アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15(D)条に基づいて提出された |
手数料書類番号
(登録者の正確な氏名はその定款に記載)
(明またはその他の司法管轄権 会社や組織) |
(税務署の雇用主 識別番号) |
適用されない |
|
(主にオフィスアドレスを実行) |
(郵便番号) |
登録者の電話番号、市外局番を含む:+
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
同法第12条(G)に基づいて登録された証券:ありません
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください
登録者が当該法第13条又は第15条(D)に従って報告書を提出する必要がないか否かを、再選択マークで示すはい、そうです ☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告を提出したかどうか、および(2)このような提出要求を過去90日以内に遵守してきたかどうかを、再選択マークで示す
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
|
☒ |
|
ファイルマネージャを加速する |
|
☐ |
|
|
|
|
|
|||
非加速ファイルサーバ |
|
☐ |
|
規模の小さい報告会社 |
|
|
|
|
|
|
|
|
|
新興成長型会社 |
|
|
|
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
証券が同法第12条(B)に基づいて登録されている場合は,登録者の財務諸表が以前に発表された財務諸表の誤り訂正を反映しているか否かを示すチェックマークを適用する
これらのエラーのより真ん中に登録者の任意のエンタルピーCER幹部が相関回復期間内に§240.10 D−1(B)に基づいて受信したインセンティブベースの補償に基づいて回復分析を行う必要があるかどうかを再選択マークで示す
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)はい、そうです ☐ 違います。
登録者の非関連会社が保有する普通株の総時価は約#ドルである
2023年2月16日現在、登録者が発行した普通株式数は
引用で編入された書類
登録者最終委託書のうち2023年株主総会に関する部分は,登録者が登録者の財政年度終了後120日以内に第14 A条に基づいて証券取引委員会に提出しようとしている2022年12月31日この報告書の第3の部分は、参照によって組み込まれる。
カタログ表
|
|
|
|
ページ |
第1部 |
|
|
|
|
第1項。 |
|
業務.業務 |
|
1 |
第1 A項。 |
|
リスク要因 |
|
53 |
項目1 B。 |
|
未解決従業員意見 |
|
102 |
第二項です。 |
|
属性 |
|
102 |
第三項です。 |
|
法律訴訟 |
|
102 |
第四項です。 |
|
炭鉱安全情報開示 |
|
102 |
|
|
|
|
|
第II部 |
|
|
|
|
五番目です。 |
|
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
|
103 |
第六項です。 |
|
保留されている |
|
106 |
第七項。 |
|
経営陣の財務状況と経営成果の検討と分析 |
|
106 |
第七A項。 |
|
市場リスクの定量的·定性的開示について |
|
119 |
第八項です。 |
|
財務諸表と補足データ |
|
119 |
第九項です。 |
|
会計と財務情報開示の変更と相違 |
|
119 |
第9条。 |
|
制御とプログラム |
|
120 |
プロジェクト9 B。 |
|
その他の情報 |
|
122 |
プロジェクト9 Cです。 |
|
検査妨害に関する外国司法管区の開示 |
|
122 |
|
|
|
|
|
第三部 |
|
|
|
|
第10項。 |
|
役員·幹部と会社の管理 |
|
123 |
第十一項。 |
|
役員報酬 |
|
123 |
第十二項。 |
|
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
|
123 |
十三項。 |
|
特定の関係や関連取引、取締役の独立性 |
|
123 |
14項です。 |
|
チーフ会計士費用とサービス |
|
123 |
|
|
|
|
|
第4部 |
|
|
|
|
第十五項。 |
|
展示品と財務諸表の付表 |
|
124 |
第十六項。 |
|
表格10-Kの概要 |
|
128 |
i
リスク要因の概要
私たちの業務は多くのリスクと不確実性の影響を受けており、私たちの業務に投資決定を下す前に、これらのリスクと不確実性を認識すべきです。これらのリスクは,本年度報告の10−K表における“リスク要因”の部分でより包括的に検討されている。これらのリスクは以下のリスクを含むが、これらに限定されない
II
本Form 10−K年度報告では,文意が別に指摘されているほか,“会社”,“CRISPR”,“CRISPR Treateutics”,“我々”,“我々”,“Our”はいずれもCRISPR治療株式会社とその合併子会社を指し,“我々の取締役会”はCRISPR治療株式会社の取締役会を指す。
“CRISPR治療学”®“標準文字マークと設計マーク、”コバルトTM“”CRISPRXTM“”CRISPR TXTM,” “CTX001TM,” “CTX110®,” “CTX112TM,” “CTX120TM,” “CTX121TM,” “CTX130TM,” “CTX131TM,” “CTX310TM,” “CTX320TM,” “CTX330TM,” “VCTX 210TM” and “VCTX211TM,” CRISPR治療株式会社の商標と登録商標です。その他すべての商標及び登録商標本年度報告に含まれる10-K表は,それぞれの所有者の財産である. 便宜上、本年度報告でForm 10−K形式で参照される商標、サービスマーク、および商号は、非使用であってもよい®あるいは…このようなどんな漏れもこのような権利を放棄することを意味しない。
前向き陳述と業界データに関する特別な説明
このForm 10−K年次報告書には,重大なリスクと不確実性に関する“前向き陳述”が含まれている。本年度報告における10-K表に含まれるすべての陳述は,歴史的事実に関する陳述を除いて前向き陳述である.これらの表現は、一般に、すべての前向き表現がこれらの識別可能な語を含むわけではないが、“予想”、“信じ”、“継続”、“可能”、“推定”、“予想”、“意図”、“可能”、“計画”、“予測”、“プロジェクト”、“潜在”、“将”、“将”またはこれらの語の否定または複数または同様の表現または変形を使用することによって識別される。本年度報告におけるForm 10−Kに関する前向きな陳述は、以下のように含まれるが、これらに限定されない
本年度報告の10-K表の任意の前向き陳述は、既知および未知のリスク、不確実性および仮定に関連する未来のイベントまたは私たちの未来の財務パフォーマンスに対する私たちの現在の見方を反映しており、これらのリスク、不確実性および仮定は、私たちの実際の結果およびいくつかのイベントの時間を、前向き陳述に明示または示唆された未来の結果と大きく異なる可能性がある。このような差異をもたらすか、または促進する可能性のある要因には、本明細書で決定された要因と、本10−K年度報告第I項第1 A項の“リスク要因”の節で議論された要因とが含まれるが、これらに限定されない。あなたは未来の事件の予測として前向きな陳述に依存してはいけない。このような展望的な陳述は本報告書までの日だけを説明する。私たちの展望的な陳述は、私たちが行う可能性がある任意の未来の買収、合併、処置、合弁、または投資の潜在的な影響を反映しない。
このForm 10-K年次報告書と、このForm 10-K年次報告書で証拠品として提出された文書を完全に読み、私たちの将来の実績、業績、または業績が私たちの予想と大きく異なる可能性があることを理解しなければなりません。法律には別に規定がある以外に、このような陳述が発表された日以降の事件や状況を反映するために、いかなる前向きな陳述も更新する義務はありません。
三、三、
このForm 10-K年度報告書には、我々自身の内部推定および研究、および業界および一般出版物および研究、調査、および第三者による研究から得られたデータから得られた統計および他の業界および市場データが含まれる。業界出版物、研究、および調査は、そのような情報の正確性または完全性を保証しないにもかかわらず、信頼できると考えられるソースから得られる一般的な声明である。私たちはこれらの研究と出版物のそれぞれが信頼できると信じているが、私たちはまだ第三者ソースからの市場と業界データを独立して確認していない。当社内の研究は信頼でき、市場定義は適切であると信じていますが、このような研究やこれらの定義はいかなる独立したメッセージ源の確認も得られていません。
四
パー?パーT I
プロジェクト1.BU無邪気ですね。
商売人
概要
我々は有力な遺伝子編集会社であり,CRISPR/Cas 9による療法の開発に専念している。CRISPR/CAS 9代表C光沢のあるR特に…IスペースSホルトP名無しさんREpeats(CRISPR)/CRISPR-a連関p回転蛋白質9(Cas 9)は革命的な遺伝子編集技術であり、ゲノムDNA特定配列を正確に変化させる過程である。我々の目標は,この技術を用いて遺伝子定義の疾患を治療し,先進的な細胞療法を設計するために,遺伝子を妨害,削除,是正,挿入することである。著者らの科学専門知識に加え、著者らの遺伝子編集方法は、希とよく見られる疾患を有する患者に新しい高効率と潜在的な治癒方法を提供する可能性があり、現在の生物製薬方法はこれらの患者に対する成功は限られていると信じている。
CRISPR/CAS 9を用いて遺伝子編集を行うことは、細菌の中で自然に発生するウイルス防御機構に由来し、著者らの科学創始者の一人であるEmmanuelle Charpentier博士が共同で発明したものであり、ドイツのベルリンマルクス·プランク病原体科学単位の代理と創始者の一人である。Charpentier博士と彼女の協力者は1つの仕事を発表し、CRISPR/Cas 9の重要な構成部分Cas 9エンドヌクレアーゼが特定の位置で二本鎖DNAを切断する機序をプログラムできることを明らかにした。チャペンティエ博士と彼女の協力者、カリフォルニア大学バークレー校のジェニファー·デュドナ博士は、彼らの先駆的な仕事で2020年のノーベル化学賞を共有した。我々はすでにCharpentier博士からCRISPR/CAS 9と関連技術を含む知的財産権の権利を獲得し,我々自身の研究と追加の許可内で努力することにより,我々の知的財産権を強化し続け,CRISPR/CAS 9に基づく療法開発における我々のリードをさらに強化している。
著者らは四つの核心特許経営領域で広範な疾病領域の治療方案の組み合わせを創立した:ヘモグロビン病、免疫腫瘍学、再生医学と体内にある近づいてきました。我々の最先端の計画は,遺伝子定義疾患輸血依存型ベータ地中海貧血(TDT)と重篤な鎌状細胞病(SCD)であり,両疾患の医療ニーズは満たされていない。我々はまた、血液病および固形腫瘍癌を治療するための同種異遺伝子キメラ抗原受容体T細胞(CAR T)、および1型糖尿病(T 1 D)の治療のための研究性、同種異体、遺伝子編集、免疫回避および幹細胞由来療法を含むいくつかの遺伝子編集の同種細胞治療計画を進めている。さらに私たちは複数の計画を進めています体内にある編集方法は,最初に心血管疾患の治療と予防に用いられた。
我々の製品開発と協力戦略は,CRISPR/CAS 9プラットフォームの潜在力を十分に発掘するとともに,我々の候補製品開発に成功する可能性を最大限に高めることを目的としている。私たちの最先端の候補製品については離体するCRISPR/Cas 9を投与する前に,CRISPR/Cas 9を用いてヒト外細胞を編集する方法である。対照的に私たちの体内にある編集プログラムにより,CRISPR/Cas 9による治療レジメンをヒト内の標的細胞に直接提供した。
ヘモグロビン病
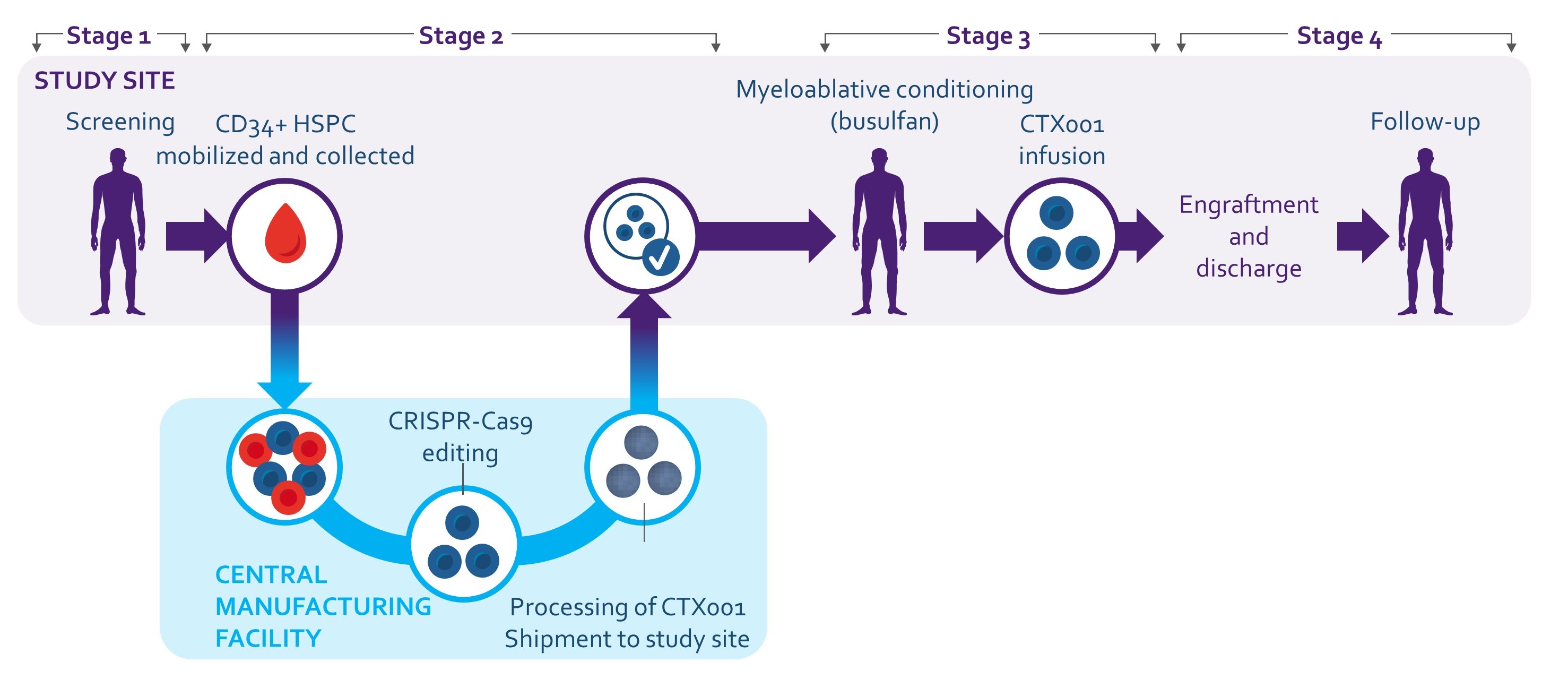
私たちの主要な候補製品、exagamglobal Autotemcel、あるいはexa-celは、以前はCTX 001と呼ばれ、調査的、自己的であった離体するCRISPR遺伝子編集の造血幹細胞療法は,TDTや重篤なSCDを有する患者の評価を行っており,患者の造血幹細胞は工学的である離体する赤血球に高レベルの胎児ヘモグロビン(HBF;ヘモグロビンF)が産生される。HBFは出生時に自然に存在する酸素運搬ヘモグロビンの一形式であり,成人形式のヘモグロビンに置換される。手術によりHbFを向上させることにより,TDT患者の輸血ニーズやSCD患者の苦痛や虚弱な血管閉塞発症を解消することが可能である。Exacelは,我々とVertex製薬会社やVertexとの共同開発と商業化プロトコルに基づいて開発された.
我々とVertexは,12歳から35歳までのTDT(GRAPH−111)やSCD(SCD−121)患者に対する単剤Exexcelの安全性と有効性を評価するための1/2/3期開放臨床試験におけるExaccel研究を2つ行っている。登る-111と登る-121の登録が完了しました。我々とVertexはまた,TDT(SCD−141)およびSCD(SCD−151)を有するTDTおよびSCD患者のための2つの追加的な3期オープン臨床試験を開始した。Exexelを受けた患者は、Exexexeの安全性と有効性を評価するために、長期的な開放ラベルフォローアップ試験に参加することを要求される。GRAPH−131は,刺激輸液後に参加者に15年間にわたる追跡調査を行うように設計されている。2022年第2四半期と第4四半期に、ヨーロッパ血液学協会大会とアメリカ血液病学会年会で、著者らはそれぞれ44名のTDT患者と31名のSCD患者がExaccel治療を受けた最新の臨床データを公表した。臨床資料の他の資料については、“を参照されたい”鉛ヘモグロビン病の候補品であるExacel.”
1
Exacelはすでにアメリカ食品と薬物管理局(FDA)の多くの監督管理称号、特に再生医学高級療法(RMAT)、快速通路、孤児薬物と稀な小児科疾患の治療称号を獲得した。Exa−celは欧州委員会の孤児薬物指定や,欧州医薬品局(EMA)の優先薬やPrime指定を受け,TDTやSCDの治療にも用いられている。規制指定の影響に関するその他の情報は、“をご覧ください”企業--政府法規 .”
2022年12月、私たちとVertexはそれぞれEUとイギリスのEMAと薬品と保健製品監督管理機関(MHRA)に輸出規制文書を提出し、EMAとMHRAはそれぞれマーケティング許可申請(MAA)を検証した。また,我々とVertexは2022年11月に米国で我々の生物製品許可申請(BLA)のスクロール提出を開始し,2023年第1四半期末に提出を完了する予定である。
最後に、私たちは既存の経験に基づいて、的確なコンディショニング療法と体内にある造血幹細胞を編集し,いずれもわれわれの療法から利益を得ることができる患者数を拡大することができる。
免疫腫瘍学
CRISPR/CAS 9は次世代CAR T細胞療法を創造する潜在力があると信じており,現在の自己療法と比較してより良い製品配置を有し,より広範な患者群の使用を可能にしている。自自離体する我々の先行計画により得られた遺伝子編集能力により,CD 19とCD 70に対する同種CAR T計画を含むいくつかの免疫腫瘍細胞治療計画を進めている。
CD 19特許経営権
CTX 110は著者らの有力な免疫腫瘍学候補製品であり、分化クラスター19或いはCD 19の健康ドナー遺伝子に対して編集した同種異体CAR研究療法である。我々は、少なくとも2つの以前の治療を受けた再発または難治性CD 19陽性B細胞悪性腫瘍の成人患者におけるCTX 110の安全性および有効性を評価するために、我々のCarbon臨床試験においてCTX 110を研究している。CTX 110は、FDAによってRMAT称号を付与されている。
第一期Carbon臨床試験は2つの部分に分けて行われる--A部分とB部分。第一段階Carbon臨床試験のA部分、即ち第一段階A部分で、患者は標準リンパ浄化方案の後に漸増用量レベルで単剤CTX 110を注入し、そして臨床治療効果に基づいて再投与を選択する。第1段階Carbon臨床試験のB部分、または第1段階B部分において、患者は、標準リンパ浄化後に用量レベル(DL)が4であるCTX 110と、初期用量後4~8週間以内に同じ用量レベルで用量を強固にするCTX 110とを、臨床的利益を示す患者に投与する。
2022年第4四半期に、我々はCTX 110治療を受けた32人の患者の第1段階A部分の最新の臨床データを公表し、これらのデータはCTX 110が長期持続的な完全緩和(CRS)を達成する潜在力があることを示し、大量の前治療を受けた患者の中で肯定的な差異安全性を有し、第1段階B部分の新興データを記述し、これらのデータは鼓舞的な治療効果概況を示し、強化用量を使用することによって治療効果を高める可能性がある。臨床資料の他の資料については、“を参照されたい”私たちがリードしている免疫腫瘍学の候補品であるCTX 110“私たちの第一段階Carbon臨床試験と監督機関との討論に現れたこれらのデータに基づいて、私たちは合併用量を含む登録可能な単一アーム、多中心、開放ラベルの第二段階臨床試験を含むCarbonの範囲を拡大した。私たちはこの重要な腕に薬を投与し始めました
CTX 110と同時に、CD 19の次世代研究、同種CAR T候補製品であるCTX 112を進めている。CTX 112は、CTX 110以外の2つの追加編集を含み、我々のCRISPR/Cas 9プラットフォームを使用して、インクリメンタル編集を次世代製品に統合することによって、継続的に革新することを可能にする。これらの編集はRegnase-1と形質転換増殖因子-β受容体2(TGFBR 2)をコードする遺伝子に対して、CAR Tの効力を増強し、CAR Tの消費を減少させることを目的としている。2022年第4四半期、CTX 112の研究新薬またはIND申請はFDAの承認を得た。
2
CD 70特許経営権
CTX 130は分化クラスター70或いはCD 70の健康ドナー遺伝子に対して編集した同種異体CAR研究療法であり、CD 70は各種の固形腫瘍と血液系悪性腫瘍に発現する抗原である。CTX 130は、成人患者におけるいくつかの用量レベルのCTX 130の安全性および有効性を評価するために、2つの独立した第1段階、単一アーム、マルチセンター、開放ラベル臨床試験を行っている。コバルト−ライム試験は、再発または難治性TまたはB細胞悪性腫瘍の治療におけるCTX 130の安全性と有効性を評価している。コバルト−腎癌試験は,再発または難治性透明細胞腎癌に対するCTX 130治療の安全性と有効性を評価している。CTX 130は、FDAのT細胞リンパ腫治療の孤児薬物指定および皮膚T細胞リンパ腫(CTCL)亜型真菌様肉芽腫およびSézary症候群(MF/SS)のRMAT指定を取得した。2022年第2四半期、ヨーロッパ血液学協会大会で、我々はCTX 130治療を受けたT細胞リンパ腫患者18人に対して少なくとも28日間のフォローアップを受けたコバルト-LYM試験の予備臨床データを公表した。また,2022年第4四半期に癌免疫治療学会年次総会において,14名の患者のコバルト−腎癌試験の予備臨床データを公表した。臨床資料の他の資料については、“を参照されたい”CTX 130です。”
CTX 130と同時に、著者らはCTX 131を進めており、これはCD 70に対する次世代研究、同種異体CAR T候補製品であり、固形腫瘍とある血液系悪性腫瘍の潜在的治療に応用できる。CTX 131は、CTX 130以外の2つの追加編集を含む。これらの編集は、CTX 112で使用されているのと同様に、Regnase-1およびTGFBR 2をコードする遺伝子について、CAR T効力を増強し、CAR T消費を減少させることを目的としており、2023年第1四半期に、CTX 131のINDがFDAによって承認される。
他の候補者
私たちはいくつかの追加的なCar T製品候補製品を発売している。2つのこのような候補に対して、我々は、新しい標的分化クラスター83またはCD 83およびGlypcan-3またはGPC 3を臨床的に検証するために、先行する癌センターと革新的なパートナーシップモデルを開発した。モフェット癌センターと協力して,急性骨髄性白血病や他の腫瘍学や自己免疫適応の治療が可能なCD 83に対する自己CAR T候補薬を発売している。ロスウェル公園総合癌センターと協力して,肝細胞癌に発現するGPC 3の遺伝子編集に対する自己CAR T候補薬を開発している。この2つの場合、著者らの学術パートナーはいずれも製造と初の人体臨床試験を行う。この構造は私たちがこのような目標の安全性と活動状況を迅速に評価できるようにするだろう。臨床結果から,これらの自己計画を国内で進めたり,同種バージョンを開発したりして,機会をさらに拡大することを選択することができる。また,CAR Tに加えて,Nkarta,Inc.やNkartaと協力関係を構築し,我々の遺伝子編集技術と細胞治療専門知識をNkartaがリードするナチュラルキラー(NK)細胞発現,開発,製造能力と組み合わせた。協力の一部として,我々はNkartaとCD 70のドナー由来の遺伝子編集されたCAR−NK細胞候補製品を共同開発し,共同商業化している。
再生医学
再生医学は、幹細胞を用いて疾病、損傷或いは年齢によって失われた組織或いは器官機能を修復或いは置換しても、稀とよく見られる疾病の治療において巨大な潜在力を持っている。私たちが作ったのは離体する遺伝子編集の専門技術として,CRISPR/Cas 9が編集した同種異遺伝子幹細胞由来療法を用いて,免疫逃避,細胞機能改善,直接細胞運命の実現に重点を置いた分野での取り組みを拡大した。我々のこの分野での最初の主な努力は糖尿病において,我々とViaCyte,Inc.は糖尿病治療のための遺伝子編集幹細胞療法を発見·開発し,それを商業化する戦略協力の一部として一連の計画を進めている。ViaCyteは2022年第3四半期にVertexに買収された。ViaCyteの幹細胞能力と我々の遺伝子編集能力の結合は、ベータ細胞代替製品を候補製品にする可能性があり、この製品は患者に持続的なメリットをもたらす可能性があり、同時に免疫抑制を行う必要がないと信じている。
私たちは、私たちのCRISPR/Cas 9プラットフォームを利用して、利益を増加させるためのインクリメンタル編集を含む複数の候補製品を推進する多段階の製品戦略を持っている。著者らの最初の候補製品VCTX 210は、T 1 Dを治療するための研究、異遺伝子、遺伝子編集、免疫回避、幹細胞由来製品であり、この製品は私たちの遺伝子編集技術をViaCyteの独自幹細胞能力に応用することによって開発された。VCTX 210は、免疫逃避および細胞健康を促進することを目的とした遺伝子編集を有する。著者らとViaCyteはT 1 D患者におけるVCTX 210の安全性、耐性と免疫逃避を評価し、この臨床試験の後続段階にあるVCTX 210を研究している第1段階の臨床試験である。我々の次世代候補製品VCTX 211は、細胞適応性をさらに増強することを意図した追加の遺伝子編集を含むT 1 Dを治療するための研究、同種遺伝子編集、幹細胞由来製品候補製品である。2022年第4四半期,VCTX 211の臨床試験申請はカナダ衛生部の許可を得ており,1/2期臨床試験が行われている。
3
生活の中で
私たちのを除いて離体する計画はいくつか進められています体内にある遺伝子編集プログラムです我々の体内にある遺伝子編集戦略の重点は遺伝子破壊と全遺伝子是正である--この2つの技術は大多数の最も一般的な深刻な単遺伝子疾患を解決するために必要である。私たちはすでに先進的なプラットフォームを構築しました体内にある遺伝子破壊は肝臓から始まります著者らはこのプラットフォームを利用して、心血管疾患或いは心血管疾患から始まる一連の稀とよく見られる疾病に関連するプロジェクトの組み合わせを推進する予定である。我々の首席調査員は体内にあるCTX 310とCTX 320プログラムはそれぞれアンギオゲニン関連蛋白3(Angptl 3)とリポ蛋白質(A)(Lp(A))に対して、これはCVDの2つの有効な標的である。遺伝子編集は自然ヒト遺伝子変異体が証明した利点を単剤形式で要約したため、心血管疾患の治療パターンを変化させる可能性がある。さらに,肝臓で脂質ナノ粒子(LNP)と腺関連ウイルスベクター(AAV)を使用することから,AAVを含まず,相同配向修復(HDR)に依存しない方法に発展した拡張可能な完全遺伝子補正プラットフォームの開発を継続した。
CRISPR-X
著者らの現在のプロジェクトの組み合わせは重大な進展を得たが、著者らは引き続き革新して、CRISPR遺伝子編集のすべての潜在力を放出し、そして変革性治療の潜在力をより多くの患者にもたらす必要があることを認識した。2022年、私たちはCRISPR-Xという新しい早期研究チームを設立し、次世代編集モデルを開発するために革新的な研究に専念した。CRISPR-Xは、HDRまたはDNAのウイルス伝達を必要とせずに全遺伝子補正および挿入を実現する技術、例えば、全RNA遺伝子補正、DNAの非ウイルス伝達、および新しい遺伝子挿入技術に専念する。
仲間関係
CRISPR/CAS 9の多くの潜在的治療応用を考慮して戦略的協力を行い,特定の技術や/あるいは疾患領域の専門知識を獲得することにより,追求できる適応を広げ,プロジェクトの開発を加速した。我々は広範なパートナー関係を保ち,特定の疾患領域で遺伝子編集に基づく療法を開発している。その中のいくつかのパートナーシップに関するより多くの情報は、“を参照してください”業務-戦略的パートナーシップと連携.”
頂点です著者らは2015年にVertexと初歩的な協力プロトコルを確立し、TDT、SCD、嚢胞性線維化と他の適応の選択に集中した。2017年12月、Vertexと共同開発と商業化協定を締結し、この合意に基づき、他の事項に加えて、TDTとSCDのExacelを共同で開発し、商業化する準備をしています。2021年4月、我々はVertexと既存の共同開発と商業化プロトコルを改訂し、再記述しました。このプロトコルによると、他の事項を除いて、Vertexとの共同開発と準備を継続してTDTとSCDのexaxを商業化していきます。2019年6月にVertexと戦略協力と許可協定を締結し,Duchenne筋ジストロフィー(DMD)と強直性筋ジストロフィー1型(DM 1)を治療する製品を開発し商業化した。
ViaCyteそれは.2018年9月、我々はViaCyteと研究と協力協定を締結し、糖尿病を治療するための遺伝子編集異遺伝子幹細胞療法の発見、開発、商業化を求めた。2021年7月、私たちはViaCyteやViaCyte JDCAと共同開発と商業化協定を締結した。ViaCyte JDCAへの加入については,我々はViaCyteの既存の研究連携プロトコルとその条項によって満了した.ViaCyte JDCAによると、私たちとViaCyteは、世界各地で1型糖尿病、2型糖尿病およびインスリン依存型/インスリンを必要とする糖尿病、またはViaCyte協力領域の治療のための候補製品および共有製品を共同開発および商業化している。ViaCyte JDCAは協力と計画管理、候補製品の臨床活動とプロトコル下の共有製品及びViaCyte協力領域の各方面の持続的な研究などに関連する条項を含む。双方に別の約束がない限り、一方で発生した研究費はその側が独自に負担する。共有製品の初商業販売日から発生した研究と連携プロトコルで最初に規定された計画費用は,60%を我々に,40%をViaCyteに割り当てた.共有製品が初めて商業販売された後、このような計画費用は私たちとViaCyteの間で平均的に分担される。共有された製品の収入は私たちとViaCyteによって分けられるだろう。Vertexは2022年第3四半期、ViaCyteとViaCyteの協力分野の権利を買収したことを発表した。
バイエル。私たちは2019年第4四半期にバイエル医療保健有限責任会社(Bayer Healthcare LLC、バイエルと略称する)とオプション合意に達し、この合意によると、バイエルは特定の時間帯内に私たちがある自己免疫疾患、眼疾患または血友病Aの診断、治療または予防のために提供した2種類の製品を共同開発、商業化する権利があり、あるいは場合によってはこのようなオプション製品を独占的に許可する権利がある。
他のパートナー関係それは.私たちは造血幹細胞、免疫腫瘍学、再生医学を支援し、補充するための追加の協力と許可協定を締結しました体内にあるNkartaと合意し、2種類のドナー由来の遺伝子編集されたCAR-NK細胞候補製品とNKとT細胞を結合した製品を共同開発と共同商業化する計画とプラットフォーム;Capsida BioTreateutics,Inc.が開発される体内にある工学AAVベクターを利用して提供した遺伝子編集療法は筋萎縮性側索硬化症とFriedreich‘s運動失調の治療に応用されている;モフェット癌センターとロスウェル公園総合癌センターは新しい標的に対して自己CAR T計画を推進する;MaxCyte,Inc離体する配達する
4
ヘモグロビン病と免疫腫瘍学プロジェクトのためのCureVac AGはmRNA構造の最適化といくつかの製造に取り組んでいます体内にあるKSQ Treateutics,Inc.とわれわれの同種異体免疫腫瘍学計画の知的財産権について協議した。
私たちの使命は深刻なヒト疾患のための変革的な遺伝子ベースの薬を作ることです。我々の経験豊富なチームは,我々の科学専門知識,製品開発戦略,パートナー関係,知的財産権に加えて,CRISPRに基づく療法開発のリーダーとなると信じている。
遺伝子編集背景
何千もの病気がDNA配列異常によって引き起こされている。伝統的な小分子と生物療法はその中の多くの疾病の治療における成功は限られており、それらは根本的な遺伝原因を解決できなかったためである。RNA療法やウイルス遺伝子療法のような新しい方法よりも、疾患に関連する遺伝子に対してより直接的に対応しているが、それぞれの方法には明らかな限界がある。RNAに基づく療法、例えばメッセンジャーリボ核酸と小干渉リボ核酸は、繰り返し投与と関連毒性の挑戦に直面している。AAVのような非統合ウイルス遺伝子治療プラットフォームは、永久的にゲノムを変化させず、産生された免疫反応のため、再投与時の治療効果が限られているため、限られた持続性を有する可能性がある。慢性ウイルスのようなウイルス遺伝子治療プラットフォームを統合し、恒久的にゲノムを変更するが、このようにすることはランダムであり、これは不良突然変異の可能性を招く。また,細胞は形質導入された遺伝子が外来であることを認識し,それらの発現を減少させることで反応し,その有効性を制限している可能性がある。そのため、ヒトゲノムマップを作成して以来、著者らは遺伝病に対する理解が増加したが、著者らはこれらの疾病を有効に治療する能力はずっと限られている。
遺伝子編集は次世代療法を実現し,正確な遺伝子修飾により多くの遺伝病に潜在的な根治療法を提供する可能性があると信じている。また,DNA配列を変化させる能力は,遺伝子定義の疾患の治療のほかにも応用されている。CRISPR/Cas 9遺伝子編集は細胞による療法の工程を可能にし,より有効,安全であり,より広範な患者群に用いることができる。細胞療法はすでにある疾病に重大な影響を与え始め、遺伝子編集は腫瘍学と糖尿病を含む異なる疾病領域の進展を加速することに役立つかもしれない。
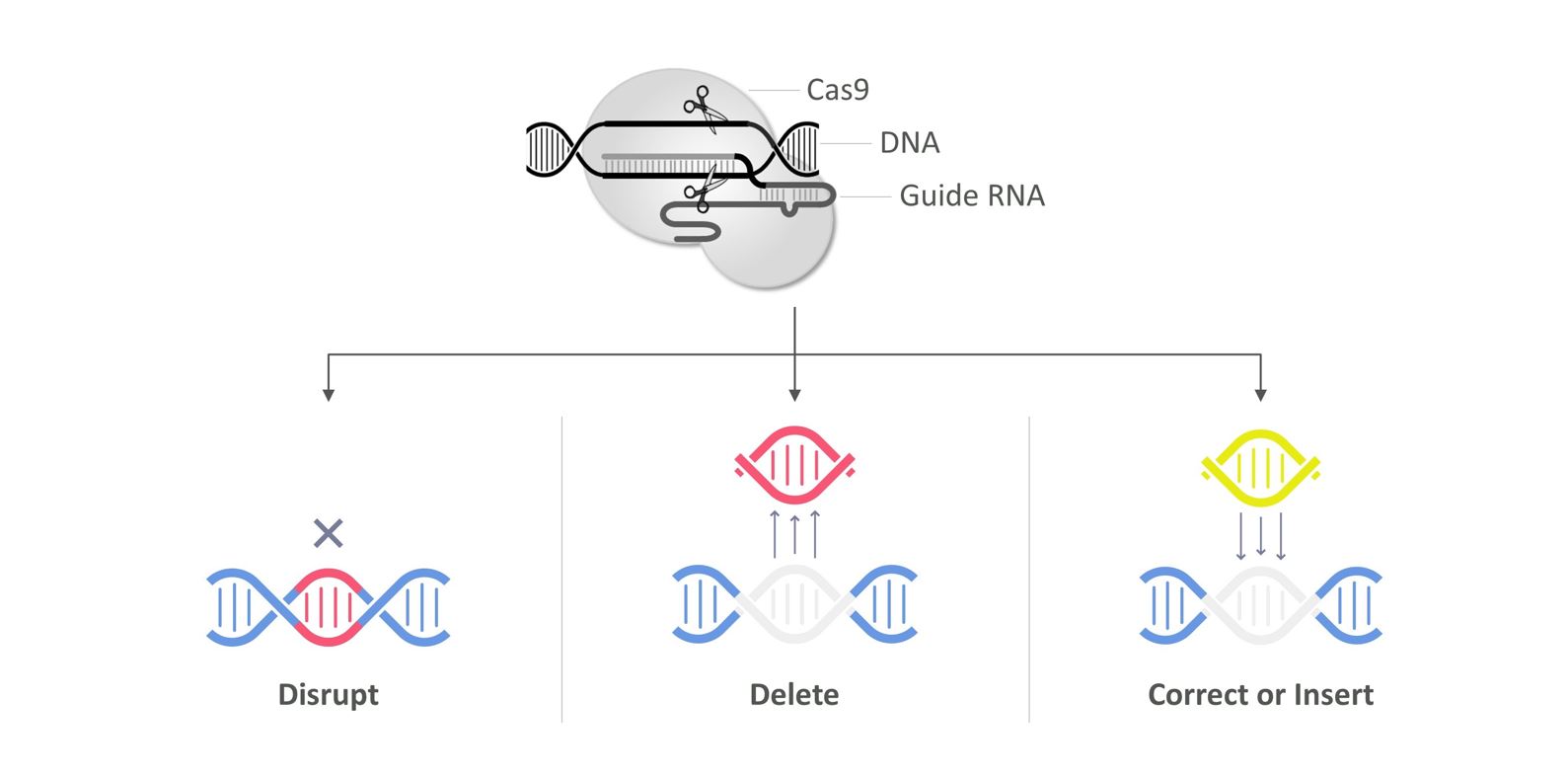
遺伝子編集のプロセスは、酵素を用いて特定の位置でDNAを切断し、細胞ゲノム中のDNA配列を正確に変化させることを含む。切断後、自然な細胞プロセスはDNAを修復して不要な配列を沈黙または修正し、潜在的にそれらの負の影響を逆転させる。重要なのは,ゲノム自体がこの過程で修正されるため,この変化は患者に恒久的であることである。初期の遺伝子編集技術、例えば亜鉛指ヌクレアーゼ(ZFN)、転写活性化物様効果ヌクレアーゼ(TALEN)とマクロヌクレアーゼは、工学蛋白質-DNA相互作用に依存して編集位置を制御する。これらのシステムは遺伝子編集の潜在力を示す重要な第一歩であるが、工学タンパク質-DNA相互作用の複雑さのため、それらの開発は実践的に挑戦的である。対照的に,CRISPR/Cas 9はRNA−DNA相互作用によって指導されており,この相互作用の方が予測可能であり,工学や応用も直接的である。したがって、私たちは様々な技術を適切に採用できるように、私たちのCRISPRプラットフォームを拡大することに投資し続けています。
CRISPR/CAS 9技術
CRISPR/Cas 9は自然発生の防御機序に進化し、細菌をウイルス感染から保護する。チャペンティエ博士と彼女の協力者はこの機序を解明し、この機序に適応と簡略化する方法を開発し、遺伝子編集に用いた。この先駆的な仕事を表彰するために,チャペンティエ博士は彼女の協力者でカリフォルニア大学バークレー校のジェニファー·デュドナ博士とともに2020年のノーベル化学賞を受賞した。彼らが説明したCRISPR/CAS 9技術は3つの基本コンポーネントから構成されているCRISPR-AS関連タンパク質9、またはCas 9、CRISPR RNA、またはcrRNA、ならびにトランス活性化CRISPR RNA、またはtrrRNA。Cas 9はこの2つのRNA分子に結合しており,選択された二本鎖DNAを特定の切断および編集が可能な“分子はさみ”として記述されている。
チャペンティエ博士と彼女の協力者はさらに遺伝子編集のためのシステムを簡略化し、crRNAとtrrRNAを誘導RNAと呼ばれる単一RNA分子に組み合わせた。RNAのCas 9への結合を誘導し,Watson−Crick塩基マッチング規則に基づいてプログラムし,Cas 9酵素を特定のDNA配列に配向させることができる。CRISPR/Cas 9技術は標的遺伝子の特定の位置でDNAを切断するために使用可能であり,遺伝子編集に基づく療法の開発に強力なツールを提供している。
DNAが切断されると,細胞は自然に発生するDNA修復機構を用いて切断された末端を再接続する。一度切断すると,非源末端結合と呼ばれる過程は塩基対の添加や欠失を招き,原始DNA配列を撹乱し,遺伝子不活化を招く可能性がある。異なる部位に対する2つの誘導RNAを用いることにより、より大きなDNA断片を削除することもできる。各部位で切断した後、非相同末端結合は、単離された末端を一緒に連結し、挿入配列を削除する。あるいは,CRISPR/Cas 9機構の近傍にDNAテンプレートを添加すれば,細胞は相同配向修復と呼ばれる過程で遺伝子を是正し,さらに新しい遺伝子を挿入することができる。
5
CRISPR/Cas 9遺伝子編集
CRISPR/Cas 9は多機能技術であり,遺伝子を干渉,削除,訂正あるいは挿入することができると考えられる。著者らはCRISPR/CAS 9システムの多機能性とモジュール化を利用して、特定の疾病応用のために個別コンポーネントを調整と迅速にカスタマイズする予定である。そのため、著者らはCRISPR/Cas 9は1種類の新しい治療方法の基礎を形成する可能性があり、稀とよく見られる疾病を治療する潜在力があると考えている。CRISPR/CASシステムの優勢を考慮して、複数の学術団体は基本編集と素数編集などのCRISPR/CAS 9に基づく新しい技術を開発した。このようなCRISPR/CASに基づく新しい技術はまだ初期段階にあるが、特定の応用領域では、CRISPR/CAS 9技術を含む既存の遺伝子編集技術よりも優勢である可能性がある。したがって、私たちは様々な技術を適切に採用できるように、私たちのCRISPRプラットフォームを拡大することに投資し続けています。
6
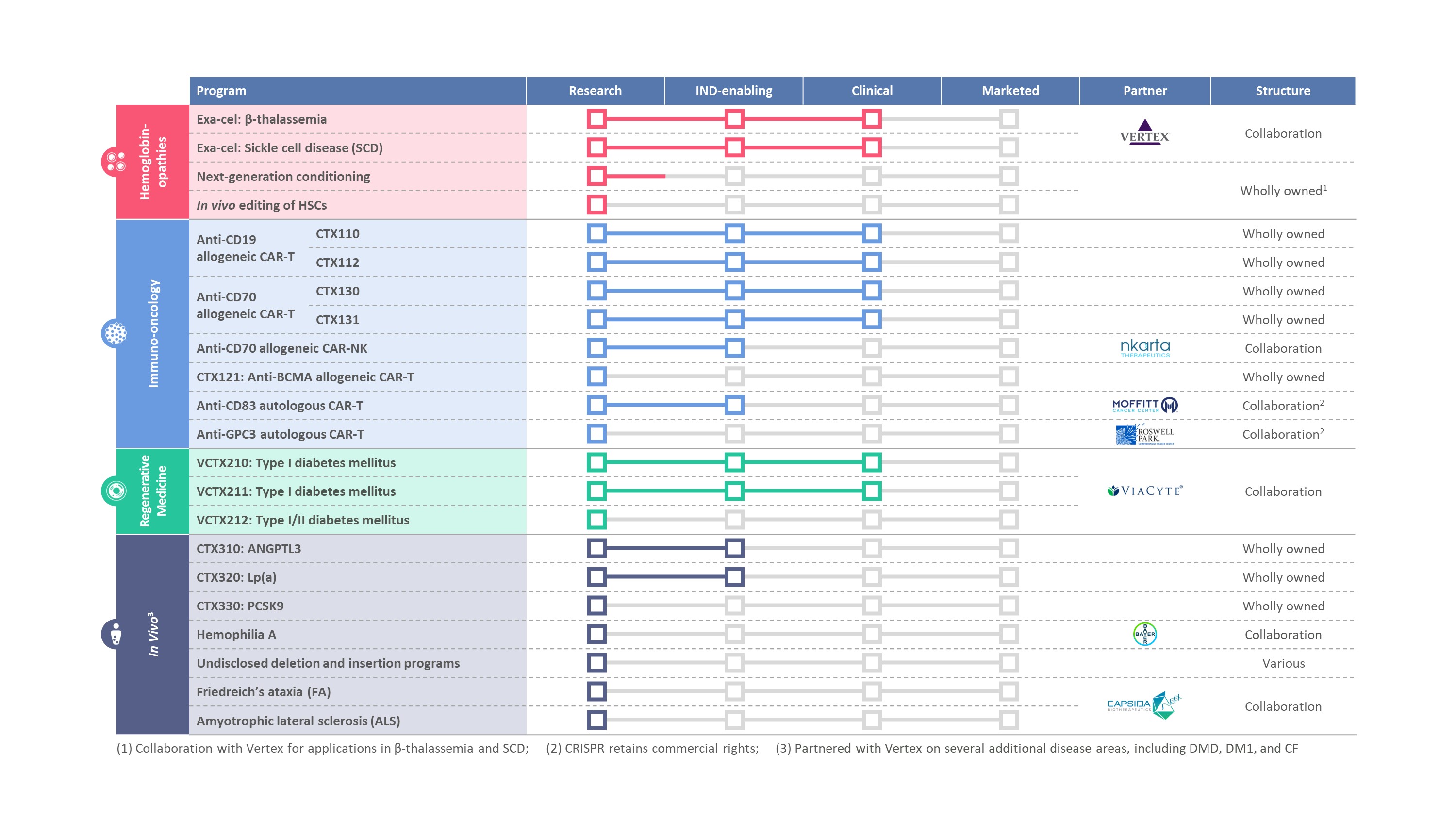
私たちのパイプは
次の表は、我々の製品開発ルートの状態をまとめています
ヘモグロビン病
私たちは主に離体する造血系に関連する疾患を治療する方法。造血系とは、骨髄、脾臓、リンパ節などの血液産生に関与する器官や組織系を指す。今日,我々が対象としている多くの造血系疾患は,同種造血幹細胞移植や同種造血幹細胞移植により治療されている。Allo−HSCTを行う際には,医師は患者の欠陥遺伝子を含む造血細胞を別の人から得られた正常遺伝子を含む細胞で置換する。不幸にも、すべての患者が適切なドナーを見つけることができるわけではない。Allo−HSCTを受けた患者は免疫抑制,移植拒絶反応や移植片対宿主病に関連する感染などの合併症の高いリスクに直面し,移植組織(移植片)中の免疫細胞はレシピエント(宿主)を“異体”と認識し,宿主の細胞を攻撃し始める。
Allo-HSCTとは異なり,患者から幹細胞を直接取得し,目的遺伝子を編集する方法である離体するこれらの同じ細胞を患者に再注入します私たちはこれを信じています離体する患者自身の細胞を用いた遺伝子編集方法はallo−HSCTよりも良い結果を提供する可能性がある。
私たちの主なプロジェクトであるヘモグロビン病は
ヘモグロビン病は1組の異なる遺伝性血液疾患であり、ヘモグロビンの合成或いは構造変化によって引き起こされる。我々とVertexのヘモグロビン疾患における先行プロジェクトは,単一の潜在的治療効果を有するCRISPR/Cas 9による療法を開発し,TDTとSCDを同時に治療することを目的としている。これらの疾患はβグロブリンをコードする遺伝子変異によるものである。ベータグロブリンはヘモグロビンの重要な構成部分であり、ヘモグロビンは赤血球中のタンパク質であり、酸素を輸送し、全身の二酸化炭素を除去することを担当する。これらの魅力的な前兆を魅力的にする要因は,(1)高度に満たされていない医療需要,(2)注目されている市場潜在力,(3)よく知られている遺伝学と(4)利用,などいくつかの要因がある離体する遺伝子破壊戦略です
7
β地中海貧血
概要
β地中海貧血はヘモグロビン生成減少に関与する血液疾患である。この疾患はβグロブリン発現不足を引き起こす突然変異によるものであり,これによる症状はヘモグロビン欠乏だけでなく,赤血球中のペアリングされていないαグロブリン蓄積にも関与している可能性がある。β地中海貧血に関連する症状の重症度は血球に存在する機能性βグロブリンのレベルに依存する。ペアリングしていないアルファグロブリン鎖は赤血球に有毒であり、赤血球寿命を短縮した。最も重篤な場合,重篤なベータ地中海貧血と記載されており,機能性ベータグロブリンは完全に欠損しているか,減少し,重篤な貧血をきたしている。これらの患者では,骨髄は赤血球の破壊と同期することができないため,定期輸血が必要である。慢性輸血は有効に症状を解決できるが、それらはよく鉄負荷が高すぎ、進行性心臓と肝臓不全を招き、最終的に早期死亡を招く。軽度β地中海貧血を有する患者は軽微な貧血を経験し,症状さえない可能性がある。推定によると、全世界の毎年のベータ地中海貧血の総発病率は60,000名の新生児であり、アメリカとEUの総罹患率は約16,000人と推定され、全世界で20万人以上が生きており、この疾病の治療を受けるために登録されている。
現在の治療法の限界は
ベータ地中海貧血の最もよく見られる治療方法は慢性輸血である。輸血に依存する患者は通常2~4週間に1回輸血を受け、長期輸血はよく体内鉄レベルの上昇を招き、これは比較的に短い時間内に器官損傷を招く可能性がある。患者は鉄錯化剤を投与されたり,血液中の鉄レベルを低下させたりすることが多く,彼ら自身の重大な毒性に関与している。発展途上国では,長期輸血が得られないため,多くの患者が小児早期に死亡している。また、ベータ地中海貧血を治療する疾患修正療法Reblozyl(luspatercept-AAMT)は2019年にFDAの承認を得た。
ベータ-地中海貧血の潜在的治療方法は同種造血幹細胞移植であるが、その関連する発病率と死亡率及び一致と自発的なドナーが不足していることを考慮すると、このような手術を選択する患者は少ない。さらに、EMAは、Zynteglo(自己CD 34)に条件付きマーケティング許可を付与した+βをコードする細胞A-T 87 Q2019年,ブルーバード生物はあるTDT患者の治療のためのレンチウイルス遺伝子療法(グロブリン遺伝子)を開発したが,2021年には衛生当局と治療価格で合意できなかったため,ブルーバード生物はヨーロッパ市場からZyntegloを撤退した。FDAは2022年8月にZyntegloを承認した。私たちは私たちの治療法がこのような壊滅的な病気に潜在的な治癒方法を提供することができると信じている。
鎌状細胞病
概要
SCDは遺伝性赤血球疾患であり、βグロブリン遺伝子の特定の突然変異によって引き起こされ、この突然変異は赤血球機能異常を招く。酸素濃度が低い場合には,異常なヘモグロビンが赤血球内に集積し,赤血球が鎌状になり,硬直する。これらの鎌状細胞は血管を閉塞し、器官への血液を制限し、最終的に深刻な痛み、感染、脳卒中、全体の生活の質の低下と早期死亡を招く。溶血増加を経験し,貧血をきたすこともある。推定によると、全世界のSCDの発病率は毎年30万人の新生児であり、全世界で2000万から2500万人がこの疾病を患っている。アメリカとEUでは総罹患率は15万人と推定されている。
現在の治療法の限界は
ベータ地中海貧血と同様に,医療インフラが支持可能な地域では,溶血レベルの高いSCD患者の標準治療には長期輸血が含まれており,キレート療法と同様の鉄過負荷や毒性に関するリスクがある。FDAおよび/またはEMAも、ヒドロキシウレア、Adakveo(crizanlizumab-TMCA)およびOxbryta(Voxelotor)を含むSCDを治療するいくつかの疾患修正療法を承認した。同種造血幹細胞移植はもう一つの潜在的な治療選択である。Allo-HSCTはSCDに唯一の治癒可能な治療経路を提供しているが、これらの患者に移植に関連する発病率と死亡率の重大なリスクが存在すること、および一致したドナーと自発的なドナーが不足していることを考慮すると、それは通常避けられている。
私たちの遺伝子編集方法は
著者らはβ地中海貧血とSCDを治療する治療方法は遺伝子編集を用いてガンマグロブリンの発現を上昇させ、これは通常新生児にしか存在しないヘモグロビンサブユニットである。βグロブリンではなくガンマグロブリンを含むヘモグロビンを胎児ヘモグロビン、あるいはHBFと呼ぶ。多くの人ではHBFは乳児期に消失しており,ガンマグロブリンは自然発生するガンマグロブリン遺伝子抑制によりβグロブリンに置換されているためである。ベータ地中海貧血とSCDの症状は通常生後数ケ月に出現し、その時HBFレベルはすでに著しく低下した。一部のβ地中海貧血或いはSCDを有する患者はHbFレベルが上昇し、成人まで持続し、この場合は遺伝性胎児ヘモグロビン持続性、或いはHPFと呼ばれる。HPFを有する患者は通常症状がないか,あるいはより軽微な疾患を経験する。
8
この保護性のHPF状況は,これらの患者のゲノムDNAが特定の変化を起こしたためか,グロブリン遺伝子領域であるか,グロブリン遺伝子発現レベルを制御するいくつかの遺伝調節エレメントにあることが証明されている。
鎌状細胞病とβ地中海貧血患者のHbFレベルと発症率の関係

CRISPR/Cas 9がヘモグロビン疾患を治療するもう一つの方法は突然変異のβグロブリン遺伝子を是正することである。著者らはHBF上昇策略を著者らの初期方法として選択した、関連する遺伝子中断策略の効率と一致性のため、この策略はベータ地中海貧血患者を含む各種の異なるβグロブリン突然変異に対する相殺能力、及びHPF患者の無症状の自然病歴データを支持する。
鉛ヘモグロビン病の候補品であるExacel
著者らの主要な候補製品ExacelはCRISPR/Cas 9を用いてHPF患者が自然に産生した高レベルHBFをシミュレーションした。この効果を達成するために,ExacelはCRISPR/Cas 9を用いて赤系特異的エンハンサーを破壊するBCL 11 Aジーン。この遺伝子はBCL 11 A蛋白をコードし、これは大多数の人のHBFレベルを低いレベルに維持させる重要な要素である。BCL 11 A赤系特異性増強子を破壊することは赤系細胞におけるBCL 11 Aの発現を低下させ、それによってγグロブリンの発現を上昇させ、HBFレベルを増加させる。
われわれの治療法は,赤血球を産生する造血幹細胞を患者から分離し,これらの細胞を治療することである離体するCRISPR/Cas 9を用いてBCL 11 A赤系特異的エンハンサーを破壊し,編集後の細胞を患者に再導入した。患者に再注入すると,これらの遺伝子組換え幹細胞は高レベルのHBFを含む赤血球を産生すると信じられている。β地中海貧血において、HbFの上昇はペアリングしていないα-グロブリン鎖の毒性を低下させ、赤血球寿命を延長する可能性がある。したがって,Exexelはこれらの患者の輸血需要を減少あるいは解消する可能性がある。SCDでは,上昇したHbFが鎌状を阻止する可能性があるため,多くの赤血球で十分に高いHbFに達することは,疾患に関連する症状を有意に減少または除去することができる。
われわれのCRISPR/Cas 9遺伝子編集戦略は,ヘモグロビン疾患治療のための遺伝子療法の開発において他の遺伝子療法よりも有意に優れている可能性が信じられている。例えば、レンチウイルスに基づく治療は、ゲノム全体にグロブリン遺伝子をランダムに統合する1つまたは複数のコピーを含む。DNAのゲノム中の正確な位置によって,新たに導入された遺伝子の発現レベルが異なり,発現レベルの不一致や可変を招く可能性がある。私たちの戦略は高比例細胞におけるグロブリンの発現をより均一にする可能性があると信じている。また、個々のランダムレンチウイルスの統合に対して突然変異が生じる可能性があり、これは発癌潜在力を含む安全問題がある可能性がある。これに対し,CRISPR/Cas 9は特定のゲノム部位を編集目標としており,これまでわれわれのExacel Guide RNAの脱標的活性は検出されていない。
臨床前研究
臨床前研究において、著者らのCRISPR/Cas 9遺伝子編集過程は大量の細胞の中で臨床規模で約80%の対立遺伝子編集効率でHSCsを編集できることを証明した。我々はすべての幹細胞亜群でこのような高い編集効率を観察し、長期再充填HSCsに含めた。その後…体外培養赤系分化では,この編集によりHbFは編集細胞で総ヘモグロビンの30%以上を占めていたが,検討した対照群ではHbFは約10%であった。各細胞に基づいて、90%以上の細胞が所望の位置で修正され、76%の細胞が標的遺伝子の2つのコピーで編集され、16%の細胞が標的遺伝子の1つのコピー上で編集された。私たちはこの後体外培養赤系分化という編集率は、標的遺伝子の2つのコピーで編集された細胞におけるHBF発現レベルが35%を超え、1つの遺伝子上で編集された細胞におけるHBF発現レベルが20%を超えることをもたらす。
9
移植の安全性をテストするために設計された臨床前マウスモデルでは,遺伝子編集のHSCは長期移植と複数の系統に分化する能力を保持している。毒理学的研究では,対照群と比較して編集細胞の生物分布に有意差は認められず,差もなかった。最後に,5,000個を超える相同系部位と2,000個を超える相同に依存しない部位を評価したところ,外部誘導RNAの脱標的活性は検出されなかった。
臨床試験
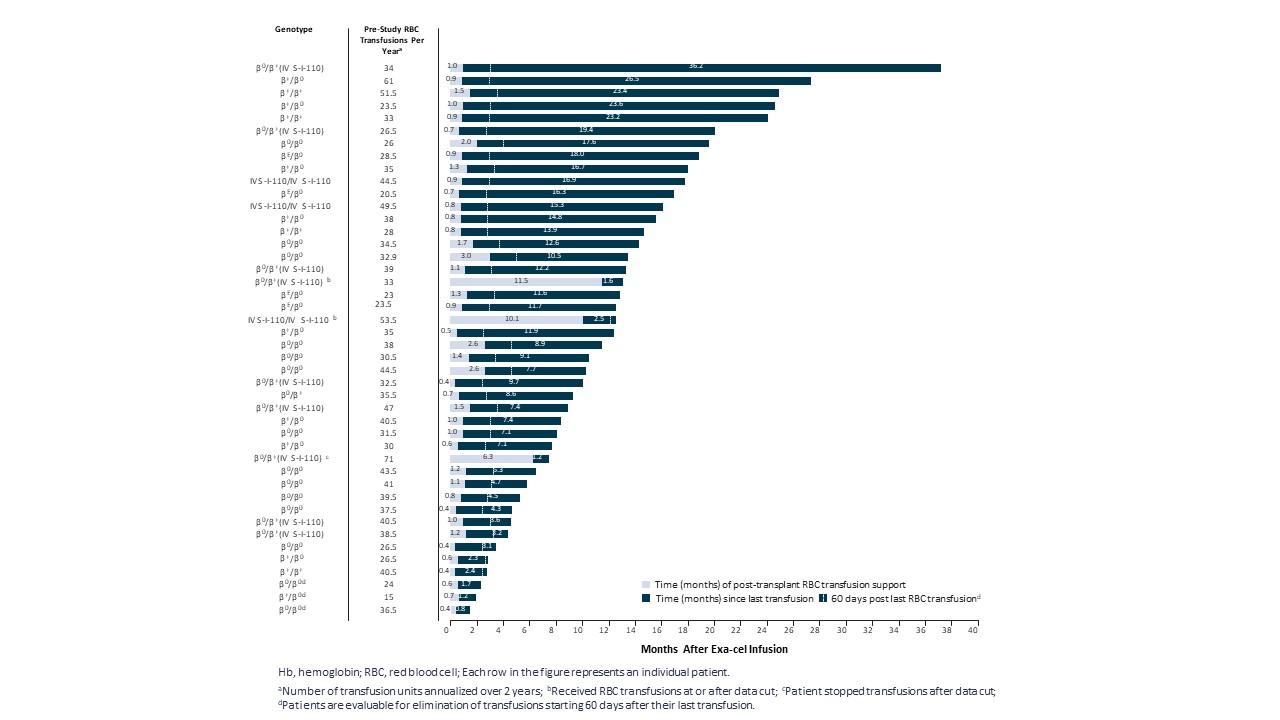
我々とVertexは,12歳から35歳までのTDT,SCD,重篤なSCD患者における単剤Exacelの安全性と有効性をそれぞれ評価することを目的とした1/2/3期開放臨床試験におけるExaccel研究を2つ行っている。いずれの臨床試験でも上位2名の患者は順に治療を行い,各臨床試験における最初の2名の患者のデータから移植が成功し許容可能な安全性があることが示唆され,同臨床試験は同時投与を開始した。両臨床試験とも輸液後約2年間の追跡調査を行うためである。登る-111と登る-121の登録が完了しました。われわれとVertexは,TDTを有する小児科患者Exacelに対する他の2つの開放ラベル臨床試験を開始し,それぞれSCD,SCD,SCDであった。Exexelを受けた患者は、Exexexeの安全性と有効性を評価するために、長期的な開放ラベルフォローアップ試験に参加することを要求される。GRAPH−131は,刺激輸液後に参加者に15年間にわたる追跡調査を行うように設計されている。
Exa-celはすでにアメリカ食品と薬物管理局(FDA)の多くの監督管理指定を獲得し、特にTDTとSCDを治療するRMAT、Fast Track、孤児薬物と稀な小児科疾患の指定を受けた。Exa−celは欧州委員会の孤児薬物称号と,TDTとSCDの治療に欧州医薬品局のPrime称号を獲得した。2022年12月,我々とVertexはそれぞれEUとイギリスのEMAとMHRAに輸出規制文書を提出し,EMAとMHRAはそれぞれMAAを検証した。また,我々とVertexは2022年11月に米国で我々のBLAのスクロール提出を開始し,2023年第1四半期末に提出を完了する予定である.
登り-111と登り-121 1/2/3期臨床試験研究プログラム模式図
TDTにおける登り-111の実験
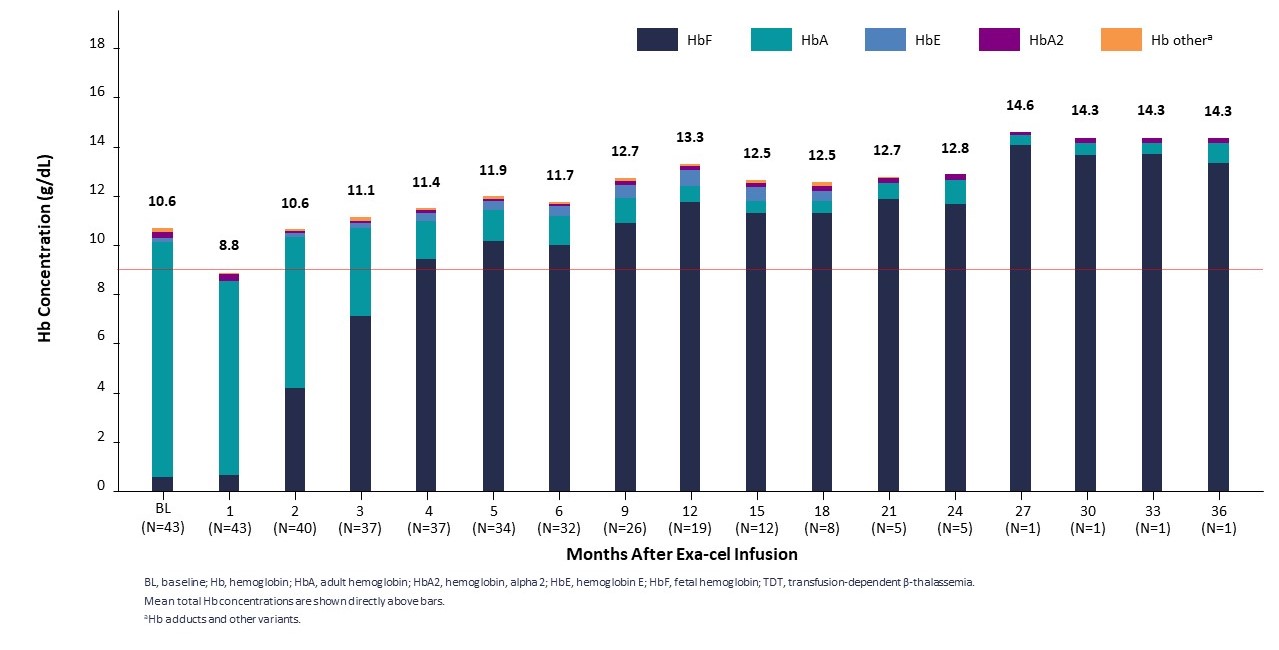
2022年第2四半期と第4四半期に,それぞれヨーロッパ血液学協会(EHA)大会と米国血液病学会(ASH)年次総会において,2022年2月までのデータ遮断点のTDT患者44例の臨床データを公表した。中位フォローアップ時間は11.9ケ月(1.2~37.2ケ月)、44例のTDT患者の中で42例は無輸血(0.8~36.2ケ月)、2例の輸血を停止しなかった患者の輸血量はそれぞれ75%と89%減少した。44例の患者はすべて類似の反応モードを示し、総ヘモグロビンとHbFは迅速に持続的に上昇し、HbFの全血中の分布は迅速に増加し、赤血球注入は減少或いは除去した。すべての12例の深刻なβ0/β0は輸血を排除した患者と評価でき、最後のフォローアップ以来輸血しなかった。また,利用可能な骨髄対立遺伝子編集データは経時的持続性を示した。この骨髄対立遺伝子編集データと一致したのは,データ締め切りまで1年以上フォローアップした全19名の患者がExexcel治療を受けた1位の患者を含めて安定と持続的な治療反応を示し,最終訪問時の総ヘモグロビンレベルは14.3 g/dLであり,Exexcel服薬3年後であった。
10
TDTは臨床的意義のあるHbFと総Hbを早期に達成し維持する
体外輸液後の輸液独立期
11
全44人の患者の安全性データは、自己幹細胞移植および清髄調節とほぼ一致した。2人の患者は、深刻な有害事象、またはSAEを経験し、調査者によってEXCESSに関連しているか、または関連する可能性があると評価された。1例は血球リンパ組織細胞貪食症(マクロファージ活性化症候群),急性呼吸窮迫症候群,頭痛に関連するSAEが3例,特発性肺炎症候群によるSAEが1例であった。すべての出来事はインプラント周囲から始まり,ヒト黄体形成ホルモンの場合に発生し,ステロイドや免疫抑制剤治療により完全に解決された。もう1例は好中球植え込み遅延と血小板減少に関与するSAEsを認めた。56日目にSAEは除去され,好中球はインプラントされ,予備細胞は使用されなかった。残りの患者はすべて体外輸液後43日以内に好中球を移植した。残りの患者では手術に関連するSAEは認められなかった。
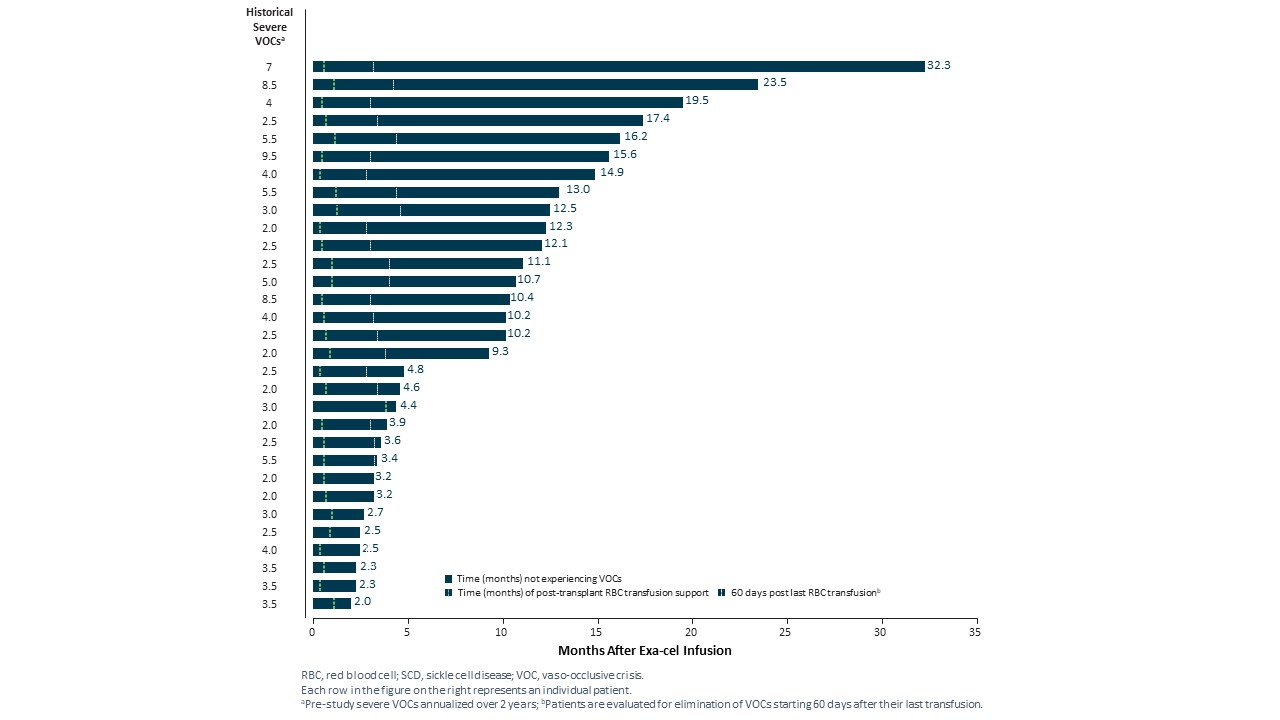
重度SCD患者のAPRACH−121試験
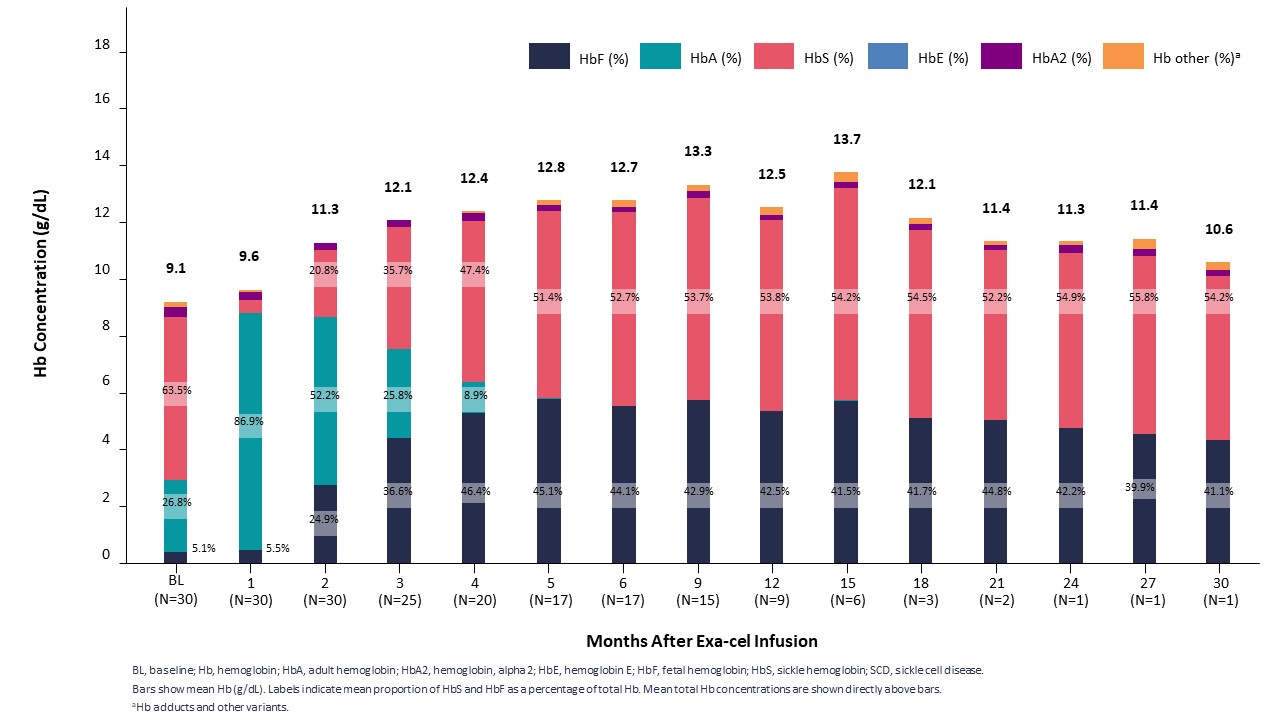
2022年第2四半期と第4四半期に,それぞれEHA大会とASH年次総会において,2022年2月までのデータ遮断点のSCD患者31名の臨床データを公表した。中位フォローアップ期間は10.2ケ月(2.0~32.3ケ月)であり、すべての31例の患者は輸液後に血管閉塞危険像(VOC)が消失し、最後のフォローアップ時にVOCは消失した。すべての患者は類似した反応パターンを示し,総ヘモグロビンとHbFは速やかに持続的に上昇し,HbFの全細胞分布を示した。4カ月までの平均総ヘモグロビンレベルは12 g/dLを超え,HbFの平均割合は40%を超えた。また,利用可能な骨髄対立遺伝子編集データは経時的持続性を示した。データ遮断日まで1年以上フォローアップした9例の患者はすべて安定と持続的な治療反応を示し、Exexelを用いて治療した1例目のSCD患者を含み、Exexel服薬30ケ月後の最後の訪問中、彼の総ヘモグロビンレベルは10.6 g/dL、HbFスコアは41%であった。
SCD患者は臨床的意義のあるHbFと総Hbを早期に達成し,維持する
12
体外輸液後VOCsから離脱する時間
全31人の患者の安全性データは、自己幹細胞移植および清髄調節とほぼ一致した。SAEはEXCESSに関与していると考えられているか,あるいは関与している可能性はなく,多くの非重篤な有害事象は軽~中等度と考えられている。2022年2月にデータが切断された後,SCDを有する成人患者がSARS−CoV−2に感染した後に肺炎と呼吸不全が出現し,死亡した。調査者はこれらの事件はSARS−CoV−2感染によるものであり,白頭翁の肺損傷に関与しており,死亡とは無関係である可能性が考えられた。
次の世代の努力
EXCESSをもとに,現在EXCEXに用いられている清髄性白丹調節レジメンよりもメリットがある可能性がある標的調節レジメンの次世代努力を行った体内にある造血幹細胞を編集し,いずれもわれわれの療法から利益を得ることができる患者数を拡大することができる。
免疫腫瘍学
免疫腫瘍学の分野では,腫瘍界への興味が急速に増加している,すなわち免疫系を用いて癌細胞を攻撃する治療法である。工学免疫細胞療法は,T細胞などの免疫系細胞を遺伝子改変することにより,癌細胞を認識·攻撃できる方法である。
工学細胞療法は鼓舞的な結果を示し、自己CAR T製品が多くの承認を得た。これらの治療法は新たな腫瘍学的療法となる可能性があるが,このすべての潜在力を実現するにはいくつかの重要な挑戦を克服する必要がある。開発されている工学的細胞療法の多くは,治療を受けた患者ごとに独自の製品を創出する必要があり,過去に腫瘍学の分野で挑戦的でコストが高いことが証明されている。このようなカスタマイズされた製造過程に時間を要し,その過程で患者の疾患が悪化する可能性があり,実行可能な製品がまったく生産できない場合がある。また,これらのバージョンの工学細胞療法は固形腫瘍の治療には能力が限られているようであり,高毒副作用を示し,複雑な管理案が必要である。対照的に、同種遺伝子工学T細胞療法は“既製品”を使用することができるので、直ちに得ることができ、より便利に、より簡単な後方勤務保障、より高い一貫性(各ロットが多くの用量を産生するため)、および用量滴定によっても再投与でも柔軟な用量分配を得ることができる。
免疫腫瘍学において最終的に成功した細胞工学戦略は様々な遺伝子修飾に関与することが予想され,CRISPR/Cas 9はこの応用において中心的な役割を果たすと信じている。他の遺伝子編集プラットフォームは、これらの目的のために使用される可能性があるが、CRISPR/CAS 9は、特に、単一の細胞内で複数の遺伝子を修飾および/または挿入する多重編集に適している。遺伝子編集技術には異なるタンパク質酵素が必要です
13
効率、細胞毒性および/または製造上の課題により、各遺伝子修正が同時に行うことができる編集回数が制限される可能性がある。これに対し,CRISPR/Cas 9は単一のCas 9蛋白と複数の小さな誘導RNA分子を用いて効率的に複数回の編集を行うことが可能である。
われわれの免疫腫瘍学的細胞療法では,CRISPR/Cas 9の多重化能を用いて同種異体投与と追加的な遺伝子編集を導入し,これらの候補製品の治療効果や安全性を向上させることを目的としている。また,我々は我々のCRISPRプラットフォームを用いて持続的な革新の過程を実現しており,その過程でインクリメンタル編集を次世代製品に統合し,治療効果のさらなる向上を図っている。私たちは、すべての腫瘍タイプ(固形腫瘍を含む)の免疫腫瘍学における工学的細胞療法のすべての潜在力を実現するために、私たちの多重輸送能力を拡大し続けている。CRISPR/CAS 9は将来の工学細胞治療において重要な役割を果たすと信じていることから、これまで、著者らは著者らの同種異体CAR T細胞計画の完全所有権を保留することを選択した。
さらに、複数のグループが、ナチュラルキラー細胞、またはNK細胞などの他の免疫細胞の免疫腫瘍学的治療における使用を証明し始めている。我々の遺伝子編集免疫細胞療法への取り組みをT細胞以外に拡張するために,Nkartaと協力関係を築き,我々の遺伝子編集技術と細胞治療専門知識をNkartaがリードするNK細胞発現,開発,製造能力と組み合わせた。我々はNkartaと共同でドナー由来の2種類の遺伝子編集CAR-NK細胞候補製品を開発し、共同商業化しており、そのうちの1つはCD 70を標的としている。また,我々は,この2つの細胞タイプの独自の利点を利用するために,NK細胞とT細胞を結合した候補製品を共同開発·共同商業化している。
私たちがリードしている免疫腫瘍学の候補品であるCTX 110
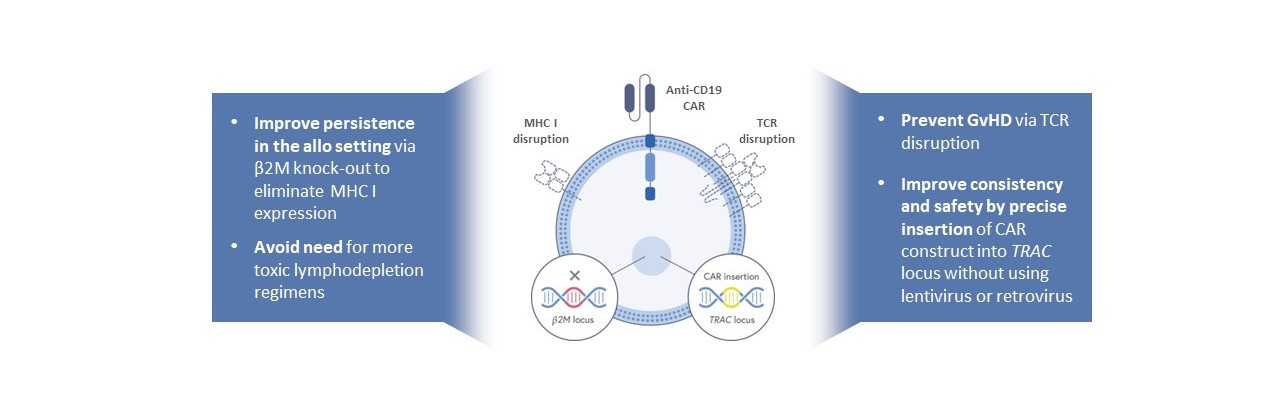
著者らの先行免疫腫瘍学候補製品CTX 110は健康なドナー由来遺伝子編集の同種異体CAR T研究療法であり、CD 19陽性の悪性腫瘍、例えばあるリンパ腫と白血病に対して。CTX 110の1つの主要な目標は、各特定の腫瘍タイプの患者のための独特な製品の低効果とコストを克服することであり、方法は、1つのロットから多くの異なる患者を治療することであり、私たちは“既製”療法と呼ぶ。CTX 110を産生するために、我々は、我々の遺伝子編集技術を使用して、健康なドナーからのT細胞を3つの修飾した:(I)T細胞受容体を除去するか、またはTCRを除去して、候補製品中の移植片対宿主病(GvHD)のリスクを低減し、(Ii)CD 19配向CARを挿入する尾行する遺伝子および(Iii)クラスI主要組織適合性複合体MHC Iは、既存の環境におけるCAR T細胞の持続性を向上させるために、細胞表面から除去される。この方法は,統合ウイルスを用いてCARを半ランダムに挿入し,MHC Iノックアウトを含まずに持続性を増加させる開発中の他の同種異体CAR T製品よりも優れていると信じている。
CTX 110:差別化CRISPR-編集の同種異体CAR-T設計
臨床試験
我々は、少なくとも2つの以前の治療を受けた再発または難治性CD 19陽性B細胞悪性腫瘍の成人患者におけるCTX 110の安全性および有効性を評価するために、我々のCarbon臨床試験においてCTX 110を研究している。私たちの第1段階Carbon臨床試験の新しいデータと規制機関との議論によると、私たちは、合併用量を含む登録された、単一アーム、マルチセンター、開放ラベルを含む臨床試験の第2段階に拡張した。私たちはこの重要な腕に薬を投与し始めましたCTX 110は、FDAによってRMAT称号を付与されている。
14
第一段階の臨床試験は二つの部分に分けて行われる--A部分とB部分。第一段階A部分では、患者は標準的なリンパ浄化方案に従って、用量レベル(DL)1(3000万個のCAR+T細胞)からDL 4(6億個のCAR+T細胞)へCTX 110を単回注入し、臨床的利益に基づいてCTX 110に再投与することを選択した。第1段階B部分では、患者は標準リンパ濾過後のDL 4でCTX 110治療を受け、臨床的利益を示す患者では、初期用量後4~8週間以内に同じ用量レベルの強固な用量CTX 110を受ける。
Carbon Part A実験設計

2022年12月、ASH年次総会で、第1段階A部分の最新の臨床データを共有した。2022年10月6日までのデータ締め切りまでに、32名の大B細胞リンパ腫(LBCL)患者が第1段階A部分でCTX 110治療を受け、分析に取り入れられた。32例の患者はすべて侵襲性LBCLがあり、DLBCL、NOS、高度悪性リンパ腫とTfLを含む。ほとんどの患者は試験に入る前に最後の治療に無効であり、47%の患者は以前に3回以上の治療を受けたことがある。患者はフルダラビン(30 mg/m)からなる標準リンパ浄化レジメンを3日後にCTX 110を単回注入した2/日)およびシクロホスファミド(500 mg/m2/日)。患者が初歩的な臨床的利益を得、その後進行した場合、彼らは追加のCTX 110注入を受けることができる。さらに、患者のセットは、35日目に2回目の計画されたCTX 110注入を受ける資格がある。
15
炭素A部分患者のベースライン特徴
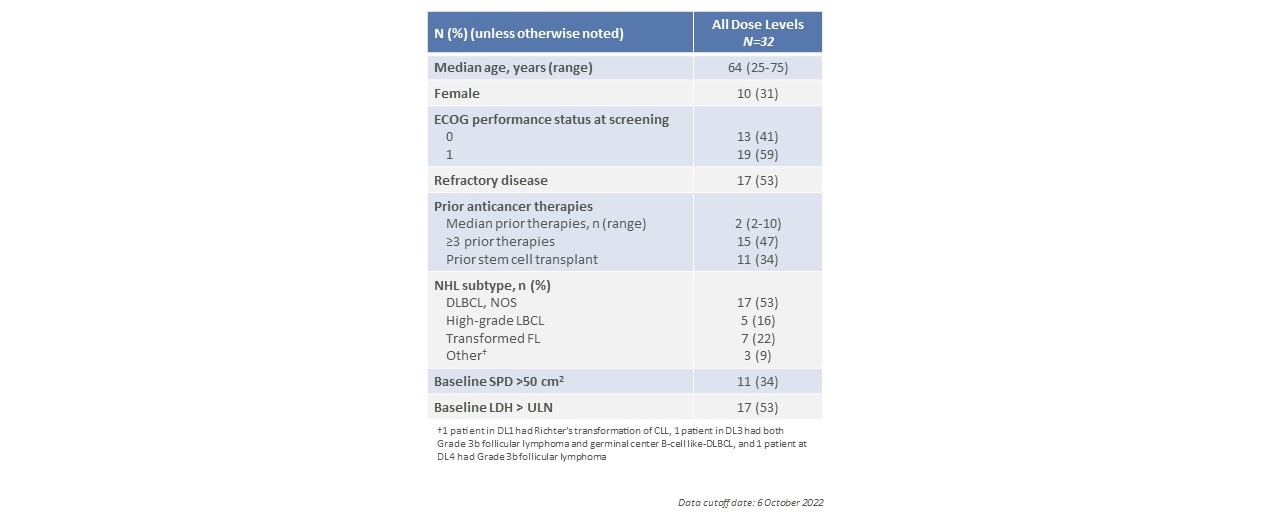
CTX 110の炭素第1段階で観察された応答率A
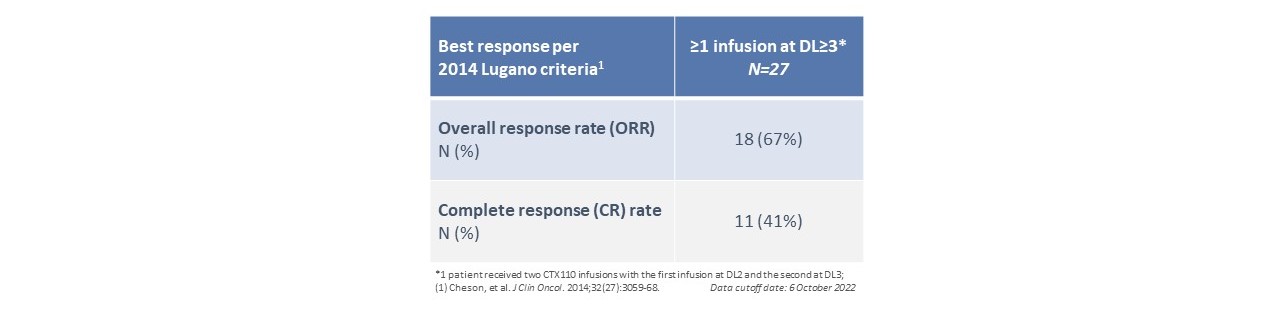
CTX 110は第1段階A部分で鼓舞的な臨床活動を示し,DL 3および以上で少なくとも1回CTX 110治療を受けた患者では,総有効率(OOR)は67%,完全応答率(CR)は41%(n=27)であった。3人の患者はすでに2年以上の完全緩和を実現し、維持し、CTX 110が持続的な緩和を産生する潜在力を示した。DL 3および以上のCTX 110単回注射6カ月の完全緩解率は19%であった。最後に,自己CAR T療法と異なり,ほとんどの登録患者がCTX 110治療を受けており,登録患者34名中2人のみがコロナウイルスと肺炎の合併感染で治療を受けていない。
16
CTX 110が炭素第1段階で観察した持続的応答A部分

CTX 110は、炭素第1段階のすべての用量レベルで良好な耐性を有する
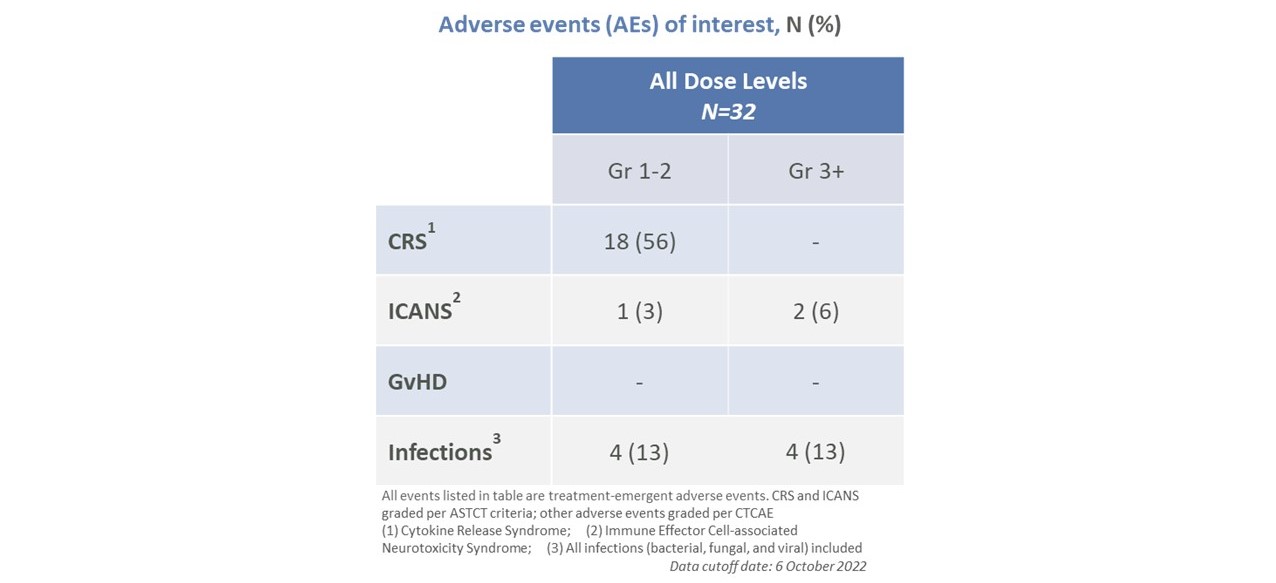
CTX 110は、第1段階A部分のすべての用量レベルで耐性が良好であり、用量制限毒性もなく、移植片対宿主病(GvHD)または任意のレベルの輸液反応もない。米国移植と細胞治療学会(ASTCT)の基準によると,すべてのサイトカイン放出症候群(CRS)症例は1級あるいは2級である。13%の患者は、HHV 6脳炎で死亡した1人の患者およびCTX 110に関連する可能性があると考えられる1人の感染を含むレベル3以上の感染を発症した。7人の患者は、CRS、免疫効果細胞関連神経毒性症候群(ICANS)、および発熱性好中球減少症を含むCTX 110の深刻な有害事象を経験した。2回目のCTX 110注入を受けた13名の患者では,全体的な安全性は変化しなかった。
17
第1段階B部分からの新しいデータサポートは、CTX 110を可能な登録試験に進める。2022年12月、私たちは上層総括を提供し、第1段階B部分からの新しいデータを説明し、以下のように観察結果を共有した:(1)著者らは(1)持続的に完全に緩和している数人の患者が6ヶ月を超えている鼓舞的な治療効果の概況を観察した;(2)明確な証拠は、用量を強固にする利点を示した;(4)初期用量と強化用量の間のピーク拡張と安定した疾患の転帰と、進行している完全緩和の部分的緩和を観察した;(3)第1段階と一致した安全性プロファイルは、強化療法の耐性を実証した;(4)初期用量と強化用量の間のピーク拡張と全体薬物動態は類似している。
CTX 112
CTX 110と同時に、CD 19の次世代研究、同種CAR T候補製品であるCTX 112を進めている。CTX 112は、CTX 110以外の2つの追加編集を含み、我々のCRISPR/Cas 9プラットフォームを十分に利用して、インクリメンタル編集を次世代製品に統合することによって、絶えず革新することを可能にする。これらの編集はRegnase-1と形質転換増殖因子-β受容体2(TGFBR 2)をコードする遺伝子に対して、CAR Tの効力を増強し、CAR Tの消費を減少させることを目的としている。編集Regnase-1はT細胞機能の内在的な“ブレーキ”を除去し、編集TGGFR 2はT細胞抗腫瘍活性の重要な外部“ブレーキ”を除去した。我々は数十個の新しい遺伝子と先に説明した遺伝子のシステムをスクリーニングすることによって、このような編集の組み合わせを決定した。これらの編集は共に、別の次世代研究計画に記載されているように、臨床前モデルの効力を著しく向上させたCTX 131“2022年第4四半期、INDのCTX 112への申請はFDAの承認を得た。
CTX 130
もう一つの免疫腫瘍学候補薬物CTX 130はCD 70を標的とする健康ドナー遺伝子編集の同種異体CAR T研究療法である。いくつかの癌はCD 70を発現し、非ホジキンリンパ腫、あるT細胞リンパ腫、腎細胞癌、神経膠芽腫及び膵臓癌、肺癌と卵巣癌を含み、正常組織は発現しない或いは極めて限られた発現を示す。この目標は、血液系悪性腫瘍、例えばT細胞リンパ腫、固形腫瘍癌、例えば腎細胞癌に移行することができる。
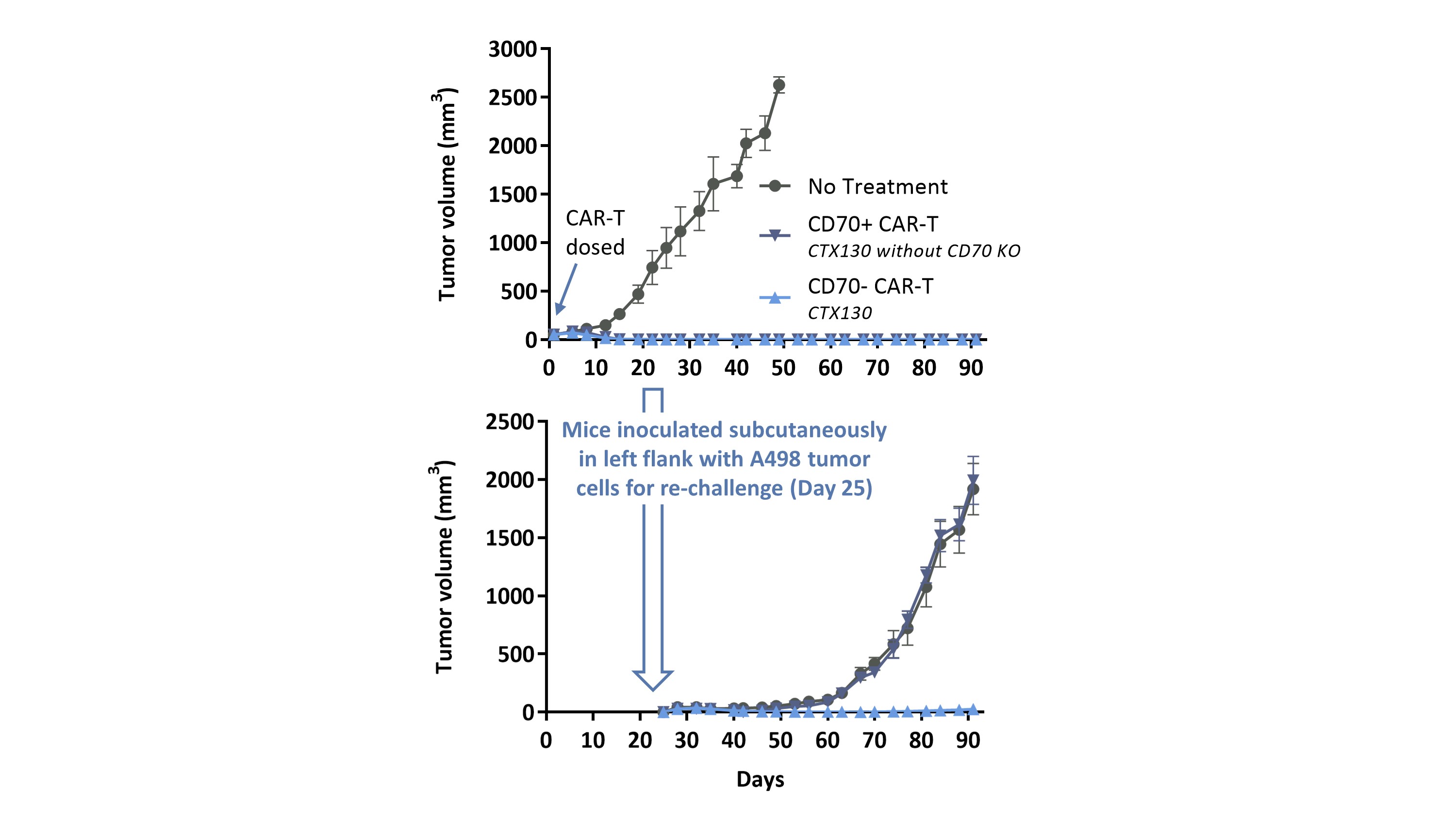
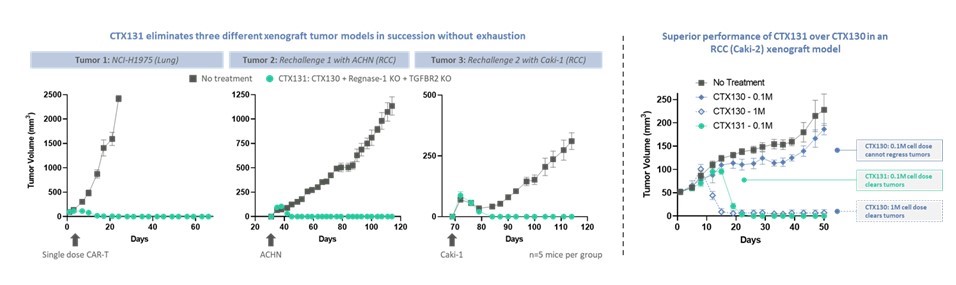
CTX 130を産生するために,CTX 110と同様の3つの修正(ただしCARはCD 19ではなくCD 70に対して)を行い,T細胞におけるCD 70遺伝子のノックアウトに加えてCAR T細胞機能を向上させた。以下の図に示すように,臨床前研究では,CTX 130はすべての治療を受けたマウスにおいて移植腎細胞癌モデルの成長を除去または重篤に抑制し,初期治療でも再挑戦でも。また,CTX 130はCD 70遺伝子が完全なCAR T細胞を保持しているよりも良好な機能を示した。
18
追加編集により、皮下A 498腎細胞癌モデルに対するCTX 130の性能が改善された
臨床試験
我々は現在、成人患者におけるいくつかの用量レベルのCTX 130の安全性および有効性を評価するために、CTX 130に対して2つの独立した第1段階、単一アーム、多中心、開放ラベルの臨床試験、すなわちコバルト−Lymおよびコバルト−RCCを行っている。CTX 130は、FDA指定のT細胞リンパ腫治療の孤児薬物および真菌症およびSézary症候群(MF/SS)の治療のRMAT指定を取得した。
コバルト-Lym試験はCTX 130による再発性或いは難治性T或いはB細胞悪性腫瘍治療の安全性と有効性を評価することを目的とした。CTX 130は、CD 70が少なくとも10%の再発または難治性T細胞リンパ腫を発現する成人患者に対して用量漸増治療を行う。患者自身が罹患したT細胞からCAR T療法を作製する固有の困難と潜在リスクを考慮すると、T細胞リンパ腫の同種異遺伝子細胞療法はこの患者群で満足されていない需要を解決する上で更に大きな潜在力を持っている。コバルトリンパ移植群の患者はフルダラビン(30 mg/m)を含む標準リンパ浄化プログラムを3日間受けた2/日)およびシクロホスファミド(500 mg/m2/日)、その後、CTX 130を単回注入した。CTX 130の初回注射は臨床的利益を示した患者は疾病の進展後に再び注射を受ける資格がある。主な終点は用量制限毒性とOORの発生率による安全性である。重要な副次的終点は無進展生存率と全体生存率を含む。
コバルト−LYM試験設計
19

2022年6月,EHA大会でコバルト−LYMの予備臨床データを共有した。2022年4月26日までのデータ遮断日まで、すでに19名のT細胞悪性腫瘍患者が入選し、その中の18名の患者はCTX 130の少なくとも28日間のフォローアップを受け、そして分析に入れた。登録前にはすべての患者が厳しい予備治療を受けており,中央値は4種類の系統治療であった。しかも、すべての患者は彼らの最後の治療に無効だった。末梢T細胞リンパ腫(PTCL)8例、皮膚T細胞リンパ腫(CTCL)10例。
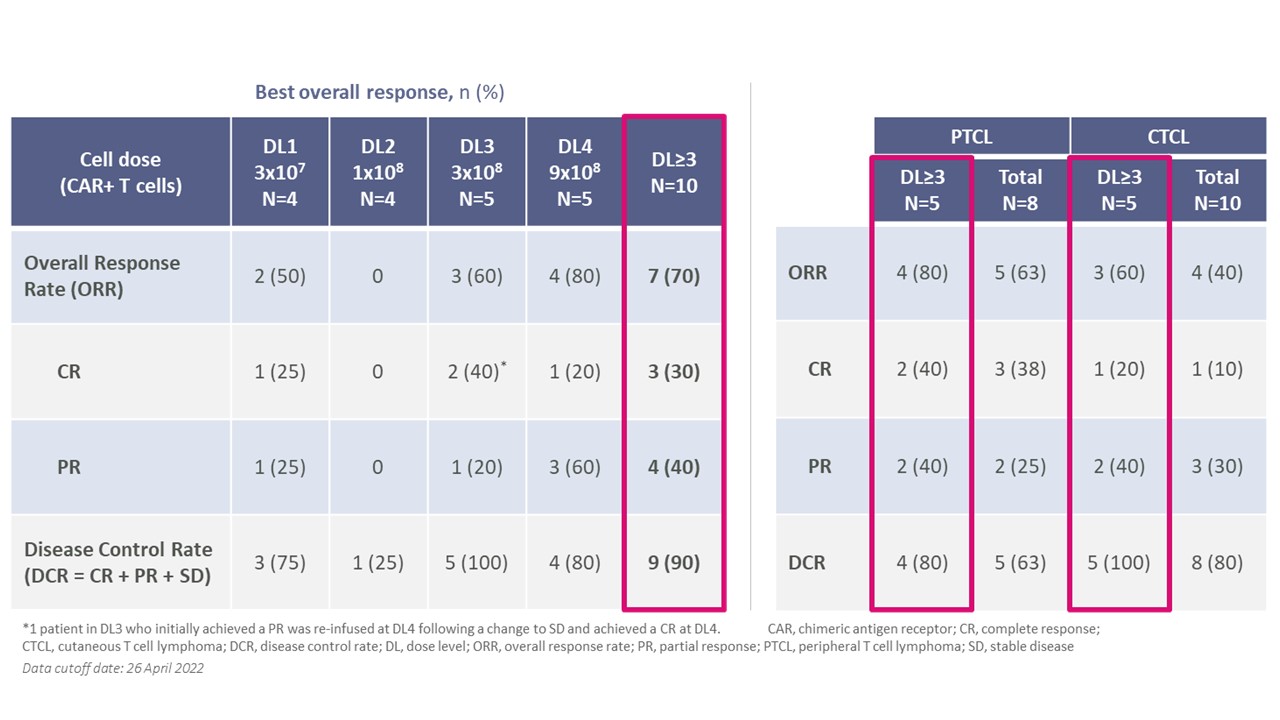
CTX 130の臨床的有意な反応は、高用量レベルの患者反応のパーセンテージがより高いことが観察された。PTCLの2014年ルガノ反応標準或いはCTCLの国際皮膚リンパ腫反応標準(Olsen標準)に基づいて、調査員の審査に基づいて疾病評価を行った。DL 3以上ではORR 70%,CR率30%であった。また、DL 3及び以上の患者の90%は臨床的メリットがあり、病状が安定或いはもっと良いと定義されている。患者ではCD 70発現の中央値は90%であったが,すべてのレベルのCD 70発現に反応が認められた。PTCLとCTCLの反応はほぼ一致し,DL 3および以上のOORはそれぞれ80%と60%であった。CTX 130治療後のCTCL患者では,すべての疾患間隔は,リンパ節,皮膚,血液を含めて広範な活動性と深さの反応を示した。
20
コバルト−リンパ球におけるCTX 130の応答率観察
CTCLのすべての車両で観察された反応
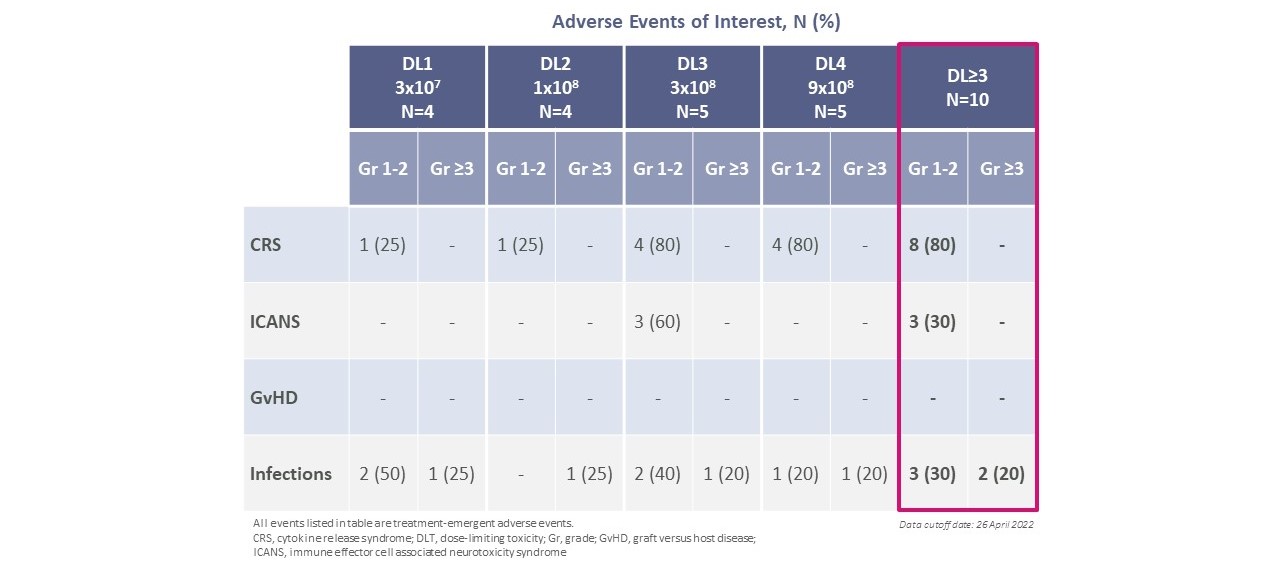
CTX 130はすべての用量レベルで耐性が良好であった。GvHD症例はなく,用量制限毒性もなく,腫瘍溶解症候群(TLS)もなかった。ASTCT規格によると,すべてのCRSとICAN症例はレベル1またはレベル2であり,特殊な介入を必要としないか,標準的なCRS治療で解決されている。CTX 130を再服用した患者では,CRSの頻度も重症度も増加しなかった。ウィリアム症候群患者が肺感染下で突然死し,CTX 130とは無関係と考えられた。試験では治療に関連した死亡例は認められなかった。
21
コバルト−ライムでは,CTX 130はすべての用量レベルに対して良好な耐性を示した
コバルト−LYMに加えて、CTX 130は、再発または難治性腎癌治療の安全性および有効性を評価することを目的としたコバルト−RCC試験で評価されている。CTX 130は切除できない或いは透明細胞分化を有する転移性腎癌患者において投与量の増加を行う。コバルト−LYMと同様にフルダラビン(30 mg/m)からなる標準リンパ浄化プログラムを3日間受けた2/日)およびシクロホスファミド(500 mg/m2/日)、その後、CTX 130を単回注入した。CTX 130の初回投与に反応した患者は、疾患が進行したときに再投与することができ、安定または進行した疾患が出現したときに臨床的利益を示す患者も再投与することができる。主な終点は用量制限毒性とOORの発生率による安全性である。重要な副次的終点は最適な全体反応、無進展生存率と全体生存率を含む。
2022年11月,癌免疫治療学会年次総会において,コバルト腎癌の初歩的な臨床データを共有した。2022年5月2日までのデータ遮断日まで、すでに14例のIV期腎透明細胞癌患者が入選し、その中のすべての患者はCTX 130治療を受け、そして安全性分析に入り、13例の患者は治療効果の評価を受けた。登録前にはすべての患者が厳しい予備治療を受けており,中央値は3種類の系統治療であった。
CTX 130はコバルト−腎癌において良好な抗腫瘍活性を示した。1人の患者は持続的な完全緩解を経験し,再発/難治性固形腫瘍患者における同種異体CAR T細胞療法を用いて初めて達成された。データ切断時,この患者は18カ月間完全緩解状態が続いていた。全体的に,大量の前処理を経た腎癌患者群では,CTX 130は77%の疾患コントロール率を実現し,9名の患者は病態安定を実現した。安定疾患に達するまでの最長持続期間は7.8カ月であり,データ切断時にも行われている。疾患安定期には、患者は他の抗癌治療を何も受けていない。CTX 130は典型的な薬物動態特性を示し,ピークは10日目の中央値に現れた。
CTX 130はすべての用量レベルで耐性が良好であった。GvHD症例はなく,用量制限毒性はなく,ICAN例もTLS例もなかった。ASTCT規格によると,CRSはすべての症例でレベル1またはレベル2であり,特別な介入を必要としないか,標準的なCRS治療で解決されている。CTX 130を再服用した患者では,CRSの頻度も重症度も増加しなかった。3例の患者は感染したSAEがあり、すべてCTX 130と関係がなく、5級肺炎合併4級呼吸困難を含み、1例が死亡した。試験では治療に関連した死亡例は認められなかった。
このCD 70標的CAR T細胞を探索して透明細胞腎癌を治療する最初のヒト臨床試験により、この薬物は耐性の安全性を有し、非標的毒性がなく、そして鼓舞的な抗腫瘍活性を有することを示した。コバルト腎癌研究のこれらの初歩的な結果は腎臓細胞癌と他のCD 70陽性悪性腫瘍中のCD 70標的CAR T細胞を更に探索するために臨床意義のある概念証明を提供し、そして更に効力を高める潜在力を強調した。
CTX 131
CTX 130と同時に、著者らはCTX 131を進めており、これはCD 70に対する次世代研究、同種異体CAR T候補製品であり、固形腫瘍とある血液系悪性腫瘍の潜在的治療に応用できる。CTX 131は、CTX 130以外の2つの追加編集を含む。これらの編集は,CTX 112で用いられているものと同様に,Regnase−1とTGGFR 2をコードする遺伝子に対して,CAR Tの効力を増強し,CAR Tの消費を減少させることを目的としている。これらの編集を組み合わせると,臨床前モデルでは協同で約10倍の効力が向上し,以下のようになる。2023年第1四半期、CTX 131のINDはFDAの承認を得た。
22
CTX 131は増強した効力を示し,Regnase-1とTGFBR 2編集は効力を約10倍増加させた
再生医学
再生医学、或いは幹細胞を用いて疾病、損傷或いは年齢によって失われた組織或いは器官機能を修復或いは置換することは、稀かつよく見られる疾患を治療する潜在力がある。この分野は臨床概念証拠が出現し始めているところに近づいている。これらの努力の多くは修正されていない幹細胞を用いており,遺伝子編集によるこれらの細胞の遺伝子工学の潜在力は大きい。我々は,CRISPR/Cas 9遺伝子で編集した異遺伝子幹細胞由来療法を用いて,免疫逃避を実現し,細胞機能を改善し,細胞運命を指導することを求めている。我々のこの分野における最初の主な努力は,糖尿病において,糖尿病治療のための遺伝子編集幹細胞療法を発見·開発し,それを商業化する戦略協力の一部として,ViaCyteと一連の計画を進めている。
糖尿病におけるViaCyte協力
ランゲルハンス島移植の臨床データにより、β細胞代替方法はインシュリンを必要とする糖尿病患者にメリットを提供する可能性があることを表明した。ViaCyteは幹細胞から膵臓系細胞を生成し、それを安全かつ効率的に患者に送達する方法を開発した。ViaCyteは以前、細胞療法を直接血管化することを可能にする非免疫防御送達デバイスを使用して未編集候補製品を評価した。この早期候補製品の臨床概念検証データは,細胞療法がT 1 D患者でインスリンを産生できることを示唆していることが鼓舞的である。しかし,患者の免疫系はこれらの細胞を外来細胞と認識するため,拒絶反応を回避するために長期的な免疫抑制が必要となる。
我々の遺伝子編集技術は,以下のように移植細胞を患者免疫系から保護する潜在力を提供している離体する膵臓細胞を産生するための幹細胞系内の免疫調節遺伝子を編集する。CRISPR/CAS 9の速度,特異性,多重化効率は,我々の技術をこのタスクに非常に適していると信じている.また、著者らのCRISPRプラットフォームは持続的な革新の過程を支持し、次世代候補製品にインクリメンタル編集を加え、治療効果を更に増加させることを目的としている。
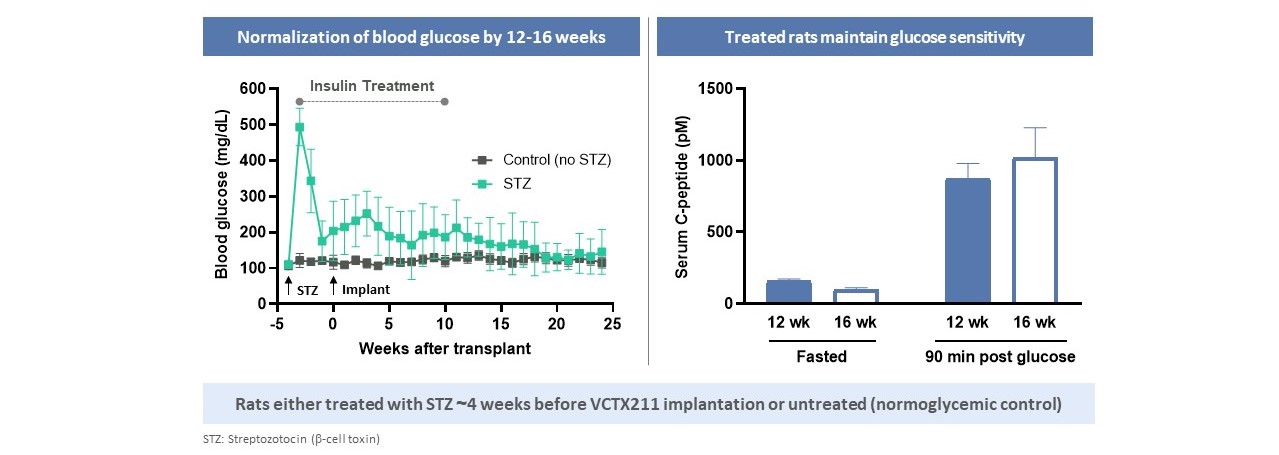
CRISPR/Cas 9プラットフォームのこの機能は,多段階製品戦略をとっている。著者らの最初の候補製品VCTX 210は、T 1 Dを治療するための研究、異遺伝子、遺伝子編集、免疫回避、幹細胞由来製品であり、この製品は私たちの遺伝子編集技術をViaCyteの独自幹細胞能力に応用することによって開発された。VCTX 210は、B 2 MおよびTXNIPのノックアウトおよびPD-L 1およびHLA-Eのノックインを促進することを目的とした4つの免疫逃避および細胞適合性を促進するための4つの遺伝子編集を含む。著者らとViaCyteはT 1 D患者におけるVCTX 210の安全性、耐性と免疫逃避を評価し、この臨床試験の後続段階にあるVCTX 210を研究している第1段階の臨床試験である。我々の次の候補研究製品VCTX 211は、移植片受容性およびβ細胞増殖を向上させ、サイトカイン誘導アポトーシスに対する保護を提供するために、MANFおよびA 20をノックダウンして移植片受容性およびβ細胞増殖を向上させるために、2つの追加の遺伝子編集をVCTX 210に加えた。つまりこれらの編集はβ細胞の免疫系からの逃避能力を向上させます体外培養そして体内にある臨床前モデルでは,以下のようになる。また,VCTX 211は糖尿病ラットモデルの高血糖を逆転させることが証明されている。2022年第4四半期,VCTX 211の臨床試験申請はカナダ衛生部の許可を得ており,1/2期臨床試験が行われている。
23
VCTX 211細胞は免疫から逃げている離体するそして生活の中で
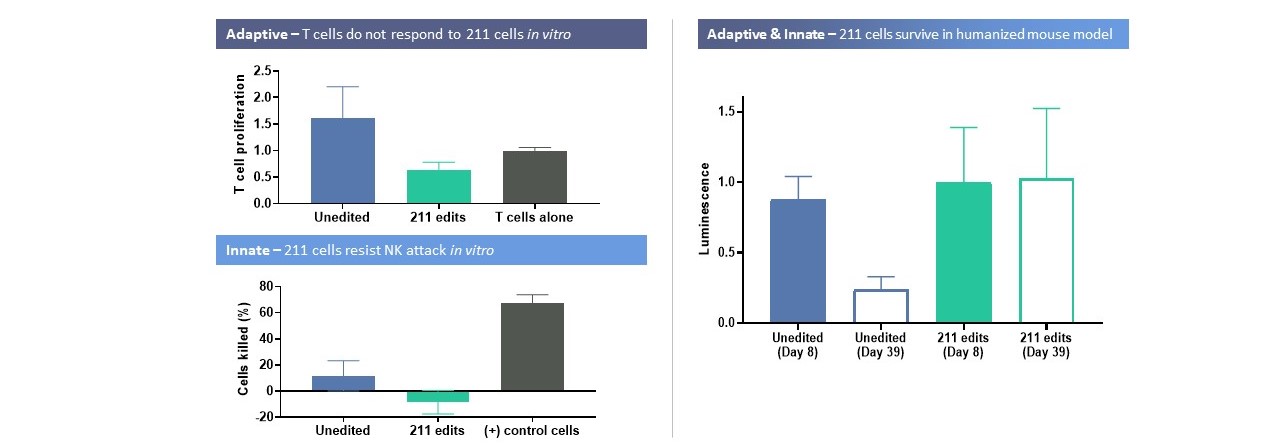
VCTX 211逆転糖尿病ラット高血糖の実験研究
Vivo手法では
私たちは信じています体内にある遺伝子編集,あるいはCRISPR/Cas 9による治療薬を直接ヒト内の組織に輸送することは,臨床翻訳のハードルに達している。そこで私たちは先進的なプラットフォームを作りました体内にある肝臓の遺伝子編集は急速に進められています体内にある珍しい病気とよく見られる臨床試験計画。私たちのリードは体内にある臨床的に確立·検証された送達技術を利用するために肝臓に対する計画が計画されており,主にLNPsであり,これらの技術は現在利用可能である。LNPにはいくつかの利点があり,CRISPR/CAS 9の配信に非常に適している体内にある効率的で安全な肝臓送達、大貨物サイズと瞬時貨物表現を含む。肝臓内では,遺伝子破壊戦略の影響を受けやすく遺伝的関連が知られている疾患,例えば以下の遺伝子標的を介した心血管疾患が研究されているAngptl 3, LPAそして、そしてPCSK 9それは.このような既存の概念証拠を利用する方法は,CRISPR/CAS 9に基づく療法の提供に関連する挑戦を減少させると信じている体内にある.
肝臓に加えて、私たちはそれを造血幹細胞、中枢神経系、および他の肝外組織に送達するために、腺関連ウイルス(AAV)ベクターおよびナノ粒子技術のさらなる進歩を含むより多くの送達技術を求めている。内部努力と外部協力を通じて、私たちは未来を支援するために新しい交付モデルを開発しています体内にある治療学です。
24
心血管と脂質異常計画
心血管疾患(CVD)は全世界の主要な死亡原因であり、2019年にすべての死亡人数の30%以上を占め、即ち1800万人近くである。心血管疾患は心不全、脳卒中、アテローム性動脈硬化性心血管疾患(ASCVD)、大動脈弁石灰化などを含む。血中脂質異常は心血管疾患を引き起こす主要な原因である。血中脂質異常症の特徴は,コレステロール,リポ蛋白,トリグリセリドを含む血中脂質レベルが異常に高いことである。最もよく見られる3種類の血中脂質異常は高コレステロール血症、高トリグリセリド血症とリポ蛋白質(A)の上昇である。
私たちは開発中です体内にある低密度リポ蛋白(LDL)-コレステロール(LDL-C)、トリグリセリドとリポ蛋白質(A)などの重要な血中脂質レベルを下げることによって、心血管疾患を治療する治療法を編集した。我々は,(1)天然ヒト遺伝学,抗体およびRNA療法に基づく実証された利点,(2)単剤,一生持続可能な編集方法により治療パラダイムを転換する機会,(3)重篤な疾患からより大きな患者集団への開発経路を拡張する能力,および(4)計画間共同治療の可能性から,この領域を追求することを選択した。
CTX 310-Angptl 3
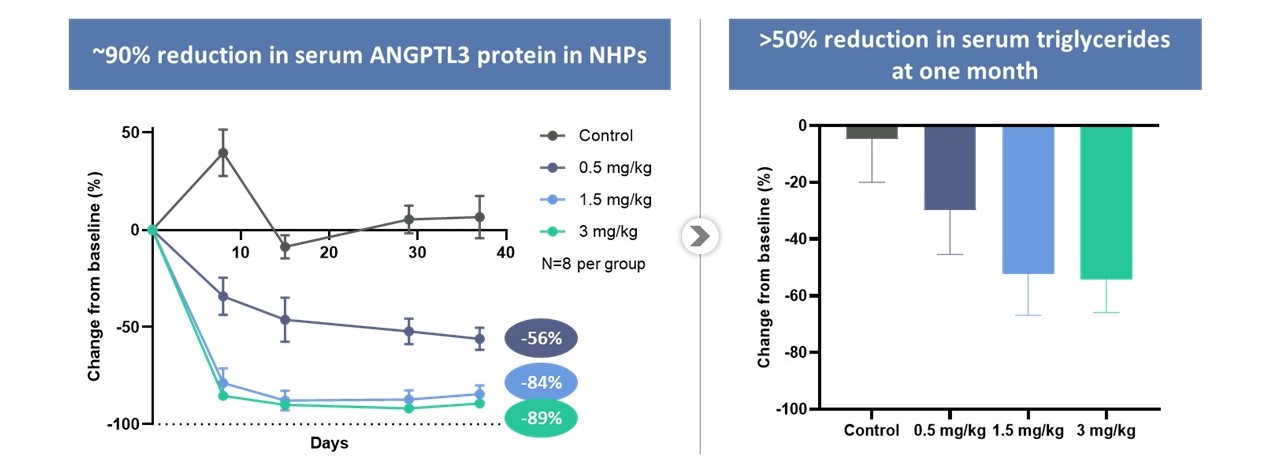
我々の首席調査員は体内にあるCTX 310計画では,現在INDイネーブル研究を行っており,アンギオゲニン関連蛋白3(Angptl 3)をコードする遺伝子の心血管疾患の治療と予防を目指している。Angptl 3はリポ蛋白質リパーゼ(LPL)と呼ばれる酵素を抑制することにより脂質代謝に重要な役割を果たしている。LPLはトリグリセリドに富むリポ蛋白質を分解する主要な酵素であり、例えば乳糜粒、極低密度リポ蛋白(VLDL)と低密度リポ蛋白質である。これらのリポ蛋白質のLPL加水分解を阻止することにより,Angptl 3の活性は循環中トリグリセリドレベルを増加させた。アンジオテンシン変換酵素を妨害することによるAngptl 3の発現抑制Angptl 3遺伝子はLPLの発現を増加させ,トリグリセリドに富むリポ蛋白と低密度リポ蛋白−Cを低下させた。この機序は自然病歴研究により検証されており、Angptl 3天然機能喪失変異を有する個体はトリグリセリドレベルが低く、低密度リポ蛋白レベルと低い冠状動脈疾患リスクを有するためである。CTX 310は,Cas 9をコードするメッセンジャーRNAとLNPを介して伝達されるAngptl 3に対する誘導RNAからなり,Angptl 3遺伝子を破壊することでこの効果を要約することを目的としている。CTX 310は非ヒト霊長類動物(NHP)のAngptl 3蛋白レベルを90%近く低下させ、血清トリグリセリドを50%以上低下させることが証明された。
CTX 310治療後の血清Angptl 3蛋白は約90%低下し、NHP患者の血清トリグリセリド低下>50%を招いた
CTX320 – Lp(a)
私たちの2回目の調査は体内にあるCTX 320プロジェクトは、心血管疾患に関連する別のタンパク質:Lp(A)を対象としている。Lp(A)は1種のリポ蛋白であり、1種の低密度リポ蛋白様粒子からアポリポタンパク質(A)或いはアポリポタンパク質(A)と呼ばれるタンパク質に共有結合する。Lp(A)は血液中でコレステロールを輸送し、高度な粥状動脈硬化作用を有する。それは大動脈弁内層と循環器系の他の領域の細胞外基質成分を浸透し、結合することができ、炎症と脂肪沈着を増加させ、時間の経過に伴い、これは大動脈弁機能の低下とその他の心血管疾患の深刻な症状を招く。Lp(A)はCVDの独立危険因子である。高濃度のLp(A)および高Lp(A)濃度に関する遺伝的変異は心血管疾患に関与している。Lp(A)レベルが50 mg/dLより高いことは大動脈弁石灰化疾患(AVCD)と直接関連している。米国では20%もの成人のLp(A)レベルが50 mg/dLを超え,100万人を超える米国成人がAVCDを患っている。また,家族性高コレステロール血症患者の30%のLp(A)レベルが上昇した。これまでLp(A)低下療法は得られていない
25
アメリカ食品医薬品局です。CTX 320はRNA標的ガイドからなるLPAApo(A)をコードする遺伝子とCas 9をコードするメッセンジャーRNAはLNPを介して伝達される。アポリポタンパク質(A)レベルを下げることによって、CTX 320は血漿Lp(A)レベルを著しく下げることができ、臨床前データにより、CTX 320治療はNHP患者のLp(A)レベルを90%以上低下させることができる。
高コレステロール血症
高コレステロール血症の定義は低密度リポ蛋白−Cであり,“悪いコレステロール”とも呼ばれ,130 mg/リットルを超え,心臓病や脳卒中のリスク増加に関与している。高コレステロール血症の中で、高レベルの低密度リポ蛋白は血管に蓄積し、粥状動脈硬化を招く。治療の目標は低密度リポ蛋白レベルを100 mg/リットル以下に低下させることであり、最終目標は70 mg/リットルであるが、一部の患者は既存の手段でこのレベルの低下を達成できない。低密度リポ蛋白レベルが200 mg/dLより高い患者は家族性高コレステロール血症(FH)を有すると考えられている。FH患者は食事と生活様式以外に、1つ以上の遺伝子変異による疾患がある。FH患者は低密度リポ蛋白-Cを有効に代謝できず、循環低密度リポ蛋白レベルが高く、ある情況下で1000 mg/リットルを超える。FHは突然変異状態によりハイブリッド型とホモ接合型家族性高コレステロール血症(HeFHとHoFH)に分類される。HoFH患者の表現型は最も深刻であり、低密度リポ蛋白-Cレベルは通常400 mg/dLを超える。HoFH患者は通常生命早期に心血管疾患を有しており,治療しなければ平均寿命は33歳である。HoFHの罹患率は20万から100万成人あたり1人であった。
高トリグリセリド血症
高トリグリセリド血症の臨床定義はトリグリセリドレベルが150 mg/dLより高いことである。最も深刻な患者のレベルは2000 mg/リットルを超えるかもしれない。高トリグリセリド血症は心血管疾患や急性膵炎に関与している。低密度リポ蛋白質コレステロールと同様にトリグリセリドレベルも食事やライフスタイル選択の影響を受け,一般療法で治療を行っている。しかし、米国では300万人以上の成人が深刻な高トリグリセリド血症(SHTG)を患っている。家族性乳糜粒微粒血症症候群(FCS)と多因子乳糜粒微粒子症症候群(MCS)を含む遺伝性疾患は自発性甲状腺機能低下を引き起こすことが知られている。FCS/MCSはHoFH/HeFHと類似している.FCSは唯一の真の単遺伝子高トリグリセリド血症であり、トリグリセリドが885 mg/dLを超える極端なレベルと関係がある。米国とEUでは,FCSの罹患率は20万人から30万人に1人がFCSを患っている。MCSは本質的に多遺伝子であり、これは疾患をもたらす遺伝的基礎が個人によって異なることを意味し、臨床的にはトリグリセリドレベルが150~885 mg/dLであると定義される。MCSの罹患率は600から1000人に1人である。
Vivo計画の他の内容は
CTX 310とCTX 320に基づいて、多くの早期調査があります体内にある肝臓における遺伝子破壊を利用してまれでよく見られる疾患を治療する計画。CTX 330が含まれていますPCSK 9これはよく知られている心血管疾患の標的であり,自然ヒト遺伝学や他の治療法からの強力なデータを有している。また、血友病Aを含む肝臓遺伝子修復に焦点を当てた項目があります。肝臓のほかに、造血幹細胞、中枢神経系、他の組織に対する項目もあります。例えばCapsidaと協力して開発しています体内にある筋萎縮性側索硬化症(ALS)とFriedreich‘s運動失調の遺伝子編集療法。Capsidaの高スループットAAV工学プラットフォームは特定の組織タイプに対して最適化されたカプシドを生成し、目標疾患とは無関係な組織と細胞タイプの形質導入を制限し、それによって潜在的に著者らの遺伝子編集研究療法の活性と耐性を高めることを目的としている。そのため、私たちの技術の結合はこれらの壊滅的な神経変性疾患に一流の治療方法を提供することができる。
血友病A
血友病Aは稀な、典型的なX連鎖劣性出血性疾患であり、凝固蛋白VIII(FVIII)の不足或いは無機能によって引き起こされる。血友病Aは最もよく見られる血友病タイプであり、血友病総人口の80%-85%を占め、全世界に90万人があり、その中に4-10000名の男児に1名がいる。血友病A患者の中で、機能性FVIII活性不足により有効な凝血機能が不足し、患者は鬱傷と腫脹しやすく、受傷後、手術後の出血時間が延長し、或いは傷口癒合前に繰り返し出血し、中、重度血友病患者に自発性出血が出現する可能性がある。
疾患の重症度は従来,血液中のFVIIIの残留量によって定義されており,軽度は>5−40%,中等度は1−5%,重度は
26
現在、血友病Aに対する看護標準は血漿由来或いは組換え凝固因子濃縮物を使用して暴走出血を防止することを含む。いくつかの遺伝子療法が臨床試験を行っており,その多くは機能的複製を提供することを目的としているF8AAVベクターを用いて遺伝子を標的細胞に導入した。遺伝子療法の一つであるRoctavianは2022年8月にEMAの条件付き承認を得た。しかし、AAVベクターが患者のゲノムに統合されていないため、形質導入細胞は分裂時に外体AAVを失う可能性があり、FVIIIレベルの低下を招き、治療効果は弱まる。さらに、AAVベクターの免疫原性は、多くの場合、患者が追加の治療注入を受けることができないことを意味する。逆に,CRISPR/Cas 9挿入機能を用いた血友病Aの治療のための候補遺伝子編集製品を開発しているF8 遺伝子を患者ゲノム中の特定の位置に導入する。このアプローチは,一度の治療法として,この場合に直接挿入することを目的としているF8 遺伝子は一生機能性FVIII蛋白の生産を招く。
CRISPR-X:遺伝子編集プラットフォームの潜在力をさらに放出します
著者らの現在のプロジェクトの組み合わせは重大な進展を得たが、著者らは引き続き革新し、CRISPR遺伝子編集のすべての潜在力を放出し、更に多くの患者に変革性の治療方法をもたらす必要があることを認識した。2022年、私たちはCRISPR-Xという新しい早期研究チームを設立し、革新研究に専念し、次世代遺伝子編集モデルを開発した。CRISPR-Xは、多くの細胞において低効率で発生する相同配向修復を必要とすることなく、またはウイルス伝達DNAテンプレートを必要とすることなく、全遺伝子補正と挿入を実現する技術に集中しており、これは毒性リスクと技術的挑戦をもたらす。これらの技術は全RNA遺伝子校正、DNAの非ウイルス伝達及び斬新な編集と挿入技術を含む。これらの努力は、RNA選択、目標上および目標外評価、および多重化を指導するなど、我々の既存のプラットフォーム能力を補完する。
Vertex協力計画
私たちはすでにDuchenne筋ジストロフィー(DMD)、強直性筋ジストロフィー1型(DM 1)、嚢胞性線維症(CF)のような他の疾患領域で私たちのいくつかのプロジェクトと協力した。我々はすでにVertexとこの3つのプロジェクトについて協力合意しており,Vertexはまれな疾患領域のグローバルリーダーであり,CF分野では広範な専門知識を有しており,DM 1を共同開発·共同商業化する製品の選択権を保持している。我々のCRISPR/Cas 9遺伝子編集技術はDMD、DM 1とCFの処理に非常に適しており、これらのすべての疾患は大量の患者群があるが、医療需要はまだ満たされていないと信じている。
杜氏筋ジストロフィー(DMD)
DMDはX連鎖劣性遺伝病であり、dystrophin遺伝子突然変異によって引き起こされ、dystrophin蛋白欠損を招く。筋ジストロフィー蛋白は筋肉繊維機能において重要な構造作用を果たしているため、筋肉細胞にこの蛋白が不足することは深刻な細胞損傷を招き、最終的に筋肉細胞の死亡と繊維化を招く。このような病気を患っている患者は筋肉退化、活動能力喪失、早死を経験する。DMDは最も一般的な深刻な遺伝病の一つであり、全世界の3300名の男児に1名がDMDを発生する。現在,米国には2つの承認されたDMD治療の疾患修飾療法があり,1つはエクソン51をスキップ可能なdystrophin遺伝子変異が確認された患者,もう1つはエクソン53をスキップ可能なdystrophin遺伝子変異が確認された患者である。これらの突然変異はそれぞれ約13%と8%のDMD群に影響した。
強直性筋ジストロフィー1型(DM 1)
DM 1は常染色体遺伝病であり、非コード領域におけるCTGトリヌクレオチド反復配列の拡張によって引き起こされるDMPKジーン。この疾患は骨格筋と平滑筋、及び他の器官系、例えば眼、心臓、内分泌系と中枢神経系に影響する。DM 1の臨床所見には軽から重への連続過程がある。これらの表現型により、DM 1は軽度、経典型、先天性の3つのいくつかの重複した形式に分類される。軽度DM 1を有する患者の寿命は正常であり,通常白内障が発生し,軽微な持続性筋収縮や筋強直を経験する。典型的なDM 1を有する患者はよく筋肉無力と痩せ、筋硬直、白内障、通常心臓伝導異常があり、身体障害、寿命が短縮される可能性がある。先天性DM 1を有する患者は通常知的障害があり,通常出生時に低張力と重篤な全身性虚弱があり,呼吸不全と早期死亡を伴うことが多い。世界では約8,000人に1人がDM 1に感染している。現在のところこの潜在的疾患を治療するための治療法は承認されておらず,逆に,これまで多くの介入措置の目標はこの疾患に対する特定の症状であった。
嚢胞性線維症(CF)
嚢胞性繊維化は進行性疾患であり、嚢胞性繊維化膜貫通調節因子遺伝子突然変異によって引き起こされ、CFTR蛋白機能の喪失或いは低下を招く。CF患者は重要な臓器,特に肺,膵,胃腸に厚い粘液を産生する。そのため、慢性閉塞性肺疾患患者は慢性深刻な呼吸器感染、慢性肺部炎症、栄養吸収不良、進行性呼吸不全と早期死亡を経験する。2017年,米国でCFで死亡した年齢の中央値は31歳であり,多くの死亡原因は呼吸不全であった。 Cfは孤児疾患であり,米国やヨーロッパでは7万人を超える患者に影響を及ぼすと推定されている。Cf患者は生涯治療を受け,毎日複数の薬物を服用し,数時間の自己ケアを行う必要がある。それらは通常頻繁な入院が必要であり,肺移植さえ必要であり,生存期間を延長することができるが,治癒できない。
27
バイエル協力プロジェクト
私たちはまたいくつかの自己免疫疾患と眼科疾患の診断、治療または予防計画を研究している。これらおよび血友病A障害に対する上述した計画について、バイエルは、私たちと共同開発および共同商業化する2つの製品を選択することができ、または場合によっては、そのようなオプションの製品を独占的に許可することができる。
戦略的パートナーシップと協力
CRISPR/Cas 9に基づく療法を独立して開発し,現在と潜在的な将来の企業パートナーと協力する予定である。私たちは戦略的パートナーシップを私たちの戦略の核心的な構成要素と見なし、私たちの治療計画を支持する能力と資源を得ることができるようにした。我々は広範な戦略的パートナーシップを保ち,特定の疾患領域で遺伝子編集に基づく療法を開発している。
頂点.頂点
我々はすでにVertexと一連のプロトコルを締結しており、様々な目標に関連するいくつかの研究、開発、製造、商業化活動を考慮している。2015年10月以降、2017年および2019年に改訂された戦略的協力、オプションおよび許可協定、または2015年の協力協定、2021年4月に改正され、再記述された共同開発および商業化協定、またはA&R Vertex JDCA、および2021年4月に改訂された戦略的協力および許可協定、または2019年の協力協定が締結された。
2015年連携協定
2015年の協力協定によると、私たちは7500万ドルの前払いと引き換えに、CRISPR/CAS技術に関連するライセンスを取得するために、Vertexに技術およびオプションを提供することに同意した。2015年、2015年の協力協定を初歩的に締結する過程で、VertexはUSに3000万ドルの株式投資も行った。
2015年のVertex協力の最初のポイントは、CRISPR/Cas 9技術を用いて、遺伝子に基づくヘモグロビン疾患および嚢胞性線維症を治療する方法を発見し、開発することである。2017年,Vertexはヘモグロビン疾患計画の共同開発と共同商業化の選択権を行使した。ヘモグロビン疾患目標に関する事項はA&R頂点JDCAで管理されており,以下のとおりである。さらなる発見作業は特定の数の他の遺伝子標的に集中している。2015年の協力協定によると、Vertexは、私たちのいくつかのプラットフォームおよび背景知的財産権に基づいて、特定の数の協力目標に対する治療を独占的に許可する権利があり、これらの協力目標は、このような各協力目標のための治療薬を開発、製造、商業化、販売および使用するために、いくつかのプラットフォームおよび背景知的財産権の下で出現する。私たちは発見活動を担当しており、関連費用はVertexによって全額援助されている。
2019年10月,Vertexは2015年の協力協定でその残りの選択権を付与し,CRISPRに基づく遺伝子編集を用いて遺伝子による治療を開発するための他の3つの目標を独占的に許可した。これらの標的は嚢胞性繊維化膜貫通伝導調節遺伝子と2つの未開示の標的を含む。2015年の協力協定の条項によると、オプション行使に関連した3,000万ドルの前金を受け取り、4.1億ドルまでの開発、規制、ビジネスマイルストーン、および3つの目標の各目標の製品純売上高の1桁から低い特許権使用料を得ることが可能だ。マイルストーンと特許使用料支払いは、2015年の協力協定に規定されている特定の条件で減少することができる。これらの目標に対して,Vertexは独自にすべての研究,開発,製造,グローバル商業化活動を担当し,Vertexはこれらの目標に関連する製品を世界で開発·商業化する独占的権利を獲得している.2015年の協力協定の研究期限が満了し、Vertexは2015年の協力協定に従って追加の目標許可を得る権利を持つことができなくなった。
いずれも他方が実質的に違約した場合に2015年の協力協定を終了することができるが、特定の通知と救済条項を遵守しなければならない。便宜上、Vertexは、任意の製品が発売承認される90日前の書面通知および製品が発売承認されてから270日後の通知後に2015年の協力協定を随時終了する権利があります。Vertexが私たちのいかなる特許権に挑戦すれば、私たちはまた2015年の協力協定を終了することができる。
事前に終了しなければ、2015年の協力協定がVertexが2015年の協力合意に基づいて負担する支払い義務が満了するまで、2015年の協力協定は有効である。
共同開発協定
二零一七年十二月に,吾らはVertexとVertex JDAを締結し,これにより,Vertex JDAが指定したExacelおよびその他の候補製品の共同開発および共同商業化に同意した。2021年4月,吾らはVertexとVertex JDAの改訂と再記述に同意し,A&R Vertex JDCAを締結することにより,双方は(A)協力の管治アーキテクチャを調整し,各者のこのアーキテクチャ下での責任を調整することに同意した,(B)輸出について双方間の純利益および純損失分配を調整するだけであり,(C)独占許可(我々のものに限定されている)である
28
特定の活動を行う権利を保持するVertexは、指定された候補製品およびこのプロトコルに従って研究、開発、製造、および商業化される可能性のある製品(Exexを含む)に関連するいくつかの知的財産権を有する。
他の事項以外に、A&R頂点JDCAは、以下に関連する規定を含む
活動を治めるそれは.私たちとVertexは、これまでに設立された連携戦略グループと、そのグループによって構築されたすべてのワーキンググループを解散し、(I)高いレベルの監視を提供するための共同監督委員会と、(Ii)そのような活動が完了するまで、フォーラムの計画、議論、および共有のための移行委員会を提供する委員会とを設立した。すべての新しい委員会はCRISPRとVertexからの同数の代表を持っている。A&R Vertex JDCAは,このようなプロトコルの条項や条件に基づいて,Vertexは指定候補製品や製品(Exacelを含む)に関するすべての研究,開発,製造,商業化活動を世界的に行う権利があるが,我々が何らかの活動を行う権利を守る必要があると規定している.私たちは双方が別の合意がなければ、このような活動のいくつかの側面に観察者として参加し続け、ある程度そうするつもりだ。
財務用語それは.2021年第2四半期、A&R Vertex JDCA計画が完了した取引に関連して、Vertexから9億ドルの前金を受け取りました。また、VertexがFDAや欧州委員会から初期候補製品の初上場承認を受けた後、一度に2億ドルのマイルストーン支払いを受ける資格がある。A&R Vertex JDCA項でA&R Vertex JDCAで指定されたすべての候補製品と製品に関する純利益と純損失(適用すれば)は我々とVertexが折半すべきである.輸出のみでは,2021年7月1日現在,A&R Vertex JDCA項で初期共有製品(すなわちExacel)に関する純利益と純損失(適用すれば適用)は我々とVertexが折半し,2021年7月1日からA&R Vertex JDCA項で発生した純利益と純損失(適用すれば適用)はCRISPRに40%,60%がVertexに割り当てられる。また,A&R Vertex JDCAは,Exexcel計画上の支出が指定された金額を超えた場合に,そのスケジュールでのコストの一部を遅らせることを許可している.いずれの繰延金額もVertexにしか支払いできず,Exacel計画の将来の収益性の相殺として,支払すべき金額の上限は毎年指定された最高額である.
端末.端末それは.いずれも、他方が実質的に違約した場合にA&R Vertex JDCAを終了することができるが、特定の通知や救済条項を遵守する必要がある場合、またはVertexの場合、指定された破産、クリア、または同様の状況の影響を受ける場合がある。A&R Vertex JDCAによってチャレンジャーに付与された任意の特許の有効性または実行可能性に疑問を提起する任意の訴訟またはプログラムに他方が開始または参加した場合、いずれの当事者もA&R Vertex JDCAを終了することができる。Vertexもあらかじめ書面で通知した後,便宜上A&R Vertex JDCAを終了する権利がある.
未治癒の重大な違約により一方がA&R Vertex JDCAを終了する権利がある場合には,A&R Vertex JDCAの効力を保持することを選択し,その違約側がその未治癒の重大な違約に関連する製品について脱退権利を行使したとみなされ(以下に述べる),違約側に支払うべき特許権使用料は指定された割合で減少する。
選択脱退権それは.A&R Vertex JDCAにより,いずれも候補製品開発の予定時点以降,候補製品の開発からの撤退を候補ごとに選択することができる.このような選択脱退の場合、退出を選択した側は、その候補製品に関する純利益と純損失を共有しなくなり、逆に、その製品が商業化されていれば、撤退を選択した側は、その製品の純売上高から高い単一から中年までの印税を得る権利がある。
2019年連携協定
2019年6月、Vertexと2019年の協力協定に署名し、合意に基づき、VertexとDMDおよびDM 1を治療する製品を協力して開発し、商業化することに同意しました。私たちとVertexは2021年4月に2019年の協力協定を改訂しました。
“2019年連携協定”には、他にも、以下に関連する条項が含まれています
統治するそれは.私たちとVertexは、2019年の協力協定でカバーされている活動に対して高いレベルの監督と調整を提供する共同諮問委員会を設立します。
発展と商業化それは.2019年の協力協定では、Vertexは開発と商業化活動を担当し、私たちの選択に応じて、指定された行使期間内に行使し、DM 1を治療する製品を共同開発し、共同商業化することができると規定されている。
財務用語それは.2019年の協力協定の締結について、Vertexから1.75億ドルの前金を受け取りました。私たちはVertexから合計7.75億ドルまでのマイルストーン支払いを受ける資格があります。これは、予定開発とビジネスマイルストーンを実現する製品の数とタイプに依存します。私たちも低い桁から低い二桁の製品販売から印税を受ける資格があります。
共同開発と共同商業化オプションそれは.DM 1を共同開発·共同商業化して治療する製品を選択すると、Vertex社DM 1の研究開発費の50%(50%)を精算する
29
このような費用の50%(50%)を負担するだろう。DM 1の開発および商業化活動に関連するさらなるマイルストーンまたは特許権使用料の支払いの代わりに、このような製品の販売利益の50%(50%)を獲得し、すべての損失の50%(50%)に責任を負う。
端末.端末それは.いずれも他方が実質的に違約した場合に2019年の協力協定を終了することができるが、特定の通知と救済条項を遵守しなければならない。Vertexが2019年の協調プロトコルに従ってVertexに付与された任意の特許の有効性または実行可能性に疑問を提起する任意の行動またはプログラムに開始または参加した場合、2019年の協調プロトコルを終了することもできる。Vertexはまた、私たちが破産したり、債務を返済しない場合に2019年の協力協定を終了したり、書面通知を出した後にいつでも協力協定を終了することもできます。
治癒していない重大な違約によりVertexが2019年の連携協定を終了する権利がある場合、Vertexは2019年の協力協定を有効に維持し、違約製品に対して支払うべき適用版税を指定パーセント削減することを選択することができます。
バイエル
2019年12月、私たちはバイエルとCRISPR/Cas 9遺伝子編集療法を発見、開発し、それを商業化し、ある疾患の遺伝的原因を治療するために、バイエルとの合弁企業を終了したオプション協定、すなわち2019年オプション協定を締結した。2019年オプション協定によれば、バイエルは、いくつかの自己免疫疾患、眼疾患、または血友病A疾患の診断、治療または予防のための2つの製品を共同開発および共同商業化するために、(将来のイベント定義の特定のトレーニング期間内に行使することができるが、いずれの場合も2019年オプション協定の発効日後5年を超えてはならない)オプションを獲得した。もしバイエルが共同開発と共同商業化製品を選択すれば、双方はその製品について交渉し、共同開発と共同商業化合意、あるいは共同商業化協定を達成することになり、バイエルは私たちが将来その製品のために発生する研究開発コストの50%を負担する。バイエルはこのような製品の販売からすべての利益の50%を獲得し、すべての損失の50%に責任を負う。
バイエルが共同開発と共同商業化製品の選択権を行使することを選択した場合、バイエルは私たちに2000万ドル、またはオプション支払いを一度に支払い、双方がオプション製品に関する共同商業化協定に署名すると、このお金は返却できないだろう。2019年のオプション合意に基づき、バイエルが初めてオプションを行使して1回のオプション支払いのみを支払います。
さらに、バイエルがオプション製品に対してその選択権を行使し、および/または共同商業化協定に署名した後、共通商業化協定の発効日から発効日までの3ヶ月の周年日または共同商業化協定の90日間の交渉過程において、より早い者を基準とした期間内に、バイエルは、そのオプション製品の開発と商業化の独占的許可について交渉する権利がある。バイエルがその権利を行使する場合、双方は双方が同意した条項に従ってそのオプション製品について独占許可協定を締結する。また、当該等のオプション製品について支払われるオプション支払いは、当該等の独占許可又は2019年オプション協定と締結された任意の他の独占ライセンスに基づいて満期になって支払われる金額に計上される。
いずれも他方が実質的に違約した場合に2019年オプション合意を終了することができるが、特定の通知と救済条項を遵守しなければならない。バイエルが任意のCRISPR特許の有効性または実行可能性に疑問を提起する行動または訴訟に開始または参加する場合、2019年オプション協定を終了することもでき、この特許は、2019年オプション協定のテーマである製品の研究、開発、製造、または商業化に必要または有用である。バイエルは、私たちが破産したり、債務を返済しない場合に2019年のオプション合意を終了したり、便宜上、書面で通知した後に随時終了したりすることもできます。
以上,我々の戦略プロトコルの記述はすべてこのようなプロトコルの全文に基づいて保持されており,これらのプロトコルのコピーは本年度報告の証拠品として10-K表形式でアーカイブされている.
知的財産権
私たちは、私たちの遺伝子編集技術および既存および計画における治療計画をカバーする特許権利(内部開発でも第三者から許可されていても)を求め、維持し、守り、守ることによって、私たちの業務に重要なビジネス的意義を持つと考えられるノウハウ、発明、ノウハウ、および改善を保護し、強化するために努力している。私たちはまた、私たちの業務において特許保護から保護されているか、または特許保護に適していないと考えている態様を保護し、持続的な技術革新と許可内の機会を求めて、遺伝子編集分野における当社の独自の地位を発展、強化、維持するために、ビジネス秘密保護および秘密保護協定に依存して、私たちのノウハウを保護しています。また,孤児薬物指定,データ独占性,市場独占性,特許期間延長(関連する場合)によって得られる商標保護,著作権保護,規制保護にも依存している。私たちの成功は、私たちの技術のために特許および他の独自保護を獲得し、維持する能力、私たちの知的財産権を擁護し実行する能力、および効果的かつ強制的に実行可能な特許および第三者の固有の権利を侵害することなく運営する能力に大きく依存するだろう。私たちも
30
私たちのオフィスの物理的なセキュリティと私たちの情報システムの物理的および電子的なセキュリティを維持することで、私たちのデータ、ノウハウ、ビジネス秘密の完全性とセキュリティを保護します。
Charpentier博士からの権限内知的財産権
2014年4月、私たちは、Charpentier博士との独占的な許可に基づいて、我々の遺伝子編集プラットフォーム技術の様々な態様、例えば、物質の組成(例えば、CRISPR/Cas 9システム)およびCRISPR/Cas 9システムを使用した遺伝子編集を含む使用方法を含むグローバル特許との組み合わせのいくつかの権利を付与した。この世界的な特許の組み合わせを“特許組合せ”と呼ぶ.例えば、これまで、この特許組み合わせは、米国、イギリス、カナダ、ドイツ、ヨーロッパ、日本、中国、インド、ウクライナ、ニュージーランド、シンガポール、オーストラリア、メキシコ、チュニジア、香港、イスラエル、ペルー、フィリピン、南アフリカで付与または許可された95件以上の特許と、米国、ヨーロッパ、カナダ、メキシコ、オーストラリアおよび中米、南アメリカ、アジアおよびアフリカの他の選択された国および地域で行われている特許出願を含む。本ライセンスは、医薬品および生物学的製品、ならびに任意の関連する随伴診断のようなヒト疾患、障害または状態を治療または予防するための治療製品に限定される。本ライセンスの詳細については、ご参照くださいビジネスライセンス契約−Dr.CharpentierとのCRISPRライセンスの締結.”
チャペンティエ博士に加えて、特許組合は、カリフォルニア大学、カリフォルニア大学、ウィーン大学、またはウィーン大学に自分の権利を譲渡する発明者をリストしている。カリフォルニアの権利は、ハワード·ヒューズ医学研究所とアメリカ政府を含む、その研究スポンサーのいくつかの圧倒的な義務に支配されている。トナカイ生物科学社は,カリフォルニア州とウィーンの独占特許権を有しているが,非営利エンティティが研究や教育目的に発明を利用することを可能にする権利を保持していると報告している。Intellia治療会社またはIntellia治療会社は、Cariouのある分野でのこのような権利の独占的ライセンスを持っていると報告している。カリフォルニア州のCharpentier博士とウィーンのCVC Groupを総称して“CVC Group”と呼ぶ.私たちは米国特許商標局(USPTO)と欧州特許庁(European Patent Office)で特許の組み合わせに関する準訴訟、当事者間の行政訴訟に直面している。これらの訴訟のリスクに関するより多くの情報は、参照されたい“リスク要因−知的財産権に関するリスク−.”
2016年12月15日、私たちはカリフォルニア州ウィーンのCharpentier博士、Intellia Treeutics、Cariou、ERSゲノム有限会社、および私たちの完全子会社TRACR血液学有限会社(TRACR)と譲渡、許可、共同所有権、および発明管理協定(IMA)を締結した。IMAによると、カリフォルニア州とウィーンはCharpentier博士が米国と世界的な範囲で私たち、TRACR、およびERSにCRISPR/Cas 9知的財産権を付与する権利に基づいてCharpentier博士に追跡的に同意する。IMAはまた、共同所有者による我々、TRACRおよび他の被許可者に付与された再許可の遡及同意、彼らが将来付与される可能性のある再許可の予想同意、ある当事者の以前の譲渡の遡及承認、および他の事項に加えて、(I)特許維持、弁護および起訴における当事者の誠実な協力、(Ii)コスト分担スケジュール、および(Iii)第三者主題特許およびCRISPR/CAS 9知的財産権のいくつかの不利なクレーム者が発生した場合の通知および調整を規定する。双方の当事者が事前に終了しない限り、IMAは、遺伝子編集技術に基づく特許の最終期限または最後の基本特許出願が放棄された日の遅い日まで有効になり続けるであろう。米国や世界の他の管轄地域における共同所有権の影響に関するさらなる情報は、“を参照されたい”リスク要因—私たちのコア遺伝子編集技術を保護する知的財産権は共通して所有されていますが、私たちの許可証はその中の一人の共通の所有者だけから来ています。これはアメリカと他の管轄地域での私たちの権利を深刻に制限しています.”
CRISPRが持つ知的財産権
特許の組み合わせに加えて、多くの特許シリーズが含まれており、多層保護を提供することを目的としたCRISPR/Cas 9技術と開発計画の重要な側面をカバーしている幅広い知的財産権を持っている。これらの特許シリーズは、我々の開発計画(例えば、物質の組成、使用方法、製造プロセス、用量および処方)、遺伝子編集におけるCRISPR/CAS 9システムの使用および改善(例えば、ヌクレアーゼおよび単一または修正された誘導RNAを含む成分系の改善)、タンパク質/核酸複合体およびRNAを細胞に供給する技術(例えば、改善されたウイルスベクターシステムおよび自己不活化システム)、ならびに幹細胞ベースの治療に関連する技術を含む。
全体的に、私たちの知的財産権には、米国、中国、ヨーロッパ、南アフリカの100以上のアクティブな特許家族および40件以上の付与または許可された特許と、米国、ヨーロッパ、オーストラリア、カナダ、中国、日本、メキシコと中米、南米、中東、アジア、アフリカの他の選択された国で出願されている特許が含まれている。これらの特許ファミリーによって付与された特許および最終的に発行可能な任意の他の特許は、適用可能な特許期間の延長を含まずに2033年から満了する予定である。
我々の米国商標権は、コバルト、CRISPRX、CRISPR Treeutics、CRISPR TX、CTX 001、CTX 130、VCTX 210およびVCTX 211のような20(20)以上の係属中の出願を含み、CRISPR Treeutics、CRISPR ateuticsロゴおよびCTX 110を含む7つの米国登録を含む。私たちの国際商標産業には、ドイツで処理されるべきCRISPR治療出願と、イギリス、イタリア、スペイン、ビホロでの4つの登録、ブラジル、ビホロ、ドイツ、香港、イタリア、南アフリカ、スペインにおける12件のCRISPR治療および設計の登録、およびブラジル、ビホルー、ドイツ、香港、イタリア、南アフリカ、スペインでの12項目のCRISPR治療および設計の登録を含む複数の処理される出願および登録が含まれている
31
香港と南アフリカです。CTX 112はEU、スイス、イギリスを指定し、CTX 131はEU、スイス、イギリスを指定し、CRISPR治療マーカーはカナダ、スイス、日本、韓国、メキシコ、ロシア、シンガポール、イギリスを指定する6つの国際登録があります。
特許譲渡協定
2014年11月,Charpentier博士,Ines Fonfara博士,ウィーンと特許譲渡協定である特許譲渡協定を締結した。特許譲渡プロトコルによれば、Charpentier博士、Fonfara博士、およびウィーンは、標的DNAまたは切断DNAにおけるそれらの使用を含む、追加のCRISPR/TRACR/Cas 9錯体および使用方法を含む、特定の物質組成に関連する一連の特許出願のすべての権利を私たちに譲渡する。
私たちに割り当てられた特許権の対価格として、前金の支払い、別の国での米国研究新薬申請または同様の出願の提出からのマイルストーン支払い、最低年間特許権使用料、その製造、使用、販売、または輸入特許権によってカバーされる製品の純販売から、より低い1桁の特許権使用料、およびより低い1桁のパーセントの許可収入を徴収することに同意する。
我々は、割り当てられた特許権によってカバーされた製品を製造、使用、販売または輸入するために、商業的に合理的な努力で規制部門の承認を得る義務があり、2021年11月までに商業的に合理的な努力を使用して米国研究新薬出願(または主要市場国/地域での同等の出願)を提出する義務に限定されない。
許可協定
Charpentier博士にCRISPRライセンスを発行します
2014年4月、私たちは私たちの共同創始者の一人であるCharpentier博士と許可協定またはCharpentier許可協定を締結し、特許組み合わせにおけるCharpentier博士の共同所有権権益に基づいて、薬物や生物製剤などの治療製品を研究、開発、商業化することができ、任意の関連する随伴診断を使用して、ヒト疾患、疾患または疾患の治療または予防に使用することができる独占的な許可を得たが、私たちはこれをCRISPR領域と呼ぶ。この許可は独占的であり,チャペンティエ博士に対しても同様であり,ただ彼女は譲渡不可能な権利を保持しており,その技術を自分の研究目的に利用し,学術や非営利パートナーと研究協力することができる。独占許可はCharpentier博士が特許出願中の利益の下でのみ付与され,独占許可はいかなる他の共通所有者の利益の下でも付与されない。さらに、Charpentier許可プロトコルは、CRISPR分野に関連する治療製品を研究、開発、生産、商業化および販売するための再許可を含む再許可の権利を、TRACRがDr.Charpentierとの許可に基づいて開発する任意の知的財産権を含む、我々が独占的に、世界的に免除する再許可を付与する。逆に,Charpentier博士に独占的な許可を与え,Charpentier博士の許可に基づいて開発した任意の知的財産権をTRACRに再許可し,鎌状細胞疾患や地中海貧血に限定されないヒトのヘモグロビン疾患の治療·予防に用いることが義務付けられている。
Charpentierライセンス契約の条項によると,許可の代償として,Charpentier博士は技術譲渡費,非実質的な年間維持費,臨床試験開始後に満期となる非実質的マイルストーン支払い,特許製品の純売上高の低い桁数パーセントの特許権使用料および再許可収入の低い桁数パーセントの特許権使用料を受け取った。私たちは、許可された治療製品を販売するために、商業的に合理的な努力を使用して規制部門の承認を得る義務がある。我々は2021年4月までに商業的に合理的な努力で米国研究用新薬申請(またはCRISPR分野での治療製品の主要市場国での等価物)を提出しなければならない。また,2024年4月までにCRISPR分野の治療製品の1つに米国研究用新薬申請(または主要市場国での同等の申請)を提出する商業的に合理的な努力をしなければならない。
事前に終了しない限り、特許者許可協定の有効期間は、各国に基づいて満了する、すなわち、その国の特許組み合わせの最後の有効な特許請求の満了時に満了する。私たちはCharpentier博士に60日間の書面通知を出した後、任意に合意を終了する権利がある。我々とCharpentier博士は,他方が90日の通知期間内に是正されなかった重大な違約であれば,90日間通知された場合にプロトコルを終了することができる.もし私たちが任意の特許の組合せの実行可能性、有効性、または範囲に疑問を提起すれば、Charpentier博士は直ちに許可プロトコルを終了することができる。
Charpentier博士が発行したTRACRライセンス
2014年4月,我々はCharpentier博士とライセンス契約を締結するとともに,TRACRとTracrの少数株主Charpentier博士は特許の組合せに基づいてライセンス契約やTRACRライセンス契約を締結した.TRACR許可プロトコルによれば、TRACRは、ヒトヘモグロビン疾患(鎌状細胞病および地中海貧血を含む)またはTRACR領域の治療および診断製品の治療および予防のための再許可の権利を含む独占的でグローバルな使用料許可を取得する。TRACRはまた、内部薬物研究を展開するために、再許可の権利を含む非独占的、世界的、免版税の許可を得ている
32
TRACR分野以外の治療製品と、TRACR分野に関連する治療製品を再許可、研究、開発、生産、商業化および販売する権利を含むグローバル範囲での、印税免除の再許可を含み、これらの製品は、CRISPRがCharpentier博士との許可の下で開発した任意の知的財産権を含む。逆に,TRACRはCharpentier博士に独占許可を付与し,TRACRとCharpentier博士が許可に基づいて開発した任意の知的財産権をCRISPRに再許可し,CRISPR分野に用いる.
ライセンスプロトコルによれば、TRACRは、ヒト疾患の予防または治療のための少なくとも1つの治療製品を研究、開発および商業化するために、商業的に合理的な努力を使用する義務がある。TRACRは,2021年4月までに商業的に合理的な努力でTRACR領域の治療製品に米国研究用新薬申請(または主要市場国の同等の申請)を提出しなければならない。また,TRACRは2024年4月までに商業的に合理的な努力でTRACR領域の治療製品に米国研究用新薬申請(あるいは主要市場国での同等の申請)を提出しなければならない。TRACRは独自にすべての臨床、規制、開発コストを負担している。
TRACR許可プロトコルによると、Charpentier博士はTRACRの商業化された各製品の非実質的な臨床と規制マイルストーン支払いを得る権利がある。TRACRはまた、その付属会社、またはその分ライセンシーによって製造された任意の承認された治療または診断製品の純売上の低桁パーセント印税、および従属許可収入の低桁パーセント印税をDr.Charpentierに支払うことを要求される。
事前に終了しない限り、ライセンス契約の期限は、各国/地域に基づいて満了し、その国/地域の特許組み合わせの最後の有効な権利要件が満了した時点で満了する。TRACRはCharpentier博士に60日間の書面通知を出した後,任意にプロトコルを終了する権利がある.TRACRおよびDr.Charpentierは、他方が90日の通知期間中に治癒する重大な違約行為がない場合、90日間通知された場合にプロトコルを終了することができる。TRACRが任意の特許権の実行可能性,有効性または範囲に疑問を提起すれば,Charpentier博士はただちに許可プロトコルを終了することができる.
技術を発揮する
私たちはまた他の多くの協力と許可協定を締結して、私たちのことを支持して補充します離体するそして体内にあるCRISPR/CAS 9を提供する技術を提供することに関連するプロトコルを含む態様離体するそして体内にある造血幹細胞や体内にあるHIV遺伝子編集療法を推進するための寄付と、私たちの免疫腫瘍学と再生医学細胞治療計画とプラットフォームを強化することが計画されている。例えば、我々はNkarta,Inc.と合意しており、ドナー由来の遺伝子編集されたCAR-NK細胞候補製品と、NKおよびT細胞に結合した2つの製品を共同開発および共同商業化している;Capsida BioTreateutics,Inc.が開発される体内にある工学AAVベクターを用いて提供した遺伝子編集療法は筋萎縮性側索硬化症とFriedreich‘s運動失調の治療に応用されている;モフェット癌センターとロスウェル公園総合癌センターは新しい標的に対して自己CAR T計画を推進する;MaxCyte Inc.on離体するヘモグロビン病と免疫腫瘍学プロジェクトにサービスを提供していますCureVac AGは特定の遺伝子構造と製造を最適化しています体内にあるKSQ Treeutics Inc.は著者らの同種異体免疫腫瘍学計画に知的財産権を提供する。
製造業
細胞や遺伝子療法の製造プロセスは複雑であり,プロジェクトや療法ごとにシステム,設備,施設,専門知識をカスタマイズする必要がある。2020年第2四半期に、私たちはマサチューセッツ州フレミンガムに私たち自身の細胞療法製造施設を設立し、私たちの候補製品とそのいくつかのコンポーネントを臨床および商業生産するための投資を発表しました。2021年第4四半期には、施設の建設を完了し、現在の良好な製造規範やcGMPに適合し、将来のヒト管理に適した細胞治療製品の供給を可能にするために必要な規制検証活動を進めている。同施設の敷地は約50,249平方フィート。
米国と海外での契約製造組織関係による外部製造能力に引き続き依存していきます。私たちはすでに第三者サプライヤーといくつかの製造と供給手配を達成して、私たちの候補製品とその部品の生産を支持しています。著者らは引き続き合格した第三者組織に依存してバルク化合物、配合化合物、ウイルスベクター或いは工学細胞を生産或いは加工し、IND活動と早期臨床試験を支持する予定である。開発された任意の化合物、担体、または工学細胞の商業的ロットが、FDAおよび他の法規に適合する施設およびプロセスで生産されることが予想される。製品開発過程の適切な時間に、私たち自身の製造施設を利用するか、引き続き第三者に依存して、私たちが開発に成功する可能性のある任意の製品の商業ロットを生産するかを決定します。
マサチューセッツ州フレミンガムでの私たちの製造能力に大きな投資を行い、第三者組織と協力してこれらのプログラムを推進し、将来的に商業化するために、私たちの遺伝子編集プログラムを開発することを期待し続けています。
33
また、候補製品が私たちのチャンネルで進展するにつれて、私たちのビジネス計画は変わるかもしれません。特に,我々のいくつかの研究計画は潜在的なより大きな適応を対象としている。データ,開発計画の規模,目標市場の規模,商業インフラの規模および製造需要は,米国,ヨーロッパ,世界の他の地域における我々の戦略に影響を与える可能性がある。アメリカやヨーロッパ以外の地域では、適切な状況で、将来的には、戦略的パートナー、流通業者、または契約販売力を利用して、私たちの製品の商業化を助けることを選択するかもしれません。場合によっては、私たちは私たち自身の商業インフラを建設することを考慮することができる。
競争
生物技術と製薬業界は、遺伝子編集、遺伝子治療と細胞治療領域を含み、その特徴は技術が迅速に進歩し、競争が激しく、そして知的財産権と独自製品を高度に重視していることである。私たちの技術、開発経験、科学知識は私たちに競争優位を提供してくれると信じていますが、私たちは現在、大型製薬、専門製薬とバイオテクノロジー会社、学術機関と政府機関、および公共および民間研究機関を含む多くの異なる源からの激しい競争に直面し、引き続き直面しています。その中のいくつかまたはすべての機関は私たちよりも資本や資源を獲得しやすいかもしれません。我々が最終的に商業化する可能性のある任意の製品については,任意の既存の療法や現在開発されている療法と競合するだけでなく,将来出現する可能性のある新しい療法とも競合するであろう。
我々は製薬,バイオテクノロジー,その他の関連市場で競争を展開しており,これらの市場では遺伝子編集,遺伝子療法,細胞療法を含むゲノム薬物をカバーする技術を用いて療法を創造している。また,我々の特定の研究開発計画に関連する分野で治療法の開発に取り組んでいる会社と競争している。
我々のプラットフォームと製品はCRISPR/Cas 9遺伝子編集技術を用いた療法の開発に重点を置いている。Intellia治療会社やEditas Medicine社を含むCRISPR/Cas 9遺伝子編集技術を用いた様々な適応の治療法の開発に専念している会社がいくつか知られている。また,いくつかの学術グループでは,CRISPR/Cas 9に基づく新しい遺伝子編集技術,例えば塩基編集やモチーフ編集が開発されており,これらの技術は治療開発において実用的な価値がある可能性がある。これらの技術に基づく療法の開発を求めている会社には,Beam TreateuticsとPrime Medicineがある。
TALEN、マクロヌクレアーゼ、ZFNなどの追加の遺伝子編集技術を用いて療法を開発している会社もある。これらの会社は270の生物会社、異遺伝子治療会社、Cellectis社、精密生物科学会社、Sangamo治療会社を含む。
いくつかの会社が我々の具体的な研究開発計画に関連した様々な分野で療法を開発していることも知られている。ヘモグロビン疾患については,これらの会社には,ビム治療会社,ブルーバード生物社,Editas Medicine社,黒鉛生物社,メルク社,ノワ製薬会社,ファイザー社,Sangamo治療会社がある。免疫腫瘍学では、これらの会社は270生物会社、アディケイト生物会社、異遺伝子治療会社、百時美施貴宝会社、Cariou生物科学会社、Cellectis社、世紀治療会社、運命治療会社、ギレド科学会社、伝説生物会社、ノワ製薬会社、ポセイダ治療会社、精密生物科学会社を含む。再生医学分野では,これらの会社にはBluerock Treateutics(2019年にバイエルに買収された),Sana Biotech,Semma Treateutics(2019年にVertexに買収)が含まれている。はい体内にあるこれらの会社にはAlnylam製薬会社,矢印製薬会社,BioMarin製薬会社,Intellia治療会社,Ionis製薬会社,Regeneron製薬会社,Verve治療会社がある。遺伝子編集は非常に活発な研究分野であり、CRISPRに関連したり、関係のない新しい技術が発見され、新しい競争が生じる可能性がある。これらの新技術はいくつかの応用においてCRISPR/Cas 9遺伝子編集よりも優れている可能性があり、他の遺伝子編集技術が製品開発において我々の技術よりも良いあるいは魅力的であるとは考えられないことは確信できない。例えば、Cas 9は、Cas 12 aまたはまだ発見されていない新規なCas酵素、または塩基エディタおよび一次エディタのような他のCRISPRタンパク質に劣る他のCRISPR関連ヌクレアーゼ変異体を決定することができる。
遺伝子編集は非常に活発な研究分野であり、CRISPRに関連したり、関係のない新しい技術が発見され、新しい競争が生じる可能性がある。これらの新技術はいくつかの応用においてCRISPR/Cas 9遺伝子編集よりも優れている可能性があり、他の遺伝子編集技術が製品開発において我々の技術よりも良いあるいは魅力的であるとは考えられないことは確信できない。例えば、Cas 9は、Cas 12 aまたはまだ発見されていない新規なCas酵素、または塩基エディタおよび一次エディタのような他のCRISPRタンパク質に劣る他のCRISPR関連ヌクレアーゼ変異体を決定することができる。
他の遺伝子編集療法または遺伝子または細胞療法からの競合に加えて、我々が開発する可能性のある任意の製品は、小分子、抗体またはタンパク質療法のような他のタイプの療法からの競合に直面する可能性もある。また,新たな科学的発見はCRISPR/Cas 9技術,あるいは全体的な遺伝子編集を招く可能性があり,劣悪な治療形態と考えられている。
34
また、私たちの多くの既存または潜在的な競争相手は、単独またはパートナーと協力しても、研究開発、製造、臨床前テスト、臨床試験、監督管理許可およびマーケティング承認を得た製品の面で、私たちよりも多くの財務資源と専門知識を持っている。製薬、生物技術、遺伝子と細胞治療業界の合併と買収はより多くの資源が私たちの少数の競争相手に集中する可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。もし私たちの競争相手が私たちが開発する可能性のあるどんな製品よりも安全で、より効果的で、副作用が少なく、より深刻ではなく、より便利で、より広い受容度とより高い販売率を開発し、あるいは私たちが開発する可能性のあるどの製品よりも安い製品であれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。また、私たちの競争相手が開発した技術は、私たちの潜在的な候補製品を経済的あるいは時代遅れにする可能性があり、私たちは私たちが開発する可能性のあるいかなる競争相手の候補製品もうまくマーケティングできないかもしれません。私たちのすべてのプロジェクトの成功に影響を与える重要な競争要素はそれらの有効性、安全性かもしれません, 利便性と精算の可用性。
私たちの現在のプロジェクトが現在計画されている臨床試験の適応に使用されていれば、遺伝子編集、遺伝子治療、細胞治療製品を含む現在開発されている他の製品と競争する可能性がある。現在開発中の他の関連製品との競争には,臨床試験地点,患者募集,製品販売の競争が含まれている可能性がある。また,遺伝子編集分野では緊張した研究や開発が行われているため,我々と我々のライバルを含めて知的財産権の構造が変化しており,競争は非常に激しい.将来的には知的財産権に関する重大な訴訟や、私たちが所有しているものや許可されていない他の第三者、知的財産権、独自の権利に関する訴訟があるかもしれない。例えば、048干渉、115干渉、および欧州野党手続きの議論を参照されたいリスク要因-知的財産権に関連するリスク-第三者が私たち、私たちのライセンシー、または私たちの協力者に提出した知的財産権侵害クレームは、私たちの製品発見と開発を阻害または延期する可能性があります。”
政府の監督管理
アメリカ連邦、州と地方の各レベル及びEUを含む他の国と司法管轄区の政府当局は他の事項を除いて、薬品の研究、開発、テスト、製造、品質管理、承認、包装、貯蔵、記録、ラベル、広告、販売促進、流通、マーケティング、承認後のモニタリングと報告及び生物製品を含む薬品の輸出入に対して広範な監督管理を行っている。アメリカ以外のいくつかの管轄区域でもこのような製品の価格設定が規制されている。米国や他の国や司法管轄区で上場承認を得る過程、その後適用される法規や法規、その他の規制機関の遵守には、多大な時間と財力が必要である。
アメリカの生物製品の許可と規制
米国では,公衆衛生サービス法(PHSA)や連邦食品,薬品·化粧品法(FDCA)とその実施条例に基づき,我々の候補製品は生物製品や生物製品として規制されている。製品開発プロセスのいずれかにおいて、非臨床試験、臨床試験、承認プロセスまたは承認後プロセスを含み、出願人が適用される米国の要求を遵守できない場合、研究の進行、規制審査および承認、および/または行政または司法制裁を遅延させる可能性がある。これらの制裁には、FDAが臨床試験の継続を許可することを拒否すること、係属中の出願の承認の拒否、免許の取り消しまたは免許の取り消し、承認の撤回、命名されていないまたは警告状、負の宣伝、製品のリコール、製品の差し押さえ、生産または流通の完全または部分的な一時停止、禁止、罰金、およびFDAまたは司法省または他の政府エンティティによって提起された民事または刑事調査および処罰を含むことができるが、これらに限定されない。
米国での新生物の販売と流通の承認を求める申請者は、通常、以下の各ステップを満足的に達成しなければならない
35
臨床前研究と探索性新薬応用
遺伝子治療候補製品を含む人体上で任意の生物候補製品をテストする前に、候補製品は臨床前テストを経なければならない。臨床前試験は製品の化学、調合と安定性の実験室評価、及び動物の治療効果と毒性潜在力の評価研究を含む。臨床前試験の進行と試験に用いる化合物調合は必ず連邦法規と要求に符合しなければならない。臨床前試験の結果および生産情報と分析データはIND申請の一部としてFDAに提出された。INDはFDAが受け取った30日後に自動的に発効し、それ以前にFDAが臨床試験の提案された製品または行為に対する懸念または問題に基づいていない限り、人類の研究対象が不合理かつ重大な健康リスクに直面することを心配して臨床保留を強制することを含む。この場合,INDスポンサーやFDAは臨床試験開始前にFDAの未解決の問題を解決しなければならない。
したがって、INDの提出は、FDAが試験開始を許可しないか、または試験を許可しないことをもたらす可能性があり、INDにおいてスポンサーが最初に指定した条項に従って開始することができる。FDAが最初の30日以内に、またはIND研究が行われている間のいつでも安全懸念または問題を提起した場合、安全懸念または規定を遵守しないことによる懸念を含む場合、一部またはすべての臨床的保留が実施される可能性がある。FDAが発表したこの命令は、提案された臨床研究を延期したり、進行中の研究の一時停止を招いたり、すべての未解決の問題が十分に解決されるまで、一部の臨床的に保留されている場合に研究を制限することができ、FDAは、調査を継続または再開することができるが、FDAによって許可された条項にのみ従うことができるように会社に通知する。これは計画中の臨床研究の適時な完成の重大な遅延や困難を招く可能性がある。
血中乳酸を支持するヒト臨床試験
臨床試験はGCP要求に基づいて、合格した首席研究者の監督の下で、研究製品候補を健康ボランティア或いは疾病患者に治療することに関連している。臨床試験は研究案に基づいて行われ,その中で研究の目標,組み入れと排除基準,安全性モニタリングのためのパラメータおよび評価すべき有効性基準が詳細に説明されている。各臨床試験の案と後続案修正案はINDの一部としてFDAに提出されなければならない。
米国国外での臨床試験のスポンサーが望ましいが,必要なくFDAの認可を得,INDによる臨床試験を行っている。米国でない臨床試験がINDに基づいて行われていなければ,スポンサーは設計と良好に行われた臨床試験のデータをFDAに提出し,BLAを支援することができ,臨床試験がGCPに従って行われていれば,FDAが現場検査で検証研究のデータを検証できる限り,FDAが必要であると考えている。
また,各臨床試験は,臨床試験を行う各機関のIRBが集中的または単独で審査·承認されなければならない。IRBは臨床試験設計,被験者のインフォームドコンセント,倫理的要因,被験者の安全などを考慮する。IRBの運営はFDAの規定に適合しなければならない。FDAまたは臨床試験スポンサーは、臨床試験がFDAの要求に従って行われていないこと、または被験者または患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由で臨床試験を随時一時停止または終了することができる。
36
同様に、1つの臨床試験が委員会の要求に従って行われない場合、または薬剤が患者に予期せぬ深刻な傷害を与えた場合、IRBは、その所在機関の臨床試験の承認を一時停止または終了することができる。臨床検査はまた広範なGCP規則とインフォームドコンセントの要求を満たさなければならない。さらに、いくつかの臨床試験は、データ安全監視委員会または委員会と呼ばれる臨床試験スポンサーによって組織された独立した合格専門家グループによって監督される。このグループは,計画的に研究を継続し,研究進行を変更したり,研究のあるデータへのアクセスに応じて,指定されたチェックポイントで研究を停止することを提案することができる.
米国で臨床試験が開始される前にFDAにINDを提出するほか,組換えや核酸分子の合成に関連するヒト臨床試験のいくつかは機関生物安全委員会(IBCs)の監督を受けなければならないことはNIHガイドライン“組換えあるいは核酸分子の合成に関する研究ガイドライン”に掲載されている。米国国立衛生研究院のガイドラインによれば、組換えおよび合成核酸は、(1)核酸分子が結合して生細胞中で複製可能な分子(すなわち、組換え核酸)、(2)化学的または他の方法で合成または増幅された核酸分子、または化学的または他の方法で修飾されたが自然に産生される核酸分子(すなわち、合成核酸)塩基対を含む分子、または(3)第(1)または(2)項に記載の分子を複製する分子を含む、と定義されている。具体的には,NIHのガイドラインによると,ヒト遺伝子転移試験の監督にはIBCによる評価と評価があり,IBCは地方機関委員会であり,組換えや合成核酸分子を用いた研究の審査·監督を担当している。IBCは、研究の安全性を評価し、公衆の健康または環境に対する任意の潜在的リスクを決定し、このような審査は、臨床試験開始前のいくつかの遅延をもたらす可能性がある。NIHガイドラインは強制的ではないが,関連研究がNIH組換えや合成核酸分子研究助成を受けた機関で行われているか,あるいはその助成によって行われていない限り,多くの会社や他のNIHガイドラインに拘束されていない機関は自発的にこれらのガイドラインに従っている。
臨床試験は通常3つの連続段階に分けて行われるが、これらの段階は重複或いは合併する可能性がある。承認された後に追加的な研究が必要かもしれない。
臨床試験結果を詳細に説明する進捗報告は少なくとも毎年FDAに提出されなければならないことが分かっている。スポンサー又はその代理人は、情報が迅速報告の条件を満たしていると判断した後、書面INDセキュリティ報告を受けてから15カレンダー日以内にFDA及び調査者に書面INDセキュリティ報告書を提出しなければならない。深刻で深刻な場合にはIND安全報告が必要です 他の研究や動物からの予期しない有害事象や体外培養テストにより、この薬物に接触した人は重大なリスクがあり、方案或いは研究者マニュアルに記載されている状況と比較して、深刻な副作用の疑いの発生率は臨床上どのような重要な増加があることが示唆された。またスポンサーは 事故、致命的、または生命に危害を及ぼす疑いのある副作用の情報を受け取った後、7つのカレンダー日にFDAに通知します。
場合によっては、FDAは候補製品のBLAを承認する可能性があるが、承認後の候補製品の安全性および有効性をさらに評価するために、スポンサーに追加の臨床試験を行うことが要求される。この承認後の試験は通常4期臨床試験と呼ばれる。これらの研究は、予期される治療適応患者の治療から追加の経験を得るために使用され、加速承認条例によって承認された生物学的製品の場合に臨床的利益を証明するために使用される。4期臨床試験での職務調査ができなかったことは,製品の承認撤回につながる可能性がある。
遺伝子治療製品ガイド
FDAは、遺伝子治療製品を、転写および/または翻訳転移による遺伝物質または宿主(ヒト)遺伝子配列を特定に変化させることによってその効果を調節する製品として定義する。遺伝子治療製品の例としては、核酸(例えば、プラスミド、インビトロ転写リボ核酸)、トランスジェニック微生物(例えば、ウイルス、細菌、真菌)、ヒトゲノム編集のための工学的部位特異的ヌクレアーゼ、および離体する遺伝子組み換えされたヒト細胞ですこの製品は細胞を修飾するために使うことができる体内にある細胞に転移したり離体する受取人を管理する前に。FDA内部では、生物製品の評価と研究センター(CBER)が遺伝子治療製品の監督管理を担当している。CBER内部では,遺伝子治療と関連製品の審査が治療製品事務室に統一されており,FDAはすでに細胞,組織,遺伝子治療を確立している
37
諮問委員会はその審査についてCBERに諮問意見を提供する。FDAとNIHは遺伝子治療案の開発と提出に関するガイドラインを発表している。
FDAは遺伝子療法に関するガイドラインに法的拘束力がないことを示しているが,我々が開発可能な任意の候補製品の承認を得るためには,これらのガイドラインを遵守する必要がある可能性が高いと考えられる。これらのガイドラインは、FDAが上述した各開発段階で考慮する他の要因を提供し、遺伝子療法の適切な臨床前評価と、INDアプリケーションに含まれるべき化学、製造、および制御情報と、INDまたはBLA適用をサポートする製品効力を測定するために正確な設計テストと、研究遺伝子療法に曝露された対象において、そのような影響リスクが高い場合の遅延副作用を観察するための措置とを提供する。さらに、FDAは、一般に、遺伝子治療に関連する潜在的遅延不良事象の対象をスポンサーに観察することを提案する。製品タイプによって、長期フォローアップは15年に達することができ、5年まで短くすることもできる。
CGMPとCGTPの要求に合致する
BLAを承認する前に、FDAは通常、製品を生産する1つまたは複数の施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に完全に適合していることを決定し、要求された仕様の下で製品が一貫して生産されることを保証するのに十分であると判断しない限り、申請を承認しないであろう。PHSAは,属性が正確に定義できない生物製品などの製品の製造制御の重要性を強調している。
遺伝子治療製品については,メーカーがCGTPに適合していなければ,FDAもこの製品を承認しない。これらの要件は、ヒト細胞、組織、および細胞および組織に基づく製品またはHCT/Pを製造するための方法、施設および制御のためのFDA法規において見つけることができ、HCT/Pは、ヒトレシピエント内に移植、移植、注入または転移するためのヒト細胞または組織である。CGTP要求の主な目的は,細胞や組織に基づく製品の製造方式の確保であり,感染症の導入,伝播,伝播を防止することを目的としている。FDAの規定はまた、組織機関がFDAに彼らのHCT/Pを登録し、適用した場合にスクリーニングとテストを通じてドナーを評価することを要求する。
製造業者や他の製品の製造·流通に参加する人、および製品、成分およびコンポーネントを提供する人は、米国市場の製品を供給するためにFDAおよびいくつかの州機関にその工場を登録し、同様の衛生規制機関に登録して、世界の他の市場の製品を供給しなければならない。米国および非米国の製造企業は、最初に生産過程に参加する際に、FDAおよび/または他の衛生規制機関により多くの情報を登録して提供しなければならない。登録されていない工場によって製造または輸入された任意の製品は、米国でも非米国でもFDCA下の誤ったブランドとされ、他の管轄区域の類似および他のコンプライアンス問題の影響を受ける可能性がある。機関はcGMPや他の法律の遵守を確保するために、政府当局の定期的な抜き打ち検査を受ける可能性がある。メーカーはまた、その工場に関する電子または実物記録の提供を要求しなければならないかもしれない。FDAまたは他の管理衛生規制機関の検査の延期、拒否、制限、または拒否は、製品が偽とみなされる可能性がある。
BLAの審査と承認
候補製品開発、臨床前試験および臨床試験の結果は、否定的または不明確な結果および積極的な発見を含み、BLAの一部としてFDAに提出され、その製品のマーケティング許可証の取得を要求する。BLAには広範な製造情報と製品組成に関する詳細な情報,アドバイスのラベル,使用料の支払いが含まれていなければならない.
FDAは、実質的な審査を可能にするために十分に完全であることを機関が決定したことに基づいて、出願を受け入れるのに十分であるかどうかを決定するために、出願を提出した後60日の間予備審査を行う。提出された申請が受け入れられると、FDAは申請の深い審査を開始する。FDAが“処方薬使用料法案”(PDUFA)によって達成された目標と政策に基づいて、FDAは、標準出願の予備審査を完了し、出願人に対応し、6ヶ月間にわたって出願を優先的に審査する10ヶ月の期間を有する。FDAは常にそのPDUFA規格と優先BLASの目標日を達成するわけではない。追加情報や明確化に対するFDAの要求はしばしば審査過程を大幅に延長するだろう。FDAが要求を出した場合、または出願人がPDUFA目標日の前の最後の3ヶ月以内に主要修正案の提出によって補足情報または提出中に提供された情報に関する明確化を提供した場合、審査プロセスおよびPDUFA目標日を3ヶ月延長することができる。
PHSAによれば、FDAが製品が安全で純粋かつ効率的であると判断し、製品を製造する施設が、その継続安全、純粋および有効性を保証するための基準に適合している場合、FDAはBLAを承認することができる。
FDAによる申請の評価および関連情報によれば、製造施設の検査結果、およびFDAがGLPおよびGCPにそれぞれ適合することを保証するために非臨床研究および臨床試験地点を監査するための任意の監査の結果を含み、FDAは承認状または完全な返信を発行する可能性がある。承認書は、製品の商業マーケティングを許可し、特定の適応に関する具体的な処方情報を提供する。申請が承認されていない場合、FDAは、申請が最終的に承認されることを確実にするために満たされなければならない条件を含み、可能な場合には、スポンサーがとりうる提案行動を概説して、出願の承認を得ることができる完全な返信を発行する。受賞のスポンサー
38
完全な応答は、FDAによって決定された質問に対する完全な応答を表す情報をFDAに提出することができる。PDUFAによると,このような再提出は1つまたは2つに分類され,再提出された分類は,申請者が行動関数に回答する際に提出された情報に基づいている.FDAがPDUFAで達成した目標と政策によると,FDAは2カ月間に1種類の再提出を審査し,6カ月の期間審査で2種類の再提出を行った。完全な返信で決定された問題が解決されるまで、FDAは申請を承認しない。代替的に、完全な返信を受信した保証人は、申請を撤回するか、または公聴会を要求することができる。
FDAはまた、申請を承認すべきかどうかを決定するために、審査、評価、および提案のために諮問委員会に申請を提出することができる。特に,FDAは新たな生物製品や安全性や有効性の問題を提起した生物製品の申請を諮問委員会に提出する可能性がある。通常,諮問委員会は臨床医や他の科学専門家を含む独立した専門家からなるグループであり,申請を審査,評価し,申請を承認すべきかどうか,どのような条件でアドバイスを提供すべきかを担当する。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。
FDAが新製品を承認した場合、それはその製品の承認適応を制限するかもしれない。それはまた製品ラベルに禁忌症、警告、または予防措置を含むことを要求するかもしれない。さらに、FDAは、承認後の製品の安全性をさらに評価するための4期の臨床試験を含む承認後の研究を要求する可能性がある。この機関はまた、製品が商業化された後にそれを監視するために、またはREMSを含む流通制限または他のリスク管理機構を含む他の条件を適用して、製品の利益が潜在的リスクよりも大きいことを保証するために、テストおよび監視計画を要求することができる。REMSは、薬物ガイドライン、医療専門家のコミュニケーション計画、および安全な使用を確保する要素、またはETASUを含むことができる。ETASUは、処方または調剤、場合によっては調剤、特殊監視、および特許登録所の使用に関する具体的または特殊なトレーニングまたは認証を含むことができるが、これらに限定されない。FDAは発売後の研究或いはモニタリングプロジェクトの結果に基づいて、製品の更なるマーケティングを阻止或いは制限することができる。承認後、新しい適応の追加、いくつかの製造変更、および追加のラベル宣言など、承認製品の多くのタイプの変更は、さらなるテスト要件およびFDAの審査および承認を受けなければなりません。
加急プロジェクト
FDAはある製品を指定して迅速な審査を行う権利があり、もしこれらの製品が深刻或いは生命に危害を及ぼす疾病或いは状況の治療中に満たされていない医療需要を解決することを目的としている場合。これらの計画は迅速チャネル指定、突破的治療指定、優先審査、再生医学高度治療指定と呼ばれる。
特に、FDAは、深刻なまたは生命に危険な疾患または状態を治療するために1つまたは複数の他の製品と単独でまたは組み合わせて使用することが意図されている場合、迅速な検討のための製品を指定することができ、そのような疾患または状態の満たされていない医療要件を満たす可能性があることを示す。Fast Track製品については,スポンサーがFDAとより多くのインタラクションを行う可能性があり,FDAは申請完了前にFast Track製品申請部分の審査を開始する可能性がある。FDAがスポンサーから提出された臨床データを初歩的に評価した後、高速チャネル製品が有効である可能性があると判断した場合、スクロール審査を行うことができる。スポンサーはまた、残りの情報を提出するスケジュールを提供しなければならず、FDAの承認を得なければならず、スポンサーは適用された使用料を支払わなければならない。しかしながら、FDAが高速チャネル申請の期間目標を検討することは、申請の最後の部分が提出されるまで開始される。また,FDAが迅速チャネル指定が臨床試験中に出現したデータの支持を得なくなったと考えた場合,あるいは指定された薬物開発計画が行われなくなった場合,FDAはその指定を撤回する可能性がある。
第二に、FDAは“画期的な療法”に指定された製品の審査を加速させる規制計画を持っている。1つの製品が、1つまたは複数の他の製品と単独で、または1つまたは複数の他の製品と組み合わせて、深刻なまたは生命に危険な疾患または状態を治療するために使用されることが意図されている場合、予備臨床証拠は、製品が1つまたは複数の臨床的重要終点において既存の療法よりも有意な改善を示す可能性があることを示しており、例えば、臨床開発早期に観察された実質的な治療効果がある場合、製品は突破的療法として指定することができる。画期的な治療法について、FDAは、開発過程全体にわたってスポンサーとの会議を行うこと、製品スポンサーに開発と承認に関する提案をタイムリーに提供すること、より多くの上級者を審査過程に参加させること、審査チームのために学際的なプロジェクト担当者を指定すること、および他のステップを取って効率的な方法で臨床試験を設計することを含むいくつかの行動をとる可能性がある。
第三に、FDAは、重篤な疾患を治療する場合、承認された場合、安全性または有効性の面で顕著な改善を提供する製品を優先的に検討することができる。FDAは具体的な状況から,他の利用可能な療法と比較して,提案された製品が有意な改善を表すかどうかを決定している。著明な改善は,ある疾患の治療の有効性の向上,制限治療の副作用の除去あるいは大幅な減少,記録のある患者のコンプライアンスの向上が重篤な結果の改善を招く可能性があること,新亜群の安全性と有効性の証拠である可能性が示唆された。優先指定の目的は、このようなアプリケーションの評価に全体的な注意とリソースを誘導し、FDAがマーケティング申請に行動する目標を10ヶ月から6ヶ月に短縮することである。
最後に,FDAは再生医学高度療法に指定された製品の審査と承認を加速することができる。もし製品が再生医学療法であれば、深刻な治療、修正、逆転または治癒を目指しています
39
あるいは生命に危害を及ぼす疾病或いは状況、初歩的な臨床証拠は、この製品がこのような疾病或いは状況が満たされていない医療需要を解決する潜在力を有することを示している。再生医学高度治療指定の利点は、開発および審査を加速するためのFDAとの早期相互作用、画期的な治療の利点、潜在的な優先審査資格、および代替または中間終点に基づく加速承認を含む。
さらに、“2022年食品·薬物総合改革法案”(“FDORA”)によれば、以下の場合、医薬品または生物製品またはそれによって使用されるプラットフォーム技術が指定されたプラットフォーム技術として指定される資格がある:(1)プラットフォーム技術がBLAによって承認された医薬品使用に組み込まれるか、またはBLAによって承認された医薬品使用;(2)承認または許可された薬物の発起人または薬物出願に提出されたデータ参照権を取得した発起人によって提出された予備的な証拠は、プラットフォーム技術が、品質、製造または安全に悪影響を与えることなく、1つ以上の薬物に組み込まれる可能性があるか、または使用される可能性があることを示し、(3)適用者が提出したデータまたは情報は、プラットフォーム技術の組み込むまたは利用が薬物開発または製造プロセスおよび審査プロセスに顕著な効率をもたらす可能性があることを示す。スポンサーは、IND出願を提出した間または後の任意の時間に、IND出願を要求対象とするプラットフォーム技術を組み込むか、または使用する指定されたプラットフォーム技術としてFDAに指定することを要求することができる。指定された場合、FDAは、プラットフォーム技術を使用または組み込まれた薬物の任意の後続の元のBLAの開発および審査を加速および検討することができる。プラットフォーム技術の状態を指定することは、薬物開発がより早くあるいはFDAの承認を得ることを保証することはできない。さらに、FDAが、指定されたプラットフォーム技術がもはやそのような指定された基準に適合していないと判断した場合、FDAは、指定を取り消すことができる。
承認ルートを加速する
FDAは、患者に既存の治療よりも意義のある治療利点を提供する深刻または生命に危険な疾患の承認を加速する可能性があり、これは、製品が臨床的利益を合理的に予測する可能性のある代替終点に影響を及ぼすことを決定することに基づく。中間臨床終点に対する製品の影響が不可逆的な発病率または死亡率またはIMMへの影響よりも早いことができ、このような状況の重症度、希少性または流行率、および代替治療が利用可能または不足している場合を考慮すると、IMMまたは他の臨床的利益への影響を合理的に予測することが可能である場合、FDAはこのような状況の加速承認を許可することもできる。加速された承認を受けた製品は、従来承認された製品と同じ安全性と有効性法定基準に適合しなければならない。
承認を加速するために、代替終点は、例えば実験室測定、放射画像、バイタルサイン、または他の臨床的利益を予測することができると考えられるが、それ自体は臨床的利益の測定基準ではない標識である。代替終点は通常、臨床終点よりも容易または迅速に測定される。中間臨床終点は治療効果の測定であり、1種の製品の臨床利益、例えばIMMに対する効果を合理的に予測することが可能であると考えられる。FDAは中間臨床終点に基づく加速承認の面で経験が限られているが、ある研究が慢性病環境において相対的に短期的な臨床利益があることを証明し、臨床利益の持続性を評価することは伝統的な承認に重要であるが、短期的な利益は長期的な利益を合理的に予測する可能性があると考えられる場合、このような終点は通常加速承認を支持することができることを示した。
加速承認経路は疾病の病気経過が比較的に長く、製品の期待される臨床利益を測定するために時間を延長する必要がある環境に最もよく用いられ、代用或いは中間臨床終点への影響は非常に速く発生した。したがって、加速承認は様々な癌を治療するための製品の開発と承認に広く使用されており、その中で治療の目標は通常、生存率を向上させること、または発病率を低下させることであり、典型的な病気経過の持続時間は長く、時には大型の試験を必要とし、臨床または生存利益を証明することである。
加速承認経路は、一般に、製品の臨床的利益を検証および説明するために、勤勉な方法で追加的な承認後の検証的研究を行うことにスポンサーが同意することに依存し、FDAは、承認前または承認が加速された製品の承認日後の特定の期間内に、そのような試験を適宜要求することを可能にする。そのため、この基礎の上で承認された候補製品は必ず厳格な発売後のコンプライアンス要求を守らなければならず、4期或いは承認後の臨床試験を完成し、臨床終点への影響を確認することを含む。必要な承認後研究を行わない場合、あるいは発売後の研究期間中に臨床利益が確認できなければ、FDAがこの製品の市場からのリコールを加速することを許可する。加速規制によって承認された候補製品のすべての販売促進材料はFDAの事前審査を経なければならない。
承認後の規則
製品発売の規制承認または既存製品の新しい適応が得られた場合、スポンサーは、すべての通常の承認後の規制要件、およびFDAが承認プロセスの一部として適用される任意の承認後要求を遵守することを要求されるであろう。スポンサーは、いくつかの不良反応や生産問題をFDAに報告し、最新の安全性および有効性情報を提供し、広告や販売促進ラベル要求に関する要求を遵守することを要求される。製造業者たちは適用された製品追跡と追跡要求を守らなければならない。製造業者およびそのいくつかの下請け業者は、FDAおよびいくつかの州機関に彼らの工場を登録し、製造業者にいくつかのプログラムおよび文書要件を適用するcGMP法規を含む現行の法規要件に適合しているかどうかを知るために、FDAおよびいくつかの州機関の定期的な抜き打ち検査を受けなければならない。
40
したがって、スポンサーおよびその第三者メーカーは、cGMP法規および他の法規要件の遵守を維持するために、生産および品質管理に時間、お金、エネルギーをかけ続けなければならない。
製品はまた正式なロット発表が必要である可能性があり、これはメーカーが製品が発表される前に、製品の各ロットに対していくつかのテストを行わなければならないことを意味する。製品が正式なバッチ発行を必要とする場合、製造業者は、各バッチのサンプルをFDAに提出し、バッチの生産履歴要約および製造業者がバッチに対して行ったすべての試験結果を示す放出スキームを提示しなければならない。さらに、FDAは、いくつかの製品のバッチに対していくつかの検証的試験を行い、その後、これらのロットを流通させるかもしれない。最後に、FDAは薬品の安全性、純度、効力と有効性に関する実験室研究を行う。
承認されると、規制要求や製品の発売後に問題が発生した場合、FDAは承認を撤回する可能性がある。その後、製品には、予期されない深刻度または頻度の不良事象、または生産プロセス、または規制要件を遵守できなかったことを含む、以前に未知の問題が存在することが発見され、新しいセキュリティ情報を追加するために承認されたラベルの改訂を招く可能性がある;新しい安全リスクを評価するために発売後研究または臨床試験を実施すること、またはREMS計画に従って流通または他の制限を実施することが可能である。規制要件を遵守できなかった他の潜在的な結果は、
FDAは、上場許可と製品のマーケティング、ラベル、広告、販売促進を厳格に規制している。薬品は承認された適応と承認されたラベルの規定でしか宣伝できない。FDAや他の機関はラベル外用途の普及を禁止する法律法規を積極的に実行しており,ラベル外用途の普及が不適切であることが発見された会社は重大な責任を負う可能性がある。
孤児薬名
米国の孤児薬物指定は,スポンサーにまれな疾患や疾患に対する製品の開発を奨励するためである。米国では、法律は、まれな疾患または疾患を、米国で20万人未満または米国で20万人を超える影響を及ぼす疾患と定義しており、米国での製品の販売からその疾患または疾患に対する生物製剤の開発および提供のコストを回収することができる合理的な期待はない。
FDAが承認すれば、孤児薬物指定は会社が製品発売許可日から7年以内に税収控除と市場排他性を得る資格を持つことになる。孤児製品に指定された出願は、当該製品の発売を承認する申請を提出する前のいつでも提出することができる。規制規定により提出された受け入れ可能な機密要求に基づいて,1つの製品がFDA孤児製品開発事務所(OOPD)の孤児薬物指定を受けた場合,その製品は孤児となる。そして、その製品は任意の他の製品のように、審査と承認手続きを通じて商業流通を行わなければならない。
スポンサーは、以前承認されていなかった製品を孤児薬として指定したり、すでに発売されている製品のために新たな孤児適応を申請することを要求することができる。さらに、1つの製品が他の態様で承認された孤児薬物と同じ製品である場合、製品が信頼できる仮定を提示することができる場合、すなわち、その製品が第1の薬剤よりも臨床的に優れている可能性がある場合、製品の発起人は、同じ稀な疾患または疾患の後続製品に対する孤児薬物名を求めることができ、取得することができる。複数のスポンサーは、同じ製品のために同じまれな疾患または疾患の孤児薬物指定を得ることができるが、孤児薬物指定を求める各スポンサーは、完全な指定申請を提出しなければならない。
専門期間はFDAが上場申請を承認した日から,この製品が指定した適応にのみ適用される。FDAは、同じ製品の第2の出願を異なる使用のために承認することができ、または同じ使用のために製品の臨床的により優れたバージョンを申請することができる。しかしながら、スポンサーの同意またはスポンサーが十分な数を提供できない限り、FDAは、他のメーカーが生産した同じ製品が市場排他期間内に同じ適応のために使用されることを許可することはできない。
小児科研究と排他性
改正された2003年の“小児科研究公平法”(PREA)によると、BLAまたはその付録は、すべての関連小児科亜群において主張される適応の安全性および有効性を評価するのに十分なデータを含む必要がある
41
この製品の安全かつ有効な各小児科亜群の投与量と投与を支持する。スポンサーはまたデータを評価する前に小児科研究計画を提出しなければならない。これらの計画は、提案された1つまたは複数の小児科研究の大綱、出願人が実施する研究、研究目標および設計、任意の延期または免除請求、および法規要件の他の情報を含む計画を含まなければならない。そして,申請者,FDA,FDAの内部審査委員会は提出された情報を審査し,相互に協議し,最終計画について合意しなければならない。FDAまたは出願人はいつでも計画の修正を要求することができる。
FDAは、成人のために製品が使用されるか、または小児科データ要件を完全にまたは部分的に免除するまで、申請者の要求に応じて、または小児科データの一部または全部の提出を延期することを許可することができる。法規が別途要求されない限り、小児科データ要件は、孤児として指定された製品には適用できないが、生物が成人癌を治療するための分子標的癌製品であり、FDAに対して小児科癌の増殖または進行に実質的に関連する分子標的を決定する場合、これらの要件は、孤児の新しい活性成分として指定されたBLAに適用されるであろう。
小児科排他性は、米国の別の非特許マーケティング排他性であり、承認された場合、任意の既存の規制排他性条項(非特許および孤児排他性を含む)の条項に追加的な6ヶ月間のマーケティング保護を追加することが規定される。BLAスポンサーから提出された小児科データがこのようなデータに対するFDAの書面要求に公平に応答すれば,この6カ月の排他性を与えることができる。これらのデータは,この製品が研究されている小児科群で有効であることを証明する必要はなく,逆に臨床試験がFDAの要求に公平に応答していると考えられれば,追加的な保護が得られる。要求された小児科研究報告が法定期限内にFDAに提出され、FDAによって受け入れられた場合、製品の法定または規制排他性または特許保護期間にかかわらず6ヶ月間延長される。これは特許期間の延長ではないが、FDAが別の出願を承認できない規制期間を効果的に延長する。
生物模倣薬と排他性
2010年3月に法律となった“患者保護·平価医療法案”(ACA)には、“2009年バイオ製品価格競争·革新法案”(BPCIA)という副題が含まれている。BPCIAは、FDAが生体模倣薬と交換可能な生物模倣薬を許可することを許可する規制方案を確立した。FDAはすでにいくつかの指導文書を発表し,生体模倣薬の審査·承認方法について概説した。
BPCIAによれば、製造業者は、以前に承認された生物製品または“参照製品”“生物学的に類似している”または“交換可能”と一致する生物製品のライセンス申請を提出することができる。FDAに生物類似製品を承認させるためには、参考製品と提案された生物類似製品が安全性、純度と効力の面で臨床的に意義のある差がないことを発見しなければならない。FDAが生物類似製品を参照製品と交換することができるようにするために、この機関は、生物学的類似製品が参照製品と同じ臨床結果を生成することが期待でき、(複数回投与された製品のための)生物学的製剤および参照生物製剤は、安全リスクを増加させることなく、または参照生物製剤の独占的使用と比較して治療効果のリスクを低下させることなく、以前の投与後に交換可能であることを発見しなければならない。
BPCIAによると,生物類似製品の申請は参考製品が承認された日から4年後にFDAに提出される。FDAは参考製品が承認された日から12年以内に生物類似製品を承認することができるかもしれない。1つの製品が独占特許を取得する資格がある参考製品と考えられていても、FDAが製品の完全なBLAを承認した場合、スポンサー自身の臨床前データと十分かつ制御された臨床試験データとを含み、その製品の安全性、純度、および効力を証明するために、別の会社も製品の競争バージョンを販売することができる。BPCIAはまた、交換可能な製品として承認された生物模倣薬のための特定の排他的期間を設定し、FDAは、それらが同じ発売初日に承認される限り、複数の“第1”交換可能製品を承認することができる。この複数の第1の交換可能製品によって共有可能な排他的期間は、(1)第1の交換可能製品の最初の商業マーケティングの1年後、(2)米国法第42編262(L)(6)条に基づいて第1の交換可能な製品出願を提出した出願人が提起した特許侵害訴訟が18ヶ月後に解決された後、訴訟中のすべての特許に関する裁判所の最終裁決に基づいて、または訴訟を損なうことなく、または損害を与えない場合に訴訟を却下する。(3)米国法第42編262(L)(6)条に基づいて第1交換可能製品出願を提出した出願人に対して提起された特許侵害訴訟は,最初の交換可能製品が承認されてから42ヶ月後である;又は(4)第42条米国法第262(L)(6)条に従って最初の交換可能製品出願を提出した出願人が起訴されていない場合は,第1交換可能製品を承認してから18ヶ月後である。この瀬戸際で, FDAが“交換可能”とされている製品が実際に州薬剤法の管轄を受けている薬局に容易に置き換えられるかどうかは不明である。
42
特許期限の回復と延長
1984年の“医薬品価格競争及び特許期間回復法”又は“ハッジ·ワックスマン修正案”によると、新生物製品を有する特許は、製品開発及びFDA規制審査中に失われた特許期間が5年間にわたる特許回復を可能にする限られた特許期間延長を得る資格がある可能性があると主張している。1つの製品をカバーする特許付与の回復期は、通常、IND発効日とマーケティング出願提出日との間の時間の半分であり、マーケティング出願提出日と最終承認日との間の時間に加えて、出願人が努力すべき時間を減算していない。特許期間回復は特許の残存期間の延長には利用できず,製品承認日から合計14年を超える。承認された製品に適用される特許は1つのみ延期する資格があり,延期出願は関連特許が満了する前に提出されなければならない。複数の製品をカバーする特許は、そのうちの1つの承認に関連して延期することしかできない。米国特許商標局は,FDAと協議した後,任意の特許期間の延長または回復の出願を審査·承認する。
ヨーロッパの薬品審査の法規と手続き
アメリカ国外で任意の製品をマーケティングするために、会社はまた他の国と司法管轄区域の品質、安全性と有効性及び製品に対する臨床試験、マーケティング許可、商業販売と流通などの方面の多くの異なる監督管理要求を守らなければならない。FDAによる製品の承認を得るか否かにかかわらず、出願人は、これらの国または司法管轄区で当該製品の臨床試験またはマーケティングを開始するために、同様の衛生監督管理機関の必要な承認を得る必要がある。具体的には,ヨーロッパの医薬製品の承認手続きは米国とほぼ同じであり,米国で1つの医薬製品を承認することは欧州での承認が保証されていないにもかかわらず,完全な承認であっても,米国の承認と同じ時間範囲で承認されることも保証されていない。この過程は、各提案に対するこの製品の適応の安全性と有効性を決定するために、臨床前研究と十分かつ良好な制御の臨床試験を満足的に完成させる必要がある。また、欧州市場管理局または関連主管当局にマーケティング許可申請を提出し、欧州市場管理局またはこれらの当局が市場許可を付与し、その後、その製品を欧州で販売·販売することを要求する。
臨床試験許可
EUで臨床試験の認可を申請した出願人は,臨床試験を行うEU加盟国の国家主管当局またはNCAの承認を得なければならず,臨床試験が複数の加盟国で行われる場合は,複数の加盟国で承認されなければならない。また,倫理委員会(EC)が臨床試験に賛成の意見を発表した後のみ,出願人は特定の研究地点で臨床試験を開始することができる。
2014年4月,EUは2022年1月31日に臨床試験指令2001/20/ECに代わる新たな臨床試験条例(EU)第536/2014号を採択した。それはEUの現在の臨床試験承認制度を徹底的に改革する。具体的には、これはすべてのEU加盟国の新しい立法に直接適用され(これは、EUの臨床試験の承認を簡略化し、簡素化することを目的として、各EU加盟国で国家立法を制定する必要がないことを意味する)。例えば、新しい臨床試験規則は、単一の入口点と厳格に定義された最終期限によって臨床試験申請を評価する簡略化された申請プログラムを規定する。
マーケティング許可
EUにおける製品の販売許可を得るためには,申請者はMAAを提出しなければならないか,EUによって管理されている中央手続きか,EU加盟国主管部門が管理する手続きの1つ(分散手続き,国家手続きまたは相互承認手続き)である。マーケティング許可は、欧州経済地域に設立された出願人(EU加盟国にアイスランド、ノルウェー、リヒテンシュタインを含む)にのみ付与されることができる。(EC)第1901/2006号条例では、欧州医薬品局のマーケティング許可を得る前に、出願人は、EMAが特定の製品の免除、カテゴリ免除、またはPIPに含まれる1つまたは複数の措置の実施を延期しない限り、欧州医薬品局によって許可された小児科人口のすべてのサブクラスをカバーする小児科調査計画(PIP)に含まれるすべての措置を遵守していることを証明しなければならない。
集中化プログラムは、欧州委員会によって欧州経済地域全体で効果的な単一マーケティング許可を付与することを規定している。(EC)第726/2004号条例によれば、特定の製品については、特定の製品については、特定の生物技術によって生産された薬剤、孤児薬物として指定された製品、高度な治療医薬製品、またはATMP、および癌、HIVまたはエイズ、糖尿病、神経変性疾患、自己免疫および他の免疫機能障害、およびウイルス性疾患の治療を含む特定の疾患の治療のための新しい活性物質を含む製品は、集中手順を強制的に実行しなければならない。 集中プログラムの使用を強制しない製品については、出願人は、他の疾患を治療するための新たな活性物質を含む場合、集中プログラムを使用することを選択することができる あるいは、出願人は、製品が重大な治療、科学的または技術革新を構成していることを証明することができ、または製品にとって、集中的なプロセスがEU一級患者の利益に適合することを証明することができる。
具体的には、EUの生存可能なヒト組織または細胞を含む製品(例えば、遺伝子治療医薬製品)のマーケティング許可は、命令と組み合わせたATMPに関する(EC)1394/2007条例によって管轄される
43
欧州議会と欧州理事会の2001/83/EC号文書,通称欧州共同体薬品法典。(EC)第1394/2007号条例は、遺伝子治療薬物製品、体細胞治療薬物製品及び組織工学製品の許可、監督及び薬物警戒について具体的な規則を制定した。先進療法薬物製品のメーカーはEMAの先進療法委員会(CAT)にその製品の品質,安全性,有効性を証明しなければならず,この委員会はMAAを科学的に評価し,ATMPにMAAに関する意見を提供する。欧州委員会はEMAによって提供された意見に基づいてマーケティング許可を承認または拒否する。
EMAに設立されたヒト用薬品委員会(CHMP)は,ATMPが要求される品質,安全性,有効性要求に適合しているかどうか,製品が積極的なメリット/リスクプロファイルを持つかどうかに関する最終意見の発表を担当している。EUの中央手続きによると、環境評価機関が重大な影響評価を評価する最長期限は、有効な重大な影響評価を受けた210日後であり、申請者がCHMPの質問に答えるために補足情報や書面または口頭解釈を提供する必要はない。タイミングを停止することは、MAAを評価する時間フレームを210日を超えるように大幅に延長する可能性がある。CHMPが肯定的な意見を与えた場合、EMAの提案を受けて67日以内にマーケティング許可の最終決定を発行するサポートファイルと共にEU委員会に意見を提供する。特殊な場合、公共健康の観点、特に治療革新の観点から、1種の医薬製品が重大な価値を有することが予想される場合、CHMPは加速評価を承認することができる。CHMPがそのような要求を受け入れる場合、210日間の評価期限は150日(クロック停止を含まない)に減少するが、CHMPが申請が加速評価にもはや適していないと判断した場合、中央プログラムの標準時限に回復する可能性がある。
イギリス(イギリスと北アイルランドを含む)がEUを離れたので、イギリスはもはや集中マーケティング許可のカバーを受けないだろう(北アイルランド議定書によれば、集中マーケティング許可は北アイルランドで認められ続けるだろう)。現在の集中マーケティング許可を有するすべての医薬製品は、2021年1月1日にイギリスのマーケティング許可に自動的に変換される。2021年1月1日から3年以内に、イギリスの薬品監督機関またはMHRAは、新しいイギリスのマーケティング許可をより迅速に承認するために、欧州委員会が集中手続きで新しいマーケティング許可を承認する決定に依存する可能性がある。しかし、まだ個別的な申請が必要になるだろう。
素数案
2016年3月、EMAは、EMAとの早期対話およびEMAの規制支援を提供することにより、現在少ないまたは治療法のない適応における候補製品の開発を促進するイニシアティブを開始した。この計画はEMAが提供した科学提案計画と加速評価プログラムに基づいて、革新を刺激し、発展を最適化し、優先薬物或いはPrimeの加速評価を可能にすることを目的としている。この計画は自発的で、資格基準を満たさなければPrimeの資格を得ることができない。
Prime計画は開発中の薬品に対して開放され、申請者は集中プログラムを通じて初歩的な発売許可申請を申請するつもりである。条件に適合する製品は、満たされていない医療需要が存在する条件(これは、EUでは満足できる診断、予防または治療方法がないことを意味し、または、ある場合、新薬は重大な治療優位性をもたらすことを意味し、それらは、新しい方法または療法を導入することによって、または既存の方法を改善することによって、満たされていない医療需要を満たす潜在力を示さなければならない。申請者は通常開発の探索的臨床試験段階にあり、そして患者の中で初歩的な臨床証拠を獲得し、この薬物の有望な活性及び満足されていない医療需要を大きく解決する潜在力を証明する。特殊な場合、関連モデルにおいて納得できる非臨床データが有望な活動の早期証拠を提供し、第1の人体研究が必要な薬物治療効果および耐性に十分な曝露があることを示す場合、学術部門または中小企業(中小企業)の申請者は、開発の早い段階で資格申請を提出することができる。
Primeプログラムのために薬を選ぶとEMA:
Prime計画に選ばれた薬品も,EMAの上場許可申請時の加速評価プログラムから利益を得ることが予想される。研究開発過程において、もし1種の薬物が資格標準に符合しなくなったら、Prime計画下の援助を取り消すことができる。
44
データと市場排他性
EUでは、改正(EC)第726/2004号条例と改正された2001/83/EC指令に基づいて、完全な独立データパケットによって承認された革新医薬製品は、8年間のデータ独占権と追加2年間の市場独占権を得る資格がある。データ排他性防止模倣薬または生物模倣薬の許可申請者は、EUが模倣薬または生物類似薬のマーケティング許可を申請する際に、参照製品が初めてEUで許可を得た日から8年以内に、この参考製品のファイルに含まれる革新者の臨床前および臨床データを参照する。追加の2年間の市場排他期間内に、模倣薬または生物類似MAAを提出することができ、革新者のデータを参照することができるが、市場排他期間が満了するまで、いかなる模倣薬または生物類似薬もEUで販売することはできない。この10年の最初の8年間に、マーケティング許可保持者が1つまたは複数の新しい治療適応の許可を得た場合、これらの適応は、許可前の科学的評価期間中に既存の療法と比較して有意な臨床的利益をもたらすと考えられ、10年全体の期間は最大11年に延長される。1つの製品がEMAによって革新的な医薬製品とみなされることは保証されず、製品はデータ独占性を獲得する資格があるかもしれない。1つの製品が革新的な医薬製品とみなされても、革新者が所定のデータ独占期間を取得し、別の会社が完全に独立した薬物試験データパケットを有するMAAに基づくマーケティング許可を取得した場合、同社はその製品の別のバージョンを販売することができる, 前臨床試験と臨床試験。
授権期間と継続期間
集中マーケティング許可は原則として5年間の有効期間であり、5年後にEMAまたはライセンスEU加盟国の主管当局が国家ライセンス製品のリスク-収益バランスを再評価することによって更新することができる。一旦更新されると、上場許可の有効期限は無期限であり、欧州委員会または主管当局が薬物警戒に関する正当な理由に基づいて、追加の5年間の継続を決定しない限り。認可後3年以内に医薬品を実際にEU市場に投入していない(集中手続きの場合)、または許可されたEU加盟国の市場での許可は有効である(いわゆる日没条項)。
孤児薬の指定と排他性
条例(EC)第141/2000号及び条例(EC)第847/2000号によると、製品は、そのスポンサーが、(1)生命又は慢性衰弱に危険な疾患の診断、予防又は治療を目的としていることを証明することができることを前提として、欧州委員会により孤児薬物として指定されることができる。(2)申請がなされたとき、(A)そのような疾患のEUにおける影響が万分の5を超えない(5);または(B)孤児の地位のメリットがなければ、その開発に必要な投資が合理的であることを証明するためにEUで十分な見返りをもたらす可能性が低い。(3)このような疾患を満足できる診断、予防または治療する方法がEUで販売されていないこと、または、そのような方法がある場合、製品は、その疾患の影響を受けている人に大きな利点を有するであろう。
孤児薬物指定は、費用の低減、規制援助、およびヨーロッパ経済地域の範囲内の集中マーケティング許可を申請することができることを含む多くの利点を提供する。孤児薬の発売許可を与えることは10年間の市場排他期につながるだろう。この市場排他期間内には、欧州委員会も加盟国も“類似医薬製品”の申請を受け入れることができず、その上場を承認することもできない。類似医薬製品“の定義は、承認された孤児医薬製品に含まれる1つ以上の類似活性物質を含む医薬製品であり、同じ治療適応のためのものである。しかしながら、5年目の終了時に、製品が指定された孤児薬の基準をもはや満たしていないと判定された場合、適応の治療を許可する市場専門期間は、例えば、製品の利益が十分に高く、市場専門性が合理的であることを証明するのに十分ではないので、6年に短縮することができる。10年間の市場排他期に基づいて、欧州委員会は同じ治療適応中の類似医薬製品に対して販売許可を与えることができ、その中にいくつかの欠陥がある:
営業許可を得た後の規制要件
医薬製品がEUで許可された場合、マーケティング許可の保持者は、医薬製品の製造、マーケティング、販売促進および販売に適した一連の要件を遵守しなければならない。これらの措置には,EUの厳格な薬物警戒や安全報告規則の遵守が含まれており,これらの規則により,認可後の研究や追加的なモニタリング義務を実施することができる。しかも、許可された製品の製造も適用されるEUの法律、法規、そして厳格に守らなければならない
45
マニュアルは、2001/83/EC番号の命令、2003/94/EC番号の命令、(EC)第726/2004号法規、および欧州委員会の良好な製造仕様ガイドラインを含む。これらの要件には,医薬製品や活性医薬成分を生産する際にEU cGMP基準を遵守することが含まれており,EU以外で活性医薬成分を製造して活性医薬成分をEUに輸入しようとすることも含まれている。最後に、EUは許可製品のマーケティングと普及、業界賛助の継続医学教育と薬品処方者及び/或いは一般大衆に対する広告を含み、すべて厳格な監督管理がある。連合は処方薬だけの広告を一般大衆に投与することを許可しない。
上記のEU規則は,EU加盟国およびノルウェー,リヒテンシュタイン,アイスランドからなるヨーロッパ経済区に一般的に適用されている。
将来的に製品の商業化上場承認を得ることを求める可能性のある他の市場や、承認を求める他の衛生規制制度については、適用される衛生規制手続きや基準、および各適用司法管轄区域の他の管理法律や法規を継続的に遵守することを確保する必要がある。
一般資料保障規程
個人健康データを含むEU個人に関する個人データを収集、使用、開示、移転、または他の方法で処理することは、2018年5月25日に施行されたEU一般データ保護条例(GDPR)によって制限される。GDPRの範囲は非常に広く、個人データを処理する会社に対して多くの要求を提出し、健康や他の敏感なデータの処理、個人データに関する個人の同意の取得、個人にデータ処理活動に関する情報を提供すること、個人データの安全と機密性を保護するための保障措置の実施、データ違反行為の通知の提供、ある責任措置の確保、第三者プロセッサの使用時に何らかの措置をとることなどの要求を含む。GDPRはまた、米国を含むEU以外の国への個人データの移転に厳しいルールを実施し、データ保護当局が2000万ユーロや世界の年収の4%に達する可能性のある罰金を含むGDPR違反行為に巨額の罰金を科すことを許可している。GDPRはまた、データ主体と消費者協会に個人訴訟権利を与え、監督当局に苦情を提出し、司法救済を求め、GDPR違反による損害について賠償を受けることができる。GDPRを守ることは厳格で時間のかかる過程となり、ビジネスコストを増加させたり、会社にそのビジネスやり方を変えて完全な遵守を確保したりする可能性がある。
イギリスの離脱とイギリスの規制枠組み
2016年6月23日、イギリスの有権者はEU離脱(通称離脱)に賛成票を投じ、イギリスは2020年1月31日にEUから正式に離脱した。EU製薬法は2020年12月31日に満了する英国に適用され続ける過渡期がある。しかし、EUとイギリスは、2021年1月1日から暫定的に適用され、2021年5月1日から正式に適用される貿易·協力協定すなわちTCAに合意した。TCAには薬品に関する具体的な条項が含まれており,その中にはGMP相互承認,医薬製品の製造施設の検査,発表されたGMP文書が含まれているが,イギリスとEUの薬品法規の大規模な相互承認は予想されていない。現在、イギリスは“2012年人類薬品条例”(改正)(北アイルランド議定書によると、EU規制枠組みは引き続き北アイルランドに適用される)を採択し、医薬製品のマーケティング、普及、販売に関するEUの立法を実施している。そのため、イギリスの規制制度は多くの点でEUの規制制度と一致しているが、イギリスの規制制度はEUから独立しており、TCAはイギリスとEUの薬品立法を相互に認めることを規定していないため、これらの制度は将来的により大きな差が生じる可能性がある。
また、英国の2018年“EU(離脱)法”第3節と2018年の“データ保護法”によると、EUのGDPR(小幅改正可能)が英国法に組み込まれ、EUのGDPRと相互補完されている。欧州委員会は2021年6月28日、4年(2025年6月27日まで)の英国への個人データの移行に関する十分性決定を採択した。同様に、連合王国は、すべてのEUと欧州経済圏加盟国がデータ保護の面で十分だと考えていることが決定された。これはイギリスとEUとヨーロッパ経済地域の間のデータフローが影響を受けないことを確実にする。
保証範囲·定価·精算
FDAや他の政府機関の規制承認が求められる可能性のある任意の候補製品のカバー範囲や精算状態には、重大な不確実性がある。米国や他国の市場では,自分の病状に応じて処方治療を受けている患者や処方サービスを提供する提供者は,通常第三者支払者に依存してすべてまたは一部の関連医療費を精算している。患者は、保険を提供し、そのような候補製品を支払うのに十分なコストの大部分を清算しない限り、私たちが開発する可能性のある任意の候補製品を使用することはあまりできない。私たちが開発する可能性のある候補商品が承認されても、これらの候補商品の販売は、連邦医療保険や医療補助、商業健康保険会社など、米国の政府医療計画を含む第三者支払者にある程度依存する
46
保健機関を管理し、これらの候補製品に保険を提供し、十分な精算レベルを確立する。支払人が精算を決定する際に考慮する要素は、製品があるかどうかに基づいている
支払者が商品に保険を提供するか否かを判定するプロセスは、保険が承認された後に支払者が製品に支払う価格または販売率を設定するプロセスと分離することができる。第三者支払者は、徴収された価格に挑戦し、医療の必要性を審査し、医療製品およびサービスの費用対効果を審査し、コストを管理するために制御を実施することが増えている。第三者支払者は、保証範囲を承認リスト上の特定の製品に制限することができ、特定の適応のすべての承認製品を含まない可能性がある配合表とも呼ばれる。
販売が許可される可能性のある任意の製品の保険·精算を確保するためには、企業は、製品の医療必要性および費用効果、およびFDAまたは他の同様の市場承認を得るために必要なコストを証明するために、高価な薬物経済学的研究を行う必要があるかもしれない。それにもかかわらず、候補製品は医学的に必要または費用効果があると考えられないかもしれない。第三者支払者は、私たちが開発する可能性のある任意の候補製品を保証しないことを決定し、承認されると、医師のこのような候補製品の使用を減少させ、私たちの販売、運営結果、財務状況に実質的な悪影響を与える可能性がある。また、支払者が製品に保険を提供することを決定することは、十分な返済率を承認することを意味するものではない。また、1人の支払人が1つの製品に保険を提供することを決定することは、他の支払人もその製品に保険や精算を提供することを保証することはできず、また、支払者によって保険や精算レベルが大きく異なる可能性がある。第三者精算や保険は、製品開発への投資の適切なリターンを実現するために、十分な価格レベルを維持することができないかもしれません。
医療費のコントロールもすでにアメリカ国内と世界の他の国の各連邦、州と/或いは地方政府及び他の支払人の優先順位になっているが、薬品価格はずっとこれらの努力の重点である。政府と他の支払人はコスト制御計画の実施に大きな興味を示し、価格制御、精算に対する制限及び後発薬代替の要求を含む。価格制御及びコスト制御措置を講じ、既存の制御及び措置を有する司法管区においてより限定的な政策をとることにより、任意の承認された製品の販売から得られる収入をさらに制限することができる。保証政策と第三者精算料率は随時変化する可能性がある。1つの会社またはその協力者がマーケティング承認を得た1つまたは複数の製品が有利な保証·精算状態を獲得しても、将来的にはあまり有利ではない保証政策および精算料率を実施することが可能である。
アメリカ以外では、私たちが開発可能な任意の候補製品に十分なカバー範囲と支払いを提供することを確保することは挑戦に直面するだろう。多くの国で、処方薬の価格設定は政府によって規制されている。政府当局との価格交渉は、製品の監督マーケティング承認を受ける範囲をはるかに超えている可能性があり、私たちが開発する可能性のある任意の候補製品の費用対効果を他の利用可能な治療法と比較するために臨床試験を要求することができるかもしれない。このような臨床試験を行うことはコストが高く、私たちの商業化努力の遅延を招く可能性がある。
連合では、様々な国の価格設定と補償プログラムの差が大きい。一部の国では、補償価格を合意した後にのみ、製品を販売することができると規定されている。いくつかの国は、精算または定価承認を得るために、特定の候補製品の費用対効果を現在利用可能な治療法(いわゆる健康技術評価、またはHTA)と比較することを追加的な研究の完了を要求する可能性がある。例えば、EUはその加盟国に様々な選択を提供し、その国の健康保険制度が精算を提供する製品範囲を制限し、人が使用する医療製品の価格を制御している。EU加盟国は製品の具体的な価格を承認することができ、その製品を市場に投入する会社の収益力を直接または間接的に制御する制度をとることもできる。他のEU加盟国は会社が自分の製品価格を固定することを許可したが、処方量を監視と制御し、医師に指導を発表して処方を制限した。最近、EUの多くの国は薬品の価格設定に必要な割引レベルを高め、各国が医療支出を管理しようとしているに伴い、これらの努力は継続する可能性があり、特にEUの多くの国が深刻な財政危機と債務危機を経験した場合には、これらの努力が継続される可能性がある。全体的には,医療コスト,特に処方薬の下り圧力が大きくなっている。
そのため、新製品の参入にはますます高い壁が設けられている。政治、経済、規制の発展は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉は継続される可能性がある。EU加盟国が使用する参考価格および平行貿易(低価格と高価な加盟国間の裁定)はさらに価格を下げることができる。特別な価格と精算規則は孤児薬品に適用されるかもしれない。孤児薬物を精算制度に入れることは往々にして患者と患者の医療有用性、需要、品質と経済効果に重点を置いている
47
どんな薬とも同じように医療システムもそうですいずれの医療製品の精算を受けるかはコスト,用途,通常の数量制限を伴う可能性があり,これらの制限も国によって異なる可能性がある。しかも、成果に基づく精算規則が適用される可能性がある。薬品に対して価格制御や精算制限を実施することが保証されていない国は、これらの国で承認されれば、私たちのいかなる製品に対しても有利な精算と定価手配を許可する。
医療保健法律法規
医療保健提供者と第三者支払人は推薦と処方が発売承認された薬品の面で主要な役割を果たしている。プロバイダ、カウンセラー、第三者支払者および顧客との配置は、広く適用される可能性のある詐欺および乱用、リベート、虚偽申告法、医師および教育医への支払いの報告、および患者のプライバシーに関する法律法規およびその他、私たちの業務および/または財務的手配を制限する可能性のある医療法令の制約を受ける可能性があります。連邦と州の医療法律や規制には
48
いくつかの州および他の法律は、製薬会社が製薬業の自発的コンプライアンスガイドラインと連邦政府が公布した関連コンプライアンスガイドラインを遵守することを要求し、また、製薬業者に医師および他の医療保健提供者またはマーケティング支出に支払うことに関する情報を報告することを要求する。州法や他の法律も健康情報のプライバシーやセキュリティを管理している場合があり,その多くの法律は互いに大きく異なり,HIPAAに先を越されず,コンプライアンス作業を複雑にしていることが多い.例えば、カリフォルニア州では、2020年1月1日に施行されたカリフォルニア消費者保護法(CCPA)が、カバーする企業のために新たなプライバシー枠組みを構築し、方法は、個人情報の定義を拡大し、カリフォルニア州の消費者のための新たなデータプライバシー権を確立し、未成年者から消費者データを収集するための特別な規則を実施し、CCPAに違反し、合理的なセキュリティ手続きを実施せず、データ漏洩を防止するために実践している企業のための新たな深刻な法定損害賠償枠組みを作成することである。HIPAAが管轄する臨床試験データや情報は現在のバージョンのCCPAに制限されていないが,他の個人情報が適用可能であり,CCPAへの変更がその範囲を拡大する可能性がある。さらに、カリフォルニア州の新しい投票イニシアティブであるカリフォルニアプライバシー権法案、またはCPRAが、2020年11月に採択された。CPRAは2023年1月1日から立法でカバーされている会社に追加の義務を課し、CCPAを大きく改正する, 特定の敏感な個人情報に対する消費者の権利を拡大することを含む。外国司法管轄区域のデータプライバシーとセキュリティ法律法規をさらに強化し、これらの法規は米国の法規よりも厳しい可能性がある(例えば、2018年5月に発効したGDPRのEUを通過した)。場合によっては、同様の州法は、健康情報のプライバシーおよびセキュリティを追加的に管理する可能性があり、多くの法律は互いに大きく異なり、同じ効果を生じない可能性がある。
医療改革
アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。過去数年間、連邦と州政府は薬品と生物製薬製品の定価制限、薬品と他の医療製品のカバー範囲と精算、政府制御及びアメリカ医療保健システムの他の改革に関する多くの提案を提出した。
例えば、米国と州政府は医療コストの削減を目的とした立法を提案し、採択し続けている。2010年3月、米国議会は、政府医療計画下の製品のカバー範囲の変更と支払い方法を含むACAを公布した。ACAにおいて私たちの潜在的な製品候補製品に重要な意味を持つ条項は、以下のことを含む
49
ACAが公布されて以来、アメリカはまた他の立法改正を提案し、採択した。例えば、2011年の“予算制御法案”(Budget Control Act)など、国会の支出削減のための措置が制定された。赤字削減合同特別委員会の任務は、2012年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これには,各年度にプロバイダに支払われる連邦医療保険総額が最高2%減少することが含まれている。これらの減税措置は2013年4月1日に施行され、その後法規の立法改正が行われたため、4%の現金現金払い制やPAYGO自動減支を除いて、これらの減税措置は2031年まで有効となる。2020年5月1日から2022年3月31日までコロナウイルスの大流行で一時停止した後、2022年4月1日から2022年6月30日まで1%の支払いを削減し、2022年7月1日に2%の支払い減免を再開する。また,2010年に法定の現金支払法,あるいはPAYGOは,連邦医療保険に対して4%の支払いを自動的に削減し,ある提供者に影響を与えることを求めている。これらの削減は2023年1月1日に発効する予定だったが、2023年の総合支出法案はさらにこれらの削減を2025年に延期した。また、2013年1月、総裁·オバマ氏は、病院、画像センター、がん治療センターなど複数の医療サービス提供者に支払う医療保険をさらに減らし、政府が医療サービス提供者に多額の支払いを取り戻す訴訟時効を3年から5年に延長する2012年の米国納税者救済法に署名した。
公布以来、ACAのいくつかの側面は多くの司法、行政、行政、立法方面の挑戦を受けており、私たちは今後もACAに対してより多くの挑戦と修正案を提出することが予想される。2021年6月17日、米国最高裁はいくつかの州がACAに対して提出した最新の司法挑戦を却下したが、ACAの合憲性を具体的に裁くことはなかった。最高裁が裁決を下す前に,総裁·バイデンはACA市場による医療保険の取得を目的とした2021年2月15日から2021年8月15日までの特殊保険期間を開始する行政命令を発表した。行政命令はまた、特定の政府機関に、医療補助モデルプロジェクトおよび免除計画の再審査、および医療補助またはACAによる医療保険カバー範囲の獲得による不必要な障害をもたらす政策を含む医療補助モデルプロジェクトおよび免除計画の再審査および見直し、医療補助またはACAによる医療保険のカバー範囲を制限する既存の政策およびルールを再検討するように指示する。バイデン政府の他の医療改革措置や他の挑戦,ACAの廃止または代替の努力(あれば)が我々の業務にどのように影響するかは不明である。
連邦レベルでは、総裁·バイデンは2021年7月9日に行政命令に署名し、政府の政策、すなわち(I)処方薬と生物製品価格を下げる立法改革を支持し、連邦医療保険の薬品価格交渉を許可し、インフレ上限を設定し、低コスト模造薬と生物模倣薬の開発と市場参入を支持すること、および(Ii)公共医療保険オプションの制定を支持することを確認した。その他の事項に加えて、行政命令は、処方薬の価格設定が高すぎ、国内の薬品サプライチェーンの強化、連邦政府の薬品支払いの価格低下、業界価格詐欺の解決のための行動を説明する報告書をHHSに提供するよう指示し、2003年の“連邦医療保険処方薬、改善と現代化法案”およびFDAの実施条例に基づいて第804条の輸入計画を制定する州とインディアン部族との協力を提案するようFDAに指示した。FDAは2020年9月24日にこのような実施条例を発表し、2020年11月30日に発効し、各州のカナダ薬品輸入計画の制定と提出に指導を提供した。2020年9月25日、CMSは、この規則に基づいて輸入された州の薬品は、社会保障法第1927条に基づいて連邦還付を受ける資格がなく、メーカーは“最適価格”や平均メーカー価格の目的でこれらの薬品を報告しないと声明した。これらの薬剤はカバーされた外来薬とは考えられないため,CMSはさらに,これらの薬剤の全国平均薬物調達コストは公表されないと述べている。実施すれば、カナダからの薬品輸入は私たちの任意の候補製品の価格に実質的かつ不利な影響を与えるかもしれない。
また、2020年11月20日、CMSは最恵国或いは最恵国モデルを実施する暫定最終規則を発表し、このモデルによると、ある薬物と生物製品の連邦医療保険B部分の販売率は1人当たりの国内総生産に類似した経済協力と発展組織国の薬品メーカーが受け取った最低価格に基づいて計算される。しかし、2021年12月29日、CMSは最恵国規則を廃止した。また、2020年12月2日、HHSは薬品メーカーがD部分でスポンサー値下げを計画している安全港の保護を取り消し、法律が値下げを要求しない限り、直接あるいは薬局福祉マネージャーを通じて値下げする規定を発表した。この規定はまた、販売時点での値下げを反映するための新しい避難港を作成し、薬局福祉マネージャーと製造業者との間のいくつかの固定料金手配のための避難港を作成する。裁判所の命令により,上記安全港の除去と増加は延期され,最近の立法は同規則の実施を2026年1月1日に一時停止した。この期限は両党のより安全なコミュニティ法案によってさらに2027年1月1日に延期され、その後、2022年インフレ削減法案(IRA)によって2032年1月1日に延期された。
また,CMSは最終的に法規制を決定し,個人や小団体市場の保険会社に基準を設定する上で各州により大きな柔軟性を与えることで,ACAがこのような市場で販売されている保険計画に要求される基本的な健康福祉を緩和する可能性がある。例えば、CMSは2019年5月にMedicare Advantage計画が2020年1月1日からB薬剤に対する階段療法の一部を選択することを許可する最終ルールを発表し、事前に許可されている。このような努力が我々の業務にどのような影響を与えるかは不明である(あれば).
50
政府はメーカーがその市場製品の価格設定の方式についてもより厳格な審査を行い、最近の国会で数回の調査を行い、製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府の薬品の精算方法を改革するための法案を提出した。アメリカの個別州もますます積極的に薬品の価格を制御するための立法と法規を制定し、実施しており、価格或いは患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示と透明性措置を含み、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。ACAへの挑戦に加えて、他の立法措置が公布され、私たちの業務に追加の価格設定と製品開発圧力をもたらす可能性がある。例えば、2018年5月30日、“裁判権法案”が署名されて法律となった。他の事項以外に、この法律はある患者に連邦フレームワークを提供し、彼らが第一段階の臨床試験を完成し、FDAの許可を得た研究用新薬製品を獲得するために調査を行っていることを許可した。場合によっては、条件を満たす患者は、臨床試験に参加することなく、FDA拡大参入計画に従ってFDA許可を得ることなく治療を求めることができる。“試用権法案”によると,薬品メーカーは条件に適合した患者に薬物製品を提供する義務はないが,メーカーは内部政策を策定し,その政策に基づいて患者の要請に応じなければならない。私たちは未来にもっと多くの外国、連邦、州医療改革措置を取ることを予想しています, いずれも連邦政府と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これはカバー範囲と精算範囲が限られている可能性があり、承認されると、私たちの製品に対する需要が減少したり、追加の価格設定圧力をもたらしたりする可能性がある。
アイルランド共和軍は2022年8月に法律に署名した。IRAは、米国政府がMedicare B部分およびD部分の価格上限を交渉および設定することを可能にする条項を含むいくつかの条項を含み、これらの条項は、模造薬または生物類似競争が存在することなく、いくつかの高コスト、単一源の薬物および生物製品の価格を対象としている;以下の態様の自己負担上限を減少させる
2025年から連邦医療保険D部分受益者は2,000ドルを獲得し,連邦医療保険部分のいわゆる“ドーナツ穴”を効果的に解消する
D;企業に、薬品価格がインフレよりも速い場合に連邦医療保険にリベートを支払うことを要求することと、薬局福祉マネージャーが受け取ることができる費用と、他の分野とを制限するリベート規則とを遅延させる。Ireland共和軍が私たちの業務と医療産業全体に及ぼす影響はまだ明確ではない。
州レベルでは、各州はますます積極的に、価格または患者の精算制限、割引、ある製品への参入の制限、マーケティングコストの開示および透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法と実施し、場合によっては、他の国からの輸入と大量購入を奨励することを目的としている。また、地域衛生保健当局や個別病院は、どの薬品やサプライヤーが彼らの処方薬や他の医療計画に含まれるかを決定するために入札プログラムを使用することが増えている。これらの措置が承認されると、私たちの製品に対する最終的な需要を下げたり、私たちの製品の価格設定に圧力をかけるかもしれません。私たちは将来、より多くの州と連邦医療改革措置を取ることが予想され、そのいずれも連邦と州政府が医療製品やサービスのために支払う金額を制限する可能性があり、これは私たちの候補製品に対する需要の減少または追加の価格設定圧力を招く可能性がある。
米国および世界の他の司法管轄地域、ならびに米国または他の司法管轄区域内のいくつかの地域、州および/または地方レベルでは、医療の獲得可能性を拡大し、医療コストを制御または低減することを目的とした立法および規制提案が継続されている可能性がある。これらの改革は、市場で承認された候補製品の予想収入の開発に成功し、我々の全体的な財務状況や候補製品を開発する能力に影響を与える可能性がある。
法規を付加する
これらの規定に加えて,環境保全や有害物質に関する州や連邦法には,“職業安全と健康法”,“資源保護·回収法”,“有毒物質制御法”が含まれており,我々の業務に影響を与える。これらの法律や他の法律は,様々な生物,化学,放射性物質の使用,処理,処分を管理しており,これらの物質は作業,作業から発生する廃棄物に用いられている。もし私たちの運営が環境汚染を招いたり、個人を危険物質に曝露させたりすれば、損害賠償と政府罰金の責任を負う可能性があります。似たような義務を加えた第3国もまた同等の法律を採択した。
人力資本
2022年12月31日現在、私たちは458人のフルタイム従業員を持っている。労働組合の代表者たちはいないし、集団交渉協定も守られていない。大多数の従業員はマサチューセッツ州のボストンにいて、ほかにいくつかの従業員はマサチューセッツ州のフレミンガム、カリフォルニア州の観瀾湾、スイスとイギリスにいます。97人の従業員が製薬博士号を持っている。医学博士号を取得しています391人は主に研究開発または技術業務に従事し、67人は商業発展、財務、情報システム、施設、人的資源、法律機能、または行政支援に従事している。私たちは私たちの従業員が仲がいいと思う。
51
私たちは企業責任の最高基準で業務を展開するために努力している。私たちの目標は、多様で情熱的な文化を構築し、患者、私たちのコミュニティ、より広い社会に積極的に影響を与えるように努力することだ。私たちの人的資本資源は、私たちの既存と新しい従業員の誘致、採用、維持、激励、統合に重点を置いている。私たちは多様性、公平、そして包容的な職場が私たちの会社が私たちの使命を最善に果たすことができると信じている。私たちは、会社全体の多様性を増加させ、私たちの従業員と私たちがサービスするコミュニティを支援する包括的な労働環境を作るために努力し続けています。私たちは会社のこの重点を拡大するために努力している多元化、公平、そして包容委員会を設立した。私たちが事業を展開しているすべての国で、私たちの政策は職場差別に関するすべての適用法律を完全に遵守することです。私たちは性別、人種、民族、年齢、障害、性指向、性別アイデンティティ、文化背景、宗教信仰を問わず、最も優秀な人材を募集することに力を入れている。
私たちの全面的な株式、現金給与と福祉計画の主な目的は、新入社員と既存従業員を誘致、激励、維持、奨励することである。私たちは一連の報酬要素を使用してこれを達成し、これらの要素は私たちの短期目標の達成と長期業績をバランスさせている。また、従業員は、医療、歯科、生命保険、障害保険計画を含む私たちの退職、健康および福祉計画など、私たちの標準従業員福祉計画に参加する資格があります。私たちはまた、従業員に納税条件に合った退職計画や401(K)計画に参加する機会を提供し、当社の業界の他の会社と競争力を持つ401(K)計画に応じて貢献することができます。
我々の人的資本資源戦略は全面的であり、私たちの核心的な仕事方式をめぐって構築されたものであると考えている:協力、勇敢、創業と結果をガイドとする。著者らは従業員の調査、訓練と発展計画及びその他の計画を通じて、技能発展課程、マネージャー訓練、リーダーシップ発展機会、授業料精算と強力なオンライン課程訓練庫を含み、各種の発展テーマに参考を提供し、それによって私たちの従業員と強固な関係を構築する。組織内の各職能部門の伝統的な昇進以外に、私たちは職能を越えた職業発展の道を支持する。私たちは私たちの発展とともに私たちの人的資本資源キットを発展させて増加させることを計画している。
コロナウイルスの大流行に関する特別な説明
進行中のコロナウイルスの大流行は引き続き全世界の社会、経済、金融市場と世界各地の商業実践に予測不可能な影響を与える。この影響の程度や持続時間はまだ不確定であり,特にウイルス変種が伝播し続ける場合には予測も困難である.私たちは私たちの反応を積極的に監視·管理し、私たちの進行中と計画中の臨床試験への影響を含む、私たちの業務運営に対する実際と潜在的な影響を評価している。私たちは引き続き第三者サプライヤー、パートナー、その他の各方面と密接に協力して、私たちの計画と候補製品のパイプを推進することを求めて、同時に私たちの従業員とその家族、パートナー、第三者サプライヤー、医療サービス提供者、患者とコミュニティの健康と安全を維持することが重要である。コロナウイルスの大流行が当社の業務及び財務業績に及ぼす影響に関する検討は,本年報10−K表第I部第1 A項の“リスク要因”及び今年度報告第II部第7項の“経営陣の財務状況及び経営業績の検討及び分析”を参照されたい。
インターネット上で提供される情報は
投資家や他の人は、投資家関係サイト(http://crisprtx.gcs-web.com/)、米国証券取引委員会の届出ファイル、プレスリリース、公開電話会議、ネットワーク放送を介して投資家に重要な情報を発表していることに注意すべきである。私たちはこれらのチャンネルとソーシャルメディアを使って私たちの会社、私たちの業務、私たちの製品候補者、その他の事項を大衆と交流します。私たちがソーシャルメディアで発表した情報は実質的な情報と考えられるかもしれない。そこで、私たちは、投資家、メディア、およびわが社に興味を持っている他の人が、投資家関係サイトに列挙されたソーシャルメディアチャネルで発表された情報を審査することを奨励します。私たちの10-Kフォームの年次報告、10-Qフォームの四半期報告、8-Kフォームの現在の報告は、証拠品、依頼書、および情報声明、および取引法第13(A)および15(D)条に基づいて提出または提供された報告書の修正案を含み、これらの材料を電子的にアーカイブまたは米国証券取引委員会ウェブサイト(http://www.sec.gov)に提供した後、合理的で実行可能な範囲内でできるだけ早く私たちのサイトで無料で取得することができる。
52
第1 A項。RISK因子です。
この報告書には危険と不確実な要素に関する前向きな陳述が含まれている。私たちの実際の結果は本報告書で議論された結果と大きく異なるかもしれない。これらの差異をもたらすか、または促進する可能性のある要因は、本報告の以下および他の部分的に説明される要素、および参照によって本報告の任意の文書に組み込まれる要素を含むが、これらに限定されない。
以下のリスク要因と、当社の財務諸表とその付記、および米国証券取引委員会に提出された他の文書を含む本報告書の他のすべての情報を慎重に考慮しなければなりません。以下のいずれかのリスク、または現在知られていないか、または現在重大でないと考えている他のリスクが実際のイベントに発展している場合、私たちの業務、財務状況、運営結果、または将来性は、重大な悪影響を受ける可能性がある。このような状況が発生すれば、私たちの普通株の市場価格は下落する可能性があり、株主は彼らの投資の全部または一部を損失する可能性がある。
私たちの財務状況と追加資本需要に関連するリスク
私たちが始めて以来、私たちは大きな運営赤字が発生しており、予測可能な未来に損失を被っていくと予想されています。
私たちは私たちの株式証券を公開と非公開で発行し、私たちの優先株を非公開で販売し、転換可能な融資と戦略パートナーとの協力協定を通じて、私たちの運営に資金を提供します。私たちは2021年12月31日までの1年間にVertexとのプリペイドにより利益を実現していますが、今後数年は利益を上げないと予想しています。私たちのこれまでの損失は、予想された将来の赤字に加え、株主赤字や運営資本に悪影響を与え続けるだろう。私たちの費用は大幅に増加すると予想されています
したがって、予測可能な未来には、重大で増加していく運営損失を受け続けることが予想される。候補遺伝子編集製品の開発に関連する多くのリスクや不確実性のため,将来の損失の程度やいつ利益を達成するかは予測できない(あれば).私たちが本当に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。
53
私たちは私たちの株主を希釈するために多くの追加資金を調達する必要があるだろう。もし私たちが必要な時に資金を調達できなければ、私たちはいくつかの製品開発計画や商業化努力を延期、減少、または廃止することを余儀なくされるだろう。
候補遺伝子編集製品の開発は資本集約型である。私たちが行っている活動に関連する費用は増加することが予想されます。特に、私たちが研究と開発を続け、臨床前研究と臨床試験を開始し、私たちの候補製品のためにマーケティング承認を求める時。さらに、もし私たちの任意の候補製品が市場承認を得た場合、このような販売、マーケティング、製造および流通は、バイエル、ViaCyte、Vertex、または他の未来のパートナーの責任ではないので、製品販売、マーケティング、製造、および流通に関連する巨額の商業化費用が生じると予想される。もし私たちが私たちの候補製品のためにより多くの兆候や地理的位置を求めることを選択したり、他の方法で私たちが現在予想しているよりも早く拡張するならば、私たちはまたもっと早く追加資金を調達する必要があるかもしれない。したがって、私たちは私たちの持続的な業務と関連した多くの追加資金を得る必要があるだろう。もし私たちが必要な時や魅力的な条件下で資金を調達できない場合、私たちはいくつかの研究開発計画や将来の商業化努力を延期、減少、または廃止することを余儀なくされるだろう。
2022年12月31日と2021年12月31日までに、それぞれ約18.684億ドルの現金、現金等価物、23.791億ドルの有価証券を持っている。2022年12月31日現在、現金、現金等価物、有価証券が手元にあります。現金、現金等価物、および有価証券は、少なくとも今後24ヶ月以内に、現在の運営計画に資金を提供するのに十分であると予想されます。
私たちの将来の資本需要は多くの要素に依存し、それによって大幅に増加するかもしれない
54
いかなる追加的な拠出努力も、私たちの経営陣の日常活動に対する関心を移すかもしれません。これは、私たちの候補製品を開発し、商業化する能力に悪影響を及ぼすかもしれません。私たちは未来の融資が十分な金額または私たちが受け入れられる条項で提供されることを保証できない。さらに、任意の融資条項は、私たちの株主の持株や権利に悪影響を及ぼす可能性があり、私たちが追加証券(株式や債務を問わず)、またはそのような証券を発行する可能性は、私たちの株式の市場価格の下落を招く可能性があります。追加の持分または変換可能な証券の売却は、清算または他の特典を含む可能性があり、株主としての権利に悪影響を及ぼす可能性がある私たちのすべての株主の権益を希釈します。債務の発生は固定支払義務の増加を招き、私たちは追加債務を発生させる能力の制限、私たちが知的財産権を得ることができるかもしれない能力の制限、私たちの業務を展開する能力に悪影響を及ぼす可能性のある他の運営制限など、いくつかの限定的な条約に同意する必要があるかもしれない。私たちはまた、他の場合ではなく、パートナーとの手配や他の方法で資金を求めることが要求される可能性があり、私たちは、私たちのいくつかの技術または候補製品の権利を放棄するか、または他の方法で私たちに不利な条項に同意することを要求されるかもしれません。いずれも、私たちの業務、運営結果、および見通しに大きな悪影響を及ぼす可能性があります。
もし私たちがタイムリーに資金を得ることができなければ、私たちは私たちの1つ以上の研究開発計画や任意の候補製品の商業化を大幅に削減、延期または停止することを要求されるかもしれないし、必要に応じて私たちの業務を拡大したり、他の方法で私たちのビジネスチャンスを利用することができなくなり、これは私たちの業務、財務状況、運営結果に大きな影響を与えるかもしれない。
私たちの運営歴史は限られていて、これは私たちの技術と製品開発能力の評価と私たちの将来の業績を予測することを困難にするかもしれません。
私たちの全体開発は進行中で、私たちの任意の候補製品の最初の臨床試験は2018年末に開始されました。私たちの計画は臨床前と臨床開発が必要である;複数の司法管轄区域で監督とマーケティングの許可を得る;製造供給、生産能力と専門知識を獲得する;商業組織を設立する;製品販売から任意の収入を得る前に、大量の投資と重大なマーケティングを行う。私たちが任意の製品を商業化する前に、私たちの候補製品はFDAまたはEMAを含むいくつかの他の衛生規制機関の承認を得なければ発売できない。
私たちの限られた運営の歴史、特に急速に発展する遺伝子編集分野を考慮すると、私たちの技術や業界を評価し、私たちの将来の表現を予測することが困難になるかもしれません。私たちの運営会社としての短い歴史は、私たちの未来の成功や生存能力のどの評価も重大な不確実性の影響を受けるようにした。私たちは急速に発展する分野でスタートアップ企業がよく直面するリスクと困難に直面するだろう。もし私たちがこのような危険にうまく対応できなければ、私たちの業務は影響を受けるだろう。同様に、様々な要因により、私たちの財務状況と経営業績は四半期ごとに毎年大幅に変動し、その多くの要素は私たちがコントロールできないと予想しています。したがって、我々の株主は、将来の経営業績の指標として、いかなる四半期や年度の業績にも依存すべきではない。
また,発展段階の会社として,予見できない費用,困難,合併症,遅延などの既知と未知の状況に遭遇した。私たちが私たちの候補製品を推進するにつれて、研究に集中している会社から、臨床開発やビジネス活動(成功すれば)を支援できる会社に移行し続ける必要があります。そのような移行で、私たちは成功しないかもしれない。
私たちはスイスで税金の繰越を使用する能力が限られているかもしれない。
スイスの法律によると、私たちは7年以内に私たちが受けた損失を繰り越す権利があり、私たちは未来の利益を相殺することができ、もしあれば、これらの損失を相殺することができる。税収損失は課税利益と相殺されたときにのみ税務機関が最終評価を行う(私たちが損失していれば、状況はそうではない)。使用しなければ、このような税金損失は発生当時の7年後に満期になるだろう。私たちの収入は限られており、繰越された税金損失は一部または全部満期の高いリスクがあるため、将来の現金税支払いを減らすために使用されない。
2020年1月1日から、ズグ州はスイスの会社税改革に関する新しい法律を発表した。この新しい法律によると、一般有効企業所得税税率は2020年に11.91%(連邦、州とコミュニティ)に低下し、その後2021年に11.85%に低下する。
55
私たちのビジネス、技術、業界に関連するリスク
私たちの候補製品を臨床開発に進めることができず、規制部門の承認を得ることができず、最終的に私たちの候補製品を商業化するか、あるいはこの点で重大な遅延があれば、私たちの業務は実質的に損害を受けるだろう。
我々の開発作業は進行中であり,これまで我々の研究と開発は主にCRISPR/CAS 9,遺伝子編集技術,我々の最初の候補製品に集中してきた.我々の将来の成功は,我々CRISPR/Cas 9候補遺伝子編集製品の成功開発に大きく依存する.私たちは私たちの現在の候補製品を決定して開発するためにほとんどの努力と財力を投入した。私たちが製品収入を作る能力は、私たちの候補製品の成功開発と最終商業化に大きく依存し、これは決して起こらないかもしれない。例えば、VertexおよびViaCyteとの協力プロトコルおよびバイエルとのオプションプロトコルを含む我々の研究計画は、様々な理由で臨床開発の潜在的候補製品を決定できない可能性があり、または臨床開発による任意の候補製品の推進に成功できない可能性がある。私たちの潜在的候補製品または私たちの潜在的候補製品は、有害な副作用を有することが証明される可能性があり、または他の特徴を有する可能性があり、候補製品が生産に適用されないか、販売できないか、または上場承認を得ることができない可能性がある。私たちは現在どんな製品の販売からも収入を得ていません。私たちは永遠に適切な製品を開発したり商業化することができないかもしれません。
著者らは監督機関にアメリカ研究用新薬(IND)申請、臨床試験申請(CTA)或いはその等価物を提出しなければ、臨床試験を開始することができない。任意の候補製品のCTAまたはINDの届出は、他の活動に加えて、許容可能な効率を有する1つまたは複数のガイドRNAを決定および選択する必要がある。また、任意の未来の臨床試験を開始するには、ヨーロッパの監督管理機関或いは同等の機関が私たちのCTA或いはFDAの私たちのINDに対する認可を受け、そして適用監督機関との討論に基づいて最終的に試験設計を確定する必要がある。欧州規制機関、FDAまたは同様の機関が追加の臨床前研究を完成させることを要求した場合、あるいは他の要求を満たすことが要求された場合、私たちの臨床試験は延期される可能性がある。私たちがこれらの規制機関の指導を受けて組み込まれた後であっても、彼らは私たちが臨床試験を開始する要求を満たしていることに同意しないかもしれないし、あるいは私たちの試験設計や選択された臨床終点に対する受容可能な立場を変えても、追加の臨床前研究や臨床試験を完成させるか、あるいは現在予想されているよりも厳しい承認条件を適用する必要があるかもしれない。
利益を実現し、維持するためには、巨大な市場潜在力を持つ候補製品を開発し、商業化しなければならず、一連の挑戦的な活動で成功することが求められる。著者らの候補製品は臨床前と臨床開発が必要である;複数の司法管轄区で監督とマーケティングの許可を得る;製造供給、生産能力と専門知識を獲得する;商業組織を構築する;大量の投資と重大なマーケティング努力を行ってこそ、製品販売から任意の収入を得ることができる。また、私たちの候補製品が商業化される前に、私たちの製品開発計画はFDA、EMA、またはいくつかの他の衛生規制機関の承認を得なければ発売できない。私たちは決してこのような活動やすべての活動で成功しないかもしれないし、たとえ私たちが成功しても、私たちは利益を達成するのに十分な収入や十分な収入を生むことができないかもしれない。もし私たちが確実に利益を達成したら、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが達成して利益を維持できなければ、私たちの価値を下げ、資金を調達し、研究開発努力を維持し、業務を拡大し、あるいは運営を継続する能力を弱める可能性がある。私たちの価値の低下はまた株主が投資の全部または一部を失うことを招くかもしれない。
私たちの候補製品の成功は以下のいくつかの要素に依存するだろう
56
また、私たちの技術は多種の細胞と組織タイプの遺伝子編集に関連しているため、私たちは遺伝子療法が直面している多くの挑戦とリスクに直面している
もし私たちがこれらの要素のうちの1つまたは複数でタイムリーまたは根本的に成功できなければ、私たちは重大な遅延に遭遇したり、私たちの候補製品を商業化することに成功しなかったりする可能性があり、これは私たちの業務に深刻な損害を与えるだろう。もし私たちの候補製品が規制部門の承認を得られなければ、私たちは運営を続けることができないかもしれない。
我々のCRISPR/Cas 9遺伝子編集製品候補製品は、比較的新しい遺伝子編集技術に基づいており、これにより、開発およびその後に規制承認を得る時間およびコスト(あれば)を予測することは困難である。遺伝子編集技術に基づく候補製品の臨床試験数は限られており,遺伝子編集製品が米国やEUで承認されていない。
CRISPR/Cas 9遺伝子編集技術は比較的に新しく、CRISPR/Cas 9或いは他の類似遺伝子編集技術に基づく製品はまだアメリカ或いはEUで承認されておらず、限られた数の遺伝子編集技術に基づく候補製品の臨床試験のみが開始された。そのため、著者らの候補製品が製品の発見或いは鑑定、臨床前研究と臨床試験過程において遭遇する可能性のある開発挑戦を正確に予測することは困難である。例えば,限られた臨床試験データしかないため,人体上の安全性を完全に評価することはできない。また,最近いくつかの他の候補製品の臨床試験が開始されたため,人体への安全性を評価することはできない。我々が開発したどの候補製品の治療も長期的な影響を与える可能性があり,現時点では予測できない。我々が開発可能などの候補製品もDNAレベルで機能し,また,動物DNAはヒトDNAとは異なるため,動物モデルで我々の候補品を試験することは,候補製品のヒト臨床試験で観察された安全性や有効性の結果を予測できない可能性がある。さらに、私たちが計画の中で追求しているいくつかの疾患を選択するためには、動物モデルは存在しないかもしれない。これらの要因により,製品候補開発の時間やコストを予測することは困難であり,我々の遺伝子編集技術や任意の類似や競合する遺伝子編集技術の応用が認識,開発につながるかどうかも予測できない, 規制当局のどんな製品に対する承認もあります我々が将来遭遇する我々の遺伝子編集技術や我々の任意の研究開発計画に関連する開発問題が重大な遅延や意外なコストを招くことは保証できず,このような開発問題が解決されることも保証されない.これらの要因のいずれも、前臨床研究または開始可能な任意の臨床試験を完了することを阻止するか、またはタイムリーまたは利益的に開発される可能性のある任意の候補製品を商業化することができる。
FDA、EMAとその他の監督機関の臨床試験要求及びこれらの監督管理機関は候補製品の安全性と有効性を決定するための標準は、候補製品のタイプ、複雑性、意外性及び期待用途と市場によって大きく異なる。現在、遺伝子編集技術に基づく製品は規制機関の承認を得ていない。したがって、私たちのような候補製品の規制承認過程は不確定で、比較できるかもしれません
57
他のより有名またはより広く研究されている技術に基づく候補製品の承認プロセス。私たちの候補製品がアメリカやEUで規制部門の承認を得るのにどのくらいの時間がかかるかを決定することは困難であり、私たちの候補製品を商業化するのにどのくらい時間がかかるかを決定することも難しい。潜在的な候補製品を市場に投入するために必要な監督管理の承認を遅延または獲得できなかった場合、または監督管理の承認を得る意外なコストは、私たちが十分な製品収入を生成する能力を低下させる可能性があり、私たちの業務、財務状況、運営結果、および見通しが損なわれる可能性がある。
著者らが設計した同種異体T細胞候補製品は癌を治療する新しい方法を代表し、これは私たちに大きな挑戦をもたらした。
我々の免疫腫瘍学プロジェクトでは、健康なドナーのT細胞から改造された一連の候補同種T細胞製品(例えば、CTX 110、CTX 112、CTX 130およびCTX 131を含む)を開発しており、特定の癌を有する任意の患者のためのキメラ抗原受容体またはCARSを発現することができる。自己CAR T療法とは異なり,同種異体CAR T療法では,健康なドナー材料に依存して候補品を生産している。健康ドナーT細胞のタイプと品質はそれぞれ異なり、このような変異は生産標準化された同種異体CAR候補製品を挑戦的にし、これらの候補製品の開発と商業化の道を不確定にした。
我々は,我々のCAR候補T細胞製品の生産に使用するT細胞の品質と整合性を向上させるためのスクリーニングプログラムを開発したが,我々のスクリーニング過程では適切なドナー材料を識別できない可能性があり,生産後に材料の故障が発見される可能性がある。私たちはまた、新しいウイルスをスクリーニングするなど、出現する可能性のある新しいリスクに対応するために、私たちの仕様を更新しなければならないかもしれない。
私たちは規制機関が要求する仕様を含むドナー材料に厳しい仕様を持っている。規格に適合したドナー材料を識別して得ることができず,規制機関と適切な仕様で合意できない場合や,ドナーT細胞の変異性を解決できない場合には,我々が生産している候補製品に不一致が存在する可能性があり,あるいは進行中の臨床試験を予期した時間で開始または継続できない可能性があり,我々の名声を損なう可能性があり,業務や将来性に悪影響を及ぼす可能性がある。
また,承認され開発されている自己CAR T療法は,サイトカイン放出症候群,神経毒性,重篤な感染,長期細胞減少と低C種グロブリン血症,その他患者死亡をきたす重篤な有害事象の発生率が高いことが示されている。私たちは私たちの同種自動車T製品候補製品にも似たような有害事象が発生すると予想しています。また,同種異体CAR T細胞療法を受ける資格があるが,侵襲性癌や自己CAR T細胞療法を待つことができず自己CAR T細胞療法の資格を満たしていない患者は,より大きな合併症や治療死亡リスクに直面している可能性がある。我々の同種CAR T細胞候補製品はまた、移植片対宿主病またはGvHD、または輸液反応など、ドナーと患者との間の差異に関連するユニークな有害事象を引き起こす可能性がある。同種異体T細胞が患者の正常組織が異体であることを認識し始めると、移植片対宿主病を招く。
著者らはCRISPR/Cas 9遺伝子編集技術を設計し、健康ドナーT細胞のT細胞受容体を除去し、著者らの候補製品GvHDのリスクを低下させ、そして細胞表面からI類の主要な組織適合性複合体を除去し、患者の免疫システムが同種異体T細胞を攻撃することを制限し、そしてCAR T細胞の持続性を高める。しかし、私たちの候補製品の遺伝子編集は患者のGvHDまたは早期拒絶のリスクを制限することに成功できないかもしれない。また,われわれの免疫腫瘍学臨床試験の結果は,副作用や予期せぬ特徴の高さと受け入れられない重症度と普遍性を示す可能性がある。
我々の候補製品を服用する際に深刻なGvHDまたは他の有害事象が観察される場合、または任意の候補製品が自己療法または他の同種療法よりも安全または有効であると考えられる場合、同種療法を開発する能力は悪影響を受ける可能性がある。
FDA,NIH,EMAは遺伝子治療治療の規制に慎重を示しており,遺伝子治療や遺伝子テストの倫理や法律への懸念は,我々の候補製品の開発や商業化に対する追加的な規制や制限を招く可能性があり,予測が困難である可能性がある。
FDA、NIHとEMAは遺伝子治療と遺伝子テストを含む生物技術をさらに規範化する興味があることを示した。例えば,EMAはリスクに基づく方法で遺伝子治療製品の開発を提唱している。米国連邦や州レベルの機関や米国議会委員会や他の政府や管理機関もバイオテクノロジー業界をさらに規制する興味を示している。そのような行動は私たちの候補製品の一部またはすべての商業化を延期または阻止するかもしれない。
58
アメリカと他の司法管轄区域の遺伝子治療製品に対する監督管理要求はよく変化し、将来も引き続き変化する可能性がある。FDAは遺伝子治療製品に関するいくつかの指導文書を発表した。FDAはその生物製品の評価と研究センター内に治療製品事務室を設置し、遺伝子治療と関連製品の審査を統合し、細胞、組織と遺伝子治療諮問委員会を設置し、この審査に提案を提供した。政府規制機関を除いて,候補製品の臨床試験を行う各機関のIBCとIRB,あるいは適切であれば,あるいは中央IRBは,提案された臨床試験を審査し,試験の安全性を評価する必要がある。さらに、他の人が行った候補遺伝子治療製品の臨床試験の不利な発展は、FDAまたは他の監督機関が私たちの任意の候補製品に対する承認要求を変更する可能性がある。同様に,EMAはEU遺伝子療法の発展を管理しており,遺伝子療法製品の開発やマーケティングライセンスに関する新しいガイドラインを発表することが可能であり,これらの新しいガイドラインの遵守が求められている。これらの規制審査機関および委員会およびそれによって公布された新しい要求またはガイドラインは、追加的な研究や試験を行うことを要求し、私たちの開発コストを増加させ、規制の立場や解釈の変化を招き、候補製品の承認と商業化を延期または阻止し、あるいは重大な承認後の制限または制限を招く可能性がある。私たちが私たちの候補製品を推進し、規制承認を求める時、私たちはこれらの規制機関や委員会と協議し、適用される要求とガイドラインを遵守することを要求される。もし私たちがそれをしなければ, 私たちはこのような候補製品の開発を延期したり停止したりすることを要求されるかもしれない。このような追加的な手続きは私たちが予想していたより長く検討と承認過程を招くかもしれない。規制承認手続きの増加または延長あるいは私たちの候補製品開発のさらなる制限による遅延はコストが高い可能性があり、私たちまたは私たちの協力者が適時に臨床試験を完成し、私たちの現在と未来の候補製品を商業化する能力にマイナスの影響を与える可能性がある。
私たちが開発する可能性のある任意の候補製品や私たちが依存する任意の管理プロセスが不良副作用を引き起こす場合、その規制承認を遅延または阻止し、商業潜在力を制限したり、任意の潜在的なマーケティング承認後に重大な負の結果を招く可能性があります。
我々が開発する可能性のある候補製品は、死または脱標的DNA切断、または標的配列以外の位置にDNA切断を導入することを含む、不良または許容できない副作用、予期しない特徴、または他の深刻な有害事象に関連する可能性がある。これらの非標的切断は、DNA中の予期されない位置の遺伝子または遺伝的調節配列の切断を引き起こす可能性があり、または、修復テンプレートとしてDNAの一部を提供する場合、非標的切断イベントの発生後に、そのような修復テンプレートからのDNAが予期されない位置でゲノムに統合され、別の重要な遺伝子またはゲノム要素を潜在的に摂動させる可能性がある。
遺伝物質や遺伝物質を運搬するための製品の他の成分の持続的生物活性により,遺伝子編集療法に曝露した後に不良事象を遅延させる潜在的リスクがある。遺伝子編集製品を用いた治療で起こりうる副作用には,投与後の免疫反応が含まれており,治療の有効性を大きく制限する可能性がある。
免疫療法とそのヒト免疫系を利用した作用方法は強力であり,重篤な副作用をきたす可能性があり,臨床試験でのみこれらの副作用を発見することができる。予測できない副作用は臨床開発過程に出現する可能性があり、あるいは、もしこのような副作用が少ない場合、著者らの候補製品が監督管理機関の許可を得て承認された製品が発売された後、意外な副作用が出現し、より多くの患者の暴露を招く可能性がある。もし我々のCRISPR/Cas 9遺伝子編集技術が類似した効果を示すならば、私たちは候補製品の臨床前開発または臨床開発を停止または延期することを決定または延期することができるかもしれない。
我々が開発する可能性のある任意の候補製品によって引き起こされる深刻な有害事象や副作用に加えて、投与過程または関連プログラムも副作用を引き起こす可能性がある。著者らの広範な臨床試験に参加した患者は彼ら自身のCRISPR/CAS 9編集後の造血幹細胞と前駆細胞があり、幹細胞移植の一部として患者に輸血し、この過程は患者に対する清髄白花丹調整を含む。幹細胞移植を受けた患者でもEXACEX使用とは無関係な副作用(軽微から重篤)に遭遇する可能性がある。著者らの免疫治療試験に参加した患者はリンパ浄化方案を受けなければならず、通常深刻な不良事件を引き起こす可能性のあるフルダラビンとシクロホスファミドを含む。これらの方案は一時的、時々長期的な免疫抑制を招くため、患者がある感染に罹患するリスクは増加し、これらの感染は患者によって除去されず、最終的に死亡を招く可能性がある。治療医療従事者たちはどんな副作用も適切に認識したり処理したりしないかもしれない。私たちまたは私たちの協力者は、私たちが開発可能な任意の候補製品を用いて医療従事者を訓練して、私たちの臨床試験およびそのような候補製品の商業化後の副作用を理解することを望んでいる。これらの候補製品に対する潜在的な副作用に対する認識不足或いは管理が不適切であり、患者の負傷或いは死亡を招く可能性がある。いかなる不良または許容できない副作用、意外な特徴、または他の深刻な有害事象が発生した場合、私たち単独またはパートナーと開発した任意の候補製品または製品の臨床試験または商業流通は一時停止または終了する可能性があり、私たちの業務および名声は重大な損害を受ける可能性がある。
59
将来私たちがこれらの有害事象が私たちの候補製品以外の要素によって引き起こされることを証明できなければ、FDA、EMA、または他の同様の衛生規制機関は、任意またはすべての標的適応のために開発できる任意の候補製品のさらなる臨床研究を停止するか、または承認を拒否するように命令することができる。将来のすべての深刻な有害事象が製品に関連していないことを証明できても,このような事件は患者募集や入選患者の試験完了能力に影響を与える可能性がある。さらに、私たちが任意の候補製品の臨床試験を延期、一時停止または終了することを選択または要求された場合、その候補製品の商業的将来性が損なわれる可能性があり、これらの候補製品から製品収入を創出する能力が延期またはキャンセルされる可能性がある。これらの状況のいずれも、候補製品を識別し開発する能力を損なう可能性があり、私たちの業務、財務状況、運営結果、および将来性を深刻に損なう可能性があります。
さらに、候補製品の開発に成功し、上場承認を得た場合、FDAは、候補製品を使用した治療の利点が各潜在的患者へのリスクよりも大きいことを確実にするために、リスク評価および緩和策(REMS)を採用することを要求するかもしれないが、製品が患者に配布されるリスクを概説する薬物ガイドライン、医療従事者向けのコミュニケーション計画、広範な患者監視、または業界の典型的な高度に制御され、制限およびコストのより高い流通システムおよびプロセスを含む可能性がある。さらに、もし私たちまたは他の人が後に私たちが開発した任意の候補製品による不良副作用が発見されれば、いくつかの潜在的な重大な負の結果を引き起こす可能性がある
さらに、他の態様で調査された候補遺伝子治療製品は、死亡を含む深刻な有害事象をもたらしており、FDAまたは他の規制機関は、私たちの候補製品と同じカテゴリに属する製品または候補製品の有害事象を意識した後、候補製品の臨床試験を強制的に一時停止する可能性がある。
これらのすべての事件は、私たちの遺伝子編集技術および私たちが決定し、開発する可能性のある任意の候補製品の受け入れの程度を達成または維持することを阻止し、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性がある。
もし私たちが臨床試験の患者登録過程で遅延や困難に遭遇した場合、必要な規制承認を受けることは延期または阻止される可能性がある。
FDAや米国国外の同様の規制機関の要求に従うことができない場合、または必要に応じて所与の試験に適切な統計データを提供することができない場合、私たちまたは私たちの協力者は、私たちが決定または開発した任意の候補製品のために臨床試験を開始または継続することができないかもしれない。私たちが未来に照準を合わせるかもしれないどんな珍しい遺伝子定義疾患に対しても、登録は特に挑戦的かもしれない。さらに、患者がバイオテクノロジー、遺伝子治療または遺伝子編集領域に関連する有害事象の負の宣伝、患者集団のような競争的臨床試験、競合製品との臨床試験または他の理由で私たちの遺伝子編集試験に参加したくない場合、私たちが開発した任意の候補製品の募集患者、研究を行い、監督管理の承認を得る時間が遅れる可能性がある。また,我々の競合他社の一部は候補製品の臨床試験を行っている可能性があり,これらの試験は我々が開発した任意の候補製品の適応と同様であり,そうでなければ我々の臨床試験に参加する資格のある患者が競争相手の候補製品の臨床試験に応募する可能性がある。
患者の入選は他の要素の影響を受けています
60
私たちの臨床試験の登録遅延は、私たちが開発する可能性のある任意の候補製品の開発コストを増加させる可能性があり、これは私たちの価値を低下させ、追加融資を得る能力を制限するだろう。もし私たちまたは私たちの協力者が計画通りに臨床試験を行うのに十分な数の患者を募集することが困難な場合、私たちは進行中または計画中の臨床試験を延期、制限または終了する必要があるかもしれないが、いずれも私たちの業務、財務状況、運営結果、および見通しに悪影響を及ぼすだろう。
我々の業務は,進行中のコロナウイルスの大流行や他の変種の出現など,感染症の大流行,流行や爆発の悪影響を受ける可能性がある。
我々の業務は,臨床試験場所や他の業務活動が集中している地域で衛生流行病の悪影響を受ける可能性があり,我々が依存している第三者契約メーカーや契約研究組織の運営や,患者を募集して臨床試験を行う能力が深刻に妨害される可能性がある。例えば,行われているコロナウイルスの大流行は,世界社会,経済,金融市場,世界各地のビジネス実践に予測不可能な影響を与え続けている。
現在のコロナウイルスの大流行はどの程度著者らの業務、業務結果と未来の成長の将来性に影響する可能性があり、これは各種の要素と未来の発展に依存し、これらの要素と未来の発展は高度に不確定であり、大流行の持続時間、範囲と深刻度、特にウイルス変種が引き続き伝播し続ける状況を含む把握できない。例えば,コロナウイルスの大流行による患者用量の一時停止や遅延,患者のICU入室,病院や医療資源の制限や減少,新たな臨床試験地点の遅延開始,各試験地点の限られた現場者の支援を含めた一時的な遅延や中断を経験した可能性もある。また、私たちのいくつかの第三者メーカーとサプライヤーは疫病初期に運営を一時停止し、コロナウイルスの大流行のより多くの波が現地のコミュニティおよび/または国家と地方法規に影響を与えたため、一部のメーカーとサプライヤーは再び運営を一時停止した。
私たちは私たちの反応を積極的に監視·管理し、私たちの進行中と計画中の臨床試験への影響を含む、私たちの業務運営に対する実際と潜在的な影響を評価している。私たちは引き続き第三者サプライヤー、パートナー、その他の各方面と密接に協力して、私たちの計画と候補製品のパイプを推進することを求めて、同時に私たちの従業員とその家族、パートナー、第三者サプライヤー、医療サービス提供者、患者とコミュニティの健康と安全を維持することが重要である。
著者らの候補製品の早期臨床前研究の積極的な結果或いは臨床試験の初歩的な結果は必ずしも著者らの候補製品の後続の臨床前研究と任意の未来の臨床試験の結果を予測するとは限らない。もし私たちが今後の臨床前研究、臨床試験と未来の臨床試験の中で著者らの候補製品の早期臨床前研究に対する積極的な結果をコピーすることができなければ、著者らは開発に成功し、監督部門の著者らの候補製品に対する許可を得て、それを商業化することができないかもしれない。
著者らの臨床前研究のいかなる積極的な結果或いは著者らの候補製品の臨床試験の初歩的な結果は必ずしも後に必要な臨床前研究と臨床試験の結果を予測するとは限らないかもしれない。より多くの患者データが利用できることに伴い、臨床試験の初歩的、中期と主要なデータは変化する可能性がある。臨床試験からの初歩的、中期またはトップラインデータは、BLAまたは米国以外の類似提出において承認を支持して提出された結果を含む最終結果を予測できるとは限らない。一時的なデータ、主要なデータ、および初歩的なデータは依然として監査と確認手続きを遵守する必要があり、これは最終データが私たちが以前発表した予備データと大きく異なる可能性がある。したがって、最終データを取得する前に、予備データ、中間データ、および主要データを慎重に見るべきである。中間データと比較して、最終データの重大な不利な変化は、我々の業務の将来性を深刻に損なう可能性がある。さらに、初期、中期、およびトップデータは、患者が研究中に成熟したときにより多くの患者データを得る場合、または候補製品の他の進行中または将来の臨床試験のさらなる発展に伴い、1つまたは複数の臨床結果が実質的に変化する可能性があるというリスクに直面する。例えば、FDAの提案と一致して、私たちのいくつかの臨床試験は15年間のフォローアップ観察期間を含み、その間私たちは引き続き患者データを収集する。
61
私たちが開示する特定の研究または臨床試験に関する情報を選択することは、一般に、より広い情報に基づいており、あなたまたは他の人は、私たちが決定した重要な情報または他の適切な情報が私たちの開示に含まれることに同意しない可能性がある。私たちが開示しないことを決定したいかなる情報も、最終的には、特定の候補製品または私たちの業務に関する未来の決定、結論、観点、活動、または他の側面に対して重要な意味を持つと考えられるかもしれない。同様に、私たちが現在の開発スケジュールに基づいて、私たちが計画した候補製品の臨床前研究あるいは任意の未来の臨床試験を完成させることができても、私たちの候補製品のこのような臨床前研究と臨床試験の積極的な結果は後続の臨床前研究あるいは臨床試験結果に複製されないかもしれない。
製薬やバイオテクノロジー業界の多くの会社が早期開発に積極的な成果をあげた後,後期臨床試験で大きな挫折を経験し,類似した挫折に直面しないことは確認できない。同様に、製薬やバイオテクノロジー業界の多くの会社は登録試験を完了したにもかかわらず、規制部門の承認を得られなかった。これらの挫折は,臨床試験が行われている間の臨床前や他の非臨床発見,あるいは前臨床研究や臨床試験で行われた安全性や有効性観察によるものであり,これまで報告されていない有害事象を含む。そのほか、臨床前、非臨床と臨床データはよく異なる解釈と分析の影響を受けやすく、多くの会社は彼らの候補製品が臨床前研究と臨床試験で満足できると考えているが、しかし依然としてFDA或いはEMAの許可を得られなかった。
私たちが必要な臨床前研究と臨床試験を完成しても、マーケティング審査過程は高価で、時間と不確定であり、私たちが開発する可能性のある任意の候補製品の商業化承認を得ることを阻止するかもしれない。もし私たちが必要な規制承認を得たり遅延したりできなければ、私たちが開発する可能性のある候補製品を商業化したり、商業化を遅延させることができなくなり、私たちの収益能力は深刻な損害を受けるだろう。
私たちが開発する可能性のある任意の候補製品と、それらの設計、テスト、製造、安全、効果、記録保存、ラベル、貯蔵、承認、広告、販売促進、販売、流通を含むそれらの開発、商業化に関連する活動は、米国FDAおよび他の規制機関、EU EMAおよび他の国の類似機関によって全面的に規制されている。候補製品のマーケティング承認が得られなければ、指定された司法管轄区域で候補製品を商業化することはできないだろう。我々は一連の疾患の臨床開発と高度臨床前開発に複数の候補製品を持っているが、私たちは私たちが完全に所有する同種異体CAR T製品候補製品のBLASをFDAに提出したり、比較可能な外国当局に類似したマーケティング申請を提出していない。2022年第4四半期、私たちとVertexはそれぞれEUとイギリスのEMAとMHRAのSCDおよびTDT潜在的治療に関する規制提出を完了し、EMAおよびMHRAはそれぞれのマーケティング許可申請を検証した。また,我々とVertexは2022年11月にFDAへのBLAのスクロール提出を開始し,我々とVertexは2023年第1四半期末に完了する予定である。しかし、私たちはどの司法管轄区域の規制機関からもいかなる候補製品の発売の承認または許可を得ていませんし、私たちの候補製品または私たちが将来単独でまたはパートナーと共同開発する可能性のある任意の候補製品は、規制部門の承認や許可を永遠に得られないかもしれません。
私たちは規制とマーケティング承認を得るために必要な申請を提出して支援する上で経験が限られている。我々は,第三者CROおよび/または規制コンサルタントに依存して我々の全額候補製品を支援する過程を希望し,我々のA&R Vertex JDCAにより,我々はVertexに依存してヘモグロビン疾患候補製品にこのような申請を提出する。BLAまたは他の同様のマーケティング申請を比較可能な外国当局に提出し、規制部門の承認を得るためには、各治療適応に対する広範な臨床前および臨床データおよび支援情報を各規制機関に提出して、各必要な治療適応の生物候補製品の安全性、純度、有効性、および効力を決定する必要があり、安全性および有効性とも呼ばれる。BLAはまた候補製品の化学,製造,制御に関する重要な情報を含まなければならない。監督管理の承認を得るためには、製品の製造過程に関する情報を関連監督機関に提出し、関連監督機関が製造施設を検査する必要がある。FDAが上場申請を承認するために検査を行わなければならないと判断し、コロナウイルスの大流行により旅行制限のために審査期間内に検査を完了できないと判断した場合、FDAは、通常、検査を完了することができるまで、申請に対して完全な返信または行動を延期することを意図していると表明している。
62
一般に、FDAは、BLAの承認を支援するために2つの重要な試験の成功を要求するが、場合によっては、1つのキー試験に基づいてBLAを承認することのみに基づいて、BLAの承認を提出して得る能力は、最終的にFDAの審査決定であり、これは、当時利用可能なデータに依存し、安全性および/または有効性の観点から、既存のデータがBLAの提出または承認をサポートするのに不十分である可能性がある。例えば、CTX 110およびCTX 130を含む、我々が行っている完全候補製品を含む、我々の任意の臨床試験が完了したときに得られるデータは、BLAへの提出を臨床的意義を有する利益を示すか、または安全性および/または有効性の観点から十分に強力であり、承認の加速または条件付き承認または完全承認をサポートするために十分に強力であることを保証することはできない。さらに、安全性および/または有効性の観点から、進行中のGRAPE−111およびSTRAPE−121臨床試験で得られ、ローリング方式でFDAに提出されたデータは、BLAの加速または条件付き承認または完全な承認をサポートするために十分にロバストであることが保証されるであろう。これらの行っている臨床試験の結果と提出されたデータのロバスト性に基づいて、一旦提出されると、FDAは、BLAの承認を提出または取得する前に追加的またはより大規模なキー試験を行うことを要求する可能性がある。さらに、何らかの不良または許容できない副作用、意外な特徴、または他の深刻な有害事象が発生した場合、このような有害事象が我々の候補製品FDA以外の要因によって引き起こされることを証明することはできない, EMAまたは他の同様の衛生規制機関は、十分な情報を収集したり、候補製品のさらなる臨床研究を中止するように命令するまで、臨床試験を一時停止することができます。このような状況が発生すれば、規制部門の承認を得るためにBLAの提出を延期する可能性が高い。私たちは外国の規制機関と似たような挑戦に直面するかもしれない。
さらに、1つまたは複数の臨床試験の失敗は、臨床試験プロセスの任意の段階で発生する可能性がある。私たちが開発した任意の候補製品は効果がないかもしれません。中程度の効果しかないかもしれません。あるいは不良または意外な副作用、毒性、あるいは他の特徴があることが証明される可能性があり、上場承認を得ることを阻止したり、商業使用を阻止したり制限したりする可能性があります。したがって、私たちの候補製品の規制方法はまだ不確定で、複雑で長く、最終的には承認されないかもしれない。たとえ我々の候補製品が臨床研究において安全性と有効性を証明したとしても、多くの要素、製品開発期間中の監督管理政策の変化を含むため、監督管理の遅延或いは拒否に遭遇する可能性がある。
アメリカと他の外国司法管轄区で上場承認を獲得する過程はコストが高く、もし追加の臨床試験が必要であれば、長年の時間を要するかもしれないが、完全に承認されれば、しかも関連する候補製品のタイプ、複雑性と新規性を含む様々な要素によって大きく変化する可能性がある。開発期間中の市場承認政策の変化、追加法規又は法規の変化、又は各提出製品申請に対する規制審査の変化は、出願の承認又は拒絶の遅延を招く可能性がある。FDAと他国の類似機関は承認過程においてかなりの自由裁量権を有しており、いかなる申請も拒否することができ、私たちのデータが承認を得るのに十分ではなく、追加の臨床前、臨床、あるいは他の研究を行う必要があると決定することもできる。そのほか、臨床前と臨床試験から得られたデータの異なる解釈は候補製品の発売許可を延期、制限或いは阻止する可能性がある。私たちが最終的に得たどのマーケティング承認も限られているかもしれないし、制限されたり、承認された後の約束は、承認された製品が商業的に実行できないようにすることができる。
もし私たちが承認を得る上で遅延に遭遇した場合、あるいは私たちが開発する可能性のある候補製品の承認を得ることができなければ、これらの候補製品の商業的な見通しが損なわれる可能性があり、私たちが収入を作る能力は実質的に損なわれるだろう。
私たちのどの候補製品もアメリカではFDAの承認を得られないかもしれません。たとえ私たちがそうしても、私たちはいかなる他の司法管轄区域でも私たちの任意の候補製品の承認を得たり、それを商業化したりすることはできません。これは私たちがそのすべての市場潜在力を達成する能力を制限するだろう。
最終的に任意の特定の管轄区域で私たちの任意の候補製品を販売するためには、各管轄区域で安全性と有効性に関する多くのおよび異なる法規要件を確立し、遵守しなければならない。米国FDAの承認を得ても、他の国や管轄区域規制機関の承認を得ることは確保できない。同様に、外国規制機関の承認はFDAの承認を保障できない。また、一国で行われた臨床試験は他の国の規制機関に受け入れられない可能性があり、一国の規制承認は他の国の規制承認を保証することはできない。承認の流れは国によって異なり、追加の製品テストと検証、追加の行政審査期間に関連する可能性があります。複数の管轄区域で規制承認を求めることは私たちに困難とコストをもたらす可能性があり、追加の臨床前研究或いは臨床試験が必要であり、これは高価で時間がかかる可能性がある。各国の規制要求には大きな違いがある可能性があり、私たちの製品が特定の国で発売されることを延期または阻止する可能性がある。米国以外の規制承認手続きはFDA承認に関連するすべてのリスクに関するものだ。私たちは国際市場を含むどの司法管轄区でも候補製品の販売が許可されておらず、会社としても国際市場で規制承認を受けた経験がない。もし私たちが国際市場の規制要求を遵守できなかったり、必要な承認を得られなかったり、あるいは国際市場の規制承認が延期された場合、私たちの目標市場は減少し、私たちの製品のすべての市場潜在力を達成する能力は達成できないだろう。
63
画期的な治療指定、迅速チャネル指定、FDAの再生医学高度治療指定または優先審査、またはEMAのPrime計画は、たとえ私たちに承認された任意の候補製品であっても、より速い開発、規制審査または承認過程を招くことができない可能性があり、私たちの任意の候補製品がマーク承認を得る可能性を増加させない可能性がある。
私たちは私たちのいくつかの候補品のために画期的な治療法の称号を求めるかもしれない。画期的な治療法は、1つまたは複数の他の療法と単独でまたは1つまたは複数の他の療法と組み合わせて重篤または生命を脅かす疾患または状態を治療することを目的とした療法として定義され、予備的な臨床的証拠は、1つまたは複数の臨床的に重要な終点において、臨床開発早期に観察される実質的な治療効果のような既存の療法よりも有意な改善を示す可能性があることを示している。画期的な治療法として指定された療法に対して,FDAと試験スポンサーとのインタラクションやコミュニケーションは,臨床開発の最も有効な経路を決定するのに役立つとともに,無効対照案中の患者数を最小限にすることができる。FDAで画期的な治療法に指定されている療法も優先審査や加速承認を得る資格がある可能性がある。画期的療法に指定されたのはFDAの裁量権である。したがって,我々の候補製品の1つが画期的療法として指定された基準に適合していると考えても,FDAは同意せず,このような指定をしないことにする可能性がある。いずれの場合も,FDAの従来のプログラムによる承認を考慮した療法と比較して,候補製品に対する突破療法指定を受けることは,より速い開発過程,審査または承認を招くことはなく,FDAの最終承認も確保できない可能性がある。さらに、我々の1つまたは複数の候補製品が画期的な療法の条件に適合していても、FDAは、これらの候補製品が資格条件を満たしていないことを後で決定するか、またはFDAの審査または承認を決定する期間が短縮されない可能性がある。
私たちは私たちのいくつかの候補製品のための高速チャネル認証を獲得し、探すことができるかもしれない。例えば、Exacelは、TDTおよびSCDを治療するための高速チャネル名をFDAによって付与されている。治療の目的が重篤または生命に危険な疾患を治療することであり、そのような疾患が満たされていない医療ニーズを解決する可能性を示す場合、治療スポンサーは、迅速なチャネル指定を申請することができる。FDAは幅広い裁量権を有しており,この称号が付与されているかどうか,したがって,ある特定の候補製品がこの称号を得る資格があると考えても,FDAがそれを付与することを決定することを保証することはできない。私たちが高速チャネル認証を受けたとしても、私たちは従来のFDAプログラムよりも速い開発過程、審査、または承認を経験しないかもしれない。Fast Track製品については,スポンサーがFDAとより多くのインタラクションを行う可能性があり,FDAは申請完了前にFast Track製品マーケティング申請の部分の審査を開始する可能性がある.FDAがスポンサーから提出された臨床データを初歩的に評価した後にFast Track製品が有効である可能性があると判断すれば,スクロール審査が可能である。スポンサーはまた、残りの情報を提出するスケジュールを提供しなければならず、FDAの承認を得なければならず、スポンサーは適用された使用料を支払わなければならない。しかしながら、FDAが出願を審査する期間の目標は、出願の最後の部分が提出されるまで開始される。FDAが我々の臨床開発プロジェクトのデータが高速チャネルの指定をサポートしなくなったと考える場合,その指定を撤回する可能性がある。高速チャネル指定だけではFDA優先審査プログラムに適合する資格は保証されない。
私たちはRMAT認証を取得して私たちのいくつかの候補製品のために求めることができる。例えば、Exacelは、TDTおよびSCDの治療のためにFDAによって許可されており、CTX 110は、再発または難治性B細胞リンパ腫の治療のために使用され、CTX 130は、真菌症およびSézary症候群(MF/SS)の治療のために使用される。2017年、FDAはRMATを“21世紀治療法”を実施する一部として指定し、以下の基準に適合する任意の薬物の検討を加速する:細胞療法、治療用組織工学製品、ヒト細胞および組織製品、またはこのような治療法または製品を使用する任意の組み合わせ製品であるRMATの定義に適合し、治療、修正、逆転または治癒を目的としている;深刻または生命に危険な疾患または状態を治療、修正または治癒することを目的としている;このような疾患または状態が満たされていない医療需要を満たす可能性があることを初歩的な臨床証拠は示す。RMAT指定は,画期的な治療指定と同様に,FDAとの会議で候補製品の開発計画をより頻繁に検討することや,スクロール審査や優先審査を行う資格があるかどうかを含む潜在的な利点を提供する。RMAT認証が付与された製品も、長期的な臨床的利益を合理的に予測する可能性のある代替物または中間終点に基づいて、またはより多くの場所に拡張することによって加速承認を得ることを含む、大量の場所から取得されたデータに依存する資格がある。FDAが私たちが開発可能な任意の候補製品が加速承認経路を通過することを可能にする保証はなく、FDAがこのような経路を確実に許可しても、このような提出または申請が受け入れられるか、または任意の加速された開発、審査、または承認がタイムリーに承認されることを保証することはできない, あるいはそうではありません加速的承認を得たRMAT指定製品は、電子健康記録のような臨床証拠、臨床試験、患者登録、または電子健康記録のような他の真の証拠源を提出することによって、より大きな検証データセットを収集することによって、または治療を承認する前に、そのような治療を受けたすべての患者を承認後に監視することによって、承認後の要求を満たすことができる。他の候補製品のRMAT認証を得ることができる保証はありません。RMAT指定はFDAの製品承認基準を変更することはなく,このような指定が審査や承認を加速する保証もなく,承認された適応が指定がカバーする適応よりも狭くならない保証もない。また,臨床データの出現に伴い資格基準を満たさなくなると,RMATの指定が取り消される可能性がある。また,加速的な承認を得ても,臨床的利益を確認·検証する必要がある承認後の研究は,この利点を示さない可能性があり,得られたいかなる承認も撤回される可能性がある。加速された承認を得ることは,製品の加速承認が最終的に従来の承認に変換される保証はない.さらに、FDORAによれば、FDAは、承認前または承認後の特定の期間内に1つまたは複数の承認後の検証的研究を行うことを適宜要求することができる
64
製品が加速的に承認された承認日。FDORAは,登録目標の実現を含めて180日ごとにFDAにこのような研究の最新状態を送信することも求められており,FDAはこれらの情報を迅速に公開しなければならない。FDORAはまた、スポンサーがこのような研究をタイムリーに行うことができなければ、必要な更新をFDAに送信するか、またはそのような承認後の研究が薬剤の期待される臨床的利益を検証できない場合、FDAは、加速的な承認を得た薬物または生物学的製剤の承認を迅速に撤回することができる。FDORAによれば、FDAは、職務調査を行っていない会社に対していかなる承認後の検証的研究を行うか、または速やかに当該機関に進捗報告を提出した会社に罰金を科すように行動する権利がある。
FDAが候補製品が重篤な疾患を治療する方法を提供すると判断し、承認された場合、製品は安全性または有効性の面で有意な改善を提供するであろう場合、FDAは候補製品を優先的に検討するように指定する可能性がある。優先審査指定は、FDA審査申請の目標が6ヶ月であり、標準的な10ヶ月の審査期間ではないことを意味する。FDAは候補製品優先審査地位を付与するか否かについて広範な裁量権を有しているため,特定の候補製品がこのような指定や地位を獲得する資格があると考えても,FDAはその資格を付与しないことを決定する可能性がある。また、従来のFDAプログラムと比較して、優先審査指定は必ずしも規制審査や承認過程を加速させるとは限らず、必ずしもいかなる承認面の優位性をもたらすとも限らない。FDAの優先審査を受けることは、6ヶ月の審査期間内に承認されることが保証されない、あるいは全くできない。
最後に,EMAからPrime計画下の候補製品資格を取得し,資格獲得を求めることが可能である。例えば、ExacelはTDTおよびSCDを治療するPrimeの称号を与えられている。Prime計画は開発中の薬物を開放し,申請者は集中プログラムで初歩的なMAAを申請しようとしている。条件に適合した製品は、満たされていない医療ニーズに対応しなければならず(EUには満足できる診断、予防または治療法がない、または、ある場合、新薬は重大な治療優位性をもたらす)、それらは、新しい方法または療法を導入することによって、既存の方法を改善することによって、満たされていない医療需要を満たす可能性を示さなければならない。私たちが他の候補製品のPrime資格を得ることができるという保証はない。Primeは製品承認の基準を変更することはなく,このような資質が承認速度を速める保証もない.また,1つの薬物が研究開発過程で資格基準を満たさなくなった場合,良質な計画下での援助は撤回される可能性がある。
私たちは、私たちのプラットフォーム技術を指定プラットフォーム技術として指定することを求めることができますが、このような指定を取得しても、そのような指定は、より速い規制審査や承認過程を招くことはありません。
私たちは私たちのプラットフォーム技術を指定されたプラットフォーム技術として指定することを求めることができる。“2022年食品·薬物総合改革法案”(“FDORA”)によると、以下の場合、(1)プラットフォーム技術がBLAによって承認されたまたは承認された薬物使用に組み込まれるか、または使用されるプラットフォーム技術が指定される資格がある。(2)承認または許可された薬物の発起人または薬物出願に提出されたデータ参照権を取得した発起人によって提出された予備的な証拠は、プラットフォーム技術が、品質、製造または安全に悪影響を与えることなく、1つ以上の薬物に組み込まれる可能性があるか、または使用される可能性があることを示し、(3)適用者が提出したデータまたは情報は、プラットフォーム技術の組み込むまたは利用が薬物開発または製造プロセスおよび審査プロセスに顕著な効率をもたらす可能性があることを示す。スポンサーは、IND出願を提出した間または後の任意の時間に、IND出願を要求対象とするプラットフォーム技術を組み込むか、または使用する指定されたプラットフォーム技術としてFDAに指定することを要求することができる。指定された場合、FDAは、プラットフォーム技術を使用または組み込まれた薬物の任意の後続の元のBLAの開発および審査を加速および検討することができる。我々のプラットフォーム技術がこのような指定の基準に適合していると考えても,FDAは同意せず,このような指定を付与しないことにした可能性がある.また,プラットフォーム技術のこのような指定を得ることは,薬物開発がより速くあるいはFDAの承認を得ることを保証することはできない。さらに何かがある, FDAが指定されたプラットフォーム技術がもはや指定された基準を満たしていないと判断した場合、FDAは指定を取り消すことができる。
私たちは孤児薬の指定や独占経営権を得ることができないかもしれない。もし私たちの競争相手が私たちの候補製品と同じ薬物と同じ適応を構成する製品を構成する孤児薬物の独占経営権を得ることができれば、私たちは長い間競争相手の製品に適用規制機関の承認を得ることができないかもしれない。
我々のいくつかのプロジェクトは、T細胞リンパ腫の治療のためのCTX 130を含むFDAの孤児薬物指定を米国で取得した。FDAと欧州委員会が指定したTDTとSCDを治療する孤児薬も獲得した。私たちは将来私たちのいくつかの他の候補製品のために孤児薬物指定を求めるかもしれないが、私たちは孤児薬物指定を維持したり、孤児薬物指定に関連するいかなる利益も得ることができないかもしれない。米国とEUを含むいくつかの司法管轄区の規制機関は、比較的少ない患者集団の治療を目的とした薬物と生物製品を孤児薬物として指定する可能性がある。1983年の“孤児薬物法案”によると、まれな疾患や状態を治療しようとする場合、FDAは候補製品を孤児薬に指定することができる。この疾患または状態の定義は、米国では患者数が20万人未満、または米国では患者数が20万人を超えるが、米国では米国の販売から薬物開発コストを回収できるという合理的な期待はない。EUでは欧州委員会は
65
EMA孤児薬物製品委員会の提案は、生命または慢性衰弱を脅かす疾患の診断、予防または治療を目的とした孤児薬物の開発を促進するために、EUの10,000人当たり5人以下に影響を与えることを提案している。さらに、生命に危害を及ぼす、深刻な虚弱または深刻かつ慢性疾患の診断、予防または治療のための製品、およびインセンティブがない場合、EUでの販売は、孤児として指定することができる薬剤または生物学的製品の開発に投資する必要があることを証明するのに十分ではない可能性が高い。孤児薬物指定は、費用の削減、規制援助、EUでの集中販売許可を申請する能力を含む多くの利点を提供する。
私たちの現在のいくつかの候補製品および将来の候補製品は、上述した数字よりも少ない患者集団を対象としている可能性がある。もし私たちが私たちの候補製品に孤児薬を指定することを要求すれば、FDAまたは欧州委員会が私たちの候補製品がそのような名前を指定することを承認する保証はありません。さらに、我々の任意の候補製品を孤立製品として指定することは、どの規制機関がその候補製品の規制審査または最終承認を加速するかを保証するものではなく、いかなる規制機関も、私たちの候補製品が独占的に発売承認される前に、私たちの候補製品と同じ適応を有する他社の候補製品に孤立薬物指定の能力を付与することを制限しない。
一般に、孤児薬物指定を有する候補製品が、その指定された適応を有する最初の発売許可を得た場合、製品は、限定された場合を除いて、FDAまたは欧州委員会が同じ薬剤を構成する製品の別のマーケティング申請を承認することを阻止する市場排他的期間を有する権利がある。もし他のスポンサーが私たちの前にこのような承認を得た場合(私たちの孤児薬物名にかかわらず)、私たちは適用された専門期間内に私たちの製品の発売許可を得ることが禁止されるだろう。適用期間は米国では7年、EUでは10年。スポンサーが提出した小児科データがこのようなデータに対するFDAの書面請求に公平に対応すれば,米国での排他期間を6カ月延長することができる。5年目の終了時に、製品が指定された孤児薬の基準を満たしていないと判定された場合、欧州連合の専門期間は、例えば、製品が十分な利益を有するため、市場独占権を得る理由がなくなることが6年に短縮されることができる。任意の規制機関が、指定された要求に重大な欠陥があると判断した場合、または製造業者が、このようなまれな疾患または疾患を有する患者の需要を満たすのに十分な数の製品を保証できない場合、孤児薬の排他性を取り消すことができる。
候補製品の孤立した薬物排他性を獲得しても,この排他性は候補製品を競合から効果的に保護できない可能性があり,異なる薬物が同じ条件で承認される可能性があるからである。米国では、孤児薬が承認された後であっても、FDAが他の薬剤が同一の薬剤ではないと結論した場合、それがより安全で、より有効であることが証明された場合、または患者ケアに大きな貢献をした場合、FDAはその後、同じ疾患の治療を別の薬剤で承認することができる。欧州連合では、以下の場合、同じ孤児適応の類似医薬製品にマーケティング許可を与えることができる
私たちが他の候補製品の孤児薬物の称号を得ることができるという保証はない。孤児薬の指定は製品承認の基準を変更することはなく,そのような指定が迅速な審査や承認につながることも保証されない。
遺伝子編集および細胞療法製品に対する公衆の否定的な見方は、私たちの候補製品の需要や規制承認に負の影響を与える可能性がある。
私たちの候補製品はヒトゲノムの編集に関するものだ。私たちの候補製品の臨床と商業成功は遺伝子編集療法を用いたヒト疾患の予防或いは治療に対する公衆の受容の程度に部分的に依存する。公衆の態度は遺伝子編集の不安全、不道徳或いは不道徳な説の影響を受ける可能性があるため、私たちの製品は公衆或いは医学界の受け入れを得ることができない可能性がある。遺伝子治療に対する公衆の一般的な負の反応は、政府が遺伝子編集製品(私たちの任意の候補製品を含む)に対するより厳格な規制とより厳しいラベル要求をもたらす可能性があり、私たちが開発する可能性のある任意の製品に対する需要の減少を招く可能性がある。公衆の悪い態度は私たちが臨床試験を募集する能力に悪影響を及ぼすかもしれない。さらに、私たちの成功は、医師が処方した処方および彼らの患者が受けることを望む治療に依存し、これらの治療は、彼らがよく知っている既存の治療方法を代替または補充するために、私たちが開発可能な候補製品を使用することに関連し、より多くの臨床データを得ることができる。
66
特に、遺伝子編集技術を人類胚胎或いは人類生殖系に応用することに関する倫理問題のため、遺伝子編集技術は公衆討論とより厳格な監督管理審査を受けている。例えば、2016年4月、ある科学者たちは、ヘモグロビンβ遺伝子を修正するために、ヒト胚のゲノムを編集しようと試みた試みを報告した。これは遺伝性血液疾患β地中海貧血患者に変異を生じる遺伝子である。この研究は生存できない胚の中で目的的に行われているにもかかわらず、この仕事はヒト卵子、精子、胚の遺伝子編集を一時停止または他のタイプに制限することを呼びかけている。また、報告によると、2018年11月、深セン南方科技大学生物系助教授、生物物理学研究員の何建奎博士は、最初のヒト遺伝子編集の赤ちゃん、双子の女の子を作ったと主張している。この説と、何博士が第二の遺伝子編集者の妊娠を助け、その後中国当局の実証を得て、公衆の負の反応、特に科学界の人を得たという説がある。新聞記事によると、2019年12月、何医師はこのような活動に関連した不法な医師行為で中国政府に懲役3年、罰金43万ドルを言い渡された。その後、世界保健機関は新しい諮問委員会を設立し、人類遺伝子編集のためにグローバル管理と監督標準を制定した。ワシントンD.C.再生医学連盟はCRISPR/Cas 9を含む遺伝子編集技術の自発的な使用停止を呼びかけている, ヒト胚やヒト生殖細胞の改変に関する研究では,治療応用に遺伝子編集を用いる原則も発表され,遺伝子編集技術を用いた会社の承認を得ている。同様に、米国国立衛生研究院は、ヒト胚におけるいかなる遺伝子編集技術の使用にも援助しないと発表し、研究目的のためのヒト胚の創造またはヒト胚の破壊のための研究への支出を禁止するディキ-ウィック修正案を含む既存の複数の立法および規制機関がこのような仕事を禁止していることを指摘した。イギリスの法律は遺伝子組換え胚胎を女性体内に移植することを禁止しているが、人類受精と胚胎学管理局の許可を得た場合、研究実験室は胚胎を変えることができる。他の多くのヨーロッパ諸国では、胚の研究はもっと厳格に統制されている。
私たちは私たちの技術を使用してヒト胚胎またはヒト生殖系を編集しないが、このようなヒト胚胎における遺伝子編集技術の使用に関する公開討論およびより厳格な規制審査は私たちの候補製品の開発を阻止または延期する可能性がある。より厳格な政府法規または負の世論は、私たちの業務や財務状況に悪影響を与え、候補製品の開発および商業化、または私たちが開発する可能性のある任意の製品の需要を遅延または損害する可能性がある。我々の前臨床研究または臨床試験または我々の競争相手または遺伝子編集技術を使用する学術研究者の有害事象は、最終的には、我々が識別および開発する可能性のある候補製品に起因することができなくても、それによって生じる宣伝は、政府規制の増加、不利な公衆認識をもたらす可能性があり、私たちが識別し開発する可能性のある潜在的候補製品テストまたは承認の潜在的規制遅延、承認された候補製品に対するより厳しいラベル要件、およびそのような候補製品のいずれかに対する需要が減少する可能性がある。
もし私たちが販売やマーケティング能力を確立できない場合、あるいは第三者と合意して私たちの技術に基づく製品を販売し、マーケティングすることができなければ、任意の候補製品が承認されれば、私たちの製品を商業化することに成功できないかもしれません。どんな収入も生まれないかもしれません。
私たちは現在販売やマーケティングインフラを持っていません。会社として、治療製品の販売、マーケティング、流通については経験がありません。販売およびマーケティング責任を保持する任意の承認された候補製品をビジネスに成功させるためには、私たちの販売、マーケティング、管理、および他の非技術的能力を確立するか、または第三者とこれらのサービスを実行することを手配しなければならない。将来的には、私たちのいくつかの候補製品を販売するために、あるいは私たちの協力者と一緒に販売活動に参加するために、重要な販売·マーケティングインフラを構築することを選択することができるかもしれません。
私たち自身の販売とマーケティング能力の確立、および第三者とこれらのサービスを実行する計画を達成することはリスクに関連している。例えば、販売チームの採用と訓練は高価で時間がかかり、どんな製品の発表も延期される可能性がある。販売チームを募集し、マーケティング能力を確立する候補製品の商業発表が何らかの理由で延期または発生していない場合、これらの商業化費用を早期または不必要に発生させる。これは費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり再配置できなければ、私たちの投資は損失します。
候補製品の商業化を阻害する可能性がある要素は
もし私たちが第三者と販売、マーケティング、流通サービスの手配を達成すれば、私たちの製品の収入あるいはこれらの収入の流れから私たちにもたらす収益力は、私たちが自分たちが開発した任意の候補製品をマーケティングして販売する場合よりも低いかもしれません。たとえば,我々のA&R Vertex JDCAにより,Vertexはすべての商業化活動を行う権利がある
67
A&R Vertex JDCAによる純利益と純損失(適用すれば)により40%をCRISPR,60%をVertexに分配した。さらに、私たちは第三者と私たちの候補製品の販売とマーケティングの手配を成功的に達成できないかもしれないし、私たちに有利な条項でそうすることができないかもしれません。私たちはこれらの第三者に対して支配権がほとんどないかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの候補製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが販売とマーケティング能力を確立することに成功できなければ、私たち自身も第三者と協力しても、私たちの候補製品を商業化することに成功しないかもしれない。しかも、私たちの業務、経営業績、財務状況、そして見通しは重大な悪影響を受けるだろう。
私たちまたは私たちが所有する可能性のある任意のパートナーが私たちが開発した任意の候補製品のためにマーケティング承認を得ても、承認条項と私たちの製品の持続的な規制は、大量の資源支出を必要とする可能性があり、私たちまたは彼らが私たちの製品を製造してマーケティングする方法を制限する可能性があり、これは私たちの収益能力を深刻に弱めるかもしれない。
私たちが発売許可を得た任意の候補製品、及びその製品の製造プロセス、承認後の臨床データ、ラベル、広告と販売促進活動は、FDAと他の監督管理機関の持続的な要求と審査を受ける。これらの要求には、安全及びその他の上場後の情報と報告の提出、登録と上場要求、現在の良好な製造規範又はcGMP、品質管理、品質保証と記録及び文書の対応する維持に関する要求、及び記録保存に関する要求が含まれる。候補製品の上場が承認されても、承認は、製品が発売される可能性のある指定用途の制限または承認条件によって制限される可能性があり、または製品の安全性または有効性を監視するために、高価な上場後のテストおよび監視の要求を含む可能性がある。FDAはまた、製品の安全な使用を保証するためにREMSを要求することを含む、承認時に他の条件を追加することができる。FDAがREMSが必要であると結論した場合,BLAのスポンサーは承認を得るために提案されたREMSを提出しなければならない。REMSは、制限された分配方法、患者登録、および他のリスク最小化ツールのような薬物ガイドライン、医師のコミュニケーション計画、または安全な使用を確保する要素を含むことができる。
したがって、私たちまたは私たちが所有する可能性のある任意の協力者が私たちが開発した1つまたは複数の候補製品のマーケティング承認を得たと仮定すると、私たちとそのような協力者と私たちと彼らの契約製造業者は、製造、生産、製品監視、品質管理を含むすべてのコンプライアンス分野で時間、お金、エネルギーをかけ続けます。また,承認されたBLAの保持者は,不良イベントやどの製品もBLAの規格に適合していない場合を監視·報告する義務がある.承認されたBLAの所有者はまた、新しい申請または追加の申請を提出し、承認された製品、製品ラベル、または製造プロセスをいくつか変更するためにFDAの承認を得なければならない。他の適用可能な連邦および州法律に加えて、広告および販売促進材料はFDAの規定に適合し、FDAの審査を受けなければならない。
もし私たちとそのような協力者が承認後の規制要求を遵守できなければ、私たちとそのような協力者は規制機関によって私たちの製品のマーケティング承認を撤回される可能性があり、私たちまたはそのような協力者が未来の製品をマーケティングする能力が制限される可能性があり、これは私たちが利益を達成したり維持したりする能力に悪影響を及ぼすかもしれない。また、承認後の法規を遵守するコストは、私たちの業務、経営業績、財務状況、見通しにマイナス影響を与える可能性があります。
私たちまたは私たちの任意のパートナーがマーケティング承認を得た任意の候補製品は、私たちまたは彼らが規制要求を遵守できなかった場合、または私たちまたは彼らの製品が予期せぬ問題に遭遇した場合、その中の任意の製品が承認された場合、私たちまたは彼らは重大な処罰を受ける可能性がある。
FDAおよび他の規制機関は、許可された適応のみおよび承認されたラベルの規定による販売を確実にするために、生物製品の承認後のマーケティングおよび販売促進を密接に規制する。FDAおよび他の規制機関は、ラベル外使用に関するメーカーのコミュニケーションに厳しい制限を加えており、もし私たちまたは私たちが所有している可能性のあるいかなるパートナーも、その承認された適応について私たちの製品をマーケティングしていない場合、私たちまたは彼らは、FDAおよび他の連邦および州法執行機関(米国司法省を含む)のラベル外マーケティング法執行行動の影響を受ける可能性がある。“虚偽請求法”を含む“連邦食品、医薬品および化粧品法”および処方製品販売促進および広告に関する他の法規に違反し、連邦および州医療詐欺および法律乱用および州消費者保護法違反を調査または告発する可能性もある。
さらに、候補製品には、予期されない深刻度または頻度の不良事象、または私たちまたは他の協力者の製造プロセス、または法規要件を遵守できなかったことを含む、以前に未知の問題が存在することが後に発見され、他のことを除いて、以下のようになる可能性がある
68
FDAの政策は変わる可能性があり、私たちの候補製品に対する規制承認を阻止、制限、または延期するための追加の政府法規が公布されるかもしれない。もし私たちまたは私たちの協力者が既存の要求の変化に緩やかに適応できない場合、または新しい要求または政策を採用することができない場合、または私たちまたは私たちの協力者が法規遵守を維持できない場合、私たちまたは私たちの協力者は、私たちまたは私たちの協力者が得る可能性のあるマーケティング承認を失う可能性があり、これは、私たちの業務、将来性、および利益を達成または維持する能力に悪影響を及ぼすだろう。
どの政府も、私たちのどのサプライヤーに対する調査も含め、違法な疑いのある調査には、対応するために多くの時間と資源を必要とする可能性があり、否定的な宣伝が生じる可能性がある。上記のいずれの事件や処罰が発生しても、私たちまたは私たちの協力者が私たちが開発する可能性のある任意の候補製品を商業化する能力を抑制し、私たちの業務、財務状況、運営結果、および将来性に悪影響を及ぼす可能性があります。
私たちの任意の候補製品の商業的成功は、それが医師、患者、第三者支払人、医療界の他の人に受け入れられる程度に依存するだろう。
遺伝子治療の倫理、社会、法律に対する懸念は、追加の法規制や私たちの製品の禁止を招く可能性がある。米国FDA、EU EMAと他の国際規制機関の必要な承認を得ても、私たちの候補製品の商業成功は医師、患者と一般遺伝子治療製品の医療保健支払い者が受け入れているかどうか、特に私たちの候補製品が医療必要性、費用効果と安全性を持っているかどうかに大きく依存する。私たちが商業化したどの製品も医師、患者、医療支払者、医学界の他の人に受け入れられないかもしれない。遺伝子治療製品、特に私たちの候補製品に対する市場の受容度は、商業販売のために承認されれば、いくつかの要素に依存する
69
1種の潜在的な製品が臨床前研究と未来の臨床試験において良好な治療効果と安全性を示しても、市場のこの製品に対する受容度はそれが発売されてから完全に知ることができる。もし私たちの候補製品が規制承認後に十分な受容度に達していなければ、もしあれば、著しい製品収入が生じないかもしれませんし、利益を上げることができないかもしれません。
技術が急速に変化する環境では、私たちは激しい競争に直面しており、私たちの競争相手は私たちの前に規制部門の承認を得たり、私たちよりも先進的で効果的な療法を開発したりすることができ、これは私たちの業務や財務状況を損なう可能性があり、私たちの候補製品のマーケティングや商業化に成功したりする能力を損なう可能性がある。
生物技術と製薬業界は、遺伝子編集、遺伝子治療と細胞治療領域を含み、その特徴は技術が迅速に進歩し、競争が激しく、そして知的財産権と独自製品を高度に重視していることである。私たちの技術、開発経験、科学知識は私たちに競争優位を提供してくれると信じていますが、私たちは現在、大型製薬、専門製薬とバイオテクノロジー会社、学術機関と政府機関、および公共および民間研究機関を含む多くの異なる源からの激しい競争に直面し、引き続き直面しています。その中のいくつかまたはすべての機関は私たちよりも資本や資源を獲得しやすいかもしれません。我々が最終的に商業化する可能性のある任意の製品については,任意の既存の療法や現在開発されている療法と競合するだけでなく,将来出現する可能性のある新しい療法とも競合するであろう。
我々は製薬,バイオテクノロジー,その他の関連市場で競争を展開しており,これらの市場では遺伝子編集,遺伝子療法,細胞療法を含むゲノム薬物をカバーする技術を用いて療法を創造している。また,我々の特定の研究開発計画に関連する分野で治療法の開発に取り組んでいる会社と競争している。
我々のプラットフォームと製品はCRISPR/Cas 9遺伝子編集技術を用いた療法の開発に重点を置いている。Intellia治療会社やEditas Medicine社を含むCRISPR/Cas 9遺伝子編集技術を用いた様々な適応の治療法の開発に専念している会社がいくつか知られている。また,いくつかの学術グループでは,CRISPR/Cas 9に基づく新しい遺伝子編集技術,例えば塩基編集やモチーフ編集が開発されており,これらの技術は治療開発において実用的な価値がある可能性がある。これらの技術に基づく治療法の開発を求めている会社はBeam TreateuticsとPrime Medicineです
TALEN、マクロヌクレアーゼ、ZFNなどの追加の遺伝子編集技術を用いて療法を開発している会社もある。これらの会社は270の生物会社、異遺伝子治療会社、Cellectis社、精密生物科学会社、Sangamo治療会社を含む。
いくつかの会社が我々の具体的な研究開発計画に関連した様々な分野で療法を開発していることも知られている。ヘモグロビン疾患については,これらの会社には,ビム治療会社,ブルーバード生物社,Editas Medicine社,黒鉛生物社,メルク社,ノワ製薬会社,ファイザー社,Sangamo治療会社がある。免疫腫瘍学では、これらの会社は270生物会社、アディケイト生物会社、異遺伝子治療会社、百時美施貴宝会社、Cariou生物科学会社、Cellectis社、世紀治療会社、運命治療会社、ギレド科学会社、伝説生物会社、ノワ製薬会社、ポセイダ治療会社、精密生物科学会社を含む。再生医学分野では,これらの会社にはBluerock Treateutics(2019年にバイエルに買収された),Sana Biotech,Semma Treateutics(2019年にVertexに買収)が含まれている。はい体内にあるこれらの会社にはAlnylam製薬会社,矢印製薬会社,BioMarin製薬会社,Intellia治療会社,Ionis製薬会社,Verve治療会社がある。
遺伝子編集は非常に活発な研究分野であり、CRISPRに関連したり、関係のない新しい技術が発見され、新しい競争が生じる可能性がある。これらの新技術はいくつかの応用においてCRISPR/Cas 9遺伝子編集よりも優れている可能性があり、他の遺伝子編集技術が製品開発において我々の技術よりも良いあるいは魅力的であるとは考えられないことは確信できない。例えば、Cas 9は、Cas 12 aまたはまだ発見されていない新規なCas酵素、または塩基エディタおよび一次エディタのような他のCRISPRタンパク質に劣る他のCRISPR関連ヌクレアーゼ変異体を決定することができる。
他の遺伝子編集療法または遺伝子または細胞療法からの競合に加えて、我々が開発する可能性のある任意の製品は、小分子、抗体またはタンパク質療法のような他のタイプの療法からの競合に直面する可能性もある。また,新たな科学的発見はCRISPR/Cas 9技術,あるいは全体的な遺伝子編集を招く可能性があり,劣悪な治療形態と考えられている。
また、私たちの多くの既存または潜在的な競争相手は、単独またはパートナーと協力しても、研究開発、製造、臨床前テスト、臨床試験、監督管理許可およびマーケティング承認を得た製品の面で、私たちよりも多くの財務資源と専門知識を持っている。製薬、生物技術、遺伝子と細胞治療業界の合併と買収はより多くの資源が私たちの少数の競争相手に集中する可能性がある。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。もし私たちの競争相手が私たちが開発する可能性のあるどんな製品よりも安全で、より効果的で、副作用が少なく、より深刻ではなく、より便利で、より広い受容度とより高い販売率を開発し、あるいは私たちが開発する可能性のあるどの製品よりも安い製品であれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちより早くfdaや他の規制機関の製品の承認を得るかもしれません。これは私たちの競争相手が私たちに入ることができるかもしれません
70
市場です。また、私たちの競争相手が開発した技術は、私たちの潜在的な候補製品を経済的あるいは時代遅れにする可能性があり、私たちは私たちが開発する可能性のあるいかなる競争相手の候補製品もうまくマーケティングできないかもしれません。私たちのすべての計画の成功に影響を与える重要な競争要素は、それらの有効性、安全性、利便性、精算の利用可能性である可能性がある。
私たちの現在のプロジェクトが現在計画されている臨床試験の適応に使用されていれば、遺伝子編集、遺伝子治療、細胞治療製品を含む現在開発されている他の製品と競争する可能性がある。現在開発中の他の関連製品との競争には,臨床試験地点,患者募集,製品販売の競争が含まれている可能性がある。また,遺伝子編集分野では緊張した研究や開発が行われているため,我々と我々のライバルを含めて知的財産権の構造が変化しており,競争は非常に激しい.将来的には知的財産権に関する重大な訴訟や、私たちが所有しているものや許可されていない他の第三者、知的財産権、独自の権利に関する訴訟があるかもしれない。例えば、048干渉、115干渉、および欧州野党手続きの議論を参照されたい知的財産権に関連するリスク-第三者が私たち、私たちのライセンシー、または私たちの協力者に提出した知的財産権侵害クレームは、私たちの製品発見と開発を阻害または延期する可能性があります。”
さらに、私たちの特許権の満期または成功の挑戦により、私たちの競争相手の製品に関連する特許の有効性および/または範囲に関するより多くの訴訟に直面する可能性があり、私たちの特許は、競争相手が競合製品を商業化することを阻止するのに十分ではないかもしれません。私たちの競争相手の製品供給は、私たちが開発し商業化する可能性のある任意の製品の需要と私たちが受け取ることができる価格を制限するかもしれません。
私たちがどんな候補製品を商業化することができても、これらの製品は不利な価格設定法規、第三者精算やり方、あるいは医療改革措置の制約を受ける可能性があり、これは私たちの業務を損なうことになる。
新生物製品の発売承認、定価と精算を管理する法規は国によって異なる。一部の国は製品の販売価格が発売前に承認されることを要求しています。多くの国で、定価審査期間はマーケティングまたは製品許可を承認した後から始まる。一部の非米国市場では、初歩的な承認を得た後も、処方薬の定価は政府の持続的なコントロールを受けている。したがって、特定の国/地域での製品のマーケティング承認を得ることができるかもしれませんが、その後、価格法規の制約を受けて、これらの法規は私たちの製品の商業発表を延期し、長い時間遅延し、その国/地域で製品を販売することによって生じる収入に悪影響を与える可能性があります。不利な価格設定制限は、私たちが開発可能な任意の候補製品が市場承認を得ても、1つ以上の候補製品への投資を回収する能力を阻害する可能性がある。
私たちがすべての製品を商業化することに成功した能力はまた、政府衛生行政部門、個人健康保険会社と他の組織によるこれらの製品と関連治療の清算程度にある程度依存する。第三者支払者、例えば個人健康保険会社、健康維持組織、政府計画、例えば連邦医療保険と医療補助は、彼らがどのような薬物を支払い、精算レベルを確立するかを決定する。アメリカの医療業界と他の地域の主な傾向の一つはコストコントロールだ。政府と個人第三者支払者は,特定の薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。ますます多くの第三者支払人は製薬会社に価格に基づいて所定の割引を提供し、医療製品の定価に挑戦することを要求している。私たちは商業化されたどの製品も精算できることを確実にすることはできません。もし精算できれば、精算レベルもそうです。精算は私たちが市場で承認された任意の候補製品の需要や価格に影響を及ぼすかもしれない。清算が得られない場合や限られたレベルに限られていれば、マーケティングの承認を得た任意の候補製品を商業化することに成功できないかもしれません。タイトルを見て“業務-カバー範囲、定価、精算” and “ビジネス-医療改革.”
新たに承認された製品は精算を得る上で重大な遅延がある可能性があり、精算範囲はFDAや米国以外の同様の規制機関がこの製品を承認する目的よりも限られている可能性がある。さらに、精算を受ける資格があるということは、どの製品もすべての場合に支払われることを意味するわけではありません。あるいは支払いの費用率は、研究開発、製造、販売、流通を含めて私たちのコストをカバーします。新製品の臨時精算水準(適用すれば)も私たちのコストを支払うのに十分ではない可能性があり、恒久的にならない可能性があります。販売率は製品の使用や臨床環境によって異なる可能性があり,すでに低コスト製品のために設定された精算レベルに基づいている可能性があり,他のサービスの既存支払いにも組み込まれている可能性がある。製品の正味価格は、政府医療計画または個人支払者が要求する強制的な割引またはリベート、および将来の法律の任意の緩和によって低下する可能性があり、これらの法律は、現在、製品が米国より低い価格で販売されている国からの製品の輸入を制限している。第三者支払者が自分の精算政策を設定する際には,通常連邦医療保険カバー政策と支払制限に依存する。私たちは、私たちが開発する可能性のある任意の承認された製品の保証範囲と利益の支払率を政府援助および個人支払者から迅速に得ることができず、これは、私たちの経営業績、製品の商業化に必要な資金を調達する能力、および私たちの全体的な財務状況に実質的な悪影響を及ぼすかもしれない。
71
私たちの第三者との関係に関するリスクは
我々のパートナーおよび戦略パートナーは、私たちの臨床試験および商業化努力の様々な側面を制御することができ、これは、私たちが提案する製品の商業化の遅延や他の障害を招き、私たちの運営結果に実質的な損害を与える可能性がある。
我々はVertexおよびViaCyte(後者は2022年にVertexに買収された)と戦略的協力と許可を達成し、将来的に他の第三者とより多くの協力と許可を達成する可能性がある。いくつかのプロジェクトについては、私たちはまた、当社の臨床試験、および任意の承認された製品の商業化を設計および実施するために、第三者パートナーおよび戦略パートナーに依存しているか、または将来的に依存する可能性がある。その中のいくつかの協力は、私たちの候補製品をより全面的に開発するために重要な技術を提供してくれます。私たちは他の会社と協力して、私たちの計画に重要な技術や資金を提供するかもしれません。このような計画の成功は私たちの協力者と許可パートナーの努力と活動に大きく依存するだろう。
協力者は通常、彼らがこれらの協力の仕事および資源に適用されることを決定する上で大きな裁量権を持ち、協力者は予想通りに彼らの義務を履行しないかもしれない。場合によっては、私たちのパートナー候補製品の開発および商業化に関する私たちのパートナーの意思決定に影響を与えることができない可能性がありますので、私たちのパートナーは、これらのパートナー候補製品の開発および商業化を最適な利益に合った方法で追求または優先しないかもしれません。さらに、協力者が競争力のある製品がより成功的に開発される可能性があると考えている場合、または我々の製品よりも経済的に魅力的な条項で商業化することができる場合、協力者は、第三者開発と直接または間接的に我々の候補製品と競合する製品を独立して開発することができ、または第三者開発と直接または間接的に競合する製品を開発することができる。協力手配当事者間の臨床開発と商業化問題における相違は開発過程の遅延或いは適用候補製品の商業化を招く可能性があり、場合によっては、協力手配の終了或いは訴訟或いは仲裁を招くことは時間がかかり、高価になる。パートナーは、候補製品や製品の開発、製造、流通、マーケティングに関する適用法規要件を遵守できない可能性もある。ライセンシーは通常、ライセンス製品に適用する努力と資源を決定する上で唯一の裁量権を持っている。
したがって、私たちは私たちが現在考えている方法やスケジュールに従ってどんな協力プロジェクトも実行できないかもしれません。これは私たちの業務運営に悪影響を及ぼすかもしれません。さらに、これらのパートナーまたは戦略パートナーのいずれかが、私たちの計画または提案された製品の支援を撤回したり、他の方法でその開発または商業化を損なう場合、私たちの業務は負の影響を受ける可能性があります。さらに、もし私たちの協力者が私たちとの合意を終了したら、私たちは新しい協力者を引き付けることがもっと難しいことを発見するかもしれませんし、ビジネスや金融界での私たちの見方は不利な影響を受けるかもしれません。
私たちはVertexと私たちのLead計画Exacelでパートナーシップを構築した;VertexはExacel計画に対して大きな支配権を持っている。
我々はすでにVertexと一連のプロトコルを締結しており、様々な目標に関連するいくつかの研究、開発、製造、商業化活動を考慮している。これらのプロトコルによると、Vertexはある活動を展開する独占的な権力を持っている。例えば,我々が2015年にVertexと達成した研究,開発,商業化ヒト疾患の潜在的遺伝原因に対する新しい療法の協力合意によると,Vertexは追求する遺伝目標を選択する唯一の権力を持っており,選択された遺伝目標に対する任意の候補製品の開発を制御することはできない。また,Vertexとの2019年の協力プロトコルによると,Vertexはこのプロトコルに従ってDMDおよびDM 1を治療する製品を開発·商業化する独占的な権力を持っている(DM 1を共同開発·共同商業化する製品を選択することに依存する).さらに、Vertexは、2022年第3四半期に、世界各地の1型糖尿病、2型糖尿病およびインスリン依存型/インスリンを必要とする糖尿病の治療のための共通の権利を有し、候補製品および共有製品を開発および商業化するViaCyteを買収したと発表した。
また,我々はVertexと連携し,A&R Vertex JDCAの下でTDTやSCD用のExaccelを商業化することを開発し準備している.A&R Vertex JDCAによると、Vertexは、このプロトコルの条項および条件に基づいて、指定された候補製品や製品(Exexcelを含む)に関するすべての研究、開発、製造、商業化活動を世界各地で行う権利があるが、いくつかの活動を行う権利を保持している。我々は、当事者が他の合意がない限り、観察者としてこのような活動のいくつかの態様に参加し続け、私たちとVertexは、共同監視委員会および移行委員会において同等の数の代表を有するが、Vertexは、ExaccelまたはA&R Vertex JDCAに制約された任意の将来の候補製品の開発を制御する。
72
Vertexとのいくつかのプロトコルでは、臨床開発、製造、規制提出および商業化活動の制御が不足しており、候補製品の開発および商業化に遅延または他の困難をもたらす可能性があり、これは、予期されるIND申請のタイムリーな完了を阻止するか、またはBLA申請の完了または遅延を阻止する可能性がある。例えば、私たちは、私たちが協力したEXACES計画から得られたデータが、臨床的意義のある利益を示すことを示すか、またはBLAの承認を支持することを示す保証はなく、安全性および/または有効性の観点から、SCRADE−111およびSCRADE−121臨床試験からのデータは、条件付き承認または完全な承認をサポートするのに十分に強力であるとは決定できない。FDAは私たちとVertexに追加的またはより大規模な重要な実験を行うことを要求し、その後、私たちとVertexはスクロール提出を完了したり、BLAの承認を得ることができるかもしれない。また,BLA提出完了条件として活性化剤の品質,純度,強度(効力を含む)を確認するための何らかの放出アッセイに関するデータを提出する必要がある。A&R Vertex JDCAにより,Vertexはこのような臨床試験と製造を担当している。Vertexが必要なデータをタイムリーに提出できない場合、我々のBLA提出の完了をさらに延期する可能性があり、米国におけるExexcelの任意の承認およびビジネス開始遅延を招く可能性がある。
また、Vertexとの合意を終了することは、このプロトコルに基づいていかなるマイルストーン、特許権使用料、他の福祉を得ることができなくなり、これは私たちの運営結果に大きな悪影響を及ぼす可能性がある。
もし私たちが私たちの協力者や戦略的パートナーと衝突すれば、これらの当事者たちは私たちに不利な方法を取り、私たちの戦略を実施する能力を制限するかもしれない。
もし私たちの企業や学術パートナーや戦略パートナーが私たちと衝突した場合、相手は私たちに不利な方法で行動し、私たちの戦略を実施する能力を制限するかもしれません。私たちのいくつかの学術協力者と戦略パートナーは私たちが協力する各分野と複数の製品開発を行っています。しかしながら、我々の協力者または戦略的パートナーは、これらの協力テーマである製品または潜在的な製品と競争力を有する関連分野の製品を単独でまたは他の人と一緒に開発することができる。例えばVertexは独自の所有を優先する可能性があります
糖尿病計画は,我々とViaCyte(同社は2022年にVertexに買収された)の糖尿病計画を損なう。協力者または戦略パートナーによって開発された競合製品であっても、協力者または戦略パートナーによって権利を有する競合製品であっても、パートナーが候補製品の支援を撤回することをもたらす可能性がある。
現在または未来のパートナーや戦略的パートナーもまた私たちの未来の競争相手になるかもしれない。私たちの協力者や戦略パートナーは競争製品を開発し、私たちがその競争相手との協力を阻止し、適時に規制の承認を得ることができず、私たちとの合意を早期に終了したり、製品の開発と商業化に十分な資源を投入することができなかった。このような進展のいずれも私たちの製品開発努力を損なう可能性がある。
私たちの協力者や戦略パートナーは、代替技術を採用することを決定するか、あるいは私たちの技術を使用して商業的に実行可能な製品を開発することができないかもしれません。これは、私たちの収入とこれらの製品を開発する戦略に負の影響を与えます。
我々の協力者や戦略的パートナーは代替技術を採用する可能性があり,これは我々のCRISPR/Cas 9遺伝子編集技術の市場性を低下させる可能性がある.例えば,Vertexも糖尿病に対する他の候補治療製品を発売している。また、私たちの現在の協力者または戦略的パートナーは、任意の未来の協力者または戦略パートナーが複数の開発プロジェクトに取り組むことが予想されているので、彼らは、彼らの資源を私たちと協力しているプロジェクト以外のプロジェクトに移すことを選択することができる。もし彼らがそうすれば、これは私たちの技術の能力をテストすることを延期し、私たちのCRISPR/Cas 9遺伝子編集技術に基づく潜在的な製品の開発を延期または終了するだろう。さらに、私たちの協力者および戦略的パートナーは、私たちの協力および戦略的協力計画によって生成された製品を開発しないか、またはこれらの製品の開発、製造、マーケティング、または販売に十分な資源を投入しないことを選択することができる。現在または将来のパートナーとの合意に基づいて候補製品を開発·商業化できなければ、将来のマイルストーンや特許権使用料の支払いを受けることができず、私たちの収入に負の影響を与えるだろう。
私たちはより多くの協力を求めるかもしれませんが、ビジネス的に合理的な条件下で協力を作ることができなければ、私たちの開発と商業化計画を変えなければならないかもしれません。
私たちの候補製品開発計画と候補製品の潜在的な商業化は費用を支払うために多くの追加の現金が必要になるだろう。私たちのいくつかの候補製品については、他の製薬やバイオテクノロジー会社と協力して、これらの候補製品を開発し、潜在的な商業化を行うことを決定するかもしれません。
73
私たちは適切な協力者を探すことで激しい競争に直面している。私たちが他の協力について最終的な合意に到達するかどうかは、他の事項に加えて、協力者の資源と専門知識の評価、協力の条項と条件、提案協力者のいくつかの要素の評価に依存する。これらの要因は、臨床試験の設計または結果、FDAまたは米国海外類似規制機関によって承認された可能性、候補製品の潜在的市場、そのような候補製品の製造および患者への送達のコストおよび複雑性、競争薬物の潜在性、挑戦の利点および全体的な業界および市場条件を考慮せずにこのような所有権に挑戦する場合、存在する可能性のある不確実性を含むことができる。協力者はまた、同様の協力可能な指示を得るための代替候補製品または技術を考慮することができ、そのような連携が、私たちと私たちとの連携よりも私たちの候補製品に魅力的であるかどうかを考慮することができる。私たちが設立する可能性のある他の協力や他の計画の条項は私たちに不利かもしれない。
既存の協力協定によれば、潜在的なパートナーと特定の条項について未来の合意を締結することができないように制限される可能性もあります。例えば、我々はVertexにいくつかの遺伝子標的の独占的権利を付与しており、協力プロトコル中に、これらの遺伝子標的に対する治療法を求めるために我々の遺伝子編集技術を使用する権利を他の当事者に付与することが制限される。この協定の非競争条項は、将来の協力者と戦略的に協力する能力を制限するかもしれない。
私たちはタイムリーで受け入れ可能な条項でもっと多くの協力を交渉することができず、交渉することさえできないかもしれない。協力の交渉と記録は複雑で時間がかかる。また,最近では大手製薬会社間の業務合併数が大きく,将来の潜在的パートナー数の減少を招いている。もし私たちが新たな協力を協議して達成できない場合、私たちは協力を求める候補製品の開発を減らし、その開発計画や私たちの1つ以上の他の開発計画を減らしたり、その潜在的な商業化を延期したり、いかなる販売やマーケティング活動の範囲を縮小したり、私たちの支出を増加させ、自費で開発または商業化活動を行わなければならないかもしれない。もし私たちが私たちの支出を増やし、私たち自身の開発や商業化活動に資金を提供することを選択すれば、私たちは追加の資本を得る必要があるかもしれないし、これらの資本は受け入れ可能な条件で私たちに提供できないかもしれないし、全く得られないかもしれない。もし私たちが十分な資金がなければ、私たちは私たちの候補製品をさらに開発したり、それらを市場に出して製品収入を作ることができないかもしれない。
私たちは第三者に頼って私たちの候補製品の臨床試験とある方面の臨床前研究を行いたい。もしこれらの第三者がその契約義務を成功的に履行し、規制要求を遵守したり、予想された最終期限までに完成できない場合、私たちは規制部門の私たちの候補製品の承認を得られないか、商業化される可能性があり、私たちの業務は深刻な損害を受ける可能性がある。
著者らは医療機関、臨床研究者、契約実験室とその他の第三者(例えばCRO)による未来の臨床試験を行う予定であり、現在著者らは第三者に依存して著者らの候補製品のいくつかの方面の臨床前研究を行っている。しかし、私たちは私たちが後援したすべての臨床前研究と任意の未来の臨床試験が適用された方案、法律と法規の要求及び科学的標準に従って行われることを保証する責任があり、私たちのCROへの依存は私たちの監督責任を軽減しない。例えば,我々のすべての臨床試験が試験の全体的な調査計画や案に沿って行われることを確保していきたい。さらに、FDAは、データおよび報告の結果が信頼性および正確であることを保証し、試験参加者の権利、完全性および機密性を保護するために、良好な臨床実践またはGCPと呼ばれる臨床試験結果を行い、記録し、報告する法規を遵守することを要求する。行われている臨床試験を一定の時間範囲で登録し,政府が援助しているデータベースClinicalTrials.gov上で完成した臨床試験結果を公表することも求められている。そうしないと罰金、否定的な宣伝、そして民事と刑事制裁につながるかもしれない。私たちが臨床前研究および臨床試験を行っている間の任意の法律および法規違反行為については、刑事起訴までの民事処罰を含む可能性がある警告状または法執行行動を受ける可能性がある。
私たちと私たちのCROはGCPを含む臨床前研究と臨床試験結果を行う、モニタリング、記録、報告する法規を遵守して、データと結果が科学的に信頼性と正確であることを保証し、試験患者は臨床試験に参加する潜在的なリスクが十分に通知され、彼らの権利は保護される。これらの規定はFDA、欧州経済区加盟国の主管当局と類似の衛生監督機関によって臨床開発中の任意の薬物に対して実行される。FDAは臨床試験スポンサー,主要研究者,試験地点の定期検査によりGCP規定を実行している。もし私たちまたは私たちのCROが適用されたGCPに従わなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられるかもしれませんが、FDAまたは同様の衛生規制機関は、私たちのマーケティング申請を承認する前に追加の臨床試験を行うことを要求するかもしれません。検査後、FDAは将来のどの臨床試験もGCPに適合しているかどうかを確認することは保証できません。また,我々の将来の臨床試験はcGMP法規に基づいて生産された候補製品を用いて行わなければならない。私たちのCROがこれらの規定を遵守できなかったかどうかは、私たちが臨床試験を繰り返す必要があるかもしれません。これは規制承認過程を延期し、法執行行動の影響を受け、支出を著しく増加させる必要があるかもしれません。
74
我々の候補製品のための臨床試験を設計する予定であるが,CROはすべての臨床試験を行う。したがって、私たちの開発計画の多くの重要な側面は、それらの実施とタイミングを含めて、私たちの直接制御範囲内ではないだろう。著者ら自身に完全に依存している従業員と比較して、著者らの第三者による未来の臨床前研究と臨床試験への依存はまた、臨床前研究と臨床試験によって開発されたデータ管理の直接制御の減少を招く。外部の当事者とのコミュニケーションも挑戦的である可能性があり、ミスや協調活動の困難を招く可能性がある。外部の当事者は
これらの要素は第三者が著者らの臨床前研究と臨床試験を行う意志或いは能力に重大な不利な影響を与える可能性があり、そして著者らのコントロール範囲を超える意外なコスト増加に直面させる可能性がある。もしCROが満足できる方法で臨床前研究と未来の臨床試験を行わなかった場合、彼らの私たちの義務に違反したり、規制要求を遵守できなかった場合、私たちの候補製品の開発、規制承認、商業化が遅れる可能性があり、私たちは規制承認を得られず、私たちの候補製品を商業化することができないかもしれません。あるいは私たちの開発計画は実質的で不可逆的な損害を受ける可能性があります。CROが収集した臨床前および臨床データに依存できなければ、私たちが行った任意の臨床試験の規模を繰り返し、延長または増加させる必要があるかもしれないが、これは商業化を著しく延期し、支出を著しく増加させる必要があるかもしれない。
医療提供者、医師、第三者支払者との関係は、適用されるリベート、詐欺、乱用、および他の医療保健法律法規の制約を受けることになり、これは、私たちを刑事制裁、民事処罰、政府医療計画から除外され、契約損害、名声損害、利益、および将来の収入の減少に直面させるかもしれない。
私たちは現在何の製品も発売されていませんが、私たちの候補製品を商業化し始めると、追加の医療法律と法規の要求、アメリカ連邦政府と州、そして私たちが業務を展開している他の司法管轄区の他の国、地域、あるいは地方政府の実行を受けます。
医療保健提供者、医師、第三者支払者は、私たちが市場で承認される可能性のある任意の候補製品を開発し、獲得することを推奨し、処方する上で主な役割を果たしている。私たちの将来の第三者支払者や顧客との手配は、私たちがマーケティング、販売、流通を制限する可能性があり、マーケティングの承認を得た候補製品の業務または財務スケジュールと関係を制限する可能性があり、広範に適用される詐欺や乱用、その他の医療法律法規に直面するかもしれません。“”というタイトルの部分を参照ビジネス−医療保健法と法規制.”
EUでは、医師の処方、推薦、裏書き、購入、供給、注文、または医薬製品の使用を誘導または奨励するために、医師に福祉または利点を提供することが禁止されている。不当な行為を誘導または奨励するために利益または利益を提供することは、一般にEU加盟国の国家反賄賂法とイギリス2010年の“収賄法”によって管轄されている。このような法律に違反することは巨額の罰金と監禁につながるかもしれない。EU指令2001/83/ECは人用薬品に関するEUの指令であり、それは更に、処方或いは薬品を供給する資格のある人に薬品を販売する場合、これらの人にいかなるプレゼント、金銭利益或いは実物利益を提供、提供或いは承諾してはならず、これらのプレゼント、金銭利益或いは実物利益が高くなく、しかも医薬或いは薬局の実践と関係がない限り、規定している。この条項は2012年の人間薬物規制に移されたので、EUから離脱したにもかかわらず、イギリスに適用された。
いくつかの連合会員国で医者に支払われた費用は公開されなければならない。また、医師との合意は、通常、医師の雇用主、その主管する専門組織、および/またはEU加盟国の監督当局に事前に通知し、承認しなければならない。これらの要件は、EU加盟国に適用される国家法律、業界規則、または専門行為規則に規定されている。このような要求を守らないことは、名声のリスク、公開非難、行政処罰、罰金、または監禁につながる可能性がある。
75
私たちが第三者の業務配置と適用される医療法律や法規に適合するように努力することは、多くのコストに関連することを確実にします。政府当局は、私たちの業務実践が現在または未来に適用される詐欺および乱用または他の医療保健法律および法規に関連する現行または未来の法規、法規または判例法に適合していない可能性があると結論するかもしれない。これらの法律の範囲が広く、法定例外状況と選択可能な避難港が限られていることから、私たちのいくつかの商業活動は1つ以上のこのような法律の挑戦を受けるかもしれない。私たちの運営には、私たちが設立する可能性のある販売およびマーケティングチームによる活動が含まれており、これらの法律または任意の他の私たちに適用される可能性のある政府法規に違反していることが発見された場合、私たちは、MedicareおよびMedicaidのような重大な民事、刑事および行政処罰、損害賠償、罰金、MedicareおよびMedicaidのような政府支援医療計画から除外され、私たちの業務を削減または再構成するかもしれない。もし私たちがそれと業務を展開することを期待している任意の医師または他の提供者または実体が適用されない法律に適合していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む刑事、民事または行政制裁を受ける可能性がある。彼らがこれらの法律に基づいて負う責任は、重大なコストや運営中断を招く可能性があり、これは私たちの業務、財務状況、運営結果、将来性に重大な悪影響を及ぼす可能性がある。
製造業に関するリスク
遺伝子編集製品は珍しいので、複雑かもしれませんが、製造が難しいです。私たちは製造問題に直面し、私たちの候補製品の開発や商業化の遅延、あるいは他の方法で私たちの業務を損なう可能性があります。
CRISPR/CAS 9に基づく候補製品を生産するための製造プロセスは、新しいので、臨床的および商業的生産の検証がなされていないため、複雑である可能性がある。いくつかの要素は新しい製造技術の開発ができない、設備故障、施設汚染、原材料不足或いは汚染、自然災害を含む生産中断を招く可能性があり、コロナウイルスの大流行、公共事業サービスの中断、人為的エラー或いはサプライヤーの運営中断を含み、第三者によるサプライヤーの買収或いは破産の発表を含む。これらの候補製品を製造するために必要な専門知識は,特定の契約製造組織独自である可能性があるため,代替契約製造組織を探すことは困難で時間がかかる.我々の製造施設または第三者製造業者の任意の関連システムにおける故障またはプロセス欠陥は、検証されると、研究、臨床、および(承認された場合)商業的に生産される候補細胞治療製品およびそれらのいくつかの構成要素を製造および供給する能力に悪影響を及ぼす可能性がある。コロナウイルスの大流行の影響に関する他の資料は、“を参照されたい”我々の業務はより多くの変種の出現を含む持続的なコロナウイルスの大流行の悪影響を受ける可能性がある.”
私たちの候補製品はほとんどの小分子薬物に必要なより複雑な加工工程を必要とするだろう。また,小分子と異なり,生物製品の物理的·化学的性質は通常完全には表現できない。したがって、完成品の検出は、製品が所望の方法で動作することを確保するのに十分ではない可能性がある。したがって、このプロセスが有効であることを保証し、そのプロセスに従って候補製品を厳密かつ一致的に製造するために、複数のステップを用いて製造プロセスを制御する。製造過程の問題は、正常過程との微小な偏差であっても、製品欠陥或いは製造失敗を招く可能性があり、それによって大量故障、製品リコール、製品責任クレーム或いは在庫不足を招く。FDA、EMAまたは他の適用基準または規範に適合する臨床レベルの材料の十分な数量と品質、および一致して許容可能な生産生産量とコストを得ることができないいくつかの問題に直面する可能性がある。
さらに、FDA、EMA、および他の衛生規制機関は、任意のロットの承認された製品のサンプルをいつでも提出し、適用試験結果を示すスキームを要求するかもしれない。場合によっては、FDA、EMA、または他の衛生規制機関は、関連機関が発行を許可する前に大量の薬物を配布しないように要求するかもしれない。製造過程中の微小な偏差は、品質属性や安定性に影響を与える偏差を含み、製品に許容できない変化を招き、大量故障や製品リコールを招く可能性がある。大量故障は私たちの製品発表或いは臨床試験を延期する可能性があります。私たちは製品リコールを行う必要があるかもしれません。これらはすべて私たちにとってコストが高いかもしれません。そうでなければ、私たちの業務、財務状況、運営結果と将来性を損なうかもしれません。私たちの製造過程の問題は私たちの製品に対する市場の需要を満たす能力を制限するかもしれない。
私たちはまた、私たちの製造プロセスを操作するために必要な経験豊富な科学、品質保証、品質管理、製造人員の問題に直接あるいは契約製造組織を通じて採用と保留することができ、これは生産遅延を招き、あるいは適用された法規要求を維持することが困難になる可能性がある。我々のサプライチェーン、製造プロセス、または施設のいずれの問題も、計画中の臨床試験の遅延およびコスト増加を招く可能性があり、潜在的なパートナー(より大きな製薬会社や学術研究機関を含む)の魅力を低下させる可能性があり、より多くの魅力的な開発計画を得ることを制限する可能性がある。私たちの製造過程における問題は、将来の潜在市場の製品に対する需要を満たす能力を制限するかもしれない。
76
私たちの候補製品の製造施設は厳格な法規によって制限されており、規制承認を得たり維持したり、既定のcGMPや国際ベスト実践と一致していなければ、候補製品を商業化する能力を延期または弱める可能性がある。
我々と我々の候補製品の第三者メーカーは,FDAが規定しているcGMPおよびEMAや他の規制機関が規定する他の規制の適用に制約されている。アメリカとヨーロッパの候補製品がFDAとEMAの承認を得るためには、私たちまたは私たちの第三者製造施設の厳格な承認前検査が必要です。私たちまたは私たちの請負業者の製造施設を検査する時、FDAまたはEMAはcGMP欠陥を列挙する可能性があり、軽微であっても重大であっても、開示を要求されないかもしれません。欠陥を修復するのは難しくて費用がかかるかもしれないし、多くの時間がかかるかもしれない。さらに、FDAまたはEMAがその検査結果に欠陥があることに気づいた場合、それは通常、欠陥が満足的に修復されたかどうかを決定するために施設を再検査する。FDAまたはEMAは、その再検査の結果におけるさらなる欠陥が、以前に決定された欠陥に関連するか、または他の態様に関連することに注目する可能性がある。もし私たちあるいは私たちの候補製品のメーカーがFDAとEMAのcGMPに対する適合性を適時に満たすことができなければ、私たちの候補製品の発売審査は深刻に遅延する可能性があり、これは逆に私たちの候補製品の商業化を延期することになる。
私たちは私たちの内部製造施設と私たちの第三者契約製造パートナーに関する規制と操作リスクに直面している。
2021年第4四半期、私たちはマサチューセッツ州フレミンガムで新しい細胞治療製造施設の建設を完了し、この施設は検証されると、私たちのいくつかの計画を満たすために、私たちの候補細胞治療製品とそのいくつかのコンポーネントの研究、臨床および商業生産を支持することができる。我々は,この施設をcGMPに適合させ,将来のヒト管理に適した細胞治療製品供給を生産できるようにするための規制検証活動を進めている。私たちは今まで私たち自身の製造施設を建設して運営したことがありません。私たちは私たちの内部製造能力を建設して運営することができますか、あるいは私たちのフレミンガム施設の必要な検証を実現することができません。この施設の設計はバイオテクノロジー施設の現行基準に基づいているが,いかなる規制機関の審査や事前承認も経ておらず,当施設の規制機関(例えばFDA)の検査も行われていない。工場の全面的な運営状態の実施を遅延させ、臨床供給の遅延や第三者契約製造パートナーの使用を延長し、計画外費用につながる可能性があります。マサチューセッツ州フレミンガムで私たちの施設が建設された時、私たちはすでに多くの支出を生み出し、将来的にこの施設を検証して運営する際に顕著な追加支出が発生すると予想されている。
私たちは第三者に頼って私たちの臨床製品を生産したいです。私たちは候補製品の製造過程で少なくとも第三者に依存するつもりです。第三者がサプライチェーン不足に遭遇し、十分な数量の製品投入品を提供できない場合、あるいは受け入れ可能な品質レベルや価格で製品を提供できない場合、私たちの業務は損害を受ける可能性があります。
マサチューセッツ州フレミンガムにある工場の建設が完了したにもかかわらず、私たちはまだ規制検証活動を完成していません。私たちは現在私たちの臨床規模の製造と加工施設として使用される可能性のある施設を持っていません。私たちは外部サプライヤーに依存して私たちの候補製品を製造し加工しなければなりません。これは私たちがこのような候補製品に対して行ったいかなる臨床試験と関係があります。私たちはまだどの候補製品にも商業規模の製造や加工を促していませんし、私たちの候補製品のためにそうすることができないかもしれません。製造過程を最適化しながら変更することに努め,過程中のわずかな変化でも安全で有効な治療法が生じることを確保することはできない。
我々の候補製品を生産するための施設は,FDAまたは他の管轄区の他の衛生規制機関がFDAまたは他の衛生規制機関に申請を提出した後に行われる検査に基づいて評価されなければならない。私たちは私たちの契約製造パートナーの製造過程を制御するのではなく、私たちの契約製造パートナーが規制要件、いわゆるcGMP要求を遵守して、私たちの候補製品を生産することに完全に依存します。もし私たちの契約メーカーが私たちの規格やFDAまたは他の規制機関の厳格な規制要件に適合した材料を生産することに成功しなかった場合、彼らはその製造施設の規制承認を確保および/または維持することができない、あるいは規制機関は彼らの欠陥を理由に、FDAまたは他の規制機関の私たちの候補製品に対する規制承認を得ることができないか、または遅延する可能性がある。しかも、私たちの契約製造業者が十分な品質管理、品質保証、合格者の能力を維持することを直接制御することはできません。FDAや同様の衛生規制機関がこれらの施設を承認しない場合、またはこれらの施設に欠陥があることを引用して私たちの候補製品を生産するか、またはそのような承認を撤回したり、将来的に欠陥を引用したりすれば、代替製造施設を探す必要があるかもしれません。これは、私たちが規制機関の承認を得たり、私たちの候補製品を販売したりする能力に深刻な影響を与えます。さらに、もし私たちの契約メーカーがFDAなどの衛生規制機関が取った行動やコロナウイルスの大流行によって、直ちに職責を履行したり、気を失ったりすることができない場合, 私たちは製造遅延に遭遇するかもしれないし、代替製造施設を探す必要があるかもしれませんが、すべての場合、これは私たちが開発、規制部門の承認を得たり、私たちの候補製品をマーケティングする能力に深刻な影響を与えます(承認されれば)。
77
限られた数の第三者製造業者への依存は、以下のリスクを含む多くのリスクに直面します
これらのリスクの各々は、私たちの臨床試験の完了またはFDAが私たちの任意の候補製品を承認することを遅延または阻止する可能性があり、コスト上昇をもたらし、または私たちの候補製品の商業化に悪影響を及ぼす可能性がある(承認された場合)。また,患者に渡す前に,第三者に依存して候補製品のいくつかの仕様テストを行う。もしこれらのテストが不適切で、テストデータが信頼できなければ、患者は深刻な傷害のリスクに直面する可能性があり、FDAは欠陥が修復されるまでわが社に重大な制限を加える可能性がある。
従業員事務に関するリスク、管理成長、および私たちの業務に関するその他のリスク
私たちの未来の成功は私たちが肝心な幹部を維持し、合格者を吸引、維持、激励する能力にかかっている。
著者らは最高経営責任者Samarth Kulkarni博士及び著者らの管理、科学と臨床チームの他の主要なメンバーの研究開発、臨床、商業と業務発展方面の専門知識に高度に依存している。私たちは私たちの幹部と雇用協定を締結したが、彼らの誰もが私たちとの雇用関係をいつでも終わらせることができる。私たちは私たちの役員や他の職員たちに“キーパーソン”保険を提供しない。また、私たちは、科学と臨床コンサルタントを含むコンサルタントとコンサルタントに依存して、私たちの研究開発と商業化戦略の制定を助けてくれます。私たちのコンサルタントやコンサルタントは、私たち以外の雇用主に雇われる可能性があり、他のエンティティと締結された相談または相談契約に基づいて約束することができ、これは、私たちが彼らを得る機会を制限するかもしれない。役員や他の重要な従業員やコンサルタントのサービスを失うことは、研究開発と商業化目標の達成を阻害し、業務戦略を成功させる能力を深刻に損なう可能性がある。もし私たちが高い素質の人材を維持できなければ、私たちが成長戦略を実施する能力は制限されるだろう。
私たちはまた、私たちの候補製品と製品ラインの開発を進めているので、合格した科学、臨床、ビジネススタッフを募集して維持する必要があります。私たちは受け入れ可能な方法でこれらの重要な人員を採用、訓練、維持、または激励することができないかもしれない
78
このような条項は、多くの製薬会社とバイオテクノロジー会社の間の類似者の競争を考慮している。私たちはまた、大学や研究機関から科学、臨床、ビジネススタッフを募集する競争に直面している。臨床試験で成功できなかったことは、合格した科学者の採用と維持をもっと挑戦的になる可能性がある。
役員報酬に関するスイスのコーポレート·ガバナンスは私たちの業務に影響を及ぼすかもしれない。
スイスの会社法は、他の事項に加えて、(A)執行管理層および取締役会メンバーの報酬について拘束力のある“報酬発言権”投票を行うことを株主に要求し、(B)執行管理層および取締役会メンバーへの解散料、前払い、取引割増および類似金の支払いを一般的に禁止し、(C)会社にその組織規約において報酬に関する様々な事項を具体的に説明し、株主投票による承認を要求する。私たちの年間株主総会では、私たちの株主は、私たちの取締役会と私たちの実行管理チームの最高総報酬の承認を要求されました。スイスの法律では、取締役や執行管理職のメンバーが賠償に関する何らかの要求を守らなければ、刑事罰を受けることも規定されている。これらの規定は私たちが経営陣と取締役会のメンバーを誘致し、維持する能力に否定的な影響を及ぼすかもしれない。
私たちの従業員、首席調査員、コンサルタント、ビジネスパートナーは、規制基準と要求を遵守しないこと、およびインサイダー取引を含む不正行為や他の不適切な活動に従事する可能性があります。
私たちは従業員、コンサルタント、ビジネスパートナー、主要な調査者詐欺または他の不適切な行為のリスクに直面している。これらの当事者の不正行為は、FDA法規またはEUおよび他の司法管轄区に適用される法規を故意に遵守しないこと、FDA、欧州委員会および他の規制機関に正確な情報を提供すること、米国および他の司法管轄区の医療詐欺および法律法規の乱用、財務情報またはデータを正確に報告すること、または不正な活動を開示することを含む可能性がある。特に、医療業界の販売、マーケティング、商業配置は、詐欺、不正行為、リベート、自己取引、その他の乱用行為を防止するための広範な法律法規によって制約されている。これらの法律法規は、広範な定価、割引、マーケティングと販売促進、販売手数料、顧客激励計画などの業務手配を制限または禁止している。このような不正行為はまた、臨床試験過程中あるいはFDA或いは他の監督機関との相互作用過程で得られた情報の不適切な使用に関連する可能性があり、これは規制制裁を招き、著者らの名声に深刻な損害を与える可能性がある。我々はすべての従業員に適用される行動基準を採択したが、常に従業員の不正行為を識別し、阻止できるわけではなく、このような行為を発見し防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御できない可能性があり、またはこれらの法律や法規を遵守できないことによる政府の調査または他の行動や訴訟から私たちを保護することができるかもしれない。また、私たちは、起きていなくても、このような詐欺や他の不正行為を告発する可能性があるというリスクに直面している。もし誰かが私たちにこのような行動を取ったら, もし私たちが私たちの権利を弁護したり、維持したりすることに成功しなかった場合、これらの行為は、民事、刑事および行政処罰、損害賠償、罰金、連邦医療保険、医療補助および他の連邦医療保健計画から除外される可能性があり、契約損害、名声損害、利益減少および将来の収益減少、および私たちの業務縮小を含む、私たちの業務、財務状況、運営結果、および将来の収益減少、および私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性がある。
もし私たちが環境、健康、安全の法律法規を守らなければ、罰金や処罰を受けたり、私たちの業務を損なう可能性のあるコストが発生するかもしれません。
私たちは多くの環境、健康と安全法律と法規の制約を受けて、それらの研究室の手続きと危険材料と廃棄物の処理、使用、貯蔵、処理と処理を管理する法律と法規を含む。私たちの行動は化学物質と生物学的材料を含む危険で燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物製品を発生させるだろう。私たちは第三者とこのような材料と廃棄物を処理する契約を締結した。私たちはこのような材料が汚染や傷害をもたらす危険をなくすことができない。もし私たちがどんな危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に責任を負うかもしれないし、どんな責任も私たちの資源の範囲を超えているかもしれない。民事や刑事罰金やこのような法律や法規を遵守しない罰に関する巨額の費用を招く可能性もある。
また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。私たちがこのような法律を守らないことはまた巨額の罰金、処罰、または他の制裁につながるかもしれない。
私たちに対する製品責任訴訟は、私たちが大量の責任を負うことを招き、私たちが開発する可能性のある任意の候補製品の商業化を制限する可能性があります。
私たちは人体臨床試験で私たちの候補製品をテストすることに関連する固有の製品責任リスクに直面し、私たちが開発可能な任意の候補製品を商業販売すれば、私たちはより大きなリスクに直面する。もし私たちが自分自身を弁護することに成功できなければ、私たちの候補製品が被害を与えたと主張すれば、私たちは重大な責任を招くかもしれない。是非曲直や最終結果にかかわらず、賠償責任は
79
私たちは製品責任保険を受けたが、それは私たちが発生する可能性のあるすべての責任をカバーするのに十分ではないかもしれない。また、もし私たちが任意の候補製品を商業化することに成功すれば、私たちは私たちの保険カバー範囲を増やす必要があると予想している。保険範囲はますます高くなっています。私たちは可能などんな責任にも対応するために、合理的な費用や十分な金額で保険範囲を維持することができないかもしれない。
財務報告書に対する適切かつ効果的な内部統制を確立し、維持することができなければ、私たちの経営業績と私たちの経営業務の能力が損なわれる可能性がある。
私たちが正確な財務諸表をタイムリーに作成できるように十分な内部財務と会計制御と手続きを持っていることを確保して、これは高価で時間のかかる仕事であり、常に再評価する必要がある。私たちは2002年のサバンズ-オキシリー法案(Sarbanes-Oxley Act、略称SOX)の要求を遵守しなければならない。この法案は私たちが財務報告書と開示統制と手続きに対して効果的な内部統制を維持することを要求する。特に、システムおよびプロセス評価を行い、我々の制御措置を記録し、財務報告の重要な制御をテストし、経営陣と我々の独立会計士事務所がSOX第404条の要求に基づいて財務報告の内部統制に対する有効性を報告できるようにしなければならない。私たちのテスト、あるいは私たちの独立会計士事務所のその後のテストは、財務報告における私たちの内部統制欠陥を明らかにするかもしれません。これらの欠陥は実質的な弱点と考えられています。もし私たちが第404条の要求を直ちに遵守できない場合、あるいは私たちまたは私たちの会計士事務所が財務報告の内部統制において重大な弱点と考えられる欠陥を発見した場合、私たちの株式の市場価格は下落する可能性があり、私たちは規制機関の訴訟、制裁、または調査を受ける可能性があり、これには追加の財務と管理資源が必要になるだろう。
私たちはこの報告書要件を管理するために、より強力な技術とより多くの資源に投資し続けている。私たちの内部統制を適切に変更することは、私たちの役人や従業員の注意を分散させる可能性があり、新しいプロセスを実施したり、既存のプロセスを修正したりすると、大量のコストを招き、完成するのに時間がかかる可能性があります。このような統制を実施するどんな困難や遅延も、私たちが財務業績をタイムリーに報告する能力に影響を及ぼす可能性がある。また、私たちは現在、いくつかの分野で人工的なプロセスに依存しており、これは、財務業績を報告する際に人為的なミスや介入に遭遇するリスクを増加させる。これらの理由により、財務業績をタイムリーかつ正確に報告することが困難になる可能性があり、これは投資家にタイムリーに情報を提供する能力に影響を与える。したがって、私たちの投資家は私たちが報告した財務情報に自信を失うかもしれないし、私たちの株価は下落するかもしれない。
また、このような変化は、私たちが内部統制の十分性を効果的に維持することを保証することはできません。このような十分性を維持できない場合は、私たちの財務業績を正確に報告することを阻止することができます。
私たちは進化するヨーロッパと他のプライバシー法を守ることができないかもしれない。
私たちは現在ヨーロッパ経済地域で臨床試験を行っている。したがって、私たちは追加的なプライバシー法の制約を受けている。GDPRは2018年5月25日に発効し、個人データの処理とこのようなデータの自由な流れに関連しています。GDPRは、GDPRに拘束されている会社に対して、個人を識別可能な個人情報を処理し、このような情報を欧州経済地域以外(米国を含む)に移転する法的根拠を有すること、これらの個人にその個人情報を処理する詳細を提供すること、個人情報を処理するセキュリティを保持すること、個人情報を処理する第三者とのデータ処理プロトコルの締結、個人の個人情報行使権利の要求への応答、主管国家データ保護機関および影響を受ける個人報告への個人データに関する安全違反行為、データ保護官の任命、データ保護影響評価、記録保存を含む、幅広い厳しい要求を課している。GDPRは、いくつかの比較的軽い犯罪に対して1000万ユーロまたは私たちの世界の年商総額の2%の罰金、より深刻な犯罪には2000万ユーロまたは世界の年商総額の4%の罰金を含む、規定を守らずに受ける可能性のある処罰を大幅に増加させます。
特に、EU加盟国の国家法律が施行されている国家法律は、GDPRから部分的に逸脱しており、国によって異なるかつより限定的な義務が加えられている可能性があるため、EUは統一された法的環境では動作しないと予想される。また、遺伝子データの処理と譲渡に関連するため、GDPRはEU加盟国が法律を公布し、より多くの具体的な要求や制限を加えることを明確に許可しているが、ヨーロッパの法律はこの分野で従来から大きな差があり、追加的な不確実性を招いている。
80
また、英国が2020年1月31日にEUを離脱したのに続き、GDPRは2020年12月31日の過渡期終了時に英国での適用を停止した。しかし、2021年1月1日現在、イギリスの2018年“EU(離脱)法案”は、GDPR(2020年12月31日に存在するGDPRと同じだが、イギリスの何らかの具体的な改正が必要)をイギリスの法律、すなわちイギリスGDPRに組み入れている。英国GDPRと2018年の英国データ保護法は,EUのデータ保護制度から独立した英国のデータ保護制度を規定しているが,EUのデータ保護制度と一致している。英国政府はそのデータ改革法案でデータ保護法の枠組みを改革する計画を発表したが,これらの計画は棚上げされている。イギリスのGDPR違反は、金額が高い者を基準に、1750万GBまたは世界収入4%の罰金を招く可能性があります。英国はEU GDPR下の第3の国とされているが、欧州委員会(以下“欧州委員会”)は現在、英国がEU GDPRで十分な保護を提供していることを認める決定を下しているため、EUからの個人データの英国への移行は制限されていない。EU GDPRと同様に,イギリスGDPRは個人データをイギリス以外の国に移しており,これらの国はイギリスが十分な保護を提供しているとはみなされていない。イギリス政府は、イギリスからヨーロッパ経済圏への個人データ転送が依然として自由に流動していることを確認した。
EU-米国とスイス-米国プライバシー盾の枠組みは、米国商務省に自己認証を許可し、特定の要求を遵守することを約束した米国会社がEUとスイスから個人データを輸入することを許可する。2020年、EU裁判所はEU-米国プライバシー盾は無効な伝送メカニズムであると判断し、これは米国会社がGDPRの国境を越えたデータ伝送制限に基づいて欧州から個人情報を輸入する主要なメカニズムの一つであり、欧州委員会の標準契約条項(SCC)が欧州から米国または多くの他の国への個人情報の送信に合法的に使用できるかどうかに対する疑問を引き起こした。標準契約条項はプライバシー盾の主要な代替案の一つである。同様に、スイス連邦データ保護·情報専門家は、スイス-米国のプライバシー盾はスイスから米国にデータを転送するのに十分ではなく、英国情報専門家事務室も、プライバシー盾の枠組みはイギリスから米国に移すのに十分ではないと述べている。また2021年6月4日には, 欧州委員会は、欧州経済地域内の制御者または処理者(または他の方法でGDPRによって拘束されている)から欧州経済区以外に設立された制御者または処理者にデータを移すための新しい形式の標準契約条項を発表した。新しい形式の標準契約条項は、以前データ保護指令に基づいて採用されていた標準契約条項に代わっている。私たちは多くの努力と費用が必要かもしれない新しい形の標準契約条項に移行することを要求されるだろう。新しい標準契約条項は、譲渡影響評価の重い要求と新標準契約条項が輸出業者に課す重大な義務を考慮して、ヨーロッパに本部を置く会社は、新しい条項を利用して個人情報を第三国に移転することを合法化したくないかもしれないので、私たちの業務に影響を与える可能性もある。2022年3月25日、欧州委員会と米国は、失効したプライバシーの盾に代わる新たな“大西洋横断データプライバシー枠組み”について政治的合意に到達することを発表した。2022年12月13日、欧州委員会は大西洋横断データプライバシー枠組みに関する十分性決定草案を発表した。もし私たちがヨーロッパデータ保護機関の調査を受けたら、私たちは罰金と他の処罰に直面するかもしれない。欧州データ保護当局のこのような調査または告発は、私たちの既存の業務および新しい顧客または製薬パートナーを引き付け、維持する能力にマイナスの影響を与える可能性がある。私たちはまたヨーロッパや多くの顧客や製薬パートナーに出会うかもしれません(特に現在, 将来的に)いくつかのデータ保護機関は、GDPRを含む現行法を解釈する際に彼らに課されるデータ保護義務を含む。これらの顧客や医薬パートナーも,任意の他のコンプライアンス方法はコストが高すぎ,負担が重すぎ,法的に不確実すぎたり反感を抱いたりする可能性があるため,我々と商売をしないことにした。上記のいずれも、我々の業務、見通し、財務状況、および経営結果に実質的な損害を与える可能性がある。
私たちの業務と運営はイギリスのEU離脱の否定的な影響を受けるかもしれない。
2016年6月23日、イギリスで国民投票が行われ、多くの適格有権者がEU離脱、すなわち通常言われている離脱を決定した。英国は2020年1月31日にEUから正式に離脱したが、2020年12月31日までEUルールと立法が適用され続ける最初の移行期間がある。英国とEUは2021年1月1日に臨時発効し、2021年5月1日に正式に発効するEU-英国貿易·協力協定に署名した。TCAには薬品に関する具体的な条項が含まれており,その中には相互承認良好製造規範(GMP),薬品生産施設の検査と発表されたGMP文書が含まれているが,イギリスとEUの薬品法規が大規模に相互承認されることは予想されていない。現在、イギリスは“2012年人類薬品条例”(改正)(北アイルランド議定書によると、EU規制枠組みは引き続き北アイルランドに適用される)を採択し、医薬製品のマーケティング、普及、販売に関するEUの立法を実施している。そのため、イギリスの規制制度は多くの点でEUの規制制度と一致しているが、イギリスの規制制度はEUから独立しており、TCAはイギリスとEUの薬品立法を相互に認めることを規定していないため、これらの制度は将来的により大きな差が生じる可能性がある。長期的に見ると、イギリスの離脱がどのようにイギリスの医療製品と器械に対する監督管理要求に影響するかは、まだ観察が必要である。
移行期間が終わったため、イギリスはもはや集中マーケティング許可のカバーを受けなくなった(北アイルランド議定書によれば、集中マーケティング許可は北アイルランドで認められ続けるだろう)。2021年1月1日からの3年間、イギリスの薬品と保健品監督機関(MHRA)は欧州委員会が集中市場の新しいマーケティング許可を承認する決定に依存する可能性がある
81
新しいイギリスのマーケティング許可をより早く付与するための手続き。しかし、まだ個別的な申請が必要になるだろう。イギリスとEUのどんな新しい規制も、私たちが業務を展開する時間と費用、そして私たちの製品がイギリス、EU、他の場所で規制の承認を受ける過程を増加させる可能性があります。イギリス離脱のこれらの比較的長期的な影響と、私たちが予測できない他の影響は、私たちの業務と運営結果に負の影響を与える可能性がある。私たちのイギリスビジネスは、EUや欧州経済地域(EEA)の他の国での業務および臨床活動(臨床活動を含むが、これらに限定されない)を現在および将来的にサポートしており、これらの業務および臨床活動は、イギリスの離脱の長期的な影響を受ける可能性がある。
私たちの業務にはかなりの国際的な足跡があり、私たちは将来さらに拡張する可能性があり、私たちの業務運営管理に挑戦しています。
私たちの本部はスイスのズグにあり、アメリカとイギリスに事務所を設置している。しかも、私たちは未来に私たちの国際業務を他の国に拡張するかもしれない。私たちはすでに多くの管理者や他の豊富な経験を持つ人員を獲得しているが、複数の国で業務を展開することは、私たちを様々なリスクと複雑さに直面させ、これらのリスクと複雑性は、私たちの業務、運営結果、財務状況、成長の見通しに実質的で不利な影響を与える可能性がある
私たちは私たちの業務を拡大し続け、私たちの会社の構造と税金構造は複雑だ。私たちの現在と未来の潜在的なパートナー関係に合わせて、私たちは技術と知的財産権の開発と応用に積極的に参加し、世界規模で製品の商業化を実現することを期待して、通常は商業化パートナーと共に。これらの活動の面で、私たちは、私たちの技術、知的財産権、その他の資産に関連し、私たちとパートナーや許可者などの他のエンティティとの間、および私たちと私たちの子会社との間の取引に関連して、複雑な国境を越えたグローバル取引に従事し続けている可能性があります。このような国境を越えてもグローバルな配置は管理が困難であり、税務処理などの分野で複雑性を引き起こす可能性があり、特に私たちは様々な税制に支配されているため、異なる税務当局も同じ国境を越えた取引や手配に対して異なる見方を持っている可能性がある。運営効率の低下、制御欠陥、または税務負担に遭遇することなく、このような増加した複雑さを効率的に管理することは保証されない。わが社の日々増加する複雑さを効果的に管理するためには、大量の管理時間と労力が必要であり、これを成功させることができなければ、私たちの業務、財務状況、運営結果、成長見通しに実質的な悪影響を与える可能性がある。
知的財産権に関するリスク
私たちが独自の遺伝子編集技術や候補製品に関連する知的財産権を獲得したり保護したりできなければ、私たちは私たちの市場で効果的に競争できないかもしれない。
私たちの成功は、私たちのCRISPR/Cas 9プラットフォーム技術と、私たちが開発した任意の独自製品および技術について、米国および他の司法管轄地域で独自または知的財産権保護を獲得し、維持する能力に大きく依存しています。私たちは特許権、商業秘密保護、秘密協定を含む一連の知的財産権によって、私たちの遺伝子編集技術と候補製品に関連する知的財産権を保護します。現在、私たちはある知的財産権の権利を持っていて、第三者の許可と私たちが持っている特許権を通じて、私たちの遺伝子編集技術および/または候補製品を開発しています。例えば、私たちの2014年のDr.Charpentierとの独占許可によって、私たちは、95(95)件を超える付与または許可された特許を含むグローバル特許の組み合わせのいくつかの権利を独占的に許可し、これらの出願は、例えば、私たちの遺伝子編集プラットフォーム技術の様々な態様をカバーしている CRISPR/CAS 9システムのような物質の組成および使用方法であって、CRISPR/CAS 9システムを用いて遺伝子編集を行うことを含む使用方法。この世界的な特許の組み合わせを“特許組合せ”と呼ぶ.さらに、私たちは、私たちの候補製品の様々な態様、例えば、物質の組成および製造および使用の方法を含む、私たちの候補製品に関する大量の特許出願を提出した。
82
私たちは、知的財産権を付与することで私たちのプラットフォーム技術をカバーし、私たちの独自の地位を保護するために、米国や他の司法管轄区域に当社の技術や製品候補に関する特許出願を提出することを求めており、これらの技術や製品は私たちの業務に非常に重要です。私たちはまた、ビジネス秘密、技術ノウハウ、持続的な技術革新によって、私たちの独自と知的財産権の地位を発展させ、維持しています。私たちまたは私たちの許可者が、CRISPR/Cas 9プラットフォーム技術および私たちが開発した任意の独自製品および技術の特許保護を取得または維持できない場合、私たちの業務、財務状況、運営結果、および見通しは深刻な損害を受ける可能性があります。
しかしながら、バイオテクノロジーおよび製薬分野、特にゲノム編集分野の特許強度は、通常、複雑な法律および科学的問題に関連しており、私たちが所有しているか、または許可されている特許権、任意のそのような特許権の広さ、または任意の発行された特許が無効であるかどうか、強制的に実行できないかどうか、または第三者の脅威を受けることを保証することはできないかもしれない。例えば、私たちがアメリカと他の国で得ることができる特許保護範囲は不確定だ。米国および他の国の特許法またはその解釈の変化は、私たちの知的財産権を保護し、私たちの知的財産権を保護し、獲得し、維持し、擁護し、実行する能力を弱める可能性があり、より広く言えば、私たちの知的財産権の価値に影響を与えたり、私たちの所有しているライセンス内の特許の範囲を縮小したりする可能性がある。ライセンス内で所有されている知的財産権については、私たちおよび私たちのライセンシーが現在求めている特許出願が任意の特定の司法管轄区域で特許として発行されるかどうか、または発行された特許の権利主張が競合相手に対して十分な保護を提供するかどうか、または私たちの競争相手が挑戦している場合、そのような特許が無効、強制執行または侵害されないことが発見されるかどうかを予測することはできません。
特許訴訟過程は高価で、時間がかかり、複雑であり、私たちは合理的なコストまたはタイムリーな提出、起訴、維持、強制執行、またはすべての必要または望ましい特許出願を許可することができないかもしれない。我々の研究開発成果における出願可能な特許の側面をタイムリーに決定できず,特許保護を得ることができない可能性もある.私たちは、私たちの研究開発成果にアクセスする権利のある機密または特許可能な当事者と、当社の従業員、会社の協力者、外部科学協力者、CRO、契約製造業者、コンサルタント、および他の第三者のような秘密および秘密協定を締結していますが、これらの当事者のいずれかは、合意に違反し、特許出願を提出する前にそのような成果を開示し、特許保護を求める能力を危険にさらしている可能性があります。また,我々が我々の遺伝子編集技術および/または候補製品を開発している分野には,第三者が所有する米国や外国から発行される特許や係属中の特許出願が多く存在する.私たちが関連する第三者特許や出願を決定できなかったかもしれない。また、科学文献で発表された発見は、米国や他の司法管轄区の実際の発見や特許出願よりも遅れがちであり、通常は出願18ヶ月後に発表され、時には全く発表されない場合もある。したがって、私たちは、私たちのライセンス特許および出願の発明者が、私たちが所有または任意のライセンス特許または係属特許出願で主張されている発明の第1の発明者であるかどうか、または私たちがそのような発明のために特許保護を申請した最初の人であるかどうかをいかなる程度でも決定することはできない。さらに何かがある, 私たちの所有および許可内の特許および特許出願に関連するすべての潜在的に関連する既存技術が発見されていることは保証されず、これらの技術は、特許を無効にするか、または係属中の特許出願から特許が発行されることを阻止する可能性がある。
任意の係属中または許可された特許出願または付与された特許の最終結果は不確定であり、特許出願において要求されるカバー範囲は、特許発行前に大幅に減少することができ、その範囲は、特許発行後に再解釈することができる。私たちが現在許可または所有している特許出願が将来特許の形態で発表されても、それらは私たちに任意の意味のある保護を提供し、競争相手または他の第三者が私たちと競争することを阻止し、または他の方法で私たちにどんな競争優位性を提供してくれるかの形で発表されることはない。
さらに、特許の発行は、その発明性、範囲、有効性、または実行可能性に対して決定的ではなく、私たちが所有している特許および許可されていない特許は、米国および他の司法管轄区の裁判所または特許庁で挑戦される可能性がある。米国特許商標局の干渉、派生、再審、その他の付与後訴訟、および外国司法管轄区の異議および他の類似訴訟を含む多くの特許に挑戦する訴訟および行政訴訟があり、バイオテクノロジーおよび製薬業界の特許および他の知的財産権に関連しており、CRISPR/CAS 9分野も同様であると予想される。参照してください“リスク要因-知的財産権に関連するリスク-第三者が私たち、私たちのライセンシー、または私たちの協力者に提出した知的財産権侵害クレームは、私たちの製品発見と開発を阻害または延期する可能性がありますもっと情報を知っています. このような挑戦は、独占的または経営の自由を失うこと、または特許主張の全部または部分的な縮小、撤回、無効、または実行不可能をもたらす可能性があり、これは、私たちが発明を実施する能力を制限するか、または他人が同様の技術および製品を使用することを阻止するか、またはそれを商業化することを阻止するか、または我々の技術および製品の特許保護期間を制限する可能性がある。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちが持っている特許と許可された特許の組み合わせは、他の会社が私たちと似ているか同じ製品を商業化することを排除するために十分な権利を提供してくれないかもしれない。
競争相手は、これらの発行された特許または特許出願において主張された発明を我々の発明者の前に発明したと主張することもでき、または我々の発明者の前に特許出願を提出した可能性がある。競争相手も私たちの製品や技術がその特許を侵害していると主張するかもしれないので、もし私たちの特許出願が発行されたら、私たちは私たちの技術を実践することができません。このようなクレームのいずれかの不利な判決は、私たちが権利を侵害せずに製品を製造または商業化することができない可能性がある
83
第三者特許権。競争相手はまた、発明が特許資格に適合していない、新規でない、明らかまたは特許主張が任意の他の特許性要件に適合していないことを証明することによって、私たちの特許に異議を唱えることができる。このような提出、訴訟、または訴訟における不利な裁決は、私たちの特許権の範囲を縮小したり、無効にしたり、第三者が私たちの技術または製品を商業化し、私たちに支払うことなく、直接私たちと競争することを可能にする可能性があります。
さらに、私たちまたは私たちのライセンシーのうちの1つは、発明の優先権または認可後の挑戦手順、例えば非米国特許庁における反対意見、発明優先権または他の特許可能な特徴に挑戦するために、米国特許商標局が発表した追加介入手順に参加しなければならない可能性がある。このような挑戦は、特許権の喪失、独占経営権の喪失、または経営の自由の喪失、または特許主張が全部または部分的に縮小され、撤回され、無効または実行できなくなる可能性があり、これは、他人が同様の技術および製品を使用または商業化することを阻止する能力を制限するか、または我々の技術および候補製品の特許保護期間を制限する可能性がある。最終的な結果が私たちに有利であっても、このような手続きは大量のコストを招く可能性があり、私たちの科学者と経営陣に多くの時間がかかる必要がある。
また、たとえそれらが挑戦されていなくても、私たちが持って許可された特許と特許出願は、私たちの知的財産権を十分に保護できず、私たちの候補製品に排他性を提供したり、私たちの声明をめぐる他の人の設計を阻止したりすることができないかもしれない。もし私たちが持っている特許出願が提供する保護の広さや強度が脅かされていれば、会社が私たちと協力して候補製品を開発することを阻止し、候補製品を商業化する能力を脅かす可能性があります。したがって、我々のゲノム編集プラットフォームの任意の進展および候補製品が、有効かつ強制的に実行可能な特許によって保護または保護されるかどうかは分からない。私たちの競争相手または他の第三者は、非侵害的に類似または代替技術または製品を開発することによって、私たちの特許を回避することができるかもしれない。例えば,第三者がCRISPR技術とCas 9以外のタンパク質を組み合わせて使用することを提案していることが知られている。私たちが持っていて許可された特許はそのような技術を含まないかもしれない。もし我々の競争相手がCRISPR技術をCas 9以外のタンパク質とともに商業化すれば、私たちの業務、財務状況、運営結果、将来性は実質的な悪影響を受ける可能性がある。また,臨床試験で遅延に遭遇すれば,特許保護された候補製品を販売できる時間が短縮される。
私たちの遺伝子編集技術および候補製品は、第三者が持っている独占権を使用する必要があるかもしれないので、私たちの業務の成長は、私たちがこれらの独占権を獲得、許可、または使用する能力にある程度依存するかもしれない。私たちは私たちが決定した第三者からこのような知的財産権を得ることができないかもしれない。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちの投資が適切な見返りを得るための条項の許可や第三者知的財産権を得ることができないかもしれない。また,業界,政府,学術界,その他のバイオテクノロジーや製薬研究の拡大に伴い,より多くの特許が発行され,我々の候補製品は他人の特許権侵害のクレームを引き起こすリスクが増加する可能性がある。私たちは私たちの遺伝子編集技術、候補製品、またはそのような候補製品を使用して第三者特許を侵害しないことを保証できない。特許権は管轄区域ごとに付与されているため、我々の候補製品を研究、開発、商業化する能力を含むいくつかの技術の自由を実施しており、国によって異なる可能性がある。
これらの結果のいずれも、第三者の競争を阻止する能力を弱める可能性があり、第三者競争は私たちの業務に悪影響を及ぼす可能性がある。私たちの未解決および将来の特許出願または私たちが許可内に配置することによって権利を獲得する特許出願は、私たちの技術または将来の候補製品の全部または一部を保護するために、または他社が競合技術および製品を商業化することを効果的に阻止するために、特許の発行を招くことができない可能性がある。
特許提供の保護に加えて、我々は、特許を出願できない独自技術、特許を実施しにくいプロセス、および私たちの候補製品発見および開発中に特許がカバーされていないノウハウ、情報または技術に関する任意の他の要素を保護するために、商業秘密保護および秘密保護プロトコルに依存して保護する。しかし、商業秘密は保護するのが難しいかもしれない。私たちは、従業員、コンサルタント、科学コンサルタント、請負業者と秘密保持協定を締結することで、私たちのノウハウやプロセスを保護することを求めています。また、私たちは私たちのビルの実体安全と、私たちの情報科学技術システムの実体と電子安全を維持して、私たちの資料と商業秘密の完全性とセキュリティを維持することに力を入れています。私たちはこのような個人、組織、そしてシステムに自信がありますが、合意や安全措置は違反される可能性があり、私たちはどんな違反にも対応する十分な救済措置がないかもしれません。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。
私たちのすべての従業員とコンサルタントが彼らの発明を私たちに譲渡することを望んでいますが、私たちのすべての従業員、コンサルタント、コンサルタント、および私たちのノウハウ、情報、または技術にアクセスできる第三者は、秘密協定を締結するために、すべてのこのような合意が正式に実行されているか、または私たちのビジネス秘密および他の機密固有情報が漏洩されないこと、または競争相手が他の方法で私たちのビジネス秘密または独立開発と実質的に同等の情報および技術を獲得しないことを保証することはできません。私たちのビジネス秘密を流用したり不正に開示したりすることは、私たちの競争地位を損なう可能性があり、私たちの業務に実質的な悪影響を及ぼす可能性があります。また,我々のビジネス秘密を守るための手順が不十分であると考えられれば,第三者による商業秘密の流用に対抗する十分な追跡権がない可能性がある.しかも、他の人たちは私たちの商業秘密と固有の情報を独立して発見するかもしれない。例えば、その透明性イニシアティブの一部として、FDAは、ビジネス秘密または他の固有情報と考えられる可能性のある情報を含む、より多くの情報を定期的に公開するかどうかを検討しており、FDAの開示政策が将来どのように変化する可能性があるかは不明である。
84
また、一部の外国の法律の専有権に対する保護程度や方式は米国の法律とは異なる。したがって、私たちはアメリカでも海外でも、私たちの知的財産権を保護して守ることで大きな問題に直面するかもしれない。もし私たちが私たちの技術に関連する非特許知的財産権を第三者に開示することを阻止できず、私たちがこのような強制的に実行可能な商業秘密保護を持つことが保証されなければ、私たちは私たちの市場で競争優位性を確立したり維持することができなくなり、これは私たちの業務に実質的な悪影響を及ぼすかもしれない。
第三者が私たち、私たちのライセンシー、または私たちの協力者に提出した知的財産権侵害疑惑は、私たちの製品発見と開発を阻害または延期する可能性があります。
私たちの商業的成功は私たちが第三者の有効な特許と独自の権利の侵害を避けることにある程度かかっている。
我々が我々の候補製品を開発している分野には,大量の米国や外国から発行された特許と,第三者が所有する未解決特許出願が存在する.業界、政府、学術界、その他のバイオテクノロジーや製薬研究の拡大、およびより多くの特許の発行に伴い、私たちの候補製品は他人の特許権侵害のクレームを引き起こすリスクが増加する可能性がある。私たちは私たちの技術、未来の候補製品、またはそのような候補製品の使用が第三者特許を侵害しないことを保証することはできない。私たちはまた関連する第三者特許や出願を決定できない可能性がある。特許権は管轄区域ごとに付与されているため、我々の候補製品を研究、開発、商業化する能力を含むいくつかの技術の自由を実施しており、国によって異なる可能性がある。
第三者は私たちが彼らの特許を侵害したと主張するかもしれないし、私たちは彼らのノウハウを不正に使用して、私たちを起訴するかもしれない。我々が発見し開発した候補製品の成分、配合、製造方法、または使用または処理方法の請求項を含む、我々が現在知らない第三者特許がある可能性がある。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、我々の候補製品が発行された特許を侵害する可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術または製造、使用、使用または販売のために私たちの候補製品がこれらの特許を侵害したと主張する可能性がある。そのような第三者特許が私たちの技術または候補製品をカバーするために管轄権のある裁判所によって所有されている場合、任意のそのような特許の所有者は、適用特許に基づいて許可されるか、またはそのような特許が満期になるまで、または最終的に無効または実行不可能と判定されない限り、適用可能な候補製品を商業化することを阻止することができるかもしれない。そのような許可は商業的に合理的な条項や根本的に存在しないかもしれない。もし私たちが商業的に合理的な条項で第三者特許の必要な許可を得ることができなければ、候補製品を商業化する能力は損害や遅延を受ける可能性があり、これは逆に私たちの業務を深刻に損なう可能性がある。
第三者の知的財産権宣言は私たちの製品発見と開発を阻害したり延期したりする可能性がある。
第三者は、私たちが所有しているか、または彼らまたは他の人から許可された知的財産権を含むか、または重複する知的財産権を要求することができる。これらの権利の範囲および所有権を決定するための法的手続きが開始される可能性があり、禁止または他の公平な救済を含む権利を失う可能性があり、これらは、私たちの候補製品のさらなる開発と商業化を効果的に阻止するかもしれない。第三者によって引き起こされるか、または米国特許商標局によって提起された干渉または派生プログラムは、我々の特許または特許出願または我々の許可者の特許または特許出願に関連する発明の優先権または正しい発明権を決定するために必要である可能性がある。不利な結果は、現在の特許権の喪失を招く可能性があり、関連技術の使用を停止したり、勝利者から許可権を得ようとしたりすることが要求される可能性があります。もし勝利者が商業的に合理的な条件で私たちにライセンスを提供しなければ、私たちの業務は損害を受ける可能性があります。訴訟、介入、または派生手続きは私たちの利益に不利な決定を招く可能性があり、私たちが成功しても、巨額のコストを招き、私たちの経営陣や他の従業員の注意を分散させる可能性がある。
例えば、第三者は、いくつかのCRISPR/CAS 9技術に権利がないと断言することができ、またはCVCグループは、CVCグループのいくつかの特許の発明権および所有権を含むいくつかのCRISPR/CAS 9技術に対する権利を有していない、またはそのような権利は限られていると断言することができる。
具体的には、ボイド研究所およびマサチューセッツ工科大学は、場合によっては、米国およびヨーロッパで発行された特許を含む一連の特許を有しており、CRISPR/CAS 9システムのいくつかの態様では、ヒト細胞中のDNAを含む真核細胞を編集することができるCRISPR/CAS 9システムのいくつかの態様を必要とする。2016年1月、米国特許商標局は、特許組合において当時審理されていた米国特許出願のうちの1つ(現在米国特許第10,266,850号として発行されている)とブロドが共同所有する12件の発行された米国特許との間に干渉(干渉番号106,048,または‘048干渉)が存在し、どの発明者が最初に発明し、米国で発明特許を取得する権利があるかを決定することを発表した。PTABの結論は、関連するクレームセットが特許上互いに異なると考えられるので、発表された干渉を停止すべきである。CVCグループが控訴した後,2018年9月10日,連邦巡回裁判所はPTAB干渉プロセスの終了決定を確認したが,どの発明者が実際にCRISPR/Cas 9ゲノム編集技術の真核細胞への使用を発明したかは決定されていない。
さらに、2019年6月、米国特許商標局は、CVCグループが共同所有する14件の係属中の米国特許出願と13(13)の特許と1つの特許出願との間に第2の干渉が存在すると発表した(干渉番号106,115または‘115
85
遠大集団が共同で所有する.遠大な特許は048妨害された特許を含む。2020年9月、臨時技術相談機関は、他の事項を除いてプログラムを優先段階に早める命令を発表した。2022年2月、PTABは優先権と判決決定を発表し、遠大が干渉標的の面でCVC集団より優先していると認定した。CVCグループはこの決定について連邦巡回控訴を行った。連邦巡回裁判所のいかなる最終決定も最高裁判所にさらに上訴することができる。
The BRoadを除いて、ビリニュス大学、ToolGen,Inc.,MillipreSigma(メルクKGaAの子会社、以前は“Sigma-Aldrich”と呼ばれていた)およびハーバード大学がCVCグループの出願後約1年以内にCRISPR/Cas 9に関連する発明を要求する特許出願を提出し、CVCグループの前にCVCグループが主張した1つまたは複数の発明を発明したと主張する(または主張することができる)The BRoadを除く他の第三者。USPTOがこれらの当事者の一方または複数のクレーム範囲がCVCグループ申請で許可されたクレームと十分に重複していると考えている場合、USPTOは、そのようなクレームの実際の発明者を決定するために、他の介入手続きを宣言することができる。例えば、2020年12月、米国特許商標局は、ヒト細胞を含む真核細胞においてDNAを編集するためにCRISPR/Cas 9システムのいくつかの態様を要求するToolGen特許出願と、‘115干渉に関与するCVC集団が共同で所有する14つの係属中の米国特許出願との間に干渉(干渉番号106,127または’127干渉)が存在すると発表した。この干渉は放置されており、連邦巡回裁判所が‘115妨害の判決を下すのを待っている。PTABの判決は連邦巡回裁判所に上訴し,最終的に最高裁に上訴することができる。さらに、2021年6月、米国特許商標局は、ヒト細胞を含む真核細胞においてDNAを編集するためにCRISPR/Cas 9システムのいくつかの態様を要求するMilLiporeSigma特許出願と‘115干渉に関与するCVC集団が共に所有する14つの係属中の米国特許出願との間に干渉(干渉番号106、132または’132干渉)が存在することを発表した。この干渉は放置されており、連邦巡回裁判所が‘115妨害の判決を下すのを待っている。最終的に,PTABの判決は連邦巡回裁判所に上訴し,最終的に最高裁に上訴する可能性がある。
CVCグループ、ブロド、ToolGen、ビリニュス大学、ミリプレシグマ、およびハーバード大学の各々は、米国および他の場所で既存または新しい特許出願を出願することができる。CVCグループおよびこれらの他の第三者は、真核細胞(ヒト細胞を含む)においてDNAを編集するためにCRISPR/Cas 9システムおよび方法の重複面を有すると主張しているので、CRISPR/Cas 9に基づくヒト療法をマーケティングおよび販売する能力は、特に競合特許の組み合わせで主張される発明の範囲および実際の所有権に依存する悪影響を受ける可能性がある。
将来を展望すると、米国特許商標局は、CVCグループまたは私たち個人に関連するCRISPR/CAS 9技術の使用に関する新しい干渉を発表することができる。さらに、我々およびCVCグループは、CRISPR/Cas 9発明に関する他の特許主張を起訴し続けており、これは、米国で許可または発行された特許を得ることにもつながる可能性がある。CVCグループおよび我々が起訴しているいくつかのクレームは、USPTOによって発見されることが許可されている場合、上に記載された特許または特許出願を含む第三者が所有する特許または特許出願に対する妨害訴訟を引き起こす可能性がある。USPTOが、これらの当事者のうちの一方または複数の特許請求の範囲が、特許の組み合わせまたは我々の特許の組み合わせ内の特許または特許出願の許容される特許請求の範囲と十分に重複していると考える場合、USPTOは、そのような特許請求の第1の発明者を決定するために、他の干渉手順を宣言することができる。私たちはこれらの結果のいずれかを決定することができず、もしあれば、実際に発生する。より多くの干渉があれば、干渉のいずれか一方は再び連邦巡回控訴に不利な決定を下すことができる。さらに、CVCグループの任意の既存または新しい特許、または私たちの既存または新しい特許の有効性または実行可能性は、他の挑戦を受ける可能性がある。このような結果が私たちと私たちの知的財産権状況に及ぼす可能性のある影響はまだ不明だ。
いずれかの第三者がその干渉で勝訴し、これらの異なる法的手続きによって、我々の候補製品または関連活動をカバーする特許主張を取得した場合、第三者は、CRISPR/CAS 9に基づく活動(商業化を含む)に基づいて発行された特許を主張することを求めることができる。私たちが特許権を主張する第三者に対して禁止または他の公平な救済を求めることができ、これは、私たちの候補製品をさらに開発し、商業化する能力を効果的に制限または阻止することができる。もし私たちが第三者の有効な知的財産権を侵害していることが発見された場合、私たちは、当社の製品や技術の開発とマーケティングを継続し、または第三者の知的財産権を無効にするために、第三者からライセンスを取得することを要求される可能性があります。これらの第三者は、私たちにそのような許可を与える義務がなく、そのような許可は、商業的に合理的な条項や全く得られないかもしれない、または非排他的である可能性がある。もし私たちがそのようなライセンスを取得して維持できなければ、私たちと私たちのパートナーは、私たちのコア遺伝子編集実践を停止し、私たちが開発する可能性のある1つ以上の候補製品の開発、製造、商業化を必要とするかもしれない。さらに、もし私たちが故意に特許を侵害したことが発見された場合、私たちは3倍の損害賠償と弁護士費を含む金銭損害賠償責任を負われる可能性がある。権利侵害発見は、1つ以上の候補製品を商業化し、侵害製品の再設計を強要したり、業務運営の一部または全部を停止させたりすることを阻止する可能性がある, いずれも私たちの業務に実質的な損害を与える可能性があり、私たちが提案した未来の候補製品のさらなる開発と商業化を阻止し、私たちに大きな被害を与える可能性があります。排他的喪失や特許主張の縮小は、他人が類似または同じ技術および製品を使用したり、それを商業化する能力を阻止することを制限する可能性がある。我々が第三者の機密情報や商業秘密を盗用したと主張することは,我々の業務に類似した負の影響を与える可能性がある.上記のいずれも、当社の業務、財務状況、経営結果、または将来性に重大な悪影響を及ぼす可能性があります。これらのクレームの弁護は、その是非にかかわらず、巨額の訴訟費用がかかり、私たちの業務の経営陣や他の従業員資源を大量に移し、私たちの名声に影響を与える可能性がある。
86
いずれにしても、最終的に優先順位を決定するのに数年かかるかもしれない。Charpentier博士とのライセンス契約条項により,Charpentier博士の特許訴訟,弁護,我々の許可内技術に関する費用の支払いまたは補償を担当する.
第三者が有するCRISPR/CAS 9に関連する知的財産権または開発、製造および商業化が可能なCRISPR/CAS 9療法に必要な他の関連技術、例えば、製品または成分の成分、治療方法、伝達技術、化学修飾、および分析および製造方法は、私たちの最終マーケティングおよび製品販売能力に悪影響を及ぼす可能性がある。第三者は、我々が実行可能な製品を開発したり、それを商業化するために必要な可能性のある製品を含む、私たちの技術および潜在的な製品をカバーするすべてまたは態様をカバーする知的財産権を有することができる。もし私たちがこのような第三者知的財産権を許可、回避、挑戦することに成功できなければ、私たちはすべてまたはいくつかの司法管轄区域で実行可能な製品を開発し、商業化することができないかもしれない。また、私たちの製品や私たちが所有したり許可したりする技術をカバーする知的財産権が法的に損傷したり失われたりすると、私たちの製品の開発や商業化を支援するのに十分な財務的リターンを達成できない可能性があります。
さらに、第三者は、通常、欧州および/または他の非米国司法管轄区域で承認されたか、または将来的に承認される可能性がある米国の出願の国際同業者に提出される。私たちと他の当事者たちはその中のいくつかの寄付に反対手続きを提起して、私たちは未来にこれらや他の申請者に他の贈与を提供することに反対するかもしれない。同様に、私たちの知的財産権は未来にヨーロッパや他の管轄区域の反対手続きに巻き込まれる可能性がある。これらの反対意見は、特許の全部または一部が撤回される可能性があり、またはクレーム範囲の縮小を招く可能性があり、欧州で競争相手に対して特許を実行する能力を弱めるまたは排除する可能性がある。例えば、2018年2月には、いくつかの方向に欧州特許庁が反対し、我々に付与された第1項のライセンス内の欧州特許に反対する。2018年遅い時期と2019年には、いくつかの締結方向が欧州特許庁に反対し、私たちに第2および第3のライセンス内の欧州特許を付与することに反対する。反対手続きは、特許のすべての撤回をもたらすことができる;付与された特許を維持するか、または修正された形態で特許を維持することができる。反対派の訴訟手続きは通常、どちらも控訴できるのに要する時間を含めて数年かかる。私たちは私たちが許可した欧州特許に対する反対結果を保証することはできず、不利な結果は欧州で第三者に対して私たちの権利を強制的に執行することを阻止するかもしれない。例えば、2020年初め、欧州特許庁は修正された形で私たちの最初の認可内の欧州特許を維持し、2021年末、彼らは私たちの2つ目の欧州特許を撤回し、その決定は控訴を待っている。
これらのイベントの結果を予測することもできず,不利な結果による損失金額や範囲を有意に見積もることもできない.将来的には、正常な業務過程で生じる法的問題やクレームの当事者になる可能性があり、これらの問題の解決は、私たちの財務状況、運営実績、またはキャッシュフローに大きな悪影響を与えないと予想されます。
私たちは私たちの候補技術と製品の権利が他の人が私たちに付与する許可証の条項と条件によってある程度制限されていることを開発し、商業化します。
私たちは第三者からのいくつかの知的財産権ライセンスに依存しており、これらの知的財産権は私たちの遺伝子編集技術と候補製品の開発に非常に重要または必要である。これらの許可および他の許可は、すべての関連使用分野でこのような知的財産権および技術を使用する独占的な権利を提供しない可能性があり、将来私たちの技術および製品の開発または商業化を望む可能性のあるすべての地域をカバーしないかもしれない。したがって、私たちは競争相手が私たちのすべてのライセンスに含まれる地域で競争製品を開発し、商業化することを阻止できないかもしれない。
さらに、私たちの2014年のDr.Charpentierとの独占ライセンス契約を含め、私たちのライセンス技術を使用して製品を販売する収入に応じて印税を支払うことが要求されることになり、これらの印税支払いは、商業化を求める可能性のある任意の製品の全体的な収益力に悪影響を及ぼす可能性があります。我々がCharpentier博士と達成したすべての許可協定によると,商業的に合理的な努力を用いて規制部門の承認を得て,許可された治療製品を開発して販売する義務がある。Charpentier博士とのライセンス契約には,2021年4月までにIND(または主要市場国に同等製品を提出)を提出する義務と,2024年4月までにIND(または主要市場国の同等製品)を提出する義務も含まれている。2021年4月までにINDを提出する義務を果たしていますが、将来的には他の余剰義務をタイムリーにまたは根本的に履行できないかもしれません。もし私たちが残りの義務を履行できなければ、Charpentier博士は私たちの許可権を終わらせる権利があるかもしれない。私たちはライセンスプロトコルがカバーする製品の臨床開発の多くの面でアウトソーシングし、第三者に依存する必要があるだろう。これらの第三者の遅延や失敗は、私たちの職務調査義務を履行する能力および第三者許可者とのライセンス契約の継続に悪影響を及ぼす可能性があります。
私たちが最善を尽くしたにもかかわらず、私たちの許可側は、許可協定に深刻に違反していると結論するかもしれませんので、許可協定を終了し、これらの許可協定がカバーする製品や技術を開発し、商業化することができません。これらの許可内で終了された場合、または基礎特許が予想される排他性を提供できなかった場合、競争相手は、規制部門の承認を求め、私たちと同じ製品を販売する権利がある。さらに、私たちは、私たちのライセンシーから追加の許可を得ることを求めることができ、このような許可を得る過程で、私たちは、第三者(私たちの競争相手を含む可能性がある)が私たちの既存のライセンスの一部のライセンスを取得することを許可する条項を含む、ライセンシーに有利な方法で私たちの既存のライセンスを修正することに同意するかもしれません
87
許可証。これらの事件のいずれも、私たちの競争地位、業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちのコア遺伝子編集技術を保護する知的財産権は共通して所有されているが、私たちの許可証はその中の一人の共通所有者だけであり、これはアメリカと他の司法管轄区における私たちの権利を深刻に制限している。
我々がCharpentier博士から独占的に許可を得た特許の組合せは,我々の遺伝子編集技術のコア特許保護である.しかし、この家族は指名された他の発明家を含み、彼らは自分の権利をカリフォルニア州やウィーンに譲渡した。したがって,特許組合せは現在カリフォルニア州のCharpentier博士とウィーンのDr.Charpentierが共同で所有している。2016年12月15日、私たちはカリフォルニア州、ウィーンおよびそのライセンシー(CariouおよびCariouを含む被許可者Intellia Treeutics)と譲渡、許可、共同所有権、および発明管理協定(IMA)に同意した。IMAによると、共通所有者は、他の共通所有者の被許可者および分被許可者に世界的な相互交差同意を提供し、費用分担協定を含むCRISPR/Cas 9遺伝子編集基本知的財産権の支援および管理に関するいくつかの他の約束および義務を合意した。以下でより詳細に説明するように、これにより、私たちは、私たちのコア遺伝子編集技術を保護する特許権の非独占または共同独占権利しか持たず、カリフォルニアとウィーンがCharpentier博士から許可を得ることに同意することに同意するために、IMA下の契約義務を引き続き履行しなければならない。
アメリカでは、すべての共通の人々がこの技術を許可して使用する自由を持っている。そのため,Charpentier博士がカリフォルニアやウィーンなどの他のエンティティと共有しているいかなる知的財産権も独占的に得ることはできない.したがって,我々とCharpentier博士の許可は,このような共有する権利に対して非排他的である.さらに、米国では、すべての共通の人々が、これらの特許権を強制的に執行するために、私たちが提起する可能性のある任意のクレームまたは訴訟の当事者に参加することを要求されている。また、米国では、非独占許可は裁判所に特許侵害訴訟を提起する資格がない。したがって,カリフォルニアとウィーンが所有している特許については,カリフォルニアとウィーンの協力がなければ,第三者侵害クレームを追及する能力がなく,彼らのライセンシーの協力がない可能性がある。第三者が特許組合における特許権を侵害した場合の通知及び調整が規定されているウィーン及びカリフォルニア州及びそのライセンシーとIMAを締結しているにもかかわらず、ウィーン及びカリフォルニア州が将来のいかなる侵害においても我々と協力することは保証されていない。Charpentier博士から取得したコア特許権を実行できなければ,第三者が我々と競合することを阻止できない可能性があり,会社に我々の技術を再許可するように説得できない可能性があり,いずれも我々の業務に実質的な悪影響を与える可能性がある.
私たちは、私たちがそこから知的財産権の許可を得た第三者または私たちが知的財産権を許可している第三者と紛争するかもしれない。これらの当事者とのどんなトラブルも私たちの業務に悪影響を及ぼす可能性があり、私たちは私たちの業務に非常に重要な許可権を失うかもしれません。
私たちは第三者から私たちの遺伝子編集技術をカバーする知的財産権許可を得ましたが、私たちの遺伝子編集技術を拡大しながら、追加の第三者知的財産権許可を得続けたいと思います。様々な理由で、私たちは私たちの知的財産権を許可した第三者と紛争するかもしれません
さらに、私たちは現在、どのプロトコルに従って第三者に知的財産権または技術を許可しているか、またはIMAに従って同意を維持しているか、これらのプロトコルのいくつかは、様々な解釈の影響を受ける可能性があり、または、私たちが1つまたは複数の合意に違反するように衝突する可能性があり、これは、私たちの1つまたは複数の許可パートナーまたはIMA当事者と長く高価な紛争を発生させることを容易にする。可能性のある契約解釈の相違の解決は、関連する知的財産権または技術に対する私たちの権利と考えられる範囲を縮小するか、または関連協定の下での私たちの財務または他の義務を増加させる可能性があり、いずれも、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。さらに、我々が許可している知的財産権をめぐる紛争が、商業的に許容可能な条項で現在の許可手配の能力を維持していることを阻害または損害すれば、影響を受けた候補製品の開発および商業化に成功することができない可能性があり、これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
88
同様に、私たちが第三者とライセンス契約、協力協定、パートナー関係を締結し続けて、私たちの開発プロジェクトを拡大するにつれて、私たちはすでに、私たちのいくつかの知的財産権をこれらの第三者にライセンスし続ける予定です。様々な理由で、私たちはこれらの第三者と紛争が発生する可能性があり、私たちはこのような任意の合意によって付与された権利範囲や他の解釈に関する問題を含む知的財産権をこれらの第三者に権限を付与します。現在または将来のパートナーまたは許可側と、このようなパートナーまたはライセンス者に付与された知的財産権の範囲について発生するいかなる紛争も、開発計画の遅延を招き、私たちのパートナーまたは許可者と長引くコストの高い紛争を容易にすることができます。
私たちは、任意の候補製品の取得または維持、または買収およびライセンス内で開発される可能性のある他の技術の必要な権利の取得または維持に成功できないかもしれません。
我々は現在知的財産権の権利を有しており,第三者の許可により候補製品を識別·開発し,他の技術を使用している。多くの製薬会社、バイオテクノロジー会社、学術機関が遺伝子編集技術の分野で私たちと競争し、私たちの業務に関連する可能性のある特許出願を提出します。例えば、公開されれば、私たちの遺伝子編集技術および候補製品をカバーすると解釈される可能性がある第三者特許出願がいくつかあることが知られている。これらの第三者特許の侵害を回避するためには、これらの第三者知的財産権所有者から許可を得ることが必要または慎重であることが発見される可能性がある。我々はまた、修正された核酸のようないくつかの遺伝子編集技術の修正または改善コンポーネントのライセンスを第三者から取得し、我々が開発可能な候補製品の配信方法を評価しているような非CRISPR/CAS 9技術を取得する必要があるかもしれない。さらに、私たちが第三者と共同で所有しているいかなる特許についても、私たちはこのような共通の所有者のこのような特許の利益を許可する必要があるかもしれない。しかしながら、我々が開発する可能性のある候補製品および遺伝子編集技術については、第三者からそのような許可を得ることができないか、または許可の任意の成分、使用方法、プロセス、または他の知的財産権を取得または取得することができない可能性がある。第三者知的財産権の許可または買収は競争分野であり、私たちよりも成熟しているか、またはより多くの資源を持っている可能性がある企業は、魅力的または必要だと思う第三者知的財産権許可または買収戦略をとっているかもしれない。より成熟した会社は規模が大きいため、私たちより競争優位になるかもしれません, 資本資源とより強い臨床開発と商業化能力。しかも、私たちを競争相手と思っている会社は私たちに権利を譲渡したり許可したりしたくないかもしれない。私たちはまた私たちが投資から適切な見返りを得ることを可能にする条項に従って許可したり、第三者知的財産権を取得することができないかもしれない。このような交渉を成功させ、最終的に買収を求める可能性のある他の候補製品や技術をめぐる知的財産権を得ることができる保証はない。もし私たちが必要な第三者知的財産権を得ることに成功したり、私たちの既存の知的財産権を維持することができなければ、関連するプログラム、技術または候補製品の開発を放棄しなければならないかもしれないし、私たちのコアCRISPR/Cas 9遺伝子編集技術の実践を停止しなければならないかもしれません。これは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
法廷やUSPTOなどの外国当局が疑問を提起した場合、我々の候補技術および製品に関連する発行された特許は、無効または実行不可能であることが発見される可能性がある。
もし私たちまたは私たちの許可者のうちの1つが第三者に対して法的訴訟を提起して、私たちが開発する可能性のある候補製品または私たちの技術(CRISPR/Cas 9を含む)をカバーする特許を強制的に執行する場合、被告はその特許を無効または実行できないと反訴することができる。米国の特許訴訟では,被告が無効または実行不可能と主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、明らかな、または実施できないことを含む、いくつかの法定要件のいずれかを満たしていないと言われているからかもしれない。主張を実行できない理由は,特許起訴に関連する者が起訴中に米国特許商標局に関連情報を隠蔽したり,誤った声明をしたりしたためかもしれない.
第三者は、我々の許可内のCRISPR/Cas 9特許出願がヨーロッパおよびオーストラリアで提出された第三者の意見および反対意見のような、我々のいくつかの許可内の特許出願の有効性に疑問を提起し、将来的には、米国または他の管轄区域の行政機関に、我々の許可内および所有する特許出願および特許に関連する同様のクレームを、訴訟範囲外に提出する可能性がある。特許庁で特許の有効性を疑問視する仕組みには、再審査、ライセンス後の再審査、各方面間審査、介入訴訟、派生訴訟、および非米国司法管轄区域の同等の訴訟(例えば、反対訴訟)。利用可能な控訴を使い切った後、そのような訴訟は、私たちの特許出願または特許の損失をもたらす可能性があり、またはそれらの範囲は、私たちの技術またはプラットフォーム、または私たちが開発する可能性のある任意の候補製品に縮小される可能性がある。法的に無効と実行不可能と断言された後の結果は予測できない。たとえば,有効性の問題については,既存技術を無効にする問題は存在しないとは限らない.もし第三者が無効または強制不可能な法的主張に勝った場合、私たちは少なくとも部分的、さらにはすべて、私たちの技術またはプラットフォーム、または私たちが開発する可能性のある任意の候補製品の特許保護を失うだろう。このような特許保護の喪失は、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼすだろう。
89
CRISPR/CAS 9を含む遺伝子編集技術をめぐる知的財産権構造は高度に動的であり、第三者が開始し、法的訴訟で勝利する可能性があり、私たちが許可または所有している特許が無効であるか、または私たちが彼らの知的財産権を侵害、流用しているか、または他の方法で彼らの知的財産権を侵害していると主張し、その結果は不確実であり、私たちの業務成功に大きな悪影響を及ぼす可能性がある。
遺伝子編集分野,特に遺伝子編集技術分野は,まだ初期段階であり,このような製品は市販されていない.我々と我々の競争相手を含むいくつかの会社がこの分野で緊張した研究·開発を行っているため、知的財産権の構造が変化しており、今後数年間も不確定である可能性がある。将来的には知的財産権に関する重大な訴訟や、私たちが所有しているものや許可されていない他の第三者、知的財産権、独自の権利に関する訴訟があるかもしれない。
私たちのビジネス成功は、第三者の知的財産権および独自の権利を侵害、流用、または他の方法で侵害することなく、私たちと私たちの協力者が開発、製造、マーケティング、販売する可能性のある任意の当社の独自技術の候補製品を開発し、使用する能力に依存します。バイオテクノロジーと製薬産業の特徴は特許と他の知的財産権に関する広範な訴訟だ。私たちの技術と私たちが開発する可能性のある任意の候補製品について、私たちは、妨害手続きの再検討、許可後の審査、および任意の候補製品の知的財産権に関する対抗性訴訟または訴訟の当事者になるか、または将来的に私たちの技術および任意の候補製品に関する対抗性訴訟または訴訟を受けることができる各方面間欧州特許庁の反対意見のような米国特許商標局の審査及び派生手続並びに他の司法管区の類似手続。第三者は、既存の特許または将来付与される可能性のある特許に基づいて、その是非曲直にかかわらず、侵害請求を私たちに提起するかもしれない。もし私たちがこれらの特許が無効であることを証明できず、私たちが商業的に合理的な条項で許可を得たり、維持することができなければ、これらの特許は私たちの業務行動に重大な悪影響を及ぼすかもしれない。もし私たちがこのような第三者特許を侵害していることが発見された場合、私たちと私たちのパートナーは、損害賠償金の支払いを要求される可能性があり、私たちのコアCRISPR/Cas 9遺伝子編集技術を含む侵害技術の商業化を停止するか、またはこれらの第三者から許可を得ることができず、これらの許可は商業的に合理的な条項では得られないかもしれない。
第三者知的財産権主張に法的根拠がないと考えても,裁判所が侵害,有効性,実行可能性,所有権や優先権などの問題で我々に有利である保証はない.管轄権のある裁判所は、これらの第三者特許が有効で、強制的に実行可能であり、侵害されていると判断する可能性があり、これは、開発可能な任意の候補製品および主張される第三者特許がカバーする任意の他の候補製品または技術を商業化する能力に実質的な悪影響を及ぼす可能性がある。連邦裁判所でこのような米国特許の有効性に挑戦することに成功するためには,有効性推定を克服する必要がある。この負担が重いため,このような米国特許主張の無効について明確で納得できる証拠を提出することが求められているため,管轄権のある裁判所がこのような米国特許の主張の無効を宣言する保証はない.もし私たちが第三者の知的財産権を侵害していることが発見され、そのような特許が無効または強制的に実行できないことを成功的に証明できなかった場合、私たちは、開発、製造、およびマーケティングの可能性のある任意の候補製品および私たちの技術を継続するために、第三者からライセンスを取得することを要求されるかもしれない。しかし、私たちは商業的に合理的な条項や必要な許可証を得ることができないかもしれない。私たちが許可を得ることができても、それは非排他的である可能性があり、私たちの競争相手や他の第三者が私たちに許可された同じ技術にアクセスできるようにするためには、大量の許可と印税を支払う必要があるかもしれない。私たちはまた裁判所の命令を含む権利侵害技術または候補製品の開発、製造、商業化を停止させることを余儀なくされる可能性がある。さらに、私たちは3倍の損害賠償と弁護士費を含む重大な金銭的損害に責任があると判断される可能性がある, もし私たちが特許や他の知的財産権を故意に侵害することが発見されたら。第三者の機密情報や商業秘密を盗用したと主張することは、当社の業務、財務状況、運営結果、見通しに類似した重大な悪影響を及ぼす可能性があります。
知的財産権訴訟は私たちに大量の資源を費やし、私たちの人員のその正常な責任に対する注意を分散させるかもしれない。
正当な理由の有無にかかわらず、知的財産権クレームに関連する訴訟や他の法的手続きは予測不可能であり、通常コストが高く、時間がかかり、私たちのコア業務から大量の資源を分流し、私たちの技術と管理者の正常な責任を分散させ、通常私たちの業務を損害する可能性がある。さらに、一部の国(米国を含む)は知的財産権訴訟において大量の情報を開示する必要があるため、私たちのいくつかの機密情報は、このような訴訟中に開示によって漏洩される可能性がある。さらに、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果が公表される可能性があり、証券アナリストや投資家がこれらの結果がマイナスであると考える場合、私たちの普通株式価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。
私たちはそのような訴訟や法的手続きを適切に行うのに十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、彼らがより多くの財政資源を持っているので、私たちよりもこのような訴訟や訴訟の費用を効率的に負担するかもしれない。したがって、私たちは努力したにもかかわらず、私たちは第三者が私たちの知的財産権を侵害したり、流用したり、私たちの知的財産権に成功したりすることを防ぐことができないかもしれない。特許訴訟または他の訴訟の開始と継続によって生じる不確実性は、市場での競争能力に重大な悪影響を及ぼす可能性がある。
90
私たちが許可したいくつかの知的財産権は、政府援助の計画によって発見される可能性があり、したがって、“パレード権利”、いくつかの報告要件、および米国メーカーの優先順位のような連邦法規の制約を受ける可能性がある。これらの規定を遵守することは、私たちの独占権を制限し、非アメリカメーカーと契約を締結する能力を制限するかもしれない。
私たちがCharpentier博士の共通利益の下で許可を得た知的財産権はカリフォルニア州によって共同所有されており、カリフォルニアは1つ以上の発明が許可番号の下で行われていることを示している。GM 081879はアメリカ国家衛生研究院から授与された。したがって、このような権利は特定の連邦規制によって制限されている。1980年の“ベイハ·ドール法案”(Bayh-Dole Act)によると、米国政府は、政府の援助計画の下で開発されたいくつかの発明の特許に対して、政府の権利をカバーするいくつかの権利を持っている。これらの権利は、任意の政府の目的のために発明を使用する非排他性、譲渡不可能、撤回不可能な世界的許可を含む。さらに、米国政府は、(I)発明を商業化するのに十分なステップが取られていないこと、(Ii)政府が公衆衛生または安全需要を満たすために行動しなければならないこと、または(Iii)政府が公衆使用に対する連邦法規の要求を満たすために行動しなければならないことを前提として、第三者に上述した任意の発明の独占的、部分的独占的または非独占的許可を付与する権利を要求する権利がある。我々又は適用される請負業者が政府に発明を開示しておらず、かつ所定の期限内に知的財産権登録申請を提出していない場合、米国政府もこれらの発明の所有権を取得する権利がある。政府援助の計画の下で生成された知的財産権もまた、いくつかの報告要件によって制約されており、これらの要求を遵守するためには、私たちまたは適用可能な請負業者が多くの資源を費やす必要があるかもしれない。また、, 米国政府は、主題発明を含む任意の製品または主題発明を使用することによって製造された製品が、実質的に米国で製造されなければならないことを要求する。知的財産権所有者が、合理的ではあるが成功しない努力をしたことを証明することができ、同様の条項で米国で大量生産される可能性のある潜在的な許可者にライセンスを付与することができる場合、またはこの場合、国内製造が商業的に不可能である場合には、製造優先権要求を免除することができる。このような米国メーカーの選好は、このような知的財産権がカバーする製品について非米国製品メーカーと契約する能力を制限する可能性がある。もし私たちが現在または未来に発明に関連する特許が米国政府の資金を使用することによって生成された場合、“ベハ-ドール法案”の条項も同様に適用される可能性がある。
私たちは世界各地で私たちの知的財産権と所有権を保護できないかもしれない。
世界各国で私たちの候補製品の出願、起訴、特許保護の費用は目を引くほど高いだろう。特許可能な要求は特定の国では、特に発展途上国では異なる可能性がある。また、私たちが知的財産権を保護·実行する能力は、世界各地の知的財産権法律の意外な変化の悪影響を受ける可能性がある。また、一部の国の特許法は米国の法律と同程度の知的財産権保護を提供していない。例えば、米国の特許法とは異なり、欧州や多くの他の管轄区域の特許法は、人体治療方法の特許性を排除し、その範囲が具体的に開示された実施形態よりも広い場合には、付与される権利請求の範囲に実質的な制限を加えている。
世界の異なる司法管轄区では、多くの会社が知的財産権の保護と保護に重大な問題に直面している。したがって、私たちは、米国以外のすべての国/地域で第三者が私たちの発明を実施したり、米国または他の管轄区域で私たちの発明を使用して製造された製品を販売または輸入することを阻止することができないかもしれない。競争相手は私たちが特許保護を申請していない司法管轄区で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っているが法執行力がアメリカに及ばない地域に輸出することもできる。これらの製品は私たちの候補製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護、特にバイオテクノロジー製品に関する保護の実行に賛成せず、私たちの特許を侵害したり、私たちの知的財産権および独自の権利に違反する競争製品を販売することを阻止することを困難にする可能性がある。世界の異なる司法管轄区域で私たちの知的財産権と独自の権利を強制的に執行する訴訟手続きは巨額のコストを招く可能性があり、私たちの努力と注意を私たちの業務の他の側面から移転させることは、私たちの特許を無効または狭義に解釈されるリスクに直面させる可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者が私たちにクレームを提起する可能性がある。私たちは私たちが起こしたどんな訴訟でも勝てないかもしれないし、判決された損害賠償や他の救済措置(もしあれば)は商業的な意味がないかもしれない。それに応じて, 私たちの知的財産権と独自の権利を世界各地で実行する努力は、私たちが開発または許可した知的財産権から顕著なビジネス的優位性を得るのに十分ではないかもしれません。
多くの国に強制許可法があり,これらの法律により特許権者は第三者に強制的に許可を付与される可能性がある。また、多くの国は、政府機関や政府請負業者を含む第三者に対する特許の実行可能性を制限している。これらの国では,特許権者の救済措置は限られている可能性があり,このような特許の価値を大幅に低下させる可能性がある。もし私たちまたは私たちの任意の許可者が第三者に私たちの業務に関連する任意の特許の許可を付与することを余儀なくされた場合、私たちの競争的地位が損なわれる可能性があり、私たちの業務、財務状況、運営結果、および見通しは不利な影響を受ける可能性があります。特許保護は最終的には国ごとに求めなければならないが,これは高価で時間のかかるプロセスであり,結果は不確定である。したがって、私たちは特定の国で特許保護を求めないことを選択することができ、私たちはこれらの国で特許保護の利点を享受しないだろう。
91
米国や他の管轄区特許法の変化は、特許の全体的な価値を低下させ、私たちの候補製品を保護する能力を弱める可能性がある。
他の生物製薬会社と同様に、私たちの成功は知的財産権、特に特許に大きく依存している。生物製薬業界で特許を獲得と実施することは技術上の複雑性と法律上の複雑性にも関連するため、コストが高く、時間がかかり、しかも内在的な不確定性を持っている。2011年9月16日に法律となった“ライシー-スミス米国発明法”(Leahy-Smith America Invents Act)を含む米国と他の国の最近の特許改革立法は、これらの不確実性とコストを増加させる可能性がある。“ライシー·スミス法案”は米国特許法を多くの重大な改正を行った。これらの条項は、特許出願起訴方式に影響を与える条項を含み、既存技術を再定義し、競争相手に特許の有効性に挑戦するために、より効果的かつ費用効果的な方法を提供する。また、“ライシー·スミス法案”は、米国の特許制度を先に申請する制度に転換した。しかし、最初の報告書の条項は2013年3月16日まで施行されなかった。したがって,Leahy-Smith法案が我々の業務運営にどのような影響を与えるかは不明である(もしあれば).しかし、Leahy-Smith法案とその実施は、私たちの発明が特許保護を得ることを難しくし、私たちの特許出願をめぐる起訴と、私たちが発表した特許の実行または保護の不確実性とコストを増加させる可能性があり、これらのすべては私たちの業務、運営結果、および財務状況を損なう可能性がある。
近年、米国最高裁判所はいくつかの特許事件に対して裁決を下し、場合によっては利用可能な特許保護範囲を縮小するか、場合によっては特許所有者の権利を弱めるかを決定している。例えばここでは分子病理学協会はMyriad Genetics社を訴えました最高裁は,“自然に産生されたDNA断片は自然の産物であり,それが分離されただけで特許を申請する資格はない”と判断し,分離したBRCA 1とBRCA 2遺伝子に対するMyriad Geneticsの主張は無効であることを宣言した。我々の特許のいくつかの請求項は、CRISPR/Cas 9遺伝子編集技術および自然に産生されたDNA配列のための誘導アセンブリに関する。このような請求項が天然製品のためのものであると考えられる場合、または孤立した天然製品の外に創造性の概念が乏しい場合、裁判所は、以下の条件に従って権利要件を無効と判断することができる何千ものそれは.また,最近では米国や他の国の特許法をより多く改正することが提案されており,採択されれば,我々のノウハウのために特許保護を受ける能力,あるいは我々のノウハウを実行する能力に影響を与える可能性がある。米国議会、米国裁判所、米国特許商標局、および他の国の関連立法機関の将来の行動によれば、特許を管理する法律および法規は予測不可能な方法で変化する可能性があり、それによって、私たちが新しい特許を獲得したり、私たちの既存の特許と将来獲得可能な特許を実行する能力を弱める可能性がある。欧州計画の統一特許裁判所は欧州競争相手の能力に対抗するために私たちの特許権を保護し実行することに特別な不確実性をもたらすかもしれません。この新しい裁判所の実施は、ヨーロッパ全体の特許法執行により多くの確実性と効率を提供するだろうが、それはまた、私たちの欧州特許を集中的に撤回するための新しいフォーラムを提供するだろう。認められる特許権の範囲と,その裁判所が提供する特許救済措置の力を知るのに数年かかるであろう。私たちは最高裁判所の最初の7年間に私たちの特許をこのシステムから除外することを選択する権利があるだろうが、そうすることは私たちが新しい統一裁判所の利点を達成することを阻止するかもしれない。
私たちの特許保護を獲得し、維持することは、政府特許機関によって適用される様々なプログラム、書類提出、費用支払い、および他の要求に準拠することに依存し、これらの要件を遵守しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
発行された特許の定期維持費は、特許有効期間内にいくつかの段階で米国特許商標局および外国特許代理機関に支払われなければならない。米国特許商標局および各種外国政府特許機関は、特許出願過程において、いくつかのプログラム、文書、費用支払い、およびその他の同様の規定を遵守することを要求する。多くの場合、適用される規則に基づいて、滞納金を支払うことによって、または他の方法で失効を是正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願の放棄または失効を招き、関連法ドメインの特許権の一部または全部を喪失させる可能性がある。特許または特許出願の放棄または失効をもたらす可能性のある不正事件には、規定された期限内に公式行動に対応できなかったこと、費用が支払われていなかったこと、および適切に合法化され、正式な文書が提出されなかったことが含まれる。この場合、私たちの競争相手が市場に参入する可能性があり、これは私たちの業務に実質的な悪影響を及ぼすだろう。
もし私たちが私たちの商業秘密の機密性を保護できなければ、私たちの業務と競争地位は損なわれるだろう。
私たちのいくつかの候補技術および製品のための特許出願に加えて、私たちは、ビジネス秘密および秘密協定によって、私たちの非特許ノウハウ、技術、および他の独自および機密情報を保護し、私たちの競争的地位を維持します。ビジネス秘密と技術的ノウハウを保護することは難しいかもしれない。特に,我々の技術プラットフォームについては,時間の経過とともに,これらのビジネス秘密や技術ノウハウが,独自に開発·記述方法を発表した定期刊行物文章や人員が学術職から業界科学職に移って業界内に伝播することが予想される.
92
私たちは、私たちの従業員、会社協力者、外部科学協力者、CRO、契約製造業者、コンサルタント、コンサルタント、および他の第三者など、これらの秘密にアクセスする権利のある当事者と秘密協定を締結することによって、これらの商業秘密および他のノウハウの保護を求めています。私たちはまた私たちの従業員とコンサルタントと秘密と発明または特許譲渡協定を締結します。私たちは私たちが可能であるか、または私たちの商業秘密またはノウハウとプロセスに接触したすべての当事者とこのような合意に到達したことを保証することができない。このような努力にもかかわらず、どちらも合意に違反し、私たちのビジネス秘密を含む私たちの固有の情報を漏洩する可能性があり、私たちは十分な救済措置を得ることができないかもしれない。強制執行側が商業秘密を不正に開示したり流用したりする主張は困難であり、高価で時間がかかり、結果は予測できない。しかも、米国国内外のいくつかの裁判所は独自の情報を保護することをあまり望まないか、または保護することを望まない。もし私たちが第三者に私たちの知的財産権を不正に開示することを阻止できない場合、あるいは第三者が私たちの知的財産権を流用することを阻止できなければ、私たちの市場で競争優位性を確立したり、維持することができないかもしれません。これは私たちの業務、経営業績、財務状況に大きな悪影響を及ぼす可能性があります。もし私たちの任意の商業秘密が競争相手または他の第三者によって合法的に取得または独立して開発された場合、私たちは彼らまたは彼らが情報を伝達する人がその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちのビジネス秘密が競争相手または他の第三者に開示された場合、またはそれによって独立して開発された場合, 私たちの競争的地位は実質的で不利な被害を受けるだろう。
もし私たちが開発する可能性のある候補製品の特許期間延長とデータ独占権を獲得しなければ、私たちの業務は実質的に損害を受ける可能性がある。
FDAが私たちが開発する可能性のある任意の候補製品の上場承認の時間、期限、および詳細によれば、私たちの1つ以上の米国特許は、1984年の“薬品価格競争および特許期限回復行動”または“ハッジ·ワックスマン修正案”に従って限られた特許期間を延長する資格がある可能性がある。ハッジ·ワックスマン修正案は、FDA規制審査過程で失われた特許期間の補償として、特許展期間を最長5年とすることを許可している。1つの特許期間の延長は、製品が承認された日から計14年の期間を超えてはならず、1つの特許を延長することしかできず、承認された薬物、その使用方法又はその製造方法に関する請求項を延長することしかできない。しかし,テスト段階や規制審査中に職務調査を行うことができなかったこと,適用の最終期限内に出願できなかったこと,関連特許の満了前に出願を提出できなかったこと,適用要件を満たしていなかったことなどにより延期が得られなかった可能性がある.しかも、特許保護の適用期間や範囲は私たちが要求しているものよりも短いかもしれない。もし私たちが特許期間の延長を得ることができない場合、あるいはもしこのような延長の期限が私たちが要求したものよりも短い場合、私たちの特許地位に依存して私たちの特許期限が満了した後に競争製品の販売を阻止することができなくなり、私たちの業務、財務状況、運営結果、および見通しが実質的に損なわれる可能性がある。
知的財産権はすべての潜在的な脅威を解決できるとは限らない。
私たちの知的財産権が提供する未来の保護の程度は不確定であり、知的財産権には限界があるため、私たちの業務を十分に保護できないか、あるいは競争優位性を維持することができるかもしれない。例えば:
93
これらの事件のいずれかが発生した場合、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性がある。
私たちは、私たちの従業員、コンサルタント、またはコンサルタントが、その現職または前任雇用主または他の第三者の機密情報を誤って使用または開示したり、私たち自身の知的財産権を持っていると主張したりする疑惑の影響を受けるかもしれない。
私たちの多くの従業員、コンサルタント、そしてコンサルタントは今あるいは以前に大学や他のバイオテクノロジーや製薬会社に雇われています。私たちは、私たちの従業員、コンサルタント、およびコンサルタントが、私たちのために働いているときに他人の固有情報またはノウハウを使用しないことを保証するために努力しているが、私たちまたはこれらの個人は、商業秘密または他の固有情報を含む任意のそのような個人の現職または前任雇用主または他の第三者の機密情報または知的財産権を使用または開示しているという疑惑を受ける可能性がある。このような疑いに対抗するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護することができなければ、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権や人員を失う可能性がある。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、私たちの経営陣と従業員の注意を分散させる可能性がある。
また、私たちの政策は、知的財産権の概念や開発に参加する可能性のある私たちの従業員と請負業者が合意に署名し、このような知的財産権を私たちに譲渡することを要求しているにもかかわらず、実際に私たちが自分の知的財産権と見なしているすべての当事者とこのような合意を実行することができないかもしれない。知的財産権の譲渡は自動的に実行されない可能性があり、あるいは譲渡協定が違反される可能性があり、私たちは第三者にクレームをつけさせられたり、私たちが私たちの知的財産権の所有権だと思うことを決定するために、私たちが提起する可能性のあるクレームを弁護したりすることができます。このようなクレームは、私たちの業務、財務状況、運営結果、および見通しに実質的な悪影響を及ぼす可能性があります。
もし私たちの商標が十分に保護されていなければ、私たちは興味のある市場で知名度を確立することができず、私たちの業務は不利な影響を受けるかもしれない。
もし私たちの商標が十分に保護されていなければ、私たちは興味のある市場で知名度を確立することができず、私たちの業務は不利な影響を受けるかもしれない。我々の未登録商標は、挑戦、侵害、回避、または汎用商標として発表されるか、または他の商標を侵害していると判断される可能性がある。私たちはこれらの商標に対する私たちの権利を保護できないかもしれません。私たちはこれらの商標が私たちの関心のある市場における潜在的なパートナーや顧客の中で知名度を確立する必要があります。時々、競争相手は私たちと似たような商標を採用して、私たちのブランド表示を確立する能力を阻害し、市場の混乱を招く可能性があります。さらに、他の登録商標または我々の未登録商標変異体を含む商標の所有者は、商標侵害クレームを提起する可能性がある。長期的には、私たちの商標の登録に成功し、私たちの商標に基づいて名称を確立することができなければ、効果的に競争できない可能性があり、私たちの業務は不利な影響を受ける可能性がある。商標、商業秘密、ドメイン名、著作権または他の知的財産権に関連する専有権を実行または保護する努力は無効である可能性があり、大量のコストおよび資源移転を招き、私たちの財務状況または運営結果に悪影響を及ぼす可能性があります。
私たちの普通株式所有権に関連するリスク
私たちは私たちの現金備蓄を使用する上で大きな自由裁量権があり、これらの現金備蓄を有効に使用できないかもしれません.
私たちの経営陣は、私たちの現金備蓄を使用するための幅広い裁量権を持っており、私たちの運営結果を改善したり、私たちの普通株価値を向上させることなく、私たちの現金備蓄を使用することができます。私たちの経営陣がこれらの資金を有効に使用できなかった場合、財務損失を招く可能性があり、これは私たちの業務に実質的な悪影響を与え、私たちの普通株価格の下落を招き、候補製品の開発を延期する可能性があります。それらが使用される前に、私たちは収入や切り下げを生じない方法で私たちの現金備蓄に投資するかもしれない。
公開市場で私たちの普通株を大量に売ることは私たちの株価を下落させるかもしれない。
公開市場で私たちの普通株を大量に販売したり、これらの売却が発生する可能性があると考えたり、私たちの普通株の市場価格を下げる可能性があり、適切な時間と価格であなたの普通株を売却することが困難になる可能性があり、追加株式証券を売却することで資金を調達する能力を弱める可能性があります。私たちは売却が私たちの普通株の現在の市場価格に及ぼす影響を予測できない。
94
私たちは予測可能な未来に配当金がないと予想している。
会社が設立されて以来、私たちは何の配当金も支払ったことがない。将来の運営が相当な分配可能な利益水準をもたらしても、私たちは現在、どんな収益も私たちの業務に再投資し、私たちが持続的な配当を支持する既定の収入源を持つまで配当金を支払わないつもりだ。また、将来の配当金を株主に支払う提案は、実際には我々の取締役会と株主が、我々の業務の見通し、現金需要、財務業績、新製品開発を含む様々な要因を考慮して適宜決定する。しかも、スイスの法律や私たちの会社の定款によると、将来の配当金の支払いはいくつかの制限を受けている。したがって、投資家は私たちの普通株の配当収入に依存することはできず、私たちの普通株に投資するいかなる見返りも、私たちの普通株価格の将来のいかなる切り上げにも完全に依存するかもしれない。私たちの普通株に支払う配当金(あれば)は、出資準備金から支払わない限り、スイス連邦源泉徴収税を納めなければなりませんカピタリンゲン.
私たちはスイスの会社です。私たちの株主の権利はアメリカ司法管轄区域の法律によって管轄されている会社の株主権利とは異なるかもしれません。
私たちはスイスの会社です。私たちの会社の事務は私たちの定款とスイスの法律によって管理されています。私たちの株主の権利と取締役会のメンバーの責任は、アメリカ司法管轄区域の法律によって管轄されている会社の株主と取締役の権利と義務とは異なる可能性があります。スイスの法律は、私たちの取締役会がその職責を履行する際に、合理的かつ公平な原則を十分に遵守した場合、会社、株主、従業員の利益を考慮することを要求している。取締役会は、あなたの株主としての利益とは異なる利益、あるいはあなたの株主としての利益以外の利益を考慮するかもしれません。スイスの会社法は私たちの株主が法廷で私たちの取締役会が下した決議や他の行動に挑戦する能力を制限する。我々の株主は通常、我々の取締役会の決定や行動を覆すために訴訟を提起することは許されず、注意義務および忠誠義務違反のために損害賠償を求めることのみが許可されている。スイスの法律によると、株主が私たちの取締役会メンバーに対して注意義務と忠誠義務に違反するというクレームはスイスのZugで提出されなければならない、あるいは私たちの取締役会関連メンバーの住所でなければならない。しかも、スイスの法律によると、私たちの株主は私たちが提起したどんなクレームもスイスのズグで完全に提起されなければならない。
スイスの会社として、私たちはスイスの法律条項によって制限されており、これらの条項は私たちが特定の計画や戦略を迅速に実施する柔軟性を制限するかもしれない。
私たちは時々私たちのスイス独立貸借対照表に示すように、関連会社への投資の帳簿価値を評価する必要がある。もしこのような投資の帳簿価値がその公正価値を超えていると判断すれば、このような投資は減少したと結論することができる。このような非現金減価に関する確認された損失は、私たちの純資産が私たちの法定株式および法定資本準備をカバーしなくなる可能性があります。スイスの法律によると、私たちの純資産が法定株式、法定資本積立金、法定収益積立金の50%をカバーできない場合、これらの積立金が株主に返済できない場合、取締役会はこのような状況を克服するために適切な措置を講じ、必要に応じて株主総会を開催し、このような資本損失を救済する措置を提出しなければならない。適切な措置は、関連状況と確認された損失の大きさに依存し、株主の承認を求め、株主の法定資本備蓄で総損失または一部の損失を相殺することを含む可能性があり、資格に適合する追加の実収資本を含み、そうでなければ、株主に割り当てたり、新しい株式を調達するために使用することができる。場合によっては、私たちの累積純損失を減らすために、条件に合った追加実収資本を使用して分配する必要があるかもしれません。この使用は、株主にスイスの源泉徴収税を支払うことなく分配する能力を低下させる可能性があります。このようなスイスの法律要求は私たちが特定の計画や戦略を迅速に施行する柔軟性を制限するかもしれない。
わが社の定款における反買収条項は、わが社の買収をさらに困難にする可能性があり、これは私たちの株主に有利になる可能性があり、私たちの株主が現在の経営陣を交換または更迭しようとすることを阻止するかもしれません。
95
わが社の定款の条項はわが社の買収やわが取締役会の構成の変化を阻害、延期または阻止する可能性があります。その他の事項を除いて、これらの条項は、株主総会で出席または投票した代表株式の少なくとも3分の2が私たちの取締役会メンバーの罷免を承認し、私たちの取締役会メンバーの最高人数を増加させることを要求する;任意の個人または実体の累積投票権を私たちの登録株式の15%に制限する;買収側が私たちの取締役会で免除を規定していない取引や一連の取引における投票権が私たちの登録株の5%を超えていることを制限することは、わが社の支配権変更を阻止または延期する可能性がある。取締役会が最長2年の期間の任意の時間に許可を得ることを規定し、私たちの現在の法定株式によると、取締役会は2023年6月10日に満期になり、株主総会が決議を通過した場合、取締役会は1つの資本区間に適用される(Kapitalband) (see “リスク要因-私たちのスイス会社としての地位は、資本管理のいくつかの面での柔軟性を制限し、私たちの株主にスイスの源泉徴収税を払わずに分配できない可能性があります)と、特定の数の株式を発行し、現在の法定株式に基づいて、商業登録簿の約49%を登録し、異なる場合に既存の株主の優先購入権を制限または撤回する。(I)転換可能な債務ツールを含む債券または同様の手段の行使に関連する株式交換および/または株式購入、および(Ii)当社またはその任意の付属会社の従業員または他のサービス提供者に付与された株式購入権の行使に関連し、(Ii)当社またはその任意の付属会社の従業員または他のサービス提供者に付与された株式の行使に関連し、合併または分割取引は、株主総会で代表される株式の少なくとも3分の2の賛成票を得なければならないと規定する。
これらの条項は,潜在的な買収者に我々の取締役会との交渉を要求することで,株主により大きな価値を得る機会を提供していると信じているが,我々の取締役会が拒否した要約が一部の株主から有益であると考えられていても,これらの条項は適用される.また、これらの規定は、株主が経営陣の任命を担当する取締役会メンバーを交代させることを困難にし、株主が現在の経営陣を交代または罷免しようとしていることを挫折または阻止する可能性がある。
私たちの普通株はスイスの法律によって発行されています。これはアメリカA州会社が提供するようなファッションの投資家を保護しないかもしれません。
私たちはスイスの法律に基づいて組織された。しかし、スイスの法律が将来変わらないことは保証されず、米国会社法の原則が提供するような方法で投資家を保護する保証もなく、投資家の権利に悪影響を及ぼす可能性がある。
私たちのスイス会社としての身分は、資本管理のいくつかの面での柔軟性を制限する可能性があり、株主にスイスの源泉徴収税を支払うことなく分配できない可能性があります。
我々の現行の定款は、追加の株主承認を必要とすることなく、取締役会によって発行された株式を株主に許可することを許可する。私たちの株主が承認した法定株式は2023年6月10日に満期になり、現行の会社定款によると、法定株式は私たちが登録した株式の49%を超えてはならない。2023年1月1日に発効したスイスの会社法改革によると、資本区間(Kapitalband)法定配当金の代替として。そのため、株主が承認した法定株式は2023年6月10日に満期になると、資本区間に置き換えられる必要がある。この資本範囲は、株主総会の決議を経て、取締役会が最大5年以内に株式を増加させることを許可するか、または株主総会がそれぞれ決議を採択した場合にも、株式を減少させることができる。いずれの場合も、許可は、既存の登録株式の50%に限定され、それぞれの期限が満了したときに株主によって継続されなければならない。特定の例外を除いて、スイス法律は既存の株主に任意の新規発行株を優先的に承認する権利を付与する。スイスの法律はまた他のいくつかの管轄区域の法律のように、異なる種類の株に適用される様々な条項に大きな柔軟性を提供している。スイスの法律はまた、取締役会が他のいくつかの管轄区域で権力を持っているある会社の行為を株主承認に保留している。例えば、配当金の支払いと在庫抹消は株主の承認を得なければならない。これらの私たちの資本管理に関連するスイスの法律要求は私たちの柔軟性を制限する可能性があり、より大きな柔軟性が私たちの株主に実質的な利益を提供することができるかもしれない。
96
スイスの法律によると、スイスの会社は、会社に十分な分配可能な利益を持っているか、または会社が分配可能な準備金(それぞれが監査された独立法定資産負債表によって証明されている)を持っており、スイスの法律と私たちの定款に要求される準備金の分配を差し引いた後に、配当金を支払うことができる。自由に分配可能な備蓄は、一般に“自由備蓄”または“資本貢献”と表記される(カピタリンゲン株主から受け取った払い込み)は“出資額積立金”にあります。資本を減らすことによってのみ、登録株である会社が登録した株式の総額面から分配することができる。私たちは出資によって配当金や準備金を増やさない限り、スイスの免税に基づいて株主に私たちの合格払込総額と登録配当金を超える配当金や他の分配を支払うことができません。私たちはまた分配可能な利益または自由に分配可能な備蓄から配当金を支払うことができるが、このような配当金はスイスの源泉徴収税を徴収されるだろう。私たちは配当金の支払いや資本削減のための十分な分配可能な利益、自由備蓄、資本出資または登録配当金からの十分な備蓄を保証することはできず、私たちの株主が私たちが提案した配当金または資本減少を承認することを保証することができないか、または資本減少による配当金の支払いまたは分配の他の法的要求を満たすことができるだろう。
会社が株主に支払う配当金と同様の現金または実物分配(清算収益および株式配当を含む)は、スイスの源泉徴収税を支払う必要がある(Verrechnungssteuer)、現在の税率は35%(課税配分総額に適用)。当社は任意の課税配分の総額からスイスの源泉徴収税を控除し、その分配満期日から30暦以内にスイス連邦税務局に税金を納める義務があります。ただし、株式額面の償還及び資格に適合した追加実収資本の返済(出資準備)オス·カピタリンを保留する)スイスの源泉徴収税は必要ありません。スイスの源泉徴収税は、当社が株式を買い戻す際の支払い(それぞれの株式および使用済み株式備蓄を超える)にも適用され、(I)自社の株式が当該等の買い戻し(償還株式)によって減少した場合、(Ii)買い戻しされた株式の総数が自社株の10%を超える場合、または(Iii)買い戻しされた株式が買い戻し後6年以内に販売されない場合。買い戻しされた株式が転換可能債券、オプション債券または従業員株式オプション計画の支払いに使用される義務が保持されている限り(従業員株式オプション計画であれば、最長休止期間は6年)、株式買い戻しの6年間の最終期限は一時停止される。課税株式を買い戻す場合、スイスは、買い戻し価格と買い戻し株式額面と買い戻し時に返済された1人当たり払込準備金の和との差額に源泉徴収税を徴収する。
個人資産として株を保有するスイス住民個人、又は住民個人株主は、原則としてスイスの源泉徴収税の所得税を全額還付又は免除する資格があり、もし彼らが所得税申告書に関連収入を適切に報告すれば。また、(I)納税目的でスイスに居住する会社株主及び個人株主、(Ii)スイスに居住していない会社株主及び個人株主、それぞれの場合、スイスで行われている貿易又は業務の一部として、納税目的でスイスに固定営業場所を有する常設機関によりその株を保有しており、(Iii)所得税目的で“専門証券取引業者”に分類されるスイス住民個人は、他の理由を除いて、株式及びその他の証券の取引又はレバレッジ投資を行うことが多い(総称して、国内商業株主“)は原則としてスイスの源泉徴収税所得税の全額還付または控除を受ける資格があり、もし彼らがその損益表や所得税申告書(場合によっては)で関連収入を適切に報告した場合。
スイス住民ではなく税務目的のため、かつ関連課税年度内に、スイス国内に固定営業場所を設けた常設機関が貿易や業務に従事しておらず、その他の理由でスイスで会社や個人所得税を納付しない株主(総称して“非住民株主”と総称する)は、スイスの源泉徴収税の全部または一部を返還することができ、受取人が税務目的で居住している国とスイスが二重課税を回避する二国間条約または税務条約を維持し、当該税務条約の他の条件を満たすことを前提としている。
米国−スイス税収条約によると、福祉を受ける資格を有する米国の株主は、15%を超える条約税率の返還(または5%を超える引き下げられた条約税率の返還を申請することができ、少なくとも10%の投票権を有する適格会社株主、または適格年金基金の場合には全額還付を申請する)の源泉徴収税に適用することができる。非住民株主は、条約給付を申請する手続き(および払い戻しを得るのに要する時間)が国によって異なる可能性があることを認識しなければならない。非住民株主は、株式の受け取り、所有権、購入、販売またはその他の処置、およびスイスの源泉徴収税の払い戻しを申請する手続きについて、自分の法律、財務、または税務コンサルタントに相談しなければならない。
97
もし私たちが支配されている外国の会社なら、あるアメリカの株主はアメリカ連邦所得税の悪影響を受けるかもしれない。
非米国会社の各“10%株主”(以下のように定義する)は、米国連邦所得税において“制御された外国会社”またはCFCに分類され、CFCsがその株主に割り当てられていなくても、米国連邦税収の収入にこのような10%の株主比率シェアを含むCFサブ部分収入と米国財産に投資する収益とが一般的に要求される。F分割収入は、一般に、配当金、利息、レンタル料および特許使用料、証券売却の収益、および関連する当事者とのいくつかの取引の収入を含む。2017年12月31日以降の納税年度では、フッ素塩化炭素の株主10%当たりに、このフルオロカーボンに関する10%株主シェアの“世界無形低税収入”を収入に計上することも求められている。さらに、フルオロカーボンの株式を売却または交換することによって収益の10%を達成する株主は、このような収益の一部を資本収益ではなく配当収入に分類することを要求される可能性がある。米国連邦所得税については、一般に、10%の株主が投票権のある会社の全カテゴリー株の総投票権またはその会社の株式の総価値の50%以上を直接または間接的に所有していれば、その会社はCFCsに分類される。“十パーセントの株主”とは、(1)投票権のあるすべてのカテゴリー株の総投票権又は(2)当該会社の全カテゴリー株の価値を有するか、又は有すると考えられる米国人(1986年に改正された米国国税法又は同法典の定義に基づく)。クロロフッ化炭素の地位の決定は複雑であり,帰属規則を含み,これらの規則の適用は完全には決定されていない。
私たちの2022納税年度に、私たちのいくつかの株主はアメリカ連邦所得税の10%の株主だと信じている。しかし,我々の2022年12月31日までの納税年度と本納税年度のフッ素塩化炭素の状況は不明であり,2022年12月31日までの納税年度のフッ化炭素,すなわち本納税年度または来年度である可能性がある。また,最近のフッ素塩化炭素の地位決定に関する帰属規則の変化は,いずれの課税年度のフッ化炭素地位の決定を困難にする可能性がある。また,本課税年度や将来の課税年度でフッ素塩化炭素でなくても,我々の非米国子会社がフッ化炭素となる可能性がある。アメリカの保有者はフッ化炭素の10%株主になる潜在的な不利なアメリカの税収結果について彼ら自身の税務顧問に相談しなければならない。私たちがCFCと受動型外国投資会社(PFIC)に同時に分類されれば、私たちがCFC期間中に10%株主定義に適合するアメリカの保有者については、通常PFICとはみなされません。
もし私たちが受動的な外国投資会社として説明されれば、いくつかのアメリカの株主は不利な税金結果を受けるかもしれない。
一般に、任意の納税年度において、総収入の少なくとも75%が受動的収入である場合、または資産価値の少なくとも50%は、受動的収入を生成するための資産または現金を含む受動的収入を生成するために保有される資産に起因することができ、米国連邦所得税目的のPFICとして説明される。これらのテストに関して、受動的収入には、配当金、利息、投資性財産の売却または交換の収益、および賃貸料および特許権使用料以外の賃貸料および特許使用料が含まれ、これらの収入は、貿易または事業を積極的に展開することに関連する関係者から得られる。PFICと同定された場合、我々普通株の米国保有者は、資本収益ではなく、普通株売却によって達成された収益を一般収入とすること、米国保有者が普通株から獲得した配当に適した優遇料率を失うこと、および私たちの分配および普通株販売収益に利息費用を徴収することを含む不利な税収結果を受ける可能性がある。
PFICとしての私たちの地位は私たちの収入の構成と私たちの資産の構成と価値に依存し、これはある程度私たちの普通株の四半期時価を参考にして決定される可能性があり、普通株の四半期時価は変動する可能性がある。私たちの地位はまた私たちがどのようにどのように速い速度で私たちの業務で以前に発行された現金収益を利用するかにかかっているかもしれません。私たちのPFICとしての地位は毎年事実に基づいて決定することであり、私たちは過去、現在あるいは未来のいかなる納税年度のPFIC地位についてもいかなる保証を提供することができない。
2021納税年度のPFICかもしれませんので、2021納税年度に合格した選挙基金やQEF選挙を行うために必要な情報を株主に提供しました。我々はこのような情報を我々のサイトで提供している(www.crisprtx.com).私たちの株についてQEF選挙を行ったアメリカの保有者は、私たちが分配するかどうかにかかわらず、現在のベースで私たちの収入シェアを比例して計上しなければなりません。2021課税年度については、良質教育基金が規則に組み込まれていることについて、当社の全社範囲内の一般収益および純資本収益は5.047億ドルおよび純資本収益は0.0ドルであり、2022課税年度または将来の課税年度に良質教育基金を規則に組み入れた場合、重大な額の一般収益および/または純資本収益がある可能性がある。我々が2021年納税年度か本納税年度のPFICかは決定されていないが,2021年納税年度および/または本納税年度のPFICである可能性もある。PFICであるか可能性のある課税年度ごとに、QEF選択に必要な情報が含まれているPFIC年度情報レポートを提供するように努力します。あるいは、米国の保有者は、我々の株式が“基準”の下の“有価証券”を構成することを前提として、時価建ての選択を行うこともでき、これは、通常、上述したPFICの地位の不利な結果を回避するが、米国の保有者は、毎年私たちの株の任意の価値を一般収入報告として増加させることを要求する(および通常、私たちの株式価値のいかなる減少も許容される)。
98
PFICと判定された場合、米国所有者は、通常、我々の任意の直接または間接子会社に所有する割合の数(価値で計算される)を有する株式とみなされ、これらの子会社もPFICであり、それぞれは、このようなより低いレベルのPFICからの分配または処置に関し、同様の不利な規則の制約を受けるであろう。いずれの場合も、米国所有者がこのような株式を直接所有しているように(米国所有者がこのような分配または処置の収益を直接受けていなくても)。TRACRとCRISPR Treateutics Ltdを含む)はまだ確認されていません前の課税年度、本課税年度、あるいは将来の課税年度の低いレベルのPFICかもしれませんが、私たちはそうするつもりはありません。アメリカの保有者がどんな低いレベルのPFICについても良質な教育基金選挙に必要な情報を提供するつもりはありませんので、良質な教育基金選挙を行うことができないことを予想すべきです。私たちはあなた自身の税務コンサルタントに相談して、私たちのPFICの地位と投資PFICに関連する税務考慮要素を理解することを促します。QEF選挙または時価計算選挙の利用可能性、実行可能性とプログラム、PFICルールは私たちの任意の子会社に適用されます。
米国の株主は、私たちまたは私たちの役員や取締役会のメンバーに対する判決や民事責任を得ることができないかもしれない。
私たちはスイスの法律に基づいて設立され、私たちの登録事務所と登録地はスイスのZugに位置している。また、私たちの一部の役員や幹部はアメリカ住民ではなく、これらの人たちの資産の全部または大部分はアメリカ以外に位置しています。したがって,投資家は米国内で我々やそのような人々に法的手続き文書を送達したり,米国連邦証券法の民事責任条項に基づく訴訟判決を含めて米国裁判所で取得した判決を実行することができない可能性がある.我々のスイス弁護士は,米国連邦と州証券法のみに基づく民事責任のスイスでの実行可能性,あるいは米国裁判所判決を執行する訴訟における実行可能性に疑問を抱いていることを教えてくれた。スイスでは,米国連邦や州証券法のみに基づいて個人に対して提起された原始訴訟は,他の事項を除いて,スイス連邦国際私法で規定されている原則によって管轄されている。この規約は、結果がスイスの公共政策と一致しない場合、スイス裁判所は非スイス法律の規定の適用を禁止しなければならない。しかも、スイスの法律の義務的な条項は他のどんな適用される法律も考慮せずに適用される可能性がある。
スイスとアメリカは民商事判決の相互承認と執行に関する条約を持っていない。米国裁判所のスイスでの判決の承認と執行は、スイス連邦国際私法法案に規定されている原則によって管轄されている。この規約は原則として、非スイス裁判所が下した判決は、以下の場合にのみスイスで実行できると規定している
一般リスク
上場企業として、私たちの運営には大きなコストがかかり、私たちの経営陣はコンプライアンスイニシアチブやコーポレートガバナンス実践に多くの時間を投じる必要があります。
上場企業として、私たちは巨額の法律、会計、その他の費用を招いた。SOX、ドッド-フランクウォール街改革と消費者保護法、ナスダック全世界市場の上場要求及びその他の適用される証券規則と法規は上場会社に対して各種の要求を提出し、有効な開示、財務制御と会社管理のやり方を確立と維持することを含む。私たちの経営陣と他の人たちはこのような要求に対する遵守を維持するために多くの時間を投じた。しかも、このような要求は私たちの法律と財務コンプライアンスコストを増加させ、いくつかの活動をもっと時間的で高価にする。
SOX第404条によれば、我々は、独立公認会計士事務所が発行した財務報告内部統制の認証報告を含む経営陣の財務報告の内部統制に関する報告書を提出しなければならない。このような点で、私たちは多くの会計費用を発生させ、多くの管理努力を費やした。私たちのテストは財務報告書に対する私たちの内部統制の欠陥を明らかにするかもしれないが、これらの欠陥は重大な弱点または重大な欠陥と考えられている
99
足りない点。1つまたは複数の重大な弱点、またはタイムリーに救済できない重大な欠陥を発見すれば、財務諸表の信頼性に対する信頼性の喪失によって金融市場に悪影響を及ぼす可能性がある。
私たち普通株の市場価格はずっと大きく変動しており、これは株主に大きな損失をもたらす可能性がある。
私たちの株価は大幅な変動の影響を受けてきたし、未来もそうかもしれない。また、株式市場全体、特にナスダック上場のバイオ製薬会社は、極端な価格と出来高変動を経験しており、これらの変動は往々にしてこれらの会社の経営業績と関係がないか比例しない。例えば、2016年10月19日、つまりナスダック世界市場に上場した初日から2022年12月31日まで、私たちの株は1株220.20ドルの高値と11.63ドルの低価格の間で取引されている。このような変動のため、私たちの株主は大きな損失を受けるかもしれない。また、私たちの普通株の市場価格は多くの要素の影響を受ける可能性があります
これらと他の市場と業界要素は私たちの普通株の市場価格と需要を大幅に変動させる可能性があり、私たちの実際の経営業績にかかわらず、投資家がいつでもその普通株を売却することを制限または阻止する可能性があり、そうでなければ、私たちの普通株の流動性にマイナスの影響を与える可能性がある。過去、1株の市場価格が変動した場合、その株の保有者は、その株を発行した会社に対して証券集団訴訟を起こす。もし私たちのすべての株主が私たちを提訴すれば、私たちは巨額の訴訟弁護費用を生むかもしれない。そのような訴訟はまた私たちの経営陣の時間と注意を移すかもしれない。
不利なグローバル経済状況は、私たちの業務、財務状況、または経営結果に悪影響を及ぼす可能性がある。
我々の業務結果は、例えば、コロナウイルスの大流行、持続的な軍事を含む、世界経済全体の状況、世界金融市場の混乱、および衰退または市場調整の悪影響を受ける可能性がある
100
ロシアとウクライナの間の紛争やロシアへの関連制裁、インフレなど他の世界的なマクロ経済要因。この状況は私たちの資本獲得能力を低下させる可能性があり、これは将来的に私たちの流動性に悪影響を与え、私たちの業務や普通株の価値に大きな影響を与える可能性がある。
もし証券アナリストが私たちの業務に関する研究や報告を発表しなければ、あるいは彼らが私たちの普通株に対するマイナス評価を発表すれば、私たちの普通株の価格は下がるかもしれません。
私たちの普通株の取引市場は、業界や金融アナリストが発表した私たちまたは私たちの業務に関する研究と報告に部分的に依存するだろう。もし私たちの業務を追跡した一人以上のアナリストが私たちの普通株に対する彼らの推定値を引き下げたら、私たちの普通株の価格は下落するかもしれない。もしこのようなアナリストの一人以上が私たちの普通株を追跡しなければ、私たちは市場での私たちの普通株の可視性を失うかもしれないが、これは逆に私たちの普通株価格の下落を招く可能性がある。
私たちの業務は国際業務に関連する経済、政治、規制、その他のリスクの影響を受けている。
私たちの業務は国際業務の展開に関連するリスクの影響を受けています。私たちと私たちの多くのサプライヤーと協力と臨床研究関係はアメリカ以外に位置している。したがって、私たちの将来の業績は様々な要素の影響を受けるかもしれない
私たちの内部コンピュータシステム、または私たちの協力者や他の請負業者やコンサルタントのシステムは、故障したり、セキュリティホールに遭遇したりする可能性があり、これは、私たちの製品開発計画が実質的に破壊される可能性があります。
私たちの内部コンピュータシステム、および私たちの現在および未来の任意の協力者および他の請負者またはコンサルタントのコンピュータシステムは、コンピュータウイルス、許可されていないアクセス、自然災害、テロ、戦争、電気通信、および電気故障の破壊を受けやすい。これまで、このような重大なシステム障害、事故、セキュリティホールを経験したことはありませんが、このような事件が発生し、私たちの運営中断を招く場合、私たちのビジネス機密や他の固有情報の損失によるものであっても、他の同様の中断によるものであっても、我々の開発計画および業務運営中断を招く可能性があります。例えば、未来の臨床試験における臨床試験データの紛失は著者らの監督管理の承認作業の遅延を招く可能性があり、そして著者らのデータの回復或いは複製のコストを著しく増加させる。もしどんな中断やセキュリティホールが私たちのデータやアプリケーションを紛失したり、破損したり、機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの競争地位が損なわれる可能性があり、私たちの候補製品のさらなる開発と商業化は延期される可能性がある。
101
会社やサプライヤーの情報システムやネットワークで維持されている情報には,我々従業員や研究対象の個人情報,および会社やサプライヤーの機密データが含まれており,流用,誤用,漏洩,改ざんや故意や意外な情報漏洩や紛失によるリスクを受ける可能性がある.さらに、外部の当事者は、私たちまたは私たちのサプライヤーのシステムを浸透させようとしたり、詐欺的な手段で私たちの人員または私たちのサプライヤーの人員に敏感な情報を開示させて、私たちのデータおよび/またはシステムにアクセスしようとするかもしれません。私たちのデータとシステムは悪意のあるコードとウイルス、ネット釣り、および他のサイバー攻撃を含む脅威にさらされる可能性がある。時間が経つにつれて、このような脅威の数と複雑さは増加している。もし私たちの情報技術システムやサプライヤーの情報技術システムが重大な破壊が発生すれば、市場の私たちのセキュリティ対策の有効性に対する見方が損なわれる可能性があり、私たちの名声と信用が損なわれる可能性がある。私たちは情報システムやネットワークを修復したり交換したりするために多くの資金と他の資源が必要かもしれない。さらに、個人訴訟における規制行動および/または個人および団体からのクレームを受ける可能性があり、これらの訴訟は、データの乱用または不正開示に対するクレーム、および不公平または詐欺的なやり方を含むデータ収集および使用慣行および他のデータプライバシー法律および法規に関連するプライバシー問題に関するものである。我々はこれらの事件の発生を防止するためのシステムと制御を開発·維持し、脅威を識別·緩和するプロセスもあるが、これらのシステム、制御、プロセスの開発と保守はコストが高く、技術の変化とセキュリティ対策を克服する努力がますます複雑になり、継続的に監視·更新する必要がある。さらに私たちが努力したにもかかわらず, このような事件が発生する可能性は完全には除去できない。より多くの情報システムをサプライヤーにアウトソーシングし、支払人や患者とより多くの電子取引を行い、クラウドベースの情報システムにより多く依存するにつれて、関連するセキュリティリスクが増加し、私たちの技術および情報システムを保護するために追加の資源が必要になるであろう。さらに、システム障害時の障害、サービス中断、データの悪化または損失から私たちを保護するのに十分なセキュリティおよび制御措置を実施するために、私たちの内部情報技術システムまたは私たちの第三者請負業者のシステム、または私たちのコンサルタントが十分なセキュリティおよび制御措置を実施するための努力を保証することは、ネットワーク攻撃、セキュリティホール、産業スパイ攻撃、または金融、法律、商業または名声被害をもたらす可能性のある内部脅威攻撃時にデータが盗まれたり破損したりすることを防止するのに十分である。
項目1 B。未解決従業員コメント。
ない。
プロジェクト2.専門家ペルテスです。
三ヶ月ごとに更新される不動産賃貸協定によると、私たちの主な執行事務所はスイスのZugにあります。私たちのアメリカ研究開発本部はマサチューセッツ州ボストン西第一街105号にあります。そこで約263,500平方フィートの実験室とオフィススペースを借りました。この施設は2034年10月までレンタルされており,レンタル期間をさらに2年5年間延長することが選択できる。
2020年5月、マサチューセッツ州フレミンガムで50,249平方フィートの建築賃貸契約を締結し、臨床生産の細胞治療製造施設として使用し、我々の研究における細胞治療候補製品の商業生産のために使用する予定である。同施設は2036年3月までレンタルされ、レンタル期間をさらに2~7年延長することが選択できる。
ケンブリッジ、マサチューセッツ州、サンフランシスコ、カリフォルニア州とイギリスロンドンで商業事務所を借りました。私たちの施設は私たちの現在の需要を満たすのに十分であり、必要であれば、私たちは適切な追加または代替空間を提供するだろうと信じている。
項目3.法律法律手続き。
通常の業務プロセスでは、私たちは時々、私たちの知的財産権(特許組合を含む)、商業配置、および他の事項に関連する訴訟、調査、法的手続き、および訴訟脅威に関連する。このような訴訟は、米国特許商標局および欧州特許庁が、私たちの知的財産権(特許組合を含む)に関連する準訴訟、当事者間の行政訴訟を含むことができる。上記のいずれの結果も,その是非曲直にかかわらず,本質的には不確実である.さらに、訴訟や関連事項の費用が高く、私たちの経営陣や他の資源の注意を分散させる可能性があり、そうでなければ、これらの資源は他の活動に使用されるだろう。もし私たちがこのような訴訟に勝つことができなければ、私たちの業務、運営結果、流動性、そして財務状況は不利な影響を受けるかもしれない。
4つ目:地雷の安全TYが披露する。
適用されません。
102
部分第2部:
項目5.登録者普通株·関連株の市場保有者は重要であり,発行者は株式証券を購入する.
市場情報
私たちの普通株はナスダック世界市場で取引され、コードは“CRSP”です
株式表現グラフ
1934年の“証券取引法”(改訂本)第18節または“取引法”については、以下の業績グラフおよび関連情報は、1933年の“取引法”(改正本)または“証券法”に基づいて提出された任意の将来の文書に、特に引用によってこのような文書に組み込まれない限り、引用によってこのような情報を“証券法”(改正本)または“証券法”に提出された任意の将来の文書に組み入れてはならない。
以下の図は、2017年12月31日から2022年12月31日までの間の私たちの株の株主累計総リターンと同期(A)ナスダックバイオテクノロジー指数と(B)ナスダック総合指数の累積総リターンを比較したものである。この図は,2017年12月31日に我々の普通株,ナスダックバイオテクノロジー指数,ナスダック総合指数への投資を100ドルと仮定し,再投資配当金(あれば)を仮定している。次の図に示す比較は履歴データに基づく.この図に含まれる株価表現は,必ずしも未来の株価表現を示唆しているとは限らない.
CRISPR治療股指、ナスダック総合指数とナスダックバイオテクノロジー指数の総リターンの比較
103
スイスの税務面の考慮
スイス源泉徴収税
スイス現行税法によると、株主に支払う満期配当金と同様の現金または実物分配(清算収益と株式配当を含む)はスイス連邦源泉徴収税を払わなければならない(Verrechnungssteuer)“源泉徴収税”),現在の税率は35%(課税配分の総額に適用)である。私たちは任意の課税分配の総額から源泉徴収税を控除し、このような分配が満期になった日から30日以内にスイス連邦税務局に税金を納める義務があります。ただし、株式額面の償還及び資格に適合した追加実収資本の返済(出資準備)オス·カピタリンを保留する)源泉徴収税を支払う必要はありません。源泉徴収税は、私たちが株式を買い戻す際の支払い(それぞれの株式および使用済み株式備蓄を超える)にも適用され、(I)当社の株式がその買い戻し(償還株式)の際に減少した場合、(Ii)買い戻しされた株式の総数が当社の株式の10%を超えた場合、または(Iii)買い戻しされた株式が買い戻し後6年以内に販売されていない場合。株式を買い戻す6年間の期間は、転換可能な債券、オプション債券または従業員株式オプション計画の支払いに使用される義務が保留されている限り(従業員株式オプション計画であれば、最長6年)停止する。課税株式を買い戻す場合には、買い戻し価格と買い戻し株式額面と買い戻し時に払戻しされた合格追加実収資本の和との差額に源泉徴収税を徴収する。
個人資産としてその株式を保有するスイス住民個人(“住民個人株主”)は、原則として源泉徴収税の所得税を全額還付または免除する資格があり、所得税申告書に関連収入を適切に報告すれば。また、(I)納税目的でスイスに居住する会社株主及び個人株主、(Ii)スイスに居住していない会社株主及び個人株主、それぞれの場合、スイスで行われている貿易又は業務の一部として、納税目的でスイスに固定営業場所を有する常設機関によりその株を保有しており、(Iii)所得税目的で“専門証券取引業者”に分類されるスイス住民個人は、他の理由を除いて、株式及びその他の証券の取引又はレバレッジ投資を行うことが多い(総称して、国内商業株主“)は原則として、その損益表または所得税申告書(場合によっては)で関連収入を適切に報告する限り、源泉徴収税所得税を全額還付または控除する資格がある。
株主は、税務目的でスイスに居住せず、かつ関連課税年度内に、スイス国内に固定営業場所を設けた常設機関が貿易や業務に従事しておらず、他の理由でスイスで会社や個人所得税(総称して“非住民株主”と呼ぶ)を納付する必要がなければ、源泉徴収税の全部または一部を返還することができ、その受取人が税務目的で居住している国とスイスが二重課税を回避する二国間条約(“税務条約”)を維持し、当該税務条約の他の条件を満たすことを前提としている。非住民株主は、条約給付を申請する手続き(および払い戻しを得るのに要する時間)が国によって異なる可能性があることを認識しなければならない。非住民株主は、株式の受け取り、所有権、購入、販売またはその他の処置、および源泉徴収税の払い戻しを申請する手続きについて、自分の法律、財務、または税務コンサルタントに相談しなければならない。
情報の自動交換
税務資料自動交換計画は経済協力及び発展組織が先頭に立つグローバル計画である。それの目的は税務情報を自動的に交換する汎用的な基準を確立し、税務透明性を増加させることだ。
AEIを実施または実施した司法管轄区(例えば、スイス、EU加盟国および世界の多くの他の司法管轄区)は、その申告金融機関に、それぞれの現地実施法律に基づいて、その口座保持者および制御者(場合によっては)の納税居住地を決定し、報告すべき口座である場合は、特定の身分情報、口座情報および財務情報(利息、配当金、他の収入および毛収入などの口座残高および関連支払いを含む)を現地税務機関に報告し、その後、現地税務機関は、受信した情報を関連する司法管轄区の税務機関と交換するように要求する。
スイスが発効したか、署名され発効したAEI協定リストは、国際金融事務局のウェブサイトで見つけることができる。
スイス連邦印紙税
発売計画および発売過程で株式および売却株式を発行するには、スイス連邦証券発行印紙税を払わなければならない(Abgabeを発射する)1%は私たちが負担します
104
その後、私たちの株を購入または販売し、常駐プライベート株主、国内商業株主、または非常駐株主(二級市場取引)によって行われても、スイス連邦証券譲渡印紙税を支払う必要があるかもしれません(ウムサザバガベ(I)スイスまたはリヒテンシュタイン銀行またはスイスまたはリヒテンシュタイン銀行またはスイス連邦印紙税法案によって定義された別のスイス証券取引業者または他のスイス証券取引業者によるこのような移転が行われ、(Ii)いかなる免除も適用されない場合、それぞれ購入価格または売却収益で計算される現在最高0.15%の税率で計算される。
スイス連邦、州、コミュニティの個人所得税と企業所得税
非住民株主s
非住民株主は配当金支払いと同様の分配についてスイス連邦、州、あるいはコミュニティ所得税を支払う必要がありません。彼らは私たちの株だけを持っているからです。株を売却する資本利益もこれに適用される。源泉徴収税の結果については、上記を参照されたい。
住民個人株主と国内商業株主
住民個人株主が配当金及び類似の現金或いは実物分配(上記清算収益及び株式配当或いは課税株式買い戻しを含む)を徴収する場合、株式額面又は合資格を償還する追加実収資本ではない場合、その個人所得税申告表に当該等の収入を申告しなければならず、関連税期の任意の課税所得額についてスイス連邦、州及びコミュニティ所得税を納付しなければならない。また、スイス連邦個人所得税の目的で、上記配当金と同様の分配は連邦レベルの70%にのみ課税される(Teilbesteuerung)であれば、投資額が少なくとも私たちの株式の10%に達した場合。州やコミュニティレベルでも類似した規定が導入されているが,居住地によって規定が異なる可能性がある。
常駐プライベート株主が第三者に株式を売却または処分する際に出現する収益または損失は、通常、免税されたプライベート資本収益または課税不可資本損失であり、具体的な状況に応じて決定される。
配当金および現金または実物割当(清算収益および赤株を含む)のような国内商業株主は、関連納税期間の損益表でその支払いを確認し、その期間に蓄積された任意の課税所得額(配当を含む)についてスイス連邦、州およびコミュニティの個人所得税または会社所得税を納付しなければならない(場合に応じて)。会社の納税者である国内商業株主は配当分配の参加減免を受ける資格があるかもしれないBeteiligungsabzug)が、保有する株式の時価が少なくとも100万スイスフランである場合、または私たちの株式の少なくとも10%を占めるか、またはそれぞれ少なくとも10%の利益および準備金を得る権利がある。
国内商業株主は、それぞれの課税期間の損益表において株式を売却する際に達成される損益を確認し、当該課税期間の任意の課税所得額(株式を売却またはその他の方法で処分することにより実現される損益を含む)についてスイス連邦、州およびコミュニティの個人所得税または会社所得税を納付しなければならない。個人納税者に属する国内商業株主に対して、株式売却による収益は連邦レベルで達成される(Teilbesteuerung)は、(I)のように、当該投資がある貿易又は業務の経営に関連して保有されているか、又は業務資産を選択する資格に適合している(グシュハフツフモルガン)スイス税法によれば、(Ii)売却された株式は、会社の株式の少なくとも10%の権益を反映し、(Iii)少なくとも1年間保有される。州やコミュニティレベルでも同様の規定が導入されているが,これらの規定は居住地によって異なる可能性がある。企業の納税者である国内の商業株主は減免に参加する権利があるかもしれませんBeteiligungsabzug)が、納税中に販売された株式(I)が会社の株式の少なくとも10%の権益を反映している場合、または売却された株式が少なくとも10%の利益および備蓄を許可する場合、および(Ii)は少なくとも1年間保有する。参加猶予は、売却収益と参加初期コストとの差額に適用される(グステヘンスコスモ)により、従来の参加減記再課税につながる。
スイスの財産税と資本税
非住民株主
私たちの株を持っている非住民株主は州とコミュニティの富税や毎年の資本税を支払う必要がありません。彼らはこれらの株だけを持っているからです。
住民個人株主と国内商業株主
住民個人株主は,その株式をその私財の一部として報告することを要求され,州やコミュニティ財税を納付しなければならない。国内商業株主は、その株式をその定義された商業財又は課税資本の一部として報告することを要求され、州及びコミュニティ財又は年間資本税を納付しなければならない。
スイス米国の“外国口座税収遵守法案”の実施促進
105
スイスは米国と“外国口座税収遵守法”の施行を促進するための政府間協定を締結した。この協定は、米国人がスイス金融機関に保有している口座が口座保持者の同意を得た場合、または行政援助の範囲内で団体要請の方法で米国税務機関に開示されることを保証する。同意なしに情報が自動的に移動するのではなく、米国とスイス間の二重課税協定に基づく行政援助の範囲内でのみ交換される。2014年10月8日、スイス連邦委員会は米国との交渉を要求し、現在の直接通知に基づく制度をスイス連邦税務局に情報を送信することに変更し、後者は米国税務当局に情報を提供することを承認した。
上の討論はスイスの重要な税金考慮事項をまとめたものだ。それは特定の株主に重要である可能性のあるすべての税務事項を含まない。各株主は株主自身の状況に応じて、税務結果についてそれ自体の税務顧問に意見を聞くべきである。
所持者
2023年2月16日現在、私たちは約19人の普通株式登録所有者がいます。この数値には、その株式が街の名義で保有されている利益所有者は含まれていない。
配当をする
設立以来、私たちは私たちの普通株について何の現金配当金も支払わず、予測可能な未来にも現金配当金を支払わないだろう。
株式補償計画に基づいて発行された証券
当社の株式報酬計画に関する情報は、ここに組み込まれ、10-K表形式の本年度報告第3部第12項を参考にします。
プロジェクト6.保留
適用されません。
項目7.経営陣の以下の問題の議論と分析財務状況と経営実績。
以下の財務状況と経営結果の検討と分析、および当社の連結財務諸表と本年度報告書の他の場所でForm 10-K形式で提供された関連説明を読むべきです。本議論および分析に含まれるまたは本Form 10-K年次報告の他の部分に記載されている情報は、リスクおよび不確定要因に関する前向きな陳述を含む、我々の業務および関連融資の計画および戦略に関する情報を含む。本年度報告10-K表の“リスク要因”の部分に列挙された要素を含む多くの要因の影響により、我々の実際の結果は、以下の議論および分析に含まれる前向き陳述に記載または示唆された結果とは大きく異なる可能性がある。
概要
我々は有力な遺伝子編集会社であり,CRISPR/Cas 9による療法の開発に専念している。CRISPR/Cas 9は革命的な遺伝子編集技術であり、ゲノムDNAの精確な、方向性の変化を許可する。CRISPR/CAS 9の遺伝子編集への応用は、我々の科学創始者の一人であるEmmanuelle Charpentier博士が共同で発明した。チャペンティエ博士と彼女の協力者は1つの仕事を発表し、細菌で発見された自然産生ウイルス防御機構CRISPR/Cas 9がどのように遺伝子編集に改造されたかを解明した。私たちはこの技術を応用して一連の稀でよく見られる疾患を潜在的に治療している。著者らは、著者らの科学専門知識に加え、著者らの遺伝子編集方法は、現在の生物製薬方法が限られた成功を得た患者を含む新たな高度な活躍と潜在的な治療方法をもたらす可能性があると信じている。
著者らは四つの核心特許経営領域で広範な疾病領域の治療方案の組み合わせを創立した:ヘモグロビン病、免疫腫瘍学、再生医学と体内にある近づいてきました。我々の最先端の計画は,遺伝子定義疾患輸血依存型ベータ地中海貧血(TDT)と重篤な鎌状細胞病(SCD)であり,両疾患の医療ニーズは満たされていない。遺伝子編集の同種細胞治療プロジェクトもいくつか行われています
異種遺伝子キメラ抗原受容体T細胞、またはCAR T、血液系および固形腫瘍癌の治療のための候補薬剤、および1型糖尿病の治療のための研究性、同種異体、遺伝子編集、免疫回避および幹細胞由来療法を含む、
106
T 1 Dである。さらに私たちは複数の計画を進めています体内にある編集方法は,最初に心血管疾患の治療と予防に用いられた。
ヘモグロビン病
私たちの主な候補製品Exexcelは調査的で自己的で離体するCRISPR遺伝子編集の造血幹細胞療法が評価されており,TDTやSCDの治療に用いられている。Exacelは,Vertex製薬会社とそのある子会社やVertexとの共同開発と商業化プロトコルに基づいて開発された。我々とVertexは,12歳から35歳までのTDT(GRAPH−111)やSCD(SCD−121)患者に対する単剤Exexcelの安全性と有効性を評価するための1/2/3期開放臨床試験におけるExaccel研究を2つ行っている。登る-111と登る-121の登録が完了しました。我々とVertexはまた,TDT(SCD−141)およびSCD(SCD−151)を有するTDTおよびSCD患者のための2つの追加的な3期オープン臨床試験を開始した。Exexelを受けた患者は、Exexexeの安全性と有効性を評価するために、長期的な開放ラベルフォローアップ試験に参加することを要求される。GRAPH−131は,刺激輸液後に参加者に15年間にわたる追跡調査を行うように設計されている。2022年第2四半期と第4四半期に、ヨーロッパ血液学協会大会とアメリカ血液病学会年会で、著者らはそれぞれ44名のTDT患者と31名のSCD患者がExaccel治療を受けた最新の臨床データを公表した。
Exa-celはすでにアメリカ食品と薬物管理局(FDA)の多くの監督管理指定を獲得し、特にTDTとSCDを治療するRMAT、Fast Track、孤児薬物と稀な小児科疾患の指定を受けた。Exa−celはまた,TDTやSCDの治療に用いられる欧州委員会の孤児薬物称号と,欧州医薬品局(EMA)の良質な称号を獲得した。2022年12月,我々とVertexはそれぞれEUとイギリスのEMAとMHRAに輸出規制文書を提出し,EMAとMHRAはそれぞれMAAを検証した。また,我々とVertexは2022年11月に米国で我々のBLAのスクロール提出を開始し,2023年第1四半期末に提出を完了する予定である.
最後に、既存の基礎の上で、私たちはターゲットを絞った条件調整と体内にある造血幹細胞を編集し,いずれもわれわれの療法から利益を得ることができる患者数を拡大することができる。
免疫腫瘍学
CRISPR/CAS 9は次世代CAR T細胞療法を創造する潜在力があると信じており,現在の自己療法と比較してより良い製品配置を有し,より広範な患者群の使用を可能にしている。自自離体する我々の先行計画により得られた遺伝子編集能力により,CD 19とCD 70に対する同種CAR T計画を含むいくつかの免疫腫瘍細胞治療計画を進めている。
CD 19特許経営権
CTX 110は著者らの有力な免疫腫瘍学候補製品であり、CD 19の健康ドナー由来遺伝子に対して編集された同種異体CAR研究療法である。我々は、少なくとも2つの以前の治療を受けた再発または難治性CD 19陽性B細胞悪性腫瘍の成人患者におけるCTX 110の安全性および有効性を評価するために、我々のCarbon臨床試験においてCTX 110を研究している。CTX 110は、FDAによってRMAT称号を付与されている。
1期炭素臨床試験は2つの部分に分けて行われる--A部分とB部分。第1段階A部分では、患者は標準リンパ濾過レジメンの後、単剤CTX 110を漸増用量レベルで注入し、臨床的利益に応じてCTX 110を再投与することを選択した。第1段階B部分では、患者は、標準リンパ濾過後に用量レベル(DL)4を受けるCTX 110と、初期用量後4~8週間以内に同じ用量レベルで用量を強固にするCTX 110とを、臨床的利益を示す患者に提供する。
2022年第4四半期に、我々はCTX 110治療を受けた32人の患者の第1段階A部分の最新の臨床データを公表し、これらのデータはCTX 110が長期持続的な完全緩和(CRS)を達成する潜在力があることを示し、大量の前治療を受けた患者の中で肯定的な差異安全性を有し、第1段階B部分の新興データを記述し、これらのデータは鼓舞的な治療効果概況を示し、強化用量を使用することによって治療効果を高める可能性がある。私たちの第一段階Carbon臨床試験と監督機関との討論に現れたこれらのデータに基づいて、私たちは合併用量を含む登録可能な単一アーム、多中心、開放ラベルの第二段階臨床試験を含むCarbonの範囲を拡大した。私たちはこの重要な腕に薬を投与し始めました
CTX 110と同時に、CD 19の次世代研究、同種CAR T候補製品であるCTX 112を進めている。CTX 112は、CAR Tの効力を向上させ、CAR T消費を低減するための追加の編集に参加し、2022年第4四半期、CTX 112のINDはFDAの承認を得た。
CD 70特許経営権
107
CTX 130はCD 70の健康ドナー遺伝子に対して編集した同種異体CAR研究療法であり、CD 70は各種の固形腫瘍と血液系悪性腫瘍に発現する抗原である。CTX 130は、成人患者におけるいくつかの用量レベルのCTX 130の安全性および有効性を評価することを目的とした2つの独立した第1段階、単一アーム、多中心、開放ラベルの臨床試験で研究されている。コバルト−ライム試験は、再発または難治性TまたはB細胞悪性腫瘍の治療におけるCTX 130の安全性と有効性を評価している。コバルト−腎癌試験は,再発または難治性透明細胞腎癌に対するCTX 130治療の安全性と有効性を評価している。CTX 130は、FDAのT細胞リンパ腫治療の孤児薬物指定および皮膚T細胞リンパ腫(CTCL)亜型真菌様肉芽腫およびSézary症候群(MF/SS)のRMAT指定を取得した。2022年第2四半期、ヨーロッパ血液学協会大会で、我々はCTX 130治療を受けたT細胞リンパ腫患者18人に対して少なくとも28日間のフォローアップを受けたコバルト-LYM試験の予備臨床データを公表した。また,2022年第4四半期に癌免疫治療学会年次総会において,14名の患者のコバルト−腎癌試験の予備臨床データを公表した。
CTX 130と同時に、著者らはCTX 131を進めており、これはCD 70に対する次世代研究同種異体CAR T候補製品であり、固形腫瘍とある血液系悪性腫瘍の潜在的治療に応用できる。CTX 131は、CAR Tの効力を向上させ、CAR Tの消費を低減するための追加の編集を含み、2023年の第1四半期、CTX 131のINDはFDAの承認を得た。
他の候補者私たちのCRISPR/Cas 9プラットフォームは、インクリメンタル編集を次世代製品に統合することで革新を続けることができるようにしています。CTX 112とCTX 131のほかに、他のいくつかの研究用CAR T製品の候補製品を発売しました。また,我々が先に開示したように,われわれが2022年6月にCTX 120の第1段階臨床試験を調査する高レベルデータを開示した後,我々はBCMAに対する次世代研究同種異体CAR Tに移行する計画を発表し,CTX 120はCAR T細胞の効力を増強し,CAR Tの消費を減少させるために特許編集を統合した。
再生医学
再生医学、或いは幹細胞を用いて疾病、損傷或いは年齢によって失われた組織或いは器官機能を修復或いは置換することは、稀かつよく見られる疾患を治療する潜在力がある。私たちが作ったのは離体する遺伝子編集の専門技術として,CRISPR/Cas 9が編集した同種異遺伝子幹細胞由来療法を用いて,免疫逃避,細胞機能改善,直接細胞運命の実現に重点を置いた分野での取り組みを拡大した。我々のこの分野における最初の主な努力は,糖尿病において,我々とViaCyte,Inc.,あるいはViaCyte,2022年第3四半期にVertexに買収され,糖尿病治療のための遺伝子編集幹細胞療法を発見·開発し,それを商業化する戦略協力の一部として一連の計画を進めている。
私たちは、私たちのCRISPR/Cas 9プラットフォームを利用して、利益を増加させるためのインクリメンタル編集を含む複数の候補製品を推進する多段階の製品戦略を持っている。著者らの最初の候補製品VCTX 210は1型糖尿病(T 1 D)を治療するための研究、異遺伝子、遺伝子編集、免疫回避、幹細胞由来製品の候補製品であり、著者らの遺伝子編集技術をViaCyteの独自幹細胞機能に応用することによって開発した。VCTX 210は、免疫逃避および細胞健康を促進することを目的とした遺伝子編集を有する。著者らとViaCyteは、T 1 D患者におけるVCTX 210の安全性、耐性および免疫逃避を評価することを目的とした第1段階臨床試験においてVCTX 210を研究しており、著者らはこの臨床試験の後続段階を行っている。我々の次世代候補製品VCTX 211は、細胞適応性をさらに増強することを意図した追加の遺伝子編集を含むT 1 Dを治療するための研究、同種遺伝子編集、幹細胞由来製品候補製品である。2022年第4四半期,VCTX 211の臨床試験申請はカナダ衛生部の許可を得ており,1/2期臨床試験が行われている。
生活の中で
我々の体内にある遺伝子編集戦略の重点は遺伝子破壊と全遺伝子是正である--この2つの技術は大多数の最も一般的な深刻な単遺伝子疾患を解決するために必要である。私たちはすでに先進的なプラットフォームを構築しました体内にある遺伝子破壊は肝臓から始まります著者らはこのプラットフォームを利用して、心血管疾患から始まる稀とよく見られる疾患に関連する一連のプロジェクトの組み合わせを推進する予定である。我々の首席調査員は体内にあるCTX 310とCTX 320プログラムはそれぞれアンギオゲニン関連蛋白3(Angptl 3)とリポ蛋白質(A)に対して、これはCVDの2つの有効な標的である。遺伝子編集は自然ヒト遺伝子変異体が証明した利点を単剤形式で要約したため、心血管疾患の治療パターンを変化させる可能性がある。また,肝臓でLNPや腺関連ウイルスベクター(AAV)を使用することから,AAVを含まずHDRに依存しない方法に発展した拡張可能な完全遺伝子補正プラットフォームの開発を継続した。
CRISPR-X
著者らの現在のプロジェクトの組み合わせは重大な進展を得たが、著者らは引き続き革新して、CRISPR遺伝子編集のすべての潜在力を放出し、そして変革性治療の潜在力をより多くの患者にもたらす必要があることを認識した。2022年、私たちはCRISPR-Xという新しい早期研究チームを設立し、次世代編集モデルを開発するために革新的な研究に専念した。CRISPR-Xは、HDRまたはDNAのウイルス伝達を必要とせずに全遺伝子補正および挿入を実現する技術、例えば、全RNA遺伝子補正、DNAの非ウイルス伝達、および新しい遺伝子挿入技術に専念する。
108
仲間関係
CRISPR/CAS 9の多くの潜在的治療応用を考慮して戦略的協力を行い,特定の技術や/あるいは疾患領域の専門知識を獲得することにより,追求できる適応を広げ,プロジェクトの開発を加速した。我々は広範なパートナー関係を保ち,特定の疾患領域で遺伝子編集に基づく療法を開発している。
頂点.頂点それは.著者らは2015年にVertexと初歩的な協力プロトコルを確立し、TDT、SCD、嚢胞性線維化と他の適応の選択に集中した。2017年12月、Vertexと共同開発と商業化協定を締結し、この合意に基づき、他の事項に加えて、TDTとSCDのExacelを共同で開発し、商業化する準備をしています。2021年4月、我々はVertexと既存の共同開発と商業化プロトコルを改訂し、再記述しました。このプロトコルによると、他の事項を除いて、Vertexとの共同開発と準備を継続してTDTとSCDのexaxを商業化していきます。2019年6月にVertexと戦略協力と許可協定を締結し,Duchenne筋ジストロフィーや強直性筋ジストロフィー1型を治療する製品を開発し商業化した。
ViaCyteそれは.2018年9月、我々はViaCyteと研究と協力協定を締結し、糖尿病を治療するための遺伝子編集異遺伝子幹細胞療法の発見、開発、商業化を求めた。2021年7月、私たちはViaCyteやViaCyte JDCAと共同開発と商業化協定を締結した。ViaCyte JDCAへの加入については,我々はViaCyteの既存の研究連携プロトコルとその条項によって満了した.ViaCyte JDCAによると、私たちとViaCyteは、世界各地で1型糖尿病、2型糖尿病およびインスリン依存型/インスリンを必要とする糖尿病、またはViaCyte協力領域の治療のための候補製品および共有製品を共同開発および商業化している。ViaCyte JDCAは協力と計画管理、候補製品の臨床活動とプロトコル下の共有製品及びViaCyte協力領域の各方面の持続的な研究などに関連する条項を含む。双方に別の約束がない限り、一方で発生した研究費はその側が独自に負担する。共有製品の初商業販売日から発生した研究と連携プロトコルで最初に規定された計画費用は,60%を我々に,40%をViaCyteに割り当てた.共有製品が初めて商業販売された後、このような計画費用は私たちとViaCyteの間で平均的に分担される。共有された製品の収入は私たちとViaCyteによって分けられるだろう。Vertexは2022年第3四半期、ViaCyteとViaCyteの協力分野の権利を買収したことを発表した。
バイエルそれは.私たちは2019年第4四半期にバイエルとオプション合意に達し、この合意によると、バイエルは特定の期間内に当社が特定の自己免疫疾患、眼疾患または血友病Aを診断、治療または予防するために予め提供している2つの製品、または場合によってはそのようなオプションの製品を独自に開発し、共同商業化する権利がある。
他のパートナー関係それは.私たちは造血幹細胞、免疫腫瘍学、再生医学を支援し、補充するための追加の協力と許可協定を締結しました体内にあるNkarta,Inc.と合意し、ドナー由来の遺伝子編集されたCAR-NK細胞候補製品とNKおよびT細胞に結合した2種類の製品を共同開発および共同商業化する計画およびプラットフォームを含む;Capsida BioTreateutics,Inc.が開発される体内にある工学AAVベクターを利用して提供した遺伝子編集療法は筋萎縮性側索硬化症とFriedreich‘s運動失調の治療に応用されている;モフェット癌センターとロスウェル公園総合癌センターは新しい標的に対して自己CAR T計画を推進する;MaxCyte,Inc離体するヘモグロビン病と免疫腫瘍学プロジェクトにサービスを提供していますCureVac AGは特定の遺伝子構造と製造を最適化しています体内にあるKSQ Treateutics,Inc.とわれわれの同種異体免疫腫瘍学計画の知的財産権について協議した。
これらの手配の主な条項に関する他の資料は、“を参照してください”業務-戦略的パートナーシップと連携".
コロナウイルスと市場状況が我々の業務に与える影響
我々はコロナウイルスの大流行の状況と世界への影響を積極的にモニタリングしてきた。2022年,2021年,2020年12月31日までの年度まで,我々の財務業績はコロナウイルス爆発の大きな影響を受けていないと信じている。私たちの混合と遠隔作業スケジュールは、2022年、2021年、2020年12月31日までの年間で内部運営を維持する能力に影響が限られていると信じています。また、コロナウイルスの大流行、ロシアとウクライナ間の持続的な軍事衝突、ロシアに対する関連制裁、インフレなどの他のグローバルマクロ経済要因は、将来的に私たちの業務と私たちの普通株の価値に負の影響を与える可能性がある。
財務概要
2013年10月の設立以来、私たちはほとんどの資源を私たちの研究·開発に投入し、薬物発見と臨床前開発活動を担当し、私たちの知的財産権の確立と保護、私たちの会社、業務計画、資金調達を組織し、これらの業務に一般的かつ行政的な支援を提供してきた。これまで、私たちは主に私募優先株、普通株の発行、転換可能な融資、戦略パートナーとの協力協定を通じて、私たちの運営に資金を提供してきました。
109
我々は数年前にVertexとの連携により純収益状況にあったが,損失を繰り返した歴史があり,予見可能な未来に損失を被ることが予想される。私たちの純損失は四半期ごとと毎年大きく変動するかもしれません。私たちは、私たちが現在の研究計画と開発活動を継続することに伴い、より多くの研究計画とより多くの候補製品を決定することを求める;臨床前研究を支持する初期薬物の応用を行い、私たちの候補製品のために臨床試験を開始する;私たちが決定し、開発した任意の他の候補製品のために臨床前テストと臨床試験を開始する;私たちの知的財産権を維持、保護、拡大すること、さらに私たちの遺伝子編集プラットフォームを開発すること、より多くの研究、臨床と科学者を募集すること、このような人員の増加に関連する施設コスト、インフラの製造と関連する監督検証活動を発展させること、および上場企業としての運営に関連する追加コストを予想する。
収入確認
これまで、私たちは製品販売から何の収入も得ておらず、近い将来も何の収入も生じないと予想されています。次の年度まで 2022年12月31日,2021年12月31日,2020年12月31日,Vertex連携協定に関する収入は40万ドル,9.131億ドル,50万ドルであることをそれぞれ確認した。
2022年、2021年、2020年12月31日までの年間で、非営利エンティティのいくつかの契約に関連する贈与収入は、それぞれ80万ドル、190万ドル、20万ドルです。
当社の収入確認ポリシーの詳細については、本年度報告書Form 10−Kに含まれる監査総合財務諸表付記2および付記8を参照されたい。
研究と開発費
研究開発費は主に私たちの研究活動によって発生するコストを含み、私たちの製品発見努力と私たちの候補製品開発を含みます
私たちの外部研究開発費用は私たちの各種の臨床前と臨床計画を支持するため、私たちは外部研究開発費用をもっと細分化することはありません。我々の内部研究開発支出には,賃金や福祉支出,施設支出,全体の研究開発活動を支援するための他の間接研究開発支出が含まれているため,特定の開発段階や治療分野には割り当てられない。研究·開発コストは発生時に費用を計上する。将来的に受け取った研究開発商品やサービスの払い戻し不可能な前払いは延期され資本化されるだろう。資本化金額は関連貨物の納入またはサービス提供時に費用を計上する。現在、私たちは、私たちが決定し、開発する可能性のある任意の候補製品を完成させるために必要な努力の性質、時間、またはコストを合理的に推定または知ることができない。これは、このような候補製品の開発に関連する多くのリスクおよび不確実性に起因しており、以下のような不確実性を含む
110
任意の候補製品の開発またはその後、私たちが開発に成功する可能性がある任意の候補製品の商業化において、これらの変数のいずれかの結果の変化は、候補製品開発に関連するコスト、タイミング、および生存能力を著しく変化させる可能性がある。
研究開発活動は私たちのビジネスモデルの核心だ。予測可能な未来には,我々の現在の開発プロジェクトの進展,新プロジェクトの増加,および規制申告書類の準備を続けるにつれ,我々の研究開発コストは大幅に増加すると予想される.これらの増加には、臨床試験場所の実施と拡大に関連するコストと、現在と未来の臨床試験の関連患者登録、モニタリング、計画管理と製造費用が含まれる可能性がある。
一般と行政費用
一般と行政費用は主に従業員に関連する費用を含み、行政、財務、会計、業務発展と人力資源職能者の賃金、福祉、公平に基づく給与を含む。その他の重大なコストには、研究開発費に含まれていない施設コスト、特許や会社事務に関する法律費用、会計およびコンサルティングサービス費用が含まれています。
我々の規模や発展段階の企業では,研究や開発に一致した一般的かつ管理費が増加し続け,将来的には持続的な研究開発活動や,我々候補製品の潜在的な商業化を支援するために増加する可能性が予想される.また,我々のいくつかの許可内知的財産権に関する第三者特許関連費用の精算に関連する継続費用が予想される.
協力費用,純額
連携費用,純額には,我々がVertexと連携した業務に関する運営費用が含まれている.A&R Vertex JDCAは、Exexcel計画上の支出が指定された金額を超えた場合に、その手配でのコストシェアの一部を遅らせることを許可しています。いずれの繰延金額もVertexにしか支払いできず,Exacel計画の将来の収益性の相殺として,支払すべき金額の上限は毎年指定された最高額である.
その他の収入
他の収入には主に投資から得られた利息収入が含まれる。
重要な会計政策と重大な判断と見積もり
私たちの財務状況と経営結果の討論と分析は私たちの財務諸表に基づいています。これらの報告書は私たちがアメリカ公認会計原則に従って作成したものです。私たちはいくつかの会計政策が私たちの歴史と未来の業績を知るために非常に重要だと思う。これらの特定の分野は、通常、推定を行う際に不確実な事項の判断および推定を要求するが、異なる推定を使用することができるはずであるが、これらの推定も合理的であるため、これらの政策をキーと呼ぶ。続いて、私たちは、以下でより詳細に説明するものを含む、私たちの推定と判断を評価する。吾らは過去の経験や当時の状況で合理的な他の特定市場や他の関連仮定に基づいて推定し、その結果が資産や負債の帳簿価値を判断する基礎となり、そのような資産や負債の帳簿価値は他の出所から容易に見えるわけではないと考えている。異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。
我々の重要な会計政策は、本年度報告10-K表の他の場所の財務諸表付記により詳細に記載されていますが、以下の会計政策は、私たちの財務状況や運営結果を十分に理解し、評価するために最も重要であると考えられます。
収入.収入
会計基準符号化主題606、取引先と契約した収入またはASC 606は、顧客とのすべての契約に適用されるが、リースおよび協調スケジュールのような他の標準範囲内に属する契約を除外する。エンティティがASC 606の範囲内のスケジュールされた収入肯定応答を決定するために、エンティティは、以下の5つのステップを実行する
1)お客様との契約の決定
顧客との契約は、(I)譲渡する商品またはサービスに対する各当事者の権利を規定し、関連する支払い条件を決定する顧客と強制的に実行可能な契約を締結し、(Ii)契約が商業的実質を有する場合に存在する
111
および(Iii)我々は,顧客の意図と支払い承諾対価格の能力に基づいて,譲渡された商品やサービスのほぼすべての対価格を受け取る可能性があることを決定した.
2)契約における履行義務の決定
契約において約束された履行義務は、顧客に譲渡される貨物およびサービスによって決定され、これらの貨物およびサービスは、いずれも異なることができ、顧客は、単独でまたは他の利用可能なリソースと共に貨物またはサービスから利益を得ることができ、契約文脈で異なるので、貨物またはサービスの譲渡は、契約内の他の約束とは別に識別することができる。1つの契約が複数の約束された貨物およびサービスを含む場合、私たちは、約束された貨物およびサービスが契約の文脈で区別できるかどうかを決定するために判断を使用しなければならない。これらの基準を満たしていなければ、約束された貨物とサービスは総合履行義務として入金されるだろう。
3)出来高決定
取引価格は、商品とサービスを顧客に移転するために、私たちが獲得する権利のある対価格によって決定されます。研究、開発、規制、ビジネスマイルストーンのような可変対価格を含む取引価格の範囲内で、このような金額を受け取る可能性があるかどうかを確認し、重大な収入逆転のリスクはありません。このような金額を受け取る可能性があると結論できない場合には,関連変数の対価格を制限し,取引対価格から除外する.取引対価格に制限されている部分を決定する際には,関連研究,開発,規制,ビジネスマイルストーンが研究と臨床開発の性質および基本計画の段階を与えた場合に実現される可能性と不確実性を考慮した。この評価は各報告書期間に行われた。この評価を行う際には,業界出版物からの情報や他の関連要因を含む入手可能な内部および外部情報を考慮した.変動による価格制限の変化は当期に確認された収入額に実質的な影響を与える可能性がある。
4)取引対価格を契約に割り当てる履行義務
契約が単一履行義務を含む場合、取引対価格全体をその単一履行義務に割り当てる。複数の履行義務を含む契約要求は、相対的に独立した販売価格に基づいて取引対価格を各履行義務に割り当て、取引対価格が可変でない限り、契約義務または単一履行義務の一部を構成する異なるサービスに完全に割り当てられる基準に適合する。受信した対価格を相対的に独立した販売価格に応じて単独の履行義務の間に割り当てる.これらの推定された独立販売価格を決定する際には,ライセンスに対して,管理層が協調開発計画ごとに確率に重み付けしてキャッシュフローを割引するという仮定を含む多くの重要な判断を行う.推定された独立販売価格は,科学的に成功する確率,割引率,予測キャッシュフローの基礎を構成する何らかの仮説など,仮説の変化に敏感である.これらの仮定を作成する際には、管理層は、同一業界内の他の基準会社からの情報や他の関連要因を含む利用可能な内部および外部情報を考慮する。これらの仮定の変更は,取引が履行義務における価格の分配や収入を確認する金額や時間に大きな影響を与える可能性がある.
5)義務を果たした場合に収入を確認する
私たちは義務を履行する性質に基づいて、時間とともに、またはある時点で義務を履行する。顧客がエンティティ業績によって提供される利益を同時に受け取り、消費する場合、エンティティ業績が資産を作成または強化する際に顧客制御資産を作成または強化する場合、またはエンティティ業績が代替エンティティ用途に代替可能な資産を作成しておらず、エンティティがこれまでに完了した実績を強制的に支払う権利を有する場合、収入は時間の経過とともに確認される。エンティティが一定期間義務を履行していない場合、ある時点で、約束された貨物またはサービスの制御権を顧客に移すことによって、関連する履行義務を履行する。
協力手配
ASC 808によると、共同運営活動を代表する連携プロトコルの要素が記録されている協力手配またはASC 808。そのため,双方を代表する積極的な参加者の活動と,双方とも活動のビジネス成功に依存した重大なリスクとリターンに直面する協力合意の要素が,協力手配として記録されている.
我々は,協調プロトコルに関するビジネス活動の適切な提示およびΣtと損失分担を評価する.ASC 808は、協力手配の当事者間の支払いが他の権威ある会計文献の範囲内にない場合、損益表の分類は、その手配の性質、その業務運営の性質、およびその手配の契約条項に基づくべきであると規定している。これらの支払いが他の権威ある会計文書の範囲内にない場合、これらの支払いの損益表分類は、権威ある会計文書との類比に基づくべきであり、または適切な類比がない場合には、合理的、合理的、および一貫して適用される会計政策選択に基づくべきである。
112
研究と開発費用を計算すべきである
財務諸表を作成する過程の一部として、私たちは私たちの計算費用を見積もる必要があります。このプロセスは、未締結契約および調達注文を審査し、私たちの担当者とコミュニケーションして、私たちに代わって実行されるサービスを決定し、インボイスを受け取っていない場合、または他の方法で実際のコストを通知している場合に、実行されたサービスレベルおよびサービスによって生じる関連コストを推定することを含む。私たちのほとんどのサービスプロバイダは毎月私たちが提供してくれるサービスや契約マイルストーンに達した時に私たちに借金の領収書を発行してくれます。私たちは、当時私たちが知っている事実と状況に基づいて、財務諸表において、各貸借対照表の日付までの課税費用を推定します。計算すべき研究および開発費用の推定例には、以下の者に支払われる費用が含まれる
我々は,我々を代表して臨床研究を行って管理している複数のCROと締結した契約に基づいて,我々が受け取ったサービスとかかる努力を見積もり,臨床研究に関する費用を発生させた。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。場合によっては、私たちのサプライヤーに支払われる費用は、提供されたサービスレベルを超え、それによって、臨床費用の前払いをもたらす可能性がある。その中のいくつかの契約によって支払われる費用は被験者の成功登録と臨床研究マイルストーンの完成などの要素に依存する。サービス料を計算する際には、サービスを提供する時間帯と各時間内の労働支出レベルを見積もり、それに応じて調整します。
株式ベースの報酬
私たちの株式ベースの報酬計画は、株式オプション、制限株式単位、および制限株式報酬を含む報酬を付与します。補助金は役員を含む従業員と非従業員に付与される。
私たちはASCテーマ718に基づいて株ベースの報酬報酬を計算した報酬--株式報酬またはASC 718。ASC 718は、従業員の株式オプションおよび制限株式単位の付与、および既存の株式オプションの修正を含む、従業員および非従業員取締役に支払われるすべての株式ベースの支払いを要求し、その公正価値に基づいて総合経営報告書および全面損失で確認しなければならない。私たちはブラック-スコアーズオプション価格モデルを使用して、付与されたオプションの公正価値を決定する。
私たちは没収が発生した時に計算して、贈与時に没収を推定し、その後の期間にこれらの推定を修正し、実際に没収することがその推定と異なるならば。財務諸表で確認された株式ベースの報酬支出は、業績やサービス条件を満たすことが期待される報酬に基づいている。
私たちの株式ベースの奨励はサービスまたは業績に基づく付与条件によって制限される。サービスを基礎とする帰属条件の従業員、取締役及び非従業員が獲得した奨励に関する補償支出は、付与日に応じて関連サービス期間(一般に帰属期限)の公正価値を直線基準で確認する。業績条件に達する可能性がある場合には、奨励業績帰属条件の従業員に関する報酬支出は、付与日が必要なサービス期間中の公正価値に基づいて確認され、加速帰属法が採用される。
私たちは奨励の公正価値に基づいて、奨励の関連サービス期間内に、直線を基礎として、従業員に制限的な株式単位の奨励を支出する。
我々はブラック·スコアーズオプション定価モデルを用いて、(I)期待株価変動、(Ii)期待奨励期間の計算、(Iii)無リスク金利、および(Iv)期待配当を含む主観的仮説の入力を要求する従業員、取締役、および非従業員に対するオプション報酬の公正価値を推定する。株式奨励全体の期待期限の完全な会社の特定の歴史と隠れた変動率データが不足しているため、私たちの予想変動率の推定は私たち自身の変動率データのほかに、代表的な上場企業のグループに基づいている。これらの分析については、企業価値、リスクプロファイル、業界における地位、および株式に基づく報酬の期待寿命を満たすのに十分な歴史的株価情報を含む、私たち自身と類似した特徴を持つ会社を選択した。計算された株の報酬に基づく期待期限の等価期間内の会社株を選定した日終値を用いて履歴変動性データを算出する。私たちは、私たち自身の株価変動に関する十分な数の履歴情報が利用できるまで、この過程を適用し続ける。我々は、“簡略化”方法を用いて、従業員株式オプションの期待期間、すなわち、十分な履歴データが不足しているため、期待期間は、オプションの帰属期限および元の契約期間の算術平均値に等しいと推定した。オプション予想期間内の無リスク金利は、満期日に関連奨励の期待期限に見合った米国債をベースとしている。私たちは配当金を支払ったことがなく、予測可能な未来に配当金を支払うことを望んでいない。
113
最近の会計公告
最近の会計声明の検討については、本年度報告書に含まれる総合財務諸表付記2の表格10-Kを参照されたい。
経営成果
以下は運営結果の構成要素についての議論である。本節では,2022年と2021年の項目および2022年と2021年の間の同比比較について議論する。当社が2022年2月15日に提出した2021年12月31日現在の10−K年度報告における“経営陣の財務状況と経営成果の検討と分析”第2部は、2021年12月31日までの財政年度報告“経営陣の財務状況と経営成果の検討と分析”に含まれていない2020年プロジェクトの検討および2021年と2020年の年度比較に掲載されている。
2022年12月31日までと2021年12月31日までの年次比較
次の表は、2022年12月31日と2021年12月31日までの年度の業務結果と、これらの項目のドル変化をまとめています
|
|
十二月三十一日までの年度 |
|
|
期限が来る |
|
||||||
|
|
2022 |
|
|
2021 |
|
|
周期変化 |
|
|||
|
|
(単位:千) |
|
|||||||||
収入: |
|
|
|
|
|
|
|
|
|
|||
協力収入 |
|
$ |
436 |
|
|
$ |
913,081 |
|
|
$ |
(912,645 |
) |
奨学金収入 |
|
|
762 |
|
|
|
1,882 |
|
|
|
(1,120 |
) |
総収入 |
|
|
1,198 |
|
|
|
914,963 |
|
|
|
(913,765 |
) |
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
461,645 |
|
|
|
340,567 |
|
|
|
121,078 |
|
一般と行政 |
|
|
102,464 |
|
|
|
99,690 |
|
|
|
2,774 |
|
協力費用,純額 |
|
|
110,250 |
|
|
|
101,178 |
|
|
|
9,072 |
|
総運営費 |
|
|
674,359 |
|
|
|
541,435 |
|
|
|
132,924 |
|
営業収入(赤字) |
|
|
(673,161 |
) |
|
|
373,528 |
|
|
|
(1,046,689 |
) |
その他の収入、純額 |
|
|
22,661 |
|
|
|
6,003 |
|
|
|
16,658 |
|
所得税前純収益 |
|
|
(650,500 |
) |
|
|
379,531 |
|
|
|
(1,030,031 |
) |
所得税の優遇 |
|
|
325 |
|
|
|
(1,870 |
) |
|
|
2,195 |
|
純収益 |
|
$ |
(650,175 |
) |
|
$ |
377,661 |
|
|
$ |
(1,027,836 |
) |
協力収入
協力収入 2022年12月31日までの会計年度は40万ドルだったが、2021年12月31日現在の会計年度は9億131億ドルだった。2021年12月31日までの年度の連携収入は,主にVertexとA&R Vertex JDCAに関する9.0億ドルの前金および2019年のVertexとの連携プロトコルにより実現された1250万ドルのマイルストーンに関連しており,そのうち1200万ドルは2021年の収入を記録している。Vertexに関する収入が確認された記述については、本年度報告に含まれる連結財務諸表付記8のForm 10−Kを参照されたい。
贈与収入
2022年12月31日と2021年12月31日までの年間の贈与収入はそれぞれ80万ドルと190万ドル。
114
研究と開発費
2022年12月31日までの1年間の研究開発費は4兆616億ドルだったが、2021年12月31日までの1年間の研究開発費は3兆406億ドルだった。次の表は、2022年12月31日と2021年12月31日までの年度の研究開発費、およびこれらの項目の変化(ドル単位)をまとめたものである
|
|
十二月三十一日までの年度 |
|
|
1号1号 |
|
||||||
|
|
2022 |
|
|
2021 |
|
|
変わる |
|
|||
外部研究開発費 |
|
$ |
197,742 |
|
|
$ |
118,698 |
|
|
$ |
79,044 |
|
従業員関連費用 |
|
|
81,683 |
|
|
|
53,100 |
|
|
|
28,583 |
|
施設費 |
|
|
114,591 |
|
|
|
99,252 |
|
|
|
15,339 |
|
株に基づく報酬費用 |
|
|
53,956 |
|
|
|
59,683 |
|
|
|
(5,727 |
) |
その他の費用 |
|
|
2,864 |
|
|
|
2,016 |
|
|
|
848 |
|
再許可と許可料 |
|
|
10,809 |
|
|
|
7,818 |
|
|
|
2,991 |
|
研究開発費総額 |
|
$ |
461,645 |
|
|
$ |
340,567 |
|
|
$ |
121,078 |
|
約1兆211億ドル増加した主な原因は以下の通りである
一般と行政費用
2022年12月31日までの年間の一般·行政費は1.025億ドルであるのに対し、2021年12月31日までの年間は9970万ドルである。280万ドル増加した主な理由は、従業員の給与、福祉、その他の従業員数に関連した支出の増加だ。
協力費用,純額
2022年12月31日までの会計年度の協力支出純額は1.103億ドルであったが,2021年12月31日現在の会計年度は1.012億ドルであった。約910万ドル増加した要因は、製造や他の商業前コストの増加である。
A&R Vertex JDCAによると、2022年、2023年、2024年12月31日までの年間で、輸出計画で1億103億ドルを超える指定コスト分を延期する権利があります。Vertexは,A&R Vertex JDCAによって毎年決定されているExacel計画の将来の収益性の相殺として,このような繰延金額のみを回収することができる.どのような繰延金額も毎年指定された最高金額を上限としている。
2022年12月31日までの年間で、A&R Vertex JDCA項で発生した私たちのシェアコストを遅らせる3610万ドルの選択権を行使しました。Vertexがこのような繰延金額を回収し,合理的に金額を見積もることが可能な場合,吾らはその遅延コストを確認する.2022年12月31日現在、このような繰延金額は確認されていない。
その他の収入、純額
2022年12月31日までの1年間、その他の純収入は2270万ドルだったが、2021年12月31日までの年間は600万ドルだった。2022年12月31日までの年度のその他の純収入には、主に現金、現金等価物、有価証券の利息収入が含まれる。
流動性と資本資源
流動資金源
2022年12月31日まで、約1,868.4ドルの現金、現金等価物、有価証券を持っています 100万ドル、うち510万ドルはアメリカ国外で保有されている。
2022年12月31日まで、私たちの手元に現金があり、現金、現金等価物、有価証券は、現在の運営計画に少なくとも今後24ヶ月の資金を提供するのに十分であると予想されます。
115
設立以来、私たちは主に運営によって損失と累積マイナスキャッシュフローが発生して、2022年12月31日まで、私たちは累計8.461億ドルの損失を出しました。私たちは少なくとも今後数年以内に、私たちが引き続き損失を受けると予想している。私たちの規模と発展段階の会社では、研究開発に合わせた運営費が引き続き増加し、将来的には、持続的な研究開発活動を支援し、私たちの候補製品の潜在的な商業化を支援する可能性があると予想される。
初公募以来、私たちは主に普通株の発行と戦略パートナーとの協力合意を通じて、私たちの運営に資金を提供してきました。最近の株式融資源には
公募する
2020年7月には、委託販売により740万株の普通株式(引受業者が追加株式選択権を行使することにより売却された株式を含む)を公開発売し、公開発売価格は1株70.00ドル、総収益純額は4.897億ドルで、株式発行コスト2,760万ドルを差し引いた。2020年12月31日までに、追加株式発行コスト490万ドルの印紙税を計上し、2021年に支払う。
市場の製品
116
流動資金源
キャッシュフロー
当社が2022年2月15日に提出した2021年12月31日現在の10−K年度報告における“経営陣の財務状況と経営成果の検討と分析”第2部は、2021年12月31日までの財政年度報告“経営陣の財務状況と経営成果の検討と分析”に含まれていない2020年プロジェクトの検討および2021年と2020年の年度比較に掲載されている。
次の表は、以下の各時期のキャッシュフローに関する情報を提供する
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
経営活動が提供する現金純額 |
|
$ |
(495,741 |
) |
|
$ |
538,972 |
|
投資活動のための現金純額 |
|
|
(258,655 |
) |
|
|
(1,035,430 |
) |
融資活動が提供する現金純額 |
|
|
38,592 |
|
|
|
250,945 |
|
為替レート変動が現金に与える影響 |
|
|
(80 |
) |
|
|
(11 |
) |
現金と制限現金の増加(減少) |
|
$ |
(715,884 |
) |
|
$ |
(245,524 |
) |
経営活動
2022年12月31日までの1年間で、経営活動に用いられた純現金は4兆957億ドルだったが、2021年12月31日までの1年間で、経営活動から提供された現金は5.39億ドルだった。経営活動で使用されている現金が増加した要因は、純損失10.278億ドル、2021年12月31日現在の純収益3.777億ドルから2022年12月31日までの純損失6.502億ドルと、営業資産と負債純変化から700万ドル減少したことである。
投資活動
2022年12月31日までの年間、投資活動のための現金純額は2.587億ドルで、2.215億ドルの有価証券の購入と、研究·開発活動のための財産·設備3720万ドルの購入が含まれている。
融資活動
融資活動が提供する現金純額は、2022年12月31日までの1年間で3860万ドルで、株式オプションの行使とESPP寄付の純収益3760万ドルを含む。
資金需要
私たちの資本の主な用途は、研究開発活動、製造活動、報酬と関連費用、実験室と関連用品、法律およびその他の規制費用、特許訴訟申請、国防と知的財産権維持コスト、および上場企業の運営に関連するコストを含む一般管理コストに引き続き使用される見通しだ。私たちの規模と発展段階の会社では、研究開発に合わせた運営費が引き続き増加し、将来的には、持続的な研究開発活動を支援し、私たちの候補製品の潜在的な商業化を支援する可能性があると予想される。
私たちのほとんどのプロジェクトはまだ開発の初期段階にあり、これらの努力の結果はまだ確定していないため、現在または将来の候補製品の開発、製造、商業化に成功するために必要な実際の金額は、承認されれば、またはいつ利益を達成することができるかどうかを推定することはできない。私たちが相当な製品収入を生み出すことができる前に、もしあれば、私たちは株式融資、債務融資、そして私たちの協力合意に関連した支払いによって私たちの現金需要を満たす予定です。私たちは、市場条件が有利な場合には、株式や債務証券を売却することで追加資金を調達する機会を考えるつもりだ。しかし、私たちの普通株と他の生物製薬会社の取引価格は非常に不安定だった。したがって、私たちは私たちの普通株を売却することで資金を調達する困難に直面するかもしれないし、このような売却は不利な条項で行われるかもしれない。さらに、コロナウイルスの持続的な伝播を含む不況、不況、または他の持続的な不利な市場事件は、私たちの業務および私たちの普通株価値に実質的な悪影響を及ぼす可能性がある。もし私たちが将来株式や債務証券を売却することで追加資本を調達すれば、私たちの株主の所有権権益は希釈され、これらの証券の条項は清算または他の既存株主の権利に悪影響を及ぼす特典を含む可能性がある。もし私たちが未来に協力計画を通じてより多くの資金を調達すれば、私たちは私たちの技術、未来の収入流、または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利になるかもしれない条項で許可証を授与しなければならないかもしれない。必要な時に株式や債務融資でより多くの資金を調達できなければ、延期、制限を要求されるかもしれません, 私たちの製品開発や将来の商業化努力を減らしたり終了したり、あるいは私たちが自分で開発·マーケティングしたい候補製品を開発·マーケティングする権利を付与したりします。
117
展望
私たちの研究開発計画とプロジェクトの進展に関連する時間予想によると、私たちの既存の現金、現金等価物、および有価証券は、Vertexと協力して得られる可能性のある追加的な収益、および私たちが達成可能な任意の他の融資取引に影響を与えることなく、少なくとも今後24ヶ月の運営費用および資本支出に資金を提供することができると予想される。私たちは間違っていることが証明される可能性があるという仮定に基づいてこの推定をして、私たちは予想よりも早く私たちの資本資源を使用することができる。私たちのプロジェクトの長期的な臨床発展を支援するために追加の資金が必要であることを考慮すると、市場条件が私たちに有利な時に追加の融資機会を考慮するつもりです。
私たちが収入を創出し、利益を達成する能力は、私たちの配信技術と私たちの遺伝子編集技術プラットフォームの開発、適切な候補製品を選択して開発すること、選択された候補製品の研究、臨床前および臨床開発を完成させること、臨床試験を達成するための候補製品の規制許可とマーケティング許可を得ること、候補製品のための持続可能かつ拡張可能な製造プロセスを開発すること、私たちが直接またはパートナーまたは流通業者と規制許可およびマーケティング許可を得る候補製品を発売し、商業化すること、私たちの候補製品に対する市場の受け入れ(承認が得られれば)、任意の相互競争を解決する技術および市場開発、を含む多くの分野での成功に大きく依存する。私たちが参加する可能性のある任意の協力、許可、または他の手配で有利な条項を交渉する;私たちの協力者およびライセンシーと良好な関係を維持し、保護し、保護し、特許、商業秘密および独自技術を含む私たちの知的財産権を維持、拡大し、そして合格者を誘致、採用、および保留する。
契約義務その他の義務
レンタルと分譲義務を経営する
私たちの運営リース義務は、主にマサチューセッツ州ボストンにあるオフィスと実験室施設のレンタル支払いと、マサチューセッツ州フレミンガムにあるバッテリ製造施設のレンタル支払いを含みます。これらの費用は、本年度報告Form 10-Kに含まれる総合財務諸表の付記7により詳細に記載されています。2022年12月31日から1年以内に満期となる経営リースと転貸義務の将来契約支払いは2930万ドル、2022年12月31日から1年後に満期となる経営賃貸と転貸義務の将来契約支払いは3.109億ドルである。
その他の義務
2016年12月15日に調印された発明管理協定によると、CRISPR/Cas 9遺伝子編集知的財産権の特許メンテナンス、弁護、起訴に関する費用を、カリフォルニア州、ウィーンおよびその許可者(CariouおよびCariouを含む被許可者Intellia Treeuticsを含む)と分担する義務がある。このようなコストは現在のところ定量化できない。
A&R Vertex JDCAによると,Exexcel計画での支出が指定金額を超えていれば,スケジュールに応じてコストシェアの一部を遅らせることができる.いずれの繰延金額もVertexにしか支払いできず,Exacel計画の将来の収益性の相殺として,支払すべき金額の上限は毎年指定された最高額である.2022年12月31日現在、輸出計画に関する繰延コストはまだ計上されていないが、将来の収益性に基づいて将来の支払いを合理的に見積もることができないためである。
正常業務過程において、著者らは臨床試験と臨床用品製造の契約研究機関及び臨床前研究とその他の運営目的サービスと製品のサプライヤーと協定を締結した。これらの契約は通常180日未満の事前書面通知なしでいつでもキャンセルできます。いくつかの合意は、本Form 10−K年次報告に含まれる総合財務諸表付記9にさらに記載されているいくつかの開発、規制、または商業マイルストーンを達成する際に、これらの第三者にマイルストーンを支払うことを要求する。マイルストーン支払いに関連する金額は、いくつかの開発、規制承認、商業マイルストーンの成功達成に依存するため、契約債務とはみなされない。これらの開発、規制承認、および商業マイルストーンは実現できない可能性がある。
私たちはまた、将来的に研究、開発、商業化協定を締結した第三者に支払うことを含む、いくつかのマイルストーンを実現した際に満期と対応する未来のお金を第三者に支払う義務があります。私たちはこのようなマイルストーンの達成と時間が固定的で確定的ではないので、このような約束を私たちの貸借対照表に含まなかった。
118
第七A項。数量化と高質市場リスクの開示について。
金利感度
私たちは金利の変化と関連した市場リスクに直面している。2022年12月31日現在、私たちは18.684億ドルの現金、現金等価物、有価証券を持っており、主にアメリカ国債と政府機関証券、社債、商業手形、通貨市場口座に投資し、アメリカ政府機関証券に投資している。これらのツールの保守的な性質のため、私たちは金利リスクに重大なリスクがあるとは思わない。金利が1%増加または減少すれば、私たちのポートフォリオの公正な価値は些細な額を増加または減少させるだろう。
外貨為替リスク
私たちの海外業務のため、私たちは外貨為替レート変動のリスクに直面しています。主にスイスフランとポンド対ドルの為替レートです。現在のリスクは主に現金、売掛金、および会社間の売掛金と支払金から来ている。為替レートの変化は私たちの総合経営報告書に影響を与え、異なる時期間の比較を歪める。これまで、外貨取引損益は私たちの財務諸表に大きな影響を与えておらず、私たちも何の外貨リッジ取引も行っていません。
インフレ率
インフレは一般的に労働コスト、臨床試験コスト、製造コストを増加させることで私たちに影響を及ぼす。2022年,2021年,2020年12月31日までの年間では,インフレは我々の業務,財務状況や運営結果に実質的な影響を与えないと考えられる。
プロジェクト8.財務諸表Sと補足データ。
本項目8の規定に基づいて提出すべき連結財務諸表は、本報告の後に添付される。これらの財務諸表のインデックスは項目15を参照されたい。
項目9.Accouとの変更と分岐会計と財務情報開示の専門。
ない。
119
第9条。制御するプログラムがあります
情報開示制御とプログラムの評価
我々のCEOおよび最高財務官は、本Form 10-K年次報告がカバーする期間終了までの間の開示制御およびプログラム(1934年の証券取引法改正ルール13 a-15(E)およびルール15 d-15(E)の定義参照)の有効性を評価した後、このような評価に基づいて、我々の開示制御およびプログラムが有効であると結論した。開示制御やプログラムを設計·評価する際には、任意の制御およびプログラムは、設計および動作がどんなに良好であっても、予想される制御目標を達成するために合理的な保証を提供することしかできないことを認識しており、我々の管理層は、可能な制御とプログラムの費用対効果関係を評価する際にその判断を適用しなければならない。
経営陣財務報告内部統制年次報告書
経営陣は財務報告書の十分な内部統制の確立と維持に責任がある。財務報告の内部統制は規則13 a-15(F)と規則15 d-15(F)で定義され、規則13 a-15(F)と規則15 d-15(F)は1934年の証券取引法(改正)に基づいて公布され、我々の主要幹部と主要財務官によって設計または監督され、公認された会計原則に基づいて財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供する。私たちの財務報告書に対する内部統制は以下の政策と手続きを含む
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
我々の経営陣は、2022年12月31日までの社内財務報告の内部統制の有効性を評価した。この評価には、テレデビル委員会後援組織委員会が発表した“内部統制--総合枠組み”(2013年枠組み)(COSO)に規定されている基準を用いた。その評価によると、我々の経営陣は、2022年12月31日現在、財務報告に対する社内統制がこれらの基準に基づいて有効であると結論している。
私たちの独立公認会計士事務所安永会計士事務所は財務報告書の内部統制に関する認証報告書を発表しました。以下を参照されたい。
財務報告の内部統制の変化
1934年に発行された証券取引法第13 a-15(F)および15(D)-15(F)条規則によると、私たちは財務報告の内部統制が2022年第4四半期に大きな影響を与えなかったか、または合理的に私たちの財務報告の内部統制に大きな影響を与える可能性がある。
120
独立公認会計士事務所報告
CRISPR治療株式会社の株主と取締役会へ
財務報告の内部統制については
我々は,トレデビル協賛組織委員会が発表した内部統制-総合枠組み(2013年枠組み)(COSO基準)で確立された基準に基づき,CRISPR治療株式会社の2022年12月31日までの財務報告内部統制を監査した。我々の考えでは,CRISPR治療株式会社(当社)はCOSO基準に基づき,すべての重要な点で2022年12月31日の財務報告に対して有効な内部統制を維持している。
また、米国上場企業会計監督委員会(PCAOB)の基準に従って、当社の2022年12月31日と2021年12月31日までの総合貸借対照表を監査し、2022年12月31日までの3年度の関連総合経営報告書と総合(損失)収益、株主権益とキャッシュフロー、および2023年2月21日までの付記と私たちの報告について保留なし意見を発表した。
意見の基礎
当社経営陣は、効果的な財務報告内部統制を維持し、添付されている“経営陣財務報告内部統制年次報告”における財務報告内部統制の有効性を評価する責任がある。私たちの責任は私たちの監査に基づいて、会社の財務報告書の内部統制に意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。
我々の監査には,財務報告の内部統制を理解すること,重大な弱点があるリスクを評価すること,評価されたリスクテストに基づいて内部統制の設計·運用有効性を評価すること,および状況下で必要と考えられる他のプロセスを実行することが含まれる。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分に関する記録の保存、(2)公認された会計原則に従って財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、会社の収入および支出は会社の経営陣と取締役の許可のみに基づいて行われる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用、または処分会社の資産を防止またはタイムリーに発見する合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
/S/安永法律事務所
ボストン、マサチューセッツ州
2023年2月21日
121
プロジェクト9 B。他にも情報です。
ない。
プロジェクト9 Cです。開示面検査が禁止されている外国司法管轄区域。
適用されません。
122
部分(三)
プロジェクト10.役員·役員職ICERSと会社管理。
本プロジェクトに必要な情報は,2022年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出された2023年度株主総会依頼書を参考にした。
プロジェクト11.行政官イー補償します。
本プロジェクトに必要な情報は,2022年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出された2023年度株主総会依頼書を参考にした。
プロジェクト12.特定の受益者の保証所有権従業員と経営陣と関連した株主問題。
本プロジェクトに要求される情報は,我々の依頼書を参考にして組み込まれており,我々の2023年株主総会は,2022年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出される。
本プロジェクトに必要な情報は,2022年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出された2023年度株主総会依頼書を参考にした。
第14項.元金口座弁護士代とサービス料です。
本プロジェクトに必要な情報は,2022年12月31日までの財政年度終了後120日以内に米国証券取引委員会に提出された2023年度株主総会依頼書を参考にした。
123
部分IV.IV
プロジェクト15.展示品と融資請求書の明細書です。
(A)(1)財務諸表。
参照してください“連結財務諸表索引“下記F-1ページを参照して、本報告書の一部として提出された財務諸表リストを参照してください。
上記別表以外の他の付表が省略されているのは、これらの付表を必要とする条件を備えていないため、あるいは財務諸表または付記に必要な資料が掲載されているためである。
(A)(2)展示品。
以下の展示品索引に記載されている展示品は,本10-K表年次報告の一部としてアーカイブまたは統合を行い,参考とする.
展示品索引
展示品 番号をつける |
|
説明する |
|
|
|
3.1 |
|
改訂·再改訂されたCRISPR治療株式会社の定款は、期日は2022年6月9日である(引用会社が2022年6月13日に提出した現在の8-K表報告書の添付ファイル3.1を本明細書に組み込む)。 |
|
|
|
4.1* |
|
資本株の説明 |
|
|
|
10.1 |
|
CRISPR Treateutics AGとEmmanuelle Marie Charpentierとの間で2014年4月15日に署名されたライセンス契約(2016年10月7日に会社が提出したS-1表登録声明の添付ファイル10.5を参照)。 |
|
|
|
10.2 |
|
ライセンス契約は、2014年4月15日に、TRACR血液病株式会社とEmmanuelle Marie Charpentierによって署名された(会社が2016年10月7日に提出したS-1表登録声明の添付ファイル10.6を参照して本明細書に組み込まれる)。 |
|
|
|
10.3 |
|
CRISPR治療株式会社、Emmanuelle Marie Charpentier、ウィーン大学とInes Fonfaraの間で締結された2014年11月7日の特許譲渡協定(2016年10月7日に会社が提出したS-1表登録声明の添付ファイル10.7を参照)。 |
|
|
|
10.4 |
|
賠償協議表(ここでは、当社が2016年10月7日に提出したS-1表の登録説明書添付ファイル10.8を参照)。 |
|
|
|
10.5# |
|
CRISPR治療株式会社とロジャー·ノバクとの間で2017年12月1日に署名された雇用協定(同社が2017年12月21日に提出した現在の8−K表の添付ファイル10.2を引用した)。 |
|
|
|
10.6# |
|
CRISPR治療株式会社とOriolus Consulting LLCの間で2019年12月27日に署名されたライセンス契約(2019年12月27日に会社が提出した現在の8-Kフォームの添付ファイル10.1を参照)。 |
|
|
|
10.7# |
|
CRISPR Treateutics AGとRodger Novakとの間で2019年12月27日に署名された終了合意(本明細書では、2019年12月27日に会社が提出した現在の8-Kフォームの添付ファイル10.2を参照)。 |
|
|
|
124
10.8# |
|
CRISPR Treateutics,Inc.とSamarth Kulkarniの間で2017年10月2日に署名された2回目の改訂および再署名された雇用協定(本明細書では、会社が2017年10月2日に提出した現在の8-K表報告書の添付ファイル10.1を参照することによって組み込まれる)。 |
|
|
|
10.9# |
|
CRISPR Treateutics,Inc.とJames R.Kasingerの間で2017年5月31日に署名された雇用協定(本明細書では、会社が2018年3月8日に提出したForm 10−K年次報告書の添付ファイル10.16を参照して組み込まれる)。 |
|
|
|
10.10# |
|
CRISPR Treateutics,Inc.とLawrence Kleinの間で2019年1月2日に署名された雇用協定(会社が2019年2月25日に提出したForm 10-K年次報告書の添付ファイル10.16を参照して本明細書に組み込まれる)。 |
|
|
|
10.11# |
|
CRISPR Treateutics,Inc.とBrendan Smithとの間の雇用協定は、2021年10月14日である(本明細書では、会社が2021年10月14日に提出した現在の8−Kフォームの添付ファイル10.1を参照)。 |
|
|
|
10.12# |
|
CRISPR Treateutics AGとPhuong Khanh Morrowとの間で2022年5月23日に署名された雇用協定(2022年8月8日に会社が提出したForm 10-Q四半期報告書の添付ファイル10.1を参照して本明細書に組み込まれる)。 |
|
|
|
10.13# |
|
上級管理職現金奨励ボーナス計画(本明細書では、会社が2018年3月8日に提出したForm 10-K年報添付ファイル10.26を参照して組み込む)。 |
|
|
|
10.14# |
|
CRISPR Treateutics AG 2015株式オプションおよび贈与計画(会社が2016年9月9日に提出したS-1表登録声明の添付ファイル10.14を参照して本明細書に組み込まれます)。 |
|
|
|
10.15# |
|
CRISPR Treateutics AGは、2016年の株式オプションおよびインセンティブ計画を修正および再起動した(本明細書は、会社が2017年6月2日に提出した現在の8−Kフォーム報告書の添付ファイル10.1を参照して組み込まれる)。 |
|
|
|
10.15.1# |
|
CRISPR治療株式会社が改正および再改訂した2016年株式オプションおよびインセンティブ計画下のインセンティブ株式オプションプロトコル表(本明細書では、会社が2017年11月8日に提出した現在の報告書10-Q表の添付ファイル10.2を参照して組み込まれる)。 |
|
|
|
10.15.2# |
|
CRISPR Treeutics AGによって改訂および再作成された2016年株式オプションおよびインセンティブ計画(2017年11月8日に会社が提出した現在の報告書10-Q表の添付ファイル10.3を参照して編入することにより)、会社員は株式オプション協定の表を保持していない。 |
|
|
|
10.15.3# |
|
CRISPR Treeutics AGに従って改訂および再作成された2016年株式オプションおよびインセンティブ計画(本明細書では、会社が2017年11月8日に提出した現在の報告書10-Q表の添付ファイル10.4を参照して組み込まれる)下の非従業員取締役非限定株式オプション協定の表。 |
|
|
|
10.15.4# |
|
CRISPR治療株式会社が改訂および再作成した2016年株式オプションおよびインセンティブ計画下の制限株式奨励プロトコル表(本明細書では、会社が2017年11月8日に提出した現在の報告書10-Q表の添付ファイル10.5を参照して組み込まれる)。 |
|
|
|
10.15.5# |
|
CRISPR治療株式会社が改訂および改訂した2016年株式オプションおよびインセンティブ計画(会社が2017年11月8日に提出した現在の報告書10-Q表の添付ファイル10.6を参照して編入することにより)下の会社従業員制限株式報酬プロトコル表。 |
|
|
|
10.15.6# |
|
CRISPR治療株式会社が改訂および改訂した2016年株式オプションおよびインセンティブ計画(会社が2017年11月8日に提出した現在の報告書10-Q表の添付ファイル10.7を参照して編入することにより)下の非従業員取締役制限株式奨励協定の表。 |
|
|
|
10.16# |
|
CRISPR治療株式会社2018年株式オプション及びインセンティブ計画及びその下の合意形態(2018年6月1日に提出された会社S-8表登録声明の添付ファイル99.1を参照して本明細書に組み込む)。 |
|
|
|
10.16.1# |
|
CRISPR治療株式会社2018年株式オプションとインセンティブ計画下のインセンティブ株式オプションプロトコル表(会社が2018年6月1日に提出したS-8表登録声明の99.2号添付ファイルを引用します)。 |
|
|
|
10.16.2# |
|
CRISPR治療株式会社2018年株式オプションおよびインセンティブ計画下の会社員非制限株式オプションプロトコル表(本明細書は、会社が2018年6月1日に提出したS-8表登録声明の99.3号添付ファイルを参照して組み込まれる)。 |
|
|
|
125
10.16.3# |
|
CRISPR治療株式会社2018年株式オプションおよびインセンティブ計画下の非従業員取締役非制限株式オプション協定の表(会社が2018年6月1日に提出したS-8表登録声明の99.4号添付ファイルを参照して本明細書に組み込まれます)。 |
|
|
|
10.16.4# |
|
CRISPR治療株式会社2018年株式オプションおよびインセンティブ計画下の制限株式奨励表(会社が2018年6月1日に提出したS-8表登録声明の99.5号添付ファイルを参照して本明細書に組み込まれます)。 |
|
|
|
10.16.5# |
|
CRISPR治療株式会社2018年株式オプションおよびインセンティブ計画下の会社従業員制限株式奨励協定のフォーマット(2018年6月1日に会社が提出したS-8表登録声明の99.6号添付ファイルを引用します)。 |
|
|
|
10.16.6# |
|
CRISPR治療株式会社2018年株式オプションおよびインセンティブ計画の下で非従業員取締役に対する制限株式奨励表(会社が2018年6月1日に提出したS-8表登録声明の99.7号添付ファイルを参照して本明細書に組み込まれます)。 |
|
|
|
10.17# |
|
2018年株式オプション及びインセンティブ計画の第1号改正案(当社が2019年4月30日に提出した付表14 A最終依頼書の付録Aを参照して本明細書に組み込む)。 |
|
|
|
10.18# |
|
2018年株式オプション·インセンティブ計画第2号修正案(ここでは、当社が2020年4月24日に提出した付表14 A最終依頼書の付録Aを引用)。 |
|
|
|
10.19# |
|
2018年株式オプション及びインセンティブ計画の第3号改正案(当社が2022年4月25日に提出した付表14 Aに関する最終依頼書の付録Aを参照して本明細書に組み込む)。 |
|
|
|
10.20# |
|
CRISPR Treateutics AG 2016従業員株式購入計画(会社が2016年9月9日に提出したS-1表登録声明の添付ファイル10.16を参照して本明細書に組み込まれる)。 |
|
|
|
10.21 |
|
CRISPR治療株式会社、カリフォルニア大学ウィーン校、Intellia治療会社、Cariou生物科学会社、ERSゲノム有限会社とTRACR血液学有限会社の間で締結されたゲノム編集のためのプログラム可能なDNA制限酵素の譲渡、許可および共同所有権および発明管理協定に同意し、日付は2016年12月15日(これに合併し、会社が2016年12月16日に提出した現在のForm 8-K表の添付ファイル10.1参照)。 |
|
|
|
10.22 |
|
CRISPR治療株式会社、CRISPR治療有限会社、CRISPR治療会社、TRACR血液学有限会社、Vertex PharmPharmticals,Inc.とVertex PharmPharmticals(ヨーロッパ)有限会社が2015年10月26日に調印した戦略協力、オプションと許可協定(ここで合併し、会社が2016年10月7日に提出したS-1表登録声明の添付ファイル10.4参照)。 |
|
|
|
10.23 |
|
Vertex PharmPharmticals Inc.とVertex PharmPharmticals(Europe)Limited間およびVertex PharmPharmticals Inc.とCRISPR Treateutics AG,CRISPR Treateutics,Inc.,CRISPR Treateutics LimitedとTRACR Hematology Ltd.の間の戦略協力、オプション、ライセンスプロトコル修正案1は、2017年12月12日(合併時参考会社が2017年12月18日に提出した現在の8-K表の添付ファイル10.2)である。 |
|
|
|
10.24 |
|
Vertex PharmPharmticals Inc.とVertex PharmPharmticals(Europe)Limited間およびVertex PharmPharmticals Inc.およびCRISPR Treateutics AG、CRISPR Treateutics Inc.,CRISPR Treateutics LimitedおよびTRACR Hematology Ltd.の間の戦略的協力、オプションおよびライセンスプロトコル修正案2は、2019年6月6日である(本明細書では、会社が2019年7月29日に提出したForm 10-Q四半期報告添付ファイル10.1を参照して組み込む)。 |
|
|
|
10.25 |
|
CRISPR Treateutics AGおよびVertex PharmPharmticals Inc.が2019年6月6日に署名した戦略的協力および許可協定(会社が2019年7月29日に提出したForm 10-Q四半期報告書の添付ファイル10.2を参照して本明細書に組み込まれる)。 |
|
|
|
10.26 |
|
CRISPR Treateutics AGおよびVertex PharmPharmticals Inc.が2021年3月17日に署名した戦略的協力および許可協定の第1の修正案(2021年4月27日に会社が提出したForm 10-Q四半期報告書の添付ファイル10.3を参照して本明細書に組み込まれる)。 |
|
|
|
10.27^ |
|
Vertex PharmPharmticals Inc.とVertex PharmPharmticals(Europe)Limited間の共同開発および商業化協定が改訂および再署名され、2021年4月16日のCRISPR Treeutics AG、CRISPR Treateutics Limited、CRISPR Treateutics,Inc.およびTRACR Hematology Ltd.である(本明細書は、参考会社が2021年4月27日に提出したForm 10-Q四半期報告の添付ファイル10.4を組み込む)。 |
|
|
|
126
10.28 |
|
CRISPR Treateutics AGとBayer Healthcare LLCが2019年12月13日に署名したオプション協定(当社が2020年2月12日に提出したForm 10-K年報添付ファイル10.31を参照)。 |
|
|
|
10.29^ |
|
レンタル日は2020年5月5日であり,CRISPR Treateutics,Inc.とCRP/Kingニューヨーク通り33号で借りられている。Owner,L.L.C.(ここでは,当社が2021年4月27日に提出したForm 10-Q四半期報告の添付ファイル10.1を参照)。 |
|
|
|
10.30^ |
|
2020年12月2日の第1改正案は、CRISPR治療会社とCRP/Kingニューヨーク通り33号の間でレンタルされている。Owner,L.L.C.(ここでは,当社が2021年4月27日に提出したForm 10-Q四半期報告の添付ファイル10.2を引用する). |
|
|
|
10.31^ |
|
CRISPR Treateutics,Inc.と33 NYA Owner(DE)LLCの間で2021年4月_に締結されたリース第2改正案は,CRP/King 33 NY Aveの権益相続人とする。Owner,L.L.C.(ここでは,当社が2021年7月29日に提出したForm 10-Q四半期報告の添付ファイル10.1を参照)。 |
|
|
|
10.32 |
|
CRISPR Treateutics,Inc.と105 W First Street Owner,L.L.C.の間の借約は,2020年7月24日(ここで合併し,会社が2020年7月27日に提出したForm 10−Q四半期報告の添付ファイル10.1参照)である。 |
|
|
|
10.33 |
|
CRISPR Treateutics,Inc.と105 W First Street Owner,L.L.C.が2022年1月6日に締結した書簡協定(社が2022年2月15日に提出したForm 10−K年度報告の添付ファイル10.37を参考に合併した)。 |
|
|
|
10.34 |
|
CRISPR Treateutics AGと105 W First Street Owner L.L.C.の間で締結されたリース開始日契約は,2022年5月1日である(社が2022年8月8日に提出したForm 10−Q四半期報告の添付ファイル10.2を引用した)。 |
|
|
|
21.1* |
|
登録者の子会社 |
|
|
|
23.1* |
|
安永法律事務所が同意した |
|
|
|
31.1* |
|
2002年のサバンズ·オキシリー法第302節で可決された1934年の証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて発行された首席執行幹事証明書。 |
|
|
|
31.2* |
|
2002年サバンズ−オキシリー法第302節で可決された1934年証券取引法第13 a−14(A)及び15 d−14(A)条に基づいて首席財務幹事が認証された。 |
|
|
|
32.1+ |
|
2002年の“サバンズ-オックススリー法案”906節で採択された“米国法典”第18編1350条による最高経営責任者と最高財務官の認証。 |
|
|
|
101.INS |
|
連結されたXBRLインスタンス文書−インスタンス文書は、XBRLタグがイントラネットXBRL文書に埋め込まれているので、相互作用データファイルには表示されない。 |
|
|
|
101.書院 |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
101.カール |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
|
|
|
101.def |
|
インラインXBRL分類拡張Linkbase文書を定義する |
|
|
|
101.介護会 |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
|
|
|
101.Pre |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
|
|
|
104 |
|
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
*アーカイブをお送りします。
+メールで提供されます。
本展示品のいくつかの部分は漏れています。それらは実質的ではないので、登録者は通常、その情報を個人または機密と見なしています。
#表格10-K第15(A)(3)項の要件に従って、証拠として提出された管理契約または補償計画またはスケジュール。
^S-K規則601項によれば、これらのプロトコルのいくつかの証拠品および添付表は省略されている。登録者は証券取引委員会に任意の展示品とスケジュールのコピーを提供することを要求しなければならない。
127
項目16.表10-Kの概要
ない。
128
サイン
1934年“証券取引法”第13又は15(D)節の要求に基づいて、登録者は、正式に許可された次の署名者が代表して本報告書に署名することを正式に手配した.
|
|
CRISPR治療会社 |
||
|
|
|
|
|
日付:2023年2月21日 |
|
差出人: |
|
/s/Samarth Kulkarni |
|
|
|
|
サマーズ·クルカルニ |
|
|
|
|
最高経営責任者 |
署名と授権書
我々は、以下に署名したCRISPR治療株式会社(“会社”)の役員および上級管理者を、それぞれロジャー·ノバク、サマース·クルカーニ、ブレンダン·スミス、ジェームズ·R·カーシンガーを構成して任命し、彼らはそれぞれ我々の真の合法的な代理人であり、彼らに対して完全な権力を有し、私たちの名義で本10-K表の任意およびすべての改訂に署名し、すべての証拠物およびこれに関連する他の文書と共に米国証券取引委員会に提出し、上記の代理人と彼らの一人に付与し、本授権書に関連するすべての必要および行わなければならないことを行う権利が完全にあり、完全に、私たち一人一人が可能であるか、または自ら行うことができるすべての意図および目的に基づいて行われ、ここで、上述したすべての受権者およびそのすべてのまたはそれらの代替者が、この授権書によって行われた、またはそれに至るすべてのことを許可および確認する権利がある。
本報告書は、改正された1934年の証券取引法の要求に基づき、以下の登録者によって登録者として指定日に署名された。
名前.名前 |
|
タイトル |
|
日取り |
|
|
|
|
|
/s/Samarth Kulkarni |
|
取締役CEO兼最高経営責任者 |
|
2023年2月21日 |
サマーズ·クルカルニ |
|
(首席行政主任) |
|
|
|
|
|
|
|
/s/ブランドン·スミス |
|
首席財務官 |
|
2023年2月21日 |
ブランドン·スミス |
|
(首席財務会計官) |
|
|
|
|
|
|
|
/s/ロジャー·ノバク |
|
総裁と役員 |
|
2023年2月21日 |
ロジャー·ノロ |
|
|
|
|
|
|
|
|
|
/s/アリ·ベヘバハニ |
|
役員.取締役 |
|
2023年2月21日 |
Ali·ベヘバハニ |
|
|
|
|
|
|
|
|
|
/s/ブラッドリー·ボルソン |
|
役員.取締役 |
|
2023年2月21日 |
ブラッドリー·ボルソン |
|
|
|
|
|
|
|
|
|
/s/マリア·ファディス |
|
役員.取締役 |
|
2023年2月21日 |
マリア·ファディス |
|
|
|
|
|
|
|
|
|
エドワード·フレミングちゃん |
|
役員.取締役 |
|
2023年2月21日 |
H.Edward Fleming,Jr |
|
|
|
|
|
|
|
|
|
/s/サイモン·J·ジョージ |
|
役員.取締役 |
|
2023年2月21日 |
サイモン·J·ジョージ |
|
|
|
|
|
|
|
|
|
/s/ジョン·グリーン |
|
役員.取締役 |
|
2023年2月21日 |
ジョン·グリーン |
|
|
|
|
|
|
|
|
|
/s/キャサリン高校 |
|
役員.取締役 |
|
2023年2月21日 |
キャサリン高校 |
|
|
|
|
|
|
|
|
|
/s/ダグラス·トレコ |
|
役員.取締役 |
|
2023年2月21日 |
ダグラス·テレコ |
|
|
|
|
|
|
|
|
|
/s/ブランドン·スミス |
|
アメリカでの許可代表は |
|
2023年2月21日 |
ブランドン·スミス |
|
|
|
|
129
130
CRISPR治療会社
連結財務諸表索引 |
|
ページのページ |
独立公認会計士事務所レポート(PCAOB ID番号) |
|
F-1 |
合併貸借対照表 |
|
F-3 |
連結経営報告書と総合報告書(赤字)収入.収入 |
|
F-4 |
合併権益表 |
|
F-5 |
統合現金フロー表 |
|
F-6 |
連結財務諸表付記 |
|
F-7 |
独立公認会計士事務所報告
CRISPR治療株式会社の株主と取締役会へ
財務諸表のいくつかの見方
CRISPR治療株式会社(当社)の2022年12月31日現在と2021年12月31日までの連結貸借対照表、2022年12月31日までの3年度の関連総合経営報告書と包括(赤字)収入、株主権益と現金流量および関連付記(総称して“総合財務諸表”と呼ぶ)を監査しました。総合財務諸表は、すべての重要な面で、2022年12月31日、2022年12月31日、2021年12月31日までの会社の財務状況、および2022年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており、米国公認会計原則に適合していると考えられる。
また、米国上場企業会計監督委員会(PCAOB)の基準に基づき、テレデビル委員会協賛組織委員会が発表した“内部統制-総合枠組み(2013枠組み)”で確立された基準に基づいて、2022年12月31日までの財務報告内部統制を監査し、2023年2月21日に発表された報告書について保留意見を発表した。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項は、監査委員会が監査委員会に伝達または要求する当期総合財務諸表監査によって生じる事項を指すものである:(1)財務諸表に対して大きな意味を有する勘定または開示に関するものであり、(2)私たちが特に挑戦的で主観的または複雑な判断に関するものである。重要監査事項の伝達は、総合財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、以下の重要な監査事項を伝達することによって、重要な監査事項またはそれに関連する勘定または開示について個別の意見を提供することはない。
F-1
|
|
進行中の協調プロトコルの可変対価格の見積り |
関係事項の記述 |
|
統合財務諸表付記8に記載されているように、同社は、いくつかの開発中のプロジェクトに関連する様々な開発、規制、およびビジネスマイルストーンが実現したときに支払う2022年12月31日までの合計約22億ドルの支払い権利を含む複数の協力協定を持っている。これらの将来支払いは,これらの提携プロトコルの取引価格に含まれる可変対価格であり,契約によって確認された累積収入が大きな収入逆転を生じない可能性が高いことを会社が確定していることを前提としている.当社が契約項での累積収入が重大な収入逆転を生じない可能性が高いと結論できない場合、当社は関連する可変対価格を制限し、取引価格から除外します。制限された可変対価格に対する会社の推定は、連結財務諸表に報告された収入と繰延収入に影響を及ぼす。
制限する取引価格部分を決定する際に,経営陣は,臨床開発の性質や基本計画の段階に鑑み,関連する開発,管理,ビジネスマイルストーンの可能性と不確実性を実現するかどうかを考慮している。この評価は各報告書期間に行われた。この評価を行う際には,管理層は,業界出版物からの情報,基礎計画の開発段階,その他の関連要因を含む入手可能な内部および外部情報を考慮している.変動対価格制限の変更は財務報告期間内に確認された収入額に実質的な影響を与える。そのため、制約を可変考慮に適用した会計を監査するには複雑な監査師の判断が必要である。
|
私たちが監査でどのようにこの問題を解決したのか |
|
我々は理解を得て,設計を評価し,会社収入確認過程の制御の操作有効性をテストした.例えば、将来の開発、法規、ビジネスマイルストーンに関連する可変対価格適用制約の制御を含む、管理層の協力プロトコルの総取引価格推定の制御をテストした。
可変価格適用制約に対する会社の判断を審査するために、会社が関連する将来の開発、規制、ビジネスマイルストーンを実現する可能性に関する判断を含む監査プログラムを実行した。同社が開発、監督と商業マイルストーンを実現する推定確率を評価するために、臨床開発の性質と基礎計画の開発段階と関連外部データを考慮し、マイルストーンを実現する可能性を現在の業界傾向、同一業界内の他の指導会社の既存情報と他の関連要素と比較した。会社の研究開発マネージャーとプロジェクトごとの開発段階に関する実現マイルストーンの可能性についても検討した。
|
/s/
2015年以来、当社の監査役を務めてきました。
2023年2月21日
F-2
CRISPR治療会社
合併B割当書
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
|
十二月三十一日 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
有価証券 |
|
|
|
|
|
|
||
売掛金 |
|
|
|
|
|
|
||
前払い費用と他の流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
非流通有価証券 |
|
|
|
|
|
|
||
無形資産、純額 |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
経営的リース資産 |
|
|
|
|
|
|
||
他の非流動資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
費用を計算する |
|
|
|
|
|
|
||
収入を繰延し,当期 |
|
|
|
|
|
|
||
課税負債 |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
|
|
|
|
||
その他流動負債 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
収入を繰延し、流動ではない |
|
|
|
|
|
|
||
賃貸負債を経営し,当期分を差し引く |
|
|
|
|
|
|
||
他の非流動負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
(注9) |
|
|
|
|
|
|
||
株主権益: |
|
|
|
|
|
|
||
普通株、スイスフラン |
|
|
|
|
|
|
||
国庫株は、原価で計算する |
|
|
( |
) |
|
|
( |
) |
追加実収資本 |
|
|
|
|
|
|
||
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
その他の総合損失を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
これらの連結財務諸表の付記を参照してください。
F-3
CRISPR治療会社
連結業務報告書純額と総合収益
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
収入: |
|
|
|
|
|
|
|
|
|
|||
協力収入 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
奨学金収入 |
|
|
|
|
|
|
|
|
|
|||
総収入 |
|
|
|
|
|
|
|
|
|
|||
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
|
|
|
|
|
|
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
協力費用,純額 |
|
|
|
|
|
|
|
|
|
|||
総運営費 |
|
|
|
|
|
|
|
|
|
|||
営業収入(赤字) |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
その他の収入: |
|
|
|
|
|
|
|
|
|
|||
その他の収入、純額 |
|
|
|
|
|
|
|
|
|
|||
その他の収入合計,純額 |
|
|
|
|
|
|
|
|
|
|||
所得税前純収益 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
所得税の優遇 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
純収益 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
外貨換算調整 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
有価証券は赤字を実現していない |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
総合収益 |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|
|||
普通株1株当たりの純収益--基本 |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
基本加重平均普通株式発行済み |
|
|
|
|
|
|
|
|
|
|||
普通株1株当たりの純収益--減額 |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
希釈加重平均普通株式発行 |
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||
これらの連結財務諸表の付記を参照してください。
F-4
CRISPR治療会社
(千単位で1株当たりおよび1株当たりのデータは含まれていない)
|
普通株 |
|
国庫株 |
|
その他の内容 |
|
|
|
|
|
|
|
||||||||||||
|
株 |
|
CHF 0.03 |
|
株 |
|
金額は、 |
|
支払い済み |
|
積算 |
|
積算 |
|
株主権益総額 |
|
||||||||
2019年12月31日の残高 |
|
|
$ |
|
|
|
$ |
( |
) |
$ |
|
$ |
( |
) |
$ |
|
$ |
|
||||||
普通株を発行し、発行コストを差し引いて#ドルです |
|
|
|
|
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
||||
株式の帰属を制限する |
|
|
|
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
|
|||
既得オプションを行使し,発行コストを差し引いて#ドル |
|
|
|
|
|
( |
) |
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
||||
ESPPにより普通株を購入する |
|
|
|
— |
|
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
株に基づく報酬費用 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
||
発行許可契約の普通株 |
|
|
|
— |
|
|
( |
) |
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
その他総合損失 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
( |
) |
|
( |
) |
純損失 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
( |
) |
|
— |
|
|
( |
) |
2020年12月31日残高 |
|
|
$ |
|
|
|
$ |
( |
) |
$ |
|
$ |
( |
) |
$ |
( |
) |
$ |
|
|||||
普通株を発行し、発行コストを差し引いて#ドルです |
|
|
|
|
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
||||
株式の帰属を制限する |
|
|
|
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
|
|||
既得オプションを行使し,発行コストを差し引いて#ドル |
|
|
|
|
|
( |
) |
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
||||
ESPPにより普通株を購入する |
|
|
|
— |
|
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
株に基づく報酬費用 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
||
その他総合損失 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
( |
) |
|
( |
) |
純収入 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
|
|
|
|
|
|||
2021年12月31日の残高 |
|
|
$ |
|
|
|
$ |
( |
) |
$ |
|
$ |
( |
) |
$ |
( |
) |
$ |
|
|||||
普通株を発行する |
|
|
|
— |
|
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
株式の帰属を制限する |
|
|
|
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
|
|||
既得オプションを行使し,発行コストを差し引いて#ドル |
|
|
|
|
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
||||
ESPPにより普通株を購入する |
|
|
|
— |
|
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
|||
株に基づく報酬費用 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
|
|
— |
|
|
— |
|
|
|
||
その他総合損失 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
( |
) |
|
( |
) |
純損失 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
( |
) |
|
— |
|
|
( |
) |
2022年12月31日の残高 |
|
|
$ |
|
|
|
$ |
( |
) |
$ |
|
$ |
( |
) |
$ |
( |
) |
$ |
|
|||||
これらの連結財務諸表の付記を参照してください。
F-5
CRISPR治療会社
合併状態現金流動額
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
経営活動 |
|
|
|
|
|
|
|
|
|
|||
純収益 |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
純(損失)収入と経営活動で使用されている現金純額を照合する |
|
|
|
|
|
|
|
|
|
|||
減価償却および償却 |
|
|
|
|
|
|
|
|
|
|||
株式ベースの報酬 |
|
|
|
|
|
|
|
|
|
|||
他の非現金プロジェクト、純額 |
|
|
|
|
|
|
|
|
|
|||
以下の変更: |
|
|
|
|
|
|
|
|
|
|||
売掛金 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
前払い費用と他の資産 |
|
|
|
|
|
( |
) |
|
|
|
||
売掛金と売掛金 |
|
|
|
|
|
|
|
|
|
|||
収入を繰り越す |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
リース資産と負債を経営する |
|
|
|
|
|
|
|
|
( |
) |
||
その他の負債、純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
経営活動が提供する現金純額 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
投資活動 |
|
|
|
|
|
|
|
|
|
|||
家屋·工場·設備を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
有価証券の満期日 |
|
|
|
|
|
|
|
|
|
|||
投資活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
融資活動 |
|
|
|
|
|
|
|
|
|
|||
普通株を発行して得られる収益は,発行コストを差し引く |
|
|
|
|
|
|
|
|
|
|||
オプション行使とESPP払込の収益は,発行コストを差し引く |
|
|
|
|
|
|
|
|
|
|||
融資活動が提供する現金純額 |
|
|
|
|
|
|
|
|
|
|||
為替レート変動が現金に与える影響 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
現金が増える |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
期初現金、現金等価物、および限定現金 |
|
|
|
|
|
|
|
|
|
|||
現金、現金等価物、制限された現金、期末 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
非現金投資·融資活動の追加開示 |
|
|
|
|
|
|
|
|
|
|||
売掛金と売掛金のうち財産·設備購入 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
売掛金と売掛金における持分発行コスト |
|
$ |
|
|
$ |
|
|
$ |
|
|||
連結貸借対照表内の金額を照合する |
|
12月31日まで |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
現金と現金等価物 |
|
|
|
|
|
|
|
|
|
|||
前払い費用と他の流動資産 |
|
|
|
|
|
|
|
|
|
|||
制限現金 |
|
|
|
|
|
|
|
|
|
|||
合計する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
これらの連結財務諸表の付記を参照してください。
F-6
CRISPR治療会社
総合備考財務諸表
1.組織と運営
CRISPR Treateutics AG(“CRISPR”または“会社”)は
同社は生物技術業界会社によく見られるリスクに直面しているが、これらに限定されない:臨床前研究と臨床試験失敗のリスク、それが確定と開発可能な任意の候補薬物は市場承認を得る必要があるリスク、その候補製品の商業化に成功し、市場の認可を得る必要があり、肝心な者への依存、ノウハウの保護、政府法規の遵守、競争相手の技術革新の開発、及び試験生産から大規模生産への移行能力。
同社の累積赤字は#ドルだ
2022年12月31日現在、会社は現金、現金等価物、有価証券を持っている
コロナウイルスの大流行が会社の業務、運営と財務業績に与える全面的な影響程度は多くの著者らが正確に予測できないかもしれない絶えず変化する要素に依存する。より詳細は、本年度報告における表10-Kの第1 A項目:“リスク要因”の節を参照されたい。現段階では、会社の業績への影響は大きくない。
2.主要会計政策の概要と列報根拠
予算の列報と使用根拠
添付されている総合財務諸表は、当社とその完全子会社が2022年12月31日までの勘定を含む米国公認の会計原則またはGAAPに従って作成されている。すべての会社間口座と取引はキャンセルされた。これらの説明における適用指導への任意の言及は、財務会計基準委員会の会計基準編纂(ASC)および会計基準更新(ASU)において発見された権威ある米国公認会計原則を意味する。
2022年から、頂点プロトコルでの連携コストはASC 808で比を占めている協力協定またはASC 808は、連結経営報告書および総合(損失)収益表における“連携費用、純額”に記載されている。そこで,2021年と2020年12月31日までの年度では,ASC 808により計算されたVertexプロトコルでの連携コストが再分類され,現在の列報方式に適合している.前期連結財務諸表の小計は影響を受けなかった。Vertexプロトコルのさらなる検討については、これらの連結財務諸表の付記8を参照されたい。
公認会計基準に従って財務諸表を作成することは、財務諸表と付記中の報告の額に影響を与えるために、管理層に推定と仮定を要求する。継続的な基礎の上で、会社経営陣は、収入確認、株式ベースの報酬支出、および報告書の研究·開発支出額を含むが、これらに限定されない推定値を評価する。これらの連結財務諸表では、収入確認と権益に基づく報酬支出が大きく推定されている。当社は過去の経験とその時点で部下が合理的と考えている他の特定の市場またはその他の関連仮定に基づいて推定しています。実際の結果は,これらの推定や仮定とは異なる可能性がある.
F-7
市場情報を細分化する
当社と当社の経営意思決定者、すなわちCEOは、当社の運営と業務管理を
外貨換算と取引
同社の業務の大部分はドルをその機能通貨とする実体で発生している。非ドル建て機能通貨子会社の資産と負債は年末時の有効為替レートでドルに換算される。収入と支出額はその期間の平均為替レートを用いて換算する。外貨換算による未実現純損益は“累計その他総合(赤字)収入”に計上されている。外国為替取引純損益は、会社の総合経営報告書の“その他の収入·純額”に計上され、影響は大きくない。
現金と現金等価物
当社は購入日から三ヶ月以下のすべての高流動性投資を現金等価物と見なしています。同社は2022年12月31日と2021年12月31日まで
制限現金
同社は2022年12月31日と2021年12月31日まで
有価証券
同社は2022年12月31日と2021年12月31日まで
評価レベルで第2レベルに分類される有価証券には、通常、米国債や政府機関証券、社債、商業手形が含まれる。債務証券は公正価値に従って帳簿を作成し、損益を他の総合(損失)収入に計上することを実現せず、株主権益の構成部分として、実現までとしている。購入時に発生した任意の割増は、最も早い償還日の間の利息支出に償却され、購入時に発生した任意の割引は、手形の有効期限内の利息収入に計上される。債務証券の実現済み収益と損失は特定の確認方法を用いて決定し、他の収入、純額に計上する。
当社は、ASU 2016−13年度に売却可能な債務証券減価モデルに基づいて、債務証券の売却可能性を評価した金融商品·信用損失(特集326):財務諸表信用損失の測定またはASC 326は、債務証券の売却確認のために使用可能な公正価値が帳簿価値よりも低い任意の低下がクレジット損失の結果であるかどうかを決定するために使用される。当社は合併経営報告書において信用損失と全面損失を他の費用純額内の信用損失費用に計上しており、純額は証券の公正価値と償却コストとの差額に限られている。現在まで、同社は売却可能な債務証券にいかなる信用損失も記録していない。
F-8
その他売掛金
信用リスクと表外リスクの集中度
会社を集中的な信用リスクに直面させる可能性のある金融商品は主に現金、現金等価物、有価証券である。同社の現金は、経営陣が信用が良いと考えている金融機関の口座に保管されている。当社は当該等の口座にいかなる信用損失も発生しておらず、当該等の資金に重大な信用リスクがあるとも考えていない。当社には表外損失リスクの金融商品はありません。
金融商品の公正価値
当社の公正価値記録に従ったいくつかの金融資産および負債は、公正価値計量会計基準に記載されている公正価値レベルにおいて、1、2または3レベルに分類される
第1レベル-アクティブな市場においてオファーされ、市場日に同じ制限されない資産または負債を得ることができる。
第2レベル-第1レベル以外の直接的または間接的に観察可能な投入、例えば、同様の資産または負債のオファー、非アクティブ市場のオファー、またはすべての重大な投入が、資産または負債の全期間にわたって観察可能な市場データによって確認され得る他の投入を観察することができる。
第三レベル-市場活動が少ないか、または市場活動支援の観察できない投入がなく、資産または負債の公正な価値に大きな意義を持っている。
ある程度、推定値は市場で観察または観察できないモデルや投入に基づいており、公正価値の決定にはより多くの判断が必要である。そのため、当社が価値を公平に判断する際に下した判断は、第3級に分類されたツールに対する判断度が最も高く、公正価値レベル内の金融商品レベルは、公正価値計量に大きな意味を持つ任意の投入の中で最低レベルに基づいている。
公正な価値に応じて恒常的に計量される項目には、有価証券がある(付記3参照有価証券注4と、公正価値計量). 2022年12月31日と2021年12月31日までに連結貸借対照表に報告された売掛金、その他の売掛金、売掛金及び売掛金の帳簿金額これらのツールの短期的な存続期間のため、約公正な価値がある。
財産と設備
財産と設備はコストから減価償却累計を引いて入金されます。対応する資産寿命を改善または延長することができないメンテナンスやメンテナンスは、発生時に運営費用を計上することができる。売却時には、関連コストや減価償却が勘定から差し引かれ、それによって生じる収益や損失はいずれも経営結果に計上される
資産 |
|
使用寿命を見込む |
コンピュータ装置 |
|
|
家具、固定装置、その他 |
|
|
実験室装置 |
|
|
賃借権改善 |
|
長期資産減価準備
事件や環境変化により資産の帳簿価値が回収できない可能性がある場合、当社は長期資産を審査します。回収能力は,資産の帳簿価値と資産予想による将来の未割引キャッシュフローを比較することで測定した。このような資産は減値とみなされ、確認すべき減値は資産の帳簿価値がその公正価値を超えた金額で計量され、公正価値は資産による予想割引に基づいて将来の純現金流量によって計測される。
F-9
収入確認
会社はASCテーマ606に基づいて収入を記録しています取引先と契約した収入またはASC 606。ASC 606は、顧客と締結されたすべての契約に適用されるが、レンタルおよび協調スケジュールのような他の標準範囲内の契約は除外される。エンティティがASC 606の範囲内のスケジュールされた収入肯定応答を決定するために、エンティティは、以下の5つのステップを実行する
1)お客様との契約の決定
(I)会社が顧客と強制的に実行可能な契約を締結した場合、当該契約は、譲渡する商品またはサービスに対する各当事者の権利を規定し、関連する支払い条項を決定し、(Ii)契約が商業的実質を有し、(Iii)会社が顧客の意図および支払い承諾の対価能力に基づいて、譲渡された商品およびサービスのほぼすべての対価格を徴収する可能性があると決定した場合、顧客との契約が存在する。
2)契約における履行義務の決定
契約において約束された履行義務は、顧客に譲渡される貨物およびサービスによって決定され、これらの貨物およびサービスは、いずれも異なることができ、顧客は、単独でまたは他の利用可能なリソースと共に貨物またはサービスから利益を得ることができ、契約文脈で異なるので、貨物またはサービスの譲渡は、契約内の他の約束とは別に識別することができる。1つの契約に複数の承諾された商品及びサービスが含まれている場合、会社は、約束された商品及びサービスが契約の背景で区別できるか否かを決定するために判断を適用しなければならない。これらの基準を満たしていなければ、約束された貨物とサービスは総合履行義務として入金されるだろう。
3)出来高決定
取引価格は、商品やサービスを顧客に移転することと引き換えに、会社が獲得する権利のある対価格によって決定される。取引価格に研究、開発、規制、ビジネスマイルストーンなどの可変対価格を含む範囲で、当社はこのような金額を受け取る可能性があるかどうかを判断し、重大な収入逆転のリスクはありません。当社が当該等の金額を受け取る可能性があると判断できない場合は、当社は関連可変対価格を制限し、取引対価から除外します。取引対価格に制限されている部分を決定する際には,同社は関連研究,開発,規制,ビジネスマイルストーンの実現の可能性と不確実性を考慮し,研究と臨床開発の性質および基本計画の段階を考慮している。この評価は各報告書期間に行われた。この評価を行う際には、会社は、業界出版物や他の関連要因からの情報を含む入手可能な内部および外部情報を考慮している。変動による価格制限の変化は当期に確認された収入額に実質的な影響を与える可能性がある。
4)取引対価格を契約に割り当てる履行義務
契約が単一履行義務を含む場合、取引対価格全体をその単一履行義務に割り当てる。複数の履行義務を含む契約要求は、相対的に独立した販売価格に基づいて取引対価格を各履行義務に割り当て、取引対価格が可変でない限り、契約義務または単一履行義務の一部を構成する異なるサービスに完全に割り当てられる基準に適合する。受信した対価格を相対的に独立した販売価格に応じて単独の履行義務の間に割り当てる.これらの推定された独立販売価格を決定する際には,ライセンスに対して,管理層が連携開発計画ごとに確率に重み付けしてキャッシュフローを割引するという仮定を含む多くの重要な判断を行う.推定された独立販売価格は,科学的に成功する確率,割引率,予測キャッシュフローの基礎を構成する何らかの仮説など,仮説の変化に敏感である.これらの仮定を作成する際には、管理層は、同一業界内の他の基準会社からの情報や他の関連要因を含む利用可能な内部および外部情報を考慮する。これらの仮定の変更は,取引が履行義務における価格の分配や収入を確認する金額や時間に大きな影響を与える可能性がある.
5)会社が契約履行義務を履行した場合または義務履行時に収入を確認する
当社は契約履行義務の性質に基づき、時間とともに又はある時点で履行義務を履行する。顧客がエンティティ業績によって提供される利益を同時に受け取り、消費する場合、エンティティ業績が資産を作成または強化する際に顧客制御資産を作成または強化する場合、またはエンティティ業績が代替エンティティ用途に代替可能な資産を作成しておらず、エンティティがこれまでに完了した実績を強制的に支払う権利を有する場合、収入は時間の経過とともに確認される。エンティティが一定期間義務を履行していない場合、ある時点で、約束された貨物またはサービスの制御権を顧客に移すことによって、関連する履行義務を履行する。
F-10
契約残高
会社が顧客が対価または満期支払いを支払う前に貨物またはサービスを顧客に譲渡する場合、会社は契約資産を確認するが、口座または他の入金として示されるいかなる金額も含まない。契約資産は、実体が顧客に譲渡する商品又はサービスの対価格権利である。契約負債または繰延収入は、主に私たちが支払いを受けたが、関連する履行義務を履行していない契約に関するものだ。2022年12月31日と2021年12月31日まで、契約資産規模は大きくない。2022年12月31日現在繰延収入として記録されている契約負債はい$です
協力手配
同社はASC 808に基づいて共同経営活動を代表する連携協定の要素を記録している。そのため,双方を代表する積極的な参加者の活動と,双方とも活動のビジネス成功に依存した重大なリスクとリターンに直面する協力合意の要素が,協力手配として記録されている.
同社は、協力協定に関連するビジネス活動の正確な陳述およびtと損失分担を評価している。ASC 808は、協力手配の当事者間の支払いが他の権威ある会計文献の範囲内にない場合、損益表の分類は、その手配の性質、その業務運営の性質、およびその手配の契約条項に基づくべきであると規定している。これらの支払いが他の権威ある会計文献の範囲内でない場合、これらの支払いの損益表分類は、権威ある会計文書との類比に基づくべきであり、適切な類比がない場合は、合理的、合理的、かつ一貫して適用される会計政策選択に基づくべきである。
VertexプロトコルによりASC 808項に入金された連携コストは,総合業務報告書と総合(損失)収入のうち“連携費用純額”にランクインしている。Vertexプロトコルのさらなる検討については、これらの連結財務諸表の付記8を参照されたい。
研究と開発費
研究及び発展活動を行うために発生したコストは、賃金及び福祉、施設コスト、間接費用、臨床研究及び関連臨床製造コスト、免許及びマイルストーン費用、契約サービス及びその他の関連コストを含み、研究及び発展コストは支出に計上される。研究·開発コストは,協力者への前払い費用やマイルストーンを含め,発生時にも費用を計上する。支払われた金額が発生したコストを超えた場合には、会社は前払い費用を記録する。当社は契約研究機関、臨床研究場所、実験室、コンサルタント或いは臨床試験活動を展開する他の臨床試験サプライヤーがまだ領収書を発行していないサービスと発生に関する費用の見積もりに基づいて、臨床試験活動のコストを計算しなければならない。同社は,パートナーの連携活動に関する精算を確認し,ASC 808により入金されたVertexプロトコルでの連携コストは含まれておらず,サービス提供期間中の研究や開発費用の削減としている。パートナーの連携活動に関するコストはASC 808項目で比を占め,研究開発費に計上されており,今年度までの年度では顕著ではなかったDecember 31, 2022, 2021 and 2020
賃貸借証書
当社はASC 842に基づいてそのリース契約を会計処理し、賃貸借証書またはASC 842。手配開始時に、当社は手配に存在する独特の事実と状況に基づいて、その手配が賃貸契約であるかどうかを決定する。レンタル期間が1年を超えるリースは、貸借対照表で使用権資産および短期·長期賃貸負債であることが確認されている(場合によっては)。その会社は融資リースを持っていない。
経営リース負債及びそれに応じた使用権資産は、当初、予想される余剰賃貸期間のリース支払い現在値に基づいて入金される。受け取った報酬などの項目については、使用権資産を何らかの調整する必要がある可能性がある。レンタル契約に隠されている金利は一般的に確定しにくい。したがって、当社はその逓増借款金利を利用して賃貸支払いを割引しており、このことは、類似経済環境下で、当社が担保に基づいて同じ通貨、類似期限で賃貸支払いの固定金利を借り入れていることを反映している。当社は、直線賃貸料支出または受信した任意の奨励について使用権資産を調整し、レンタル開始または移行日に有効な同じ増量借款金利を使用して純現在値で賃貸負債を再計量することが予想される。
F-11
当社は貸借対照表でオリジナル期限が1年以下であることを確認しない賃貸を選択しました。同社はレンタルスケジュールを評価する際には通常初期レンタル期間のみを含む。合理的な継続確実性がない限り、契約更新の選択は当社の評価に含まれません。
当社が発効日に作成した仮定は、何らかの事件(改訂賃貸借契約を含む)が発生した後に再評価されます。レンタル変更がテナントにオリジナルリースに含まれない追加使用権を付与し、レンタル支払いが追加使用権の独立価格に適合する場合、レンタル修正は別個の契約を生成する。1つのリース変更が別個の契約を生成した場合、その会計処理方式は新規契約と同じである。
権益に基づく報酬費用
同社の株式ベース報酬計画は、株式オプション、制限株式単位、制限株式報酬を含む報酬を付与する。補助金は役員を含む従業員と非従業員に付与される。
当社はASCテーマ718に基づいて株式に基づく報酬報酬を計算している報酬--株式報酬またはASC 718。ASC 718は、従業員株式オプションおよび制限株式単位の付与、および既存の株式オプションの修正を含む、従業員および非従業員取締役に支払われるすべての株式ベースの支払いを要求し、その公正価値に基づいて総合経営報告書および包括的(損失)収入で確認しなければならない。同社はブラック·スコアーズオプション定価モデルを使用して、付与されたオプションの公正価値を決定した。
当社は没収発生時に会計処理を行い、付与時に没収を推定するのではなく、実際の没収がその推定と異なる場合は、その後の期間にその推定を改訂する。財務諸表で確認された株式ベースの報酬支出は、業績やサービス条件を満たすことが期待される報酬に基づいている。
同社の株式奨励はサービスまたは業績に基づく付与条件によって制限される。サービスを基礎とする帰属条件の従業員、取締役及び非従業員が獲得した奨励に関する補償支出は、付与日に応じて関連サービス期間(一般に帰属期限)の公正価値を直線基準で確認する。業績条件に達する可能性がある場合には、奨励業績帰属条件の従業員に関する報酬支出は、付与日が必要なサービス期間中の公正価値に基づいて確認され、加速帰属法が採用される。
会社の支出は株式単位の奨励を従業員に制限し、奨励の公正価値は奨励の関連サービス期間内の直線に基づいている。
会社は、(I)期待株価変動、(Ii)期待奨励期間の計算、(Iii)無リスク金利、および(Iv)期待配当を含む主観的仮定の入力を要求する従業員、取締役、および非従業員に付与されたオプションの公正価値を推定するために、ブラック-スコアーズオプション定価モデルを使用する。株式を基礎とした全体の期待期間を奨励する完全な会社の特定の歴史と隠れた変動率データが不足しているため、当社は自分の変動率データのほかに、代表的な上場企業の予想変動率の推定に基づいている。これらの分析について、当社は、企業価値、リスクプロファイル、業界における地位、および株による報酬の期待寿命を満たすのに十分な歴史的株価情報を含む、自身と類似した特徴を有する会社を選択した。当社は、算出された株式奨励予想期間の同値期間に会社株を選定した1日当たりの市価を用いて履歴変動率データを算出する。同社は、その株価変動に関する十分な数の履歴情報が利用可能になるまで、このプロセスを適用し続ける。十分な履歴データが不足しているため、当社は、“簡略化”方法を使用して、その従業員株式オプションの期待期間、すなわち、期待期間がオプションの帰属期限および元の契約期間の算術平均値に等しいと推定する。オプション予想期間内の無リスク金利は、満期日に関連奨励の期待期限に見合った米国債をベースとしている。同社は配当金を支払ったこともなく、予見可能な未来に配当金を支払うことも望んでいない。
特許費用
特許出願を取得·起訴するコストおよび当社の知的財産権の保護に関する他の法的コストは、発生時に費用を計上し、当社の総合経営報告書では一般と行政費用に分類される。
F-12
所得税
所得税はASC主題740に基づいて記録されている所得税またはASC 740は、貸借対照法を使用して繰延税金を規定する。この方法によれば、繰延税金資産と負債は、資産と負債の財務報告と税務報告基準との差に基づいて決定され、制定された税率および法律を用いて計量され、これらの税率および法律は、差が逆転したときに発効することが予想される。既存の証拠の量に基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合、推定値減値準備が提供される。当社は既存の証拠を評価し、当社は繰延税金資産の全利益を実現できない可能性があると結論したため、当社が実現不可能と考えている繰延税金資産金額については、推定値準備を設けている。
ASC 740の規定によると、当社は不確定な税務頭寸を会計処理する。不確定な税収頭寸が存在する場合、当社は税収頭寸の税収割引を確認し、より実現可能にする。税収優遇がより実現可能かどうかの決定は,税収状況に基づく技術的利点および既存の事実や状況への考慮である。2022年12月31日と2021年12月31日まで会社はそうします
総合収益
普通株主は1株当たり純収益を占めなければならない
1株当たり基本純(損失)収益の算出方法は、普通株株主に帰属する純(損失)収益を当期発行普通株の加重平均で割る。1株当たりの純(損失)収益の算出方法は、普通株株主が占めるべき純(損失)収入を当期発行普通株の加重平均数で割ったものであり、在庫株方法を用いた既発行株式オプションや制限株式単位によるいかなる希薄化影響も含む。詳細は付記12を参照されたい。
新会計公告
指定された発効日から、財務会計基準委員会または会社が採用している他の基準作成機関は、時々新しい会計声明を発表する。当社は最近発表された指針を採用することがその総合財務諸表や開示に重大な影響を与える可能性があるとは考えていません。
3.有価証券
会社の現金等価物と有価証券の概要2022年12月31日と2021年12月31日公正価値記録(#ドルは含まれていません
F-13
2022年12月31日 |
|
償却する |
|
|
毛収入 |
|
|
毛収入 |
|
|
公正価値 |
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
会社債務証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
預金証書 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
|
|||
現金等価物合計 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
|
|
|
|
|||
会社債務証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
預金証書 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
政府支援企業証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
商業手形 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
有価証券総額 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
現金等価物と有価証券総額 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
2021年12月31日 |
|
償却する |
|
|
毛収入 |
|
|
毛収入 |
|
|
公正価値 |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
貨幣市場基金 |
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
|
||
会社債務証券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
預金証書 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
商業手形 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
現金等価物合計 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
会社債務証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
預金証書 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
政府支援企業証券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
商業手形 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
|
||
有価証券総額 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
現金等価物と有価証券総額 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
2022年12月31日と2021年12月31日まで未実現損失状態にある12カ月未満の有価証券の公正価値合計は$である
当社は、2022年、2022年、2021年12月31日まで、上記投資に重大な信用リスクがないと認定した。当社は取り戻すまでこのような証券を保有する意欲と能力があります。したがって,2022年12月31日および2021年12月31日までに,当社はその有価証券について信用に関する減価費用を計上していない.
4.公正価値計測
以下の表に当社の公正価値によって日常的に計量された金融資産の資料を記載し、この等公正価値の分類を以下のように示す2022年12月31日および2021年12月31日(単位:千):
F-14
|
|
公正価値に応じて計量する |
|
|||||||||||||
|
|
2022年12月31日 |
|
|||||||||||||
|
|
合計する |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
||||
現金と現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
貨幣市場基金 |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
||
会社債務証券 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
預金証書 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
会社債務証券 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
預金証書 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
政府支援企業証券 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
他の非流動資産 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
|
|
公正価値に応じて計量する |
|
|||||||||||||
|
|
2021年12月31日 |
|
|||||||||||||
|
|
合計する |
|
|
レベル1 |
|
|
レベル2 |
|
|
レベル3 |
|
||||
現金と現金等価物: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
現金 |
|
$ |
|
|
$ |
|
|
$ |
— |
|
|
$ |
— |
|
||
貨幣市場基金 |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
||
会社債務証券 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
預金証書 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
有価証券: |
|
|
|
|
|
|
|
|
|
|
|
|
||||
アメリカ国債 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
会社債務証券 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
預金証書 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
政府支援企業証券 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
商業手形 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
||
他の非流動資産 |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
評価レベルで第2レベルに分類される有価証券には、通常、米国債や政府機関証券、社債、商業手形が含まれる。同社は第三者価格源から得られた推定値を考慮することで、これらの有価証券の公正価値を推定している。
当社は3級に分類された株式証券を保有しており、当社の財務状況に大きな影響はありません。
5.財産と設備、純額
財産と設備の純額は以下の部分からなる(千計)
|
|
12月31日まで |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
コンピュータ装置 |
|
$ |
|
|
$ |
|
||
家具、固定装置、その他 |
|
|
|
|
|
|
||
実験室装置 |
|
|
|
|
|
|
||
賃借権改善 |
|
|
|
|
|
|
||
建設中の工事 |
|
|
|
|
|
|
||
財産と設備総額(毛額) |
|
|
|
|
|
|
||
減価償却累計 |
|
|
( |
) |
|
|
( |
) |
財産と設備の合計 |
|
$ |
|
|
$ |
|
||
2022年12月31日現在、2021年と2020年12月31日までの年間減価償却費用はい$です
F-15
6.課税料金
計算すべき費用には、以下の項目が含まれる
|
|
12月31日まで |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
賃金総額と従業員に関するコスト |
|
$ |
|
|
$ |
|
||
研究コスト |
|
|
|
|
|
|
||
許可料 |
|
|
|
|
|
|
||
専門費 |
|
|
|
|
|
|
||
知的財産権コスト |
|
|
|
|
|
|
||
課税財産と設備 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
合計する |
|
$ |
|
|
$ |
|
||
7.賃貸証書
当社は2015年6月、マサチューセッツ州ケンブリッジ市にある研究施設スペースのレンタルについて賃貸契約を締結し、レンタル開始日は
二零一六年五月、当社はマサチューセッツ州ケンブリッジ市にある主要なオフィス及び研究施設について分譲契約を締結し、開始日は
当社は2019年5月、マサチューセッツ州ケンブリッジ市でオフィス施設スペースレンタル契約を締結し、レンタル開始日は
2019年12月,Casebia治療有限責任組合(Casebia)は当社の完全子会社となった。そのため,Casebiaはマサチューセッツ州ケンブリッジ市に位置するオフィスと研究施設の分譲を当社に貸した。転貸後2021年に改訂されました
2020年5月、同社はマサチューセッツ州フレミンガムまたはフレミンガムリース会社に細胞療法製造工場を設立し、当社の研究用細胞療法候補製品を臨床および商業生産するためのレンタル契約を締結した。フレミンガム賃貸契約は#年で満期になります
2020年7月、当社はマサチューセッツ州ボストンにあるオフィスと実験室施設の賃貸契約を締結し、レンタル開始日は2021年6月1日、すなわち2020年レンタルとなった。レンタル開始時、会社は#ドルの使用権資産を記録した
当社はまた、選定された実際の方便に基づいて、スイスZugで資産や負債を記録していないオフィススペースを短期的に借りている。
会社はいくつかの研究と許可契約にレンタルを埋め込み、会社はこれらのプロトコルに対して使用権、資産、負債を記録した。会社の経営リース資産や負債総額と比較して、これらの手配は重要ではない。また、選択された実際の方便に基づいて、当社はその製造契約内のいくつかの短期賃貸が当社の貸借対照表に記録されていないことを確認した。
F-16
当社は、使用権資産と相応の負債を確認する際に、以下の推定を確認·評価した
下表は現在までの賃貸資産と負債をまとめたものです2022年12月31日および2021年12月31日(単位:千):
|
|
12月31日まで |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
資産 |
|
|
|
|
|
|
||
経営的リース資産 |
|
$ |
|
|
$ |
|
||
リース資産総額 |
|
|
|
|
|
|
||
負債.負債 |
|
|
|
|
|
|
||
現在のところ |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
|
|
|
|
||
当面ではない |
|
|
|
|
|
|
||
賃貸負債を経営し,当期分を差し引く |
|
|
|
|
|
|
||
リース総負債 |
|
$ |
|
|
$ |
|
||
研究と開発,一般と行政費用および12カ月までの分譲収入に含まれる経営リースコストをまとめた2022年12月31日、2021年、2020年(単位:千):
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
リースコストを経営する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
短期賃貸コスト |
|
|
|
|
|
|
|
|
|
|||
可変リースコスト |
|
|
|
|
|
|
|
|
|
|||
転貸収入 |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
純賃貸コスト |
|
$ |
|
|
$ |
|
|
$ |
|
|||
次の表は、賃貸負債の下で満期になった未割引支払いの満期日とこれらの負債の現在値をまとめています2022年12月31日(千):
|
|
合計する |
|
|
2023 |
|
|
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
その後… |
|
|
|
|
合計する |
|
$ |
|
|
現在価値調整 |
|
|
( |
) |
賃貸負債現在価値 |
|
$ |
|
|
次の表は,以下の日までのレンタル期間(年単位)と経営的リースの割引率をまとめている2022年12月31日および2021年12月31日:
|
|
12月31日まで |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
加重平均残余レンタル期間 |
|
|
|
|
|
|
||
加重平均割引率 |
|
|
% |
|
|
% |
||
F-17
下表は賃貸負債を計量する際に支払った現金をまとめたものであるDecember 31, 2022, 2021 and 2020 (単位:千):
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
賃貸負債に含まれる金額を計量するために支払う現金: |
|
|
|
|
|
|
|
|
|
|||
経営的リース使用の経営的キャッシュフロー |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
レンタル非現金プロジェクトを運営しています |
|
|
|
|
|
|
|
|
|
|||
使用権資産は賃貸借契約の見直しと見直しにより増加する |
|
|
|
|
|
( |
) |
|
|
|
||
経営性リース負債と引き換えに使用権資産 |
|
|
— |
|
|
|
|
|
|
|
||
レンタル改善は大家さんが直接支払います |
|
|
|
|
|
|
|
|
— |
|
||
8.重要な契約
Vertex PharmPharmticals Inc.とそのある子会社とのプロトコル
要約.要約
2015年10月26日、会社はVertexと戦略的協力、オプション、許可協定、または2015年の協力協定を締結した。2015年の協力協定は,同社のCRISPR/Cas 9遺伝子編集技術を用いてヒト疾患の潜在的遺伝原因に対する潜在的な新しい療法を発見·開発することに重点を置いている。
2017年12月12日、当社はVertexと2015年協力協定の第1号改正案(すなわち第1号改正案)と共同開発協定(JDA)を締結した。第1号改正案は、それ以外にも、2015年の協力協定のいくつかの定義と規定を改正し、共同司法審査と一致させ、2015年の協力協定で規定されたいくつかの目標に対してどれだけの選択権を行使したかを明らかにした。第1号修正案はまた、満期条項を含む2015年の協力協定の他の条項を改正した。
2015年の協力協定について、Vertexは払戻不可能な$を前払いした
2019年6月、当社はVertexと戦略的協力と許可協定、または2019年の協力合意を含む一連の合意を達成し、Duchenne筋ジストロフィー(DMD)と強直性筋ジストロフィー1型(DM 1)を治療する製品を開発し、商業化した。2019年の協力協定の条項によると、同社は払戻不可能な前払い$を受け取った
♪the the the会社には2019年の協力協定で発生する可能性のある任意の製品の将来の純売上高で等級別印税を受ける資格があります。DMD計画については,Vertexはすべての研究,開発,製造,商業化活動,およびすべての関連コストを担当している.DM 1プロジェクトについては,同社は特定のGUIDE RNA研究を行い,Vertexは他のすべての研究,開発,製造,商業化コストを担当している。新薬またはIND申請の提出を検討した後、同社はDM 1マイルストーンと特許権使用料を放棄することを選択することができ、共同開発と
F-18
共同商業化世界のすべてのDM 1製品は
二零一九年協力協定を履行するために、当社はVertexと二零一五年協力協定の第二次改訂、すなわち第二号改訂を締結した。このうち、第二号改訂は二零一五年協力協定のいくつかの定義及び条文を修正し、二零一九年協力協定と一致させ、二零一五年協力協定の下での協力目標の数及び身分を明らかにした。The CompanyとVertexは同意した
2019年10月、Vertex行使残り
2021年4月に、当社はVertexと“共同開発及び商業化協定”の改訂及び再記述に同意し、改訂及び再予約された共同開発及び商業化プロトコル、又は“A&R Vertex JDCA”を締結し、この合意により、双方は(A)協力の管理構造を調整し、それに基づいて各方面の責任を調整することに同意し、これにより、Vertexは指導者及び輸出計画に関するすべての意思決定(すなわち、制御)を持ち、(B)双方は輸出(前CTX 001と呼ぶ)の純利益及び純損失の分配を調整する限られていますが、
A&R Vertex JDCA計画が完了した取引について、当社は1ドル受け取りました
頂点プロトコルに関する記帳
F-19
“2015年協力協定”、“修正案1”、“JDA”を総称して“2015合意”、“2019年協力合意”、“修正案2”を総称して“2019年合意”と呼ぶ。2015年連携プロトコル、第1号修正案、第2号修正案、JDA、A&R Vertex JDCA、2019年連携プロトコルを総称してVertexプロトコルと呼ぶ
頂点プロトコルは、ASC 606によって定義されるようなクライアント·プロバイダ関係のコンポーネント、ASC 808によって定義されるような協調スケジュール、およびASC 730によって定義される研究開発コストを含む研究と開発, or ASC 730.
ASC 606での会計分析
A&R頂点JDCAの計算
契約的識別
A&R Vertex JDCAはJDAに対する契約修正を代表している.会計目的でA&R Vertex JDCAは単独の契約とされている。
義務を果たすことを確定する
同社は、A&R Vertex JDCAは、Vertexに追加のグローバル独占ライセンスを付与する単一の材料承諾を含むと結論した
成約価格の決定
取引価格は前金#ドルで構成されています
取引価格と履行義務の分配
履行義務の販売価格は会社のESSPによって決定されます。同社はExacel独自ライセンスのESSPを制定し、このような物品を定期的に独立して販売すれば、どのような価格で販売されるかを決定することを目的としている。
Exa-cel独占ライセンスのESSPは約$と決定されました
収入の確認
同社はExacelが機能性知的財産権を代表することを独占的に許可していると認定しているが、知的財産権はVertexが研究開発、製造と商業化の形式で機能或いは任務を実行できるようにしているからである。したがって,Exa−cel独占ライセンスに関する収入は2021年第2四半期譲渡時に確認される。
2019年の合意についての説明
2019年の合意は2015年の合意の契約修正です。したがって、会計目的で2019年協定と2015年協定が統合され、単一の手配とみなされている。2022年12月31日と2020年12月31日までの12ヶ月間、2019年の合意での取引は重要ではない。2021年12月31日までの12ヶ月間、会社は確認しました
当社は、2022年12月31日現在、2019年協議で上記で議論されたマイルストーンと特許使用料により残りの他のすべての可能な可変対価格が完全に制限されることを決定しました。会社は各報告期間内に取引価格を再評価するだろう。
F-20
Vertexプロトコルに関する収入が確認された
Vertexプロトコルにより確認された2022年12月31日までの年度収入重要ではありません。Vertexプロトコルにより確認された2021年12月31日までの年間収入は#ドル
2022年12月31日と2021年12月31日まであります
頂点合意下の未来の一里塚
その会社はVertex協定と関連したマイルストーンを受信する可能性があることを評価した。上述したように、同社は最高$を取得する資格があります
その会社は追加の潜在的な支払いを得る資格があり、最高で$に達する
その会社は追加の潜在的な支払いを得る資格があり、最高で$に達する
2022年12月31日まで、残りのすべてのマイルストーンは完全に制限されている。臨床開発の性質とCRISPR/CAS 9技術の段階に鑑み,研究と開発マイルストーンを獲得したイベントが実現するかどうかは不確定である。残りの研究、開発、規制マイルストーンは、著しい収入逆転が生じない可能性が高いまで制限されるだろう。ビジネスマイルストーンと使用料は主に知的財産権許可に関連しており、販売または使用に基づく敷居によって決定される。商業マイルストーンと特許権使用料は特許権使用料確認制限の下で入金され、制限された可変対価として入金される。同社は事業マイルストーンごとに印税確認制限を適用しており、その後のライセンス製品の販売(各マイルストーン実現)まで、一里塚ごとの収入は確認されていない。
ASC 808での会計分析
Vertexプロトコルについては,同社は,ASC 808項に,(I)A&R Vertex JDCAにおけるExacelに関する任意の移行サービス,(Ii)後続製品の研究開発サービス,および(Iii)委員会参加を含む共有製品の開発と商業化サービスを計上する連携要素を決定した.費用分担に関する影響は,合併業務表と総合(赤字)収入における連携費用純額に計上されている。
2022年、2021年、2020年12月31日までの年間で、会社は$を確認しました
F-21
A&R Vertex JDCAによると,会社は輸出計画で$$を超える指定コスト分を延期する権利がある
9.支払いの引受およびその他の事項
知的財産権協定
Charpentier許可プロトコル
2014年4月、当社はEmmanuelle Charpentier博士といくつかの技術許可協定を締結し、この協定に基づき、当社はCharpentier博士といくつかの知的財産権を共同で持ち、ヒト疾患の治療または予防製品を開発し、商業化した。これに関連して,Charpentier博士は,名義臨床マイルストーン支払い,第三者と達成された任意の再許可合意に基づいて受信した再許可支払いのより低い桁数パーセント,および当社とその関連会社および再許可譲受人許可製品およびサービスの年間純売上高に基づいて計算されたより低い桁数パーセントの特許使用料を得る権利がある。
特許譲渡協定
2014年11月、会社はCharpentier博士、Ines Fonfara博士、およびウィーン(総称して“譲渡者”と呼ぶ)と特許譲渡協定を締結し、この協定に基づいて、会社は米国特許出願第61/905,835号に要求される特定の特許権利のすべての権利、所有権、および権益を譲渡された。そのため,譲渡者はいくつかの低い1桁の臨床マイルストーン支払いと低い1桁の特許権使用料を得る権利があり,これは当社,その連属会社,特許所有者の許可製品とライセンスサービスの年間純売上高に基づいて計算される。
当社は2022年,2021年および2020年12月31日までの年度中に,Charpentier許可協定に基づいてCharpentier博士および特許譲渡協定に基づいて譲渡者に非実質金額の費用を支払い,これらの費用を研究および開発費と記す。
研究、製造、許可協定
同社はすでにいくつかの研究機関や会社を招聘し、同社の遺伝子編集技術の新たな交付戦略と応用を決定している。同社も多くのライセンス契約の当事者であり、これらの合意は大量の前払いを必要とし、将来の特許権使用料と潜在的なマイルストーン支払いを時々支払う必要があるかもしれない。また、同社は知的財産権協定の一方でもあり、これらの協定はメンテナンスとマイルストーン費用を時々支払う必要がある。また、同社は将来のサービス性能の前払いを要求するいくつかの製造協定の締約国である。
これらの合意に関連して、個々の製品に基づいて、取引相手は具体的な研究、開発、規制マイルストーンに基づいて最高8桁の低潜在的支払いを獲得する資格がある。また、個々の製品に基づいて、取引相手は、指定された年間販売敷居に基づいて潜在的なビジネスマイルストーン支払いを得る資格がある。潜在的な支払いは指定された年間売上高の敷居の一桁パーセントだ。取引相手には将来の純売上高から低い一桁の特許権使用料を得る資格があります。
場合によっては、将来いくつかのイベントが発生したり、イベントがあったりする場合、Vertexは最高$を得る資格があります
A&R Vertex JDCAによると,会社は$を延期した
F-22
その他の事項
2016年12月15日、会社はウィーン大学、Charpentier博士、Intellia治療会社、Cariou生物科学会社、ERSゲノム有限会社と会社の子会社と譲渡、許可、共同所有権と発明管理協定(“発明管理協定”)を締結した。発明管理協定によると、会社は特許維持、弁護、起訴に関連する費用を分担する義務がある。当社は2022年12月31日まで、2021年12月31日及び2020年12月31日までに発生しますエド$
訴訟を起こす
通常の業務過程において、当社は時々、当社の知的財産権(ある許可内の知的財産権を含む)、商業手配及びその他の事項に関連する訴訟、調査、法律手続き及び訴訟脅威に関連する。このような訴訟には準訴訟が含まれているかもしれません各方面間米国特許商標局および欧州特許庁の行政訴訟は、特定の認可内の知的財産権を含む会社の知的財産権に関連する。T.T上記のいずれの場合の結果も,是非曲直にかかわらず,本質的には不確実である.また、訴訟や関連事項はコストが高く、会社の経営陣や他の資源の注意を分散させる可能性があり、そうでなければ、これらの資源は他の活動に従事することになる。もし会社がこのような訴訟で勝訴できなければ、会社の業務、運営結果、流動資金、財務状況は不利な影響を受ける可能性がある。
10.配当金
その会社は所有している
|
|
|
|
12月31日まで |
|
|||||
株式タイプ |
|
条件資本 |
|
2022 |
|
|
2021 |
|
||
普通株 |
|
登録株 |
|
|
|
|
|
|
||
普通株 |
|
法定株 |
|
|
|
|
|
|
||
普通株 |
|
条件付き株式--債券または同様の債務ツール |
|
|
|
|
|
|
||
普通株 |
|
条件付き株式-従業員福祉計画 |
|
|
|
|
|
|
||
|
|
合計する |
|
|
|
|
|
|
||
登録株に含まれる
普通株発行
最近の公募株
2020年7月に同社は
市場の製品
2019年8月に、当社は公開市場販売協定を締結しましたSMJefferies LLCまたはJefferiesと協力し、この合意によれば、当社は時々Jefferiesをその販売代理、その普通株、または2019年8月の販売契約として、適宜提案および販売することができる。2019年8月、当社は米証券取引委員会に目論見書補充書類を提出し、総収益総額がドルを超えない普通株を随時発売した
F-23
2020年12月、2019年8月の販売協定について、当社は米証券取引委員会に目論見書補充書類を提出し、総収益総額がドルを超えない普通株を随時発売した
2021年1月に、当社は2020年にATM機の発行と販売に合わせて
2021年1月、2019年8月の販売協定について、当社は米証券取引委員会に株式募集説明書の補編を提出し、総収益総額がドル以下の普通株を随時発売した
当社は2021年12月31日までに発行·販売しております
2022年12月31日まで当社は共同で発行及び販売しております
普通株式は以下のような特徴を持つ
投票権
普通株保有者には権利がある
配当をする
株主総会が取締役会それぞれの提案に基づいて決議を下した場合、普通株式保有者は配当金を得る権利があり、会社が十分な自由に備蓄を分配できることが条件である。2022年12月31日まで,
清算する
普通株式保有者は、当社で任意の自発的または非自発的な清算、解散または清算または清算とみなされる事件が発生した場合、当社が株主に割り当てることができる資産を比例的に共有する権利がある。
11.株式ベースの報酬
オプションと贈与計画
2015年4月、会社株主は2015年株式オプション及び付与計画、又は2015年計画を承認し、2016年7月、会社株主は2016年株式オプション及びインセンティブ計画、又は2016年計画を承認した。2018年5月、会社株主は、2018年株式オプションおよびインセンティブ計画、または2018年計画(総称して“計画”と呼ぶ)を承認した。IPOの後
F-24
株式ベースの報酬費用
会社が確認した株式ベースの報酬支出総額は#ドル
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
合計する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
2022年12月31日まで一ドルあります
株式オプション
従業員に発行される各オプションの公正価値は、付与された日にブラック·スコアーズオプション定価モデルを用いて以下の加重平均仮定の下で推定される
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
付与したオプション |
|
|
|
|
|
|
|
|
|
|||
加重平均行権値 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
加重平均は日公正価値を付与する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
仮定: |
|
|
|
|
|
|
|
|
|
|||
予想変動率 |
|
|
% |
|
|
% |
|
|
% |
|||
予想期限(年単位) |
|
|
|
|
|
|
|
|
|
|||
無リスク金利 |
|
|
% |
|
|
% |
|
|
% |
|||
期待配当収益率 |
|
|
% |
|
|
% |
|
|
% |
|||
以下の表は、会社持分奨励計画における株式オプション活動(内的価値千計):
|
|
株 |
|
|
重み付けの- |
|
|
重み付けの- |
|
|
骨材 |
|
||||
2021年12月31日現在の未返済債務 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
キャンセルまたは没収 |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2022年12月31日に返済されていません |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日に行使できます |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
すでに帰属しており,2022年12月31日に帰属する予定である |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年,2021年および2020年12月31日までの年度内に行使される株式オプションの総内的価値(公平時価が行権価格を超える金額)はい$です
2022年12月31日まで購入可能なオプション
F-25
2022年12月31日まで購入可能なオプション
“会社”ができた
制限株 職場.職場
以下の表は、会社の持分奨励計画における制限的な株式単位活動をまとめたものである
|
|
株 |
|
|
重み付けの- |
|
||
2021年12月31日現在の未帰属残高 |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
|
|
||
既得 |
|
|
( |
) |
|
|
|
|
キャンセルまたは没収 |
|
|
( |
) |
|
|
|
|
2022年12月31日の未帰属残高 |
|
|
|
|
$ |
|
||
2022年、2021年、2020年12月31日までの年間で,帰属制限株式単位の総公正価値は$である
2022年に同社は
“会社”ができた
修正奨励
退職従業員が2022年、2021年、2020年12月31日までに保有する特定の持分奨励に対する持分奨励改正は、会社の株式ベースの報酬支出を修正することは重要ではない。
従業員株購入計画
F-26
12.普通株主が1株当たり純収益を占めるべき
1株当たり基本純(損失)収益の算出方法は、普通株株主に帰属する純(損失)収益を当期発行普通株の加重平均で割る。1株当たりの純(損失)収益の算出方法は、普通株株主が占めるべき純(損失)収入を当期発行済み普通株等価物の加重平均で割ることであり、在庫株方法を用いた既発行株式オプションと引受権証によるいかなる償却影響も含む。会社の純(赤字)収入とは、すべての列報期間中に普通株株主が占めるべき純(赤字)収入のことである。
次の表に基本純値と希釈純値の計算方法を示す(損失)締切日の1株当たり収益(単位は千で、1株および1株当たりの金額は含まれていない):
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
純収益 |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
基本加重平均普通株式発行済み |
|
|
|
|
|
|
|
|
|
|||
潜在的希釈証券の影響: |
|
|
|
|
|
|
|
|
|
|||
未平倉オプション |
|
|
— |
|
|
|
|
|
|
— |
|
|
無帰属制限普通株 |
|
|
— |
|
|
|
|
|
|
— |
|
|
希釈加重平均普通株式発行 |
|
|
|
|
|
|
|
|
|
|||
普通株1株当たりの純収益--基本 |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
普通株1株当たりの純収益--減額 |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
1株当たり純(損失)収益を計算する際、同社は次の表の証券を計算していないが、時期ごとに逆希釈の影響が生じるからである
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
未平倉オプション |
|
|
|
|
|
|
|
|
|
|||
無帰属制限普通株 |
|
|
|
|
|
|
|
|
|
|||
ESPP |
|
|
|
|
|
|
|
|
|
|||
合計する |
|
|
|
|
|
|
|
|
|
|||
13.401(K)貯蓄計画
当社は2016年11月に国税法第401(K)節(“401(K)計画”)に基づいて固定払込貯蓄計画を構築した。401(K)計画は、規定された最低年齢およびサービス要件に適合するすべての従業員をカバーし、参加者が税引前に年間給与の一部の支払いを延期することを可能にする。その会社は$を貢献した
14.所得税
当社はアメリカ連邦と各州の企業所得税、及び外国親会社と外国子会社が設立された外国司法管轄区の税収を納めなければなりません。
税引き前純収益
ここ数年で2022年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2020年12月31日、2020年12月31日、2022年12月31日、2020年12月31日、2022年12月31日、2022年12月31日、2021年12月31日、2020年12月31日、2021年12月31日、2021年12月31日、2022年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2020年12月31日、2022年12月31日、2021年12月31日、2020
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
国内では |
|
$ |
|
|
$ |
|
|
$ |
|
|||
外国.外国 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
合計する |
|
$ |
( |
) |
|
$ |
|
|
$ |
( |
) |
|
所得税の収益(計上)には、以下の内容(千計)が含まれる
|
|
十二月三十一日までの年度 |
|
F-27
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
当期所得税: |
|
|
|
|
|
|
|
|
|
|||
連邦制 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
状態.状態 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
外国.外国 |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
当期所得税総額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
繰延所得税: |
|
|
|
|
|
|
|
|
|
|||
連邦制 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
状態.状態 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
外国.外国 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
繰延所得税総額 |
|
|
|
|
|
( |
) |
|
|
( |
) |
|
所得税優遇総額 |
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
法定企業所得税税率で計算される所得税費用と締切年度の実際所得税税率との入金2022年12月31日、2021年12月31日、2020年12月31日:
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
法定税率下の所得税費用 |
|
|
% |
|
|
% |
|
|
% |
|||
州所得税、連邦福祉を差し引いた純額 |
|
|
% |
|
|
( |
)% |
|
|
% |
||
差し引かれない費用 |
|
|
( |
)% |
|
|
% |
|
|
% |
||
外貨利回り |
|
|
( |
)% |
|
|
% |
|
|
( |
%) |
|
法律とアメリカで公認されている会計基準は永久的に違います |
|
|
% |
|
|
% |
|
|
% |
|||
株に基づく報酬 |
|
|
( |
)% |
|
|
( |
%) |
|
|
% |
|
遅延金利変動の影響 |
|
|
( |
)% |
|
|
% |
|
|
% |
||
研究単位 |
|
|
% |
|
|
( |
)% |
|
|
% |
||
評価免除額を変更する |
|
|
( |
)% |
|
|
( |
)% |
|
|
( |
%) |
有効所得税率 |
|
|
|
|
|
% |
|
|
( |
%) |
||
連邦法定料率はスイスの混合会社のサービス料率を反映する。
繰延税項は、財務諸表の資産と負債ベースと所得税との一時的な差異で確認されている。同社の繰延税金資産の重要な構成要素には、以下の項目(千計)が含まれる
|
|
十二月三十一日までの年度 |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失が繰り越す |
|
$ |
|
|
$ |
|
||
課税項目と準備金 |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
|
|
|
|
||
その他繰延税金資産 |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
研究単位 |
|
|
|
|
|
|
||
繰延税金資産総額 |
|
|
|
|
|
|
||
推定免税額を差し引く |
|
|
( |
) |
|
|
( |
) |
繰延税項目純資産 |
|
|
|
|
|
|
||
繰延税金負債: |
|
|
|
|
|
|
||
減価償却 |
|
|
( |
) |
|
|
( |
) |
経営的リース資産 |
|
|
( |
) |
|
|
( |
) |
無形資産 |
|
|
( |
) |
|
|
( |
) |
その他繰延税金負債 |
|
|
( |
) |
|
|
( |
) |
繰延税金負債総額 |
|
|
( |
) |
|
|
( |
) |
長期繰延税金 |
|
$ |
( |
) |
|
$ |
( |
) |
F-28
当社はすでにその繰延税金資産の現金化に影響するプラスと負の証拠を評価した。同社の世界的な経営赤字の歴史によると、同社は、米国と非米国繰延税金資産の利益が実現できない可能性が高いと結論した。2022年と2021年12月31日までに当社はスイスやイギリスでの繰延税項目純資産について全額評価を提供する準備をしている。同社はまた、既存の繰延税金負債が現金化できない米国の繰延税金資産に評価準備金を提供しており、これらの計画がいつ打ち切られるかに基づいている。推定免税額は#ドル増加した
2022年12月31日まで会社が所有しています
2022年12月31日までこの会社はアメリカ国内の連邦研究と開発信用繰越$を持っています
米国会計基準第740条は、企業財務諸表で確認された所得税の不確実性について会計説明し、納税申告書に採用されているまたは予想される納税立場の最低確認敷居と計量を規定している。
2022年12月31日まで同社には未確認の税収割引総額$があります
税収優遇総額の変化が確認されていない場合は以下のとおりである(千計)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
年初残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
当期税収が増加する |
|
|
|
|
|
|
|
|
|
|||
前の期間の税収が増加する |
|
|
|
|
|
— |
|
|
|
— |
|
|
当期税収頭寸減少額 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
前期税収頭寸減少額 |
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
年末残高 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
同社はアメリカ連邦司法管轄区、マサチューセッツ州、カリフォルニア州、一部の非アメリカ司法管轄区に所得税申告書を提出した。同社は2018年12月31日までの納税年度に米国連邦、マサチューセッツ州、カリフォルニア州、非米国所得税当局の審査を受けている。以前の納税年度に発生した研究は閉鎖審査を免除した後、すでにまたは将来の期間に使用されれば、未来の審査時に調整することができる。同社はすべての年に米国管轄区域の主管部門ではない所得税審査を受けなければならない。
F-29