN患者のミダメチニブを評価した。この臨床試験では,42%の患者が12カ月の治療で客観的な反応を経験した(彼らの目標PN腫瘍体積は少なくとも20%減少したと定義されている)。これらのデータとFDAとの相互作用に基づいて、著者らは2019年10月にNF 1-PN患者におけるミダミニーの潜在的登録単一アーム開放ラベル2 b期ReNeu臨床試験を開始した。ReNeu試験の主要な終点はORRであり,客観的反応は体積MRI評価により決定された腫瘍体積がベースラインより少なくとも20%減少すると定義されている。臨床試験の結果が良好であれば、私たちはアメリカでミダミニーの発売許可を申請し、具体的な国はまだ最終的に確定していないにもかかわらず、国際市場を選択する予定だ。
我々のNF 1−PNにおける単一治療計画に加えて,ミダメチニブは腫瘍学的多標的併用治療や他の遺伝子定義腫瘍の治療への応用が期待できると信じている。我々の最初の仕事は,ミダミニと百済神州のRAFダイマー阻害剤リフェラファニー(BGB−283)の併用を評価することである。2019年5月,百済神州によるこの組み合わせの適応1 b/2期臨床試験の開始を発表した。この試験は現在米国とオーストラリアの末期あるいは難治性固形腫瘍患者を募集しており、これらの患者はMAPK経路に関連する遺伝子変異が存在し、著者らは2022年6月に会社が協賛した研究開発日にこの試験の初歩的な臨床データを報告した。また,スローン·キャトリン癌センター後援,SpringWorksにより支持されている1 b/2 a期臨床試験では,ミダミニとfulvestrantの併用によるエストロゲン受容体陽性転移性乳癌患者,MEK 1またはMEK 2活性化変異の存在のための単一療法として固形腫瘍患者を評価している。聖徳児童研究病院と協力して、著者らは1/2期の臨床試験を行い、ミダミニによる児童と若い低レベルグリオーマ患者の治療効果を評価している。
米デルタチニブによるNF 1−PNの治療
疾患背景
NF 1は稀な常染色体優性遺伝性腫瘍感受性疾患であるNF 1この遺伝子は神経フィブリンをコードし、それはMAPK経路の重要な負の制御因子である。NF 1は最もよく見られる神経線維腫症であり、全世界の出生発病率は約3000人に1人と推定されている。著者らはアメリカに約100,000人のNF 1患者がいると推定している。NF 1は臨床異質性を有し、異常色素沈着、骨格奇形、腫瘍成長と神経合併症、例えば認知機能障害などの多くの器官系の各種症状として表現されている。一般群と比較して,NF 1患者の平均期待寿命は15年短縮された。
NF 1患者の叢状神経線維腫或いはPNの生涯リスクは約30%~50%であり、PNは末梢神経鞘に沿って浸潤性モードで成長する腫瘍であり、深刻な毀容、疼痛と機能障害を招くことができる;ごく少数の情況下で、NF 1-PNは致命的である可能性がある。NF 1−PNの最も一般的な診断は生命の20年前であり,通常のイメージング技術で確認できる。これらの腫瘍の特徴は侵襲性成長であり,通常小児期にはより速く成長する。NF 1−PNは通常自発的に回帰しない。
NF 1−PNは良性であるが,これらの腫瘍は悪性形質転換を起こし,悪性末梢神経鞘腫,あるいはMPNSTをきたす可能性がある。NF 1患者のMPNSTの生涯リスクは8%から15%であり、診断結果はその12ケ月の生存率は50%より低いことを示した。MPNST以外に、NF 1患者の他の悪性腫瘍に罹患するリスクは増加し、乳癌とグリオーマを含む。
NF 1−PNの治療の現状
MEK阻害剤であるKoselugo(Selumetinib)は2020年4月にFDAにより2歳以上の有症状,手術不能のPNを有するNF 1小児患者の治療に許可され,症状があり手術できない成人NF 1患者の治療に用いられる第3段階臨床試験が行われている。手術切除はNF 1−PN患者のもう一つの治療選択であるが,腫瘍を切除するには広い切縁が必要であり,これはNF 1−PN患者ではあまり達成できない結果である。これはNF 1-PNが神経細胞に起源し、浸潤性モードで成長するため、永久性神経損傷と毀容などの深刻な合併症が出現しないため、腫瘍の切除に成功し、挑戦性があるからである。手術条件を満たしていない患者や術後再発の患者は通常採用されています
様々なラベル外療法ですこれらのラベル外の療法には,化学療法や免疫療法など様々な全身療法があるが,持続的な臨床的メリットは示されていない。
手術の不足はシステム療法の改善の必要性を示唆している。NF 1−PNはMAPK経路の過活性化によって駆動されることから,MEK阻害剤はNF 1−PNの治療が期待される治療法となっており,MEK阻害剤が治療の基準となる可能性が信じられている。
Koselugoに加えて,少なくとも他の2つのMEK阻害剤が研究者が支援しているこの適応の第2段階臨床試験が行われており,他の腫瘍学的適応のために承認されたMEK阻害剤を含み,NF 1−PN患者のタグ以外に使用されることがあることが知られている。NF 1−PNの終身性と破壊性,若年からの治療が必要であることから,最適なMEK阻害剤は長期投与に適した耐性特徴を有し,腫瘍成長を阻止あるいは逆転すると考えられる。
米デルタチニブによるNF 1−PNの治療
ミダミニーは経口MEK 1とMEK 2の小分子阻害剤であり,NF 1−PNの単一療法として開発されている。先の臨床試験の結果から,NF 1−PN第二段階臨床試験の用量とスケジュールを用いたミダミニーは,他のMEK阻害剤と比較して,この患者集団に必要な長期治療を可能にするために,潜在的な同種の最適なプロファイルを提供する可能性が考えられる。これまでのNF 1−PN臨床試験で観察されたミダミニーの臨床活性と耐性を考慮し,FDAと検討した後,行われている潜在登録2 b期臨床試験またはReNeu試験を設計し,小児および成人NF 1−PN患者の承認を支援するのに十分なデータを生成できると信じている。ReNeu試験の結果が有利であれば、米国でミダミニーの上場承認を申請し、具体的な国が最終的に確定していないにもかかわらず、国際市場を選択する予定だ。
作用機序
神経フィブリンはRASシグナルの重要な阻止物であり、突然変異しているNF 1遺伝子は,MAPK経路の構造的活性化を引き起こす。MEK阻害剤はMAPK経路の活性を低下させ,NF 1−PNの成長を阻止または逆転させることが臨床的に観察されており,ミダミニーを含むいくつかのMEK阻害剤が観察されている。
米デルタチニブによるNF 1−PN治療の臨床研究進展
ミダミニーは神経線維腫症臨床試験連盟が行った前の第2段階臨床試験で臨床活性を示し,青少年19名と成人NF 1−PN患者を募集し,この臨床試験のデータを2021年の“臨床腫瘍学雑誌”に発表した。患者は2 mg/mの経口投与を受けた2 BIDは,最大投与量4 mg,BID(食物を考慮しない),4週間を1周期とし,3週間から1週間休む。8人の患者(42%)は第12周期で客観的反応を達成し、目標PN容量が少なくとも20%減少し、10人の患者(53%)の病状が安定していると定義されることが予想される。この試験では,ミダミチニブの耐性は一般に良好であり,4級や5級の副作用の報告はない。同一患者に発生した2つの治療関連3級事件を報告した。どのレベルでも最もよく見られる副作用はざ瘡様皮疹(95%)、疲労(58%)と吐き気(53%)である。この臨床試験におけるミダミニーの活性と耐性を考慮して、著者らは潜在的な登録2 b期ReNeu試験において同じ用量とスケジュールを使用した。また,FDAとの検討により,青少年や成人患者のほかに,小児NF 1−PN患者を募集した。
ミダミニーはすでに広範な用量範囲(1 mg qdから30 mg bid)の単一療法臨床試験で試験されており,最大耐容量(MTD)は15 mg bidと決定され,推奨される第2段階用量は10 mg bidと決定され,5日間,2日間休講されている。
これまでミダミニーの投与量は10 mg以下であり,2回/dの進行癌患者における間欠療法の安全性の特徴は毒性が制御可能で可逆的であることが多く,ミダミニー全体の耐性が良好であることが認められた。これらの副作用の中で最もよく見られるのは皮疹,嘔気,嘔吐,下痢と疲労である。
NF 1−PNの2 b期ReNeu試験
これまでのミダミニー第2段階臨床試験で観察されたNF 1−PN患者の臨床的利益の程度を考慮し,FDAとの検討から,2019年第4四半期に登録可能なReNeu臨床試験を開始したことが分かった。ReNeu試験は2 b期縦方向開放ラベル臨床試験であり、少なくとも2歳のNF 1-PN患者におけるミダミニーの有効性、安全性と耐性を評価することを目的とし、この患者のNF 1-PNは手術できず、明らかな発病率を招く
深刻な奇形でもありますReNeu試験は北米の臨床地点で行われている。これまでにNF 1−PN患者に対して行った第2段階臨床試験と同様に,ミダミニーの経口速度は2 mg/mであった2 BID用量は,最大投与量4 mg,BID(食物を考慮しない)であった。投与周期は4週間で,3週間ごとに始まり,1週間で終了する。24サイクルまで続くと予想されている。これまでの2期臨床試験とは異なり,ReNeu試験を設計し,介入期間は最適化されていると考えられ,NF 1−PN患者におけるミダミニーの全面的な抗腫瘍活性を示した。
2021年11月、我々は2 b期ReNeu試験の全面登録を発表し、50人以上の成人と50人以上のNF 1-PNを有する小児科患者が参加した。主な終点は3次元MRI体積分析を用いてOORを測定し,ブラインド法による独立中央審査(BICR)による評価である。客観的反応は,先の2期臨床試験と同様に,BICRにより標的NF 1−PN中の腫瘍体積を少なくとも20%減少させると定義されている。重要な副次的な終点は、反応持続時間と健康に関連する生活の質測定を含む。2023年下半期にReNeu試験の背線データを報告する予定である。
中期臨床データ
2021年2月,2 b期ReNeu試験に参加した最初の20名の成人患者の中期臨床データを報告し,2021年6月に小児腫瘍基金会NF会議でこの20名の患者の最新データを公表した。これらの患者の人口統計データとベースライン特徴を以下の表に示す。
| | | | | |
| 特徴.特徴 | n (%) |
| 登録された患者 | 20 |
| 入学時の中位年齢[日程を測る]-年だ | 33.5 [19 – 69] |
性 男性 女性は |
4 (20)
16 (80) |
標的神経線維腫の定位 頭と首 四肢.四肢 胸壁 脊椎のそば 上肢.上肢 他にも |
9 (45)
6 (30)
1 (5)
1 (5)
1 (5)
2 (10) |
神経線維腫関連合併症のタイプ 痛みがある 重大な奇形 運動機能障害/虚弱 下肢.下肢 上肢.上肢 入院時PNの進展 視神経膠腫 気道機能障害 他にも |
20 (100)
10 (50)
10 (50)
7 (35)
3 (15)
6 (30)
2 (10)
1 (5)
3 (15) |
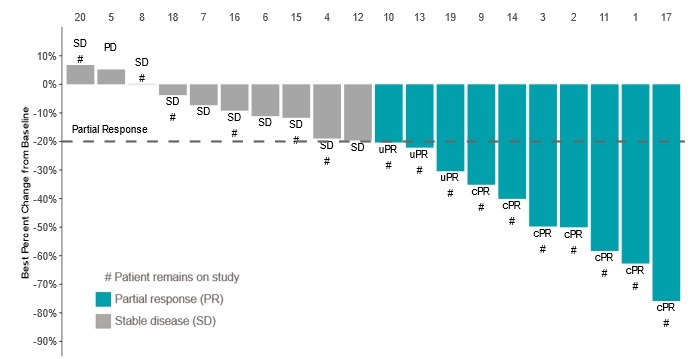
2021年3月23日までのデータ締め切りでは,この20名中10名(50%)が客観的緩解を実現し,BICR評価により腫瘍体積は少なくとも20%減少した。下のグラフは上位20名の成人患者の腫瘍体積のベースラインと比較した最適百分率変化を示している。初歩的な客観的反応が得られた10名中7名に部分反応が確認された
______________________________
PD:進行性疾患;PR:部分寛解(腫瘍体積20%縮小と定義);CPR:確定診断部分寛解;UPR:確定診断されていない部分寛解;SD:安定期
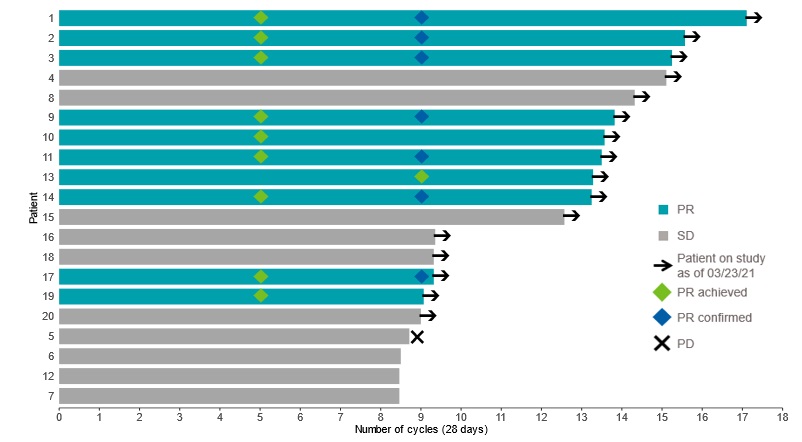
評価を受けた20名の患者の治療コースを以下に表に示す。2021年3月23日までのデータ遮断日まで,16名の患者(80%)が治療を受けており,4名の患者は治療を中止し,そのうち1名の患者は疾患が進行し,1名の患者が1級下痢の不良事象が出現し,1名の患者が決定し,1名の患者は非治療関連の脊柱側弯の悪化によりチタン棒を移植して必要なMRI画像を受けられなかった。データ切断時には,この20名の患者の中位治療期間は13周期(約12カ月)であり,客観的な反応を得た10名すべての患者が治療を受けている。
______________________________
PR:部分的寛解(腫瘍体積20%減少と定義);SD:病状安定
2021年3月23日までのデータ遮断日まで,ミダメチニブは全体的な耐性が良好であった。治療に関連する有害事象(TRAE)の多くはレベル1または2であり,レベル3皮疹は1回しか報告されておらず,レベル4やレベル5の有害事象の報告はない。1人の患者はグレード3の皮疹が出現したため、用量を減少させる必要がある。表に最もよく見られるTEAEを示し,評価を受けた患者20名中15%が発生し,データ締め切りまでの3級TEAEとTRAEであった。
| | | | | | | | | | | | | | | | | |
| 治療−救急AEs(患者の15%を占める) | 治療に関連する副作用 |
| 全学年 n (%) | 3年生 n (%) | 4年生 n (%) | 3年生 n (%) | 4年生 n (%) |
不良事件 |
少なくとも1つのAE | 20 (100) | 3 (15) | — | 1 (5) | — |
ざ瘡皮膚炎/皮疹/ 斑丘性皮疹 | 18 (90) | 1 (5) | — | 1 (5) | — |
吐き気がする | 12 (60) | — | — | — | — |
腹をくだす | 10 (50) | — | — | — | — |
疲れている | 6 (30) | — | — | — | — |
腹痛がする | 6 (30) | — | — | — | — |
吐く | 5 (25) | — | — | — | — |
肌が乾燥している | 4 (20) | — | — | — | — |
駆出点が下がる | 4 (20) | — | — | — | — |
便秘する | 3 (15) | — | — | — | — |
呼吸が苦しい | 3 (15) | 1 (5) | — | — | — |
胃食道逆流症 | 3 (15) | — | — | — | — |
関節が痛い | 3 (15) | — | — | — | — |
耳が痛い | 3 (15) | — | — | — | — |
尿路感染 | 3 (15) | — | — | — | — |
コロナウイルス感染 | — | 1 (5) | — | — | — |
コロナウイルス試験は陽性であった | — | 1 (5) | — | — | — |
頭が痛い | — | 1 (5) | — | — | — |
非心源性胸痛 | — | 1 (5) | — | — | — |
脊柱側弯 | — | 1 (5) | — | — | — |
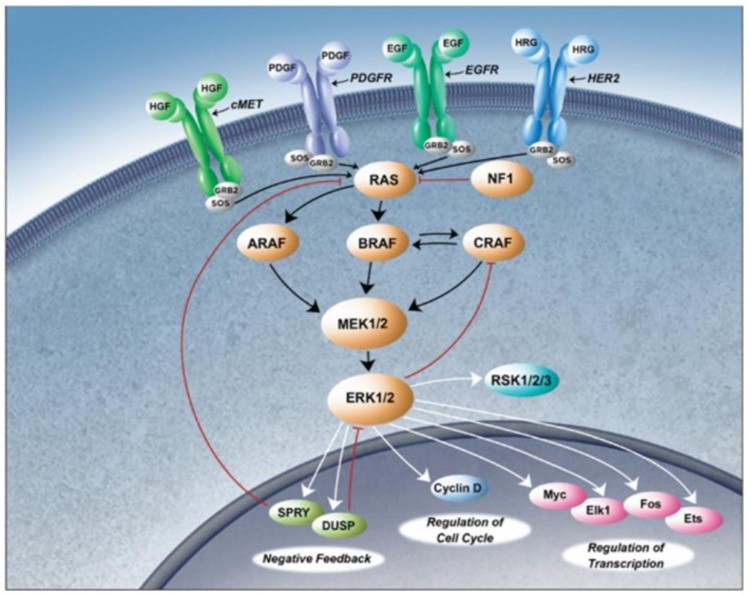
MAPK経路計画
概要
われわれはいくつかの単一療法や併用療法が開発されており,MAPK経路変異の固形腫瘍の治療に用いられている。これらの方法は、ミダミニを百済神州のRAFダイマー阻害剤リフェラフェニと組み合わせて使用すること、およびBGB-3245との単一治療方法を含み、BGB-3245は、研究における経口選択性小分子活性化モノマーおよび二量体阻害剤であるBRAF百済神州が所有するMapKureとの変異による固形腫瘍患者の治療RASあるいは…。ロイヤル空軍突然変異です。
他のMAPK異常を含む固形腫瘍に対するミダミニーの治療作用も学術機関と協力して評価している。SpringWorksによって支持されている学術賛助試験は、聖徳児童研究病院が後援した低レベルグリオーマを有する児童と若者に対する1/2期臨床試験と、スローン·キャトリン癌センターが後援した1 b/2 a期プラットフォーム研究を含み、2つの患者行列がある:最初の評価ミダミニー連合Fulvestrantの臨床試験:最初の評価ミダミニー連合Fulvestrantは、エストロゲン受容体陽性の転移性乳癌MAPK変化(特にMAPK遺伝子不活化変異)のための選択的エストロゲン受容体分解剤であるNF 1)、および第2の評価ミダメチニブは、単一療法として腫瘍原性を有する進行固形腫瘍を治療するMEK 1あるいは…MEK 2突然変異です。
疾患背景
MAPKシグナル経路の研究進展
MAPK経路はRAS-RAF-MEK-ERKシグナル経路の下落に依存し、すべてのヒト細胞の中で1本の中央生物経路を代表し、細胞の転写、増殖と生存の調節を担当する。この経路の全体構造は1つの小さいGTP酵素RASと3つの下流のプロテインキナーゼRAF、MEKとERKからなる。また、RASのレベルでは、この経路は神経フィブリンを含むいくつかのタンパク質の負の制御を受けており、このタンパク質はNF 1ジーン。それは直接ERKを調節し、ERKはMAPK経路を通じて直接に下流シグナルを制御するため、MEKはこのシグナル伝達経路の中で重要な地位を占め、そして1つの合理的な治療標的を代表し、MAPK経路の過剰活性化は疾病の発生と/或いは進展と密接に関連する適応を処理するために用いられる。
ヒト癌の約25%では,MAPK経路が結腸癌,肺癌,乳癌,膵癌,卵巣癌,腎癌を含む構造的に活性化されることが報告されている。経路活性化の原因は多種多様であり、組織特有であるが、以下の1つまたは複数の機構によって駆動され、各機序は、以下の図に示すように、(I)EGFRのような1つまたは複数の受容体チロシンキナーゼの上流活性化、(Ii)RAS異性体、例えばKRAS(Iii)経路内の他の変異または異常、例えば、(Iii)BRAFそしてNF 1.
MAPK経路を垂直に抑制する2つの重要な隣接成分,ミダミニーとRAF阻害剤,例えばlifirafenibやBGB−3245の組み合わせにより,抵抗機構やフィードバックループを解決する可能性があり,これらの機構やフィードバックループは,MAPK経路遺伝子変異を持つ多くの破壊的癌の治療開発を阻止していると信じているRAS, ロイヤル空軍そしてNF 1.
現在承認されているRAF阻害剤は、モノマー形態によるBRAFのシグナル、例えば、腫瘍を治療するために設計されているBRAFV 600変異、転移性黒色腫によく見られるMAPK異常のサブセット。この場合,BRAF V 600阻害剤にMEK阻害剤を添加し,単一治療BRAF阻害よりも有意な臨床活性を示した。RAFを標的とする単量体および二量体形態、RAF二量体阻害剤、例えばリフェラファニーなどの、腫瘍を治療するために設計されたものだけでなく、BRAFV 600は変異しています
より広範な遺伝的定義を解決するための患者集団ですこれにはRAS主に異種およびホモ二量体RAFによってシグナルを発する変異癌であって、両方のコンホメーションがlifirafenibによって解決される可能性がある。
RAS突然変異
RAS突然変異は人類癌の中で発見された最もよく見られる遺伝異常の一つであり、これらの駆動突然変異はすべての固形腫瘍の中で約25%を占め、毎年アメリカで新しく診断された20万人以上の患者に相当する。RAS蛋白はKRAS、HRASとNRASの3種類の亜型から構成され、受容体チロシンキナーゼシグナル伝達において中心的な役割を果たし、そしてMAPK経路中の典型的なRAF-MEK-ERKシグナル低下の下流活性化を招く。
効果的な治療法は患者を助けると信じていますRAS突然変異は重要な臨床需要を代表する。これまで単一療法として使用されてきたMEKやRAF阻害剤は,腫瘍患者では限られた臨床活性のみを示すのが一般的であるRAS突然変異です。これらの腫瘍は標的治療に対する反応が一般的に悪くRAS変異は通常、癌タイプによって結果が異なるかもしれないが、不良な予後をもたらすRAS突然変異です。
RAF変異
ロイヤル空軍すべての固形腫瘍の7%に変異が存在することが報告されており,その中で最も広く記述されているのはBRAFV 600変異は、通常転移性黒色腫患者で発見される。MEK-RAF標的併用療法は承認されていますがBRAFV 600突然変異、患者は最終的にこれらの治療に進展を得て、重大な満足されていない臨床需要を代表している。
また多くの非V 600がありますBRAF記載された変異は現在承認されている治療には反応しないが、既存の治療の使用は、腫瘍細胞とこれらの非V 600の能力を矛盾的に増加させることが証明されているBRAF突然変異が広がります
他のMAPK像差
MAPK経路に変異と異常が存在する患者RASそしてロイヤル空軍すでに承認された治療方法が不足しているため、突然変異はまた大量の満足されていない臨床需要を代表している。このような腫瘍にはNF 1MPNSTのような突然変異です
現在の治療景観
現在2つの承認された固形腫瘍を治療する方法があり、その中には1つの特殊なものが含まれているRAS-突然変異です。2021年5月、安進のLUMAKRAS(ソトラシブ)、RAS GTP酵素ファミリー阻害剤の一種であり、アメリカ食品と薬物管理局の加速許可を得て、成人患者の治療に応用したKRASG 12 C変異は局所末期または転移性非小細胞肺癌であり、これらの患者は以前に少なくとも1回の系統的治療を受けたことがある。2022年12月、Mirati治療会社のRas GTPaseファミリー阻害剤KRAZATI(Adagrasib)はFDAの加速承認を得て、罹患の治療に使用したKRASG 12 C変異は局所末期または転移性非小細胞肺癌であり、これらの患者は以前に少なくとも1回の系統的治療を受けたことがある。LUMAKRASやKRAZATI以外にも多くのプロジェクトが行われていますRAS変異固形腫瘍、様々な作用機序が評価されており、特定のRAS突然変異です。
適用することができますロイヤル空軍変異は現在FDAが非V 600ウイルスを持っている患者を治療するための治療法を承認していることはわかりませんBRAF突然変異です。いくつかの承認された治療法は以下のような場合に適用されるロイヤル空軍突然変異は頻繁ですが解決するためのものはありませんがロイヤル空軍標的治療以外の変異はBRAFV 600変異は,これらの変異に対しても満たされていない医療ニーズが存在し,最終的にこれらの治療法が進展するためである。
腫瘍に他のMAPK異常を有する患者では,現在FDAがどのような治療法を承認しているかは知られていない。このMAPK経路異常が頻繁と考えられる適応には,これらの異常を解決するために専用に設計されているわけではないにもかかわらず,いくつかの承認された治療法がある。
ミダメチニブとリフェラフィニの併用によるMAPK変異固形腫瘍の治療
ミダミニとリフェラファニーの併用による抗腫瘍活性を示す臨床前データRAS突然変異癌モデルは2015年アメリカ癌研究協会(AACR)会議で発表された。この臨床前研究において、多種のMEK阻害剤とリフェラファニーの連合応用を評価し、ミダミニは協同作用が最も強く、抗腫瘍活性が最も強いMEK阻害剤の一つであることが観察された。より多くの前臨床データ
2020年AACR仮想年会で発表されたミダミニとリフェラファニーの共同応用の研究により、体外と体内の1組の癌モデルの中で、ミダミニとリフェラフィニは有効かつ協同的な活性を有し、このモデルは多種の種類を含むことが示されたRAS突然変異です。百済神州のリフィニは完成した一期臨床試験で抗腫瘍活性を示したRASそしてロイヤル空軍変異した固形腫瘍は,一般的な耐性が良好であった。
2019年5月,我々は適応1 b/2期臨床試験を開始し,ミダミニとリフェラファニーの組み合わせを評価することを発表した。この臨床試験はMAPK経路に関連遺伝子変異が存在する末期あるいは難治性固形腫瘍患者を募集した。この臨床試験は百済神州と私たちがアメリカやオーストラリアで協力して行った。臨床試験は2段階に分けられる。第1段階では、併用治療のMTDおよび推奨される第2段階用量を決定する予定である。第2段階では、対象の腫瘍タイプおよび変異背景を有する患者からなる拡大キューに組み込まれることが予想され、非小細胞肺癌および子宮内膜癌が含まれる可能性があるKRAS推奨される第2段階用量の併用治療の抗腫瘍有効性、安全性および耐性を評価するための変異。行っている試験の予備臨床データは2022年6月にわが社が協賛した研究開発日に公表され,概念検証が示された。
遺伝子定義BRAF変異型固形腫瘍におけるBGB-3245の役割
2019年6月、私たちと百済神州が共同で所有するMapKureの設立を発表しました。百済神州はMapKureにBGB-3245の独占使用権を与え、BGB-3245は新型、試験的経口、選択性単量体と二量体活性化形式の小分子阻害剤であるBRAFV 600を含む変異はBRAF突然変異、非V 600 BRAF突然変異とRAF融合。MapKureは固形腫瘍患者の臨床開発によりBGB−3245を進めているBRAF駆動因子の変異とBRAF臨床前研究ではこの化合物に対する敏感な融合が観察された。2020年2月、MapKure、百済神州とSpringWorksは1つの第1段階用量の増加と拡張臨床試験を開始することを発表し、成年末期或いは難治性固形腫瘍患者におけるBGB-3245の応用を評価し、これらの腫瘍は特定の遺伝子突然変異を有し、臨床前の結果により、これらの突然変異はBGB-3245治療に敏感であることが予想される。行っている試験の予備臨床データは2022年6月にわが社が協賛した研究開発日に公表され,概念検証が示された。2023年上半期の医学会議では,1期試験からの単一療法データがより多く公表される予定である。2023年2月、第1の患者は、1/2 a期開放ラベル、用量増加、および拡大試験において用量治療を受け、ミダミニーとBGB-3245の併用によるMAPK活性化変異を含む末期固形腫瘍患者の治療効果を評価した。
MapKureにおける私たちの重要だが持ち株ではない持株を除いて、私たちはMapKureの共同指導委員会と取締役会にそれぞれ1つの席を持っている。我々はまた,MapKureとのサービスプロトコルによりBGB−3245の臨床開発や他の業務活動に貢献している。
臨床前データから,BGB−3245はそのBRAF結合や解離特性において唯一無二である可能性が考えられ,他の既知のRAF阻害剤と比較して差別化された抗腫瘍活性を有する可能性がある。BGB−3245を臨床開発に位置づけ,特定のバイオマーカーとして定義された患者集団の単一療法に位置づけられる可能性があると信じている。これらのバイオマーカーには最初から2つ目の種類がありますBRAF突然変異して最初から始めるBRAF融合とBRAFBRAF V 600阻害剤による治療後の薬剤耐性変異。
これまでに約200個のユニークな変異体がありますBRAFヒト腫瘍に対立遺伝子が発見されています活性化していますBRAF突然変異は3種類に分類される:1種類の変異体、V 600 E変異などの活性単量体から構成される変異体、2種類の変異体、活性二量体を構成する3種類の変異体、キナーゼ損傷或いは死亡した3種類の変異体。今日、1種類のBRAF変異のみが、BRAF V 600 E/K変異の治療のための転移性黒色腫のウィモラファニブ、ダプラファニー、およびエンケラフィニなどの承認された標的治療レジメンを有する。2017年までの科学文献に記載されているBRAF変異の分布を表にまとめた。
承認されたBRAF阻害剤は1種類の患者において臨床活性を有するにもかかわらずBRAF変異、新たに出現した証拠は、薬剤耐性は、通常、p 61のようなタンパク質の二量化によってリガンド独立なシグナル伝達を達成する変異によって形成されることを示しているBRAFV 600 EおよびBRAFV 600 E/L 514 Vは、満たされていない医療需要領域を表している。BGB-3245はすでにこれらの突然変異に対する臨床前活性を証明した。
また,BRAF融合蛋白は最近癌細胞成長の駆動力として記述されており,現在では臨床環境で患者のこのような融合スクリーニングが可能となっている。最近の文献により、これらの突然変異はすべての人類癌の0.3%を占める可能性があり、現在12種類の異なる腫瘍タイプの中で20種類の新しいBRAF融合が発見され、そして特定の癌の中で豊富である。BGB−3245は,これらのBRAF融合患者にも適用可能であると考えられる。
米デルタチニブは他のMAPK異常固形腫瘍を治療した
単一療法や併用療法を含む他のMAPK異常が存在する固形腫瘍に対するミダミニーの治療も評価されている。2021年6月、私たちはミダミニによる児童と若者の低レベルグリオーマ治療の1/2期臨床試験を開始することを発表した。この研究は聖ユダ児童研究病院が後援し,SpringWorksが支援している。第一段階用量増加研究の初期データは2022年小児科神経腫瘍学国際シンポジウムで公表された。2021年8月,我々はスローン·キャトリン癌センター後援,SpringWorksによって支持された1 b/2 a期プラットフォーム研究におけるミダミニーの評価を発表し,この化合物を単一療法および併用療法としてMAPK活性化変異を含む進行固形腫瘍の治療を探索した。この試験は2021年第3四半期にスタートし、2つの患者列におけるミダミニーの応用を初歩的に探索した:最初の連合投与fulvestrant、1種の選択性エストロゲン受容体分解剤、エストロゲン受容体陽性の転移性乳癌MAPK変化(特に不活化突然変異)に応用したNF 12つ目は単一療法として末期発癌固形腫瘍を治療することですMEK 1あるいは…MEK 2突然変異です。
許可と協力協定
ファイザー許可協定
我々は最初にファイザー社から構想され,サービス不足の患者に大きな希望を持つ可能性のある研究療法を革新的な方式として進めてきた。ファイザーは最初に株式投資を行い、特許使用料とマイルストーンの意味を持つ製品許可証を提供し、私たちの2つの主要な候補製品であるナイルキャストとミダメチニブを含む。
Niroacestatライセンスプロトコル
2017年8月、私たちはファイザー社とライセンス契約、あるいはナイルシタン許可協定を締結し、これにより、すべての疾患の治療、診断、予防のための研究、開発および生産、ならびにアルツハイマー病、乳癌および前立腺癌以外のすべての疾患の治療、診断および予防のためのナイルキセチンの世界的独占的再許可権を獲得した。また,ファイザー社は10年以内に靭帯様腫を治療するガンマ分泌酵素阻害剤の臨床試験を行わないことに同意した。ファイザー社はアルツハイマー病、乳癌、前立腺癌を治療するナイルキセチンを商業化する権利を保持している。その後、私たちは2019年7月に知的財産権に関連するいくつかの条項の“ナイルカシタ許可協定”を改正した。
改正されたNiRogacestatライセンス協定によると、ビジネス的に合理的な努力で米国で少なくとも1つの製品の規制承認を求める義務があり、規制承認を受けた場合、米国で製品を商業化する義務がある。米国で規制承認を受けた後、特定の国/地域である程度の補償を受けることが合理的に予想される場合には、その国/地域で製品開発と規制承認を求める義務があり、規制承認を受けた場合は、その国/地域で製品を商業化する義務がある。
私たちはいくつかの商業マイルストーン事件を完了した後、ファイザーに合計2.325億ドルを支払うことを要求された。
ファイザーに中位数桁から20桁以上の割合でファイザー等級特許権使用料を支払うことになり,有効クレーム満期,第三者許可満期金額,後発薬競争の控除を受ける可能性がある。
事前に終了しない限り、ナイルカスター許可協定はすべての印税義務が満了した時に無効になるだろう。特許権使用料期間は、(I)最初の商業販売から10年、(Ii)すべての法規またはデータ排他性満了、および(Iii)最後に満了した有効特許請求の期限が満了する後に満了する。ある国·地域に適用される印税期限が満了した後,その国/地域の印税期間内に許可を得たすべての知的財産権に永久的で全額支払われる非独占許可を付与し続ける。便宜上、30日前にNiroacestatライセンス契約を終了する権利があります。ファイザーは便宜のために合意を終わらせないかもしれない。一方が合意に深刻に違反し,指定された治癒期間内に是正されていない場合には,我々もファイザーもNiRogacestatライセンスプロトコルを終了することができる。また,他方に関連する特定の破産事件が発生した場合,我々もファイザーもNiogue acestat許可プロトコルを終了することができる。ファイザーが私たちの治癒していない重大な違約または私たちの破産によって合意を終了した場合、ファイザーは選択権を行使した目標に対する許可証を保持する(ファイザーに他の選択がない限り)、支払い義務は減少する。
米デルタチニブ許可協定
2017年8月、ファイザーとライセンス契約またはMirdametinibライセンス契約を締結し、この合意に基づき、すべての疾患を治療するためのミダミーニのグローバル独占再許可権(ファイザーを含む)を取得しました。さらに、ファイザー社は、10年以内に、NF 1に対してMEK阻害剤の臨床試験を行わないが、ファイザーによって買収または買収された第三者によって所有または制御されるMEK阻害剤は含まれないことに同意した。私たちはその後、知的財産権に関連するいくつかの条項に関連するMirDametinib許可協定を2019年8月に修正した。
改訂された“ミダミチニブ許可協定”によると、米国で少なくとも1つの製品を開発し、規制部門の承認を求める義務があり、規制部門の承認を得たら、米国で製品を商業化する義務がある。米国で規制部門の承認を得た後、特定の国である程度精算されることが合理的に予想されている場合は、その国/地域で開発し、規制部門の承認を求め、規制部門の承認を得たら、その国/地域で製品を商業化する。
私たちはいくつかの商業マイルストーン事件を完了した後、ファイザーに合計2億298億ドルを支払うことを要求された。
ミダメチニブの販売のためにファイザーに分級特許権使用料を支払い、百分率の中央値から一桁まで、有効クレーム満期、第三者許可満期金額、模造薬競争の控除を受ける可能性があります。
事前に終了しない限り、Mirdametinib許可協定はすべての印税義務が満了した時に無効になるだろう。特許権使用料期間は、(I)最初の商業販売から10年、(Ii)すべての法規またはデータ排他性満了、および(Iii)最後に満了した有効特許請求の期限が満了する後に満了する。ある国·地域に適用される印税期限が満了した後,その国/地域の印税期間内に許可を得たすべての知的財産権に永久的で全額支払われる非独占許可を付与し続ける。便宜上、私たちはMidametinib許可プロトコルを30日前に書面で通知する権利があります。ファイザーは便宜のために合意を終わらせないかもしれない。一方が合意に深刻に違反し,指定された治癒期間内に是正されなければ,我々もファイザーもMirdametinib許可プロトコルを終了することができる。さらに、他方に関連する特定の破産事件が発生した場合、私たちまたはファイザーはMirdametinib許可プロトコルを終了することができる。ファイザーが私たちの治癒していない重大な違約または私たちの破産によって合意を終了した場合、ファイザーは選択権を行使した目標に対する許可証を保持する(ファイザーに他の選択がない限り)、支払い義務は減少する。
百済神州臨床協力協定
2018年8月、著者らは百済神州と臨床協力協定、即ち百済神州協力協定を締結し、百済神州で研究されているRAFダイマー阻害剤リフィニブ(BGB-283)とミダミニーの併用による末期或いは難治性固形腫瘍患者の1 b期臨床試験の安全性、耐性と初歩的な治療効果を評価した。
私たちと百済神州は商業的に合理的な努力で私たちそれぞれの臨床試験活動を達成する義務があります。百済神州は臨床試験の管理を担当し,固定用量処方活動を担当し,費用は私たちが負担している。いずれも単独で臨床試験に用いられる化合物の製造や供給に関するコストを担当する。臨床試験が完成した後、もし双方がある事前に定義した標準がすでに満たされたことに同意すれば、双方は誠実に1つの最終合意を交渉し、臨床協力の拡大と特定の原則に基づく商業関係を規定するため、双方がこのような最終合意を達成する義務がないことを前提とする。
臨床試験に関する費用を百済神州と折半する。協力は私たちと百済神州で構成された平等代表の共同指導委員会で管理されている。
指定された排他期間内には、いずれも相手の化合物を開発または商業化してはならない。さらに、協定の発効日から一定期間内に、いずれの当事者も、臨床開発(または臨床開発の準備)または商業化の任意の形態の特定の阻害剤の組み合わせ、または任意のそのような組み合わせを含む任意の製品を使用してはならないが、百済神州協力協定によって許可されるものは除外される。
早期に終了しない限り、百済神州協力協定は百済神州が臨床試験の最終臨床試験報告を提供した日から1周年で終了する。いずれか一方は、(I)いずれか一方がその化合物のすべての開発を完全に停止すること、(Ii)いずれか一方が患者の安全問題があると合理的に結論を出すこと、または(Iii)規制機関がいずれか一方の化合物または臨床試験の承認を撤回する場合、以下の方法で百済神州協力協定を終了することができる。他方に実質的な違約が存在し,指定された治癒期間内に治癒が得られなければ,いずれか一方も百済神州協力協定を終了することができる。
グラクソ·スミスクライン臨床協力協定と修正案
2019年6月、著者らはGSK協力協定、すなわちGSK協力プロトコルをグラクソ·スミスクラインと締結し、RRMM患者の適応性1 b期臨床試験におけるナイルシタンとGSKのBCMA ADC belantamab mafodotin(Belamaf)の併用を評価した。
2021年10月、我々は、ナイルキャストとベラマフとの組み合わせを評価するために、進行中の臨床試験において追加の用量レジメンを追加し、他の薬剤との組み合わせ治療再発および難治性多発性骨髄腫を評価するために、グラクソ·スミスクライン協力協定を修正した。この改訂により、私たちは、第1の組み合わせ用量レベルから第2の段階キューを拡大することを宣言し、第1の段階キューで観察された鼓舞的な予備データに基づいて、0.95 mg/kg Q 3 Wベラマフガニダゾシターを評価した。2.5 mg/kgのQ 3 W Belamaf単一治療対照群と比較して,拡大した2期行列はさらに安全性と有効性の概況を探索しており,FDAが承認したBelamaf単一治療用量とスケジュールと同様である。著者らはまた、2つの新しいサブ研究を追加し、ベラマフとナイル西特、ポマドアミンとデキサメタゾンの併用及びレナドアミン+デキサメタゾンの併用によるRRMM患者の治療を探索することを発表した。これらのサブ研究からのデータは,将来の多発性骨髄腫早期臨床試験を可能にする可能性が信じられている。修正案は締約国の業務や財政的責任を修正または修正しない。
2022年9月、我々は、早期治療シリーズにおいてベラマフとニグリスタットの組み合わせを開発し、新たな診断のための多発性骨髄腫患者を含む、グラクソ·スミスクライン協力協定を拡大した。拡大された合意に基づいて、私たちはナイルキャストの彼のすべての商業権を維持し続けた。また,今後のベラマフ臨床試験にナイル西塔を供給し続け,グラクソ·スミスクラインとベラマフとの併用の承認が求められている市場での商業化を図る。グラクソ·スミスクラインは併用療法研究に関するすべての開発費に資金を提供しているが,ニロシット供給に関する費用や知的財産権に関する何らかの費用は除外している。共同療法の一部として、私たちはナイルキャストを商業化する費用を担当している。
その他の臨床協力協定は、ナイルキャストとBCMA指導の共同治療開発に関連している
グラクソ·スミスクラインの協力協定以外に、著者らは業界パートナーと他のいくつかの臨床試験協力と供給プロトコルを締結し、ニロシタンとBCMA指導の各種方式の治療を評価し、CAR T細胞治療、二重特異性抗体とモノクロナル抗体のRRMM患者における連合治療を含む
各パートナーはニトロクロロシタと併用したBCMA指導療法を評価し,直接臨床試験に関連するすべての費用を担当する臨床試験の管理を担当しているが,ナイル西特の生産と供給および知的財産権に関するいくつかの費用は除外されている。毎回の協力は私たちの代表とそれぞれのパートナーの代表で構成された共同委員会によって管理される
事前に終了しない限り、各協力協定は臨床試験で期待される分析が完了した後に無効になる。我々またはそれぞれの取引相手は,連携プロトコルに規定されている他の理由で連携プロトコルを終了することができる.
Jazz PharmPharmticals資産購入と独占ライセンス契約
2020年10月、我々はJazz PharmPharmticalsアイルランド有限会社と資産購入と独占許可協定であるJazz合意を達成することを発表し、この合意に基づいて、JazzはPF-04454845を含む我々の脂肪酸アミド加水分解酵素(FAAH)阻害剤計画を買収した。Jazzは私たちに3500万ドルを前払いし、ある臨床開発、監督と商業マイルストーンの実現によって、将来3.75億ドルを支払うかもしれない。また,ジャズは将来PF−04457845の純売上高に販売ベースの印税を支払う義務がある。
Jazz協定によると、Jazzは、商業的に合理的な努力を用いて米国で少なくとも1つの製品を開発し、規制部門の承認を求める義務があり、規制部門の承認を得た場合、Jazzは米国でこのような製品を商業化する義務がある。
Jazzプロトコルは、事前に終了しない限り、Jazzプロトコルで定義されている製品がその国の印税期間が満了するまで、製品および国/地域によって有効に維持される。どちらか一方がジャズプロトコルに違反し,一定時間是正されなければ,どちらか一方がジャズプロトコルを終了することができる.Jazzはどのような理由でもJazzプロトコルを終了することができます。Jazzがこのプロトコルの規定に従って事前に書面で私たちに通知すればいいです。
TEAD阻害剤組合せ許可プロトコル
2021年5月、私たちはルモール大学およびフランダースバイオテクノロジー研究所と世界的に独占的な許可協定を達成することを発表し、合意に基づいて、カバ経路異常シグナルによって駆動されるバイオマーカーによって定義される固形腫瘍を潜在的に治療することを目的とした転写因子ファミリーTEAドメインまたはTEADファミリーの一連の新しい小分子阻害剤の許可を得た。合意条項によると、私たちはKUルモールとVIBに1100万ドルを前払いした。協定条項によると、KUルモールとVIBには、2.85億ドルまでの開発、規制、ビジネスマイルストーン、およびライセンス内技術に基づいて開発された製品の将来の任意の純売上に基づく1桁分のパーセント特許権使用料を取得する資格がある。
EGFR阻害剤組合せ許可プロトコルとスポンサー研究プロトコル
2021年10月、我々はDana-Farber癌研究所(Dana-Farber)と独占的な世界的許可協定を達成することを発表し、この合意に基づいて、EGFR変異肺癌の治療のための新しい上皮増殖因子受容体(EGFR)小分子阻害剤の組み合わせの許可を得た。合意条項に基づいて、私たちはDana-Farberに前金を支払った。合意条項によると、Dana-Farberには、開発とビジネスマイルストーン、ライセンス内技術に基づいて開発された製品の将来の任意の純売上に基づく特許権使用料を取得する資格もあります。
この許可協定と同時に、著者らはスタンフォード医学と長年賛助する研究協定を締結し、スタンフォード医学の実験室とダナ-ファーバーの協力実験室の持続的な研究と開発に資金を提供した。この賛助の研究協定はEGFR阻害剤の組み合わせが開発候補指名に向かって進む過程で行われたリードした最適化と転化生物学的努力を支持することを目的としている。
製造業
私たちは私たちのすべての資産を作るために第三者に依存する。私たちはすでに契約製造機関(CMO)と合意し、ナイルカスタット、ミダデチニブ、TEADのために薬物物質と薬物製品の生産を計画している。我々はすべてのCMOが現行の良好な製造規範やcGMP要求に従って生産活動を行うことを要求している。著者らは現在完全にこれらのCMOに依存して拡大と技術開発を行い、そして十分な数量の著者らの候補製品を生産し、臨床前研究と臨床試験に応用している。これらのCMOは臨床供給と商業規模の生産を支援する能力があると予想される。私たちはニトロクロロキセチン、薬物物質、完成品を商業的に供給する協定を締結した。私たちは予想された商業需要に基づいて、予備供給源の手配を含む、このような手配を適時に行うつもりだ。
販売とマーケティング
もし私たちの任意の候補製品が承認されれば、私たちは単独でまたは他の会社と協力して、アメリカでこれらの製品をマーケティングし、商業化し、国際市場を選択するつもりです。
多くの靱帯様腫瘍とNF 1-PN患者は専門医師によって管理され、腫瘍学者、医学遺伝学者と神経科医師を含むため、著者らは的確な販売チームを通じて実現できると信じている。
我々が他のエージェントと連携して開発している候補製品や高度に流行している疾患については,潜在的なマーケティング承認に近い場合には,我々のパートナーと連携して,候補製品ごとに商業化戦略を策定し,それぞれのビジネスインフラ,能力,特定の国/地域の専門知識を考慮して責任を分担する予定である。
教育と患者計画
著者らは一連の計画を通じて硬繊維腫とNF 1-PNメンバーと積極的に協力し、患者会議と教育計画に参与することを含む。これらの成分の例としては,硬線維腫研究基金,COG,小児腫瘍基金がある。これらの活動を展開するのは,これらの患者が直面している負担や満たされていない需要をよりよく理解し,我々の製品候補製品(承認されれば)を獲得するのをより効果的に支援するためである。各疾患領域において、私たちは情報を提供し、医師の意識を高め、より効果的な回診経路を作成することによって、確定された患者に対する疾病認識と診断およびその後の治療を支持する。これらの目標を推進するために,広範な多チャンネル会社が後援する硬線維腫に対する疾患教育活動を開始し,患者,医師,介護者を支援した。
競争
製薬業界の特徴は技術の進歩が迅速で、競争が激しいことだ。私たちの候補製品、技術、知識、経験、科学資源は私たちに競争優位を提供してくれると信じていますが、私たちは主要な製薬とバイオテクノロジー会社、学術機関、政府機関、公共と個人研究機関などからの競争に直面しています。
私たちが開発と商業化に成功した任意の候補製品は、ラベル外療法および将来発売される可能性のある新しい療法を含む承認された治療案と競争するだろう。私たちが他の療法と有効に競争する能力を影響する重要な要素は私たちの製品の有効性、安全性、投与方法、コスト、販売促進活動レベルと知的財産権保護を含む。私たちが競争する可能性のある多くの会社は研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング許可を得た製品の面で私たちより多くの財務資源と専門知識を持っている。
ナイル西司は硬線維腫を治療しています
FDAが承認した靭帯様腫の治療法は現在のところない。他の会社はAyala PharmPharmticals,Inc.,Bayer Corporation,Cellstia Biotech AG,Iterion Treateutics,Inc.を含むが,これらに限定されないこの適応の製品を開発していることが知られているが,いくつかの療法が知られており,その中のいくつかは
後発薬は,ラベル外の硬線維腫治療に用いられる。これらの療法は、リポソームアドリアマイシンおよびビンクリスチン/メトトレキサートなどの化学療法薬、非ステロイド性抗炎症薬、抗ホルモン療法およびチロシンキナーゼ阻害剤、例えば、ソラフェニブ、イマチニブおよびパゾパニを含む。
NF 1-PNのミダメチニブ
アスリカンのKoselugoは現在FDAによって許可されている唯一のNF 1-PNを治療する方法である。他の会社は、アレイバイオ製薬会社(ファイザー社の子会社)、第一三共株式会社、Exelixis社、Infix ion Bioscience社、NFlect治療会社、ノバ国際株式会社、パシシア治療会社、復星医薬(グループ)有限会社を含むが、この適応のための製品を開発していることが知られている。非特許薬がNF 1−PNの治療に使用されていることもいくつか知られている。これらの治療法には,化学療法や免疫療法などの放射線治療や各種全身治療がある。
多発性骨髄腫のBCMA標的治療とGSI併用療法
多発性骨髄腫の治療には,他社が他のガンマ分泌酵素阻害剤とBCMA標的治療を組み合わせた薬剤を開発しているか,あるいは開発されている可能性があることが知られている。これには百時美施貴宝会社が含まれているが、これらに限定されない。
MAPK異常癌における米デルタチニブとBGB−3245の役割
我々のバイオマーカーで定義された固形腫瘍製品の組み合わせについては,他の会社が特定の変異や異常を有する固形腫瘍を治療するための製品を開発しているか,あるいは開発されている可能性があり,これらの製品が我々の計画の目標であることが知られている。複数の製品が開発目標に向かっているRAS突然変異はロイヤル空軍変異およびその他のMAPK異常は、安進、アスリーカン、ブラックダイヤモンド治療会社、バーリンガー-インゲルハイム国際会社、中外製薬会社、初日バイオ製薬会社、礼来社、Erasca社、F.ホフマン-ラロー社、Fore BioTreateutics社、韓米製薬会社、Kinnate Biophma社、メルク社、Mirati治療会社、Moderna、ノバ国際社、輝瑞社、革命薬物会社、WellSpring Biosciences社を含むが、これらに限定されない。これらの患者集団の解決に適した計画を持っている会社もあるかもしれないが,これらの会社は我々の努力と競争力を持っている可能性があるが,具体的な臨床開発計画は開示されていない。
TEAD抑制プログラム
他の早期開発段階にあるTeadパルミチン化阻害剤は,Cedilla Treeutics,Ikena Oncology,Invenva S.A.,Kyowa Kirin Co.,Ltd.,Genentech,Inc.およびVivace Treeutics,Inc.の候補品を含むがこれらに限定されないことが知られている
腫瘍学に集中した治療会社を含め、規模が小さいか早い段階にある会社は、特に大型·成熟会社との連携により重要な競争相手であることが証明されている可能性もある。これらの会社はまた、合格した科学と管理者を募集と維持し、臨床試験場を設立し、患者を臨床試験に参加させ、著者らの計画と相互補完或いは必要な技術を獲得する方面で私たちと競争する可能性がある。
政府と個人支払者の精算も私たちの製品の定価と競争力に大きな影響を与えるだろう。私たちの競争相手は私たちよりも早くFDAや他の規制機関のその製品の承認を得るかもしれませんが、これは私たちの競争相手が私たちの候補製品を商業化する前に強力な市場地位を確立することができるかもしれません。
知的財産権
私たちの成功は、私たちの候補製品、製造と加工発見、および他の独自技術のために独自保護を獲得し、維持する能力があるかどうかにある程度依存し、他人の独占権を侵害することなく運営し、他の人が私たちの独占権を侵害することを防止する。私たちは、独自技術、発明、および改善に関連する米国および外国特許出願を含む様々な方法を使用して、私たちのビジネスの発展および実施に非常に重要であると考えられる物質成分および使用方法を含む様々な方法を使用する予定である。例えば、私たち、私たちの許可者、または私たちの協力者は現在、私たちの候補製品をカバーする物質からなる特許を持っているか、申請しています。私たちは、1つ以上の臨床プロジェクトの使用方法をカバーする特許保護を一般的に求める予定です。私たちはまた、ビジネス秘密、商標、技術ノウハウ、持続的な技術革新、潜在的な許可内機会に依存して、私たちの独自の地位を発展させ、維持しています。
特許
2017年8月に設立された際、私たちはファイザーとリーディング候補製品についてライセンス契約を締結し、この合意に基づいて、リーディング候補製品を開発、製造、商業化するためにファイザー特許と独自技術のグローバル独占権利を獲得しました。
“ナイルキャスト許可協定”によると、私たちはアメリカや多くの外国司法管区にナイルキャストに関連する特許権の独占許可を持っている。Niroacestatライセンス契約に基づいて付与される特許権には、米国で付与された6つの特許(最初に3つのこのような特許が含まれており、その後、私たちが行った作業でファイザーの同意を得て付与された他の3つの特許)と、オーストラリア、カナダ、中国、フランス、ドイツ、スペイン、イギリス、日本などの外国司法管轄区域で付与された25件を超える特許が含まれている。物質組成物をカバーする米国特許の法定期限は2025年であり、多結晶ナイルタンを含む米国特許は2039年に満了し、臨床開発中の形態を含む米国特許は2039年に満了し、医薬組成物を含む米国特許は2042年に満了し、いずれの場合も特許期限調整や規制延長を含まず、外国の同業者が待っている。もし私たちが硬線維腫治療のためのニューノカルスの規制承認を得ることに成功すれば、私たちは孤児薬の独占性に依存することが予想され、これは通常アメリカで7年間の市場独占経営権を与え、ヨーロッパで10年間の市場独占権を与える。NiRogacestatライセンスプロトコルにおける我々の権利の他の情報については、上記の“ライセンスおよび連携プロトコル-ファイザーライセンスプロトコル”を参照されたい。Niroacestatはアメリカで硬繊維腫を治療する孤児薬物の称号を獲得し、そしてEU委員会の軟組織肉腫治療の称号を獲得した。
ミダミチニブ許可協定によると、私たちは米国や多くの外国司法管轄区域でミダミチニブに関連する特許権の独占許可を持っている。MirDametinibライセンス契約に基づいて許可された特許は、我々およびファイザーによる作業に基づいて付与された特許を含み、4つの米国特許、現在臨床開発されている形態、多形体の治療方法、および医薬組成物を含む米国特許を含む多形体をカバーする4つの米国特許を含み、いずれの場合も規制延長は含まれておらず、外国の同業者が待っている。NF 1治療のためのミダミニーの規制承認に成功すれば、米国での7年間の市場独占とヨーロッパでの10年間の市場独占に依存する孤児薬独占に依存することが予想される。Mirdametinibライセンスプロトコルにおける我々の権利の他の情報については、上記の“ライセンスと連携プロトコル-ファイザーライセンスプロトコル”を参照されたい。FDAはNF 1−PNのミダミニー孤児薬名を承認しており,欧州委員会はNF 1のミダミチニブ孤児薬名を承認している。
ナイルシタンやミダメチニブに関連した併用療法については,われわれの特許地位を高める機会がある可能性があり,探索を行う。このような努力のいずれも特許が発行されることは保証されない。
商業秘密
特許に加えて、私たちは商業秘密と技術ノウハウに依存して、私たちの競争地位を発展させ、維持しています。私たちは通常ビジネス秘密に依存して私たちの業務を特許保護から保護したり特許保護に適していないと考えています。私たちは、従業員、コンサルタント、科学コンサルタント、請負業者、およびパートナーと秘密協定および発明譲渡協定を確立することによって、商業秘密およびノウハウを保護します。これらのプロトコルは,一般に,個人やエンティティと我々との関係過程で開発または公表されるすべての機密情報は,関係期間および後に秘密にしなければならないことを規定している.これらのプロトコルはまた、一般に、私たちが履行している仕事または私たちの業務に関連するすべての発明、ならびに雇用または譲渡中に構想または完了したすべての発明を、私たちの固有財産としなければならないと規定している。さらに、私たちの独自の情報が第三者に盗用されることを防止するために、物理的および技術的セキュリティ対策のような他の適切な予防措置を講じた。
保証範囲·定価·精算
新薬製品の商業化成功は政府衛生行政当局、私営健康保険会社とその他の組織によるこれらの薬品の清算程度にある程度依存する。政府当局や他の第三者支払人、例えば個人健康保険会社や健康維持組織は、彼らがどの薬品のために支払うかを決定し、精算レベルを確立する。政府と個人支払者の獲得性と精算範囲は大多数の患者が薬品を負担できるキーポイントである。薬品の販売は国内外の薬品費用が健康維持、管理保健、薬局福祉と類似の医療管理組織が支払う程度、あるいは政府衛生行政当局、個人健康保険会社と他の第三者支払人が精算する程度に大きく依存する。
アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。政府当局や他の第三者支払人は、特定の薬品のカバー範囲や精算金額を制限することでコストを抑制しようとしている。はい
多くの国では、薬品価格は国家衛生システムの一部として、異なる価格制御メカニズムの制約を受けている。一般的に、この制度での薬品価格はアメリカを大きく下回っています。他の国は会社が自分の薬品価格を固定することを許可していますが、会社の利益を監視してコントロールしています。そのため、米国以外の市場では、薬品の精算額が米国を下回る可能性がある。米国では、新薬製品の精算に関する主な決定は、医療保険や医療補助サービスセンター(CMS)が行うのが一般的であり、衛生·公衆サービス部(HHS)の機関である。CMSは新薬製品を決定するかどうか及びどの程度連邦政府医療保健計画(例えば連邦医療保険)によってカバーと精算されるかを決定し、個人支払者はよくCMSに大きく従う。しかし、第三者支払者の間には統一された薬品保険と精算政策がなく、支払者間の薬品保険と精算レベルに大きな差がある可能性がある。米国では、第三者支払い者が生物製品に保険を提供するかどうかを決定するプロセスは、一般に、そのような製品の価格を設定すること、または保険が承認された後に支払者が製品に支払うべき償還率を決定するプロセスから分離される。バイオ製品に関しては、第三者支払者は、承認リスト上の特定の製品にカバー範囲を制限することができ、処方表とも呼ばれ、FDAによって承認された特定の適応のすべての製品を含まない可能性があるか、または製品をいくつかの処方レベルに配置することにより、より低い精算レベルおよびより高いコスト分担義務を患者に課すことができる。第三者支払人が私たちの候補製品を保証しないと決定すれば、医師の製品の使用を減らす可能性があります。さらに何かがある, 第三者支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。製造業者がその製品開発投資の適切なリターンを達成するために、十分な価格レベルを維持することができるように、十分な第三者補償を得ることができない可能性がある。また、製品の保証範囲と精算範囲は支払人によって異なる。第三者支払者は,ある特定の医療製品を保証することを決定し,他の支払者もその医療製品に保険を提供することを確保することができないか,あるいは適切な販売率で保険を提供することになる。そのため、保証範囲の確定過程は通常、メーカーがその製品の使用のためにそれぞれ各支払人に科学と臨床支持を提供する必要があり、これは時間のかかる過程である。支払人が精算を決定する際に考慮する要素は、製品があるかどうかに基づいている
•健康計画の下で保障された福祉
•安全で効果的で医学的に必要なものです
•特定の患者に適しています
•費用対効果があります
•実験的でも調査的でもない。
保証政策と第三者精算料率は随時変化する可能性がある。規制部門の承認を得た1つまたは複数の製品が有利な引受·精算状態を獲得しても、将来的にはあまり有利ではない引受政策や精算料率が実施される可能性がある。第三者支払者は,安全性と有効性を疑問視するほか,医療製品やサービスの価格に挑戦し,医療の必要性を検査し,薬品のコスト効果を審査することが増えている。第三者支払者が、1つの製品が他の利用可能な療法と比較して費用対効果があると思わない場合、彼らはFDA承認後に製品をカバーしないかもしれない、または、彼らがそうした場合、支払いレベルは、製造業者に利益を得るためにその製品を販売させるのに十分ではない可能性がある。
また、多くの外国の国では、薬品の提案価格は必ず承認されなければならず、合法的に発売されることができる。各国の薬品定価と精算に対する要求は大きく異なる。EUでは,各国政府はその定価と精算規則や国家医療システムの制御により製品価格に影響を与え,これらのシステムは消費者にこれらの製品の大部分のコストを支払っている。一部の司法管轄区域はプラスリストとネガティブリスト制度を実施しており、この制度の下で、製品は政府が価格を精算することに同意した後にのみ販売することができる。精算或いは定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の製品のコスト効果を現在利用可能な治療法と比較する。他の会員国たちは会社が自分の薬品価格を固定することを許可するが、会社の利益を監視する。薬品に対して価格制御や精算制限を実行する国が私たちのいかなる製品にも有利な精算と定価手配を許可することは保証されません。全体的には,医療コスト,特に処方薬の下り圧力が非常に大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。また、一部の国では、低価格市場からの国境を越えた輸入が一国国内の定価に商業圧力をかけている。
政府の監督管理
アメリカ連邦、州と地方の各レベル及びEUを含む他の国と司法管轄区の政府当局は薬物製品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、承認後の監視と報告、マーケティング及び輸出入などの薬品に対して広範な監督管理を行い、ナイルガタ、ミダメチニブと私たちの他の候補製品を含む。一般的に、1つの新薬が発売される前に、その品質、安全性と有効性を証明するデータを大量に獲得し、それを各監督管理機関特有のフォーマットに組織し、監督管理機関の審査と承認を提出しなければならない。
臨床試験
臨床開発段階は、合格した研究者の監督の下で、良好な臨床実践(GCP)に基づいて健康ボランティア或いは患者に研究製品を提供することに関連し、通常は試験スポンサー或いは試験スポンサーの制御下に雇われていない医師であり、その中にはすべての研究対象が任意の臨床試験に参加することについてインフォームドコンセントを提供することを含む。臨床試験は,臨床試験の目標,用量プログラム,被験者の選択と排除基準,および被験者の安全性をモニタリングし,治療効果を評価するためのパラメータを詳細に説明した場合に行われる。INDの一部として、すべての議定書とその後の議定書のいかなる修正もFDAに提出されなければならない。また,各臨床試験は,臨床試験を行う各機関の機関審査委員会(IRB)によって審査·承認されなければならず,臨床試験に参加する個人が直面するリスクが最小限に低下し,期待される利益に対して合理的であることを保証しなければならない。IRBはまた、各臨床試験対象またはその法律代表に提供されなければならないインフォームドコンセントを承認し、完成まで臨床試験を監視しなければならない。行っている臨床試験や完成した臨床試験結果を公的登録機関に報告することも求められている。多くの臨床試験に関する情報は,www.Clinicaltrials.govサイトで発表するために,特定の時間枠内で提出されなければならない。製品·患者群·調査段階に関する情報, 臨床試験登録の一部として,臨床試験の試験地点や調査者,その他が公開されている。スポンサーも完成後に彼らの臨床試験結果を検討する義務がある。場合によっては、これらの裁判結果の開示は、裁判が完了した日から2年に延期されることができる。競争相手はこれらの公開された情報を用いて開発計画の進捗状況を知ることができる.人体臨床試験は通常3つの連続段階に分けて行われ、この3つの段階は重なる可能性があり、合併する可能性もある
•第1段階の臨床試験は、一般に、一部の健康ボランティアまたは疾患の影響を受ける患者に関連し、彼らは最初に単剤に接触し、その後、多剤候補製品に接触する。これらの臨床試験の主な目的は薬物の代謝、薬理作用、副作用耐性と安全性を評価することである。
•第二段階の臨床試験は、予想される利益を産生するために必要な投与量を決定するために、疾患の影響を受ける患者を研究することに関する。同時に、安全性と更なる薬物動態学と薬効学情報を収集し、可能な副作用と安全リスクを識別し、そして初歩的な治療効果の評価を行った。
•第三段階臨床試験は、通常、必要なデータを提供して、その予期される用途に対する製品の有効性、使用中の安全性を証明し、製品の全体的な利益/リスク関係を確立し、製品の承認に十分な基礎を提供することを目的としている複数の場所の大量の患者に関連する。これらの試験は、プラセボおよび/または他の対照治療との比較を含むことができる。治療の持続時間が常に延長され、製品のマーケティング期間中の実際の使用をシミュレートする。
登録試験は1種の臨床試験であり、それは監督管理機関の候補薬物に対する治療効果と安全性の評価要求を十分に満たし、それによってこの薬物の承認が合理的であることを証明することができる。一般に,登録試験は第3段階試験であるが,試験設計が臨床的利益の信頼性の高い評価を提供しており,特に満たされていない医療ニーズが存在する場合には,第2段階試験である可能性がある。
承認後試験は,4期臨床試験と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの試験は,治療適応が予想される患者の治療から追加的な経験を得るために用いられ,特に長期的な安全なフォローアップのために用いられている。場合によっては、FDAは、生物製品ライセンス申請またはBLAを承認する条件として、第4段階臨床試験を強制的に要求することができる。
臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出しなければならず,深刻な有害事象が発生すればより頻繁に提出される。FDAまたはスポンサーは、臨床試験を随時一時停止または終了することができ、またはFDAは、研究患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由に基づいて他の制裁を適用することができる。同様に、ある臨床試験が委員会の要求に従って行われていない場合、またはその薬剤が患者に予期せぬ深刻なダメージを与えた場合、委員会は、その所在機関の臨床試験の承認を一時停止または終了することができる。ある臨床試験と完成した臨床試験結果の登録と報告に対しても関連する要求がある。
米国−FDA規制
審査の流れ
アメリカでは、医薬品はFDAによって広く規制されている。“連邦食品、薬品と化粧品法”あるいは“連邦薬品と化粧品法”及びその他の連邦と州の法規と条例は薬品の研究、開発、テスト、製造、貯蔵、記録保存、承認、ラベル、販売促進とマーケティング、流通、承認後のモニタリングと報告、サンプリング及び輸出入などを管理する。適用される米国の要求を遵守しないことは、FDAが未解決の新薬申請またはNDAの承認を拒否し、警告または無タイトル手紙、製品リコール、製品差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、民事処罰、および刑事起訴のような様々な行政または司法制裁を受ける可能性がある。
米国では、新製品または承認製品のいくつかの変更に対する医薬製品の開発は、一般に、FDA承認を求める各適応に対する薬剤の安全性および有効性を決定するために、臨床前実験室および動物試験、IND(臨床試験開始前に発効しなければならない)および十分かつ制御された臨床試験をFDAに提出することに関連する。FDA上場前の審査要求を満たすには通常長年の時間を要し、実際の所要時間は製品或いは疾病のタイプ、複雑性と新規性によって大きく異なる可能性がある。
臨床前試験は製品の化学、調合と毒性に対する実験室評価、及び製品特性と潜在安全性と有効性を評価する動物研究を含む。臨床前試験の進行は必ず良好な実験室実践を含む連邦法規と要求に符合しなければならない。臨床前試験の結果はINDの一部として他の情報とともにFDAに提出され,製品化学,製造と制御に関する情報および提案された臨床試験案が含まれている。IND提出後,生殖毒性や発ガン性の動物試験など,長期的な臨床前試験を継続する可能性がある。
ヒト臨床試験を開始する前に,各INDの提出後30日間の待機期間が求められている。FDAがこの30日間INDにコメントもINDにも疑問を提起しなければ,INDで提案された臨床試験が開始される可能性がある。
臨床試験は合格した研究者の監督の下で、健康ボランティア或いは患者に研究用新薬を提供することに関連する。臨床試験は、(I)連邦法規に適合する;(Ii)患者の権利と健康を保護し、臨床試験発起人、管理者および監督者の役割を定義するための国際基準であるGCPに適合する;および(Iii)試験目標を詳細に説明し、安全性を監視するためのパラメータおよび評価すべき有効性基準のプロトコルである。米国患者のテストに関する各々のプログラムおよび後続のプログラム修正案は、INDの一部としてFDAに提出されなければならない。
FDAが臨床試験がFDAの要求に沿って行われていないと考えている場合,あるいは臨床試験患者に受け入れられないリスクとなっている場合,FDAはいつでも臨床試験の一時的または永久的な停止を命じたり,他の制裁を加えたりすることができる。臨床試験における患者の試験案やインフォームドコンセント情報もIRB承認に提出しなければならない。IRBはまた、IRBの要求を遵守できなかったために、現場の臨床試験を一時的または永久的に停止することを要求することができ、または他の条件を適用することができる。ニューノミンの発売承認を支持する臨床試験は通常3つの連続段階で行われるが、これらの段階は重なる可能性がある。第1段階、すなわち、最初に健康なヒト対象または患者に薬物を導入した場合、新陳代謝、薬物動態、薬理作用、用量増加に関連する副作用、および可能であれば有効性を評価するための早期証拠を評価するために、薬物が試験される。第2段階は、一般に、特定の適応、用量耐性、および最適用量での薬剤の有効性を決定し、よく見られる副作用および安全リスクを決定するために、限られた患者集団で試験を行うことに関連する。化合物が第2段階評価において有効性および許容可能な安全性を証明する場合、第3段階試験は、FDAが薬剤の全体的利益-リスク関係を評価し、薬物のラベルに十分な情報を提供することを可能にするために、より多くの患者の臨床的有効性および安全性に関する追加の情報を得るために実施される。ほとんどの場合, FDAはこの薬物の治療効果を証明するために、2つの十分かつ制御された良好な3期臨床試験が必要である。少数の場合、試験が内部一致性を示す大型多中心試験であり、統計学的に非常に説得力のある発見が死亡率、不可逆的な発病率或いは疾病の予防に臨床意義があり、潜在的な深刻な結果を持っているが、第二回の試験で結果が実際に或いは倫理的に不可能であることを確認すれば、他の確実な証拠を持つ単一の第三段階試験で十分であるかもしれない。
2016年12月13日に公布された“21世紀治療法”によると、深刻または生命に危険な疾患を治療する研究薬の製造業者は、例えば、そのウェブサイト上でその評価政策を発表することによって提供しなければならない
拡張アクセスの要求に応答する.この要求は,研究薬物の公布日または初めて2期または3期試験を開始した日から60日後に適用される。必要な臨床試験が完了した後,NDAを用意してFDAに提出する。この製品が米国市場で販売されるようになる前に、FDAが製品を承認する必要がある。NDAは、すべての臨床前、臨床および他の試験の結果、および製品の薬理、化学、製造および制御に関連するデータアセンブリを含まなければならない。秘密協定を準備して提出する費用は巨大だ。NDAの提出の多くは高額な申請使用料を支払う必要があり、現在は2023年度の3,242,026ドルであり、ただ承認されたNDA下のメーカーおよび/またはスポンサーも条件に合った製品のための年間計画費を支払う必要があり、現在は2023年度の393,933ドルである。
FDAはNDAを受信した日から60日の時間があり,当該機関の敷居に基づいて申請が届出を受けているかどうかを決定し,申請が十分完全であると考え,実質的な審査を行うことができる。提出された申請が受け入れられると、FDAは深い検討を始めた。FDAは新薬申請審査におけるいくつかの業績目標に同意した。このような標準審査薬製品の申請の多くは10~12ヶ月以内に審査され、優先審査薬の申請の多くは6~8ヶ月以内に審査される。優先審査は、FDAが治療において大きな進展を得るか、または適切な治療方法がない場合に治療を提供する薬剤を決定するのに適用することができる。FDAは、いくつかの遅延された情報を考慮するために、または提出中に提供された情報の情報を明確にするために、標準審査および優先審査の審査手続きをさらに3ヶ月延長することができる。
FDAはまた、新薬製品の申請、または安全性または有効性の問題を提起する医薬製品の申請を、諮問委員会に提出することもできる--通常、臨床医および他の専門家を含むグループであり、審査、評価を行い、申請を承認すべきかどうかについて提案することができる。FDAは諮問委員会の提案によって制限されていないが、それは一般的にそのような提案に従っている。NDAを承認する前に、FDAは、通常、GCPに適合することを確実にするために、1つまたは複数の臨床場所を検査する。さらに、FDAは薬物を製造する1つ以上の施設を検査するだろう。FDAは、cGMPに適合しない限り、この製品を承認しないであろうし、NDAに含まれるデータは、研究された適応において安全かつ有効であることを証明する多くの証拠を提供するであろう。
FDAがNDAと製造施設を評価した後、それは承認状または完全な返信を発行するだろう。完全な応答文は、一般に、提出中の不足点を概説し、FDAが出願を再検討するために、大量の追加のテストまたは情報を必要とする可能性がある。NDAが再提出されたとき、またはいつ、これらの欠陥がFDAによって満足的に解決された場合、FDAは承認書を発行するであろう。FDAは、含まれる情報タイプに依存して、そのような再提出を2ヶ月または6ヶ月以内に検討することを約束している。
この薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。NDA承認の条件として、FDAは、薬物の利点が潜在的リスクよりも大きいことを確実にするために、リスク評価および緩和戦略、またはREMSを必要とする可能性がある。REMSは、薬物ガイドライン、医療専門家のコミュニケーション計画、および安全な使用を確保する要素、またはETASUを含むことができる。ETASUは、処方または調剤のための特別なトレーニングまたは認証、場合によっては調剤、特殊な監視、および患者登録簿の使用を含むことができるが、これらに限定されない。REMSに対する要求はこの薬物の潜在的な市場と収益力に重大な影響を与える可能性がある。さらに、製品承認には、薬物の安全性または有効性を監視するために、大量の承認後の試験および監視が必要となる可能性がある。承認されると、規制基準が守られていない場合、または最初のマーケティング後に問題が発見された場合、製品承認は撤回される可能性がある。
承認申請において決定されたいくつかの条件は、適応、ラベルまたは製造プロセスまたは施設の変更を含み、新しいNDAまたはNDA付録を提出し、FDAの承認を得て、変更を実施する必要がある。新適応のNDAサプリメントは通常,オリジナル申請と類似した臨床データが必要であり,FDAがNDAサプリメントを審査する際に使用するプログラムや行動は,NDAを審査する際に使用するプログラムや行動と同じである。
排他性
NDAが新しい化学物質またはNCEを承認すると、すなわち、FDAが任意の他のNDAで承認された活性部分を含まない薬剤は、5年間の市場排他性を得るであろうが、その間、FDAは、その薬剤を承認する模倣薬を求める任意の略語新薬出願(ANDA)を受け入れることができない。薬物のいくつかの変化は,パッケージ挿入に新たな適応を増加させるように,3年間の排他期に関連しており,その間,FDAはこのような変化を含む後発薬のANDAを承認することはできない。
第4項の認証が提出された場合、ANDAはNCE排他性満了の1年前に提出することができる。Orange Bookに記載されている特許がなければ,4段目の認証がない可能性があるため,排他期間が満了するまでANDAを提出することはできない.
特許期間を延長する
NDA承認後,関連薬物特許の所有者は,1つの特許出願のために最大5年間の特許延期を行うことができる。許容される特許期間延長は、薬物試験段階の半分−INDとNDA提出との間の時間、およびすべての審査段階−NDA提出と承認との間の時間として計算され、最長5年である。FDAが出願人が職務調査を経て承認を求めていないと判断した場合、時間を短縮することができる。展示期間後の総特許期間は承認日から14年を超えてはならない。
出願段階で満了する可能性のある特許については,特許権者は臨時特許延期を請求することができる。臨時特許の延期は特許期間を1年間延長し,最大4回延長することができる.暫定特許の承認が延期されるごとに、承認後の特許延期は1年減少する。米国特許商標局の取締役は,特許延期を申請している特許に含まれる薬物が承認される可能性が高いことを確認しなければならない。秘密保護協定が提出されていない薬物は一時的な特許延期を受けることができない。
孤児薬
孤児医薬品法によれば、FDAは、まれな疾患または疾患の治療に使用される薬物または生物製品を孤児薬の称号を付与することができる。孤児薬は、通常、米国で20万人未満または米国で20万人を超える影響を与える疾患または疾患であり、米国では米国の販売から薬物または治療用生物製剤を回収するコストは合理的に期待されていない。秘密協定を提出する前に、孤児薬の称号を申請しなければならない。孤児薬物指定の他の利点は、いくつかの研究の税金控除およびNDA申請使用料の免除を含む。FDAが孤児薬物指定を承認した後、FDAは、この薬剤の模倣薬識別情報およびその潜在的孤児用途を開示する。孤児薬物指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の持続時間を短縮することもない。FDAによって承認された特定の疾患の特定の活性成分を治療するための最初のNDA申請者は、FDA孤児薬物の称号を有し、米国でこの製品のこの適応に7年間の独占営業期間を提供する権利がある。7年間の独占期間内に、FDAは、限定された場合、例えば、孤児薬物に対して排他的な製品に対する臨床的優位性を示すことができない限り、同じ疾患のために同じ薬物を販売する他のいかなる他の出願も承認しない可能性があり、またはFDAは、指定された薬物の疾患または状態に罹患している患者の需要を満たすのに十分な数の孤児製品を保証することができることを証明していない。孤児薬物排他性は、FDAが同じ疾患または状態に対する異なる薬物、または異なる疾患または状態に対する同じ薬物を承認することを阻止しない。場合によっては、FDAはそのような指定を取り消すこともできる, 例えば、機関が申請者の指定請求が“孤児薬品法”及びその実施条例に要求される重要な情報を見落としていることを発見した場合。
迅速な承認指定と承認の加速
FDAは、深刻または生命に危険な疾患または疾患を治療するための薬物の開発に便利さを提供し、審査を加速することが要求されており、これらの薬物または条件は有効な治療方法を有さず、このような疾患が満たされていない医療需要を解決する潜在力を示している。迅速チャネル計画によると,新薬候補のスポンサーは,薬剤候補のINDの届出と同時にまたはその後,特定の適応の候補薬を迅速チャネル薬として指定することをFDAに要求することができる。FDAはスポンサーからの要請を受けて60日以内に候補薬剤が迅速チャネル指定を受ける資格があるかどうかを決定しなければならない。
FDAとのより頻繁なインタラクションが可能であるなどの他の利点に加え,FDAは申請完了前にFast Track薬のNDA部分の審査を開始することができる。申請者が余剰情報を提出するスケジュールを提供し、申請者が適用された使用料を支払うことができれば、スクロール審査を行うことができる。しかしながら、FDA審査申請の期間目標は、NDAの最後の部分が提出されてから開始される。また,FDAが高速チャネルの指定が臨床試験中に出現したデータの支持を得なくなったと考えると,FDAはその指定を撤回する可能性がある。
FDAの加速承認規定によれば、FDAは、患者に既存の治療よりも意義のある治療利益を提供する深刻または生命に危険な疾患に対する薬剤を承認することができ、その基礎は、臨床利益を合理的に予測する可能性のある代替終点、または不可逆的な発病率または死亡率よりも早く測定することができる臨床終点であり、不可逆的な発症率または死亡率または他の臨床的利益への影響を合理的に予測することができ、同時に、病状の深刻性、希少性または流行率、および代替療法の利用可能性または不足を考慮することである。
臨床試験において、代替終点は1種の疾病或いは状況の実験室或いは臨床症状の測定であり、それは患者の感覚、機能或いは生存方式の直接測定の代わりになる。代替終点は通常、臨床終点よりも容易または迅速に測定される。これに基づいて承認された候補薬物は厳格な発売後審査を経なければならない
臨床終点への影響を確認するために、4期または承認後の臨床試験を完了することを含むコンプライアンス要件は、2022年の食品·薬物総合改革法案によれば、FDAは、承認前または承認が加速された製品の承認日後の特定の期間内に行うことを適宜要求することが許可されている。必要な承認後研究を行わない場合や,発売後研究期間中に臨床的利益が確認できなければ,FDAが迅速に市場から撤回することを許可し,FDORAによりFDAがより大きな権力を持って迅速にプログラムすることができる。さらに、承認の加速を検討している製品については、FDAは、一般に、機関が別の通知がない限り、上場承認後120日以内に伝播または出版しようとするすべての広告および販売促進材料を、承認前の審査中に機関に提出しなければならないことを要求する。
突破的治療指定
FDAの画期的な治療法はFDA高級従業員により広範な開発相談機会を提供し、審査薬物の承認申請を転動することを可能にし、申請を提出する時に深刻な或いは生命に危害を及ぼす疾患或いは疾病の臨床データ支持を治療しようとする場合、この製品は優先審査を受ける資格がある可能性があり、初歩的な臨床証拠は、この薬物が1つ以上の臨床重要な終点で既存の治療法よりも実質的な改善を示す可能性があることを示している。画期的療法計画によると,新薬候補のスポンサーは,薬剤候補のIND提出と同時にあるいはその後,特定適応の候補薬を画期的療法として指定することをFDAに求めることができる。FDAはスポンサーの申請を受けてから60日以内に候補薬が突破療法指定を受ける資格があるかどうかを決定しなければならない。
臨床試験情報の開示
FDA規制製品(薬品を含む)の臨床試験スポンサーはいくつかの臨床試験情報を登録し、開示しなければならない。そして,臨床試験の製品,患者群,調査段階,試験地点と調査者およびその他に関する情報が公開され,臨床試験登録の一部となった。スポンサーも完成後にその臨床試験の結果を開示する義務がある。場合によっては、これらの裁判結果の開示は、裁判が完了した日から最大2年に延期されることができる。競争相手はこれらの公開された情報を用いて開発計画の進捗状況を知ることができる.
承認後に要求する
FDAによって生産または流通を許可された薬品は、記録保存、定期報告、製品サンプリングおよび流通、広告および販売促進、製品不良経験の報告、および適用された追跡および追跡要求の遵守に関連する要求を含むFDAの普遍的かつ持続的な規制を受けなければならない。承認後、承認された製品の大多数の変更は、新たな適応または他のラベル宣言を追加するなど、FDAの審査および承認を事前に受けなければならない。どの市場製品に対しても、持続的で毎年の使用料要求がある。
FDAはNDAを承認するための条件として、いくつかの承認後の要求を加えるかもしれない。例えば、FDAは、第4段階の臨床試験を含む上場後試験を要求し、製品の商業化後の安全性と有効性をさらに評価し、監視するために監視を行う可能性がある。
FDAの規定は,製品が特定の施設で生産され,cGMP規定に適合することを要求しており,この規定は他を除いて,品質管理と品質保証,記録やファイルの維持,cGMPとの任意の偏差を調査·是正する義務を要求している。また,医薬品メーカーや他の薬品の製造·流通に関与するエンティティ,およびその製品,成分や成分を提供するエンティティは,FDAや州機関にその機関を登録し,FDAやこれらの州機関の定期的な抜き打ち検査を受け,cGMP要求を遵守することを確保しなければならない。製造プロセスの変更は厳しく規制されており,通常FDAが事前に承認して実施する必要がある。FDA法規はまた、cGMP要求との任意の偏差を調査および是正し、スポンサーおよびスポンサーが使用を決定する可能性のある任意の第三者製造業者に報告および文書要求を提出することを要求する。そのため、メーカーはcGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、労力をかけ続けなければならない。
1つの薬剤が承認されると、規制要件や基準が守られていない場合、または製品が発売された後に問題が発生した場合、FDAは承認を撤回する可能性がある。製品は後に、予期されない重症度または頻度の有害事象、または製造プロセス、または法規要件を遵守できなかったことを含む、以前に未知の問題を発見し、新しいセキュリティ情報を追加するために承認されたラベルの強制改訂をもたらす可能性がある;新しい安全リスクを評価するために発売後研究または臨床試験を実施すること、または流通またはその他を実施すること
RMS計画下の制限。承認またはマーケティングに関するこれらの制限は、製品の商業普及、流通、処方、または配布を制限する可能性がある。他の他の潜在的な結果には
•製品の販売や製造を制限し、市場から製品を完全に撤回したり、製品をリコールしたりする
•承認後の臨床試験には罰金、警告状、一時停止を科す
•FDAが承認すべきNDAまたは承認されたNDAの補充剤の承認を拒否するか、または製品承認を一時停止または撤回すること;
•製品を差し押さえたり差し押さえたり、製品の輸入または輸出を許可することを拒否したり;
•民事または刑事処罰を禁令または適用する。
FDAは市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。製造業者および製造業者を代表する任意の第三者は、承認された適応に対してのみ、製品の承認されたラベルと一致する方法で薬物を普及させることができる。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。
EU法規
連合では、私たちの候補製品もまた広範囲な規制要求を受ける可能性がある。アメリカと同様に、医薬製品は主管監督機関のマーケティング許可を得た後にのみ発売されることができる。
アメリカと類似して、EUの臨床前と臨床研究の各段階は重要な監督管理によって制御されている。
2014年4月,新たな臨床試験条例(EU)第536/2014号が採択され,2022年1月31日に施行され,臨床試験指令2001/20/ECが廃止された。臨床試験条例はすべてのEU加盟国に直接適用され、これは各EU加盟国で国家立法を制定する必要がないことを意味する。新しい臨床試験条例の一時的な条項は、以前にこの指示によって許可された進行中の臨床試験をその指示の下に保持することができ、またはそれらを条例に移行することができると規定している。2025年1月31日までに行われているすべての臨床試験は新たな規定に移行しなければならない。
新しい臨床試験条例はEUの臨床試験の審査を簡略化と簡素化することを目的としている。この条例の主な特徴は、臨床試験情報システム(CTIS)による単一入口点による申請プログラムの簡略化、申請のための単一文書の準備と提出、および臨床試験スポンサーの報告手続きの簡略化、臨床試験申請評価の調整プログラムは2つの部分に分けられる(第1部分は科学と医薬製品文書を含み、第2部分は国と患者レベルの文書を含む)。第1部は、加盟国が作成した報告書を参照して臨床試験許可申請(関係加盟国)が提出されたすべてのEU加盟国主管当局の協調審査によって評価される。二番目の部分は関連する各会員国によって個別的に評価される。臨床試験申請の評価には厳しい期限が設定されている。評価手続きにおける道徳管理委員会の役割は、EU加盟国に関する国家法律によって引き続き管轄されるだろう。しかし、全体的に関連するスケジュールは臨床試験条例によって定義されるだろう。
EUでの薬物の発売許可を得るために、私たちはいわゆる集中型または国家認可プログラムの下でMAASを提出することができる。
集中手順
集中化プログラムは、欧州薬品管理局(EMA)の有利な意見に基づいて、すべてのEU加盟国及び欧州経済区の他の加盟国(アイスランド、リヒテンシュタイン、ノルウェー)で有効な単一マーケティング許可を付与することを規定している。特定のバイオテクノロジー技術によって生産された薬物、孤児薬物として指定された製品、先進的な治療薬(例えば、遺伝子治療、体細胞治療または組織工学薬物)および新しい活性を有する製品では、集中手順は強制的である
HIV/エイズ、癌、糖尿病、神経変性疾患または自己免疫疾患、ならびに他の免疫機能障害およびウイルス性疾患などの特定の疾患を治療するための物質。他の疾患のために告発された新しい活性物質を含む製品、または重大な治療、科学的または技術的革新を表す製品、またはその許可が公衆の健康に有利になる製品については、集中手順が任意である。集中プログラムによると,環境保護局がMAAを評価する最長期限は210日であり,タイマを含まず,申請者は人が薬品委員会やCHMPからの質問に答えるために追加の書面や口頭情報を提供する。CHMPは特殊な状況下で加速評価を承認することができ、特に治療革新の角度から見ると、1種の医薬製品が重大な公衆衛生利益を有することが予想される場合。加速評価プログラムによる重大な影響評価の制限時間は150日であり,クロックポーズは含まれていない.
国家認可手続き
いくつかのEU諸国では、他の2つの可能な方法が医薬製品を許可することができ、これらの経路は集中プログラム範囲に属さない研究用医薬製品に使用することができる
•分散したプログラム。分散プロセスを使用して、出願人は、1つ以上のEU諸国において、どのEU諸国でも許可されていない薬品を同時に許可することができ、集中手続きの強制範囲に属さない。
•プログラムを相互認識する。相互認識手続きでは、EU加盟国の国家手続きに基づいて、薬物がまずその国で許可される。その後、元の国家マーケティング許可を認めることに関係国が同意する手続きにおいて、他のEU諸国にさらなるマーケティング許可を求めることができる。
上記の手順により、販売許可を付与する前に、EU市場管理局又はEU加盟国主管当局は、製品の品質、安全性及び有効性に関する科学的基準に基づいて、製品のリスク−利益バランスを評価する。
EU規制排他性
EUでは、発売を許可された新製品(すなわち参考製品)は、マーケティング許可を得た後、8年間のデータ独占と追加2年間の市場独占を得る資格がある。データ独占期間防止後発薬申請者はEUが模倣薬の発売許可を申請する時に参考製品ファイルに含まれる臨床前と臨床試験データに依存し、参考製品が初めてEUの許可を得た日から8年以内である。市場排他期は成功した後発薬申請者がその製品をEUで商業化することを禁止し、参考製品がEUで最初の許可を得た10年後まで。10年の最初の8年間に、マーケティング許可保持者が1つまたは複数の新しい治療適応の許可を得た場合、許可前の科学的評価期間中に、これらの適応は既存の療法と比較して有意な臨床的利益をもたらすことができると考えられる場合、10年間の市場専門期間は最大11年に延長することができる。
EU孤児指定と排他性
EUでは、孤児医薬品を指定する基準は原則として米国と類似している。条例(EC)141/2000第3条によると、生命または慢性衰弱にかかわる疾患の診断、予防または治療に使用されている場合、孤児として指定することができる;(Ii)申請を行う際には、EUで10,000人中5人以下の影響を与えるか、または(B)孤児の身分のメリットがなければ、投資が合理的であることを証明するためにEUで十分な見返りを生じることはない。および(Iii)このような疾患を満足できる診断、予防または治療する方法がEU市場で販売されていないか、またはそのような方法が存在する場合、製品は、(EC)847/2000規制によって定義されているこのような疾患の影響を受ける人に大きな利益をもたらすであろう。孤児医薬製品は費用の低減や費用の免除などの財政奨励を受ける資格があり、マーケティング許可を得た後、承認された治療適応の10年間の市場排他性を得る権利がある。上場許可を申請する前に、孤児指定申請を提出しなければならない。孤児が孤児として指定された場合、出願人はマーケティング許可申請の費用減免を受けるが、マーケティング許可を提出したときにその指定が待っている場合は、そうではない。孤児指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の継続時間を短縮することもない。
5年目の終了時に製品が孤児指定の基準を満たしていないと判断された場合、例えば、製品が証明するのに十分な利益がなく、証明されていない場合、EUにおける10年間の市場独占権は6年に減少することができる
市場の排他性を守る。さらに、以下の場合、ライセンス孤児製品と同じ適応を有する類似医薬製品には、いつでもマーケティング許可を付与することができる
•第2の出願人は、その製品は類似しているが、許可された孤児製品よりも安全で、より効果的であり、または臨床的に良いことを証明することができる
•許可された孤児製品の販売授権者は、2回目の孤児薬品の申請に同意する;または
•孤児製品を許可する販売授権者は十分な孤児薬品を供給することができない。
上記のEU規則は、EU加盟国にノルウェー、リヒテンシュタイン、アイスランドを加えた欧州経済地域(EEA)に一般的に適用されている。
EU小児科調査計画
EUでは、新しい医薬製品を開発する会社はEMAの小児科委員会或いはPDCOと小児科調査計画或いはPIPについて合意しなければならず、EMAがPIPに含まれる1つ以上の措置について製品の免除、分類免除或いは延期を許可しない限り、このPIPに基づいて小児科臨床試験を行わなければならない。会社が認可された薬物のために新たな適応,薬物形式あるいは投与経路を増加させようとした場合にも,この要求は適用される。PIPは,市販認可が求められている薬物の小児科適応を支援するためのデータ生成の時間とアドバイスを規定している。PDCOは、成人における製品の有効性および安全性を証明するのに十分なデータがあるまで、PIPの一部または全ての措置の実施を延期する義務を許可することができる。さらに、小児科臨床試験データが不要または不適切に提供される場合、PDCOは、子供に無効または安全でない可能性があるので、これらのデータを提供する義務を免除することができ、この製品は、治療のために使用される疾患または状態が成人集団でのみ発生することが予想される、または小児科患者の既存の治療に対して有意な治療利益がない場合である。PIPによる小児科臨床試験結果の発売許可を得た製品(結果が否定的であっても)6カ月の補充保護証明書の取得延期は,SPC製品申請の提出と同時に,あるいはSPC満期2年前のいつでも延期を申請することを前提としている。孤児の医療製品の場合, 孤児市場の独占権は2年間延長されるかもしれない。この小児科奨励は特定の条件の制約を受け,PIPに適合したデータを開発·提出する際に自動的に獲得されない。
承認後にコントロールする
上場許可所持者は薬物警戒システムを構築·維持し,薬物警戒に従事する資格のある個人を指定し,このシステムの監督を担当しなければならない。主な義務は深刻な副作用が疑われる報告を加速させ、安全更新報告書を定期的に提出することを含む。
すべての新しいMAAは、企業が実施するリスク管理システムを記述し、製品に関連するリスクを最小限に防止または低減するための措置を記録したリスク管理計画またはRMPを含まなければならない。規制部門はまた、上場許可の条件として特定の義務を規定することができる。このようなリスク最小化措置または認可後の義務は、追加の安全監視、PSURsのより頻繁な提出、または追加の臨床試験または認可後の安全性研究を含む可能性がある。RMPおよびPSURsは通常,アクセスを要求する第三者に利用可能であるが,限られた編集を行う必要がある.
製品のすべての広告および販売促進活動は、承認された製品特性要約と一致しなければならないため、すべてのラベル外の販売促進活動は禁止されます。連合はまた消費者に直接向けた処方薬の広告を禁止する。EU指令は医薬製品の広告と販売促進の一般的な要求を規定しているが、詳細は各加盟国の法規によって管理されており、各国は異なる可能性がある。
他の規制は世界の他の地域では
EUや米国以外の他の国、例えば東欧、ラテンアメリカ、アジアの国では、臨床試験、製品許可、定価、精算の要求は司法管轄区域によって異なる。また,臨床試験はCGCP要求および“ヘルシンキ宣言”からの適用法規要求と倫理原則に従って行わなければならない。
もし私たちが適用される外国監督管理要求を遵守できない場合、私たちは罰金、規制許可の一時停止または撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などに直面する可能性がある。
他の医療保険法
製薬会社は、連邦政府およびそれらが業務を展開している州と外国の司法管轄区域当局の追加的な医療監督管理と法執行を受けており、これは、私たちが研究、販売、マーケティング、流通し、マーケティングの許可を得た任意の製品の財務的配置と関係を制限するかもしれない。このような法律には、州および連邦反リベート、詐欺および乱用、虚偽声明、ならびに薬品の価格設定および支払い、ならびに医師および他の医療提供者への他の価値移転に関連する透明性のある法律および法規が含まれているが、これらに限定されない。もし私たちの業務がこのような法律または任意の他の適用可能な政府法規に違反していることが発見された場合、私たちは行政、民事と刑事処罰、損害、罰金、返還、削減または再構成業務、誠実な監督と報告義務、連邦および州医療計画から除外され、責任個人が監禁される可能性があるが、処罰を受ける可能性がある。
FDAのほか、製品承認後の製造、販売、販売促進、その他の活動も米国の多くの規制機関によって規制されており、CMS、HHS監察長事務室、HHS民権事務室、HHSの他の部門、司法省も含まれている。
医療提供者、医師、第三者支払者は、私たちが市場で承認された任意の製品の推薦と処方において主な役割を果たすだろう。私たちの現在と未来の第三者支払者、医療提供者、医師との手配は、私たちが市場で承認された任意の薬物の業務または財務的配置と関係を制限するかもしれない幅広い適用された詐欺と乱用、および他の医療法律と法規に直面するかもしれません。米国では、これらの法律は、州および連邦反リベート、虚偽声明、医師透明性、および患者データプライバシーおよびセキュリティ法律法規を含むが、これらに限定されないが、これらに限定されない。
米国連邦反リベート法規(AKS)は、任意の個人またはエンティティが、任意の個人またはエンティティが、任意の商品、施設、物品またはサービスを購入、レンタル、注文、発注、手配または推薦、購入、レンタル、または任意の商品、施設、物品またはサービスを購入、レンタルまたは注文する見返りとして誘導または提供するために、任意の個人またはエンティティが、任意の商品、施設、物品またはサービスを直接または間接的に提供、開示または隠蔽することを禁止し、これらの商品、施設、物品またはサービスはMedicare、Medicaidまたは他の連邦医療保健計画の下で全部または部分的に精算することができる。“報酬”という言葉は価値のあるものを含むと広く解釈されている。AKSは、一方が医薬品および医療機器製造業者である一方で、処方者、購入者、処方マネージャー、および受益者の間の手配に適していると解釈される。いくつかの法定例外状況と規制安全港保護のいくつかの一般的な活動は起訴されないが、例外状況と安全港の範囲は狭い。特定の適用された法定例外や安全港を規制するすべての要求を満たしていないことは、AKSによって、このような行為自体が不法であることを意味するわけではない。代わりに、そのすべての事実と状況の累積審査に基づいて、この手配の合法性を逐案的に評価する。いくつかの裁判所は、この法規の意図要求を、報酬の手配に関連する任意の目的が連邦医療カバーの業務への移行を誘導することである場合、法規が違反されたと解釈する。また,個人や実体は,法規や法規違反の具体的な意図を実際に知る必要がなく,違反行為を実施することができる.また、連邦民事虚偽請求法によると、AKS違反による物品やサービスを含むクレームは、虚偽または詐欺的クレームを構成する。
支払人に直接請求を提出しないにもかかわらず、連邦虚偽請求法案によれば、医薬品製造業者は、民事告発者または準訴訟を含む個人またはエンティティ(製造業者を含む)に民事罰を適用する可能性があり、その理由は、連邦計画(連邦医療保険および医療補助を含む)に意図的に提出されるか、または連邦計画(連邦医療保険および医療補助を含む)に虚偽または詐欺的な物品またはサービスクレーム、クレームに従って提供されていない物品またはサービスのクレーム、または医療上不要な物品またはサービスのクレームを引き起こすことを含む。政府は、製造業者が顧客に不正確な請求書や符号化情報を提供したり、ラベル外で製品を宣伝したりすることによって、虚偽または詐欺的なクレームの提出を“招く”と考える可能性がある。いくつかのバイオ製薬、医療機器、その他の医療保険会社は、顧客に製品を無料で提供する疑いがあり、顧客がその製品の連邦計画に料金を徴収することが予想されるため、連邦虚偽クレームと民事罰金法律で起訴されている。他社は未承認(例えば、またはラベル外)の製品を販売し、虚偽の声明を提出したことで起訴された。さらに、民事罰金法規は、連邦健康計画にクレームを提起することが決定されたまたは原因となった任意の者に処罰を加え、その人は、クレームに従って提供されていない項目またはサービスまたは虚偽または詐欺的な項目またはサービスであることを知っているか、または知っているべきである。
私たちの将来のマーケティングと報告卸売業者や私たちの製品の推定小売価格に関する活動(承認されれば)、医療補助フィードバック情報を計算するための価格報告、その他私たちの製品に影響を与える連邦、州、第三者精算の情報、および候補製品の販売とマーケティングは、これらの法律の審査を受けています。
1996年に連邦“健康保険可搬性と責任法案”(HIPAA)は追加の連邦刑法を制定し、他の行動以外に、計画を知り知り、故意に実行または実行しようとし、虚偽または詐欺的な言い訳、陳述または約束の方式で、詐欺或いは虚偽或いは詐欺的な口実、陳述或いは約束を通じて、任意の医療福祉計画が所有或いは制御或いは保管している任意の金銭或いは財産を獲得し、医療福祉計画を故意に流用又は窃取し、医療保険違法行為に対する刑事調査を故意に阻害し、故意に重要な事実を偽造、隠蔽又は隠蔽し、又は任意の重大な虚偽、虚偽の行為を行うことを明らかにしている。医療福祉、プロジェクトまたはサービスの交付または支払いに関する架空または詐欺的陳述。AKSと同様に,個人や実体は法規や法規違反の具体的な意図を実際に知る必要がなく違反を実施することができる.
また,最近では連邦や州政府が医師や他の医療提供者に支払う費用の規制を強化する傾向が見られている。その他の事項を除いて、“平価医療法案”(ACAと略称する)は“医師支払い日光法案”を可決し、医師(医師、歯科医師、検眼師、足科医師と脊椎マッサージ師を含む)と教育病院に提供されるいくつかの支払いと“価値移転”、及び医師及びその直系親族が持つ所有権と投資権益に対して、新しい年間報告要求を規定した。これらの報告義務は,2022年1月1日から,医師アシスタントや看護師従業員などのある非医師提供者への価値移転を含むように拡大された。すべての支払い、価値譲渡および所有権または投資利益に必要な情報を適時、正確かつ完全に提出できなかった場合、民事罰金を招く可能性がある。カバーされた製造業者は、その後の各例年の90日目までにレポートを提出しなければならず、レポートの情報は検索可能なウェブサイト上で公開される。
私たちは連邦政府価格報告法および連邦消費者保護法と不正競争法の制約を受ける可能性があり、連邦政府価格報告法は複雑な価格指標を正確かつタイムリーに計算し、それを政府プロジェクトに報告することを要求し、連邦消費者保護法と不正競争法は市場活動を広く規制し、消費者の活動を損なう可能性がある。
私たちはまた連邦政府と私たちが業務を展開している州のデータプライバシーと安全規制の制約を受ける可能性がある。“健康情報技術促進経済と臨床健康法案”(HITECH)改正されたHIPAA及びそのそれぞれの実施条例は、2013年1月25日に発表された最終HIPAA総合規則を含み、保証実体及びその業務パートナーが持つ単独で識別可能な健康情報のプライバシー、安全と伝送に対して具体的な要求を提出した。他の事項に加えて、HITECHは、HIPAAのセキュリティ基準を“ビジネスパートナー”に直接適用し、すなわち、カバーエンティティまたはカバーエンティティに代わってサービスを提供する際に、保護された健康情報を作成、受信、維持、または送信する独立した請負者またはエージェントを、通常の業務中に“ビジネスパートナー”とみなされるかどうかは不明であるが、保護された健康情報を作成、受信、維持、または送信する。HITECHはまた、実体、商業パートナー、および可能な他の人に適用される民事と刑事罰を増加させ、州総検察長に新たな権力を与え、連邦裁判所に民事訴訟を提起し、連邦HIPAA法を実行するために損害賠償または禁令を要求し、連邦民事訴訟の提起に関連する弁護士費と費用を求めることができる。また、州法は、場合によっては健康情報のプライバシーやセキュリティを管理しており、その多くの法律は互いに大きく異なり、同じ要求がなく、コンプライアンス作業を複雑にしている可能性がある。EUのデータプライバシーとセキュリティに関する議論は、以下の“欧州データ収集”を参照されたい。
例えば、カリフォルニア州の“消費者プライバシー法”(Consumer Privacy Act、CCPAと略称する)は2020年1月に施行され、その後カリフォルニア州総検察長は最終法規を公布した。この法律はカリフォルニアの消費者にその個人情報を収集して使用する広範な権利を提供し、ある企業にデータ保護義務を課している。CCPAは、HIPAAによって制約された保護された健康情報または連邦法律定義の研究において収集、使用または開示された個人情報には適用されないが、CCPAは依然として我々の商業活動に影響を与える可能性がある。また、2020年11月3日、カリフォルニア州有権者は、CPRAと略称する投票イニシアティブの下でカリフォルニアプライバシー権法案を可決した。CPRAは既存のCCPAを改正し,新たな消費者権利と追加のデータ保護義務を加えた。CPRAにおける新しいデータ保護要件は,2022年1月1日以降に収集された情報に適用される.最終法規の公布に伴い、カリフォルニア州の総検察長はCCPAに違反した人に対する法執行行動を開始した。CCPAとCPRA改正案の実施をめぐる不確実性は,我々の業務が個人データや保護された健康情報に関する変化する規制環境の影響を受けやすいことを示している。カリフォルニア州の法律は、プライバシーとプロセスの強化に対する需要をさらに拡大し、リソース投入を約束してコンプライアンスを支援している。また、昨年は10以上の州でCCPAやCPRAに類似した条項の法案が提出された。可能性が高い
他の州では近い将来CCPAやCPRAのような法律が成立し,連邦データ保護法も登場する可能性がある。
同様の州および外国詐欺および法律法規の乱用、例えば州反リベートおよび虚偽請求法は、医療プロジェクトまたはサービスに関連する販売またはマーケティング配置およびクレームに適用される可能性がある。このような法律は一般的に広く、様々な国家機関と個人行動によって実行される。また,連邦医療補助や他の州が計画して精算するプロジェクトやサービスを除いて,多くの州で類似した詐欺や法律や法規の乱用があり,その範囲は支払者にかかわらず適用可能である可能性がある。いくつかの州の法律は、製薬会社が製薬業の自発的コンプライアンスガイドラインと関連する連邦政府コンプライアンスガイドラインを遵守することを要求し、医師および他の医療保健提供者への支払いおよび他の価値移転、マーケティング支出または薬品定価に関する情報を報告するように製薬業者に要求する。
製品を商業的に流通させるためには、州の法律を遵守しなければならず、ある州で製品をその州のメーカーおよびディーラーに輸送することを含む州で薬品および生物製品の製造業者および卸売業者の登録を要求しなければならない。一部の州はまた、メーカーと流通業者が流通チェーン中で製品の系統を確立することを要求しており、いくつかの州はメーカーと他の州に流通チェーン中の製品の流れを追跡し追跡できる新しい技術を採用することを要求している。いくつかの州はすでに立法を公布し、製薬と生物技術会社にマーケティングコンプライアンス計画を確立し、州政府に定期報告を提出し、販売、マーケティング、定価、臨床試験およびその他の活動を定期的に公開し、および/またはその販売代表を登録し、薬局および他の医療保健実体が製薬および生物技術会社にいくつかの医師処方データを販売およびマーケティングのために提供することを禁止し、いくつかの他の販売およびマーケティング行為を禁止する。私たちのすべての活動は連邦と州消費者保護と不正競争法によって制限されるかもしれない。
これらの法律の範囲も執行も不確定であり,現在の医療改革環境下では,特に適用の前例や法規が乏しい場合には,急速な変化が生じる可能性がある。連邦と州法執行機関は最近、医療保険会社と医療保健提供者の間の相互作用の審査を強化し、医療保健業界の一連の調査、起訴、有罪判決と和解を招いた。政府当局は、私たちの業務実践が現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関連する現行または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちの業務がこれらの法律または任意の他の私たちの政府法規に適用される可能性があることが発見された場合、私たちは重大な民事、刑事と行政処罰、損害賠償、罰金、返還、契約損害、名誉損害、利益減少と将来の収益、監禁、メディケアおよびMedicaidのような政府援助の医療計画から医薬品を排除し、私たちの業務を削減または再編し、もし私たちが会社の誠実な合意や他の合意に制約された場合、これらの法律違反に関する告発、および追加の報告義務と監督を解決することができます。いずれも私たちの業務運営能力と財務業績に悪影響を及ぼす可能性がある。もし私たちがそれと業務を展開することを期待している任意の医師または他の医療提供者または実体が適用されない法律に適合していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む重大な刑事、民事または行政制裁を受ける可能性がある。業務手配が適用される医療保険法に適合することを確保する, 政府当局が可能な調査に応えるほか、時間や資源がかかる可能性があり、会社の業務への注意を分散させる可能性がある。
ヨーロッパデータ収集
EU域内またはEUから生成された個人健康データの収集と使用は、データ保護指令および一般データ保護法規(GDPR)の規定によって管轄される。この指令は,個人データに関する個人の同意,個人への情報提供,主管国家データ保護機関へのデータ処理義務の通知,個人データの安全と秘密についていくつかの要求を行っている。データ保護指令やGDPRは、個人データをEUから米国に移すことにも厳しいルールを実施しています。データ保護指令、GDPR、EU加盟国の関連国家データ保護法の要求に違反すると、罰金やその他の行政処罰を招く可能性があります。GDPRはEUに新たなデータ保護要求を導入し,データ保護規則違反に巨額の罰金を科した。GDPR規制は、私たちが処理している個人データ(臨床試験を含む)に追加的な責任と責任を課す可能性があり、新しいデータ保護ルールの遵守を保証するための追加のメカニズムを確立する必要があるかもしれません。これは激務であり、私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性がある。
現在と未来の立法
米国や他の管轄地域では、医療システムに関する立法や法規の変更、提案された変更は、候補製品の上場承認を阻止または延期し、承認後の活動を制限または規制し、マーケティング承認された候補製品を収益的に販売する能力に影響を与える可能性がある。現在の法律、および将来取られる可能性のある他の医療改革措置は、より厳しいカバー基準をもたらす可能性があり、私たちまたは任意の協力者が受ける可能性のある任意の承認された製品の価格に追加の下振れ圧力をかける可能性があると予想しています。
国内でも海外でも,政府も個人も,支払側は医療コストを抑えるための複雑な方法を開発しているが,これらの方法は,遺伝子療法やまれな疾患に対する療法,例えば我々が開発しているような新しい技術に特化しているわけではない。米国や一部の外国司法管轄地域では、医療システムの立法や規制に多くの変化が生じており、製品販売の収益性に影響を与える可能性がある。特に、2010年に“医療·教育調整法”により改正された“患者保護·平価医療法案”(または総称して“ACA”)が公布され、他の事項を除いて、生物製品を低コストの生体模倣薬の潜在的競争を受けさせ、医療補助薬物リベート計画に基づいてメーカーが医療補助薬物リベート計画の下で不足しているリベートを計算する新しい方法を解決し、医療補助薬物リベート計画の下で不足している最低医療補助リベートを大部分のメーカーが増加させ、医療補助医薬品リベート計画を医療補助管理のケア組織に登録された個人処方に拡大する。メーカーはあるブランドの処方薬に対して新しい年間費用と税収を支払うことを要求した;新しいMedicare Part D保証不足割引計画を作成し、メーカーは保証不足中に条件を満たす受益者に50%(2018年の両党予算法に従って70%に増加)の販売時点割引を提供することに同意しなければならず、メーカーの外来薬物としてMedicare Part Dの条件を組み込む;そして連邦政府の比較的有効性研究の計画を増加させるために激励を提供しなければならない
公布以来、反腐敗法のある方面は司法、行政、行政と立法方面の挑戦を受けた。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、作業要求を含む医療補助モデル項目および免除計画の再検討、医療補助またはACAによる医療保険カバー範囲の獲得による不必要な障害をもたらす政策の再検討、医療補助またはACAによる医療保険の取得を制限する既存の政策および規則の見直しを指示する。バイデン政府の他の医療改革措置や他の挑戦,ACAの廃止または代替の努力(あれば)が我々の業務にどのように影響するかは不明である。
また、ACAが公布されて以来、アメリカは他の連邦医療改革措置を提案し、採択した
•2011年の“予算制御法案”には、他の内容に加え、各年度に提供者に支払われる医療保険の総減少幅が2%となっている。これらの削減は2013年4月1日に施行され,その後の法規制改正により2030年まで有効であったが,新冠肺炎の大流行により2020年5月1日から2022年3月31日までの一時停止は除外された。一時停止後、2022年4月1日から2022年6月30日までに1%の支払いを下げ、2022年7月1日に2%の支払い減免を再開する。
•2012年の“米国納税者救済法”は、いくつかの医療サービス提供者への医療保険支払いを減らし、政府が提供者に多額の金を取り戻す訴訟時効を3年から5年に延長した。
•2017年4月13日、CMSは、各州に個人や小団体市場に保険会社のための基準を設定する上でより大きな柔軟性を与える最終ルールを発表し、このような市場で販売されている保険計画に対してACAが要求する基本的な健康福祉を緩和する可能性がある。
•2018年5月30日、“裁判権法案”が法律に署名された。他の事項以外に、この法律はある患者に連邦フレームワークを提供し、彼らが第一段階の臨床試験を完成し、FDAの許可を得た研究用新薬製品を獲得するために調査を行っていることを許可した。場合によっては、条件を満たす患者は、臨床試験に参加することなく、FDA拡大参入計画に従ってFDA許可を得ることなく治療を求めることができる。“試用権法案”によると,製薬業者はその薬品を条件に該当する患者に提供する義務はない
•CMSは2019年5月23日、Advantage計画が2020年1月1日からB薬剤の一部に階段療法を使用することを許可する最終ルールを発表した。
•2019年12月20日、トランプ前総裁は“さらなる総合支出法案”(H.R.1865)に署名し、ケディラク税、医療保険提供者税、医療機器消費税を廃止した。未来に似たような税金が発行されるかどうかを確定することは不可能だ。
•2021年3月11日、総裁·バイ登は“2021年米国救援計画法案”に署名し、2024年1月1日から、単一源と革新多源薬物に対する法定医療補助薬品還付上限を廃止し、現在この上限は薬品メーカー平均価格の100%である。
•2022年8月16日、総裁·バイデンは“2022年インフレ率低減法案”に署名し、処方薬のコスト低減のためのいくつかの措置と関連する医療改革を含む。これらの提案、提案、および法規は、現行の所得税フレームワークの変更と、新しいタイプの非所得税(例えば、収入のパーセンテージで課税するか、またはデジタルサービスに適用される税金)とを含む。
また、米国の薬品価格決定方法に対する立法と法執行への関心もますます大きくなっている。 具体的に、政府はメーカーがその販売製品に価格を設定する方式に対して更に厳格な審査を行い、アメリカ議会はいくつかの調査を行い、連邦と州立法を提出し、公布し、薬品定価の透明性を高め、連邦医療保険制度下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査した。 連邦レベルでは、総裁·バイデンは2021年7月9日に行政命令に署名し、政府の政策、すなわち(I)処方薬と生物製品価格を下げる立法改革を支持し、連邦医療保険の薬品価格交渉を許可し、インフレ上限を設定し、低コスト模造薬と生物模倣薬の開発と市場参入を支持すること、および(Ii)公共医療保険オプションの制定を支持することを確認した。その他の事項に加えて、行政命令は、処方薬の価格設定が高すぎ、国内の薬品サプライチェーンの強化、連邦政府の薬品支払いの価格低下、業界価格詐欺の解決のための行動を説明する報告書をHHSに提供するよう指示し、2003年の“連邦医療保険処方薬、改善と現代化法案”およびFDAの実施条例に基づいて第804条の輸入計画を制定する州とインディアン部族との協力を提案するようFDAに指示した。FDAは2020年9月24日にこのような実施条例を発表し、2020年11月30日に発効し、各州のカナダ薬品輸入計画の制定と提出に指導を提供した。2020年9月25日、CMSは、この規則に基づいて輸入された州の薬品は、社会保障法第1927条に基づいて連邦還付を受ける資格がなく、メーカーは“最適価格”や平均メーカー価格の目的でこれらの薬品を報告しないと声明した。これらの薬剤はカバーされた外来薬とは考えられないため,CMSはさらに,これらの薬剤の全国平均薬物調達コストは公表されないと述べている。実施すれば、カナダからの薬品輸入は私たちの任意の候補製品の価格に実質的かつ不利な影響を与えるかもしれない。また、2020年11月20日、CMSは最恵国待遇を実施する臨時最終規則、すなわち最恵国待遇を発表した, このようなモデルの下で、ある薬品と生物製品の医療保険B部分の販売率は1人当たりの国内総生産に類似した経済協力と発展組織国の薬品メーカーが獲得した最低価格に基づいて計算される。 しかし、2021年12月29日、CMSは最恵国規則を廃止した。また、2020年11月30日、HHSは薬品メーカーがD部分でスポンサー値下げを計画している安全港の保護を廃止し、法律が値下げを要求しない限り、直接あるいは薬局福祉マネージャーを通過する法規を発表した。この規定はまた、販売時点での値下げを反映するための新しい避難港を作成し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための避難港を作成する。裁判所の命令により,上記安全港の除去と増加は延期され,最近の立法は同規則の実施を2026年1月1日に一時停止した。いくつかの措置および他の提案された措置は発効するために追加の立法によって許可される必要があるかもしれないが、バイデン政府はこれらの措置を撤回または他の方法で変更する可能性があるが、バイデン政府と国会は薬品コストを制御するための新しい立法措置を求め続けると表明している。
私たちは将来的により多くのアメリカ連邦医療改革措置を取ることが予想され、そのいずれも米国連邦政府が医療薬やサービスのために支払う金額を制限する可能性があり、これは私たちの候補薬物に対する需要の減少または追加の価格設定圧力を招く可能性がある。
アメリカ各州もますます立法を通じて薬品の価格を制御するための法規を実施しており、価格或いは患者の精算制限、割引、ある製品への参入とマーケティングコスト開示の制限及び透明性措置を含み、場合によっては、ある状況では、
他の国と大口調達。法律で規定されている第三者支払者の支払金額の価格制御またはその他の制限は、私たちの業務、財務状況、運営結果、将来性を損なう可能性があります。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これは私たちの薬品の最終需要を減少させ、あるいは私たちの薬品の価格設定に圧力を与える可能性があり、これは私たちの業務、財務状況、運営結果と将来性に負の影響を与えるかもしれない。
人力資本
2022年12月31日現在、私たちは227人のフルタイム従業員を持っており、そのうち113人は研究開発計画の推進に集中し、36人は商業運営に集中し、78人は戦略業務発展、財務、その他の技術専門家、および一般および行政サービスを提供している。私たちの職員たちは労働組合代表もなく、集団交渉協定のカバー範囲もない。 予想される未来には、従業員数は引き続き増加し、特に製品や商業化能力の開発を継続している状況であると予想される。
私たちは従業員がわが社の最も重要な資産の一つであり、私たちの目標と期待を達成する鍵だと信じている。したがって、私たちは人材を誘致して維持することを非常に重視している。私たちの管理チームと機能担当者は従業員の尊敬度と満足度調査を定期的に審査し、従業員の流出率を監視します。
報酬と福祉
私たちは最も優秀な人材を誘致して維持するために競争力のある報酬を提供する。私たちの総給与プログラムには市場競争力のある賃金、ボーナス、そして株式が含まれている。私たちは常勤従業員が招聘された時に株式を提供し、自由に支配可能な年間持分によって付与されています。会社の所有権株式を所有し、長期的な成功に取り組んでほしいからです。医療、家庭、金融、コミュニティ、有給休暇などの分野をカバーし、医療や健康福祉、401(K)計画、法的サービスを受ける機会、育児休暇など幅広い福祉を提供しています。
多様性と包括性
多様性と包括性は私たちの文化の重要な構成要素だ。私たちの重点は私たちの多様性と包括性の利点と機会を理解し、さらなる進歩を支援する戦略を実行することだ。私たちは、性別、人種、性指向、または他の共通属性のような多様性をめぐる対話および参加を促進するために、多くの従業員リソースグループを作成し、これらの属性は、コミュニティを構築し、発展の機会をもたらすのに役立つと考えられる。私たちは人材パイプの構築に集中し続け、職場の多様化のためにより多くの機会を創出し、社内のより大きな代表性を支援しています。
会社の歴史と情報
私たちは2017年8月にデラウェア州に設立され、2019年3月29日までデラウェア州の有限責任会社SpringWorks Treateutics,LLCを介して業務を展開しています。2019年3月29日に完成した会社再編·合併の条項によると、SpringWorks Treateutics,LLCのすべての持分がSpringWorks Treateutics,Inc.が新たに発行された同じ数量と種類の証券に交換されるため、SpringWorks Treateutics,LLCはSpringWorks Treateutics,Inc.の完全子会社となる。
2019年9月17日、私たちは初公募株式(IPO)を完了し、これにより、引受業者が彼らの選択権を全面的に行使することを含む10,350,000株の普通株を発行·売却し、1株18.00ドルの公開発行価格で1,350,000株の追加普通株を購入し、引受割引と手数料およびその他の発行費用を差し引いた純収益は1.697億ドルだった。初公募終了時、私たちの発行済み転換可能優先株は自動的に普通株に変換されます。
2020年10月13日、私たちは後続発行を完了し、それに基づいて、私たちは5,637,254株の普通株を発行·売却し、引受業者が彼らの選択権を全面的に行使し、1株51.00ドルの公開発行価格で735,294株の追加の普通株を購入し、引受割引と手数料およびその他の発行費用を差し引いた後、純収益は2.695億ドルだった。
2022年12月31日までの12ヶ月間、市場発行計画やATM計画に基づいて2247,500株の普通株を売却し、総収益は6970万ドル、手数料とその他の費用を引いた190万ドル、純収益は6780万ドルだった。ATM計画では2022年12月31日現在も約1億303億ドルが利用可能だ
2022年9月6日、私たちはグラクソ·スミスクライン社(前身はグラクソ·スミスクライン、またはグラクソ·スミスクライン)と拡大した世界的な非独占的許可と協力協定を締結し、niroacestatとbelantamab mafodotin(Belamaf)を組み合わせて使用した。この合意を実行すると同時に、グラクソ·スミスクラインの関連会社グラクソ·スミスクライングループ有限会社またはGGLと株式購入協定を締結し、この合意によると、GGLは私募取引で2,050,819株の普通株を購入することに同意し、総購入価格は約7,500万ドル、1株当たり36.57ドルであった。
2022年9月7日、吾らはいくつかの認可投資家或いは投資家と証券購入協定を締結し、この合意に基づいて、吾らは私募取引或いは私募方式で投資家に8,650,520株の普通株を売却·発行することに同意し、購入価格は1株26.01ドルであった。指向性増発では,約2.25億ドルの総収益を得ており,手数料と発行コストを差し引くと,純収益は約2.168億ドルである。
私たちは私たちの会社名とロゴを含む様々なアメリカ連邦商標申請と未登録商標を持っています。本年度報告書で言及されている他のすべての商標または商号は、そのそれぞれの所有者の財産である。便宜上、本明細書の付録に記載された商標および商品名は、記号および使用されていないが、そのような言及は、そのそれぞれの所有者が、適用法に従ってその権利を最大限に主張しないいかなる指示も解釈されないと解釈されるべきではない。
上記取引に関するより多くの情報は、第2部である項目7−経営層の財務状況及び業務成果の検討及び分析、並びに第2部である項目8に掲げる連結財務諸表付記1を参照されたい。
私たちの主な実行事務室はコネチカット州スタンフォードワシントン通り100号にあります。郵便番号:06902、電話番号は(2038839490)。私たちのサイトはhttp://www.springworkstx.comです。当社のサイトに掲載されているか、当社のサイトから取得可能な資料は本年度報告に含まれていませんので、閣下はこれを本年度報告の一部と見なすべきではありません。
利用可能な情報
私たちのインターネットアドレスはwww.springworkstx.comです。我々の10-Kフォーム年次報告、10-Qフォーム四半期報告、8-Kフォーム現在の報告は、証拠品、依頼書、および情報声明、および取引法第13(A)および15(D)条に基づいて提出または提供されたレポートの修正案を含み、材料を電子的にアーカイブまたは米国証券取引委員会に提供した後、合理的で実行可能な範囲内でできるだけ早く私たちのサイトの“投資家”によって部分的に無料で取得することができる。私たちのウェブサイト上の情報は、参照によって特に本明細書に組み込まれない限り、本Form 10-K年次報告または私たちの任意の他の証券届出ファイルの一部ではない。また、私たちがアメリカ証券取引委員会に提出した書類は、アメリカ証券取引委員会の相互情報電子申請システムを介して調べることができます。URLはHttp://www.sec.govそれは.私たちが任意の証券届出文書で下したすべての陳述は、すべての前向きな陳述または情報を含み、その陳述を含む文書の日付で行われ、法律が私たちにそうすることを要求されない限り、私たちはこれらの陳述または文書を更新するいかなる義務も負わない。
新冠肺炎が大流行する
2019年12月、武漢で1種の新しいコロナウイルス株--深刻な急性呼吸症候群コロナウイルス2型を発見し、SARS-CoV-2と略称し、中国。2020年3月11日、世界保健機関はSARS-CoV-2に関連する疾病新冠肺炎の発生を全世界大流行とした。この疾患は,新たに出現した新冠肺炎変異株を含め,我々が業務を展開している地域に伝播し続けている。新冠肺炎の蔓延を緩和するため、世界各国の政府と企業は現地避難令、隔離措置、旅行に対する重大な制限、および多くの従業員の出勤禁止の制限を含む未曽有の行動を取った。新冠肺炎疫病発生から、著者らは一連の業務連続性措置を開始し、著者らの業務に対する潜在的な中断を減少し、そして著者らの研究開発計画の完全性を維持した。新冠肺炎の流行期間中、私たちの対応は変化し、私たちは基本的に正常な運営を回復した。これまで、私たちは現在行われている研究開発活動のいかなる実質的な中断も経験したことがない;しかし、新冠肺炎の大流行或いは新しく出現した新冠肺炎変異株のいかなる影響により、私たちは私たちの研究開発、商業化スケジュールと結果に影響を与える可能性のある中断を経験する可能性がある。私たちは進行中の新冠肺炎の流行と新興変種が私たちの業務に与える影響を評価し続ける。現在の新冠肺炎大流行が著者らの未来の結果に与える影響程度は未来の発展に依存するが、大流行及びその関連影響は、大流行の持続時間、伝播と強度(任意の再発を含む)、新冠肺炎ウイルス新変異株の影響及び新冠肺炎ワクチンの発売を含む, これらすべては依然として不確定で予測困難であり、私たちの業務、将来の見通し、将来の財務状況、運営結果、およびキャッシュフローに実質的な影響を与える可能性がある。
第1 A項。リスク要因
当社及び当社の業務を評価する際には、本Form 10−K年次報告書及び米国証券取引委員会(SEC)又は米国証券取引委員会に提出された他の文書に記載されている他の情報に加えて、以下のリスク要因を慎重に考慮しなければならない。私たちの普通株に投資することは高い危険と関連がある。もし実際に以下のいかなるリスクと不確定要素が発生すれば、私たちの業務、将来性、財務状況と経営結果は重大な不利な影響を受ける可能性がある。以下に述べるリスクは詳細ではなく、同社が直面している唯一のリスクでもない。新しいリスク要素は時々出現する可能性があり、いかなる要素或いは要素の組み合わせが私たちの業務、将来性、財務状況と運営結果に与える影響を予測できない。
会社特有の重大なリスク要因の概要
私たちはSpringWorksに特定された重大なリスクと考えられる要約を含めている。この要約は、私たちのビジネスに関連するすべての重大なリスクを含まず、私たちのリスク要因に対する決定的な順位や優先順位でもありません。また,その中のいくつかのリスクを要約部分に置くことは,他のリスクではなく,我々の証券への投資を考慮する際に,要約に含まれるリスク要因が考慮すべき唯一の重大なリスクの指導意見とはならない.この10-K表の年次報告書に記載されているすべてのリスク要因は、私たちの会社を知るために重要であり、慎重に考慮すべきだと信じています。また、企業固有の重大なリスクの概要には、これらのリスクを十分に理解するために必要な適切な詳細さは含まれておらず、その後の対応するリスク要因は、当社の業務に関連するこれらの企業固有のリスクを十分に理解して理解するために必要な詳細および背景を提供する。
私たちの研究開発に関わるリスクは
•私たちの業務は私たちの主要候補製品niroacestatとmirdametinibの成功と、私たちが開発している他の候補製品に強く依存しています。もし私たちが候補製品の臨床開発を成功させ、監督部門の許可を得たり、それを商業化することができなければ、もし私たちがこの点で遅延に遭遇したら、私たちの業務は実質的な損害を受けるだろう。
•私たちは主要候補製品の早期開発に参加しておらず、私たちの候補製品と一緒に開発された第三者エージェントの開発にも参加していないので、私たちは私たちの候補製品のためのいくつかの臨床前および臨床試験データを正確に生成、収集、解釈、報告する第三者に依存している。
•もし著者らの臨床試験が著者ら或いは第三者が行った早期臨床前研究或いは臨床試験の陽性結果を複製できなければ、著者らは開発に成功し、監督部門の許可を得たり、著者らの候補製品を商業化することができないかもしれない。
•私たちが時々発表したり公表したりする臨床試験の一時的な“バックライン”と初歩的なデータは、より多くのデータの獲得に伴って変化する可能性があり、必ずしも完成した研究の最終結果あるいは他の進行中あるいは未来の研究の結果を予測するとは限らず、重大な変化を招く可能性のある監査と検証手続きの制約を受ける。
•我々はDEFI試験の二重盲検部分を完成させることに成功したが、これは登録段階3であり、世界的、無作為、二重盲検、プラセボ対照の臨床試験であり、その開放ラベル拡張部分はまだ行われているが、私たちが登録臨床試験を完成した経験は限られており、私たちは私たちが開発可能な他の候補製品のためにこのようにすることができないかもしれない。
•私たちは、他の療法と組み合わせて新しいノキシットおよびミダメチニブ、および将来可能な候補製品を開発することが予想され、併用製品の安全または供給問題は、そのような候補製品の開発および承認を延期または阻止する可能性がある。
•いずれの臨床試験で患者を募集する際に困難に遭遇すれば,われわれの臨床開発活動は延期されたり,他の悪影響を受けたりする可能性がある。
•硬線維腫の治療のためのナイルシタンおよびNF 1−PNの治療のためのミダメチニブの標的患者数は少なく、最終的には決定されておらず、治療可能な患者数の推定値が予想以下であれば、候補製品の販売潜在収入(承認されれば)が影響を受け、収益性を達成することも影響を受けるであろう。
私たちが第三者に依存するリスクは
•著者らは第三者による臨床前研究と臨床試験のいくつかの方面に依存している。これらの第三者がその契約義務を成功的に履行し、予想される期限までに規制要求を完了または遵守できない場合、私たちは規制部門の任意の潜在的候補製品の承認を得ることができないか、または商業化することができないかもしれない。
•私たちは第三者の製造と供給パートナーに依存しているため、私たちの臨床前および臨床開発材料の供給は限られたり中断されたりする可能性があり、あるいは数量や品質が満足できない可能性があり、これは私たちの開発や商業化努力を延期、阻止、または損害する可能性がある。
•ニゲシスタットの有効な薬物成分と完成薬の供給に関する商業製造と供給協定を締結したにもかかわらず、私たちはまだ商業生産に投入していない
私たちはまた私たちの他の候補製品について商業供給計画を達成していません。承認されれば、第三者に候補製品の商業ロットを生産し、加工することに依存すると予想されます。
•私たちは少数のサプライヤーに依存して私たちの候補製品を生産するためのいくつかの材料を提供し、1つの会社に依存して私たちのすべての候補製品のための活性医薬成分を生産する。
•私たちの既存と未来の協力は私たちの業務に非常に重要だ。既存の連携を維持できない場合や新たな連携に入ることができなければ,あるいはこれらの連携が成功しなければ,我々の業務は悪影響を受ける可能性がある.また,我々の協力者は,協調活動を実行する多くの点で広範な裁量権を持っており,彼らは我々が同意しない行動をとるかもしれない.
私たちの知的財産権に関するリスクは
•我々の主要候補製品は、第三者(ファイザーまたはファイザーを含む)から取得された知的財産権許可に依存し、これらの許可のいずれを終了しても重大な権利の損失をもたらす可能性があり、これは私たちの業務を損なうことになる。
•もし私たちが私たちと第三者の特許許可義務を履行できなかったら、私たちは私たちの業務に重要な許可権を失うかもしれない。
政府の規制に関連するリスク
•私たちはニトロシタンとミダメチニブの孤児薬物の称号を獲得し、他の候補製品のための孤児薬物の称号を求める可能性があるが、私たちは潜在的な市場排他性を含む、そのような称号を獲得したり維持したりすることができないかもしれないが、これは私たちの財務業績に否定的な影響を与えるかもしれない。
•私たちの主な候補製品の一部は中国で生産され、追加の生産能力はインドから来て、第三者メーカーを通じて。これらのメーカーの運営が深刻な妨害を受けた場合、貿易戦争や政治的動揺は、私たちの業務、財務状況、および運営結果に実質的な悪影響を及ぼす可能性がある。
私たちの業務や運営に関するリスクを管理する
•私たちは私たちの組織の規模を拡大する必要があり、私たちはこのような成長を管理する時に困難に直面するかもしれない。
•私たちは製品を商業的に販売した歴史がなく、私たちはまだ私たちの商業化運営を実施していない。私たちは多くの時間とお金を投入することでこのような能力を建設し、商業化に準備している。私たちが私たちの商業化能力を確立することに成功するという保証はない。
•私たちは現在、新しい候補製品を独立して発見するために必要な内部研究能力がありません。私たちは私たちの成長戦略を実行し、他の発見され、最初に開発された候補製品を識別し、許可したり、獲得したりすることで実現する予定です。私たちは私たちの成長戦略を成功的に実行できないかもしれないし、そのような成長戦略は予想された結果を生むことができないかもしれない。
•私たちの現在の業務は2つの場所に集中しており、私たちまたは私たちが依存している第三者は、気候変動に関連する可能性のある災害や他の予見できないまたは制御できない事件を含む自然災害の悪影響を受ける可能性があり、私たちの業務連続性および災害復旧計画は、深刻な災害から私たちを十分に保護できない可能性がある。
私たちの財務状況と追加資本需要に関連するリスク
•設立以来、私たちは重大な純損失が発生し、将来的に純損失が出ると予想されている。
•私たちの運営の歴史は限られていて、これは私たちの見通しと成功の可能性を評価するのを難しくするかもしれない。
•私たちは私たちの運営に資金を提供するための追加の資本が必要であり、必要な資本を得ることができなければ、私たちは候補製品の開発と商業化を達成できないだろう。
•追加資本の調達は、私たちの既存の株主を希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません。
私たちの普通株に関するリスクは
•私たちは私たちの普通株に配当金を支払うつもりはないので、どんな見返りも私たちの株の価値に限定されるだろう。
•私たちの主要株主と経営陣は私たちのかなりの割合の株を持っており、株主の承認が待たれる事項に大きな制御を加えることができるだろう。
•私たちの定款文書とデラウェア州法律における反買収条項は、制御権の変更を延期または阻止する可能性があり、これは私たちの普通株の市場価格を制限し、私たちの株主の現在の経営陣の変更を阻止または罷免する試みを阻止または挫折させる可能性がある。
•私たちの定款は、私たちと株主との間のいくつかの紛争の唯一かつ排他的な法廷として特定の裁判所を指定し、これは、私たちまたは私たちの役員、役員、または従業員との紛争において有利な司法フォーラムを獲得する私たちの株主の能力を制限するかもしれない。
会社に特化した重大なリスク要因
私たちの研究開発に関わるリスクは
私たちの業務は私たちの主要候補製品niroacestatとmirdametinibの成功と、私たちが開発している他の候補製品に強く依存しています。もし私たちが候補製品の臨床開発を成功させ、監督部門の許可を得たり、それを商業化することができなければ、もし私たちがこの点で遅延に遭遇したら、私たちの業務は実質的な損害を受けるだろう。
私たちの将来の成功と私たちの候補製品から収入を生み出す能力(数年以内には起こらないと予想される)は、私たちが開発に成功し、規制部門の承認を得て、1つ以上の候補製品を商業化する能力に依存する。2020年7月、われわれはナイルシタン3期臨床試験を完全に登録することを発表し、2019年10月にミダメチニブが可能な2 b期臨床試験を開始することを発表した。2022年5月,我々はニューノカル司の3期臨床試験の陽性結果を発表し,2022年9月にヨーロッパ医学腫瘍学会大会で3期試験のより多くのデータを公表した。2022年12月、米国食品医薬品局(FDA)に新薬申請(NDA)を提出した。FDAは2023年2月にNDA申請を受け,優先審査を付与し,処方薬ユーザ費用行動(PDUFA)を割り当て,目標行動日は2023年8月27日であった。もし私たちのすべての主要な候補製品が安全或いは治療効果の問題、開発遅延或いは監督管理問題或いはその他の問題に遭遇した場合、持続的な新冠肺炎疫病を含む場合、私たちの発展計画と業務は深刻な損害を受ける。
私たちの他のすべての候補製品は初期開発段階にあり、1つ以上の司法管轄区域の臨床前開発、臨床開発、監督審査と承認に大量の追加投資が必要である。
規制当局が候補製品を承認したり、商業化したりすることを遅延させたり阻害したりする問題に遭遇した場合、候補製品を開発し続ける財力がない場合や、既存の候補製品を修正したり、新たな協力を行う財力がないかもしれません
•私たちはFDAや似たような外国の規制機関に私たちの候補製品が安全で効果的であることを証明することができない
•私たちはビジネスプロセスと製品供給計画を確立する能力を持っています
•私たちの財政と他の資源が不足していて、必要な臨床前研究と臨床試験を完成できません
•我々の前臨床研究、臨床試験、または我々と類似した他の候補製品の臨床試験の陰性または不確定な結果は、追加の臨床前研究または臨床試験または計画放棄の決定または要求をもたらす
•私たちの臨床試験における対象または私たちの候補製品と同様の薬物または治療用生物学的製剤を使用した個人的経験の製品に関連する有害事象;
•新薬研究出願またはINDまたは同様の外国出願の提出を遅延させるか、または臨床試験を開始するために規制機関の必要な承認を得ることができなかったか、または臨床試験の開始後に臨床試験を一時停止または終了すること;
•FDA、欧州医薬品局、EMAなどの外国の監督管理機関が私たちの臨床試験の範囲または設計について適用した条件
•私たちの候補製品は臨床試験中に効果がありませんでした
•対照群の表現は期待よりも良く、例えばプラセボ群であり、これは臨床試験の陰性または不確定な結果を招く可能性がある
•被験者の臨床試験への参加を延期しました
•臨床試験の被験者の中退率は高い
•臨床試験に必要な候補製品または他の材料の供給または品質不足
•予想以上の臨床試験や製造コスト
•FDA、EMA或いは類似の監督機関の臨床試験場の検査と審査に不利である
•私たちの第三者請負業者または調査者は、監督要求をタイムリーにまたは根本的に遵守しなかったか、またはその契約義務を履行しなかった
•一般的な臨床試験または特に私たちの治療法のための追加的な規制監視を含む、規制要件、政策、およびガイドラインの遅延および変更;または
•FDA、EMA、そして似たような外国の規制機関のデータの異なる解釈。
私たちは主要候補製品の早期開発に参加しておらず、私たちの候補製品と一緒に開発された第三者エージェントの開発にも参加していないので、私たちは私たちの候補製品のためのいくつかの臨床前および臨床試験データを正確に生成、収集、解釈、報告する第三者に依存している。
私たちは、私たちの主要な候補製品、または私たちの候補製品と組み合わせて開発されている第三者エージェントの初期臨床前および臨床開発に参加または制御していません。著者らは第三者が適用されたプロトコルと法律、法規と科学標準に基づいて研究と開発を行うことに依存し、このような候補製品に関連するすべての臨床前研究と臨床試験の結果を正確に報告し、これらの試験からのデータを正確に収集し、解釈する。もしこれらの活動が不正確、不正確、あるいは不正確であれば、私たちの候補製品の臨床開発、監督管理許可或いは商業化は不利な影響を受ける。
もし著者らの臨床試験が著者ら或いは第三者が行った早期臨床前研究或いは臨床試験の陽性結果を複製できなければ、著者らは開発に成功し、監督部門の許可を得たり、著者らの候補製品を商業化することができないかもしれない。
候補製品に対する臨床前研究あるいは早期臨床試験は,われわれが行っても第三者が行っても,われわれが行った後続の臨床試験の結果を予測できるとは限らない。同様に,候補製品の計画臨床試験を完成させることができても,これらの臨床試験の積極的な結果は,われわれのその後の臨床前研究や臨床試験で複製されない可能性がある。
製薬やバイオテクノロジー業界の多くの会社が早期開発に積極的な成果をあげた後,後期臨床試験で大きな挫折を経験し,類似した挫折に直面しないことは確認できない。その他を除いて,これらの挫折は,以前に報告されていない有害事象を含む臨床試験進行期間中の臨床前発見や臨床前研究や臨床試験で行われた安全性や有効性観察によるものである。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、依然としてFDA、EMA或いは類似の外国の監督管理機関の許可を得られなかった。さらに、FDA、EMA、または同様の外国規制機関の承認政策または法規に大きな変化が生じる可能性があり、私たちの臨床データが承認を得るのに十分ではなく、FDA、EMAまたは同様の外国規制機関が私たちの候補製品の承認を延期、制限、または拒否する可能性がある。
私たちが時々発表したり公表したりする臨床試験の一時的な“バックライン”と初歩的なデータは、より多くのデータの獲得に伴って変化する可能性があり、必ずしも完成した研究の最終結果あるいは他の進行中あるいは未来の研究の結果を予測するとは限らず、重大な変化を招く可能性のある監査と検証手続きの制約を受ける。
私たちは時々私たちの臨床試験の一時的なTOPLINE或いは初歩的なデータ、例えばReNeu試験における成年患者の中期データの更新、2021年2月と2021年6月に発表された私たちのミダミチニ2 b期の臨床試験、DEFI試験二重盲検部分のTOPLINE陽性結果、およびDEFI試験の追加データを公開するかもしれない。一時更新は,その時点で入手可能なデータの初歩的な分析に基づいており,特定の研究や実験に関するデータをより網羅的に審査した後,データおよび関連する結論や結論が変化する可能性がある.私たちはまた、私たちのデータ分析の一部として、すべてのデータを全面的かつ詳細に評価する機会がないか、または受け取る機会がないかもしれないという仮説、推定、計算、および結論を出した。したがって、私たちが報告した任意のバックライン結果は、同じ研究の将来の結果と異なる可能性があり、またはより多くのデータを受信して十分な評価を行うと、異なる結論または考慮要因がこれらの結果を合格させる可能性がある。例えば,われわれのReNeu試験の一時的なデータは,試験に参加した最初の成人患者の結果を反映しているが,すべての患者のこの試験の最終データは報告されておらず,これらの結果は成人におけるデータと大きく異なる可能性がある。一時的なデータや初歩的なデータはまだ監査とチェック手続きを守らなければならず、これは最終データが以前に公表された予備データと大きく異なる可能性がある。したがって、最終データが利用可能になる前に、中間バックラインまたは予備データを慎重に見なければならない。また、, 私たちはすべての端末の中間分析ではなく、いくつかの端末の中間分析のみを報告することができる。著者らが完成する可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、1つ以上の臨床結果が実質的に変化する可能性があるというリスクに直面している。したがって,中期データは同一研究の最終結果を予測できない可能性があり,進行中や将来研究の結果を予測することもできない.初期または中期データと最終データとの違いは、私たちの業務の将来性を深刻に損なう可能性があり、私たちの普通株の取引価格を大幅に変動させる可能性があります。
さらに、規制機関を含む他の人は、私たちの仮定、推定、計算、結論または分析を受け入れないか、またはデータの重要性を異なる方法で解釈またはトレードオフする可能性があり、これは、特定の計画の価値、特定の候補製品または製品の承認または商業化、およびわが社の全体的な状況に影響を与える可能性がある。さらに、開示された特定の研究または臨床試験に関する情報は、一般に、より広い利用可能な情報から選択される。あなたまたは他の人は、私たちが開示すべき重要な情報または他の適切な情報を決定することに同意しないかもしれませんが、私たちが開示しないことを決定したいかなる情報も、最終的には、特定の製品、候補製品、または私たちの業務に関する未来の決定、結論、観点、活動、または他の側面に対して大きな意味を持つと考えられるかもしれません。もし私たちが報告した一時的または初歩的なデータが後期、最終的または実際の結果と異なるなら、あるいは規制機関を含む他の人が結論を出すことに同意しなければ、私たちは得ることができる
研究中の候補製品または私たちの他の任意の候補製品の承認および商業化は損害を受ける可能性があり、これは私たちの業務、財務状況、運営結果、および将来性を損なう可能性がある。
DEFI試験の二重盲検部分の完成に成功したにもかかわらず、その開放ラベル拡張部分はまだ行われているが、私たちが登録臨床試験を完成した経験は限られており、私たちが開発可能な他の候補製品のためにそうすることはできないかもしれない。
著者らは登録臨床試験を成功に完成して、FDA、EMA或いは類似の外国監督管理機関の許可を得て、任意の候補製品を販売することができる。後期登録臨床試験を含む臨床試験を展開することは、複雑な過程である。我々は2022年5月にヨーロッパ医学腫瘍学会大会でDEFI二重盲検部分からの陽性背線試験データと2022年9月にヨーロッパ医学腫瘍学大会で行ったDEFI試験の追加データを報告したが、これらのデータは2022年12月に提出したniroacestatに対するNDA申請を支持し、この申請は2023年2月にFDAに受け入れられ優先審査を受け、割り当てられたPDUFA目標作用日は2023年8月27日であるが、組織として登録臨床試験を完成する経験は限られている。私たちは引き続き私たちの臨床開発と規制能力を建設し、拡大する必要があり、私たちは合格した人員を募集し、訓練することができないかもしれない。また,第三者に依存した臨床試験を継続する予定である。これらの第三者がその契約義務を成功的に履行し、予想される期限までに規制要求を完了または遵守できない場合、私たちは規制部門の任意の潜在的候補製品の承認を得ることができないか、または商業化することができないかもしれない。したがって,必要な臨床試験を成功的かつ効率的に実行·完了することができず,NDAの提出と候補製品の承認につながる可能性がある。私たちは私たちの競争相手よりも多くの時間とより多くのコストを必要とするかもしれないし、規制部門が私たちが開発した任意の候補製品の承認を得ることに成功できないかもしれない。私たちが計画した臨床試験を開始または完了または延期することができず、候補製品の商業化を阻止または延期する可能性がある。
私たちは、他の療法と組み合わせて新しいノキシットおよびミダメチニブ、および将来可能な候補製品を開発することが予想され、併用製品の安全または供給問題は、そのような候補製品の開発および承認を延期または阻止する可能性がある。
私たちは、ナイル西およびミダメチニブを開発し、癌または他の疾患の治療のための1つまたは複数の他の承認または承認されていない合理的な療法と組み合わせて、他の将来の候補製品を開発することが可能である。例えば,我々は現在,このような療法を開発した業界トップとの協力により,ミダミニとリフェラフィニ,百済神州株式会社のあるいは百済神州の,RAFダイマー阻害剤,ナイルシットといくつかのBCMA指導療法との併用治療を評価している。
承認されていない癌療法が最終的に単独または私たちの製品と組み合わせてマーケティング承認を得ることができない場合、私たちはナイルシタン、ミダデチニブ、または私たちが開発した承認されていない合理的な療法と組み合わせて癌を治療する任意の候補製品をマーケティングして販売することができないだろう。また,われわれが現在開発·臨床試験している候補製品については,未承認癌療法は,重篤な副作用の可能性,臨床試験遅延,FDAの承認不足など,他の場所と同様のリスクに直面している。
他の既存療法と組み合わせて使用するために開発された任意の候補製品が市販承認または商業化されていても、FDA、EMAまたは米国以外の同様の外国規制機関が、我々の製品と組み合わせて使用される治療法の承認を取り消す可能性があるか、またはこれらの既存療法のいずれかが安全性、有効性、製造、または供給の問題が生じる可能性があるというリスクに直面するであろう。もし私たちが候補製品と組み合わせて使用する療法が任意の候補製品のために選択された適応の看護基準に置き換えられれば、FDA、EMAまたは同様の外国の規制機関は追加の臨床試験を要求するかもしれない。このようなリスクの発生は、私たち自身の製品を招く可能性があり、承認されれば、市場から撤退されたり、商業的にあまり成功しないだろう。
FDA、EMA、または同様の外国の規制機関がこれらの他の薬剤を承認しない場合、または私たちが開発した候補製品に関連して評価する薬剤を選択した場合、安全性、有効性、品質、製造または供給の問題が生じた場合、私たちはそのような併用療法の承認や販売を得ることができないかもしれない。
いずれの臨床試験で患者を募集する際に困難に遭遇すれば,われわれの臨床開発活動は延期されたり,他の悪影響を受けたりする可能性がある。
臨床試験方案に基づいて適時に臨床試験を完成し、他の事項以外に、試験が終了するまで十分な数の患者を募集する能力があるかどうかに依存する。様々な理由から、臨床試験で患者を募集する際に困難に遭遇する可能性があります
•協定に規定されている患者資格と排除基準
•臨床試験の主な終点に必要な患者数を分析した
•私たちの研究プロジェクトや臨床サプライチェーンは新冠肺炎の疫病に関連する要素によって遅延している
•患者と臨床試験場所の距離
•臨床試験の設計
•適切な能力と経験を持つ臨床試験研究者の能力と、これらの研究者が適切な患者を識別して募集する能力を募集します
•私たちの候補製品の安全状況を感じています
•患者の同意を得て維持する能力は
•臨床試験に参加した患者が試験完了前に試験を終了するリスク。
例えば,硬線維腫治療のためのネオノキセチンとNF 1−PNの治療に用いられるミダメチニブが開発されており,いずれもまれな疾患であり,患者数は少ない。したがって,DEFIとReNeu試験の登録が完了しているにもかかわらず,これらの候補製品のための被験者を募集する他の臨床試験では困難である可能性があり,一部の原因はこれらの患者数が少ないためである。また,我々の臨床試験は,我々の試験に参加することを選択する可能性のある患者のうちの1つによる臨床試験への参加を選択する可能性があるため,我々の候補製品と同じ治療分野での製品を他の臨床試験と争うことになる。合格した臨床研究者の数は限られているため、著者らはいくつかの競争相手が使用している同じ臨床試験地点で著者らのいくつかの臨床試験を行い、このような臨床試験地点で臨床試験を行うことができる患者数を減少させることが予想される。また,ミダミニーの場合,医師や患者の不良耐性を感知するため,登録困難に直面する可能性がある。
患者登録の遅延はコスト増加を招く可能性があり,あるいはわれわれの臨床試験の時間や結果に影響を及ぼす可能性があり,これらの試験の完了を阻止し,候補製品開発を進める能力に悪影響を及ぼす可能性がある。
硬線維腫の治療のためのナイルシタンおよびNF 1−PNの治療のためのミダメチニブの標的患者数は少なく、最終的には決定されておらず、治療可能な患者数の推定値が予想以下であれば、候補製品の販売潜在収入(承認されれば)が影響を受け、収益性を達成することも影響を受けるであろう。
私たちの目標疾患を有する患者数およびこれらの疾患患者サブセットの推定(承認されれば)は、私たちの信念および推定に基づいており、これらの推定は正しくないことが証明されるかもしれない。これらの推定は各種の源から来ており、科学文献、著者らの目標疾患患者を治療する医師の意見、患者基金会と二級市場研究データベースを含む。さらに、新しい研究は、これらの疾患の推定発症率または流行率を変化させる可能性があり、私たちが得られる可能性のある任意の候補製品の規制承認は、使用または禁忌症を制限し、アドレス指定可能な患者数を減少させることを含む可能性がある。したがって、対象患者数が予想を下回る可能性があり、この場合、候補製品が承認されれば、潜在的な販売収入は予想を下回ることになる。
私たちが第三者に依存するリスクは
著者らは第三者による臨床前研究と臨床試験のいくつかの方面に依存している。これらの第三者がその契約義務を成功的に履行し、予想される期限までに規制要求を完了または遵守できない場合、私たちは規制部門の任意の潜在的候補製品の承認を得ることができないか、または商業化することができないかもしれない。
われわれは第三者による臨床前研究のいくつかの面に依存し,独立研究者を含む第三者に依存して大学,医療機関,契約研究組織やCRO,戦略パートナーなどとの合意による臨床試験を行っている。これらの第三者と予算や契約を交渉することが予想され、これにより、私たちの開発スケジュールが延期され、コストが増加する可能性があります。
私たちは2017年8月に運営を開始し、インフラ建設を継続し、運営計画を実行するために必要な人員を募集しています。われわれの臨床試験では,われわれは特に第三者に強く依存しているため,臨床研究者のコントロールが限られている可能性があり,彼らの日常活動への可視性も限られており,承認された臨床計画を遵守する場合も含めて限られている。しかし、私たちは私たちのすべての臨床試験が適用された方案、法律と法規の要求、そして科学的な基準に従って行われ、私たちの第三者への依存が私たちの規制責任を免除しないことを確実にする責任がある。我々およびこれらの第三者は、FDAおよび同様の外国規制機関が臨床開発における候補製品に対して実行する法規およびガイドラインである良好な臨床実践またはGCP要件を遵守しなければならない。監督管理機関は定期的に臨床試験スポンサー、臨床研究者と臨床試験地点を検査することによって、これらのGCP要求を実行する。もし私たちまたはこれらの第三者のいずれかが適用されたGCP要件を遵守できなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられる可能性があり、FDAまたは同様の外国の規制機関は、これらの要求を一時停止または終了することを要求するかもしれない
マーケティング申請を承認する前に、臨床試験を行ったり、追加の臨床前研究や臨床試験を行ったりすることができます。検査後、これらの規制機関が私たちの臨床試験がGCP要求に適合しているかどうかを確認することはできません。また,われわれの臨床試験は現在の良好な生産規範やcGMP要求に応じて生産された製品を用いて行わなければならず,大量の患者が必要となる可能性がある。
私たちの失敗やこれらの第三者がこれらの規定を遵守できなかった場合、私たちは臨床試験を繰り返す必要があるかもしれません。これは規制承認過程を延期します。さらに、これらの第三者のいずれかが連邦または州詐欺および乱用または虚偽クレーム法律法規または医療プライバシー·セキュリティ法に違反した場合、私たちの業務は悪影響を受ける可能性がある。
私たちの臨床前研究または臨床試験を実行する任意の第三者は私たちの従業員ではなく、また、私たちとこのような第三者との合意によって私たちに提供される可能性のある救済措置を除いて、彼らが私たちの臨床前研究と臨床計画に十分な時間と資源を投入しているかどうかを制御することができない。これらの第三者はまた、私たちの競争相手を含む他の商業実体と関係があるかもしれません。彼らはまた、これらの実体のための臨床試験や他の製品開発活動を行っているかもしれません。これは、彼らが私たちを代表する表現に影響を与えるかもしれません。これらの第三者がその契約の義務または義務を成功裏に履行できなかった場合、または予想される期限内に完了した場合、彼らが交換する必要がある場合、または彼らが得た臨床前または臨床データの品質または正確性が、私たちの方案または法規の要求または他の理由を遵守できなかった場合、臨床開発スケジュールを含む我々の開発スケジュールは、延長、延期または終了される可能性があり、私たちは候補製品の開発を完了し、規制部門の承認を得たり、商業化に成功したりすることができないかもしれない。したがって、私たちの財務業績と候補製品のビジネス見通しは損なわれ、私たちのコストは増加する可能性があり、私たちの収入を創出する能力は延期されたり完全に排除されたりする可能性がある。
もし私たちがこれらの第三者契約研究機関またはCROまたは他の機関との任意の関係が終了した場合、私たちはCROまたは他の第三者と合意することができないか、または商業的に合理的な条項でそうすることができないかもしれない。
追加のCROを交換または増加させることは、管理職の時間と労力を必要とする追加的なコストに関連する。しかも、新しいCROが仕事を始める時、自然な過渡期がある。そのため,遅延が生じる可能性があり,期待される開発スケジュールを満たす能力に大きな影響を与える可能性がある.持続的な新冠肺炎の大流行と政府の対応措置も著者らのCROに重大な影響を与え、著者らは新冠肺炎及び新たに出現した変異株の灰色再発、最近新冠肺炎がもっと伝播しやすい変異株の著者らの業務のある地区での加速伝播、ワクチン接種率の停滞及び関連要素により、著者らのCROは更なる中断に直面する可能性があり、これらの要素は著者らの臨床前研究と臨床試験を開始と完成する能力に影響する可能性がある。CRO、調査者、その他の第三者との関係を慎重に処理しているにもかかわらず、将来的に挑戦や遅延に遭遇しない保証はなく、これらの遅延や挑戦が私たちの業務、財務状況、および見通しに実質的な悪影響を与えないことを保証することはできない。
私たちは第三者の製造と供給パートナーに依存しているため、私たちの臨床前および臨床開発材料の供給は限られたり中断されたりする可能性があり、あるいは数量や品質が満足できない可能性があり、これは私たちの開発や商業化努力を延期、阻止、または損害する可能性がある。
私たちは第三者契約メーカーに依存して私たちのすべての臨床前と臨床試験製品の供給を生産します。私たちは製品供給の製造施設を何も生産していない。著者らの臨床前と臨床開発製品の供給が制限されないこと、中断され、品質が満足できる或いは受け入れ可能な価格で供給を継続することを保証することはできない。特に、私たちのメーカーのどの交換にも大量の努力と専門知識が必要であり、合格した交換数は限られている可能性があるからです。
候補製品の製造過程はFDA,EMA,類似の外国規制機関の審査を受けなければならない。サプライヤーと製造業者は、適用された製造要求を満たし、cGMPのような規制基準に適合するために、監督機関の要求された厳格な施設とプロセス検証テストを受けなければならない。もし私たちのどの製造業者もこのような要求を遵守できなかった場合、または品質、時間、または他の側面での私たちの義務を履行できなかった場合、または私たちの部品や他の材料の供給が他の理由で限られたり中断されたりした場合、私たちは自分で材料を製造することを余儀なくされるかもしれませんが、私たちは現在能力や資源を持っていない、あるいは他の第三者と合意している場合、私たちは合理的な条項でそうすることができないかもしれません。いずれの場合も,代替供給源の確立に伴い,われわれの臨床試験供給は著しく遅延する可能性がある。場合によっては、私たちの候補製品を製造するために必要な技術的スキルまたは技術は、元の製造業者固有または独自である可能性があり、困難に遭遇する可能性があり、またはそのようなスキルまたは技術を他方に譲渡することを禁止する契約制限が存在する可能性があり、実行可能な代替案が存在しない可能性がある。これらの要素は、私たちのこれらの製造業者への依存を増加させ、あるいは他の第三者が私たちの候補製品を生産するために、これらのメーカーからライセンスを取得することを要求するだろう。もし私たちが何らかの理由でメーカーの交換を要求された場合、新しいメーカーの施設とプログラムが品質基準とすべての適用された法規とガイドラインに適合しているかどうかを確認するように要求されます。新メーカー検証に関する遅延は我々の開発能力に悪影響を及ぼす可能性がある
候補製品はタイムリーにまたは予算の範囲内で提供される。また、メーカーは、当該メーカーが独立して所有する我々の候補製品の製造に関する技術を持つことができる。これは私たちのこれらのメーカーへの依存を増加させ、あるいは他のメーカーが私たちの候補製品を生産するために、これらのメーカーからライセンスを取得することを要求するだろう。また,メーカーの変化は通常,製造プロセスやプロセスの変化に関連しており,従来の臨床供給と任意の新しいメーカーの供給との間での移行研究が求められている可能性がある。臨床用品の比較可能性の証明には成功しない可能性があり,追加の臨床試験が必要かもしれない。
私たちまたは第三者が私たちの製造要求を実行し、cGMPを遵守できなかった場合、様々な態様で私たちのビジネスに悪影響を及ぼす可能性があります
•開発中の候補製品の臨床試験を開始または継続できない
•規制申請の提出を遅延させるか、規制部門の候補製品の承認を受ける
•既存または未来の協力者の協力を失うこと
•第三者製造施設に規制機関の追加検査を受けさせる
•私たちの候補製品ロットの流通を停止したり、リコールしたりすることを要求する;
•候補製品の市場化と商業化が承認された場合、私たちの製品のビジネスニーズを満たすことはできません。
また、適切な専門知識、施設、規模を持つ包装サプライヤーと契約を締結し、私たちのニーズを満たしています。CGMPを維持できなかったことは,請負業者がFDAの制裁を受ける可能性があり,我々の運営能力に影響を与えたり,臨床開発計画の遅延を招いたりする可能性がある。私たちの現在の包装請負業者はcGMPに従って運営されていると信じているが、FDA、EMA、または同様の外国の規制機関がコンプライアンスに欠ける結論を出さない保証はない。さらに、パッケージサービス契約の任意の遅延、または契約製造業者が必要に応じてサービスを履行できなかった場合、臨床試験、登録、および発表が延期される可能性があり、これは私たちの業務に負の影響を与える可能性がある。現在の新冠肺炎の大流行は著者らが臨床前と臨床試験製品の供給を獲得する能力にどの程度の影響があり、ウィルス伝播の重症度と持続時間(及び新しく出現した変異株、最近新冠肺炎がもっと伝播しやすい変異株は著者らが運営する地区で伝播を加速し、及びワクチン接種率が停滞している)及び新冠肺炎を制御或いはその影響を治療するための行動に依存し、そして遅延を招く可能性がある。もし私たちの現在の第三者契約製造業者が合意通りに履行できなければ、私たちはこれらのメーカーの交換を要求されるかもしれません。私たちは彼らをタイムリーにあるいは交換できないかもしれません。私たちの現在と未来の他人が私たちの候補製品や製品を生産することへの依存は、私たちの将来の利益率と、マーケティングの承認をタイムリーかつ競争力的に獲得する製品の商業化能力に悪影響を及ぼす可能性があると予想されています。
私たちの候補製品と私たちが開発する可能性のあるどんな薬物も他の候補製品や薬物と生産施設を競争する可能性があります。私たちはcGMP規制によって運営され、私たちが生産しているメーカーと同様のビジネス計画を達成することができる他の保証はありません。私たちの既存または未来のメーカーのどんな業績失敗も、臨床開発やマーケティング承認を延期する可能性がある。
ナイルキャストの活性医薬成分や完成薬製品の供給に関する商業製造·供給協定が締結されているにもかかわらず、ビジネス規模の生産は行われておらず、他の候補製品についても商業供給スケジュールが達成されておらず、承認されれば、候補製品の商業ロットの生産と加工に依存することが予想される。*
もし私たちの候補製品が規制部門の承認を受けたら、私たちは引き続き第三者メーカーに依存すると予想される。私たちの候補製品については、私たちは限られた製造と供給協定だけだ。ナイルキャストの活性薬物成分と完成品に商業供給を提供する協定があるが、他の候補製品への供給スケジュールは非商業的、開発段階の製造と供給に限られている。したがって、私たちはまだこのような他の候補製品に対する長期供給計画を持っていない。私たちが第三者と私たちの候補製品の商業供給について未来の製造手配を達成する限り、承認されれば、私たちはこれらの第三者に依存して、品質管理と保証に関する要求を含む、契約と法規の要求に符合する方法でその義務を適時に履行する。
私たちの既存または未来の第三者メーカーのいかなる不振も、ニロシタンに関連する問題を含む臨床開発、市場承認、または商業供給を延期する可能性がある。私たちの現在のサプライヤーまたは未来の第三者製造業者が約束通りに合意を履行できない場合、またはこれらの契約メーカーが私たちとの合意を終了することを選択した場合、私たちはこれらのメーカーの交換を要求されるだろう。私たちは、任意のそのような代替製造業者または任意のそのような代替製造業者と合意することを決定し、同定する際に、追加のコスト、遅延、および困難を生じる可能性がある。我々はまた、例えば比較可能な研究を製造することによって、任意の新しい供給者が、以前にFDA、EMAまたは他の同様の規制機関に提出された仕様に基づいて、私たちの候補製品または製品を生産するかどうかを検証する必要がある。はい
また、サプライヤーの変化は通常、製造プロセスとプロセスの変化に関連しており、これは、臨床試験で使用されていた以前の臨床供給と任意の新しいサプライヤーの供給との間の移行研究が要求される可能性がある。新規サプライヤーまたは新製造プロセスの比較可能性の検証に関連する遅延は、候補製品をタイムリーにまたは予算内に開発したり、製品を商業化する能力に悪影響を及ぼす可能性があります。
私たちの契約メーカーが私たちの候補製品を生産するための施設はまた、FDA、EMAなどの外国規制機関の承認を経なければなりません。検査は、私たちがFDA、EMAまたは同様の外国規制機関に申請した後に行われます。私たちは私たちの契約製造パートナーの製造過程を直接制御せず、私たちの候補製品の製造に完全に依存してcGMP要求に適合するだろう。もし、私たちの契約製造業者が、私たちの規格およびFDA、EMA、または外国規制機関のような厳格な規制要件に適合した材料を生産することに成功しなければ、彼らはその製造施設の規制承認を確保および/または維持することができないだろう。しかも、私たちの契約製造業者が十分な品質管理、品質保証、合格者の能力を維持することを直接制御することはできません。FDA、EMA、または同様の外国規制機関が、これらの施設が私たちの候補製品を生産するために承認されていない場合、または将来的に承認を撤回すれば、代替製造施設を探す必要があるかもしれません。これは、承認されれば、規制機関の承認を得たり、私たちの候補製品をマーケティングする能力に深刻な影響を与えます。
私たちは少数のサプライヤーに依存して私たちの候補製品を生産するためのいくつかの材料を提供し、1つの会社に依存して私たちのすべての候補製品のための活性医薬成分を生産する。
私たちは現在少数のサプライヤーに依存して私たちの候補製品に使用するいくつかの材料と開発に必要な技術を提供しています。私たちは、これらのサプライヤーやサービスプロバイダが経営を継続すること、または私たちの需要を満たすのに十分な能力や供給を持っていることを保証することができず、私たちの競争相手や他の私たちと協力し続けることに関心のある会社によって購入されないことを確実にすることはできない。私たちは少量のサプライヤーを使用して、供給中断、価格上昇、または納品遅延を含むいくつかのリスクに直面させます。一般的に、代替材料の代替供給源は比較的少ない。私たちの現在のサプライヤーは未来の臨床試験或いは商業販売に対する需要を満たすことができないか、或いは満足したくないかもしれない。適切な代替サプライヤー、材料と技術を探すには大量の時間が必要かもしれないし、監督管理要求に符合する代替サプライヤーを構築することは難しいかもしれない。供給の中断や遅延は、候補製品を開発し、最終的に商業化する能力を損なう可能性がある。
私たちの既存と未来の協力は私たちの業務に非常に重要だ。既存の連携を維持できない場合や新たな連携に入ることができなければ,あるいはこれらの連携が成功しなければ,我々の業務は悪影響を受ける可能性がある.また,我々の協力者は,協調活動を実行する多くの点で広範な裁量権を持っており,彼らは我々が同意しない行動をとるかもしれない.
私たちの戦略の重要な部分は評価であり、適切と考えられる場合には、現在のパートナー関係を延長したり、主要なバイオ製薬会社と構築可能なパートナーシップを含む将来的により多くのパートナー関係を構築したりする。私たちの製品開発能力は限られていて、現在私たちの臨床前研究開発と商業能力を建設しています。したがって、私たちは他の会社と協力して、私たちの候補製品をより全面的に開発するために重要な技術を提供してくれました。私たちはまた、他社と協力して、私たちの計画に重要な技術や資金を提供することができます。
私たちが現在または未来に拡張または加入する可能性のある任意の協力は、以下のリスクを含む多くのリスクをもたらす可能性がある
•協力者は彼らが適用する努力と資源を決定する上で大きな自由裁量を持っている
•協力者は期待通りに義務を履行していないかもしれない
•協力者は、規制部門の承認を得た任意の候補製品の開発および商業化を行ってはならず、臨床試験結果、協力者の戦略的重点または利用可能な資金の変化または外部要因(例えば、資源の移転や競争優先度を作成する可能性のある戦略取引)に基づいて、開発または商業化計画または許可スケジュールを継続または更新しないことを選択してはならない
•協力者は臨床試験を延期し、臨床試験計画に資金不足を提供し、臨床試験を停止或いは候補製品を放棄し、新しい臨床試験を繰り返し或いは行うことができ、或いは臨床試験候補製品の新しい調合を要求することができる
•協力者は、競争力のある製品がより成功する可能性があると考えているか、または私たちよりも経済的に魅力的な条項で商業化できることを前提として、第三者開発と直接または間接的に我々の製品および候補製品と競合する製品を独立して開発または間接的に開発することができる
•まだ一緒にテストされていない併用療法の協力は、緊急有害事象の治療は予見できない可能性があり、私たちの候補製品の単一療法開発に悪影響を及ぼす可能性がある
•私たちと協力して発見された候補製品は、私たちの協力者によって彼ら自身の候補製品や製品と競争されるかもしれません。これは、協力者が資源を投入して私たちの候補製品を商業化することを停止する可能性があります
•協力者は、候補製品または製品の開発、製造、流通、またはマーケティングに関する適用法規要件を遵守できない可能性がある
•私たちの1つまたは複数の候補製品にマーケティングおよび分配権を持ち、規制承認を得た協力者は、そのような製品または製品をマーケティングおよび流通するのに十分な資源を投入していない可能性がある
•特許権、契約解釈、または第一選択開発プロセスにおける相違を含む協力者との相違は、候補製品の研究、開発または商業化の遅延または終了をもたらす可能性があり、候補製品に対して追加的な責任を負うこと、または訴訟または仲裁を引き起こす可能性があり、いずれも時間的で高価である可能性がある
•協力者は、私たちの知的財産権を正確に維持したり、守ったりすることができないかもしれないし、何らかの方法で私たちの固有の情報を使用して、それによって訴訟を引き起こし、それによって、私たちの知的財産権または固有の情報を無効にしたり、私たちを潜在的な訴訟に直面させたりすることができる
•協力者は第三者の知的財産権を侵害する可能性があり、これは私たちを訴訟と潜在的な責任に直面させるかもしれない
•協力は協力者によって終了することができ、終了すれば、適用候補製品に対する許可権を失う可能性があり、または適用候補製品をさらに開発または商業化するために追加資金を調達する必要がある可能性がある。
百済神州との協力合意に基づき,ミダミニとリフェラファニーの組み合わせで1 b/2期臨床試験が行われている。また,業界をリードするBCMAガイド療法開発者との様々な協力合意に基づき,再発や難治性多発性骨髄腫患者にナイルキャストとこのような開発者ごとのBCMAガイド療法を併用して評価している。これらの既存の協力手配により,関連する臨床試験を完了した後,我々は我々のパートナーと誠実に交渉する機会があり,それぞれの臨床協力と構築可能なビジネス関係を拡大するために準備していく。しかし,我々のパートナーは,適用される臨床試験結果にかかわらず,共同製品の開発を継続する義務はない。BGB 3245の開発のために百済神州と共同でMapKure,LLCまたはMapKureを設立し,臨床開発や他の業務活動に貢献し,MapKureの取締役会や共同指導委員会に代表を持っているにもかかわらず,開発過程を制御することはできない。MapKureは私たちの予想とは違う開発計画を追求するかもしれないし、これは成功するかもしれないし、成功しないかもしれない。
もし私たちの協力が候補製品の発見、開発、商業化に成功しなかった場合、または私たちの協力者が未来の発展を追求するために協力協定を締結しないことを選択した場合、私たちはそのような協力の下で未来の資金、マイルストーン、または印税支払いを受けないかもしれない。本報告に記載されている製品開発、規制承認、商業化に関するリスクは、我々の協力者の活動にも適用可能である。
さらに、もし私たちの協力者が私たちとの合意を終了したら、私たちは新しい協力者を引き付けることがもっと難しいことを発見するかもしれませんし、ビジネスや金融界での私たちの見方は不利な影響を受けるかもしれません。
また、私たちは私たちの候補製品のために適切なパートナーを探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑である。私たちが私たちの候補製品との協力を成功させるためには、潜在的なパートナーは、私たちが求めている条項と他の会社が許可することができる製品に基づいて、私たちの候補製品が彼らが魅力的だと思う市場で経済的価値があると考えなければならない。また,最近では大手バイオ製薬会社間の大量の業務合併により将来の潜在的パートナー数が減少している。我々が協力について最終的な合意を達成できるかどうかは,他の事項に加えて,協力者の資源や専門知識の評価,協調の条項や条件,提案された協力者のいくつかの要因の評価に依存する.もし私たちが適時に、受け入れ可能な条項によって、または適切なパートナーと合意できない場合、私たちは候補製品の開発を削減し、その開発計画または私たちの1つまたは複数の他の開発計画を減らしたり、その潜在的な商業化を延期したり、販売やマーケティング活動や計画の範囲を縮小したり、私たちの支出を増加させ、自費で開発または商業化活動を行わなければならないかもしれない。もし私たちが私たちの支出を増やし、私たち自身の開発や商業化活動に資金を提供することを選択すれば、私たちはより多くの専門知識や資本を得る必要があるかもしれません。これらは私たちが受け入れられない条件であるか、あるいは全く得られないかもしれません。もし私たちが協力を達成できなかったり、必要な開発と商業化活動を展開するのに十分な資金や専門知識がなければ、私たちの候補製品をさらに開発することができないかもしれない, それらを市場に出して薬品販売から収入を得るか、あるいは私たちの技術を開発し続けることで、私たちの業務は実質的な悪影響を受ける可能性があります。我々が新たな戦略的パートナーシップの構築に成功したとしても、我々が合意した条項は、例えば、候補製品の開発や承認が延期または承認された製品販売に失望した場合、このような戦略的パートナーシップを維持できない可能性がある。もし何か遅延があれば
我々の候補製品との新たな戦略パートナー協定の締結は、私たちの候補製品の開発と商業化を延期し、市場に進出しても競争力を低下させる可能性がある。
私たちの知的財産権に関するリスクは
我々の主要候補製品はファイザーを含む第三者許可の知的財産権に依存しており、これらの許可のいずれを終了しても重大な権利の損失を招く可能性があり、これは私たちの業務を損なうことになる。
私たちは特許、ノウハウ、そして独自技術に依存しており、私たち自身のものもあれば、他の人が許可しているものもある。製品ライセンスのいかなる終了も重大な権利の喪失を招く可能性があり、候補製品を商業化する能力に実質的な悪影響をもたらすだろう。
ライセンス契約によると、私たちとライセンシーとの間で知的財産権紛争が発生する可能性もあります
•ライセンス契約に従って付与された権利範囲および解釈に関連する他の問題;
•私たちの技術とプロセスが、ライセンス契約に拘束されていないライセンス側の知的財産権をどの程度侵害しているかどうか
•私たちは協力開発関係の下で特許と他の権利を第三者に再許可する権利;
•私たちの候補製品の開発と商業化に関する使用許可技術の職務義務、およびどのような活動がこれらの職務義務を満たしているか
•私たちの許可者、私たちと私たちのパートナーが共同で知的財産権を創造または使用することによって生成された発明およびノウハウの所有権。
私たちが許可している知的財産権をめぐる紛争が許容可能な条項で現在の許可手配の能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた候補製品の開発に成功し、商業化することができない可能性がある。
私たちは通常、私たちが私たちのために許可してくれた知的財産権が直面しているリスクのように、私たちが持っている知的財産権を保護するのと同じすべてのリスクに直面している。もし私たちまたは私たちの許可者がこの知的財産権を十分に保護できなければ、私たちが製品を商業化する能力は深刻な影響を受ける可能性がある。
もし私たちが私たちと第三者の特許許可義務を履行できなかったら、私たちは私たちの業務に重要な許可権を失うかもしれない。
私たちはライセンス契約の側であり、これらの合意に基づいて、私たちは私たちの候補製品にキー特許の許可を提供します。2017年8月に運営を開始した際、私たちはファイザーと4つのライセンス契約を締結しましたが、そのうちの3つは依然として有効で、すべての主要な候補製品niroacestatとmirdametinibのライセンス契約を含み、この2つの合意は2019年に修正され、再説明されました。また,2021年には,ルモール大学やフランダースバイオテクノロジー研究所とTEAD阻害剤計画のライセンスを署名し,Dana−Farber癌研究所と上皮増殖因子受容体小分子阻害剤を組み合わせたライセンスを署名した。私たちの既存のすべての許可証は私たちに様々な勤勉さ、マイルストーン支払い、特許権使用料、保険、その他の義務を課している。もし私たちがこれらの義務を履行しなければ、私たちの許可者は許可を終了する権利があるかもしれません。この場合、私たちはこのような許可知的財産権に含まれる製品を開発またはマーケティングすることができません。我々は2020年10月にJazz PharmPharmticalsアイルランド株式会社またはJazzに我々FAAH阻害剤計画をカバーするファイザー許可協定を譲渡したが、Jazzがこのような許可の条項を遵守することは保証されず、これはその終了を招く可能性があり、Jazzのこのような販売に関連する潜在的な実質的な違反を補うために資産を取り戻すことができない。
私たちは、これらの許可内の権利、活動、または私たちの許可内の知的財産権に関連する任意の他の知的財産権の維持および起訴に限られた支配権を持っている可能性があります。例えば、私たちは、これらのライセンシーのこのような活動が適用された法律および法規に準拠しているか、または効果的かつ強制的に実行可能な特許および他の知的財産権が生成されるだろうと判断することはできない。私たちは、ライセンシーが知的財産権の第三者侵害者に対して侵害訴訟を提起したり、私たちに許可されたいくつかの知的財産権を弁護する方法に限られた支配権を持っています。ライセンシーの権利侵害訴訟や弁護活動は私たち自身が行ったように活発ではないかもしれない。
政府の規制に関連するリスク
私たちはニトロシタンとミダメチニブの孤児薬物の称号を獲得し、他の候補製品のための孤児薬物の称号を求める可能性があるが、私たちは潜在的な市場排他性を含む、そのような称号を獲得したり維持したりすることができないかもしれないが、これは私たちの財務業績に否定的な影響を与えるかもしれない。
アメリカとヨーロッパを含むいくつかの司法管轄区の監督機関は、比較的少ない患者集団の薬物と治療用生物製品を孤児薬物として指定する可能性がある。孤児医薬品法によれば、薬物または治療生物製剤がまれな疾患または疾患を治療するための薬物または治療生物製剤である場合、FDAは、通常、米国では年間患者数が20万人未満であるか、または米国での患者数が20万人を超えると定義されており、米国では薬剤または治療生物薬の開発コストが米国の販売から回収されることが合理的に予想できない。米国では、孤児薬物指定は、臨床試験費用、税金優遇、およびユーザ費用減免のための贈与資金を提供する機会などの財政的インセンティブを得る権利がある。しかしながら、場合によっては、FDAは、例えば、FDAが出願人の指定要求が孤児医薬品法およびその実施条例によって要求される重要な情報を見落としていることを発見した場合、このような指定を取り消す可能性がある。孤児薬物指定を有する製品がその後、そのような指定された疾患に対するFDAの最初の承認を得た場合、この製品は、孤児薬物排他性を得る権利があり、これは、FDAが完全なNDAまたは生物製品ライセンス申請またはBLAを含む他の申請を承認しない可能性があり、限られた場合、例えば、孤児薬物に対して排他的な製品に対する臨床的利点を示すか、または製造業者が十分な製品数を保証できない限り、7年以内に同じ適応で同じ製品を販売することを意味する。
2018年6月、FDAは硬線維腫を治療するナイルキャストの孤児薬物名を承認し、2019年9月、欧州委員会は軟組織肉腫を治療するナイルキャスト彼の孤児薬物名を承認した。FDAは2018年10月、NF 1を治療するミダミニーの孤児薬物名を承認し、2019年7月、欧州委員会はNF 1を治療するミダミニー孤児薬名を承認した。私たちは他の適応や私たちの他の候補製品のためにニロシタンとミダメチニブの孤児薬の名前を求めるかもしれない。私たちがそのような称号を得ることができるという保証はない。
将来のいずれかの特定の適応候補製品の孤児薬物指定を獲得しても,医薬品開発に関連する不確実性により,市販承認された孤児指定適応のためのナイルキャスト,ミダメチニブ,あるいは他の任意の他のこのような候補品ではない可能性がある。さらに、孤児によって指定された適応よりも広い適応の承認を求める場合、またはFDAが後に指定要求に重大な欠陥があると判断した場合、または製造業者がまれな疾患または疾患を有する患者の需要を満たすのに十分な数の製品を保証できない場合、米国での独占営業権が制限される可能性がある。
さらに、私たちが米国で製品の孤児薬物排他性を獲得しても、この排他性は、異なる活性部分を有する異なる薬物または治療用生物製品が同じ疾患のために許可されることができるので、競合から製品を効果的に保護することができない可能性がある。孤児製品が承認された後であっても、FDAが、後の薬剤または治療生物がより安全で、より効率的で、または患者ケアに重大な貢献があると結論した場合、FDAは、その後、同じ活性部分を有する同じ薬剤または同じ疾患の治療のための生物学的製剤を承認することができる。ヨーロッパでは、私たちが求めている同じ適応によって類似した医薬製品が孤児薬物の称号を獲得すれば、私たちの製品の販売を阻止されるかもしれない。承認されると、少数の例外を除いて、EU加盟国、欧州医薬品局、または欧州委員会の主管当局は、同じ治療適応を有する他の同様の医薬製品の申請を受け入れることができないか、またはマーケティング許可を与えることができる。同じ孤児の適応を持つ類似薬品が元の孤児薬品より安全で、もっと有効で、あるいは臨床上もっと良いなら、その発売を許可することもできる。
物質組成物をカバーする米国特許の法定期限は2025年であり、多結晶ナイルタンを含む米国特許は2039年に満了し、現在臨床開発されている形態の特許は2039年に満了し、医薬組成物を含む米国特許は2042年に満了し、いずれの場合も特許期限調整や規制延長を含まず、外国の同業者が待っている。現在臨床開発されている形態、多形体の治療方法、および医薬組成物を含む4つの米国特許が2041年に満了する多形体ミダミニーをカバーする4つの米国特許は、いずれの特許も規制延長を含まず、外国の同業者が待っている。特許有効期間が予想されているにもかかわらず、孤児薬の独占性がこれらの製品を競争から保護できない場合、私たちの業務および財務状況は実質的な悪影響を受ける可能性がある。孤児薬物指定は薬物或いは治療生物の開発時間或いは監督審査時間を短縮することもなく、監督審査或いは承認過程において薬物或いは治療生物にいかなる利点をもたらすこともない。しかも、私たちは未来の候補製品のために孤児薬物の称号を求めるかもしれないが、私たちは決してそのような称号を得ないかもしれない。
私たちの主な候補製品の一部は中国で生産され、追加の生産能力はインドから来て、第三者メーカーを通じて。これらのメーカーの運営が深刻な妨害を受けた場合、貿易戦争や政治的動揺は、私たちの業務、財務状況、および運営結果に実質的な悪影響を及ぼす可能性がある。
私たちは現在、製造業務を第三者にアウトソーシングしています。私たちの主要候補製品の臨床数は、これらのアメリカ以外の第三者によって生産されており、中国を含め、追加の生産能力はインドから来ています。私たちはこのような第三者製造業者を候補製品として引き続き使用する予定だ。自然災害でも他の理由でも、これらの国/地域の製造業者のどのような生産が中断されても、私たちの需要を満たすのに十分な数の製品を生産することができなくても、私たちの日常的な業務を運営し、私たちの候補製品を開発し続ける能力を損なう可能性があります。また、これらのメーカーのいくつかが中国に位置しているため、米国や中国政府の政策が変化し、政治的動揺や中国の経済状況が不安定であれば、製品供給中断やコスト増加の影響を受ける可能性がある。例えば、貿易戦争は、私たちが使用している中国製の化学中間体に関税をかける可能性がある。これらの事項のいずれも私たちの業務と経営結果に実質的な悪影響を及ぼす可能性がある。臨床試験で使用される我々の候補製品の任意の生産ロットまたは同様の行動の任意のリコールは、試験を延期するか、または試験データの完全性を損なう可能性があり、および将来の規制申告文書におけるその潜在的な使用を損なう可能性がある。さらに、これらのメーカーのいずれかの生産中断または規制要件を遵守できないことは、潜在的製品の臨床開発を著しく遅らせる可能性があり、提案試験に対する第三者または臨床研究者の興味および支持を低下させる可能性がある。このような中断や故障はまた私たちの候補製品の商業化を阻害し、私たちの競争地位を損なう可能性がある。さらに進む, 私たちは中国とインドの現地通貨価値の変動の影響を受けるかもしれない。未来の現地通貨の上昇は私たちのコストを増加させるかもしれない。また、これらの国では熟練労働力に対する需要が増加し、熟練労働力の獲得性が低下し、賃金率が上昇しているため、我々の労働力コストは上昇し続ける可能性がある。
私たちの業務や運営に関するリスクを管理する
私たちは私たちの組織の規模を拡大する必要があり、私たちはこのような成長を管理する時に困難に直面するかもしれない。
2022年12月31日現在、私たちは227人のフルタイム従業員がいる。私たちの臨床開発と商業化計画と戦略の発展に伴い、私たちはより多くの管理、臨床、製造、医療、監督、販売、マーケティング、財務、法律、その他の人員が必要になると予想される。今後の成長は、経営陣のメンバーにより多くの重大な責任を負わせるだろう
•より多くの従業員を募集し、統合し、維持し、激励する
•私たちの候補製品の臨床、製造、および品質審査プロセスを含む、私たちの開発作業を効果的に管理し、同時に、請負業者、パートナー、および他の第三者に対する契約義務を遵守し、
•私たちの業務、財務、管理制御、報告システム、そして手続きを改善する。
私たちの将来の財務業績と候補製品を商業化する能力(承認されれば)は、将来のどんな成長の能力を効果的に管理するかにある程度依存し、私たちの経営陣はまた、これらの成長活動を管理するために、不比例な注意を日常活動から移しなければならないかもしれない。
現在、予測可能な未来には、独立した組織、コンサルタント、コンサルタントを含む第三者に大きく依存して、私たちの業務を支援し、実行するためのいくつかのサービスを提供するだろう。私たちはこのような第三者のサービスが必要な時に適時に提供されることを保証することができず、私たちが合格した代替者を見つけることができるという保証もない。さらに、私たちのアウトソーシング活動を効率的に管理することができない場合、または提供されるサービスの品質、正確性、または数が何らかの理由で影響を受ける場合、私たちの臨床試験は延期または終了される可能性があり、私たちは規制当局の承認を得たり、私たちの候補製品を承認したり、他の方法で私たちの業務を推進する上で大きな遅延が生じる可能性があります。既存のコンサルタント会社を経済的に合理的な条件で管理したり、他の適切な外間引受業者やコンサルタント会社を見つけることができる保証はありません。
新入社員の雇用やコンサルタントや請負業者チームを拡大することで効率的に組織を拡大することができなければ,我々の候補製品をさらに開発·商業化するために必要な任務を成功させることができない可能性があり,我々の開発や商業化目標を達成できない可能性がある.
私たちは製品を商業的に販売した歴史がなく、私たちはまだ私たちの商業化運営を実施していない。私たちは多くの時間とお金を投入することでこのような能力を建設し、商業化に準備している。私たちが私たちの商業化能力を確立することに成功するという保証はない。
私たちは現在、単独でも他の会社と組み合わせても、私たちの候補製品を販売できるように、私たちの商業化能力を建設しています。商業化能力を確立するには大量の時間とお金の投入が必要であり、大量の管理重点と資源を移転する可能性がある。また、より大きなバイオ製薬やバイオテクノロジー会社と競争し、これらの会社は成熟した商業化·マーケティング能力を有しており、適切な人員の募集を求めている。したがって、私たちが商業化能力を確立する努力が必ず成功するという保証はない。
私たちは現在、新しい候補製品を独立して発見するために必要な内部研究能力がありません。私たちは私たちの成長戦略を実行し、他の発見され、最初に開発された候補製品を識別し、許可したり、獲得したりすることで実現する予定です。私たちは私たちの成長戦略を成功的に実行できないかもしれないし、そのような成長戦略は予想された結果を生むことができないかもしれない。
著者らは現在内部発見と臨床前研究開発能力を確立しているが、著者らが独立した発見と初歩的な新製品候補製品の開発に成功する能力を保証することはできない。我々はまた、我々の既存の候補製品と相補的である可能性のある候補製品を含む、内部許可によって、または他の会社、学術機関、または他の資産発信者から新しい候補製品を買収する予定である。もし私たちが候補製品を識別、許可、買収、統合できなければ、私たちが成長戦略を実施する能力は制限されるだろう。
新製品候補製品の研究計画と業務開発を確定するには大量の技術、財政と人力資源が必要であるが、著者らの現在の内部薬物発見と臨床前研究と開発能力は限られている。ライセンス内や候補製品の取得や開発計画は通常,多額の金や費用を支払う必要があり,貴重な資源を消費する可能性がある。私たちの既存の候補製品の開発と商業化に加えて、許可や取得された技術や製品候補を開発し、商業化するために、多くの時間と人員を投入する必要があります。私たちの業務開発努力や買収は、臨床開発と商業化のためにより多くの補充または成功の候補製品を生み出すことができないかもしれない
•私たちの識別や業務開発方法や検索基準およびプロセスは、開発進行中に成功する確率の高い潜在的な候補製品を識別することができない可能性がある
•私たちは、識別および許可またはより多くの候補製品を取得するために、十分なリソースまたは専門知識を集めることができないか、または集めたくないかもしれない
•私たちが許可や買収を求めている候補製品については、これらの候補製品の許可者または所有者と受け入れ可能な条項を達成できないかもしれない
•私たちが許可したり獲得した候補品は臨床前研究や臨床試験では成功しないかもしれません
•私たちはこのような許可されていないまたは獲得された候補製品を作成または開発することに成功しないかもしれない
•これらの許可されていない、または許可された候補製品は、有害な副作用を有することが証明される可能性があり、または製品が規制部門の承認を得ることができないか、または承認された後に販売できないように、他の特徴を有する可能性がある
•競争相手は代替製品を開発し、これらの許可された候補製品を時代遅れや魅力を低下させる可能性がある
•許可されていないまたは取得された候補製品は、第三者特許または私たちがアクセスできない可能性のある他の独占的権利によって保護される可能性がある
•私たちが開発した未許可または獲得された候補製品は、現在予想されているように私たちの専門知識と私たちの開発と商業インフラを最善に利用することができないかもしれません
•私たちが候補製品を開発する過程で、私たちが許可または獲得した候補製品の市場が変化する可能性があるため、その候補製品は不合理になり、開発を続けることができなくなる可能性がある
•私たちが許可または獲得した候補製品は、許容可能なコストで商業的に量産できないかもしれないし、生産できないかもしれない
•私たちが許可または獲得した候補製品は、患者、医学界、または第三者支払人によって安全かつ効果的に受け入れられないかもしれない。
もしこのような事件のいずれかが発生すれば、私たちは私たちの成長戦略を成功的に実行できないかもしれないし、私たちの成長戦略は予想された結果を生むことができないかもしれない。
私たちの現在の業務は2つの場所に集中しており、私たちまたは私たちが依存している第三者は、気候変動に関連する可能性のある災害や他の予見できないまたは制御できない事件を含む自然災害の悪影響を受ける可能性があり、私たちの業務連続性および災害復旧計画は、深刻な災害から私たちを十分に保護できない可能性がある。
私たちの現在の本部はコネチカット州のスタンフォードにあります。私たちの開発業務は現在ノースカロライナ州のダラムにあります。私たちは現在私たちの製造業務を第三者にアウトソーシングしています。私たちの臨床ロット候補製品はカナダ、中国、フランス、インドを含むアメリカ以外のこれらの第三者によって生産されています。どんなものでも
洪水、火災、爆発、地震、極端な天気条件、医療流行病、電力不足、電気通信故障、または他の自然または人為的事故または事件のような意外な事件は、私たちの施設や私たちの第三者契約メーカーの製造施設を十分に利用できなくなり、私たちの業務運営能力に重大かつ不利な影響を与える可能性があり、特に日常生活において、私たちの財務と運営状況に重大なマイナス影響を与える可能性がある。
これらの施設を使用できないとコスト増加、候補製品の開発遅延、私たちの業務運営が中断する可能性があります。地震やその他の自然災害は、私たちの運営をさらに混乱させ、私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性がある。自然災害、停電、または他の事件が発生した場合、本社または私たちの開発業務の全部または大部分を使用することができなくなり、第三者契約メーカーの製造施設のような重要なインフラを破損したり、他の方法で運営を中断したりすることは困難かもしれませんし、場合によっては、かなり長い間私たちの業務を継続することはできません。深刻な災害または同様のイベントが発生した場合、災害復旧およびトラフィック連続性計画は十分ではないことが証明される可能性がある。私たちの災害復旧と業務連続計画の限られた性質のため、私たちは大量の費用を発生する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性があります。我々のリスク管理方法の一部として,保険カバー範囲を我々の業務に適していると考えられるレベルに維持している。しかし、これらの施設で事故や事件が発生した場合、保険金額がどんな損害や損失を補うのに十分な保証はできません。もし私たちの工場や私たちの第三者契約メーカーの製造施設が事故や事件あるいは他のいかなる理由でも作動できなければ、短い時間であっても、私たちのいかなる研究開発プロジェクトも損害を受ける可能性があります。どの業務中断も、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
私たちの財務状況と追加資本需要に関連するリスク
設立以来、私たちは重大な純損失が発生し、将来的に純損失が出ると予想されている。
私たちが設立されて以来、私たちはすべての報告期間に重大な純損失を発生させた。今まで、私たちは主に株式融資を通じて私たちの運営に資金を提供してきた。私たちのすべての収入と繰延収入は、Jazz資産購入および許可協定、およびグラクソ·スミスクラインとの非独占的許可および協力協定に従って受信された払い戻し不可能な前払いから来ています。私たちは商業販売や経常的な収入源のために承認された製品は何もない。もし私たちの候補製品が開発と承認に成功しなかったら、私たちは決してこれらの製品から何の収入も得られないかもしれない。私たちは私たちの持続的な運営に関連した巨額の研究開発と他の費用を発生させ続けている。したがって、私たちは利益を上げておらず、設立以来毎年赤字が発生している。2022年12月31日、2021年12月31日、2020年12月31日までの会計年度の純損失はそれぞれ2.774億ドル、1.739億ドル、4560万ドルだった。2022年12月31日と2021年12月31日までの累計赤字はそれぞれ5兆699億ドルと2兆925億ドルだった。私たちは予測可能な未来に引き続き大きな損失を受けることが予想され、私たちが引き続き私たちの候補製品を研究·開発し、規制部門の承認を求め、その商業化に備えて、私たちの主要候補製品ナイルキャストとミダメチニブ、そして任意の未来の候補製品を含むこれらの損失は増加すると予想される。
私たちは次の場合、私たちの費用が大幅に増加すると予想している
•登録臨床試験と他の適応に使用可能な臨床試験を含む後期臨床試験を通じて、著者らの主要な候補製品ナイルシターとミダメチニブの開発を推進した
•臨床開発を通じて我々の他の候補製品の開発計画を進め,後期臨床開発に入る
•臨床試験に成功した候補製品のために市場承認を求める
•投資は、さらに臨床前および臨床開発の他の技術または製品候補に使用することができるかもしれない
•臨床、品質管理、科学、医療、業務発展、財務人員を含め、より多くの人員を招聘し、私たちのインフラを建設し続ける
•私たちの臨床開発、製造、商業化の努力、上場企業としての私たちの運営を支援する人員を含む、私たちの運営、財務、管理システムを拡大し、人員を増やす
•私たちの知的財産権の組み合わせを維持し、拡大し、保護する
•販売、マーケティング、流通インフラを構築し、マーケティングの承認を得ることができ、単独または第三者と連携して商業化しようとしている任意の製品を商業化する。
利益を維持し、利益を維持するためには、私たちまたは任意の潜在的な未来のパートナーは、巨大な市場潜在力を持つ製品を開発し、最終的に商業化しなければならない。これは私たちが一連の挑戦的な活動の中で成功することを要求します。臨床前研究と臨床試験を完成させ、候補製品の発売許可を得て、製造、精算許可を得て、マーケティングと販売はマーケティングの許可を得ることができて、任意の後を満たすことができます
市場需要。私たちは決してこのような活動やすべての活動で成功しないかもしれないし、たとえ私たちが成功しても、私たちは利益を達成するために十分な大きさまたは十分な収入を生むことができないかもしれない。もし私たちが確実に利益を達成したら、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱める可能性がある。わが社の価値の低下は株主損失の全部または一部の投資を招く可能性もあります。新冠肺炎の疫病或いはその他の要素による市場変動もまた私たちが必要な時に資本を獲得する能力に不利な影響を与える可能性がある。
私たちが私たちの1つ以上の候補製品を商業化することに成功しても、私たちはより多くの候補製品を開発、登録、マーケティングするために、大量の研究開発と他の支出を生成し続ける。私たちは予測できない費用、困難、合併症、遅延、および私たちの業務に悪影響を及ぼす可能性のある他の未知の要素に直面するかもしれない。私たちの未来の純損失の規模は私たちの未来の支出の成長率と私たちが収入を作る能力にある程度依存するだろう。私たちの以前の損失と予想された未来の損失はすでに私たちの株主権益と運営資本に悪影響を与え続けるだろう。
私たちの運営の歴史は限られていて、これは私たちの見通しと成功の可能性を評価するのを難しくするかもしれない。
私たちは臨床段階の生物製薬会社で、運営の歴史は限られている。私たちは2017年8月に設立され、これまで私たちの運営は、私たちの候補製品の準備と臨床試験の実行、私たちのインフラの建設、資金調達、パートナー関係の実行に集中してきました。そのため、私たちが評価するための業務の運営は限られており、より長い運営歴史や開発成功と商業化医薬製品の歴史があれば、私たちの将来の成功や生存能力の予測はそれほど正確ではないかもしれない。生物製薬製品開発への投資は非常に高い投機性があり、それは大量の前期資本支出を必要とし、重大なリスクが存在するため、即ちいかなる潜在的な候補製品は十分な活動或いは受け入れ可能な安全状況を証明できず、監督管理の承認を得ることができず、市場参入と精算を確保することができず、商業上実行可能ではない。
著者らは2022年5月にノロカスタットの登録3期臨床試験-DEFI試験のTOPLINE結果を発表し、2022年9月にヨーロッパ医学腫瘍学会大会でDEFI試験からのより多くのデータを提出したが、これらのデータは2022年12月に提出されたNDA申請を支持し、この申請は2023年2月にFDAによって受け入れられ、優先審査を受け、分配されたPDUFA目標行動日は2023年8月27日であるが、監督部門のいかなる候補製品の承認に成功したかは証明されておらず、私たちはまだ製品の商業化販売が許可されておらず、私たちはこれまで製品販売から何の収入も発生していない。また、経営歴史が限られている企業として、早期生物製薬会社が急速に発展している分野でよく遭遇する予見不可能な費用、困難、合併症、遅延および他の既知および未知の要素およびリスク、または他の既知または未知の要素およびリスクに遭遇する可能性があり、これらの要素およびリスクは稀またはユニークである可能性がある。
また、開発に専念している会社からビジネス活動を支援する能力のある会社に移行するための商業化能力を建設しており、このような移行では成功しない可能性がある。
私たちは私たちの運営に資金を提供するための追加の資本が必要であり、必要な資本を得ることができなければ、私たちは候補製品の開発と商業化を達成できないだろう。
設立以来、私たちの業務は大量の現金を消費した。私たちは引き続き大量の現金を使って、私たちの候補製品のさらなる研究開発と臨床試験を行い、規制機関の私たちの候補製品の承認を求め、規制の承認を得た任意の製品を発売し、商業化する予定だ。2022年12月31日現在、私たちは5.97億ドルの現金、現金等価物、有価証券を持っている。私たちの現在の運営計画によると、私たちは私たちの現金、現金等価物、有価証券が2026年までの運営費用と資本支出要求を支払うのに十分だと信じている。しかし、私たちの将来の資本需要と私たちの既存資源が私たちの運営を支持する期限は私たちが予想していたのとは大きく異なる可能性があり、私たちはどうしても臨床開発を完成させ、規制機関の候補製品の承認を得るための追加の資本が必要になるだろう。私たちの毎月の支出水準は新しいものと進行中の開発や会社活動によって異なります。我々の候補製品開発に関連する時間や活動の長さは非常に不確定であるため、開発および承認されたマーケティングおよび商業化活動のためにどれだけの実際の資金が必要かを見積もることはできない。
私たちの未来の資金需要は多くの要素に依存するだろうが、これらに限定されない
•私たちの候補製品の臨床試験の開始、進行、時間、コストと結果;持続的な新冠肺炎の大流行、ロシアとウクライナの衝突、または他の原因による臨床試験の遅延による可能性のある任意の予測不可能なコストを含む
•私たちはこれらの候補製品の臨床と臨床前開発と製造計画を策定しました
•私たちが開発または許可した候補製品の数量と特徴
•臨床前活動または臨床活動のコストを含む潜在的候補製品を決定し、評価するためのコスト;
•私たちは締結された任意の協力または許可協定の条項を選択することができる
•FDA、EMA、および他の類似外国規制機関が制定した規制要求の結果、時間、コストを満たす
•私たちの特許主張と他の知的財産権の提出、起訴、弁護、そして実行のコスト
•知的財産権紛争の弁護コストは、第三者が私たちまたは私たちの候補製品に対して提起した特許侵害訴訟を含む
•競争の技術と市場発展の影響
•ビジネス規模のアウトソーシング製造活動を完了するコストと時間
•私たちが単独でまたは第三者と協力して私たちの製品を商業化することを選択した地域では、規制の承認を得る可能性のある任意の候補製品のための販売、マーケティング、および流通能力を確立する
•候補製品の開発と監督管理審査活動に成功した後に得られた商業成功程度。
もし私たちが受け入れられる条件で十分な追加資本を調達できない場合、私たちは私たちの1つまたは複数の候補製品または1つまたは複数の他の研究開発計画の開発または商業化を大幅に延期、削減または停止しなければならないかもしれない。上記のどの事件も、私たちの業務、将来性、財務状況と経営結果を深刻に損害し、私たちの普通株価格の下落を招く可能性があります。
追加資本の調達は、私たちの既存の株主を希釈し、私たちの運営を制限したり、私たちの技術や候補製品に対する権利を放棄することを要求するかもしれません。
私たちの開発努力のために、私たちは外部資金源や他の支援を約束していないし、私たちは受け入れ可能な条件で追加的な資金を提供するかどうか、あるいは全くできないかどうかを決定することもできない。私たちが十分な製品または特許使用料収入を生成して私たちの現金需要を満たすことができる前に、私たちは公開または私募株式発行、債務融資、協力、戦略連合、許可手配、および他のマーケティングまたは流通手配を通じて、将来の現金需要に資金を提供する予定です。公開または私募株式発行によってより多くの資金を調達する場合、これらの証券の条項には、清算または他の株主権利に悪影響を及ぼす特典が含まれている可能性があります。さらに、普通株または転換可能または普通株に交換可能な証券を売却することによって追加資本を調達する場合、既存株主の所有権権益は希釈される可能性がある。さらに、いかなる債務融資も、追加債務を招く、資本支出を行う、または配当を発表するなど、私たちが具体的な行動を取る能力を制限または制限する固定支払義務と契約を負担させる可能性がある。もし私たちがマーケティングおよび流通手配、または第三者との他の協力、戦略連合、または許可手配によって追加の資本を調達する場合、私たちは、私たちの候補製品、技術、将来の収入フロー、または研究計画のいくつかの価値のある権利を放棄しなければならないか、または私たちに不利になる可能性のある条項で許可を付与しなければならないかもしれない。私たちはまた、私たちの主導的な製品または任意の未来の候補製品のために商業または開発パートナーを事前に探したり、候補製品や技術に対する私たちの権利を放棄したりすることを要求されるかもしれません。そうでなければ、私たちは自分自身の開発または商業化を求めます。
私たちの普通株に関するリスクは
私たちは私たちの普通株に配当金を支払うつもりはないので、どんな見返りも私たちの株の価値に限定されるだろう。
私たちは現在、事業の発展、運営、拡張のために未来の収益を保留し、予測可能な未来に現金配当金を発表または支払うことはないと予想している。さらに、将来の債務または他の融資計画には、私たちの普通株が発表または支払い可能な配当金の額を禁止または制限する条項が含まれている可能性がある。したがって、株主のどんな見返りもその株の増価に限定されるだろう。
私たちの主要株主と経営陣は私たちのかなりの割合の株を持っており、株主の承認が待たれる事項に大きな制御を加えることができるだろう。
2022年12月31日現在、私たちの役員、取締役及びその付属会社と5%を超える普通株保有者は、議決権付き株の62.8%を保有しています。したがって、このような株主たちはこのような所有権地位を通じて私たちに影響を与えることができるだろう。この株主たちは株主の承認を必要とするすべての事項を決定することができるかもしれない。例えば、これらの株主は、取締役選挙を制御し、私たちの組織文書を修正し、任意の合併、資産の売却、または他の重大な会社取引を承認することができるかもしれない。これは阻止されるかもしれない
あるいは我々普通株に対する能動的買収提案や要約を阻止したり,これらの提案や要約は株主が我々の株主の1つとしての利益に最も適していると考える可能性がある.
私たちの定款文書とデラウェア州法律における反買収条項は、制御権の変更を延期または阻止する可能性があり、これは私たちの普通株の市場価格を制限し、私たちの株主の現在の経営陣の変更を阻止または罷免する試みを阻止または挫折させる可能性がある。
当社の会社登録証明書又は会社登録証明書の改訂及び再記載、さらに改正された改訂及び再記載された定款又は定款には、わが社の支配権変更又は当社の株主が有利と考えられる取締役会変更を遅延又は阻止する可能性のある条項が含まれている。いくつかの規定には
•取締役会は3つのレベルに分かれており、任期が3年交錯しており、すべての取締役会メンバーが1回の選挙で生まれたわけではない
•株主の書面同意による行動を禁止することは、すべての株主の行動が私たちの株主会議で行われなければならないことを要求する
•株主特別会議は取締役会長、最高経営責任者、認可役員総数の過半数しか開催できないことが求められている
•株主指名と指名が取締役会に入る事前通知要求;
•法律で規定されている他の投票の理由であり、当時取締役選挙で投票する権利のある投票権のある株の3分の2以上の流通株が承認された場合でなければ、私たちの株主は、私たちの取締役会のメンバーを罷免しないことを要求する
•議決権のある株の3分の2以上の全流通株を承認し、株主行動で任意の定款または会社登録証明書の特定の条項を修正することを要求し、取締役会は株主の承認なしに取締役会が決定した条項に従って転換可能な優先株を発行する権利があり、転換可能な優先株は普通株式保有者の権利よりも高い権利を含む可能性がある。
また、私たちはデラウェア州で登録されているので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、株主が議決権を持って発行された株の15%以上のいくつかの業務統合を禁止する可能性があります。これらの反買収条項やわが社の登録証明書や定款の他の条項は、株主や潜在的な買収者が私たちの取締役会に対する支配権を得ることを困難にしたり、当時の取締役会が反対する行動を起こしたりして、わが社の合併、要約買収、または代理権競争に関連することを延期または阻害する可能性もあります。これらの規定はまた、代理権競争を阻止し、株主が彼らが選択した取締役を選出することを難しくしたり、彼らが取りたい他の会社の行動を取らせたりする可能性がある。制御権の変更、取引、取締役会の変動を遅延または阻止することは、私たちの普通株の市場価格の下落を招く可能性がある。
私たちの定款は、私たちと株主との間のいくつかの紛争の唯一かつ排他的な法廷として特定の裁判所を指定し、これは、私たちまたは私たちの役員、役員、または従業員との紛争において有利な司法フォーラムを獲得する私たちの株主の能力を制限するかもしれない。
私たちの規約は、私たちが書面で代替裁判所を選択することに同意しない限り、デラウェア州衡平裁判所または衡平裁判所は、州法律クレームの唯一および独占裁判所であり、これらの訴訟は、(I)私たちを代表して提起された任意の派生訴訟または訴訟に関し、(Ii)私たちの取締役、上級管理職または他の従業員の私たちまたは私たちの株主に対する受託責任に違反すると主張するいかなる訴訟も、(Iii)デラウェア州一般会社法、当社の会社登録証明書または私たちの定款の任意の規定に基づいてクレームを提起する任意の訴訟、(Iv)任意の解釈訴訟、当社の登録証明書又は定款の有効性を適用、強制又は決定し、又は(V)内部事務原則又はデラウェアフォーラム条項によって管轄されるクレームを主張する任意の訴訟。デラウェアフォーラム条項は、1933年の“証券法”(改正された)、“証券法”、“1934年証券取引法”(改正された)または“取引法”によって発生したいかなる訴訟原因にも適用されない。私たちの規約はさらに、私たちが書面で代替裁判所を選択することに同意しない限り、アメリカコネチカット州地域裁判所は、証券法または連邦フォーラム条項に基づいて提起された任意の訴えを解決する唯一の独占的なフォーラムとなるだろう。私たちの規約では、私たちの株式の権益を購入または他の方法で獲得した個人またはエンティティは、前述のデラウェアフォーラム条項および連邦フォーラム条項を通知し、同意したとみなされます。しかし、株主が連邦証券法およびその下の規則および法規の遵守を放棄するとはみなされません。
デラウェアフォーラム条項と連邦フォーラム条項は、株主が上記のクレームを提出する際に追加の訴訟費用を負担させる可能性があり、特に株主がデラウェア州またはコネチカット州またはその近くに住んでいなければならない。また、デラウェア州フォーラム条項と連邦フォーラム条項は、司法フォーラムにおいて、私たちまたは私たちの役員、幹部、または他の従業員と紛争が発生することに有利であると考えるクレームを株主が提出する能力を制限する可能性がある
これはこのような訴訟を阻止するかもしれない。また、デラウェア州最高裁判所は2020年3月に、デラウェア州法律によると、証券法に基づいて請求を要求する連邦裁判所選択条項は表面的に有効であると判断したが、他の裁判所が我々の連邦フォーラム条項を実行するかどうかには不確実性がある。連邦フォーラムの規定が訴訟で実行できないことが発見された場合、私たちはこのような訴訟の解決に関連した追加費用を発生させるかもしれない。連邦フォーラムの条項はまた、その条項が実行不可能または無効だと主張する株主に追加的な訴訟費用を適用するかもしれない。衡平裁判所または米国コネチカット州地域裁判所もまた、訴訟の株主が私たちの株主よりも有利または不利である可能性がある訴訟を考慮するか、または訴訟を提起する裁判所を選択する可能性があることを含む、他の裁判所とは異なる判決または結果を行う可能性がある。
一般リスク因子
研究開発やバイオ製薬業界に関わるリスク
臨床開発は長くて高価な過程に関連しており、結果は不確定である。私たちは私たちの候補製品の開発と商業化を完了したり、最終的に完成できなかったりする過程で追加のコストが発生したり、遅延が生じる可能性があります。
任意の候補製品を商業化するために必要な監督管理許可を得るために、著者らは広範な臨床前研究と臨床試験を通じて、この候補製品が人体に対して安全かつ有効であることを証明しなければならない。臨床試験費用は高価であり,完成まで数年かかる可能性があり,その結果自体も確定していない。著者らは適用された監督管理機関が臨床意義があると考える臨床終点を構築できない可能性があり、臨床試験はテストのどの段階でも失敗する可能性がある。
早期臨床試験と後期臨床試験の試験設計上の差異は早期臨床試験の結果を後期臨床試験に外挿することが困難である。そのほか、臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床試験で満足できると考えているが、しかし依然としてその製品の発売許可を得られなかった。また,患者や研究者が研究製品候補,あるいは既存の承認薬やプラセボを受け入れているかどうかを知っているオープンラベル試験を行って計画している。最も典型的な場合は,オープンラベル臨床試験は候補の研究製品のみをテストし,異なる用量レベルで試験を行う場合がある. 開放ラベル臨床試験は様々な制限を受けており,これらの制限はいかなる治療効果も誇張する可能性があり,これらの試験中の患者は治療を受ける際に知られているからである。オープンラベル臨床試験は“患者偏見”の影響を受ける可能性があり,すなわち患者が症状が改善したと考えているのは,実験的治療を受けていることを意識しているだけである。また,オープンラベル臨床試験は,“調査者偏見”の影響を受ける可能性があり,すなわち臨床試験結果を評価·審査する人は,どの患者が治療を受けているかを知り,その知識を知っている場合に治療群の情報をより有利に解釈することが可能である。ランダム、プラセボ対照の臨床試験が、登録された対象が治療群に交差することを可能にするように設計されている場合、交差する前に被験者が無意識に失明するリスクが存在する可能性があり、これは、これらのデータの臨床的意義を制限する可能性があり、追加の臨床試験を行う必要がある可能性がある。したがって,オープンタグ試験の結果は,我々の任意の候補製品の将来の臨床試験結果を予測できない可能性があり,制御された環境でプラセボや能動対照を用いて研究を行う場合には,オープンタグ臨床試験を含む。
臨床試験の成功は、NDAをFDAに提出すること、EMAにマーケティング許可申請またはMAAを提出すること、および同様の外国規制機関に各候補製品の類似マーケティング申請を提出することであり、それによって、最終的に任意の候補製品を承認し、商業マーケティングを行うための前提条件である。
われわれが行っている,あるいは計画中のいずれの臨床試験も,ナイルシスタットとミダメチニブを用いた併用療法の試験が,予定通りに完了するかどうか,あれば,あるいは場合によっては,そのような臨床試験が開始されるかどうかは分からない。
私たちは臨床試験の開始または完了と規制提出の準備に遅延があるかもしれない。私たちが行う可能性のある任意の未来の臨床試験の間、または結果として、私たちはまた、上場許可を得ることを延期または阻止するか、または私たちの現在の候補製品または任意の未来の候補製品を商業化する能力を遅延または阻止する可能性がある多くの予測不可能なイベントに遭遇する可能性がある
•私たちの臨床試験と臨床前計画は新冠肺炎の疫病に関連する要素によって遅延した
•ロシア-ウクライナ紛争に対応するために実施された制裁および他の措置、または紛争による世界的な商業中断は、収入およびサプライチェーンに影響を与える可能性がある
•規制機関、機関審査委員会または倫理委員会は、私たちまたは私たちの研究者が予想される試験場所で臨床試験を開始したり、臨床試験を行ったりすることを許可してはならない
•私たちは、予想される臨床試験場所および予想されるCROと受け入れ可能な条項の合意に遅延または合意できない可能性があり、これらの条項は広範な交渉を必要とする可能性があり、異なるCROと臨床試験地点の間に有意差がある可能性がある
•任意の候補製品の臨床試験は、許容可能な安全性または有効性を示すことができないか、または陰性または不確実な結果を生じる可能性があり、私たちは、決定または監督機関が、追加の臨床前研究または臨床試験を要求するか、または製品開発計画を放棄することを決定することができるかもしれない
•候補製品の臨床試験に必要な被験者の数は、私たちが予想していたよりも多かったかもしれません。これらの臨床試験の登録速度は、私たちが予想していたよりも遅いかもしれません。または被験者は、これらの臨床試験を脱退するか、または予想以上の速度で戻って治療後のフォローアップを行うことができないかもしれません
•私たちの第三者請負業者は、法規の要求を直ちに遵守することができないか、または彼らの契約義務を履行することができないか、または全く遵守しないか、または臨床試験案から逸脱したり、試験を脱退する可能性があり、新たな臨床試験場所または調査者を増加させる必要があるかもしれない
•私たちは、規制要件を遵守しないこと、または参加者が受け入れられない健康リスクにさらされていることを含む、様々な理由で臨床研究または試験を一時停止または終了することを、規制機関、IRBs、または道徳委員会が要求することができるか、または私たちの調査者が様々な理由で臨床研究または試験を一時停止または終了することを要求することができる
•候補製品の臨床試験コストは予想以上に高いかもしれません
•私たちの候補製品の供給または品質、または候補製品の臨床試験に必要な他の材料は、所与の臨床試験を開始または完了するのに十分ではないかもしれない
•私たちの候補製品は副作用または他の予期しない特徴がある可能性があり、私たちまたは私たちの調査者、監督機関、IRBs、または倫理委員会が臨床試験を一時停止または終了させる可能性がある
•他の療法の臨床試験報告は,我々の候補製品の安全性や有効性に対する懸念を引き起こす可能性がある
•FDA、EMA、または同様の規制機関は、長期毒理学研究のような追加のデータの提出を要求するか、または臨床試験を開始することを可能にする前に他の要求を適用するかもしれない。
臨床試験が我々、臨床試験を行っている機関のIRBs、FDA、EMAまたは同様の規制機関によって一時停止または終了された場合、またはデータ安全監視委員会またはデータ安全監視委員会によって一時停止または終了が提案された場合、遅延に遭遇する可能性もある。臨床試験の一時停止または中止は、監督管理の要求または著者らの臨床規程に従って臨床試験を行うことができなかった、FDA、EMAまたは類似の外国の監督機関が臨床試験操作または臨床試験場所を検査することによる臨床一時停止、予見できない安全問題または副作用の実施、製品または治療のメリットを証明できなかった、臨床意義のある試験終点、政府法規または行政行動の変化、または十分な資金の不足を含む、臨床試験の一時停止または終了は多種の要素による可能性がある。臨床試験の開始または完了遅延をもたらす多くの要因は、最終的には、私たちの候補製品が規制部門の承認を得ることを拒否される可能性もある。また、FDA、EMAまたは同様の外国の規制機関は、私たちの臨床試験設計と臨床試験データの解釈に同意しないかもしれないし、あるいは彼らが私たちの臨床試験設計を審査してコメントした後であっても、承認要求を変更する可能性がある。
もし私たちが臨床テストやマーケティング承認の面で遅延に遭遇したら、私たちのコストは増加するだろう。われわれのいずれの臨床試験が計画通りに開始されるかどうか,再分配が必要かどうか,予定通りに完成するかどうか,あるいは全く知られていない。重大な臨床試験遅延は、候補製品を商業化する独占的な権利を持つ可能性のある任意の期限を短縮することも可能であり、私たちの競争相手が私たちの前に製品を市場に出すことを可能にすることができ、これは候補製品を商業化することに成功する能力を弱化させ、私たちの業務と運営結果を損なう可能性がある。著者らの臨床開発計画のいかなる遅延も著者らの業務、財務状況と運営結果を深刻に損害する可能性がある。我々のパートナー支援の臨床試験は,我々の候補製品および我々パートナーの療法と組み合わせて,同様の開発リスクとなっている。
バイオ製薬の成功開発には大きな不確実性がある。
生物製薬の成功開発は高度に不確定であり、多くの要素に依存し、その中の多くの要素は私たちがコントロールできない。開発の初期段階で有望そうな候補製品は市場に進出できない可能性がある
•臨床試験結果は、候補製品の有効性が予想よりも低いこと(例えば、臨床試験がその主要または重要な副次的終点を満たすことができない可能性がある)または許容できない副作用または毒性を有することを示す可能性がある
•必要な規制承認や遅延がこのような承認を受けることができなかった。他の要素以外に、この遅延は患者が試験スクリーニング過程を通過できなかったこと、臨床試験登録が遅いこと、患者が試験から退出し、患者がフォローアップを失ったためかもしれない
•試験終点に達する時間の長さ、データ分析またはNDA準備の追加の時間要件、FDAとの議論、FDAは追加の臨床前または臨床データ(例えば、長期毒理学研究)または意外な安全または製造問題を必要とする
•臨床前の研究結果は候補製品が予想された有効あるいは有害な副作用に及ばないことを示す可能性がある
•私たちの候補製品を他の治療法とうまく組み合わせることができないことを含め、供給問題、製造コスト、処方の問題
•上場後の承認要求;
•他人とその競争製品と技術の専有権は、私たちの候補製品の商業化を阻止するかもしれない。
臨床試験の完成と監督機関の最終決定のための上場承認申請提出に要する時間長は候補製品によって国によって大きく異なり、予測が困難かもしれない。
私たちが市場承認を得ることに成功したとしても、承認された製品のビジネス成功は、第三者支払者の保証範囲と十分な補償に大きく依存し、第三者支払者は、MedicareやMedicaid計画、米国の管理医療組織や外国の特定の国の政府組織などの政府支払者を含み、これらの組織は、既存および将来の医療コストを低減するための医療改革措置の影響を受ける可能性がある。第三者支払者は、製品のコスト効果に関連する上場後の研究を含む他の研究を要求する可能性があり、精算を受ける資格があり、これはコストが高く、私たちの資源を移転する可能性がある。政府や他の医療支払者が承認されると,我々の製品に保険と十分な補償を提供しなければ,市場受容度や商業成功は低下するであろう。
また、任意の候補製品が上場承認された場合、安全及び他の上場後の情報及び報告及び登録の提出に重大な規制義務を受け、承認後に行われた任意の臨床試験のcGMP及びGCPを引き続き遵守(又は第三者提供者が遵守することを確保する)が必要となる。また、私たち、監督管理機関或いは第三者は常にこのようなリスクが存在し、即ち私たち、監督管理機関或いは第三者は製品審査後に以前未知の問題、例えば意外な深刻或いは頻度の高い不良事件を発見する可能性がある。これらの要求を守るコストが高く、私たちの候補製品が承認後に遵守できなかったり、他の問題が発生したりすれば、私たちの業務、財務状況、運営結果に悪影響を及ぼす可能性があります。
私たちの資源と追加資金を得るルートが限られているため、私たちは特定の計画と候補製品を優先的に開発しなければならない;これらの決定は間違っていることが証明され、私たちの業務に悪影響を及ぼす可能性がある。
いくつかの理由で、私たちは購入することで実行可能な臨床開発候補新製品を識別し、獲得することができないかもしれない。もし私たちがより多くの候補製品を識別して得ることができなければ、私たちの業務は実質的な損害を受けるかもしれない。
最終的に成功するかどうかにかかわらず、新しい候補製品と疾病目標を確定し、追求する努力は大量の技術、財政と人的資源を必要とする。私たちは現在と未来の協力者を含めて第三者に依存しており、私たちのすべての研究と臨床前活動を実行しています。方案は最初に臨床前研究で希望を示す可能性があるが、臨床開発過程で積極的な結果が得られなかった
•使用される方法は、潜在的な適応および/または候補製品の識別に成功しない可能性がある;または
•さらなる研究により、候補製品は有害な悪影響や他の特徴を有することが証明される可能性があり、それらが有効な製品である可能性が低いことを示した。
私たちの財力と人的資源が限られているため、私たちは最初に有限適応の計画と製品候補に重点を置くつもりだ。したがって、私たちは他の候補製品を求める機会を放棄または延期したり、私たちの既存の候補製品の他の兆候を求めたりすることができ、これらの製品は後でより大きな商業潜在力またはより大きな成功可能性を有することが証明される可能性がある。私たちは私たちの努力と資源を潜在的な候補製品または最終的に成功しないことが証明された他の潜在的な計画に集中するかもしれない。
私たちの未来の臨床試験或いは私たちの未来のパートナーの臨床試験は以前の臨床前研究或いは臨床試験で見たことのない重大な不良事件を掲示し、そして安全状況を招く可能性があり、監督管理部門の許可或いは市場が私たちの任意の候補製品を受け入れることを阻害するかもしれない。
いずれの臨床試験においても重大な有害事象や他の副作用が観察された場合,患者をわれわれの臨床試験に参加することは困難である可能性があり,患者はわれわれの試験を脱退するか,あるいは試験やわれわれの開発を放棄することが要求される可能性がある
1つまたは複数の候補製品の共同努力。例えば,先のミダミニーの2期臨床試験は終了し,1/2期臨床試験の2期部分の登録は,1日2回15 mg以上の用量のミダミニー使用間欠および連続投与計画の有害事象が観察されたため一時停止された。これらの不良事件は眼疾患(視力障害、視力不明瞭と網膜静脈閉塞)、神経系疾患(意識障害、思考遅延、話し方曖昧と幻覚)、筋肉骨格と結合組織疾患(全身無力と頚部筋肉無力、そしてクレアチンホスホキナーゼ軽度と中度の上昇を伴う)と心臓疾患(左心室駆出率の低下とうっ血性心不全)を含む。これらの用量は,我々が行っているNF 1−PNにおけるミダミニーの2 b期臨床試験で許容される最大用量4 mgよりも有意に高いにもかかわらず,この試験で患者を24カ月治療する予定であり,これまでの試験ではどの被験者がミダミニー治療を受けていたよりも長くなるであろう。われわれが行っている2 b期臨床試験では,潜在的な治療時間の延長やその他の要因により,先の臨床試験で高用量のミダメチニブに類似した有害事象が観察される可能性がある。また,この試験の登録対象にはNF 1−PN小児科患者も含まれている。ミダミニーの16歳以下児での安全性データは限られており,この患者群では思わぬ有害事象が観察される可能性がある。
もし私たちが私たちが開発した任意の候補製品の臨床試験を延期、一時停止、または終了することを選択または要求された場合、これらの候補製品の商業的将来性は損なわれ、これらの候補製品から製品収入を創出する能力は延期またはキャンセルされる。臨床試験で観察された深刻な不良事件或いはその他の不良事件、及び耐性問題は、市場が論争のある候補製品の受け入れを阻害或いは阻止する可能性がある。
我々、FDA、EMA、または同様の外国の規制機関またはIRBは、そのような試験の対象が許容できない健康リスクまたは副作用に直面していると考えることを含む、様々な理由で候補製品の臨床試験を随時一時停止することができる。生物技術業界で開発されたいくつかの潜在療法は最初に早期試験で治療の将来性を示したが、その後副作用が発生することが発見され、それらの更なる発展を阻害した。副作用が候補製品の発売承認または保持を妨げなくても、承認に制限を加えることができ、または承認された製品が枠警告を受ける可能性があり、これは安全問題に関するFDAの最も顕著な警告であり、承認製品の耐性が他の療法よりも高いため、不良副作用は市場の承認を受ける製品を阻害する可能性がある。
私たちの候補製品に対する同情的な使用の需要を増加させることは私たちの名声にマイナスの影響を与え、私たちの業務を損なうかもしれません。
我々は,現在限られているか,または利用可能な治療選択がない適応を治療するための候補製品を開発している。個人や団体は会社を目指し,破壊的なソーシャルメディア活動により,重大な満足されていない医療ニーズを有する患者のための未承認薬物の獲得を要請することに関与している可能性がある。もし私たちがアクセス権限拡大政策の下で現在または未来の候補製品のアクセス権限を提供または提供しないことを決定した場合、私たちの名声はマイナスの影響を受ける可能性があり、私たちの業務は損なわれる可能性がある。
最近のメディアによる個別患者参入要求の拡大への関心は、2018年5月30日に法律となった2017年連邦裁判権利法案に署名するなど、地方レベルおよび国家レベルの立法が“試用権利”と呼ばれる法律の導入と公布をもたらし、患者が従来の拡大参入計画よりも早く未承認治療を受けることを可能にすることを目的としている。この分野の急進主義と立法の可能な結果の一つは、予期しない拡大参入計画を開始するか、あるいは予想よりも早く私たちの製品候補製品をより広く提供する必要があるということかもしれない
さらに、一部の患者は、商業的承認を得る前に、慈悲によって使用し、計画を獲得し、または薬物の権利を獲得しようと試みることによって薬物を獲得し、彼らは生命に危険な疾患を有し、他のすべての利用可能な治療方法を使い切っている。これらの患者群の中で、深刻な不良事件が発生するリスクが高く、もし私たちがこれらの患者にこれらの製品を提供すれば、私たちの候補製品の安全性に負の影響を与える可能性があり、これは重大な遅延を招き、あるいは私たちの候補製品を商業化することに成功できず、これは私たちの業務に実質的な損害を与える可能性がある。もし私たちが拡大参入計画の下で患者に私たちの任意の候補製品を提供する場合、私たちは将来、様々な理由で任意の同情使用を再構成または一時停止する必要があり、および/または参入計画を拡大する必要があり、これは、そのような計画の既存または潜在的参加者に関連する負の宣伝または他の中断を引き起こす可能性がある。
私たちは他の生物製薬会社からの激しい競争に直面しており、私たちが効果的に競争できなければ、私たちの経営業績は影響を受けるだろう。
生物製薬業界の特徴は競争が激しく、革新が迅速だということだ。我々の競争相手は他の化合物や薬物を開発することができ、類似またはより良い効果を達成することができるかもしれない。我々の潜在的競争相手には,大手多国籍製薬会社,老舗バイオテクノロジー会社,専門製薬会社,大学などの研究機関がある。私たちの多くの競争相手は財務、技術、その他の面で大きな優勢を持っています
より多くの研究開発者や経験豊富なマーケティング·製造組織、成熟した販売チームなどの資源。規模が小さいか早い段階にある企業も重要な競争相手であることが証明される可能性があり,特に疾患を治療する新しい方法を開発する際には,我々の候補製品も治療に集中している。老舗製薬会社も,新療法の発見と開発を加速させたり,我々が開発した候補製品を時代遅れにする可能性のある新しい療法を認可したりするために投資を大挙する可能性がある。バイオテクノロジーと製薬産業の合併と買収は、私たちの競争相手により多くの資源を集中させる可能性がある。技術の商業適用性の進歩やこれらの業界に投資する資本の増加により、競争はさらに激化する可能性がある。我々の競争相手は、単独であっても、パートナーと協力しても、我々の候補製品よりも効率的で、安全で、商業化しやすい、またはコストの低い薬物または生物学的製品の開発、買収、または独占ライセンスに成功するか、または我々が技術および製品を開発するために必要なノウハウを開発するか、または特許保護を受けることが可能である。私たちは候補製品の開発と商業成功に影響を与える重要な競争要素は治療効果、安全性、耐性、信頼性、使用利便性、価格と精算であると信じている。
私たちの候補製品に対する規制機関の承認を得ても、競争相手の製品の供給と価格は、私たちの候補製品に対する需要と価格を制限するかもしれません。もし価格競争や医者が既存の治療方法から私たちの候補製品に転換したくない場合、あるいは医者が他の新薬や生物製品に変更したり、私たちの候補製品を保留することを選択した場合、私たちの商業計画を実施できないかもしれません。
我々が開発した任意の候補製品が市場承認を得ても,医師,患者,第三者支払者,医学界の他の人がビジネス成功に必要な市場受容度を達成できない可能性がある。
我々が開発した任意の将来の候補製品が市場承認を得ていれば,単一エージェントとしても他の療法と併用しても,医師,患者,第三者支払人,医学界の他の人の十分な市場受容度を得ることができない可能性がある。もし私たちが開発した候補製品が十分な受容度に達していなければ、著しい製品収入が生じないかもしれませんし、利益を上げることができないかもしれません。すべての候補製品が商業販売のために承認された場合、市場の受け入れ度は多くの要素に依存する
•他の治療法と比較した治療効果と潜在的優位性
•私たちの製品を提供することができ、承認されたら競争力のある価格で販売することができる
•他の治療法と比較して,投与の利便性と簡易性;
•対象患者群が新たな療法を試みる意欲と,医師がこれらの治療法を処方する意欲
•有力なマーケティングと流通支援
•十分な第三者カバー、市場アクセス、および適切な補償を得ることができる
•どんな副作用の流行率と重症度でも。
候補製品の製造または処方を変更する方法は、追加のコストや遅延を招く可能性がある。
候補製品が臨床前研究から後期臨床試験まで、潜在的な承認と商業化を得るために、製造方法や調合などの開発計画の様々な方面に伴い、この過程で変更を行い、過程と結果の最適化に努力することはよく見られる。このような変化はこのような期待された目標を達成できない可能性がある。これらの変化のいずれも、我々の候補製品の表現が異なることを招き、計画中の臨床試験またはプロセスを変えて製造された材料を用いた他の将来の臨床試験の結果に影響を与える可能性がある。このような変更はまた、生産変更、FDA通知、またはFDA承認後に臨床試験で得られた臨床データの有効性を証明するために、ブリッジまたは比較可能な試験を含む追加の試験を必要とする可能性がある。
これまでのすべてのナイルシターとミダメチニブの臨床試験は第三者によって行われていたため,任意の新しい薬物製品材料がすべての面で効力を含むことを証明するために分析や他の試験を行う必要があり,これらの早期臨床試験で使用されている製品に相当する。このような製品が必要な比較可能なテストに合格する保証はありませんし、私たちが招聘した任意の未来の第三者メーカーが私たちの候補製品の生産に成功することを保証することはできません。また、私たちが招聘したいかなる第三者メーカーが生産したいかなる材料も患者に対して、私たちが以前の臨床試験で使用した材料と同じ効果を観察したことを保証することはできません。
すべての上述の情況は臨床試験の完成を遅らせる可能性があり、移行臨床試験或いは1つ或いは複数の臨床試験を繰り返し、臨床試験コストを増加し、著者らの候補製品に対する承認を延期し、そして著者らの販売開始と収入を創造する能力を脅かす必要がある。
また、私たちはまだ商業規模の製造や加工をしていません。承認されれば、私たちの候補製品のためにそうすることができないかもしれません。私たちは私たちの製造プロセスを最適化するために努力する時に変更するかもしれませんが、私たちのプロセスのわずかな変化に対しても安全で効果的で、商業販売のための治療法が承認されることを保証することはできません。
私たちに製品責任訴訟を提起すれば、私たちは多くの責任を負い、候補製品の商業化を制限することを要求されるかもしれません。
臨床試験で私たちの候補製品をテストするため、私たちは固有の製品責任リスクに直面しています。もし私たちがどんな製品を商業化すれば、私たちはもっと大きなリスクに直面します。例えば、私たちの候補製品が臨床試験、製造、マーケティング、または販売中に傷害をもたらすと思われるか、または他の態様では不適切であることが発見された場合、私たちは起訴されるかもしれない。このような製品責任クレームは、製造欠陥、設計欠陥、製品固有の危険について警告、不注意、厳格な責任、または保証違反の告発を含む可能性がある。州消費者保護法によると、クレームも主張することができる。もし私たちが製品責任クレームで自分自身を弁護することに成功できなければ、私たちは重大な責任を招いたり、私たちの候補製品の商業化を制限することを要求されるかもしれません。成功的な防御であっても、多くの財政的で管理的な資源が必要だ。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
•候補品を市場に出すことはできません
•私たちの製品への需要が減少しました
•私たちの名声を損なう
•臨床試験参加者は脱退し、臨床試験を継続することができない
•規制当局が調査を開始しました
•関連訴訟の弁護費用
•経営陣の時間と資源を移転する
•臨床試験参加者や承認製品を受けた患者に巨額の金銭的報酬を与える
•製品のリコール、撤回またはラベル付け、マーケティング、または販売促進制限;
•収入損失
•利用可能なすべての保険と私たちの資本資源を枯渇させる
•承認された場合、候補製品を商業化することはできない
•私たちの株価は下落しています。
潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を得ることができず、これは、私たち単独またはパートナーと開発した製品の商業化を阻止または阻害する可能性がある。たとえ私たちが現在または未来の会社のパートナーと合意しても、私たちは損害賠償を受ける権利があります。もし何かクレームがあれば、この賠償は利用できないか十分かもしれません。私たちは現在臨床試験保険を受けていますが、私たちが保険を受けている保険金額は十分ではないかもしれませんし、将来的にはこの保険範囲を維持できないかもしれません。あるいは合理的な費用で追加または代替保険を得ることができないかもしれません(もしあれば)。私たちの保険証書にも様々な例外があります。私たちは製品責任クレームの影響を受けるかもしれません。私たちは保険範囲を持っていません。私たちは、裁判所によって裁決されたり、和解協定で協議された、私たちの保険範囲の制限を超えたり、私たちの保険カバー範囲内にない金額を支払わなければならないかもしれません。私たちは、これらの金額を支払うために十分な資本を得ることができないか、または得ることができません。
知的財産権に関するリスク
私たちの成功は私たちの知的財産権を保護する能力にある程度かかっているが、特許条項は私たちの競争地位を保護するのに十分ではないかもしれない。私たちの固有の権利と技術を保護することは難しくて高価で、私たちはそれらの保護を保障できないかもしれない。
私たちのビジネスの成功は、私たちの特許技術および私たちの候補製品、それらのそれぞれの成分、処方、共同療法、それらを製造するための方法および治療方法のための特許、商標および商業秘密保護の獲得と維持、およびこれらの特許を第三者の挑戦から保護することに大きく依存するだろう。私たちが許可されていない第三者が私たちの候補製品を製造、使用、販売、提供、または輸入することを阻止する能力は、これらの活動をカバーする有効かつ実行可能な特許の権利の程度に基づいて、私たちの影響を受ける。もし私たちの特許が満期になった場合、あるいは私たちが開発した任意の製品や技術の特許保護を確保して維持することができない場合、あるいは獲得した特許保護範囲が十分でなければ、私たちの競争相手は私たちと同じ製品や技術を開発し、商業化する可能性があり、私たちが開発する可能性のある任意の候補製品の商業化能力は悪影響を受ける可能性がある。特許の寿命は限られている。米国では、すべての維持費が適時に支払われる場合、特許の自然失効時間は、通常、米国で最初の非臨時出願日から20年である。特許期限調整および/または延長などの様々な延長は利用可能である可能性があるが、特許の有効期間およびその提供される保護は限られている。私たちの現在の物質成分特許にはナイルキャストとミダメチニブが含まれています。これらの特許はファイザー社のわが社の設立に関する許可です。ナイルシタンを物質組成物とした米国特許の法定期限は
2025年、多形体ナイル西タンをカバーする米国特許は2039年に満了し、現在臨床開発されている形態を含み、医薬組成物をカバーする米国特許は2042年に満了し、いずれの場合も特許期間調整またはいかなる規制延長も含まれておらず、外国の同業者が待っている。現在臨床開発されている形態、多形体の治療方法、および医薬組成物を含む4つの米国特許が2041年に満了する多形体ミダミニーをカバーする4つの米国特許は、いずれの特許も規制延長を含まず、外国の同業者が待っている。私たちの最初の特許は候補製品がアメリカまたは外国の管轄区域で上場承認される前または直後に満期になる可能性があります。既存の特許が満期になった後、私たちは現在、孤児薬の独占経営権に依存して私たちの主導的な製品を販売するつもりです。特許の有効期限が切れると、私たちは模造薬を含む競争製品からの競争に直面するかもしれない。したがって、私たちの特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために、私たちに十分な権利を提供していないかもしれない。私たちの主要候補製品をカバーする特許が満期になり、追加の特許保護を受けることができず、私たちの業務、運営結果、財務状況、将来性に大きな悪影響を及ぼす可能性もあります。
特許出願プロセスは高価で時間がかかり、私たちはすべての必要または望ましい特許出願を合理的なコストまたはタイムリーに提出して起訴することができないかもしれない。しかも、私たちはすべての関連市場で特許保護を求めたり、獲得しないかもしれない。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.さらに、場合によっては、私たちは、第三者から許可を得て、第三者に与えられ、私たちのライセンシーまたはライセンシーの技術に依存することができるかもしれないことを含む、特許出願の準備、提出および起訴を制御する権利がないかもしれない。
生物製薬分野の特許の強度は複雑な法律や科学的問題に関連しており,不確実である可能性がある。私たちが現在または将来所有または許可している特許出願は、私たちの候補製品または米国または他の国/地域でのその使用をカバーすることを必要とする発行された特許を生成できないかもしれない。特許が確実に発行されたとしても、第三者はその有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、このような特許が縮小され、無効にされ、または強制的に実行されない可能性がある。また,それらが挑戦されていなくても,我々の候補製品をカバーする特許や特許出願は,我々の知的財産権を十分に保護したり,我々の主張をめぐる他の人の設計を阻止したりすることができない可能性がある.もし私たちが持っている特許が私たちの候補製品に提供する保護の広さや強度が脅かされれば、会社が私たちと協力して私たちの候補製品を開発することを阻止し、私たちがそれを商業化する能力を脅かすかもしれない。また、臨床試験で遅延に遭遇すれば、特許保護の下で候補製品を販売できる時間が短縮されるだろう。
米国およびほとんどの他の国の特許出願は、提出されてから一定期間秘密であるため、候補製品に関連する任意の特許出願が最初に提出されたかどうかは不明である。さらに、2013年3月16日までに優先日を有する少なくとも1つの請求項がある米国出願については、誰が出願の特許権利要件によってカバーされる任意の主題を最初に発明したかを決定するために、第三者によって介入手続きを開始するか、または米国特許商標局(USPTO)によって訴訟を提起することができる。
私たちは私たちが最初の発明の任意の未解決特許出願がカバーしている発明の会社であることを確認することができません。もし私たちがそうでなければ、私たちは優先権紛争の影響を受ける可能性があります。私たちは、特定の特許の一部または全期間または特定の特許出願の全期間を放棄することを要求されるかもしれない。私たちが知らないいくつかの既存技術は、特許請求の範囲の有効性または実行可能性に影響を及ぼす可能性があるかもしれない。我々が知っているかもしれないが,クレームの有効性や実行可能性に影響を与えるとは考えられない従来技術が存在するが,最終的にはクレームの有効性や実行可能性に影響を与えることが発見される可能性がある。もし挑戦されれば、私たちの特許は裁判所によって有効または強制執行可能であると宣言されるか、または有効かつ強制的な執行が発見されても、競争相手の技術または製品は裁判所によって私たちの特許を侵害したと認定されることを保証することはできない。私たちは、私たちの活動に関連すると考えられる競争相手の特許または特許出願を分析し、私たちの候補製品に対して自由に運営できると考えているかもしれませんが、私たちの競争相手は、私たちが関係ないと思う特許を含むクレームを出すかもしれません。これは、私たちの努力を阻害したり、私たちの製品候補や私たちの活動がこのようなクレームを侵害したりする可能性があります。他の会社は、私たちの製品と同じ効果を持つ製品を独立した上で開発するかもしれませんが、これらの製品は、私たちの特許や他の知的財産権を侵害することなく、私たちがすでに発表した私たちの製品をカバーする特許の権利要件を中心に設計されます。また、, 私たちのいくつかの特許出願と特許は、私たちと私たちの協力者が共同で持っている発明をカバーするかもしれない。我々と我々の協力者が有効な特許戦略を実行するために必要な特許出願や起訴戦略に関する事項について合意する保証はなく,我々の協力者の決定が我々独自の知的財産権を保護する目標と一致することは保証されない.
最近または将来の特許改革立法は、私たちの特許出願をめぐる起訴と、私たちが発表した特許の実行または保護の不確実性とコストを増加させるかもしれない。2013年に公布された“ライシー·スミス米国発明法”(Leahy-Smith America Invents Act、略称“米国発明法”)によると、米国は“先発明”から“先申請”制度に転換した。“先提出書類”制度の下で、他の特許性要件が満たされていると仮定すると、最初に特許出願を提出した発明者は、通常、他の発明者がいるか否かにかかわらず、その発明の特許を取得する権利がある。アメリカの発明家は
ACTには,米国特許法の他のいくつかの重大な変化が含まれており,特許出願起訴方式に影響を与える条項を含み,既存技術を再定義し,新たなライセンス後審査制度を確立する。これらの変化の影響は不明であるが,米国特許商標局は最近“米国発明法”に関する新しい法規やプログラムを制定したが,特許法の多くの実質的な変化は,“まず出願を提出する”条項を含めて2013年3月に発効したからである。また,裁判所はこれらの条項の多くを扱っておらず,この法案や本明細書で議論されている特定特許に関する新条例の適用性はまだ決定されておらず,審査が必要である。しかしながら、米国発明法およびその実施は、任意の特許出願をめぐる起訴および我々が発行した特許の実行または保護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務および財務状況に実質的な悪影響を及ぼす可能性がある。
法的手段は限られた保護しか提供できず、私たちの権利を十分に保護できないか、あるいは私たちの競争優位性を獲得または維持することができるかもしれないので、未来の私たちの所有権の保護の程度は不確定だ。例えば:
•他の人は、私たちの候補製品の成分と類似しているが、私たちの特許請求の範囲内ではない化合物を製造または使用することができるかもしれない
•私たちの現在の候補製品中の活性成分は最終的に模倣薬製品で商業化され、製剤または使用方法に関する特許保護がない可能性がある
•ある会社またはその許可者(場合によっては)は、米国政府の任意の許可内特許および米国政府によって資金援助された特許出願の義務を履行できず、特許権の喪失を招く可能性がある
•その会社またはその許可者(場合によっては)は、これらの発明のために特許を出願した最初の会社ではない場合がある
•他の会社は類似または代替技術を独立して開発したり、私たちの任意の技術をコピーしたりすることができる
•係属中の特許出願は、発行された特許を生成しない可能性がある
•私たちまたは私たちのライセンシーの特許(場合によっては)または私たちまたは彼らの一部の特許は、事前の開示によって無効になる可能性があります
•他の人たちは私たちが持っている特許や許可された特許を迂回するかもしれない
•未公表の出願または特許出願が秘密にされている可能性があり、今後、我々の製品または技術と同様の権利要件が提示される可能性がある
•外国の法律はアメリカの法律のように私たちまたは私たちのライセンシーの固有の権利を保護しないかもしれない
•私たちが所有または許可されていない許可された特許または特許出願の権利要件、および発行された場合、私たちの候補製品が含まれていない可能性がある;
•私たちが所有しているまたは許可されていない特許は、競争優位性を提供してくれないかもしれないし、範囲を縮小したり、第三者の法的挑戦によって無効または実行不可能と認定されたりする可能性がある
•特許または特許出願が可能かもしれない発明者は、競争相手と付き合うことができ、私たちの特許を中心に設計された製品または方法を開発するか、または私たちまたは彼らが発明者として指定された特許または特許出願に敵意を持っているかもしれない
•可能性のある特許または特許出願を有することは、発明者として登録されるべき個人または発明者として登録されてはならない個人を含むことを見落としている可能性があり、これにより、これらの特許またはこれらの特許出願から発行された特許が無効または強制的に実行できないと認定される可能性がある
•私たちは過去に科学協力に参加して、未来もそうし続けるだろう。このような協力者は私たちと隣接したり競合したりする製品を開発するかもしれませんこれらの製品は私たちの特許の範囲内ではありません
•特許保護を受けることができる他のノウハウを開発することはできない
•私たちが開発した候補製品は、第三者特許または他の独占権によって保護される可能性がある
•他の人たちの特許は私たちの業務に悪影響を及ぼすかもしれない。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
特許保護に加えて、私たちは、特に特許保護が不適切であると考えられている場合に、当社の機密および独自の情報を保護するために、当社の従業員、コンサルタント、および第三者と締結された秘密協定および発明譲渡協定に深刻に依存している。契約措置に加えて、私たちは物理的および技術的セキュリティ措置を使用して、私たちの固有の情報の機密性を保護しようと努力している。例えば、従業員またはアクセス許可権限を有する第三者が商業秘密を盗用する場合、このような措置は、私たちの独自の情報に十分な保護を提供しない可能性がある。私たちの安全対策は、従業員やコンサルタントが私たちのビジネス秘密を盗用して競争相手に提供することを阻止できないかもしれませんが、このような不正行為に対する追跡権は、私たちの利益を十分に保護するための十分な救済措置を提供できないかもしれません。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかる可能性があり、結果は予測できない。また,ビジネス秘密は,我々の法的追跡を阻止するように他の人によって独立して開発されている可能性がある.もし私たちのビジネス秘密のような機密や独自の情報が漏洩されたり
このような情報が盗用された場合、またはそのような情報が競合他社によって独立して開発された場合、我々の競争的地位が損なわれる可能性がある。
しかも、アメリカ以外の裁判所は商業秘密を保護することをあまり望まないことがある。もし私たちが第三者が私たちのどんな商業秘密を使用するのを阻止するために法廷に訴えることを選択すれば、私たちは巨額の費用を生むかもしれない。私たちが勝訴しても、このような訴訟は私たちの時間と他の資源を消費するかもしれない。私たちは、当社の従業員やコンサルタントと契約を締結することを含む、当社の独自情報およびビジネス秘密を保護する措置をとっていますが、第三者は、実質的に同じ独自の情報および技術を独立して開発したり、他の方法で私たちのビジネス秘密を取得したり、当社の技術を開示したりすることができます。
したがって、私たちは私たちの商業秘密を意味的に保護することができないかもしれない。私たちの政策は、私たちの従業員、コンサルタント、外部科学協力者、協賛研究者、および他のコンサルタントに、私たちとの雇用や相談関係を開始する際に秘密協定を実行することを要求します。これらの合意は、当方との関係過程において、関係個人又は実体に開示されたすべての当方の業務又は財務に関連する機密情報を秘密にしなければならず、特定の場合を除き、第三者に開示してはならないと規定している。従業員の場合、合意は、個人的に構想された、私たちの現在または計画中の業務または研究開発に関連するもの、または通常の勤務時間内、私たちのオフィス内で行われる、または私たちの設備または独自の情報を使用するすべての発明が、私たちの固有財産であることを規定している。また、第三者が私たちのノウハウを盗用することを防止するために、物理的および技術的セキュリティ対策のような他の適切な予防措置を講じている。
知的財産権侵害に対する第三者のクレームは、私たちの製品発見と開発を阻害または延期する可能性があります。
私たちのビジネス成功は、私たちの候補製品を開発、製造、マーケティング、販売し、第三者の固有の権利を侵害することなく、当社の独自技術を使用する能力にある程度依存します。バイオテクノロジーおよび製薬業界には、干渉、派生、当事者間の審査、許可後の再審、米国特許商標局における再審手続き、または外国司法管轄区域における異議および他の同様の手続きを含む特許および他の知的財産権に関する訴訟、および特許に挑戦する行政訴訟が多くある。私たちは、私たちの候補製品および/またはノウハウが彼らの知的財産権を侵害していると主張する特許または他の知的財産権を持つ第三者の将来の訴訟の脅威に直面するか、または受ける可能性がある。我々が我々の候補製品を開発している分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する.バイオテクノロジーや製薬業界の拡張や特許の発行に伴い,我々の候補製品は他人の特許権侵害のクレームを引き起こすリスクが増加する可能性がある。さらに、私たちを含む業界参加者は、どの特許が様々なタイプの薬剤、製品、またはそれらの使用または製造方法をカバーしているかを常に明確にしているわけではない。したがって,我々の分野で発行される特許や提出された特許出願数が多いため,第三者は我々の候補製品,技術または方法を含む特許権を持っていると主張する可能性がある.
もし第三者が私たちが知的財産権を侵害していると主張すれば、私たちはいくつかの問題に直面するかもしれませんが、これらに限定されません
•権利侵害と他の知的財産権のクレームは、事件の状況にかかわらず、訴訟を提起するのは高価で時間がかかる可能性があり、そして私たちの管理層の核心業務に対する注意力を移す可能性がある
•権利侵害の実質的な損害賠償は、もし裁判所が紛争製品または技術侵害または第三者の権利を侵害すると判断した場合、私たちは支払わなければならないかもしれない。もし裁判所が侵害が故意であることを発見した場合、私たちは3倍の損害賠償金と特許権者の弁護士費の支払いを命じられる可能性がある
•裁判所は、第三者がその製品の権利を私たちに許可しない限り、私たちの候補製品を開発、製造、マーケティング、または販売することを禁止し、第三者はそうする必要はありません
•第三者から許可を得た場合、大量の使用料、前払い費用、および/または私たちの製品の知的財産権にクロスライセンスを付与する必要があるかもしれません
•私たちの候補製品やプロセスを再設計して、彼らが権利を侵害しないようにすることは不可能かもしれないし、大量のお金の支出と時間が必要かもしれない。
私たちのいくつかの競争相手は、彼らがより多くの資源を持っているので、私たちよりも複雑な特許訴訟の費用を効果的に受けるかもしれない。さらに、いかなる訴訟の開始および継続に生じるいかなる不確実性も、運営を継続するために必要な資金を調達する能力に重大な悪影響を及ぼす可能性があり、または私たちの業務、運営結果、財務状況、および見通しに重大な悪影響を及ぼす可能性がある。
第三者は私たちが許可されていない状況で彼らのノウハウを使用していると主張するかもしれない。一般に,米国で行われている臨床試験やその他の開発活動は,“米国法典”第35編271節に規定されている安全港の免除によって保護されている。もし私たちの候補製品がfdaの承認を得たら、第三者は提出することができます
私たちの特許侵害訴訟に対して。私たちは候補製品の商業化に実質的な悪影響を及ぼす可能性のある特許主張が(承認されれば)有効で実行可能であると信じていないが、私たちはこのような見方が正しくないかもしれないし、訴訟でこれを証明できないかもしれない。この点で,米国で法に基づいて発行された特許は有効性推定を有しており,証拠が“明確で納得できる”場合にのみ,この推定を覆すことができる,より高い証明基準である.我々が現在知らない第三者特許、すなわち、私たちの候補製品の使用または製造に関連する材料、製剤、製造方法、または治療方法の請求項が存在する可能性がある。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、我々の候補製品が発行された特許を侵害する可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。管轄権のある裁判所が任意の第三者特許を保有し、私たちの候補製品、製造過程で使用または形成された構造または分子の製造プロセス、または任意の最終製品自体をカバーする場合、任意のそのような特許の所有者は、適用特許に基づいて許可を得るか、またはそのような特許が満了するまで、無効または強制的に実行されない限り、候補製品を商業化する能力を阻止することができるかもしれない。同様に、任意の第三者特許が管轄権のある裁判所によって所有されている場合、私たちの処方、製造プロセス、または使用方法の様々な態様をカバーします, このような特許の所有者は、ライセンスを取得したか、またはその特許が満了するまで、または最終的に無効または実行不可能と判定されない限り、候補製品を開発および商業化する能力を阻止することができるかもしれない。いずれの場合も、そのような許可は商業的に合理的な条項や全く存在しないかもしれない。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができない場合、あるいは根本的にできなければ、候補製品を商業化する能力が損なわれたり、延期されたりする可能性があり、逆に私たちの業務を深刻に損なう可能性がある。我々が許可を得ても,我々の競争相手が我々に許可された同じ技術にアクセスできるように非排他的である可能性がある.さらに、もし私たちの特許および任意の特許出願によって提供される保護の広さまたは強度が脅かされている場合、会社が私たちと協力し、現在または将来の候補製品を許可、開発、または商業化することを阻止するかもしれない。
私たちにクレームを出した当事者は、禁止令や他の公平な救済を求めて獲得する可能性があり、これは、私たちの候補製品のさらなる開発と商業化を効果的に阻止することができるかもしれない。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。もし私たちに対する侵害クレームが成功した場合、私たちまたは私たちの許可者は、故意の侵害の3倍の損害賠償と弁護士費、第三者から1つ以上の許可を得ること、印税を支払うこと、または私たちの権利侵害製品を再設計することを含む大量の損害賠償を支払わなければならないかもしれないし、多くの時間とお金の支出が必要かもしれない。私たちはそのようなライセンスがあるかどうか、あるいは商業的に合理的な条項で提供されるかどうかを予測できない。また、訴訟がない場合であっても、第三者からライセンスを取得して、私たちの研究を進めたり、候補製品の商業化を許可したりする必要があるかもしれません。私たちはもしあれば、合理的な費用または合理的な条項でこのような許可証のいずれかを得ることができないかもしれない。この場合、私たちは私たちの候補製品をさらに開発して商業化することができなくなり、これは私たちの業務を深刻に損なう可能性があります。
第三者は、我々の従業員、コンサルタント、協力者、またはパートナーが機密情報を誤って使用または漏洩したり、商業秘密を流用したりすると主張する可能性がある。
バイオテクノロジーや製薬業界でよく見られるように、私たちが雇った個人は以前、私たちの競争相手や潜在的な競争相手を含む大学や他の生物製薬や製薬会社に雇われていた。現在、私たちに対するクレームが解決されていないにもかかわらず、私たちの従業員およびコンサルタントが、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを確実にするために努力しているにもかかわらず、私たちまたは私たちの従業員、コンサルタントまたは独立請負業者は、商業秘密または他の独自情報を含む、以前の雇用主または他の第三者の知的財産権を不注意または他の方法で使用または開示する可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権や人員を失う可能性がある。このようなクレームに対抗することに成功しても、知的財産権クレームに関連する訴訟や他の法的手続きは、巨額の費用を発生させる可能性があり、私たちの技術や管理者の正常な責任を分散させる可能性があります。また、公聴会、動議、その他の一時的な手続きや事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果をマイナスと考える場合、, これは私たちの普通株の価格に実質的な悪影響を及ぼすかもしれない。このような種類の訴訟や訴訟は、私たちの運営損失を大幅に増加させ、開発活動に利用できる資源を減少させる可能性がある。私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らの財政資源がはるかに大きいので、私たちよりもこのような訴訟や訴訟の費用を効果的に負担するかもしれない。特許訴訟または他の知的財産権関連訴訟の開始および継続によって生じる不確実性は、市場での競争能力に悪影響を及ぼす可能性がある。
私たちは許容可能な条項で任意の未来の候補製品を開発するために必要な権利を獲得または維持することができないかもしれない。
私たちの計画は他の候補製品に関連する可能性があり、第三者が保有する独占権を使用する必要があるかもしれないので、私たちの業務の成長は、私たちがこれらの独占権を獲得、許可、または使用する能力にある程度依存するかもしれない。
私たちの候補製品はまた効果的に働くために特定の処方が必要かもしれません。これらの権利は他の人が持っているかもしれません。私たちは私たちの化合物と既存の医薬化合物を含む製品を開発するかもしれない。私たちは、当社の業務運営に必要または重要であると考えられる任意の成分、使用方法、プロセス、または他の第三者知的財産権を第三者から得ることができないかもしれません。私たちは合理的なコストまたは合理的な条項でこれらのライセンスのいずれかを得ることができないかもしれません。もしあれば、これは私たちの業務を損なうかもしれません。私たちは、このような第三者知的財産権がカバーする成分や方法の使用を停止する必要があるかもしれませんし、このような知的財産権を侵害しない代替方法の開発を求める必要があるかもしれません。たとえこのような不可能な代替方法を開発することができても、追加のコストと開発遅延を招く可能性があります。我々が許可を得ることができても,非排他的である可能性があり,我々の競争相手が我々に許可された同じ技術にアクセスできるようにする.この場合、私たちは代替技術を開発または許可するために多くの時間と資源を必要とするかもしれない。
また,学術機関と協力し,これらの機関と合意した書面により,われわれの臨床前研究や開発を加速する場合がある。場合によっては、これらの機関は、協力によって得られた任意の技術的権利の許可を交渉するためのオプションを提供してくれるかもしれない。このようなオプションにかかわらず、私たちは指定された時間範囲内で、または私たちが許容できる条項の下でライセンスを交渉することができないかもしれない。もし私たちがそれができなければ、その機関は他の人たちに知的財産権を提供するかもしれないし、私たちの計画を実行し続けることを阻止するかもしれない。必要な第三者知的財産権を成功させたり、既存の知的財産権を維持したりすることができなければ、このようなプロジェクトの開発を放棄しなければならない可能性があり、私たちの業務や財務状況が影響を受ける可能性がある。
第三者知的財産権の許可·買収は競争分野であり、私たちよりも成熟したり、より多くの資源を持っている企業も、私たちの候補製品を商業化するために必要または魅力的だと思う第三者知的財産権許可または買収戦略をとることができるかもしれない。より成熟した会社は私たちより競争優位にあるかもしれません。それらの規模、現金資源、そしてより強い臨床開発と商業化能力のためです。このような交渉を成功させ、最終的に買収を求める可能性のある他の候補製品をめぐる知的財産権を得ることができる保証はない。
私たちは私たちの特許または私たちの許可者の特許を保護または強制する訴訟に巻き込まれるかもしれないが、これは高価で、時間がかかり、成功しないかもしれない。
競争相手は私たちの特許や私たちの許可側の特許を侵害するかもしれない。権利侵害や不正使用に対抗するために、私たちは費用が高く時間がかかるかもしれない侵害請求を要求されるかもしれない。さらに、侵害訴訟では、裁判所は、私たちの1つまたは複数の特許が無効または強制執行できないと判断するか、または私たちの特許が関連技術をカバーしていないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続きにおける不利な結果は、私たちの1つまたは複数の特許を無効にするか、強制的に実行できない、または狭い解釈に直面させるリスクに直面させ、任意の特許出願を発行できないリスクに直面させる可能性がある。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。
私たちは、米国特許商標局に、一方的な再審、当事者間審査または許可後の審査手続きにおいて特許権利要件を審査することを要求することによって、第三者の米国特許における特許請求の可能性に挑戦することを選択することができる。このような手続きは高価で、私たちの時間や他の資源を消費するかもしれない。我々は,欧州特許庁または欧州特許庁または他の外国特許庁の特許反対手続において第三者の特許に挑戦することを選択することができる.このような訴訟に反対する費用は巨大かもしれないし、私たちの時間や他の資源を消費するかもしれない。もし私たちが米国特許商標局、ヨーロッパ特許庁、または他の外国特許庁で有利な結果を得ることができなかった場合、私たちは私たちの候補製品または独自技術が私たちの特許を侵害している可能性があると非難する第三者の訴訟に直面する可能性がある。
さらに、米国のいくつかの特許出願は、特許発行前に秘密にされている可能性があるため、米国および多くの外国司法管轄区域の特許出願は、通常、出願後18ヶ月以内に発行される可能性があり、科学文献中の出版物は、実際の発見よりも遅れていることが多く、他の人が発行された特許または任意の係属中の技術について特許出願を提出していないか、または適用可能であれば、最初に技術を発明した人であることを決定することはできないかもしれない。私たちの競争相手もすでに提出されている可能性があり、将来的に特許出願を提出することができ、私たちと類似した製品または技術をカバーすることができる。このような任意の特許出願は、私たちの特許または任意の特許出願よりも優先される可能性があり、これは、そのような技術をカバーする発行された特許を取得する権利を要求することができる。もし他方が私たちが所有しているか、または私たちに許可された発明にアメリカ特許出願を提出した場合、私たちは、または、許可された場合に
もし私たちが特許技術を使用している場合、許可者は米国特許商標局が発表した妨害プログラムに参加して、米国における発明の優先度を決定しなければならないかもしれない。もし私たちまたは私たちのライセンシーが妨害プログラムの側であれば、私たちが私たちに与えることができるかもしれない発明を持っているアメリカ特許出願に関連して、私たちが成功しても、私たちは巨額のコストを招き、管理職を移転する時間と他の資源を費やす可能性がある。
第三者によって開始されるか、または米国特許商標局によって提起される干渉プログラムは、我々の特許または任意の特許出願または我々の許可者の発明に関する優先度を決定するために必要である可能性がある。不利な結果は、現在の特許権の喪失を招く可能性があり、関連技術の使用を停止したり、勝利者から許可権を得ようとしたりすることが要求される可能性があります。もし勝利者が商業的に合理的な条件で私たちにライセンスを提供しなければ、私たちの業務は損害を受ける可能性があります。訴訟や介入手続きは、私たちの利益に不利な決定を招く可能性があり、私たちが成功しても、巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性があります。私たちは私たちの商業秘密や機密情報の盗用を単独でまたは許可者と一緒に防ぐことができないかもしれないが、特に法律では米国のようにこれらの権利を十分に保護していない国があるかもしれない。
さらに、知的財産権訴訟は大量の開示を必要とするため、私たちのいくつかの機密情報は、このような訴訟中に開示によって漏洩される可能性がある。さらに、公聴会、動議、または他の一時的な手続き、または事態の発展の結果が発表される可能性がある。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
発行された特許の定期維持費は、特許有効期間内にいくつかの段階で米国特許商標局および外国特許代理機関に支払われなければならない。米国特許商標局および各種外国政府特許機関は、特許出願中および特許発行後に、いくつかのプログラム、文書、費用支払い、およびその他の規定を遵守することを要求する。多くの場合、適用規則に従って滞納金を支払うか、または他の方法で不注意を是正することによって失効を是正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効され、関連法ドメインの特許権の一部または全部の喪失をもたらす可能性がある。特許または特許出願の放棄または失効をもたらす可能性のある不正事件には、規定された期限内に公式行動に応答できなかったこと、費用が支払われていなかったこと、および適切に合法化され、正式な文書が提出されなかったことが含まれるが、これらに限定されない。この場合、私たちの競争相手が市場に参入する可能性があり、これは私たちの業務に実質的な悪影響を及ぼすだろう。
法廷または米国特許商標局で疑問が提起された場合、私たちの候補製品に関連する発行された特許は、無効または実行不可能と認定される可能性がある。
私たちまたは私たちの許可パートナーのうちの1つが第三者に対して法的訴訟を提起して、私たちの候補製品のうちの1つをカバーする特許を強制的に執行する場合、被告は、私たちの候補製品をカバーする特許が無効であり、および/または実行不可能であることを反訴することができる。米国の特許訴訟では、被告が特許の無効および/または強制執行できないと主張する反訴はありふれており、第三者は様々な理由に基づいて特許が無効または強制的に実行できないと断言することができる。第三者も米国や海外の行政機関に類似したクレームを出すことができ、訴訟範囲外でも同様である。このようなメカニズムには、再審、贈与後審査、外国司法管轄区の同等の訴訟手続(例えば、反対訴訟手続)が含まれる。このような訴訟は私たちの特許がこれ以上私たちの候補製品をカバーしないように撤回したり修正したりするかもしれない。法的に無効と実行不可能と断言された後の結果は予測できない。有効性に関しては、例えば、無効な以前の技術がないことは確認できませんが、私たち、私たちの特許弁護士、特許審査員は起訴中にこれを知りません。もし被告が無効および/または強制できない法的主張で勝った場合、または私たちが他の方法で私たちの権利を十分に保護できなかった場合、私たちは私たちの候補製品の少なくとも一部またはすべての特許保護を失うだろう。このような特許保護の喪失は、私たちの業務および私たちの技術および候補製品を商業化または許可を得る能力に実質的な悪影響を及ぼす可能性がある。
米国と旧米国司法管区特許法の変化は、特許の全体的な価値を低下させ、製品を保護する能力を弱める可能性がある。
他の生物製薬会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存している。生物製薬業界で特許を獲得と実施することは技術上の複雑性と法律上の複雑性にも関連するため、コストが高く、時間がかかり、しかも内在的な不確定性を持っている。また、米国は最近、広範囲な特許改革立法を公布し、実施している。米国最高裁の最近の裁決は特許の範囲を縮小した
場合によっては保護を受けることができ、場合によっては特許権者の権利を弱める。我々の将来の特許取得能力に関する不確実性の増加に加えて,このようなイベントの結合は,いったん特許を取得する価値に関する不確実性をもたらしている.米国議会、連邦裁判所、および米国特許商標局の決定によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それにより、私たちが新しい特許を獲得したり、私たちの既存の特許と将来獲得可能な特許を強制的に執行する能力を弱める可能性がある。これらの裁決や裁判所、米国議会、または米国特許商標局の将来の任意の裁決が私たちの特許価値にどのように影響するかを予測することはできません。同様に、他の管轄区域特許法のいかなる不利な変化も、私たちの業務や財務状況に重大な悪影響を及ぼす可能性がある。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
全世界のすべての国で候補製品特許を申請、起訴、擁護する費用は高い。私たちの主要候補製品の特許を含むいくつかのライセンス特許は、主要市場や他の国で発行されていますが、アメリカ以外のいくつかの国では知的財産権が米国の知的財産権を広く持っていない可能性があります。また、いくつかの外国の国の法律は、米国の連邦や州法よりも知的財産権の保護の程度が低いです。そのため、私たちは、第三者がアメリカ以外のすべての国で私たちの発明を実施したり、アメリカや他の司法管轄区で私たちの発明を使用して製造した製品を販売したり、輸入したりすることができないかもしれません。競争相手は、私たちが特許保護を受けていない司法管轄区で私たちの技術を使用して彼ら自身の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っているが、法執行力がアメリカに及ばない地域に輸出することができるかもしれない。これらの製品は、私たちが発行した特許を持っていない司法管轄区域で私たちの製品と競争する可能性があり、私たちの特許主張や他の知的財産権は彼らの競争を効果的にまたは阻止することができないかもしれない。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護、特に生物製薬製品に関する保護の強制執行を支持しておらず、これは、私たちまたは私たちのライセンシーが、私たちの特許の侵害や第三者の競争製品の販売に対する行為を阻止することを困難にする可能性があり、これらの行為は、全体的に私たちの独占権を侵害する。第三者は、外国管轄区域における特許権の範囲または有効性に挑戦するために訴訟を開始し、巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移転させ、私たちの特許を無効または狭義に解釈されるリスクに直面させる可能性があり、どの特許出願も発表できないリスクに直面し、第三者が私たちまたは私たちの許可者にクレームを提起する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
もし私たちが開発する可能性のある候補製品のために特許期間の延長とデータ独占権を得なければ、私たちの業務は実質的に損害を受ける可能性がある。
FDAが私たちが開発する可能性のある任意の候補製品の上場承認の時間、期限、および詳細によれば、私たちの1つ以上の米国特許は、1984年の“薬品価格競争および特許期限回復行動”または“ハッジ·ワックスマン修正案”に従って限られた特許期間を延長する資格がある可能性がある。ハッジ·ワックスマン修正案は、FDA規制審査過程で失われた特許期間の補償として、特許展期間を最長5年とすることを許可している。1つの特許期間の延長は、製品が承認された日から計14年の期間を超えてはならず、1つの特許を延長することしかできず、承認された薬物、その使用方法又はその製造方法に関する請求項を延長することしかできない。しかし,テスト段階や規制審査中に職務調査が行われていないこと,適用の最終期限内に出願できなかったこと,関連特許が満了する前に出願できなかったこと,適用要求を満たしていなかったことなどの理由で延期が得られなかった可能性がある。しかも、特許保護の適用期間や範囲は私たちが要求しているものよりも短いかもしれない。もし私たちが特許期間の延長やどのような延長の期限も私たちが要求したより短いことができなければ、私たちの競争相手は私たちの特許が満期になった後に競争製品の承認を受けるかもしれません。私たちの業務、財務状況、運営結果、見通しは実質的に損なわれる可能性があります。
もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は悪影響を受けるかもしれません。
私たちの商標または商号は、挑戦、侵害、回避、または汎用商標として発表されるか、または他の商標を侵害していると判断される可能性がある。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれないし、これらの名前の使用を停止させることができないかもしれません。私たちは関心のある市場で潜在的なパートナーや顧客の名前の承認を得るために必要です。もし私たちが私たちの商標と商号に基づいて名称を確立することができなければ、私たちは効果的に競争できない可能性があり、私たちの業務は不利な影響を受けるかもしれない。
政府の規制に関連するリスク
私たちはアメリカ、EUと他の司法管轄区域で私たちの候補製品に対する監督管理審査過程は現在まだ確定しておらず、しかも長く、時間と内在的に予測できないものになり、私たちは私たちの候補製品の臨床開発と監督審査(もしあれば)の方面で重大な遅延に遭遇するかもしれない。
薬品の研究、テスト、生産、ラベル、承認、販売、輸入、輸出、マーケティングと流通はすべてアメリカFDA、EU EMA或いはEU及び類似外国の監督管理機関の広範な監督管理を受けている。対応する規制機関のマーケティング承認を得るまで、いかなる司法管轄区域でもいかなる製品も販売することは許されない。我々が2022年12月にFDAに提出した成人硬線維腫治療のためのndaを除いて、2023年2月にFDAによって優先審査を受け、PDUFA目標行動日を2023年8月27日と指定したNDAを除いて、私たちはこれまでFDAにndaを提出したことがなく、MAAまたは比較可能な外国規制機関に同様のマーケティング申請をEMAに提出したことがある。米国では、NDAは、候補製品が各必要な適応に対して安全、純粋かつ有効であることを決定するために、広範な臨床前および臨床データおよび支持情報を含まなければならない。セキュリティプロトコルはまた、製品の化学、製造、および制御に関する重要な情報を含まなければならず、製造施設は成功した事前承認検査を完了しなければならない。
FDAはまた、承認を支援する安全性および有効性データの十分性を審議する専門家グループ、すなわち諮問委員会を要求することができる。諮問委員会の意見には拘束力はないが,完成した臨床試験に基づいて開発された任意の候補製品が承認される能力に大きな影響を与える可能性がある。
また、臨床試験は様々な原因で遅延または終了する可能性があり、以下の項目に関連する遅延または失敗を含む
•適用されれば臨床試験開始の規制を受けることができます
•計画の実験を始めて完成させるための財政資源があるかどうか
•将来のCROや臨床試験地点で受け入れられる条項と合意し,これらの条項は広範な交渉が可能であり,CROと臨床試験地点の条項は大きく異なる可能性がある
•各臨床試験場所で独立したIRBまたは倫理委員会の承認を得る
•適切な患者を速やかに臨床試験に参加させる
•患者に臨床試験を完成させ、或いは治療後のフォローアップを行った
•臨床試験場が臨床試験方案から外れ、GCP要求を満たしていない、あるいは試験を脱退した者
•ロシアとウクライナの間の持続的な衝突とそれによる米国からのより厳しい経済制裁のため、私たちは臨床試験に必要な製品の材料または製造槽の利用可能性に直面する可能性があり、私たちはアメリカ、EU、および他の管轄区域の候補製品のより高いコストまたはより少ない供給、材料、部品、またはサービスに直面する可能性がある
•臨床試験中に出現した任意の患者の安全問題を解決する
•新しいまたは既存の法律または法規とのいかなる衝突も解決する;
•新たな臨床試験場所を増やすこと
•CGMP法規により臨床試験のための合格材料を生産した。
患者の入選は臨床試験時間の重要な要素であり、多くの要素の影響を受ける。さらに、臨床試験は、法規要件または我々の臨床規程に基づいて臨床試験を行うことができない、FDA、EMAまたは同様の外国の規制機関による臨床試験操作または臨床試験場所の検査によって、臨床試験操作または臨床試験場所の検査を実施することによって、臨床保留、予見不可能な安全問題または副作用の実施、候補製品の使用の利点を証明できないことを含む、我々、そのような臨床試験を行っている機関のIRBs、FDA、EMAまたは同様の外国の規制機関によって一時停止または終了される可能性がある。政府法規や行政行為の変化或いは臨床試験を継続するのに十分な資金が不足している。もし私たちが任意の候補製品の臨床試験が終了したり、完成が遅れたりする場合、私たちの候補製品のビジネスの将来性は損なわれ、私たちが製品収入を作る能力は延期されるだろう。また、どんな臨床試験を完成するいかなる遅延も私たちのコストを増加させ、私たちの製品開発と審査過程を緩和し、そして私たちの製品販売と収入を創造する能力を危険にさらす。
2020年3月に国内外の施設に対する検査は基本的に保留されて以来、FDAは常規のモニタリング、生物研究モニタリングと審査前検査を含む大流行前の検査活動の回復に努力している。FDAが承認を得るために検査を行う必要があると判断し、旅行制限のために審査期間内に検査を完了することができず、FDAが遠隔相互作用評価が不十分であると判断した場合、FDAは、場合によっては、検査を完了することができるまで、申請に完全な返信または実行を延期することを意図していることを示している。新冠肺炎突発公衆衛生事件の間、多くの家が
その会社は、FDAがその申請の必要な検査を完了できなかったため、完全な返信を受け取ったと発表した。米国以外の規制機関は、進行中の新冠肺炎の大流行に対応するために、類似した制限や他の政策措置をとる可能性があり、規制活動に遅延が生じる可能性がある
FDA、EMA、または同様の外国規制機関は、私たちの候補製品に対する規制計画に同意しないかもしれない。
FDAが新薬を承認する一般的な方法は関連患者群の中で候補製品に対して1つ或いは複数の制御された良好な3期臨床試験を行うことである。3期の臨床試験は通常大量の患者に関連し、コストが高く、完成するまで数年かかる。
私たちの臨床試験結果は私たちの候補製品が承認されることを支持しないかもしれない。また、私たちの候補製品は規制承認を得られない可能性があり、あるいは規制承認が延期される可能性があります。理由はいくつかあります
•FDA、EMA、または同様の外国の規制機関は、私たちの臨床試験の用量方案、設計、または実施に同意しないかもしれない
•私たちの候補製品がその提案のどの適応に対しても安全かつ有効であることをFDA、EMA、または同様の外国の規制機関に証明することはできないかもしれない
•私たちは持続的な新冠肺炎の流行による安全や治療効果の問題に直面するかもしれない
•臨床試験結果はFDA、EMA或いは類似の外国監督管理機関が許可した統計的意義レベルに符合しない可能性がある
•候補製品の臨床的および他の利益が安全リスクを超えていることは証明できないかもしれません
•FDA、EMAあるいは同様の外国の監督管理機関は、臨床前研究または臨床試験データの解釈に同意しないかもしれない
•我々の候補製品の臨床試験から収集されたデータは、FDA、EMAまたは同様の外国の規制機関を満足させるのに不十分である可能性があり、外国の管轄区域で秘密協定または他の同様の提出を提出することを支持するのに十分ではないか、または米国または他の場所で規制の承認を得るのに十分ではないかもしれない
•FDA、EMAまたは同様の外国の規制機関は、私たちと臨床および商業用品契約を締結する第三者メーカーの製造プロセスまたは施設を承認できない可能性がある
•FDA、EMA或いは類似の外国監督管理機関の承認政策或いは法規は重大な変化が発生する可能性があり、著者らの臨床データは承認を得るのに十分ではない。
登録試験に対する中間分析に基づいて、規制機関が私たちの候補製品を承認することを求める可能性があり、特に中期分析が主要な終点に対して統計的意義があり、安全データが許容可能な安全性と耐性プロファイルを示す可能性がある。このような中期分析の結果は、NDA前の会議でFDAと議論され、データがNDAの提出をサポートするのに十分であるかどうかを評価するが、FDAが中間分析が規制承認に十分な基礎を提供することに同意しない場合、このような登録試験が終了するまでNDAを提出しないであろう。
FDAの画期的な治療指定または迅速チャネル指定は、実際には、より速い開発または規制審査または承認プロセスをもたらすことがない可能性がある。
FDAは、進行性、切除不可能、再発または難治性硬線維腫症または深層線維腫症を有する成人患者の迅速チャネル指定および突破的療法指定のためのニューノシドの使用を許可し、NF 1に関連する少なくとも2歳の手術不可能PNを治療するためのミダミニーの迅速チャネル指定を許可しており、これらの患者は進行しているか、または顕著な発症率をもたらしている。私たちは私たちの他の候補製品のための突破的な治療認証または迅速なチャネル認証を求めるかもしれない。
1つの製品が深刻なまたは生命に危険な疾患を治療することを意図しており、そのような疾患が満たされていない医療需要を解決する潜在力を示す場合、製品スポンサーは迅速なチャネル認証を申請することができる。FDAは広い自由裁量権を持っており、この称号が付与されているかどうか、したがって、私たちの候補製品がこの称号を得る資格があると信じていても、FDAがそれを付与することを決定することを保証することはできません。私たちが高速チャネル認証を受けたとしても、私たちは従来のFDAプログラムよりも速い開発過程、審査、または承認を経験しないかもしれない。FDAが我々の臨床開発プロジェクトのデータが高速チャネルの指定をサポートしなくなったと考える場合,その指定を撤回する可能性がある。
画期的な治療法は、重篤または生命に危険な疾患または状態を治療するために、単独でまたは1つまたは複数の他の製品と組み合わせることを目的とした製品として定義され、初期臨床証拠は、製品が1つまたは複数の臨床的重要終点において、既存の治療法よりも有意な改善を示す可能性があることを示す
臨床発展早期に治療効果が観察された。画期的な治療法として指定された製品に対して,FDAと試験スポンサーとの相互作用やコミュニケーションは,臨床開発の最も有効な方法を決定するのに役立つとともに,無効対照案中の患者数を最小限にすることができる。
画期的療法に指定されたのはFDAの裁量権である。したがって,我々の候補製品の1つが画期的療法として指定された基準に適合していると考えても,FDAは同意せず,このような指定をしないことにする可能性がある。いずれの場合も、FDAの従来の手順に従って承認を考慮した製品と比較して、画期的な治療指定を受けることは、より速い開発プロセス、審査または承認をもたらすことはなく、FDAの最終承認を確保することもできない可能性がある。また,候補製品が画期的な治療法となる資格があっても,FDAは後でその製品が資格条件を満たしていないことを決定し,画期的な療法の指定を取り消すことが可能である。
米国以外の臨床試験地点で行われた臨床試験の結果はFDAに受け入れられない可能性があり,外国司法管轄区以外で開発されたデータも同様にこのような外国の規制機関に受け入れられない可能性がある。
私たちの候補製品の前のいくつかの臨床試験はアメリカ以外で行われていて、私たちはアメリカ以外でより多くの臨床試験を行うつもりです。FDA、EMAなどの外国の監督管理機関は関連する司法管轄区域以外で行われた臨床試験データを受け入れるかもしれませんが、これらのデータの受け入れはいくつかの条件に依存します。例えば,FDAは臨床試験は合格した研究者が倫理原則(例えばIRBや倫理委員会の承認やインフォームドコンセント)に基づいて行わなければならないことを要求しており,試験群は米国人口を十分に代表しなければならず,データはFDAが臨床的意義を持つと考えられる方法で米国人口や米国医療実践に適用されなければならない。また,これらの臨床試験は適用される現地法に制限されているが,FDAがこれらのデータを受け入れるかどうかは,試験の進行がすべての適用された米国の法律や法規に適合しているかどうかに依存する。FDAが米国国外で行った試験データを受け入れる保証はなく,マーケティング応用の十分な支援となっている。同様に,外国規制機関に提出されたどのデータもその臨床試験の基準や要求に適合していることを確保しなければならず,類似した外国規制機関がその管轄外からの試験のデータを受け入れる保証もない。
医療保健提供者、医師、第三者支払者との関係は、適用される反リベート、詐欺および乱用、および他の医療法律法規の制約を受けることになり、これは、私たちを刑事制裁、民事処罰、契約損害、名声損害、および利益と将来の収入の減少に直面させる可能性がある。
米国や他の地域の医療提供者,医師,第三者支払者は薬品の推奨と処方に主な役割を果たしている。第三者支払者および顧客との配置は、連邦反リベート法規(AKS)および連邦虚偽クレーム法案(FCA)を含むが、これらの企業の販売、マーケティング、および流通薬品の業務または財務配置および関係を制限する可能性があるが、これらの製薬業者を広く適用される詐欺および乱用、および他の医療法律および法規に直面させる可能性がある。特に、私たちの候補製品の研究、および医療製品やサービスの普及、販売とマーケティング、および医療業界のいくつかのビジネス配置は、詐欺、リベート、自己取引、および他の乱用を防止するための広範な法的制約を受けている。これらの法律および法規は、広範な価格設定、割引、マーケティングおよび販売促進、構造および手数料、特定の顧客インセンティブ計画、および他のビジネススケジュールを制限または禁止する可能性があります。これらの法的拘束を受けた活動は,患者を募集して臨床試験を行う過程で得られた情報の不正使用に関するものである。私たちの運営能力に影響を与える可能性のある連邦、州、外国の医療法規は含まれていますが、これらに限定されません
•AKSは、任意のリベート、賄賂またはリベートを含む任意の報酬(任意のリベート、賄賂またはリベートを含む)を知りながら、故意に請求、または故意に請求し、または提供し、または任意の商品、施設、物品またはサービスを購入、レンタル、または推薦して、連邦医療保険および医療補助計画のような連邦医療計画(例えば、連邦医療保険および医療補助計画)に従って全部または部分的に支払いを行うために、直接的または間接的に、現金または実物の形態で誘導または見返りとして、個人を紹介することを禁止する。法規又は法規違反の具体的な意図を実際に理解することなく、一人又は実体が法規違反罪に問われる可能性がある。違反は民事と刑事罰金に処せられ、違反ごとに処罰され、最高3倍の報酬、監禁、そして政府医療計画から除外される。また、AKS違反による物品やサービスのクレームを含み、FCAについては、虚偽または詐欺的クレームを構成する
•FCAを含む連邦民事および刑事虚偽申告法および民事罰金法であって、個人または実体が虚偽または詐欺的な支払い申請の提出または提出を意図的に禁止するか、またはMedicare、Medicaidまたは他の連邦医療保健計画によって承認され、虚偽記録または報告書の作成、使用または作成または使用をもたらすことは、虚偽または詐欺的クレームまたは連邦政府への資金の支払いまたは移転の義務に重要な意味を持ち、または故意に隠蔽または承知して、連邦政府に金銭を支払う義務を不当に回避または減少または隠蔽することを含む、連邦民事および刑事虚偽申告法および民事罰金法。メーカーは責任を問われる可能性がある
FCAでは、政府支払人に直接クレームを提出していなくても、虚偽または詐欺的なクレームを“原因”とみなされている場合。政府は、製造業者が顧客に不正確な請求書や符号化情報を提供したり、ラベル外で製品を宣伝したりすることによって、虚偽または詐欺的なクレームの提出を“招く”と考える可能性がある。FCAはまた、“密告者”としての個人代表が連邦政府を代表して訴訟を起こし、FCA違反を告発し、任意のお金を共有して取り戻すことを許可している。実体がFCA違反と判断された場合、政府は民事罰金と罰金を科し、3倍の損害賠償を科し、その実体とその製品を連邦医療保険、医療補助、その他の連邦医療保健計画から除外することができる
•1996年の連邦“健康保険携帯および責任法案”、またはHIPAAは、支払人(例えば、公共または個人)にかかわらず、支払人(例えば、公共または個人)にかかわらず、任意のトリックまたは詐欺的な言い訳、陳述または約束によって、支払人(例えば、公共または個人)にかかわらず、任意の医療福祉計画が所有または保管または制御する任意の金銭または財産を故意にまたは意図的に実行または実行しようとすることを禁止する追加の連邦刑法を制定し、任意のトリックまたは装置を故意に偽造、隠蔽または隠蔽し、または医療福祉の交付または支払いに関連する任意の重大な虚偽陳述を行う。医療事項に関するプロジェクトやサービス。AKSと同様に、一人またはエンティティは、法規または法規違反の具体的な意図を実際に理解していない場合に、HIPAA違反罪に判決されることができる
•2009年に“健康情報技術促進経済および臨床健康法案”(HITECH)によって改正されたHIPAAおよびそのそれぞれの実施条例は、他にも、特定の医療保健提供者、健康計画および医療保健情報交換所(保険エンティティと呼ばれる)およびそのそれぞれの業務パートナー、保証エンティティにサービスを提供する独立請負業者に要求を提出し、これらのサービスは、個人が健康情報を識別することができるプライバシー、安全、および個人識別可能な健康情報の使用または開示に関する要求を提出する。HITECHはまた新しい民事罰金等級を作成し、HIPAAを改訂し、民事と刑事処罰を商業パートナーに直接適用し、州総検察長に新しい権力を与え、連邦裁判所に民事訴訟を提起し、損害賠償または禁止令を要求して連邦HIPAA法律を執行し、連邦民事訴訟の提起に関連する弁護士費と費用を求めることができる。さらに、他の連邦、州、非米国法は、場合によっては健康および他の個人情報のプライバシーおよびセキュリティを管理している可能性があり、多くの法律は互いに大きく異なり、コンプライアンス作業を複雑化させるために同じ効果を生じない可能性がある
•改正された患者保護及び平価医療法案(ACA)によって作成された連邦医師支払い陽光法案及びその実施条例によると、連邦医療保険、医療補助又は児童健康保険計画(いくつかの例外)によって支払うことができる薬品、設備、生物製品及び医療用品の製造業者は、毎年米国衛生公衆サービス部(HHS)の医療保険及び医療補助サービスセンター(CMS)に医師(医師、歯科医師、検眼士、足科医師及び脊医を含むと定義される)及び教育病院支払い又は他の価値移転に関する情報を報告することを要求する。医師とその直系親族が所有している所有権と投資権益です2022年1月1日から,これらの報告義務は,医師アシスタントや看護師従業員(医師アシスタント,看護師従業員,臨床看護師専門家,登録看護師麻酔科医,麻酔科医アシスタントおよび登録看護師助産師)のようないくつかの非医師提供者への価値移転を含むように拡大されている。
また、他の事項以外にも、上記の各医療法律と法規を遵守する州と国外等価物が必要であり、その中のいくつかの法律と法規の範囲はもっと広く、支払者が誰であっても適用できる可能性がある。アメリカの多くの州は連邦反リベート法規と虚偽クレーム法案のような法律を採用しており、研究、流通、販売またはマーケティング手配、および非政府支払人(個人保険会社を含む)によって精算される医療項目またはサービスに関するクレームを含むが、私たちの商業実践に適用可能である。また、いくつかの州は、製薬会社に、2003年4月の総監察長室の製薬メーカーに関するコンプライアンス計画ガイドラインおよび/または米国の製薬研究および製造業者の医療専門家との相互作用に関するガイドラインを遵守することを求めている。いくつかの州はまた他のマーケティング制限を実施し、あるいは製薬会社に州政府にマーケティング或いは価格開示を要求し、薬品販売代表の登録を要求した。2018年5月に施行された欧州連合一般データ保護条例を含む国家法律および外国法は、健康情報のプライバシーやセキュリティを管理する場合もあり、その多くの法律は互いに大きく異なり、HIPAAに先を越されず、コンプライアンス作業を複雑化させることが多い。このような州の要求を守るために何が必要なのかは曖昧で、もし私たちが適用された州の法律要求を守らなければ、私たちは処罰されるかもしれない。最後に,健康情報のプライバシーや安全を管理する国や外国の法律もあり,その多くの法律は互いに大きく異なり,HIPAAに先を越されず,コンプライアンス作業を複雑にしていることが多い。また、, 一部の州ではすでに法律が成立しており,医療機器会社にHHS監察長事務室や先進医療技術協会が発表した指導意見の遵守を求めている。
医薬製品の流通は広範な記録保存、許可、貯蔵と安全要求を含む追加の規定と条例を遵守し、許可されていない医薬製品の販売を防止しなければならない
製薬会社はまた連邦消費者保護や不正競争法の制約を受ける可能性があり,これらの法律は市場活動や消費者を損なう可能性のある活動を広く規制している。
これらの法律の範囲も執行も不確定であり,現在の医療改革環境下では,特に適用の前例や法規が乏しい場合には,急速な変化が生じる可能性がある。連邦と州法執行機関は引き続き医療会社と医療提供者の間の相互作用を密接に検討し、これは医療保健業界のいくつかの調査、起訴、有罪判決、和解を招いた。ビジネス手配が適用される医療保険法に適合することを確保することや,政府当局が行う可能性のある調査に対応することは,時間や資源がかかる可能性があり,会社の業務への関心を分散させる可能性がある。
政府および法執行当局は、私たちの業務実践が、詐欺および乱用または他の医療保健法律および法規を適用する現在または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちにこのような訴訟を提起し、私たちが私たちの権利を弁護したり、維持することに成功しなかった場合、これらの行動は、民事、刑事と行政処罰、損害賠償、罰金、返還、監禁、連邦と州政府が援助した医療計画から除外され、契約損害と削減または私たちの業務を制限し、もし私たちが会社の誠実な合意または他の合意の制約を受けた場合、これらの法律違反に関する告発、および追加の報告義務と監督を解決することを含む、私たちの業務に重大な影響を与えるかもしれない。さらに、私たちがそれと業務を展開することを期待している任意の医師または他のヘルスケア提供者またはエンティティが、適用された法律に適合していないことが発見された場合、彼らは、政府が援助する医療計画から除外されることを含む重大な刑事、民事または行政制裁を受ける可能性がある。これらの法律に違反するいかなる行為も、正当化に成功しても、バイオ製薬メーカーが巨額の法的費用を招き、経営陣の業務運営への注意をそらす可能性がある。今後発売される製品の販売を禁止または制限または撤回することは、不利な方法で業務に大きな影響を与える可能性がある。
一つの管轄区域で私たちの候補製品に対する規制承認を獲得し、維持することは、私たちが他の管轄区域で私たちの候補製品の規制承認を得ることに成功するという意味ではない。
1つの管轄区域で私たちの候補製品の規制承認を獲得し、維持することは、他の管轄区域で規制承認を得ることができるか、または維持できる保証はなく、1つの管轄区で規制承認を得ることができなかったり、遅延したりすることは、他の管轄区の監督審査過程に悪影響を及ぼす可能性がある。例えば、FDAが候補製品の発売を承認しても、EMAまたは同様の外国規制機関は、候補製品のこれらの国での製造、マーケティング、および普及を承認しなければならない。承認手続きは司法管轄区域によって異なり、1つの管轄区で行われる臨床試験は他の管轄区の監督機関によって受け入れられない可能性があるため、米国と異なるか、または米国の要求と行政審査期限よりも大きい場合があり、追加の臨床前研究または臨床試験を含む。米国以外の多くの管轄区では、候補製品は先に精算許可を得てから、その管轄区で販売を許可することができる。場合によっては、私たちが私たちの製品のために受け取る価格もまた承認されなければならない。
私たちはまた他の国でマーケティング申請を提出することができる。米国以外の管轄区域の監督管理機関は候補製品の承認に要求があり、私たちはこれらの管轄区が発売される前にこれらの要求を守らなければならない。外国の監督管理の承認を得て、外国の規制要求を遵守することは、私たちに重大な遅延、困難、コストをもたらす可能性があり、私たちの製品がある国/地域で発売されることを延期または阻止する可能性がある。もし私たちが国際市場の規制要求を遵守できず、および/または適用されたマーケティング承認を得られなければ、私たちの目標市場は減少し、私たちの候補製品の市場潜在力を十分に発揮する能力は損なわれるだろう。
私たちがどんな候補製品の規制承認を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性があり、もし私たちが規制要求を遵守できなかったり、私たちの候補製品が予期しない問題に遭遇したら、私たちは処罰を受けるかもしれない。
もし私たちの任意の候補製品が承認されたら、それらは製造、ラベル、包装、貯蔵、広告、販売促進、サンプリング、記録保存、発売後研究及び安全、治療効果とその他の発売後情報を提出することを含む持続的な法規要求を遵守し、アメリカの連邦と州要求及び比較可能な外国監督機関の要求を含む。また,われわれが承認後に行ったいずれの臨床試験についても,cGMPやGCP要求を遵守していく。
メーカーとメーカーの工場は、品質管理と製造プロセスがcGMP法規と適用される製品追跡と追跡要求に適合することを確保することを含む、FDA、EMAおよび類似の外国規制機関の広範な要求を遵守しなければならない。したがって、私たちと私たちの契約製造業者は、cGMPの遵守状況と任意の秘密協定で約束された遵守状況を評価するために、持続的な審査と検査を受けるだろう
BLAまたは他のマーケティング申請と、検査意見に対する以前の応答。したがって、私たちと私たちと協力している他の人たちは、製造、生産、品質管理を含むすべての規制コンプライアンス分野で時間、お金、エネルギーをかけ続けなければならない。
私たちの候補製品のために得られた任意の規制承認は、その製品が発売される可能性のある承認指示用途の制限または承認条件によって制限される可能性があり、または候補製品の安全性および有効性を監視する第4段階の臨床試験および監視を含む、コストの高い上場後試験の要求を含む可能性がある。さらに、FDORAによれば、承認された薬物および生物製品のスポンサーは、市場状態が任意に変化したときに、薬物の撤回などの6ヶ月の通知をFDAに提供しなければならず、そうしなければ、FDAは製品を生産停止製品リストに登録する可能性があり、これは製品の発売能力をキャンセルするであろう。FDAはまた、我々の候補製品を承認する条件としてリスク評価および緩和戦略またはREMS計画を要求することができ、これは、患者の長期フォローアップ、薬物使用ガイドライン、医師コミュニケーション計画、または分配方法の制限、患者登録、および他のリスク最小化ツールのような安全な使用を保証する他の要素の要求を必要とする可能性がある。さらに、FDA、EMA、または同様の外国規制機関が私たちの候補製品を承認した場合、安全および他の上場後の情報、報告、登録の提出を含む要求を遵守しなければなりません。
規制要件や基準が守られていない場合、あるいは製品発売後に問題が発生した場合、FDAは同意法令を強制的に実施したり、承認を撤回したりする可能性がある。その後、私たちの候補製品には、意外な深刻性や頻度の有害事象、または私たちの第三者製造業者または製造プロセスを含む以前に未知の問題が存在することが発見され、あるいは規制要件を遵守できなかった場合、承認されたラベルを修正して新しいセキュリティ情報を追加し、発売後の研究または臨床試験を実施して新しいセキュリティリスクを評価するか、またはREMS計画に従って流通制限または他の制限を実施することにつながる可能性がある。他の他の潜在的な結果には
•私たちの製品の販売や製造を制限し、製品を市場からリコールするか、または自発的または強制的に製品をリコールする
•罰金、警告状、または臨床試験を一時停止する者
•FDAは、処理すべき出願の承認を拒否するか、または私たちが提出した承認された出願の追加を拒否するか、またはライセンス承認を一時停止または撤回する
•製品を差し押さえたり抑留したり、私たちの候補製品の輸入または輸出を許可することを拒否したり;
•民事または刑事処罰を禁令または適用する。
FDAは市場に投入された製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。製品は承認された適応と承認されたラベルの規定でしか販売促進できません。承認された製品ラベルに含まれるいくつかの終点データも、患者報告の結果測定のような探索的または二次終点データを含むこのようなラベルにも現れない可能性があり、これは、承認された製品を普及させる能力に影響を与える可能性があることを期待する。FDA、EMA、および同様の外国規制機関の政策は変わる可能性があり、追加の政府法規を公布して、私たちの候補製品に対する規制承認を阻止、制限、または延期するかもしれない。米国や海外の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、または規制適合性を維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、利益を達成したり維持することができないかもしれない。
承認されれば、いくつかの細分化された市場における私たちの候補製品のカバー範囲と精算範囲が限られているか、利用できないかもしれません。これは、任意の候補製品を利益的に販売することを困難にするかもしれません。
もし私たちの候補製品が承認されれば、その成否は保証範囲の可用性と第三者支払者の十分な精算に依存する。私たちの候補製品が保証と精算を受けることを保証することはできません。あるいは私たちの候補製品の潜在的な収入を正確に推定することはできません。また、私たちが開発する可能性のあるどの製品も保証と精算を受けることは保証されません。
自分の病状に医療サービスを提供する患者は通常,第三者支払者によってその治療に関連する費用の全部または一部を精算する。カバー範囲と政府医療計画(例えば連邦医療保険や医療補助)および商業支払者からの十分な補償は、新製品の受容度に重要である。
政府当局や他の第三者支払人、例えば個人健康保険会社や健康維持組織は、どのような薬物や治療および精算金額をカバーするかを決定する。第三者支払者の製品使用状況の決定を含む第三者支払人の保証範囲と精算は多くの要素に依存する可能性がある
•健康計画の下で保障された福祉
•安全で効果的で医学的に必要なものです
•特定の患者に適しています
•費用対効果があります
•実験的でも調査的でもない。
米国では、第三者支払者の間に統一された製品保証や精算政策はない。したがって、政府や他の第三者支払人から製品の保証と精算承認を得ることは時間がかかり、費用がかかるプロセスであり、これは、各支払人に科学的、臨床的、費用効果的なデータを提供して、個々の支払人に基づいて私たちの製品を使用することを支援する必要があるかもしれないが、保証され、十分な精算を得ることは保証されないかもしれない米国では,新薬精算に関する主な決定は通常CMSによって行われる。CMSは新薬がどの程度連邦医療保険の下でカバーと精算されるかを決定し、個人支払者はよくCMSに大きく従う特定の製品の保険を受けても,それによる精算支払率は利益を実現したり維持したりするのに不十分である可能性があり,あるいは患者が受け入れられないと考える高い共済額が必要となる可能性がある。さらに、承認されると、第三者支払者は、候補製品を使用した後に必要な長期的な後続評価をカバーすることができず、十分な補償を提供しない可能性がある。患者が承認されると、保険を提供し、彼らの費用の大部分を支払うのに十分な費用を精算しなければ、私たちの候補製品を使用することはあまり不可能だ。新たに承認された製品の保険カバーと精算に関する不確実性が大きい。第三者決済者が私たちの候補製品の保証範囲と精算についてどのように決定するか予測するのは難しいです。
支払い方法は医療立法と規制措置の影響を受けるかもしれない。例えば、2012年の“中産階級減税と雇用創出法案”は、連邦医療保険計画を管理するCMSに2013年に連邦医療保険臨床実験室費用表を2%削減することを求めており、2014年以降の基礎となっている。また,CMSは2014年1月1日より,患者が病院外来でサービスを受ける際に注文したある実験室テストの連邦医療保険支払いをバンドルし始めた。将来的にはより多くの州および連邦医療改革措置が取られることが予想され、いずれも連邦および州政府が医療製品およびサービスのために支払う金額を制限する可能性があり、これはいくつかの薬品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
医薬品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、および将来、米国価格よりも低い価格で販売される可能性のある国から医薬品を輸入することを制限する法律の緩和によって低下する可能性がある。ますます多くの第三者支払人は製薬会社に価格に基づいて所定の割引を提供し、医療製品の定価に挑戦することを要求している。私たちは私たちが商業化されたどの候補製品も精算できることを確実にすることはできない。もし精算できるなら、精算のレベルもそうだ。そのほか、多くの薬品メーカーは平均メーカー価格、平均販売価格と最適価格などのいくつかの価格報告指標を計算し、政府に報告しなければならない。場合によっては、これらの指標が正確かつタイムリーに提出されていない場合には、処罰が適用される可能性がある。また,これらの薬品の価格は,政府医療計画が要求する強制的な割引やリベートによって低下する可能性がある。支払い方法は医療立法と規制措置の影響を受けるかもしれない。
また,米国や海外の政府や他の第三者支払者が医療コストの制限や低減に力を入れ,これらの組織が新承認製品のカバー範囲や精算レベルを制限する可能性があるため,候補製品の支払いや十分な支払いを提供できない可能性がある。アメリカでは、特殊薬物の価格決定に関する立法と法執行の関心が増加している。
具体的には、米国議会は最近数回の調査を行い、薬品定価の透明性を高め、連邦医療保険下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査し、政府計画の薬品精算方法を改革するための連邦と州立法を提出し、公布した。私たちは、管理型ヘルスケアの傾向、ヘルスケア組織のますます増加する影響力、コスト制御措置、追加の立法変化により、私たちの任意の候補製品の販売に関連する価格設定圧力に直面すると予想される。
また、2020年12月2日、HHSは薬品メーカーがD部分でスポンサー値下げを計画している安全港の保護を取り消し、法律が値下げを要求しない限り、直接あるいは薬局福祉マネージャーを通じて値下げする規定を発表した。この規定はまた、販売時点での値下げを反映するための新しい避難港を作成し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための避難港を作成する。米国コロンビア特区地域裁判所の命令によると、連邦医療保険D部分によると、メーカーから計画発起人への薬品の販売または購入に関連するいくつかのリベートを廃止する安全港保護の一部は、2023年1月1日に延期された。2020年12月31日,CMSは2023年1月1日から施行され,メーカーに自己援助の全価値が患者に転嫁されることを確保するように要求する新しいルールを発表し,そうでなければこれらのドルは薬物の平均メーカー価格と最適価格計算(“医療補助アキュムレータルール”)に計上される。五月十七日
2022年、米国コロンビア特区地域裁判所は、無効医療補助アキュムレータルールの決定を要求する米国製薬研究およびメーカー(PhRMA)の動議を承認した。また、バイデン政府は現在、この変化の実施状況を検討しており、処方薬製品や薬局福祉マネージャーサービス料の販売所値下げに新たな安全港を提供し、改訂または廃止する可能性がある。いくつかの措置および他の提案された措置は、発効するために追加の立法によって許可される必要があるかもしれないが、バイデン政府はこれらの措置を撤回または他の方法で変更する可能性があるが、国会は薬品コストを制御するための新しい立法措置を求め続けると表明している。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品への参入およびマーケティングコスト開示の制限および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量調達を奨励するための法規も実行されている。
将来的にとりうる医療改革措置は,より厳しいカバー基準を招き,承認された任意の製品の価格に追加の下り圧力を与える可能性が予想される。コスト抑制措置や他の医療改革を実施することは、私たちの収入の創出、利益の実現、あるいは私たちの製品の商業化を阻止するかもしれない。承認後の要求を拡大し、医薬品の販売や販売促進活動を制限するための立法·規制提案がなされている。私たちは、より多くの立法変化が公布されるかどうか、あるいはFDAの法規、ガイドラインまたは解釈が変わるかどうか、またはこれらの変化が私たちの候補製品の上場承認または許可にどのような影響を与える可能性があるかどうかを決定することはできない
また、一部の外国の国では、薬品の提案価格は必ず承認されなければならず、合法的に発売されることができる。各国の薬品定価に対する要求は大きく異なる。例えば、欧州連合は、その加盟国に様々な選択を提供し、その国の健康保険制度が補償を提供する医療製品の範囲を制限し、人が使用する医療製品の価格を制御する。精算または定価の承認を得るために、その中のいくつかの国は臨床試験の完成を要求する可能性があり、特定の候補製品のコスト効果を現在利用可能な治療法と比較する。加盟国は医薬製品の具体的な価格を承認することができ、医薬製品を市場に投入する会社の収益力に対して直接或いは間接的に制御制度をとることもできる。薬品に対して価格制御や精算制限を実行することを保証できない国は、私たちの任意の候補製品に対して有利な精算と定価手配を許可する。歴史的に見ると、EUで発売された製品はアメリカの価格構造に従わず、通常価格ははるかに低くなることが多い。
また、2017年の“試用権利法案”などは、ある患者に連邦フレームワークを提供し、第1段階の臨床試験が完了し、FDAの承認を得るために調査を受けているいくつかの研究用新薬製品を使用できるようにしている。場合によっては、条件を満たす患者は、臨床試験に参加することなく、FDA拡大参入計画に従ってFDA許可を得ることなく治療を求めることができる。2017年の“試用権法案”によると、製薬業者は条件を満たす患者に薬品を提供する義務はない。
行われている医療立法や規制改革措置は、私たちの業務や運営結果に実質的な悪影響を及ぼす可能性がある。
規制、法規、または既存の規制の解釈の変化は、(I)私たちの製造スケジュールを変更すること、(Ii)製品ラベルを追加または修正すること、(Iii)私たちの製品をリコールまたは停止すること、または(Iv)追加の記録保存要件を要求することなど、私たちの将来の業務に影響を与える可能性がある。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。
国内でも海外でも,政府も個人も,支払側は医療コストを抑えるための複雑な方法を開発しているが,これらの方法は,遺伝子療法やまれな疾患に対する療法,例えば我々が開発しているような新しい技術に特化しているわけではない。米国や一部の外国司法管轄地域では、医療システムの立法や規制に多くの変化が生じており、製品販売の収益性に影響を与える可能性がある。特に、2010年に“医療·教育調整法”により改正された“患者保護·平価医療法案”(または総称して“ACA”)が公布され、他の事項を除いて、生物製品を低コストの生体模倣薬の潜在的競争を受けさせ、医療補助薬物リベート計画に基づいてメーカーが医療補助薬物リベート計画の下で不足しているリベートを計算する新しい方法を解決し、医療補助薬物リベート計画の下で不足している最低医療補助リベートを大部分のメーカーが増加させ、医療補助医薬品リベート計画を医療補助管理のケア組織に登録された個人処方に拡大する。製造業者にあるブランドの処方薬に新しい年間費用と税金を支払うことを要求する;新しいMedicare Part D保険カバーギャップ割引計画を作成し、メーカーはカバーギャップ中に条件を満たす受益者に50%(2018年の両党予算法に従って70%に増加し、2019年1月1日から発効)のPOS割引を提供することに同意しなければならない
医療保険D部分がカバーする外来薬物;そして連邦政府の比較有効性研究の項目を増加させるために激励を提供する。
ACAのいくつかの側面は公布以来、司法、国会、行政面の挑戦を受けてきた。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、特定の政府機関に、医療補助モデルプロジェクトおよび免除計画の再審査、および医療補助またはACAによる医療保険カバー範囲の獲得による不必要な障害をもたらす政策を含む医療補助モデルプロジェクトおよび免除計画の再審査および見直し、医療補助またはACAによる医療保険のカバー範囲を制限する既存の政策およびルールを再検討するように指示する。バイデン政府の他の医療改革措置や他の挑戦,ACAの廃止または代替の努力(あれば)が我々の業務にどのように影響するかは不明である。
バイデン政権に先立ち、2017年10月13日、トランプ前総裁はACAによる保険会社への精算のコスト分担補助金を中止する行政命令に署名した。前トランプ政権は、ACAが保険会社に支払うコスト分担削減やCSRが国会の必要な支出を受けていないことを要求し、これらの支出が完了するまで直ちに支払いを停止すると発表した。いくつかの州の検事総長は政府による補助金の中止を阻止するよう提訴したが、彼らが提出した制限令請求は2017年10月25日にカリフォルニア州の連邦裁判官によって却下された。2020年8月14日、米国連邦巡回控訴裁判所は2つの異なる事件の中で、連邦政府はこれまで数年(2017年を含む)支払われていなかったCSRに対して全額責任を負うと判断した。健康保険会社が2018年以降に提出した企業社会責任クレームについては,満期額(あれば)を決定するためのさらなる訴訟が必要となる。また、2018年6月14日、米国連邦巡回控訴裁判所は、連邦政府は第三者支払者に120億ドルを超えるACAリスク回廊に支払う必要がないと判断し、これらの支払人はこれらの支払いが彼らに不足していると弁明した。2020年4月27日、米国最高裁判所は米国連邦巡回控訴裁判所の裁決を覆し、事件を米国連邦クレーム裁判所に送り、政府は関連式に基づいてこれらのリスク回廊支払いを支払う義務があると結論した。このような判決が私たちの業務にどのような影響を与えるのかまだ分からない。
また,CMSは2020年から各州が個人や小団体市場の保険会社に基準を設定する上でより大きな柔軟性があり,ACAがこのような市場で販売されている保険計画に要求される基本的な健康福祉を緩和する可能性があるという最終規則を発表した。
ACAが公布されて以来、アメリカはまた他の立法改正を提案し、採択した。
•2011年の“予算制御法案”(Budget Control Act)などの法案は国会の支出削減に向けた措置を策定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、各年度に医療保険提供者に支払う合計2%の削減を含むいくつかの政府プロジェクトの自動削減をトリガしている
•2013年1月2日、2012年に米国納税者救済法が署名され、いくつかのタイプの提供者に支払われる医療保険をさらに削減し、政府が提供者に多額の支払いを取り戻す訴訟時効が3年から5年に延長された。
•2017年4月13日、CMSは、各州に個人や小団体市場に保険会社のための基準を設定する上でより大きな柔軟性を与える最終ルールを発表し、このような市場で販売されている保険計画に対してACAが要求する基本的な健康福祉を緩和する可能性がある。
•2018年5月30日、“裁判権法案”が法律に署名された。他の事項以外に、この法律はある患者に連邦フレームワークを提供し、彼らが第一段階の臨床試験を完成し、FDAの許可を得た研究用新薬製品を獲得するために調査を行っていることを許可した。場合によっては、条件を満たす患者は、臨床試験に参加することなく、FDA拡大参入計画に従ってFDA許可を得ることなく治療を求めることができる。“試用権法案”によると,製薬業者はその薬品を条件に該当する患者に提供する義務はない
•CMSは2019年5月23日、Advantage計画が2020年1月1日からB薬剤の一部に階段療法を使用することを許可する最終ルールを発表した
•2019年12月20日、トランプ前総裁は“さらなる総合支出法案”(H.R.1865)に署名し、ケディラク税、医療保険提供者税、医療機器消費税を廃止した。未来に似たような税金が発行されるかどうかを確定することは不可能だ。
•2021年の総合支出法案は、2021年3月31日まで停止期間を延長する。支出やその他の目的を全面的に直接削減することを防ぐ法案が2021年4月14日に法律に署名し、一時停止期間を2021年12月31日に延長した
上述したように、米国は特殊薬品の価格決定方法に対する立法と法執行への興味がますます大きくなっている。承認された場合、いくつかの細分化された市場における我々の候補製品のカバー範囲および精算範囲は、限られているか、または利用できない可能性があり、これは、任意の候補製品を利益的に販売することを困難にする可能性があることを参照されたい。連邦レベルでは、前トランプ政権の2021年度予算提案には、薬品価格の引き下げ、競争の増加、患者の自己負担薬品コストの低減、患者の低コスト模造薬と生体類似薬の獲得を求める立法提案を支援するための1350億ドルの手当が含まれている。2020年3月10日、前トランプ政権は薬品定価の“原則”を国会に提出し、連邦医療保険Dの一部受益者の自己負担薬局費用を制限し、連邦医療保険D部分受益者の毎月の自己負担費用を制限するオプションを提供し、薬品価格の上昇を制限する立法を呼びかけた。また、前トランプ政権はこれまでに、メーカー競争の増加、ある連邦医療保健計画の交渉力の増加、メーカーの製品価格の引き下げ、消費者の支払いの薬品自己負担コストの低減を奨励する追加提案が含まれている薬品価格の引き下げと薬品自己負担コストの低減の“青写真”を発表した。衛生·公衆サービス部はすでにその中のいくつかの措置についてフィードバック意見を求め始めており、同時に、その現有の権力に基づいて直ちに他の措置を実施している。例えば,2019年5月,CMSはMedicare Advantage計画が2020年1月1日からB薬剤に対する階段療法の一部を選択することを可能にする最終ルールを発表した。しかしバイデン政権が挑戦し逆転するかどうかは不明です, このような実行と行政操作を撤回したり修正したりする。
また,340 B薬品定価計画も数回変化しており,薬品メーカーがある医療機関に売却する薬品の価格に上限を設定している。2018年12月27日、コロンビア特区地区裁判所は340 B薬品定価計画下の精算式の変更が無効であることを発表し、CMSはその後、2019年度と2018年度の特定保険外来薬の精算式を変更した。裁判所は、この変化は部長が適宜決定した“調整”ではなく、補償計算の根本的な変化であると判断した。しかし、最近では2020年7月31日、米コロンビア特区巡回控訴裁判所が地域裁判所の裁決を覆し、これらの変化が長官の権力範囲内にあることが分かった。2020年9月14日,原告−被控訴者はEN Bancの再審(すなわち全裁判所へ)の請願書を提起し,2020年10月16日に却下された。原告−被控訴者は2021年2月10日に最高裁に移審令の請願書を提出し,2021年7月2日に請願書を承認した。2022年6月15日、最高裁はHHS 2018年と2019年の340 B病院の精算料率が法規に違反し、不法であるとする控訴裁判所の裁決を全会一致で覆した。私たちは340 B計画の発展に影響を及ぼすことを検討し続ける
2020年、トランプ前総裁は処方薬の定価に関するいくつかの行政命令を発表し、前政府のいくつかの提案を実施しようとした。これに応じてFDAは2020年9月24日に最終規則を発表し,2020年11月30日に発効し,各州がカナダからの薬品輸入計画の策定と提出を指導した。また、2020年11月20日、CMSは最恵国或いは最恵国モデルを実施する暫定最終規則を発表し、このモデルによると、ある薬物と生物製品の連邦医療保険B部分の販売率は1人当たりの国内総生産に類似した経済協力と発展組織国の薬品メーカーが受け取った最低価格に基づいて計算される。最恵国待遇模範条例は、確定されたB部分提供者の参加を要求し、2021年1月1日から2027年12月31日までの7年間、米国のすべての州·地域に適用される。しかし、2021年12月29日、CMSは最恵国規則を廃止した。
2021年7月9日、総裁·バイデンは、政府の政策、すなわち(I)連邦医療保険交渉薬品価格を許可し、インフレ上限を設定し、低コスト模造薬および生物模倣薬の開発と市場参入を支援することを含む立法改革を支持する行政命令に署名し、(Ii)公共医療保険オプションの制定を支持した。その他の事項に加えて、行政命令は、処方薬の価格設定が高すぎ、国内の薬品サプライチェーンの強化、連邦政府の薬品支払いの価格低下、業界価格詐欺の解決のための行動を説明する報告書をHHSに提供するよう指示し、2003年の“連邦医療保険処方薬、改善と現代化法案”およびFDAの実施条例に基づいて第804条の輸入計画を制定する州とインディアン部族との協力を提案するようFDAに指示した。FDAは2020年9月24日にこのような実施条例を発表し、2020年11月30日に発効し、各州のカナダ薬品輸入計画の制定と提出に指導を提供した。2020年9月25日、CMSは、この規則に基づいて輸入された州の薬品は、社会保障法第1927条に基づいて連邦還付を受ける資格がなく、メーカーは“最適価格”や平均メーカー価格の目的でこれらの薬品を報告しないと声明した。これらの薬剤はカバーされた外来薬とは考えられないため,CMSはさらに,これらの薬剤の全国平均薬物調達コストは公表されないと述べている。実施すれば、カナダからの薬品輸入は私たちの任意の候補製品の価格に実質的かつ不利な影響を与えるかもしれない。
また、2020年11月30日、HHSは、直接または薬局福祉マネージャーを通じて、薬品メーカーからD部分の計画発起人に値下げの安全港保護を廃止しなければならないという規定を発表した
値引きは法律で定められています。この規定はまた、販売時点での値下げを反映するための新しい避難港を作成し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための避難港を作成する。裁判所の命令により,上記安全港の除去と増加は延期され,最近の立法は同規則の実施を2026年1月1日に一時停止した。この最終期限は、両党の“より安全なコミュニティ法案”によって2027年1月1日に延期された。2022年のインフレ削減法案はさらにこの規定の実施を2032年1月1日に延期する。
2022年8月16日にCMSが連邦医療保険B部分とD部分によって精算された単一由来薬物と生物製品の価格について交渉することを許可した“2022年インフレ削減法案”が可決され、まず2026年から連邦医療保険D部分から支払われた10種類の高コスト薬物、次いで2027年の15種類のD部分薬、2028年の15種類のB部分またはD部分薬、および2029年以降の20種類のB部分またはD部分薬である。 この立法は、製薬業者に民事罰金と潜在的な消費税を受けることを要求し、その理由は、この立法を遵守できなかったため、提供された価格が法律で規定された協議の“最高公平価格”以下でないか、または値上げ幅がインフレを超えているからである。 この法案は連邦医療保険受益者の年間自己払い薬品費用を2000ドルに限定している。“2022年インフレ削減法案”が我々の業務や医療業界全体に与える影響は不明である。
私たちは未来に取られる可能性のある計画を予測できない。政府、保険会社、医療組織、その他の医療サービスを管理する支払人は、医療コストの抑制または低減、および/または価格規制の実施に努力し続けており、悪影響を及ぼす可能性がある
•私たちの候補製品の需要は、もし私たちが規制部門の承認を得たら
•私たちは承認された製品のために公平だと思う価格を設定することができます
•私たちは収入を作ったり利益を達成したり維持したりします
•私たちが払わなければならない税金レベル
•資金の入手可能性
連邦医療保険や他の政府計画精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性があり、これは私たちの将来の収益性に悪影響を及ぼす可能性がある。
また、2020年12月31日、CMSは2023年1月1日から発効し、メーカーに自己援助のすべての価値を患者に転嫁することを確保するように新規定を発表し、そうでなければ、これらのドルは薬物の平均メーカー価格と最適価格計算に計上される。2022年5月17日,米国コロンビア特区地域裁判所は米国薬物研究·メーカー(PhRMA)の簡易判決動議を承認し,アキュムレータ調整ルールの無効を宣言した。
アメリカ各州もますます立法を通じて薬品の価格を制御するための法規を実施しており、価格或いは患者の精算制限、割引、ある製品への参入とマーケティングコスト開示の制限及び透明性措置を含み、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。承認された場合、いくつかの細分化された市場における我々の候補製品のカバー範囲および精算範囲は、限られているか、または利用できない可能性があり、これは、任意の候補製品を利益的に販売することを困難にする可能性があることを参照されたい
これらの法律および将来の州および連邦医療改革措置は将来的に採択される可能性があり、いずれも連邦医療保険および他の医療保険資金のさらなる減少をもたらし、他の方法で規制の承認を得る可能性のある任意の候補製品の価格、またはそのような候補製品の処方または使用頻度に影響を与える可能性がある。
ラベルの外で使用したり、私たちの製品を誤用したりすることは、私たちの市場での名声を損なったり、ダメージを招いたりして、コストの高い製品責任訴訟を引き起こす可能性があります。
われわれは,硬線維腫の治療のためのニューロキセチンと,NF 1−PNの治療のためのミダミニーを開発している。もし私たちの候補製品がFDAの承認を受けたら、私たちはその特別に承認された適応に対して、承認されたラベルと一致する方法で私たちの候補製品を宣伝またはマーケティングすることしかできません。私たちのマーケティングと販売チームを訓練し、承認された使用適応以外の候補用途、いわゆる“非ラベル用途”を宣伝することを防止します。しかし、もし医者が彼や彼女の独立した専門的な医学判断が適切だと思うなら、私たちは医者がラベルの外で私たちの製品を使用することを阻止することはできない。また,われわれの製品をFDA承認以外の適応に用いることは,このような疾患の治療に有効ではない可能性がある。私たちの候補製品に対するどんなこのようなラベルの外使用は医者と患者における私たちの市場名声を損なう可能性があります。もし医者が私たちの製品をいかなるラベル外の用途に使用しようとすれば、患者の負傷のリスクを増加させる可能性があり、これは製品責任訴訟を招く可能性があり、これは大量の資金が必要かもしれません
資源を管理し、これは私たちの名声を損なうかもしれない。さらに、FDAは、ラベル外使用に関する製造業者のコミュニケーションに厳しい制限を加えており、もし私たちまたは私たちの協力者が承認されたラベルと一致した方法で私たちの製品を普及させなければ、私たちまたは彼らはラベル外マーケティングによって警告されたり、法執行行動を受けたりするかもしれない。FCAを含む“連邦食品、薬物および化粧品法”および他の処方薬普及および広告に関連する法規に違反することは、連邦および州医療詐欺および法律乱用および州消費者保護法違反を調査または告発する可能性がある。
食品·医薬品局、米国証券取引委員会および他の政府機関の資金不足や世界的な健康懸念による中断は、重要な指導部および他の人員の雇用および保持能力を阻害したり、新たなまたは修正された製品がタイムリーまたは完全に商業化されて開発、承認または商業化されることを阻止したり、またはこれらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを他の方法で阻止することができ、私たちの業務に負の影響を与える可能性がある。
FDAが新製品を審査·承認する能力は、政府予算と資金レベル、キーパーソンの雇用と維持、ユーザー費用の支払いを受け入れる能力、法律、法規と政策の変化、およびFDAの通常の機能を履行する能力に影響を与える可能性のある他の事件を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また、政府が米国証券取引委員会や我々の業務に依存する可能性のある他の政府機関に提供する資金は、研究開発活動に資金を提供する機関を含め、政治プロセスの影響を受けており、政治プロセス自体が不安定で予測不可能である。
FDAや他の機関の中断も、新薬が必要な政府機関によって審査および/または承認されるのに要する時間を遅らせる可能性があり、これは私たちの業務に悪影響を及ぼすだろう。例えば、近年、2018年や2019年を含め、米国政府は何度も閉店しており、米国食品·医薬品局や米国証券取引委員会などのある規制機関は、キー従業員を休暇させ、キー活動を停止せざるを得ない。現在行われている新冠肺炎突発公共衛生事件の期間中、アメリカ食品と薬物管理局はその申請に対する規定検査を完成できないため、多くの会社は完全な返信を受け取ることを発表した。政府が長期的に停止している場合、または世界的な健康問題がFDAや他の規制機関の定期検査、審査、または他の規制活動を阻害した場合、FDAが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に大きな悪影響を及ぼす可能性がある。また、当社の上場企業としての運営では、将来的に政府の閉鎖や遅延が公開市場に参入し、必要な資本を得る能力に影響を与える可能性があり、適切に業務を資本化して運営を継続しています。
EUの薬品マーケティングと清算法規は私たちのヨーロッパ加盟国の製品マーケティングと保証能力に重大な影響を与えるかもしれない。
私たちはアメリカと選定された外国司法管轄区で私たちの候補製品を販売することを承認することを求めるつもりです。もし私たちの候補製品が1つ以上の外国司法管轄区で承認されたら、私たちはこれらの管轄区域の規則によって制限されるだろう。一部の外国国家、特にEU諸国では、薬品の価格設定は政府のコントロールと他の市場によって規制されており、これは私たちの候補製品の定価と使用に圧力をかける可能性がある。これらの国では、候補製品の市場承認を得た後、政府当局との定価交渉にかなりの時間がかかる可能性がある。また、私たちの候補製品に対する市場の受け入れと販売は、私たちの候補製品が十分な保証範囲と第三者支払者の精算を持っているかどうかに大きく依存し、既存と将来の医療改革措置の影響を受ける可能性がある。
米国のAKS禁止と非常に類似しており、処方、提案、裏書き、購入、供給、医療製品の注文または使用を誘導または奨励するために医師に福祉または利点を提供することはEUでも禁止されている。医師への福祉または利益の提供はEU加盟国の国家反賄賂法や他の法律によって管轄され、連合王国での業務は連合王国“2010年反賄賂法”を含む連合王国関連法律によって拘束される。このような法律に違反することは巨額の罰金と監禁につながるかもしれない。
いくつかの連合会員国で医者に支払われた費用は公開されなければならない。また、医師との合意は、通常、医師の雇用主、医師の主管専門組織、および/またはEU加盟国の監督当局に事前に通知し、承認しなければならない。これらの要件は、EU加盟国に適用される国家法律、業界規則、または専門行為規則に規定されている。このような要求を守らないことは、名声のリスク、公開非難、行政処罰、罰金、または監禁につながる可能性がある。
また、多くの外国の国では、ヨーロッパ経済区(European Economic Area、略称EEA)を含み、薬物の提案価格は合法的に発売されるために承認されなければならない。各国の薬品定価と精算に対する要求は大きく異なる。例えば、EUは、その加盟国に様々な選択を提供し、その国の健康保険制度が補償を提供する医療製品の範囲を制限し、人が使用する医療製品の価格を制御する。複数のEU加盟国が使用する参考価格と平行分配、または低価格と高価格の間で裁定を行う-
価格設定された会員国は、もっと安くすることができる。加盟国は医薬製品の具体的な価格を承認することができ、医薬製品を市場に投入する会社の収益力に対して直接或いは間接的に制御制度をとることもできる。ある国では、私たちは臨床研究や他の研究を行う必要があるかもしれません。私たちの任意の候補製品の費用効果を他の利用可能な治療法と比較して、精算または価格設定の承認を得たり維持したりする必要があります。生物製薬製品に対して価格制御や精算制限を実施することを保証できない国は、私たちのいかなる製品に対しても有利な精算と定価手配を許可する。歴史的に見ると、EUで発売された製品はアメリカの価格構造に従わず、通常価格ははるかに低くなることが多い。第三者支払者や主管当局が割引を公表することは、公布国や他の国の価格や補償レベルにさらなる圧力を与える可能性がある。もし価格設定が満足できないレベルに設定されている場合、あるいは私たちの製品が精算できない場合、範囲や金額が限られていれば、私たちの販売収入およびこれらの国/地域における私たちの候補製品の潜在的な収益力はマイナスの影響を受けるだろう。また、現在ロシアとウクライナの間の紛争がこの地域に拡大すれば、コスト上昇やEUや他の管轄区域候補製品の原材料や製造槽の減少という問題に直面する可能性がある。
私たちは、変化するグローバルデータ保護法律や法規を遵守しようと努力する過程で巨額のコストが生じる可能性があり、私たちがこのような法律や法規を遵守できないことは、私たちの業務と運営を損なう可能性があると考えられています。
法律で保護されている健康情報またはPHI、個人識別情報、知的財産権、および独自のビジネス情報を含む敏感なデータを収集、保存、処理、送信します。私たちが私たちの業務を拡大することを求めているにつれて、私たちはますます多くの州、連邦と外国の法律、法規と標準、契約義務の制約を受けています。これらの法律、法規、標準は私たちの管轄区域内の敏感で個人情報の収集、使用、保存、安全、開示、移転、その他の処理に関連しています。多くの場合、これらの法律、法規、基準は第三者取引だけでなく、当社の子会社と、私たちとビジネス関係にある他の当事者との間の情報伝達にも適用されます。これらの法律、法規、および基準は、時間の経過および司法管轄区域によって異なるように解釈され、適用される可能性があり、それらの解釈および適用は、私たちの業務、財務状況、および運営結果に実質的な悪影響を及ぼす可能性がある。世界のデータプライバシー、データセキュリティ、データ伝送の規制枠組みは急速に変化しており、人々はますますプライバシーとデータ保護問題に注目しており、これは私たちの業務に影響を与える可能性があるため、予測可能な未来には、基準の解釈と実施および法執行のやり方は依然として不確定である可能性がある。これらの法律および法規を遵守しないことは、罰金、監禁会社の役人と大衆の非難、影響を受けた個人が損害賠償を要求し、私たちの名声と名誉を損なうことを含む、私たちに対する法執行行動を招く可能性があり、これらはいずれも私たちの業務に実質的な悪影響を及ぼす可能性がある。
しかも、私たちが運営している多くの州は敏感で個人情報のプライバシーと安全を保護する法律を持っている。いくつかの州の法律は、敏感かつ個人情報の面で、連邦、国際、または他の州の法律よりも厳格または範囲が広いか、またはより多くの個人権利を提供する可能性があり、これらの法律は互いに異なる可能性があり、これはコンプライアンス作業を複雑化させる可能性がある。州法がHIPAAよりも保護的なところでは,HIPAAに加えて,我々が受けている州法を守らなければならない。場合によっては、このようなより厳しい州法に適合するために、私たちが計画した操作と手続きを修正する必要があるかもしれない。また,複数の州からの個人の敏感さや個人情報を扱う場合には,任意の情報に適用される最も厳しい州法を遵守する必要があることが分かるかもしれない.カリフォルニアは最近、“カリフォルニア消費者プライバシー法”(California Consumer Privacy Act、略称CCPA)を公布し、カリフォルニアの消費者のために新しいプライバシー権(法律で定義されているような)を創造し、消費者または家庭の個人データを処理する実体に対してより多くのプライバシーと安全義務を規定した。CCPAは、カバーする会社に、そのデータ収集、使用、および共有アプローチに関するいくつかの開示を消費者に提供し、影響を受けたカリフォルニア住民に、特定の個人情報販売または移転から撤退することを選択する方法を提供する。CCPAは2020年1月1日に発効し、カリフォルニア州総検察長は2020年6月2日に最終法規を提出し、これらの法規は最終的に決定され、現在すでに発効している。カリフォルニア州総検察長は2020年7月1日から違反者に対する法執行行動を開始した。また、カリフォルニアの有権者は2020年11月3日にCPRAと略称する新しいカリフォルニアプライバシー法であるカリフォルニアプライバシー権法案を可決した。CPRAは2023年1月1日に発効, 個人情報の処理と保存に追加的な義務が生じる。CPRAに関連する発展を引き続き監視し、CPRAコンプライアンスに関連する追加コストと費用を予想する。他の4州では包括的なプライバシー法が成立しており,米国の他の州でも総合的なプライバシー立法の制定が考えられている.CCPAおよびCPRAには、HIPAAの下で公衆衛生施設の活動に関連するいくつかの例外が含まれているが、CCPA、CPRA、または他のこのような将来の法律、法規、および基準が私たちの業務に及ぼす可能性のある影響を決定することはできない。
我々の米国での業務は、医療および他の健康情報および他の個人情報のプライバシーおよびセキュリティに関する法律によって制約される可能性があるほか、EEAでの臨床試験が求められ、追加のヨーロッパデータプライバシー法律、法規、ガイドラインの制約を受ける可能性がある。もし私たちが臨床試験を行うか、私たちが行っているまたは未来の臨床試験で被験者を募集することを決定した場合、私たちは追加のプライバシー制限を受ける可能性があります。個人健康データを含むEU個人に関する個人データを収集、使用、記憶、開示、移転、または他の方法で処理し、2018年5月25日に施行されたEU一般データ保護法規(GDPR)によって制限される。GDPRの範囲は広く,個人データを扱う会社には多くの要求があり,要求を含めている
健康や他の敏感なデータの処理に関連して,個人データ関連個人の同意を得て,個人にデータ処理活動に関する情報を提供し,個人データの安全と機密性を保護する保障措置を実施し,データ漏洩の通知を提供し,第三者プロセッサを使用する際に何らかの措置をとる。GDPRはまた、米国を含むEU以外の国への個人データの移転に厳しいルールを実施し、データ保護当局が2000万ユーロや世界の年収の4%に達する可能性のある罰金を含むGDPR違反行為に巨額の罰金を科すことを許可している。GDPRはまた、データ主体と消費者協会に個人訴訟権利を与え、監督当局に苦情を提出し、司法救済を求め、GDPR違反による損害について賠償を受けることができる。さらに、GDPRは、国境を越えたデータ転送の制限を含む。GDPR[5月.]増す[d]我々の責任と責任は,我々が扱う個人データに関連しており,このような処理がGDPRによって制約されていれば,個別国で実施されるメカニズムを含めてGDPRの遵守を確保するための追加的なメカニズムを構築する必要があるかもしれない.GDPRを遵守することは厳格で時間のかかる過程となり、私たちの業務コストを増加させたり、私たちに業務やり方の変更を要求したりする可能性があり、私たちはこれらの努力をしたにもかかわらず、私たちは私たちのヨーロッパ活動に関連した罰金と処罰、訴訟、名声損害のリスクに直面するかもしれない。また、イギリスがEU離脱を決定したのは、一般にイギリスの離脱と呼ばれ、イギリスのデータ保護規制に不確実性をもたらした。特に,イギリスがEUを離れた以上,イギリスに出入りするデータ転送がどのように規制されるかは不明である。
特に,EU加盟国の国家法律は,GDPRの要求に応じてGDPRから部分的に逸脱する可能性のある国家法律を実施し,異なる国に異なる義務を課すように調整されているため,欧州経済圏は統一された法的環境では動作しないと予想される。また、遺伝子データの処理と譲渡に関連するため、GDPRは特に国家法律がより多く、より具体的な要求や制限を加えることを許可しているが、ヨーロッパの法律はこの分野で従来から大きな違いがあり、追加的な不確実性を招いている。また、英国が2020年1月31日にEUを正式に離脱することは、英国への影響は不確定であり、現時点でも予測できない。
ヨーロッパ経済区で臨床試験を開始すれば,欧州データ保護法に従って個人データをヨーロッパ経済区以外,特に米国に移行できるように十分な保障措置を維持しなければならない。私たちは、私たちが欧州プライバシー法で規定されているいかなる義務を遵守する努力が十分かどうかという不確実性に直面し続けると予想する。もし私たちがヨーロッパデータ保護機関の調査を受けたら、私たちは罰金と他の処罰に直面するかもしれない。欧州データ保護当局のこのような調査または告発は、私たちの既存の業務および新しい顧客または生物製薬パートナーを引き付け、維持する能力にマイナスの影響を与える可能性がある。私たちはまた、いくつかのデータ保護機関が現行法(GDPRを含む)を説明する際に彼らに課せられた現在(特に未来)のデータ保護義務による潜在的なリスクのため、ヨーロッパまたは多国籍顧客または生物製薬パートナーが、私たちの製品および解決策の継続を躊躇し、または拒否することに遭遇する可能性がある。これらの顧客や生物製薬パートナーも,任意のコンプライアンスの代替方法はコストが高すぎ,負担が重すぎ,法的に不確実すぎたり,その他の面で反感を抱いている可能性があるため,我々と商売をしないことにした。上記のいずれも、我々の業務、見通し、財務状況、および経営結果に実質的な損害を与える可能性がある。
国際業務を管理する追加の法律法規は、私たちの業務に否定的な影響や制限を与える可能性がある。
私たちがアメリカ以外での業務をさらに拡大すれば、私たちは各管轄区域で事業を展開することを計画している多くの法律と法規を遵守するために追加の資源を投入しなければならない。米国の“反海外腐敗防止法”は、いかなる米国の個人または企業がいかなる外国人官僚、政党または候補者に任意の価値のあるものを支払い、提供、許可、または提供することを禁止し、個人または企業が業務を獲得または保留するのを助けるために、任意の外国エンティティの任意の行為または決定に影響を与えることを目的としている。“海外腐敗防止法”はまた、米国に上場する証券会社にある会計条項を遵守することを要求しており、これらの条項は、企業(国際子会社を含む)のすべての取引の帳簿と記録を正確かつ公平に反映し、国際業務のために十分な内部会計制御システムを設計し、維持することを要求している。
“反海外腐敗法”を遵守することは高価で困難であり、特に腐敗は公認問題である国である。また、海外腐敗防止法は製薬業に特別な挑戦をもたらしており、多くの国では病院が政府によって運営されているため、医師や他の病院従業員は外国人官僚とみなされている。臨床試験やその他の仕事に関連して病院に支払われた何らかの金は、政府関係者に支払われた不正金と考えられ、“海外腐敗防止法”の法執行行動につながった。
様々な法律、法規、および行政命令はまた、国家安全目的のために秘密にされた情報および特定の製品およびこれらの製品に関連する技術データ、またはいくつかの非米国国民との共有を米国国外で使用および伝播することを制限する。もし私たちがアメリカ以外での業務を拡大すれば、これらの法律を遵守するためにより多くの資源を投入する必要があります。これらの法律は、私たちがアメリカ以外で特定の製品や候補製品を開発、製造、販売することを阻止するかもしれません。これは、私たちの成長潜在力を制限し、私たちの開発コストを増加させるかもしれません。
国際ビジネス慣行に関する法律を遵守しなければ、重大な民事·刑事罰を受け、政府契約の資格を一時停止または廃止する可能性がある。米国証券取引委員会は、発行者が海外腐敗防止法の会計規定に違反したため、発行者の米国取引所での証券取引を一時停止または禁止する可能性もある。
私たちはアメリカと外国のいくつかの反腐敗、反マネーロンダリング、輸出規制、制裁、その他の貿易法律法規の制約を受けている。このような法律法規に違反しても、私たちは深刻な結果に直面するだろう。
その他の事項を除いて、米国および外国の反腐敗、反マネーロンダリング、輸出規制、制裁およびその他の貿易法律法規は、総称して貿易法と呼ばれ、会社およびその従業員、代理人、臨床研究組織、法律顧問、会計士、コンサルタント、請負業者および他のパートナーの許可、約束、提供、提供、誘致、直接または間接的に公共または民間部門の受取人の腐敗または不当な支払い、または任意の他の価値のあるものを受け入れることを禁止する。貿易法違反は、巨額の刑事罰金と民事処罰、監禁、貿易特権の喪失、資格取り消し、税収の再評価、契約違反と詐欺訴訟、名誉損害、その他の結果を招くことができる。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。私たちはまた、アメリカ以外での私たちの活動がタイムリーに増加すると予想している。私たちは、第三者を招いて臨床試験および/または必要な許可、許可、特許登録、および他の規制承認を得ることを計画しており、私たちが明確な権限を持っていなくても、私たちの人員、代理またはパートナーの腐敗や他の不正活動を事前に知っていても、私たちは責任を要求される可能性がある。
管理業務と運営に関するリスク
公衆衛生の発生、流行病と流行病、例えば行われている新冠肺炎の大流行は、著者らの臨床前研究と臨床試験を含む著者らの業務に不利な影響を与える可能性がある。
公衆衛生暴発、流行病、流行病は私たちの業務に悪影響を及ぼすかもしれない。例えば、2019年12月に武漢で発見された新型コロナウイルス株の重症急性呼吸症候群コロナウイルス2型、あるいはSARS-CoV-2と呼ばれ、中国が2019年12月に発見し、それによって引き起こされた疾病はすでに世界的な大流行になっている。この疾患は,最近伝播が加速しているより伝播しやすい新冠肺炎変種を含め,我々が業務を展開している地域で伝播し続けている。疫病と政府が取った対応措置は直接と間接的に世界各地の商業と商業に重大な影響を与えた:労働者の不足、サプライチェーンの中断、施設と生産の一時停止、医療サービスと用品などのある商品とサービスに対する需要は急増し、旅行などの他の商品とサービスに対する需要は低下した。本報告日までに、我々が現在行っている研究開発活動は、新たに出現した新冠肺炎変異株の影響を含む今回の疫病の実質的な影響を受けていないが、最近より伝播しやすい変異株の著者らの業務のある地域での伝播速度の加速、新冠肺炎ワクチンの供給と使用、および未来のいかなる流行病も含まれているが、これらは依然として不確定かつ予測困難であるが、私たちは引き続き妨害を受け、研究開発、商業化スケジュール、結果に深刻な影響を与える可能性があるが、これらに限定されない
•患者を臨床試験に参加させるのは遅延したり困難です
•臨床サイト起動の遅延または困難は、臨床サイト調査員と臨床サイトスタッフを募集する上での困難を含む
•医療資源を臨床試験の進行から移し,われわれの臨床試験場所である病院や臨床試験を支援してくれた病院スタッフを他の場所に移すことを含む
•連邦、州または外国政府、雇用主および他の人が強要または提案した旅行制限または臨床試験被験者の訪問および研究プログラムの中断(例えば、法律、法規または機関政策によって不必要とされるプログラム)により、被験者のデータおよび臨床研究終点の完全性に影響を与える可能性があり、患者が試験場所に行くことができない、あるいは予定の研究訪問を完了することができず、臨床試験地点のデータ監視などの重要な臨床試験活動の中断を招く
•FDAまたは他の規制機関の動作中断または遅延は、審査および承認スケジュールに影響を与える可能性がある
•人員不足、生産減速または停止、および交付システムの中断のため、私たちの契約製造組織から私たちの候補製品の供給を受けることを中断または遅延した
•私たちが契約した研究機関の制限や限られた操作のため、臨床前研究を中断した
•私たちは持続的な新冠肺炎の大流行の影響によって引き起こされる可能性のある予見できないコスト、仕事のコストを緩和することを含む
•世界的な信用と金融市場が悪化し、これは私たちが外部融資を受けて私たちの業務と資本支出に資金を提供する能力を制限する可能性がある
•現在の市場状況や不利な投資業績により投資の清算が困難であることを含む投資に関するリスク
•従業員資源の制限、そうでなければ、従業員またはその家族が病気になったか、または従業員が大勢との接触を避けることを望んでいることを含む、私たちの研究開発活動に集中するだろう
•臨床試験を実行する契約研究組織およびスポンサーが、候補製品または他の方法で参加する臨床試験を提供するパートナーを提供することを含む、我々のサービスプロバイダおよびパートナーに影響を与える上記のタイプの中断または制限。
新冠肺炎の大流行が始まって以来、新冠肺炎のいくつかのワクチンはすでにアメリカ食品と薬物管理局の緊急使用許可を得ており、その中のいくつかのワクチンは後に発売許可を得た。未来にはより多くのワクチンが許可されたり承認されるかもしれない。これにより生じるワクチンの需要や1950年の“国防生産法案”や同様の外国立法によって徴用された製造施設や材料の可能性は,我々の臨床試験に必要な製品が材料や製造槽を得ることを困難にする可能性があり,これらの試験の遅延を招く可能性がある。また,新冠肺炎の影響により,他の生物製薬会社の普通株の取引価格も高度に変動している。新冠肺炎の疫病は引き続き変化しており、最近の新冠肺炎を含むもっと伝播性のある変種は著者らが業務を展開している地区で加速的に伝播し、その持続と長期的な影響は予測しにくい。大流行の負の影響は軽減されているようであり,ワクチンは米国で広く配布され,配布され続けているが,多くの他の国ではワクチンが全く開発されたり配布されたりしていないため,新冠肺炎ウイルスの広範な影響が見られ続ける可能性がある。一般経済体への負の経済的影響、それによる株式市場の変動、および多くの業界、労働力、小売業者への負の影響が続いている。また,すでに多くの新冠肺炎ウイルスの変異株が出現しており,我々の既存のワクチンはこれらの変異株を防ぐことができない可能性や,ワクチン接種率の停滞への懸念があり,最近では新冠肺炎ウイルスが伝播しやすい変異株が我々の運営地域で加速的に伝播している, そして大流行前の活動の回復と消費者自信レベルの進展を阻害し続ける関連要素。現在の大流行および未来の任意の潜在的な再発または爆発が私たちの業務、臨床前研究および臨床試験に与える影響の程度は未来の事態の発展に依存し、これらの事態の発展は高度な不確実性を有し、例えば疾病の最終的な地理的伝播と分布、大流行の持続時間、アメリカと他の国の旅行制限と社会距離、企業閉鎖または商業中断、新冠肺炎ウイルスに対抗するための治療およびワクチンの成功、および米国および他の国が疾患の診断、制御および治療のために取った他の行動の有効性を予測することができない。もし私たちが私たちと接触している任意の第三者が閉鎖や他の業務中断に遭遇した場合、私たちが現在計画している方法とスケジュールに従って業務および開発活動を展開する能力は実質的で否定的な影響を受ける可能性がある。このような中断や遅延が、私たちの業務、運営結果、財務資源の取得、および私たちの財務状況に実質的な悪影響を与えないという保証はありません。
もし私たちが重要な管理職を失った場合、あるいは私たちがより多くの高技能者を募集できなければ、私たちの業務戦略を実行する能力が損なわれ、市場シェアや市場シェアを失い、私たちの競争力を低下させる可能性があります。
競争の激しい生物製薬業界における競争能力は私たちが高い素質管理、科学と医療人員を誘致し、維持する能力にかかっている。私たちは、最高経営責任者サチブ·イズラム、フランク·ペリル最高財務責任者、Bhavesh Ashar、最高経営責任者Badreddin Edris、L.Mary Smith、最高医療責任者James Cassidyを含む、私たちの管理者、科学者、医療従事者に高度に依存している。私たちの役員、他の重要な従業員、他の科学と医療コンサルタントのサービスを失うこと、そして私たちはこの人たちのために適切な代替者を見つけることができず、私たちの業務を損なうかもしれません。
私たちの業界は技術人材に対する競争が非常に激しく、私たちが受け入れられる条件で適時あるいは根本的に素質の高い人材を採用し、維持する能力を制限するかもしれない。価値のある従業員をわが社に引き付けるために、給料や現金インセンティブのほか、時間の経過とともに付与された株式インセンティブを提供しており、当時の会社の状況や市場状況に応じて他の形態のインセンティブを時々考慮しています。時間が経つにつれて、制限された株式単位および奨励および株式オプションの従業員に対する価値は、私たちの株価変動の大きな影響を受ける可能性があり、これらの変動は私たちの制御範囲を超え、他社が提供するより利益のあるオファーを相殺するのにいつでも十分ではない可能性がある。私たちは価値のある従業員を引き留めるために努力しているにもかかわらず、私たちの管理、科学、開発チームのメンバーは勝手な従業員であり、彼らは短時間で私たちとの雇用関係を終了するかもしれない。私たちはこのような個人や私たちの他のどんな従業員のためにも“キーパーソン”保険証書を維持しない。私たちの計画の段階的な拡大と業務の計画を考慮すると、私たちの成功はまた、組織全体の高技能初級、中級、上級者を引き続き誘致、維持、激励できるかどうかにかかっている。
私たちの業務はサイバーセキュリティの脅威によって否定的な影響を受けるかもしれない。
ネットワーク攻撃または同様のイベントは、情報の盗難、データの破損、運営の中断、私たちの名声の損傷、または経済的損失をもたらす可能性があります。私たちはますます情報技術システムやインフラに依存しています
私たちの業務を運営するためのモバイル技術です当社の技術、システム、ネットワークまたは他の固有情報、ならびに当社のプロバイダ、プロバイダ、および他のビジネスパートナーの技術、システム、ネットワーク、または他の固有情報は、ネットワーク攻撃または情報セキュリティホールの目標となる可能性があり、これらの攻撃または情報セキュリティホールは、許可されていない発行、収集、監視、誤用、損失、または独自情報および他の情報を破壊することができ、または他の方法で私たちのビジネス運営を中断させる可能性があります。ネットワーク攻撃はますます複雑になっており、監視のようないくつかのネットワークイベントは、長い間発見されていない可能性があり、重要なシステムが機密または他の保護された情報を中断または許可していないことをもたらす可能性がある。これらのイベントは、救済措置、業務損失、運営中断、名声被害、または潜在的な責任によって財務損失を招く可能性がある。私たちのネットワークセキュリティリスクを防ぐシステムと保険カバー範囲はまだ十分ではないかもしれない。さらに、サイバー攻撃の継続的な発展に伴い、私たちは、私たちの保護措置を修正したり強化したり、ネットワーク攻撃を受けやすい脆弱性を調査し、修復するために、多くの追加資源を必要とするかもしれません。
私たちはますます重要で複雑で依存している情報技術(IT)システムとデータに依存して私たちの業務を運営しています。この技術の任意の障害、不足、中断、またはセキュリティホールは、セキュリティ攻撃、イベント、および/または脆弱性を含み、トラフィックを効率的に運営する能力を損なう可能性があります。
我々はITや業務インフラの重要な部分を第三者プロバイダにアウトソーシングしており,現在これらのプロバイダを用いて重要なITや業務サービスを提供している.したがって,我々は関連するネットワークやシステムのネットワークセキュリティ攻撃やイベントを受けやすく,それらが直接管理されているか,我々と契約した第三者が管理しているかにかかわらず,我々は将来このようなネットワークセキュリティの脅威や攻撃を経験している可能性がある.新冠肺炎の大流行を背景に、仮想と遠隔作業がより広く使用されることや、安全性の低い家庭環境で働く従業員が敏感なデータにアクセスすることにより、このような脅威や攻撃のリスクが増加する。我々の動作方式は,業務の大部分に重要な遠隔コンポーネントを含めて持ち続けることが可能であり,この要因を我々のネットワークセキュリティリスク管理戦略に取り入れていきたい.さらに、第三者プロバイダへの私たちの依存により、将来的にITサービス利用可能性に関連する中断、遅延または中断を経験する可能性があり、これらの第三者プロバイダが経験したり、原因となる技術障害、自然災害、詐欺、またはセキュリティ攻撃を含む、我々が制御できない様々な要因による中断、遅延または中断を経験することができる。これらの第三者プロバイダが提供するサービス中断は,我々がキータスクを実行する能力に影響を与える可能性がある.
世界的な製薬会社として、私たちのシステムはしばしばサイバー攻撃を受けている。いくつかの攻撃の性質のため、それらは一定期間検出されないリスクを維持する可能性がある。データや情報技術の保護に投資しているが,サービス中断やセキュリティホール(例えば,恐喝ソフトウェア攻撃)を防ぐことができない可能性がある.私たちのシステムのこのような中断または破壊は、私たちのビジネス運営に悪影響を及ぼす可能性があり、および/または、キーまたは敏感な機密情報または知的財産権の損失をもたらし、財務、法律、ビジネス、および名声の損害をもたらす可能性があります。私たちはネットワーク責任保険を維持している;しかし、この保険は、私たちのシステムの中断または破壊によって引き起こされる可能性のある財務、法律、業務、または名声損失をカバーするのに十分ではないかもしれない。
セキュリティ技術および組織対策が実装されているにもかかわらず、我々の内部コンピュータシステムおよび我々と契約した第三者のコンピュータシステムは、セキュリティイベント、侵入および/または攻撃(例えば、恐喝ソフトウェア、コンピュータウイルス、ワームおよび他の破壊または破壊ソフトウェア)、不正アクセス、自然災害、テロ、戦争、および電気通信および電気故障の破壊を受けやすい。システム障害、事故または安全攻撃および/または当社システムの破壊は、運営中断および/または私たちの臨床および商業化活動および業務運営の実質的な中断をもたらす可能性があり、また、修復のために大量の資源が必要となる可能性がある。臨床試験データの紛失或いは完全性は著者らの監督管理の審査作業の遅延を招く可能性があり、そして著者らのデータの回復或いは複製のコストを著しく増加させる。任意のシステム中断、セキュリティイベント、またはセキュリティホールが、私たちのデータまたはアプリケーションの損失、破損または完全性の損傷、または機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの製品研究、開発、および商業化努力は中断または遅延する可能性があります。しかも、私たちはこのような事件に関連したどんな損失も補償するのに十分な保険範囲を持っていないかもしれない。
私たちは、会社の情報システムおよびネットワークで維持されている情報(私たち従業員の個人情報を含む)の流用、誤用、漏洩、改ざん、または故意または意外な開示または損失によるリスクに直面する可能性があります。さらに、外部の当事者は、私たちまたは私たちのサプライヤーのシステムを浸透させたり、私たちの人員またはサプライヤーの人員に敏感な情報を開示させて、私たちのデータにアクセスするように詐欺的に誘導しようとするかもしれません。他の会社のように、私たちのデータとシステムは悪意のあるコードやウイルス、他のセキュリティ事件、侵入、攻撃を含む脅威にさらされることがある。時間が経つにつれて、このような脅威の数と複雑さは増加している。私たちは上記のいくつかの事件を経験したにもかかわらず、今まで、それらは私たちの運営に実質的な影響を与えていない。それにもかかわらず、将来的に上記のいかなる事件が発生しても、私たちの業務運営を混乱させ、可能な罰金と処罰、損害クレーム、株主訴訟を含む法執行行動または責任を引き起こす可能性がある。
安全事件はまたサプライチェーン攻撃を含む可能性があり、成功すれば、私たちの製品または候補薬物の生産遅延を招く可能性がある。私たちの主要業務パートナーは似たようなリスクに直面しており、彼らのシステムのいかなるセキュリティホールも私たちのセキュリティ態勢に悪影響を及ぼす可能性がある。さらに、クラウド技術をますます使用することは、これらおよび他の運営リスクを増加させる可能性があるが、クラウド技術サービスプロバイダは、そのシステムを十分に保護することができず、ネットワーク攻撃を防止することができず、私たちの運営を混乱させ、機密または独自の情報を流用、腐敗、または失わせる可能性がある。
最後に、私たちがビジネス活動を増やすにつれて、私たちのブランドはより広く知られ、認められるようになり、私たちは悪意のある第三者のより魅力的な目標になるかもしれない。もし私たちまたは第三者サプライヤーの安全が重大な破壊を受けたら、市場の私たちの安全措置の有効性に対する見方が損なわれる可能性があり、私たちは業務を失う可能性があり、私たちの名声と信頼性は損なわれる可能性がある。私たちは、情報資産および/または情報システムを修復または交換するために、大量の資金および他の資源を必要とするかもしれない。私たちはまた、第三者プロバイダおよび/または製品を高いコストで交換することを要求されるかもしれない。これらの事件の発生を防止するためのシステムや制御を開発·維持し、脅威を識別·緩和する流れもあるが、これらのシステム、制御、プロセスの開発と保守はコストが高く、技術の変化とセキュリティ対策の克服に伴う努力がより複雑になり、継続的に監視·更新する必要がある。しかも、私たちが努力したにもかかわらず、このような事件が発生する可能性を完全になくすことはできない。第三者行為、従業員の不注意および/またはエラー、汚職、欠陥、または私たちのデータの機密性、完全性、または利用可能性を損なうことによって、私たちのセキュリティ対策に違反する可能性があります:
•私たちの名声やブランドを深刻に損なうか、または情報の機密性、完全性、可用性を保護するために取られた技術および組織措置の全体的な市場認知に重大な悪影響を及ぼす
•個人および/または集団訴訟は、私たちに不利な経済判決をもたらす可能性があり、法的費用および費用を招く可能性がある
•法律または法執行行動を規制することは、罰金および/または処罰を招き、法的費用およびコストを発生させる可能性がある;および/または
•業務中断またはセキュリティイベントおよび/または違反に対応することに関連する追加のコスト、例えば、調査および修復コスト、個人および/またはデータ所有者に違反通知を提供するコスト、法的費用、任意の追加の詐欺またはネットワーク検出活動のコスト、または長期システム中断または閉鎖のコスト。
このような事件のいずれも、私たちの業務と運営結果に実質的な悪影響を及ぼす可能性がある。
当社の従業員、独立請負業者、コンサルタント、学術協力者、パートナーおよびサプライヤーは、法規基準および要件を遵守しないことを含む不適切な行為または他の不適切な活動に従事する可能性があります。
私たちは、従業員、独立請負業者、コンサルタント、学術協力者、パートナー、サプライヤーが従業員詐欺や他の不正活動を行うリスクに直面している。これらの当事者の不正行為には、故意、無謀、および/または不注意な行為が含まれている可能性があり、FDA、EMAおよび同様の外国規制機関の法律に準拠できず、FDA、EMAおよび類似の外国規制機関に真実、完全かつ正確な情報を提供し、私たちが制定した製造基準を遵守し、米国の医療詐欺および乱用法律および同様の外国詐欺不正行為法律を遵守し、または財務情報またはデータを正確に報告したり、許可されていない活動を開示したりすることができる。もし私たちの候補製品がFDAの承認を得て、アメリカでこれらの製品を商業化し始めたら、私たちのこのような法律の下での潜在的なリスクは著しく増加し、私たちはこのような法律を遵守することに関連するコストも増加する可能性がある。これらの法律は私たちの現在の主要な研究者と患者の活動、提案と未来の販売、マーケティングと教育計画に影響を与える可能性がある。私たちはビジネス行為と道徳的規範を通過しましたが、私たちの従業員、独立請負業者、コンサルタント、学術協力者、パートナー、およびサプライヤーの不当な行為を常に識別し、阻止することができるわけではありません。私たちがこのような活動を発見し、防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御できないか、またはこれらの法律や法規を遵守できないことによる政府の調査または他の行動または訴訟から私たちを保護することができないかもしれません。もし私たちにどんな行動を取っても、私たちが私たちの権利を弁護したり、維持したりすることに成功しなかったら、これらの行動は民事、刑事、そして行政処罰、損害賠償、罰金の適用につながるかもしれない, これらの法律を遵守しない疑惑、契約損害、名声損害、利益減少、および将来の収益減少、および私たちの業務縮小を解決するために、会社の誠実な合意または他の合意の制約を受けた場合、監禁、返還、政府医療計画への参加、追加の報告義務、監督から除外される可能性がある。
もし私たちが環境、健康、安全の法律法規を守らなければ、罰金や処罰を受けたり、私たちの業務を損なう可能性のあるコストが発生するかもしれません。
私たちは危険材料と廃棄物の処理、使用、貯蔵、処理、処分に関する法律と法規を含む多くの環境、健康、安全な法律と法規によって制約されている。私たちの開発活動は生物と危険材料を使用し、危険廃棄物製品を発生させる可能性がある。これらの材料の汚染や傷害リスクを除去することはできません。これは、私たちの商業化努力、研究開発努力、業務運営の中断、高価な清掃作業の環境破壊を招き、これらの材料と指定廃棄物の使用、貯蔵、処理、処理を管理する適用法律と法規に規定された責任を招く可能性があります。我々の第三者メーカーがこれらの材料を処理·処分する際に使用するセキュリティプログラムは,全体的にこれらの法律法規に規定されている基準に適合していると信じているが,状況が確かにそうである保証はなく,これらの材料の意外な汚染や傷害のリスクを解消することもできない。この場合、私たちはそれによって生じるいかなる損害に対しても責任を負うことができ、このような責任は私たちの資源範囲を超える可能性があり、州、連邦、または他の適用機関は、いくつかの材料の使用を制限し、および/または私たちの業務運営を中断するかもしれない。
また,環境法律法規は複雑で変化が頻繁であり,より厳しくなる傾向にある。私たちはこのような変化の影響を予測することもできないし、私たちの未来のコンプライアンス状況を決定することもできない。また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。このような法律法規を遵守しないことはまた巨額の罰金、処罰、または他の制裁につながる可能性がある。
危険材料の使用や他の作業に関連するダメージにより生じる可能性のある費用や支出を支払うための労働者補償保険を維持しているが、潜在的な責任に対応するには不十分である可能性がある。私たちは生物学的または危険な廃棄物の暴露または汚染による損害と罰金を含む特定の生物廃棄物または危険廃棄物保険、労働者補償または財産と死傷者および一般責任保険を保証しない。
税法の変化は私たちの業務と財政状況に悪影響を及ぼすかもしれない。
米国連邦、州、地方所得税に関する規則は立法過程に参加する人員およびアメリカ国税局とアメリカ財務省の審査を受け続けている。税法の変化(これらの変化は追跡力を持つ可能性がある)は、私たちまたは私たちの普通株の所有者に悪影響を及ぼすかもしれない。近年、このような変化は多く発生しており、未来も変化し続けるかもしれない。将来の税法の変化は、私たちの業務、キャッシュフロー、財務状況、または運営結果に実質的な悪影響を及ぼす可能性があります。株主は税法の潜在的な変化が私たちの普通株に投資する影響について彼らの法律と税務顧問に相談しなければならない。
私たちが純営業損失の繰越と特定の税収控除を使う能力は制限される可能性があります。
2022年12月31日まで、私たちの連邦、州と都市の純営業損失はそれぞれ3.683億ドル、2.609億ドル、370万ドルに転換し、将来の課税収入を減らすことができる。2018年から2022年にかけて発生した連邦純営業損失3.64億ドルは、完全利用まで一定期間、課税収入の80%を相殺するように制限される。2017年に報告された連邦純運営損失は430万ドルで、州と市の純運営損失の繰越は2040年まで異なる日に満期となる。私たちは未来の税金負担を相殺するために2270万ドルの連邦税金免除を持っている。このような税金の繰越免除は2038年に始まる様々な日に満了されるだろう。
1986年の国税法または改正された国税法第382条と383条によると、私たちの所有権の変化は、将来の課税所得額を相殺するために毎年使用できる純営業損失の繰越と税収控除の金額を制限する可能性があります。この制限は一般的にわが社の所有権が三年間で累積的に50%を超える場合に適用されます。このような制限は、純営業損失の繰越と税収控除の満期までにそれらを利用する能力を大幅に低下させる可能性がある。守則第382及び383条によると、当社設立以来発生した私募その他の取引、及び当社の初公募により、このような所有権変更がトリガとなる可能性があります。このような制限は、初公募、以前の私募、私たちの既存株主が私たちの普通株を売却したこと、または私たちが追加的に私たちの普通株を売却したことによっても、今後数年間の経営業績に重大な悪影響を及ぼす可能性があります。一般的に、現行法によると、2017年12月31日以降に発生した連邦純営業損失は満期の影響を受けず、これまでの納税年度にさかのぼることもできません。しかし、コロナウイルス援助、救済、経済安全法案、またはCARE法案は、2018年、2019年、2020年に発生する純営業損失の80%の課税所得額制限を停止しており、これらの損失が特殊な5年間の繰り越し期間または2018、2019または2020納税年度に枯渇することを前提としている。また、上述したように、2020年12月31日以降の納税年度については、CARE法案の条項は適用されなくなり、このような連邦純営業損失の控除額は、今後いずれの納税年度の課税所得額の80%に制限される。
不利なグローバル経済状況は、私たちの業務、財務状況、または経営結果に悪影響を及ぼす可能性がある。
私たちの経営業績は世界経済と世界金融市場の全体的な状況の悪影響を受ける可能性があります。われわれの将来の一部の臨床試験は米国以外で行われる可能性があり,不利な経済条件によりドルが弱くなり,これらの臨床試験の操作コストが高くなる。また、現在のロシア-ウクライナ紛争については、ウクライナやロシアでは臨床試験場所や業務は何もありませんが、現在の紛争がこの地域に拡大し続けていれば、米国や国際社会に経済制裁を増大させ、環境規制に加えて、臨床試験に必要な製品の調達やいくつかの材料の使用やスロットマシンの製造能力を制限する可能性があり、これらの試験の遅延を招く可能性があります。また、最近の世界金融危機は資本·信用市場の極度な変動と中断を招き、近年、米国や海外の経済不確実性の増加が観察されている。深刻または長期的な経済低迷(インフレや政治的暴力や混乱による不確実性を含む)は、必要に応じて許容可能な条件で追加資本を調達する能力の低下(あれば)を含む様々なリスクを私たちの業務にもたらす可能性がある。疲弊したり衰退したりする経済や国際貿易紛争も私たちのサプライヤーに圧力を与える可能性があり、その中のいくつかのサプライヤーはアメリカ以外に位置し、更新不足を含む供給中断を招く可能性がある。上記のいずれも私たちの業務を損なう可能性があり、現在の経済気候や金融市場の状況が私たちの業務に悪影響を及ぼす可能性のあるすべての方法を予見することはできません。
環境、社会およびガバナンスまたはESG、政策および実践に対するますます厳格な審査および政府の期待の変化は、追加のコストを発生させたり、追加のリスクに直面させたりする可能性がある。
投資家、政府、非政府組織は企業のESGやり方に対する大衆の関心と審査がますます多くなっている。私たちのESG実践は、私たちのすべての株主の基準に適合していないかもしれません。提唱団体は、さらなる変更を要求する動きを開始するかもしれません。関連する予想に応えられなかったか、または対応できなかったと考えられることは、私たちの業務や名声に損害を与え、私たちの証券の市場価格に悪影響を及ぼす可能性がある。新しい政府条例は、新しい条例および新しいまたはより厳しいESG監視および開示形態をもたらす可能性があり、これは、持続可能な開発イニシアティブの支出増加をもたらす可能性がある。
企業の財務状況と追加資本需要に関連するリスク
私たちの将来の赤字額は不確定であり、私たちの四半期と年度の経営業績は大幅に変動する可能性があり、あるいは投資家や証券アナリストの予想を下回る可能性があり、どれも私たちの株価変動や下落を招く可能性がある。
私たちの四半期と年度経営業績は未来に各種の要素によって大幅に変動する可能性があり、その中の多くの要素は私たちがコントロールできないもので、以下の要素を含む予測が難しいかもしれない
•私たちの候補製品または競争候補製品の臨床試験のタイミングおよび成功または失敗、または私たちの競争相手またはパートナー間の統合を含む業界競争構造における任意の他の変化;
•私たちは臨床試験の被験者の能力と、このような努力の困難によるいかなる遅延も募集して維持することに成功した
•私たちの候補製品のために市場の承認を得る能力と、私たちがこのような承認を得る可能性のある時間と範囲
•私たちの候補製品に関する研究や開発活動の時間、コスト、投資レベルは時々変化する可能性がある
•私たちの候補製品を製造するコストは、生産数量と私たちが製造業者と合意した条項によって変化するかもしれない
•私たちは合格した人材を引きつけ、採用し、維持する能力を持っている
•私たちはより多くの候補製品を開発するための支出が発生するか起こりうる
•もし私たちの候補製品が承認されれば、彼らの需要レベルに大きな差があるかもしれない
•規制の承認を得る前と後に、候補製品の商業化を支援する努力に投資する時間とレベル
•私たちの候補製品に関連するリスク/収益プロファイル、コストおよび補償政策(承認されれば)、ならびに私たちの候補製品と競争する既存および潜在的な未来療法;および
•未来の会計宣言や私たちの会計政策の変化。
これらの要因の累積影響は我々の四半期や年度経営業績に大きな変動と予測不可能を招く可能性がある。したがって、異なる時期に私たちの経営業績を比較することは意味がないかもしれない。このような変化性および予測不可能性はまた、業界や金融アナリスト、または投資家の任意の時期に対する期待を満たすことができない可能性がある。もし私たちの収入や経営業績がアナリストや投資家の予想よりも低い場合、または私たちが市場に提供するいかなる予測よりも低い場合、または私たちが市場に提供する予測がアナリストや投資家の予想よりも低い場合、私たちの普通株の価格は大幅に下落する可能性がある。私たちが提供する可能性のある以前の公開声明の指導を満たしていても、このような株価下落は起こる可能性がある。
普通株関連リスク
私たちの普通株の活発な取引市場は持続できないかもしれない。
我々の普通株は2019年9月13日にナスダック世界ベスト市場で取引を開始した。私たちの普通株の取引歴史が限られていることから、私たちの株の活発な取引市場は持続できないかもしれません。これは私たち普通株の市場価格に下振れ圧力を与え、株主の株売却能力に影響を与える可能性があります。また、活発でない市場は、普通株を売却することで資金を調達する能力を弱める可能性があり、戦略的パートナーシップを達成したり、私たちの普通株を対価格で会社や製品を買収する能力を弱める可能性があります。
私たちの株価は変動し続けている可能性があり、株主は彼らの投資の全部または一部を損失するかもしれない。
私たちの普通株の取引価格はずっと高度に変動し続ける可能性があり、様々な要素の広範な変動を受ける可能性があり、その中のいくつかの要素は私たちがコントロールできないことであり、往々にして私たちの財務表現に関係がないか比例しない、限られた取引量を含む。この“リスク要因”の節と本報告の他の部分で議論されている要因のほかに、これらの要因には、以下のような要因が含まれている
•私たちが行っているナイルキャストの登録臨床試験の完成と、私たちの潜在的なミダミチニブの登録臨床試験の開始、登録、または結果
•私たちの候補製品に対する規制届出の任意の遅延、およびこのような届出に対する規制機関の適用に関するいかなる不利な発展、または不利とされる事態の発展は、FDAが発行した“届出拒否”手紙またはより多くの情報を提供することを要求する要求を含むが、これらに限定されない
•将来の臨床試験の不良結果や遅延
•私たちは臨床試験を開始し、臨床試験を開始しないか、既存の臨床試験を中止することにした
•私たちの製品候補または任意の未来の製品候補の規制承認を得ることができなかったことを含む不利な規制決定
•承認された臨床試験要件を含むが、これらに限定されないが、私たちの候補製品または任意の将来の候補製品に適用される法律または法規の変化
•医療支払い制度の構造を変え
•私たちのメーカーの不利な発展について
•私たちは承認された製品のために十分な製品を供給することができないし、受け入れ可能な価格でそうすることができない
•必要であれば、私たちは協力やパートナーシップを作ることができない
•もし承認されれば、私たちの候補製品は商業化されなかった
•重要な医療科学管理者の増減
•私たちの候補製品の使用に関連した意外な深刻な安全問題
•私たちや競争相手が提供する新製品やサービスを紹介します
•私たちの候補製品と競争できる他の候補製品の臨床試験結果
•私たちまたは私たちの競争相手は、重大な買収、戦略的パートナーシップ、合弁企業、または資本約束を発表します
•私たちは成長を効果的に管理することができます
•四半期の経営業績の実際または予想変化
•私たちの現金は
•投資界の推定と予測を達成できなかったか、または私たちが一般に提供する可能性のある推定および予測を達成できなかった
•私たちまたは私たちの業界に関する研究報告、または特定の製品候補報告、または証券アナリストのプラスまたは負の推薦または研究報告の撤回;
•同じ会社の市場予想が変化しています
•株式市場の全体像
•私たちまたは株主は将来私たちの普通株を売却します
•当社の普通株式出来高
•会計慣行の変化
•内部統制に力が入らない
•特許、訴訟事項、および私たちの技術のための特許保護を得る能力を含む、専有権に関する論争または他の発展;
•特許または株主訴訟を含む重大な訴訟;
•一般的な政治的·経済的条件
•他の事件や要素、その多くは私たちがコントロールできない。
また、持続的な新冠肺炎疫病と他のマクロ経済要素(ロシア-ウクライナ紛争を含む)、株式市場、特に生物製薬会社の市場は、極端な価格と出来高の変動を経験し、しかも往々にしてこれらの会社の経営業績と関係がないか比例しない。私たちの実際の経営業績にかかわらず、広範な市場と業界要素は私たちの普通株の市場価格にマイナス影響を与える可能性がある。もし私たちの普通株の市場価格が株主の購入価格を超えなければ、その株主は私たちの投資で何の見返りも達成できず、投資の一部または全部を損失する可能性がある。従来、証券集団訴訟は、会社証券の市場価格が変動した後に会社に提起されることが多かった。このような訴訟を提起すれば、巨額の費用と経営陣の注意と資源の移転を招く可能性があり、これは私たちの業務、経営業績、あるいは財務状況を損なうことになる。
上場企業としては、私たちの運営コストが高く、私たちの経営陣は、新しいコンプライアンスと既存のコンプライアンスを実施するために多くの時間を投入する必要があります。
上場企業として、私たちは巨額の法律、会計、その他の費用を招いた。私たちは、私たちの業務および財務状況に関する年間、四半期、および現在の報告書をアメリカ証券取引委員会に提出することを要求する取引法の報告要件を遵守しなければなりません。また、サバンズ-オキシリー法案および米国証券取引委員会とナスダックが後にサバンズ-オクスリ法案の条項を実施するために採択された規則は、有効な情報開示と財務制御の確立と維持を要求し、コーポレートガバナンスのやり方を変更することを含む上場企業に重大な要求を提出した。また、2010年7月には“ドッド·フランクウォール街改革·消費者保護法案”、あるいは“ドッド·フランク法案”が公布された。テレス·フランク法案には重要な会社管理と役員報酬に関する条項があり、米国証券取引委員会はこれらの分野で“報酬発言権”や代理アクセスのような追加的な規則を取ることを要求している。
株主急進主義、現在の政治環境、および現在の高度な政府介入と規制改革は、大量の新しい法規と開示義務を招く可能性があり、これは追加のコンプライアンスコストを招き、現在予見できない方法で業務を運営する方法に影響を与える可能性がある。
私たちの既存株主が公開市場で大量の普通株を売ることは私たちの株価を下落させる可能性があります。
もし私たちの既存株主が公開市場で私たちの普通株を大量に売却する意向を示したり、私たちの普通株の取引価格が低下する可能性があります。2022年12月31日まで、私たちは62,423,129株の普通株を発行しており、その中の893,713株は制限株であり、未来に帰属する必要がある。
2022年12月31日現在、普通株の約62.8%が役員、役員、5%を超える普通株を保有する保有者が実益で保有しており、証券法第144条の何らかの制限を受ける。
また、我々の既存持分補償計画によれば、各種ホームスケジュール及び証券法第144条及び第701条の規定により許容される範囲内で、我々の既存持分補償計画に基づいて、未償還オプション制限又は将来発行のために予約された普通株は、公開市場で販売する資格がある。これらの追加的な普通株が公開市場で販売されている場合、またはそれらが売却されると考えられている場合、私たちの普通株の取引価格は低下する可能性がある。また、2019年の株式オプションと配当インセンティブ計画によると、発行のために予約された普通株式数は毎年1月1日に自動的に増加し、その中で2020年1月1日が初めて増加し、2030年1月1日(2030年1月1日を含む)まで続き、前年の12月31日に発行された株式総数の5%や、取締役会が決定した少ない数の株が増加する。我々の取締役会が将来付与可能な株式数を毎年増加させないことを選択しない限り、私たちの株主は追加的な希釈を経験する可能性がある。
証券や業界アナリストが我々の業務に関する研究報告を発表しない場合、あるいは不正確または不利な研究報告を発表しなければ、私たちの株価や取引量は低下する可能性がある。
私たちの普通株の取引市場は、証券や業界アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存している。私たちの1人以上のアナリストを追跡して私たちの株式格付けを引き下げたり、私たちの業務に関する不正確または不利な研究報告を発表したりすれば、私たちの株価は下落するかもしれない。これらのアナリストのうちの1人以上がわが社への報道を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちの株に対する需要が減少する可能性があり、これは私たちの株価や取引量を低下させる可能性がある。
もし私たちが効果的な財務報告内部統制制度を維持できなければ、私たちは私たちの財務結果を正確に報告したり、不正を防止することができないかもしれない。したがって、株主は私たちの財務や他の公開報告書に自信を失う可能性があり、これは私たちの業務と私たちの普通株の取引価格を損なうだろう。
財務報告に対する効果的な内部統制は、信頼できる財務報告を提供するために必要であり、適切な開示制御や手順とともに詐欺を防止することを目的としている。必要な新しい制御措置や改善された制御措置を実施できなかったり、実行中に遭遇した困難は、私たちの報告義務を履行できない可能性があります。さらに、サバンズ·オキシリー法案第404条または第404条および米国証券取引委員会および米国上場企業会計監督委員会の関連規則および規定に基づいて行われる任意のテスト、または私たちの独立公認会計士事務所がその後に行う任意のテストは、財務報告書の内部統制に重大な欠陥があることを明らかにすることができ、または私たちの財務諸表を前向きまたは追跡的に変更する必要があるか、またはさらなる関心または改善を必要とする他の分野を発見する可能性がある。悪い内部統制はまた、投資家が私たちが報告した財務情報に自信を失う可能性があり、これは私たちの株式の取引価格にマイナス影響を与える可能性がある。
私たちは四半期ごとに内部統制と手続きの変化を開示することを要求され、私たちの経営陣は毎年これらの統制の有効性を評価することを要求されるだろう。
第404条によると、私たちの独立公認会計士事務所は、財務報告書の内部統制に対する私たちの有効性を証明しなければならない。我々の財務報告の内部統制の有効性の独立した評価は、我々の経営陣が発見していない問題を発見し、重大な弱点を含む可能性があることをより多くの発見に導く可能性がある。私たちは財務報告書の内部統制に重大な欠陥があり、私たちの財務諸表の再説明を招く可能性があり、救済費用が発生することを要求します。
私たちの開示統制と手続きはすべてのミスや詐欺を阻止したり検出できないかもしれない。
私たちは取引法の特定の報告書の要求事項を守らなければならない。我々の開示制御および手続きは、取引所法案に基づいて提出または提出された報告書において、開示を要求する情報が蓄積され、米国証券取引委員会規則および表で指定された期間内に管理層、記録、処理、まとめおよび報告に伝達されることを合理的に確保することを目的としている。任意の開示制御およびプログラムまたは内部制御およびプログラムは、発想および動作がどのように完全であっても、絶対的な保証ではなく、合理的な保証を提供することしかできず、制御システムの目標が達成されることを確保することしかできないと信じている。これらの固有の限界は,意思決定過程における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという現実を含む.さらに、ある人の個人的な行動、2人以上の結託、または無許可超越制御は、制御を回避することができる。したがって、我々の制御システムの固有の制限により、エラーや詐欺によるエラー陳述や開示不足が発見されることなく発生する可能性がある。
項目1 B。未解決従業員意見
ない。
項目2.財産
私たちの本部はコネチカット州スタンフォードにあります。2028年4月に満期になった賃貸契約によると、私たちはそこで約24,000平方フィートのオフィス空間を借りました。2つの5年更新オプションあるいは10年更新オプションがあります。私たちの開発業務はノースカロライナ州のダラムに本部を置いています。2023年に満期になった賃貸契約によると、私たちはそこで約10,350平方フィートのオフィススペースを借りて、2つの5年間の契約更新オプションがあります。私たちの既存および計画中の施設は、私たちの予測可能な未来の需要を満たすのに十分であり、必要であれば、私たちの業務の任意のこのような拡張を満たすために適切な追加空間を提供すると信じています。
項目3.法的訴訟
私たちは現在どんな実質的な法的手続きの当事者でもない。
プロジェクト4.鉱山安全情報開示
適用されません。
第II部
項目5.登録者普通株式市場、関連株主事項、発行者による株式証券の購入
市場情報
私たちの普通株は2019年9月13日からナスダック世界ベスト市場に上場し、取引コードはSWTXです。これまで、私たちの普通株は公開取引市場を持っていなかった。
私たち普通株保有者
我々普通株の登録株主は2023年2月22日現在で約168人である。
配当政策
私たちは普通株に現金配当金を支払ったことがなく、予測可能な未来にも何の現金配当金も支払わないだろう。
株式表現グラフ
本業績グラフは、取引法第18条の規定に基づいて“届出”が行われたとみなされてはならず、このような届出文書にこれが明確に規定されていない限り、証券法又は“取引法”に基づいて提出された任意の届出文書に引用的に組み込まれてはならない。
次の図は2019年9月13日、つまり私たちの普通株がナスダック世界ベスト市場で取引を開始した日から、2022年12月31日まで、私たちの普通株、標準プール500指数、ナスダックバイオテクノロジー指数、ナスダック総合指数に対する100ドルの投資価値を示している。業績図に示す我々普通株の歴史的株価表現は,必ずしも将来の株価表現を示唆しているとは限らない.
持分補償計画
表格10-K第5項に要求される持分補償計画に関する情報は、本年度報告表格10-K第3部第12項を参照することにより本報告に組み込まれる。
最近売られている未登録証券
ない。
株式証券を購入する
ない。
プロジェクト6.保留
第七項。 経営陣の財務状況と経営成果の検討と分析
以下の財務状況および経営結果の議論および分析、ならびに本年度報告書Form 10-Kまたは年次報告書の他の部分に“選択された財務データ”と題する部分、および連結財務諸表および関連注釈を読まなければなりません。 文意が別に指摘されている以外に、すべて“私たち”、“SpringWorks”あるいは“会社”に言及すると、すべてSpringWorks Treateutics,Inc.とその子会社を指す。本議論と分析は、現在の予想に基づく展望的陳述を含み、リスクと不確実性に関連している。展望的陳述は、将来の業績の保証ではなく、私たちの実際の運営結果、財務状況および流動性、および私たちが経営している業務および業界の発展は、本年度報告に含まれる展望的陳述に含まれる議論または予測の結果と実質的に異なる可能性があることを想起させる。私たちは、本年度報告の他の部分で、第1 A項の下に含まれるこれらの潜在的な差異をもたらすか、または促進する可能性のあるリスクおよび他の要因について議論する。“リスク要因”と“前向き陳述に関する特別な説明”の下で。また、我々の経営業績、財務状況、流動性、および当社の業務および当社が経営している業界の発展が本年度報告に含まれる前向きな陳述と一致していても、将来の結果や発展を予測することができない可能性がある。我々は読者に,我々が行ったいかなる前向き陳述にも過度に依存しないように注意し,これらの陳述はそれらの発表日の状況のみを反映している.私たちは、法律および米国証券取引委員会(米国証券取引委員会)の規則が、私たちの予想またはそのような声明に基づく可能性のあるイベント、条件、または状況の任意の変化を反映するために、または実際の結果および前向き声明に記載されている内容の可能性に影響を与える可能性があるために、任意のそのような声明を公開的に更新または修正することを明確に要求しない限り、いかなる義務も負わない。
概要
著者らは臨床段階の生物製薬会社であり、精確な医学方法を用いて、壊滅的な稀な疾病と癌を受けた十分なサービスを受けていない患者群のために生活を変える薬物を獲得、開発、商業化している。著者らは差別化された小分子標的腫瘍学候補製品の組み合わせがあり、そして2つの稀な腫瘍タイプに対する末期臨床試験、1つの登録と1つの潜在登録、及び他のいくつかの高度に流行している遺伝子に対して癌を定義する計画を進めている。我々の研究,転化科学,臨床開発における戦略的方法と卓越した運営は,2023年2月にFDAに受け入れられ,優先審査を受け,指定された処方薬使用料法案(PDUFA)の目標行動日を2023年8月27日とするとともに,業界リーダーと複数の共有価値のパートナー関係を構築し,我々の製品組合せを拡大することができるように,我々の2つの先行候補を後期臨床試験に迅速に進めることができるようにしている。その上で、著者らは引き続き差別化された全集を生物製薬会社に構築し、患者とその疾病を理解することに集中し、変革性の標的薬物を開発した。
第1部第1項で述べたとおりである.“ビジネス”、私たちは現在臨床開発に3種類の候補製品があります。第1項第1項を参照されたい。“ビジネス”は私たちの臨床計画の概要を得る。
2022年9月7日、吾らはいくつかの認可投資家或いは投資家と1つの証券購入協定を締結し、この合意に基づいて、吾らは私募取引或いは私募方式で投資家に8,650,520株の普通株を売却及び発行することに同意し、1株当たり額面は0.0001ドル、買収価格は1株26.01ドルである。私募完了時には,約2.25億ドルの総収益を得ており,手数料と発売コストを差し引いた純収益は約2.168億ドルであった。私募配給については、吾らは投資家とも登録権協定を締結し、当社の普通株の登録転売について規定している
株です。これらの株式は,我々が2020年10月6日に米国証券取引委員会に提出したS-3表登録声明または登録声明と,2022年9月26日に米国証券取引委員会に提出された目論見補編に基づいて登録転売されたものである.
2022年9月6日、我々は、ナイルキャストをbelantamab mafodotin(Belamaf)またはB細胞成熟抗原に対する任意の他の細胞毒性抗体-薬物結合体(BCMA)と組み合わせて使用し、BCMAがGSK制御belantamabから誘導され、単独で併用療法として、または他の薬剤と組み合わせて使用する拡大された世界的非独占的許可および協力プロトコル、またはGSK許可プロトコルをグラクソ·スミスクラインと締結した。グラクソ·スミスクライン許可協定を実行すると同時に、吾らはグラクソ·スミスクライン、グラクソ·スミスクラインまたはGGLの連属会社と株式購入プロトコルを締結し、この合意によると、GGLは私募取引方式で2,050,819株の私たちの普通株を購入し、1株当たり額面0.0001ドル、総購入価格は約7,500万ドル、あるいは1株当たり36.57ドルであった。同等株式は、株式購入契約前の特定30日の間、普通株出来高より加重平均株価割増25%の価格で販売されている。私たちはいくつかの開発と商業マイルストーンを達成した上で5億5千万ドルの追加支払いを受ける資格がある。私たちはNIOROCASTATのすべての商業権を維持した。また,今後のベラマフ臨床試験へのナイルシタの供給を継続し,グラクソ·スミスクラインがベラマフとの併用を求めている市場でのニロシタの商業化を図る。グラクソ·スミスクラインは引き続きすべての開発コストに資金を提供するが、ニダゾール供給に関連する費用と知的財産権に関するいくつかの費用は除外される。
2021年2月25日、Cowen and Company、LLCまたはCowenと販売契約を締結し、この協定によると、Cowenを介して私たちの販売エージェントとして2億ドルまでの総発売収益の普通株または株を時々発行して販売することができる。配給通知を出した後、販売契約の条項及び条件を満たした場合には、コーエンは、改正された1933年の“証券法”により公布された第415(A)(4)条の規則に基づいて定義された“市場発売”とみなされる法律で許可された任意の方法で株を売却することができる。吾等は販売契約の条項及び条件に応じて、時々決められた金額及び時間に応じて株式を売却することができるが、吾等は販売契約に基づいていかなる株式も売却する義務はない。私らまたはコーエンは、他方に通知した後、株式発売を一時停止または終了し、他の条件によって制限されることができる。2022年8月、私たちはこれに基づいて市場での発行計画やATM計画に基づいて224.75万株の普通株を売却し、総収益は6970万ドルで、190万ドルの手数料を引いて、純収益は6780万ドルだった。ATM計画では2022年12月31日現在も約1億303億ドルが利用可能だ。
2020年10月13日、私たちは普通株式の後続公開を完了した。今回の発行では、1株51.00ドルで5637,254株の普通株を一般公開·売却した。1720万ドルの引受割引と手数料および約80万ドルの発売費用を差し引くと、今回発行された純収益は2億695億ドル。
2017年8月の設立以来、私たちはほとんどの資源を私たちの候補製品の研究と開発活動に投入し、私たちの業務発展戦略の実行、私たちの知的財産権の組み合わせの構築、組織と当社の会社の配備、商業化能力の確立、業務計画、資金調達、これらの活動のための一般的かつ行政的な支援を提供しています。
今まで、私たちのすべての収入と繰延収入は、私たちがJazz PharmPharmticalsアイルランド株式会社またはJazzと合意した資産購入と独占許可協定に基づいて受け取った払い戻し不可能な前払い、Jazz協定、およびGSK許可協定から来ました。私たちは商業販売や経常的な収入源のために承認された製品は何もない。私たちは2022年12月31日と2021年12月31日まで、それぞれ5.97億ドルと4.327億ドルの現金、現金等価物、販売可能な有価証券を持っている。設立以来,我々の運営資金は主に証券売却収益に由来しており,2020年10月の後続融資の純収益2.695億ドル,2022年8月のATM計画の純収益6780万ドル,2022年9月にGSKライセンス契約と同時に締結された株式購入契約の総収益約7500万ドル,および2022年9月の私募純収益2.168億ドルである。私たちは私たちの現金、現金等価物、そして有価証券が2026年までの運営費用と資本支出需要に資金を提供できると信じている。
設立以来、私たちは重大な運営損失が発生した。2022年12月31日、2021年12月31日、2020年12月31日までの年度の純損失はそれぞれ2.774億ドル、1.739億ドル、4560万ドルだった。2022年12月31日と2021年12月31日までの累計赤字はそれぞれ5兆699億ドルと2兆925億ドルだった。私たちは予測可能な未来に巨額の費用と運営損失が続くと予想している。また、私たちは私たちが行っている活動に関連する費用が大幅に増加すると予想している
•登録臨床試験と他の適応に使用可能な臨床試験を含む後期臨床試験を通じて、著者らの主要な候補製品ナイルシターとミダメチニブの開発を推進した
•販売、マーケティング、流通インフラを構築し、市場の承認を得ることができ、単独または第三者と連携して商業化しようとしている任意の製品を商業化する
•私たちの他の候補製品の開発計画を推進し、臨床前と臨床開発を通じて、後期臨床開発に入る
•臨床試験に成功した候補製品のために市場承認を求める
•投資は、さらに臨床前および臨床開発の他の技術または製品候補に使用することができるかもしれない
•臨床、品質管理、科学、医療、業務発展、財務、その他の技術者を含め、より多くの人員を招聘し、私たちのインフラを建設し続ける
•私たちの臨床開発、製造、業務発展、商業化努力、上場企業としての私たちの運営を支援する人員を含む、私たちの運営、財務、管理システムを拡大し、人員を増加させる
•私たちの知的財産権の組み合わせを維持し、拡大し、保護する。
私たちが臨床開発に成功し、規制部門から候補製品の承認を得ない限り、私たちは製品販売から収入を得ません。また、私たちがナイル西やミダメチニブの規制部門の承認を得たら、私たちの商業化能力の開発に関連する巨額の費用が発生し、製品販売、マーケティング、流通活動を支援し、単独でも他の会社とも協力することが予想される。
私たちの許可と協力協定は
ファイザー許可協定
2017年8月、ファイザー社とNIOROGACEATライセンス契約を締結し、この合意に基づき、NIOROCASTATのグローバル独占使用権を取得しました。その後、私たちは2019年7月に知的財産権に関連するいくつかの条項のNiroacestat許可協定を修正しました。改正されたNiRogacestatライセンス契約によると、いくつかのビジネスマイルストーン事件が完了したときに、合計2.325億ドルのお金をファイザーに支払う必要がある。ニトロキセチンを販売する分級特許権使用料を中央値−1桁から20%以下の割合でファイザー社に支払うことになり,有効クレーム満期,第三者許可満期金額,後発薬競争の控除を受ける可能性がある。
2017年8月、ファイザーとライセンス契約、またはMirdametinibライセンス契約を締結し、総称して“ファイザーライセンス契約”と呼ばれ、この合意に基づき、Mirdametinibのグローバル独占使用権を取得しました。私たちはその後、知的財産権に関連するいくつかの条項に関連するMirDametinib許可協定を2019年8月に修正した。改訂されたMirdametinibライセンス契約によると、いくつかのビジネスマイルストーン事件が完了したときに、ファイザーに合計2億298億ドルを支払う必要があります。ミダメチニブの販売のためにファイザーに分級特許権使用料を支払い、百分率の中央値から一桁まで、有効クレーム満期、第三者許可満期金額、模造薬競争の控除を受ける可能性があります。
Teadライセンス契約
2021年5月、私たちはKatholieke University Leuven(KU Luuven)とフランダースバイオテクノロジー研究所(VIB)と世界的に独占的な許可協定を締結し、プロトコルに従って、TEAドメイン(TEAD)ファミリー転写因子の新しい小分子阻害剤の組み合わせの許可を得て、カバ経路異常シグナルによって駆動されるバイオマーカーによって定義される固形腫瘍を潜在的に治療することを目的とした。合意条項によると、私たちはKUルモールとVIBに1100万ドルを前払いした。協定条項によると、KUルモールとVIBには、2.85億ドルまでの開発、規制、ビジネスマイルストーン、およびライセンス内技術に基づいて開発された製品の将来の任意の純売上に基づく1桁分のパーセント特許権使用料を取得する資格がある
EGFR許可協定
2021年10月、私たちはDana-Farberと独占的なグローバル許可協定に署名し、Stanford MedicineまたはStanfordとEGFR変異癌の新しい上皮増殖因子受容体(EGFR)小分子阻害剤の組み合わせを治療するための賛助研究協定を署名した。ダイナーとのライセンス契約条項によると-
私たちはDana-Farberに前金を支払い、Dana-Farberは許可内技術による任意の将来の純売上高の開発と商業マイルストーンおよび特許使用料を得る資格があるだろう
Dana-Farberとライセンス契約を締結すると同時に、私たちはスタンフォード大学と長年賛助の研究協定を達成し、スタンフォード医学の研究室とDana-Farberの協力実験室の持続的な研究と開発に資金を提供した。スタンフォード大学との協賛研究協定は、EGFR阻害剤の組み合わせが開発候補指名に向かっているため、リードした最適化と転化生物学的努力を支持することを目的としている。協賛研究協定によると、我々は交渉許可のさらなる知的財産権の選択権を付与されており、これらの許可は賛助研究の表現に生じる可能性がある。
百済神州臨床協力協定
2018年8月、著者らは百済神州有限会社或いは百済神州と臨床協力協定を締結し、リフィニブとミダメチニブの併用による末期或いは難治性固形腫瘍患者の1 b期臨床試験の安全性、耐性と初歩的な治療効果を評価した。プロトコルにより,それぞれ単独で臨床試験に用いられる化合物の製造や供給に関するコストを担当している。臨床試験に関する他の費用を百済神州と折半した。
グラクソ·スミスクラインの非独占的許可と協力協定の拡大
2022年9月,グラクソ·スミスクラインとの非排他的臨床協力の拡大を発表し,最初に2019年6月に開始した。この公告は、グラクソ·スミスクラインとのライセンス契約に当たり、グラクソ·スミスクラインとの抗体-薬物結合体belamafまたはADC、GSKによって制御されるbelantamab由来BCMAを標的とするBCMAを標的とする任意の他の細胞毒性ADCが、単独で併用療法として、または他の医薬製剤と共に使用される可能性がある。
グラクソ·スミスクライン許可協定の条項に基づき、この協定に署名するとともに、吾らは株式購入協定を締結し、この合意により、GGLは私募取引方式で2,050,819株の我々の普通株を購入し、総購入価格は約7500万ドル、あるいは1株当たり36.57ドルであった。株式購入契約を締結する前の特定30日間の間、当該株式は、当社の普通株出来高よりも加重平均株価割増25%の価格で販売されている。
グラクソ·スミスクライン許可協定の条項によると、いくつかの開発とビジネスマイルストーンに達したら、5億5千万ドルまでの追加支払いを受ける資格がある。私たちはNIOROCASTATのすべての商業権を維持し続けている。また,SpringWorksは将来のbelamaf臨床試験にniRogacestatを提供し,GSKがbelamafとの合併承認を求めた市販niRogacestatを求める。グラクソ·スミスクラインは引き続きすべての開発コストに資金を提供するが、ニダゾール供給に関連する費用と知的財産権に関するいくつかの費用は除外される。
その他の臨床協力協定は、ナイルキャストとBCMA指導の共同治療開発に関連している
グラクソ·スミスクラインの協力協定以外に、著者らは業界パートナーと他のいくつかの臨床試験協力と供給プロトコルを締結し、ニロシタンとBCMA指導の各種療法との併用治療を評価し、CAR T細胞療法、二重特異性抗体とモノクロナル抗体を含み、再発或いは難治性多発性骨髄腫患者の治療に用いられる
各パートナーはニトロクロロシタと併用したBCMA指導療法を評価し,直接臨床試験に関連するすべての費用を担当する臨床試験の管理を担当しているが,ナイル西特の生産と供給および知的財産権に関するいくつかの費用は除外されている。すべての協力は私たちとそれぞれのパートナーで構成された共同委員会によって管理される
事前に終了しない限り、各協力協定は臨床試験で期待される分析が完了した後に無効になる。私たちまたは双方は協力協定に規定されている他の理由で協力協定を終了することができる。
ジャズ.ジャズ 製薬会社の資産購入と独占許可協定
2020年10月,JazzとJazzはPF−04457845を含む我々の脂肪酸アミド加水分解酵素(FAAH)阻害剤計画を買収したJazzプロトコルを発表した。Jazzは私たちに3500万ドルを前払いし、ある臨床開発、監督と商業マイルストーンの実現によって、将来3.75億ドルを支払うかもしれない。また,ジャズは将来PF−04457845の純売上高に販売ベースの印税を支払う義務がある。
我々のライセンスおよび連携プロトコルの詳細については、“業務-ライセンスおよび連携プロトコル”を参照されたい。
新冠肺炎による影響
2019年12月、武漢で1種の新しいコロナウイルス株--深刻な急性呼吸症候群コロナウイルス2型を発見し、SARS-CoV-2と略称し、中国。2020年3月11日、世界保健機関はSARS-CoV-2に関連する疾病新冠肺炎の発生を全世界大流行とした。この疾患は,新たに出現した新冠肺炎変異株を含め,我々が業務を展開している地域に伝播し続けている。新冠肺炎の蔓延を緩和するため、世界各国の政府と企業は現地避難令、隔離措置、旅行に対する重大な制限、および多くの従業員の出勤禁止の制限を含む未曽有の行動を取った。新冠肺炎疫病発生から、著者らは一連の業務連続性措置を開始し、著者らの業務に対する潜在的な中断を減少し、そして著者らの研究開発計画の完全性を維持した。新冠肺炎の流行期間中、私たちの対応は変化し、私たちは基本的に正常な運営を回復した。これまで、私たちは何の実質的な中断も経験していません。私たちは現在研究と開発活動を行っています。しかし、新冠肺炎の大流行の結果、あるいは新しく出現した新冠肺炎ウイルス変異株のいかなる影響により、ワクチン接種率の停滞と関連要素は、私たちは中断を経験し、私たちの研究開発と商業化に影響を与えるかもしれません スケジュールと結果です。私たちは進行中の新冠肺炎の流行と新興変種が私たちの業務に与える影響を評価し続ける。現在の新冠肺炎大流行が著者らの未来の業績に与える影響程度は未来の発展に依存するが、今回の大流行と関連影響は、大流行の持続時間、拡散と強度(任意の再発を含む)、新冠肺炎ウイルス新変異株の影響及び新冠肺炎ワクチンの発売を含み、これらはすべてまだ不確定と予測困難であり、著者らの業務、将来性、未来の財務状況、運営結果とキャッシュフローに実質的な影響を与える可能性がある。
私たちの運営結果の構成要素は
収入.収入
今まで、私たちのすべての収入および繰延収入は、JazzプロトコルおよびGSKライセンスプロトコルに従って受信された払い戻し不可能な前払いから来ています。私たちは販売製品から商業収入を何も得ていない。現在の候補製品や将来開発可能な他の候補製品の開発に成功し、商業化できれば、将来的に承認された製品の販売から収入を得ることができるかもしれない。しかも、私たちは時々協力と許可協定を締結し、私たちに支払われるべきいくつかのお金を規定するかもしれない。したがって、私たちは未来にこのような協力や許可協定から収入を得るかもしれない。
研究開発費
私たちの研究開発費には私たちの候補製品開発に関する費用が含まれています。これらの費用には
•研究開発者の賃金、福祉、株式給与を含む従業員関連の費用
•私たちの研究開発計画に直接関連したサービス費用をコンサルタントに支払います
•私たちまたは私たちと協力して研究·開発活動を行っている第三者契約研究機関やCRO、調査性臨床試験場所、学術機関、コンサルタントとの合意による費用
•臨床前研究や臨床試験に関連する費用
•前臨床試験および臨床試験のための医薬物質および完成品の製造に関連するコスト;
•技術や知的財産権ライセンスに関連するコスト;
•施設と施設関連費用の分担分には、レンタル料や他の施設関連費用や他用品の費用が含まれている。
研究開発費の外部コストは個々の計画に基づいて追跡されている。臨床開発支出は、我々の候補製品に関する前払い許可料とマイルストーン支払いを含めて、計上されます
発生した研究と開発費用。これらの費用には、賃金および福祉、材料および用品、臨床前費用、臨床試験および関連臨床製造費用、設備減価償却、契約サービス、および他の外部費用が含まれる開発活動を行うことによって生じる費用が含まれる。製造および臨床試験のようないくつかの開発活動のコストは、特定のタスクを達成する進捗の評価に基づいており、実際に完了した活動または生成されたコストに関する情報を、我々のサプライヤーが提供するような時間ベースの測定基準またはデータを使用して提供される。
将来的には,我々の候補製品の開発や臨床前計画に関する活動に投資し続けるとともに,いくつかの候補製品が後期開発段階に入るにつれて,2022年12月に提出された守秘協定Niroacestatや,我々が登録可能なミダミーニ2 b期臨床試験ReNeu試験を含めて,我々の研究·開発費が増加することが予想される。必要な臨床試験を行って監督部門の許可を得る過程は高価で時間がかかり、著者らの候補製品の開発成功も非常に不確定である。そのため、私たちの研究開発プロジェクトの持続時間と完成コストを決定することもできませんし、いつ、どの程度私たちの候補製品の商業化と販売から収入を得るかを決定することもできません。
一般と行政費用
一般的および行政費用は、主に株式ベースの報酬を含む行政、財務、会社、商業、業務発展および行政機能者の賃金と関連費用を含む。一般および行政費用には、特許および会社の事務に関連する法律費用、会計、監査、税務および行政相談サービスの専門費用、保険料、行政出張費用、直接減価償却コスト、施設賃貸料および維持分配費用、および他の運営コストが含まれる施設関連費用も含まれる。
候補製品の持続的な開発を支援し、運営を拡大して組織(商業化を含む)を支援するための従業員の増加に伴い、将来的には一般的かつ管理費が増加することが予想される
利子とその他の収入
利息と他の収入は主に利息収入で構成されている。利息収入には、私たちの現金、現金等価物、および販売可能な有価証券から稼いだ利息が含まれています。
株式投資損失
株式投資損失はMapKure投資損失における著者らのシェアを代表し、株式会計方法を用いて計算を行う。
所得税
所得税は貸借対照法を用いて計算される。繰延税項資産及び負債は、既存資産及び負債の帳簿金額及びそれぞれの計税基礎と営業損失及び税項相殺繰越との差異による将来の税務結果を確認する。税金資産及び負債を繰延して税率計量を策定し、その等の一時的な差額を回収又は決済する予定の年度の課税収入に適用されることが予想される。税率変動が繰延税金資産や負債に及ぼす影響は、公布日を含む期間中に収入として確認された。繰延税金資産と負債の変動を所得税に計上する準備。
私たちは繰延税金資産の程度が、これらの資産がより現金化される可能性があると考えていることを確認した。この決定を下す際には、管理層は、既存の課税の一時的な差異の将来の輸出、将来の課税収入の予想、税務計画戦略、最近の業務の結果を含む、利用可能なすべてのプラスおよび負の証拠を考慮する。既存の証拠の重みに基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合には、推定免税額を計上する。経営陣が将来的に私たちの繰延税金資産が私たちの純記録金額を超えることができると判断すれば、管理層は繰延税金資産の推定値を調整し、所得税の支出を減らすことになる。
我々は、会計基準編纂またはASC主題740に基づいて、2段階に分けて行われるプロセスである不確定な税務頭寸を記録し、(1)管理層は、頭寸の技術的利点に基づいて、税務頭寸をより維持する可能性があるかどうかを決定し、(2)より確認可能性の高い税務頭寸に適合するかどうかを決定する
最低課税点では、経営陣は最終的に関連税務機関と和解した後に実現可能な最大税収割引額が50%を超えることを確認した。
私たちは不確定な税収状況に関連する様々な税務機関の潜在的な納税のために準備している。これらの準備金は、私たちが提出した書類または手がかりで得られた税金優遇がどれだけあるか、税金優遇に関連する任意の潜在的または有事事項を解決した後に達成される可能性があるかどうかに基づいて決定される。所得税の少納に関連する潜在的利息は所得税費用の1つの構成要素に分類され、任意の関連する処罰は経営報告書中の所得税費用に分類される。
2022年12月31日現在、連邦、州、都市の純運営損失はそれぞれ3.683億ドル、2.609億ドル、370万ドルであり、将来の課税収入の減少に利用できる。2018年から2022年までに発生した連邦純営業損失は3.64億ドルで、完全利用まで一定期間にわたって課税収入の80%を無期限に相殺する。2017年に報告された連邦純運営損失は430万ドルで、州と市の純運営損失の繰越は2040年まで異なる日に満期となる。私たちは未来の税金負担を相殺するために2270万ドルの連邦税金免除を持っている。このような税金の繰越免除は2038年に始まる様々な日に満了されるだろう。
行動の結果
2022年12月31日までと2021年12月31日期との比較
次の表は,2022年12月31日と2021年12月31日までの年間の運営結果をまとめたものである。
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 12月31日までの12ヶ月間 | | | | |
| (単位:千) | | 2022 | | 2021 | | $Change | | 変更率 |
| 運営費用: | | | | | | | | |
| 研究開発 | | $ | 146,122 | | | $ | 101,676 | | | $ | 44,446 | | | 44 | % |
| 一般と行政 | | 134,552 | | | 71,792 | | | 62,760 | | | 87 | % |
| 総運営費 | | 280,674 | | | 173,468 | | | 107,206 | | | 62 | % |
| 運営損失 | | (280,674) | | | (173,468) | | | (107,206) | | | 62 | % |
| 利息とその他の収入: | | | | | | | | |
| その他の収入,純額 | | (138) | | | (152) | | | 14 | | | (9) | % |
| 利子収入,純額 | | 6,285 | | | 698 | | | 5,587 | | | 800 | % |
| 利子とその他の収入総額 | | 6,147 | | | 546 | | | 5,601 | | | 1026 | % |
| 株式投資損失 | | (2,890) | | | (988) | | | (1,902) | | | 193 | % |
| 純損失 | | $ | (277,417) | | | $ | (173,910) | | | $ | (103,507) | | | 60 | % |
研究開発費
2022年12月31日までの1年間で、研究開発支出は4440万ドル増加し、2021年12月31日現在の1.017億ドルから1兆461億ドルに増加し、44%増加した。
私たちの研究開発費は以下のようにまとめられた
| | | | | | | | | | | | | | | | | | | | |
| | 12月31日までの12ヶ月間 | | |
| (単位:千) | | 2022 | | 2021 | | $Change |
| 関係者の | | $ | 70,876 | | | $ | 39,102 | | | $ | 31,774 | |
| 許可、試験、薬物製造 | | 66,447 | | | 57,181 | | | 9,266 | |
| 施設に関するものやその他 | | 8,799 | | | 5,393 | | | 3,406 | |
| 研究開発費総額 | | $ | 146,122 | | | $ | 101,676 | | | $ | 44,446 | |
研究·開発費が増加した要因は,内部コストが3180万ドル増加したことであり,在庫ベースの増加を含む人員数の増加に関連する従業員コストの増加が原因である
報酬支出は2260万ドル増加し,薬物製造,臨床試験,その他の研究に関連する外部コストは2260万ドル増加したが,2021年5月にTEAD阻害剤計画の内部許可によりKUルモールやVIBに支払われた払戻不可能な前払いに関する許可コストが1100万ドル減少したことと部分的に相殺された。
一般と行政費用
2022年12月31日終了年度の一般費用と行政費用はそれぞれ1兆346億ドルと7180万ドルであり、2021年12月31日終了年度の一般費用と行政費用は以下の通り
| | | | | | | | | | | | | | | | | | | | |
| | 12月31日までの12ヶ月間 | | |
| (単位:千) | | 2022 | | 2021 | | $Change |
| 関係者の | | $ | 81,441 | | | $ | 44,861 | | | $ | 36,580 | |
| 専門と相談料 | | 43,996 | | | 20,923 | | | 23,073 | |
| 施設に関するものやその他 | | 9,115 | | | 6,008 | | | 3,107 | |
| 一般と行政費用総額 | | $ | 134,552 | | | $ | 71,792 | | | $ | 62,760 | |
一般的かつ行政費用の増加は、主に人員数の増加に関連する従業員コストの増加による内部コストの36.6ドルの増加であり、組織を支援するための業務の拡大に伴い、株式ベースの報酬費用の増加と、ビジネス能力、情報技術コスト、コンサルティングおよび専門サービスを含む新たな能力の確立に伴い、法律、法規、コンプライアンスを含めて2310万ドル増加した。
その他の収入
2021年12月31日までの年度と比較して、2022年12月31日までの1年間で、他の収入の増加は利息収入純額の増加によるものである。この成長は金利の著しい向上によるものであり、これは2022年12月31日現在の年間現金、現金等価物、および有価証券のより高いリターンを推進する。
2021年12月31日までと2020年12月31日までの年度比較
2021年12月31日と2020年12月31日までの年間運営結果を表にまとめた。
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 12月31日までの12ヶ月間 | | | | |
| (単位:千) | | 2021 | | 2020 | | $Change | | 変更率 |
| 許可収入 | | $ | — | | | $ | 35,000 | | | $ | (35,000) | | | (100) | % |
| 運営費用: | | | | | | | | |
| 研究開発 | | 101,676 | | | 51,859 | | | 49,817 | | | 96 | % |
| 一般と行政 | | 71,792 | | | 29,465 | | | 42,327 | | | 144 | % |
| 総運営費 | | 173,468 | | | 81,324 | | | 92,144 | | | 113 | % |
| 運営損失 | | (173,468) | | | (46,324) | | | (127,144) | | | 274 | % |
| 利息とその他の収入: | | | | | | | | |
| その他の収入,純額 | | (152) | | | 25 | | | (177) | | | (708) | % |
| 利子収入,純額 | | 698 | | | 1,330 | | | (632) | | | 100 | % |
| 利子とその他の収入総額 | | 546 | | | 1,355 | | | (809) | | | (60) | % |
| 株式投資損失 | | (988) | | | (605) | | | (383) | | | 63 | % |
| 純損失 | | $ | (173,910) | | | $ | (45,574) | | | $ | (128,336) | | | 282 | % |
収入.収入
私たちは2021年12月31日までの年間の何の収入も確認していない。2020年12月31日までの年間収入は3,500万ドルで、2020年10月のJazzと私たちとの間の資産購入と独占ライセンス契約に関する払戻不可能な前払いによるものです。
研究開発費
2021年12月31日までの1年間で、研究開発支出は4980万ドル増加し、2020年12月31日現在の5190万ドルから1.017億ドルに増加し、96%に増加した。
研究·開発費が増加した理由は,ライセンス,医薬品製造,試験費用に関する外部費用が2540万ドル増加し,内部コストが2320万ドル増加したのは,人員数の増加や非現金シェア報酬支出に関する従業員コストの増加と,施設関連120万ドルやその他の雑部門費用の増加によるためである。外部コストの増加は,薬品製造や試験コストに関する外部コストの1,440万ドルの増加と,TEAD阻害剤計画の内部許可のためにKU LeuvenとVIBに支払われた1100万ドルの払戻不可能な前金によるものである。
私たちの研究開発費は以下のようにまとめられた
| | | | | | | | | | | | | | | | | | | | |
| | 12月31日までの12ヶ月間 | | |
| (単位:千) | | 2021 | | 2020 | | $Change |
| 関係者の | | $ | 39,102 | | | $ | 15,900 | | | $ | 23,202 | |
| 試験と薬物製造 | | 57,181 | | | 31,766 | | | 25,415 | |
| 施設に関するものやその他 | | 5,393 | | | 4,193 | | | 1,200 | |
| 研究開発費総額 | | $ | 101,676 | | | $ | 51,859 | | | $ | 49,817 | |
一般と行政費用
2021年12月31日現在と2020年12月31日終了年度までの一般料金と行政費はそれぞれ7180万ドルと2950万ドルで、詳細は以下の通り
| | | | | | | | | | | | | | | | | | | | |
| | 12月31日までの12ヶ月間 | | |
| (単位:千) | | 2021 | | 2020 | | $Change |
| 関係者の | | $ | 44,861 | | | $ | 16,476 | | | $ | 28,385 | |
| 専門と相談料 | | 20,923 | | | 10,437 | | | 10,486 | |
| 施設に関するものやその他 | | 6,008 | | | 2,552 | | | 3,456 | |
| 一般と行政費用総額 | | $ | 71,792 | | | $ | 29,465 | | | $ | 42,327 | |
一般的かつ行政的費用の増加は、主に一般的かつ行政的機能においてより多くの人員を雇用しているためであり、商業化能力、非現金株式ベースの報酬費用の増加を含む本組織を支援するための業務を拡大しているからである。さらに、一般的および行政的費用には、法律、規制、およびコンプライアンスを含む1050万ドルの情報技術費用の増加、コンサルティングおよび専門サービスが含まれています
その他の収入
その他の収入の減少は,2021年12月31日までの年度が2020年12月31日までの年度に比べて利息収入純額が減少したためである。この低下は新冠肺炎疫病の経済影響による金利の大幅な低下であり、2020年の一部の時間と2021年12月31日までの年間現金、現金等価物と有価証券のリターン率が低い。
流動資金と資本資源
流動資金源
設立以来、私たちの運営資金は主に証券売却の収益から来ており、2020年10月の後続融資の純収益2.695億ドル、2022年8月のATM計画の純収益6780万ドル、締結された株式購入契約の総収益約7500万ドルを含む
2022年9月のグラクソ·スミスクライン許可協定と同時に行われ、2022年9月の私募純収益は2兆168億ドル。
設立以来,運営損失が発生し,運営キャッシュフローは負であり,少なくとも予見可能な未来には損失を続けることが予想される。2022年12月31日、2021年12月31日、2020年12月31日までの年度の純損失はそれぞれ2.774億ドル、1.739億ドル、4560万ドルだった。2022年12月31日と2021年12月31日までの累計赤字はそれぞれ5兆699億ドルと2兆925億ドルだった。
資金需要
私たちの現金の主な用途は、私たちの研究開発計画と、私たちの商業化活動と会社運営を含む運営費用に資金を提供することです。運営費用に資金を提供するための現金は、これらの費用を支払う時間の影響を受けており、これは私たちの未済売掛金、売掛金、前払い費用の変化に反映されている。
私たちは2022年12月31日まで、私たちの現金、現金等価物、有価証券残高は私たちの2026年までの運営費用と資本支出要求を満たすのに十分になると信じている。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは現在予想されているよりも早く私たちが利用できる資本資源を利用することができる。私たちの有価証券には、会社債務証券、アメリカ政府証券、非アメリカ政府証券、商業手形を含む高品質で流動性の高い売却可能な債務証券が含まれています。
私たちの未来の支出需要は多くの要素に依存するだろう
•著者らの候補製品の臨床前研究と臨床試験の開始、進捗、時間、コストと結果
•私たちが私たちの製品を商業化することを選択した地域では、規制の承認を得ることができる任意の候補製品の販売、マーケティング、流通能力を確立するコスト
•候補製品の開発と監督管理審査活動に成功した後に得られた商業成功程度。
•私たちの候補製品のための臨床開発計画は
•私たちが開発した候補製品の数量と特徴は
•アメリカ食品と薬物管理局、ヨーロッパ薬品管理局と他の類似外国監督管理機関が制定した監督管理要求の結果、時間とコストを満たす
•私たちは、事前支払い、マイルストーン、および印税義務の金額を含む、既存および任意の将来の許可または協力協定の条項を選択することができます
•研究開発および人員コストの増加など、新しい技術許可内に関連する他のコスト
•私たちの特許主張と他の知的財産権の提出、起訴、弁護、そして実行のコスト
•知的財産権紛争の弁護コストは、第三者が私たちまたは私たちの候補製品に対して提起した特許侵害訴訟を含む
•競争の技術と市場発展の影響
•ビジネス規模のアウトソーシング製造活動を完了するコストと時間
私たちは臨床試験、その他の研究開発支出、商業活動と業務発展努力の運営需要と資本要求を満たすために追加の資金が必要である。私たちの候補製品の開発や商業化に関連する多くのリスクと不確実性のため、現在および予想されている臨床研究に関連する増加した資本支出および運営支出の金額を見積もることができない。
その前に、相当な製品収入を生み出すことができれば、株式発行、債務融資、協力、戦略連合とマーケティング、流通、または許可の組み合わせを通じて、私たちの運営に資金を提供したいです
手配ができました。私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、現在の所有権権益は希釈され、これらの証券の条項は清算または普通株主の権利に悪影響を及ぼす他の特典を含む可能性がある。債務融資および優先株融資に関与する可能性のある協定は、追加債務を招く、買収または資本支出を行う、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限または制限する契約を含む。もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配を通じてより多くの資金を調達するならば、私たちは私たちの技術、将来の収入源、研究計画、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資や他の手配を通じて追加資金を調達できない場合、私たちは私たちの研究、製品開発、または将来の商業化努力を延期、制限、減少または終了する必要があるかもしれません。あるいは私たちが自分で開発し、マーケティングすることをより望んでいた候補製品の権利を与える必要があります。
キャッシュフロー
次の表は、列挙された各期間の現金源と用途をまとめています
| | | | | | | | | | | | | | | | | | | | |
| | 12月31日までの12ヶ月間 |
| (単位:千) | | 2022 | | 2021 | | 2020 |
| 経営活動のための現金純額 | | $ | (161,563) | | | $ | (127,877) | | | $ | (32,191) | |
| 投資活動提供の現金純額 | | (215,597) | | | 83,592 | | | (418,832) | |
| 融資活動が提供する現金純額 | | 340,702 | | | 1,157 | | | 270,485 | |
| 現金と現金等価物の純減少 | | $ | (36,458) | | | $ | (43,128) | | | $ | (180,538) | |
経営活動に使われている現金流量
2022年12月31日現在、2021年12月31日と2020年12月31日までの年間、経営活動で使用されている純現金はそれぞれ1.616億ドル、1億279億ドル、3220万ドルだった。
2022年12月31日現在の年度において、経営活動に用いられる現金純額は、主に今年度の純損失2.774億ドル、7780万ドルの非現金費用調整後、純営業資産と負債の純変化3810万ドルによるものである。非現金費用には、主に7300万ドルの株式ベースの給与支出、110万ドルの非現金運営リース費用の償却、290万ドルのMapKureへの投資に関連する株式投資損失が含まれる。私たちの純営業資産と負債の変化は主に売掛金と売掛金の純増加により1750万ドル、繰延収入は1950万ドル増加し、前払い費用やその他の非流動資産は190万ドル増加したが、経営リースの現金支払いにより賃貸負債は90万ドル減少し、この増加を部分的に相殺した。
2021年12月31日までの年度において、経営活動に使用されている現金純額は、主に今年度の純損失1億739億ドルにより、4090万ドルの非現金費用と510万ドルの純営業資産と負債の変化により調整されている。非現金費用には、主に株式ベースの給与支出3840万ドル、非現金運営リース支出100万ドル、MapKureへの投資に関連する株式投資損失100万ドルが含まれています。私たちの純営業資産と負債の変化は主に売掛金と売掛金の純増加で1200万ドル増加したが、前払い費用と他の非流動資産の530万ドルの減少とレンタル負債の140万ドルの減少によって部分的に相殺されたのは、経営リースの現金支払いによるものである。
2020年12月31日までの年度において、経営活動に用いられる現金純額は、主に今年度の純損失4560万ドル、1200万ドルの非現金費用と130万ドルの純営業資産と負債変動調整後の純損失によるものである。非現金費用には、主に株式ベースの報酬支出1,000万ドル、非現金運営リース支出100万ドル、MapKureへの投資に関連する持分投資損失60万ドルが含まれている。私たちの純営業資産と負債の変化は主に売掛金と売掛金の純増加によって460万ドル増加したが、前払い費用と他の非流動資産の200万ドルの増加によって部分的に相殺され、レンタル現金支払いの経営に推進されてレンタル負債は140万ドル減少した。
投資活動によるキャッシュフロー
2022年12月31日までの1年間、投資活動のための現金純額は2.156億ドルで、4.811億ドルの売却可能な債務証券を購入し、資本支出は1,020万ドルであり、2022年6月のMapKureへの投資は420万ドルであり、売却と満期に債務証券を売却できる収益279.8ドルで相殺されたからである
百万ドルです。2021年12月31日までの年間で、投資活動が提供する現金純額は8360万ドルであり、これは主に売却可能な有価証券の純売上高が8560万ドルであるためである。2020年12月31日までの1年間、投資活動のための現金純額は4.188億ドル、4.427億ドルの売却可能な債務証券を購入するために使用され、私たちの2020年6月のMapKureへの投資は350万ドル、資本支出は60万ドルであり、債務証券の売却と満期の売却可能な収益2800万ドルによって相殺された。
融資活動が提供するキャッシュフロー
2022年12月31日までの1年間で、融資活動が提供する現金純額は3兆407億ドルで、これは普通株発行の純収益3.401億ドルに後押しされた。2021年12月31日までの1年間、融資活動が提供した現金純額は120万ドルであり、株式オプションの収益を行使した結果となった。2020年12月31日までの年間で、融資活動が提供する現金純額は2兆705億ドルで、普通株発行収益、発行コスト2億696億ドル、株式オプション90万ドルの行使を含む。
契約義務その他の約束
私たちは正常業務過程で臨床試験、臨床前研究、製造、その他の運営目的のためのサービスと製品の契約を締結した。これらの契約は一般に通知後の一定期間で契約を終了することが規定されているため,これらの合意の下での撤回不可能な義務は実質的ではないと考えられる.
2022年12月31日まで、私たちは不確定な税金のための準備金を何も記録していない。
重要な会計政策と試算
経営陣の財務状況と経営結果の議論と分析は、米国公認の会計原則に基づいて作成された我々の総合財務諸表に基づいている。これらの連結財務諸表を作成する際には、連結財務諸表の日付の報告済み資産および負債額、または有資産および負債の開示、および報告期間内に発生した報告済み費用に影響を与える推定および仮定を行う必要がある。我々の見積もりは,我々の歴史的経験と,当時の状況では合理的な様々な他の要因に基づいており,これらの要因の結果は資産や負債の帳簿価値を判断する基礎を構成しているが,これらの資産や負債の帳簿価値は他の源からは明らかではないように見える。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。
我々の主な会計政策は、本年報の他の部分の総合財務諸表付記3により詳細に記載されているが、以下の会計政策が総合財務諸表の作成に使用する判断と推定に最も重要であると信じている。
収入.収入
我々は,我々の内部の開発,第三者との共同開発や協力協定,製品権利の販売や対外許可,その他を含む医薬品開発や商業化に関する収入を確認した.これらの手配および合意の条項は、ライセンス、ノウハウ、医薬品、関連プロトコル、および他の交付可能な成果を含むことができる複数のコミットメントされた貨物およびサービスを含むことができる。これらの手配に基づいて私たちに支払われるお金は、前払い許可料、マイルストーン支払い、および将来の製品販売の印税のうちの1つまたは複数を含むことができる。
ASC 606の範囲内の手配、顧客との契約収入
我々は、ASC 606に従って収入を確認し、これは、顧客とのすべての契約に適用されるが、協調手配およびレンタルのような他の標準範囲内の契約に属するものを除く。
ASC 606によれば、顧客が約束された商品またはサービスの制御権を取得すると、収入を確認し、金額は、私たちが受信したいこれらの商品またはサービスの対価格を決定することを反映している。ASC 606の範囲内のスケジュールの収入確認を決定するために、(I)顧客との契約を決定するステップ、(Ii)契約における履行義務を決定するステップ、(Iii)取引価格を決定するステップ、(Iv)契約に取引価格を割り当てる履行義務、および(V)契約義務を履行する際に収入を確認するステップの5つのステップを実行する。これらの手配の会計計算の一部として、契約中の履行義務の決定、取引価格に含まれる可変対価格金額の推定、取引価格を各履行義務に割り当てることなど、重大な判断が必要かもしれません。
契約がASC 606の範囲内にあると決定されると、契約で約束された貨物またはサービスを評価し、それらが義務を履行していると判断する。
私たちは契約の履行義務を決定するために、各約束された貨物またはサービスが異なるかどうかを評価する。このような評価は、管理層に個別に約束された貨物またはサービスの判断、およびこれらの貨物またはサービスが契約関係の他の態様から分離可能であるかどうかを要求することができる主観的決定に関するものである。約束された貨物およびサービスは異なると考えられ、条件は、(1)顧客が単独で、または顧客がいつでも入手可能な他のリソースと共に貨物またはサービスから利益を得ることができる(すなわち、貨物またはサービスが異なることができる)、(2)エンティティが顧客に貨物またはサービスを譲渡する約束は、契約内の他の約束とは別に識別可能である(すなわち、貨物またはサービスを譲渡する約束は契約範囲内で異なる)ことである。約束された商品やサービスがユニークであるかどうかを評価する際には,顧客の研究,製造,商業化能力,関連専門知識の一般市場での可用性などを考慮する.約束された貨物またはサービスが契約中の他の約束とは別に識別できるかどうかを評価する際には、契約の予想利益も考慮します。約束された商品またはサービスが異なる場合、エンティティは、異なる商品またはサービスを識別するまで、その商品またはサービスを他の約束された商品またはサービスと統合することが要求される。
もし契約で約束された価格に可変金額が含まれている場合、約束された商品やサービスを顧客に移転するために、私たちが獲得する権利があると推定します。我々は,期待値手法や最尤数法を用いることで変数の対価格額を決定する.私たちは制限されない推定可変対価金額を取引価格に計上します。取引価格に含まれる金額は,確認された累積収入が大きく逆転しない可能性の高い金額に制限される.その後の各報告期間が終了した場合,吾らは取引価格に含まれる推定変動の価格および任意の関連制限を再評価し,必要に応じて全体の取引価格の見積りを調整する.いずれの調整も調整期間内に累積追跡方式で記録されている.
そして、各履行義務がある時点または一定期間にわたって履行された場合には、時間が経過するにつれて、1つの産出または投入方法の使用に基づいて、該当履行義務に割り当てられた取引価格の金額を収入として確認する。
知的財産権ライセンス:我々のライセンス契約の条項には、知的財産権の機能性が私たちが行っている活動によって実質的に変化しないことが予想されるため、機能性知的財産権の許可が含まれている。他のコミットメントとバンドルされたライセンス(すなわち、手配中の他のコミットメント商品およびサービスと区別されていないライセンス)については、合併履行義務が時間の経過とともに履行されているか、またはある時点で履行されているかを決定するために、判断を利用して、合併履行義務が時間とともに履行されているかどうかを決定し、時間の経過とともに収入を確認するための進行を測定する適切な方法が決定される
前払い費用:ライセンス契約が手配中に決定された他の履行義務とは異なると判定された場合、許可が被許可者に譲渡され、被許可者が使用許可を使用して利益を得ることができる場合には、許可に割り当てられた取引価格の収入を確認する。他の承諾とバンドルされたライセンスについては、ライセンスが主要項目とみなされているか否か、および合併履行義務が一定期間またはある時点で履行されているか否かを決定するために、合併履行義務の性質を評価するために使用される。
マイルストーン支払い:マイルストーン支払い(可変対価格)を含むそれぞれの手配の開始時に、マイルストーンが達成可能であると考えられるかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定する。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。臨床試験と規制承認の成功の不確実性により、関連事件が発生する前に、重大な収入逆転は不可能と考えられないため、開発と規制承認マイルストーン支払いは通常、関連活動が完了するまで実現不可能であると考えられる。
印税:販売ベースの特許使用料を含む手配については、販売レベルに基づく商業マイルストーン支払いを含み、許可が特許使用料に関連する主な項目とみなされている場合には、(I)関連販売が発生した場合、または(Ii)特許権使用料の一部または全部が割り当てられた履行義務が履行された(または部分的に履行されている)ときに収入を確認する。
精算、費用分担、利益共有支払い:いくつかの手配によると、私たちは一部の研究開発費の精算、あるいはそのような研究開発費のコスト分担に参加することを得ている。この精算と費用分担手配は私たちの研究開発費の減少に反映されています
総合業務報告書は、研究開発サービスの精算を提供することが私たちが行っている主要あるいは中央業務の一部だとは思わないからです。
研究と開発コストを計算すべきである
研究と開発費用は発生時に研究と開発費用を計上した。これらの費用には、賃金と福祉、持分補償費用、臨床前費用、臨床試験および関連臨床製造費用、契約サービスおよびその他の外部費用が含まれる開発活動を行うために発生する費用が含まれる。いくつかの研究および開発活動によって生じる費用は、契約研究組織、臨床試験研究場所、および契約製造組織によって実行される特定の活動に関連する費用を含み、特定のタスクの進捗または完了状況の評価に基づいて確認され、これらの評価は、供給者によって提供される実際の発生コストに関する情報のような時間に基づく測定基準またはデータを使用する。これらの活動の支払いは個別手配の条件に基づいており、これは費用確認のモデルとは異なる可能性がある。関連活動を行うサプライヤーが領収書を発行していない研究開発活動費用は、連結財務諸表に課税費用として反映される。今後研究·発展活動のために受け取った貨物やサービスの前払いは延期され資本化される。資本化金額は関連貨物の納入またはサービス提供時に費用を計上する。
私たちは私たちの推定が実際に発生した金額と実質的に差がないと予想する。本報告に掲げる期間中、私たちの計上費用と実費の間に実質的な差はありません。
最近の会計声明
詳細については、当社の連結財務諸表付記3“重要会計政策の概要--最近発表された会計公告”を参照されたい。
第七A項。 市場リスクの定量的·定性的開示について
私たちの投資活動の主な目標は流動性を保障して資本を保存することだ。私たちは正常な業務過程で市場リスクに直面している。このような危険は金利敏感性を含む。2022年12月31日と2021年12月31日現在、私たちはそれぞれ5.97億ドルと4.327億ドルの現金、現金等価物、および有価証券を持っており、銀行預金、高流動性通貨市場基金、高品質、高流動性が債務証券を売却できる投資を含む。金利の歴史的変動は私たちにとってそれほど大きくない。2022年12月31日まで、私たちは未済債務を持っていない。私たちの現金等価物の短期満期日と、私たちが販売可能な債務取引可能証券の高品質、高流動性の性質のため、金利が直ちに1ポイント変化することは、私たちの現金等価物の公平な市場価値に実質的な影響を与えない。将来のリスクを最小限に抑えるために、米国財務省と米国財務省が支援する買い戻し協定、短期米国国債、および会社債務証券、政府支援企業証券、商業手形を含む機関市場基金における現金等価物と有価証券のポートフォリオを維持するつもりだ。インフレ、金利の変化、あるいは為替レートの変動が、本稿で述べた任意の時期の経営業績に大きな影響を与えるとは思いません。
私たちは正常な業務過程で市場リスクに直面している。このような危険は主に金利敏感性を含む。
項目8.財務諸表と補足データ
連結財務諸表索引
| | | | | |
独立公認会計士事務所レポート(PCAOB ID:42) | 116 |
| |
合併貸借対照表 | 118 |
| |
連結業務報告書 | 119 |
| |
合併全面損失表 | 120 |
| |
株主権益合併報告書/(損失) | 121 |
| |
統合現金フロー表 | 122 |
| |
連結財務諸表付記 | 123 |
独立公認会計士事務所報告
SpringWorks治療会社の株主と取締役会に。
財務諸表のいくつかの見方
添付SpringWorks治療会社(“当社”)2022年12月31日現在と2021年12月31日までの総合貸借対照表、2022年12月31日現在の3年度に関する総合経営報告書、全面赤字、株主権益/(赤字)と現金流量および関連付記(総称して“総合財務諸表”と呼ぶ)を監査しました。総合財務諸表は、すべての重要な面で、2022年12月31日、2022年12月31日、2021年12月31日までの会社の財務状況、および2022年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
また、米国上場企業会計監督委員会(PCAOB)の基準に基づき、テレデビル委員会協賛組織委員会が発表した“内部統制-総合枠組み(2013枠組み)”で確立された基準に基づいて、2022年12月31日までの財務報告内部統制を監査し、2023年2月28日に発表した報告書に対して保留意見を発表した。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、監査委員会に伝達または要求が監査委員会に伝達された当期財務諸表監査によって生じる事項である:(1)財務諸表に対して重大な意義を有する勘定または開示に関するものであり、(2)特に挑戦的で主観的または複雑な判断に関するものである。重要な監査事項の伝達は、総合財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、以下の重要な監査事項を伝達することによって、重要な監査事項またはそれに関連する勘定または開示について個別の意見を提供することもない。
| | | | | |
| 研究と開発コストを計算すべきである |
| |
関係事項の記述
| 2022年12月31日現在、同社の研究·開発コストは総額1230万ドルである。総合財務諸表付記3に記載されているように、複数の研究開発活動による支出は、契約研究機関による特定活動、臨床試験に関する調査地点及び契約製造組織が行う特定活動に関する支出を含み、特定のタスクの進捗や完了状況の評価に基づいて確認され、この等の評価は、供給者が当社に提供する実コスト資料のような時間に基づく計量やデータを採用することができる。これらの活動の支払いは個別手配の条件により、発生した費用パターンとは異なる可能性がある。
監査会社の研究開発すべき項目は特に複雑であり、管理層は重大な判断を行う必要があるため、臨床試験の数量と第三者サービスプロバイダの使用程度によって各報告期間に発生したものとまだ請求書のコストに計上されていないと推定される。また,臨床試験の継続時間および第三者から領収書を受信した時間のため,監査報告日までに実際に発生した金額は通常不明である。 |
| |
私たちが監査でどのようにこの問題を解決したのか
| 著者らは会社の研究開発コストの内部制御について理解し、設計を評価し、計算に応じた研究開発費用の完全性と推定値の制御を含む内部制御の操作有効性をテストした。
会社の研究開発すべきプロジェクトをテストするために、私たちの監査プログラムは、管理層が記録すべきプロジェクトを推定するための重要な仮定を評価することと、基礎データの完全性と正確性をテストすることを含む。重大な仮説を検証するために、著者らは会社と第三者サービスプロバイダとの契約と任意の関連改訂を検査し、会社とこれらの活動を監督する研究開発者と臨床試験と他の研究開発プロジェクトの進展状況を確認し、第三者からこれまでに発生したコストの見積もりを含む直接第三者から情報を得た。また,後に第三者から受け取った伝票をテストし,記録された計算すべき項目の完全性を評価した. |
| |
| GSK plcとの非独占許可と連携プロトコル(“GSKプロトコル”)との収入確認 |
| |
関係事項の記述
| 総合財務諸表付記10で述べたように、同社は2022年12月31日現在、そのGSKプロトコルに関する1950万ドルの繰延収入を確認している。2022年12月31日までの1年間、この合意に関連した収入は取るに足らない
手配中に異なる履行義務を決定することは高い判断性を持ち,管理層は承諾した財やサービスおよびこれらの財やサービスが契約関係と区別できるかどうかを主観的に判断する必要がある.監査管理層は異なる履行義務を決定することはまた監査人の重大な判断を必要とする。 |
| |
私たちが監査でどのようにこの問題を解決したのか
| 監査会社の履行義務の決定のために、収入確認プロセスに関連する制御の有効性を試験すること、異なる履行義務を識別するための制御、GSKプロトコルを読んで承諾した商品及びサービスを決定すること、会社の会計結論文書を審査すること、および会計指導の適用を評価することを含む監査プログラムを実行する。GSK協定に関連した活動を監督する会社研究開発者および社内法律顧問と契約の理解および契約における当事者の権利と義務を確認しました |
/s/ 安永法律事務所
2018年以来、当社の監査役を務めてきました。
ニューヨーク、ニューヨーク
2023年2月28日
| | | | | | | | | | | |
| 十二月三十一日
2022 | | 十二月三十一日
2021 |
| (千単位で、共有および1株当たりのデータは含まれていない) | | | |
| 資産 | | | |
| 流動資産: | | | |
| 現金と現金等価物 | $ | 67,490 | | | $ | 103,961 | |
| 有価証券 | 524,722 | | | 269,540 | |
| 前払い費用と他の流動資産 | 7,548 | | | 9,409 | |
| 流動資産総額 | 599,760 | | | 382,910 | |
| 長期有価証券 | 4,794 | | | 59,230 | |
| 財産と設備、純額 | 13,571 | | | 3,187 | |
| 経営的リース使用権資産 | 4,698 | | | 1,010 | |
| 株式投資 | 4,193 | | | 2,883 | |
| 制限現金 | 578 | | | 565 | |
| その他の資産 | 2,648 | | | 2,709 | |
| 総資産 | $ | 630,242 | | | $ | 452,494 | |
| 負債と株主権益 | | | |
| 流動負債: | | | |
| 売掛金 | $ | 8,010 | | | $ | 3,429 | |
| 費用を計算する | 39,242 | | | 25,378 | |
| 賃貸負債を経営し、流動 | 483 | | | 1,162 | |
| 収入を繰延し,当期 | 3,314 | | | — | |
| 流動負債総額 | 51,049 | | | 29,969 | |
| 長期経営賃貸負債 | 4,768 | | | 129 | |
| 収入を繰延し,長期 | 16,233 | | | — | |
| 総負債 | 72,050 | | | 30,098 | |
| 引受金とその他の事項 | | | |
| 株主権益: | | | |
優先株 $0.0001額面は 10,000,000株式を許可して 違います。発行済みまたは発行済み株式 卓越した 2022年12月31日と2021年12月31日に。 | — | | | — | |
普通株、$0.0001額面は150,000,000株式を許可して62,453,328そして49,247,985発行済みおよび発行済み株式62,423,129そして49,247,9852022年12月31日と2021年12月31日にそれぞれ発行された株。 | 6 | | | 5 | |
| 追加実収資本 | 1,130,224 | | | 715,216 | |
| 赤字を累計する | (569,930) | | | (292,513) | |
在庫株は,コストで計算する30,199そして0それぞれ2022年12月31日と2021年12月31日の普通株)。 | (1,341) | | | — | |
| その他の総合損失を累計する | (767) | | | (312) | |
| 株主権益総額 | 558,192 | | | 422,396 | |
| 総負債と株主権益 | $ | 630,242 | | | $ | 452,494 | |
連結財務諸表の付記を参照。
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| (千単位で、共有および1株当たりのデータは含まれていない) | 2022 | | 2021 | | 2020 |
| 許可収入 | $ | — | | | $ | — | | | $ | 35,000 | |
| 運営費用: | | | | | |
| 研究開発 | 146,122 | | | 101,676 | | | 51,859 | |
| 一般と行政 | 134,552 | | | 71,792 | | | 29,465 | |
| 総運営費 | 280,674 | | | 173,468 | | | 81,324 | |
| | | | | |
| 運営損失 | (280,674) | | | (173,468) | | | (46,324) | |
| 利息とその他の収入: | | | | | |
| その他の収入,純額 | (138) | | | (152) | | | 25 | |
| 利子収入,純額 | 6,285 | | | 698 | | | 1,330 | |
| 利子とその他の収入総額 | 6,147 | | | 546 | | | 1,355 | |
| 株式投資損失 | (2,890) | | | (988) | | | (605) | |
| 純損失 | $ | (277,417) | | | $ | (173,910) | | | $ | (45,574) | |
| | | | | |
| 1株当たり基本と希釈して純損失 | $ | (5.21) | | | $ | (3.59) | | | $ | (1.05) | |
| 加重平均発行済み普通株式、基本普通株式、希釈後普通株 | 53,290,528 | | | 48,497,790 | | | 43,300,063 | |
| | | | | |
連結財務諸表の付記を参照。
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| (単位:千) | 2022 | | 2021 | | 2020 |
| 純損失 | $ | (277,417) | | | $ | (173,910) | | | $ | (45,574) | |
| その他の全面的な収益変動: | | | | | |
| 有価証券が収益を実現せず,純額 | (455) | | | (353) | | | 41 | |
| その他総合収益変動総額 | $ | (455) | | | $ | (353) | | | $ | 41 | |
| 総合損失 | $ | (277,872) | | | $ | (174,263) | | | $ | (45,533) | |
連結財務諸表の付記を参照。
| | |
SpringWorks治療会社
株主権益合併報告書/(損失) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | 2020年、2021年、2022年12月31日までの年度 |
| | | | | | | | | | | | | | 追加実収資本 | | その他の総合収益を累計する | | 赤字を累計する | | 合計する |
| | | | | | ごく普通である | | 財務局 | | | | |
| (千単位、共有および単位データを除く) | | | | | | | | | | 株 | | 金額 | | 株 | | 金額 | | | | |
| 2019年12月31日の残高 | | | | | | | | | | 43,006,077 | | | $ | 4 | | | — | | | $ | — | | | $ | 395,097 | | | $ | — | | | $ | (73,029) | | | $ | 322,072 | |
| 株に基づく報酬費用 | | | | | | | | | | | | | | | | | | 10,034 | | | | | | | 10,034 | |
| 後続発行終了時に普通株式を発行し、発行コストを差し引く | | | | | | | | | | 5,637,254 | | | 1 | | | | | | | 269,591 | | | | | | | 269,592 | |
| 制限株奨励を没収する | | | | | | | | | | (14,224) | | | | | | | | | | | | | | | — | |
| 株式オプションの行使 | | | | | | | | | | 190,484 | | | | | | | | | 893 | | | | | | | 893 | |
| その他の総合収益、税引き後純額 | | | | | | | | | | | | | | | | | | | | 41 | | | | | 41 | |
| 純損失 | | | | | | | | | | | | | | | | | | | | | | (45,574) | | | (45,574) | |
| 2020年12月31日残高 | | | | | | | | | | 48,819,591 | | | $ | 5 | | | — | | | $ | — | | | $ | 675,615 | | | $ | 41 | | | $ | (118,603) | | | $ | 557,058 | |
| 株に基づく報酬費用 | | | | | | | | | | | | | | | | | | 38,444 | | | | | | | 38,444 | |
| 制限株式奨励を発行する | | | | | | | | | | 332,226 | | | — | | | | | | | | | | | | | — | |
| 制限株奨励を没収する | | | | | | | | | | (55,475) | | | — | | | | | | | | | | | | | — | |
| 株式オプションの行使 | | | | | | | | | | 151,643 | | | — | | | | | | | 1,157 | | | | | | | 1,157 | |
| その他総合損失、税引き後純額 | | | | | | | | | | | | | | | | | | | | (353) | | | | | (353) | |
| 純損失 | | | | | | | | | | | | | | | | | | | | | | (173,910) | | | (173,910) | |
| 2021年12月31日の残高 | | | | | | | | | | 49,247,985 | | | $ | 5 | | | — | | | $ | — | | | $ | 715,216 | | | $ | (312) | | | $ | (292,513) | | | $ | 422,396 | |
| 株に基づく報酬費用 | | | | | | | | | | | | | | | | | | 72,965 | | | | | | | 72,965 | |
| グラクソ·スミスクラインに普通株を発行する | | | | | | | | | | 2,050,819 | | | — | | | | | | | 55,454 | | | | | | | 55,454 | |
| 私募で普通株を発行し,発行コストを差し引く | | | | | | | | | | 8,650,520 | | | 1 | | | | | | | 216,830 | | | | | | | 216,831 | |
| 市価で発行される普通株の発行は,発行コストを差し引く | | | | | | | | | | 2,247,500 | | | — | | | | | | | 67,782 | | | | | | | 67,782 | |
| 制限株式奨励を発行する | | | | | | | | | | 36,625 | | | — | | | | | | | | | | | | | — | |
| 制限株奨励を没収する | | | | | | | | | | (27,957) | | | — | | | | | | | | | | | | | — | |
| 帰属制限株式単位 | | | | | | | | | | 24,369 | | | — | | | | | | | | | | | | | — | |
| 株式オプションの行使 | | | | | | | | | | 223,467 | | | — | | | | | | | 1,977 | | | | | | | 1,977 | |
| 源泉徴収代行税を履行するための普通株 | | | | | | | | | | | | | | 30,199 | | | (1,341) | | | | | | | | | (1,341) | |
| その他の総合収益、税引き後純額 | | | | | | | | | | | | | | | | | | | | (455) | | | | | (455) | |
| 純損失 | | | | | | | | | | | | | | | | | | | | | | (277,417) | | | (277,417) | |
| 2022年12月31日の残高 | | | | | | | | | | 62,453,328 | | | $ | 6 | | | 30,199 | | | $ | (1,341) | | | $ | 1,130,224 | | | $ | (767) | | | $ | (569,930) | | | $ | 558,192 | |
連結財務諸表の付記を参照。
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| (単位:千) | 2022 | | 2021 | | 2020 |
| 経営活動 | | | | | |
| 純損失 | $ | (277,417) | | | $ | (173,910) | | | $ | (45,574) | |
| 純損失と経営活動で使用される現金純額の調整: | | | | | |
| 減価償却費用 | 765 | | | 490 | | | 349 | |
| 非現金でレンタル料金を扱っております | 1,131 | | | 993 | | | 1,049 | |
| 株補償費用 | 72,965 | | | 38,444 | | | 10,034 | |
| 株式投資損失 | 2,890 | | | 988 | | | 605 | |
| 経営性資産と負債の変動 | | | | | |
| 前払い費用と他の流動資産 | 1,861 | | | (4,609) | | | (1,205) | |
| その他の資産 | 61 | | | (707) | | | (843) | |
| 売掛金 | 4,168 | | | 2,074 | | | (1,304) | |
| 費用を計算する | 13,325 | | | 9,912 | | | 5,945 | |
| リース責任 | (859) | | | (1,388) | | | (1,411) | |
| 収入を繰り越す | 19,547 | | | — | | | — | |
| その他負債 | — | | | (164) | | | 164 | |
| 経営活動のための現金純額 | $ | (161,563) | | | $ | (127,877) | | | $ | (32,191) | |
| 投資活動 | | | | | |
| 資本支出 | (10,196) | | | (2,016) | | | (642) | |
| 株式投資 | (4,200) | | | — | | | (3,500) | |
| 有価証券を購入する | (481,050) | | | (305,423) | | | (442,690) | |
| 満期債務証券の売却と収益 | 279,849 | | | 391,031 | | | 28,000 | |
| 投資活動提供の現金純額 | $ | (215,597) | | | $ | 83,592 | | | $ | (418,832) | |
| 融資活動 | | | | | |
| 後続発行終了時に普通株を発行して得られた収益は,発行コストを差し引く | — | | | — | | | 269,592 | |
| GSKに普通株で得た金を発行する | 55,454 | | | — | | | — | |
| 私募で普通株を発行して得られる収益は,発行コストを差し引く | 216,830 | | | — | | | — | |
| 市価で普通株を発行して得られた収益は,発行コストを差し引く | 67,782 | | | — | | | — | |
| 在庫株 | (1,341) | | | — | | | — | |
| 株式オプションを行使して得られる収益 | 1,977 | | | 1,157 | | | 893 | |
| 融資活動が提供する現金純額 | $ | 340,702 | | | $ | 1,157 | | | $ | 270,485 | |
| 現金と現金等価物の純減少 | (36,458) | | | (43,128) | | | (180,538) | |
| 制限された現金、期初を含む現金と現金等価物 | 104,526 | | | 147,654 | | | 328,192 | |
| 制限された現金、期末を含む現金と現金同等物 | $ | 68,068 | | | $ | 104,526 | | | $ | 147,654 | |
| | | | | |
| ASC 842の採用のため、上記の補足非現金項目は含まれていない | | | | | |
| 経営的リース使用権資産の初歩的確認 | $ | — | | | $ | — | | | $ | 2,879 | |
| 賃貸負債の初歩的確認 | — | | | — | | | (4,030) | |
| | | | | |
| 非現金投資活動 | | | | | |
| 経営性賃貸義務と引き換えに使用権資産 | $ | 5,580 | | | $ | — | | | $ | 31 | |
連結財務諸表の付記を参照。
SpringWorks治療会社
連結財務諸表付記
1.運営の性質
SpringWorks治療会社、あるいは当社と呼ばれ、2017年8月18日にデラウェア州に設立されました。
同社は臨床段階の生物製薬会社であり、精確な医学方法を応用して、壊滅的な稀な疾病と癌を受けた十分なサービスを受けていない患者群のために生活を変える薬物を獲得、開発、商業化している。同社は異なる小分子標的腫瘍学候補製品の組み合わせがあり、そして2つの稀な腫瘍タイプに対する末期臨床試験、1つの登録と1つの潜在登録、及び他のいくつかの高度に流行している遺伝子に対して癌を定義する計画を進めている2点これらの方案には末期臨床候補製品がある:ナイルシットとミダメチニブ。2022年12月、同社はアメリカ食品·薬物管理局に新薬申請を提出し、成人硬線維腫の治療を要求した。FDAは2023年2月にNDA申請を受け,優先審査を承認し,処方薬ユーザ費用法案(PDUFA)を割り当て,目標行動日は2023年8月27日であった。
私募する
二零二二年九月七日に、当社はいくつかの認可投資家又は投資家と証券購入協定を締結し、これにより、当社は私募取引又は私募方式で投資家に売却及び発行することに同意した8,650,520普通株、買い取り価格は$26.01一株ずつです。方向性増発では、同社は約#ドルの毛収入を受けた225100万ドルで、手数料と発行コストを差し引いて、純収益は約#ドルです216.8百万ドルです。方向性増発については、当社は投資家と登録権協定を締結し、期日は2022年9月7日で、転売株式の登録を規定している。2022年9月26日に米国証券取引委員会に提出された株式に関する登録説明書と募集説明書補編によると、当該等の株式は転売を登録する。
2022年9月6日、当社はグラクソ·スミスクライン、plc、元グラクソ·スミスクライン、またはグラクソ·スミスクラインとniroacestatとbelantamab mafodotin(Belamaf)について拡大したグローバル非独占許可および協力協定を締結し、この協定に署名するとともに、グラクソ·スミスクラインの関連会社グラクソ·スミスクライン、グラクソ·スミスクグループ株式会社またはグラクソ·スミスクラインと株式購入協定を締結し、この合意に基づき、グラクソ·スミスクラインは私募取引で当社に買収することに同意した2,050,819普通株、総購入価格は約$です75.0百万ドル、あるいはドル36.57一株ずつです。これらの株は安値で売っている25当社の普通株式出来高加重平均株価割増率30日間株式購入契約を締結するまでの間。
市場で製品を提供する
2021年2月25日、当社はCowen and Company,LLCと販売契約を締結し、この合意により、当社は普通株式株式を発行および売却することができ、総発売金は最高$に達する200.0万元は時々コーエンを通じて私たちの販売代理とします。配給通知を出した後、販売契約の条項及び条件を満たした場合、コーエンは、改正された1933年に証券法が公布された第415(A)(4)条の規則に基づいて定義された“市場発売”とみなされる法律で許可された任意の方法で株を売却することができる。当社は販売契約に基づいて普通株株式を売却することができ、売却金額及び時間は販売契約の条項及び条件に依存しますが、当社は販売契約に基づいていかなる株式も売却する義務はありません
2022年12月31日までの12ヶ月間、当社は販売しました2,247,500ATM機計画下の普通株、総収益は#ドルです69.7百万ドル、手数料と他の費用を引いて$1.9純収益は百万ドルである67.8百万ドルです。2022年12月31日までに130.3現金自動支払機計画によると、まだ100万人が利用できます
後続サービス
2020年10月13日会社が完成しました5,637,254普通株式を含む公開発行された普通株式735,294引受業者が追加株式を購入するオプションを十分に行使して売却する普通株により、発行価格は$となる51.001株当たり、会社に純収益$をもたらす269.5百万ドルです。
新冠肺炎が大流行する
2020年3月11日、世界保健機関は新型コロナウイルス新冠肺炎株に関連する疾病の爆発を全世界の大流行とした。この病気は新たに出現したCOVID-19変異株を含めて
会社が運営している地域に広がり続けている。新冠肺炎の蔓延を緩和するため、世界各国の政府と企業は現地避難令、隔離措置、旅行に対する重大な制限、および多くの従業員の出勤禁止の制限を含む未曽有の行動を取った。新冠肺炎疫病発生から、同社は一連の業務連続性措置を開始し、その運営に対する潜在的な中断を減少し、そしてその研究開発計画の完全性を維持した。新冠肺炎の流行期間中、会社の対応措置は変化し、会社はすでに基本的に正常な運営を回復した。これまで、会社が現在行っている研究開発活動はまだ実質的な妨害を受けていない;しかし、新冠肺炎の大流行、あるいは新冠肺炎ウイルスが新たに出現した変異株のいかなる影響により、ワクチン接種率の停滞及び関連要素により、会社はその研究開発と商業化に影響を与える可能性のある中断を経験する可能性がある スケジュールと結果です。同社は引き続き発生している新冠肺炎の流行および新興変種のその業務への影響を評価する。現在の新冠肺炎大流行が当社の未来の業績に与える影響程度は未来の発展に依存するが、今回の大流行及びその関連影響は、大流行の持続時間、蔓延と強度(任意の再発を含む)、新冠肺炎ウイルス新変異株の影響及び新冠肺炎ワクチンの発売を含み、これらはすべてまだ不確定かつ予測困難であり、当社の業務、将来性、未来の財務状況、運営結果とキャッシュフローに実質的な影響を与える可能性がある。
2.リスクと流動性
会社は設立以来損失と経営キャッシュフローを負にし、累計損失#ドルを計上した569.9百万ドルとドル292.5100万ドル運営資本は$548.7百万ドルとドル352.9それぞれ2022年12月31日と2021年12月31日である。今まで、私たちのすべての収入および繰延収入は、JazzプロトコルおよびGSKライセンスプロトコルに従って受信された払い戻し不可能な前払いから来ています。その会社は商業販売のために許可された製品や経常的な収入源を持っていない。同社はいずれのバイオ製薬会社にも関連するリスクに直面しており、これらの会社は開発に大量の支出を抱えている。会社の開発プロジェクトが成功することは保証されず、開発された製品は必要な規制承認を受けるか、あるいは任意の承認された製品は商業的可能性があるだろう。また,同社は技術が急速に変化する環境で運営されており,その従業員,コンサルタント,コンサルタント,サプライヤーのサービスに大きく依存している。
会社は現金、現金等価物、有価証券#ドルを持っている597.0百万ドルとドル432.7それぞれ2022年12月31日と2021年12月31日まで。会社の2022年12月31日の現金、現金等価物、および有価証券によると、経営陣は、その現在の流動資金は、財務諸表の発表日後少なくとも12ヶ月以内に運営費用を支払うことができるようになると推定している。
3.重要会計政策の概要
陳述の基礎
これらの連結財務諸表は米国公認会計原則または米国公認会計原則に基づいて作成された。
合併原則
連結財務諸表には、SpringWorks治療会社とその子会社(総称して当社と呼ぶ)の勘定が含まれる。すべての会社間取引と残高は合併で販売された。当社はコントロール権が不足しているが、確かに経営と財務政策に重大な影響を与える商業実体への投資は、権益会計方法を用いて入金することができる。
予算の使用
米国公認会計原則に従って財務諸表を作成することは、財務諸表と付記中の報告書の金額に影響を与えるために、管理層に推定と仮定を要求する。これらの連結財務諸表に反映される重大な推定および仮定には、研究および開発費用、株式に基づく報酬報酬の推定値が含まれるが、これらに限定されない。当社は過去の経験、既知の傾向、その他その当時の状況にあると考えられる合理的な市場特定または関連要素に基づいて推定します。実際の結果はこれらの推定とは異なる可能性がある。継続的な基礎の上で、経営陣はその推定を評価し、事実や状況が変化した場合にこれらの推定および仮定を調整する。推定された変化は、それらが知られている期間に記録される。
市場情報を細分化する
業務部門は、エンティティの構成要素として定義され、そのエンティティに関する個別離散情報は、首席業務意思決定者または意思決定グループが、リソースをどのように割り当てるかを決定し、業績を評価する際に評価することができる。会社は以下の点でその運営·管理業務を確認します1つは運営部門です。
現金と現金等価物
当社は買収時満期日が三ヶ月以下のすべての高流動性手形を現金等価物と見なしています。2022年12月31日と2021年12月31日まで、会社の現金と現金等価物はドルです67.5百万ドルとドル104.0それぞれ100万ドルです
有価証券
売却可能な債務証券は公正価値で報告され、損益計上は他の総合収益を計上していない。各報告期間内に、会社は公正価値が償却コストよりも低下したかどうかを評価し、これらの低下は信用損失によるかどうか、および企業が予想回復まで投資する能力と意図を持っている。この2つの基準がいずれも保有意向または保有能力に適合している場合、信用損失による公正価値の低下は、会社総合経営報告書上の他の収入(費用)によって計上されるが、公正価値は償却コストベースの金額よりも小さいという制限を受ける。上記のいずれかの基準に適合できなかった場合、以前に記録された信用損失準備および公正な価値を超える任意の償却コスト基準は、当社の総合経営報告書の他の収入(支出)に記入される。2022年12月31日まで及び2021年12月31日までの年度内に、当社はその有価証券の信用損失或いは減値について準備していない。
信用リスクが集中する
会社を集中的な信用リスクに直面させる可能性のある金融商品は、主に現金、現金等価物、および販売可能な有価証券を含む。会社はその現金、現金等価物残高および有価証券残高と高品質の金融機関を維持し、会社の有価証券は会社の債務証券、米国政府証券、商業手形を含む高品質で流動性の高い債務証券に投資する。
財産と設備
財産と設備はコンピュータ設備、ソフトウェア、家具、レンタル改善を含み、コストで入金される。財産と設備はその推定耐用年数内に直線的に減価償却される。
収入.収入
同社が受け取った薬品開発や商業化に関する対価格収入を確認することは,社内開発,第三者との共同開発や協力協定,製品権利の売却や対外許可など様々な方式で行われている。これらの手配および合意の条項は、ライセンス、ノウハウ、医薬品、関連プロトコル、および他の交付可能な成果を含むことができる複数のコミットメントされた貨物およびサービスを含むことができる。これらの手配に基づいて会社に支払われるお金は、前払い許可料、マイルストーン支払い、および将来の製品販売の特許権使用料のうちの1つまたは複数を含むことができる。
ASC 606の範囲内の手配、顧客との契約収入
同社は、ASC 606に従って収入を確認し、これは、顧客とのすべての契約に適用されるが、協力手配およびレンタルのような他の標準範囲内の契約は含まれていない。
ASC 606によれば、その顧客が約束された商品またはサービスの制御権を取得すると、会社は収入を確認し、その金額は、会社がこれらの商品またはサービスと交換することが予期されている対価格を決定することを反映する。会社がASC 606の範囲内に属すると認定した手配の収入確認を決定するために、会社は、(I)顧客との契約を決定するステップ、(Ii)契約中の履行義務を決定するステップ、(Iii)取引価格を決定するステップ、(Iv)契約に取引価格を割り当てる履行義務、および(V)会社がその履行義務を履行する際に収入を確認するステップの5つのステップを実行する。これらの手配の会計作業の一部として、会社は、契約中の履行義務の確定、取引価格に含まれる可変対価格金額の推定、取引価格を個々の履行義務に割り当てることを含む重大な判断を行う必要があるかもしれない。
契約がASC 606の範囲内にあると決定されると、会社は、契約において約束された商品またはサービスを評価し、どれが義務を履行するかを決定する。
契約における履行義務を決定するために、会社は各約束された貨物またはサービスが異なるかどうかを評価する。このような評価は、管理層に個別に約束された貨物またはサービスの判断、およびこれらの貨物またはサービスが契約関係の他の態様から分離可能であるかどうかを要求することができる主観的決定に関するものである。約束された貨物およびサービスは異なると考えられ、条件は、(1)顧客が単独で、または顧客がいつでも入手可能な他のリソースと共に貨物またはサービスから利益を得ることができる(すなわち、貨物またはサービスが異なることができる)、(2)エンティティが顧客に貨物またはサービスを譲渡する約束は、契約内の他の約束とは別に識別可能である(すなわち、貨物またはサービスを譲渡する約束は契約範囲内で異なる)ことである。承諾された商品やサービスが独特であるかどうかを評価する際には,会社は顧客の研究,製造と商業化能力,関連専門知識の一般市場での可用性などを考慮する.約束された貨物またはサービスが契約中の他の約束とは別に識別できるかどうかを評価する際に、会社はまた、契約の予想利益を考慮する。約束された商品またはサービスが異なる場合、エンティティは、異なる商品またはサービスを識別するまで、その商品またはサービスを他の約束された商品またはサービスと統合することが要求される。
契約で約束された対価格に可変金額が含まれている場合、会社は、約束された貨物またはサービスを顧客に移転するために、獲得する権利のある対価格金額を推定する。当社は期待値法または最大可能値法を用いて可変対価金額を決定します。同社は制限されない推定可変対価金額を取引価格に計上している。取引価格に含まれる金額は,確認された累積収入が大きく逆転しない可能性の高い金額に制限される.その後報告期間ごとに終了すると,当社は取引価格に含まれる推定変動コストおよび任意の関連制限を再評価し,必要に応じて全体の取引価格の推定を調整する.いずれの調整も調整期間内に累積追跡方式で記録されている.
そして、各履行義務がある時点または一定期間にわたって履行され、時間が経過するにつれて、顧客への貨物やサービスの産出または投入方法の使用が最もリアルに記述されている場合には、当社は、各履行義務に割り当てられた取引価格の金額を収入として確認する。
知的財産権ライセンス:会社ライセンス契約の条項には機能的知的財産権の許可が含まれており,知的財産権の機能は会社が行っている活動によって大きく変化しないと予想されるためである。当社は、他の承諾とバンドルされたライセンス(すなわち、手配中の他のコミットメント商品およびサービスと区別されていないライセンス)について、合併履行義務が時間の経過とともに履行されているか、またはある時点で履行されているかを決定するために、判断を利用して、合併履行義務が時間の経過とともに履行されているかを決定し、時間の経過とともに収入を確認するための進捗を測定するための適切な方法を決定する
前払い費用:ライセンス契約が手配中に決定された他の履行義務とは異なると判定された場合、ライセンスが被許可者に譲渡され、被許可者が使用許可を使用して利益を得ることができる場合、会社は、許可に割り当てられた取引価格の収入を確認する。他の承諾とバンドルされたライセンスについては、会社は、ライセンスが主要項目とみなされているかどうか、および合併履行義務が一定期間またはある時点で履行されているかどうかを決定するために、合併履行義務の性質を評価するために判断を利用する。
マイルストーン支払い:マイルストーン支払い(可変対価格)を含む各手配の開始時に、会社はマイルストーンが達成可能であると考えられているかどうかを評価し、最も可能な金額法を用いて取引価格に含まれる金額を推定する。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。臨床試験と規制承認の成功の不確実性により、関連事件が発生する前に、重大な収入逆転は不可能と考えられないため、開発と規制承認マイルストーン支払いは通常、関連活動が完了するまで実現不可能であると考えられる。
印税:販売に基づく特許権使用料を含む手配については、販売レベルに基づく商業マイルストーン支払いを含み、許可が特許権使用料に関連する主要項目とみなされ、当社は、(I)関連販売が発生した場合、または(Ii)特許権使用料の一部または全部が割り当てられた履行義務が履行された(または部分的に履行されている)ときに収入を確認する。
精算、費用分担、利益共有支払い:ある手配によると,その会社は一部の研究·開発費用の補償を受けているか,あるいはこのような研究·開発に参加する費用を分担している
料金です。この精算とコスト分担手配は、会社が精算を行う研究や開発サービスがその継続主要または中核業務の一部であるとは考えていないため、会社の総合経営報告書の研究や開発費用に反映されている。
研究と開発
ASC 730“研究·開発”によると,臨床開発支出は,米国食品·医薬品局の承認を得ていない製品に関する前払い許可料とマイルストーン支払いを含み,発生時に研究·開発費用を計上している。これらの費用には、賃金と福祉、持分補償費用、臨床前費用、臨床試験および関連臨床製造費用、契約サービスおよびその他の外部費用が含まれる開発活動を行うために発生する費用が含まれる。ある研究開発活動によって生じる費用は、契約研究機関、臨床試験に関連する調査場所および契約製造組織が行う特定の活動に関する費用を含み、特定のタスクの進捗または達成状況の評価、またはサプライヤーが実際に完了した活動または発生したコストに関する情報を当社に提供するような時間に基づく措置またはデータを使用して確認される。これらの活動の支払いは個別手配の条件に基づいており、これは費用確認のモデルとは異なる可能性がある。関連活動を行うサプライヤーが領収書を発行していない研究開発活動費用は、連結財務諸表に課税費用として反映される。今後研究·発展活動のために受け取った貨物やサービスの前払いは延期され資本化される。資本化金額は関連貨物の納入またはサービス提供時に費用を計上する。
一般と行政
一般と行政費用は主に賃金と関連費用を含み、行政、財務、会社、商業、業務発展と行政機能者の株式報酬を含む。一般および行政費用には、特許および会社の事務に関連する法律費用、会計、監査、税務およびコンサルティングサービスの専門費用、保険料、行政出張費用、直接減価償却コストおよび施設の賃貸料および維持費用、ならびに他の運営費用が含まれる施設関連費用も含まれる。
株式ベースの報酬費用
同社は、ASC 718“報酬-株式報酬”に基づいて従業員株式報酬を会計処理し、これは、従業員および非従業員取締役のすべての持分報酬について、運営報告書において、付与日報酬の公正価値に基づいて費用として確認することを要求する。会社の株式ベースの奨励は通常授与されます三つあるいは…4年.
株式補償費用は,付与日公正価値をもとに,奨励に必要なサービス期間内(通常は帰属期限)に直線法で確認する。
業績条件に制限された奨励と、市場と業績条件を同時に含む報酬については、経営陣がマイルストーンを実現する可能性があると判断した場合、企業は残りのサービス期間内に加速確認方法を用いて持分奨励補償費用を確認する。管理層は現在までの報告日の業績条件に対する期待満足度に基づいて、いつ業績を基礎とするマイルストーンを実現することが可能かを評価する。
当社は“会計基準更新指針”(ASU、番号2016-9)の採択に基づき、実際の没収事件発生時に没収を確認します。
モンテカルロシミュレーション方法を用いて市場条件によって付与された業績奨励の授与日の公正価値を推定し、この方法は授与日までの業績条件が満たされる確率を組み入れた。
発行された株式オプションについて、会社は、ブラック·スコアーズオプション定価モデルを使用して、付与日の公正価値と、それによって生成される株式ベースの補償費用とを推定する。
ブラック·スコアーズオプション定価モデルは、株式報酬公正価値を決定するいくつかの主観的仮定を使用することを要求する。ブラック·スコアーズオプション定価モデルで使用される投入は以下の通り
•普通株の公正価値、すなわち会社普通株の現在の取引価格である。
•予想期間-予想期間は、株式に基づく奨励予想未償還期間を表す。当社が使用できる当社固有の歴史資料は限られているため、当社は簡略化方法を用いて期待期限を計算しています。
•予想変動率-同社は会社固有の歴史と隠れた変動率情報が不足している。そのため、主に1組の上場同業者会社の履歴変動率に基づいて予想される株式変動率を推定し、それ自身の取引株変動性に関する十分な履歴データを有するまで継続することが予想される。
•無リスク金利-無リスク金利は、ゼロ金利米国国庫券を付与する際に有効な米国国庫券収益率に基づいており、その満期日は奨励の期待期限にほぼ等しい。
•期待配当-同社はその普通株または株式に配当を支払ったことがなく、その普通株に配当を支払う計画もない。したがって期待配当収益率はゼロ.
1株当たり純損失
1株あたりの基本純損失の計算方法は,純損失を当期に発行された加重平均株式数で割ることである。1株当たりの純損失を希釈するには、会社の純損失により、それらの影響が逆薄になるため、未帰属制限株や株式オプションの潜在的な影響は含まれない。当社は報告の期間ごとに純損失があるため、1株当たりの基本純損失は希釈後の純損失と同じです。
所得税
所得税は貸借対照法を用いて計算される。繰延税項資産及び負債は、既存資産及び負債の帳簿金額及びそれぞれの計税基礎と営業損失及び税項相殺繰越との差異による将来の税務結果を確認する。税金資産及び負債を繰延して税率計量を策定し、その等の一時的な差額を回収又は決済する予定の年度の課税収入に適用されることが予想される。税率変動が繰延税金資産や負債に及ぼす影響は、公布日を含む期間中に収入として確認された。繰延税金資産と負債の変動を所得税に計上する準備。
当社は繰延税金資産の範囲を確認し、これらの資産がより現金化する可能性があると考えている。この決定を下す際には、管理層は、既存の課税の一時的な差異の将来の輸出、将来の課税収入の予想、税務計画戦略、最近の業務の結果を含む、利用可能なすべてのプラスおよび負の証拠を考慮する。既存の証拠の重みに基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合には、推定免税額を計上する。経営陣が当社が将来その記録純額を超える繰延税金資産を実現できると判断した場合、管理層は繰延税金資産の推定値を調整し、所得税の支出を減らすことになる。
当社は、米国会計基準第740条の規定に基づき、2段階の手順で不確定な税務倉位を記録し、その中で、(1)管理層は、税務倉位の技術的利点に基づいて税務倉位を維持する可能性が高いかどうかを決定し、(2)確認の可能性が高い敷居に適合した税務倉位について、管理層は、最終的に関連税務機関と和解した後に実現可能な50%を超える最大税務優遇額を確認する。
当社は不確定な税務状況に関連して各税務機関に支払うことが可能な税金のための準備金を保留しています。これらの準備金は、当社がその申告書類または頭寸で得た税金優遇があるかどうか、および税収割引に関連する任意の潜在的または有事項を解決した後に達成される可能性が高いかどうかの決定に基づいている。所得税の少納に関連する潜在的利息は所得税費用の1つの構成要素に分類され、任意の関連する処罰は経営報告書中の所得税費用に分類される。
最近採用と最近発表された会計公告
最近採用された会計声明は会社の財務諸表に大きな影響を与えておらず、最近発表された会計声明も会社の財務諸表に大きな影響を与えていないと予想される。
4.有価証券
次の表は、会社が2022年12月31日までと2021年12月31日までの帳簿純価値で販売可能な有価証券をまとめています
| | | | | | | | | | | | | | | | | | | | | | | |
| 2022年12月31日まで |
| (単位:千) | 原価を償却する | | 未実現収益総額 | | 未実現損失総額 | | 公正価値を見積もる |
| 有価証券: | | | | | | | |
| 短期投資: | | | | | | | |
| アメリカ政府証券 | $ | 232,229 | | | $ | — | | | $ | (690) | | | $ | 231,539 | |
| 非アメリカ政府証券 | 9,388 | | | — | | | (31) | | | 9,357 | |
| 会社債務証券 | 45,710 | | | — | | | (44) | | | 45,666 | |
| 商業手形 | 238,160 | | | — | | | — | | | 238,160 | |
| 長期投資: | | | | | | | |
| 会社債務証券 | 4,796 | | | | | (2) | | | 4,794 | |
| 合計する | $ | 530,283 | | | $ | — | | | $ | (767) | | | $ | 529,516 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| 2021年12月31日まで |
| (単位:千) | 原価を償却する | | 未実現収益総額 | | 未実現損失総額 | | 公正価値を見積もる |
| 有価証券: | | | | | | | |
| 短期投資: | | | | | | | |
| アメリカ政府証券 | $ | 105,043 | | | $ | 3 | | | $ | (79) | | | $ | 104,967 | |
| 会社債務証券 | 78,729 | | | — | | | (52) | | | 78,677 | |
| 商業手形 | 85,896 | | | — | | | — | | | 85,896 | |
| 長期投資: | | | | | | | |
| アメリカ政府証券 | 59,414 | | | — | | | (184) | | | 59,230 | |
| 合計する | $ | 329,082 | | | $ | 3 | | | $ | (315) | | | $ | 328,770 | |
同社の有価証券は売却可能な証券であり、高品質、高流動性の債務証券からなり、会社債務証券、米国政府証券、非米国政府証券、商業手形を含む。
当社が総合貸借対照表で短期有価証券に分類している売却可能証券は、貸借対照表の日から1年以下で満期になります。貸借対照表の日から1年以上満期となる有価証券は長期証券に分類される。当社は2022年12月31日現在、5年以上の投資を保有していません。
5.公正価値計量
経常的な基礎の上で計量した会社の金融資産の公正価値は公正価値等級によって分類され、公正価値等級は以下の3つのレベルからなる
第1レベル-アクティブ市場の未調整見積は、計量日に同じ、制限されていない資産又は負債の見積を得ることができる。
第2レベル--アクティブ市場における同様の資産および負債のオファー、非アクティブ市場におけるオファー、または実質的にツール期間全体にわたって直接または間接的に観察可能な投入。
第3レベル--価格または推定技術は、公正な価値計量に重大な意義があり、観察できない投入(すなわち、市場活動の支持が少ないかないか)を必要とする。
公正価値等級は観察可能または観察不可能な公正価値を計量するための推定技術に基づく投入である。観察可能な投入は、資産または負債に価格を設定する際に市場参加者が使用するという仮定を反映している
独立したソースから得られた市場データであり,観察できない投入は,報告実体がそれ自身の市場仮定に基づいた定価を反映している.
以下の表は、企業が2022年12月31日まで、2021年12月31日までに経常的に計測した金融資産と負債の公正価値レベルを示している
| | | | | | | | | | | | | | | | | | | | | | | |
| 2022年12月31日まで |
| | | 公正価値階層構造 |
| (単位:千) | 合計する | | レベル1 | | レベル2 | | レベル3 |
| 公正な価値で勘定された金融商品(資産頭寸): | | | | | | | |
| 現金等価物: | | | | | | | |
| 貨幣市場基金 | $ | 22,494 | | | $ | 22,494 | | | $ | — | | | $ | — | |
| | | | | | | |
| 短期投資: | | | | | | | |
| アメリカ政府証券 | 231,539 | | | 231,539 | | | — | | | — | |
| 非アメリカ政府証券 | 9,357 | | | — | | | 9,357 | | | — | |
| 会社債務証券 | 45,666 | | | — | | | 45,666 | | | — | |
| 商業手形 | 238,160 | | | — | | | 238,160 | | | — | |
| 長期投資: | | | | | | | |
| | | | | | | |
| 会社債務証券 | 4,794 | | | — | | | 4,794 | | | — | |
| 合計する | $ | 552,010 | | | $ | 254,033 | | | $ | 297,977 | | | $ | — | |
| | | | | | | | | | | | | | | | | | | | | | | |
| 2021年12月31日まで |
| | | 公正価値階層構造 |
| (単位:千) | 合計する | | レベル1 | | レベル2 | | レベル3 |
| 公正な価値で勘定された金融商品(資産頭寸): | | | | | | | |
| 現金等価物: | | | | | | | |
| 貨幣市場基金 | $ | 89,905 | | | $ | 89,905 | | | $ | — | | | $ | — | |
| 短期投資: | | | | | | | |
| アメリカ政府証券 | 104,967 | | | 104,967 | | | — | | — | |
| 会社債務証券 | 78,677 | | | — | | 78,677 | | | — | |
| 商業手形 | 85,896 | | | — | | 85,896 | | | — | |
| 長期投資: | | | | | | | |
| アメリカ政府証券 | 59,230 | | | 59,230 | | | — | | | — | |
| 合計する | $ | 418,675 | | | $ | 254,102 | | | $ | 164,573 | | | $ | — | |
同社の金融資産は公正な価値によって経常的な基礎に従って計量され、現金等価物、通貨市場基金と有価証券を含む。
同社の通貨市場基金は容易に現金に変換することができ、各基金の本四半期最終日の資産純資産値は公正価値を決定するために使用される。米国政府債券は1級に分類され、市場見積もりを利用して評価される。同社の会社債務証券、非米国政府証券、商業手形は2級に分類され、様々な市場や業界投入を利用して評価されている。
現金等価物、売掛金、売掛金の短期満期日により、会社総合貸借対照表に反映される帳簿金額は公正価値に近い。
6.財産と設備
財産と設備、純額は:
| | | | | | | | | | | | | | | | | | | | |
| | 十二月三十一日 | | |
| (単位:千) | | 2022 | | 2021 | | 使用寿命 |
| 賃借権改善 | | $ | 826 | | | $ | 1,357 | | | レンタル期間またはレンタル期間5年、短い者を基準とします |
| コンピュータ装置 | | 368 | | | 157 | | | 3-5年.年 |
| 家具.家具 | | 437 | | | 360 | | | 5年.年 |
| ソフトウェア | | 6,048 | | | 361 | | | 3-10年.年 |
| 建設中の工事 | | 7,137 | | | 1,971 | | | |
| 財産と設備、毛額 | | 14,816 | | | 4,206 | | | |
| 減価償却累計を差し引く | | (1,245) | | | (1,019) | | | |
| 財産と設備、純額 | | $ | 13,571 | | | $ | 3,187 | | | |
減価償却費用は$765,000, $490,000、と$349,0002022年12月31日まで、2021年12月31日および2020年12月31日まで。
7.賃貸借証書
賃貸借契約を経営する
同社の経営リースは不動産と関係があります。
2018年10月、同社はコネチカット州スタンフォードにある会社本社の賃貸契約を締結した。同社は$を受け取りました1.5賃貸契約の負担に関連した上位テナントの百万ドル。同社は#ドルの保証金を設立した0.5百万ドル、信用状の形で。2022年1月、当社は本レンタル契約を改訂し、レンタル期間を2028年4月に延長した二つ5年制更新オプションまたは1つは10年オプションを更新する。レンタル料が増えた2.52022年12月1日から毎年の割合。
2018年8月に当社が締結しました5年制ノースカロライナ州ダラムでの運営リースは二つ5年制オプションを更新する。レンタル料が増えた2.75次のどれもが4年のです5年制レンタル期間を経営する。継続期間のレンタル料は現在の市場価格で支払います。
当社の総合経営報告書に記録されているリースコスト構成は以下の通りです
| | | | | | | | | | | | | | | | | | | | |
| | 12月31日までの12ヶ月間 |
| (単位:千) | | 2022 | | 2021 | | 2020 |
| リースコストを経営する | | | | | | |
| 据え置き | | $ | 1,131 | | | $ | 993 | | | $ | 1,049 | |
| 変数.変数 | | 336 | | | 548 | | | 486 | |
| 総賃貸コスト | | $ | 1,467 | | | $ | 1,541 | | | $ | 1,535 | |
同社の賃貸は、その総合貸借対照表に含まれている
| | | | | | | | | | | | | | |
| (単位:千) | | 2022年12月31日まで | | 2021年12月31日まで |
| 賃貸借契約を経営する | | | | |
| 経営的リース使用権資産 | | $ | 4,698 | | | $ | 1,010 | |
| リース資産総額を経営する | | $ | 4,698 | | | $ | 1,010 | |
| | | | |
| 賃貸負債を経営し、流動 | | $ | 483 | | | $ | 1,162 | |
| 長期経営賃貸負債 | | 4,768 | | | 129 | |
| リース負債総額を経営する | | $ | 5,251 | | | $ | 1,291 | |
2022年12月31日まで、会社がASC 842に規定する経営リース債務満期日は以下の通り
| | | | | | | | |
| (単位:千) | | 賃貸借契約を経営する |
| 2023 | | $ | 783 | |
| 2024 | | 1,155 | |
| 2025 | | 1,184 | |
| 2026 | | 1,213 | |
| 2027 | | 1,244 | |
| その後… | | 424 | |
| 賃貸支払総額 | | 6,003 | |
| 差し引く:推定利息 | | (752) | |
| 賃貸負債現在価値 | | $ | 5,251 | |
当社の賃貸に関する加重平均残存期間と割引率は以下のとおりである
| | | | | | | | | | | | | | |
| | 2022年12月31日まで | | 2021年12月31日まで |
| 加重平均残存賃貸年限(年) | | | | |
| 賃貸借契約を経営する | | 5.2 | | 1.1 |
| 加重平均割引率 | | | | |
| 賃貸借契約を経営する | | 4.2 | % | | 3.5 | % |
会社のレンタルに関する補足キャッシュフロー情報は以下のとおりである
| | | | | | | | | | | | | | | | | | | | |
| (単位:千) | | 2022年12月31日 | | 2021年12月31日 | | 2020年12月31日 |
| 賃貸負債に含まれる金額を計量するために支払う現金: | | | | | | |
| レンタル経営からの経営キャッシュフロー | | $ | 859 | | | $ | 1,388 | | | $ | 1,411 | |
| 新しい経営リース負債と引き換えに使用権資産 | | 5,580 | | | — | | | 31 | |
8.費用を計算する
計算すべき費用には以下が含まれている
| | | | | | | | | | | | | | |
| | 十二月三十一日 | | 十二月三十一日 |
| (単位:千) | | 2022 | | 2021 |
| 専門費用を計算する | | $ | 1,780 | | | $ | 1,108 | |
| 報酬と福祉に計上すべきである | | 19,142 | | | 12,081 | |
| 計画に応じて研究·開発する | | 12,321 | | | 10,069 | |
| その他の措置を講じる | | 5,999 | | | 2,120 | |
| 費用総額を計算する | | $ | 39,242 | | | $ | 25,378 | |
9. 権益に基づく報酬
会社は次の期間に株式ベースの報酬支出総額を記録した
| | | | | | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 |
| (単位:千) | | 2022 | | 2021 | | 2020 |
| 研究開発 | | $ | 29,373 | | | $ | 14,664 | | | $ | 3,055 | |
| 一般と行政 | | 43,592 | | | 23,780 | | | 6,979 | |
| 株式報酬費用総額 | | $ | 72,965 | | | $ | 38,444 | | | $ | 10,034 | |
2019年持分インセンティブ計画
“2019年持分インセンティブ計画”は、会社幹部、従業員、取締役などのキーパーソン(コンサルタントを含む)に奨励性株式オプション、非制限株式オプション、株式付加価値権、制限株式単位、制限株式奨励、非制限株式奨励と配当権の同値権を付与することを規定している。2019年の持分インセンティブ計画によると発行可能な株式数は、毎年1月1日から2030年1月1日まで(2030年1月1日を含む)累計増加する512月31日の直前に、会社は普通株式数の%または会社報酬委員会が決定したより少ない数の株を発行した。2022年12月31日までに3,162,2052019年の株式インセンティブ計画によると、将来発行可能な株。
株式オプション及び制限株式奨励の条項は、帰属要件を含み、2019年株式インセンティブ計画の規定に適合する取締役会又はその代表によって決定される。会社が従業員に付与する株式オプションと制限株式奨励は一般的に三つあるいは…4年そして、会社が取締役に付与する株式オプション及び制限株式奨励は、一般に1年から3年以内に付与される。
2019年従業員株購入計画
2019年8月30日、株主は2019年9月12日の会社のIPOに関する登録声明の発効直前に発効する2019年従業員の株式購入計画を承認した。合計する442,153普通株式はESPPによる発行のために保留されている。また、ESPPにより発行可能な普通株式数は、以下の小者を基準として、毎年1月1日から2028年1月1日(2028年1月1日を含む)まで自動的に増加する663,229普通株式、(二)1(三)株主特別引出権管理人が定めたより少ない株式数。2022年12月31日までに1,852,890ESPPにより発行保留株式である.2022年12月31日現在、ESPPでのいかなる引受期間も開始されていない。
株式オプション
A 本報告で述べた期間における、会社株式オプションの変化の概要は、以下のとおりである
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 株 | | 加重平均学習
値段 | | 重みをつける
平均値
余剰契約
命
(年) | | 原料を徴収する
価値がある |
| 2019年12月31日現在返済しておりません | | 3,225,725 | | | $ | 5.08 | | | 9.4 | | $ | 107,771,472 | |
| 授与する | | 1,580,788 | | | 35.90 | | | — | | | — | |
| 鍛えられた | | (190,484) | | | 4.69 | | | — | | | — | |
| 没収/キャンセルされる | | (110,483) | | | 23.49 | | | — | | | — | |
| 2020年12月31日現在返済していません | | 4,505,546 | | | 15.51 | | | 8.7 | | 256,860,405 | |
| 授与する | | 2,547,813 | | | 73.37 | | | — | | | — | |
| 鍛えられた | | (151,643) | | | 7.62 | | | — | | | — | |
| 没収/キャンセルされる | | (188,303) | | | 37.34 | | | — | | | — | |
| 2021年12月31日現在の未返済債務 | | 6,713,413 | | | 37.03 | | | 8.2 | | 196,012,147 | |
| 授与する | | 3,207,347 | | | 43.28 | | | — | | | — | |
| 鍛えられた | | (223,467) | | | 8.84 | | | — | | | — | |
| 没収/キャンセルされる | | (520,478) | | | 68.33 | | | — | | | — | |
| 2022年12月31日に返済されていません | | 9,176,815 | | | 38.13 | | | 8.0 | | 53,719,297 | |
| 2022年12月31日に行使できます | | 4,392,905 | | | 28.15 | | | 7.1 | | 48,765,645 | |
総内的価値の計算方法は,会社普通株の出来高日の終値からオプションの行権価格を減算し,オプションごとの株式数を乗じたものである.
2022年に付与された株式オプションの公正価値を決定する際に使用される仮定は1.46% – 4.23%、期待配当率は0.00%、期待期間(年数)5.5数年-6.1年と予想変動率71.2% - 76.4%.
2019年度CEO業績賞
2019年6月、会社のCEOが獲得176,411株式オプション、または2019年CEO業績賞。2019年のCEOパフォーマンス賞は48毎月の分割払いの根拠は4年サービス、業績状況(IPOのような流動性イベント)、および市場状況は、各ホーム日中に雇用およびサービスが継続されると仮定する。帰属期間中4年各授与日に市場条件が達成されない限り、2019年のCEO業績賞は受賞されません。市場状況が帰属日に到達するのではなく、将来の帰属日に達成される場合、最終的に市場状況に到達した最終日からの全期間にわたって報酬が得られる。全てまたは一部の報酬は最初の4年制その後次に市場状況に達した場合、サービス期間は4年制就役中もイズラムさんは続投していた。会社の普通株はアメリカ国家証券取引所に上場し、60-取引日の平均終値は少なくとも1ドル28.491株当たり(株式分割、資本再編、類似事件に応じて調整)。
2022年12月31日までの年間で33,077最高経営責任者業績賞のオプションは、その賞に適用される市場条件に満足した後に行使することができる。
2022年12月31日現在,未帰属株式オプションに関する未確認補償支出総額は#ドルである137.4百万ドル、会社は加重平均残りの期間内に約を確認する予定です2.58何年もです。2022年12月31日までの株式オプションの株式報酬支出総額は$54.3百万ドルです。
制限株式賞
上述した期間における当社限定株式報酬の変化の概要は、以下のとおりである
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | 番号をつける
の株 | | 重みをつける
平均値
授与日
公正価値 |
| 2019年12月31日現在の未帰属と未返済 | | | | | | 1,289,437 | | | $ | 1.21 | |
| 既得 | | | | | | (588,345) | | | 1.14 | |
| 没収される | | | | | | (14,224) | | | 1.45 | |
| 2020年12月31日現在の未帰属と未返済 | | | | | | 686,868 | | | 1.26 | |
| 授与する | | | | | | 332,226 | | | 73.31 | |
| 既得 | | | | | | (493,309) | | | 1.19 | |
| 没収される | | | | | | (55,475) | | | 13.75 | |
| 2021年12月31日現在の未帰属と未返済 | | | | | | 470,310 | | | 50.76 | |
| 授与する | | | | | | 36,625 | | | 61.74 | |
| 既得 | | | | | | (257,219) | | | 32.03 | |
| 没収される | | | | | | (27,957) | | | 79.25 | |
| 2022年12月31日現在の未帰属と未返済 | | | | | | 221,759 | | | 70.71 | |
2022年12月31日現在、未帰属制限株式奨励に関する未確認補償支出総額は#ドルである10.7百万ドル、会社は加重平均残りの期間内に約を確認する予定です1.46何年もです。2022年12月31日までの年間限定株式奨励補償支出総額は$7.9百万ドルです。
限定株単位
次の表は、2022年12月31日までの年間限定株式単位活動をまとめたものである
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | 番号をつける
の株 | | 重みをつける
平均値
授与日
公正価値 |
| 2021年12月31日現在の未帰属と未返済 | | | | | | — | | | $ | — | |
| 授与する | | | | | | 738,508 | | | 49.77 | |
| 既得 | | | | | | (24,369) | | | 24.62 | |
| 没収される | | | | | | (42,185) | | | 58.76 | |
| 2022年12月31日現在の未帰属と未返済 | | | | | | 671,954 | | | 50.11 | |
2022年12月31日現在、未帰属制限株式単位に関する未確認補償支出総額は#ドルである23.6百万ドル、会社は加重平均残りの期間内に約を確認する予定です2.12何年もです。制限株式単位報酬支出総額は、2022年12月31日までの年度10.7百万ドルです。
10.許可と協力協定
ファイザー。
2017年8月と10月に当社が締結しました四つファイザーやファイザーといくつかの技術の権利について合意することは、合意できるかもしれない。ライセンス契約によると、同社はナイルシタンおよびミダメチニブを含む特定の製品を使用、研究、開発、製造、商業化する権利をファイザー社から得た。ライセンス契約について,同社は発表した6,437,500初級系列A交換可能単位はファイザーより優先される(注1参照)違います。会社はこれらの職場の現金を受け取りました。
同社はファイザーマイルストーン社に総額$までの支払いを要求された232.5100,000,000,000,000,000,000ドル229.8いくつかのビジネスマイルストーン事件を終えた後、一人当たり100万ドルのミダミーニを得るだろう。各ライセンス契約に基づいて、純売上高の特定のパーセント(1桁の中央数百分から低い20%)に基づいて印税を支払わなければならず、各ライセンス契約下の印税支払いは、その製品に適用されるライセンス特許が最後に満了するまで継続されるが、それ以下ではない10年初めて国ごとの商業販売の後です。
百済神州株式会社です。
2018年8月、同社は百済神州有限会社または百済神州と臨床協力協定を締結し、ミダミニとリフェラファニーに指定された百済神州化合物の組み合わせについて臨床研究を行った。協定条項によると、当社は百済神州と臨床研究に関する費用を折半する。百済神州は百済神州複方を提供する必要があり,会社はミダメチニブを提供して臨床研究を行う必要がある。協力は共同指導委員会によって指導される。具体的な開発分野は共同指導委員会のすべての会員たちの同意を得る必要がある。
同社が記録した費用は#ドルです1.12022年12月31日と2021年12月31日までの年間100万ドル、0.92020年12月31日までの年間で,本協力協定に関する費用は100万ドルであり,会社の運営報告書では研究開発費に分類されている。
TEAD阻害剤組合せ許可プロトコル
2021年5月、同社は、カバ経路異常シグナルによって駆動されるバイオマーカーによって定義される固形腫瘍を潜在的に治療するために、転写因子ファミリーTEAドメインまたはTEADファミリーの新しい小分子阻害剤の組み合わせの許可を得た、ルモール大学およびフランダースバイオテクノロジー研究所と世界的に独占的なライセンス契約を締結したことを発表した。合意条項によると、同社は#ドルを前払いした11KUルモールとVIBに100万ドルを支払い、合併経営報告書に研究開発費として入金した。合意条項によると、KUルモールとVIBも最高$を得る資格があります285ライセンス内技術に基づいて開発された製品の任意の将来の純売上高に基づいて、開発、規制、ビジネスマイルストーンと階層桁数パーセントの特許権使用料で百万ドルの収入を取得します。
EGFR阻害剤組合せ許可プロトコルとスポンサー研究プロトコル
2021年10月、同社は、EGFR変異肺癌の治療のための新規上皮増殖因子受容体(EGFR)小分子阻害剤の組み合わせの許可を得たDana-Farber癌研究所(Dana-Farber)との独自の世界的許可協定を達成することを発表した。契約条項によると、同社はDana-Farberに前払金を支払い、この金は総合経営報告書に研究·開発費用と記されている。合意条項によると、Dana-Farberには、開発とビジネスマイルストーン、ライセンス内技術に基づいて開発された製品の将来の任意の純売上に基づく特許権使用料を取得する資格もあります。
この許可協定に署名すると同時に、同社はスタンフォード医学会社と長年賛助の研究協定を締結し、スタンフォード医学会社の一つの実験室とダナ-ファーバーの協力実験室の持続的な研究と開発に資金を提供した。この賛助の研究協定はEGFR阻害剤の組み合わせが開発候補指名に向かって進む過程で行われたリードした最適化と転化生物学的努力を支持することを目的としている。
グラクソ·スミスクラインの非独占的許可と協力協定の拡大
同社は2022年9月、グラクソ·スミスクラインとの非排他的臨床協力の拡大を発表し、最初に2019年6月に開始した。この公告は、ちょうど当社がグラクソ·スミスクラインと改訂および再記載された協力および許可プロトコル、またはグラクソ·スミスクライン許可プロトコルを締結し、ベランタマブ(Belamaf)、グラクソ·スミスクラインとの抗体薬物結合体(Belamaf)、B細胞成熟抗原またはBCMAを標的とするADC、またはGSKによって制御されるbelantamabから誘導されるBCMAに対する任意の他の細胞毒性ADCを開発および商業化する可能性があり、単独で併用療法として、または他の医薬製剤と共に使用される。
GSKライセンス契約の条項によると、この協定に署名すると同時に、会社はGGLと株式購入協定を締結し、GGLはこの合意に基づいて購入した2,050,819私募取引中の会社普通株、総購入価格は約$75.0百万ドル、あるいはドル36.57一株ずつです。これらの株は安値で売っている25当社の普通株式出来高加重平均株価割増率30日間株式購入契約を締結するまでの間。株式購入協定発効日前日の普通株終値から計算される普通株公正価値は$である55.5100万ドルを株式に計上していますこれは1ドルです19.51,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,同社は$を記録した19.5当社は2022年9月に収入を繰延収入として確認し、それに応じた履行義務がGSKライセンス契約に関連する支出(臨床供給や研究開発費を含む)に比例して履行されているため、収入を確認する。この1年の
2022年12月31日現在,GSKライセンスプロトコルから受信した前払い対価に関する収入は重要ではない.
グラクソ·スミスクライン許可協定の条項によると、同社には最高$を取得する資格がある550.0特定の開発と商業マイルストーンに到達すれば、100万ドルの追加支払いを受けるだろう。同社はNIOROGACEATの全商業権を保持し続けている。また,グラクソ·スミスクラインがbelamafとの合併承認を求める市場では,SpringWorksはniRogacestatの商業化を求める。
Jazz PharmPharmticals資産購入と独占ライセンス契約
2020年10月、会社とJazzは、PF-4457845を含む会社の脂肪酸アミド加水分解酵素(FAAH)阻害剤計画を買収した資産購入および独占許可協定を発表した。FAAH阻害剤計画は、同社が2018年に取得したライセンス契約の一部である。Jazzは前払いした$35会社に100万ドルを支払い、将来の潜在的な支払い金額は最高$に達する375百万ドルは、ある臨床開発、規制、商業マイルストーンの成果に基づいている。また,Jazzは将来のPF−4454845の1桁範囲の純売上高に応じて等級別販売に基づく特許権使用料を会社に支払う義務がある。
Jazz協定によると、Jazzは、商業的に合理的な努力を用いて米国で少なくとも1つの製品を開発し、規制部門の承認を求める義務があり、規制部門の承認を得た場合、Jazzは米国でこのような製品を商業化する義務がある。
ジャズ契約によると移籍金は
同社は、ASC 606に従ってJazzプロトコルを評価し、単一の履行義務が存在することを決定し、ライセンスは異なる商品バンドルにおける主要な項目であると判断した。同社は2020年12月31日までの期間内にライセンスを含むバンドル商品中のすべての物品を譲渡した。ライセンスは機能的知的財産権であり,知的財産権の機能性は会社の継続活動によって大きく変化しないことが予想されるため,収入はある時点で確認すべきである。同社は2020年12月31日までの間に前金を収入として確認している。関連活動が達成されるまで、会社は開発または規制承認のマイルストーンを確認しない;販売レベルに基づく商業マイルストーン支払いを含む特許権使用料は、(I)関連販売が発生した場合、または(Ii)特許使用料の一部または全部が履行された(または部分的に履行された)ときに確認され、ジャズ協定の場合、これは関連販売が発生する期間となる。
11.投資する
MapKure
2019年6月、当社は百済神州と共同所有するエンティティMapKureの設立を発表しました。百済神州はMapKureにBGB-3245の独占経営権を付与し、BGB-3245は1種の試験的経口小分子選択性阻害剤であり、特定のBRAFドライバ突然変異と遺伝子融合を抑制できる。MapKureはBGB-3245の臨床開発を進めており、BRAFドライバ突然変異と遺伝子融合を携帯する固形腫瘍患者に用いられ、臨床前研究でこれらの突然変異と遺伝子融合はこの化合物に敏感であることが観察された。MapKureでの会社の持分のほか、会社はMapKureの共同指導委員会と取締役会にメンバーを任命した。同社はまた,MapKureとのサービスプロトコルにより,BGB−3245の臨床開発や他の業務活動に貢献している。
2019年6月にMapKureを設立すると同時に、会社は購入しました3,500,000Aシリーズの第一選択のMapKure単位、または25.0%所有権、$3.5100万ドルで2020年6月に会社は追加の3,500,000AシリーズMapKure第一選択単位、販売価格$3.5百万所有権を増加させます38.9%は、最初の単位調達プロトコルの条項によって要求されます。
2022年6月、会社はMapKureに追加投資を行い、購入した4,200,000BシリーズはMapKureデバイスが第一選択で、価格は$4.2100万ドルです。Bシリーズ優先単位によって合意の条項を購入します。会社は追加のを買う義務がある2,800,000BシリーズはMapKureデバイスが第一選択で、価格は$2.8このような合意によると、2回目の成約時は100万ドルだった。2022年12月31日現在、MapKureにおける会社の所有権は38.9%です。MapKureでの会社の持分のほか、会社はMapKureの共同指導委員会と取締役会にメンバーを任命した。同社はまた,MapKureとのサービスプロトコルにより,BGB−3245の臨床開発や他の業務活動に貢献している。
その会社はMapKureが可変利益実体だと確信している。当社は主な受益者ではありません。当社にはMapKureの経済業績に最も影響を与える活動を指導する権限がないからです。そのため、当社は当該実体の財務諸表を統合せず、権益会計方法を用いてこの投資を会計処理する。当社は2022年12月31日までの評価を再確認します。ASC 323-10-35-6によれば、同社は、1四半期の遅延時間に基づいて、MapKureにおける収益または損失部分を記録する。
2022年12月31日までに、当社は1ドルを確認しました2.9MapKure損失に占めるシェアの損失。MapKureへの会社の投資は、総合貸借対照表の“権益法投資”に含まれている。2022年12月31日現在、会社がMapKureへの参加により直面する最大損失リスクは$7.0百万ドルは投資の帳簿価値を表します#ドル4.2100万ドルと資金源のない債務#ドル2.8百万ドルです
2023年1月、Bシリーズ優先単位購入契約の条項により、当社は購入した1つの追加の2,800,000BシリーズはMapKureデバイスが第一選択で、価格は$2.8百万ドルです。
12.引受金とその他の事項
同社の債務は2022年12月31日現在、施設の運営リースを含む。脚注7:賃貸借証書より多くの情報を得るために。
同社は正常業務過程において臨床試験、臨床前研究、製造と運営目的のための他のサービスと製品について契約を締結した。これらの契約は一般に通知後一定時間後に終了することが規定されているため、会社はこれらの合意下の取消不能債務は実質的ではないと考えている。
また、会社は、そのような義務の時間及び金額が未知又は不確定であるため、マイルストーン又は特許権使用料支払い又はその他の契約支払い義務を排除している。
事件があったり
当社は時々正常な業務過程で発生した紛争や規制調査に関与する可能性があります。当社が損失が可能で合理的に推定可能であると判断した場合、その金額が財務諸表全体に重大な意味を有する場合、負債を記録して開示する。重大な意外損失が発生した場合、当社は負債を記録するのではなく、クレームの性質及び金額、並びに損失推定又は損失範囲(合理的に推定できれば)を開示する。
2022年12月31日と2021年12月31日まで、訴訟やアクシデントが少なくとも重大な損失をもたらす合理的な可能性はない。
13.所得税
会社は設立以来赤字が発生したため、2022年12月31日と2021年12月31日まで、当期や繰延所得税の支出や福祉がない。
2022年12月31日現在、同社の連邦、州、都市の純営業損失は$に転換している368.3百万、$260.9百万ドルとドル3.7それぞれ100万ドルで、将来の課税収入を減らすために使うことができる。2018年から2022年までに発生した連邦純営業損失は364.0100万ドルで相殺できます80課税所得額のパーセンテージは完全に使用されるまで一定期間不確定である。連邦純営業損失はドルに繰り越す4.32017年に報告された純営業損失は100万ドルで、州と市の純営業損失の繰越は2040年まで異なる日に満期となる。その会社はまた#ドルの連邦税金免除を受けている22.7100万ドル、これは未来の納税義務を相殺するために使用されることができる。このような税金の繰越免除は2038年に始まる様々な日に満了されるだろう。
純営業損失及び税額控除の繰越はアメリカ国税局及び国家税務機関が審査及び調整する可能性がある。主要株主の所有権権益が3年間の累計変動が50%を超えた場合(国税法第382条及び第383条及び国税法に類似した国の規定及びその他の規定をそれぞれ参照)、純営業損失及び税項相殺繰越は年次制限を受ける可能性がある。これは、将来の課税収入または納税義務を相殺するために毎年使用できる税金属性の数を制限することができるかもしれない。年間限度額の金額は、所有権変更直前の会社価値に基づいて決定される。その後の所有権変更は今後数年間の制限にさらに影響を及ぼすかもしれない。
当社は2022年12月31日または2021年12月31日まで、不確定な税務状況に準備金を記録していません。当社は2021年12月31日までの研究開発信用繰越研究を完了しており、2022年12月31日までの研究開発信用繰越についてはまだ研究を行っていない。2022年の研究が完成すると、会社の研究開発信用の転換の調整につながる可能性がある。すでに当社の研究及び発展信用について全数推定免税額を準備し、もし調整が必要であれば、この調整は推定免税額の調整によって相殺される。したがって、調整が必要であれば、貸借対照表や業務表や全面損失に影響を与えることはない。
未確認の税収割引に関する利息および懲罰的費用(ある場合)は、添付されている経営報告書および全面赤字報告書のうち所得税費用に分類される。2022年12月31日までに会社は違います。不確定な税務状況に関連するものは利息または罰金を計算しなければならない。
当社は赤字繰越状況にあるため、赤字繰越が存在するすべての納税年度において、同社は通常、米国連邦、州、地方所得税当局の審査を受けている。当社は現在、いかなる納税年度にも米国国税局又はその他の管轄区域の審査を受けていません。
繰延税金資産と負債の主な構成要素は以下のとおりである
| | | | | | | | | | | | | | |
| | 12月31日まで |
| (単位:千) | | 2022 | | 2021 |
| 繰延税金資産: | | | | |
| 純営業損失が繰り越す | | $ | 93,032 | | | $ | 63,418 | |
| 研究開発単位 | | 3,422 | | | 2,581 | |
| 孤児薬品信用 | | 19,401 | | | 14,198 | |
| 費用を計算する | | 136 | | | 18 | |
| 減価償却 | | — | | | 128 | |
| 資本化研究と開発 | | 21,262 | | | — | |
| 株の報酬 | | 20,286 | | | 8,476 | |
| リース負債を経営する | | 1,103 | | | 271 | |
| 共同投資 | | 286 | | | 356 | |
| 繰延税金資産総額 | | 158,928 | | | 89,446 | |
| 繰延税金負債: | | | | |
| 経営的リース使用権資産 | | (987) | | | (212) | |
| 減価償却 | | (113) | | | — | |
| 他にも | | (2) | | | (2) | |
| 推定免税額 | | (157,826) | | | (89,232) | |
| 繰延税項目純資産 | | $ | — | | | $ | — | |
既存の証拠の重みに基づいて、繰延税金資産の一部または全部が現金化されない可能性が高い場合、ASC 740は、報告された繰延税金資産を低減するために推定準備金を計上する必要がある。すべての積極的かつ消極的な証拠を考慮した後、会社は2022年12月31日と2021年12月31日に繰延税金資産に全額推定準備金を計上したが、会社経営陣が確定しているため、これらの資産はさらに現金化できない可能性がある。評価免税額は#ドル増加した68.62022年の100万ドルは主に同社で発生した純損失と発生した連邦研究と孤児薬物の免除に関連している。
当社の2022年12月31日まで、2021年12月31日及び2020年12月31日までの実質税率はゼロ百分率以下は、連邦法定税率で計算された所得税費用と会社の有効所得税税率との入金である
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 法定税率 | 21.00 | % | | 21.00 | % | | 21.00 | % |
| アメリカ州と地方所得税、アメリカ連邦所得税の割引を差し引く | 2.33 | | | 5.15 | | | — | |
| 株に基づく報酬 | (0.77) | | | (0.03) | | | 0.55 | |
| 信用を研究開発する | 0.27 | | | 0.50 | | | 2.12 | |
| 孤児薬品信用 | 1.87 | | | 4.99 | | | 11.95 | |
| 他にも | — | | | — | | | 0.02 | |
| 評価免除額を変更する | (24.70) | | | (31.61) | | | (35.64) | |
| 実際の税率 | — | % | | — | % | | — | % |
14.401(K)計画
2017年、同社は税務条件に適合した従業員貯蓄と退職計画、すなわち401(K)計画を採択し、21歳以上の全従業員をカバーした。401(K)計画によれば、参加者は、連邦許可税引前収入最高限度額を401(K)計画に支払うことを選択することができる。2021年1月1日まで、当社はいかなるセット出資もしていません。2022年12月31日までの年度,等額寄付の費用総額は$1.0100万ドルは経営報告書に含まれています。
15.関係者取引
当社は2022年12月31日まで、2021年12月31日または2020年12月31日まで関連先取引は何もありません。
16.1株当たり純損失
1単位当たりと1株当たりの基本と償却純損失は以下のように計算される
| | | | | | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 |
| (千単位で、共有および1株当たりのデータは含まれていない) | | 2022 | | 2021 | | 2020 |
| 分子: | | | | | | |
| 純損失 | | $ | (277,417) | | | $ | (173,910) | | | $ | (45,574) | |
| 普通株主は純損失を占めなければならない | | (277,417) | | | (173,910) | | | (45,574) | |
| 分母: | | | | | | |
| 加重平均流通株、基本株、希釈株 | | 53,290,528 | | | 48,497,790 | | | 43,300,063 | |
| 1株当たり基本と希釈して純損失 | | $ | (5.21) | | | $ | (3.59) | | | $ | (1.05) | |
各償却計算に含まれていない潜在的希薄化証券は以下の通りである
| | | | | | | | | | | |
| 12月31日まで |
| 2022 | | 2021 |
| 発行済みと未償還普通株式オプション | 9,176,815 | | | 6,713,413 | |
| 未来帰属制限株式単位 | 671,954 | | | — | |
| 将来の帰属制限を受けた限定的な株式奨励 | 221,759 | | | 470,310 | |
| 潜在的希薄化証券総額 | 10,070,528 | | | 7,183,723 | |
項目9.会計·財務開示面の変更と会計士との相違
ない。
第9条。制御とプログラム
情報開示制御とプログラムの評価
我々の経営陣は、本報告で述べた期間終了までの間、開示制御及びプログラムの設計及び動作の有効性を評価し、これらの制御及びプログラムの定義は、取引法第13 a−15(E)及び15 d−15(E)条を参照されたい。
私たちの評価によると、私たちのCEOと財務官は、2022年12月31日まで、私たちの開示制御と手続きが有効であり、取引所法案に基づいて提出または提出された報告書で開示を要求する情報が、米国証券取引委員会規則および表が指定された期間内に記録、処理、まとめ、報告されることを合理的に保証することができ、これらの情報は、タイムリーに必要な開示について決定するために、私たちの経営陣に伝達されると結論した。
財務報告の内部統制
財務報告の内部統制に関する経営陣の報告
我々の経営陣は、財務報告の十分な内部統制の確立と維持を担当しており、この用語は、“取引法”第13 a-15(F)条において、会社の主要幹部および主要財務官が設計またはその監督の下で、会社の取締役会、管理職、および他の者によって実施される過程と定義されている。公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供し、以下の政策と手続きを含む:(1)合理的で詳細、正確かつ公平に会社の資産を反映する取引と処分に関する記録の保存;(Ii)公認会計原則に従って財務諸表を作成するために必要に応じて取引を記録することを保証する合理的な保証を提供し、当社の収支は、会社の管理層および取締役の許可のみに基づいて行われ、(Iii)財務諸表に重大な影響を与える可能性のある不正買収、使用または処分について合理的な保証を提供する。
本年度報告書を作成する際には、我々の経営陣、最高経営責任者及びCEOを含め、以下の基準に基づいて、2022年12月31日までの財務報告内部統制の有効性を評価した内部統制であるテレデビル委員会が組織委員会が発表した総合的な枠組みを後援する(2013年フレームワーク)(“COSO標準”)。その評価によると、我々の経営陣は、財務報告書に対する内部統制は2022年12月31日から有効であると結論した。
独立公認会計士事務所認証報告
2022年12月31日現在、財務報告の内部統制の有効性を独立公認会計士事務所安永会計士事務所が監査していることは、本年度報告Form 10−Kの他の部分の報告で説明されている。
制御措置の有効性に対する制限
私たちの経営陣は、私たちのCEOやCEOを含めて、私たちの開示制御や手続き、あるいは財務報告に対する私たちの内部統制がすべてのミスやすべての詐欺を防ぐことができることを期待していません。発想や動作がどんなに良くても、絶対的な保証ではなく、合理的な保証しか提供できず、制御システムの目標が実現されることを確保する制御システム。また,制御システムの設計は,資源制約が存在し,そのコストに対する制御の利点を考慮しなければならないという事実を反映しなければならない.すべての制御システムの固有の限界により,どの制御評価もすべての制御問題や不正イベントが発見されたことを絶対に保証することはできない.また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
財務報告の内部統制の変化
2022年12月31日までの第4四半期において、財務報告の内部統制(取引法第13 a-15(F)条参照)は、取引法規13 a-15(D)段落の要求の評価に関連しており、変化がなく、財務報告の内部統制に大きな影響を与えたり、財務報告の内部統制に大きな影響を与えたりする可能性が高い。
独立公認会計士事務所報告
SpringWorks治療会社の株主と取締役会に
財務報告の内部統制については
我々は,トレデビル協賛組織委員会が発表した内部統制-総合枠組み(2013年枠組み)(COSO基準)で確立された基準に基づき,SpringWorks Treeutics,Inc.2022年12月31日までの財務報告内部統制を監査した。COSO規格によれば,SpringWorks Treateutics,Inc.(“当社”)は2022年12月31日にすべての重要な面で財務報告に対して有効な内部統制を維持していると考えられる。
また、米国上場企業会計監督委員会(PCAOB)の基準に従って、当社の2022年12月31日、2022年12月31日、2021年12月31日までの総合貸借対照表を監査し、2022年12月31日までの3年度の関連総合経営報告書、総合損失、株主権益/(損失)とキャッシュフロー、および2023年2月28日の関連付記と私たちの報告について保留のない意見を発表した。
意見の基礎
当社経営陣は、効果的な財務報告内部統制を維持し、添付されている“経営陣財務報告内部統制報告”に掲載されている財務報告内部統制の有効性を評価する責任がある。私たちの責任は私たちの監査に基づいて、会社の財務報告書の内部統制に意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。
我々の監査には,財務報告の内部統制を理解すること,重大な弱点があるリスクを評価すること,評価されたリスクテストに基づいて内部統制の設計·運用有効性を評価すること,および状況下で必要と考えられる他のプロセスを実行することが含まれる。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分に関する記録の保存、(2)公認された会計原則に従って財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、会社の収入および支出は会社の経営陣と取締役の許可のみに基づいて行われる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用、または処分会社の資産を防止またはタイムリーに発見する合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
/S/安永法律事務所
ニューヨーク、ニューヨーク
2023年2月28日
プロジェクト9 B。その他の情報
ない。
プロジェクト9 Cです。検査妨害に関する外国司法管区の開示
適用されません。
第三部
プロジェクト10.取締役、上級管理者、および企業管理
本プロジェクトに要求される情報は、我々が米国証券取引委員会に提出した2023年年次総会に関する最終委託書に含まれ、参照されて本明細書に組み込まれる。
プロジェクト11.役員報酬
本プロジェクトに要求される情報は、我々が米国証券取引委員会に提出した2023年年次総会に関する最終委託書に含まれ、参照されて本明細書に組み込まれる。
プロジェクト12.特定の実益所有者の保証所有権及び管理職及び株主に関する事項
本プロジェクトに要求される情報は、我々が米国証券取引委員会に提出した2023年年次総会に関する最終委託書に含まれ、参照されて本明細書に組み込まれる。
第13項:特定の関係及び関連取引、並びに取締役独立性
本プロジェクトに要求される情報は、我々が米国証券取引委員会に提出した2023年年次総会に関する最終委託書に含まれ、参照されて本明細書に組み込まれる。
プロジェクト14.主な会計費用とサービス
本プロジェクトに要求される情報は、我々が米国証券取引委員会に提出した2023年年次総会に関する最終委託書に含まれ、参照されて本明細書に組み込まれる。
第4部
プロジェクト15.証拠品および財務諸表の添付表
財務諸表
本報告の一部として提出された連結財務諸表は、115ページの財務諸表インデックスに記載されている。
財務諸表付表
米国証券取引委員会が適用する会計条例に規定されているすべての付表は、関連指示が要求されているか適用されないものではないため、省略されている。
陳列品
S-K条例第601項および本年度報告テーブル10-K第15(B)項で要求される証拠物は、本年度報告テーブル10-K署名ページ前の添付ファイルインデックスに記載される。展示品索引に記載されている展示品は、引用されて本明細書に組み込まれる。
項目16.表格10-Kの概要
その会社は要約情報を含まないことを選択した。
展示品索引
| | | | | | | | |
展示品
番号をつける | | 説明する |
| 3.1 | | 改正され再印刷された登録者は現在有効な登録証明書を発行する。(添付ファイル3.1を参照して登録者に組み込まれ、2019年9月17日に米国証券取引委員会の8-Kフォームの現在の報告書に提出されます)。 |
| 3.2 | | 登録者は現在有効な規定を持っている。(添付ファイル3.2を参照して登録者に組み込まれ、2019年9月17日に米国証券取引委員会の8-Kフォームの現在の報告書に提出されます)。 |
| 3.3 | | 登録者定款修正案は、現在有効である。(添付ファイル3.1を参照して登録者に組み込まれ、2020年5月27日に証券取引委員会の8-Kフォームの現在の報告書に提出される)。 |
| 4.1 | | 普通株式株式を証明する株式サンプル証明書(2019年9月12日に米国証券取引委員会に提出された登録者登録声明S-1/A表(第333-233351号書類)添付ファイル4.1合併を参照)。 |
| 4.2 | | 登録者とその一部株主が2018年8月30日に締結した“投資家権利協定”(登録者が2019年9月12日に米国証券取引委員会に提出したS-1表登録説明書添付ファイル4.2(第333-233351号文書)が会社として設立された)に改訂·再署名された。 |
| 4.3 | | 登録者証券説明(登録者が2020年3月12日に米国証券取引委員会に提出した2019年12月31日までの10-K表年次報告添付ファイル4.3登録が成立します。) |
| 4.4 | | 改訂·改訂された投資家権利協定は、期日は2021年2月25日である(参照登録者が2021年2月25日に証券取引委員会に提出した2020年12月31日までの10−K表年次報告書の添付ファイル4.4により成立)。 |
| 10.1 | | 2019年株式オプション及びインセンティブ計画及びその奨励協定のフォーマット(2019年9月12日に米国証券取引委員会に提出された登録者登録声明S−1/A表(文書番号333−233351)添付ファイル10.1を参照して編入)。 |
| 10.2 | | 2019年の株式オプションおよび持分インセンティブ計画およびその下の奨励協定フォーマットを改正して再起動する(参照登録者が2022年2月24日に米国証券取引委員会に提出された2021年12月31日現在の10-K表年次報告書添付ファイル10.2に組み込む)。 |
| 10.3 | | 2019年従業員株購入計画(2019年9月12日に米国証券取引委員会に提出された登録者登録説明書S-1/A表(書類番号333-233351)添付ファイル10.3を参照して成立)。 |
| 10.4 | | 上級管理者現金奨励金計画(登録者は2019年9月12日に米国証券取引委員会のS-1/A表登録説明書(書類番号333-233351)添付ファイル10.4)に提出する。 |
| 10.5 | | 第2回改正·再策定された非従業員役員報酬政策(参照登録者が2022年2月24日に米国証券取引委員会に提出した2021年12月31日現在の10−K表年次報告書の添付ファイル10.5により成立)。 |
| 10.6 | | 賠償協定表は、登録者とその取締役毎の間で提供される(登録者が2019年9月12日に米国証券取引委員会に提出したS−1/A表登録説明書添付ファイル10.6(第333-233351号書類)は法団)。 |
| 10.7 | | 賠償協定表は,登録者とその上級者ごとの間で記入される(登録者が2019年9月12日に米国証券取引委員会に提出したS-1/A表登録説明書(第333-233351号文書)添付ファイル10.7は法団)。 |
10.8§ | | 登録者ファイザー、SpringWorks子会社2,Inc.及びファイザー製品会社が2019年7月31日に改訂及び再署名したライセンス契約(合併により、参照登録者が2019年9月12日に米国証券取引委員会のS−1/A表登録声明の添付ファイル10.8(書類番号333−233351)に提出する)。 |
10.9§ | | 登録者ファイザー、SpringWorks子会社3,Inc.とワーナー·ランバート社との間で2019年8月7日に改訂·再署名されたライセンス契約 (添付ファイル10.9を参照して登録者に組み込まれ、2019年9月12日に米国証券取引委員会のS-1/Aフォーム登録説明書(文書番号333-233351))に提出されます。 |
10.10§ | | SpringWorks子会社3、中国人民銀行、百済神州有限公司が2018年8月16日に調印した臨床協力協定(2019年9月12日に米国証券取引委員会に提出された登録者S-1/A表登録声明(書類番号333-233351)添付ファイル10.10合併を参照することにより)。 |
10.11§ | | 登録者とグラクソ·スミスクラインとの間の臨床試験協力·供給協定は、2019年6月25日(2019年9月12日に米国証券取引委員会に提出された登録者S-1/A表登録声明(書類番号333-233351)添付ファイル10.11を参照して合併する)。 |
10.11.1§ | | 2021年10月22日、GlaxoSmithKline LLCとSpringWorks Treateutics,Inc.との間の日付は、2019年6月25日の臨床試験協力および供給協定の第1号修正案である(登録者を参照して2021年10月27日に米国証券取引委員会に提出された8-K表の現在報告されている添付ファイル1.1(文書番号001-39044)を統合する)。 |
10.12§ | | 譲渡とリースを負担し、日付は2018年10月10日で、研究開発子会社と構造ポートフォリオ管理有限責任会社(登録者は2019年9月12日に米国証券取引委員会に提出されたS-1/A表登録声明(書類番号333-233351)添付ファイル10.12合併)。 |
| | | | | | | | |
| 10.13# | | 改正·再署名された雇用協定は、日付が2021年7月30日であり、登録者とサチブ·イズラムとの間で締結される(2021年8月4日に米国証券取引委員会に提出された登録者10-Q四半期報告添付ファイル10.1に従って会社として設立される)。 |
| 10.14# | | 改正·再署名された雇用協定は、2021年7月30日に、登録者と小フランシス·I·ペリールによって署名された。(添付ファイル10.2を参照して登録者に組み込まれ、2021年8月4日に証券取引委員会に提出されたForm 10-Q四半期報告書)。 |
| 10.15# | | 改正·再署名された雇用協定は、日付が2021年7月30日であり、登録者とBadreddin Edrisとの間の合意(登録者が2021年8月4日に米国証券取引委員会に提出された10-Q表四半期報告添付ファイル10.3が法団として設立された)。 |
| 10.16# | | 改正·再署名された雇用協定は、日付が2021年7月30日であり、登録者とBhavesh Asharとの間の合意(登録者が2021年8月4日に米国証券取引委員会に提出された10-Q表四半期報告添付ファイル10.4が法団として設立された)。 |
| 10.17# | | 改正および再署名された雇用協定は、日付が2021年7月30日であり、登録者とL.Mary Smithとの間の合意(登録者が2021年8月4日に証券取引委員会に提出された10-Q表四半期報告添付ファイル10.6が法団として設立された)。 |
| 10.18# | | 改正·再署名された雇用協定は、日付が2021年7月30日であり、登録者とDaniel J.Pichlとの間で締結される(登録者は2021年8月4日に米国証券取引委員会に提出された10-Q表四半期報告添付ファイル10.7が法団として設立される)。 |
| 10.19# | | 改正および再署名された雇用協定は、2021年7月30日に、登録者とHerschel S.Weinsteinとの間の合意(登録者が2021年8月4日に米国証券取引委員会に提出された10-Q表四半期報告添付ファイル10.8合併)である。 |
| 10.20#* | | 登録者とジェームズ·カシディとの雇用協定は、2021年8月16日となっている。 |
| 10.21 | | 販売契約は,日付は2021年2月25日であり,SpringWorks Treeutics,Inc.とCowen and Company,LLC(参照登録者により2021年2月25日に米国証券取引委員会に提出された8−Kテーブル現在報告の添付ファイル1.1(文書番号001−39044)を統合する)。 |
| 10.22 | | 第二借約修正協定は、2022年1月31日に、Two Harbor Point Square LLCとSpringWorks Treateutics,Inc.(2022年5月5日に証券取引委員会に提出された登録者四半期報告Form 10-Qの添付ファイル10.1を参照して統合される)である。 |
| 10.23# | | 保留協定は,2022年5月2日にSpringWorks治療会社とL.Mary Smithによって署名された。(添付ファイル10.3を参照して登録者に組み込まれ、2022年5月5日に証券取引委員会に提出された10-Q表四半期報告)。 |
| 10.24 | | SpringWorks治療会社とグラクソ·スミスクライン知的財産権開発有限公司が2022年9月6日に署名した臨床試験協力と許可協定の改訂と再署名 (添付ファイル10.4を参照して登録者に組み込まれ、2022年11月3日に証券取引委員会に提出されたForm 10-Q四半期報告書)。 |
| 10.25** | | 登録権利協定は、2022年9月7日にSpringWorks治療会社と投資家の双方が署名した。(添付ファイル10.2を参照して登録者に組み込まれ、2022年9月8日に証券取引委員会の現在のテーブル8-K報告書に提出される)。 |
| 21.1 | | 登録者の子会社(参照登録者により2019年9月12日に米国証券取引委員会に提出されたS−1/A表登録説明書(書類番号333−233351)添付ファイル21.1が成立)。 |
| 23.1* | | 独立公認会計士事務所安永法律事務所同意。 |
| 24.1* | | 授権書(本年報の署名ページに表格10-K形式で掲載されている)。 |
| 31.1* | | 2002年サバンズ-オキシリー法第302節で可決された1934年“証券取引法”第13 a-14(A)条または第15 d-14(A)条に基づいて発行された最高経営責任者証明書。 |
| 31.2* | | 2002年サバンズ-オキシリー法302節に基づいて可決された1934年の証券取引法規則13 a-14(A)または規則15 d-14(A)に基づいて首席財務官を認証する。 |
| 32.1† | | 2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条に規定されている最高経営責任者証明書。 |
| 32.2† | | 2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による首席財務官の認証。 |
| 101.INS | | XBRLインスタンスドキュメントを連結する. |
| 101.書院 | | インラインXBRL分類拡張アーキテクチャ文書. |
| 101.カール | | インラインXBRL分類拡張はリンクベース文書を計算する. |
| 101.def | | XBRLソート拡張を連結してLinkbase文書を定義する. |
| 101.介護会 | | XBRL分類拡張ラベルLinkbase文書を連結する. |
| 101.Pre | | XBRL分類拡張プレゼンテーションLinkbaseドキュメントを内部接続する. |
| 104 | | 表紙相互データファイル(添付ファイル101に含まれるイントラネットXBRLのフォーマット) |
__________________________________________
* 本局に提出します。
**S-K規則601(A)(5)項により,表と証拠物が省略されている.任意の漏れたスケジュールおよび/または証拠品のコピーは、米国証券取引委員会に提供することを要求しなければならない。
#は、管理契約または任意の補償計画、契約、またはスケジュールを表します。
† 改正された1934年の証券取引法第18条の規定によると、本証明書は提出されたものとみなされず、当該条項の責任にも拘束されない。このような証明は、引用によってそのような出願が明示的に組み込まれない限り、改正された1933年の証券法に基づいて提出されたいかなる出願にも組み込まれているとはみなされない。
§ 本展示品の編集部分に対しては,秘匿処理が与えられている.この展示品の編集部分は単独でアメリカ証券取引委員会に提出された。
サイン
1934年の証券取引法の要求によると、登録者はすでに正式に本報告を正式に許可した署名者がそれを代表して署名することを促した。
| | | | | | | | | | | |
| | スプリングウォークス治療会社は |
| | |
| 日付:2023年2月28日 | | 差出人: | /s/ザチブ·イズラム |
| | | ザチブ·イズラム |
| | | 最高経営責任者 |
授権依頼書
以下の個人署名の各々は、サチブ·イズラムおよび小フランシス·I·ペリルをここで許可し、任命し、それぞれその真および合法的な事実受権者および代理人として、完全な代替および再代替の権力を有し、相手なしに行動し、本人の名義、場所、代理で行動し、各人の名義および代表、個別および以下に述べる身分証明書を作成し、本年度報告の任意およびすべての修正を表格10-Kの形で提出し、本年度報告およびすべての証拠物およびそれに関連する他の文書を証券取引委員会に提出する。上記の事実代理人および代理人およびその各々に、物事の全ての権力および権力としてのすべての権利および権限を付与し、上記の事実代理人および代理人またはそれらのいずれかまたはそれらの代替者が、合法的にまたはそれに至るすべての行為および事柄を合法的に行うことができることを承認および確認する。
本報告は、1934年の証券取引法の要求に基づき、以下の者によって登録者として指定日に署名された。
| | | | | | | | | | | | | | |
| サイン | | タイトル | | 日取り |
| | | | |
| /s/ザチブ·イズラム | | 取締役CEO兼最高経営責任者
(首席行政主任) | | 2023年2月28日 |
| ザチブ·イズラムJ.D. | | | |
| | | | |
| フランシス·I·ペリル | | 首席財務官
(首席財務官) | | 2023年2月28日 |
| フランシス·I·ペリル | | | |
| | | | |
| /s/マイケル·P·ノフィー | | 首席会計官
(首席会計主任) | | 2023年2月28日 |
| マイケル·P·ノフィー | | | |
| | | | |
| /S/Daniel S.リンチ | | 議長.議長 | | 2023年2月28日 |
| ダニエル·リンチM.B.A. | | | | |
| | | | |
| /s/Carlos Albán | | 役員.取締役 | | 2023年2月28日 |
| カルロス·アルベイン | | | | |
| | | | |
| /s/エレン·フォルマン | | 役員.取締役 | | 2023年2月28日 |
| エレン·フォルマン | | | | |
| | | | |
| /s/ジュリーHambleton | | 役員.取締役 | | 2023年2月28日 |
| ジュリー·ハンブルトン医学博士です | | | | |
| | | | |
| /s/フレダ·ルイス·ホール | | 役員.取締役 | | 2023年2月28日 |
| Freda Lewis-Hall医学博士DFAPA | | | | |
| | | | |
| /s/ジェフリー·シュワルツ | | 役員.取締役 | | 2023年2月28日 |
| ジェフリー·シュワルツ商工管理修士 | | | | |
| | | | |