morf-2022123100016793632022会計年度誤りHttp://Fasb.org/us-GAAP/2022#PrepaidExpenseAndOtherAssetsCurrentHttp://Fasb.org/us-GAAP/2022#AccountsPayableAndAcruedLiabilitiesCurrentAndNonCurrentHttp://Fasb.org/us-GAAP/2022#AccountsPayableAndAcruedLiabilitiesCurrentAndNonCurrent00016793632022-01-012022-12-3100016793632022-06-30ISO 4217:ドル00016793632023-02-21Xbrli:共有00016793632022-12-3100016793632021-12-31ISO 4217:ドルXbrli:共有00016793632021-01-012021-12-3100016793632020-01-012020-12-310001679363アメリカ-アメリカ公認会計基準:普通株式メンバー2019-12-310001679363US-GAAP:AdditionalPaidInCapitalMembers2019-12-310001679363アメリカ-公認会計基準:前払いメンバーを保留2019-12-310001679363アメリカ公認会計原則:他の総合収入メンバーを累計2019-12-3100016793632019-12-310001679363US-GAAP:AdditionalPaidInCapitalMembers2020-01-012020-12-310001679363アメリカ-アメリカ公認会計基準:普通株式メンバー2020-01-012020-12-310001679363Morf:AtTheMarketOfferingMember2020-01-012020-12-310001679363アメリカ-アメリカ公認会計基準:普通株式メンバーMorf:AtTheMarketOfferingMember2020-01-012020-12-310001679363US-GAAP:AdditionalPaidInCapitalMembersMorf:AtTheMarketOfferingMember2020-01-012020-12-310001679363アメリカ公認会計原則:他の総合収入メンバーを累計2020-01-012020-12-310001679363アメリカ-公認会計基準:前払いメンバーを保留2020-01-012020-12-310001679363アメリカ-アメリカ公認会計基準:普通株式メンバー2020-12-310001679363US-GAAP:AdditionalPaidInCapitalMembers2020-12-310001679363アメリカ-公認会計基準:前払いメンバーを保留2020-12-310001679363アメリカ公認会計原則:他の総合収入メンバーを累計2020-12-3100016793632020-12-310001679363US-GAAP:AdditionalPaidInCapitalMembers2021-01-012021-12-310001679363アメリカ-アメリカ公認会計基準:普通株式メンバー2021-01-012021-12-310001679363Morf:AtTheMarketOfferingMember2021-01-012021-12-310001679363アメリカ-アメリカ公認会計基準:普通株式メンバーMorf:AtTheMarketOfferingMember2021-01-012021-12-310001679363US-GAAP:AdditionalPaidInCapitalMembersMorf:AtTheMarketOfferingMember2021-01-012021-12-310001679363Morf:Second DaryOfferingMember2021-01-012021-12-310001679363Morf:Second DaryOfferingMemberアメリカ-アメリカ公認会計基準:普通株式メンバー2021-01-012021-12-310001679363US-GAAP:AdditionalPaidInCapitalMembersMorf:Second DaryOfferingMember2021-01-012021-12-310001679363アメリカ公認会計原則:他の総合収入メンバーを累計2021-01-012021-12-310001679363アメリカ-公認会計基準:前払いメンバーを保留2021-01-012021-12-310001679363アメリカ-アメリカ公認会計基準:普通株式メンバー2021-12-310001679363US-GAAP:AdditionalPaidInCapitalMembers2021-12-310001679363アメリカ-公認会計基準:前払いメンバーを保留2021-12-310001679363アメリカ公認会計原則:他の総合収入メンバーを累計2021-12-310001679363US-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001679363アメリカ-アメリカ公認会計基準:普通株式メンバー2022-01-012022-12-310001679363Morf:AtTheMarketOfferingMember2022-01-012022-12-310001679363アメリカ-アメリカ公認会計基準:普通株式メンバーMorf:AtTheMarketOfferingMember2022-01-012022-12-310001679363US-GAAP:AdditionalPaidInCapitalMembersMorf:AtTheMarketOfferingMember2022-01-012022-12-310001679363アメリカ公認会計原則:他の総合収入メンバーを累計2022-01-012022-12-310001679363アメリカ-公認会計基準:前払いメンバーを保留2022-01-012022-12-310001679363アメリカ-アメリカ公認会計基準:普通株式メンバー2022-12-310001679363US-GAAP:AdditionalPaidInCapitalMembers2022-12-310001679363アメリカ-公認会計基準:前払いメンバーを保留2022-12-310001679363アメリカ公認会計原則:他の総合収入メンバーを累計2022-12-310001679363Morf:Second DaryOfferingMember2022-01-012022-12-310001679363Morf:Second DaryOfferingMember2020-01-012020-12-310001679363Morf:AtMarketOfferingProgramメンバMorf:OpenMarketSaleAgreementメンバ2020-07-012020-07-31Xbrli:純0001679363Morf:AtMarketOfferingProgramメンバMorf:OpenMarketSaleAgreementメンバ2021-08-112021-08-110001679363Morf:AtMarketOfferingProgramメンバ2022-01-012022-12-310001679363Morf:AtMarketOfferingProgramメンバ2022-12-310001679363Morf:FollowOnPublicOfferingMember2021-03-012021-03-310001679363Morf:FollowOnPublicOfferingMember2022-03-310001679363SRT:最小メンバ数2022-12-3100016793632021-08-310001679363米国-GAAP:デバイス構成員2022-01-012022-12-310001679363Morf:ComputerEquipmentandSoftwareMembersSRT:最小メンバ数2022-01-012022-12-310001679363Morf:ComputerEquipmentandSoftwareMembersSRT:最大メンバ数2022-01-012022-12-31Morf:セグメントMorf:プロトコル0001679363アメリカ公認会計基準:副次的事件メンバーUS-GAAP:PrivatePlacementMembers2023-02-132023-02-130001679363アメリカ公認会計基準:副次的事件メンバーUS-GAAP:PrivatePlacementMembers2023-02-130001679363アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001679363アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001679363アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001679363アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値投入レベル3メンバー2022-12-310001679363アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001679363アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001679363アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001679363アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値投入レベル3メンバー2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するMorf:アメリカ政府スポンジ企業のセキュリティメンバー2022-12-310001679363アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するMorf:アメリカ政府スポンジ企業のセキュリティメンバー2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバーMorf:アメリカ政府スポンジ企業のセキュリティメンバー2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値投入レベル3メンバーMorf:アメリカ政府スポンジ企業のセキュリティメンバー2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定する米国-GAAP:ビジネス紙のメンバー2022-12-310001679363アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する米国-GAAP:ビジネス紙のメンバー2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー米国-GAAP:ビジネス紙のメンバー2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値投入レベル3メンバー米国-GAAP:ビジネス紙のメンバー2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定する米国-公認会計基準:社債証券メンバー2022-12-310001679363アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する米国-公認会計基準:社債証券メンバー2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー米国-公認会計基準:社債証券メンバー2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値投入レベル3メンバー米国-公認会計基準:社債証券メンバー2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001679363アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値投入レベル3メンバー2022-12-310001679363アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001679363アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001679363アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2021-12-310001679363アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値投入レベル3メンバー2021-12-310001679363アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001679363アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001679363アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2021-12-310001679363アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値投入レベル3メンバー2021-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定する米国-GAAP:ビジネス紙のメンバー2021-12-310001679363アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する米国-GAAP:ビジネス紙のメンバー2021-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー米国-GAAP:ビジネス紙のメンバー2021-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値投入レベル3メンバー米国-GAAP:ビジネス紙のメンバー2021-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定する米国-公認会計基準:社債証券メンバー2021-12-310001679363アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する米国-公認会計基準:社債証券メンバー2021-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー米国-公認会計基準:社債証券メンバー2021-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値投入レベル3メンバー米国-公認会計基準:社債証券メンバー2021-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001679363アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2021-12-310001679363アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値投入レベル3メンバー2021-12-310001679363アメリカ-公認会計基準:アメリカ証券メンバー2022-12-310001679363Morf:アメリカ政府スポンジ企業のセキュリティメンバー2022-12-310001679363米国-GAAP:ビジネス紙のメンバー2022-12-310001679363アメリカ-公認会計基準:会社債務証券メンバー2022-12-310001679363アメリカ-公認会計基準:アメリカ証券メンバー2021-12-310001679363米国-GAAP:ビジネス紙のメンバー2021-12-310001679363アメリカ-公認会計基準:会社債務証券メンバー2021-12-310001679363米国-GAAP:デバイス構成員2022-12-310001679363米国-GAAP:デバイス構成員2021-12-310001679363Morf:ComputerEquipmentandSoftwareMembers2022-12-310001679363Morf:ComputerEquipmentandSoftwareMembers2021-12-310001679363アメリカ-公認会計基準:リース改善メンバー2022-12-310001679363アメリカ-公認会計基準:リース改善メンバー2021-12-310001679363オフィスと実験室空間のメンバー2021-08-31Utr:SQFTMorf:投票0001679363住宅ローン:2900元株式計画メンバー2022-01-310001679363住宅ローン:2900元株式計画メンバー2022-12-310001679363米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001679363米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001679363米国-公認会計基準:従業員株式オプションメンバー2020-01-012020-12-310001679363米国-公認会計基準:制限された株式メンバー2022-01-012022-12-310001679363米国-公認会計基準:制限された株式メンバー2021-01-012021-12-310001679363米国-公認会計基準:制限された株式メンバー2020-01-012020-12-310001679363米国-GAAP:制限株式単位RSUメンバー2022-01-012022-12-310001679363米国-GAAP:制限株式単位RSUメンバー2021-01-012021-12-310001679363米国-GAAP:制限株式単位RSUメンバー2020-01-012020-12-310001679363アメリカ公認会計基準:従業員ストックメンバー2022-01-012022-12-310001679363アメリカ公認会計基準:従業員ストックメンバー2021-01-012021-12-310001679363アメリカ公認会計基準:従業員ストックメンバー2020-01-012020-12-310001679363米国-公認会計基準:研究·開発費メンバー2022-01-012022-12-310001679363米国-公認会計基準:研究·開発費メンバー2021-01-012021-12-310001679363米国-公認会計基準:研究·開発費メンバー2020-01-012020-12-310001679363アメリカ-公認会計基準:一般と行政費用メンバー2022-01-012022-12-310001679363アメリカ-公認会計基準:一般と行政費用メンバー2021-01-012021-12-310001679363アメリカ-公認会計基準:一般と行政費用メンバー2020-01-012020-12-310001679363住宅ローン:2900元株式計画メンバー米国-公認会計基準:制限された株式メンバー2021-12-310001679363住宅ローン:2900元株式計画メンバー米国-公認会計基準:制限された株式メンバー2022-01-012022-12-310001679363住宅ローン:2900元株式計画メンバー米国-公認会計基準:制限された株式メンバー2022-12-310001679363米国-公認会計基準:制限された株式メンバー2022-12-310001679363米国-公認会計基準:制限された株式メンバー2021-12-310001679363米国-公認会計基準:制限された株式メンバー2020-12-310001679363住宅ローン:2900元株式計画メンバー米国-GAAP:制限株式単位RSUメンバー2021-12-310001679363住宅ローン:2900元株式計画メンバー米国-GAAP:制限株式単位RSUメンバー2022-01-012022-12-310001679363住宅ローン:2900元株式計画メンバー米国-GAAP:制限株式単位RSUメンバー2022-12-310001679363米国-GAAP:制限株式単位RSUメンバー2022-12-310001679363米国-GAAP:制限株式単位RSUメンバー2021-12-310001679363米国-公認会計基準:従業員株式オプションメンバー2021-12-310001679363米国-公認会計基準:従業員株式オプションメンバー2022-12-310001679363住宅ローン:2900元株式計画メンバー米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001679363住宅ローン:2900元株式計画メンバー米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001679363住宅ローン:2900元株式計画メンバー米国-公認会計基準:従業員株式オプションメンバー2020-01-012020-12-310001679363Morf:EmployeStockPurche ePlanMember2019-12-310001679363Morf:EmployeStockPurche ePlanMember2021-01-010001679363SRT:最大メンバ数Morf:EmployeStockPurche ePlanMember2022-01-012022-12-310001679363Morf:EmployeStockPurche ePlanMember2022-01-012022-12-310001679363米国-GAAP:国内/地域メンバー2022-12-310001679363アメリカ-公認会計基準:州と地方法律法規のメンバー2021-12-310001679363米国-GAAP:国内/地域メンバー2022-01-012022-12-310001679363アメリカ-公認会計基準:州と地方法律法規のメンバー2021-01-012021-12-310001679363アメリカ-公認会計基準:州と地方法律法規のメンバー2022-12-310001679363アメリカ公認会計基準:ライセンスメンバーMorf:AbbVieMember2018-10-012018-10-310001679363アメリカ公認会計基準:ライセンスメンバーMorf:AbbVieMember2018-10-31Morf:計画Morf:オプション0001679363アメリカ公認会計基準:ライセンスメンバーMorf:AbbVieMember2022-12-310001679363アメリカ公認会計基準:ライセンスメンバーMorf:AbbVieMember2018-12-310001679363アメリカ公認会計基準:ライセンスメンバーMorf:AbbVieMember2020-08-252020-08-250001679363アメリカ公認会計基準:ライセンスメンバーMorf:AbbVieMember2020-07-012020-09-300001679363アメリカ公認会計基準:ライセンスメンバーMorf:AbbVieMember2022-01-012022-12-310001679363アメリカ公認会計基準:ライセンスメンバーMorf:AbbVieMember2021-01-012021-12-310001679363アメリカ公認会計基準:ライセンスメンバーMorf:AbbVieMember2020-01-012020-12-310001679363Morf:AbbVieMember2021-12-310001679363担保ローン:前置支払収入メンバーMorf:JanssenPharmticalsIncMember2019-02-012019-02-28Morf:目標0001679363担保ローン:前置支払収入メンバーMorf:JanssenPharmticalsIncMember2019-02-280001679363担保ローン:前置支払収入メンバーMorf:JanssenPharmticalsIncMember2021-12-310001679363Morf:JanssenPharmticalsIncMember2021-01-012021-12-310001679363担保ローン:前置支払収入メンバーMorf:JanssenPharmticalsIncMember2022-12-310001679363Morf:JanssenPharmticalsIncMemberMorf:前期支払収入の最初の2つの計画メンバー2022-01-012022-12-310001679363Morf:JanssenPharmticalsIncMemberMorf:前期支払収入の最初の2つの計画メンバー2022-12-310001679363Morf:前置支払い収入第3計画のメンバーMorf:JanssenPharmticalsIncMember2022-01-012022-12-310001679363担保ローン:前置支払収入メンバーMorf:JanssenPharmticalsIncMember2022-01-012022-12-310001679363Morf:ReimbursementRevenueMemberMorf:JanssenPharmticalsIncMember2022-01-012022-12-310001679363Morf:ReimbursementRevenueMemberMorf:JanssenPharmticalsIncMember2021-01-012021-12-310001679363Morf:ReimbursementRevenueMemberMorf:JanssenPharmticalsIncMember2020-01-012020-12-310001679363担保ローン:前置支払収入メンバーMorf:JanssenPharmticalsIncMember2021-01-012021-12-310001679363担保ローン:前置支払収入メンバーMorf:JanssenPharmticalsIncMember2020-01-012020-12-310001679363Morf:JanssenPharmticalsIncMemberアメリカ公認会計基準:ライセンスメンバー2022-01-012022-12-310001679363Morf:JanssenPharmticalsIncMemberアメリカ公認会計基準:ライセンスメンバー2021-01-012021-12-310001679363Morf:JanssenPharmticalsIncMemberアメリカ公認会計基準:ライセンスメンバー2020-01-012020-12-310001679363担保ローン:前置支払収入メンバーMorf:JanssenPharmticalsIncMember2022-10-012022-12-310001679363Morf:JanssenPharmticalsIncMemberアメリカ公認会計基準:ライセンスメンバー2022-12-310001679363Morf:JanssenPharmticalsIncMemberアメリカ公認会計基準:ライセンスメンバー2021-12-310001679363米国-公認会計基準:制限された株式メンバー2022-01-012022-12-310001679363米国-公認会計基準:制限された株式メンバー2021-01-012021-12-310001679363米国-公認会計基準:制限された株式メンバー2020-01-012020-12-310001679363米国-GAAP:制限株式単位RSUメンバー2022-01-012022-12-310001679363米国-GAAP:制限株式単位RSUメンバー2021-01-012021-12-310001679363米国-GAAP:制限株式単位RSUメンバー2020-01-012020-12-310001679363米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001679363米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001679363米国-公認会計基準:従業員株式オプションメンバー2020-01-012020-12-310001679363Morf:EmployeStockPurche ePlanMember2022-01-012022-12-31
アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表10-K
| | | | | |
| (マーク1) | |
| ☒ | 1934年証券取引法第13条又は第15条に基づいて提出された年次報告 |
本財政年度末まで十二月三十一日, 2022
あるいは…。 | | | | | |
| ☐ | 1934年証券取引法第13条又は15条に基づいて提出された移行報告 |
そこからの過渡期について
依頼書類番号: 001-38940
モニック·ホールディングス
(登録者の正確な氏名はその定款に記載)
| | | | | |
デラウェア州 (明またはその他の司法管轄権 会社や組織) | 47-3878772 (税務署の雇用主 識別番号) |
門楼通り35号, A2 ウォルザム, 体積量 (主な行政事務室住所) |
02451 (郵便番号) |
登録者の電話番号は市外局番を含んでいます (781) 996-0955
取引法第12条(B)に基づいて登録された証券:
| | | | | | | | |
| クラスごとのタイトル | 取引コード | 登録された各取引所の名称 |
| 普通株は、1株当たり0.0001ドルの価値があります | モルフ | ナスダック世界市場 |
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。 はい、そうです ☒ No ☐
登録者がこの法第13節または第15節(D)節に基づいて報告を提出する必要がないかどうかを再選択マークで示す。 Yes ☐ 違います。 ☒
再選択マークは、登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13条または15(D)節に提出されたすべての報告書を提出したかどうか、および(2)過去90日以内にそのような提出要件に適合しているかどうかを示すはい、そうです☒違います☐
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各対話データファイルを電子的に提出したか否かを示すはい、そうです☒違います☐
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
| | | | | |
| 大型加速ファイルサーバ☐ | 加速ファイルサーバ☐ |
非加速ファイルサーバ ☒ | 規模の小さい報告会社☒ |
| 新興成長型会社☐ |
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する☐
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる☐
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する ☐
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す ☐
登録者が空殻会社であるか否かをチェックマークで表す(取引法規則12 b−2で定義されるように)。はい、そうです☐ No ☒
2022年6月30日(登録者が最近完了した第2四半期の最終営業日)のナスダック世界市場における報告によると、非関連会社が保有する登録者普通株の総時価(約)は$である559,358,817.
2023年2月21日現在、登録者普通株の流通株数は39,534,138.
引用で編入された書類
登録者は、その2023年株主総会で提出される最終委託書の内容の一部を参照して本文書の第3部に組み込む。このような依頼書は、本年度報告書に含まれる10-K表に含まれる財政年度終了後120日以内に米国証券取引委員会に提出される。
カタログ
| | | | | |
| ページ |
| |
第1部 | 2 |
前向きに陳述する | 2 |
プロジェクト1.ビジネス | 2 |
第1 A項。リスク要因 | 32 |
項目1 B。未解決従業員意見 | 76 |
項目2.財産 | 76 |
項目3.法的訴訟 | 76 |
プロジェクト4.鉱山安全情報開示 | 76 |
第II部 | 77 |
項目5.登録者普通株式市場、関連株主事項、発行者による株式証券の購入 | 77 |
プロジェクト6.保留 | 77 |
プロジェクト7.経営陣の財務状況と経営成果の検討と分析 | 78 |
項目8.財務諸表と補足データ | 92 |
項目9.会計·財務開示面の変更と会計士との相違 | 93 |
第9条。制御とプログラム | 93 |
プロジェクト9 B。その他の情報 | 93 |
プロジェクト9 Cです。検査妨害に関する外国司法管区の開示 | 93 |
第三部 | 94 |
プロジェクト10.取締役、上級管理者、および企業管理 | 94 |
プロジェクト11.役員報酬 | 94 |
プロジェクト12.特定の実益所有者の保証所有権及び管理職及び株主に関する事項 | 94 |
第13項:特定の関係及び関連取引、並びに取締役独立性 | 94 |
プロジェクト14.主な会計費用とサービス | 94 |
第IV部 | 95 |
プロジェクト15.展示品 | 95 |
項目16.表格10-Kの概要 | 97 |
サイン | 98 |
第1部
前向きに述べる。
このForm 10−K年次報告又は年次報告書には、1934年“証券取引法”(改正)第21 E節又は“取引法”及び改正された1933年“証券法”第27 A条又は“証券法”の意味に適合する前向きな陳述が含まれている。歴史的事実に関する陳述を除いて、本年度報告に含まれるすべての陳述は、会社の将来の経営業績と財務状況、業務戦略、市場規模、潜在的成長機会、非臨床および臨床開発活動、候補製品の有効性と安全性、候補製品が受信したいくつかの指定された利益を維持し、認識する能力、非臨床研究および臨床試験の時間と結果、第三者との協力および潜在的な規制指定、候補製品の承認および商業化の受信と時間に関する声明を含み、いずれも前向き声明である。すべての前向き表現がこれらの識別語を含むわけではないが、“信じる”、“可能”、“推定”、“継続”、“予想”、“意図”、“可能”、“将”、“プロジェクト”、“計画”、“予想”、および未来のイベントまたは結果の不確実性を表す同様の表現は、前向き表現を識別することを意図している。
これらの展望的な陳述は主に私たちの現在の未来の事件と傾向の予想と予測に基づいており、私たちはこれらの事件と傾向が私たちの財務状況、運営結果、業務戦略、短期と長期業務運営、目標と財務需要に影響を与える可能性があると考えている。これらの展望的陳述は、“リスク要因”および本年度報告の他の部分で説明されたリスク、不確実性、および仮説を含む多くのリスク、不確実性および仮説の影響を受ける。また、私たちは競争が激しく、めまぐるしく変化する環境の中で運営しており、新たなリスクが時々発生している。私たちの経営陣はすべてのリスクを予測することはできませんし、すべての要素が私たちの業務に与える影響を評価することもできません。あるいは任意の要素や要素の組み合わせは、実際の結果が私たちが行う可能性のある任意の前向きな陳述に含まれる結果と大きく異なる程度をもたらす可能性があります。これらのリスク、不確実性、および実現されていない仮定は、本年度報告で議論または提案された前向きイベントおよび状況が発生しない可能性があり、実際の結果は前向き陳述とは大きく異なる可能性があることを意味する可能性がある。
あなたは未来の事件の予測として前向きな陳述に依存してはいけない。私たちは展望性陳述が作成時に合理的であると考えているが、私たちは展望性陳述に反映された未来の結果、活動レベル、業績或いは事件、仮説と状況が実現或いは発生することを保証できない。私たちは、これらの陳述が法的要件を満たさない限り、実際の結果または私たちが予想する変化と一致するように、本年度報告書の発行日後に、任意の理由で任意の前向き陳述を公開更新する義務はありません。本年度報告書を読む時、あなたは私たちの実際の未来の結果、活動レベル、業績、事件と状況が私たちの前向きな陳述とは大きく異なるかもしれないことを理解しなければなりません。
文意が別に指摘されているほか、本年度報告で使用される用語“私たち”、“会社”は、他の説明がない限り、デラウェア州モルフェイホールディングスおよびその子会社を全体として指す。“モルフィー”、“モルフェイ療法”、モルフェイ標識、およびすべての製品名は、私たちの一般法商標です。本年度報告書には,他社の他の商号,商標,サービスマークが含まれており,これらはそれぞれの所有者の財産である。私たちは、これらの他社との関係、またはこれらの他の会社の私たちへの支援または賛助を示唆するために、他社の商標、商標またはサービスマークを使用または展示するつもりはありません。
プロジェクト1.ビジネス
概要
我々はバイオ製薬会社であり,インテグリンに対する独自の知見を潜在的に一流の経口小分子インテグリン療法のパイプラインの発見と開発に応用している。インテグリンは1種類の標的であり、多種の承認された重ポンド爆弾薬物を有し、自己免疫、心血管と代謝性疾患、繊維化と癌を含む深刻な慢性疾患の治療に用いられる。これまで,経口小分子インテグリン療法は米国食品·薬物管理局(FDA)の承認を得ていない。それにもかかわらず、私たちの独特なプラットフォームは特定のインテグリン標的に対して確実に高品質の経口分子を産生する潜在力を放出できると信じている。著者らのインテグリン構造と機能に対する独特な理解を利用してMorphyインテグリン技術プラットフォーム或いはMINTプラットフォームを作成し、新しい候補製品を開発し、経口投与に必要な効力、高選択性と薬学特性を実現することを目的とした。我々の主要候補製品Morf−057,炎症に関与するα4β7特異的インテグリン阻害剤を含む我々のチューブを進めており,炎症性腸疾患の治療の臨床開発に用いられている。我々は2020年7月にMorf−057の新薬研究申請を提出したか,あるいはIND,FDAがINDによる研究を2020年8月に継続することを許可した。2020年9月,われわれは健康ボランティアの中でMorf−057の第1段階臨床試験を開始し,われわれの臨床計画を確立し,われわれのIBD第2段階計画のための投与量を選択し,最初の重点は潰瘍性大腸炎(UC)であった
Morf-057第1段階研究は、単一用量上昇用量(SAD)、多用量上昇用量(MAD)および食物効果(FE)を含み、Morf-057安全性、薬物動態学および薬効学的行列を評価する。健康な対象は、3:1にランダムに分割され、SAD群において単回用量の25、50、100、150および400 mgのMorf−057または一致プラセボを受け、狂ったキューで1日2回またはBIDの25、50および100 mgのMorf−057または一致プラセボを14日間受け入れた。条件に適合した健常者67名が研究に参加し,そのうち36名がSAD群,9名がFE群,22名がMAD行列であった。66人の被験者が研究治療を完了し、50 mg Bid狂ったプラトゥーンのうちの1人が個人的な理由で同意を撤回した。
MORF−057はすべてのキューで耐性が良好であり,セキュリティ信号は認められなかった。MORF−057は良好なPKプロファイルを示し,その中で目標交戦が確認され,明確なPKとPD関係が確立された。Morf−057は迅速に吸収され,全身曝露はほぼ用量割合で増加することが確認された。食物効果研究では,高脂肪食服用時に曝露量はやや減少したが,谷濃度には影響が認められなかった。その結果,食物摂取量は低地Morf−057レベルに影響を与えず,計画した患者研究では,食物を考慮せずにMorf−057を使用することが可能であった。
α4β7受容体占有率(RO)は用量と研究日数の増加とともに増加し、14日目に飽和(>99%RO)に達した。100 mgのBID行列では、α4β7受容体は飽和した(平均RO>99%)。特定のα4β7高発現免疫細胞群を含むバイオマーカーの用量と時間依存の変化が観察され,Morf−057の生物学的証拠が増加した。これらの変化は,IBDの治療に承認された抗体薬vedolizumabを含む他のインテグリン阻害剤の報告と一致している。
別のMORF-057第1段階研究では、被験者は1日2回服用し、毎回の用量は200 mgであり、プラセボと比較して、100 BIDまたは200 mg BIDを服用した被験者はα4β7受容体飽和を示し、循環中央記憶、効果記憶Tリンパ細胞およびスイッチ記憶Bリンパ細胞数は明らかに増加した。25 mgと50ミリグラムBIDの探索的用量では,キーPD測定でも方向性が増加する傾向が認められた。全用量の耐性は良好であり,安全信号は認められず,良好なPK曲線が認められた。Morf−057は単剤200 mgMorf−057および200 mgBidの14日間でα4β7受容体がCにあることを示した低い谷それは.リンパ球亜群とCCR 9 mRNAの統計学的有意な変化が認められ,これは先行研究と一致している。
第1段階研究の結果,2022年3月にMorf−057の第2段階臨床試験を開始した。Emerald-1(Morf-057-201)は、中重度UCを有する成人に対するMorf-057の有効性、安全性と耐性を評価するための開放ラベルのマルチセンター2 a期試験であり、2022年10月に方向性登録を完了し、30名の患者がこの研究に参加した。また,方向性登録完了時にスクリーニングを受けている患者が研究に組み込まれ,計35名の患者が主要キューに登録されている。これまでvedolizumabで治療に失敗した10名までの患者への探索的キューの募集が行われている。Emerald−1研究に参加した患者は米国とポーランドの地点でBid 100 mgの治療を受けている。試験の主な終点はロバツ組織病理学的指数(RHI)の変化であり,ベースラインと比較して12週における潰瘍性大腸炎の組織学的疾患活動を測定できる検証ツールである。その後,患者は追加40週間の維持治療を継続し,52週間の評価を行う。Emerald-1研究中の副次的およびその他の結果指標は修正されたメオ臨床採点、安全性、PKパラメータと重要なPD指標を含み、α4β7受容体占有率とリンパ細胞亜群輸送を含む。Emerald−1ステージ2 a試験からの主要終点データは,中から重度UC患者のMorf−057を対象とした2023年第2四半期に報告されると予想される。Emerald-2(Morf-057-202)はMorf-057の全世界2 b期無作為対照試験であり、2022年11月に患者への投与量を開始した。Emerald-2研究に登録された患者は、ランダムに3つの有効群またはプラセボ群に分けられた:100 mg Bid、200 mg Bid、Qd(1日に1回)群, あるいはプラセボ群は,12週の誘導段階後に交差してMorf−057に入る。試験の主な終点は臨床緩解率であり,12週間で修正したMayoスコアで測定した。副次的な終点は、RHI、PK、およびPD対策の変化、およびセキュリティパラメータを含む。12週の誘導期の後,40週間の維持期に入る。2025年上半期にMorf−057が中~重度UC患者で行ったEmerald−2期2 b試験の主な終点を完成させると信じている。Morf-057の進展を考慮して、著者らは2022年に第二世代開発候補薬物の前IND段階を停止し、重点的にMorf-057の第二段階臨床計画による進展である。著者らは引き続き私たちのα4β7製品の組み合わせを拡大し、未来の臨床研究のために新世代α4β7小分子の候補製品を開発した。
2022年6月、私たちは便宜上、私たちがエバーヴィバイオテクノロジー株式会社またはエバービーと2018年10月に締結した協力またはエバービー協定が終了したと報告した。今回の協力のテーマは,特発性肺線維化を含む線維化疾患の治療のために開発した選択的経口αとβ6特異的インテグリン阻害剤,およびAbbVieと連携した他の適応である。2020年8月,AbbVieは我々のαvβ6特定インテグリン阻害剤計画の選択権を許可し,一度に2000万ドルを支払った。AbbVieは,臨床前試験でαvβ6を介した安全シグナルを標的とする疑わしいシグナルが観察されたため,αvβ6特異的インテグリン阻害剤を選択的に経口投与することを意図していないと述べ,詳細な情報は2022年12月の毒理学学会誌に発表された。AbbVie協定は2022年12月に終了したので、私たちはこの合意に従って支払われた追加金を受け取ることはできないだろう
2019年2月、私たちはJanssen PharmPharmticals、Inc.またはJanssenと合意し、新しいインテグリン療法、またはJanssenプロトコルを開発した。2021年2月,Janssenは500万ドルを支払い,特定のインテグリン標的に対する抗体活性化剤を発見するための第3の研究計画を開始した。2021年12月、Janssenは、最初の2つのインテグリン目標に対して選択権を行使しないことを決定し、インテグリン抗体活性化剤の潜在的開発を含む第3のインテグリン研究計画に重点を置いたことを通知した。最初の2つのインテグリンの標的は rJanssenに興味のある特定の疾患の標的検証が乏しいため,この報告を受けた。2023年1月、私たちはJanssenから通知を受け、彼らは合意を終了することを選択した。終了は2023年1月13日から60日以内に発効しますので、ヤンソン協定の下での追加支払いは受けないと予想されますが、終了前に2023年に完了した研究サービスに関連する潜在的な名目金額は除外されます。
われわれの主要目標Morf−057に加えて,われわれのMINTプラットフォームを用いて様々な治療分野で広範な臨床前計画パイプラインを進めており,これらすべての計画はインテグリン受容体の抑制や活性化の潜在力を利用することを目的としている。他の全額所有プロジェクトは発見の先行最適化段階に近づいているか、または段階に入っている。われわれは2021年4月の米国癌研究協会年次総会でわれわれのαvβ8計画の積極的な臨床前データを示し,チェックポイント難治性癌モデルの抗腫瘍活性を示し,我々のαvβ8計画の推進に専念している。肺動脈高圧(PAH)を含む肺動脈高圧疾患や他の治療分野におけるフィブロネクチン統合標的に対する追加研究段階計画も行っている。インテグリンは細胞増殖,生存,肥大成長,線維化を促進することが知られており,これらはPAH進展の重要な因子である。
私たちはハーバード医学院とボストン児童病院のティモシー·A·スプリンガー博士が2014年に創立したもので、世界的に有名な免疫学者と生物物理学者で、インテグリンを発見した。彼は疾患活動の調節におけるインテグリン配座の重要性を実証した。今日、児童医学センター会社或いはシュプリンガー実験室の独占許可により、著者らのMINTプラットフォームはこれらの初歩的な知見及び統合素コンホメーション、親和性調節と動力学に関する著者らの独自知識によって支持を提供した。つまり,新たな候補製品を発見し,疾患特定のインテグリンコンホメーションを結合して非疾患の生理状態に回復させることができる。
著者らは経験豊富な管理チーム、取締役会と科学顧問委員会を結成し、インテグリン治療に関する専門知識を持っている。彼らは共同で治療薬の発見、開発と商業化の豊富な経験をもたらし、ArQule社、Biogen社、Cubist製薬会社、ジョンソン、ファイザー、Pharmacia社、武田製薬有限会社とTheravance Biophma社で働いたことがある。
設立から2022年12月31日まで、私たちは主に株式を発行することで、私たちの転換可能な優先株証券、私たちの初公募株、2021年3月の私たちの公募株、ATMによる私たちの普通株の売却、私たちの協力合意に基づいて受け取った支払いを含む合計約7.43億ドルの総収益を集めました。
私たちの戦略
我々の目標は,我々のMINTプラットフォームを用いて潜在的な一流経口小分子インテグリン療法を発見·開発することである。私たちのプラットフォームは様々な深刻な慢性病を患っている患者の治療モードを変える可能性があると信じている。この目標を達成するために策定された業務戦略の主な原則は、
•自己免疫、心血管と代謝性疾患、繊維化と癌を含む経口インテグリン調節剤を深刻な慢性疾患の新しい治療法として確立した。我々は、インテグリン調節不全を治療し、経口治療の潜在的利点が存在する疾患を治療するために、我々のMINTプラットフォームを使用して、新しい経口インテグリン標的治療薬を作成している。臨床終点とバイオマーカーが確立された疾患の初歩的な開発を優先し,臨床概念検証をより迅速に実現できると信じている。我々は,α4β7特異的インテグリン阻害剤である先行全資候補製品MORF−057を進めており,IBD治療の臨床開発を行っている。
•独自のミントプラットフォームと知識ベースを利用して、私たちの新しい統合素療法ルートを拡大します。私たちの全面的なミントプラットフォームは、私たちの開発能力に加えて、インテグリン失調による慢性疾患に対する一連の新しい候補製品を構築することができる。私たちは4つのインテグリン亜群の治療潜在力を放出することで、私たちのルートを拡大し、高度に満足されていない医療需要の疾病を治療し、私たちの現在の候補製品を新しい適応に拡張することが可能である。
•私たちのミントプラットフォームで革新を推進し続けている。著者らは引き続きプラットフォームの革新を開発と統合することを通じて、インテグリン薬物領域における著者らのリード的な地位を拡大するつもりであり、これらの革新は更に著者らの口腔インテグリン計画の潜在的な治療範囲を拡大することができる。著者らの主要な重点領域は技術投資を通じて結晶学方面の構造知識の広さを繰り返し拡大し、著者らのコンホメーション特異性統合素化学型バンクを拡大し、インテグリン疾患生物学に対する基本的な理解を深めることを含む。私たちが知識ベースをさらに拡大するにつれて、私たちは私たちのプラットフォームを繰り返し発展させ、より多くのインテグリン標的に対する理解を深めることができると信じています。
•私たちの製品を独立して商業化し、承認されれば、私たちが最大の価値を達成できると思う標識と地理的位置。私たちは、明確な臨床と規制承認経路を持っていると思う候補製品を独立して推進する予定で、承認されれば、商業化に成功できると信じています。私たちはまた、いくつかの目標、候補製品、または疾患分野をめぐる戦略的協力を求めることができ、これらの目標、候補製品または疾患分野は、より大きな生物製薬会社または特定の分野の専門会社の資源から利益を得ることができると考えられる
私たちの焦点はインテグリン受容体です
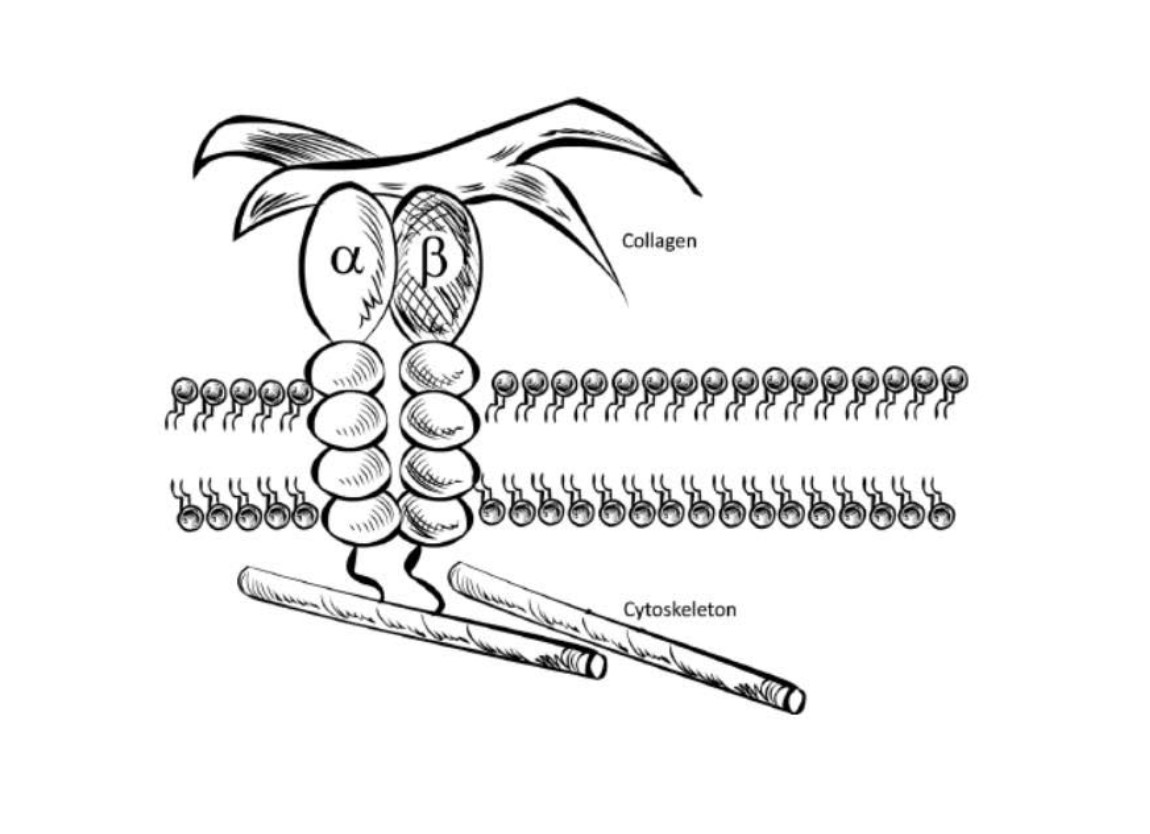
インテグリンは,ヒト内で唯一細胞内と細胞外リガンドを用いて細胞内から細胞外にシグナルを伝達し,細胞外から細胞内に伝達する受容体である。逆に,細胞外リガンドや細胞内細胞骨格に結合すると,これらの状態はインテグリン伝達の張力によって調節される。このような双方向シグナル伝達能力はインテグリンをほとんど細胞と器官の動態バランスの各方面に影響することができる。そのため、インテグリンシグナルの失調は多くの人類疾病と関係があり、自己免疫、心血管と代謝性疾病、繊維化と癌を含む。
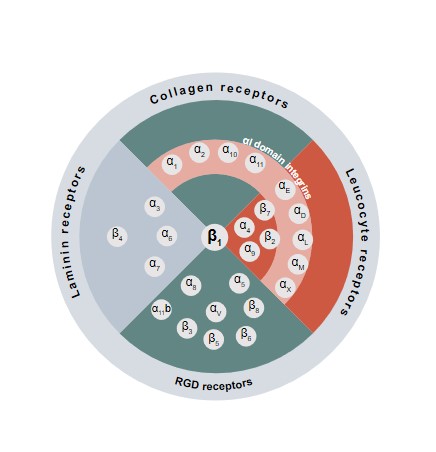
インテグリン受容体は進化的に保守的である。インテグリンは18個のαと8つのβサブユニットの対の組み合わせで存在し、24個の既知のヘテロ二量体がある。これらのペアリングは、インテグリンがそのリガンドを認識し、特定の方法で細胞機能を調節する独自の能力を有するようにする。インテグリンは白血球上のインテグリンとRGD−ペプチド,コラーゲン,ラミニンリガンドを認識するインテグリンに分類される。それらは細胞生物学と生理学の多くの方面を調節し、白血球の輸送、血小板と白血球の活性化、成長因子、例えば形質転換増殖因子βの活性化、細胞と基底膜と細胞外基質との粘着、及び細胞の組織内の滞留或いは粘着強化を含む。この異なる機能はそれらを臨床前モデリング或いは臨床確立に基づく広範な人類疾患に対する操作可能な目標にする。次の図は24人のメンバーのインテグリンファミリーと臨床関連領域をまとめた
治療の標的となるインテグリンファミリー
インテグリンは長い間薬物標的とされてきた。1980年代、インテグリンの治療問い合わせはRGDインテグリン、αIIbβ3に集中した。血小板上のαIIbβ3が活性化されると、フィブリンに結合し、フィブリンが隣接する血小板に結合し、血栓形成を招く。血小板凝集におけるαIIbβ3の重要な作用を決定する分子詳細の出現に伴い,そのリガンド結合機能を抑制することが抗血栓作用を有することが明らかになった。1994年、アキシマブ(市場名ReoPro)は経皮的冠動脈形成術患者のインテグリン療法を受けることが最初に許可され、その後、チロフィバン(市場名はaggraastat)とエプフェナペプチド(市場名はINTEGRILIN)が承認された。
薬物標的であるインテグリンの次の段階開発は白血球上のインテグリン受容体に集中する。これらの療法は,T細胞を含む活性化された免疫細胞が慢性炎症組織に入る能力を抑制することで自己免疫を調節する。4つの承認されたインテグリン薬はこのカテゴリに属しています
•エファリズマブ(以前の名称はRaptiva、後に市場から撤退した)、αLβ2の注射可能な抗体阻害剤は、2003年に食品と薬物管理局によって慢性中から重度乾癬の治療に許可された
•タズマブ(市販名Tysabri)はα4β1の融合不可能な抗体阻害剤であり、アメリカ食品と薬物管理局は2004年に再発性多発性硬化症の治療に使用することを許可し、2008年に中度から重度の活動性クローン病の治療に使用することを許可した
•Vedolizumab(市場名Entyvio)、α4β7の非融合抗体阻害剤であり、2014年にアメリカ食品と薬物管理局の許可を得て、重度活動性潰瘍性大腸炎或いはクローン病の治療に用いられた;
•Lifitegrast(市場名)は、局所的に使用されているαLβ2小分子阻害剤であり、2016年にアメリカ食品と薬物管理局によってドライアイの治療に許可された。
Global Dataのデータによると,これらの自己免疫療法はそれぞれの2021年度に約72億ドルの年間売上高を達成していると推定されている。
口腔インテグリン調節剤の開発課題
これらの療法の不溶性、注射可能、または局所使用の性質は、それらの使用を制限する。これらの限界を解決するために,製薬会社は経口系インテグリン療法を発見·開発するために大量の資源を投入した。αIIbβ3だけでは、6種類の異なる化合物(ロシフェバン、シブラフェバン、オポフェバン、サイミロフィバン、レフォダ非バン、ロトリフェバン)が登録第三段階臨床試験に入った。これらの試験の結果,これらのαIIbβ3を経口投与する系阻害剤が急性冠症候群患者の血管死を増加させたことは失望的であった。これらの失敗を理解した10年後,すべての失敗した経口阻害剤は活性インテグリンのコンホメーションを安定させ,リガンドシグナル伝達を促進し,完全な活性結合を維持するのに不十分であれば知られている。これらの欠陥はより大きな血小板凝集とより高い不良事象の発生率を招く。
また,α4β1とα4β7を経口投与する非選択的阻害剤Firategrastの第2段階開発では,白血球インテグリン阻害剤を経口投与すると意外な疾患活性化活性が認められ,Firategrastを不飽和用量で投与すると多発性硬化症患者の症状が進行した。これは損傷の増加を招き、不良事件の発生率は増加する。この化合物の開発はその後中止された。
私たちのプラットフォームと方法は
私たちのMINTプラットフォームは私たちが第一世代経口統合ホルモン標的治療薬物開発者が直面している挑戦に対応し、克服できると信じている。
インテグリンモデル
私たちの発見プラットフォームは私たちを24人のメンバー全体のインテグリン目標家族で仕事を展開する唯一の会社になると信じています。我々のMINTプラットフォームには以下のようなユニークな機能が含まれている
•インテグリン構造の固有能力を決定する。著者らのタンパク質構造、細胞系とノウハウを利用して、著者らはすでにインテグリン類の臨床重要な標的のために500種類以上の特許構造を解明した。
•計算を支援する製品候補設計エンジン。我々はすでに16,500個を超える最適化化合物のライブラリーを構築し、複雑な薬物と計算化学機能および各インテグリンに対する生物分析を用いて、効率的かつ選択的なインテグリン阻害剤と活性化剤を臨床前と臨床開発の候補製品に調整することができるようにした。著者らはシュレーディンガーとの独占計算協力は可変製品エンジンから候補製品を生成する能力を大幅に加速し、シュレーディンガーは先進的な物理ベースのモデリングと機械学習を用いて優れた経口候補薬物を作成した。我々は最近,特殊なシュレーディンソフトウェアクライアントとしてのアクセス範囲を拡大し,そのソフトウェアキットの使用がインテグリンの範囲を超えている
•我々の独特な能力を利用して特定のインテグリンコンホメーション構造を発現と分離することによって、抗体候補を活性化と抑制する新興能力を発見した。
•生物学と病気の転化能力です我々の複雑で包括的なバイオツールキットは,遺伝子とタンパク質発現プロファイル,ヒト組織の単細胞分解能プロファイルおよびバイオマーカーの開発を含み,目標参加と興味のある疾患に対する薬効学的活性を評価できるようにした。
私たちは最初に、高度に満たされていない医療ニーズ分野の目標カテゴリに対する候補製品の開発に専念しました
•α4β7およびα4β1は、自己免疫疾患の既定の標的である;それらの作用機序およびそれらの利点とリスクを抑制することはよく理解されている
•あるαvインテグリンは転化成長因子βの活性化を通じて臨床前の良好な作用機序を有し、転化成長因子は1種の臨床上重要な抗炎症性サイトカインであり、多くの人類病理において失調を調節する。
これまでわれわれは臨床研究でのみα4β7特異的インテグリン阻害剤Morf−057を試験してきたが,現在われわれの他の候補製品の経口バイオアベイラビリティに関する臨床前データのみである。
著者らはインテグリン受容体活性機序の理解は唯一無二であり、それは複雑なコンホメーションとシグナルによって制御され、インテグリン受容体標的ファミリー中の阻害因子と活性化因子を発見することができる。我々の能力は,α4β7,αvβ8,および他のインテグリン計画の改善によって検証されている。私たちのMINTプラットフォームは3つの主要なコンポーネントで構成されています
•インテグリン構造の固有能力を決定すること
•調整可能な製品候補設計エンジン;
•生物学と病気の転化能力です
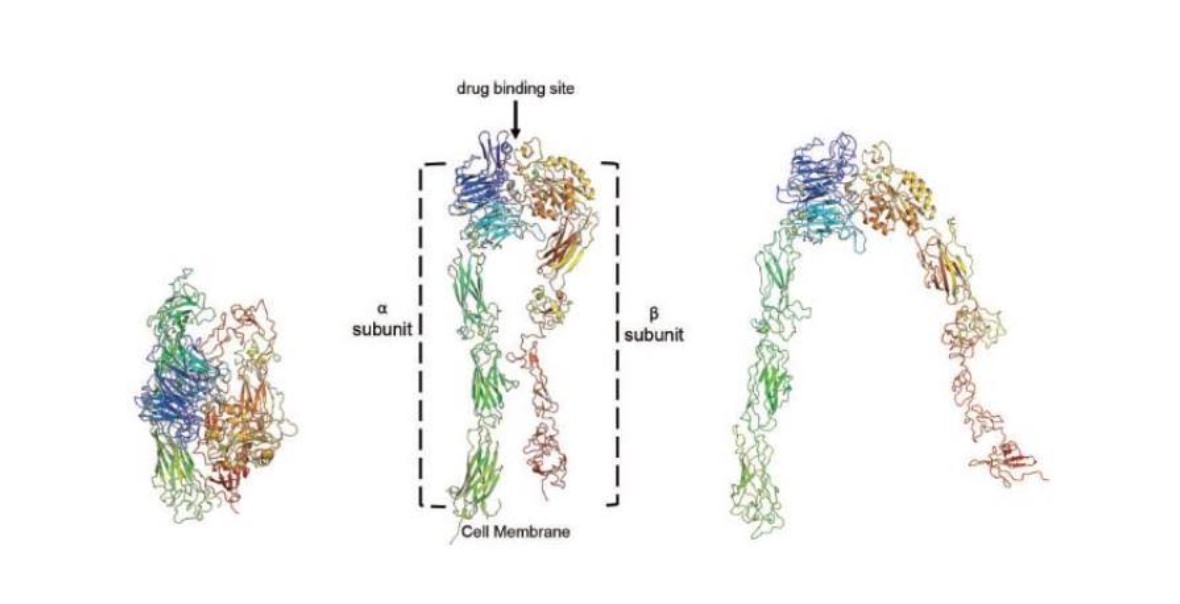
著者らのインテグリンコンホメーションと分子作用モードに対する深い理解を利用することは、著者らが候補製品戦略を確定する重要な要素である。これらの受容体は,図1に示すように,受容体の不活性(屈曲閉鎖と延長閉鎖)と活性化状態(延長開放)をもたらす大量のコンホメーション変化を経験している。屈曲閉鎖の形式では,インテグリンの上半分はαとβサブユニットで形成され,半分は折り畳まれ,上半分と下半分が互いに関連し(図1左側),インテグリンを無効にする。活性化されるインテグリンの場合、延長−閉鎖状態(図1中)は、細胞表面のαおよびβ中段に延長開放状態を示すように延在する(図1右側)。多種のインテグリンと比べ、屈曲閉鎖コンホメーションと伸展閉鎖コンホメーションはリガンドとの親和性が低いが、伸展開放コンホメーションとリガンドの親和性はインテグリンの違いによって700~5000倍向上する。インテグリンコンホメーションと親和性のこれらの変化は双方向シグナルを伝達し、その表面インテグリンを発現する細胞を他の細胞上の細胞外基質或いはリガンドとコミュニケーションさせることができる。
図1:インテグリン動的コンホメーション状態。インテグリン異性二量体対の左弯閉鎖不活性形態、中伸長閉鎖不活性形態、および右伸長開放活性形態。
著者らの新型MINTプラットフォームは複雑な統合素コンホメーション状態制御に対する深い見解に基づく構造生物学的能力に基づいている。スプリンガー博士は特定のインテグリンコンホメーションをロックする初期小分子のセットを述べ、私たちはすでにこの知識を利用して改善し、私たちの経口インテグリンの薬理学を最適化した。私たちは病気における生理的失調のインテグリン立体構造状態を識別するために化合物を設計した。著者らの化合物はインテグリンと結合し、インテグリンの健康組織特有の構造の採用を促進し、そして疾病特異的インテグリンシグナルを阻止する。インテグリンに対する小分子を過去に開発する試みは失敗しており,一部の原因は十分な理解が不足していると考えられる
これらのコンホメーション変化と疾患への影響。われわれのMINTプラットフォームは,インテグリン機能障害に関連する疾患の生物学的基礎の深い理解を応用し,一連の新規インテグリン療法を開発できると信じている。
Morphyインテグリン技術(MINT)プラットフォーム
インテグリン標的ファミリーが構造と機能に関連するタンパク質から構成されていることから、MINTプラットフォームの各周期は私たちの興味のある計画中に化学資産と生体データを産生し、同時に新しいインテグリン複合体の構造と機能の理解を促進する。造幣工場の設計周期のすべての転換において、これは知識と資産の迅速な戦略的組み合わせをもたらすと信じている。我々のα4β7計画は,計画起動後3年以上の間に最初の開発プロジェクト候補を生成した.我々のαvβ6プロジェクトがわずか2年で同様の目標を達成したのは,経口バイオアベイラビリティ,除去性,代謝安定性の化学特性を最適化する上で一定の知見が得られたためと考えられる。我々のαvβ8計画は,候補プロファイル化合物の開発により速い進展を遂げている.我々が発見した化学タイプと最初の薬物化学打撃はツールや化合物となり,さらに個々のインテグリンに関する知識ベースを得ることができ,これも関連するインテグリンにまで伸びている。たとえば,αvβ6における発見作業は,高い選択性を持つαvβ8の高度なセールス手がかりや他の目標の起点をもたらし,新たな全額計画を直接支援し,連携努力を支援する.
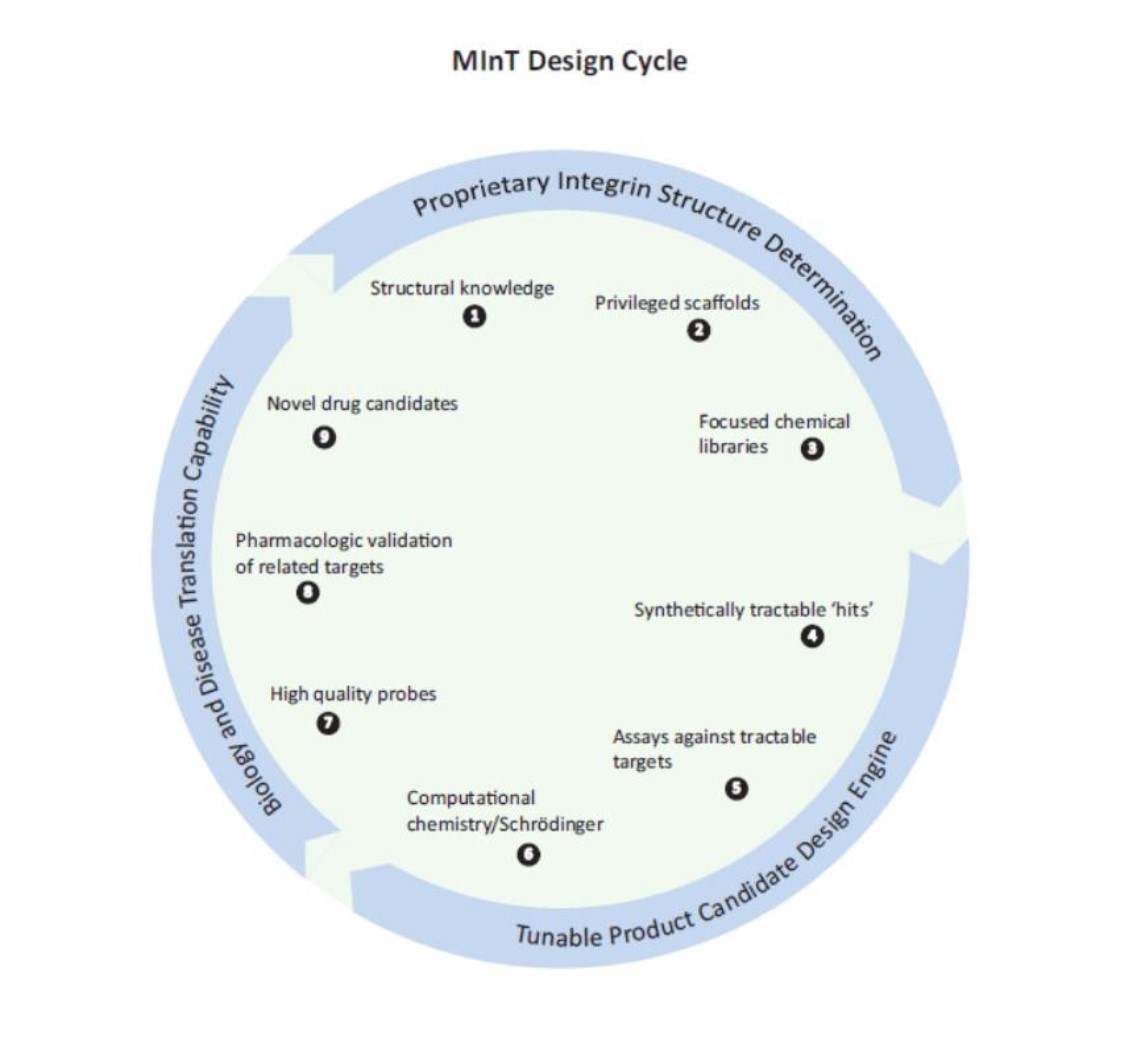
次の図に示すように、私たちのミントプラットフォームの3つの支柱に基づいて、反復ミント設計サイクルは、インテグリン構造の固有能力、私たちの調整可能な製品候補設計エンジン、および私たちの生物学的および疾患変換能力を決定する9つのステップを含む。
インテグリン構造の固有能力を決定する
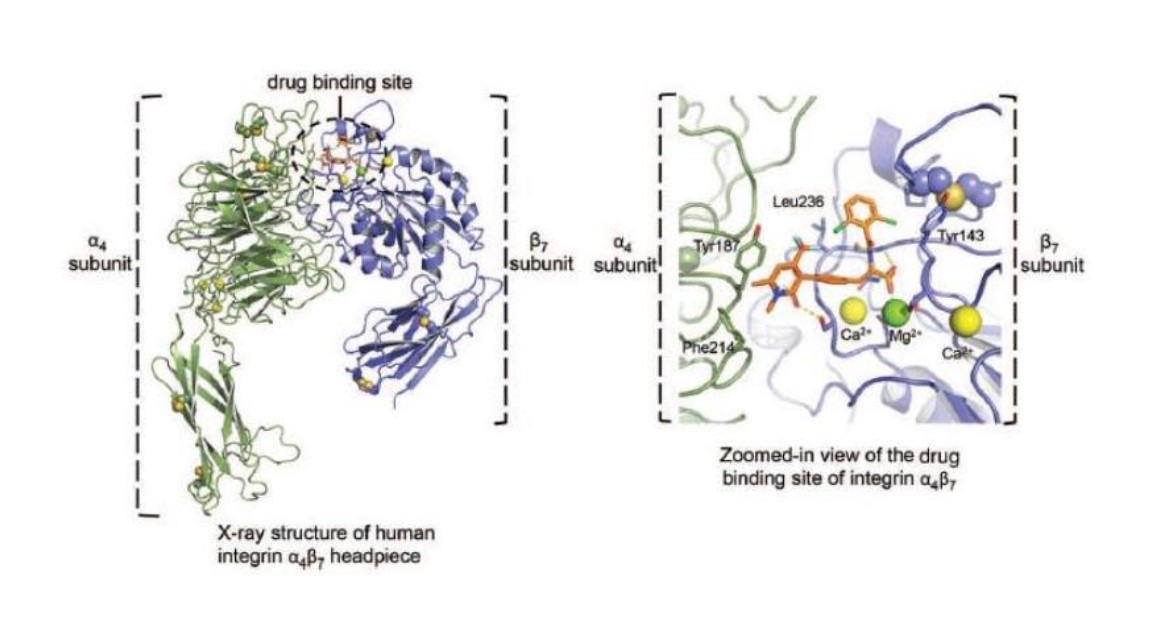
タンパク質の結晶構造を理解することは,製品候補設計をより有効にすることができると信じている。インテグリンの構造は、多くの柔軟なドメインとドメイン間リンカーからなるため、特徴づけが困難である(図1参照)。著者らはインテグリン構造知識と細胞系における独特な地位、及び半分のインテグリン標的結晶構造、特許タンパク質試薬と技術ノウハウを獲得し、500以上の臨床重要標的の独自構造を解明できるようにした。著者らの新しい方法は構造生物学に対する著者らの深い理解とインテグリン蛋白コンホメーションに基づいてどのように疾病中の機能を調節するかである。この点の一例は,我々のα4β7プログラムにおいて,薬物結合部位の結晶構造が,αとβサブユニットの界面結合を設計できる新たなリガンドである(図2)。分子レベルでのこの重要な情報は,この受容体ファミリーの潜在力を放出し,特定のコンホメーションに対するインテグリン受容体の小分子を開発するために我々の研究を指導した。
図2:ヒトα4β7インテグリン受容体ヘテロ二量体或いは頭部頂部の左X線結晶構造、左側はαサブユニット、右側はβサブユニットである。この受容体の薬物結合部位はαとβサブユニットの境界に位置する。右図は薬物結合部位を示し,このインテグリン中のタンパク質コンホメーションの調節を担う重要な相互作用を示している。構造レンダリングのためのデータは,Yu,Y.,朱,J.,Mi,L.Z,Walz,T.,Sun,H.,Chen,J.-F.,Springer,T.A.(2012)からである.α4β7は転がり癒着を調節するインテグリンの構造特化である.J.細胞生物学。196,131-146。
調整可能製品候補設計エンジン
特許化学:著者らは特定のインテグリン受容体コンホメーションの分子開発を安定させる上で重要な技術ノウハウを有しており、経口インテグリン抑制製剤を同定する新しい方法を支持している。今日、私たちの小分子化学バンクは16,500個を超える独特な設計のインテグリン調節剤(阻害剤と活性化剤)を含み、私たちの薬物設計技術はインテグリン標的動力学の独自の理解を利用している。特定のインテグリン分子作用モデルに対する深い理解を結合して、私たちはすべてのインテグリン機能のために適切な化学タイプを設計できると信じている。ライブラリー化合物のさらなる最適化、薬物化学の優れた発現を結合させ、同定に有効な、選択的な経口小分子製品を提供することが可能となる。
シュレーディンガーの計算化学協力化学シミュレーションと電子薬物発見分野の先駆者であるシュレーディンガーと協力し,インテグリンの独占的な協力である。この協力により,様々な物理に基づく次世代計算技術を利用して,設計,反復,最適化の手がかりにより薬物発見を加速させることができると信じている。シュレーディンガーとの協力により、先進的な構造誘導薬物設計技術を利用して、原子精度で分子を設計することができるようになった。我々は最近,特殊なシュレーディンソフトウェアクライアントとしてのアクセス範囲を拡大し,そのソフトウェアキットの使用がインテグリンの範囲を超えている
私たちの体外インテグリン分析チームは疾患関連受容体コンホメーションを安定化する新しい阻害剤を決定するために、著者らはインテグリンファミリーの各メンバーをカバーする強力な体外テストを確立した。これらの独自の内部スクリーニング分析はインテグリンファミリー中の効力と選択性に対して生化学と機能表現を行うことができ、薬物設計過程の異なる段階で強力なツールとして機能する。
生物学と病気の転化能力は
MINTプラットフォームはヒト疾患におけるインテグリン生物学に対する深い理解に基づいて、インテグリン組織と細胞発現マップを含む。著者らはすでに一連の複雑かつ全面的な体外、体外と疾病特異性体内試験を確立し、インテグリン調節の薬理作用を評価し、そしてその作用機序についてもっと理解することを目的とした。これらの検出から得られた生物学的知識は,複数のインテグリン発見計画における我々の作業を加速させる可能性がある。われわれは戦略的に臨床前観察を著者らの臨床発展計画に転換することを望んでいる
これらに加え,薬物動態学や薬効学モデリングにおいて日々増加している能力に加え,インテグリン阻害剤を発見することができ,これらの薬物はヒト自己免疫,心血管や代謝疾患,線維化や癌などの疾患に影響する可能性がある。
我々のルート計画は
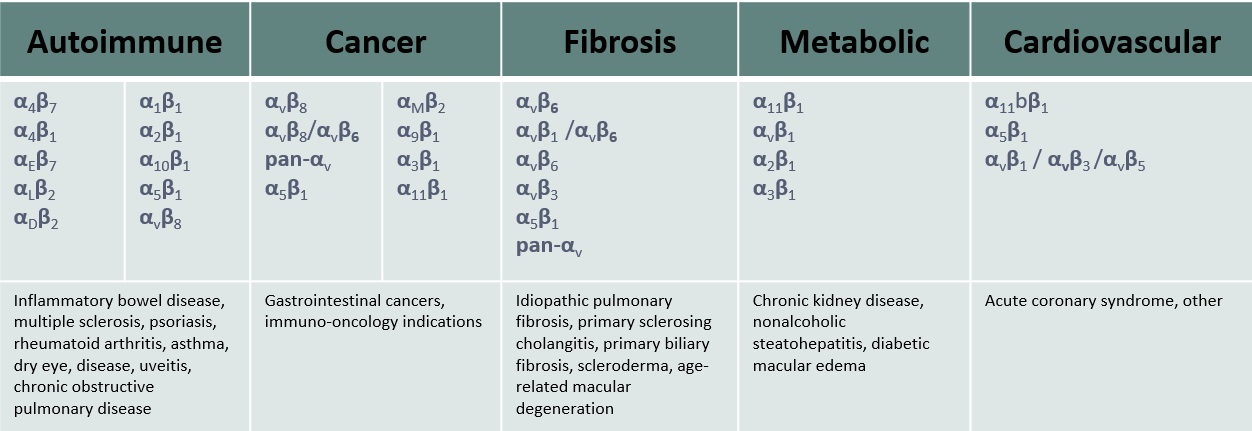
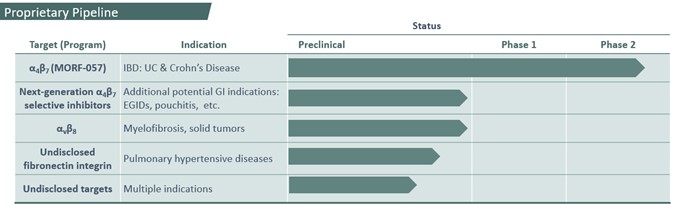
著者らは生物学、安全性、技術準備と開発の実行可能性を検証した上で、インテグリンが人類疾病中に抑制する機会について分析を行った。著者らはすでにすべての4つのインテグリンファミリーの中でいくつかの操作可能なインテグリン標的を確定し、著者らの最初の重点は自己免疫、心血管と代謝性疾患、繊維化と癌などの高度に満足されていない医療需要領域である。次の表は、現在の候補製品に関する重要な情報をまとめています
私たちの主な候補製品
MORF-057:我々のα4β7特異的インテグリン阻害剤による炎症性腸疾患の治療
最も一般的な炎症性腸疾患である潰瘍性大腸炎とクローン病を潜在的に経口治療する薬剤として、我々の先行するα4β7インテグリン阻害剤Morf−057を進めている。現在、IBDの医療管理策略はコルチコステロイド、免疫調節剤と注射可能なモノクロナル抗体治療による疾病緩和の誘導と維持に重点を置いている。もし承認されれば、私たちの経口インテグリンはIBD患者に的確で、安全で、有効かつ便利な治療方法を提供する可能性があると信じている。
炎症性腸疾患の背景
IBDはいくつかの自己免疫と免疫介在性疾患を含み、その特徴は胃腸の慢性炎症である。潰瘍性大腸炎では炎症は結腸内壁に限られているが,クローン病では炎症は胃腸の任意の部分にセグメント的に関与し,腸壁厚全体に広がっている。これらの症状には,持続性下痢,腹痛,直腸出血,体重減少,疲労がある。承認された治療中の重度IBDの生物薬物の初期誘導無反応率は30%に達し、時間の経過とともに45%の患者が反応を失った;そのため、比較的新しい薬物であっても多くの病状の重い患者の組織炎症或いは症状を十分に制御できない可能性があるため、一部の患者は合併症が出現し、結腸と直腸を手術切除する必要がある。クローンや大腸炎財団のデータによると,米国には約170万人のIBD患者がいる
数年来IBD治療の主要な薬物は内服と外用水楊酸塩とグルココルチコイドである 免疫抑制剤と抗体療法です。炎症性腸疾患を治療する抗インテグリン抗体療法は、多発性硬化症の最初の承認後に承認されたα4インテグリン阻害剤ナタズマブによるクローン病の治療の初めての承認を得た。そのナズマブ治療は進行性多病巣性白質脳症に関連し,ブラックボックス警告を持ち,そのα4β1阻害活性に関与しており,クローン病への応用を制限している。PMLは稀かつよく致命的なウイルス性疾患であり、その特徴は多数の部位の脳白質進行性損害である。Vedolizumabはインテグリンα4β7のモノクロナル抗体阻害剤であり、重度の活動性潰瘍性大腸炎およびクローン病の治療に許可され、ブラックボックス警告を伴わない。
経路と標的生物学的概要
インテグリンα4β7は粘膜アドレス細胞接着分子と結合し,ほとんど腸管内皮細胞にのみ高レベルで発現している。この相互作用を遮断することで免疫細胞の腸管への炎症組織への進入を防ぎ,IBD治療に有効であることが証明されており,vedolizumabの承認が証明されている。
私たちの解決策は
われわれはすでにα4β7に対する経口小分子インテグリン療法を生じ,潰瘍性大腸炎やクローン病患者の治療を目指している。われわれの戦略は,経口療法を発見する能力と,α4β1を抑制する目標リスクをいかに最小限にするかという知識によって駆動され,PMLはPMLに関与している。この計画は目標階層の例を代表しており,我々のMINTプラットフォームを用いて既存の療法と区別する機会があると信じている。安全で有効な経口療法は,(I)早期の治療法として,(Ii)IBD領域の他の薬剤との併用が2つの異なる方法でIBD患者の生活を変える可能性があると信じている。
臨床前データ薬理学バイオマーカーデータ
我々の独自のMINTプラットフォームを用いて,Morf−057を含むα4β7小分子阻害剤を設計し,一連の体外試験により評価したが,これらの小分子阻害剤は他のインテグリンに対してα4β1とαEβ7を含み,高効率かつ高選択性であった。以下の表1に我々の細胞接着分析において評価されたMorf−057の効力を示し,参考品vedolizumab,Natalizumabおよびetrolizumabおよび第三者により開発された候補製品AJM 300と比較した。私たちは実験室でこれらすべての可能性を測定した。体外細胞接着実験はα4β7とそのリガンドMadcam、α4β1とそのリガンドVcam、αEβ7とそのリガンドEカルシウムアドヘシンの結合能力を測定した。これらの検出法はIBD候補薬を探す上で有用であることが証明されている。IC 50値は一般的に受け入れられている薬物効力の測定基準である。
われわれの細胞接着試験では,MORF−057は効率的なα4β7阻害剤であり,α4β1やαEβ7と比較して選択性>3,000倍であることが観察された。
| | | | | | | | | | | | | | | | | | | | |
| α4β7集積回路50a | | α4β1集積回路50a | | | |
| 抑制者 | RPMI 8866 | α4β1集積回路50aユルカーター | RPMI 8866 | | αEβ7集積回路50a | |
| MAdCAMの50% | VCAMの50% | VCAMの50% | α4β7/α4β1しわ | K562- αEβ7 E- | α4β7/αEβ7しわ |
| 血清.血清 | 血清.血清 | 血清.血清 | 選択性 | カルシウムアドヘシン | 選択性 |
| MORF-057 | 1.2 ± 0.8 nM | >50 µM | 4,290 ± 670 nM | >3,000 | 52 µM | >143,000 |
| ウェドーズマブ | 0.035 ± 0.020 nM | >180 nM | >1,000 nM | >3,000 | ネオジム | -- |
| じゃあ彼は単抗体です | 0.166 nM | 1.8 nM | 0.14 nM | 1 - 12 | ネオジム | -- |
AJM 300b | 93 ± 66 nM | 4200 nM | 779 ± 261 nM | 8 - 45 | ネオジム | -- |
| 依曲利珠単抗 | 0.0185 nM | ネオジム | >1,000 nM | >106 | 1.2 nM | 14 |
表1
*RPMI 8866、JurkatおよびK 562-αEβ7は、それぞれα4β7、α4β1およびαEβ7細胞に使用される。
ND=未確定
“変形治療”ジェイミー·Wong ePoster Tul 1283
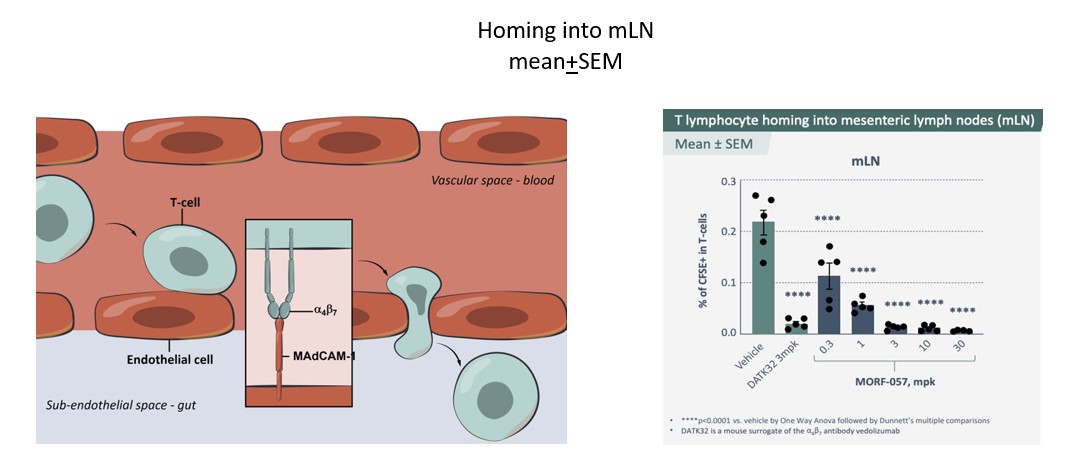
われわれはまた,単用量急性薬効学モデルにおいてわれわれのα4β7阻害剤の体内活性を評価し,このモデルにおいて,α4β7インテグリンの腸管への輸送遮断への影響を評価した。Tリンパ球ホーミング過程は蛍光標識TK 1細胞を用い,この細胞表面はα4β7インテグリンを高レベルに発現し,各群n 5匹の動物であった。我々が開発した候補化合物Morf−057を含むいくつかの化合物は、用量応答を評価するために本試験で評価されている(図3)。測定したすべての用量で統計的に有意な反応が認められ,試験の3つの最高用量でα4β7抗体vedolizumabのマウス代替品DATK 32と同様に有効であることが観察された。以下の図3において,右側パネルは,我々の小分子α4β7阻害剤とDATK 32(試験中のVedolizumabのマウス代替品)で観察された標識T細胞を腸間膜リンパ節に帰巣したカルボキシフルオレセインスクシンイミドまたはcFSEの用量依存抑制を示している。各治療群間に統計学的有意差があった(*p
図3.左側パネルは,炎症性腸疾患におけるα4β7のリンパ球発現機序を示している。α-4-β-7発現リンパ球は腸管に入り、MAdCAMと接着し、その後炎症部位に滲出と遷移する。右のパネルは,我々の候補製品の活性を検出し,vedolizumabのマウス代替品と比較するための体内試験の結果を示している。
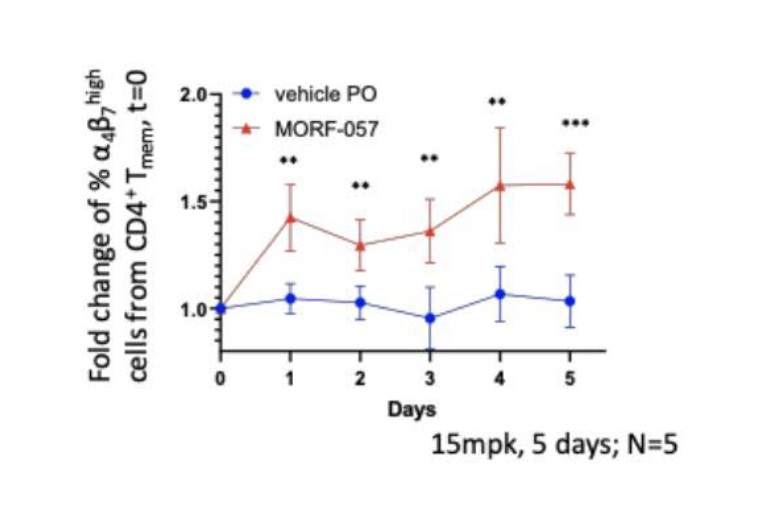
非ヒト霊長類モデルでは,Morf−057はα4,CD 4,β+T細胞の粘膜部への輸送を抑制することも証明されている。このモデルでは,細胞の体循環中の増加を観察することにより,細胞の腸管輸送抑制を間接的にモニタリングした。次に図4にMorf−057を1日2回経口投与し,循環中のT記憶細胞のレベルを統計的に有意に増加させたことを示す。
図4:非ヒト霊長類動物におけるMorf−057経口投与後の%α4β7高T記憶細胞の倍数変化。各個体が時点0 h(第1用量投与)で正規化されたデータの平均値および標準偏差。各時点でt検定分析を行った.Holm−Sidak法を用いて統計学的有意差を決定し,α=0.05であった。**ページ
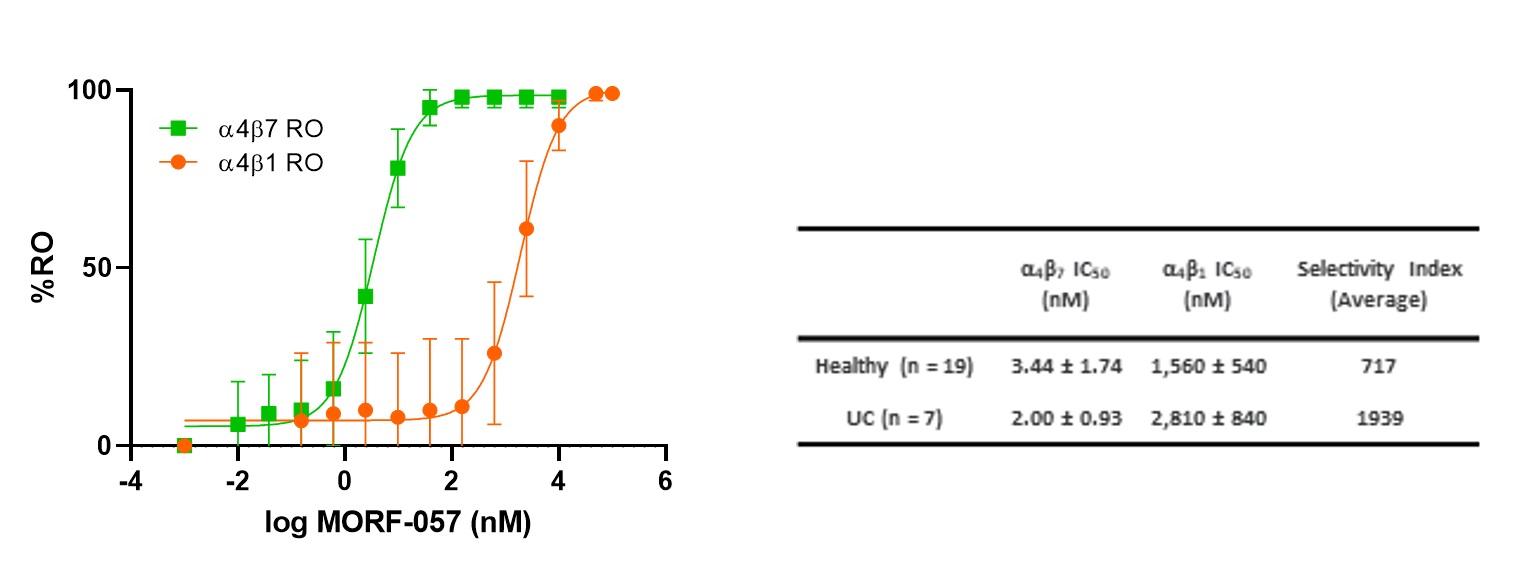
受容体占拠、またはROなどのバイオマーカーの翻訳は、vedolizumabの臨床前研究および早期臨床試験においてPDマーカーとして確認されている。1つの候補産物がα4β7に結合する時、それはインテグリンリガンド結合部位を占め、MadCAM結合免疫細胞を妨害し、免疫細胞の炎症腸管組織への凝集を促進する能力を有する。循環血中のリンパ球中のα4β7に結合する候補産物の測定方法であって、血液に基づくα4β7 RO分析と呼ばれる分析方法。遊離α4β7とα4β1信号強度は濃度の増加とともに低下した。図5の結果はまた、Morf−057はα4β7に対して高い効力および選択性を有することを示しており、RO試験は健常被験者およびUC患者においてほぼ同様の発現を示した
図5左:健常被験者と潰瘍性大腸炎患者から分離した血液から計算したα4β7とα4β1の異なる濃度での占有率。26例の献血者のデータは平均値±標準偏差であった。表5右:MORF−057は体外ヒト全血中のα4β7のα4β1に対する有効かつ選択的阻害剤であり,正常健常ボランティア,UC患者ともに同様であった。値は平均値±標準偏差であった。
臨床発展の概要
ステップ1
2020年9月、著者らは多用量Morf-057の安全性とPK状況を評価するために、SAD、FEとMADキューを含む健康ボランティアの第1段階臨床試験を開始することを発表した
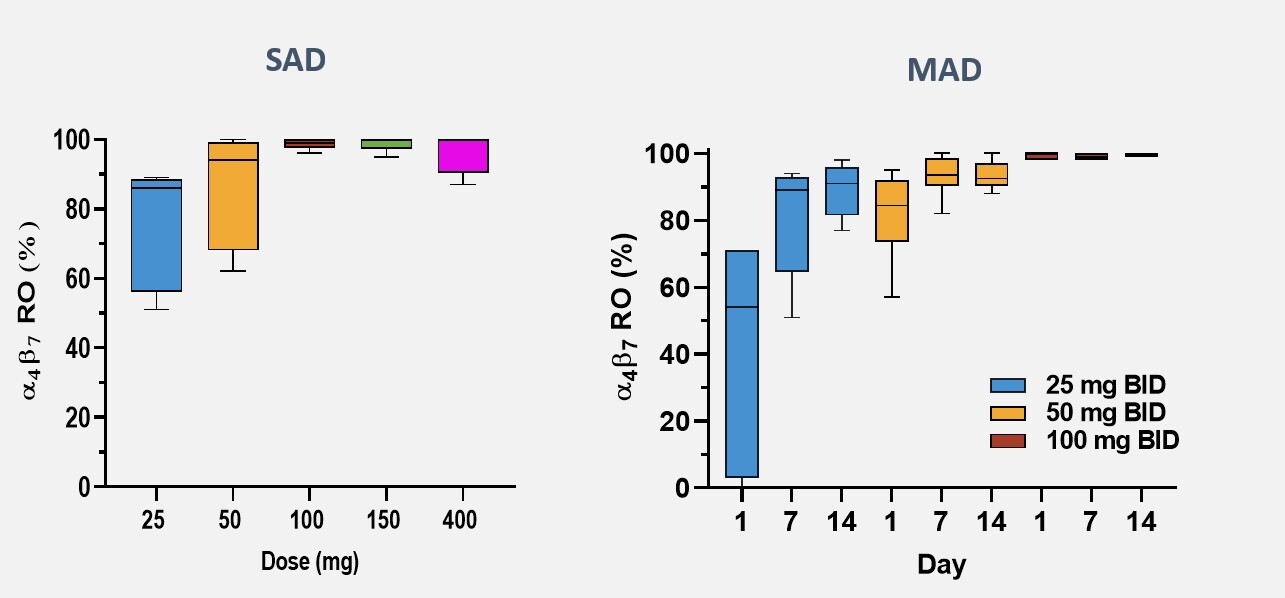
2021年3月,Morf−057第1期SAD臨床試験の初歩的な結果を公表し,Morf−057は25 mgから400 mgまでのすべての用量列で良好な耐性を有し,3つの最高用量レベルでα4β7インテグリンの平均受容体占有率が95%を超え,飽和α4β7受容体の経口投与の潜在力を証明した
2021年7月,Morf−057臨床計画のMADおよび食物効果部分が完了した後,欧州クローンおよび大腸炎組織(ECCO)2021年仮想大会で第1段階臨床試験の完全なデータセットを報告した。完全なMorf−057第1段階研究は、SAD、MAD、およびFEキューを含み、Morf−057の安全性、PK、およびPDを評価する。健康な対象は、ランダムに3:1に分けられ、SAD群において単回用量の25、50、100、150および400 mgのMorf−057または一致するプラセボを受け、狂ったキューで1日2回の用量の25、50および100 mgのMorf−057または一致するプラセボを14日間受けた。条件に適合した健常者67名が研究に参加し,そのうち36名がSAD群,9名がFE群,22名がMAD行列であった。66人の被験者が研究治療を完了し、50 mg Bid狂ったプラトゥーンのうちの1人が個人的な理由で同意を撤回した。
MORF−057はすべてのキューで耐性が良好であり,セキュリティ信号は認められなかった。MORF−057は良好なPKプロファイルを示し,その中で目標交戦が確認され,明確なPKとPD関係が確立された。Morf−057は迅速に吸収され,全身曝露はほぼ用量割合で増加することが確認された。FEの結果,食物摂取量は低地Morf−057レベルに影響を与えず,計画した患者研究では,食事を考慮せずにMorf−057を使用することが可能であった。
図6:受容体占有率は用量と研究日数に応じて変化し,1列あたり25 mgを超える個体で飽和度(>99%)に達している。
α4β7受容体の占有率は用量と研究日数の増加とともに増加し,14日目には25 mgを超える行列中のすべての患者が飽和(>99%RO)に達した(図6).Morf−057は100 mgBID行列ですべての測定時点でα4β7受容体(平均RO>99%)を飽和させた。特定のα4β7高発現免疫細胞群を含むバイオマーカーの用量と時間依存の変化が観察され,Morf−057の生物学的証拠が増加した。これらの変化は,IBDの治療に承認された抗体薬vedolizumabを含む他のインテグリン阻害剤の報告と一致している
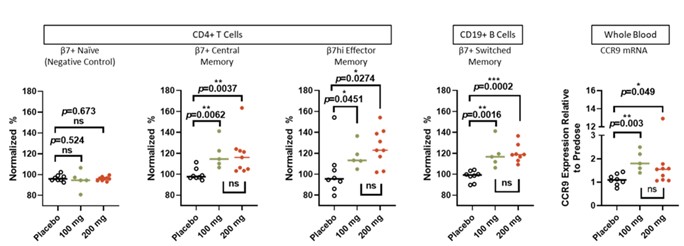
MORF-057の第1段階研究では、MORF-057を1日2回200 mg服用した被験者はα4β7受容体飽和を示し、循環中央記憶、効果記憶Tリンパ球、および切替記憶Bリンパ球数はプラセボと比較して有意に増加した(図7)。25 mgと50ミリグラムBIDの探索的用量でも,キー薬効学的指標の方向性が増加する傾向が認められた。いずれの用量も耐性が良好であり,安全信号は認められず,良好な薬物動態学的特徴が認められた。Morf−057は単剤200 mgMorf−057および200 mgBidの14日間でα4β7受容体がCにあることを示した低い谷それは.リンパ球亜群とCCR 9 mRNAの統計学的有意な変化が認められ,これは先行研究と一致している。完全な第1段階データセットはMORF-057の第2段階研究への進展を強力に支持した.
図7:多色フローサイトメトリーを用いてリンパ球亜群を測定した。Morf−057を服用した被験者は100 mg,1日2回と200 mg,1日2回,循環リンパ球亜群の有意な増加を統計的に示し,機械学的期待と一致した。
第二段階 エメラルドはIBDで
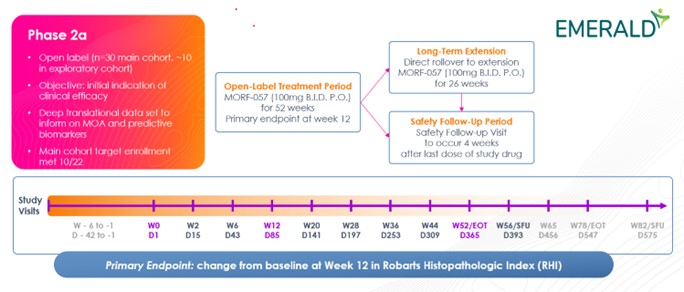
第1段階研究の結果,2022年3月にMorf−057の第2段階臨床試験を開始した。Emerald-1(Morf-057-201)は、中重度UCを有する成人に対するMorf-057の有効性、安全性と耐性を評価するための開放ラベルのマルチセンター2 a期試験であり、2022年10月に方向性登録を完了し、30名の患者がこの研究に参加した。また,方向性登録完了時にスクリーニングを受けている患者が研究に組み込まれ,計35名の患者が主要キューに登録されている。これまでvedolizumabで治療に失敗した10名までの患者への探索的キューの募集が行われている。Emerald−1研究に参加した患者は,米国とポーランドの地点でBid 100 mg(1日2回)の治療を受けている。試験の主な終点はロバツ組織病理学的指数(RHI)の変化であり,ベースラインと比較して12週における潰瘍性大腸炎の組織学的疾患活動を測定できる検証ツールである。その後,患者は追加40週間の維持治療を継続し,52週間の評価を行う。Emerald-1研究中の副次的およびその他の結果指標は修正されたメオ臨床採点、安全性、PKパラメータと重要なPD指標を含み、α4β7受容体占有率とリンパ細胞亜群輸送を含む。Emerald−1ステージ2 a試験からの主要終点データは,中から重度UC患者のMorf−057を対象とした2023年第2四半期に報告されると予想される
図8.Emerald-1フェーズ2テスト設計
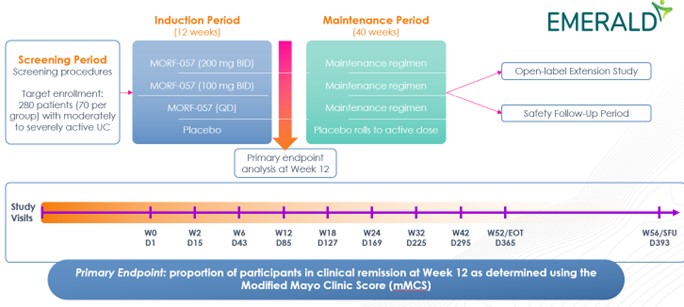
Emerald-2(Morf-057-202)はMorf-057の世界2 b期無作為対照試験であり、2022年11月に開始された。Emerald−2試験に参加した患者は、100 mg Bid(1日2回)、200 mg Bid、およびQD(1日1回)群、または12週間誘導段階後にMorf−057に入るプラセボ群の3つの有効群またはプラセボ群にランダムに分類される。試験の主な終点は臨床緩解率であり,12週間で修正したMayoスコアで測定した。副次的な終点は、RHI、薬物動態および薬効学的指標、および安全パラメータの変化を含むであろう。12週の誘導期の後,40週間の維持期に入る。2025年上半期にMorf−057が中~重度UC患者で行ったEmerald−2期2 b試験の主な終点を完成させると信じている。
図9.EMREALD-2ステージ2 b試験設計
MORF-057による進展に基づいて、私たちは2022年に第二世代α4β7候補開発をIND前段階で休止し、MORF-057の第二段階臨床計画による進展に集中した。しかし、私たちは引き続き私たちのα4β7製品の組み合わせを拡大し、未来の臨床研究のために次世代α4β7小分子の候補製品を開発した
より多くの臨床前と発見の仕事
胃腸疾患治療の新世代α4β7インテグリン阻害剤
よく知られているα4β7阻害生物学とMorf−057計画におけるわれわれの進展に基づき,Morphiは新世代α4β7阻害剤ファミリーの臨床前開発を発見·推進しており,予備治療は好酸性胃腸疾患(EGID),膀胱炎,その他の適応を含む炎症性腸疾患以外の胃腸徴候の潜在的開発に重点を置いている。
骨髄線維化と免疫腫瘍学のためのαvβ8インテグリン阻害レジメン
著者らは臨床前開発により、著者らのαvβ8インテグリン阻害剤を骨髄繊維化と固形腫瘍の治療に進展させた。骨髄線維化の進展経路と商業的操作性に基づいて,固形腫瘍のための免疫腫瘍学的計画ではなく,骨髄線維化のためのαvβ8阻害剤計画を優先し,免疫腫瘍学に対する外部αvβ8標的計画の臨床と競争パターンの発展を待っているからである。
インテグリンαvβ8は骨髄繊維化経路調節因子形質転換増殖因子-β1/3の細胞タイプ特異性と組織定位活性化を媒介する。このプロジェクトは臨床前開発段階にある
骨髄繊維化(MF)は骨髄増殖性腫瘍(MPN)の亜型であり、血球(赤血球、白血球或いは血小板)の過剰生成を表現する血液系悪性腫瘍である。MPNには真性赤血球増加症(PV),本態性血小板血症(ET)と原発性骨髄線維化(PMF)の3つの亜型がある。PVあるいはETは骨髄繊維化(それぞれPV後MFとET後MF)に進展する。JAK-STATシグナルの構造的活性化はMFの発病機序において中心的な役割を果たしている。早期MPNからMFへの進展は異常造血幹細胞(HSCs)の累積拡張と関係があり、HSCsは疾病が現れる前の数十年前に得られた体細胞突然変異を持っている。 JAK 2、CALRとMPL遺伝子の表現型突然変異は多くのMF症例と関係がある。MFの特徴は,骨髄間質線維化変化やサイトカイン異常に対する反応として,細胞減少,負担の重い症状,脾腫大,進行性骨髄(BM)線維化と髄外造血である。全体的な生存(OS)は悪く,平均5−7年であった。
αの基本原理Vβ8MFに対する抑制作用は形質転換成長因子β活性化における作用に基づく。抑制作用はαvβ8を介した転化成長因子-β1と転化成長因子-β3の活性化を遮断でき、それによって組織特異性と局部抑制転化成長因子-βシグナル伝達を実現する。転化成長因子-βは骨髄増殖性腫瘍と骨髄繊維化(MF)の発生発展に重要な役割を果たす可能性がある。それはMF患者で発現が上昇し、コラーゲン沈着と骨髄繊維化を促進することはMFの特徴である。巨核球産生に直接影響し,巨核球成熟や血小板産生を抑制することが報告されている。また,正常ではあるがMFではない造血幹細胞の休眠を促進した。形質転換増殖因子-βシグナル経路を抑制することは、突然変異前駆細胞の数ではなく野生型を増加させることによって、MFの繊維化刺激を減少させ、巨核細胞の成熟と正常造血を回復させることが期待できる。αVβ8拮抗は転化成長因子-β1と3を選択的に抑制する利点を提供し、それによって転化成長因子-βシグナルの全体的な抑制を避け、そして心血管と癌促進を含む副作用を防止した。αVβ8MF発病機序に関連する細胞タイプに発現することは、骨髄中の形質転換増殖因子-β活性化の重要なメディエーターである。したがって,αVβ8抑制剤は局部と細胞タイプ特異性の転化増殖因子-βシグナルを抑制し、発病した生態位内にあることを可能にする。唯一承認された治療法はJAK阻害剤であり,これらの薬剤は線維化,赤血球,血小板の改善は報告されていない。
転化成長因子-βも炎症と免疫の重要な調節因子であり、腫瘍の形成、発展と転移において重要な役割を果たしている。腫瘍微小環境において、転化成長因子-β発現が上昇し、癌細胞を免疫監視から脱出させ、免疫系の抗癌反応を阻害する。転化成長因子-βの過剰発現は不良な臨床結果とチェックポイント抵抗と関係がある。一致により、転化増殖因子-βを抑制することは動物モデル中のT細胞の抗腫瘍免疫を促進することによって、免疫検査点抑制剤の治療効果を増強することを表明した。現在、他の人は検査点阻害剤の阻害と転化成長因子-β抑制を結合する多くの臨床試験が行われており、各種の固形腫瘍を治療する。しかし、転化成長因子-βは組織間で重要な動態平衡作用を発揮しており、この経路の広範な阻害剤は臨床で安全問題に直面している。
αvβ8は腸管免疫動態バランスの維持によく知られている役割を果たしている。樹状細胞(DC)と他の免疫細胞上のαvβ8インテグリンは形質転換増殖因子βを活性化し、免疫寛容を誘導し、腸管制御性T細胞(Treg)の産生を促進する。Treg上のαvβ8はTregを介した炎症性T細胞抑制に重要な役割を果たしている。また,α−v−β−8は単球炎症反応や腸管マクロファージの恒常性を調節する。全体的に言えば、αvβ8は異なるタイプの免疫細胞に作用し、獲得性免疫の負の制御と免疫動態バランスの促進に重要である。
記載された生物学によれば、αvβ8は、腫瘍免疫耐性を含む耐性を系統的に調節することが可能である。この標的は固形腫瘍と腫瘍間質細胞に発現し、免疫と非免疫細胞を含む。インテグリン調節剤は多種の作用機序を有する可能性があり、腫瘍の内因性と外因性形質転換成長因子-βを抑制し、Treg、DC刺激抗原提示を抑制し、腫瘍関連線維芽細胞の活性を低下させることによって腫瘍浸潤性リンパ細胞を増加させることを含む。このプロジェクトでは,化学物質は協同構造活性関係やSARにより,他のインテグリン調節剤プロジェクトとともにスクリーニングが進んでいる。我々のMINTプラットフォームを用いて目的インテグリンの結晶構造を初めて現場で明らかにした。私たちのいくつかの化合物は、駆動化合物の選択性および有効性の特徴の理解を促進するために共結晶されている。標的検証と翻訳生物学の取り組みは小分子阻害剤を用いて行われている。
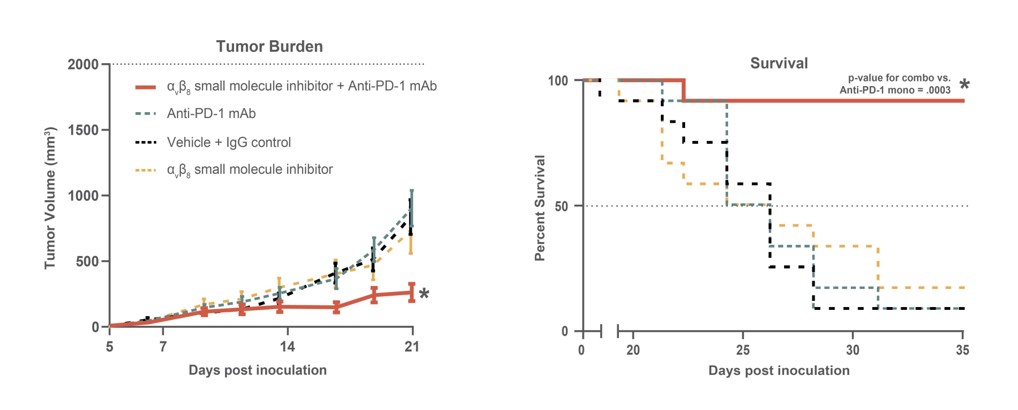
多くの同遺伝子マウス腫瘍モデルにおいて治療効果が確立されている。例えば,免疫拒絶の乳癌モデル(EMT 6)では,Morphical小分子阻害剤が免疫チェックポイント遮断に対する不感受性を逆転させている(ICB;図10)。腫瘍負担と生存率の著明な改善はT細胞浸潤率の増加と腫瘍耐性低下を伴う証拠である。高級免疫検査点遮断難治性腫瘍モデルにおいて、インテグリンαvβ8と低用量放射線の併用を抑制することは抗腫瘍効果を誘導できる(図11)。これらの結果は,αvβ8標的抑制物の経口投与が免疫シナプスに作用することにより抗腫瘍免疫反応を調節する可能性を示唆している。
図10:EMT 6同遺伝子マウス乳癌モデルの腫瘍移植後6日目から27日目までの投与後の治療効果。左のパネルは,賦形剤や単一薬物よりもこの組み合わせによる腫瘍体積の縮小幅が大きいことを示している。右のパネルは、このような組み合わせで処理された動物は、薬物または単一製剤で治療された動物よりも生存時間が長いことを示している。
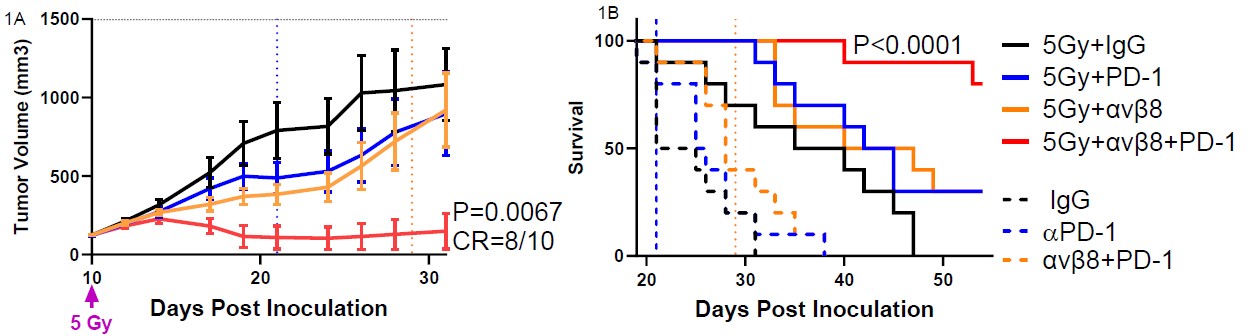
図11:(1 A)腫瘍負荷を指標として、低用量放射線治療(小動物放射線研究プラットフォーム)、PD-1 mAb(週2回、計10 mg/kg、計2週間)とαvβ8 mAb(7 mg/kg、週3回、計3週間)の連合治療の効果を用いた。5 Gy+PD-1および5 GyvIg G-8は腫瘍成長に対する抑制作用が最も小さく、単純放射線治療(5 GyIg G)と比較してやや改善した。αvβ8拮抗薬(5 Gy+αvβ8+PD-1)を添加することは10匹のマウス中8匹にCRが出現した抗腫瘍反応を改善できる。(1 B)進行時間を示すKaplan-Meier曲線。αvβ8やPD−1モノクロナル抗体と比較して、5 GY+Ig Gはマウスの生存率を向上させることができる。5 Gyvαvβ8+PD-1治療群の生存率は、他の治療群よりも有意に高かった。
肺動脈高圧疾患治療のインテグリン阻害剤計画
臨床前研究により、フィブロネクチンインテグリンの抑制は高血圧疾患中の多数の重要な独立過程を推進でき、肺血管再構築の逆転、右室繊維化の予防と心筋細胞の代謝効率の向上を含む
肺動脈高圧(PAH)は稀な進行性疾患であり、その特徴は肺血管再構築であり、肺動脈高圧と進行性右室(RV)の機能障害を招く。現在、プロスタサイクリン、エンドセリン-1或いは一酸化窒素経路に対する治療方法は疾病の進展を緩和した。しかし,約60%の5年生存率は肺血管再構築経路に代わる治療の必要性を強調している。癌細胞と同様に細胞外基質(ECM)再構築によるPA硬直の程度が増加し,PASMCは過増殖と抗アポトーシスを示すことが認識されている。インテグリンは細胞増殖,生存,肥大成長と線維化を促進すると考えられているが,細胞増殖,生存,肥大成長と線維化はPAH進展の重要な因子であるため,インテグリンの発現及びPA再構築とRV不全における作用(Bonnet Et)を検討した。エルAHA 2021で発表, Circulation. 2021; 144:A10717).
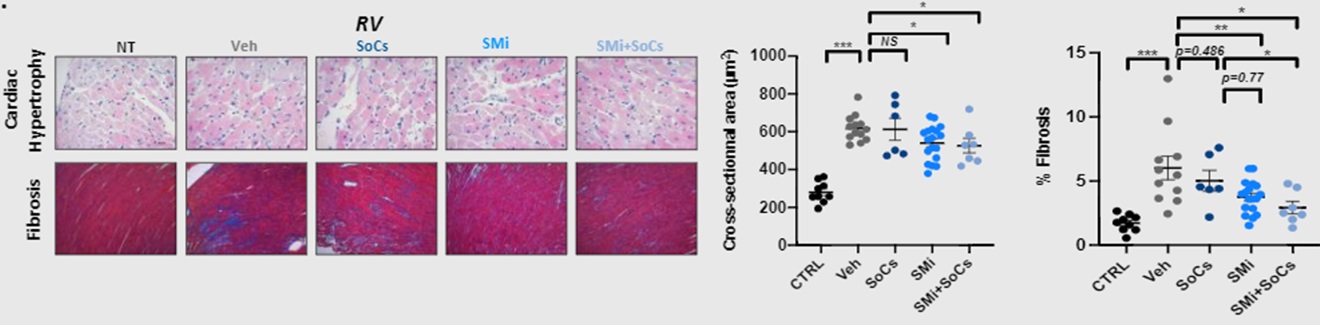
PAH患者の遠位肺動脈、肺動脈平滑筋細胞と右心室におけるフィブロネクチン結合インテグリンの発現は明らかに増加した。また,野生百合塩基(MCT)と肺動脈結紮術(PAB)ラット肺組織にも異なる程度の発現があった。体外薬物によるフィブロネクチン結合インテグリンの抑制はPAH肺血管平滑筋細胞の増殖と抗アポトーシス能力を低下させることができ、これはインテグリン下流シグナル経路の活性化減少と関係がある。心筋細胞やヒト右室線維芽細胞では,これらのインテグリンを抑制することで肥大や右室線維芽細胞の活性化や増殖を減少させることができる。心エコーと右心カテーテル術により、フィブロネクチン結合インテグリンを抑制し、すでに樹立した肺動脈高圧のMCTラット体内の血管再構築と右心機能を改善した。広域インテグリン抑制は血管再構築とRV不全を改善でき、インテグリンはPAHの病理生理学において重要な役割を果たしていることを表明した(図12)。 このようなフィブロネクチン結合インテグリンの理解はMorphyがどのインテグリンがPAH病理生理学において重要であるかを決定することを指導し、これらの結果を臨床開発方法に転化する。
[図12]広域インテグリン小分子阻害剤(SMI)を用いて、標準ケア(SOC)がある場合となしでフィブロネクチン結合インテグリンの薬理阻害を用いて、確立されたPAHのMCT-ラットモデルにおけるPAHを逆転させる図である。SOCとSMIの併用はRV機能を保持している.
他の新しいインテグリン計画は
我々の戦略は,複数のインテグリン標的の小分子を認識することができ,これらの機構を深く問い合わせることを可能にしている。NASH,線維狭窄,胆道線維化,肺動脈高圧などの線維化に関する指標に,より多くのインテグリン調節剤計画が求められている。形質転換増殖因子-βの活性化、機械的形質導入、細胞遷移と細胞増殖におけるインテグリンの作用により、異なる病理生理状態下で、インテグリンは異なる経路の起動或いは繊維化を誘発する可能性があり、その阻害剤は異なる器官システムの治療に非常に適している可能性がある。これらのプロジェクトは異なる発見段階にあり、少なくとも1つのプロジェクトは今後12~18ヶ月以内にリーダーシップ最適化に移行すると予想される。
許可協定
AbbVie協定
2018年10月、我々は線維化関連適応に対するαvβ6特異的インテグリン計画を含む一連の経口インテグリン療法を推進するためにAbbVieと研究開発協力を達成した。
AbbVieはその後、標的であることが疑われるので、αvβ6特異的インテグリン阻害剤を選択的に経口投与することを意図していないことを私たちに通知した αvβ6前臨床試験において観察された媒介安全信号の詳細が2022年12月の毒理学学会誌に発表された。2022年6月、AbbVieは私たちに、便宜のために、2022年12月に発効する権利を行使してAbbVie協定を終了することを決定したので、私たちはAbbVie協定の下でいかなる追加支払いも受けないだろうと通知した
ヤンソン協定
2019年2月,われわれはJanssenと合意し,既存療法が十分に解決できなかった疾患患者のために新たなインテグリン療法を発見·開発した。Janssenの協力は3つのインテグリン標的に集中しており、各標的は研究計画のテーマであり、私たちが探索したことのない2つまでのインテグリン標的を置き換えることができる
2023年1月、Janssenは、その権利の行使を決定し、便宜上Janssen協定を終了することを決定したが、60日間の通知期間が必要であることを私たちに通知した。もし私たちとJanssenが同意すれば、Janssen協定は2023年3月またはそれ以上に終了するだろう
シュレーディンガー協定
2015年6月、私たちはシュレーディンガーと協力協定(改訂された)を達成し、私たちが選択した薬物標的を探索した。協力により、シュレーディンガーはその技術プラットフォームを用いて仮想スクリーニングを行い、著者らとシュレーディンガーは協力して目標の優先順位を確定し、目標検証と分析を実行し、手がかりを識別し、手がかりの最適化を行う。合意条項によると、Schrödingerは合意期間内に私たちとインテグリン目標について独占的に協力するだろう。Schrödingerは協力の下で行われた活動を考慮して,約340万シリーズの種子優先株を受け取った。さらに、協力の一部として決定された化合物については、シュレーディンガーは、開発マイルストーンに関連するいくつかの支払いを私たちから得る資格があり、総額310万ドル以下であり、目標ごとに計算し、鉛最適化および化合物1つずつを開始した上でより低い6桁の支払いを支払い、このような化合物を含む製品のより低い桁の特許使用料を販売する資格がある可能性がある。さらに、私たちは、そのような化合物の採掘権を許可またはそのような第三者に譲渡することに関連する第三者から受信されたいくつかの支払いの中央桁パーセントと、2019年に支払われた使い捨て費用100万ドルとをSchrödingerに支払うことに同意した。シュレーディンガーは特定の時間枠内で一定の数の発展マイルストーンを実現できなかった場合を含む“シュレーディンガー協定”を中止することができる。
2022年12月には,特殊なシュレーディンソフトウェアクライアントとしてのアクセス範囲を拡大し,そのソフトウェアキットの使用がインテグリンの範囲を超えている
児童医療センター会社協定
2015年10月、私たちは、シュプリンガー博士がボストン児童病院で働いている間に開発されたインテグリンを抑制する技術に関するCMCCまたはCMCC協定と独占的な許可協定(改訂)を締結した。本協定によれば、我々は、任意の治療または診断用途のための製品を世界的に開発および商業化するために、いくつかの特許権の下で独占的許可を有し、特定の独自技術の下で非独占的許可を有する
人間と獣医の応用ですまた、Springer博士の研究室で生成された新しい特許権およびノウハウを、公平な市場価値に一致する追加支払いを得るために、プロトコルの発効日後の特定の時間に追加することを選択することができる。許可証を授与する代償として、中国移動協定に署名した後、私たちは完全償却基準に従って中国にいくつかの普通株を移動発行し、当時発行されたおよび発行された単位の6%に相当する。私たちはまた中国に50,000ドルの前払い許可証発行費を支払い、中国移動にいくつかの特許訴訟費用を精算した。また、CMCC協定の発効日後の最初の3年間に許可維持費、いくつかの開発マイルストーン、私たちが入手可能な再許可収入の一定の割合、およびライセンス製品の純売上高でより低い桁の使用料を支払うことに同意した。
“中国移動通信協定”によると、我々は、商業的に合理的に1つまたは複数の許可製品を市場に投入し、合意が規定する時間範囲内で開発計画における活動を実施するように努力することに同意する。また、私たちが1つ以上の特定の発展マイルストーンに到達できず、適切な是正措置が取られていない場合、中国移動は合意を終了する権利がある。
知的財産権
私たちの成功は、(I)私たちの候補製品および関連方法に関連する知的財産権、および(Ii)インテグリン構造およびこれらの構造の調節剤を生成するためのMINTプラットフォームの能力にある程度依存する。私たちの成功はまた、私たちの候補製品が商業化され、承認されれば、他の人が私たちの特許権を侵害することを防止するために、運営の自由を持つことにかかっている。私たちは商業秘密、ノウハウ、そしてごく少数の場合の特許を使用して私たちのミントプラットフォームを保護する。私たちは特許を使用して私たちの小分子製品を保護し、私たちの政策は重要な司法管轄区で製品特許保護を求めることであり、アメリカ、主要ヨーロッパ諸国、その他の私たちが適切だと思う司法管轄区、あるいは私たちの協力協定が要求する他の司法管轄区を含む。
我々は、インテグリン活性を調節する小分子阻害剤を含む組成物、化合物自体、そのような化合物の疾患治療のための使用、および関連する製造方法について特許を出願している。
特許権
我々は、修飾インテグリンポリペプチドおよび修飾インテグリンポリペプチド二量体に関連する権利主張を含む、中国移動が発行した米国特許および出願中の米国特許を独占的に許可した。ライセンスを取得した米国特許と、係属中の米国特許出願から発行される可能性のある他のいかなる米国特許も2035年に満了する予定であり、いかなる調整や延長も行われない。また,我々のMINTプラットフォームは商業秘密保護に広く依存しており,これは最初に中国移動から許可を得たインテグリン技術を超えている.
2022年12月31日現在、我々は、米国および世界の多くの他の主要な司法管轄区域(ヨーロッパ、日本、および中国を含む)において、α4β7インテグリンに関連する治療適応を有する物質成分および使用方法に関する様々な特許および承認すべき特許を発行している。我々のα4β7号化合物に対する特許(または特許出願、付与されている場合)の予想満了日は、国の法律に従って利用可能な任意の延長または調整期間に加えて、2039年から2041年である
知的財産権保護
我々が追求している特許出願が任意の特定の管轄区域で特許として発行されるかどうか、または発行された特許の権利主張が競合相手に対する特許保護を提供するかどうかを予測することはできない。さらに、特許訴訟中に取られた行動のため、発行された任意の特許は、ある日以降に特許を提出する期間のような免責声明を提出するなど、上記で開示された予期された期限の前に満了する可能性がある。私たちの未解決特許出願が発行された特許として付与されても、これらの特許および第三者から許可された任意の特許は、第三者によって挑戦、回避、または無効にされる可能性がある。上述したいずれの特許出願に関連する論争のある訴訟や第三者クレームは現在のところないが、遅い時期または一旦特許が付与されると、このような訴訟や第三者クレームは行われない保証はない。
特許の期限は,特許を取得した特定国の特許の法的期限に依存する。私たちが出願したほとんどの国では,特許期間は非臨時特許出願が提出された最初の日から20年である。米国では、FDAによって承認された薬物をカバーする特許期限は、FDA規制審査中に失われた特許期限の補償として、場合によっては特許期限を回復する資格がある可能性がある。場合によっては、“ハッジ·ワックスマン法案”は、米国特許が延長されていない期限の後に最大5年間の特許期間の延長を許可する。特許期間の延長の長さは,承認された薬物が規制審査を受ける時間の長さと関係がある。特許期間の延長は,製品が承認された日から14年間の余剰特許期間を超えてはならず,承認された薬物に適用される特許を延長することしかできない。ヨーロッパでは供給があります
他の外国司法管轄区域は、承認された薬物をカバーする特許の期間を延長するか、または特許が満了した後に承認された薬物に追加の保護期間を提供する。将来、私たちの製品がFDAの承認を得たら、私たちはこれらの製品をカバーする特許出願のために特許期間を延長する予定です。私たちは、特許を得ることができる任意の管轄区域で、私たちが発行した任意の特許のために特許期間の延長を求めることを計画しているが、米国特許商標局と欧州国家特許庁を含む適用当局は、このような延長を承認すべきかどうか、承認された場合、期限を延長すべきかどうかの評価に同意するであろう。
私たちの発明、候補製品、および研究プロジェクトは特許保護に依存するほか、私たちは商業秘密に依存して私たちの機密および独自の情報を保護する。例えば、私たちのミントプラットフォームのいくつかの要素は、開示されていない非特許商業秘密に基づくことができる。私たちは、当社の従業員やコンサルタントと契約を締結することを含む、当社の独自情報およびビジネス秘密を保護する措置をとっていますが、第三者は、実質的に同じ独自の情報および技術を独立して開発したり、他の方法で私たちのビジネス秘密を取得したり、当社の技術を開示したりすることができます。したがって、私たちは私たちの商業秘密を意味的に保護することができないかもしれない。私たちの政策は、私たちの従業員、コンサルタント、外部科学協力者、協賛研究者、および他のコンサルタントに、私たちとの雇用や相談関係を開始する際に秘密協定を実行することを要求します。これらの合意は、当方との関係過程において、関係個人又は実体に開示されたすべての当方の業務又は財務に関連する機密情報を秘密にしなければならず、特定の場合を除き、第三者に開示してはならないと規定している。従業員の場合、合意は、個人的に構想された、私たちの現在または計画中の業務または研究開発に関連するもの、または通常の勤務時間内、私たちのオフィス内で行われる、または私たちの設備または独自の情報を使用するすべての発明が、私たちの固有財産であることを規定している。また、第三者が私たちのノウハウを盗用することを防止するために、物理的および技術的セキュリティ対策のような他の適切な予防措置を講じている。私たちはまた政策を制定し、訓練を行い、ビジネス秘密を保護する期待とやり方を指導してくれた。
製造業
現在、著者らはすべての臨床薬物の製造、貯蔵、流通或いは品質測定のための臨床製造施設はすべて第三者メーカーにアウトソーシングしている。我々の開発計画の進展と新たなプロセス効率の構築に伴い,登録試験の需要を満たし,承認されれば,商業製品の製造,販売,流通を満たすことを目指してこの戦略を評価し続ける予定である
競争
生物技術と医薬業界は技術進歩が速く、競争が激しく、知的財産権保護が強いという特徴がある。私たちのMINTプラットフォームと私たちの知識、経験と科学資源は私たちに競争優位を提供してくれると信じていますが、私たちは主要な製薬とバイオテクノロジー会社、学術機関、政府機関、公共と個人研究機関などからの競争に直面しています。
我々が開発·商業化に成功したどの候補製品も,現在承認されている療法や将来発売される可能性のある新しい療法と競争するであろう。私たちが他の療法と効果的に競争する能力に影響を与える重要な製品機能には、私たちの製品の有効性、安全性、利便性があります。
バイオ製薬業界は大量の投資を行っているにもかかわらず、アメリカとヨーロッパでは経口インテグリン療法が承認されていない。経口小分子α4β7特異的インテグリン阻害剤Morf−057を開発している IBDの治療に用いられている。現在承認されているIBD療法には,武田製薬有限会社が販売している注射可能なα4β7モノクロナル抗体Entyvio(Vedolizumab)と,AbbVie,ジョンソン,UCB,Biogen Inc.,ファイザーや百時美施貴宝などの製薬会社が販売している異なる作用機序を有する療法があり,承認されれば候補製品が競合する可能性がある。また,主役治療会社,ジリッド科学社,EA製薬有限会社が臨床開発した経口α4β7療法や,エバービー,ジョンソン,ファイザー社,礼来社,百時美施貴宝などの製薬会社が臨床開発において異なる作用機序を有する療法が知られている。
我々のαvβ8特異的小分子インテグリン阻害剤計画は,骨髄線維化や固形腫瘍の治療に開発されている。現在承認されている骨髄線維化治療薬には,Incell社とノワール国際社が販売している経口JAK阻害剤Jakafi(Ruxolitinib),百時美施貴宝社が販売しているInrebic(Fedratinib),CTI Biophma社が販売しているVonjo(パリチニブ)が含まれており,グラクソ·スミスクライン社,MorPhoSys AG,Incell Corp,Geron社,Abbvie,百時美施貴宝社が臨床開発している骨髄線維化療法が知られている。現在,αvβ8阻害剤のいかなる適応も承認されていない。ファイザー社は固形腫瘍治療のための抗αvβ8モノクロナル抗体を開発していることが知られている。また,Venn TreeuticsやCorbus PharmPharmticals Holdings,Inc.固形腫瘍に対する臨床前段階抗αvβ8モノクロナル抗体計画,Pplant Treeuticsの小分子計画も知られている。また、
ノワール国際会社、エバーヴィ社、羅氏ホールディングス会社、メルク社、百時美施貴宝会社と学者岩石会社及びその他の製薬会社はすべて転化成長因子-β経路に対する多種の抗体と小分子療法を開発し、固形腫瘍の治療に応用している。
私たちの多くの競争相手は研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング許可を得た製品の面で私たちより多くの財務資源と専門知識を持っている。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。また,我々の競争相手には,自己免疫,心血管および代謝性疾患,線維化および癌を含む,我々が目指している同じ治療分野で治療法を開発している会社も含まれている可能性がある。
政府の監督管理
その他の事項以外に、アメリカ連邦、州と地方各級及びその他の国家と司法管轄区の政府当局は薬品の研究、開発、テスト、製造、品質管理、承認、包装、貯蔵、記録、ラベル、広告、販売促進、流通、マーケティング、承認後のモニタリングと報告及び輸出入などの方面に対して広範な監督管理を行った。米国や他の国や管轄区域で規制の承認を得る手続きや、その後適用される法規や条例、その他の規制当局の遵守には、多くの時間と財力が必要だ。
FDA承認プロセス
アメリカでは、医薬品はFDAによって広く規制されている。連邦食品、薬物と化粧品法案或いはFD&C法案及びその他の連邦と州法規と規定は、他の事項以外に、薬品の研究、開発、テスト、製造、貯蔵、記録保存、承認、ラベル、販売促進とマーケティング、流通、承認後の監視と報告、サンプリングと輸出入を管理する。適用されない米国の要求を遵守しないことは、臨床封印、FDAが未解決のNDAの承認を拒否すること、警告または無タイトル手紙、製品リコール、製品差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、民事処罰、および刑事起訴のような様々な行政または司法制裁を受ける可能性がある。
米国では、新製品または承認製品のいくつかの変更に対する医薬製品の開発は、一般に、FDA承認を求める各適応に対する薬剤の安全性および有効性を決定するために、臨床前実験室および動物試験、試験新薬申請またはIND(臨床試験開始前に発効しなければならない)および十分かつ制御された臨床試験をFDAに提出することに関連する。FDA上場前の審査要求を満たすには通常長年の時間を要し、実際の所要時間は製品或いは疾病のタイプ、複雑性と新規性によって大きく異なる可能性がある。
臨床前試験は製品の化学、調合と毒性に対する実験室評価、及び製品特性と潜在安全性と有効性を評価する動物試験を含む。臨床前試験の進行は必ず良好な実験室実践を含む連邦法規と要求に符合しなければならない。臨床前試験の結果はINDの一部として他の情報とともにFDAに提出され,製品化学,製造,制御に関する情報,提案された臨床試験案が含まれている。IND提出後,生殖毒性や発ガン性の動物試験など,長期的な臨床前試験を継続する可能性がある。ヒト臨床試験を開始する前に,各INDの提出後30日間の待機期間が求められている。FDAがこの30日間INDにコメントもINDにも疑問を提起しなければ,INDで提案された臨床試験が開始される可能性がある。臨床試験は合格した研究者の監督の下で、健康ボランティア或いは患者に研究用新薬を提供することに関連する。臨床試験は、(I)連邦法規に適合する;(Ii)良好な臨床実践またはGCPに適合し、これは、患者の権利および健康を保護し、臨床試験発起人、管理者および監督者の役割を定義するための国際基準であり、(Iii)試験目標を詳細に説明し、安全性を監視するためのパラメータおよび評価すべき有効性基準のプロトコルである。米国患者のテストに関する各々のプログラムおよび後続のプログラム修正案は、INDの一部としてFDAに提出されなければならない。
FDAが臨床試験がFDAの要求に沿って行われていないと考えている場合,あるいは臨床試験患者に受け入れられないリスクとなっている場合,FDAはいつでも臨床試験の一時的または永久的な停止を命じたり,他の制裁を加えたりすることができる。臨床試験における患者の研究案やインフォームドコンセント情報も機関審査委員会やIRBや倫理委員会に提出しなければならない。IRBはまた、IRBの要求を遵守できなかったために、現場の臨床試験を一時的または永久的に停止することを要求することができ、または他の条件を適用することができる。
ニューノミンの発売承認を支持する臨床試験は通常3つの連続段階で行われるが、これらの段階は重なる可能性がある。第1段階、すなわち、最初に健康なヒト対象または患者に薬物を導入した場合、新陳代謝、薬物動態、薬理作用、用量増加に関連する副作用、および可能であれば有効性を評価するための早期証拠を評価するために、薬物が試験される。第2段階は、一般に、特定の適応、用量耐性、および最適用量での薬剤の有効性を決定し、よく見られる副作用および安全リスクを決定するために、限られた患者集団で試験を行うことに関連する。薬剤が第2段階評価において有効性および許容可能な安全性を証明する場合、第3段階試験は、FDAが薬剤の全体的な利益-リスク関係を評価し、薬剤のラベルに十分な情報を提供することを可能にするために、より多くの患者の臨床治療効果および安全性に関する追加の情報を得るために実施される。多くの場合、FDAはこの薬物の治療効果を証明するために、十分かつ良好にコントロールされた2つの3期臨床試験を必要とする。ごく少数の場合、単一の3期試験で十分である可能性があり、(1)研究は大規模な多中心試験であり、内部一致性を示し、統計学的に非常に説得力があり、死亡率、不可逆的な発病率または疾患の予防に臨床的意義のある影響を発見し、潜在的な深刻な結果を有し、第2の試験で結果を確認することは実際的にまたは倫理的に不可能であるか、または(2)他の確認性証拠と結合することを含む。
深刻または生命に危険な疾患の2期または3期臨床試験を行う研究薬剤の製造業者は、例えば、そのウェブサイト上に掲示することによって、その評価およびアクセス拡大要求に応答するためのその政策を提供しなければならない。
必要な臨床試験が完了した後,NDAを用意してFDAに提出する。この製品が米国市場で販売されるようになる前に、FDAが製品を承認する必要がある。NDAは、すべての臨床前、臨床および他の試験の結果、および製品の薬理、化学、製造および制御に関連するデータアセンブリを含まなければならない。秘密協定を準備して提出する費用は巨大だ。NDAの提出の多くは高額な申請使用料を支払う必要があり、現在2023年度には3,200,000ドルを超えており、承認されたNDAによると、製造業者および/またはスポンサーは年間計画費用を支払う必要があり、現在2023年度には、処方薬1個あたりの年会費は39,000ドルを超えている。これらの費用は通常毎年増加します。孤児薬の指定を受けた薬物を申請した発起人はこれらの使用料を免除することができる。
FDAがNDAを受信してから60日後には、実質的な審査を可能にするために、機関の敷居に基づいて申請が十分に完全であるかどうかが決定されるかどうかを決定するために60日がある。文書が提出されると、FDAは深い検討を始めた。FDAはNDAを審査する際にいくつかの業績目標を設定し、即時性を奨励することに同意した。標準審査薬の申請の多くはFDAにNDAを提出した日から10~12カ月以内に審査を行い,多くの優先審査薬品の申請はFDAにNDAを提出した日から6~8カ月以内に審査を行う。優先審査は、FDAが治療において大きな進展を得るか、または適切な治療方法がない場合に治療を提供する薬剤を決定するのに適用することができる。FDAは、いくつかの遅延された情報を考慮するために、または提出中に提供された情報の情報を明確にするために、標準審査および優先審査の審査手続きをさらに3ヶ月延長することができる。
FDAはまた、新薬製品の申請、または安全性または有効性の問題を有する医薬製品の申請を外部諮問委員会に提出することができる−通常、臨床医および他の専門家を含むグループである−審査、評価を行い、申請を承認すべきかどうかについて提案することができる。FDAは諮問委員会の提案によって制限されていないが、それは一般的にそのような提案に従っている。
NDAを承認する前に、FDAは、通常、GCPに適合することを確実にするために、1つまたは複数の臨床場所を検査する。さらに、FDAは薬物を製造する1つ以上の施設を検査するだろう。FDAは、現在の良好な生産実践またはcGMPに適合しない限り、この製品を承認せず、NDAに含まれるデータは、研究された適応において安全かつ有効であることを証明する大量の証拠を提供する。
FDAがNDAと製造施設を評価した後、それは承認状または完全な返信を発行するだろう。完全な応答文は、一般に、提出中の不足点を概説し、FDAが出願を再検討するために、大量の追加のテストまたは情報を必要とする可能性がある。NDAが再提出されたとき、またはいつ、これらの欠陥がFDAによって満足的に解決された場合、FDAは承認書を発行するであろう。FDAは、含まれる情報タイプに応じて、そのような再提出された出願を2ヶ月または6ヶ月以内に検討することを約束している。この薬物の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。NDA承認の条件として、FDAは、薬物の利点が潜在的リスクよりも大きいことを確実にするために、リスク評価および緩和戦略、またはREMSを必要とする可能性がある。REMSは、薬物ガイドライン、医療専門家のコミュニケーション計画、および安全な使用を確保する要素、またはETASUを含むことができる。ETASUは、処方または調剤のための特別なトレーニングまたは認証、場合によっては調剤、特殊な監視、および患者登録簿の使用を含むことができるが、これらに限定されない。REMSに対する要求はこの薬物の潜在的な市場と収益力に重大な影響を与える可能性がある。さらに、製品承認には、薬物の安全性または有効性を監視するために、大量の承認後の試験および監視が必要となる可能性がある。承認されると、規制基準が守られていない場合、または最初のマーケティング後に問題が発見された場合、製品承認は撤回される可能性がある。
承認された出願において確立されたいくつかの条件の変更は、適応、ラベルまたは生産プロセスまたは施設の変更を含み、変更を実施するためには、新しいNDAまたはNDA付録を提出し、FDAの承認を得る必要がある。新適応のNDAサプリメントは通常,オリジナル申請と類似した臨床データが必要であり,FDAがNDAサプリメントを審査する際に使用するプログラムや行動は,NDAを審査する際に使用するプログラムや行動と同じである。
迅速な承認指定と承認の加速
FDAは、深刻または生命に危険な疾患や疾患を治療するための薬剤の開発と審査の加速を促進することが求められており、これらの薬剤には有効な治療法がなく、このような疾患が満たされていない医療ニーズを解決する潜在力を示している。迅速チャネル計画によると,新薬候補のスポンサーは,薬剤候補のINDの届出と同時にまたはその後,特定の適応の候補薬を迅速チャネル薬として指定することをFDAに要求することができる。FDAはスポンサーからの要請を受けて60日以内に候補薬剤が迅速チャネル指定を受ける資格があるかどうかを決定しなければならない。提出された申請が高速チャネル指定を受けた場合、スポンサーはFDAとより頻繁に相互作用することができ、FDAは申請が完了する前にNDAの内容の一部を審査する可能性がある。申請者が余剰情報を提出するスケジュールを提供し、申請者が適用された使用料を支払うことができれば、スクロール審査を行うことができる。しかしながら、FDA審査申請の期間目標は、NDAの最後の部分が提出されてから開始される。また,FDAが高速チャネルの指定が臨床試験中に出現したデータの支持を得なくなったと考えた場合,FDAはその指定を撤回する可能性がある。
FDAの加速承認規定によると、FDAは深刻または生命に危害を及ぼす疾患に対する薬物を許可することができ、この薬物は患者に既存の治療よりも意義のある治療利益を提供し、その基礎は臨床利益を合理的に予測する可能性のある代替終点、あるいは不可逆的な発病率或いは死亡率よりも早く測定できる臨床終点であり、深刻さを考慮して、不可逆的な発病率或いは死亡率或いは他の臨床利益への影響を合理的に予測する可能性が高い。このような状況の稀または一般的、および代替治療の獲得可能性または欠如。承認経路を加速させることは、製品の臨床的利益を検証および説明するために、追加の承認後の検証的研究を行うことにスポンサーが同意することに依存する。これらの検証試験は、職務調査の場合に完了しなければならず、多くの場合、FDAは、承認前に設計、起動、および/または完全に試験に組み込むことを要求する可能性がある。必要な承認後研究を行わない場合、あるいは発売後の研究期間中に臨床利益が確認できなければ、FDAがこの製品の市場からのリコールを加速することを許可する。
最近“食品·薬物総合改革法案”(FDORA)が公布され、承認経路の加速に関する条項が含まれている。FDORAによれば、FDAは、承認前または承認後の特定の期間にわたって承認後の検討を要求することを許可される。FDORAはまた、研究完了の目標日のようなマイルストーンを含むことができ、必要な承認後の研究の進捗報告書の提出をスポンサーに要求するとともに、承認後180日よりも遅くなく、研究完了または終了後180日毎の頻度を下回ることなく、FDAに必要な承認後研究の条件を規定することを要求する。FDORAは、FDAがFDAが規定したいかなる要求条件を満たしていないか、または適時に報告を提出できなかったことを含む、職を全うするために必要な承認後の研究ができなかった状況に対して法執行行動をとることを可能にする。
臨床試験において、代替終点は1種の疾病或いは状況の実験室或いは臨床症状の測定であり、それは患者の感覚、機能或いは生存方式の直接測定の代わりになる。代替終点は通常、臨床終点よりも容易または迅速に測定される。この基礎の上で承認した候補薬物は必ず厳格な発売後のコンプライアンス要求を遵守しなければならず、4期或いは承認後の臨床試験を完成し、臨床終点への影響を確認することを含む。必要な承認後研究を行わない場合や,発売後の研究期間中に臨床的利益が確認できなければ,FDAが市場からのリコールを加速させることが可能である。加速規制により承認された候補薬物の宣伝材料はすべてFDAの優先審査を経なければならない。
突破的治療指定
FDAはまた、重症または生命に危険な疾患または状態を治療するための薬物承認申請の開発および検討を加速することを要求されており、初歩的な臨床証拠が示されている場合、薬剤は1つまたは複数の臨床的に重要な終点で既存の治療法よりも実質的に改善されている可能性がある。画期的療法計画によると,新薬候補のスポンサーは,薬剤候補のIND提出と同時にあるいはその後,特定適応の候補薬を画期的療法として指定することをFDAに求めることができる。FDAはスポンサーの申請を受けてから60日以内に候補薬が突破療法指定を受ける資格があるかどうかを決定しなければならない。
孤児薬
孤児医薬品法によると、FDAは、まれな疾患や状態を治療するための薬剤に孤児薬物の称号を付与することができる。米国では、通常、20万人の病気や状況に影響を与えない。NDAを提出する前に、孤児薬物の称号を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、この薬剤の模倣薬識別情報およびその潜在的孤児用途を開示する。孤児薬物指定は、規制審査および承認過程においていかなる利点も伝達されず、規制審査および承認過程の持続時間を短縮することもない。FDAによって承認された特定の疾患の特定の活性成分を治療するための最初のNDA申請者は、FDA孤児薬物の称号を有し、米国でこの製品のこの適応に7年間の独占営業期間を提供する権利がある。7年間の排他的期間内に、FDAは、限定された場合、例えば、孤児薬物に対して排他的な製品と比較した臨床的利点を示さない限り、同じ疾患のために同じ薬物を販売する他のいかなる出願も承認しない可能性がある。孤児薬物排他性は、FDAが同じ疾患または状態に対する異なる薬物、または異なる疾患または状態に対する同じ薬物を承認することを阻止しない。孤児薬物指定の他の利点は、いくつかの研究の税金控除およびNDA申請使用料の免除を含む。
臨床試験情報の開示
FDA規制製品(薬品を含む)の臨床試験スポンサーはいくつかの臨床試験情報を登録し、開示しなければならない。製品、患者集団、調査段階、研究場所と研究者、および臨床試験の他の方面に関する情報は、その後、登録の一部として公開される。スポンサーも完成後に彼らの臨床試験結果を検討する義務がある。場合によっては、これらの裁判結果の開示は、裁判が完了した日から最大2年に延期されることができる。競争相手はこれらの公開された情報を用いて開発計画の進捗状況を知ることができる.
小児科情報
“小児科研究公平法”によると、新薬または新薬補充剤は、すべての関連小児科亜群で薬物が主張する適応の安全性と有効性を評価し、薬物に対して安全かつ有効な各小児科亜群の投与量および投与を支持するためのデータを含まなければならない。FDAは、提出されたデータの全部または一部を免除または延期することができる。PREAは,いくつかの例外を除いて,孤児の称号が付与されたいかなる適応薬にも適用できない。
小児最適医薬品法案、またはBPCAは、いくつかの条件が満たされる場合、任意の薬物の排他的-特許または非特許-であるNDA保持者に6ヶ月の延長を提供する。排他的条件は、FDAが小児科集団における新薬の使用に関連する情報がその集団の健康に利益をもたらす可能性があることを決定すること、FDAが小児科研究の書面請求を提出すること、および申請者が法定時間内に要求された研究を行うことに同意し、報告することを含む。BPCA下の出願は優先出願とみなされ,与えられたすべての利点を指定することができる.
承認後に要求する
機密協定が承認されると、製品はいくつかの承認後に要求される制約を受けるだろう。例えば、FDAは薬品の承認後のマーケティングと販売促進を密接に監督し、消費者向けの広告、ラベル外販売促進、業界賛助の科学と教育活動及びインターネットに関連する販売促進活動の基準と法規を含む。薬品は承認の適応と承認されたラベルの規定でしか販売できない。
FDAがNDAを承認した後,有害事象報告と定期報告を提出する必要がある。FDAはまた、上場後試験、いわゆる第4段階試験、リスク評価および緩和策、またはREMS、ならびに承認製品の効果を監視することを要求することができ、またはFDAは、承認時に条件を追加して、製品の流通または使用を制限する可能性がある。また,品質管理,薬品製造,包装,ラベルプログラムは承認された後もcGMPに適合し続けなければならない。医薬品製造業者と彼らのいくつかの下請け業者はFDAといくつかの州機関に彼らの工場を登録することを要求された。FDAの登録要求エンティティはFDAの定期抜き打ち検査を受け,その間,FDAは製造施設を検査し,cGMPの遵守状況を評価する。そのため,メーカーはcGMPの遵守を維持するために,生産や品質管理の分野で時間,お金,精力をかけ続けなければならない。ある企業が規制基準を遵守できなかった場合、初期マーケティング後に問題に遭遇した場合、または後に以前に意識されていなかった問題が発見された場合、監督管理機関は製品の承認を撤回したり、製品のリコールを要求したりすることができる。
“ハッジ·ワックスマン法案”
オレンジ図書リスト
秘密協定を介して薬物の承認を求める場合、出願人は、その特許請求が出願人製品をカバーする各特許をFDAにリストすることを要求される。1つの薬剤が承認されると、医薬出願に列挙された各特許は、一般にオレンジブックと呼ばれるFDAによって承認された治療同等性評価を有する医薬製品に開示される。逆に、オレンジブックに記載されている薬物は、簡略化された新薬申請、すなわちANDAの承認を支持するために、潜在的な模倣薬競争相手によって参照されることができる。ANDAが規定して販売されている医薬製品は,列挙した薬剤と同じ強度と剤形の同じ活性成分を有し,生物学的同等性試験により治療上列挙された薬剤と同じであることが証明されている。生物学的同等性テストの要求以外に、ANDA申請者は臨床前或いは臨床テスト結果を行ったり提出したりする必要がなく、その薬物製品の安全性或いは有効性を証明する。このようにして承認された薬物は一般に市販薬の“模倣等価物”と呼ばれ,通常薬剤師が元の市販薬のために処方された処方に基づいて代替できる。
ANDA申請者はFDAが承認した製品がFDAのオレンジブックに記載されている任意の特許をFDAに証明しなければならない。具体的には、出願人は、(I)要求された特許情報がまだ提出されていないこと、(Ii)に記載されている特許が満了していること、(Iii)に記載されている特許が満了していないが、特定の日に満了し、特許が満了した後に承認を求めること、または(Iv)に記載された特許が無効であるか、または新製品の侵害を受けないことを証明しなければならない。ANDA出願人はまた、列挙された使用方法特許を証明するのではなく、第VIII節の声明を提出し、その提案されたANDAタグが特許使用方法に関するいかなる言語も含まない(または彫刻)ことを証明することを選択することができる。出願人が列挙された特許に挑戦していない場合、ANDA出願は、参照製品を必要とするすべての特許が満了するまで承認されないであろう。新製品が承認された製品の上場特許又はそのような特許を侵害しない無効な認証を第4項認証と呼ぶ。ANDA出願人が第4段落の認証をFDAに提供している場合、FDAがANDAの届出を受けると、出願人はまた、NDAおよび特許所有者に第4段落の認証の通知を送信しなければならない。そして、NDA及び特許所有者は、第4項の認証の通知に対して特許侵害訴訟を提起することができる。第4項の認証を受けてから45日以内に特許侵害訴訟を提起すると、特許満了、訴訟和解または侵害事件におけるANDA申請者に有利な裁決の30ヶ月前まで、FDAによるANDAの承認が自動的に阻止される。
ANDA出願もオレンジブックに記載されている引用製品の任意の適用の非特許排他性が満了するまで承認されないであろう。
排他性
NDAが新しい化学物質またはNCEを承認すると、すなわち、FDAが任意の他のNDAで承認された活性部分を含まない薬剤は、5年間の市場排他性を得ることになり、その間、FDAは、薬剤模倣薬バージョンの承認を求めるANDAを得ることができない。第4項の認証が提出された場合、ANDAはNCE排他性満了の1年前に提出することができる。Orange Bookに記載されている特許がなければ,4段目の認証がない可能性があるため,排他期間が満了するまでANDAを提出することはできない.薬物のいくつかの変化は、例えば、パッケージ挿入において新たな適応を増加させ、スポンサーによるまたはスポンサーによる承認申請に重要な新しい臨床研究(バイオアベイラビリティ研究を除く)の報告を含む場合、3年間の排他的期間の主題である可能性がある。FDAは排他期間内に変化した後発薬を含むANDAを承認することはできない。
特許期間を延長する
NDA承認後,関連薬物特許の所有者は最長5年間の特許延期を申請することができる。許容される特許期間延長は、薬物試験段階の半分(IND出願とNDA提出との間の時間)とすべての審査段階(NDA提出と承認との間の時間)として計算され、最長5年である。FDAが出願人が職務調査を経て承認を求めていないと判断した場合、時間を短縮することができる。継続後の特許の総期限は14年を超えてはならず,1つの特許しか更新できない.出願段階で満了する可能性のある特許については,特許権者は臨時特許延期を請求することができる。臨時特許の延期は特許期間を1年間延長し,最大4回延長することができる.暫定特許の承認が延期されるごとに、承認後の特許延期は1年減少する。米国特許商標局の取締役は,特許延期を申請している特許に含まれる薬物が承認される可能性が高いことを確認しなければならない。秘密保持協定を提出していない薬物は一時的な特許延期を受けることができない。
他の医療保険法
FDAの薬品マーケティングに対する制限以外に、近年、他のいくつかのタイプの州と連邦法律を応用して製薬業界のいくつかの一般的な商業とマーケティングやり方を制限している。これらの法律には反リベート法規、虚偽請求法規、および他の医療保健法律法規が含まれている。
他の事項に加えて、連邦反バックル法規は、Medicare、Medicaid、または他の連邦によって援助された医療計画に従って精算可能な任意の医療項目またはサービスを誘導または見返りとして購入、レンタル、注文または購入、レンタル、またはMedicaid、または他の連邦によって援助された医療計画に従って精算することを誘導または発注することを意図的に提供、支払い、請求または受け取ることを禁止する。“保健と教育和解法案”によって改正された“患者保護と平価医療法案”は、総称してACAと呼ばれ、連邦法規の意図部分を改正し、個人或いは実体が法規或いは法規違反の具体的な意図を実際に理解する必要がなく、違反行為を実施できるようにした。この法規は,薬品メーカーと処方者,購入者,処方管理人などの間の手配に適用されると解釈されている。いくつかの法定例外と規制避風港はいくつかの一般的な活動を起訴や他の規制制裁から保護しているが、例外と避風港の範囲は狭く、処方、購入または推薦を誘導するための報酬に関するやり方が例外や避風港の資格に適合していなければ、審査される可能性がある。
連邦民事虚偽請求法案を含む連邦民事および刑事虚偽請求法は、任意の個人または実体が虚偽請求を意図的に連邦政府に提出するか、または虚偽請求を故意に行うか、または虚偽請求を支払うために虚偽陳述を引き起こすことを禁止する。これには,連邦政府が連邦供給スケジュール外で購入した場合のように,連邦政府が直接購入者である項目(例えば連邦医療保険や医療補助など)の精算項目に対するクレームが含まれる.最近,いくつかの製薬や他の医療保険会社がこれらの法律に基づいて訴訟を起こしているが,価格設定サービス機関に報告されている薬品価格が上昇しているのに対し,定価サービス機関は連邦医療保険や医療補助の販売率を設定するために使用されており,顧客に製品を無料で提供している疑いがあり,顧客はその製品の連邦計画に課金することを希望している。さらに、ラベル外販売促進を含むいくつかのマーケティング行為は、虚偽クレーム法律に違反する可能性もある。また,ACAは連邦反リベート法規を改正し,この法規に違反した行為を連邦民事虚偽クレーム法案の規定の責任基盤とすることができるようにした。ほとんどの州にも連邦反リベート法規や民事虚偽請求法案のような法規や法規があり、医療補助や他の州で計画されている精算プロジェクトやサービスに適用されたり、いくつかの州では支払者にかかわらず適用されています。
医療詐欺および乱用に関連する他の連邦法規には、医療補助または医療保険受益者への報酬の提供または支払いが禁止された民事罰金法規が含まれており、これが受益者に特定のサプライヤーから精算可能な物品またはサービスを得るように受益者に命令することに影響を与える可能性があることを知っているか、または知るべきである場合、1996年に作成された追加連邦刑法(HIPAA)は、任意の医療福祉計画または虚偽または詐欺的な言い訳によって得られた計画を故意かつ故意に実行または実行しようと試みる追加連邦刑法を禁止し、任意の医療福祉計画が、医療福祉、プロジェクトまたはサービスの交付または支払いに関連する任意の金銭または財産を所有または制御することを陳述または承諾する。
また、2009年に発表された“健康情報技術促進経済·臨床健康法案”(HITECH)により改正されたHIPAA及びそのそれぞれの実施条例は、2013年1月25日に発表された最終総合規則を含み、特定のヘルスケア提供者、健康計画、医療保健情報交換所(カバーエンティティと呼ぶ)及びその業務パートナーが、個人が識別可能な健康情報のプライバシー、安全、及び伝送を保護する上で義務を負い、これらのサービスは、個人識別可能な健康情報(強制契約条項を含む)を記憶、使用、又は開示し、特定の個人識別可能な健康情報の安全に違反した場合に影響を受けた個人及び規制機関に通知することを要求する。HITECHは、実体、商業パートナー、および可能な他の人に適用される可能性のある民事および刑事罰を増加させ、州総検察長に新たな権力を与え、連邦裁判所に民事訴訟を提起し、連邦HIPAA法を実行するために損害賠償または禁令を要求し、連邦民事訴訟の提起に関連する弁護士費と費用を求めることができる。また、多くの州の法律は、場合によっては健康情報のプライバシーやセキュリティを管理しており、その多くの法律は互いに大きく異なり、同じ効果が生じない可能性があり、HIPAAの先制が得られていないことが多い。
また、ACAによると、医療保険と医療補助サービスセンター(CMS)は最終規則を発表し、処方薬メーカーにいくつかの医師、医師アシスタント、看護師勤務者或いは臨床看護師専門家、登録看護師麻酔師、登録看護師助産師と教育病院への支払い或いは移転価値の情報、及び医師及びその直系親族が持つ投資権益を収集し、報告することを要求した。最初の報告書は2014年に提出されなければならず、年に1回提出されなければならない。報告されたデータは毎年公共サイト上で検索可能な形で提供される。必要な情報を提出できなかったことは民事罰金を招く可能性がある
また、いくつかの州は現在、処方薬会社に薬品マーケティングや販売促進に関連するいくつかの費用を報告し、これらの州が個人保健従事者に支払うプレゼントと支払いを報告することを要求している。他の州では、いくつかのタイプのプレゼントや食事を提供するなど、マーケティング関連の様々な活動が禁止されている。臨床研究とその結果に関する情報の公表を要求する州もある。一部の州は、値上げに関する情報や値上げを正当化する情報、または処方薬価格詐欺を禁止することを含む、いくつかの薬品定価情報の報告を要求している。さらに、いくつかの州は、製薬会社にコンプライアンス計画および/またはマーケティングコードを実施することを要求する。ある州と地方司法管轄区域はまた薬品販売と医療代表の登録を要求する。これらの法律を守ることは困難で時間がかかり、これらの州の法律を守らない会社は民事処罰に直面する。
第三者の業務配置に適合した医療法や法規を確保する努力は大量のコストに及ぶ。製薬会社の運営がこのような要求に違反していることが発見された場合、民事、刑事および行政処罰、損害賠償、罰金、返還、監禁、削減または再構成、その業務の削減または再構築、FDAの承認を得た資格を失うこと、連邦医療保険と医療補助、誠実監督と報告義務、監禁、名声損害を含む、民事、刑事·行政処罰、損害賠償、罰金、罰金、または州政府医療保健計画などの重大な処罰を受ける可能性がある。有効なコンプライアンス計画は,これらの法律違反による調査·起訴のリスクを低減することができるにもかかわらず,これらのリスクを完全に解消することはできない。容疑や違反の疑いのあるいかなる行動も製薬会社に巨額の法的費用を招き、このような行動が成功しても経営陣の業務運営への注意を移す可能性がある。
アメリカの医療改革
米国では、連邦政府、州政府、監督機関、第三者支払者は、医療コストの増加を制御または管理するために引き続き提案を提出し、より広く言えば、米国の医療システムを改革する。製薬産業はこのような努力の特別な重点であり、重大な立法計画の重大な影響を受けてきた。例えば、2010年3月にACAが公布され、医療保険の許容性を拡大し、医療支出の増加を減少または制限し、詐欺や乱用に対する救済措置を強化し、医療保健と医療保険業界の新たな透明性要求を増加させ、医療業界に新たな税費を徴収し、追加の医療政策改革を実施し、医療保健が政府と民間保険会社によって融資される方式を大きく変え、米国製薬業に大きな影響を与えた
また、ACAが公布されて以来、米国は他の立法改正を提案し、採択した。2021年9月9日、バイデン政府は範囲の広い政策提案リストを発表し、その大部分は薬品価格と薬品支払いを下げるために国会が実行する必要がある。これらの提案は最近ピークに達してインフレ低減法案が公布されたり 2022年8月、IRAは、CMSが連邦医療保険B部分およびD部分に基づいて精算されるいくつかの薬物および生物製品の販売価格を交渉することを可能にするが、CMSは少なくとも7年(生物製品11年)が承認された高支出単一由来薬物のみを選択して交渉することしかできないが、交渉価格は選択年度後2年以内に発効する。交渉価格は2026年に初めて発効し、法定最高価格を上限とする。2023年1月から連邦医療保険B部とD部はそれぞれ2023年1月と2022年10月から開始され,IRAはインフレ率よりも高い速度で連邦医療保険B部とD部分の薬品価格を向上させた製薬業者を処罰する。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。IRAを守らないメーカーは民事罰金を含めて様々な処罰を受ける可能性がある。アイルランド共和軍はまた、ACA市場で医療保険を購入した個人に2025年まで強化された補助金を提供する。このような規定は法的挑戦を受ける可能性があるにもかかわらず、2023年から段階的に施行されるだろう。
また、支払い方法は医療立法と規制措置の影響を受ける可能性がある。例えば、2003年の“連邦医療保険処方薬、改善と現代化法案”(MMA)は連邦医療保険カバーと薬品の支払い方式を変えた。この立法は高齢者が薬品を購入する医療保険のカバー範囲を拡大し、医師が管理する薬品の平均販売価格に基づく新しい精算方法を導入した。しかも、この法案はどんな治療カテゴリーでもカバーされる薬物の数を制限するための権力を提供する。MMAは連邦医療保険受益者の薬品福祉にのみ適用されるが,個人支払者は自分の精算料率を設定する際に連邦医療保険カバー政策や支払い制限に従うことが多い。したがって、MMAによるいかなる償還減少も、個人支払者支払いのような減少をもたらす可能性がある。
また、2018年5月30日、2017年Trickett Wendler、Frank Mongiello、Jordan McLinn、Matthew Bellina Right to Trial Actが法律に署名しました。他の事項以外に、この法律はある患者に連邦フレームワークを提供し、彼らが第一段階の臨床試験を完成し、FDAの許可を得た研究用新薬製品を獲得するために調査を行っていることを許可した。場合によっては、条件に適合する患者は、臨床試験に参加することなく、FDA拡大参入計画に従ってFDAの許可を得ることなく治療を求めることができるが、現在の連邦試用権法によれば、メーカーは試験的新薬製品を提供する義務がない。
人力資本
従業員
2022年12月31日までに102人のフルタイム従業員がいます。これらの従業員のうち、43人が医学博士や博士号を持っています。私たちは時々独立請負業者を招いて私たちの組織を支援します。私たちの従業員は労働組合代表もなく、集団交渉合意のカバー範囲もなく、私たちは従業員との関係が良いと信じている。
多様性と包括性
私たちは肌の色、人種、性別、国籍、民族、宗教、年齢、障害、性的指向、性別同意または表現、または法律によって保護された任意の他の地位に基づいた差別または嫌がらせのない職場を創造し、維持するために努力している。私たちの管理チームと従業員は職場で誠実、道徳、そして他人を尊重する行動を見せて促進しなければならない。私たちのすべての従業員は適切な行動のために基準を設定した行動基準を遵守しなければならず、どんな種類の差別や嫌がらせを防止、識別、報告、制止するのを助けるために年間訓練に参加しなければならない。わが社の採用、採用、発展、訓練、給与と昇進は、経歴、業績、技能、経験を基礎としており、性別、人種、民族を問わない。
競争力のある報酬と福祉
私たちは給与、総合的な福祉、サービスを提供し、従業員の異なる需要を満たすように努力している。私たちの総奨励プログラムには、競争力のある報酬、従業員の全面的な医療福祉、家庭病休暇、柔軟な勤務時間手配が含まれている。また、私たちは、株式オプションと私たち従業員の株式購入計画を付与することにより、免税および非免税従業員を含むフルタイム従業員毎に会社の株式のメリットを提供する。私たちは401(K)計画を賛助し、従業員の支払いを一定の上限に一致させる。
従業員の発展と訓練
我々は優秀な人材を引きつけ,引き留め,育成することを重視している.私たちは、幅広いオンラインと講師の指導の発展と持続的な学習計画を提供することで、従業員の発展と訓練を強調します。従業員が科学、臨床と技術会議に参加することを奨励し、そして成功に必要な広範な資源を獲得する機会がある。
安全問題
私たちの職員たちの安全、健康、そして健康は重要なことだ。我々は引き続き衛生流行病や新冠肺炎を含む他の懸念に関する事態の発展を監視し、将来必要となる可能性のある任意の安全協定を実施する。
企業情報
我々はデラウェア州の法律に基づいて2014年8月に成立したもので,名称はIntegrin Rock,LLCである。その後、2014年10月にMorphy Rock Holding,LLCと改名し、2016年6月にMorphy Holding,LLCと改名した。2018年12月5日、私たちは一連の取引または再編を完了し、これらの取引に基づいて、Morphy Holding LLCは免税再編方式でMorphy Holding,Inc.に変換し、3つの完全子会社Lazuli,Inc.,Tourmarine,Inc.とPhyllite,Inc.を別の完全子会社Morphy治療会社に統合した。私たちの主な実行オフィスはマサチューセッツ州ウォルザムGatehouse Drive 35号にあり、郵便番号:02451、電話番号は(781)996-0955。私たちのサイトの住所はwww.morictx.comです。当社のウェブサイトに掲載されているか、本サイトで取得可能な情報は、本年度報告の一部ではなく、引用で本年度報告に組み込まれることもありません。
利用可能な情報
我々は、改正された1934年証券取引法又は取引法に基づいて、年度、四半期及び現在の報告、委託書及びその他の書類を米国証券取引委員会又は米国証券取引委員会に提出する。米国証券取引委員会は、報告書、依頼書、および情報声明、および発行者(我々を含む)に関する他の情報を含む相互接続サイトを維持し、これらの情報は、米国証券取引委員会に電子的に報告される。公衆はwww.sec.govで私たちがアメリカ証券取引委員会に提出した任意の書類を得ることができる。報告書および修正案が米国証券取引委員会または米国証券取引委員会に電子的に提出された後、米国証券取引委員会に提出された各文書のコピーを、当社のウェブサイトwww.Morictex.comで無料で表示してダウンロードすることもできます。
第1 A項リスク要因
私たちの普通株に投資することは高い危険と関連がある。私たちの普通株への投資を決定する前に、以下に述べるリスクと、本年度報告書Form 10-Kに含まれる他の情報、ならびに我々の財務諸表および関連説明、ならびに“経営陣の財務状況および経営業績の検討および分析”をよく考慮しなければなりません。以下に説明するリスクと不確実性は私たちが直面している唯一の危険と不確実性ではない。私たちは意識していない、あるいは私たちは現在実質的な他のリスクや不確実性ではないと考えており、私たちに影響を与える重要な要素になる可能性もある。私たちはあなたに次のように議論されたどんな事件も起こらないという保証がありません。これらの事件は、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすかもしれない。もしこのような状況が発生したら、私たちの普通株の取引価格は下がるかもしれません。あなたはあなたの全部あるいは一部の投資を損失するかもしれません。
リスク要因の概要
以下のリスク要約は、業務活動の正常な過程において我々が直面している多くのリスクについて概説する。したがって、以下のリスク要約には、あなたにとって重要である可能性のあるすべての情報は含まれていません。リスク要約および本節の後の“リスク要因”のタイトルの下でのリスクのより詳細な議論、および本Form 10-K年次報告における“経営陣の財務状況および運営結果の議論および分析”のタイトルの他の部分を読むべきです。以下の概要または“リスク要因”および“経営層の財務状況および経営結果の議論および分析”で議論されるリスクに加えて、他のリスクは、現在または将来行う可能性のある活動または経営、または私たちが経営または将来経営する可能性のある市場に適用される可能性がある。このような状況と一致して、私たちは以下のようなリスクを含む様々なリスクに直面している
•私たちは臨床段階の生物製薬会社で、運営歴史が限られていて、商業化販売が許可されている製品は何もありません。私たちは重大な損失の歴史があり、予測可能な未来に引き続き重大な損失を受けることが予想される。
•私たちは私たちの候補製品の開発を進めるために多くの追加資金が必要になるだろうが、これは受け入れ可能な条項で提供されているのではないか、あるいは全くそうではないかもしれない。必要な時に必要な資本を得ることができなければ、私たちは私たちの製品開発計画、商業化努力、または他の運営を延期、制限、または終了させることを余儀なくされる可能性がある。
•われわれの候補製品はまだ開発の初期段階にある 開発に失敗したり、遅延を受けたりする可能性があり、それによってその商業生存能力に重大な悪影響を与える可能性がある。もし私たちまたは私たちの協力者が私たちの候補製品の開発や商業化を達成できない場合、あるいはそうする過程で重大な遅延に遭遇すれば、私たちの業務は実質的な損害を受けるだろう。
•私たちの現在と未来の臨床試験あるいは任意の協力者の臨床試験は私たちの臨床前研究に見られない重大な不良事件を掲示するかもしれない そして安全状況を招く可能性があり、規制部門の承認や市場が私たちの任意の候補製品を受け入れることを阻害するかもしれない。
•私たちは歴史的に協力してきて、将来的には第三者との協力を求めて、私たちの候補治療薬を発見し、開発するかもしれません。もし私たちの未来の協力者が協力協定に従って開発を停止した場合、またはこれらの合意が終了した場合、協力は商業製品を生成できない可能性があり、私たちは合意の下で記念碑的支払いや未来の印税を決して受けないかもしれない。
•私たちおよび/または私たちのパートナーは、アメリカまたは外国の規制当局の承認を得ることができないか、または遅延する可能性があります したがって、私たちの候補製品を商業化することはできない
•合格した肝心な管理と技術者を引き付け、維持することができなければ、私たちの業務計画を実施する能力を弱めることになる
•私たちの主要株主と経営陣は私たちのかなりの割合の株を持っていて、株主の承認を待つ事項をコントロールすることができるだろう。
•私たちは、新しい治療法や技術プラットフォームを開発することを含む、自己免疫、心血管および代謝性疾患、線維化および癌の候補製品を開発または開発することが可能な実体からの競争に直面している。もしこれらの会社が技術や候補製品を開発する速度が私たちよりも速い場合、あるいは彼らの技術がより効果的であれば、私たちが候補製品を開発し、成功させる能力は悪影響を受ける可能性がある。
•私たちの定款文書とデラウェア州法律によると、反買収条項は私たちの買収を阻止または延期する可能性があり、これは私たちの株主に有利であり、私たちの株主の試みを阻止することができるかもしれない。
•私たちが再記述した会社登録証明書の独占裁判所条項は、株主が司法フォーラムで、私たちまたは私たちの任意の取締役、上級管理職、または他の従業員と紛争することに有利であると考えるクレームを提出する能力を制限する可能性があり、これは、このようなクレームに関連する訴訟を阻害する可能性がある。
私たちの業務と運営に関するリスク
私たちは私たちの組織を発展させる必要があり、私たちは私たちの成長を管理し、私たちの業務を拡大する上で困難に直面する可能性があり、これは私たちの業務に悪影響を及ぼすかもしれない。
2022年12月31日までに102個あります fフルタイムの従業員。私たちの発展と商業化計画と戦略の発展に伴い、私たちは私たちの管理、運営、財務、その他の資源の従業員基盤を拡大したい。しかも、製品開発での私たちの経験は限られている。私たちの候補製品が臨床前研究と臨床試験に入って進展することに伴い、著者らは私たちの開発と監督能力を拡大し、そして他の組織と契約を締結し、私たちに製造とその他の能力を提供する必要がある。将来的に、私たちは協力者やパートナー、サプライヤー、および他の組織とのより多くの関係を管理しなければならないと予想される。私たちが運営と未来に成長する能力を管理することは、私たちの運営、財務、管理制御、報告システム、手続きを引き続き改善することを要求するだろう。私たちは、我々の管理情報および制御システムを効果的またはタイムリーに改善することができず、既存のシステムおよび制御における不足点を発見することができるかもしれない。私たちは私たちの成長を管理し、私たちの業務を拡大することに成功できず、私たちの業務、財務状況、運営結果、将来性に実質的な悪影響を及ぼすかもしれない。
合格した肝心な管理と技術者を引き付け、維持することができなければ、私たちの業務計画を実施する能力を弱めることになる。
私たちの成功は私たちの最高経営責任者Praveen P.Tipirneni医学博士と私たちの管理チームの他のメンバーと他の重要な従業員とコンサルタントの持続的なサービスに大きく依存する。私たちは現在このような個人に重要な人物保険を提供していない。1人以上の管理チームのメンバーまたは他の重要な従業員やコンサルタントを失うことは、私たちの研究開発計画を延期し、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性があります。私たちの主要な管理者たちが私たちの業界で築いた関係は、私たちが特に彼らに依存して私たちと協力し続けるようにした。私たちは、私たちの候補製品と私たちのMINTプラットフォームに関連する技術が高い技術性、および規制承認過程の専門性を持っているため、私たちの技術者の持続的なサービス、特に統合素結晶に関連する者に依存している。私たちの管理チームとキーパーソンは私たちに持続的なサービスを提供する義務がないので、彼らは罰を受けることなく、いつでも私たちとの雇用関係を終わらせることができます。
私たちはマサチューセッツ州ウォルザムの工場で業務を展開しています。この地域は多くの他の生物製薬会社と多くの学術·研究機関の本部である。私たちの市場は技術者に対する競争が非常に激しく、私たちが受け入れられる条件で高い素質の人員を採用し、維持する能力を制限する可能性があり、甚だしきに至っては根本的にはできない。私たちは他の会社、大学、公共、民間研究機関、政府実体、その他の組織からの人員競争に直面している。私たちの未来の成功は私たちが引き続き他の高い素質の科学、技術と管理者、及び臨床テスト、製造、政府監督と商業化の専門知識を持つ人員を誘致し、維持することに大きく依存する。もし私たちが引き続き高い素質の人材を誘致し、維持することができなければ、私たちは候補製品を発見し、開発する速度と成功率が制限され、これは私たちの業務、財務状況、運営結果と将来性に実質的な不利な影響を与える可能性がある。
私たちの将来の成長は海外市場における私たちの運営能力にある程度依存するかもしれませんが、そこでは追加の規制負担や他のリスクや不確実性の影響を受けることになります。
私たちの将来の成長は、海外市場での私たちの候補製品の開発と商業化および/または普及能力にある程度依存するかもしれませんが、私たちは第三者との協力に依存するかもしれません。外国市場関連規制機関の規制承認を得る前に、私たちは海外市場で私たちの任意の候補製品をマーケティングしたり普及させたりしてはいけません。私たちの任意の候補製品は決してこのような規制承認を受けないかもしれません。多くの他の国で単独の規制承認を得るためには、これらの国の安全性と有効性に関する多くのおよび異なる規制要求、および私たちの候補製品の臨床試験や商業販売、定価、流通などに対する規制要求を守らなければならず、これらの司法管轄区域で成功するかどうかを予測することはできない。もし私たちが国際市場の規制要求を遵守し、適用されたマーケティング承認を得ることができなければ、私たちの目標市場は減少し、私たちの候補製品の市場潜在力を十分に発揮する能力が損なわれ、私たちの業務は悪影響を受けるだろう。もしあれば、私たちは外国の規制部門の承認をタイムリーに得られないかもしれない。私たちの任意の候補製品は他の国の監督管理機関の承認を得られず、この候補製品の商業見通しと私たちの業務、財務状況、運営結果とを著しく低下させる可能性があります
将来性は実質的で不利な影響を受けるかもしれない。また、私たちの候補製品の承認を得て、最終的に私たちの候補製品を海外市場で商業化しても、複雑で変化する外国の監督管理、税収、会計、法律要求を遵守する負担、一部の外国の知的財産権保護の減少など、リスクと不確定要素の影響を受けるだろう。
私たちの業務には重大な製品責任リスクがあり、私たちが十分な保険を得る能力は、私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性があります。
私たちの候補製品に対して臨床試験を行う時、私たちは治療療法の開発、テスト、製造とマーケティング過程に固有の重大な製品責任リスクに直面するかもしれない。製品責任クレームは私たちの開発計画の完了を延期または阻止する可能性があります。もし私たちがマーケティング製品の面で成功した場合、このようなクレームは、FDAが私たちの製品、私たちの製造プロセスおよび施設、または私たちのマーケティング計画の安全性と有効性を調査し、私たちの製品をリコールすること、またはより深刻な法執行行動を取り、承認の適応を制限すること、または承認を一時停止または撤回することを招く可能性がある。是非曲直あるいは最終結果にかかわらず、責任クレームはまた、臨床試験で参加者を募集する難度の増加、臨床試験場所或いは全体の試験計画の終了、臨床試験参加者の脱退、私たちの名声損傷とメディアの重大な負の関心、関連訴訟を弁護するための巨大なコスト、管理層の時間と私たちの資源の私たちの業務運営からの移転、試験参加者や患者への巨額の金銭的奨励、収入損失、商業化できないおよび私たちが開発する可能性のある製品、そして私たちの株価の下落を招く可能性がある。私たちは現在一般責任保険を維持しており、保険金額は最高1,000万ドルに達しています。しかし、私たちは、私たちの任意の候補製品の後期段階を臨床開発またはマーケティングするために、より高いレベルの製品責任保険を得る必要があるかもしれない。私たちが持っているか得ることができるどんな保険も潜在的な責任に十分な保険を提供できないかもしれない。しかも、臨床試験と製品責任保険はますます高くなっている。その結果は, 私たちは製品責任クレームによる損失から私たちを保護するために合理的なコストで十分な保険を得ることができないかもしれません。これらの損失は私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性があります。
当社の従業員、独立請負業者、コンサルタント、ビジネスパートナー、およびサプライヤーは、規制基準および要件を遵守しないことを含む、不適切な行為または他の不適切な活動に従事する可能性があります。
私たちは、従業員、独立請負業者、コンサルタント、ビジネスパートナー、サプライヤーが従業員詐欺や他の不正活動を行うリスクに直面している。これらの当事者の不正行為は、故意、無謀、および/または不注意な行為を含む可能性があり、FDAの規定を遵守できず、FDAおよび他の同様の外国の規制機関に真実、完全かつ正確な情報を提供し、私たちが制定する可能性のある製造基準を遵守し、医療詐欺および法律法規を遵守し、財務情報またはデータを正確に報告し、または不正な活動を開示してくれるかもしれない。もし私たちの候補製品がFDAの承認を得て、米国でこれらの製品の商業化を開始すれば、私たちはこれらの法律によって直面する可能性のあるリスクが著しく増加し、私たちはこれらの法律を遵守することに関連するコストが増加するかもしれない。特に、医療業界の販売、マーケティング、商業配置は、詐欺、リベート、自己取引、その他の乱用を防止するための広範な法律法規によって制約されている。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネス計画を制限または禁止する可能性があります。従業員の不当行為はまた臨床試験過程で得られた情報を不当に使用する可能性があり、これは規制制裁と著者らの名声に深刻な損害を与える可能性がある。また、私たちは、起きていなくても、このような詐欺や他の不正行為を告発する可能性があるというリスクに直面している。従業員の不正行為を識別し阻止することは常に可能ではない, このような活動を検出し防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはそのような法律または法規を遵守できないことによる政府の調査または他の行動または訴訟から私たちを保護することができない可能性がある。もし私たちにこのような訴訟を提起し、私たちが私たちの権利を弁護または維持することに成功しなかった場合、これらの行動は、重大な民事、刑事および行政処罰、損害、罰金、返還、監禁、削減または再編成、FDAの承認を得る資格を失った、政府契約、医療清算または他の政府計画(MedicareおよびMedicaidを含む)、誠実な監督と報告義務、または名声被害から除外されるかもしれない。
私たちは、私たちの情報技術システム、これらのシステムの任意の故障、または私たちのCROまたは私たちが利用する可能性のある他の請負業者またはコンサルタントの故障に依存して、私たちの業務を損なう可能性があります。セキュリティホール、ネットワーク攻撃、データ損失、および他の中断は、私たちの業務に関連する敏感な情報を危険にさらしたり、重要な情報へのアクセスを阻止し、私たちに責任を負わせたりする可能性があり、これは私たちの業務、運営結果、財務状況、および見通しに悪影響を及ぼす可能性があります。
業務を展開するために必要なデジタル形式の情報を収集·維持し,情報技術システムやインフラに依存して業務を運営するようになってきている.私たちの日常生活では
ビジネス面では、知的財産権、独自の商業情報、個人データを含む大量の機密情報を収集、記憶、送信します。重要なのは、私たちはこのような機密情報の機密性と完全性を維持するために、安全な方法でそうしなければならないということだ。私たちは、データ漏洩を防止し、商業的に利用可能なシステム、ソフトウェア、ツール、および監視に依存して、私たちの情報技術システムおよびデジタル情報の処理、送信、および記憶にセキュリティを提供するために、私たちのシステムを保護し、保護するための物理的、電子的、および組織的措置を確立している。私たちはまた、私たちの情報技術インフラの構成要素をアウトソーシングしているので、一部の第三者サプライヤーは私たちの機密情報にアクセスできるかもしれません。我々の内部情報技術システムおよびインフラ、ならびに私たちの現在および未来の任意の協力者、請負業者、コンサルタント、ならびに私たちが依存している他の第三者のシステムおよびインフラは、盗難または推定された証拠を使用して第三者が従業員アカウント、コンピュータウイルス、ネットワーク釣り攻撃にアクセスするなど、ネットワーク事件の破壊を受けやすい 脅迫ソフトウェア攻撃、迷惑メール、マルウェア、ネットワーク攻撃、またはインターネット上のネットワーク侵入、電子メール添付ファイル、私たちの組織内部の人、または私たちの組織内部システムにアクセスできる人、およびコンピュータシステムおよびネットワークへの不正アクセスを試みる人。我々の内部情報技術システムやインフラも、自然災害、テロ、戦争、電気通信、電力故障の破壊を受けやすい。システムの故障や停止は、これらの機能をタイムリーに実行する能力に影響を与える可能性があり、これは、業務を展開する能力を損なうか、または財務報告を延期する可能性があります。このような失敗は私たちの経営業績や財務状況に実質的な悪影響を及ぼす可能性がある。
世界各地からの未遂攻撃と侵入の数、強度と複雑性の増加に伴い、セキュリティホール或いは中断或いはデータ損失のリスクは普遍的に増加し、特にコンピュータハッカー、外国政府とネットワークテロリストを含むネットワーク攻撃或いはネットワーク侵入を介している。地政学的緊張および間欠的戦争が米国国外で継続またはエスカレートするにつれて、これらおよび他のより複雑または国家によって支持される攻撃行動の発生は、例えば、ロシアとウクライナの衝突のために増加する可能性がある。さらに、機密情報にアクセスするモバイルデバイスの一般的な使用は、機密情報または他の知的財産権の損失をもたらす可能性があるデータセキュリティホールのリスクを増加させる。我々はネットワークセキュリティ問題、エラー、ウイルス、ワーム、マルウェアプログラム、セキュリティホールを緩和するコストが高い可能性があり、データセキュリティと情報技術システムを保護するためのセキュリティ対策を実施していますが、これらの問題を解決する努力は成功しないかもしれません。これらの問題は、予期せぬ中断、遅延、サービス停止、私たちの業務と私たちの競争地位に他の損害をもたらす可能性があります。このような事件が発生して私たちの運営が中断されると、私たちの製品開発計画が実質的に中断される可能性があります。例えば、完了または進行中または計画中の臨床試験における臨床試験データの損失は、我々の規制承認作業を遅延させる可能性があり、データを回復または複製するコストを著しく増加させる可能性がある。さらに、コンピュータセキュリティホールが私たちのシステムに影響を与えたり、個人識別情報を不正に発行したりした場合、私たちの名声は大きな被害を受ける可能性があります。さらに、このような違反は政府機関に通知する必要があるかもしれない, メディアまたは個人は、1996年の健康保険携帯性および責任法案、またはHIPAA、2009年の医療情報技術促進経済および臨床健康法案、またはHITECH、その実施規則および条例、ならびに連邦貿易委員会によって公布された条例および州違反通知法を含む様々な連邦および州プライバシーおよびセキュリティ法律に基づいて適用される。さらに、このようなネットワーク攻撃、データ漏洩、またはデータ破壊または損失は、適用される国際プライバシー、データ保護、および他の法律に違反する可能性があり、重大な民事および/または刑事責任を負う。さらに、私たちの一般責任保険および会社リスク計画は、私たちが直面しているすべての潜在的なクレームを含まない可能性があり、私たちが適用する可能性のあるすべての責任を補償するのに十分ではないかもしれません。そして、私たちの業務、運営結果、財務状況、および見通しに実質的な悪影響を及ぼす可能性があります。さらに、ネットワーク攻撃または他のデータセキュリティホールによって名声被害を受けたり、訴訟または不利な規制行動に直面したりする可能性があり、さらなるデータ保護措置の実施によって重大な追加費用を招く可能性があります。
もし私たちが環境、健康、そして人間の安全を保護する法律を守らなければ、私たちの業務は不利な影響を受けるかもしれない。
私たちの研究と開発活動は放射性材料を含む危険化学品と材料の使用を含む。私たちはマサチューセッツ州ウォルザムの施設に大量の可燃性と有毒化学品を保持しています。これらの化学品は私たちの研究と開発活動に必要です。これらの危険化学品と材料の使用、製造、貯蔵、運搬と処置について、私たちは連邦、州と地方の法律と法規を守らなければならない。私たちが施設でこのような材料を貯蔵し、処理し、処理する手続きは一致していると信じているにもかかわらず 法規やガイドラインに規定されている基準を適用すると,これらの材料による意外な汚染や傷害のリスクは解消できない。もし事故が発生したら、私たちはそれによる損害に責任を負うかもしれないし、損失は大きいかもしれない。著者らはまた多くの環境、健康と職場の安全法律と法規の制約を受け、それらの管理実験室プログラム、血液伝播病原体の接触及び動物と生物危険材料を処理する法律と法規を含む。私たちは労災保険を維持していますが、この保険は私たちの従業員がこれらの材料を使用することによる潜在的な責任を保障するのに十分ではないかもしれませんが、私たちはそれによって重大な損失を招くかもしれません
コストと支出です。もし私たちが健康と人間の安全、および危険な化学品や材料の使用、製造、貯蔵、処理、処分に関連する適用法律と法規に違反すれば、巨額のコストを招き、巨額の罰金や処罰を受ける可能性もある。
私たちの現在の業務は1つの場所に集中しており、私たちまたは私たちが依存している第三者は極端な天気事件や他の自然災害の悪影響を受ける可能性があり、私たちの業務の連続性や災害復旧計画は深刻な災害から私たちを十分に保護できないかもしれません。
私たちの現在の業務はマサチューセッツ州のウォルザムに集中しています。洪水、火災、爆発、地震、極端な天気条件(例えば、ハリケーンや大雪)、医療流行病や大流行、電力不足、電気通信故障、または他の自然または人為的事故や事件などの計画外事件は、私たちの施設や私たちの第三者契約製造業者の製造施設を十分に利用できず、特に日常生活において、私たちの業務運営能力に重大かつ悪影響を与える可能性があり、私たちの財務および運営状況に大きなマイナス影響を与える可能性がある。当社の施設や第三者契約メーカーの製造施設を使用できないことは、コスト増加、候補製品の開発遅延、あるいは私たちの業務運営中断を招く可能性があります。自然災害、停電、または他の事件が発生した場合、本社の全部または大部分を使用することができなくなり、私たちの研究施設や私たちの第三者契約製造業者の製造施設のような重要なインフラを損傷させたり、他の方法で運営を中断したりすることは、困難かもしれませんし、場合によっては、かなり長い間私たちの業務を継続することはできません。深刻な災害や同様の事件が発生した場合、我々の既存の災害復旧および業務連続計画は十分ではないことが証明される可能性がある。私たちの災害復旧と業務連続計画の限られた性質のため、私たちは大量の費用を発生する可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性があります。また,気候変動が一般経済条件,特に製薬業に及ぼす長期的な影響は不明であり,既存の自然災害リスクを増加あるいは悪化させる可能性がある。私たちのリスク管理政策の一部として, 私たちは私たちの業務に適していると思うレベルで保険範囲を維持する。しかし、私たちはそのような保険範囲が私たちが遭遇する可能性のある損害と損失を補償するのに十分であるという保証はない。もし私たちの施設や私たちの第三者契約メーカーの製造施設がどんな理由でも稼働できなければ、短い時間であっても、私たちのいかなる研究開発プロジェクトも損害を受ける可能性があります。どの業務中断も、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
私たちは私たちの業務に関連する複雑な税務規則の制約を受けて、どんな監査、調査、あるいは税務手続きも私たちの業務、運営結果、財務状況に重大な悪影響を及ぼす可能性があります。
私たちはアメリカで所得税と非所得税を払わなければならない。所得税会計は往々にして複雑な問題に関連しており、私たちの所得税や他の税金負債の準備を決定する際に判断する必要がある。私たちは未来に他の非アメリカ司法管轄区で事業を展開するかもしれない。私たちはまた非アメリカ司法管轄区で所得税と非所得税を支払うことができる。また、多くの管轄区域には詳細な譲渡定価規則があり、非住民関連側とのすべての取引は、このような規則が指す公平定価原則に従って定価することが求められている。源泉徴収税、商品およびサービス税、販売税、および他の非所得税の適用は常に明確ではなく、このような源泉徴収税または非所得税に関連する税務監査を受ける必要があるかもしれない。私たちは私たちの納税状況が合理的だと信じている。私たちは現在どんな税務監査も受けていない。しかし、アメリカ国税局や他の税務機関は私たちの立場に同意しないかもしれない。国税局や他の税務機関が私たちの地位に挑戦することに成功した場合、私たちはそのために設立された任意の準備金を超える追加税金と罰金、それに関連する利息または他の税金を負担する必要があるかもしれません。これは私たちの業績と運営、そして未来のキャッシュフローに大きな影響を与えるかもしれません。
私たちの純営業損失の繰越といくつかの他の税務属性を利用する能力は限られているかもしれません。
2022年12月31日現在、連邦と州所得税における純営業損失(NOL)はそれぞれ1.803億ドルと1兆915億ドルで、2037年に満期となる。2022年12月31日現在、連邦と州所得税の目的に使用できる税収控除金も持っており、それぞれ1260万ドルと570万ドルで、この2つの繰り越しは2032年から満期になる。私たちの課税収入が今年度のどんな営業赤字を超えている限り、私たちは私たちの繰り越しで本来課税されていた収入を相殺する予定です。しかし、2017年12月31日以降に開始された納税年度で生じる繰越の使用率は最高で同年度の課税所得額の80%に制限されており、このような繰越は考慮されていない。また、国内税法(以下、税法)第382条によると、私たちの所有権の変更は、将来の課税所得額を相殺するために毎年使用できる純営業損失の繰越と税収控除の繰越額(あれば)を制限する可能性がある。この制限は一般的にわが社の所有権が三年間で累積的に50%を超える場合に適用されます。我々は,規則382節によって所有権変更が行われたかどうかを決定するために分析を行っていない.このような制限は、純営業損失の繰越と税収控除を利用する能力を著しく低下させる可能性がある
満期までに繰り越す。米国国税法第382条の規定によると、私募、当社の初公募株、および会社設立以来発生または将来発生する可能性のある他の取引は所有権変更を招く可能性があります。このような制限は、私たちの初公募株、以前の私募、私たちの既存株主が私たちの普通株を売却したこと、または私たちが追加的に私たちの普通株を売却したことによっても、今後数年間の経営業績に重大な悪影響を及ぼす可能性があります。もう一つのリスクは、NOLの使用停止や他の予見できない理由のような州レベルの規制変化のため、私たちの既存のNOLが満期になるか、または将来の所得税債務を相殺するために使用できない可能性があるということだ
私たちは不利な立法や税金の変化を規制する影響を受けるかもしれないが、これは私たちの財務状況に否定的な影響を及ぼすかもしれない。
米国連邦、州、地方所得税に関する規則は立法過程に参加する人員およびアメリカ国税局とアメリカ財務省の審査を受け続けている。税法の変更(これらの変更はトレーサビリティを持つ可能性がある)は、私たちの株主や私たちに悪影響を及ぼす可能性があります。私たちは各税制改革提案と現行税務条約改正のすべての司法管轄区域における影響を評価して、私たちの業務に対する潜在的な影響、そして未来の課税収入に対する私たちのいかなる仮定も決定します。具体的なアドバイス,そのようなアドバイスの条項が作成されるかどうか,あるいはこれらのアドバイスが通過すれば,我々の業務にどのような影響を与えるかを予測することはできない.2022年から、2017年の“減税·雇用法案”(略称“税法”)は、現在研究開発支出を差し引くために利用できる選択肢を廃止し、一般的に5年以内に研究開発支出を償却することを納税者に求めている。アメリカ議会は立法を考慮して、現在の研究開発支出控除額を回復していますが、この条項が廃止されたり、他の方法で修正される保証はありません.
私たちの財務状況と資金需要に関連するリスク
私たちは臨床段階の生物製薬会社で、運営歴史が限られていて、商業化販売が許可されている製品は何もありません。私たちは重大な損失の歴史があり、予測可能な未来に引き続き重大な損失を受けることが予想される。
私たちは臨床段階の生物製薬会社で、運営の歴史は限られている。生物製薬製品の開発は高度な投機的な仕事であり、それは大量の前期資本支出を必要とし、重大なリスクが存在するため、即ち任意の潜在的な候補製品は十分な効果或いは受け入れ可能な安全性を証明できず、監督部門の許可を得られない、或いは商業上実行可能ではない。
我々の主要候補製品Morf−057は健康ボランティアにおける第1段階の臨床試験を完了した。われわれはすでにMorf−057の第2段階計画を開始し,最初に潰瘍性大腸炎に用いられた。これまで商業販売のための製品は承認されておらず,商業製品販売から何の収入も得られておらず,我々の臨床開発や継続運営に関する大量の研究開発やその他の費用が発生し続けている。2022年12月31日現在の会計年度では、純損失5900万ドルを報告している。2022年12月31日までの累計赤字は約2.971億ドル。私たちのほとんどの損失は私たちの研究や開発計画に関する費用と私たちの業務に関する一般的かつ行政的コストによるものです。私たちは予測可能な未来に大きな損失を受けることが予想され、私たちが私たちの候補製品の研究と開発を続けるにつれて、これらの損失は増加すると予想される。
私たちは次の場合、私たちの費用が大幅に増加すると予想している
•現在と未来の候補製品の臨床試験を行っています
•新しい候補製品を発見と開発し、研究開発活動、臨床前研究と臨床試験を展開した
•私たちの候補製品の臨床的、臨床的、商業的供給を製造または製造しました
•私たちの候補品や未来の候補品のための規制承認を求める
•私たちの現在の候補製品または任意の未来の候補製品を商業化し、承認されれば
•販売、マーケティング、流通インフラの構築など、研究に専念している会社からビジネス活動を支援できる会社に転換しようとしている
•臨床、科学、管理職を増やす
•国際業務を含む業務、財務、および管理情報システムおよび人員を増加させる;
•他の化合物または候補製品を決定し、ライセンスを介して第三者からこれらの化合物または候補製品を取得する権利;
•新冠肺炎あるいはサプライチェーン中断の影響(新冠肺炎の大流行の影響、ウクライナの持続的な衝突、インフレ、金利上昇、持続的な労働力不足或いはその他の原因による)により、私たちの臨床前或いは臨床研究及び私たちの候補製品に対する規制承認はいかなる遅延がある。
1つ以上の候補製品の商業化に成功しても、より多くの候補製品を開発·マーケティングするために、大量の研究開発や他の支出を生み出し続けることが可能である。私たちは予測できない費用、困難、合併症、遅延、および私たちの業務に悪影響を及ぼす可能性のある他の未知の要素に直面するかもしれない。私たちの未来の純損失の規模は私たちの未来の支出の成長率と私たちが収入を作る能力にある程度依存するだろう。私たちの以前の損失と予想された未来の損失はすでに私たちの株主権益と運営資本に悪影響を与え続けるだろう。
私たちは製品販売から収入を得たことがなく、永遠に利益を上げないかもしれない。
私たちが利益を達成して利益を維持する能力は私たちが収入を作る能力にかかっている。私たちは、私たち単独またはパートナーと規制部門の承認を得ることができず、私たちのα4β7計画の主要候補製品または私たちが開発する可能性のある他の候補製品の商業化に成功しない限り、相当な収入は発生しないと予想される。成功した商業化は、臨床試験において安全性と有効性を証明し、マーケティングを含む規制を獲得し、これらの候補製品を承認し、私たちまたは現在または未来の任意のパートナーが規制の承認を得る可能性のある製品を製造、マーケティング、販売し、任意の上場後の要求を満たし、個人保険や政府支払者から私たちの製品の精算を得ることを含む多くの重要なマイルストーンを実現する必要があるだろう。これらの活動に関連する不確実性とリスクのため、私たちは将来の任意の収入の時間および金額、いかなるさらなる損失の程度、または私たちがいつ利益を達成する可能性があるかどうかを正確かつ正確に予測することができない。私たちと現在または未来の協力者たちは決してこのような活動で成功しないかもしれないし、たとえ私たちまたはどんな協力者が成功しても、私たちは利益を達成するのに十分な収入を生むことができないかもしれない。たとえ私たちが確実に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。
もし私たちが利益を上げて利益を維持できなければ、私たちの普通株の市場価格を下げ、資金を集めたり、業務を拡大したり、運営を継続する能力を弱めるかもしれません。もし私たちが過去のように損失を被り続けると、投資家の投資は何の見返りも得られず、すべての投資を失う可能性がある。
私たちは私たちの候補製品の開発を進めるために多くの追加資金が必要になるだろうが、これは受け入れ可能な条項で提供されているのではないか、あるいは全くそうではないかもしれない。必要な時に必要な資本を得ることができなければ、私たちは私たちの製品開発計画、商業化努力、または他の運営を延期、制限、または終了させることを余儀なくされる可能性がある。
バイオ製薬候補製品の開発は資本集約型である。もし私たちの候補製品が臨床前研究と臨床試験を通じて入って進展すれば、私たちは私たちの開発、監督、製造、マーケティングと販売能力を拡大または創出するために大量の追加資金が必要になるだろう。私たちはすでに私たちの候補技術と製品を開発するために大量の資金を使用して、私たちの候補製品のさらなる研究と開発および臨床前試験と臨床試験を行い、私たちの候補製品のための規制承認を求め、商業販売が許可された製品の製造と販売を必要としている(もしあれば)
設立以来、私たちは研究開発活動に大量の精力と財力を投入した。2022年12月31日現在、私たちは3.482億ドルの現金、現金等価物、有価証券を持っている。私たちの現在の運営計画によると、私たちの利用可能な現金、現金等価物、有価証券に加えて、2023年2月に普通株と事前融資承認株式証を非公開で発行して調達した1.0億ドルは、2026年下半期までの運営費用と資本支出需要を支払うのに十分であると信じている。しかし、私たちの将来の資本需要と既存資源が私たちの運営をサポートすると予想される期間と将来の資本需要の期限は私たちが予想しているのとは大きく異なる可能性があり、私たちは計画よりも早く追加資金を求める必要があるかもしれない。私たちの候補製品の成功した研究や開発に関連する時間や活動の長さは非常に不確定であるため、承認された製品のマーケティングや商業化活動を開発·開発するためにどれだけの実際の資金が必要かを見積もることはできない。私たちの将来の短期的かつ長期的な資金需要は多くの要素に依存するだろうが、これらに限定されない
•臨床前と臨床開発活動の時間、コストと進展
•私たちは臨床前と臨床プロジェクトの数と範囲を決定しました
•私たちと締結または将来的に協力を締結する可能性があり、および/または開発協定の締約国との開発努力の進展状況を研究する
•私たちの協力協定によると、私たちは支払いのマイルストーンと他の支払いの時間と金額を受け取ることができます
•私たちは現在のライセンスと研究開発計画を維持し、新しい協力計画を立てることができる
•特許と他の知的財産権主張に関連する費用を起訴して実行する
•第三者が私たちの候補製品を生産するコストは
•規制提出の費用と規制承認の時間;
•もし私たちの候補製品または任意の未来の候補製品が販売を許可された場合、マーケティング、販売、および流通コストを含む商業化活動のコスト
•私たちは、私たちの候補製品開発を支援する人員を含め、運営システムの強化とより多くの人員の雇用に努力している
•上場企業としての私たちの義務を果たすために、財務·報告システムを含むより多くの内部システムやインフラを実施する必要があります。
我々のより多くの資金を調達する能力は、新冠肺炎の流行、インフレの激化、金利上昇、持続的な労働力不足、グローバルサプライチェーンの中断、ウクライナの持続的な衝突、およびそれに応じて実施された世界的な制裁を含む、世界経済状況の悪化および米国と世界各地の信用と金融市場の中断と変動の悪影響を受ける可能性がある。もし私たちが適時あるいは受け入れ可能な条件下で資金を得ることができなければ、私たちは私たちの研究開発計画と臨床前研究或いは臨床試験を延期、減少或いは中止し、戦略機会を制限したり、リストラ或いはその他の会社の再編活動を行わなければならないかもしれない。これまで、私たちは主に協力協定に基づいて受け取った支払い、株式証券の売却、債務融資を通じて、私たちの運営に資金を提供してきました。
私たちは将来的に追加の資金を求めることを要求され、現在は公開または私募株式発行または債務融資、追加の協力および/または許可協定、信用または融資手配、またはこれらの資金源のうちの1つまたは複数の組み合わせによってこれを行う予定だ。もし私たちが株式証券を発行することで追加資金を調達すれば、私たちの現在有効なS-3表登録声明に基づいて、私たちの株主は希釈され、いかなる融資条項も私たちの株主の権利に悪影響を及ぼす可能性があります
また、私たちに追加資金を提供する条件として、将来の投資家は、既存の株主よりも高い権利を要求し、付与される可能性がある。私たちの将来の債務融資は、もしあれば、私たちが将来の業務活動を行う柔軟性を制限する制限契約に関連する可能性があり、破産が発生した場合、債務保有者は私たちの持分証券所有者がわが社の資産の任意の分配を受ける前に返済されるだろう。もし私たちが許可や第三者との協力計画を通じてより多くの資金を調達すれば、私たちは私たちの候補製品に価値のある権利を放棄したり、私たちに不利な条項で許可を付与しなければならないかもしれない。私たちはまた、より早い段階で候補製品のためのパートナーを探すか、あるいは候補製品または技術に対する私たちの権利を放棄することを要求されるかもしれません。そうでなければ、私たちは自分自身の開発または商業化を求めます。必要に応じて受け入れ可能な条項で資金を得ることができない場合、現在または将来の候補製品の製品開発および商業化を延期、制限、または終了させる可能性があり、これは私たちの業務、財務状況、運営結果、および見通しに重大な悪影響を及ぼす可能性がある。
発見·開発·商業化に関するリスク
私たちの業務はα4β7の主要な候補製品を含む現在と未来の候補製品の成功に大きく依存します プログラムです。これらの候補製品は既存と未来の臨床前研究と臨床試験は成功しない可能性があり、もし私たちがこれらの候補製品を商業化或いは商業化過程で重大な遅延に遭遇できなければ、私たちの業務は実質的な損害を受ける。
私たちは私たちのα4β7特定インテグリン阻害剤計画を開発するために多くの精力と財力を投入した。私たちは商業製品の収入を作る能力があって、これは何年も起こらないと予想して、もしあれば、私たちのα4β7の主要な候補製品の成功と最終商業化に大きく依存します プログラムです。私たちは以前にFDAに候補製品の新薬申請やNDAを提出したこともなく、比較可能な外国当局に同様の規制承認文書を提出したこともなく、私たちの候補製品が臨床試験で成功したり、監督部門の承認を得るかどうかを決定することはできない。また,我々の候補製品は臨床試験で成功しても,規制部門の承認を得られない可能性がある。さらに、規制機関が審査過程をタイムリーに完了できない可能性があり、またはFDA諮問委員会または他の規制機関が承認または制限を提案しない場合、追加の遅延を招く可能性がある。さらに、将来の立法や行政行動における追加的な政府規制、または製品開発、臨床試験、審査過程における規制機関の政策の変化による遅延や拒否に遭遇する可能性がある。規制当局はまた、要求よりも限られた適応を有する候補製品を承認するか、または以下のラベルを有する候補製品を承認することができる
使用条件に関する警告、禁忌症または予防措置。監督管理機関はまたリスク評価と緩和策略、あるいはREMS、あるいは高価な発売後の臨床試験の表現を要求する可能性がある。もし私たちの候補製品が規制部門の承認を得られなければ、私たちは運営を続けることができないかもしれない。私たちが規制部門の承認を得て候補製品をマーケティングすることに成功したとしても、私たちの収入は、私たちが規制部門の承認を得て商業権を持つ地域の市場規模にある程度依存するだろう。もし私たちが狙っている患者亜群市場が私たちが予想しているほど大きくなければ、承認されれば、このような製品の販売から大量の収入が発生しないかもしれません。
私たちは規制部門の承認を求め、私たちの候補製品をアメリカと選定された外国で商業化する予定です。他の国で単独の規制承認を得るためには、これらの国の安全性と有効性に関する多くのかつ異なる規制要件を遵守しなければならない。他の事項以外にも、他の国にも独自の法規管理臨床試験や商業販売、そして私たちの候補製品の価格設定と流通があり、規制承認(これは成功しないかもしれない)を得るために多くの資源が必要かもしれませんし、これらの管轄区域で行われている法規を遵守する必要があります。
私たちの現在と未来の候補製品の成功は、私たちまたは私たちの協力者が取る以下の行動を含む多くの要素に依存するだろう
•臨床試験を開始するために必要な臨床前研究を成功させた
•患者の募集に成功し、著者らの臨床試験を完成し、良好な結果を得た
•私たちの候補製品の商業化に必要な規制許可を得て承認します
•第三者製造業者との手配の確立と維持
•私たちの候補製品とその構成要素の特許および商業秘密保護および非特許専有権を獲得し、維持する
•私たちの知的財産権と主張を実行して擁護します
•私たちの候補製品の期待適応のために理想的な治療特性を実現する
•もし私たちの候補製品が承認されれば、単独でも第三者と協力しても、商業販売を開始することができる
•患者、医療界、第三者支払者が私たちの製品候補を承認した場合、その製品を受け入れます
•他の治療法と効果的に競争し
•臨床試験と監督管理の承認を通じて、私たちの候補製品の受け入れ可能な安全性を維持する。
もし私たちがこれらの要素のうちの1つまたは複数をタイムリーにまたは根本的に達成できなければ、私たちは重大な遅延に遭遇したり、私たちの候補製品を商業化することに成功しなかったりする可能性があり、これは私たちの業務に深刻な損害を与えるだろう。
我々の候補製品は開発の初期段階にあり,開発に失敗したり遅延に遭遇したりして,その商業生存能力に実質的な悪影響を与える可能性がある.もし私たちまたは私たちの協力者が私たちの候補製品の開発や商業化を達成できない場合、あるいはそうする過程で重大な遅延に遭遇すれば、私たちの業務は実質的な損害を受けるだろう。
私たちはまだ製品が発売されていません。私たちのすべての候補製品は初期開発段階にあります。また,早期発見や臨床前開発段階にある一連の目標や計画があり,決して臨床段階の開発には入らない可能性がある。私たちが利益を達成し維持する能力は、規制機関の私たちの候補製品の承認を得て、私たちの候補製品を商業化することに成功しています。単独でも第三者と協力しても、私たちのどの候補製品も規制部門の承認を得ることを保証することはできません。著者らはFDAの承認を含む規制承認を得るために必要な臨床試験の実施と管理に必要な経験が限られている。規制機関が私たちの候補製品の商業流通を承認する前に、私たちまたは未来のパートナーは、私たちの候補製品の人体における安全性と有効性を証明するために、広範な臨床前試験および臨床試験を行わなければならない。
規制当局の承認や候補製品の商業化を遅延させたり阻害したりする問題に遭遇した場合、候補製品の開発を継続したり、既存の候補製品を修正したり、新たな協力を行う財力がないかもしれません
•臨床前の研究結果は、候補製品が予想された有効性に及ばないか、または有害または問題のある副作用があることを示す可能性がある
•米国国外で行われた臨床前研究は、米国や他の国政府が実施した関税や輸出入制限の影響を受ける可能性がある
•我々の臨床試験または他の類似候補製品の臨床試験の陰性または不確定な結果は、追加の臨床前試験または臨床試験または計画放棄を決定または要求することをもたらす
•私たちの臨床試験中の患者または私たちの候補製品に類似した薬物または治療用生物学的製剤を使用した個人が経験した製品に関連する副作用;
•私たちの第三者メーカーは私たちの製品を作ることに成功しなかった
•いかなる第三者契約メーカーも、臨床試験または商業販売の需要を満たすために、私たちの候補製品と私たちの協力者の製品の生産規模を拡大することができません
•INDまたは同様の外国出願の提出を遅延させるか、または規制機関から臨床試験を開始するために必要な承認を得ることができないか、または臨床試験が開始されると一時停止または終了する
•FDAや同様の外国当局が私たちの臨床試験の範囲または設計について適用した条件
•患者の臨床試験への参加を延期しました
•臨床試験患者の高中退率は
•臨床試験を行うために必要な候補製品コンポーネントまたは材料または他の供給品の供給または品質が不足している
•候補製品の部品や材料源の代替供給源を得ることはできません
•臨床試験期間中、著者らの候補製品は有害な副作用が出現し、或いは治療効果の終点に達しなかった
•FDAまたは他の規制機関が許容できる利益-リスクプロファイルを証明できなかった
•FDAまたは他の規制機関は、私たちの任意の候補製品を試験および製造するための1つまたは複数の臨床試験場所または製造施設の不利な検査および審査を行う
•私たちの第三者請負業者または調査者は、監督要求をタイムリーにまたは根本的に遵守しなかったか、またはその契約義務を履行しなかった
•一般的な臨床試験または特に私たちの技術に追加の規制監視を適用することを含む、規制要件、政策、およびガイドラインの遅延および変更;または
•FDAと似たような外国の規制機関は私たちのデータについて違う解釈を持っている。
私たちまたは私たちのパートナーは、私たちの候補製品の開発または商業化を達成できないか、またはこれらの要因のうちの1つまたは複数による製品開発または商業化の重大な遅延は、私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を及ぼす可能性があります。
もし私たちが発表し、予想された時間枠内で予想された開発目標を達成しなければ、私たちの製品の商業化は遅れる可能性があるので、私たちの株価は下落するかもしれません。
私たちは時々各種の科学、臨床、法規と他の製品開発目標が達成される予定の時間を見積もり、私たちはそれをマイルストーンと呼ぶことがある。これらのマイルストーンは科学研究と臨床試験の開始または完成、そして規制文書の提出を含むかもしれない。私たちは時々いくつかのマイルストーンの予想時間を公開するかもしれない。このすべてのマイルストーンは今と未来に無数の仮定に基づいている。私たちの推定と比較して、このようなマイルストーンの実際の時間は大きく異なる可能性があり、場合によっては、原因は私たちの統制を超えている。もし私たちが公開発表されたこれらのマイルストーンに達しなかったり、これらのマイルストーンに達していなければ、私たちの製品の商業化は延期されたり、永遠に実現されないかもしれないので、私たちの株価は下落するかもしれない。
我々が発見·開発した治療法は,未検証の新技術に基づいており,適切な製品が生じない可能性がある。
私たちは私たちのミントプラットフォームを使って一連の候補製品を開発している。歴史的に見ると、他社が後期臨床試験に入った数十種類のインテグリン標的経口小分子候補薬はすべてFDAやEMAを通過できなかった
承認された薬品。他の会社は経口インテグリン方法の開発努力と臨床結果が成功しない可能性があり、経口インテグリンに対する否定的な見方を招き、そして著者らの候補製品の監督管理審査過程に負の影響を与え、これは著者らの業務に実質的かつ不利な影響を与える。我々のMINTプラットフォームによって決定された候補製品は、コンホメーション指向次世代の物理ベースの技術および機械学習および人工知能を利用することによって、発見中に設計、反復、および最適化の手がかりを提供することができると信じている。しかし,我々が我々のMINTプラットフォームを用いて候補製品を開発する基礎を構成する科学研究が行われており,実行可能な候補製品が生じない可能性がある.
最終的に、私たちのミントプラットフォームとそれによって生成された任意の候補製品は、特定のインテグリンコンホメーションをロックする能力を含む治療効果に必要ないくつかの特性を備えていないことが発見されるかもしれない。私たちの候補製品はまた、薬物が標的組織に到達するのに必要な期間内に人体を安定させることができない可能性があり、あるいはそれらは免疫反応を誘発し、候補製品が標的組織に到達する能力を抑制し、あるいはヒトへの副作用を引き起こす可能性がある。また,我々のミントプラットフォームに基づく候補製品は,患者において実験室研究とは異なる化学的および薬理学的特性を示す可能性がある。私たちのミントプラットフォームとそれによって生成された任意の候補製品は、ヒトにおいて異なる化学的および薬理特性を示す可能性があり、予測不可能、無効または有害な方法でヒト生物システムと相互作用する可能性がある。例えば、AbbVieは、臨床前試験において疑わしい標的/αvβ6媒介安全シグナルが観察されたため、その任意の選択的経口インテグリン阻害剤を推進することを意図しておらず、その後、2022年12月に施行されるAbbVieプロトコルを終了する権利の行使を容易にするために、AbbVieプロトコルを行使することを示唆している。
私たちのような新製品候補製品の規制承認プロセスは、他のより有名で広く研究されている候補製品と比較して、より高価で、時間がかかる可能性がある。我々の知る限り,規制機関は小分子インテグリン阻害剤の経口投与を許可していない。FDAはインテグリンによる治療法の経験が限られており,候補製品の規制承認過程の複雑さ,不確実性,長さを増加させる可能性があると考えられる。私たちと私たちの既存または未来のパートナーは、マーケティングおよび商業化のいかなる候補製品の承認も決して得られないかもしれない。私たちまたは既存または未来の協力者が規制の承認を得ても、承認された対象、疾患適応、または患者集団は、私たちが期待しているまたは期待しているほど広くないかもしれないし、重大な使用または配布制限または安全警告を含むラベルが必要になる可能性がある。私たちまたは既存または未来の協力者は、承認を得るために追加的または予期しない臨床試験を行うことを要求されるか、または規制承認を維持するために上場後試験要求を受ける可能性がある。もし私たちのミントプラットフォームと研究プロジェクトによって生成された製品が無効で安全ではないことが証明され、または商業的に不可能であれば、私たちのミントプラットフォームとパイプラインはほとんど価値がなく、これは私たちの業務、財務状況、運営結果、そして見通しに実質的で不利な影響を与えるだろう。
臨床前と臨床開発は長く高価な過程に関連し、結果は不確定であり、早期研究と試験の結果は未来の試験結果を予測できない可能性がある。私たちは、現在の候補製品または任意の未来の候補製品の開発および商業化を完了する過程で追加コストを発生させたり、遅延に遭遇したり、最終的には達成できない可能性がある。
我々のすべての候補製品は臨床前あるいは臨床開発段階にあり,すべての計画が失敗するリスクが高い。私たちは私たちのすべての候補製品がいつ、あるいは規制部門の承認を受けるかどうかを正確に予測できない。任意の候補製品を商業化するために必要な監督管理許可を得るために、著者らは広範な臨床前研究と長く、複雑かつ高価な臨床試験を通じて、私たちの候補製品が人体で安全かつ有効であることを証明しなければならない。臨床テストは完成するまで数年かかる可能性があり、その結果自体も確定していない。臨床試験では,いつでも失敗する可能性がある。著者らの候補製品の臨床前研究と早期臨床試験の結果は後期臨床試験の結果を予測できないかもしれない。著者らは適用された監督管理機関が臨床意義があると考える臨床終点を構築できない可能性があり、臨床試験はテストのどの段階でも失敗する可能性がある。早期臨床試験と後期臨床試験の試験設計上の差異は早期臨床試験の結果を後期臨床試験に外挿することが困難である。そのほか、臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床試験で満足できると考えているが、しかし依然としてその製品の発売許可を得られなかった。生物製薬業界のいくつかの会社は高級臨床試験で重大な挫折を経験し、原因は治療効果の不足或いは安全性が良くないことである, 早期の実験で好ましい結果が得られたにもかかわらず。通常,臨床試験による候補製品の失敗率は高い。大多数の臨床試験を開始した候補製品は製品として承認されたことがなく、私たちの未来のいかなる臨床試験が最終的に成功或いは私たちの現在或いは任意の未来の候補製品の臨床開発を支持することを保証することはできない。
臨床試験の開始は、最終的に試験設計を決定し、INDまたは同様の提出をFDAまたは同様の外国規制機関に提出することに依存する。私たちが他の管轄区域で私たちのINDや同様の提出書類を提出したとしても、FDAや他の規制機関は私たちが彼らの最初の要求を満たしたことに同意しないかもしれない
私たちの臨床試験は、私たちの研究設計に同意しないか、追加の臨床前研究を完成させたり、私たちの方案を修正したり、臨床試験の開始時により厳しい条件を加える必要があるかもしれません。
私たちまたは私たちの協力者は臨床試験を開始したり完成したりするのに遅延があるかもしれない。私たちまたは私たちのパートナーはまた、現在または未来の臨床試験中、または私たちが行う可能性のある臨床試験のために多くの予見不可能な事件に遭遇する可能性があり、これらのイベントは、私たちの上場承認を延期または阻止するか、または私たちのインテグリン阻害剤計画または任意の将来の候補製品を商業化することを含むかもしれない
•規制機関または機関審査委員会、FDAまたは道徳委員会は、私たちまたは私たちの調査者が予想される試験場所で臨床試験を開始するか、または臨床試験を行うことを許可してはならない
•私たちは、潜在的な試験地点および潜在的なCROと受け入れ可能な条項との合意に遅延または合意できない可能性があり、これらの条項は、広範な交渉を必要とする可能性があり、異なるCROと試験地点との間に大きな差があるかもしれない
•臨床試験場所は試験案から外れたり、試験を終了したりする可能性がある
•任意の候補製品の臨床試験は安全性または有効性を示すことができず、陰性または不確定な結果をもたらす可能性があり、私たちは決定または監督機関が追加の臨床前研究または臨床試験を要求するか、または製品開発計画を放棄することを決定するかもしれない
•候補製品の臨床試験に必要な被験者の数は、私たちが予想していたよりも多かったかもしれません。これらの臨床試験の登録速度は、私たちが予想していたよりも遅いかもしれません。または被験者は、これらの臨床試験を脱退するか、または予想以上の速度で戻って治療後のフォローアップを行うことができないかもしれません
•私たちの第三者請負業者は、法規の要求を直ちに遵守することができないか、または彼らの契約義務を履行することができないか、または全く遵守しないか、または臨床試験案から逸脱したり、試験を脱退する可能性があり、新たな臨床試験場所または調査者を増加させる必要があるかもしれない
•私たちは選択するかもしれません、あるいは規制機関、IRBs、または倫理委員会は、規制要件を遵守していないこと、または私たちの試験の参加者が受け入れられない健康リスクに直面していることを含む、様々な理由で臨床研究または試験を一時停止または終了することを要求するかもしれません
•私たちの候補製品の臨床試験コストは予想以上に高いかもしれません
•私たちの候補製品の品質または候補製品の臨床試験に必要な他の材料は、所与の臨床試験を開始または完了するのに十分ではないかもしれない
•臨床試験のために十分な数の候補製品を生産できないかもしれません
•他の療法の臨床試験報告は,我々の候補製品の安全性や有効性の懸念を引き起こす可能性がある
•候補製品の臨床的または臨床的前データおよび候補製品と同じクラスの他の分子からのデータに基づいて、候補製品のための適切な安全性プロファイルを確立することができない可能性がある
•FDA、EMA、あるいは他の規制機関は、長期毒理学研究や他の要求を加えて、その後、臨床試験を開始することを可能にするように、追加のデータを提出することを要求するかもしれない。
患者登録は臨床試験時間スケジュール中の1つの重要な要素であり、多くの要素の影響を受け、患者群の大きさと性質、著者らが登録した臨床地点の数量と位置、患者と臨床場所の接近程度、試験の資格と排除基準、臨床試験の設計、患者の同意を得られない、登録した参加者が完成する前に退出するリスク、競争する臨床試験と臨床医師、および研究中の候補製品の他の利用可能な治療法に対する潜在的な優位性に対する患者の見方は、私たちが調査している適応のための任意の新薬或いは治療生物製剤を承認される可能性がある。また、著者らは著者らの協力者、CROと臨床試験サイトに依存して、著者らの現在或いは未来の臨床試験が正確かつ適時に行われることを確保し、患者登録過程を含み、そして著者らのそれらの表現に対する影響は限られている。また,治療医が患者を我々の製品候補に参加する現在あるいは将来の臨床試験に参加することに関連する未解決倫理問題に遭遇した場合,安全性と有効性の概況が確立された既存の治療案を処方するのではなく,遅延に遭遇する可能性がある。
臨床試験が我々、そのような試験を行う機関のIRBs、FDA、EMAまたは他の規制機関によって一時停止または終了された場合、または臨床試験がデータ安全監視委員会またはデータ安全監視委員会によって一時停止または終了を提案された場合、遅延に遭遇する可能性もある。一時停止または終了は、法規の要求または著者らの臨床規程に従って臨床試験、FDA、EMAまたは他の法規機関の臨床試験操作または試験場所の検査を行うことができなかったことによる臨床休止、予見できない安全問題または副作用の実施、製品または治療の利益を証明できなかった、臨床的意義のある試験を確立または実現できなかったことを含む多くの要素による可能性がある
終点、政府法規或いは行政措置の変化、或いは臨床試験を継続するのに十分な資金が不足している。中期結果が不明確あるいは否定的であるため、臨床研究も延期または終了される可能性がある。臨床試験の開始または完了遅延をもたらす多くの要因は、最終的には、私たちの候補製品が規制部門の承認を得ることを拒否される可能性もある。また、FDA、EMAあるいは他の監督機関は、私たちの臨床試験設計と臨床試験データの解釈に同意しないかもしれないし、あるいは承認要求を変更する可能性があり、たとえ彼らが私たちの臨床試験設計を審査し、コメントしたとしても。
もし私たちが臨床テストやマーケティング審査の面で遅延に遭遇したら、私たちの製品開発コストは増加します。われわれのいずれの臨床試験が計画通りに開始されるかどうか,再構成が必要かどうか,予定通りに完成するかどうか,あるいは全く知られていない。重大な臨床試験遅延は、候補製品を商業化する独占的な権利を持つ可能性のある任意の期限を短縮することも可能であり、私たちの競争相手が私たちの前に製品を市場に出すことを可能にすることができ、これは候補製品を商業化することに成功する能力を弱化させ、私たちの業務と運営結果を損なう可能性がある。著者らの臨床開発計画のいかなる遅延も著者らの業務、財務状況と運営結果を深刻に損害する可能性がある。
臨床前研究と早期臨床試験の結果は後の臨床試験結果を予測できないかもしれない。
臨床前研究と早期臨床試験の結果は,その後の臨床試験の成功,臨床試験の中期結果を予測できない可能性がある。製薬やバイオテクノロジー業界の多くの会社が早期開発に積極的な成果をあげた後,後期臨床試験で大きな挫折を経験し,類似した挫折に直面する可能性がある。早期試験のように、著者らは多様性を考慮せずにいくつかの治療効果指標を検査することができ、早期臨床試験における積極的な結果は、名目的に統計学的意義のある結果を含み、異なる設計の未来試験或いは他の未来試験で複製しないかもしれない。臨床試験の設計はその結果が製品の承認を支持するかどうかを決定することができるが,臨床試験設計における欠陥は臨床試験の進展が良好になるまで明らかにならない可能性がある。著者らは設計臨床試験の経験が限られており、臨床試験を設計と実行して上場承認を支持できないかもしれない。そのほか、臨床前と臨床データはよく異なる解釈と分析の影響を受けやすい。多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると思っているが、まだ候補製品のマーケティング許可を得られなかった。私たちまたは未来のパートナーが私たちの候補製品の臨床試験結果が発売承認に値すると思っていても、FDAや同様の外国の規制機関は同意しない可能性があり、私たちの候補製品の発売を承認しないかもしれない。
場合によっては、多くの要素のため、同じ候補製品の異なる臨床試験間の安全性或いは有効性結果は有意差が存在する可能性があり、方案中に規定された試験プログラムの差異、終点、患者群の大きさと特徴の差異、投与方案と他の臨床試験方案の差異とコンプライアンス、及び臨床試験患者の中退率を含む。もし私たちが候補製品の臨床試験で積極的な結果を得なければ、私たちの候補製品の開発スケジュールと規制承認および商業化の将来性は負の影響を受け、それに応じて、私たちの業務と財務の将来性も負の影響を受けるだろう。
私たちが時々発表したり公表したりする臨床試験の中期と初歩データ或いはバックラインデータはより多くの患者データの出現に伴い変化する可能性があり、監査と検証手続きの制限を受け、これは最終データの重大な変化を招く可能性がある。
時々、私たちは私たちが予想している臨床試験の中期と初歩的なデータを公表するかもしれない。私たちが完成する可能性のある臨床試験で予め指定された中期分析データは、患者登録の継続とより多くの患者データの獲得に伴い、1つまたは複数の臨床結果が実質的に変化する可能性のあるリスクに直面する可能性がある。予備データまたはバックラインデータは依然として監査および確認手続きを受ける必要があり、これは最終データが私たちが以前に公表した予備データまたはバックラインデータと実質的な差がある可能性がある。したがって、最終データが利用可能になる前に、中間データおよび予備データまたはバックラインデータは慎重に表示されなければならない。中間、予備、またはバックラインデータと最終データとの間の不利な差は、私たちの名声およびビジネスの将来性を深刻に損なう可能性があります。
私たちの現在と未来の臨床試験あるいは私たちの現在と未来の協力者の臨床試験は私たちの臨床前研究に見られなかった重大な不良事件を掲示し、そして安全状況を招く可能性があり、監督部門の許可或いは市場が私たちの任意の候補製品を受け入れることを阻害するかもしれない。
私たちの任意の臨床試験において重大な有害事象または他の副作用が観察された場合、私たちは患者を私たちの臨床試験に参加することが困難になる可能性があり、患者は私たちの試験から撤退するかもしれない、または1つ以上の候補製品の試験または開発作業を完全に放棄することが要求される可能性がある。例えば,進行性多病巣性白質脳症,あるいはpmlは,α非融合抗体阻害剤として臨床開発された晩期副作用が他に観察されている4β1インテグリン、彼は珠単抗である。この副作用は前臨床研究では観察されなかった、あるいは
彼のジュマズマブの早期臨床開発では我々、FDA、EMA、または他の適用可能な規制機関またはIRBは、そのような試験における対象または患者が許容できない健康リスクまたは副作用に直面していると考えることを含む、様々な理由で候補製品の臨床試験を随時一時停止することができる。生物技術業界で開発されたいくつかの潜在療法は最初に早期試験で治療の将来性を示したが、その後副作用が発生することが発見され、それらの更なる発展を阻害した。副作用が候補製品の発売承認を阻止したり保持したりしなくても,他の療法と比較して承認製品の耐性により,副作用が市場の受け入れを阻害する可能性がある。このような事態のどのような発展も、私たちの業務、財務状況、そして見通しに実質的な損害を与える可能性がある。
私たちのミントプラットフォームを利用して私たちの候補製品ルートを拡大し、適切な製品を開発する努力は成功しないかもしれません。
私たちの業務の成功は私たちのMINTプラットフォームに基づく製品の発見、開発、商業化の能力にある程度依存します。我々のα4β7リーダー計画や我々の研究計画,あるいは我々の協力者の研究計画は,様々な理由で他の潜在的な臨床開発候補製品を決定できない可能性がある。我々の研究方法は、潜在的な候補製品の識別に成功しない可能性があるか、または私たちの潜在的候補製品が有害な副作用を有することが証明される可能性があるか、または製品を販売できないか、または発売承認を得ることができない可能性がある他の特徴を有する可能性がある。これらのいずれかが発生した場合、私たちは、1つまたは複数の計画のための開発作業を放棄することを余儀なくされる可能性があり、これは、私たちの業務に実質的な損害を与え、運営を停止させる可能性があります。新製品候補製品を確定する研究プロジェクトには大量の技術、財力と人的資源が必要である。
私たちは、より利益的またはより成功する可能性の高い候補製品を利用することなく、特定の候補製品を追求するために限られた資源を使うかもしれない。
私たちの財務と管理資源が限られているため、私たちはいくつかの特定の候補製品に研究開発に重点を置いている。例えば、私たちのα4β7特定インテグリン阻害剤計画では、私たちは最初に私たちの主要な候補製品MORF-057に焦点を当てた。したがって、私たちは他の候補製品を探す機会を放棄したり延期したりするかもしれないが、これらの製品は後により大きな商業潜在力を持っていることが証明された。私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。現在および将来の研究開発計画および特定の適応の候補製品への支出は、いかなる商業的に実行可能な候補製品も生じない可能性がある。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価しなければ、私たちは協力、許可、または他の印税手配によって候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利だ。
私たちは、新しい治療法や技術プラットフォームを開発することを含む、自己免疫、心血管および代謝性疾患、線維化および癌の候補製品を開発または開発することが可能な実体からの競争に直面している。もしこれらの会社が技術や候補製品を開発する速度が私たちよりも速い場合、あるいは彼らの技術がより効果的であれば、私たちが候補製品を開発し、成功させる能力は悪影響を受ける可能性がある。
薬品の開発と商業化競争は激しい。私たちの候補製品が承認されれば、激しい競争に直面し、効果的な競争ができなければ、重大な市場浸透を達成することを阻止するかもしれない。私たちのほとんどの競争相手は私たちよりずっと大きい資源を持っていて、私たちは競争に成功できないかもしれない。私たちは様々な多国籍生物製薬会社、専門バイオテクノロジー会社、新興バイオテクノロジー会社と競争し、大学や他の研究機関が開発している技術や候補製品とも競争している。私たちの競争相手は、私たちの候補製品およびプロセスと競争する候補製品およびプロセスを開発し、開発しています。競争的治療には、市場に参入する新しい技術プラットフォームに基づく治療方法を含む、医学界によって承認され受け入れられた治療方法および任意の新しい治療方法が含まれる。相当な数の製品が現在開発中であり、将来的に商業的に使用される可能性があり、私たちが開発を試みているか、または開発を試みる可能性のある候補製品を治療するために使用される可能性があると信じている。生物技術、生物製薬、インテグリンと免疫調節療法領域に激しいと迅速に変化する競争が存在している。多くの源からの競争がすでに存在しているか、あるいは未来に現れるかもしれない。私たちの競争相手には、より規模が大きく、資金が豊富なバイオ製薬、バイオテクノロジーと治療会社、自己免疫、心血管と代謝性疾患、繊維化と癌治療に集中する会社、多くの小さな会社が含まれている。また,大学や他の研究機関が開発した現在と将来の療法と競争している。その中の一部の会社は資本が十分で、私たちとは対照的に, 私たちの既存または未来の協力者を含むかもしれない豊富な臨床経験を持っている。しかも、これらの会社は科学と管理人材の募集で私たちと競争している。
私たちの成功は競争相手の製品よりも安全で効果的な治療薬を開発し商業化する能力に部分的に依存するだろう。もし競争相手の製品が私たちが開発した治療法よりも安全で、より効果的で、あるいは安いなら、私たちのビジネス機会と成功は減少または消滅するだろう。
バイオ製薬業界は大量の投資を行っているにもかかわらず、アメリカとヨーロッパでは経口インテグリン療法が承認されていない。経口小分子α4β7特異的インテグリン阻害剤Morf−057を開発している IBDの治療に用いられている。現在承認されているIBD療法には,武田製薬有限会社が販売している注射可能なα4β7モノクロナル抗体Entyvio(Vedolizumab)と,AbbVie,ジョンソン,UCB,Biogen Inc.,ファイザーや百時美施貴宝などの製薬会社が販売している異なる作用機序を有する療法があり,承認されれば候補製品が競合する可能性がある。また,主役治療会社,ジリッド科学社,EA製薬有限会社が臨床開発した経口α4β7療法や,エバービー,ジョンソン,ファイザー社,礼来社,百時美施貴宝などの製薬会社が臨床開発において異なる作用機序を有する療法が知られている。
我々のαvβ8特異的小分子インテグリン阻害剤計画は,骨髄線維化や固形腫瘍の治療に開発されている。現在承認されている骨髄線維化治療薬には,Incell社とノワール国際社が販売している経口JAK阻害剤Jakafi(Ruxolitinib),百時美施貴宝社が販売しているInrebic(Fedratinib),CTI Biophma社が販売しているVonjo(パリチニブ)が含まれており,グラクソ·スミスクライン社,MorPhoSys AG,Incell Corp,Geron社,Abbvie,百時美施貴宝社が臨床開発している骨髄線維化療法が知られている。現在,αvβ8阻害剤のいかなる適応も承認されていない。ファイザー社は固形腫瘍治療のための抗αvβ8モノクロナル抗体を開発していることが知られている。また,Venn TreeuticsやCorbus PharmPharmticals Holdings,Inc.固形腫瘍に対する臨床前段階抗αvβ8モノクロナル抗体計画,Pplant Treeuticsの小分子計画も知られている。また,ノワール国際社,エバービー社,ロ氏ホールディングス社,メルク社,百時美施貴宝社と学者岩石会社,その他の製薬会社は形質転換増殖因子−β経路に対する多様な抗体や小分子療法を開発し,開発中の固形腫瘍の治療にも用いられている。
その中の多くの競争相手は私たちよりも多くの財務、技術、製造、マーケティング、販売、供給資源または経験を持っている。もし私たちが任意の候補製品の承認を得ることに成功したら、私たちは私たちの製品の安全性と有効性、私たちの製品の管理の容易さ、患者が比較的新しい投与経路を受け入れる程度、これらの製品が規制の承認を受ける時間と範囲、製造、マーケティングと販売能力の可用性とコスト、価格、精算範囲、特許地位を含む多くの異なる要素に基づく競争に直面するだろう。私たちと競争する製品は、私たちが開発する可能性のある任意の製品よりも効率的で、より安全で、より安価で、またはより効率的なマーケティングおよび販売を含む、より良い治療代替案を提供することができます。我々が候補製品を開発·商業化する費用を回収する前に、競争力のある製品は、私たちが開発したいかなる製品も時代遅れまたは競争力を持たないかもしれない。これらの競争相手はまた私たちの従業員を募集する可能性があり、これは私たちの専門レベルと業務計画を実行する能力にマイナスの影響を与える可能性がある。
承認された場合、私たちの現在の候補製品または任意の未来の候補製品は、医師、患者、ヘルスケア第三者支払者、および医療コミュニティにおいて商業成功のために必要な他の人において十分な市場受容度を得ることができない可能性があり、販売可能かもしれない候補製品から未来の収入を得ることはできないかもしれない。
規制機関による候補製品の承認を得ても,製品が競争力のあるコストで販売できるかどうか,市場に受け入れられるかどうかなどの要因により,製品の販売から収入を生じたり維持したりすることができない可能性がある。歴史的に見ると、FDAはいくつかの注射可能なインテグリン阻害剤を炎症性腸疾患、多発性硬化症、乾癬、急性冠症候群とドライアイの治療に許可した。しかし,われわれの候補品は新しい経口インテグリン療法に基づいており,インテグリンはよく知られている受容体ファミリーであるが,これまで経口小分子インテグリン療法はFDAの承認を得ていない。新しい治療法の受容度に大きな影響を与える市場参加者、例えば医師や第三者支払者は、私たちの新技術に基づく経口生物利用型製品を採用しない可能性があり、医学界および第三者支払者が、私たちまたは私たちの既存または未来のパートナーが開発した任意の候補製品を受け入れて使用するように説得することができないかもしれません。私たちの候補製品に対する市場の受け入れ度は他の要素に依存するだろう
•マーケティングや商業化の承認を受けた時間は
•承認された条項や承認された国
•私たちの候補製品の臨床試験における安全性と有効性
•私たちの候補品に関連するいかなる不良副作用の流行率と重症度
•FDAまたは他の規制機関によって承認された任意のラベルに含まれる制限または警告;
•私たちの候補製品は比較的便利で管理しやすい
•患者が新しい投与方法を受けたいかどうか
•私たちの現在の候補製品または未来の候補製品への不利な宣伝;
•私たちの医師教育プログラムは成功しました
•販売とマーケティング努力の有効性
•政府と第三者支払者が提供する保険と適切な補償
•私たちの製品の価格は特に代替療法と比較して
•我々の候補製品は、治療のための疾患適応の代替的に有効な治療方法の利用可能性、およびこれらの治療方法の相対的リスク、収益およびコストを目的としている。
医療製品の販売は医師が治療処方を出す意思にも依存し、これはこれらの医師に基づいて製品が安全で、治療が有効であり、費用効果があることを確定した可能性が高い。また,異なる医師団体による治療ガイドラインや影響力のある医師の観点から製品を取り入れたり排除したりすることは,他の医師が治療処方を処方する意思に影響する可能性がある。医師、医師組織、病院、他の医療提供者、政府機関、あるいは個人保険会社が、私たちの製品が競争相手の治療法と比較して安全で、治療が有効で、費用効果があるかどうかを予測することはできません。任意の候補製品が承認されたが、このような各当事者の十分な受容度に達していない場合、候補製品から十分な収入を生成または得ることができず、利益を達成または維持することができない可能性がある。
我々の候補製品は新技術に基づいているため、広範な研究と開発が必要であり、相当な製造と加工コストが必要であると予想される。また、私たちのいかなる兆候の潜在的な市場規模の推定は、私たちが製品の商業化(あれば)を開始した時に発見された状況と大きく異なる可能性があり、これは私たちの業務計画に大きな変化をもたらし、私たちの業務、財務状況、運営結果、見通しに大きな悪影響を及ぼす可能性があります。また、私たちが商業化した任意の候補製品が市場の受け入れを得られなければ、私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性がある。
将来私たちが米国や世界で販売およびマーケティング能力を確立することができない場合、または第三者と合意して私たちが承認された任意の候補製品を販売およびマーケティングすることができなければ、このような候補製品を商業化することに成功できないかもしれないし、いかなる収入も生まれないかもしれない。
私たちは現在、規制部門の承認を得ることができる候補製品をマーケティング、販売、流通するためのマーケティングや販売チームを持っていません。承認後に任意の候補製品を商業化するには、各地域に基づいてマーケティング、販売、流通、管理、その他の非技術的能力を確立したり、第三者にこれらのサービスを提供するように手配しなければなりませんが、成功しないかもしれません。もし私たちの候補製品が規制機関の承認を得たら、私たちの候補製品を商業化するために、技術的な専門性と流通能力を支援する内部販売やマーケティングチームを設立することに決定するかもしれません。これは高価で時間がかかり、私たちの幹部が非常に注意して管理する必要があります。例えば、一部の州と地方司法管轄区は薬品販売代表に対して許可証と継続教育要求があり、これには時間と財力が必要である。私たちの内部販売、マーケティング、および流通能力の発展のいかなる失敗や遅延も、私たちが市場の承認を得た任意の候補製品の商業化に悪影響を及ぼすだろう。
規制の承認を得た候補製品の商業化については、世界的にまたは地域ごとに、直接販売チームや構築された流通システムを有する第三者と連携して、自分たちの販売チームや流通システムを強化したり、自分たちの販売チームや流通システムの代わりにしたりすることを選択することができる。もし私たちが必要な時に受け入れ可能な条項でそのような計画を達成できない場合、あるいは根本的にできない場合、私たちは規制部門の承認を得た候補製品を商業化することに成功できないかもしれないし、そのような商業化は遅延や制限に遭遇する可能性がある。もし私たちが私たちの候補製品を商業化することに成功できなければ、私たち自身も1つ以上の第三者と協力することで、私たちの未来の製品収入は影響を受け、私たちは重大な追加損失を受けるかもしれない。
もし私たちの任意の候補製品がマーケティング承認を得て、私たちまたは他の人が後にその候補製品が不良な副作用をもたらしていることを発見した場合、私たちはマーケティングと候補製品から収入を得る能力が影響を受ける可能性がある。
私たちの候補製品による不良副作用は、規制機関の臨床試験の中断、延期、または停止を招く可能性があり、より厳しいラベルやFDAまたは他の規制機関が規制承認を延期または拒否する可能性がある。私たちの臨床試験結果は深刻で受け入れられない副作用の深刻さと流行率を示すかもしれません
効果がある。この場合、私たちの将来の臨床試験は一時停止または終了する可能性があり、FDAまたは同様の外国規制機関は、私たちの任意またはすべての目標適応を承認する候補製品のさらなる開発を停止または拒否するように命令するかもしれない。このような副作用はまた、患者が患者の臨床試験を開始または完了する能力に影響を与えるか、または潜在的な製品責任クレームを引き起こす可能性がある。これらのいずれも、私たちの業務、財務状況、経営結果、および見通しに重大な悪影響を及ぼす可能性があります。
また,臨床試験の本質は潜在的な患者集団のサンプルを利用することである。患者数と暴露時間が限られているため、私たちの候補製品は希で深刻な副作用は候補製品に接触する患者数が著しく増加した時にのみ暴露される可能性がある。
もし私たちの候補製品が監督部門の許可を得て、私たちまたは他の人がこの製品による不良副作用を発見したら、以下のいずれかの不良事件が発生する可能性があります
•規制部門は製品の承認を取り消したり、製品を差し押さえたりすることができる
•私たちは製品をリコールしたり患者に投与する方法を変更することを要求されるかもしれません
•特定の製品の販売またはその製品またはその任意の構成要素の製造プロセスに追加の制限を加えることができる
•私たちは罰金、禁止、あるいは民事または刑事処罰を受けるかもしれない
•規制当局は、ボックス警告または禁忌症のようなラベル宣言の追加を要求するかもしれない
•患者に配布するためにこのような副作用のリスクを概説するための薬物ガイドラインを作成する必要があるかもしれません
•私たちは起訴され、患者への傷害に責任を負うかもしれない
•製品の競争力が低下する可能性があります
•私たちの名声は損なわれるかもしれない。
このようなすべての事件は、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちのいくつかの候補製品は第三者薬物と結合して研究する可能性があり、その中のいくつかはまだ開発中である可能性があり、私たちはこのような薬物の供給、監督管理状態、あるいは規制承認の制御が限られているか、あるいはコントロールされていないと予想している。
私たちの候補製品のいくつかは第三者薬物と組み合わせて研究されるかもしれない。別の製品または候補製品と共に使用される候補製品の開発は、単一のエージェント候補製品が直面しない課題をもたらす可能性がある。FDAまたは他の規制機関は、任意の観察された効果に対する各製品および候補製品の貢献を評価するために、より複雑な臨床試験設計を使用することを要求するかもしれない。これらの試験の結果は,従来のいずれの積極的な試験結果も,われわれの候補品ではなく併用療法によるべきであることを示唆している可能性がある。さらに、製品承認後、FDAまたは他の規制機関は、共同使用のために相互に使用される製品交差標識を要求する可能性がある。私たちが他の製品に権利がない範囲では、これはこのような要求を満たすために第三者と協力する必要があるかもしれない。また、私たちが発売許可を得たら、別の製品に関連する開発は、この組み合わせの臨床試験および私たちのビジネスの将来性に影響を与える可能性があります。このような発展には、他の製品の安全性または有効性を変更すること、承認製品の利用可能性を変更すること、および看護基準を変更することが含まれる可能性がある。
もし私たちがこの連合療法を追求すれば、私たちはこの薬が安定した商業供給があるかどうかを確認することができない。このようなビジネス関係を構築できなかったり、市場で治療法を購入する費用は、私たちの開発スケジュールを延期し、私たちのコストを増加させ、私たちの候補製品を商業的に実行可能な併用療法として開発する能力を危うくする可能性がある。いずれの場合も、私たちの業務、運営結果、および財務状況に悪影響を及ぼす可能性があります。
もし将来の協力者やサプライヤーが商業的に合理的な条項や彼らの製品を提供することができなくなったり、彼らの製品を提供したくない場合、私たちはそのような製品を得るための代替案を探す必要があるだろう。また、任意の協力者やサプライヤーの製品供給が中断、遅延、あるいは私たちに提供できない場合、私たちの臨床試験は延期される可能性がある。もし私たちがどんな代替療法の供給を得ることができない場合、あるいは商業的に合理的な条項でそうすることができなければ、私たちの業務、運営結果、財務状況は不利な影響を受ける可能性がある。
私たちの第三者への依存に関するリスクは
私たちは歴史的に協力してきて、将来的には第三者との協力を求めて、私たちの候補治療薬を発見し、開発するかもしれません。もし私たちの未来の協力者が協力協定に従って開発を停止した場合、またはこれらの合意が終了した場合、協力は商業製品を生成できない可能性があり、私たちは合意の下で記念碑的支払いや未来の印税を決して受けないかもしれない。
私たちは過去に、将来的に第三者との協力合意を求めて、いくつかのインテグリンに基づく治療法を発見または開発する可能性があり、このような協力は私たちの製品ラインの重要な部分を占めるかもしれない。これまで、私たちのほとんどの収入は第三者との協力協定から来ており、私たちの将来の収入の一部は協力協定や私たちが将来締結する可能性のある他の類似した合意から来ているかもしれません。研究開発協力の収入は、協力の持続的、研究開発サービスの支払い、およびそれによって生成された成功候補製品許可証を取得するオプション、および私たちの研究開発された将来の製品から得られるマイルストーン、または支払いおよび特許権使用料(ある場合)に依存する。もし私たちが候補製品の開発を成功的に進めたり、マイルストーンを達成することができない場合、または私たちの協力が他の方面で成功しなければ、私たちが達成する可能性のある任意の協力合意に基づいて、マイルストーン支払いからの収入および現金資源は予想を大幅に下回るだろう。AbbVieは、臨床前試験において疑わしい標的/αvβ6媒介安全シグナルが観察されたため、αvβ6特異的インテグリン阻害剤の選択的経口投与を推進することを意図しておらず、2022年6月に私たちに通知され、便宜上、その権利を行使してAbbVieプロトコルを終了することが決定され、このプロトコルは2022年12月に施行される。さらに、2021年12月、Janssenは、Janssen協定の下で最初の2つのインテグリン目標のオプションを行使しないことを決定したことを通知しました Janssenに興味のある特定の疾患の標的検証が不足しているため,2023年1月に通知され,便宜上Janssenプロトコルを終了する権利を行使することが決定され,インテグリン抗体活性化剤の潜在的開発を含む第3のインテグリン研究計画に重点を置いた。もし私たちとJanssenが同意すれば、Janssen協定は2023年3月またはそれ以上に終了するだろう。
また、将来的には、他の治療技術や候補製品の研究、開発、商業化のために第三者パートナーを探すことができるかもしれません。バイオ製薬会社は、私たちの任意のマーケティング、流通、開発、許可、またはより広範な協力計画の以前で可能な未来のパートナーです。もし私たちが商業的に合理的な条項で未来の協力を行うことができなかった場合、または全く協力が達成されなかった場合、またはこのような協力が成功しなかったか、または終了されなかった場合、最近終了したエバービ合意およびヤンソン合意を含む場合、私たちは、より大きなバイオ製薬会社または特定の関連分野の専門資源から利益を得ることができると考えられるいくつかの目標、候補製品、または疾患分野を開発するために、私たちの戦略を実行できないかもしれない。
我々の既存の連携プロトコル,および将来予想される任意の連携プロトコルについては,我々の協力者が我々の候補製品の開発や商業化に取り組んでいる資源の数と時間を限定的に制御し続けることが予想される.さらに、私たちがこれらの計画から収入を創出する能力は、私たちの協力者がこれらの手配の中で彼らに割り当てられた機能を成功的に履行する能力に依存するだろう。
私たちの候補製品に関する協力は現在、私たちに以下のリスクをもたらし続けるだろう
•協力者は彼らがこれらの協力の努力と資源に適用することを決定する上で大きな自由裁量を持っている
•パートナーは、我々の候補製品を開発および商業化してはならない、または臨床前研究または臨床試験結果、パートナー戦略の重点または利用可能な資金の変化または外部要因(例えば、資源の移転または競争優先度の創造の買収)に基づいて、開発または商業化計画を継続しないか、または継続しないか、または継続するか、または継続しないか、または商業化計画を選択することができる
•協力者は臨床試験を延期し、臨床試験計画に資金不足を提供し、臨床試験を停止或いは候補製品を放棄し、新しい臨床試験を繰り返し或いは行うことができ、或いは臨床試験候補製品の新しい調合を要求することができる
•提携者が競争力のある製品がより成功的に開発される可能性があると考えている場合、または私たちの製品よりも経済的に魅力的な条項で商業化できる場合、協力者は、第三者開発と直接または間接的に私たちの候補製品と競合する製品を独立して開発することができ、または第三者開発と直接または間接的に競合する製品を開発することができる
•1つまたは複数の製品のマーケティングおよび流通権利を有する協力者は、これらの製品または製品をマーケティングおよび流通するために十分なリソースを投入していない可能性がある
•協力者は、私たちの知的財産権を正確に維持したり、守ったりすることができないかもしれないし、何らかの方法で私たちの固有の情報を使用して、それによって訴訟を引き起こし、それによって、私たちの知的財産権または固有の情報を危険にさらしたり、無効にしたり、私たちを訴訟または潜在的な責任に直面させたりすることができる
•協力者は第三者の知的財産権を侵害する可能性があり、これは私たちを訴訟と潜在的な責任に直面させるかもしれない
•協力者と私たちの間で紛争が発生する可能性があり、私たちの候補製品の研究、開発または商業化の遅延または終了、または高価な訴訟や仲裁を招き、管理職の関心と資源を分散させる;
•協力は終了する可能性があり、終了すれば、適用可能な候補製品をさらに開発するために追加の資金が必要になるか、または商業化される可能性がある。
このような理由で、私たちと未来の任意の協力協定は、最も効果的な方法で、または私たちの任意のまたはすべての候補製品の開発または商業化を招くことができないかもしれない。もし私たちのパートナーが業務合併に参加すれば、製品開発や商業化計画の継続的な追求と重視は延期、減少、または終了される可能性がある。私たちの現在または未来の協力協定によれば、私たちの候補製品の開発に成功しなかったり、それを商業化したりすることができなかった場合は、私たちの業務、財務状況、運営結果、および将来性に重大で不利な影響を与える可能性があります。
さらに、私たちの既存または未来の任意のパートナーがAbbVieプロトコルおよびJanssenプロトコルの終了を含む協力協定を終了した場合、私たちは、臨床前研究または臨床試験の支援、マーケティングおよび流通コストを負担し、知的財産権を保護すること、または場合によっては候補製品を完全に放棄することを含むこれらの候補製品を独立して開発することを余儀なくされる可能性があり、いずれも、私たちの業務計画を変化させ、私たちの業務、財務状況、運営結果、および見通しに重大な悪影響を及ぼす可能性があります。
私たちとシュレーディンガーの既存の発見協力は私たちの業務に非常に重要だ。もし私たちがこのような協力を維持できない場合、あるいはこのような協力が成功しなければ、私たちの業務は不利な影響を受ける可能性がある。
2015年6月、2018年3月と2019年5月に改訂、またはシュレーディンガー協定を締結したシュレーディンガーと協力協定を締結した。この協力により、シュレーディンガーはその技術プラットフォームを用いて目標クラスの人類インテグリンメンバーに対して仮想スクリーニングを行い、著者らは薛定騰と協力して、目標の優先順位を促進し、目標の検証と分析を実行し、手がかりを識別し、手がかりの最適化を行う。シュレーディンガーは私たちの候補製品にすべての知的財産権の独占許可を与えた。
私たちは現在シュレーディンガーの発見能力に大きく依存しているため、もしシュレーディンガーがシュレーディンガー合意の下での義務の履行を遅延したり履行できなかったりすれば、もしシュレーディンガー協定の下での義務を履行できなかったら、私たちの協力条項或いは私たちの発見計画の解釈に同意しない、あるいはシュレーディンガー合意を中止すれば、私たちの候補製品ラインは不利な影響を受ける。シュレーディンガーも、私たちが彼らから得た知的財産権を適切に維持したり、守ったりすることができないかもしれないし、私たちの知的財産権を侵害して、私たちの知的財産権を無効にしたり、私たちを訴訟や仲裁に直面させたりすることができないかもしれません。これらはすべて時間と費用がかかります。また、シュレーディンガー協定の条項によると、どちらか一方が協力を中止する権利がある。もし私たちとシュレーディンガーとの協力が中止されたら、特に私たちの発見段階で、私たちの候補製品の開発は実質的に延期または損害されるだろう。
私たちは私たちの協力者と衝突する可能性があり、これは私たちの候補製品の開発や商業化を遅延または阻止するかもしれない。
私たちは、臨床前または臨床データの解釈、マイルストーンの実現、契約義務の解釈、サービス支払い、開発義務、または私たちの協力中に開発された知的財産権の所有権に関する紛争など、私たちの協力者と衝突する可能性がある。もし私たちのどの協力者とも衝突すれば、その協力者の行動は私たちの最大の利益に反するかもしれない。どのような相違も、すべての状況が私たちの候補製品の開発または商業化を遅延または阻止し、逆に私たちの収入の発生を阻止する可能性がある:協力者は私たちに記念碑的な支払いや特許権使用料を支払うことを望まず、これは私たちの追加的な資本の調達を必要とするかもしれない;私たちの協力活動によって生じる知的財産権の不確実性は、私たちの追加的な協力を阻止するかもしれない;協力者は製品の開発や製造で私たちに製品データや材料を提供することを含む、製品の開発や製造で協力することを望まない。協力者は、その開発および商業化活動の進展状況を随時私たちに通報することを望まない、またはこれらの活動の結果の開示を許可すること;いずれか一方が論争を解決するために訴訟または代替論争解決策を開始すること、または任意の一方が合意を終了しようと試みること。
私たちは協力協定を含む戦略取引に参加する可能性があり、これは私たちが候補製品を開発·商業化する能力に悪影響を与え、私たちの現金状況に影響を与え、私たちの費用を増加させ、私たちの経営陣に大きな妨害をもたらすかもしれない。
私たちは、協力協定、会社買収、資産購入、および私たちの既存業務の候補製品または技術の許可を補充または拡大することなど、戦略的取引を時々考慮するかもしれない。特に、戦略的に魅力があれば、主要なバイオテクノロジーやバイオ製薬会社を含む第三者との協力合意を求める評価を行います。パートナーの競争は非常に激しく、交渉過程は時間がかかって複雑だ。例えば、候補製品の開発または承認が延期され、承認された候補製品の販売が期待に達しない場合、または協力者が協力を終了することができない場合、私たちはいかなる新しい連携も維持できない可能性がある。また、最近の大手製薬会社間の大量の業務合併により、将来の潜在的戦略パートナーの数が減少している。私たちのパートナーは、似たような協力可能な兆候を得るために代替候補製品や技術を考慮することができ、そのような協力が私たちと協力する協力よりも私たちの候補製品に魅力的であるかどうかを考慮するかもしれない。私たちが協力について最終的な合意に到達できるかどうかは、他にも、戦略パートナーの資源と専門長の評価、提案協力の条項と条件、提案された戦略パートナーのいくつかの要素の評価に依存する。これらの要因は、臨床試験の設計または結果、FDAまたは米国国外の類似規制機関によって承認された可能性、候補被験者製品の潜在的市場を含む可能性がある, このような候補製品の製造と配送のコストと複雑性,競合製品の潜在力,我々の技術所有権に関する不確実性の存在は,挑戦の利点や業界や市場条件を考慮せずにこのような所有権に挑戦すれば,このような不確実性が存在する可能性がある。また、将来性のある市場や技術の資産を買収すれば、既存の技術と組み合わせることに成功しなければ、これらの資産を買収するメリットを実現できない可能性があります。私たちは戦略買収によって発生した任意の新製品を開発、テスト、製造、マーケティングする時に多くの困難に直面するかもしれません。これらの新製品は私たちが期待した利益を達成したり、私たちの業務を強化したりすることを延期したり阻止したりすることができます。
このような協力や他の戦略的取引の後に、取引が合理的であることを証明するために、予想される相乗効果を達成することを保証することはできません。例えば、このような取引は、非日常的または他の費用を発生させ、私たちの最近および長期的な支出を増加させ、統合または実施に重大な挑戦を構成し、または私たちの管理または業務を混乱させることを要求するかもしれない。これらの取引は、未知の債務、業務中断、および管理層の移転に暴露された時間および注意を、協力または開発買収の製品、候補製品または技術、取引対価格またはコストの支払いのために生成された巨額の債務または希釈発行された株式証券、予想以上の協力、買収または統合コスト、資産減記または営業権または減価費用、償却費用の増加、任意の買収業務の協力または合併業務および人員の困難およびコスト、主要サプライヤーとの関係が損なわれることを含む多くの運営および財務リスクをもたらす。経営陣と所有権の変化、買収された企業のキー従業員、買収された企業のメーカーや顧客を維持することができない。
したがって、上記の性質の任意の取引を行うか、または成功することは保証されないが、私たちが確実に達成した任意の取引は、前述または他のリスクの影響を受ける可能性があり、私たちの業務、財務状況、運営結果、および見通しに重大な悪影響を及ぼすであろう。逆に、私たちに有利な追加協力や他の戦略取引に参加しなければ、候補製品の開発や潜在的な商業化を延期し、市場に進出する任意の候補製品の競争力に悪影響を及ぼす可能性がある。
著者らは引き続き第三者に依存していくつかの臨床前研究或いは臨床試験を行うことを予想している。これらの第三者が契約要件に従って履行されていない場合、法律または法規の要求を満たしていない、予想締め切りを逃したり、協力関係を終了したりする場合、私たちの開発計画は延期され、私たちの業務、財務状況、運営結果、および見通しに潜在的な実質的かつ不利な影響を与える可能性がある。
著者らは未来に第三者臨床研究者、CRO、臨床データ管理組織と顧問に依存し、著者らの候補製品の臨床前研究と臨床試験の設計、実施、監督とモニタリングを協力或いは提供するつもりである。著者らはこれらの第三者に依存して、独立にすべての臨床前研究或いは臨床試験を行う能力がないため、著者ら自身が臨床前研究と臨床試験を行うことと比べ、著者らは臨床前研究と臨床試験の時間、品質とその他の方面の制御は比較的に少ない。これらの調査者、CRO、コンサルタントは私たちの従業員ではなく、私たちのプロジェクトに使用される時間と資源を制限します。これらの第三者は他のエンティティと契約関係にある可能性があり、その中のいくつかは私たちの競争相手である可能性があり、これは私たちの計画から時間と資源を消費するかもしれない。著者らはそれと契約した第三者が著者らの臨床前研究或いは臨床試験を行う時に勤勉でない、慎重或いは適時でない可能性があり、臨床前研究或いは臨床試験の遅延或いは不成功を招く可能性がある。
商業的に合理的な条項で受け入れ可能な第三者と契約を結ぶことができない場合、またはこれらの第三者が彼らの契約義務を履行していない場合、臨床前研究または臨床試験を行うための法律および法規の要件を満たしていない場合、または予想される期限内に達成できない場合、私たちの臨床開発計画は延期され、他の方法で悪影響を受ける可能性がある。いずれにしても,われわれのすべての臨床前研究と臨床試験が試験の一般的な調査計画や案および適用された法律や法規の要求に沿って行われることを確保する責任がある。FDAは一般的に臨床前研究は良好な実験室実践に従って行わなければならないことを要求し、臨床試験は必ず良好な臨床実践に従って行わなければならず、設計、進行、記録と臨床前研究と臨床試験の結果を報告し、データと報告の結果が信頼性と正確であることを保証し、そして臨床試験参加者の権利、完全性とセキュリティを保護する。私たちがコントロールできない第三者への私たちの依存は私たちのこのような責任と要求を解除しないだろう。著者らの第三者への依存により、著者らの臨床前研究或いは臨床試験はいかなる不利な発展或いは遅延が出現し、すべて著者らの業務、財務状況、運営結果と将来性に実質的な不利な影響を与える可能性がある。
もし私たちがこれらの第三者CROまたは他の人との任意の関係が終了した場合、私たちはCROまたは他の第三者と合意することができないか、または商業的に合理的な条項でそうすることができないかもしれない。CROを交換または追加することは、追加のコストをもたらし、管理時間と労力を必要とします。しかも、新しいCROが仕事を始める時、自然な過渡期がある。したがって,遅延が生じる可能性があり,期待される臨床開発スケジュールを満たす能力に実質的な影響を与える可能性がある。
私たちは第三者製造業者とサプライヤーに依存して私たちの候補製品に部品を提供する。私たちの第三者製造業者またはサプライヤーの損失、または私たちまたは彼らが適用可能な法規要件を遵守できなかったか、または許容可能な品質レベルまたは価格で十分な数の製品を供給できなかったか、または全くできなかったことは、私たちの業務に実質的な悪影響を与えるだろう。
私たちは薬品の製造、貯蔵、流通、または品質検査施設を持っていないし、経営していない。私たちは現在依存しており、イギリスと中国を含む第三者契約メーカーが原料薬、薬品、原材料、サンプル、成分または他の材料と報告を生産することに依存し続ける可能性がある。第三者メーカーに依存することは、私たち自身が候補製品を作るのではなく、私たちを違うリスクに直面させるかもしれない。われわれの臨床前と臨床開発製品の供給が制限されないこと、中断、終了或いは品質が満足できること、或いは受け入れ可能な価格で供給を継続することを保証することはできない。特に、当社のメーカーやサプライヤーのどの交換にも、合格した交換数が限られている可能性があるため、多くの努力や専門知識が必要となる可能性があります。もし私たちの第三者メーカーとサプライヤーやサプライチェーンのいずれかの第三者が不利な影響を受けた場合、新冠肺炎の流行やグローバルサプライチェーンの中断の結果を含めて、私たちの臨床前研究所に必要な候補製品の供給を確保できないかもしれません。
候補製品の製造過程はFDAと外国規制機関の審査を受けなければならない。私たちおよび私たちのサプライヤーとメーカーは、適用された製造要求を満たし、現行の良好な製造規範やcGMPのような規制基準に適合するために、監督機関の要求された厳格な施設とプロセス検証テストを受けなければならない。上場承認を得るには、製品製造過程に関する情報をFDAや外国規制機関に提出し、FDAと外国規制機関が製造施設を検査する必要がある。もし私たちの契約メーカーが私たちの規格やFDAなどの外国規制機関の厳しい規制要件に合った材料を作ることに成功しなければ、私たちの候補製品の要素を製造するために彼らの製造施設に依存できないかもしれません。また、私たちは契約製造業者の製造過程を制御せず、現在の規制要件を遵守するために彼らに完全に依存している。もし私たちのどの製造業者も、そのような要求を遵守できなかった場合、または品質、時間、または他の側面での義務を履行できなかった場合、または私たちのコンポーネントまたは他の材料の供給が他の理由で制限または中断された場合、私たちは別の第三者との合意を余儀なくされる可能性があり、私たちは合理的な条項でそうすることができないかもしれない。場合によっては、我々の候補製品を製造するために必要な技術的スキルまたは技術は、元の製造業者固有または独自である可能性があり、他の第三者に譲渡することは困難である可能性がある。これらの要素は私たちの製造業者への依存を増加させ、あるいは私たちが他の第三者を持つことができるように、またはその製造業者からライセンスを取得することを要求するだろう, 私たちの候補製品を作ります。もし私たちが何らかの理由でメーカーの交換を要求された場合、私たちは新しいメーカーの施設とプログラムが品質基準とすべての適用された法規とガイドラインに適合しているかどうかを検証することを要求されます。また、いくつかの開発手順を繰り返すことが要求される可能性があります。新メーカー認証に関する遅延は、タイムリーまたは予算内で候補製品を開発する能力に悪影響を及ぼす可能性があります。
もし私たちが規制部門の任意の候補製品の承認を得たら、私たちは引き続き第三者メーカーに依存すると予想される。私たちが第三者とすでにまたは将来製造手配を達成した範囲内で、私たちはこれらの第三者に依存して、品質管理と保証に関する要求を含む契約と監督管理の要求に適合する方法でその義務を適時に履行する。私たちの製品を生産するためのどの製造施設も、引き続きを含むfdaと外国監督機関の定期審査と検査を受けます
CGMP要求、品質管理、品質保証、およびそれに応じた記録とファイルメンテナンスを遵守します。もし私たちが候補製品の第三者製造を獲得したり維持したり、商業的に合理的な条項でそうすることができなければ、私たちは候補製品の開発と商業化に成功できないかもしれない。私たちまたは第三者が私たちの製造要件を実行できず、cGMPを遵守しない場合、またはFDAまたは外国規制機関が許容可能なコンプライアンス状態を維持しない場合、様々な態様で私たちの業務に悪影響を及ぼす可能性があります
•開発中の候補製品の臨床試験を開始または継続できない
•規制申請の提出を遅延させるか、規制部門の候補製品の承認を受ける
•既存または未来の協力者の協力を失うこと
•第三者製造施設に規制機関の追加検査を受けさせる
•私たちの候補製品ロットの流通を停止したり、リコールしたりすることを要求する;
•候補製品の市場化と商業化が承認された場合、私たちの製品のビジネスニーズを満たすことはできません。
また、私たちの契約メーカーは、資源制限や労使紛争、不安定な政治環境、流行病または流行病(例えば、新冠肺炎の大流行)やグローバルサプライチェーンの中断によって製造困難に遭遇する可能性がある。もし私たちの契約製造業者がこれらの困難に遭遇した場合、私たちは臨床前および臨床試験中の患者に私たちの候補製品を提供したり、承認された後に患者に治療製品を提供する能力が脅かされる。
例えば、イギリスは2020年1月31日に正式にEUを離脱し、通常はイギリスの離脱と呼ばれ、過渡期は2020年12月31日に終了する。しかし、EUとイギリスは、イギリス議会、欧州理事会、欧州議会の承認を得た貿易·協力協定、すなわちTCAに合意した。TCAには薬品に関する具体的な条項が含まれており,その中にはGMP相互承認,医薬製品の製造施設の検査,発表されたGMP文書が含まれているが,イギリスとEUの薬品法規が大規模に相互承認されることは予想されていない。そのため、会社は現在、イギリス(イングランド、ウェールズ、スコットランド)で医薬製品を商業化するために、単独のイギリス規制法の枠組みを遵守する必要がある。現在、イギリスは“2012年人類薬品条例”(改正)(北アイルランド議定書によると、EU規制枠組みは引き続き北アイルランドに適用される)を採択し、医薬製品のマーケティング、普及、販売に関するEUの立法を実施している。したがって,現在イギリスの規制制度はEUの規制制度と大きく一致しているにもかかわらず,イギリスの規制制度はEUから独立しており,TCAはイギリスとEUの薬品立法を相互に認めることを規定していないため,これらの制度は将来的に異なる可能性がある。例えば、新しい臨床試験条例は2022年1月31日にEUで施行され、複数のEU加盟国をカバーする簡略化された臨床試験申請と評価手続きが規定されているが、イギリスの法律では実施されておらず、イギリスで単独の臨床試験許可申請を提出する必要がある。いかなる遅延も、上場承認を得ることができない場合もある, 貿易·協力協定の結果やその他の理由として、任意の候補製品をイギリスおよび/またはEUで商業化することを阻止し、収入を創出し、利益を達成し、維持する能力を制限することができる。上記のいずれかの結果が生じた場合、私たちは、イギリスおよび/またはEUで任意の候補製品の規制承認を求める努力を制限または延期することを余儀なくされる可能性があり、これは、私たちの業務に重大で実質的な損害を与える可能性がある。現在、イギリスの離脱実施に関する詳細や解決策が不足しており、私たちの候補製品のイギリスでの部品製造と供給中断を招く可能性があり、この破壊が欧州規制枠組みに与える影響を自信を持って予測することはできない。私たちはイギリスの離脱によって業務と運営のどのような調整も重大な遅延と追加費用を招く可能性があります。上記のいずれの要因も、我々の業務、経営結果、または財務状況に重大な悪影響を及ぼす可能性がある。
私たちまたは私たちの第三者契約研究機関は、新冠肺炎の大流行を含む衛生流行病、流行病、その他の爆発に関連するリスクに直面しており、これは私たちの運営を深刻に混乱させる可能性がある
私たちの業務は新冠肺炎の大流行あるいは他の流行病あるいは流行病の影響を受けるかもしれません。もし私たちや私たちのメーカーとサプライヤーが運営している場所が閉鎖やその他の制限に関連していれば、私たちの運営は妨害される可能性があります。私たちはすでに未来に新冠肺炎疫病または他の衛生流行病または疫病を経験して、1つ以上のこれらの場所で私たちのいくつかのサプライヤーへの影響を発生する可能性があり、これは私たちの業務、財務状況、および運営結果に実質的な悪影響を及ぼすかもしれない。また、私たちの運営は過去も未来も中断される可能性があり、私たちのサプライヤーのオフィスの一時閉鎖やサービス停止に関する中断を含めて、私たちのためにしなければならないかもしれません
代替サプライヤーからの候補製品は、私たちの開発スケジュールおよび私たちの業務、財務状況、および運営結果に実質的な悪影響を及ぼす可能性があります
私たちの分子の製造は複雑で、私たちの第三者製造業者は生産で困難に直面するかもしれない。もし私たちまたは私たちの任意の第三者メーカーがそのような困難に遭遇した場合、私たちが臨床試験に候補製品を提供する能力、私たちが市場の承認を得る能力、または私たちが患者に製品を提供する能力(承認されれば)が延期または停止される可能性がある。
著者らの候補製品は生物製薬であり、生物製薬の製造過程は複雑で、時間がかかり、監督管理が厳しく、そして多重リスクの影響を受ける。我々の契約メーカーは,臨床試験や市販製品(承認されれば)のためのバイオ医薬品製造のための法的要求,cGMP,ガイドラインを遵守しなければならない。私たちの契約メーカーのcGMPロットの生産に関する経験は限られているかもしれない。
汚染、設備故障、設備設置或いは操作が不適切、サプライヤー或いはオペレータの誤り、生産量の不一致、製品特性の多変性及び生産技術の困難のため、生物製薬生産は製品損失の影響を受けやすい。正常製造プロセスとの微小な偏差でも生産量の低下、製品欠陥、その他の供給中断を招く可能性がある。もし私たちの第三者メーカーの施設で微生物、ウイルス、または他の汚染が発見された場合、これらの施設は汚染を調査および修復するために長い時間閉鎖する必要があるかもしれないが、これは臨床試験を延期し、私たちの業務に悪影響を及ぼす可能性がある。さらに、FDAが、我々の第三者製造業者の工場がcGMPを管理する法律および法規を含むFDAの法律および法規に適合していないと判断した場合、FDAは、欠陥が是正されるまで、またはNDA内の製造業者を法規に適合するメーカーに置き換えるまで、NDAの承認を拒否する可能性がある。
そのほか、臨床試験或いは商業規模の大規模生産はリスクが存在し、その中にコスト超過、技術拡大の潜在問題、技術再現性、安定性問題、cGMP、ロット一致性と原材料の適時獲得性を含む。私たちの協力者が私たちの任意の候補製品の規制承認を得ても、メーカーがFDAや他の規制機関が許容できる規格に従って承認された製品を生産して、製品が発売される可能性のある要求または将来の潜在的な需要を満たすのに十分な数の製品を生産することができる保証はない。もし私たちのメーカーが臨床試験や商業化のために十分な数量を生産できなければ、商業化努力は損害を受け、これは私たちの業務、財務状況、運営結果、将来性に不利な影響を与えるだろう。
生物製薬製造プロセスを拡大することは困難かつ不確定な任務であり、私たちの第三者メーカーは実施、製造と開発プロセスを完成するために必要な能力を備えていないかもしれない。既存のメーカー工場の製造プロセスを十分に検証したり拡大したりすることができなければ、別のメーカーに移転して製造検証過程を完成させる必要があり、長い可能性がある。もし私たちが契約メーカーと私たちの候補製品の製造プロセスを十分に検証し、拡大することができれば、私たちは依然として契約製造業者と商業供給協定を交渉する必要があり、私たちが受け入れられる条項について合意できるかどうかは定かではありません。
将来、私たちの候補製品や製品の製造に関連するいかなる安定性や他の問題も発生しないことを保証することはできません。もし私たちの第三者メーカーがこれらの困難に遭遇すれば、計画された臨床試験で患者に任意の候補製品を提供する能力が脅かされ、承認されると、患者に製品を提供する能力が脅かされる。臨床試験供給のいかなる遅延或いは中断も計画の臨床試験の完成を遅らせる可能性があり、臨床試験計画の維持に関連するコストを増加させ、そして遅延した時間帯に基づいて、追加費用で新しい臨床試験を開始するか、或いは臨床試験を完全に中止することを要求する。私たちの候補製品または製品の臨床的または商業的製造に影響を与える不利な発展は、出荷遅延、在庫不足、ロット故障、製品撤回またはリコール、または私たちの候補製品または製品供給の他の中断を招く可能性があります。また、在庫抹消を行い、製品候補または規格に適合していない製品に他の費用や支出を発生させ、高価な救済努力を行ったり、より高価な製造代替案を求めたりしなければならない可能性もある。したがって、私たちのサプライチェーンのどのレベルで遭遇した故障や困難は、私たちの業務に悪影響を及ぼす可能性があり、私たちの任意の候補製品や製品の開発および商業化を遅延または阻害し、承認されれば、私たちの業務、見通し、財務状況、および運営結果に悪影響を及ぼす可能性があります。
我々のプロセス開発作業の一部として、コストのコントロール、規模の実現、加工時間の短縮、製造成功率の向上など、様々な理由で、開発過程における異なる時間点で製造プロセスを変更することも可能である。これらの変化は予想される目標を達成できない可能性があり、これらの変化はすべて私たちの候補製品の表現の違いを招き、著者らが行っている臨床試験あるいは未来の臨床試験の結果に影響を与える可能性がある。場合によっては、製造過程の変更は実行する必要があるかもしれません離体するより高度な臨床試験を行う前に,比較可能な研究を行い,患者からより多くのデータを収集する。例えば臨床開発では私たちの過程の変化を示す必要があるかもしれません
臨床早期又は試験早期に使用される製品と臨床後期又は試験後期に使用される製品との比較可能性。
知的財産権に関するリスク
もし私たちの技術や候補製品のために特許保護を獲得、維持、実施することができなければ、私たちの候補製品の開発と商業化は不利な影響を受ける可能性がある。
私たちの成功は、私たちの候補製品のために他人の知的財産権を獲得し、維持する能力と、私たちのビジネス秘密を保護し、第三者が私たちの独占権を侵害することを防止し、他人の固有の権利を侵害することなく運営する能力を含む、私たちの特許および他の形態の知的財産権を取得し、維持する能力にある程度依存する。2022年12月31日現在、私たちは、米国および世界の多くの主要な司法管轄区域(ヨーロッパ、日本、中国を含む)に、我々のインテグリン治療化合物を保護するために、複数のプロジェクト(私たちの候補製品を含む)の様々な特許および出願されている特許を発行している。さらに、私たちは、児童医学センター会社またはCMCCプロトコルと、いくつかの米国特許および修飾インテグリンポリペプチド、修飾インテグリンポリペプチドを含む結晶性二量体および関連方法に関連する米国特許出願について独自のグローバルライセンスプロトコルを有する。私たちは私たちの候補製品のいくつかの側面について特許を申請することができないか、または根本的に解決できないかもしれない。さらに、私たちは、必要または望ましいすべての特許出願を合理的なコストまたはタイムリーに起訴することができないか、またはそのような特許出願から発行される可能性のある任意の特許を維持、実行および許可することができないかもしれない。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.私たちは、第三者から許可を得たすべての特許出願の準備、提出および起訴を制御する権利、または第三者に許可された特許の権利を維持する権利がないかもしれない。だから…, このような特許と出願は、私たちの業務の最高の利益に合った方法で起訴されてはならない。私たちが将来獲得した特許はまだ十分ではないかもしれませんが、他の人が私たちの技術を使用したり、それと競争する製品や技術を開発するのを阻止するのには十分ではありません。私たちの任意の未解決特許出願が発行または特許付与をもたらすことは保証されず、私たちが将来発行または付与された任意の特許が後で無効または強制執行されないことが発見されないことを保証することはできず、将来発行または付与された任意の特許が、私たちの候補製品をカバーしたり、競合相手から効果的な保護を提供するのに十分な広範なクレームを含むことも保証されない。さらに、バイオテクノロジーと生物製薬会社の特許地位は複雑な法律と事実の問題に関連しているため、非常に不確定な可能性がある。私たちは、私たちの現在および未来のノウハウおよび製品候補が効果的かつ実行可能な特許によってカバーされているか、またはビジネス秘密として効果的に保護されている範囲内でのみ、第三者による許可されていない私たちの固有の権利を保護することができる。もし第三者が私たちの独占権を開示したり盗用したりすれば、私たちの市場地位に実質的な悪影響を及ぼすかもしれない。
私たちの係属中の特許出願は、特許がこのような出願から発行されるまで、そのような出願において要求される技術を実施する第三者に対して強制的に実行することはできない。特許可能性の他の要件が満たされていると仮定すると,現在,最初に特許出願を提出した発明者は通常特許を取得する権利がある。しかし、2013年3月16日までに、米国では、最初に保護された発明を発明した者が特許を有している。科学文献で発表された発見は往々にして実際の発見に遅れており、米国と他の司法管轄区の特許出願は通常、申請18ヶ月後に発表され、時には全く発表されないこともある。したがって、私たち、私たちのライセンシーまたは協力者、または任意の未来の戦略パートナーが、私たちが所有または許可した特許または係属中の特許出願に要求された最初の発明者であるか、または私たち、私たちのライセンシーまたは協力者または任意の未来の戦略パートナーが、そのような発明のために特許保護を申請する最初の人であることを決定することはできない。
米国特許商標局(USPTO)および各種外国政府特許機関は、特許過程において大量のプログラム、文書、費用支払い、その他の規定を遵守することを要求している。場合によっては、規定を遵守しないことは、特許または特許出願の放棄または失効をもたらす可能性があり、それにより、関連する管轄区域の特許権の一部または全部が失われる可能性がある。この場合、競争相手は他の場合よりも早く市場に参入するかもしれない。米国特許商標局と外国特許庁が特許を付与する際に採用する基準は,常に統一的または予測可能ではない。例えば、バイオテクノロジーやバイオ製薬特許で許可されている特許標的または特許請求の範囲については、世界的に統一された政策はない。したがって、私たちは私たちの独自製品と技術に対する未来の保護の程度を知らない。特許を取得する過程は時間がかかって高価で、時々予測できないこともある。
付与されると、特許は、許可または特許付与後の所与の期間内に、反対、干渉、再審、付与後審査を継続して受けることができる各方面間裁判所または特許庁または同様の手続における審査、廃止または派生訴訟は、その間、第三者は、そのような初期ライセンスに異議を唱えることができる。このような訴訟は長い間続く可能性があり、そのような訴訟における不利な裁決は、そのような攻撃の許容または承認クレームの範囲を縮小することができるかもしれないし、または私たちの特許の全部または一部を無効にする可能性があり、または実行不可能とみなされる可能性があり、これは、第三者が私たちの候補製品を商業化し、私たちに支払うことなく直接私たちと競争することを可能にするかもしれない。また、保証はできない
•他の会社は、私たちの候補製品と同じまたは類似しているが、私たちが所有または許可している特許請求の範囲内ではない化合物を製造、使用または販売することができないかもしれない
•私たちまたは私たちのライセンス者、または私たちの既存または未来の協力者は、私たちが所有または許可している各発行特許および係属特許出願によってカバーされている発明を最初にした者である
•私たちまたは私たちのライセンシー、または私たちの既存または未来の協力者は、私たちの発明のいくつかの態様に関する特許出願を最初に提出した人である
•他の会社は、私たちの知的財産権を侵害することなく、類似または代替技術を独立して開発したり、私たちの技術をコピーしたりしません
•第三者は私たちの特許に挑戦してはいけません。もし挑戦されたら、裁判所は私たちの特許が有効で、強制的に実行可能で、侵害されていると判断します
•私たちが許可しているか、または将来許可されている可能性のある任意の発表された特許は、いかなる競争優位性を提供してくれるか、または第三者の挑戦を受けないだろう
•特許を申請できる技術をより多く開発することができます
•他人の特許は、私たちの業務、財務状況、経営結果、および将来性に実質的または不利な影響を与えないだろう
•私たちの競争相手は、特許権を強制的に実行できる国で研究や開発活動を行うことはなく、これらの活動から学んだ情報を利用して競争力のある製品を開発し、私たちの主な商業市場で販売します。
もし私たちまたは私たちの許可者または協力者が私たちの候補製品をカバーする特許および特許出願を維持できなかった場合、私たちの競争相手は市場に参入する可能性があり、これは私たちの業務、財務状況、運営結果、および将来性に重大で不利な影響を与える可能性がある。さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、結果にかかわらず、会社が現在または将来の候補製品を許可、開発、または商業化することを阻止することができる。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの商業と競争の地位は損なわれるだろう。
私たちの候補製品のいくつかの側面のために特許保護を求めるほか、機密と非特許技術を含む商業秘密は、私たちの競争地位を維持するために重要だと思います。私たちの第三者への依存は、私たちのビジネス秘密を共有することを要求し、これは、競争相手がこれらの秘密や私たちのビジネス秘密が流用または開示されていることを発見する可能性を増加させる。私たちは、企業秘密および機密および非特許ノウハウの保護を求めており、方法の1つは、私たちの従業員、企業協力者、外部科学協力者、CRO、契約製造業者、コンサルタント、コンサルタント、および他の第三者のような秘密および秘密を取得する権利を有する当事者と秘密および秘密協定を締結することである。私たちはまた、私たちの従業員とコンサルタントと秘密と発明または特許譲渡協定を締結し、彼らに秘密にして彼らの発明を私たちに譲渡することを要求します。このような努力にもかかわらず、どちらも合意に違反し、私たちのビジネス秘密を含む私たちの固有の情報を漏洩する可能性があり、私たちは十分な救済措置を得ることができないかもしれない。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかり、結果は予測できない。また、米国のいくつかの裁判所と一部の外国司法管轄区は商業秘密を保護することをあまり望んでいないか、または保護したくない。もし私たちのどんなビジネス秘密も競争相手によって合法的に取得または独立して開発されたなら、私たちは彼らがその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。また,時間の経過とともに,我々のビジネス秘密,ノウハウ,ノウハウ,独自の情報が独立した発展によって業界内に伝播する可能性が予想される, 定期刊行物論文の発表及び学術と工業科学職場の人員流動。したがって、私たちのノウハウを保護するために高い努力をしなければ、私たちは他の人がこの技術を利用することを阻止できないかもしれないが、これは国内と国際市場での私たちの拡張能力に影響を及ぼすかもしれない。もし私たちのビジネス秘密が競争相手に漏れたり、競争相手によって独立して開発されたりすれば、私たちの競争地位は損なわれ、これは私たちの業務、財務状況、運営結果、および将来性に実質的な悪影響を及ぼす可能性がある。
承認された場合、他の会社または組織は、私たちまたは私たちの許可側の特許権に挑戦するか、または私たちが開発および商業化した製品の特許権を阻止することを主張する可能性があります。
経口インテグリンによる繊維化と炎症性腸疾患或いは他の疾患領域の治療は比較的に新しい科学領域である。私たちはすでに第三者に申請して、私たちのMINTプラットフォームに関連するアメリカ特許出願の独占許可を得ました。アメリカと世界各地の重要な市場での他の未解決特許出願
小分子インテグリン阻害剤および他の療法の発見、開発および製造に関連する多くの異なる方法、組成物、およびプロセスが要求されるかもしれない。
小分子インテグリン抑制製剤による治療分野の成熟に伴い,世界各地の国家特許庁が特許出願を処理している。どの特許が発行され、彼らが発行された場合、いつ発行され、誰に授与され、どのような権利要求が提出されるかには、不確実性がある。しかも、第三者は私たちの知的財産権を無効にしようと努力するかもしれない。たとえ私たちの権利が直接挑戦されなくても、紛争は私たちの知的財産権を弱化させる可能性がある。私たちは第三者が私たちの知的財産権を回避または無効にしようとするいかなる弁護も私たちにとってコストが高いかもしれません。私たちの経営陣が多くの時間と精力を投入する必要があるかもしれません。私たちの業務、財務状況、運営、将来性、または私たちの成功競争の能力に重大で不利な影響を与えるかもしれません。もし私たちが第三者の知的財産権を侵害していることが発見された場合、裁判所の命令を含めて、権利侵害の候補製品や製品の開発、製造、または商業化を停止させられる可能性がある。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
米国および世界の他の司法管轄区域での私たちの技術に関する特許の申請、起訴、弁護、実行は極めて高価になるだろうし、私たちまたは私たちのライセンシーや協力者の知的財産権はアメリカ以外のいくつかの国には存在しないかもしれないし、いくつかの国ではアメリカほど広くないかもしれない。私たちまたは私たちのライセンシーまたは協力者が特許保護を受けていない司法管轄区域では、競争相手は、私たちまたは私たちのライセンシーまたは協力者の技術を使用して競合製品を開発することを求めることができ、さらに、特許保護を有しているが、米国よりも特許を実施することが困難な地域に他の侵害製品を輸出することが可能である。競争相手の製品は、私たちが特許を発行または付与していない司法管轄区、または私たちまたは私たちのライセンシーまたは協力者が発行または付与した特許声明または他の知的財産権が、これらの管轄区域での競争相手の活動を阻止するのに十分ではない司法管轄区で、将来の製品と競争することができないかもしれない。ある国、特にある発展途上国の法律制度は特許の強制執行を困難にし、これらの国は他のタイプの知的財産権保護、特に薬品や生物製薬に関連する知的財産権保護を認めない可能性がある。これは私たちまたは私たちのライセンシーや協力者が特定の管轄区域で私たちまたは彼らの特許またはマーケティング競争製品を侵害し、私たちまたは彼らの独占権を侵害することを防止することを困難にするかもしれない。外国の管轄区域で私たちの特許権を強制的に執行する訴訟は大きなコストを招く可能性があり、私たちと私たちのライセンシーまたは協力者の努力と注意を私たちの業務の他の側面から移すことができます, 私たちと私たちのライセンシーまたはパートナーの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちおよび私たちのライセンシーまたはパートナーの特許出願が発行できないリスクに直面し、第三者が私たちまたは私たちのライセンシーまたはパートナーにクレームを付ける可能性がある。私たちまたは私たちのライセンシーまたは協力者は、私たちまたは私たちのライセンシーまたは協力者が起こしたいかなる訴訟でも勝利しないかもしれないし、判決された損害賠償または他の救済措置(ある場合)は商業的意味がないかもしれない。
私たちが発明を特許保護することを選択した時、私たちは通常、まず米国特許商標局に米国仮特許出願(優先権出願)を提出する。特許協力条約に基づいて出願された国際特許出願及び/又は非特許協力条約に基づいて出願された国家特許出願は,優先権出願後12ヶ月以内に提出することができる。PCTの出願によれば、1つまたは複数のPCT加盟国で国および地域特許出願を提出することができる。今まで、私たちは特許保護を提供する可能性のあるすべての国と地域の司法管轄区域で特許保護を申請していません。しかも、私たちは付与される前に国と地域の特許出願を放棄することに決定するかもしれない。最後に、各国または地域特許庁の付与手続は、特許出願が特定の法域で関係登録当局によって拒否され、他の法域によって承認される可能性がある独立した手続である。同様に一般的な場合は、国によっては、同じ候補製品または技術に異なる範囲の特許保護が付与される可能性があることである。
いくつかの司法管轄区の法律は知的財産権の保護程度はアメリカの法律に及ばず、多くの会社はこれらの司法管轄区の保護とこのような権利を保護する時に重大な困難に直面している。私たちまたは私たちのライセンシーや協力者が知的財産権の保護に困難に直面したり、他の理由でこれらの管轄地域の業務に重要な知的財産権を効果的に保護できない場合、これらの権利の価値が低下する可能性があり、私たちはこれらの管轄区域の他の人からの追加的な競争に直面する可能性がある。多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は政府機関や政府請負業者に対する特許の実行可能性を制限している。これらの国では、特許所有者の救済措置が限られている可能性があり、このような特許の価値を大幅に低下させる可能性がある。もし私たちまたは私たちの任意のライセンシーまたは協力者が私たちの業務に関連する任意の特許について第三者に許可を付与することを余儀なくされた場合、関連する司法管轄区における私たちの競争地位が損なわれる可能性があり、私たちの業務、財務状況、運営結果、および見通しは不利な影響を受ける可能性がある。
もし私たちがいかなる許可、協力、または他の合意の下での義務を履行できなかった場合、私たちは損害賠償金の支払いを要求され、私たちの候補製品の開発と保護に必要な知的財産権を失う可能性があり、あるいは再許可を付与するいくつかの権利を失う可能性がある。
私たちは特許、ノウハウ、そして独自技術に依存しており、私たち自身のものもあれば、他の人が許可しているものもある。私たちのライセンスのいかなる終了も重大な権利の喪失を招き、候補製品を開発する能力を損なう可能性がある。私たちの現在の許可、そして私たちが未来に達成したどんな許可も、私たちに様々な開発、商業化、資金、マイルストーン、印税、勤勉、再許可、保険、特許起訴、強制執行、および/または他の義務を課すかもしれない。もし私たちがこれらの義務に違反したり、許可されていない方法で許可された知的財産権を使用したりする場合、私たちは損害賠償金の支払いを要求される可能性があり、許可側は許可を終了する権利がある可能性があり、これは、許可技術がカバーする任意の未来の製品を開発、製造、販売することができない、あるいは競争相手が許可技術を得ることができるようにすることができるかもしれない。さらに、私たちの許可者は私たちに許可されていない知的財産権を持っているか、またはコントロールしている可能性があり、したがって、その是非にかかわらず、私たちは私たちが許可者の権利を侵害しているか、または他の方法で侵害しているというクレームを受ける可能性がある。また、将来の製品の販売にどのくらいの印税義務が支払われるかは確定できませんが、金額は大きいかもしれません。私たちの将来の印税義務の金額は、開発や商業化に成功した製品で使用される技術や知的財産権にかかっています(あれば)。したがって、私たちが製品を開発して商業化することに成功しても、私たちは利益を達成したり維持することができないかもしれない。
さらに、ライセンス契約によると、知的財産権に関する紛争が生じる可能性があります
•ライセンス契約に従って付与された権利範囲および解釈に関連する他の問題;
•私たちの候補製品、技術、およびプロセスは、ライセンス契約に拘束されていないライセンス側の知的財産権をどの程度侵害しているか
•私たちの協力開発関係に基づいて、特許と他の権利を再許可する
•私たちのライセンス契約の下での義務と、どのような活動がこれらの職務義務を満たしていますか
•私たちのライセンス者、私たちおよびパートナーが知的財産権を共同で創造または使用することによって生成された発明およびノウハウの発明および所有権;
•特許技術発明の優先権。
また,我々が現在第三者から知的財産権や技術許可を得ているプロトコルは複雑であり,このようなプロトコルのいくつかは様々な解釈の影響を受ける可能性がある.可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。さらに、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で現在の許可手配の能力を維持していることを阻害または損害すれば、影響を受けた候補製品の開発および商業化に成功することができない可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たち、私たちの許可者または協力者、または任意の未来の戦略的パートナーは、私たちの特許または他の独占権を保護または強制するために訴訟に訴える必要があるかもしれません。これらは、コストが高く、時間がかかる可能性があり、私たちの候補製品の開発および商業化を延期または阻止するか、または私たちの特許が付与されたときに特許出願および他の固有の権利を危険な状況に置くことができます。
競争相手は私たちが持っているか許可された特許、特許出願、または他の知的財産権を侵害するかもしれない。もし私たちが第三者に対して法的訴訟を提起して、私たちの候補製品や技術をカバーする特許を強制的に執行する場合、被告は私たちの特許が無効または強制執行できないと反訴することができる。米国の特許訴訟では,被告が無効または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、適切な書面記述の欠如、明らかまたは実施できないことを含む、いくつかの法定要件のいずれかを満たすことができなかったと言われている可能性がある。主張を実行できない理由は,特許訴訟に関連する個人が起訴期間中に米国特許商標局に関連情報を隠蔽したり,誤った陳述をしたりしたためであろう.特許訴訟の間、法律が無効または実行不可能と断言した後の結果は予測できない。例えば、有効性の問題については、私たちは無効な以前の技術がないとは確信できないが、私たちと特許審査員は起訴中にこれを知らない。もし被告が無効または強制不可能な法的主張に勝った場合、私たちは少なくとも部分的、さらにはすべて、私たちの1つまたは複数の候補製品または私たちのプラットフォーム技術のいくつかの態様の特許保護を失うだろう。このような特許保護の喪失は、我々の業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。第三者によって引き起こされるか、または私たちによって提起されるか、またはUSPTOによって宣言される干渉または派生プログラムは、
我々の特許または特許出願については、発明の発明権または優先権を決定することが必要である。不利な結果は、私たちが関連技術の使用を停止することを要求するか、または勝利者から許可を得ようとすることを要求するかもしれない。もし勝利者が商業的に合理的な条項で私たちに許可を提供しない場合、あるいは私たちに許可を全く提供しない、あるいは非独占的な許可を提供し、私たちの競争相手が同じ技術を獲得した場合、私たちの業務は損害を受ける可能性がある。また、訴訟に関連する不確実性は、私たちの臨床試験を継続するために資金を調達し、私たちの研究計画を継続し、第三者から必要な技術的許可を得たり、開発パートナー関係を構築して、私たちの候補製品を市場に出す能力に実質的な悪影響を与える可能性があります。さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。聴聞、動議、または他の一時的手続き、または事態発展の結果を公開することもできる。もし証券アナリストや投資家がこれらの結果がマイナスだと思っていれば、私たちの普通株の価格に実質的な悪影響を及ぼす可能性がある。もし競争相手が私たちの特許や他の知的財産権を合法的に侵害することなく、私たちが保護された技術を中心に設計すれば、特許や他の知的財産権は私たちの技術を保護することができないだろう。
第三者の知的財産権は、候補製品を商業化する能力に悪影響を及ぼす可能性があり、私たち、私たちのライセンシーまたはパートナー、または任意の将来の戦略パートナーは、特許または他の専有権の侵害を告発し、または特許または他の固有の権利を無効にしようとする第三者のクレームまたは訴訟を受ける可能性がある。私たちの候補製品を開発またはマーケティングするためには、第三者に訴訟を提起したり、許可を得たりする必要があるかもしれません。そのような訴訟や許可証は費用が高いかもしれないし、商業的に合理的な条項では得られないかもしれない。
私たち、私たちのライセンシーまたは協力者、または任意の未来の戦略パートナーは、特許または他の独占権の侵害または流用によって第三者からクレームを受ける可能性がある。アメリカ国内と国外では、特許侵害訴訟、妨害、派生、反対とを含む生物技術と製薬業界の特許とその他の知的財産権に関連する大量の訴訟がある各方面間米国特許商標局及び対応する外国特許庁の手続を審査する。発行された特許および係属中の特許出願は、私たちの目標、MINTプラットフォーム、または私たちの候補製品のいくつかの態様を必要とし、私たちは私たちの候補製品の修正に適用する必要があるかもしれません。インテグリン阻害剤が要求される特許がある可能性があり,これらの特許は我々が開発したい製品に関連している可能性がある。したがって、1つ以上の組織が特許権を持つ可能性があり、私たちは許可を得る必要がある。もしこれらの組織が合理的な条項でこのような特許権の許可を与えることを拒否した場合、私たちはこれらの特許がカバーする製品を販売したり、研究開発や他の活動を行うことができない可能性があり、これは私たちの業務、財務状況、運営結果、および将来性に大きな悪影響を及ぼす可能性がある。もし私たち、私たちのライセンシーまたはパートナー、または任意の未来の戦略パートナーが第三者特許または他の知的財産権の侵害を発見された場合、私たちは損害賠償金の支払いを要求されるかもしれません。もし私たちまたは彼らが故意の侵害が発見された場合、損害賠償金と弁護士費の3倍が含まれるかもしれません。さらに、私たち、私たちのライセンシーまたは協力者、または任意の未来の戦略パートナーは、第三者の許可を求めるか、または要求されることを選択することができ、これらのライセンスは、全くない場合には、受け入れ可能な条項で提供できない可能性がある。許容可能な条項で許可を得ることができても,これらの権利は非排他的である可能性があり,これは我々の競争相手が我々に許可する同じ技術や知的財産権を獲得する可能性がある.もし私たちが必要な許可を得ることができなければ、私たちまたは私たちの既存または未来のパートナーは、私たちの技術に基づいて候補製品を効率的に販売することができないかもしれません。これは、私たちが収入を創出したり、利益を達成する能力を制限し、私たちの運営を維持するのに十分な収入を生成することを阻止するかもしれません。また、, 私たちは、私たちの特許や他の知的財産権を保護または実行するために、クレームを出したり、訴訟を提起したりする必要があることを発見するかもしれない。私たちが特許や他の専有権に関連する任意の訴訟や他の手続きを弁護したり、訴訟を提起するコストは巨大かもしれません。たとえ解決策が私たちに有利であっても、訴訟は私たちの経営陣の注意をそらすかもしれません。私たちのいくつかの競争相手は、彼らがより多くの資源を持っているので、私たちよりも複雑な特許訴訟の費用を効果的に受けるかもしれない。特許訴訟または他の訴訟の開始と継続による不確実性は、私たちの研究開発努力を延期し、私たちが運営を継続する能力を制限する可能性がある。
インテグリンによる療法の将来性が進んでいるため,第三者の権利を侵害することなく操作の自由を最終的に評価することは困難である。多くの会社が未解決の特許出願を持っており、私たちが追求している目標と同じまたは類似した目標に対するインテグリン、または私たちの候補製品と類似した化合物をカバーするインテグリンを広くカバーする特許を発行している。もし許可が得られなければ、私たちの候補製品の商業化が延期されるかもしれない。第三者に発行される特許または他の第三者知的財産権が、私たちの製品、候補製品またはその要素、または私たちの開発計画に関連する製造または使用をカバーする場合、私たちの競争地位は影響を受ける可能性がある。この場合、私たちは、そのような特許が満了するまで、製品または候補製品を開発または商業化することができない場合があり、または関連する第三者知的財産権を無効または無効にするか、または知的財産権所有者とライセンス契約を締結することができない場合がある(商業的に合理的な条項で利用可能であれば)。私たちが知らない発行された特許が存在する可能性があり、これらの特許は第三者が保有しており、発見されて有効かつ強制的に実行可能であれば、私たちのミントプラットフォームおよび候補製品によって侵害される可能性がある。私たちが知らない未解決の特許出願は、発行された特許を招く可能性もあります。これらの特許は、私たちのミントプラットフォームや候補製品によって侵害される可能性があります。もしこのような1つが
侵害請求が提起されて成功した場合、私たちは故意に侵害された可能性のある3倍の損害賠償と弁護士費を含む大量の損害賠償を要求される可能性があり、私たちは私たちの候補製品を放棄したり、任意の特許所有者に許可を求めたりすることを余儀なくされるかもしれない。許可証が商業的に合理的な条項で提供されることを保証することはできない。
私たちはまた関連する第三者特許や出願を決定できない可能性がある。例えば、2000年11月29日までに提出された米国出願と、その日以降に提出されたいくつかの米国出願は、該当する特許が取得されない限り、米国国外に提出されないであろう。米国および他の地方の特許出願は、優先権を要求する最初の出願の約18ヶ月後に発行され、このような最も早い出願日は、一般に優先権日と呼ばれる。したがって、私たちの候補製品またはミントプラットフォームに関する特許出願は、私たちが知らずに他の人に提出されたかもしれない。さらに、いくつかの制限の下で、公表された係属中の特許出願は、私たちのMINTプラットフォーム、私たちの候補製品、または私たちの候補製品の使用をカバーするために、後で修正することができる。第三者知的財産権者たちもまた私たちに権利侵害請求を積極的に提起するかもしれない。私たちは私たちがこのような侵害請求を成功的に解決したり、他の方法で解決できるという保証はない。もし私たちが受け入れられる条項で未来のクレームを成功的に解決できなければ、私たちは費用が高く、予測できない、時間のかかる訴訟を行うことを要求されるかもしれません。もし承認されれば、私たちの製品を販売する上で重大な遅延に遭遇するかもしれません。私たちにクレームを出した当事者は、彼らがより多くの資源を持っているので、私たちよりも複雑な特許訴訟の費用を効果的に維持することができるかもしれない。さらに、知的財産権訴訟や行政訴訟に関連する大量の開示要求により、私たちのいくつかの機密情報が開示によって漏洩される可能性がある。また、, いかなる訴訟の開始および継続によって生じるいかなる不確実性も、私たちが追加資金を調達する能力に重大な悪影響を及ぼすか、または私たちの業務、運営結果、財務状況、および見通しに重大な悪影響を及ぼす可能性がある。もし私たちがこのような紛争で失敗すれば、損害賠償金の支払いを余儀なくされるほか、私たちの権利侵害と思われる任意の候補製品の商業化は一時的または永久的に禁止される可能性がある。可能であれば、私たちは第三者の知的財産権を侵害しないように、候補製品の再設計を余儀なくされる可能性もあります。これらのすべての事件は、私たちが最終的に勝っても、私たちが大量の財務と管理資源を移転する必要があるかもしれません。そうでなければ、私たちは私たちの業務に投入することができ、私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性があります。
知的財産権訴訟は私たちに大量の資源を費やし、私たちの人員の正常な義務に対する注意を分散させるかもしれない。
正当な理由があるかどうかにかかわらず、知的財産権クレームに関連する訴訟や他の法律手続きは予測できず、通常コストが高く、時間がかかり、私たちの核心業務から大量の資源を分流し、私たちの技術と管理者の正常な職責を分散することを含むかもしれない。さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。また、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスだと思う場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。さらに、このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。
私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源とより成熟して発展した知的財産権の組み合わせを持っているので、このような訴訟や法的手続きの費用を私たちよりも効率的に負担するかもしれない。したがって、私たちは努力したにもかかわらず、私たちは第三者が私たちの知的財産権を侵害したり、流用したり、私たちの知的財産権に成功したりすることを防ぐことができないかもしれない。特許訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争能力に重大な悪影響を及ぼす可能性がある。
私たちは、私たちまたは私たちの従業員またはコンサルタントが、私たちの従業員またはコンサルタントの元の雇用主またはその顧客のいわゆる商業機密を誤って使用または開示したと非難されるかもしれない。これらのクレームの弁護コストは高いかもしれませんが、私たちがそうしなければ、私たちは金銭損害賠償の支払いを要求され、貴重な知的財産権や人員を失う可能性があります。
私たちの経営陣を含め、私たちの多くの従業員は、以前、私たちの競争相手や潜在的な競争相手を含む大学やバイオテクノロジーやバイオ製薬会社に雇われていました。その中の一部の従業員は以前の仕事に関連した所有権、秘密とスポーツ禁止協定に署名した。現在、私たちのクレームについて解決されていないにもかかわらず、私たちは、これらの従業員、私たちの許可者または協力者の従業員、または私たちが意図していない、または他の方法でその前の雇用主の商業秘密または他の固有情報を使用または漏洩した疑いを受ける可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちや私たちの許可者や協力者がこのようなクレームを弁護できなかったら、金銭損害賠償を支払う以外に、私たちは貴重な知能を失う可能性があります
財産権や人員。重要な研究者や彼らの作業製品を失うことは、私たちの候補製品の開発と最終的な商業化の能力を阻害したり、私たちの候補製品の開発と商業化を阻止したりする可能性があり、これは私たちの業務を深刻に損なう可能性があります。私たちや私たちの許可者や協力者がこれらのクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。
特許条項は候補製品に対する私たちの競争地位を十分に長く保護するのに十分ではないかもしれない。
特許の寿命は限られている。米国では、すべての維持費が適時に支払われる場合、特許の自然失効時間は、通常、米国で最初の非臨時出願日から20年である。様々な特許期間の調整または延長を行うことができるが、特許の有効期限およびその提供される保護は限られている。私たちの候補製品が特許を取得しても、特許有効期限が切れると、私たちは模造薬や生体模倣薬を含む競争製品からの競争に直面するかもしれない。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
特許および/または出願の定期維持費、継続費、年会費、および様々な他の政府費用は、特許および/または出願の有効期間内にいくつかの段階で米国特許商標局および米国以外の様々な政府特許機関に支払われる。私たちは私たちにこれらの費用を支払うことをシステム的に想起させ、私たちはコンサルタントと外部会社を招聘し、および/または私たちの外部弁護士に依存してUSPTOと非米国特許機関にこれらの費用を支払う。米国特許商標局および様々な非政府特許機関は、特許出願中にいくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。私たちは評判の良い法律事務所や他の専門家を招いて私たちの遵守を助けてくれました。多くの場合、不注意は滞納金を支払うことによって、あるいは規則を適用する他の方法で是正することができます。しかしながら、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効される可能性があり、それにより、関連法ドメインの特許権の一部または全部が失われる可能性がある。この場合、私たちの競争相手が市場に参入する可能性があり、このような状況は私たちの業務に実質的な悪影響を及ぼすだろう。
米国特許と前米国特許法の変化は、特許の全体的な価値を低下させ、私たちの候補製品を保護する能力を弱める可能性がある。
米国又は他の管轄区域における特許法又は特許法解釈の変化は、特許出願をめぐる起訴及び発行された特許の実行又は弁護の不確実性及びコストを増加させる可能性がある。米国では,最近の特許法の大量修正やUSPTOルールの提案修正は,我々の技術を保護し,知的財産権を実行する能力に大きな影響を与える可能性がある。私たちはあなたに後続の裁決が私たちの特許や特許出願に悪影響を与えないということを保証できません。このような事件の結合は,我々の将来の特許取得能力の不確実性の増加に加えて,いったん特許が付与された価値に関する不確実性をもたらしている.例えば、米国最高裁の審査を受けているにもかかわらず、この事件ではアンはセノフィを訴えたしかし、連邦巡回裁判所は、広範な機能抗体主張は無効であり、使用可能性が乏しいためであると考えている。さらにここではジュノは凧事件を訴えたしかし,連邦巡回裁判所は広範な抗体やキメラ抗原受容体を主張しており,支持する例は少なく,書面記述が乏しいため無効である。米国議会、連邦裁判所、USPTO、および特許保護を求める可能性のある他の国の同様の立法および規制機関の決定によれば、特許を管理する法律および法規は、予測不可能な方法で変化する可能性があり、特に医薬品特許保護において、新しい特許を取得したり、私たちまたは私たちの許可者または協力者の既存の特許を実行したりする能力を弱めることになる。
もし私たちの商標と商号が十分に保護されていなければ、私たちは興味のある市場で知名度を作ることができないかもしれません。私たちの業務は悪影響を受けるかもしれません。
我々の一般法商標または商号は、挑戦、侵害、回避、または汎用商標として発表されるか、または他の商標を侵害すると判断される可能性がある。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれないし、これらの名前の使用を停止させることができないかもしれません。私たちは関心のある市場で潜在的なパートナーや顧客の名前の承認を得るために必要です。もし私たちが私たちの商標と商号に基づいて名称を確立することができなければ、私たちは効果的に競争できないかもしれません。これは私たちの業務、財務状況、運営結果、将来性に重大で不利な影響を与えるかもしれません。
政府の規制に関連するリスク
私たちおよび/または私たちの協力者は、アメリカや外国の規制機関の承認を得ることができないか、遅延する可能性があるので、私たちの候補製品を商業化することができません。
私たちの候補製品は広範な政府法規の制約を受けており、これらの法規は薬品の研究、テスト、開発、製造、承認、記録保存、報告、ラベル、貯蔵、包装、広告と販売促進、定価、承認後の監視、マーケティングと流通に関連する。新薬が発売される前に、アメリカと多くの外国司法管轄区で厳格な臨床前試験と臨床試験、及び広範な監督管理許可手続きを成功に完成する必要がある。これらと他の規制要件を満たすことは高価で、時間がかかり、不確定であり、予期しない遅延が生じる可能性がある。私たちが単独で開発したり、私たちの協力者と協力して開発した候補製品は、私たちまたは私たちの既存または未来の協力者がそれらを販売するために必要な規制承認を得ないかもしれません。
われわれはこれまで臨床試験を行って管理した経験がなく,これらの試験はFDAの承認を含めて規制部門の承認を得る必要がある。FDAおよび他の承認を得るのに要する時間は予測不可能であるが、一般に、候補製品のタイプ、複雑性、および新規性に依存する臨床試験の開始後数年後に必要である。FDAとその外国の同業者が私たちを監督する時に使用する基準は判断する必要があり、しかも変化する可能性があり、これはそれらの応用を確定的に予測することを困難にする。著者らは臨床前と臨床活動データに対するいかなる分析も監督管理機関の確認と解釈を得る必要があり、これは監督部門の承認を延期、制限或いは阻止する可能性がある。私たちまたは私たちの協力者はまた、将来の立法または行政行動、または製品開発、臨床試験、およびFDA規制審査中のFDA政策の変化など、新しい政府法規による予期しない遅延またはコスト増加に遭遇する可能性がある。また、新冠肺炎に関連する感染と死亡は世界のある医療と医療監督システムを乱している。このような干渉は,FDAや類似の外国規制機関から医療資源を分流したり,その審査を実質的に延期したりする可能性がある。このような干渉が発生した場合、これらの干渉がどのくらい続くかは不明である。このような中断による臨床前研究或いは臨床試験のいかなる延期或いは優先順位の取り消し、或いは監督審査の遅延は、著者らの候補製品の開発と研究に重大な影響を与える可能性がある。より多くの立法変化が公布されるかどうか、あるいはFDAや外国の法規、ガイドラインまたは解釈が変わるかどうか、あるいはこれらの変化の影響(あれば)は予測できない。
私たちが開発している候補製品を考慮して、単独開発でも、私たちのパートナーと共同開発しても、新しい治療方法を代表しており、FDAとその外国の同業者はまだこれらの候補製品についていかなる明確な政策、やり方或いはガイドラインを制定していないかもしれない。さらに、FDAは、私たちが提出する可能性のある任意の秘密協定に応答して、私たちが予想していなかった要求を定義するかもしれない。このような反応は私たちの候補製品の臨床開発を延期するかもしれない。また,ある疾患の承認を求める可能性があるため,規制部門の承認を得るためには,これらの疾患を治療するための候補製品(あれば)が安全かつ有効であるだけでなく,既存製品よりも安全または効率的であることを臨床試験により証明する必要があるかもしれない。また、近年、FDAは新薬承認手続きとFDA標準の面で、特に製品安全の面で、ますます大きな公衆と政治的圧力に直面している。
必要な承認を得る上でのどんな遅延や失敗も、私たちが承認を求めている特定の候補製品から収入を創出する能力に重大で不利な影響を与える可能性があります。さらに、どんな販売製品の規制承認も、私たちがその製品を販売する承認用途やラベルまたは他の制限によって制限される可能性があります。
私たちはまた未来に多くの外国の監督管理要求の制約を受け、これらの要求は臨床試験の進行、製造とマーケティング許可、定価と第三者精算を管理している。外国の監督管理審査の流れは国家/地区によって異なり、上述のFDA審査プロセスに関連するすべてのリスクと、外国司法管轄区の現地法規を満たすことによるリスクを含む可能性がある。さらに、承認を得るのに要する時間は、FDA承認を得るのに要する時間とは異なる可能性がある。FDAの承認は米国以外の規制機関の承認を得ることを保証することはできず、その逆も同様である。米国や外国の監督管理機関による候補製品の承認が遅延されたり、得られなかったりすることは、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性がある。
私たちの候補製品が規制機関の承認を得ても、私たちは持続的な規制義務と持続的な規制審査の制約を受けることになり、これは多くの追加費用を招く可能性がある。また、私たちの候補製品が承認されれば、ラベルや他の制限、市場撤退の影響を受ける可能性がある。もし私たちが規制要求を守らなかったり、私たちの製品が予期しない問題に遭遇したら、私たちはまた処罰されるかもしれない。
私たちまたは私たちの既存または未来のパートナーが私たちの候補製品のために獲得した任意の規制承認は、製品上場によって承認された指定用途の制限または承認条件によって制限される可能性があり、または候補製品の安全性および有効性を監視するために、コストの高い上場後のテストおよび監督要件を含む可能性がある。
さらに、FDAまたは同様の外国規制機関が私たちの任意の候補製品を承認する場合、製品の製造過程、ラベル、包装、流通、承認後の監視および不良事象報告、貯蔵、輸入、輸出、広告、販売促進および記録は、広く持続的な規制要件を受けるだろう。FDAは重要な上場後の権力を持ち、新しいセキュリティ情報に基づいてラベルを変更することを要求する権力と、発売後の研究或いは臨床試験に製品使用に関連する安全リスクの評価を要求すること、或いは市場からその製品をリコールすることを要求する権力を含む。FDAはまた、承認された薬物の流通または使用にさらなる要求または制限を加える可能性がある承認後のREMS計画を要求する権利がある。我々は将来の製品を製造するための製造施設であり,あればFDAや他の規制機関の定期審査や検査も受け,引き続きcGMP要求に適合しているかどうかを含む。もし私たちの第三者製造業者、製造プロセス、または施設が任意の新しいまたは以前に未知の問題を発見した場合、製品を市場から引き下げることを含む製品、製造業者、または施設の制限を引き起こす可能性がある。もし私たちが第三者メーカーに依存すれば、私たちはこのような製造業者が適用された規制を遵守することを制御できないだろう。どんな製品の普及と広告もまた規制要求と持続的な規制によって検討されるだろう。FDAはメーカーがその製品を使用する通信に厳しい制限を加えている。FDAによって承認されたラベルと一致しないか、またはFDA規定に適合しない方法で候補製品を宣伝すれば, 私たちは法執行行動の影響を受けるかもしれない。もし私たちまたは私たちの既存または未来のパートナー、製造業者、またはサービスプロバイダが、私たちが製品を販売するアメリカまたは外国の管轄区域に適用される持続的な法規要件を遵守できない場合、私たちまたは彼らは罰金、警告状、臨床試験の一時停止、FDAまたは同様の外国の規制機関が未解決の申請または承認された申請の承認を遅延または拒否する補充、一時停止または監督許可の撤回、製品のリコールと差し押さえ、製品の行政拘留、製品の輸出入の許可、経営制限、禁止、禁止を拒否する可能性がある。民事処罰と刑事起訴。
その後、予想されていない深刻度または頻度の有害事象、または当社の第三者製造業者または製造プロセス、または規制要件を遵守できなかったことを含む製品に以前に未知の問題が存在することが発見され、以下のことが含まれる可能性がある
•製品の販売または製造を制限し、製品を市場からリコールまたは自発的または強制的にリコールすること
•臨床試験に罰金、警告、または無見出し手紙または一時停止を科す
•FDAは、私たちまたは私たちの戦略的パートナーが提出した係属中の出願または承認された出願を承認することを拒否する
•製品の許可承認の一時停止または取り消し;
•輸入または輸出の許可を差し押さえ、差し押さえたり、または拒否したり;
•民事または刑事処罰を禁令または適用する。
FDAの政策は変わる可能性があり、政府は私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加法規を公布するかもしれない。例えば2016年12月21日ST世紀治療法案、あるいは治療法案、署名が法律となる。その他の事項以外に、“治療法案”は薬品と生物製品の監督管理を現代化し、革新を刺激することを目的としている。私たちが既存の要求の変化に緩やかに適応できない場合や、新しい要求や政策を採用することができない場合、または規制適合性を維持できない場合、私たちが得る可能性のあるマーケティング承認を失う可能性があり、収益性を実現または維持できない可能性があり、これは私たちの業務に悪影響を及ぼすだろう。
米国や海外の将来の立法や行政や行政行動によって生じる可能性のある政府規制の可能性、性質、程度も予測できない。FDA人員配置の変化は、FDAの応答遅延、またはその審査提出または申請、発表法規または指導、タイムリーまたは規制要件を実質的に実施または実行できない能力に遅延をもたらす可能性がある。もし連邦政府が再び大幅に停止すれば、2018年12月22日に発生したような同様の結果も生じるだろう
2019年1月25日まで。これらの要求がどのように実施されるか、およびそれらがFDAがその規制権力を行使する能力にどの程度影響するかを予測することは困難である。いかなる立法、行政命令、あるいは機関の資金ミスがFDAが正常な過程で監督と活動に参加する能力に制限を与える場合、私たちの業務はマイナスの影響を受ける可能性がある。
私たちは医療立法改革措置の困難に直面するかもしれない。
既存の規制政策は変わる可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。米国や海外の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが既存の要求の変化に緩やかに適応できない場合、あるいは新しい要求や政策を採用することができない場合、または規制適合性を維持できない場合、私たちは私たちが得る可能性のあるマーケティング承認を失う可能性があり、利益を達成したり維持することができないかもしれない。
アメリカでは、医療費用を統制するための多くの立法計画が続いているだろう。例えば、2010年3月には、“医療·教育和解法案”により改正された“患者保護·平価医療法案”、または“医療保険·平価医療法案”が公布され、医療保険を獲得する機会を拡大し、医療支出の増加を減少または制限し、詐欺や乱用に対する救済措置を強化し、医療·医療保険業界の新たな透明性要求を増加させ、医療業界に新たな税費を徴収し、追加の医療政策を実施することを目的としている。
トランプ政権中に取られた措置を含むACAのすべてまたはいくつかの条項を修正、廃止、または他の方法で無効にするための立法および司法努力が行われた。他にも、税法には、2019年1月1日からACAが1年の全部または一部の期間に合格医療保険を維持できなかった個人に対して実施された税収ベースの分担責任支払いを廃止する条項が含まれており、これは一般に“個人強制”と呼ばれる。2021年6月17日、米国最高裁は、ACAは全体的に違憲であり、“個人権限”が国会で廃止されたため、プログラム理由に基づく挑戦を却下した。したがって、ACAは現在の形態で継続的に有効であるだろう。ACAは未来に司法や国会で挑戦される可能性がある。ACAまたはその実施条例またはその内容の一部を廃止、置換、修正または廃止するためのいかなる努力、およびバイデン政府の医療改革措置がACAまたは我々の業務にどのように影響するかは不明である。また,2020年1月1日から永久廃止された2020年連邦支出案では,ACAが規定しているある高コスト雇用主が後援する保険計画に徴収される“キャデラック”税や,非免除医療機器に対する医療機器消費税,2021年1月1日からも健康保険会社税が廃止されている。また,2018年に両党予算法案(BBA)などがACAを改正し,2019年1月1日から施行され,連邦医療保険D部分に参加する製薬メーカーが不足している販売時点割引を50%から70%に引き上げ,多くの連邦医療保険薬物計画のカバーギャップを縮小し,通常は“ドーナツ穴”と呼ばれている。また、CMSは各州により大きな柔軟性を与え、2020年1月1日から発効する最終規則を発表した, これは,個人や小団体市場の保険会社に基準を設定する上で,ACAがこのような市場で販売されている保険計画に要求される基本健康福祉を緩和する可能性がある。
また,ACAが公布されて以来,米国は医療支出を削減するために他の立法改正を提案し,採択した。2011年8月2日、それ以外にも、2011年予算統制法案は国会支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これには、2013年4月1日に施行された各年度に提供者に支払われる医療保険総額が2%減少することが含まれており、その後の法規制の立法改正により、BBAやコロナウイルス援助、救済、経済安全法案、またはCARE法案が含まれ、2031年まで有効となる。また、2012年の“米国納税者救済法”は、病院、画像センター、癌治療センターを含むいくつかの医療サービス提供者への医療保険支払いをさらに減少させ、政府が提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長した。連邦支出がさらに減少すれば、予想される予算不足は、FDAや国家衛生研究院が現在のレベルで運営し続ける能力のような関連機関にも影響を与える可能性がある。連邦贈与と契約に割り当てられた金額は減少またはキャンセルされる可能性がある。これらの削減は、関連機関が研究開発、製造、マーケティング活動を適時に審査·承認する能力にも影響を与える可能性があり、私たちが開発、マーケティング、販売する可能性のある任意の製品を開発、販売する能力を遅らせる可能性があります。
また、支払い方法は医療立法と規制措置の影響を受ける可能性がある。例えば、2003年の“連邦医療保険処方薬、改善と現代化法案”(MMA)は連邦医療保険カバーと薬品の支払い方式を変えた。この立法は高齢者が薬品を購入する医療保険のカバー範囲を拡大し、医師が管理する薬品の平均販売価格に基づく新しい精算方法を導入した。しかも、この法案は薬品の数量を制限する権限を規定する
心理治療の授業です。MMAは連邦医療保険受益者の薬品福祉にのみ適用されるが,個人支払者は自分の精算料率を設定する際に連邦医療保険カバー政策や支払い制限に従うことが多い。したがって、MMAによるいかなる償還減少も、個人支払者支払いのような減少をもたらす可能性がある。
最近、政府はメーカーがその販売する製品に価格を設定する方式をより厳格に審査し、これはいくつかの大統領行政命令、国会調査、提案と公布された連邦と州立法を招き、これらの立法は製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険(Medicare)下の薬品コストを下げ、政府計画の薬品の精算方法を改革することを目的としている
また、2020年11月、HHSは薬品メーカーの値下げに対する安全港保護を廃止し、直接或いは薬局福祉マネージャーを通じて、値下げした避風港保護をD部分下の計画スポンサーに移し、法律が値下げを要求しない限り、法規を決定した。この規定はまた、販売時点での値下げを反映するための新しい安全港を創出し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための安全港を作成する。この最終規則の実施はバイデン政府によって2023年1月1日に延期され、その後インフレ削減法案(IRA)によって2032年1月1日に延期された。 2020年12月、CMSは医療補助薬品返却計画に基づいて、薬局福祉マネージャーアキュムレータ計画の影響を受けるメーカー支援計画の規定、およびいくつかの価値に基づく調達手配に関する最適価格報告を含む重大なメーカー価格報告変化を実施した最終規則を発表した。2021年1月1日に施行される“2021年米国救援計画法案”によると、メーカーが州医療補助計画に支払う医療補助薬品還付計画還付の法定上限が撤廃される。この上限を廃止することは、販売製品よりも多くのリベートを支払うように製薬業者に要求するかもしれない。これらの新しい要求がどの程度実施されるか、およびこれらの法規やバイデン政府の将来の任意の立法または法規が、私たちの収入の創出と利益を達成する能力を含む、私たちの業務にどの程度影響を与えるかは不明である
最近,医療改革の取り組みは最終的に2022年8月に“アイルランド共和法”が公布され,CMSがMedicare B部分とD部分で精算されたいくつかの薬物や生物製品の販売価格についてHHSが交渉することが許可されるが,CMSは少なくとも7年(生物製品は11年)の高支出単一由来薬物しか承認されていないが,交渉価格は選択年度後2年で発効する。交渉価格は2026年に初めて発効し、法定最高価格を上限とする。2023年1月から連邦医療保険B部とD部はそれぞれ2023年1月と2022年10月から開始され,IRAはインフレ率よりも高い速度で連邦医療保険B部とD部分の薬品価格を向上させた製薬業者を処罰する。アイルランド共和軍は衛生·公衆サービス部長官が最初の数年に規制ではなく指導によってその多くの規定を実施することを許可した。IRAを守らないメーカーは民事罰金を含めて様々な処罰を受ける可能性がある。アイルランド共和軍はまた、2025年まで“平価医療法案”市場で医療保険を購入した個人に強化補助金を提供する。このような規定は法的挑戦を受ける可能性があるにもかかわらず、2023年から段階的に施行されるだろう。アイルランド共和軍のすべての経済的影響はまだ明らかにされていないが、この法律の採択は私たちの製品と候補製品の価格に影響を及ぼす可能性がある。新しい司法管轄区で制限的な価格規制を採用し、既存の司法管轄区でより厳格な規制を実施したり、適時或いは十分な価格設定を取得したり維持できなかったりすることも、収入に不利な影響を与える可能性がある。私たちは価格設定圧力が世界的に持続されると予想する。
州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入およびマーケティングコスト開示の制限、および透明性措置を含む、価格または患者の精算制限、割引、特定の製品参入およびマーケティングコスト開示の制限および透明性措置を含む医薬品および生物製品の定価を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することが目的である。私たちは将来的により多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品およびサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品やセット診断に対する需要の減少または追加の価格設定圧力を招く可能性がある。
ACAとIRA,および将来とりうる他の医療改革措置は,より厳しいカバー基準を招き,我々が受け取った任意の承認された製品の価格に追加の下り圧力をもたらす可能性が予想される。コスト抑制措置や他の医療改革を実施することは、私たちの収入の創出、利益の実現、あるいは私たちの製品の商業化を阻止するかもしれない。
医療提供者、医療機関、顧客および第三者支払者との業務および関係は、適用される反賄賂、リベート、詐欺および乱用、透明性および他の医療およびプライバシーの法律法規の制約を受けることになり、これは、法執行行動、刑事制裁、民事処罰、契約損害、名声損害、行政負担、利益および将来の収入減少などの問題に直面する可能性があります。
私たちの現在と未来の医療提供者、医療機関、第三者支払者、顧客との手配は、私たちの製品の研究、マーケティング、販売、流通の業務または財務配置と関係を制限する可能性がある広範な反賄賂、詐欺、乱用、および他の医療保健法律と法規に直面させています
候補者です。また,米国連邦政府および我々が業務を展開している国の州や外国政府による患者データのプライバシーや安全の規制を受ける可能性がある。適用される連邦と州の反賄賂と医療に関する法律法規による制限は、
•連邦反リベート法規は、他の事項に加えて、個人および実体が任意の商品またはサービスを誘導または奨励するために、現金または実物形態で直接または間接的に報酬を請求、提供、受け入れ、または提供することを禁止し、個人の推薦または購入、注文または推薦を奨励し、これらの商品またはサービスは、連邦および州医療保健計画(例えば、連邦医療保険および医療補助)に従って支払うことができる。個人またはエンティティは、法規または法規違反の具体的な意図を実際に知る必要がなく、違反を実施することができる
•連邦刑事および民事虚偽クレームおよび民事罰金法律であって、民事告発者または個人または実体に対する訴訟によって強制的に執行することができ、他の事項に加えて、虚偽または詐欺的な支払いクレームの提出または提出を意図的に禁止し、虚偽または詐欺的クレームに関連する虚偽記録または報告書を故意に作成、使用または使用させること、または連邦政府への支払いの義務を回避、減少または隠蔽するための虚偽陳述を意図的に行うことを禁止する連邦刑事および民事虚偽クレームおよび民事罰金法律。さらに、ラベル外販売促進を含むいくつかのマーケティング行為は、虚偽クレーム法律に違反する可能性もある。また、連邦“虚偽申告法”によると、連邦“反リベート条例”違反による物品やサービスのクレームが虚偽または詐欺的クレームを構成していると断言できる
•HIPAAは、刑事および民事責任を規定し、他の事項に加えて、任意の医療福祉計画を詐欺の任意の医療福祉計画を知りながら実行または意図的に実行または実行しようとすることを禁止し、または医療福祉、プロジェクトまたはサービスの提供または支払いに関連する重大な事実を知り、または故意に隠蔽または隠蔽し、または任意の重大な虚偽陳述を行うことを禁止する;連邦反リベート法規と同様に、個人またはエンティティは、法規または法規違反の具体的な意図を実際に理解する必要がなく、違反行為を実施することができる
•HITECHによって改正されたHIPAAおよびそのそれぞれの実施条例は、2013年1月25日に発表された最終総合規則を含み、特定のヘルスケア提供者、健康計画およびヘルスケア交換所(カバーエンティティと呼ぶ)およびその事業パートナーが、個人が識別可能な健康情報のプライバシー、セキュリティおよび伝送を保護する上で、個人が識別可能な健康情報を記憶、使用または開示することに関するいくつかのサービス(強制契約条項を含む)の義務を規定し、個人が健康情報の安全を識別できる特定の違反に違反した場合に影響を受けた個人および規制当局を通知することを要求する。連邦立法は、ACAおよびその実施条例の一部として一般的に呼ばれ、ACAおよびその実施条例の一部として、Medicare、Medicaidまたは児童健康保険計画に従って精算可能な保険薬品、設備、バイオ製品および医療用品のいくつかの製造業者に、医師(医師、歯科医師、検眼師、足科医師および脊椎マッサージ師を含むと定義される)および教育病院のいくつかの支払いおよび他の価値移転に関する情報、および上述の医師およびその直系親族が所有する所有権および投資権益を毎年CMSに報告し、検索可能なサイト上でこれらの情報を開示することを要求する
•カリフォルニア州、バージニア州、コロラド州、コネチカット州、ユタ州のような州プライバシー法律および法規、これらの法律および法規は、健康情報およびHIPAAに制約されない他の敏感な個人情報の使用および開示に制限的な要件を加えている。例えば、2018年6月、カリフォルニア州は2020年1月1日に施行されたカリフォルニア消費者プライバシー法と、CCPAを改正し、2022年1月1日から追加義務を設定するカリフォルニアプライバシー権法案を公布し、カリフォルニア住民に個人情報のアクセスと削除の権利を付与し、敏感な個人情報の処理を制限し、特定の個人情報共有から退出し、その個人情報がどのように使用されるかに関する詳細な情報を取得し、違反行為に対する民事処罰を規定し、データ漏洩訴訟の個人訴訟権を増加させることを規定し、コンプライアンスコストと潜在的責任の増加を招く。バージニア州の消費者データ保護法案は、消費者の心身健康診断を開示する情報と、消費者を識別することができる遺伝子および生体識別情報を含む消費者の選択加入同意を得ることを要求する2023年1月1日に施行された
•1977年に改正された米国の“海外腐敗防止法”は、他の事項に加えて、米国企業およびその従業員および代理人が、外国政府関係者、国際公共組織および外国政府の所有または付属実体の従業員、外国政治職候補者、外国政党または官僚に腐敗または不正な支払いまたは任意の他の価値のあるものを提供、または間接的に提供することを禁止している
•同様の州および外国の法律、例えば、州反リベートおよび虚偽請求法は、民間保険会社によって精算される保健項目またはサービスを含む非政府第三者支払者に関する販売またはマーケティング手配およびクレームに適用可能である
•いくつかの州の法律は製薬会社が製薬業界の自発的なコンプライアンス基準と連邦政府が公布した関連するコンプライアンスガイドラインを遵守することを要求し、そのほか、薬品メーカーに医師と他の医療保健提供者への支払い金或いはマーケティング支出と薬品定価情報に関する情報を報告することを要求し、州と地方の法律は薬品販売代表の登録を要求し、そしてある場合の健康情報のプライバシーと安全を管理する州法律であり、その中の多くの法律は互いに大きく異なり、しかもよくHIPAAによって先制されず、それによってコンプライアンス仕事を複雑化させる。
もし私たちまたは私たちの協力者、製造業者、またはサービスプロバイダが適用された連邦、州、または外国の法律または法規に従わなかった場合、私たちは法執行行動の影響を受ける可能性があり、これは私たちの製品の開発、マーケティング、販売に成功する能力に影響を与え、私たちの名声を損なう可能性があり、私たちの製品に対する市場の受容度を低下させる可能性がある。これらの法執行活動には
•政府が援助している医療計画から除外されました
•私たちの製品に付与された政府契約の資格は排除された。
プライバシー法、規則、法規はしばしば変化し、それらの範囲は新しい立法、既存の立法の改正と法執行の変化に従って絶えず変化する可能性があり、異なる司法管轄区域間で一致しない可能性がある。アメリカ、欧州連合と他のところでは、消費者、健康とデータ保護法の解釈と適用、特に遺伝子サンプルとデータの解釈と適用については、しばしば不確定、相互矛盾と絶えず変化している。したがって、予測可能な未来には、基準と法執行慣行を実施することはまだ確定していない可能性があり、私たちはこれらの未来の法律、法規、基準が私たちの業務にどのような影響を与える可能性があるかを決定することができない。私たちは、現在または未来の法律が、私たちが個人データを生成または維持することを阻止しないこと、または患者が(必要に応じて)彼らの個人データを使用することに同意することを保証することはできない;これらの場合のいずれも、必要な研究開発、製造、および商業化を阻止または発表することができ、これは、私たちの業務、運営結果、財務状況、および見通しに実質的な悪影響を及ぼす可能性がある。
我々の現在と将来の第三者の業務配置が適用される医療法律や法規に適合していることを確保するために努力しており、巨額のコストがかかる可能性がある。政府当局は、私たちの業務やり方が現在または未来に適合していないと結論するかもしれないが、詐欺および乱用または他の医療保健法律および法規の適用に関する現行または将来の法規、機関指導または判例法に関連している。もし私たちの業務がこのような要求に違反していることが発見された場合、私たちは、民事、刑事および行政処罰、損害賠償、罰金、返還、監禁、削減または再構成、私たちの業務の削減、FDAの承認を得る資格を失ったこと、政府契約への参加、医療清算または他の政府計画(MedicareおよびMedicaidを含む)、誠実な監督および報告義務、または名声損害を含む重大な処罰を受ける可能性があり、これらはいずれも私たちの財務業績に悪影響を及ぼす可能性がある
このような危険が完全に除去されることは不可能だ。私たちの不正の疑いや疑いのある行動は、私たちの巨額の法的費用を招く可能性があり、私たちの弁護が成功しても、私たちの経営陣の業務運営への関心を移す可能性があります。さらに、お金、時間、そして資源の面で、適用された法律と法規を達成し、継続的に遵守することは私たちにとって高価かもしれない。
私たちがどんな候補製品を商業化することができても、これらの候補製品は不利な価格設定法規や第三者保険·清算政策の制約を受ける可能性があり、これは私たちの業務を損なうだろう。
新薬を管理する監督管理の審査、定価と精算の規定は国によって異なる。一部の国は薬品の販売価格が承認されてから発売されることを要求している。多くの国で、定価審査期間は発売が承認された後から始まる。一部の国外市場では、初歩的な承認を得た後であっても、処方薬の生物薬品の定価は依然として政府の持続的な制御を受けている。したがって、特定の国で製品の規制承認を受ける可能性がありますが、その後、価格規制の制約を受け、これらの規制は私たちの製品の商業発表を延期し、長い間遅延し、その製品をその国/地域で販売することによって生じる収入にマイナスの影響を与える可能性があります。不利な価格設定制限は、私たちの候補製品が規制部門の承認を得ても、1つ以上の候補製品への投資を回収する能力を阻害する可能性がある。
私たちが任意の製品を商業化することに成功した能力はまた、MedicareおよびMedicaid、個人健康保険会社および他の組織などの政府当局を含む、これらの製品および関連治療の保証範囲および十分な補償が第三者支払者からどの程度得られるかにある程度依存するであろう。患者に医療サービスを提供する患者は一般的に第三者支払人によって全部または一部の費用を精算する
彼らの治療と関係がある。保証範囲と第三者支払者の十分な精算は新製品の受け入れ度に重要である。たとえ私たちが1つ以上の製品を市場に出すことに成功しても、これらの製品は費用効果があると思われない可能性があり、どの製品の精算金額も競争に基づいて私たちの製品を販売させるのに十分ではないかもしれません。我々の計画は開発の初期段階にあるため,現在のところそのコスト効果を決定することもできず,カバーや精算の可能なレベルや方法を決定することもできない.患者や医療保健提供者(政府や個人保険計画のような)に精算する第三者支払者は、製薬会社に価格に基づいて所定の割引を提供することを要求し、バイオ製薬製品の課金や精算金額の低減を求めている。私たちが開発した任意の製品のための価格、あるいはこれらの製品に提供される保険と補償ができれば、私たちの開発や他のコストに比べて不十分であり、私たちの投資リターンは悪影響を受ける可能性があります。
新たに承認された薬物の精算には大きな遅延がある可能性があり,カバー範囲はFDAや同様の外国規制機関がこの薬剤を承認する目的よりも限られている可能性がある。さらに、精算を受ける資格があるということは、任意の薬物または治療用生物製剤がすべての場合に精算されることを意味するわけではなく、または精算の費用率は、研究、開発、製造、販売、および流通を含む私たちのコストを補うのに十分である。
適用すれば、新薬の一時精算レベルも私たちのコストを支払うのに十分ではないかもしれませんし、恒久的にはならないかもしれません。販売率は、精算されたより低いコストの薬物によって許容される支払いに基づく可能性があり、他のサービスの既存の支払いに統合される可能性があり、予算制限または連邦医療保険データの欠陥を反映する可能性がある。医薬品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、および将来、米国価格よりも低い価格で販売される可能性のある国から医薬品を輸入することを制限する法律の緩和によって低下する可能性がある。また、米国には統一された保険や精算政策がなく、支払者によって引受や精算が大きく異なる可能性がある。したがって、第三者支払人から製品の保証と精算承認を得ることは時間がかかり、高価な過程であり、これは各支払人に科学的、臨床的、費用効果的なデータを提供し、個々の支払人に基づいて私たちの製品を使用するために必要かもしれないが、保証され、十分な精算を得ることは保証されないかもしれない。新たに承認された製品の保険カバーと精算に関する不確実性が大きい。第三者支払者は,自分の精算料率を設定する際には通常連邦医療保険カバー政策や支払い制限に依存するが,連邦医療保険確定に加えて,独自の方法や承認プロセスもある。私たちが開発し、規制承認を受けた新薬については、政府援助や個人支払者から保険と十分な販売率を迅速に得ることができず、これは私たちの業務、財務状況、運営結果、見通しに実質的な悪影響を及ぼす可能性がある。
FDAの迅速なチャネル指定を求めることにした場合、より速い開発や規制審査や承認プロセスを招くことはないかもしれません。
私たちは私たちの1つ以上の候補製品のための迅速なチャネル認証を求めるかもしれない。重症または生命に危険な疾患の治療に使用され、そのような疾患が満たされていない医療需要を解決する可能性を示す薬剤がある場合、製品スポンサーはFDA迅速チャネル認証を申請することができる。FDAは幅広い裁量権を有しており,この称号が付与されているかどうか,したがって,ある特定の候補製品がこの称号を得る資格があると考えても,FDAがそれを付与することを決定することを保証することはできない。私たちが高速チャネル認証を受けたとしても、私たちは従来のFDAプログラムよりも速い開発過程、審査、または承認を経験しないかもしれない。FDAが我々の臨床開発プロジェクトのデータが高速チャネルの指定をサポートしなくなったと考える場合,その指定を撤回する可能性がある。
もし私たちが私たちのいくつかの候補製品のために孤児の薬物指定を求めることを決定すれば、私たちは成功しないかもしれないし、孤児の薬物指定に関連する利点を維持できないかもしれない。
私たちの業務戦略の一部として、私たちは私たちの1つ以上の候補製品のために孤児薬物認証を求めるかもしれませんが、私たちは成功しないかもしれません。米国やヨーロッパを含むいくつかの管轄区の規制機関は、患者数が比較的少ない薬物を孤児薬に指定する可能性がある。孤児医薬品法によれば、1つの薬剤がまれな疾患または疾患を治療するための薬剤である場合、FDAは孤児薬として指定することができる。米国では、まれな疾患または疾患の定義は、通常、患者数が20万人未満であること、または米国では患者数が20万人を超えることを意味するが、米国では、米国の販売から薬剤開発コストを回収することができる合理的な期待はない。米国では,孤児薬物指定は,税収割引やユーザ料金減免などの経済的インセンティブを当事者に与える権利がある。さらに、孤児薬物指定を有する製品がその後、このような指定された疾患を有するFDAの最初の承認を得た場合、この製品は、孤児薬物排他性を得る権利があり、これは、FDAが7年以内に任意の他の出願を承認することができず、限られた場合、例えば孤児薬物に対して排他的な製品に対する臨床的利点を示すか、または製造業者が十分な製品数を保証できない限り、同じ適応で同じ製品を販売することを意味する。
特定の適応の中で我々の候補製品のために孤児薬物指定を獲得しても,薬品開発に関する不確実性により,これらの孤児指定適応候補製品の上場承認を得た最初の会社ではない可能性がある。さらに、孤児によって指定された適応よりも広い適応の承認を求める場合、またはFDAが指定要求に重大な欠陥があると後に決定した場合、または製造業者がまれな疾患または疾患を有する患者の需要を満たすのに十分な数の製品を保証できない場合、米国での独占営業権が制限される可能性がある。また,異なる活性部分を有する異なる薬剤が同じ条件で許可されるため,製品の孤児薬物排他性を獲得しても,この排他性は競合から製品を効率的に保護できない可能性がある。孤児製品が承認された後であっても、FDAが後の薬剤がより安全で、より効率的で、または患者ケアに大きな貢献があると結論した場合、FDAはその後、同じ疾患に対して同じ活性部分を有する同じ薬剤を承認することができる。孤児薬物指定は薬物の開発時間や監督審査時間を短縮することもなく、監督審査或いは承認過程において薬物にいかなる利点をもたらすこともない。しかも、私たちは私たちの候補製品のために孤児薬物の称号を求めるかもしれないが、私たちは決してそのような称号を受けないかもしれない。
2017年12月22日に法律となる税改正立法に署名し、スポンサーが相殺と主張する可能性のある指定孤児製品の合格臨床研究費の金額を50%から25%に引き下げた。これは優位性をさらに制限し、将来的に孤児薬の称号を求める私たちのビジネス戦略に影響を与えるかもしれない。
私たちはアメリカとある外国の輸出入規制、制裁、禁輸、反腐敗法、反マネーロンダリング法律法規の制約を受けている。このような法律基準を遵守することは国内と国際市場での私たちの競争能力を弱めるかもしれない。私たちは違反によって刑事責任と他の深刻な結果に直面するかもしれないし、これは私たちの業務を損なうかもしれない。
我々は、米国輸出管理条例、米国税関条例、米国財務省外国資産規制弁公室によって実施された様々な経済·貿易制裁条例、1977年に改正された米国反海外腐敗法またはFCPA、米国連邦法典第18編201節に含まれる米国国内賄賂法規、米国旅行法、米国愛国者法、および私たちが活動している国の他の州と国の反賄賂および反マネーロンダリング法を含む輸出規制と輸入法律および法規の制約を受けている。腐敗防止法は広く解釈され、会社およびその従業員、代理人、請負業者、および他の協力者の許可、約束、提供、直接的または間接的に公共または民間部門の受給者に不当なお金または任意の他の価値のあるものを支払うことを禁止する。私たちは第三者を招いて私たちの製品を販売し、アメリカ国外で私たちの製品を販売し、臨床試験を行い、および/または必要な許可、許可証、特許登録、その他の規制承認を得ることができる。私たちは政府機関や政府付属病院、大学、その他の組織の役人や従業員と直接または間接的な相互作用を持っている。私たちは、私たちが明確に権限を持っていなくても、または実際にこれらの活動を理解していなくても、従業員、代理、請負業者、および他の協力者の腐敗や他の不法活動に責任を負わなければならないかもしれない。上記の法律および法規に違反するいかなる行為も、重大な民事および刑事罰金および処罰、監禁、輸出入特権の喪失、資格取り消し、税収の再評価、契約違反および詐欺訴訟、名誉損害、およびその他の結果を招く可能性がある。
米国以外の政府は厳しい価格制御を実施する傾向があり、これがあれば私たちの収入に悪影響を及ぼす可能性がある。
いくつかの国、特にEU加盟国では、処方薬の価格設定は政府によって統制されている。これらの国では、製品の発売許可を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。また、費用抑制措置の一部として、各国政府や他の利害関係者は価格や補償レベルにかなりの圧力をかける可能性がある。政治、経済、規制面の事態は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉が継続される可能性がある。EUの各加盟国が採用している参考定価と平行分配、あるいは低価格と高価な加盟国の間で裁定を行うことで、価格をさらに下げることができる。いくつかの国で保険範囲および精算または価格設定承認を得るために、私たちまたは現在または未来の協力者は、臨床試験または他の研究を行うことを要求される可能性があり、私たちの候補療法の費用対効果を他の利用可能な治療法と比較して、精算または価格設定承認を得るか維持することができる。第三者支払者や主管当局が割引を公表することは、公布国や他の国の価格や補償レベルにさらなる圧力を与える可能性がある。マーケティングを許可された候補製品が入手できない場合、または範囲または金額に制限されている場合、または定価が満足できないレベルに設定されている場合、私たちの業務、財務状況、運営結果、または見通しは実質的な悪影響を受ける可能性がある。また、イギリスの離脱により、イギリスとEUの間には2020年末までに包括的な貿易協定が交渉されるまでの過渡期がある
ヨーロッパのデータ収集は,個人情報の使用,処理,国境を越えた移行に関する限定的な規定によって制約されている.
EUでは,個人健康データの収集と使用は一般データ保護条例(GDPR)によって規制されている。GDPRは,個人データに関する個人の同意,個人への情報提供,国家データ保護主管部門へのデータ処理義務の通知,個人データのセキュリティと秘密についていくつかの要求を行っている。GDPRはまた,個人データをEUから米国に移行するための厳しいルールを実施している。GDPRは欧州経済区にある臨床試験地点から米国や他の司法管轄区への個人データの審査を強化しており,欧州委員会はこれらの司法管轄区域には“十分な”データ保護法がないとしている。例えば、EU裁判所の2015年10月の判決によると、米国安全港計画のメンバーと認証された米国会社への個人データの移転は無効と発表された。2016年7月、欧州委員会は米国の安全港計画に代わるEU-米国プライバシー盾枠組み、すなわちプライバシー盾枠組みを採択した。2020年7月16日、EU裁判所は、プライバシー遮蔽枠組みの無効を宣言し、標準契約条項を実施する会社に追加のコンプライアンス義務を負担させ、個人データをヨーロッパ以外に移す有効な基礎を確保する判決を発表した。2020年11月10日, 欧州データ保護委員会は標準契約条項の発効に必要な追加保障措置について提案した。個人データをEUからアメリカに移す能力が制限される可能性がある。私たちと他の多くの会社は、EUから米国および他の第三者国への個人データの送信と受信の合法的な手段を達成し、維持するために、追加の措置を取ることを要求されるかもしれない。データ保護命令、GDPR、およびEU加盟国の関連国データ保護法の要件を遵守しないことは、罰金(例えば、最高罰金2000万ユーロまたは前期(高い者を基準とする)の世界年商の4%)および他の行政処罰をもたらす可能性がある。GDPR条例は、私たちが処理している個人データに追加の責任と責任を課す可能性があり、新しいデータ保護ルールを遵守することを保証するために追加のメカニズムを確立することを要求されるかもしれません。これは激務であり、私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性がある。GDPRの実施により、新しいデータ保護ルールの遵守を保証するために追加のメカニズムを確立する必要があるかもしれません。データ保護当局がGDPRの強制遵守を求める方式に関する重大な不確実性は不明である。たとえば,当局がEUで業務を行っている会社をランダムに監査するかどうか,あるいは当局がその権利が侵害されていると主張する個人の訴えを待つかどうかは不明である.法執行の不確実性とGDPRコンプライアンスの確保に関連するコストは重く、私たちの業務、財務状況に悪影響を与えます, 経営成果と将来展望。また、英国の離脱は英国のデータ保護規制に不確実性をもたらした。英国はGDPRを直接実行するための2018年データ保護法を公布した。
私たちの普通株式所有権に関連するリスク
私たちの四半期の経営業績は大幅に変動する可能性があり、あるいは投資家や証券アナリストの予想を下回る可能性があり、すべての状況は私たちの株価の変動や下落を招く可能性がある。
私たちの経営業績は四半期変動の影響を受けると予想されています。私たちの純損失と他の経営業績は様々な要素の影響を受けます
•私たちのMINTプラットフォーム、候補製品、または将来の開発計画の持続的な開発に関する費用レベルの変化
•前臨床試験および臨床試験の結果、または既存または将来の協力者または許可パートナーによって、既存または将来の臨床試験または資金支援を増加または終了すること;
•私たちは、任意の追加の協力、許可、または同様のスケジュールを実行し、既存または将来のスケジュールに従って支払いまたは支払いを受信する時間を手配するか、またはそのような任意の既存または将来のスケジュールを終了または修正することができます
•私たちが巻き込まれる可能性のある知的財産権侵害訴訟や異議、妨害、または撤回手続き
•キーパーソンの増減
•買収、剥離、剥離、合弁企業、戦略投資、または業務戦略の変化など、私たちのパートナーまたは私たちの競争相手の戦略決定
•もし私たちの候補製品が規制部門の承認を得たら、その承認された条項と市場の候補製品に対する受け入れと需要
•私たちの候補製品や競争相手の規制発展に影響を与える;
•一般市場と経済状況の変化は、新冠肺炎などの全世界流行病、ウクライナの持続的な衝突、インフレ、金利上昇と持続的な労働力不足を含む。
もし私たちの四半期の経営業績が投資家や証券アナリストの予想を下回れば、私たちの普通株の価格は大幅に低下する可能性がある。また、私たちの経営業績のどの四半期の変動も、私たちの普通株の価格を大幅に変動させる可能性があります。私たちの財務業績を四半期比較することは必ずしも意味があるわけではなく、私たちの将来の業績としての指標に依存すべきではないと考えられます。
私たちの株の市場価格は変動するかもしれません。あなたはあなたの投資の全部または一部を失うかもしれません。
私たちの普通株の取引価格は様々な要素によって高度に変動し、広範な変動の影響を受ける可能性があり、その中のいくつかの要素はコントロールできません。新冠肺炎の流行などにより、我々の普通株と他の生物製薬会社の普通株の取引価格は非常に不安定になってきた。私たちの普通株の市場価格は、本節と本報告の他の部分、および以下に説明する他のリスクを含む多くの要素の影響を受ける可能性がある
•私たちの候補製品、競争相手、私たちの既存または未来のパートナーの臨床前研究、および臨床試験の結果
•アメリカや他の国の法規や法律の発展、特に私たちの候補製品に適用される法律または法規の変化
•競争力のある製品や技術の成功
•私たち、私たちの未来の商業化パートナーまたは私たちの競争相手の新製品の紹介と公告、およびこれらの紹介または公告の時間;
•規制当局が私たちの製品、臨床研究、製造技術、または販売とマーケティング条項に対する行動
•私たちの財務業績または私たちに似ていると思われる会社の財務業績の実際または予想変化
•私たちは他の技術、製品、または候補製品の努力が成功したかどうかを獲得または許可します
•開発および商業化パートナーとの協力を含むが、これらに限定されない未来の協力の発展について
•製薬とバイオテクノロジー部門の市場状況
•私たちまたは私たちの競争相手は、重大な買収、戦略協力、合弁企業、または資本約束を発表します
•特許、訴訟事項、および私たちの候補製品および製品のための特許保護を得る能力を含む、特許または他の所有権に関連する発展または紛争;
•私たちが追加資本を調達する能力や能力と資本調達の条件は
•キーパーソンの採用や退職
•医療支払い制度の構造を変え
•収益推定の実際または予想変化または株式市場アナリストの私たちの普通株、他の比較可能な会社または私たちの業界全体に対する提案の変化
•私たちまたは私たちの競争相手が私たちまたは私たちの競争相手が市場のアナリストに与える予測や導きに到達できなかった
•投資家は会社の評価の変動は私たちと互角だと思っています
•資金調達の努力を発表し期待しています
•ジャーナリズムや投資界の投機行為
•普通株の株価と取引量の変動は、私たちの取引流動性と公衆流通株に影響を与える可能性がある
•私たち、内部者、または私たちの株主は私たちの普通株を売却します
•私たちの普通株の集中所有権
•会計原則の変化
•権利株主や他の人が提起した訴訟
•ウクライナで続く紛争のようなテロ行為、戦争行為、または広範な内乱期
•新冠肺炎などの世界的な流行病を含む自然災害やその他の災害
•全体的な経済、業界と市場状況は、インフレ、金利上昇、持続的な労働力不足を含む。
また、株式市場全体、特に製薬、生物製薬と生物技術株式市場は、新冠肺炎の大流行、インフレ激化、金利上昇、グローバルサプライチェーンの中断、ウクライナの持続的な衝突及びそれによって実施された世界的な制裁を含む極端な価格と出来高の変動を経験し、これらは往々にして発行者の経営業績と関係がない或いは比例しない。これらの広範な市場と業界要素は、私たちの実際の経営業績にかかわらず、私たちの普通株の市場価格を深刻に損なうかもしれない。本“リスク要因”部分に記載されたリスクを含む、上記の任意のリスクまたは任意の広範な他のリスクを達成することは、私たちの普通株の市場価格に重大かつ不利な影響を与える可能性がある。また、株主にとっては、株主が少ない数の株を売却できるのと同じ価格で大量の株を売却することがより困難になる可能性がある。
従来、証券集団訴訟は、ある会社の証券市場価格が下落した後に提起されることが多かった。このリスクは生物製薬会社と特に関連しており,これらの会社は近年大幅な株価変動を経験している。また、もし私たちが彼らが私たちの株の内在的価値を反映していないと考えている市場推定値を経験すれば、新冠肺炎の流行による市場変動は株主の急進主義的な感情を高める可能性がある。我々の戦略方向と競争したり衝突したり、取締役会構成の変更を求める過激な活動は、私たちの経営業績や財務状況に悪影響を及ぼす可能性があります。
私たちの普通株を売る大量の株は私たちの普通株の価格を下落させるかもしれない。
公開市場で私たちの大量の普通株を売ることはいつでも起こる可能性がある。もし私たちの株主や市場が私たちの株主が公開市場で私たちの普通株を大量に販売しようとしていると思うなら、私たちの普通株の市場価格は大幅に低下する可能性があります
公開市場で私たちの株や売却可能な株が私たちの普通株の市場価格にどのような影響を与えるか予測できません(あれば)。しかし、将来的には、私たちの発行された株式承認証やオプションを行使する際に発行された株を含む、私たちの普通株を公開市場で大量に売却したり、このような売却が発生する可能性があると考えられたりして、私たちの普通株の市場価格に悪影響を及ぼす可能性があります。
私たちはまた、未来に私たちが計画している業務を継続するために多くの追加資本が必要かもしれないと予想している。資本を調達するために、私たちは1回または複数回の取引で時々決定された価格および方法で普通株、転換可能証券、または他の株式証券を販売することができる。株式または他の株に変換可能な証券を売却して発行することで追加資本を調達すれば、我々の株主は希釈されるだろう。これらの売却、あるいは市場で大量の株式保有者が株を売却しようとしているとの見方は、我々の普通株の市場価格を低下させる可能性がある。
私たちとジェフリーの間で時々改訂された販売協定によると、私たちは普通株の“市場”発売に参加しました。販売契約のいくつかの制限及び適用法律を遵守する場合には、販売契約期間内のいつでもジェフリーに配給通知を出すことができる。私たちが配給通知を出した後、ジェフリーが販売する株式数は、販売期間内の普通株の市場価格と、私たちがジェフリーと設定した制限によって変動します。販売された1株当たりの株価は販売期間中に我々の普通株の市場価格によって変動するため,販売プロトコルにより最終的に発行される株式数(あれば)を予測することはできない.販売契約に基づいて販売される任意の株式の発行は、私たちの既存の株主に希釈効果を与えます。さらに、S-3表での棚上げ登録宣言に基づいて、他の取引で普通株、優先株、転換可能証券、および他の株式証券を販売すれば、既存投資家はその後の売却によって深刻に希釈される可能性があり、新しい投資家は私たちの既存株主よりも高い権利を得る可能性がある。
私たちの主要株主と経営陣は私たちのかなりの割合の株を持っていて、株主の承認を待つ事項をコントロールすることができるだろう。
2022年12月31日現在の私たちの普通株の実益所有権によると、私たちの役員、取締役、5%以上の株式の所有者と、それぞれの関連会社の実益は、私たちの発行済み議決権株の約50%を持っています。したがって、この株主たちが一緒に行動すれば、彼らは会社の結果を統制し続けるだろう
株主の承認を必要とする行動は、取締役の選挙、私たちの組織文書の修正、任意の合併、合併、または私たちのすべての資産またはほとんどの資産の売却、および任意の他の重大な会社取引を含む。これらの株主の利益はあなたの利益とは違うかもしれないし、あなたの利益と衝突する可能性もある。例えば、これらの株主は、わが社の支配権の変更を延期または阻止することができ、たとえこのような制御権の変更が私たちの他の株主に利益をもたらすとしても、私たちの株主が私たちの会社や私たちの資産を売却する際に普通株のプレミアムを得る機会を奪い、私たちの普通株の現行の市場価格に影響を与える可能性がある。投資家は利益衝突が存在または発生する可能性があると考えているため、株式所有権の著しい集中は私たちの普通株の取引価格に悪影響を及ぼす可能性がある。
私たちの定款書類やデラウェア州法律によると、反買収条項は私たちの買収を阻止または延期する可能性があり、これは私たちの株主に有利になる可能性があり、私たちの株主が現在の経営陣を交換または更迭しようとすることを阻止することができるかもしれない。
私たちが再記述した会社登録証明書と私たちの改訂と再記述の定款には、わが社の制御権変更を延期または阻止する可能性のある条項が含まれています。これらの規定は、我々の取締役会の現職メンバーが指名した取締役や他の会社の行動ではなく、株主が我々の経営陣を変動させることを含めて、株主を選挙することを困難にする可能性もある。これらの規定には
•私たちの取締役会のすべてのメンバーが選挙で生まれないように分類された取締役会を作ります
•取締役会が取締役数を確定し、取締役会の空きを埋めることだけを許可する
•取締役は“理由がある”場合にのみ免職され、三分の二の株主の承認を得なければならないと規定されている
•絶対多数票で、私たちが再記載した会社登録証明書の修正および改訂および再記載された定款のいくつかの条項を要求する
•“空白小切手”優先株の発行を許可し、私たちの取締役会は株主権利計画を実施することができます
•株主が株主特別会議を開催する能力を廃止し
•株主の書面同意による行動を禁止することは、すべての株主の行動が私たちの株主会議で行われなければならないことを要求する
•累積投票を禁止します
•指名が取締役会に入るか、株主が年次株主総会で行動できる事項の事前通知要求を作成する。
私たちが再記述した会社登録証明書の独占裁判所条項は、株主が司法フォーラムで、私たちまたは私たちの任意の取締役、上級管理職、または他の従業員と紛争することに有利であると考えるクレームを提出する能力を制限する可能性があり、これは、このようなクレームに関連する訴訟を阻害する可能性がある。
我々が再記述した会社登録証明書は、法律が許容する最大範囲内で、デラウェア州衡平裁判所が以下の独占裁判所であることを規定している:私たちを代表して提起された任意の派生訴訟または訴訟、受託責任違反を主張するいかなる訴訟、デラウェア州会社法またはDGCL、私たちが再記載した会社登録証明書、または私たちが改正して再説明した定款に基づいて私たちにクレームを提起した任意の訴訟、または内部事務原則に基づいて私たちにクレームを提起した任意の訴訟である。この排他的裁判所条項は,取引法で規定されている義務や責任を実行するための訴訟には適用されない.しかし、排他的裁判所の規定によって列挙された1つまたは複数のカテゴリに属する訴訟に適用することができる。
このような裁判所条項の選択は、株主が司法裁判所において、私たちまたは私たちの任意の取締役、上級管理者、または他の従業員と紛争することに有利であると考えられるクレームを提出する能力を制限する可能性があり、これは、このようなクレームに関連する訴訟を阻害する可能性がある。代替的に、裁判所が、私たちが再記載した会社登録証明書に含まれる裁判所条項の選択が訴訟において適用されないか、または実行できないことを発見した場合、私たちは、他の司法管轄区域でこのような訴訟を解決することに関連する追加費用を生じる可能性があり、これは、私たちの業務、運営結果、および財務状態を損なう可能性がある。
また,DGCLの第203条はわが社への制御権変更を阻害,延期,阻止する可能性がある。第203条私たちと15%以上の普通株式を保有する者との間の合併、業務合併、その他の取引には、いくつかの制限が加えられている。
改正された1933年証券法又は証券法第22条によると、連邦裁判所及び州裁判所は、証券法又はその下の規則及び条例を執行するために生じた任意の義務又は責任について提起されたすべてのクレームに対して同時管轄権を有する。2020年3月、私たちはアメリカ合衆国の連邦地域裁判所が法的許容の最大レベルでいかなる訴えを解決するための唯一のフォーラムになることを規定する添付例を改正し、再確認した
証券法や連邦フォーラム条項による訴訟因を主張する。私たちが連邦フォーラムの規定を採択することを決定する前に、デラウェア州最高裁判所は、デラウェア州の法律によると、これらの規定は事実上有効であると判断した。連邦または州裁判所がデラウェア州最高裁判所の判決に従うこと、または特定の事件で連邦フォーラム条項を実行することを決定することは保証されないが、連邦フォーラム条項の適用は、私たちの株主が証券法を実行するために発生したいかなる義務または責任のために提起された訴訟は、州裁判所に提起することができず、連邦裁判所に提起しなければならないことを意味する。
取引法第27条は,連邦政府が取引法又はその下の規則及び条例を実行するために生じた任意の義務又は責任に対して提起されたすべてのクレームに対して排他的連邦管轄権を有すると規定されている。また,排他的裁判所条項も連邦裁判所条項も,取引法で規定されているいかなる義務や責任を実行するための訴訟にも適用されない.したがって、私たちの株主は、取引法またはその下の規則および条例によって生じる任意の義務または責任を強制的に執行するために、連邦裁判所に訴訟を提起しなければならない
私たちの株主は連邦証券法とそれに基づいて公布された法規の遵守を放棄したとみなされないだろう。
任意の個人またはエンティティが、私たちの任意の証券の任意の権益を購入または他の方法で取得または保有することは、連邦フォーラム条項を含む、私たちの独占フォーラム条項を通知し、同意したとみなされなければならない。これらの条項は、株主が私たちまたは私たちの役員、役員、または他の従業員との紛争について司法裁判所でクレームを提起する能力を制限する可能性があり、これは私たちと私たちの役員、役員、および他の従業員に対する訴訟を阻止するかもしれない。
私たちは予測可能な未来に私たちの株に現金配当金を支払わないと予想されているので、資本増加(あれば)があなたの唯一の収益源になるだろう。
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、私たちの未来のすべての収益(もしあれば)を維持し、私たちの業務の成長と発展、運営、拡張に資金を提供し、予測可能な未来にいかなる現金配当金も発表または支払うことはないだろう。したがって、私たちの普通株の資本付加価値は(もしあれば)あなたが予測可能な未来に唯一の収益源になるだろう。
一般リスク因子
証券や業界アナリストが私たちの業務に関する研究や報告を発表しない場合、または彼らが私たちの株に不利または誤った意見を発表した場合、私たちの株価や取引量は低下する可能性がある。
私たちの普通株の取引市場は、業界または証券アナリストが発表した私たちまたは私たちの業務に関する研究と報告の影響を受けるだろう。私たちは産業や証券アナリストを統制することもできないし、彼らの報告書の内容と意見を統制することもできない。もし私たちのどのアナリストが私たち、私たちのビジネスモデル、私たちの知的財産権、あるいは私たちの株式表現に否定的または誤った意見を発表した場合、あるいは私たちの臨床前研究、臨床試験、運営結果がアナリストの期待に達しなかったら、私たちの株価は下落するかもしれない。1人以上のそのようなアナリストが私たちへの報告を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちは金融市場で可視性を失う可能性があり、これは逆に私たちの株価や取引量を低下させる可能性がある。
もし私たちが将来有効な財務報告内部統制システムを維持できなければ、私たちは私たちの業務、運営結果、財務状況とキャッシュフロー、将来の見通しを正確に報告できないかもしれません。これは投資家に私たちの自信に悪影響を与え、私たちの普通株の価値に影響を与えるかもしれません。
サバンズ-オキシリー法案は、財務報告および開示統制および手続きに対して有効な内部統制を維持し、財務報告の内部統制に対する私たちの有効性に関する報告書を管理層に提出することを要求する。この評価には、財務報告書の内部統制で発見された私たちの経営陣の重大な弱点を開示する必要がある。重大欠陥とは、財務報告内部統制の欠陥又は欠陥の組み合わせであり、年度又は中期財務諸表の重大な誤報が適時に防止又は発見されない可能性が合理的な可能性を超えている
私たちは404条を遵守して、私たちに多くの会計費用を発生させ、多くの管理努力をすることを要求するだろう。我々には現在内部監査チームはなく、限られた会計·財務者によって必要なシステム·プログラムファイルを作成し、第404条に必要な年間評価を実行する。私たちは私たちの年間評価、テスト、そしてどんな必要な救済措置もタイムリーに達成できないかもしれない。評価およびテスト中に、発見される可能性のある重大な欠陥や重大な弱点を発見して修正できなかった場合、あるいは財務報告内部統制評価に問題や遅延があった場合、私たちの財務報告内部統制が有効であるとは断言できないだろう。私たちの財務内部統制に重大な欠陥や重大な欠陥が発生しないことを保証することはできません
未来の記事。財務報告を内部統制できなかったいかなる行為も、財務状況、運営結果、またはキャッシュフローを正確に報告する能力を深刻に抑制する可能性がある。もし私たちが財務報告の内部統制に有効な結論を出すことができない場合、あるいは私たちの独立公認会計士事務所が私たちの財務報告の内部統制に重大な弱点や重大な欠陥があると判断した場合、私たちは投資家が私たちの財務報告の正確性と完全性に自信を失う可能性があり、私たちの普通株の市場価格は下落する可能性があり、私たちはナスダック株式市場、ナスダック、米国証券取引委員会、または他の規制機関の制裁や調査を受ける可能性がある。財務報告の内部統制のいかなる重大な欠陥を補うことができなかったり、上場企業に必要な他の効果的な制御システムを維持できなかったり、将来の資本市場への参入機会を制限する可能性もある。
私たちの開示統制と手続きはすべてのミスや詐欺を阻止したり検出できないかもしれない。
我々は、1934年に改正された証券取引法または“取引法”の定期報告要求を遵守しなければならない。我々の開示制御および手続きは、取引所法案に基づいて提出または提出された報告書において、開示を要求する情報が蓄積され、米国証券取引委員会規則および表で指定された期間内に管理層、記録、処理、まとめおよび報告に伝達されることを合理的に確保することを目的としている。任意の開示制御およびプログラムまたは内部制御およびプログラムは、発想および動作がどのように完全であっても、絶対的な保証ではなく、合理的な保証を提供することしかできず、制御システムの目標が達成されることを確保することしかできないと信じている。
これらの固有の限界は,意思決定過程における判断が誤りである可能性があり,簡単な誤りや誤りによって故障が発生する可能性があるという現実を含む.さらに、ある人の個人的な行動、2人以上の結託、または無許可超越制御は、制御を回避することができる。したがって,我々の制御システム固有の制限により,誤りや詐欺による誤った陳述や開示不足が発見されることなく発生する可能性がある.
項目1 B未解決従業員意見
ない
第二項です属性
私たちの主な実行事務室はマサチューセッツ州のウォルザムにあります。2021年8月、私たちは2025年5月に一度に延期する権利を行使し、行政、研究開発、その他の活動のための3つの建物のうち、合計約32,405平方フィートのオフィスと実験室空間を獲得した
第三項です。 法律訴訟
時々、私たちは正常な業務過程で法的訴訟に巻き込まれるかもしれない。私たちは現在、経営陣が私たちの業務に重大な悪影響を及ぼすと考えているいかなる法的手続きにも参加していません。結果にかかわらず、訴訟は弁護と和解費用、管理資源の移転、負の宣伝と名声損害などの要素によって私たちに不利な影響を与える可能性がある。
第四項です。 炭鉱安全情報開示
適用されません。
第2部-その他の資料
五番目です登録者普通株市場、関連株主事項及び発行者による株式証券の購入
市場情報
私たちの普通株はナスダックの世界市場で取引され、コードは“Morf”だ。私たちの初公募株の完成に伴い、私たちの普通株は2019年6月26日に取引を開始しました。これまで、私たちの普通株はまだ公開取引市場を設立していなかった。
株主.株主
私たちの普通株は2023年2月21日までに登録された株主16人です。この数値には、その株式が街の名義で保有されている利益所有者は含まれていない。
配当をする
設立以来、私たちは株主に何の配当金も発表したり、支払ったりしたことはなく、予測可能な未来に、私たちは現金配当金を発表したり支払うつもりもない。私たちは現在、私たちの業務運営と拡張のために、すべての利用可能な資金と未来の任意の収益を維持すると予想している。将来の任意の配当政策に関連する決定は私たちの取締役会が適宜決定し、私たちの経営結果、財務状況、資本要求、契約制限、業務見通しと取締役会が関連する他の要素に依存するだろう。投資家は現金配当金を得ることを期待して私たちの普通株を購入してはいけない。
登録証券を使って収益を得る
2019年6月26日、我々のS-1表(文書番号333-231837)のうち、我々の普通株初公募株に関する登録声明が米国証券取引委員会によって発効されたことが発表された。この登録声明によると,合計6,900,000株の普通株を1株15.00ドルで売却し,引受割引と手数料および発売コストを差し引いた現金収益総額は約9,330万ドルであった。
初公募株の余剰純収益を,我々の経口小分子インテグリン療法のさらなる開発に用い,さらに我々のプラットフォームを開発し,我々の候補製品ルートを広げ,運営資金や一般企業用途に利用する予定である。
株式補償計画に基づいて発行された証券
当社の株式補償計画及びこの計画に基づいて認可された証券の発行に関する資料は、本年度報告表格10-Kの第3部第12項に記載されている。
株式表現グラフ
S−K法規第10項で定義された“小さな報告会社”として,この情報を提供する必要はない。
発行人が株式証券を購入する
ない。
第六項です保留します。
第七項。 経営陣の財務状況と経営成果の検討と分析
私たちの財務状況と経営結果に関する以下の議論、および当社の連結財務諸表および本年度報告書の他の場所にForm 10-K形式で含まれる関連説明および他の財務情報を読むべきです
歴史的財務情報に加えて、この議論は、私たちの計画、目標、期待、意図、および信念の陳述のような、現在予想されているリスクおよび不確実性に関連する前向きな陳述を含む。様々な要因の影響により、私たちの実際の結果は、第1部1 A項の“リスク要因”と題する章で述べた要因を含む、これらの前向き陳述で予想される結果と大きく異なる可能性がある。これらの展望的声明は、私たちの将来の経営業績と財務状況、新冠肺炎疫病の影響、業務戦略、市場規模、潜在的成長機会、臨床前と臨床開発活動、候補製品の治療効果と安全性、私たちが提供する製品の純収益の使用、候補製品がいくつかの指定された利益を受ける能力、臨床前研究と臨床試験の時間と結果、第三者とのビジネス協力、および潜在的規制指定、候補製品の承認と商業化の受け入れとタイミングを維持し、認識することを含むことができるが、これらに限定されないかもしれない。“信じる”、“可能”、“そうなる”、“可能”、“推定”、“継続”、“予想”、“予測”、“目標”、“計画”、“可能”、“プロジェクト”、“計画”、“予想”、および未来のイベントまたは結果の不確実性を表す同様の表現は、これらの識別可能な言葉を含むわけではないが、前向き表現を識別することを意図している。
これらの陳述は,本年度報告10-K表までの日に我々に提供された情報に基づいており,これらの情報はこのような陳述の合理的な基礎を構成していると考えられるが,このような情報は限られているか不完全である可能性があり,我々の陳述は,入手可能なすべての関連情報について詳細な調査や検討が行われていることを示していると解釈されてはならない.このような陳述は本質的に不確実であり、投資家たちにこのような陳述に過度に依存しないように想起させる。
概要
我々はバイオ製薬会社であり,インテグリンに対する独自の知見を潜在的に一流の経口小分子インテグリン療法のパイプラインの発見と開発に応用している。インテグリンは1種類の標的であり、多種の承認された重ポンド爆弾薬物を有し、自己免疫、心血管と代謝性疾患、繊維化と癌を含む深刻な慢性疾患の治療に用いられる。これまで,経口小分子インテグリン療法は米国食品·薬物管理局(FDA)の承認を得ていない。それにもかかわらず、私たちの独特なプラットフォームは特定のインテグリン標的に対して確実に高品質の経口分子を産生する潜在力を放出できると信じている。著者らのインテグリン構造と機能に対する独特な理解を利用してMorphyインテグリン技術プラットフォーム或いはMINTプラットフォームを作成し、新しい候補製品を開発し、経口投与に必要な効力、高選択性と薬学特性を実現することを目的とした。我々の主要候補製品Morf−057,炎症に関与するα4β7特異的インテグリン阻害剤を含む我々のチューブを進めており,炎症性腸疾患の治療の臨床開発に用いられている。我々は2020年7月にMorf−057の新薬研究申請を提出したか,あるいはIND,FDAがINDによる研究を2020年8月に継続することを許可した。2020年9月,われわれは健康ボランティアの中でMorf−057の第1段階臨床試験を開始し,われわれの臨床計画を確立し,われわれのIBD第2段階計画のための投与量を選択し,最初の重点は潰瘍性大腸炎(UC)であった
Morf−057第1段階研究は、Morf−057安全性、薬物動態またはPKおよび薬効学またはPDのキューを評価するSAD、MADおよびFEを含む。健康な対象は、3:1にランダムに分割され、SAD群において単回用量の25、50、100、150および400 mgのMorf−057または一致プラセボを受け、狂ったキューで1日2回またはBIDの25、50および100 mgのMorf−057または一致プラセボを14日間受け入れた。条件に適合した健常者67名が研究に参加し,そのうち36名がSAD群,9名がFE群,22名がMAD行列であった。66人の被験者が研究治療を完了し、50 mgのBid狂った列のうちの1人が個人的な理由で同意を撤回しました
MORF−057はすべてのキューで耐性が良好であり,セキュリティ信号は認められなかった。MORF−057は良好なPKプロファイルを示し,その中で目標交戦が確認され,明確なPKとPD関係が確立された。Morf−057は迅速に吸収され,全身曝露はほぼ用量割合で増加することが確認された。FE研究では,高脂肪食服用後,曝露量はやや減少したが,谷濃度には影響が認められなかった。その結果,食物摂取量は低地Morf−057レベルに影響を与えず,計画した患者研究では,食物を考慮せずにMorf−057を使用することが可能であった。
α4β7 ROは用量と研究日数の増加とともに増加し、14日目に飽和(>99%RO)に達した。100 mgのBID行列では、α4β7受容体が飽和した(平均RO>99%)。特定のα4β7高発現免疫細胞群を含むバイオマーカーの用量と時間依存の変化が観察され,Morf−057の生物学的証拠が増加した。これらの変化は,IBDの治療に承認された抗体薬vedolizumabを含む他のインテグリン阻害剤の報告と一致している。
別のMORF-057第1段階研究では、被験者は1日2回服用し、毎回の用量は200 mgであり、プラセボと比較して、100 BIDまたは200 mg BIDを服用した被験者はα4β7受容体飽和を示し、循環中央記憶、効果記憶Tリンパ細胞およびスイッチ記憶Bリンパ細胞数は明らかに増加した。25歳の時
重要なPD対策では,mgと50 mg Bid探査用量の方向性が増加する傾向が認められた。全用量の耐性は良好であり,安全信号は認められず,良好なPK曲線が認められた。Morf−057は単剤200 mgMorf−057および200 mgBidの14日間でα4β7受容体がCにあることを示した低い谷それは.リンパ球亜群とCCR 9 mRNAの統計学的有意な変化が認められ,これは先行研究と一致している。
第1段階研究の結果,2022年3月にMorf−057の第2段階臨床試験を開始した。Emerald-1(Morf-057-201)は、中重度UCを有する成人に対するMorf-057の有効性、安全性と耐性を評価するための開放ラベルのマルチセンター2 a期試験であり、2022年10月に方向性登録を完了し、30名の患者がこの研究に参加した。また,方向性登録完了時にスクリーニングを受けている患者が研究に組み込まれ,計35名の患者が主要キューに登録されている。これまでvedolizumabで治療に失敗した10名までの患者への探索的キューの募集が行われている。Emerald−1研究に参加した患者は米国とポーランドの地点でBid 100 mgの治療を受けている。試験の主な終点はRHIの変化であり,ベースラインと比較して12週間の潰瘍性大腸炎の組織学的疾患活動を測定した有効なツールである。その後,患者は追加40週間の維持治療を継続し,52週間の評価を行う。Emerald-1研究中の副次的およびその他の結果指標は修正されたメオ臨床採点、安全性、PKパラメータと重要なPD指標を含み、α4β7受容体占有率とリンパ細胞亜群輸送を含む。Emerald−1ステージ2 a試験からの主要終点データは,中から重度UC患者のMorf−057を対象とした2023年第2四半期に報告されると予想される。Emerald-2(Morf-057-202)はMorf-057の全世界2 b期無作為対照試験であり、2022年11月に患者への投与量を開始した。Emerald-2研究に登録された患者は、ランダムに3つの有効群またはプラセボ群に分けられた:100 mg Bid、200 mg Bid、Qd(1日に1回)群, あるいはプラセボ群は,12週の誘導段階後に交差してMorf−057に入る。試験の主な終点は臨床緩解率であり,12週間で修正したMayoスコアで測定した。副次的な終点は、RHI、PK、およびPD対策の変化、およびセキュリティパラメータを含む。12週の誘導期の後,40週間の維持期に入る。2025年上半期にMorf−057が中~重度UC患者で行ったEmerald−2期2 b試験の主な終点を完成させると信じている。Morf-057の進展を考慮して、著者らは2022年に第二世代開発候補薬物の前IND段階を停止し、重点的にMorf-057の第二段階臨床計画による進展である。著者らは引き続き私たちのα4β7製品の組み合わせを拡大し、未来の臨床研究のために新世代α4β7小分子の候補製品を開発した。
2022年6月、私たちは便宜上、私たちがエバーヴィバイオテクノロジー株式会社またはエバービーと2018年10月に締結した協力またはエバービー協定が終了したと報告した。今回の協力のテーマは,特発性肺線維化を含む線維化疾患の治療のために開発した選択的経口αとβ6特異的インテグリン阻害剤,およびAbbVieと連携した他の適応である。2020年8月,AbbVieは我々のαvβ6特定インテグリン阻害剤計画の選択権を許可し,一度に2000万ドルを支払った。AbbVieは,臨床前試験でαvβ6を介した安全シグナルを標的とする疑わしいシグナルが観察されたため,αvβ6特異的インテグリン阻害剤を選択的に経口投与することを意図していないと述べ,詳細な情報は2022年12月の毒理学学会誌に発表された。AbbVie協定は2022年12月に終了したので、私たちはこの合意に従って支払われた追加金を受け取ることはできないだろう
2019年2月、私たちはJanssen PharmPharmticals、Inc.またはJanssenと合意し、新しいインテグリン療法、またはJanssenプロトコルを開発した。2021年2月,Janssenは500万ドルを支払い,特定のインテグリン標的に対する抗体活性化剤を発見するための第3の研究計画を開始した。2021年12月、Janssenは、最初の2つのインテグリン目標に対して選択権を行使しないことを決定し、インテグリン抗体活性化剤の潜在的開発を含む第3のインテグリン研究計画に重点を置いたことを通知した。最初の2つのインテグリンの標的は rJanssenに興味のある特定の疾患の標的検証が乏しいため,この報告を受けた。2023年1月、私たちはJanssenから通知を受け、彼らは合意を終了することを選択した。終了は2023年1月13日から60日以内に発効しますので、ヤンソン協定の下での追加支払いは受けないと予想されますが、終了前に2023年に完了した研究サービスに関連する潜在的な名目金額は除外されます。
われわれの主要目標Morf−057に加えて,われわれのMINTプラットフォームを用いて様々な治療分野で広範な臨床前計画パイプラインを進めており,これらすべての計画はインテグリン受容体の抑制や活性化の潜在力を利用することを目的としている。他の全額所有プロジェクトは発見の先行最適化段階に近づいているか、または段階に入っている。われわれは2021年4月の米国癌研究協会年次総会でわれわれのαvβ8計画の積極的な臨床前データを示し,チェックポイント難治性癌モデルの抗腫瘍活性を示し,我々のαvβ8計画の推進に専念している。肺動脈高圧(PAH)を含む肺動脈高圧疾患や他の治療分野におけるフィブロネクチン統合標的に対する追加研究段階計画も行っている。インテグリンは細胞増殖,生存,肥大成長,線維化を促進することが知られており,これらはPAH進展の重要な因子である。
2021年3月、私たちは1株70.00ドルで350万株の普通株を一般に発行し、引受割引、手数料、私たちが支払った他の発行費用を差し引いた純収益は約2.3億ドルであることを発表した。
2020年7月、私たちはJefferies LLCまたはJefferiesと市場発売計画または以前のATMについて公開市場販売プロトコルまたは元の合意を締結し、この合意によると、私たちは時々Jefferiesを介して販売代理として私たちの普通株の株式を発売し、販売することができ、総発売金額は75,000,000ドルに達し、配給株式と呼ばれる。2021年8月に旧憲法の第1号改正案に署名した
JefferiesやNew ATMと合意し,発売·販売された配給株の総価値を最高150,000,000ドルの総発売金額に増加させた。従来のATMと新しいATMを総称してATMと呼ぶ.2022年12月31日までの年間で,新ATMにより1,000,000株の株が発行され,発売手数料と費用を差し引いた純収益は3,920万ドルであった。2022年12月31日まで、私たちは新しいATMで約9720万ドルの普通株が販売できます。当社は以前のATM機では何の配給株も販売しません
設立以来、私たちの業務は組織とわが社を配備し、業務計画、資金を調達し、私たちの知的財産権の組み合わせを構築し、経口小分子統合素療法を発見と開発するために研究を行ってきた。これまで,創設活動はAbbVieやJanssenとの連携協定から受け取った支払いに限られており,本年度報告Form 10−Kの他の部分に添付されている連結財務諸表の付記12でさらにこの問題が検討されてきた。今まで、私たちは何の製品も販売を許可されていませんし、製品販売から何の収入も得ていません。設立から2022年12月31日まで、私たちは主に株式を発行することで合計約7.43億ドルの総収益を集めました。その中には、転換可能な優先株証券、私たちの初公開株、2021年3月の公募株の引受、ATMによる私たちの普通株の売却、私たちの協力合意に基づいて受け取った支払いが含まれています。
設立以来、私たちは重大な運営損失が発生した。2022年12月31日までの累計赤字は2.971億ドル。私たちは、予測可能な未来に、私たちが臨床前と臨床開発を通じて私たちの現在と未来の候補製品を推進することに伴い、監督部門の承認を求め、私たちの知的財産権の組み合わせを維持と拡大し、より多くの研究開発と業務人員を招聘し、上場企業として運営し、私たちは引き続き大量かつ増加している費用と運営損失を招くことを予想している。
私たちが臨床開発に成功し、規制部門から候補製品の承認を得ない限り、私たちは製品販売から収入を得ません。また、規制部門の候補製品の承認を得て、第三者商業化パートナーシップに加入していなければ、製品販売、マーケティング、製造、流通活動を支援するために私たちの商業化能力を発展させることに関連した巨額の費用が生じることが予想されます。
したがって、私たちは私たちの持続的な運営を支援し、私たちの成長戦略を実施するために多くの追加資金を必要とするだろう。私たちは、製品販売から相当な収入を得ることができる前に、公開または私募株式発行および債務融資または他のソース(例えば、追加の協力協定)によって、私たちの運営に資金を提供する予定です。私たちは必要な時に受け入れ可能な条件で追加資金を調達することができないかもしれないし、そのような他の合意や手配を達成することができないかもしれない。私たちが必要な時に資金を調達したり、このような合意に達しなかったりすることは、私たちの業務、経営結果、財務状況に実質的な悪影響を及ぼす可能性があります。
2022年12月31日現在、私たちは3.482億ドルの現金、現金等価物、有価証券を持っている。私たちは、私たちの既存の現金、現金等価物、有価証券に加えて、2023年2月に普通株と事前融資権証を非公開で発行して調達した1.0億ドルを加えて、2026年下半期の運営費用と資本支出需要に資金を提供できると信じている
新冠肺炎の大流行の影響
持続的な新冠肺炎疫病は引き続き世界各地の公衆衛生と経済に挑戦をもたらし、そして私たちの従業員、患者、コミュニティと業務運営、及びアメリカ経済と金融市場に影響を与えている。新冠肺炎が私たちの運営と財務業績に与える持続的な影響(サプライチェーン中断を含む)及びその他の経済状況と傾向(消費者価格上昇、インフレ、金利上昇と持続的な労働力不足を含む)が私たちの運営と財務業績に与える影響の程度は、疫病の持続時間と蔓延、既存のワクチンにあまり敏感でない変種の台頭、私たちの臨床と臨床前研究への影響、従業員或いは業界事件及び私たちのサプライヤーとメーカーへの影響を含むいくつかの発展に依存し、これらはすべて不確定で予測できない。私たちは現在、私たちの業務に大きな影響を与えていませんが、もし私たちまたは私たちのサプライヤーが仕事や輸送供給の場所を閉鎖したり、他の制限をしたりすれば、私たちの候補製品の供給制限、あるいは私たちの臨床と前研究あるいは計画の臨床試験の遅延を招く可能性があり、私たちの業務、運営結果と未来の期間の全体的な財務表現は重大な不利な影響を受ける可能性があります。また、新冠肺炎疫病のため、私たちと全世界各地の会社が業務を展開する方式は変化し、旅行と対面会議の制限、未来のサイト活性化と未来の臨床試験登録の遅延、病院資源の新冠肺炎疫病に対する努力の優先順位、及び食品と薬物管理局と類似外国の監督機関の審査の遅延を含むが、これらに限定されない。本表格10-Kまでの提出日, 新冠肺炎と他の経済状況と傾向或いは地政学的行動は、現在のウクライナの武力衝突を含み、どの程度私たちの財務状況、運営或いは指導結果に影響を与える可能性があり、その影響は今後一定期間内に私たちの運営結果と全体の財務業績に完全に反映されない可能性がある。新冠肺炎の流行,全体的な経済状況と地政学的行動がわれわれの業務に及ぼす可能性のある影響のさらなる検討については,本年度報告10−K表の他の部分の“リスク要因”を参照されたい。
財務運営の概要
協力収入
私たちは販売を許可された製品は何もありません。したがって、私たちは製品販売から何の収入も発生せず、予測可能な未来に製品販売から何の収入も発生することを期待しません。
今まで、私たちのすべての協力収入はAbbVieとJanssenとの協力協定から来てきた。私たちのAbbVieとJanssenとの協力は現在終了しており、未来を展望して、私たちは依然としていくつかの候補治療薬、地理市場、あるいは疾病分野をめぐる日和見的な評価と戦略的パートナーシップの構築に開放的である。私たちは、私たちが発売製品を持つ前に、私たちの収入は主に私たちが未来に締結する可能性のある協力と許可協定の下での支払いから来ると予想しています。
共同収益-AbbVie
2018年10月,AbbVieと協力し,AbbVieは投資家であり,合意時に約5%の普通株を保有し,線維化関連適応に対するいくつかの経口インテグリン療法を推進することを目的とした。AbbVie協定の条項によると、AbbVieは私たちに1.00億ドルの研究開発活動の前払いを支払い、私たちはAbbVieに複数の目標の候補製品に対する独占的な許可オプションを提供した。2020年8月,AbbVieは選択性αvβ6特定インテグリン阻害剤計画の選択権を許可し,2000万ドルを一度に支払った。
2022年6月、AbbVieは、便宜上AbbVie協定を終了する権利を行使することを決定したことを私たちに通知し、この協定は2022年12月に発効した。
コラボレーション収入-Janssen
2019年2月,既存療法が十分に解決できなかった疾患患者のために新たなインテグリン療法を発見·開発するヤンソン協定に署名した。Janssenプロトコルは3つのインテグリン標的に集中しており、各標的は研究計画のテーマであり、私たちが探索したことのない2つまでのインテグリン標的を置き換えることができる。ヤンソン協定によって付与されたある権利を考慮すると,2019年の間,ヤンソンは前の2つの研究項目にそれぞれ1,000万ドルの起動費を前払いし,2021年2月,ヤンソンは第3の研究プロジェクトに500万ドルの起動費を前払いした。2021年12月、Janssenは、最初の2つのインテグリン目標に対して選択権を行使しないことを決定したことを私たちに通知し、これは“Janssen協定”の下での私たちの責任範囲を縮小させた。2023年1月、Janssenは、その権利の行使を決定し、便宜上Janssen協定を終了することを決定したが、60日間の通知期間が必要であることを私たちに通知した。もし私たちとJanssenが同意すれば、Janssen協定は2023年3月またはそれ以上に終了するだろう
費用.費用
研究と開発
研究と開発費用は主に私たちの研究と開発活動によるコストを含み、私たちの研究計画下の製品候補発見仕事と臨床前研究を含み、その中には:
•研究開発者の賃金、福祉、株式給与費用を含む従業員関連の費用
•研究開発や臨床前活動を行う第三者を代表した研究に資金を提供するコスト
•現在または将来の候補製品に関連する臨床用品の製造コスト
•契約研究組織または臨床試験を行うCROと研究サイトとの合意による費用;
•現在または未来の候補製品の臨床前研究のコストは
•非従業員への持分補償を含む研究·開発活動に関する相談費および専門費
•臨床前研究で使用されている実験室用品および非資本設備を購入する費用;
•臨床法規の遵守に関連するコスト
•施設賃貸料および修理費用、保険、減価償却および他の用品費用を含む施設費用および他の分配費用
•メンテナンス許可の費用と、私たちの第三者ライセンス契約に基づいて支払わなければならない他の金額です。
研究·開発コストは発生時に費用を計上する。ある活動のコストは,特定のタスクを達成する進捗の評価に基づいており,我々のサプライヤーが提供してくれた情報などのデータを用いて,我々の臨床前研究や提供された他のサービスの進捗を分析している.任意の報告期間終了時の課税費用残高を確定する際に重大な判断と推定を行う。将来的に第三者から受信した研究開発商品又はサービスの払戻不可能な前払いは、関連商品の交付又はサービス提供時に資本化して費用を計上する。
私たちの候補製品が開発に成功するかどうかは大きな不確実性を持っている。したがって、現在、私たちは私たちの未来の候補製品を完成させるために必要な仕事の性質、時間、コストを合理的に推定したり、知ったりすることはできない。承認されれば,我々の候補製品を販売してから,いつ(あれば)大量の現金純流入が開始されるかも予測できない.これは、以下のような不確実性を含む、候補製品の開発に関連する多くのリスクと不確実性のためである
•私たちが行っている研究活動と任意の追加的な臨床前研究と臨床試験、その他の研究と開発活動の範囲、進捗、費用
•適切なセキュリティプロファイルを構築すること
•臨床試験の登録に成功しました
•私たちの候補製品が臨床試験で安全性と有効性を示しているかどうか
•関連規制機関の上場承認を受ける(もしあれば)
•商業製造能力を確立するか、または第三者製造業者との手配を行う
•私たちの候補製品のために特許と商業秘密保護および規制排他性を獲得し、維持する
•単独でまたは他人と協力して、承認された後に候補製品を商業化すること;および
•すべての規制が承認された後、製品の持続的に許容可能な安全状況。
これらの変数のいずれも、現在および将来の候補製品の開発における結果が変化し、これらの候補製品開発に関連するコストおよび時間を著しく変化させるであろう。
研究開発活動は私たちのビジネスモデルの核心だ。臨床開発後期段階にある候補製品は通常,臨床開発早期段階の候補品よりも高い開発コストを有しており,これは主に後期臨床試験の規模と持続時間が増加しているためである。我々が我々の候補製品を開発し続けるにつれて,予見可能な将来,研究·開発コストが大幅に増加することが予想される.しかし、私たちは現在、商業化によって特定の計画の総費用を正確に予測することは不可能だと思う。私たちの任意の候補製品の商業化成功に関連する要素はたくさんあり、未来の試験設計と各種法規要求を含み、その中の多くの要素は現在私たちの開発段階に基づいて正確に確定できない。また、私たちがコントロールできない未来のビジネスと規制要因は、私たちの臨床開発計画と計画に影響を及ぼすだろう。
一般と行政
一般費用と行政費用は主に従業員に関連する費用を含み、幹部、財務、会計、業務発展、法律と人力資源職能者の給料、福祉、株式ベースの給与費用を含む。その他の重大な一般及び行政費用には、研究開発費に含まれていない施設費用、特許及び会社事務に関する法律費用、会計及びコンサルティングサービス費用が含まれる。
事業の拡張に伴い、将来的には、今後の臨床プロジェクトを含む研究開発活動の期待成長を支援するために、将来的に一般的かつ管理費が増加することを予想しています。これらの増加には、より多くの人を雇うことに関連する費用の増加や外部相談者の費用、その他の費用が含まれる可能性がある。また、米国証券取引委員会規則の遵守に関する監査、法律、規制、税務関連サービスの費用、ナスダック上場会社に適用される上場基準、取締役上場会社の役員報酬と保険料、投資家関係費用を含む上場会社に関する費用も発生している。また、いずれかの候補製品が規制部門の承認を得て第三者の商業化協力に参加していなければ、製品販売、マーケティング、流通活動の支援に関連した巨額の一般的かつ管理費が発生することが予想される。
利子収入,純額
利息収入、純額は主に私たちの現金、現金等価物、有価証券が得た利息収入からなります。
所得税の支出から利益を得る
私たちは今年度の実際の税率に基づいて税引き前収入または赤字の所得税の支出や福祉を記録します。本年度の税務支出のその他の詳細については、本年度報告書の他の表格10−Kに記載されている総合財務諸表付記を参照されたい
経営成果
2022年と2021年12月31日終了年度比較
次の表は、2022年12月31日と2021年12月31日までの年間経営実績をまとめたものです
| | | | | | | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 | | 変わる |
| 2022 | | 2021 | | $ | | % |
| | | | | | | |
| (百分率を除いて千単位) |
| 協力収入 | $ | 70,808 | | | $ | 19,794 | | | $ | 51,014 | | | 258 | % |
| 運営費用: | | | | | | | |
| 研究開発 | 102,062 | | | 87,789 | | | 14,273 | | | 16 | % |
| 一般と行政 | 32,142 | | | 27,811 | | | 4,331 | | | 16 | % |
| 総運営費 | 134,204 | | | 115,600 | | | 18,604 | | | 16 | % |
| 運営損失 | (63,396) | | | (95,806) | | | 32,410 | | | (34) | % |
| その他の収入: | | | | | | | |
| 利子収入,純額 | 4,567 | | | 272 | | | 4,295 | | | 1,579 | % |
| その他の費用 | (145) | | | (8) | | | (137) | | | 1,713 | % |
| その他の収入合計,純額 | 4,422 | | | 264 | | | 4,158 | | | 1,575 | % |
| 所得税受益前損失 | $ | (58,974) | | | $ | (95,542) | | | $ | 36,568 | | | (38) | % |
| 所得税支給 | (67) | | | — | | | (67) | | | — | |
| 純損失 | $ | (59,041) | | | $ | (95,542) | | | $ | 36,501 | | | (38) | % |
協力収入
協力収入が5 100万ドル増加したのはAbbVie協定が締結され、Janssen協定の範囲が縮小されたためだ。2022年第2四半期に、AbbVieプロトコルにおける残り履行義務を達成する推定を下方修正し、AbbVieに基づいて2022年12月からAbbVieプロトコルを終了する選択権を行使することを通知し、累計収入の追跡状況を記録した。2022年第4四半期に、我々は予想作業量の変化と第3のインテグリン研究計画の研究サービス時間の変化に基づいて、ヤンソン合意での余剰業績義務を達成するために、私たちの推定を下方修正した
研究と開発費
2021年12月31日までの1年間で、研究開発支出は1,430万ドル増加し、16%増加し、2021年12月31日現在の8780万ドルから2022年12月31日現在の1.021億ドルに増加した。著者らの研究開発コストの大部分は外部臨床と臨床前CROコストであり、候補臨床製品が確定すると、プロジェクトごとにこれらのコストを追跡する。私たちの内部研究開発コストは主に人員関連のコスト、減価償却、その他の間接コストです。次の表は、2022年12月31日と2021年12月31日までの年間研究開発費をまとめています
| | | | | | | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 | | 変わる |
| 2022 | | 2021 | | $ | | % |
| | | | | | | |
| (百分率を除いて千単位) |
| 計画的に列挙された外部コスト: | | | | | | | |
| MORF-057 | $ | 37,826 | | | $ | 31,467 | | | $ | 6,359 | | | 20 | % |
| | | | | | | |
| AbbVieプロトコル計画 | 155 | | | 2,759 | | | (2,604) | | | (94) | % |
| ヤンソン協定プロジェクト | 1,263 | | | 1,751 | | | (488) | | | (28) | % |
| | | | | | | |
| αvβ8計画 | 11,899 | | | 8,924 | | | 2,975 | | | 33 | % |
| | | | | | | |
| 他の初期開発候補や未分配のコストは | 10,149 | | | 6,516 | | | 3,633 | | | 56 | % |
| 外部総コスト | 61,292 | | | 51,417 | | | 9,875 | | | 19 | % |
| 内部コスト: | | | | | | | |
| 従業員補償と福祉 | 36,673 | | | 32,150 | | | 4,523 | | | 14 | % |
| 施設やその他 | 4,097 | | | 4,222 | | | (125) | | | (3) | % |
| 内部総コスト | 40,770 | | | 36,372 | | | 4,398 | | | 12 | % |
| 研究と開発費用総額 | $ | 102,062 | | | $ | 87,789 | | | $ | 14,273 | | | 16 | % |
研究と開発費の変化は主に以下の点に起因する
•外部コストは2021年12月31日までの年度から2022年12月31日までの年度まで990万ドル増加し,主にMorf−057が行っている第2段階臨床研究や他の開発活動に関するコスト,および我々の協力者による開発マイルストーン費用,αvβ8を含む我々の早期開発候補プロジェクトを支援する他の外部研究コストが増加している。これらの増加はAbbieプロトコルやヤンソン合意下での活動の減少分によって相殺されている。
▪2021年12月31日までの年度から2022年12月31日までの年度までに内部コストが440万ドル増加したのは,主に非現金持分報酬支出,購読,幹部検索の増加により,Morf−057およびわれわれの早期候補プロジェクトの進行中の臨床活動を支援している
一般と行政費用
一般·行政費は430万ドル、あるいは16%増加し、2021年12月31日現在の年度の2780万ドルから2022年12月31日までの年度の3210万ドルに増加した。一般と行政費用の増加は、主に非現金持分による補償費用が420万ドル増加したことと、法律や特許コストが80万ドル増加したことによるものであるが、それぞれコンサルティング費用と監査および税金の50万ドル減少と20万ドルの減少によって部分的に相殺された。
利子収入,純額
2021年12月31日までの年度と比較して,2022年12月31日までの1年間で,現金等価物や有価証券収益の増加および投資有価証券の増加により利子収入が430万ドル増加した。
2021年12月31日までと2020年12月31日までの年度比較
次の表は、2021年12月31日現在と2020年12月31日までの年度の経営成果をまとめたものである
| | | | | | | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 | | 変わる |
| 2021 | | 2020 | | $ | | % |
| | | | | | | |
| (百分率を除いて千単位) |
| 協力収入 | $ | 19,794 | | | $ | 44,945 | | | $ | (25,151) | | | (56) | % |
| 運営費用: | | | | | | | |
| 研究開発 | 87,789 | | | 73,630 | | | 14,159 | | | 19 | % |
| 一般と行政 | 27,811 | | | 18,495 | | | 9,316 | | | 50 | % |
| 総運営費 | 115,600 | | | 92,125 | | | 23,475 | | | 25 | % |
| 運営損失 | (95,806) | | | (47,180) | | | (48,626) | | | 103 | % |
| その他の収入: | | | | | | | |
| 利子収入,純額 | 272 | | | 1,630 | | | (1,358) | | | (83) | % |
| その他の費用 | (8) | | | (19) | | | 11 | | | (58) | % |
| その他の収入合計,純額 | 264 | | | 1,611 | | | (1,347) | | | (84) | % |
| 所得税受益前損失 | $ | (95,542) | | | $ | (45,569) | | | $ | (49,973) | | | 110 | % |
| 所得税から利益を得る | — | | | 570 | | | (570) | | | (100) | % |
| 純損失 | $ | (95,542) | | | $ | (44,999) | | | $ | (50,543) | | | 112 | % |
協力収入
共同収入が2520万ドル増加したのは、主にAbbVie協定の下での活動が減少したからだ。前年の間,AbbVieは選択的αvβ6特定インテグリン阻害剤計画のオプションを行使し,AbbVieは2000万ドルのオプション行使支払いを支払い,2020年12月31日までの年度内に確認された。また,AbbVieオプション行使後,αvβ6特定インテグリン阻害剤計画下での余剰実績義務を達成するために我々の推定を改訂し,前年期間の収入増加を招いた。協力収入が前年より減少した影響を相殺するために、最近の科学的結果に基づいて、予想される仕事と研究サービス時間の変化に基づいて、AbbVieプロトコル下の余剰業績義務を達成する推定値を下方修正し、2021年第4四半期に620万ドルの収入を確認した。我々の連携の収入は,連携計画によるコストの時間と大きさおよび余剰業績義務を達成した場合の見積りの変化に応じて変動する.Janssen協定の契約修正は協力収入に実質的な影響を与えない
研究と開発費
研究開発費は1420万ドル増加し、2020年12月31日までの年度の7360万ドルから2021年12月31日までの年度の8780万ドルに増加し、19%増となった。著者らの研究開発コストの大部分は外部臨床と臨床前契約研究組織(“CRO”)コストであり、候補臨床製品を確定すると、著者らは項目ごとに候補臨床製品に関連するコストを追跡する。私たちの内部研究開発コストは主に人員関連のコスト、減価償却、その他の間接コストです。次の表は、2021年12月31日と2020年12月31日までの年間研究開発費をまとめたものです
| | | | | | | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 | | 変わる |
| 2021 | | 2020 | | $ | | % |
| | | | | | | |
| (百分率を除いて千単位) |
| 計画的に列挙された外部コスト: | | | | | | | |
| MORF-057 | $ | 31,467 | | | $ | 24,122 | | | $ | 7,345 | | | 30 | % |
| | | | | | | |
| AbbVieプロトコル計画 | 2,759 | | | 8,679 | | | (5,920) | | | (68) | % |
| ヤンソン協定プロジェクト | 1,751 | | | 2,680 | | | (929) | | | (35) | % |
| | | | | | | |
| αvβ8計画 | 8,924 | | | 4,489 | | | 4,435 | | | 99 | % |
| | | | | | | |
| 他の初期開発候補や未分配のコストは | 6,516 | | | 4,566 | | | 1,950 | | | 43 | % |
| 外部総コスト | 51,417 | | | 44,536 | | | 6,881 | | | 15 | % |
| 内部コスト: | | | | | | | |
| 従業員補償と福祉 | 32,150 | | | 25,158 | | | 6,992 | | | 28 | % |
| 施設やその他 | 4,222 | | | 3,936 | | | 286 | | | 7 | % |
| 内部総コスト | 36,372 | | | 29,094 | | | 7,278 | | | 25 | % |
| 研究と開発費用総額 | $ | 87,789 | | | $ | 73,630 | | | $ | 14,159 | | | 19 | % |
研究と開発費の変化は主に以下の点に起因する
•2020年12月31日までの1年間から2021年12月31日までの1年間で、外部コストは690万ドル増加し、主にMorf-057二期臨床研究を支援するための製造コストや他の開発活動、および我々の早期開発候補薬(αvβ8を含む)を支援するための他の外部研究コストが増加した。これらの増加は、AbbVieの2020年8月のオプション行使によるαvβ6計画に関連する支出の減少とAbbVieおよびJanssenの他の計画に関連する活動の減少分によって相殺される。
▪2020年12月31日までの1年から2021年12月31日までの1年間に内部コストが730万ドル増加したのは,主にMorf−057の持続的な臨床活動およびわれわれの早期候補製品を支援するために増加した従業員数である。
一般と行政費用
一般·行政費は930万ドル、すなわち50%増加し、2020年12月31日までの年度の1850万ドルから2021年12月31日までの年度の2780万ドルに増加した。一般·行政費の増加は,主に非現金株による報酬支出が640万ドル増加し,人員コストが120万ドル増加し,相談費が80万ドル増加し,専門費が60万ドル増加したのは,上場企業の行政コストに関する法律や会計費用の増加と,D&O保険料の40万ドル増加によるものである。
利子収入,純額
2021年12月31日までの1年間で有価証券の収益率が低下したため、利息収入は140万ドル減少した。
所得税の割引を受ける
2020年3月27日、CARE法案が法律に署名された。CARE法案には、純営業損失の繰越許可、利息減額の拡大、支出の加速を含むいくつかの所得税改革が含まれている
いくつかの資本面の改善。2019年12月31日の納税期間に支払われた連邦所得税の予想回収に基づき、2020年12月31日までの年間60万ドルの所得税割引を確認しました
流動性と資本資源
流動資金源
設立から2022年12月31日まで、私たちは主に株式発行を通じて合計約7.43億ドルの総収益を集めました。私たちの転換可能な優先株証券、私たちの初公開株と後続株式発行、ATMで私たちの普通株を売却し、協力協定に基づいて受け取った支払いを含めています。
以下の表は、我々の現金、現金等価物、および有価証券総額に関する情報を提供し、いずれも2022年12月31日および2021年12月31日までの公正価値で述べている
| | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| | | |
| (単位:千) |
| 現金 | $ | 458 | | | $ | 292 | |
| 現金等価物 | 58,814 | | | 171,142 | |
| 有価証券 | 288,976 | | | 236,701 | |
| 現金、現金等価物、有価証券総額 | $ | 348,248 | | | $ | 408,135 | |
キャッシュフロー
次の表は、2022年12月31日、2021年12月31日、2020年12月31日までの年間キャッシュフロー情報を提供しています
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| | | | | |
| (単位:千) |
| 経営活動のための現金純額 | $ | (100,977) | | | $ | (83,461) | | | $ | (45,990) | |
| 投資活動が提供する現金純額 | (56,451) | | | (113,180) | | | 8,181 | |
| 融資活動が提供する現金純額 | 45,266 | | | 266,313 | | | 38,297 | |
| 現金、現金等価物、および制限的現金純増加 | $ | (112,162) | | | $ | 69,672 | | | $ | 488 | |
経営活動に使われている現金純額
すべての期間の現金使用は主に非現金費用と運営資本構成要素の変化調整後の純損失によるものである。2022年12月31日までの年度の経営活動用現金純額は1.01億ドルであったが,2021年12月31日現在の年度の経営活動用現金純額は8350万ドルであった。経営活動のための現金増加は主に経営資産と負債が6180万ドル減少したためであり、主な原因は繰延収入が5280万ドル減少し、売掛金が320万ドル増加し、売掛金が250万ドル減少したが、純損失が3650万ドル減少したことと株式ベースの報酬が790万ドル増加したことであり、この減少を部分的に相殺した
2020年12月31日までの1年間、経営活動に用いられた現金純額は4600万ドル。2021年12月31日までの年度と2020年12月31日までの年間で、経営活動で使用される現金が増加したのは、主に2021年12月31日までの年度純損失が5050万ドル増加し、一部が運営資産や負債の変化によって相殺され、株式ベースの報酬が1010万ドル増加したためである。純損失の増加は,主にAbbVieが2020年に2000万ドルのオプション行権支払いを支払ったことと,2021年の運営費が2350万ドル増加したためである。
投資活動が提供する現金純額
2022年12月31日までの年度の投資活動用現金純額は5650万ドルであったが、2021年12月31日現在の年度の投資活動用現金純額は1兆132億ドルであった。投資活動で使用される現金の変化は、主にその間に有価証券または有価証券を購入する満期日に基づく。2022年12月31日までの年度内に、投資活動のための現金は、主に満期日を超える有価証券の購入と有価証券の売却に由来する。2021年12月31日までの年間投資活動のための現金増加は
2021年3月に完成した引受公開で得られた金の形で有価証券への投資を増加させる。
2020年12月31日までの1年間、投資活動が提供する現金は、主に有価証券の満期日が有価証券購入より870万ドル多い収益に由来している。
融資活動が提供する現金純額
融資活動が提供する現金純額は2022年12月31日までの1年間で4530万ドルで、現金自動支払機による普通株売却の純収益3920万ドルと、2019年の従業員株式購入計画(ESPP)と株式オプション行使による普通株発行収益から610万ドルとなった。2021年12月31日までの1年間に、融資活動が提供した現金純額は2億663億ドルで、主に2021年3月に完成した引受公開発売から受け取った2.30億ドルの純収益、ATMで普通株を売却した2570万ドルの純収益、および特別引出権と株式オプションに基づいて普通株を発行した1060万ドルの収益から来ている
2020年12月31日までの年間で、融資活動が提供する現金純額は3830万ドルで、うち3370万ドルはATMで普通株を売却する純収益から、460万ドルはESPPと株式オプション行使によって普通株を発行する収益純額から来ている
資金需要
私たちが行っている活動に関連する費用は増加すると予想され、特に私たちが研究と開発を続け、臨床試験を行い、私たちの現在と未来の任意の候補製品のために市場承認を求める時に。また、現在または将来の任意の候補製品がマーケティング承認を得た場合、製品販売、マーケティング、製造、流通に関連した巨額の商業化費用が発生することが予想され、第三者との協力協定を締結することで、これらのコストを相殺することができる。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。もし私たちが必要な時に受け入れ可能な条件で資金を調達できない場合、あるいは新冠肺炎に関連するマクロ経済要素、ウクライナの持続的な衝突、インフレや金利上昇を含むが、資金を調達できない場合、私たちは研究開発計画や将来の商業化努力の延期、減少、または廃止を余儀なくされるだろう。私たちは、既存の現金、現金等価物、有価証券に加えて、2023年2月に普通株と事前融資権証を非公開で発行して調達した1.0億ドルに加えて、2026年下半期の運営費と資本支出需要に資金を提供できるようになると予想している。
2023年2月13日、私たちは既存の投資家と私募証券購入契約を締結し、1株35.35ドルで848,655株の私たちの普通株を売却して発行することに同意し、35.3499ドルの購入価格で1,980,198株の普通株の事前資本金権証(“予資権証”)を購入することに同意し、1部当たりの資本権証の行使価格は1株当たり0.0001ドルである。私たちはコストと私たちが支払うべき費用を提供する前に、合計約1.00億ドルの純収益を受けた。事前資金調達権証は原始発行後いつでも行使でき、失効することはない。彼らの条項によると、保有者の実益所有権総額が私たちの普通株発行済み株式と発行済み株式総数の9.99%を超えた場合、会社普通株株式を購入した発行済み事前融資承認持分証は一般的に行使できない。事前資本権証プロトコルに記載されているように、任意の株式配当および分割、資本再編、再編または同様の取引が発生した場合、予備資本金権証株式を行使する際に普通株を発行することができる1株当たりの行使価格および株式数が調整される可能性がある
私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは現在予想されているよりも早く私たちが利用できる資本資源を使用するかもしれない。私たちの将来の資本需要は多くの要素に依存します
•追加的な臨床と臨床前研究と将来の臨床試験を行う費用
•将来の製造コストは
•私たちが開発する可能性のある他の潜在的候補製品の発見、臨床前開発、実験室テストと臨床試験の範囲、進捗、結果、コスト
•私たちの候補製品に対する規制審査のコスト、時間、結果
•私たちが有利な条件で協力を確立し維持する能力があれば
•マイルストーンに到達したり、他の事態が発生したりして、私たちが当時達成する可能性のある任意の協力合意での支払いをトリガします
•将来の商業化活動のコストと時間は、製品販売、マーケティング、製造、流通を含み、私たちが市場の承認を得た任意の候補製品について
•もし私たちの候補製品が発売承認されたら、私たちの候補製品の商業販売から得られた収入(もしあれば)
•特許出願を準備し、提出し、起訴し、私たちの知的財産権を取得、維持し、実行し、知的財産権に関連するクレームを弁護するコスト;
•私たちは、経済制裁および/または米国と国際会社を敵視しているいくつかの国で特許出願を提出し、起訴し、私たちの知的財産権を獲得、維持、実行し、知的財産権に関連するクレームを弁護することができる
•私たちの業務運営と研究開発活動の拡大に伴い、私たちの従業員の増加と関連コスト
•新冠肺炎の大流行の影響(サプライチェーン中断を含む)或いは地政学的行動の影響、戦争(例えば現在のウクライナの武力衝突)を含むため、著者らの臨床前研究、開発計画及び現在と計画中の臨床試験は遅延する可能性がある
•インフレ、金利上昇、持続的な労働力不足を含む全体的な経済状況と傾向
•上場企業の運営コストとして。
これまで、相当な製品収入を生み出すことができれば、株式発行、債務融資、協力、戦略連合、許可手配の組み合わせで現金需要を満たす予定です。
重要な会計政策と重大な見積もり
経営陣の議論と分析は、米国公認会計原則に従って作成された我々の連結財務諸表に基づいている。これらの連結財務諸表を作成する際には、連結財務諸表で報告されている資産、負債、収入および費用、または資産および負債の開示に影響を及ぼす判断および推定を行う必要がある。我々は,歴史的経験,既知の傾向,事件,および当時の状況で合理的と考えられる様々な他の要因から推定した。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。持続的な基礎の上で、私たちは環境、事実、経験の変化に基づいて私たちの判断と推定を評価する。重大な改訂が予想される場合、その影響は推定変動の日から連結財務諸表に反映されることになる。
我々の重要会計政策は、本年度報告10−K表に含まれる総合財務諸表付記により詳細に記載されているが、総合財務諸表を作成する際に用いられる以下の会計政策には、最も重要な判断と推定が必要であると考えられる。
取引先と契約した収入
2022年12月31日まで、私たちの今までのすべての収入はAbbVie協定とJanssen協定から来ている。我々は、ASC 606、顧客との契約収入、またはASC 606に基づいて収入計算を行い、ASC 606は、エンティティが顧客との契約から生成された収入を計算し、特定の業界の指導を含む現在の収入確認指導の大部分を置換するための単一の統合モデルである。ASC 606関連会計ポリシーの詳細については、本年度報告書10−K表の他の部分の総合財務諸表付記2および12を参照されたい。
連結財務諸表作成過程の一部として、連携収入で確認する金額を決定するための試算を行う必要がある。特に,AbbVieやJanssenとの連携については,これまでに発生したコストとこれらの業績義務を果たすことが期待される総コストの割合から,研究サービスを提供する際に研究サービスの収入に起因することを確認した。余剰作業に関する見積りコストは判断し,収入確認に影響を与える必要がある。余剰作業に関する見積費用における不確実性は,主にこれらの費用推定数が新たな研究への期待努力に基づいており,科学的結果に部分的に依存しているためである。研究と科学成果は余剰履行義務を達成する時間と費用の大きさに影響する。そのため、見積もり費用を大きく改訂したため、収入が異なる時期に変動し、履行債務の進展測定基準が変化する可能性がある。 私たちがAbbVie協定に基づいて提供したサービスは2022年12月に完了した。私たちはAbbVieプロトコルと関連した追加的な収入を確認しないと予想される。2023年1月、Janssenは、その権利の行使を決定し、便宜上Janssen協定を終了することを決定したが、60日間の通知期間が必要であることを私たちに通知した。もし私たちとJanssenが同意すれば、Janssen協定は2023年3月またはそれ以上に終了するだろう。
研究と開発費用を計算すべきである
連結財務諸表作成過程の一部として、貸借対照表毎の日付までの計上費用を見積もる必要がある。この流れは未決済契約と購入注文の審査と私たちの
領収書を受け取っていない場合や他の方法で実際のコストを通知している場合には、スタッフに提供されているサービスを決定し、提供されたサービスレベルおよびサービスによって生じる関連コストを推定してください。私たちのほとんどのサービスプロバイダは毎月私たちが提供してくれるサービスや契約マイルストーンに達した時に私たちに借金の領収書を発行してくれます。私たちは、私たちが当時知っていた事実と状況に基づいて、各貸借対照表の日付までの課税費用を推定した。私たちは定期的にサービスプロバイダと私たちの推定の正確性を確認し、必要に応じて調整します。私たちの計算すべき研究開発費用の大きな見積もりには、私たちのサプライヤーが提供する研究開発活動に関連するサービスによるコストが含まれていますが、領収書は受け取っていません。場合によっては、私たちは将来提供されるサービスのために前払い費用を支払うつもりだ。これらの金額はサービス実行時に料金を計上します。
我々は、我々を代表して研究·開発を行っているサプライヤーとのオファーや契約に基づいて、受信したサービスや費用の見積もりに基づいて、研究·開発活動に関する費用を計算する。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われるお金は、提供されたサービスレベルを超え、研究開発費の前払いにつながる可能性があります。サービス料を計算する際に、サービスを提供する時間帯と各時間の努力度を見積もります。サービス実行の実際の時間または努力の程度が私たちの推定と異なる場合、私たちはそれに応じて計算すべきまたは前払い残高を調整します。将来の研究·開発活動のための貨物·サービスの前金は払い戻しできず、活動が行われた場合や貨物を受け取った場合には、支払い時ではなく費用を計上する。
発生した金額と実質的に差がないと予想されているにもかかわらず、提供されたサービスの状態および時間の推定が提供されたサービスの実際の状態および時間と異なる場合、これは、任意の特定の時期に報告された金額が高すぎたり、低すぎたりする可能性がある。今まで、このような費用の見積もりと発生した金額の間に実質的な差はありませんでした。
株式ベースの報酬
我々は、日持分奨励を付与する公正価値に基づいて従業員と非従業員の持分報酬を計量し、奨励に必要なサービス期間(通常は相応の奨励の獲得期間)内に直線的に基づいて持分報酬支出を確認する。私たちは罰金が発生しなかった時に確認します。
私たちは、通常株の公正価値と、予想される株価変動、予想される奨励期間、無リスク金利、および予想配当を含む主観的仮定を使用して、Black-Scholesオプション定価モデルを使用して、付与された株式オプションの公正価値を推定する。企業固有の履歴データが不足しているため、代表的な上場企業の歴史変動率に基づいて期待変動率を推定し、我々自身の履歴変動率に加えて、これらの会社の歴史情報も得ることができる。履歴変動率は,一般に期待期限仮定に見合った一定期間に基づいて計算される.私たちは単純化された方法を使用して従業員と取締役に付与されたオプションの期待期間を計算する。私たちがこの方法を使用したのは、予想期間を推定するための合理的な基礎を提供するために十分な履歴演習データがないからである。非従業員に付与されたオプションについて、私たちは契約条項を使用する。無リスク金利は、株式オプションの予想期限と一致する米国債ツールに基づいている。期待配当収益率はゼロと仮定しています。私たちは配当金を支払ったことがないので、今のところ私たちの普通株に何の配当も支払う計画もありません。
契約義務
二零二一年八月に、吾らはレンタル条項に基づいて既存のレンタル32,405平方尺オフィスと実験室空間から二零二五年五月までの一度の継続権利を行使し、本年報10-K表の他の部分の総合財務諸表付記6“レンタル”には将来のレンタル料が420万ドルであることを反映し、その他の追加レンタル料は2023年に30万ドルを支払うことになる。2022年12月31日現在、私たちの主要オフィスと実験室空間に対する契約義務は、2021年12月31日までの年次報告Form 10-Kで開示された義務と一致しています。
私たちは多くの第三者と契約を締結しました。その中には外部CROが含まれています。これらの契約は私たちに前払い金を要求します。その中のいくつかは返却できないかもしれません。第三者との様々な許可や関連協定により、記念碑的な支払いと印税を第三者に支払うことに同意しました。2018年に改訂された児童医療センター会社との独占ライセンス契約によると、協定が終了するまで毎年80,000ドルを支払う必要があります。また、ライセンス製品の初回規制承認による130万ドルまでの開発マイルストーン支払いを担当し、ライセンス契約でカバーされている製品の販売を実現した場合には、ライセンス製品の純売上高の下位桁の割合で等級別ライセンス使用料を支払い、中国移動権利の再許可による非ライセンス使用料収入の10%~20%を支払うことも担当する。中国移動に支払われた金額は、経営報告書に研究·開発費用と記されている。
シュレーディンガーとの協力協定によると、シュレーディンガーにいくつかの開発マイルストーンを支払い、目標に応じて総額の6桁以下の低報酬と、鉛最適化開始時に支払われる低6桁の報酬と、化合物ごとに支払われる310万ドルと、このような化合物を含む製品を販売する低い桁の特許権使用料を1つずつ支払うことが要求される可能性がある。さらに、私たちは、2019年に支払われる使い捨て費用100万ドルを含む、第三者から受け取った許可またはそのような化合物の採掘権をそのような第三者に譲渡することに関連するいくつかの支払いの中央値パーセントをSchrödingerに支払うことに同意した。
著者らは正常業務過程中に臨床前研究、臨床前と臨床供給及び製造サービスサプライヤー、専門家コンサルティング専門顧問及びその他の運営サービスサプライヤーと協定を締結した。私たちはこれらの支払いを上記の契約義務表に入れていません。これらの契約には最低調達約束が含まれていないので、いつでもキャンセルすることができます。通常は30日前に書面で通知された場合ですので、これらの合意の下でのキャンセル不可義務は実質的ではないと思います。
最近の会計公告
私たちは最近発表されたすべての基準を検討し、これらの基準が私たちの財務諸表に実質的な影響を与えないか、または私たちの業務に適用されないと判断した。
モニック·ホールディングス
連結財務諸表索引
| | | | | |
| ページ |
| |
独立公認会計士事務所報告(PCAOB ID:42) | F-1 |
合併貸借対照表 | F-2 |
合併経営報告書と全面赤字 | F-3 |
株主権益合併報告書 | F-4 |
統合現金フロー表 | F-5 |
連結財務諸表付記 | F-6 |
独立公認会計士事務所報告
Morphy Holding,Inc.の株主と取締役会へ。
財務諸表のいくつかの見方
添付のMorphy Holding,Inc.(当社)2022年12月31日と2021年12月31日までの総合貸借対照表、2022年12月31日までの年間ごとの関連総合経営報告書と全面赤字、株主権益と現金流量および関連付記(総称して“総合財務諸表”と呼ぶ)を監査しました。総合財務諸表は、すべての重要な面で、2022年12月31日、2022年12月31日、2021年12月31日までの会社の財務状況、および2022年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。 私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性に対する意見を表明するためではありません。 したがって、私たちはそのような意見を表現しない。
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
重要な監査事項とは、財務諸表を当期監査する際に生じる、監査委員会に伝達または要求された事項である:(1)財務諸表に対して重大な意義を有する勘定または開示に関連する;(2)私たちが特に挑戦的で、主観的または複雑な判断に関連する。 私たちは重要な監査事項が存在しないと確信する。
/s/ 安永法律事務所
2017年以来、当社の監査役を務めてきました。
ボストン、マサチューセッツ州
2023年2月23日
モニック·ホールディングス
合併貸借対照表
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
| | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 資産 | | | |
| 流動資産: | | | |
| 現金と現金等価物 | $ | 59,272 | | | $ | 171,434 | |
| 有価証券 | 288,976 | | | 236,701 | |
| 売掛金 | 455 | | | 2,307 | |
| 前払い費用と他の流動資産 | 13,479 | | | 7,892 | |
| 流動資産総額 | 362,182 | | | 418,334 | |
| 経営的リース使用権資産 | 3,514 | | | 4,806 | |
| 財産と設備、純額 | 2,119 | | | 2,583 | |
| 制限現金 | 560 | | | 560 | |
| その他の資産 | 214 | | | 7 | |
| 総資産 | $ | 368,589 | | | $ | 426,290 | |
| | | |
| 負債.負債 | | | |
| 流動負債: | | | |
| 売掛金 | $ | 3,475 | | | $ | 4,798 | |
| 費用を計算する | 13,181 | | | 12,838 | |
| 繰延収入,当期分 | 470 | | | 20,628 | |
| 流動負債総額 | 17,126 | | | 38,264 | |
| | | |
| 長期負債: | | | |
| 賃貸負債を経営し,当期分を差し引く | 2,344 | | | 3,838 | |
| 繰延収入,当期分を差し引く | — | | | 47,489 | |
| 総負債 | 19,470 | | | 89,591 | |
| | | |
| 引受金及び又は有事項(付記11) | | | |
| | | |
| 株主権益 | | | |
優先株、$0.0001額面は10,000,000株式を許可して違います。2022年12月31日と2021年12月31日までの発行済株式 | — | | | — | |
普通株、$0.0001額面は400,000,000株式を許可して38,584,6782022年12月31日までに発行·発行された株式と37,085,3972021年12月31日現在の発行済み株式と発行済み株 | 4 | | | 4 | |
| 追加実収資本 | 649,549 | | | 575,231 | |
| 赤字を累計する | (297,095) | | | (238,054) | |
| その他の総合損失を累計する | (3,339) | | | (482) | |
| 株主権益総額 | 349,119 | | | 336,699 | |
| 総負債と株主権益 | $ | 368,589 | | | $ | 426,290 | |
付記はこれらの連結財務諸表の構成要素である。
| | |
モニック·ホールディングス
合併経営報告書と全面赤字
(千単位で1株当たりおよび1株当たりのデータは含まれていない) |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | 十二月三十一日までの年度 |
| | | | | 2022 | | 2021 | | 2020 |
| 協力収入 | | | | | $ | 70,808 | | | $ | 19,794 | | | $ | 44,945 | |
| 運営費用: | | | | | | | | | |
| 研究開発 | | | | | 102,062 | | | 87,789 | | | 73,630 | |
| 一般と行政 | | | | | 32,142 | | | 27,811 | | | 18,495 | |
| 総運営費 | | | | | 134,204 | | | 115,600 | | | 92,125 | |
| 運営損失 | | | | | (63,396) | | | (95,806) | | | (47,180) | |
| その他の収入: | | | | | | | | | |
| 利子収入,純額 | | | | | 4,567 | | | 272 | | | 1,630 | |
| その他の費用、純額 | | | | | (145) | | | (8) | | | (19) | |
| その他の収入合計,純額 | | | | | 4,422 | | | 264 | | | 1,611 | |
| 所得税受益前の損失(準備金) | | | | | (58,974) | | | (95,542) | | | (45,569) | |
| 所得税から利益を得る | | | | | (67) | | | — | | | 570 | |
| 純損失 | | | | | $ | (59,041) | | | $ | (95,542) | | | $ | (44,999) | |
| | | | | | | | | |
| 1株当たり基本と希釈して純損失 | | | | | $ | (1.55) | | | $ | (2.67) | | | $ | (1.47) | |
| | | | | | | | | |
| | | | | | | | | |
| 加重平均発行済み普通株式、基本普通株式、希釈後普通株 | | | | | 38,112,498 | | | 35,797,969 | | | 30,594,897 | |
| | | | | | | | | |
| | | | | | | | | |
| 総合的な損失: | | | | | | | | | |
| 純損失 | | | | | $ | (59,041) | | | $ | (95,542) | | | $ | (44,999) | |
| その他の全面的な損失: | | | | | | | | | |
| 取引可能証券の未実現保有損失、税引き後純額 | | | | | (2,857) | | | (461) | | | (65) | |
| その他総合損失合計 | | | | | (2,857) | | | (461) | | | (65) | |
| 総合損失 | | | | | $ | (61,898) | | | $ | (96,003) | | | $ | (45,064) | |
付記はこれらの連結財務諸表の構成要素である。
| | |
モニック·ホールディングス
合併株主権益報告書
(単位:千、共有データを除く) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | 普通株 | | その他の内容
支払い済み
資本 | | 積算
赤字.赤字 | | 積算
他にも
総合収益(赤字) | | 合計する
株主の
権益 |
| 株 | | 金額 |
| 2019年12月31日の残高 | | 30,110,251 | | | $ | 3 | | | $ | 238,384 | | | $ | (97,513) | | | $ | 44 | | | $ | 140,918 | |
| 株式ベースの報酬費用 | | — | | | — | | | 11,053 | | | — | | | — | | | 11,053 | |
| 株式の帰属を制限する | | 252,890 | | | — | | | — | | | — | | | — | | | — | |
| 株式オプション行使時に普通株を発行する | | 431,554 | | | — | | | 3,532 | | | — | | | — | | | 3,532 | |
| 従業員の株式購入計画に基づいて普通株を発行する | | 85,298 | | | — | | | 1,112 | | | — | | | — | | | 1,112 | |
市場で普通株を発行することにより、発行コスト$を差し引く1.5百万 | | 1,157,693 | | | — | | | 33,646 | | | — | | | — | | | 33,646 | |
| 有価証券は保有損失を実現していない | | — | | | — | | | — | | | — | | | (65) | | | (65) | |
| 純損失 | | — | | | — | | | — | | | (44,999) | | | — | | | (44,999) | |
| 2020年12月31日残高 | | 32,037,686 | | | $ | 3 | | | $ | 287,727 | | | $ | (142,512) | | | $ | (21) | | | $ | 145,197 | |
| 株式ベースの報酬費用 | | — | | | — | | | 21,184 | | | — | | | — | | | 21,184 | |
| 株式の帰属を制限する | | 127,335 | | | — | | | — | | | — | | | — | | | — | |
| 株式オプション行使時に普通株を発行する | | 827,830 | | | — | | | 9,536 | | | — | | | — | | | 9,536 | |
| 従業員の株式購入計画に基づいて普通株を発行する | | 35,563 | | | — | | | 1,095 | | | — | | | — | | | 1,095 | |
市場で普通株を発行することにより、発行コスト$を差し引く1.2百万 | | 556,983 | | | — | | | 25,708 | | | — | | | — | | | 25,708 | |
二次発行で普通株を発行し、発行コストを差し引く$15.0百万 | | 3,500,000 | | | 1 | | | 229,981 | | | — | | | — | | | 229,982 | |
| 有価証券は保有損失を実現していない | | — | | | — | | | — | | | — | | | (461) | | | (461) | |
| 純損失 | | — | | | — | | | — | | | (95,542) | | | — | | | (95,542) | |
| 2021年12月31日の残高 | | 37,085,397 | | | $ | 4 | | | $ | 575,231 | | | $ | (238,054) | | | $ | (482) | | | $ | 336,699 | |
| 株式ベースの報酬費用 | | — | | | — | | | 29,048 | | | — | | | — | | | 29,048 | |
| 株式の帰属を制限する | | 9,171 | | | — | | | — | | | — | | | — | | | — | |
| 株式オプション行使時に普通株を発行する | | 459,839 | | | — | | | 5,175 | | | — | | | — | | | 5,175 | |
| 従業員の株式購入計画に基づいて普通株を発行する | | 30,271 | | | — | | | 885 | | | — | | | — | | | 885 | |
市場で普通株を発行することにより、発行コスト$を差し引く1.3百万 | | 1,000,000 | | | — | | | 39,210 | | | — | | | — | | | 39,210 | |
| 有価証券は保有損失を実現していない | | — | | | — | | | — | | | — | | | (2,857) | | | (2,857) | |
| 純損失 | | — | | | — | | | — | | | (59,041) | | | — | | | (59,041) | |
| 2022年12月31日の残高 | | 38,584,678 | | | $ | 4 | | | $ | 649,549 | | | $ | (297,095) | | | $ | (3,339) | | | $ | 349,119 | |
付記はこれらの連結財務諸表の構成要素である。
| | |
モニック·ホールディングス
統合現金フロー表
(単位:千) |
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 経営活動のキャッシュフロー: | | | | | |
| 純損失 | $ | (59,041) | | | $ | (95,542) | | | $ | (44,999) | |
| 純損失と経営活動で使用される現金純額の調整: | | | | | |
| 減価償却および償却 | 1,005 | | | 1,030 | | | 1,126 | |
| 有価証券の割増償却と割引増価 | 800 | | | 1,215 | | | 450 | |
| 株式ベースの報酬 | 29,048 | | | 21,184 | | | 11,053 | |
| 有価証券売却の損失 | 154 | | | — | | | — | |
| 設備処分損失 | — | | | 4 | | | — | |
| | | | | |
| 営業資産と負債の変動: | | | | | |
| 売掛金 | 1,852 | | | 5,007 | | | (3,847) | |
| 前払い費用と他の流動資産 | (5,529) | | | (4,035) | | | (767) | |
| その他の資産 | (207) | | | 59 | | | 75 | |
| 経営的リース使用権資産 | 1,292 | | | 1,098 | | | — | |
| 売掛金 | (1,553) | | | 970 | | | (1,071) | |
| 費用を計算する | 60 | | | 1,467 | | | 3,521 | |
| 収入を繰り越す | (67,647) | | | (14,821) | | | (11,466) | |
| 賃料を繰延する | — | | | — | | | (65) | |
| リース負債を経営する | (1,211) | | | (1,097) | | | — | |
| | | | | |
| 経営活動のための現金純額 | (100,977) | | | (83,461) | | | (45,990) | |
| 投資活動によるキャッシュフロー: | | | | | |
| 有価証券を購入する | (238,274) | | | (244,160) | | | (145,275) | |
| 有価証券満期日収益 | 175,042 | | | 132,000 | | | 154,000 | |
| 有価証券を売却して得た金 | 7,146 | | | — | | | — | |
| 財産と設備を購入する | (365) | | | (1,020) | | | (544) | |
| 投資活動が提供する現金純額 | (56,451) | | | (113,180) | | | 8,181 | |
| 資金調達活動のキャッシュフロー: | | | | | |
| | | | | |
| 従業員の株式購入計画に基づいて普通株を発行して得た金 | 885 | | | 1,095 | | | 1,112 | |
| 市場で発行された収益は,発行コストを差し引く | 39,210 | | | 25,700 | | | 33,653 | |
| 二次発行の収益は,発行コストを差し引く | — | | | 229,982 | | | — | |
| 株式オプション行使時に普通株で得られた金を発行する | 5,171 | | | 9,536 | | | 3,532 | |
| 融資活動が提供する現金純額 | 45,266 | | | 266,313 | | | 38,297 | |
| 現金、現金等価物、および制限的現金純増加 | (112,162) | | | 69,672 | | | 488 | |
| 期初現金、現金等価物、および限定現金 | 171,994 | | | 102,322 | | | 101,834 | |
| 現金、現金等価物、制限された現金、期末 | $ | 59,832 | | | $ | 171,994 | | | $ | 102,322 | |
| | | | | |
| 非現金活動: | | | | | |
| 売掛金と売掛金に掲げる財産と設備の購入 | $ | 176 | | | $ | — | | | $ | 11 | |
| 前払い費用及びその他の流動資産に含まれる株式オプション行使の金額 | $ | 4 | | | $ | — | | | $ | — | |
| 売掛金に含まれる発行コスト | $ | — | | | $ | — | | | $ | 7 | |
| 経営性リース負債と引き換えに使用権資産 | $ | — | | | $ | 4,434 | | | $ | — | |
| キャッシュフロー情報の追加: | | | | | |
| 税金の現金を納める | $ | 50 | | | $ | 170 | | | $ | 480 | |
付記はこれらの連結財務諸表の構成要素である。
連結財務諸表付記
1.業務的性質
組織と流動性
Moric Holding,Inc.(“会社”)は2014年8月にデラウェア州法律に基づいて設立された。同社はバイオ製薬会社であり,独自の知見をインテグリン薬に応用し,一流の経口小分子インテグリン療法を発見·開発している。インテグリンは1種類の有効な標的薬物であり、多種の許可された薬物は深刻な慢性病の治療に応用されている。生物製薬業界は大量の投資を行ったが、アメリカ食品と薬物管理局(FDA)はまだ小分子インテグリン療法の経口投与を許可していない。同社はインテグリン構造と生物学に対する独特な理解を利用して、Morphyインテグリン技術プラットフォーム或いはMINTプラットフォームを創立し、一連の新しい候補製品を開発し、効力、高選択性と経口投与に必要な薬物特性を実現することを目的とした。
同社は生物技術業界の早期会社によく見られるリスクと不確定要素の影響を受け、競争相手の新技術革新の開発、肝心な者への依存、ノウハウの保護、政府法規の遵守及び追加資本を獲得して運営に資金を提供する能力を含むが、これらに限定されない。現在開発されている候補製品は、広範な臨床前と臨床試験及び商業化前の監督管理承認を含む大量の追加的な研究と開発作業が必要となる。このような努力は多くの追加資本、十分な人員とインフラ、そして広範囲なコンプライアンス報告能力を必要とする。同社の薬物開発努力が成功しても,同社がいつ(あれば)製品販売から相当な収入を得るかは定かではない。同社は、予見可能な将来、運営が引き続き赤字になると予想している。同社は、その現金、現金等価物、および有価証券は、これらの財務諸表の発行日から少なくとも今後12ヶ月以内に、その運営費用および資本支出需要に資金を提供するのに十分であると予想している。
二零二年七月、当社はJefferies LLC(“Jefferies”)と市場発売計画について公開市場販売協定(“元合意”)を締結し、この計画により、当社は時々その普通株株式を適宜発売することができ、総発行価格は最高$に達する75.0ジェフリーを通じてその販売代理として配給株式と呼ばれている。その会社がJefferiesに支払った手数料は3.0元の合意によれば、Jefferiesによって販売された任意の配給株の総販売収益の%は、Jefferiesに慣用的な賠償および供給権も提供される。2021年8月11日、会社はジェフリーと元協定の第1号改正案を締結し、新たな市場発行(“新ATM”)を設立し、総発行価格は最高$に達する150.0百万ドルも払っています3.0ジェフリーを通じて販売された配給株は総販売収入のパーセントを占めています。新しいATM機によると、会社はいつでもジェフリーを介してその販売代理としてその普通株の株式を提供·売却することができ、配給株式と呼ばれる。
当社は2022年12月31日までに発行·販売します1,000,000純収益$の株式39.2発売手数料および発売費用を差し引いた後、当社は新ATM機による発売費用を支払います。2022年12月31日までに同社は97.2新しいATMによると、100万株の普通株が販売されている。
2021年3月,当社は引受の後続公開発行を完了した3,500,000普通株が一般公開されている価格は$です70.00一株ずつです。二次発行の総収益は約#ドルである245.0百万ドル、引受割引、手数料、その他の発行費用を差し引いて約$です15.0100万ドル、会社が支払い、純収益は約#ドル230.0百万ドルです。
2.列報根拠と重大な会計政策
陳述の基礎
連結財務諸表には、モルフェイホールディングス及びその完全子会社、モルフェイ治療会社、モルフェイ治療イギリス有限会社、モルフェイ安全会社の勘定が含まれる。2022年3月2日、同社はイギリスロンドン(以下、“イギリス”と称する)にモフィー治療イギリス有限会社を登録し、米国以外での会社の業務を支援する。すべての会社間残高はすでに合併中に販売された.
同等の総合財務諸表は、米国公認会計原則(“公認会計原則”)に基づいて作成されている。本付記内の適用指針に対するいかなる言及も、財務会計基準委員会(“FASB”)の会計基準編纂(“ASC”)及び“会計基準更新”(“ASU”)に掲載されている権威あるアメリカ公認会計原則を指す。
主要会計政策の見積もりと要約の使用
公認会計原則に基づいて財務諸表を作成することは、管理層に推定および判断を要求し、これらの推定および判断は、財務諸表の日に報告された資産および負債額、または有資産および負債の関連開示および報告期間内の収入および費用に関する報告に影響を与える可能性がある。これらの連結財務諸表に反映される重大な会計推定は、収入確認、計算すべき研究および開発費用、株式ベースの報酬推定値および所得税に関する推定を含むが、これらに限定されない。実際の結果はこれらの推定とは異なる可能性がある
上述したように、同社は2022年3月2日、米国以外での会社の機能を支援するために、イギリスロンドンにモフィー治療イギリス有限会社を登録設立した。その会社のすべての長期資産の地理的位置は依然としてアメリカだ。
モフィー治療イギリス有限会社の機能報告通貨はドルです。外貨再計量は会社の総合経営報告書の他の収入(費用)に計上される。
信用リスクと表外リスク集中
会社を集中的な信用リスクに直面させる可能性のある金融商品には、主に現金と現金等価物、有価証券、売掛金が含まれる。同社の現金と現金等価物は認められた金融機関に保管されており、金額は連邦保険限度額を超えている。当社は、商業銀行関係に関連する正常信用リスクを除いて、異常信用リスクの影響を受けないと考えている。
同社のポートフォリオの主な目標は資本の保存と流動性の維持だ。会社の投資政策は、資金を銀行口座の外に保有することを許可しているが、市場価値が確定しやすく、ドル建ておよび支払いが容易な固定収益ツールにしか投資できず、米国政府、米国政府が支持する企業、米国政府機関、格付けの高い会社債務、商業手形や社債、1940年の“投資会社法”第2 a-7条に基づいて登録された通貨市場基金を含む。通貨市場基金の投資は承認されたツールと一致しなければならず、管理されている資産は少なくとも#ドルでなければならない1.0十億ドルです。
売掛金は通常、ヤンソン製薬会社の売掛金を代表する。同社は経済状況を監視し、任意の売掛金が回収リスクに直面している可能性がある事実や状況を確定する。現在まで、同社は売掛金の受け取りに何の損失も出ていない。
当社には外為契約、オプション契約、その他の外国為替ヘッジ手配などの表外リスクはありません。
現金および現金等価物および限定現金
当社は購入満期日が三ヶ月以下の高流動性投資を現金等価物と見なしています。現金と現金等価物には、2022年12月31日現在、銀行当座預金、主に米国政府証券に投資される通貨市場基金、米国債が含まれる。2021年12月31日現在、現金および現金等価物には、米国政府証券に主に投資されている銀行当座預金と通貨市場基金が含まれている。現金等価物はコストごとに列報され、公正価値に近い。
制限現金には信用状を担保とした現金が含まれており、金額は#ドルです560,000事業主に2022年12月31日と2021年12月31日までの会社施設賃貸借契約を出す。それを担保にした信用状と現金が#ドルに増加した560,0002021年8月、付記6で経営リース延期がさらに検討されたため。信用状の条項は超えています1年. 次の表は、貸借対照表中の現金、現金等価物、および制限現金をキャッシュフロー表と照合する(千で計算)
| | | | | | | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 | | 2020 |
| 現金と現金等価物 | $ | 59,272 | | | $ | 171,434 | | | $ | 102,047 | |
| 制限現金 | 560 | | | 560 | | | 275 | |
| 現金総額、現金等価物、制限された現金 | $ | 59,832 | | | $ | 171,994 | | | $ | 102,322 | |
有価証券
同社は、米国政府債務証券、米国政府が支援する企業債務証券、購入日の元満期日が3カ月を超える会社債務証券に資金を投資し、有価証券としている。有価証券は販売可能に分類され、公正価値に基づいて入金される。当社は,売却可能な証券は,満期日が12カ月を超える証券を含め,現在の運営流動資金需要を支援するために利用可能であると考えているため,購入日満期日が3カ月を超えるすべての証券を流動資産に分類した。有価証券の公正価値変動は他の全面収益(損失)に計上され,有価証券としての未実現純収益(損失)である。同社は純損失#ドルが実現していないことを確認した2.9百万、$0.5百万ドルとドル0.12022年12月31日まで、2021年、2020年12月31日までの年度はそれぞれ100万ドル。
当社は、各報告日に、ASC 326における売却可能債務証券減価モデルに従って、売却可能債務証券確認のための公正価値が帳簿価値よりも低い任意の低下がクレジット損失に起因するかどうかを決定するために、売却可能債務証券を評価する。当社は合併経営報告書において信用損失と全面損失を他の費用純額内の信用損失費用に計上しているが、有価証券の公正価値と償却コストとの差額に限られている。現在まで、同社は売却可能な債務証券にいかなる信用損失も記録していない。
会社が債務証券を売却することができることに関する課税利息は前払い費用と他の流動資産会社の総合貸借対照表にあります。当社はすでに債務証券を売却できる公正価値と余剰コスト基準から計算すべき利息を除去し、任意の減価を識別し、計量することを選択した。資産が現金にならないと確定すると、会社は解約して利息を計算しなければなりません。計算すべき利息の任意のログアウトは、利息収入を打ち消すこと、信用損失費用を確認すること、または両者の組み合わせによって記録される。現在まで、同社はその有価証券に関連する利子を計上していない。
投資利息収入
同社は通貨市場基金と債務証券投資を売却できる利息収入を計算制で確認し、割増償却や逓増割引を含む。2022年、2021年及び2020年12月31日まで、当社は確認しました4.6百万、$0.3百万ドルとドル1.6利息収入はそれぞれ百万ドルです。
利子収入純額は他の収入、総合経営報告書、総合損失の純額に計上される。
財産と設備、純額
財産と設備はコストで入金される。財産と設備の使用寿命を延長する重大な更新または改築支出は資本化に記入し、維持·修理支出は発生した費用に記入する減価償却は関連資産の推定耐用年数内に直線的に計算される。財産と設備の減価償却は以下の通り
| | | | | | | | |
| | 使用寿命(年)を見込む |
| 実験室装置 | | 5 |
| コンピュータとソフトウェア | | 3-5 |
| 賃借権改善 | | 賃貸借年数または残存期間の短い者 |
賃貸借証書
当社は、ASC 842賃貸(“ASC 842”)に基づいてそのリース義務を計測し、この条項は、テナントに大多数のリース手配の使用権(“ROU”)資産と賃貸負債を確認することを要求する。当社は2021年1月1日からASC 842を採用し、改正された遡及移行法を採用し、発効日をその初適用日とした。したがって,2020年12月31日までの年度はASC 840賃貸借契約における先の指針に基づいて記載されており,これにより先の比較可能期間を再分類することはない.
手配開始時に、当社は手配に存在する独特の事実と状況に基づいて、その手配が賃貸契約であるかどうかを決定する。レンタル期間が1年を超えるリースは貸借対照表で純収益資産および短期·長期賃貸負債であることが確認された(場合によっては)。
当社は貸借対照表でオリジナル期限が1年以下であることを確認しない賃貸を選択しました。同社はレンタルスケジュールを評価する際には通常初期レンタル期間のみを含む。会社は更新を検討している
当社は、賃貸借期間の延長を合理的に決定した場合には、当該等の選択権を含む。
当社が発効日に作成した仮定は、何らかの事件(改訂賃貸借契約を含む)が発生した後に再評価されます。レンタル変更がテナントにオリジナルリースに含まれない追加使用権を付与し、レンタル支払いが追加使用権の独立価格に適合する場合、レンタル修正は別個の契約を生成する。1つのリース変更が別個の契約を生成した場合、その会計処理方式は新規契約と同じである。レンタル修正に単独の契約が生じていない場合は、契約修正に計上する。
契約修正は、レンタルの分類を再評価して、レンタルを経営するか融資リースを経営するかを決定することを会社に要求する。当社には融資リースは何もありません。
エンティティはレンタルと非レンタル構成要素を分離しないことを選択することができる。当社は賃貸と非リース構成要素を一緒に単一賃貸構成要素として会計処理することを選択しました。
当社は、レンタル期間内の将来の賃貸支払いの現在値に基づいて、移行日または契約修正日までのROU資産と賃貸負債を確認します。ASC 842は、金利が容易に決定できない限り、テナントがレンタル中の暗黙的な金利を使用することを要求し、その後、将来の最低レンタル支払いを割引するためにその増分借入金利(IBR)を使用することができる。同社の賃貸手配は隠れた金利を提供していないため、同社はそのIBRを使用して将来の最低賃貸支払いを割引している。当社は、その信用格付け、移行日まで、または修正日までの現在の経済情報、および決定されたリース期限に基づいてIBRを決定します。会社のROU資産や関連リース負債は会社IBRの変化に敏感ではない。
当社は、移行日または契約修正日が有効な同じIBRを用いて現在値で賃貸負債を再計測し、直線賃貸料支出に対してROU資産を調整することが予想される。
長期資産減価準備
長期資産は主に財産と設備で構成されている。保有·使用待ちの長期資産が,事件やビジネス環境の変化が発生して資産の帳簿価値が完全に回収できない可能性があることを示した場合には,回収可能テストを行う。当社がいつ減値審査を行うかを決定する際に考慮する要素は、業務パフォーマンスが予想に関連する重大な不良、業界或いは経済傾向の重大なマイナス影響、及び資産用途の重大な変化或いは計画変更を含む。長期資産の回収可能性を評価するために減値審査を行うと、当社は長期資産の使用と最終処分による未割引キャッシュフローの予測をその帳簿価値と比較する。1つの資産の使用が予想される推定未割引将来の現金流量がその帳簿金額よりも少ない場合には、減価損失が確認される。減価損失は、減価資産の帳簿価値がその公正価値を超え、割引現金流量に基づいて決定される。現在まで、当社は長期資産について何の減価損失も記録していない。
公正価値計量
ASCテーマ820“公正価値計量”(“ASC 820”)は、公正価値計量のためのツールのための公正価値レベルを構築し、市場データに基づく仮説(観察可能な投入)と会社自身の仮説(観察不可能な投入)とを区別する。観察できる投入は,市場参加者が当社以外のソースから得られた市場データに基づいて資産や負債を定価する際に使用する投入である。観察できない投入は、市場参加者が資産や負債の価格設定のために使用されるという会社の仮定を反映し、その時点で入手可能な最適な情報に基づいて制定される。ASC 820は、公正価値を交換価格または退出価格として決定し、市場参加者間の秩序ある取引において資産を売却するか、または負債を移動させることによって受信された金額を表す。公正価値計量において市場参加者の仮説を考慮する基礎として、ASC 820は以下の項目を区別した三級公正価値階層構造を構築した
第1レベル-同じ資産または負債の活発な市場見積もり。
第2レベル-第1レベル以外の直接または間接的に観察可能な投入、例えば見積市場価格、金利、収益率曲線。
第3レベル-会社が作成した仮説の見積りを用いた観察不可能な入力であり,これらの仮説は市場参加者が用いる仮説を反映している.
2022年12月31日現在、投資には、米国債、米国政府が支援する企業証券、および会社債務証券、社債および商業手形を含み、これらの証券の推定値は、最近の非アクティブ市場の証券取引に基づくか、同様のツールの見積市場価格および観察可能な市場データまたは観察可能な市場データから確認される他の重要な投入に基づいている。
ある程度、推定値は市場で観察または観察できないモデルや投入に基づいており、公正価値の決定にはより多くの判断が必要である。そのため、当社が価値を公平に判断する際に下した判断は、第3級に分類されたツールに対する判断度が最も高く、公正価値レベル内の金融商品レベルは、公正価値計量に大きな意味を持つ任意の投入の中で最低レベルに基づいている。
当社は、当社の総合金融商品の帳簿は、前払い支出及びその他の流動資産、売掛金、売掛金及び売掛金を含み、当該等のツールの短期的な性質のため、その帳簿価額は公正価値と一致すると信じている。
市場情報を細分化する
業務部門は、エンティティの構成要素として定義され、そのエンティティに関する個別離散情報は、首席業務意思決定者または意思決定グループが、リソースをどのように割り当てるかを決定し、業績を評価する際に評価することができる。会社の最高経営責任者は会社の経営意思決定者で、運営状況を見て会社の業務を管理します1つはアメリカとイギリスで運営されている運営部門
収入確認
これまで、当社とAbbVieおよびJanssenのすべての収入は、2018年10月に署名され、2022年12月に終了し、Janssenは2019年2月に署名し、2020年12月に改訂されました。ASC 606が当社とAbbVieおよびJanssenとの合意に適用される詳細については、以下の付記12を参照されたい。
会社は、まず、契約に基づいて各当事者のリスク、報酬、および活動を手配し、そのスケジュール(またはスケジュールの一部)がASCテーマ808の“協力スケジュール”に適合する協力スケジュールを代表するかどうかを決定するために協力スケジュールを評価する。ASC 808によると、当社は契約中に顧客関係ではなく協力手配とみなされる任意の協力手配や要素を計算する。2022年12月31日までに会社は二つ--AbbVieおよびJanssenとのプロトコル--ASC 606に従って入金されました。
ASC 606によれば、エンティティは、そのクライアントが約束された商品またはサービスの制御権を取得したときに収入を確認し、その金額は、エンティティがこれらの商品またはサービスと交換するために予期される対価格を反映する。ASC 606の範囲内で確認すべき適切な収入額が決定されたことを決定するために、会社は、(I)顧客との契約を決定するステップ、(Ii)契約で約束された貨物またはサービスを決定し、約束された貨物またはサービスが履行義務であるかどうかを決定するステップ、(Iii)取引価格を測定するステップ、(Iv)取引価格を履行義務に割り当てるステップ、および(V)会社が各履行義務を履行するときに収入を確認するステップの5つのステップを実行する。エンティティが顧客に譲渡された商品またはサービスと交換するために獲得する権利のある対価格を受け取る可能性がある場合にのみ、会社は5ステップモードを契約に適用する。
以下のすべての基準を満たす場合、当社が顧客と締結する契約は、ASC 606の範囲に属する:(I)手配は当事者の承認を得ており、各当事者はそれぞれの義務を履行することを承諾し、(Ii)譲渡する商品またはサービスに関する各当事者の権利を決定することができ、(Iii)譲渡する商品またはサービスの支払い条件を決定することができる。(Iv)この手配は、商業的実質を有し、(V)顧客に譲渡された商品またはサービスと交換するために、当社が請求する権利のある実質的にすべての代価を受け取る可能性がある。
履行義務は、契約において約束された独自の商品又はサービスを顧客に移転する義務である。約束された貨物またはサービスは、(1)顧客が単独でまたは他の既製の資源と共に貨物またはサービスから利益を得ることができ、(2)約束された貨物またはサービスを契約内の他の約束とは別に識別することができる場合とは異なると考えられる。追加商品又はサービスの選択権を購入することはマーケティング要約とみなされ、顧客がこのような選択権を選択する際に個別の契約として入金され、会社が選択権が契約を締結していない場合には提供されない実質的な権利を提供しているとみなされない限り、提供されない。しかしながら、オプションが契約が締結されていない場合に提供されない実質的な権利を提供すると判定された場合、取引価格の一部はオプションに割り当てられる。
当社は譲渡契約で約束された貨物またはサービスにより予想される対価格金額に基づいて取引価格を推定します。対価は固定対価格と変動対価を含むことができます。可変価格を含む各手配の開始時に、会社は潜在的な支払いの金額と支払いを受ける可能性を評価する。当社は最高額法または期待値法を用いて取引価格を推定し、どの方法によって予想される対価格額を予測することができる。大きな収入逆転が起こらない可能性が高い場合、可変対価は取引価格に含まれる。
当社はまた、顧客支払い時間と義務履行時間とが一致しない場合に融資要因が含まれているか否かを評価し、融資要因の影響に応じて取引価格(あれば)を調整する。
その会社の顧客との取引には開発と規制マイルストーン支払いが含まれるかもしれない。当社はマイルストーンが達成可能とされているかどうかを評価し、可能な金額法を用いて取引価格に計上される金額を推定しています。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。規制承認のような会社や顧客統制の範囲内での一里塚的な支払いは、承認を受ける前に実現可能であるとは考えられない。各報告期間終了時に、当社は、このようなマイルストーンを実現する可能性や任意の関連制限を再評価し、必要に応じて全体の取引価格の見積もりを調整します。いずれの調整も累積追跡に基づいて記録されており,これは調整期間中の連携収入や純収入(損失)に影響を与える.
販売ベースの特許使用料の場合、販売レベルに基づくマイルストーン支払いを含む場合、会社は、特許使用料に関連する唯一または主要な項目がライセンスであると判断する。ライセンスが販売に基づく特許使用料に関連する唯一または主要な項目である場合、当社は、(I)関連販売が発生した場合、または(Ii)特許使用料の一部または全部が割り当てられた履行義務が履行された(または部分的に履行された)とき、以下の遅い時間に収入を確認する。
当社は履行責任が確認された推定独立販売価格に基づいて取引価格を分配します。会社は仮説を立てなければならず,契約で決定された契約義務ごとの独立販売価格を決定する必要がある。同社は、他の比較可能な取引、交渉取引において考慮される価格、および推定コストを含む可能性がある独立販売価格を決定するためにキー仮説を使用する。可変対価格の条項が義務の履行状況に関連している場合、ある可変対価格は、契約のうちの1つまたは複数の履行義務に専用的に割り当てられ、各履行義務に割り当てられた結果の金額は、会社が各履行義務から得られると予想される金額と一致する。
当社は、各履行義務がある時点又は一定時間以内に履行された場合に、それぞれの履行義務に割り当てられた取引価格の金額を収入として確認する。特に,会社とAbbVieやJanssenとの協力については,終了前にこれまでに発生したコストにより,研究サービスの収入によりこれらのサービスが提供されていることが確認された。
その会社は各契約で決定された請求書スケジュールに基づいて顧客から支払いを受ける。前払い金及び費用は、当社が当該等の手配の義務を履行するまで、受領又は満了時に繰延収入と記す。会社の対価格権利が無条件である場合,金額は売掛金として記録される.
研究と開発費
研究及び発展支出はすでに発生した時に計上され、研究及び発展活動を行うことによるコスト、研究及び発展人員の報酬関連支出、臨床前及び臨床活動(供給コストを含む)、間接費用(施設費用、材料及び供給品を含む)、コンサルタント及び外部サービス供給者に支払う金額、及び設備減価償却を含む。技術的可能性や将来的に代替用途がない取得技術に関する前期ライセンス支払いも研究·開発費に含まれている。
契約と開発コスト及び対策プロジェクトを検討する
当社は各種研究開発サービス手配を締結しており、これらの手配に基づき、サプライヤーが当社を代表して各種サービスを提供しています。当社はその手配による見積もりコストの計上費用を記録しています。計算すべき費用の十分性を評価する際に、会社は、受信された請求書および契約費用を含む研究、試験、または他のサービスの進捗状況を分析する。各報告期間終了時の課税費用残高を決定した場合に判断と推定を行う。
株式ベースの報酬
会社は、取締役を含む特定の従業員および非従業員に株式オプション、制限普通株および制限株式単位を発行する。当社は、付与日公正価値計算制限普通株と制限株式単位費用に基づいて、公正価値は通常、付与日会社普通株の市場価格であり、付与に必要なサービス期間(通常は帰属期間)内で直線的に確認する。会社の株式オプション補償費用は,付与日をもとに,ブラック·スコアーズオプション定価モデルにより決定された各報酬の公正価値計算により,奨励に必要なサービス期間(通常は授権期間)内で直線的に確認される。会社は株式に基づく報酬費用を
総合業務表と総合損失表は,受賞者の給料と関連費用を同様に分類したり,受賞者のサービス支払いを分類したりする.会社は没収行為が発生した場合に確認します。
詳細は注9を参照されたい。
総合損失
総合損失とは、企業が一定期間内に非所有者源の取引とその他の事件と状況によって発生した権益変化である。総合損失には当期純損失と累積その他総合損失の変動が含まれる。2022年、2021年、2020年12月31日まで、総合損失はドルを含む2.9百万、$0.5百万ドルとドル0.1それぞれ未実現持株損失、取引可能証券純額である。
所得税
設立以来、同社は、資産および負債方法を使用して繰延税金を規定するFASB ASC主題740−所得税(“ASC 740”)に従って所得税を記録した。この方法によれば、繰延税金資産および負債は、資産および負債の財務諸表基準と課税基準との間の一時的な差異および純営業損失と信用繰越との間の一時的な差に基づいて決定され、予想差を使用して償却された年間の現行税率が適用される。税率変動が繰延税金資産や負債に及ぼす影響は、公布日を含む期間の収入で確認されている。一部の繰延税金資産が現金化できない可能性が高い場合には、推定免税額が確立される。
会社は財務諸表で確認された所得税の不確実性を計算し、確認すべき税収割引額を決定するために2ステップ法を採用した。まず、税務機関が外部審査後にこのような状況を維持する可能性を決定するために、税収状況を評価しなければならない。税務状況がより継続する可能性があると考えられる場合、税務状況は、財務諸表で確認される利益金額を決定するために評価される。確認可能な利益額は、最終和解時に50%以上の実現可能性がある最大額である。2022年12月31日、2022年12月31日および2021年12月31日まで、当社には重大な不確定税務状況は見られませんでした。
後続事件
当社は、総合財務諸表において確認または開示を可能にするために、貸借対照表の日後であるが、総合財務諸表の発表前に発生したイベントまたは取引を考慮している。これらの連結財務諸表が印刷された日前に、連結財務諸表において確認または開示するために、後続のイベントが評価された。
当社は2023年2月13日に既存投資家と証券購入協定を締結し、これにより、当社は私募での販売及び発行に同意しました848,655普通株の価格は$です35.351株および前払い資金引受権証1,980,198普通株、買い取り価格は$35.3499事前資本金権証であり、その中で各資本金権証の行使価格は$である0.0001(“事前出資株式証”)。同社が受け取った純収益総額は約#ドルだった100.0当社が支払うべきコストと提供費用を差し引く前に、当社が支払うべき費用。事前資金調達権証は原始発行後いつでも行使でき、失効することはない。彼らの条項によると、所有者の総実益所有権が9.99この権力を行使した後、会社の普通株は発行され、発行された株式総額の割合。事前計画資承認株式を行使する時に普通株の1株当たり使用価格及び配当数を発行することができ、事前計画資承認株式証に記載されている任意の配当金及び分割、資本再編、再編或いは類似取引が発生した場合、調整しなければならない。
3.金融資産と負債の公正価値
次の表は,2022年12月31日までと2021年12月31日までの公正価値で恒常的に計量された会社金融資産の情報(千単位)を提供し,公正価値レベルにおける計量ごとの分類レベルを示している。
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | |
| 2022年12月31日の公正価値計測 |
| 合計する | | レベル1 | | レベル2 | | レベル3 |
| 資産: | | | | | | | |
| 現金等価物 | $ | 58,814 | | | $ | 38,942 | | | $ | 19,872 | | | $ | — | |
| 有価証券: | | | | | | | |
| アメリカ国債 | 177,932 | | | — | | | 177,932 | | | — | |
| アメリカ政府が支援する企業証券 | 12,396 | | | — | | | 12,396 | | | — | |
| 商業手形 | 9,860 | | | — | | | 9,860 | | | — | |
| 社債 | 88,788 | | | — | | | 88,788 | | | — | |
| | | | | | | |
| | | | | | | |
| 総資産 | $ | 347,790 | | | $ | 38,942 | | | $ | 308,848 | | | $ | — | |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | |
| 2021年12月31日の公正価値計測 |
| 合計する | | レベル1 | | レベル2 | | レベル3 |
| 資産: | | | | | | | |
| 現金等価物 | $ | 171,142 | | | $ | 171,142 | | | $ | — | | | $ | — | |
| | | | | | | |
| 有価証券: | | | | | | | |
| アメリカ国債 | 16,212 | | | — | | | 16,212 | | | — | |
| 商業手形 | 99,898 | | | — | | | 99,898 | | | — | |
| 社債 | 120,591 | | | — | | | 120,591 | | | — | |
| 総資産 | $ | 407,843 | | | $ | 171,142 | | | $ | 236,701 | | | $ | — | |
現金等価物には、2022年12月31日現在の通貨市場基金と米国債、および2021年12月31日現在の通貨市場基金が含まれる。上の表に含まれている通貨市場基金は市場見積もりで評価されたアメリカ政府証券に投資しています。したがって、通貨市場基金は2022年12月31日と2021年までの1段階に分類される。以上の表に含まれる取引可能証券には、米国債、米国政府が支援する企業証券、商業手形、社債が含まれており、これらの証券は2022年12月31日現在、2021年12月31日現在2級証券に分類されている。2022年12月31日と2021年12月31日までの年度内に、公正価値階層構造カテゴリ間で移行する資産はない。その会社は所有している違います。2022年12月31日と2021年12月31日に公正価値で恒常的に計量された負債。
4.有価証券
次の表は、販売可能な有価証券に分類された同社への投資(千計)をまとめた
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 2022年12月31日まで |
| 成熟性 | | 償却する
コスト | | 毛収入
実現していない
収益を保有する | | 毛収入
実現していない
持株損失 | | 骨材
見込みを立てる
公正価値 |
| 有価証券: | | | | | | | | | |
| アメリカ国債 | 1年もたたないうちに | | $ | 179,854 | | | $ | — | | | $ | (1,922) | | | $ | 177,932 | |
| アメリカ政府が支援する企業証券 | 2年以内 | | 12,500 | | | — | | | (104) | | | 12,396 | |
| 商業手形 | 1年もたたないうちに | | 9,860 | | | — | | | — | | | 9,860 | |
| 社債 | 2年以内 | | 90,076 | | | — | | | (1,288) | | | 88,788 | |
| | | | | | | | | |
| | | | | | | | | |
| 有価証券総額 | | | $ | 292,290 | | | $ | — | | | $ | (3,314) | | | $ | 288,976 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 2021年12月31日まで |
| 成熟性 | | 償却する
コスト | | 毛収入
実現していない
収益を保有する | | 毛収入
実現していない
持株損失 | | 骨材
見込みを立てる
公正価値 |
| 有価証券: | | | | | | | | | |
| アメリカ国債 | 1年もたたないうちに | | $ | 16,224 | | | $ | — | | | $ | (12) | | | $ | 16,212 | |
| 商業手形 | 1年もたたないうちに | | 99,900 | | | — | | | (2) | | | 99,898 | |
| 社債 | 2年以内 | | 121,034 | | | — | | | (443) | | | 120,591 | |
| 有価証券総額 | | | $ | 237,158 | | | $ | — | | | $ | (457) | | | $ | 236,701 | |
当社のすべての投資は売却可能なものに分類され、公正価値に基づいて計算され、損益計上は他の全面的な損失を累積する構成要素に計上されていない。当社は,満期日が12カ月を超える証券を含むすべての売却可能な証券は,現在の運営流動資金需要をサポートできると考えているため,売却可能なすべての証券を流動資産に分類している。
当社は、上記投資が2022年12月31日および2021年12月31日までの年度の信用リスクに大きな変化はないと認定している。したがって、信用損失準備金は確認されなかった。当社は2022年12月31日現在、このような証券を売却するつもりはなく、当社が償却コストを回収した上でこれらの証券を売却することを要求される可能性は低い。
同社が債務証券を売却できる受取利息総額は#ドルである1.4百万ドルとドル1.2それぞれ2022年12月31日と2021年12月31日まで。
5. 財産と設備、純額
財産·設備の純額には、2022年12月31日と2021年12月31日まで、以下が含まれる(千計)
| | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 実験室装置 | $ | 6,094 | | | $ | 5,608 | |
| コンピュータとソフトウェア | 596 | | | 546 | |
| 賃借権改善 | 611 | | | 606 | |
| | | |
| 7,301 | | | 6,760 | |
| 減算:減価償却累計と償却 | (5,182) | | | (4,177) | |
| $ | 2,119 | | | $ | 2,583 | |
減価償却と償却費用は#ドルです1.0百万、$1.0百万ドルとドル1.12022年12月31日まで、2021年、2020年12月31日までの年度はそれぞれ100万ドル。
6. 賃貸借証書
同社はマサチューセッツ州ウォルザムにある会社本社のためにレンタル手配を締結した。2021年8月に、当社は既存のレンタルの一次継続権を行使します32,405レンタル条項によると、2025年5月までのオフィスと実験室空間は平方フィートです。既存の賃貸の一次延長権を行使して単独賃貸入金としての資格を満たしていないため、当社は一度の延長権を行使して契約修正入金とします。
当社は、改訂された条項及び条件及び当該日までの事実及び状況に基づいて、改訂発効日までの会社本社の借約種別を再評価します。これには,対象資産を用いたその日の残存経済寿命,その日レンタルの割引率,およびその日契約における残存対価格の再計量と再分配が含まれる。当社は,このリースは引き続き経営リースに分類され,改正発効日までのIBRを再評価し,リース負債とROU資産を再計測したと結論した。
貸借対照表情報を補完する
補足経営リース貸借対照表情報の概要は以下の通り(千計)
| | | | | | | | |
| | 2022年12月31日 |
| 経営的リース使用権資産 | | $ | 3,514 | |
| | |
費用を計算する(1) | | $ | 1,494 | |
| リース負債を経営する | | 2,344 | |
| リース負債総額を経営する | | $ | 3,838 | |
(1)賃貸負債を経営する短期部分は総合貸借対照表の課税費用に計上される。
その他補足資料
その他の補足経営リース情報は以下のようにまとめられている(年と割引率を除いて、千計)
| | | | | | | | |
| | 2022年12月31日までの年度 |
| レンタル負債の金額を計上するために支払った現金 | | $ | 1,507 | |
| 加重平均残余レンタル期間 | | 2.4年.年 |
| 加重平均割引率 | | 6.5 | % |
賃貸承諾の満期日
2022年12月31日までの経営賃貸約束期限は以下の通り(千単位)
| | | | | | | | |
| 十二月三十一日までの年度 | | 合計する |
| 2023 | | $ | 1,700 | |
| 2024 | | 1,732 | |
| 2025 | | 728 | |
| | |
| 賃貸支払総額 | | $ | 4,160 | |
| 差し引く:推定利息 | | 322 | |
| リース負債現在価値を経営する | | $ | 3,838 | |
当社は、2022年、2021年および2020年12月31日までの年間経営リース支出を確認しました1.6百万、$1.3百万ドルとドル1.1それぞれ100万ドルです
7. 費用を計算する
12月31日、2022年、2021年までの費用には、以下の内容が含まれています(千計)
| | | | | | | | | | | |
| | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 給与明細及び関連費用 | $ | 6,569 | | | $ | 6,396 | |
| 研究と開発活動 | 4,265 | | | 4,268 | |
| 賃貸負債の当期部分を経営する | 1,494 | | | 1,211 | |
| その他の費用 | 853 | | | 963 | |
| $ | 13,181 | | | $ | 12,838 | |
8. 株主権益
普通株
普通株式は以下のような特徴を持つ
投票する.
普通株保有者には権利がある1つは保有するすべての普通株に投票する。
配当をする
もし会社の取締役会が発表した場合、普通株式保有者は配当金を得る権利がある。発行された優先株のすべての未支払配当金がその条項に従って支払われる前に、普通株式保有者に現金配当金を支払うことを宣言したり、または支払ってはならない。2022年と2021年12月31日までに違います。優先株が際立っている違います。普通株式発行以来、会社は普通株式保有者に配当金を発表または支払いしてきた。
清算する
普通株式保有者は、会社の清算又は解散の場合には、任意の優先株主よりも優先的な清算優先を含む現金分配を得る権利がある。任意の優先株株主がそれぞれの優先株割当を受けた後、任意の残りの分配すべき資産は、換算に基づいて決定された優先株または普通株の保有者に分配されなければならない。
9. 株式ベースの報酬
会社の2019年7月の初公開について、会社は2019年6月に2019年持分インセンティブ計画(2019年予定)を採択し、2018年の株式インセンティブ計画に代わりました。取締役会は2022年4月27日に改訂及び再予約された2019年株式激励計画(“A&R 2019計画”及び2019年計画“2019年計画”)を採択し、2022年6月8日に当社の株主の承認を得て、この計画に基づいて当社の非従業員取締役に付与できる年間報酬総額を改訂することができる。A&R 2019では、会社役員、上級管理者と従業員、および会社コンサルタントやコンサルタントに株式オプション、制限株式配当、株式配当金、現金奨励、株式付加価値権、制限株式単位、業績奨励を付与する計画です。2019年計画の自動増資により、A&R 2019計画で発行可能な普通株式数が増加しました1.52022年1月は100万株。2022年12月31日までに1.82019年の計画によると、将来の奨励金に利用可能な株は100万株。
会社は総合経営報告書と総合赤字報告書の中で奨励タイプ別に株式に基づく補償費用を確認し、具体的には以下の通り(単位:千)
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | 十二月三十一日までの年度 | | |
| | | | | 2022 | | 2021 | | 2020 | | | | |
| 株式オプション | | | | | $ | 25,828 | | | $ | 19,894 | | | $ | 9,418 | | | | | |
| 制限普通株 | | | | | 6 | | | 283 | | | 955 | | | | | |
| 制限株式単位 | | | | | 2,764 | | | 403 | | | 187 | | | | | |
| 従業員株購入計画 | | | | | 450 | | | 604 | | | 493 | | | | | |
| 合計する | | | | | $ | 29,048 | | | $ | 21,184 | | | $ | 11,053 | | | | | |
統合業務表と総合損失表における権益に基づく報酬費用の分配状況(単位:千):を費用別にまとめた
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | 十二月三十一日までの年度 | | |
| | | | | 2022 | | 2021 | | 2020 | | | | |
| 研究開発費 | | | | | $ | 13,518 | | | $ | 9,866 | | | $ | 6,114 | | | | | |
| 一般と行政費用 | | | | | 15,530 | | | 11,318 | | | 4,939 | | | | | |
| 合計する | | | | | $ | 29,048 | | | $ | 21,184 | | | $ | 11,053 | | | | | |
制限普通株
次の表は、2022年12月31日までの年間制限普通株活動をまとめたものである
| | | | | | | | | | | |
| 株式数 | | 重みをつける
平均公平である
1株当たりの価値
発行時に |
| 2021年12月31日現在の非帰属制限普通株式 | 2,171 | | | $ | 4.32 | |
| 授与する | — | | | — | |
| 既得 | (2,171) | | | 4.32 | |
| 没収される | — | | | — | |
| 2022年12月31日現在の非帰属制限普通株式 | — | | | $ | — | |
2022年12月31日までに会社は違います。制限された普通株式に関連する未確認持分補償費用。2022年,2021年および2020年12月31日までの年間で,帰属の制限的普通株奨励の総公平価値は約$である0.1百万、$5.0百万ドルと$5.6それぞれ100万ドルです
限定株単位
次の表は、2022年12月31日までの年間限定株式単位活動をまとめています
| | | | | | | | | | | |
| 株式数 | | 重みをつける
平均公平である
1株当たりの価値
発行時に |
| 2021年12月31日現在の未帰属制限株式単位 | 7,000 | | | $ | 57.73 | |
| 授与する | 291,970 | | | 43.98 | |
| 既得 | (7,000) | | | 57.73 | |
| 没収される | (42,880) | | | 44.75 | |
| 2022年12月31日現在の未帰属制限株式単位 | 249,090 | | | $ | 43.85 | |
2022年12月31日現在、会社の未確認株式報酬支出は$8.3限定株式単位に関する百万ドルは、#年加重平均期間中に確認される予定です3.1何年もです。2022年12月31日までおよび2021年12月31日までの年間帰属制限株式単位の総公平価値は約$である0.2百万ドルとドル2.1それぞれ100万ドルです
株式オプション
下表は、2022年12月31日までの年間における会社の株式オプション活動をまとめたものである
| | | | | | | | | | | | | | | | | | | | | | | |
| 量
株 | | 重みをつける
平均値
行権価格 | | 重みをつける
平均値
残り
契約条項 | | 骨材
内在的価値 |
| | | | | (単位:年) | | (単位:千) |
| 2021年12月31日現在の未返済債務 | 4,781,565 | | | $ | 20.03 | | | | | |
| 授与する | 1,498,918 | | | 39.70 | | | | | |
| 鍛えられた | (459,839) | | | 11.26 | | | | | |
| 没収または期限切れ | (557,091) | | | 33.86 | | | | | |
| 2022年12月31日現在の未返済債務 | 5,263,553 | | | $ | 24.94 | | | 7.60 | | $ | 38,525 | |
| 2022年12月31日までに行使可能なオプション | 2,990,254 | | | $ | 19.19 | | | 6.96 | | $ | 32,522 | |
次の表は、2022年、2022年、2021年、2020年12月31日までの年度内に付与、付与、行使された株式オプションのいくつかの情報を提供しており、単位は千であり、オプションごとの価値は除く
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 重み付け-オプションごとに付与されたオプションの平均公正価値 | $ | 28.88 | | | $ | 26.16 | | | $ | 10.98 | |
| 株式オプションを行使して受け取った現金総額 | $ | 5,175 | | | $ | 9,536 | | | $ | 3,532 | |
| 株式オプションの総内在時価を行使する | $ | 8,507 | | | $ | 39,335 | | | $ | 7,752 | |
次の表は,2022年,2022年,2021年,2020年12月31日終了年度に付与されたオプションの公正価値を決定する際に用いる重み付き平均仮定をまとめたものである
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 無リスク金利 | 2.02% | | 1.20% | | 0.46% |
| | | | | |
| 予想期限(年単位) | 6.0年.年 | | 6.0年.年 | | 6.0年.年 |
| 予想変動率 | 86.81% | | 87.09% | | 80.85% |
オプション付与の期待期限は、当社が期待期限を得るのに十分な履歴情報がないため、簡略化方法によって決定される。この方法では、従業員と取締役株式オプションの期待期間は、行権期間と契約期間の加重平均との間の中間点である。当社は代表的な上場企業の歴史変動率に基づいてオプション付与の変動率を決定し、当社自身の履歴変動率に加えて、これらの会社は歴史情報を得ることができる。履歴変動率は,一般に期待期限仮定に見合った一定期間に基づいて計算される.無リスク金利はゼロ金利米国債ツールに基づいており、その条項は株式オプションの期限と一致している。当社はまだ支払っておらず、普通株の現金配当金の支払いも期待されていません。そのため、期待配当率はゼロと仮定しています。
2022年12月31日現在、会社の未確認株式報酬支出は$50.0従業員と非従業員に発行された株式オプションに関する百万ドルは、加重平均期間中に確認される予定です2.2何年もです。
従業員株購入計画
2019年、会社は“2019年従業員株購入計画”を採択し、2019年6月26日に発効した。その会社は最初に保留した300,000ESPPによって販売されている普通株です。ESPPの自動増額により,ESPPによって発行可能な普通株式数が増加した0.42022年1月1日、百万株。国税法第423条によると,ESPPはほぼすべての従業員に最高可達$の購入を提供する適格な補償計画である25,000毎年普通株の15割引率は発売期初め価格または発売期末価格の中で低い者である。
ESPP割引により購入した補償費用は,Black−Scholesモデルを用いて回顧準備金と購入割引の公正価値を計算し,要件期間内に補償費用として確認した。
10. 所得税
2022年12月31日、2021年12月31日、2020年12月31日までの年度所得税前損失分(千計)は以下の通り
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| アメリカです。 | $ | (59,036) | | $ | (95,542) | | $ | (45,569) |
| 外国.外国 | 62 | | — | | — |
| 合計する | $ | (58,974) | | $ | (95,542) | | $ | (45,569) |
2022年12月31日、2021年12月31日、2020年12月31日までの年間所得税(福祉)支出の構成要素(千単位)は以下の通り
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 現在: | | | | | |
| 連邦制 | $ | — | | $ | — | | $ | (511) |
| 状態.状態 | 55 | | — | | (59) |
| 外国.外国 | 12 | | — | | — |
| 当期税収総額 | 67 | | — | | (570) |
| 延期: | | | | | |
| 連邦制 | — | | — | | — |
| 状態.状態 | — | | — | | — |
| 外国.外国 | — | | — | | — |
| 繰延税金(福祉)を総額に充てる | — | | — | | — |
| 所得税(給付)総額 | $ | 67 | | $ | — | | $ | (570) |
実際の所得税率は、連邦法定税率を会社所得税前の損失に適用して計算した金額とは異なり、以下のようになる
| | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 法定税率で課税する | 21.00 | % | | 21.00 | % | | 21.00 | % |
| 州税 | 7.84 | | | 9.31 | | | 8.38 | |
| 株式ベースの報酬 | (1.02) | | | 5.33 | | | 0.76 | |
| 差し引かれない費用 | (3.87) | | | (0.01) | | | (0.01) | |
| 連邦研究開発信用 | 6.83 | | | 3.34 | | | 6.21 | |
| 評価免除額を変更する | (30.89) | | | (38.97) | | | (35.09) | |
| (0.11) | % | | — | % | | 1.25 | % |
繰延税金資産と負債には、2022年12月31日と2021年12月31日まで、以下の内容が含まれています(千単位)
| | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 繰延税金資産: | | | |
| 純営業損失が繰り越す | $ | 49,969 | | $ | 44,269 |
| 資本化研究と実験支出 | 22,147 | | — |
| 研究開発信用繰り越し | 17,101 | | 10,843 |
| 株式ベースの報酬 | 5,649 | | 4,958 |
| 固定資産と無形資産 | 3,332 | | 3,077 |
| 準備金と課税項目 | 1,764 | | 1,790 |
| 賃貸負債 | 1,049 | | 1,380 |
| 収入を繰り越す | 128 | | 18,610 |
| 繰延税金資産総額 | 101,139 | | 84,927 |
| 推定免税額 | (99,417) | | (82,703) |
| 小計 | 1,722 | | 2,224 |
| 使用権資産 | (960) | | (1,313) |
| 固定資産 | (472) | | (572) |
| 前払い費用 | (290) | | (339) |
| 繰延税項目純資産総額 | $ | — | | $ | — |
ASC 740の要求によると、当社は、その繰延税金項目資産の現金化能力に影響を与えるプラスおよび負の証拠を評価しており、これらの繰延税金資産は、主に課税と確認されたが年末帳簿報告に繰延された協力収入と繰越された純営業損失から構成されている。当社は、連邦と州繰延税金資産の収益を実現できず、#ドルの推定手当を得ることができない可能性が高いことを確認しました99.4百万ドルとドル82.72022年と2021年にはそれぞれ百万人が設立された。推定手当の変動は#ドルである16.7百万ドルとドル37.12022年12月31日と2021年12月31日までの年間は100万ドル。
2022年から減税·雇用法案(TCJA)が第174条を改正し,現在米国と非米国での研究·実験(R&E)支出をそれぞれ5年または15年以内に資本化·償却することが求められており,これらの支出は2021年12月31日以降の納税年度に支払われている。TCJA改正案の前に、第174条は、納税者が直ちに支払いまたは発生した年間にR&E支出を差し引くことを許可する。 同社は2022年から会計方法をこの必要な変更を行っており、計算方法は将来の米国国税局の指導の下で調整される可能性がある。
会社は設立以来純営業損失(“NOL”)が発生している。2022年12月31日現在,同社の連邦と州NOL繰り越し額は約$である180.3百万ドルとドル191.5将来の課税所得を減らすための100万ドルは2037年から満期になります176.4何百万人もの連邦NOLは無限の持続寿命を持っている。2022年12月31日までに、同社は連邦と州の研究開発税収控除繰越約$を保有している12.6百万ドルとドル5.7それぞれ百万人 2032年12月に満期になる将来の所得税を相殺するために。当社のNOL繰越は適切な税務機関が審査及び調整可能でなければなりません。同社は、NOL年間使用を制限する可能性のあるIRC 382制限が存在するかどうかを決定するために、その歴史的所有権の変化を検討し続ける
同社は、財務諸表において不確定な税収状況の税収利益をどのように確認、計量、記録するか、不確定税収事項の特定の開示を要求すること、不確定税収状況を規定する準備金が貸借対照表上でどのように分類されるべきか、過渡期および中期指導、および他の規定を提供することを規定するASC 740-10“所得税不確実性会計”の規定に従う。2022年12月31日と2021年12月31日現在、会社には未確認の税収割引がない。当社は信用計算について何の不確定なポジションも確定していません。同社の政策は、その経営報告書において、いかなる不確定な税収状況から生じる利息及び罰金を所得税費用の構成要素として確認することである(あれば)。
2022年12月31日と2021年12月31日までの年度違います。見積もり利息や罰金は不確定な税収頭寸として確認されています。同社はその不確定な税務状況が今後12ヶ月以内に大きな変化はないと予想している。
同社は米国連邦と州所得税申告書を提出し、2018年12月31日以降のすべての納税年度は通常、これらの当局の所得税審査を受ける。現在、連邦や州所得税申告書はそれぞれの所得税当局の審査を受けていない。
所得税支給
会社が記録した所得税支出は#ドルです0.12022年12月31日までの年間所得税支出は0.02021年12月31日までの年間収入は100万ドル、所得税収益は0.62020年12月31日までの1年間で2020年に記録された福祉は、注目法で許可された連邦純営業損失繰越クレームによる
協力収入にもかかわらず、会社はすべての繰延税金資産に対して評価手当を維持し続けている。同社は、会社の研究計画は引き続き大量の投資を必要としており、将来の収入は不確定要素の影響を受けているため、会社はこれらの属性の将来の税収割引を実現しない可能性が高いと考えている。いかなる繰延税金資産の最終的な現金化は、繰延期間(ある場合)が満了する前に、当社が適切な税務管区区で将来課税すべき収入を生成するのに十分な能力に依存する。
11. 引受金とその他の事項
保証と補償
再記載された会社の登録証明書及び改訂及び重述の定款に規定されている賠償のほか、当社は取締役、高級管理者及びいくつかの主要従業員と締結し、引き続き単独の賠償協定を締結する予定である。他に加えて、これらの合意は、弁護士費、判決、罰金、および和解金額を含む取締役、高級管理者、および主要従業員のいくつかの費用を会社に賠償することを要求し、これらの個人は、会社またはその任意の子会社またはこれらの個人が、会社がサービスを提供することを要求する任意の他の会社または企業にサービスを提供する際に、任意の訴訟または訴訟で実際に引き起こされる和解金額を含む。いくつかの制限を受けた場合、賠償協定はまた、賠償が必要または許可された任意の訴訟を弁護するために、会社の前借り役員、高級管理者、およびキー従業員に発生する費用を要求する。
当社はその実験室とオフィススペース賃貸契約に標準的な賠償手配があり、家主が当社の賃貸契約中のいくつかの行為、違反行為、違反行為、または義務を履行しないことによるいかなるクレーム、訴訟、訴訟または費用による傷害、損失、事故または損害のいかなる責任も賠償することを要求する。
当社は2022年12月31日現在、これらの賠償義務に関する損失を経験しておらず、重大なクレーム待ちもない。当社はこれらの賠償義務に関する重大なクレームはないと予想しており,これらの債務の公正価値は無視でき,関連準備金も確立されていないと結論した。
法律訴訟
当社は現在、いかなる重大な法的手続きの当事者でもありません。
12. オプションとライセンス契約
AbbVieプロトコルの概要
2018年10月に当社が締結しました5年間エバーヴィバイオテクノロジー株式会社(“エバーヴィ協定”)との協力とオプション協定(“エバービー協定”)は,エバービーは研究に基づく世界的なバイオ製薬会社であり,エバービー協定調印時に会社のAシリーズとBシリーズ転換可能優先株(“エバービー協定”)を保有している。この合意によると、AbbVieは会社に払い戻しできないお金#ドルを前払いした100.0百万ドルです。交換として,当社:(I)は,多様な線維化に固有の化合物を決定·開発するための研究·開発活動の義務を担っている(分類は四つ新薬(IND)の研究を完成させることにより、及び(Ii)はAbbVieに研究開発結果の選択権を付与し、単独の前払い選択権使用料と交換する。2022年6月、AbbVieは当社に通知し、便宜上、AbbVieプロトコルを中止する権利を行使することを決定し、AbbVieプロトコルは2022年12月に終了した。したがって、会社はAbbVie協定の下で追加的な支払いを受けないと予想される。
AbbVieプロトコルが終了する前に、AbbVieは、会社に書面通知を提供し、以下のオプション使用料を支払うことによって、選択された薬物プロファイルを有する分子に対する許可オプションを行使する権利がある
$20.0選択権を行使するごとに百万ドル三つ)を集計する。AbbVieプロトコルの終了後に発効し、協定によって付与されたすべての権利およびライセンスは直ちに終了し、当社に返却されます。
AbbVieプロトコル会計分析
同社の結論は,合意における履行義務は四つ研究プロジェクトです。当社の結論は、行使されていない許可選択権はマーケティング要約であり、選択権はいかなる割引や他の権利も提供しないため、これらの権利は手配中の重大な権利とみなされる。他のすべての義務は契約の範囲内で重要ではないと決定された。
会社は研究を行う内部と外部コストに合理的な利益率を加えて各研究プロジェクトの独立販売価格を推定している10%です。研究·開発サービスの総見積りコストは,提供するサービスの性質および会社のサービス提供に要する時間の最適な見積りを反映している.会社が収入を確認したのは,研究·開発サービスがこれまでに発生したコストに基づいて提供されているためであり,これらのコストはAbbVieに対する履行義務の履行における会社の努力と進展に直接関係しているからである。内部と外部開発サービス見積り費用の変化は変動期間内に累積的な追跡調整として確認された
同社は、取引価格には払い戻し不可能な前金#ドルのみが含まれていると判断した100.0100万ドルで、2018年12月31日までの繰延収入として記録されています。当社は合意費用が当該オプションの公正価値を代表すると認定しているため、オプション行使支払いは取引価格には計上されていない。AbbVieが任意のオプションを行使するかどうか、およびいかなる関連マイルストーンが実現されるかどうかの不確実性により、マイルストーン支払いは完全に制限されている。AbbVieプロトコルが終了するまで、取引価格は変化しなかった。
AbbVieプロトコルのプリペイド構造を考慮して、同社はまた、この合意に重要な融資部分が存在するかどうかを考慮している。この評価によると、会社は、前払金は、融資を提供するためではなく、正当な商業的理由で提供されていると結論した。したがって、会社はAbbVie協定の前払い構造が重大な融資構成要素の存在を招いていないと結論した。
2020年8月25日,AbbVieは特発性肺線維化を含む線維化疾患や他の適応の治療のために,Morphiのαvβ6特定インテグリン抑制製剤のさらなる開発と商業化を許可·制御する選択権を行使した。選択権の行使について、AbbVieは会社に#ドルを支払った20.0百万ドルです。選択権を行使する際に、同社は、契約の変更が現行手配の継続または単独の合意として扱われるべきかどうかを評価した。追加の履行義務は異なると考えられ、定価は当該義務の独立販売価格と一致するため、会社は、許可証及び任意の追加の履行義務は別個の契約として入金されなければならないと結論した。手配に含まれる潜在的履行義務には,(1)αvβ6特定インテグリン抑制製剤の研究,開発,商業化の許可証と,(2)オプション行使前に製造された材料の購入選択権(“材料選択権”)がある。同社は,材料選択権は物質的権利ではなく,材料購入価格が独立した販売価格に近いためであると結論している。この結論に基づいて、ドルのすべての出来高は20.0ライセンスに100万ドルが割り当てられ、2020年第3四半期の交付時に確認された。ライセンスはAbbVieプロトコルの終了時に終了するため、会社はAbbVieから本計画の追加支払いを受けることはないと予想される。
AbbVieプロトコルを終了する権利を行使する通知を受けた後、会社は、AbbVieプロトコルを終了する通知によって会社がASC 606の下の既存の契約を契約修正したと結論した三つその下の残りの研究計画。AbbVie協定終了通知の条項には追加的な約束された貨物やサービスは含まれていない。AbbVieの通知により、会社は収入が#ドルであることを確認した57.7累積追跡の上で、最新の進捗測定方法を用いて、研究と開発サービスの業績義務を履行する上で進展を得た
2022年12月31日までの年間で、会社はAbbVie合意項目での履行義務を完了した次の表は、2022年12月31日、2021年12月31日、2020年12月31日までの年間で、AbbVie合意下での会社の業績に関する研究開発コストと確認収入(単位:千)をまとめたものです
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | 十二月三十一日までの年度 | | |
| | | | | 2022 | | 2021 | | 2020 | | | | |
| 収入が確認された | | | | | $ | 60,500 | | | $ | 11,197 | | | $ | 36,056 | | | | | |
| 招いた費用 | | | | | $ | 631 | | | $ | 5,376 | | | $ | 13,474 | | | | | |
2021年12月31日現在、同社は60.5繰延収入は1000万ドルで、連結貸借対照表で契約改正前に収入が確認される予定の期間を当期または当期純額に分類する。この繰延収入残高は、2021年12月31日現在の一部未返済の履行債務に割り当てられた取引価格総額を表す。2022年12月31日現在、会社はAbbVie協定に対してこれ以上履行されていない履行義務を持っていない。
ヤンソン協定の概要
2019年2月,当社はジョンソン(“ヤンソン”)の子会社であるヤンソン製薬株式会社(“ヤンソン協定”)と既存療法で十分に解決できなかった疾患の患者に適した新規インテグリン療法を発見·開発するための研究協力·オプション協定を締結した。ヤンソン協定のポイントは三つインテグリン標的、各標的は研究計画のテーマであり、もし研究結果が良くなければ、会社が探索していない他のインテグリン標的を限られた能力で代替する。合意条項によると、Janssenは会社に費用#ドルを前払いする10.0初めて百万ドル二つ2019年と2020年12月、会社はヤンソンと合意し、第3の研究計画の仕事を開始し、ヤンソンは$を支払うことに同意した5.0三番目の研究プロジェクトの開始費は100万ドルだった。また、Janssenは合意された契約料率で合意期間内に発生したすべての内部および外部コストと支出を会社に返済する。同社は四半期ごとにJanssenに領収書を発行し、支払い期限は60何日ですか
Janssenは2021年12月、最初の2つのインテグリン目標に対してオプションを行使しないことを決定したことを会社に通知したため、この2つの目標も停止した二つ研究プロジェクトです。同社はインテグリン抗体活性化剤の潜在的開発を含む第3のインテグリン研究計画に重点を置いている。2023年1月、Janssenは当社に通知し、便宜上、会社はJanssen協定を終了する権利を行使することを決定しましたが、遵守しなければなりません60期日通知期間。当社とヤンソンが同意すれば、ヤンソン協定は2023年3月またはそれ以上に終了する
ジェーン·ソン·プロトコル会計分析
当社の結論は,合意における履行義務は三つ研究計画と三つこれらの研究プロジェクトの成果を許可するオプションは、Janssenに物質的権利を提供すると決定された。他のすべての義務は契約の範囲内で重要ではないと決定された。
同社は研究を行う内部と外部コストに合理的な利益率を加えて各研究プロジェクトの独立販売価格を推定している。研究開発サービスの総見積もりコストは、実行するサービスの性質および会社のサービス実行に要する時間の最適な推定を反映している。当社は、比較可能オプションの推定独立販売価格と比較して提供される割引を決定し、研究作業に成功した適切な可能性を含む適切な行使可能性を適用することにより、各材料の権利の独立販売価格を推定する。決定された独立販売価格に基づいて、会社は番組と材料権利との間に取引総価格を分配する。
会社が収入を確認したのは,研究サービスはこれまでに発生したコストに基づいて提供されているためであり,これらのコストはヤンソンに対する業績義務の履行における会社の努力と進展に直結しているからである。重大な権利に割り当てられた取引価格が繰延され、Janssenのオプション行使またはオプション期限が満了したときに収入として確認される。発生すると予想される内部と外部総費用推定数の変化は変動期間内に累積追跡調整であることが確認された
同社が決定した取引価格には、払い戻しができない前払い#ドルが含まれている10.0初めて百万ドル二つ計画、$5.0第三項目の前払金100万ドルの払い戻しはできず、会社が行っている研究サービスは、Janssenから合意された契約料率で支払われる推定精算金を受け取る。オプション支払は取引価格に含まれていません。いかなるオプションの行使も継続とみなされるだろう
Janssenが書面で行使通知を交付した場合、現在の契約は有効です。ヤンソンがいかなるオプションを行使するかどうか、どのような関連マイルストーンが実現されるかどうかは定かではないため、マイルストーン支払いは完全に制限されている。
ヤンソン協定の前金構造を考慮して、当社はヤンソン協定に重要な融資構成要素が存在するかどうかも考慮した。この評価によると、会社は、前払金は、融資を提供するためではなく、正当な商業的理由で提供されていると結論した。したがって、当社は、ヤンソン協定の前払い構造が重大な融資構成要素の存在を招くことはないと結論した。
契約変更の会計処理
終了が通知されたとき二つ研究計画と関連オプションを考慮して、会社はASC 606により既存契約を契約修正し、2021年12月の通知によりヤンソン合意下での会社の責任範囲が縮小したと結論した。2021年12月の通知には、追加のコミットメント商品やサービスは含まれていません。改正後、ヤンソン協定によると、残りの研究計画に研究サービスを提供することと、ヤンソンの余剰許可選択権に材料権利を提供するという2つの履行義務がある。同社の結論は,第3の研究計画を満たすために提供される残りの研究サービスは,既存の研究サービスと変わらないため,修正を元の契約の一部(単独の契約ではなく)と見なすことである。そこで、当社は、研究サービスの残りの繰延収入と可変対価格を推定し、契約修正日の推定独立販売価格に基づいて、更新された取引価格を残りの履行義務に割り当てる取引価格を更新した
研究サービスについては,契約改正が取引価格に与える影響と,#ドル履行義務の履行における進展があった1.9累積追跡をもとに100万ドルを確認した。余剰物権に割り当てられた取引価格は$0.5100万ドルは延期され、行使または満期まで延期されるだろう。契約改正日までに履行されたいかなる履行義務の会計処理も修正の影響を受けない
次の表は、2022年12月31日、2021年12月31日、2020年12月31日までの年度内に、ヤンソン合意の業績による研究開発コストと確認収入(千単位)をまとめたものである
| | | | | | | | | | | | | | | | | | | | | |
| | | 十二月三十一日までの年度 |
| | | | | 2022 | | 2021 | | 2020 |
| 収入を清算する | | | | | $ | 3,159 | | | $ | 4,944 | | | $ | 6,601 | |
| 前払収入 | | | | | 7,149 | | | 3,653 | | | 2,288 | |
| 確認された総収入 | | | | | $ | 10,308 | | | $ | 8,597 | | | $ | 8,889 | |
| 招いた費用 | | | | | $ | 2,637 | | | $ | 4,426 | | | $ | 5,865 | |
2022年第4四半期に会社が確認した追加収入は3.0第3の研究計画の研究サービスを完成させるために必要な余剰作業に要する見積もり費用が変化するため,累計百万ドルを追う。予想作業の変化と研究サービスのスケジュールにより、将来の予想費用見積もり数は減少した。
同社は2022年12月31日と2021年12月31日まで0.5百万ドルとドル2.3売掛金にはJanssenの満期100万ドルがそれぞれ記録されている。2022年12月31日と2021年12月31日までに0.5百万ドルとドル7.6予想される収入が確認されている期間に応じて、繰延収入の当期または純額は、それぞれ添付の総合貸借対照表において当期または当期純額に分類される。2022年12月31日と2021年12月31日までの繰延収入残高とは、2022年12月31日現在および2021年12月31日現在の履行されていない履行債務の一部または全部に割り当てられた前払い部分を指す。同社は2023年第1四半期までに余剰業績義務に関する収入を確認する予定だ。
もし会社とヤンソンが同意すれば、会社の実質的な権利に関する持続的な義務は2023年3月またはそれ以上に終了するだろう。
13. 1株当たり純損失
1株あたりの基本純損失の計算方法は,普通株株主に割り当てられた純損失を当期に発行された加重平均普通株で割ったものであり,普通株等価物は考慮しない。
純利益がある期間については、1株当たりの純収入は、在庫株方法を用いて決定された期間の株式オプションおよび制限された普通株および発行された株式単位を含む、普通株等価物の希釈効果に基づいて加重平均流通株を調整することによって算出される。
1株当たり純損失を希釈する際に、普通株等価物の影響が逆薄であれば、計算には含まれない。したがって,普通株株主に適用される1株当たり基本純損失と希釈後の1株当たり純損失は純損失期間中と同じである。
次の表は、各新聞期間ごとの1株当たり基本損失と希薄損失の決定(株式データを除く千計)について説明した
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | 十二月三十一日までの年度 | | |
| | | | | 2022 | | 2021 | | 2020 | | | | |
| 純損失 | | | | | $ | (59,041) | | | $ | (95,542) | | | $ | (44,999) | | | | | |
| 加重平均発行済み普通株式、基本普通株式、希釈後普通株 | | | | | 38,112,498 | | | 35,797,969 | | | 30,594,897 | | | | | |
| 1株当たり基本と希釈して純損失 | | | | | $ | (1.55) | | | $ | (2.67) | | | $ | (1.47) | | | | | |
| | | | | | | | | | | | | |
以下の表は、期間中の希釈1株当たり純損失の計算から除外された各期末の発行済み金額に基づいて報告された発行された普通株等価物を示し、これらの等価物は、赤字(適用される普通株等価株に適用される)に計上されるからである
| | | | | | | | | | | | | | | | | | | | | |
| | | 十二月三十一日までの年度 |
| | | | | 2022 | | 2021 | | 2020 |
| 制限普通株 | | | | | — | | | 2,171 | | | 100,989 | |
| 制限株式単位 | | | | | 249,090 | | | 7,000 | | | 66,216 | |
| 株式オプション | | | | | 5,263,553 | | | 4,781,565 | | | 4,352,095 | |
| | | | | 5,512,643 | | | 4,790,736 | | | 4,519,300 | |
上の表に記載されている証券を除いて、2022年12月31日まで、当社は予約しました1,142,204ESPPが売却した普通株によると、発行すれば、2022年12月31日までの年度希釈後の1株当たり純損失の計算に含めると、逆薄となる。
14. 従業員福祉計画
同社は2016年、従業員納付と雇用主マッチング納付により、条件を満たす従業員に退職収入を提供する適格退職計画であるMorphy Treateutation,Inc.401(K)計画を採択した。ペアリング寄付総額は#ドル0.6百万、$0.6百万ドルとドル0.52022年12月31日まで、2021年、2020年12月31日までの年度はそれぞれ100万ドル。
第九項です会計と財務情報開示の変更と相違
第9条。 制御とプログラム
(A)開示制御およびプログラムの有効性
我々は,我々の最高経営責任者と最高財務官(それぞれ我々の最高経営責任者とCEO)の監督と参加の下で,2022年12月31日に評価を行い,我々の開示制御と手順(1934年証券取引法改正後の規則13 a-15(E)または規則15 d-15(E)の定義)が2022年12月31日から有効であり,取引法に基づいて提出または提出された報告書で開示を要求した情報が記録,処理,処理されることを確保することを目的としている.米国証券取引委員会規則及び表で指定された期間内にまとめて報告し、これらのデータを蓄積し、必要な開示に関する決定をタイムリーに行うために、我々のCEO及び最高財務官を含む経営陣に伝達する。
(B)規制効力の内在的制限
私たちの経営陣は、CEOやCEOを含め、財務報告に対する内部統制や私たちの内部統制がすべてのエラーやすべての詐欺を防止または発見することを期待していません。設計および動作がどんなに良好であっても、絶対的な保証ではなく、合理的な保証しか提供できず、保証制御システムの目標が実現される制御システム。いずれの制御システムの設計も、将来のイベント可能性のいくつかの仮定にある程度基づいており、どの設計もすべての潜在的な未来条件でその目標を成功的に達成することが保証されていない。
(C)財務報告の内部統制に関する経営陣の年次報告
私たちの経営陣は財務報告書の十分な内部統制を確立して維持する責任がある。最高経営責任者と最高財務責任者の監督の下、経営陣は、テレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み(2013)”の枠組みに基づいて、2022年12月31日までの財務報告の内部統制の有効性を評価した。この評価の結果、経営陣は、財務報告に対する内部統制は2022年12月31日から有効であると結論した
(D)財務報告の内部統制の変化
2022年12月31日までの四半期内に、財務報告の内部統制(“取引法”第13 a-15(F)および15 d-15(F)規則の定義による)に大きな影響を与えなかったか、または合理的に財務報告の内部統制に大きな影響を与える可能性のある変化はなかった。
プロジェクト9 Bその他の情報
ない。
プロジェクト9 Cです検査妨害に関する外国司法管区の開示
適用されません。
第三部
プロジェクト10.役員、役員、および企業管理
第10項の要件の情報は、2022年12月31日後120日後に提出され、参照によって本明細書に組み込まれる2023年株主総会に関する我々の最終委託書に含まれる。
第11項.行政職報酬
第11項の要件の情報は、2022年12月31日後120日後に提出され、参照によって本明細書に組み込まれる2023年株主総会に関する我々の最終委託書に含まれる。
プロジェクト12.特定の実益所有者の保証所有権および管理職および株主に関する事項。
第12項の要件の情報は、2022年12月31日後120日後に提出され、参照によって本明細書に組み込まれる2023年株主総会に関する我々の最終委託書に含まれる。
第13項:特定の関係及び関連取引、並びに取締役の独立性。
第13項の要件の情報は、2022年12月31日後120日後に提出され、参照によって本明細書に組み込まれる2023年株主総会に関する我々の最終委託書に含まれる。
プロジェクト14.主な会計費用とサービス
第14項の要件の情報は、2022年12月31日後120日以内に提出され、参照によって本明細書に組み込まれる2023年株主総会に関する我々の最終委託書に含まれる。
第4部
第十五項。 展示品と財務諸表の付表
(1)財務諸表:
我々の連結財務諸表は、本年度報告の表格10-Kの第2部分第8項に記載され、参照によって本明細書に組み込まれる。
(2)財務諸表付表:
スケジュールは必要ではないか,適用されないか,あるいは情報が他の方法で本明細書に含まれるため省略される.
(3) Exhibits
本年度報告10−K表の一部として提出された証拠物は以下のとおりである。
S-Kルール601項(本章229.601節)に要求される証拠物を提供する.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
展示品
番号をつける | | 説明する | | 表 | | 書類番号. | | 展示品 | | 証拠品保存
日取り | | 同封のアーカイブ/提供 |
| 3.1 | | 再記載の会社登録証明書 | | 10-Q | | 001-38940 | | 3.1 | | 2020年8月13日 | | |
| 3.2 | | 付例を改訂および再制定する | | 8-K | | 001-38940 | | 3.1 | | 2023年2月13日 | | |
| 4.1 | | 普通株の書式 | | S-1/A | | 333-231837 | | 4.1 | | June 14, 2020 | | |
| 4.2 | | 投資家権利協定は、日付が2018年12月5日であり、登録者及びその特定の株主によって署名される。 | | S-1/A | | 333-231837 | | 4.2 | | June 14, 2020 | | |
| 4.3 | | 改正された1934年証券取引法第12節に基づいて登録された登録者証券説明 | | 10-K | | 001-38940 | | 4.3 | | March 1, 2021 | | |
| 4.4 | | 債務担保の形式 | | S-3 | | 333-258435 | | 4.3 | | 2021年8月4日 | | |
| 4.5 | | 義歯の形式 | | S-3 | | 333-258435 | | 4.4 | | 2021年8月4日 | | |
| 4.6 | | あらかじめ出資して株式証の書式を承認する | | 8-K | | 001-38940 | | 4.1 | | 2023年2月13日 | | |
| 10.1* | | 契約書のフォーマットを完済する。 | | S-1/A | | 333-231837 | | 10.1 | | June 14, 2020 | | |
| 10.2* | | 2018年株式インセンティブ計画および奨励プロトコルフォーマット | | S-1/A | | 333-231837 | | 10.2 | | June 14, 2020 | | |
| 10.3* | | 2019年持分インセンティブ計画の改訂と再策定 | | 10-Q | | 001-38940 | | 10.1 | | 2022年8月3日 | | |
| 10.4* | | 株式オプション奨励プロトコルフォーマット(2019年持分インセンティブ計画を含む) | | S-1/A | | 333-231837 | | 10.3 | | June 14, 2020 | | |
| 10.5* | | 制限株式奨励プロトコルフォーマット(2019年持分インセンティブ計画を含む) | | S-1/A | | 333-231837 | | 10.3 | | June 14, 2020 | | |
| 10.6* | | 2019年従業員株購入計画と奨励協定フォーマット | | S-1/A | | 333-231837 | | 10.4 | | June 14, 2020 | | |
| 10.7* | | 登録者とメリーランド州Praveen P.Tipirneniの間の招待状は、2019年6月10日となります | | S-1/A | | 333-231837 | | 10.5 | | June 14, 2020 | | |
| 10.8* | | 登録者とブルース·N·ロジャース博士の間の招聘状は、2019年6月10日となっています. | | S-1/A | | 333-231837 | | 10.6 | | June 14, 2020 | | |
| 10.9* | | 登録者とウィリアム·ドウォルの間の招待状は、2019年6月10日となっております | | 10-K | | 001-38940 | | 10.7 | | 2021年2月27日 | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 10.10 | | 賃貸借契約は,日付は2015年8月5日であり,登録者とアスリーカン製薬有限責任組合企業との間で締結され,改訂された。 | | S-1/A | | 333-231837 | | 10.9 | | June 14, 2020 | | |
| 10.11† | | 研究協力とオプション協定は,2019年2月15日であり,Janssen PharmPharmticals,Inc.と登録者の間で署名されている。 | | S-1/A | | 333-231837 | | 10.10 | | June 14, 2020 | | |
| 10.12† | | 協力協定は,2015年6月10日にMorphy Rock治療会社とSchrödinger,LLCの間で署名され,改訂された。 | | S-1/A | | 333-231837 | | 10.12 | | June 14, 2020 | | |
| 10.13† | | 児童医療センター会社と登録者との間の独占許可協定は,日付が2015年10月7日であり,改訂された。 | | S-1/A | | 333-231837 | | 10.13 | | June 14, 2020 | | |
| 10.14 | | 証券登録契約の書式 | | S-1/A | | 333-231837 | | 10.17 | | June 14, 2020 | | |
| 10.15* | | 招待状は,日付は2020年2月3日,Marc SchegerinとMorphy Holding,Inc. | | 10-Q | | 001-38940 | | 10.1 | | 2020年8月10日 | | |
| 10.16 | | 楊森製薬会社と登録者の間で2020年12月30日に署名された研究協力と選択協定の第1号改正案。 | | 10-K | | 001-38940 | | 10.19 | | March 1, 2021 | | |
| 10.17 | | 登録者とヤンソン製薬会社との間の研究協力及び選択協定の第2号改正案は、期日は2021年6月18日である。 | | 10-Q | | 001-38940 | | 10.1 | | 2021年8月4日 | | |
| 10.18 | | 借書の5回目の改訂 | | 10-Q | | 001-38940 | | 10.1 | | 2021年11月4日 | | |
| 10.19 | | 支配権および離職契約のフォーマットを変更する | | 10-K | | 001-38940 | | 10.19 | | 2022年2月24日 | | |
| 10.20 | | 証券購入協定は、2023年2月13日に、Morphy Holding,Inc.と署名ページに記載された投資家との間で署名される | | 8-K | | 001-38940 | | 10.1 | | 2023年2月13日 | | |
| 10.21 | | 2023年2月13日にMorphy Holding,Inc.と署名ページに記載された投資家との間で署名された登録権利協定 | | 8-K | | 001-38940 | | 10.2 | | 2023年2月13日 | | |
| 21.1 | | 登録者の子会社。 | | | | | | | | | | X |
| 23.1 | | 独立公認会計士事務所が同意します。 | | | | | | | | | | X |
| 24.1 | | 授権書。ここのサインページを参考にしてください。 | | | | | | | | | | X |
| 31.1 | | 2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 | | | | | | | | | | X |
| 31.2 | | 2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 | | | | | | | | | | X |
| 32.1** | | 2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編1350条に規定されている最高経営責任者証明書。 | | | | | | | | | | X |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 32.2** | | 2002年サバンズ·オキシリー法第906条で可決された“米国法典”第18編1350条による首席財務官の証明 | | | | | | | | | | X |
| 101.INS | | XBRLインスタンスドキュメントを連結する | | | | | | | | | | X |
| 101.書院 | | イントラネットXBRL分類拡張アーキテクチャ文書 | | | | | | | | | | X |
| 101.カール | | インラインXBRL分類拡張計算リンクライブラリ文書 | | | | | | | | | | X |
| 101.def | | インラインXBRL分類拡張Linkbase文書を定義する | | | | | | | | | | X |
| 101.介護会 | | XBRL分類拡張ラベルLinkbase文書を連結する | | | | | | | | | | X |
| 101.Pre | | インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント | | | | | | | | | | X |
| 104 | | 表紙インタラクションデータファイル(イントラネットXBRLに埋め込む) | | | | | | | | | | X |
*役員報酬計画またはプロトコル。
**本契約添付ファイル32.1および32.2に提供される証明は、本10-Kフォームと共に提供されるものとみなされ、取引法第18条の目的に従って“アーカイブされた”とみなされるものとはみなされず、この条項の責任を受けているとみなされるべきでもなく、参照によって証券法または取引法下のいずれの出願に組み込まれているともみなされてはならない。
イ登録者が見落としたS-K条例第601(B)(10)項に許可された部分展示品
項目16.表格10-Kの概要
ない。
サイン
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、正式に許可された署名者がその代表を代表して本報告に署名することを正式に手配した。
| | | | | | | | |
| モニック·ホールディングス |
| | |
| | |
| 2023年2月23日 | 差出人: | /s/Praveen P.Tipirneni |
| | Praveen P.Tipirneni,M.D. |
| | 取締役CEO兼最高経営責任者
(首席行政主任) |
| | |
| 差出人: | /s/Marc Schegerin |
| 2023年2月23日 | | Marc Schegerin医学博士 |
| | 首席財務官兼首席運営官
(首席財務官) |
| | |
| 2023年2月23日 | 差出人: | ロバート·E·ファレル |
| | ロバート·E·ファレル公認会計士 |
| | 首席財務官兼補佐財務担当者
(首席会計主任) |
授権依頼書
このような陳述によってすべての人を認識し、以下の署名の各々は、Praveen P.TipirneniおよびWilliam D.DeVaulを構成し、任命し、それぞれ完全な代替および再代替の権力を有し、相手なしにその真および合法的な事実受権者および代理人として、その名義、場所および代理として行動し、各個人の名義および以下に説明する各身分で署名し、本年度報告の任意およびすべての修正をテーブル10-Kの形で提出し、それをすべての証拠物およびそれに関連する他の文書と共にアーカイブし、米国証券取引委員会に、上記の事実弁護士および代理人、ならびに彼ら一人一人が各行為および事柄を行うすべての権力および権力を付与し、上述したすべての事実弁護士および代理人または彼らのいずれかまたは彼らまたはそれらの代替者が、行われたすべての行為および事柄を合法的に行うことができるか、または結果として生じるすべての行為およびことを承認および確認することができる。
1934年の証券法の要求によると、本報告は、以下の者によって登録者の身分で指定日に登録者名で署名された。
| | | | | | | | | | | | | | |
| サイン | | タイトル | | 日取り |
| /s/Praveen P.Tipirneni | | CEOと役員(最高経営責任者) | | 2023年2月23日 |
| Praveen P.Tipirneni,M.D. | | | | |
| | | | |
| /s/Marc Schegerin | | 首席財務官と首席運営官(首席財務官) | | 2023年2月23日 |
| Marc Schegerin医学博士 | | | | |
| | | | |
| /s/ロバート·E·ファレル | | 首席会計幹事と財務担当補佐(首席会計幹事) | | 2023年2月23日 |
| ロバートE.Farrell Jr.CPA | | | | |
| | | | |
| /s/Norbert Bischofberger | | 役員.取締役 | | 2023年2月23日 |
| Norbert Bischofberger博士 | | | | |
| | | | |
| /s/グスタフ·クリステンセン | | 役員.取締役 | | 2023年2月23日 |
| グスタフ·クリステンソン | | | | |
| | | | |
| マーティン·エドワーズ | | 役員.取締役 | | 2023年2月23日 |
| マーティン·エドワーズ | | | | |
| | | | |
| /s/Susannah Gray | | 役員.取締役 | | 2023年2月23日 |
| スザンナ·グレイ | | | | |
| | | | |
| /s/Nisha Nanda | | 役員.取締役 | | 2023年2月23日 |
| Nisha Nanda博士 | | | | |
| | | | |
| /s/アミール·ナサウジ | | 役員.取締役 | | 2023年2月23日 |
| アミール·ナサウジ | | | | |
| | | | |
| /s/ジョセフ·P·スリトリ | | 役員.取締役 | | 2023年2月23日 |
| ジョセフ·P·スリトリー公認会計士 | | | | |
| | | | |
| /s/Timothy A.Springer | | 役員.取締役 | | 2023年2月23日 |
| ティモシー·A·スプリンガー博士 | | | | |