アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
|
本財政年度末まで |
|
あるいは…。 |
|
1934年証券取引法第13条又は15条に基づいて提出された移行報告 |
|
そこからの過渡期について |
|
依頼文書番号
(登録者の正確な氏名はその定款に記載)
|
|
|
|
登録者の電話番号は市外局番を含んでいます(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください
登録者がこの法第13節または第15節(D)節に基づいて報告を提出する必要がないかどうかを再選択マークで示す。はい。☐
再選択マークは、登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13条または15(D)節に提出されたすべての報告書を提出したかどうか、および(2)過去90日以内にそのような提出要件に適合しているかどうかを示す
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
☒ |
|
ファイルマネージャを加速する |
☐ |
|
非加速ファイルサーバ |
☐ |
|
規模の小さい報告会社 |
|
|
|
|
新興成長型会社 |
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したか否かを再選択マークで示し、その経営陣が“サバンズ·オクスリ法案”(“米国連邦法典”第15編726(B)節)第404(B)条に基づいて財務報告書の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意のエンタルピーCER幹部が相関回復期間内に§240.10 D−1(B)に基づいて受信したインセンティブベースの補償に基づいて回復分析を行う必要があるかどうかを再選択マークで示す
登録者が空殻会社であるか否かをチェックマークで示す(同法第12 b-2条で定義される)。はい、違います
登録者の普通株2022年6月30日のナスダック世界精選市場における終値計算によると、登録者の非関連会社が保有する投票権と無投票権普通株の総時価は約$である
2023年2月16日
引用で編入された書類
登録者最終委託書の特定部分は、登録者2023年株主総会とともに発表され、登録者財政年度終了後120日以内に提出される予定です2022年12月31日本年度報告第III部に引用して組み込む。引用によって登録者の委託書に明示的に組み込まれていない限り、登録者の委託書は、本10−K年度報告の一部とみなされてはならない。
BRIDGEBIO製薬会社
2022年Form 10-K年次報告
カタログ表
|
|
ページ |
|
第1部 |
|
第1項。 |
業務.業務 |
1 |
第1 A項。 |
リスク要因 |
57 |
項目1 B。 |
未解決従業員意見 |
129 |
第二項です。 |
属性 |
129 |
第三項です。 |
法律訴訟 |
129 |
第四項です。 |
炭鉱安全情報開示 |
129 |
|
第II部 |
|
五番目です。 |
登録者普通株,関連株主事項及び発行者が株式証券を購入する市場 |
130 |
第六項です。 |
[保留されている] |
131 |
第七項。 |
経営陣の財務状況と経営成果の検討と分析 |
132 |
第七A項。 |
市場リスクの定量的·定性的開示について |
151 |
第八項です。 |
財務諸表と補足データ |
153 |
第九項です。 |
会計と財務情報開示の変更と相違 |
216 |
第9条。 |
制御とプログラム |
216 |
プロジェクト9 B。 |
その他の情報 |
218 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
218 |
|
第三部 |
|
第10項。 |
役員·幹部と会社の管理 |
219 |
第十一項。 |
役員報酬 |
219 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
219 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
219 |
14項です。 |
チーフ会計士費用とサービス |
219 |
|
第4部 |
|
第十五項。 |
展示品と財務諸表の付表 |
220 |
第十六項。 |
表格10-Kの概要 |
221 |
陳列品 |
221 |
|
サイン |
226 |
|
このForm 10-K年次報告では、他の説明または文脈要件がない限り、言及された“BridgeBio”、“会社”、“私たち”、“私たち”または同様の名称は、BridgeBio Pharma,Inc.およびそれらの合併子会社を意味する。
i
前向き陳述に関する特別説明
このForm 10−K年度報告書には,改正後の1933年証券法第27 A節と改正後の1934年証券取引法第21 E節に該当する前向き陳述が含まれている。このような展望的陳述は重大な危険、不確定要素、そして仮定と関連がある。本年度報告には、歴史的事実に関する記述を除いて、本年度報告におけるすべての記述は、我々の戦略、将来運営、将来運営費用、将来財務状況、将来収入、予想コスト、見通し、計画、意図、期待、目標、目的に関する記述を含むが、これらに限定されず、前向き表現である可能性がある。“予想”、“信じる”、“可能”、“設計”、“推定”、“予想”、“目標”、“意図”、“可能”、“目標”、“計画”、“プロジェクト”、“追求”、“将”、“将”、および同様の表現(その否定を含む)は、これらの識別語を含む前向き表現を識別することを目的としている。本報告書の前向きな陳述は、以下の態様に関する陳述を含むが、これらに限定されない
II
私たちは私たちが展望的陳述で開示した計画、意図、期待、または目標を実際に実現できないかもしれないが、私たちの展望的陳述の背後にある仮説は正しくないことが証明されるかもしれない。しかも、もし私たちの展望的な陳述が不正確であることが証明されたら、この不正確さは実質的である可能性がある。したがって、あなたは私たちの展望的な陳述に過度に依存してはいけません。あなたはこのような陳述を私たちまたは他の誰もが私たちが任意の特定の時間枠内で私たちの目標と計画の陳述または保証を達成するか、または根本的にできないと思ってはいけません。実際の結果またはイベントは、私たちが前向きな陳述で開示した計画、意図、期待、および目標とは大きく異なるかもしれない。実際の結果やイベントが前向き表現と大きく異なる可能性のある重要な要素としては、“第I部”管理層の財務状況や経営結果の検討と分析“第I部第1 A項”管理層の財務状況や経営成果の検討および分析“リスク要因”項に列挙された要因、および本10−K年度報告に記載されている他の部分に挙げられる前向き表現が含まれるが、これらに限定されないと考えられる。
三、三、
リスク要因の概要
以下は,我々の普通株投資に投機的あるいはリスクを持たせる要因の概要である.この結論は私たちが直面しているすべての危険を解決していない。本リスク要因要約でまとめられたリスクおよび我々が直面している他のリスクに関するより多くの議論は、以下の“リスク要因”のタイトルで見つけることができ、我々の普通株について投資決定を行う前に、本年度報告Form 10-Kおよび米国証券取引委員会または米国証券取引委員会に提出された他の文書の他の情報をよく考慮しなければならない。
四
v
部分 I
第1項商売人
概要
BridgeBio Pharma,Inc.は商業段階の生物製薬会社であり、設立の目的は発見、創造、テストと変革性薬物を提供し、明確な遺伝駆動要素を有する遺伝病と癌患者の治療に用いることである。BridgeBioの開発プロジェクトの範囲は早期科学から高度臨床試験までである。BridgeBioは2015年に設立され、経験豊富な薬物発見者、開発者と革新者からなるチームは遺伝子医学の進展を応用してできるだけ早く患者を助けることに取り組んでいる。設立以来、BridgeBioはすでに15個の調査性新薬申請またはINDを作成し、2つの製品がアメリカ食品·薬物管理局の許可を得た。私たちは20以上の異なる発展段階にある疾病州で仕事をしている。私たちのいくつかの計画は潜在力があると思う兆候を狙っています。承認されれば、私たちの候補製品は年間売上高が少なくとも10億ドルの一部の市場機会を狙っています。
我々は高度に満たされていない患者の需要と処理しやすい生物学的交差点に存在するため、遺伝疾患に注目した。我々の方法は,学術実験室とリーディング医療機関が初めて開発した研究を,最終的に患者に触れたい製品に変換することである。著者らは一連の科学進歩を通じてこの機会を実現することができ、(1)費用効果の高いゲノムとエクソン群のシークエンシングの出現に伴い、疾病の遺伝基礎を決定する;(2)分子生物学的進展;(3)遺伝子を疾病と結びつけることができる縦方向データと遡及性研究の発展と成熟を含む。私たちはこの初期革新が新薬を作る最大の実際の源の一つだと信じている。
私たちはすでに世界的な製品プラットフォームを開発し、わが社の持続的な成長と私たちの生産ラインの推進を支持していると信じています。
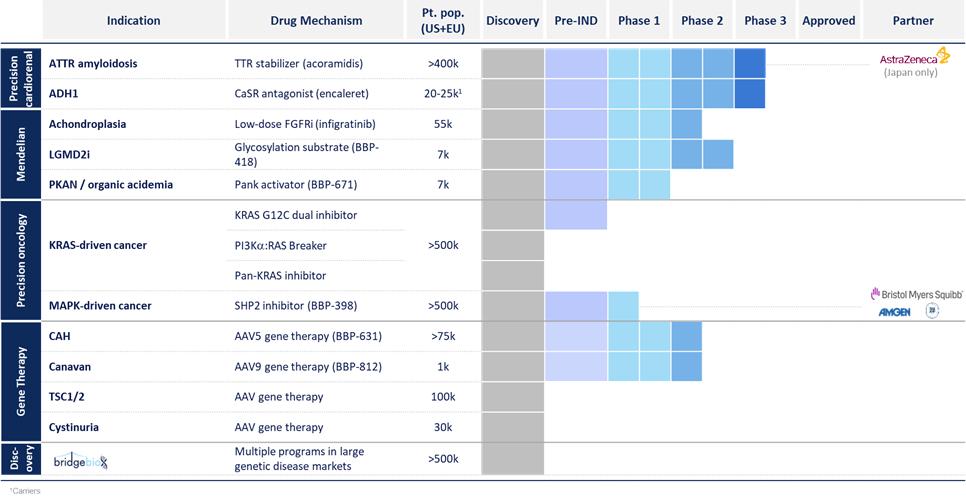
私たちのパイプは
私たちのパイプラインは今後5年以内に8つの潜在的な第3段階読み取り値を提供する可能性があり、私たちはアコラミ、低用量infigraitinib、アングリタット、BBP-418、およびBBP-631の市場価値が10億ドル以上に達すると予想する。以下の表に,我々の開発計画,推定患者数,治療様式,開発状況をまとめた
1
私たちの研究計画の中で、以下の要素が最も潜在力がわが社に顕著な短期的価値をもたらすと考えられ、これは、それらの発展段階、加速発展経路の潜在的可用性、未満足の医療需要程度、および適用目標適応における潜在市場規模を含む一連の要素の組み合わせによるものと考えられる
Acoramidis(Eidos):TTR型アミロイドーシス
要約.要約
AG 10とも呼ばれるAcoramidis,TTRアミロイドーシスの治療,あるいはATTRの経口小分子TTR安定剤の開発を行っている。632名のATTR心筋症患者に対する3期臨床試験が現在行われており,ATTRCM研究と呼ばれている。ATTRIBUTE-CM研究B部分のトップ30カ月の結果データを2023年に報告する予定である.著者らは依然としてacoramidisが30ケ月の分級複合主要終点で利点を示す可能性があり、その中にはすべての原因の死亡率と心血管に関連する入院治療を含むと信じている。
2021年12月27日、私たちは、12ヶ月の6分間の歩行距離において、Attribute-CMがそのA部分の主要な変化終点に達していないことを報告した。12ヶ月の時、アコラミ治療を受けた参加者は、N-末端前駆体BNP、またはNT-proBNP、心臓バイオマーカー、および血清TTR濃度、TTR安定性を測定する指標、およびカンザスシティ心筋症の総スコア、またはKCCQ-OS、生活の質測定を含む重要な副次的および探査終点において、プラセボ治療を受けた参加者よりも改善したことを観察した。
市場のチャンス
予見可能な未来には,疾患意識の向上と非侵襲的診断技術の広範な採用に伴い,確定診断患者数の増加に伴い,ATTR治療介入の総市場は引き続き増加すると信じている。米国で確定診断されたATTR−CM患者数は2019年の5,000人未満から2021年の30,000人以上に増加していると推定されている。したがって,アコラミが承認されれば,かなりの数の新診断患者が以前に疾患修正療法の治療を受けず,アコラミで治療可能である可能性が考えられる。もし承認されれば、私たちはアコラミが大きなビジネス潜在力を持つかもしれないと信じている。また,承認されれば,アコラミはATR治療の最適安定剤になる可能性があると信じている。
2
病気の概要
ATTRはTTR 4量体の不安定による進行性アミロイド沈着による疾患である。TTRは4量体の形式で自然に存在する蛋白質であり、4つの同じサブユニット或いは単量体から構成され、多種の生理作用を有し、輸送に必要なホルモンとビタミンを含む。Attrでは,TTR 4量体は遺伝子変異により不安定になるTtr遺伝子や自然老化過程の一部として不安定なTTRは単量体に解離し,自己集積し,線維に組み立てられ,主に心臓や神経系に沈着し,疾患の病理生理学を推進する。
心筋症ATTRは通常,その遺伝子型によって野生型ATTR型心筋症(ATTRv−CM)と変異型ATTRv−CM(ATTRv−CM)に分類される。この二つの病気は進行性と致命的だ。ATTRUT−CMやATTRv−CM患者は通常晩年(50歳以上)に症状が出現し,治療しなければ確定診断からの中位予想寿命は2~5年である。この2種類の疾病の発展はすべて深刻な障害を招き、生産力と生活の質を影響し、そして患者の支持性看護に関連するコストによって重大な経済負担を招く可能性がある。病態の進展に伴い,ATTRUT−CMやATTRV−CM患者は入院と重複介入を繰り返す可能性がある。
世界的にATTRUT−CMとATTRV−CMの流行率はそれぞれ40万と4万を超えると推定されている。今日の心筋症のATTRは大きく過小評価されていると考えられる。例えば,最近の文献では,心不全と診断されたが駆出率が保持されている患者のうち,10%から13%の患者が未診断のATTR−CMを有する可能性が示唆されている。駆出率節保留の心不全患者はアメリカの心不全患者の推定人数(600万から700万人)の約半分を占める。疾患修正療法の普及に伴い,疾患に対する認識が高まっている。
正確で確実な非侵襲的診断イメージング技術への移行により,確定診断されたATTR−CM患者の数も急速に増加していると信じている。歴史的には,ATTR−CMの診断には心臓生検が必要である。しかし、最近の研究により、単光子放出コンピュータ断層撮影(SPECT)と結合したオスミウム標識放射性トレーサーシンチグラフィ、CT画像は高精度、非侵襲性とコスト効果の高いATTR-CM診断方法であることが明らかになった。疾患意識の向上とこのような非侵襲的診断イメージング技術の獲得性は,ATTR−CM患者の早期診断と以前誤診された患者の識別を可能にしたと考えられる。
設計規範
Acoramidisは臨床段階の経口小分子TTR安定剤であり,ATRを源から治療するために開発されている。我々が設計したacoramidisは2つの主要な基準である保存循環中の天然TTRに適合し,有毒TTRモノマーの形成を最大限に減少させることでアミロイド沈着を減少させる。
TTRは全進化過程において高度に保存されたタンパク質であり、持続的な代謝エネルギー消費を必要とする血漿中に含有量が豊富であり、比較的に速い回転が必要である。そこで,除去ではなくTTR 4量体の最大安定を実現することを求めた。
Acoramidisはすでに臨床前研究と臨床試験で四量体TTRの単量体への解離を防止できることが証明され、臨床前研究でアミロイド線維形成の速度を低下させることが証明された。また,4量体TTR 4量体の循環レベルを増加させることが証明されている。Acoramidisは1種の方法でTTRと結合し、TTRのコンホメーション構造を特徴の良好なT 119 M変異体を模倣させるように設計され、T 119 M変異体は自然に発生する救命突然変異であり、TTR四量体を超安定させることができる。T 119 M変異体はTTR四量体の単量体への解離を防止することが観察されており,生化学分析ではT 119 M四量体の解離速度は野生型四量体より40倍遅い。トランス対立遺伝子トランス抑制遺伝子として、T 119 M救助性突然変異とTTR破壊安定突然変異を持つ個体はATTRの発生を予防することができる。
3
もう一つの小分子TTR安定剤タファミディを経口投与する第三者臨床試験では、全原因死亡率と心血管関連入院時間の測定により、TTR安定度を増加させる介入方法はこの疾患の予後を改善し、血清TTRの増加に関連する。また,遺伝データによると,TTR値安定レベル,血清TTR値レベルと疾患重症度との間には相関が認められた。したがって,血清TTR値は疾患予後を予測するバイオマーカーであり,より有効なTTR値が安定しており,血清TTR値と臨床結果の改善との間に関係がある可能性が考えられる。比較非臨床研究の結果から,アコラミは他のTTR安定剤よりもTTRを安定させる潜在力が大きいと考えられた。
臨床資料
第2段階データ
2018年11月,症状のあるATTR−CM患者におけるアコラミの第2段階データを公表した。ランダム、プラセボ対照、用量範囲の異なるこの臨床試験は、49人の症状を有するATTR−CM患者を含み、そのうちの14人の患者がATTR-CMを有する。条件に適合する患者は、プラセボまたは400 mgまたは400 mgまたは800 mgのアコラミを1:1:1の割合でランダムに添加し、28日以内に1日2回服用した。全体的に言えば,アコラミは症状のあるATTR−CM被験者では耐性が良好であり,薬物の潜在的な臨床注目の安全シグナルを研究していない。コナジラミは血清TTR値を有意に上昇させた(p
2019年11月、私たちはアコラミの長期耐性とATTR-CM疾患対策の安定性を示す第2段階開放ラベル拡張(OLE)のデータを発表しました。AcoramidisはOLEでは耐性が良好であり,潜在的な臨床的関心のない安全シグナルは研究薬物によるものである。OLE参加者の探索的分析で観察された全原因死亡率(死亡または心臓移植を含む、8.5%)および心血管関連入院(少なくとも1回の事象を経験した割合、25.5%)は、ATTR−ACT研究においてプラセボ治療を受けた患者が15カ月後に観察された(死亡または心臓移植を含むすべての原因死亡率、15.3%;心血管関連入院、41.8%)よりも低かった。
2022年4月,われわれの第2段階OLEの最新結果を公表し,アコラミの持続長期耐性とATTR−CM疾患対策の安定性を証明した。連続治療の中央値は38ケ月であり、OLEにおける阿可拉米の耐性は通常非常に良く、薬物の研究には潜在的な臨床関心の安全シグナルがない。
4
ステップ3データ
2019年2月,ATTRCMの世界3期無作為プラセボ対照アコラミ臨床試験ATTRCMを開始した。ATTRIBUTE−CMは,野生型あるいは変異型TTRやニューヨーク心臓協会またはNYHA I−III症状に関連する症状を有するATTR−CM被験者632名を募集した。2つの部分からなる試験では、被験者は、1日2回、2:1の治療群(アコラミ800 mg)およびプラセボ群にランダムに分けられた。 A部では,治療群とプラセボ群の12カ月時の6 MWDの変化を比較し,潜在的な登録終点とした。B部では,全因死亡率と心血管入院を含む分級複合主要終点を30カ月で治療群と対照群との間で比較する。二次終点は、KCCQ-OS評価の生活の質、安全パラメータ、血清TTRレベル(TTR安定性を測定する指標)、およびNT-proBNPレベル(心臓バイオマーカー)を含む。B部分では、彼のファミディの同時使用が許可されている。実験模式図は以下のとおりである

2021年12月27日,6 MWDベースライン変化の主な終点に達していないATTRIBUTE−CM試験A部分の背線データを報告した(p=0.76)。アコラミド群とプラセボ群の6 MWDの平均低下はそれぞれ9 mと7 mであった。観察されたATTRIBUTE−CM両腕機能低下は,12カ月時の健常高齢者の期待機能低下と類似していた。12カ月でアコラミを服用した参加者は,プラセボを服用した参加者よりも副次的および探査終点(NT−proBNP,血清TTR濃度およびKCCQ−OSを含む)の改善が観察された。
Acoramidisは全体的に耐性が良好であり,臨床的に注目されている安全シグナルは認められなかった。B部分の完全性を保護するために,スポンサーとして,A部分の非盲目的有害事象データへのアクセスは,心血管入院(調査者による決定)を引き起こす有害事象は含まれておらず,死亡結果を有するイベントは除外した。85.3%のプラセボ服用参加者および91.9%のアコラミ服用参加者が有害事象または副作用を起こした。プラセボ群と積極治療群では,副作用の重症度の多くが軽度から中等度であった。プラセボ治療を受けた参加者の23.2%に重篤な有害事象が発生したが,アコラミ治療を受けた参加者の20.2%に重篤な有害事象が発生した。プラセボ治療を受けた参加者の6.2%およびアコラミ治療を受けた参加者の4.5%がアンギオテンシン変換酵素で死亡した。
著者らは依然としてacoramidisが30ケ月の分級複合主要終点で利点を示す可能性があり、その中にはすべての原因の死亡率と心血管に関連する入院治療を含むと信じている。ATTRIBUTE−CM試験B部分のトップラインの30カ月間の結果を2023年に報告する予定である。
5
競争
アコラミがATTR−CM治療薬として承認されれば,Vyndaqel/VynDamax(タファミディス/タファミディ)からの競争に直面することが予想され,後者は米国,EU,日本を含むATTR−CM治療薬としていくつかの地域で承認されている。そのほか、現在いくつかのRNAi、アンチセンスオリゴヌクレオチド、抗体と遺伝子編集製品候補薬物が開発されており、ATTR-CMの潜在的な治療法となっている。
低用量 Infigratinib:軟骨発育不全
要約.要約
我々は,軟骨発育不全児の治療の選択肢として,FGFR 1−3選択性チロシンキナーゼ阻害剤またはTKIを経口投与する低用量infigratinibを開発している。著者らは現在患者を募集してPROPELに参加しており、これは軟骨発育不全児童に対する展望性観察研究であり、PROPEL 2は軟骨発育不全児童に対する小用量infigratinibの2期投与量の増加と拡大研究である。2022年7月,我々が報告した初歩的なデータによると,PROPEG 2の4回目の用量増加行列(0.125 mg/kg,1日1回)では,5歳または5歳以上の軟骨発育不全児の年化増加速度はベースラインより+1.5 cm/年増加した。私たちは第5剤(1日1回、0.25 mg/kg)のアップグレードキューを登録し、2023年第1四半期に初歩的な早期結果を共有する予定です。
市場のチャンス
軟骨発育不全と他のFGFRによって駆動される骨発育不良は潜在的な50億ドルを超える世界市場機会を代表すると信じている。2021年末以来、軟骨発育不全市場だけで着実に増加しており、これは1種の新しい利用可能な治療方法が児童の治療を促進し、及び小児科内分泌科医師の新しい治療方法に対する認識が日々高まっているためである。承認されれば、低用量のinfigratinibは重要な商業潜在力を持ち、一流の治療効果、および多くの患者の第一選択の差別化経口投与経路を示すことができると信じている。
条件の概要
軟骨発育不全は不割低をきたす最もよく見られる原因であるが,FGFR 3遺伝子の変異はこのような場合の分子源であることが証明されている。軟骨発育不全の米国とEUでの罹患率は55000人を超えており,全世界の未熟児発症率は10,000人から30,000人に1人と推定されている。このような状況は比例しない小柄、骨格発育異常を招き、そして後頭大孔狭窄、椎管狭窄、心血管合併症と肥満を招く可能性がある。男性の平均身長は約4フィート4インチ,女性軟骨発育不全患者の平均身長は約4フィート1インチである。寿命と知能は一般的に正常だ。
軟骨発育不全は常染色体優性遺伝病であり、FGFR 3遺伝子の機能獲得点突然変異によるものである。約97%の症例がG 380 R変異によるものであり,80%の症例が原因であった初めからやり直す突然変異です。FGFR 3は骨芽細胞と軟骨細胞に発現し,MAPK経路とSTAT 1経路により骨成長を制御し,MAPK経路は肥大分化を駆動し,STAT 1経路は軟骨細胞増殖を駆動する。日本で承認された成長ホルモン以外に、1種類の薬物のみがFDA、ヨーロッパ薬品管理局(European Medicines Agency、EMAと略称する)と薬品と医療機器局(PharmPharmticals and Medical Devices Agency、PMDAと略称する)の許可を得て発売され、軟骨発育不全の治療に応用されている:Voxzogo(Vosoritie)、1種のC型ナトリウム利尿ペプチド、或いはCNP、類似物はMAPK経路を活性化することができるが、STAT 1経路を活性化しない。
設計規範
著者らは2つの重要な設計原則に基づいて低用量のinfigratinibを開発している-著者らは根源から軟骨発育不全(FGFR 3機能獲得突然変異)を求め、臨床活性を最大限に高め、このような疾病のすべての表現に対抗し、身長だけではなく、児童及びその家族の注射治療負担を軽減するために、耐えられる経口治療方案を提供することを求めている。低用量のinfigratinibは,この2つの設計原則を組み合わせた唯一の研究療法であると信じている。
6
低用量のinfigratinibはFGFR 3機能に対して直接突然変異を獲得するように設計され、これらの突然変異は軟骨発育不全病理生理学の背後の駆動要素である。FGFR 1−3の阻害剤として,低用量のinfigratinibがFGFR 3下流の病理シグナルを減少させ,軟骨発育不全を源から治療する可能性が考えられる。潜在的な競争的CNPシミュレーション法とは異なり,われわれの方法はMAPKシグナルのみを抑制し,我々の方法もSTAT 1シグナルを抑制することを目的としている。
低用量のinfigratinibも経口投与経路として設計されている。盲目的な市場研究により、経口投与は軟骨発育不全児童を治療する医療保健提供者の第一選択投与方式である。
我々の第2段階用量アップグレード試験で完成した4つの用量列、および2023年2月9日までの第5回用量列では、低用量infigratinibは全体的に耐性が良好であり、深刻な有害事象の報告はなかった。2023年2月9日現在,有害事象による中止はなく,用量依存性のリン酸塩上昇もなく,眼部有害事象もなかった。
臨床前データ
小用量のinfigratinibはすでに軟骨発育不全のマウスモデルにおいて臨床前研究を行い、このモデルは成長板、椎骨と椎間板の異常を再現した。調査者は少量のinfigratinibが救助されたことを観察しました離体する突然変異マウス胚胎大腿骨治療6日後の骨成長。さらに15日間の治療では体内にある骨の成長は、多くの方面で人類の軟骨発育不全を模倣している。この研究では,アタッチメントと軸方向骨格パラメータへの影響を観察した。
下の図は、野生型、未治療モデルおよび低用量infigratinib(2 mg/kg)モデルマウス大腿骨成長および椎間板幅回復の程度を示している

7
生体内同一実験室では低用量(0.2 mg/kgと0.5 mg/kg)でさらに骨成長が確認された。つまり、すべての用量の臨床前研究により、治療と未治療の突然変異マウス間の骨格成長パラメータは以下のように有意に増加した
未処理マウスと比較してマウスの長さ(%)が増加した
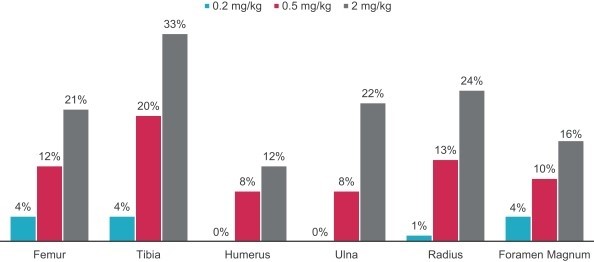
注意すべきことは,低用量のinfigratinib治療は成長板肥大区FGFR 1の発現を変化させなかったことである。これらの効果は主にFGFR 3が抑制されたためであり,これらの臨床前研究では他に重篤な副作用は認められなかった。また,低用量infigratinib治療マウスの15日後の生存率は,未治療マウスと比較して投与量にかかわらず向上した。
臨床発展計画
われわれは現在PROPELに参加する患者を募集しており,軟骨発育不全児に対する前向きな観察研究である。この研究は各子供のために少なくとも6ヶ月の年化成長速度またはAGVを確立するだろう。PROPELは児童にベースライン測定を提供することを目的としており、これらの児童はPROPEL 2、低用量infigratinibの第2段階研究、またはその後に行われる予想される第3段階試験に参加することが予想される。
PROPEL 2は、板閉鎖前に軟骨発育不良を有する児童の成長に使用するための開放ラベル、用量増加と拡大試験として設計されている。この研究の主な目的は軟骨発育不全児童の安全性と耐性を評価することである。副次的な目標には、PK分析、成長速度の変化、生活の質の評価が含まれる。
2022年7月に我々が報告した初歩的なデータによると、PROPEG 2の4回目の用量増加行列では、5歳または5歳以上の軟骨発育不全児の年化増加速度がベースラインより+1.52 cm/年増加した。私たちはすでに第5回用量アップグレードキューを募集し、2023年第1四半期に初歩的な結果を共有する予定だ。
FDAのフィードバックにより、2023年に低用量infigratinibによる軟骨発育不全治療の3期の重要な試験を開始する予定である。
8

主な競争相手
低用量infigratinibは、軟骨発育不全を治療するために唯一開示された経口直接FGFR 1-3阻害剤である。また,他の3社が代替機械法を用いて軟骨発育不全を治療する化合物を開発している:Ascendis Pharma A/S(Transcon CNP),セノフィ社(SAR 442501),リボソーム社(RBM−007)。また,BioMarin製薬会社は軟骨発育不全の治療にVoxzogoというCNP類似体を開発した。
Encaleret:常染色体優性遺伝性1型低カルシウム血症と副甲状腺機能低下症
要約.要約
Encaleretは経口カルシウム誘導受容体小分子拮抗薬であり,常染色体優性遺伝性1型低血カルシウム症(ADH 1)の治療に開発されている。われわれはADH 1患者の潜在的治療法として,現在行われている2 b期と3期臨床試験におけるENALERETを検討している。私たちは2022年に2 b段階研究の結果を報告した。2 b期試験の参加者13名のうち,Enlaretを用いた治療は正常ミネラル安定を迅速かつ持続的に回復させ,治療5日目の血中カルシウム,尿カルシウム,血液副甲状腺ホルモン(PTH)の平均値は正常範囲内であり,24週間持続し,耐性は良好であり,重篤な有害事象の報告はなかった。EncaleretはすでにFDAから常染色体優性低血カルシウム症を治療する孤児薬物と高速チャネルが指定されている。Encaleretは欧州委員会から孤児にも指定されており,ADH 1を含む副甲状腺機能低下症の治療に用いられている。
9
市場のチャンス
私たちはADH 1が巨大な商業的潜在力を持つ市場だと信じている。ADH 1はCaSR遺伝子の機能獲得変異によるものであり,一般集団遺伝データセットの独立した研究では,EUや米国では25,000名のADH 1が変異を引き起こすキャリアが推定されている。承認された場合、EnlaretはADH 1の最初の標的治療となる可能性がある。
設計規範
Encaleretは研究中の経口CASRの小分子拮抗薬である。これまでの研究では,1200人以上のヒト被験者で研究されており,1用量依存的に血清カルシウムが増加することが観察されている。ADH 1患者の潜在的治療法としてEnlaretを開発した理由は,非臨床的·臨床的エビデンスに基づいているからである。CASRの拮抗者は証明されています体外培養異常なCASR“設置点”を正常ICに戻す50 カルシウム上と体内にある副甲状腺ホルモンの分泌を促進し、血中カルシウム濃度を高め、尿カルシウム排泄を減少させる。CaSRを選択的に拮抗することにより,EnlaretはADH 1を有する患者の正常なCaSR機能を回復し,低カルシウム血症や高カルシウム尿に関連する症状を緩和することが可能である
臨床資料
2022年6月13日,われわれがADH 1患者で行ったEnlaret 2 b期臨床試験の陽性データを報告した。13例の成人ADH 1は9種類のユニークなCASR変異体は3つの時期の2 b期開放ラベル、用量範囲の臨床試験に参与した。カルシウム剤と活性ビタミンDサプリメントの経口投与は服用開始前に中止した。ステップ1とフェーズ2はそれぞれ5入院日でEnlaretを評価し,ステップ3には24週間の外来評価を含む。24週間の外来データに基づいて観察されました
研究を終えた第3段階の参加者は、25カ月にわたるオープンラベル延期を継続する資格がある。
2022年にADH 1でEnlaretの第3段階登録試験を開始することも発表した。
10

主な競争相手
EnlaretはADH 1の治療に特化して公開されている唯一の開発中の分子であると考えられる。他にも,Ascendis Pharma A/S(TransconPTH),Amolyt Pharma(AZP−3601),EnteraBio Ltd.(EB612),Extended Biosciences Inc.(EXT 607)が,組換え副甲状腺ホルモン類似体や副甲状腺受容体アゴニストを用いて副甲状腺機能低下症を治療する化合物が開発されている。
BBP−418:肢体筋萎縮症2 I型
BBP−418は研究,経口,小分子基質代替療法であり,LGMD 2 Iの治療に用いられており,LGMDR 9とも呼ばれる。著者らは現在LGMD 2 I患者に対する開放ラベル投与量増加第二段階臨床試験を行い、BBP-418を研究している。私たちは2022年10月に第一線のデータを発表した。我々は2023年に世界3期臨床試験を開始する予定である。
病気の概要
LGMD 2 Iは遺伝性神経筋疾患であり、特徴は下肢無力と行動不便であり、肺と心臓機能障害もある可能性がある。米国およびヨーロッパでは、LGMD 2 Iおよび他の潜在的にアドレス指定可能な血糖ジストロフィーを含むBBP−418の潜在的アドレス指定可能人口は7,000人である。現在,利用可能な疾患修正治療法はない。看護の基準は支持性看護であり、終末臓器機能障害を軽減する。
11
設計規範
LGMD 2 Iの潜在的治療法としてBBP−418を開発する基本原理は,我々の疾患機序の理解に基づいている。健康組織において、機能的に正常なフォチン関連タンパク質、またはFKRP、グリコシル化α-ジストロフィーグリカン、またはαDG。このグリコシル化は細胞外リガンドを結合させることで筋細胞の安定化を助ける。LGMD 2 Iでは,変異したFKRp機能が正常でなく,筋細胞における機能障害をきたす低グリコシル化αDGは,αDGの筋線維としての“ダンパ”の能力を制限し,細胞の損傷に対する感受性を増加させている。
BBP-418は、上流に超生理学的レベルのBBP-418を提供することによって、変異FKRP酵素の残留活性を駆動し、αDGのグリコシル化レベルを潜在的に増加させ、それによってLGMD 21の疾患機序を標的とすることを意図している。
臨床資料
2022年10月14日,我々はLGMD 2 I患者で行われているBBP−418の第2段階臨床試験の積極的な中期データを第27回世界筋協会国際交雑年次総会で共有した。この開放ラベル、用量増加の第2段階試験は、3つのキュー中の非外来診察と非外来LGMD 2 I患者を含む14名の参加者を募集した。2022年8月16日現在、治療12カ月後のデータによると、
臨床発展計画
われわれはLGMD 2 IでBBP−418を検討する第二段階臨床試験を行っている。著者らの第二段階試験の主要なデータが公表された後、著者らはすでに監督機関と交渉して、第三段階試験設計を調整し、2023年に第三段階臨床試験を開始する予定である。
主な競争相手
BBP−418はLGMD 2 I潜在疾患修飾治療の唯一の臨床段階経口療法であり,臨床開発に公開されていると考えられる。現在,他に既知の口腔療法は開発されていない。また,他の2社がLGMD 2 Iを治療する遺伝子療法:Asklepiosバイオ製薬会社(LION−101)とAtamayo治療会社(ATA−001−FKRP/ATA−100)を開発している。
BBP−631:先天性副腎増殖症
要約.要約
我々は、21 OHDによるCAHの治療のための候補AAV遺伝子転移製品BBP−631を開発している。BBP−631は2021年にFDAにより先天性副腎増殖症21−ヒドロキシ酵素欠乏症を治療する迅速チャネル薬として承認された。2023年2月22日までに、4人の患者が前の2つの用量レベル(1.5 e 13 Vg/kgと3 e 13 Vg/kg)の治療を受けた。私たちは2023年末までに第三用量レベル(6 e 13 Vg/kg)で患者の最新状況を治療する予定である。
12
市場のチャンス
21−ヒドロキシ酵素や21 OH欠乏によるCAHは5つの最大の患者集団の1つを有しており,遺伝子治療に適している可能性があり,重要なビジネス機会であると考えられる。米国やEUでは75,000人以上がアドレス指定可能なCAH人口に属すると推定されており,また,現在の看護治療基準に関連する副作用を考慮すると,満たされていない需要は巨大である。
病気の概要
CAHは人を虚弱にし、生命に危害を及ぼす疾患であり、アメリカのどの州でも新生児疾患のスクリーニングを行っているにもかかわらず、利用可能な治療法はない。この疾患はコルチゾールやアルドステロンが産生されないこと,テストステロンの過剰産生が定義されている。コルチゾールの欠乏はブドウ糖代謝と身体の圧力に対する正常な反応を乱し、潜在的な致命的な副腎危機を招き、アルドステロンの欠乏はナトリウム滞留を乱し、低血圧、不整脈と脱水を招く。また,過剰なテストステロンは女性の男性化を招き,通常生殖器不明瞭や出生時の男性化特徴を引き起こす。思春期のホルモン変化はCAHの欠陥を悪化させた。女性は通常生育力が限られており,妊娠前,妊娠期間,妊娠後に強化治療が必要であり,成人男性の40%が副腎休憩腫瘍を有しており,性腺機能障害や不妊をきたす可能性があり,まれに手術が必要である。
CAH症例の90%以上が21 OHDによるものであり,21 OH酵素をコードするCYP 2 1 A 2遺伝子の遺伝的欠陥である。21 OH酵素活性の喪失を招く突然変異はプロゲステロンの11-デオキシコルチゾンへの転化と17-ヒドロキシプロゲステロン(17 OHP)の11-デオキシコルチゾールへの転化を阻止し、この2種類の物質はそれぞれアルドステロンとコルチゾールの前駆体である。
遺伝子突然変異のタイプによって、21 OHDを有するCAH患者は2種類に分類できる:典型的と非経典型。著者らは主に典型的な患者の治療に集中し、彼らは更に深刻な表現型を持ち、アルドステロン欠乏の重症度と残留した21 OH酵素活性レベルによって、それを単純男性化(約25%)と塩消費(約75%)に分けることができる。塩消費型疾患患者の残存酵素活性は正常の0%~1%であり、単純性雄化表現型患者の酵素活性は1%~10%である。すべての典型的なタイプの患者は出生時に治療が必要であり,コルチゾール欠乏は周囲の生命の中で副腎危機を招き,すぐに死亡する可能性があるからである。塩消費型新生児の発病率は20,000名の新生児に1例であるが、単純男性化型新生児の発病率は60,000名の新生児に1例である。合わせて、毎年600人の典型的な患者がアメリカとヨーロッパで生まれたと推定されている。米国やヨーロッパのアドレス可能患者総数のうち,7.5万人を超える患者が想定されている。
現在の標準看護治療は患者を治愈することができず、欠損したグルココルチコイド(例えばコルチゾール)と鉱質コルチコイド(例えばアルドステロン)を置換し、及び過剰なアンドロゲン分泌を減少させる。グルココルチコイドはCAH治療の主要な薬物であるが、一人一人の反応方式はそれぞれ異なり、児童と成人はグルココルチコイドを長期的に使用するには慎重に管理する必要があり、これらの薬物のよく知られている副作用、例えばクシンゴッド特徴、代謝性疾患、肥満、高血圧、成長遅延、グルコース耐性、電解質の乱れ、骨格脱灰/骨折の増加と思春期遅延のリスクである。典型的なCAHの臨床治療はよく高アンドロゲン血症と高コルチゾール血症の間でバランスをとることは困難である。
13
設計規範
BBP-631はAAV 5遺伝子転移製品の静脈注射であり、副腎皮質中の21 OH酵素を置換することによって21 OHDによるCAHを治療することを目的としている。酵素機能の代替は,この経路を介した流量を正常化するとともに,コルチゾールやアルドステロン欠乏やテストステロンや他のアンドロゲン過剰の問題を解決する可能性がある。CAH中の遺伝子型-表現型相関性研究により、通常無症状かつ治療を必要としない非典型的な患者の酵素活性は正常人の10%~20%であることが分かった。AAV遺伝子療法は,単純な男性化や塩消費疾患を有するCAH患者のこのレベルの酵素活性を回復し,実質的な臨床影響を提供し,外因性ステロイド治療の需要を潜在的に除去することができると信じている。BBP-631は、2018年に21 OHDによるCAHの治療のためのFDAおよびEMA孤児薬物指定を取得した。
臨床前データ
AAVRH 10を用いてCyp21ノックアウトマウスモデルにおいて最初の臨床前活性を探索した。担体ゲノムの静脈または静脈注射は、体重増加、尿プロゲステロン(21 OHの主要基質)の減少、レニン発現の増加(塩保持能力の増加を示す)を含む15週間のウィンドウ内で様々な疾患関連因子を改善することができることが観察された。
非ヒト霊長類動物(NHP)で行われた研究では,評価されたAAV血清型1,5と6を比較し,AAV 5が最適な血清型であることを確認した。合成21 OHの副腎で有意な転染性が認められた。また,AAV 5は集団での血清陽性率が相対的に低く,潜在的な免疫原性の問題を制限する可能性がある。
著者らはすでに2群のNHP研究を完成し、発現の持続性、用量/遺伝子組換え発現関係と初歩的な安全性を評価することを目的とした。第1グループの実験では、より低い用量の3 x 10が評価された12 1 kgあたりのベクターゲノムでは,Cyp21 mRNAレベルの持続的な増加が6カ月まで観察された。ベクターゲノム数とmRNAレベルは1.5から6カ月の間に副腎細胞の回転により急速に低下することは認められず,持続的な遺伝子組換え発現に初歩的な支持を提供した。
第2群の実験では、合計20名のNHPがBBP-631の3種類の静脈注射用量のうちの1つの治療を受けた。低、中用量群の投与後4、12週、高用量群の投与後12週間、24週の副腎組織中のベクターコピー数と遺伝子組換えmRNAの発現を分析した。試験された任意の用量では、どの時点でも用量に関連する副作用は認められなかった。
全体的にBBP−631で処理すると副腎中のベクターコピー数(VCN)とmRNA発現が増加し,BBP−631は副腎に対して強い走性と摂取作用を有することが示唆された。高用量群ではCNsは12−24週に維持された。また,中用量群のmRNAレベルは4−12週で増加したが,高用量群のmRNAレベルは12−24週で一致していた。また,試験した3種類の用量のうち,CNsとmRNAレベルはいずれも用量の増加とともに増加することが分かった。
主な競争相手
CAH治療の2種類の代替治療種別が検討されている。1つ目は副腎皮質刺激ホルモン放出因子1型、またはCRF 1、受容体拮抗薬である。CRF 1受容体拮抗剤は下垂体副腎皮質刺激ホルモン或いはACTHの放出を調節し、副腎アンドロゲンとコルチゾールの合成を刺激する。健康な個体において、内因性コルチゾールはACTHの放出に対して負のフィードバックを提供し、アンドロゲン合成の良好な調節を維持する。この負のフィードバックはCAH患者で深刻に障害されているため,これらの患者のアンドロゲン合成を正常化するためには超生理量の外因性ステロイドが必要である。CRF 1受容体拮抗薬はアンドロゲンの合成を調節できるが、それらはこれらの患者のコルチゾール或いはアルドステロン産生不足の問題を解決できない。したがって,CRF 1受容体拮抗薬は依然としてステロイドを補充する必要がある。2種類のCRF受容体拮抗薬Crinecerfont(Neurocrine Biosciences,Inc.が開発中)TildacerFont(Spruce Biosciences,Inc.開発)は現在,それぞれ第3段階と第2段階の臨床試験にある。
14
第二の代替治療はACTH受容体拮抗薬である。CRF 1経路下流を抑制する経路は副腎アンドロゲンやコルチゾール合成の抑制にもつながる。しかし,CRF 1受容体拮抗薬と同様に,ACTH阻害剤はこれらの患者のコルチゾールやアルドステロン産生不足の問題を解決できない。CRN 04904は経口ACTH拮抗薬であり、現在Crinetics製薬会社の1期臨床開発中である。
これらの代替治療機序は,外因性ステロイドの需要を潜在的に減少させることで疾患の有意な側面を解決しようとしているが,いずれの機序も疾患の完全な特徴を定義することで根源的に疾患を解決することはできない。特に,これらの機序はステロイド投与の必要性を解消できず,体がコルチゾールやアルドステロンを合成できないという問題を解決できないためである。逆に,遺伝子治療による酵素置換は,ホルモン経路中の適切な流量を回復させることで疾患の様々な側面を同時に解決し,前駆体分子がコルチゾールやアルドステロンに変換する代替経路を提供することでアンドロゲン産生を減少させる可能性が考えられる。
KRAS駆動の癌
我々のKRAS阻害剤の組み合わせは、KRAS駆動癌の新しい選択的機序を治療することによって発癌RASシグナルを抑制する方法に集中している。この仕事には3つの主要な仕事が含まれています
15
KRASはRAS癌遺伝子ファミリーのメンバーであり、このファミリーはまたHRASとNRASを含み、共にいくつかの最も有名な癌単遺伝子駆動要素を構成した。NRAS突然変異は白血病と黒色腫によく見られ、HRAS突然変異は膀胱癌、甲状腺癌と頭頚部扁平上皮癌によく見られる。KRAS遺伝子突然変異は高度に医療需要を満たしていない最大癌適応を有する多くのよく見られる駆動要素であり、30%の非小細胞肺癌、98%の膵臓癌と45%の結腸直腸腺癌を含む。最もよく見られるKRAS変異は、タンパク質12位のグリシンからアスパラギン酸(G 12 D、すべてのKRAS変異の36%)、バリン(G 12 V、24%)とシステイン(G 12 C、15%)への変化を含むが、グリシン13とグルタミン61の変異も含む。全体的に言えば、アメリカとヨーロッパでは毎年50万人を超える患者がKRAS駆動癌と診断されている。
BBP−398:多様な腫瘍学的適応を狙う
BBP−398はSHP 2の小分子阻害剤であり,過剰に活性化した受容体チロシンキナーゼ(RTK)やMAPKシグナルによって駆動される癌の治療に開発されている。我々は2021年7月に百時米施貴宝(Bristol-Myers-Squibb)或いは百時米施貴宝(BMS)とBBP-398とPD-1阻害剤nivalumabの共同開発協定を締結し、2022年5月にBMSと独占許可協定を締結し、この合意に基づいて、NavreはBMSに独占権利を付与し、BBP-398を世界的に開発と商業化したが、人民病院のRepublic of China、マカオ、香港、台湾、シンガポール、韓国(アジア地域)を除く。
われわれは現在,3つの独立した一期試験(NAV−1001,NAV−1003およびNAV−1004)への患者の参加を募集しており,BMSとのライセンス契約および2022年1月に安進と締結された安進のKRAS G 12 C阻害剤(Sotarasib)との共同試験に関する臨床試験連携と供給プロトコルの条項の完成を担当している。
NAV-1001はRASとRTK突然変異患者に対する単一治療用量のアップグレードと拡張臨床試験を行っている。
2022年には、NAV-1003およびNAV-1004を開始し、それぞれBBP-398およびKRAS G 12 C阻害剤(sotorasib;Amgen)およびPD-1(nivolumab;BMS)阻害剤との併用試験を行った。
また,LianBioは現在,我々が2020年8月に締結した独占許可協定に基づき,アジア地域で患者を募集して一期単一療法用量逓増試験に参加している。BBP-398と表皮成長因子受容体阻害剤(Osimertinib)を研究する一期臨床試験は連合生会社が中国で行い、2023年にスタートする予定である。
BBP−671:PKANと有機アシドーシス
BBP-671は我々が開発している経口、小分子、変構造汎酸キナーゼ活性化剤であり、パントニンキナーゼ関連神経変性(PKAN)と有機アシドーシス(OAS)の治療に用いられている。BBP−671は現在第1段階で開発されている。第一段階臨床試験の健康ボランティア部分はすでに完成し、BBP-671の初歩的な安全性と耐性、標的参加と適切なPKを示した。第一段階臨床試験のOAキューは現在募集中である。BBP-671は、プロピオン酸血症(PA)の治療のための孤児薬指定を受け、PKANの治療に米国およびEUで使用されている。BBP−671は、PKANおよびPAの治療のためのまれな小児科疾患を治療する薬剤としても指定されている。
PKANは稀な遺伝性疾患であり、進行性神経変性を伴う。早期発症患者は通常6歳以内に運動障害を示し,網膜変性により視力問題が出現する可能性がある。遅く発症する疾患は異質性であり,精神症状と進行性パーキンソン病は児童後期から成人までの間に進行する。PKANの罹患率は約百万分の1であり,米国とEUには800~850名の患者がいる。PKANを治療する方法は現在承認されていない。
OASは酵素の変異によるものであり,これらの酵素はアミノ酸代謝を撹乱し,入院が必要な急性代償不全や,心臓,膵,腎臓,肝臓,脳など多くの臓器系に及ぶ長期合併症である。OASの発病率は約10万人の新生児に5名である。看護基準は飲食制限と補充を含むが、代謝失調と長期合併症のため、満足されていない需要は依然として高い。
16
BBP-812:カナダ犬病
BBP−812は我々が開発しているCanavan病の治療のためのAAV遺伝子治療候補製品である。BBP−812は2021年にFDAからCanavan病を治療する迅速チャネルの称号を授与され,Canavan病を治療するBBP−812 1/2期臨床試験が行われている。私たちは2022年10月13日に3人の患者の低用量(1.32 e 13 Vg/kg)での予備バイオマーカーデータを公表し、2023年末までに私たちの高用量(3 e 14 Vg/kg)で第1の患者に投与量を提供する予定である。
Canavan病は乳児期から致命的な進行性神経変性疾患である。この疾患は脳白質ジストロフィー症であり,脳中の白質退化によるものである。患者は通常予想された発育マイルストーンを逃し、頭囲は迅速に増加し、次第に運動コントロールが不足し、しかもよく10代にしか生きていない。全世界の加那文病の発病率は約10万人の新生児に1人である。 カナダ病の治療法は承認されていない;看護の重点は症状管理である。
計画に関する他の情報
製造業
私たちは所有したり経営したりしないし、現在は製造施設を設立する計画もない。著者らは現在第三者契約製造組織、即ちCMOに依存して、著者らの臨床前研究と行っている候補製品の臨床試験が原材料、薬物物質と薬物製品に対するすべての要求を満たす。2019年12月に私たちの子会社で締結された製造協定を通過し、2022年5月に双方の合意によって終了したほか、既存のCMOと長期合意を締結していません。私たちはCMOに依存して私たちの候補製品の後期開発と商業化を継続して、私たちが決定する可能性のある他の任意の候補製品を含むつもりです。私たちはCMOに依存していますが、私たちは私たちの契約メーカーとの関係を監督するために豊富な製造経験のある人員と第三者コンサルタントがいます。私たちのいくつかの開発候補は、短期的に余分で重複した薬物物質および薬物製品サプライチェーンを持っているか、または予想されている。
17
販売とマーケティング
ある地域が規制承認を受ける可能性があると考えた場合,米国に商業インフラを構築し,現在の候補製品の商業化を支援するために他の地域を選択する予定である。我々の目標適応の多くは稀な疾患であり、集中的な処方受容者と少数の関連患者集団の処方治療に影響を与える重要なオピニオンリーダーがあるため、現在、内部販売者、内部マーケティンググループ、流通支援の支持の下で、私たちは自分の的確な専門販売とマーケティング組織を通じて各市場の需要を効果的に満たすことができると信じている。私たちはこれまでに開発した2つの製品の規制部門の承認を得ており、TRUSELTIQ(Infigratinib)はtに使用されている以前に治療された局所末期または転移性胆管細胞癌、またはFGFR 2融合または再編成が存在するCCA患者を治療し、NULIBRY(ホスホトレキサート)とMoCD型AA患者の死亡リスクを低下させる第一選択の治療法として注射Entynl治療会社、あるいはSentynlと呼ばれ、2022年第1四半期にNULIBRYの世界的権利を獲得しました現在,NULIBRYの米国での持続的な開発と商業化,世界的な開発,製造,商業化を担当しているFDAが2021年5月にTRUSELTIQ(Infigratinib)を承認した後,米国における主要販売先となった。2022年1月から,残りのTRUSELTIQ過渡的供給をHelsinn Healthcare S.A.とHelsinn Treateutics(U.S.),Inc.,あるいは総称してHelsinn,Helsinnと総称して主要販売先とした。ヘルシンヌは、2023年3月31日までTRUSELTIQの供給を停止すると発表した。2022年12月、私たちはヘルシンと腫瘍学的適応におけるInfigratinibの商業化を段階的に終了することを含む相互終了合意に達した。(当社の連結財務諸表付記11参照。)
私たちはすべての候補製品の臨床開発と規制承認を推進する過程で、私たちの商業化戦略を評価します。私たちが確定する可能性のあるアメリカ以外のどのコア市場でも、承認されれば、戦略パートナー、流通業者、または契約販売チームを利用して、私たちの候補製品の商業供給を拡大することができるかもしれない。我々の任意の候補製品が承認されれば,いくつかの地域や適応や他の戦略的目的での協力を考慮する可能性があるにもかかわらず,大規模な製薬パートナーが商業化する必要はないと予想されている。
優先協力計画
2022年1月には、開発計画の優先順位の再決定などを含む再編計画の実施を約束した。この再編計画の一部として,潜在的な新しいパートナーシップのみでいくつかのプロジェクトを進める予定である。以下にその中のいくつかの調査項目を示す。
そのほか、著者らはもう一つの臨床前発見計画(BBP-954)を発見し、不可逆的なグルタチオンペルオキシダーゼ4(Gpx 4)阻害剤を探し、固形腫瘍と血液病の治療に用いた。2022年12月、私たちの共同開発パートナーヘルシンは、この発見計画に対する私たちのパートナー関係を終了した。
18
知的財産権
概要
私たちは、私たちの製品候補および構成、それらの使用方法および製造過程、私たちのプラットフォーム技術、および私たちの業務発展に商業的重要性を有する任意の他の発明態様をカバーするために、特許および特許出願を求めて維持することを含む、様々な方法によって、私たちの業務に重要であると考えられるノウハウを保護するために努力している。私たちは、米国での特許出願の提出と起訴に加えて、このような外国出願が有益である可能性があると考えている他の国/地域に、オーストラリア、カナダ、ヨーロッパ、中国、日本、メキシコを含む対応する特許出願を提出することを求めている。私たちはいくつかの特許を私たちの候補製品の開発と商業化に使用する権利を得るために様々なライセンス協定を締結している。“私たちの実質的な合意”を参照してください。私たちはまたビジネス秘密に依存して私たちの業務を特許保護から保護したり特許保護に適していないと考えています。
私たちの成功は、私たちが特許および他の固有の権利を取得して維持する能力があるかどうかに依存し、私たちの業務に関連する商業的重要性を有する技術、発明、およびノウハウを保護し、私たちの現在および将来に発表された特許を擁護し、実行し、私たちのビジネス秘密の機密性を保護し、第三者が効果的かつ強制的に実行可能な特許および独自の権利を侵害することなく運営される。私たちは、私たちの知的財産戦略を評価し、改善して、私たちの地位を最大限に強化し、私たちの知的財産戦略がこのような申請を提出する必要がある場合には、より多くの特許出願を提出します。私たちはまた、独自技術、持続的な技術革新、潜在的な許可内機会に依存して、私たちの知的財産権の組み合わせを開発し、維持しています。国内外の特許保護を獲得し、商業価値のある新発明に対して適時に特許申請を提出することを目指している。
私たちのような生物製薬会社の特許地位は通常不確定であり、複雑な法律、科学、事実の問題に関連している。また,特許出願において要求されるカバー範囲は特許発行前に大幅に縮小することができ,特許範囲は発行後に裁判所によって再解釈することができる.さらに、多くの司法管轄区域は、第三者が行政訴訟において発行された特許に挑戦することを可能にし、これは、特許主張のさらなる縮小またはキャンセルをもたらす可能性がある。私たちが現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるかどうか、または任意の特許の権利主張が発行されれば、競争相手からの攻撃から十分な保護を提供するかどうかを予測することはできない。
米国および他のいくつかの司法管轄区域の特許出願は18ヶ月以上秘密にされており、科学的または特許文献における発見は、実際の発見よりも遅れていることが多いため、係属中の特許出願によってカバーされる発明の優先度を決定することができない。さらに、私たちは、発明の優先権を決定するために、米国特許商標局(USPTO)が発表した干渉訴訟または派生訴訟に参加しなければならないかもしれない。
2023年2月13日現在、私たちの知的財産権の組み合わせには、学術および研究機関および他の第三者から許可を得た135件の発行された特許および81の特許出願、ならびに私たちが所有または共同所有している33個の発行された特許および374件の係属中の特許出願が含まれています。これらの特許および特許出願は、通常、米国と世界各地で私たちの候補製品を開発する権利を提供してくれる。我々のコア価値駆動要因である各項目の知的財産権の組み合わせは以下でさらに説明すると考えられる.
私たちの子会社QED治療会社について、私たちはノ華会社から発行された2つの米国特許の権利、ならびにオーストラリア、カナダ、中国、ヨーロッパ、日本とメキシコ、ならびにアジアと南アメリカの他の国の関連して申請および発行された外国特許および特許出願を取得しており、これらの特許および出願はinfigratinibの組成物を対象としている。外国特許や特許出願が発表されれば、2025年から2030年の間に満期になると予想される。発行された米国特許は2028年から2029年の間に満期になると予想されており、これは、米国特許商標局が付与した特許期限の調整と、1つの発行された特許の別の米国特許に対する最終免責声明とを考慮している。Infigratinibの予備承認後、QEDは、infigratinib化合物をカバーする米国特許出願のために1,516日間の特許期間延長、すなわちPTEを延長した;PTE出願が承認されたと仮定すると、この特許の有効期限は2029年8月25日から2033年10月19日に延長される可能性がある。
19
我々はまた、オーストラリア、カナダ、中国、ヨーロッパ、日本とメキシコ、ならびにアジアおよび南アメリカの他の国および地域で2つの発行された米国特許、出願中の米国特許、および関連する出願および発行されている外国特許および特許出願を許可しており、これらの特許および特許出願は、infigratinibを含む医薬製剤を対象としている。発行された特許と特許出願が発行されれば,2034年に満了する予定である。さらに、我々は、オーストラリア、カナダ、中国、ヨーロッパ、日本およびメキシコ、ならびにアジアの他の国および地域で発行された米国特許、承認されるべき米国特許出願、および関連する出願および発行されている外国特許および特許出願をノーワーズ社に許可し、これらの特許および出願は、低リン血症を治療する方法を対象としている。発行された特許と特許出願が発行されれば,2033年に満了する予定である。
また、Inserm Transfer t ESAとフランスパリ公共援助会社から、infigratinibを用いて骨発育不良および軟骨発育不全を治療する方法を対象とした2つの発表された米国特許と、未解決の米国特許出願と、ヨーロッパで付与された特許とを取得した。発行された米国特許,欧州で付与されている特許,及び出願中の特許は,発行されれば2032年に満了する予定である。
さらに、QED治療会社は、オーストラリア、カナダ、中国、ヨーロッパ、日本およびメキシコ、ならびにアジアの他の国および地域に、3つの係属中の米国特許出願、係属中の特許協力条約またはPCT特許出願、およびinfigratinibを使用して様々な癌または骨疾患を治療する方法に関する関連する係属中の外国特許出願を有する。これらの特許出願から任意の特許を取得した場合、これらの特許は2040年から2043年の間に満了すると予想される。
我々の子会社Eidos Treateutics,Inc.については、アラン·スタンフォード初級大学またはスタンフォード大学の取締役会から、発行された10つの米国特許、2つの係属中の米国特許出願、欧州特許、および日本特許の権利を取得しており、これらの特許は、アコラミに関連する物質組成および使用方法に関する。これらの特許は2031年または2033年に満期になると予想される。
さらに、私たちは、オーストラリア、カナダ、ヨーロッパ、中国、日本、メキシコなどの異なる司法管轄区域に、4つの発表された米国特許、3つの被審査米国特許出願、51件の関連外国特許出願を有しており、アコラミに関連する塩および固体形態、製造方法、投与方法および処方に関する。発行された米国特許は2038年または2039年に満期になる予定だ。未解決の米国と外国特許出願が発表されれば、2038年または2039年にも満期になると予想される。
我々の子会社Adrenas Treateutics,Inc.については、係属中の米国特許出願、係属中のPCT特許出願、およびカナダ、中国、ヨーロッパ、日本および韓国を含む複数の司法管轄区域の6つの関連外国特許出願を有しており、これらの出願は、BBP−631に関連する組換えAAV担体およびその用量に関する。これらの特許出願が発行されれば,2039年または2042年に満了する予定である。
我々の子会社theras,Inc.は、Leidos Biomedical Research,Inc.,またはLeidos、ならびにLawrence Livermore National Security,LLC、またはlivermoreと共に6つの係属中の米国特許出願、4つの係属中のPCT出願、およびK-RAS調節器に対する8つの未解決外国出願を有し、調節剤が物質の組成であると主張すること、および癌治療を含む治療における使用を含む。これらの出願から発行されるいずれの特許も2042年から2044年の間に満期になると予想される。さらに、THERASは、レドースおよびリバモアと共に、係属中の米国特許出願、係属中のPCT出願、およびPI 3 Kαと小GTP酵素との間の相互作用を撹乱する可能性がある化合物に関する2つの係属中のPCT出願を有し、これらの化合物は、癌の治療を含む物質の組成およびその治療における使用であると主張することを含む。このような出願によって発行されたどの特許も2043年に満期になると予想される。
20
我々の実質的な合意は
アクラミディス
Alexionとのライセンス契約
2019年9月、私たちの子会社Eidos Treateutics、Inc.またはEidosを通じて、私たちはAlexion製薬会社の子会社Alexion Pharma国際運営無限会社または共同で、日本でAcoramidisを開発および商業化するためのライセンス契約またはAlexionライセンス契約に署名した。また、2019年9月、EidosはAlexionと株式購入契約を締結し、この合意によると、EidosはAlexionに556,173株の普通株を売却し、現金収益総額は2,500万ドルだった。Alexionライセンス契約の条項によると、EidosはAlexionに、私たちが日本で開発、製造、商業化したacoramidisのある知的財産権の独占許可を授与した。ライセンス付与の対価格として,Eidosは2500万ドルの前金を受け取り,規制マイルストーンの実現により,さらに3000万ドルを一度に支払う可能性がある。また,EidosはAlexion of acoramidisによる日本での純売上高の2桁の低使用料を得る権利がある。Alexionが日本でacoramidisを開発、製造または商業化するために第三者から知的財産権を取得する必要がある場合、または市場に模倣薬競争を導入する場合、特許権使用料料率が低下する可能性がある。
リラン·スタンフォード初級大学取締役会とのライセンス契約です
2016年4月、私たちはEidosを通じて、スタンフォード大学と独占的な許可協定を達成し、新しいトランスサイレチン凝集阻害剤に関する権利を獲得した。私たちの合意によると、スタンフォード大学は許可特許権でカバーされた製品を製造、使用、販売するグローバル独占許可を授与してくれました。このライセンスは最後のライセンス特許が満了した時点で満期になる。ライセンスに基づいて独占的に付与された特許権は、上記のタイトル“知的財産権-Eidos治療会社”の下でより詳細に記載されている
スタンフォード大学は、特許権の下で勤務する任意の非営利目的(協賛研究および協力を含む)のために、自分および他のすべての非営利学術研究機関を代表する権利を保持する。特許権がカバーする製品の開発や商業化に積極的に取り組んでいる限り、第三者に再許可を与えることができます。場合によっては、私たちが潜在的な市場や地域にサービスを提供したくないか、または提供できない場合、第三者がその市場または地域の許可者になることを望む場合、私たちはまた、スタンフォード大学の要求の下で合意下での私たちの権利を再許可することを要求されるかもしれない。
契約期間内にスタンフォード大学にライセンス維持費を毎年支払う義務がありますが、維持費はその年度の純売上高で稼いだ印税支払いと相殺することができます。スタンフォードは特許製品の純売上高の特許権使用料を得る権利があり、割合は低い1桁である。私たちは、私たちが許可された人から得られた非特許権使用料収入の一定の割合をスタンフォード大学に支払うことに同意し、私たちが適用されるライセンス契約を締結した時間によって、私たちに借りた金額は3年以内に毎年減少する。また、特定の知的財産権、臨床、規制のマイルストーン事件を終えた後、スタンフォード大学に最高約100万ドルを支払う義務がある。Eidosに関する制御権変更取引が発生した場合,250,000ドルの制御権変更費用をStanfordに支払うことが義務付けられており,これはEidosの購入者に許可プロトコルを譲渡することに関係している.
スタンフォード大学とのライセンス協定によると、私たちは商業的に合理的な努力で少なくとも1つのライセンス製品を開発、製造、商業化すること、このようなライセンス製品のための市場を開発すること、そして私たちがスタンフォードと達成したいくつかの開発マイルストーンを満たす義務がある。
上記ライセンス付与が満了するまでは、当該プロトコルには指定された期限はない。スタンフォード大学に事前に書面通知を出して合意を終了することができます。もし私たちがいくつかのマイルストーンに達しなかった場合、あるいは合意に基づいてお金を支払うことができなかった場合、あるいは私たちが積極的に許可製品を開発していなかった場合、または他の方法で合意に深刻に違反し、指定された猶予期間内にこのような違反を是正できなかった場合、スタンフォード大学は合意を終了する権利があります。
21
Infigratinib:ノワ国際製薬株式会社とライセンス契約
2018年1月、私たちの子会社QED治療会社を通じて、私たちはノワール国際製薬有限会社またはノワール社と許可協定を達成し、CCA、UCおよび軟骨発育不全患者を含むFGFR駆動の疾患を治療するための特許とノウハウを含むinfigratinibに関連するいくつかの知的財産権を獲得した。私たちはこの合意をノワール許可と呼ぶ。
ノワールのライセンスによれば、我々は、研究、開発、製造、製造、使用、輸入、販売、および他の方法でinfigratinibを商業化するライセンス、およびinfigratinibを含む治療製品を取得し、ライセンスの付与がない場合、これらの製品は、世界のすべての使用分野でノワールのライセンス特許権を侵害するか、またはノワールのライセンス技術を使用または包含または具現化する。私たちに付与された許可には、複数のレベルで再許可される権利が含まれる。第三者と締結された材料譲渡協定によると、ノワール付属会社に知的財産権を付与するいくつかの権利も有している。
ノワールの許可は、第三者臨床試験の既存の義務を支援するためにノワールが第三者にinfigratinibを提供するという制約を受けており、私たちまたは私たちの分被許可者は、規制部門の様々な適応に対するinfigratinibの承認を求めて商業化することを意図しており、その後、規制部門の承認と商業化を求めないと決定した場合、あるいはノワール認定は規制部門の承認と商業化を十分に求めることができず、ノバはこのような第三者に限られたinfigratinibの開発と商業化の権利を付与する権利がある。
ノバ許可の条項によると、私たちはノワールに1,500万ドルを一度に支払い、ノバにQEDのAシリーズ優先株を発行することに同意し、総価値は約170万ドルだった。しかも、私たちは特定の規制マイルストーンを達成する時に合計6000万ドルを支払うことやマイルストーンの支払いを義務している。私たちはまたinfigratinibを含む治療製品がいくつかの販売マイルストーンを達成した時に合計3500万ドルまたはマイルストーンの支払いを支払う義務があります。QEDはinfigratinibを含む治療製品の純販売のためにノワールに2桁の階層的低特許権使用料を支払うことにも同意した。FDAが2021年5月にTRUSELTIQを承認した後、私たちはノワールに2000万ドルの一次規制マイルストーン支払いを支払った。
ノワール社の許可によると、私たちは商業的に合理的な努力を使用してinfigratinibを開発し、アメリカとEUで監督部門のinfigratinibの承認を得て商業化しなければならない。
60日前にノバ社に書面で通知した場合、ノーファ社のすべてのライセンスを随時終了したり、製品または国/地域によってライセンスを終了したりすることができます。QEDが継続的に経営している企業として動作しなくなった場合、いくつかの破産または同様のプログラムの対象であるか、または他の方法で業務を終了または停止する場合、ノワールは終了する可能性がある。いずれか一方が重大違約通知を受けた後の所定期間内に,他方がまだ是正されていない場合は,当該重大な違約を解除することができる。そうでなければ、ノワール社のライセンスは製品および国/地域によって終了し、終了期間は遅くともライセンス特許権の満了、法規排他的満期、またはその国での初の商業販売終了10周年となる。
Fosdenopterin:Alexion Pharma Holding無限会社と資産購入合意
2018年6月、我々は、我々の子会社Origin Biosciences,Inc.を通じて、Alexion Pharma Holding UnLimited CompanyまたはAlexion Pharmaと資産購入合意に達し、この合意に基づいて、Fosdenopterinに関連する特定の資産に対するAlexionの権利、所有権、および権益を特許および他の知的財産権を含むAlexionを取得した。
22
FDAが私たちに優先審査券を付与した場合、私たちはPRVを第三者に売却した任意の収益の約15%の割合をAlexion Pharmaに支払うことに同意した。もし私たちがPRVを受け取ってから180日以内にPRVを第三者に販売しなかった場合、私たちはAlexion Pharmaに1880万ドルを支払う義務があり、この金額は私たちが将来PRVを販売する時、前の文に従ってAlexion Pharmaに支払われるべき任意の金額から差し引かれるだろう。ある開発マイルストーンを実現する際に合計300万ドルまたはマイルストーン支払いを義務付け,ある販売マイルストーンを実現した場合にはリントレキサート分子を含む製品に1,700万ドルを支払うことが義務付けられている。リントレキサート分子を含む製品の純売上高の分級特許権使用料をAlexion Pharmaに支払うことにも同意し,低さから中程度まで様々であった。
2021年、FDAがNULIBRYを承認した後、Alexionに200万ドルの規制マイルストーンを支払いました。これは販売された100万ドルのマイルストーンに基づいて支払い、PRVをEidosに売却した1500万ドルです。
我々は,ビジネス上の合理的な努力を用いてPRVを獲得し,特定のマイルストーン事件を実現し,規制部門の承認を得た後に少なくとも1種のホスホトレキサート分子を含む製品を商業化する義務がある。
BBP-398:百時米施貴宝社とのライセンス開発と商業化協定
2022年5月12日、BridgeBioと我々の子会社NAVRE Pharma,Inc.,またはNAVREは、百時米施貴宝社(BMS)と独占的な開発と商業化協定を締結し、この合意に基づいて、NAVREはBMSに独占的権利を付与し、NAVREの候補製品BBP-398、人民Republic of China、マカオ、香港、台湾、タイ、シンガポール、韓国、または総称してアジア地域と呼ばれる。このプロトコルをNAVRE-BMSライセンスプロトコルと呼ぶ.
NAVERE-BMS許可協定の条項によると、NAVEREは2022年6月に全額徴収された9000万ドルの払戻不可能な前金を受け取る権利がある。また,NAVAREは合計約8.15億ドルの追加支払いを受ける資格があり,これは開発,規制,ビジネスマイルストーンの実現,およびBMSがアジア地域以外の世界で販売されている特許製品調整後の純売上高のパーセンテージで計算されるミドルエンド階層特許使用料に依存する。NAVIREは、登録研究開始時の開発コストの一部を支援するために、米国でより高い印税を得る選択権を保持する。NAVRE−BMS許可協定の条項によると、NAVREは第1段階の単一療法と併用療法試験をリードし続け、BMSは他のすべての開発と商業化活動をリードし、援助する。
政府の監督管理
アメリカ連邦、州と地方の各レベル及びその他の国の政府当局は他の以外に、薬物と生物製品の研究、開発、製造、テスト、品質管理、承認、ラベル、包装、保存、記録保存、販売促進、広告、流通、承認後のモニタリングと報告、マーケティングと輸出入、遺伝子療法及び診断、及び任意の未来の候補製品を含む監督管理を行う。通常、新薬、生物または診断薬が発売される前に、その品質、安全性と有効性を証明するデータを大量に取得し、各監督管理機関の特定のフォーマットに組織し、審査を提出し、適用される監督管理機関によって承認、許可または承認されなければならない。
23
アメリカ政府の薬品と生物製品の規制
アメリカでは、FDAは連邦食品、薬物と化粧品法案(FDCA)及びその実施条例に基づいて薬品を監督し、FDCAと公衆衛生サービス法(PHSA)及びその実施条例に基づいて生物製品を監督する。薬品や生物製品も他の連邦、州と地方法規の制約、例えば競争に関連する法規によって制限されている。規制の承認を得て、その後適切な連邦、州、地方法規と条例を遵守する過程には多くの時間と財力が必要だ。製品開発過程,承認過程または承認後のいずれかの場合,出願人が適用された米国の要求を遵守できなかった場合,行政行為や司法制裁を受ける可能性がある。これらの行動および制裁には、FDAによる承認保留申請の拒否、承認撤回、免許取り消し、臨床封印、無タイトルまたは警告状、自発的または強制的な製品のリコールまたは市場撤回、製品差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、政府契約の拒否、原状回復、返還、返還、および民事または刑事または罰金または処罰が含まれる可能性がある。どの機関または司法法執行行動も、私たちの業務、私たちの候補製品の市場受容度(承認されれば)、および私たちの名声に実質的な悪影響を及ぼす可能性がある。
私たちの候補製品は新薬申請、NDA或いは生物製品許可証申請或いはBLAプログラムを通じてFDAの承認を得なければならず、その後アメリカで合法的に発売することができる。このプロセスは、一般に以下のことを含む
臨床前と臨床試験と承認過程は大量の時間、精力と財政資源を必要とし、薬物と生物製品の監督管理方案は変化しており、そしていつでも変化が発生する可能性がある。私たちは私たちの候補製品がタイムリーに承認されるかどうか、あるいは承認されないかどうかを確認することができない。
臨床前研究
24
人体で任意の薬物、生物或いは遺伝子治療候補薬物をテストする前に、候補製品は厳格な臨床前テストを経なければならない。臨床前研究には製品化学と処方の実験室評価があります体外培養安全性を評価し、場合によっては治療使用の理由を確立するための動物研究を提供する。臨床前研究の進行はGLPの安全/毒理学研究に関する法規を含む連邦と州の法規と要求を遵守しなければならない。
INDスポンサーは,臨床前試験の結果を生産情報,分析データ,任意の利用可能な臨床データや文献,臨床試験計画などとともにFDAに提出し,INDの一部としなければならない。INDはFDAがヒトに研究製品の使用を許可する要求であり,ヒト臨床試験が開始される前に発効しなければならない。いくつかの長期的な臨床前試験、例えば生殖不良反応と発ガン性の動物試験は、IND提出後も継続する可能性がある。INDはFDAが受領してから30日後に自動的に発効し、それ以前に、FDAは1つまたは複数の提案された臨床試験に対して懸念または問題を提起し、試験を臨床保留状態に置かなければならない。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。したがって,INDの提出はFDAが臨床試験の開始を許可しない可能性がある。さらに、INDによって提出された情報の審査は、FDAに既存のINDまたは任意の上場製品を審査させる可能性があり、他の候補製品または計画に関する情報または臨床的に封印された要求を生成する可能性がある。
臨床試験
臨床開発段階は、合格した研究者の監督の下で、GCP要求に基づいて健康ボランティアまたは患者に研究製品を提供することであり、通常は試験スポンサーに雇用されない、または試験スポンサーの制御下にある医師であり、すべての研究対象に任意の臨床試験への参加についてインフォームドコンセントを提供することを含む。臨床試験は,臨床試験の目標,用量プログラム,被験者の選択と排除基準,および被験者の安全性をモニタリングし,治療効果を評価するためのパラメータを詳細に説明した場合に行われる。INDの一部として、すべての議定書とその後の議定書のいかなる修正もFDAに提出されなければならない。また,各臨床試験は,臨床試験を行う各機関の内部審査委員会によって審査·承認されなければならず,臨床試験に参加する個人が直面するリスクが最小限に減少することを保証し,期待される利益については合理的である。IRBはまた、各臨床試験対象またはその法律代表に提供されなければならないインフォームドコンセントを承認し、完成まで臨床試験を監視しなければならない。行っている臨床試験や完成した臨床試験結果を公的登録機関に報告することも求められている。臨床試験結果を含むいくつかの臨床試験に関する情報は、www.Clinicaltrials.govサイト上で発表するために、特定の時間枠内で提出されなければならない。
米国で臨床試験が開始される前にINDがFDAに提出されるほか、組換えまたは核酸分子の合成に関連するいくつかのヒト臨床試験は、機関生物安全委員会(IBC)の監督を受けなければならないことは、NIHガイドライン“組換えまたは核酸分子の合成に関連する研究ガイドライン”に記載されている。米国国立衛生研究院のガイドラインによれば、組換えおよび合成核酸は、(1)核酸分子が結合して生細胞中で複製可能な分子(すなわち、組換え核酸)と、(2)化学的または他の修飾されているが自然に産生される核酸分子(すなわち、合成核酸)塩基対と結合することができる分子、または(3)(1)または(2)項に記載の分子を複製する分子を含む化学的または他の方法で合成または増幅された核酸分子と定義されている。具体的には,NIHのガイドラインによると,ヒト遺伝子転移試験の監督にはIBCによる評価と評価があり,IBCは地方機関委員会であり,組換えや合成核酸分子を用いた研究の審査·監督を担当している。IBCは、研究の安全性を評価し、公衆の健康または環境に対する任意の潜在的リスクを決定し、このような審査は、臨床試験開始前のいくつかの遅延をもたらす可能性がある。NIHガイドラインは強制的ではないにもかかわらず、関連研究がNIH組換えまたは合成核酸分子研究助成を受けた機関で行われているか、またはその助成によって行われていない限り、多くの会社および他のNIHガイドラインに拘束されていない機関は、自発的にこれらのガイドラインに従っている。
25
米国国外で臨床試験を行うスポンサーはFDAの認可を得ることができるが,INDによる臨床試験を希望している。海外の臨床試験がINDに基づいて行われていなければ,スポンサーはNDAやBLAを支援するために臨床試験のデータをFDAに提出することができる。研究がGCP要求に基づいて行われれば,FDAはINDではない設計良好で良好な外国臨床研究を受け,必要であればFDAは現場検査でデータを検証することができる。
臨床試験は通常3つの連続段階で行われ,第1段階,第2段階と第3段階と呼ばれ,重なる可能性がある。
承認後試験は,4期臨床試験と呼ばれることがあり,最初の上場承認後に行われる可能性がある。これらの試験は、通常、臨床環境において製品を使用するための追加の安全データを生成するために、予期される治療適応下で患者の治療から追加の経験を得るために使用される。場合によっては,FDAはNDAまたはBLAを承認する条件として4期臨床試験を強制的に実行することができる。
その他の情報に加えて、臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出されなければならず、書面IND安全報告は、試験スポンサーにおいて、これらの情報に深刻かつ意外な疑わしいAEs、他の研究または動物からの発見、またはこれらの情報を報告する資格があることを確認しなければならない体外培養方案或いは研究者マニュアルに記載されたテストと比べ、人類被験者に重大なリスクがあるテスト、及び任意の臨床上重要な深刻な疑似副作用の発生率の増加を表明した。スポンサーはまた、FDAに任意の意外、致命的、あるいは生命に危害を及ぼす疑いのある副作用をできるだけ早く通知しなければならないが、いずれの場合も、スポンサーが初めて情報を受け取った後の7つの日数に遅れてはならない。
第1段階、第2段階、第3段階、および他のタイプの臨床試験は、あれば、任意の指定された期間内に成功しない可能性がある。FDA或いはスポンサーはいつでも様々な理由で臨床試験を一時停止或いは中止することができ、研究対象或いは患者が受け入れられない健康リスクに直面していることを発見することを含む。同様に、臨床試験がIRBの要求に従って行われない場合、または薬物または生物が患者に予期せぬ深刻な傷害を受けることに関連している場合、IRBは、その機関の臨床試験の承認を一時停止または終了することができる。さらに、いくつかの臨床試験は、データ安全監視委員会または委員会と呼ばれる臨床試験スポンサーによって組織された独立した合格専門家グループによって監督される。このグループは,実験のあるデータへのアクセスにより,許可試験が指定されたチェックポイントで行えるかどうかを決定する.臨床試験と同時に,会社は通常追加の動物研究を完成させ,薬物や生物の化学的および物理的特性に関する追加情報を開発し,cGMP要求に基づいて商業量産製品のプロセスを最終的に決定しなければならない。製造過程は一貫して高品質の製品ロットを生産できる必要があり、他の以外にも、会社は最終製品の特性、強度、品質、純度をテストする方法を開発しなければならない。また,適切な包装を選択·テストし,候補製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
26
FDA審査プログラム
臨床試験が完了した後、研究製品が提案された1つまたは複数の指示用途に対して安全に有効であるかどうかを評価するために、データを分析する。臨床前研究および臨床試験の結果は、その後、NDAまたはBLAの一部としてFDAに提出され、製品品質および他の関連データを確保するために、提案されたラベル、化学および製造情報が提示される。NDAまたはBLAは、1つまたは複数の適応を指定するための薬物または生物学的薬物の承認要求であり、薬物の安全性および有効性または生物学的薬物の安全性、純度および有効性の証拠を含まなければならない。応用は臨床前研究と臨床試験の陰性とファジィ結果、及び陽性結果を含む可能性がある。データは、製品使用の安全性および有効性を試験するために、または研究者によって開始された研究を含む多くの代替源からの臨床試験からのものである可能性がある。上場承認を支持するためには、提出されたデータは研究製品の安全性と有効性を確定し、FDAを満足させるために、品質と数量で十分でなければならない。薬物や生物製剤が米国で発売される前に,NDAやBLAに対するFDAの承認を得なければならない。
改正された処方薬使用料法案(PDUFA)によると,NDAまたはBLAごとに使用料が付与されなければならない。FDAは毎年PDUFAユーザ料金を調整する。場合によっては、小企業が初めて出願した出願料を免除することを含む、費用を免除または減免することができる。さらに,孤児薬として指定された製品については,この製品が孤児適応も含まれていない限り,NDAまたはBLAに対して使用料を評価しない。
FDAは、提出されたすべてのNDAおよびBLAを受け入れる前にそれらを検討し、NDAまたはBLAの提出を受け入れるのではなく、追加の情報の提供を要求する可能性がある。FDAはNDAまたはBLAを受信してから60日以内に決定し,NDAまたはBLAの届出を受けなければならず,このような決定にはFDAが届出を拒否することが含まれる可能性がある。提出された申請が受け入れられると、FDAはNDAまたはBLAの深い審査を開始する。FDAがPDUFAで合意した目標および政策によれば、FDAの目標は、提出日から10ヶ月以内に新しい分子実体NDAまたは元のBLAの予備審査を完了し、出願人に応答し、優先審査のために指定された新しい分子エンティティNDAまたは元のBLAの提出日から6ヶ月間である。FDAは、そのPDUFA規格および優先NDAまたはBLAの目標日を常に満たすわけではなく、審査プロセスは、FDAがより多くの情報を提供または明確にすることを要求することによって延長されることが多い。
FDAはすでに腫瘍学卓越センターリアルタイム腫瘍学審査(RTOR)試験計画を開発し、ある腫瘍学候補製品に対するより有効な審査過程を促進する。この計画は、FDAが完全なNDAまたはBLAを提出する前に臨床データの審査を開始することを可能にするが、この計画はPDUFA審査スケジュールを変更するつもりはない。
27
NDAまたはBLAを承認する前に、FDAは、それらがcGMP要件に適合しているかどうかを決定するために、新製品の製造施設を承認前に検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分でない限り、この製品を承認しないであろう。FDAはまた、GCP要求に適合することを確保するために、臨床試験のデータを監査することが可能である。さらに、FDAは、新製品の申請または安全性または有効性の問題を提起する製品の申請を諮問委員会に提出することができ、一般に、申請が承認されるべきかどうか、およびどのような条件下であるか(ある場合)を審査、評価および提案するための臨床医および他の専門家を含むグループである。FDAは諮問委員会の提案に制限されていないが、承認決定を下す際にこれらの提案を考慮する。FDAは臨床試験データを再分析する可能性があり,FDAや出願人の審査過程で広く議論される可能性がある。FDAがNDAまたはBLAを評価した後、それは承認書または完全な返信を発行する。医薬または生物学的薬物の商業マーケティングを許可し、特定の適応の特定の処方情報を提供する。完全な返信は、申請の審査期間が終了し、現在の申請が承認されないことを示している。完全な応答文は、一般に、FDAによって決定されたNDAまたはBLA内のすべての特定の欠陥を記述する。完全な返事は、申請者が追加的な臨床データを得ることを要求するかもしれない, 追加の重要な第3段階臨床試験を行う必要がある可能性があり、および/または臨床試験に関連する他の重要で時間のかかる要件を達成するか、または追加の臨床前研究または生産活動を行う必要がある可能性がある。完全な返信が発行された場合、出願人は、機密協定またはBLAを再提出し、手紙で発見されたすべての不足点を解決するか、または申請を撤回するか、または聴聞機会を要求することができる。このようなデータや情報を提出しても,FDAはNDAやBLAが承認基準を満たしていないと決定する可能性がある.臨床試験から得られたデータはつねに決定的ではなく,FDAのデータ解釈は我々の同じデータに対する解釈とは異なる可能性がある。
孤児薬の指定と排他性
孤児医薬品法によれば、FDAは、米国では20万人未満、または米国では20万人以上に影響を与える疾患または疾患であり、米国でそのような疾患または疾患を治療する製品を開発および提供するコストは、その製品の販売から回収されるという、まれな疾患または疾患の治療のための医薬または生物製品に孤児薬物の称号を付与することができる。
NDAやBLAを提出する前に,指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の識別およびその潜在的な孤児の使用を開示する。指定孤児薬は、規制審査と承認過程でいかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない。
孤児薬物指定を有する製品がその後、そのような指定された疾患または状況に対するFDAの最初の承認を得た場合、この製品は、孤児薬物排他性を得る権利があり、これは、FDAが承認日から7年以内に、限定された場合を除いて、例えば、より効果的、より安全、または患者ケアまたは医薬品供給問題に大きな貢献を提供することによって、孤児に対して排他的な製品に対する臨床的利点を示す他の他の同じ適応のための同じ薬物の販売の申請を許可しないことを意味する。しかしながら,競争相手は同一適応の異なる製品や異なる適応の同じ製品の承認を得る可能性があるが,これはラベル外で孤立した適応に用いられる可能性がある。もし競争相手が私たちの前にFDAで定義された同じ製品の承認を得た場合、すなわち私たちが承認を求めている同じ適応、または私たちの製品が競争相手の製品の範囲に含まれていると決定された場合、孤立薬物排他性はまた7年以内に私たちの製品の承認を阻止する可能性がある。市販承認の適応が私たちが獲得した孤児薬物指定よりも広い適応を求めれば,孤児薬物の排他性を得る資格がないかもしれない。EUの孤児薬の地位は似たような要求と利益を持っているが、同じではない。
28
まれな小児科疾患指定と優先審査証明書
改訂されたFDCAによると、FDAは“稀な小児科疾患”の定義に符合する薬物と生物製品の開発を奨励し、“稀な小児科疾患”の定義は1種の深刻な或いは生命に危害を及ぼす疾患であり、このような疾患の中で、深刻な生命危害の表現は主に出生から18歳までの個人に影響を与え、このような疾患はアメリカで20万人を超え、アメリカでは20万人を超え、しかも合理的な期待はなく、アメリカでこのような疾病或いは疾病を治療する薬物の開発と製造のコストはアメリカでこのような薬物を販売することから来る。稀な小児科疾患候補製品のスポンサーはクーポン券を獲得する資格がある可能性があり、このクーポン券は稀な小児科疾患薬物製品が承認された日後に後続のヒト薬物或いは生物応用を獲得する優先審査に使用することができ、優先審査証明書或いはPRVと呼ばれる。スポンサーは、NDAまたはBLAを提出する前に、FDAにまれな小児科疾患の指定を要求することができる。まれな小児科疾患指定は、スポンサーがそのNDAまたはBLA承認後にPRVを受信することを保証しない。さらに、まれな小児科疾患指定要求を提出しないスポンサーを選択し、最初のマーケティング申請においてそのような証明書を要求し、すべての資格基準を満たしている場合、そのマーケティング申請が承認された後もPRVを受信することができる。PRVを受信した場合、それは何度も販売または譲渡される可能性がある。国会はPRV計画を2024年9月30日まで延長し、2026年9月30日までPRVを承認する可能性がある。
開発と審査計画を加速する
スポンサーは、開発加速、FDA審査、特定基準に適合した新薬や生物製品の承認を目的とした計画に基づいて、その候補製品の開発と承認を求めることができる。例えば、FDAは新薬と生物製品の審査過程を加速或いは促進することを目的とした迅速なチャンネル計画を持っており、これらの新薬と生物製品は深刻な或いは生命に危害を及ぼす疾患或いは疾病を治療することを目的とし、そしてこのような疾病が満たされていない医療需要を解決する潜在力を示す。高速チャネル指定は製品にも,検討中の特定の適応にも適用可能である。高速チャネル指定製品の場合、FDAは、完全な出願を提出する前に、NDAまたはBLAを考慮した部分をスクロールして審査することができ、スポンサーが申請部分の提出スケジュールを提供した場合、FDAは、申請の部分を受け入れることに同意し、スケジュールが許容可能であると判断し、スポンサーは、申請の第1の部分を提出する際に任意の必要な使用料を支払うことができる。スポンサーは、NDAまたはBLAの承認を得る前の任意の時間に、FDAに製品を高速チャネル状態として指定することを要求することができるが、NDA前またはBLA前の会議よりも遅くないことが好ましい。
迅速なチャネル計画の下で、優先審査および承認の加速など、FDAの開発または審査を加速させるための他のタイプの計画に参加する資格がある可能性があるFDA上場製品を提出する。優先審査は、新しい分子実体または元のBLAについて、FDAが上場申請に対して行動する目標日を、10ヶ月ではなく、申請を受けてから6ヶ月とすることを意味する。1つの製品が重篤または生命に危険な疾患を治療するために設計されており、承認された場合、既存の治療法と比較して、その安全性および有効性が著しく向上する場合、優先審査を受ける資格がある。FDAは、審査を促進するために、優先審査として指定された新薬または生物学的出願を評価するために追加のリソースを使用しようと試みるであろう。優先審査の基準を満たしていない場合、新しい分子実体または元のBLAの出願は、FDAが申請を受けてから10ヶ月の標準FDA審査期間によって制限される。優先審査の指定は、承認された科学/医学基準または承認を支援するために必要な証拠の質を変更しません。
29
“2022年食品·薬物総合改革法案”(FDORA)によれば、(1)プラットフォーム技術がNDAまたはBLAによって承認された薬物使用に組み込まれるか、または許可された薬物の発起人またはそのような薬物出願に提出されたデータ参照権が付与された発起人によって提出された初期証拠は、品質、製造または安全に悪影響を及ぼすことなく、許可または許可された薬物の発起人またはそのような薬物出願に提出されたデータ参照権が付与された発起人によって提出される可能性があることを示す。(3)適用者が提出したデータまたは情報は、プラットフォーム技術の導入または利用が薬物開発または製造過程および審査過程に顕著な効率をもたらす可能性があることを示している。スポンサーは、IND出願を提出した間または後の任意の時間に、IND出願を要求対象とするプラットフォーム技術を組み込むか、または使用する指定されたプラットフォーム技術としてFDAに指定することを要求することができる。指定された場合、FDAは、プラットフォーム技術を使用するか、または使用する薬物の任意の後続の元のNDAまたはBLAの開発および検討を加速することができる。プラットフォーム技術状態を指定することは、薬物開発がより速く、またはより速いFDA審査プロセスまたは最終FDA承認を得ることを保証するものではない。さらに、FDAが、指定されたプラットフォーム技術がもはやそのような指定された基準に適合していないと判断した場合、FDAは、指定を取り消すことができる。
1つの製品が深刻または生命に危険を及ぼす疾患または状態を治療するために設計され、一般に既存の療法よりも有意な利点を提供する場合、製品は、製品が代替または中間臨床終点に有効であることを決定する資格がある可能性もあり、代替臨床終点または中間臨床終点は、臨床的利益を合理的に予測することができるか、または不可逆的な発症率または死亡率またはIMMよりも早く測定可能な臨床終点で有効であり、IMMまたは他の臨床的利益の影響を合理的に予測することができ、同時に、疾患または病状の深刻性、希少性または流行率、ならびに代替治療を使用することができるか、または代替治療を欠くことができることを考慮しながら、適切である。承認の条件として、FDAは、承認を加速させる薬物または生物のスポンサーが、職務調査の場合に十分かつ良好に制御された上場後の検証臨床試験を行うことを要求することができ、FDAは、FDORAに従って、適切な場合に、そのような検証的研究を承認前または承認加速後の特定の期間内に行うことを要求することができる。さらに、FDAは、機関が別途通知しない限り、販売促進材料を事前に承認しなければならず、製品の商業発売時間に悪影響を及ぼす可能性があることを要求している。FDORAによれば、FDAは、例えば、検証試験が製品の予期される臨床的利益を検証することができない場合、承認の加速下で承認された薬物または適応の承認を撤回することができるように、手続きを加速させる権限を増加させる。
さらに、1つの医薬または生物学的製剤が、1つまたは複数の他の薬剤または生物学的製剤と共に、重篤または生命に危険な疾患の治療のために単独でまたは1つまたは複数の他の医薬または生物学的製剤と共に使用されることが意図されている場合、その製品が、1つまたは複数の臨床的重要終点において、現在承認されている療法よりも実質的に改善されている可能性があり、例えば、臨床開発早期に観察された実質的な治療効果を示す可能性がある場合、医薬または生物学的製剤は、画期的な療法として指定される資格がある可能性があることを示す。FDAが画期的な治療法を指定した場合、申請の開発と審査を加速するために適切な行動をとることができ、これは治療開発全体の過程でスポンサーと審査グループとの会議を行うことを含む可能性がある;薬物の開発についてスポンサーに適時なアドバイスを提供し、それと相互作用して、承認に必要な非臨床および臨床データを収集する開発計画が可能な限り有効であることを保証する;高度管理者と経験豊富な審査者を適宜協力、学際的な審査に参加させることができる。FDA審査チームに1人の学際プロジェクト担当者を割り当てて、開発計画の有効な審査を促進し、審査チームとスポンサーとの間の科学的連絡人として機能する;そして科学的に適切な場合に臨床試験設計の代替を考慮することは、より小さい試験或いはより有効な試験を招く可能性があり、これらの試験はより少ない時間を必要とし、潜在的に低い治療効果の治療に直面する患者の数を最大限に減少させる可能性がある。画期的な治療指定は、高速チャネル指定のすべての利点を伴い、これは、いくつかの条件が満たされれば、スポンサーがBLAに提出された部分をスクロールして審査することができることを意味する, FDAと一部の申請を提出する提案されたスケジュールについて合意することと、FDAが審査を開始する可能性がある前に適用される使用料を支払うことを含む。
30
21世紀治療法案の一部として、国会は、再生医学高度療法(RMATs)の有効な開発計画を促進し、細胞および遺伝子療法、治療用組織工学製品、ヒト細胞および組織製品、および任意のこのような治療法または製品を使用した組み合わせ製品を含むRMATsの審査を加速するためにFDCAを改訂した。RMATsは、PHSA第361条および21 CFR Part 1271によってのみ規制されるヒト細胞、組織、および細胞および組織製品を含まない。この計画は再生医学療法の有効な開発と審査の加速を促進することを目的とし、これらの治療法は深刻な或いは生命に危害を及ぼす疾病或いは状況を治療、修正、逆転或いは治愈し、そしてRMAT資格を獲得する資格がある。スポンサーは、INDの提出と同時にまたはその後の任意の時間に、FDAにRMATとして候補製品を指定するように要求することができる。FDAは60個のカレンダー日があり、候補製品が標準に符合するかどうかを確定し、初歩的な臨床証拠があるかどうかを含み、候補製品が深刻或いは生命に危害を及ぼす疾病或いは状況の未満足医療需要を満たす可能性があることを表明した。RMAT指定された候補製品を取得したBLAは、長期的な臨床的利益を合理的に予測する可能性のある代替物または中間エンドポイントを使用することによって、または大量のサイトから取得されたデータに依存して、優先審査または加速承認を得る資格がある可能性がある。RMATを指定する利点はまた、承認の加速をサポートするための任意の潜在的代替品または中間終点を検討するために、FDAとの早期相互作用を含む。加速承認を得て承認された後に要求されるRMAT認証を有する候補製品は、臨床研究の臨床証拠を提出することによって、これらの要求を満たすことができる, 患者登録または他の実世界証拠源、例えば電子健康記録、より大きな検証データセットを収集すること、または承認前にそのような治療を受けたすべての患者を承認した後に監視すること。
FDAはまた、既存の療法よりも実質的に改善される可能性のある腫瘍学的候補製品のためのRTORパイロット計画を提供することを発表し、これには、以前に同じまたは他の適応に画期的な治療指定を付与した薬剤、および迅速チャネルおよび優先審査のような他の加速計画の他の基準に適合する候補薬剤が含まれている可能性がある。RTORに提出された考慮文書にも、単刀直入な研究設計および説明しやすい終点(例えば、全体的な生存または無進行生存)があるべきである。RTOR計画の受け入れは、FDA審査員によって一般的に行われる利益-リスク評価に依存するが、この計画は、FDAが出願人が完全な出願を正式に提出する前にデータをより早く審査することを可能にする、出願の承認性を保証または影響するものではない。RTOR計画はFDAのPDUFAスケジュールに影響を与えない.
1つの製品が1つまたは複数のこれらの計画の条件に適合していても、FDAは、製品がもはや資格条件を満たしていないと後で決定することができ、またはFDAが審査または承認する期間が短縮されない可能性がある。さらに、迅速チャネル指定、優先審査、加速承認、突破的治療、およびRMAT指定は、承認の基準を変更しない。
小児科情報と小児科排他性
“小児科研究公平法”によれば、いくつかのNDAおよびBLAならびにいくつかのNDAまたはBLAのサプリメントは、すべての関連する小児科亜集団において主張される適応の安全性および有効性を評価し、安全で有効な各小児科亜群に対する製品の用量および投与をサポートするためのデータを含まなければならない。FDAは小児科データの提出を延期することを許可するか、またはすべてまたは部分的な免除を与える可能性がある。食品·薬物管理局安全·革新法案(FDASIA)はFDCAを改正し、新しい活性成分、新適応、新剤形、新投与案または新投与経路を含む薬物マーケティング申請のスポンサーに第2段階会議終了後60日以内に予備小児科研究計画を提出することを要求し、そのような会議がなければ、第3段階または第2/3段階研究が開始される前に可能な限り早く提出する。最初のPSPは、研究目標および設計、年齢群、関連する終点および統計方法、またはそのような詳細な情報を含まない理由、ならびに小児科研究データおよび支援情報の提供を延期または完全または部分的に免除することを要求する任意の要件を含む、スポンサー計画によって行われる1つまたは複数の小児科研究の概要を含まなければならない。FDAとスポンサーはPSPについて合意しなければならない。臨床前研究,早期臨床試験および/または他の臨床開発計画から収集したデータに基づいて小児科計画の変化を考慮する必要があれば,スポンサーは合意した初期PSPに対する修正案を随時提出することができる。
31
1つの薬物または生物製品はまた、米国で小児科市場の排他性を得ることができる。小児科専有権が付与された場合、既存の専有期間と特許条項を6ヶ月増加させる。この6カ月間の排他性は,他の排他的保護や特許期間終了時から,FDAが発表したこのような研究の“書面請求”によって小児科研究を自発的に完成させることができる。
発売後要求
新製品が承認された後、製造業者および承認された製品は、監視および記録保存活動、適用された規制機関に不良経験を報告すること、販売促進および広告要件を遵守すること、消費者向けの広告基準、インターネット上の販売促進活動の要求、承認されていない用途への製品の使用を制限すること、または製品承認されたラベルに記載されていない患者群(“ラベル外使用”と呼ばれる)を制限すること、および業界支援の科学的および教育活動を制限することを含むFDAによって規制され続けるであろう。医師は合法的に利用可能な処方を出すかもしれませんがラベル外で使用される製品については、製造業者は、このようなラベル外用途を販売または普及させてはならない。FDAは医師が治療を選択する行為を規範化していないが,FDAの規定は確かにメーカーの非ラベル使用に関するコミュニケーションに厳しい制限を加えている。FDAや他の機関はラベル外用途の普及を禁止する法律や法規を積極的に実行しており、ラベル外用途を不当に普及させていないことが発見された会社は、連邦や州当局の調査を含む重大な責任に直面している可能性がある。処方薬宣伝材料は、最初の使用または最初の発表時にFDAに提出されなければならず、加速された承認を得た製品のような事前審査が必要となる場合がある。
さらに、適応、ラベルまたは製造プロセスまたは施設の変化を含む医薬または生物学的に何らかの修正がある場合、出願人は、新しいNDA/BLAまたはNDA/BLAサプリメントの承認を得るために提出および提出を要求される可能性があり、これは、追加のデータまたは臨床前研究および臨床試験を開発する必要があるかもしれない。このような規制審査は、計画の変更を拒否または修正すること、または追加のテストまたは評価を要求することをもたらす可能性があり、これは、計画変更のコストを大幅に延期または増加させる可能性がある。
FDAはまた、製品の安全な使用を保証するためにREMSを要求することを含む承認時に他の条件を追加することができる。FDAがREMSが必要であると結論した場合,NDAまたはBLAのスポンサーは提案したREMSを提出しなければならない。必要に応じて、FDAは、承認されていないREMSなしにNDAまたはBLAを承認しないであろう。REMSは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全な使用を確保する要素を含むことができる。承認またはマーケティングに関するこれらの制限は、製品の商業普及、流通、処方、または配布を制限する可能性がある。初期マーケティング後に問題が発生した場合、またはFDAが製品がもはや安全または有効でないと判断した場合、製品承認は規制要件を満たしていないために撤回される可能性がある。
FDAの規定は,製品は特定の承認された施設で生産され,cGMP規定に適合しなければならないことを要求している。私たちは依存し、cGMP法規に従って任意の承認された製品を生産する第三者の臨床的および商業的数量に依存し続けることが予想される。契約製造業者、実験室または包装業者を使用するNDAおよびBLA所有者は、合格した会社を選択および監視する責任があり、場合によっては、これらの会社の合格サプライヤーも担当する。これらのメーカーはcGMP法規、これらの法規要求、他の事項を除いて、品質管理と品質保証、記録とファイルの維持、cGMPからの逸脱を調査し、是正する義務を守らなければならない。承認された薬品または生物製品の生産および流通に参加する製造業者および他のエンティティは、FDAおよびある州機関にその機関を登録し、cGMP要求および他の法律を遵守する状況を理解するために、FDAおよび特定の州機関の定期的な抜き打ち検査を受けなければならない。そのため、メーカーはcGMPコンプライアンスを維持するために、生産や品質管理の分野で時間、お金、労力をかけ続けなければならない。CGMP規定に適合しないことを含む違反が発見され、法執行行動を引き起こす可能性があり、承認後に製品が発見された問題は、製品のリコールまたは市場からの撤回を含む製品、製造業者または承認されたNDAまたはBLAの所有者の制限をもたらす可能性がある。
32
処方薬、生物製品、薬品サンプルのどの流通も米国処方薬販売法(PDMA)とPHSAを遵守しなければならない。また、2013年には、米国で流通されているいくつかの処方薬や生物製品を識別して追跡するための電子システムを構築することを目的とした“医薬品サプライチェーン安全法案”が公布された。DSCSAは薬品メーカー,卸,流通業者に10年間の段階的と資源集約型の義務を要求し,2023年11月に終了する予定である。この法律の要求には、疑わしい製品に対して検疫と迅速な調査を行い、不正製品であるかどうかを決定すること、貿易相手およびFDAに任意の不正製品を通報すること、および製品追跡および追跡要求を遵守することが含まれる。
承認されると、規制要件および基準の遵守が維持されていない場合、または薬物または生物が市場に進出した後に問題が発生した場合、FDAは強制実行書を発行したり、製品の承認を撤回したりすることができる。是正行動は薬物や生物の分配を遅らせる可能性があり、多くの時間と財政支出を必要とするかもしれない。予期されなかった重症度または頻度の副作用、または製造プロセス、または規制要件を遵守できなかった場合、承認されたラベルを修正して、新しい警告および禁忌症を含む新たなセキュリティ情報を追加することを含む、薬物または生物学的態様の以前に未知の問題が後に発見された場合、新しい安全リスクを評価するために発売後研究または臨床試験を実施すること、またはREMS計画に従って流通または他の制限を実施することが必要となるかもしれない。他の他の潜在的な結果には
仲間診断学の規制
私たちのある候補製品の成功はセット診断ソフトウェアの開発と商業化にある程度依存する可能性があると考えられる。診断に伴い、特定の治療製品から利益を得る可能性が最も高い患者を決定すること、特定の治療製品を使用することによって治療によって深刻な副作用のリスクを増加させる可能性がある患者を決定すること、または特定の治療製品の治療に対する反応を監視することより良い安全性または有効性を達成するために治療を調整するための治療製品。診断に伴いFDAは医療機器として規制されている。アメリカでは、医療機器の設計と開発、臨床前と臨床試験、発売前の承認或いは承認、登録と発売、製造、ラベル、貯蔵、広告と販売促進、販売と流通、輸出と輸入及び発売後の監督などの事項は、すべてアメリカ食品薬品監督管理局及びその実施条例及びその他の連邦と州法規と条例によって管理されている。免除またはFDAが法執行自由裁量権を行使することを得ない限り、診断テストは、一般に、商業化前に市場許可またはFDAの承認を得る必要がある。医療機器に適用される2つの主要なタイプのFDAマーケティング許可は、510(K)承認、および発売前承認、またはPMA承認とも呼ばれる上場前通知である。
33
医療デバイスの510(K)許可を得るか、または510(K)の許可を得たデバイスをいくつか修正するためには、製造業者は、提案されたデバイスが以前に承認された510(K)デバイスと実質的に等しいか、または1976年5月28日以前に商業販売中の改訂前デバイス、またはFDAがPMAの述語デバイスの提出を要求していないことを証明する発売前通知を提出しなければならない。装置が述語装置と実質的に等しいと判断した場合、FDAは、提案された装置を1つまたは複数の述語装置と比較し、予期される用途、技術、設計、および安全性および有効性に影響を与える可能性のある他の特徴において、主語装置が述語装置または述語装置に匹敵するかどうかを評価する。FDAが、エージェントデバイスが1つまたは複数の述語デバイスに実質的に等しいと判断した場合、エージェントデバイスは市販されることを許可することができる。510(K)の発売前通知経路は、通常、申請完了日から3~12ヶ月を必要とするが、より長い時間を要する場合がある。
PMAの申請は有効な科学的証拠支持がなければならず、これは通常、技術、臨床前、臨床と製造データを含む大量のデータを必要とし、この装置の安全性と有効性を証明し、FDAを満足させる。診断テストの場合、PMAアプリケーションは、一般に、分析および臨床検証研究に関するデータを含む。PMA審査の一部として、FDAは、製造業者が設計、テスト、制御、文書、および他の品質保証手順に従うことを要求する品質システム法規(QSR)に適合することを保証するために、1つまたは複数の製造施設を承認前に検査する。法規によると、FDAの最初のPMA申請の審査には6~10ヶ月を要するが、この過程は通常より長い時間を要するが、完成には数年かかるかもしれない。PMA出願および製造施設の評価にFDAが有利である場合、FDAは、PMAの最終承認を保証するために満たされなければならないいくつかの条件を一般的に含む承認状または承認状を発行する。PMAまたは製造施設に対するFDAの評価が有利でない場合、FDAは、PMAの承認を拒否するか、または承認できない手紙を発行する。承認できない手紙は、申請の不足点を概説し、可能な場合には、PMAを承認するために必要な条件を決定する。承認されると、承認後の要求、承認条件、または他の規制基準が遵守されていない場合、または最初のマーケティング後に問題が発見された場合、FDAはPMAの承認を撤回する可能性がある。
FDAは2014年7月31日、“体外随伴診断装置”の開発と承認手順を述べた最終指導文書を発表した。ガイドラインによれば、診断テストに依存して使用される新しい治療製品のために、診断装置が対応する治療製品を安全かつ有効に使用するために不可欠である可能性がある場合、FDAは同時開発が不可能である可能性があることを認識しているにもかかわらず、治療の同時開発および承認または承認の前に適用されなければならない。しかしながら、1つの薬剤がセット診断なしに安全または有効に使用できない場合、FDAのガイドラインは、診断装置の承認または許可がない場合、通常はその薬剤を承認しないことを示している。FDAは2016年7月にガイドライン草案を発表し、体外随伴診断設備と治療製品の共同開発の原則を述べた。ガイドライン草案は治療製品及びそれに対応する体外随伴診断の開発と同時マーケティング許可を指導する原則を記述した。
承認或いは承認を得ると、セット診断設備はFDA品質体系法規の要求、不良事件報告、リコールと是正及び製品マーケティング要求と制限を含む上場後の要求を守らなければならない。医薬品やバイオメーカーと同様に,キット診断メーカーはFDAで製品や会社施設を監査し,その権威規定に適合したいつでもFDAの抜き打ち検査を受ける。
34
生物模倣薬と排他性
私たちのいくつかの候補製品は生物製品として規制されている。ACAの一部として、2009年の“生物製品価格競争と革新法”(BPCIA Act)は、生物製品のためにFDA許可の参考生物製品と類似または交換可能な簡略化された承認経路を作成した。PHSAのこの修正案は繰り返し検出を最小限にしようとある程度努力している。生物類似性は分析研究、動物研究と1つ或いは複数の臨床試験を通じて証明することができ、それは生物製品が参考製品と高度に類似していることを要求し、臨床では活性成分の微小な差異がないにもかかわらず、しかも安全性、純度と効力の面で、この製品と参考製品の間に臨床上意義のある差異がない。互換性は、生物学的製品と参照製品とが生物学的に類似していることを必要とし、任意の所与の患者において、参照製品と同じ臨床結果を生成することが期待でき、複数回投与された製品の場合、以前の投与後、製品および参照製品は、安全リスクを増加させることなく、またはこのような代替または切り替えを行うことなく、安全リスクを増加させることなく、または参照生物製品の独占的使用に対して治療効果のリスクを低減することができる。小分子薬物に比べて生物製品の構造が大きく,複雑であることが多く,このような製品を生産するプロセスは,FDAが策定している実施に重大な障害となっている。
参考生物製品は製品が初めて許可を得た日から4年と12年の専門期間が付与された。FDAは、参照製品が初めて許可された日から4年まで、基準生物製品に基づく生物学的類似または交換可能製品の申請を受け入れず、FDAは、参照製品が最初に許可された日から12年まで、基準生物製品に基づく生物学的類似または交換可能製品の申請を承認しないであろう。“初許可”とは、一般に、米国で特定の製品が許可された初期日を意味する。最初に許可された日は、生物製品の許可日を含まず(かつ、新しい特定期間は適用されない)、許可が生物製品のためのサプリメントである場合、またはバイオ製品のための同じ発起人または製造業者(もしかしたら人、利害関係者または他の関連エンティティ)の後続の出願である場合、新たな適応、投与経路、投与スケジュール、投与スケジュール、剤形、投与システム、投与装置または強度の変化(バイオ製品の構造の変更を含まない)、または安全性、純度、純度、強度の変化をもたらすことなく、バイオ製品のための構造の変更を行う。あるいは効力である。したがって、新製品が以前の許可製品の構造の修正を含むかどうかを決定し、それにより、安全性、純度、または効力の変化をもたらし、新製品の許可がそれ自身の排他期間をトリガする最初の許可であるかどうかを評価する必要がある。その後の出願は、承認された場合には、生物製品としての“第一次許可”の排他性が保証されるか否かは、具体的な状況やスポンサーが提出したデータに依存する。
その他の規制事項
FDA以外にも、製品承認後の製造、販売、販売促進およびその他の活動も米国の多くの監督機関によって規制されており、医療保険と医療補助サービスセンター(CMS)を含み、監察長事務室と公民権事務室、HHS部門の他の部門、司法省、薬品監督管理局、消費財安全委員会、連邦貿易委員会、職業安全·健康管理局、環境保護局、州と地方政府を含む。
医療提供者、医師、第三者支払者は、私たちが市場で承認された任意の製品の推薦と処方において主な役割を果たすだろう。私たちの現在と未来の医療提供者や医師との手配、および第三者支払者との任意の未来の手配は、広く適用される詐欺や乱用、その他の医療法律や法規に直面する可能性があり、これらの法律と法規は、私たちがマーケティング、販売、流通、上場承認を得た任意の薬物の業務または財務的配置と関係を制限するかもしれません。米国では、これらの法律には、連邦“反リベート法令”、“虚偽請求法”、連邦“1996年健康保険移行性と責任法案”(HIPAA)がある。
35
逆控除条例には、処方薬製造業者(またはそれを代表する者)を含む任意の人が、インフォームドコンセントおよび故意の場合、現金または実物の形態で、連邦医療保険または医療補助などの連邦医療計画に従って支払いを行うことができる、特定の薬物の処方を購入、推薦、注文または発行することを含む、直接または間接的に請求、受け入れ、提供、または支払うことができると規定されている。この法律に違反した行為は、監禁、刑事罰金、行政民事罰金、連邦医療計画から除外された罰を受けるだろう。さらに、個人または実体は、法規や法規違反の具体的な意図を実際に理解する必要はない。また、ACA規定は、政府は、連邦民事虚偽クレーム法案又は連邦民事罰金の目的に基づいて、連邦反リベート法規違反による物品又はサービスのクレームが虚偽又は詐欺的クレームを構成することを含むと主張することができる。
支払人に直接クレームを提出することはありませんが、連邦民事および刑事虚偽請求法および民事金銭罰法によると、医薬品製造業者は、個人またはエンティティ(製造業者を含む)に民事告発者または準訴訟を含む個人またはエンティティ(製造業者を含む)に故意に提出すること、または連邦計画(MedicareおよびMedicaidを含む)に虚偽または詐欺的な項目またはサービスクレームを提出すること、または医療上不要な項目またはサービスに対するクレームを意図的に連邦計画(MedicareおよびMedicaidを含む)に提出することを含む責任を問われる可能性がある。虚偽請求法案違反の処罰には,政府が実際に受けた被害の3倍,個々の虚偽クレームに対する強制民事罰が含まれており,連邦医療保健計画への参加から除外される可能性があり,連邦虚偽クレーム法案は民事法規であるにもかかわらず,虚偽クレーム法案違反を招く行為には様々な連邦刑事法規が関与している可能性がある。政府は、製造業者が顧客に不正確な請求書や符号化情報を提供したり、ラベル外で製品を宣伝したりすることによって、虚偽または詐欺的なクレームの提出を“招く”と考える可能性がある。“虚偽申告法”については,連邦“反リベート条例”違反による物品やサービスを含むクレームは虚偽または詐欺的クレームである。私たちの将来のマーケティングと、報告卸売業者または私たちの製品の推定小売価格に関する活動(承認されれば)、医療補助フィードバック情報を計算するための価格報告やその他、私たちの製品に影響を与える連邦、州、第三者の精算情報(承認されれば)、および候補製品の販売とマーケティングは、この法律の審査を受けています。
HIPAAは、計画を知りながら故意に実行または実行しようとすることを禁止し、虚偽または詐欺的な口実、陳述または約束の方法で任意の医療福祉計画(プライベート第三者支払者を含む)が所有または制御または保管している任意の金銭または財産を詐欺または取得し、故意に医療福祉計画を流用または盗み取り、医療保健違法行為の刑事調査を故意に阻害し、悪巧み、計画または装置によって重要な事実を故意に偽造、隠蔽または隠蔽し、あるいは医療福祉の提供または支払いに関連するいかなる重大な虚偽、架空または詐欺的な陳述を行うことを禁止する連邦刑法を制定した。物やサービスです。連邦“反リベート法令”と同様に,個人や実体は,その法令やその法令に違反する具体的な意図を実際に知ることなく違法行為を実施することができる.
民事罰金法規は、任意の個人またはエンティティに処罰を加え、その個人またはエンティティは、連邦健康計画へのクレームを出したか、または結果として判断され、そのクレームがクレームに従って提供されていないプロジェクトまたはサービス、または虚偽または詐欺的であることを知っているか、または知るべきである。
36
私たちは連邦政府と私たちが業務を展開している州のデータプライバシーと安全規制によって制限されるかもしれない。2009年の“健康情報技術促進経済および臨床健康法案”(HITECH)によって改正されたHIPAAおよびその実施条例は、2013年1月に発表された最終総合規則を含み、この規則は、特定の保険医療提供者、健康計画および医療保健情報交換所およびそれらのそれぞれの業務パートナー、独立請負業者または健康保証エンティティエージェントに要求を提出し、これらのエンティティは、彼らが提供するサービスの作成、維持、受信、使用、または個人識別可能な健康情報に関連する個人識別可能な健康情報を提供する。他の事項に加えて、HITECHは、HIPAAのセキュリティ基準を商業パートナーに直接適用し、商業パートナーは、カバーエンティティまたはカバーエンティティにサービスを提供することに関連する保護された健康情報を作成、受信または取得する特定の医療保健プロバイダ、健康計画、および医療チケット交換所を含むエンティティをカバーする独立した請負者またはエージェントとして定義される。HITECHはまた新しい民事罰金等級を作成し、HIPAAを改訂し、民事と刑事処罰を商業パートナーに直接適用し、州総検察長に新しい権力を与え、連邦裁判所に民事訴訟を提起し、損害賠償または禁止令を要求して連邦HIPAA法律を執行し、連邦民事訴訟の提起に関連する弁護士費と費用を求めることができる。また、他の連邦、州、非アメリカ法律は、場合によっては健康と他の個人情報のプライバシーとセキュリティを管理しているかもしれませんが、その中の多くの法律は互いに大きく異なり、同じ効果を与えないかもしれません, コンプライアンスを複雑にしていますこのような法律を守らなければ、重大な民事と刑事罰を受けるかもしれない。
しかも、私たちが運営している多くの州は敏感で個人情報のプライバシーと安全を保護する法律を持っている。いくつかの州の法律は、敏感かつ個人情報の面で、連邦、国際、または他の州の法律よりも厳格または範囲が広いか、またはより大きな個人権利を提供する可能性があり、これらの法律は互いに異なる可能性があり、これはコンプライアンス作業を複雑化させる可能性がある。州法がHIPAAよりも保護的なところでは,HIPAAに加えて,我々が受けている州法を守らなければならない。場合によっては、このようなより厳しい州法に適合するために、私たちが計画した操作と手続きを修正する必要があるかもしれない。また,複数の州からの個人の敏感さや個人情報を扱う場合には,任意の情報に適用される最も厳しい州法を遵守する必要があることが分かるかもしれない.カリフォルニアは最近、“カリフォルニア消費者プライバシー法”(California Consumer Privacy Act、略称CCPA)を公布し、カリフォルニアの消費者のために新しいプライバシー権(法律で定義されているような)を創造し、消費者または家庭の個人データを処理する実体に対してより多くのプライバシーと安全義務を規定した。CCPAは、カバーする会社に、そのデータ収集、使用、および共有アプローチに関するいくつかの開示を消費者に提供し、影響を受けたカリフォルニア住民に、特定の個人情報販売または移転から撤退することを選択する方法を提供する。CCPAは2020年1月1日に発効し、カリフォルニア州総検察長は2020年6月2日に最終法規を提出し、これらの法規は最終的に決定され、現在すでに発効している。カリフォルニア州総検察長は2020年7月1日から違反者に対する法執行行動を開始した。また、カリフォルニアの有権者は11月3日にCPRAと略称する新しいカリフォルニアプライバシー法であるカリフォルニアプライバシー権法案を可決した, 2020年です。CPRAは、個人情報の処理および記憶に追加的な義務を追加し、2023年1月1日から施行される(特定の条項は2022年1月1日まで遡及効力を有する)。CPRAに関連する発展を引き続き監視し、CPRAコンプライアンスに関連する追加コストと費用を予想する。米国の他の州でも総合的なプライバシー立法の制定が考えられており,業界組織はこれらの分野で新基準を採用し提唱していることが多い.CCPAおよびCPRAには、HIPAAの下で公衆衛生施設の活動に関連するいくつかの例外が含まれているが、CCPA、CPRA、または他のこのような将来の法律、法規、および基準が私たちの業務に及ぼす可能性のある影響を決定することはできない。
また、一部の観察者は、CCPAとCPRAは、米国のより厳格なプライバシー立法傾向の開始を示している可能性があり、これは私たちの潜在的な責任を増加させ、私たちの業務に悪影響を及ぼす可能性があると指摘している。アメリカで、私たちは州レベルの重大な発展を目撃した。例えば、2021年3月2日、バージニア州は消費者データ保護法、またはCDPAを公布し、2021年7月8日、コロラド州知事はコロラド州プライバシー法、またはCPAに署名し、法律にした。CDPAとCPAはいずれも2023年1月1日に発効した。CDPAとCPAはCCPAとCPRAの多くの類似した概念を含んでいるが、範囲、適用と法執行の面でもいくつかの重要な違いがあり、これらの法律は規制された企業の運営方法を変える。また、2022年3月24日、ユタ州知事はユタ州消費者プライバシー法案、またはUCPAに署名し、法律にした。“海外腐敗防止法”は2023年12月31日に施行される。また、2022年5月、コネチカット州知事のネッド·ラモントはコネチカット州データプライバシー法案、CTDPAと略称し、法律にすることに署名した。UCPAとCCTPAはバージニア州とコロラド州の先輩に大きく依存しています。CTDPAがあれば、コネチカット州は全面的なプライバシー法を公布した5番目の州となった。
37
他のいくつかの州もまた、上述した最近採択された法律と類似した新しいプライバシー法を提案した。このような提案された立法が通過されると、追加の複雑性、要求変化、制限、および潜在的な法的リスクが増加する可能性があり、コンプライアンス計画、影響戦略、および以前の有用なデータの利用可能性に追加のリソースを投入する必要があり、コンプライアンスコストの増加および/またはビジネス実践および政策の変化をもたらす可能性がある。アメリカの異なる州に全面的なプライバシー法が存在することは、私たちのコンプライアンス義務をより複雑で費用を高くし、私たちが法執行行動を受けたり、他の方法で規則に合わないことで責任を負う可能性を増加させるかもしれない。
さらに、ACA内の連邦医師が日光法案または陽光法案およびその実施条例を支払うことは、連邦医療保険、医療補助または児童健康保険計画(いくつかの例外)に従って支払うことができる薬品、器具、生物および医療用品のいくつかのメーカーが、医師および教育病院、または医師、いくつかの他の医療専門家および教育病院への要求またはその指定された実体または個人を代表してまたは割り当てられたいくつかの支払いまたは他の価値移転に関する情報をCMSに毎年報告し、医師、いくつかの他の医療保健専門家およびその直系親族が所有するいくつかの所有権および投資権益を毎年報告することを要求する。これらの報告義務は,2022年1月1日から,医師アシスタントや看護師従業員などのある非医師提供者への価値移転を含むように拡大された。また、多くの州では支払いや他の価値移転の報告も管理されており、その多くは互いに大きく異なり、往々にして先制されておらず、“陽光法案”よりも尻込み的な効果が生じ、遵守努力をさらに複雑化させる可能性がある。
同様の連邦、州、および外国詐欺および州反リベートおよび虚偽クレーム法律のような法律法規の乱用は、医療プロジェクトまたはサービスに関連する販売またはマーケティング手配およびクレームに適用される可能性がある。このような法律は一般的に広く、様々な国家機関と個人行動によって実行される。アメリカの多くの州は連邦反リベート法規と虚偽クレーム法案のような法律を採用しており、研究、流通、販売またはマーケティング手配、および非政府支払人(個人保険会社を含む)によって精算される医療項目またはサービスに関するクレームを含むが、私たちの商業実践に適用可能である。さらに、一部の州は、2003年4月の医薬品メーカー監察長オフィスコンプライアンス計画ガイドラインおよび/または米国の製薬研究および製造業者の医療専門家との相互作用に関するガイドラインを遵守することを要求する法律を採択している。また,Medicaidや他の州が計画して精算するプロジェクトやサービスに加えて,上記の各医療法律や法規を遵守しなければならない州や海外等価物も必要であり,その中には支払者にかかわらず適用可能な法律や法規の範囲が広い可能性がある。いくつかの州の法律は、製薬会社が製薬業の自発的コンプライアンスガイドラインおよび関連する連邦政府コンプライアンスガイドラインを遵守することを要求し、医師および他の医療保健提供者またはマーケティング支出への支払いおよび他の価値移転に関する情報を報告することを製薬業者に要求する。いくつかの州では他のマーケティング制限や製薬会社にも要求されています 国にマーケティングまたは価格開示を行い、薬品販売代表の登録を要求する。2018年5月に施行された欧州連合一般データ保護条例を含む国と外国の法律は、健康情報のプライバシーやセキュリティを管理する場合もあり、その多くの法律は互いに大きく異なり、HIPAAに先を越されず、コンプライアンス作業を複雑化させることが多い。このような州の要求を守るために何が必要なのかは曖昧で、もし私たちが適用された州の法律要求を守らなければ、私たちは処罰されるかもしれない。最後に,健康情報のプライバシーや安全を管理する国や外国の法律もあり,その多くの法律は互いに大きく異なり,HIPAAに先を越されず,コンプライアンス作業を複雑にしていることが多い。
38
製品を商業的に流通させるためには、州の法律を遵守しなければならず、ある州で製品をその州のメーカーおよびディーラーに輸送することを含む州で薬品および生物製品の製造業者および卸売業者の登録を要求しなければならない。いくつかの州はすでに立法を公布し、製薬と生物技術会社にマーケティングコンプライアンス計画を確立し、州政府に定期報告を提出し、販売、マーケティング、定価、臨床試験およびその他の活動を定期的に公開し、および/またはその販売代表を登録し、薬局および他の医療保健実体が製薬および生物技術会社にいくつかの医師処方データを販売およびマーケティングのために提供することを禁止し、いくつかの他の販売およびマーケティング行為を禁止する。私たちはまた、複雑な価格指標を正確かつタイムリーな方法で計算し、政府プロジェクトに報告することを要求する連邦政府価格報告法を遵守しなければならない。私たちのすべての活動は連邦と州消費者保護と不正競争法によって制限されるかもしれない。
これらの法律の範囲も執行も不確定であり,現在の医療改革環境下では,特に適用の前例や法規が乏しい場合には,急速な変化が生じる可能性がある。連邦と州法執行機関は最近、医療保険会社と医療保健提供者の間の相互作用の審査を強化し、医療保健業界の一連の調査、起訴、有罪判決と和解を招いた。政府当局は、私たちの業務実践が現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関連する現行または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。私たちの運営が上記の任意の法律または他の私たちに適用される可能性のある任意の政府法規に違反していることが発見された場合、私たちは重大な民事、刑事および行政処罰、損害賠償、罰金、契約損害、名声損害、利益減少および将来の収入減少、個人監禁、医療資金援助の医療計画(例えばMedicareおよびMedicaid)から医薬品を除外し、私たちの業務を削減または再編することに直面する可能性があり、これらはいずれも私たちの業務運営能力および財務業績に悪影響を及ぼす可能性がある。しかも、このような行動を防御するのは高価で時間がかかる可能性があり、大量の財政と人的資源が必要となる可能性がある。したがって、私たちが私たちに提起される可能性のあるいかなる訴訟も防ぐことに成功しても、私たちの業務は損害を受ける可能性がある。もし私たちがそれと業務往来があることを期待している任意の医師や他の保健提供者や実体が適用されない法律を遵守していないことが発見された場合、彼らは刑事、民事、または行政処罰を受ける可能性がある, 政府の援助を含む医療計画から除外される。業務手配が適用される医療保険法に適合することを確保することや,政府当局が行う可能性のある調査に対応することは,時間や資源がかかる可能性があり,会社の業務への関心を分散させる可能性がある。
米国では,患者が承認した製品を負担できるようにするために,患者支援計画や条件に適合した患者の自己負担クーポン計画など,様々な計画を利用して支援する可能性がある。PAPはCMS OIGの規制と指導を受けている。さらに、少なくとも1つの保険会社は、そのネットワーク薬局が、保険会社が決定した特定の特殊薬物の共同支払い券を受け入れないように指示している。私たちの自己負担クーポン計画は保険会社の似たような行動の目標になるかもしれない。また,2013年11月,CMSはACA市場で販売されている合格健康計画の発行者に指導意見を発表し,このような計画が第三者が提供する患者費用分担支援を拒否することを奨励し,CMSがこのような支援の提供を監視し,将来的に規制行動をとる可能性があることを示した。CMSはその後、個人市場に合格した健康計画がある政府関連実体の支払いを受ける第三者保険料と費用分担を要求する規定を発表した。2014年9月、HHSのOIGは、Dの受益者の一部が共同支払いクーポンを使用することを適切な措置を講じなければ、連邦反リベート法規および/または民事罰金法律の制裁を受ける可能性があると警告する特別諮問公告を発表した。したがって、会社はこれらのD部分受益者を自己払いクーポンの使用から除外した。
現在と未来の立法
米国や一部の外国司法管轄地域では、医療システムに関する複数の立法·規制改革および提案された改革が継続して存在する可能性があり、医療の獲得性を拡大し、医療の質を向上させ、医療のコストをコントロールまたは低減することを目的としている。
39
例えば、2010年3月、アメリカはACAを公布した。ACAに含まれる措置は、政府や民間保険会社が医療資金を融資する方法を大幅に変更し続けることが予想される。ACAの製薬業界にとって最も重要な条項は以下のとおりである
40
公布以来、ACAのいくつかの方面に対して多くの司法、行政、行政と立法方面の挑戦を提出した。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、特定の政府機関に、医療補助モデルプロジェクトおよび免除計画の再審査、および医療補助またはACAによる医療保険カバー範囲の獲得による不必要な障害をもたらす政策を含む医療補助モデルプロジェクトおよび免除計画の再審査および見直し、医療補助またはACAによる医療保険のカバー範囲を制限する既存の政策およびルールを再検討するように指示する。バイデン政府の他の医療改革措置や他の挑戦,ACAの廃止または代替の努力(あれば)が我々の業務にどのように影響するかは不明である。バイデン政権に先立ち、2017年10月13日、トランプ前総裁はACAによる保険会社への費用分担補助金を中止する行政命令に署名した。前トランプ政権は、ACAが保険会社に支払うコスト分担削減やCSRが国会の必要な支出を受けていないことを要求し、これらの支出が完了するまで直ちに支払いを停止すると発表した。いくつかの州の検事総長は政府による補助金の中止を阻止するよう提訴したが、彼らが提出した制限令請求は2017年10月25日にカリフォルニア州の連邦裁判官によって却下された。2020年8月14日, 米国連邦巡回控訴裁判所は2つの異なる事件の中で、連邦政府は2017年前の数年(2017年を含む)に支払われなかったCSRに対して全額責任を負うべきだと判断した。健康保険会社が2018年以降に提出した企業社会責任クレームについては,満期額(あれば)を決定するためのさらなる訴訟が必要となる。また、2018年6月14日、米国連邦巡回控訴裁判所は、連邦政府は第三者支払者に120億ドルを超えるACAリスク回廊に支払う必要がないと判断し、これらの支払人はこれらの支払いが彼らに不足していると弁明した。2020年4月27日、米国最高裁判所は米国連邦巡回控訴裁判所の裁決を覆し、事件を米国連邦クレーム裁判所に送り、政府は関連式に基づいてこれらのリスク回廊支払いを支払う義務があると結論した。このような判決が私たちの業務にどのような影響を与えるのかまだ分からない。
また,CMSは2020年から各州が個人や小団体市場の保険会社に基準を設定する上でより大きな柔軟性があり,ACAがこのような市場で販売されている保険計画に要求される基本的な健康福祉を緩和する可能性があるという最終規則を発表した。
また、ACAが公布されて以来、米国は他の立法と規制改革を提案し、採択した
41
また,340 B薬品定価計画も数回変化しており,薬品メーカーがある医療機関に売却する薬品の価格に上限を設定している。2018年12月27日、コロンビア特区地区裁判所は340 B薬品定価計画下の精算式の変更が無効であることを発表し、CMSはその後、2019年度と2018年度の特定保険外来薬(SCOD)の精算式を変更した。裁判所は、この変化は部長が適宜決定した“調整”ではなく、補償計算の根本的な変化であると判断した。しかし、最近では2020年7月31日、米コロンビア特区巡回控訴裁判所が地域裁判所の裁決を覆し、これらの変化が長官の権力範囲内にあることが分かった。2020年9月14日,原告−被控訴者はEN Bancの再審請願書(すなわち全裁判所まで)を提出したが,2020年10月16日に却下された。原告-被控訴人は2021年2月10日に最高裁に移審令の請願書を提出した。2021年7月2日金曜日、最高裁はこの請願書を承認した。これらの発展が将来の製品を購入する保証病院にどのように影響するか、将来承認された製品にそのような施設のレートを受け取る可能性があるかどうかは不明である(あれば)。2022年6月15日、最高裁はHHS 2018年と2019年の340 B病院の精算料率が法規に違反し、不法であるとする控訴裁判所の裁決を全会一致で覆した。私たちは340 B計画の発展に影響を及ぼすことを検討し続ける。
42
また、米国の薬品価格決定方法に対する立法と法執行への関心もますます大きくなっている。具体的に、政府はメーカーがその販売製品に価格を設定する方式に対して更に厳格な審査を行い、アメリカ議会はいくつかの調査を行い、連邦と州立法を提出し、公布し、薬品定価の透明性を高め、連邦医療保険制度下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査した。連邦レベルでは、総裁·バイデンは2021年7月9日に行政命令に署名し、政府の政策、すなわち(I)処方薬と生物製品価格を下げる立法改革を支持し、連邦医療保険の薬品価格交渉を許可し、インフレ上限を設定し、低コスト模造薬と生物模倣薬の開発と市場参入を支持すること、および(Ii)公共医療保険オプションの制定を支持することを確認した。その他の事項に加えて、行政命令は、処方薬の価格設定が高すぎ、国内の薬品サプライチェーンの強化、連邦政府の薬品支払いの価格低下、業界価格詐欺の解決のための行動を説明する報告書をHHSに提供するよう指示し、2003年の“連邦医療保険処方薬、改善と現代化法案”およびFDAの実施条例に基づいて第804条の輸入計画を制定する州とインディアン部族との協力を提案するようFDAに指示した。FDAは2020年9月24日にこのような実施条例を発表し、2020年11月30日に発効し、各州のカナダ薬品輸入計画の制定と提出に指導を提供した。2020年9月25日, CMSは,この規則により,各州から輸入された薬物は社会保障法第1927条に基づいて連邦還付を得る資格がなく,メーカーは“最適価格”や平均メーカー価格の目的でこれらの薬剤を報告しないと述べている。これらの薬剤はカバーされた外来薬とは考えられないため,CMSはさらに,これらの薬剤の全国平均薬物調達コストは公表されないと述べている。実施すれば、カナダからの薬品輸入は私たちの任意の候補製品の価格に実質的かつ不利な影響を与えるかもしれない。また、2020年11月20日、CMSは最恵国或いは最恵国モデルを実施する暫定最終規則を発表し、このモデルによると、ある薬物と生物製品の連邦医療保険B部分の販売率は1人当たりの国内総生産に類似した経済協力と発展組織国の薬品メーカーが受け取った最低価格に基づいて計算される。しかし、2021年12月29日、最恵国規則が廃止された。
また、2020年12月2日、HHSは薬品メーカーがD部分でスポンサー値下げを計画している安全港の保護を取り消し、法律が値下げを要求しない限り、直接あるいは薬局福祉マネージャーを通じて値下げする規定を発表した。この規定はまた、販売時点での値下げを反映するための新しい避難港を作成し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための避難港を作成する。裁判所の命令により,上記安全港の除去と増加は延期され,最近の立法は同規則の実施を2026年1月1日に一時停止した。この最終期限は、両党の“より安全なコミュニティ法案”によって2027年1月1日に延期された。2022年のインフレ削減法案はさらにこの規定の実施を2032年1月1日に延期する。また、2020年12月31日、CMSは2023年1月1日から発効し、メーカーに自己援助のすべての価値を患者に転嫁することを確保するように新規定を発表し、そうでなければ、これらのドルは薬物の平均メーカー価格と最適価格計算に計上される。2021年5月21日,PhRMAは米国コロンビア特区地域裁判所でHHSを起訴し,医療補助リベートに関する連邦法に違反していると主張した。2022年5月17日,米国コロンビア特区地域裁判所はPhRMAが提出した即決判決動議を承認し,医療補助アキュムレータルールの無効を宣言した。私たちはこの規則の施行とどんなさらなる変化が私たちの業務にどのように影響するのか予測できない。その中のいくつかの措置と他の提案された措置は発効するために追加の立法許可を必要とするかもしれないが、バイデン政府はこれらの措置を撤回または他の方法で変更するかもしれない, バイデン政府も国会も、彼らは薬品コストを抑えるための新たな立法措置を求め続けると表明した。
私たちは将来的により多くのアメリカ連邦医療改革措置を取ることが予想され、そのいずれも米国連邦政府が医療薬やサービスのために支払う金額を制限する可能性があり、これは私たちの候補薬物に対する需要の減少または追加の価格設定圧力を招く可能性がある。
43
アメリカの個別州もますます積極的に立法と実施を通じて、価格或いは患者の精算制限、割引、ある薬品への参入の制限、マーケティングコストの開示と透明性措置を含む薬品と生物製品の定価を制御するための法規を実施し、他の国からの輸入と大量購入を奨励することを目的としている。法律で規定されている第三者支払者の支払金額の価格制御またはその他の制限は、私たちの業務、財務状況、運営結果、将来性を損なう可能性があります。また,地域医療当局や個別病院では,どの薬品やサプライヤーが処方薬や他の医療計画に含まれるかを決定するために入札プログラムが使用されるようになってきている。これは私たちの薬品の最終需要を減少させ、あるいは私たちの薬品の価格設定に圧力を与える可能性があり、これは私たちの業務、財務状況、運営結果と将来性に負の影響を与えるかもしれない。
私たちは未来にどのような医療改革措置が取られるのか予測できない。連邦、州、外国の立法と規制はさらに発展する可能性があり、私たちが行っている措置は薬品価格の圧力を増加させると予想される。これらの改革は、候補製品の予想収入に悪影響を及ぼす可能性があり、私たちの全体的な財務状況や候補製品を開発する能力に影響を与える可能性がある。
アメリカの包装と流通
もし私たちの製品が承認されると、連邦供給スケジュールの許可ユーザーに提供することができます。他の法律と要求が適用されます。製品はアメリカの“毒物防止包装法”に適用される児童保護包装要求に適合しなければならない。製造、販売、販売促進、その他の活動はまた、連邦と州消費者保護および不正競争法によって制限される可能性がある。
医薬製品の流通は広範な記録保存、許可、貯蔵と安全要求を含む追加の規定と条例を遵守し、許可されていない医薬製品の販売を防止しなければならない。
これらの法律または規制要件のいずれかを守らない場合、会社は可能な法律または規制行動に直面するだろう。場合によっては、適用される規制要件を満たさないことは、刑事起訴、罰金またはその他の処罰、禁止、連邦医療計画から除外されること、リコールの要求、製品の差し押さえ、生産の完全または部分的な一時停止、製品の承認の拒否または撤回、または政府契約を含む会社の供給契約の締結を拒否する可能性がある。これらの法律に違反して私たちにとった行動は、たとえ私たちが弁護に成功しても、巨額の法的費用を招き、私たちの経営陣の業務運営への注意をそらす可能性があります。私たちが販売している未来の製品の販売を禁止または制限または撤回することは、不利な方法で私たちの業務に大きな影響を与えるかもしれません。
規制、法規、または既存の規制の解釈の変化は、(I)私たちの製造スケジュールを変更すること、(Ii)製品ラベルを追加または修正すること、(Iii)私たちの潜在的製品をリコールまたは停止すること、または(Iv)追加の記録保存要件を要求することなど、私たちの将来の業務に影響を与える可能性がある。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。
他のアメリカの環境、健康、安全の法律法規
私たちは多くの環境、健康と安全法律と法規によって制約される可能性があり、それらの管理実験室手続きと危険材料と廃棄物の処理、使用、貯蔵、処理と処理の法律と法規を含むかもしれない。時々、将来、私たちの業務は、化学品および生物学的材料を含む危険かつ可燃性材料の使用に関連する可能性があり、危険な廃棄物製品を生成する可能性もある。これらの材料や廃品を処分する契約を第三者と締結しても、これらの材料による汚染や傷害のリスクを完全に解消することはできません。私たちの危険な材料を使用または処分して汚染または損傷をもたらす場合、私たちはそれによるいかなる損害に責任を負う可能性があり、いかなる責任も私たちの資源範囲を超える可能性がある。民事や刑事罰金やこのような法律や法規を遵守しない罰に関する巨額の費用を招く可能性もある。
私たちは従業員の負傷によって生じる可能性のある費用と費用を支払うために労働者補償保険を維持していますが、この保険は潜在的な責任に対応するのに十分ではないかもしれません。しかし、私たちは私たちが提起した環境責任や有毒侵害請求に保険を提供することはできない。
44
また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。現在または未来の環境法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。しかも、このような法律法規を遵守しないことは巨額の罰金、処罰、または他の制裁につながる可能性がある。
米国特許期間延長と市場排他性
FDAが承認した薬物または生物の時間、期限および詳細によれば、1984年の“薬品価格競争および特許期限回復法”(通常はハッチ·ウェクスマン法と呼ばれる)によれば、いくつかの米国特許は、限られた特許期間を延長する資格がある可能性がある。ハッジ·ワックスマン法は、FDA規制審査中に失われた特許期間の補償として、特許期間を特許正常期限日を超えて最大5年まで延長することを可能にする。しかしながら、特許期間の延長は、特許の残り期間を製品が承認された日から合計14年まで延長することはできず、その承認された医薬製品、その使用方法、または製造方法に関連する権利要件のみを延長することができる。承認された薬物に適用される特許は1つのみ延期する資格があり,延期出願は特許が満期になる前に提出されなければならない。米国特許商標局はFDAと協議し,任意の特許期限延長の出願を審査·承認する。秘密協定またはライセンス契約の出願人は、臨床試験の期待長および関連する秘密協定または許可協定の提出に関連する他の要因に依存する現在の満了日後の特許寿命を延長するために、現在所有または許可されている特許の特許期間を延長することを申請することができる。
FDCAにおけるマーケティング排他的条項はまた、いくつかの申請の提出または承認を延期する可能性がある。FDCAは新しい化学実体秘密協定の承認を得た最初の申請者に5年間の米国内の非特許マーケティング排他性を提供した。FDAが以前に同じ活性部分を含む他の新薬を承認していなければ,薬物は新しい化学実体であり,活性部分は薬物物質の作用を担う分子やイオンである。排他期間内に、出願人が承認に必要なすべてのデータを所有または合法的に参照しない場合、FDAは、薬剤の別のバージョンのために別の会社が提出したANDAまたは505(B)(2)NDAの審査を受け入れない可能性がある。しかしながら、出願が特許無効または非侵害の証明を含む場合、4年後に提出することができる。FDAが、出願人が行っているまたは後援する新しい臨床研究(バイオアベイラビリティ研究を除く)が承認申請に不可欠であると考えている場合、FDCAはまた、NDA、505(B)(2)NDAまたは既存のNDAの補充のために、既存の薬剤の新しい適応、用量または強度のような3年間の市場排他性を提供する。この3年間の排他性には,新たな臨床研究に関する使用条件のみが含まれており,FDAが原始活性物質を含む薬物のANDAを承認することは禁止されていない。5年と3年の排他性は完全な秘密協定の提出や承認を延期したり承認したりしないだろう。しかしながら、完全なセキュリティプロトコルを提出する出願人は、安全かつ有効であることを証明するために、必要なすべての臨床前研究および十分かつ良好に制御された臨床試験を参照する権利を行うか、または得ることを要求されるであろう。
EU薬物開発
連合や連合では、私たちの未来の製品もまた広範囲な規制要求を受けるかもしれない。アメリカと同様に、医薬製品は主管監督機関のマーケティング許可を得た後にのみ発売されることができる。
アメリカと類似して、EUの臨床前と臨床研究の各段階は重要な監督管理によって制御されている。
2014年4月、EUは2022年1月31日に臨床試験指令2001/20/ECまたは指令に代わる新しい臨床試験条例(EU)第536/2014号、またはEU臨床試験条例を採択した。新たなEU臨床試験条例の一過性条項は,2025年1月31日までに行われているすべての臨床試験を新たなEU臨床試験条例に移行しなければならないと規定している。
45
新しいEU臨床試験条例はEU以前の臨床試験審査制度を徹底的に改革した。具体的には、新しいEU臨床試験条例は、EUの臨床試験の承認を簡略化し、簡素化することを目的として、すべての加盟国に直接適用される(これは、各EU加盟国で国家立法を制定する必要がないことを意味する)。例えば,単一入口点(臨床試験情報システムによる)により簡略化された申請手順を提供し,臨床試験申請評価の最終期限を厳密に規定している。
EUの薬品審査と承認
EUでは、医薬製品はマーケティング許可やMAを得た後にのみ商業化されることができる。二つの種類のマーケティング許可があります。
46
上記の手順により、MAを付与する前に、欧州市場管理局又はEU加盟国主管当局は、製品の品質、安全性及び有効性に関する科学的基準に基づいて、製品のリスク−利益バランスを評価する。
連合王国(大ブリテンおよび北アイルランド連合王国からなる)がEUを離脱したため、大ブリテンは中央集権のMAに属さなくなった(北アイルランド議定書によると、中央集権のMAは北アイルランドで引き続き認められている)。現在集中しているMAを有するすべての医薬製品は2021年1月1日に自動的にイギリスMAに変換された。2021年1月1日からの3年間、イギリスの薬品監督機関またはMHRAは、欧州委員会が集中手続きで新しいMAを承認する決定に依存し、新しいイギリスMAをより迅速に承認する可能性がある。しかし、まだ個別的な申請が必要になるだろう。2023年1月24日、MHRAは、欧州医薬品管理局および他のいくつかの規制機関のMA承認に関する決定を考慮する2024年1月1日から新たな国際認可枠組みを構築すると発表した。MHRAはまた、連合王国または大ブリテンでMAをより迅速に授与するために、EU加盟国が分散または相互承認手続きによって承認されたMAを考慮する権利がある。
EUの新しい化学実体排他性
EUでは、完全かつ独立したデータパケットによって承認された革新医薬製品はマーケティング許可を得た後、8年間のデータ独占経営権を獲得する資格があり、他の2年間の市場独占経営権を有する。データ排他性が付与された場合、参照製品が初めてEUで許可された日から8年以内に、EUで模倣薬または生物類似MAを申請した場合、模倣薬または生物類似申請者は、製品アーカイブに含まれる革新者の臨床前および臨床試験データを参照することができない。追加の2年間の市場排他期間内に、模倣薬または生物類似MAAを提出することができ、革新者のデータを参照することができるが、市場排他性が満了するまで、いかなる模倣薬や生物類似製品もEU市場に投入することはできない。この10年の最初の8年間に、マーケティング許可保持者が1つまたは複数の新しい治療適応の許可を得た場合、10年全体の期間は最大11年に延長され、これらの新しい治療適応は、認可前の科学的評価において、現在承認されている治療法と比較して有意な臨床的利益をもたらすと決定される。1つの製品がEMAによって革新的な医薬製品とみなされることは保証されず、しかも製品はデータ独占性を得る資格がない可能性がある。しかしながら、1つの製品が革新的な医薬製品とみなされても、革新者が所定のデータ独占期間を取得し、別の会社がMAAベースのMAを取得し、完全かつ独立した薬物試験、臨床前試験、および臨床試験パケットを有する場合、同社はその製品の別のバージョンを販売することができる。
EU孤児指定と排他性
EUでは、EMAの孤児薬物製品委員会は、そのスポンサーが、(1)生命または慢性衰弱に危険な疾患を診断、予防または治療することを目的としていること、(2)申請されたとき、そのような疾患がEUで10,000人中5人以下に影響を与えることを証明することができることを証明できることを前提としている。および(3)このような疾患を満足できる診断、予防または治療する方法がEUで販売されていないことを許可しなければならない、または、そのような方法があれば、製品はその疾患の影響を受けている人に大きな利益を与えるであろう。
47
EUでは、孤児指定は、費用の削減や費用の免除など、一方が経済的インセンティブを得る権利があり、マーケティング許可を付与した後、10年間の市場排他性を付与する。この市場排他期間内に、欧州医薬品管理局、欧州委員会またはEU加盟国のどの主管機関も申請を受け入れることができず、“類似医薬製品”の販売許可を承認することもできない。類似医薬製品“の定義は、承認された孤児医薬製品に含まれる1つ以上の類似活性物質を含む医薬製品であり、同じ治療適応のためのものである。この製品が市場排他性を維持するのが合理的であることを証明するのに十分な利益があることを証明するのに十分な利益があることを含む孤児指定基準をもはや満たしていない場合、この期限は6年に短縮される可能性がある。非常に特別な場合、市場排他性は、例えば、(I)類似の医薬製品が認可製品よりも安全で、効率的で、または臨床的に良いと判断した場合、(Ii)マーケティング許可保持者がこのような撤回に同意する場合、または(Iii)マーケティング許可保持者が十分な孤立した医薬製品を供給できないと判断する場合もある。上場承認申請を提出する前に、孤児としての指定を要求しなければならない。孤児指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の継続時間を短縮することもない。
英国では2021年1月1日より単独の孤児指定手続きの採用が開始された。英国には現在発売前の孤児指定(EUと同様)がなく,孤児指定の申請はイギリスまたはイギリスMAのMAA時にMHRAによって審査される。孤児を指定する基準はEUと同様であるが,それらはイギリスにのみ適用される(例えば,EUとは逆に,イギリスでは関連疾患を診断,予防または治療する方法はない必要がある)。
ヨーロッパ小児科調査計画
EUでは、新しい医療製品のMAAは、EMAが特定の製品の免除、カテゴリ免除、またはPIPに含まれる1つまたは複数の措置を承認しない限り、EMA小児科委員会(PDCO)と合意された小児科調査計画(PIP)に適合する小児科集団で行われた研究結果を含まなければならない。会社が認可された薬物のために新たな適応,薬物形式あるいは投与経路を増加させようとした場合にも,この要求は適用される。PIPは,市販認可が求められている薬物の小児科適応を支援するためのデータ生成の時間とアドバイスを規定している。PDCOは、成人における製品の有効性および安全性を証明するのに十分なデータがあるまで、PIPの一部または全ての措置の実施を延期する義務を許可することができる。さらに、小児臨床試験データを必要としないか、または提供するのに適していない場合、PDCOは、子供に無効または安全でない可能性があるので、これらのデータを提供する義務を免除することができ、この製品は、治療のために使用される疾患または状態が成人集団でのみ発生することが予想される場合、または小児科患者の既存の治療に対して有意な治療利益がない場合。MAを取得し,製品情報に実験結果を含めると,否定的であっても,その製品は6カ月の補足保護証明書を取得して延期する資格がある.孤児薬品の場合、孤児市場の独占権は2年間延長することができる。この小児科奨励は特定の条件の制約を受け,PIPに適合したデータを開発·提出する際に自動的に獲得されない。
営業許可を得た後の規制要件
もし1つの医薬製品がEUで許可された場合、MAの保持者は医薬製品の製造、マーケティング、普及と販売に適用される一連の要求を守らなければならない。これらの措置には
48
上記のEU規則は一般に欧州経済圏(EEA)に適用され、この地域はEU加盟国およびノルウェー、リヒテンシュタイン、アイスランドからなる。
イギリスの離脱とイギリスの規制枠組み
イギリスは2020年1月31日に正式にEUを離脱し、EUはイギリスと貿易·協力協定を締結し、TCAと略称し、2021年1月1日から暫定的に適用され、2021年5月1日から正式に適用される。TCAには薬品に関する具体的な条項が含まれており,その中にはGMP相互承認,医薬製品の製造施設の検査,発表されたGMP文書が含まれているが,イギリスとEUの薬品法規の大規模な相互承認は予想されていない。現在、イギリスは“2012年人類薬品条例”(改正)(北アイルランド議定書によると、EU規制枠組みは引き続き北アイルランドに適用される)を採択し、医薬製品のマーケティング、普及、販売に関するEUの立法を実施している。そのため,新しいEU臨床試験規制を除いて,イギリスの規制制度は多くの点でEUの現行の薬物規制制度と一致しているが,イギリスの規制制度はEUから独立しているため,“イギリス薬物規制法”はイギリスとEUの薬物法の相互承認について規定されていないため,これらの制度は将来的により大きな差がある可能性がある。
49
ヨーロッパデータ収集
欧州経済圏における個人健康データの収集と使用は,2018年5月25日に施行された“一般データ保護条例”(GDPRまたはEU GDPR)の規制を受けている。GDPRは、欧州経済地域に設立された任意の会社および欧州経済区以外に設立された欧州経済地域内のデータ主体に商品またはサービスを提供すること、または欧州経済地域内のデータ主体の行動に関する個人データを監視することを処理する会社に適用される。GDPRは,データ当事者の同意に関する厳しい要求,個人データがどのように使用されるかに関するより広範な開示,“高リスク”処理に対するプライバシー影響評価の要求,保持個人データの制限,強制的なデータ漏洩通知,“プライバシー設計”要求を含む個人データ制御者のデータ保護義務を強化し,データ処理者であるサービス提供者に直接義務を規定している.GDPRはまた,個人データをヨーロッパ経済圏以外に移転する国に対して厳しいルールを実施しており,これらの国では米国のような十分な保護レベルを確保できていない。GDPRの要求や欧州経済圏加盟国の関連国データ保護法を遵守しないことは、2000万ユーロにのぼる罰金や会社の前財政年度の世界年収の4%を招き、高い者を基準とする可能性がある。また,GDPRはデータ主体にGDPR侵害による物質や非物質被害の権利を付与する。データ保護義務の変化の広さと深さを考慮して、GDPRの遵守を維持するには大量の時間、資源、および費用が必要となり、新たなデータ保護規則の遵守を保証するための追加のメカニズムを確立する必要があるかもしれない。これは激務であり、私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性がある。
また、連合王国が2020年1月31日にEUを離脱したのに続き、GDPRは2020年12月31日の移行期間終了時に連合王国への適用を停止した。しかし、2021年1月1日現在、イギリスの2018年“EU(離脱)法案”は、GDPR(2020年12月31日に存在するGDPRと同じであるが、イギリスの何らかの具体的な改正が必要)をイギリスの法律に組み入れ、イギリスGDPRと呼ばれている。英国GDPRと2018年の英国データ保護法は,EUのデータ保護制度から独立した連合王国のデータ保護制度を規定しているが,EUのデータ保護制度と一致している。英国政府はデータ改革法案でデータ保護法の枠組みを改革する計画を発表したが,これらの計画は棚上げされている。イギリスのGDPR違反は、金額が高い者を基準に、1750万GBまたは世界収入4%の罰金を招く可能性があります。
イギリスのGDPRは国境を越えたデータ転送の制限を含む。EUとイギリスのデータ保護法によると,個人データがEUやイギリス以外の地域,特に米国に移行できるように十分な保障措置が施行されなければならない。2021年6月4日、欧州委員会(EC)は、欧州経済地域内のコントローラまたはプロセッサ(または他の方法でEU GDPRによって拘束されている)から欧州経済領域外に設立されたコントローラまたはプロセッサ(EU GDPR制約を受けない)からデータを送信するための新しい形態の標準契約条項を発表した。新しい標準契約条項は、以前データ保護指令に基づいて採用されていた標準契約条項に代わっている。イギリスはEUの新しい標準契約条項の制約を受けないが、2022年3月21日に発効し、イギリスからの移転を許可する“国際データ転送協定”と呼ばれる独自バージョンの標準条項を発表した。これらの新しいメカニズムによる移転は、受け入れ国の法律が欧州経済地域と“ほぼ同じ”保護を提供して移転した個人データを保護することを確保するためにケースベースで評価する必要があり、この基準に適合しなければ、企業は補充措置を要求される。英国GDPRによる制限されたデータ転送時にこれらの新たな保障措置を実施することが要求され,多大な努力とコストが必要となる。
英国はEU GDPR下の第3の国とされているが、欧州委員会は現在、英国がEU GDPRで十分な保護を提供していることを認める決定を発表しているため、EU由来の個人データの英国への移行は依然として制限されていない。EU GDPRと同様に,イギリスGDPRは英国以外の国への個人データの移行を制限しており,これらの国はイギリスから十分な保護を提供しているとはみなされていない。イギリス政府は、イギリスからヨーロッパ経済圏への個人データ転送が依然として自由に流動していることを確認した。
50
世界の他の地域の規制
EUや米国以外の他の国,例えば東欧,ラテンアメリカ,アジアの国では,臨床試験,製品許可,定価,精算の要求は国によって異なる。また,臨床試験はGCP要求および“ヘルシンキ宣言”からの適用法規要求と倫理原則に基づいて行わなければならない。
もし私たちが適用される外国監督管理要求を遵守できない場合、私たちは罰金、規制許可の一時停止または撤回、製品のリコール、製品の差し押さえ、経営制限、刑事起訴などに直面する可能性がある。
国際業務を管理するその他の法律·法規
私たちがアメリカ以外での業務をさらに拡大すれば、私たちは各管轄区域で事業を展開することを計画している多くの法律と法規を遵守するために追加の資源を投入しなければならない。“反海外腐敗法”(FCPA)は、個人または企業が業務を獲得または保持するのを助けるために、任意の米国人または企業が、任意の外国人官僚、政党または候補者に直接的または間接的に支払い、提供、許可支払い、または任意の価値のあるものを提供することを禁止する。“海外腐敗防止法”はまた、米国に上場する証券会社にある会計条項を遵守することを要求し、これらの条項は、会社(国際子会社を含む)のすべての取引の帳簿と記録を正確かつ公平に反映し、国際業務のために適切な内部会計制御システムを設計し、維持することを要求する。
“反海外腐敗法”を遵守することは高価で困難であり、特に腐敗は公認問題である国である。また、海外腐敗防止法は製薬業に特別な挑戦をもたらしており、多くの国では病院が政府によって運営されているため、医師や他の病院従業員は外国人官僚とされている。臨床試験やその他の仕事に関連して病院に支払われた何らかの金は、政府関係者に支払われた不正金と考えられ、“海外腐敗防止法”の法執行行動につながった。
様々な法律、法規、および行政命令はまた、米国国外での使用および伝播を制限するか、または国家セキュリティ目的のために秘密にされた情報と、特定の製品およびこれらの製品に関連する技術データとを特定の非米国国民と共有する。もし私たちがアメリカ以外での業務を拡大すれば、これらの法律を遵守するためにより多くの資源を投入する必要があります。これらの法律は、米国以外の地域でいくつかの候補製品を開発、製造、販売することを阻止するかもしれません。これは、私たちの成長潜在力を制限し、私たちの開発コストを増加させるかもしれません。
国際ビジネス慣行に関する法律を遵守しなければ、重大な民事·刑事罰を受け、政府契約の資格を一時停止または廃止する可能性がある。米国証券取引委員会(Securities and Exchange Commission,略称米国証券取引委員会)も、発行者が“反海外腐敗法”の会計条項に違反したため、発行者の米国取引所での証券取引を一時停止または禁止する可能性がある。
51
保証と精算を請け負う
新薬製品の商業化成功は政府衛生行政部門、私営健康保険会社とその他の組織によるこれらの薬物製品の清算程度にある程度依存する。米国や他の国の市場では,患者は通常第三者支払者によって治療に関連する費用の全部または一部を精算する。政府医療保健計画(例えば連邦医療保険や医療補助)や商業支払者の十分なカバーと精算は新製品の受容度に重要である。私たちが私たちの候補製品を商業化することに成功できるかどうかは、政府衛生行政部門、個人健康保険会社、その他の組織がこれらの製品と関連治療に提供する保険範囲と十分な補償にある程度依存する。保険を提供しても、承認された精算金額が十分に高くない可能性があり、十分な投資リターンを実現するために十分な価格設定を確立したり維持したりするのに十分ではないかもしれません。政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、どのような薬剤を支払うかを決定し、精算レベルを確立する。政府当局と第三者支払人、例えば個人健康保険会社や健康維持組織は、彼らがどの薬品のためにお金を払い、精算レベルを確立するかを決定する。政府と個人支払者の獲得性と精算範囲は大多数の患者が薬品を負担できるキーポイントである。薬品の販売は健康維持、管理保健、薬局福祉と類似の保健管理組織が薬品費用を支払う程度に大きく依存する, あるいは政府衛生行政部門、個人健康保険会社、その他の第三者支払人によって精算される。
アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。政府当局と第三者支払者は、特定の薬品のカバー範囲や精算金額を制限することでコストを抑制しようとしている。多くの国では、国家衛生システムの一部として、薬品価格は異なる価格制御メカニズムの制約を受けている。一般的に、この制度での薬品価格はアメリカよりずっと低い。他の国は会社が自ら薬品価格を確定することを許可したが、会社の利益を監視し、コントロールした。そのため、米国以外の市場では、薬品の精算が米国よりも減少する可能性がある。
T新たに承認された製品の保険カバー範囲や精算に関する不確実性も大きく,カバー範囲はFDAや同様の外国規制機関がこの薬剤を承認する目的よりも限られている可能性がある。アメリカでは、新薬製品の精算に関する主な決定は通常衛生と公衆サービス部に属するCMS機構によって行われる。CMSは新薬製品がどの程度連邦医療保険の下でカバーと精算されるかを決定し、個人支払者はよくCMSに大きく従う。しかし、第三者支払者の間には統一された薬品保険と精算政策がなく、支払者間の薬品保険と精算レベルに大きな差がある可能性がある。支払人が精算を決定する際に考慮する要素は、製品があるかどうかに基づいている
52
第三者支払者は、承認されたリストまたは処方内の特定の製品に保証範囲を制限することができ、これは、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。さらに、より低い価格の模倣薬または他の代替薬が利用可能である場合、第三者支払者は、その処方に特定のブランド薬を含むことを拒否するか、または他の方法で患者がブランド薬を得ることを制限する可能性がある。医薬品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、および将来、米国価格よりも低い価格で販売される可能性のある国から医薬品を輸入することを制限する法律の緩和によって低下する可能性がある。 第三者決済者は、医療製品やサービスの価格に挑戦し、医療の必要性を審査し、医療製品やサービスの費用便益を審査し、コストを管理するための制御を実施することが増えている。私たちは私たちが商業化されたどの候補製品も精算できることを確実にすることはできない。もし精算できるなら、精算のレベルもそうだ。また、多くの製薬業者は平均販売価格(ASP)と最適価格のようないくつかの価格報告指標を計算し、政府に報告しなければならない。場合によっては、これらの指標が正確かつタイムリーに提出されていない場合には、処罰が適用される可能性がある。また,これらの薬品の価格は,政府医療計画が要求する強制的な割引やリベートによって低下する可能性がある。
2003年の“連邦医療保険処方薬、改善と現代化法案”(MMA)は連邦医療保険D部分計画を創立し、連邦医療保険受益者に自発的な処方薬の福祉を提供した。D部によると、連邦医療保険受益者は、個人実体が提供する処方薬計画に参加することができ、これらの計画は外来処方薬の保険を提供する。連邦医療保険A部やB部と異なり,D部のカバー範囲は標準化されていない。D部分処方薬計画発起人はすべてのD部分薬物に費用を支払う必要はなく、各薬物計画は自分の薬物処方を制定することができ、それがどの薬物及びレベル或いはレベルをカバーするかを決定することができる。すべての連邦医療保険薬物計画は少なくとも連邦医療保険が設定した標準保険レベルを提供しなければならないが、D部分の処方薬計画発起人はすべての保険を受けたD部分の薬物に費用を支払う必要はなく、各薬物計画は自分の薬物処方を開発し、それがどの薬物および被覆のレベルまたはレベルをカバーするかを決定することができる。しかしながら、D部分処方薬処方は、必ずしも各カテゴリまたはカテゴリのすべての薬剤を含むとは限らないにもかかわらず、各治療カテゴリおよびカバーされたD部分薬剤カテゴリの薬剤を含む必要がある。D部分の処方薬計画に使用されるどの処方も薬局と治療委員会が開発·審査しなければならない。政府が処方薬の費用の一部を支払うことは、私たちが発売許可を得た薬の需要を増加させるかもしれない。Dの一部の処方薬計画がカバーしている私たちの未来の製品のいかなる交渉価格も私たちが獲得する可能性のある価格より低いかもしれません。また,MMAは連邦医療保険受益者の薬品福祉にのみ適用されるが,個人支払者は自分の支払率を設定する際に連邦医療保険カバー政策や支払制限に従うことが多い。MMAによる任意の支払い減少は、非政府支払者支払いの同様の減少をもたらす可能性がある。
MedicaidまたはMedicare Part B計画に従って連邦補償を獲得するか、または米国政府機関に直接販売する薬品の場合、製造業者は割引を340 B薬品定価計画に参加する資格のあるエンティティに拡大しなければならない。製品を与えるために必要な340 B割引は,メーカー平均価格(AMP)とメーカーから報告された医療補助返金金額から計算される。ACAは2010年から、現在の法律状況に応じて、これらの新しい条件に適合するエンティティ(児童病院を除く)は、孤児薬品340 B割引定価を取得する資格がないことにもかかわらず、340 B割引定価を取得する資格があるエンティティタイプを拡大した。340 B医薬品定価はAMPと医療補助返却点データに基づいて決定されるため,上記の医療補助返却式とAMP定義の改訂は必要な340 B割引を増加させる可能性がある。2009年の“米国回復·再投資法案”は、同一疾患の異なる治療法の有効性を比較するために連邦政府に資金を提供した。
この研究の計画は2012年に衛生·健康サービス部、医療保健研究と品質局と国家衛生研究院によって公表され、定期的に研究状況と関連支出の報告を国会に提出する。比較有効性研究の結果,公共または個人支払者の保証政策を許可するためのものではないにもかかわらず,いずれかのこのような薬物や治療しようとしている場合が試験のテーマであれば,候補薬物の販売にどのような影響を与えるかは不明である(あれば)。比較有効性研究では,競合他社の薬物が利点であり,候補薬物の販売に悪影響を及ぼす可能性もある。もし第三者支払者が私たちの薬物が他の利用可能な療法と比較して費用効果があると思わない場合、彼らは承認後に私たちの薬物をその計画の下の福祉としてカバーしないかもしれない、または、もし彼らがそう思う場合、支払いレベルは利益に基づいて私たちの薬を販売するのに十分ではないかもしれない。
53
これらの法律および将来の州および連邦医療改革措置は将来的に採択される可能性があり、いずれも連邦医療保険および他の医療保険資金のさらなる減少をもたらし、他の方法で規制の承認を得る可能性のある任意の候補製品の価格、またはそのような候補製品の処方または使用頻度に影響を与える可能性がある。
米国以外の多くの国では,薬品や医療機器の定価は政府によって制御されている。例えば、連合では、様々な国の価格設定と補償プログラムの差が大きい。いくつかの会員国は、製品は提案された価格設定が承認された後にのみ販売できると規定している。一部の加盟国は、ある特定の治療法の費用対効果を既存の治療法またはいわゆる保健技術評価と比較して、精算または価格設定の承認を得ることを要求する可能性がある。加盟国は医薬製品の具体的な価格を承認することができ、医薬製品を市場に投入する会社の収益力に対して直接或いは間接的に制御制度をとることもできる。会員国は会社が自分の製品価格を決定することを許可することができるが、製品数を監視·制御し、処方を制限するために医師に指導意見を発表する。各国が医療支出を管理しようとするにつれ,薬品や医療機器の価格や使用を抑える努力が続く可能性がある。薬品に対して価格制御や精算制限を実行することを保証できない国は、私たちの任意の候補製品に対して有利な精算と定価手配を許可する。歴史的に見ると、EUで発売された製品はアメリカの価格構造に従わず、通常価格ははるかに低くなることが多い。
人的資本管理
私たちの人的資本理念は持続的に優れたチームメンバーを吸引し、維持することに依存する。私たちの文化と私たちの人材に対する態度は採用、専門発展、業績管理と全面的な奨励を含むこの理念を強化した。私たちのいくつかの核心的な人的資源の流れについて、以下ではより多くの詳細を提供します。
2022年12月31日現在、392人のフルタイム従業員と4人のアルバイト従業員がいます。うち、297社は私たちの付属会社を通じて研究開発計画を推進することに集中しており、99社は私たちの付属会社で働き、戦略業務発展、財務と行政指導者の専門知識、および私たちの付属会社の一般的かつ行政サービスを提供している。私たちは一度も労働を停止したことがなく、私たちの従業員の中の一つも労働機関やどんな集団交渉の手配によって代表されていない。私たちは私たちの従業員が仲がいいと思う。
募集する
2020年には、当社の付属会社が適切な時期に適切な人材を採用することを支援する人材獲得能力を確立しました。私たちは経験豊富な人材獲得専門家からなるチームは採用マネージャーと密接に協力し、職位を公開するために必要な技能と能力を理解し、そして応募者の面接過程と評価を支援する。私たちはトップ人材の採用に努力しているため、質の高い採用プロセスと応募者の経験が必要です。私たちは各ポストを最も合格した候補者で埋めるために努力しており、これは時々外部採用機関と協力する必要がある。私たちはBridgeioで働く才能のある人を募集するための新しい機会と方法を探してきた。
2023年の人材買収チームの重点は、BridgeBioおよびその付属会社の求人需要を満たすことである。私たちは、他のバイオテクノロジーや製薬会社、研究·学術機関、政府実体、コンサルティング·投資会社を含む、私たちの既存かつ潜在的な未来のチームメンバーに雇用機会の選択があることを認識している。最もパフォーマンスの良いチームメンバーを吸引し、維持するために、私たちは自主性、専門成長と影響力を許容する環境を作ることに集中し、同時に競争力のある総奨励方案を提供する。
専門的な発展と業績管理
著者らは定期的なフィードバックと指導及び的確な学習と発展機会を通じて、私たちのチームメンバーの専門発展に投資し、証明された需要を満たす。著者らは5種類の核心品質を構築し、私たちはすべてのBridgeBioチームのメンバーが自分の職責を履行する時に表現することを望んでいる:患者のチャンピオン、企業家経営者、真理探索者、卓越と高品質の実行者を激励する。
54
BridgeBioはすべてのチームメンバーのために半年に1回の業績審査を行い、業績を評価し、これらの属性に対するフィードバックを提供します。フィードバックは優勢と改善機会に集中し、すべてのチームメンバーの専門発展を実現する。年末には、業績評価には自己、同僚、マネージャーのフィードバックが含まれ、正式な格付けも含まれ、業績ボーナス、賃金調整と昇進を含む報酬決定に情報を提供する。
核心的価値観と倫理的道徳
全世界で数百万人が遺伝疾患に悩まされているが,患者数が少なく,業界が早期開発を望まないことは,多くの人にとって治療が到来していないことを意味する。私たちはこのギャップを埋めるために努力している:ビジネス事例と科学的可能性の間、忍耐と希望の間。これは私たちの最初の核心的価値から始まります病人を第一に置くそれは.まだ努力しています独立して考えるそれは.私たちの目標は、他人の考えや意見を事実として簡単に受け入れるのではなく、“なぜ?”と聞くことです。そして“何がいけないの?”私たちは私たちが扱っているすべての問題に厳格で第一原則の考え方をもたらすために努力している。私たちは私たちのすべてのチームのメンバーが考えやフィードバックを持って共有する時に言いたいことを言って、私たちの文化を誇りに思うことを奨励します完全に透明だいつですか それは考えに対する討論と関連がある。独立思考に対する約束は私たちが他の人の考えを考慮することを要求し、もしそれらが最高であることが証明されたら、それらを採択する。私たちはどんな考えも考慮して検証する価値があるという文化を維持するために努力している。知っています1分ごとに重要ですそれは.我々の分散モデルは,個々の資産に集中した専門家チームを利用することにより,発見から治療までの治療を可能な限り速い速度で患者に提供することに取り組んでいる。重大な決定は、不必要なサイクルに時間を無駄にすることなく、それらを最も理解する能力のある人によって行うことができる。そして私たちは科学に話をさせるそれは.私たちのモデルは私たちの計画に対する合理的な評価を促進することを目的としている。1つのプロジェクト運命に関する決定は客観的基準に対する表現によって決定され,潜在薬物ごとの科学的価値の最後の決定権を与える。すべての従業員はこれらの価値観とBridgeBioの商業行為と道徳基準を維持する責任があり、これは私たちの政策と実践の基礎を構成している。
総奨励
トップレベルの人材を誘致し、維持するために、私たちは競争力のある全体的な奨励プログラムを提供する。私たちはすべての直接補償を市場のハイエンドに固定した。私たちは業績ボーナス計画を通じて各従業員の報酬の一部を業績にリンクさせた。所有権意識を創造し、従業員インセンティブを私たちの長期成功と一致させるために、私たちは株式オプションまたは制限株式単位付与および私たちの従業員株式購入計画を通じて、条件を満たす従業員に会社持分を提供する。我々はまた、INDまたはNDA承認のような特定のマイルストーンを達成するために、支店レベルの従業員がコア価値転換点で特定のマイルストーンを達成するように奨励する計画を設計した。
私たちは私たちの福祉を従業員とその直系親族の健康と効率性を維持する重要な分野に集中している。私たちは毎週平均少なくとも30時間働くすべての従業員に心身健康福祉を提供する。私たちはチームメンバーが彼らが必要な時に彼らが必要な時間を利用できるように柔軟な有給休暇政策を持っている。
多様性公平性包括性
私たちは多様性、公平、そして包容的な文化がBridgeBioの成功に必須的だと信じている。私たちは、多様性と包容性のある従業員チームが企業の急務であり、長期的な成功の鍵でもあることを知っているため、私たちの組織内外で独特の声を広めることができ、他人の経験を学ぶことを渇望している。
2022年には,我々の多様性,公平性,包摂性,あるいはDE&I,実行·指導委員会が影響力のある作業を継続し,BridgeBioのDE&Iビジョンを実現する.DE&Iグループ委員会は,関連する重点グループや調査によって発生した問題を解決するための解決策を設計·実施した。委員会の年間組織と開催された多くのDE&I活動の中で,DE&Iランチと学習シリーズ活動を主催し,ヘルスケア臨床試験において代表不足を影響する少数民族の突出した問題に注目し,企業としてどのように自分の力を尽くしてこれらの違いの解決を支援している。
従業員資源グループ(ERG)は依然として私たちのDE&I仕事の基本要素である。2022年、Bridge ERGの女性は会社全体に影響を与え続けた。私たちは講演者シリーズに基づいて、科学界の影響力のある女性を招いて講演を行い、その中に私たちの取締役会のメンバーを含めた。私たちの指導者計画もImpactを推進し続けている。
55
女性のBridgeでの成功は、Asians@Bridgeと呼ばれる2021年第3四半期に第2の従業員資源グループを出現させる。アジア人@Bridgeは2022年に一連の文化,教育,コミュニティ活動を開催し,参加者や参加者から特殊なフィードバックを得た.LGBTQコミュニティを支援する従業員リソースグループの計画も開始しましたPride@Bridgeと呼ばれ2023年1月にスタートしました
新冠肺炎への対応
新冠肺炎の全世界疫病の持続に伴い、私たちは引き続き追加の予防措置を取って、すべての従業員のウィルス暴露リスクを下げ、従業員の健康と安全を第一の位置に置く。2022年の大流行に対する著者らの反応は3つの主要な部分を中心にしている:(1)適切な安全規程;(2)検査要求;(3)ワクチン接種要求。
会社やその他の情報
私たちは2019年にデラウェア州会社として設立されました。名前はBridgeBio Pharma,Inc.です。私たちの主な実行オフィスはカリフォルニア州パロアルト94304号ポート路3160号Suite 250にあります。私たちの電話番号は(650)391-9740です。
私たちのホームページの住所はHttp://Bridge geBio.comそれは.私たちの投資家関係サイトはHttps://Investor.Bridge gio.comそれは.我々は、我々の投資家関係サイト上の“米国証券取引委員会アーカイブ”の下で、我々のForm 10-K年次報告、Form 10-Q四半期報告、Form 8-K現在の報告を無料で提供し、展示品、私たちの役員および上級管理者の第16条の報告、および米国証券取引委員会にこのような材料を準備または提供した後、これらの報告の任意の修正を含む。私たちのウェブサイトのアドレスへの参照は、参照によってウェブサイト上に組み込まれた情報を構成するものではなく、ウェブサイトに含まれる情報は、本ファイルまたは米国証券取引委員会に記録または提供される任意の他のファイルの一部でもない。
56
第1 A項。R.RISK要因
私たちの業務は重大な危険と関連があり、その中のいくつかの危険は以下のように説明される。以下に述べるリスクおよび不確定要因、ならびに本10-K年度報告書に含まれる他のすべての情報は、“経営陣の財務状況および経営成果の検討および分析”および連結財務諸表および関連付記を含むすべての情報を慎重に考慮しなければならない。実際に以下のいずれかのリスクが発生すれば、私たちの業務、見通し、経営業績と財務状況および将来の見通しを損なう可能性があります。この場合、私たちの普通株の市場価格は下落するかもしれません。あなたはあなたの全部または一部の投資を失うかもしれません。私たちは今知らないか、あるいは私たちが現在どうでもいいと思っている他のリスクと不確実性はまた私たちの業務運営を損なう可能性がある。このForm 10-K年次報告書には、リスクと不確実性要因に関する前向きな陳述も含まれている。本年度報告の以下および他の部分に記載されている要因により、我々の実際の結果は、前向き陳述で予想される結果と大きく異なる可能性がある。
私たちの財務状況と成長戦略に関連するリスク
薬物開発は高度に不確実な仕事であり、大きなリスクに関連している。設立以来、私たちは大きな被害を受けており、予測可能な未来には、引き続き大きな被害を受けると予想されています。私たちは設立以来著しい収入が生まれていません。それに、私たちの限られた運営歴史に加えて、私たちの将来の生存能力を評価することが難しくなるかもしれません。
製薬と生物製薬製品開発は投機性の強い仕事であり、大きなリスクに関連している。私たちは新しいビジネス段階のバイオ製薬会社で、運営履歴が限られていますので、それに基づいて私たちの業務や将来性を評価することができます。私たちの子会社は主に初期段階にあるバイオ製薬会社であり、私たちはそれらの成功に大きく依存している。今まで、私たちは主に子会社レベルで私たちの候補製品を識別、獲得し、私たちの製品を開発することに集中して、ほとんどの製品は発見、リード最適化、臨床前或いは臨床開発中です。私たちの候補製品パイプラインは、広範な臨床研究と資源を含む多くの追加の開発時間を必要とし、その後、私たちは追加の規制承認を申請または獲得し、承認されれば、これらの候補製品の販売から収入を発生させることができる。
私たちは利益を上げていません。2015年4月に設立されて以来、毎年赤字になっています。2022年、2021年、2020年12月31日までの年度の純損失はそれぞれ4.847億ドル、5.865億ドル、5.055億ドルだった。2022年12月31日現在、私たちの累計赤字は19億ドルです。私たちは2種類の製品が商業販売、すなわちNULIBRYとTRUSELTIQのために許可されていますが、製品販売は何の重大な収入も発生せず、株式証券の売却、債務融資、特定の資産の売却だけで運営に資金を提供しています。Sentynlは2022年3月にNULIBRYのグローバル権利を購入し,ヘルシンはTRUSELTIQの主要販売先であり,2023年3月31日までTRUSELTIQの販売を中止する。私たちは引き続き大量の研究開発や研究開発、持続的な運営に関する他の費用を発生させ、予測可能な未来に損失が出ることを予想している。また,持続的な新冠肺炎の流行により,我々が行っている臨床試験や計画中の臨床試験および我々が行っている臨床試験に関連するいくつかの研究手順の調整,例えば予備サイト,遠隔医療,家庭訪問および訪問投与を可能にすることは,我々の支出を増加させたり,最初の予想よりも長い時間で支出を枯渇させたりする可能性があると考えられる。さらに、私たちは非臨床実験室活動のために契約研究組織(CRO)と契約製造組織(CMO)と協力して、私たちのサプライチェーンに対する任意の潜在的な短期的影響の変化を軽減することを選択し、最初の予想ではなく、私たちの支出を増加させるかもしれない。私たちはこのような損失が未来に大幅に増加すると予想する。
薬物開発や商業化に関連する多くのリスクや不確実性のため、私たちが支出した時間や金額を予測することはできず、私たちがいつどんな意味のある収入を生み出すことができるのか、あるいは利益を達成したり維持したりすることも予測できない。さらに、米国食品医薬品局、FDAまたは同様の外国の規制機関が、現在予想されている基礎の上で非臨床または臨床前研究または臨床試験を行うことを要求する場合、または現在の上場承認申請の支援または臨床開発の継続を支援するために必要と考えられるデータを他の方法で提供するか、または私たちまたは将来の協力者の臨床試験または私たちが決定する可能性のある候補製品の開発に何らかの遅延が生じた場合、私たちの費用は現在の予想を超える可能性がある。将来のいずれかの候補製品が承認され,コンプライアンス努力が行われていれば,商業化に関連した巨額のコストが生じると予想される。
57
私たちは絶対に適切な薬物を商業化したり、利益を達成することに成功できないかもしれない。すべての製品を販売する収入は、私たちが規制の承認を得たか、または許可を得る可能性のある地域の市場規模、製品の許容可能な価格、任意の価格で補償を受ける能力、および私たちがその地域の商業権を持っているかどうかに部分的に依存するだろう。私たちの成長戦略は私たちが収入を作る能力にかかっている。私たちが未来に利益を達成しても、私たちはその後の時期に利益を維持できないかもしれない。私たちが持続的な利益を達成できなければ、会社の価値を下げ、資金を調達し、業務を拡大し、私たちの研究開発ルートを多様化し、私たちの候補製品をマーケティングする能力を弱めることができます(承認されたら)私たちは私たちの業務を識別して追求したり、継続することができます。私たちのこれまでの損失は、予想された将来の赤字に加え、株主赤字や運営資本に悪影響を与え続けるだろう。
もし私たちが未来にもっと多くの会社の持株権を獲得すれば、私たちの経営業績と普通株価値に悪影響を与え、私たちの業務を乱すかもしれません。
私たちの戦略の一部として、より多くの完全子会社と可変利益実体を設立し、それに投資する予定です。私たちの既存と未来の子会社への投資は多くのリスクに関連しているが、必ずしも限定されるものではない
新たな研究開発計画の作成や既存の研究開発計画の維持に関連する潜在的な買収、許可、投資、または他の取引を正確に評価できない場合、このような取引の予想される収益を達成することができない可能性があり、予想を超えるコストが生じる可能性があり、管理資源および注意が他の必要または価値のある活動から移行する可能性がある。例えば、2021年1月に、私たちはEidos普通株のすべての流通株の買収を完了しました。これらの普通株は以前は私たちまたは私たちの子会社が所有していなかったもので、私たちはEidos合併と呼んでいます。Eidosとの合併やEidosの歴史的業務を我々の業務に統合する際には、各会社の経営陣の一部のメンバーや1社あたりの資源の注意が日常業務運営から移行している。また、私たちはEidosの株主に普通株を発行し、私たちが取引に関連するEidosのいくつかの配当金を負担するため、私たちの株主の利益は希釈された。私たちは未来に他の可能な取引について似たような議論をするかもしれません。これらの取引は私たちが行っている業務の時間と資源を移すかもしれません。また,プロジェクトに関連するコストやリスクの成功可能性の継続的な評価によると,我々は時々,将来的には我々の完全子会社やVIEを通じて,最終的に進めない可能性のある研究·開発計画を実施することも可能である。
58
私たちの候補製品開発に関するリスク
われわれは臨床試験において重大な遅延に遭遇する可能性があり,あるいは予想されるスケジュール内で臨床試験を行うことや完成できない可能性があり,まったくなければ。
臨床テストは高価で時間がかかり、不確実性も存在する。私たちが進行中で計画中のどの臨床試験も計画通りに行われるか、予定通りに完成することは保証できません。もしあれば。また,これらの試験がタイムリーに起動あるいは行われても,このような臨床試験の一時停止や終了を招く可能性があるという問題がある。1つ以上の臨床試験の失敗は、試験の任意の段階で発生する可能性があり、私たちが行っている臨床試験および将来の臨床試験は成功しない可能性がある。成功またはタイムリーな臨床試験の開始または完了を妨げる可能性のあるイベントは、
59
臨床試験の開始や完了に成功できない場合は、私たちの追加コストをもたらしたり、収入を創出する能力を弱める可能性があります。さらに、私たちの候補製品を生産または処方変更した場合、私たちは、修正された候補製品から得られたデータを、これらの候補製品の早期バージョンを使用して行われた非臨床研究および臨床研究によって得られたデータに関連付けるために、追加の非臨床研究または臨床試験を行うことを要求または選択される可能性がある。臨床試験遅延はまた、私たちの候補製品の特許保護の任意の期限を短縮する可能性があり、私たちの競争相手が私たちの前に製品を市場に出すことを可能にすることができ、これは候補製品を商業化することに成功する能力を弱める可能性があり、私たちの業務と運営結果を損なう可能性がある。
進行中または計画中の臨床試験が我々、データ安全監視委員会またはデータ安全監視委員会によって一時停止または終了されれば、我々が行っているacoramidis臨床試験、低用量infigratinibの進行中の第2段階および計画されている第3段階の臨床試験、我々が行っているBBP−418の第2段階および計画されている第3段階の臨床試験、我々が行っているBBP−631の第1/2段階の臨床試験、および我々が行っているEnlaretの2 bおよび第3段階の臨床試験、またはFDAまたは他の規制機関によって行われている遅延に遭遇する可能性もある。または、そのような試験を行っている機関のIRBsが、その臨床研究者およびその審査を受けている場所の参加を一時停止または終了する場合。このような機関は、様々な要素のために臨床試験を一時停止または終了する可能性があり、これらの要素は、監督管理要求または著者らの臨床規程に従って臨床試験を行うことができなかったこと、FDAまたは他の監督機関の臨床試験操作または試験場所の検査の実施による臨床休止、予見できない安全問題または副作用、候補製品の使用のメリット、政府法規または行政措置の変化、または十分な資金が不足して臨床試験を継続することを含む例えば,ATTRIBUTE−CM研究の12カ月目では,アコラミはその主要な終点に到達できなかったにもかかわらず,ATTRIBUTE−CM独立データ監視委員会は,盲目でないデータ審査に基づいて30カ月目まで継続することを提案した。私たちは過去に、将来的にFDAまたは他の規制機関からの臨床保留通知の一部または全部を受信する可能性があり、これらの通知は、将来的により多くの研究、より多くのデータを生成すること、私たちの臨床試験レジメンを修正すること、および/または私たちの臨床試験の開始または継続を延期または停止することを要求する可能性がある。様々な理由で、私たちは、将来的に私たちの1つまたは複数の候補製品を臨床的に放置することを要求または自発的に決定される可能性があり、これは、私たちの臨床開発努力および規制部門の任意のそのような候補製品の承認を得る能力を遅延または他の方法で損なう可能性がある。また,FDAはIND提出を審査した後,被験者のリスクを評価するために必要な十分な情報を提供していないか,あるいは我々が提出したINDが臨床試験の開始をサポートするのに十分ではないと判断する可能性がある。FDAがわれわれの反応に同意することは十分であることは保証されず,FDAが提案した臨床試験の継続を許可する前に,追加の臨床前研究や生産手順が必要となる可能性がある。
60
また,われわれの臨床試験の首席研究員は時々私たちの科学コンサルタントやコンサルタントを務め,このようなサービスに関する報酬を得る可能性がある。場合によっては、私たちはFDAまたは同様の外国規制機関にいくつかの関係を報告することを要求されるかもしれない。FDAや同様の外国の規制機関は結論を出す可能性があり、私たちと主要な研究者との財務関係は利益の衝突をもたらしたり、他の方法でこの研究の解釈に影響を与えたりする。したがって,FDAや同様の外国の規制機関は,適用された臨床試験地点で発生するデータの完全性を疑問視する可能性があり,臨床試験自体の効用が脅かされる可能性がある。これは、FDAまたは同様の外国規制機関が私たちの上場申請の承認を遅延または拒否することを招き、最終的に私たちの1つ以上の候補製品が上場承認を拒否することにつながる可能性がある。
候補製品の開始、進行、または完了を遅延させる任意の臨床試験は、私たちのコストを増加させ、候補製品の開発および承認プロセスを緩和し、製品販売を開始し、そのような候補製品から収入を得る能力を遅延または危険にさらす可能性がある(承認された場合)。さらに、臨床試験の開始または完了遅延をもたらす多くの要因は、最終的には、私たちの候補製品が規制部門の承認を得ることを拒否される可能性もある。もし私たちが他の候補製品を決定した場合、INDやCTAの提出がFDAまたは同様の外国の監督管理機関に直ちに臨床試験を開始することを許可するかどうかを決定することはできない。これらの事件のいずれも、私たちの業務、見通し、財務状況、および運営結果に重大な悪影響を及ぼす可能性があります。
早期研究或いは臨床試験の結果は未来の臨床試験結果を予測できない可能性があり、初歩的な研究或いは臨床試験は著者らの候補製品のために十分な安全性或いは有効性概況を確立できない可能性があり、それによって高度な臨床試験に入る或いは監督管理の許可を申請することは合理的であることを証明する。
非臨床と臨床前研究と臨床試験の結果は後期臨床試験の結果を予測できない可能性があり、臨床試験の中期結果は必ずしも最終結果を予測できるとは限らない。また,我々が獲得したいくつかの候補製品については,われわれ自身が臨床前研究や臨床試験を行っていない。1群の患者や疾患適応で行われた臨床前研究や臨床試験の結果,あるいはわれわれが先頭を切っていない臨床前研究や臨床試験からの結果は,別の群の患者や疾患適応で得られた結果を予測できない可能性がある。いくつかの場合、多くの要素のため、同一候補製品の異なる臨床試験間の安全性或いは有効性結果は有意差が存在する可能性があり、方案中に規定された試験プログラムの変化、患者群の大きさとタイプの差異、投与方案と他の臨床試験方案の変化と遵守状況及び臨床試験参加者の退学率を含む。そのほか、臨床前と臨床データはよく各種の解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると思っているが、まだ発売許可を得られなかった。非臨床研究と初歩的な臨床試験で進展を得たが、臨床試験後期段階の候補製品は必要な安全性と有効性を示すことができないかもしれない例えば、属性−CM研究の12カ月目には、アコラミは、アコラミ群とプラセボ群の平均的に観察された6分間歩行距離または6 MWDがそれぞれ9メートルおよび7メートル低下したため、これら2つの低下幅は、健康な高齢者と同様に、これまで治療を受けていなかったATTR−CMキューよりも低かったが、属性−CM独立データ監視委員会は、非盲目的データ審査に基づいて、30カ月目の終点まで継続すべきであると提案した。製薬と生物製薬業界のいくつかの会社は高度な臨床試験において治療効果の不足或いは不良安全性のために重大な挫折を受け、早期の研究で良好な結果を得たにもかかわらず、私たちが類似した挫折に直面しないことを確定することはできない。早期臨床試験が成功しても、私たちはより多くの患者集団または異なる治療条件下で私たちの候補製品に対して追加の臨床試験を行う必要があるかもしれないし、その後、FDAとアメリカ以外の規制機関の許可を求めて、これらの候補製品をマーケティングし、販売することができる。もし私たちの製品が商業的に実行可能な適応候補製品のマーケティング承認を得られなかった場合、あるいは全く承認されなかった場合、私たちの業務、将来性、財務状況、運営結果に深刻な損害を与えます。
61
また,これまで,候補製品に対するいくつかの臨床試験は開放ラベル研究の形で設計されており,限られた数の臨床地点で限られた数の患者に対して行われてきた。オープンタグ“臨床試験”とは、患者および研究者の両方が、患者が研究製品候補を受け入れているかどうかを知っているかどうか、または既存の承認薬またはプラセボを意味する。最も典型的には,オープンラベル臨床試験は候補の研究製品のみをテストし,異なる用量レベルで試験を行う可能性がある。開放ラベル臨床試験は様々な制限を受けており,これらの制限は任意の治療効果を誇張する可能性があり,開放ラベル臨床試験中の患者が治療を受ける際に知られているからである。オープンラベル臨床試験は“患者偏見”の影響を受ける可能性があり,すなわち患者が症状が改善したと考えているのは,実験的治療を受けていることを意識しているだけである。また,早期臨床試験に選ばれた患者には,通常最も重篤な患者が含まれており,新たな治療法が採用されているにもかかわらず,症状は必ず改善する可能性がある。また,オープンラベル臨床試験は,“調査者偏見”の影響を受ける可能性があり,すなわち臨床試験の生理結果を評価·審査する人は,どの患者が治療を受けているかを知り,その知識を知っている場合に治療群の情報をより有利に解釈することが可能である。我々のアコラミの第二段階臨床試験は、開放ラベル臨床試験の拡張と、軟骨発育不全児童を治療する小用量infigratinibの第二段階用量増加と拡大研究、またはPROPEL 2を含むことを考慮すると、開放ラベル試験として設計されている, これらの臨床試験の結果は、これらまたは他の候補製品の将来の臨床試験結果を予測できない可能性があり、制御された環境においてプラセボまたは能動対照を用いて研究を行う場合、われわれは開放ラベル臨床試験を含む。
患者を募集して臨床試験に参加することは困難である可能性があるため,臨床開発活動が延期されたり,他の悪影響を受けたりする可能性がある。
臨床試験方案に基づいて適時に臨床試験を完成し、他の事項以外に、試験が終了するまで十分な数の患者を募集する能力があるかどうかに依存する。我々は現在の候補製品の適応を評価する予定であり、1種の稀な疾病或いは疾病を代表し、患者の人数は限られており、これらの疾病或いは疾病の中から臨床試験の参加者を引き付けることができる。メンデル疾患および遺伝子駆動癌を治療する候補製品の開発に焦点を当てているため、その多くはまれな疾患であるため、十分な数の患者、または必要なまたは必要な特徴および標準を有する患者をタイムリーに識別し、募集することができない可能性がある。
様々な理由から、臨床試験で患者を募集する際に困難に遭遇する可能性があります
計画通りの臨床試験を行うのに十分な数の患者を募集することが困難であれば,進行中や計画中の臨床試験を延期または終了する必要がある可能性があり,いずれも業務に悪影響を及ぼす。
62
私たちの候補製品の使用は、副作用、有害事象または他の特性または安全リスクに関連する可能性があり、これはその臨床開発を遅延または停止し、その規制承認を阻止し、私たちが臨床試験を一時停止または停止させ、製品または候補製品を放棄し、候補製品の商業潜在力を制限し、承認された場合、あるいは他の深刻な負の結果を招く可能性があり、私たちの業務、将来性、運営結果、および財務状況を深刻に損なう可能性がある。
薬品の一般的な場合と同様に,我々の候補製品の使用に関する副作用や有害事象や有害事象がある可能性がある.われわれの臨床試験結果は副作用や予期せぬ特徴の重症度と流行度を示す可能性がある。私たちの候補製品による副作用は、私たちまたは規制機関の臨床試験の中断、延期、または停止を招く可能性があり、より厳しいラベル、またはFDAまたは同様の外国の規制機関が規制承認を延期または拒否する可能性がある。私たちの候補製品の薬物関連副作用は、患者の募集或いは患者の試験完了能力に影響を与えるか、あるいは潜在的な製品責任クレームを招く可能性がある。このようなどんな状況でも、私たちの業務、財政状況、そして見通しに大きな被害を及ぼす可能性がある。
さらに、私たちの候補製品が臨床前研究または臨床試験において副作用に関連している場合、または予期しない特徴を有する場合、それらの開発を放棄することを選択するか、またはそれらの開発をより狭い用途または集団に制限することを選択する可能性があり、有害副作用または他の特徴は、リスク効果の観点からは、それほど一般的ではなく、それほど深刻ではなく、またはより容易に受け入れられ、これは、候補製品の商業的期待を制限する可能性がある。私たちはまた、私たちの臨床前研究または臨床試験の結果に基づいて、私たちの研究計画を修正または中止することを要求されるかもしれない。例えば,我々のinfigratinibがFGFR駆動癌を治療する第2段階臨床試験では,任意のレベルの緊急AEを治療する最も一般的な治療は高リン血症であり,電解質障害であり,血中リン酸塩レベルが上昇する。候補製品の実施および将来の臨床前研究および臨床試験で観察される可能性のあるこれらおよび他の副作用は、影響を受けた候補製品または影響を受けた候補製品と同じ特性を有する他の候補製品の開発計画を延期、修正または放棄することを要求するかもしれない。早期テストでは最初に希望を示した候補製品の多くが,その後副作用が発見され,さらなる開発を阻害する可能性がある。既存の候補製品を推進し、新たな候補製品を決定しようと努力すると、早期試験において最初に所望の後続試験または候補製品を示す試験が、類似または異なる許容できない副作用をもたらすことが発見されず、それらのさらなる開発を阻害することは決定できない。
私たちがより大きく、より長い時間、そしてより広い臨床試験で私たちの候補製品を試験する場合、または私たちの候補製品の使用(規制部門の承認を得た場合)がより一般的になるにつれて、被験者は、早期試験で観察された疾患、傷害、不快感、および他の副作用、および以前の試験で発生しなかったか、または検出されなかったことを報告する。これらの副作用が開発後期または承認後に知られれば、これらの発見は私たちの業務、財務状況、および将来性に大きな損害を与える可能性がある。
また、他の人が行った薬物や生物製薬製品の臨床試験の不利な発展は、FDAまたは他の監督監督機関が私たちの臨床試験を一時停止または中止させ、私たちの任意の候補製品に対する承認要求を変更する可能性がある。
候補製品による副作用に加えて,投与過程や関連プログラムも副作用を引き起こす可能性がある。もしこのような副作用が発生した場合、候補製品に対する臨床試験は一時停止または終了する可能性がある。もし私たちがどんな副作用が管理手続きまたは関連手続きによって引き起こされたかを証明できない場合、FDA、欧州委員会、EMA、または他の規制機関は、任意またはすべての目標適応の候補製品のさらなる開発を停止するか、または承認を拒否するように命令することができる。将来のすべての深刻な有害事象やSAEが製品に関連していないことを証明できても,このような事件は患者の募集や患者の試験完了能力に影響を与える可能性がある。さらに、もし私たちが私たちの任意の候補製品の任意の将来の臨床試験を開始、延期、一時停止、または終了しないことを選択または要求された場合、その候補製品の商業的将来性が損なわれる可能性があり、これらの候補製品から製品収入を創出する能力は延期またはキャンセルされる可能性がある。これらの状況のいずれも、他の候補製品を開発する能力を損なう可能性があり、私たちの業務、財務状況、および将来性を深刻に損なう可能性があります。
63
さらに、私たちの任意の候補製品が発売承認された場合、FDAは、私たちの製品ラベルにボックス警告を適用することができ、リスク評価および緩和戦略、またはREMSを採用することを要求することができ、製品の利益がそのリスクよりも大きいことを保証するために安全使用を保証する要素を適用することができ、その中には、患者に配布し、医療従事者に計画を伝播するための製品リスクを概説する薬物ガイドラインが含まれている可能性がある。さらに、もし私たちまたは他の人が後に私たちの候補製品が承認されると不良な副作用が発生することを発見すれば、いくつかの潜在的な重大な負の結果を招く可能性がある
これらの状況のいずれも、特定の製品または候補製品に対する市場受容度を達成または維持することを阻止することができ、承認されれば、私たちの業務、財務状況、および将来性を深刻に損なう可能性がある。
我々のいくつかの候補製品は、重篤な合併症を有する患者集団の治療のために開発されており、これらの患者は、死亡または深刻な不良または許容できない副作用を引き起こす可能性があり、私たちの臨床開発活動を放棄または制限する必要がある。
我々は、FGFR駆動癌に対するinfigratinib臨床試験、およびRASおよびRTK変異患者に対するBBP-398単一療法用量の増加および拡大臨床試験、および私たちが開発する可能性のある他の候補製品の治療を受ける可能性のある患者を含むいくつかの候補遺伝子駆動癌臨床試験を行っており、彼らの疾患の治療中にも化学療法、放射線および/または他の高用量または清髄治療を受ける可能性があり、したがって、死亡を含む私たちの候補製品とは無関係な副作用や副作用に遭遇する可能性がある。これらの副作用或いは副作用は著者らの候補製品と関係がないかもしれないが、それらは依然として著者らの臨床試験の成功に影響する可能性がある。重篤な患者を著者らの臨床試験に組み入れることも死亡或いはその他の不良医療事件を招く可能性があり、原因は潜在疾病或いはこのような患者が受ける可能性のある他の治療或いは薬物である。これらの事件のいずれも、私たちが臨床開発を通じて私たちの候補製品を推進し、規制部門の承認を得ることを阻止し、候補製品を商業化する能力を弱めるかもしれない。臨床開発を通じて私たちの候補製品を推進できないいかなる状況も、私たちの業務、財務状況、運営結果と将来性を損なう可能性があります。
私たちが時々発表したり公表したりする臨床試験の一時的、“トップライン”および初歩的なデータは、より多くの患者データの取得またはより多くの分析に伴って変化する可能性があり、これらのデータは監査と検証プログラムの影響を受け、これは最終データの重大な変化を招く可能性がある。
私たちは時々私たちの臨床試験の中期、“頂線”あるいは初歩的なデータを公表するかもしれない。著者らが完成する可能性のある臨床試験の中期データは、患者登録の継続とより多くの患者データの獲得に伴い、1つ以上の臨床結果が実質的に変化する可能性があるというリスクに直面している。予備データや“トップライン”データはまだ監査と確認手続きを受ける必要があり、これは最終データが以前に公表された予備データと大きく異なる可能性がある。したがって、最終データを得る前に、中期と予備データを慎重に見なければならない。初歩的、“トップライン”または中間データと最終データとの間の重大な不利な変化は、私たちの業務、財務状況、運営結果、および将来性を深刻に損なう可能性がある。
64
私たちの候補製品の審査と承認に関連するリスク
私たちの候補製品は臨床前或いは臨床開発段階にあり、これは長くて高価な過程であり、結果が確定せず、重大な遅延が出現する可能性がある。私たちは私たちのどの候補製品も規制部門の承認を得ることができるという保証はありません。これはそれらが商業化できる前に必要です。
規制部門の許可を得て私たちの候補製品を販売する前に、私たちは広範な臨床試験を行い、候補製品の人体上の安全性と有効性を証明しなければならない。今まで、著者らのほとんどの努力と財政資源は著者らの候補製品を確定、獲得と開発に集中し、手がかり最適化、非臨床研究、臨床前研究と臨床試験を行い、そしてこれらの業務に一般と行政支持を提供する。実際にあれば,どの臨床試験が予定通りに行われているかどうかは確認できない。著者らは臨床前と臨床開発に成功できず、著者らの追加コストを招き、著者らの創造能力に負の影響を与える可能性がある。私たちの将来の成功は、私たちが開発に成功し、規制部門の承認を得て、候補製品を商業化する能力にかかっている。私たちは以前2種類の製品の販売が許可されましたが、私たちは薬品販売から相当な収入を得ていません。私たちは決して適切な薬の商業化に成功できないかもしれません。
私たちのすべての候補製品は追加的な開発;臨床前、臨床、そして製造活動の管理、そして規制部門の承認が必要だ。さらに、私たちは十分な製造供給を得ること、商業組織の建設を完成させること、候補製品に対するマーケティング努力を開始すること、そのような候補製品の商業製品販売から任意の重大な収入を得る前に、精算を得る必要があるだろう。私たちの多くの候補製品は早期研究や開発の転換段階にあり、これらの計画が失敗するリスクが高い。私たちは私たちの候補製品が臨床試験で成功したり、監督部門の許可を得たりするかどうかを確認できない。また,我々の候補製品は臨床試験で成功しても,規制部門の承認を得られない可能性がある。もし私たちの候補製品が規制部門の承認を得られなければ、私たちと私たちの子会社は運営を継続できない可能性があり、これは私たちが子会社を徐々に閉鎖·解散し、許可技術を売却したり、あるいは代替戦略を求めたりする可能性がある。
私たちが識別し開発する可能性のある任意の候補製品に対する1つ以上の司法管轄区域の規制承認を得ることができなければ、私たちの業務は深刻な損害を受けるだろう。
適切な規制機関が候補製品を審査して承認するまで、私たちは製品を商業化することができない。FDAと同様の外国規制機関の承認は長く予測不可能であり、規制機関のかなりの自由裁量を含む多くの要因に依存する。承認政策、法規、または承認を得るために必要な非臨床または臨床データのタイプおよび数量は、候補製品の開発中に変化する可能性があり、司法管轄区域によって異なる可能性があり、これは、承認の遅延または承認申請の不承認の決定をもたらす可能性がある。FDAの規制を受けていましたがEUropean Medicines Agency、またはEMA、およびイスラエル保健省、NULIBRY(Fosdenopterin)、モリブデン補助因子欠乏、またはMoCD、A型患者の死亡リスクを低減するための、FDAとカナダ保健省、TRUSELTIQ(Infigratinib)、過去の治療のための、切除できない局所末期または転移性胆管癌、FGFR 2融合または他の再配置、私たちの現在の候補製品と私たちが将来開発する可能性のある任意の他の候補製品は、監督部門の許可を得ることができないだろう。私たちは私たちのすべての候補製品が規制部門の承認を受けるか、あるいは承認されれば、私たちの任意の候補製品が商業化に成功すると確信できない。
上場承認を得ることは広く、長く、高価で、本質的に不確定な過程であり、監督管理機関は様々な理由で私たちの候補製品の承認を延期、制限、または拒否する可能性があるが、これらに限定されない
65
さらに、NDA、BLA、または他の提出された規制承認が提出されて審査を受けても、FDAまたは同様の規制機関は、審査または承認プロセスを延期するか、または様々な理由で規制承認の承認を拒否する可能性がある。長い承認過程と、臨床試験結果の予測不可能性と変化する規制要求は、米国や他の場所で求められる可能性のある候補市場製品に対する規制機関の承認を得ることができず、これは私たちの業務、将来性、財務状況、運営結果を深刻に損なう可能性がある。
私たちの臨床試験は、私たちが識別し、追求する可能性のある候補製品の安全性と有効性の実質的な証拠を証明できないかもしれないが、これは規制承認と商業化の範囲を阻止、延期、または制限するだろう。
私たちの候補製品の商業化販売が規制の承認を得る前に、私たちは長い、複雑で高価な非臨床研究、臨床前研究、臨床試験を通じて、適用された候補製品が各目標適応で使用されることが安全で有効であり、もし私たちの候補製品が生物製品として規制されていれば、その候補製品は安全で純粋かつ有効であり、その目標適応に使用することができることを証明しなければならない。各候補製品は、その目標患者集団およびその目標用途において十分なリスクおよび収益状況を証明しなければならない。
臨床試験費用は高価であり,完成まで数年かかる可能性があり,その結果自体も確定していない。失敗は臨床開発過程のいつでも起こるかもしれない。臨床試験を開始した候補製品の多くは規制機関の商業化承認を得たことがない。著者らは設計臨床試験の経験が限られており、臨床試験を設計と実行して未来の市場承認を支持できないかもしれない。
66
私たちの現在の臨床試験あるいは他の未来の臨床試験が成功するかどうかは確認できない。さらに、我々の目標適応のいずれの臨床試験で観察される任意の安全問題も、これらの適応および他の適応において規制部門の承認を得る見通しを制限する可能性があり、これは、私たちの業務、財務状況、および運営結果に大きな悪影響を及ぼす可能性がある。また,このような臨床試験が成功しても,FDAや同様の外国規制機関が我々のように結果を解読することは保証されず,承認のための候補製品を提出する前に,より多くの試験が必要となる可能性がある。特定の適応の臨床試験で成功することは,候補品が他の適応でも成功することは確保できない。同様に,特定の適応で1つの候補を承認することは,その候補が他の適応でも成功することを確保することはできない.さらに、一方の法域が許容可能な支持承認の結果は、別の規制機関によって、別の法域の規制承認を支持するのに十分ではないと考えられる可能性がある。試験結果がFDAまたは同様の外国規制機関を満足させてマーケティング申請を支援することができない場合、私たちの候補製品の潜在的な承認を支援するために、追加の試験を行うために、入手できない可能性のある大量の資源を費やしてしまう可能性がある。候補製品の規制承認を得ても、このような承認された条項は、特定の候補製品の範囲や用途を制限する可能性があり、その商業的潜在力を制限する可能性がある。
米国以外の候補製品に対して臨床試験を行っており,FDAや類似の外国規制機関はこのような試験のデータを受け入れていない可能性がある。
私たちは現在ヨーロッパを含めてアメリカ以外で臨床試験を行っている。例えば,われわれのアコラミ3期臨床試験には米国以外の患者が含まれている。FDAや同様の外国規制機関が米国や他の管轄地域以外で行われている臨床試験を受けた研究データは,何らかの条件によって制限される可能性があり,まったく受け入れられない可能性もある。外国の臨床試験からのデータが米国の上場承認の基礎となることを意図している場合、FDAは通常、(I)このデータが米国の人口および米国の医療実践に適用されない限り、外国のデータのみに基づいて申請を承認することはない。(Ii)この試験は公認能力を有する臨床研究者によって行われ、GCP規定に適合する。および(Iii)データは、FDAによる現場検査を必要とすることなく、有効であると考えることができ、または、FDAがそのような検査を行う必要があると考える場合、FDAは、現場検査または他の適切な手段でデータを検証することができる。そのほか、十分な患者群と統計能力を含むFDAの臨床試験要求を満たさなければならない。多くの外国の規制機関もまた似たような承認要求を持っている。また、このような外国裁判は、裁判を行う外国司法管轄区域に適用される現地法によって管轄される。FDAや同様の外国規制機関が米国や適用司法管轄地域以外で行われている試験からのデータを受け入れることは保証されておらず,我々が行っていることや計画中のアコラミ臨床試験のデータを含めて,米国以外でキューを募集している。FDAまたは同様の外国の規制機関がそのようなデータを受け入れなければ、追加の試験が必要になるだろう, これは高価で時間がかかり、私たちの業務計画を遅延させる側面であり、私たちが開発する可能性のある候補製品が適用される司法管轄区域で商業化の承認や許可を得られない可能性がある。
現在米国での候補製品がFDAの承認を得ても,これらの候補製品のいずれかを米国以外の場所で商業化することは決して承認されない可能性があり,すべての市場潜在力を実現する能力を制限するであろう。
米国以外の市場で任意の製品を販売するためには、安全と有効性に関する他の国の多くの規制要件を確立し、遵守しなければならない。一国で行われる臨床試験は、他の国の規制部門に受け入れられない可能性があり、一国の規制承認は、他のどの国でも規制承認を受けることを意味するものではない。承認の流れは国/地域によって異なり、1つの管轄区域で行われる臨床試験は、他の管轄区域の規制機関によって受け入れられない可能性があるので、追加の製品試験および検証、および米国とは異なる追加または異なる行政審査期間が関連する可能性がある。米国以外の多くの管轄区では、候補製品は先に精算許可を得てから、その管轄区で販売を許可することができる。場合によっては、私たちの製品のための価格を取るつもりで、承認されると、承認される必要もあります。
67
外国の監督管理機関の承認を求めることは困難とコストを招く可能性があり、追加の非臨床研究或いは臨床試験が必要であり、これは高価で時間がかかる可能性がある。各国の規制要求には大きな違いがある可能性があり、私たちの候補製品がこれらの国で発売されることを延期または阻止する可能性がある。外国規制機関の承認過程には、FDA承認の取得に関連するすべてのリスクが含まれる可能性がある。以前は2つの製品がアメリカでの販売が許可されていましたが、国際市場での販売を許可された候補製品は何もありませんでしたし、国際市場で規制の承認を受けた経験もありません。もし私たちが国際市場の規制要求を遵守できなかったり、必要な承認を得られなかったり、あるいは国際市場の規制承認が延期された場合、私たちの目標市場は減少し、私たちは任意の承認された製品のすべての市場潜在力を達成する能力が損なわれるだろう。
私たちの候補製品に孤児薬物指定を申請することができても、私たちはこのような指定や孤児薬物状態に関連する福祉を維持することができず、孤児薬物マーケティング排他性を含む可能性がある。
私たちの業務戦略は遺伝病を治療する候補製品の開発に集中しており、これらの製品はFDAまたはEMA孤児薬物の指定資格に適合する可能性がある。米国とEUを含むいくつかの司法管轄区の規制機関は、比較的少ない患者集団の薬物または生物製品を孤児薬物として指定する可能性がある。孤児医薬品法によれば、薬物がまれな疾患または疾患の治療を目的としている場合、FDAは孤児薬として指定することができる。このような疾患または疾患は、一般に、米国では年間患者数が20万人未満であるか、または米国では患者数が20万人を超えると定義されているが、米国では、米国での販売によって薬剤開発コストを回収することは合理的に期待されていない。孤児薬物指定を得るためには,秘密保持プロトコルやBLAを提出する前に申請しなければならない。米国では,臨床試験費用に贈与資金を提供する機会,税収割引,ユーザ費用減免など,孤児薬を指定して一方が財政的インセンティブを得る権利がある。FDAが孤児薬物指定を承認した後、FDAは、この薬剤の模倣薬識別情報およびその潜在的孤児用途を開示する。孤児薬物指定は規制審査と承認過程でいかなる利点も伝達されず、規制審査と承認過程の持続時間を短縮することもない
孤児薬物の称号を有する製品がその後、孤児薬物の称号を有する疾患に対するFDAの最初のFDA承認を得た場合、この製品は、孤児製品の排他性を得る権利があり、これは、FDAがNDAまたはBLAを含む他の出願を7年以内に承認しない可能性があり、同じ適応で同じ薬物または生物学的製剤を販売することを意味する。限定された場合でなければ、孤児薬物排他性を有する製品に対する臨床的優位性を示すように、またはFDAは、指定された薬物の疾患または状態を有する患者の需要を満たすために十分な数の孤児薬を得ることができることを証明していないことを発見する。したがって、私たちの候補製品が孤児専門権を獲得しても、FDAは、同じ適応または疾患の治療のための、または異なる適応または疾患を治療するための他の医薬または生物学的製品を専門期間内に使用することを許可することができる。さらに、私たちが十分な製品供給を生産できない場合、FDAは孤児の独占特許を放棄することができ、あるいはその後の申請者が私たちの製品よりも良い臨床的優位性を示す場合、孤児薬物独占特許を克服することができる。
EUでは、EMAの孤児医薬製品委員会は、(1)生命または慢性衰弱に危険な疾患の診断、予防または治療を目的とした製品に孤児の称号を付与する場合、(2)申請を行うと、EUで10,000人中5人以下の影響を与える場合、または(2)孤児のアイデンティティによるメリットがなければ、その開発に必要な投資が合理的であることを証明するためにEUで十分な見返りを生じる可能性が低い。および(3)このような疾患を満足できる診断、予防または治療方法がEUで販売されていないことを許可しなければならないか、または、そのような方法があれば、製品はその疾患の影響を受けている人に大きな利益を与えるであろう。
68
甲状腺ホルモンアミロイドーシスの治療のためのアコラミ、軟骨発育不全の治療のための小用量infigratinib、常染色体優性遺伝性低血カルシウム症(ADH 1型およびADH 2型を含む)の治療のためのEnlaret、21 OHDによるCAHの治療のためのBBP-631、Canavan病の治療のためのBBP-812、PKANおよびPAの治療のためのBBP-671を含む孤児医薬名がFDAから取得されている。我々は、ATTRアミロイドーシスの治療のためのアコラミ、軟骨発育不全の治療のための小用量infigratinib、21 OHDによるCAHの治療のためのBBP−631、肢体帯状筋ジストロフィーの治療のためのBBP−418、Canavan病の治療のためのBBP−812、PKANおよびPAを治療するためのBBP−671、副甲状腺機能低下症(ADH 1を含む)を治療するためのEnlaretをEMAおよび欧州委員会から取得した。私たちは他の候補製品のための孤児薬物の称号を求めるかもしれない。孤児薬物指定を受けても,孤児指定適応よりも広い適応の承認を求めると,米国での独占営業権が制限される可能性があり,FDAが後に指定請求に重大な欠陥があると判断した場合,まれな疾患や疾患を有する患者の需要を満たすのに十分な数の製品が保証されない場合,あるいは後続申請者がその臨床的優位性がわれわれの製品を超えていることを証明した場合,独占営業権を失う可能性がある。また、私たちは他の候補製品のための孤児薬の指定を求めるかもしれないが、私たちは決してそのような指定を受けないかもしれない。私たちの候補製品を獲得、維持、または他の方法で認識できなかった孤児薬物指定の利点は、私たちの将来性に実質的な悪影響を及ぼす可能性がある。
2017年8月3日、国会はFDA 2017年再認可法案、FDARAと略称することを可決した。その他の事項以外に、FDARAはFDAの以前に存在した監督管理解釈をスポンサーに1種の孤児薬物の臨床優位性を証明することを要求し、この薬物は他の方面で以前に許可された同じ稀な疾病を治療する薬物と同じであって、孤児薬物の排他性を得ることができる。この立法は以前の前例を覆し、即ち“孤児薬品法”はその臨床優勢にかかわらず、FDAに孤児排他期を認めることを明確に要求した。また、2021年の“総合支出法案”では、FDARA編纂の解釈がFDARA発行前に孤児指定が発行されたが、製品承認がFDARA公布後に適用されることが明らかになったため、国会はこの解釈をさらに変更しなかった。FDAは孤児薬物法とその規制と政策をさらに再評価するかもしれない。FDAがいつ、どのように孤児薬の法規や政策を変えるかどうかもわかりませんし、どのような変化が私たちの業務に影響を与える可能性があるかどうかもわかりません。FDAがその孤児の薬物法規や政策に変化する可能性があることによって、私たちの業務は悪影響を受ける可能性がある。
FDAはPKANとPAのまれな小児科疾患の治療にBBP-671を許可し、軟骨発育不全の治療に小用量infigratinib、BBP-812をCanavan病の治療に応用した。しかしながら、BBP−671のマーケティング申請が承認された場合、優先審査証明書の資格基準を満たしていない可能性がある。
FDAはまれな小児科疾患を BBP-671はPKANおよびPAの治療に使用され、小用量のinfigratinibは軟骨発育不全の治療に使用され、BBP-812はCanavan病の治療に使用される。稀な小児科疾患を治療する薬剤として薬物を指定することは、申請が承認されたときに、小児科まれな疾患の優先審査証明書を得る資格基準に適合することを保証することができない。連邦食品、薬品、化粧品法案(FDCA)によると、私たちは元のNDAでBBP-671の珍しい小児科疾患優先審査証明書を申請する必要がある。FDAは、BBP−671、低用量infigratinib、またはBBP−812のいずれかのNDAが承認された場合、優先審査証明書の資格基準に適合しないことを決定することができる
69
FDAが2024年9月30日または以前に小児科稀疾患指定を取得した薬物および生物製品に対して稀な小児科疾患優先審査券を付与する権限は現在、2024年9月30日または以前に小児科稀疾患指定を獲得した候補者に限られており、FDAは2026年9月30日までに稀な小児科疾患優先審査券を付与することしかできない。しかし、議会はFDAが稀な小児科疾患に資格証明書を優先的に審査する権限をさらに延長する可能性がある。そのような延期がない場合、BBP−671、低用量infigratinib、またはBBP−812のNDAがいかなる理由でも2026年9月30日までに承認されなければ、小児科まれな疾患優先審査券の基準に適合するか否かにかかわらず、優先審査券を取得する資格がないであろう。
FDAの加速承認は、私たちのどの候補製品も承認されても、より速い開発や規制審査や承認過程をもたらさない可能性があり、私たちの候補製品が上場承認される可能性を増加させることはありません。
私たちはFDAの加速された承認手続きを使用して私たちの候補製品の承認を求めるかもしれない。FDAの承認手続きを加速させることにより、適用された場合により多くの候補製品の承認を求めることができる。このアプローチは、より速い開発、規制審査、あるいは承認過程をもたらすことができず、私たちの候補製品が上場承認される可能性を増加させることもないかもしれない。1つの製品が深刻または生命に危険な疾患を治療する場合、通常、既存の治療方法よりも有意な利点を提供し、臨床的利益を合理的に予測する可能性のある代替終点に影響を与える場合、製品は加速承認を得る資格がある可能性がある。承認の一つの条件として,FDAは承認を加速させた製品のスポンサーに十分かつ良好な制御を行う上場後検証性臨床試験を要求する可能性がある。このような検証的な実験は職務調査の方法で行われなければならない。FDAは、2022年の食品·薬物総合改革法案によれば、承認前または承認加速日後の特定の期間内に1つまたは複数の承認後検証試験を行うことを適宜要求することが許可されている。FDORAは,登録目標の実現を含めて180日ごとにFDAにこのような研究の最新状態を送信することも求められており,FDAはこれらの情報を迅速に公開しなければならない。また、FDORAによれば、FDAは、例えば、職務調査を行っていない会社に対していかなる承認後の確認試験を行ったり、速やかに当該機関に進捗報告を提出した会社に罰金を科すかのような行動をとる権利がある。また、承認の加速を検討している製品については、FDAは、その機関が別の要求をしない限り、現在要求している, 審査期間内に、上場承認後120日以内に伝播または出版しようとしている宣伝材料を機関に提出し、製品商業発売の時期に悪影響を及ぼす可能性がある。したがって,加速的な承認経路の利用を求めても,加速的な承認を得ることができない可能性があり,加速された承認を得ても,その製品のより速い開発,規制審査,承認過程を体験できない可能性がある.また,加速承認を得ることは,製品の加速承認が最終的に従来の承認に変換される保証はない.
FDAが画期的な治療、高速チャネル、または再生医学高度治療によって指定された候補製品に使用可能な任意の加速開発または規制審査および承認手続きを選択または使用できない可能性がある。
私たちは、私たちの候補製品がどんな加速開発経路を得る資格があるかどうか、あるいは規制機関が関連する合格指定を付与または維持することができるかどうかを決定することができないにもかかわらず、FDAと規制戦略について継続的な議論を続けていきたい。私たちが追求できる潜在的な加速発展の道は、突破的治療、迅速チャネル指定および/または再生医学高度治療、またはRMATを含む。
70
画期的な治療指定は、深刻または生命を脅かす疾患を治療するための候補製品の設計の開発および検討を加速することを目的としており、“初歩的な臨床証拠は、薬物が1つまたは複数の臨床的に重要な終点で、例えば臨床開発早期に観察される実質的な治療効果のような既存の療法よりも実質的な改善を示す可能性があることを示す”としている。候補製品を画期的な治療法に指定することは,FDAとより頻繁な会議を行い,候補製品の開発計画を検討し,承認を支援するために必要な適切なデータの収集を確保すること,臨床試験の設計やバイオマーカーの使用を提案することなどについてFDAとより頻繁に書面通信を行うこと,効率的な薬物開発計画の密集指導,最初に第1段階から開始すること,高度管理者に関わる組織約束,審査と優先審査の資格を転動することを含む潜在的な利点を提供している。
迅速チャネル指定は、深刻なまたは生命を脅かす疾患または状態を治療するための候補製品のために設計されており、この場合、非臨床的または臨床的データは、このような疾患または状態が満たされていない医療需要を満たす可能性があることを示す。
私たちは私たちの1つ以上の候補製品のためにRMAT認証を求めるかもしれない。2017年、FDAはRMATを“21世紀治療法”を実施する一部として指定し、以下の基準に適合する任意の薬物の検討を加速する:細胞療法、治療用組織工学製品、ヒト細胞および組織製品、またはこのような治療法または製品を使用する任意の組み合わせ製品であるRMATの定義に適合し、治療、修正、逆転または治癒を目的としている;深刻または生命に危険な疾患または状態を治療、修正または治癒することを目的としている;このような疾患または状態が満たされていない医療需要を満たす可能性があることを初歩的な臨床証拠は示す。RMAT指定は,画期的な治療指定と同様に,FDAとの会議で候補製品の開発計画をより頻繁に検討することや,スクロール審査や優先審査を行う資格があるかどうかを含む潜在的な利点を提供する。RMAT認証が付与された製品も、長期的な臨床的利益を合理的に予測する可能性のある代替物または中間終点に基づいて、またはより多くの場所に拡張することによって加速承認を得ることを含む、大量の場所から取得されたデータに依存する資格がある。加速的承認を得たRMAT指定製品は、電子健康記録のような臨床証拠、臨床試験、患者登録、または電子健康記録のような他の真の証拠源を提出することによって、より大きな検証データセットを収集することによって、または治療を承認する前に、そのような治療を受けたすべての患者を承認後に監視することによって、承認後の要求を満たすことができる。
我々のいくつかの候補製品は、FDAの高速チャネル認証を取得した製品を含むが、他の候補製品に対していかなる画期的な治療、迅速チャネル、またはRMAT認証を行わないかを選択することができ、FDAは広範な自由裁量権を有し、これらの認証を付与するかどうかを決定することができる
ある特定の候補製品が画期的な治療,迅速チャネル指定,RMATを得る資格があると信じていても,FDAがそれを承認することを決定する保証はない。画期的な治療指定、迅速チャネル、およびRMAT指定は、製品承認の基準を変更することはなく、そのような指定または資格が迅速審査または承認をもたらすことを保証することもできないし、承認された適応が突破的治療、高速チャネルまたはRMAT指定によってカバーされる適応範囲よりも狭くならないことを保証することはできない。したがって,画期的な治療,迅速チャネル,RMAT指定を受けても,従来のFDAプログラムよりも速い開発過程,審査,承認を経験しない可能性がある。FDAがこの製品がもはや資格基準を満たしていないと思う場合、突破的治療、迅速チャネル、またはRMAT指定を撤回する可能性がある。もし私たちがこのような他の開発と規制を加速させる方法を利用できなければ、私たちの業務は損害を受けるかもしれない。
そのほか、ある腫瘍学候補製品はリアルタイム腫瘍学審査(RTOR)試験計画の審査を受ける資格がある可能性があり、この計画はFDA腫瘍学卓越センターの提案であり、患者への安全かつ有効な癌治療の提供を加速することを目的としている。この計画は、FDAがより早くデータを審査することを可能にするが、申請者が完全な申請を正式に提出する前に、RTORパイロットの受け入れは、FDA審査員が通常行われる利益-リスク評価に依存し、FDAのPDUFAスケジュールにも影響を与えない。
71
私たちは、私たちのプラットフォーム技術を指定プラットフォーム技術として指定することを求めるかもしれませんが、私たちはこのような指定を受けないかもしれません。たとえそうしても、そのような指定は、より速い開発や規制審査や承認過程をもたらすことはありません。
私たちは私たちのプラットフォーム技術を指定されたプラットフォーム技術として指定することを求めることができる。FDORAによれば、以下の場合、薬物または生体内に組み込まれるか、またはそれによって使用されるプラットフォーム技術は、指定されたプラットフォーム技術として指定される資格がある:(1)プラットフォーム技術がNDAまたはBLAによって承認された薬物使用に組み込まれているか、または許可された薬物の発起人によって提出された予備証拠、または薬物出願において提出されたデータの参照権を付与された発信者によって提出された予備証拠は、プラットフォーム技術が品質、製造または安全に悪影響を与えることなく、1つまたは複数の薬物使用に組み込まれる可能性があることを示す。(3)適用者が提出したデータまたは情報は、プラットフォーム技術の導入または利用が薬物開発または製造過程および審査過程に顕著な効率をもたらす可能性があることを示している。スポンサーは、IND出願を提出した間または後の任意の時間に、IND出願を要求対象とするプラットフォーム技術を組み込むか、または使用する指定されたプラットフォーム技術としてFDAに指定することを要求することができる。指定された場合、FDAは、プラットフォーム技術を使用するか、または使用する薬物の任意の後続の元のNDAまたはBLAの開発および検討を加速することができる。我々のプラットフォーム技術がこのような指定の基準に適合していると考えても,FDAは同意せず,このような指定を付与しないことにした可能性がある.さらに、プラットフォーム技術のこのような指定を受けることは、薬物開発がより速く、またはより速いFDA審査プロセスまたは最終FDA承認を得ることを保証することはできない。さらに何かがある, FDAが指定されたプラットフォーム技術がもはや指定された基準を満たしていないと判断した場合、FDAは指定を取り消すことができる。
もし私たちがこのようなテストから利益を得る候補薬剤の診断テストの必要性、またはそのようなテストから利益を得るための規制機関の承認を得ることができない場合、あるいはそのような点で大きな遅延に遭遇した場合、私たちはこれらの候補製品のすべての商業的潜在力を実現することができないかもしれない。
私たちのいくつかの適応候補製品の臨床開発については、協力者と協力して開発したり、獲得したりする可能性があります体外培養疾患分類の中で著者らの候補薬物から選択性と意義のある利益を得る可能性のある患者亜群を決定するために、セット診断テストを行った。例えば,我々は基礎医学(FMI)と協力してTRUSELTIQと同時にFDAの承認を得たCCA患者のinfigratinibを治療するためのキット診断薬を開発した。このようなセット診断技術は我々の臨床試験および候補製品の商業化に使用される。成功するためには、私たちまたは私たちの協力者はいくつかの科学、技術、監督と後方勤務方面の挑戦を解決しなければならない。FDAと同様の外国規制機関は体外培養医療機器のセット診断としては,この規制の枠組みの下で,我々が開発可能な任意の診断方法の安全性と有効性を証明するための臨床試験が必要となる可能性があり,商業化前には,単独の規制承認や承認が必要となると予想される。
72
私たちは第三者が私たちの候補治療製品のための診断テストを設計、開発、製造することに依存するかもしれません。これらの製品はこのようなテストが必要かもしれません。もし私たちがこのような協力合意に到達すれば、私たちはこれらの随伴診断の承認を開発し、獲得するための私たちの将来の協力と努力に依存するだろう。開発と監督管理の審査過程において、関連診断の選択性/特異性、分析検証、再現性或いは臨床検証などの問題を解決する必要があるかもしれない。また,前臨床研究や早期臨床試験からのデータが候補製品開発に伴う診断を支持しているようであっても,その後の臨床試験で発生したデータは診断に伴う分析や臨床検証を支援できない可能性がある。私たちと未来の協力者は、規制許可の獲得、規制の承認、製造、商業化が私たちの候補治療薬や治療薬自体と類似した随伴診断薬の開発に困難に直面する可能性があり、規制許可や承認を得ること、商業規模と適切な品質基準で十分な数の製品を生産すること、市場承認を得ることなどの問題に直面する可能性がある。もし私たちがこれらの候補治療製品のための診断を開発することに成功しなかった場合、または開発を遅延させることができなければ、これらの候補治療製品の開発は不利な影響を受ける可能性があり、これらの候補治療製品はマーケティング承認を得ることができない可能性があり、マーケティング承認を得ることができる任意の治療薬のすべての商業的潜在力を達成することができないかもしれない。したがって、私たちの業務、運営結果、そして財務状況は実質的な被害を受ける可能性がある。また、, 私たちと契約した診断会社は、当社の候補製品の開発と商業化に期待されている診断テストの販売中止や製造を決定するか、またはその診断会社との関係が終了する可能性があります。私たちは、代替診断テストの供給を得るために、私たちの候補製品の開発および商業化のために、または商業的に合理的な条項でそうすることができない可能性があり、これは、私たちの候補治療製品の開発または商業化に悪影響および/または遅延をもたらす可能性がある。
承認されれば、生物製品規制の研究製品として、規制経路の簡略化によって承認された生物模倣薬の競争に直面する可能性がある。
“患者保護および平価医療法案”は、2010年の“医療および教育調整法案”によって改正され、または総称してACAと呼ばれ、“2009年生物製品価格競争および革新法案”、またはBPCIAという副題を含み、FDA許可の参照生物製品と類似しているか、または交換可能な生物製品のための短い承認経路を作成する。BPCIAによると,生物類似製品の申請は,参考製品が初めてFDA許可を得た4年後にFDAに提出されなければならない。また,FDAによる生物類似製品の承認は,参考製品が初めて許可された日から12年後に発効する可能性がある。この12年間の独占期間内に、FDAが競合製品のBLAを承認した場合、競合製品は、スポンサー自身の臨床前データと、他の会社の製品の安全性、純度、および効力を証明するために十分かつ制御された良好な臨床試験データとを含む場合、別の会社は、参照製品の競合バージョンを販売する可能性がある。この法律は複雑であり、FDAはまだ説明して施行している。したがって、その最終的な影響、実施、そして意味には不確実性がある。
BLAによりバイオ製品として承認されたいずれの我々の候補製品も12年の専門期間を得る資格があるべきであると考えられる。しかしながら、国会の行動や他の理由により、このような排他性が短縮される可能性があるか、またはFDAが我々の研究薬を競合製品の参考製品とみなさないことは、予想よりも早く後発薬競争の機会を創出する可能性がある。BPCIAの他の面では,そのいくつかがBPCIAの排他的条項に影響を与える可能性があり,最近の訴訟のテーマでもある.さらに、許可を得ると、生物類似体が私たちのいずれかの参考製品をどの程度置換するか、その方法は非生物製品の伝統的な模倣薬代替に類似しており、これはまだ発展中のいくつかの市場および規制要因に依存するかどうかは不明である。
もし競争相手が私たちの任意の候補製品に関連する生物模倣薬のマーケティング承認を得ることができれば、私たちの製品はこのような生物模倣薬の競争を受ける可能性があり、これは私たちがこのような製品を商業化し、販売から収入を得ることに成功する能力を弱めるだろう。
73
私たちの候補製品が承認されれば、持続的な規制義務と持続的な規制審査の制約を受けることになり、これは大量の追加費用を招く可能性があり、もし私たちが規制要求を遵守できなかったり、私たちの候補製品が予期しない問題に遭遇したら、私たちは処罰を受けるかもしれない。
私たちの候補製品が承認されれば、FDAと他の適用規制機関の製造、ラベル、包装、貯蔵、広告、販売促進、サンプリング、記録保存、上場後の研究及び安全性、有効性とその他の上場後の情報の提出に関する持続的な監督要求と審査を受けることになり、アメリカ連邦と州の要求及び外国の監督管理機関のような要求を含む。
製造業者および製造業者の工場は、品質管理および製造プロセスが現在の良好な製造実践またはcGMP法規に適合することを保証することを含む、FDAおよび同様の外国規制機関によって適用される広範な要件を遵守するように要求されている。したがって、私たちと私たちのCMOは、cGMPの遵守状況、および任意のNDA、BLAまたはマーケティング許可申請またはMAAで行われたコミットメントの遵守状況を評価するために、持続的な審査および検査を受ける。したがって、私たちと私たちと協力している他の人たちは、製造、生産、品質管理を含む規制コンプライアンスのすべての分野に時間、お金、エネルギーをかけ続けなければならない。また、医薬品サプライチェーン安全法によると、ある商業処方薬製品については、製造業者およびサプライチェーンに関連する他の当事者も流通チェーン要求を満たし、製品を追跡および追跡するための電子的、相互運用可能なシステムを構築し、偽、移転、窃盗、故意に混入した製品または米国で流通するのに適していない他の製品をFDAに通報しなければならない。また、サンプルを含む処方薬製品の流通は、連邦レベルの薬品および薬品サンプルの流通を規制する“処方薬販売法”(PDMA)によって制限されている。そして、各州の薬品流通業者に対する登録と監督管理に最低基準を設定した。PDMA,州法ともに処方薬製品サンプルの配布を制限し,配布中の責任の確保を求めている。処方薬製品はまたアメリカの“毒物予防包装法”に適用される児童保護包装要求に符合しなければならない。もし承認後の法規の要求を守ることができなければ, 私たちは規制機関によって任意の承認された製品のマーケティング承認を撤回される可能性があり、私たちがこのような製品をマーケティングする能力が制限される可能性があり、これは私たちが利益を達成したり維持したりすることに悪影響を及ぼす可能性があり、重大な処罰を受ける可能性があります。そのため、承認後の法規を遵守するコストは、私たちの経営業績や財務状況に悪影響を及ぼす可能性があります。
私たちの候補製品のために得られる可能性のある任意の規制された承認は、製品がマーケティングおよび普及のために使用される可能性のある承認された表示用途によって制限されるか、または承認条件によって制限されるか、または製品の安全性および有効性を監視する第4段階の臨床試験および監視を含む、コストの高い上場後試験要件を含むかもしれない。私たちはFDAと似たような外国の規制機関にいくつかの副作用と生産問題を報告することを要求されるだろう。薬物安全問題を解決するいかなる新しい立法も、製品開発や商業化の遅延を招き、あるいはコンプライアンスを確保するコストを増加させる可能性がある。さらに、FDORAによれば、承認された薬物および生物製品のスポンサーは、市場状態が任意に変化したときに、薬物の撤回などの6ヶ月の通知をFDAに提供しなければならず、そうしなければ、FDAは製品を生産停止製品リストに登録する可能性があり、これは製品の発売能力をキャンセルするであろう。
FDAおよび司法省を含む他の機関は、製品の承認後のマーケティング、ラベル、広告および販売促進活動を密接に規制し、監督して、製品の製造、マーケティング、流通が承認の適応にのみ適用され、承認されたラベルの規定に適合することを保証する。承認される可能性のある製品については、私たちは広告と販売促進に関する要求を守らなければならない。処方薬に関する販売促進情報は様々な法律や法規によって制限されており,製品が承認されたラベル上の情報と一致しなければならない。したがって、私たちは承認されていない適応や用途のためにこのような製品を普及させないかもしれない。
74
承認されたセキュリティプロトコル、BLAまたはMAAの所有者は、新しいまたは追加の申請を提出し、承認された製品、製品ラベル、または製造プロセスのいくつかの変更の承認を得なければならない。我々の製品が一般的または特定の患者亜群で承認されれば、我々の製品の安全性と有効性を検証するために、発売後の臨床試験を行うことも要求されることができる。もし最初の上場承認が承認経路を加速することによって得られた場合、著者らは成功した発売後の臨床試験を行い、これらの製品の臨床治療効果を確認する必要があるかもしれない。
規制当局が、予期しない深刻さや頻度の副作用、または製品を生産する施設に問題があるような以前に未知の問題があることを発見した場合、または製品の販売促進、マーケティング、またはラベルを提供することに同意しない場合、規制機関は、製品の市場からの撤退を要求することを含む、製品または私たちに制限を加えることができる。もし私たちが適用される規制要求を遵守できなければ、規制機関や法執行当局は他の措置を取るかもしれない
政府は違法の疑いのあるいかなる調査にも対応するために多くの時間と資源を必要とする可能性があり、否定的な宣伝が生じる可能性がある。現行の法規の要求を守らないいかなる行為も、私たちの製品の商業化と創造能力に重大な悪影響を及ぼす可能性がある。規制制裁を実施したり、規制承認を撤回したりすれば、わが社の価値と私たちの経営業績は悪影響を受けるだろう。FDAおよび他の規制機関の政策は変更される可能性があり、追加の政府法規が公布される可能性があり、私たちの製品候補製品に対する規制承認を阻止、制限、または延期するか、または私たちの製品に対する規制承認を一時停止、撤回、または修正する可能性がある。
FDAと他の規制機関は非ラベル使用の普及を禁止する法律法規を積極的に実行している。
もし私たちの現在のすべての候補製品が承認され、私たちが私たちの製品のラベル外用途を不正に普及させたことが発見されれば、私たちは重大な責任を負うかもしれない。FDAや他の規制機関は、処方薬の販売促進を行う可能性のある声明を厳格に規制している。特に、FDAは、承認された製品に関する真の情報および非誤解性の情報を伝播することを可能にするが、スポンサーは、FDAまたは他の規制機関によって承認されていない使用のための製品を宣伝してはならず、これらの用途は、製品の承認ラベルに反映されている。もし私たちがこのようなラベル外の使用を普及させることを発見されたら、私たちは重大な責任を負うかもしれない。連邦政府はラベル外使用の不当な普及の疑いがある会社に巨額の民事と刑事罰金を科し、いくつかの会社がラベル外販売促進に従事することを禁止している。FDAはまた、会社に同意法令、会社誠実協定、または永久禁止を締結することを要求しており、これらの禁止に基づいて、特定の販売促進行為を変更または制限しなければならない。もし私たちが私たちの候補製品の普及を成功的に管理できなければ、承認されれば、私たちは重大な責任を負う可能性があり、これは私たちの業務や財務状況に実質的な悪影響を及ぼすだろう。
75
製品普及に関連する規定があるにもかかわらず、FDAとその他の監督機関は会社がその製品について真実、非誤解性と非販売性の科学交流を行うことを許可した。医学教育活動に従事し,すべての適用された法律や法規を遵守して医療提供者とコミュニケーションをとる予定である。
私たちの候補製品の新規性に関するリスク
私たちのいくつかの候補製品は、私たちのタンパク質治療と遺伝子治療製品の候補製品を含み、すべて斬新で、複雑で製造が困難である。私たちは製造問題に直面し、私たちの開発や商業化計画の遅延や他の方法で私たちの業務を損なう可能性があります。
私たちのCMOは、私たちのタンパク質治療および遺伝子治療製品を含む私たちの候補製品を生産するために使用され、その製造プロセスは複雑で、斬新であり、商業用途の検証されていない。多種の要素はすでに未来の生産中断を招く可能性があり、ある製造業務が制限され、及び新冠肺炎疫病による政府の“留守”指令によるCMO製造施設の現場人員不足、設備故障、施設汚染、原材料不足或いは汚染、自然災害、公共事業サービス中断、人為ミス或いはサプライヤー運営中断を含む。
我々のいくつかの小分子候補製品は特に複雑であり、場合によっては、必要なステップの数量、プロセスの複雑性及び末端或いは中間段階製品の毒性のため、製造が特に困難である。我々のタンパク質治療および遺伝子治療候補製品は、ほとんどの小分子薬物に必要な処理工程よりも複雑な処理工程を必要とする。また,小分子とは異なり,我々のいくつかの候補生物製品の物理的および化学的性質は通常完全には表現できない。したがって、完成品の分析は、製品が各ロット間で一貫性を維持するか、または予期された方法で動作することを保証するのに十分ではない可能性がある。したがって、私たちのCMOは、プロセスが反復可能であり、候補製品がこのプロセスに厳密かつ一貫して適合することを保証するために、製造プロセスを制御するために複数のステップを使用しなければならない。製造過程中の問題は、正常過程との微小な偏差であっても、製品欠陥或いは製造失敗を招く可能性があり、それによって大量故障、製品リコール、製品責任クレーム或いは在庫不足を招き、臨床試験或いは商業市場の供給ができない。私たちは問題に直面する可能性があり、十分な数量と品質の臨床レベルの材料を得ることができず、FDA、EMA或いは他の適用される標準或いは規範を満たし、一致と受け入れ可能な生産生産量とコストを持っている。
さらに、FDA、EMA、および他の外国規制機関は、任意のロットの承認された製品のサンプルをいつでも提出し、適用試験結果を示すプロトコルを要求することができるかもしれない。場合によっては、FDA、EMA、または他の外国規制機関は、機関が発行を許可するまで大量の製品を配布しないように要求するかもしれません。製造過程中の微小な偏差は、品質属性や安定性に影響を与える偏差を含み、製品に許容できない変化を招き、大量故障や製品リコールを招く可能性がある。大量失敗や製品リコールは製品の発表や臨床試験を延期する可能性があり、これは高い代価を払わせるかもしれません。そうでなければ、私たちの業務、財務状況、運営結果、将来性を損なうことになります。
私たちのCMOはまた私たちの製造プロセスを操作するために必要な経験豊富な科学、品質保証、品質管理と製造人員を採用と維持する上で問題に直面する可能性があり、これは生産遅延或いは適用の法規要求を維持することを維持することを招く可能性がある。
我々CMOの製造プロセスや施設における任意の問題は、計画の臨床試験の遅延およびコスト増加を招く可能性があり、潜在的なパートナー(より大きなバイオテクノロジー会社や学術研究機関を含む)の魅力の低いパートナーになる可能性があり、これは、より多くの魅力的な開発プロジェクトを制限する可能性がある。私たちの製造過程における問題はまた、将来承認される可能性のある製品に対する潜在的な市場需要を満たす能力を制限する可能性がある。
76
著者らのいくつかの候補製品は1種の新型の腺関連ウイルス或いはAAV遺伝子治療技術に基づいており、これまで、臨床或いは監督管理経験は限られており、これにより、製品候補開発とその後監督管理許可を得る時間とコストを予測することは困難である。
私たちのいくつかの候補製品は遺伝子治療技術に基づいており、私たちの将来の成功はこの新しい治療方法の成功開発にかかっている。私たちや他の遺伝子治療会社が将来遭遇する遺伝子治療技術に関するいかなる開発問題も、私たちの候補製品開発に重大な遅延や意外なコストをもたらすことはなく、このような開発問題が解決される保証はありません。そのほか、FDA、EMAとその他の監督管理機関の臨床研究要求及びこれらの監督機関は候補製品の安全性と有効性を決定するための標準は、潜在製品のタイプ、複雑性、意外性と期待用途と市場によって大きく異なる。私たちのような候補新製品の規制承認過程は、他のより有名で、広く研究されている治療法よりも高価で、時間もかかるかもしれない。また,我々は疾患のために新たな治療法を開発しているため,これらの疾患では,新たな終点や方法に対する臨床経験が限られており,FDA,EMAあるいは類似した外国の規制機関は臨床試験終点を考慮して臨床的意義のある結果を提供するリスクが増加している可能性があり,それによる臨床データや結果の分析が困難である可能性がある。これまで、遺伝子治療製品がFDAや同様の外国規制機関の承認を得ることは少なく、私たちの候補製品が米国、EU、または他の管轄区域で規制承認を得るのにどのくらい時間がかかるか、またはいくらかかるかを決定することは困難であった。しかも、一つの規制機関の承認は他の規制機関が何を承認する必要があるかを代表しないかもしれない。
遺伝子治療製品開発に対する監督管理要求は常に変化し、将来も変化し続ける可能性がある。FDAは2016年、遺伝子治療および関連製品の審査を統合し、その審査についてCBERに提案を提供するために、その生物製品評価·研究センター(CBER)内に組織および高度治療オフィス(OTAT)を設立した。2022年9月、FDAはOTATを治療製品オフィス(OTP)に変更し、OTPを“スーパーオフィス”に向上させ、日々増加する細胞および遺伝子治療仕事量を満たすことを発表した。また,米国国立衛生研究院(NIH)が発表したガイドラインによると,遺伝子治療臨床試験も機関生物安全委員会(IBC)の審査と監督を受け,IBCは地方機関委員会であり,組換えや合成核酸分子を用いた研究の審査·監督を担当している。どの機関が臨床試験を開始する前に、機関審査委員会(IRB)およびそのIBCは、研究の安全性を評価し、公衆衛生または環境に対する任意の潜在的リスクを決定する。NIHガイドラインは強制的ではないが,関連研究がNIH組換えや合成核酸分子研究助成を受けた機関で行われているか,あるいはその助成によって行われていない限り,多くの会社や他のNIHガイドラインに拘束されていない機関は自発的にこれらのガイドラインに従っている。さらに、候補遺伝子治療製品の臨床試験において他の人が発生する深刻な有害事象または進展は、FDAまたは他の規制機関が私たちの臨床試験を一時停止し始めるか、または他の方法で私たちの任意の候補製品に対する承認要求を変更する可能性がある。FDAは個々の細胞や遺伝子治療を継続できるかどうかを決定していますが, 他の審査機関の審査過程や決定は,FDAがこの試験を審査して起動を許可したとしても,臨床試験の開始を阻害または延期する可能性がある。
同様に,EMAはEUの遺伝子療法の承認を管理しており,遺伝子療法製品の開発やマーケティングライセンスに関する新しいガイドラインを発表することが可能であり,これらの新しいガイドラインの遵守が求められている。
これらの規制審査委員会や諮問グループおよびその発表された新しいガイドラインは、追加的な研究や試験を行うことを要求し、私たちの開発コストを増加させ、規制の立場や解釈の変化を招き、候補製品の承認と商業化を延期または阻止し、あるいは重大な承認後の制限または制限を招くことができるかもしれない。私たちが私たちの候補製品を推進する時、私たちはこれらの規制や諮問グループと協議し、適用されるガイドラインを遵守することを要求されるだろう。もし私たちがこれをできなかったら、私たちはこのような候補製品の開発を延期または停止することを要求されるかもしれない。このような追加的な手続きは私たちが予想していたより長く検討と承認過程を招くかもしれない。規制承認手続きの増加または延長あるいは私たちの候補製品開発のさらなる制限による遅延はコストが高い可能性があり、私たちが臨床試験を完成し、適時に私たちの現在と未来の候補製品を商業化する能力(あれば)の能力にマイナス影響を与える可能性がある。
77
著者らは遺伝子治療技術に基づく候補製品は不良と予見できない副作用を招く可能性があり、あるいは公衆に安全ではないと思われる可能性があり、これはそれらが臨床試験或いは監督管理許可に入ることを延期或いは阻止し、臨床一時停止を実施し、商業潜在力を制限し、或いは重大な負の結果を招く可能性がある。
公衆の態度は、遺伝子療法が新しい技術として安全でない、非道徳的、または不道徳であるという説の影響を受ける可能性があり、したがって、私たちの候補製品は公衆または医学界の受け入れを得ることができないかもしれない。公衆の悪い態度は私たちが臨床試験を募集する能力に悪影響を及ぼすかもしれない。そのほか、FDAは近年、遺伝子治療候補に対してより多くの臨床制限を加えている。さらに、私たちの成功は、医師が処方した処方および彼らの患者が受けることを望む治療に依存し、これらの治療は、彼らがよく知っている既存の治療方法を代替または補充するために、私たちが開発可能な候補製品を使用することに関連し、より多くの臨床データを得ることができる。例えば,これまでの候補遺伝子治療製品の臨床試験では,白血病や死亡例が報告されていることを含め,他のベクターを用いた他の試験ではいくつかの著明な副作用がみられている。これらの副作用を減少させるための新たなAAVベクターが開発されているが,遺伝子治療は比較的新しい疾患治療法であり,より多くの副作用が生じる可能性がある。遺伝物質や遺伝物質を運搬するための製品の他の成分の持続的生物活性により,遺伝子治療製品に曝露した後に遅延した副作用の潜在的リスクがある。
遺伝子治療製品の治療に出現する可能性のある副作用は、投与後の早期の免疫反応を含み、これは、患者の健康を損なうか、または治療の有効性および持続性を大幅に制限する可能性がある。例えば、AAV遺伝子治療の1つのますます期待される副作用はT細胞免疫反応の発展であり、最もよく見られるのは肝臓に影響することである。我々のAAV遺伝子治療候補製品または第三者が開発している製品の任意の実際または予想される負の影響は、これらの候補製品を継続して開発する能力を弱める可能性があり、私たちの将来性に悪影響を及ぼす可能性がある。
私たちの第三者への依存に関するリスクは
我々は第三者に依存して著者らの臨床試験および著者らの研究と臨床前テストのいくつかの方面を行うことが予想されるが、これらの第三者の表現は締め切り前にこのような試験、研究或いはテストを完成できないことを含む満足できないかもしれない。
著者らは現在、CRO、臨床データ管理組織、医療機関と臨床研究者などの第三者に依存して、ある方面の研究、臨床前試験と臨床試験を行う予定である。これらの第三者のいずれも、私たちとの契約関係を終了したり、その契約義務を履行できない可能性があります。もし私たちがこのような第三者とのいかなる関係も終わらなければ、私たちは商業的に合理的な条項で代替の第三者と合意できないかもしれない。もし私たちが代替計画を達成する必要があれば、これは私たちの製品開発活動を延期するだろう。
我々のこれらの第三者の研究開発活動への依存はこれらの活動の制御を減少させたが,我々の責任を軽減していない.例えば、私たちは、私たちそれぞれの臨床試験が試験の全体的な研究計画と方案、適用された法律、法規、科学的基準に従って行われることを保証する責任があり、私たちの第三者への依存が私たちの規制責任を免除しないことを保証する責任がある。さらに、FDAおよび同様の外国の規制機関は、データおよび報告の結果が信頼性、反復可能かつ正確であることを保証し、試験参加者の権利、完全性、および機密性を保護するために、GCPを遵守し、臨床試験結果を記録、報告することを要求する。規制当局は,試験スポンサー,主要調査者,試験地点を定期的に検査することでこれらのGCPを実行している。私たちまたはこれらの第三者のいずれかが適用されるGCP法規を遵守できない場合、私たちの臨床試験で生成された臨床データの一部または全部は信頼できないと考えられる可能性があり、FDAまたは同様の外国の規制機関は、追加の非臨床または臨床試験を要求するか、または私たちのマーケティング申請を承認する前により多くの患者を募集することができる。検査後、これらの規制機関が私たちの臨床試験がGCP規定に適合しているかどうかを確認することはできません。臨床試験中はいかなる法律法規にも違反します, 私たちは民事処罰が含まれている可能性があり、最高で刑事起訴に達する可能性のある無タイトルの警告状や法執行行動を受けるかもしれない。現在行われている臨床試験を登録し,完了した臨床試験の結果を一定時間範囲で政府後援のデータベースに発表することも求められている。そうしないと罰金、否定的な宣伝、そして民事と刑事制裁につながるかもしれない。
78
これらの第三者が規制要件や私たちが規定した規程に従って契約責任を成功裏に履行し、期待された期限内に臨床試験を完了または行うことができない場合、私たちは私たちが開発する可能性のある任意の候補製品の発売承認を得ることができないか、遅延する可能性があり、私たちの薬物の商業化に成功する努力を遅らせることができないか、または延期することができるだろう。私たちの失敗やこれらの第三者が適用された規制要件を遵守できなかったことや、私たちが宣言した合意は、私たちを法執行行動の影響を受ける可能性もある。
私たちはまた他の第三者に依存して私たちの臨床試験のための薬品の貯蔵と配布を望んでいる。私たちのディーラーのどんな業績ミスも、私たちが開発する可能性のある任意の候補製品の臨床開発やマーケティング承認を延期したり、私たちの薬品を商業化して、追加の損失をもたらし、私たちの潜在的な製品収入を奪う可能性があります。
私たちの現在の候補製品あるいは私たちが臨床前研究および臨床試験のために開発する可能性のある他の候補製品の製造は完全に第三者に依存している。もしこれらの第三者が十分な量の薬品を提供できない場合、あるいは許容可能な品質レベルや価格で提供できない場合、私たちの業務は損害を受ける可能性があります。
私たちは今のところありませんし、私たちが行っている臨床試験や私たちが行う可能性のある未来の臨床試験のための薬物供給のインフラや能力を内部で得るつもりもありませんし、私たちの候補製品を生産する資源が不足しています。承認されれば、商業規模の生産を行います。私たちは依存し、引き続き第三者メーカーに依存して、私たちの現在の候補製品を生産するか、または臨床試験のための他の候補製品、および上場承認を得る可能性のある任意の候補製品の商業生産を決定する可能性があります。私たちは通常臨床試験を開始しませんが、試験を完了するのに十分な候補製品供給があると信じない限り、第三者メーカーを交換する必要があるため、行われている臨床試験の候補製品やその原材料供給のいかなる重大な遅延や中断も、臨床開発と候補製品の潜在的な規制承認を大幅に遅らせる可能性があり、これは私たちの業務と運営結果を損なう可能性があります。私たちはまた、承認されれば、私たちの候補製品の商業供給は主に第三者製造に依存すると予想している。
私たちは第三者製造業者を識別し、適切な資格鑑定を行うことができないかもしれないし、第三者製造業者と合意を確立したり、許容可能な条項でそうすることもできないかもしれない。たとえ第三者製造業者と合意できても、第三者メーカーに依存することは、追加的なリスクをもたらす
79
また、私たちのすべてのCMOは他の会社と交渉して、これらの会社に材料や製品を供給および/または製造し、これは私たちのメーカーがこのような材料と製品を生産する時に規制リスクに直面させます。私たちの契約メーカーが私たちの候補製品を生産するための施設はFDAの審査を受け、検査は私たちがFDAに機密協定またはBLAを提出した後に行われます。私たちは薬品と生物製品の生産過程をコントロールせず、私たちの契約生産パートナーが法規要求、即ちcGMP要求に完全に依存している。もし私たちの契約メーカーが私たちの規格やFDAや他の機関の厳格な規制要件に合った材料を生産することに成功しなければ、私たちはこれらの製造施設で生産された私たちの候補製品の規制承認を確保または維持することができません。しかも、私たちの契約製造業者が十分な品質管理、品質保証、合格者の能力を維持することを制御することはできません。FDAや同様の外国規制機関が我々の候補製品を生産するためにこれらの施設を承認しない場合、またはどの機関が将来その承認を撤回すれば、代替製造施設を探す必要があるかもしれないが、これは、私たちの候補製品の開発、規制承認またはマーケティング(承認された場合)を得る能力に悪影響を及ぼすだろう。
2020年3月27日、トランプ総裁は新冠肺炎の流行に対応するCARE法案に署名し、法律にした。全新冠肺炎の大流行期間中、公衆は肝心な医療製品の入手可能性と可及性に対してずっと懸念しており、CARE法案は薬品不足措置におけるFDAの現有の権威を強化した。CARE法案によると、特定の重篤な疾患の承認薬物供給または薬剤または有効薬物成分の生産を許可する各機関の条件が直面するリスクを決定し、評価するためのリスク管理計画を策定しなければならない。リスク管理計画は検査中にFDAの審査を受ける予定だ。もし私たちが上場承認された候補製品の供給が不足すれば、私たちの結果は実質的な影響を受けるかもしれない。
私たちの候補製品は他の候補製品と販売されている薬品と生産施設を競争するかもしれない。例えば、新冠肺炎の大流行が始まって以来、新冠肺炎のいくつかのワクチンはすでにアメリカ食品と薬物管理局の緊急使用許可を得ており、その中のいくつかは後に発売許可を得た。未来にはより多くのワクチンが許可されたり承認されるかもしれない。これによって生じるワクチンの需要や1950年の“国防生産法案”や同様の外国立法によって徴用された製造施設や材料の可能性は,臨床試験に必要な製品の材料や製造槽を得ることを困難にする可能性があり,これらの試験の遅延や商業供給の問題を招く可能性がある。私たちの既存または未来のメーカーのどんな業績失敗も、臨床開発、市場承認、または商業化を延期する可能性がある。私たちの現在と予想されている未来の他の候補製品製造への依存は、私たちの将来の利益率と、私たちが適時かつ競争力のあるマーケティング承認の候補製品を商業化する能力に悪影響を及ぼすかもしれない。
私たちのいくつかの候補製品の薬物物質と薬品は現在単一源の供給者から得られている。これらの供給者を失ったり、彼らが私たちに薬物や薬品を供給できなかったりすることは、私たちの業務に実質的な悪影響を及ぼすかもしれない。
私たちのいくつかの候補製品の薬物物質と薬物製品は単一源供給者またはCMOによって開発と製造契約、サービス、品質協定と調達注文によって製造された。私たちは現在、これらの候補製品の薬物物質や薬品の他のいかなる供給者もいません。私たちの臨床的および商業的需要を満たす他の代替供給源があると信じていますが、代替源を探し、これらの源との関係を確立することは、私たちの候補製品の開発を著しく遅延させないことを保証することはできません。
私たちの単一ソース供給者への依存は、以下のリスクを含むいくつかのリスクに直面させます
80
また、私たちは商業的に合理的な条項で代替サプライヤーと供給スケジュールを達成できないかもしれないし、供給スケジュールを全く達成できないかもしれない。私たちの候補製品の開発を遅延させたり、既存のサプライヤーとよりも優遇された条項で異なる第三者と新しい合意を締結しなければならない場合、私たちの業務に実質的な悪影響を及ぼす可能性があります。
もし私たちが依存している代行施設が規制要求を満たし続けられない場合、あるいは私たちの供給需要を満たすことができなければ、私たちの業務は損害を受けるだろう。
臨床試験または商業販売のための候補製品を準備するすべてのエンティティ、我々の既存のすべての候補製品を含むCMOは、広く規制されている。商業販売または末期臨床試験のために許可された完成治療製品の成分は、cGMPまたは米国以外の類似した法規要件に従って生産されなければならない。これらの規定は、調査製品及び承認販売された製品の品質を制御及び確保するために、記録保存、並びに品質システムの実施及び実行を含む生産プロセス及びプログラムを管理する。生産過程の不良な制御は汚染物質の導入を招くか、あるいは無意識に私たちの候補製品の性能または安定性を変化させる可能性がある。私たちまたは第三者製造業者が適用された法規を遵守できないことは、臨床保有、罰金、禁止、民事処罰、遅延、承認の一時停止または撤回、許可証の取り消し、生産の一時停止、候補製品の差し押さえまたはリコールまたは上場薬物、運営制限、および刑事起訴を含む制裁を実施する可能性があり、いずれも私たちの候補製品の臨床的または商業的供給に重大な悪影響を及ぼす可能性がある。
私たちまたは私たちのCMOは、NDA、BLA、またはMAAをサポートするすべての必要な文書をタイムリーに提供し、FDAおよび他の規制機関がその施設検査計画によって実行される法規を遵守しなければならない。私たちのCMOは商業的に許可された薬品を生産したことがないので、必要な規制機関の許可を得ていません。私たちの第三者請負業者の一部または全部の施設および品質システムは、適用法規に適合する承認前検査によって、規制部門として私たちの候補製品または任意の他の潜在的製品の条件を承認しなければならない。さらに、規制当局は、私たちの候補製品または私たちの他の潜在的製品または関連品質システムを準備することに関連する製造施設が、進行中の活動に適用される法規に適合しているかどうかを随時審査または検査することができる。CMOを監視しているが,CMOパートナーの製造過程を制御することはできず,CMOパートナーの法規遵守要求に完全に依存している。これらの施設が承認前の工場検査を通過していない場合には、適用製品候補の規制承認が承認されない場合や、どのような違反が是正され、規制当局が満足する程度に達するまで大幅に延期される可能性がある。
規制当局はまた、製品の販売を承認した後のいつでも、当社の第三者請負業者の製造施設を監査することができます。そのような検査または審査が適用法規に準拠していないことが発見された場合、またはそのような検査または審査以外に私たちの製品仕様または適用法規に違反する場合、私たちまたは関連規制機関は、臨床試験または商業販売の一時的または永久的な一時停止、または施設の一時的または永久的な閉鎖を含む可能性がある高価および/または時間がかかる可能性のある救済措置を我々または関連する規制機関に要求する可能性がある。私たちまたは私たちと契約を締結した第三者に課せられたどのような救済措置も、私たちの業務に実質的な損害を与える可能性があります。
81
さらに、承認された製造業者の供給が中断された場合、別の代替製造業者は、セキュリティプロトコル、BLA補充またはMAA変更または同等の外国規制申請によって資格を取得する必要があり、これは、さらなる遅延をもたらす可能性がある。新しいメーカーに依存して商業生産を行えば、規制機関は追加の研究を要求する可能性もある。場合によっては、私たちの候補製品を製造するために必要な技術的スキルは、元のCMO固有または独自のものである可能性があり、困難に遭遇する可能性があり、またはそのようなスキルを予備または代替サプライヤーに譲渡することを禁止する契約制限が存在する可能性があり、またはそのようなスキルを全く譲渡できない可能性があります。また,我々が何らかの理由でCMOの交換を要求された場合,新たなCMOが品質基準とすべての適用法規に適合する施設やプログラムを保持していることを確認するように要求される.私たちはまた、例えば比較可能な研究を製造することによって、任意の新しい製造プロセスが、FDAまたは他の規制機関に以前に提出された、またはFDAまたは他の規制機関によって承認された仕様に従って、私たちの製品または候補製品を生産することを検証する必要がある。新しいCMO検証に関連する遅延は、私たちが候補製品を開発したり、承認されたり、適時または予算内でこれらの候補製品を商業化する能力に悪影響を及ぼす可能性があります。また、メーカーの変化は通常、製造プロセスやプロセスの変化に関連しており、これは、臨床試験で使用されている以前の臨床供給と任意の新しいメーカーの供給との間の過渡的な研究が要求されるかもしれない。臨床用品の比較可能性の証明には成功しない可能性があり,追加の臨床試験が必要かもしれない。それに応じて, 製造業者の交換は大量のコストを伴う可能性があり、私たちが期待する臨床およびビジネススケジュールの遅延を招く可能性がある。
これらの要素は、私たちにより高いコストをもたらす可能性があり、臨床試験、規制提出、必要な承認、または私たちの候補製品の商業化遅延または終了を招く可能性がある。さらに、私たちのサプライヤーが契約要件を満たしておらず、実質的に同じコストで生産できる1つ以上の代替サプライヤーを得ることができない場合、私たちの臨床試験は延期される可能性があり、あるいは潜在的な収入を失う可能性があります。
第三者との協力関係は、私たちに大量の資源を費やし、財務的リターン保証なしに大量の業務リスクを招く可能性があります。
私たちは私たちの既存の候補製品をマーケティングして商業化するために戦略的協力に依存する予定だ。例えば、EidosはAlexion Pharma International Operations UnLimited CompanyまたはAlexionと許可協定を締結し、この協定によると、私たちはAlexionに依存して日本でacoramidisの臨床開発と商業化を行い、QEDは以前にヘルシン医療保健会社とヘルシン治療(米国)会社と許可と協力協定を締結し、私たちは総称してHelsinnと呼ばれ、QEDはHelsinnに独占的な許可を与え、開発、製造、商業化QEDは腫瘍学と軟骨発育不良あるいは任意の他の骨格発育不良以外のすべての他の適応性の候補製品infigratinib、全世界範囲内で(Republic Of China)香港とマカオ,QEDはすでにどの地域でLianBioと協力関係を結んでいるか).私たちはその後、ヘルシンヌが商業的考慮を理由に協力を中止するという通知を受けた。2022年12月、私たちは相互終了協定に署名した。また、私たちは戦略協力に依存して他の候補製品を開発することができるかもしれませんが、製薬やバイオテクノロジー会社との戦略的パートナー関係を通じて他の製品を供給することができるかもしれません。
もし我々が製品開発の初期段階で研究開発協力を行えば、成功は研究協力者の表現にある程度依存する。我々は,研究協力者が候補製品に関する活動に投入する資源の数や時間を直接制御しない.研究協力者は私たちの研究開発計画に十分な資源を投入していないかもしれない。任意の研究協力者が十分な資源を投入できない場合、協力に関連する臨床前または臨床開発計画は延期または終了される可能性がある。さらに、協力者は、我々と協力して開発した製品または代替技術ではなく、既存または他の開発段階の製品または代替技術を優先的に開発することができる。最後に、私たちが協力者に必要なマイルストーンまたは特許使用料を支払うことができなかった場合、またはそれと達成された合意における他の義務を遵守できなかった場合、協力者は、これらの合意の履行を終了または停止する権利がある可能性がある。
82
戦略的協力を構築することは難しくて時間がかかる。私たちと潜在協力者との議論は、もしあれば、有利な条件で協力関係を築くことにはつながらないかもしれない。潜在的な協力者は、私たちの財務、法規、または知的財産権の状況に対する彼らの評価に基づいて協力を拒否するかもしれない。また,最近では大手製薬会社間の業務合併数が大きく,将来の潜在的パートナー数の減少を招いている。たとえ我々が協力関係を構築することに成功しても、これらの関係は候補製品の開発や商業化に成功することはなく、販売収入も生じない可能性がある。私たちが協力手配を達成した程度では、関連製品の収入は私たちの直接マーケティングと販売のこのような製品の収入よりも低いかもしれません。そのような協力者はまた、同様の協力可能な兆候を得るために候補製品または技術を代替することと、任意の将来の候補製品に対して、そのような協力が私たちとの協力よりも魅力的であるかどうかを考慮することができる。
協力者との関係を管理する必要があります
もし私たちが将来私たちに有利な条項でこのような戦略的協力を構築したり維持したりすることができなければ、私たちの研究開発努力と収入創出の潜在力は制限されるかもしれない。
私たちは他の協力、許可、その他の類似手配の締約国であり、他の協力、許可、その他の類似手配への加入を求める可能性があり、既存の手配を維持したり、新しい手配に加入したりすることは成功できない可能性があり、私たちが締約国であっても、このような関係の利点を実現できない可能性がある。
私たちの協力計画の成功は私たちの協力者の努力と活動に大きく依存するだろう。協力は多くのリスクに直面しており、以下のリスクが含まれている可能性がある
83
また、候補製品の開発や商業化に要する資本コストや製造制限により、我々の候補製品を開発または商業化するために、他の協力、合弁企業、許可、その他の同様の合意を求めることが可能である。私たちの候補製品のためのこのような協力を構築する努力は成功しないかもしれません。私たちの研究開発ルートが不足している可能性があるので、私たちの候補製品は協力努力の開発段階が早すぎると思われるかもしれません。あるいは第三者は私たちの候補製品が安全性と有効性や重大なビジネス機会を示す必要な潜在力を持っていると思わないかもしれません。また、適切な戦略的パートナーを探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑かもしれない。また、未来の協力協定は、潜在的な協力者と他の合意を締結することを制限することができます。私たちは戦略的取引や許可証の後、私たちが経済的利益を得て、このような取引が合理的であることを証明することを確信できない。
私たちがこのような協力を成功させたとしても、私たちが合意した条項は私たちに不利になるかもしれないが、例えば、候補製品の開発や承認が延期され、候補製品の安全性が疑問視されたり、承認された候補製品の販売が満足できない場合、私たちはこのような協力を維持できないかもしれない。例えば、2022年8月23日、私たちはヘルシンーングループから書面通知を受け、商業的考慮のため、便宜上、改訂された許可と協力協定(“改訂QED-ヘルシン許可と協力協定”)によってヘルシンヌグループを終了し、2022年12月にヘルシンーンと相互終了協定を締結する予定であることを示した。
しかも、どんな潜在的な未来の協力も私たちの戦略的パートナーによって中止されるかもしれないし、私たちはこのような合意の下で私たちの権利を十分に保護できないかもしれない。また、戦略的パートナーは、(承認されれば)候補製品の開発および商業化に関する決定を制御するために、いくつかの権利について交渉することができ、これらの活動を私たちと同じ方法で行わない可能性がある。もし私たちが将来協力を終了したり、私たちの候補製品に関する協力を遅延させたりすれば、候補製品の開発と商業化を延期し、それらの競争力を低下させる可能性があり、それらが市場に進出すれば、私たちの業務、財務状況、運営結果に大きな悪影響を及ぼす可能性がある。
私たちの知的財産権に関するリスクは
もし私たちが私たちの候補製品のために十分な知的財産権保護を得ることができなければ、アコラミ、低用量infigratinib、BBP-418、BBP-631、アナリート、および任意のKRAS候補阻害剤を含む、または得られた知的財産権保護範囲が十分でない場合、私たちの競争相手は私たちと類似または同じ製品または候補製品を開発し、商業化する可能性があり、私たちの候補製品を商業化することに成功する能力は損なわれる可能性がある。
他の製薬やバイオ製薬会社の場合と同様に、私たちの成功は、私たちの候補製品や技術について私たちが単独で他の人と共同で所有する可能性のある知的財産権、特にアメリカと他の国の特許を獲得し、維持する能力があるかどうかに大きくかかっている。私たちは、私たちの候補品に関する特許出願をアメリカと海外に提出することで、私たちの独自の地位を保護することを求めています。
84
製薬および生物製薬特許の取得および実行は高価で、時間がかかり、複雑であり、必要または望ましいすべての特許出願を合理的なコストで、またはそのような特許出願から発行される可能性のある任意の特許を維持、実行および許可することができない可能性がある。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.私たちは、特許出願の準備、提出および起訴を制御する権利がないか、または第三者に許可された特許の権利を維持する権利がないかもしれない。したがって、このような特許および出願は、私たちの業務の最適な利益に合った方法で起訴され、強制されてはならない。
生物技術と製薬会社の特許地位は通常高度に不確定であり、複雑な法律、技術と事実問題に関連し、近年ずっと多くの訴訟のテーマである。しかも、外国の法律はアメリカの法律のように私たちの権利を保護しないかもしれないし、その逆も同様である。しかも、私たちは私たちの候補製品に関連する可能性のあるすべての第三者知的財産権を知らないかもしれない。科学文献で発表された発見は往々にして実際の発見に遅れており、米国および他の司法管轄区の特許出願は通常、出願18ヶ月後に発表され、場合によっては全く発表されない。したがって、私たちは、私たちが最初に私たちの特許または係属中の特許出願で主張された発明を提出したのか、最初にそのような発明のために特許保護されたのかを正確に知ることができない。さらに、特許請求の範囲は、法律解釈、特許における書面開示、および特許の起訴履歴によって決定され、専門家の意見のような他の要因に関連する可能性がある。私たちのこれらの問題の分析は、特許または保留出願における特許請求の関連性または範囲を説明すること、そのような特許請求の範囲の独自技術、候補製品への適用性を決定すること、第三者未定特許出願が関連範囲の権利要件を提出するかどうかを予測すること、および関連すると考えられる任意の米国または海外特許の満了日を決定することが正しくない可能性があり、これは、私たちの候補製品を開発およびマーケティングする能力に悪影響を及ぼす可能性がある。私たちはいつも第三者が未解決の特許出願と特許を独立して審査するわけではない。そのため、発行、範囲、有効期限, 私たちの特許権の実行可能性と商業的価値は高い不確実性を持っている。私たちの未決および将来の特許出願は、私たちの候補製品の全部または一部を保護するために、または他社が競争製品を商業化することを効果的に阻止するために、特許の発行を招くことができないかもしれない。私たちの特許出願が特許の形で発表されても、それらの発表形態は私たちに何の意味のある保護を提供してくれず、競争相手が私たちと競争することを阻止したり、他の方法で私たちにどんな競争優位性を提供してくれたりしないだろう。私たちの競争相手は、非侵害的な方法で類似または代替の候補製品を開発することによって、私たちの特許を迂回することができるかもしれない。
私たちが特許権を執行する能力はまた私たちが侵害行為を検出する能力にかかっている。その製品やサービスに関するコンポーネントや方法を宣伝しない侵害者を検出することは困難である可能性がある.さらに、競争相手または潜在的な競争相手の製品またはサービス侵害の証拠を得ることは困難または不可能である可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、もし私たちが勝てば、得られた損害賠償や他の救済措置は商業的な意味がないかもしれない。もし私たちが私たちの特許を保護または強制するために訴訟を提起したり、第三者のクレームを訴訟したりすれば、そのような訴訟は費用が高く、私たちの経営陣と技術者の注意をそらすだろう。このような訴訟はまた、私たちの1つまたは複数の特許の一部または全部のクレームが無効であるか、または他の方法では実行できないことを含む、第三者からのクレームを引き起こす可能性がある。
さらに、既存技術の第三者事前発行を米国特許商標局または米国特許商標局に提出したり、反対、派生、再審に参加したりする可能性がある各方面間私たちの特許権または他人の特許権の介入手続きを審査、許可後に審査または挑戦します。このような提出、訴訟、または訴訟における不利な裁決は、私たちの特許権の範囲を縮小したり、無効にしたりして、第三者が私たちの候補製品を商業化し、私たちに支払うことなく、直接競争することを可能にしたり、第三者の特許権を侵害することなく薬物を製造または商業化することができなくなる可能性があります。さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、結果にかかわらず、会社が現在または将来の候補製品を許可、開発、または商業化することを阻止することができる。
85
さらに、特許の発行は、その発明性、範囲、有効性、または実行可能性に対して決定的ではなく、我々の特許は、米国および海外の裁判所または特許庁で挑戦される可能性がある。このような挑戦は、独占的または経営の自由を失うこと、または特許主張が全体的または部分的に縮小され、無効であるか、または実行できないことをもたらす可能性があり、これは、他人が私たちと類似または同じ候補製品の使用または商業化を阻止する能力を制限するか、または私たちの候補製品の特許保護期間を制限する可能性がある。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちの特許の組み合わせは、他社が私たちと類似しているか、または同じ薬物を商業化することを排除するために、十分な権利を提供してくれないかもしれない。
しかも、私たちの知的財産権は1つ以上の第三者の権利によって維持されるかもしれない。例えば、私たちの特定の特許権と技術をもたらす研究部分はアメリカ政府によって援助されています。そのため、政府はこれらの特許権や技術に対して進行権を含む一定の権利を持っている。政府資金を用いて新しい技術を開発する場合、政府は、政府が発明を使用することを許可すること、または他の人が政府を代表してその発明を使用させることを許可することを含む、生成された任意の特許のいくつかの権利を得ることができる。これらの権利は、政府が第三者に私たちの情報を開示し、第三者が私たちの技術を使用することを使用または許可する先行権を行使することを可能にするかもしれない。政府が政府援助の技術の実用化を実現できなかったため、健康や安全需要を緩和し、連邦法規の要求を満たすために行動する必要があると考えている場合、あるいは米国工業を優先しなければならないため、政府はそのデモ権利を行使することができる。さらに、このような発明に対する私たちの権利は、そのような発明を含む製品を米国で製造するいくつかの要件によって制約される可能性がある。政府または任意の第三者がその権利を保持するいかなる行使も、私たちの競争地位、業務、財務状況、経営結果、および将来性を損なう可能性があります。
私たちが私たちの候補製品を開発して商業化する権利は、他人が私たちに付与する許可の条項と条件にある程度制限されていますが、私たちのいくつかの候補製品の特許保護、起訴、実行は私たちの許可者に依存するかもしれません。
私たちは現在、候補製品に関連する技術を含む、当社の独自技術の開発に重要または必要な第三者からのいくつかの知的財産権およびノウハウの許可に依存している。これらのライセンスおよび将来締結される可能性のある他のライセンスは、すべての関連する使用分野において、または将来的に技術および製品を開発または商業化することを望むすべての地域でそのような知的財産権および独自技術を使用するのに十分な権利を提供しないかもしれない。我々の開発計画に必要な他の第三者ノウハウまたは知的財産権の許可は、将来的に提供されない場合があり、または商業的に合理的な条項で提供されない可能性がある。この場合、私たちは、私たちの独自技術または候補製品を再設計するために、多くの時間およびリソースを必要とするかもしれないし、代替技術を開発または許可することは、技術的または商業的に不可能である可能性がある。もし私たちがそれができなければ、私たちはこのようなライセンスによって権利を獲得していない使用分野や地域で技術や製品候補製品を開発·商業化することができないかもしれません。これは、私たちの競争地位、業務、財務状況、運営結果、そして将来性を深刻に損なう可能性があります。
場合によっては、私たちは、第三者から許可された技術を含む特許出願の準備、提出、起訴、および実行を制御する権利がないかもしれない。また、私たちとライセンシーとのいくつかの合意は、特許権を実行する前にライセンシーの同意を得ることを要求し、私たちのライセンス者は、このような同意を拒否するか、または直ちに提供しない可能性がある。したがって、私たちのライセンシーまたは協力者が、独自技術および商業秘密の機密性を保護するための合理的な措置をとること、または知的財産権登録に関連するすべての適用可能な起訴および維持費を支払うことを含む、私たちの業務の最適な利益に適合する方法で起訴、保守、実行および擁護するかどうかを決定することはできません。私たちはまた、私たちの許可者が適用された法律法規に従って私たちに付与された特許および特許出願を起草または起訴したかどうかを決定することができず、これは、そのような特許またはそのような出願が発行される可能性のある任意の特許の有効性および実行可能性に影響を与える可能性がある。これは私たちが許可中の任意の知的財産権を適用する権利を失う可能性があるため、私たちが候補製品を開発して商業化する能力は不利な影響を受ける可能性があり、競争相手が競合製品を製造、使用、販売することを阻止できないかもしれない。
86
さらに、私たちの許可者は私たちに許可されていない知的財産権を所有したりコントロールしたりする可能性がありますので、その是非にかかわらず、私たちは私たちが許可者の権利を侵害しているか、または他の方法で侵害しているというクレームを受ける可能性があります。また、将来の製品の販売にどのくらいの印税義務が支払われるかは確定できませんが、金額は大きいかもしれません。私たちの将来の印税義務の金額は、私たちが開発と商業化に成功した候補製品で使用される技術と知的財産権に依存するだろう。したがって,我々はこれまでに2種類の承認された製品を開発して商業化してきたにもかかわらず,これらの製品の販売から相当な収入を得ていないため,将来商業化する可能性のある任意の候補製品の収益性を実現したり維持することができない可能性がある.さらに、我々は、我々の許可者からより多くの許可を得ることを求めることができ、このような許可を得る際には、第三者(我々の競争相手を含む可能性がある)が私たちの既存の許可によって制限されている知的財産権の一部の許可を得ることを許可する条項を含む、許可者に有利な方法で既存の許可を修正することに同意するかもしれない。これらの事件のいずれも、私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります
私たちが第三者に知的財産権を許可する契約の義務を履行できなかった場合、あるいはこれらの合意が終了されたり、許可者との業務関係が妨害されたりすると、私たちは私たちの業務に重要な知的財産権を失う可能性があります。
私たちは様々な合意の締約国であり、私たちはこれらの合意に依存して私たちの業務を運営しており、私たちは現在許可されている知的財産権または将来許可される知的財産権の権利を使用しているか、またはこれらの合意条項の継続と遵守によって制約されるだろう。例えば、私たちは、ラン·スタンフォード初級大学またはスタンフォード大学取締役会と締結された独占的な許可協定の一方であり、acoramidisまたは私たちが決定して追求する可能性のある他の候補製品の商業化を可能にするために、他の人から追加の許可を得る必要があるかもしれない。私たちはスタンフォード大学との許可協定で、私たちは未来の許可協定が様々な開発、勤勉、商業化、その他の義務を私たちに強要すると予想している。特に,スタンフォード大学とのライセンス契約によると,ライセンス製品に関する様々な開発や商業化活動に従事し,特定のマイルストーンや印税支払い義務を満たさなければならない商業的に合理的な努力をしなければならない.私たちもノ華国際製薬有限会社と署名したinfigratinib許可協定の一方であり、この合意によると、商業的に合理的な努力を使用してinfigratinibを開発し、米国とEUで監督管理部門の少なくとも1つのinfigratinibを含む治療製品の承認を得て商業化しなければならない。我々は2021年5月に米国でTRUSELTIQの規制承認を得た。
我々は努力しているにもかかわらず、私たちの許可側は、このような許可協定の下での義務に深刻に違反しているため、許可協定を終了し、これらの許可協定がカバーする製品および技術を開発または商業化する能力を廃止または制限する可能性があると結論する可能性がある。例えば、スタンフォード大学とのライセンス契約が終了すれば、競争相手や他の第三者は、規制部門の承認を求め、acoramidisと同じ製品を市場に投入する権利があり、acoramidisの開発と商業化を停止することが要求されるかもしれない。上記のいずれも、我々の競争地位、業務、財務状況、経営結果、および見通しに重大な悪影響を及ぼす可能性がある。
さらに、ライセンス契約によると、知的財産権に関する紛争が生じる可能性があります
87
しかも、私たちの許可協定のいくつかの条項は様々な解釈の影響を受けるかもしれない。可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または合意下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。また、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で現在の許可スケジュールの能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた候補製品の開発および商業化に成功することができない可能性があり、これは、私たちの競争地位、業務、財務状況、運営結果、および見通しに大きな悪影響を及ぼす可能性がある。
知的財産権侵害に対する第三者のクレームは、私たちの開発と商業化努力を阻害または延期する可能性がある。
私たちの商業的成功は私たちが第三者の特許と固有の権利の侵害を避けることにある程度かかっている。しかし、私たちの研究、開発、および商業化活動は、第三者が所有または制御している特許または他の知的財産権を侵害または他の方法で侵害される可能性がある。アメリカ国内と国外では、裁判所の特許侵害訴訟、妨害、反対、および生物技術と製薬業界の特許とその他の知的財産権に関連する大量の訴訟がある各方面間米国特許商標局及び対応する外国特許庁の再審手続。我々が開発候補を求めている分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する.バイオテクノロジーおよび製薬業界の拡張およびより多くの特許の発行に伴い、アコラミ、低用量infigratinib、BBP-418、BBP-631、アングリタゾン、任意のKRAS阻害剤候補薬物、または私たちが決定する可能性のある他の候補製品が第三者特許権侵害のクレームを受ける可能性があるリスクが増加する。
他の第三者は私たちが許可されていない状況で彼らのノウハウを使用していると断言するかもしれない。我々の候補製品の使用または製造に関連する材料、配合、製造方法、または治療方法を必要とする他の第三者特許または特許出願がある可能性がある。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、我々の候補製品が発行された特許を侵害する可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使ってこれらの特許を侵害したと主張するかもしれない。管轄権のある裁判所が任意の第三者特許を持ち、私たちの候補製品の製造過程、製造過程で形成された任意の分子または任意の最終製品自体をカバーする場合、そのような特許の所有者は、適用特許に基づいて許可を得るか、またはそのような特許が満了するまで、その製品または候補製品を商業化する能力を阻止することができるかもしれない。
同様に、管轄権のある裁判所が任意の第三者特許を保有し、共同療法を含む私たちの処方、製造プロセス、または使用方法の様々な態様をカバーする場合、そのような特許の所有者は、許可または特許の満了を取得しない限り、適用可能な製品または候補製品を開発し、それを商業化する能力を阻止することができるかもしれない。いずれの場合も、そのような許可は、商業的に合理的な条項では得られないかもしれないし、全く存在しないかもしれないし、非排他的である可能性があり、これは、私たちの競争相手が同じ知的財産権を獲得することをもたらす可能性がある。
私たちにクレームを出した当事者たちは禁止または他の公平な救済を受ける可能性があり、これは私たちの候補製品のさらなる開発と商業化を効果的に阻止することができるかもしれない。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、当社の業務における従業員資源を大量に移転することになる。もし私たちに対する侵害クレームが成功した場合、私たちは故意に侵害された3倍の損害賠償と弁護士費、特許権使用料の支払い、私たちの侵害製品の再設計、または第三者から1つまたは複数の許可を得ることを含む大量の損害賠償を支払わなければならないかもしれないし、多くの時間とお金の支出が必要かもしれない。
88
私たちにクレームを出した当事者は、彼らがより多くの資源を持っているので、私たちよりも複雑な特許訴訟の費用を効果的に維持することができるかもしれない。さらに、知的財産権訴訟や行政訴訟に関連する大量の開示要求により、私たちのいくつかの機密情報が開示によって漏洩される可能性がある。さらに、任意の訴訟の開始および継続によって生じる任意の不確実性は、追加資金を調達する能力に重大な悪影響を及ぼすか、または私たちの業務、運営結果、財務状態、および将来性に重大な悪影響を及ぼす可能性がある。
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許の寿命は限られている。米国では、すべての維持費が直ちに支払われる場合、特許の自然失効時間は、通常、その最初の米国非一時的または国際特許出願提出日から20年である。様々な延期があるかもしれないが、特許の有効期限とその提供される保護は限られている。私たちの候補製品が特許を取得しても、特許有効期限が切れると、私たちは模造薬や生体模倣薬を含む競争製品からの競争に直面するかもしれない。製品または新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、そのような製品または候補製品を保護する特許は、そのような製品または候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
もし私たちが“ハッジ-ワックスマン法案”に基づいてアメリカと同様の法律に基づいて外国で取得したり、適用された場合に特許期間を延長したり、非特許独占を維持することができなければ、私たちの候補製品のマーケティング独占期間を延長する可能性があり、私たちの業務は実質的に損害を受ける可能性がある。
FDAが我々の候補製品の発売を承認した期間、期限、および具体的な状況によると、“ハッジ·ワックスマン法案”によると、このような各製品または候補製品またはその使用をカバーする米国特許は、最長5年間の特許期間延長を得る資格がある可能性がある。ハッジ·ワックスマン法は、FDA規制審査中に失われた特許期間の補償として、各FDAが承認した製品に対して最大1つの特許を延長することを可能にする。特許期間の延長は、製品承認日から計14年の期間を超えてはならず、当該承認された薬品、その使用方法又はその製造方法に関連する請求項のみが延長することができる。私たちの候補製品が規制部門の承認を得られれば、特定の国/地域でも特許期間を延長することができる。しかしながら、例えば、試験段階または規制審査中に職務調査を行うことができなかったこと、適用された最終期限内に出願を提出できなかったこと、関連特許が満了する前に出願を提出することができなかったこと、または適用要件を満たすことができなかったため、米国または他のどの国/地域でも特許期間を延長する許可を得ない可能性がある。また、政府当局が提供する延期期限およびそのような延期期間のいずれかの特許保護範囲は、私たちが要求しているものよりも短い可能性がある。
もし私たちが特許期間の延長を得ることができない場合、あるいはこのような延長の期限が私たちが要求したものよりも短い場合、私たちは承認される可能性のある製品を独占的に販売する権利があり、私たちの競争相手は私たちの特許が満期になった後により早く競争製品の承認を得るかもしれません。私たちの収入は減少する可能性があり、実質的な可能性があります。
“ハッジ·ワックスマン法案”によると、ある候補製品をカバーする米国特許のために“ハッジ·ワックスマン法案”に基づいて特許期間を延長しない可能性があり、その特許が特許期間を延長する資格があっても、またはそのような延長を受けた場合、その期間は私たちが求めているよりも短い可能性がある。さらに、私たちが許可したいくつかの特許については、“ハッジ·ワックスマン法案”に基づいて米国特許商標局に特許期間の延長を申請することを含む起訴を制御する権利はありません。したがって,我々のライセンス特許が“ハッジ·ワックスマン法案”によって特許期間延長を得る資格がある場合,特許期間延長を取得した出願を提出したか否か,または米国特許商標局から特許期間延長の出願を取得したかどうかを制御することができない可能性がある。
89
さらに、治療同等性評価を有する医薬製品またはオレンジマニュアルに列挙するために、承認された治療同等性評価を有する医薬製品またはオレンジマニュアルに列挙するために、特許に関する詳細な規則および要求がある。私たちはオレンジブックの発売要件を満たす1つ以上の特許を含む私たちの候補製品をカバーする特許を得ることができないかもしれない。私たちがオレンジブックに特許を提出しても、FDAはその特許のリストを拒否するかもしれないし、模倣薬製造業者は発売に疑問を提起するかもしれない。現在の候補製品のうちの1つが承認され、候補製品をカバーする特許がオレンジマニュアルに記載されていない場合、模倣薬製造業者は、そのような製品の模倣薬バージョンを販売する許可を得るために、FDAに提出された任意の簡略化された新薬申請またはANDAを事前に通知する必要がないであろう。
当製品の発売承認の時間と詳細によると、FDAおよび他の適用される規制機関は、いくつかの非特許排他性を付与する可能性があります。我々が開発している候補製品のために新たな化学実体排他性を求め,他の排他性を求めることが可能であるにもかかわらず,我々はそれに成功しない可能性がある.さらに、これらの非特許専有権が付与されていれば、これらの専有権は限られており、他社は私たちが予想していたよりも早くマーケティング申請を提出し、承認される可能性がある。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの技術的価値は実質的な悪影響を受ける可能性があり、私たちの業務は損害を受けるだろう。
従業員、コンサルタント、科学コンサルタント、請負業者、協力者と秘密協定や発明譲渡協定を締結することで、私たちの機密独自情報をある程度保護することを求めています。このような協定は私たちの固有の情報を保護することを目的としている。しかし、私たちはすべての関係者とこのような合意を締結したことを確認することはできず、私たちの商業秘密や他の機密固有情報が開示されないか、あるいは競争相手が私たちの商業秘密を取得したり、実質的に同等の情報や技術を独立して開発しないかを決定することもできない。例えば、これらの当事者のいずれかは、合意に違反し、商業秘密を含む独自の情報を開示する可能性があり、私たちは、このような違反について十分な救済措置を得ることができない可能性がある。私たちはまた、私たちの住宅地の実体セキュリティと私たちの情報技術システムの実体と電子セキュリティを維持することで、私たちの機密固有の情報の完全性と機密性を維持しようとしているが、これらのセキュリティ措置は破壊される可能性がある。もし私たちの任意の機密固有情報が競争相手によって合法的に取得または独立して開発された場合、私たちはその競争相手がその技術または情報を使用して私たちと競争することを阻止する権利がなく、これは私たちの競争地位を損なう可能性がある。
許可されていない当事者たちはまた、私たちの候補製品のいくつかを複製または逆工学しようと試みるかもしれないが、私たちは固有の側面だと思う。もしこのような不正な使用が発生したら、私たちは十分な救済措置を得ることができないかもしれない。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかる可能性があり、結果は予測できない。しかも、米国国内外のいくつかの裁判所は商業秘密をあまり望んでいないか、または保護したくない。時間の経過とともに、商業秘密はまた、独立して定期刊行物文章を開発、発表し、芸術技能を持つ人員を1つの会社から別の会社に移転すること、あるいは学術界から業界科学職に移転することによって業界内に伝播する。私たちが第三者と合意した合意は、通常、私たちのコンサルタント、従業員、協力者、ライセンシー、サプライヤー、第三者請負業者、およびコンサルタントが、私たちのビジネス秘密に関連する可能性のあるデータを発行する能力を制限しますが、私たちの合意には、いくつかの限られた発行権が含まれている可能性があります。さらに、もし私たちの任意のビジネス秘密が競争相手によって合法的に取得または独立して開発された場合、私たちはその競争相手がその技術や情報を使用して私たちと競争することを阻止する権利がなく、これは私たちの競争地位を損なう可能性がある。上述した契約および他の安全予防措置がとられているにもかかわらず、商業秘密を共有する必要は、そのような商業秘密が私たちの競争相手に知られ、意図せずに他の人の技術に組み込まれるか、または開示されるか、またはこれらの合意に違反するリスクを増加させる。もしこのような事件が発生した場合、あるいは私たちが私たちのビジネス秘密の保護を失った場合、これらの情報の価値は大幅に低下する可能性があり、私たちの競争地位、業務、財務状況、運営結果、および将来性は損なわれるだろう。
90
もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は悪影響を受けるかもしれません。
我々の登録または未登録商標または商号は、疑問、侵害、回避、または汎用商標として発表されるか、または他の商標を侵害していると認定される可能性がある。私たちはこれらの商標と商品名に対する私たちの権利を保護できないかもしれません。私たちは私たちが関心のある市場における潜在的な協力者や顧客の中で知名度を確立するために必要です。時々、競争相手は私たちと似たような商品名や商標を採用して、ブランド表示を確立する能力を阻害し、市場の混乱を招く可能性があります。さらに、他の商標または商標の所有者は、我々の登録または未登録商標または商号の変異体を含む商号または商標侵害クレームを提起することができる。長期的には、私たちの商標や商号に基づいて名称を確立することができなければ、効果的に競争できない可能性があり、私たちの業務は悪影響を受ける可能性があります。私たちは私たちの商標と商品名を流通業者のような第三者に許可するかもしれない。これらのライセンス契約は、私たちの商標や商号をどのように使用するかに指針を提供する可能性がありますが、ライセンシーは、これらの合意に違反したり、私たちの商標およびビジネス番号を乱用したりすることで、私たちの権利を危険にさらしたり、私たちの商標や商号に関連する商標の名誉を弱める可能性があります。商標、商業名、商業秘密、ドメイン名、著作権、または他の知的財産権に関連する独自の権利を実行または保護する努力は無効である可能性があり、大量のコストおよび資源移転をもたらす可能性があり、私たちの競争地位、業務、財務状況、運営結果、および将来性に悪影響を及ぼす可能性があります。
私たちは私たちの特許や他の知的財産権を保護したり強制したりする訴訟に巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
競争相手は私たちの特許や他の知的財産権を侵害するかもしれない。もし私たちが第三者に対して法的訴訟を提起して、私たちの1つ以上の候補製品をカバーする特許を強制的に執行する場合、被告は私たちの候補製品をカバーする特許を無効および/または実行できないと反訴することができる。米国の特許訴訟では,被告が無効および/または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性、不可視性、書面記述、または有効化を含むいくつかの法定要件のいずれかを満たしていないと言われているからかもしれない。主張を実行できない理由は,特許起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った声明をしたりしたためかもしれない.法的に無効と実行不可能と断言された後の結果は予測できない。第三者によって引き起こされるか、または我々によって提起されるか、または米国特許商標局によって発表される干渉または派生プログラムは、我々の特許または特許出願に関連する発明の優先権を決定するために必要である可能性がある。不利な結果は、私たちが関連技術の使用を停止することを要求するか、または勝利者から許可を得ようとすることを要求するかもしれない。もし勝利者が商業的に合理的な条項で私たちに許可を提供しない場合、あるいは私たちに許可を全く提供しない、あるいは非独占的な許可を提供し、私たちの競争相手が同じ技術を獲得した場合、私たちの業務は損害を受ける可能性がある。私たちの訴訟、介入、または派生訴訟の弁護は失敗する可能性があり、成功しても巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性がある。また,訴訟に関する不確実性は,臨床試験を継続し,研究計画を継続するために必要な資金を調達する能力に実質的な悪影響を及ぼす可能性がある, 第三者から必要な技術許可を得て、開発パートナー関係を構築し、候補製品の市場進出を支援してくれます。さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に実質的な悪影響を及ぼす可能性がある。
私たちは私たちの特許と他の知的財産権発明権のクレームに疑問を受けるかもしれない。
私たちと従業員との合意と私たちの人事政策規定は、個人が私たちにサービスを提供する過程で構想されたどの発明も私たちの独自の財産になるだろう。私たちの政策はこのすべての個人がこれらの合意を締結させることですが、私たちはいかなる状況でもこれらの合意を獲得しないかもしれません。私たちとこれらの合意を持っている個人は彼らの条項を守らないかもしれません。知的財産権の譲渡は、発明創造時に自動的に行うことはできず、このような合意にもかかわらず、このような発明は第三者に譲渡される可能性がある。私たちの商業秘密または独自の情報を不正に使用または開示する場合、これらの合意を取得しても、特に私たちの商業秘密または他の機密情報を提供することができない可能性がある。
91
私たちまたは私たちの許可者は、元従業員、協力者または他の第三者が、発明者または共同発明者として、私たちの所有または許可内の特許、商業秘密、または他の知的財産権において権益を有するというクレームを受ける可能性がある。例えば、私たちまたは私たちの許可者は、私たちの候補製品の開発に参加している従業員、コンサルタント、または他の人の義務紛争によって発明権紛争を生じる可能性があります。これらのまたは他の挑戦在庫または私たちまたは私たちの許可者による私たちの所有または認可内の特許、商業秘密、または他の知的財産権の所有権のクレームに対抗するために、訴訟を提起する必要があるかもしれない。もし私たちまたは私たちの許可者がこのようなクレームを弁護できなかった場合、金銭損害賠償を支払うことに加えて、私たちの候補製品に非常に重要な知的財産権の独占所有権や使用権のような貴重な知的財産権を失う可能性があります。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
上記のいずれも、我々の競争地位、業務、財務状況、経営結果、および見通しに重大な悪影響を及ぼす可能性がある。
私たちの候補製品をカバーする発行された特許が法廷で疑問視されれば、無効または実行不可能と認定される可能性がある。
私たちまたは私たちの許可パートナーのうちの1つが第三者に対して訴訟を起こし、私たちの1つまたは複数の候補製品をカバーする特許の強制執行を要求する場合、被告は関連する候補製品をカバーする特許の無効および/または強制執行を反訴することができる。米国の特許訴訟では,被告が無効および/または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性、不可視性、書面記述、または有効化を含むいくつかの法定要件のいずれかを満たしていないと言われているからかもしれない。主張を実行できない理由は,特許起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った声明をしたりしたためかもしれない.第三者も米国や海外の行政機関に類似したクレームを出すことができ、訴訟範囲外でも同様である。このようなメカニズムには、再審、贈与後審査、外国司法管轄区の同等の訴訟手続(例えば、反対訴訟手続)が含まれる。このような訴訟は、私たちの製品や候補製品をカバーしないように、私たちの特許が撤回または修正される可能性がある。法的に無効と実行不可能と断言された後の結果は予測できない。例えば、有効性の問題については、私たちは無効な以前の技術がないとは確信できないが、私たちと特許審査員は起訴中にこれを知らない。もし被告が無効および/または強制執行できない法的主張に勝った場合、私たちは私たちの候補製品の少なくとも一部またはすべての特許保護を失うだろう。このような特許保護の喪失は私たちの業務に実質的な悪影響を及ぼすだろう。
私たちは、私たちの従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示したか、または私たちの従業員がその前の雇用主によって言われた商業秘密を誤って使用または開示したという疑惑の影響を受けるかもしれない。
バイオテクノロジーや製薬業界でよく見られるように、私たちが雇った個人は以前、私たちの競争相手や潜在的な競争相手を含む大学や他のバイオテクノロジーや製薬会社に雇われていた。私たちは、私たちの従業員、コンサルタント、および独立請負業者が、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちは、商業秘密または他の固有情報を含む、私たちまたは私たちの従業員、コンサルタントまたは独立請負業者が、私たちの従業員の任意の元雇用主または他の第三者の知的財産権を無意識にまたは他の方法で使用または開示しているというクレームを受ける可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償の支払いに加えて、貴重な知的財産権や人員を失う可能性があり、これは私たちの業務に悪影響を及ぼす可能性がある。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
92
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
特許および/または出願の定期維持費、継続費、年会費、および様々な他の政府費用は、特許および/または出願の有効期間内にいくつかの段階で米国特許商標局および米国以外の様々な政府特許機関に支払われる。私たちはこれらの費用を支払うようにシステム的に注意して、私たちは外部会社を招聘し、外部弁護士に依存して非アメリカの特許代理機関にこれらの費用を支払います。しかし、私たちは私たちの許可者がこのような費用を支払うために似たようなシステムと手続きを持っているという保証はない。米国特許商標局および様々な非政府特許機関は、特許出願中にいくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。私たちは評判の良い法律事務所や他の専門家を招いて私たちの遵守を助けてくれました。多くの場合、不注意は滞納金を支払うことによって、あるいは規則を適用する他の方法で是正することができます。しかしながら、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効される可能性があり、それにより、関連法ドメインの特許権の一部または全部が失われる可能性がある。この場合、私たちの競争相手が市場に参入する可能性があり、このような状況は私たちの業務に実質的な悪影響を及ぼすだろう。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
世界のすべての国で私たちの候補製品のために特許を申請、起訴、保護する費用は目を引くほど高く、私たちのアメリカ以外のいくつかの国での知的財産権はアメリカほど広くないかもしれない。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使って自分の製品を開発することができ、私たちが特許保護を持っている地域に侵害製品を輸出することもできるが、法執行力はアメリカに及ばない。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護の強制執行、特にバイオテクノロジーおよび医薬製品に関する保護を支持しておらず、これは、私たちの特許を侵害したり、私たちの独占権を侵害する方法で競争製品を販売することを阻止することを困難にするかもしれない。外国の管轄区域で私たちの特許権の訴訟を強制的に執行することは、成功するかどうかにかかわらず、巨額のコストを招く可能性があり、業務の他の方面への私たちの努力と注意力を移転させ、私たちの特許を無効または狭義に解釈されるリスクに直面させる可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者が私たちにクレームを提起する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。したがって、私たちが世界各地で私たちの知的財産権を実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
93
米国特許法の変化は特許の全体的な価値を低下させ、製品を保護する能力を弱める可能性がある。
米国特許法または特許法解釈の変化は、特許出願をめぐる起訴および発行された特許の実行または弁護の不確実性およびコストを増加させる可能性がある。特許性の他の要求が満たされていると仮定すると,2013年3月までに,米国では,最初に発明して保護された発明を要求した者が特許を取得する権利があるが,米国以外では,最初に特許出願を提出した者が特許を取得する権利がある。2013年3月以降、2011年9月に公布された“ライシー·スミス米国発明法”または“米国発明法”によれば、米国は、第1の発明者が特許を出願する制度に移行し、すなわち、他の特許性要件を満たすと仮定して、最初に特許出願を提出した発明者は、要求された発明が第三者が最初に発明したものであるか否かにかかわらず、発明の特許を取得する権利がある。2013年3月以降に米国特許商標局に特許出願の第三者を提出したが、我々の前に、当該第三者が発明を行う前に本発明を作成したとしても、我々の発明をカバーする特許を付与することができる。これは私たちが発明から特許出願までの提出時間を認識することを要求するだろう。米国およびほとんどの他の国/地域の特許出願は、提出後または発行前の一定期間秘密であるため、私たちまたは私たちのライセンス者が、私たちの候補製品に関連する任意の特許出願または(Ii)発明または私たちのライセンシーの特許または特許出願に要求される任意の発明を最初に提出することを決定することはできない。
米国発明法にはいくつかの重大な変化も含まれており、これらの変化は特許出願の起訴方法に影響を与え、特許訴訟に影響を与える可能性もある。これらの措置には、第三者が特許訴訟中に米国特許商標局に以前の技術を提出することを可能にすることと、米国特許商標局によって管理されたライセンス後のプログラム(ライセンス後審査を含む)が特許有効性を攻撃することを可能にする追加のプログラムとが含まれる各方面間審査と派生手続き。USPTO手続きの証拠基準は、米国連邦裁判所が特許請求の無効を宣言するために必要な証拠基準よりも低いため、第三者は、USPTO手続において、USPTOが権利請求を無効にするのに十分な証拠を提供する可能性があり、同じ証拠が最初に連邦地域裁判所訴訟で提出された場合には、権利請求を無効にするのに十分ではない。したがって,第三者は米国特許商標局の手続きを用いて我々の特許主張の無効を宣言しようとする可能性があり,第三者が連邦地方裁判所の訴訟で最初に被告として疑問を提起すれば,我々の特許主張は無効ではない.したがって、米国発明法およびその実施は、私たちの所有または許可をめぐる特許出願の起訴、および私たちが所有または許可した発行された特許の実行または弁護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
また、薬品研究開発と商業化における企業の特許地位は特に不確定である。米国最高裁判所の最近の裁決は、場合によっては入手可能な特許保護範囲を縮小し、場合によっては特許所有者の権利を弱める。この一連の事件は特許取得後の有効性と実行可能性の面で不確実性をもたらしている。米国議会、連邦裁判所、および米国特許商標局の将来の行動によると、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、私たちの既存の特許の組み合わせおよび私たちの将来の知的財産権の保護と実行能力に重大な悪影響を及ぼす可能性がある。
94
商業化に関連するリスク
我々の候補製品が承認されれば,医師,患者,医療支払者,医学界の他の人がビジネス成功に必要な市場受容度を得ることができない可能性がある。
私たちの候補製品の商業成功は、承認されれば、彼らが医師、患者、第三者支払人と医学界の他の人に受け入れられる市場の程度に依存する。私たちの候補製品は、承認されれば、依然として医師、患者、医療支払人、医学界の他の人の十分な市場受容度を得ることができないかもしれない。もし私たちが開発した候補製品が商業販売に使用されることが許可されれば、市場のその製品に対する受け入れ度は多くの要素に依存する
医療製品の販売は医師が治療処方を出す意思にも依存し、これはこれらの医師に基づいて製品が安全で、治療が有効であり、費用効果があることを確定した可能性が高い。また,異なる医師団体による治療ガイドラインや影響力のある医師の観点から製品を取り入れたり排除したりすることは,他の医師が治療処方を処方する意思に影響する可能性がある。私たちは医師、医師組織、病院、他の医療提供者、政府機関、あるいは個人保険会社が、私たちの製品が競争相手の治療と比較して安全で、治療が有効で、費用効果があるかどうかを決定することができません。もし私たちが開発したすべての候補製品が十分な受容度に達していなければ、私たちは著しい製品収入が生じないかもしれません。私たちは利益を上げることができないかもしれません。
95
販売およびマーケティング能力を確立することができない場合、または我々が開発する可能性のある任意の候補製品を販売およびマーケティングするために第三者と合意できない場合、これらの候補製品が承認された場合、商業化に成功できない可能性がある。
私たちが販売とマーケティング責任を保留する任意の承認された製品を商業的に成功させるためには、販売とマーケティング組織を発展させたり、これらの機能を第三者にアウトソーシングしたりしなければならない。将来、私たちの候補製品が承認されれば、私たちの重点販売、マーケティング、商業支援インフラを拡大して、私たちの候補製品をマーケティングして販売することを選択するかもしれません。TRUSELTIQが承認された後にHelsinnに対して行ったように、TRUSELTIQが承認された後にHelsinnに対して行ったように、特定の候補製品、標識、または地理的地域(米国以外の地域を含む)の商業化活動に参加することを選択することもできる。
私たち自身のビジネス能力の確立と第三者とのこれらのサービス提供の合意はリスクに関連している。例えば、販売員の募集と訓練や専門員の精算は高価で時間がかかり、どんな製品の発表も延期される可能性がある。販売チームを募集し、マーケティングや他の商業化能力を確立する候補製品の商業発表が何らかの理由で延期または発生していない場合には、これらの商業化費用を早期または不必要に発生させる。これは費用がかかるかもしれないし、もし私たちが商業化者を維持したり再配置できなければ、私たちの投資は損失するだろう。
私たち自身が承認された製品を商業化するのを阻害するかもしれません
もし私たちが第三者と合意して、販売、マーケティング、商業支援、流通サービスを行えば、私たちの製品収入または製品収入の収益力は、私たちの内部で開発された任意の製品をマーケティングし、販売する場合よりも低いかもしれません。さらに、私たちは第三者との合意に成功し、私たちの候補製品を商業化することができないかもしれません(承認されれば)、または私たちまたは彼らに有利な条項でそうすることができないかもしれません。私たちはこのような第三者に対して統制権がほとんどないかもしれませんが、どの第三者も必要な資源や注意力を投入して私たちの製品を効果的に販売し、マーケティングすることができないかもしれません。あるいは処方薬製品の販売と普及に関する規制要件や制限(ラベル外販売を制限する要求や制限を含む)に従わないことで、法律と規制リスクに直面する可能性があります。もし私たちが私たちの商業化能力を成功させ続けなければ、私たち自身も第三者と協力して、私たちの候補製品が承認されれば、私たちはそれを商業化することに成功しないだろう。
96
新ロット製品の保険カバー範囲と精算状況はまだ確定していない。私たちの候補製品は不利な価格設定法規、第三者保険、清算やり方、あるいは医療改革措置の影響を受ける可能性があり、これは私たちの業務を損なうことになります。新製品や既存製品のために十分な保険や精算を得ることができない場合、これらの製品をマーケティングする能力を制限し、収入を創出する能力を低下させる可能性があります。
新薬の発売審査、定価、カバーと精算を管理する規定は国によって異なる。米国では、最近公布された立法が承認要求を大幅に変更する可能性があり、これは追加のコストに関連し、承認の遅延を招く可能性がある。一部の国は薬品の販売価格が承認されてから発売されることを要求している。多くの国で、定価審査期間はマーケティングまたは製品許可を承認した後から始まる。一部の外国市場では、処方薬の定価は初歩的な承認を得た後も、政府の持続的なコントロールを受けている。したがって、特定の国/地域での製品のマーケティング承認を得ることができるかもしれませんが、その後、価格法規の制約を受けて、これらの法規は私たちの製品の商業発表を延期し、長い時間遅延し、その国/地域で製品を販売することによって生じる収入に悪影響を与える可能性があります。不利な価格設定制限は、これらの候補製品が市場承認を得る可能性があっても、1つ以上の候補製品への投資を回収する能力を阻害する可能性がある。タイトルを見て“企業-政府規制-保険範囲と補償.”
私たちの候補製品の商業化に成功できるかどうかは、政府衛生行政部門、個人健康保険会社、その他の組織がこれらの製品と関連治療に提供する保険範囲と十分な補償にある程度依存する。政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、どのような薬剤を支払うかを決定し、精算レベルを確立する。政府や個人支払者が提供する保険範囲や精算範囲は,多くの患者が遺伝子治療製品などの治療を負担できるために重要である。承認された場合、私たちが決定する可能性のあるこれらの候補製品または他の候補製品の販売は、私たちの候補製品のコストが、健康維持、管理医療、薬局福祉および同様の医療管理組織によってどの程度支払われるか、または政府衛生行政当局、個人健康保険会社、および他の第三者支払者によってどの程度精算されるかに大きく依存する。保険や十分な精算が得られない場合、あるいは限られたレベルに限られていれば、候補製品を商業化することに成功できないかもしれません。保険を提供しても、承認された精算金額が十分に高くない可能性があり、十分な投資リターンを実現するために十分な価格設定を確立したり維持したりするのに十分ではないかもしれません。
また、私たちは私たちの候補製品と一緒に使用するセット診断テストを開発するかもしれない。私たちまたは私たちの協力者は、これらのテストのために単独で保険と精算を受けることを要求されるかもしれません。承認されると、私たちの候補製品のために求められる保険と精算を除いて。このようなセット診断に対する規制機関の承認や許可を得ても、私たちの候補製品に適用されるのと同じ理由で、私たちが保険と十分な補償を受ける能力にはまだ大きな不確実性がある。連邦医療保険精算方法は、A部分、B部分、あるいは臨床実験室費用表に基づいて、時々修正される可能性があり、これらの方法の任意の変更が私たちが承認された任意の製品或いは製品候補或いは随伴診断にどのような影響を与えるかを予測することができない。私たちが開発し、規制の承認を得たセット診断テストでは、第三者支払者から保険と十分な補償を迅速に受けることができず、私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性があります。
97
もし私たちが医療保険法を守らなければ、私たちは巨額の処罰に直面する可能性があり、私たちの業務、運営、財務状況は不利な影響を受けるかもしれない。
米国や他の地域の医療提供者,医師,第三者支払者は薬品の推奨と処方に主な役割を果たしている。第三者支払者および顧客との手配は、製薬業者を広範に適用される詐欺および乱用、および他の医療法律および法規に直面させる可能性があり、これらの法律および法規は、このような会社の販売、マーケティング、および薬品の流通業務または財務手配および関係を制限する可能性がある。特に、医療製品やサービスの普及、販売およびマーケティング、および医療業界のいくつかの商業的配置は、詐欺、リベート、自己取引、および他の乱用行為を防止するための広範な法律によって制限されている。これらの法律および法規は、広範な所有権、定価、割引、マーケティングおよび販売促進、構造および手数料、特定の顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。これらの法的拘束を受けた活動は,患者を募集して臨床試験を行う過程で得られた情報の不正使用に関するものである。タイトルを見て“企業−政府規制−その他規制事項.”
これらの法律の広さ、および法定例外状況および既存の規制安全港の狭さのため、私たちのいくつかの商業活動は、株式または株式オプションで医師に報酬を支払うことを含み、遵守しようと努力しているにもかかわらず、1つ以上のそのような法律の挑戦を受ける可能性がある。さらに、FDAまたは外国の規制機関は、臨床試験において研究者または機関に提供される支払いまたは株式によって生じる任意の偏見リスクを軽減することに同意しない可能性があり、これは、マーケティング申請を支援するために、規制当局がこれらの臨床試験データを受け入れることを制限する可能性がある。また,我々の業務手配が適用される医療保険法に適合することを確保する努力は巨額のコストに及ぶ可能性がある。政府および法執行当局は、私たちの業務実践が、詐欺および乱用または他の医療保健法律および法規を適用する現在または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。もし私たちにこのような訴訟を提起した場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの訴訟は、違反疑惑、契約損害、名声損害、利益減少および将来の収益を解決するために、重大な民事、刑事および行政処罰、損害賠償、返還、罰金、連邦医療保険、医療補助および他の連邦医療保健計画から除外された重大な影響を与えることができ、いずれも、私たちの業務運営能力および運営結果に悪影響を及ぼす可能性があります。また、, 我々の米国以外の任意の候補製品の承認と商業化は,上記の医療保健法や他の外国法の外国等価物の制約を受ける可能性もある。
98
会社の資金支援を受ける第三者患者援助計画は、政府や規制機関の審査強化の対象となっている。政府の法執行機関は、精算支援サービスを含む製薬会社の製品や患者支援計画にますます大きな興味を示しており、これらの計画のいくつかの調査は、重大な民事·刑事和解につながっている。米国政府はすでにガイドラインを策定しており,製薬業者が医療保険患者に共同支払い援助を提供する慈善団体への寄付が合法であることを提案しており,これらの組織が真の慈善機関であり,メーカーから完全に独立しており,メーカーに支配されず,一致した財務基準に基づいて先着で申請者に援助を提供し,援助をドナーの製品の使用にリンクさせないことを前提としている。しかし、患者支援プロジェクトへの寄付はいくつかの否定的な宣伝を受け、政府の複数の法執行行動の対象となったが、これらの寄付は他のコストの低い代替製品ではなく、ブランド薬品を普及させるために使用されたという疑惑があるからである。具体的には,近年,様々な連邦や州法により,政府がその患者援助計画の合法性を疑問視し,複数の和解を招いている。私たちは独立した慈善基金に寄付金を提供し、経済的に困難な患者が保険料、共同支払い、共同保険の義務を履行するのを助けるかもしれない。もし私たちがそうすることを選択した場合、私たちまたは私たちの供給者または寄付受給者がこれらの計画の運営中に関連する法律、法規、または変化する政府指導を遵守できなかったと考えられた場合、私たちは損害賠償、罰金、処罰、または他の刑事、民事罰を受ける可能性がある, 行政処罰や法執行行動です私たちは私たちのコンプライアンス統制、政策、そして手続きが私たちの従業員、業務パートナー、またはサプライヤーの行為が私たちの管轄区域の法律や法規に違反することを防ぐのに十分であることを確実にすることはできない。私たちが法律を遵守しているかどうかにかかわらず、政府の調査は私たちのビジネス行動に影響を与え、私たちの名声を損ない、経営陣の注意を移し、私たちの費用を増加させ、助けを必要とする患者に基礎的な支援を提供する可能性を減らすことができる。さらに、保険会社の自己クーポン政策の変更および/または新しい立法または規制行動の導入および発行は、これらの患者支援計画に制限または他の方法で悪影響を及ぼす可能性があり、これは、影響を受けた製品をより少ない患者に使用させる可能性があり、それによって、私たちの販売、業務、および財務状態に実質的な悪影響を及ぼす可能性がある。いくつかの措置および他の提案された措置は、発効するために追加の立法許可を必要とする可能性があり、現米大統領政府はこれらの措置を撤回または他の方法で変更する可能性があるが、現在の米国大統領政府も国会も、薬品コストを制御するための新しい立法措置を求め続けると表明している。私たちはこの規則の施行とどんなさらなる変化が私たちの業務にどのように影響するのか予測できない。
健康およびデータ保護法律法規を遵守しないことは、政府の法執行行動(民事または刑事罰を含む可能性がある)、個人訴訟および/または負の宣伝をもたらす可能性があり、私たちの経営業績および業務に負の影響を与える可能性がある。
私たちと任意の潜在的な協力者は、連邦、州、および外国のデータ保護法律および法規(すなわち、プライバシーおよびデータセキュリティに関する法律および法規)によって制約される可能性がある。米国では、健康関連情報および他の個人情報の収集、使用、開示および保護を管理する多くの連邦および州法律法規、連邦健康情報プライバシー法、州データ漏洩通知法、州健康情報プライバシー法および連邦および州消費者保護法(例えば、連邦貿易委員会法第5条)は、私たちの運営または私たちの協力者の運営に適用される可能性がある。また,HIPAAのプライバシーやセキュリティ要求に制約された第三者(臨床試験データを取得した研究機関を含む)から健康情報を取得することが可能であり,HIPAAは2009年の“経済·臨床健康情報技術法案”(HITECH)により改正されている。事実および状況によると、もし私たちがHIPAAによって許可されていないまたは許可されていない方法でHIPAAによってカバーされているエンティティによって維持されている個別に識別可能な健康情報を取得、使用、または開示する場合、私たちは民事、刑事、および行政処罰を受ける可能性がある。
99
カリフォルニアは最近、“カリフォルニア消費者プライバシー法”(California Consumer Privacy Act、略称CCPA)を公布し、カリフォルニアの消費者のために新しいプライバシー権(法律で定義されているような)を創造し、消費者または家庭の個人データを処理する実体に対してより多くのプライバシーと安全義務を規定した。また、カリフォルニアの有権者は2020年11月3日にCPRAと略称する新しいプライバシー法であるカリフォルニアプライバシー権法案を可決した。CPRAは、2023年1月1日から個人情報に関する義務を増加させる(ある条項は遡及効力を有する2022年1月1日まで)。立法および提案された法規には、CCPAおよびCPRAがHIPAAによって制約された活動の例外を含むことが含まれているが、CCPA、CPRA、または他のこのような将来の法律、法規、および基準が私たちの業務に及ぼす可能性のある影響を決定することはできない。CCPA、CPRA、その他の管轄区域で採用される可能性のある他の類似の法律、法規と標準の実施には不確実性があり、これは私たちの業務が個人データと保護された健康情報に関する変化する規制環境の影響を受けやすいことを示している。米国と国際データ保護法律法規を遵守することは、私たちがデータを収集、使用、開示する能力を制限し、または場合によってはいくつかの司法管轄区域で運営する能力に影響を与える契約でより重い義務を負うことを要求するかもしれない。これらの法律法規を遵守しないことは、政府の法執行行動(民事、刑事および行政処罰を含む可能性がある)、個人訴訟および/または負の宣伝をもたらす可能性があり、私たちの経営業績および業務に負の影響を与える可能性がある。さらに、私たちまたは潜在的な協力者が取得した臨床試験被験者、従業員、他の個人の個人情報、およびこの情報を共有する提供者は, 私たちが情報を収集、使用し、開示する能力を制限するかもしれない。私たちがプライバシー権を侵害していると主張し、データ保護法を遵守できなかったり、私たちの契約義務に違反したりして、私たちが責任を負わなくても、弁護の費用が高く、時間がかかり、否定的な宣伝を招き、私たちの業務を損なう可能性があります。
また、一部の観察者は、CCPAとCPRAは、米国のより厳格なプライバシー立法傾向の開始を示している可能性があり、これは私たちの潜在的な責任を増加させ、私たちの業務に悪影響を及ぼす可能性があると指摘している。アメリカで、私たちは州レベルの重大な発展を目撃した。例えば,2021年3月2日,バージニア州でCDPAが公布され,2021年7月8日,コロラド州知事がCPAに署名し,法律とした。CDPAとCPAはいずれも2023年1月1日に発効した。CDPAとCPAはCCPAとCPRAの多くの類似した概念を含んでいるが、範囲、適用と法執行の面でもいくつかの重要な違いがあり、これらの法律は規制された企業の運営方法を変える。その他の事項を除いて、新しい法律は、規制された企業が個人の敏感なデータを収集して処理し、データ保護評価を行い、個人データを付属会社に移転し、消費者の権利要求に応答する方法に影響を与える
他のいくつかの州もまた、上述した最近採択された法律と類似した新しいプライバシー法を提案した。このような提案された立法が通過されると、追加の複雑性、要求変化、制限、および潜在的な法的リスクが増加する可能性があり、コンプライアンス計画、影響戦略、および以前の有用なデータの利用可能性に追加のリソースを投入する必要があり、コンプライアンスコストの増加および/またはビジネス実践および政策の変化をもたらす可能性がある。アメリカの異なる州に全面的なプライバシー法が存在することは、私たちのコンプライアンス義務をより複雑で費用を高くし、私たちが法執行行動を受けたり、他の方法で規則に合わないことで責任を負う可能性を増加させるかもしれない。
最近や新興の州プライバシー法律,法規,基準の実施には不確実性があり,これらの法律,法規,基準は他の管轄区で採用される可能性があり,我々の業務が個人データや保護された健康情報に関する変化する規制環境の影響を受けやすいことを示している。米国と国際データ保護法律法規を遵守することは、私たちがデータを収集、使用、開示する能力を制限し、または場合によってはいくつかの司法管轄区域で運営する能力に影響を与える契約でより重い義務を負うことを要求するかもしれない。これらの法律法規を遵守しないことは、政府の法執行行動(民事、刑事および行政処罰を含む可能性がある)、個人訴訟および/または負の宣伝をもたらす可能性があり、私たちの経営業績および業務に負の影響を与える可能性がある。さらに、私たちまたは私たちの潜在的な協力者が個人情報を取得する臨床試験被験者、従業員および他の個人、およびこの情報を共有する提供者は、情報を収集、使用、および開示する能力を制限する可能性がある。私たちがプライバシー権を侵害していると主張し、データ保護法を遵守できなかったり、私たちの契約義務に違反したりして、私たちが責任を負わなくても、弁護の費用が高く、時間がかかり、否定的な宣伝を招き、私たちの業務を損なう可能性があります。
100
ヨーロッパのデータ収集は,個人情報の使用,処理,国境を越えた移行に関する限定的な規定によって制約されている.
もし私たちが追加の臨床試験を行うことを決定したり、EUやイギリスで被験者を募集して私たちが行っているまたは未来の臨床試験に参加し続けると、追加のプライバシー制限を受ける可能性があります。EUでは,個人健康データの収集と使用はイギリスGDPRの規定によって管轄されている。GDPRは,個人データに関する個人の同意,個人への情報提供,国家データ保護主管部門へのデータ処理義務の通知,個人データのセキュリティと秘密についていくつかの要求を行っている。英国GDPRはまた,個人データをEUから米国に移行するための厳しいルールを実施している。英国GDPRの要求やEU加盟国の関連国データ保護法を遵守しないことは罰金やその他の行政処罰を招く可能性がある。イギリスのGDPR規制は、私たちが処理している個人データに追加の責任と責任を課す可能性があり、これらおよび/または新しいデータ保護ルールの遵守を保証するための追加のメカニズムを確立する必要があるかもしれません。これは激務であり、私たちの業務、財務状況、見通し、運営結果に悪影響を及ぼす可能性がある。
医療コストを低減するための医療立法措置は,我々の業務や運営結果に実質的な悪影響を及ぼす可能性がある。
米国や多くの外国司法管轄区域は、医療システムに影響を与える立法や規制改革を策定または提案しており、これらの改革は、候補製品や将来の候補製品の上場承認を阻止または延期し、承認後の活動を制限または規制し、マーケティングの承認を得た任意の製品を収益的に販売する能力に影響を与える可能性がある。規制、法規、または既存の規制の解釈の変化は、(I)私たちの製造スケジュールを変更すること、(Ii)製品ラベルを追加または修正すること、(Iii)任意の製品をリコールまたは停止すること、または(Iv)追加の記録保存要件を要求することなど、私たちの将来の業務に影響を与える可能性がある。このような変化を強制的に実施すれば、私たちの業務運営に悪影響を及ぼす可能性がある。タイトルを見て“企業−政府規制−現在と将来の立法.”
さらに、2019年には、承認されたNDAおよびBLAのスポンサーに、商業的に合理的で市場に基づく条項で、模倣薬および生物類似生物製品を開発する実体に十分な数の製品サンプルを提供することを要求する“創造と回復平等獲得同等のサンプル法”が公布された。この法律は、開発者が、その申請を支援するために必要な製品サンプルの売却を拒否したアプリケーション所有者を起訴することを可能にする個人的な訴権を確立する。この法律によれば、私たちは、そのような要求または任意の法的挑戦に応答するために製品サンプルを提供するか、または追加のリソースを割り当てることを要求された場合、私たちの業務は悪影響を受ける可能性があります。
外国、連邦、州の各レベルはすでに医療コストをコントロール或いは低減するための立法と監督管理提案を提出し続ける可能性がある。コスト抑制措置や他の医療改革を実施することは、承認されれば、収入を創出し、利益を達成することができ、あるいは候補製品を商業化することができるかもしれない。これらの改革は、私たちが開発に成功し、規制承認を得る可能性のある候補製品の予想収入に悪影響を及ぼす可能性があり、私たちの全体的な財務状況や候補製品を開発する能力に影響を及ぼす可能性がある。私たちは未来に取られる可能性のある計画を予測できない。政府、保険会社、医療組織を管理し、他の医療サービス支払者が医療コストのコントロールまたは低減に努力し続け、および/または価格制御を実施することは悪影響を及ぼす可能性がある
101
ACAおよび将来とりうる他の医療改革措置は,連邦医療保険や他の医療保険資金のさらなる削減,より厳しいカバー基準,より低い精算,新たな支払い方法につながる可能性が予想される。これは私たちが受け取ったすべての承認された製品の価格を下げるかもしれない。連邦医療保険または他の政府援助計画の支払いを拒否または減少させるいかなる精算も、同様の私的支払者の支払いを拒否または減少させる可能性があり、これは、十分な収入を生成することができ、利益を達成することができるか、または候補製品を商業化することができることを阻止することができるかもしれない(承認されれば)。
最近の連邦立法や州や地方政府の行動は、それらの薬品販売価格が米国より低い外国を含む外国からの薬品の再輸入を許可する可能性があり、これは私たちの経営業績に実質的な悪影響を及ぼす可能性がある。
もし私たちの候補製品が承認されれば、私たちはアメリカで外国からの療法の競争に直面するかもしれません。これらの療法はすでに薬品の価格コントロールを実施しています。米国では、“連邦医療保険現代化法案”に含まれる条項が米国の輸入法を変更する可能性があり、カナダ政府の価格規制があるため、薬剤師や卸売業者がカナダから承認された薬物や競合製品の安価なバージョンを輸入する能力を拡大することができる。米国輸入法のこれらの変化は発効しないが、HHS長官がこれらの変化が公衆の健康や安全に追加的なリスクを構成しないことを証明しなければ、消費者の製品コストを著しく低下させるだろう。また、MMAは、HHS長官がこれらの変化が公衆の健康および安全に追加的なリスクを構成しないことを証明しなければ、米国輸入法のこれらの変化は発効しないと規定し、消費者の製品コストを著しく低下させる。2020年9月23日、HHS大臣は国会にこのような認証を行い、2020年10月1日、FDAはカナダからある処方薬の輸入を許可する最終規則を発表した。最終規則によると,各州とインディアン部族,および将来的には薬剤師や卸がFDAに輸入計画提案を提出し,その審査と認可を行うことができる。2020年11月23日に最終規則が発表されて以来、いくつかの業界団体は米国コロンビア特区地域裁判所に連邦訴訟を提起し、禁止救済を求め、この規則の実施を阻止した。また、カナダ当局はカナダの薬品供給を不足から保護するための規定を採択した。2020年9月25日, CMSは,この規則により,各州から輸入された薬品は社会保障法第1927条に基づいて連邦還付を得る資格がなく,メーカーは“最適価格”や平均メーカー価格の目的でこれらの薬品を報告しないと述べている。これらの薬剤はカバーされた外来薬とは考えられないため,CMSはさらに,これらの薬剤の全国平均薬物調達コストは公表されないと述べている。さらに、FDAは、製造業者がFDAによって承認された薬物のために追加の国家医薬品コード(NDC)を得る方法を概説し、最初に外国で販売され、外国で販売されることを許可された最終指導文書を発表した。実施すれば、カナダからの薬品輸入は私たちの任意の候補製品の価格に実質的かつ不利な影響を与えるかもしれない。最終規則と指導意見が規制と市場に与える影響はまだ明らかにされていない。麻薬再輸入の支持者たちは立法を通じて、場合によっては再輸入を直接許可しようと努力するかもしれない。立法や規制が薬物の再輸入を許可すれば、私たちが開発する可能性のある任意の製品の価格を低下させ、将来の収入や利益の見通しに悪影響を及ぼす可能性がある。私たちは引き続き事態の発展と私たちの業務に潜在的な影響に集中するつもりだ。
技術や科学が急速に変化する環境では、私たちは激しい競争に直面しており、私たちの競争相手は、私たちの前に規制部門の承認を得たり、私たちよりも安全で、より先進的で、より効果的な療法を開発する可能性があり、これは、私たちが開発する可能性のある任意の候補製品の能力に悪影響を与え、最終的には私たちの財務状況を損なう可能性がある。
新薬製品の開発と商業化競争は激しい。私たちは将来開発や商業化を求める任意の候補製品の面で、世界各地からの主要な製薬会社、専門製薬会社、バイオテクノロジー会社の競争に直面するかもしれない。潜在的な競争相手はまた学術機構、政府機関とその他の公共と個人研究組織を含み、これらの組織は研究を展開し、特許保護を求め、研究、開発、製造と商業化のための協力手配を確立する。
102
現在、多くの大手製薬とバイオテクノロジー会社が私たちの核心価値駆動要素が追求している適応を治療するための製品を開発し、商業化している。これらの会社は、TTR四量体安定剤タファミディ(現在ファイザー社によって販売されており、VynDamaxとVyndaqel)、アコラミの競争相手;ボルソペプチド、CNP類似物(現在BioMarin製薬会社によって販売されており、Voxzogoと呼ばれている)、軟骨発育不全を治療する低用量infigratinibの潜在的な競争相手;crinecerfont、CRF 1受容体アンタゴニスト、BBP-631の競争相手、Natpara、副甲状腺ホルモンの競争相手として含まれているが、これらに限定されない。もしこれらまたは他の競争相手が、私たちの他の候補製品の競争相手を含めて、私たちの前にFDAの承認を得たら、私たちの候補製品は市場で最初の治療薬ではなく、私たちの市場シェアが制限される可能性がある。我々の目標適応に対する他社からの競争に加えて,我々が開発する可能性のあるどの製品も他のタイプの療法からの競争に直面する可能性がある。
私たちの多くの既存または潜在的な競争相手は、単独または彼らとの戦略パートナーであっても、研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング承認を得た製品の面で、私たちよりも多くの財務資源と専門知識を持っている。
製薬とバイオテクノロジー産業の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。さらに、現在承認されている製品は、我々の目標疾患適応または同様の適応の治療に適用されることが発見される可能性があり、これにより、このような製品は、我々の候補製品と比較して顕著な規制および市場タイミング利点を有する可能性がある。私たちの競争相手も、私たちよりも早くFDAや他の規制機関の製品の承認を得ることができ、私たちの目標によってFDAから孤立した製品独占経営権を得ることができ、これは、私たちの競争相手が市場に入る前に強力な市場地位を確立することができるかもしれません。また、私たちの競争相手が開発した製品や技術は、私たちの候補製品を経済的あるいは時代遅れにする可能性があり、私たちはこれらの候補製品のマーケティングに成功できないかもしれません。承認されると、競争相手に対抗する可能性があります。
さらに、私たちは、私たちの競争相手の製品に関連する特許の範囲、所有権、有効性、および/または実行可能な訴訟または他の訴訟に直面する可能性があり、私たちの競争相手は、私たちの製品の侵害、流用、または他の方法で彼らの知的財産権を侵害することを告発するかもしれない。私たちの競争相手の製品供給は、私たちが開発し商業化する可能性のある任意の製品の需要と私たちが受け取ることができる価格を制限するかもしれません。“私たちの知的財産権に関連したリスク”を見てください
103
もし私たちの候補製品の市場機会が私たちが思っているより小さいなら、私たちの収入は不利な影響を受けるかもしれません。私たちの業務は影響を受けるかもしれません。私たちが患者を識別することに成功し、相当な市場シェアを得る能力は、私たちが利益と成長を達成するために必要になるだろう。
我々はメンデル疾患や遺伝子駆動癌の治療研究や製品開発に焦点を当てており,その多くはまれあるいは孤立した適応である。我々の目標疾患適応の影響を受け,候補製品治療から利益を得る可能性のある個人数の予測は,我々の信念と推定に基づいている。これらの見積りは科学文献を含む様々なソースから得られており,正しくないことが証明されている可能性がある.また、新しい研究はこれらの疾病の推定発病率或いは流行率を変える可能性がある。患者数は予想より少ないかもしれない。我々が治療を求めている疾患を識別する患者の努力はまだ早期段階であり,治療が得られる可能性のある患者数を正確に予測することはできない。さらに、私たちの候補製品の潜在的にアドレス指定可能な患者集団は限られているかもしれないし、または私たちの製品または候補製品の治療を受け入れられない可能性があり、新しい患者はますます識別または接触が困難になる可能性があり、これは私たちの運営結果および業務に悪影響を及ぼすだろう。また、潜在的なターゲット層が少ないため、私たちの重要な価値駆動計画で開発中の候補製品のためにかなりの市場シェアを獲得しても、私たちはこのような巨大な市場シェアを獲得しているにもかかわらず、決して利益を達成しないかもしれない。さらに、市場シェアは、Vyndamax(タファミディス)およびVyndaqel(タファミディスグルコサミン)を含む他の療法の獲得可能性によって制限される可能性があり、ファイザー社は、ATTR−CMの治療に米国および日本(Vyndaqelのみ)で使用することを許可されている。したがって,承認されてもAcoramidisは市場で初めてATTR−CMを治療する薬剤ではなく,その市場シェアや収入創出の潜在力は限られている可能性がある。
私たちのビジネスや産業に関するリスクは
わが社の再編計画と2022年1月に発表された関連リストラは、予想されるすべての節約を実現できず、運営を混乱させる可能性がある。
2022年1月には、当社のビジネスプロセス、効率、コスト節約の運営変化を推進し、当社の戦略と発展計画を推進するための再編計画を実施することを約束しました。再編計画には,我々の施設の統合と合理化,開発計画の優先順位の再決定,従業員の削減が含まれている。予見できない困難、遅延或いは意外なコストのため、私たちは私たちの再構成努力による予想されるメリット、節約とコスト構造の改善を十分に実現できない可能性があり、再構成の費用は予想よりも大きいかもしれない。もし私たちが再編から予想されるコスト節約を実現できなければ、私たちの経営業績や財務状況は不利な影響を受ける可能性がある。しかも、私たちは開発計画の優先順位を再決定することが私たちの運営を混乱させるかもしれない。例えば、売上高が計画を超えたリストラや日常運営における困難な増加など、私たちのリストラは予期せぬ結果をもたらす可能性がある。私たちのリストラはまた私たちの業務に重要な合格者を引き付けて維持する能力を損なうかもしれない。合格者を引き付けたり引き留めたりできなかった場合は、私たちが重要な技術的措置を成功的に実行することを妨げるかもしれない。
新冠肺炎の疫病は私たちの業務、運営結果と財務状況に不利な影響を与える可能性がある。
持続的な新冠肺炎疫病は全世界の健康と経済環境に巨大な影響を与え、数百万の確定診断例と死亡、企業の減速或いは閉鎖、労働力不足、サプライチェーン挑戦、政府要求変化、監督管理挑戦、インフレ圧力と市場変動を含む。これまで、私たちの業務の大部分を継続しようとしてきましたが、将来の事態を予測することはできませんし、その経済的影響を含めて、この世界的な大流行病が、私たちの業務、業務結果、財務状況に大きな悪影響を与えない保証はありません。新冠肺炎の私たちへの影響程度は多種の要素と未来の事態発展に依存し、これらの要素と未来の事態発展は非常に高い不確定性を持っており、疫病の持続時間、範囲と深刻度を含む自信に満ちて予測することができず、アメリカと他の国が旅行制限と社会距離の持続時間と程度を実施し、企業閉鎖或いはその他の商業中断、サプライチェーン中断と労働力不足、及びアメリカと他の国が新冠肺炎の抑制と治療のために行った行動の有効性、全世界のワクチン接種仕事を含む。
104
全世界が新冠肺炎の疫病に対応するために取った公衆衛生行動は、隔離、家にいる、行政命令と類似の政府命令及び医療資源の優先順位を含み、すでに私たちの業務、運営結果と財務状況に不利な影響を与える可能性がある。これらの公衆衛生行動のため、私たちはすでに経験し、未来に私たちの業務、臨床試験、臨床前研究および運営の中断を深刻に影響する可能性がある
持続的な新冠肺炎の大流行により、著者らの業務、運営結果及び臨床前と臨床開発プロセスはすでにマイナスの影響を受け続ける可能性があり、監督管理機関が引き続き適時な審査と申請を許可する能力を確保することを含む。新冠肺炎突発公共衛生事件の期間中、アメリカ食品薬品監督管理局はその申請に対する規定検査を完成できなかったため、多くの会社は完全な回復状を受け取ることを発表した。現在計画されている方式とスケジュールに従って業務を展開する能力は、私たちの業務、運営結果、財務状況に重大な悪影響を及ぼす可能性があります。
現在の新冠肺炎の大流行が私たちの業務や財務業績に悪影響を及ぼす場合、それはまた、“リスク要因”部分に記載されている多くの他のリスク、例えば、私たちの臨床開発業務に関連するリスク、私たちが行っており、計画中の臨床試験および商業販売のサプライチェーン、政府および規制機関が私たちの臨床試験場を検査し、私たちが提出した支援を検討し、私たちの候補製品の承認と承認を申請することを支持し、私たちの運営を支援し、私たちの債務を返済するためにより多くの資本を集めることができるかどうかを悪化させる可能性がある。また、新冠肺炎症例の任意の再発或いはその後の“波”は、それらの新しい変種による症例を含め、他の広範あるいはより深刻な影響を招く可能性があり、具体的には感染率が最も高い地域に依存する。新冠肺炎のあるワクチンと治療方法はすでに使用が許可されており、その中のいくつかは緊急時であるが、これらの措置が新冠肺炎の進展を適時或いは根本的に阻止或いは緩和することを保証することはできない。
105
私たちの未来の成功は私たちが肝心な従業員、役員、顧問と顧問を維持する能力、及び合格者を吸引、維持、激励する能力にかかっている。
著者らは幹部、取締役、管理委員会及び科学と臨床チームの他のメンバーの管理、研究開発、臨床、財務と業務発展方面の専門知識に高度に依存している。
もし私たちがクマール博士や私たちの他の幹部や重要な人たちを失ったら、私たちは適切な後継者をタイムリーに見つけることができないかもしれない。また、私たちの従業員の一部は、複数の子会社にまたがる集中的な支援源を提供しているため、これらの従業員のいずれかの流出は、影響を受けた子会社の運営に悪影響を及ぼす可能性があり、私たちの財務状況や運営結果は大きな悪影響を受ける可能性がある。
しかも、私たちのすべての幹部は私たちとの雇用関係をいつでも終わらせることができる。私たちは私たちのどんな幹部や従業員にも“キーパーソン”保険を提供しない。合格した科学と臨床人員を募集と維持し、もし私たちが薬物導管の発展の方面で進展を得て、商業化、販売とマーケティング人員の規模を拡大すれば、私たちの成功のキーポイントにもなる。幹部や他の重要な従業員のサービスを失うことは、研究開発と商業化目標の実現を阻害し、業務戦略を成功させる能力を深刻に損なう可能性がある。また、幹部やキースタッフの交換は困難かもしれませんし、私たちの業界では開発に成功し、規制部門の承認を得て、私たちの候補製品を商業化するために必要なスキルや経験を持っている個人数が限られているので、時間がかかるかもしれません。私たちの業界の中で、合格者を募集する競争は非常に激しく、多くの製薬と生物技術会社の類似者に対する競争を考慮して、私たちは受け入れ可能な条件でこれらの肝心な人員を採用、訓練、維持或いは激励することができないかもしれない。さらに、もし私たちが競争相手から人員を雇用すれば、私たちは彼らが不当に要求されたり、彼らが独自または他の機密情報を漏らしたり、彼らの元雇用主が彼らの研究成果を持っているという告発を受けるかもしれない。私たちはまた、大学や研究機関から科学や臨床人を募集する競争に直面している。
また、私たちは、科学と臨床コンサルタントを含むコンサルタントとコンサルタントに依存して、私たちの研究開発と商業化戦略の制定を助けてくれます。私たちのコンサルタントやコンサルタントは、私たち以外の雇用主に雇われる可能性があり、他のエンティティと締結された相談または相談契約に基づいて約束することができ、これは、私たちが彼らを得る機会を制限するかもしれない。もし私たちが引き続き高い素質の人材を誘致し、維持することができなければ、私たちが成長戦略を推進する能力は制限されるだろう。
私たちは限られた数の従業員からなる中央チームに依存しており、彼らは組織全体で様々な管理、研究開発、その他のサービス、および子会社レベルの専門チームを提供しており、運営に挑戦をもたらし、私たちの業務に悪影響を及ぼす可能性がある。
2022年12月31日まで、私たちは392人の従業員を持っている。このような構造は私たちがいくつかのインフラコストを下げることができると信じていますが、私たちの中央チームの規模は比較的に小さく、私たちは私たちのすべての子会社の運営を支持するために十分な人員、時間、資源を投入することができないかもしれません。それらの研究開発活動、従業員の採用と維持努力、管理層の財務と会計、報告事項を含む。我々の中央チームのメンバーは,我々の子会社の業務や運営に関する十分な情報を得ることができず,これらの事務を十分に管理できない場合がある.また、当社の子会社レベルの従業員や経営陣は主に子会社レベルでインセンティブを行っているため、これらの従業員や管理チームのメンバーは、私たちの組織全体の価値を最大化するのに十分なインセンティブを得られない可能性があります。私たちのコアチームが組織全体で十分な行政、研究開発、または他のサービスを提供できなかった場合、あるいは私たちの子会社レベルの従業員と経営陣のパフォーマンスが私たちの組織全体の利益に合致しない場合、私たちの業務、財務状況、運営結果は損なわれる可能性があります。
106
アメリカ食品·薬物管理局、アメリカ証券取引委員会と他の政府機関の資金変化或いは運営中断は、それらの重要な指導部と他の人員の採用と維持能力を阻害し、新製品とサービスの適時な開発或いは商業化を阻止し、あるいは他の方法でこれらの機関が私たちの業務運営が依存する可能性のある正常な機能を履行することを阻止し、それによって私たちの業務に負の影響を与える可能性がある。
米国食品医薬品局、米国証券取引委員会および他の政府機関の資金不足は、政府の閉店やこれらの機関の運営の他の中断を含め、重要な指導部や他の人員の能力を雇用·保留し、新製品やサービスのタイムリーな開発や商業化を阻止するか、あるいはこれらの機関が私たちの業務運営に依存する可能性のある正常な業務機能を履行することを阻止することを阻害する可能性があり、これは私たちの業務に負の影響を与える可能性がある。
食品·薬物管理局が新製品を審査·承認し、或いは他の規制事項に対して行動する能力は、政府予算と資金レベル、肝心な人員の雇用と維持及び使用料の支払いを受ける能力、新冠肺炎疫病に対応する政府の“家にいる”命令によって人員とその他の資源を提供する能力、及び法規、法規と政策の変化を含む様々な要素の影響を受ける可能性がある。したがって、その機関の平均検討時間は近年変動している。また、政府が米国証券取引委員会や我々の業務に依存する可能性のある他の政府機関に提供する資金は、研究開発活動に資金を提供する機関を含め、政治プロセスの影響を受けており、政治プロセス自体が不安定で予測不可能である。
FDAおよび他の機関の中断は、関連政府機関が新薬を審査および/または承認したり、他の行動を取るのに要する時間を遅らせる可能性もあり、これは私たちの業務に悪影響を及ぼすだろう。例えば、過去数年間、2018年12月22日から35日間を含めて、米国政府は何度も閉店しており、食品·医薬品局や米国証券取引委員会などの規制機関は、食品·医薬品局、米国証券取引委員会、その他の政府従業員を休暇させ、重要な活動を停止しなければならない。2020年3月に国内外の施設に対する検査は基本的に保留されて以来、FDAは常規のモニタリング、生物研究モニタリングと審査前検査を含む大流行前の検査活動の回復に努力している。 FDAが、上場申請を承認するために検査を行わなければならないと判断し、旅行制限のために審査期間内に検査を完了することができず、FDAが遠隔相互作用評価が不十分であると判断した場合、FDAは、場合によっては、検査が完了するまで、申請に完全な返信または実行を延期することを意図していることを示している。新冠肺炎疫病に対して、アメリカ以外の監督管理機関は類似の制限或いは他の政策措置をとる可能性があり、監督活動の遅延に遭遇する可能性がある。政府が長期的に停止したりFDAの運営が中断されたりすれば、FDAが私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。同様に、政府の長期的な停止や米国特許商標局の運営中断は、私たちの特許出願のタイムリーな審査を阻害する可能性があり、これは、私たちが本来獲得する権利がある可能性のある任意の米国特許の発行を延期する可能性がある。今後の政府の停止や同様の事件は、資本を適切に利用し、事業を継続するために、公開市場に参入し、必要な資本を得る能力に影響を与える可能性がある。
私たちは私たちの組織を拡大する必要があり、私たちはこのような成長を管理することに困難に直面するかもしれないし、これは私たちの運営を混乱させるかもしれない。
2022年12月31日までに、当社の全会社で392人のフルタイム従業員がいます。私たちの成熟に伴い、私たちは私たちの全従業員基盤を拡大し、より多くのコンサルタントと請負業者を雇いたい。私たちの経営陣は私たちの日常活動から不比例な注意を移し、これらの成長活動を管理するために多くの時間を投入する必要があるかもしれない。私たちは私たちの業務の拡張を効果的に管理できないかもしれません。これは私たちのインフラが弱く、操作ミス、ビジネス機会の喪失、従業員の流失、残りの従業員の生産性の低下を招く可能性があります。私たちの予想成長は大量の資本支出を必要とする可能性があり、他のプロジェクトから財務資源、例えば私たちの候補製品の商業化、承認されれば、より多くの候補製品を開発する可能性がある。私たちの経営陣が私たちの成長を効果的に管理できなければ、私たちの費用は予想以上に増加する可能性があり、私たちは収入を創出および/または成長する能力が低下する可能性があり、私たちは私たちの業務戦略を実施できないかもしれない。私たちの将来の財務業績と私たちが候補製品を商業化し、効果的に競争する能力は、私たちが将来のどんな成長を効果的に管理する能力にある程度依存するだろう。
107
我々は複数の計画や候補製品が開発されており,様々な標的適応や治療方式が求められているため,より利益や成功可能性の高い開発機会や候補製品を利用することなく,特定の候補製品を追求するために限られた資源を利用する可能性がある。
我々は,メンデル疾患や遺伝子駆動癌を解決するための候補製品の開発に焦点を当て,治療法やこの分野の特定の標的適応にかかわらず。私たちの財力と人的資源が限られているため、潜在的な目標適応や候補製品を持つ機会の追求を放棄または延期する可能性があり、これらの兆候や製品は、後に、私たちの現在および計画中の開発計画および候補製品よりも大きなビジネス潜在力を持っていることが証明された。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。現在および将来の研究開発計画および他の特定の適応の将来の候補製品への支出は、いかなる商業的に実行可能な将来の候補製品も生じない可能性がある。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければならない場合、私たちは協力、許可、または他の印税手配を通じてその候補製品に価値のある権利を放棄する必要があるかもしれないが、この場合、私たちはこれらの未来の候補製品の独占開発権と商業化権利を維持することがより有利になるだろう。
さらに、追加的な許可内や開発段階の資産や計画を買収することが求められる可能性があり、これは私たちに追加的なリスクをもたらすだろう。将来性のある候補製品を確定、選別、獲得するには大量の技術、財政と人的資源の専門知識が必要である。このような努力は、成功するかもしれない候補製品を実際に獲得することを招くことはなく、何のメリットも生じることなく、私たちの経営陣の時間と資源支出の分流を招く可能性がある。例えば、最終的に製品の承認につながる計画を決定できなければ、最終的に投資収益を提供できない製品を評価、買収、開発するために大量の資本や他の資源がかかる可能性がある。
私たちに対する製品責任訴訟は、私たちが重大な責任を負うことを招き、私たちが開発する可能性のある任意の候補製品の商業化を制限する可能性があります。
承認された薬品の商業販売では,人体臨床試験における候補製品の試験に関連する製品責任曝露の固有のリスクに直面している。もし私たちが自分自身を弁護することに成功できなければ、私たちの候補製品や薬物による傷害のクレームに反対すれば、私たちは重大な責任を招くかもしれない。是非曲直や最終結果にかかわらず、賠償責任は
私たちが協賛する臨床試験と私たちの商業製品販売に保険を提供することを含む製品責任保険を維持していますが、私たちが発生する可能性のあるすべての責任をカバーするのに十分ではないかもしれません。より多くの臨床試験を開始し、承認される可能性のある候補製品の商業化に伴い、保険カバー範囲を増やす必要があると予想されます。保険カバー市場はますます高くなり、私たちの臨床プロジェクトと商業化努力の規模拡大に伴い、保険カバーのコストは増加する。私たちは可能などんな責任にも対応するために、合理的な費用や十分な金額で保険範囲を維持することができないかもしれない。
108
当社の従業員、独立請負業者、コンサルタント、ビジネスパートナー、およびサプライヤーは、規制基準および要件を遵守しないことを含む、不適切な行為または他の不適切な活動に従事する可能性があります。
私たちは従業員、独立請負業者、コンサルタント、ビジネスパートナー、サプライヤーの詐欺、不正行為、または他の不正活動のリスクに直面している。これらの当事者の不正行為は、FDAおよび同様の外国規制機関の法律に準拠できなかった故意、無謀、および不注意な行為を含む可能性があり、FDAおよび類似の外国規制機関に真実、完全かつ正確な情報を提供すること、私たちが制定した製造基準を遵守すること、米国の医療詐欺および乱用法律、および同様の外国詐欺的不正行為法律を遵守すること、または財務情報またはデータを正確に報告すること、または不正な活動を開示することを含むことができる。もし私たちがFDAの候補製品の承認を得て、アメリカでこれらの製品を商業化し始めたら、私たちはこのような法律の下で潜在的なリスクが著しく増加すると信じており、私たちはこのような法律を遵守することに関連するコストも増加する可能性がある。特に、医療業界の研究、販売、マーケティング、教育およびその他のビジネス配置は、詐欺、リベート、自己取引、および他の乱用行為を防止するための広範な法的制約を受けている。これらの法律法規は、幅広い価格設定、割引、教育、マーケティングおよび販売促進、販売および手数料、特定の顧客インセンティブ計画、および他のビジネスプランを制限または禁止する可能性があります。これらの法律に拘束された活動はまた、患者を募集して臨床試験を行う過程で得られた情報を不適切に使用することに関連し、これは規制制裁を招き、著者らの名声に深刻な損害を与える可能性がある。私たちはすでに私たちの従業員と役員に適用される商業行為と道徳基準を通過しましたが、従業員と第三者の不正行為を常に識別し、阻止できるわけではありません, このような活動を発見し防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはそのような法律を遵守しないことによる政府の調査または他の行動または訴訟から私たちを保護することができない可能性がある。もし私たちにこのような行動を取って、私たちが自分の権利を弁護したり、維持することに成功しなかったら、これらの行動は巨額の罰金や他の制裁を加えることを含めて、私たちの業務に大きな影響を与えるかもしれない。
私たちの国際業務は、アメリカ国外での業務展開に関連するビジネス、規制、政治、運営、財務、定価と精算、経済リスクに直面するかもしれません。
我々はグローバルCROを通じて国際的に臨床試験を行っており,我々の業務戦略には潜在的な国際拡張が含まれており,米国以外の患者集団を狙っている。米国以外の患者集団で規制部門の承認を得て任意の候補製品を商業化すれば,販売代表を招聘し,米国以外の場所で医師や患者協会の外連活動を行うことができる。国際的な業務展開は多くのリスクに関連しているが、これらに限定されない
109
これらの要素のいずれも、私たちの潜在的な国際拡張と運営を深刻に損害し、それによって私たちの運営結果を損なう可能性がある。
110
有効な財務報告開示制御および内部制御システムを維持できなければ、タイムリーで正確な財務諸表を作成したり、適用法規を遵守する能力が損なわれたりする可能性があり、これは私たちの普通株価格に悪影響を及ぼす可能性がある。
上場企業として、財務報告書の内部統制を維持し、このような内部統制におけるいかなる重大な弱点も報告することが求められている。2002年のサバンズ-オキシリー法案またはサバンズ-オキシリー法案は、財務報告に対する私たちの内部統制の有効性を評価し、決定することを要求し、サバンズ-オキシリー法案404(B)条または404条に基づいて、私たちが株式を初めて公募した後の第2の年間報告書から、財務報告の内部統制に関する管理報告書を提供する。また、私たちは、私たちの独立公認会計士事務所が発行した財務報告内部統制を含む認証報告書が必要です。有効な制御を開発または維持できなかった場合、または実施または改善の過程でいかなる困難に遭遇しても、我々の経営業績を損なう可能性があり、報告義務を履行できず、従来の財務諸表を再記述したり、米国証券取引委員会に提出された定期報告に含まれる財務報告内部統制の管理評価および独立公認会計士事務所監査の結果に悪影響を及ぼすことが最終的に要求される。
また、私たちがより多くの連結子会社やVIEを買収または設立する限り、これらのエンティティの財務諸表は最初に私たちが作成したものではない可能性があり、それらの財務諸表作成を直接制御することはありません。したがって、私たちの財務報告は、これらの実体が私たちに報告した内容に依存し、これは、私たちの監視と監査手続きを増加させ、将来の私たちの財務プロセスと報告書の実施と十分な統制を維持することの難しさを増加させる可能性があり、これは私たちの外部報告の遅延を招くかもしれない。特に、複雑な財務会計プロセスのないパートナーとそのようなエンティティを構築している場合、あるいは私たちが新たな関係に急速に入っており、私たちの統合能力を緊張させている場合には、このような状況が発生する可能性があります。私たちは過去に、私たちが財務報告書の内部統制に重大な弱点を持っているということを発見した。これらの実質的な弱点は救済されているが、将来的にはより多くの実質的な弱点を発見することができ、重大な欠陥の組み合わせは実質的な弱点を招く可能性がある。このような追加的な重大な弱点を発見した場合、または任意の追加期間に以前に発表された財務諸表を再説明する必要がある場合、私たちの名声は損なわれ、投資家の信頼喪失を招き、私たちの業務、経営業績、および財務状況に悪影響を及ぼす可能性があります。さらに、統合子会社または制御されたVIEからの情報をタイムリーに受信しなければ、外部報告の遅延を招く可能性があります。無効な開示統制と手続きおよび財務報告の内部統制はまた、投資家が私たちの報告書の財務および他の情報に自信を失う可能性がある, これは私たちの普通株の取引価格に否定的な影響を及ぼすかもしれない。
歴史的には、私たちは引き続き第三者契約サービスプロバイダに依存しており、財務報告を支援することが予想されています。サバンズ-オクスリ法案を遵守するために必要な財務報告の内部統制を設計、実施、維持する過程は、時間がかかり、高価で複雑である。もし私たちが財務報告の内部統制に有効であると断言できない場合、あるいは将来必要に応じて、私たちの独立公認会計士事務所が私たちの財務報告の内部統制の有効性に否定的な意見を発表すれば、投資家は私たちの財務報告の正確性と完全性に自信を失う可能性があり、私たちの普通株式の市場価格は悪影響を受ける可能性があり、私たちは証券取引所、米国証券取引委員会、または他の規制機関の調査または制裁を受ける可能性があり、これには追加的な財務·管理資源が必要となる可能性がある。
111
私たちは財務報告書に対する私たちの開示統制や内部統制がすべてのミスと詐欺を防ぐことを期待していない.
設計および動作がどんなに良好であっても、絶対的な保証ではなく、合理的な保証を提供することしかできず、制御システムの目標が実現されることを確保する制御システム。また,制御システムの設計は,資源制約が存在し,そのコストに対する制御の利点を考慮しなければならないという事実を反映しなければならない.すべての制御システムの固有の限界により,どの制御評価もすべての制御問題や不正イベントが発見されたことを絶対に保証することはできない.これらの固有の限界は、意思決定過程における判断が誤っている可能性があり、簡単なエラーまたはエラーによって故障が発生する可能性があるという現実を含む。また、ある人の個人的な行動、2人以上の結託、または制御の管理によって、制御を回避することができる。任意の制御システムの設計も、将来のイベント可能性のいくつかの仮定に部分的に基づいており、どの設計も、すべての可能な未来の条件でその目標を成功的に達成できる保証はなく、時間の経過とともに、制御が条件の変化によって不十分になる可能性があり、またはポリシーまたはプログラムを遵守する程度が悪化する可能性がある。コスト効果のある制御システムの固有の制限により,誤りや詐欺による誤った陳述が発生する可能性があり,発見されない可能性がある.
私たちの負債に関するリスクは
私たちは多くの借金を背負っており、未来にはもっと多くの借金が生じるかもしれない。債務の返済には大量の現金が必要であり、私たちの業務には私たちの巨額の債務を支払うための十分なキャッシュフローがないかもしれない。
2022年12月31日現在、私たちと私たちの子会社の総合債務総額は17億ドルで、2027年満期の無担保2.50%転換可能優先手形(すなわち2027年手形)項目の5.5億ドルの未償還債務、2029年満期の2.25%転換可能優先手形(2029年手形)項の未償還債務7.475億ドル、米銀行協会、ある貸手、BridgeBioの借り手としてのある子会社、保証人であるBridgeBioのある子会社(または融資契約)の4.452億ドルの債務を含む。当社の既存および将来の債務条項の制限を受けて、当社およびその付属会社は追加の債務、既存または将来の債務を保証したり、私たちの債務を再融資したりする可能性があります。私たちは債務の利息と元本を支払うために、運営キャッシュフローの大部分を使用する必要があるかもしれない。私たちが計画通りに債務元金を支払う能力があるかどうか、利息を支払うこと、あるいは債務を再融資する能力があるかどうかは、私たちの未来の表現と、私たちが業務から十分なキャッシュフローを生み出す能力にかかっており、これらは経済、金融、競争、そして他の私たちがコントロールできない要素の影響を受けている。このような支払いは、運営資本、資本支出、および他の企業用途に利用可能な資金を減少させ、運営資本、資本支出、拡張計画、および他の投資のための追加融資を得る能力を制限し、逆に業務戦略を実施する能力を制限し、業務、業界または全体的な経済低迷時の脆弱性を増加させ、事業および業界の変化に計画または対応する柔軟性を制限し、ビジネスチャンスが発生したときにそれらを利用することを阻止する可能性がある。また…, もし私たちが私たちの債務を返済し、私たちの運営に資金を提供するのに十分なキャッシュフローを生成できない場合、私たちは資産の売却、再編債務、または煩雑または高度に希釈される可能性のある条項で追加の株式を得るような1つまたは複数の代替案を採用することが要求されるかもしれない。私たちはこのような活動のいずれにも従事できないかもしれないし、理想的な条件でこれらの活動に従事することができないかもしれません。これは私たちの債務不履行をもたらす可能性があります。
112
私たちは、私たちの転換可能な優先手形の下で債務を発生させ、私たちの業務や融資活動を制限することができる経営と財務契約を含むローンと保証プロトコルの一方である。
2020年3月、2027年債を発行しましたが、この債券によると、半年ごとに延滞利息を支払い、年利は2.50%です。2027年債券は2027年3月15日に満期になります。事前に転換または買い戻ししない限り、私たちの選択に応じて、2027年債券の任意の転換を現金、普通株、または両方の組み合わせで決済します。2021年1月と2月に、2029年債を発行し、この債券に基づいて、半年ごとに延滞利息を支払い、年利率は2.25%です。2029年債券は、事前に転換または買い戻ししない限り、2029年2月1日に満期になり、自分の選択に応じて、現金、普通株、または両者の組み合わせで2029年債券の任意の転換を決済する。場合によっては、2027年債券および2029年債券の保有者、または総称して債券保有者は、それぞれの満期日までに現金で債券項の下で返済されていない元本および利息の全部または一部を請求することが可能であり、これは我々の財務業績に悪影響を及ぼす可能性がある。
二零二一年十一月に、吾等は融資契約を締結し、これにより吾等は、(I)第1回前払4.5億ドル、又は第1回前払、及び(Ii)第2回前払300,000,000ドル、又は第2回前借り、又は合わせて定期借款を含む元金総額7.5億ドルの定期融資を受けた。最初の前払いは2021年11月17日に資金を獲得した。2022年5月には、第2弾の前払金を1.00億ドルに削減することを含む融資協定を改訂した。2022年11月には、私たちが第2回目の前払いを抽出する能力を終了することを含む融資協定をさらに修正しました。定期ローンは2026年11月16日に満期になる。ローン協定は私たちの能力を制限するかもしれません
また、融資協定の規定により、様々な運営契約や違約条項を遵守しなければなりません。これらの条項は、私たちの業務融資、業務活動に従事したり、私たちの業務戦略を拡大したり、全面的に推進したりする能力を制限する可能性があります。このような契約または条項に違反することは、融資協定下の違約を招く可能性があり、これは、融資項目下のすべての未償還債務が直ちに満期になり、支払われる可能性がある。
ローン契約によると、私たちなどにも私たちの付属会社の株式を拘留する責任があります。また、私たちのいくつかの非運営子会社も共同協定を締結する義務があり、協定によると、それらも融資協定の条項を守らなければなりません。これらの子会社は主に私たちの開発計画を進める子会社ではありません。私たちの融資協定違反のいかなる行為や違約事件は、私たちの業務、財務状況、経営業績に重大な悪影響を及ぼす可能性があります。
113
手形の条件付き変換機能がトリガされると、私たちの財務状況や経営業績に悪影響を及ぼす可能性があります。
債券の条件付転換機能がトリガされると、債券保有者は、指定期間内に自己の選択に応じて随時債券を変換する権利がある。もし1人以上の所持者が彼らの手形を転換することを選択した場合、私たちが普通株のみを渡すことで私たちの転換義務(細かい株式を渡すのではなく現金の支払い)を履行することを選択しない限り、私たちは現金を支払うことで私たちの転換義務の一部または全部を返済することを要求され、これは私たちの流動資金に悪影響を及ぼす可能性がある。また、所有者が彼らの手形を転換することを選択しなくても、適用される会計規則により、手形のすべてまたは一部の未返済元金を長期負債ではなく流動負債に再分類する必要がある可能性があり、これにより、私たちの運営資本の純額が大幅に減少する。
私たちが追加資本を必要とするリスク
私たちは私たちの業務目標を達成するために多くの追加資金が必要になるだろう。もし私たちが必要な時に受け入れ可能な条件でこの資金を得ることができなければ、私たちは私たちの製品開発と商業化努力を延期、制限、または中止させることを余儀なくされるかもしれない。
生物製薬製品の開発と商業化は高価で時間がかかり、著者らは研究、臨床前試験と人体研究を行うために大量の追加資金が必要であると予想し、中試規模と商業規模の製造技術と施設を構築し、品質管理、監督、マーケティング、販売と管理能力を確立し、発展させ、私たちの現有の計画と潜在的な追加計画を支持するかもしれない。また、第三者への費用の支払いを担当しており、これらの費用には、これらの機関または会社からそれぞれの授業の知的財産権を取得した機関または会社とのいくつかの合意を含むマイルストーン支払い、ライセンス維持費、および印税が含まれている可能性があります。いかなる臨床前或いは臨床開発と監督管理審査過程の結果は非常に不確定であるため、著者らは確定する可能性のある任意の未来の候補製品の開発、監督管理審査の流れと商業化に必要な実際の金額を合理的に推定することができない。
2022年12月31日まで、私たちの運営資金は4.274億ドルで、その中で現金、現金等価物と有価証券は4.283億ドル、制限的な現金は3790万ドル、株式証券投資は4370万ドルだった。私たちの現金および現金等価物、有価証券、制限された現金および株式証券投資は、本報告書の発表日から少なくとも12ヶ月以内に私たちの運営に資金を提供するのに十分であると予想される。しかし、私たちの現在知られていない多くの要素、新冠肺炎疫病が私たちの研究開発と商業化活動に与える影響を含むため、私たちの運営計画はそれによって変わる可能性があり、私たちは計画よりも早く公共または私募株式や債務融資や他の出所、例えば戦略協力や許可と開発協定などを通じて、追加の資金を求める必要があるかもしれない。私たちの任意の追加拠出努力は、私たちの経営陣の彼らの日常活動に対する関心を移す可能性があり、これは、私たちが決定し、追求する可能性のある候補製品の能力を開発し、商業化することに悪影響を及ぼすかもしれない。また、このような融資は、株主への希釈、債務契約や償還義務の強制実施、あるいは我々の業務の他の制限に影響を及ぼす可能性がある。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画のために十分な資金があると考えても、追加の資本を求めることができる。
私たちの未来の資金需要は多くの要素に依存するだろうが、これらに限定されない
114
私たちが必要な時、受け入れられる条件で、あるいは追加的な資金が全くない。もし私たちが十分な資金をタイムリーに得ることができない場合、私たちは1つ以上の研究または開発計画または任意の候補製品の商業化の延期、制限、または終了を要求されるかもしれないし、必要に応じて業務を拡大したり、他の方法でビジネスチャンスを利用することができず、これは私たちの業務、見通し、財務状況、および運営結果に大きな影響を与える可能性がある。
追加資本の調達は、私たちの既存の株主に希釈し、私たちの運営を制限するか、または現在の候補製品または任意の未来の候補製品に対する権利を不利な条項で放棄することを要求するかもしれません。
私たちは公共およびプライベート·エクイティ発行、債務融資、戦略的パートナーシップ、連合、および許可スケジュールを含む、任意の利用可能なソースを通じてより多くの資金を求めることができます。私たちおよび間接的な私たちの株主は、任意のこのような証券の発行とサービス、および任意のこのような戦略的パートナーシップまたは他の手配の確立および維持のコストを負担するだろう。私たちが将来債務や株式証券を発行する任意の決定は、市場状況および他の私たちがコントロールできない要素に依存するため、私たちは将来のいかなる融資取引の金額、時間、または性質を予測または推定することができない。もし私たちが追加の株式または債務証券を売却することによって追加資本を調達する場合、あなたの所有権権益は希釈され、条項は清算または株主としての権利に悪影響を及ぼす他の特典を含む可能性がある。追加債務は、固定支払義務の増加を招き、追加債務を発生させる能力の制限、知的財産権を得ることができるかもしれない能力の制限、および私たちの業務を展開する能力に悪影響を及ぼす可能性のある他の経営制限など、追加的な制限契約に関連する可能性がある。さらに、私たちが第三者と達成した任意の未来の協力は短期的に資金を提供する可能性があるが、私たちの未来の潜在的なキャッシュフローと収入を制限するだろう。もし私たちが戦略的パートナーシップと連合、および第三者との許可手配を通じてより多くの資金を調達すれば、私たちは私たちの技術または候補製品に対する貴重な権利を放棄しなければならないか、または不利な条項で許可証または他の権利を授与しなければならないかもしれない。
115
また、我々の子会社が第三者に株式証券を発行することで資金を調達すれば、当社の株主の当該子会社での損失権益は大幅に減少する可能性がある。もし私たちの子会社が協力と許可手配を通じて追加資金を調達する場合、私たちの技術または候補製品に対するいくつかの権利を放棄するか、または私たちに不利な条項で許可を付与する必要があるかもしれない。
もし私たちが他の買収や戦略協力を行えば、これは私たちの資本金要求を増加させ、私たちの株主を希釈し、私たちに債務を発生させたり、負債を負担したり、他のリスクに直面させたりする可能性がある。
私たちは将来、相互補完製品、知的財産権、技術、または業務の許可または買収を含む様々な買収と戦略的パートナーシップに従事するかもしれません。買収や戦略的パートナーシップは多くのリスクをもたらす可能性があります
また、私たちがこのような取引を行うと、希釈証券の発行、債務の負担または発生、巨額の使い捨て費用の発生、および買収が重大な将来の償却費用を招く可能性のある無形資産を発行する可能性があり、これらはいずれも私たちの業務、将来性、財務状況、および運営結果に大きな悪影響を及ぼす可能性がある。例えば、Eidosの合併は私たちの現金を減少させ、私たちが前にEidos株主が発行した株式証券は希釈された。将来的に私たちが現金や株の対価格を提供する必要がある類似の取引は、私たちの財務状況を損なう可能性があり、私たちの既存の株主にマイナスの影響を与える可能性がある。
最近の資本市場の変動やわが証券の市場価格の下落は、普通株の売却や債務発行によって新たな資本を得る能力に影響を与える可能性があり、これは私たちの流動性を損なう可能性があり、私たちの成長業務の能力を制限し、私たちの運営インフラの買収や改善を求め、市場で競争する能力を制限することができる。
私たちの運営は大量の現金を消費し、私たちは引き続き私たちの業務成長を支援し、業務挑戦やチャンスに対応し、新しい候補製品を開発し、現在の人員レベルを維持または拡大し、既存の候補製品を改善し、私たちの運営インフラを強化し、相補的な業務と技術を買収することが可能です。私たちの未来の資本需要は私たちの現在の見積もりと大きく異なるかもしれません。必要を含む多くの要素に依存します
116
したがって、私たちは私たちの資本需要を満たすために株式、債務、または他の融資を求める必要があるかもしれない。資本市場の不確実性と他の要素のため、このような融資は私たちに有利な条項で提供されないかもしれないし、根本的にはないかもしれない。もし私たちが株式または転換可能な債務証券をさらに発行することでより多くの資金を調達すれば、私たちの既存の株主は重大な希釈を受ける可能性があり、私たちが発行したいかなる新しい株式証券も普通株式保有者よりも高い権利、優遇、特権を持っている可能性がある。また、吾等が担保した任意の債務融資は、私等に固定支払義務やチノを負担させ、資金調達活動、追加債務を招く、資本支出を招く、または配当を発表するなどの特定の行動をとる能力を制限または制限する可能性があり、潜在的な買収を含む、吾などの資金調達活動および他の財務および運営に関する追加的な制限的な契約に関連する可能性があり、吾等は潜在的な買収を含む追加資本を得ることを困難にする。もし私たちがマーケティングと流通手配または他の協力、戦略連合、または第三者との許可手配によって追加資本を調達する場合、私たちは私たちの候補製品、技術、将来の収入流、または研究計画のいくつかの価値のある権利を放棄しなければならないかもしれないし、私たちに不利になる可能性のある条項で許可を付与しなければならないかもしれない。また、最近の金利の上昇は、銀行の信用限度額や公開または個人売却債務証券などの借入によって運営資金を獲得する能力に影響を与える可能性があり、これは流動性の低下、運営資本の減少、私たちの業務に他の悪影響を及ぼす可能性がある。もし私たちが十分な資金を得られなければ、私たちが満足した条件で融資を受けることができなければ, 私たちの投資運営能力は大きく制限されるかもしれません。そうでなければ、私たちの業務に損害を与えます。
私たちの普通株に関するリスクは
私たちの普通株の市場価格はずっと不安定で、非常に不安定かもしれません。私たちの普通株を購入した人は大きな損失を受ける可能性があります。
私たちの普通株の市場価格はずっと変動し続ける可能性がある。私たちの株価は様々な要因で大幅に変動している可能性があります
117
また、株式市場で取引されている会社、特にナスダックでは、極端な価格や出来高変動を経験しており、これらの変動はこれらの会社の経営業績に関係なく、あるいは比例しないことが多い。広範な市場と業界要素は、新冠肺炎の大流行の影響、ウクライナの持続的な衝突、及び世界経済状況が世界経済に与える影響を含み、私たちの実際の経営業績にかかわらず、私たちの普通株の市場価格にマイナスの影響を与える可能性がある。
私たちはかつて、証券集団訴訟や他のタイプの株主訴訟を受ける可能性もあった。
株式市場全体、特にナスダック世界市場とバイオテクノロジー会社は、極端な価格と出来高変動を経験しており、これらの変動は往々にしてこれらの会社の経営業績と関係がないか比例しない。従来、証券集団訴訟は、会社証券の市場価格が変動した後に会社に提起されることが多かった。私たちはまた、当社の取締役または上級管理者が会社の資産/資源または利益の衝突の乱用/管理によって受託責任に違反するという他のタイプの訴訟に直面する可能性があります。どのような訴訟も、提起されれば、巨額の費用と経営陣の注意と資源の移転を招く可能性があり、これは私たちの業務、経営業績、または財務状況を損なうことになる。また、役員や上級管理職の責任保険費用の急激な増加は、巨額の弁護費用、和解、原告が得た損害賠償を支払うために、より低い全体保険限度額を選択したり、私たちが依存していた可能性のある保険を放棄したりする可能性があります。
将来、私たちの株式インセンティブ計画に基づいて、私たちの株主の所有率をさらに希釈し、私たちの株価を下落させる可能性があることを含む、私たちの普通株を売却して発行する権利、または普通株を購入する権利。
私たちは未来に私たちが計画している業務を継続するために追加的な資金が必要になるだろう。ある程度、私たちは株式証券を発行することで追加資本を調達し、私たちの株主は深刻な希釈を経験するかもしれない。私たちは1回または複数回の取引で、私たちが時々決定した価格および方法で普通株、転換可能証券、または他の株式証券を販売することができる。もし私たちが1つ以上の取引で普通株、転換可能証券、または他の株式証券を販売すれば、投資家はその後の売却によって深刻に希釈されるかもしれない。これらの売却はまた、私たちの既存株主の実質的な希釈をもたらす可能性があり、新しい投資家は私たちの既存株主よりも優れた権利を得る可能性がある。
118
私たちが2021年に改訂し、再編成した株式オプションおよびインセンティブ計画、またはA&R 2021計画によると、私たちは、従業員、取締役、コンサルタントに株式オプションおよび他の株式ベースの奨励を付与することを許可されています。また、2019年のインセンティブ株式計画によると、私たちは、現在当社または当社の子会社に雇用されていない将来の上級管理者および従業員に株式オプションおよび他の株式ベースの報酬を付与することを許可されています。もし私たちの取締役会または取締役会が将来的に付与可能な株式数を増加させることを選択し、A&R 2021計画の場合、私たちの株主がこのようなさらなる増加を承認すれば、私たちの株主は追加的な希釈を経験する可能性があり、私たちの株価は下落する可能性がある。
私たちの総流通株の大部分を市場に売却することは、私たちの普通株の市場価格を大幅に低下させる可能性があります。
公開市場で私たちの大量の普通株を売ることはいつでも起こる可能性がある。これらの売却や市場で大量の普通株を持っていると思っている人が株を売却しようとしているという見方は、私たちの普通株の市場価格を下げる可能性があります。
2019年までに再編された非帰属制限株および発行済み普通株および発行済み株式は、当該株式が帰属した直後に売却することができる。我々の株式インセンティブ計画又はこれらの計画に基づいて付与された将来奨励は、適用されるホームスケジュール、任意の適用される市場対峙協定の規定、並びに改正された1933年証券法又は証券法の下の第144条及び第701条の許容範囲内で、我々の株式インセンティブ計画又はこれらの計画に基づいて付与された未来奨励に基づいて、発行された株式を公開市場で販売することができる。
条件によると、私たちの普通株式のいくつかの所有者は、彼らの株式に関する登録声明を提出することを要求する権利があり、または彼らの株式を私たち自身または他の株主のために提出する可能性のある登録声明に含める権利がある。2020年7月、私たちはS-3 ASR表に提出時から自動的に発効する登録声明書を提出した。この登録声明によると、私たちは販売中に最大3.5億ドルの普通株を発行することを許可され、証券法で定義された“市場発行”とみなされ、私たちが証券法第405条で定義されている“有名な経験豊富な発行者”になる資格がある限り、私たちの普通株式、優先株、債務証券、引受権証および/または単位の株式数は制限されない。2020年7月、私たちはS-3 ASR表または販売株主表S-3に登録声明を提出し、私たちの一部の株主が時々提出した要約と転売、合計65,121,374株の私たちの普通株に関連した。2021年2月、私たちの株主の一人は、売却株主表S-3に従って3,450,000株の私たちの普通株の包売公開を完了しました。私たちはまた、将来的に私たちの普通株の大量の株を登録し、これらの登録声明に基づいて任意の追加の株を売却するか、あるいはそれらが公開市場で販売されると考えると、私たちの普通株の市場価格が低下する可能性がある。また、S-8表の登録声明を提出し、私たちの株式補償と持分激励計画に基づいて未来に発行する普通株の発行または予約を登録しました。これらのS-8表により登録された株式は発行時に公開市場で自由に販売することができ、一旦付与されると、関連会社に適用される数量制限を受ける。また、普通株の販売を実現するために、我々の一部の役員、従業員及び付属会社は、改正された1934年の証券取引法第10 b 5−1条に基づいて売却計画を策定しているか、又は将来的に計画されている可能性がある。これらの追加株のいずれかが公開市場で販売されている場合、またはそれらが売却されると考えられている場合、私たちの普通株の市場価格は低下する可能性がある。
もし証券アナリストが私たちの業務に関する研究や報告を発表しなければ、あるいは彼らが私たちの株に対するマイナス評価を発表したら、私たちの株価は下落するかもしれません。
私たちの普通株の取引市場は、業界や金融アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存している。もし私たちの業務を追跡している一人以上のアナリストが私たちの株に対する彼らの評価を引き下げたら、私たちの株価は下落するかもしれない。もしこのようなアナリストの一人以上が私たちの株を追跡しなければ、私たちは市場での私たちの株の可視性を失うかもしれないし、これは逆に私たちの株価を下落させるかもしれない。
119
私たちの主要株主と私たちの経営陣の一部のメンバーは私たちのかなりの割合の株を持っていて、株主の承認が必要な事項に大きな制御を加えることができるだろう。
2022年12月31日までの発行済み普通株によると、利益株主、取締役、役員実益は、私たちが発行した普通株の58.4%を持っています。この株主たちは彼らの所有権地位を通じて私たちに影響を与えることができるだろう。この株主たちは株主の承認を必要とするすべての事項を決定することができるかもしれない。例えば、これらの株主が共同で行動することは、取締役選挙を制御し、私たちの組織文書を修正し、任意の合併、資産売却、または他の重大な会社取引を承認することができるかもしれない。逆に、これは私たちの普通株の市場価格に悪影響を及ぼすかもしれない。これは私たちの普通株に対する能動的な買収提案や要約を阻止または阻止する可能性がありますが、これらの提案や要約は私たちの株主としての最良の利益に合致していると考えられるかもしれません。場合によっては、これらの株主の株主としての利益は、私たちの株主の利益とは異なり、衝突する可能性もある。
当社の会社登録証明書の改訂及び再記述の定款の条項、並びにデラウェア州法律における条項は、第三者が我々を買収したり、買収コストを増加させたりすることを困難にする可能性があり、そうしても、私たちの株主が現在の経営陣を利益または罷免することになります。
当社の会社登録証明書の改正および再記述の法律およびデラウェア州法律に含まれる条項は、私たちの統制権の変更や私たちの経営陣の変更を遅延または阻止する可能性があります。当社の会社登録証明書の改訂と再記述の定款には、以下の条項が含まれています
これらの条項は単独でまたは一緒に敵意の買収、統制権の変更、または私たちの経営陣の変動を延期または阻止する可能性がある。
また、私たちはデラウェア州で登録されているので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、私たちが発行した議決権株の15%以上の株主が私たちと合併または合併する能力を持つことを制限しています。私たちが改正して再記載した会社登録証明書または改正および再記述された定款またはデラウェア州法律の遅延または抑止権変更を有するいかなる条項も、私たちの株主が彼らが保有する普通株から割増の機会を得ることを制限する可能性があり、一部の投資家が私たちの普通株に支払う価格に影響を与える可能性もある。
120
私たちの改正と再記述の定款は、私たちの株主によって開始される可能性のあるいくつかの訴訟の独占フォーラムとして特定の裁判所を指定し、これは、私たちの株主が有利な司法フォーラムを得ることが私たちとの紛争を処理する能力を制限することができる。
我々が書面で代替裁判所を選択することに同意しない限り、デラウェア州衡平裁判所は、(I)私たちを代表して提起された任意の派生訴訟または訴訟に関連する任意の州法クレームの唯一および独占裁判所であり、(Ii)私たちの現職または前取締役、上級管理職または従業員が私たちまたは私たちの株主に負う信頼された責任を主張する任意の訴訟、(Iii)デラウェア州会社法または私たちの改正および再記載された会社定款の任意の規定に基づいて、私たちにクレームを提出する任意の訴訟、(Iii)デラウェア州会社法または私たちの改正および再説明された会社定款に基づく任意の訴訟、(Iv)私たちが改正および再記載した会社登録証明書または改正および再記載された法律の有効性を決定する任意の訴訟、または(V)デラウェア州内政原則によって管轄されるクレームを主張する任意の訴訟。私たちが書面で代替裁判所を選択することに同意しない限り、アメリカ連邦地域裁判所は、証券法に基づいて提起された任意の訴因を解決する唯一かつ独占的な裁判所であることをさらに規定し、再記述する。私たちの改正と再記述の定款における裁判所選択条項は、私たちとの紛争において私たちの株主が有利な司法フォーラムを得る能力を制限するかもしれない。
私たちの経営業績は大幅に変動する可能性があり、これは私たちの将来の経営業績を予測しにくくし、私たちの経営業績が予想を下回ったり、私たちの指導を招いたりする可能性があります。
私たちの四半期と年間経営業績は将来的に大きく変動する可能性があり、将来の経営業績を予測することは困難です。私たちの経営業績は様々な要素によって変動する可能性があり、その中の多くの要素は私たちがコントロールできないもので、予測が難しいかもしれませんが、以下の要素に限定されません
121
これらの要因の累積影響は我々の四半期や年度経営業績に大きな変動と予測不可能を招く可能性がある。したがって、異なる時期に私たちの経営業績を比較することは意味がないかもしれない。このような変化性および予測不可能性はまた、業界や金融アナリスト、または投資家の任意の時期に対する期待を満たすことができない可能性がある。もし私たちの経営業績がアナリストや投資家の予想よりも低い場合、または私たちが市場に提供するいかなる予測よりも低い場合、または私たちが市場に提供する予測がアナリストや投資家の予想よりも低い場合、私たちの普通株の価格は大幅に下落する可能性がある。
私たちが将来純営業損失の繰越といくつかの他の税務属性を利用する能力は限られているかもしれない。
私たちの歴史の中で、私たちは大きな損失を受けて、私たちは近い将来利益が出ないと予想して、私たちは永遠に利益を達成しないかもしれない。我々が課税損失を継続した場合,未使用損失が利用されなければ無期限繰り越しとなるが,2018年前に発生した連邦純営業損失が繰越された場合には,このような繰越満期に制限される。また、改正後の1986年の国税法第382条及び383条、又はこの法典及び州法の該当条項によると、ある会社が“所有権変更”を経験した場合、一般に3年間の間にその持分所有権の変化が50ポイントを超える(価値で計算される)と定義され、同社は変更前の純営業損失の繰越又はNOL及びその他の変更前の税収属性を用いて変更後の収入又は税金を相殺する能力は限られている可能性がある。私たちの現在の純営業損失或いは信用は以前の所有権変更による制限を受ける可能性があります。もし私たちが未来に所有権変更が発生すれば、その中の多くは私たちの制御範囲内にない可能性があり、私たちの純運営損失或いは信用を利用する能力は規則382と383条の更なる制限を受けるかもしれません。したがって、私たちは私たちの純営業損失や信用の大部分を利用できないかもしれない。また、2022年12月31日から開始される納税年度は、控除が認められた2017年以降の不良債権額は、当該年度の課税所得額の80%を超えてはなりません。
税法の変化は私たちや私たちの投資家に悪影響を及ぼすかもしれない。
米国連邦、州、地方所得税に関する規則は立法過程に参加する人員およびアメリカ国税局とアメリカ財務省の審査を受け続けている。税法の変化(これらの変化は追跡力を持つ可能性がある)は、私たちまたは私たちの普通株の所有者に悪影響を及ぼすかもしれない。例えば、守則第174条によれば、2021年12月31日以降の納税年度には、米国で発生した研究開発費が資本化·償却され、我々のキャッシュフローに悪影響を及ぼす可能性がある。近年、このような変化は多く発生しており、将来も変化し続ける可能性がある。税務法律、法規、および裁決がいつ、どのような形態、または発効日に公布、公布または発表されるかどうかは予測できません。これは、私たちまたは私たちの株主の納税責任を増加させるか、または税法の変化のいかなる悪影響を最大限に減少または軽減するために、経営方式を変更することを要求する可能性があります。
私たちは普通株に配当金を支払うつもりはありません。したがって、私たちの株主が投資リターンを達成する能力は私たちの普通株の価格上昇にかかっています。
私たちは私たちのどんな株にも現金配当金を支払ったことがなく、今は予測可能な未来に私たちの普通株に現金配当金を支払うつもりもありません。さらに、融資協定によれば、限られた状況を除いて、吾らは、任意の現金配当金を発表または支払いしたり、任意のカテゴリの配当金または任意の他の持分について現金割り当てを行ったりしてはならない。私たちは現在、将来の収益(あれば)を投資に使って、私たちの成長に資金を提供するつもりです。したがって、予測可能な未来では、あなたはあなたの普通株式から何の配当も得ることはあまりできない。私たちは配当金を支払うつもりがないので、あなたはあなたの投資からリターンを得ることができるかどうかは、私たちの普通株の将来の市場価値が高くなるかどうかにかかっています。私たちの普通株が値上がりし、保有者が購入した時の価格を維持する保証はありません。
122
上場企業としては、巨大なコストが発生し続けており、我々の経営陣は新たなコンプライアンスを実施するために多くの時間を投入する必要がある。私たちは財務報告書と他の要求に制約されており、私たちの会計と他の管理システムと資源はこれらの要求に対して十分な準備ができていないかもしれない。
上場企業として、私たちは多くの法律、会計、その他の費用を負担していますが、これは私たちが民間会社として起きていないことです。私たちは取引法の報告要件を遵守しなければなりません。その中で、私たちの業務と財務状況、私たちの合併子会社の年間、四半期、現在の報告書をアメリカ証券取引委員会に提出することを要求します。また、“サバンズ-オキシリー法案”と、米国証券取引委員会とナスダックが後に“サバンズ-オクスリ法案”を実施するために採択された規則は、当社の業務や財務状況に関する年間、四半期、イベント駆動報告書の提出を要求し、有効な開示と財務統制の確立と維持、およびコーポレートガバナンスの変更を含む上場企業に大きな要求を出している。
第404条によれば、我々は、独立公認会計士事務所が発行した財務報告内部統制の認証報告を含む経営陣の財務報告の内部統制に関する報告書を提出しなければならない。私たちは監査人の証明要件を含めて404条を遵守しているので、大量の会計費用を発生させ、多くの管理努力をかける必要があります。内部資源を投入し続け、外部コンサルタントを招聘することが可能であり、詳細な作業計画により、財務報告内部制御の十分性を評価して記録し、適宜手順をとって制御プログラムを改善し、テスト検証制御が文書記録の役割を果たしていることを検証し、財務報告内部制御の継続報告及び改善手順を実施する。我々は努力したにもかかわらず,我々も我々の独立公認会計士事務所も,規定された時間枠内で結論を出すことが可能であり,財務報告の内部統制に有効であり,第404条の要件に適合していることを証明している。これは、私たちの財務諸表の信頼性に自信を失ったため、金融市場に不利な反応をもたらす可能性がある。
また、2010年7月には“ドッド·フランクウォール街改革·消費者保護法案”、あるいは“ドッド·フランク法案”が公布された。テレス·フランク法案には、米国証券取引委員会がこれらの分野で“報酬発言権”や代理アクセスのような追加的な規制をとることを要求する重要な会社管理と役員報酬に関する条項がある。株主急進主義、現在の政治環境、および現在の高度な政府介入と規制改革は、大量の新しい法規と開示義務を招く可能性があり、これは追加のコンプライアンスコストを招き、現在予見できない方法で業務を運営する方法に影響を与える可能性がある。
また、上場企業の環境、社会、ガバナンスやESG活動に関する公共利益や立法圧力が増加し続けている。多様性と包括性、環境管理、地域コミュニティへの支援、コーポレートガバナンスおよび透明性、および私たちの運営においてESGおよび人的資本要因を考慮することを含む、いくつかの重要な分野で責任ある行動を取らなければ、代理コンサルティングサービス、および私たちのブランドや名声が損なわれることを含む株主の負の反応に直面する可能性がある。ますます多くの州要求組織もその取締役会の構成を報告したり、ニューヨークとカリフォルニア州を含む性別多様性と代表的な不足のコミュニティを強制的に要求したりしている。
上場企業としての私たちに適用される規則や法規は、私たちの法律と財務コンプライアンスコストを大幅に増加させ、いくつかの活動をより時間的で高価にすることを予想しています。これらの要求が私たちの経営陣と従業員の注意を他の業務から移すと、それらは私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼす可能性がある。増加したコストは私たちの純収益を減少させ、あるいは私たちの純損失を増加させ、私たちの子会社を含む他の業務分野のコストを下げることを要求するかもしれません。例えば、私たちは上場企業としての身分により、取締役や上級管理者責任保険を獲得することがより困難で高価になり、上場企業に適していると考えられる保証レベルを維持するために多くのコストが発生する可能性があります。私たちは、進行中の要件を満たすために、またはこれらの要件の任意の変更に応答して生じる可能性のある追加コストの金額または時間を予測または推定することができない。これらの要求の影響は、私たちの取締役会、取締役会委員会、または役員に参加することをより難しくし、合格した人を引き付け、維持することを可能にするかもしれない。
123
私たちのサービスは、企業市民権および環境、社会および企業統治またはESG事項、および/またはそのような事項の報告に対する私たちの負の影響を受ける可能性がある。
一部の投資家、消費者、および他の利害関係者はますます企業市民と持続可能な発展問題に注目している。私たちはこのような問題について責任を持って行動しないと思われるかもしれない。私たちの業務はこのような事件の否定的な影響を受けるかもしれない。このような問題、または関連する企業市民および持続可能な開発の問題は、私たちの業務に実質的な悪影響を及ぼす可能性がある。
Eidos合併後のリスク
私たちはEidos合併と関連した訴訟を受けるかもしれない。
Eidos合併に関連した訴訟は、私たち、私たちのいくつかの子会社、Eidos、そしてEidosの役員たちを対象にした。さらに、私たち、Eidos、私たちの子会社、または私たちのそれぞれの役員または役員は、Eidosの合併と関連取引によって追加的な訴訟が提起される可能性があり、将来の訴訟とその影響を正確に予測することはできないが、このような訴訟はBridgeioとEidosに巨額の費用を支払う可能性がある。
任意の法的訴訟または将来の訴訟の弁護または和解は、時間も高価である可能性があり、BridgeBio管理層および/またはEidos管理層の日常業務に対する注意を移動させる可能性があり、これらの法律訴訟または任意の未来の訴訟のいずれかがBridgeBioまたはEidosに不利に解決された場合、それらのそれぞれの財務状況、運営結果、または流動資金に重大な悪影響を及ぼす可能性がある。
一般リスク因子
もし私たちが環境、健康、安全の法律法規を守らなければ、罰金や処罰を受けたり、私たちの業務を損なう可能性のあるコストが発生するかもしれません。
私たちは多くの環境、健康と安全法律と法規の制約を受けて、それらの研究室の手続きと危険材料と廃棄物の処理、使用、貯蔵、処理と処理を管理する法律と法規を含む。私たちの行動は化学物質と生物学的材料を含む危険で燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物製品を発生させるだろう。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。私たちはこのような材料が汚染や傷害をもたらす危険を除去することができない。もし私たちが危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に責任を負い、いかなる責任も私たちの資源の範囲を超える可能性がある。民事や刑事罰金やこのような法律や法規を遵守しない罰に関する巨額の費用を招く可能性もある。
危険材料の使用による従業員の負傷により生じる可能性のあるコストや支出を支払うために労働者賠償保険を維持しているにもかかわらず、潜在的な責任に対応するには不十分である可能性がある。私たちは私たちが生物、危険または放射性物質を貯蔵したり処分したりすることによって、私たちが提起した環境責任や有毒侵害に対して保険を維持することはできません。
また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。私たちがこのような法律を守らないことはまた巨額の罰金、処罰、または他の制裁につながるかもしれない。
124
不利なグローバル経済状況は、私たちの業務、財務状況、または経営結果に悪影響を及ぼす可能性がある。
私たちは、私たちの業務を投資して拡張し、私たちの財務義務を履行し、第三者請負業者とパートナーを誘致し、維持し、より多くの資本を調達することができるかどうかは、私たちの経営と財務業績に依存し、これはまた、失業率、アメリカの未加入者数、政治的影響、インフレ圧力など、現在の経済と政治的条件、金融、商業、その他の私たちがコントロールできない要素を含む多くの要素に依存する。例えば、高い失業率(特に新冠肺炎が大流行した結果)、雇用不足或いはACAのいくつかの条項の廃止により、アメリカの個人保険カバー面の全面的な減少或いは喪失は、医療サービスや薬品に対する需要を減少させる可能性がある。そのほか、新冠肺炎が大流行したため、現在医療サービスと資源の獲得性は制限されている。より少ない患者が保険カバー範囲がないために医療を求めたり、ヘルスケアシステムの資源制限により医療を得ることができない場合、最終的に候補製品や私たちの業務を商業化する際に困難に遭遇し、運営結果、財務状況、キャッシュフローが悪影響を受ける可能性があります。
また、私たちの経営結果は、世界経済や世界金融市場の一般的な状況の悪影響を受ける可能性があり、私たちのような製薬やバイオ製薬会社はこれらの市場に依存して資金を獲得している。過去、世界金融危機は資本と信用市場の極端な変動と混乱をもたらした。新冠肺炎疫病を含む深刻または長期的な経済低迷は、必要に応じて許容可能な条件で追加資本を調達する能力の低下(あれば)、候補製品に対する需要の減弱など、私たちの業務に様々なリスクをもたらす可能性がある。経済が疲弊したり下落したりすることは、私たちのサプライヤーに圧力を与え、供給中断を招く可能性もある。上記のいずれも私たちの業務を損なう可能性があり、私たちは新冠肺炎の疫病、現在の経済気候、金融市場の状況が私たちの業務に悪影響を及ぼす可能性のあるすべての方法を予見できない。
ロシアとウクライナの間の紛争による世界的な経済状況は、私たちの業務、財務状況、株価、運営結果に悪影響を及ぼす可能性がある。
2022年2月、ロシアはウクライナへの軍事侵入を開始し、この地域は衝突と破壊が続く可能性がある。これまで、この衝突は私たちの業務にほとんど直接的な影響を与えていなかったにもかかわらず、この衝突による不確実性と連鎖反応は未知の間接的な影響を与える可能性がある。侵入の結果、米国や他の一部の国はロシアに制裁を科し、さらなる制裁を実施する可能性があり、国際商業や世界経済を損害または混乱させる可能性がある。この紛争またはこれまで実施されてきた制裁のより広範またはより長期的な結果は予測できず、これらの制裁は、さらなる制裁、禁輸、地域不安定、報復ネットワーク攻撃、地政学的転換、およびマクロ経済条件、安全条件、通貨為替レート、および金融市場への悪影響を含む可能性がある。紛争の潜在的影響には,我々の業務に影響を与える法律法規の変化,外国為替市場の変動,潜在的なサプライチェーンの中断,および我々の業務や運営に影響を与える可能性のあるマクロ経済要因に悪影響を及ぼす可能性のある市場変動性や不確実性が含まれる.
125
我々の内部コンピュータシステム、または我々の第三者研究機関協力者、CROまたは他の請負業者またはコンサルタントが使用するシステムは、故障したり、セキュリティホールに遭遇したりする可能性があり、これにより、私たちの製品開発計画が実質的に破壊される可能性があります。
セキュリティ対策が実施されているにもかかわらず、私たちの内部コンピュータシステムおよび私たちの現在および未来のCROおよび他の請負者およびコンサルタントのコンピュータシステムは、コンピュータウイルス、許可されていないアクセス、自然災害、テロ、ネットワークセキュリティ脅威、戦争、および電気通信および電気故障の破壊を受けやすいかもしれない。我々の知る限り,これまでこのような重大なシステム障害やセキュリティホールは経験していないが,このようなイベントが発生して我々の運営が中断されると,開発計画や業務運営の重大な中断を招く可能性がある.例えば、完了した、行われている、または将来の臨床試験における臨床試験データの損失は、私たちの規制承認作業を遅延させ、データを回復または複製するコストを著しく増加させる可能性がある。同様に,我々は第三者に依存して研究·開発を行い,薬品や薬物を生産·供給し,臨床試験や商業化活動を行っている。我々はこれらの第三者に依存して適切な制御と保障措置を実施し、ネットワーク事件を防止し、報告する。もし彼らがそうしなければ、私たちは私たちの情報、運営、業績、名声を含む財務と他の被害を受けるかもしれない。もし任意の中断またはセキュリティホールが私たちのデータやシステムを紛失したり、破損したり、機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの候補製品のさらなる開発と商業化は延期される可能性がある。我々はまた、第三者サービスプロバイダによる財務報告の内部統制に依存しており、これらのサービスプロバイダは、重大なシステム障害に遭遇したり、他の義務を履行できない可能性があり、正確かつタイムリーな財務諸表を作成する能力に影響を与える可能性がある, それによって私たちの経営業績、私たちの業務を経営する能力、そして私たちの投資家の私たちに対する見方を損害しました。
内部およびクラウド内のネットワーク脅威は、マルウェア、破壊マルウェア、恐喝ソフトウェア、不正アクセスシステムまたはデータ、運営中断、キーシステムまたはサービス拒否攻撃、機密、個人または他の保護された情報の不正発行、データ、ネットワークまたはシステムの損傷、個人への損害、および資産損失を含むが、これらに限定されないが、進化している。さらに、ネットワークの脅威または第三者が提供してくれる製品またはサービスで発見された他の中断または脆弱性の影響を受ける可能性があります。犯罪者がコンピュータシステムを攻撃するための技術は複雑で、変化が頻繁で、世界で規制の少ない遠隔地に由来する可能性がある。したがって、私たちはこのような技術を自発的に解決したり、十分な予防措置を施行することができないかもしれない。これらの事件を予防あるいは効果的に緩和しなければ、私たちの名声を損なう可能性があり、救済措置をとる必要があり、業務損失、監督管理行動、潜在的責任とその他の財務損失を招く。
いくつかのデータ漏洩はまた、HITECHによって改正されたHIPAA条項、他の米国連邦および州法律、非米国司法管轄区域の要求(EU GDPRおよびEU関連加盟国法律および他の外国法律を含む)に基づいて、影響を受けた個人および様々な政府および/または規制機関に報告されなければならず、場合によってはメディアに報告されなければならず、経済的処罰が適用される可能性がある。
私たちの保険証書は、インフラが破壊されたり、故障したり、中断されたり、悲劇的な事件と災害、または他の原因で発生した私たちの潜在的な損失を補償するのに十分ではないかもしれません。しかも、私たちは未来に経済的に合理的な条項でこのような保険を受けないかもしれないし、根本的にできないかもしれない。さらに、私たちの保険は、私たちに対するすべてのクレームをカバーすることができず、訴訟を弁護することができず、その価値にかかわらず、費用が高く、経営陣の注意をそらすことができるかもしれない。
126
私たちまたは私たちが依存している第三者は、気候変動、地震、病気の発生、または他の自然災害の悪影響を受ける可能性があり、私たちの業務の連続性と災害復旧計画は、深刻な災害から私たちを十分に保護できないかもしれない。
気候変化、地震、病気の爆発、または他の自然災害は、嵐、洪水、干ばつ、火災、温度変化のような極端な天気事件と絶えず変化する天気パターンを含み、これらはより一般的になり、私たちの運営を深刻に混乱させ、私たちの業務、運営結果、財務状況、および見通しに実質的な悪影響を及ぼす可能性がある。自然災害、極端な天気リスク、停電、ネットワークセキュリティ攻撃、または他の事件が発生した場合、本部の全部または大部分を使用することができず、当社の第三者CMOの製造施設のような重要なインフラを破損させたり、他の方法で運営を中断したりすることは難しいかもしれません。場合によっては、長い間私たちの業務を継続することはできません。例えば、全世界の新冠肺炎疫病により、著者らのいくつかの第三者CMOの製造施設の運営が中断され、著者らは臨床試験の薬物製品の供給遅延に遭遇する可能性がある。政府当局や企業が新冠肺炎の伝播を抑制するために持続的またはその後に講じた任意の措置、または将来再び爆発する場合にはこのような措置が必要である可能性があるとの見方は、我々のCMO製造製品の能力を制限し、依存する施設を閉鎖させたり、候補製品の臨床的または商業的供給を得ることに関連するコストを増加させ、それによって、私たちの業務、財務状況、または運営結果に悪影響を及ぼす可能性がある。新冠肺炎の大流行が著者らの結果に与える影響程度は未来の発展に依存し、これらの発展は高度な不確定性があり、正確な予測もできず、出現する可能性のある新冠肺炎の大流行の重症度と持続時間に関する新しい情報、及び大流行の制御或いはその影響を治療する行動を含む, ほかにもあります。我々が現在策定している災害復旧·業務継続計画は限られており、深刻な災害や同様の事件が発生した場合に十分であることはあまり証明されていない。また,ネットワークセキュリティ責任保険は入手が困難であり,コンピュータセキュリティプロトコル違反や他のネットワークセキュリティ攻撃による我々の被害は含まれていない可能性がある.私たちの災害復旧と業務連続計画の性質が限られているため、大量の費用が発生する可能性があり、特に地震保険が不足している場合には、私たちの業務に実質的な悪影響を及ぼす可能性があります。
気候変動や気候変動に対応する法律、法規、あるいは市場措置は私たちの業務と運営結果にマイナスの影響を与える可能性がある。
大気中の二酸化炭素や他の温室効果ガス濃度の増加による気候変動は,世界の気温,天気パターン,極端な天気や自然災害の頻度や重症度に悪影響を及ぼすことを含めて我々の業務にリスクとなる可能性がある。ハリケーン、竜巻、地震、野火、洪水のような自然災害と極端な天気条件は、私たちの施設に物理的なリスクを構成し、私たちのサプライチェーンの運営を乱す可能性があります。気候変動の水資源への影響は水資源不足を招く可能性があり,ある場所で十分な良質な水を得る能力を制限することは,運営コストを増加させる可能性がある。気候変動への懸念はまた、温室効果ガス排出の削減および/または気候変動の環境への影響を軽減するための新しいまたは追加の法律または法規要件をもたらす可能性がある。これらの法律または法規が現在の法律または法規義務よりも厳しい場合、私たちは候補製品の調達、製造、流通に関連するコストの中断または増加に遭遇する可能性があり、これは私たちの業務、運営結果、または財務状況に悪影響を及ぼす可能性があります。また、気候変動の影響は顧客の選好に影響を与え、気候友好型製品を提供できなければ、市場シェアの喪失を招く可能性がある。
127
ソーシャルメディアプラットフォームの使用はますます多くなり、新たなリスクと挑戦をもたらしている。
ソーシャルメディアは、私たちの研究、候補製品、研究薬、そして私たちの候補製品と研究薬を交流するためにますます使用されています。バイオ製薬業界のソーシャルメディア実践は発展し続けており,このような使用に関する法規は常に明確ではない。この変化は不確実性と私たちの業務に適用される法規を遵守しないリスクをもたらし、それによって私たちに対する規制行動を招く可能性がある。例えば、患者は、進行中の盲目的な臨床研究における彼らの経験をレビューするためにソーシャルメディアチャネルを使用するか、またはいわゆる有害事象を報告するかもしれない。このような開示が発生した場合、適用される有害事象報告義務を監視し、遵守できないリスクがあり、またはソーシャルメディアによって生じる政治的および市場的圧力の下で、私たちの業務または公衆の合法的な利益を守ることができない可能性があります。これは、私たちの開発候補薬、研究薬、および承認された製品に対する発言が制限される可能性があるからです。いずれのSNSにおいても、敏感な情報または否定的または不正確な投稿またはコメントを不適切に開示するリスクがある。もしこのような事件が発生したり、私たちが適用された法規を遵守できなかった場合、私たちは責任を負い、規制行動に直面したり、私たちの業務に他の損害を与える可能性があります。
私たちの業務運営は私たちを紛争、クレーム、訴訟に直面させるかもしれません。これらは高価で時間がかかる可能性があり、私たちの財務状況と運営結果に実質的な悪影響を及ぼす可能性があります。
時々、私たちは私たちの業務運営に関する紛争、クレーム、訴訟に巻き込まれるかもしれない。例えば、私たちは時々知的財産権問題、雇用問題、または商業紛争に関するクレームに直面したり、提起したりすることができる。いかなる紛争、クレーム、または訴訟も、経営陣の私たちの業務への関心を移す可能性があり、私たちは、任意の紛争、クレームまたは訴訟を処理または弁護する際に巨額の費用を発生する可能性があり、損害賠償金の支払いや和解、または私たちの運営や財務業績に悪影響を及ぼす可能性のある公平な救済を要求される可能性があります。これらの紛争に関連する訴訟は高価で時間がかかる可能性があり、私たちに不利であれば、私たちの財務状況や運営結果に大きな悪影響を及ぼす可能性がある。また、訴訟に関連する不確実性は、私たちの株価の変動性を増加させる可能性がある。
128
項目1 B。未解決問題教育署職員コメント
ない。
項目2.Pサーカス.サーカス
2022年12月31日現在、以下は私たちが占めている材料の属性です
属性 |
|
位置 |
|
正方形 |
|
|
持っているか |
|
頭文字をとる |
|
賃貸借延期 |
|
事務スペース |
|
カリフォルニア州パロアルト |
|
|
3,900 |
|
|
レンタルする |
|
2020 |
|
賃貸借契約は2023年4月に満期になります |
オフィススペースと実験室施設 |
|
ノースカロライナ州ローリー |
|
|
13,809 |
|
|
レンタルする |
|
2024 |
|
5年延長の選択肢 |
オフィススペースと実験室施設 |
|
カリフォルニア州パロアルト |
|
|
9,789 |
|
|
レンタルする |
|
2023 |
|
1年延長可能な2つのオプション |
事務スペース |
|
カリフォルニア州サンフランシスコ |
|
|
52,604 |
|
|
レンタルする |
|
2026 |
|
2年間延長するオプション |
実験室施設 |
|
ケベック州モントリオール |
|
|
20,039 |
|
|
レンタルする |
|
2032 |
|
5年延長の選択肢 |
事務スペース |
|
カリフォルニア州サンフランシスコ |
|
|
10,552 |
|
|
レンタルする |
|
2026 |
|
ありません |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
項目3.法律法律手続き
本年度報告Form 10−Kの日付まで,当社は重大な法的訴訟には関与していない。将来的には、私たちは法的手続きの一方と、正常な業務過程で提起されたクレームになるかもしれない。訴訟や請求の結果は正確には予測できないが、吾らは吾らがいかなる請求や訴訟の側であるとは信じておらず、その結果が個別または全体的に決定されて吾等に不利であれば、吾等の財務状況、運営業績やキャッシュフローに重大な悪影響を及ぼすことが合理的に予想される。結果にかかわらず,弁護や和解コスト,管理資源分流などにより,訴訟は我々に悪影響を与える可能性がある。
プロジェクト4.地雷安全情報開示
適用されません。
129
部分第2部:
項目5.登録者普通株の市場、関連するSTOCKHOLDER Mattersと発行者による株式証券の購入
市場情報
私たちの普通株式は2019年6月27日にナスダック世界ベスト市場またはナスダックで取引を開始し、取引コードはBBIOです。これまで、私たちの普通株は公開取引市場を持っていなかった。
所持者
私たちの普通株は2023年2月16日までに47名の登録株主がいます。我々の多くの普通株は仲介人や他の機関代表株主が保有しているため,これらの記録保有者が代表する株主総数を見積もることはできない.
配当政策
私たちはどんな配当金も発表したり支払ったりしたことがなく、予測可能な未来にも私たちの普通株にどんな配当金も支払わないだろう。さらに、融資協定によれば、限られた状況を除いて、吾らは、任意の現金配当金を発表または支払いしたり、任意のカテゴリの配当金または任意の他の持分について現金割り当てを行ったりしてはならない。
株式補償計画に基づいて発行された証券
本年度報告書10-K表第3部第12項における当社の株式報酬計画に関する情報は、参照されて本明細書に組み込まれる。
株式表現グラフ
次の図は、2019年6月27日(私たちの普通株がナスダックで取引を開始した)から2022年12月31日までの間の私たちの普通株の累積株主総リターンと同期のナスダック総合指数とナスダックバイオテクノロジー指数の累積総リターンを比較したものである。このグラフは、2019年6月27日に私たちの普通株1株100.00ドル、ナスダック総合指数、ナスダックバイオテクノロジー指数に17.00ドルの初公募価格で投資したと仮定し、配当金の再投資を仮定しています。
次の図に示す比較は履歴データに基づく.我々は,次の図に示す株価表現は,必ずしも我々の普通株の潜在的な未来表現を予測するつもりもないことを示していると警告する.グラフで使用されている情報は,ナスダック,ブルームバーグ,ロイター通信などから信頼できるソースとされているが,我々はこのような情報の誤りや見落としについては何の責任も負わない.
私たちは、改正された1933年の証券法または改正された1934年の“証券取引法”によって提出された任意の以前または未来の文書に相反する規定があり、本Form 10-K年度報告またはこれらの法規に従って提出された未来の文書を組み込む可能性があるが、本株式表現グラフ部分は“募集材料”ではなく、米国証券取引委員会に提出されたものとみなされてはならず、引用によってこれらの法規に従って提出された任意の以前の文書または未来の文書に組み込まれてはならないとみなされてはならない。
130
累積総リターン比較*
BridgeBio Pharma,Inc.ではナスダック総合指数とナスダックバイオテクノロジー指数:
*2019年6月27日に配当金の再投資を含む100ドルで普通株式または指数株を購入します。
未登録の証券を売却する
2022年12月31日までの年度内に、未登録証券は発行または売却していません。
発行人が株式証券を購入する
2022年12月31日までの年間で、吾らはどの会社の株式証券も買い戻していない。
第六項です[R保存された]
131
プロジェクト7.経営陣の議論と分析F財務状況と業務成果
私たちの財務状況と経営結果の議論と分析、本年度報告書の他の場所にForm 10-K形式で含まれている財務諸表と関連付記を読むべきです。
このForm 10−K年次報告書には、“1933年証券法”(改正)第27 A条又は“証券法”及び“1934年証券取引法”(改正)第21 E条又は“取引法”が指す“前向き陳述”が含まれている。場合によっては、これらの陳述は、“可能”、“予想”、“予想”、“信じる”、“予想”、“意図”、“可能”、“すべき”、“推定”または“継続”などの前向き語彙、および同様の表現または変形によって識別することができる。このような前向き表現は、リスク、不確定要素、および他の要素の影響を受けることができ、これらの要素は、実際の結果およびいくつかのイベントの時間が、このような前向き表現が明示的または示唆する未来の結果とは大きく異なる可能性がある。このような差異をもたらすか、または促進する可能性のある要因には、以下の決定された要因と、本年度報告10−K表に“リスク要因”と題する節で議論された要因とが含まれるが、これらに限定されない。本Form 10−K年次報告における前向き陳述は,我々のForm 10−K年次報告発表日までの観点を代表している。法的に別の要求がない限り、私たちはこれらの前向きな陳述を更新する義務を負いませんし、更新結果がこれらの前向きな陳述とは異なる可能性がある理由も負いません。したがって、あなたはこのような前向きな陳述に依存して、本年度報告書10-K表の日付までの私たちの任意の日付の観点を代表してはいけません。
132
概要
BridgeBio Pharma,Inc.(“私たち”または“会社”)は商業段階の生物製薬会社であり、設立の目的は明確な遺伝駆動因子を有する遺伝病と癌を有する患者を治療するために、革新性薬物を発見、創造、テスト、提供することである。BridgeBioの開発プロジェクトの範囲は早期科学から高度臨床試験までである。BridgeBioは2015年に設立され、経験豊富な薬物発見者、開発者と革新者からなるチームは遺伝子医学の進展を応用してできるだけ早く患者を助けることに取り組んでいる。設立以来、BridgeBioはすでに15個の調査性新薬申請またはINDを作成し、2つの製品がアメリカ食品·薬物管理局の許可を得た。私たちは20以上の異なる発展段階にある疾病州で仕事をしている。私たちのいくつかの計画は潜在力があると思う兆候を狙っています。承認されれば、私たちの候補製品は年間売上高が少なくとも10億ドルの一部の市場機会を狙っています。
我々は高度に満たされていない患者の需要と処理しやすい生物学的交差点に存在するため、遺伝疾患に注目した。我々の方法は,学術実験室とリーディング医療機関が初めて開発した研究を,最終的に患者に触れたい製品に変換することである。著者らは一連の科学進歩を通じてこの機会を実現することができる:(1)費用効果の高いゲノムとエクソン群のシークエンシングの出現に伴い、疾病の遺伝基礎を確定する;(2)分子生物学方面の進展;(3)遺伝子と疾病を結びつけることができる縦方向データと遡及性研究の発展と成熟。私たちはこの初期革新が新薬を作る最大の実際の源の一つだと信じている。
私たちは2015年に設立されて以来、すべての努力と財務資源を製品および技術権利の獲得と開発、私たちの知的財産権の組み合わせの構築、および私たちの完全子会社および制御エンティティ(私たちが考えている多数の制御権に基づいて決定された可変利益エンティティまたはVIEモデルまたは投票権エンティティまたはVOEモデルを含む)内で私たちの候補製品の研究および開発活動に集中しています。これらの活動を支援するために、我々は、我々の完全子会社BridgeBio Services,Inc.(I)新しいプロジェクトの安全を決定し、確保すること、(Ii)新しい完全子会社または制御されたエンティティを確立すること、(Iii)キー管理チームのメンバーを募集すること、(Iv)ポートフォリオ全体で資本を調達および分配すること、および(V)会計、法律、情報技術および人的資源、および作業空間を含む特定の共有サービスを提供すること。私たちは製品販売から相当な収入を得ていない。FDAによって承認された二人の製品の販売活動が私たちのそれぞれのパートナーに移行したか、または移行したので、短期的には私たちの製品販売から何の収入も得られないと予想されます. これまで、私たちの運営資金は、私たちの株式証券の売却、転換可能な手形の発行、債務借金、ある資産の売却の収益、および少ない程度の許可手配収入から来ています。
設立以来、私たちは重大な運営損失を受けた。2022年、2021年、2020年12月31日までの当社の純損失はそれぞれ4.847億ドル、5.865億ドル、5.055億ドルだった。私たちが利益を達成するために十分な製品収入を生み出すことができるかどうかは、私たちの完全子会社と制御された実体が私たちの候補製品の開発と最終商業化に大きく依存するだろう。私たちは少なくとも今後数年以内に営業と純損失が続くと予想しています。
133
臨床前と臨床開発の本質的に予測不可能であるため,我々の新しい治療法やわれわれの候補製品の開発段階を考慮すると,我々が必要とするスケジュールや候補製品開発コストを見積もることもできない。各種の要素のため、臨床と臨床前開発のスケジュールとコスト及び開発成功の潜在力は期待と大きく異なる可能性がある。例えば,新冠肺炎の持続的な影響や医療提供者や病院のウイルスとその変種への関心から,我々子会社が行っている臨床試験の患者募集に遅延や一時停止が生じている。そのほか、著者らは計画中の臨床試験、非臨床試験とINDを支持する良好な実験室実践毒理学研究の開始を含むいくつかの進行中の活動中に遅延に遭遇する可能性がある。遅延の持続時間とそれが私たちの業務に与える全体的な影響はまだ不明で、私たちは状況を監視し続けている。新冠肺炎の持続的な伝播により政府は世界的に重大な措置を講じた。これらの措置は業務、供給、薬品製造の中断を招き、運営の減少を招く可能性があり、そのいずれも私たちの業務、財務状況、運営結果に重大な影響を与える可能性がある。したがって、私たちは連邦、州、あるいは地方当局の要求に基づいて、あるいは私たちが最も公共の健康と安全に最も適合していると考え、私たちの患者コミュニティ、従業員、パートナー、サプライヤー、株主の利益に基づいて、さらなる予防と先制行動を取ることができる。このような行動、新冠肺炎の大流行の持続時間、あるいはその持続的な影響が私たちの業務や戦略に与える影響を予測することはできません, 我々が行っており計画中の臨床開発活動や将来性への影響,あるいは我々の財務·運営結果への影響を含む。
2022年1月には、当社のビジネスプロセス、効率、コスト節約の運営変化を推進し、当社の戦略と発展計画を推進するための再編計画を実施することを約束しました。再編計画には,我々の施設の統合と合理化,開発計画の優先順位の再決定,従業員の削減が含まれている。2022年12月31日までの財政年度中、私たちの再編、減値、関連費用は4380万ドルに達し、その中には主に段階的清算コスト、脱退とその他の関連コスト、長期資産の減価とログアウト、解散費と従業員関連コストが含まれている。2023年までの事業年度の再編計画を引き続き評価しており、発生する総費用は約600万~900万ドルと推定されている。我々のコスト範囲の見積りは何らかの仮説の影響を受けており,実際の結果はこれらの見積りや仮定とは異なる可能性がある.業務プロセス、効率、コスト節約の運営変化を推進するために、私たちの再構成代替案を評価し続けるにつれて、現在予測できない追加コストが生じる可能性もあります
2022年8月23日、私たちはヘルシンーングループから書面通知を受け、商業的考慮のため、便宜上、改訂と再署名の許可と協力協定(“改訂QED-ヘルシン許可と協力協定”)を終了する意向を示した。2022年12月21日から、我々の子会社QED治療会社またはQEDは、ヘルシンまたはヘルシンーンの双方と相互終了協定またはMTAを締結し、改訂されたQED-ヘルシン許可および協力協定およびその下のすべての権利と義務を終了した。ヘルシンヌ双方は、QEDが潜在的な非腫瘍学的適応の治療方法として、例えば世界的(中国、香港およびマカオを含まない)軟骨発育不良のような潜在的な非腫瘍学的適応の治療方法として、QEDがinfigratinibの開発、製造および商業化を継続することを可能にすることに同意した。終了により、QEDはもはや将来の規制または販売ベースのマイルストーン支払いを得る権利がないだろう。QEDは、ヘルシンヌが2023年3月31日までライセンス製品を販売しなくなるまで、TRUSELTIQの純売上高に印税を課す。ヘルシンヌ締約国はすでにFDAに通知を出し、TRUSELTIQの流通が永久的に停止し、関連IND項目下のすべての臨床研究が停止したことを通知した。私たちはライセンス製品が2023年第1四半期に販売されると予想している。MTAの定義によると、ヘルシンヌの各方面は終了計画を立てた。最初の1100万ドルの費用の後、計画内の活動を終えて均等に分担するのはQEDの責任だ。終了計画内の活動は2023年に完了する予定だ。
134
列報と合併の基礎
設立以来、私たちは完全子会社を作成したり、VOEモデルに従って多数決権権益を持っていたり、VIEモデルに従って私たちが主要な受益者である部分が所有している子会社を含めて、総称して私たちの合併エンティティと呼ばれています。私たち以外の実体が保有する合併実体の所有権権益は、私たちの合併貸借対照表で償還可能、転換可能な非持株権益と非持株権益として報告されている。償還可能な転換可能な非持株資本と非持株権益の損失は私たちの総合経営報告書で単独で報告することができる。
経営成果
2022年と2021年12月31日終了年度比較
私たちは2022年の財務業績と2021年の財務業績を比較した。2020年12月31日までの年度について,第7項で要求される他の情報は,2022年2月25日に米国証券取引委員会または米国証券取引委員会に提出された2021年12月31日までのForm 10−K年度報告第7項で見つかり,引用により本明細書に組み込むことができる。
以下の表に示した時期における我々の業務成果をまとめる
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
ライセンスとサービス収入 |
|
$ |
76,094 |
|
|
$ |
65,923 |
|
|
$ |
10,171 |
|
製品販売 |
|
|
1,554 |
|
|
|
3,793 |
|
|
|
(2,239 |
) |
ライセンス収入と製品販売のコスト |
|
|
3,434 |
|
|
|
3,114 |
|
|
|
320 |
|
研究開発 |
|
|
399,462 |
|
|
|
451,024 |
|
|
|
(51,562 |
) |
販売、一般、行政 |
|
|
143,189 |
|
|
|
192,210 |
|
|
|
(49,021 |
) |
再編成·減価·関連費用 |
|
|
43,765 |
|
|
|
— |
|
|
|
43,765 |
|
運営損失 |
|
|
(512,202 |
) |
|
|
(576,632 |
) |
|
|
64,430 |
|
優先審査クーポン券の収益、純額 |
|
|
107,946 |
|
|
|
— |
|
|
|
107,946 |
|
純損失 |
|
|
(484,652 |
) |
|
|
(586,454 |
) |
|
|
101,802 |
|
BridgeBio普通株株主は純損失を占めるべきです |
|
|
(481,183 |
) |
|
|
(562,539 |
) |
|
|
81,356 |
|
現金、現金等価物、有価証券 |
|
|
428,269 |
|
|
|
787,515 |
|
|
|
(359,246 |
) |
制限現金 |
|
|
37,930 |
|
|
|
177 |
|
|
|
37,753 |
|
株式証券投資 |
|
|
43,653 |
|
|
|
49,148 |
|
|
|
(5,495 |
) |
2022年12月31日と2021年12月31日までの年度の経営結果は、2023年12月31日までの年度または任意の他の未来年度または中期の予想結果を必ずしも示しているとは限らない。
現金、現金等価物、有価証券、限定現金および持分証券投資
2022年12月31日まで、私たちは現金、現金等価物、有価証券4.283億ドル、制限的な現金3790万ドル、株式証券投資4370万ドルを持っている。制限された現金とは、主に、付記10に記載された自社融資·担保協定第2改正案に基づいて設立された制御口座内の資金を指す。融資協定を基本的に改正する条項によると、このような無利子現金の使用は制限されており、NAVRE−BMSライセンス契約の履行に関する義務の履行に直結するいくつかの研究·開発費用にしか利用できず、付記11はさらにこの点を説明している。 私たちは株式証券への投資が私たちの流動性の一つの源だと思います。必要であれば、これらの株を清算し、現在の業務に資金を提供する可能性があるからです。
135
2022年3月、我々の子会社Origin Biosciences,Inc.とSentynl Treateutics,Inc.(“Sentynl”)またはOrigin-Sentynl APA間の資産購入プロトコル(“APA”)が完了した後、1,000万ドルの前払いを受けた。
2022年6月、私たちは、当社の子会社NAVERE Pharma,Inc.(“NAVRE”)と百時米施貴宝社(“BMS”)と締結したライセンスおよび協力協定(NAVRE-BMSライセンス契約と略す)から9,000万ドルの前金を受け取りました。BMSの事前支払いを受けたことは、次の章でさらに説明される修正された融資協定のいくつかの強制的な前払い条項をトリガし、したがって、私たちは融資者に2,050万ドルを支払い、2022年12月31日までに3,780万ドルを個別に制御口座に入金する。
2022年6月、我々の優先審査証明書(“PRV”)を売却して1.1億ドルの毛収入を得、2022年12月31日までの年間取引コストを差し引いた純収益1.079億ドルを確認した。
2022年11月、私たちが提出した2020年の棚登録の一部として、市場で普通株を発行することで490万ドルの純収益を得た。
収入.収入
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
収入: |
|
|
|
|
|
|
|
|
|
|||
ライセンスとサービス収入 |
|
$ |
76,094 |
|
|
$ |
65,923 |
|
|
$ |
10,171 |
|
製品販売 |
|
|
1,554 |
|
|
|
3,793 |
|
|
|
(2,239 |
) |
総収入 |
|
$ |
77,648 |
|
|
$ |
69,716 |
|
|
$ |
7,932 |
|
許可とサービス収入には、主に、私たちの許可と連携協定に関する前払い許可、マイルストーン支払い、サービス収入の確認が含まれています。ライセンスとサービス収入の確認レベル部分は、継続業績義務、マイルストーンや他またはイベントの前金に割り当てられた予定確認期限、研究開発契約サービスによる努力レベル、および新たな協力協定(あれば)に依存する。
2022年12月31日までの年間、ライセンスおよびサービス収入は7610万ドルで、うち7470万ドルは、NavireとBMSが2022年5月に締結したライセンスと連携協定またはNAVERRE-BMSライセンス契約に関連する前期ライセンスおよびサービス収入を確認するために使用されます。残りの金額は、主に私たちのパートナーに移転した商業製品と、私たちのパートナーに提供された製品を販売することで受け取った印税収入を確認するために使われます。
ライセンスおよびサービス収入は、2021年12月31日までの年間6590万ドル
連合博が2021年11月に初めて公募されるまで、連合博との取引は関連側と取引されていたが、当時私たちは1人の取締役を任命または罷免して聯博取締役会に入る権利は終了した(私たちの合併財務諸表付記7参照)。連合博は初公募後に関連側とみなされなくなった。
136
運営コストと支出
研究と開発費
|
|
十二月三十一日までの年度 |
|
|
|
||||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|||||
|
|
(単位:千) |
|||||||||||
研究開発 |
|
$ |
399,462 |
|
|
$ |
451,024 |
|
|
$ |
(51,562 |
) |
|
2022年の研究開発費が2021年に比べて5160万ドル減少したのは、主に我々の再編計画に基づいて開発計画の優先順位を再配置し、外部コストと株式ベースの報酬コストを削減したためである。2022年に研究開発費に記録された株式ベースの報酬は3800万ドルだったが、2021年には5620万ドルだった。この低下の主な原因は、2022年12月31日までに実現され、可能なマイルストーンとして決定されたのと比較して、2021年12月31日までに実現され、可能な監督管理と発展マイルストーンとして決定された業績マイルストーン補償スケジュールの数が多いためである。以下に議論するQED−Helsinn許可と協力協定により、2022年度の研究開発費の減少分は、2021年度に比べて外部コストの減少を相殺している。
QED−Helsinn許可と連携プロトコルにより,Helsinn分担プロトコルで規定されている何らかの適応のinfigratinib開発コストの60%である。改訂されたQED−Helsinn許可と連携協定が発効した日からHelsinnは独自に何らかの適応を担当するinfigratinibの開発費を,過渡期に発生した費用は全額精算することができる。中の概要部分で議論されているように項目2.経営陣の財務状況と経営結果の検討と分析Helsinnは2022年8月23日に改訂されたQED-Helsinn許可と連携協定を終了する予定であることを私たちに通知した。ヘルシンヌ双方は2022年12月21日からMTAを締結し、改訂されたQED-ヘルシン許可と協力協定及びその下でのすべての権利と義務を終了した。ヘルシンヌ双方は、QEDが潜在的な非腫瘍学的適応の治療方法として、例えば世界的(中国、香港およびマカオを含まない)軟骨発育不良のような潜在的な非腫瘍学的適応の治療方法として、QEDがinfigratinibの開発、製造および商業化を継続することを可能にすることに同意した。すべての清算コストは“再編·減価·関連費用”の一部として我々の総合経営報告書に記載されている。
QED−Helsinnライセンスと連携プロトコル、改訂されたQED−Helsinnライセンスと連携プロトコル、およびQED−Helsinnライセンスおよび連携プロトコルの終了に関するより多くの情報は、我々の合併財務諸表の付記11を参照されたい。
研究開発コストは主にコンサルタント、請負業者、契約製造組織(CMO)と契約研究組織(CRO)に支払う費用など、著者らの臨床前と臨床開発活動に関連する外部コストを含み、計画通りに追跡する。候補製品を指定した後に発生する候補製品に直接関連するライセンス料やその他のコストは特定の計画費用に含まれる。候補製品を指定する前に発生するライセンス料とその他のコストは,次の表に示すように他の開発計画に含まれる。
137
以下の期間に計画的に発生した研究と開発費用を表にまとめた
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
Acoramidis(従来AG 10と呼ばれていた) |
|
$ |
91,901 |
|
|
$ |
107,806 |
|
少量のイフェラチニブによる軟骨発育不全の治療 |
|
|
32,387 |
|
|
|
40,650 |
|
BBP−418による2 I型肢体筋ジストロフィーの治療、またはLGMD 2 I型 |
|
|
22,372 |
|
|
|
15,202 |
|
Encaleret |
|
|
27,485 |
|
|
|
15,739 |
|
BBP-631 |
|
|
34,009 |
|
|
|
46,035 |
|
KRAS阻害剤組み合わせ |
|
|
33,216 |
|
|
|
15,554 |
|
その他の発展計画 |
|
|
90,492 |
|
|
|
147,303 |
|
他の研究プロジェクトは |
|
|
67,600 |
|
|
|
62,735 |
|
合計する |
|
$ |
399,462 |
|
|
$ |
451,024 |
|
販売、一般、行政費用
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
販売、一般、行政 |
|
$ |
143,189 |
|
|
$ |
192,210 |
|
|
$ |
(49,021 |
) |
2021年と比較して,2022年の販売,一般,行政費が4900万ドル減少したのは,主に我々の再編措置がコストを簡略化したためである。2022年に販売、一般、行政費に記録された株式ベースの報酬は5470万ドルだったが、前年は4940万ドルだった。この増加は主に2022年第1四半期と2021年末に近い時点で支給される贈与が増加したためだ。
QED-ヘルシンエン許可と協力協定によると、ヘルシンは米国でTRUSELTIQを共同商業化し、50:50の割合で利益と損失を共有している。改訂されたQED−Helsinn許可と協力協定が発効した日から,ヘルシンーンはTRUSELTIQの商業化を独自に担当し,過渡期に発生した費用は全額精算できる。中の概要部分で議論されているように項目2.経営陣の財務状況と経営結果の検討と分析ヘルシンヌは2022年8月23日に、改訂されたQED-ヘルシン許可と協力協定を終了しようとしていると私たちに通知した。ヘルシンヌ双方は2022年12月21日からMTAを締結し、改訂されたQED-ヘルシン許可と協力協定及びその下でのすべての権利と義務を終了した。ヘルシンヌ双方は、QEDが潜在的な非腫瘍学的適応の治療方法として、例えば世界的(中国、香港およびマカオを含まない)軟骨発育不良のような潜在的な非腫瘍学的適応の治療方法として、QEDがinfigratinibの開発、製造および商業化を継続することを可能にすることに同意した。すべての清算コストは“再編·減価·関連費用”の一部として我々の総合経営報告書に記載されている。
QED−ヘルシン許可と協力協定でのヘルシンーンの130万ドルの商業化損失におけるシェアを,2022年12月31日までの年間販売,一般,行政費の減少に計上した。2021年12月31日までの1年間で、比較可能額は890万ドル。
再編成·減価·関連費用
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
再編成·減価·関連費用 |
|
$ |
43,765 |
|
|
$ |
— |
|
|
$ |
43,765 |
|
138
私たちの合併財務諸表付記17で述べたように、2022年1月には、当社のビジネスプロセス、効率、コスト節約の運営変化を推進し、当社の戦略と発展計画を推進するための再編計画の実施を約束しました。再編計画には,我々の施設の統合と合理化,開発計画の優先順位の再決定,従業員の削減が含まれている。2022年12月31日までの年間で発生する費用には、主に、コストの段階的な削減、脱退やその他の関連コスト、長期資産の減価とログアウト、解散費、従業員関連コストが含まれています。私たちは2023年度の再編計画を評価し続けており、発生した総費用は約600万~900万ドルと推定されている。我々のコスト範囲の見積りは何らかの仮説の影響を受けており,実際の結果はこれらの見積りや仮定とは異なる可能性がある.業務プロセス、効率、コスト節約における運営変化を推進するために、我々の再構成代替案を評価し続けるにつれて、現在予測できない追加コストが生じる可能性もある。
その他の収入,純額
利子収入
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
利子収入 |
|
$ |
7,542 |
|
|
$ |
1,133 |
|
|
$ |
6,409 |
|
利息収入には現金等価物と有価証券から稼いだ利息収入が含まれている。利息収入の増加は私たちの有利子現金口座の平均残高とより高い金利によるものです。
利子支出
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
利子支出 |
|
$ |
(80,438 |
) |
|
$ |
(46,778 |
) |
|
$ |
(33,660 |
) |
2022年の利息支出には、主に2021年1月に発行された2029年手形、2020年3月に発行された2027年手形、および2021年11月17日の融資協議に基づいて各貸主に提供される定期融資による利息支出が含まれています。
2021年の利息支出には、主に、私たちの2029年手形、2027年手形、2018年6月19日の私たちの日付に基づく融資および保証協定(時々改訂された)とHercules Capital、Inc.またはHerculesによって提供される現在全額返済されている定期融資、および2019年11月13日の日付に応じた融資と保証契約またはSVBおよびHercules融資プロトコルとシリコンバレー銀行(SVB)およびHerculesによって提供される現在全額返済されている定期融資が含まれています。
2021年に比べて2022年に3370万ドル増加したのは,主に我々の債務元金が増加し,定期融資が2021年11月に抽出され,2022年の実物支払利息の選択権を行使したためである。
優先審査クーポン券の収益、純額
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
優先審査クーポン券の収益、純額 |
|
$ |
107,946 |
|
|
$ |
— |
|
|
$ |
107,946 |
|
139
2022年5月、私たちはPRVを1.1億ドルで売る最終合意に達したことを発表した。我々は2021年2月に米国食品·薬物管理局の計画に基づいてPRVを受け取り、この計画は稀な小児科疾患の治療方法の開発を奨励することを目的としている。私たちの子会社OriginがNULIBRYの承認を得た時、私たちはPRVを獲得した。PRVの売却は慣例成約条件の制約を受け、適用された米国の反独占承認要求が満期になった後、2022年6月に完成する。2022年12月31日までの年間で1.1億ドルの総収益を受け取り,2022年12月31日までの年度の純収益1.079億ドルを確認した。
その他の収入,純額
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
その他の収入,純額 |
|
$ |
(7,500 |
) |
|
$ |
35,823 |
|
|
$ |
(43,323 |
) |
その他の収入(費用)、2022年の純額は主に私たちの株式証券投資公正価値の変化による実現済みと未実現純損失8.2ドルからなる Origin資産の売却損失630万ドルと,Origin規制マイルストーンに関する支出350万ドルは,改訂されたQED−Helsinn許可と協力協定によりヘルシンからの1,250万ドルの売掛金の収益部分が相殺されることが確認された。
その他の収入(支出)、2021年の純額は主に2021年11月のLianBio初公募時にLianBioへの株式方法投資を持分証券投資の未実現収益6850万ドルに変換し、一部は2021年12月31日までのこのような株式証券投資公開価値が変化した未実現損失3770万ドル(私たちの合併財務諸表付記7参照)と早期返済我々の債務330万ドルの損失によって相殺される。その他の収入(支出)は、年内純額にもレオコールオプション負債の公正価値変動が含まれている。2021年3月、レオはレオコールオプションを終了することを選択し、レオコールオプション負債560万ドルを終了確認した。
所得税
会社として、私たちはアメリカ連邦、州、そして外国所得税を払わなければならない。米国連邦所得税については、合併条例の規定の要求に適合する合併実体に合併の米国連邦所得税申告書を提出する必要がある。合併申告に組み込まれたハードルを満たしていない実体は、単独の米国連邦所得税申告書を提出し続ける。当社が税務目的で会社期間中に営業損失が発生したとみなされている範囲では、適用税法により、当社は一般的に繰り越した純営業損失を利用して将来課税収入の現金税を相殺することができます。
2022年から2017年の減税·雇用法案は174条を改正し、今年度の研究·実験(R&E)支出とソフトウェア開発コスト(総称してR&E支出と呼ぶ)の控除を廃止し、彼らのR&E支出を資本口座に計上し、5年以内に償却するよう納税者に要求した(15年は米国国外で行われたR&E活動の支出によることができる)。我々は2022年12月31日までの年度に資本化R&E支出の繰延税金資産を実現し、この資産は完全に推定準備金によって相殺された。
140
2022年12月31日現在、連邦と州所得税における純運営損失はそれぞれ約14億ドルと2.554億ドルであり、将来の課税収入の減少に利用できる(あれば)。2018年までに発生した連邦純営業損失3,750万ドルは2035年に満期となり、2018年以降に発生した14億ドルの損失は無期限に繰り越され、使用年度には80%の課税所得額が制限される。国の純営業損失は一般的に2038年に満期になります。2022年12月31日まで、私たちは9270万ドルの連邦研究開発と孤児薬物信用の繰越があり、使用しなければ、これらの資金は2036年に満期になります。2022年12月31日まで、私たちは1720万ドルの国家研究開発信用繰越があります。州の研究開発税収控除は異なる日に期限が切れるが、カリフォルニアの研究開発税収控除は無期限に続く。
繰延税金資産の回収可能性が不確定な場合、推定値を計上する。評価免税額を提供する決定は、将来、繰延税金資産を利用するために十分な将来課税所得額を生成する可能性が高いかどうかを評価することに依存する。既存の証拠(私たちの合併実体の歴史的経営損失と未来の損失の予測を含む)の重みに基づいて、私たちはアメリカ連邦と州の繰延税金資産に税金損失と相殺繰越による推定値を提供した。私たちは2020年に2027年手形を発行したため、一時的な差異の償却を審査する際に、私たちの既存の繰延税金資産は繰延税金負債を完全に相殺することはできないと確定した。これにより、2020年12月31日までの年度に確認された繰延税金負債は110万ドルとなった。私たちは2021年1月1日にASU 2020-06年度を事前に採用した後に繰延税金負債の確認を取り消し、所得税の支出に影響を与えていない。2022年12月31日までと2021年12月31日までの年間推定免税額はそれぞれ1億1千万ドルと1億995億ドル増加した。
純営業損失及び貸記繰越の使用が重大な年次制限を受ける可能性があるのは、1986年に改正された“国内税法”第382条又は当該法規及び類似の国の規定により規定された所有権変更制限によるものである。年間限度額は純営業損失や使用前の信用満期になる可能性があります。もし私たちの所有権が変化すれば、純営業損失と税収控除の使用が制限される可能性があります。
償還可能な非制御的権益と非制御的権益は純損失を占めなければならない
当社の連結経営報告書のうち転換可能な非持株権益及び非持株権益を償還可能な純損失には、当社に割り当てられていない当該等の合併実体の純損失部分が含まれています。非持株権益は純損失金額の変化を占めるべきであり、直接に著者らの合併実体の純損失変化の影響を受け、しかも所有権のパーセンテージ変化の結果である。私たちの連結財務諸表の付記6を参照してください。
2022年、転換可能な非持株権益と非持株権益の純損失は350万ドルだったが、2021年は2390万ドルだった。
流動性と資本資源
私たちはこれまで主に株式証券の売却、転換可能な手形の発行、債務借金、ある許可手配の収入とある資産の売却を通じて、私たちの業務に資金を提供してきた。2022年12月31日まで、私たちの現金、現金等価物と有価証券は4.283億ドル、制限現金は3790万ドル、株式証券投資は43.7ドルです 百万ドルです。私たちは株式証券への投資が私たちの流動性の一つの源だと思います。必要であれば、これらの株を清算し、現在の業務に資金を提供する可能性があるからです。融資プロトコルの下でNAVERRE−BMSライセンスプロトコルに関する制限的現金は3,780万ドルであり,総合貸借対照表における“制限的現金”の一部として示されている。私たちの完全子会社と制御された実体が持っている資金は特定の実体に使用することができますが、限られた場合は除外します。2022年12月31日現在、私たちの未返済債務は17億ドルで、債務発行コストと債務蓄積を差し引いている。
141
設立以来、私たちは重大な運営損失を受けた。2022年、2021年、2020年12月31日までの当社の純損失はそれぞれ4.847億ドル、5.865億ドル、5.055億ドルだった。運営から419.5ドルの現金純流出が発生しました 同期はそれぞれ4.979億ドル、3.997億ドル、3.997億ドルだった。2022年12月31日現在、私たちの累計赤字は19億ドルです。業務プロセス、効率、コスト節約における運営変化を推進するための再構成計画を採用しているが、今後数年間、薬物開発や発見作業に資金を提供し、後期計画の商業投入準備に関連したコストを継続していくため、今後数年は運営および純損失が続くことが予想される。特に,パートナーなしで計画を進め,後期臨床試験を通過すれば,多大な費用が発生する。我々の現在の業務計画も、我々が利益を得るのに十分な製品販売を実現する能力を生成することを含む重大な不確実性やリスクの影響を受けており、これは、私たちの合併実体候補製品の開発成功と最終商業化、そしていくつかの臨床プロジェクトを協力して開発する能力に大きく依存するであろう。
私たちの短期·長期流動資金需要には、2029年手形、2027年手形、定期融資に関する契約支払い(私たちの総合財務諸表付記10参照)、および私たちの不動産賃貸項目下の債務(私たちの総合財務諸表付記14参照)と、私たちの再編計画下の残りの負債(私たちの総合財務諸表付記17参照)が含まれています。
また、いくつかの従業員やコンサルタントとパフォーマンスに基づくマイルストーン報酬スケジュールを作成し、その帰属は様々な規制と発展マイルストーンに依存し、最初に知られている固定金額は、私たちが唯一の選挙時に現金または株式の形でそれぞれまたはマイルストーンを達成したときに支払うことができる(私たちの総合財務諸表付記9参照)。
また、様々な許可や協力協定に基づいて、特定の知的財産権、臨床、規制、販売マイルストーンの完成と実現に向けたマイルストーンの支払いを要求する支払い義務があるかどうかが求められています。私たちはまた、正常な業務過程でCROと他の臨床試験サプライヤーおよび臨床前研究と他の運営目的のためのサービスと製品サプライヤーと協定を締結し、これらの協定は一般に書面通知後にキャンセルすることができ、終了費用が発生する可能性がある。
現在の運営計画と財務予測によると、私たちの現金と現金等価物、有価証券、制限された現金、および株式証券への投資は少なくとも今後12ヶ月以内に私たちの運営に資金を提供すると予想される。もし私たちの現在の運営計画や財務予測が変化すれば、一般市場と経済状況、インフレ圧力、サプライチェーン問題及び持続的な新冠肺炎疫病が私たちの研究開発活動に与える影響を含む場合、私たちは公募株或いは私募株式発行、債務融資或いは追加協力と許可手配などの形式の追加資金がもっと早く必要になるかもしれない。しかし、未来の融資は、もしあれば、私たちが受け入れられる金額や条項で提供されないかもしれない。
また、私たちは持続的な新冠肺炎の疫病とインフレ圧力に関連する持続的な事態の発展に密接に注目しており、これは私たちの財務と運営業績にマイナスの影響を与えるかもしれない。私たちは引き続き私たちの運営コストと支出と私たちの現金と現金等価物を評価します。もし必要であれば、私たちの運営計画を適切に調整します。
142
流動資金源
初公開と市場で株を発行する
2020年7月7日、我々は米国証券取引委員会にS-3 ASR表または2020年棚登録声明を提出し、普通株、優先株、債務証券、権証と単位またはそれらの任意の組み合わせの登録に関連した。吾らも同時にJefferies LLCやSVB Leerink LLCや販売エージェント(総称)と公開市場販売プロトコルや2020年販売プロトコルを締結し,吾らが時々2020年の棚下の“市価”で35,000,000ドルにのぼる普通株を発売,発行および販売することを規定し,その制限を受けている。2020年の販売協定によると、適用される販売エージェントに普通株販売総収益の3.0%の現金手数料を支払う。2022年12月31日までの年間で、会社は1株10.90ドルの平均価格で45.58万株を売却し、純収益は490万ドルだった。2022年12月31日現在、2020年の販売協定に基づき、会社は最大3.45億ドルの普通株を売却する資格がある。
債務
2022年12月31日現在、2029年手形、2027年手形、融資協議下の借入金がありますが、以下ではこれらの借金について検討します。2021年12月31日現在、私たちの債務には、2029年手形、2027年手形下の借金、改正されたHerculesローンと保証協定、シリコンバレー銀行とHerculesローンと保証協定下の定期ローンが含まれており、この2つのローンは2021年に全額返済される。
2029年ノート
2021年1月、証券法第144 A条によれば、受託者である米国銀行全国協会又は2029年手形受託者との間の2021年1月28日に発行された2029年手形元金総額7.475億ドルの債券、又は2021年手形発行。
2029年に発行された債券は半年ごとに配当され、毎年2月1日および8月1日に配当され、2021年8月1日から年利2.25厘となる。事前に両替、償還、または買い戻ししない限り、2029年に発行された債券は2029年2月1日に満期になる。2029年の手形は、私たちの選択に応じて現金、普通株式、または現金と普通株の組み合わせに変換することができます。
2029年債券初期購入者割引を差し引くと、2021年債発行から約7.314億ドルの純収益を得た。私たちは2029年の債券の直接発売費用を負担しない。2021年手形発行純収益のうち約6130万ドルを用いて2021年上限コール取引のコストを支払い、約5000万ドルを使用して普通株の株式買い戻しを支払います。
2029年債券保有者は、場合によっては、2028年11月1日前の営業日取引終了直前の任意の時間に、その2029年債券の全部または任意の部分を選択的に変換することができる。
2028年11月1日以降、満期日直前の第2の予定取引日の取引が終了するまで、保有者は、その2029年債の全部または任意の部分を随時変換することができる。
143
私たちは2026年2月6日までに2029年の手形を償還しないかもしれない。場合によっては、2026年2月6日以降の償還日と、満期日直前の第41回予定取引日または前に、私たちの選択に応じて、2029年債券の全部または任意の部分を償還することができる。2029年に発行された債券は債務返済基金を設けない。当行が基本変動を行う場合(定義は“2029年債券契約”参照)、所持者は吾などにその全部または任意の部分2029年手形を現金で購入することを要求することができ、買い戻し価格は購入予定の2029年手形の本金額の100%に等しく、また基本変動買い戻し日(ただし基本変動購入日を含まない)の任意の課税および未払い利息を加えることができる。2029年債券契約には、ある違約事件が発生及び継続した場合、2029年債券受託者又は当時の未償還債券元金総額の25%以上の保有者を含む慣用条項及び契約が記載されており、すべての債券の全ての元金が応算特別利息(あればある)とともに即時満期及び対応することが宣言できる。2029年手形は私たちの一般的な無担保債務であり、支払権は私たちのすべての債務よりも優先され、2029年手形の支払権は私たちのすべての債務に明らかに従属する;私たちのすべての債務(私たちの2027年手形を含む)と同等の支払権である;このような債務を保証する資産価値の範囲内で、実際には私たちの任意の保証債務よりも低い;構造的には私たちの子会社のすべての債務および他の負債(貿易支払いを含む)よりも低い。
私たちの今後の2029年の手形項目の最低支払いを含む、他の詳細を理解するために、総合財務諸表の付記10を参照してください。
2027年ノート
2020年3月,証券法第144 A条に基づき,BridgeBioと米国銀行全国協会(受託者または受託者として)との間の日付が2020年3月9日のIndentureまたはIndentureに基づき,2027年手形の元金総額5.5億ドルを発行し,受託者として適格機関の買手への非公開発行,または2020年手形発行を行った.
2027年債はBridgeBio社の優先無担保債務で、2020年9月15日から半年ごとに利息を支払い、2020年9月15日から半年ごとに支払い、金利は2.50%となる。事前に両替したり買い戻ししたりしない限り、2027年に発行された債券は2027年3月15日に満期になる。転換後、2027年の手形は私たちの選択に応じて現金、普通株または現金と普通株の組み合わせに変換することができる。
最初の購入者の割引と発売費用を差し引いた後、2020年のチケット発売から約5.37億ドルの純収益を得た。2020年手形発行純収益のうち約4,930万ドルを用いて上限コール取引のコストを支払い,2020年手形発行に関する普通株買い戻しを約7,500万ドル用いた.
2027年債券保有者は、場合によっては、2026年12月15日前の営業日取引終了直前の任意の時間に、その2027年債の全部または任意の部分を選択することができる。
2026年12月15日またはその後、満期日直前の第2の予定取引日の取引が終了するまで、保有者は、その2027年債の全部または任意の部分を随時変換することができる。
私たちは満期日までに2027年の債券を償還しないかもしれないし、2027年の債券に債務超過基金を設立することもないかもしれない。吾らが基本的な変動(契約で定義されているように)を行うと、所持者は吾などにその全部または任意の部分2027年手形を現金で購入することを要求することができ、買い戻し価格は購入しようとした2027年手形の本金額の100%に等しく、また基本変動買い戻し日(ただし基本変動購入日を含まない)の任意の課税および未払い利息を加えることができる。この契約は、ある違約事件が発生及び持続した場合、受託者又は当時の未償還債券元金総額の25%以上の保有者が、すべての債券の全ての元金が応算特別利息(あればある)とともに即時満期及び対応することを宣言することができることを含む慣用条項及び契約を掲載している。2027年手形は私たちの一般的な無担保債務であり、支払権は私たちのすべての債務よりも優先され、2027年手形の支払権は2027年手形に明らかに従属する;私たちがこのように従属していないすべての債務の支払権と等しい;このような債務を保証する資産価値については、実際には私たちの任意の保証債務よりも低く、構造的には私たちの子会社のすべての債務および他の負債(貿易債権を含む)よりも低い。
144
私たちの今後の2027年の手形項目の最低支払いを含む他の詳細を理解するために、私たちの総合財務諸表の付記10を参照してください。
融資と保証協定
2021年11月に、吾らは(I)アメリカ銀行全国協会(行政代理として)及び担保代理(担保代理として)、(Ii)いくつかの貸手又は貸手、(Iii)BridgiBioを借款人及び(Iv)BridgiBioを保証人又は保証人とするいくつかの付属会社と融資協定を締結した。
融資協議の元の条項及び条件に基づいて、貸金者は、(I)第1回前借り4500百万ドル、又は第1回前借り、及び(Ii)第2回前借り300,000,000ドル、又は第2回前借り又は集団前借り定期融資を含む元金総額7500百万ドルに達する定期融資を吾等に提供することに同意する。融資協議下の第1回前払金は2021年11月17日に資金を獲得した。2022年12月31日まで、第2弾の前払いは援助に使用することができ、私たちの臨床試験データに関連するいくつかのマイルストーン事件が発生した後、私たちの選挙でこの資金を得ることができる。第2回前払いに関連する条項は、第1改正案及び第2改正案で修正され、以下さらに議論される。第1修正案は、第2期前金の総額を3.00億ドルから1.00億ドルに減少させることを含む。二番目の修正案は1.00億ドルの第2回前払いをキャンセルした。第2修正案の結果として,融資の元金総額は450.0ドルであった 強制的に返済する前に、まず100万ドルを支払う。
我々の融資プロトコルの義務の保証として、BridgeBioと保証人はそれぞれ融資者の利益のために担保エージェントにBridgeBioと保証人のほとんどの資産の持続的な保証権益(BridgiBioと保証人が所有またはその後に買収したすべての持分を含む)を付与したが、いくつかの慣例の例外に適合している。いくつかの投資と処置の限界値を超えると、BridgeBioのより多くの子会社は保証人として加入することを要求されるだろう。
定期ローン立て替え金のいずれの未返済元金も固定金利で利下げされ、年利は9.0%に相当し、うち3.0%は2025年1月1日まで実物または実物で支払うことができる。利息支払いは定期ローンの前金を提供した後四半期ごとに支払います。私たちは2025年1月2日から始まる定期ローンの前払金や定期ローンの償却日の未返済残高に元金を支払い、9四半期に分けて支払い、利息を加算することを要求されます。もし私たちが2025年1月1日までにAcoramidis臨床試験データに関連するいくつかのマイルストーン、またはAcoramidisマイルストーンを完成させた場合、定期ローン償却日は2026年1月2日に自動的に延長される。定期ローン前払い金のいずれの未返済額も2026年11月17日または満期日に満期になって支払います。
当行は定期借款立て替え金の未返済元本金額(すべてが部分的ではない)を随時前払いし,課税および未払い利息,および支払時間に応じて定められた未返済元金の1.0%から3.0%の割増前払い(2022年11月17日または前に前払いする場合は,通常の補充金額を別途加算することができる)。
貸手の選択では、私たちはまた、買い戻しまたは償還質担保、特定の特許権使用料取引の達成、他の資産または子会社の処分、許可および他の金銭化取引に関連するいくつかの前払い事件(すべてこのような事件を前払い事件と呼ぶ)が発生した場合に、事前支払いを強制することを要求され、これは、このような取引現金純収益の50%または75%であり、特にAcoramidisマイルストーンの完了状況に依存する可能性がある。
145
あるプリペイド事件の強制的な前金要求の規定の下で、融資プロトコルは、私たちの能力を制限すること、(I)追加の債務を生成すること、(Ii)配当金を支払うこと、または特定の分配を行うこと、(Iii)私たちの資産を処分すること、留置権を付与すること、私たちの資産を許可または担保すること、または(Iv)私たちの業務特性を根本的に変えることを含む慣用的な肯定的および限られた負の約束を含む。BridgeBioと保証人は、私たちの知的財産権を許可し、他の資産を処分し、貨幣化と特許権使用料取引を行うことができる幅広い能力を持っているが、いずれの場合も、上記の強制的な前払いを支払わなければならない。融資協定は、ある制限の場合、BridgeBioおよび保証人は、(X)BridgeBioの株式およびその任意の子会社の株式を買い戻すことができること、(Y)任意の合弁企業または同様の投資を行うこと、および(Z)他の投資および買収を行うことができることを規定する。上記の強制的事前返済要求の制約の下で、BridgeBioが所有する非融資協定当事者側のポートフォリオ会社は、ある例外的な場合を除いて、融資合意下のいかなる契約または制限の制約も受けない。
ローン協定にも慣用的な違約事件が含まれており、吾らが満期時にいかなる元金或いは利息を支払うことができなかったか、ある破産或いは債務返済不能事件が発生したか、或いはローン協定に違反した場合の契約を含む。約束違反事件が発生すると、他の事項を除いて、貸手は融資協議の下での私たちの義務を加速させることができる。
債務割引と発行コスト1,870万ドルを差し引いた後、最初の前払いから純収益4.313億ドルを獲得し、そのうち約110万ドルの債務発行コストはKKR Capital Markets LLCが提供する専門サービスである。KKR Capital Markets LLCはKKR Genetic Disorder L.P.の付属会社であり,KKR Genetic Disorder L.P.はBridgeBioの主要株主の一つである。
2022年5月に第1修正案に署名しました
2022年6月に、NAVERE-BMS許可協定項目の下の前払いを受け取り(付記11参照)、改訂された融資協定のいくつかの強制前払い条項をトリガした。したがって、私たちは貸主に2,050万ドルを支払い、そのうちの2,010万ドルはそれぞれ元金と脱退費用に使用した。
融資協議及び改訂された融資協定の条項に基づき、吾らは選択権を行使し、2022年、2022年及び2021年12月31日までの年度の課税利息を元金に転換し、金額はそれぞれ1,530万ドル及びゼロとなる。
2022年11月に第2修正案に署名しました
146
私たちの融資合意による将来の最低支払いを含む他の詳細を理解するために、私たちの総合財務諸表の付記10を参照してください。
キャッシュフロー
次の表は私たちが示した期間のキャッシュフローをまとめています
|
|
十二月三十一日までの年度 |
|
|
|
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
|
|
(単位:千) |
|
|||||||||
経営活動のための現金純額 |
|
$ |
(419,494 |
) |
|
$ |
(497,934 |
) |
|
$ |
78,440 |
|
投資活動提供の現金純額 |
|
|
453,147 |
|
|
|
(200,826 |
) |
|
|
653,973 |
|
融資活動が提供する現金純額 |
|
|
(13,134 |
) |
|
|
736,446 |
|
|
|
(749,580 |
) |
現金が純増し,現金が純増する |
|
$ |
20,519 |
|
|
$ |
37,686 |
|
|
$ |
(17,167 |
) |
|
|
|
|
|
|
|
|
|
|
|||
経営活動に使用されているキャッシュフロー純額
2022年の経営活動のための現金純額は4.195億ドルで、主に非現金プロジェクトで調整された4.847億ドルの純損失を含み、我々のPRVを売却して得られた1.10億ドルの収益(取引コストを含まない)、株式ベースの給与支出9160万ドル、定期融資元金が増加した実物支払利息1360万ドル、長期資産減額1270万ドルを含み、改訂されたQED-ヘルシン許可と協力協定によりヘルシンの売掛金が獲得した1250万ドルの収益、債務が860万ドル増加することが確認された。株式証券のある投資の純損失820万ドル、減価償却·償却損失680万ドル、Origin-Sentynl APA関連のある資産を売却する損失630万ドル、ライセンス契約により発行された株の非現金リース費用520万ドルと公平価値460万ドル、経営資産や負債の変化に関する現金純流入2810万ドル。運営資産や負債変動に関する2,810万ドルの現金純流入は,主にNAVERE−BMS許可プロトコルによる繰延収入の1,530万ドルの増加,許可および協力協定からの受取金の1,520万ドルの減少,その他の資産の1,100万ドルの減少であるが,専門サービスの500万ドルの減少,計算すべき研究開発負債の430万ドルの減少,その他の計上その他の長期負債の270万ドルの減少,主に支払時間による報酬や福祉の240万ドルの減少により部分的に相殺される。
2021年、経営活動に使用される現金純額は4.979億ドルで、主に非現金プロジェクトで調整された5.865億ドルの純損失を含み、株式報酬支出9,950万ドル、株式証券投資純収益2,990万ドル、減価償却および償却580万ドル、債務増加580万ドル、非現金リース支出560万ドルおよびレオコールオプション負債の廃止確認による収入560万ドル、および運営資産および負債変化に関する現金純流出440万ドルを含む。業務資産と負債の変化に関する440万ドルの現金純流出は、主に許可証と協力協定の受取金が1 970万ドル増加し、その他の資産が980万ドル増加し、経営リース負債が610万ドル減少したが、他の計算すべき負債とその他の長期負債が1200万ドル増加し、計算すべき研究開発負債が1120万ドル増加し、給与と福祉が740万ドル増加したため、部分的に相殺された。
投資活動が提供するキャッシュフロー純額
2022年に、投資活動が提供する現金純額は4.531億ドルで、主に4.797億ドルの有価証券満期日、1.10億ドルのPRV売却の毛収入、52.8百万ドルの株式証券投資および1,000万ドルのいくつかの資産売却による金を含むが、一部は1.375億ドルの有価証券および5560万ドルの株式証券投資を購入して相殺された。
147
2021年、投資活動のための現金純額は2.08億ドルで、主に5.899億ドルの有価証券の購入、5340万ドルの株式証券の購入、3,500万ドルの無形資産の買収、1,320万ドルの財産と設備の購入を含むが、この部分は有価証券の満期·販売の3.802億ドルと6,270万ドル、株式証券売却投資の3,420万ドル、PellePharmの合併により増加した現金と現金等価物1,370万ドルに相殺される。
融資活動が提供するキャッシュフロー
2022年の融資活動のための現金純額は1,310万ドルで、主に2,050万ドルの定期融資前払いを含み、一部はATMで普通株を発行した純収益490万ドルで相殺されている。
2021年の融資活動で提供される現金純額は7.364億ドルで、主に2029年の手形を発行した純収益7.314億ドル、私たちの定期融資項目の純借款4.563億ドルと株式オプション1660万ドルを行使したが、私たちの普通株の2.0億ドルの買い戻し、前払い改訂されたHercules定期融資とA部分融資の合計1.241億ドルと購入上限6,130万ドルの部分によって相殺された。私たちはまた8,510万ドルの現金でEidosの非持株権益を買い戻し、関連する直接取引コストを支払った。
重要な会計政策と試算
私たちの経営陣は私たちの財務状況と経営結果の討論と分析は私たちの総合財務諸表に基づいています。これらの報告書はアメリカ公認会計原則あるいはアメリカ公認会計原則に基づいて作成されています。これらの連結財務諸表を作成する際には、連結財務諸表日報告の資産、負債、または有資産および負債の開示、および報告期間内に発生する収入および費用に影響を与える推定および仮定を行う必要がある。我々の見積もりは,我々の歴史的経験と,当時の状況では合理的な様々な他の要因に基づいており,これらの要因の結果は資産や負債の帳簿価値を判断する基礎を構成しているが,これらの資産や負債の帳簿価値は他の源からは明らかではないように見える。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。
本報告で述べた期間の総合財務諸表の作成に用いられる判断と推定には,以下の会計政策が最も重要であると考えられる。
協力手配
私たちはパートナーと協力計画を達成し、これらの手配に基づいて、私たちの医薬化合物および/または製品のさらなる開発、製造、商業化の許可を与えることができる。私たちはまた、私たちの協力協定に基づいて研究、開発、製造、商業化、供給活動を行うことができる。これらの手配の考慮には、前金、開発と規制マイルストーン、費用精算、商業製品の純売上に基づく特許権使用料、商業販売マイルストーン支払いが含まれる可能性がある。
協力協定を締結する際には、これらのスケジュールが会計基準編纂(ASC)808の範囲に属するかどうかを評価する協力して計画し手配が共同経営活動に関連しているかどうか、及び双方が手配に積極的に参加し、重大なリスクとリターンに直面しているかどうかによって。このスケジュールがASC 808の範囲に属する場合、私たちと私たちのパートナーとの間の支払いが他の会計文書の範囲に属するかどうかを評価します。パートナーが私たちに支払ったお金は、ライセンス料、契約製造、研究開発活動など、顧客の対価格を表していると結論すれば、これらの支払いをASC 606の範囲に計上している取引先と契約した収入それは.しかし、私たちのパートナーが、いくつかの協力研究、開発、製造、および商業活動のようないくつかの活動および関連支払いの顧客ではないと結論した場合、私たちの基本費用に基づいて、このような支払いを研究開発費または販売、一般および管理費用の減少として記録します。また,これらの活動を我々の協力パートナーに精算すれば,このような精算を研究開発費や販売,一般,管理費として記録し,具体的には基礎費用の性質に依存する.
148
もし私たちの協力計画が商業化活動に利益と損失を共有する規定を提供した場合、利益共有構造における私たちのシェアの待遇は販売者が誰であるかにかかっている。私たちが販売側であり、依頼者とみなされている場合は、パートナーの利益シェアを販売、一般、行政費用以外の追加費用として記録し、パートナーの損失シェアを販売、一般、管理費用の減少として記録します。もし私たちのパートナーが販売側であり、依頼人とみなされた場合、私たちの利益シェアを協力収入として記録し、私たちの損失シェアを販売、一般、管理費用以外のシェアとして記録します。
収入確認
ASC 606に従って計算すべきと考えられる要素または取引については、(I)顧客との契約を決定するステップ、(Ii)契約中の履行義務を決定するステップ、(Iii)取引価格を決定するステップ、(Iv)契約に取引価格を割り当てる履行義務、および(V)契約義務を履行する際に収入を確認するステップの5つのステップを実行する。私たちは五段階モデルを契約に適用して、顧客に転送する商品やサービスと交換するために、私たちが獲得する権利のある価格を受け取るかもしれません。
手配開始時に、私たちは契約における履行義務を決定するために、約束された貨物またはサービスを評価する。そして、履行義務を履行する際(または一定期間内)に、それぞれの履行義務に割り当てられた取引価格の金額が収入であることを相対的に独立した販売価格で確認する。履行義務が時間の経過とともに履行されれば,入力法の使用により収入を確認する.これらの手配の会計計算の一部として、契約で決定された契約義務ごとの独立販売価格を決定するための判断が必要な仮説を立てた。これらの重要な仮定は、予測された収入或いはコスト、開発スケジュール、割引率、及び臨床と監督管理が成功する確率を含む可能性がある。
許可証料:私たちの知的財産権許可の付与を含む手配については、許可付与が手配に含まれる他の履行義務とは異なるかどうかを考慮します。一般に、顧客が利用可能なリソースを利用してライセンスから利益を得ることができれば、ライセンスは異なると結論することができる。異なるライセンスについては、ライセンス期限の開始時に許可に割り当てられた払戻不可能な前払い許可料と他の対価格の収入を確認し、基本知的財産権に関するすべての必要な情報を顧客に提供しており、これは、通常、手配開始時または手配に近いときに発生する。他の承諾とバンドルされたライセンスについては、合併の履行義務が時間の経過とともに満たされるか、ある時点で満たされるかを決定する。時間の経過とともに総合的な履行義務が履行された場合,前払い許可料からの収入を確認するために,判断を用いて進行を測定する適切な方法を決定する。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。
発展と規制マイルストーン支払い:開発と規制マイルストーン支払いを含む各手配の開始時に、マイルストーンが実現可能であると考えられているかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定します。私たちは通常、これらの記念碑的な支払いを実現する際にそれを含めています。私たちの合意によると、これらの支払いを触発する研究開発過程にはかなりの不確実性があるからです。同様に、製品が関連規制機関の承認を受けると、私たちは承認マイルストーン支払いを取引価格に含めるつもりだ。その後各報告期間が終了した時点で,我々はこのような発展や規制マイルストーンや任意の関連規制を達成する可能性を再評価し,必要に応じて全体の取引価格の見積もりを調整している。どのような調整も累積追跡に基づいて記録されている.
販売のマイルストーン支払いと特許使用料に基づいて: 販売ベースの特許使用料を含む手配については、販売量に基づくマイルストーン支払いを含めて、許可が特許使用料または販売ベースのマイルストーンに関連する主要項目とみなされているか否かを判断し、そうであれば、(I)関連販売が発生した場合、または(Ii)特許権使用料分配の履行義務の一部または全部が履行された(または部分的に履行されている)ときに収入を確認する。
149
製品供給サービス:許可された人によって、臨床開発または商業供給のための医薬品を将来的に提供する約束を適宜決定することを含む手配は、一般に代替方法とみなされる。私たちは、これらのオプションが被許可者に実質的な権利を提供しているかどうかを評価し、そうであれば、個別の履行義務として入金され、オプションに関連する将来の商品またはサービスが提供されるか、またはオプションが満了したときに確認する。
研究と開発サービス:研究開発サービスを含むスケジュールについては,時間の経過とともに収入を確認し,合意期間内に活動を行う際に移行する商品やサービスを代表する入力法を用いる.
計算すべき研究と開発負債
我々は,臨床前研究,臨床試験,契約製造活動を含む第三者サービスプロバイダによる研究と開発活動の見積りコストの計上項目を記録した。我々は,提供されているが領収書が発行されていない推定サービス金額に基づいて研究開発活動の推定コストを記録し,これらのコストを合併貸借対照表における計算すべき研究·開発負債,総合経営報告書における研究·開発費用に計上する.このような費用は私たちの研究開発費の重要な構成要素だ。
私たちが計算すべき推定研究と開発費用の例としては
我々は,我々を代表して臨床試験を行って管理している複数の研究機関とCROと締結した契約による受信サービスとかかる努力の見積もりに基づいて,臨床試験に関する費用課税を計算した。このような合意の財政的条項は契約によって異なり、支払いの不均衡を招く可能性がある。その中のいくつかの契約下の支払いは患者の成功登録と臨床試験マイルストーンの完成などのいくつかの要素に依存する。私たちのサービス提供者は通常毎月私たちが提供するサービスのために借金の領収書を発行する。サービス料を計算する際に、サービスを提供する時間帯と各時間の努力度を見積もります。もし私たちが発生し始めたコストを確定しなければ、あるいは提供されたサービスのレベルやこれらのサービスのコストを過小評価したり、過大評価したりすれば、私たちの実際の支出は私たちの推定とは異なるかもしれない。私たちはサービス提供者への前金を前払い資産として記録する。
第三者によって実行される契約製造活動の見積もりコストの計上項目を記録します。これらの合意の財務条項は協議が必要で、契約によって異なり、私たちのサプライヤーへの支払いの不均衡を招く可能性があります。契約下の支払いには、プリペイドやマイルストーン支払いが含まれており、これは製造過程のある段階の完了状況などに依存する。費用を確認するために,生産プロセスの定義が貨物の配送やサービス提供と考えられるのに十分であると考えられるかどうかを評価し,これらのプロセスや生産量が発展しており,あまり確定していない.もし私たちがこの過程が商品の納品だと思ったら、薬品交付時に費用を確認します。そうでなければ、損失のリスクを負うことになります。もし私たちがこの過程がサービスの交付だと思うなら、契約メーカーが契約の各段階の進捗を完了する最適な見積もりに基づいて費用を確認します。私たちの推定はその時得られる最高の情報に基づいている。しかし、私たちはもっと多くの情報を得るかもしれないし、これは私たちが未来にもっと正確な推定をすることを可能にするかもしれない。この場合、実際の活動レベルがより確実になった場合には、研究·開発費の調整を今後の時期に記録する必要があるかもしれない。コストの任意の増加または減少は、一般に推定された変化と考えられ、決定された期間の研究および開発費用に反映される。
150
これまで,我々の対応研究と開発負債の見積もりは報告期間後に大きな変化はなかった。しかし,推定の性質から,我々の臨床研究や他の研究活動の状態や進行に関するより多くの情報が分かった場合には,将来的に我々の推定を変えることは保証されない。
一里塚の補償手配に応じて計算する
私たちはいくつかの従業員やコンサルタントと業績に基づくマイルストーン報酬スケジュールを持っており、その帰属は様々な規制と発展マイルストーンを満たすことに依存し、最初に知られている固定通貨金額は、私たちが唯一選択した時に、(I)現金、(Ii)BridgeBioの株式、または(Iii)BridgeBioの現金または株式の形で、私たちが唯一の選挙時に現金またはBridgeBioの株式の形で支払うことができる。私たちの唯一の選挙でBridgeBioの現金または株式決済に関する手配については、可能な固定金額和解結果によって実現可能な場合には、マイルストーン補償スケジュールを責任分類報酬に分類する。株式決済を選択すれば、これらの手配は、それぞれまたはマイルストーン実現日の当時の株価に基づく可変数の株式決済にもつながる。
特定または開発マイルストーンが実現可能である場合には、各開発マイルストーンによって生成される補償費用の計算すべき項目を記録し、各報告期間においてそのような計算すべき項目を計測する。著者らは関連する臨床計画の進展と期待結果からこれらのマイルストーンを実現する可能性を推定した。私たちの推定はその時得られる最高の情報に基づいている。しかし、私たちはもっと多くの情報を得るかもしれないし、これは私たちが未来にもっと正確な推定をすることを可能にするかもしれない。この場合、私たちは未来の間にマイルストーンの報酬費用の調整を記録しなければならないかもしれない。このような費用の任意の増加または減少は、一般に、推定数の変化とみなされ、決定された期間に反映されるであろう。
これまで,報告期間終了後,計マイルストーン補償費用の見積もりに大きな変化はなかった。しかし,評価の性質上,われわれの臨床計画の進展や期待結果に関するより多くの情報が分かった場合には,将来的に我々の評価を変更することは保証されない。
最近の会計公告
より多くの情報は、我々の連結財務諸表第2部第8項の付記2、“重要会計政策概要--最近採択された会計公告”を参照されたい。
第七A項。定量と定性IVE市場リスクに関する開示
2022年12月31日と2021年12月31日現在、私たちはそれぞれ現金、現金等価物、有価証券、制限現金4.662億ドル、7.877億ドルを持っている。私たちの現金等価物には、通貨市場基金や短期商業手形のような通貨市場口座に投資される金額が含まれている。私たちの有価証券は高投資レベルの固定収益証券を含み、主に商業手形、社債、アメリカ政府証券に投資します。私たちは取引や投機目的に投資しないし、派生金融商品を使って私たちの金利リスクを管理していません。私たちは金利の変化によって大きなリスクに直面しているわけでもなく、金利の変化によって大きなリスクに直面するとも思わない。私たちは私たちの現金、現金等価物、または有価証券に重大な違約リスクや流動性不足があるとは思わない。
2022年12月31日まで、私たちは変動金利の未返済債務を持っていない。2022年12月31日現在、我々の2029年手形、2027年手形、定期融資の元金残高はそれぞれ7.475億ドル、5.5億ドル、4.452億ドルであり、固定金利を負担している。2021年12月31日まで、私たちは変動金利の未返済債務を持っていない。2021年12月31日現在、我々の2029年手形、2027年手形、定期融資の元金残高はそれぞれ7.475億ドル、5.5億ドル、4.5億ドルであり、固定金利を負担している。このような債務に対する私たちのキャッシュフローは金利変化の影響を受けない。金利が上記のいずれの期間に100ベーシスポイント変動しても、我々の財務諸表に実質的な影響を与えないと仮定する。
151
私たちの株式証券投資の公正な価値は変化するだろう。2022年12月31日現在、上場企業の株式証券を含む株式証券への投資残高は4370万ドルである。当該等株式は、報告期間最終取引日に保有株式の収市価に応じて公正価値で我々の総合貸借対照表に計上される。株式入札の変動は重大な損益を招く可能性がある。2021年12月31日現在、上場企業の株式証券を含む株式証券への投資残高は4910万ドルである。当該等株式は、報告期間最終取引日に保有株式の収市価に応じて公正価値で我々の総合貸借対照表に計上される。
インフレと変化する価格が私たちの業務、財務状況、または本報告で述べた任意の時期の経営結果に大きな影響を与えるとは思いません。未来のインフレと物価の重大な不利な変化は大きな損失を招くかもしれない。
152
プロジェクト8.財務統計員TSと補足データ
|
ページ |
独立公認会計士事務所レポート(PCAOB ID番号) |
154 |
2022年12月31日現在と2021年12月31日現在の連結貸借対照表 |
157 |
2022年12月31日終了の3年度ごとの連結業務報告書 |
158 |
2022年12月31日までの3年間の各年度の総合全面損失表 |
159 |
2022年12月31日までの3年度の償還可能非持株権益と株主権益(損失)連結報告書 |
160 |
20年12月31日までの3年間の連結現金フロー表22 |
161 |
連結財務諸表付記 |
163 |
153
独立地域登録所の報告書イギリス特許会計士事務所
BridgeBio Pharma,Inc.株主と取締役会へ
財務諸表のいくつかの見方
我々はBridgeBio Pharma,Inc.及びその子会社と制御実体(“当社”)を2022年12月31日と2021年12月31日までの連結貸借対照表、2022年12月31日までの3年度の関連総合経営表、全面赤字、償還可能転換可能非持株権益と株主権益(赤字)と現金流量、及び関連付記(総称して“財務諸表”と呼ぶ)を監査した。財務諸表は,すべての重要な面で,会社の2022年12月31日と2021年12月31日までの財務状況,および2022年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており,米国公認の会計原則に適合していると考えられる。
我々はまた、米国上場会社会計監督委員会(PCAOB)の基準に従って、会社が2022年12月31日までの財務報告の内部統制を監査し、根拠を監査した内部統制--統合フレームワーク(2013)テレデビル委員会は、組織委員会が発表した報告書と2023年2月23日の私たちの報告書を後援し、会社の財務報告の内部統制について保留のない意見を表明した。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、財務諸表を当期監査する際に生じる事項であり、監査委員会に伝達または要求され、(1)財務諸表に対して大きな意味を有する勘定または開示に関し、(2)特に挑戦的、主観的、または複雑な判断に関するものである。重要監査事項の伝達は、財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、次の重要な監査事項を伝達することによって、重要な監査事項又はそれに関連する勘定又は開示について個別の意見を提供することもない。
154
研究と開発費用と計上研究·開発負債−財務諸表付記2と9を参照−
重要な監査事項の説明
当社は、臨床前研究や臨床試験に関する研究開発サービスを提供するために、契約研究組織(CRO)と契約製造組織(CMO)と締結された第三者サービス協定を含む研究開発活動コストに関する研究開発費用を発生させ、これらの費用を報告期間毎に見積もる。2022年12月31日までの会社の累計研究開発費は4,000万ドルで、そのうち3,400万ドルはCROとCMOと関係がある。また,会社の前払い費用と他の流動資産総額は2200万ドル,他の資産は2000万ドルであり,その中にはそれぞれ700万ドルと1000万ドルの前払い額が含まれており,これらの金額はCROとCMOがサービスを提供する前に支払われている。2022年12月31日までの年間で,会社は3.99億ドルの研究開発費を発生させ,そのうち1.5億ドルはCROやCMOが提供するサービスに関係している。同社は,署名された契約の規定に基づいてこれまでに完了したサービスや活動の推定数に基づいて,これまでに領収書や支払いを発行した金額に対して,これらの費用を記録し,期末に課税負債や前払い費用残高を発生させる。
これらの第三者研究開発コストの計算費用を重要な監査事項として決定したのは、管理層が提供しているが領収書が発行されていないサービスのコスト、取引量が大きいこと、および異なる仕入先が獲得した監査証拠の性質が異なるからである。確認された費用額および対応する計算および前払い残高は、各スケジュールにおける固有の条項および条件に基づいて記録され、しばしば、仕入先によって提供される報告日までのサービス進捗状況に関する限られた情報に依存する。供給者から提供される情報の数や可変性を手配するためには、広範な監査作業が必要であり、監査管理層が総費用、計上、前払い残高の推定を行い、これらの手続の結果を評価する際には、監査員の高度な判断力が必要となる。
監査で重要な監査事項をどのように処理するか
他にも、私たちの研究·開発費用見積もり数および関連する計算および前払い残高に関する監査プログラムは、以下のように含まれる
— 主サービス協定、変更書、作業説明書、および修正案を含むが、これらに限定されないが、これまでに発生した推定費用の分析を満たすために、スケジュール、予算、および関連料率を含む合意のキー条項について合意した。
155
— プロトコルの完全性および受信した支払い、請求書および請求書が発行されていない請求書、およびこれまでに発生したコストを確認するために、CROまたはCMOに書面確認を直接送信し、状態報告を含む彼らから直接受信された通信をチェックし、これらの情報を会社の推定で使用された金額と比較する。
— 会社の分析に使用した他の第三者情報に同意し、会社の見積もり費用、計上、前払い残高を再計算する。
— 2021年12月31日までの推定課税残高と、2021年12月31日以降に受信した伝票とを回顧的に分析することにより、会社が計上すべき残高を推定する能力を評価する。
/s/
2023年2月23日
2018年以来、当社の監査役を務めてきました。
156
BRIDGEBIO製薬会社
強固な基礎スプレー銃のシーツ
(千単位で、株や1株当たりの金額は含まれていない)
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
有価証券 |
|
|
|
|
|
|
||
株式証券投資 |
|
|
|
|
|
|
||
許可と連携協定からの売掛金 |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
経営的リース使用権資産 |
|
|
|
|
|
|
||
無形資産、純額 |
|
|
|
|
|
|
||
その他の資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債、償還可能な転換可能な非持株権益、 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
報酬と福祉に計上すべきである |
|
|
|
|
|
|
||
計算すべき研究と開発負債 |
|
|
|
|
|
|
||
専門サービスに応じる |
|
|
|
|
|
|
||
賃貸負債を経営し、今期の部分 |
|
|
|
|
|
|
||
繰延収入,当期分 |
|
|
|
|
|
|
||
その他負債を計算すべき |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
2029年手形、純額 |
|
|
|
|
|
|
||
2027年手形、純額 |
|
|
|
|
|
|
||
定期ローン,純額 |
|
|
|
|
|
|
||
賃貸負債を経営し,当期分を差し引く |
|
|
|
|
|
|
||
その他長期負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
償還可能な転換可能非持株権益 |
|
|
( |
) |
|
|
|
|
株主赤字: |
|
|
|
|
|
|
||
非指定優先株式、$ |
|
|
|
|
|
|
||
普通株、$ |
|
|
|
|
|
|
||
在庫株は、コストで計算する |
|
|
( |
) |
|
|
( |
) |
追加実収資本 |
|
|
|
|
|
|
||
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
BridgeBio株主の総損失 |
|
|
( |
) |
|
|
( |
) |
非制御的権益 |
|
|
|
|
|
|
||
株主総損失額 |
|
|
( |
) |
|
|
( |
) |
総負債、償還可能な転換可能な非持株資本、 |
|
$ |
|
|
$ |
|
||
付記はこれらの連結財務諸表の構成要素である。
157
ブリッチEBIO製薬会社
連結業務報告書
(千単位で、株や1株当たりの金額は含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
収入: |
|
|
|
|
|
|
|
|
|
|||
ライセンスとサービス収入 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
製品販売 |
|
|
|
|
|
|
|
|
|
|||
総収入 |
|
|
|
|
|
|
|
|
|
|||
運営コストと支出: |
|
|
|
|
|
|
|
|
|
|||
ライセンス収入と製品販売のコスト |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
|
|
|
|
|
|
|
|||
販売、一般、行政 |
|
|
|
|
|
|
|
|
|
|||
再編成·減価·関連費用 |
|
|
|
|
|
|
|
|
|
|||
総運営コストと費用 |
|
|
|
|
|
|
|
|
|
|||
運営損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他の収入(費用)、純額: |
|
|
|
|
|
|
|
|
|
|||
利子収入 |
|
|
|
|
|
|
|
|
|
|||
利子支出 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
優先審査クーポン券の収益、純額 |
|
|
|
|
|
|
|
|
|
|||
その他の収入,純額 |
|
|
( |
) |
|
|
|
|
|
|
||
その他の収入を合計して純額 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
純損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
転換社債の償還は純損失を占めなければならない |
|
|
|
|
|
|
|
|
|
|||
普通株主は純損失を占めなければならない |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
普通株主は1株当たり純損失を占めるべきである |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
純損失の加重平均シェアを計算する |
|
|
|
|
|
|
|
|
|
|||
付記はこれらの連結財務諸表の構成要素である。
158
BRIDGEBIO製薬会社
合併状態全面損失分割払い
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
その他の全面的な損失: |
|
|
|
|
|
|
|
|
|
|||
証券売却可能な未実現損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
総合損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
転換株の総合損失を償還する |
|
|
|
|
|
|
|
|
|
|||
普通株主は総合損失を占めなければならない |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
付記はこれらの連結財務諸表の構成要素である。
159
BRIDGEBIO製薬会社
変換可能N連結レポートを償還可能持株権益と株主権益(損失)について
(千単位で、株や1株当たりの金額は含まれていない)
|
|
償還可能である |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
積算 |
|
|
|
|
|
合計する |
|
|
|
|
|
|
|
|||||||||||
|
|
オープンカー |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
その他の内容 |
|
|
他にも |
|
|
|
|
|
橋接生物 |
|
|
|
|
|
合計する |
|
|||||||||||
|
|
非制御性 |
|
|
|
普通株 |
|
|
在庫株 |
|
|
支払い済み |
|
|
全面的に |
|
|
積算 |
|
|
株主の |
|
|
非制御性 |
|
|
株主の |
|
|||||||||||||||||
|
|
利益. |
|
|
|
株 |
|
|
金額 |
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
収入(損) |
|
|
赤字.赤字 |
|
|
権益(赤字) |
|
|
利益. |
|
|
権益(赤字) |
|
|||||||||||
2019年12月31日現在の残高 |
|
$ |
|
|
|
|
|
|
$ |
|
|
|
— |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|
$ |
|
|||||||||
権益の下で株式を発行する |
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
2020年の株式発行によると |
|
|
— |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
|
|
— |
|
||||
株に基づく報酬 |
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
2027年債券の権益部分、純額 |
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
購入上限のあるコール |
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
普通株買い戻し |
|
|
— |
|
|
|
|
( |
) |
|
|
— |
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
ESPPにより普通株式を発行する |
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
源泉徴収税を満たすために普通株を買い戻す |
|
|
— |
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
証券売却可能な未実現損失 |
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
非制御的権益を発行する |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|||
非制御的権益から移行する |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|||
純損失 |
|
|
( |
) |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
2020年12月31日までの残高 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||||||||
ASU 2020−06に採用された累積効果 |
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
Eidosの非持株権を現金と株で買い戻し |
|
|
— |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
||
購入上限のあるコール |
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
合併時の非制御的権益の薬膜公正価値 |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
普通株買い戻し |
|
|
— |
|
|
|
|
( |
) |
|
|
— |
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
株式補償計画に基づいて株式を発行する |
|
|
— |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||
株に基づく報酬 |
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
ESPPにより普通株式を発行する |
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
源泉徴収税を満たすために普通株を買い戻す |
|
|
— |
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
証券売却可能な未実現損失 |
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
非制御的権益を発行する |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|||
非持株権から移転する |
|
|
( |
) |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
|
|
|
|
||
純損失 |
|
|
( |
) |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
2021年12月31日現在の残高 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
( |
) |
||||||
株式補償計画に基づいて株式を発行する |
|
|
— |
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||||
源泉徴収税を満たすために普通株を買い戻す |
|
|
— |
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
株に基づく報酬 |
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
|||
ESPPにより普通株式を発行する |
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
普通株を市場で発行することで純額 |
|
|
— |
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||||
証券売却可能な未実現損失 |
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
非制御的権益を発行する |
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
非制御的権益から移行する |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
|
|
|
( |
) |
||
純損失 |
|
|
( |
) |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
2022年12月31日現在の残高 |
|
$ |
( |
) |
|
|
|
|
|
$ |
|
|
|
|
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|||||
付記はこれらの連結財務諸表の構成要素である。
160
BRIDGEBIO製薬会社
合併状態キャッシュフロープロジェクト
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
経営活動 |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営活動で使用される現金純額の調整: |
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬 |
|
|
|
|
|
|
|
|
|
|||
減価償却および償却 |
|
|
|
|
|
|
|
|
|
|||
非現金レンタル費用 |
|
|
|
|
|
|
|
|
|
|||
株式証券投資純損失 |
|
|
|
|
|
( |
) |
|
|
|
||
取引コストは含まれていない優先審査証明書の収益 |
|
|
( |
) |
|
|
|
|
|
|
||
定期ローンの受取利息を支払う |
|
|
|
|
|
|
|
|
|
|||
ライセンスと連携プロトコルからの入金の収益を確認する |
|
|
( |
) |
|
|
|
|
|
|
||
ライセンス契約に基づいて発行された株式の公正価値 |
|
|
|
|
|
|
|
|
|
|||
債務の増加 |
|
|
|
|
|
|
|
|
|
|||
権証の公正価値調整 |
|
|
|
|
|
|
|
|
( |
) |
||
ある資産を売却した損失 |
|
|
|
|
|
|
|
|
|
|||
長期資産減価準備 |
|
|
|
|
|
|
|
|
|
|||
レオオプション費用(収入) |
|
|
|
|
|
( |
) |
|
|
|
||
債務損失を繰り上げ返済する |
|
|
|
|
|
|
|
|
|
|||
買収している研究開発資産は |
|
|
|
|
|
|
|
|
|
|||
その他の非現金調整 |
|
|
|
|
|
|
|
|
|
|||
経営性資産と負債変動状況: |
|
|
|
|
|
|
|
|
|
|||
許可と連携協定からの売掛金 |
|
|
|
|
|
( |
) |
|
|
|
||
関係者の売掛金 |
|
|
|
|
|
|
|
|
|
|||
前払い費用と他の流動資産 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
その他の資産 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
売掛金 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
報酬と福祉に計上すべきである |
|
|
( |
) |
|
|
|
|
|
|
||
計算すべき研究と開発負債 |
|
|
( |
) |
|
|
|
|
|
|
||
専門サービスに応じる |
|
|
( |
) |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
収入を繰り越す |
|
|
|
|
|
|
|
|
|
|||
その他の負債とその他の長期負債 |
|
|
( |
) |
|
|
|
|
|
|
||
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
投資活動 |
|
|
|
|
|
|
|
|
|
|||
有価証券を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券の満期日 |
|
|
|
|
|
|
|
|
|
|||
有価証券の販売 |
|
|
|
|
|
|
|
|
|
|||
株式証券投資を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
株式証券投資 |
|
|
|
|
|
|
|
|
|
|||
PellePharmの合併により増加した現金と現金等価物 |
|
|
|
|
|
|
|
|
|
|||
無形資産の支払い |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
優先審査クーポン券の収益は、取引コストは含まれておりません |
|
|
|
|
|
|
|
|
|
|||
ある資産を売却して得られる収益 |
|
|
|
|
|
|
|
|
|
|||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他の投資活動 |
|
|
|
|
|
|
|
|
( |
) |
||
投資活動提供の現金純額 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
融資活動 |
|
|
|
|
|
|
|
|
|
|||
2021年および2020年の発行2029年および2027年の債券発行による金 |
|
|
|
|
|
|
|
|
|
|||
2029年と2027年の債券発行に関する発行コストと割引 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
購入上限のあるコール |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
普通株買い戻し |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
非制御的権益所得金を発行する |
|
|
|
|
|
|
|
|
|
|||
直接取引コストを含むEidos非制御的権益の買い戻し |
|
|
|
|
|
( |
) |
|
|
|
||
定期融資収益、発行コストを差し引く |
|
|
|
|
|
|
|
|
|
|||
定期ローンを返済する |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
ESPPにより普通株式を発行して得た金 |
|
|
|
|
|
|
|
|
|
|||
源泉徴収税を満たすために普通株を買い戻す |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
株式オプションを行使した収益は,買い戻し後の純額を差し引く |
|
|
|
|
|
|
|
|
|
|||
Eidosは非制御的権益の収益、純額を市場で発行している |
|
|
|
|
|
|
|
|
|
|||
普通株を市場で発行することで得られた純額 |
|
|
|
|
|
|
|
|
|
|||
非制御的権益を買い戻す |
|
|
|
|
|
|
|
|
( |
) |
||
その他の融資活動 |
|
|
|
|
|
|
|
|
|
|||
融資活動が提供する現金純額 |
|
|
( |
) |
|
|
|
|
|
|
||
現金、現金等価物および制限現金純増加(マイナス) |
|
|
|
|
|
|
|
|
( |
) |
||
年初の現金、現金等価物、制限現金 |
|
|
|
|
|
|
|
|
|
|||
年末現金、現金等価物、制限現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
付記はこれらの連結財務諸表の構成要素である。
161
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
キャッシュフロー情報の補足開示: |
|
|
|
|
|
|
|
|
|
|||
利子を支払う現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
非現金投融資情報の追加開示: |
|
|
|
|
|
|
|
|
|
|||
定期融資元金を計上した前年度は実物で利子を支払う |
|
$ |
|
|
$ |
|
|
$ |
|
|||
売掛金と売掛金専門サービスに含まれる繰延合併取引コスト |
|
$ |
|
|
$ |
|
|
$ |
|
|||
レンタル改善費用は大家さんが払います |
|
$ |
|
|
$ |
|
|
$ |
|
|||
非持株権を譲渡する(付記6) |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|
以前他の資産に帰属していた財産と設備を確認する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
非制御的権益の非現金出資 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
未払い財産と設備 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
他の計上すべき負債および他の長期負債に記録されている確認された無形資産 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
未済債務発行コスト |
|
$ |
|
|
$ |
|
|
$ |
|
|||
Eidos非持株権益の非現金純額を買い戻す |
|
$ |
|
|
$ |
|
|
$ |
|
|||
買い戻しEIDOの直接取引コストは,従来前払い料金や他の流動資産に分類されていた追加実収資本に記録されている |
|
$ |
|
|
$ |
|
|
$ |
|
|||
現金、現金等価物、および限定現金の入金 |
|
|
|
|
|
|
|
|
|
|||
現金と現金等価物 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
制限現金 |
|
|
|
|
|
|
|
|
|
|||
限られた現金-他の資産に含まれています |
|
|
|
|
|
|
|
|
|
|||
年末現金、現金等価物、制限現金総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
付記はこれらの連結財務諸表の構成要素である。
162
BRIDGEBIO製薬会社
Solidについての説明財務諸表を列記した
BridgeBio Pharma,Inc.またはBridgeBioまたは同社は商業段階の生物製薬会社であり、明確な遺伝的駆動因子を有する遺伝病および癌を有する患者の治療のための変異薬を発見、創造、テスト、および提供することを目的としている。BridgeBioの開発プロジェクトの範囲は早期科学から高度臨床試験までである。BridgeBioは2015年に設立され、経験豊富な薬物発見者、開発者と革新者からなるチームは遺伝子医学の進展を応用してできるだけ早く患者を助けることに取り組んでいる。
設立以来、BridgeBioは完全子会社を設立するか、BridgeBioが多数決権権益を持つ部分が所有する子会社、すなわちBridgeBioが主要な受益者であるVIE、または私たち、私たち、または私たちの全体を含むいくつかの制御されたエンティティに投資してきた。BridgiBioの本社はカリフォルニア州のパロアルトにあります。
列報根拠と合併原則
連結財務諸表には、BridgeBio Pharma,Inc.,その完全子会社、および制御されたエンティティの勘定が含まれており、これらの勘定は基本的にドル建てである。すべての会社間の残高と取引はすでに合併中に販売されている。私たちが100%以下の経済リスクを持っているか、または直面している合併実体については、私たちは私たちの合併経営報告書に“償還可能な非制御権益と非制御資本の占めるべき純損失”を記録し、それぞれの非制御者がそのような実体の経済または所有権権益に保持している割合に相当する。
エンティティが制御対象とされているかどうかを決定する際には,VIEとVotting Interest EntityやVOEモデルを適用した.私たちは、VIEの主な受益者であるかどうかを評価し、VIE活動を指導する権限に基づいて、これらの活動がVIEの経済表現に最も大きな影響を与える権利と、損失の義務またはVIEからVIEに大きな影響を与える可能性のある利益を得る権利を負担する。VOEモードでは、VIE資格を満たしていないエンティティを統合評価する。VOEモードではBridgeBioがそれが大きいと判断すれば
総合財務諸表はアメリカ公認会計原則あるいはアメリカ公認会計原則に基づいて作成され、管理層は、それはすべての調整を反映していると考えており、その中には、私たちの財務状況、私たちの経営業績と全面的な損失、及び私たちが列挙した期間の現金流量の公正な陳述に必要な正常な経常的な調整しか含まれていない。2022年·2021年·2020年12月31日までの経営実績必ずしも2023年12月31日までの年度または今後の任意の他の年度または中期の予想結果を表すとは限らない。
再分類する
2020年12月31日までの年度の総合キャッシュフロー表を何らかの再分類した本年度の陳述に適合するように。前述したように,これらの変更は経営,投資,融資活動によるキャッシュフローに純影響を与えていない.
163
可変利益主体と議決権利益主体
BridgeBioは、VIEモードまたはVOEモードに従って、財務的利益を直接または間接的に制御するエンティティを統合する。
VIEとは、設計上、(I)エンティティが他の当事者の追加的な従属財務支援なしにその活動に資金を提供することを可能にするのに十分な持分が不足していること、または(Ii)株式投資家が投票権によってエンティティの運営について大きな決定を下す能力がないか、または予期された損失を吸収する義務がないか、またはエンティティの残りのリターンを得る権利がないことを意味する。
VIEの主な受益者はVIEの資産と負債を統合する必要がある。主な受益者は、(I)VIEの活動を指導し、VIEの経済表現に最も大きな影響を与える権利がある側、および(Ii)VIEの損失またはVIEの利益を介してVIEからVIEに大きな影響を与える可能性のある利益を得る権利がある側である。
BridgeBioがVIEの活動を指導する権利があるかどうかを評価するために、VIEの経済表現に最も大きな影響を与えるために、BridgeBioは、VIEを確立する上でのその役割およびその継続的な権利および責任を含むすべての事実および状況を考慮する。この評価には,VIEの経済表現に最も影響を与える活動を決定することと,どちらが(あれば)これらの活動を制御する権利があるかを決定することがある.一般に、VIE(取締役会管理および代表)に影響を与える最も重要な決定を行い、これらの意思決定者を一方的に罷免する権利を有する当事者は、VIEの活動を指導する権利があるとみなされる。
BridgeBioがVIEの損失を吸収する義務があるかどうか、あるいはVIEからVIEに大きな影響を与える可能性のある収益を得る権利があるかどうかを評価するために、BridgeBioはそのすべての経済的利益を考慮し、主に優先株および普通株への株式投資、および優先株に変換可能な手形を発行することを含み、これらはVIEの可変利益と考えられる。この評価は、BridgeBioが、これらの利益が全体的にVIEに潜在的な重要性があると考えられるかどうかを決定する際に判断を適用することを要求する。重要性を評価する際に考慮される要因は、その資本構造、利息従属関係、支払い優先順位、VIE資本構造における異なるカテゴリにわたる資本の相対シェア、およびその資本がBridgeBioによって所有される理由を含むVIEの設計である。
VIE成立時には,BridgeBioは主な受益者であるかどうか,VIEが事実や状況に応じて統合すべきかどうかを決定する.BridgeBioを主な受益者とする統合VIEはそれぞれ業務の定義に適合していることが確認された.BridgeBio統合後のVIEの資産と負債には大きな制限はない。BridgeBioは、その後、再議イベントに基づいてVIEを継続的に再評価し、各報告期間において、統合および開示結論を変更する必要があるかどうかを再評価する。
VOEモードでは、VIE資格を満たしていないエンティティを統合評価する。VOEモデルでは、BridgeBioがそれが直接または間接的に超過していると判断した場合
権益法とその他の持分投資
コントロールではなく、被投資者の経営と財務決定に重大な影響を与える能力がある場合、私たちは権益法を使って投資を計算する。一般的に投資家が所有する資産が
164
権益法を適用する際には、初期確認が子会社であることが連結を解除した結果でない限り、コストに応じて投資を計上し、この場合、投資は公正価値で入金される。その後、私たちは、それぞれの報告期間内の普通株式所有権における割合に基づいて、被投資先の純収益または損失と他の総合収益の割合に応じて、投資の帳簿価値を増加または減少させる。被投資者への支払い、例えば追加投資、融資及び被投資者を代表して発生した費用、及び被投資者の支払い、例えば配当、分配及び返済融資は、いずれも投資帳簿価値の調整に計上される。被投資先の純損失が帳簿金額をゼロに低下させた場合、被投資先に他の投資が権益法に計上されていない場合や、被投資先の担保債務がある場合、または被投資先にさらなる財務支援を提供することを約束した場合、追加の純損失を記録する可能性がある。
私たちは被投資対象に大きな影響がない場合、公正な価値で投資を会計処理します。いつでも決められる公正な価値が足りない場合、私たちはコストから減値を引いて可視価格変動(あればある)を加えたり引いたりして投資を計量します。私たちは被投資者の収益分配から発表された任意の配当金の収入を確認する。
2020年12月31日まで、私たちはPellePharmに対して持分方法と株式安全投資を行った。ペライパムの持分証券投資には確定しやすい公正な価値がなく、コストから減値を差し引いて観察可能な価格変化を加算または減算する。PellePharmは2021年4月に合併後のVIEとなった。2020年12月31日まで、私たちはLianBioに株式方法投資を行い、BridgeBio Pharma、LLCが保有する普通株を代表しています ("2021年11月、私たちはLianBioに大きな影響力を持たなくなったため、ASC 321によるLianBioへの投資の会計処理を開始した投資--株式証券それは.PellePharmとLianBioの投資をさらに検討するために付記7を参照してください。この2つの投資は2021年12月31日に権益法投資として入金されなくなりました。2022年12月31日と2021年12月31日まで、私たちはこれ以上権益法投資はありません。
権益会計方法により、著者らの投資は各報告期間に減値指標に基づいて審査を行い、そして証拠が非一時的な価値損失を表明した時に公正価値に減記する。減値を反映する可能性のある要素には、被投資者の一連の経営損失、投資を回収する能力がない帳簿価値、被投資者が収益力を維持できないこと、投資の現在の公正価値がその帳簿価値よりも低いことがある。価値低下が一時的でない可能性のある指標は、公正価値または時価が帳簿価値より低い時間の長さと程度、被投資者の財務状況と最近の見通しを推定することを含み、私たちは市場価値と一般市場状況が任意の予想の回復を達成できるように、十分な時間内に被投資者への投資の意図と能力を保留することを含む。公正価値の推定および非一時的減値が発生したかどうかは重大な判断を適用する必要があり、未来の結果は現在の仮定と異なる可能性がある。2021年12月31日および2020年12月31日までに,権益法投資に関する減価費用は確認されていない。
信用リスクその他のリスクと不確定要因が集中している
私たちが高度に集中的な信用リスクに直面させる金融商品は主に現金、現金等価物、および限定的な現金を含む。私たちのほとんどの現金、現金等価物、そして制限された現金はアメリカの金融機関に保管されている。預金金額は連邦保険の限度額を超える場合があります。経営陣は、金融機関の財務状況が良好であるため、金融機関の信用リスクが最も低いと考えている。
私たちはいくつかのリスクと不確定要素の影響を受け、以下のいずれの変化も、将来の財務状況や経営結果に重大な悪影響を及ぼす可能性があると考えられる:将来の融資を得る能力、規制部門の候補製品の承認と市場受け入れ程度、清算状況、私たちが依存する第三者契約研究機関やメーカーの表現、販売ルートの発展、私たちの知的財産権の保護、知的財産権、特許、製品、法規、臨床またはその他の要素による私たちの訴訟やクレーム、そして私たちの成長を支持する従業員の能力を引き付け、維持する。
165
私たちは第三者メーカーに依存して私たちの計画中の研究開発活動に製品を提供します。特に,少数のメーカーがこれらの計画に関連する活性医薬成分や処方薬の要求を提供してくれることに依存し,依存し続けることが予想される。これらのプロジェクトは活性薬物成分や製剤薬物供給の深刻な中断の悪影響を受ける可能性がある。
2020年3月、世界保健機関はSARS-CoV-2の爆発を発表し、これは1種の新しいコロナウイルス株であり、コロナウイルス病19、或いは新冠肺炎を招き、全世界で大流行した。以来,医療提供者や病院は抗ウイルスとその変種に大量の資源を集中させ,子会社が行っている臨床試験における患者募集の遅延や一時停止を経験した。そのほか、著者らはいくつかの進行中の肝心な計画活動の中で遅延に遭遇する可能性があり、計画中の臨床試験の開始、及び非臨床試験と研究性新薬申請--良好な実験室実践毒理学研究を可能にする。遅延の正確な時間と私たちの業務に対する全体的な影響はまだ不明であり、進行している新冠肺炎の大流行を監視している。ますます多くの人が新冠肺炎ワクチンを接種することに伴い、世界のある地区のある措置はすでにリラックスしたが、他の措置は依然として有効であり、いくつかの地区は引き続き新しい疫病の発生と感染率の急上昇を経験している。これらの措置を廃止したり、新しい措置を実施する程度は大流行の変化、および現有のワクチンの分配、接種速度とウイルス新変種の出現に依存する。私たちは状況を積極的に監視し続けており、連邦、州または地方当局の要求または私たちが最も公衆衛生と安全に最も適合していると考え、私たちの患者コミュニティ、従業員、パートナー、サプライヤー、株主の最適な利益に基づいて、さらなる予防と先制行動をとる可能性がある。このような行動や新冠肺炎が世界の業務運営や経済状況に与える影響が私たちの業務や戦略に与える影響は予測できません, 我々が行っており計画中の臨床開発活動や将来性への影響,あるいは我々の財務·運営結果への影響を含む。
予算の使用
公認会計原則に従って連結財務諸表を作成することは、経営陣に推定および仮定を要求し、これらの推定および仮定は、連結財務諸表の日付の資産、負債およびまたは負債の報告金額および報告期間内に報告された費用金額に影響を与える。添付の連結財務諸表に作成された重大な推定および仮定は、これらに限定されない
私たちの推定は歴史的経験と様々な他の合理的な仮定に基づいている。実際の結果は,これらの推定や仮定とは異なる可能性がある.
現金、現金等価物、有価証券
元の満期日に購入したすべての高流動性投資は
166
私たちの有価証券は高投資レベルの固定収益証券を含み、主に商業手形、社債、アメリカ政府証券に投資します。私たちは私たちの有価証券を売却可能な証券に分類し、現金等価物または有価証券の公正価値を総合貸借対照表に報告し、関連する未実現収益と損失を株主損失の構成要素として計上する。私たちは各ツールの基礎契約満期日に基づいて私たちの有価証券を短期または長期に分類します。債務証券の償却コストは割増の償却や満期割引の増加に応じて調整され、これらの割引額は総合経営報告書の利息収入に計上される。売却可能証券の実現損益と非一時的と判定された価値低下(あれば)は“その他の収入(費用),純額”に計上される。証券売却のコストは具体的な識別方法によって決定される。証券を売却可能な利息と配当金に分類されて利息収入に計上される。
私たちの現金、現金等価物、および有価証券は、そのような資産の第三者の違約を保有または発行する際に信用リスクに直面している。私たちの現金、現金等価物、有価証券は、経営陣が高い信用品質を持つと考えている金融機関が持っています。私たちの投資政策は、社債、会社商業手形、米国政府債券、通貨市場基金のようなドル建ておよび支払いされた固定収益証券に投資を制限し、タイプおよび発行者によって期限と集中度を制限する。
以下の表では、合併貸借対照表内に報告されている現金、現金等価物、および制限現金を照合し、これらの現金の合計は、統合現金フロー表に記載されている金額の総和である
|
|
十二月三十一日 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
現金と現金等価物 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
制限現金 |
|
|
|
|
|
|
|
|
|
|||
流動現金は含まれていません資産“ |
|
|
|
|
|
|
|
|
|
|||
現金総額、現金等価物、および限定現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
制限現金
制限された現金とは、主に、付記10に記載されている会社ローンと保証協定第2改正案に基づいて設立された制御された口座内の資金のことである。融資協定の基本改正条項によると、このような無利子現金の使用は制限されており、NAVRE-BMSライセンス契約の履行に関する義務の履行にのみ使用されることができるが、付記11はこれをさらに記載している。2022年12月31日現在本プロトコルに関連する制限された現金は$である
さらに、いくつかの賃貸契約と信用状によると、私たちは現金と現金等価物を担保として抵当しました。2022年12月31日までこのようなプロトコルに関連する制限された現金は#ドルである
167
株式証券投資
私たちは2021年から上場企業の株式証券に投資します。私たちはASC 321に基づいて各報告期間に株式証券投資の公正な価値を測定しました投資--株式証券それは.観察可能な価格変化による公正価値変化は、我々の総合経営報告書の“その他の収入(費用)、純額”に含まれている。株式証券を売却する際には、実現した収益や損失はいずれも私たちの総合経営報告書で確認されます。私たちは一般的に株式証券に対する私たちの投資を非流動資産に分類する。必要に応じてこれらの株を現金化し、現在の業務に資金を提供しようとする場合、株式証券への投資を流動資産に分類する。
公正価値計量
総合貸借対照表では,公正価値で常時記録されている資産と負債は,その公正価値を計測するための投入に関する判断レベルに基づいて分類される.公正価値は、計量日に市場参加者間の秩序ある取引において資産または負債の元本または最も有利な市場の負債を移動させるために、受信される交換価格または支払いされる退出価格として定義される。公正価値を計量するための推定技術は,観察可能な投入を最大限に利用し,観察不可能な投入を最大限に減少させなければならない。“公正価値計量に関する権威指針”は公正価値計量の開示のために三級公正価値等級を確立し、具体的には以下の通りである
第1レベル-観察可能な投入、例えば、計量日に同じ資産または負債のアクティブ市場における未調整オファー。
第2レベル-資産または負債の投入を直接または間接的に観察することができる(第1レベルに含まれる見積を除く)。これらのオファーは、アクティブ市場における同様の資産または負債の見積もりと、非アクティブ市場における同じまたは同様の資産または負債の見積もりとを含む
第三レベル-市場活動が少ないか、または市場活動支援の観察できない投入がなく、資産または負債の公正な価値に大きな意義を持っている。
ある程度、推定値は市場で観察または観察できないモデルや投入に基づいており、公正価値の決定にはより多くの判断が必要である。したがって、公正価値を決定する際の判断度が最も大きいのは、第3レベルに分類されたツールであり、公正価値レベルにおける金融商品のレベルは、公正価値計測に重要な意味を有する任意の投入の中で最低レベルに基づいている。
現金及び現金等価物、制限的現金、許可及び協力協定からの売掛金、前払い支出及びその他の流動資産、支払すべき帳簿及び売掛金の短期的性質により、付随する合併貸借対照表に反映される帳簿金額はその公正価値に近い。
財産と設備、純額
財産と設備は減価償却と償却後のコスト別に列記する。財産や設備の減価償却と償却は直線法を用いてそれぞれの資産の推定耐用年数を計算する。資産寿命の改善や延長のないメンテナンスやメンテナンスは、発生時に費用を計上する。資産を売却または廃棄する際には、コストおよび減価償却および償却は、総合貸借対照表から除外され、それによって生じる任意の収益または損失は、実現された期間の総合経営報告書に反映される。
私たちの財産と設備の推定使用年数は以下の通りです
|
|
|
家具と事務設備 |
|
|
実験室と機械装置 |
|
|
賃借権改善 |
|
財産と設備の減価償却と償却費用は#ドルです
168
賃貸借証書
私たちのレンタルグループには、会社本部、オフィススペース、実験室施設のレンタルが含まれています。私たちは契約の開始時に一つの手配がレンタルかどうかを確認します。私たちの経営リースの資産部分は“経営賃貸使用権資産”として記録され、負債部分は私たちの総合貸借対照表に“経営賃貸負債、当期部分”と“経営賃貸負債、当期部分を差し引く”と記録されている。融資リースの資産部分には“財産と設備純額”が計上され、流動および非流動融資リース負債はそれぞれ“その他の計上すべき負債”および“その他の長期負債”として総合貸借対照表に示されている。融資リース項目における資産の減価償却方式は、他の財産や設備と類似している。
使用権資産及びリース負債はリース開始日レンタル期間内のリース支払い現在値で確認します。レンタル支払いの現在値は、レンタル中の暗黙的な金利(この金利が容易に決定されやすい場合)に基づいて決定され、そうでなければ、レンタル開始日に取得可能な情報に基づく増分借入金金利を使用して、レンタル支払いの現在値を決定する。使用権資産は、予想されたレンタル奨励金額に応じて調整されます。賃貸開始日には,吾らは将来のイベントに基づくリースインセンティブ金額を任意に推定して我々の賃貸支払いに計上しており,条件は,(1)イベントが吾らの支配下にあること,および(2)インセンティブを得る権利をトリガするイベントが合理的に確実に発生することである。受け取ったリース報酬がレンタル開始時に確認された金額よりも大きい場合、差額が使用権資産および/またはレンタル負債の調整であることを確認します(場合によっては)。
使用権資産およびリース負債は、レンタルが若干改正された場合に、残りの賃貸支払いの現在値およびリース改訂時の推定増額額で借入金金利を再計測する。経営リースコストはレンタル期間内に直線法で確認され、短期賃貸に関する金額が含まれています。融資リースについては、賃貸期間や使用権資産の使用年数の短い時間で使用権資産を償却するほか、賃貸負債の利息支出も記録し、通常は直線償却である。私たちは可変賃貸支払いが当該等支払いの債務が発生している間の運営費用であることを確認した。可変レンタル支払いには主に公共エリアのメンテナンス、光熱費、不動産税、保険、その他の運営コストが含まれています。これらのコストはレンタル者が私たちがレンタルした空間に比例して転嫁されています。
資産買い入れ
我々は、買収資産のコスト(取引コストを含む)に基づいて、企業合併とはみなされない資産買収を計量·確認する。営業権は資産買収で確認されていません。資産買収では,買収が行われている研究·開発やIPR&Dに割り当てられたコストは買収日に研究·開発費用に計上されるが,将来的には他の用途はない。
長期資産減価準備
長寿資産は毎年あるいはイベントや環境変化がある資産の帳簿価値が回収できない可能性があることを示すたびに,減値を検討する。回収能力は,資産グループの帳簿価値と資産予想による将来の未割引キャッシュフローを比較することで測定した。ある資産グループの帳票金額がその推定された将来のキャッシュフローを超えていれば、その資産グループの帳票金額がその資産グループの公正価値を超えた金額の中で減価費用を確認する。2022年12月31日までに年度確認されたいくつかの長期資産の減値については、付記14を参照されたい。
細分化市場
我々は単一の運営と報告可能な部門であり,治療患者の変革性薬物を識別·推進する業務に従事している。私たちの業務は
169
主に許可と協力計画からの総収入は、パートナー本部がある地域に起因する。
2022年12月31日と2021年12月31日までアメリカとカナダの資本財産と設備の約
上限のコール取引
2021年1月と2020年3月に、BridgeBioはそれぞれ2029年債券と2027年債券(付記10参照)の発行についていくつかの上限催促取引、あるいは上限催促取引を締結した。一般に、上限のあるコールオプション取引は、任意のチケット変換時のBridgeBio普通株式所有者の潜在的な希薄化を減少させ、および/またはBridgeBioを相殺するには、変換されたチケットの本金額を超える任意の現金支払いを支払う必要があり、これらの減少および/または相殺は、上限価格に基づく上限に制限されなければならない(付記10参照)。カプセル化された呼は、ASC 815−40で概説された条件を満たす派生ツールおよびヘッジ株主資本の中で追加実収資本の減少に分類され、持分分類の条件が引き続き満たされている限り、その後再計量されることはない。
起債コスト
債務発行コストは実際の利子法に従って関連債務の推定年限内に利息支出に償却する。ASC 835によると利子我々は、関連債務から直接差し引かれるように、総合貸借対照表に債務発行コストを示している。
在庫株
買い戻しの在庫株はコストで入金され、任意の手数料と手数料が含まれています。
協力協定
私たちはパートナーと協力計画を達成し、これらの手配に基づいて、私たちの医薬化合物および/または候補製品のさらなる開発、製造、商業化のライセンスを付与することができる。私たちはまた、私たちの協力協定に基づいて研究、開発、製造、商業化、供給活動を行うことができる。これらの手配の考慮には、前金、開発と規制マイルストーン、費用精算、商業製品の純売上に基づく特許権使用料、商業販売マイルストーン支払いが含まれる可能性がある。
170
協力協定を締結する際には、これらのスケジュールがASC 808の範囲に属するかどうかを評価します協力して計画し手配が共同経営活動に関連しているかどうか、及び双方が手配に積極的に参加し、重大なリスクとリターンに直面しているかどうかによって。このスケジュールがASC 808の範囲に属する場合、私たちと私たちのパートナーとの間の支払いが他の会計文書の範囲に属するかどうかを評価します。パートナーが私たちに支払ったお金は、許可料、契約製造、および研究開発活動のような顧客の対価格を表していると結論した場合、これらの支払いをASC 606の範囲で処理します。しかし、私たちのパートナーが、いくつかの協力研究、開発、製造、および商業活動のようないくつかの活動および関連支払いの顧客ではないと結論した場合、私たちの基本費用に基づいて、このような支払いを研究開発費または販売、一般および管理費用の減少として記録します。また,これらの活動を我々の協力パートナーに精算すれば,このような精算を研究開発費や販売,一般,管理費として記録し,具体的には基礎費用の性質に依存する.
もし私たちの協力計画が商業化活動に利益と損失を共有する規定を提供した場合、利益共有構造における私たちのシェアの待遇は販売者が誰であるかにかかっている。私たちが販売側であり、依頼者とみなされている場合は、パートナーの利益シェアを販売、一般、行政費用以外の追加費用として記録し、パートナーの損失シェアを販売、一般、管理費用の減少として記録します。もし私たちのパートナーが販売側であり、依頼人とみなされた場合、私たちの利益シェアを協力収入として記録し、私たちの損失シェアを販売、一般、管理費用以外のシェアとして記録します。
収入確認
ASC 606に従って計算すべきと考えられる要素または取引については、(I)顧客との契約を決定するステップ、(Ii)契約中の履行義務を決定するステップ、(Iii)取引価格を決定するステップ、(Iv)契約に取引価格を割り当てる履行義務、および(V)契約義務を履行する際に収入を確認するステップの5つのステップを実行する。私たちは五段階モデルを契約に適用して、顧客に転送する商品やサービスと交換するために、私たちが獲得する権利のある価格を受け取るかもしれません。
手配開始時に、私たちは契約における履行義務を決定するために、約束された貨物またはサービスを評価する。そして、履行義務を履行する際(または一定期間内)に、それぞれの履行義務に割り当てられた取引価格の金額が収入であることを相対的に独立した販売価格で確認する。履行義務が時間の経過とともに履行されれば,入力法の使用により収入を確認する.これらの手配の会計計算の一部として、契約で決定された契約義務ごとの独立販売価格を決定するための判断が必要な仮説を立てた。これらの重要な仮定は、予測された収入或いはコスト、開発スケジュール、割引率、及び臨床と監督管理が成功する確率を含む可能性がある。
許可証料:私たちの知的財産権許可の付与を含む手配については、許可付与が手配に含まれる他の履行義務とは異なるかどうかを考慮します。一般に、顧客が利用可能なリソースを利用してライセンスから利益を得ることができれば、ライセンスは異なると結論することができる。異なるライセンスについては、ライセンス期限の開始時に許可に割り当てられた払戻不可能な前払い許可料と他の対価格の収入を確認し、基本知的財産権に関するすべての必要な情報を顧客に提供しており、これは、通常、手配開始時または手配に近いときに発生する。他の承諾とバンドルされたライセンスについては、合併の履行義務が時間の経過とともに満たされるか、ある時点で満たされるかを決定する。時間の経過とともに総合的な履行義務が履行された場合,前払い許可料からの収入を確認するために,判断を用いて進行を測定する適切な方法を決定する。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。
171
発展と規制マイルストーン支払い:開発と規制マイルストーン支払いを含む各手配の開始時に、マイルストーンが実現可能であると考えられているかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定します。私たちは通常、これらの記念碑的な支払いを実現する際にそれを含めています。私たちの合意によると、これらの支払いを触発する研究開発過程にはかなりの不確実性があるからです。同様に、製品が関連規制機関の承認を受けると、私たちは承認マイルストーン支払いを取引価格に含めるつもりだ。その後各報告期間が終了した時点で,我々はこのような発展や規制マイルストーンや任意の関連規制を達成する可能性を再評価し,必要に応じて全体の取引価格の見積もりを調整している。どのような調整も累積追跡に基づいて記録されている.
販売のマイルストーン支払いと特許使用料に基づいて: 販売ベースの特許使用料を含む手配については、販売量に基づくマイルストーン支払いを含めて、許可が特許使用料または販売ベースのマイルストーンに関連する主要項目とみなされているか否かを判断し、そうであれば、(I)関連販売が発生した場合、または(Ii)特許権使用料分配の履行義務の一部または全部が履行された(または部分的に履行されている)ときに収入を確認する。
製品供給サービス:許可された人によって、臨床開発または商業供給のための医薬品を将来的に提供する約束を適宜決定することを含む手配は、一般に代替方法とみなされる。私たちは、これらのオプションが被許可者に実質的な権利を提供しているかどうかを評価し、そうであれば、個別の履行義務として入金され、オプションに関連する将来の商品またはサービスが提供されるか、またはオプションが満了したときに確認する。
研究と開発サービス:研究開発サービスを含むスケジュールについては,時間の経過とともに収入を確認し,合意期間内に活動を行う際に移行する商品やサービスを代表する入力法を用いる.
許可および協力協定からの売掛金は、請求書を発行していない売掛金と、許可会社の技術により第三者が支払うべき特許権使用料とを含む、我々のパートナー、顧客、生物製薬会社に対する有効なクレームである。未開票売掛金には,我々のバイオ製薬顧客の開発サービスに関する売掛金および移行に関する売掛金が含まれており,これらの売掛金は協力プロジェクトのコストが発生した場合には契約発行権を実現する前に確認される。2022年12月31日まで2021年には未開請求書の売掛金$があります
同社は、履歴入金傾向、支払いパートナー、顧客とバイオ製薬会社の財務状況及び外部市場要因に基づいて、許可及び協力協定から得られた売掛金の回収可能性を評価し、管理層による可能な信用損失金額の最適な推定に基づいて、潜在的な信用損失に備える。同社は2022年12月31日と2021年12月31日まで
研究と開発費
研究·開発コストは発生時に費用を計上する。研究開発費は賃金、福祉及びその他の人事関連コストを含み、株式給与支出、実験室用品、臨床前研究、臨床試験及び関連する臨床製造コスト、生産製剤に関連するコスト、他の実体に当社を代表していくつかの研究開発活動を行う費用、及び分配された施設及びその他の関連コストを含む。将来の研究および開発活動のために使用される商品またはサービスの払戻不可能な前払いは延期され、関連商品またはサービスが交付または提供されるまで前払い費用として資本化される。
172
計算すべき研究と開発負債
我々は,臨床前研究,臨床試験,契約製造活動を含む第三者サービスプロバイダによる研究と開発活動の見積りコストの計上項目を記録した。我々は,提供されているが領収書が発行されていない推定サービス金額に基づいて研究開発活動の推定コストを記録し,これらのコストを合併貸借対照表における計算すべき研究·開発負債,総合経営報告書における研究·開発費用に計上する.このような費用は私たちの研究開発費の重要な構成要素だ。
私たちが計算すべき推定研究と開発費用の例としては
我々は,我々を代表して臨床試験を行って管理している複数の研究機関とCROと締結した契約による受信サービスとかかる努力の見積もりに基づいて,臨床試験に関する費用課税を計算した。このような合意の財政的条項は契約によって異なり、支払いの不均衡を招く可能性がある。その中のいくつかの契約下の支払いは患者の成功登録と臨床試験マイルストーンの完成などのいくつかの要素に依存する。私たちのサービス提供者は通常毎月私たちが提供するサービスのために借金の領収書を発行する。サービス料を計算する際に、サービスを提供する時間帯と各時間の努力度を見積もります。もし私たちが発生し始めたコストを確定しなければ、あるいは提供されたサービスのレベルやこれらのサービスのコストを過小評価したり、過大評価したりすれば、私たちの実際の支出は私たちの推定とは異なるかもしれない。私たちはサービス提供者への前金を前払い資産として記録する。
第三者によって実行される契約製造活動の見積もりコストの計上項目を記録します。これらの合意の財務条項は協議が必要で、契約によって異なり、私たちのサプライヤーへの支払いの不均衡を招く可能性があります。契約下の支払いには、プリペイドやマイルストーン支払いが含まれており、これは製造過程のある段階の完了状況などに依存する。費用を確認するために,生産プロセスの定義が貨物の配送やサービス提供と考えられるのに十分であると考えられるかどうかを評価し,これらのプロセスや生産量が発展しており,あまり確定していない.もし私たちがこの過程が商品の納品だと思ったら、薬品交付時に費用を確認します。そうでなければ、損失のリスクを負うことになります。もし私たちがこの過程がサービスの交付だと思うなら、契約メーカーが契約の各段階の進捗を完了する最適な見積もりに基づいて費用を確認します。私たちの推定はその時得られる最高の情報に基づいている。しかし、私たちはもっと多くの情報を得るかもしれないし、これは私たちが未来にもっと正確な推定をすることを可能にするかもしれない。この場合、実際の活動レベルがより確実になった場合には、研究·開発費の調整を今後の時期に記録する必要があるかもしれない。コストの任意の増加または減少は、一般に推定された変化と考えられ、決定された期間の研究および開発費用に反映される。
資産買収、許可内その他の合意に基づいてマイルストーンと特許使用料を支払う
私たちの資産買収、内部許可、その他の合意によると、いくつかの実質的なマイルストーンに達したら、開発、規制、販売に基づくマイルストーン支払いを要求される可能性があります。私たちは一般的に開発マイルストーンの費用を発生した費用に計上する。資産承認に関連する規制または販売ベースのマイルストーンについては、資産購入に関連するマイルストーン支払い資本を有限寿命無形資産に資本化し、マイルストーン支払いが予想されるキャッシュフローに基づいて回収され、資産に代替的な将来の用途がある場合を前提としている。このような無形資産は直線的にその推定耐用年数内に償却され、資産を取得した日から計算され、この日は通常監督管理機関が承認した日である。事件や環境変化が帳簿価値を完全に回収できない可能性があることを示した場合、有限年限無形資産の帳簿価値を評価して減値する。有限年限無形資産の回収可能能力は、資産の帳簿価値と資産予想による将来の未割引キャッシュフローを比較することで測定される。
ライセンス内契約や資産買収の実純売上高に応じて特許権使用料を支払うことも要求される可能性があります。この特許使用料は製品の販売期間内に支出されます。
173
非金融資産を売却する
通常、ASC 610-20に規定されている通常の活動範囲を超えた非金融資産の販売を計算しますその他の収入−非金融資産の損益確認の取り消しそれは.ASC 610−20によれば、我々は、ASC 606における指示を適用して、契約が存在するか否かを決定し、異なる非金融資産を識別し、制御権がいつ移行するかを決定し、それにより、非金融資産の識別がいつキャンセルされるかを決定する。また、吾らは、ASC 606の計量原則を適用して、非金融資産を売却する損益を計算する際に計上すべき対価金額(あれば)を特定する。
再編成·減価·関連費用
長期資産は毎年またはイベントや状況変化(再編や脱退活動を含む)がある資産の帳簿価値が回収できない可能性があることを示すたびに、長期資産を減値審査する。回収能力は,資産グループの帳簿価値と資産予想による将来の未割引キャッシュフローを比較することで測定した。ある資産グループの帳票金額がその推定された将来のキャッシュフローを超えていれば、その資産グループの帳票金額がその資産グループの公正価値を超えた金額の中で減価費用を確認する。
将来の福祉のない契約に関連する費用または契約終了費用は、契約終了日または使用停止日の早い日に確認される。従業員解散費は一般的に支払う可能性があり、金額が合理的に見積もることができる場合に確認します。他の清算と脱退関連費用は発生したことが確認された。
株に基づく報酬
株式ベースの報酬スケジュールには、株式オプション付与、制限株式報酬またはRSA、制限株式単位または我々の株式インセンティブ計画下のRSU報酬、および我々の従業員株式購入計画またはESPPによって発行された株が含まれており、従業員はこれらの株を介して市場価格よりも低い価格で私たちの普通株を購入することができる。
私たちはブラック·スコアーズオプション定価モデルを使用して、私たちの株式インセンティブ計画によって付与されたオプションの公正価値と、私たちのESPPによって付与された株を買収する権利を推定する。ブラック·スコアーズオプション評価モデルは、期待される付与期限と予想される株価変動を含む仮説を使用する必要がある。私たちは期待オプション期間を推定するために“簡略化”の方法を使用する。
株式ベースの報酬は、付与日に報酬の公正価値に基づいて、従業員および非従業員のすべての株式ベースの報酬を計量する。ESPPにより調達された補償費用は見積日の奨励公正価値に基づいて確認されます。株式による補償は、必要なサービス期間内の直線に基づく費用として確認され、これは通常授権期間である。
業績マイルストーンに達する可能性があると判断されると、業績条件を含む株式奨励の推定公正価値は、奨励期間内に加速方法を用いて支出される。業績条件を含む株式分類奨励の報酬費用は、奨励の付与日公正価値に基づいて計量される。業績条件を含む責任分類奨励の報酬支出は、最初に奨励の付与日に基づいて公正価値を計量し、決算日まで報告日ごとに公正価値で再計量する。補償支出は、管理層が付与された株式に対して帰属可能であるか否かの最適な推定に基づいて、必要なサービス期間内に入金する。私たちは業績のマイルストーンに達する確率を評価し続けるつもりだ。
没収が発生した同時期に株式ベースの補償を減らすことで、実際の没収を確認することを選択しました。
株式ベースの報酬は、通常、研究開発費および販売、一般、行政費用に記録されており、これは従業員および非従業員の機能を適用することによって決定される。
174
一里塚の補償手配に応じて計算する
私たちは、いくつかの従業員やコンサルタントとパフォーマンスに基づくマイルストーン報酬スケジュールを持っています。その帰属は、様々な規制と発展マイルストーンに依存しており、最初に知られている固定通貨金額は、私たちが唯一の選挙で解決することができます。私たちがそれぞれまたはマイルストーンを達成したとき、私たちはそれぞれまたはマイルストーンがあるときに、私たちが唯一の選挙で解決することができます。私たちの唯一の選挙でBridgeBioの現金または株式決済に関する手配については、可能な固定金額和解結果によって実現可能な場合には、マイルストーン補償スケジュールを責任分類報酬に分類する。株式決済を選択すれば、これらの手配は、それぞれまたはマイルストーン実現日の当時の株価に基づく可変数の株式決済にもつながる。
特定または開発マイルストーンが実現可能である場合には、各開発マイルストーンによって生成される補償費用の計算すべき項目を記録し、各報告期間においてそのような計算すべき項目を計測する。著者らは関連する臨床計画の進展と期待結果からこれらのマイルストーンを実現する可能性を推定した。私たちの推定はその時得られる最高の情報に基づいている。しかし、私たちはもっと多くの情報を得るかもしれないし、これは私たちが未来にもっと正確な推定をすることを可能にするかもしれない。この場合、私たちは未来の間にマイルストーンの報酬費用の調整を記録しなければならないかもしれない。このような費用の任意の増加または減少は、一般に、推定数の変化とみなされ、決定された期間に反映されるであろう。
所得税
所得税は貸借対照法で計算される。繰延税項資産及び負債は、既存資産及び負債の帳簿金額及びそのそれぞれの税ベースと営業損失純額及び税額相殺繰越との差額による将来の税項影響を確認することができる。繰延税項資産及び負債は、総合財務諸表帳簿額面と資産及び負債の課税基準との差額に基づいて決定され、予想差額に予想される年度に適用される課税収入の制定税率を用いて計量される。繰延税金資産の一部または全部が現金化できない可能性が高い場合、繰延税金資産減価準備。
アメリカ連邦所得税の場合、私たちは合併条例の規定の要求に符合する合併実体に合併のアメリカ連邦所得税申告書を提出する必要がある。合併申告に組み込まれたハードルを満たしていない実体は、単独の米国連邦所得税申告書を提出し続ける。私たちは、連結財務諸表にそれらの財務業績を統合しても、各エンティティの独立した評価免税額を個別に評価することを要求された。私たちはほとんどの管轄区域で合併された州納税申告書を提出し続けた。したがって、私たちはこのような管轄区域の推定免税額の国家部分を総合的に基づいて評価し続けている。当社も複数の海外司法管轄区で経営しており、各海外経営実体の独立評価免除額をそれぞれ評価している。
私たちは定期的に私たちの繰延税項資産を評価して、事実や状況の変化による、例えば未来の税前収益、税法、税務機関との相互作用及び判例法の発展を期待し、推定値準備の調整が適切であるかどうかを決定する。この評価を行う時、私たちは私たちの最近の税引前収益の歴史に依存する。私たちの重大な仮定は、私たちの未来の税引前収益の予測と、繰延税金資産と負債に代表される将来の控除と収入の性質と時間であり、これらはすべて重大な判断の行使に関するものだ。私たちは私たちの推定が合理的だと信じているが、私たちは繰延税金資産記録に対する適切な推定支出金額を決定するために重要な判断を使用しなければならない。
私たちは最大額の不確定所得税の頭寸を確認し、関連税務機関の監査を経て、当該頭寸は更に維持される可能性がある。確認や計測の変化は判決が発生した期間に反映される.私たちの政策は所得税の少納に関連する利息と罰金を所得税支給の構成要素として確認することだ。これまで、未確認の税収割引に関する利息や罰金は記録されていない。
175
レオオプション責任
このオプションを行使すれば、私たちは私たちのPellePharm株をあらかじめ決められた価格でLeoに売却する義務があるので、私たちはLeoのコールオプションを流動負債に計上した。レオ引受オプション負債は、レオ協定に署名したときに公正価値で入金されます。レオ引受オプション負債は、レオ引受オプションが行使され、終了するか、または権益分類資格に適合しないために満期になるまで、各貸借対照表の日ごとに公正価値で再計量されなければならない。連結業務報告書では、レオコールオプション負債の公正価値のどの変化も“他の収入(費用)、純額”の構成要素として確認されている。レオのコールオプションは2021年にレオに中止されたため、2022年12月31日現在、未返済のオプションはなくなっている。さらなる議論のために注釈3および7を参照されたい。
BridgeBio普通株主1株当たり純損失
BridgeBio普通株株主が1株当たりの基本純損失を占めるべき計算方法は:BridgeBio普通株株主が純損失を占めるべきであり、この期間のBridgeBio普通株の加重平均流通株数で割って、潜在的な普通株希釈株、例えば株式オプション、付与されていない制限的な株式単位と奨励、業績に基づくマイルストーン補償奨励、従業員の株式購入計画に基づいて発行可能な株及び2029年と2027年手形の仮定転換を考慮しない。報告期末の業績条件が満たされた場合にのみ、業績一里塚報酬スケジュールの普通株式等価物が潜在的希薄化株式に計上される。買い戻しすべき普通株式は加重平均株式以外に含まれていない。我々が報告したすべての時期は赤字状態であるため,BridgeBio普通株株主の1株当たりの基本純損失はBridgeBio普通株株主の希釈後の1株当たり純損失と同様であり,潜在的希薄化証券の影響が逆希釈されているためである。
最近採用された会計公告
最近採択または発表された会計声明は、米国証券取引委員会に提出された2021年12月31日現在の10-K表年次報告書の“財務諸表および補足データ”と題する一部開示された財務諸表および補足データと比較して、大きな変化はない私たちは最近発表されたすべての会計声明を検討し、これらのまだ発効していない基準が私たちの財務諸表に実質的な影響を与えないか、または私たちの業務に適用されないと判断した。
以下の表は、公正価値によって日常的に計量された金融資産と負債の情報を示し、評価の公正価値レベルを示している
|
|
2022年12月31日 |
|
|||||||||||||
|
|
合計する |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
||||
|
|
(単位:千) |
|
|||||||||||||
資産 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
商業手形 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物合計 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
商業手形 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
有価証券総額 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
株式証券投資 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
LianBio保証書 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
金融資産総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
負債.負債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
埋め込み導関数 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
176
|
|
2021年12月31日 |
|
|||||||||||||
|
|
合計する |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
||||
|
|
(単位:千) |
|
|||||||||||||
資産 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
商業手形 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物合計 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国庫券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
商業手形 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
会社債務証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
超国家債務証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
有価証券総額 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
株式証券投資 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
LianBio保証書 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
金融資産総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
負債.負債 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
埋め込み導関数 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
いくつありますか
観察不可能な投入とこれらの観察不可能な投入との相互関係により,第3レベルに分類されたツールの公正価値計測には不確実性があり,公正価値計測が高いか低い可能性がある.
有価証券
私たちがレベル2に分類した有価証券の公正価値は、観察可能な入力に基づいており、これらの入力は、基準収益率、報告の取引、ブローカー/取引業者オファー、発行者価格差、二国間市場、基準証券、入札、オファー、および市場研究出版物を含む参照データを含む可能性がある。
株式証券投資
我々は上場企業の株式証券に投資しており、これらの証券は既製のオファーで活発な取引を行っており、これらの証券を売却する能力に制限はない。したがって、これらは第1級に分類されます。私たちの株式証券への投資総公正価値は#ドルです
2022年12月31日と2021年12月31日までLianBioに投資をしていますその公正な価値は$です
本報告に記載されている間の株式証券投資に関連する実現済みおよび未実現損益総額は、以下のことを含む
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
権益法投資から権益証券投資への収益への転換 |
|
$ |
|
|
$ |
|
||
売却持分証券投資確認の純収益 |
|
|
|
|
|
|
||
期末までに保有している権益系証券投資確認の未実現純損失 |
|
|
( |
) |
|
|
( |
) |
純収益合計は“その他収入(費用),純額”に計上される |
|
$ |
( |
) |
|
$ |
|
|
177
LianBio保証書
2022年12月31日と2021年12月31日まで、我々の子会社QED Treateutics,Inc.またはQEDは、QEDにLianBioの株を購入する権利があるか、またはLianBio株式権証を承認する権利を持たせ、注7を参照する。この株式証券の公正価値は、活発な市場オファーのような観察可能な投入から来るので、LianBio株式証を一級に分類する。
レオオプション責任
2022年12月31日と2021年12月31日まで私たちは、私たちが以前合併貸借対照表で負債として負担していたレオコールオプションを確認しない。2018年11月、Leo PharmaまたはLeoは、当社の子会社PellePharma,Inc.またはPellePharmを買収する独占的で撤回不可能なオプションを取得しました。レオコールオプションは、ペライパム社の臨床開発計画に関連するいくつかの事件が発生した時または前に行使され、遅くとも2021年7月30日より遅くない。このオプションを行使すれば、PellePharmでの私たちの株式をあらかじめ決められた価格でLeoに売却する義務があるので、私たちはLeoコールオプションを流動負債に計上した。私たちは、レオコールオプションが行使、終了、または満了されるまで、レベル3投入に分類された観察不可能な入力を使用して、その後の各貸借対照表日にレオコールオプションを公正価値として再計量する。2021年3月30日、レオは2021年4月15日に発効したレオコールオプションを終了する通知を提供した。そこで、2021年3月31日までに存在する事実と状況に基づいて、リオが上記オプションを行使する可能性がわずかであることを評価し、レオコールオプション負債を再測定した
備考
当社の2029年債券および2027年債券、あるいは総称して債券(付記10参照)と呼ばれる公正価値は、それぞれの帳簿価値と異なり、市場取引で観察された債券価格によって決定される。債券の売買市場は活発な市場とはみなされていないため、公正価値の推定は第二級投入に基づいている。2022年12月31日まで2029年債券と2027年債券の推定公正価値、総額面は$です
定期ローン
我々の2022年12月31日と2021年12月31日までの未返済定期ローン(注10参照)の公正価値は、支払いの純現在価値を用いて推定され、市場金利と一致する金利で割引され、これは二次投入である。2022年12月31日現在、定期ローンを返済していない推定公正価値はい$です
私たちは現金等価物に分類されたいくつかのアメリカ政府通貨市場基金に投資している。取引可能証券には高投資級固定収益証券が含まれ、主に商業手形、社債、米国政府証券に投資される。
178
販売可能な現金等価物および有価証券に分類されることは、
|
|
2022年12月31日 |
|
|||||||||||||
|
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
推定数 |
|
||||
|
|
(単位:千) |
|
|||||||||||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
商業手形 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
現金等価物合計 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
商業手形 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
有価証券総額 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
現金等価物総額と |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
|
|
2021年12月31日 |
|
|||||||||||||
|
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
公正価値 |
|
||||
|
|
(単位:千) |
|
|||||||||||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
商業手形 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
現金等価物合計 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国庫券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
商業手形 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
会社債務証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
超国家債務証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
有価証券総額 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
現金等価物総額と |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
あったことがある
BridgeBioが最初に投資した日から
179
開ける
開ける
合併取引を完了することで,我々が発生する取引コストの合計は$となる
Eidosとの合併取引が完了した後、Eidosは私たちの完全子会社となった。2021年1月26日、EIDOSの普通株はナスダック世界ベスト市場の寄り付き前に取引を停止し、2021年2月5日、EIDOSは取引法第12(G)節に基づいてEIDOSの登録終了証明と通知を米国証券取引委員会に提出した。
2022年12月31日と2021年12月31日まで、私たちは合併後の一部のすべての実体の中で償還可能な転換可能な非持株権益と非持株権益を持っており、BridgeBioはVIEモードでの主要な受益者である。これらの残高は、連結貸借対照表における“償還可能転換可能非制御権益”のうち、株主損失の一部として株主損失の一部として単独で報告されている。
合併部分が実体を持つ非持株株主がそれぞれの報告期間内に所有権が変化した場合に占めるべき帳簿価値を反映するように、非持株権益の帳簿価値を調整する。このような調整総額は,2022年12月31日,2021年12月31日,2020年12月31日までの年度である(
180
共同生物
2019年10月、BBP LLCはLianBioと排他的合意を締結し、この合意により、BBP LLCはLianBioの持分を獲得し、LianBioに相当する
私たちの帳簿価値
2020年12月31日現在、総合貸借対照表における権益法投資連生の帳簿金額は、私たちの聯博投資に関する最大の損失を代表している。いくつありますか
2021年11月1日、共同生物はIPOを完了した。LianBioの初公募が完了した後、LianBioにおけるBBP LLCの所有権は約2%に減少しました
QEDとLianBioが2019年10月に締結したライセンス契約、またはQED-LianBioライセンス契約(注11参照)によると、QEDもQED購入を許可する引受権証を受け取っている
2021年10月、QEDが保有するLianBioの子会社株式を購入する権利証はLianBio引受権証に変換され、QEDを購入する権利がある
181
錠剤.錠剤
2018年11月19日、PellePharm,Inc.はLeoプロトコルに署名し、この合意に基づいて、Leo PharmaまたはLeoは、PellePharmを買収するための排他的選択権を付与された。レオコールオプションは、ペライパム社の臨床開発計画に関連するいくつかの事件が発生した時または前に行使され、遅くとも2021年7月30日より遅くない。私たちはLeoコールオプションを流動負債、またはレオコールオプション負債に計上し、BridgeBioはオプションを行使した場合にPellePharmの株式を所定価格でLeoに売却する義務があるので、財務諸表の付記3を統合する。私たちはその後の各貸借対照表の日付にレオコールオプションを公正価値として再計量し、レオコールオプションが行使、終了、または満期になるまで再計量した。
Leoと合意する前に,BridgeBioはPellePharmをVIEモードに統合する.“LEO協定”の締結日はVIE再議イベントとして決定された。私たちの評価によると、私たちは事件を再考した後、PellePharmはまだVIEであり、十分なリスク持株がないので、追加的な従属財務支援なしにその活動に資金を提供すると結論した。しかし、当時のリオ合意によるPellePharmの管理構造と取締役会構成の変化によると、BridgeBioはもはや主要な受益者ではなく、PellePharm経済表現に最も重要な重要な意思決定の権力を持たなくなったからである。これにより,BridgeBioは2018年11月19日にPellePharmの統合を解除した.2018年11月の統合解除後,PellePharmはBridgeBioの関連先と考えられている.
2018年にPellePharmが合併を解除した後、私たちの留保普通株投資を権益法投資に計上し、私たちの留保優先株投資をコスト投資に計上します。会計基準更新“ASU”2016-01が通過した後、金融商品−全体(825−10特別テーマ):金融資産と金融負債の確認と計量2019年、私たちはPellePharm優先株への投資を株式証券に計上しますが、確定しやすい公正な価値はありません。2020年12月31日までにPellePharmに投資した帳簿価値総額は
182
下表は本年度までの確認済み無形資産をまとめたものである2022年12月31日と2021年12月31日は、以下の各節で述べた予定である
|
|
|
|
|
|
|
|
|
|
||
|
2022年12月31日 |
|
|
2021年12月31日 |
|
||||||
|
加重平均 |
|
金額 |
|
|
加重平均 |
|
金額 |
|
||
|
|
|
(単位:千) |
|
|
|
|
(単位:千) |
|
||
総金額 |
|
$ |
|
|
|
$ |
|
||||
差し引く:累計償却 |
|
|
|
( |
) |
|
|
|
|
( |
) |
合計する |
|
|
$ |
|
|
|
|
$ |
|
||
ノバ社許可協定
2018年1月、QEDは、FGFR駆動疾患患者のinfigratinibの治療に関連するいくつかの知的財産権を取得し、特許およびノウハウを含む、ノワール国際製薬会社またはノワール社とライセンス契約を締結した。QEDは取引を資産買収と見なし、買収した総資産の推定公正価値は基本的にすべて単一確定資産、進行中の研究開発或いはIPR&Dに集中しているため、ASU 2017-01におけるスクリーニングテストの要求を満たす企業合併(主題805)について、企業の定義を明らかにするそれは.取引で取得した資産と負担する負債は,その公正価値に基づいて計測される.買収した知的財産権研究開発の公正な価値は研究と開発費用に計上されており、買収時にこの知的財産権の研究開発には他の未来の用途がないからである。
いくつかの重要なマイルストーンに達したら、QEDは$までの支払いを要求される可能性があります
Alexionと締結した資産購入契約
2018年6月、我々の子会社Origin Biosciences,Inc.,またはOriginは、Alexion Pharma Holding UnLimited Company,またはAlexionと、特許権、技術的ノウハウ、および契約を含むALXN 1101分子に関連する知的財産権を得るための資産購入協定に合意した。買収した総資産の見積もり公正価値のほぼすべてが確認された資産や知的財産権研究開発に集中しているため,ASU 2017−01年度スクリーニングテストの要求を満たすため,Originは取引を資産買収として会計処理した。取引で取得した資産と負担する負債は,その公正価値に基づいて計測される.買収した知的財産権研究開発の公正な価値は研究と開発費用に計上されており、買収時にこの知的財産権の研究開発には他の未来の用途がないからである。
資産購入契約により、Originは最高$の支払いを要求される可能性があります
OriginとSentynl Treateutics,Inc.,またはSentynlが2022年3月に締結した資産購入プロトコル,またはOrigin-Sentynl APA(付記12参照)によれば,SentynlはOrigin-Sentynl APAの満了後にAlexionに販売に基づくマイルストーン支払いおよび特許権使用料を支払う義務を負う.欧州医薬品局(EMA)が初めて定価承認を得た後、Originは引き続き規制に基づくマイルストーン支払いを担当し、最高で$に達する
183
診断学と基礎医学のプロトコル
2018年11月,QEDとFoundation Medicine,Inc.(FMIと略す)はQEDの薬物発見と開発計画についてキット診断合意を達成した。契約により、QEDは#ドルの支払いを要求される可能性があります
記念碑的な報酬スケジュール
私たちはある従業員やコンサルタントと業績に基づくマイルストーン報酬スケジュールを持っています。その帰属は様々なマイルストーンに達し、最初に知られている固定金額に依存して、私たちが自分で現金や株式の形で決済することができます。2020株式·持分奨励交換計画または交換計画の一部として、一部の従業員やコンサルタントと業績に基づくマイルストーン報酬スケジュールを達成しました。注16を参照してください。交換計画下の報酬スケジュールは株式のみで決済されます。株式形式で決済された業績に基づくマイルストーン奨励は、完全に既得の制限的株式奨励またはRSAの形式で満たされた。関連するマイルストーンが達成され、総合貸借対照表の当期部分の“計上すべき補償および福祉”、および非流動部分の“他の長期負債”に計上される場合、私たちはこのようなまたは補償があることを計算しなければならない。1種類あります
|
|
潜在固定通貨 |
|
|
応策 |
|
||
決済タイプ |
|
(単位:千) |
|
|||||
現金 |
|
$ |
|
|
$ |
|
||
在庫品(2) |
|
|
|
|
|
|
||
現金や株は私たちが自分で決めます |
|
|
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
||
他の研究開発やビジネス協定は
我々は、正常業務過程において臨床試験の契約研究機関、臨床用品の契約製造機関及び商業及び運営目的のための臨床前研究、供給及びその他のサービス及び製品の他のサプライヤーと契約を締結することもできる。これらの契約は一般的に通知後に終了することが規定されており、終了費用を徴収する可能性がある。2022年12月31日まで私たちは研究開発プロジェクトの優先順位の再決定に関するいくつかの費用に約$を持っています
184
賠償する
通常の業務プロセスでは、サプライヤー、レンタル者、業務パートナー、取締役会メンバー、上級管理者、および他の当事者に、このような合意違反、私たちが提供するサービス、私たちの不注意または故意の不正行為、法律違反または第三者による知的財産権侵害クレームによる損失を含むが、これらに限定されない異なる範囲および条項の賠償を提供する可能性があります。また、取締役や特定の高級職員や従業員と賠償協定を締結しており、取締役、高級職員、または従業員の身分やサービスとして生じる可能性のある責任について賠償することを要求します。当該等の合意によれば、吾等は賠償を要求されていないため、吾等の知る限り、当社の連結財務諸表に重大な影響を与える可能性のあるクレームは何もない。
私たちは取締役と上級職員保険も維持しています。これは私たちが取締役義務を賠償することによるいくつかの責任をカバーしているかもしれません。現在まで、私たちはいかなる重大なコストも発生しておらず、このような準備のために総合財務諸表に負債を計上すべきでもない。
事件があったり
備考
2029年ノート
開ける
2021年の債券発行の純収益は約$を受け取りました
185
2029年債券保有者は、2028年11月1日前の営業日取引終了直前の任意の時間に、2029年債券の全部または任意の部分を選択し、換算倍数を$とすることができる
2028年11月1日またはその後、満期日直前の第2の予定取引日の取引が終了するまで、保有者は、前述の規定にかかわらず、その2029年債の全部または任意の部分を随時転換することができる。
変換率は最初に
転換率は場合によっては調整される可能性があるが、任意の課税利息や未払い利息については調整されない。また期日前に発生したある会社の事件はあるいは、償還通知を出した場合、場合によっては、このような企業活動に関連する保有者が2029年債の転換率を転換することを選択する。株式交換比率が増加すれば、発行可能株式の最高数は
私たちは2026年2月6日までに2029年の手形を償還しないかもしれない。場合によっては、2026年2月6日またはその直後、満期直前の第41回予定取引日またはそれ以前の償還日の全部または任意の部分2029年債券を選択することができる。債券には債務返済基金が用意されていない。もし私たちが根本的な変化(2029年債券契約で定義されているような)を経験すれば、所有者は私たちに2029年の債券の全部または任意の部分を現金で買い戻すことを要求するかもしれません。基本的な変化は買い戻し価格に相当します
2029年の債券発行については約$を生み出しています
186
2027年ノート
2020年3月9日に元金総額$を発行しました
2020年の手形発行の純収益は約$を受け取りました
2027年債券保有者は、2026年12月15日前の営業日取引終了直前のいつでも、ドルの倍数で、2027年債券の全部または任意の部分を選択することができます
2026年12月15日またはその後、満期日直前の第2の予定取引日の取引が終了するまで、保有者は、前述の規定にかかわらず、その2027年債の全部または任意の部分を随時転換することができる。
2021年3月31日と2021年6月30日までの各カレンダー四半期においてのみ、2027年債券は保有者の選択の下で転換する資格があり、その間に転換価格条件を満たしているからである。2022年と2021年の財政年度のどの他の期間も割引価格条件を満たしていない。
変換率は最初に
転換率は場合によっては調整される可能性があるが、任意の課税利息や未払い利息については調整されない。また、期日前に発生したいくつかの企業イベントの後、場合によっては、このような企業イベントに関連して2027年のチケットを変換する所持者を選択する変換率を向上させる。株式交換比率が増加すれば、発行可能株式の最高数は
187
私たちは満期日までに2027年の債券を償還しないかもしれないし、2027年の債券に債務超過基金を設立することもないかもしれない。もし私たちが根本的な変化(2027年債券契約で定義されているような)を経験したら、保有者は私たちに現金で2027年の債券の全部または任意の部分を買い戻すことを要求するかもしれません。基本的な変化は買い戻し価格に等しいです
2020年にASC 470-20によって発行された2027年手形を計算する際には、債務:転換やその他の選択肢を持つ債務私たちは現金、BridgeBio普通株、または私たちが選択した現金とBridgeBio普通株の組み合わせで2027年の手形を決済することができるため、私たちは負債部分と埋め込まれた転換オプションまたは株式部分の間に収益を分配することで、2027年の手形の負債と株式部分をそれぞれ会計処理した。2021年1月1日からASU 2020-06を採用しています債務--転換および他のオプションを有する債務(主題470-20)および派生ツールおよびヘッジ--エンティティ自己持分契約(主題815-40):エンティティ自己持分変換可能ツールおよび契約の会計、したがって、私たちは2027年債の負債と権益部分を単独で計算するのではなく、2027年債券を完全に債務として計算する。
2027年の債券発行については約$が生まれました
付記に関するその他の情報
未償還手形残高には以下の項目が含まれる
|
2022年12月31日 |
|
2021年12月31日 |
|
||||||||||
|
2029年ノート |
|
|
2027年ノート |
|
2029年ノート |
|
|
2027年ノート |
|
||||
|
(単位:千) |
|
(単位:千) |
|
||||||||||
元金 |
$ |
|
|
$ |
|
$ |
|
|
$ |
|
||||
未償却債務割引と発行コスト |
|
( |
) |
|
|
( |
) |
|
( |
) |
|
|
( |
) |
帳簿純額 |
$ |
|
|
$ |
|
$ |
|
|
$ |
|
||||
188
次の表には、債券に関する確認された利息支出総額および実金利を記載します
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
2022年12月31日までの年度 |
|
|
2021年12月31日までの年度 |
|
|
2020年12月31日までの年度 |
|
|
|||||||||||||||||||
|
|
2029年ノート |
|
|
2027年ノート |
|
|
合計する |
|
|
2029年ノート |
|
|
2027年ノート |
|
|
合計する |
|
|
2027年ノート |
|
|
|||||||
|
|
(単位:千) |
|
|
|||||||||||||||||||||||||
契約利子支出 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
|||||||
債務の償却 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
総利息及第 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
|||||||
実利率 |
|
|
% |
|
|
% |
|
|
|
|
|
% |
|
|
% |
|
|
|
|
|
% |
|
|||||||
2022年12月31日まで2029年と2027年の債券の支払利息は$
債券項目の次の最低支払日2022年12月31日、詳細は以下の通り
|
|
2029年ノート |
|
|
2027年ノート |
|
|
合計する |
|
|||
|
|
(単位:千) |
|
|||||||||
12月31日までの年度: |
|
|
|
|
|
|
|
|
|
|||
2023 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
2024 |
|
|
|
|
|
|
|
|
|
|||
2025 |
|
|
|
|
|
|
|
|
|
|||
2026 |
|
|
|
|
|
|
|
|
|
|||
2027 |
|
|
|
|
|
|
|
|
|
|||
その後… |
|
|
|
|
|
|
|
|
|
|||
将来支払総額 |
|
|
|
|
|
|
|
|
|
|||
利子を表す金額が少ない |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
元金総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
189
債券に関する上限引受及び株式買い戻し取引
2021年1月25日及び2020年3月4日に、それぞれ2029年債券及び2027年債券の定価と同時に、吾等はそれぞれいくつかの金融機関又は上限償還取引相手と単独の私的協議上限償還取引(それぞれ“2021年上限償還取引”及び“2020上限償還取引”)を締結し、又は共に上限償還取引を締結する。私たちは約$を使いました
これらのカバー付き呼装置は、ASC 815−40に要約された条件に適合する派生ツールやヘッジ株主権益に分類され、持分分類の条件を満たし続ける限り、その後再計量されることはない。私たちが記録した追加実収資本は約#ドル減少した
さらに私たちは約$を使って
定期ローン
融資と保証協定
2021年11月に、吾らは、(I)米国銀行全国協会(行政エージェントとして、または行政エージェントおよび担保エージェントとして、または担保エージェントとして)、(Ii)ある貸手、または貸手、(Iii)BridgeBio、および(Iv)BridgeBioのいくつかの子会社として、保証人、または保証人として、融資とセキュリティ協定、または融資協定を締結した。2022年5月には“融資協定第1修正案”を締結し、2022年11月には以下のように“融資協定第2改正案”を締結した。
190
PU融資協議の元の条項と条件により、貸手は当社に元金総額最大#ドルまでの定期融資を提供することに同意しました
我々の融資プロトコルの義務の保証として、BridgeBioと保証人はそれぞれ融資者の利益のために担保エージェントにBridgeBioと保証人のほとんどの資産の持続的な保証権益(BridgiBioと保証人が所有またはその後に買収したすべての持分を含む)を付与したが、いくつかの慣例の例外に適合している。いくつかの投資と処置の限界値を超えると、BridgeBioのより多くの子会社は保証人として加入することを要求されるだろう。
定期ローン立て替えのいかなる未返済元金も固定金利で利息を計算し,金利は同じである
当行は任意の時間に定期ローン立て替え金の未返済元金(すべてが部分的ではない)を前払いすることができ,課税及び未払い利息及び前払い保険料,保険料は
貸手の選択では、私たちはまた、買い戻しまたは償還質担保、特定の特許権使用料取引への参入、他の資産または子会社の処分、参入許可および他の金銭化取引に関連するいくつかの前金事件が発生した場合に、事前支払いを強制することを要求される(このような事件はすべて前払い事件と呼ばれる)。これらの事件は、
あるプリペイド事件の強制的な前金要求の規定の下で、融資プロトコルは、私たちの能力を制限すること、(I)追加の債務を生成すること、(Ii)配当金を支払うこと、または特定の分配を行うこと、(Iii)私たちの資産を処分すること、留置権を付与すること、私たちの資産を許可または担保すること、または(Iv)私たちの業務特性を根本的に変えることを含む慣用的な肯定的および限られた負の約束を含む。BridgeBioと保証人は、私たちの知的財産権を許可し、他の資産を処分し、貨幣化と特許権使用料取引を行うことができる幅広い能力を持っているが、いずれの場合も、上記の強制的な前払いを支払わなければならない。融資協定は、ある制限の場合、BridgeBioおよび保証人は、(X)BridgeBioの株式およびその任意の子会社の株式を買い戻すことができること、(Y)任意の合弁企業または同様の投資を行うこと、および(Z)他の投資および買収を行うことができることを規定する。上記の強制的事前返済要求の制約の下で、BridgeBioが所有する非融資協定当事者側のポートフォリオ会社は、ある例外的な場合を除いて、融資合意下のいかなる契約または制限の制約も受けない。
ローン協定にも慣用的な違約事件が含まれており、吾らが満期時にいかなる元金或いは利息を支払うことができなかったか、ある破産或いは債務返済不能事件が発生したか、或いはローン協定に違反した場合の契約を含む。約束違反事件が発生すると、他の事項を除いて、貸手は融資協議の下での私たちの義務を加速させることができる。
191
私たちは最初の前払いの純収益1ドルを受け取った
2022年5月に第1修正案に署名しました
2022年6月に、NAVERE-BMS許可協定項目の下の前払いを受け取り(付記11参照)、改訂された融資協定のいくつかの強制前払い条項をトリガした。そこで私たちは$を支払いました
融資協定及び改訂された融資協定の条項に基づいて、本行は選択権を行使し、計算すべき利息を元金に変換する
2022年11月に第2修正案に署名しました
2022年12月31日までの年度私たちは、ローン協定に関連した利息支出#ドルを確認した
次の表は私たちの定期ローンをまとめています
|
|
2022年12月31日 |
|
|
2021年12月31日 |
|
||
|
|
(単位:千) |
|
|||||
定期融資元金価値 |
|
$ |
|
|
$ |
|
||
PIKはエージェントに追加されました |
|
|
|
|
|
|
||
債務割引、発行コスト、脱退費用の増加 |
|
|
( |
) |
|
|
( |
) |
定期ローン,純額 |
|
$ |
|
|
$ |
|
||
192
ローン契約の下での将来最低支払い、現在まで2022年12月31日、詳細は以下の通り
|
|
金額 |
|
|
|
|
(単位:千) |
|
|
12月31日までの年度: |
|
|
|
|
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
将来支払総額 |
|
|
|
|
利子を表す金額が少ない |
|
|
( |
) |
より少ない退場料 |
|
|
( |
) |
定期ローンで元金総額を支払う |
|
$ |
|
|
力持ちローンと保証契約
私たちはHercules Capital,Inc.またはHercules Capital,Inc.またはHercules Term Loanと融資と安全協定を締結し、時々修正し、この合意に基づいて私たちは元金#ドルを借りた
2021年1月には、2029年手形の発行を許可し、2021年上限償還と株式買い戻し取引を行うことを可能にするための融資·担保協定第5改正案に署名した。
2021年4月には、融資·担保協定第6修正案、または改正大力神定期融資を実施した。
T彼はHercules定期融資を改訂し、2021年11月に上記融資協定で規定された第1段階前払いの純収益の一部を全額前払いし、債務損失#ドルを早期返済した
シリコンバレー銀行とHercules融資と保証協定
Eidosシリコンバレー銀行(SVB)とHercules Capital,Inc.と融資とセキュリティ協定、またはSVBとHercules融資協定を締結し、この合意に基づいてEidosは元金#ドルを借入する
♪the the theA部分ローンは2021年4月に全額返済され、金額は#ドルです
193
BMSとライセンス開発と商業化協定を締結
2022年5月12日、BridgeBioと我々の子会社NAVARE Pharma,Inc.,またはNAVAREは、BMSと独占ライセンス開発と商業化協定、またはNAVRE-BMS許可協定を締結し、この合意に基づいて、NAVREはBMS独占権利を付与し、NAVREの候補製品BBP-398、人民病院、中国人民健康保険協会、中国人民解放軍Republic of China、マカオ、香港、台湾、タイ、シンガポール、韓国またはアジア地域を除外した。BBP−398のアジア地域における開発および商業化は、NAVERRE−LianBioライセンスプロトコルによって制限される(以下に述べる)。NAVRE-BMS許可プロトコルは、KRAS変異を有する末期固形腫瘍を治療する連合治療試験においてBBP-398、または2021年NAVIRE-BMSプロトコルを研究するために、2021年7月に署名されたNAVREとBMSとの間の早い時期に署名されたプロトコルを拡張する。NAVRE-BMSライセンスプロトコルは、2021年のNAVERE-BMSプロトコルの条項を変更しません。
NAVERE-BMSライセンス契約の条項によると、NAVEREは、払戻不可能な前金#ドルを受け取る権利がある
NAVERE−BMSライセンスプロトコルは、BMSがプロトコル内のクライアントであるので、ASC 606の範囲に属すると判断し、プロトコルにおいて以下の義務を履行することを決定した
上記権利及び活動が互いに独立していることを考慮して、上記履行義務は、契約範囲内で異なるものであってもよいことを決定する。BMSは、研究開発サービスなしにこのライセンスを使用することができる。同様に、これらのサービスは、ライセンスに関係なく、契約範囲内でBMSに明らかな利点を提供し、これらのサービスは、我々の助けを必要とすることなく、BMSまたは他の第三者によって提供されることができるからである。追加の貨物またはサービスの選択権は、NAVRE-BMS許可プロトコルの開始時に、これらの貨物またはサービスが割引で提供されないため、履行義務としても決定されないので、実質的な権利とはみなされない。
194
NAVRE-BMSライセンスプロトコル開始時の初期取引価格は$であることを確認した
私たちが割り当てた出来高は$です
ライセンスのSSPは、ライセンス譲渡に関連する割引を考慮してキャッシュフローを確率的に重み付けする方法を使用して決定される。研究·開発されているサービスのSSPは,これらのサービスに関する作業とコストの見積り数に基づいて計算され,類似契約予想に基づいて実現される合理的な毛金利に基づいて調整されている。
この2つの履行義務の収入は以下のとおりであることを確認した
2022年12月31日までの年度我々は認めました$
195
ヘルシンヌとの許可と協力協定
2021年3月29日、QEDはHelsinn Healthcare S.A.またはHHC、Helsinn Treateutics(U.S.)、Inc.またはHTU、およびHHC、HelsinnまたはQED-Helsinnと協力協定集団と許可と協力協定を締結し、この合意に基づいて、QEDはHHCに独占的な許可を与え、QEDの候補製品infigratinibを開発、製造および商業化し、腫瘍学および軟骨発育不良または任意の他の骨格発育不良以外のすべての他の適応、人民Republic of China、香港とマカオまたは大中国を除外するために使用された。このプロトコルにより,QEDは米国で許可された適応で共同商業化infigratinibの共同独占許可を得た。この合意によると、ヘルシンヌはまた、事件が発生したり、事件があったりした場合に特定の領土について最初の交渉を行う権利がある。このプロトコルの一部として、QEDはまた、QED-Helsinnライセンスおよび協調プロトコルで説明されているように、過渡期内に在庫を転送することを要求される。QED−Helsinn許可と連携協定は2021年4月16日に発効した。QED−Helsinn許可と協力協定の条項によると,QEDは総額約$を獲得する資格がある
2022年2月28日、QEDとヘルシンは、QED-ヘルシン許可と協力協定、または改訂されたQED-ヘルシン許可と協力協定を改訂し、2022年3月1日から発効する。改訂されたQED-ヘルシン許可と協力協定の条項によると、ヘルシンはアメリカでinfigratinibを商業化する独占的な許可を持ち、そしてinfigratinibの開発、製造と商業化を担当し、軟骨発育不良或いは任意の他の骨格発育不良以外の世界各地の腫瘍学適応に使用され、中国大区を含まない。QEDは骨発育不良(軟骨発育不良を含む)を開発、製造、商業化治療するinfigratinibのすべての権利を保持する。
改訂されたQED−Helsinn許可と連携プロトコルにより,QEDは認可適応におけるinfigratinibの商業化を共有しなくなり,認可適応におけるinfigratinibのいかなるグローバル開発コストも担当しなくなった。
また、改正されたQED-Helsinn許可と協力協定によると、QEDは規制と販売に基づくマイルストーン支払いを受ける資格があり、金額は最高$に達する
改正されたQED−ヘルシン許可と協力協定は、発効日から2022年8月31日まで延長され、その間、QEDは研究開発と商業化活動に協力する契約を締結された。過渡期内に発生したQED契約活動に関する費用はヘルシンによって全額返済され,過渡期終了後にQEDに支払われる。ヘルシンヌはFMIに対するQEDの返済義務にも同意しており,改訂されたQED−ヘルシン許可と協力協定の一部として付記8を参照されたい。売掛金を記録する際には、2022年12月31日現在の年度総合経営報告書に“その他の収入(費用)、純額”の一部として記録されている収益を確認した。
196
2022年8月23日、私たちはヘルシンヌから書面で通知を受け、商業的な考慮のため、便宜上、改訂されたQED-ヘルシン許可と協力協定を終了するつもりだと表明した。QEDとヘルシンエンまたはヘルシンエンの双方は2022年12月21日から相互終了協定またはMTAを締結し、改訂されたQED-ヘルシン許可および協力協定およびその下でのすべての権利と義務を終了した。MTAの条項によると、ヘルシン側はQEDがinfigratinibの開発、製造、商業化を継続できるように、潜在的な非腫瘍学的適応の治療方法として、例えば世界的に(中国、香港およびマカオを含まない)軟骨発育不良を治療することを担当する。さらに、QEDは、将来の規制または販売ベースのマイルストーン支払いを得る権利がなくなり、ヘルシンーンが2023年3月31日までライセンス製品を販売しなくなるまで、TRUSELTIQの純売上高の印税を取得し続ける。ヘルシン側はTRUSELTIQの流通が永久的に停止することを通知し,関連IND項下のすべての臨床研究が停止することをFDAに通知した。
“多国間貿易協定”によると、改正された“QED-ヘルシン許可証と協力協定”に規定されている過渡期間内に契約サービスに関連するすべての未清債務が満期になった。これには返済可能な契約研究と開発と商業活動#ドルが含まれている
過渡期以降に発生したすべての費用は清算費用とみなされ、ヘルシンーン各当事者間の責任は“中期戦略協定”で定義されているように“清算計画”で概説されている。終了計画内の活動は最初の$の後に割り勘になります
MTAに署名する前に、QED−Helsinn許可および連携協定、ならびに改訂されたQED−Helsinn許可および連携協定は、双方が積極的な参加者であり、協力活動の重大なリスクおよびリターンに直面し、ASC 606の範囲内であり、Helsinnが顧客の会計単位として決定される部分がASC 808の範囲内であると考えられる。協力手配の中でサプライヤー-顧客関係を代表しない課金単位に対して、協力研究開発と商業化サービスの業績を含むに対して、著者らはASC 606が応用を類推すべきではないことを確定し、そして合理的かつ合理的な会計政策選択を適用し、この選択は業績を推定する間に協力パートナーにサービスを移転する状況を忠実に描写した。ヘルシンーンが支払った協力研究開発や商業化サービスに関する精算は関連費用として確認され、取引価格には含まれていない基礎費用の相殺に分類されている。
QED−Helsinn許可と協力協定の条項を評価し,Helsinnは以下の2つの異なる履行義務を有する顧客であることを決定した:(1)基礎製品の開発,製造,商業化の独占的許可,および(2)移行供給期間内に在庫を移転する。改正されたQED-Helsinn許可と協力協定は追加的な履行義務を生じなかった。
QED-Helsinn許可と連携プロトコル開始時の初期取引価格は$であることを確認した
197
変数.変数その後,可変対価格に関する不確実性を解決する際に大きな収入逆転を招く可能性が高い場合を考慮する。ASC 606内の特許使用料例外に従って後続販売が発生した場合、ライセンスは、ライセンスが特許使用料または販売ベースのマイルストーンに関連する主要項目であるので、販売ベースのマイルストーンおよび特許権使用料に関連する対価格を確認する。2021年第4四半期に、私たちはEMAからinfigratinibマーケティング許可の検証を受けました。規制マイルストーンに関する可変対価格の不確実性が解決されたため、この対価格を含む取引価格を更新したため、取引価格を$上昇させた
私たちは$を割り当てた
2021年12月31日現在、私たちは、許可条項の下で許可を受けることができ、在庫の移転を完了することができるように、ヘルシンヌに必要なすべての情報を提供しました。2021年12月31日までの年度内我々は認めました$
QED−Helsinn許可と連携プロトコルによると,ASC 808の範囲に属する連携研究開発サービスに関する計算単位については,Helsinnの研究開発費におけるシェアが#ドルであることが確認された
FDAが2021年5月にTRUSELTIQを承認した後,米国での主要販売先となり,総合経営報告書で製品の販売を確認した。2022年1月から,残りの過渡的TRUSELTIQをヘルシンに供給し,ヘルシンが主な売り手となった。したがって,2022年からTRUSELTIQに関する製品販売は確認されなくなり,分担し続けているにもかかわらず
LianBioとのライセンス契約
航海する
はい2020年8月、NAVAREはLianBioと独占ライセンス契約、またはNAVRE-LianBioライセンス契約を締結した。NAVERE-LianBio許可プロトコルによれば、NAVREは、許可された特許権および独自技術に基づいて、RASおよび受容体チロシンキナーゼ変異駆動腫瘍のためのSBP 2阻害剤BBP-398またはBBP-398を開発、製造および商業化するために、LianBioに独占的、再許可可能な許可を付与する。下にある
198
条項NAVRE-LianBio許可協定によると、LianBioは中国と一部のアジア市場の商業権を獲得し、BBP-398の臨床開発活動に参与する。LianBioに付与された権利の補償として、私たちは払い戻しできないドルを受け取った
我々は、ASC 606に基づいてNAVER−LianBioライセンスプロトコルを説明し、LianBioは、独自の資源を使用して基盤製品を開発し、それを商業化することによって、許可から独立して利益を得ることができるので、独占許可を明確な履行義務として決定した。しかも、私たちはカードを持っている地域のために臨床と商業供給協定を締結するつもりだ。私たちはこのような供給協定に基づいて、未来の製品の選択可能な権利が物質的権利を代表しないと確信する。2022年7月、NAVEREとLianBioは臨床数量の許可製品の生産と供給について臨床供給協定を締結した。NAVREは2022年12月31日までの1年間に,臨床供給協定の一部としてLianBioに些細な金額を出し,このような金額を許可収入として確認した。
2021年第2四半期、発展マイルストーンが可能になった。開発マイルストーンに関する可変対価格の不確実性を解決したため,この対価格を含むように取引価格を更新したため,取引価格を$増加させた
将来の潜在的な発展マイルストーンや販売に基づく特許権使用料は可変的な考慮要因であると考えられる。将来可能なマイルストーン支払いは、ASC 606によって完全に制約されていると決定されるので、取引価格には含まれない。このような開発マイルストーンの成果は将来の臨床試験と規制承認の成功にかかっており、これらは私たちのコントロール範囲内ではなく、現段階でも確定していないことを確認した。我々は、ASC 606の下での販売ベースの特許使用料例外に基づいて、販売または販売ベースのマイルストーンが発生した場合、ライセンスが特許権使用料または販売ベースのマイルストーンに関連する主要項目であるので、特許使用料スケジュールおよび販売ベースのマイルストーンが確認されると予想する。各報告期間内および不確定なイベントが解決された場合や状況が他に変化した場合に取引価格を再評価します。
QED
2019年10月、QEDはLianBioと独占ライセンス契約(“QED-LianBioライセンス契約”)を締結した。QED-LianBio許可プロトコルによれば、QEDは、米国以外のいくつかの地域でinfigratinibを開発、製造および商業化するために、許可された特許権およびノウハウに基づいてLianBioに独占的かつ再許可可能な許可を付与し、すべての癌適応の任意およびすべてのヒト予防および治療用途(他の療法との組み合わせを含む)のために使用される。QED-LianBioライセンス契約によると、QEDは払い戻し不可能な前払い$を受け取りました
我々はQED-LianBioライセンスプロトコルとLianBio独占プロトコルをASC 606の下の取引と見なし、LianBioは自分の資源を利用して基礎製品を開発し、それを商業化することができ、それによって許可から独立して利益を得ることができるので、独占許可を独自の履行義務と見なしている。しかも、私たちはカードを持っている地域のために臨床と商業供給協定を締結するつもりだ。私たちはLianBioがこのような供給協定に基づいて未来の製品の選択権を実質的な権利とみなさないと確信する。2021年第4四半期に臨床供給協定が締結された。QEDは臨床供給協定の一部としてLianBioに些細な金額を提供し,2021年12月31日までの年度中に許可収入としてこのような金額を確認した
199
将来の潜在的な発展マイルストーンや販売に基づく特許権使用料は可変的な考慮要因であると考えられる。将来可能なマイルストーン支払いは、ASC 606によって完全に制約されていると決定されるので、取引価格には含まれない。このような開発マイルストーンの成果は将来の臨床試験と規制承認の成功にかかっており、これらは私たちのコントロール範囲内ではなく、現段階でも確定していないことを確認した。販売が発生した場合、ライセンスは、特許使用料または販売ベースのマイルストーンに関連する主要項目であるので、特許使用料スケジュールおよび販売ベースマイルストーンが確認されるか、またはライセンスが特許使用料または販売ベースマイルストーンに関連する主要項目であるので、ASC 606下の販売ベース特許使用料例外に従ってマイルストーンが達成されると予想される。各報告期間内および不確定なイベントが解決された場合や状況が他に変化した場合に取引価格を再評価します。
Alexionとのライセンス契約
2019年9月、EidosはAlexion製薬会社の子会社Alexion Pharma国際運営無限会社または共同Alexionと独占ライセンス契約を締結し、日本でacoramidis(以前AG 10と呼ばれていた)と呼ばれる化合物およびその任意の化学形態および任意のacoramidisを含む薬品、またはEidos-Alexionライセンス契約を開発、製造した。契約によると、Eidosは返金できない前金#ドルを受け取りました
EidosはAlexionと株式購入契約を締結し、協定によりEidosはAlexionに売却される
Eidosも$を得る資格がある
Eidosは、ASC 606でのライセンスプロトコルを説明し、Alexionは、自分のリソースを使用してベース製品を開発し、それを商業化することによって、自分がライセンスから利益を得ることができるので、独占ライセンスを独自の履行義務として決定する。また,Eidosは臨床供給協定を締結し,許可地域の商業供給協定を締結する。Eidosは、これらの供給プロトコルに基づいて、未来の製品に対する選択可能な権利は実質的な権利とはみなされないと認定している。Eidosはドルを認めました
Eidosは将来の潜在的な規制マイルストーンは約5ドルに達すると考えています
Eidosは2020年7月にAlexionと臨床供給協定を決定し、この協定は許可証とは別の履行義務として決定された。臨床供給プロトコルの一部として,EidosがAlexionに出荷した金額はわずかであり,これらの金額を許可収入として確認した。
200
優先審査券を販売します
2022年5月、私たちはPRVを#ドルで売る最終合意に達したことを発表した
Sentynlとの資産購入協定
2022年3月4日、OriginとSentynlは、Origin-Sentynl APAを締結し、この合意に基づいて、SentynlはNULIBRYの世界的権利およびOriginの特定の資産を取得し、米国におけるNULIBRYの持続的な開発および商業化、およびフォースデンプリンの世界的な開発、製造および商業化を担当する。この取引は2022年3月31日、つまり取引終了日に完了した。Origin-Sentynl APAの条項によると、Originは#ドルの前金を受け取りました
私たちはASC 610-20に従ってこの取引を計算した。決算日に販売損失#ドルを確認しました
OriginによるOrigin-Sentynl APAの売却に含まれる資産は、融資契約条項に基づいて資産能力を処分する我々の制限を受けません(付記10参照)。
201
13.内部ライセンスプロトコル
スタンフォード大学許可協定
2016年4月、Eidosは、Eidosの薬物発見と開発計画に関連するライセンス契約を、ラン·スタンフォード初級大学、スタンフォード大学またはスタンフォード大学取締役会と締結した。この協定によると、Eidosはすでにある世界的な独占許可を得ており、許可された特許権でカバーされた製品を製造、使用、販売することができる。2017年3月、Eidosは1ドルの許可料を支払った
また、スタンフォード大学とのライセンス契約に基づき、スタンフォード大学に許可院落下の再許可に起因するすべての非特許権使用料再許可対価格の一部を支払います。許可協定はこの事件が3年目に起きたら
Leidos生物医学研究許可証と共同研究開発協定
2017年3月、THERASはLeidos Biomedical Research,Inc.またはLeidosと協力研究開発協定、またはLeidos CRADAを締結した。2018年12月、TherasとLeidosは、ある世界的に独占的な許可を得て、許可された化合物を使用することができる初期Leidos許可というライセンス契約を締結した。LeidosプロトコルはTherasの薬物発見と開発計画と関連がある。最初のLeidosライセンスは2021年に終了した。サーラスとレドスは
診断学と基礎医学のプロトコル
付記8で述べたように,QEDとFMIはQEDの薬物発見·開発イニシアティブに関する診断プロトコルを締結している。2022年まで、2021年および2020年12月31日まで年度,QED確認の研究と開発費用は$
その他の許可と連携協定
上記の合意に加えて、上記の条項と類似した条項に従って、様々な機関や商業エンティティと他の許可および協力協定を締結しており、これらの協定のうち、単独または総合的な重大な合意はない。
202
14.レンタル証書
会社本部、オフィススペース、実験室施設の運営賃貸契約があります。私たちのオフィススペースの一つは融資レンタル部分があります。レンタル人が提供する家具とオフィス設備を代表します。私たちの融資リースは私たちの総合貸借対照表に“財産と設備純額”の一部として示されており、実質的ではない。
いくつかのレンタルオプションは、私たちの選択に更新オプションが含まれています。更新オプションが行使されると合理的に判断された場合、更新オプションが含まれます。賃貸負債は、それぞれの賃貸負債を算出する際の最新の借入金金利に基づいて、残存賃貸期間と賃貸総金額に応じて調整する加重平均割引率計量を用いる。
レンタル料金の構成は以下のとおりである
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
直線運営リースコスト |
|
$ |
|
|
$ |
|
|
$ |
|
|||
融資リースコスト |
|
|
|
|
|
|
|
|
|
|||
可変リースコスト |
|
|
|
|
|
|
|
|
|
|||
総賃貸コスト |
|
$ |
|
|
$ |
|
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
レンタルに関する補足キャッシュフロー情報は以下のとおりである
|
|
十二月三十一日 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
賃貸負債の金額を計上するための現金 |
|
|
|
|
|
|
|
|
|
|||
レンタル経営キャッシュフロー |
|
$ |
|
|
$ |
|
|
$ |
|
|||
融資リースの経営キャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
賃貸義務を交換することで得られた使用権資産 |
|
|
|
|
|
|
|
|
|
|||
賃貸借契約を経営する |
|
|
|
|
|
|
|
|
|
|||
融資リース |
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||
残存期間と割引率に関する補足資料は以下のとおりである
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
加重平均残存賃貸年限(年) |
|
|
|
|
|
|
||
賃貸借契約を経営する |
|
|
|
|
|
|
||
融資リース |
|
|
|
|
|
|
||
加重平均割引率 |
|
|
|
|
|
|
||
賃貸借契約を経営する |
|
|
% |
|
|
% |
||
融資リース |
|
|
% |
|
|
% |
||
|
|
|
|
|
|
|
||
203
自分から2022年12月31日、私たちがキャンセルできない経営賃貸の将来の最低賃貸支払いは以下の通りです。私たちの融資リースによると、未来の最低賃貸支払いは重要ではない。
|
|
金額 |
|
|
|
|
|
(単位:千) |
|
|
|
12月31日までの年度: |
|
|
|
|
|
2023 |
|
$ |
|
|
|
2024 |
|
|
|
|
|
2025 |
|
|
|
|
|
2026 |
|
|
|
|
|
2027 |
|
|
|
|
|
その後… |
|
|
|
|
|
将来の最低賃貸支払い総額 |
|
|
|
|
|
利子を推定する |
|
|
( |
) |
|
合計する |
|
$ |
|
|
|
|
|
|
|
|
|
2022年12月31日までの報告 |
|
|
|
|
|
賃貸負債を経営し、今期の部分 |
|
$ |
|
|
|
賃貸負債を経営し,当期分を差し引く |
|
|
|
|
|
リース負債総額を経営する |
|
$ |
|
|
|
割引キャッシュフローモデル(収益法)を用いて推定されたある資産グループの減価損失を確認した$
製造協定
2019年12月、BridgeBioのある子会社の候補製品の大量有効医薬成分の臨床的および商業規模製造能力を確保するために、サプライヤーと製造協定を締結した。製造協定が許可された方法で終了しない限り、協定は無効になるだろう
私たちは建設中の資産#ドルを記録した
2022年3月、私たちはサプライヤーと共同で製造協定を終了することに同意した。終了協定は2022年5月に正式に施行される。契約終了により$を支払いました
204
15.株式買い戻し計画および棚登録
2021年株式買い戻し計画
2021年5月、取締役会は株式買い戻し計画を承認し、承認しました。この計画によると、私たちは最大$を購入することができます
2020年棚登録
2020年7月7日、我々は米国証券取引委員会にS-3表棚上げ登録書を提出し、普通株、優先株、債務証券、権利証と単位あるいはそれらの任意の組み合わせの登録に関連した。私たちはまた、Jefferies LLCとSVB Leerink LLCまたは販売エージェントと共同で公開市場販売契約を締結し、私たちが発行、発行、販売した総金額が最高$に達することを規定しています
16.株ベースの報酬
各法人エンティティの持分計画の下で、従業員と非従業員の総合運営報告書において、株式ベースの報酬を以下の費用カテゴリに記録する
|
|
2022年12月31日までの年度 |
|
||||||||
|
|
橋接生物 |
|
他にも |
|
|
合計する |
|
|||
|
|
(単位:千) |
|
||||||||
研究開発 |
|
$ |
|
$ |
|
|
$ |
|
|||
販売、一般、行政 |
|
|
|
|
|
|
|
|
|||
再編成·減価·関連費用 |
|
|
|
|
|
|
|
|
|||
株に基づく報酬総額 |
|
$ |
|
$ |
|
|
$ |
|
|||
|
|
2021年12月31日までの年度 |
|
|||||||||
|
|
橋接生物 |
|
|
他にも |
|
|
合計する |
|
|||
|
|
(単位:千) |
|
|||||||||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
販売、一般、行政 |
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
205
|
|
2020年12月31日までの年度 |
|
|||||||||||||
|
|
橋接生物 |
|
|
Eidos |
|
|
他にも |
|
|
合計する |
|
||||
|
|
(単位:千) |
|
|||||||||||||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
販売、一般、行政 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
株に基づく報酬総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
記録しました$
BridgeBioの配当金は
2019年6月22日、2019年株式オプションとインセンティブ計画、または2019年計画を採択し、2019年6月25日に発効します。2019年には、普通株式オプションや他の株式奨励を含む株式インセンティブ奨励を付与する予定です。私たちは発行を許可された
2019年計画では、2020年1月1日から、毎年1月1日に予約と発行可能な株式数が自動的に増加することが規定されている
2019年11月13日、私たちは2019年株式誘導計画、または2019年誘導計画を採択しました。2019年インセンティブ計画は、現在BridgeBioまたはその子会社に雇用されていない高素質の潜在的幹部と従業員の就職を誘導し、普通株式オプションおよび他の株式ベースの報酬を含むBridgeBioの所有権権益を提供することを規定している。私たちは発行を許可された
2022年12月31日まで,
206
2020年株式·持分奨励交換計画(交換計画)
2020年4月22日、特定の子会社に対する2020年株式および株式奨励交換計画を完了し、これは、いくつかの発展および規制マイルストーンの実現に関連するBridgeBayes株式(普通株式、既得および未付与株式オプションおよびRSAを含む)および/または業績に基づくマイルストーンの報酬と交換する機会を提供する。交換計画は、BridgeBioグループ会社の従業員やコンサルタントに提供する奨励的な報酬構造を、わが社全体の目標の実現と一致させます。コミュニケーション計画に合わせて、2019年のA&R計画下のBridgio持分奨励金を発表しました
2020年11月18日、私たちは私たちの交換計画に基づいて子会社の株式と株式奨励を完成させた。私たちは2019年のA&R計画でBridgio持分奨励金を発表しました
ASC 718によって制御されたエンティティの未償還普通株式と配当金をBridgeBio報酬に交換する修正を評価した株式ベースの支払いそれは.ASC 718によれば、修正は、株式ベースの報酬報酬の条項または条件の変更である。会計処理を評価する時、吾らは制御された実体権益を交換前の公正価値、帰属条件及び分類を権益或いは責任奨励と見なし、交換の一部として受信したBridgeBio権益と比較して、改訂会計を適用しなければならないかどうかを決定する。会計修正を適用する際には、2020年4月22日及び11月18日に確認される適切な株式ベースの補償コスト(各修正日)を決定するために修正の種類を考慮する日付を修正した後です。
我々は、各制御エンティティの各参加者が保有する普通株式および株式報酬の総シェアを、既得または非既得にかかわらず、会計単位とする。制御されたエンティティは、各会計単位における普通株式および株式報酬は、Bridgeioの普通株式、時間ベースのホーム持分報酬、および/または業績に基づくマイルストーン報酬の組み合わせとして交換される。制御された実体持分奨励を業績に基づくマイルストーン奨励に交換する以外、他のすべての交換されたBridgio持分奨励はすべて元の帰属条件を保留する。したがって、時間に基づく持分報酬の交換による株式計算による増分報酬支出は存在しない。
交換計画が完了した時、私たちは$を確定しました
2022年12月31日まで,2021年12月31日と2020年12月31日までの年度我々は認めました$
207
表現に基づくマイルストーン賞
上記で議論した交換計画に加えて、一部の従業員やコンサルタントとパフォーマンスに基づくマイルストーン報酬スケジュールを持っており、その帰属は、様々な規制や発展マイルストーンに達しているかどうか、および最初に知られている固定金額に依存しており、それぞれまたはマイルストーンを実現する際に、個別に現金や株式の形で決済することができます。マイルストーンが達成されるか、またはマイルストーンがある場合、業績に基づくマイルストーン報酬が株式の形態で決済される場合、これらの報酬は完全に帰属するRSAの形態で満たされる。マイルストーンが達成される可能性がある時、私たちはこれや株式報酬支出があることを確認する。2022年12月31日までの年度2021年までに私たちは
BridgeBioの株式オプション付与
BridgeBioの今年度までの計画での株式オプション活動を表にまとめた2022年12月31日:
|
オプション |
|
|
重み付けの- |
|
|
重み付けの- |
|
|
骨材 |
|
||||||||
2021年12月31日現在の未返済債務 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
定期持分計画 |
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
Eidos大賞取引所 |
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
交換計画 |
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
定期持分計画 |
|
|
|
|
|
|
$ |
|
|
|
|
|
|
|
|||||
鍛えられた |
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
||||
Eidos大賞取引所 |
|
( |
) |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
交換計画 |
|
( |
) |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
キャンセルします |
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
|
||||
定期持分計画 |
|
( |
) |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
Eidos大賞取引所 |
|
( |
) |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
交換計画 |
|
( |
) |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
2022年12月31日現在の未返済債務 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
定期持分計画 |
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
Eidos大賞取引所 |
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
交換計画 |
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
2022年12月31日から行使可能 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
定期持分計画 |
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
Eidos大賞取引所 |
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
交換計画 |
|
|
|
|
|
|
$ |
|
|
|
|
|
$ |
|
|||||
従業員と非従業員に付与されたオプションは、BridgeBio社がそれぞれの付与日の普通株式価格で行使することができる。付与されたオプションにはサービス条件があり,通常一定期間以内に付与される
2022年12月31日までに年度内に授与されるオプションの加重平均授受日公正価値はい$です
上の表の2022年12月31日までの未償還と行使可能オプションの総内在価値は、BridgeBio普通株の行使価格と現在の公正価値との差額から計算される。2022年12月31日までに年度内に行使されるオプションの内的価値総額はい$です
2022年12月31日まで,2021年12月31日と2020年12月31日までの年度株式ベースの報酬支出が$であることを確認しました
208
BridgeBioの限定株式単位(RSU)
次の表にBridgeBioの今年度までの計画でのRSU活動をまとめた2022年12月31日:
|
|
未帰属の |
|
|
重み付けの- |
|
||
2021年12月31日現在の残高 |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
$ |
|
||
既得 |
|
|
( |
) |
|
$ |
|
|
キャンセルします |
|
|
( |
) |
|
$ |
|
|
2022年12月31日現在の残高 |
|
|
|
|
$ |
|
||
RSUにはサービス条件があり,一般に2年から4年の期間で付与される.
2022年12月31日まで,2021年12月31日と2020年12月31日までの年度株式ベースの報酬支出が$であることを確認しました
BridgeBioの限定株式賞(RSA)
2019年、BBP LLCは、再編および初公募前に存在するすべての帰属されていない非帰属管理インセンティブユニットおよび一般ユニットがログアウトされ、BridgeBioのRSA株式に変換される。
本年度までの計画でのRSA活動を以下の表にまとめる2022年12月31日:
|
|
未帰属の |
|
|
重み付けの- |
|
||
2021年12月31日現在の残高 |
|
|
|
|
$ |
|
||
交換計画が付与されました |
|
|
|
|
$ |
|
||
既得権益交換計画 |
|
|
( |
) |
|
$ |
|
|
既得権益−定期持分計画 |
|
|
( |
) |
|
$ |
|
|
通常の株式計画は廃止されました |
|
|
( |
) |
|
$ |
|
|
2022年12月31日現在の残高 |
|
|
|
|
$ |
|
||
ここ数年で2022年、2021年、2020年12月31日、RSAに関する株式ベースの報酬支出は以下のようになることを計画で確認しました
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
交換計画 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
他のRSA |
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
2022年12月31日まで一ドルあります
209
BridgeBio 2019年従業員株購入計画(ESPP)
2019年6月22日、2019年6月25日から施行され、2019年12月12日から改訂された2019年ESPPを採択しました。ESPPは最初に最大合計の発行を保留して許可しました
ESPPによると、条件を満たした従業員は賃金控除でBridgeBio普通株を購入することができ、価格は価格に相当する
2022年12月31日までの年度我々は認めました$
推定値仮定
我々はBlack-Scholesモデルを用いて、ESPPにおける株式オプションと株式購入権の公正価値を推定する。ブラック·スコイルズの計算には以下の重み付き平均仮定を用いた
|
|
十二月三十一日までの年度 |
|
|||||||||||||||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||||||||||||||
|
|
株式オプション |
|
|
ESPP |
|
|
株式オプション |
|
|
ESPP |
|
|
株式オプション |
|
|
ESPP |
|
||||||
予想期限(年単位) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
予想変動率 |
|
|
% |
|
|
|
|
|
|
|
|
|
|
|||||||||||
無リスク金利 |
|
|
% |
|
|
|
|
|
|
|
|
|
|
|||||||||||
配当率 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
付与された株式奨励金の加重平均公正価値 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||||
Eidosの株式賞
合併取引の前に、EidosはEidos 2016持分激励計画とEidos 2018株式オプションと激励計画に基づいて自分の株式奨励を発表し、あるいは総称してEidos計画と呼ぶ。合併取引が完了した後、私たちは発行します
Eidos計画によると、2021年1月1日から合併取引終了まで、株式ベースの報酬は重要ではないEidosは、2020年12月31日までの年間で、株式ベースの報酬支出が#ドルであることを確認した
210
この年度までに2020年12月31日、Eidos株式オプション報酬の公正価値は、付与された日にBlack-Scholesモデルを使用して以下の仮定の下で推定される
|
|
|
2020年12月31日までの年度 |
|
|
予想期限(年単位) |
|
|
|
|
|
予想変動率 |
|
|
|
% |
|
無リスク金利 |
|
|
|
% |
|
配当率 |
|
|
|
||
Eidosも同様にEidos計画に基づいてRSUを発表し,その2018年の従業員株式購入計画に基づいてESPP計画を策定した。2020年12月31日まで年度このような株式奨励項目の下での株式ベースの給与支出は重要ではない。
17.再構成、減価、および関連費用
2022年1月には、当社のビジネスプロセス、効率、コスト節約の運営変化を推進し、当社の戦略と発展計画を推進するための再編計画を実施することを約束しました。再編計画には,我々の施設の統合と合理化,開発計画の優先順位の再決定,従業員の削減が含まれている。2022年12月31日までの財政年度では、再編、減価、関連費用は#ドル
数年前に再構成された計画はなかった
|
|
2022年12月31日までの年度 |
|
|
|
|
(単位:千) |
|
|
清算、脱退、その他の関連費用 |
|
$ |
|
|
長期資産減価とログアウト |
|
|
|
|
解散費と従業員関連の費用 |
|
|
|
|
合計する |
|
$ |
|
|
211
本年度までの再編計画に関する再編負債活動を表にまとめる2022年12月31日:
|
|
2022年12月31日までの年度 |
|
|
|
|
(単位:千) |
|
|
2021年12月31日現在の残高 |
|
$ |
|
|
製造に関する最終支払い義務の再分類 |
|
|
|
|
再編成·減価·関連費用 |
|
|
|
|
現金払い |
|
|
( |
) |
非現金活動 |
|
|
( |
) |
2022年12月31日現在の残高 |
|
$ |
|
|
|
|
|
|
|
2022年12月31日までの報告 |
|
(単位:千) |
|
|
売掛金 |
|
$ |
|
|
報酬と福祉に計上すべきである |
|
|
|
|
計算すべき研究と開発負債 |
|
|
|
|
合計する |
|
$ |
|
|
18. 所得税
次の表に所得税前の純損失の構成要素を示します
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
国内では |
|
$ |
|
|
$ |
|
|
$ |
|
|||
外国.外国 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
所得税前総損失 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
あったことがある
次の表に法定連邦税率と私たちの有効税率との調節関係を示します
|
|
十二月三十一日までの年度 |
|
|
|||||||||||
|
|
2022 |
|
|
|
2021 |
|
|
|
2020 |
|
|
|||
法定連邦税率で課税する |
|
|
|
% |
|
|
|
% |
|
|
|
% |
|||
評価免除額を変更する |
|
|
( |
) |
|
|
|
( |
) |
|
|
|
( |
) |
|
研究開発単位 |
|
|
|
|
|
|
|
|
|
|
|
|
|||
株に基づく報酬 |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
||
他にも |
|
|
( |
) |
|
|
|
( |
) |
|
|
|
( |
) |
|
有効所得税率 |
|
|
|
% |
|
|
|
% |
|
|
|
% |
|||
212
私たちの繰延税金資産と負債の重要な構成要素は以下の通りです
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
繰延税金資産: |
|
|
|
|
|
|
||
営業純損失繰り越し |
|
$ |
|
|
$ |
|
||
償却する |
|
|
|
|
|
|
||
課税項目と準備金 |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
税金控除 |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
|
|
|
|
||
資産売却繰延収益 |
|
|
|
|
|
|
||
資本化研究と実験支出 |
|
|
|
|
|
|
||
利子支出を繰延する |
|
|
|
|
|
|
||
財産と設備 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
繰延税項目総資産 |
|
|
|
|
|
|
||
推定免税額を差し引く |
|
|
( |
) |
|
|
( |
) |
繰延税金資産は,推定準備後の純額を差し引く |
|
|
|
|
|
|
||
繰延税金負債: |
|
|
|
|
|
|
||
財産と設備 |
|
|
|
|
|
( |
) |
|
経営的リース使用権資産 |
|
|
( |
) |
|
|
( |
) |
未実現損益 |
|
|
( |
) |
|
|
( |
) |
繰延税金負債 |
|
|
( |
) |
|
|
( |
) |
繰延税項目純資産(負債) |
|
$ |
|
|
$ |
|
||
2022年12月31日まで、純営業損失の繰越があり、将来の課税収入を減らすことができ、もしあれば、連邦と州所得税の目的で、約Ly$
2022年12月31日まで私たちは連邦研究開発と孤児薬物信用繰越$を持っています
2022年から2017年に減税·雇用法案は174条が改正され、今年度の研究·実験(R&E)支出とソフトウェア開発コスト(総称してR&E支出)の控除が廃止され、納税者にR&E支出を資本口座に計上することが求められた
213
繰延税金資産の回収可能性が不確定な場合、推定値を計上する。評価免税額を提供する決定は、将来、繰延税金資産を利用するために十分な将来課税所得額を生成する可能性が高いかどうかを評価することに依存する。既存の証拠(私たちの歴史的経営損失と将来の損失の予測を含む)の重みに基づいて、私たちはアメリカ連邦と州の繰延税金資産に税金損失と相殺によって繰越された評価準備金を提供した。私たちは2020年に2027年手形を発行したため、一時的な差異の償却を審査する際に、私たちの既存の繰延税金資産は繰延税金負債を完全に相殺することはできないと確定した。これは税金負債#ドルの繰延につながる
純営業損失と貸記繰越の使用が重大な年次制限を受ける可能性があるのは,1986年に改正された“国内税法”第382条及び類似の国が規定する所有権変更制限によるものである。年間限度額は純営業損失や使用前の信用満期になる可能性があります。もし私たちの所有権が変化すれば、純営業損失と税収控除の使用が制限される可能性があります。
2022年12月31日まで、大量の非米国子会社の未分配収益があり、これらの収益のために非米国源泉徴収税と州税を用意していません。これらの収益は無期限に国際業務に再投資するつもりですから。もしこのような収入が分配されたら、支払われるべき税金の額はどうでもいいだろう。したがって、私たちは永久再投資を収益する非米国子会社のためにアメリカ州税と外国源泉徴収税を割り当てていない。
未確認税収割引の期初と期末金額の入金は以下の通り
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
年初残高 |
|
$ |
|
|
$ |
|
||
前年のポストを増やす |
|
|
|
|
|
|
||
前年のポストを減らす |
|
|
|
|
|
( |
) |
|
以下の項目に関連する納税額に基づく付加額 |
|
|
|
|
|
|
||
年末の残額 |
|
$ |
|
|
$ |
|
||
2022年12月31日と2021年12月31日まで私たちはあります
繰延税金資産について推定免税額を計上しておりますので、未確認の総税額割引は確認後に年間有効税率を下げることはありません。
私たちは連邦と各種所得税申告書を提出します。私たちは現在連邦や州税務審査を行っていない。すべての年は連邦と州当局の検討を受けることを開放した。
2020年3月27日、コロナウイルス援助、救済、経済安全法案またはCARE法案の署名が法律となった。CARE法案には、純営業損失の繰越期間、最低税収相殺払い戻しの代替、純利息控除制限の改正、合格改善物件の税収減価償却方法の技術修正に関する所得税条項が含まれる。CARE法案はまた、雇用主賃金税の納付を延期することを許可しており、この負債は私たちの連結財務諸表に計上されています。CARE法案の規定は私たちの連結財務諸表に実質的な影響を与えない。
214
2022年8月16日、総裁·バイデンは“2022年インフレ降下法案”、あるいは“インフレ法案”と署名し、法律にした。インフレ法には、会社が最低税率を代替することを含むいくつかの税金措置が含まれている
19.BridgeBio普通株主1株当たり純損失
BridgeBio社の普通株株主が1株当たり基本純損失を占めるべき計算方法は、BridgeBio社の普通株株主が占めるべき純損失を発行済み普通株の加重平均株式数で割ることである。BridgeBio社の普通株株主が1株当たりの純損失を占めるべき計算方法は、純損失を発行済み普通株の加重平均株式数で割って、発行すべきすべての追加普通株を加えて、他の希釈性証券の潜在的希薄普通株が発行されたと仮定する。2022年,2021年,2020年12月31日までの年度では,BridgeBio普通株株主の希釈後の1株当たり純損失は基本純損失と同じであり,潜在的な普通株は計算から除外されており,それらの影響が逆希釈されているためである。
以下の普通株等価物は、BridgeBio普通株株主の1株当たりの純損失の計算から除外され、それらを計上することは逆希釈されるからである
|
|
12月31日まで |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
帰属しないRSA |
|
|
|
|
|
|
|
|
|
|||
帰属しないRSU |
|
|
|
|
|
|
|
|
|
|||
無許可市場ベースのRSU |
|
|
|
|
|
|
|
|
||||
許可されていないパフォーマンスベースRSU |
|
|
|
|
|
|
|
|
|
|||
無許可の業績ベースのRSA |
|
|
|
|
|
|
|
|
||||
発行済みと未償還普通株式オプション |
|
|
|
|
|
|
|
|
|
|||
業績マイルストーンの見積もりに基づいて株を発行できる |
|
|
|
|
|
|
|
|
|
|||
ESPPに基づいて発行可能株式を推定する |
|
|
|
|
|
|
|
|
|
|||
2027年の債券の両替を想定する |
|
|
|
|
|
|
|
|
|
|||
2029年の債券の両替を想定する |
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|||
適用された転換率によると、私たちの2029年手形と2027年手形は、私たちの選択に応じて現金、普通株式、またはそれらの組み合わせに変換することができる。
付記9および16に記載されているように、私たちは、各規制および発展マイルストーンに適合することによって帰属される表現に基づくマイルストーン報酬スケジュールを持っており、最初に知られている固定金額は、それぞれまたはマイルストーンを達成するために、私たちが唯一選択したときに現金または株式形式で決済することができる。このような手配の普通株の同値を見積もる際には、報告日にマイルストーンが実現されたか、またはマイルストーンがあったかのように、これらの手配は権益に応じて決済されている。
215
項目9.ACCOとの変化と相違会計と財務開示に関するUNTANTS
適用されません。
第9条。制御するSとプログラム
情報開示制御とプログラムの評価
我々は、1934年の“取引法”(改正)に基づいて米国証券取引委員会(米国証券取引委員会)に提出された定期的および現在の報告で開示すべき情報が、米国証券取引委員会規則および表に指定された期間内に記録、処理、集計および報告され、そのような情報が蓄積され、必要な開示に関する決定を迅速に行うために、我々のCEOおよび最高財務官を含む、開示制御および手続きを維持することを目的としている。
我々の経営陣は、最高経営責任者及び最高財務責任者(それぞれ最高経営責任者及び最高財務官)の参加の下、2022年12月31日までの開示統制及び手続の有効性を評価し、その日までに、我々の開示統制及び手続が合理的な保証レベルで有効であると結論した。取引法規則13 a-15(E)および15 d-15(E)に定義されている用語“開示制御および手順”は、取引法に従って提出または提出された報告において開示を要求する会社の情報が米国証券取引委員会規則および表によって指定された期間にわたって記録、処理、集約および報告されることを保証するための会社の制御および他のプログラムを意味する。開示制御及び手続は、取引所法に基づいて提出又は提出された報告書に開示を要求する情報が蓄積されていることを確保し、その主要幹部及び主要財務官を含む会社管理層に適宜伝達し、開示要求に関する決定を直ちに行うことを目的としているが、制御及び手続に限定されない。経営陣は、どのような制御やプログラムが、どんなに設計や操作が良くても、その目標を実現するために合理的な保証を提供するしかないことを認識しており、管理部門は、可能な制御とプログラムのコスト-利益関係を評価する際にその判断を運用しなければならない。
財務報告の内部統制の変化
我々は財務報告の内部統制に何の変化も生じておらず、これらの変化は取引所法案規則13 a-15(D)および15 d-15(D)が要求する評価と関連しており、本10-Q表四半期報告がカバーする期間に発生し、私たちの財務報告の内部統制に大きな影響を与え、あるいは合理的な可能性が財務報告の内部統制に大きな影響を与える。
経営陣財務報告内部統制年次報告書
我々の経営陣は、取引法ルール13 a-15(F)で定義されているように、財務報告の十分な内部統制の確立と維持を担当している。我々の財務報告の内部統制は、我々の最高経営責任者と最高財務官の監督の下で設計され、財務報告の信頼性を合理的に保証し、米国公認会計原則に基づいて外部目的のために私たちの財務諸表を作成することを目的としている。
2022年12月31日現在、私たちは、CEOや最高財務責任者を含む経営陣の監督と参加の下、テレデビル委員会後援組織委員会(COSO)が発表した2013年の“内部統制-総合枠組み”下の財務報告書の有効な内部統制基準に基づいて、財務報告の内部統制の有効性を評価した。この評価に基づき、我々の経営陣は、2022年12月31日現在、財務報告に対して有効な内部統制を維持していると結論した。
徳勤会計士事務所は独立した公認会計士事務所であり、2022年12月31日までの財務報告書の内部統制に対する有効性を監査しており、これは彼らの認証報告書に記載されており、この報告書は本稿に含まれている。
216
独立公認会計士事務所報告
BridgeBio Pharma,Inc.株主と取締役会へ
財務報告の内部統制については
我々はBridgeBio Pharma,Inc.、その子会社と制御エンティティ(“当社”)の2022年12月31日までの財務報告内部統制、根拠を監査した内部統制--統合フレームワーク(2013)テレデビル委員会(COSO)が主催して組織委員会が発表した。2022年12月31日現在、当社はすべての重要な面で財務報告に対する有効な内部統制を維持しており、その根拠は内部統制--統合フレームワーク(2013)COSOから発表されます。
また、米国上場企業会計監督委員会(PCAOB)の基準に従って、当社の2022年12月31日までおよび2022年12月31日までの年度の総合財務諸表および2023年2月23日までの報告を監査し、このような財務諸表に対して保留のない意見を表明した。
意見の基礎
当社の経営陣は、財務報告を効率的に内部統制し、財務報告の内部統制の有効性を評価する責任がある。私たちの責任は私たちの監査に基づいて、会社の財務報告書の内部統制に意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。我々の監査には,財務報告の内部統制を理解すること,重大な弱点があるリスクを評価すること,評価されたリスクテストに基づいて内部統制の設計·運用有効性を評価すること,および状況下で必要と考えられる他のプロセスを実行することが含まれる。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、米国公認会計原則(“公認会計原則”)に基づいて、財務報告の信頼性と外部目的の財務諸表作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分に関する記録の保存、(2)公認された会計原則に従って財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、会社の収入および支出は会社の経営陣と取締役の許可のみに基づいて行われる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用、または処分会社の資産を防止またはタイムリーに発見する合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
/s/徳勤法律事務所
カリフォルニア州サンフランシスコ
2023年2月23日
217
プロジェクト9 B。他にもR情報
ない。
プロジェクト9 Cです。開示するウレ·レガディ検査禁止の外国司法管区
ない。
218
部分(三)
プロジェクト10.取締役、執行役員会社法人と会社管理
以下に述べる以外に、本プロジェクトに必要な情報は、2022年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出される2023年株主年次総会に提出された最終依頼書に参考に組み込まれる。
私たちは、私たちのCEO、最高財務官、最高会計官、財務総監、または同様の機能を実行する者を含む、私たちの役員、上級管理者、および従業員に適用される書面ビジネス行動および道徳的基準を通過しました。規則の最新版は当社の会社管理部に掲示されています。サイトはHttps://Investor.Bridge gio.com/静的ファイル/e 15 fa 82 d-1 c 86-4951-96 a 1-0676 f 0 a 6 bb 3 dそれは.もし私たちの最高経営責任者、最高財務責任者、最高会計責任者、財務総監、または類似の機能を実行する者、または任意の上級管理者または取締役の商業行為および道徳基準を実質的に修正または免除する場合、私たちは、このような修正または免除の性質を、私たちのウェブサイト上または現在のForm 8-K報告書に開示する。
第11項.実行VE補償
本プロジェクトが要求する情報は,2022年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出される2023年株主年次総会に提出された最終依頼書に引用される。
プロジェクト12.利益OWの特定の保証所有権所有者と経営陣および関連株主の件
本プロジェクトが要求する情報は,2022年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出される2023年株主年次総会に提出された最終依頼書に引用される。
本プロジェクトが要求する情報は,2022年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出される2023年株主年次総会に提出された最終依頼書に引用される。
プロジェクト14.依頼人ACCO料金やサービスは請求しません
本プロジェクトが要求する情報は,2022年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出される2023年株主年次総会に提出された最終依頼書に引用される。
219
部分IV.IV
プロジェクト15.展示品と国際水泳連盟NCIAL文スケジュール
登録者の次の財務諸表および付表は、本年度報告シートの第10-K第2部第8項“財務諸表および補足データ”に掲載されている
|
|
ページ |
独立公認会計士事務所報告 |
|
154 |
2022年と2021年12月31日までの連結貸借対照表 |
|
157 |
2022年12月31日までの3年間の総合経営報告書 |
|
158 |
2022年12月31日までの3年間の各年度の総合全面損失表 |
|
159 |
2022年12月31日までの3年度の償還可能非持株権益と株主権益(損失)連結報告書 |
|
160 |
2022年12月31日までの3年度の連結現金フロー表 |
|
161 |
連結財務諸表付記 |
|
163 |
これらの付表が必要である条件がないため、または必要な資料が財務諸表、財務諸表、または補足財務資料に必要な資料が表示されているため、すべての付表は省略されている。
添付ファイルインデックスに記載されている証拠は、本年度報告書の一部として提出されるか、または引用によって本年度報告書の表10 Kに組み込まれる。
220
項目16.表10-Kの概要
ない。
展示会ITS.ITS
展示品 番号をつける |
|
展示品名 |
|
表 |
|
書類番号. |
|
展示品 |
|
提出日 |
|
|
|
|
|
|
|
|
|
|
|
2.1 |
|
協定と合併計画は,期日は2020年10月5日であり,BridgeBio Pharma,Inc.,Eidos治療会社,Globe Merge Sub I,Inc.とGlobe Merger Sub II,Inc.の間で署名·実施される(合併内容はBridgeBioを引用して2020年10月6日に米国証券取引委員会の8−K表に現在報告されている添付ファイル2.1)である。 |
|
8-K |
|
001-38959 |
|
2.1 |
|
2021年1月26日 |
|
|
|
|
|
|
|
|
|
|
|
3.1 |
|
改正され再印刷された現行登録者登録証明書. |
|
8-K |
|
001-38959 |
|
3.1 |
|
July 3, 2019 |
|
|
|
|
|
|
|
|
|
|
|
3.2 |
|
改正·再改訂された登録者現行規約. |
|
S-4 |
|
333-249944 |
|
3.2 |
|
2020年11月6日 |
|
|
|
|
|
|
|
|
|
|
|
4.1 |
|
普通株式証明書サンプル。 |
|
S-1 |
|
333-231759 |
|
4.1 |
|
June 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
4.2 |
|
登録者及びその一部株主間の登録権協定フォーマットは、日付が2019年6月26日である。 |
|
S-1 |
|
333-231759 |
|
4.3 |
|
June 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
4.3 |
|
証券説明。 |
|
10-K |
|
001-38959 |
|
4.3 |
|
2022年2月25日 |
|
|
|
|
|
|
|
|
|
|
|
4.4 |
|
契約は,日付は2020年3月9日であり,BridgeBio Pharma,Inc.と米国銀行全国協会との間で受託者としている. |
|
8-K |
|
001-38959 |
|
4.1 |
|
March 10, 2020 |
|
|
|
|
|
|
|
|
|
|
|
4.5 |
|
BridgeBio Pharma,Inc.を代表するグローバルチケット形式は、2027年に満了する2.50%の変換可能優先チケット(添付ファイルAとして添付ファイル4.1に含まれる). |
|
8-K |
|
001-38959 |
|
4.2 |
|
March 10, 2020 |
|
|
|
|
|
|
|
|
|
|
|
4.6 |
|
契約は,日付は2021年1月28日であり,BridgeBio Pharma,Inc.と米国銀行全国協会の間で受託者としている. |
|
8-K |
|
001-38959 |
|
4.1 |
|
2021年1月29日 |
|
|
|
|
|
|
|
|
|
|
|
4.7 |
|
グローバルチケットの形態は、BridgeBio Pharma,Inc.を代表して、2029年に満了する2.25%変換可能優先チケット(添付ファイルAとして添付ファイル4.1として提出された契約に含まれる)。 |
|
8-K |
|
001-38959 |
|
4.2 |
|
2021年1月29日 |
|
|
|
|
|
|
|
|
|
|
|
10.1# |
|
2021年の株式オプションとインセンティブ計画とその奨励協定のフォーマットを改正し、再策定した。 |
|
10-K |
|
001-38959 |
|
10.1 |
|
2022年2月25日 |
|
|
|
|
|
|
|
|
|
|
|
10.2# |
|
2019年の従業員の株式購入計画を修正し、再策定した。 |
|
10-Q |
|
001-38959 |
|
10.1 |
|
2021年11月4日 |
|
|
|
|
|
|
|
|
|
|
|
221
10.3# |
|
上級管理職現金奨励ボーナス計画。 |
|
S-1 |
|
333-231759 |
|
10.3 |
|
June 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.4# |
|
登録者とその各取締役との間の賠償協定フォーマット。 |
|
S-1 |
|
333-231759 |
|
10.4 |
|
June 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.5# |
|
登録者とその各執行者との間の賠償協定フォーマット。 |
|
S-1 |
|
333-231759 |
|
10.5 |
|
June 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.6 |
|
BridgeBio Pharma LLCとMichael J.Harbourの間のレンタル契約は、2017年3月23日。 |
|
S-1 |
|
333-231759 |
|
10.8 |
|
May 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.7 |
|
独占(株式)協定は、Eidos治療会社とリランスタンフォード初級大学取締役会が署名し、2016年4月10日から発効し、2017年9月25日に施行された改正案第1号により改正された。 |
|
S-1 |
|
333-231759 |
|
10.9 |
|
May 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.8 |
|
QED治療会社とノワ国際製薬株式会社との間のライセンス契約は,2018年1月29日である。 |
|
S-1 |
|
333-231759 |
|
10.10 |
|
May 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.9 |
|
BridgeBio Pharma LLC,Origin Biosciences,Inc.とAlexion Pharma Holding無限会社間の資産購入契約は,2018年6月7日である。 |
|
S-1 |
|
333-231759 |
|
10.11 |
|
May 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.10 |
|
Leidos Biomedical Research,Inc.が運営し,国立癌研究所が協賛したFrederick国立癌研究実験室とTheas,Inc.の間の独占特許許可協定は,2018年12月14日である。 |
|
S-1 |
|
333-231759 |
|
10.16 |
|
May 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.11 |
|
Life Technologies CorporationとBridgeBio Services,Inc.により署名された細胞線許可プロトコルは,2018年11月15日から発効した。 |
|
S-1 |
|
333-231759 |
|
10.17 |
|
May 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.12# |
|
BridgeBio Services,Inc.とNeil Kumarの間の招待状は,2017年12月14日である. |
|
S-1 |
|
333-231759 |
|
10.19 |
|
June 11, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.13# |
|
BridgeBio Services,Inc.とBrian Stephensonの間の招待状は,2018年10月28日である. |
|
S-1 |
|
333-231759 |
|
10.20 |
|
June 11, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.14# |
|
BridgeBio Services,Inc.とCharles Homcyの間の招待状は,2019年2月20日である. |
|
S-1 |
|
333-231759 |
|
10.22 |
|
June 11, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.15# |
|
BridgeBio Services,Inc.とRichard Schellerの間の招待状は,2019年4月5日である. |
|
S-1 |
|
333-231759 |
|
10.23 |
|
June 11, 2019 |
|
|
|
|
|
|
|
|
|
|
|
222
10.16# |
|
フランク·マッコミックと登録者との間の諮問協定は,2021年1月1日から発効する。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
10.17# |
|
フランク·マッコーミックと登録者との間の諮問協定の第1号改正案は,2022年3月3日から発効する。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
10.18# |
|
フランク·マッコーミックと登録者との間の諮問協定第2号改正案は,2023年3月3日から発効する。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
10.19 |
|
登録者とその各子会社との間の税金共有協定形態。 |
|
S-1 |
|
333-231759 |
|
10.27 |
|
June 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.20 |
|
BridgeBio Pharma LLCとKKR Genetic Disorder,L.P.との間の賠償協定は,2016年3月26日であった。 |
|
S-1 |
|
333-231759 |
|
10.28 |
|
June 24, 2019 |
|
|
|
|
|
|
|
|
|
|
|
10.21 |
|
ライセンス契約は、Eidos Treateutics,Inc.とAlexion Pharma国際運営無限会社によって署名され、日付は2019年9月9日です. |
|
10-Q |
|
000-38959 |
|
10.1 |
|
2019年11月8日 |
|
|
|
|
|
|
|
|
|
|
|
10.22# |
|
BridgeBio Pharma,Inc.2019年誘導株式計画。 |
|
S-8 |
|
333-234803 |
|
99.1 |
|
2019年11月20日 |
|
|
|
|
|
|
|
|
|
|
|
10.23# |
|
BridgeBio Pharma,Inc.2019年インセンティブ持分計画下の制限株式奨励プロトコルフォーマット。 |
|
S-8 |
|
333-234803 |
|
99.2 |
|
2019年11月20日 |
|
|
|
|
|
|
|
|
|
|
|
10.24# |
|
BridgeBio Pharma,Inc.2019年インセンティブ持分計画下の非制限株式オプションプロトコルフォーマット。 |
|
S-8 |
|
333-234803 |
|
99.3 |
|
2019年11月20日 |
|
|
|
|
|
|
|
|
|
|
|
10.25# |
|
BridgeBio Pharma,Inc.2019年インセンティブ株式計画下の制限株式単位報酬プロトコルのフォーマット。 |
|
S-8 |
|
333-234803 |
|
99.4 |
|
2019年11月20日 |
|
|
|
|
|
|
|
|
|
|
|
10.26# |
|
役員報酬政策を修正して再定義した。 |
|
10-K |
|
001-38959 |
|
10.30 |
|
2022年2月25日 |
|
|
|
|
|
|
|
|
|
|
|
10.27 |
|
公開市場販売協定SM日付は2020年7月7日. |
|
S-3 |
|
333-239734 |
|
1.2 |
|
July 7, 2020 |
|
|
|
|
|
|
|
|
|
|
|
10.28 |
|
上限のコール取引の確認表. |
|
8-K |
|
001-38959 |
|
10.1 |
|
March 10, 2020 |
|
|
|
|
|
|
|
|
|
|
|
10.29 |
|
購入契約日は2021年1月25日であり、BridgeBio Pharma,Inc.とモルガン大通証券有限責任会社とみずほ証券米国有限責任会社がいくつかの初期購入者の代表として署名された。 |
|
8-K |
|
001-38959 |
|
10.1 |
|
2021年1月26日 |
|
|
|
|
|
|
|
|
|
|
|
223
10.30 |
|
上限が設定された呼取引の確認テーブル。 |
|
8-K |
|
001-38959 |
|
10.1 |
|
2021年1月29日 |
|
|
|
|
|
|
|
|
|
|
|
10.31 |
|
米国全国銀行協会は、行政代理と担保代理、融資先、登録者と登録者のある子会社との間の融資·担保協定として、2021年11月17日としている。 |
|
10-K |
|
001-38959 |
|
10.39 |
|
2022年2月25日 |
|
|
|
|
|
|
|
|
|
|
|
10.32 |
|
融資·担保協定第1修正案は、期日が2022年5月12日であり、米国銀行全国協会の後継者である米国銀行信託会社全国協会、その行政代理と担保代理の身分、その貸手、登録者と登録者のある子会社との間で合意されている。 |
|
10-Q |
|
001-38959 |
|
10.2 |
|
2022年8月4日 |
|
|
|
|
|
|
|
|
|
|
|
10.33 |
|
“融資·担保協定第二修正案”は、米国銀行信託会社全国協会(米国銀行全国協会の後継者)、融資先、登録者及び登録者のある子会社が行政代理及び担保代理として署名し、期日は2022年11月30日である。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
10.34 |
|
許可、開発、商業化協定は、期日は2022年5月11日であり、登録者、NAVE Pharma,Inc.と百時美施貴宝社が署名した。 |
|
10-Q |
|
001-38959 |
|
10.1 |
|
2022年8月4日 |
|
|
|
|
|
|
|
|
|
|
|
21 |
|
登録者の子会社リスト。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
23.1 |
|
独立公認会計士事務所が同意します。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
24 |
|
授権書(ここの署名ページを参照)。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
31.1 |
|
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
224
31.2 |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
32.1* |
|
2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条に規定されている最高経営責任者証明書。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
32.2* |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による首席財務官の認証。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
101.INS |
|
内連XBRLインスタンスドキュメント。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
101.SCH |
|
内連XBRL分類拡張アーキテクチャドキュメント。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
101.CAL |
|
内連XBRL分類拡張はリンクベース文書を計算する. |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
101.DEF |
|
内連XBRL分類拡張はLinkbase文書を定義する. |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
101.LAB |
|
内連XBRL分類拡張タグLinkbase文書. |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
101.PRE |
|
内連XBRL分類拡張プレゼンテーションLinkbaseドキュメント. |
|
— |
|
— |
|
— |
|
同封アーカイブ |
|
|
|
|
|
|
|
|
|
|
|
104 |
|
カバーインタラクションデータファイル(フォーマットは、添付ファイル101に含まれる適用分類拡張情報を含むイントラネットXBRLである)。 |
|
— |
|
— |
|
— |
|
同封アーカイブ |
* 本証明書はForm 10−K形式で本年度報告とともに提出されたものとみなされ,改正された1934年の“証券取引法”(以下,“取引法”と略す)第18節の規定により提出されたものとはみなされず,同節の責任の制約も受けない。このような証明は、参照によってそのような出願が明示的に組み込まれない限り、改正された1933年の証券法または“取引法”に基づいて提出された任意の出願に引用によって組み込まれているとはみなされない。
# 契約または任意の補償計画、契約または手配を管理することを指す。
米国証券取引委員会の規則によれば、いくつかの秘密部分(括弧および星番号で表される)は、このような情報(I)が実質的ではなく、(Ii)が登録者が個人または機密とみなすタイプであるので、本展覧会から省略されている。
225
登録する解決策
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、正式に許可された以下の署名者がその代表を代表して本報告に署名することを正式に手配した。
|
|
BridgiBio製薬会社は |
|
|
|
|
|
日付:2023年2月23日 |
|
差出人: |
/s/ニール·クマール |
|
|
|
ニール·クマール博士です |
|
|
|
取締役最高経営責任者 |
|
|
|
(首席行政主任) |
権力があるのです弁護士
このような陳述を通じて、私はすべての人が、以下に署名したすべての人がニール·クマールとブライアン·ステファソンを構成し、彼らの真の合法的な事実受権者と代理人のために、彼または彼女の名義、場所、代理、任意およびすべての身分で、Form 10-K年報の任意およびすべての修正に署名し、その年報をすべての証拠物とこれに関連する他の書類と共に証券取引委員会に提出することを知っている。上記事実受権者及び代理人に完全な権力及び権限を付与し、当該所内及び周囲で必要及び行わなければならないすべてのことを行い及び実行し、ここで上記事実を承認及び確認する権利及び代理人又はその代理人が合法的に又は手配することができるすべてのことを行うことができる。
本報告は、1934年の証券取引法の要求に基づき、以下の者によって登録者として指定日に署名された。
サイン |
|
タイトル |
|
日取り |
|
|
|
|
|
/s/ニール·クマール |
|
取締役最高経営責任者総裁 (首席行政主任) |
|
2023年2月23日 |
ニール·クマール博士です |
|
|
|
|
/s/ブライアン·ステファソン |
|
首席財務官 (首席財務官と首席会計官) |
|
2023年2月23日 |
ブライアン·ステファソンCFA博士 |
|
|
|
|
/s/エリックAguiar |
|
役員.取締役 |
|
2023年2月23日 |
エリック·アジアル医学博士 |
|
|
|
|
|
|
|
|
|
ジェニファー·E·クック |
|
役員.取締役 |
|
2023年2月23日 |
ジェニファー·E·クック |
|
|
|
|
|
|
|
|
|
/s/Douglas A.Dachille |
|
役員.取締役 |
|
2023年2月23日 |
ダグラス·A·ダジル |
|
|
|
|
|
|
|
|
|
ロナルド·J·ダニルス |
|
役員.取締役 |
|
2023年2月23日 |
ロナルド·J·ダニルス |
|
|
|
|
|
|
|
|
|
アンドレア·ジョン·エリス |
|
役員.取締役 |
|
2023年2月23日 |
アンドレア·ジョン·エリス |
|
|
|
|
|
|
|
|
|
/s/フレッド·ハサン |
|
役員.取締役 |
|
2023年2月23日 |
フレッド·ハサン |
|
|
|
|
|
|
|
|
|
226
/s/Charles Homcy |
|
役員.取締役 |
|
2023年2月23日 |
チャールズ·ホムシー |
|
|
|
|
|
|
|
|
|
/s/Andrew W.Lo |
|
役員.取締役 |
|
2023年2月23日 |
アンドリュー·W·ロー博士 |
|
|
|
|
|
|
|
|
|
/s/フランクP.McCormick |
|
役員.取締役 |
|
2023年2月23日 |
フランク·P·マッコミック |
|
|
|
|
|
|
|
|
|
/s/ジェイムズC.Momtazee |
|
役員.取締役 |
|
2023年2月23日 |
ジェームズ·C·モンタジ |
|
|
|
|
|
|
|
|
|
/s/Ali衛星 |
|
役員.取締役 |
|
2023年2月23日 |
アリサヴァト |
|
|
|
|
|
|
|
|
|
/s/Brenton L.Saunders |
|
役員.取締役 |
|
2023年2月23日 |
ブレントン·L·サンダース |
|
|
|
|
|
|
|
|
|
/s/ランデル·スコット |
|
役員.取締役 |
|
2023年2月23日 |
ランデル·スコット博士 |
|
|
|
|
|
|
|
|
|
/s/ハンナ·ヴァランディン |
|
役員.取締役 |
|
2023年2月23日 |
ハンナ·ヴァランディン医学博士 |
|
|
|
|
|
|
|
|
|
227