sgmo-20221231誤り2022会計年度0001001233P 3 YHttp://Fasb.org/us-GAAP/2022#AcruedLiabilitiesCurrentHttp://Fasb.org/us-GAAP/2022#AcruedLiabilitiesCurrentP 5 YHttp://Fasb.org/us-GAAP/2022#AcruedLiabilitiesCurrentP 3 Y00010012332022-01-012022-12-3100010012332022-06-30ISO 4217:ドル00010012332023-02-17Xbrli:共有00010012332022-12-3100010012332021-12-31ISO 4217:ドルXbrli:共有00010012332021-01-012021-12-3100010012332020-01-012020-12-310001001233アメリカ-アメリカ公認会計基準:普通株式メンバー2019-12-310001001233US-GAAP:AdditionalPaidInCapitalMembers2019-12-310001001233アメリカ-公認会計基準:前払いメンバーを保留2019-12-310001001233アメリカ公認会計原則:他の総合収入メンバーを累計2019-12-310001001233アメリカ公認会計基準:非制御的利益メンバー2019-12-3100010012332019-12-310001001233アメリカ-アメリカ公認会計基準:普通株式メンバー2020-01-012020-12-310001001233US-GAAP:AdditionalPaidInCapitalMembers2020-01-012020-12-310001001233アメリカ公認会計基準:非制御的利益メンバー2020-01-012020-12-310001001233アメリカ公認会計原則:他の総合収入メンバーを累計2020-01-012020-12-310001001233アメリカ-公認会計基準:前払いメンバーを保留2020-01-012020-12-310001001233アメリカ-アメリカ公認会計基準:普通株式メンバー2020-12-310001001233US-GAAP:AdditionalPaidInCapitalMembers2020-12-310001001233アメリカ-公認会計基準:前払いメンバーを保留2020-12-310001001233アメリカ公認会計原則:他の総合収入メンバーを累計2020-12-310001001233アメリカ公認会計基準:非制御的利益メンバー2020-12-3100010012332020-12-310001001233アメリカ-アメリカ公認会計基準:普通株式メンバー2021-01-012021-12-310001001233US-GAAP:AdditionalPaidInCapitalMembers2021-01-012021-12-310001001233アメリカ公認会計基準:非制御的利益メンバー2021-01-012021-12-310001001233アメリカ公認会計原則:他の総合収入メンバーを累計2021-01-012021-12-310001001233アメリカ-公認会計基準:前払いメンバーを保留2021-01-012021-12-310001001233アメリカ-アメリカ公認会計基準:普通株式メンバー2021-12-310001001233US-GAAP:AdditionalPaidInCapitalMembers2021-12-310001001233アメリカ-公認会計基準:前払いメンバーを保留2021-12-310001001233アメリカ公認会計原則:他の総合収入メンバーを累計2021-12-310001001233アメリカ公認会計基準:非制御的利益メンバー2021-12-310001001233アメリカ-アメリカ公認会計基準:普通株式メンバー2022-01-012022-12-310001001233US-GAAP:AdditionalPaidInCapitalMembers2022-01-012022-12-310001001233アメリカ公認会計原則:他の総合収入メンバーを累計2022-01-012022-12-310001001233アメリカ-公認会計基準:前払いメンバーを保留2022-01-012022-12-310001001233アメリカ-アメリカ公認会計基準:普通株式メンバー2022-12-310001001233US-GAAP:AdditionalPaidInCapitalMembers2022-12-310001001233アメリカ-公認会計基準:前払いメンバーを保留2022-12-310001001233アメリカ公認会計原則:他の総合収入メンバーを累計2022-12-310001001233アメリカ公認会計基準:非制御的利益メンバー2022-12-310001001233SGMO:AtTheMarketOfferingMembers2022-01-012022-12-310001001233SGMO:AtTheMarketOfferingMembers2021-01-012021-12-310001001233SGMO:AtTheMarketOfferingMembers2020-01-012020-12-310001001233SGMO:生物遺伝子協力プロトコルのメンバー2022-01-012022-12-310001001233SGMO:生物遺伝子協力プロトコルのメンバー2021-01-012021-12-310001001233SGMO:生物遺伝子協力プロトコルのメンバー2020-01-012020-12-310001001233アメリカ公認会計基準:従業員ストックメンバー2022-01-012022-12-310001001233アメリカ公認会計基準:従業員ストックメンバー2021-01-012021-12-310001001233アメリカ公認会計基準:従業員ストックメンバー2020-01-012020-12-310001001233アメリカ-公認会計基準:連携性手配メンバーSGMO:ChangeInCollaborationAgreementスコープメンバSGMO:ファイザーSB 525メンバー2021-01-012021-12-310001001233アメリカ-公認会計基準:連携性手配メンバーSGMO:ChangeInCollaborationAgreementスコープメンバSGMO:ファイザーSB 525メンバー2020-01-012020-12-310001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:ノワール生物医学研究所IncMember2022-01-012022-12-31Xbrli:純0001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:ノワール生物医学研究所IncMember2021-01-012021-12-310001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:ノワール生物医学研究所IncMember2020-01-012020-12-310001001233国家薬品監督管理局:KtePharmaIncMember米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバ2022-01-012022-12-310001001233国家薬品監督管理局:KtePharmaIncMember米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバ2021-01-012021-12-310001001233国家薬品監督管理局:KtePharmaIncMember米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバ2020-01-012020-12-310001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:BigenMAIncMembers2022-01-012022-12-310001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:BigenMAIncMembers2021-01-012021-12-310001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:BigenMAIncMembers2020-01-012020-12-310001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:Sanofiembersメンバー2022-01-012022-12-310001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:Sanofiembersメンバー2021-01-012021-12-310001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:Sanofiembersメンバー2020-01-012020-12-310001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:PfizerMember2022-01-012022-12-310001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:PfizerMember2021-01-012021-12-310001001233米国-GAAP:顧客と契約した収入SGMO:協調プロトコルからの収入付与と許可集中度リスクメンバSGMO:PfizerMember2020-01-012020-12-310001001233SGMO:売掛金連携プロトコルコスト補償集中度リスクメンバからアメリカ公認会計基準:売掛金メンバーSGMO:ノワール生物医学研究所IncMember2022-01-012022-12-310001001233SGMO:売掛金連携プロトコルコスト補償集中度リスクメンバからアメリカ公認会計基準:売掛金メンバーSGMO:ノワール生物医学研究所IncMember2021-01-012021-12-310001001233国家薬品監督管理局:KtePharmaIncMemberSGMO:売掛金連携プロトコルコスト補償集中度リスクメンバからアメリカ公認会計基準:売掛金メンバー2022-01-012022-12-310001001233国家薬品監督管理局:KtePharmaIncMemberSGMO:売掛金連携プロトコルコスト補償集中度リスクメンバからアメリカ公認会計基準:売掛金メンバー2021-01-012021-12-310001001233SGMO:売掛金連携プロトコルコスト補償集中度リスクメンバからアメリカ公認会計基準:売掛金メンバーSGMO:BigenMAIncMembers2022-01-012022-12-310001001233SGMO:売掛金連携プロトコルコスト補償集中度リスクメンバからアメリカ公認会計基準:売掛金メンバーSGMO:BigenMAIncMembers2021-01-012021-12-310001001233SGMO:売掛金連携プロトコルコスト補償集中度リスクメンバからアメリカ公認会計基準:売掛金メンバーSGMO:Sanofiembersメンバー2022-01-012022-12-310001001233SGMO:売掛金連携プロトコルコスト補償集中度リスクメンバからアメリカ公認会計基準:売掛金メンバーSGMO:Sanofiembersメンバー2021-01-012021-12-310001001233SRT:最小メンバ数2022-01-012022-12-310001001233SRT:最大メンバ数2022-01-012022-12-31SGMO:細分化市場0001001233アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:アメリカ政府スポンジ企業債務証券メンバー2022-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:アメリカ政府スポンジ企業債務証券メンバー2022-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-公認会計基準:アメリカ政府スポンジ企業債務証券メンバー2022-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:アメリカ政府スポンジ企業債務証券メンバー2022-12-310001001233米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバー米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバー米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-公認会計基準:会社債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:会社債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-公認会計基準:会社債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:会社債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233米国-公認会計基準:資産認可証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバー米国-公認会計基準:資産認可証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233米国-公認会計基準:資産認可証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバー米国-公認会計基準:資産認可証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:アメリカ証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ公認会計基準:預金メンバー資格認証2022-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ公認会計基準:預金メンバー資格認証2022-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ公認会計基準:預金メンバー資格認証2022-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ公認会計基準:預金メンバー資格認証2022-12-310001001233SGMO:米政府スポンジ債券代理メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーSGMO:米政府スポンジ債券代理メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233SGMO:米政府スポンジ債券代理メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2022-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーSGMO:米政府スポンジ債券代理メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2022-12-310001001233アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2021-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ公認会計基準:MoneyMarketFundsMembersアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2021-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:アメリカ政府スポンジ企業債務証券メンバー2021-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:アメリカ政府スポンジ企業債務証券メンバー2021-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ-公認会計基準:アメリカ政府スポンジ企業債務証券メンバー2021-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:アメリカ政府スポンジ企業債務証券メンバー2021-12-310001001233米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバー米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2021-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバー米国-GAAP:ビジネス紙のメンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ-公認会計基準:会社債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-公認会計基準:会社債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ-公認会計基準:会社債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2021-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-公認会計基準:会社債務証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233米国-公認会計基準:資産認可証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバー米国-公認会計基準:資産認可証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233米国-公認会計基準:資産認可証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバー2021-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバー米国-公認会計基準:資産認可証券メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定する2021-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ公認会計基準:預金メンバー資格認証2021-12-310001001233アメリカ-公認会計基準:公正価値入力レベル1メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ公認会計基準:預金メンバー資格認証2021-12-310001001233アメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ-公認会計基準:公正価値入力レベル2メンバーアメリカ公認会計基準:預金メンバー資格認証2021-12-310001001233アメリカ-公認会計基準:公正価値投入レベル3メンバーアメリカ-GAAP:公正価値は再帰的メンバーを測定するアメリカ公認会計基準:預金メンバー資格認証2021-12-310001001233アメリカ公認会計基準:MoneyMarketFundsMembers2022-12-310001001233アメリカ公認会計基準:現金等価物メンバー2022-12-310001001233アメリカ-公認会計基準:アメリカ政府スポンジ企業債務証券メンバー2022-12-310001001233米国-GAAP:ビジネス紙のメンバー2022-12-310001001233アメリカ-公認会計基準:会社債務証券メンバー2022-12-310001001233米国-公認会計基準:資産認可証券メンバー2022-12-310001001233アメリカ-公認会計基準:アメリカ証券メンバー2022-12-310001001233アメリカ公認会計基準:預金メンバー資格認証2022-12-310001001233SGMO:米政府スポンジ債券代理メンバー2022-12-310001001233アメリカ公認会計基準:MoneyMarketFundsMembers2021-12-310001001233アメリカ公認会計基準:現金等価物メンバー2021-12-310001001233アメリカ-公認会計基準:アメリカ政府スポンジ企業債務証券メンバー2021-12-310001001233米国-GAAP:ビジネス紙のメンバー2021-12-310001001233アメリカ-公認会計基準:会社債務証券メンバー2021-12-310001001233米国-公認会計基準:資産認可証券メンバー2021-12-310001001233アメリカ公認会計基準:預金メンバー資格認証2021-12-310001001233SGMO:連携と許可プロトコルメンバSGMO:ノワール生物医学研究所IncMember2020-08-012020-08-310001001233SRT:最大メンバ数SGMO:連携と許可プロトコルメンバSGMO:指定された臨床前開発と最初の商業販売マイルストーンのメンバーSGMO:ノワール生物医学研究所IncMember2020-07-272020-07-270001001233SRT:最大メンバ数SGMO:連携と許可プロトコルメンバSGMO:ノワール生物医学研究所IncMemberSGMO:ビジネスマイルストーンのメンバー2020-07-272020-07-270001001233SGMO:連携と許可プロトコルメンバSGMO:ノワール生物医学研究所IncMember2020-07-272020-07-270001001233SGMO:連携と許可プロトコルメンバSGMO:ノワール生物医学研究所IncMember2022-12-310001001233SGMO:連携と許可プロトコルメンバSGMO:ノワール生物医学研究所IncMember2021-12-310001001233SGMO:ノワール生物医学研究所IncMember2022-01-012022-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:ノワール生物医学研究所IncMember2022-01-012022-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:ノワール生物医学研究所IncMember2021-01-012021-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:ノワール生物医学研究所IncMember2020-01-012020-12-310001001233SGMO:ノワール生物医学研究所IncMember国家気象局:研究サービスメンバー2022-01-012022-12-310001001233SGMO:ノワール生物医学研究所IncMember国家気象局:研究サービスメンバー2021-01-012021-12-310001001233SGMO:ノワール生物医学研究所IncMember国家気象局:研究サービスメンバー2020-01-012020-12-310001001233SGMO:ノワール生物医学研究所IncMember2021-01-012021-12-310001001233SGMO:ノワール生物医学研究所IncMember2020-01-012020-12-310001001233SGMO:連携と許可プロトコルメンバSGMO:ノワール生物医学研究所IncMember2020-12-310001001233SGMO:連携と許可プロトコルメンバSGMO:ノワール生物医学研究所IncMember2020-01-012020-12-310001001233SGMO:連携と許可プロトコルメンバSGMO:ノワール生物医学研究所IncMember2022-01-012022-12-310001001233SGMO:連携と許可プロトコルメンバSGMO:ノワール生物医学研究所IncMember2021-01-012021-12-310001001233SGMO:株式購入プロトコルメンバーSGMO:BigenMAIncMembers2020-04-012020-04-300001001233SGMO:株式購入プロトコルメンバーSGMO:BigenMAIncMembers2020-04-300001001233SGMO:連携と許可プロトコルメンバSGMO:BigenMAIncMembers2020-05-012020-05-310001001233SGMO:連携と許可プロトコルメンバSGMO:BigenMAIncMembers2020-04-300001001233SGMO:連携と許可プロトコルメンバSGMO:BigenMAIncMembersSGMO:PreApprovalMilestoneMember2020-04-300001001233SGMO:連携と許可プロトコルメンバSGMO:BigenMAIncMembersSGMO:販売ベースのマイルストーンメンバー2020-04-300001001233SGMO:連携と許可プロトコルメンバSGMO:BigenMAIncMembers2020-04-012020-04-30SGMO:製品_目標0001001233SRT:最大メンバ数SGMO:連携と許可プロトコルメンバSGMO:BigenMAIncMembers2020-04-012020-04-300001001233SGMO:連携と許可プロトコルメンバSGMO:BigenMAIncMembers2022-01-012022-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:BigenMAIncMembers2020-04-300001001233US-GAAP:ライセンスとサービスメンバーSGMO:BigenMAIncMembers2022-12-31SGMO:マテリアル_権限0001001233SGMO:株式購入プロトコルメンバーSGMO:BigenMAIncMembers2022-12-310001001233SGMO:株式購入プロトコルメンバーSGMO:BigenMAIncMembers2021-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:BigenMAIncMembers2021-12-310001001233SGMO:BigenMAIncMembers2022-01-012022-12-310001001233アメリカ公認会計基準:ライセンスメンバーSGMO:BigenMAIncMembers2022-01-012022-12-310001001233アメリカ公認会計基準:ライセンスメンバーSGMO:BigenMAIncMembers2021-01-012021-12-310001001233アメリカ公認会計基準:ライセンスメンバーSGMO:BigenMAIncMembers2020-01-012020-12-310001001233SGMO:BigenMAIncMembers国家気象局:研究サービスメンバー2022-01-012022-12-310001001233SGMO:BigenMAIncMembers国家気象局:研究サービスメンバー2021-01-012021-12-310001001233SGMO:BigenMAIncMembers国家気象局:研究サービスメンバー2020-01-012020-12-310001001233SGMO:BigenMAIncMembers2021-01-012021-12-310001001233SGMO:BigenMAIncMembers2020-01-012020-12-310001001233SGMO:連携と許可プロトコルメンバSGMO:BigenMAIncMembers2020-12-310001001233SGMO:連携と許可プロトコルメンバSGMO:BigenMAIncMembers2022-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:BigenMAIncMembers2022-01-012022-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:BigenMAIncMembers2021-01-012021-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:BigenMAIncMembers2020-01-012020-12-310001001233SGMO:株式購入プロトコルメンバーSGMO:BigenMAIncMembers2020-01-012020-12-310001001233国家薬品監督管理局:KtePharmaIncMemberSGMO:連携と許可プロトコルメンバ2018-04-012018-04-300001001233SRT:最大メンバ数国家薬品監督管理局:KtePharmaIncMemberSGMO:連携と許可プロトコルメンバ2018-04-012018-04-300001001233国家薬品監督管理局:KtePharmaIncMemberSGMO:連携と許可プロトコルメンバ国家薬品監督管理局:得られた成果指定の研究臨床開発監督と第一商業販売マイルストーンのメンバー2018-04-012018-04-300001001233国家薬品監督管理局:KtePharmaIncMemberSGMO:連携と許可プロトコルメンバSGMO:完了数指定売上高マイルストーンに基づいて世界的に純売上高許可数製品が指定レベルのメンバーに達する場合2018-04-012018-04-30SGMO:マイルストーンSGMO:オプション0001001233国家薬品監督管理局:KtePharmaIncMemberUS-GAAP:ライセンスとサービスメンバー2018-04-300001001233国家薬品監督管理局:KtePharmaIncMemberUS-GAAP:ライセンスとサービスメンバー2019-09-30SGMO:パフォーマンス_義務0001001233国家薬品監督管理局:KtePharmaIncMemberUS-GAAP:ライセンスとサービスメンバー2019-09-012019-09-300001001233国家薬品監督管理局:KtePharmaIncMemberSGMO:連携と許可プロトコルメンバ2022-12-310001001233国家薬品監督管理局:KtePharmaIncMemberSGMO:連携と許可プロトコルメンバ2021-12-310001001233国家薬品監督管理局:KtePharmaIncMemberUS-GAAP:ライセンスとサービスメンバー2022-12-310001001233国家薬品監督管理局:KtePharmaIncMemberUS-GAAP:ライセンスとサービスメンバー2021-12-310001001233国家薬品監督管理局:KtePharmaIncMember2022-01-012022-12-310001001233国家薬品監督管理局:KtePharmaIncMemberアメリカ公認会計基準:ライセンスメンバー2022-01-012022-12-310001001233国家薬品監督管理局:KtePharmaIncMemberアメリカ公認会計基準:ライセンスメンバー2021-01-012021-12-310001001233国家薬品監督管理局:KtePharmaIncMemberアメリカ公認会計基準:ライセンスメンバー2020-01-012020-12-310001001233国家薬品監督管理局:KtePharmaIncMember国家気象局:研究サービスメンバー2022-01-012022-12-310001001233国家薬品監督管理局:KtePharmaIncMember国家気象局:研究サービスメンバー2021-01-012021-12-310001001233国家薬品監督管理局:KtePharmaIncMember国家気象局:研究サービスメンバー2020-01-012020-12-310001001233国家薬品監督管理局:KtePharmaIncMember2021-01-012021-12-310001001233国家薬品監督管理局:KtePharmaIncMember2020-01-012020-12-310001001233SGMO:PfizerMember2017-05-012017-05-310001001233SGMO:SBFiveTwoFiveAndOtherProductsMemberSGMO:PfizerMember2017-05-012017-05-310001001233SGMO:成果指定臨床開発知的財産権と規制マイルストーンのメンバーSGMO:SBFiveTwoFiveAndOtherProductsMemberSGMO:PfizerMember2017-05-012017-05-310001001233SGMO:最初の商業販売マイルストーンの成果メンバーSGMO:SBFiveTwoFiveAndOtherProductsMemberSGMO:PfizerMember2017-05-012017-05-310001001233SGMO:SBFiveTwoFiveAndOtherProductsMemberSGMO:PfizerMember2017-05-310001001233SGMO:その他の製品メンバーSGMO:PfizerMember2017-05-310001001233SRT:最小メンバ数US-GAAP:ライセンスとサービスメンバーSGMO:PfizerMember2017-05-310001001233SRT:最大メンバ数US-GAAP:ライセンスとサービスメンバーSGMO:PfizerMember2017-05-310001001233SGMO:SBFiveTwoFiveAndOtherProductsMemberSGMO:ファイザーSB 525メンバー2017-05-012022-12-31SGMO:製品SGMO:印税_費用0001001233SGMO:ファイザーSB 525メンバー2017-05-012022-12-310001001233SGMO:ファイザーSB 525メンバー2022-01-012022-12-310001001233SGMO:PfizerGiroctocogene FitelparvoveMembers2019-12-012019-12-310001001233SGMO:AmendedCollaborationAndライセンスプロトコルのメンバーSGMO:PfizerGiroctocogene FitelparvoveMembers2019-12-012020-12-310001001233SGMO:AmendedCollaborationAndライセンスプロトコルのメンバーSGMO:PfizerGiroctocogene FitelparvoveMembers2020-01-012020-12-310001001233SGMO:第3段階臨床試験メンバーSGMO:PfizerGiroctocogene FitelparvoveMembers2020-09-012020-09-300001001233SGMO:第3段階臨床試験メンバーSGMO:PfizerGiroctocogene FitelparvoveMembers2020-12-310001001233SGMO:PfizerGiroctocogene FitelparvoveMembers2022-01-012022-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:PfizerGiroctocogene FitelparvoveMembers2022-01-012022-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:PfizerGiroctocogene FitelparvoveMembers2021-01-012021-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:PfizerGiroctocogene FitelparvoveMembers2020-01-012020-12-310001001233SGMO:記念碑的業績メンバーSGMO:PfizerGiroctocogene FitelparvoveMembers2022-01-012022-12-310001001233SGMO:記念碑的業績メンバーSGMO:PfizerGiroctocogene FitelparvoveMembers2021-01-012021-12-310001001233SGMO:記念碑的業績メンバーSGMO:PfizerGiroctocogene FitelparvoveMembers2020-01-012020-12-310001001233SGMO:PfizerGiroctocogene FitelparvoveMembers2021-01-012021-12-310001001233SGMO:PfizerGiroctocogene FitelparvoveMembers2020-01-012020-12-310001001233SGMO:PfizerC 9 ORF 72メンバー2017-12-012017-12-310001001233SRT:最大メンバ数SGMO:指定された臨床前開発と最初の商業販売マイルストーンのメンバーSGMO:PfizerC 9 ORF 72メンバーSGMO:CNINOまたはRFSINGの2人のメンバー2017-12-012017-12-310001001233SRT:最大メンバ数SGMO:PfizerC 9 ORF 72メンバーSGMO:CNINOまたはRFSINGの2人のメンバーSGMO:ビジネスマイルストーンのメンバー2017-12-012017-12-310001001233SRT:最小メンバ数US-GAAP:ライセンスとサービスメンバーSGMO:PfizerMember2017-12-310001001233SRT:最大メンバ数US-GAAP:ライセンスとサービスメンバーSGMO:PfizerMember2017-12-310001001233SGMO:PfizerC 9 ORF 72メンバー2017-12-012021-12-310001001233SGMO:PfizerMemberSGMO:CNINOまたはRFSINGの2人のメンバー2017-12-012017-12-310001001233SGMO:PfizerC 9 ORF 72メンバーSGMO:CNINOまたはRFSINGの2人のメンバー2017-12-012017-12-310001001233SGMO:PfizerMemberSGMO:CNINOまたはRFSINGの2人のメンバー2020-09-012020-09-300001001233SGMO:PfizerMemberSGMO:CNINOまたはRFSINGの2人のメンバー2020-01-012020-12-310001001233SGMO:PfizerC 9 ORF 72メンバーSGMO:CNINOまたはRFSINGの2人のメンバー2022-01-012022-12-310001001233SGMO:PfizerC 9 ORF 72メンバーSGMO:CNINOまたはRFSINGの2人のメンバー2021-01-012021-12-310001001233SGMO:PfizerC 9 ORF 72メンバー2022-01-012022-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:PfizerC 9 ORF 72メンバー2022-01-012022-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:PfizerC 9 ORF 72メンバー2021-01-012021-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:PfizerC 9 ORF 72メンバー2020-01-012020-12-310001001233SGMO:記念碑的業績メンバーSGMO:PfizerC 9 ORF 72メンバー2022-01-012022-12-310001001233SGMO:記念碑的業績メンバーSGMO:PfizerC 9 ORF 72メンバー2021-01-012021-12-310001001233SGMO:記念碑的業績メンバーSGMO:PfizerC 9 ORF 72メンバー2020-01-012020-12-310001001233SGMO:PfizerC 9 ORF 72メンバー2021-01-012021-12-310001001233SGMO:PfizerC 9 ORF 72メンバー2020-01-012020-12-310001001233SGMO:PfizerMember2020-01-012020-12-310001001233SGMO:Sanofiembersメンバー2014-01-012014-01-31SGMO:計画0001001233SGMO:Sanofiembersメンバー2014-01-012022-06-280001001233SGMO:Sanofiembersメンバー2014-01-012022-12-310001001233SGMO:連携と許可プロトコルメンバSGMO:Sanofiembersメンバー2014-01-012022-12-310001001233SGMO:Sanofiembersメンバー2022-09-062022-09-060001001233SGMO:連携と許可プロトコルメンバSGMO:Sanofiembersメンバー2014-01-012022-06-280001001233SGMO:連携と許可プロトコルメンバSGMO:Sanofiembersメンバー国家気象局:マイルストーン3メンバー2022-06-280001001233SGMO:連携と許可プロトコルメンバSGMO:Sanofiembersメンバー2022-12-310001001233SGMO:連携と許可プロトコルメンバSGMO:Sanofiembersメンバー2021-12-310001001233アメリカ-GAAP:前払い費用と他の現在の資産メンバー2022-01-012022-12-310001001233SGMO:Sanofiembersメンバー2022-01-012022-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:Sanofiembersメンバー2022-01-012022-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:Sanofiembersメンバー2021-01-012021-12-310001001233US-GAAP:ライセンスとサービスメンバーSGMO:Sanofiembersメンバー2020-01-012020-12-310001001233SGMO:Sanofiembersメンバー国家気象局:研究サービスメンバー2022-01-012022-12-310001001233SGMO:Sanofiembersメンバー国家気象局:研究サービスメンバー2021-01-012021-12-310001001233SGMO:Sanofiembersメンバー国家気象局:研究サービスメンバー2020-01-012020-12-310001001233SGMO:記念碑的業績メンバーSGMO:Sanofiembersメンバー2022-01-012022-12-310001001233SGMO:記念碑的業績メンバーSGMO:Sanofiembersメンバー2021-01-012021-12-310001001233SGMO:記念碑的業績メンバーSGMO:Sanofiembersメンバー2020-01-012020-12-310001001233SGMO:Sanofiembersメンバー2021-01-012021-12-310001001233SGMO:Sanofiembersメンバー2020-01-012020-12-310001001233SGMO:カリフォルニア再生医療協会のメンバー2018-05-012018-05-310001001233アメリカ-公認会計基準:メンバーに授与SGMO:カリフォルニア再生医療協会のメンバー2020-01-012020-12-310001001233SGMO:カリフォルニア再生医療協会のメンバー2020-12-310001001233SGMO:カリフォルニア再生医療協会のメンバー2021-12-310001001233アメリカ-公認会計基準:メンバーに授与SGMO:カリフォルニア再生医療協会のメンバー2021-11-012021-11-300001001233アメリカ-公認会計基準:メンバーに授与SGMO:カリフォルニア再生医療協会のメンバー2021-11-300001001233SGMO:カリフォルニア再生医療協会のメンバー2022-12-310001001233SGMO:SigmaAldrichCorporationメンバー2007-01-012007-12-310001001233SGMO:SigmaAldrichCorporationメンバー2009-10-310001001233SGMO:SigmaAldrichCorporationメンバー2009-10-012009-10-310001001233アメリカ公認会計基準:ライセンスメンバーSGMO:SigmaAldrichCorporationメンバー2009-10-310001001233US-GAAP:ライセンスプロトコル用語メンバSGMO:SigmaAldrichCorporationメンバー2009-10-012009-10-310001001233SGMO:SigmaAldrichCorporationメンバー2022-01-012022-12-310001001233SGMO:SigmaAldrichCorporationメンバー2021-01-012021-12-310001001233SGMO:SigmaAldrichCorporationメンバー2020-01-012020-12-310001001233US-GAAP:ライセンスプロトコル用語メンバSGMO:DowAgroScienceメンバー2005-01-012005-12-310001001233US-GAAP:ライセンスプロトコル用語メンバSGMO:DowAgroScienceメンバー2005-12-310001001233US-GAAP:RoyaltyMemberUS-GAAP:ライセンスプロトコル用語メンバSGMO:DowAgroScienceメンバー2005-01-012005-12-310001001233SRT:最小メンバ数US-GAAP:ライセンスプロトコル用語メンバSGMO:DowAgroScienceメンバー2005-01-012005-12-310001001233SRT:最大メンバ数US-GAAP:ライセンスプロトコル用語メンバSGMO:DowAgroScienceメンバー2005-01-012005-12-310001001233US-GAAP:ライセンスプロトコル用語メンバSGMO:DowAgroScienceメンバー2020-10-012020-10-300001001233アメリカ公認会計基準:ライセンスメンバーUS-GAAP:ライセンスプロトコル用語メンバSGMO:DowAgroScienceメンバー2022-01-012022-12-310001001233アメリカ公認会計基準:ライセンスメンバーUS-GAAP:ライセンスプロトコル用語メンバSGMO:DowAgroScienceメンバー2021-01-012021-12-310001001233アメリカ公認会計基準:ライセンスメンバーUS-GAAP:ライセンスプロトコル用語メンバSGMO:DowAgroScienceメンバー2020-01-012020-12-310001001233SGMO:SangamoFranceMemberSGMO:共有購入プロトコルと見積プロトコルメンバ2018-12-310001001233SGMO:SangamoFranceMemberSGMO:共有購入プロトコルと見積プロトコルメンバ2018-07-200001001233SGMO:SangamoFranceMemberSGMO:共有購入プロトコルと見積プロトコルメンバ2022-12-310001001233SGMO:SangamoFranceMember2018-10-012018-10-010001001233SGMO:SangamoFranceMember2018-10-010001001233SGMO:SangamoFranceMemberSGMO:共有購入プロトコルと見積プロトコルメンバ2021-12-310001001233SGMO:実験室装置のメンバー2022-12-310001001233SGMO:実験室装置のメンバー2021-12-310001001233アメリカ-公認会計基準:リース改善メンバー2022-12-310001001233アメリカ-公認会計基準:リース改善メンバー2021-12-310001001233アメリカ-GAAP:家具と固定機器のメンバー2022-12-310001001233アメリカ-GAAP:家具と固定機器のメンバー2021-12-310001001233SGMO:製造設備メンバ2022-12-310001001233SGMO:製造設備メンバ2021-12-310001001233アメリカ-アメリカ公認会計基準:建設中のメンバー2022-12-310001001233アメリカ-アメリカ公認会計基準:建設中のメンバー2021-12-310001001233国家気象局:事務室と実験室のメンバーSGMO:カリフォルニア州ブリスベンのメンバー2022-12-31Utr:SQFT0001001233国家気象局:事務室と実験室のメンバー国家気象局:2031年8月満期国家気象局:リッチモンド·カリフォルニア州のメンバー2022-12-310001001233国家気象局:事務室と実験室のメンバー国家気象局:リッチモンド·カリフォルニア州のメンバー国家気象局:2026年8月満期2022-12-310001001233SGMO:ResearchAndOffice空間のメンバーSGMO:ValbonneFranceMembers2022-12-310001001233国家気象局:リッチモンド·カリフォルニア州のメンバー2020-05-310001001233国家気象局:リッチモンド·カリフォルニア州のメンバー2021-01-310001001233国家気象局:リッチモンド·カリフォルニア州のメンバー2021-02-010001001233SGMO:ValbonneFranceMembers2021-01-310001001233SGMO:ValbonneFranceMembers2021-01-2900010012332021-10-310001001233国家気象局:1201年11月~8月31日国家気象局:リッチモンド·カリフォルニア州のメンバー2021-10-310001001233国家気象局:リッチモンド·カリフォルニア州のメンバー2021-10-010001001233SRT:最小メンバ数2022-12-310001001233SRT:最大メンバ数2022-12-310001001233SGMO:ランサオランダB 11月2022-12-3100010012332020-06-2900010012332020-06-300001001233SGMO:AtTheMarketOfferingAgreementメンバーSGMO:JefferiesLLCMメンバー2020-08-012020-08-310001001233SGMO:AtTheMarketOfferingAgreementメンバーSGMO:JefferiesLLCMメンバー2022-12-012022-12-310001001233SGMO:AtTheMarketOfferingAgreementメンバーSGMO:JefferiesLLCMメンバー2022-01-012022-12-310001001233SGMO:AtTheMarketOfferingAgreementメンバーSGMO:JefferiesLLCMメンバー2021-01-012021-12-310001001233SGMO:2018 Stock IncentivePlanMembersを再起動2020-05-310001001233SGMO:2018 Stock IncentivePlanMembersを再起動2022-05-310001001233SGMO:二千八百十八株インセンティブ計画メンバー2022-01-012022-12-310001001233SRT:最大メンバ数SGMO:二千八百十八株インセンティブ計画メンバー2022-01-012022-12-310001001233SRT:最小メンバ数SGMO:二千八百十八株インセンティブ計画メンバー2022-01-012022-12-310001001233SGMO:二千八百十八株インセンティブ計画メンバー2022-12-310001001233SGMO:EmployeStockPurachePlanMember2021-05-310001001233SGMO:EmployeStockPurachePlanMember2021-05-012021-05-310001001233米国-GAAP:制限株式単位RSUメンバー2022-01-012022-12-310001001233米国-GAAP:制限株式単位RSUメンバー2021-01-012021-12-310001001233米国-GAAP:制限株式単位RSUメンバー2020-01-012020-12-310001001233米国-公認会計基準:研究·開発費メンバー2022-01-012022-12-310001001233米国-公認会計基準:研究·開発費メンバー2021-01-012021-12-310001001233米国-公認会計基準:研究·開発費メンバー2020-01-012020-12-310001001233アメリカ-公認会計基準:一般と行政費用メンバー2022-01-012022-12-310001001233アメリカ-公認会計基準:一般と行政費用メンバー2021-01-012021-12-310001001233アメリカ-公認会計基準:一般と行政費用メンバー2020-01-012020-12-310001001233米国-GAAP:制限株式単位RSUメンバー2022-12-310001001233SRT:最小メンバ数米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001001233SRT:最大メンバ数米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001001233SRT:最小メンバ数米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001001233SRT:最大メンバ数米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001001233SRT:最小メンバ数米国-公認会計基準:従業員株式オプションメンバー2020-01-012020-12-310001001233SRT:最大メンバ数米国-公認会計基準:従業員株式オプションメンバー2020-01-012020-12-310001001233米国-公認会計基準:従業員株式オプションメンバー2022-01-012022-12-310001001233米国-公認会計基準:従業員株式オプションメンバー2021-01-012021-12-310001001233米国-公認会計基準:従業員株式オプションメンバー2020-01-012020-12-310001001233SGMO:EmployeStockPurachePlanMemberSRT:最小メンバ数2022-01-012022-12-310001001233SRT:最大メンバ数SGMO:EmployeStockPurachePlanMember2022-01-012022-12-310001001233SGMO:EmployeStockPurachePlanMemberSRT:最小メンバ数2021-01-012021-12-310001001233SRT:最大メンバ数SGMO:EmployeStockPurachePlanMember2021-01-012021-12-310001001233SGMO:EmployeStockPurachePlanMemberSRT:最小メンバ数2020-01-012020-12-310001001233SRT:最大メンバ数SGMO:EmployeStockPurachePlanMember2020-01-012020-12-310001001233SGMO:EmployeStockPurachePlanMember2022-01-012022-12-310001001233SGMO:EmployeStockPurachePlanMember2021-01-012021-12-310001001233SGMO:EmployeStockPurachePlanMember2020-01-012020-12-310001001233米国-GAAP:他の非現在の資産メンバ2022-12-310001001233米国-GAAP:他の非現在の資産メンバ2021-12-310001001233アメリカ-GAAP:経済金融と業界フランスのメンバーのミクロ構造2022-12-31 アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
_______________________________________________________________________________________________
表10-K
_______________________________________________________________________________________________ | | | | | |
| ☒ | 1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで十二月三十一日, 2022
あるいは…。 | | | | | |
| ☐ | 1934年証券取引法第13条又は15条に基づいて提出された移行報告 |
そこからの過渡期について
依頼書類番号:000-30171
_______________________________________________________________________________________________
Sangamo治療会社
(登録者の正確な氏名はその定款に記載)
_______________________________________________________________________________________________ | | | | | | | | | | | | | | |
| デラウェア州 | | | | 68-0359556 |
(明またはその他の司法管轄権
会社や組織) | | | | (税務署の雇用主
識別番号) |
| | | | |
| マリナ通り7000番地です。 | | | | |
ブリスベン, カリフォルニア州 | | | | 94005 |
| (主にオフィスアドレスを実行) | | (郵便番号) |
(510) 970-6000
(登録者の電話番号、市外局番を含む)
同法第12条(B)に基づいて登録された証券: | | | | | | | | |
| クラスごとのタイトル | 取引コード | 登録された各取引所の名称 |
| 普通株は、1株当たり0.01ドルです | SGMO | ナスダック世界ベスト市場 |
同法第12条(G)により登録された証券:なし
______________________________________________________________________________________________
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。はい。☐違います。 ☒
登録者が取引法第13節又は第15節(D)節に基づいて報告書を提出する必要がない場合は,再選択マークで示してください。はい。☐違います。 ☒
再選択マークは、登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13条または15(D)節に提出されたすべての報告書を提出したかどうか、および(2)過去90日以内にそのような提出要件に適合しているかどうかを示すはい、そうです ☒ No ☐
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示すはい、そうです ☒ No ☐
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。 | | | | | | | | | | | | | | |
| 大型加速ファイルサーバ | ☒ | | ファイルマネージャを加速する | ☐ |
| 非加速ファイルサーバ | ☐ | | 規模の小さい報告会社 | ☐ |
| | | 新興成長型会社 | ☐ |
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる☒
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意の実行者が関連回復中に第240.10 D−1(B)条に従って受信されたインセンティブベースの補償に従って回復分析を行う必要があるかどうかを再選択マークで示す
登録者が空殻会社であるか否かをチェックマークで示す(同法第12 b-2条で定義される)。はい、違います☒
ナスダック世界精選市場によると、登録者の非関連会社が保有する普通株の総時価は、2022年6月30日(登録者が最近完成した第2四半期の最終営業日)の普通株の終値に基づいて$と計算される632,920,753それは.この計算については、登録者の役員と役員は連属会社とみなされている。他の目的に対して,このような関連地位の決定は必ずしも決定的な決定であるとは限らない.
2023年2月17日までに168,483,317普通株の1株当たり額面は0.01ドルで、発行された。
引用で編入された書類
この表格10−Kの第III部第10−14項に要求されるいくつかの情報は、登録者が2023年株主総会に提出した最終委託書を参照して編入されたものであり、この委託書は、当表格10−Kがカバーする財政年度終了後120日以内に第14 A条の規定に従って証券取引委員会に提出されるが、当該委託書がその期間内に提出されていない場合、そのような情報は、120日以内に提出される予定である本表格10−Kに対する修正案に含まれる.
カタログ | | | | | | | | |
| | ページ |
| 第1部 | |
| | |
第1項。 | 業務.業務 | 6 |
第1 A項。 | リスク要因 | 62 |
項目1 B。 | 未解決従業員意見 | 90 |
第二項です。 | 属性 | 90 |
第三項です。 | 法律訴訟 | 90 |
第四項です。 | 炭鉱安全情報開示 | 90 |
| | |
| 第II部 | |
| | |
五番目です。 | 登録者普通株市場、関連株主事項及び発行者による株式証券の購入 | 91 |
第六項です。 | [保留されている] | 92 |
第七項。 | 経営陣の財務状況と経営成果の検討と分析 | 92 |
第七A項。 | 市場リスクの定量的·定性的開示について | 101 |
第八項です。 | 財務諸表と補足データ | 103 |
第九項です。 | 会計と財務情報開示の変更と相違 | 140 |
第9条。 | 制御とプログラム | 140 |
プロジェクト9 B。 | その他の情報 | 142 |
プロジェクト9 Cです。 | 検査妨害に関する外国司法管区の開示 | 142 |
| | |
| 第三部 | |
| | |
第10項。 | 役員·幹部と会社の管理 | 142 |
第十一項。 | 役員報酬 | 142 |
第十二項。 | 特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 | 142 |
十三項。 | 特定の関係や関連取引、取締役の独立性 | 143 |
14項です。 | 最高料金とサービス | 143 |
| | |
| 第4部 | |
| | |
第十五項。 | 展示品と財務諸表の付表 | 144 |
第十六項。 | 表格10-Kの概要 | 148 |
| | |
| サイン | |
前向き陳述に関する特別説明
本報告に含まれるいくつかの陳述は、“1933年証券法”(改正された)第27 A節又は“証券法”及び“1934年証券取引法”(改正された)第21 E節又は“取引法”が指す“前向き陳述”に属する。これらの陳述は私たちの未来の事件と関係があり、私たちが予想している運営、研究、開発、製造と商業化活動、臨床試験、経営結果と財務状況を含む。これらの展望性陳述は既知と未知のリスク、不確定要素とその他の要素に関連し、私たちの実際の結果、表現或いは成果と展望性陳述の明示或いは暗示の任意の未来の結果、表現或いは成果とは大きく異なる可能性がある。前向きな陳述は、以下の態様に関する陳述を含むことができるが、これらに限定されない
•私たちの戦略は
•候補製品の予期される研究および開発、およびそれによって生成される任意の承認された製品の潜在的商業化;
•私たちの協力者または戦略パートナーとの臨床前研究と臨床試験の開始、範囲、進展速度、登録、用量、予想結果と時間
•治療効果の持続性を含む候補製品の治療と商業的潜在力
•我々の遺伝子治療および細胞治療技術、亜鉛指(ZF)技術プラットフォーム、亜鉛指ヌクレアーゼ(ZF)および亜鉛指転写調節因子(ZF-TRs)を含む候補製品で使用される技術の治療および商業的潜在力、亜鉛指阻害剤(ZF-Rs)および亜鉛指活性化因子(ZF-AS)を含む
•私たちは、BIVV 003計画のための潜在的な新しいパートナーを見つける能力を含む、協力および戦略的パートナーシップを確立し、維持し、そのような計画の予想される利点を達成する能力がある
•既存と新しい協力の期待収入とその時間;
•私たちの業務と運営、私たちの協力者の業務と運営(臨床試験と製造を含む)に対する新しい冠肺炎疫病の影響と、私たちがこのような影響を管理する能力の推定
•私たちの研究開発や他の費用は
•私たちは既存で潜在的な新しいサプライヤーと製造業者から、あるいは私たち自身の内部製造施設から十分な臨床前および臨床候補製品供給を得ることができます
•Sangamoと私たちのパートナーおよび戦略パートナーは、候補製品の規制承認を獲得し、維持する能力と、規制承認を得ることに関連する時間およびコスト;
•私たちが法規の要求、義務、制限を遵守する能力と私たちの業務と運営に与える影響
•私たちは、他人の知的財産権を侵害することなく、私たちの知的財産権を保護し、私たちの業務を運営する能力、私たちの候補製品を開発し、商業化するために必要な技術の権利を獲得し、維持する能力を含む
•競合製品と候補製品が私たちの競争地位に与える影響と、私たちがこのような競争に対応すべき能力を含む競争発展
•私たちは現金資源と支出、資本需要、追加融資需要の推定、そして私たちが追加融資を得る能力
•私たちが経営を続ける能力があるかどうかを疑わせる条件や事件
•私たちはビジネスの成長を管理しています
•私たちが予想している経営と財務業績は
•私たちの運営と法的リスクは
•私たちの計画、目標、期待、そして意図、そして他の非歴史的事実の陳述。
場合によっては、将来の日付または以下の用語を使用することによって、“予想”、“信じる”、“継続”、“可能”、“推定”、“予想”、“可能”、“可能”、“計画”、“求める”、“すべき”、“会議する”、および前向き陳述を識別するための同様の表現を識別することによって、前向きな陳述を識別することができる。これらの陳述は、私たちの現在の未来の事件に対する見方を反映しており、仮説に基づいており、リスクと不確定要素の影響を受けている。このような危険と不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。我々は、この10-K表年次報告書の“リスク要因”および“経営陣の財務状況および経営結果の議論および分析”のタイトルの下で、その中の多くのリスクをより詳細に議論し、法的要件がない限り、新たな情報または将来のイベントまたは発展を反映するために、いかなる前向きな陳述を更新または修正する義務はない。前向き陳述に過度に依存しないように注意し,これらの前向き陳述は本年度報告がForm 10−Kの形で発表された日にのみ発表された。
リスク要因の概要
私たちの業務は重大な危険と関連がある。以下は,我々の普通株への投資に投機的かつリスクを持たせる我々の業務が直面している重大なリスクの概要である.この要約はこのようなすべての危険を扱っていない。以下、本年度報告表格10−K第I部第1 A項の“リスク要因”というタイトルの下で、これらのリスクについてより全面的に説明する。私たちの普通株に投資決定を下す前に、あなたはこのような危険を慎重に考慮しなければならない。以下に述べる任意のイベントまたは開発の発生は、当社の業務、経営結果、財務状況、見通し、および株価に重大な悪影響を及ぼす可能性があります。この場合、私たちの普通株の市場価格は下落するかもしれません。あなたはあなたの全部または一部の投資を失うかもしれません。また、私たちが現在知らない、あるいは私たちが現在どうでもいいと思っている他のリスクもあり、これらのリスクは私たちの業務、運営、あるいは私たちの普通株の市場価格に大きな影響を与える可能性もある。
•私たちは臨床段階のバイオテクノロジー会社で、承認された製品や製品の収入を得ていない。著者らの成功は臨床試験結果に大きく依存し、この結果は著者らの候補製品の安全性と有効性を証明し、監督管理機関を満足させた。積極的な臨床試験結果と監督管理の承認を得ることは高価で、長く、挑戦性と予測不可能であり、いかなる候補製品に対しても永遠に発生しない可能性がある。
•研究や臨床前研究の成功あるいは早期臨床試験の結果は,以降の試験で得られた結果を代表しない可能性がある。同様に、臨床試験の初歩的、初歩的或いは中期データは最終データと大きく異なる可能性がある。
•私たちの多くの候補製品は新しいZF技術に基づいており、これらの技術はまだ承認された商業的に実行可能な治療製品を生産していない
•設立以来、重大な運営損失が発生しており、予測可能な未来に損失が続くと予想されている。私たちは永遠に利益を上げないかもしれない。
•私たちは私たちの運営計画を実行し、継続的に経営する企業として運営するために多くの追加資金が必要になるだろう。私たちは割引条項でもっと多くの資金を集めることができないかもしれません。もしあれば、これは私たちの技術と候補製品を開発する能力を損害したり排除したりして、私たちの計画の一部または全部を延期または終了するかもしれません。将来の株式証券の売却と発行はまた、私たちの株主の株式を大幅に希釈する可能性がある。
•私たちは大手バイオ製薬会社との協力に深刻に依存して収入を創出し、私たちの多くの候補製品の開発、監督管理の承認を得て商業化している。もし私たちの協力者と衝突した場合、あるいは協力が何らかの理由で終了すれば、私たちの収入と製品開発はマイナスの影響を受けるだろう。
•バイオテクノロジーと遺伝子医学は競争の激しい産業だ。私たちの競争相手は私たちの競争相手よりも優れた技術と製品を開発したり、私たちの候補技術や製品よりも早く商業化するかもしれません。
•遺伝子薬物を製造することは複雑で、高価で、高度な規制と危険がある。私たちは現在第三者に強く依存しています‑党のメーカーは、私たち自身が製品を製造する経験が限られている。製造挑戦は予期せぬコスト、供給中断、そして私たちの製品開発作業への損害と遅延を招く可能性があります。
•私たちの候補製品が規制部門の承認を得ても、私たちが承認した製品は医師や患者の市場承認を得ることができない可能性があり、第三者支払者から十分な保険と補償を受けることができず、その商業的可能性を証明できない可能性もある。
•私たちはすべての予想される司法管轄区域で私たちの技術および候補製品のために必要かつ理想的な知的財産権保護を獲得、維持、実行することができない可能性があり、これは私たちの技術および製品開発作業の価値に悪影響を与え、高価で冗長で注意を分散させる訴訟のリスクを増加させ、予測できない結果をもたらす可能性がある。
•第三者は競争相手でもないかもしれませんが、彼らは私たちが不正な方法で彼らの特許または他の独占権を侵害、流用、または他の方法で行使していると主張するかもしれません。このような告発は、侵害訴訟、他の公金流用訴訟、または脅威がこのような行動をとる可能性があり、これらすべては、高価で冗長で注意を分散させる訴訟のリスクを増加させ、予測不可能な結果をもたらす可能性がある。
•私たちの成功は採用、統合とより多くの素質の高い熟練従業員を維持し、そして現在の肝心な幹部と従業員を維持することにかかっており、これらの人に対する激しい競争を考慮すると、これは挑戦性があるかもしれない
•新冠肺炎の流行は、私たちの業務と運営、そして私たちの協力者、メーカー、その他の商業パートナーの業務と運営に悪影響を与え続ける可能性がある。もしこれらの影響が実質的になれば、私たちの収入と製品開発努力は否定的な影響を受けるかもしれない。
•私たちの普通株の市場価格はずっと変動し続ける可能性があります。あなたは私たちの普通株への全部あるいは一部の投資を損失するかもしれません。
第1部
プロジェクト1--ビジネス
概要
我々は臨床段階の遺伝子薬物会社であり,画期的な科学を薬物に変換し,重篤な疾患を有する患者と家庭の生活を変えることに取り組んでいる。私たちは私たちの臨床と臨床前候補製品を開発することでこの使命を実現し、私たちの新しい科学と私たちの内部製造能力を利用する予定です。
私たちの候補製品
今日、私たちは診療所で私たちの第1波遺伝子療法と自己細胞療法候補者と一緒にいる。私たちの第2の長期発展戦略は、私たちの最適化された亜鉛指やZF技術の利用に集中することであり、これは差別化されたツールであり、私たちはそれを使用して、自己および同種細胞療法を含むゲノム薬を開発している体内にあるゲノム工学療法です
私たちの現在の臨床段階の候補は
•我々が所有しているファーブリー病を治療する遺伝子治療製品ST−920は,Isaralgagene Ciparvovecとも呼ばれ,現在われわれの1/2期STAAR臨床研究で評価されており,潜在的な3期臨床試験計画を行っている
•TX 200、我々は完全に所有しているキメラ抗原受容体、あるいはCAR、工学制御性T細胞、あるいはCAR-Treg、細胞治療製品、ヒト白血球抗原A 2不整合腎移植免疫を介した拒絶反応を予防するために、現在私たちの1/2期の堅固な臨床研究で評価されている
•Giroctocogene fitelparvovecはSB−525とも呼ばれ,重症血友病Aを治療する候補遺伝子治療製品であり,現在登録されている3期後発臨床試験が行われている。私たちは協力者のファイザーやファイザーと一緒にgiroctocogene fitelparvoecを開発しています
•BIVV 003は我々の亜鉛指ヌクレアーゼまたはZFヌクレアーゼであり、鎌状細胞病(SCD)を治療する候補遺伝子編集細胞治療製品であり、現在我々の1/2期PRECIZN-1臨床研究で評価されている。BIVV 003は、Sangamoが2022年6月にSanofi S.A.またはSanofiからSangamoに移行した後に完全所有するプロジェクトである。以下に述べるように,我々は最近,FabryとTX 200計画の資源を優先的に配置するために,1/2期PRECIZN-1研究が完了した後,BIVV 003計画へのさらなる実質的な投資を停止する戦略決定を行った.
われわれの臨床前開発は2つの革新の優先分野に集中している:(I)自己免疫疾患用CAR−Treg細胞療法と(Ii)神経疾患用ゲノム工学。著者らの臨床前プロジェクトの適応は神経発達障害、癌、炎症性腸疾患或いはIBD、神経病変と神経変性疾患、例えば筋萎縮性側索硬化症、多発性硬化症とハンチントン病を含み、その中のいくつかの疾患は著者らがパートナーのBiogen MA、Inc.とBiogen International GmbHと共同開発したものであり、著者らは総称してBiogen、ノワール生物医学研究研究所或いはノワール、輝瑞、武田製薬有限会社とKite Pharma,Inc.と呼ばれる。
私たちは生物製薬会社との何度もの協力は私たちに重要な財務と戦略的利益をもたらし、私たちの研究開発努力と私たちのZF技術プラットフォームの潜在力を強化した。彼らは著者らの協力者の治療と臨床専門知識及び商業資源を利用して、私たちの薬物を患者にもっと早くもたらすことを期待している。私たちはこれらの協力が私たちのZF技術プラットフォームの価値を反映し、潜在的に候補製品の潜在的な市場を拡大すると信じている。私たちはこれまでに約8.15億ドルの前払い許可料、マイルストーン支払い、私たちの普通株を協力者に売却する収益を受けており、潜在的な製品印税に加えて、私たちの協力から67億ドルまでの未来のマイルストーン支払いを稼ぐ機会がある。
私たちの小説“科学”は
著者らは亜鉛指蛋白(ZFP)の研究と開発においてリードしており、亜鉛指蛋白は大量に存在するヒト蛋白であり、すでにDNAと調節蛋白との相互作用を通じてゲノムを調節するように進化した。著者らはすでに(I)ゲノム編集とエピジェネティック調節において潜在的な臨床応用価値を有する独自の合成ZF技術プラットフォームを開発と最適化し、著者らは総称してゲノム工学、および(Ii)遺伝子編集細胞治療と呼ばれ、私たちは細胞治療と呼ばれる。
私たちの戦略は私たちの差別化と多機能なZF技術プラットフォームを最適或いは一流の臨床潜在力を持つ候補製品に転化することである。例えば、ZFPはZFヌクレアーゼに改造することができ、ZFヌクレアーゼは使用可能なタンパク質である
選択された遺伝子をノックアウトまたはノックアウトすることにより,DNA配列を専門的に修正してゲノムを編集する。ZFPはまた、亜鉛フィンガー転写調節剤、またはZF-TRsに改変することができ、遺伝子発現を選択的に増加または減少させることによってゲノムを調節することができるタンパク質である。
これらのゲノム工学技術を開発する過程で,多くの科学,製造,開発能力,遺伝子治療分野に広く適用される関連技術が蓄積され,我々の遺伝子治療候補製品の開発に利用されている。
最後に、著者らはまた、著者らのZF技術プラットフォームと買収によって得られた技術を利用して、腎臓移植拒絶反応、多発性硬化症とIBDを含む広範な患者集団の自己免疫性と炎症性疾患の治療に用いられるCAR-Treg候補製品の研究と開発の先頭になった。CARが導入した抗原特異性により,CAR−TregsはポリクローナルTregsよりも強い抑制機能を有すると考えられている
私たちの内部製造は
私たちは私たちの内部製造能力が私たちに競争優位を提供すると信じている。我々は現在カリフォルニア州ブリスベンの本社に腺関連ウイルス(AAV)製造工場を設置し,カリフォルニア州ブリスベンとフランスバル州に細胞療法メーカーを設置している。我々の製造戦略は,内部製造·契約製造組織(CMO)パートナーシップによるバランスと必要な能力を構築し,製造プロセスや分析に投資し,強力なサプライチェーンを発展させ,より大きな柔軟性,品質,制御を提供することである。我々のCMOは2000リットルまでのバイオリアクタ規模のAAV製造能力を提供し,この柔軟性を実現した。
商業動態
伊氏·ファブリー病
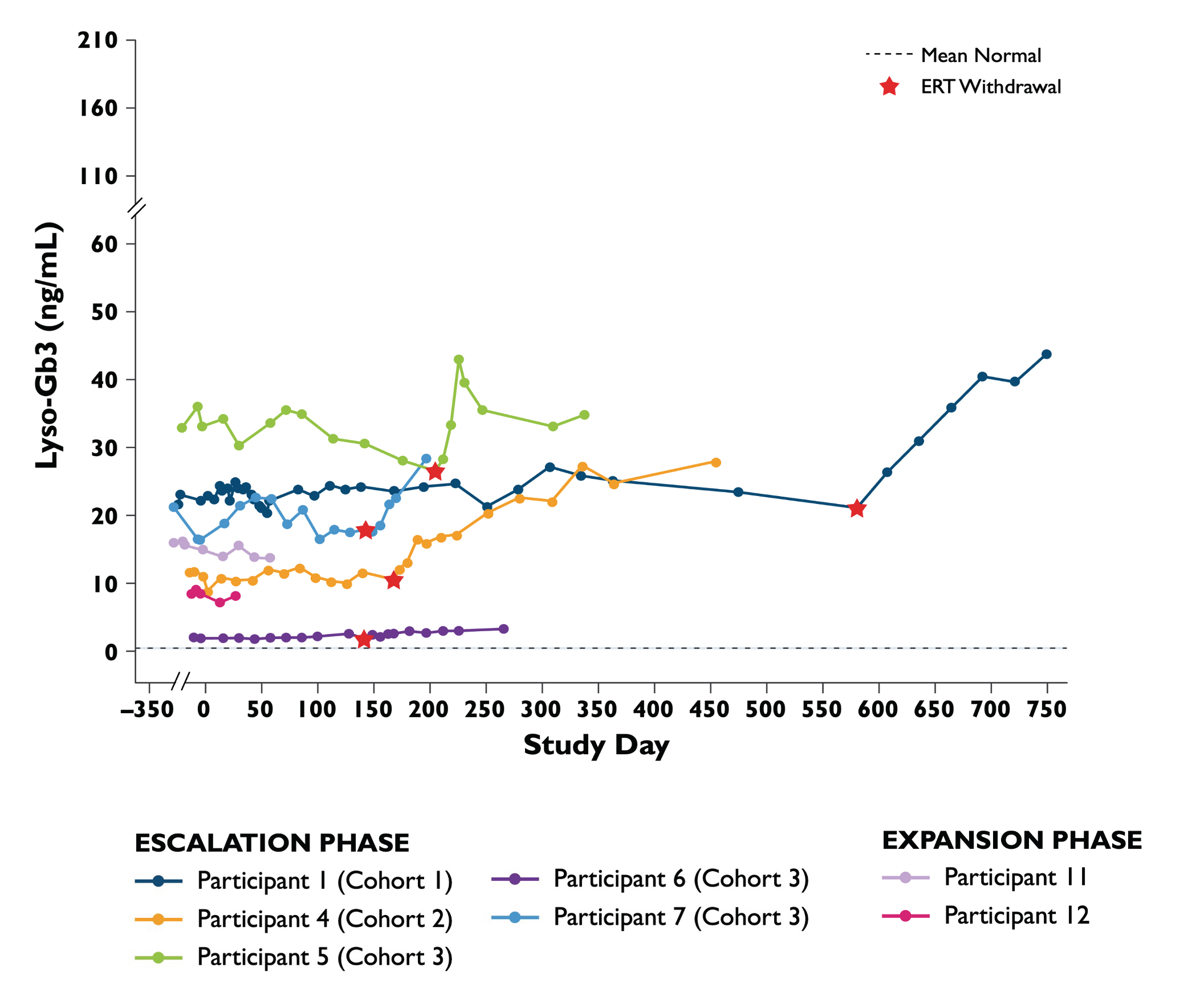
2023年2月22日、私たちは、Fabry病を治療するための完全な遺伝子治療製品候補製品であるIsaralgagene Cirobvovec、またはST-920を評価したSTAAR研究からの最新の臨床データを発表しましたこれは…。年度世界シンポジウム2023年2月24日。以下にデータの要約を示す.この声明には,2022年10月20日までにプロポフォール治療を受けた13名の患者のデータが含まれており,2人の患者の腎臓生検データが含まれている。締め切り以来,1/2期STAAR研究では4名の患者が薬物治療を受けており,これまでに計17名の患者が薬物治療を受けてきた。現在、20のサイトが活発に求人を行っている。より多くの男性や女性患者が現在スクリーニングを行っていることに伴い,この研究の進展は続いている。
1/2期STAAR研究拡張段階が進行中であり,潜在的な3期臨床試験の準備が積極的に行われている。監督部門の相互作用により,2023年末に第3段階試験が開始される予定であり,1人目の患者の投与量は早くても2024年上半期に開始される可能性がある。第1/2段階拡大段階の調剤は2023年末に完了する予定であり,第3段階試験を開始するゲートコントロール要因とはならないと予想される。
2022年12月,研究拡張段階の患者が輸液後14日に3級重篤な不良事象,すなわちSAEが出現し,入院治療が必要となった。それ以来,事件は完全に解決され,患者はまだ研究中である。この研究の首席調査員と安全監視委員会はSAEが治療に関与している可能性を評価し,規制機関にSAEを報告した。安全監視委員会はその後、この研究を修正せずに行うことができ、この事件を理解のために他の調査者に報告することができると判断した。
2023年2月22日に発表されたIsaralgagene Civparvovecの1/2期STAAR研究に関する最新の臨床データの概要これは…。年度世界シンポジウム2023年2月24日
•STAAR研究は進行中の1/2期多中心、開放ラベル、用量範囲の臨床研究であり、18歳のFabry病患者におけるイソプロテレノール単回投与の安全性と耐性を評価することを目的としている。患者は単回静脈注射を受け、52週間のフォローアップを行った。この研究で治療を受けた患者の治療後5年間にわたって監視するための単独の長期追跡研究が行われている。この研究設計は、各用量キュー内の少なくとも2人の患者が用量を受け、各キュー内の潜在的な拡張を規定する。安定した酵素代替療法(ERT)を受けている患者は,治療後に制御されモニタリングされた方法でERTを中止することを患者や研究者が自ら決定することができる。
•投与量増加段階は典型的なファブリック病を有する男性を含む。その後の研究拡張段階は2022年下半期に開始され、女性およびより深刻なFabry関連心臓または腎臓疾患を有する患者も治療される。この研究の主な終点は突発性不良事件(AES)の治療の発生率である。その他の安全性評価は常規の血液学、化学と肝臓検査;バイタルサインモニタリング;心電図;超音波心電図;肝臓の連続A胎児蛋白測定と磁気共鳴画像(Mri)を含む
肝臓腫瘍の潜在的な形成を監視しています二次終点は、1年間の研究中の特定の時点とベースラインとの変化を含み、α-ガラクトシダーゼAまたはα-GalA、血漿中の活性、球三糖セラミドまたはGb 3およびLyso-Gb 3レベルの変化;ERT注入頻度;および心臓核磁気共鳴およびrAAV 2/6ベクターによる測定された腎機能および心機能(左心室質量)の変化を含む。肝心な研究終点は生活の質、ファブリー症状と神経病理性疼痛採点、及びAAV 6カプシドとα-GalAに対する免疫反応を含む。
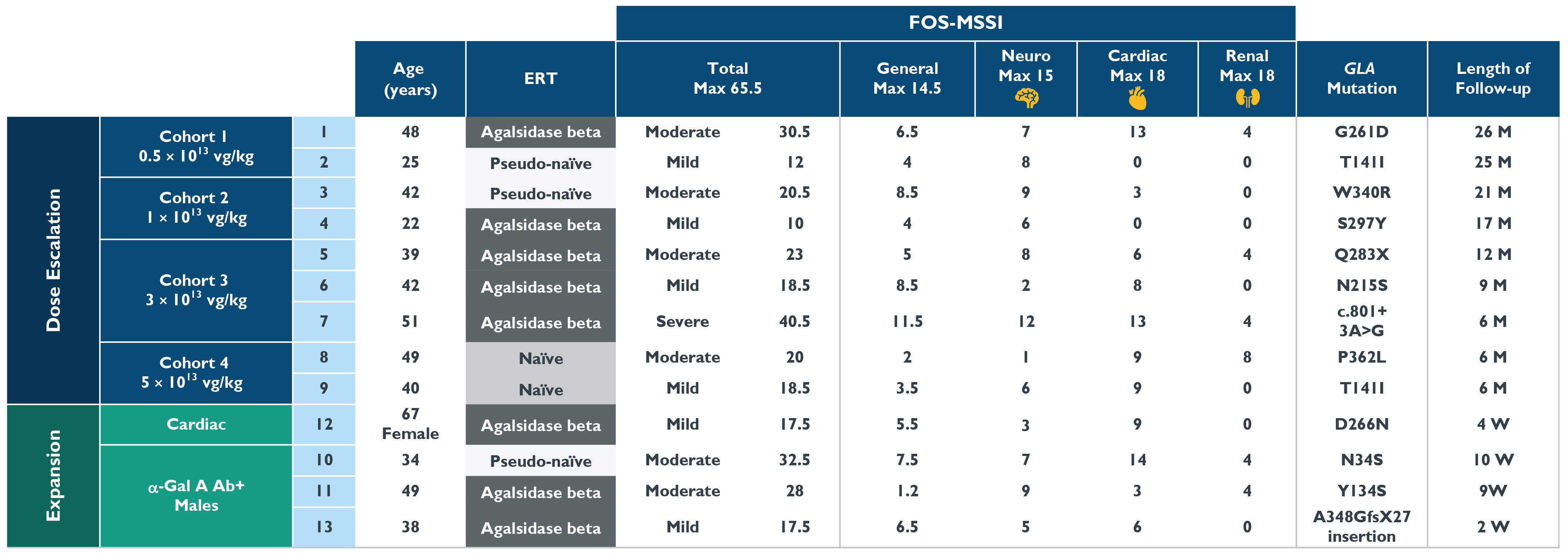
•2022年10月20日までに13名の患者,年齢は22歳から67歳まで様々であり,イシャラゲニ民事治療を受け,9名の患者は投与量増加段階にあり,4名の患者は研究の拡張段階にある。以下の表1にこの13名の患者のベースライン特徴を示す。用量増加段階では、キュー1内の2人の患者が0.5 x 10の用量を投与される13Vg/kg、2人の患者は列2で1×10用量で投与した13Vg/kg,列3中3例の患者を3 x 10用量で投与した13Vg/kg、2人の患者は列4で5×10用量で投与した13Vg/kg。拡張段階では、4人の患者が5 x 10投与された13心臓列中の女性患者1人およびα-GalA抗体陽性キュー中の男性患者3人を含むVg/kg。2022年10月20日までに,最初に治療を受けた患者は服薬後少なくとも26カ月フォローアップし,最近治療を受けた患者は服薬後2週間フォローアップした。
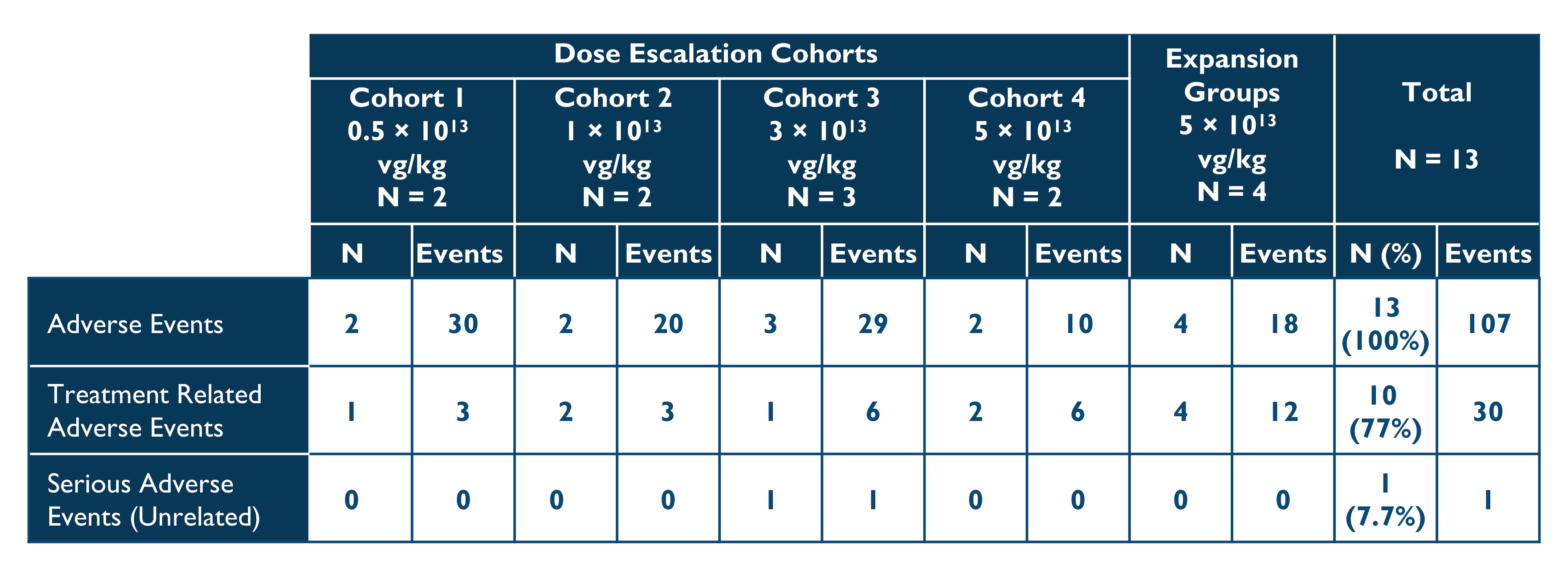
•2022年10月20日現在,治療を受けた患者13名のうち,イサラギは全用量列で良好な耐性を維持し続けており,予防的コルチコステロイドや他の免疫調節剤は使用されていない。2022年10月20日締め切りまでに報告された治療に関する有害事象の概要を以下の表2に示す。キュー1中1例,キュー2中2例,キュー3中1例,キュー4中2例と拡張期4例の計30件の治療関連有害事象が発生し,いずれも軽度(1級)または中等度(2級)であった。治療に関するSAE報告はない。遺伝子治療に関連する副作用は認められず,コルチコステロイドを用いてトランスアミナーゼを上昇させず,臨床的に著明な血小板低下もなく,心臓イベントもなかった。1例の拡張期患者は1級アレルギー反応が出現し、ジフェンヒドラミンで治療した
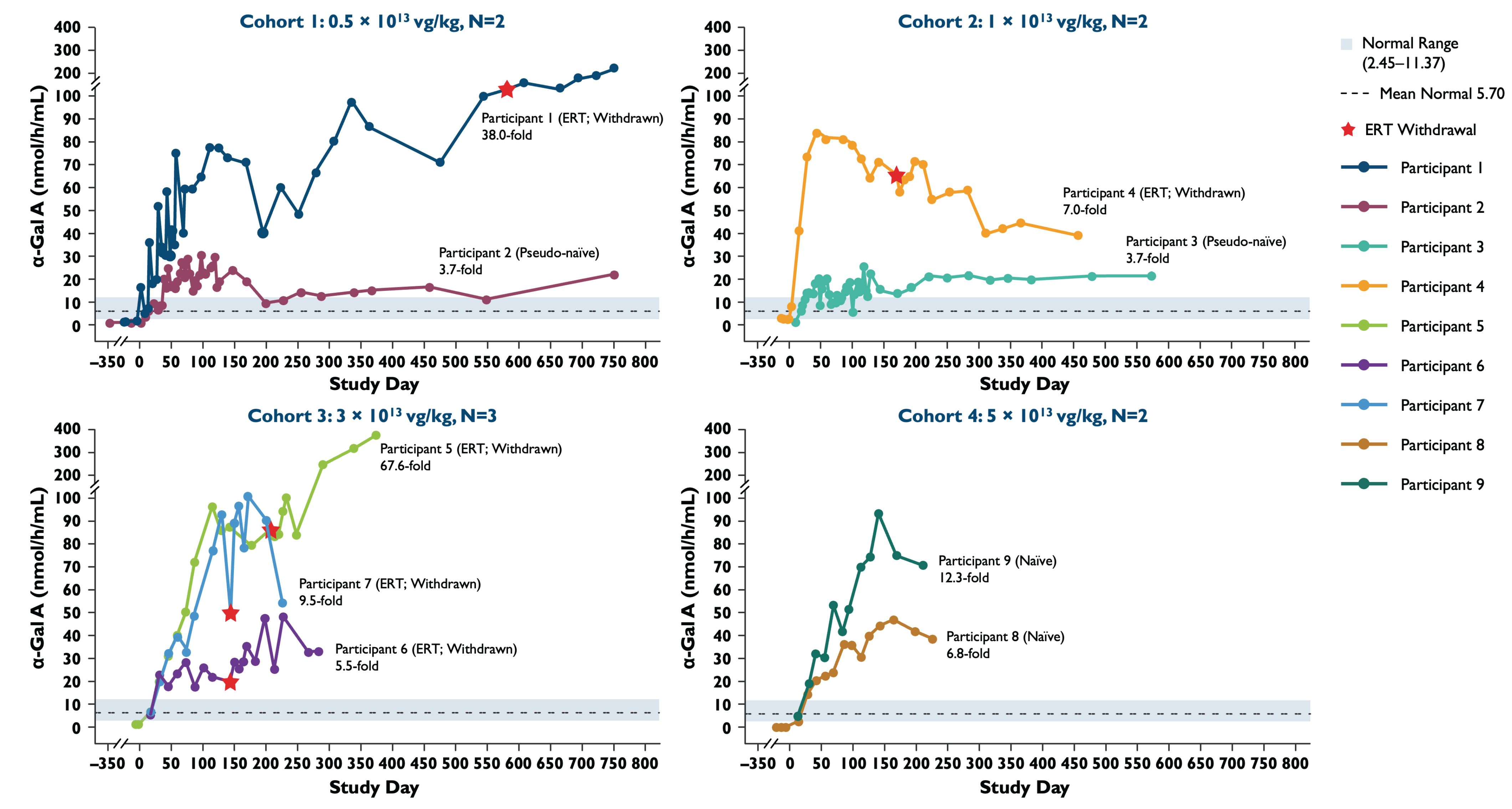
•α−GalA活性の補充締め切りは2022年11月15日であり,投与量増加段階にある治療患者9名と拡大段階にある治療患者4名の血漿α−GalA活性の結果を表3と表4に示し,以下により詳細に説明する。2022年11月15日までに治療締め切りを補充し,治療期間が最も長い患者のうち13名で2年を超える間にα−GalA活性の持続的な上昇が認められた。
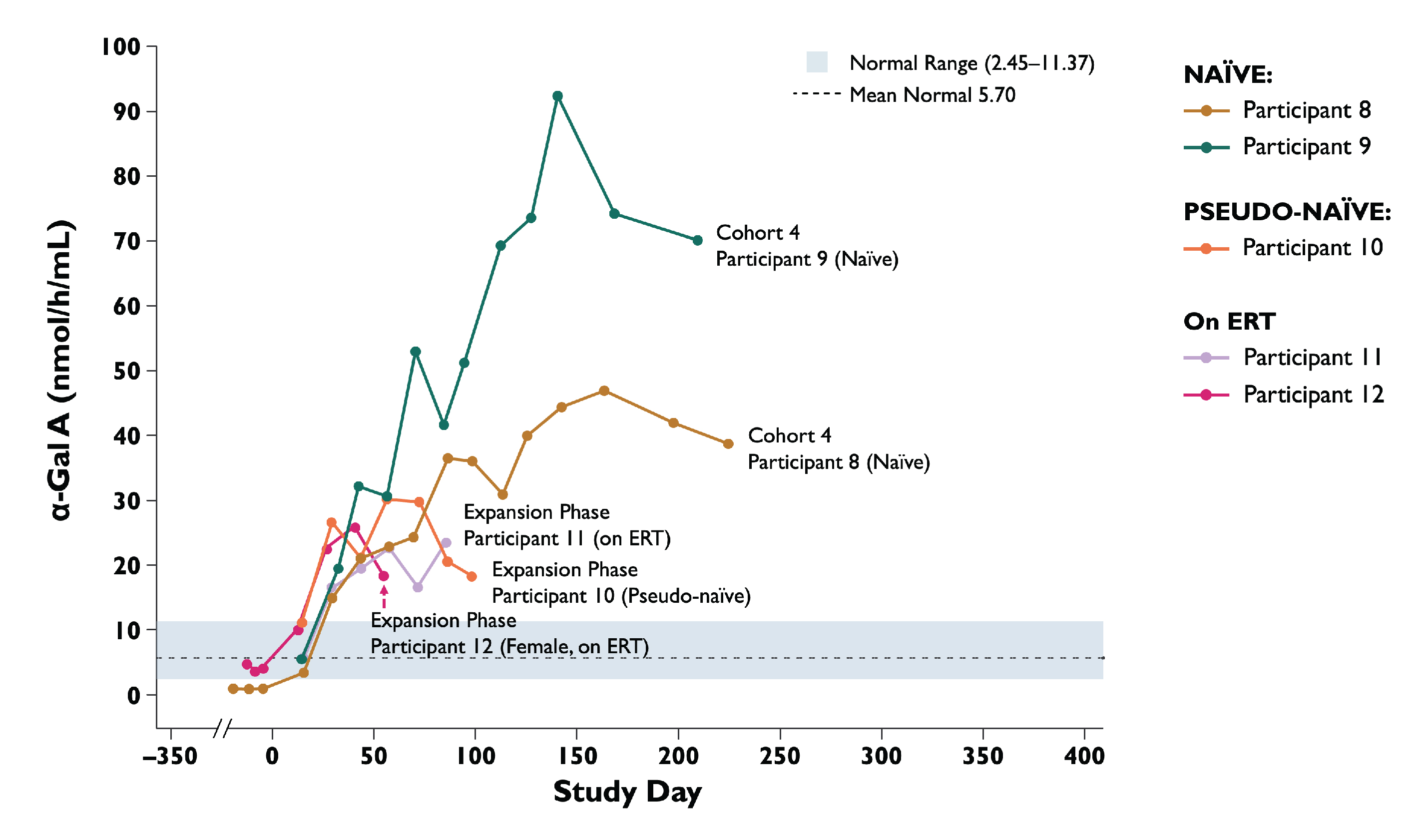
•2022年11月15日までの補充締め切りは、用量増加段階で治療を受けた9名の患者のα-GalA活性が持続的に上昇し、最後の測定日の平均正常レベルの3.7倍から67.6倍であった。これらの患者では,ST−920投与後4~8週間でα−GalA活性が急速に増加した。ERT研究を開始した5名すべての患者がERTを完了し,ERT停止後,α−GalA活性は超生理学的レベルで持続した。2023年2月22日現在,これらの患者のうちERTの回復を必要とする人は一人もいない。投与量増加段階の幼稚と偽幼若患者に対して、第4群患者のα-GalA活性レベルは低用量群患者より明らかに高かった。補充締め切りまで、すべての患者のα-GalA活性レベルは持続的に上昇した。
•2022年11月15日までに補充締め切り,上位3名が5 x 10の拡張段階で服薬13Vg/kg用量は投与後α−GalA活性が急速に増加し,最終測定日に14週間にわたって維持された。4名目の患者は服薬4週間後に正常範囲内に増加した。研究中の1人目の女性患者は,締め切りを補充する際に男性に類似した反応特徴を示した。
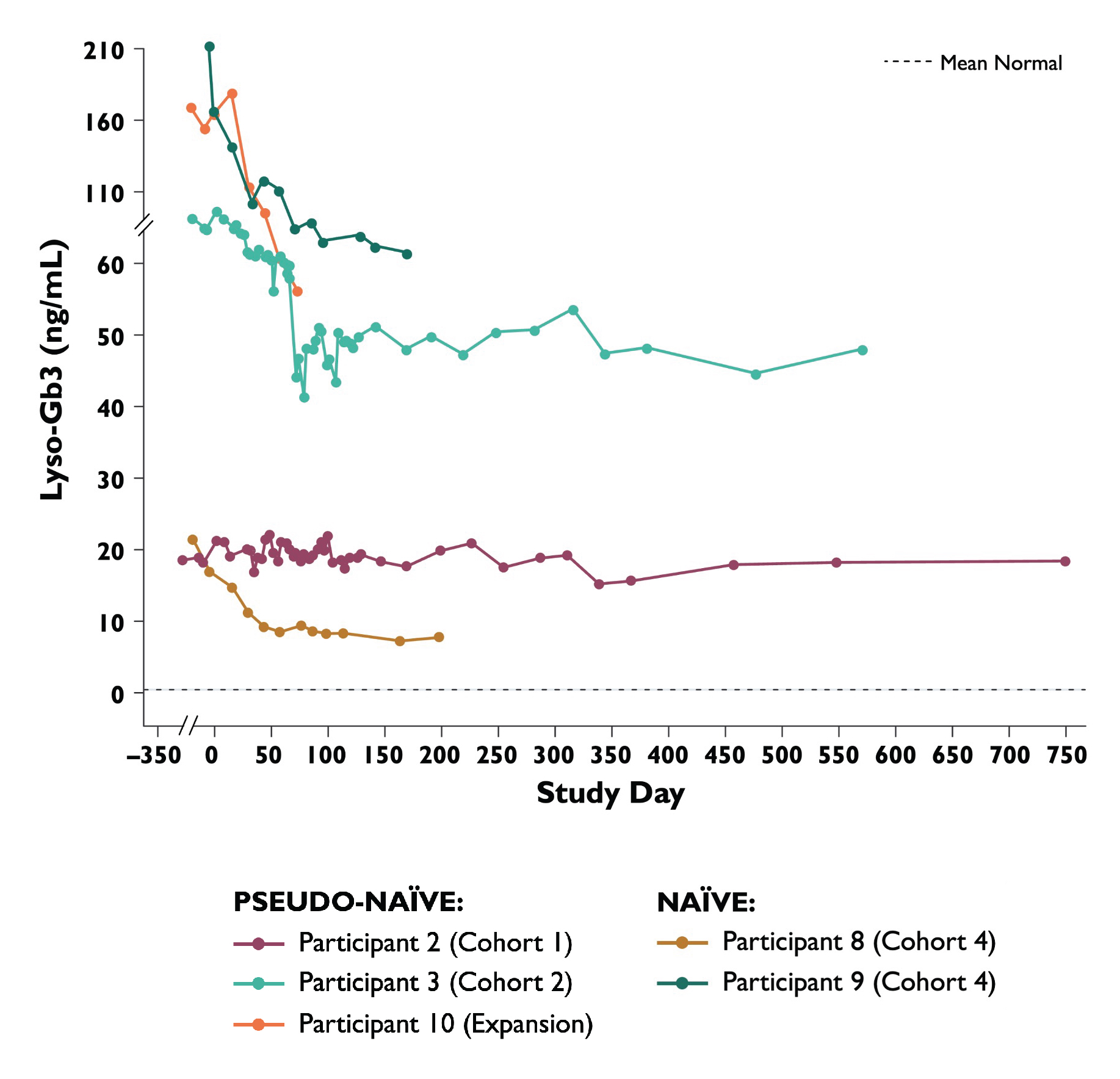
•次に表5と表6に治療を受けた患者13名の2022年10月20日時点のGlybotriaosylsphingosine(lyso−Gb 3)レベルの結果を示す。投与量の拡大とアップグレード段階において、ベースラインlyso-Gb 3レベルは80 ng/mlより高い無邪気と偽無邪気患者のレベルは40%-65%低下した。ベースラインlyso-Gb 3レベルが25 ng/mL未満のナイーブおよび仮性ナイーブ患者は5 x 10を受け入れた13Vg/kg用量はlyso−Gb 3レベルを54%低下させた。2022年10月20日現在,2名の患者のLyso−Gb 3は低下し続けている。低用量レベル(0.5 x 10)の無邪気で偽無邪気な患者のために13Vg/kgおよび1 x 1013Vg/kg),Lyso−Gb 3レベルは25カ月と安定していた。ERT研究を開始した用量アップグレード段階にある患者では,ERT中止後のlyso−Gb 3レベルは,ERT治療を受けた患者で一般的に観察されるレベルおよび変異性の範囲内に保たれている。これらの参加者のうち,α−GalA活性は上昇しており,締め切りまでERTの回復を必要とする症状は出現しなかった。ERT研究を開始した拡張期患者では,2022年10月20日時点でERTは中止されていない。
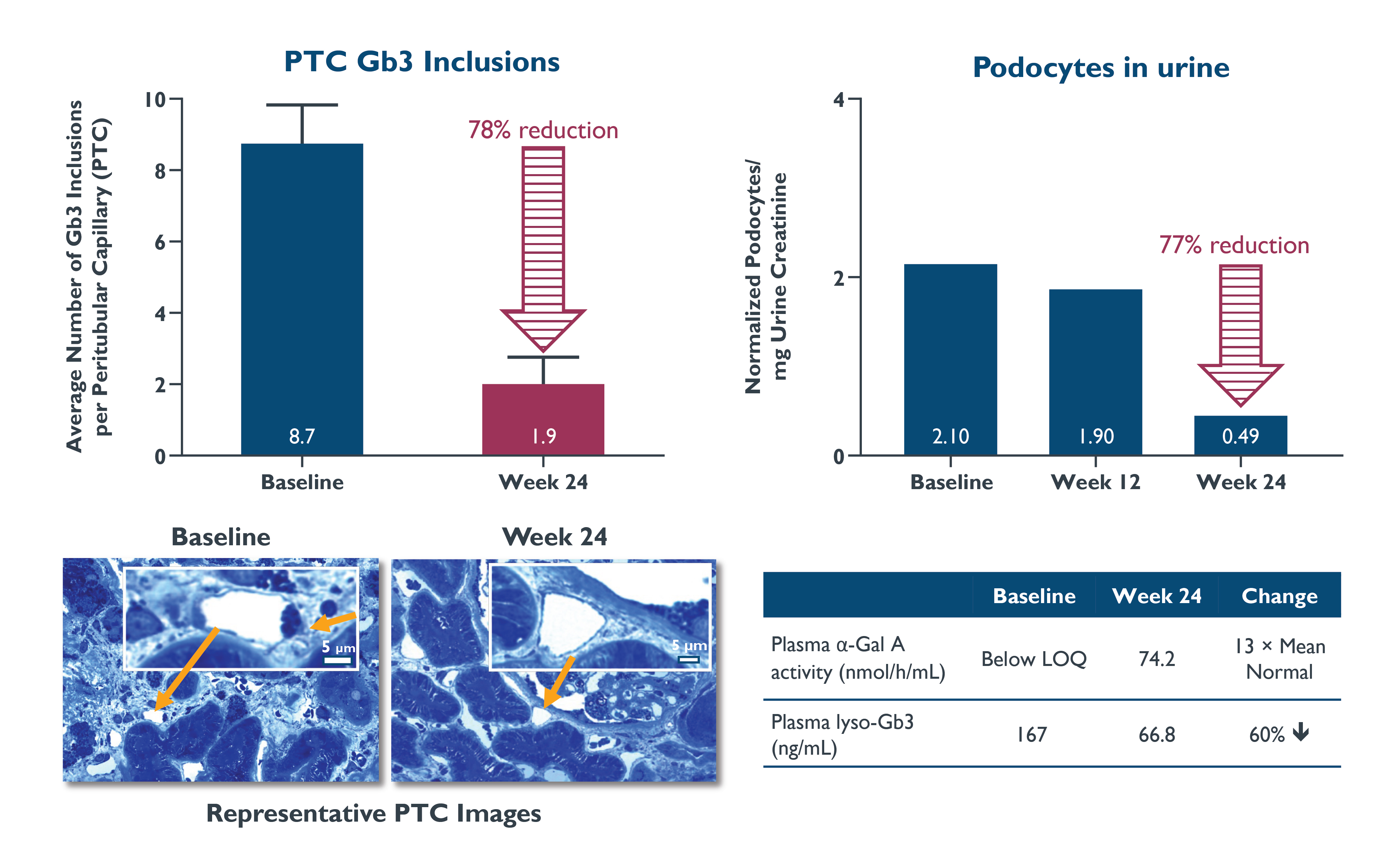
•2022年10月20日現在,患者8と9の腎臓生検と尿足細胞の結果を次の表7と8に示す。球三糖セラミド(Gb 3)は脂肪基質であり、Fabry病患者の細胞に蓄積し、腎臓、心臓と中枢神経系を含む多数の器官の損害を招くことができる。2022年10月20日までの締め切りまでの患者9の腎臓生検では,ベースラインレベルのGb 3封入体数と血漿Lyso−Gb 3レベルが高く,末梢毛細血管(PTC)あたりのGb 3封入体の除去率は78%,ベースライン時の平均1管週毛細血管内Gb 3封入体8.7個から24週目までのPTCあたりのGb 3封入体は平均1.9個であった。この評価は2人の視覚障害者病理学者が作成しましたベースラインと6ヶ月間の生検腎臓切片のデジタル画像を独立して採点しました
3つ目の独立した病理学者によって裁かれましたまた,6カ月後,尿足細胞損失は77%減少した。患者8の腎臓生検はGb 3封入体の数量と血漿Lyso-Gb 3レベルはベースラインレベルが比較的に低く、服用6ケ月後に安定したPTC包有物を示し、ベースライン時の平均PTC包有物は1個3.5個、24週目は各PTC包有物が3.7個であった。6カ月後、患者の尿足細胞損失は97%減少した。
•SF-36一般健康調査或いはSF-36の測定によると、用量増加段階で治療を受けたすべての患者の平均一般健康採点は臨床的意義と統計的に顕著な増加があった。SF-36は、健康に関連する生活の質を全面的に評価するために、十分に検証され、広く使用されている汎用アンケートである。この36項目は,健康に関する8つの分野(身体機能,身体役割,身体痛,一般健康,活力,社会機能,感情役割,心理健康を含む)を評価し,これらの領域は身体成分得点と心理成分得点をまとめたものである。臨床定義の患者群間の横断面差異の研究により、任意のSF-36尺度表上の3点から5点の変化はすべて最小の臨床重要な差異、即ちMCIDであることを表明した。2022年10月20日の締め切りまで、SF-36の測定によると、用量増加段階にある患者は安定または改善された一般的な健康採点を示した。この領域得点はベースラインの平均改善より有意な統計学的有意差を示した[mean=19.6, 95% CL: [7.8, 31.4],p=0.0100(ペアt検定)]52週目にMCIDが変化した。
表1:ベースライン患者の特徴
データ削減:2022年10月20日
≤-MSSI総得点分類:軽度18点、中等度(MOD)=19~38点、重度>38点
FOS-MSSI,Fabry結果調査,Mainz重症度スコア指数,kg,kg;M,月;最大,最大;VG,ウイルスゲノム,W,週
表2:治療に関する有害事象
データ削減:2022年10月20日
フォローアップ時間は2週間から26ケ月まで様々であった
Vg/kg総体重1キロあたりのベクターゲノム数
表3:用量増加行列におけるα−GalA活性の発現
データ削減:2022年11月15日
血漿α−GalA活性を3時間反応時間で測定した。健康な男性の正常な範囲。最後に測定した時点で折り畳み率と正常平均値の変化を計算した
長期フォローアップデータ:データ点>学習日365。α−GalA,α−ガラクトシダーゼA;ERT,酵素代替療法
表4:キュー4と拡張フェーズα-GalAアクティビティ
データ削減:2022年11月15日
被験者13(拡張期):6週目,3.9 nmol/h/mL。α−GalA活性は3時間反応時間で測定した。健康な男性の正常な範囲。
長期フォローアップデータ:データ点>学習日365。α−GalA、α−ガラクトシダーゼA、ERT、酵素代替療法
表5:用量増加と拡張段階の幼稚と仮性幼若患者のlyso−Gb 3
データ削減:2022年10月20日
健常男性と女性のLyso−Gb 3正常範囲。男性と女性の正常範囲は0.32~0.63 ng/mlであった
長期フォローアップデータ:データ点>学習日365。Lyso−Gb 3,球三糖スフィンゴシン
表6:ERT治療用量の増加と拡張期患者におけるlyso−Gb 3
データカット日:2022年10月20日
被験者13:2週目34.5 ng/m L。健常男性と女性のLyso−Gb 3正常範囲。男性と女性の正常範囲は0.32~0.63ミリグラム/ml
長期フォローアップデータ:データ点>学習日365。Lyso−Gb 3,球三糖スフィンゴシン;ERT,酵素代替療法。
表7:患者9尿中PTC Gb 3封入体と足細胞
データカット日:2022年10月20日
足細胞定量は免疫蛍光により行い,尿クレアチニンは正常化した。Barisoni Lipid小包評価システム(BLISS)は3名の独立した病理学者がブラインド法を用いてPTC Gb 3包有物を定量化した。ライン上方のラインは標準偏差を表す.
α−GalA,α−ガラクトシダーゼA;ERT,酵素代替療法;PTC,尿細管周囲毛細血管;Lyso−Gb 3,球三糖スフィンゴシン;Gb 3,球三糖セラミド
表8:患者8尿中PTC Gb 3封入体と足細胞
データカット日:2022年10月20日
足細胞定量は免疫蛍光により行い,尿クレアチニンは正常化した。Barisoni Lipid小包評価システム(BLISS)は3名の独立した病理学者がブラインド法を用いてPTC Gb 3包有物を定量化した。ライン上方のラインは標準偏差を表す.
α−GalA,α−ガラクトシダーゼA;ERT,酵素代替療法;PTC,尿細管周囲毛細血管;Lyso−Gb 3,球三糖スフィンゴシン;Gb 3,球三糖セラミド
TX 200−ヒト白血球抗原−A 2不整合腎移植拒絶反応
2022年3月,われわれはTX 200を評価する1/2期堅固な臨床研究において,1人目の患者にTX 200を投与し,TX 200はわれわれが完全に所有している自己CAR−Treg細胞治療製品候補薬であり,生体ドナーのHLA−A 2不適合腎移植に対する免疫媒介拒絶反応を防止し,2名目の患者は2022年9月に治療を受けた。この候補製品は2人の患者における耐性は全体的に依然として良好である。3人目の患者は腎臓移植を受けており、彼らの個性化TX 200細胞療法はすでに製造されており、投与量は2023年第2四半期初めに予定されている。この3人目の患者の用量は、1/2期STESTATE研究の最初の完全行列の完成を示す。第2列の製造と臨床活動が行われており,第4患者の投与量は2023年夏に予定されている。より多くの患者が予備スクリーニングを行っており,この研究に参加する予定である。用量アップグレード計画を加速させる機会を規制機関と検討している。
巨細胞遺伝子Fitelparvovec−血友病A
2021年11月、いくつかの治療を受けた患者のFVIIIレベルが150%を超えることを観察した後、私たちとファイザーは、FVIIIレベルの上昇した第3段階模倣臨床試験における追加の患者のスクリーニングおよび用量を自発的に停止したことを発表し、これは、我々が治療中の重度から重度の血友病Aを治療する研究遺伝子療法であり、FVIIIレベルの上昇に臨床管理指導を提供するためのプロトコル修正案を実施することを発表した。その後、2021年11月3日、アメリカ食品医薬品局(FDA)はファイザーに通知し、方案修正案と関連文書を審査している間、この試験は臨床で保留された。FDAは2022年3月に臨床制限を解除した
2022年9月、ファイザーによる自発的な一時停止は廃止され、募集再開を試行し、募集を再開した。初歩的な分析を支持する具は2022年11月に回復し、2022年末に完成する予定です
2023年第1四半期。2024年上半期には重要なデータが発表される予定で、ファイザーは2024年下半期に生物製品許可証申請を提出する予定だ。
Affineは世界的な3期開放、マルチセンター、単一腕試験であり、60人以上の成人(年齢18-64歳)で中重度から重度の血友病Aを有する男性患者におけるgirococogene fitelparvoecの単回注入の有効性と安全性を評価する。主な終点は、girococogene fitelparvovec治療12カ月後のABRへの影響であり、第3段階導入研究期間中に収集したFVIII代替療法を用いたABRと比較することである。著者らとファイザーは、この試験の重要なデータ読み取りは、第1陣の50名の患者が12ケ月後にFVIII発現の安定状態に達したとき、すべての研究参加者の全面的な分析に基づくと予想している
私たちは未来の臨床、監督と商業マイルストーン支払いで2.4億ドルに達する収入を稼ぐことができ、未来の潜在製品販売の14%から20%の等級は特許権使用料を増加させ、商業販売のために許可された場合、特許満期、生物類似製品の市場への参入、およびある第三者知的財産権許可証による費用の支払いの制限を受ける可能性がある。
2022年12月、我々とファイザー社は原癌遺伝子fitelparvovecに対する1/2期ALTA研究の最新の後続データを公表した。計11名の男性患者がこの研究に参加し,そのうち5名は3 E 13−VG/kg最高用量群に属していた。ベースライン患者の人口統計データについては,次の表9を参照されたい
2022年9月6日まで、すべての患者は153~263週間フォローアップされ、すべての患者は少なくとも35ケ月のフォローアップを完成した。2022年9月6日までに,11名中6名が最高用量列を含む5名中4名の治療に関する副作用を経験した。最もよく見られる治療関連副作用は肝酵素上昇と輸液関連反応を含む:アラニンアミノトランスフェラーゼ上昇(5/11(45.5%)全体;3/5(60.0%)最高用量列中)、アスパラギン酸トランスアミナーゼ或いはAST(全体3/11(27.3%)、最高用量列中2/5(40.0%)、発熱(全体3/11(27.3%)、最高用量列中3/5(60.0%)、及び頻脈(2/11(18.2%)。最高用量群では2/5(40.0%)であった。最高用量のコホートでは,原癌遺伝子fitelparvovec注入約6時間後に3級低血圧と発熱が出現した患者が報告されており,これらの事件は治療により完全に軽快し,注入後翌日の退院を遅らせることはなかった。治療に関連する副作用の詳細については,以下の表10を参照されたい。2022年9月6日までの締め切りでは,確定診断されたFVIII阻害剤の開発はなく,血栓事象,腫瘍事象,異常甲胎蛋白および/または肝臓腫瘍の報告もない。
最高用量群の5名の患者全員がFVIII活性を示し,156週目まで以下の表11に示すようにした。中心実験室発色凝固法で測定し、156週目のFVIII平均活性は正常の25.5%であった。この投与量が最も高いキューでは,年間出血率,すなわち輸液後3週間からの全出血回数を年単位の観察期間で割ったところ,輸液後1年目はゼロであり,2022年9月6日までのフォローアップ期間全体の平均年間出血率は1.2であった。この最高用量のコホートでは,2名の患者が外因性FVIII治療を必要とする出血事件を経験し,1名が17回の出血事件(侵襲性8名,自発性5名,原因不明4名)を経験し,1名が1回の目標関節の出血事件を経験しており,状況は不明である。この最高用量列には締め切りまでに予防を回復する患者はいなかった。原癌遺伝子fitelparvovecの治療効果や他の長期効果の持続性,例えば患者全体の肝臓健康への影響を評価するためにはさらなるフォローアップが必要である。
表9:Giroctocogene Fitelparvovec用量列のベースライン患者人口統計
データ切断:2022年9月6日,最大値=最大値,最小値=最小値,SD=標準偏差,VG=ベクターゲノム
表10:Giroctocogene Fitelparvovec用量キューにおける治療に関連する有害事象
データ削減:2022年9月6日
(a)1例の患者は3級低血圧が出現し、研究薬物と関係があると考えられ、治療後に緩和した
AE=有害事象,ALT=アラニントランスアミナーゼ,AST=アスパラギン酸アミノトランスフェラーゼ,VG=ベクターゲノム
表11:Giroctocogene Fitelparvovec最高用量群(3 e 13 Vg/kg,第4群)患者のFVIII活性レベル(発色分析法測定)
2022年9月6日の最新利用可能なFIII値データ、FVIII=第VIII因子、VG=ベクターゲノム
BIVV 003−鎌状細胞病
2022年1月、私たちはサイノフェイがBIVV 003に関連する権利と義務を私たちに移管することを発表し、BIVV 003はSCDの治療に使用されているZFヌクレアーゼ遺伝子編集細胞療法候補製品である。私たちはセノフィと秩序ある移行について協力し、2022年6月に完成した。この移行に関するより詳細な情報は、“-開発中の候補製品-独自計画-BIVV 003-鎌状細胞疾患”を参照されたい
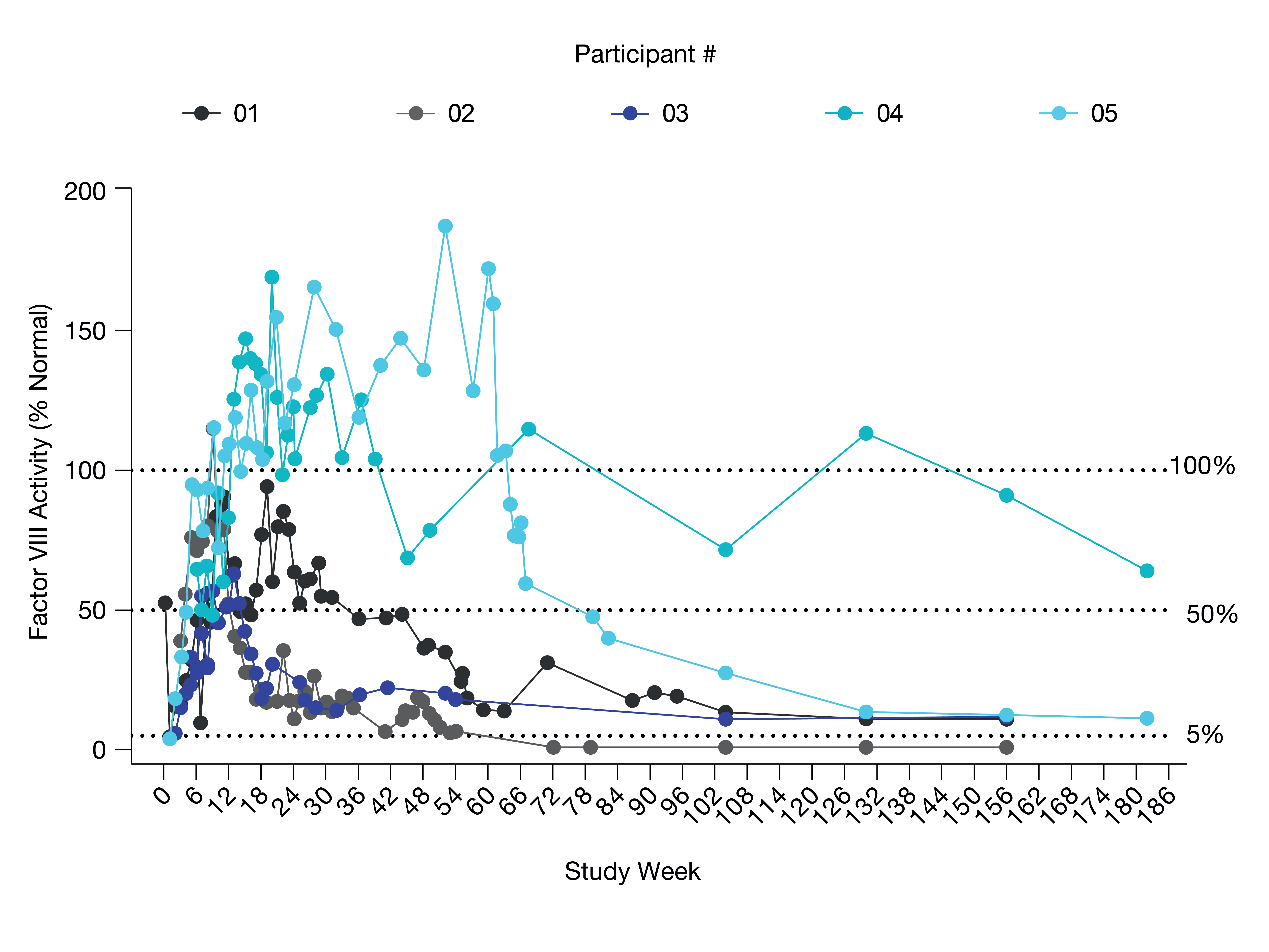
2022年12月、我々は、我々の1/2期PRECIZN-1研究からの初期概念検証臨床データを提供し、64でBIVV 003を評価しましたこれは…。アメリカ血液病学会2022年年次総会と博覧会、ASHと略称する。以下にデータの要約を示す
2022年12月にASHに最新データを提出して以来、著者らはすでに患者7号の投与量を準備する臨床と製造活動において進展を得ており、潜在的な3期試験の試験設計と必要な製造技術についてFDAと合意した。また,より多くの製造改善が得られており,これらの改善は潜在的な3期試験において臨床結果をさらに強化し,製造コストを低減する可能性があると信じている。
私たちは最近、1/2段階PRECIZN-1研究が完了した後、BIVV 003計画へのさらなる実質的な投資を停止して、私たちのFabryとTX 200計画の資源を優先的に配置するための戦略決定を下した。我々はBIVV 003の1/2段階PRECIZN−1研究を完成させることに取り組んでおり,約束された資金を用いてこの研究を完成させる予定である。私たちはこの計画を潜在的な第3段階実験に進めることができる潜在的な協力パートナーを探し始めるつもりだ。
2022年12月10日にASHで発表されたBIVV 003 1/2期PRECIZN-1研究の最新の安全性、耐性と有効性の結果の概要
•条件を満たした患者はプリシャ福の動員と分離を受けた。ZFヌクレアーゼメッセンジャーリボ核酸をin vitroで自家造血幹/前駆細胞に導入し,BIVV 003を作製した。ブチルチオダンプレコンディショニング後少なくとも72時間に1回の静脈輸液を行った。BIVV 003注入後、患者の幹細胞移植と造血回復、AEs、臨床と実験室溶血マーカー、総ヘモグロビン(Hb)と胎児ヘモグロビン(HBF)、F細胞パーセンテージとSCD関連イベントをモニタリングした。6例の患者はHSPCの目標生産量を達成することに成功した。2022年9月30日現在,HSPC成功目標収量を達成した6名の患者のうち,5名にBIVV 003が注射されている。以下の表12にこれら5名の患者のベースライン特徴を示す
•初期製造プロセスを用いて生産したBIVV 003の上位4人の患者を以下の表12で第1群と呼び,第1群の患者は輸液後30カ月間のフォローアップを行った。次の表12に記載の第2のグループの患者は、最終製品中長期前駆細胞の数を増加させるために、内部実験で示された改良された方法を用いて製造されたBIVV 003を受けた。締め切り,第2群治療の第1位患者,あるいは5人目の患者は,すでに5カ月間フォローアップしていた。第2群の2人目の患者は2022年9月30日の締め切り後に服用した。BIVV 003投与から2022年9月30日までに第1群と第2群の計5名中4名の臨床症状が改善したのを以下の表13に示す。
第1グループでは:
•BIVV 003注入による総HbとHbFレベルへの影響は30カ月まで維持された。
•4名の患者のうち,3名の患者は安定してZFヌクレアーゼ修飾HSPCを移植し,HBFレベルの30%以上の持続的な上昇をもたらし,BIVV 003注射後に重篤な血管閉塞危険像やVOCsはなかった。
第2グループでは
•患者5は、最終製品中長期前駆細胞の数を増加させるために、内部実験において示された改良された方法を用いて製造されたBIVV 003を受け入れた。
•患者5は注入後26週のHbFレベル45%,総Hb 12.4 g/dLであり,締め切り後に最近採取したサンプルでは,HbFレベルは群1の26週時のレベルより高かった。
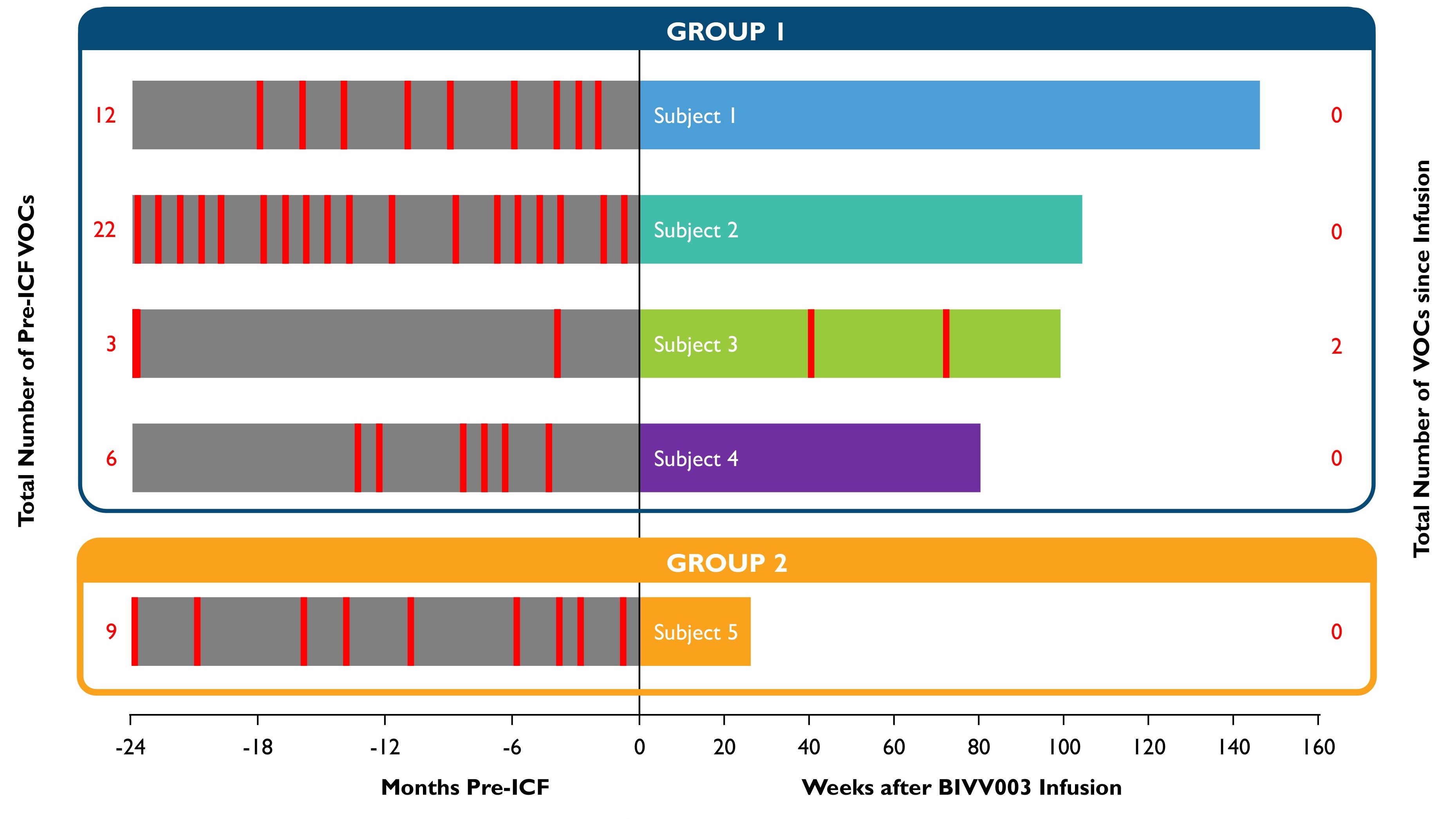
•2022年9月30日の締め切りまで,BIVV 003の全体的な耐性は良好であり,スクリーニング,動員,分離,調整期報告の多くのAEsはSCD関連イベントであった。プリシャフォードに関連する鎌状細胞性貧血合併VOCのSAE 2例と,ブチルチオダンに関連する嘔気SAEの1例が報告されている。BIVV 003投与後に報告された副作用では,半数近くが白花丹に関与していた。鎌状細胞性貧血患者2例は輸液後9カ月と16カ月にVOCが出現し,そのうち1例はHBFレベルが低かった(11−14%)と報告されている。輸液後のSCDに関するSAE報告は他になかった。調査員やスポンサーはBIVV 003と関連したAEsを報告しなかった。BIVV 003注射前後に報告されたVOCsについて,次の表14を示す。
表12:上位5名の服薬患者のベースライン特徴
VOC=血管閉塞発症,ICF=インフォームドコンセント,RBC=赤血球
第1のグループは、初期製造プロセスを使用して生産されたBIVV 003を使用したBIVV 003を服用した最初の4人の患者を含む。
第2のグループは、改善された方法を使用して生産されたBIVV 003を受けた患者5を含み、内部実験は、この方法が最終製品中長期前駆細胞の数を増加させることができることを示している。
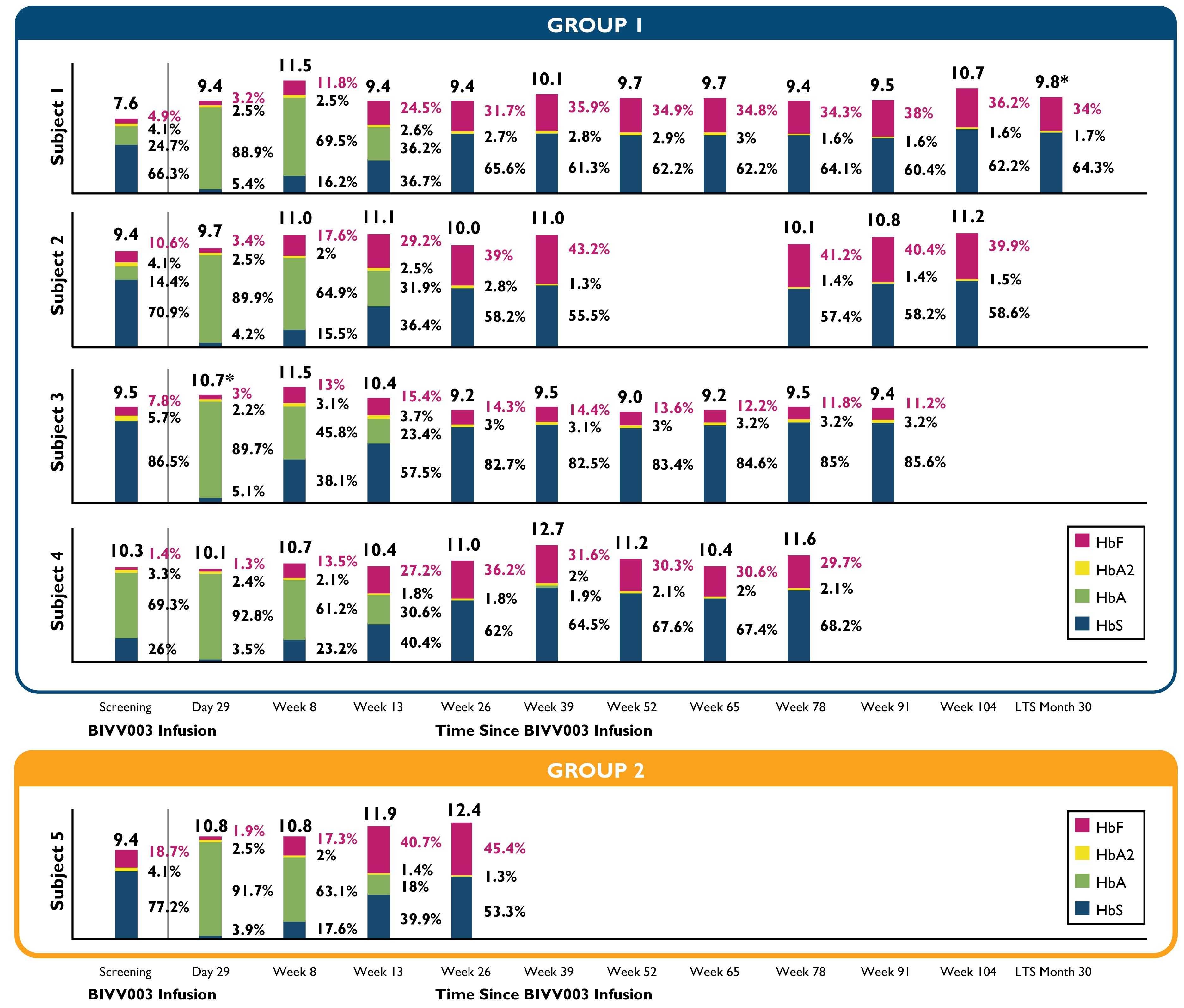
表13:総HbとHb分類
時間の経過とともにスクリーニングとBIVV 003注入後の総ヘモグロビンとヘモグロビン分級。
HbA=成人ヘモグロビン,HbA 2=変異成人ヘモグロビン,HbF=胎児ヘモグロビン,HbS=鎌状ヘモグロビン
LTS=長期フォローアップ研究
(*)中心実験室の値が収集されていないので、地元の実験室からのHb値を表します
第1のグループは、初期製造プロセスを使用して生産されたBIVV 003を使用したBIVV 003を服用した最初の4人の患者を含む。
第2のグループは、最終製品中長期前駆細胞の数を増加させるために内部実験で示された改善された方法を使用してBIVV 003を産生するBIVV 003治療を受けた患者5を含む。
表14:BIVV 003注入後のVOC発生率
研究インフォームドコンセント(ICF)に署名するまでの24カ月とBIVV 003注入後24カ月以内に報告された重篤な血管閉塞性発症(VOC)の数。
赤い線は深刻なVOCsを表している;同じ月に発生した2つの深刻なVOCsは1つの赤い線として示された
第1のグループは、初期製造プロセスを使用して生産されたBIVV 003を使用したBIVV 003を服用した最初の4人の患者を含む。
第2のグループは、最終製品中長期前駆細胞の数を増加させるために内部実験で示された改善された方法を使用してBIVV 003を産生するBIVV 003治療を受けた患者5を含む。
私たちの技術は
私たちの戦略は私たちの差別化と多機能なZF技術プラットフォームを最適或いは一流の臨床潜在力を持つ候補製品に転化することである。我々の技術プラットフォームの多機能性と柔軟性は、潜在的な遺伝或いは細胞疾患の原因を解決するために治療方法を設計することができ、このような治療を提供するのに最適な任意の技術を使用することができると信じている。著者らは現在臨床前研究における革新領域は著者らのZF技術プラットフォームを利用して中枢神経系(CNS)におけるエピジェネティック制御、及び自己免疫性疾患に対するCAR-Treg細胞療法を含む。
ZFPS:ヒトに天然に存在する配列特異的DNA結合タンパク質
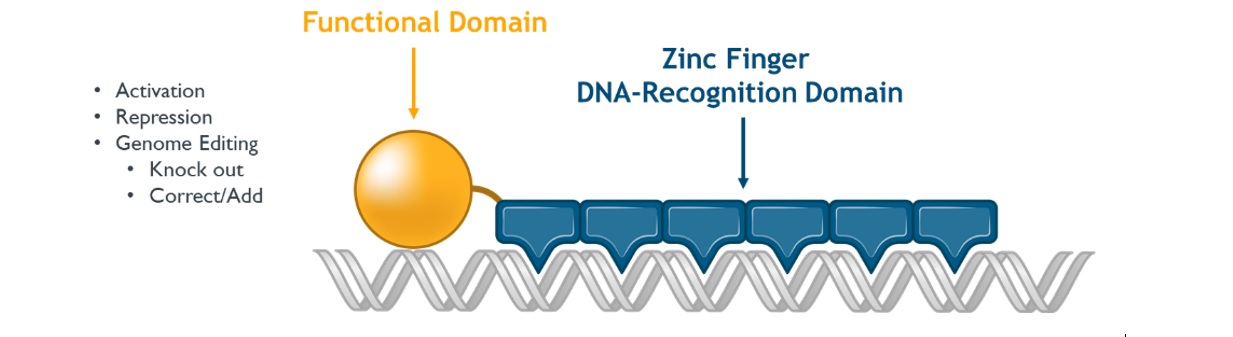
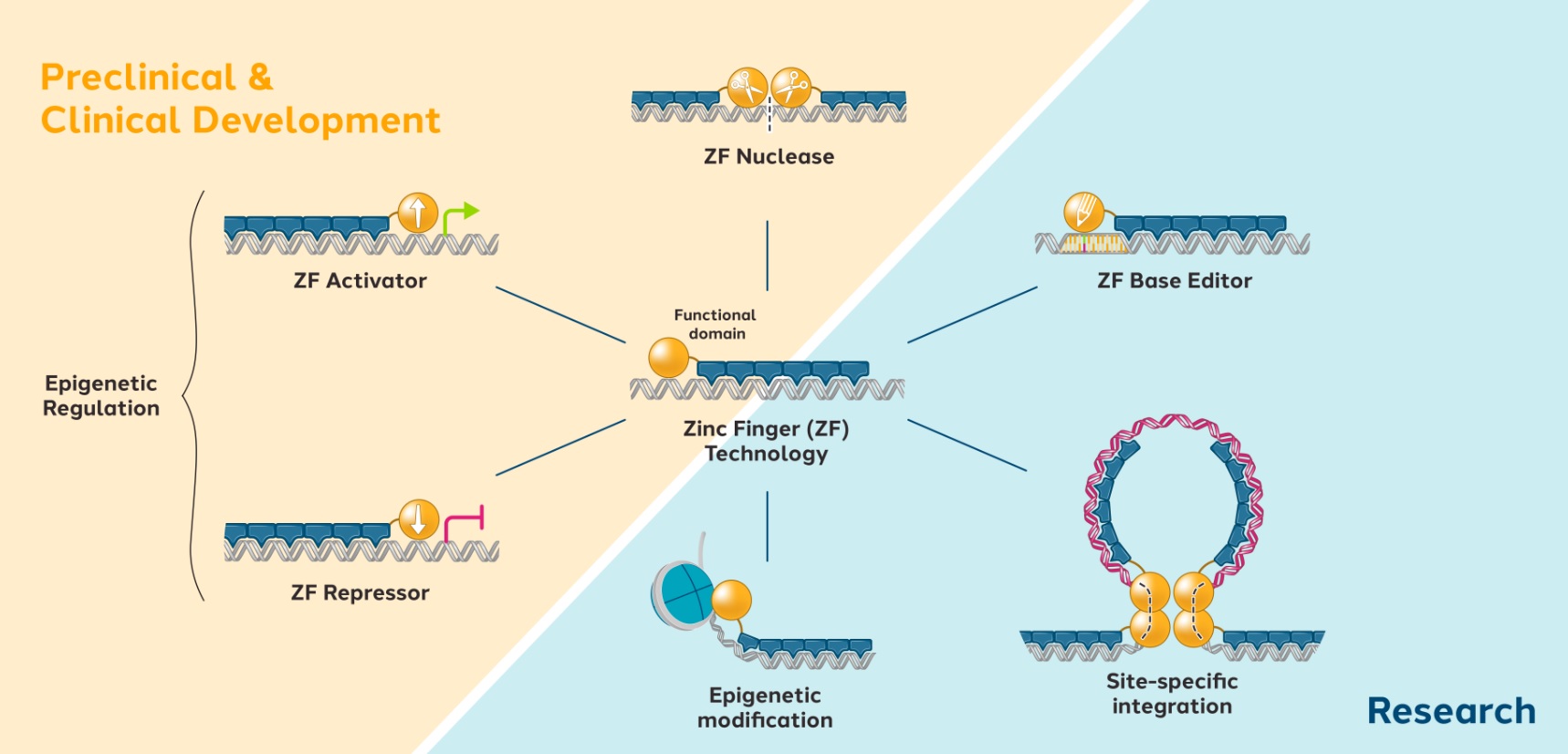
ZFPは、ヒトが自然に産生する配列特異的DNA結合タンパク質であり、特定の遺伝子内または近傍の特定のDNA配列を認識し、結合し、その遺伝子の発現を“オン”(活性化)または“閉鎖”(抑制)させる。ZFPは酵母からヒトまでの各種生体の中で最もよく見られる自然産生タンパク質である。機能ドメインは、ZFP DNAによって決定される特定のゲノム位置でゲノム編集(ヌクレアーゼまたはインテグラーゼなどの酵素を使用する)またはエピジェネティック調節(活性化剤および阻害剤を使用する)を行うことができるように、ZFPに添加することができる‑結合ドメイン。
図1:亜鉛指DNA結合ドメインとその機能ドメインの二重ドメイン構造図
天然ZFPの構造と一致し,モジュール化した方法で設計したタンパク質を設計した。私たちが設計したタンパク質のDNA認識部分は通常4~6つの亜鉛フィンガーで構成されています各個々の指は、3つまたは4つの塩基対のDNA配列を認識して結合し、複数の指を一緒に連結することができ、より長いDNA断片を識別する亜鉛フィンガーアレイを形成し、それによって特異性を向上させることができる。ZFPのアミノ酸配列を修正することにより,新たな亜鉛フィンガーアレイを設計することができ,選択されたゲノム標的のユニークなDNA配列を識別することができる。
[図2]ZFPおよび6つのZFPからなる亜鉛フィンガーアレイの概略図
そして設計されたDNA結合亜鉛フィンガーアレイを機能ドメインに連結した。DNA結合亜鉛フィンガーアレイは,この機能ドメインを興味のある標的にバンドする。高度に特異的なZFPを用いてDNA配列を興味のある遺伝子に正確に定位することができ、様々な細胞タイプに適用可能な一連のゲノム編集およびエピジェネティック調節機能を提供する。
図3:ZFプラットフォームが提供できるゲノム工学ツールの例
我々が設計した亜鉛指は制限エンドヌクレアーゼの切断領域に結合することができ,この酵素はDNAを切断してZFヌクレアーゼを産生することができる。一対のZFヌクレアーゼが正しい方向および間隔でDNAに結合すると、ZF結合部位間のDNA配列が切断される。2つのZFヌクレアーゼとDNAの結合は切断に必要であり、エンドヌクレアーゼの2つの部分は正確な方向に存在しなければ、相互作用してこそ、DNA切断を媒介することができる。DNAのこの切断は細胞内DNA修復の自然過程を触発する。このような内因性DNA修復プロセスは、治療に有用である可能性のあるいくつかの結果のうちの1つを達成するために利用される可能性がある(図2)。細胞をZFヌクレアーゼのみで処理すると,修復過程は切断DNAの両端を連結し,断裂部位の少量の遺伝物質の喪失(欠失)や増加(挿入)を招くことが多い。これらの挿入と削除イベントを総称して“INDELS”と呼ぶ.それらは標的DNA配列を撹乱し,標的遺伝子の切断や無機能蛋白の発現を招き,遺伝子機能を有効に“ノックアウト”した。ZFヌクレアーゼを介したゲノム編集は、疾患の病理に関連する遺伝子を妨害するために使用することができる。我々はZFヌクレアーゼを介したBCL 11 A赤系特異的エンハンサーやESEのゲノム編集を用いており,CD 34陽性の造血幹細胞やHSPCにおいて,潜在的な長期効果と一次治療SCDの基礎となっている(BIVV 003)。
逆に,特定の遺伝子で変異した細胞をZFヌクレアーゼで処理するだけでなく,正しい遺伝子配列をコードする追加のDNA配列(“ドナー”DNAと呼ぶ)と,変異の両側配列を認識し結合するZFヌクレアーゼで処理すれば,細胞の修復機構はドナーDNAをテンプレートとして用いて変異遺伝子を修正することができる。このZFヌクレアーゼを介した遺伝子補正は、補正された遺伝子をその自然染色体環境で発現させることができ、特定の単遺伝子疾患を引き起こすDNA配列変異を正確に修復するために新しい方法を提供することができる。完全な遺伝子をコードするドナー配列を提供することに加えて、遺伝子の新しいコピーを特定の位置でゲノムに正確に添加することができる。遺伝子サイズのDNA断片をゲノム中の所定の位置に正確に位置決めする能力は、遺伝子変異の範囲を拡大し、単一ステップでこれらの変異を修正することができる。
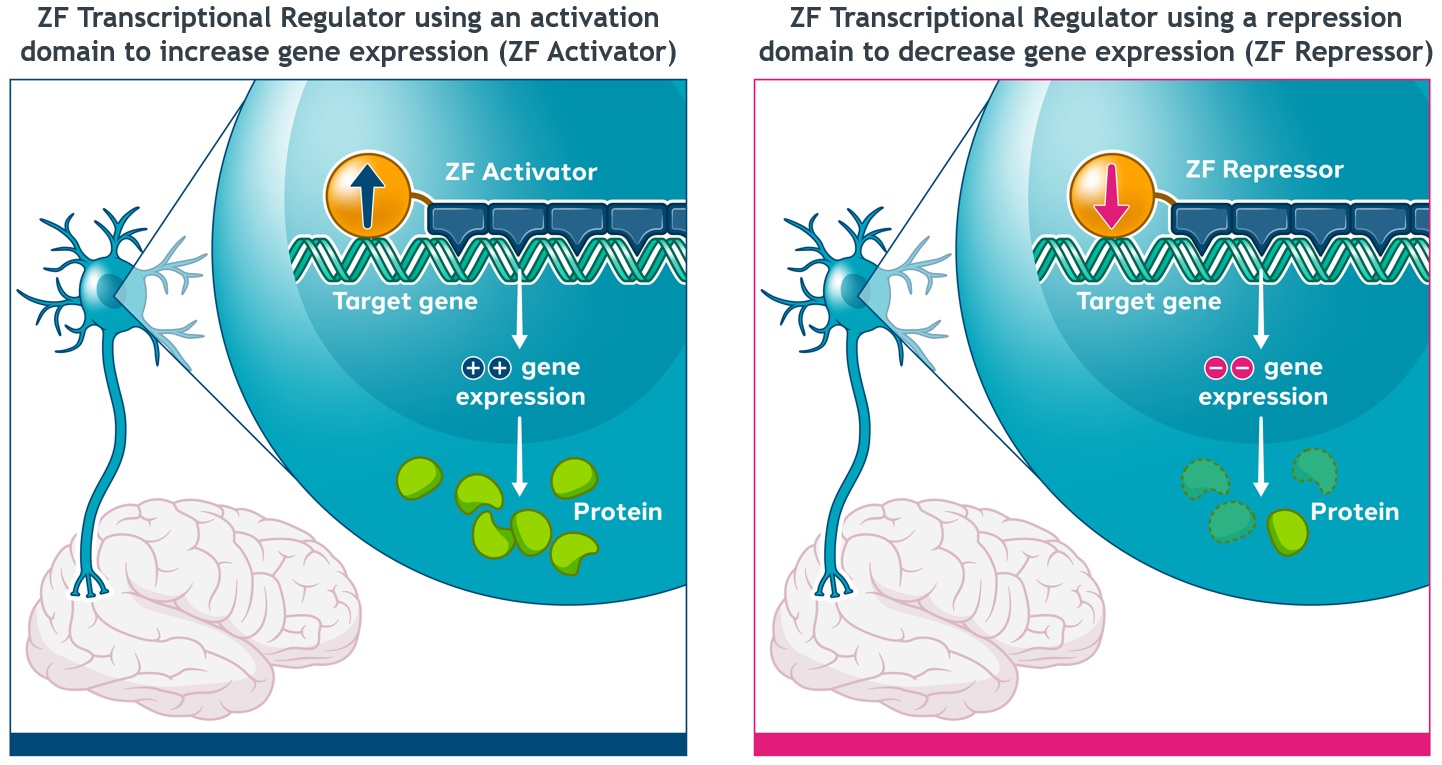
標的遺伝子の発現を調節する可能性があるZF−転写調節因子、またはZF−TRsも評価している(図4)。ZF活性化剤、またはZF-ASは、未処理細胞と比較して標的遺伝子の発現を増加させることを目的として、亜鉛フィンガーアレイを活性化ドメインに連結することによって作成される。代替的に、ZF阻害子またはZF-Rsは、遺伝子を下方制御または完全にオフにするために、亜鉛フィンガーアレイを阻害ドメインに接続することによって生成される。ZF-Rsはまた、健康対立遺伝子の発現を可能にしながら、変異対立遺伝子の発現を選択的に抑制するように設計されてもよい。著者らはいくつかの臨床前プロジェクトがZF-Rsの潜在力を評価し、これらのプロジェクトは遺伝子発現を低下させることを目的とし、中枢神経系疾患の潜在的な治療方法として、Biogenと達成したタウリンとパーキンソン病の治療の協力協定、武田社とハンチントン病に対する協力協定、及びファイザー社とALSに対する協力協定を含む。著者らはまたノワール社と臨床前協力を行い、ZF-ASが遺伝子発現を上昇させる潜在力を評価し、自閉症スペクトラム障害と知的障害の潜在的な治療方法とした
図4:ZF−TRsは標的遺伝子発現を調節する潜在力を有する
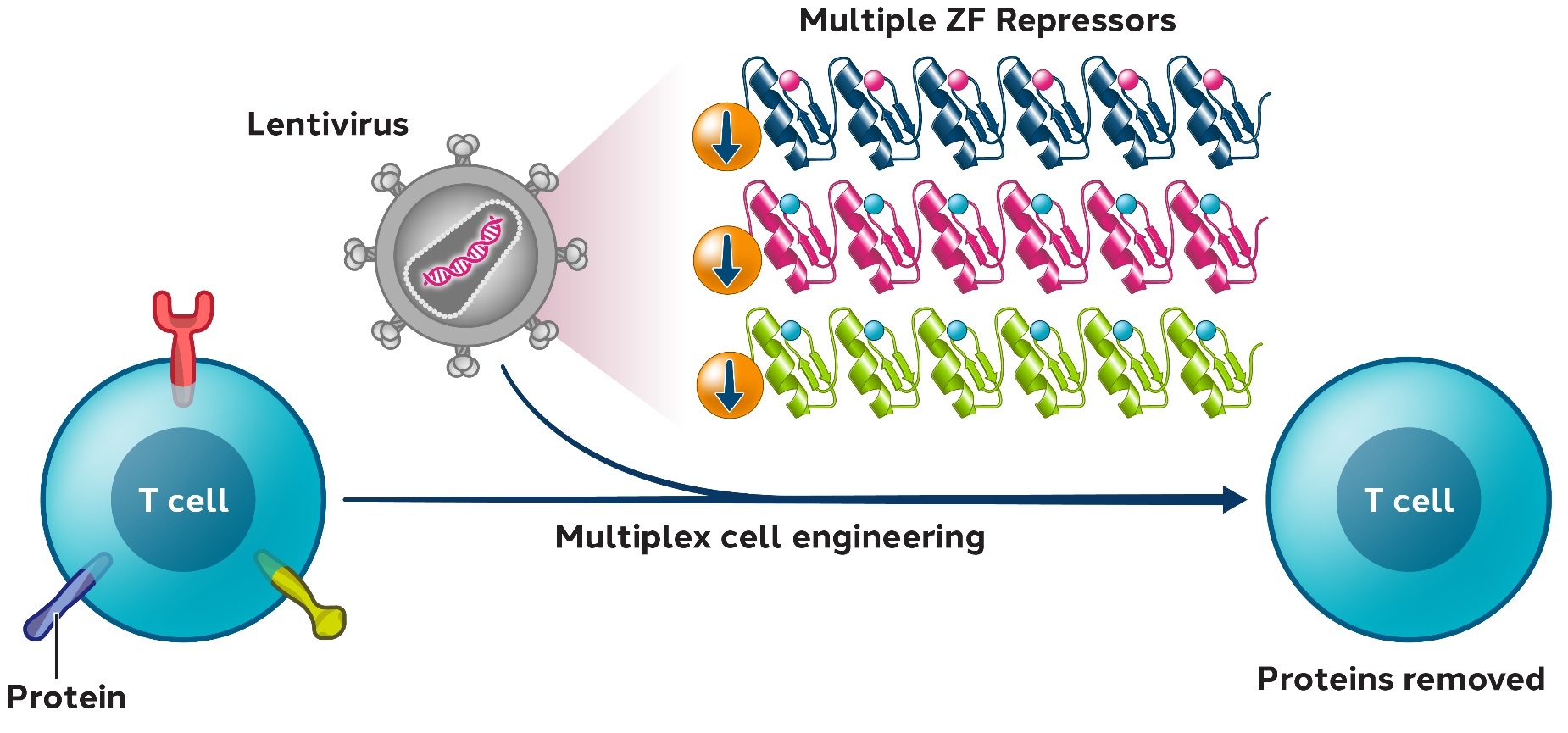
ZF−リプレッサーを用いた多細胞工学
ZF転写調節剤は、単一細胞中のいくつかの遺伝子を阻害するために使用されてもよい。そこで,いくつかのZF-Rを設計した.規制の程度は調整可能であり、部分的または完全にノックダウンする可能性を提供する。そのコンパクトなサイズのため、複数のZF-Rは1つの単一のウイルス構築物に結合することができ、単一の形質導入イベントにおいて有効な多遺伝子制御を実現し、二本鎖切断を必要としない。レンチウイルスの交付は成熟した方法を利用しており、既存の製造プロセスを大きく変更する必要はない。
この新しいプラットフォームの概念証明として、著者らは単一のレンチウイルスにコードされた複数のZF-Rを使用して、CARを含む初代ヒトT細胞を設計し、いくつかの同種工学標的またはチェックポイント阻害剤の発現を抑制するためにCARを含まない。ZF−RsはRNAとタンパク質レベルで選択された標的遺伝子に対して効率的かつ特異的な作用を有することを証明した。
ZF-RプラットフォームはT細胞核酵素編集方法の有効な代替或いは補充である可能性があり、最適化T細胞製品を産生する選択を著しく拡大する潜在力があると考えられる。
図5:ZF−Rsを用いた多重細胞工学
中枢神経系(CNS)を目指した工学化AAV
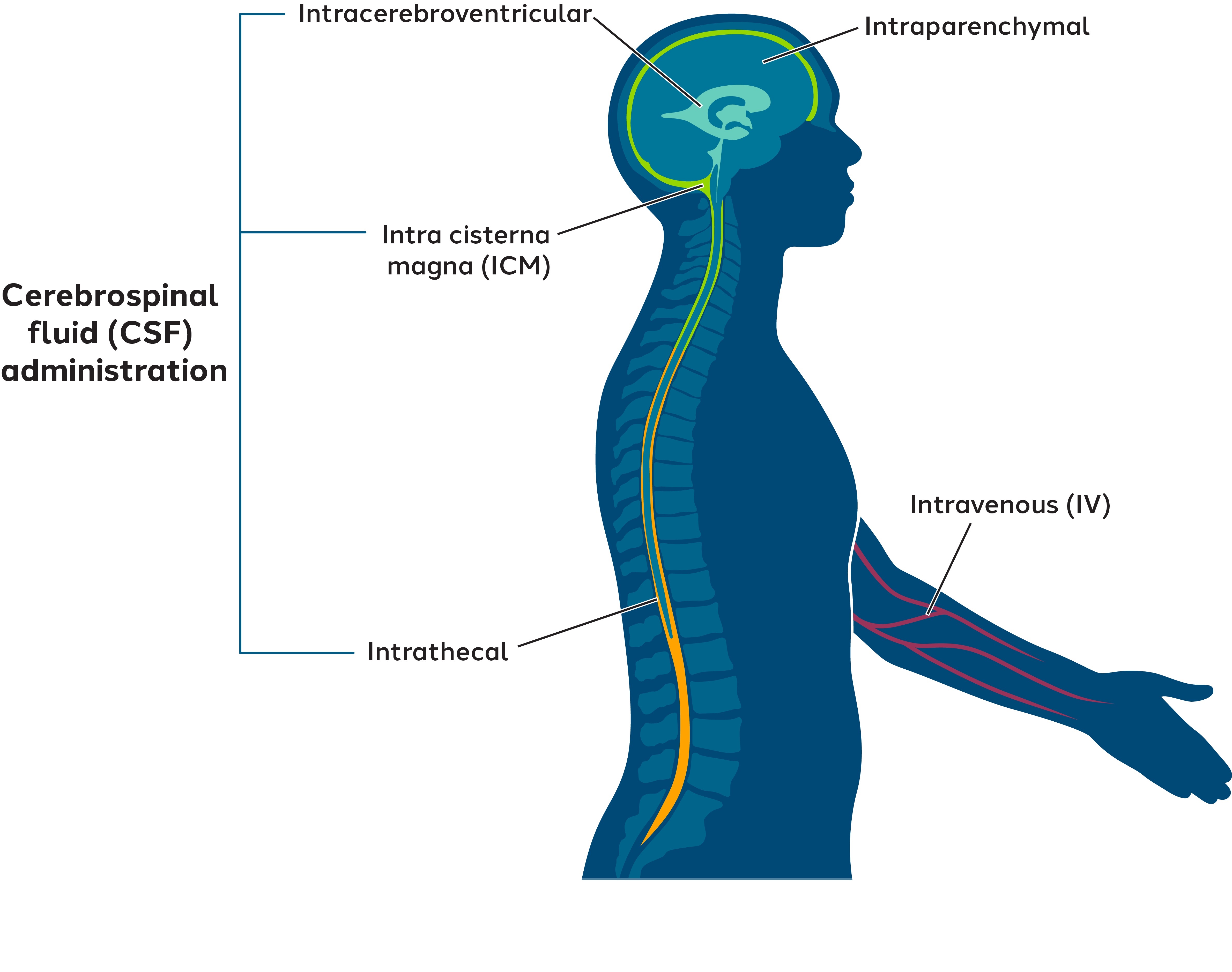
我々は,中枢神経系へのゲノム薬の送達が中枢神経系疾患を治療する潜在療法の開発の大きな障害であるため,われわれの中枢神経系標的研究療法のいくつかの可能な投与経路を評価している。
図6:標的中枢神経系の潜在的投与経路
いくつかのAAV血清型は,最も有名なのはAAV 9であり,脳に分布しているが,限られた発現を実現するためには高用量が必要である。著者らはすでに独自のAAVカプシド発見プラットフォーム、Siefter(体内形質導入と発現リボ核酸を選択する)を開発し、改善された中枢神経系形質導入を利用してカプシドを設計することを目的とした。著者らはいくつかの殻を識別するために、数千万計の独特な殻をスクリーニングするためにスクリーニング器を応用しており、これらの殻は中枢神経系に対する優れた輸送を媒介している。必要な治療プロファイルを繰り返し表示できるカプシドを見つけるために、連続数回のスクリーニングを行った
図7:Sangamo独自のSITFERプラットフォームは、中枢神経系のための新しいAAVカプシドを開発するためのものである
2022年5月,われわれの科学者はSIFTERプラットフォームを用いて静脈(IV)と脳脊髄液(CSF)投与で得られた結果を公表した。このプラットフォームは、AAV 9:STAC−102およびSTAC−103(STAC=Sangamo Treateutics AAVキャプシド)に対してより良い送達を示す新しいカプシドを識別することができることに留意されたい。
全体的に言えば、著者らはもっと高い標的組織送達効率と特異性を有するAAVカプシドを改良することは安全かつ有効なゲノム薬物を創造して中枢神経系疾患を治療する強力な潜在力があると考えられる。
ゲノム工学-塩基編集
我々のZFプラットフォームはまた塩基編集を実行するために使用することができ、これはゲノム医学分野の新しい方法であり、特定の標的DNA塩基を二本鎖切断を必要とすることなく、別のDNA塩基に変換することを可能にする。塩基編集は,DNA一本鎖中の特定の塩基を変化させるデアミナーゼなどのDNA配列を直接変化させる酵素の使用に依存する
我々は,ZFを用いて高精度かつ特異的に標的を行うことができ,十分小さく,関連するウイルスベクターにカプセル化し,高レベルの編集を実現することができるコンパクトな塩基エディタアーキテクチャを開発し,治療応用に適している可能性がある
図8:コンパクトZFインフラストラクチャ
ZF塩基編集は複数の遺伝子を一度にノックアウトするのに非常に適しており,同時にDNA二本鎖切断の間に染色体転座事象が発生する可能性が低いためである。注目すべきは、ZFベースのエディタのコンパクトな構造は、すべての3つのコンポーネントを単一のAAVキャリアにカプセル化することを可能にし、潜在的な治療用途を示唆することである体内にある.
ZFプラットフォームは新しい人間治療学の開発に機会を提供した
我々のZFプラットフォームは広範な新型薬物に独特な特許基礎を提供し,これらの薬物は小分子薬物,タンパク質薬物,RNAによる療法,伝統的な遺伝子療法および他の遺伝子やゲノム編集プラットフォームと比較した差別化技術的優位性を有しており,広範な医療ニーズを満たしていない治療法を開発できる可能性があると信じている。われわれのZF遺伝子薬は重篤な疾患の治療戦略を症状管理から持続的な治療に転換する潜在力があると信じている。
我々は、一連の独自の方法を使用して、ゲノム編集のための高度に特異的なZFヌクレアーゼおよびエピジェネティック調節のためのZF−TRsを生成することができる。我々は,mRNA,AAV,アデノウイルス,プラスミド,脂質ナノ粒子の使用,脳組織や脳脊髄液への直接注入など,これらの療法を応用した送達戦略を開発している。より多くの遺伝子とDNA配列が特定の疾病に関連することに伴い、私たちはZF治療試薬の臨床範囲と範囲が引き続き拡大すると信じている。
CAR−Tregsは自己免疫性と炎症性疾患を治療する潜在力を有する
われわれの臨床前計画の重要な領域の一つは,我々が研究している自己免疫性と炎症性疾患のCAR−Treg計画である。Tregは白血球であり、免疫系の重要な調節器である。それらの天然作用は免疫バランスを維持し、自己抗原(自己免疫)或いは正常に耐性の抗原(食物抗原、吸入抗原、接触抗原と細菌菌群抗原)に対する不良免疫反応を防止することである。Tregは,他のT細胞が生体に傷害を与える前にそれらを含め,免疫系が健康な臓器を誤って攻撃しないようにするとともに,ウイルスや細菌から身体を保護する役割を果たしている
私たちはTregsを遺伝子再プログラミングしています離体するTregsが抗原と呼ばれる特定のタンパク質を標的にできるように車を添加しましたしたがって,CAR−Tregsは特定の組織に識別され蓄積するように再プログラミングされ,これらの組織では抗原が発現しており,免疫を介した障害が発生している。われわれの臨床前研究により,CAR−Tregsは体内の過剰活性な免疫細胞を抑制することが示唆された。また,それらは長期免疫寛容である免疫系が特定の自己抗原に反応しない状態を誘導する可能性がある。広範な炎症性や自己免疫疾患に対応するために,免疫寛容を誘導·回復できる療法の開発を目指している。
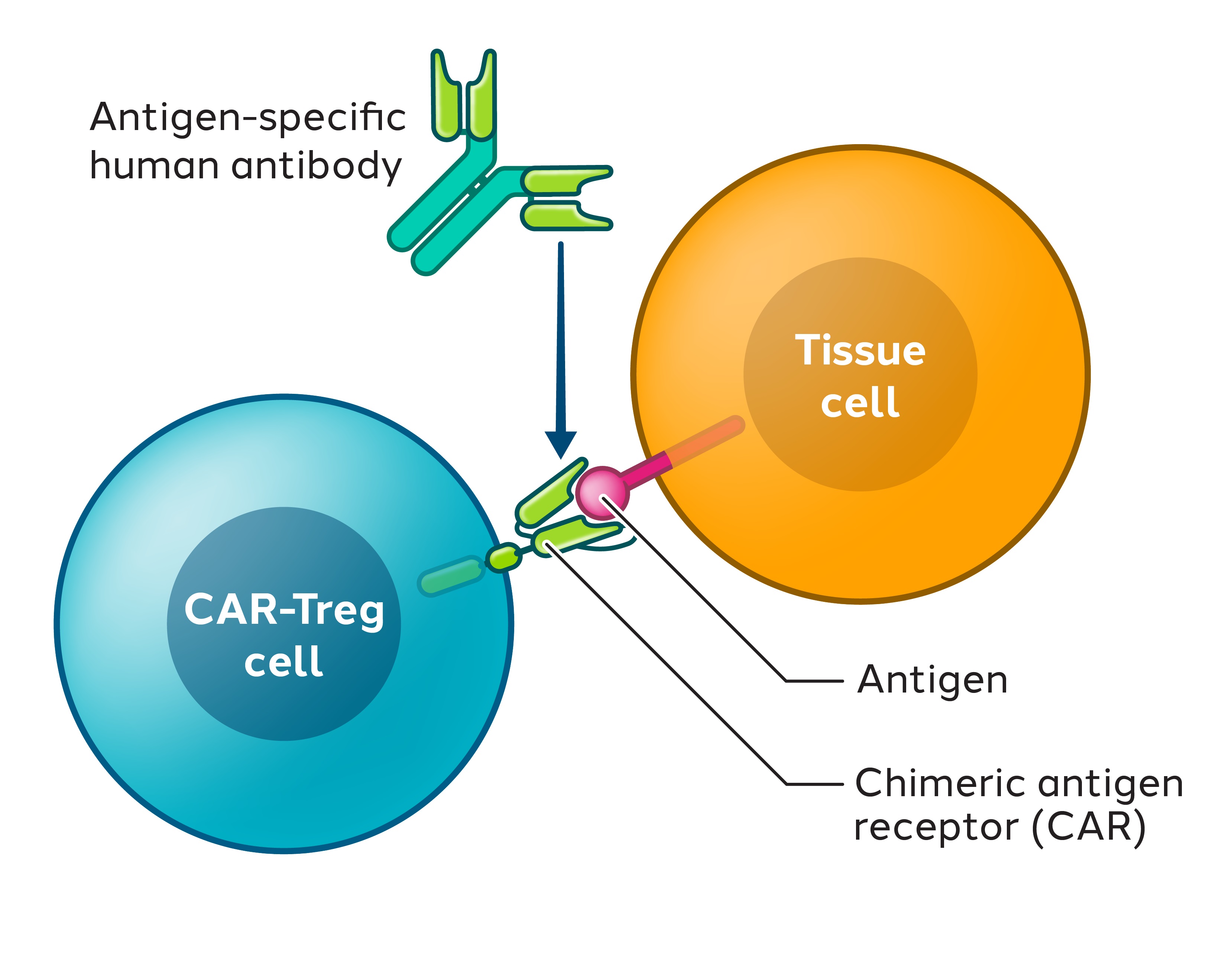
自動車は3つの主要部分から構成されている(図5参照)
•細胞外部分は単鎖可変領域(ScFv)とスペーサーまたはヒンジから構成され、一本鎖可変領域は通常モノクロナル抗体から由来し、標的抗原を識別し、スペーサーまたはヒンジは空間柔軟性を増加させる
•膜貫通ドメインはCARを質膜に固定する。
•細胞内部分はシグナルドメインと共刺激ドメインからなり,単鎖抗体が抗原を認識する際に細胞内シグナルを伝達する。
図9:CAR−Treg細胞認識組織細胞上抗原の模式図
自己免疫性あるいは炎症性適応ごとにCAR標的抗原を慎重に選択した。われわれのCAR−Treg細胞は炎症部位でのみ活性化し,特定と選択的役割を確保するように設計されている。例えば,多発性硬化症のような中枢神経系疾患では,標的抗が中枢神経系に位置することを確保したい。場合によっては、標的抗原は疾病の病因学と関係があるかもしれない
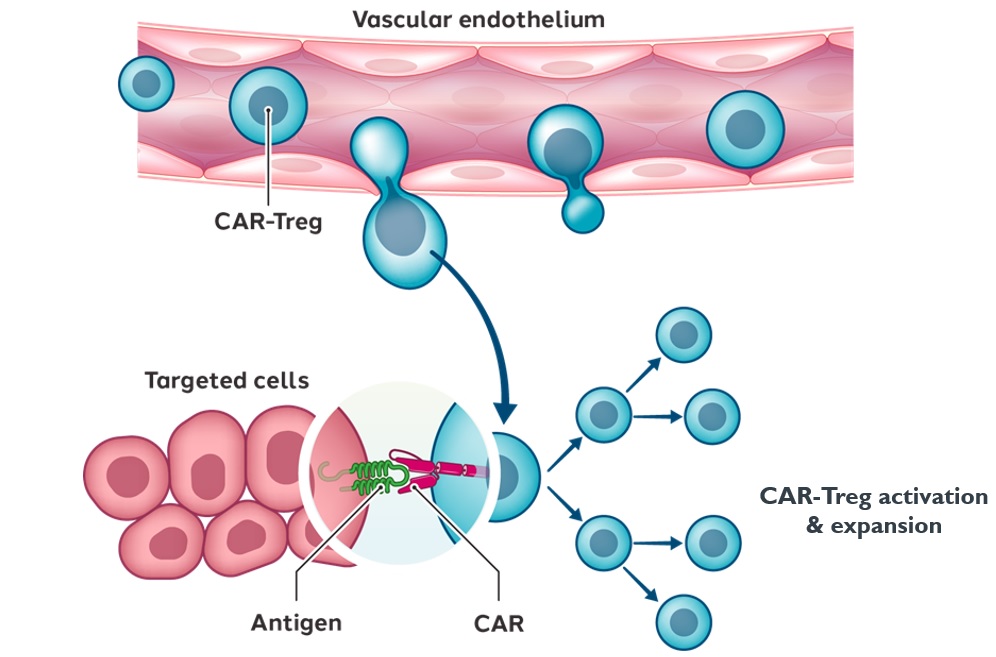
Tregsの主な特徴の一つは、それらが様々なメカニズムで弾圧を調整することができるということだ。それらの作用機序は、細胞接触、可溶性因子、新陳代謝中断および/または細胞溶解によって調節することができる。
•静注後,Tregsの炎症組織への遊走の自然能により,Car−Tregsは炎症組織への移行が期待される。
•その後,CAR−Tregはその特定の抗原に結合し,CAR−Treg細胞の増殖と活性化をもたらすことが期待される
•この活性化はTregに天然の抗炎症や免疫抑制活性を発揮させ,複数の分子や細胞標的を介して作用することが期待される。
図10:CAR−Tregsの期待作用機序
我々の最先端のCAR−Treg候補製品TX 200は,生体ドナーによるヒト白血球抗原A 2不適合腎移植後の免疫媒介拒絶反応を予防するために研究されている。TX 200は自己CAR-Treg細胞治療候補製品である。自家細胞療法は,細胞受容者と同様のヒト由来の細胞を用いて行ったものを図11に示す
図11:我々の自己CAR−Treg手法の概略図
TX 200では,患者のTregsは移植前に収集され,自動車で遺伝子工学を行い,同一患者に注入される。この詳細な経過の結果として,患者は登録後数カ月で用量分配を行うことが予想される。TX 200中のCARは、移植腎臓上に存在するヒト白血球抗原A 2タンパク質を認識するように設計されている。
前の2人の患者はすでに著者らの揺るぎない1/2期の臨床研究で薬物を服用しており、これは私たちが人類におけるCAR-Tregsの動作原理を理解することに役立ち、そしてTregsを用いた遺伝子組換え細胞治療にもっと広範な概念証拠を提供する可能性があると予想される
我々のCAR−Treg法の根本的な影響を信じ,より多くの患者集団に使用できるように次の目標を開始している。そこで,ZFヌクレアーゼが編集した同種異体Treg療法を開発している。異遺伝子細胞療法はドナー由来であり,自己体細胞療法ではなく,異なるヒト由来細胞を細胞の受容者として用いる。同種療法は細胞療法の将来である可能性があり,規模や製造などの自家法の挑戦を克服できると信じている。自己TX 200の概念検証を証明できれば,後続の自己と同種異体プログラムが期待される.その時から、巨大な潜在力が多くの他の大型自己免疫指標、例えば関節リウマチ或いは糖尿病に入る。
遺伝子療法は遺伝子を患者細胞に導入して遺伝病を治療する
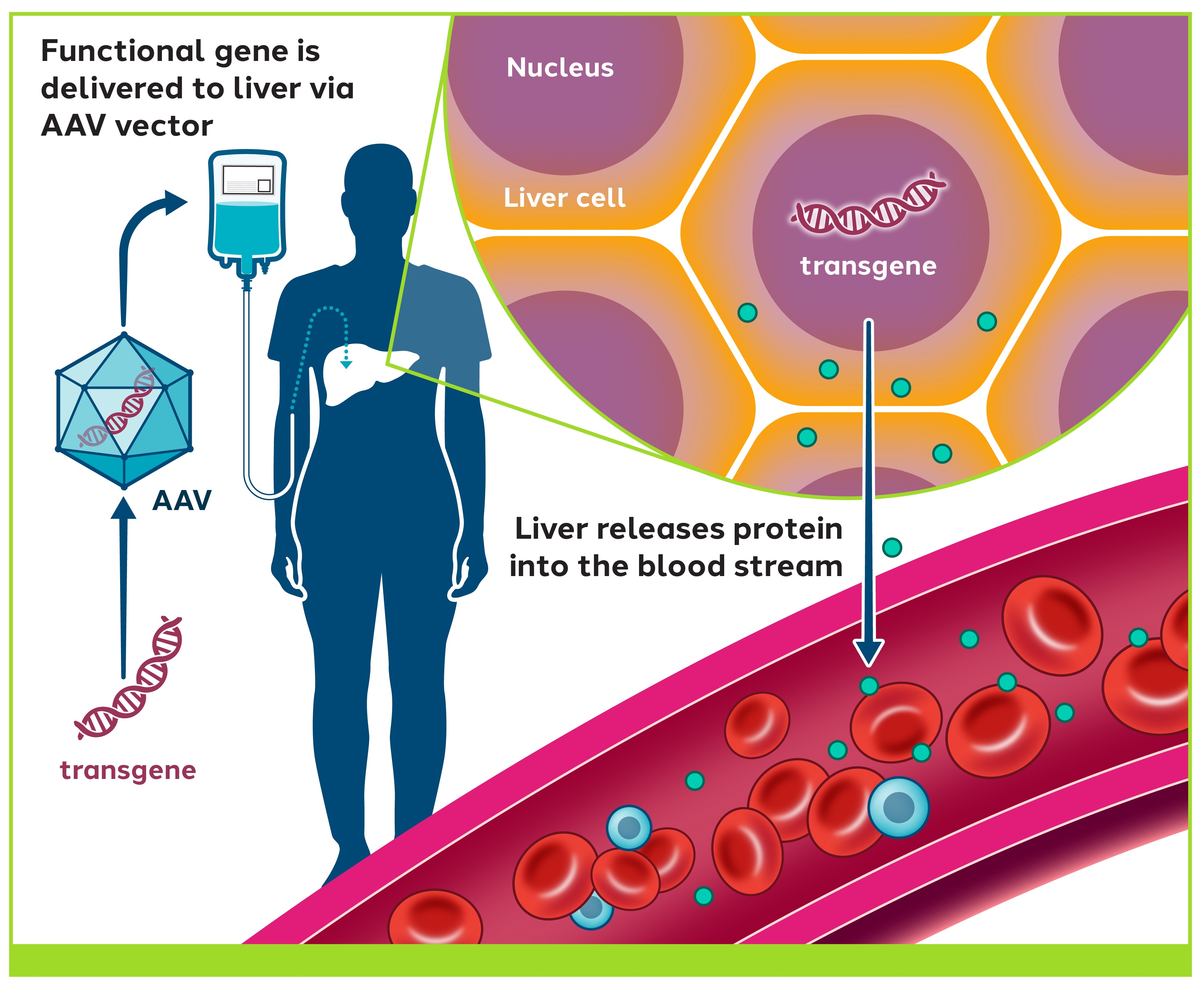
ZF技術を開発する過程で,遺伝子療法の理解を完全にした。遺伝子治療は,新たな遺伝子を患者の細胞に注入することで,誤った遺伝子や損傷した遺伝子を置換することで疾患を治療する方法である。多くの場合、遺伝子治療の動作原理は、DNAを除去または修正することなく、欠陥遺伝子の正確なコピーを患者の細胞に導入することである。遺伝子治療の目標は遺伝病を治療または潜在的に治癒することであり、方法はこのような疾患を引き起こす正常な遺伝子コピーを再添加することである。
遺伝子治療において、著者らは不活化ウイルスを設計することによって、ウイルス蛋白ではなく、ヒト治療性蛋白のDNAを伝達するために治療用遺伝子を伝達することができる。遺伝子治療によく使われるウイルスの一つはAAVである。AAVは自然に産生されるウイルスであり,ヒトに感染するが,疾患を引き起こすことは知られていない。アメリカとヨーロッパの多くの臨床試験において、工学AAVは遺伝子治療の送達方法として使用されており、これまで、この方法の普遍的な耐性は良好であり、重大な副作用はないことが発見された。治療用タンパク質をコードする遺伝子はAAVに包装され,肝臓,眼,脳あるいは心臓などの組織の細胞に輸送されることができる。細胞に入ると遺伝子がウイルスの殻や殻から解き放たれ
治療用タンパク質を産生させることができますAAVはヒト治療に十分な規模で製造できる。
図12:我々の遺伝子治療技術は
開発中の候補治療製品
独自のプログラム
伊氏·ファブリー病
Isaralgagene Citizparvovecは我々が開発しているFabry病を治療するための遺伝子治療製品候補品であり,Fabry病はまれな遺伝性代謝性疾患である。STAARは現在行われている1/2期多中心、開放ラベル、用量範囲の臨床研究であり、18歳のファブリ病患者のイソプロテレノール単回注入の安全性と耐性を評価することを目的としている。≥患者は単回静脈注射を受け、52週間のフォローアップを行った。この研究で治療を受けた患者の治療後5年に及ぶ期間を監視して、安全性、耐久性をさらに評価するための単独の長期追跡研究が行われている
薬の効果がありますこの研究設計は、各用量キュー内の少なくとも2人の患者が用量を受け、各キュー内の潜在的な拡張を規定する。安定したERT治療を受けた患者は,患者や研究者が自ら決定した場合,制御されモニタリングされた方法で治療後にERTをキャンセルすることができる。
投与量増加段階は典型的なファブリック病を有する男性を含む。この研究はその後,女性の治療や,Fabryに関連する心臓や腎臓疾患患者の治療に拡大した。この研究の主な終点は緊急不良事件の治療の発生率である。その他の安全性評価は常規の血液学、化学と肝臓検査;バイタルサインモニタリング;心電図;超音波心電図;肝臓系メチルフェトプロテイン検査と磁気共鳴画像(MRI)を含み、任意の肝臓腫瘍の潜在的形成をモニタリングする。二次終点は1年間の研究期間中の特定の時点のα-GalA活性、血漿Gb 3とLyso-Gb 3レベルとベースラインの変化;ERT注入頻度;心臓磁気共鳴とrAAV 2/6担体除去測定された腎機能と心機能(左心室質量)の変化を含む。肝心な研究終点は生活の質、ファブリー症状と神経病理性疼痛採点、及びAAV 6カプシドとα-GalAに対する免疫反応を含む。
本研究の目的は,ヒトα遺伝子をコードする組換えAAV 2/6ベクターを用いてERTの必要性を除去することである‑Gal Aは,αの長期発現を招く‑肝臓ガイドの遺伝子療法として,Iaralgagene ciphopvovecは一度の静脈内注入として設計されており,患者への事前適応を必要としない。イシャラゲニは腎機能を温存し,心臓発症率や神経病変を減少させた場合に有効である可能性が信じられている
Isaralgagene ciciparvovecの最新情報については、ご参照ください商業動態上です。
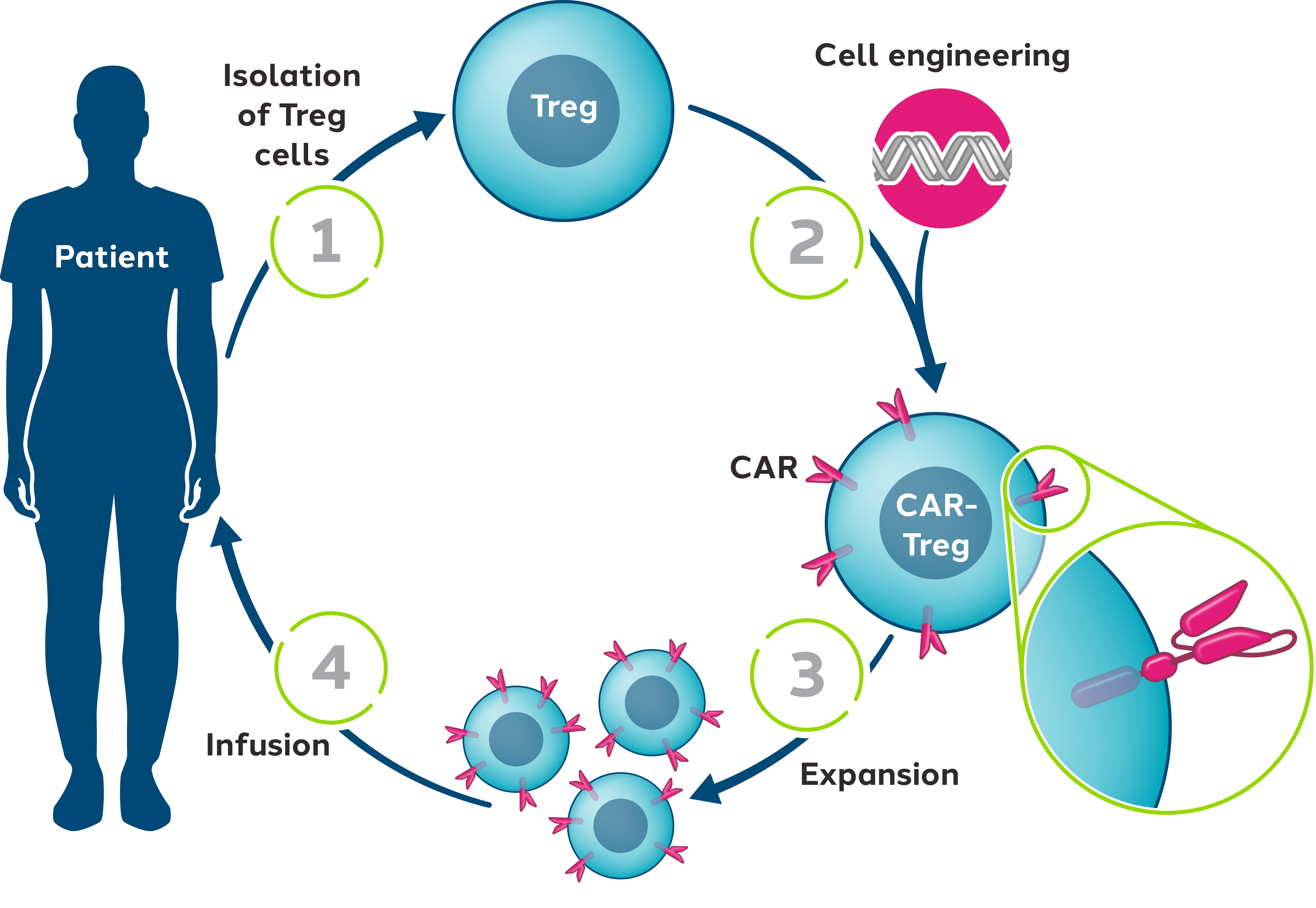
CAR−Treg細胞治療−TX 200−HLA−A 2不適合腎移植拒絶反応−
TX 200は著者らがヒト白血球抗原A 2不合腎移植後の免疫介在性拒絶反応を防止するために開発した自己ヒト白血球抗原A 2特異性CAR-Treg細胞治療候補製品である。我々は現在,我々の1/2期におけるTX 200の強固な臨床研究を評価している。この揺るぎない研究は,CAR−Treg薬理学と患者生物学を理解し,プロセス開発と製造技術を確立するために重要であると信じている
TX 200は末期腎症または末期腎症患者のために開発され、腎臓移植を受ける患者のために開発され、その中で腎臓受容者のヒト白血球抗原A 2は陰性であり、ドナーのヒト白血球抗原A 2陽性である。腎臓移植は末期腎症(ESRD)の最適な治療選択と考えられ、ESRDは慢性腎臓疾患の最終段階であり、一人の腎臓が仕事をしなくなる時である。ヒト白血球抗原不一致はABO血液型不一致後の移植成功の最も重要な障害であり、約21-26%の移植器官はヒト白血球抗原A 2不一致である。ヒト白血球抗原A 2陽性の腎臓がヒト白血球抗原A 2陰性患者に移植された場合,レシピエントの免疫系はこの不整合を認識することができ,長期免疫抑制薬がない場合には,ヒト白血球抗原A 2蛋白を持つ新しい腎臓を攻撃し,移植拒絶反応を招く。生涯免疫抑制治療は顕著な発病率と死亡率と関係があり、全身感染、悪性腫瘍と心血管疾患の発展を含み、これらはすべてこの患者群の主要な死亡原因である。そのため、誘導免疫寛容は安定かつ許容可能な移植片機能と定義され、免疫抑制を必要としないことは依然としてこの医学領域の重要な優先順位である。
TX 200は自己Treg細胞からなり、工程処理後にHLA-A 2 CARを発現することができ、腎臓移植に定位し、HLA-A 2抗原を認識した後に活性化することができる。TX 200はHLA−A 2陽性腎臓と結合することにより免疫寛容を誘導し,腎拒絶反応を予防する作用があると考えられる
他の遺伝子工学細胞治療法と類似しており,白血球分離プログラムを受け,その中からTreg細胞を分離し,工学的処理を行い,凍結保存する必要がある。ヒト白血球抗原A 2陰性の患者はその後移植手術を受け、そのヒト白血球抗原A 2陽性生体ドナーからの腎臓を受けた。回復期を経て、移植者は個性化されたTX 200候補薬を受け取る。この詳細な経過の結果として,患者の投与量は登録後数カ月で行われることが予想される。
われわれの目標は,TX 200が自己免疫性肝炎,クローン病,神経脊髄炎,関節リウマチ,全身性硬化症,1型糖尿病および潰瘍性大腸炎などの主要な自己免疫適応症のCAR−Tregs製品の組み合わせに基礎を築くことである。同種療法は細胞療法の将来である可能性があり,規模や製造などの自家法の挑戦を克服できると信じている。自己TX 200の概念検証が証明できれば,現在臨床前開発中の他の自己と同種異体後続適応を進めていきたい
TX 200の最新更新については、ご参照ください商業動態上です。
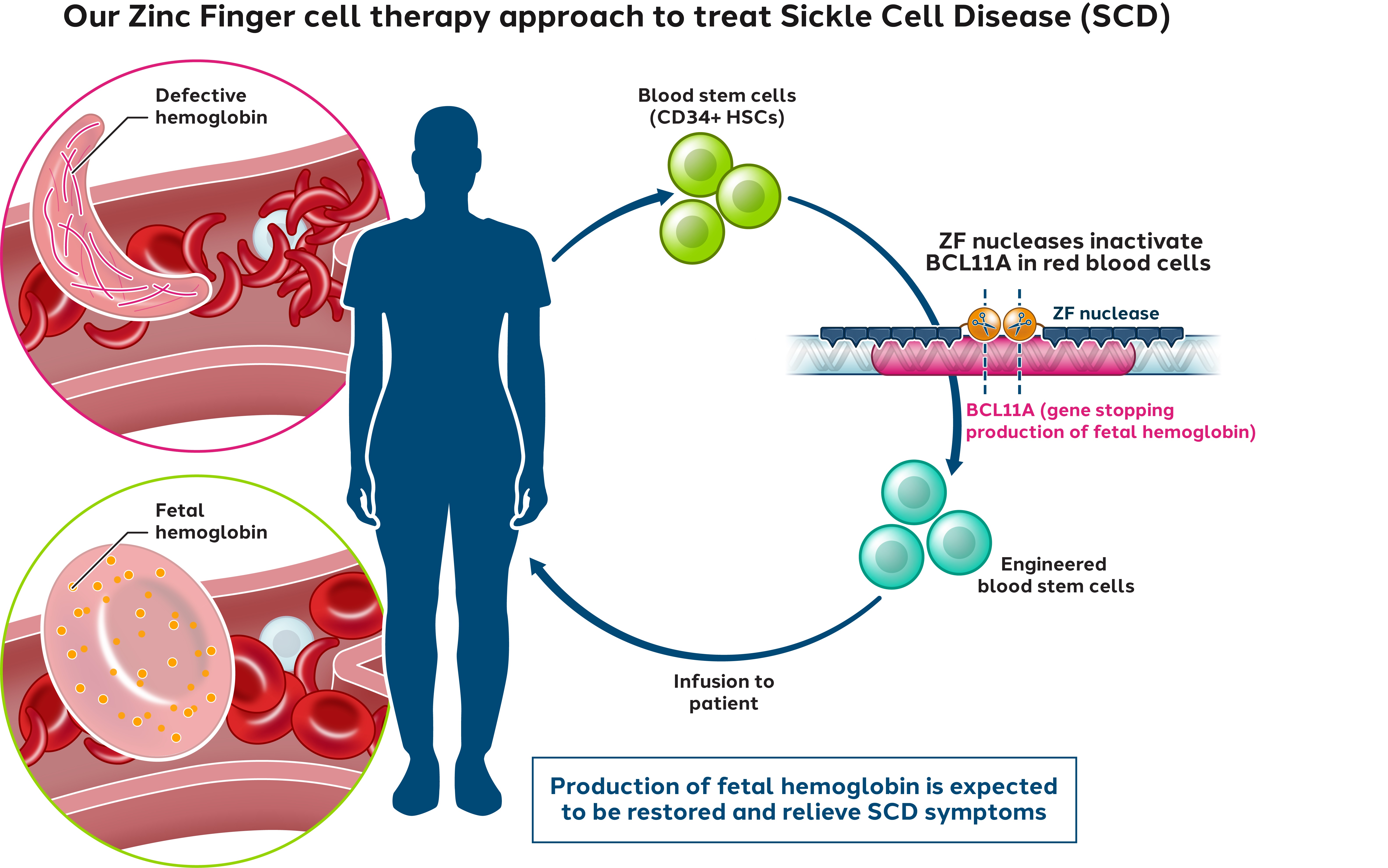
BIVV 003−鎌状細胞病
我々は現在1/2期PRECIZN−1研究でBIVV 003を評価しており,これは我々のZFヌクレアーゼ遺伝子編集細胞療法製品であり,SCDに適している
BIVV 003は,胎児ヘモグロビン合成を誘導するためのZFヌクレアーゼ技術を用いた非ウイルス伝達に関し,患者自身の造血幹細胞前駆細胞のゲノム編集を行う。これはBCL 11 A遺伝子の赤系特異的エンハンサーを遺伝子編集ノックアウトすることにより実現され,BCL 11 A遺伝子はガンマグロブリン遺伝子をコードする強い抑制子である。SCDにおいて、胎児ヘモグロビン合成増加は患者に機能性ヘモグロビンを提供し、異常な鎌状ヘモグロビンの下方制御を助ける可能性があり、それによって苦しい鎌状細胞の危険像と他の疾患特徴を招く。
図1:SCDに対するZF細胞療法の治療
2022年1月、私たちとセノフィは、2022年6月28日または終了日からBIVV 003に関連する権利と義務を返してくれることを発表しました。私たちとセノフィは協力して秩序ある移行を完了し、双方は2022年9月6日に終了と移行協定に署名し、この協定によると、セノフィはSangamoに独占、世界、全額支払い、印税免除、永久的、撤回不可能な許可を授与し、複数のレベルでそのある知的財産権に再許可を付与し、開発、製造、製造、使用、販売、提供販売、輸入、その他の方法でBIVV 003を商業化する権利がある。我々は,行われている臨床試験の完成や関連する長期追跡研究を含め,BIVV 003に関連するすべての臨床試験の責任を負うことに同意した。私たちはまたBIVV 003と関連したすべての規制責任を負っている。セノフィは、BIVV 003に関連するファイル、材料、および第三者との契約を譲渡し、BIVV 003に関連する特定のセノフィが所有またはレンタルしたデバイスを使用する権利を付与します
BIVV 003の最新更新については、ご参照ください商業動態上です。
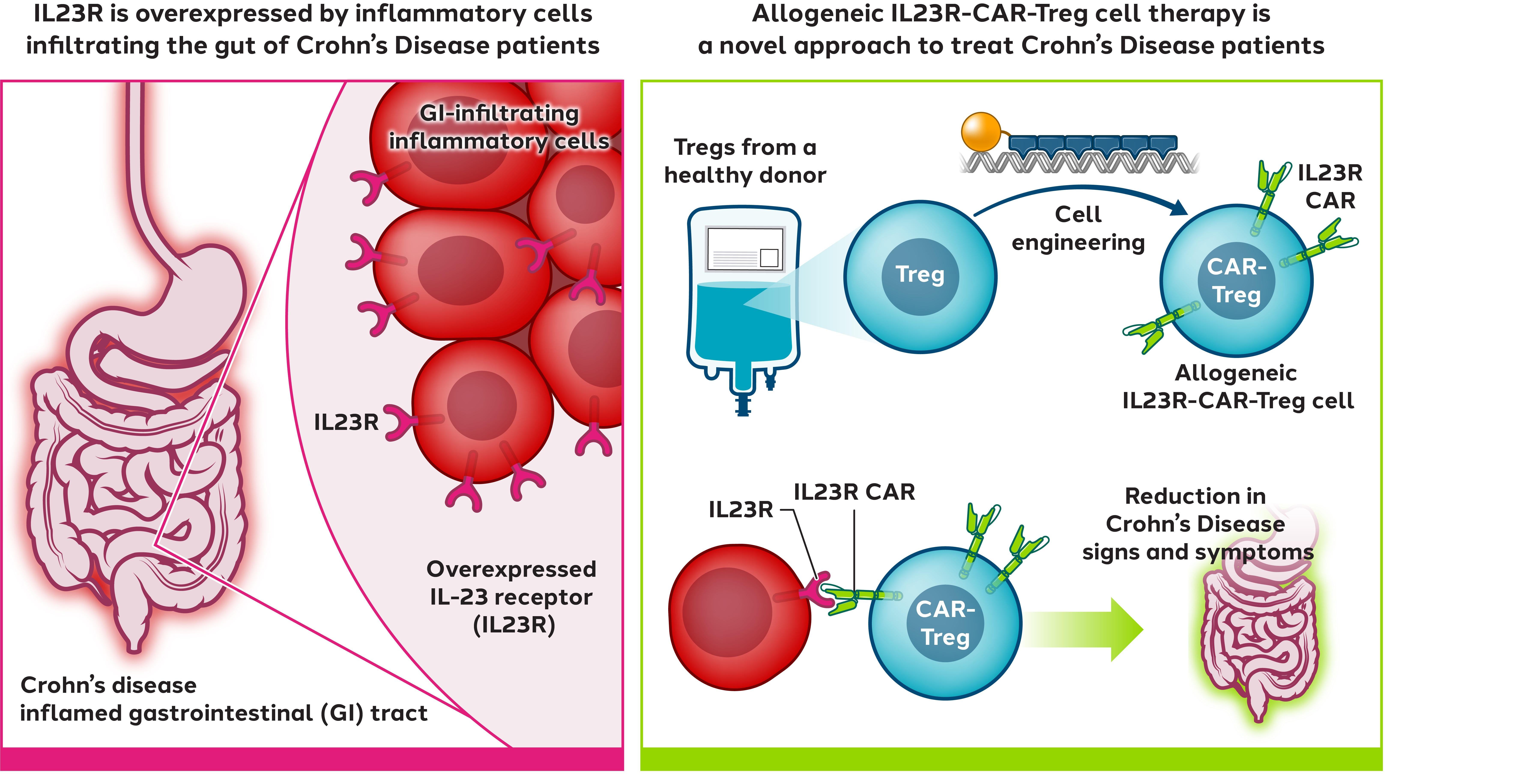
CAR−Treg細胞療法−IBD−
われわれの全資本が有するIBD治療のCAR−Treg計画の臨床前開発を進めていく。IBDは潰瘍性大腸炎とクローン病を含む慢性消化管炎症に関連する衰弱疾患を含む。IBDを治療する候補製品は、IBDに関連する抗原を認識することを目的としたCARを発現するように設計された自己Treg細胞からなり、それにより、産生されたCAR−Treg細胞の腸管における局在および活性化を可能にする。
図2:クローン病の治療のためのIL 23 R Car-Treg候補
CAR−Treg細胞療法−MS
我々はMS、中枢神経系の自己免疫疾患を治療するために、我々の全資本が持つCAR−Treg計画の臨床前開発を進めていく。我々のIBD計画と同様に、我々の治療MSの候補製品は自己Treg細胞からなり、これらの細胞は改造された後、MSに関連する抗原を識別するためのCARを発現することができ、したがって産生されたCAR−Tregsは中枢神経系において局在および活性化することができる。
ゲノム工学であるPrion病
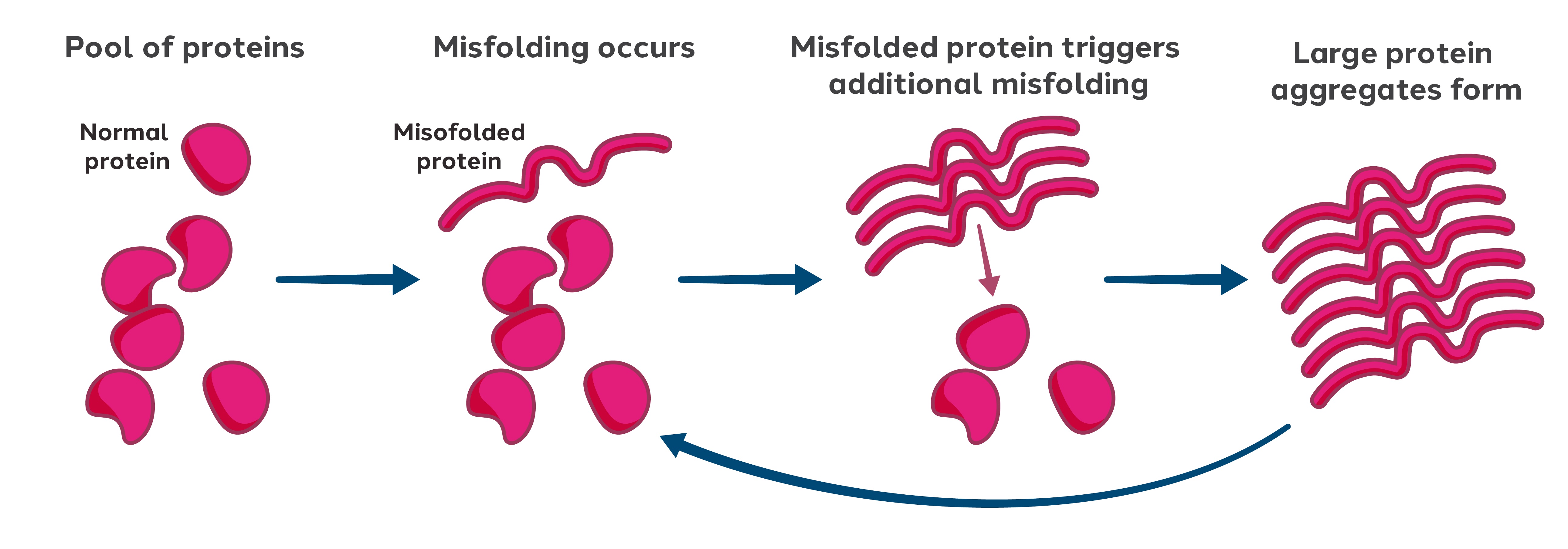
著者らは引き続き著者らが所有する臨床前ゲノム工学計画を推進し、PrNP病に応用し、これは致命的で治愈できない神経変性疾患であり、PrnP遺伝子がコードするPrionタンパク質の誤った折り畳みによって引き起こされる
誤って折り畳まれたPrionタンパク質は、他の正常に折り畳まれたPrionコピーミスフォールディングを引き起こす可能性がある。これは大量に集積したタンパク質プールを引き起こす可能性があり,これは連鎖反応のように,さらに誤って折り畳まれたPrionの誤ったフォールディング,集積,拡散を招く可能性がある。
図3:誤って折り畳まれたPrionタンパク質重合鎖反応
この過程はニューロンに強い毒性を有し,誤って折り畳まれたプーンタンパク質の毒性からそれらを保護するためにニューロンから一部のプーンタンパク質を除去することを目標としている。これは誤って折り畳まれたPrionの拡散や増殖を阻止する可能性があるため,神経変性や疾患の進展を緩和あるいは阻止する可能性があると考えられる。
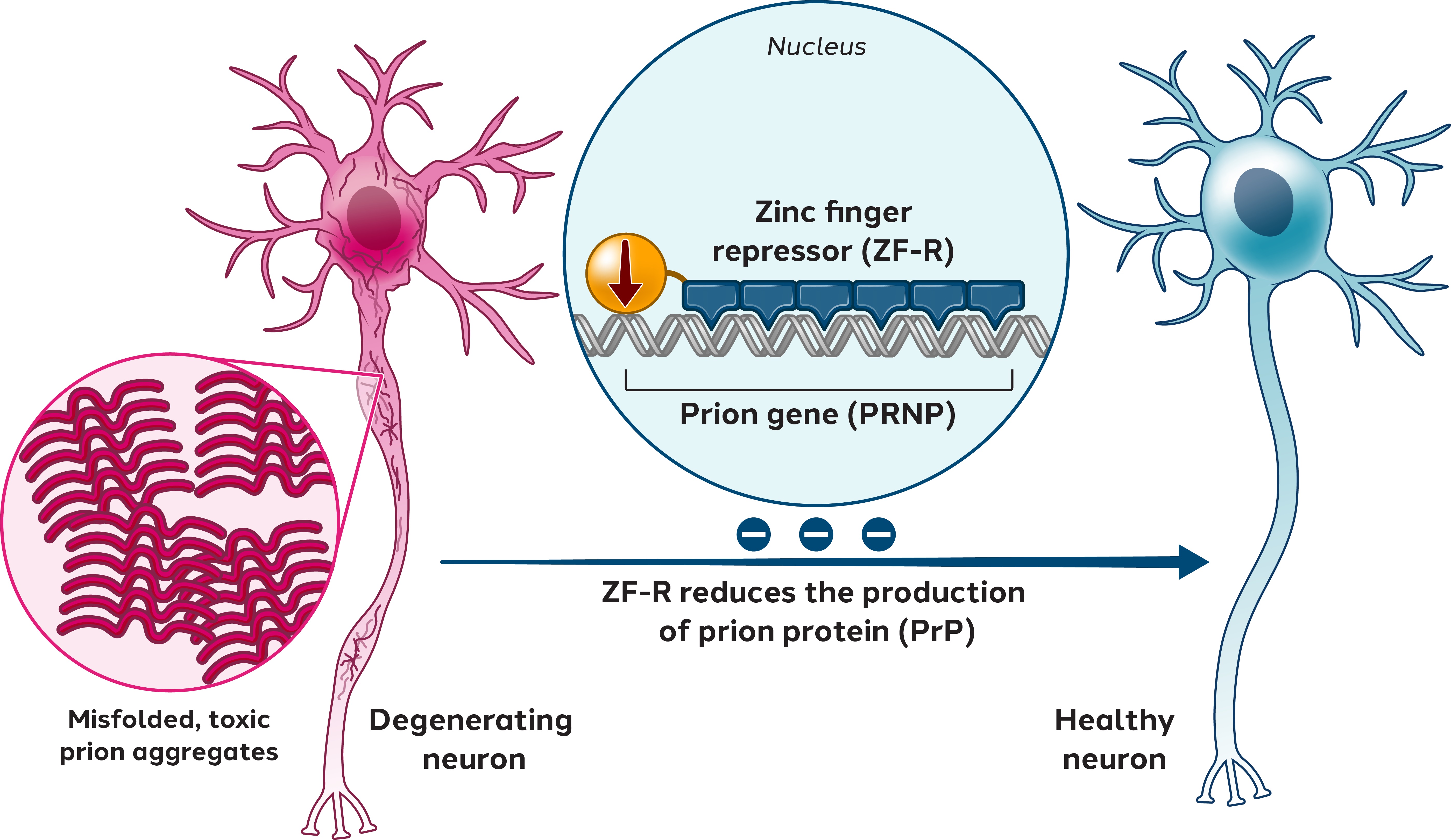
Prion病を解決するために,PRNP遺伝子に対するZF−Rsを開発し,抑制ドメインをその機能ドメインとしている。
図4:ZF-Rsを用いてPron病を治療する方法
私たちは2022年9月のPrion会議でこのプロジェクトの最初の臨床前データを発表した。我々のZF−Rsは体外と体内でマウスのプルーンを特異的に抑制し,これらの接種マウスの生存時間を延長したことを証明した。全体的に、これらの早期の臨床前データはAAV伝達のZF-Rの更なる発展を支持し、獲得性、遺伝性と散発性形式を含む潜在的なPron疾患の治療に用いられると考えられる。
2023年末までに、私たちは次の完全所有のCNSエピジェネティック制御計画の新しいデータを共有する予定です。2024年末までに、私たちは私たちの第2弾CARの2つの新しいIND申請-TregとCNS計画を提出する予定だ
協力計画
巨細胞遺伝子Fitelparvovec−血友病A
我々とファイザー社は原癌遺伝子Fitelparvovec,あるいはSB−525を開発し続けている。著者らとファイザーの協力協定に基づいて、著者らは1/2期ALTA臨床研究とある製造活動を行い、ファイザーはその後の全世界の開発、製造、マーケティングと商業化を担当し、3期生体模倣臨床試験を含む。
Affineは世界的な3期開放、多中心、単一腕試験であり、中重度から重度の血友病Aを有する成人男性患者60名以上(年齢18-64歳)における原癌遺伝子fitelparvovecの単回注入の有効性と安全性を評価する。主な終点は、第3段階先導研究期間中に収集したFVIII代替療法のABRと比較して、原癌遺伝子fitelparvovec治療12カ月後の年間出血率(ABR)への影響である
ALTA研究の初歩的な結果によると、FDAは再生医学高度治療、あるいはRMATを許可し、原癌遺伝子fitelparvovecに指定した。RMATは深刻な疾病を治療、修正、逆転或いは治愈するための再生医学療法を授与され、初歩的な臨床証拠により、この薬物は未満足の医療需要を解決する潜在力があることを表明した。RMAT指定は、FDAとの早期相互作用を含む迅速チャネルおよび画期的な治療指定計画を含むすべての利点を含む。FDAはまたgiroctocogene fitelparvovec孤児薬物と快速チャンネルの称号を与え、ヨーロッパ薬品管理局(European Medicines Agency,EMA)はその孤児薬品の称号を授与した。
Giroctocogene fitelparvovecの最新情報については、ご参照ください商業動態上です。
Kite-037-癌
我々とKite Pharma,Inc.あるいはKiteはGilead Sciences,Inc.の完全子会社であり,我々の研究を用いて癌を治療する細胞療法を開発し続け,ZFヌクレアーゼとウイルスベクターを設計し,T細胞とナチュラルキラー細胞(NK細胞)にCARRS,T細胞受容体あるいはTCRとNK細胞受容体(NKRs)をコードする遺伝子を挿入することを含む選択された遺伝子を妨害·挿入し,これらの遺伝子は共通に合意された標的を指す。Kiteはすべての臨床開発、製造、マーケティング、商業化を担当している。2021年5月、私たちは、最近のポートフォリオ審査の一部として、Kite-037の研究新薬申請またはINDを提出しないことを決定したと発表した。Kite-037の開発計画はまだ進行中であり、私たちとKiteは1つ以上の新しい候補製品の開発に取り組んでいくつもりだ。
ST−501−筋萎縮性側索硬化症,ST−502−シナプス核症と1型強直性筋ジストロフィー症(DM 1)
我々と生物遺伝研究社は,副甲状腺疾患の治療のためのST−501候補品,パーキンソン病を含む同核病変の治療用ST−502候補,神経筋疾患DM 1に対する候補品を含む臨床前ゲノム工学療法の開発を継続している。我々の協力合意によると,Biogenはまた未開示の第4の神経疾患遺伝子目標を選択し,この目標に対する療法の早期研究活動を開始している。私たちの生物遺伝会社との協力協定によると、それは5年間の目標選択期間内に最大8つの追加目標を指名する独占的な権利を持っている。この協力はZF転写制御因子を利用して、神経疾患に関連する重要な遺伝子の発現を調節することを目的としている。
2021年3月、著者らは“科学進展”雑誌で臨床前データを発表し、tau標的ZF転写阻害子はtauメッセンジャーRNAとタンパク質を選択的に50%から80%まで減少させ、11ケ月間持続し、脱標的事象は検出されなかったことを示した
2021年上半期に15ヶ月間に前臨床データを提出しましたこれは…。アルツハイマー病とパーキンソン病国際会議(AD/PD)とアメリカ遺伝子と細胞治療学会(ASGCT)年次総会での研究により、αシヌクレインに対するZF転写阻害物はヒトアルツハイマー病とパーキンソン病を著しく抑制でき、しかも耐性が良好であることが明らかになった体内にある.
ゲノム工学−自閉症スペクトラム障害と神経発達障害
我々とノワーズ社は引き続き3つの神経発育目標のために自閉症スペクトラム障害と知的障害に関連する遺伝子を含む臨床前ゲノム工学療法を開発している。このコラボレーションは我々のZFを利用している‑転写調節因子は神経発達障害に関連する重要な遺伝子の発現を上昇させることを目的としている。
ゲノム工学−筋萎縮性側索硬化症と前頭葉変性−
我々はファイザー社と協力協定を結び,対立遺伝子特異的ZFを用いた臨床前ゲノム工学候補製品を開発した‑転写調節剤はALSと前頭側頭葉変性、またはFTLDを治療し、遺伝子変異に関連するC 9 ORF 72ジーン。ALSの最も一般的な遺伝的原因は、最初のイントロンにおける6ヌクレオチド反復配列またはG 4 C 2反復配列の拡張であるC 9 ORF 72ジーン。我々の方法は,疾患対立遺伝子からの特定の原因遺伝子の発現を抑制しながら,健康対立遺伝子の発現を保持するためにZFインヒビターを設計することである
2020年9月、私たちはこの協力に関する研究義務を達成して、私たちに識別、表現、臨床前のZFの開発を要求します‑あらかじめ合意された基準を満たすRS.ファイザーは現在、後続の研究開発活動と後続の開発、製造、マーケティング、商業化を担当している。
2021年5月にASGCT年次総会で臨床前データを提出し、ZFを示しました‑RSは大量の範囲内で疾病正義とアンチセンス異性体の発現を選択的に抑制でき、同時に患者の正常異性体の発現を維持することができる‑誘導された神経細胞です検出可能な脱標的遺伝子制御は認められなかった。
武田-ハンチントン病
我々と武田は引き続き潜在的な臨床前ゲノム工学製品を開発してハンチントン病を治療し、この製品はZF-Rを使用して、変異の原因ハンチントン遺伝子、あるいはHTT遺伝子を差異的に下方制御し、この遺伝子の正常バージョンの発現を維持することを目的としている。
これらの連携計画に基づく連携のより多くの情報については,次の“-連携”を参照されたい.
レガシー臨床研究プロジェクト
われわれは以下の臨床研究プロジェクトの開発を中止した。我々は引き続き研究案に従って遺産研究に対して適切な長期的な後続行動と終了活動を行った。
ST−400−ベータ地中海貧血
2021年11月、私たちとセノフィは、SCD計画に資源を集中させるために、ベータ地中海貧血適応症の開発を中止するビジネス決定を下したと発表した。5人の患者を1/2期テリス試験で服用し、ST 400の安全性および有効性を評価するための開放ラベルの単腕臨床研究である。結果は最後にアメリカ血液学会年次総会と2021年博覧会で発表された
SB−728−ヒト免疫不全ウイルスまたはHIV
SB-728は、我々のZFヌクレアーゼ媒介性早期ゲノム編集技術を使用した最初の臨床候補細胞のうちの1つである。我々はSB−728を評価するいくつかの臨床研究を行い,このプラットフォームの安全性を証明し,一部の患者の免疫反応を示したが,これらの研究はわれわれの臨床期待に達しておらず,HIVの開発を中止している。
SB−318−MPS I,SB−913−MPS IIおよびSB−FIX−血友病B
我々は,MPS I,MPS IIおよび血友病Bを治療する候補ゲノム編集製品であるSB−318,SB−913およびSB−FIXの開発を中止した。
協力する
私たちはすでにより大きな生物製薬会社と私たちのいくつかの治療計画について戦略的協力を達成し、私たちの技術のいくつかの非治療応用について他のパートナー関係を構築した。我々は、適切な時期に引き続きさらなる協力を求め、内部研究·開発活動に資金を提供し、製品開発、製造、規制承認、商業化に協力する。協力するかどうかの決定は、私たちの内部資源、機関知識、商業的考慮事項の審査に基づくだろう。
ノバ会社
2020年7月、著者らはノワール社と遺伝子制御療法の研究、開発と商業化について協力と許可協定を達成し、3種類の神経発育障害を治療した。協定によると、我々は、我々の関連特許および技術に基づいて、自閉症スペクトラム障害および知的障害を含む神経発達障害に関連する3つの未開示遺伝子について、我々のいくつかのZF転写調節剤を開発、製造および商業化するために、ノワールに独占的、印税、および世界的許可を付与した。我々は協力中に遺伝子標的ごとに早期研究活動を行い,このような研究所に必要なZF転写調節剤を製造し,そのコストはノワール社から資金を提供した。ノワール社は他の研究活動、INDの研究支援、臨床開発、監督管理許可、臨床前、臨床と承認製品の製造及び全世界の商業化を担当している。協定に規定されているいくつかの例外を除いて、私たちは、協力テーマの任意の3つの遺伝子のいずれかの治療製品の開発、製造、または商業化を禁止されている。ノワール社はまた、開発、製造、商業化の協力によるライセンス製品の開発、製造、商業化のために、当社のいくつかの独自AAVを許可することもできます
協定によると、ノファ社は2020年8月に7500万ドルの前払い許可料を支払ってくれた。また、ノバ社から4億2千万ドルにのぼる開発マイルストーンと3.00億ドルにのぼるビジネスマイルストーンを取得する資格がある。ノバから協力によるライセンス製品の潜在的な商業純売上高からノワールの高さから十代以下の二桁の印税を稼ぐ資格もあります。特許の満期、市場独占性の喪失、および第三者知的財産権のいくつかの許可による支払いにより、これらの印税支払いは減少する可能性がある。この協定は、適用される特許権使用料の期限が満了するまで、個々の製品および国ごとに継続して行われる。ノワール社は、規定された通知期間の後、任意の理由で完全または一つずつ合意を終了する権利がある。いずれにしても、他方の破産や重大な違約行為によって合意を終了する権利がある。
生物遺伝研究
2020年2月、我々は生物遺伝会社と全世界許可協力協定を締結し、神経系疾患を治療するための遺伝子制御療法を研究、開発し、それを商業化し、この協定は2020年4月に発効した。私たちの協力は、AAVによって提供された独自のZF技術を利用して、神経疾患に関連するキー遺伝子の発現を調節することを目的としている。協力協定を実行すると同時に、Biogen MA,Inc.と株式購入契約を締結し、この合意に基づいて、Biogen MA,Inc.は我々の普通株であるBiogen株を24,420,157株購入し、総購入価格は2.25億ドルであった。
協力協定によると、Biogenは私たちに1.25億ドルの前払い許可料を支払った。私たちはまだ研究、開発、規制、商業マイルストーンの支払いを得る資格があります。Biogen選択協定で許可されているすべての目標と合意に規定されているすべての指定されたマイルストーンがあれば、総金額は約24億ドルに達するかもしれません
その中には、9.25億ドルまでの承認前マイルストーン支払いと、15億ドルにのぼる初の商業販売とその他の販売ベースのマイルストーン支払いが含まれている。また,連携による許可製品の潜在的な商業純売上高から段階的に桁数から10代以下の印税を得る資格もある。特許の満了、生物類似製品の市場進出、およびいくつかの第三者知的財産権ライセンスに基づいて支払われる費用により、これらの特許権使用料は減少する可能性がある。
協力協定によれば、我々は、Biogenのために選択された特定の神経疾患遺伝子標的のいくつかのZFおよび/またはAAVベースの製品を開発、製造および商業化するために、我々の関連特許および技術に基づいて、Biogenに独占的、特許使用料および世界的許可を付与する。Biogenは、緊張性疾患の治療のためのST-501製品候補、パーキンソン病を含む同型核病の治療のためのST-502製品候補、神経筋疾患DM 1に対する3つ目の製品候補、および4つ目の不開示神経疾患遺伝子標的のうちの4つを選択している。生物遺伝研究会社は、協力協定が発効した日から5年以内(すなわち2025年4月まで)に、最大7つの追加目標を指名する独占的権利を持っている。生物遺伝会社が選択した遺伝子標的ごとに早期研究活動を行い,費用は両社が分担し,独自のCNS送達ベクターと治療関連遺伝子に対するZF−TRs(または潜在的な他のZF製品)の組み合わせを開発することを目的とした。生物遺伝会社はIND使能研究、臨床開発、関連する監督管理相互作用と全世界商業化の責任とコストを担っている。我々は主に協力した上位3製品の初期臨床試験の製造活動を担当し,現在開発中の内部製造能力を適切な状況で利用する予定である。生物遺伝研究会社は上位3製品の初臨床試験以外の生産活動を担当している。我々の任意の目標に対する研究活動は,協力協定が発効した日から7年以下(すなわち2027年4月まで)に行われる。協力協定に規定されているいくつかの例外を除いて、開発は禁止されています, 生物遺伝会社が選択した標的のための任意の治療製品を生産または商業化する。
協力協定は、適用されるすべての印税条項が満期になるまで、製品や国/地域で継続して実行されます。生物遺伝会社は、規定された通知期間の後、任意の理由で完全または1つずつ協力協定を終了する権利があり、最大8つの目標を交換する権利がある。いずれにしても、他方の破産または重大な違約行為によって本合意を終了する権利がある。さらに、Biogenが私たちがBiogenに許可した任意の特許に異議を唱えたら、私たちは協力協定を終了するかもしれない
株式購入契約の条項によると、生物遺伝はすでに同意しており、吾等の事前書面同意及び特定の条件及び例外を受けていない場合には、吾等の発行された普通株の株式を直接又は間接的に買収することなく、買収又は提案募集又は交換要約又は双方間の合併を求め、任意の事項について依頼書又は同意を求めるか、又は潜在的買収吾等の追加持分に関する他の指定行動をとることができない。このような停滞制限は、協力協定の発効3年記念日と生物遺伝会社が私たちの普通株の5%未満の日(早い者を基準に)の満期を受けることになる。
株式購入協定はまた,ある制限に適合した場合には,生物遺伝の要求に応じて,吾等は,すべての残りの生物遺伝株が証券法下で公布された第144条規則に従って売却できるまで,米国証券取引委員会に提出された登録声明に任意の生物遺伝株を登録しなければならないと規定している。
たこ
2018年2月、2018年4月に発効し、2019年9月に改正および再記述されたKiteと協力·許可協定を締結し、癌工学細胞療法の研究、開発、商業化に使用した。Kiteはすべての臨床開発と任意の結果製品の商業化を担当している。
本協定の条項によれば、我々は、我々の関連特許及び技術に基づいて、癌治療のための特定の細胞治療製品を開発、製造、商業化するために、特許権使用料付き、世界的に再許可可能な許可をKiteに付与し、これらの製品は、研究計画から生成され、改造されている可能性がある離体する候補標的に対するCAR、TCRまたはNKRsは、選択されたZFヌクレアーゼおよびこの研究計画の下で開発された最終ベクター(すなわち、AAVs、RVV)を用いて発現される。
研究計画期間内に、本プロトコルにより、本プロトコルの規定に適合しない限り、癌治療の目的のための任意の細胞治療製品の研究、開発、製造、商業化が禁止される離体するヒト癌細胞上またはその中に発現する標的に対するCAR、TCRまたはNKRを発現するゲノム編集。研究計画期間終了後,本プロトコルに基づき,ある例外を除いて,癌治療の目的のための任意の細胞治療製品の開発,製造,商業化を禁止した結果,離体するゲノム編集は,候補標的に対するCAR,TCRまたはNKRを発現する。
協定が2018年4月に施行された時、私たちはKiteから1.5億ドルの前払いを受けた。また,Kiteは我々が行う共同研究プロジェクトの直接費用を精算し,Kiteはすべての後続ライセンス製品の開発,製造,商業化を担当している.開発や販売に基づくマイルストーン支払いを受ける資格もありますが、本協定に規定されているすべての指定マイルストーンが実現すれば、総額は30億ドルに達する可能性があります。その中で、約13億ドルは特定の研究、臨床開発、監督管理と初の商業販売マイルストーンの実現と関係があり、約18億ドルは特許製品が全世界の年間純売上高が特定のレベルに達した時に特定の販売マイルストーンを実現することと関係がある。各開発および販売に基づくマイルストーン支払い(I)は、許可製品が関連マイルストーンイベントを達成した回数にかかわらず、許可製品毎に1回しか支払われず、(Ii)は、そのようなマイルストーンイベントを実現することが可能な許可製品の数にかかわらず、関連マイルストーンイベントを実現する前の10回のみである。また、ライセンス製品の将来の世界における潜在的な年間純売上高に応じて、桁数の割合で段階的に増加する等級版税を得る権利がある。特許の満了、生物類似製品の市場進出、およびいくつかの第三者知的財産権ライセンスに基づいて支払われる費用により、これらの特許権使用料は減少する可能性がある。
Kiteは、規定された通知期間の後、任意の理由で本プロトコルを完全に終了するか、または許可された製品または候補目標に従って本プロトコルを終了する権利がある。いずれにしても、他方の破産または重大な違約行為によって本合意を終了する権利がある。
ファイザー社
ファイザーとは2つの独立した協力協定があります
Giroctocogene Fitelparvovec協力
2017年5月、Giroctocogene fitelparvovecを研究、開発、商業化するためのファイザー社と独占的なグローバル協力と許可協定を締結し、SB-525、血友病A候補遺伝子治療製品、および密接に関連する製品を2019年12月に改訂した。
このプロトコルによると、著者らは原癌遺伝子fitelparvovecの1/2期臨床研究とある製造活動を担当し、ファイザーはその後全世界範囲で原癌遺伝子fitelparvovecの開発、製造、マーケティングと商業化を担当する。我々はまた,他のAAVに基づく血友病A遺伝子治療製品を研究·開発することも可能である。
7,000万ドルの前払い許可料を受け取り,2019年12月にgiroctocogene fitelparvovecのINDのファイザーへの移行を完了して2,500万ドルのマイルストーンを実現し,2020年10月に3,000万ドルのマイルストーンを実現し,鍵となる3期アフィン試験の最初の患者が投与量を服用したためである。私たちは特定の臨床開発、知的財産権、監督、および最初の商業販売マイルストーンを実現した上で、girococogene fitelparvovecと可能な他の製品のための更なる開発マイルストーン支払いを得る資格がある。本プロトコルで指定されたすべてのマイルストーンを実現すると仮定すると、潜在的な臨床開発、知的財産権、規制、および第1回商業販売マイルストーン支払いの総金額は4.75億ドルであり、その中には最大3,000万ドルのgiroctocogene fitelparvovecおよび最高1.75億ドルのプロトコルによって開発可能な他の製品が含まれているが、特定のライセンスによって第三者に支払われるお金は減少するであろう‑党の知的財産権。また、ファイザーは、協定に基づいて開発されたすべての潜在的な許可製品のために米国に印税を支払うことに同意し、これらの印税は、このような製品の世界の年間純売上高の14%~20%に相当し、特許満了、生物類似製品の市場への参入、およびいくつかの許可に基づいて第三者に費用を支払うことによって減少する‑党の知的財産権。
合意条項に適合した場合,我々はファイザーに独占的,世界的,印税の許可を付与し,再許可を付与する権利があり,我々が制御しているいくつかの技術をgirococogene fitelparvovecおよび関連製品の開発,製造,商業化に利用している.ファイザーは我々に非独占的で世界的な印税免除の全額許可を与え,再許可を付与し,プロトコルに基づいて開発されファイザーによって制御されたいくつかの製造技術を用いてAAV交付システムを使用した製品を製造する権利がある。特定の期間内に、私たちもファイザーも、協力以外の臨床でいくつかのAAVベースの血友病A遺伝子治療製品の開発または商業化を許可しない。
以前に終了しない限り、協定の有効期間は、各製品及び各国を基礎として、(I)一国での製品の特許権満了まで継続し、(Ii)一国での製品の規制排他性満了、及び(Iii)一国で初めて商業販売されてから15年以内である。ファイザーは、理由なしに合意全体を終了するか、製品または国/地域で合意を終了する権利がある。合意は、他方が治癒していない実質的な違約または他方の破産に基づいていずれか一方によって終了することもできる。すべての理由で終了すると、ファイザー社にgiroctocogene fitelparvovecおよび関連製品の開発、製造、商業化のライセンスが自動的に終了します。私たちやファイザーがどの国や地域でも理由で終わったときにファイザーは
ファイザー制御の特定の技術に基づいて、終了した1つまたは複数の国でgiroctocogene fitelparvovecを開発、製造、および商業化するために、独自の印税の許可を自動的に付与する必要がある。
C 9 ORF 72 協力する
2017年12月、私たちはファイザー社と単独の独占的なグローバル協力と許可協定を締結し、潜在的な遺伝子治療製品を開発し、それを商業化し、これらの製品はZF転写調節因子を使用して遺伝子変異に関連するALSとFTLDを治療するC 9 ORF 72ジーン。この合意によると、私たちは、変異形態の識別、同定、および臨床前に結合し、特異的に減少するZF転写阻害物質を識別、同定、および臨床的に開発するために、ファイザー社と協力する研究計画に同意するC 9 ORF 72ジーン。
ファイザーから1200万ドルの前金を得て2020年9月にすべての研究活動の完成に関連した500万ドルの記念碑的支払いを完了しましたC 9 ORF 72協力する。私たちは特定の臨床前開発、臨床開発、初商業販売マイルストーンの実現状況に基づいて、ファイザー社から最高6000万ドルの開発マイルストーン支払いを獲得する資格があり、ライセンス製品の世界的な年間純売上高が指定レベルに達したら、最高9000万ドルのビジネスマイルストーン支払いを得る資格がある。また、ファイザーは特許製品の世界の年間純売上高の14%~20%の印税を支払う。特許の満了により、バイオ類似製品の市場参入や、特定のライセンスに基づいて第三者に支払われる費用により、これらの特許権使用料が減少する可能性がある‑党の知的財産権。すべての側は研究計画を実行する費用に責任を負わなければならない。ファイザーは運営と財務においてライセンス製品の後続開発、製造、商業化を担当している。
協定条項によると、著者らは著者らの関連特許と技術に基づいて、予め合意された標準に適合するZF転写制御因子の遺伝子治療製品の開発、製造および商業化のための全世界独占特許と技術許可をファイザーに付与した。特定の期間内に、私たちもファイザーも、協力以外の研究、開発、製造、または商業化は許されませんC 9 ORF 72ジーン。
以前に終了しない限り、協定の期限は、各ライセンス製品および各国/地域をベースに、(I)ライセンス製品の国/地域での特許権満了をカバーし、(Ii)ライセンス製品の国/地域での規制排他的満了、および(Iii)ライセンス製品が主要市場国で初めて商業販売された後15年まで継続される。ファイザーは、理由なしに合意全体を終了するか、製品または国/地域で合意を終了する権利がある。合意は、他方が治癒していない実質的な違約または他方の破産に基づいていずれか一方によって終了することもできる。指定された期間内に任意の開発のリード候補を決定することができない場合、またはファイザーが指定された期間内に特定の開発マイルストーンを超えてリード候補を推進しないことを選択した場合、プロトコルも終了する。何らかの理由で終了すると、合意に基づいてファイザーに製品の開発、製造、商業化の許可を与える許可は自動的に終了します。私たちまたはファイザーが任意の国または地域の任意の許可製品によって製品を理由なく終了することができるかもしれないとき、私たちはファイザーと交渉し、ファイザーが制御する特定の技術に基づいて、中止された1つまたは複数の国/地域で製品を開発、製造、および商業化することができるかもしれないために、非独占的で印税付きの許可を得る権利がある
ファイザーの重大な違反により契約を終了した後、いずれの側も研究、開発、製造、あるいは商業化してはならないC 9 ORF 72遺伝子にはしばらく時間があります
セノフィ
2014年1月、私たちはベータ地中海貧血およびSCDに対する治療法を開発するために、独占的なグローバル協力と許可協定、または2014年の協力協定を達成した。2014年の協力協定は最初にBiogen MA,Inc.と署名され、後者はその後Bioverativ Inc.に譲渡され、後者はその後サイノフィによって買収された。2014年の協力協定によると,我々は最初に2021年第3四半期に停止するベータ地中海貧血計画と,SCD計画によりSAR 445136(現在BIVV 003)を開発し,SCD治療のためのZFN遺伝子編集細胞療法候補製品であるSAR 445136(現在BIVV 003)の開発を行った。セノフィは2021年12月、便宜上終了を通知し、2014年の協力協定の2022年6月28日の終了日から発効します。双方は2022年9月6日に終了と移行協定に署名し、この協定によると、セノフィは独占的、全世界的、全額支払い、印税免除、永久的、撤回不可能な許可を与え、複数のレベルでそのある知的財産権に再許可を付与する権利があり、開発、製造、製造、使用、販売、要約、輸入、その他の方法で商業化されたBIVV 003であり、この製品はSCD計画下で開発された候補製品である。我々は,行われている臨床試験や関連する長期的な後続研究の完了を含めて,BIVV 003に関連するすべての臨床試験の責任を負うことに同意した。私たちはまたBIVV 003と関連したすべての規制責任を負っている。セノフィは、BIVV 003に関連する文書、材料、および第三者契約、ならびにBIVV 003に関連するサイノフィが所有またはレンタルしたいくつかのデバイスの使用権を米国に譲渡し、譲渡する。
セノフィは,進行中のBIVV 003臨床試験の費用や,2023年12月31日までの長期後続研究費の返済にも同意し,最高700万ドルに達している。また、2023年12月31日以降にBIVV 003の開発を継続しないことを選択した場合、セノフィは2023年以降に発生した長期後続研究の費用を精算する義務があり、最大530万ドルに達する。サイノフェイの清算義務は、BIVV 003に関する第三者との協力、協力、販売、許可または剥離が契約を締結する場合、またはFDAが臨床試験および/または長期後続研究を早期閉鎖することを許可する場合を含む、いくつかのトリガイベントが発生したときに終了する。
武田さん
2012年1月、武田の完全子会社Shire International GmbHと協力·許可協定を締結し、2015年9月にこの協定を改訂·再記述し、我々のZF技術に基づく単遺伝子疾患のヒト療法と診断を研究·開発し、商業化した。私たちは2012年に1300万ドルの前払い許可料を受け取り、2014年に100万ドルのマイルストーンを実現した。改訂と再確認の合意によると、武田はZFがハンチントン病を治療する世界的な独占ライセンスを持っている。
改訂と重述の合意によると、武田はハンチントン病計画を完全にコントロールし、ハンチントン病計画の費用を完全に担当しているが、ZF設計、最適化、評価サービスを実行する義務を保留し、このようなサービスの費用を返済することを含むいくつかの義務がある。武田には記念碑的な支払い義務はありませんが、ハンチントン病を治療するZF治療製品の商業販売については、指定された最高上限に達する1桁パーセントの印税を支払う必要があります。改訂および再記載されたプロトコル期間内に、いくつかのHTT遺伝子のためのいくつかの製品をプロトコル外で研究、開発、または商業化することは許可されない。
改正と重述の合意により、武田が私たちに返還した血友病AとB計画の費用を完全にコントロールし、その費用に対して全責任を負うが、何らかの職務義務を守らなければならない。武田には、場合によってはこのようなプロジェクト許可証を取得する優先交渉権も付与されている。武田が特定の血友病AとB計画に返還してくれる事業化を続けていれば、計画中の治療製品の商業販売に1桁パーセント印税を武田に支払うことを要求され、最高上限は指定上限となる。改正と再記述の合意に基づいて、私たちは武田に記念碑的なお金を支払う義務はありません。
改訂および再記載された合意は、(I)一方または武田による他方の治癒されていない実質的な違約行為をすべてまたは部分的に終了すること、(Ii)一方または武田の他方の破産または他の破産手続きを終了すること、および(Iii)武田がすべて終了し、少なくとも90日前に書面通知を発効させることによって終了することができる。
他のパートナー関係
私たちは人間の治療応用の開発におけるパートナー関係に加えて、遺伝子組換え動物や細胞線工学の生産を含む植物農業や研究試薬など、他のいくつかの分野で私たちの技術を承認した。これらのライセンスパートナーには,Corteva Agriscience(前身はDow AgroSciences LLCまたはDAS),Sigma−Aldrich Corporation(現在米国国内のMillipreSigmaと米国国外のMerck KGaA),Genentech,Inc.,Open Monoclon Technology,Inc.(現在Ligand製薬会社)がある。F.Hoffmann-La Roche LtdとHoffmann-La Roche Inc.
知的財産権
特許、商業秘密、技術ノウハウ、ライセンス技術は私たちの業務に非常に重要です。私たちの戦略は、私たちの技術および候補製品の研究、開発、商業化に非常に重要だと思う技術、発明、改善を保護するために、提出、取得、維持、許可を含み、必要に応じて私たちの特許および特許出願を保護することを含む。我々はすでに米国特許商標局(USPTO)と複数の外国司法管轄区の特許庁に大量の特許出願を提出した。著者らの独自の知的財産権は亜鉛フィンガー蛋白、転写活性化因子様効果器或いはTALE、蛋白質とクラスター規則間隔短回文反復配列(CRISPR/CA)、編集システム、ゲノム編集技術の治療応用、Treg細胞治療プラットフォームとウイルスベクター伝達プラットフォームに関連する方法を含み、著者らのプラットフォームに関連する技術を可能にし、そして各種の応用においてゲノム編集を使用する。私たちは、特許、著作権、商標、ノウハウ、持続的な技術革新および商業秘密保護、および秘密保護プロトコル、材料譲渡プロトコル、研究プロトコル、およびライセンスプロトコルに依存して、私たちの固有の権利を確立し、保護します。
許可を得た技術
著者らは関連領域で多くの学術機関の設計、選択と使用を許可し、ZFP、ZFヌクレアーゼとZF-転写阻害物をゲノム編集とエピジェネティック制御のある知的財産権に応用した。私たちのZFPおよびZFヌクレアーゼプラットフォームの全面的な保護に重要な役割を果たしている個人的な許可はないが、これらの許可内では、私たち自身の技術的ノウハウ、特許出願、および特許を組み合わせることで、私たちの製品または技術を複製または使用しようとする可能性のある許可されていない第三者から私たちを保護することができると信じている。
さらに、私たちの細胞治療製品について、私たちの子会社フランスのSangamoはブリティッシュコロンビア大学と許可協定に署名し、この協定によると、Sangamo Franceは関連分野で自動車が私たちのTX 200候補製品で使用されることを独占的に許可した。本ライセンスは、2038年9月に満了すると予想される特許シリーズを含み、特許期限調整またはPTA、特許期間延長またはPTEまたは免責宣言はない。
我々の知的財産権は
私たちが許可した特許の組み合わせに加えて、私たちは、ZFP、ZFヌクレアーゼ、ZF転写阻害剤、TALLタンパク質およびCRISPR/CAS編集システム、Treg細胞治療プラットフォーム、ウイルスベクター送達プラットフォーム、および私たちの計画に関連する他の技術の設計、構成および使用に関する約170個の特許シリーズを含む多くの承認された特許および出願されている特許を有する。
亜鉛指技術の20年以上の歴史を考慮すると、私たちのポートフォリオの中のいくつかの最初の亜鉛指特許は2015年に満期になった。しかし、私たちはこの特許の組み合わせに基づいて発展を続け、より多くの特許を取得し、私たちのZF技術を保護する出願を出願している。しかも、私たちの議決権が発行される可能性のある特許は、私たちの特許財産の特許独占権を延長するだろう。
私たちは、私たちが許可して所有している特許と特許出願に加えて、私たちの技術的ノウハウと商業秘密に加えて、私たちの遺伝子治療、細胞治療、およびゲノム工学計画の商業開発に実質的な保護と専門権を提供すると信じている。この点で、以下のタイプの発明、プロセス、および製品を含む、私たちが出願または独占的および非独占的に許可してくれた特許は、以下のタイプの発明、プロセス、および製品を含むビジネス関連技術をカバーしています
•ZFPおよびZFヌクレアーゼ設計、エンジニアリングヌクレアーゼおよび組成物(複数の特許発行、2029年から2036年までの有効期間が予想され、PTA、PTEまたは免責宣言はありません)これらの特許がカバーする発明は、DNA標的選択、亜鉛フィンガー結合ドメイン設計、ヌクレアーゼドメイン設計、リンカー設計、DNAニッケル酵素、ZFPライブラリーデータベースおよび構築方法、および亜鉛フィンガー結合特異性を向上させる方法を含む(例えば、US 9982245、US 10066242、US 10113207参照)
•ZFP Treeutics(複数の特許発行、満期日は2028年から2031年まで、PTA、PTE、または免責声明はありません):これらの特許がカバーする発明は、内因性遺伝子の活性化および阻害に関連する方法、ハンチントン病の治療、HIV、癌治療、心臓収縮能の調節およびグルココルチコイド受容体の調節方法(例えば、US 9943565参照)を含む染色体内到達可能領域の識別を含む
•ヌクレアーゼ治療(複数の特許が発行される予定日は2031年から2036年まで、PTA、PTE、または免責宣言はありません)これらの特許がカバーする発明は、HIV、β地中海貧血およびSCD、血友病遺伝性代謝性疾患、ゲノム編集、パーキンソン病、PD 1発現の調節;免疫調節療法;嚢胞性線維症;中枢神経系疾患;ヒト白血球抗原遺伝子ノックアウトおよび幹細胞編集方法を含む深刻な併用免疫不全、修飾T細胞(例えば、US 9877988、US 9963715、US 10072066、US 10081661、US 10143760参照)、および;
•ZFPおよびヌクレアーゼの非治療的使用(発行された複数の特許の予想期限は2028年から2035年まで、PTA、PTEまたは免責宣言はない):これらの特許がカバーする発明は、調節配列の同定、遺伝子調節、構造および生物機能の分析、農業バイオテクノロジーの方法、細胞分化状態を変化させる方法、タンパク質生産を改善するための細胞系の開発、トランスジェニック動物開発の方法、幹細胞工学、ゲノム編集方法(例えば、US 9890395参照)を含む。
生物製薬会社の特許地位は、私たちの特許地位を含め、不確定であり、複雑な法律と事実問題に関連し、重要な法律原則はまだ大きく解決されておらず、行政、司法と規制解釈と完備が必要である。米国や他の国で特許保護を獲得,維持,実行することは依然として不確定であり,これらの国の特許庁,裁判所,行政機関,立法者の決定にある程度依存する。私たちはまた未来に特許を申請できない独自製品や技術を開発する可能性がある。特許出願は特許の発行を招くことはない可能性があり、特許が発行される前に、特許出願に要求されるカバー範囲が大幅に減少する可能性がある。場合によっては、特許出願は拒否される可能性があり、私たちはその後このような申請を放棄するだろう。関連コストを考慮すると、私たちは、発行された特許または係属中の特許出願が私たちに提供する競争優位性が少ないか、または全くないことを決定する可能性があり、この場合、私たちは、その特許または特許出願の失効を放棄または許可することができる。私たちは私たちの技術のいくつかの側面について特許を申請したにもかかわらず、これらの係属中の出願が特許を取得するか、またはすでに発行されているか、または発表される可能性のあるいかなる特許も支持されることを保証することはできない。私たちの既存の特許、または私たちが後で獲得する可能性のある特許は、成功的な挑戦、全部または部分的に無効にされるかもしれない、または実行不可能とみなされるかもしれない。いくつかの外国の法律はアメリカの法律のように私たちの所有権を保護しないかもしれない。
私たちの特許保護の獲得と維持は、政府特許機関によって提出された様々なプログラム、書類提出、費用支払い、および他の要求を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。特許および/または出願の定期維持費、継続費、年会費、および様々な他の政府費用は、特許および/または出願の有効期間内にいくつかの段階で米国特許商標局および米国以外の様々な政府特許機関に支払われる。私たちはこれらの費用を支払うようにシステム的に注意して、私たちは外部会社を招聘し、私たちの外部弁護士に依存して非アメリカの特許代理機関にこれらの費用を支払います。米国特許商標局および様々な非政府特許機関は、特許出願中にいくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。私たちは専門家を招いて私たちの遵守を助けてくれます。多くの場合、不注意は滞納金を支払うことによって、あるいは規則を適用する他の方法で修復することができます。しかしながら、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効される可能性があり、それにより、関連法ドメインの特許権の一部または全部が失われる可能性がある。この場合、私たちの競争相手が市場に参入する可能性があり、このような状況は私たちの業務に実質的な悪影響を及ぼすだろう。私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
世界各国で候補製品特許を出願、起訴、擁護する費用は目を引くほど高く、米国以外のいくつかの国での知的財産権は米国ほど広くないかもしれない。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っている地域に輸出することもできるが、法執行力はアメリカに及ばない。結局、各国に基づいて特許保護を求めなければならないが、これは高価で時間のかかる過程であり、結果は不確定である。したがって、私たちは特定の国で特許保護を求めないことを選択することができ、私たちはこれらの国で特許保護の利点を享受しないだろう。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護、特にバイオテクノロジー製品に関する保護の実行に賛成しておらず、これは、私たちの特許の侵害を阻止したり、私たちの専有権を侵害する方法で競争製品を販売することを困難にする可能性がある。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは、巨額のコストを招き、私たちの努力と注意を私たちの業務の他の側面から移転させる可能性があり、私たちの特許が無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者からのクレームを引き起こす可能性がある。私たちは、特に米国のようにこれらの権利を十分に保護していないかもしれない国では、私たちの知的財産権の盗用を単独でまたは許可者と一緒に防ぐことができないかもしれない。多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は、政府機関や政府請負業者を含む第三者に対する特許の実行可能性を制限している。一部の国では、特許権者の救済措置は限られている可能性があり、これはこのような特許の価値を大幅に低下させる可能性がある。もし私たちまたは私たちの任意の許可者が第三者に私たちの業務に関連する任意の特許の許可を付与することを余儀なくされた場合、私たちの競争的地位が損なわれる可能性があり、私たちの業務、財務状況、運営結果、および見通しは不利な影響を受ける可能性があります
将来、第三者は、私たちの業務に非常に重要な技術に対して特許、著作権、商標、および他の知的財産権を主張するかもしれません。いかなる潜在的な侵害、無効、および実行不可能な法律主張の結果は予測できない。私たちの製品が第三者の固有の権利を侵害しているか、または第三者の固有の権利を侵害する可能性があると主張する任意のクレームは、私たちに不利であると判断された場合、私たちの業務を深刻に損なう可能性があります。“リスク要因”を見てください私たちの知的財産権と関連した危険。”
競争
我々と我々のバイオ製薬パートナーは,ZF DNA結合蛋白を用いた遺伝子療法,細胞療法,ゲノム工学療法の研究と開発のリーダーである
遺伝子の編集や遺伝子発現の調節に専念する他のいくつかの会社や,少数の商業·学術団体がZFゲノム工学技術の開発に取り組んでいることが知られている。遺伝子治療、細胞治療とゲノム工学領域の競争は激しく、私たちは多くの異なる源からの競争が持続的かつ激化することを予想し、他の生物製薬会社;学術と研究機関;およびTALEタンパク質とCRISPR-CAS編集システムのようなZFおよび私たちのZF技術プラットフォームと競争する技術を開発する政府機関を求める。
したがって、私たちの競争相手は私たちの前に特許保護を得ることに成功し、FDAの承認を得たり、競合製品を商業化することに成功するかもしれない。もし私たちが商業製品の販売を始めるなら、私たちはマーケティング、販売、流通、製造能力のより強い会社と競争するかもしれませんが、これらの分野での経験は限られているか、あるいは経験がありません。はい
また、我々が開発に成功したどの候補製品も、長期的で安全かつ有効な使用履歴を有する既存製品と競合する可能性がある。
私たちは運営の臨床開発段階にあり、現在治療製品の販売はありませんが、以下の会社、製品、および/または技術は私たちの技術や私たちが開発している候補製品と競争する可能性があると信じています
•製薬とバイオテクノロジー会社はF.Hoffman-LaRoche Ltd.,Protalix BioTreateutics,Inc.,Sanofi S.A.および多くの他の生物製薬会社などのタンパク質薬物を開発している。
•遺伝子治療会社は,BioMarin製薬会社,F.Hoffman−LaRoche株式会社,その完全子会社Spark Treateutics,Freeline Treateutics Holdings plc,4 D分子治療会社,および他の多くの遺伝子治療会社のような遺伝子ベースの製品を臨床試験で開発した。
•細胞製品を開発した細胞治療会社は,ABATA治療会社,異遺伝子治療会社,AZ治療会社,ビーム治療会社,ブルーバード生物社,Cellectis S.A.,Cellenkos社,Cova治療社,CRISPR治療社,Editas Medicines社,GentiBio社,Graphite Bio社,Kyverna社,Precision BioSciences社,Sonoma生物治療社,Tera免疫社,Quell治療社,Vertex製薬会社,多くの他の細胞治療会社である
•ゲノム修正のための治療応用が開発されているヌクレアーゼおよび塩基編集技術は,Cariou Biosciences,Inc.,CRISPR Treateutics AG,Editas Medicine,Inc.およびBeam TreeuticsがCRISPR/Cas編集システムを開発し,Cellectis S.A.開発ストーリーヌクレアーゼと巨ヌクレアーゼ,Bluebird Bio,Inc.居場所エンドヌクレアーゼとMegaTALs,Precision BioSciences,Inc.開発巨大ヌクレアーゼや多くの他の遺伝子編集会社を開発している。
•RNAiとmicroRNAを含むアンチセンス治療とRNA干渉技術は、遺伝子発現を調節することによって作用する新しい治療製品の開発において私たちと競争する可能性がある。これらの技術はいくつかの会社によって開発されており,Alnylam製薬会社,Ionis製薬会社,Moderna社,Regulus治療会社,Voyager治療会社,浪潮生命科学社,他の多くの会社が含まれている。
•生物遺伝会社,ファイザー社,Vertex製薬会社や多くの他社などの製薬会社が開発している小分子である。
バイオ製薬会社との協力手配,学術·研究機関との関係,特許技術のライセンス,まれな疾患治療の臨床試験の対象など,他社からの激しい競争に直面することが予想される。これらの競争相手は、単独でも、彼らのパートナーとの協力でも、私たちよりも効率的またはコストの低い技術または製品の開発に成功する可能性がある。
私たちの成功競争の能力は私たちが能力があるかどうかにある程度かかっています
•安全で効果的でビジネス的に魅力的な独自製品を開発すること
•ビジネス上の合理的な条件で遺伝子転移技術を得ることができます
•必要な規制の承認を得る
•承認された適応に基づいて私たちの製品の精算を受けました
•合格した科学と製品開発者を引きつけて維持する
•私たちの競争相手を含む他社と協力と戦略的パートナーシップを構築して、私たちの技術と候補製品を開発します
•私たちの製品および技術のために特許、ライセンス、または他の独自の保護を取得し、実行する
•私たちが開発した製品を開発し、製造し、マーケティングし、販売する
•最初に市場に進出し、市場の他の製品よりも技術的に優れているか、またはそれ以下の製品を開発し、維持すること
•直ちに被験者を募集して私たちの臨床試験に参加します。
製造業
著者らは現在CMOに深刻に依存しており、FDAとEMAの規定によって著者らの臨床前と臨床候補製品を生産し、これらの規定は現在の良好な製造規範、或いはcGMPとも呼ばれる。プロセス開発、分析開発、品質管理、品質保証、サプライチェーン、
プロジェクト管理と製造は、私たちのCMOの適切な監督を促進し、私たちの監督文書と臨床試験の実行を支持する
私たちは内部製造能力が競争優位を提供することができると信じている。そのため,我々はすでにカリフォルニア州ブリスベンの工場でAAV cGMPの生産を開始し,我々の遺伝子治療パイプラインのための1/2期臨床研究用品の生産を目指している。カリフォルニア州ブリスベンでの細胞治療製造資格認証も完了し,フランスバルバンにある工場で細胞治療製造資格認証活動を開始した。
我々は,いずれの第3段階臨床試験のためにもCMOに依存して我々の候補製品を生産し続ける予定であり,承認されれば商業供給にも利用される。このバランスのとれた製造方法は、内部生産能力と能力に投資すると同時に、外部生産能力との約束を強化し、私たちが期待しているパイプ需要を満たすことができると信じています。
著者らは現在三つの異なる製造プラットフォームを利用している:著者らのゲノム工学と遺伝子治療候補製品のためにAAVベクターを生産し、著者らのいくつかの細胞治療候補製品のためにHSPCを改造し、及び工程T細胞療法を改造した。我々は、ゲノム編集および遺伝子治療のためのAAVベクターを製造するために、商業規模のバキュロウイルス製造プラットフォームを使用して、各AAVベクターは、標的またはZFヌクレアーゼに対する異なるトランスジェニックを包装する。われわれのHSPC細胞治療候補製品の製造過程は患者自身のHSPCを用いた。これらのHSPCは,メッセンジャーリボ核酸を用いて特定のDNA部位に対するZFヌクレアーゼを産生することにより,修飾HSPCを産生する。第三のプラットフォームは、我々のZFヌクレアーゼ技術を使用してCAR-Tregsを自己および同種細胞治療に変換した。炎症や自己免疫疾患の治療に用いられる治療量の制御性T細胞を作製する能力があると信じられている。
政府の監督管理
私たちは厳格に監督管理されている生物製薬業界で運営しており、私たちの大部分の業務は非臨床と臨床試験、開発、製造、商業化、マーケティングと精算を含めて、すべて監督部門の許可を得る必要がある。関連する規制機関はFDA、EMA、欧州委員会、EUまたはEUの国家主管機関、加盟国とイギリスの薬品と保健品監督機関(MHRA)を含むが、これらに限定されない。
製品の監督管理
米国では,FDAは連邦食品,薬物と化粧品法案(FDCA),公衆衛生サービス法案(Public Health Service Act)やPHSA,およびこれらの法律を実施する法規やガイドラインに基づいて,遺伝子治療やヒト細胞治療製品を含む生物製品を規制している。その他を除いて、FDCA、PHSAおよびその対応する法規は、生物製品に関連するテスト、製造、安全、効果、ラベル、包装、貯蔵、記録保存、流通、報告、広告およびその他の販売促進方法を管理する。バイオ製品のヒト臨床試験を行う前にFDAに申請する必要がある。生物製品が発売される前に、FDAの承認を受けなければならない。EUでは、臨床試験を開始する前に、EU加盟国の主管部門の承認を得なければならない。そのほか、医薬製品は主管監督機関のマーケティング許可或いはMAを獲得した後にのみ発売することができる
規制承認を得る過程と、その後適切な連邦、州、地方、外国の法律、法規、適用指導を遵守する過程には多大な時間と財力がかかり、必要な規制承認を得ることができない可能性がある。
アメリカの生物製品開発プロセス
私たちの候補製品はアメリカで合法的に発売されるためにFDAの承認を受けなければならない。FDAがバイオ製品候補製品が米国で発売される前に必要とされるプログラムは、一般に以下のような態様を含む
•臨床前の実験室テストと体内にあるFDAの現行の良好な実験室操作規範(GLP)、法規と実験動物の人道使用の適用要求或いは他の適用法規に基づいて研究を行った
•FDAが30日以内に反対しない限り、IND申請をFDAに提出し、ヒト臨床試験の開始を許可する
•各臨床試験を開始する前に、各臨床場所を独立した機関審査委員会またはIRBによって審査する
•FDAの良好な臨床実践あるいはGCP法規による十分かつ良好な制御を行うヒト臨床試験、および人体研究対象と彼らの保護
その予期される用途のために使用される候補生物製品の安全性および有効性を決定するための健康情報;
•非臨床試験および臨床試験の結果に基づいて提供される安全性および有効性の実質的な証拠と、適用される場合)使用料の支払いとを含む、上場承認のためにBLAをFDAに提出する準備がなされている
•適用される場合、製品はFDA諮問委員会によって検討される
•CGMP要件に適合することを評価し、施設、方法、および候補生物製品を保存するのに十分な識別、安全、強度、品質、効力、および純度を保証するために、FDAの候補生物製品を生産する1つまたは複数の製造施設の検査を満足的に完了させる;
•FDAは、BLAを支持するデータが生成された非臨床および臨床試験地点を検査することができる
•FDAによるBLAの審査および承認、またはライセンス。
遺伝子治療候補製品を含む人体上で任意の生物候補製品をテストする前に、候補製品は臨床前テストを経なければならない。臨床前試験は、非臨床研究とも呼ばれ、製品の化学、毒性と調合の実験室評価、及び体内にある候補製品の潜在的安全性と活性の研究を評価し、治療使用の理論基礎を構築する。臨床前試験の進行はGLPを含む連邦法規と要求に適合しなければならない。
臨床試験と同時に、会社は通常、生殖不良事象や発ガン性の動物試験を含む可能性がある追加の臨床前試験を完了しなければならず、薬物化学および物理的特徴に関する追加情報を開発し、cGMP要求に基づいて商業的な大量生産プロセスを最終的に決定しなければならない。製造過程は一貫して高品質の候補薬物ロットを生産することができなければならず、他の以外に、メーカーは最終薬物製品の特性、強度、品質と純度をテストする方法を開発しなければならない。また,適切な包装を選択·試験し,候補薬物が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
ヒト遺伝子転移プロトコルはFDAの監督と他の臨床試験法規,およびアメリカ国立衛生研究院(NIH)ガイドラインに規定されている地方レベルの監督に支配されている。具体的には,NIHのガイドラインによると,ヒト遺伝子転移試験の監督には,機関生物安全委員会(IBC)による評価と評価があり,IBCは地方機関委員会であり,組換えや合成核酸分子を用いた研究の審査·監督を担当している。IBCは、研究の安全性を評価し、公衆の健康または環境に対する任意の潜在的リスクを決定し、このような審査は、臨床試験開始前のいくつかの遅延をもたらす可能性がある。NIH資金を受けて組換えDNA研究に関与する機関の調査者にとっては,NIHガイドラインの遵守は強制的である。しかし,多くの会社や他機関は他の面でNIHガイドラインの制約を受けず,自発的にこれらのガイドラインに従っている。
臨床試験スポンサーは臨床前試験の結果を生産情報、分析データ、任意の利用可能な臨床データ或いは文献、提案された臨床方案と共にFDAに提出し、INDの一部としなければならない。IND提出後も,いくつかの臨床前試験が継続される可能性がある。INDはFDAが臨床試験を保留しない限り、INDがINDを受け取った30日後に自動的に発効する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。FDAはまた、臨床試験の前または期間のいつでも、安全考慮または規定に適合しない理由で、生物候補製品の臨床保留を実施することができる。FDAが臨床一時停止を強制した場合、試験はFDA許可なしに再開されず、その後、FDA許可の条件下でのみ再開される可能性がある。したがって,INDの提出がFDAが臨床研究の開始を許可するか,あるいは開始すると,このような研究を一時停止または終了させるという問題はないとは判断できない。
EUの薬品開発プロセス
アメリカと類似して、EUの臨床前と臨床研究の各段階は重要な監督管理によって制御されている。いくつかの臨床前(“非臨床”とも呼ばれる)データは、臨床試験を行い、その後、市場許可申請またはMAAのアーカイブに使用されるために必要である。すべての研究はGLPとすべての適用されたEMA、欧州委員会とヨーロッパ薬典の臨床前研究に関連するガイドラインに基づいて行われ、遺伝子組換え細胞を含む薬品の品質、非臨床と臨床方面に関するガイドラインを含む。
必要な臨床前データ量は臨床試験の設計を行うことができ,第1段階(ヒト初例臨床試験)から第2段階と第3段階まで,いずれも品質,安全性,有効性の研究である。米国と類似した制限や要求は臨床前データにも適用され,ウイルスベクターを用いた試験を支援している。臨床前試験は毒性、薬効学と薬物動態学特性などのパラメーター、及び遺伝子治療薬物製品の品質を確定すべきである。遺伝子治療薬製品の特殊な性質により、人々は常に可能ではないことを認識している
GLP規則による重要な非臨床安全性研究がなければ,GLP原則に適合した非臨床安全性研究を提出し,適切な理由を提供すべきである。
臨床研究は必要なデータを獲得するキーポイントであり、以下に更に臨床試験の要求を分析指導する。
すべての薬品や高級療法薬,あるいはATMPは,GMPガイドラインに従って生産され,GMP許可施設で生産されなければならず,これらの施設はGMP検査を受けることができる。
人体臨床試験
臨床試験は合格調査者の監督下で患者に生物候補製品を服用することに関連し、合格調査員は通常試験スポンサー或いは試験スポンサーの制御下に雇われない医師である。臨床試験は書面の研究案に基づいて行われ、臨床試験の目標、用量手順、被験者の選択と排除基準、および被験者の安全性を監視するためのパラメータは、いくつかの有害事象が発生した場合に臨床試験を停止する停止規則を保証することを含む詳細に説明されている。各スキームおよびスキームの任意の修正は、INDの一部としてFDAに提出されなければならない。臨床試験は,すべての研究対象にインフォームドコンセントを要求することを含むFDAのGCP要求を含む規定に基づいて行われなければならない。
また,各臨床試験は,臨床試験を行う機関またはそれにサービスを提供する機関のIRBによって審査·承認されなければならない。IRBは試験参加者の福祉や権利の保障を担当し,臨床試験に参加する個人のリスクが最低に低下するかどうか,期待利益と比較して合理的かどうかなどの項目を考慮している。IRBはまた、各臨床試験対象またはその法律代表によって署名されなければならないインフォームドコンセントの形式および内容を承認し、研究案を審査および承認し、完成まで臨床試験を監視しなければならない。
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複或いは合併する可能性がある
•第一段階です。この生物候補製品は最初に少量の人体被験者に導入され、安全性、用量耐性、吸収、代謝、分布、排泄をテストし、可能であれば、その有効性を早期に理解する。遺伝子や細胞療法の第1段階臨床試験は通常,健常ボランティアではなく患者で行われる。
•第二段階です。生物候補製品は限られた患者集団で評価を行い、可能な副作用と安全リスクを確定し、特定の目標疾患に対するこの製品候補製品の治療効果を初歩的に評価し、用量耐性、最適用量と用量計画を決定する。
•第三段階です。第三段階臨床試験は一般に“キー”研究と呼ばれ、通常は研究を指し、FDAまたは他の関連規制機関が生物製品のデータを承認するかどうかを決定することを提案している。第3段階研究では、生物学的候補製品は、一般に、十分かつ制御された臨床試験における複数の地理的に分散された臨床試験地点において、製品の有効性および安全性を統計的に確認するのに十分なデータを生成するために、拡大された患者集団に使用される。これらの臨床試験は候補製品の全体的なリスク/収益比を確定し、製品ラベルに十分な基礎を提供することを目的としている。
承認後の臨床試験は,4期臨床試験と呼ばれることがあり,予備承認後に行うことができる。これらの臨床試験は,治療適応が予想される患者の治療から追加的な経験を得るためのものであり,特に長期安全なフォローアップのためである。時々、製品の承認は発売後の臨床研究の完成にかかっている。
臨床開発のすべての段階において、監督管理機関(例えばFDA、EMA、EU加盟国の国家主管機関とその他の類似の監督機関)はすべての臨床活動、臨床データと臨床試験研究者に対して広範なモニタリングと監査を行う必要がある。臨床試験結果を詳細に説明する年次進展報告はFDAに提出しなければならない。
書面のINDセキュリティ報告書は、深刻で予期せぬ有害事象、その他の実験のいかなる発見も、FDAと調査者に提出しなければならない体内にある実験室テストや体外培養人類被験者に対して重大なリスクがあることを表明したテスト;或いは方案或いは研究者マニュアルに記載されたテストと比べ、深刻な不良反応の疑いの発生率は臨床上任意の重要な増加がある。スポンサーは15日以内にINDセキュリティ報告書を提出し,スポンサーがその情報有資格報告を確定した後でなければならない。スポンサーはFDAに事故や死や命を通知しなければなりません‑発起人が最初にメッセージを受け取ってから7日以内に脅威が出現する可能性のある副作用。
FDA或いはスポンサー或いはそのデータ安全監督委員会はいつでも様々な理由で臨床試験を一時停止することができ、研究対象或いは患者が受け入れられない安全リスクに直面していることを発見することを含む。同様に1つは
臨床試験がIRBの要求に従って行われない場合、あるいは候補生物製品が患者が意外な深刻な損傷を受けることに関連している場合、IRBはその機関の臨床試験の承認を一時停止または終了することができる。
FDAは通常、スポンサーが遺伝子治療に関連する潜在的遅延不良事象を観察する被験者を15年間観察することを提案している。
EUでは,臨床試験は2022年1月31日に施行された臨床試験条例(EU)第536/2014号またはCTRによって管轄されている。CTRは臨床試験の許可を調整と簡略化し、不良事件報告プログラムを簡略化し、臨床試験の監督を改善し、臨床試験の透明性を増加させることを目的としている。具体的には,すべてのEU加盟国に直接適用されるCTRは,単一入口点である臨床試験情報システム(CTIS)を介して,申請のために準備·提出された文書のセットであり,臨床試験スポンサーの報告手続きを簡略化した簡略化された申請手続きが導入されている。臨床試験申請を評価する統一プログラムが採用されており,このプログラムは2つに分類されている。第1部評価は,試験スポンサーが選択した参考加盟国の主管当局が指導し,EU加盟国間で科学的に協調していると考えられる臨床試験に関連している。そして、その評価報告書はその検討のためにすべての会員国の主管当局に提出されるだろう。二番目の部分はEU加盟国に関する各主管当局と道徳委員会によってそれぞれ評価される。個別のEU加盟国はその領土で臨床試験を行うことを許可する権限を保持している。行われている臨床試験がCTRによってどの程度管理されるかは,個別臨床試験の持続時間に依存する。臨床試験が2022年1月31日以降3年以上継続すれば,CTRはこの3年満了後に臨床試験への適用を開始する。臨床試験がCTRフレームワークに移行した場合、CTRはより早い日の臨床試験に適用される。
もし医薬製品が遺伝子組換え生物或いは遺伝子組換えと考えられている場合、EU加盟国国家遺伝子組換え主管部門の遺伝子組換え許可を得る必要があるかもしれない。EU加盟国間で遺伝子組換え承認の方法とスケジュールが一致していないことは、EU加盟国間の要求が異なる可能性がある。また,EU加盟国国家主管当局に遺伝子組換え生物承認申請を提出し,CTIが提出しなければならない臨床試験承認申請と同時に提出したわけではない。したがって、単独承認を必要とするトランスジェニック生物を含む臨床試験の発起人は、臨床試験およびその後のEU加盟国の同時応答を承認するために単一の申請ファイルを提出することから利益を得ることができない。これは特定の国の研究開始に影響を及ぼすかもしれない。
臨床試験の進行は承認された臨床試験方案、インフォームドコンセント要求に従うべきであり、患者のインフォームドコンセント、このような研究のために設計と許可されたプログラムと制御プログラム、公認された標準医学と科学研究プログラムを含み、そしてGCPの関連原則とすべての適用する法律法規に基づいて行うべきである。また,遺伝子治療薬製品はATMPに対するGCPルールを遵守しなければならず,このルールは具体的な追加的保障措置と要求を概説している。ATMPに対する記録保留要求は,関連する長期追跡および人の安全とトレーサビリティ要求があるため向上した。
CGMPの要件に合致する
生物製品メーカーは適用される現行の良好な製造規範或いはcGMP法規を遵守しなければならず、品質管理と品質保証及び記録とファイルの維持を含む。製造者やそのような製品の製造·流通に参加している他の人も、FDAと特定の国の機関、EU加盟国の主管当局を含む外国当局に彼らの機関を登録しなければならない。国内·海外製造企業が初めて製造過程に参加する際には、FDAおよびEU加盟国主管当局を含む外国当局に登録して追加情報を提供しなければならない。承認された製造場所の製造設備、プロセスまたは位置の任意の実質的な変更は、関連機関/当局に報告されなければならない。機関は、cGMP要求や他の法律の遵守を確保するために、政府当局(規制機関を含む)の定期的な抜き打ち検査を受ける可能性がある。問題の発見は、政府エンティティが承認されたBLAの製品、製造業者または所有者または臨床試験許可に制限を加えることをもたらす可能性があり、製品が市場からの撤回、警告または同様の手紙を発行することを要求するか、またはその会社に民事、刑事または行政制裁を実施することを求めるまで延長する可能性がある。FDAとEU加盟国の主管当局を含む外国当局は、製造プロセスや施設がcGMP要求に適合していると判断しない限り、要求された規格内で製品が持続的に生産されることを保証するのに十分である。
臨床試験と同時に、会社は候補製品の物理的および生物学的特徴に関するより多くの情報を開発し、cGMP要求に基づいて候補製品を商業的に量産するプロセスを最終的に決定する。PHSAは,外来製剤の導入や生物製品の使用による他の有害事象のリスク低減を支援するために,属性が正確に定義できない製品の製造制御の重要性を強調している。製造過程は一致しなければならない
高品質の候補製品ロットを生産し、他の要求以外に、スポンサーは最終生物製品の特性、強度、品質、効力と純度をテストする方法を開発しなければならない。また,適切な包装を選択·試験し,候補生物製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
ヒト細胞あるいは組織製品である候補製品に対して、FDAは現在の良好な組織実践、すなわちcGTPを遵守することも要求されている。これらは、ヒト細胞、組織、および細胞および組織に基づく製品またはHCT/Pを製造するための方法、施設および制御を管理するFDAおよびEUの法規であり、HCT/Pは、ヒトレシピエントに移植、移植、注入または転移するためのヒト細胞または組織である。GTP要求の主な目的は,細胞や組織に基づく製品の製造方式の確保であり,感染症の導入,伝播,伝播を防止することを目的としている。FDAとEU法規はまた、ティッシュ機関がFDAまたはEU加盟国の主管当局にそのHCT/Pを登録し、リストし、適用時にスクリーニングとテストを通じてドナーを評価することを要求する。
アメリカの審査と承認の流れ
前臨床試験や臨床試験の結果は,製品CMCに関する詳細な情報,アドバイスのラベルなどとともにBLAの一部としてFDAに提出され,1つまたは複数の適応で販売されることの承認が求められている。
改正された処方薬使用料法案(PDUFA)によると、各BLAは相当な使用料を伴わなければならない。FDAは毎年PDUFAユーザ料金を調整する。PDUFAは承認された生物製品に年間計画費も徴収している。場合によっては、小企業が提出した最初の出願または孤児疾患の製品適応を免除する申請料を含む費用を免除または減免することができる。
FDAは、提出後60日以内にBLAを審査し、機関がその届出を受ける前に実質的に完了したかどうかを決定する。FDAは、それが不完全であるか、または提出時に適切に審査できないと考えられる任意のBLAの提出を拒否することができ、より多くの情報の提供を要求することができる。この場合、BLAと補足情報を再提出しなければならない。再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると,FDAはBLAの深い,実質的な審査を開始する。
FDAは、提案された候補製品が安全かつ有効であるかどうか、その期待される用途に適しているかどうか、および候補製品がcGMPに従って生産されているかどうかを決定して、候補製品の識別、安全性、強度、品質、効力および純度を確保および保存するためにBLAを審査する。FDAは、新しい生物製品または安全性または有効性の問題を提起する生物製品の出願を諮問委員会に提出することができ、一般に、申請が承認されるべきかどうか、およびどのような条件下で承認されるべきかを審査、評価および提案するための臨床医および他の専門家を含むグループである。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。製品承認中に、FDAはまた、候補製品の安全な使用を保証するために、リスク評価および緩和戦略、またはREMSが必要であるかどうかを決定する。REMSは、製品の利点が潜在的なリスクよりも大きいことを確実にするために、専門ラベル以外のリスク最小化戦略を使用する。REMSが必要かどうかを決定するために、FDAは、製品を使用する可能性のある集団の大きさ、疾患の重症度、製品の予期される利益、予期される治療持続時間、既知または潜在的有害事象の重篤性、および製品が新しい分子実体であるかどうかを考慮するであろう。REMSは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、および安全な使用を確保する要素を含むことができる。FDAがREMSが必要であると結論した場合,BLAのスポンサーは提案したREMSを提出しなければならず,必要であればFDAはREMSのないBLAを承認しないであろう。
BLAを承認する前に、FDAは候補製品を生産する施設を検査するだろう。FDAは、製造プロセスおよび施設がcGMP要件に適合していることを決定し、候補製品が要求された仕様で一貫して生産されることを保証するのに十分でない限り、候補製品を承認しないであろう。さらに、BLAを承認する前に、FDAは通常、IND試験要求およびGCP要求に従って臨床試験が行われることを確実にするために、1つまたは複数の臨床場所を検査する。
BLAと付随する情報に基づいて、製造施設に対する検査結果を含めて、FDAは承認状または完全な返信を発行することができる。許可書はこの生物製品の商業マーケティングを許可し、特定の適応の具体的な処方情報を提供する。完全な応答文は、一般に、提出文書の不足点を概説し、FDAが出願を再検討するために、多くの追加のテストまたは情報を必要とする可能性がある。これらの不足点がBLAの再提出時にFDAによって満足的に解決された場合、FDAは承認書を発行する。
候補製品が規制部門の承認を得た場合、承認は特定の疾患や投与量に明らかに制限される可能性があり、または使用の適応が制限される可能性がある。さらに、FDAは、いくつかの禁忌症、警告、または予防措置を製品ラベルに含めることを要求する可能性がある。FDAは製品に制限と条件を加える可能性がある
RMSの形態で配布、処方または配布、または他の方法で任意の承認の範囲を制限する。そのほか、FDAは発売後の臨床試験を要求する可能性があり、時々第四段階の臨床試験と呼ばれ、生物製品の安全性と有効性を更に評価することを目的とし、そして商業化された承認製品の安全性を監視するためにテストと監督計画を要求する。
FDAはPDUFA下でBLASを審査する際に具体的なパフォーマンス目標を設定することに同意した。1つの目標は,FDAがBLA届出を受けてから10カ月以内に基準BLASを審査し,6カ月以内にBLASを優先的に審査し,審査決定を行うことである。FDAは常にそのPDUFA標準や優先BLASの目標日を達成するわけではなく,その審査目標は時々変更される可能性がある。FDAがPDUFA目標日の前の最後の3ヶ月以内に提出材料で提供された情報に関する追加の情報または明確化を要求またはBLAスポンサーが要求する場合、審査プロセスおよびPDUFA目標日を3ヶ月延長することができる。
EUの審査と承認手続き
EUでは、医薬製品は関連するMAを獲得してこそ、商業化することができる。欧州経済区または欧州経済区(EU 27加盟国にノルウェー、アイスランド、リヒテンシュタインからなる)の製品でMAを獲得するためには、申請者はMAAを提出しなければならないか、EMAによって管理されている中央手続きか、EU加盟国主管当局が管理する手続きのうちの1つ(分散手続き、国家手続き、または相互承認手続き)でなければならない。MAはEUで設立された申請者にのみ付与されることができる。
中央手続きは欧州委員会によってすべてのEU加盟国に効果的な単一M&Aを付与することを規定している。(EC)第726/2004号条例によれば、特定の製品については、(I)バイオテクノロジー製品、(Ii)孤児医薬品に指定された製品、(Iii)抗精神病薬、および(Iv)HIV/エイズ、癌、神経変性疾患、糖尿病、自己免疫および他の免疫機能障害、ならびにウイルス性疾患を治療するための新しい活性物質を含む製品は、集中的に行われなければならない。他の疾患の治療のための新しい活性物質を含む製品、および高度な革新性または集中プロセスを有する患者の利益に有利な製品については、関連する承認時に、集中手順による許可が任意である。
集中プログラムの下で、EMAの人は薬品委員会(CHMP)を用いて製品の初歩的な評価を行った。CHMPはまた、既存のMAの修正または拡張の評価のようないくつかの許可された後および保守活動を担当する。評価MAAの最長期限は210日であり、申請者がCHMP質問に回答する際に補足情報または書面または口頭解釈を提供する時間は含まれていない。特別な場合、CHMPは加速評価を承認することができる。CHMPが加速評価の要求を受けた場合、210日間の制限時間は150日(クロックポーズを含まない)に減少する。しかしながら、CHMPが加速評価を行うのにもはや適していないと考える場合、集中プログラムの標準期限に回復することができる。
集中権限プログラムとは異なり、分散MAプログラムは、製品販売が存在するEU加盟国毎の主管当局に個別に申請し、単独で承認する必要がある。この申請は,中央プログラムを介して環境管理機関に提出して許可する申請と同様である。EUは加盟国が有効な申請を受けてから120日以内に評価草案と関連材料の草稿を作成することを参考にする。これにより生成された評価報告は関係EU加盟国に提出され、これらの加盟国は、評価報告および関連材料を受け取ってから90日以内に、その評価報告および関連材料を承認するか否かを決定しなければならない。もしEU加盟国が公共健康の潜在的深刻なリスクに対する懸念から評価報告や関連材料を承認できなければ、論争内容は薬品機関の責任者相互承認と委譲手続き調整グループ-人に審査に提出される可能性がある。欧州委員会のその後の決定はすべての連合会員国に拘束力がある。
相互承認手続きは、他のEU加盟国の主管当局の承認を得るために、EU加盟国で許可された医薬製品を所有する会社がこの許可を申請することを可能にする。権限委譲手続きと同様に、相互承認手続きの基礎は、EU加盟国の主管当局が他のEU加盟国の主管当局の医薬製品の認可を受け入れることである。国MAの所有者は、EU加盟国の主管当局に申請することができ、その主管当局に、別のEU加盟国の主管当局によって交付されたMAを認めるように要求することができる。
原則として,MAの初期有効期間は5年である.5年後、欧州金融管理局または最初にこの合意を付与したEU加盟国の主管当局によるリスク·収益バランスの再評価に基づいて、協定を更新することができる。申請をサポートするために、MA所有者は、MAの故障の少なくとも9ヶ月前に、MAが付与されてから導入されたすべての変化を含む電子汎用技術文書の統合バージョンをEMAまたは主管当局に提供しなければならない。欧州委員会またはEU加盟国の主管当局は、薬物警戒に関する正当な理由に基づいて、MAのために5年間の継続期間を延長することを決定することができる。その後一度
最終更新後、金融管理専門家の有効期限は無期限となる。いかなる許可も、認可が失効してから3年以内に、医薬製品をEU市場(集中型MAの場合)またはEU加盟国の市場(非集中型MAの場合)に実際に投入していない(いわゆる日没条項)。
満たされていない医療需要に対して、重大な公共衛生利益を有すると予想される革新製品は、優先薬品或いはPrime指定のようないくつかの加速開発と審査計画を獲得する資格がある可能性がある。Prime条件に適合する製品は満たされていない医療需要がある場合に対して、新しい治療方法の導入或いは既存の方法を改善することによって、満たされていない医療需要を満たす潜在力を示しなければならない。Primeの称号を持つ候補製品のスポンサーはメリットを得ることができ、しかし限らないが、早期にEMAと積極的な監督対話を行い、臨床試験設計とその他の開発計画要素を頻繁に討論し、及びファイルを提出した後にMAA評価を加速する可能性がある。
EUでは,必要なすべての安全性や有効性データが得られていない場合には,“条件付き”MAが付与される可能性がある.以下のすべての基準を満たすことが証明された場合、欧州委員会は医薬製品の条件付きM&Aを承認することができる:(I)医薬製品の利益-リスクバランスは正であり、(Ii)申請者は許可後に全面的なデータを提供することができる可能性が高い、(Iii)医薬製品は満たされていない医療需要を満たし、(Iv)患者が直ちに医薬製品を得ることができる利点はまだ追加のデータが必要であるという事実に固有のリスクよりも大きい。条件付きMAは,失われたデータの生成やセキュリティ対策の増加を確保する条件を満たさなければならない.それの有効期間は1年で、すべての関連条件が満たされるまで毎年更新しなければならない。任意の未解決の研究が提供されると、条件MAは従来のMAに変換されることができる。しかしながら、これらの条件がEMAが設定した時間枠内で満たされず、欧州委員会の承認を得なければ、MAは更新を停止する。
“特別な場合”では、出願人が、製品が許可され、特定のプログラムを実施した後であっても、正常な使用条件下での治療効果及び安全性に関する包括的なデータを提供できないことを証明することができれば、MAを付与することができる。これらのことは,特に予想される適応では非常にまれであり,当時の科学的知識状況では,網羅的な情報を提供することは不可能である場合や,データを生成することが一般的に受け入れられている倫理原則に違反する可能性がある場合である。条件付きMAと同様に、特別な場合に付与されたMAは、まれな疾患または満たされていない医療需要を治療するために許可されることが意図された医薬製品であり、出願人は、標準MAを付与するために必要な完全なデータセットを有さない。ただし,条件付き買収と異なるのは,特殊な場合に認可を申請した出願人がその後紛失データを提供する必要がないことである.“特殊な場合”のMAは最終的に承認されているが,医薬製品のリスク−収益バランスは毎年審査されており,リスク−収益比が有利でなければMAは撤回される
EUの製造業規制
様々な要求は医薬製品の製造とEU市場への投入に適用される。EUで医薬製品を製造するには製造許可が必要であり、EUへの医薬製品の輸入は輸入を許可する製造許可が必要である。製造許可保持者は、EU cGMP規格を含む、適用されるEUの法律、法規、およびガイドラインに規定されている様々な要件を遵守しなければならない。同様に、EU域内での医薬製品の流通は、EU加盟国の主管当局に付与された適切な流通許可を要求することを含む、適用されるEUの法律、法規、および基準を遵守しなければならない。EUまたはEU加盟国が医薬製品製造に適用される要件を遵守しない場合、マーケティング許可保持者および/または製造および輸入許可、またはMA所有者および/または流通許可保持者は、製造許可の一時停止を含む民事、刑事または行政制裁を受ける可能性がある。
承認後に要求する
FDAとEUの生物製品に対する厳格かつ広範な規制は、特にcGMP要求の面で承認された後も継続している。製造業者は、品質管理と品質保証、記録とファイルのメンテナンスを含むcGMP法規に適用される要求を遵守しなければならない。他の生物製品に適用される承認後の要求は、配布製品の身分、効力、純度と全体的な安全性に影響を与える可能性のあるcGMP偏差、記録保存要求、不良反応の報告、最新の安全と治療効果情報の報告及び電子記録と署名要求の遵守を含む。BLAが承認された後、この製品はまた正式なロット発表が必要になる可能性がある。製品がFDAによって正式に発表されなければならない場合、製造業者は、各製品のサンプルをFDAに提出し、バッチの生産履歴およびバッチについて行われたすべての試験結果の要約を示す発表プロトコルを発行する。FDAはまた、流通のためのロットを発表する前に、いくつかの製品に対していくつかの検証的テストを行う可能性がある。そのほか、FDAは生物製品の安全性、純度、効力と有効性について監督管理標準に関連する実験室研究を行った。FDA承認後の規定を守らないことは、製品の承認と許可の撤回につながる可能性がある。
スポンサーはまた、FDAまたはEMA、欧州委員会および/または適用可能なEU加盟国主管規制機関の広告および販売促進要件、例えば、製品の承認のためのラベル(またはEU製品特性要約)に記載されていない用途または患者群の製品の普及(“ラベル外使用”と呼ばれる)を禁止しなければならない。以前未知の問題が発見されたり、適用された規制要求を遵守できなかったりすると、製品の販売を制限したり、その製品を市場から引き揚げたりし、民事または刑事制裁を受ける可能性がある。さらに、製造プロセスまたは施設の変更は、通常、新たな適応および追加のラベル宣言を追加するような承認された製品の他のタイプの変更を実施するためにFDAの承認を得る必要があり、FDAのさらなる審査および承認を受ける必要がある。
孤児とRMAT指定
米国では,20万人未満のまれな疾患の治療に用いたり,20万人を超える影響を与えたりしているが,治療薬の開発やマーケティングコストを回収できない製品は,孤児に指定される資格がある可能性がある。EUでは,これらのまれな疾患の定義は,EUの10,000人あたり5人以下の流行率であるか,このような疾患を治療する医薬製品であり,孤児身分によるメリットがなければ,EUでは十分な見返りが生じず,その医薬製品を開発するための必要な投資が合理的であることを証明するのに十分ではないということである。孤児に指定された薬品が発売承認されると、米国で承認された期間は7年、EUで承認された期間は最長10年である特定の孤児適応のマーケティング排他期から利益を得ることができる。製造業者がこの製品が孤児指定基準に適合しているか、または十分な数を提供できないと断言できない場合、それは孤児市場の排他性を失う可能性がある。
重篤なまたは生命の疾患の治療、修正、逆転または治癒を目的とした、細胞療法、治療用組織工学製品、ヒト細胞および組織製品、またはこれらの療法または製品を使用する任意の組み合わせ製品の検討を加速することを目的とする、再生医学高度療法、またはRMAT‑脅威性を有する疾病或いは状況は、その初歩的な臨床証拠はこのような疾病或いは状況が満たされていない医療需要を解決する可能性があることを表明した
RMATの指定は、FDAとより頻繁に会議を開いて候補製品の開発計画を検討することと、関連するBLAのスクロール審査および優先審査を行う資格があることとを含む潜在的な利点を提供する。RMAT認証が付与された製品も、長期的な臨床的利益を合理的に予測する可能性のある代替物または中間終点に基づいて、またはより多くの場所に拡張することによって加速承認を得ることを含む、大量の場所から取得されたデータに依存する資格がある。しかし、RMATの指定はFDAの製品承認基準を変更しないだろう。また,臨床データの出現に伴い資格基準を満たさなくなると,RMATの指定が取り消される可能性がある。
臨床試験データ開示
多くの司法管轄区域はスポンサーに対して強制的な臨床試験情報義務を持っている。EUでは,CTRは臨床試験情報に関する透明性要求を確立し,個人データや商業機密情報を保護し,EU加盟国間の評価報告準備に関する機密通信を保護したり,EU加盟国の臨床試験を効果的に監視する理由を確保したりする理由で秘密にされていない限り,CTIに含まれる情報を公開すべきである一般的な原則を確立している。もしすべてを圧倒する公共利益が存在することを開示すれば、この秘密例外は覆されるかもしれない。臨床試験に関するデータや文書の公表は特定のスケジュールで行う。スケジュールはEMAによって決定され,文書と臨床試験の分類によって決定される。そのほか、臨床試験の発起人は初歩的な臨床試験申請を提出する時にいくつかの書類の公表を延期することを申請することができる。公表延期申請は正当な理由に基づいており、合理的な提案延期期限を含まなければならない。公表を延期する申請は関連EU加盟国の承認を受けなければならない
さらに、文書を閲覧することに関する第1049/2001号条例、または“ATD条例”、および文書の閲覧に関するEUの政策0043は、環境事務管理局によって把握されているいくつかの情報を取得するために、EUの利害関係者に広範な権利を提供し、EU関係者に閲覧文書の要求を提出することができる。非常に限られた情報のみが開示を免れた(すなわち、その解釈がますます狭くなり保護された個人データを説明する商業機密情報)。これらのデータが公共分野に入ると,競争相手は世界のどこの自分の研究開発プロジェクトでこれらのデータにアクセスして使用することが可能となる.
私たちの業務に対する規制
私たちは現在市場には何の製品もありませんが、連邦政府と私たちが業務を行っている州と外国の司法管轄区域当局の追加医療法規と法執行の制約を受けるかもしれません。これらの法律には限定されません
•連邦医療保健反減税法規は、他の事項に加えて、個人および実体が直接または間接的に、公開的または公開的に故意に報酬を請求、提供、受け入れ、または提供することを禁止する
秘密的には、個人の推薦または購入、注文または推薦または推薦の任意の商品またはサービスを現金または実物の形態で誘導または奨励し、これらの商品またはサービスは、連邦医療保険および医療補助計画に従って支払うことができる
•個人または実体が連邦政府(連邦医療保険および医療補助計画を含む)に意図的に提出することを禁止する連邦虚偽申告法および民事金銭罰法を含む連邦民事および刑事虚偽申告法は、虚偽または詐欺的な支払いまたは承認請求を連邦政府に提出するか、または虚偽陳述によって連邦政府への支払い義務を回避、減少または隠蔽することを禁止する
•1996年の連邦“健康保険携行性と責任法案”、あるいはHIPAAと呼ばれ、詐欺の任意の医療福祉計画を実施する計画に刑事と民事責任を適用し、連邦刑法を制定し、他に加えて、重大な事実を故意に偽造、隠蔽または隠蔽したり、医療福祉、プロジェクトまたはサービスの提供または支払いに関連する任意の重大な虚偽陳述を行うことを禁止した
•HIPAAは、“健康情報技術促進経済·臨床健康法”及びその実施条例の改正により、特定の医療保健提供者、健康計画及び医療情報交換が保有する個別に識別可能な健康情報のプライバシー、安全及び伝送を保護する義務を規定し、強制契約条項、保証実体と呼ばれる義務、及び個別に識別可能な健康情報に関するサービスを提供する個人及び実体、商業パートナー及び保証下請け業者と呼ばれる
•2010年に患者保護および平価医療法案に基づいて作成された連邦医師支払い陽光法案は、2010年の医療保健および教育調整法案によって改正され、または総称してACAと呼ばれ、いくつかの薬品、設備、バイオ製品および医療用品メーカーが毎年医療保険および医療補助サービスセンター(CMS)に医師(現在、医師、歯科医師、視光師、足科医師および脊医を含むと定義されている)、他の医療保健専門家(例えば、医師アシスタントおよび看護師従業員)および教育病院、医師およびその直系親族が持つ所有権および投資利益への支払いおよびその他の価値移転に関する情報を報告することが要求される
•同様の州および外国の法律および条例、例えば、州反リベートおよび虚偽請求法は、民間保険会社によって精算された保健項目またはサービスの販売またはマーケティング手配およびクレームを含む非政府第三者支払者に適用可能であり、いくつかの州法律は、製薬会社に製薬業の自発的コンプライアンス基準および連邦政府が発行した関連するコンプライアンス基準を遵守することを要求し、医薬品製造業者に、他の保健提供者および保健エンティティへの支払いおよび他の方法での価値移転、マーケティング支出または薬品定価に関する情報を報告することを要求する;および/または販売者の登録を確保する;および/または販売者の登録を確保する;
•国や外国の法律は,特定の場合に健康情報のプライバシーやセキュリティを管理しており,その多くの法律は互いに大きく異なり,HIPAAに先を越されず,コンプライアンス作業を複雑にしていることが多い
私たちの業務が、私たちに適用される任意のこのような法律または任意の他の政府法規に違反していることが発見された場合、私たちは、行政、民事および刑事罰、損害賠償、罰金、返還、削減または再構成、連邦および州医療保健計画への参加、監禁、商業承認製品に関するマーケティングおよび商業化の一時停止または撤回、およびこれらの法律に準拠していない疑惑を解決するために会社の誠実な合意または同様の合意の制約を受けた場合、追加の報告要件および監督を受ける可能性があり、いずれも私たちの業務運営能力および財務業績に悪影響を及ぼす可能性があります。調査への対応に時間と資源がかかる可能性があり、経営陣の業務への関心を分散させる可能性がある。このような調査や和解は、私たちのコストを増加させるか、または他の方法で私たちの業務に悪影響を及ぼす可能性がある。
医療改革
米国や一部の外国司法管轄区域は、医療システムを変更するための追加立法·規制提案を検討または公布しており、承認されれば、これらの提案は候補製品の販売収益性に影響を与える可能性がある。米国や他地域の政策立案者や支払者の中では,医療システムの改革を推進することに大きな興味があり,医療コストの抑制,質の向上,参入拡大を目指している。米国では、製薬業はこれらの努力の重点であり、その中にはACAのような主要な立法措置が含まれており、医療システムの変化を通じて医療コストを低減し、薬品と生物製薬製品の価格設定、カバー範囲と精算を制限し、特に政府が援助する医療保健計画の下で、政府の薬品価格の制御を強化することを含む。ACAとその実施細則
他の事項に加えて、ある薬品と生物製品(私たちの候補製品と類似した製品を含む)を計算する医療補助薬品還付計画の下でメーカーが不足している税金還付の新しい方法を解決し、医療補助薬品還付計画の下でメーカーが不足している医療補助最低税還付を増加させ、医療補助薬品還付計画を医療補助管理に参加する看護組織を使用する個人の処方に拡大し、メーカーにあるブランドの処方薬に対して新しい年会費と税収を徴収することを要求し、新しい患者を中心とした結果研究所を作成し、連邦政府の比較研究の有効性を増加させる計画に激励を提供し、新しい連邦医療保険D部分ギャップ割引計画を構築し、その中で,メーカーは現在,保証間隔期間内に条件を満たす受益者に適用ブランド薬品交渉価格の70%の販売時点割引を提供することに同意しなければならず,メーカーの外来薬として連邦医療保険D部分保険の条件に組み入れ,後続生物製品のための許可枠組みを作成した。
ACAのいくつかの態様は、法律および政治的挑戦、ならびにACAのいくつかの態様を廃止または置換するための努力を受ける。例えば、2021年6月17日、米国最高裁は、ACAが“個人権限”が国会によって廃止されたため、全体的に違憲であると弁明する手続き理由に基づく挑戦を却下した。米国の最高裁が裁決を下す前に、総裁·バイデンは2021年1月28日に行政命令を発表し、ACA市場を通じて医療保険を得ることを目的とした特殊な保険加入期間を開始した。行政命令はまた、特定の政府機関に、医療補助モデルプロジェクトおよび免除計画の再審査、および医療補助またはACAによる医療保険カバー範囲の獲得による不必要な障害をもたらす政策を含む医療補助モデルプロジェクトおよび免除計画の再審査および見直し、医療補助またはACAによる医療保険のカバー範囲を制限する既存の政策およびルールを再検討するように指示する。また、2022年8月16日、総裁·バイデンは2022年インフレ削減法案に署名し、個人がACA市場で医療保険を購入する強化補助金を2025年に延長した。2025年からアイルランド共和軍は新たに設立されたメーカー割引計画により、受益者の最大自己負担コストを著しく低減し、連邦医療保険D部分計画下の“ドーナツの抜け穴”を解消した。ACAは未来に司法や国会で挑戦される可能性がある。バイデン政府の追加的な挑戦と医療改革措置がACAにどのように影響するかは不明である。
ACAが公布されて以来、アメリカはまた他の立法改正を提案し、採択した。2011年8月、“2011年予算制御法案”(Budget Control Act Of 2011)などの法案は国会のための支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これには、2013年4月に施行された各年度に提供者に支払われる医療保険総額が2%減少することが含まれており、その後の法規の立法改正により、2018年の両党予算法が含まれており、追加の国会行動がとられない限り、2031年まで有効となる。しかし、新冠肺炎の大流行救済立法によると、新冠肺炎が大流行したため、これらの連邦医療保険の自動減支措置は2020年5月1日から2022年3月31日まで停止される。現在の立法によると、医療保険支出の実際の減少幅は2022年の1%から本自動減額の最終年度の4%まで様々になる。また、総裁·バイデンは2021年3月11日に“2021年米国救援計画法案”に署名し、2024年1月1日から単一源と革新多源薬に対する法定医療補助薬品還付上限を廃止し、現在この上限は薬品メーカー平均価格の100%である。2013年1月、“2012年米国納税者救済法”(American納税者救済法、略称ATRA)が法律に署名し、病院やがん治療センターを含むいくつかの医療サービス提供者に支払う医療保険をさらに減少させ、政府が医療サービス提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長した。しかも、議会は追加的な医療改革措置を考慮している。
また、処方薬や生物製品価格の上昇を受けて、政府は最近、薬品の価格設定の審査を強化している。このような審査は、最近のいくつかの大統領行政命令、国会調査、提案と公布された連邦と州立法を招き、これらの立法は製品定価の透明性の向上、価格設定とメーカー患者計画との関係の審査、政府計画の薬品精算方法の改革を目的としている。また、2021年7月、バイデン政府は“米国経済における競争を促進する”という行政命令を発表し、その中には処方薬に対する条項が複数ある。バイデン行政命令への対応として,2021年9月9日,米国衛生·公衆サービス部(HHS)は高薬価に対応する総合計画を発表し,その中で薬品定価改革の原則を概説し,国会がとりうる各種潜在立法政策と,HHSがとることができるこれらの原則を推進する潜在行政行動を示した。このような原則を施行するための立法や行政行動はまだ最後に決定されていない。このような措置や似たような措置が未来に施行されるかどうかはまだ分からない。また,アイルランド共和軍(IRA)は他の事項を除いて,(1)HHSに連邦医療保険(Medicare)で覆われたある単一由来薬物と生物製品の価格について交渉するよう指示し,(2)連邦医療保険B部分とD部分にリベートを徴収し,インフレを超える価格上昇を懲罰するよう指示した。これらの規定は法的挑戦を受ける可能性があるにもかかわらず、2023年度から段階的に施行されるだろう。アイルランド共和軍がどのように実施されるかは不明であるが,製薬業に大きな影響を与える可能性がある。さらに進む, バイデン政府は2022年10月14日に追加の行政命令を発表し、HHSに90(90)日以内に報告書を提出するよう指示し、医療保険と医療補助革新センターをさらに利用して新しいモデルをテストする方法を説明した
医療保険と医療補助受益者の薬品コストを低減する。この行政命令や同様の政策措置が将来的に実施されるかどうかは不明である。州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入の制限、マーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。
米国、EU、および我々の候補製品の他の潜在的に重要な市場では、政府当局および第三者支払者は、医療製品およびサービスの価格、特に新しいおよび革新的な製品および療法を制限または規制しようとするようになっており、これはより低い平均販売価格をもたらしている。また、米国の管理型医療保健に対する日々の重視及びEUの国家と地域の定価と精算制御の日々の重視は製品の定価、精算と使用に追加の圧力をもたらし、これは私たちの未来の製品販売と運営結果に不利な影響を与える可能性がある。これらの圧力は,管理型医療集団のルールと実践,司法裁決および連邦医療保険,医療補助と医療改革,薬品精算政策と一般定価に関する政府法律法規から来る可能性がある。また、政府は新冠肺炎の流行に対応するためにより多くの行動をとる可能性がある。
定価、カバー範囲、精算
治療製品の定価や精算は,その製品の負担性を大きく決定し,その製品が発行されて患者に供給されるかどうか,民間保険会社は政府の精算方法を考慮する可能性がある。これらの提案と公布された法律およびその他の行動により、私たちが監督管理によって承認された任意の候補製品のカバー範囲と精算状態には重大な不確実性があり、特に新製品については重要な不確実性がある。国内外の市場では、どの承認された製品の販売と精算も、政府医療計画、商業保険と信託医療組織がこのような製品に保険を提供する程度、十分な精算レベルを確立するなど、第三者支払い者にある程度依存する。第三者決済者は、医療製品やサービスの価格に挑戦し、コストを管理するための制御を実施することが増えている。第三者支払者は、承認リスト上の特定の製品に保証範囲を制限することができ、処方表とも呼ばれ、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。しかも、私たちは私たちの製品の費用効果を証明するために高価な薬物経済学研究を行う必要があるかもしれない。もし第三者支払者が私たちの製品が他の治療法に比べて費用効果がないと思った場合、これらの支払人は彼らの計画の下で福祉後に私たちの製品をカバーすることが承認されないかもしれません。あるいはもし彼らがそう思うなら、清算レベルは利益に基づいて私たちの製品を販売するのに十分ではないかもしれません。
連合では、様々な国の価格設定と補償プログラムの差が大きい。EU加盟国は製品の具体的な価格を承認することができ、製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることもできる。他のEU加盟国は会社が自分の製品価格を固定することを許可したが、処方量を監視と制御し、医師に指導を発表して処方を制限した。最近,EUの多くの国が薬品割引要求を高め,各国が医療支出を管理しようとしていることに伴い,これらの努力は継続される可能性がある。衛生技術評価、又はHTAとは、特定の医療製品が個別の国の国家医療システムで使用される公共健康影響、治療影響及び経済及び社会的影響を評価するプログラムである。特定の医薬製品に関するHTAの結果は、EU個別加盟国主管当局がこれらの医薬製品の価格設定と補償地位を与えることに影響を与えることが多い。EUは2021年12月、EU全体のHTA臨床利益評価を調整するために2025年に施行されるHTA条例を採択した。“リスク要因”を見てくださいたとえ承認された製品を商業化することができても、これらの製品は米国と私たちが商業化を求める他の国/地域の第三者支払者から保険と十分な補償を受けることができない可能性があり、これは私たちの業務を損なう可能性があります.”
環境規制
安全作業条件,環境保全と有害物質に関する米国連邦と州法は,“職業安全と健康法”,“資源節約と回収法”,“有毒物質制御法”を含め,我々の業務に影響を及ぼす。これらの法律と他の法律は私たちの様々な生物、化学、放射性物質の使用、処理と処理、これらの物質は私たちの行動、そして私たちの行動によって生成された廃棄物を規範化している。私たちが今または未来にこのような法律と法規を遵守することは大きな費用をもたらすかもしれない。もし私たちの運営が環境汚染を招いたり、個人を危険物質に曝露させたりすれば、損害賠償と政府罰金の責任を負う可能性があります。私たちは、私たちが適用される環境法律と法規を実質的に遵守し、これらの法律と法規を遵守し続けることは、私たちの業務に実質的な影響を与えないと信じている。しかし、私たちはこのような法律の変化が私たちの未来の運営にどのように影響するか予測できない
プライバシー規制
我々は、改正された2018年EU(離脱)法案第3節または英国GDPRおよび2018年カリフォルニア消費者プライバシー法案(CCPA)によって英国法律の一部を構成するため、適用される連邦貿易委員会法案、EU一般データ保護法規、またはEU GDPR、EU GDPRを含む、米国および他の外国司法管轄区域の多くのプライバシーおよびデータセキュリティ法律および法規によって制約される可能性がある。
欧州経済地域の個人に関する個人データは、EU GDPRによって制限されている個人健康データを含む、収集、使用、開示、移転、または他の方法で処理される。EU GDPRの範囲は広く,個人データに関連する個人の個人データの制御,個人にその個人データの処理に関する通知,我々が維持しなければならない文書,個人データのセキュリティ,データ漏洩通知,個人データを処理する際に第三者プロセッサを使用する必要があるいくつかの要求を提示している.EU GDPRはまた、個人データを欧州委員会が十分なレベルのプライバシーやデータセキュリティを提供できないと考えている国(米国を含む)に厳格なルールを実施している。欧州委員会は最近、2025年6月27日までの4年間、個人データを欧州経済圏からイギリスに限定せずに移行させる決定を発表したが、この決定は撤回または更新することができ、このような移転を継続するための追加的なメカニズムを実施することが求められる。EU GDPRは、2000万ユーロに達する可能性のある罰金または違反会社の全世界の年収の4%を含む違反行為に巨額の罰金とその他の是正行動を科すことを許可し、より大きな者を基準に、最終的にデータ処理または法的許可によってその利益を表す様々なデータ主体または消費者保護組織が提出した個人データに関する個人訴訟の処理を禁止する。国際データ転送に関するEU GDPR要求は、第三者取引だけでなく、私たちの子会社(例えばフランスSangamo)との間の情報伝送にも適用されます, 従業員情報も含まれている。EU GDPRは、これまでのEU法と比較して、個人データ処理における私たちの責任と潜在的責任を増加させ、特にSangamo Franceを買収したことを考慮すると、EU GDPRの遵守を確保するための追加的なメカニズムを構築する必要があるかもしれません。これは、経営陣の注意をそらし、私たちの業務コストを増加させる可能性があります。
米国では、連邦、州、地方政府はデータ漏洩通知、個人データプライバシー、消費者保護に関する法律を含む多くのプライバシーとデータセキュリティ法律を制定した。例えば、CCPAは、企業がプライバシー通知において詳細な開示を提供することを要求し、カリフォルニア州住民がその個人データに関連するいくつかのプライバシー権を行使する要求(その個人データを削除し、その個人データを販売しないことを選択する権利を含む)を尊重する。CCPAは、違反行為ごとに最高7500ドルの民事罰金を科すことができ、あるデータ漏洩の影響を受けた個人訴訟当事者が巨額の法定損害賠償を取り戻すことを許可すると規定している。CCPAはいくつかの臨床試験で処理されたデータを免除しているにもかかわらず、CCPAはコンプライアンスコストを増加させ、私たちが保持する可能性のあるカリフォルニア住民の他の個人データに関する責任を増加させる可能性がある。2020年カリフォルニアプライバシー権法案、またはCPRAは、2023年1月1日に施行され、CCPAの要求を商業代表や従業員に適用される個人情報に拡大し、法律を実施し実行し、行政罰金を科すための新たな規制機関を設立した。バージニア州、コロラド州、ユタ州、コネチカット州などの他の州でも全面的なプライバシーとデータセキュリティ法律が可決され、他のいくつかの州や連邦や地方レベルでも同様の法律が考えられている。これらの開発はコンプライアンス作業をさらに複雑化させ、私たちと私たちが依存する第三者の法的リスクとコンプライアンスコストを増加させる可能性がある。
これらおよび他の任意の適用可能なプライバシーおよびデータセキュリティ法律法規を遵守することは、厳格で時間のかかるプロセスであり、新しいデータ保護ルールの遵守を保証するために追加のメカニズムを確立する必要があるかもしれません。もし私たちがこのような法律や法規を守らなければ、私たちは巨額の罰金と処罰に直面する可能性があり、これは私たちの業務、財務状況、運営結果に悪影響を及ぼすかもしれない。また,法律は一致しておらず,広範囲のデータ漏洩が発生すると,法律を遵守するコストも高い。“リスク要因”を見てください私たちの現在と将来の医療提供者、顧客、第三者支払者との関係は、適用されるリベート、詐欺、乱用、プライバシー、データセキュリティ、および他の医療に関する法律と法規を遵守することを要求しています。もし私たちがこれらの規定を守らなければ、私たちは規制機関の調査や行動、訴訟、巨額の罰金と処罰に直面する可能性があり、私たちの業務、名声、経営業績、財務状況、見通しは不利な影響を受けるかもしれない。”
人的資本管理
私たちの使命と従業員は
Sangamoでは,画期的な科学を患者の生活を変えるゲノム薬物に変換することに取り組んでいる。著者らは情熱的なバイオテクノロジー専門家であり、本部はアメリカ、フランスとイギリスに設置され、長年の経験と技術専門を持ち、一流のゲノム薬物の開発に取り組んでいる。私たちは協力、規律、そして効率性を受け入れ、同時に新しい考えと個人の発展を刺激することを歓迎する。私たちは励まして
多様性、公平、そして包容を抱き、これは私たちの共通の目標に向かっての仕事を強化すると信じている:私たちがサービスを目的としている患者の生活を変える。
私たちは職員たちが使命を履行する最も価値のある資産の中の一つだと思っている。私たちは競争の激しいバイオテクノロジー業界で競争し、才能のある従業員を誘致、維持、育成することは、私たちの戦略と私たちの効果的な競争の能力に重要である。私たちは、私たちの研究、製品開発、製造、規制の努力、および私たちの計画、すなわち承認されれば、私たちが所有する候補製品を商業化するために、私たちの従業員チームを発展させ、保留することに取り組んでいる。ゲノム薬物の発見,開発,製造経験を持つスキルのある個人が依然として不足しており,この状況が続く可能性が高い。そのため,生物製薬会社と学術機関の間には,これらのスキルを持つ個人を争う競争が続いている
我々の価値観は
私たちの核心的価値観と私たちの使命を結びつけると、成功が来ると信じています。私たちの使命は、今日の対症療法の代わりにゲノム薬を提供し、患者の生活を変えることです。私たちの核心的価値観は
•病人に正しいことをする:
◦私たちは目的を持って協力し、患者に利益をもたらす結果を原動力にしている。
◦私たちは患者の安全と看護の質を第一にしようと努力している。
◦患者の需要は私たちに薬を送る緊急感を駆り立てている。
◦我々は生物倫理学の観点からゲノム医学分野を開拓する責任を担っている。
◦私たちは私たちの薬物開発を指導するために包括的な方法を取る。
•チームワークで成功する:
◦私たちの共通のビジョンは、ゲノム医学が患者の生活や医療分野を変えることだ。
◦私たちは情熱と尊敬に満ちた個人であり、彼らは積極的で開放的に協力し、私たちの業務を実行し、発展させる。
◦私たちは私たちの優先順位を明確に定義し、コミュニケーションを行い、集団責任を取って、すべての利害関係者に成果を渡す。
◦患者が私たちに依存しているので、私たちは粘り強く、一緒に成功しようと決心した。
•賢明な意思決定を通じて革新を実現する:
◦我々は邁進し、たゆまず、ゲノム医学領域で成功した革新の旅を切実に追求している。
◦私たちは科学的な可能性を発掘し、目標は深刻な疾病の新しい治療方案をロック解除することだ。
◦私たちは柔軟で包容的で効率的な意思決定を通じて私たちの業務目標を達成するために努力している。
◦著者らは数十年の科学経験から学び、成長し、医学の最前線の治療法を開発した。
◦私たちは失敗から教訓を得て、突破を実現する旅の一部として業績の向上を求めています。
•帰属感を養う:
◦私たちは共通の目標を立てて帰属感を作る。
◦私たちは会社で、ここでは、違う個人が盛んに発展し、彼らの専門知識を成長させ、同時に彼らの本当の自分を仕事に持っていくことができます。
◦私たちは地域コミュニティ、私たちが生活している環境、私たちがサービスする患者コミュニティと関連があると感じている。
◦我々は一堂に会し,我々の科学知識を理解し,我々の業務の発展を推進する.
◦私たちは多様性、公平、そして包容を擁護する。
◦私たちは医療の公平性を促進するために、多様で包括的な環境の育成に取り組んでいる。
わが国の人的資本管理
私たちの首席人事官は、私たちの人的資源機能を指導し、候補者の誘致と採用、リーダーシップ訓練と発展、多元化と包括的な努力、報酬と福祉を含む総奨励案、従業員の尊敬度と維持に集中している。私たちのグローバル人材機能は、2022年12月31日までにフルタイムの人材専門家12人で構成されています。
2022年12月31日現在、私たちはアメリカ、フランス、イギリスに478人のフルタイム従業員を持っています。これらの従業員のうち、375人はアメリカに位置し、主にサンフランシスコ湾区に位置し、94人はフランスバル州に位置し、残りの9人はイギリスロンドンの近くに位置している。これらの従業員のうち、206人は主に研究と開発活動に従事し、176人は主に技術業務と製造に従事し、96人は主に一般と行政活動に従事している。また、特別または一時的なプロジェクトまたは特定の専門知識の必要に応じて、独立請負業者およびコンサルタントを招いてサービスを提供します
私たちの人的資源を管理するために、業務部門と国家/地域別の従業員数、歴史人数の増加、売上高、新入社員と退職人数、開放役割と従業員人口統計データ(性別、人種、民族を含む)を含む重要な人材指標を内部で追跡し、報告します。私たちの上級管理者はこれらの指標を使用して、資源計画、採用と維持計画、そして私たちの報酬と福祉計画の設計を助けます。私たちは、四半期ごとに私たちの取締役会の報酬委員会とこれらの指標を共有して、それがその役割を果たすのを助け、私たちの企業の報酬理念を確立し、私たちの報酬と福祉計画を管理し、私たちの役員と重要な従業員のパフォーマンスを評価し、経営陣の発展と後継計画を審査し、監督します。
私たちの従業員は年に二回の従業員敬業度調査に参加します。この調査の結果は引き続き従業員の文化、仕事の動態、全体的な約束をよりよく理解することを助け、従業員全体の従業員の尊敬度を高める重点分野を決定する。私たちは74%の参加率に満足しており、これは従業員の尊敬度と忠誠度が有利なレベルにあることを示している。
多様性、公平、そして包容に対する私たちの約束は
私たちは多様な労働環境の中で、Sangamoのすべての従業員が差別、嫌がらせ、偏見、偏見のない包容環境ですくすくと成長できると信じている。私たちの目標はすべての人を尊重し、尊重し、すべてのSangamo従業員に平等な機会と利点に基づく公平な待遇を提供することだ。多様性と包括性を抱擁することで、私たちの価値観に一致したSangamo使命を支援するために、革新的な解決策の協力開発に取り組む組織を作りました。Sangamoでは,異なる背景や観点が尊重されて耳を傾けるだけでなく,抱擁やお祝いも得られる文化や環境を育成している。多様化、公平と包容の心理状態と文化は積極的に参加し、責任を果たす職場に重要であるだけでなく、患者に革新的な解決方案を提供する上でも重要である。
以下は、2022年12月31日現在、Sangamoの自己認識に基づく米国人従業員の統計データである
人的資源チームには、最高のプロジェクトマネージャーがいて、彼の時間は、最高人事官や首席運営官との協力の多様性、公平さ、包摂性、またはDEI努力をリードするために完全に使用されています。会社が多元化計画を実施する重要な重点分野を決定する作業部会もある。私たちは6つの従業員資源グループ(ERG)があり、従業員と実行スポンサーが協力して指導し、各ERGのために専門的な予算を制定し、コミュニティの構築と強化、リーダーシップの発展、人材誘致に具体的な影響を与えることができるようにした。私たちは生命科学配慮組織と様々なパートナー関係を構築しています。これは非営利団体であり、生命科学会社の資源を利用して貧困の影響を減らすことを使命としています。私たちの首席運営官D.Mark McClungは取締役会のメンバーです。私たちはまたブルームバーグ性別平等指数に参加して、私たちの投資と計画を私たちの従業員とよりよく一致させるために参加した。
私たちの報酬と福祉は
私たちの業界の高度な競争性と従業員の採用と維持が私たちの成功に対する重要性を考慮して、私たちは私たちの従業員に、報酬、福祉、発展機会を含む、非常に競争力があると考えられる全面的な報酬プランを提供するように努力しています。このプログラムは、市場レベル以上の報酬、従業員および家族の医療福祉、条件に適合した米国人従業員の健康貯蓄口座、雇用主が市場レベルよりも高い賃金を支払うこと、手厚い有給休暇福祉、帰省休暇、親戚訪問休暇、柔軟な勤務時間、退職および/または年金計画の支払い、長期障害計画を補充すること、精神健康福祉および現場ジム参入を含む。さらに、私たちは従業員たちに健康と福祉の支出のための毎月手当を提供する。また、株式オプションおよび/または限定株式単位を付与することにより、全世界の常勤従業員一人ひとりにSangamo持分のメリットを提供する。私たちのアメリカ人従業員には、少なくとも15%の割引で私たちの普通株を購入する機会を提供する従業員の株式購入計画に参加する資格があります。
新冠肺炎が大流行する
従業員の安全と福祉はどの年も私たちにとって重要であり、新冠肺炎疫病の持続を考慮して、2022年の従業員の安全と福祉は依然として私たちが特に注目している問題である。疫病に対応するために、著者らは安全、コミュニケーションと資源方面の努力を通じて、私たちの従業員と政府が新冠肺炎の疫病を抑制するための努力を支持した。これらの努力は、大流行病による外部と内部環境の変化を待つために、必要に応じて継続されるだろう。
環境.環境
Sangamoはカリフォルニア州ブリスベンに本部を置き,カリフォルニア州リッチモンドに研究機関,フランスとイギリスバルバンにヨーロッパ機関を設置している。内部製造業務はブリスベンとバルバンの工場内にある。Sangamoはブリスベンにある本社がLEED認証を取得したことは,米国グリーン建築委員会が制定したグリーン建築要求に適合していることを意味している。
商標と商号
SangamoおよびBetter Treateutics by Designは米国における当社の登録商標であり、Sangamo TreateuticsおよびSiefterは私たちの商標です。本年度報告でForm 10−K形式で言及されている他のすべての商標または商号は、そのそれぞれの所有者の財産である。
利用可能な情報
私たちのウェブサイトはWwwww.sangamo.comそれは.本年度報告Form 10-K、当社のForm 10-Q四半期報告、当社の現在のForm 8-K報告、および取引法第13(A)または15(D)節に提出または提供されたこれらの報告書の修正案は、米国証券取引委員会または米国証券取引委員会にこれらの資料を電子的に提供した後、合理的で実行可能な範囲内でできるだけ早く無料で提供します。我々のサイトで見つけたり,我々のサイトを介してアクセスしたりする情報は,本10-Kフォーム年次報告の一部ではなく,本年度報告にも盛り込まれない.また、米国証券取引委員会には、米国証券取引委員会に電子的に提出された報告書、依頼書、情報声明、その他の発行者に関する情報が含まれたサイトwww.sec.govが設置されている。
プロジェクト1 A--リスク要因
私たちの業務は重大な危険と関連があり、その中のいくつかの危険は以下のように説明される。私たちの普通株に投資決定を下す前に、これらのリスクと、当社の10-K年度報告書の他の情報、私たちの財務諸表と関連説明、および“経営陣の財務状況と経営業績の検討と分析”をよく考慮すべきです。以下に述べる任意のイベントまたは開発の発生は、当社の業務、経営結果、財務状況、見通し、および株価に重大な悪影響を及ぼす可能性があります。この場合、私たちの普通株の市場価格は下落するかもしれません。あなたはあなたの全部または一部の投資を失うかもしれません。また、以下に説明しない他のリスクもあり、これらのリスクは私たちが現在知らないことか、現在重要ではないと考えているかであり、これらの追加リスクはまた、私たちの業務、運営、または私たちの普通株の市場価格に大きな損害を与える可能性がある
私たちの候補製品と技術の研究、開発、規制承認と商業化に関するリスク
著者らの成功は臨床試験結果に大きく依存し、この結果は著者らの候補製品の安全性と有効性を証明し、監督管理機関を満足させた。私たちは私たちのすべての候補製品のために肯定的な臨床試験結果と規制承認を得ることができないかもしれない
私たちは臨床段階のバイオテクノロジー会社で、承認された製品もなく、製品収入もありません。臨床試験を行い使用を評価しています我々は遺伝子治療と細胞治療におけるプラットフォーム技術であり、将来的に他の候補製品のより多くの臨床試験を開始する予定である。我々はこれらの臨床試験の結果に大きく依存しており,現在あるいは未来に我々の候補製品に対する臨床試験の最終結果が我々の任意の候補製品の安全性と有効性を証明することは保証されない。しかも、私たちの候補製品は規制部門の承認を受けなかった。積極的な臨床試験結果と監督管理の承認を得ることは高価で、長く、挑戦性と予測不可能であり、私たちのいかなる候補製品にとっても永遠に起こらない可能性がある。もし私たちの候補製品のために積極的な臨床試験結果と監督管理の承認を得ることができなければ、候補製品からの期待収入と私たちの利益の見通しは不利な影響を受け、私たちの普通株の市場価格を大幅に低下させる可能性がある。
臨床試験を行うことと監督管理の許可を得ることは複雑であり、そして著者らの業務を多くのリスクに直面させ、潜在的な意外なコストと遅延を含む
私たちは、商業化に必要な規制承認を得るために、規制機関を満足させるために、候補製品の安全性と有効性を証明するために、広範な臨床試験を行わなければならない。われわれは後期臨床試験を行う上での経験が限られており,このような試験を遂行するために必要な資源や専門知識を備えていない可能性がある。臨床試験費用は高く、時間がかかり、しかも予測できない。もしあれば、どんな臨床試験も計画通りに行われるか、予定通りに完成することは保証できません。1つまたは複数の臨床試験の失敗は任意の段階で起こる可能性がある。臨床開発と規制承認の成功またはタイムリーな完了を延期または阻止する可能性があるイベントは、
•遅延と監督部門は臨床試験設計について合意した
•遅延と予想される臨床研究組織或いはCROと臨床試験場所は受け入れられる条項について合意した
•開放臨床試験サイトを遅延させるか、または各臨床試験サイトで必要な機関審査委員会、倫理委員会または国家主管部門の許可を得る、例えば、新冠肺炎の大流行のために、私たちはイギリスで私たちのSTAAR臨床研究のために1/2期臨床研究開放臨床試験サイトを評価する時に遭遇する遅延;
•(I)患者の募集、スクリーニング、および募集において遭遇した遅延、および1/2期STAAR臨床研究でisaralgagene Citizopvocを評価する患者の投与において遭遇し、経験し続ける遅延、例えば、(I)新規冠肺炎検出が陽性であること、研究に参加しているかどうかおよびスクリーニング場所が限られているかどうかを再検討すること、および(Ii)ファイザーが2022年3月に実施され、2022年9月に廃止されたgiroctocogene fitelparvec第3段階アフィン試験における追加患者の投与休止;
•規制当局は、2021年11月に実施され、2022年3月に廃止されたgirococogene fitelparvovec 3期模倣試験に適用された臨床休止のような、私たちまたは著者らの協力者の臨床試験に臨床休止を適用した
•新冠肺炎の全世界大流行による臨床試験活動の遅延は、ある患者が研究或いは服用に参加する前に新冠肺炎の測定が陽性を呈したことと関連する遅延を含み、これ以前に著者らのFABRYとTX 200計画の臨床試験スケジュールを影響したことがある
•鎌状細胞疾患の治療のための我々のZFN遺伝子編集自己細胞治療製品BIVV 003の最終患者の募集および用量の点で遅延または困難に遭遇する可能性があることを、私たちの1/2期PRECIZN-1研究で評価した
•私たちが招いたCROや他の第三者は臨床試験の要求を守ることができませんでした
•FDAの“良好な臨床操作規範”および“良好な実験室操作規範”に準拠していない、またはEUおよび他の国に適用される比較可能な外国規定に従って実行されていない
•私たちと契約していくつかの機能を実行する第三者の遅延、または私たちの候補製品の製造または配合変更の結果を含む、私たちの候補製品のテスト、検証、製造、および臨床現場に渡される遅延
•患者を完全に試験或いは治療後のフォローアップに参加させることを遅延させた
•臨床試験場所や患者が試験を脱退した者
•より長い時間を要する臨床観察または結果データ分析の臨床終点を選択する
•候補製品に関連する深刻な有害事象や他のセキュリティ問題の発生は、それらの潜在的な利点を超えていると考えられ、承認遅延や他の規制制限を招き、または私たちの名声を損なう
•他の発起人が行った同類の薬物臨床試験において深刻な不良事件或いは他の安全問題が発生した
•候補製品がその提案に対する適応が安全かつ有効であることは証明できなかった
•新しい臨床プログラムの規制要件およびガイドラインの変更を修正または提出する必要がある
•予想外のコストと支出と私たちの候補品を開発するのに十分な資金が不足しています
•重要な知的財産権の許可損失。
私たちは、特定の候補製品の完全な開発経路について規制機関と合意していません。これらの規制機関は、特にこれらの分野の技術が発展し続けている場合に、承認可能なエンドポイントに関する決定や指導を変更する能力があります。例えば、他の会社が血友病Aを治療する遺伝子療法を開発していることが知られており、FDAは、同社がその第3段階の研究を完了し、すべての研究参加者の2年間のフォローアップ安全性および有効性データを提出することを提案しているが、同社は、FDAが以前に生物製品ライセンス申請(BLA)を支援するために必要なデータ範囲と一致していると弁明している。我々とファイザーは、我々の3期アフィン試験の重要なデータ読み取り値がすべての研究参加者の全面的な分析に基づくと予想しているが、第1陣の50人の患者が12ヶ月後にFVIII発現の安定状態に達した場合、FDAまたは他の同様の外国の規制機関は
この試験で予想よりも多くの患者を治療する必要があることを確認するか、または所望のデータを生成するために患者を追跡する時間が必要であるか、または試験の他の修正が必要である可能性があり、いずれも、試験の完了および規制機関のgirococogene fitelparvecの承認を求める能力に悪影響を及ぼす可能性があり、これは、逆に、その競争地位および商業生存能力に大きな悪影響を与え、それによって、私たちの業務、将来性、および株式市場価格に影響を与える可能性がある。
いくつかの候補製品とその技術的新規性のため、規制承認を支援するために必要な端末は、最初に予想されていたものとは異なる可能性がある。候補製品の臨床前および臨床開発を成功させることができない場合、または予想される時間内にこのような試験を完了することができなければ、追加コストをもたらしたり、製品販売からの収入を獲得したり、規制および商業化マイルストーンおよび特許使用料を実現する能力を弱めるか、または独占経営権を持つ可能性のある任意の期限を短縮する可能性がある。
候補製品がFDAおよび同様の外国規制機関の承認を得ることに成功したとしても、どの承認も、特定の年齢層の使用制限、警告、予防措置または禁忌症に関連する重大な制限を含むことができ、または重い承認後の研究またはリスク管理要件の影響を受ける可能性がある。さらに、私たちの候補製品に対するいかなる規制承認も、一度承認されると、撤回、変更、または一時停止される可能性がある。もし私たちが1つ以上の管轄区域で私たちの候補製品のために規制承認を得ることができない場合、または任意の承認に重大な制限が含まれていれば、予想される収入を生み出すことができず、利益を達成することが困難になる可能性があり、これは私たちの業務運営や財務状況に悪影響を及ぼすだろう。
研究や臨床前研究の成功あるいは早期臨床試験の結果は,以降の試験で得られた結果を代表しない可能性がある。同様に、臨床試験の初歩的、初歩的或いは中期データは最終データと大きく異なる可能性がある
研究と臨床前研究或いは早期臨床試験の結果は必ずしも未来の臨床試験結果を暗示するとは限らず、臨床試験の初歩的、初歩的と中期結果も必ずしも最終結果を代表するとは限らない。著者らの候補製品は臨床試験において期待される安全性と有効性を示すことができない可能性があり、臨床前研究で積極的な結果を証明したにもかかわらず、或いは初歩的な臨床試験或いは臨床試験の初歩的な段階に成功した。私たちは時々予備、予備、または中間データを発表したり報告したりするつもりだ。著者らおよび著者らの協力者からの臨床試験の予備、初期または中期データは、試験の最終結果を示すことができず、患者登録の継続および/またはより多くの患者データの出現に伴って、1つまたは複数の臨床結果が重大に変化する可能性のあるリスクに直面する可能性がある。この点で、これらのデータは臨床的利益の初歩的な証拠を示す可能性があるが、患者の持続的な追跡およびより多くの患者データの獲得に伴い、任意の治療効果が患者に持続的ではなく、および/または時間の経過とともに減少または完全に停止するリスクが存在する。初期データ、初期データ、または一時データは依然として監査とチェック手続きを遵守しなければならず、これは最終データがこれらの予備データ、初期データ、または一時データと大きく異なる可能性がある。したがって、最終データを取得する前に、予備データ、初期データ、または一時データを慎重に考慮すべきである。例えば、ファイザーおよび我々が2022年12月に発表したgiroctocogene fitelparvovecに関する1/2期ALTA研究の更新データに示されるFVIIIレベルは、将来の後続研究またはALTA研究または3期アフィン試験からの任意の他のデータ中に継続的に存在することは保証されない。ALTA研究で示されたFVIII平均は,最初のピーク後に, 治療時間から毎週のフォローアップまで低下傾向にあった。時間の経過とともに,この傾向が低下し続けるかどうか,どの程度低下するかは予測できない.同様に、STAAR研究で観察された患者におけるα-GalAレベルの持続的な上昇またはLyso-Gb 3レベルの低下は、将来のフォローアップ中またはSTAAR研究の任意の他のデータにおいて持続的に存在する保証はなく、これらの患者または研究中の任意の他の未来にERTを脱退する患者がERTを停止し続けることも保証されない。さらに、STAAR研究中の患者は、より深刻なAEs、またはSAEsを経験する可能性がある。この原因と潜在的な他の原因により、適用されたALTA研究、3期模倣臨床試験或いは1/2期STAAR研究の最終結果において、giroctocogene fitelparvovecとisaralgagene ciciparvovecは最終的に監督機関を満足させる持続的、安全かつ有効な臨床利益を証明できない可能性があり、そして監督当局が満足していても、このような利益は商業的に実行可能な製品を生産するのに十分ではない可能性がある。
未解決の臨床試験が成功する保証はありません。私たちの多くの候補製品は現在、ZFヌクレアーゼおよびZF転写制御技術を含むZF技術プラットフォームを使用しており、この技術はまだいかなる承認された治療製品も生成していない。そのほか、著者らの多くの候補製品はすべて臨床前製品であり、いかなる臨床利益も示したことがない。また,我々のウイルス伝達システムは発展し続けており,まだ承認された製品には使用されていない.ZF技術プラットフォームおよびウイルス伝達システムの候補製品を使用して、私たちが見たい安全、有効かつ持続的な結果を臨床試験で示すことができない場合、候補製品の一部または全部の開発を一時停止または終了させるか、または候補製品を開発または交付するための代替技術を求めることができるかもしれない。
また,臨床試験による候補製品の失敗率が高かった。生物製薬業界の多くの会社は後期臨床試験で重大な挫折を経験し、臨床前試験と早期臨床試験においても奮い立つ結果を得た。臨床前と臨床活動から得られたデータは異なる解釈を受ける可能性があり、これは監督部門の承認を延期、制限或いは阻止する可能性がある。このようなどんな挫折も、私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性がある。
私たちの候補製品は承認を求めるすべての司法管轄区域で長く予測できない規制承認過程を経験しなければならない。
いかなる人類治療製品もFDA、欧州委員会或いは類似の外国監督管理機関などの監督機関の許可を得なければ、その管轄する司法管轄区域内で販売することができる。監督部門の承認を得る過程は長く予測できず、候補製品はこの過程の厳格なテストに耐えられない可能性がある。米国でヒト臨床試験が開始される前に,INDをFDAに提出しなければならない。米国以外のある国にも類似したプログラムがあり,ヒト臨床試験開始前に臨床試験申請を提出することが求められており,INDに似ている。例えば,EUでは,各臨床試験はスポンサーが臨床試験を希望するEU加盟国の各国の主管当局や関連倫理委員会に臨床試験承認申請を提出しなければならない。INDの発効及び/又は臨床試験の許可を得た後にのみ、臨床試験を開始することができる。我々の候補製品に適用される規制承認の流れの詳細については、“企業-政府規制条例”を参照されたい。いくつかの重複があるが、臨床試験と上場承認を求める監督管理要求は司法管轄区によって異なる。生成された安全性研究および他のデータは、計画または一旦生成されたすべての司法管轄区域での臨床試験を可能にするのに十分であり、このような臨床試験データが、このような承認を求めるすべての司法管轄区で上場承認されるのに十分であることは保証されない。もし私たちの臨床試験を行い、私たちの候補製品を商業化するために必要な規制承認を得ることができなければ、あるいはこのような承認が延期または一時停止された場合、私たちの業務は, 私たちの普通株の見通しと市場価格は不利な影響を受けるだろう。
私たちの臨床試験のために十分な患者を識別、鑑定、募集することができないかもしれないし、適時に私たちの臨床試験を完成できないかもしれません。これは私たちの候補製品の開発を延期または阻止するかもしれません。
患者を識別、鑑定と募集して著者らの候補製品の臨床試験に参加し、そしてこれらの臨床試験を完成することは、著者らの成功のキーポイントである。患者登録と試験完了に影響する要素は以下のとおりである
•患者集団の規模と患者を識別するプログラム
•実験案の設計
•資格と排除基準
•研究を受けた製品候補製品のリスクと収益を感知する
•ゲノム治療法のリスクと利点を認識しています
•競争療法と臨床試験の有用性
•新冠肺炎の全世界大流行に関連する潜在的な追加遅延は、ある患者が登録或いは薬を飲む前に新冠肺炎測定が陽性である影響を含む
•2022年3月に以前に自発的に募集を一時停止し、より多くの患者にgiroctocogene fitelparvovec第3段階アフィン試験に参加し、2022年9月に解除され、試験サイトの活性化を一時停止するなど、私たちの臨床試験または私たちの協力者の自発的な一時停止に関連する遅延または中断
•規制当局は、giroctocogene fitelparvovecの3期模倣試験に適用された以前の臨床一時停止のような、著者らの臨床試験または著者らの協力者の臨床試験に臨床休止を適用し、この一時停止はその後キャンセルされ、Sangamoと私たちの協力者は、監督機関によって適用された臨床一時停止をタイムリーまたは許容可能な条件で解除できないかもしれない、または根本的に解除できない可能性がある
•私たちの1/2期PRECIZN-1研究では、私たちはより多くの患者を募集し、投与する際に遅延または困難に遭遇する可能性がある
•調査中の病気の重症度は
•潜在的な患者に対する遺伝子検査の利用可能性;
•潜在患者に臨床試験場所の近似性と可用性を提供する
•患者の必要と希望の特徴;
•患者の同意を得て維持する能力
•試験に参加した患者が試験終了前に退出するリスク;
•医師の患者への転職方法と
•治療期間と治療後に患者を十分にモニタリングする能力がある。
われわれの臨床試験の時間は,患者の参加を募集する能力と完成に必要なフォローアップ期間に依存する。私たちの競争相手も他の候補製品を開発しています。彼らは同じ限られた患者集団のために競争しています。もし私たちが適時に必要な数量の患者を募集できなければ、私たちは私たちが希望するスケジュール内で臨床試験を完成することができず、甚だしきに至っては臨床試験を完成できない可能性があり、これは私たちの候補製品の競争地位と商業実行可能性にマイナス影響を与えるか、あるいは候補製品から稼ぐことが予想される製品収入、マイルストーン支払いまたは特許権使用料を延期または減少させるかもしれない。例えば,新冠肺炎の大流行に関する挑戦,新冠肺炎検出陽性の患者,この研究への参加の見直しやスクリーニングサイト数が限られていることなどから,STAAR 1/2期臨床研究(我々の全資本が持つFabry病治療の遺伝子治療製品候補薬isaralgene cinparvovecを評価する)を募集,スクリーニング,募集,投与する患者において遅延や挑戦に遭遇している。TX 200の1/2期堅固な臨床研究も同様の遅延と挑戦を経験したことを評価した
また、著者らとファイザーは以前、原癌遺伝子fitelparvovecの3期アフィン試験で治療を受けた一部の患者は治療後にFVIII活性が150%を超え、ファイザーはすでに自発的に今回の試験中の追加患者のスクリーニングと投与量を一時停止することを決定し、提案した方案修正案を実施し、FVIIIレベルが上昇した臨床管理にガイドラインを提供することを目的とした。自発的に一時停止した後,FDAはこの試験を臨床放置し,その後2022年3月に解除した。ファイザーによる自発的な一時停止は中止され,試験が再開され,募集,登録,服薬が回復したが,服薬が速やかに完了すること,あるいは全く不可能であることを保証することはできず,このような試験のデータプレゼンテーションが速やかに公表されることも保証されない。第3段階生体模倣試験の持続的な遅延または追加的な一時停止は、試験の実施と完了、および規制機関のgiroctocogene fitelparvovecの承認を求める予想スケジュールに悪影響を及ぼす可能性があり、これは逆に、giroctocogene fitelparvovecの競争地位と商業可能性、ならびに私たちの業務、将来性、および私たちの普通株の市場価格に大きな悪影響を及ぼす可能性がある。
また,遺伝子薬物に関連する有害事象の負の宣伝,患者群のような競争的臨床試験やその他の理由でわれわれの臨床試験に参加したい患者が少ない場合には,われわれの候補製品に対する臨床試験や臨床データの提出時間が遅れる可能性がある。これらの遅延は,コスト増加,臨床試験の制限や終了,製品開発スケジュールの遅延を招く可能性がある。もし私たちがこのような挑戦に対応するためにより多くの司法管轄区域に拡大することを強要すれば、追加的な費用、遅延、そして危険をもたらすかもしれない。もし私たちが計画通りに臨床試験を成功できなければ、私たちの業務、財務状況、運営結果、将来性と私たちの普通株の市場価格に不利な影響を与えるだろう。
候補製品を研究項目から臨床前·臨床開発に進めることは困難である可能性がある。
我々の候補製品を研究計画から臨床前開発に進め,新たなIND,臨床試験承認申請と他の司法管轄区でのヒト臨床試験評価に必要な同等の書類を提出する予定である。臨床試験の申請の準備と提出には,厳格かつ時間のかかる臨床前試験と研究を行い,候補製品の毒性,安全性,製造,化学,臨床案などに関する書類を用意する必要がある。私たちは予測できない困難に直面するかもしれないが、このような困難は私たちがこの戦略を成功的に実行することを延期したり、他の方法で阻害するかもしれない。例えば、候補製品を製造する際に問題に遭遇する可能性があり、候補製品配合の整合性を証明できない可能性がある。われわれの前臨床試験は陰性または不確定な結果をもたらす可能性があり、これは私たちの決定を招く可能性があり、あるいは監督機関が追加の臨床前試験を要求する可能性がある。前臨床試験で積極的な結果が得られなければ,候補製品を完全に放棄することに決定するかもしれない。また,このような申請を完了して提出して臨床試験を行う能力は,我々の協力者の支援や,関連する協力協定に基づいて速やかに義務を履行している場合に依存する可能性がある。もし我々の協力者がこのような義務を履行できない場合,あるいは候補製品の開発を遅らせることを選択した場合,臨床試験申請を速やかに提出できず,提出できない可能性もある。また、臨床試験を行う申請の提出は巨大なコストと人力に関連しており、すべての予想される申請の提出を完了するための十分な資源と人員がないかもしれない, これは,アプリケーションの数を削減したり,将来性が明るいと考えている潜在的なアプリケーションを放棄したりする可能性がある.著者らは臨床前と臨床開発戦略のいかなる遅延、一時停止或いは減少は私たちの業務に不利な影響を与え、私たちの普通株の市場価格の下落を招く可能性がある。
我々の候補製品は、RMATや孤児薬物指定のような特別な規制指定を得ることができない場合があり、またはより速い開発または規制審査または承認プロセスを引き起こさない可能性がある。
私たちは深刻な血友病Aを治療する候補製品のRMAT認証を取得しました。また、私たちのいくつかの候補製品は、私たちのFabry病治療の候補製品を含み、FDAの孤児薬物指定も得られ、いくつかの製品はEMAによって孤児薬に指定されています。アメリカとEUを含むいくつかの管轄区域の規制機関は、比較的少ない患者集団の薬物を
孤児は麻薬を使う。これらの特殊規制指定の他の情報については、“企業-政府規制”を参照されたい
現在または将来の他の候補製品がこのような指定を受けることを要求する場合、FDA、欧州委員会、または同様の外国規制機関が、私たちの任意の候補製品がそのような指定を受けることを承認することは保証されない。また、このような指定は、どの規制機関がこれらの候補製品の規制審査または最終承認を加速させるかを保証するものではなく、私たちの候補製品が発売承認される前に、このような指定を他の会社の候補製品に付与する能力も制限されず、これらの会社は、私たちの候補製品が発売承認される前に、私たちの候補製品に対して同じ適応を持っている。このような指定はまた撤回されることができる。臨床データの出現に伴い資格基準を満たさなくなれば,RMATの指定は撤回されることができる。任意の規制機関が、指定された要求に重大な欠陥があると判断した場合、または製造業者が、このようなまれな疾患または疾患を有する患者の需要を満たすのに十分な数の製品を保証できない場合、孤児薬の排他性を取り消すことができる
私たちの候補製品は不良副作用或いは他の特性を招く可能性があり、その規制承認を延期或いは阻止し、承認された適応或いは商業潜在力を制限するか、あるいは任意の潜在的な上場承認後に重大なマイナス結果を招く可能性がある。
臨床試験を行っている間、患者は彼らの研究医に疾病、傷害、不快感を含む彼らの健康変化を報告した。通常,研究中の候補製品がこれらの状況を招いているかどうかを決定することは不可能であり,特に我々が研究している多くの疾患に複雑な共病があるためである。もし臨床経験が候補製品に副作用があることを表明したり、深刻な或いは生命に危害を及ぼす副作用を引き起こした場合、候補製品の開発は失敗或いは遅延する可能性があり、あるいは、候補製品が監督部門の許可を得た場合、撤回される可能性があり、これは私たちの業務、将来性、経営業績、財務状況を深刻に損なうことになる。
従来,遺伝子治療にはいくつかの著明な副作用があり,報告されている白血病や死亡例を含め,他のゲノム療法を用いた他の試験で見られた。遺伝子治療は依然として比較的に新しい疾病治療方法であり、より多くの副作用を産生する可能性がある。遺伝物質あるいは遺伝物質を携帯する製品の他の成分の持続的生物活性のため、遺伝子治療製品に接触した後も不良事象を著しく遅延させる潜在的なリスクが存在する。遺伝子治療製品の治療に出現する可能性のある副作用には,投与後早期の免疫反応が含まれており,必ずしも患者の健康に不利ではないが,治療の有効性を大きく制限する可能性がある。例えば,STAAR研究の拡張段階の患者の1人は2022年12月にレベル3 SAEが出現し,入院治療が必要であり,イソプロピルエピネフリン服用後14日後に発生した。この研究の首席調査員と安全モニタリング委員会は,SAEがイソプロピルエピネフリン治療に関与している可能性を評価し,規制当局にSAEを報告した。安全監視委員会は、その後、研究を修正せずに行うことができると判断したが、患者はSTAAR研究に登録されており、この事件を他の調査者に報告して理解のために報告しているが、この研究または他の研究において出現する可能性のある副作用は、追加のSAEを含み、将来的に生じる可能性があり、これは、適用候補製品の任意のさらなる開発または潜在的な商業化を延期または停止する可能性がある。
我々の製品開発が成功しても,必要な規制承認を得ても,我々の製品は医師,患者,医療支払者,医療界の市場認可を得ることができない可能性がある。
私たちが将来開発または買収する可能性のある任意の候補製品が監督部門の承認を得ても、承認された製品は医師、医療保険支払人、患者、あるいは医学界の市場で受け入れられない可能性がある。承認された製品に対する市場の受容度は、多くの要素に依存する
•臨床試験はこの製品の有効性と安全性を証明した
•この製品が承認された臨床適応と患者の人々
•医師、治療センター、および患者はこの製品を安全かつ有効な治療方法である
•医師、病院、第三者支払者は新しいゲノム療法を採用しています
•代替療法と比較して、製品の潜在的かつ知覚可能な利点
•承認された適応以外の使用を含む、より広範な患者集団における製品の安全性
•他の薬と一緒に使用するための制限はありません
•副作用の流行率や重症度は
•FDAまたは他の外国規制機関のような製品ラベルまたは製品挿入要件;
•製品と競争製品が市場に参入するタイミング
•製品製造と流通プロセスの発展
•代替治療に関連する治療費
•保険と適切な補償を提供し、第三者支払者と政府当局がカバーまたは補償不足がない場合、患者が自己負担費用を支払うことを望むかどうか
•相手が便利で管理しやすいこと
•私たちの販売とマーケティング努力と私たちの協力者の努力の有効性。
もし私たちの候補製品が承認されたが、医師、患者、医療支払者、あるいは治療センターの市場で受け入れられなければ、承認された製品から大量の収入を生み出すことができず、収益性を損なうことになる。
たとえ承認された製品を商業化することができても、これらの製品は、米国と、それを商業化する他の国/地域の第三者支払者から保険と十分な補償を得ることができない可能性があり、これは私たちの業務を損なう可能性があります。
私たちが任意の製品を商業化することに成功した能力は、これらの製品と関連する治療のカバー範囲と適切な補償の程度にある程度依存する。政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、どの薬剤をカバーするかを決定し、精算レベルを決定することは、任意の承認された製品の需要または価格に影響を与える可能性がある。我々が開発している候補製品の性質を考慮すると,一部の患者は1回の治療(例えば,単剤投与)のみを必要とする可能性があり,このような製品の価格設定構造や患者の一生における慢性治療と比較して,単剤治療へのカバー範囲や精算レベルには大きな不確実性がある。もし他の会社が長期的な治療効果の証明に基づく支払いを含む新しい価格設定構造または商業モデルを確立した場合、私たちは製品の価格設定または補償を得る能力が悪影響を受ける可能性がある。もしこのような定価構造やビジネスモデルが私たちの研究開発、製造、商業化努力に十分な資金を提供できなければ、私たちの業務は不利な影響を受ける可能性がある。
私たちのある候補製品の潜在的な価格設定構造に不確定性がある以外、コスト制御は医療保健業界の繰り返し出現する傾向である。政府当局と第三者支払者は,ある薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。商業化されたどの製品にもカバー範囲と十分な精算があることを確実にすることはできません。もし精算があれば、精算レベルはいくらですか。精算が得られない場合や限定精算だけでは、規制部門の承認を得た任意の候補製品を商業化することに成功しない可能性がある。私たちは、私たちが開発した承認された製品の保証範囲と利益の収益率を政府援助や個人支払者から迅速に得ることができず、私たちの経営業績、製品の商業化に必要な資金を調達する能力、および私たちの全体的な財務状況に悪影響を及ぼす可能性があります。
多くのEU加盟国はその医薬製品の精算手続きを定期的に審査しており、これは精算状況に悪影響を及ぼす可能性がある。EU加盟国の立法者、政策立案者、医療保険基金は、最高価格の低減、精算範囲の低下、または精算範囲の低下などのコスト制御措置を引き続き提案し、実施し、より安く、通常模倣された製品をブランド製品の代替品として使用することを奨励し、および/または平行輸入によって得られたブランド製品は、医療コストを低減するために、特にEU加盟国の医療システムにもたらす財政的圧力を考慮する。これらの措置には、私たちが開発に成功する可能性のある候補製品に対する価格制限と、規制部門の承認を得る可能性のある候補製品の価格、または政府当局または第三者支払人がこれらの製品に提供する清算レベルが含まれる可能性がある。また、多くのEUや他の外国国は、自国内の製品価格の決定を助けるために、他国で制定された医薬製品価格を“参考価格”として使用するようになっている。そのため、一部の国の医薬製品価格の低下傾向は他の国に類似した低下傾向を招く可能性がある
また、いくつかのヨーロッパ諸国(いくつかのEU加盟国を含む)で私たちの製品の精算を得るためには、追加のデータをまとめ、私たちの製品の費用対効果を他の既存療法と比較する必要があるかもしれません。いくつかのEU加盟国では、より大きな市場を代表する国を含め、医療製品のHTAは、価格設定および補償手続きにおいてますます一般的な一部となっている。HTAの結果は、EU個別加盟国の主管当局がこれらの医薬製品の価格設定と補償地位を与えることに影響を与えることが多い。現在、EU加盟国間の定価と補償決定は特定の医薬製品のHTAの影響を受ける程度がそれぞれ異なる。EUは2021年12月、EU全体のHTA臨床利益評価を調整するために2025年に施行されるHTA条例を採択した。しかし、個別のEU加盟国は、衛生技術の非臨床(例えば、経済、社会、倫理)の評価を担当し続け、定価と精算について決定する。もし私たちがEU加盟国で候補製品のために有利な価格設定と精算の地位を維持できなければ、私たちは開発に成功し、可能性があるかもしれない
規制部門の承認が得られれば、これらの製品はEUのどのような予想収入と成長見通しにおいてもマイナスの影響を受ける可能性がある。
最近公布された将来の立法は、不利になる可能性のある価格設定法規や他の医療改革措置を含み、規制部門の承認を得て、私たちの候補製品を商業化する難しさとコストを増加させ、入手可能な価格に影響を与える可能性がある。
新薬製品を管理する監督管理審査、カバー範囲、定価と精算などの方面の法規は国によって異なる。米国や一部の外国司法管轄地域では、医療システムに関する立法や規制変更、提案された変更は、候補製品の規制承認を阻止または延期し、承認後の活動を制限または規制し、規制承認を得た任意の候補製品の販売に成功する能力に影響を与える可能性がある。また、処方薬や生物製品コストの上昇を受けて、政府は最近、バイオ医薬品の価格設定の審査を強化している。このような審査は、最近のいくつかの大統領行政命令、国会調査、および提案と公布された連邦と州立法を招き、これらの立法は製品定価の透明性の向上、価格設定とメーカー患者計画との関係の審査、および生物製薬製品に対する政府の計画精算方法の改革を目的としている。州レベルでは、立法機関は、価格または患者の精算制限、割引、特定の製品参入の制限、およびマーケティングコスト開示および透明性措置を含む、医薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入および大量購入を奨励することを目的としている。医療改革活動と現在の価格設定枠組みの議論については、“企業-政府監督--医療改革”と“企業-政府監督--定価、カバー範囲と精算”を参照されたい
政府、保険会社、医療組織、その他の医療サービスを管理する支払人は、医療コストの抑制または低減、および/または価格規制の実施に努力し続けており、悪影響を及ぼす可能性がある
•私たちの候補製品の需要は、もし私たちが規制部門の承認を得たら
•私たちは私たちの製品に公平だと思う価格を設定することができます
•私たちは収入を作ったり利益を達成したり維持したりします
•私たちが払わなければならない税金レベル
•資金の入手可能性。
連邦医療保険や他の政府計画精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性があり、これは私たちの将来の収益性に悪影響を及ぼす可能性がある。
また、FDA、EU加盟国の主管機関、欧州薬品管理局、欧州委員会、その他の同様の規制機関の臨床試験における政策は変化する可能性があり、追加の政府法規が公布される可能性がある。例えば,EUの臨床試験に関する規制構造が最近変化している。CTRは2014年4月に採択され,EU臨床試験指令が廃止され,2022年1月31日に施行された。CTRは、スポンサーが各EU加盟国の主管当局と道徳委員会に文書を提出し、統一的な評価手続きを採用することを可能にし、各EU加盟国が単一の決定を下すことを可能にする。我々と我々の第三者サービスプロバイダ(例えばCRO)がCTR要求を遵守することは、我々の開発計画に影響を与える可能性がある。イギリスが未来にどの程度その法規をEUと統合することを求めているのかは不明だ。イギリスはその法規をEUが採用する新しい方法と緊密に結合しないことを決定し、イギリスでの臨床試験のコストに影響する可能性があり、及び/或いは私たちの候補製品をイギリスで行われた臨床試験に基づいてEUでマーケティング許可を求めることを困難にする。
もし私たちが既存の要求の変化にゆっくりあるいは適応できない場合、あるいは新しい要求を採用したり、臨床試験を管理する政策を採用すれば、私たちの発展計画は影響を受ける可能性がある。
私たちの候補製品が規制部門の承認を得ても、私たちの製品は規制部門の審査を受けるだろう。
たとえ私たちが司法管轄区域の監督管理の許可を得ても、主管監督機関は私たちの候補製品の指定用途やマーケティングに重大な制限を加えたり、コストの高い承認後の研究、上場後の監視或いは患者或いは薬物制限に持続的な要求を提出する可能性がある。例えば、FDAは通常、遺伝子治療を受けた患者に15年間の潜在的有害事象の追跡観察を受けることを提案する。また、承認されたBLAの所有者は、FDAのルールを遵守し、FDAの審査および定期検査、およびcGMPの遵守とBLAにおける約束の遵守を確保するために、他の適用可能な連邦および州法律を受け入れなければならない。
我々または規制機関が、意外な重大性または頻度の不良事象など、以前に未知の問題があることを発見した場合、または製品の製造施設に問題がある場合、規制機関は、市場からの製品のリコールまたは撤回または生産の一時停止を要求することを含む、製品または製造施設に制限を加える可能性がある。さらに、承認された製品の製品ラベル、広告、および販売促進活動は、規制要件および持続的な規制審査の制約を受ける。もし適用された場合にこれらの要求を守らなければ、私たちは警告状から製品差し押さえや巨額の罰金、その他の行動まで一連の行動に直面する可能性がある。
政府は法律や法規違反の疑いのあるいかなる調査にも対応するために多くの時間と資源を必要とする可能性があり、否定的な宣伝が生じる可能性がある。上記のいずれの事件や処罰の発生も、候補製品を商業化し、収入を創出する能力を抑制することができる。
上場許可を付与する前および後に、臨床試験、生産承認、医薬製品の上場許可およびそのような製品のマーケティングを行うのに適したEUおよびEU加盟国の法律、または他の適用される法規要件を遵守しない場合、行政、民事または刑事罰を受ける可能性がある。これらの処罰には、臨床試験の遅延または拒否、またはマーケティング許可の付与、製品の撤回およびリコール、製品の差し押さえ、一時停止、マーケティング許可の撤回または変更、生産の完全または部分的な一時停止、流通、製造または臨床試験、経営制限、禁止、免許停止、罰金、および刑事罰が含まれる可能性がある。
詳細については、“企業-政府法規-承認後要求”を参照されたい。
私たちの従業員または請負業者は、研究、開発、製造、または規制基準と要求を遵守しないことを含む、不適切な行為または他の不適切な活動に従事する可能性があり、これは私たちに重大な責任をもたらし、私たちの名声を損なう可能性がある。
我々は、FDAの規定または同様の外国規制機関の同様の規定に故意に準拠できなかったこと、FDAまたは同様の外国規制機関に正確な情報を提供すること、私たちが制定した製造基準の遵守、連邦および州医療詐欺および乱用法律法規の遵守、および同様の外国規制機関によって制定され実行された類似の法律法規、財務情報またはデータを正確に報告すること、または許可されていない活動を開示することを含む、従業員および請負業者の詐欺または他の不正行為のリスクに直面している。私たち従業員と請負業者の不当な行為はまた、臨床試験の過程で得られた情報の不適切な使用に関連する可能性があり、これは規制制裁と私たちの名声に深刻な損害を与える可能性がある。このような不正行為を常に識別し、阻止できるわけではなく、私たちがそのような活動を発見し、防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはそのような法律や法規に準拠していないことによる政府の調査または他の行動または訴訟から私たちを保護することができないかもしれない。もし私たちにこのような訴訟を提起した場合、私たちは私たちの権利を弁護したり、維持することに成功しませんでした。これらの行動は、重大な民事、刑事および行政処罰、損害賠償、罰金、返還、個人監禁、MedicareおよびMedicaidなどの政府援助の医療計画から除外された医療計画や同様の外国計画から除外され、もし私たちが会社の誠実協定や同様の合意の制約を受けて、これらの法律違反に関する疑惑を解決し、私たちの業務を削減または再編することを含むかもしれません。
私たちは、私たちの財務および人的資源を使用して、特定の研究計画や候補製品を追求することができ、他のより利益的または成功可能性の高い他の計画または候補製品を利用することができないかもしれない。
私たちの資源は限られており、いくつかの後により大きなビジネス潜在力が証明された研究プロジェクトや候補製品の追求を放棄または延期する可能性がある。私たちの資源配分決定は、私たちが実行可能な商業製品や利益のある市場機会を利用できない、あるいは唯一の開発責任を維持するのではなく、協力を求めることになるかもしれない。私たちの現在と未来の候補製品に対する研究と開発計画はどんな商業的に実行可能な製品も発生しないかもしれません。特定の候補製品の商業潜在力または目標市場の評価は前向きであり、例えば市場変化、疾病看護標準の進歩、競争と精算などの仮定に基づいているが、これらに限定されない。この仮説への依存は、もし私たちの仮説が不正確または不完全であることが証明された場合、私たちは最終的に私たちの候補製品よりも先進的な競争相手を持つ機会を求めることができるかもしれないし、あるいは戦略協力、許可、または他の印税手配によって候補製品に対する貴重な権利を放棄する可能性があり、候補製品の独占開発と商業化権利を保持すれば、より有利になることを意味する。例えば、私たちは最近、1/2段階PRECIZN-1研究が完了した後、BIVV 003計画へのさらなる実質的な投資を停止して、私たちのFabryとTX 200計画の資源を優先的に配置するための戦略決定を下した。この計画を潜在的な第3段階実験に進めることができる協力パートナーを探し始めたいと考えているが,タイムリーで受け入れ可能な条項やまったく成功していない可能性があるため,BIVV 003計画の潜在力を利用する貴重な機会を逃す可能性がある。私たちは
治療分野の候補製品に内部資源を割り当てることも可能であり,その分野で協力した方が有利であるか,あるいは実行可能なビジネス機会がないことが証明されている。私たちの財政と人的資源を有効に活用できなかったいかなるものも、私たちの業務と運営を損なう可能性がある。
ZF技術は斬新で、いかなる承認された、商業的に実行可能な治療製品の開発に使用されたことがない。
我々のZF技術は新しい技術であり,これまで承認された商業的に実行可能な治療製品は何も生成されておらず,ZF技術を用いた製品開発努力が効果的であることは保証されていない。私たちはこの技術の開発に大量の資金を投入して、もし私たちがZF技術を利用して許可された商業的に実行可能な製品を開発できなければ、私たちの業務と将来性を大きく制限し、私たちの普通株の市場価値に悪影響を与えるだろう。
製造業に関するリスク
私たちは最近いくつかの臨床試験用品施設の建設を完了した。我々がバイオ製薬製品を製造する経験は限られており,コンプライアンスの製造施設を維持し,より多くの施設を建設し,期待どおりに候補製品を生産できる保証はない。
私たちは契約製造組織やCMO、および私たち自身の施設を使用して、臨床試験供給に対する予想される需要を満たしたい。私たちはカリフォルニア州ブリスベンでAAV製造工場を経営しており、私たちの遺伝子治療候補製品のために1/2期の臨床研究用品を生産し、2021年にカリフォルニア州ブリスベンとフランスバル邦で細胞治療製造工場の建設を完成し、私たちの細胞治療候補製品のために用品を生産した。これらの新施設を運営するためには,我々の候補製品の製造プロセスや技術ノウハウを我々のCMOから自分たちの施設に移す必要がある。製造プロセスおよびノウハウを移転することは複雑であり、時間の経過とともに変化する可能性のある記録および記録の有無の審査および組み込まに関するプロセスである。さらに、生産を異なる施設に移転するためには、特定の施設の具体的な要件を満たすために、新しいプロセスまたは異なるプロセスを利用する必要がある場合がある。いくつかの製造プロセスの移転およびプロセス改善を支援するために、追加の研究が必要かもしれない。CMO以前に生産された材料とわが工場で生産された材料との比較性を証明するための研究や評価が完了するまで,すべての関連技術ノウハウやデータが製造過程に十分に組み込まれているとは判断できなかった。私たちの一部の従業員は以前他の会社で働いたことがあるため、生物製薬製品の製造経験を持っていますが、会社として、私たちはこれまで生物製薬製品の製造経験がなく、これらの施設を運営することは私たちに複雑な法規を遵守し、経験豊富な科学、品質管理、品質保証と製造人員を採用し、保留し続けることを要求します。また、, 私たちは製造施設を運営するために政府の承認が必要であり、これらの施設を獲得して維持するのに非常に時間がかかる。バイオ製薬製品のメーカーとしても,cGMPのコンプライアンスを証明し維持することが求められる。これらの要求は、他にも、品質管理、品質保証、および記録およびファイルの維持を含む。また、製造業務を確立するには、他の資源、特に私たちの上級管理職の時間と注意力を再分配する必要があります。私たちが自分の製造能力を確立することができても、cGMP、法規、または他の適用要件に適合して製造施設を運営する際に挑戦に直面し、規制行動を含む潜在的な負の結果を招く可能性があり、これらの施設を使用して自身の製造需要を満たす能力を弱める可能性がある。私たちの製造能力開発のどんな失敗や遅延も私たちの候補製品の開発に悪影響を及ぼす可能性があります。
私たちの候補製品の製造、貯蔵、輸送は複雑で、高価で、高度な規制とリスクであり、これはそれらの商業生存能力を阻害するかもしれない
我々の候補製品の製造、貯蔵および輸送には、cGMPコンプライアンス、コスト超過、プロセス拡大の技術問題、専用施設、プロセス再現性、安定性の問題、ロット一致性、良率、および高度な特定の原材料のタイムリーな可用性を含むが、これらに限定されない重大なリスクがある。それでも…臨床試験のための製品ロットを発表するにはサンプリングテストを行い、いくつかの欠陥は発表後に発見できるかもしれない。さらに、プロセス偏差または承認されたプロセス変更の予期しない影響は、これらの中間製品が安定性要件または仕様に適合しない可能性がある。しかも、私たちの候補製品は一定の範囲の温度で貯蔵されて輸送されなければならない。これらの環境条件がずれていれば、私たちの候補製品の残り賞味期限が損なわれる可能性があり、あるいはそれらの有効性と安全性が不利な影響を受け、使用に適していない可能性がある。また、生物製品の候補製品としては、細胞系または細胞系を開発して生物製剤を製造し、顕著な変異性の挑戦に直面することを含む複雑なプロセスに関連する。このような細胞を大量に培養し、あるタイプの細胞を持続的かつ十分に分離し、それらによって産生された生物を収穫と精製することは困難である。生物製品を製造するコストは通常,従来の小分子化合物よりはるかに高く,製造過程の複製が困難である。
しかも、私たちの候補製品の製造、貯蔵、輸送は厳格な規制基準を受けており、これは追加的な生産リスクを増加させる。私たちの臨床試験の有効性と安全性データ支援でも
製品候補が規制承認を受けた場合、我々または我々のCMOがFDAや他の同様の外国規制機関の規制承認を支援または維持するために必要な仕様に従って、我々の候補製品を生産できる保証はない
したがって、私たちの候補製品のためにより大規模な商業製造プロセスを構築したり、必要な製造能力を得ることに成功する保証はありません。これらの製造課題のため、私たちのいくつかの候補製品は在庫中断、名声損害と製品責任リスクに直面し、追加の費用と臨床試験と商業化の遅延を招く可能性がある。供給中断や不足は私たちの業務、将来性、私たちの普通株の市場価格に潜在的なマイナス影響を与える可能性がある
もし私たちが化学、生物、あるいは危険材料を使用して傷害や法律違反をしたら、私たちは損害賠償責任を負うかもしれない。
我々の研究·開発活動は,潜在的有害生物材料および危険材料,化学品および各種通常分子や細胞生物学研究に用いられる放射性化合物の制御使用に関する。培養や遺伝子伝達ベクターに細胞を使用し,トレーサー実験では少量の放射性同位体を用いることが多い。私たちは最新の許可と訓練計画を維持しているにもかかわらず、これらの材料の使用、貯蔵、処理、または処分による意外な汚染や傷害のリスクを完全に除去することはできない。汚染や傷害が発生した場合、私たちはそれによる損害に責任を負う可能性があり、どんな責任も私たちの資源範囲を超える可能性がある。私たちは現在このような材料の使用によって引き起こされたいくつかのクレームのために保険をかけている。しかし、もし私たちが合理的な費用と十分な保険範囲で私たちの保険範囲を維持できなければ、私たちの保険は出現する可能性のあるいかなる責任もカバーしないかもしれない。私たちは連邦、州と地方の法律法規に支配されて、これらの材料と特定の廃棄物製品の使用、貯蔵、運搬と処分を管理します。このような法律法規を遵守しないことは、罰金、処罰、そして追加的な責任と私たちの業務に対する制限を招く可能性がある。
著者らは現在第三者に依存して臨床前と臨床開発候補製品の一部或いはすべての方面の製造を行っている。もし私たちのある第三者メーカーが私たちの需要を十分に発揮したり、満足したりできなければ、私たちは巨額のコストを負担し、新しいサプライヤーやメーカーを探すことに多くのエネルギーを投入することが要求されるかもしれない。
著者らは現在臨床規模で著者らの候補製品を生産する経験は限られており、著者らは第三者CMOが著者らの臨床前研究と臨床試験のために薬物製品を製造と供給することに大きく依存している。カリフォルニア州ブリスベンとフランスバル州に内部製造工場を持っているにもかかわらず、これらの工場は私たちの早期臨床試験の限られた数の候補製品しか生産しないだろう。我々は,第三者生産後期臨床試験の候補製品と,任意の承認された製品の商業規模生産に引き続き依存していく予定である。FDAのcGMPあるいは類似の外国GMP法規に従って生物製薬製品を生産するには、先進的な製造技術と技術制御の開発を含む大量の専門知識と資本投資が必要である。生物製薬製品メーカーは生産中に常に困難に直面し、生産コストと生産量方面の困難、品質管理方面の困難、候補製品の安定性と品質保証テスト、合格者不足、及び厳格に実行されたcGMP要求、その他の連邦と州監督管理要求及び外国法規を含む。もし私たちのメーカーがこれらの困難に遭遇したり、彼らの私たちへの義務を履行できなかったり、法規を適用することによって、私たちが後期臨床試験を行う能力が脅かされる可能性がある。臨床試験材料供給のいかなる遅延或いは中断も著者らの臨床試験の完成を延期し、著者らの候補製品の開発に関連するコストを増加させ、遅延した時間帯に基づいて、私たちに新しい臨床試験を開始し、大量の追加費用を支払うか、あるいは臨床試験を完全に終了することを要求する。
我々と我々のCMOは,FDAがその施設検査計画や類似の外国規制機関によって実行しているcGMP要求を遵守しなければならない。これらの要求は、他にも、品質管理、品質保証、および記録およびファイルの維持を含む。私たちと私たちのCMOは、これらのcGMP要件および他のFDA、州、および同様の外国規制要件を遵守できないかもしれない。FDAまたは同様の外国規制機関は、新しい基準を随時実施したり、製品製造、包装、または試験の既存の基準の解釈および実行を変更したりすることもできる。私たちは製造業者たちにこのような規制と基準を遵守する統制が限られている。これらの要件を遵守しないことは、罰金や民事処罰、生産停止、一時停止、製品承認の変更または延期、製品の差し押さえまたはリコール、または製品承認の撤回につながる可能性があります。もし私たちのメーカーが適用された法律や他の理由を遵守できなかった場合、提供されたいかなる製品の安全が損なわれた場合、私たちは規制機関の私たちの製品の承認を得ることができないか、または商業化に成功できない可能性があり、それによるいかなるダメージにも責任を負わなければならないかもしれない。これらの要素のいずれも、私たちの候補製品の臨床試験、規制提出、承認または商業化の遅延を招き、コスト上昇または私たちの名声を損なう可能性がある。
我々が現在CMOと達成している合意は,すべての予想される臨床試験や全面的な商業化に必要なすべての薬物製品を提供することを規定していない。もし私たちと私たちのCMOが彼らが私たちの臨床と商業供給需要に必要な薬品の条項と条件を提供することに同意できなければ、私たちは生産できないかもしれません
合格した代替メーカーを決定する前に、候補製品を使用していますが、これも候補製品の開発を延期し、候補製品を商業化する能力を弱める可能性があります。
必要な製造や規制の専門知識や施設を持つ第三者CMOの数は限られており,CMOの代替を手配するコストが高く,多大な時間を要する可能性があり,我々の業務に悪影響を及ぼす可能性がある。任意の候補製品の新しい製造業者は、適用される法規要件に基づいて資格を取得することを要求され、適用される知的財産権法に基づいて候補製品を製造する方法に対して十分な権利を有する必要がある。適用される法規要件に基づいて必要な承認または他の資格を取得し、第三者の知的財産権を侵害しないことを保証することは、供給の深刻な中断を招く可能性があり、新しいメーカーが私たちに転嫁される可能性のある大量の追加コストを負担する必要があるかもしれない。
私たちと私たちが依存している第三者は、自然災害や悲劇的な事件、または私たちがコントロールできない他の事件の悪影響を受けるかもしれませんが、私たちの業務の連続性と災害復旧計画は、深刻な災害や事件から私たちを十分に保護できないかもしれません。
自然災害は私たちの現在のカリフォルニア州ブリスベンとフランスバル州の製造施設、私たちのCMOの製造施設を含め、私たちの運営と施設を深刻に混乱させる可能性があり、どんな中断も私たちの業務、財務状況、運営結果、見通しに悪影響を及ぼす可能性があります。もし自然災害、大流行、または流行病、新冠肺炎の大流行、政治的危機、停電、または私たちがコントロールできない他の任意の事件を含む場合、私たちまたは私たちが依存している第三者が私たちまたは彼らが依存しているすべてまたは大部分の施設を使用することができなくなり、重要なインフラを損傷したり、他の方法で私たちまたは彼らの運営を妨害したりすることは、私たちが難しいかもしれないし、場合によっては、長い間私たちの業務と運営を続けることはできない。我々が現在策定している災害復旧·業務連続計画は限られており、深刻な災害や同様の事件が発生した場合には十分ではないことが証明されている可能性がある。私たちの災害復旧と業務連続計画の限られた性質のため、私たちは大量の費用を発生する可能性があり、これは私たちの業務、財務状況、運営結果、将来性に悪影響を及ぼす可能性があります。私たちが依存している第三者施設で発生したこのような災害や事件は、私たちの業務や運営にも悪影響を及ぼす可能性があります。
私たちの業界に関わるリスクは
私たちの候補製品は新しいゲノム薬物技術に基づいており、開発とその後に規制承認を得る時間とコストを予測することは困難である。
私たちは遺伝子治療、遺伝子編集細胞治療、ゲノム工学を含むゲノム医学の研究と開発に集中している。我々のような新製品候補製品の規制承認過程はまだ不明であり、他のより有名あるいはより広く研究されている製品候補製品の審査過程よりも長く、より高価である可能性がある。
規制審査委員会および諮問グループおよびその発表された任意の新しいガイドラインは、追加の臨床前研究または臨床試験を行うことを要求し、私たちの開発コストを増加させ、規制の立場や解釈の変化を招き、現在または未来の候補製品の承認および商業化を延期または阻止し、または重大な承認後の制限または制限を招く可能性がある。私たちが私たちの候補製品を推進する時、私たちはこれらの規制や諮問グループと協議し、適用されるガイドラインを遵守することを要求されるだろう。もし私たちがこれをできなかったら、私たちは私たちの候補製品の開発を延期または停止することを要求されるかもしれない。このような追加的な手続きは私たちが予想していたより長く検討と承認過程を招くかもしれない。潜在的な製品を市場に出すために必要な監督管理の承認を遅延したり、許可を得られなかったりする意外なコストは、私たちが十分な製品収入を生成する能力を低下させる可能性があり、私たちの業務、財務状況、運営結果、および将来性は損なわれるだろう。私たちの候補製品が承認されても、FDAなどの外国規制機関は、承認された後に、私たちの臨床試験患者に関する数年間の追跡データを提出することを要求すると予想される。これらのフォローアップデータがこれらの患者の長期安全性または治療効果の結果が陰性であることを示す場合、FDAまたは同様の外国の規制機関は、彼らの承認を撤回したり、私たちの製品のラベルを変更したりする可能性があり、それによって私たちの業務に悪影響を及ぼす可能性がある。
さらに、他の人が行った遺伝子薬物臨床試験の不利な発展は、FDAまたは他の同様の外国規制機関が私たちの候補製品に対する承認要求を変更する可能性がある。FDAと欧州委員会は体内遺伝子治療製品の承認において非常に新しいと限られた経験しかない。したがって、私たちの候補製品が規制部門の承認を得るのにどのくらいの時間またはどのくらいのコストがかかるかを決定することは難しい。
もし私たちまたは私たちの競争相手が私たちよりも効果的な技術や製品を開発、獲得、またはマーケティングすれば、私たちの財務状況および私たちの候補製品のマーケティングまたは商業化に成功したり、利益を達成する能力は不利な影響を受けるだろう。
生物製薬業界は競争が激しく、重大かつ迅速な技術変革の影響を受けている。いくつかの会社が細胞の編集、遺伝子の編集、遺伝子発現の制御に集中している他の方法を知っています
ますます多くの商業と学術団体がゲノム工学技術の発展に取り組んでいる。ゲノム医学領域の競争は激しく、未来には生物製薬会社、学術と研究機関及び私たちのZF技術プラットフォームと競争する製品と技術の開発を求める政府機関を含む複数の異なる源からの競争が持続的かつ激化することを予想している。例えば、ゲノム工学および遺伝子治療製品において、我々の製品開発の重点に関連する相互競合のノウハウは、組換えタンパク質、他の遺伝子治療/cDNA、ヌクレアーゼおよび塩基編集技術、アンチセンス治療およびRNA干渉技術、siRNA、RNAiおよびmicroRNA方法、エクソンスキップ、小分子薬、クローン抗体、CRISPR/Cas技術およびストーリータンパク質、巨ヌクレアーゼおよびMegaTALsを含むが、これらに限定されない。ますます多くの会社がそれと競争する細胞治療技術や候補製品を開発している。私たちが直面する可能性のある競争に関するより多くの情報は、“ビジネス-競争”を参照されたい。
私たちまたは私たちのパートナーや戦略パートナーが開発したどの製品も競争の激しい市場に入るだろう。たとえ私たちがその期待用途に対して安全かつ有効な製品を生産することができても、競争技術はより効果的またはより安いことが証明される可能性があり、これはある程度、これらの競争技術が市場に受け入れられ、私たちの収入機会を制限するだろう。場合によっては、相互競争の技術が有効であることが証明され、コストがより低い。これと競合する技術は、遺伝子発現を調節するか、または遺伝子を修正する他の方法を含むことができる。ZFヌクレアーゼとZF-TRsは生命科学業界で広範な応用があり、そして多くの会社が遺伝子研究に応用している一連の新しい技術と方法と競争している。
競争相手の技術に加えて、私たちの競争相手には、以下のような特徴を持つバイオ製薬会社が含まれています
•私たちの資本資源よりもはるかに多い
•私たちよりも多くの研究者や施設を持っています
•製品開発と規制承認と特許保護についてもっと多くの経験がある
これらの組織はまた、合格者を誘致し、各当事者が買収、合弁または他の協力を行い、私たちの技術と競争する学術·研究機関のノウハウを許可するために競争しており、同様の機会を求めることを阻止する可能性がある。したがって、私たちの競争相手は私たちの前に特許保護を受けることに成功したり、製品を商業化することに成功するかもしれない。私たちの候補製品がもっと効果的であっても、それが最初に市場に出されなければ、それは不利な立場にあるかもしれない。さらに、私たちが開発したどの製品も、市場で成熟した既存製品やサービスと競争する可能性がある。しかも、私たちが開発しているいくつかの候補製品は使い捨てのために設計されている。使い捨て療法の開発のどの成功も,患者の一生のための治療法を設計する潜在的経常的収入を失う可能性がある。
新型肺炎の流行は、私たちの業務と運営、そして私たちの協力者、メーカー、その他の業務パートナーの業務と運営に悪影響を与え続ける可能性があります。
私たちはすでに新冠肺炎の大流行が私たちの業務と運営に与える影響を経験し続け、大流行のアメリカ、フランス、イギリスと私たちの臨床研究と試験地点(カナダ、イタリア、オーストラリアでの新しい研究地点を含む)の発展に伴い、私たちはこれらのあるいはもっと深刻な影響を経験し続けるかもしれない。例えば,臨床試験患者における新冠肺炎陽性例を扱う場合,われわれの現場運営は周期的な短期中断を経験しており,臨床試験患者に新冠肺炎が発生すれば,我々の運営は将来より長期的な中断を経験する可能性がある。また、私たちは時々私たちの資源の再構成と優先順位を要求されて、新冠肺炎の影響による適度な供給制限を緩和する。私たちの計画が長期中断に遭遇すれば、これまでの短期中断がこのような影響をもたらしたとは考えられないにもかかわらず、協力協定で想定されているバイオ製薬パートナーを支援する能力に影響を与える可能性があります。
また,われわれの1/2期STAAR臨床研究は以前にスケジュールの遅延を経験しており,一部の原因は新冠肺炎の影響と医療資源が大流行に対抗するために移行されたことである。例えば,新冠肺炎がイギリスで広く流行しているため,この研究はイギリスでの最初の臨床試験地点で2021年4月に開業したが,約1年遅れている。そのほか、著者らはこの研究のための募集、募集と投与に遅延があり、一部の原因は患者が疫病大流行期間中に飛行機で車で距離以外の試験地点に行きたくないことと医療機関に入ることであり、一部の原因は試験地点が新冠肺炎臨床看護をSTAAR研究などの研究活動の優先順位に置くことである。ある患者が登録或いは薬を飲む前に新冠肺炎ワクチンの接種或いは新冠肺炎の検査が陽性であることを決定した時、この研究も遅延する。また、新冠肺炎の影響により、著者らはこれまでに必要な原材料を調達してSTAAR研究用品の製造と臨床試験材料の輸送にいくつかの短期遅延に遭遇した。私たちはこれらの挑戦が私たちの2021年のSTAAR研究スケジュールを約3~6ヶ月延期させたと推定する。もし新冠肺炎がワクチン接種遅延、新たな新冠肺炎変異或いは意外事件により著者らの募集、スクリーニング、登録と投与及び本研究の原材料源に与える影響が激化すれば、本研究の臨床スケジュールは再改訂される可能性がある。
また,われわれの揺るぎない研究は以前,われわれのCMOの製造や技術移転挑戦に関連した新冠肺炎の影響,患者とドナーの新冠肺炎検出が陽性であったためタイムラインを延期した。私たちはこのような挑戦が私たちの臨床研究スケジュールを約3ヶ月延期したと推定する。新冠肺炎の影響は将来の投与スケジュールの指導に影響する可能性がある。
我々の協力プロジェクトについて、これらの研究および実験がSTAARおよびSTESTRATE研究において経験し、経験し続ける類似の課題を経験した場合、我々の協力者によって管理される研究および実験のスケジュールも将来的に遅延される可能性がある。
新冠肺炎疫病がどの程度直接或いは間接的に著者らの業務、運営と財務状況に影響するかは、現在も高度に不確定な未来の発展に依存する。これらの動態は、疫病の最終持続時間と重症度、新冠肺炎の新変種の影響、旅行制限、アメリカ、フランス、イギリス、オーストラリア、台湾およびその他の国の公衆衛生制限、企業閉鎖または商業中断、およびアメリカ、フランス、イギリス、オーストラリア、台湾と他の国が疾病をコントロールと治療するために取った行動の有効性と適時性を含み、ワクチン接種計画の有効性を含む。このウイルスの新しい変種の急増はすでに引き起こされ、将来的には以前の命令と制限を新たに回復または回復させる可能性がある。私たちの運営の中断と、将来、私たちが運営する場所や業界に一般的に適用される制限や、私たちと私たちの施設に対する制限によって生じる可能性のあるより深刻な中断は、私たちのタイムリーな研究能力を阻害し、協力者に対する研究義務を履行し、私たちの治療計画の発展を推進するかもしれません。このような遅延と中断は、私たちの業務、運営業績、財務状況に実質的な影響を及ぼす可能性がある。私たちの事件に対する理解が絶えず発展し、更に多くの情報を得ることに伴い、著者らは著者らの収入、費用と製造、臨床試験と研究開発スケジュールに関連する指導を大幅に変えるかもしれない。
また,新冠肺炎の流行が我々の業務や運営結果に悪影響を与え続ける場合,本“リスク要因”の節で述べた多くの他のリスクや不確実性を増加させる可能性もある。
否定的な世論と遺伝子薬物の規制審査の強化は、私たちの候補製品の安全性に対する大衆の見方を損なう可能性があり、私たちが業務を展開したり、規制機関が私たちの候補製品を承認してくれる能力に悪影響を及ぼす可能性がある。
遺伝子組換え製品は現在大衆の討論とより厳格な規制によって検討されている。遺伝子治療はまだ2つの新しい技術です体内にあるこれまでアメリカで承認された遺伝病の遺伝子治療製品はいくつかしかありませんでした体内にある今まで、EUは遺伝病のための遺伝子治療製品を承認した。公衆の認知は遺伝子療法が安全でないという説の影響を受ける可能性があり,遺伝子療法は公衆や医学界の受け入れを得られない可能性がある。例えば、フランスがX連鎖深刻連合免疫不全(X連鎖SCID)乳児に対するレトロウイルス遺伝子転移試験の深刻な不良事件の発生に関する報告、およびFDAがその後アメリカで行った関連試験の一時停止行動は、遺伝子治療に参与するある会社の公衆認知と株価に重大な負の影響を与え、特定の会社がレトロウイルス遺伝子転移に参与するかどうかにかかわらず、あるいは特定の会社の臨床試験がこれらの事件によって保留されているかどうか。他の有害事象は遺伝子薬物の分野で発生する可能性があり、規制審査の強化、潜在的な監督管理の遅延、あるいは公衆認識の遺伝子薬物への負の影響を招く可能性があり、これは私たちの株価の下落を招く可能性がある。
特に、私たちの成功は、私たちの候補製品が対象とする遺伝病を専門的に治療する医師に依存し、彼らが開発した治療レジメンは、彼らがよく知っている、より多くの臨床データを得ることができる既存の治療方法を代替または補充するために、私たちの候補製品を使用することに関する
私たちの技術を利用して開発された遺伝子組換え製品が監督部門の許可を得ても、私たちの成功は薬品、植物と植物製品を含む遺伝子組換え製品の使用に対する大衆の受け入れ度に依存する。遺伝子組換え製品は安全に食べられない、あるいは環境に危険であると主張し、公衆の態度に影響を与える可能性がある。私たちの遺伝子組み換え製品は大衆に受け入れられないかもしれない。より厳格な政府法規または否定的な世論は、私たちの業務、財務状況、運営結果、および将来性に悪影響を与え、私たちの候補製品の開発および商業化、または私たちが開発する可能性のある任意の製品の需要を遅延または損害する可能性がある。例えば,早期の遺伝子治療試験は,他のベクターを用いた他の試験で出現した白血病や死亡例など,いくつかのよく知られている有害事象を招いている。我々の臨床試験あるいは遺伝子治療製品または競争相手製品に関する他の臨床試験における深刻な有害事象は、最終的に関連候補製品によるものでなくても、それによって生じる宣伝は、政府の監督管理の増加を招く可能性があり、公衆は私たちの否定的な見方に不利であり、私たちの候補製品のテストまたは承認は規制遅延が生じる可能性があり、承認された候補製品に対してより厳しいラベル要求を提出し、そのような任意の候補製品に対する需要が減少する可能性がある。
私たちの現在と将来の医療提供者、顧客、第三者支払者との関係は、適用されるリベート、詐欺、乱用、プライバシー、データセキュリティ、および他の医療に関する法律と法規を遵守することを要求しています。もし私たちがこれらの規定を守らなければ、私たちは規制機関の調査や行動、訴訟、巨額の罰金と処罰に直面する可能性があり、私たちの業務、名声、経営業績、財務状況、見通しは不利な影響を受けるかもしれない。
医療提供者は、医師や第三者支払者を含め、規制の承認を得た任意の候補製品を推薦·処方する上で主な役割を果たす。医療保健提供者、第三者支払者、顧客との手配は、私たちの製品のマーケティング、販売、流通の業務または財務配置と関係を制限する可能性があり、広範に適用される詐欺や乱用、その他の医療法律や法規に直面する可能性があります。バイオテクノロジー会社としては、医療サービスの転転を制御したり、連邦医療保険、医療補助、または他の第三者支払者に課金することはありませんが、詐欺や乱用、透明性、健康プライバシーと安全、ならびに患者の権利に関連する連邦および州医療法律、および同様の外国法律は現在も将来も私たちの業務に適用されています。米国以外でも、製薬会社と医療専門家との相互作用は、EU加盟国の国家反賄賂法律、国家陽光規則、法規、業界自律行為規則、医師職業行為規則など、厳格な法律によって制限されている。もし私たちがこのような規定を守らなかったり、このような規定を十分にまたは適切に遵守しなかったら、私たちは重罰を受けるかもしれない。
私たちの運営能力に影響を及ぼす可能性のある適用連邦および州医療保健法律および法規下の規制の詳細については、“企業−政府法規-付加法規”を参照されたい
これらの法律の範囲も執行も不確定であり,現在の医療改革環境下では,特に適用の前例や法規が乏しい場合には,急速な変化が生じる可能性がある。審査はまた強化され、これは医療産業のいくつかの調査、起訴、有罪判決、和解につながった。調査への対応に時間と資源がかかる可能性があり、経営陣の業務への関心を分散させる可能性がある。私たちが第三者の業務配置と適用される医療法律や法規に適合するように努力することは、多くのコストに関連することを確実にします。私たちの業務または私たちと業務の往来が予想される任意の医師または他のヘルスケア提供者またはエンティティが、適用された法律または適用法規に適合していないことが発見された場合、私たちおよび彼らは、重大な民事、刑事、および行政法執行行動を受ける可能性がありますので、“企業-政府法規-追加法規”を参照してください
さらに、EU GDPRおよびCCPAのような国内および国際プライバシーおよびデータセキュリティ法律を遵守しなければならず、これらの法律は、私たちが収集した臨床試験に関連する試験参加者のデータを含む個人データの収集、使用、開示、送信、または他の処理に適している。バージニア州、コロラド州、ユタ州、コネチカット州などの他の州でも全面的なプライバシーとデータセキュリティ法律が可決され、他のいくつかの州や連邦や地方レベルでも同様の法律が考えられている。ある司法管轄区はすでにデータ現地化と国境を越えたデータ転送法を公布しており、これは司法管轄区を越えた情報移転を更に困難にする可能性がある。個人データの国境を越えた移動に便利になる可能性のある既存のメカニズムは変更または廃止される可能性がある。特に、欧州経済圏やイギリスは、プライバシーやデータセキュリティ法が不足していると考えている国への個人データの移転を米国や他の彼らが大幅に制限している。もし私たちがヨーロッパ経済区やイギリスからの個人データの移転が合法であることを保証するための法的メカニズムを実施できない場合、私たちはより多くの規制行動、巨額の罰金、個人データの処理または移転の禁止を含む不良な結果に直面する可能性があり、巨額の費用でヨーロッパ経済地域、イギリス、または他の場所でのデータ処理能力を向上させることが要求される可能性がある。EEA、イギリス、または他の場所から個人データを送信する能力の制限は、EEAまたはイギリスでの臨床試験活動に影響を与え、CROや他の第三者と協力する能力を制限する可能性がある。これらの法規に関するより多くの情報は、“企業-政府法規-プライバシー規制”を参照されたい
プライバシーやデータセキュリティに関する私たちの義務は急速に変化しており、ますます厳しくなっており、規制の不確実性をもたらしている。これらの義務は、異なる適用および解釈によって制約される可能性があり、これらの適用および解釈は、法ドメイン間で不一致または衝突する可能性がある。このような義務を準備して履行するには私たちが多くの資源を投入する必要がある。これらの義務はまた、私たちの情報技術、システム、そしてやり方、そして私たちが依存する第三者の情報技術、システム、そしてやり方を変える必要があるかもしれない。また、私たちが努力しているにもかかわらず、私たちが依存している私たちの人員や第三者がこのような義務を履行できない可能性があり、これは私たちの業務運営やコンプライアンス状態に悪影響を及ぼす可能性があります。
私たちまたは私たちの第三者パートナーは、プライバシーまたはデータセキュリティに関連する法律、法規、政策、法律または契約義務、業界標準または規制指導意見(私たちのプライバシー、データセキュリティ、マーケティングまたは通信に関する政策、プログラムまたは措置に欠陥があることを含む)を遵守できなかったと言われており、重大な結果を招く可能性があります。これらの結果は、政府の調査および法執行行動、訴訟(階級に関連するクレームを含む)、追加の報告要件および/または監督、罰金および処罰、個人データの処理の禁止、個人データの廃棄または使用の禁止、民事および刑事責任、および否定的な宣伝を含むことができるが、これらに限定されない。これらのイベントのいずれも、業務運営中断または休止(臨床試験を含む)、個人データを処理できないことを含むが、これらの業務運営中断または休止(臨床試験を含む)を含む、当社の名声、ビジネスまたは財務状態に重大な悪影響を及ぼす可能性があります
あるいはある司法管轄区域で業務を展開し、私たちの製品を開発したり、それを商業化する能力が限られていて、任意のクレーム、問い合わせ、修正、再構成のために時間と資源をかけて私たちの業務を弁護します。
私たちに対する製品責任訴訟は、私たちが重大な責任を負い、私たちが開発する可能性のある任意の製品の商業化を制限する可能性があります。
私たちは私たちの候補製品が人体臨床試験でテストを行うことに関連する固有の製品責任リスクに直面しています。もし私たちが商業的に許可された製品を販売すれば、私たちはより大きな製品責任リスクに直面します。私たちの臨床試験に参加した被験者、患者、医療提供者、あるいは私たちの製品を使用、管理、販売した人は、私たちに製品責任クレームを出すかもしれません。もし私たちが私たちの候補製品や製品に被害を与えるクレームを自己弁護することに成功できなければ、私たちは重大な責任を招くかもしれない。是非曲直や最終結果にかかわらず、賠償責任は
•すべての候補製品や私たちが開発する可能性のある製品の需要が減少した
•臨床試験場所や試験項目全体を中止し
•私たちの名声とメディアの深刻な否定的な関心を損なう
•臨床試験参加者の脱退
•関連訴訟の巨額の抗弁費用
•臨床試験患者に大量のお金の報酬を提供します
•収入損失
•私たちのビジネス運営から管理と科学的資源を分流し
•私たちが開発する可能性のあるどんな製品も商業化できない。
私たちは現在、製品責任保険のレベルを持っていて、これは似たような場合の会社の慣例であり、予測可能なリスクを提供するのに十分な保険だと思いますが、私たちが生じる可能性のあるすべての責任をカバーするのに十分ではないかもしれません。保険範囲はますます高くなっています。私たちは可能などんな責任にも対応するために、合理的な費用や十分な金額で保険範囲を維持することができないかもしれない。もし私たちが開発している候補製品が規制部門の承認を得たら、製品の保険範囲を拡大し、商業製品の販売を含めるつもりですが、規制部門の承認を受けた製品のために商業的に合理的な商品責任保険を得ることができないかもしれません。予期せぬ副作用を有する薬物に基づく集団訴訟では,すでに多くの判決が下されている。成功した製品責任クレームまたは一連の私たちに対するクレーム、特に判決が私たちの保険範囲を超えた場合、私たちの現金を減少させ、私たちの業務に悪影響を及ぼす可能性があります。
不利な世界経済状況は私たちの業務にマイナスの影響を与える可能性があり、これは私たちの業務、財務状況、運営結果、将来性、私たちの普通株の市場価格に実質的な悪影響を及ぼす可能性がある。
政治的不安定と衝突(ロシアとウクライナ間の持続的な衝突を含む)は、米国と他の国の金融不安定や経済状況の普遍的な低下、および新冠肺炎などの一般的な健康危機による経済挑戦は、大口商品価格の大幅な変動、信用と資本市場の不安定、およびサプライチェーンの中断を含む市場混乱を招き、これらは全世界の記録的なインフレを招いた。これらのマクロ経済要素は、私たちの業務、運営、経営結果、財務状況、および私たちの普通株の価格と、必要に応じて許容可能な条件で追加資本を調達する能力に悪影響を及ぼすかもしれない。有利な条件で任意の必要な融資をタイムリーに得ることができなければ、我々の株価に実質的な悪影響を及ぼす可能性があり、臨床開発計画の延期または放棄を要求する可能性がある。さらに、これらのすべての影響は、私たちと私たちの協力者のサプライチェーンを乱す可能性があり、私たちと私たちの協力者が私たちの候補製品が進行しており、将来の臨床試験の能力に悪影響を及ぼす可能性がある。軍事行動、制裁、それによる経済、市場、その他の混乱の程度や持続時間は予測できないが、巨大かもしれない。このような中断のいずれも、本明細書で説明される他のリスクの影響を増幅する可能性がある。
私たちの財務に関するリスクは
設立以来、重大な運営損失が発生しており、予測可能な未来に損失を被っていくことが予想される。
1995年に運営を開始して以来、私たちは運営赤字を発生させた。私たちの未来の損失の程度と利益の時間は不確定で、私たちは予測可能な未来に損失が出ると予想しています。私たちは最初から私たちのZF技術の開発に取り組んでいますが、これはすでに大量の研究開発支出を必要とし続けています。これまで、私たちの資金は、株式証券の発行、協力協定からの収入、私たちの技術の非治療応用における他の戦略的パートナー関係、連邦政府の研究支出、付与された贈与から来ています
研究財団が提供しています私たちが引き続き私たちの候補製品を開発することに伴い、今後数年以内に追加の運営損失が発生することが予想されます。大量の製品収入が発生し、利益を達成するのに要する時間が現在予想されているよりも長い場合、あるいは株式融資や他の資金源を通じて流動性を発生させることができない場合、私たちは私たちの業務を縮小または一時停止させることを余儀なくされる可能性がある。
私たちは私たちの運営計画を実行し、継続的に経営する企業として運営するために多くの追加資金が必要になるだろう。私たちは割引条項でもっと多くの資金を集めることができないかもしれません。もしあれば、これは私たちの技術と候補製品を開発する能力を損害したり排除したりして、私たちの計画の一部または全部を延期または終了するかもしれません。将来の株式証券の売却と発行はまた、私たちの株主の株式を大幅に希釈する可能性がある。
設立以来、深刻な運営赤字とマイナス運営キャッシュフローが発生しており、まだ利益を達成していない。インフラや研究·製品開発活動の拡大に伴い,今後数年間の資本支出と運営支出が増加すると予想される。2022年12月31日までに、私たちの利用可能な現金、現金等価物、および有価証券は、現在計画されている運営に資金を提供するのに十分であると信じていますが、本年度報告書10-K表の合併財務諸表の発表日から今後12ヶ月、私たちの将来の生存能力は、私たちが大量の追加資本を集めて私たちの運営に資金を提供する能力にかかっています。既存の現金、現金等価物、および有価証券が私たちの運営に資金を提供し続けることができるという私たちの推定は、間違っていることが証明される可能性があるという仮定に基づいており、私たちは現在予想されているよりも早く利用可能な資本資源を使用するかもしれない。さらに、変化する状況-そのいくつかは私たちの制御を超えているかもしれない--私たちの資本消費速度は私たちの現在の予想よりも大きく速く、私たちは計画よりも早く追加資金を求める必要があるかもしれない。
私たちが経営を続ける能力があるかどうかに対する人々の疑いを軽減するために、私たちは私たちの運営に資金を提供するために大量の追加資本を調達することを要求される。このような点で、私たちは、公共またはプライベート·エクイティまたは債務融資、特許使用料融資、または戦略的協力のような他のソースを含むより多くの資本を積極的に求めている。しかし、私たちは追加的な資本を得ることができないかもしれないし、条件は受け入れられる、あるいは根本的にはできない。もし私たちが十分な資金をタイムリーに得ることができない場合、あるいは十分な資金が全くない場合、運営費用の削減、延期、縮小、停止、または私たちの研究·開発活動の変更など、私たちの流動性需要を満たすための追加の行動を取ることが要求されます。
私たちが公開または私募株式発行によってより多くの資本を調達すれば、Jefferies LLCとの市場発売計画による販売を含めて、私たちの既存株主の所有権権益は希釈され、この希釈は巨大である可能性があり、任意の新しい株式証券の条項は、私たちの普通株式よりも優先し、私たちの普通株よりも高い権利を含むかもしれない。もし私たちが特許使用料融資または他の協力、戦略連合、または第三者との許可手配によって追加資本を調達する場合、私たちは私たちの候補製品、技術、将来の収入流、または研究計画にいくつかの価値のある権利を放棄するか、または有利ではないかもしれない条項で許可を付与する必要があるかもしれない。もし私たちが債務融資を通じて追加資本を調達すれば、私たちは特定の金融契約や契約の制限を受けたり、追加債務を招いたり、資本支出を行ったり、いくつかの取引を行うなど、特定の行動をとる能力を制限することができ、いずれも候補製品を商業化したり、企業として運営する能力を制限したりする可能性がある。
さらに、私たちが独自の人間療法に重点を置く場合、私たちの候補製品に対するFDAや他の同様の外国規制機関の規制承認を求める必要があり、この過程は各製品のコストを数億ドル以上にする可能性がある。新興生物科学技術会社の株式市場の変動や、米国や海外の全体的な経済·市場状況など、我々がコントロールできない外部要因により、資本市場に参入する際に困難に直面する可能性がある。例えば、我々の追加資本調達能力は、最近経験した状況のような世界経済状況および米国と世界各地の信用および金融市場の中断および変動の悪影響を受ける可能性があり、一部の原因は新冠肺炎疫病とロシアとウクライナの間の持続的な衝突の影響である。私たちは私たちが受け入れられる条項で融資を受けることができるかどうか、あるいは根本的にできないかどうかを確認することができない。もし私たちが適時に十分な資金を得ることができなければ、私たちの業務および候補技術や製品を開発する能力に悪影響を及ぼすだろう。
私たちは純営業損失を利用して将来の課税収入を相殺する能力が制限されるかもしれません。
私たちが繰り越した一定額の連邦純営業損失(ただしパーセント制限)ですが、私たち連邦と全州が繰り越したかなりの部分の純営業損失はそれぞれ2024年と2029年から満期になります。使用しなければ。満期繰越の純営業損失は未使用期限が切れる可能性があり、将来の所得税負債を相殺することができない。また、改正された1986年の国内税法第382条及び383条、及び州法の規定によると、ある会社が“所有権変更”を経験した場合、これは通常、3年間の間にその持分所有権価値の変化が50ポイントを超えると定義され、同社は変更前の純営業損失の繰越及び他の変更前の税収属性を用いて変更後の収入又は税収を相殺する能力が限られている可能性がある。私たちは過去に所有権の変化を経験して、未来に私たちはもっと多くの所有権の変化を経験するかもしれません。これは私たちの株式の所有権がその後変化した結果です
これは私たちがコントロールできることではないかもしれない。もし所有権変更が発生したら、私たちの純営業損失の繰越能力を使用して実質的に制限されて、これは私たちの未来の経営業績を損なうことになります。それは実際に私たちの未来の納税義務を増加させます。また、州レベルでは、一定期間使用を一時停止したり、純営業損失の繰越を制限したりする可能性があり、州政府の課税額を加速または永久的に増加させる可能性がある。したがって、課税収入の純額を稼ぐと、私たちの純営業損失の全部または大部分の繰越と他の税務属性を使うことができないかもしれません。これは私たちの将来の納税義務を増加させ、私たちの将来のキャッシュフローに悪影響を及ぼす可能性があります。
証券や業界アナリストが我々の業務に関する研究報告を発表しない場合、あるいは不正確または不利な研究報告を発表しなければ、私たちの株価や取引量は低下する可能性がある。
私たちの普通株の市場価格は、証券や業界アナリストが発表した私たちまたは私たちの業務に関する研究と報告にある程度依存するだろう。もし私たちの証券や業界アナリストが私たちの株式格付けを引き下げたことを報道したり、私たちの業務の不正確または不利に関する研究報告を発表したりすれば、私たちの株価は下落する可能性がある。これらのアナリストのうちの1人以上がわが社への報道を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちの株に対する需要が減少する可能性があり、これは私たちの株価や取引量を低下させる可能性がある。
私たちの第三者への依存に関するリスクは
もし私たちの請負業者、協力者、または他のビジネスパートナーと衝突すれば、これらの衝突は私たちの戦略を実施する能力を制限し、私たちの業務と将来性を損なう可能性があります。
私たちの請負業者、協力者、または他の業務パートナーと衝突すれば、相手は自分の利益で行動する可能性が高く、戦略を実施する能力を制限する可能性があります。例えば、私たちのいくつかの協力者は、彼らが私たちと協力している各分野で複数の製品開発を行っている。私たちの協力者は単独でまたは他の人と一緒に関連分野の候補製品を開発することができ、これらの候補製品は彼らが私たちと協力している候補製品と競争力を持っている。競合製品は、協力者によって開発されたものであっても、協力者によって開発されていても、権利を有するものであっても、我々の候補製品に対する支援を撤回させる可能性がある。
私たちのいくつかの協力者たちはまた未来に私たちの競争相手になるかもしれない。私たちの協力者は競争製品を開発または投資し、私たちがその競争相手との協力を阻止し、適時に規制の承認を得ることができず、意外にも早期に終了したり、彼らと私たちとの合意に違反したり、協力がカバーする候補製品の開発と商業化に十分な資源を投入することができなかった。
さらに、私たちと私たちの協力者との間には、適用プロトコルの下での私たちまたは私たちの協力者または戦略的パートナーに関する表現によって紛争が生じる可能性があり、私たちと私たちの協力者との合意違反の疑いによる論争が生じる可能性があります。
これらの紛争のいずれも、私たちの製品開発努力を損なうか、あるいは私たちの業務や将来性に悪影響を及ぼす可能性があります。
私たちの協力者は私たちのいくつかの臨床試験を含めて、私たちの製品開発のいくつかの側面を制御しています。これは私たちの候補製品の商業化に予期せぬ遅延と他の障害を引き起こす可能性があります。
著者らは著者らのいくつかの候補製品の設計といくつかの臨床試験を協力者に依存している。したがって,これらの臨床試験はわれわれの希望する方式やスケジュールでは行われない可能性があり,我々の製品開発に悪影響を及ぼす可能性がある。例えば,ファイザーは原癌遺伝子fitelparvovec第3段階アフィン試験の試験スポンサーであり,我々はファイザーの努力により,第3段階アフィン試験の臨床棚上げを解除し,試験を回復することを求めている。アフィン試験の投与量は現在回復しているが,この試験で将来の遅延に遭遇しないことや,試験が予想される時間枠内で完了するか,あるいは全く保証されないことは保証されない。
私たちはノワール、Biogen、Kite、武田、ファイザーとの合意で製品開発の制御が不足しており、これは私たちの候補製品の開発と商業化の遅延や他の困難を招く可能性があり、これは私たちが期待したINDファイルを適時に完成させることを阻止し、合意によって任意のマイルストーン、特許権使用料、他の福祉を得ることができるかもしれない。また、私たちのそれぞれの合意によると、私たちの第三者パートナーは、合意を終了することを事前に通知する権利がありますので、これらの合意によって受信可能な実際のマイルストーン支払いは、これらの合意によって規定されるすべての金額を大幅に下回る可能性があります。
我々のZF技術を認可したパートナーは、代替技術または製品を採用することを決定するか、または私たちのZF技術を使用して商業的に可能な製品を開発することができないか、またはそれを使用することができないかもしれません。これは、私たちの収入およびZF技術を使用する候補製品を開発する私たちの戦略に負の影響を与えます。
私たちのいくつかの協力は私たちのZF技術プラットフォームを利用した。これらの協力者は将来的に代替技術を採用することを選択する可能性があり、これは私たちのZF技術プラットフォームの価値を低下させ、そのプラットフォームを使用する候補製品の開発を阻害する可能性がある。また、私たちの多くの協力者は1つ以上の開発プロジェクトに従事しているかもしれないので、彼らは彼らの資源を私たちと協力しているプロジェクト以外のプロジェクトに移すことを選択することができる。もし…
もし彼らがそうすれば、これは私たちがZF技術プラットフォームの能力をテストして開発することを延期し、プラットフォームを使用する候補製品の開発を延期または終了するだろう。さらに、私たちの協力者は、私たちの協力によって生成された候補製品を開発しないか、またはこれらの候補製品の開発、製造、マーケティング、または販売に十分な資源を投入しないことを選択することができる。もし彼らが私たちとの協力を中止し、私たちは候補製品を引き続き開発したいなら、私たちは他の協力者の支持や自分で製品を開発することを要求されるだろう。適切なパートナーを決定したり、有利な連携プロトコルを協議することができない可能性があり、私たちの内部には十分な資源や専門知識がない可能性があり、これらの候補製品を開発し続けることができないかもしれない。
私たちの技術の商業化は他社との協力にある程度依存するだろう。もし私たちが未来に協力者が見つからなければ、あるいは私たちの協力者が製品開発に努力しなければ、私たちは私たちの技術や候補製品を開発できないかもしれません。これは私たちの成長を減速させ、私たちの普通株の市場価値を下げるかもしれません。
私たち自身は私たちの候補製品の承認を得て商業化するための十分な開発、規制部門の資金がない。私たちは、臨床前研究と臨床テストを含む、他の生物製薬会社との協力に大きく依存して、私たちの研究と開発努力に資金を提供し、このような協力に大きく依存して、私たちの候補製品の商業化に必要な長い規制承認過程に資金を提供することが予想される。
例えば、我々はノワール社と協力して、自閉症および知的障害を含む特定の神経発達障害の治療のための候補製品を開発し、アルツハイマー病、アルツハイマー病などのα-シヌクレインに関連する疾患および他の神経系疾患の治療のための生物遺伝研究会社と協力して候補製品を開発し、Kiteと協力して癌を治療する候補製品を開発し、ファイザー社と協力してC 9 ORF 72遺伝子変異に関連する血友病Aおよび筋萎縮性側索硬化症および前頭側頭葉変性の治療のための候補製品を開発した。
2022年6月、これまでの協力協定により、セノフィ社(Sanofi S.A)の権利と義務の受け渡しが完了しました。我々がSCDを治療する候補製品BIVV 003の1/2期PRECIZIN−1研究を完成させたいが,この研究をタイムリーまたは根本的に達成できない保証はない。また,我々のSCD計画では追加的な実質的な投資は行われないと予想されるため,本研究が完了した後,BIVV 003の開発を継続するつもりはない.BIVV 003の開発を推進するための潜在的なパートナーを探しているにもかかわらず、タイムリーで許容可能な条項で、またはこのような協力を確実にすることができない保証はありません。この場合、BIVV 003の継続開発はさらに延期または完全に排除される可能性があり、この場合、BIVV 003計画を停止することが選択される可能性がある。この計画のどのようなさらなる遅延または中断も、私たちの業務、運営結果、財務状況、および見通しに悪影響を及ぼす可能性があります。
もし私たちがより多くの協力を得ることができない場合、あるいは私たちの協力者が私たちの候補製品の開発、規制承認、商業化を推進するために努力できない場合、私たちの成長は減速し、私たちの技術や候補製品の開発のために資金を作る能力に悪影響を及ぼすかもしれない。また,我々の協力者は,事前にほとんど通知されずに開発計画を再許可したり放棄したりしたり,我々の協力者と食い違いやトラブルが発生したりする可能性があり,関連製品の開発の減速や停止を招く可能性がある.また、私たちの協力者の業務や運営は、再編、買収、その他の戦略取引によって大きく変化する可能性があり、これらの取引は、彼らが私たちの計画を進める能力に悪影響を及ぼす可能性があります
典型的な協力では、特定のマイルストーンの成果に基づく候補製品研究開発収入と、任意の商業化製品の売上パーセントに基づく印税を獲得したい。このようなマイルストーンを達成することは私たちの協力者の努力にある程度かかっており、私たちはこのような努力と私たち自身の努力をコントロールすることができない。さらに、業務合併、協力者業務戦略の変化、および財務的困難または他の要因は、協力者が協力者との連携協定に含まれる任意の候補製品の開発を放棄または遅延させる可能性がある。例えば,サイノフェイはBIVV 003に関する権利や義務を返納してくれたことや,サイノフェイは便宜上我々のこれまでの協力合意を終了し,これまでサイノフィの戦略方向が変化し,自己個人化細胞療法ではなく同種異体汎用ゲノム医学的手法に重点を置いてきた。さらに、もし私たちが失敗した場合、または任意の協力パートナーが特定のマイルストーンに到達できなかった場合、協力協定は終了される可能性があり、これは、その協力協定に基づいて任意の追加のマイルストーン支払いを稼ぐことができず、私たちの収入を減少させるだろう。また、協力候補製品の開発に成功し、関連規制部門のマーケティング承認を得ても、商業化製品の販売が期待に達していなければ、予想を下回る印税を得ることが可能である。いずれの場合も、私たちの協力に関連するマイルストーンと印税支払い機会は、実現するために大きなリスクに関連しており、永遠に得られないかもしれない。それに応じて, 投資家は私たちの協力によって提供されたすべての潜在的なマイルストーン支払いを受けると仮定してはならず、私たちはさらなる重大なマイルストーン支払いや私たちの協力によって支払われたいかなる特許権使用料も決して受け取っていないかもしれない。
私たちの知的財産権に関するリスクは
私たちの技術および候補製品のための特許保護の取得、維持および実行は困難であり、時間がかかり、高価であり、第三者は私たちと同様の発明をしている可能性があるので、私たちの技術および候補製品に最適な特許保護を提供することを保証することができないかもしれない。
私たちのビジネス成功は、私たちの技術および候補製品の特許保護を獲得、維持、実行することにある程度依存し、私たちが挑戦を受ける可能性のある任意の特許を保護することに成功するかもしれない。生物製薬特許の取得、維持および実行は高価で、時間がかかり、複雑であり、私たちはすべての所望の司法管轄区域ですべての必要または望ましい特許出願を提出して起訴することができないか、または合理的なコストまたはタイムリーまたは根本的に維持、実行および許可できない任意のそのような特許出願から発行される可能性のある特許を提出し、起訴することができないかもしれない。我々は,特許保護を得るのが遅くなるまで,我々の研究開発成果で特許を申請できることを確認できない可能性もある.生物製薬会社の特許地位は非常に不確定である可能性があり、複雑な法律と事実問題に関連する可能性がある。バイオテクノロジー特許で許容される特許請求の範囲の広さについては,これまで一貫した政策は現れていない.さらに、将来の特許法律、法規、規則、および裁判所判決は、私たちの現在および未来の特許権利要件の範囲、有効性、実行可能性、および関連する救済措置に影響を与える可能性がある。したがって,我々が所有または許可している任意の特許出願が発行される可能性のあるクレームの広さを予測することはできず,第三者特許が我々の候補製品や技術に関連したクレームを発行する可能性があるかどうかも予測できない.特許が確実に発行されても、これらの特許が私たちの技術および候補製品をカバーしていても、第三者はその有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、このような特許が縮小され、無効にされ、または実行不可能とみなされる可能性がある。私たちの特許や特許出願に関連するすべての潜在的に関連する既存技術が発見されたことを保証することはできない, その存在は、特許を無効にするか、または係属中の特許出願から特許が発行されることを阻止する可能性がある。さらに、サードパーティも同様の発明を行った場合、それらは、様々な方法で私たち自身のアプリケーションのカバー範囲に影響を与える可能性がある。
私たちは様々なライセンス契約の当事者であり、これらのライセンスプロトコルは私たちに特定の特許と特許出願の権利を付与します。我々も様々なライセンス契約の一方であり,これらの合意に基づいて,第三者に特定特許及び特許出願の下での権利を付与する。私たちの現在のライセンスには履行義務が含まれている。もし私たちがこのような義務を履行できなければ、許可証は終了されるかもしれない。もし私たちが商業的に合理的な条項でこれらの技術を許可し続けることができなければ、私たちは私たちの製品開発と研究活動のいくつかの側面を延期または終了させることを余儀なくされるかもしれない。
我々は,我々の内部開発した知的財産権行使のように,我々が第三者から権限を与えた知的財産権を同程度に制御することはできない.私たちは私たちが第三者から許可されたいくつかの特許出願の起訴を統制しない;したがって、特許出願は私たちが望むように、またはタイムリーに起訴されないかもしれない。
未来の私たちの所有権の保護の程度は不確定で、私たちは保証できない
•私たちまたは私たちの許可者は、私たちのすべての未解決特許出願がカバーする発明を実施することを最初に考え、および/または減少させる人である
•私たちまたは私たちのライセンシーは最初にこれらの発明のために特許を提出しました
•他人の特許は私たちの経営能力に悪影響を与えない
•他の会社は、類似または代替技術を独立して開発したり、私たちの任意の製品、プロセス、または技術を逆工程したりしない
•私たちの任意の未解決特許出願は発行された特許をもたらすだろう
•私たち、私たちの協力者、または戦略パートナーに発行または許可された任意の特許は、商業的に実行可能な製品に基礎を提供するか、または任意の競争優位性を提供してくれるだろう
•私たちに発行されたり許可されたいかなる特許も第三者の疑問と無効にされない
•アメリカと外国の法律、法規、規則、または裁判所の判断は、私たちの特許権を修正したり、特許権を実行または維持する能力に影響を与えると解釈したりしません
•私たちは特許を申請できるより多くの製品、プロセス、または技術を開発するつもりだ。
他の会社はすでに特許出願を提出しており、将来的にも私たちのような特許出願を提出するかもしれない。亜鉛フィンガー,TALE,CRISPR/CA,その他のDNA結合タンパク質を用いた技術を開発しようとしている学術団体や他の会社が知られており,これらの団体や会社が特許出願を提出している。私たちは現在、主張された発明を使用する計画はないにもかかわらず、この技術に対するいくつかの特許が発行されている。これらまたは他の特許出願が特許として発行された場合、任意の特許またはこれらの出願について付与された特許の所有者は、損害賠償を要求し、損害賠償を請求するために、私たち、協力者または戦略パートナーに侵害訴訟を提起する可能性がある
影響を受けた製品やプロセスに関する研究,開発や商業活動の禁止が求められている。結果がどうであっても、訴訟を提起する費用は巨大かもしれない。しかも、私たちは私たち、私たちの協力者、または戦略的パートナーがどんな行動で勝つか予測できない。さらに、関連する特許主張が有効かつ実行可能に維持され、私たちの製品または方法が1つまたは複数の特許を侵害していることが発見された場合、私たちまたは私たちの協力者は、意図的に侵害された3倍の損害賠償および弁護士費を含む大量の損害賠償を支払わなければならない可能性があり、私たちは、私たちまたは私たちの協力者が許可を得ることができない限り、または特許主張をバイパスして設計することができる限り、製造、使用、販売、販売または関連製品または方法を米国に輸入することを阻止される可能性がある。私たちまたは私たちの協力者が商業的に合理的な条項でそのような許可を得ることができることを保証することはできませんし、関連する特許請求の範囲を中心に設計に成功する保証はありません。ゲノム学や細胞治療業界では、特許や他の知的財産権に関する重大な訴訟がある可能性があり、これはコストが高く、冗長で、注意力を分散させる訴訟に直面する可能性があり、結果は予測できない
私たちは私たちが特許保護に適していないと思う技術を商業秘密に依存して保護する。しかし、商業秘密は保護することが難しい。従業員、学術協力者、コンサルタントにセキュリティ協定を締結することを要求していますが、私たちのビジネス秘密や他の固有情報を十分に保護したり、これらのセキュリティ協定を実行したりすることはできないかもしれません。
私たちの協力者、戦略的パートナー、科学コンサルタントは、私たちが得る権利があるかもしれないデータと情報を発表する権利がある。もし私たちが私たちの技術と私たちの協力および戦略的パートナーシップに関連する他の機密情報を秘密にすることができなければ、私たちは特許保護や私たちの固有の情報を保護することができないかもしれない。
特許条項は候補製品での私たちの競争地位を十分に保護するのに十分ではなく、管轄区域によって異なる可能性があるかもしれない。
特許の寿命は限られている。米国では、特許の自然失効時間は、通常、その最初の米国非一時出願日または対応する国際出願の出願日から20年である。この予想された期限を延長するための様々な方法が利用可能かもしれない。とにかく、特許の有効期間と提供される保護は限られている。私たちの候補製品をカバーする特許を取得しても、製品の特許有効期限が切れると、私たちは模倣薬からの競争に直面するかもしれない。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの技術的価値は不利な影響を受ける可能性があり、私たちの業務も損害を受けるかもしれない。
我々は、特許を出願できない独自技術、特許を強制的に実行することが困難な方法、および私たちの候補製品発見および開発中に特許がカバーされていない独自技術、情報または技術に関する任意の他の要素を選択するために、商業秘密保護および秘密協定に依存して保護する。しかし、商業秘密は保護するのが難しいかもしれない。私たちは、従業員、コンサルタント、科学コンサルタント、協力者、パートナー、請負業者と秘密保護協定を締結することで、私たちのノウハウとプロセスを保護することを求めています。また、私たちは私たちのビルの実体安全と、私たちの情報科学技術システムの実体と電子安全を維持して、私たちの資料と商業秘密の完全性とセキュリティを維持することに力を入れています。私たちはこのような個人、組織、そしてシステムに自信がありますが、合意や安全措置は未来に違反される可能性があり、私たちはどんな違反に対応するのに十分な救済措置がないかもしれません。また、“私たちの情報技術システムまたはデータまたは私たちが依存する第三者のシステムまたはデータが損害を受けた場合、私たちは、規制調査または行動、訴訟、罰金および処罰、私たちの業務運営中断および名声損害を含むが、規制されていないが、これらに限定されない不利な結果を経験する可能性がある”と題するリスク要因を見る。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。
私たちのすべての従業員とコンサルタントが彼らの発明を私たちに譲渡することを望んでいますが、私たちのすべての従業員、コンサルタント、コンサルタント、協力者、パートナー、および私たちのノウハウ、情報、または技術にアクセスできる第三者は、秘密協定を締結するために、このようなすべての合意が正式に実行されているか、または私たちのビジネス秘密や他の機密ノウハウが漏洩しないこと、または競争相手が他の方法で私たちのビジネス秘密や独立開発と実質的に同等の情報および技術を得ることができないことを保証することはできません。私たちのビジネス秘密を流用したり不正に開示したりすることは、私たちの競争地位を損なう可能性があり、私たちの業務に悪影響を及ぼす可能性があります。また,我々のビジネス秘密を守るための手順が不十分であると考えられれば,第三者による商業秘密の流用に対抗する十分な追跡権がない可能性がある.しかも、他の人たちは私たちの商業秘密と固有の情報を独立して発見するかもしれない。
また、一部の外国の法律の専有権に対する保護程度や方式は米国の法律とは異なる。したがって、私たちは知識人たちを保護して守ることで重大な問題に直面するかもしれない
アメリカと海外での財産です。もし私たちが私たちの技術に関連する非特許知的財産権の第三者への実質的な開示を阻止できず、私たちがこのような強制的に実行可能な商業秘密保護を持っている保証がなければ、私たちは私たちの市場で競争優位性を確立したり、維持することができないかもしれません。これは私たちの業務、運営結果、および財務状況に悪影響を及ぼすかもしれません。
私たちは、買収およびライセンス内の許可によって、私たちのプロセスを開発するために必要な製品コンポーネント、プラットフォーム、プロセスの権利を取得または維持することができないかもしれません。
現在、私たちは、第三者の許可と私たちが持っている特許を通じて、私たちの遺伝子と細胞治療製品候補製品を開発する知的財産権を持っていると信じています。我々の計画は、TX 200および潜在的な将来のCAR−Treg療法のような他の候補製品に関連する可能性があり、第三者が保有する独占権を使用する必要がある可能性があるので、私たちのビジネスの成長は、これらの独占権を取得、許可、または使用する能力にある程度依存する可能性がある。さらに、私たちの候補製品は、効率的かつ効率的に動作するために特定の配合物を必要とする可能性があり、これらの権利は他の人によって保持されている可能性がある。私たちは商業的に合理的な条項で第三者から得ることができないかもしれませんが、私たちが決定した任意の成分、使用方法、プロセス、または他の第三者知的財産権。第三者知的財産権のライセンス及び買収は競争分野であり、一部の成熟した企業も、第三者知的財産権を許可又は買収するための戦略をとっており、魅力的であると考えられる他の企業や学術機関を含む。他社はその規模、現金資源、そしてより強い臨床開発と商業化能力のため、私たちより競争優位を持っているかもしれない。私たちが望む知的財産権が他の会社に許可されると、私たちはそのような権利を得る許可から除外されるかもしれない。
しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちの投資が適切な見返りを得るための条項の許可や第三者知的財産権を得ることができないかもしれない。もし私たちが必要な第三者知的財産権を得ることに成功しなければ、私たちの業務、財務状況、成長の見通しは影響を受ける可能性がある。
もし私たちが第三者に知的財産権を許可する合意の義務を履行できなかった場合、あるいは私たちとライセンス者との業務関係が妨害された場合、私たちは私たちの業務に非常に重要な許可権を失う可能性があります。
私たちは、私たちの業務に非常に重要な知的財産権ライセンス協定の多くの締約国であり、将来的により多くのライセンス契約を締結したいと思っています。私たちの既存の許可協定は、私たちの未来の許可協定が私たちに様々な職務調査、マイルストーン、特許権使用料、その他の義務を課すと予想している。もし私たちがこれらの合意の下で私たちの義務を履行できなかった場合、あるいは私たちが破産に直面した場合、許可側は許可を終了する権利があるかもしれません。この場合、許可がカバーする製品を販売することができません。
私たちは第三者から許可を得て、私たちの研究を進めたり、私たちの候補製品の商業化を許可する必要があるかもしれません。私たちはよくそうします。私たちはもしあれば、合理的な費用または合理的な条項でこのような許可証のいずれかを得ることができないかもしれない。この場合、私たちは代替技術を開発または許可するために多くの時間と資源を必要とするかもしれない。もし私たちがそれができなければ、影響を受けた候補製品を開発したり商業化することができないかもしれません。これは私たちの業務を深刻に損なう可能性があります。現在の候補製品や将来の製品に対して強制的に執行される可能性のある第三者特許が存在しないことを保証することはできません。それによって、私たちの販売を禁止したり、私たちの販売に関しては、第三者に印税および/または他の形態の賠償を支払う義務があります。
多くの場合、私たちが許可した技術の特許訴訟は完全に許可者によって統制されている。もし私たちのライセンス者が彼らから私たちが許可した独自知的財産権の特許や他の保護を獲得し、維持できなかった場合、私たちは知的財産権に対する私たちの権利やこれらの権利に関する排他性を失う可能性があり、私たちの競争相手はこれらの知的財産権を使用して競争製品をマーケティングするかもしれない。場合によっては、私たちは許可技術によって作られた特許の起訴を統制する。もし私たちがこのような起訴に関連したいかなる義務にも違反したら、私たちは私たちの許可パートナーに重大な責任を負うかもしれない。知的財産権許可は私たちの業務に重要であり、複雑な法律、商業と科学問題に関連し、そして私たちの業界の科学発見の迅速な発展によって複雑になった
我々は現在、第三者から知的財産権や技術許可を得るプロトコルは複雑であり、このようなプロトコルのいくつかの条項は様々な解釈の影響を受ける可能性がある。可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼす可能性があります。私たちが許可している知的財産権をめぐる紛争が許容可能な条項で現在の許可手配の能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた候補製品の開発や商業化に成功できない可能性があり、これは私たちの業務、財務状況、運営結果、見通しに悪影響を及ぼす可能性がある。例えば、Sangamo Franceはブリティッシュコロンビア州大学(UBC)からTX 200でこの車を独占的に使用する権利を獲得した。UBCが本ライセンス契約を終了すれば、適切な車を開発または購入しなければならないかもしれません。これは私たちのを延長します
予想される開発スケジュールと増加した費用は、フランスのSangamo買収の期待的なメリットを実現できない可能性がある。
私たちは、私たちがコントロールしている特許または第三者が侵害特許を主張する特許または知的財産権訴訟または同様の紛争に巻き込まれる可能性があり、これらの訴訟は、高価で時間がかかり、開発および商業化活動を損害または阻止する可能性がある。
私たちの商業的成功は私たちが第三者の特許と固有の権利の侵害を避けることにある程度かかっている。米国国内外において、生物技術と製薬業界には特許とその他の知的財産権に関連する訴訟が大量にあり、特許侵害訴訟、宣言的判決訴訟、無効訴訟、妨害、異議、一方的或いは当事双方の再審、付与後再審、及び米国特許庁及び対応する外国特許庁に提起された当事審査手続を含む。我々が開発候補を求めている分野には,第三者が所有する米国や外国から発行された特許や係属中の特許出願が多く存在する.バイオテクノロジーや製薬業界の拡張や特許の発行に伴い、我々の候補製品が第三者特許権侵害の告発を受けるリスクが高まる可能性がある。
第三者は、私たちが許可されていない場合に彼らのノウハウを使用していると断言するかもしれないが、これらの当事者は、彼らがより多くの資源を持っているので、複雑な特許訴訟の費用を私たちよりも効果的に受けることができる。我々の候補製品の使用または製造に関連する第三者特許または材料、配合、製造方法または治療方法の請求項に記載の特許出願が存在する可能性がある。例えば、第三者が保有するいくつかの特許は、いくつかのキャリアおよびキャリア製造方法に関連しており、これらの特許は、私たちのいくつかの候補製品に関連していることが知られている。私たちはまだ私たちの候補製品のために商業規模の製造プロセスを決定していない。私たちの商業規模製造プロセスがこれらのベクトル製造方法を使用し、これらの第三者特許が商業化時に有効かつ有効である場合、これらの特許に挑戦し、非侵害代替製品を使用または開発するか、またはこれらの特許の許可を求める必要があるかもしれない。いずれの場合も、管轄権のある裁判所が、私たちの候補製品、製造過程で形成された任意の分子または任意の最終製品自体をカバーするために、任意の第三者特許を保有している場合、そのような特許の所有者は、適用特許によって許可されたか、またはそのような特許が満了するまで、その候補製品を商業化する能力を阻止または阻害することができるかもしれない。同様に、管轄権のある裁判所が、私たちの処方、製造プロセス、または使用方法のあらゆる面をカバーするために、任意の第三者特許を持っている場合、そのような特許の所有者は、許可証を取得しない限り、またはそのような特許が満了するまで、私たちが適用可能な候補製品を開発および商業化する能力を阻止することができるかもしれない。また、特許出願は発表まで数年かかる可能性があるため, 現在処理されている特許出願が存在する可能性があり、これらの出願は、私たちの候補製品が発行された特許を侵害する可能性がある。
場合によっては、第三者は、競争相手でなくても、関連したビジネスであっても、私たちが彼らの特許または他の固有の権利を侵害していると主張するかもしれない。これらの訴訟人は、高価で時間のかかる訴訟ではなく、私たちから和解金を得ることを目的とした侵害訴訟や訴訟の脅威を提起するだろう。
これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、独自の情報を暴露し、私たちの業務における従業員資源を大量に移転する可能性がある。もし私たちに対する侵害クレームが成功した場合、私たちは故意に侵害された3倍の損害賠償と弁護士費、特許権使用料の支払い、私たちの侵害製品の再設計、または第三者から1つまたは複数の許可を得ることを含む大量の損害賠償を支払わなければならないかもしれないし、多くの時間とお金の支出が必要かもしれない。
競争相手はまた私たちの特許や私たちの許可側の特許を侵害するかもしれない。権利侵害や不正使用に対抗するために、私たちは費用が高く時間がかかるかもしれない侵害請求を要求されるかもしれない。さらに、侵害訴訟では、裁判所は、私たちまたは私たちのライセンシーの特許が無効であり、全部または部分的に強制的に実行できないと判断するか、または私たちの特許が関連技術をカバーしていないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。任意の訴訟または弁護手続きにおける不利な結果は、私たちの1つまたは複数の特許を無効が宣言されるか、強制的に実行できないか、または狭い解釈されるリスクに直面させる可能性があり、私たちの特許出願を発表できないリスクに直面させる可能性がある。さらに、私たちまたは私たちの許可パートナーのうちの1つが第三者に対して法的訴訟を提起して、私たちの候補製品のうちの1つをカバーする特許を強制的に執行する場合、被告は、私たちの候補製品をカバーする特許の無効および/または強制執行ができないことを反訴することができる。このような訴訟は私たちの特許がこれ以上私たちの候補製品をカバーしないように撤回したり修正したりするかもしれない。法的に無効と実行不可能と断言された後の結果は予測できない。例えば、有効性の問題については、私たちは無効な以前の技術がないとは確信できないが、私たちと特許審査員は起訴中にこれを知らない。もし被告が無効および/または強制執行できない法的主張に勝った場合、私たちは私たちの候補製品の少なくとも一部またはすべての特許保護を失うだろう。このような特許保護の喪失は私たちの業務に悪影響を及ぼすかもしれない。
我々の特許または特許に関連する発明または他のリスト事項の優先度を決定するために、第三者によって引き起こされるか、または米国特許商標局によって発表された干渉または派生手続が必要である可能性がある
アプリケーションや私たちの許可側のアプリケーションです不利な結果は、私たちを重大な金銭的損失に直面させ、貴重な知的財産権の損失を招く可能性があり、関連技術の使用を停止したり、勝利者から許可権利を得ようとしたりすることが要求される。もし勝利者が商業的に合理的な条項で私たちに許可を提供しない場合、あるいは私たちに許可を全く提供しない、あるいは非独占的な許可を提供し、私たちの競争相手が同じ技術を獲得した場合、私たちの業務は損害を受ける可能性がある。私たちの訴訟、妨害、派生、または他の手続きの弁護は失敗する可能性があり、成功しても巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性がある。私たちは、特に米国のようにこれらの権利を十分に保護していないかもしれない国では、私たちの知的財産権の盗用を単独でまたは許可者と一緒に防ぐことができないかもしれない。
私たちは、私たちの従業員、コンサルタント、または独立請負業者が第三者の機密情報を誤って使用または開示したか、または私たちの従業員がその前の雇用主によって言われた商業秘密を誤って使用または開示したという疑惑の影響を受けるかもしれない。
私たちが雇った人は以前、私たちの競争相手や潜在的な競争相手を含む大学や他のバイオテクノロジーや製薬会社に雇われていた。私たちは、私たちの従業員、コンサルタント、および独立請負業者が、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちは、商業秘密または他の固有情報を含む、私たちまたは私たちの従業員、コンサルタントまたは独立請負業者が、私たちの従業員の任意の元雇用主または他の第三者の知的財産権を無意識にまたは他の方法で使用または開示しているというクレームを受ける可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭損害賠償の支払いに加えて、貴重な知的財産権や人員を失う可能性があり、これは私たちの業務に悪影響を及ぼす可能性がある。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣や他の従業員の注意を分散させる可能性がある。
私たちは遺伝子転移技術を許可できないかもしれませんが、承認されれば、私たちのZF技術と潜在製品を商業化するためにこれらの技術が必要かもしれません。
細胞中の遺伝子を調節または修飾するためには,ZFPを効率的に細胞に輸送する必要がある。我々はすでに研究しているZFPのためにAAVおよびmRNA技術を含むいくつかの遺伝子転移技術の許可を得ており、これらのシステムおよび他のZFPを細胞に送達するための技術が体外およびインビボで応用するために必要である可能性があることを評価している。私たちはまだ私たち自身の遺伝子転移技術を完全に開発しておらず、私たちは許可合意を達成する能力によって必要な遺伝子転移技術を提供する権利を提供している。私たちの方法は必要に応じて適切な技術を許可してきた。例えば、私たちが現在候補製品で使用している私たち自身のベクトル製造方法に加えて、第三者が持っているいくつかの特許は、いくつかのベクトル製造方法に関連しており、これらの方法は、現在、いくつかの候補製品で使用されていることが知られている。私たちはまだ私たちの候補製品のために商業規模の製造プロセスを決定していない。我々の商業規模製造プロセスがこれらのベクトル製造方法を使用し、これらの第三者特許が商業化時に有効である場合、非侵害製造方法を使用または開発するか、またはこれらの特許の許可を求める必要があるかもしれない。しかし,合理的な条項で遺伝子転移技術を許可することはできない可能性があり,もしあれば,我々の候補製品を開発して商業化するために必要である。合理的な商業条項でこのような技術を持つ実体と遺伝子転移技術を使用する許可証を得ることができない場合、私たちの候補治療製品の臨床前評価、薬物開発協力、臨床試験および/または商業化を延期または阻止する可能性がある。
私たちの業務運営に関するリスク
もし私たちの情報技術システムまたはデータまたは私たちが依存する第三者のシステムまたはデータが破壊された場合、私たちは、規制調査または行動、訴訟、罰金および処罰、私たちの業務運営の中断、名声被害、および他の不利な結果を含むが、これらに限定されないこのような損害によって生じる不利な結果を経験する可能性がある。
私たちはますます情報技術システムやインフラに依存して私たちの業務を運営しています。これらのシステムやインフラは膨大で複雑です。私たちの通常のビジネスプロセスでは、私たちおよび私たちが依存する第三者は、知的財産権、商業秘密、および個人データ(例えば、健康に関連する情報)を含む大量の独自、機密および敏感な情報を収集、受信、記憶、処理、生成、使用、開示、取得、保護、保護、処置、共有および送信することができる。重要なのは、私たちはこのような敏感な情報の機密性、完全性、そして利用可能性を維持するために、安全な方法でそうしなければならないということだ。我々はまた,我々の業務要素(情報技術インフラの要素を含む)を第三者にアウトソーシングするため,多くの第三者プロバイダを管理しており,我々のコンピュータネットワークや我々の機密情報にアクセスできる可能性がある.これらの第三者の多くは、彼らのいくつかの責任を他の第三者に委託またはアウトソーシングする。第三者の情報セキュリティアプローチを監視する能力は限られており、これらの第三者には十分な情報セキュリティ対策がない可能性がある
すべての情報技術業務は生まれつき無意識あるいは故意のセキュリティホール、事件、攻撃と暴露の影響を受けやすいが、私たちの情報技術の規模、複雑性、獲得性と分散性質
これらのシステムおよびこれらのシステムに格納された大量の敏感な情報は、これらのシステムが我々の技術環境に対する意図的または悪意のある内部および外部攻撃を容易に受けることを可能にする。この性質の攻撃は頻度,持続性,複雑性,強度の面で増加している.情報システムとデータに対する脅威はますます検出が困難になり、その源は多種多様であり、その出所は多種多様であり、伝統的なコンピュータ“ハッカー”、脅威行為者、“ハッカー活動家”、組織的犯罪脅威行為者、人員(例えば窃盗または乱用によって)、複雑な民族国家と民族国家によって支持される行為者を含む。一部の行為者は現在、地政学的な理由で軍事衝突と防御活動を組み合わせた民族国家行為者を含むサイバー攻撃に参加し続けると予想されている。戦争と他の重大な衝突の間、私たちと私たちが依存している第三者は、報復的なネットワーク攻撃を含むこれらの攻撃を受けやすいリスクが増加する可能性があり、これは私たちのシステム、運営、およびサプライチェーンを深刻に混乱させる可能性がある。我々と私たちが依存する第三者は、社会工学攻撃(ネットワーク釣り攻撃を含む)、悪意コード(例えば、ウイルスおよびワーム)、マルウェア(高度な持続的脅威侵入の結果を含む)、サービス拒否攻撃(証明書充填のような)、証拠取得、人員不正行為またはエラー、恐喝ソフトウェア攻撃、サプライチェーン攻撃、ソフトウェア脆弱性、サーバ障害、ソフトウェアまたはハードウェア障害、データまたは他の情報技術資産の損失、広告ソフトウェア、電気通信障害、自然災害(例えば地震·火災·洪水)·戦争, テロと他の似たような脅威。脅迫ソフトウェア攻撃はますます一般的で深刻になっており、私たちの運営の深刻な中断、データと収入損失、名声損害、資金移転を招く可能性がある。恐喝支払いは恐喝ソフトウェア攻撃の否定的な影響を軽減するかもしれないが、例えば、適用された法律または法規によってそのような支払いが禁止されているため、私たちはそのような支払いを望んでいないか、または支払うことができないかもしれない。また、新冠肺炎疫病の影響と私たちの最新の在宅勤務政策は私たちの情報技術システムへの依存を悪化させ、私たちのネットワーク安全リスクを増加させる可能性がある。なぜなら、私たちの多くの重要な業務活動は現在遠隔で行われているので、私たちの住宅地やネットワーク以外のネットワーク接続、コンピュータと設備を利用して、私たちはますます家で、途中で、公共の場所で働く人に依存している。
以前に識別されたまたは同様の脅威は、データセキュリティイベントまたは私たち、第三者プロバイダおよび/またはビジネスパートナーの情報技術システムの他の中断をもたらす可能性があり、これは、私たちの業務運営に悪影響を及ぼす可能性があり、および/または、敏感な情報の損失、流用、および/または不正アクセス、使用または開示をもたらし、またはアクセスを阻止することができ、これは、私たちに財務的および名声的損害をもたらす可能性がある。データセキュリティ事故防止のためのセキュリティ対策を実施しているが,これらの措置が有効である保証はない。我々は過去につねにできるわけではなく,将来的には我々の情報技術システムにおける脆弱性を検出できない可能性があり,これらの脅威や技術は常に変化し,性質はしばしば複雑であり,データセキュリティ事件が発生してから発見される可能性がある.例えば、2018年4月、私たちは役員の会社の電子メールアカウント漏洩に関連したデータセキュリティ事件を発表した。私たちの事件の調査では、私たちのシステムが他の側面で事件に関連しているか、または患者や幹部以外の他の個人に関する個人データがアクセスまたは開示されているという証拠は何も発見されなかった。しかし、私たちと他のエンティティの固有、機密、および他の敏感な情報がアクセスされ、そのイベントによって漏洩される可能性がある。この事件に関連した意外な事態が発生する可能性があり、これは私たちにさらなる悪影響を及ぼす可能性がある。この事件によって引き起こされたいかなる訴訟や規制審査や調査は、私たちを重大な法的リスクに直面させる可能性がある。セキュリティイベントや他の中断は,我々の開発計画や業務運営の実質的な中断を招く可能性もある.例えば, 完成或いは未来の臨床試験中の臨床試験データの紛失は著者らの監督管理の審査作業の遅延を招く可能性があり、そして著者らのデータの回復或いは複製のコストを著しく増加させる。
上記の会社の電子メールイベントは知っているが,まだ発見されていない他のデータセキュリティイベントを経験しているかどうかは不明である.状況がこうなると信じる理由はないが,攻撃者はシステムのアクセス権限を隠蔽する方式で非常に老練になっており,攻撃を受けた多くの会社は自分が攻撃されたことを知らない.攻撃のどんな遅延を発見することは費用の増加を招き、私たちの名声を損なう可能性がある。私たちまたは私たちが依存している第三者が経験した任意のセキュリティイベントまたは中断(上述した会社の電子メールイベントを含む)は、政府の法執行行動(例えば、調査、罰金、処罰、監査および検査)、追加の報告要件および/または監督、データ(個人データを含む)の処理の制限、訴訟(類クレームを含む)、賠償義務、私たちの名声に対する損害、通貨資金移転、および経済的損失を含む不良な結果をもたらす可能性がある。適用されるデータプライバシーとセキュリティ義務は、関連利害関係者にセキュリティイベントを通知することを要求する可能性があります。このような開示は費用が高く、そのような要求を開示または遵守しないことは悪い結果をもたらすかもしれない。また、セキュリティホールやネットワークセキュリティに関する有効な内部会計制御を維持できず、タイムリーかつ正確な財務諸表を作成する能力に影響を与え、規制機関の審査を受ける可能性があります。私たちは、セキュリティイベントや他の中断を防ぐために、大量のリソースを費やしたり、私たちの業務活動を修正したりする可能性があります。さらに、私たちはこのような決定された抜け穴を解決するための救済措置を制定して配置することに遅延があるかもしれない。私たちの契約には責任制限が含まれていないかもしれない, 彼らがそうしても、私たちの契約における責任制限が、私たちのデータのプライバシーと安全義務に関連する責任、損害、またはクレームから私たちを保護するのに十分である保証はありません。もし私たちの第三者パートナーがプライバシーやデータセキュリティに関する義務を履行できなかった場合、損害賠償を受ける権利があるかもしれませんが、どの賠償も私たちの損害を補うのに十分ではないかもしれません。あるいはそのような賠償を取り戻すことができないかもしれません。
さらに、私たちの保険範囲が、私たちのプライバシーおよび安全慣行によって生じる責任から私たちを保護または軽減するのに十分か、または軽減するのに十分かどうかを決定することはできず、このような保険が商業的に合理的な条項で提供され続けるか、または根本的に確定できないか、またはそのような保険が将来のクレームを支払うと判断できない。
私たちはフランスとイギリスで業務運営をしており、これは私たちを追加的な費用と危険に直面させる
私たちのフランスとイギリスでの業務運営は、アメリカ以外での業務に関連するいくつかの追加コストとリスクに直面しています
•地理的位置の異なる場所で国際業務を管理することに固有の複雑さおよびコストの増加
•様々な法規、財務、法律要求を遵守する挑戦は、いつでも変化する可能性がある
•適用される税金の法律と法規の変化を含む潜在的な不利な税金の結果
•潜在的コストの高い貿易法、関税、輸出割当量、関税または他の貿易制限、およびそれらのいかなる変化も
•外国に住んだり旅行したりする従業員は税収、雇用、移民、労働法を遵守する
•私たちの国際業務の活動やそれに関連した責任
•システム、政策、福祉、およびコンプライアンス計画を異なる労働者および他の法規に適応させる必要があることを含む、異なる地理的位置を効率的に管理する従業員固有の挑戦
•自然災害、政治と経済不安定、戦争、テロと政治動乱、ロシアとウクライナ間の衝突、衛生流行病の発生、新冠肺炎の大流行、及びそれによる全世界の経済と社会影響を含む
•労働騒乱がアメリカよりも一般的な国では労働力の不確実性
•データセキュリティおよび不正使用または商業および個人情報へのアクセスに関連する異なる法律法規。
しかも、私たちのフランスとイギリスでの国際業務は私たちにユーロとドル、ポンドとドルの通貨為替レートの変動の影響を受けた。通貨レートの変動性を考慮して、通貨取引および/または両替リスクを効率的に管理できる保証はない。これまで、為替変動の影響を相殺する派生ツールを締結していません。為替変動は私たちの財務状況や経営業績に悪影響を及ぼす可能性があるからです。いずれにしても、これらのリスクや米国以外の業務に関連する他のリスクによる困難は、私たちをより高い費用に直面させ、私たちの発展努力を損ない、私たちの財務状況や運営結果に悪影響を与え、私たちの競争的地位を損なう可能性があります。
私たちは合格した熟練従業員を採用、統合、維持する上で困難に直面しており、引き続き困難に直面する可能性がある
私たちの組織の成長と安定は私たちの戦略目標を成功的に達成する能力に必須的だ。私たちは適切な経験と技能を持つ十分な数の合格従業員を採用し、統合し、維持することができないかもしれない。
ゲノム薬物の発見,開発,製造経験を持つ熟練した個人が不足しており,この状況が続く可能性がある。したがって、この人たちに対する競争は非常に激しく、離職率が高いかもしれない。これらのスキルを持つ個人に対する多くの生物製薬会社と学術機関との競争を考慮すると,これらのスキルを持つ従業員を受け入れ可能な条件で募集,統合,保持する困難を経験し続けている可能性がある。また、私たちの臨床前或いは臨床試験或いはマーケティング承認申請中の任意のマイナス或いは予期しない結果は、合格した熟練従業員を雇用と維持することをより挑戦的にする。もし私たちが私たちの成長目標を達成しなければ、私たちの研究開発、製造、規制作業の進捗は減速し、これは私たちの業務、財務状況、運営結果、将来性に悪影響を及ぼすだろう。
私たちは私たちの管理チームのいくつかの重要なメンバーと私たちのいくつかの科学、臨床開発と製造者に依存して、彼らのサービスを失うことは私たちの研究、開発、製造と監督管理の進展を阻害するかもしれない。例えば、2022年には、我々の前開発主管上級副社長がSangamoから辞任し、近年、他の様々なバイオテクノロジー会社での機会を求めるために、高級財務や法律従業員の中でも同様の退職を経験している。バイオテクノロジー業界の人材に対する激しい競争を考慮すると,特に旧金山湾区では,将来的には他の幹部や従業員が辞任する状況に遭遇する可能性がある
より多くの辞任は私たちの成長と安定にもっと深刻な妨害と脅威をもたらすかもしれない。私たちは私たちのすべての幹部と雇用協定を締結しましたが、彼らの誰でもいつでも私たちの仕事を離れることができます。私たちのすべての従業員は“勝手”な従業員だからです。私たちは誰の従業員にも“キーパーソン”保険に加入していない
私たちは新しい潜在的な製品候補の発見、許可、または獲得に成功しないかもしれないより大きなビジネス機会や成功する可能性のある候補品を利用できないかもしれません.
もし私たちの既存の候補製品が規制部門の承認を得なかったり、商業化に成功しなかったりすれば、私たちの業務の成功は、私たちが発見、許可、または買収を通じて私たちの製品ラインを拡大し続ける能力にかかっている。私たちはそれができないかもしれない。もし私たちが許可または買収のための潜在的な製品候補を決定した場合、私たちは許可者または売り手と受け入れ可能な条項を達成できないかもしれない。また、候補製品に関連するリスクと責任が存在する可能性がありますが、私たちの職務調査は発見されていない、私たちに開示されていない、私たちは十分に評価されていない、あるいは私たちは効果的に管理できません。さらに、様々な理由で、候補製品が臨床試験で安全でないことまたは有効であることが証明され、候補製品を私たちの運営に統合することに成功できないこと、または予想される収益が予想される時間内に達成できないことを含む、このような許可または買収の期待収益を達成することができない可能性がある
さらに、私たちは資源が限られているので、私たちはいくつかの候補製品または後により大きな商業潜在力の兆候が証明された機会の追求を放棄または延期するかもしれない。私たちの現在と未来の研究開発プロジェクトへの支出はいかなる商業的に実行可能な製品も発生しないかもしれない。もし私たちが特定の候補製品の商業的潜在力を正確に評価しなければ、私たちは戦略協力、許可、または他の手配を通じて候補製品に価値のある権利を放棄するかもしれないが、この場合、候補製品の独占開発権と商業化権利を維持することは私たちに有利だ。あるいは,ある治療領域の候補製品に内部資源を割り当てることができ,その分野では協力手配を達成した方が有利である。
私たちの普通株や会社組織に関するリスク
私たちの株価はずっと変動していて、引き続き変動する可能性があり、これは投資家に重大な損失をもたらし、遺伝子薬物と生物技術業界に対する大衆の見方の影響を受ける可能性がある。
私たちの株価はずっと変動していて、引き続き変動する可能性があり、これは株主に大きな損失をもたらす可能性がある。私たちの普通株の活発な公開市場は持続できないかもしれませんし、私たちの普通株の市場価格は変動し続けるかもしれません。私たちの普通株の市場価格は様々な要素によって大幅に変動します。その中のいくつかの要素は制御できません。以下の要素を含んでいますが、これらに限定されません
•私たちまたは協力者によって発表された公告は、候補製品の最新の進展または開発状態または臨床試験データを提供する
•臨床試験を開始または終了します
•同じ会社の市場予想が変化しています
•新興バイオテクノロジー会社の株式市場を含む市場全体と経済状況
•私たちの経営結果は私たちが与えたガイドラインとの偏差である
•私たちまたは私たちの競争相手は、新しいまたは強化された製品または技術または重大な契約、買収、戦略関係、合弁企業または資本約束を発表する
•私たちの協力者の業務と運営における変化、または私たちの既存の協力協定の変化を発表します
•遺伝子薬物世論の変化
•遺伝子薬物の規制審査を強化することを含む規制動態
•私たちの株式に対する1人以上の証券アナリストの推薦、格付け、またはカバー範囲の変化
•キーパーソンの増減
•私たち、上級管理者、または取締役は、私たちの普通株または他の証券、清算機関基金を売却し、これは私たちが持っている大量の株を構成し、私たちの現金残高を減少させます。
また、株式市場は最近、極端な価格や出来高変動を経験しており、多くの会社の株式証券の市場価格に影響を与え続けており、基本的な業務モデルや見通しが根本的に変わっていないにもかかわらず、多くの企業の株価下落を招いている。これらは
変動は往々にしてこれらの会社の経営業績に関係なくまたは比例しない。我々の実際の経営業績にかかわらず、広範な市場と業界要素は、絶えず悪化するマクロ経済条件と新冠肺炎疫病に関連する他の不利な影響或いは事態の発展、及び政治、地政学、監督管理とその他の市場条件を含み、すべて私たちの普通株の市場価格にマイナス影響を与える可能性がある。
実際または潜在的に私たちの普通株を市場に売却する大量の株は、私たちの普通株の市場価格の下落や様々な理由で上昇を阻止する可能性があります。
公開市場で私たちの大量の普通株を売ることはいつでも起こる可能性がある。これらの売却、あるいは市場で大量の株式保有者が株を売却しようとしているとの見方は、我々の普通株の市場価格を低下させる可能性がある。我々が発行した普通株は、1933年の証券法(改正本)または証券法第144および701条に規定する許容範囲に適合すれば、またはこのような株式の発行が証券法に基づいて登録され、当社の非関連会社が保有している限り、いつでも公開市場で自由に販売することができる。2022年には、我々が生物遺伝研究会社に発行した株の売却制限が失効し、それに応じて公開市場で制限なく販売することができる。また、いくつかの制限を受けた場合、証券法に基づいて、私たちが生物遺伝会社に発行した任意の株を転売することに同意します。私たちはまた登録声明を提出して、私たちが株式補償計画に従って発行可能な普通株を登録しました。このような株は発行後は公開市場で自由に販売することができるが,数量制限や付属会社への適用禁止期間の制限を受けている。また、Jefferies LLCと販売協定を締結し、時々公開市場で現在の市場価格で最大3.25億ドルの普通株を適宜販売することを可能にした。2023年2月22日現在、販売契約により22,953,199株の普通株が売却されており、純収益は約1兆177億ドル。
また、改正された1934年の証券取引法規則10 b 5-1に規定されたガイドラインと、株式取引に関する私たちの政策によると、私たちの従業員、役員、取締役は株式取引計画を採用し続ける可能性があり、これらの計画によると、彼らは今後時々私たちの普通株を売却するように手配されている。一般的には、我々の役員や役員がこのような計画に基づいた販売を行うには書類を公開提出する必要がある。我々の従業員、役員、取締役、関連株主も、重大·非公開情報を把握することなく、ルール10 b 5-1計画以外の追加株を購入または売却することができる。これらの人たちの私たちの普通株に対する実際または潜在的な販売は他の投資家によってマイナスとみなされる可能性があり、私たちの普通株の価格が下落したり、上昇を阻止したりする可能性がある。
私たちは私たちの普通株に配当金を支払うつもりはないので、どんな見返りも私たちの株の価値に限定されるだろう。
私たちは普通株式に対するどんな現金配当金も発表したり支払ったりしたことがない。私たちは現在、事業の発展、運営、拡張のために未来の収益を保留し、予測可能な未来に現金配当金を発表または支払うことはないと予想している。したがって、株主のどんな見返りもその株の増価に限定されるだろう。
当社の登録証明書、デラウェア州法律、当社の定款における反買収条項は、当社を買収することをより困難にする可能性があり、当社の株主が既存の経営陣を更迭または交換しようとすることを阻止する可能性があります。
デラウェア州法律の反買収条項とわが社の登録証明書と私たちの定款はわが社の支配権の変更を阻止、延期、あるいは阻止する可能性があり、たとえ制御権の変更が私たちの株主に有利になるかもしれません。また、これらの規定は、株主が取締役会のメンバーを交代させることをより困難にし、株主が現在の経営陣を交代または罷免しようとしていることを挫折または阻止する可能性がある。特に、当社の登録証明書によると、我々の取締役会は最大5,000,000株の優先株を発行することができ、その権利および特権は、普通株式保有者の同意を必要とすることなく、私たちの普通株よりも優先する可能性があります。また、株主がこれ以上投票したり、何か行動を取ったりしない場合、取締役会は、優先株の価格、権利、特典、特権、制限を決定する権利がある。このような優先株が発行されれば、普通株式保有者より優先され、彼らの権利を損なう可能性がある。優先株発行は可能な買収や他社の目的に関する柔軟性を提供してくれるが、今回の発行は第三者が発行した議決権のある株の大部分を買収することを難しくする可能性がある。
同じように、私たちは発行されていない普通株式を未来に発行することができ、株主の承認を必要としない。私たちの会社の登録証明書は、株主が書面の同意の下で行動してはならないとさらに規定している。
さらに、私たちが改訂して再説明した付例は以下のとおりである
•取締役会への指名又は株主会議で行動可能な事項を規定する事前通知要求;
•株主による株主特別会議の開催を禁止する。
私たちはまた、ある例外的な場合を除いて、誰かが私たちの投票権のある株の15%を獲得し、その人が“利害関係のある株主”であると規定しているデラウェア州会社法第203条の制約を受けている
私たちと“業務合併”を行い、期限は3年で、その人が15%以上の株式または私たちの議決権のある株を獲得した日から計算します。場合によっては、第203条の適用は、わが社の制御権の変更を阻止または阻止する可能性があり、このような変更がわれわれの株主に有利である可能性がある。
私たちの改正と再記述の定款は、いくつかの紛争の裁決に排他的フォーラムを指定しており、これは、私たちまたは私たちの役員、役員、または従業員との紛争に有利であると考えられる司法フォーラムでの株主のクレーム能力を制限する可能性があります。
私たちが書面で代替裁判所を選択することに同意しなければ、デラウェア州衡平裁判所、またはその裁判所に管轄権がない場合、デラウェア州連邦地域裁判所は以下の唯一で独占的な裁判所になるだろう
•私たちが提起した任意の派生訴訟や法的手続きを代表する
•取締役、上級職員、または他の従業員または株主が私たちまたは私たちの株主に対して負う受託責任に違反すると主張するいかなる訴訟も;
•デラウェア州“会社法総則”、我々の定款又は本定款のいかなる規定に基づいて、デラウェア州“会社法総則”に基づいてデラウェア州衡平裁判所に管轄権を与えるいかなる訴訟、及び
•内政原則に支配されているという主張を主張するいかなる行為も。
私たちの改正と重述の付例は、米国連邦地域裁判所が改正された“1933年証券法”に基づいて提出した任意の訴えの唯一かつ独占的なフォーラムであることをさらに規定している。これらの規定はさらに規定されており、任意の個人または実体が私たちの株式株式の任意の権益を取得することは、これらの規定に了承され、同意されたものとみなされる。
これらの条項は、株主が司法法廷で、私たちまたは私たちの役員、役員、または他の従業員との紛争に有利だと思うクレームを出す能力を制限する可能性があり、これは、私たちと私たちの役員、役員、および他の従業員に対する訴訟を阻止するかもしれません。もし裁判所がこれらの条項のいずれかが訴訟で適用されないか、または実行できないことを発見した場合、私たちは他の管轄区域で紛争を解決するために追加費用が発生する可能性があり、これは私たちの業務を深刻に損なう可能性がある。
プロジェクト1 B--未解決スタッフの意見
ない
プロジェクト2--財産
2029年5月に期限が切れる賃貸契約によると、当社の本社はカリフォルニア州ブリスベンに約87,700平方フィートのオフィスと研究開発実験室施設を持っています。2031年8月に満期になった賃貸契約によると、約59,485平方フィートの研究とオフィススペースもレンタルし、2026年8月に満期になった賃貸契約によると、カリフォルニア州リッチモンドで約7,700平方フィートのオフィススペースをレンタルした。私たちはフランスのヴァル州で約26,600平方フィートのオフィスと研究開発空間をレンタルし、レンタル契約は2025年6月から2030年1月まで満期になった。私たちは私たちの現在の施設が私たちの必要性を満たすのに十分だと信じている。私たちが事業を拡大し続けるにつれて、私たちはもっと多くの施設をレンタルしたり購入したりする必要があるかもしれない。
プロジェクト3−法的訴訟
私たちは法的手続きを待ついかなる重大な事項でもない。時々、私たちは正常な業務過程で発生した法的訴訟に巻き込まれるかもしれない。
プロジェクト4−鉱山安全開示
適用されません。
第II部
項目5−登録者普通株市場,関連株主事項,発行者による株式証券の購入
市場情報
私たちの普通株はナスダック世界の精選市場で取引され、コードは“SGMO”です
所持者
2023年2月17日現在、私たち普通株の記録保持者は60人です。この数字には、銀行、ブローカー、金融機関、および他の指名者が保有する“街頭名前”または受益者の株式は含まれていない。
配当をする
私たちは普通株の配当金を支払っていないし、現在予測可能な未来に現金配当金を支払うつもりもない。
株式表現グラフ
上述した株式表現グラフおよび関連情報は、“募集材料”または米国証券取引委員会によって“保存されている”とみなされてはならないし、参照によってこのような申告文書に明示的に組み込まれない限り、証券法または取引法(両方とも改正)に規定された任意の将来の申告文書に引用によって格納されてはならない。
ITEM 6 – [保留されている]
米国証券取引委員会第33−10890号プレスリリースに含まれるS−K規則第301項の修正によると、第6項に応答するデータは提供されていない。
プロジェクト7−経営陣の財務状況と経営成果の検討と分析
“経営陣の財務状況及び経営結果の検討及び分析”における議論には、動向分析、推定及びその他の前向きな陳述が含まれており、改正証券法第27 A条及び改正取引法第21 E条の意味に適合している。これらの前向き陳述は、“予想”、“信じる”、“継続”、“可能”、“推定”、“予想”、“意図”、“可能”、“計画”、“求める”、“すべき”、“会する”などの語彙を含む表現、およびこれらの語彙または表現の他の類似した意味または否定的な意味を含むが、これらに限定されない。このような展望的陳述は、既知および未知のリスク、不確実性、推定および他の要素の影響を受けることができ、これらのリスク、不確実性、推定および他の要素は、私たちの実際の結果、表現または成果または業界結果をもたらす可能性があり、そのような前向き陳述が明示的または暗示する任意の未来の結果、表現または成果とは大きく異なる。しかし、本年度報告10−K表第I項第1 A項に記載されている“リスク要因”に限定されないため、実際の結果は、このような前向き陳述に記載されている結果とは大きく異なる可能性がある。以下の議論および分析、および連結財務諸表およびこれらのレポートに添付された付記を読むべきであり、これらのレポートは、本報告の他の場所に含まれています。
また,“経営陣の財務状況と経営成果の検討と分析”の節では,2022年と2021年の項目,および2022年と2021年の同比比較を一般的に検討した。2020年プロジェクトの検討および2021年と2020年の年度比較は本10−K年報には含まれていないが,2022年2月24日に米国証券取引委員会に提出された2021年12月31日現在の会計年度10−K年報第2部の“経営陣の財務状況と経営成果の検討と分析”で見つけることができる。
概要
我々は臨床段階の遺伝子薬物会社であり,画期的な科学を薬物に変換し,重篤な疾患を有する患者と家庭の生活を変えることに取り組んでいる。私たちは私たちの臨床と臨床前候補製品を開発することでこの使命を実現し、私たちの新しい科学と私たちの内部製造能力を利用する予定です。
私たちの現在の臨床段階の候補は
•我々が所有しているファーブリー病を治療する遺伝子治療製品ST−920は,Isaralgagene Ciparvovecとも呼ばれ,現在われわれの1/2期STAAR臨床研究で評価されており,潜在的な3期臨床試験計画を行っている
•TX 200、我々は完全に所有しているキメラ抗原受容体、あるいはCAR、工学制御性T細胞、あるいはCAR-Treg、細胞治療製品、ヒト白血球抗原A 2不整合腎移植免疫を介した拒絶反応を予防するために、現在私たちの1/2期の堅固な臨床研究で評価されている
•Giroctocogene fitelparvovecはSB−525とも呼ばれ,重症血友病Aを治療する候補遺伝子治療製品であり,現在登録されている3期後発臨床試験が行われている。私たちは協力者のファイザーやファイザーと一緒にgiroctocogene fitelparvoecを開発しています
•BIVV 003は我々の亜鉛指ヌクレアーゼまたはZFヌクレアーゼであり、鎌状細胞病(SCD)を治療する候補遺伝子編集細胞治療製品であり、現在我々の1/2期PRECIZN-1臨床研究で評価されている。BIVV 003は、Sangamoが2022年6月にSanofi S.A.またはSanofiからSangamoに移行した後に完全所有するプロジェクトである。以下に述べるように,我々は最近,FabryとTX 200計画の資源を優先的に配置するために,1/2期PRECIZN-1研究が完了した後,BIVV 003計画へのさらなる実質的な投資を停止する戦略決定を行った.
われわれの臨床前開発は2つの革新の優先分野に集中している:(I)自己免疫疾患用CAR−Treg細胞療法と(Ii)神経疾患用ゲノム工学。著者らの臨床前プロジェクトの適応は神経発達障害、癌、炎症性腸疾患或いはIBD、神経病変と神経変性疾患、例えば筋萎縮性側索硬化症、多発性硬化症とハンチントン病を含み、その中のいくつかの疾患は著者らがパートナーのBiogen MA、Inc.とBiogen International GmbHと共同開発したものであり、著者らは総称してBiogen、ノワール生物医学研究研究所或いはノワール、輝瑞、武田製薬有限会社とKite Pharma,Inc.と呼ばれる。
私たちは生物製薬会社との何度もの協力は私たちに重要な財務と戦略的利益をもたらし、私たちの研究開発努力と私たちのZF技術プラットフォームの潜在力を強化した。彼らは私たちの
パートナーの治療と臨床専門知識とビジネス資源は,我々の薬物をより迅速に患者にもたらすことを目標としている。私たちはこれらの協力が私たちのZF技術プラットフォームの価値を反映し、潜在的に候補製品の潜在的な市場を拡大すると信じている。私たちはこれまでに約8.15億ドルの前払い許可料、マイルストーン支払い、私たちの普通株を協力者に売却する収益を受けており、潜在的な製品印税に加えて、私たちの協力から67億ドルまでの未来のマイルストーン支払いを稼ぐ機会がある。
私たちは私たちの内部製造能力が私たちに競争優位を提供すると信じている。我々は現在カリフォルニア州ブリスベンの本社に腺関連ウイルス(AAV)製造工場を設置し,カリフォルニア州ブリスベンとフランスバル州に細胞療法メーカーを設置している。我々の製造戦略は,内部製造·契約製造組織(CMO)パートナーシップによるバランスと必要な能力を構築し,製造プロセスや分析に投資し,強力なサプライチェーンを発展させ,より大きな柔軟性,品質,制御を提供することである。
当社業務のその他の資料については、本年度報告表格10-K第I部分第1項の“業務”を参照されたい。
最近の業務のハイライト
ファブリック病
•2023年2月22日、私たちは、Fabry病を治療するための完全な遺伝子治療製品候補製品であるIsaralgagene Cirobvovec、またはST-920を評価したSTAAR研究からの最新の臨床データを発表しましたこれは…。年度世界シンポジウム2023年2月24日。この声明には,2022年10月20日までにプロポフォール治療を受けた13名の患者のデータが含まれており,2人の患者の腎臓生検データが含まれている。締め切り以来,1/2期STAAR研究では4名の患者が薬物治療を受けており,これまでに計17名の患者が薬物治療を受けてきた。現在、20のサイトが活発に求人を行っている。より多くの男性や女性患者が現在スクリーニングを行っていることに伴い,この研究の進展は続いている。
•1/2期STAAR研究拡張段階が進行中であり,潜在的な3期臨床試験の準備が積極的に行われている。監督部門の相互作用により,2023年末に第3段階試験が開始される予定であり,1人目の患者の投与量は早くても2024年上半期に開始される可能性がある。第1/2段階拡大段階の調剤は2023年末に完了する予定であり,第3段階試験を開始するゲートコントロール要因とはならないと予想される。
•2022年12月,研究拡張段階の患者が輸液後14日に3級重篤な不良事象,すなわちSAEが出現し,入院治療が必要となった。それ以来,事件は完全に解決され,患者はまだ研究中である。この研究の首席調査員と安全監視委員会はSAEが治療に関与している可能性を評価し,規制機関にSAEを報告した。安全監視委員会はその後、この研究を修正せずに行うことができ、この事件を理解のために他の調査者に報告することができると判断した
腎移植拒絶反応
•2022年3月、我々はTX 200を評価する1/2期STEST研究中の1人目の患者に対して薬物治療を行い、TX 200は我々が完全に所有している自己ヒト白血球抗原A 2 CAR Treg細胞治療候補製品であり、生体ドナーからヒト白血球抗原A 2不整合腎臓を受けた患者を治療し、2人目の患者は2022年9月に治療を受けた。3人目の患者は腎臓移植を受けており、彼らの個性化TX 200細胞療法はすでに製造されており、投与量は2023年第2四半期初めに予定されている。第2列の製造と臨床活動が行われており,第4患者の投与量は2023年夏に予定されている。より多くの患者が予備スクリーニングを行っており,この研究に参加する予定である。用量アップグレード計画を加速させる機会を規制機関と検討している。
血友病A
•2022年9月,ファイザー社による我々の重症血友病A治療に対する研究的遺伝子療法Fitelparvovecの3期アフィン臨床試験の自発的休止は中止され,募集を再開し募集を再開した。予備分析を支持する投与量は2022年11月に回復し、2023年第1四半期末に完成する予定である。2024年上半期には重要なデータが発表される予定で、ファイザーは2024年下半期に生物製品許可証申請を提出する予定だ。
•2022年12月、我々とファイザー社はASHが原癌遺伝子fitelparvovecに対して行った1/2期ALTA研究の最新の後続データを公表した。2022年9月6日締め切り:
◦156週の最高用量3 e 13 Vg/kg列の5名の患者の発色凝固法による第8因子(FVIII)の平均活性は25.5%であった
◦このキューでは,輸液後1年目の平均年間出血率は0.0,フォローアップ期間全体で1.2であった。すべての出血事件は輸液後69週間後に発生した。2名の患者は出血イベントが出現し、外因性FVIII治療が必要である。最高用量の列では、参加者が予防を回復しなかった。
◦Girococogene Fitelparvovecは全体的に良好な耐性を継続していた。
鎌状細胞病
•2022年12月、我々はASH上でBIVV 003の1/2期PRECIZN-1研究の最新の初期概念検証臨床データを発表した。BIVV 003はZFヌクレアーゼが編集した細胞治療候補薬であり、セノフィと共に開発されている。2022年9月30日現在,HSPC目標収量に達した6名の患者のうち,5名にBIVV 003が注射されている。
•BIVV 003を服用した最初の4人の患者については、最初の製造プロセスを使用して生産された:
◦BIVV 003注入による総HbとHbFレベルへの影響は30カ月まで維持された。
◦4名の患者のうち,3名の患者は安定してZFヌクレアーゼ修飾HSPCを移植し,HBFレベルの30%以上の持続的な上昇をもたらし,BIVV 003注射後に重篤な血管閉塞危険像やVOCsはなかった。
•患者5は、最終製品中長期前駆細胞の数を増加させるために、内部実験において示された改良された方法を用いて製造されたBIVV 003を受け入れた
•患者5は注入後26週のHbFレベル45%,総Hb 12.4 g/dLであり,締め切り後に最近採取したサンプルでは,HbFレベルは群1の26週時のレベルより高かった。
•自在ASHに最新データを提出して以来,患者7号投与のための臨床と製造活動,3期試験設計,CMCパッケージとその他の要求はFDAと一致した。また,より多くの製造改善が得られており,潜在的な3期試験で臨床結果をさらに強化し,製造コストを低減することが可能である。
•私たちは最近、1/2段階PRECIZN-1研究が完了した後、BIVV 003計画へのさらなる実質的な投資を停止して、私たちのFabryとTX 200計画の資源を優先的に配置するための戦略決定を下した。我々はBIVV 003の1/2段階PRECIZN−1研究の完成に取り組んでおり,すでに約束された資金を用いて研究中の投与量を完成させる予定である。我々は,この計画を可能な第3段階実験に進めることができる協力パートナーを探し始める予定である。
製造業
•我々は現在カリフォルニア州ブリスベンの本社に腺関連ウイルス(AAV)製造工場を設置し,カリフォルニア州ブリスベンとフランスバル州に細胞療法メーカーを設置している。
新冠肺炎の大流行の影響
私たちはすでに新冠肺炎の大流行が私たちの業務と運営に与える影響を経験し続け、大流行のアメリカ、フランス、イギリスと私たちの臨床研究と試験場所の発展に伴い、例えばカナダ、イタリア、オーストラリアの新しい研究地点で、私たちはこれらのあるいはもっと深刻な影響を経験し続けるかもしれない。例えば,臨床試験患者における新冠肺炎陽性例を扱う場合,われわれの現場運営は周期的な短期中断を経験しており,新冠肺炎が爆発すれば,我々の運営は将来より長期的な中断を経験する可能性がある。また、私たちは時々私たちの資源の再構成と優先順位を要求されて、新冠肺炎の影響による適度な供給制限を緩和する。私たちの計画が長期的に中断されれば、私たちの協力協定で想定されている私たちのバイオ製薬パートナーを支援する能力に影響を与え、スケジュールを調整することにつながる可能性があります。
また,イサラギの1/2期STAAR臨床研究が評価され,そのスケジュールの遅延を経験し続け,一部の原因が新冠肺炎の影響である。例えば、ある患者が研究や服薬に参加する前に新冠肺炎の検査が陽性であった場合、研究が遅延する。そのほか、新冠肺炎の影響により、著者らはSTAAR研究に必要な原材料を提供する方面でいくつかの短期遅延に遭遇し、臨床試験材料の輸送においてもいくつかの短期遅延に遭遇した。これらの課題は私たちの最初のSTAAR研究スケジュールを約3~6ヶ月後退させたと推定される。もし新冠肺炎がワクチン接種遅延、新たな新冠肺炎変異或いは意外事件により著者らの募集、スクリーニング、登録と投与及び本研究の原材料源に与える影響が激化すれば、本研究の臨床スケジュールは再改訂される可能性がある
また,われわれはわれわれの全資本が有する腎移植拒絶反応を治療するCAR−Treg細胞療法候補品であり,われわれCMOの製造や技術移転挑戦に関連する新冠肺炎の影響,患者とドナーの新冠肺炎検出が陽性であったため,そのスケジュールが延期されているTX 200を確固として評価した。もし新冠肺炎の影響で追加的な遅延があれば、私たちがこの研究を行うスケジュールは調整されるかもしれない。
未来を展望して、著者らは引き続き新冠肺炎が著者らの運営、研究承諾と臨床試験及び著者らの協力者、臨床試験地点とCMOに与える影響に注目する。これらの操作の中断、および将来的には、私たちが運営する場所や業界に一般的に適用される制限、あるいは特に私たちと私たちの施設に適用される制限によって生じる可能性のあるより深刻な中断は、私たちのタイムリーな研究能力を阻害し、協力者に対する研究義務を履行し、私たちの治療計画の発展を推進することができるかもしれない。このような遅延と中断は、私たちの業務、運営業績、財務状況に実質的な影響を及ぼす可能性がある。
私たちは、新冠肺炎の疫病が私たちの2023年の財務状況に実質的な負の影響を与えないと予想している。私たちは現在、新冠肺炎の疫病は私たちの貸借対照表上の金融資産あるいは商業権の評価にいかなる実質的な損害を与えないと予想している。私たちは、オフィス従業員のために実施された遠隔職場配置が、私たちの財務報告や制御システムに影響を与えているとは思いません。
新冠肺炎疫病がどの程度直接或いは間接的に著者らの業務、運営と財務状況に影響するかは、現在まだ高度で不確定な未来の事態の発展に依存する。これらの動態は疫病の最終持続時間と重症度、新しい新冠肺炎変種の影響、旅行制限、アメリカ、フランス、イギリスとその他の国の新しい公共衛生制限、商業中断及び疾病の制御と治療行動の有効性と適時性を含み、ワクチン接種計画の有効性とタイミングを含む。私たちの事件に対する理解が絶えず発展し、更に多くの情報を得ることに伴い、著者らは著者らの収入、費用と製造、臨床試験と研究開発スケジュールに関連する指導を大幅に変えるかもしれない。
新冠肺炎大流行のリスクと不確実性に関するより多くの情報は,本年度報告の10−K表第I部第1 A項の“リスク因子”と題する部分を参照されたい。
経営成果の一部は
私たちの収入は主に前払い許可料、研究サービス精算、マイルストーン成果と研究支出援助の収入を含む。我々は収入が異なる時期に変動し続けることを予想しており,新たなパートナーやパートナー精算がその初期条項の後に継続することは保証されず,これらの合意に規定されたマイルストーンを達成できる保証もない.
設立以来,我々は純損失を出しており,少なくとも今後数年以内には,我々の研究·開発活動を継続するにつれて,損失が予想される。これまで、私たちは主に株式証券の発行や協力と助成金の収入を研究することで、私たちの業務に資金を提供してきました。
今後も研究開発に大量の資源を投入することが予想され,我々の候補製品を研究段階から臨床試験に進めることに成功すれば,今後数年間の研究開発費が増加することが予想される。Biogen、Kite Pharma、Inc.またはKiteとNovartisとの私たちとの協力協定、およびセノフィとの終了および移行協定の条項によると、研究開発活動に関連するいくつかの費用は私たちに精算される。Biogen,Kite,Novartisから受け取った精算資金は収入として確認され,関連コストが発生しており,入金が合理的に保証されているためである。セノフィが提供した精算資金は私たちの研究開発費を減らすだろう。
一般及び行政費用は主に行政人員、財務及び行政人員の給与及び人事関連費用、株式給与費用、専門費用、分配済み施設費用、特許訴訟費用及びその他の一般会社費用を含む。私たちの候補製品を診療所に普及させ、診療所を通過することに伴い、私たちの業務の成長には一般的な費用と管理費用が増加することが予想される。
重要な会計政策と試算
私たちの総合財務諸表と関連開示はアメリカ公認会計原則に基づいて作成されています。これらの連結財務諸表を作成する際には、連結財務諸表や付記中の報告金額に影響を与える見積もり、仮説、判断を行う必要があります。我々は過去の経験や当時の状況では合理的な様々な他の仮定を推定していると考えられるが,これらの仮定の結果は資産や負債の帳簿価値を判断する基礎となっており,そのような資産や負債の帳簿価値は他の出所から容易に見られるわけではない.異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。私たちは、本質的に不確実な事項の推定、仮説、判断を要求するため、以下の政策が私たちの財務状況と経営結果を知るために最も重要であると考えている。
私たちは、収入確認と長期資産(営業権や無形資産を含む)の評価に関する重要な会計政策と推定は、総合財務諸表を作成する際に使用される最も重要な推定と仮定であると信じている。
私たちの重要な会計政策の完全な説明については、注1を参照してください重要会計政策の組織·列報根拠と要約本年度報告Form 10−K第2部第8項“財務諸表と補足データ”に掲載されている連結財務諸表に付記されている
収入確認
私たちの収入は主に協力手配から来ており、主に知的財産権の許可と研究開発サービスの提供を含む。私たちの顧客が契約で約束された商品やサービスのコントロール権を獲得した場合、私たちは収入を確認し、その金額は私たちがこれらの商品やサービスから得たい対価格を反映しています
我々の多くの手配に対して,知的財産権を付与する許可は,関連する研究開発サービスの提供と変わらず,この結合の履行義務は時間とともに履行される.このようなプロトコルはまた、実質的な権利とみなされる追加の貨物およびサービスのオプションを含むことができる。これらのプロトコルに対しては,取引価格を推定し,履行義務ごとの推定独立販売価格に応じて取引価格を割り当てる必要がある.私たちの業績義務の大部分は時間の経過とともに交付された。我々は,入力方法に基づく進捗メトリックを用いて収入(たとえば,実際の作業量を累積し,我々の研究者が実際に発生した時間に第三者コスト精算の総推定作業量に対する価値を加えたり,総推定時間数に対する実際の発生時間数を積算したりすることを含む)を用いており,関連業績義務に対する満足度を最も示していると考えられる.私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。
物質的権利の独立販売価格を見積もり,その行使の可能性を含めて重大な判断が必要である。進展を見積もる措置も複雑であり,重大な判断に触れ,それぞれの義務履行に要する総コストの見積もりに影響されている。このような推定値の変化は私たちの収入確認に実質的な影響を及ぼすかもしれない。
私たちの収入確認のさらなる説明については、付記4-を参照されたい大顧客、パートナーシップ、戦略同盟本年度報告Form 10−K第2部第8項“財務諸表と補足データ”に掲載されている連結財務諸表に付記されている
営業権と無形資産を含む長期資産の推定値
私たちは、イベントや環境変化がその資産の公正価値をその帳簿価値よりも低くする可能性が高い場合、少なくとも毎年またはより頻繁に営業権および無期限無形資産の減値を検討する。2022年12月31日現在、営業権減額や無期限無形資産減値は確認されていない。
長寿資産は、財産及び設備及び有限年限無形資産を含み、いかなる事件や状況で当該等の資産の帳簿価値が回収できない可能性があることを示す場合には、当該等の資産が減値可能であるか否かを検討する。評価は,キャッシュフローが他の資産および負債キャッシュフローと実質的に独立していることが確認できる最低レベルで行った.これらの資産の回収可能性は,帳簿金額と資産予想が使用および最終処分から生じる将来の未割引現金流量との比較によって測定される。当該等の審査により、物件及び設備及び無形資産の帳簿額面が回収できないことが示された場合、当該等の資産の帳簿額面は公正価値に減少する。提出された年度内に、私たちは何の重大な減価費用も記録していません。
経営成果
2022年、2021年、2020年12月31日までの年度
収入.収入 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| (単位は千、100%比は除く) |
| 2022 | | 2021 | | 変わる | | % | | 2021 | | 2020 | | 変わる | | % |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
| 収入.収入 | $ | 111,299 | | | $ | 110,701 | | | $ | 598 | | | 1 | % | | $ | 110,701 | | | $ | 118,192 | | | $ | (7,491) | | | (6) | % |
収入には私たちの協力協定から稼いだ金額が含まれている。今後数年間の収入は主に生物遺伝会社、ノバ社、Kite社、ファイザー社との協力協定から来ると予想されています。
2022年の収入は2021年と横ばいだ。私たちとKiteとNovartisとの協力協定はそれぞれ1310万ドルと180万ドルの収入増加をもたらした。これらの成長は部分的に相殺されました
Biogenの協力プロトコルに関する収入は1390万ドル減少し,Sigma−Aldrich Corporationのライセンスプロトコルに関する収入は20万ドル減少し,我々のDow AgroSciences LLCのプロトコルに関する再許可料収入は20万ドル減少した。
運営費 | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| (単位は千、100%比は除く) |
| 2022 | | 2021 | | 変わる | | % | | 2021 | | 2020 | | 変わる | | % |
| 運営費用: | | | | | | | | | | | | | | | |
| 研究開発 | $ | 249,898 | | | $ | 230,819 | | | $ | 19,079 | | | 8 | % | | $ | 230,819 | | | $ | 180,647 | | | $ | 50,172 | | | 28 | % |
| 一般と行政 | 62,682 | | | 63,219 | | | (537) | | | (1) | % | | 63,219 | | | 67,097 | | | (3,878) | | | (6) | % |
| 総運営費 | $ | 312,580 | | | $ | 294,038 | | | $ | 18,542 | | | 6 | % | | $ | 294,038 | | | $ | 247,744 | | | $ | 46,294 | | | 19 | % |
研究と開発費
研究開発費は主に報酬関連支出を含み、株式給与、実験室用品、臨床前と臨床研究、生産臨床用品、契約研究及び分配された施設と情報技術費用を含む。
2021年に比べて2022年の研究開発費が1910万ドル増加したのは施設と情報技術費駆動者全体的なコスト増加とプロジェクトの進展により、より多くの空間を研究開発部門に再分配することになります私たちの試験のスケジュールと主に私たちの協力と研究計画による活動の増加により、臨床前、臨床と実験室の供給費用は710万ドル増加し、私たちの計画、臨床試験と製造業務を支持する従業員の数を増加させたため、報酬と他の人員のコストは190万ドル増加し、出張と娯楽費用は130万ドル増加した。これらの成長は210万ドルで相殺されていますサイノフィは研究開発費を精算しています. 2022年12月31日と2021年12月31日までの年間の研究開発費に含まれる株式報酬支出はそれぞれ1840万ドル、1950万ドルだった。
下表にわれわれの臨床,臨床前,その他の研究開発計画に関する研究開発費を示す。以下の表に示すように,我々が増加した研究開発費のうち,臨床計画は3420万ドル貢献しているが,2021年に比べて2022年にはわれわれの臨床前·研究計画が1400万ドル減少しており,これは主に我々が所有する計画に関する活動のスケジュールによるものである。 | | | | | | | | | | | | | | | | |
| | 十二月三十一日までの年度 |
| | (単位:千) |
| 番組 | | 2022 | | 2021 | | |
| 臨床計画: | | | | | | |
| ファブリ臨床プロジェクト | | $ | 67,351 | | | $ | 58,880 | | | |
| | | | | | |
| TX 200臨床プログラム | | 26,185 | | | 14,557 | | | |
| | | | | | |
| | | | | | |
| | | | | | |
| 鎌状細胞臨床プログラム | | 14,547 | | | 483 | | | |
| 小計 | | 108,083 | | | 73,920 | | | |
| 臨床前と研究計画: | | | | | | |
| 全資本が持つプロジェクトと早期研究活動 | | 103,160 | | | 99,297 | | | |
| CNSパートナシップ計画 | | 28,247 | | | 47,418 | | | |
| 腫瘍学パートナー計画 | | 2,222 | | | 894 | | | |
| 他の人は | | — | | | 38 | | | |
| 小計 | | 133,629 | | | 147,647 | | | |
| 他の研究と開発計画 | | 8,186 | | | 9,252 | | | |
| 研究開発費総額 | | $ | 249,898 | | | $ | 230,819 | | | |
(*)この額はCIRMのST−400臨床計画に関する贈与の償還義務解除に関連しており,2021年に停止する予定である。注4-を参照大顧客、パートナーシップ、戦略同盟本年度報告Form 10−K第2部第8項“財務諸表と補足データ”に掲載されている連結財務諸表に付記されている。
今後も研究·開発に多くの資源を投入し続ける予定であり,われわれの臨床計画を進めることに成功すれば,われわれの早期候補製品を臨床試験に入れることができれば,今後数年間の研究·開発費が増加することが予想される。
我々の開発計画の完成に要する時間の長さとこれらの計画の開発コストは,我々の候補製品が臨床試験に参加する範囲と時間,他の治療分野での開発計画の決定,パートナーや協力者とともに我々の候補製品を独自に開発するか,の影響を受ける可能性がある。例えば,我々の候補製品は複数の治療領域が開発されており,その中のいくつの治療領域を追求し続けるかは不明である。また、各追求された治療分野の規制承認を得るために必要な臨床試験の範囲および数は、適用規制機関の意見に依存しており、我々が展開する可能性のあるすべての潜在的治療分野についてこのような意見を求めていないが、このような意見を提供した後であっても、適用される規制機関は、当社または他社によって生成された新しいデータに基づいて、または私たちが制御できない他の理由で規制を承認する前に、追加の臨床研究を要求する可能性がある。いかなる監督管理許可の条件として、私たちはまた上場後の開発承諾の制約を受ける可能性があり、追加の臨床試験要求を含む。以上で議論した不確実性のため,我々の開発計画に関する継続時間や完了コストを決定することはできない.
私たちの潜在的な治療製品は長く不確実な規制過程の影響を受けており、これは私たちが必要な規制承認を得ることにつながらないかもしれない。必要な規制承認が得られなければ、私たちは影響を受けた候補製品を商業化することができないだろう。また,我々の候補製品の臨床試験は安全性と有効性を証明できない可能性があり,規制部門の承認を阻止または著しく遅らせる可能性がある。我々の研究·開発活動に関するリスク·不確実性の検討は、我々の候補製品の開発完了、および我々の業務、財務状況、成長見通しへの影響を含み、本年度報告10-K表第I部の第1 A項の“リスク要因”で見つけることができる。
一般と行政費用
一般及び行政支出は主に給与に関連する支出を含み、行政、法律、財務及び行政人員の株式給与、専門費用、分配された施設及び情報科学技術支出、及びその他の一般会社の支出を含む。
2021年と比較して、2022年の一般·行政費用が50万ドル減少したのは、主により多くの空間を我々の研究開発部門に再分配したため、分配コストが740万ドル減少したためである。従業員数の増加により給与やその他の人件費が330万ドル増加し、法律·専門費が180万ドル増加し、施設·情報技術費が130万ドル増加し、出張·娯楽費が40万ドル増加し、この減少額を相殺した。2022年12月31日と2021年12月31日までの年間で、一般および行政費用に含まれる株式ベースの報酬支出はそれぞれ1320万ドル、1340万ドル。
利息とその他の収入,純額
2022年12月31日と2021年12月31日までの年度の利息とその他の収入純額はそれぞれ940万ドルと530万ドルだった。2022年に2021年に比べ410万ドル増加したのは、主に利息収入が350万ドル増加したためであり、市場金利の上昇を反映しているコロナウイルス援助、救済、経済安全法案によると、300万ドルの従業員が留任して免除されているフランスのSangamo社が得た研究税控除は50万ドル増加した。これらの増加は,外貨レート変動に関する190万ドルの減少と,2021年にカリフォルニア再生医学研究所のST−400計画停止に関する贈与の償還義務解除により記録された120万ドルの純収益によって部分的に相殺された。
所得税費用
2022年、2021年、2020年の所得税の支出はそれぞれ40万ドル、30万ドル、30万ドルとなる。すべての年度の所得税支出は外国所得税によるものであり、外国繰延税収割引部分によって相殺される。
2022年から2017年に減税·雇用法案は174条が改正され、今年度の研究·実験(R&E)支出とソフトウェア開発コスト(総称してR&E支出と呼ぶ)の控除が廃止され、納税者にR&E支出を資本口座に計上し、5年以内に償却することが求められた(米国国外で行われたR&E活動による支出は15年)。私たちは2022年12月31日までの年間資本化R&E支出のために繰延税金資産を発生し、この資産は推定手当によって完全に相殺された。
2022年12月31日現在、我々の連邦と州所得税の純営業損失繰越額はそれぞれ約6.897億ドルと3.12億ドルである。2018年までに発生した連邦純運営損失は2023年に満期になり、利用しなければ2037年まで満期が続く。2018年に発生した連邦純運営損失は
無期限に前進する。利用しなければ、繰り越しの国の純営業損失は2029年に満期になる。私たちはまた連邦と州研究税収免除を繰り越して、それぞれ3680万ドルと2610万ドルです。連邦研究単位は2022年から満期になるが、州研究単位は満期日がない。“国税法”や国が定めるような所有権変更制限により、私たちの純営業赤字の繰越と検討税収控除の利用は重大な年度制限を受ける可能性がある。年度限度額は繰り越しの純営業損失と使用前の研究税収控除満期を招く可能性がある。純営業損失や研究開発税収控除に関する繰越により、近い将来、収入に関する米国連邦税は一切支払われないと予想される。
流動性と資本資源
流動性
設立以来、私たちは大きな純損失が発生し、私たちは主に株式証券、会社協力者、戦略パートナーの支払い、研究支出を発行することで私たちの運営に資金を提供しています。
自分から2022年12月31日合計3.075億ドルの現金、現金等価物、有価証券を持っています2021年12月31日。私たちの最も重要な資本使用は従業員の給与と外部研究開発費用、例えば製造、臨床試験と著者らの治療計画に関連する臨床前活動のためのものである。私たちの現金と投資残高は、米国政府が支援する実体債務証券、商業手形証券、通貨市場基金、会社債務証券、資産支援証券、預金を含む様々な利息ツールの形で保有されている。即時需要を超えた現金は、資本保証と流動性を実現するために、私たちの投資政策に基づいて投資されるだろう。
2020年8月、Jefferies LLCと公開市場販売契約または販売協定を締結し、既存の棚登録声明によると、最大1.5億ドルの普通株を時々市場で販売し、2022年12月31日現在も3,500万ドルが利用可能となっている。2022年12月に、吾らは公開市場販売協定第2号改正案を締結し、販売協定項下の総発行価格を1.75億ドル追加引き上げた。2022年12月31日までの年間で,販売契約により19,300,743株の普通株を売却し,純収益は約8,490万ドルであった。2023年1月1日から本年度報告Form 10−Kの日までに,販売契約により1,644,524株の普通株を売却し,純収益は約570万ドルであった。
会計基準に基づいて編集され、またはASC、主題205-40に基づいて財務諸表の列報−継続経営−これらの債務は、本Form 10-K年次報告書に含まれる総合財務諸表の発行日から12ヶ月以内に満了するため、条件および/またはイベントが将来の財務義務を履行する能力を評価する責任があります。私たちは、私たちの流動性の需要を満たすために、タイムリーに起動されるコスト保存措置を含むいくつかの可能な行動を決定しました
•いくつかの研究開発計画の優先順位を延期して再決定することは、計画と従業員の支出を減らすことに関連する
•新しい求人を一時停止し、出張や招聘費用などの補助費用を削減する
•追加設備、実験室改善、効率プロジェクト、および業務支援支出を含む非重要資本および運営支出を削減します。
経営陣が上述したような計画は、本年度報告書10-K表に含まれる連結財務諸表の発表日から少なくとも12ヶ月以内に経営を継続する能力があるかどうかに対する重大な疑いのリスクを軽減するのに十分であると信じています。
私たちは追加資金を調達し、私たちの運営に資金を提供し、私たちの製品開発努力を支援することを要求されるだろう。このような点で、私たちは、公共またはプライベート·エクイティまたは債務融資、特許使用料融資、または戦略的協力のような他のソースを含む多くの追加資本を積極的に求めている。しかし、私たちは追加的な資本を得ることができないかもしれないし、条件は受け入れられる、あるいは根本的にはできない。もし私たちが十分な資金をタイムリーに得ることができない場合、あるいは十分な資金が全くない場合、運営費用の削減、延期、縮小、停止、または私たちの研究·開発活動の変更など、私たちの流動性需要を満たすための追加の行動を取ることが要求されます。私たちが公開または私募株式発行によってより多くの資本を調達すれば、Jefferies LLCとの市場発売計画による販売を含めて、私たちの既存株主の所有権権益は希釈され、この希釈は巨大である可能性があり、任意の新しい株式証券の条項は、私たちの普通株式よりも優先し、私たちの普通株よりも高い権利を含むかもしれない。もし私たちが特許使用料融資または他の協力、戦略連合、または第三者との許可手配によって追加資本を調達する場合、私たちは私たちの候補製品、技術、将来の収入流、または研究計画にいくつかの価値のある権利を放棄するか、または有利ではないかもしれない条項で許可を付与する必要があるかもしれない。より多くの資金を集めると
債務融資によって、私たちは、追加債務を招く、資本支出を行う、またはいくつかの取引を行うなど、特定の金融契約または契約によって制限されるか、または特定の行動をとる能力を制限することができ、いずれも候補製品を商業化するか、または企業として運営する能力を制限する可能性がある。また、経営陣計画のコスト削減は、私たちの運営費用を削減し、私たちの現金資源を最適化することを目的としています。これらのコスト削減計画のスケジュールによると、2023年第3四半期から私たちの努力のメリットを実現することが予想されていますが、予想されたスケジュールでコスト削減計画のメリットを実現している保証はありません。
キャッシュフロー
経営活動
2022年の経営活動のための現金純額は2.236億ドルで、主に私たちの純損失が1.923億ドル、繰延収入が9130万ドル減少し、前払い費用とその他の資産が490万ドル増加し、レンタル負債が220万ドル減少したことを反映している。株式補償、減価償却、償却、リース使用権資産の償却と有価証券の割増(割引)の純償却に関する非現金支出は5100万ドル、売掛金やその他の負債は1330万ドル増加し、売掛金は230万ドル増加し、この減少額を部分的に相殺した。
2021年の経営活動のための現金純額は2.333億ドルで、主に私たちの純損失は1.783億ドル、繰延収入は8420万ドル減少し、売掛金とその他の負債は770万ドル減少し、前払い費用とその他の資産は720万ドル増加し、贈与終了に関するCIRM奨励調整負債は640万ドル減少し、リース負債は430万ドル減少した。株式補償、減価償却、償却、リース使用権資産の償却と有価証券の割増(割引)純償却に関する非現金支出5340万ドル、および非流動負債が120万ドル増加し、これらの減少額を部分的に相殺した。
投資活動
2022年、投資活動が提供する現金純額は5930万ドルで、主に3.56億ドルの有価証券の満期日と230万ドルの有価証券販売と関係があるが、2.774億ドルの有価証券と2020万ドルの不動産と設備の購入によって部分的に相殺される。
2021年、投資活動が提供する現金純額は2.482億ドルで、主に6.029億ドルの有価証券満期日と690万ドルの有価証券販売と関係があるが、3.382億ドルの有価証券購入と2330万ドルの不動産·設備購入部分によって相殺される。
融資活動
2022年、融資活動が提供する現金純額は8470万ドルで、主に8710万ドルの市場発行収益に関係しており、220万ドルの発売費用を差し引くと、わが従業員の株式購入計画で発行された普通株の収益は180万ドル増加し、株式純決済配当金奨励に関する税収は210万ドル減少した
2021年、融資活動が提供する現金純額は3290万ドルで、主に市場発行収益2790万ドルに関連しており、80万ドルの発売費用を差し引くと、560万ドルの増加は株式オプション行使の収益に関連しており、我々の従業員の株式購入計画に基づいて普通株を発行して得られた収益は340万ドル増加し、株式純決済配当金奨励に関する税収は330万ドル減少して相殺される。
運営資本および資本支出要求
私たちは少なくとも今後数年以内に営業赤字が続くと予想し、大量の追加資本を調達する必要がある。現在のマクロ経済環境の影響は、新冠肺炎の大流行、ウクライナ戦争の影響、インフレ、気候変化、金利上昇とその他の経済不確定性と波動性を含み、すでに世界金融市場の深刻な混乱を招き続ける可能性があり、これは受け入れ可能な条項または根本的に受け入れられない条項で資本を獲得する能力を弱める可能性があり、更に私たちの流動性に負の影響を与える可能性がある。今後12ヶ月後の将来の資本需要は巨大になり、私たちは大量の追加資本を集め、候補製品の開発、製造、潜在的な商業化に資金を提供する必要がある。このような点で、私たちは、公共またはプライベート·エクイティまたは債務融資、特許使用料融資、または戦略的協力のような他のソースを含む多くの追加資本を積極的に求めている。しかし、私たちは追加的な資本を得ることができないかもしれないし、条件は受け入れられる、あるいは根本的にはできない。もし私たちが十分な資金をタイムリーに得ることができない場合、あるいは十分な資金が全くない場合、私たちは、運営費用の削減や延期、縮小、停止、または私たちの研究·開発活動の変更など、コスト節約措置を含む追加の行動を要求され、私たちの業務に大きな悪影響を及ぼす可能性があります。
私たちが特許人間療法に集中する時、私たちは私たちの候補製品に対するFDAの承認を求める必要があり、この過程は各製品のコストを数億ドル以上にするかもしれない。利用可能な十分な資金がない場合、または潜在的な資金源の条項が不利であれば、私たちの業務と私たちの候補製品パイプラインを推進する能力は損なわれるだろう。私たちの未来の資本需要は多くの展望的な要素に依存するだろう
•私たちの候補製品および潜在的候補製品の臨床試験の開始、進展、スケジュール、および完了
•規制承認の結果、時間、コスト
•私たちの協力協定の成功は
•規制要件の変化がもたらす可能性のある遅延;
•私たちが求めている候補製品の数は
•特許出願の提出及び起訴、並びに特許請求の執行及び弁護に係る費用;
•将来の許可内と許可外取引の時間と条項
•販売、マーケティング、製造、流通能力のコストとタイミングを確立する
•私たちの候補製品の臨床と商業用品を調達するコストは
•私たちは、このような買収および投資に関連するコストを含む企業、製品または技術に買収または投資する程度である
•潜在的な紛争と訴訟の費用。
契約義務
2022年12月31日現在、私たちの契約義務は、主に(I)カリフォルニア州ブリスベン、カリフォルニア州リッチモンド、フランスバル州の施設における基本賃貸料、(Ii)製造、施設、設備に関する購入義務、および(Iii)許可可能内特許協定に関する持続的許可維持費の許可義務を含む運営リースに関するものである。これらの協定は、強制的かつ法的拘束力があり、使用される固定または最低サービス、固定、最低または可変価格、および契約に従って行動する実質的な時間を含むすべての重要な条項が規定されている。2022年12月31日現在の契約義務と約束に関するより多くの情報については、付記7-を参照されたい引受金とその他の事項本年度報告Form 10−K第2部第8項“財務諸表と補足データ”に掲載されている連結財務諸表に付記されている。
プロジェクト7 A−市場リスクの定量的かつ定性的開示について
私たちの市場リスクの開放は私たちの現金、現金等価物、有価証券と関係があります。私たちの投資政策の目標は、元本を保証し、流動性需要を満たすことと、私たちの投資政策パラメータと市場状況に基づいて市場収益率を得ることです。私たちはこのような指針の下で利息収入を最大限に増加させる投資を選択した。我々の目標を達成するために、期待される現金需要を満たすために、高い信用品質と異なる満期日を有する現金等価物と証券ポートフォリオとを維持している。
私たちのポートフォリオの証券はレバレッジ化されておらず、販売可能に分類されている。これらの売却可能な証券の多くは短期証券であり、金利リスクはわずかである。私たちの投資には現在、米国政府がサポートしている実体債務証券、商業手形証券、会社債務証券、資産保証証券、預金証書が含まれている。私たちの投資政策は取締役会によって承認され、任意の種類の投資発行者に投資できる金額を制限し、信用リスク集中度を低下させます。すべての投資は市価で計算されており、市価はコストにほぼ相当する。私たちはポートフォリオで派生金融商品を使用しない。市場金利が100ベーシスポイント上昇または低下した場合、私たちポートフォリオの公正価値はわずかな額だけ増加または低下するだろう。
外貨両替リスク
私たちはアメリカとヨーロッパで業務をしています。各外資系子会社の本位貨幣は本貨幣である.私たちは外貨リスクに直面しています。主にヨーロッパの子会社の業務を通じて、これらの子会社は主にユーロで業務を展開しています。子会社の財務諸表をドルに換算したため、株主権益に収益と損失を記録した。
わが海外子会社の業績を転換するための為替レート向上/(弱める)10%は、2022年12月31日までの年度純損失を約330万ドル増加させ、私たちの運営損失に実質的な影響を与えません。
また,米国とヨーロッパ間の活動レベルに関する外貨取引収益や損失も生じている。2022年、私たちは310万ドルの外国為替取引損失が発生した。2022年12月31日、ユーロとドルの為替レートに10%の悪影響があれば、2022年の外貨取引損失に無形の影響を与える。
2022年12月31日まで、私たちはアメリカで外貨建ての現金残高が何もありません。
プロジェクト8--財務諸表と補足データ
Sangamo治療会社
連結財務諸表索引 | | | | | |
| ページ |
| |
独立公認会計士事務所報告(PCAOB ID:42) | 104 |
合併貸借対照表 | 106 |
連結業務報告書 | 107 |
合併全面損失表 | 108 |
株主権益合併報告書 | 109 |
統合現金フロー表 | 110 |
連結財務諸表付記 | 111 |
独立公認会計士事務所報告
Sangamo治療会社の株主と取締役会に
財務諸表のいくつかの見方
Sangamo治療会社(当社)の2022年12月31日現在と2021年12月31日までの連結貸借対照表、2022年12月31日までの3年間の年間関連総合経営報告書、全面赤字、株主権益と現金流量および関連付記(総称して“総合財務諸表”と呼ぶ)を監査しました。総合財務諸表は、すべての重要な面で、2022年12月31日、2022年12月31日、2021年12月31日までの会社の財務状況、および2022年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
また、米国上場企業会計監督委員会(PCAOB)の基準に基づき、テレデビル委員会協賛組織委員会が発表した“内部統制-総合枠組み(2013枠組み)”で確立された基準に基づいて、2022年12月31日までの財務報告内部統制を監査し、2023年2月22日に発表された報告書について保留意見を発表した。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、(1)財務諸表に対して重大な意味を有する勘定又は開示に関し、(2)特に挑戦的、主観的、又は複雑な判断に係る財務諸表を監査して生じた事項である。重要監査事項の伝達は、財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、次の重要な監査事項を伝達することによって、重要な監査事項又はそれに関連する勘定又は開示について個別の意見を提供することもない。
| | | | | | | | |
| | 連携手配の収入確認 |
| | |
| 関係事項の記述 | | 同社の契約収入は、主に知的財産権の許可と研究と開発サービスの提供を含む協力手配から来ている。総合財務諸表付記1で述べたように、当社がその合意に規定された義務を履行する際に確認すべき収入金額を決定する際に、当社は、(I)契約で約束された貨物又はサービスを決定するステップと、(Ii)約束された貨物又はサービスが履行義務であるか否かを決定するステップと、(Iii)可変対価格を含む取引価格を計量するステップと、(Iv)推定販売価格に基づいて取引価格を履行義務に割り当てるステップと、(V)会社が履行義務を履行する際に収入を確認するステップとを実行する。投入方法を用いて関連履行義務履行の進捗状況(すなわち累積実コスト対推定総コスト)を測定し,一定期間収入を確認する 監査会社の協力手配下での収入会計は複雑であり、主に物質的権利の独立販売価格(行使の可能性を含む)を評価することに係る重大な判断と、履行義務の達成に要する総コストの会社の推定と、履行義務の達成に予想される総コスト推定の定期的な再評価によるものである。これらの見積もりの変化は確認された収入に実質的な影響を及ぼす可能性がある。
|
| 私たちが監査でどのようにこの問題を解決したのか | | 設計を評価し、社内会計処理の新たな連携スケジュールや既存の手配に関連する修正に対する内部制御の操作有効性をテストした。例えば、権利独立販売価格を決定するための管理層の制御をテストし、行使の可能性と、履行義務の達成に要する総コストの会社の推定と、履行義務の達成に期待される総コスト推定の定期的な再評価を行った。 それに加えて、私たちの監査手続きは検査協定と履行義務の評価の決定を含む。具体的な開発プロジェクトを担当する研究·開発者に問い合わせ,関連四半期合同指導委員会会議の記録を調べ,管理層が顧客に対して材料権利を行使する可能性を評価する仮説を評価することにより,決定された材料権利履行義務に対する会社の独立販売価格の推定を試験した。また,仮定の大きな変化による推定独立販売価格の変化を評価するために,独立販売価格を推定するためのこれらの仮定を感度分析した。各業績義務を達成するために必要な総コストの初期とその後の再計量をテストするために、著者らは具体的な開発プロジェクトを担当する研究と開発者に問い合わせ、関連四半期共同指導委員会会議の記録を調べ、プロジェクトごとに区分された予想総コストの推定に使用された経営陣の仮定を評価し、プロジェクトによって区分された総予想コストの変化を評価した。また,総期待コストの変化に対して感受性分析を行い,重大な変化を仮定した総期待コストの変化を評価した。我々は,従来の項目ごとに計算した予想総コストと実際に発生したコストを比較し,予測の正確性を評価した.項目ごとに累積実コストをテストし,会社モデルにより確認されたそれによる収入を再計算した。
|
/s/ 安永法律事務所
1997年以来、当社の監査役を務めてきました。
カリフォルニア州サンマテオ
2023年2月22日
Sangamo治療会社
合併貸借対照表
(単位は千で、1株当たりおよび1株当たりの金額は含まれていない) | | | | | | | | | | | |
| 十二月三十一日
2022 | | 十二月三十一日
2021 |
| 資産 | | | |
| 流動資産: | | | |
| 現金と現金等価物 | $ | 100,444 | | | $ | 178,872 | |
| | | |
| 有価証券 | 177,188 | | | 197,676 | |
| 受取利息 | 794 | | | 349 | |
| 売掛金 | 3,678 | | | 6,013 | |
| 前払い費用と他の流動資産 | 18,223 | | | 15,859 | |
| 流動資産総額 | 300,327 | | | 398,769 | |
| 非流通有価証券 | 29,845 | | | 88,169 | |
| 財産と設備、純額 | 63,531 | | | 51,523 | |
| 無形資産 | 50,729 | | | 53,760 | |
| 商誉 | 37,552 | | | 39,702 | |
| 経営的リース使用権資産 | 62,002 | | | 73,181 | |
| 他の非流動資産 | 17,023 | | | 15,319 | |
| 制限現金 | 1,500 | | | 1,500 | |
| 総資産 | $ | 562,509 | | | $ | 721,923 | |
| 負債と株主権益 | | | |
| 流動負債: | | | |
| 売掛金 | $ | 22,418 | | | $ | 9,759 | |
| その他負債を計算すべき | 16,007 | | | 11,577 | |
| 報酬と従業員の福祉に計上しなければならない | 21,506 | | | 20,840 | |
| 収入を繰り越す | 51,780 | | | 85,711 | |
| 流動負債総額 | 111,711 | | | 127,887 | |
| 繰延収入、非流動収入 | 109,377 | | | 166,776 | |
| 賃貸負債の長期部分 | 38,986 | | | 44,055 | |
| 所得税を繰延する | 6,270 | | | 6,645 | |
| 他の非流動負債 | 1,207 | | | 1,217 | |
| 総負債 | 267,551 | | | 346,580 | |
| 引受金とその他の事項 | | | |
| 株主権益: | | | |
優先株、$0.01額面は5,000,000ライセンス株、そして違います。発行済みまたは発行済み株式 | — | | | — | |
普通株、$0.01額面価値320,000,000ライセンス株;166,793,320そして145,921,530それぞれ2022年12月31日及び2021年12月31日に発行及び発行された株式 | 1,668 | | | 1,459 | |
| 追加実収資本 | 1,450,239 | | | 1,334,138 | |
| 赤字を累計する | (1,148,545) | | | (956,267) | |
| その他の総合損失を累計する | (8,404) | | | (3,987) | |
| | | |
| | | |
| 株主権益総額 | 294,958 | | | 375,343 | |
| 総負債と株主権益 | $ | 562,509 | | | $ | 721,923 | |
連結財務諸表付記を参照してください。
Sangamo治療会社
連結業務報告書
(千単位で、1株当たりを除く) | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| | | | | |
| | | | | |
| | | | | |
| 収入.収入 | $ | 111,299 | | | $ | 110,701 | | | $ | 118,192 | |
| 運営費用: | | | | | |
| 研究開発 | 249,898 | | | 230,819 | | | 180,647 | |
| 一般と行政 | 62,682 | | | 63,219 | | | 67,097 | |
| 総運営費 | 312,580 | | | 294,038 | | | 247,744 | |
| 運営損失 | (201,281) | | | (183,337) | | | (129,552) | |
| 利息とその他の収入,純額 | 9,432 | | | 5,346 | | | 8,775 | |
| 所得税前損失 | (191,849) | | | (177,991) | | | (120,777) | |
| 所得税費用 | 429 | | | 306 | | | 345 | |
| 純損失 | (192,278) | | | (178,297) | | | (121,122) | |
| 非持株権益は純損失を占めなければならない | — | | | (11) | | | (126) | |
| Sangamo治療会社の株主は純損失を占めるべきです | $ | (192,278) | | | $ | (178,286) | | | $ | (120,996) | |
| Sangamo治療会社の株主の1株当たり基本と希釈後の純損失 | $ | (1.25) | | | $ | (1.23) | | | $ | (0.90) | |
| Sangamo治療会社の株主の1株当たりの基本的かつ希釈後の純損失の株式を計算するための | 154,345 | | | 144,568 | | | 134,449 | |
連結財務諸表付記を参照してください。
Sangamo治療会社
総合総合損失表
(単位:千) | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 純損失 | $ | (192,278) | | | $ | (178,297) | | | $ | (121,122) | |
| 外貨換算調整 | (4,606) | | | (8,351) | | | 8,345 | |
| 年金純収益 | 786 | | | (716) | | | (193) | |
| 有価証券は赤字変動を実現せず,税引き後純額 | (597) | | | (339) | | | (284) | |
| 総合損失 | (196,695) | | | (187,703) | | | (113,254) | |
| 非持株権益は総合損失を占めなければならない | — | | | (11) | | | (126) | |
| Sangamo治療会社の全面的な損失。 | $ | (196,695) | | | $ | (187,692) | | | $ | (113,128) | |
連結財務諸表付記を参照してください。
Sangamo治療会社
合併株主権益報告書
(単位は千で、シェアは含まれていない) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 普通株 | | その他の内容
支払い済み
資本 | | 積算
赤字.赤字 | | 積算
他にも
総合収益 | | -ではない
制御管
利子 | | 合計する
株主の
権益 |
| 株 | | 金額 | | | | | |
| 2019年12月31日の残高 | 115,972,708 | | | $ | 1,160 | | | $ | 1,090,828 | | | $ | (656,985) | | | $ | (2,449) | | | $ | 185 | | | $ | 432,739 | |
| | | | | | | | | | | | | |
株式オプションを行使して制限株式単位に関連する場合に普通株を発行し,税金を差し引く | 1,395,956 | | | 14 | | | 8,545 | | | — | | | — | | | — | | | 8,559 | |
従業員株購入計画による普通株の発行 | 274,382 | | | 3 | | | 2,012 | | | — | | | — | | | — | | | 2,015 | |
生物遺伝研究協力協定に関する普通株発行,発行コストを差し引く | 24,420,157 | | | 244 | | | 142,282 | | | — | | | — | | | — | | | 142,526 | |
| 株に基づく報酬 | — | | | — | | | 25,708 | | | — | | | — | | | — | | | 25,708 | |
Sangamoの追加株式を買収する
フランス | — | | | — | | | — | | | — | | | — | | | (927) | | | (927) | |
| | | | | | | | | | | | | |
| 外貨換算調整 | — | | | — | | | — | | | — | | | 8,345 | | | | | 8,345 | |
| 年金純損失 | — | | | — | | | — | | | — | | | (193) | | | — | | | (193) | |
有価証券は純損失を実現せず,税引き後の純額 | — | | | — | | | — | | | — | | | (284) | | | — | | | (284) | |
| 純損失 | — | | | — | | | — | | | (120,996) | | | — | | | (126) | | | (121,122) | |
| 2020年12月31日の残高 | 142,063,203 | | | 1,421 | | | 1,269,375 | | | (777,981) | | | 5,419 | | | (868) | | | 497,366 | |
市場発行に関する普通株を発行し、発行費用を差し引いた純額 | 2,007,932 | | | 20 | | | 27,079 | | | — | | | — | | | — | | | 27,099 | |
株式オプションを行使して制限株式単位に関連する場合に普通株を発行し,税金を差し引く | 1,417,288 | | | 14 | | | 2,375 | | | — | | | — | | | — | | | 2,389 | |
従業員株購入計画による普通株の発行 | 433,107 | | | 4 | | | 3,366 | | | — | | | — | | | — | | | 3,370 | |
| 株に基づく報酬 | — | | | — | | | 32,956 | | | — | | | — | | | — | | | 32,956 | |
Sangamoの追加株式を買収する
フランス | — | | | — | | | (70) | | | — | | | — | | | (64) | | | (134) | |
| 外貨換算調整 | — | | | — | | | — | | | — | | | (8,351) | | | — | | | (8,351) | |
| 年金純損失 | — | | | — | | | — | | | — | | | (716) | | | — | | | (716) | |
有価証券は純損失を実現せず,税引き後の純額 | — | | | — | | | — | | | — | | | (339) | | | — | | | (339) | |
| 買断非持株権益 | — | | | — | | | (943) | | | — | | | — | | | 943 | | | — | |
| 純損失 | — | | | — | | | — | | | (178,286) | | | — | | | (11) | | | (178,297) | |
| 2021年12月31日の残高 | 145,921,530 | | | 1,459 | | | 1,334,138 | | | (956,267) | | | (3,987) | | | — | | | 375,343 | |
市場発行に関する普通株を発行し、発行費用を差し引いた純額 | 19,300,743 | | | 193 | | | 84,676 | | | — | | | — | | | — | | | 84,869 | |
株式オプションを行使して制限株式単位に関連する場合に普通株を発行し,税金を差し引く | 994,097 | | | 10 | | | (1,990) | | | — | | | — | | | — | | | (1,980) | |
従業員株購入計画による普通株の発行 | 576,950 | | | 6 | | | 1,765 | | | — | | | — | | | — | | | 1,771 | |
| 株に基づく報酬 | — | | | — | | | 31,650 | | | — | | | — | | | — | | | 31,650 | |
| | | | | | | | | | | | | |
| 外貨換算調整 | — | | | — | | | — | | | — | | | (4,606) | | | — | | | (4,606) | |
| 年金純収益 | — | | | — | | | — | | | — | | | 786 | | | — | | | 786 | |
有価証券は純損失を実現せず,税引き後の純額 | — | | | — | | | — | | | — | | | (597) | | | — | | | (597) | |
| | | | | | | | | | | | | |
| 純損失 | — | | | — | | | — | | | (192,278) | | | — | | | — | | | (192,278) | |
| 2022年12月31日の残高 | 166,793,320 | | | $ | 1,668 | | | $ | 1,450,239 | | | $ | (1,148,545) | | | $ | (8,404) | | | $ | — | | | $ | 294,958 | |
連結財務諸表付記を参照してください。
Sangamo治療会社
統合現金フロー表
(単位:千) | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 経営活動: | | | | | |
| 純損失 | $ | (192,278) | | | $ | (178,297) | | | $ | (121,122) | |
| 純損失と経営活動から提供される現金純額の調整: | | | | | |
| 減価償却および償却 | 12,108 | | | 9,439 | | | 5,682 | |
| 有価証券(割引)を割増償却する | (1,242) | | | 2,844 | | | (825) | |
| 経営的リース使用権資産の償却その他の変動 | 8,454 | | | 8,199 | | | 7,687 | |
| 株に基づく報酬 | 31,650 | | | 32,956 | | | 25,708 | |
| 無料株収益 | — | | | (18) | | | (63) | |
| | | | | |
| 財産と設備の純損を処分する | — | | | (52) | | | 222 | |
| 贈与終了に関するCIRM決裁責任の調整 | — | | | (6,427) | | | — | |
| | | | | |
| | | | | |
| | | | | |
| 営業資産と負債の純変化: | | | | | |
| 受取利息 | (445) | | | 686 | | | (353) | |
| 売掛金 | 2,335 | | | (789) | | | 31,685 | |
| 前払い費用と他の資産 | (4,909) | | | (7,175) | | | (10,411) | |
| | | | | |
| 支払すべき帳簿その他の負債 | 13,348 | | | (7,664) | | | 10,703 | |
| 報酬と従業員の福祉に計上しなければならない | 941 | | | 373 | | | 6,877 | |
| 収入を繰り越す | (91,331) | | | (84,202) | | | 216,546 | |
| 賃貸負債 | (2,249) | | | (4,340) | | | (3,761) | |
| 他の非流動負債 | (9) | | | 1,216 | | | 1,300 | |
| 経営活動が提供する現金純額 | (223,627) | | | (233,251) | | | 169,875 | |
| 投資活動: | | | | | |
| | | | | |
| 有価証券を購入する | (277,391) | | | (338,159) | | | (570,779) | |
| 有価証券の満期日 | 354,587 | | | 602,885 | | | 314,570 | |
| 有価証券の販売 | 2,260 | | | 6,870 | | | — | |
| 財産と設備を購入する | (20,171) | | | (23,278) | | | (14,714) | |
| Sangamoフランス株を追加購入します | — | | | (119) | | | (704) | |
| | | | | |
| | | | | |
| 投資活動提供の現金純額 | 59,285 | | | 248,199 | | | (271,627) | |
| 融資活動: | | | | | |
| 市場で発行された収益は,発行費用を差し引く | 84,869 | | | 27,099 | | | — | |
| | | | | |
| 生物遺伝研究会社との連携協定に関する普通株発行による収益は,発行コストを差し引く | — | | | — | | | 142,526 | |
| 配当金の株式純額決済に関する支払済み税 | (2,104) | | | (3,258) | | | (765) | |
| 従業員の株式購入計画に基づいて普通株を発行して得た金 | 1,771 | | | 3,369 | | | 2,015 | |
| 株式オプションを行使して得られる収益 | 124 | | | 5,648 | | | 9,324 | |
| 融資活動が提供する現金純額 | 84,660 | | | 32,858 | | | 153,100 | |
| 現金と現金等価物および制限現金に及ぼす為替レート変動の影響 | 1,254 | | | (263) | | | (447) | |
| 現金、現金等価物、および制限的現金純増加 | (78,428) | | | 47,543 | | | 50,901 | |
| 期初現金、現金等価物、および限定現金 | 180,372 | | | 132,829 | | | 81,928 | |
| 現金、現金等価物、制限された現金、期末 | $ | 101,944 | | | $ | 180,372 | | | $ | 132,829 | |
| 補足キャッシュフロー開示: | | | | | |
| | | | | |
| 未払い負債に計上する財産と設備 | $ | 6,539 | | | $ | 1,535 | | | $ | 4,569 | |
| テナント改善手当は反賃貸負債に含まれています | $ | 243 | | | $ | — | | | $ | — | |
| 買断非持株権益 | $ | — | | | $ | 943 | | | $ | — | |
| 賃貸義務と引き換えに使用権資産 | $ | — | | | $ | 10,418 | | | $ | 1,333 | |
連結財務諸表の付記を参照
Sangamo治療会社
連結財務諸表付記
NOTE 1 – 重大会計政策の組織·列報根拠とまとめ
業務の組織と記述
Sangamo治療会社(“Sangamo”または“当社”)は1995年6月にデラウェア州に登録設立され、2017年1月にSangamo生物科学会社から改名された。Sangamoは臨床段階のゲノム薬物会社であり、画期的な科学を深刻な疾病患者の生活を変える薬物に転化することに力を入れている。
陳述の基礎
添付されている総合財務諸表は、当社及びその付属会社の勘定を含む米国公認会計原則(“米国公認会計原則”)に基づいて作成されている。すべての会社間残高と取引は連結財務諸表から抹消されました。当社が100%以下の経済リスクを持っているか、または直面している合併実体については、当社はその総合経営報告書に非持株権益を純損失を占めるべきであり、各非制御側が当該などの実体に保持している経済或いは所有権権益のパーセンテージに相当する。
流動性資本資源経営陣の計画
Sangamoは現在,実験技術に関するいくつかの長期開発プロジェクトを行っている.このようなプロジェクトは達成するために数年と多くの支出がかかるかもしれないし、最終的には成功しないかもしれない。近年、同社の運営資金は主に協力と戦略パートナーシップ、研究支出、株式証券の発行から来ている。2022年12月31日までの会社の資本資源は307.5現金、現金等価物、そして有価証券を含む百万ドル。経営陣は、会社の既存の現金、現金等価物、および有価証券は、これらの総合財務諸表の発行日から少なくとも12ヶ月以内にその運営に資金を提供するのに十分であると信じている。
会計基準編集(“ASC”)テーマ205−40では、財務諸表の列報−継続経営−(“ASC主題205-40”)企業は、これらの債務が合併財務諸表の発行日から1年以内に満了するので、条件および/またはイベントが将来の財務義務を履行する能力を有するかどうかを評価する責任がある。ASCテーマ205-40の要求によると、経営陣の評価は、当初、連結財務諸表の発表日までに完全に実施されていない経営陣計画の潜在的緩和効果を考慮すべきではない
実質的な疑いを引き起こした
第1ステップ評価を行ったところ、同社は、持続経営企業としての持続経営能力に大きな疑いを抱かせたと結論した
•純損失$192.3百万ドルとドル178.32022年12月31日と2021年12月31日までの年度はそれぞれ100万ユーロ、経常純損失の歴史となっている
•累計赤字は#ドル1,148.5百万ドルとドル956.3それぞれ2022年12月31日と2021年12月31日まで。
経営陣の計画に対する考え方
この評価の第2ステップを行う際には、当社は、その計画が総合財務諸表発行後1年以内に有効に実施される可能性があるかどうか、およびその継続的な経営能力に対する外部の実質的な疑いを解消する可能性があるかどうかを評価する必要がある。
会社は、総合財務諸表の発表日から12ヶ月間の会社の流動性需要を満たすために、適時に起動されるコスト保存措置を含むいくつかの可能な行動を決定した
•いくつかの研究開発計画の優先順位を延期して再決定することは、計画と従業員の支出を減らすことに関連する
•新しい求人を一時停止し、出張や招聘費用などの補助費用を削減する
•追加設備、実験室改善、効率プロジェクト、および業務支援支出を含む非重要資本および運営支出を削減します。
持続的経営能力の管理評価
当社は、経営陣の計画は、上述したように、その財務義務を履行するのに十分な流動資金を提供し、総合財務諸表の発表日から12ヶ月間流動資金レベルを維持すると信じている。したがって、経営陣は、これらの計画は、同社が総合財務諸表発表日から少なくとも12ヶ月以内に経営を継続する能力に対する大きな疑いを緩和したと結論している。
添付されている総合財務諸表の作成仮説会社は、正常業務過程で資産と負債を清算することを考えている継続的な経営企業として経営している。総合財務諸表は、将来的に資産の回収可能性および分類に及ぼす可能性のある影響、または企業の持続的な経営企業としての能力に関連する不確実性に起因する可能性のある負債額を反映するための調整を含まない。
未来の計画と考慮事項
同社は追加の資本を調達し、その運営に資金を提供し、その製品開発努力を支援することが求められる。このような点で、会社は、公共またはプライベート·エクイティまたは債務融資、特許使用料融資、または戦略的協力のような他のソースを含む多くの追加資本を積極的に求めている。しかし、受け入れ可能な条項または全く受け入れられない条項の下で、会社は追加的な資本を得ることができないかもしれない。会社が十分な資金をタイムリーに得ることができない場合、あるいは十分な資金が全くない場合、会社は運営費用の削減、延期、縮小、停止、またはその研究開発活動のコスト保存措置を含む追加の行動を取ってその流動性需要を満たすことを要求されるだろう。もし同社が特許使用料融資または他の協力、戦略連盟、または第三者との許可手配によって追加資本を調達する場合、それは、その候補製品、技術、将来の収入流、または研究計画のいくつかの価値のある権利を放棄するか、または不利になる可能性のある条項で許可を付与する必要があるかもしれない。会社が公開またはプライベートエクイティ発行によってより多くの資本を調達する場合、Jefferies LLCとの市場発売計画による販売を含む場合、その既存株主の所有権権益は希釈され、このような希釈は巨大である可能性があり、任意の新しい株式証券の条項は、その普通株式よりも優先し、その普通株よりも高い権利を含む可能性がある。もし会社が特許使用料融資または他の協力、戦略連盟、または第三者との許可手配によって追加資本を調達する場合、それはいくつかの価値のある権利をその候補製品、技術に放棄する必要があるかもしれない, 将来の収入源や研究計画や許可証を付与する条項はあまり有利ではないかもしれない。企業が債務融資によって追加資本を調達する場合、それは、追加債務を招く、資本支出を行う、または何らかの取引を行うなど、特定の財務契約またはチノによって制限されるか、または特定の行動をとる能力を制限する可能性があり、これらのいずれも、その候補製品を商業化するか、または業務として運営する能力を制限する可能性がある。また、経営陣が計画しているコスト削減は、会社の運営費用を削減し、その現金資源を最適化することを目的としている。これらのコスト削減計画のスケジュールによると、同社は2023年第3四半期からその努力のメリットを実現すると予想されているが、会社が予想されるスケジュールでコスト削減計画のメリットを実現する保証はない、あるいは全く保証されていない。
重要会計政策の概要
予算の使用
アメリカ公認会計原則に従って連結財務諸表を作成することは、合併財務諸表と付記中の報告の金額に影響を与えるために、経営陣に推定と仮定を要求する。管理層は、重要な会計政策または収入確認、臨床試験課税項目、所得税、資産および負債の公正価値(買収所得を含む)および株式報酬に関する推定を含む、その推定値を継続的に評価する。過去の経験や当社が当時の状況で部下が合理的だと考えていた様々な他の特定の市場やその他の関連仮定に基づいて行われたものであり、その結果は資産や負債の額面を判断する基礎となり、そのような資産や負債の帳簿価値は他の出所から容易に現れるわけではないと思われる。実際の結果はこれらの推定とは異なる可能性がある。
同社は2021年12月31日までの年間で、セノフィ社(“セノフィ”)提携協定に関する見積もり変化に関する収入調整を記録している。これらの見積り数が変動した原因は,2021年9月にプロジェクト範囲や関連プロジェクト費用が変化したことと,その後2022年6月28日に連携協定の終了通知が出され,比例累積実行状況の測定基準が変化したためである.これらの調整は収入を#ドル減少させた1.6百万ドル純損失増加$1.6100万ドルで会社の1株当たり基本的に希釈して純損失を増加させました0.012021年12月31日までの年度。
2020年12月31日までの年度内に、セノフィやファイザーとの協力協定(“ファイザー”)の見積もりが変化したため、同社は収入に関する調整を記録した。これらの推定数の変化は,項目範囲や関連項目費用の変化によるものであり,比例積算実行状況計測の変化を招いている.このような調整は収入をドル増加させた8.9百万ドル純損失が減少しました8.9百万ドル、会社の1株当たり基本と希釈後の純損失を#ドル減少させます0.062020年12月31日までの年度。
収入確認
当社は会計基準編纂(“ASC”)テーマ606の規定に基づいてその収入を会計処理している取引先と契約した収入(“ASCトピック606”)。同社の契約収入は、許可手配や研究サービスを含む協力協定から来ている。研究およびライセンス契約には、一般に、払戻不可能な前署名可能費用、契約料率による会社の研究者にかかる時間の支払い、第三者コスト補償、追加の目標選択費用、再許可費用、持続可能な開発および製品商業化に関連するマイルストーン支払い、および将来のライセンシー製品販売の印税が含まれる。会社のパートナーから受け取ったすべての資金は一般的に返金されません。払い戻しできない前払い料金は契約開始時に確定します。他のすべての費用は契約書の可変対価格を代表します。同社の契約の1つには、私たちがその顧客に発生したいくつかの費用を補償する条項が含まれています。これらの費用は、このような支払いと交換するために異なる商品やサービスを得ていないので、契約取引価格の減価に計上されています。繰延収入とは、主に受け取ったが稼いでいない払戻不可能な前払いまたはマイルストーン支払いの部分を指す。
会社がその合意に規定された義務を履行する際に確認すべき適切な収入額を決定する場合、会社は、(1)契約で約束された貨物またはサービスを決定するステップと、(2)契約で異なるか否かを含む約束された貨物またはサービスが履行義務であるかどうかを決定するステップと、(3)可変対価格の制限を含む取引価格を測定するステップと、(4)推定販売価格に基づいて取引価格を履行義務に割り当てるステップと、(5)会社が各履行義務を履行する場合(または義務を履行するとき)に収入を確認するステップとを実行する
同社のその協力協定における大部分の履行義務は異なる知的財産権許可と研究開発サービスバンドルを代表しており、これらの構成部分は単独で異なる。私たちの知的財産権および/または研究開発サービスの取得を許可するオプションは、顧客に実質的な権利を付与する際の義務を表しており、例えば、顧客が既存の契約に従って私たちのサービスを購入していない場合、彼らは割引の権利を得ない
知的財産権と研究開発サービスバンドル許可からの収入は比例業績法を用いて経時的に確認した。この方法によれば,収入は関連履行義務の履行の進捗状況を測定することで確認され,採用された測定基準は履行に関する義務履行の進捗状況を最も反映している。会社のほとんどの合意について、進捗の測定基準は、会社の研究者の実際の時間価値に第三者コスト補償を加えた、生成された努力レベルに基づく投入測定基準である
実質的な権利を含むオプションに割り当てられた対価格は、オプション行使または満期に延期されてから行われる。このオプションの行使は契約更新と記載され,目標選択費用と推定可変対価格はそのときの取引価格に含まれ,それぞれの目標の履行義務に特化して割り当てられる
一つの手配を決定するために必要な努力の程度と、当社がその手配の下でその履行責任を達成することを期待している間には、重大な経営陣の判断が必要である。これらの見積もりの変化は確認された収入に実質的な影響を及ぼす可能性がある。会社が義務履行がいつ完了したか、どうでもいいと合理的に見積もることができない場合、会社が合理的にその等の推定を行うことができるまで収入確認が延期される。可変対価については,取引価格に含まれる金額が確認された累積収入に制限され,大きな逆転が生じない可能性が高い金額である.その後報告期間ごとに終了すると,当社は取引価格に含まれる推定変動コストおよび任意の関連制限を再評価し,必要に応じて全体の取引価格の推定を調整する.そして今期記録で累計追跡し、更新された取引価格と更新の進捗測定基準を反映する。会社の研究者の時間価値と第三者コストを含む業績期間と作業レベルを推定し、四半期ごとに審査し、必要に応じて調整して、会社の現在の期待を反映させる。
これらの手配に対する会計処理の一部として、会社は仮説を立てなければならず、これらの仮定は、契約で決定された個々の履行義務の独立販売価格を決定するために判断する必要がある。同社は重要な仮定を使用して独立販売価格を決定し、その中には予測収入、開発スケジュール、割引率と技術行使と監督管理が成功する確率、および研究開発サービスの期待努力レベルが含まれている可能性がある
我々の収入確認および主要顧客、パートナー、および戦略連盟に関連するいくつかの開示は、前の期間に関連する非実質的な変化に適合するように更新された。
主な協力協定と研究活動の贈与の収入が総収入に占める割合は以下の通りである | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| | | | | |
| ノワール生物医学研究所です | 36 | % | | 34 | % | | 4 | % |
| 凧製薬会社 | 35 | % | | 23 | % | | 24 | % |
| 生物遺伝研究会社 | 26 | % | | 38 | % | | 24 | % |
| セノフィ社 | 3 | % | | 3 | % | | 5 | % |
| ファイザー。 | — | % | | — | % | | 40 | % |
売掛金
売掛金には、会社のパートナーに支払う研究サービス費用の精算金額が含まれています。協力からの売掛金は通常無担保であり、バイオ製薬業界に集中している。したがって、同社は、通常、生物製薬会社に関連しているか、またはその協力協定に関連する信用リスクに直面している可能性がある。当社は信用損失を差し引いて準備した売掛金を記録します。当社は帳簿年齢方法を用いて信用損失を推定し、その歴史損失資料(現在の状況を考慮するように調整された)、及びその顧客に影響する未来の経済状況の合理的かつ支持可能な予測を考慮した。2022年12月31日現在、会社では当該等の売掛金に関する損失は何も発生していない。2022年12月31日と2021年12月31日まで、売掛金の10%以上を単独で占める提携パートナーの売掛金比率は以下の通り
| | | | | | | | | | | |
| 12月31日まで |
| 2022 | | 2021 |
| ノワール生物医学研究所です | 59 | % | | 32 | % |
| 凧製薬会社 | 19 | % | | 2 | % |
| 生物遺伝研究会社 | 14 | % | | 46 | % |
| | | |
| セノフィ社 | — | % | | 11 | % |
| | | |
| | | |
商誉と無形資産
営業権とは、企業合併で取得した資産と負担した負債が移転した価格が公平な価値を超えた部分である。使用年数が不確定な無形資産は購入した進行中の研究開発(“IPR&D”)プロジェクトと関係があり、買収日それぞれの公正価値によって計量される。耐用年数不確定の営業権と無形資産は償却しない。知的財産権研究開発プロジェクトに関連する無形資産は無期限とされている−関連研究や開発が完了または放棄されるまで存在している。開発が完了した場合、一般に、規制機関の許可を得て製品を販売する場合、関連資産は有限寿命とみなされ、その後、その時点でのそれぞれの推定使用寿命に基づいて償却される。当社は毎年、営業権および無期限無形資産に対して減値テストを行い、当社が任意の事件や環境変化が当該などの資産の公正価値がそれぞれの帳簿価値よりも低いことを示すように、当社は年次テストの間で当該などの資産の減値をテストする。2022年12月31日までに違います。営業権の減価または無期限無形資産が確認された。
長寿資産の評価
内部または外部の事実または状況が資産の帳簿価値が回収できない可能性があることを示唆する可能性がある場合、長期資産、財産および設備、および限られた寿命を含む無形資産は、減値が検討される。これらの資産の回収可能性は,個々の資産の帳簿価値と資産使用とその最終処分が予想される将来の未割引現金流量とを比較することで測定される。この資産が減値とみなされる場合、いずれの減値金額も、減値資産の帳簿価値と公正価値との差額で計量される。2022年12月31日までに違います。長期資産の減値を確認した。
公正価値計量
現金及び現金等価物,売掛金,売掛金及びその他の計上すべき負債からなる金融商品の帳簿金額は,その短期満期日により公正価値に近い。有価証券はその見積もりに基づいて公正価値を列報する.
現金、現金等価物、制限された現金
Sangamoは、購入日に購入された元の満期日が3ヶ月以下であるすべての高流動性投資を現金等価物とみなす。現金と現金等価物には現金、普通通貨市場口座の預金と米国政府が支援する実体債務証券。制限された現金は#ドルの信用状で構成されている1.5百万ドルで、カリフォルニア州ブリスベン社の本社を借りる保証金に相当します。
統合貸借対照表で報告されている現金、現金等価物、および限定的な現金と合併キャッシュフロー表で報告されている金額の入金状況は以下のとおりである(千計) | | | | | | | | | | | | | | | | | | | |
| 12月31日まで |
| 2022 | | 2021 | | 2020 | | |
| 現金と現金等価物 | $ | 100,444 | | | $ | 178,872 | | | $ | 131,329 | | | |
| | | | | | | |
| 非流動制限現金 | 1,500 | | | 1,500 | | | 1,500 | | | |
| 現金フロー表で報告されている現金、現金等価物、および限定的な現金 | $ | 101,944 | | | $ | 180,372 | | | $ | 132,829 | | | |
有価証券
Sangamoはその有価証券を販売可能に分類し、ほぼ同じ資産の見積市場価格または観察可能な市場投入に基づいて、推定公正価値に従ってその投資を記録し、保有損益を累積他の全面収益(損失)“AOCI”)に計上することは実現されていない。当社は、現在の業務で使用する必要がない、12ヶ月後に満期となる投資を、添付の総合貸借対照表中の非流動有価証券に分類します。
当社の投資は定期減価審査を受けなければなりません。当社は、減価費用を確認するか否かを決定する際に、公正価値が当社のコスト基準を下回る時間の長さと程度、被投資者の財務状況及び最近の見通し、及び当社が投資に関する意向及び能力を持っており、市場価値をどのような期待を回復させるのに十分であるかを考慮する。有価証券の実現済み収益と損失を利子や他の収入純額に計上し、特定の確認方法を用いて決定する。有価証券に関連する信用損失は利子やその他の収入(費用)を計上し、信用損失により証券償却コストベースの減値ではなく、総合経営報告書純額に計上する準備をしている。
信用リスクとその他のリスクの集中度
現金、現金等価物、および有価証券は、総合貸借対照表に記録された公正価値に依存する会社に集中的な信用リスクを受ける可能性のある金融商品を含む。同社は不要な現金を主にリスクが最小の高流動性ツールに投資する。会社は証券の品質、多様化、満期日に関する政策を制定し、会社がその信用リスクを管理できるようにした。当社の現金、現金等価物および総合貸借対照表に記録されている投資を持つ金融機関や発行者が違約すれば、当社は信用リスクに直面します。
会社が運営に使用しているいくつかの材料と重要な部品は単一のサプライヤーによって獲得された。肝心な部品と材料のサプライヤーはアメリカ食品·薬物管理局(FDA)に提出した調査性新薬申請(IND)にリストしなければならないため、新しいサプライヤーの資格が必要であれば、重大な遅延が生じる可能性がある。もし会社のサプライヤーの材料納入がいかなる原因で中断すれば、会社はそのいかなる候補製品を供給して臨床試験を行うことができないかもしれない。
財産と設備
財産と設備はコストから減価償却累計と償却を差し引く。減価償却は関連資産の推定耐用年数から直線法で計算され、推定耐用年数は一般的に三つ至れり尽くせり5年それは.賃貸改善については、使用年数またはレンタル期間の短い1つに基づいて、直線法を用いて償却を計算する。事件や状況の変化がある資産の帳簿金額が回収できない可能性があることを示すたびに、当社はその物件や設備の減価状況を審査します。
研究と開発費
研究と開発費用は主に人員費用を含み、賃金、福祉と株式給与、契約研究組織による臨床研究、材料と用品、各種支援と施設に関連する費用からなる間接費用分配を含む。研究·開発コストは発生時に費用を計上する。
一般と行政費用
一般的で行政的費用には財務、人的資源、法律、および他の行政活動が含まれる。これらの費用には、主に人件費が含まれ、賃金、福祉、株式ベースの補償、施設と間接費用、法律費用、その他の一般的および行政費用が含まれる。
株に基づく報酬
当社は、従業員株式オプション、制限株式単位(“RSU”)および従業員株式購入計画(“ESPP”)に関連する従業員株式購入を含む、奨励付与日の推定公正価値計量と確認に基づいて、Sangamo従業員および取締役に支払われたすべての株式に報酬の補償費用を支払う。株式奨励の公正価値は、奨励の帰属中に直線法で償却される。
報酬の公正価値を見積もるために、同社はブラック·スコアーズオプション定価モデルを使用している。このモデルは期待寿命、予想変動率、株式期待配当収益率と無リスク金利を入力する必要がある。これらの投入は主観的であり,通常大量の分析と判断が必要である.期待寿命と変動性の推定は主に同社の歴史データによるものであるが、無リスク金利は付与時に有効な米国債収益率曲線に基づいており、期待寿命仮定に見合っている。当社は発生期間の没収を精算します。
所得税
所得税費用は貸借対照法で計算されます。繰延税項資産及び負債は、財務諸表と資産及び負債の税ベースとの差額に基づいて決定され、この差額は、これらの差額を返送する際に発効した制定された税率によって計量される。既存の証拠によると、繰延税金資産が現金化される可能性があまりない場合、当社は繰延税金資産の純資産に推定値を提供する準備をする。
当社は、税務機関が税務状況の技術的価値に基づいて審査を行った後、その税務状況を維持する可能性が高い場合にのみ、不確定税務状況からの税務利益を確認します。企業の総合財務諸表で確認されたこれらのポストからの税金優遇は、実現可能性が50%を超える最大割引に基づいて測定される。当社は、税務関連の利息及び罰金が所得税の一部であることを確認し、計上すべき利息及び罰金を、関連する所得税負債とともにその総合貸借対照表に計上すべき他の計算すべき負債に計上する。当社は、不確定な税務状況を定期的に評価し、事実や状況が変化した場合に、税務監査を終了したり、見積もりを改善したりするなど、これらの課税項目を調整します。
賃貸借証書
当社は、1つのスケジュールに確認された資産が含まれているかどうか、および確認された資産を制御する権利があるかどうかを評価することによって、そのスケジュールが開始時にレンタルまたはレンタルを含むかどうかを決定する。使用権(“ROU”)資産代表会社がリース期間内に対象資産を使用する権利は、リース負債代表会社がリースにより発生したリース金を支払う義務を有する。レンタル負債はレンタル開始日にレンタル期間内の将来のレンタル支払いの現在値に基づいて確認します。純収益資産は、リース負債の計量に基づいており、レンタル開始前またはレンタル開始時に支払われる任意のレンタル支払いも含まれており、レンタル報酬および最初に生じる直接コストは含まれていない(場合によっては)。
当社の借款の暗黙的金利は一般的に未知であるため、当社は借款開始日に得られる資料に基づいて、逓増借款金利を採用して余剰賃貸支払いの現在値を決定している。逓増借款金利は、当社が賃貸開始時に類似経済環境下での賃貸期限内に担保をもとに賃貸支払いに相当する金額を借り入れることによる金利の見積もりである。当社はその逓増借入金利を計算する際に、その信用リスク、レンタル期間および賃貸支払い総額を考慮し、必要がある場合には担保の影響に応じて調整します。レンタル条項は、会社が任意のこのような選択権を行使することを合理的に決定したときに、リース契約を延長または終了する選択権を含むことができる。当社が経営している賃貸料支出はレンタル期間内の直線的な基礎で確認します。
当社はその不動産とコピー機リースのリースと非レンタル構成要素を分離しないことを選択したため、任意のレンタル構成要素と非レンタル構成要素を単一リース構成要素として会計処理を行う。当社もいずれの年間にも適用されない12ヶ月以下の賃貸契約を選択しており、当社が合理的に必ず行使する標的資産を購入する選択権も含まれていません。
外貨換算
会社の海外子会社の本位貨幣は主にユーロです。外貨建ての資産と負債を貸借対照表の日の為替レートでドルに換算する。外貨換算調整は株主権益内でAOCIの構成要素と表記されている。当社の海外子会社の収入と支出は#年期間の毎月平均為替レートに換算します
取引が起こった位置。外貨取引損益は当社の総合経営報告書に利息とその他の収入純額を計上します。
1株当たり純損失
Sangamo治療会社の株主が1株当たりの基本純損失を占めるべき計算方法は:Sangamo治療会社株主は純損失をこの期間に発行された普通株の加重平均で割るべきである。Sangamo治療会社の株主が1株当たりの純損失を占めるべき計算方法は:Sangamo治療会社の株主は純損失を占め、普通株の加重平均にその間に発行した潜在的な希薄化証券を加えるべきである。
Sangamo治療会社株主の希釈1株当たり純損失を計算する際には,株式オプションや発行済み株式単位に拘束された株式総数および発行のために保留されたESPP株は含まれておらず,これらの株は逆希釈されている。2022年、2021年、2020年12月31日現在、発行済み株式オプションと発行済みRSUおよび予約発行ESPP株は18,560,755, 15,159,908そして、そして14,237,871それぞれ,である.
細分化市場
同社は以下の地域で運営している1つは市場を細分化する。経営陣は収益性を測る指標を用いており、その業務を分けて内部報告を行っていない。2022年12月31日と2021年12月31日まで、会社の財産や設備の大部分は米国に残されている。2022年12月31日、2021年12月31日、2020年12月31日までの年間で、会社のすべての収入が米国で発生している。
最近の会計公告
ない。
NOTE 2 – 公正価値計量
当社は公正価値によって一定の基礎に基づいていくつかの金融資産と負債を計量し、現金等価物と有価証券を含む。公正価値は公正価値計量と開示の権威指導の下で三級階層構造によって確定され、公正価値計量に使用される投入優先順位は以下の通りである
レベル1:同じ、制限されていない資産または負債が計量日に得られるアクティブ市場の調整されていないオファー;
第2レベル:資産または負債の全期間にわたって、非アクティブな市場オファーまたは直接または間接的に観察可能な投入;および
第3級:価格や推定技術は公正な価値計量に重大な意義があるが観察できない投入が必要である(つまり、市場活動の支援は少ないか全くない)。
同社の現金等価物および有価証券の公正価値計量は、公正価値システム内の以下のレベルで決定される(千で計算) | | | | | | | | | | | | | | | | | | | | | | | |
| 2022年12月31日 |
| 公正価値計量 |
| 合計する | | レベル1 | | レベル2 | | レベル3 |
| 資産: | | | | | | | |
| 現金等価物: | | | | | | | |
| 貨幣市場基金 | $ | 50,820 | | | $ | 50,820 | | | $ | — | | | $ | — | |
| | | | | | | |
| 合計する | 50,820 | | | 50,820 | | | — | | | — | |
| 有価証券: | | | | | | | |
| アメリカ政府が支援する実体債務証券 | 18,417 | | | — | | | 18,417 | | | — | |
| 商業手形証券 | 101,165 | | | — | | | 101,165 | | | — | |
| 会社債務証券 | 11,670 | | | — | | | 11,670 | | | — | |
| 資産支援証券 | 24,792 | | | — | | | 24,792 | | | — | |
| アメリカ国庫券 | 7,938 | | | | | 7,938 | | | |
| 預金証書 | 37,461 | | | — | | | 37,461 | | | — | |
| 機構債券 | 5,590 | | | | | 5,590 | | | |
| 合計する | 207,033 | | | — | | | 207,033 | | | — | |
| 現金等価物と有価証券総額 | $ | 257,853 | | | $ | 50,820 | | | $ | 207,033 | | | $ | — | |
| | | | | | | |
| | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
| 2021年12月31日 |
| 公正価値計量 |
| 合計する | | レベル1 | | レベル2 | | レベル3 |
| 資産: | | | | | | | |
| 現金等価物: | | | | | | | |
| 貨幣市場基金 | $ | 119,919 | | | $ | 119,919 | | | $ | — | | | $ | — | |
| | | | | | | |
| 合計する | 119,919 | | | 119,919 | | | — | | | — | |
| 有価証券: | | | | | | | |
| アメリカ政府が支援する実体債務証券 | 30,614 | | | — | | | 30,614 | | | — | |
| 商業手形証券 | 105,757 | | | — | | | 105,757 | | | — | |
| 会社債務証券 | 33,682 | | | — | | | 33,682 | | | — | |
| 資産支援証券 | 70,701 | | | — | | | 70,701 | | | — | |
| 預金証書 | 45,091 | | | — | | | 45,091 | | | — | |
| 合計する | 285,845 | | | — | | | 285,845 | | | — | |
| 現金等価物と有価証券総額 | $ | 405,764 | | | $ | 119,919 | | | $ | 285,845 | | | $ | — | |
| | | | | | | |
| | | | | | | |
現金等価物と有価証券
当社では一般に有価証券を2段階に分類しています。あまり活発でない市場で取引されている同じ証券の観察可能な市場価格を使用する場合、ツールは2段階に分類されます。同じ証券の観察可能な市場価格が得られない場合、このようなツールは、基準曲線、同種の証券の基準、業界グループ、行列定価、および推定モデルを用いて価格設定を行う。これらの評価モデルは、価格設定プロバイダまたはブローカー固有であり、基準収益率、報告の取引、ブローカー/取引業者見積、発行者価格差、二国間市場、基準証券、入札、見積、および市場研究出版物を含む参照データを含む多くの情報を含む。いくつかのセキュリティタイプでは、他の入力が使用される場合があり、またはいくつかの標準入力は適用されない場合があります。評価者は、市場状況に応じて任意の日に任意の証券の投入を異なる優先順位付けすることができ、列挙されたすべての投入が任意の日の各証券評価の評価プロセスに使用できるわけではない。
NOTE 3 – 現金等価物と有価証券
次の表は、同社の現金等価物と有価証券(単位:千)をまとめた | | | | | | | | | | | | | | | | | | | | | | | |
| 償却する
コスト | | 毛収入
実現していない
収益.収益 | | 毛収入
実現していない
損 | | 推定数
公正価値 |
| 2022年12月31日 | | | | | | | |
| 資産 | | | | | | | |
| 現金等価物: | | | | | | | |
| 貨幣市場基金 | $ | 50,820 | | | $ | — | | | $ | — | | | $ | 50,820 | |
| | | | | | | |
| 合計する | 50,820 | | | — | | | — | | | 50,820 | |
| 有価証券: | | | | | | | |
| アメリカ政府が支援する実体債務証券 | 18,710 | | | — | | | (293) | | | 18,417 | |
| 商業手形証券 | 101,336 | | | 22 | | | (193) | | | 101,165 | |
| 会社債務証券 | 11,760 | | | — | | | (90) | | | 11,670 | |
| 資産支援証券 | 24,970 | | | 2 | | | (180) | | | 24,792 | |
| アメリカ国庫券 | 7,950 | | | — | | | (12) | | | 7,938 | |
| 預金証書 | 37,599 | | | 4 | | | (142) | | | 37,461 | |
| 機構債券 | 5,598 | | | — | | | (8) | | | 5,590 | |
| 合計する | 207,923 | | | 28 | | | (918) | | | 207,033 | |
| 現金等価物と有価証券総額 | $ | 258,743 | | | $ | 28 | | | $ | (918) | | | $ | 257,853 | |
| | | | | | | |
| 2021年12月31日 | | | | | | | |
| 資産 | | | | | | | |
| 現金等価物: | | | | | | | |
| 貨幣市場基金 | $ | 119,919 | | | $ | — | | | $ | — | | | $ | 119,919 | |
| | | | | | | |
| 合計する | 119,919 | | | — | | | — | | | 119,919 | |
| 有価証券: | | | | | | | |
| アメリカ政府が支援する実体債務証券 | 30,700 | | | 1 | | | (87) | | | 30,614 | |
| 商業手形証券 | 105,792 | | | 7 | | | (42) | | | 105,757 | |
| 会社債務証券 | 33,723 | | | 1 | | | (42) | | | 33,682 | |
| 資産支援証券 | 70,807 | | | 1 | | | (107) | | | 70,701 | |
| 預金証書 | 45,116 | | | 1 | | | (26) | | | 45,091 | |
| 合計する | 286,138 | | | 11 | | | (304) | | | 285,845 | |
| 現金等価物と有価証券総額 | $ | 406,057 | | | $ | 11 | | | $ | (304) | | | $ | 405,764 | |
契約満期日に計算される有価証券の公正価値は以下のとおりである(千で計算) | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 1年以下の期間で満期になる | $ | 177,188 | | | $ | 197,676 | |
| 1年から5年後に期限が切れる | 29,845 | | | 88,169 | |
| 合計する | $ | 207,033 | | | $ | 285,845 | |
2022年、2021年、2020年12月31日までの年間で、投資販売の実現損益は重要ではない。2022年12月31日までの1年間、証券の未実現収益総額と他の全面収益を累計した純収益は実質的ではない。
同社はその投資政策によりそのポートフォリオに関する信用リスクを管理し,購入を高品質の発行者に制限し,そのポートフォリオが単一発行者に投資できる金額を制限している。“会社”ができた違います。Tは、2022年、2021年または2020年12月31日までの年度内に、その有価証券に関する信用損失やその他の減価費用準備を記録する。
同社は2022年、2021年、2020年12月31日までの年度内に、その有価証券に関する未実現損失を計上している。2022年12月31日と2021年12月31日まで12カ月連続で赤字を達成していない有価証券については、会社に個別と全体の重大な未実現損失はない。その投資予定期限に基づき、当社はこれらの投資を一定期間保有する可能性が高く、その償却コスト基盤を回収するのに十分であると判断した。これらの未実現損失は信用リスクによるものではなく、市場状況の変化と関係がある。同社は定期的に有価証券を審査し、信用損失の兆候を探している。当社が考慮している要因には,持続時間,価値低下の幅や原因,潜在的な回収期間,証券発行者の信用とその売却意向などがある。有価証券については、委員会も、(I)当社がその償却コスト基準を回収する前に債務証券を売却する必要があるかどうか、および(Ii)信用損失により余剰コスト基準を回収できない可能性があるかどうかを考慮する。当社の保有証券の発行者の信用に重大な悪化があることを示す重大な事実や状況はありません。当社は、当社の当該等証券の審査に基づいて、未実現損失の持続時間と深刻度の評価、及び当社が当該等の投資を満期まで保有する能力及び意図を含めて、当社が決定する違います。有価証券関連の信用損失準備金は2022年12月31日または2021年に要求される。
NOTE 4 – 大顧客、パートナー関係、戦略同盟
ノワール生物医学研究所です
2020年7月27日、同社はノワール生物医学研究所(“ノワール”)と遺伝子制御療法の研究、開発と商業化について協力と許可協定を達成し、3種類の神経発育障害を治療する。署名された協定によると、同社は、その特定の亜鉛指(“ZF”)転写調節剤(“ZF-TRs”)を開発、製造および商業化するために、ノファ社の関連特許および技術に基づいて、特定の神経発達障害(自閉症スペクトラム障害および知的障害を含む)に関連する3つの未開示遺伝子について、ノバ社に独占的、特許使用料および世界的許可を付与する。同社は協力中に遺伝子標的ごとに早期研究活動を行い,このような研究所に必要なZF−TRsを生産し,その費用はノヴァ社から資金を提供している。ノワール社は他の研究活動、INDの研究、臨床開発、監督管理許可、臨床前、臨床と承認製品の製造及び全世界の商業化を担当している。協定に規定されているいくつかの例外を除いて、同社は、協力対象となる3つの遺伝子のいずれかに対する任意の治療製品を開発、製造または商業化してはならない。ノワール社はまた、同社の特定のアデノ関連ウイルス(“AAV”)を許可することを選択することができ、その唯一の目的は、協力して生成された許可製品を開発、製造、商業化することである。
協定によると、ノファ社は同社に$を支払った75.02020年8月の前払い許可料は100万ドルである。この費用と早期研究活動の費用補償のほかに、同社にはノバ社から最高#ドルの収入を得る資格がある420.0百万ドルの発展の一里塚で、最高で300.0百万ドルのビジネスマイルストーンです。同社にはノファ社から協力によるライセンス製品の潜在的な商業純売上高からノファ社等級の高い桁数から10代以下の2桁の印税を稼ぐ資格がある。特許の満了、市場独占性の喪失、および第三者知的財産権のいくつかのライセンスに基づいて支払われる費用により、これらの特許権使用料支払いは減少するだろう。この協定は、適用される特許使用料の期限が満了するまで、個々の製品および国ごとに継続される。ノワール社は、規定された通知期間の後、任意の理由で完全または一つずつ合意を終了する権利がある。双方はもう一方の破産や重大な、未治癒の違約によって合意を中止する権利がある。
本プロトコルにより受け取ったすべての支払いは、稼いだ後、返却できないと貸記できません。成約価格は1ドルです95.1前払い許可料が含まれています75.0100万ドルで研究コストは$と推定されます20.1推定された研究期間内に100万ドルが提供されるだろう。協定の開始時に、すべての臨床的または規制的なマイルストーンの金額は完全に制限されていると考えられる。制限の評価の一部として、会社は現在のマイルストーンの実現が不確定であり、可変対価格に関する不確実性が解決される将来の時期に依存するなど、多くの要因を考慮している。各報告期間内に、不確定イベントが解決された場合、または他の状況が変化した場合、当社は、取引価格に含まれる推定可変対価格およびすべての制限された金額を含む取引価格を再評価する。
同社はASCテーマ606に基づいてノファ社との合意を評価し、ノファ社が顧客であると結論した。同社はこの手配の中で、技術と持続的な研究サービスの許可として単一の履行義務を決定した。同社の結論は、ライセンスは離散的ではなく、合意に基づいて行われる研究サービスを除いて、ノファ社に対して独立した価値がないからである。そこで,会社は継続研究サービスの予想研究期間内の比例表現に基づいて前金の収入を確認した。履行義務やプロジェクトコストの進捗推定は四半期ごとに審査され,必要に応じて調整され,会社の現在の業績時間の仮定を反映している
義務です。2022年12月31日と2021年12月31日までの同社の売掛金は2.2百万ドルとドル1.9100万ドルと繰延収入は1ドルです9.6百万ドルとドル40.9百万ドルは、それぞれこの合意と関連がある。これらの金額は2023年までに確認される予定です。
プロトコルにより確認された収入は以下のとおりである(千計) | | | | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 | | | |
| ノワール協定に関連した収入: | | | | | | | | |
| 前払い許可証料を確認する | $ | 31,344 | | | $ | 29,945 | | | $ | 4,143 | | | | |
| 研究サービス | 8,384 | | | 7,999 | | | 1,109 | | | | |
| 合計する | $ | 39,728 | | | $ | 37,944 | | | $ | 5,252 | | | | |
その会社は$を支払った1.52020年12月31日までの年間財務顧問費は百万ドルで、2% of $75.0ノワール社の協力とライセンス契約に関する前払い許可料は100万ドルを得た。同社は$を確認した1.5契約資産として百万ドルは、このような金額が合意を得るコストを表すからだ。この残高は、ASC主題340によるノワールへのサービス転送に一致するシステムに基づいて償却され、一般的および管理費用に含まれるその他の資産と延期コスト(“ASC主題340”)。その会社は$を償却した0.6百万ドルとドル0.62022年12月31日と2021年12月31日までの年間で、それぞれ100万ドル。
生物遺伝研究会社
2020年2月、会社はBiogen MA、Inc.(“BIMA”)とBiogen International GmbH(BIMAと共に、“Biogen”)と協力と許可協定を締結し、神経系疾患を治療するための遺伝子制御療法を研究、開発し、商業化した。両社は同社がAAVが提供する独自のZF技術を利用して神経疾患に関連する重要な遺伝子の発現を調節する計画である。提携契約を実行するとともに、当社はBIMAと株式購入プロトコルを締結し、これによりBIMAは購入に同意します24,420,157会社普通株(“生物遺伝株”)、1株当たり価格は$9.2137購入総価格は約$です225.0百万ドルです。
協力協定は、改正された1976年の“ハート-スコット-ロディノ反トラスト改良法”に基づいて待機期間を終了した後、2020年4月に発効し、#ドルの支払いを含む他の慣例成約条件を満たした225.0100万ドルは生物遺伝会社の株式を購入するために使われています
協力協定に基づき,生物遺伝会社は同社に#ドルのライセンス料を前払いした125.02020年5月までに、この数字は100万だ。その会社には研究、開発、監督と商業マイルストーンの支払いを得る資格があり、総額は約ドルに達する可能性があります2.410億ドル、Biogenがプロトコルによって許可されたすべての目標を選択し、合意に規定されたすべての指定されたマイルストーンを達成した場合、最高可達$を含む925.0百万ドルの承認前のマイルストーン支払いとガンダム1.5最初の商業販売と他の販売ベースのマイルストーン支払いは10億ドルだった。また、同社は協力によるライセンス製品の潜在的な商業純売上高から1桁から10代以下の等級別高額印税を得る資格がある。特許の満了、生物類似製品の市場進出、およびいくつかの第三者知的財産権ライセンスに基づいて支払われる費用により、これらの特許権使用料は減少する可能性がある。
協力協定によれば、同社は、生物遺伝会社によって選択された特定の神経疾患遺伝子標的のためのZFおよび/またはAAV製品を開発、製造および商業化するために、その関連する特許および技術に基づいて、生物遺伝会社に独占的、特許使用料および世界的ライセンスを付与する。生物遺伝会社は、筋萎縮性側索硬化症の治療のためのST-501、パーキンソン病を含む同型核病の治療のためのST-501、DM 1に対する第3の候補製品、神経筋疾患、および第4の不開示神経疾患遺伝子標的のうちの4つを選択している。生物遺伝会社は最も多く指名する権利があります7人残りの時間内の追加目標5年協力協定の発効日から始まります。生物遺伝会社が選択した各遺伝子目標について、同社は早期研究活動を行い、費用は両社が分担し、独自の中枢神経系送達ベクターと治療関連遺伝子に対するZF-TRs(または潜在的な他のZF製品)との組み合わせを開発することを目的としている。生物遺伝会社はIND使能研究、臨床開発、関連する監督管理相互作用と全世界商業化の責任とコストを担っている。同社は初の臨床試験の製造活動を担当している三つ同社は引き続き協力製品を開発し、現在開発中の内部製造能力を適宜利用する予定だ。生物遺伝研究会社は初回臨床試験以外の生産活動を担当している三つ製品です。会社のいかなる目標に対する研究活動も一定期間以内に行われるだろう7年になる協定が発効した日から(すなわち2027年4月まで)。協力協定に規定されているいくつかの例外を除いて、同社は、生物遺伝会社が選択した目標のための任意の治療製品の開発、製造、または商業化を禁止されている。
協力協定は,適用されるすべての印税条項が満了するまで製品や国/地域で継続して行われる.Biogenは目標全体または個々の協力協定を終了する権利がある
規定された通知期間の後、いかなる理由で、最大交換する権利があります8人目標です。いずれにしても、他方の破産または重大な違約行為によって本合意を終了する権利がある。また,生物遺伝会社が当社が生物遺伝研究会社に付与した任意の特許に異議を唱えた場合,同社は協力協定を終了することができる。
株式購入契約の条項によると、生物遺伝は、当社の事前書面による同意を得ていない場合及び特定の条件及び例外を満たす場合には、当社が発行した普通株の株式を直接又は間接的に買収すること、要約買収又は交換要約又は双方間の合併を求めるか、任意の事項について依頼書又は同意を求めるか、又は当社の追加株式を潜在的に買収することに関連する他の指定行動を行うことに同意している。このようなポーズ制限は,次の日付の早い時間に満了する3年制協力協定発効記念日とBiogen実益は5会社普通株の%を占めています。
同社はASCテーマ606に基づいて生物遺伝会社との協力協定を評価し,生物遺伝会社が顧客であると結論した。取引価格には$の前払い許可料が含まれている125.0100万ドルと株式購入による追加対価格$79.6百万ドルはドルと225.0生物遺伝株を購入した100万ドルと145.4発行された株式の推定公正価値は百万ドルである。生物遺伝会社に発行される株式はオプション定価モデルを用いて推定され、ある保有期間制限を反映する。いかなる臨床的または規制的マイルストーンも取引価格に含まれていない。何のマイルストーンも達成されていないので、これらのすべての金額は完全に制限されている。制限の評価の一部として、当社は、現在のマイルストーンの実現が不確定であり、可変対価格に関する不確実性が解決される将来の期間に依存する多くの要因を考慮している。取引価格には,生物遺伝会社が会社研究者の仕事のために支払う実際と見積もりの費用分担,会社と第三者で発生した費用の精算も含まれている。生物遺伝研究会社の資源を使用するために生物遺伝研究会社に支払う金額と期待される金額とその費用は顧客への掛け値である。当社は異なる商品やサービスを購入することでこれらの支払いを交換しないため、取引価格を下げ、収入減少と記録されている。当社は期待値手法を用いてコスト分担支払いを推定し,制約の影響を考慮している。可変対価は、確認された累積収入が大きく逆転しない可能性が高い場合にのみ取引価格に含まれます。関連する標的のオプションが行使された場合,入札対象選択費用は取引価格に含まれる.不確定事件が解決された場合や状況が他に変化した場合、会社は取引価格を再評価する。
当社の結論は,その知的財産権の許可は関連する研究や開発活動と変わらず,当社が合意に基づいて提供する研究サービスがない場合には,許可された技術はBiogenと共有できず,Biogenでも使用できないからである。一方、特定の目標に適用される会社の知的財産権ライセンスと関連する研究開発活動の各組み合わせは、任意の他の目標のプロジェクトとは異なる独立した研究プロジェクトである。生物遺伝会社が将来選択可能な目標は,オプションの行使に増量許可権の価値に見合った費用を支払う必要がないため,生物遺伝研究会社に材料権利のオプションを提供することである。したがって、このような選択はまた約束履行義務を代表する
契約開始時に,会社は固定対価#ドルを割り当てた204.6初期取引価格は、既存目標の許可と研究サービス履行義務と、材料権利のオプションを含む履行義務とを含み、その相対的に独立した販売価格に基づく。2022年12月31日まで1つは物権が満期になり,かつ7人物質的権利は未解決のままであり、2023年または2025年に行使されなければ、これらの権利は満了するだろう。
2022年12月31日と2021年12月31日までの同社の売掛金は0.5百万ドルとドル2.8100万ドルと繰延収入は1ドルです132.2百万ドルとドル154.0百万ドルは、それぞれこの合意と関連がある。繰延収入残高の変化は主に履行債務の交付進捗と関係がある。なお確認されている取引価格金額は$151.3百万ドルとドル182.2それぞれ2022年12月31日と2021年12月31日まで。これらの金額は2027年までに確認される予定です。承認された期間は,合意で規定された年間活動量およびBiogenがいつ追加目標の選択権を行使するかどうかの影響を受け,大きな変化の影響を受ける可能性がある。
プロトコルにより確認された収入は以下のとおりである(千計) | | | | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 | | | |
| Biogen協定に関連した収入: | | | | | | | | |
| 許可証と他の固定対価格を認める | $ | 21,820 | | | $ | 29,224 | | | $ | 21,356 | | | | |
| 研究サービスの費用は支払いを分担して可変に考えている | 6,599 | | | 13,076 | | | 6,545 | | | | |
| 合計する | $ | 28,419 | | | $ | 42,300 | | | $ | 27,901 | | | | |
その会社は$を支払った7.02020年12月31日までの年間財務顧問費は百万ドルで、2% of $225.0株式を売却して得た百万元と2% of $125.0前払い費用は100万ポンドです。発生する費用は以下の点と関係がある
バイオ遺伝会社との協力協定でも、株式売却に関する株式購入協定でも。当社は、相対的に公正な価値で2つの合意の間に費用を分配することが合理的だと考えている。同社は$を確認した4.1百万というのは2初期価格$のパーセントは204.6契約コスト資産として百万ドルです。ASC主題340によれば、この残高は、生物遺伝研究会社にサービスを譲渡する場合に応じて、一般的および行政費用に体系的に計上される。会社は費用を確認しました$0.4百万、$0.6百万ドルとドル0.42022年12月31日、2021年、2020年12月31日までの年間で、それぞれ100万ドル。同社は$を確認した2.9百万ドルというのは2ドルのパーセント145.41,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,000,
凧製薬会社
2018年2月、同社はギリッド科学社の子会社Kite Pharma,Inc.(“Kite”)と2018年4月5日(“発効日”)に発効し、潜在的な癌工学細胞療法を研究·開発し、商業化するための2019年9月に改訂·再記述したグローバル協力および許可協定を締結した。今回の協力で、SangamoはKiteと協力して研究計画を展開しており、この計画によると、両社はキメラ抗原受容体(CARS)、T細胞受容体(TCR)とNK細胞受容体(NKRs)をコードする遺伝子を挿入して挿入することを含む、T細胞およびナチュラルキラー細胞(NK細胞)のいくつかの遺伝子を妨害して挿入するために、亜鉛指ヌクレアーゼ(ZFN)とウイルスベクターを設計している。Kiteはすべての臨床開発、製造、そして任意の最終製品の商業化を担当している。
この協定の条項によると、会社は、会社の関連特許および独自技術に基づいて、癌の治療を目的として、特定の細胞治療製品を開発、製造、商業化するために、Kite独自の印税付き、世界的に再許可可能な許可証を付与し、これらの製品は、研究計画によって生成される可能性があり、研究計画に従って開発された選定されたZFNおよびウイルスベクターを用いて体外で工学を行い、候補目標に対するCAR、TCRまたはNKRを表現する。
研究計画期間中、ある例外的な状況を除いて、当社は癌を治療する目的で任意の細胞治療製品を研究、開発、製造、商業化してはならず、この製品は体外ゲノム編集の結果であり、ヒト癌細胞上またはその中に発現する標的に対するCAR、TCRまたはNKRを発現する。研究計画期間が終了した後、会社はいくつかの例外を除いて、癌治療の目的のための任意の体外ゲノム編集によって候補目標に対するCAR、TCRまたはNKRを発現する細胞治療製品の開発、製造、商業化を禁止される。
発効日後、当社は1ドルを受け取りました150.0Kiteは百万ドル前払いした。また,Kiteは会社が共同研究プロジェクトを展開している直接費用を精算している。Sangamoには開発や販売に基づくマイルストーン支払いを受ける資格があり、総額は$に達する可能性があります3.0この協定で規定されたすべての具体的なマイルストーンが達成されれば10億ドル。このお金のうち約$は1.3特定の研究、臨床開発、規制、および初の商業販売マイルストーンのための10億ドル、および約ドル1.8特許製品の世界的な年間純売上高が特定の水準に達すると、10億ドルは特定の販売に基づくマイルストーンの実現につながる。各開発·販売に基づくマイルストーン支払い(I)は、当該ライセンス製品が関連するマイルストーンイベントを達成した回数にかかわらず、許可製品毎に1回のみ支払われ、(Ii)は1回目のみである10このようなマイルストーンイベントを実現することが可能なライセンス製品の数を考慮することなく、関連マイルストーンイベントの回数を実現する。また、同社は将来のライセンス製品の世界的な年間純売上高に応じて、桁数の割合で段階的に増加する特許使用料を得る権利がある。特許の満了、生物類似製品の市場進出、およびいくつかの第三者知的財産権ライセンスに基づいて支払われる費用により、これらの特許権使用料は減少する可能性がある。
合意における初期研究条項は6年発効日からです。Kiteは研究期間を延長する権利がある二つその他の内容1年制期間中は別途前払い料金をいただきます10.0年間百万ドルです。本プロトコル項の下のすべてまたは支払いがあり、稼いだ後、返却および貸記できません。2019年9月の合意の修正および再説明によって、会社およびKiteは、協力計画の範囲を拡大することに同意し、Kiteが提供するレンチウイルスまたはレトロウイルスベクターの使用を組み込む。Kiteは、指定された通知期間の後、本プロトコルのすべての内容または各ライセンス製品または各候補ターゲットを任意の理由で終了する権利がある。いずれにしても、他方の破産または重大な違約行為によって本合意を終了する権利がある。
会社はASCテーマ606に基づいてKiteとのプロトコルを評価し、Kiteが顧客であると結論した。取引価格には$の前払い許可料が含まれている150.0実行期間内の研究プロジェクトの百万ドルとサービス料の返済が予想されます。どんな臨床的または規制的マイルストーンも取引価格に含まれていません。まだ何のマイルストーンも達成されていないので、すべての金額は完全に制限されています。制限の評価の一部として、会社は現在のマイルストーンの実現が不確定であり、可変対価格に関する不確実性が解決される将来の時期に依存するなど、多くの要因を考慮している
取引価格には,Kiteが会社の研究者の作業のために支払った実際と見積もり費用,会社と第三者が発生した費用の精算も含まれている。会社は期待値手法を用いて会社の研究者の仕事に関する報酬を推定し,制約の影響を考慮している.可変対価は、確認された累積収入が大きく逆転しない可能性が高い場合にのみ取引価格に含まれます。各報告期間内に、不確定イベントが解決された場合、または他の状況が変化した場合、会社は、取引価格に含まれる推定可変対価格およびすべての制限された金額を含む取引価格を再評価する
その会社は確定した四つKiteプロトコルにおける履行義務は,(1)技術ライセンスおよび会社の技術をKite選定目標に適用した研究·開発サービスの義務,(2)研究材料の生産,および(3−4)である二つ物質的権利、それぞれが研究期間を延長するための別の項目1年制学期です。このような延期は、それらの行使が増分研究期間の価値に見合った費用を支払う必要がないので、実質的な権利を含む。会社の知的財産権への許可は関連する研究や開発活動と変わらず,会社が研究サービスを提供していない場合には,許可された技術はKiteと共有されず,Kiteでも利用できないためである
会社は、ASCトピック606における割り当て目標を満たすので、可変対価格(Kiteが会社の研究者のために行った仕事および第三者コストによって支払われたお金、および任意の将来のマイルストーンおよび特許権使用料)を、それらに関連する特定の業績義務に割り当てる。会社が割り当てた固定コストは#ドルです150.0その相対的に独立した販売価格に応じて、義務履行に百万ドルを支払う。オプション研究年度の独立販売価格は最初の1年の価格と類似しているが,割引の内在的価値や行使の可能性は別途考慮されている
実質的な権利を有するオプションに割り当てられた費用は、オプション行使または満期時に支払われることになる。オプションの行使は契約継続入金として,目標選択費用と推定可変対価格はそのときの取引価格に含まれ,その目標の履行義務に特化して割り当てられる
総合許可と研究サービス履行義務の収入は時間の経過とともに確認され,Kiteは会社が履行しているこのようなサービスのメリットを消費しているためである.研究開発サービスの義務履行と組み合わせたライセンスについては,会社は継続研究サービスの会社がサービスを提供している間の割合に基づいて収入を確認している。この業績義務とプロジェクトコストの進捗の見積もりを四半期ごとに審査し,必要な活動推定数に対する会社の仮定を反映するように必要に応じて調整した。会社はこれらのサービスについてKiteに伝票を発行する権利があり,その金額はサービスの価値に直接対応するため,生産研究材料の履行義務は実際に方便の権利項に入金される
2022年12月31日と2021年12月31日までの同社の売掛金は0.7百万ドルとドル0.1100万ドルと繰延収入は1ドルです19.4百万ドルとドル56.5百万ドルは、それぞれこの合意と関連がある。繰延収入残高の変化は主に履行債務の交付進捗と関係がある。確認すべき取引価格金額(生産研究材料の履行義務のために確認された伝票金額は含まれていない)は#ドルである19.7百万ドルとドル51.4それぞれ2022年12月31日と2021年12月31日まで。これらの金額は2024年までに確認される予定です。認可された期間は,合意下の年間活動量やKiteがいつ追加サービス年限の選択権を行使するかどうかの影響を受け,大きな変化の影響を受ける可能性がある。
プロトコルにより確認された収入は以下のとおりである(千計) | | | | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 | | | |
| 凧協定に関連した収入: | | | | | | | | |
| 確認許可料固定対価 | $ | 37,032 | | | $ | 24,977 | | | $ | 25,046 | | | | |
| サービス可変考慮要素を研究する | 1,560 | | | 476 | | | 3,562 | | | | |
| 合計する | $ | 38,592 | | | $ | 25,453 | | | $ | 28,608 | | | | |
ファイザー。
Giroctocogene Fitelparvovecグローバル連携とライセンスプロトコル
2017年5月、当社はファイザーと独占的なグローバル協力及び許可協定を締結し、これにより、当社は血友病Aを治療する遺伝子治療製品Fitelparvovec及び密接な関連製品の研究、開発及び商業化について協力関係を構築した。
この協定によると、同社は1/2期臨床試験とある癌原遺伝子fitelparvovecの製造活動を担当し、ファイザー社はその後全世界範囲で原癌遺伝子fitelparvovecの開発、製造、マーケティングと商業化を担当している。Sangamoはまた、他のAAVに基づく血友病A遺伝子治療製品を協力して研究開発することも可能である。
協定条項によると、会社はファイザーに独占的なグローバル特許使用料許可を付与し、会社が制御するいくつかの技術を使用してgirococogene fitelparvovecおよびその関連製品を開発、製造、商業化する権利がある。ファイザーは同社に非独占的で世界的に、印税免除、全額支払いの許可を付与し、プロトコルに基づいて開発されファイザーによって制御されたいくつかの製造技術を用いてAAV交付システムを使用した会社製品を生産する権利がある。特定の時期に、同社とファイザー社は協力以外の臨床でいくつかのAAVに基づく血友病A遺伝子治療製品を開発或いは商業化してはならない。
以前に終了しない限り、本協定の期限は、各製品及び各国を基礎として、(I)一国で製品をカバーする特許権が満了するまで、(Ii)一国における製品の規制排他性が満了するまで、及び(Iii)15年ある国の製品が初めて商業販売された後。ファイザーは、理由なしに合意全体を終了するか、製品または国/地域で合意を終了する権利がある。合意は、他方が治癒していない実質的な違約または他方の破産に基づいていずれか一方によって終了することもできる。いかなる理由でも終了すると、当社がファイザー社にgiroctocogene fitelparvovecおよび関連製品の開発、製造、商業化を付与するライセンスは自動的に終了します。会社が任意の1つまたは複数の国または地域で原因またはファイザー会社の契約を終了するとき、ファイザー社は、終了された1つまたは複数の国/地域でgiroctocogene fitelparvovecを開発、製造、および商業化するために、ファイザー社が制御するいくつかの技術に従って自動的に会社に独占的、印税付きの許可を付与する。
契約を実行する際、会社は前払い費用#ドルを受け取る70.0100万ドルで最高$を得る資格があります208.5特定の臨床開発、知的財産権、規制マイルストーンの実現に基づいて百万ドルを支払い、最高で$に達する266.5Giroctocogene fitelparvovecと可能な他の製品の最初の商業販売マイルストーンに100万ドルを支払います。契約に規定されているすべてのマイルストーンを実現すると仮定して、潜在的な臨床開発、知的財産権、規制、および最初の商業販売マイルストーンの支払い総額は最大で$に達することができます475.0100万ドルに達しています300.0Giroctocogene fitelparvovecに100万ドルを提供し、最高$に達する175.0協定によって開発可能な他の製品の費用は100万ドルであるが、第三者知的財産権のいくつかのライセンスによって支払われる費用は減少する。また,ファイザーは,合意に基づいて開発された潜在的許可製品ごとの特許権使用料を会社に支払うことに同意した14% - 20特許の満期、生物類似製品の市場進出、およびある第三者知的財産権許可に基づいて費用を支払うことにより、このような製品の世界的な年間純売上高は低下するだろう。今までのところ二つマイルストーン:$55.0しかし合計100万ドルを支払いました違います。製品が承認されたので違います。合意に基づいて、特許使用料を稼いだ。
同社はASC主題606に基づいてファイザーとの合意を評価し、ファイザーは顧客であると結論した。本契約項の下の取引総価格は$である134.0百万ドル、その中に前払い費用と研究サービス料が含まれています#ドル79.0百万元と以下に関連する費用二つ取得したマイルストーンの総金額は#ドル55.0百万ドルです。Sangamoは内部と外部の研究費用を担当し、前払い費用の一部として、ある条件を満たしていれば、Sangamoはファイザー社に追加精算を要求する権利がある。取引価格には制限された臨床的または規制的なマイルストーンはない。規制の評価の一部として、会社は当時のマイルストーンの実現が不確定であり、可変対価格に関する不確実性が解決された将来の期間に依存するなど、多くの要因を考慮している。
同社は協定における履行義務を技術と持続的な研究サービスのライセンスとして決定している。同社は、契約に基づいて会社が実行する研究サービスを除いて、ファイザー社に対して独立した価値がないため、ライセンスは独立していないと結論している。そこで,会社は継続研究サービスの割合表現に基づいて前金の収入を確認し,2020年までの間に会社が研究サービスを提供している.その履行義務やプロジェクトコストの進捗状況の見積りを四半期ごとに審査し,必要に応じて企業の成果交付時間の仮定を反映するように調整した
2020年12月、会社は手配内の成果の交付と研究サービスの責任を果たした。そこで、会社は2020年12月に前払いの余剰繰延収入を確認し、違います。2022年12月31日と2021年12月31日までの年度内に収入が確認された。
2019年12月、当社は提携協定修正案に署名し、この合意に基づき、当社はgiroctocogene fitelparvovecのINDをファイザーに譲渡した。今回の移転で、会社は$を獲得しました25.0100万個のマイルストーンが、マイルストーンに到達するための条件が満たされたからだ。年間確認の累計収入
このマイルストーンとのつながりは$です25.02020年12月31日まで1.32020年12月31日までの年間で確認された百万ドル。
2020年9月、当社は1ドルを実現する可能性が高いことを確認しました30.0ファイザーは原癌遺伝子fitelparvovecに対する百万マイルストーンである。このマイルストーンはその後、2020年10月初めの第三段階臨床試験中の最初の被験者の投与量で実現された。このマイルストーンに関する累計確認収入は#ドル30.02020年12月31日までの年間で
プロトコルにより確認された収入は以下のとおりである(千計) | | | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 | | |
ファイザー赤カビ遺伝子Fitelparvovecプロトコルに関連する収入: | | | | | | | |
| 前期費用と研究サービスの確認 | $ | — | | | $ | — | | | $ | 3,111 | | | |
| 記念碑的業績 | — | | | — | | | 31,338 | | | |
| 合計する | $ | — | | | $ | — | | | $ | 34,449 | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
同社は2020年3月,ファイザーと達成したgiroctocogene fitelparvoecとの連携協定の推定変化に関する収入調整を記録した。この調整は減少項目範囲とそれに応じたコストを決定する直接結果であり,giroctocogene fitelparvoveec候補製品をファイザーへの移行に成功させた後,両決定とも比例累積業績測定基準の向上を招いたためである。この調整は収入をドル増加させた2.4百万ドル純損失が減少しました2.4100万ドルで会社の1株当たりの純損失を$に減らします0.022020年12月31日まで年度を終了する。
C 9 ORF 72研究連携とライセンスプロトコル
2017年12月、当社はファイザーと潜在遺伝子治療製品の開発と商業化について単独の独占的なグローバル協力と許可協定を締結し、これらの製品はZF-TRsを用いて筋萎縮性側索硬化症と遺伝子変異に関連する前頭側頭葉変性を治療したC 9 ORF 72ジーン。この合意によると、同社は、変異形態のZF-TRsの発現を決定、表現、および臨床前開発と結合し、特に減少させるために、ファイザー社と協力して研究計画に同意したC 9 ORF 72ジーン。
本協定の条項によると、会社は、予め合意された基準を満たすZF−TRsの遺伝子治療製品の開発、製造、商業化のために、会社の関連特許及びノウハウに基づいてファイザー社に独占的な特許権使用料のグローバルライセンスを付与している。特定の期間内に、当社とファイザーはいずれも協力以外に研究、開発、製造、または商業化のいずれもしてはならないC 9 ORF 72ジーン。
以前に終了しない限り、本協定の期限は、(I)国/地域におけるライセンス製品をカバーする特許請求の期限が満了するまで、(I)国/地域におけるライセンス製品の排他的規制が満了するまで、(Ii)ライセンス製品の国/地域における排他的規制が満了するまでである15ライセンス製品が主要市場国で初めて商業販売された数年後。ファイザーはまた、理由なしに合意全体を終了するか、製品または国/地域で合意を終了する権利がある。合意は、他方が治癒していない実質的な違約または他方の破産に基づいていずれか一方によって終了することもできる。特定の期間内に任意の主要候補者を決定して開発を行うことができない場合、またはファイザー社が特定の期間内にリード候補を特定の開発マイルストーンを超えて引き上げないことを選択した場合、合意も終了する。いかなる理由でも終了すると、当社が合意に基づいてファイザー社に開発、製造、商業化許可製品を付与するライセンスは自動的に終了します。任意の特許製品または特許製品によっていずれか1つまたは複数の国または地域が会社または輝瑞によって無断で終了された場合、会社は、終了された1つまたは複数の国/地域で製品を開発、製造、および商業化するために、ファイザーによって制御される特定の技術下の非独占的で印税の許可を得る権利があるであろう。
ファイザーが重大な違約で会社に中止された後、ファイザーは研究、開発、製造、或いは商業化専門に許可されないC 9 ORF 72遺伝子にはしばらく時間がありますファイザーが会社の重大な違約によって終了した後、会社は研究、開発、製造或いは商業化の専門とすることを許可されないC 9 ORF 72遺伝子にはしばらく時間があります
その会社は1ドルを受け取った12.0ファイザーから100万ドルの前金を得て最高$を得る資格があります60.0ファイザー社は特定の臨床前開発、臨床開発と最初の商業販売マイルストーンの実現状況に基づいて100万ドルの開発マイルストーン支払いを支払い、最高で90.0ライセンス製品の世界的な年間純売上高が指定水準に達した場合、百万ドルの商業マイルストーン支払いを受けることができる。またファイザーは会社にも支払います14% - 20ライセンス製品の世界の年間純売上高の%です。これらの特許権使用料は特許によって減少する可能性があります
生物類似製品の満期、生物類似製品の市場への参入、およびある第三者知的財産権許可証による支払い。すべての当事者たちはそれが研究計画を実行する費用に責任があるだろう。ファイザー社はライセンス製品の後続開発、製造、商業化の運営と財務責任を担当する。これまでのマイルストーンは$5.0しかし100万ドルを達成して支払いました違います。製品が承認されたので違います。特許使用料はC 9 ORF 72ファイザーの合意。
同社はASC主題606に基づいてファイザーとの合意を評価し、ファイザーは顧客であると結論した。同社は,本契約での取引総価格は$であると結論した17.0百万ドルは前払い費用に相当します12.0百万ドルとマイルストーンの実現に関連した費用は$です5.0百万ドルです。取引価格には制限された臨床的または規制的なマイルストーンはない。規制の評価の一部として、会社は当時のマイルストーンの実現が不確定であり、可変対価格に関する不確実性が解決された将来の期間に依存するなど、多くの要因を考慮している。
同社は本協定における履行義務を技術と継続研究サービスのライセンスとして決定している。同社は、契約に基づいて会社が提供するサービスを除いて、ファイザー社に対して独立した価値がないため、ライセンスは離散的ではないと結論した。そこで,会社は継続研究サービスの割合に基づいて前金の収入を確認し,2020年まで,すなわち会社が研究サービスを提供するまでの期間を示した
同社は2020年9月に手配中の成果と研究サービスの責任を果たしたため、1ドルを稼いだ5.0百万マイルストーンは、会社が2020年12月31日までの年間累計でこの数字を確認している。また、当社は2020年9月に前払いの残り繰延収入を確認し、違います。2022年12月31日と2021年12月31日までの年度内に収入が確認された
プロトコルにより確認された収入は以下のとおりである(千計) | | | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 | | |
ファイザー関連の収入C 9 ORF 72プロトコル: | | | | | | | |
| 前払い料金を確認する | $ | — | | | $ | — | | | $ | 7,985 | | | |
| 記念碑的業績 | — | | | — | | | 5,000 | | | |
| 合計する | $ | — | | | $ | — | | | $ | 12,985 | | | |
二零年十二月三十一日までに当社は合格したC 9 ORF 72ファイザーとの協力協定。これらの調整はプロジェクト範囲の縮小と方案推進による相応の費用を決定する直接結果であり、比例累積業績評価基準の増加を招いた。このような調整は収入をドル増加させた8.8百万ドル純損失が減少しました8.8100万ドルで会社の1株当たりの純損失を$に減らします0.062020年12月31日までの年度。
セノフィ社
同社は2014年1月、ベータ地中海貧血と鎌状細胞病(“SCD”)に対する治療法を開発するための独占的なグローバル協力と許可協定(“2014協力協定”)を締結した。2014年の協力協定は最初にBIMAと署名され、BIMAはその後Bioverativ Inc.に譲渡され、後者はその後サイノフィによって買収された。2014年の協力協定によると,同社は最初に共同で行った二つ研究計画:2021年第3四半期に中止されたベータ地中海貧血計画と,SAR 445136(現在はBIVV 003)の開発につながるSCD計画であり,SAR 445136はSCD治療のためのZFN遺伝子編集細胞治療候補製品である。セノフィは2021年12月、便宜上2014年の提携契約の終了を当社に通知し、2022年6月28日(“終了日”)から発効します。双方は2022年9月6日に終了と移行協定(“終結と移行協定”)に署名した
SCD計画では,同社とセノフィはIND申請前に共同で研究·開発活動を担当しているが,セノフィはその後プロトコルに従って開発された特許製品の世界的な臨床開発,製造,商業化を担当している。協定条項に適合する場合、同社は、同社が制御するいくつかのZFおよび他の技術を使用して、当該合意に基づいて開発された特許製品を研究、開発、製造および商業化するために、セノフィに独占的、特許権使用料を徴収する権利を付与した。同社はまた、セノフィの世界規模での非独占的、免版税の全額許可を付与し、同社が同協定に基づいて開発したいくつかの他の知的財産権の権益に基づいて再許可を付与する権利がある。契約期間内に、契約を除いて、会社は、許可製品に関連する遺伝子に対するいくつかの遺伝子治療製品を研究、開発、製造、または商業化してはならない。
2014年の協力協定によると、同社は前払い許可料$を受け取った20.0指定された臨床開発、規制マイルストーンと販売マイルストーンに達した時に追加支払いを受け、各許可製品の純売上高に応じて特許権使用料を支払う資格がある。セノフィはまた,SangamoがSangamoと行った研究や開発活動に関する合意費用を返済する。終了日まで、合計$13.5臨床開発マイルストーンの成果に基づいて100万部を受け取った違います。製品が承認されたので違います。2014年の協力協定によると、すでに印税費用を稼いでいるか、または徴収される。
サイノフェイは同社への終了通知で,その終了はサイノフェイの戦略方向の変更に関連しており,自己個人化細胞療法ではなく同種異体汎用ゲノム薬物法に重点を置いていると述べている。終了日から2014年の協力協定はすべて終了し、終了日から、当社はセノフィから記念碑的な支払いや特許使用料を得る権利がない。終了日まで、セノフィは、2014年の協力協定に基づいて、2014年の協力協定の下で任意の協力研究プロジェクトの発展を策定または支援する義務がありません。2014年の協力協定によりセノフィに付与されたライセンスは終了し、ライセンス権は会社に回復された。
終了および移行協定の一部として、セノフィは、その特定の知的財産権に再許可、開発、製造、使用、販売、提供販売、輸入、および他の方法で商業化されたBIVV 003を独占的、世界的に、全額支払い、永久的、取消不可能なライセンスを同社に付与し、SCD計画下で開発された候補製品である。同社は,行われている臨床試験や関連する長期後続研究の完成を含め,BIVV 003に関連するすべての臨床試験の責任を負うことに同意した。同社はまたBIVV 003に関するすべての規制責任を担っている。サイノフィは、BIVV 003に関連するすべての文書、材料、および第三者と締結された契約、およびサイノフィがBIVV 003に関連するいくつかのデバイスを所有またはレンタルする権利を譲渡し、譲渡する。
セノフィは,行っているBIVV 003臨床試験の費用および2023年12月31日までの長期後続研究の費用を同社に補償することにも同意し,最高で#ドルに達する7.0百万ドルです。また,会社が2023年12月31日以降にBIVV 003の開発を継続しないことを選択した場合,セノフィは2023年後に発生した長期後続研究費を会社に返済する義務があり,最高で$に達する5.3百万ドルです。サイノフェイの清算義務は、BIVV 003との第三者との協力、協力、販売、許可または剥離について締結された契約を含む、いくつかのトリガイベントが発生したときに終了するか、またはFDAが臨床試験および/または長期後続研究の早期閉鎖を許可する場合を含む。
同社はASCテーマ606に基づいて2014年の協力協定を評価し、この計画によると、セノフィは顧客であると結論した。同社はこの手配における履行義務を,継続的な研究サービス活動と組み合わせた技術許可として決定している。同社の結論は、ライセンスは明らかではなく、研究サービスがなければ、セノフィにとって独立した価値がないからである。そこで,会社は進行中の研究サービスの業績進展に基づいて前金とマイルストーンの収入を確認した。履行義務やプロジェクトコストの履行に進展した見積もりは四半期ごとに審査され,必要に応じて会社の当時の成果交付時間の現在の仮定を反映するように調整されている。これらのスケジューリングに関するコストと支出は従来から確認されている収入に近い.セノフィ2021年12月に発行された2014年の協力協定の終了通知は、会社が予想するサービス範囲を縮小し、取引価格を推定し、残りの履行スケジュールを短縮する改正である。この変化と一致して、会社が2014年の協力協定に基づいて提供したすべてのサービスは2022年6月28日に完了し、最終的に取引価格に計上されたすべての金額がこの日に確認された。最終成約価格は$96.3前払い許可料が含まれています20.0百万、二つの記念碑的な支払い、合計金額は$です13.5100万ドルで研究費を精算します62.8百万ドルです。2022年12月31日と2021年12月31日までの会社の売掛金はゼロそして$0.62014年の協力協定にそれぞれ関連した100万ドル。2014年の協力協定に関する繰延収入はゼロそして$1.1それぞれ100万ドルです
当社の結論は,終了と移行プロトコルにより,セノフィは顧客ではなく,セノフィが得られなくても進行中の臨床試験や長期後続研究の結果を用いることができないためである。この関係はASCトピック808の範囲内の連携でもない協力手配それは.同社の結論は、セノフィから買収された資産は事業を代表するものではなく、これらの資産のほとんどの価値が取得または再取得された知的財産権許可に集中しているからである。同社にはセノフィが行っている臨床試験や長期後続研究費の返済義務はない。そこで,同社はセノフィの補償をその研究開発費の減少として確認した。2022年12月31日までの年間で,会社は研究開発費を削減した$2.1100万ドルのうち1.1100万ドルは、2022年12月31日現在の統合アセットバランスシート上の前払い費用およびその他の流動資産に含まれる。
プロトコルにより確認された収入は以下のとおりである(千計) | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| セノフィ協定に関連した収入: | | | | | |
| 前払い料金を確認する | $ | 677 | | | $ | 34 | | | $ | 298 | |
| 研究サービス | 2,126 | | | 3,057 | | | 4,823 | |
| 記念碑的業績 | 457 | | | 23 | | | 201 | |
| 合計する | $ | 3,260 | | | $ | 3,114 | | | $ | 5,322 | |
同社は2021年12月31日までの年間で、セノフィ協力協定に関する推定変化に関する収入調整を記録している。これらの見積り数が変化した原因は,2021年9月にプロジェクト範囲と関連プロジェクト費用が変化し,その後連携プロトコルの終了が通知され,比例累積実行状況計測が変化したためである.これらの調整は収入を#ドル減少させた1.6百万ドル純損失増加$1.6100万ドルで会社の1株当たり基本的に希釈して純損失を増加させました0.012021年12月31日までの年度。
同社は2020年12月31日までの年間で、セノフィとの協力協定推定の変化により、収入に関する調整を記録している。この調整は2020年3月にプロジェクト範囲の拡大と相応の費用を決定した直接的な結果であり、両決定とも実行状況を比例的に累積する測定基準の減少を招いている。この調整は収入を1ドル減少させた2.2百万ドル純損失増加$2.2100万ドルで会社の1株当たりの純損失を増加させます0.022020年12月31日までの年度。
カリフォルニア州再生医学研究所
2018年5月、カリフォルニア再生医学研究所(CIRM)は戦略パートナー賞を授与し、ドルを奨励した8.0SangamoのZFヌクレアーゼゲノム編集技術を応用した上で、潜在的な治療効果を有するZF療法によるβ地中海貧血治療の臨床研究を援助した。この贈与はST-400を支援するための一致した資金を提供し、ST-400は輸血依存型ベータ地中海貧血患者に対する遺伝子編集細胞治療候補薬である。CIRM助成金の条項によると、同社は、CIRM助成の候補製品またはCIRM助成技術の純売上高の低い桁の特許使用料パーセンテージに基づいて印税と許可料を支払う義務がある。当社はCIRMが付与したいかなる金額もすべての金額を拒否する権利があり、収入共有の代替案として、当社は奨励をローンに転換する権利があるが、2020年12月31日までこのような選択はしていない。その会社は#ドルを受け取った5.22020年12月31日現在、同賞の100万ドル。同社はすでに$を記録している6.4百万ドル、課税利息を含めて#ドル1.22020年12月31日現在の総合貸借対照表上の他の非流動負債のうち、この奨励に関する融資として100万ドル
2021年11月にST−400の開発停止が決定したため,他の候補製品を優先的に開発するため,贈与は終了した。この案の終了と中止について、会社は奨励を融資に転換しないことを決定し、返金されない奨励金額#ドルを確認した5.2研究開発費を削減するための百万ドル1.2会社が2021年12月31日までの年度総合経営報告書では、奨励された受取利息を利息やその他の収入純額としている違います。この奨励に関連する金額は、2022年12月31日と2021年12月31日までに総合貸借対照表に含まれる。
Sigma-Aldrich社と合意しました
2007年、SangamoはSigma-Aldrich Corporation(“Sigma”)とSangamoのZFノウハウをSigmaに提供し、この技術を独占的に使用して研究分野の研究試薬製品およびサービスを開発し、それを商業化する権利を締結し、Sangamo以前に陶氏化学会社の完全子会社Dow AgroSciences LLC(“DAS”)に許可されていたいくつかの農業研究用途を含まない。SangamoはそのZF技術を用いて1年以上の間に実験室研究試薬を開発した3年制研究サービス期間。それ以来,SangamoはZF製造技術をSigmaに譲渡した。
2009年10月、SangamoはSigmaとの許可協定を拡大した。許可プロトコルの元の条項に加えて、Sigmaは、タンパク質薬物の商業生産および商業応用のためのいくつかのZFトランスジェニック動物の開発および流通のためのZFトランスジェニック細胞株の開発および流通の独占的権利を獲得した。協定条項によると,Sigmaは#ドルの現金を前払いした20.0$を含む百万ドル4.9100万を購入しました636,133Sangamo普通株、価値$4.9100万ドルと1ドルです15.1百万ドルの前払い許可料です。Sangamoには#ドルの商業許可料を得る資格もある5.0百万ドル、純売上高と再許可収入のパーセンテージで計算して、その後、印税税率は10.5純売上高と分許可収入の%です。また、いくつかの累積的なビジネスマイルストーンに達した後、SigmaはSangamoに記念碑的な支払いを支払い、総額は$に達する25.0百万ドルです。協定によると、Sangamoには追加的な継続的な業績義務がない。
シグマとの合意によると、2022年、2021年、2020年12月31日までの年度確認収入は0.9百万、$1.1百万ドルとドル0.5それぞれ100万ドルです
DASと合意に達する
2005年にSangamoはDASと独占的なビジネスライセンスを締結しました3年制研究用語。このプロトコルによれば、Sangamoは、その固有のZF技術をDASに提供し、この技術を使用して植物細胞、植物または植物細胞培養物のゲノムを修正するか、または核酸またはタンパク質の発現を変化させる独占的権利を有する。Sangamoは、診断、治療または予防目的のために、植物または植物由来製品を使用してZF-TRsまたはZFNをヒトまたは動物に送達する権利を保持する。2008年、DASはその選択権を行使し、会社のZF技術を使用して製造された植物細胞または植物細胞由来製品を販売する商業ライセンスを取得した。選択権の行使は一度の商業許可料#ドルを触発する6.0100万ドルで残りの$を支払います2.3この前合意した百万ドル4.0百万ドルの研究マイルストーン、各製品の開発と商業化マイルストーンの支払い、製品販売の印税。2010年12月、会社はDASとの合意を改正し、試薬製造サービスと研究サービスの期限を2012年12月31日に延長した。
DASとの協定では、DASは、会社ZF技術由来のZF製品(“許可計画”)を使用するために第三者と何らかの分割許可を締結する権利があり、許可計画がDASによって終了されないことを前提として、毎年10月にSangamoに支払う最低年次支払い義務が規定されている。年会費は1ドルから1ドルまで様々です0.3百万ドルから百万ドルまで3.0百万ドル総額$25.3100万人以上115年、最後の支払いは2020年10月で、金額は#3.0百万ドルです。その会社はこの手配における履行義務をこの技術のライセンスとして決定した。2021年7月、DASは会社に許可計画の終了通知を出し、2021年9月から発効した。しかし,SangamoはDASの再許可は依然として有効であり,DASプロトコル自体も同様である.プロトコルが終了した場合、会社のZF技術を使用するすべての権利はSangamoに回復され、DASは、SangamoのZF技術を実行することをもはや許可されないか、または限られた場合には、会社のZF技術からの任意の製品を商業化することが許可されるであろう。
DASとの合意によると、2022年、2021年、2020年12月31日までの年度確認収入はゼロ, $0.2百万ドルと$3.0それぞれ100万ドルです
NOTE 5 – フランスのSangamo社を買収する
2018年、Sangamoは様々な合意を達成し、最終目標は買収です100%フランスSangamo社の株式と約477,000Sangamo Franceの自由株式は、これにより、当社は保有者に当該等の株式(引受オプション)を購入する権利があり、当該等保有者は、当該等の株式を2021年中(承認オプション)まで時々当社に売却する権利がある(総称して“自由株式オプション”と呼ぶ)。2021年12月31日までに会社はすべてを買収しました477,000無料株は100フランスSangamoの%所有権。
Sangamo Franceの買収は、ASCテーマ805による業務統合とみなされる企業合併交換として、総費用は約$です45.9買収の日に百万ドルです。フランスのSangamo社の買収日後の経営業績は、会社の総合経営報告書に含まれている。あったことがある違います。2022年、2021年または2020年12月31日までの年間営業権減額。
非制御的権益
すべての自由株式を買収する前に、残りの非持株権益の公正価値は、非持株権益およびドルを含む発行された自由株式の数に基づいて決定される2.99買収日までの1株当たりの買い入れ価格。非持株権益は、会社が2020年12月31日までの総合貸借対照表に株主権益の一部として示されている。2022年と2021年12月31日までに買収100普通株、非持株権益の帳簿価値は追加支払いとして記録されています‑会社の総合貸借対照表に資本を計上する。
NOTE 6 – その他の資産負債は細かいことを示している
財産と設備、純額
財産と設備の純額には、以下の項目が含まれる(千計) | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 実験室装置 | $ | 39,080 | | | $ | 31,988 | |
| 賃借権改善 | 26,559 | | | 21,970 | |
| 家具と固定装置 | 9,744 | | | 9,080 | |
| 製造設備 | 9,908 | | | 8,781 | |
| 建設中の工事 | 14,770 | | | 4,729 | |
| 100,061 | | | 76,548 | |
| 減算:減価償却累計と償却 | (36,530) | | | (25,025) | |
| 財産と設備、純額 | $ | 63,531 | | | $ | 51,523 | |
減価償却と償却費用は#ドルです12.12022年には百万ドル9.42021年には100万ドルです5.72020年までに100万に達するだろう。
無形資産
無形資産の変動状況は以下のとおりである(千計) | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 年初残高 | $ | 53,760 | | | $ | 58,128 | |
| | | |
| 外貨換算調整 | (3,031) | | | (4,368) | |
| 年末残高 | $ | 50,729 | | | $ | 53,760 | |
商誉
営業権の変化は以下の通り(千計) | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 年初残高 | $ | 39,702 | | | $ | 42,798 | |
| | | |
| 外貨換算調整 | (2,150) | | | (3,096) | |
| 年末残高 | $ | 37,552 | | | $ | 39,702 | |
その他負債を計算すべき
その他の負債には、以下の(千計)が含まれる | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 研究と開発費用を計算すべきである | $ | 7,115 | | | $ | 4,878 | |
レンタル負債を経営しています--流動負債 | 4,122 | | | 4,026 | |
| 専門費用を計算する | 1,704 | | | 869 | |
| | | |
| 他にも | 3,066 | | | 1,804 | |
| その他の負債総額を計算すべき | $ | 16,007 | | | $ | 11,577 | |
NOTE 7 – 引受金とその他の事項
賃貸借証書
Sangamoが約半数を占めています87,7002029年5月に期限が切れる賃貸契約によると、カリフォルニア州ブリスベンに1平方フィートのオフィスと研究開発実験室施設がある。Sangamoは約20%を占めています59,4852平方フィートの研究とオフィススペース、賃貸借契約は2031年8月に満期になり、約7,700オフィススペース、レンタル契約は2026年8月に満期になり、カリフォルニア州リッチモンドに位置する。また、同社は約10万人をレンタルしました26,600フランスのヴァル州にあるオフィスと研究開発空間は平方フィートで、賃貸契約は2025年6月から2030年1月まで満期となる。
2020年5月、当社は既存の賃貸契約を改訂し、約を買収した8,500カリフォルニア州リッチモンドにある1平方フィートの追加研究とオフィススペースです改訂されたリースは2020年10月1日に施行され、会社は賃貸負債とそれに応じた使用権資産#ドルを記録した1.3この改訂賃貸契約が開始された時、レンタル料は百万元だった。
2021年1月に、当社は既存の賃貸契約を改訂し、約を買収した5,000カリフォルニア州リッチモンドにある1平方フィートの研究とオフィス空間ですこの改正があれば、既存の賃貸契約は2026年8月に満期になるだろう。本改訂リース契約の有効期限内で、レンタル支払い総額は約$となります0.9百万ドルです。可変賃貸支払いには、建物の運営·管理において所有者が発生するコストと支出における会社分担のシェアが含まれる。2021年2月1日、すなわちレンタル開始日に、会社は経営的賃貸使用権資産とそれに応じた賃貸負債#ドルを記録した0.7百万ドルです。
2021年1月に、当社も新規賃貸契約を締結し、契約を買収した5,800フランスのヴァル州にある1平方フィートの研究と事務空間は、2030年1月に満期になる。本改訂リース契約の有効期限内で、レンタル支払い総額は約$となります0.8百万ドルです。可変賃貸支払いには、建物の運営·管理において所有者が発生するコストと支出における会社分担のシェアが含まれる。2021年1月29日、すなわちレンタル開始日に、会社は経営的賃貸使用権資産とそれに応じた賃貸負債#ドルを記録した0.6百万ドルです。
2021年10月に、当社はカリフォルニア州リッチモンドに位置する研究及びオフィススペースのレンタル期間を延長する協定を締結した5年2031年8月までですその会社は追加のを借りました7,9972021年11月から2031年8月までの同一地点のオフィス空間面積は2平方フィートである。改正された賃貸は2021年10月1日に施行され、会社は賃貸負債とそれに応じた使用権資産を調整し、金額は#ドルとなった9.1この改訂賃貸契約が開始された時、レンタル料は百万元だった。改訂された賃貸契約条項によると、所有者は会社に最大#ドル賠償することに同意した2.6100万は、テナント改善手当と関連がある。
その中のいくつかのレンタル契約には、会社で賃貸借契約の更新または延長を選択して増加させることが含まれています5人至れり尽くせり10年それは.当該等リースに関する投資リターン資産やリース負債を特定する際には、当該等のオプション期間は計算されていないが、当社は当該等選択権を行使することを合理的に肯定するとは考えていないからである。
同社はその契約を評価し、確定した賃貸契約ごとに経営的賃貸契約であることを確定した。経営リースの構成要素は以下の通り(千計)
| | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| リースコストを経営する | $ | 11,029 | | | $ | 10,839 | |
| 可変リースコスト | 3,305 | | | 2,831 | |
| 合計する | $ | 14,334 | | | $ | 13,670 | |
可変リース費用は、会社営業ROU資産と賃貸負債の計量には計上されていません。この可変費用は主に会社が比例して分担する営業費用,財産税,保険からなり,会社はリースと非レンタル部分を分離しないことを選択しているため,リース費用に分類される。
2022年、2021年、2020年12月31日までの年度、賃貸負債を計上して計量した現金は#ドル10.1百万、$6.9百万ドルと$6.4当社の総合キャッシュフロー表は、それぞれ経営活動で使用されている現金純額と現金フロー表に計上されています。
賃貸契約に関連するレンタル料は#ドルです11.0百万、$10.8百万ドルと$10.42022年12月31日まで、2021年、2020年12月31日までの年度はそれぞれ100万ドル2022年12月31日現在、レンタル義務下の将来最低支払いには、以下のものが含まれています(千単位) | | | | | |
| 合計する |
| 2023 | $ | 6,832 | |
| 2024 | 7,318 | |
| 2025 | 7,552 | |
| 2026 | 7,533 | |
| 2027 | 7,480 | |
| その後… | 15,807 | |
| 賃貸支払総額 | 52,522 | |
| もっと少ない: | |
| 利子を推定する | (9,171) | |
| テナント改善手当は反賃貸負債に含まれています | (243) | |
| 合計する | $ | 43,108 | |
| |
2022年12月31日までの記事: | |
| 賃貸負債の短期部分(総合貸借対照表の他の計算すべき負債に含まれる) | $ | 4,122 | |
| 賃貸負債の長期部分 | 38,986 | |
| 合計する | $ | 43,108 | |
2022年12月31日までの加重平均残存レンタル期間は7.0年、経営リース負債を決定するための加重平均増量借入金金利は5.6会社の経営賃貸契約に%を使用します。
契約承諾
2022年12月31日現在、会社は製造関連のサプライヤーの手配の下で、オランダの龍沙会社に関連する取消不能材料契約約束金額は$です7.5100万ドルで2023年12月に満期になりますその会社は$を持っている0.62022年12月31日現在、知的財産権に関する許可義務は100万ドルに達している。
事件があったり
Sangamoは未解決の法的手続きに参加しない。時々、Sangamoは正常な業務過程で発生する法的手続きに巻き込まれる可能性がある。
NOTE 8 – 株主権益
優先株
当社の会社の登録証明書は当社が最も多く発行しています5,000,000優先株は、会社の取締役会が適宜発行することができる。2022年12月31日までに違います。その会社の優先株は発行されたか発行された。
普通株
2020年6月、会社株主は、会社普通株の授権発行株式総数を増加させるために、会社登録証明書の改正案を承認した160,000,000共有する320,000,000株式です。2022年12月31日までに166,793,320同社の普通株は発行された。
本連結財務諸表付記4で述べたBIMAとの連携協定について、当社はBIMAと株式購入協定を締結し、これにより、BIMAはBiogen株を1株$#の価格で買収することに同意した9.2137購入総価格は$です225.0百万ドルです。当社は2020年4月に生物遺伝株式の売却を完了した。
市場で協定を発売する
二零二年八月、当社はJefferies LLC(“Jefferies”)と市場発売計画について公開市場販売協定を締結し、この計画によると、当社は時々適宜、発行価格の高い当社の普通株式を適宜発売することができる150.0ジェフリーを通じて会社の販売代理や依頼人とします。2022年12月、会社は“公開市場第2号改正案”を締結
販売契約は市場発売計画下の総発行価格を追加ドル増加させた175.0百万ドルです。売却契約によると、当社はいかなる株式も売却する義務はありません。2022年12月31日までに会社が販売19,300,743普通株で、純収益は約$です84.9百万ドルです。2021年12月31日現在、会社が販売している2,007,932普通株で、純収益は約$です27.1百万ドルです
2018年株式インセンティブ計画
2020年5月に、当社の株主は、2018年度株式インセンティブ計画(“2018年度計画”)の改訂および再記述を承認し、2018年度計画の下で発行する自社普通株株式総数を増加させる9,900,000株式です。また、2022年5月、会社株主は、2018年計画に基づいて発行保留会社の普通株式総数を増加させることを含む2018年計画の改訂と再記述を承認した7,900,000株式です
2018年計画に基づいて付与された株式オプションの行権価格は下回ってはならない100株式オプションを付与した日は,会社普通株が時価の%を公正にし,オプション期限を超えない10何年もです。株式オプションを取得した人は10%株主は、付与された株式オプションがインセンティブ株式オプションの条件を満たしている場合は、1株当たりの権権価格が下回らない110付与日会社普通株式公正時価の%は,オプション期限を超えない5年それは.一般的に、2018年計画によって付与された株式オプションは三つあるいは…4年期限が切れました10付与された日の数年後、または会社への雇用またはサービスを終了する際には、より早い。
2018年計画に基づいて予約発行される普通株式数は減少する:(I)1−計画に従って株式引受権または株式付加価値権を付与する普通株1株当たり1対1の基準;(2)固定比率1.33この計画に基づいて付与された全価値奨励発行された1株当たり普通株を普通株と交換する。
2018年に計画されている未償還株式オプションまたは他の奨励制約を受けた株式は、発行前に満期または他の方法で終了した場合、そのような株式オプションまたは報酬に制限された株式は、2018年計画に従って後続発行するために使用することができる。当社はその後、2018年計画下の買い戻し権により購入した2018年計画に基づいて発行されたいかなる未帰属株式も、2018年計画に基づいて予約して発行された株式数に日ごとに再加入します1-1ベースまたはa1.33-1の基本(元の報酬における株式備蓄のデビットの比率に依存して)、それに応じて2018年に計画された条項による後続の発行に使用することができる。
2022年12月31日までに9,869,961会社の2018年計画に基づいて将来のために予約した会社の普通株式を奨励する。
2020年従業員株購入計画
2021年5月、会社株主は会社の2020年従業員株購入計画(“ESPP”)を承認した。ESPPは全部で5.0発行に供する普通株百万株を保留する。条件に合った従業員は以下の位置で普通株を購入することができます85適用初日には、会社普通株式公開時価の小さい者の割合2年制提供期限または適用日の最終日6か月購入期限。2022年12月31日までに4,205,502ESPPにより将来発行のために保留されている会社普通株。
株式オプション活動
同社の株式オプション活動の概要は以下の通り | | | | | | | | | | | | | | | | | | | | | | | |
| 量
株 | | 重み付けの-
平均値
すべてのトレーニング
株価.株価 | | 加重平均
残り
契約条項 | | 骨材
固有の
価値がある |
| | | | | (単位:年) | | (単位:千) |
| 2021年12月31日現在の未返済オプション | 11,963,277 | | | $ | 10.36 | | | | | |
| 付与したオプション | 3,399,360 | | | $ | 5.75 | | | | | |
| 行使のオプション | (28,354) | | | $ | 4.40 | | | | | |
| オプションはキャンセルされました | (2,159,288) | | | $ | 10.09 | | | | | |
| 2022年12月31日未償還オプション | 13,174,995 | | | $ | 9.22 | | | 6.96 | | $ | — | |
| | | | | | | |
| 2022年12月31日に行使可能なオプション | 7,823,318 | | | $ | 10.30 | | | 5.83 | | $ | — | |
オプション行使の内在的価値はゼロ, $2.8百万ドルとドル5.42022年12月31日、2021年、2020年12月31日までの年間で、それぞれ100万ドル。
限定株単位
当社は、2022年、2021年及び2020年12月31日までの年間で、4,349,795, 2,140,785そして、そして2,517,101それぞれRSUである.2022年12月31日,2021年,2020年12月31日までの年度内に付与されたRSUの平均付与日公正価値は5.52, $11.16そして$8.06それぞれ,である.この報酬は通常授与されます3年それは.2022年まで,2021年および2020年12月31日までの年間帰属の総公平価値は13.1百万、$9.0百万ドルとドル3.7それぞれ100万ドルです
同社のRSU活動の概要は以下の通り | | | | | | | | | | | | | | | | | |
| 量
株 | | 加重平均
残り
契約条項 | | 元征を集約する
価値がある |
| | | (単位:年) | | (単位:千) |
| 2021年12月31日までの未返済RSU | 3,139,594 | | | | | |
| 受賞したRSU | 4,349,795 | | | | | |
| 解放されたRSU | (1,346,660) | | | | | |
| 没収されたRSU | (898,831) | | | | | |
| 2022年12月31日までの未返済RSU | 5,243,898 | | | 1.03 | | $ | 16,466 | |
| | | | | |
2022年まで、2022年、2021年及び2020年12月31日までに年度帰属するRSUは株式純額で決済され、当社は従業員が所得税及びその他の雇用税項を適用する最低法定責任に相当する株式を差し押さえ、現金を適切な税務機関に送金する。差し押さえられた株式の総数は約380,917, 293,120そして、そして90,617二零二二年、二零二年、二零二一年及び二零二年十二月三十一日までの年度は、当社株式市場価格を決定した各発行日のRSU価値で計算します。従業員が税務機関に納めた税金の総額は#ドルだ2.1百万、$3.3百万ドルとドル0.82022年12月31日、2021年、2020年12月31日までの年間で、それぞれ100万ユーロであり、融資活動として添付されている合併現金フロー表に反映されている。これらの株式の純額決済は、帰属により発行すべき株式数を減少·抹消し、当社に対して支出を構成しないため、当社が株式を買い戻す効果がある。
NOTE 9 – 株に基づく報酬
添付の統合作業報告書で確認された在庫による報酬費用総額(千単位)を表に示す | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 研究開発 | $ | 18,404 | | | $ | 19,534 | | | $ | 13,523 | |
| 一般と行政 | 13,246 | | | 13,422 | | | 12,185 | |
| 株式に基づく報酬総支出 | $ | 31,650 | | | $ | 32,956 | | | $ | 25,708 | |
2022年12月31日現在、未帰属株式オプションに関する今後の期間に確認される株式ベースの報酬支出総額は$23.3100万ドルは加重平均の間に2.07何年もです。2022年12月31日現在、RSUに帰属していないことに関連する今後の期間に確認される株式ベースの報酬支出総額は$23.7100万ドルは加重平均の間に1.90何年もです。あったことがある違います。2022年、2021年または2020年12月31日までの資本化株式従業員給与支出。
推定値仮定
従業員持株計画に基づいて、株式オプションと従業員株式購入にBlack-Scholesオプション推定モデルを用いて従業員株式報酬支出を決定する。オプション推定モデルは主観仮説を入力する必要があるが,これらの仮定は時間とともに変化する可能性がある.RSUの公正価値は,日関連普通株に付与された終値に基づいている。
同社はその普通株の履歴変動性を評価することで期待変動率を決定している。当社は、これらのオプションの公正価値を決定するために、その歴史的行使と既存の終了活動に依存して、その期待期間を推定する。
二零二二年、二零二一年及び二零年十二月三十一日までに、購入持分の加重平均一株当たりの公正価値を$とした3.73, $7.34、と$5.25ブラック·スコアーズの評価に用いられた仮説に基づいています
モデルです従業員の株式オプション公正価値を推定するための仮定は以下のとおりである | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 無リスク金利 | 2.15-3.69% | | 0.95-1.22% | | 0.34-0.61% |
| 予想期限(年単位) | 5.46-5.49 | | 5.46-5.52 | | 5.51-5.57 |
| 株式期待配当収益率 | — | | | — | | | — | |
| 予想変動率 | 72.38-76.01% | | 77.30-79.77% | | 77.61-80.32% |
購入した社員576,950, 433,107そして274,382普通株式はESPPにより加重平均行権価$である3.07, $7.78、と$7.34それぞれ2022年、2021年および2020年12月31日までの年間1株当たり利益を上げている。2022年,2021年および2020年12月31日までに,当社株主特別引出権計画に基づいて購入した株式の加重平均推定公正価値は1.85, $4.48そして$8.02ブラック-スコアーズ推定モデルで用いた仮定にそれぞれ基づく.
ESPP購入権公正価値を推定するための仮定は以下のとおりである | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 無リスク金利 | 1.62-4.61% | | 0.01-2.80% | | 1.53-2.80% |
| 予想期限(年単位) | 0.5-2.0 | | 0.0-2.0 | | 0.5-2.0 |
| 株式期待配当収益率 | — | | | — | | | — | |
| 予想変動率 | 57.97-72.14% | | 32.54-97.88% | | 51.02-91.96% |
NOTE 10 – 従業員福祉計画
当社は国税法第401(K)節の規定に基づき、全常勤従業員を対象とした固定払込貯蓄計画(略称Sangamo 401(K)計画)を開始した。Sangamo 401(K)計画は、国内税法401条の規定に適合することを目的としている。
その会社は従業員の支払いと100% in 2022 and 2021 and 501回目は%8%、最高限度$4,0002022年、2021年、2020年。セット資金は出資時に完全に帰属する。同社のSangamo 401(K)計画への貢献は$1.5百万、$1.5百万ドルと$1.22022年12月31日まで、2021年、2020年12月31日までの年度はそれぞれ100万ドル。
NOTE 11 – 所得税
所得税前に赤字となった国内と海外の部分は以下の通り(千計) | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 国内では | $ | (216,573) | | | $ | (185,216) | | | $ | (126,624) | |
| 外国.外国 | 24,724 | | | 7,225 | | | 5,847 | |
| 所得税前損失 | $ | (191,849) | | | $ | (177,991) | | | $ | (120,777) | |
I所得税支出は以下の部分からなる(千計) | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 所得税支出: | | | | | |
| 現在: | | | | | |
| 連邦制 | $ | — | | | $ | — | | | $ | — | |
| 状態.状態 | — | | | — | | | 133 | |
| 外国.外国 | 500 | | | 886 | | | 686 | |
| 小計 | 500 | | | 886 | | | 819 | |
| 延期: | | | | | |
| 連邦制 | — | | | — | | | — | |
| 状態.状態 | — | | | — | | | — | |
| 外国.外国 | (71) | | | (580) | | | (474) | |
| 小計 | (71) | | | (580) | | | (474) | |
| 所得税費用 | $ | 429 | | | $ | 306 | | | $ | 345 | |
所得税支出と所得税前損失に対して連邦法定所得税税率を適用して算出した金額との差を以下のように解釈する(千単位) | | | | | | | | | | | | | | | | | |
| 十二月三十一日までの年度 |
| 2022 | | 2021 | | 2020 |
| 連邦法定税率で課税する | $ | (40,288) | | | $ | (37,372) | | | $ | (25,363) | |
| 州税、純額 | (6,895) | | | (6,734) | | | (3,168) | |
| 外貨利回り | 309 | | | 362 | | | 376 | |
| 世界の無形低税収入 | 1,002 | | | 637 | | | 1,335 | |
| 差し引かれない株の報酬 | 3,545 | | | 2,770 | | | 4,232 | |
| 研究単位 | (6,694) | | | (5,230) | | | (3,657) | |
| 評価免除額を変更する | 44,005 | | | 45,373 | | | 26,537 | |
| 譲渡定価決済 | 4,343 | | | — | | | — | |
| 他にも | 1,102 | | | 500 | | | 53 | |
| 所得税費用 | $ | 429 | | | $ | 306 | | | $ | 345 | |
繰延所得税は、財務報告目的のための資産および負債の帳簿金額と所得税目的のための金額との間の一時的な差異の純税影響を反映する会社の繰延税金資産と負債の重要な構成要素は以下の通り(千計)
| | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 資産: | | | |
| 繰延税金資産: | | | |
| 純営業損失が繰り越す | $ | 174,129 | | | $ | 159,740 | |
| 研究と開発税収は繰越免除 | 44,264 | | | 35,260 | |
| 株に基づく報酬 | 7,695 | | | 6,691 | |
| 収入を繰り越す | 38,700 | | | 61,114 | |
| 資本化研究 | 37,985 | | | — | |
| 固定資産 | 10,087 | | | 10,130 | |
| リース責任 | 10,074 | | | 11,279 | |
| 課税項目と準備金 | 1,603 | | | 1,119 | |
| 他にも | 283 | | | 106 | |
| 繰延税金資産総額 | 324,820 | | | 285,439 | |
| 推定免税額 | 301,840 | | | 259,820 | |
| 繰延税金資産 | 22,980 | | | 25,619 | |
| 負債: | | | |
| 無形資産 | (13,512) | | | (13,856) | |
| 経営的リース使用権資産 | (14,620) | | | (17,348) | |
| 繰延税金負債 | (28,132) | | | (31,204) | |
| 繰延納税純負債総額 | $ | (5,152) | | | $ | (5,585) | |
税務管区に基づく繰延税金資産と負債は、総合貸借対照表に以下のように示されている(千計) | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 |
| 繰延税金資産(総合貸借対照表上の他の非流動資産に含まれる) | $ | 1,118 | | | $ | 1,060 | |
| 繰延税金負債 | (6,270) | | | (6,645) | |
| 繰延税金純負債 | $ | (5,152) | | | $ | (5,585) | |
繰延所得税資産のすべてまたは一部が現金化できない可能性が高い場合には、推定準備を計上する。当社はその繰延所得税資産計について評価する必要があるかどうかを定期的に評価し、当社の繰延所得税資産の現金化の可能性が大きいかどうかに関するプラスおよび負の証拠を考慮する方法である。繰延所得税資産を生成する司法管轄区域内で繰延所得税資産を回収する能力を評価する際に、会社は、繰延所得税負債の予定沖販売、将来の課税収入、税務計画戦略、および最近の経営結果を含むすべての利用可能な積極的かつ消極的な証拠を考慮する。そのため、当社のこれらの要因の分析によると、繰延税項目の純資産は推定値から大幅に相殺されている。推定免税額は#ドル増加した42.0百万、$45.5百万ドルとドル26.62022年12月31日まで、2021年、2020年12月31日までの年度はそれぞれ100万ドル。
Sangamoの連邦と州所得税における純営業損失は2022年12月31日現在約$に転換している689.7百万ドルとドル312.0それぞれ100万ドルです2018年までに発生した連邦純運営損失は2023年に満期になり、利用しなければ2037年まで満期が続く。2018年に発生した連邦純運営損失は無期限繰り越しとなる。利用しなければ、繰り越しの国の純営業損失はそれぞれ2029年に満期になる。同社のフランス純営業損失繰越残高は#ドル115.6100万ドル無期限に続いていますその会社は連邦と州研究税の繰越免除を持っています36.8百万ドルとドル26.1それぞれ100万ドルです連邦研究単位は2023年に満期になり、2042年まで続くが、州研究単位は満期日がない。国税法や国が定めるような所有権変更制限により、当社の純営業損失繰越や税収控除繰越の使用が重大な年度制限を受ける可能性がある。年度限度額は繰り越しの純営業損失と使用前の研究税収控除満期を招く可能性がある。
同社の政策は、その非米国子会社の収益をこれらの事業に再投資することだ。同社は外国子会社の収益に米国税を計上しておらず、同社はこれらの収益を無期限に海外に再投資しようとしているからだ。しかし、これらの資金が国内に送金された場合、会社は適用されたアメリカ国税と源泉徴収税を計算して支払うことを要求されるだろう。海外で発生した累積損失により、国内に送金できる収益はありません。
同社は連邦と州所得税申告書を提出したが、異なる制限法規がある。純営業損失や税収控除の繰越により、2002年からの納税年度はまだ審査が必要である。当社もイギリスおよびフランスの所得税申告表を提出し、2008年からその後の課税年度はイギリスでも開放されているが、2018年とその後のフランスでの課税年度はまだ審査が必要である。
当社のやり方は、所得税支出において所得税事項に関する利息及び/又は罰金を確認することです。2022年12月31日現在、同社は0.2百万ドルは利息と/または罰金を計算します。通常業務中に出現する項目については、未確認の税収割引が来年度に変化する可能性がある。未確認の税収割引が確認されれば、実際の税率に影響を与える金額は#ドルとなる1.2百万、$1.2百万ドルと$0.6それぞれ2022年、2021年、2020年12月31日まで。
以下の表は、同社が確認していない税収割引に関する活動(単位:千):をまとめています | | | | | | | | | | | | | | | | | |
| 十二月三十一日 |
| 2022 | | 2021 | | 2020 |
| 期初残高 | $ | 15,062 | | | $ | 12,892 | | | $ | 11,630 | |
| 本年度に関連する納税状況に基づいて計算される増加額 | 3,177 | | | 2,454 | | | 2,834 | |
| 数年前の納税状況を増やす | 278 | | | 130 | | | 1,982 | |
| 先日の減税状況 | (338) | | | (414) | | | (3,554) | |
| 期末残高 | $ | 18,179 | | | $ | 15,062 | | | $ | 12,892 | |
NOTE 12 – 関係者取引
2022年12月31日および2021年12月31日までに重大な関連者取引はなかった。
項目9−会計·財務開示面の変更と会計士との相違
ない。
プロジェクト9 A--制御とプログラム
情報開示制御とプログラムの評価
我々は、我々の取引所法案報告において開示が必要な情報が、米国証券取引委員会規則および表で指定された期間内に記録、処理、集約および報告されることを確実にするための合理的な保証を提供し、これらの情報が、必要な開示に関する決定をタイムリーに行うために、我々の経営陣に蓄積されて伝達されることを目的とする開示制御および手続きを維持する。
我々の最高経営責任者と最高財務責任者の監督の下で、2022年12月31日までの開示制御および手順(取引所法案規則13 a-15(E)および15 d-15(E)で定義されるような)の有効性を評価した。この評価に基づき、2022年12月31日現在、我々の最高経営責任者および最高財務官は、我々の開示制御および手続きが合理的な保証レベルで有効であると結論した。
制御やプログラムへの固有の制限
私たちの経営陣は、CEOやCEOを含め、私たちの開示統制や手続き、および財務報告に対する私たちの内部統制がすべてのミスやすべての詐欺を防ぐことを期待していません。制御システムの設計や動作がどんなに良くても,合理的な保証を提供し,制御システムの目標を実現することしかできない.制御システムの設計は資源の制限を反映しており,制御の利点はそのコストに対して考慮しなければならない.すべての制御システムには固有の限界があるため、どの制御評価もわが社のすべての制御問題や不正事件がすでにまたは検出されることを絶対に保証することはできません。これらの固有の制限は、開示および財務報告手続きの既知の特徴であるため、これらのリスクを除去するのではないにもかかわらず、保障措置を設計することが可能である。これらの固有の限界には,意思決定における判断が誤りである可能性があり,故障発生は簡単な誤りや誤りによる現実がある.制御はまた、ある人の個人的な行動、2人以上の結託、または制御の管理優先によって回避することができる。いずれの制御システムの設計も,将来のイベントの可能性に対する何らかの仮定にある程度基づいている.私たちの開示制御と手続き、および財務報告に対する私たちの内部統制は、その目標を達成するための合理的な保証を提供することを目的としているが、どの設計もすべての未来の条件でその目標を達成することに成功する保証はない。時間の経過とともに,条件の変化や政策やプログラムに対する遵守度の悪化により,制御が不十分になる可能性がある.費用対効果のある制御システムの固有の限界から, ミスや詐欺による誤った陳述は発生する可能性があり、発見されないだろう。
財務報告の内部統制に関する経営陣の報告
私たちの経営陣は、取引規制13 a-15(F)および15 d-15(F)で定義されている十分な財務報告内部統制を確立し、維持する責任があります。私たちの経営陣は、CEOや最高財務責任者を含め、テレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み”(2013年枠組み)で提案された枠組みに基づいて、財務報告の内部統制の有効性を評価しました。この枠組みの下での評価によると、我々の経営陣は、2022年12月31日現在、財務報告の内部統制を合理的な保証水準で有効であると結論している。
2022年12月31日現在、財務報告の内部統制の有効性は、独立公認会計士事務所安永会計士事務所が監査しており、この報告書は本稿に含まれている。
財務報告の内部統制の変化
2022年12月31日までの3ヶ月間、取引所法案規則13 a-15(D)および15 d-15(D)に要求された評価によると、財務報告の内部統制には何の変化もなく、これは私たちの財務報告の内部統制に大きな影響を与え、あるいは合理的な可能性が私たちの財務報告の内部統制に大きな影響を与える。
独立公認会計士事務所報告
Sangamo治療会社の株主と取締役会に
財務報告の内部統制については
我々は、トレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み”(2013年枠組み)(COSO基準)で確立された基準に基づき、Sangamo治療会社が2022年12月31日までの財務報告内部統制を監査した。COSO基準によると,2022年12月31日現在,Sangamo治療会社(当社)はすべての重要な面で財務報告に対して有効な内部統制を行っていると考えられる。
我々はまた、米国上場会社会計監督委員会(PCAOB)の基準に従って当社の2022年総合財務諸表を監査し、2023年2月22日の報告書に保留のない意見を発表した。
意見の基礎
当社経営陣は、効果的な財務報告内部統制を維持し、添付されている“経営陣財務報告内部統制報告”に掲載されている財務報告内部統制の有効性を評価する責任がある。私たちの責任は私たちの監査に基づいて、会社の財務報告書の内部統制に意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。
我々の監査には,財務報告の内部統制を理解すること,重大な弱点があるリスクを評価すること,評価されたリスクテストに基づいて内部統制の設計·運用有効性を評価すること,および状況下で必要と考えられる他のプロセスを実行することが含まれる。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分に関する記録の保存、(2)公認された会計原則に従って財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、会社の収入および支出は会社の経営陣と取締役の許可のみに基づいて行われる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用、または処分会社の資産を防止またはタイムリーに発見する合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
/s/安永法律事務所
カリフォルニア州サンマテオ
2023年2月22日
プロジェクト9 B-その他の資料
ない。
プロジェクト9 C−妨害検査に関する外国司法管区の開示
ない。
第三部
私たちは、改正された1934年証券取引法第14 A条、または2023年の委託書に基づいて、本10-K年度報告書に含まれる財政年度終了後120日以内に、我々の最終委託書または2023年委託書を次の株主年次総会に提出する予定であるため、本報告の表格10-Kは、第III部分に要求されるいくつかの情報を省略し、2023年委託書に含まれるいくつかの情報を引用して本明細書に組み込む。
プロジェクト10--取締役、執行幹事、およびコーポレートガバナンス
本プロジェクト要求の情報は,我々の2023年依頼書に含まれ,以下のようになる
•私たちの役員と取締役の有名人の指名に関する情報は、“取締役選挙”というタイトルの部分に含まれます
•実行幹事に関する情報は、“実行幹事”と題する部分に含まれます
•私たちの監査委員会と監査委員会の財務専門家に関する情報は“役員選挙-監査委員会”の節に含まれる
•私たちの道徳基準に関する情報は、“取締役選挙--商業行為と道徳基準”と題する部分に含まれる
•株主が取締役会に著名人を推薦するプログラムに関する情報は、“これらのエージェント材料と投票に関する質疑応答”と題する部分に含まれる
•取引法第16条(A)条の遵守に関する情報は、“延滞第16条(A)報告書”と題する章に含まれる
これらの情報は,2023年依頼書が本年度報告に含まれる10-K表に含まれる財政年度終了後120日以内に提出されていなければ,漏れた情報が当該120日の期間終了前に提出された本10-K表年次報告の修正案に含まれることを前提としている.
プロジェクト11--役員報酬
本プロジェクトに要求される資料は、“役員報酬”、“役員報酬”、“役員選挙-報酬委員会連動と内部者参加”および“報酬委員会報告”というタイトルの2023年依頼書に盛り込まれ、参考にして組み込まれるが、2023年依頼書が本年度報告書に含まれる10−K表に含まれていない財政年度終了後120日以内に提出される場合、漏れた資料は、120日の期間終了前に提出される10−K表年次報告修正案に含まれる。
プロジェクト12--特定の実益所有者の保証所有権及び経営陣及び株主に関する事項
本条項に要求される持分補償計画に関する情報は、2023年委託書の“持分補償計画情報”の節に含まれ、本条項が要求する特定の利益所有者及び経営層の保証所有権に関する情報は、2023年委託書に含まれる“特定の利益所有者及び経営層の保証所有権”という節に含まれ、いずれの場合も引用により本明細書に組み込まれ、2023年委託書が本年度報告に含まれる10−K表に含まれていない財政年度終了後120日以内に提出されなければ、漏れた資料は、120日以内の期限終了前に提出された本10-K表年次報告の修正案に含まれる。
プロジェクト13--特定の関係および関連取引、および取締役の独立性
このプロジェクトに要求される資料は,我々の2023年依頼書に含まれ,そのタイトルは“若干の関係および関連取引”および“選挙役員−取締役会独立性”の節であり,参考にして当社の2023年依頼書に組み込まれるが,本年度報告がForm 10−K表でカバーされている財政年度終了後120日以内に2023年依頼書が提出されていない場合には,漏れた資料は当該120日の期間終了前にForm 10−K表で提出された本年報修正案に含まれる。
プロジェクト14--主に課金とサービス
このプロジェクトに要求される情報は、2023年の委託書の“独立公認会計士事務所の承認”と題する部分に含まれ、引用によって本明細書に組み込まれるが、2023年の委託書が本年度報告10-K表に含まれる財政年度終了後120日以内に提出されていない場合、漏れた情報は、120日以内に終了する前に提出された本10-K表年次報告書の修正案に含まれる。
第4部
プロジェクト15--証拠品と財務諸表の添付表
(A)本年報の表格10-Kは、以下のファイルを含む:
1.財務諸表--項目8の連結財務諸表インデックスを参照。
2.財務諸表の添付表--は適用されません。
3.展示品
| | | | | |
展示品 番号をつける | 書類説明 |
| 2.1 | 当社と署名ページに記載されているTxCell売却株主との間で2018年7月20日に締結された株式購入契約(2018年7月23日に提出された会社の現在報告8-K表の添付ファイル2.1を参照して組み込む)。 |
| 2.2 | 会社とTxCell S.A.が2018年10月1日に締結した株式購入協定改訂協定(2018年11月6日に提出された会社の現在報告8-K表の添付ファイル2.2を参照して統合された)。 |
| 2.3 | 会社とTxCell S.A.が2018年7月20日に署名した買収要約契約(2018年7月23日に提出された会社の現在報告8-K表の添付ファイル2.2を参照して統合された)。 |
| 2.4 | 会社とTxCell S.A.が2018年10月1日に締結した入札要約プロトコル第1号修正案(2018年11月6日に提出された会社現在報告8-K表の添付ファイル2.4を参照して統合された)。 |
| 3.1 | 7番目の改訂および再改訂された会社登録証明書(2017年8月9日に提出された会社四半期報告10-Q表の添付ファイル3.1を参照して編入)。 |
| 3.2 | 7回目の改訂後の会社登録証明書の第4回改訂証明書(当社が2020年5月22日に提出した8−K表の現在報告書の添付ファイル3.1を参照して編入)。 |
| 3.3 | 第5回改正及び再改訂の定款(2022年12月19日に当社が提出した8−K表の現在の報告書の添付ファイル3.1を参照して編入)。 |
| 4.1 | 株本説明 |
| 4.2 | 普通株式証明書サンプルフォーマット(2017年1月6日に提出された会社の現在の報告書8-K表の添付ファイル4.1を参照して組み込まれます)。 |
| 10.1(+) | 2013年度株式インセンティブ計画(“2013計画”)の改訂および再作成(当社が2018年5月10日に提出した10-Q表四半期報告添付ファイル10.2参照)。 |
| 10.2(+) | 2018年株式インセンティブ計画(“2018年計画”)を改訂し、再記述する(添付ファイル10.1を参照して2022年5月25日に提出された現在の8-K表報告書に組み込まれる)。 |
| 10.3(+) | 2018年株式インセンティブ計画フランス株式オプションサブ計画(“フレンチオプションサブ計画”)(会社が2019年3月1日に提出した10-Kフォーム年次報告書の添付ファイル10.3を参照して編入)。 |
| 10.4(+) | 2018年株式インセンティブ計画フランス限定株式単位奨励子計画(“フランスRSU子計画”)(会社が2019年3月1日に提出したForm 10-K年報添付ファイル10.4を参照して編入)。 |
| 10.5(+) | 2020年従業員株購入計画(2020年10月15日に提出された会社S-8表登録説明書添付ファイル99.1を参照)。 |
| 10.6(+) | 2013年計画下の制限株式報酬プロトコル表(当社が2013年6月14日に提出した現在の8-K表の添付ファイル10.2を参照して組み込まれます)。 |
| 10.7(+) | 2013年度計画は、株式オプション通知書表を付与する予定です(当社が2013年6月14日に提出した現在の8-K表添付ファイル10.3を参照)。 |
| 10.8(+) | 2013年に計画された株式オプション協定表(当社が2013年6月14日に提出した現在の8-K表の添付ファイル10.4を参照して組み込む)。 |
| 10.9(+) | 2013年に株式オプション通知フォーム-取締役初期付与を計画しています(会社が2013年6月14日に提出した現在の8-Kフォームの添付ファイル10.5に合併)。 |
| 10.10(+) | 2013年に株式オプション通知表-取締役年次付与表(会社が2013年6月14日に提出した現在の8-K表に統合された添付ファイル10.6)を付与する予定です。 |
| 10.11(+) | 2013年に計画された自動株式オプション協定表(当社が2013年6月14日に提出した現在の8-K表の添付ファイル10.7を参照して編入することにより)。 |
| 10.12(+) | 2018年計画下の株式オプション付与通知フォーマットおよびオプションプロトコルフォーマット(添付ファイル99.2を参照して、2018年6月15日に提出された現在の8-Kレポートの添付ファイル99.2に組み込まれます)。 |
| | | | | |
展示品 番号をつける | 書類説明 |
| 10.13(+) | 2018年に計画されている株式オプション付与通知フォームおよびオプションプロトコルテーブル(非従業員取締役)(2018年6月15日に提出された会社の現在の8-Kフォームの添付ファイル99.3を参照して組み込まれています)。 |
| 10.14(+) | 2018年に計画された株式オプション付与通知フォームおよびオプションプロトコルテーブル(イギリス人従業員)(添付ファイル99.4を参照して2018年6月15日に提出された現在の8-Kフォームレポートの添付ファイル99.4に組み込まれます)。 |
| 10.15(+) | 2018年計画およびフレンチオプション二次計画(2019年3月1日に提出された企業10-Kフォーム年次報告添付ファイル10.14を参照して編入することにより)下の株式オプション付与通知用紙(フランス人従業員)。 |
| 10.16(+) | 2018年計画およびフランスオプション二次計画(2019年3月1日に提出された企業10-Kフォーム年次報告書添付ファイル10.15を参照して編入することにより)下の株式オプション契約表(フランス人従業員)。 |
| 10.17(+) | 2018年計画下の制限株式単位付与通知テーブルおよび制限株式単位報酬プロトコルテーブル(米国人従業員)(添付ファイル99.5を参照して、2018年6月15日に提出された8-Kフォームの現在の報告書に組み込まれています)。 |
| 10.18(+) | 2018年に計画された制限株式単位授出通知書表および制限株式単位奨励協定(非従業員取締役)表(添付ファイル99.6を参照して、2018年6月15日に提出された会社の現在報告の表格8-Kに組み込まれます)。 |
| 10.19(+) | 2018年計画下の制限株式単位授出通知表および制限株式単位奨励協定(英国人従業員)表(添付ファイル99.7を参照して2018年6月15日に提出された8-K表の現在の報告書に組み込まれています)。 |
| 10.20(+) | 2018年計画及びフランスRSUサブ計画下の制限株式単位付与通知及び制限株式単位奨励協定(フランス人従業員)のフォーマット。(当社が2019年3月1日に提出したForm 10-K年次報告書の添付ファイル10.19を参照)。 |
| 10.21(+) | 改正·再策定された離職計画(当社が2019年3月1日に提出した10-K表年次報告書の添付ファイル10.20を引用して組み込む)。 |
| 10.22(+) | インセンティブ報酬計画が改訂および再作成された(会社が2018年5月10日に提出した10-Q表四半期報告書の添付ファイル10.2を参照して組み込まれた)。 |
| 10.23(+) | 賠償プロトコル表(当社が2020年5月11日に提出したForm 10-Q四半期報告の添付ファイル10.3参照)。 |
| 10.24(+) | 会社とAlexander(Sandy)Macraeの雇用契約は、2016年5月17日となっている(会社が2016年8月4日に提出したForm 10-Q四半期報告の添付ファイル10.1を参考にして組み込まれている)。 |
| |
| |
| |
| 10.25(+) | 会社とロルフ·アンドリュー(アンディ)·ラメルマイヤーとの雇用協定が2017年11月1日に施行された(添付ファイル10.26を参照して2020年2月28日に提出されたForm 10-K年次報告書の添付ファイル10.26に組み込まれる)。 |
| 10.26(+) | アンドリュー·ラメルマイヤー特別ボーナスに関する書面協定(当社が2020年8月5日に提出した10-Q表四半期報告書の添付ファイル10.2を引用して組み込む)。 |
| 10.27(+) | 当社とJason Fontenotが2019年1月28日に締結した書簡協定(当社が2021年5月4日に提出したForm 10-Q四半期報告添付ファイル10.3を引用して当社に組み込まれています)。 |
| 10.28(+) | 当社とRobert J.Schottが2021年1月6日に締結した書簡協定(当社が2021年5月4日に提出したForm 10-Q四半期報告添付ファイル10.4を引用)。 |
| 10.29(+) | 会社とPrahyusha DuraiBabuが2021年5月21日に署名した書簡協定(会社が2021年8月5日に提出したForm 10-Q四半期報告添付ファイル10.1を参照して編入)。 |
| 10.30(+) | 会社とScott Willoughbyとの間の日付は、2021年8月2日の書簡合意である(2021年11月4日に提出された会社10-Q四半期報告添付ファイル10.1を参照して組み込まれる)。 |
| 10.31(+) | 当社はDavidと2021年11月1日に締結した書簡協定(当社が2022年2月24日に提出したForm 10-K年報添付ファイル10.34を参考に当社に組み込まれています)。 |
| 10.32(+) | 会社とNathalie Dubois-StringFloorが2022年9月28日に署名した書簡協定(会社が2022年11月3日に提出したForm 10-Q四半期報告の添付ファイル10.1を参照して組み込まれている)。 |
| 10.33(+) | 会社とPoint Richmond R&D Associates II,LLCとの間のTriple Net実験室リースは,1997年5月23日(会社が2000年2月24日に提出したS-1表登録声明の添付ファイル10.5を引用して合併した)である. |
| | | | | |
展示品 番号をつける | 書類説明 |
| 10.34 | 2004年3月12日にPoint Richmond R&D Associates II,LLCと締結されたTriple Net実験室賃貸借契約の第1の修正案(社が2005年2月23日に提出したForm 10−K年次報告書の添付ファイル10.20を参照して組み込まれた)。 |
| 10.35 | 2007年3月15日、会社とPoint Richmond R&D Associates II,LLCとの間のTriple Net実験室賃貸契約の第2の改訂(会社が2013年11月4日に提出したForm 10-Q四半期報告書の添付ファイル10.1を参照することによって組み込まれる)。 |
| 10.36 | 2013年8月1日に会社とPoint Richmond R&D Associates II,LLCとの間のTriple Netラボリース第3回修正案(2013年11月4日に提出されたForm 10-Q四半期報告書の添付ファイル10.2を参照して組み込まれた)。 |
| 10.37 | 2016年6月10日、会社とPoint Richmond R&D Associates II,LLCとの間のTriple Netラボリース第4の修正案(会社が2019年3月1日に提出したForm 10-K年間報告書の添付ファイル10.33を参照して組み込まれます)。 |
| 10.38 | 同社とPoint Richmond R&D Associates II,LLCが2017年7月10日に締結したTriple Netラボリース第5修正案(2019年3月1日に提出された会社年次報告Form 10-Kの添付ファイル10.34を引用することにより統合)。 |
| 10.39 | 社とPoint Richmond R&D Associates II,LLCが2018年5月11日に締結したTriple Netラボリース第6修正案(社が2018年8月8日に提出したForm 10-Q四半期報告書の添付ファイル10.9を参照して組み込まれている)。 |
| 10.40 | 2020年5月20日に同社とPoint Richmond R&D Associates II,LLCとの間のTriple Netラボ賃貸の第7回修正案(会社が2020年8月5日に提出したForm 10-Q四半期報告書の添付ファイル10.4を引用して組み込む)。 |
| 10.41 | 2020年5月29日、同社とPoint Richmond R&D Associates II,LLCとの間のTriple Netラボリースの8回目の改訂(会社が2020年8月5日に提出したForm 10-Q四半期報告書の添付ファイル10.5を参照して組み込まれる)。 |
| 10.42 | 会社とPoint Richmond R&D Associates II,LLCが2021年1月4日に締結したTriple Netラボ賃貸借契約第9修正案(会社が2021年5月4日に提出したForm 10-Q四半期報告書の添付ファイル10.2を引用して組み込む)。 |
| 10.43 | 社とPoint Richmond R&D Associates II,LLCとの間で2021年10月18日に締結されたオフィスと実験室賃貸契約を改訂·再締結した(社が2022年2月24日に提出したForm 10−K年度報告書の添付ファイル10.34を参考に合併)。 |
| 10.44 | 会社とMarina Boulevard Propertyのリース契約は、2017年11月3日となっています(2018年3月1日に提出された会社年次報告Form 10-Kの添付ファイル10.21を参照して合併)。 |
| 10.45 | 会社とMarina Boulevard Propertyのリース契約第1修正案は、2019年1月1日となっている(2019年3月1日に提出された会社年次報告Form 10-Kの添付ファイル10.37を参照して編入)。 |
| 10.46 | 会社とJefferies LLCの公開市場販売協定は、2020年8月5日(会社が2020年8月5日に提出したForm 10-Q四半期報告書の添付ファイル1.1を参照して組み込まれています)。 |
| 10.47 | 当社とJefferies LLCが2021年5月5日に締結した公開市場販売協定の第1号改正案(2021年5月5日に提出された会社S-3表登録声明の添付ファイル1.3を参照して合併したもの)。 |
| 10.48 | 当社とJefferies LLCが2022年12月23日に締結した公開市場販売協定第2号改正案(当社が2022年12月23日に提出した8-K表現在報告の添付ファイル1.1を参照して組み込む)。 |
| 10.49† | 当社とShire International GmbHが2015年9月1日に締結した協力及び許可協定を改訂及び再予約する(合併内容は当社が2015年10月30日に提出した10-Q表四半期報告添付ファイル10.1を参照)。 |
| 10.50† | 会社とBiogen MA Inc.(Bioverativ Inc.)が2014年1月8日に署名した世界的な研究、開発および商業化協力および許可協定(会社が2014年5月7日に提出したForm 10-Q四半期報告書の添付ファイル10.1を参照することによって組み込まれる)。 |
| 10.51† | “グローバル研究、開発および商業化協力と許可協定の書簡修正案”は、会社とBiogen MA Inc.(Bioverativ Inc.)であり、日付は2015年12月14日(会社が2016年2月18日に提出したForm 10-K年度報告書の添付ファイル10.63を参照して編入された)。 |
| 10.52† | 会社とBiogen MA Inc.(Bioverativ Inc.)との間の書簡合意および免除は、2016年3月24日(会社が2016年5月5日に提出したForm 10-Q四半期報告書の添付ファイル10.1を参照して組み込まれる)である。 |
| | | | | |
展示品 番号をつける | 書類説明 |
| 10.53⁑ | 当社がファイザーと締結した協力·許可協定は、2017年5月10日となっています。 |
| 10.54⁑ | 2019年12月17日の会社とファイザーの協力と許可協定の改訂状は、2017年5月10日(会社が2020年2月28日に提出したForm 10-K年報添付ファイル10.45を引用して組み込まれています)。 |
| 10.55† | 同社とファイザーが締結した研究協力·許可協定は、2017年12月28日(会社が2018年3月1日に提出したForm 10-K年報添付ファイル10.40を引用して編入)となっている。 |
| 10.56† | 2019年3月21日にファイザーと締結した研究協力·許可協定第1号改正案(2019年5月8日に提出された会社の四半期報告10-Q表の添付ファイル10.3を引用して組み込む)。 |
| 10.57⁑ | 当社とファイザーが2020年7月31日に締結した“研究協力と許可協定”第2号改正案(当社が2020年11月4日に提出したForm 10−Q四半期報告の添付ファイル10.3を引用して組み込む)。 |
| 10.58† | 当社がKite Pharma,Inc.と2019年9月11日に締結した協力とライセンス契約を改訂し、再署名しました(当社が2019年11月6日に提出したForm 10-Q四半期報告書の添付ファイル10.1を引用して合併します)。 |
| 10.59⁑ | 会社、Biogen MA、Inc.とBiogen International GmbHとの間の協力および許可協定は、2020年2月26日(会社が2020年5月11日に提出したForm 10-Q四半期報告書の添付ファイル10.1を参照して組み込まれる)である。 |
| 10.60 | 会社とBiogen MA,Inc.が2020年2月26日に締結した株式購入協定(当社が2020年5月11日に提出したForm 10-Q四半期報告書の添付ファイル10.2を引用して組み込む)。 |
| 10.61⁑ | 同社とノワール生物医学研究所が2020年7月27日に締結した協力·許可協定(2020年11月4日に会社が提出したForm 10−Q四半期報告書の添付ファイル10.2を引用して組み込む)。 |
| 21.1 | 当社の付属会社です。 |
| 23.1 | 独立公認会計士事務所が同意します。 |
| 24.1 | 授権書(署名ページに含まれる)。 |
| 31.1 | 第13 a-14(A)条特等に対して幹事の認証を行う。 |
| 31.2 | 細則13 a-14(A)首席財務幹事の証明。 |
| 32.1* | “米国法典”第18編第1350条に基づく認証。 |
| |
| 101.INS | XBRLインスタンス文書−インスタンス文書は、そのXBRLタグがイントラネットXBRL文書に埋め込まれているので、対話データファイルには表示されない。 |
| 101.書院 | XBRL分類拡張アーキテクチャドキュメント |
| 101.カール | XBRL分類拡張計算リンクライブラリ文書 |
| 101.def | XBRL分類拡張Linkbase文書を定義する |
| 101.介護会 | XBRL分類拡張タグLinkbaseドキュメント |
| 101.Pre | XBRL分類拡張プレゼンテーションLinkbaseドキュメント |
| 104 | Sangamo 2022年12月31日までの年間Form 10-K年次報告の表紙はイントラネットXBRL形式で添付ファイル101に掲載されている |
_______________________† 米国証券取引委員会の命令により,本稿に含まれるいくつかの情報に対して秘匿処理を与える.このような情報は省略され、米国証券取引委員会に個別に提出された。
本展示品のある部分を“”で表す[*]“)は、”連邦判例アセンブリ“第17章229.601(B)項により省略されている。
(+)は、管理契約または補償計画またはスケジュールを示します。
* “米国法典”第18編の規定によると,本年度報告は添付ファイル32.1に添付されている証明書を10−K表の形で添付する 1350通過後 2002年の“サバンズ-オキシリー法案”第906節によると、改正された1934年の証券取引法第18節に基づいて当社が提出した書類とみなされてはならない。
本年度報告書が10-K表の形式で提出された合意および他の文書は、合意または他の文書自体の条項に加えて、事実情報または他の開示を提供することを意図しておらず、これらに依存してこの目的を達成してはならない。特に、私たちがこのような合意や合意で下した任意の陳述と保証は
他の文書は、協定または文書に関する具体的な範囲内でのみ作成され、その締結日まで、または任意の他の時間における実際の状況を記述することはできない。
項目16--表格10-Kの概要
ない。
サイン
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、2023年2月22日に、次の署名者がその代表として本報告に署名することを正式に許可した。 | | | | | | | | | | | |
| 日付:2023年2月22日 | | | |
| | Sangamo治療会社 |
| | 差出人: | / S / ALEXANDER D。 MACRAE |
| | | アレクサンダー·D·マクレレ 社長と最高経営責任者 |
以下の署名の各個人は、このような身分で構成され、Alexander D.MacraeおよびScott Willoughbyをその真の合法的な事実代理人および代理人に委任し、その名前、職または代理人の名義、職または代替の十分な権限を有し、本Form 10-K年次報告に対する任意およびすべての修正案(発効後の修正案を含む)に任意およびすべての修正案を署名し、これを証拠物および他の関連文書と共に米国証券取引委員会に提出し、上記事実代理人および代理人を付与する。そして、彼らの各々は、その場所およびその周囲で必要かつ行わなければならないすべてのことを行い、実行する権利が完全にあり、可能であるか、または自ら行うことができるすべての意図および目的を尽くし、ここで上記のすべての事実弁護士および代理人を承認し、確認することができ、またはそれらの1人または複数の代理人は、本条例によってなされたすべてのことおよび事柄として合法的にまたは手配することができる。
1934年の証券取引法の要求によると、本報告は、次の日に登録者として次の日に署名された | | | | | | | | |
| サイン | タイトル | 日取り |
| | |
/S/Alexander D.Macrae | CEO社長
(CEO)と役員 | 2023年2月22日 |
| Alexander D.Macrae,M.B.,Ch.B.,Ph.D. |
| | |
| /S/PRATHYUSHA DURAIBABU | 上級副総裁と首席財務官(首席財務会計官) | 2023年2月22日 |
| パトスシャ·デュレバブ |
| | |
/S/H.スチュアート·パーカー | 取締役と取締役会長 | 2023年2月22日 |
| スチュアート·パーカー |
| | |
/S/コットニービール | 役員.取締役 | 2023年2月22日 |
| コットニー·ビルス博士 |
| | |
| /S/ロバート·F·ケリー | 役員.取締役 | 2023年2月22日 |
| ロバート·F·ケリー |
| | |
/S/Kenneth J.Hillan | 役員.取締役 | 2023年2月22日 |
| ケネス·J·ヒランM.B,Ch.B. |
| | |
/ Sマーガレット·A·ホーン | 役員.取締役 | 2023年2月22日 |
| マーガレット·ホーンJ.D |
| | |
/S/John H.Markels | 役員.取締役 | 2023年2月22日 |
| ジョン·H·マルクス博士です |
| | |
/S/ジェームズ·R·マイルズ | 役員.取締役 | 2023年2月22日 |
| ジェームズ·R·マイエス |
| | |
/ S/カレン·L·スミス | 役員.取締役 | 2023年2月22日 |
| Karen L.Smith,M.D.,Ph.D.,M.B.A.,L.L. |