アメリカです
アメリカ証券取引委員会
ワシントンD.C.,20549
表
(マーク1)
1934年証券取引法第13条又は15条に基づいて提出された年次報告 |
本財政年度末まで
あるいは…。
1934年証券取引法第13条又は15条に基づいて提出された移行報告 |
移行期になります 至れり尽くせり
手数料書類番号
(登録者の正確な氏名はその定款に記載)
|
||
(登録設立又は組織の国又はその他の管轄区域) |
|
(国際税務局雇用主身分証明書番号) |
|
|
|
(主にオフィスアドレスを実行) |
|
(郵便番号) |
登録者の電話番号は市外局番を含んでいます(
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル |
|
取引 記号 |
|
登録された各取引所の名称 |
|
|
同法第12条(G)により登録された証券:なし
登録者が証券法第405条に規定する有名な経験豊富な発行者である場合は、再選択マークで表示してくださいはい、そうです ☐ ☒
登録者がこの法第13又は15(D)条に従って報告書を提出する必要がないか否かを、再選択マークで示すはい、そうです ☐
登録者が(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に1934年の証券取引法第13条または15(D)条に提出されたすべての報告書を再選択マークで示すかどうか、および(2)このような提出要求を過去90日以内に遵守してきた
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
|
☒ |
|
ファイルマネージャを加速する |
|
☐ |
|
|
|
|
|
|||
非加速ファイルサーバ |
|
☐ |
|
小型報告会社 |
|
|
|
|
|
|
|
|
|
新興成長型会社 |
|
|
|
|
|
|
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する☐
登録者が報告書を提出したかどうかを再選択マークで示し、その経営陣が“サバンズ-オクスリ法案”(“米国連邦法典”第15編、第7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する。この評価は、その監査報告書を作成または発行する公認会計士事務所によって行われる
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)はい、そうです ☐
登録者の非関連会社が保有する普通株の総時価は,当該株の2022年6月30日の終値に基づくナスダックによると、1ドルです
登録者2023年1月31日までの普通株流通株数かつては…
引用で編入された書類
Atara生物治療会社
カタログ
|
|
ページ |
第1部 |
|
|
第1項。 |
業務.業務 |
6 |
第1 A項。 |
リスク要因 |
33 |
項目1 B。 |
未解決従業員意見 |
77 |
第二項です。 |
属性 |
77 |
第三項です。 |
法律訴訟 |
77 |
第四項です。 |
炭鉱安全情報開示 |
77 |
|
|
|
第II部 |
|
|
五番目です。 |
登録者普通株市場、関連株主事項及び発行者による株式証券の購入 |
78 |
第六項です。 |
[保留されている] |
79 |
第七項 |
経営陣の財務状況と経営成果の検討と分析 |
80 |
第七A項。 |
市場リスクの定量的·定性的開示について |
91 |
第八項です。 |
財務諸表と補足データ |
92 |
第九項です。 |
会計と財務情報開示の変更と相違 |
125 |
第9条。 |
制御とプログラム |
125 |
プロジェクト9 B。 |
その他の情報 |
128 |
プロジェクト9 Cです。 |
検査妨害に関する外国司法管区の開示 |
128 |
|
|
|
第三部 |
|
|
第10項。 |
役員·幹部と会社の管理 |
129 |
第十一項。 |
役員報酬 |
129 |
第十二項。 |
特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 |
129 |
十三項。 |
特定の関係や関連取引、取締役の独立性 |
129 |
14項です。 |
最高料金とサービス |
129 |
|
|
|
第4部 |
|
|
第十五項。 |
展示·財務諸表明細書 |
130 |
第十六項。 |
表格10-Kの概要 |
134 |
2
前向きに陳述する
このForm 10-K年度報告書には、“1995年米国個人証券訴訟改革法”安全港条項に適合する前向きな陳述が含まれている。このような展望性陳述は私たちの意図、信念或いは現在の予想を代表し、リスク、不確定要素及びその他の要素に関連し、実際の結果とある事件の時間はこのような展望性陳述の明示或いは暗示の未来の結果とは大きく異なる可能性がある。場合によっては、これらの陳述は、“信じる”、“可能”、“予想”、“推定”、“継続”、“予想”、“意図”、“可能”、“将”、“プロジェクト”、“予測”、“計画”、“予想”またはこれらの言葉の否定または複数の形態または同様の表現によって識別することができる。前向きな陳述は、以下の態様に関する陳述を含むが、これらに限定されない
3
これらの陳述は現在の予測に過ぎず、既知および未知のリスク、不確実性の影響を受ける可能性があり、高価で時間のかかる薬品開発プロセスおよび臨床成功の不確実性に関連するリスクおよび不確実性を含むが、これらのリスクおよび不確実性は、(I)南カリフォルニアおよびコロラド州での私たちの運営、および臨床試験場所での私たちの運営、ならびに私たちの第三者メーカー、契約研究組織、または私たちと業務を往来する他の第三者の業務または運営、(Ii)私たちが資本を得る能力、および(Iii)私たちの普通株の価値を含む、私たちの業務、研究、臨床開発計画および運営に著しく影響する可能性がある。私たちの資本資源の充足性と追加資本に対する需要、およびその他は私たちまたは私たちの業界の実際の結果、活動レベル、業績、あるいは成果を展望性陳述で予想されているのとは大きく異なる要素を招く可能性がある。私たちはこの報告書のタイトル“1 a”の下でその中の多くの危険についてより詳細に議論した。危険要素“とこの報告書の他の部分。あなたは未来の事件の予測として前向きな陳述に依存してはいけない。新たなリスク要因や不確定要因が時々出現する可能性があり、経営陣がすべてのリスクや不確定要因を予測することは不可能である。
この10-K表年次報告では、文意が他に指摘されているほか、“Atara”、“Atara BioTreateutics”、“Company”、“We”、“Our”、“Us”はいずれもAtara BioTreateutics、Inc.およびその子会社を指す。
リスク要因をまとめる
私たちの業務は多くのリスクと不確定要素の影響を受けており、これらのリスクと不確定要素は私たちの業務、財務状況、あるいは経営結果に重大な悪影響を及ぼす可能性がある。このような危険はタイトル“1 A”の下でより包括的に描写されている。リスク要因“および本報告書の他の部分は、他の部分を含む:
4
5
部分 I
項目1.B有用性
概要
Atara BioTreateuticsはT細胞免疫療法の先駆者であり、その新型同種異体EBウイルス(EBV)T細胞プラットフォームを用いて癌と自己免疫疾患患者のための変革性療法を開発した。Tabelecleucelは私たちがアメリカで第三段階の臨床開発の主要なプロジェクトであり、すでに欧州委員会(EC)のマーケティング許可(MAA)を獲得し、独自名のEbvalloで欧州連合(EU)で商業販売を行っている。われわれは最先端の同種異体T細胞免疫治療会社であり,高度に満たされていない医療ニーズの高い患者に迅速に既製の治療を提供する予定である。著者らのプラットフォームはEBV T細胞の独特な生物学を利用し、そして統合工学キメラ抗原受容体(CARS)或いはT細胞受容体(TCR)を通じて広範なEBV駆動疾患或いはその他の深刻な疾患を治療する能力がある。Ataraは、強力な導管を作成するためにTCRまたはHL A遺伝子編集を必要としないこのプラットフォームを適用している。私たちの戦略的なポイントは
上述の戦略重点以外に、著者らは多くの臨床と臨床前プロジェクトがあり、ATA 2271を含み、腫瘍抗原間硫黄蛋白を発現する固形腫瘍に対する自己CAR T免疫療法は、現在第一段階に開発されている;及びATA 3271は、間硫黄蛋白に対する同種異体CAR T免疫療法であり、現在臨床前開発段階にある。
我々のT細胞免疫治療プラットフォームは同種異体と自己プログラムを行う能力を含み、広範な標的と疾患に適用可能である。我々の既存の同種異体T細胞プラットフォームは、患者が必要とする前に製造され、在庫に貯蔵されたT細胞免疫治療製品を迅速に提供することができ、各製造されたバッチ細胞は無数の潜在的な患者に治療を提供することができる。これは自己治療とは異なり,自己治療では,個々の患者自身の細胞が抽出されなければならず,体外で遺伝子改変を行い,患者に返送するには複雑な物流ネットワークが必要である。われわれの同種移植計画では,患者独自の免疫状況に応じて適切な細胞集を選択して使用した。私たちの契約製造組織(CMO)はTABCELの商業生産鑑定活動を完了しており、私たちの他の契約製造組織は現在TABCELの商業生産鑑定活動を完了しており、同時に私たちは私たちの商業製品供給戦略に基づいて在庫を構築している。
2021年10月、我々はPierre Fabreと商業化協定(Pierre Fabre商業化協定)を締結し、これにより、Pierre Fabreに独占的、限られた分野の許可を付与し、規制承認後にヨーロッパでEbvalloを商業化·流通し、中東、アフリカ、東欧、中央アジアで新興市場を厳選した。私たちは北米、アジア太平洋地域、ラテンアメリカのTabcelを含む他の主要市場に完全な権利を保持している。Pierre Fabre商業化プロトコルの想定によると,Pierre Fabreと(I)製造·供給プロトコル(Ii),薬物警戒プロトコル(Iii),品質プロトコルをそれぞれ締結し,Pierre Fabreとの協力関係をさらに進める。2022年9月、EBV+PTLDのためのEbvalloの使用をECが承認し、その後、Pierre FabreにMAA移転申請を提出した後、Pierre Fabreから3,000万ドルのマイルストーン支払いを追加的に取得し、(I)領土純売上高に対するEbvalloの資格のある特許使用料の割合、および(Ii)Pierre Fabreが購入したEbvalloの供給価格値上げと引き換えに、Pierre Fabre商業化協定を改正した。また,Pierre Fabre商業化プロトコルに従ってPierre Fabreに何らかのサービスを提供する期限を延長することにも同意した.2022年12月、私たちはPierre Fabre商業化協定に従って、Ebvalloで特許使用料といくつかのマイルストーンの権利の一部をHCR Molag Fund L.P(HCRx)に売却し、総投資額は3100万ドル、上限はHCRx総投資額の185%~250%である。
2020年12月に,吾らはバイエルと研究,開発および許可協定(バイエル許可協定)を締結し,これにより,吾らは当社とその連属会社が所有または制御しているATA 2271およびATA 3271またはATA 3271に関する適用特許およびノウハウに基づいて,バイエルに独占的な限られた分野許可を付与した。2021年3月には
6
バイエル許可協定によると,我々はバイエルと(I)製造·供給協定(バイエル製造協定),(Ii)薬物警戒協定,(Iii)品質協定,および(Iv)技術移転協定を締結し,バイエルとの協力をさらに進めている。バイエル許可プロトコル,製造とプロビジョニングプロトコルおよび技術移転プロトコルを総称してバイエルプロトコルと呼ぶ.2022年5月、バイエルはベイヤ協定の終了決定を通知し、2022年8月2日にバイエルと終了、改訂、計画譲渡協定(バイエル終了協定)を締結し、ベイヤ協定を終了し、ATA 2271とATA 3271の全製品開発権をAtaraに返還し、2022年7月31日から発効した。
著者らはまた、スローン-キャトリン癌センター(MSK)、クイーンズランド医学研究所理事会(QIMR Berghofer)とH.Lee Moffitt癌センターと研究所(Moffitt)を記念するなど、有力な学術機関と研究協力を行い、これにより、斬新かつ独自の技術とプロジェクトの権利を獲得した。
我々のカリフォルニア州千樫市(ARC)とコロラド州オロラ市の研究機関には,我々の翻訳と臨床前科学,分析開発と過程科学機能が含まれている。これらの施設は私たちの製品ライン、技術開発を支持し、私たちの同種異体細胞治療プラットフォームを利用して革新を推進する。
2022年1月、富士フイルムカリフォルニアバイオテクノロジー社(FDB)および富士フイルムホールディングス米国(Fujifilm Holdings America Corporation)と資産購入協定を締結し、カリフォルニア州千どん市にあるAtara T細胞運営·製造施設(ATOM施設)関連資産のすべての権利、所有権、権益を1億ドルの現金で売却し、資産購入協定(富士フイルム取引)に基づいて可能な取引後調整を行った。富士フイルム取引は2022年4月4日に完了し,ATOM施設のリースをFDBに譲渡したことが富士フイルム取引の完了につながっていた。また、取引終了時に発効し、最長10年間延長できるFDB(Fujifilm MSA)とプライマリサービスとプロビジョニング協定を締結しました。富士フイルムMSAによれば、FDBはcGMP規格に従って特定の数の細胞治療製品(承認された場合)および候補製品を提供する。富士フイルムMSAはFDBから製品と候補製品だけを購入することを要求していません。患者に対する私たちの期待とEUの製品に対する需要によると、私たちの現在のEbvallo在庫は2023年末までのEUの商業需要を満たすのに十分だと信じている。
我々はまた,2019年12月に締結したビジネス製造サービスプロトコル(CRL MSA)に基づいてCharles River実験室(CRL)と連携している.CRL MSAにより、CRLは私たちの製品といくつかの候補製品に製造サービスを提供します。2023年2月、CRL MSAを修正し、期限を2023年9月30日の早い時期に延長するか、またはいくつかのロットの製品および候補製品を受信します。
我々は製品やサービスに対してキャンセル不可能な最低約束があるが,臨床研究組織やCMOと1年以上の契約を締結しなければならない。
2022年8月、私たちの活動を研究開発を中心としたより簡素化された組織として、私たちの革新的なパイプをさらに推進し、現金消費を削減するために、約20%のリストラを発表しました。リストラには約600万ドルの総再編費用が含まれる予定で、主に解散費、カリフォルニア労働者調整·再訓練通知法に基づく60日間の通知期間の賃金、退職後しばらくの持続医療保険が含まれる。ほとんどの場合、解散費は2022年10月に一度に支払われる。通知された従業員の中には、昇給の形で離職福祉を提供することが規定されている雇用協定があり、これらの福祉は2022年10月から2023年11月までの間に支払われる。すべての解散費は現金支出です。
2022年12月、我々はデラウェア州有限組合企業HCR Molag Fund,L.P.(HCRx)と購入契約(HCRx協定)を締結した。HCRx協定の条項によると、HCRxがEbvalloの部分階層、販売に基づく特許権使用料、金額の中央値の1桁から重要な2桁まで、いくつかの記念碑的な支払いと引き換えに、総投資額3,100万ドルを受け取り、両者ともPierre Fabre商業化協定に従ってPierre Fabreによって支払われている。HCRx協定によると、HCRxに支払われる特許使用料およびマイルストーン総額の上限は、HCRx総投資額の185%~250%であり、これは、このような特許使用料およびマイルストーンのスケジュールに依存する。
7
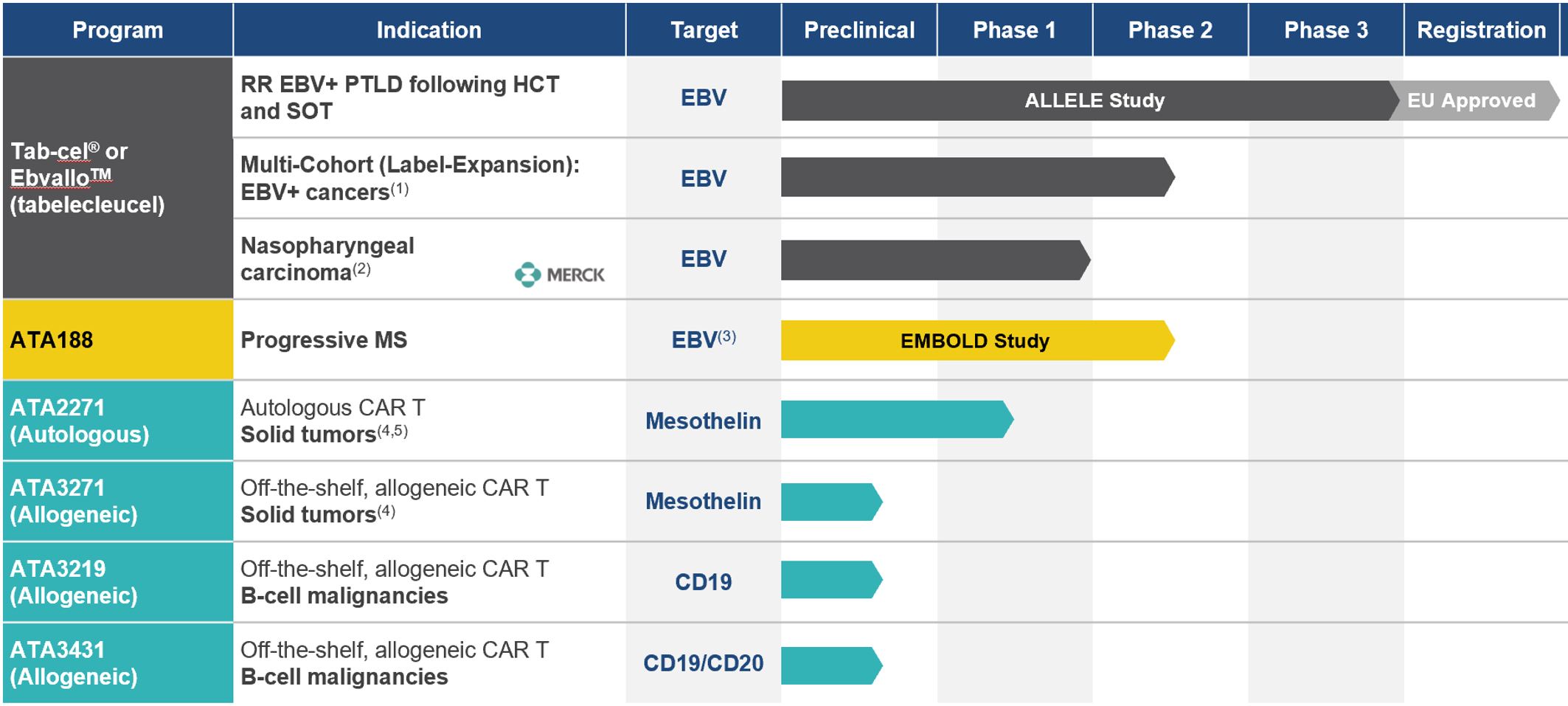
パイプ.パイプ
私たちの販売ルートの概要は以下の通りです
EUのEbvalloを除いて、このような調査機関はどんな規制機関の承認も受けていない。治療効果と安全性はまだ確定されていない。
EBV+PTLD:EBV関連性移植後リンパ増殖性疾患;RR:リツキシマブ再発/難治;HCT:異遺伝子造血細胞移植;SOT:実体臓器移植
私たちはピエール·ファブレと合意してTab-celを商業化しました®ヨーロッパ、中東、アフリカ、その他の選りすぐりの新興市場のためのEBV+癌。
その他の計画:EBVワクチン、その他の固形腫瘍、感染症計画
Ebvallo(Tab-cel®)
EBV+PTLD
最初のヒト腫瘍ウイルスとして発見されて以来、EBVはすでにリンパ腫と他の癌を含む一連の疾病の発展と関係がある。EBVは人の群れの中に広く存在し、そして一生、無症状の感染として持続的に存在する。健康な個体では,T細胞の一部がEBVの制御に取り組んでいる。対照的に、免疫機能が低下した患者、例えば造血細胞移植(HCT)や固形臓器移植(SOT)を受けた患者は、EBVを制御する能力が低下する。もし適切な免疫モニタリングがなければ、EBV変換した細胞はいくつかの患者で増殖する可能性があり、そしてEBV+PTLDと呼ばれる侵襲性、生命に危害を及ぼす癌を招く。HCT後に発生したPTLD症例のほとんどがEBV陽性であったが,SOT後に発生したPTLD症例の約60%がEBV陽性であった。
8
歴史研究により、EBV+PTLD患者の中で、リツキシマブ或いはリツキシマブプラス化学療法に失敗した患者に対して、治療を改善する医療需要は非常に高く、約40%~60%の患者は第一線の治療に対して反応或いは進展がない。リツキシマブによる一次治療に失敗したHCT後EBV+PTLD患者では,予想される中位総生存期間は約1.7カ月であったが,リツキシマブによる第一線治療に失敗したSOT後EBV+PTLD患者では,中位総生存率は約3.3カ月であった。メロワ治療に失敗したEBV+PTLD患者の中で、患者の虚弱と化学療法に関連する深刻な毒副作用のため、化学療法の使用はよく治療に関連する死亡率と著しい相関がある。我々の市場研究によると、2019年にアメリカでは数百名のEBV+PTLD患者がメロワ或いはメロワプラス化学療法を通過できなかったと推定されている。
Tabcell-cell®(Ebvallo)EBV+PTLD用
2015年6月、独占ライセンス契約に基づき、MSKから特定の特許権、技術的ノウハウ、およびEBVに対するT細胞バンクおよび細胞株を取得した。ライセンス契約により、許可製品を商業的に合理的な努力で商業化し、許可計画について記念碑的な支払いを支払い、MSKに印税を支払うことに同意し、協力による候補製品が商業化されていることを前提としている。私たちの最初の商業製品EbvalloはMSK協力の一部であり、EBVを目標としている。
Tabcell-cell®(Ebvallo)は同種異体EBV特異的T細胞免疫療法であり、EUで承認され、現在アメリカで第三段階に開発され、メロワ或いはメロワ連合化学療法に失敗したEBV+PTLD患者の治療に用いられる。2020年第3四半期に開始された第2段階多コホート研究により,他のEBV+疾患に対するTabcelも開発されており,これらの疾患の医療ニーズは満たされていない。
Tabcelはアメリカ食品と薬物管理局(FDA)の突破的治療称号(BTD)を獲得し、HCT後のEBV+PTLD患者の治療に用いられ、これらの患者はアメリカと欧州連合(EU)でリツキシマブ治療を通過せず、孤児にも指定され、HCT或いはSOT後のEBV+PTLD患者の治療に応用されている。
MSKによる臨床研究では,HCTとSOT後EBV+PTLD患者を募集しており,これらの患者の歴史データよりも単剤治療後の方が奏効率が良い。2つの独立した臨床研究において、HCT後のEBV+PTLD患者はリツキシマブ治療とTAABCEL治療を受けることができず、その2年の総生存率は約83%であった。リツキシマブ失敗患者のSOT後EBV+PTLDの設置でも類似した結果が認められ,Tabcel治療を受けた患者では2年の総生存率は約86%であった。これらの研究では,HCTやSOT患者の応答率が50%以上認められた。
2017年12月、著者らはTABCELに対する2つの3期研究を開始し、2つの独立適応の承認、即ちHCT後のEBV+PTLDの治療(マッチング研究と呼ばれる)とメロワ失敗患者のSOT治療(対立遺伝子研究と呼ぶ)を支持することを目的とした。2019年、規制機関との検討と調整後、私たちはMatchと対立遺伝子を単一の研究(現在は対立遺伝子研究と呼ぶ)に合併し、この研究は現在メロワ治療を通過できなかったEBV+PTLD患者に対するHCT行列と、以前のメロワ治療に失敗したEBV+PTLD患者に対する単一SOT行列を含む。また,対立遺伝子研究を地理的に拡張し,ヨーロッパとカナダの臨床地点を含めた。
2020年第3四半期に対立遺伝子研究の中期分析を完了した。中期分析からのデータでは,HCTあるいはSOT後再発難治性EBV+PTLD患者では,独立腫瘍学と放射線学的評価(IORA)によるTABCELの客観応答率(OOR)は50%であり,OOR評価後少なくとも6カ月のフォローアップが得られた。このOORは先に発表した研究者の評価データと一致している。Tabcelのセキュリティプロファイルもこれまでに公表されたデータと一致しており,新たなセキュリティ信号はない.2022年12月、著者らは2022年アメリカ血液学会年会で対立遺伝子研究からの最新の中期分析と安全性結果、及びEBV+平滑肉腫患者に対する2つの単中心、開放ラベルと多中心拡大参入計画からの最新の治療効果と安全性データを公表した。
2021年10月、私たちはPierre Fabre商業化協定を締結し、この協定に基づいて、ヨーロッパと一部の新興市場での商業化とEbvalloの流通を可能にする独占的で分野の限られた許可をPierre Fabreに付与した。私たちは北米、アジア太平洋地域、ラテンアメリカのTabcelを含む他の主要市場に完全な権利を保持している。2022年9月、私たちはPierre Fabreが購入したEbvalloの特許使用料と供給価格の値上げと引き換えに、Pierre Fabre商業化協定を修正し、Pierre Fabreから3000万ドルの記念碑的支払いを追加した。より詳細については、以下の“いくつかの許可および連携協定の条項”の節を参照されたい。2022年12月、私たちはPierre Fabre商業化協定に従って、Ebvalloで特許使用料と特定のマイルストーンの権利の一部をHCRxに売却し、総投資額は3100万ドルで、上限はHCRx総投資額の185%~250%です。
9
2021年11月、我々はEBV+PTLD患者のためのTabcelのEUマーケティング許可申請(MAA)を提出した。2022年12月、欧州委員会は、2歳以上の再発または難治性EBV+PTLDの成人および小児患者の治療のための単一療法として、“特殊な状況”規制経路下でのEbvalloのマーケティング許可を承認し、これらの患者は、以前に少なくとも1つの治療を受けた。SOT患者では,化学療法が適切でない限り,これまでの治療には化学療法が含まれている。私たちはEbvalloのマーケティング許可をPierre Fabreに譲渡することを要求し、2023年2月に欧州委員会によって採択された。Pierre Fabreは2023年第1四半期に第1陣のヨーロッパ諸国でEbvallo発射活動を開始する予定だ。“特殊事情”のマーケティング許可によると、ピエール·ファブレにはEbvalloのメリットを確認し続けるための継続的な上場後義務がある。Ebvalloは特別な状況で承認されているため、Ebvalloマーケティング許可の継続は毎年再評価される必要がある。年次見直しは,Pierre Fabreが上場後の義務を履行している場合とEbvalloのリスク/収益状況に基づいて,Ebvalloのマーケティング許可を維持,変更または一時停止すべきかどうかを決定する。
2022年10月,イギリス(UK)の医薬品·保健製品監督局(MHRA)にEbvalloのMAAを提出し,2023年3月までにイギリスでMAAを承認する可能性があると予想した。
著者らは広範な研究を行い、キー対立遺伝子研究のためのラベル製造プロセスバージョンと商業化のためのラベル製造プロセスバージョンとの間の分析可能性を証明した。全面的な比較可能性分析は、効力、純度、および同種異体反応性の21個の重要な属性をカバーしている。我々は、成熟した統計方法と国際調整理事会(ICH)ガイドラインの応用に基づいて、TAB-CELプロセスバージョン間の分析の比較可能性が実証され、重要で一致した臨床経験の更なる支持を得たと信じている。これらの比較可能なデータ分析は,我々のMAA届出を介してEMAに提出された.EMAはその評価報告書で,欧州委員会がTAABCELのMAAを承認した後に発表され,予想される商業製品は展示する臨床使用製品と比較可能であると考えられている。
我々は、(I)化学、製造および制御(CMC)モジュール3の内容およびキー対立遺伝子研究で使用される製品と商業化しようとする製品との比較性評価、および(Ii)臨床データパッケージ要件を含む、潜在的な生物製品ライセンス申請(BLA)提出米国のラベルについてFDAと議論してきた。
2022年2月、我々はFDAとB型CMC会議を開催し、予想される商業と肝心な臨床試験プロセスのバージョンとの間の比較可能性を討論した。今回の会議では比較性については合意されておらず,FDAは最初に商業化製品の臨床研究を提案したが,FDAはキー対立遺伝子研究で使用されている製品と期待される商業化製品との間に比較可能性が証明されていることに同意しなかったからである。更なる討論を経て、FDAは新しい臨床研究を必要とせずにBLAを提出できる潜在的な方法を提案した。
その後,FDAと別の会議を行い,CMCに関するテーマを検討し,最終的に潜在的BLAが提出したCMCモジュール3の具体的な要求について明確な指導を提供し,合意した。今回の会議の後、FDA要求の追加CMC情報を提供するために、TAB−CELの研究新薬(IND)申請に対する修正案を提出した。
2023年2月にFDAと臨床上の問題について会議を行い,BLAをTABCELに提出する可能性がある。今回の議論の後,我々とFDAは別の会議を開催する予定であり,さらにCMCが異なるプロセスバージョンを集約する可能性のある臨床データの比較可能性を支援することを含めて潜在的なBLA提出に関する問題について検討する予定である。2023年第2四半期にTAB−CELに提出可能なBLAのさらなる更新を提供する予定である。
TAB−CELマルチコホート研究
著者らは引き続き多くの患者群の中でTABCELを開発し、主に免疫不全関連リンパ増殖性疾患(IA-LPD)に注目し、それらの免疫低下患者におけるEBV駆動の疾病機序は共通であり、高度に満たされていない医療需要とTAABCELのこれまで陽性の臨床データである。以前の治療に失敗した患者のうち,AID−LPD群の客観的有効率(完全反応を含む)は33.3%(9例中3例)であったのに対し,PID−LPD群は37.5%(8例中3例)であった。TAABCELは全体的な耐性が良好で、良好な安全性を有し、先に発表した臨床研究と一致した。これらの臨床データは、Tabcel耐性が良好であり、この患者群の中で鼓舞的な臨床活動を示し、客観応答率は50%(4分の2の患者)から80%(5分の4の患者)まで様々であることを示している。EAP-201研究で治療したEBVウイルス血症患者の1年総生存率(OS)は100%であり、平均14.6ケ月(最小12.2ケ月、最大17.8ケ月)フォローアップした。
2020年第3四半期には,第2段階マルチコホート研究を開始し,サイトを積極的に開設し,米国とEUで6名の患者群を募集しており,そのうち4名はIA−LPDのうち,2名は他のEBV駆動疾患であった。私たちはまだ学生募集を続けている
10
この研究中の患者です私たちはこのマルチコホート研究を通じてより多くのラベル拡張機会を調査する予定だ。この研究のデータは2023年に発表される予定だ。
NPC用タグユニット
鼻咽頭癌(NPC)は主にEBVに関連する頭頚部癌である。鼻咽頭癌の標準治療は通常放射線治療、白金に基づく化学療法或いは両者の組み合わせを含む。手術介入は使用が少なく,通常は選定された早期症例でのみ使用される。現在まだ承認されていない治療薬は再発/難治性鼻咽頭癌の治療に応用可能であり、多種の薬物がこのような患者群のために開発されているにもかかわらず。
著者らの1 b期研究は2018年にスタートし、いくつかの患者の中で安全終点と安定した疾患を実現した。EBV駆動鼻咽頭癌(NPC)の治療構造は絶えず変化しているため、著者らは著者らの方法及び白金耐性或いは再発に対するEBV駆動鼻咽頭癌患者の発展と制御経路を再評価する時、積極的にいかなる開発活動を展開していない。
ATA 188
多発性硬化症
EBV抗原に対する同種異体T細胞免疫療法であるATA 188も開発されており,多発性硬化症(MS)の潜在的治療に重要であると考えられている。MSは1種の慢性中枢神経系(CNS)自己免疫性疾患であり、炎症と組織損失を通じて脳、視神経と脊髄の髄鞘形成と正常機能を破壊する。多発性硬化症の発展は生理と認知(例えば、記憶)機能の日々の喪失を招く。これは全世界で約230万人の確定診断され多発性硬化症に生活している患者に実質的な負の影響を与えており,そのうち約100万人の患者が進行性多発性硬化症を患っている。
多発性硬化症は進行型多発性硬化症(PMS)と再発緩解型多発性硬化症(RRMS)に分類される。RRMSはMSの一形態であり、新しいまたは悪化したバイタルサインまたは症状(再発)が出現し、その後回復期と静止期であり、その間に疾患は進行しないことを特徴とする。経前症候群は重篤な多発性硬化症であり,時間の経過とともに多発性硬化症症状や身体障害の持続的な進展と悪化を特徴とし,治療案が少ない。経前症候群は2種類のタイプがある:原発進行性多発性硬化症(PPMS)と続発性進展性多発性硬化症(SPMS)。PPMSは患者の経過中に発生し,発症後の病態が安定し,徐々に悪化することが特徴である。SPMSは最初はRRMSで始まったが,患者の疾患が持続的に進行するとSPMSに進展する。
科学と臨床研究結果はEBVとMSの間の潜在的な生物学的関係を支持する。ほとんどのMS患者はEBVが存在する。MS病気経過はすでにEBV活動の測定と内因性EBV特異性T細胞群の枯渇と関係があることが証明された。また,単独研究では,非多発性硬化症患者と多発性硬化症患者の脳では,EBV感染B細胞と形質細胞の位置や頻度が有意に異なり,EBV感染B細胞と形質細胞は活発な脱髄領域に非常に近い。更なるデータにより、中枢神経系中のEBV陽性のB細胞と形質細胞は自己免疫反応を触媒する潜在力があり、典型的なMS病理生理学を招く。多発性硬化症患者では,彼らのT細胞はEBV陽性のB細胞や形質細胞を制御できない可能性があり,B細胞や形質細胞が脳に蓄積し,抗原提示細胞の役割を果たし,髄鞘を攻撃·破壊する抗体を産生し,髄鞘は脳と脊髄神経を隔離する保護層である。この髄鞘の喪失は最終的に多発性硬化症の症状を引き起こす。FDAはPPMSにおけるocriszumabの使用を許可し、MSにおけるB細胞の作用を支持し、このモノクロナル抗体はCD 20と呼ばれる細胞表面マーカーを発現することによって、中枢神経系外のB細胞(形質細胞ではなく)に対して広く標的となっている。
業界データの分析および一流治療の治療率と市場シェア増加の仮定によると、2025年までに、米国における前症候群の潜在的な年間市場機会は少なくとも35億ドルになる可能性があると推定される。
ATA 188、MSに適用
我々は、標的抗原認識を使用して、様々な疾患(多発性硬化症などの自己免疫疾患を含む)に適した既製T細胞免疫療法製品候補製品を作成するために、QIMR Berghoferのいくつかの独自技術および技術の許可を得た。私たちは、QIMR Berghoferとのライセンス契約に基づいて、この協力によって生成された製品の売上(できれば)に基づいて、QIMR Berghoferに様々なマイルストーンおよび特許権使用料を支払うことを要求している。QIMR Berghoferと連携してEBV標的や他のウイルス標的T細胞を開発している。この技術により,T細胞による免疫療法の役割を腫瘍やウイルス感染から自己免疫疾患に拡大している。
11
我々がこの技術を用いたT細胞免疫治療製品ATA 188は,MS特異的標的抗原認識技術を用いて,我々が管理するT細胞がEBV抗原を発現する細胞を選択的に認識できるようにした既製のEBV特異的T細胞製剤であり,これらの細胞はMSの潜在的治療に重要であると考えられる。ATA 188は,EBV陽性の細胞ではなく,EBV陽性の細胞のみを選択的に対象とすることを目的としている。最新の研究は科学そして自然界新しい疫学データを提供し、EBVがMSの主要な原因であることを表明し、機序データは、EBV感染はMS脳に対する自己免疫攻撃を起動し、伝播することができることを表明した。私たちはEBV陽性のB細胞と形質細胞を除去するだけでいくつかの経前症候群と経前症候群患者に利益を得る可能性があると信じている。
2017年第4四半期、著者らは異遺伝子ATA 188を用いて経前症候群患者を治療する開放ラベル、単腕、多中心、多国1期研究を開始した。この第一段階研究の主な目標は、ATA 188が最初の服薬後に少なくとも1年間フォローアップした患者の安全性を評価することである。研究中の肝心な副次的終点は臨床改善措置を含み、公認されたMS症状、機能と障害標準を使用し、拡張障害状態尺度(EDSS)、疲労重症度採点、MS Impact Scale-29(身体)、時間測定25フィート歩行(T 25 FW)、9-Hole Pegテスト、MS歩行12項目尺度(MSWs-12)と視覚鋭敏度を含む。
この研究1 a段階の第4回も最後の用量増加キューの登録は2019年第3四半期に完了し、著者らはMSVirtual2020:8でこの研究の最新の治療効果と安全性結果を示したこれは…。ACTRIMS-ECTRIMS共同会議,2020年9月。データによると、ATA 188はすべての4つの用量群で耐性が良好であり、用量制限性毒性もなく、致命的な有害事象もなかった。さらに、任意の時点で持続的障害改善(SDI)を示す患者は、将来のすべての時点で改善を維持し、用量の増加に伴い、より高い割合の患者はSDI(キュー3および4(より高い用量)42%、キュー1および2(より低い用量)17%)が出現する。SDIは,連続する2つの時点で観察されたEDSSまたはT 25 FWの臨床的有意な改善と定義されている。ATA 188治療はサイトカインレベルに臨床的意義の影響はなく,用量に関する安全傾向も認められなかった。鼻漏(流鼻水)は、複数の被験者において唯一発生する治療関連事象である。用量制限毒性と致命的な有害事件の報告はない。セキュリティプロファイルは前に報告されたデータと一致している.われわれはまた,ACTRIMS−ECTRIMSに臨床前翻訳データを示し,さらにEBV感染B細胞に対するATA 188の提案した作用機序を支持した。これらのATA 188を含むT細胞の併用解析は,提案したEBV感染を標的とするB細胞の機序と一致し,定義されたTCRによりこれらの細胞上のMSに関連するEBV抗原を認識する。これらのデータは、二重盲検、プラセボ対照の無作為研究で実証される必要があるが、それらは、前症候群の第1の治療レジメンが疾患の発展を阻止または逆転する可能性があることを示している。これらの結果は,EBウイルス感染を支持するB細胞のMSの慢性自己免疫病理における重要な役割のエビデンスと一致していると考えられる。
われわれは現在,原発と二次性経前症候群患者のためのATA 188第一段階研究の開放ラベル拡張(OLE)を行っている。OLEからの2年間の長期臨床データと2021年10月37時の第1段階研究からの変換データを示したこれは…。ヨーロッパ多発性硬化症治療と研究委員会大会。臨床データによると,多くの患者は持続的な障害改善を示すか,病態安定を示している。中枢神経系の髄鞘形成状態を反映していると考えられる新たなイメージングバイオマーカーデータを磁化転移率(MTR)と呼ぶことも報告されている。ATA 188に対するわれわれの臨床評価では,MTRはEDSS改善の機序に重要な知見を提供する可能性がある。
2020年6月に第2段階の1人目の患者を募集しましたランダム、二重盲検、プラセボ対照用量膨張試験(EMBOLD)ATA 188による経前症候群治療の治療効果と安全性評価私たちはこの研究に患者を募集し続けていますそれは.研究1 a段階のデータに基づき,第2段階EMBOLD研究に参加するキュー4用量を選択した。この研究には,障害測定とベースラインとの比較の変化,特にSDIの経時的変化のほかに,患者機能の複数の測定や様々なバイオマーカーが含まれている。
2022年3月、我々は更新された第1段階とOLEデータを公表し、24名の患者のうち20名が研究中にEDSSの改善またはEDSS安定を認めたことを示した;高用量キューでは、33%の患者が12ヶ月の時点で確認された拡張障害状態尺度(EDSS)の改善を得た。
2021年1月、EMBOLD研究設計の更新をFDAと検討し、いくつかのポイントおよび潜在的な登録研究において一致した:(I)障害改善終点は適切であり、FDAはEDSSを改善する傾向があることを明確に示した;(Ii)SPMSおよびPPMS研究集団に組み込むための基準が適切であり、(Iii)第2段階試験を少なくとも12ヶ月継続すべきであり、中期分析を適切に行うことが適切である。患者数を80人に増やし,研究の主な終点をEDSS障害改善に変更し,生物と機能終点を維持する案修正案をFDAに提出した。
12
2022年6月には,計画中のEMBOLD研究の中期分析(IA)を完了し,研究のサンプル量調整や修正は行わないことを決定した。報告実行時に得られたEMBOLDデータの分析から,改善12カ月のEDSSに対する6カ月のEDSSの予測価値に関する十分なデータセットが得られなかった。独立データ·安全監視委員会(IDSMC)は,6カ月の一時的な終点がこの場合のこのような介入の可能性を正確に測定していない可能性があるとしている。IDSMCは,サンプル量を調整または修正することなく検討を継続することを提案している.7月末にEMBOLDに参加した人数に基づき,12カ月で約90名の患者を拡張障害状況尺度(EDSS)に組み入れて障害改善を確認する主要な研究終点を予定している。これらのデータのコミュニケーション計画は2023年10月に適切なフォーラムで行われる。
2022年10月、我々は、進行性MSにおけるATA 188の第1段階研究の新しい磁気共鳴イメージング(MRI)バイオマーカーイメージングおよびOLE臨床データを、2022年ヨーロッパ多発性硬化症治療·研究委員会(ECTRIMS)会議で公表した。
ATA 188はFDA指定のPPMSとSPMSを治療する迅速なチャネルを獲得した.
我々は、2つの高速チャネル指定に基づいてFDAと相互作用し、独自の大型バイオリアクタ製造プロセスをさらに開発することを含む第3段階の準備を継続する。
ATA 3219
我々はまたATA 3219を開発しており,これは潜在的な同種最適な同種異体CD 19 CAR T免疫療法であり,B細胞悪性腫瘍に対して,我々の次世代1 XX CAR共刺激ドメインとEBV T細胞プラットフォームを利用して,TCRやヒト白血球抗原(HLA)遺伝子編集を必要としない。ATA 3219の臨床前研究データにより、T細胞幹細胞の製造技術を重視することにより、CD 19が発現した腫瘍細胞の体外と体内での機能持続性、多機能表現型と有効な標的性はすべて増強された。
ある臨床研究の学術データによると、EBV T細胞プラットフォームは既製の同種異体CAR T免疫療法を産生する可能性があり、高い応答率、持続的な反応と低毒性リスクを有し、迅速に患者に渡すことができる。
我々は引き続きATA 3219の製造技術の面で進展を得て、規模を拡大した。我々は現在,我々のCMOのGMP製造キットがプロセス最適化と製造運転を完了した後,2023年第2四半期にATA 3219計画のINDを提出する予定である。著者らのEBV CD 19 CAR T計画は記憶性T細胞表現型を豊富にし、臨床前研究において引き続き強い活性を示した。
その他の計画やプラットフォーム拡張活動
上述した優先項目以外にも、私たちは他にも多くの臨床と臨床前プロジェクトがある。
我々のCAR T免疫治療パイプラインは、Mesothelinに対する自己ATA 2271および同種異体ATA 3271を含み、Mesothelinは中皮腫、卵巣癌、膵臓癌、非小細胞肺癌、およびMesothelinを過剰発現する他の腫瘍を含む多くの固形腫瘍に発現する腫瘍抗原である。この2つのプロジェクトはいずれも2020年12月に独占有限領域許可(バイエル許可協定)によってバイエルに許可された。2022年5月、バイエルはベイヤ協定の終了決定を通知し、2022年8月2日にATA 2271とATA 3271の全製品開発と商業化権利を返還し、2022年7月31日から発効するバイエル終了協定を締結した。より詳細については、以下の“いくつかの許可および連携協定の条項”の節を参照されたい。
2018年にはMSKといくつかの合意を達成し,MSKとの連携をCAR T免疫療法の開発に拡張し,2018年5月に複数の連携目標に関するライセンスを取得し,2018年12月に我々の次世代CAR T計画に関するライセンスを取得し,間コルチゾンを目標とした。これらのCAR T協定によると、私たちは商業的に合理的な努力を使用して開発、規制承認を得ることに同意し、承認されれば、いくつかの協力目標を商業化し、いくつかのマイルストーンと特許権使用料を支払うことに同意する。
13
CD 28/CD 3 zに基づくCARSと比較して、ATA 2271は、PD-1優性負性受容体(DNR)を有する新規1 XX CAR共刺激シグナルドメインおよび細胞内チェックポイント抑制技術を使用することによって、治療効果の持続性および応答持続性を向上させることを意図している。ATA 2271からの研究性新薬応用(IND)により研究可能なデータが2020年6月の米国癌研究協会(AACR)仮想会議IIで公表された。これらのデータ支持は初めて1 XX共刺激ドメインと細胞内チェックポイント抑制技術をPD-1 DNRと結合し、第一世代CD 28/CD 3 zに基づくinterothelin CARと比べ、PD-1 DNRは更に少ない細胞消耗、機能持続性改善、連続細胞殺傷と体内治療効果を有し、そして複数回の腫瘍再攻撃を通じてこの効果を維持した。FDAは2020年8月にMSKの協力者が提出したIND申請を受け,2020年9月に晩期間中皮腫患者の開放ラベル,単腕1期臨床研究を開始した。2021年12月、ヨーロッパ医学腫瘍学会(ESMO)免疫腫瘍学大会の小型口頭会議で、本研究の最低用量列の最初の臨床前、臨床と翻訳データを示し、ATA 2271の早期安全性と持続性を証明した。MSKはこの研究の第3のコホートに薬を登録して投与した。2022年2月、MSKは、本研究における3番目のより高用量キューで治療を受けた患者に関連する致命的な深刻な有害事象をFDAに通報した。MSKは自発的にこの研究における新患者の募集を一時停止するとともに,この症例に関するより多くの情報を収集·検討した。2022年10月にMSKはFDAに評価を伝えました, その後、自発的に一時停止した後、最近この研究の登録が再開された。2022年12月、最新の発見は、臨床と安全性観察を含め、ESMO免疫腫瘍学大会の会議で公表された。
ATA 3271は既製の同種異体CAR T療法であり、著者らのEBV T細胞プラットフォームを通じて、PD-1 DNRと1 XX CAR共刺激シグナルドメインを用いてMesothelinを標的とした。ATA 3271の臨床前データでは,機能持続と著明な生存益を示すと考えられる抗腫瘍活性が認められ,体内に異体細胞毒性の証拠は認められず,同種異遺伝子MSLN−CAR工学EBV T細胞がMSLN陽性癌を治療する有望な方法であることが示唆された。ベイヤプロトコル終了後,我々はATA 3271の開発を一時停止した.
我々はまたATA 3431,B細胞悪性腫瘍に対する多標的同種異体CAR T免疫療法を開発している。著者らはまた、早期にB細胞のEBVに対する反応のみに焦点を当てたEBVワクチンの努力とは異なる潜在的な次世代EBVワクチンをQIMR Berghoferと協力して開発した。
私たちは、私たちのプラットフォームが現在の目標セットを超える効用を持つと信じている。私たちはパートナーとの協力によって派生した製品を含む他の候補製品を評価し続けるつもりだ。私たちはまた、私たちの既存のプラットフォームの機会を強化するために、許可を評価したり、より多くの候補製品や技術を取得したりし続けるつもりだ。
競争
生物技術と製薬業界の特徴は技術進歩が迅速で、競争が激しく、そして独自製品を高度に重視していることである。私たちは多くの製薬とバイオテクノロジー企業の競争と、学術機関、政府機関、民間、公共研究機関の現在の候補製品に対する競争に直面している。その中のいくつかの競争相手や潜在的な競争相手の市場での地位、財力、技術専門長は私たちよりずっと大きい。もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少ない、またはより安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消滅するだろう。
もし私たちのすべての候補T細胞製品が使用を許可されたら、私たちは激しい競争に直面するだろう。現在患者に対する看護標準以外に、免疫治療領域のいくつかの方面は商業と学術臨床研究を行っている。これらの研究の早期結果は,T細胞免疫療法に対する持続的な興味を刺激した。また,承認されれば,我々のT細胞計画は,現在市販されている適応治療に用いられている薬物や療法と競合し,現在開発されている同じ適応の候補製品と競合する可能性がある。
EBV+PTLD
FDAが承認したEBV+PTLD治療の製品は現在のところなく,EbvalloのほかにECが承認したこの適応の製品もない。しかし、いくつかの市場の製品および治療法は、いくつかの医療専門家および機関によってラベルの外でEBV+PTLD、例えばリツキシマブおよび連合化学療法レジメンの治療に使用されていることを知っている。そのほか、いくつかの会社と学術機構はEBV+PTLDと他のEBV駆動の疾病のために候補製品を開発しており、Viracta Treateutics,Inc.を含み、それは重要な第二段階の臨床研究を行っており、Nanatinostat(以前はTractinostat、あるいはVRX-3996と呼ばれていた)を抗ウィルス薬物valganciclovirと併用して再発/難治性EBV+リンパ腫を治療している。アロビル(前身はウイルス細胞)は、異体多ウイルスT細胞製品である異体多ウイルスT細胞製品である異体卵巣癌(ALVR 105)に対する第2段階臨床研究を完成し、この製品はEBウイルスを含む同種異遺伝子造血幹細胞移植レシピエントの6種類のウイルスに対して、そしてウイルス関連性出血性膀胱炎に対する2つの第3段階臨床試験、及びウイルス予防の第3段階試験を行っている
14
CMV,ADV,EBV,HHV 06とJCVは異遺伝子HSCT患者とTessa Treeutics Pte Ltd.で再発した難治性CD 30陽性リンパ腫に対する同種異体CD 30−CAR EBVST候補製品の1期研究を行っている。
多発性硬化症
多発性硬化症は市場競争が激しく、米国およびEUは、臨床隔離症候群、再発緩和多発性硬化症(RRMS)、二次性進展性多発性硬化症(SPMS)、および原発進行性多発性硬化症(PPMS)を含む4つの模倣薬または生物学的等価物を含む少なくとも20の療法を承認した。MS市場には主要な国際的に完全に統合された製薬会社と老舗のバイオテクノロジー会社を含む多くの競争相手がいる。最近,TG Treeuticsで販売されているBriumvi(Ublituximab),ジョンソンから販売されているPonvory(S 1 P調節剤)とKesimpta®米国および/またはEUで再発形態のMSの治療のために許可されたノ華社によって販売されている(抗CD 20モノクロナル抗体)。
第3段階研究では、再発および/または進行型多発性硬化症に対する多くの候補開発薬があり、将来的には、KGaAのBruton‘sチロシンキナーゼ(BTK)阻害剤evobrutinib、羅氏のBTK阻害剤evobrutinib、サイノフェナントレンのBTK阻害剤fenebrutinib、tlebrutinibおよびAB Scienceのチロシンキナーゼ阻害剤Masitinibを含む2つまたは2つの適応のうちの1つまたは2つでより多くの新薬が承認される可能性がある。MediciNovaは、不活性SPMSにおけるPDE阻害剤イソブチスト(MN 166)の第3段階研究を開始することを計画している。
CAR T計画
現在6つの自己CAR T療法がアメリカおよび/またはEUで承認されていますノワールのKymriah®(Tisagenlecleucel)、Gilead/KiteのYescarta®(Axicabagene Cilolucel)およびTecartusTM百時美施貴宝のBrexucabagene autolucel)と®(リソカタエン)およびアベマ(アダカタエン白血球)、ならびにジョンソンおよびレジェンド生物のカリクティ(シラカタエン自生白血球)。多くのCARを介した細胞療法が開発されており,多くは自己であるにもかかわらず,同種異体や既製細胞療法も含まれている。現在、複数の異遺伝子CARプラットフォームが開発されており、方法が異なり、ドナー細胞が患者の身体を異体あるいは患者の身体でドナー細胞を拒絶することを最大限に減少させる。これらの方法は、遺伝子編集を使用してTCRを除去または阻害することと、TCRを含まない細胞タイプを使用することとを含む。多くの臨床段階の同種異体CAR計画は細胞型としてα−βT細胞を用い,T細胞受容体やヒト白血球抗原の遺伝子編集を第一選択の技術的方法としているが,他の戦略も開発されている。これらの他の方法のいくつかは、私たちが使用する方法よりも有利な特徴を有する可能性があり、これは、我々の製品よりも潜在的なパートナーまたは顧客に好まれることをもたらすであろう。我々の将来の疾患によれば,関心のある適応において,自己および異体CAR療法および他の方法(例えば,小分子,抗体,二重特異性)からの競争に直面する可能性がある。
特定の許可と協力協定の条項
外注許可
ピエール·ファブレ商業化協定
2021年10月、私たちはPierre Fabre商業化協定を締結し、この協定に基づいて、私たちはPierre Fabreに独占的で場所の限られた許可を与え、ヨーロッパでのEbvalloの商業化と流通を許可し、規制の承認を得た後に領土内の新興市場を選択することを許可した。Ataraは北米、アジア太平洋地域、ラテンアメリカなどの他の主要市場でTabcelを販売するすべての権利を保持している。2022年9月、Pierre Fabre商業化協定(PF改正案)の第1号改正案を締結した。PF修正案の条項によると、欧州委員会がEBV+PTLDのためのEbvalloの使用を許可し、その後、Pierre Fabreにマーケティング許可申請を提出した後、私たちは他の事項と引き換えに追加の3,000万ドルのマイルストーン支払いを得る権利がある:(I)この地域でのEbvalloの純売上高の割合を占めるEbvalloの権利使用料と、(Ii)Pierre Fabreが購入したEbvalloの供給価格が上昇する。また,Pierre Fabre商業化プロトコルに従ってPierre Fabreに何らかのサービスを提供する期限を延長することにも同意した.2022年12月、私たちはPierre Fabre商業化協定に従って、Ebvalloで特許使用料といくつかのマイルストーンの権利の一部をHCR Molag Fund L.P(HCRx)に売却し、総投資額は3100万ドル、上限はHCRx総投資額の185%~250%である。
著者らは進行中の3期対立遺伝子臨床研究と2期多列臨床研究の結論に責任を持っている。また,ピエール·ファブレ欧州商業化協定の条項に基づき,規制部門の承認を得るためのEBV陽性リンパ増殖性疾患TAB−CELの他の活動の費用を担当する。Pierre Fabreは、他のすべての規制承認、承認後の義務、およびこの領土でのEbvalloの商業化と流通を獲得し、維持する責任があるだろう。私たちはこの協定に従って私たちが単独で開発した任意の知的財産権を持つつもりだ。
15
ピエール·ファブレは2021年第4四半期に4500万ドルの前払い現金を支払って独占許可を得た。2022年12月、特定の規制マイルストーンに基づいて4000万ドルのマイルストーン支払いを得る契約権を実現しました。“HCRx協定”の条項によると、いくつかの規制および商業マイルストーンに達した後、合計3.08億ドルに達する残りのマイルストーン支払いを得る権利があり、また、製品が国で初めて商業販売され、特許権が満期または指定されたすべての規制排他性が国/地域で計算される12年後まで、2桁の等級別使用料がEbvalloの純売上高の割合を占める権利がある。
2022年12月、私たちはPierre Fabreと単独の製造と供給協定を締結し、2023年12月31日までの固定価格と2024年1月1日からのコストと保証金に基づいて、Pierre Fabreをこの地域で使用するためにEbvalloを生産した。私たちは領土内で商業化するためにPierre FabreのEbvalloの製造と供給を担当し、費用はPierre Fabreによって支払われ、領土内のEbvalloの最初の商業販売(Pierre Fabre商業化協定で定義されているように)から少なくとも7年間。その後,製造責任や関連製造技術を第三者CMOに譲渡することを選択することができ,Pierre Fabreも製造責任を直接負担して関連製造技術を得ることを選択することができる。
双方がこの日までに関連するセル選択技術をPierre Fabreに譲渡することに同意しない限り、私たちはまた、一定期間内にセル選択サービスを自費で提供する責任がある。この期間の後、私たちがセル選択サービスを継続することに同意すれば、費用はピエール·ファブレによって完全に負担されるだろう。
バイエル許可と連携協定
2020年12月,われわれは固形腫瘍を治療するための間コルチゾールガイドのCAR T細胞療法を開発するためのバイエル許可協定を締結し,これにより,われわれおよびその付属会社が所有または制御しているカバーまたはATA 2271およびATA 3271(ライセンス製品)に関する適用特許およびノウハウを含む独占的,領域的に限られた許可をバイエルに付与した。
バイエル許可協定の条項によると、著者らはMSKと協力して、ATA 2271のすべての共通合意された臨床前と臨床活動を担当し、ヒト第一段階の臨床研究の最初まで、その後バイエルはATA 2271の更なる開発を担当し、費用は自費である。バイエルはATA 3271の開発を担当し、費用はバイエルが負担するが、著者らはATA 3271に関連するいくつかの双方が合意した臨床前、翻訳、製造とサプライチェーン活動は除外した。バイエルは独自にライセンス製品を商業化し、費用はバイエルが負担する
2022年5月、バイエルはバイエル協定の終了決定を通知し、2022年8月2日、私たちはバイエル終了協定を締結し、発効日は2022年7月31日です。終了発効日から、ライセンス製品に関連する全製品開発権はAtaraに返却されます。Ataraが発効日を終了する前に行ったいくつかの活動への見返りとして、バイエルは2022年9月にAtaraに420万ドルを支払った。
In-許可
MSKプロトコル
2015年6月,MSKと3種類の臨床段階T細胞療法の独占ライセンス契約を締結した。私たちは、特定の規制と販売関連マイルストーンの完成状況に応じてMSKに支払い、将来の開発許可製品候補製品(ある場合)の製品販売状況に応じてMSKに1桁の中央値パーセントの階層印税を支払うことを要求された。また、場合によっては、MSKに一定の最低年間印税を支払うことを要求され、これらの費用は、同じ年の期間に支払われた当然の印税を免除することができる。私たちはまた、再許可によって許可された権利のために私たちが受け取った任意の代価の低い2人の数百分を支払うことを要求された。ライセンス契約は、製品及び国/地域の最終期限で満了する:(I)各ライセンス製品に関連する最終ライセンス特許権の満了、(Ii)ライセンス製品毎に法的に付与された任意の市場排他期間の満了、及び(Iii)各国·地域における初の商業販売ライセンス製品後の指定年数。許可協定が満了した後、Ataraは許可製品に対する非独占的な権利を保持するだろう。
2018年5月と12月、私たちはMSKから追加の技術許可を得た。特定の開発、法規、販売に関するマイルストーンの達成に応じて追加のマイルストーン支払いと、将来の開発許可製品候補製品による製品販売(あれば)に応じて中桁数百分比分級印税を支払う義務があります。
2021年3月、MSKとのライセンス契約を修正し、再確認し、いくつかの権利の許可を終了し、既存のプロトコルではカバーされていない他の独自技術的権利を許可しました。
16
QIMR Berghoferプロトコル
2015年10月、QIMR Berghoferと独占ライセンス契約と研究開発協力協定を締結した。ライセンス契約の条項により,我々は独自の世界的許可を得て,QIMR Berghoferが開発した技術とノウハウを用いて同種異体T細胞治療プロジェクトを開発·商業化した。2016年9月、独占ライセンス協定と研究開発協力協定の改正と再記述が行われた。改訂·再記述されたプロトコルによると、より多くのT−cellプロジェクトの開発と商業化の独占的なグローバルライセンスを取得し、2018年6月により多くの技術の選択権を許可した。2019年8月、我々は、サイトメガロウイルス(CMV)に関連するいくつかの権利の許可を終了するために、QIMR Berghoferとの許可プロトコルおよび研究開発協力協定をさらに修正し、再確認した。また、私たちは2020年8月にさらにQIMR Berghoferとの許可プロトコルと研究開発協力プロトコルを修正し、再確認して、BKポリオーマウイルスとJCポリオーマウイルスに関する権利の許可を終了した。2021年12月、私たちは、HPV関連癌に関連するいくつかの権利の許可を終了するために、QIMR Berghoferとの許可プロトコルおよび研究開発協力協定をさらに修正し、再確認した。我々は,2021年12月のQIMR Berghoferとの4回目の改訂と再記述許可プロトコルをQIMR許可プロトコルと呼び,QIMR Berghoferとの2021年12月の第4回改訂と再記述した研究開発協力協定を我々のQIMR協力協定と呼ぶ.
QIMRライセンスプロトコルは,将来の製品売上に応じて,様々な記念碑的かつ中央桁以下の特許権使用料をQIMR Berghoferに支払うことを規定している.QIMR連携協定の条項により,連携して開発したプロジェクトに関する合意開発活動のコストを返済する必要がある.QIMR協力協定はまた,ある発展と規制マイルストーンの実現に応じて,QIMR Berghoferに様々な記念碑的な金を支払うことを規定している。
知的財産権
私たちのビジネスの成功は、私たちの候補製品のために独自の保護を獲得し、維持し、他人の固有の権利を侵害することなく運営し、他の人が私たちの固有の権利を侵害することを防止する能力があるかどうかにある程度依存する。私たちは、米国および非米国特許出願や、私たちのノウハウ、発明、改善に関連する他の規制出願などの方法を提出することで、私たちの独自の地位を保護することを求めており、これらは私たちの業務の発展と実施に非常に重要である。私たちはまた、商標、商業秘密、技術ノウハウ、持続的な技術革新、潜在的な許可内の機会に依存して、私たちの独自の地位を発展させ、維持しています。私たちが依存しているいくつかの特許、商標、商業秘密、ノウハウ、および他の知的財産権は私たちの所有物であり、他は私たちのパートナーから得られた内部許可だ。私たちが“私たちの”技術、発明、特許、特許出願、または他の知的財産権を言及するとき、私たちは私たちが所有または所有する権利、および私たちが許可する権利を意味する。また,生物類似分子や孤児薬物の地位規制に関する米国,ヨーロッパ,その他の国の様々な法的枠組みから利益を得ることが予想される。これらの法定フレームワークは条件を満たす分子に一定の規制排他期を提供した。“政府規制”を参照されたい
特許
我々は、重要な治療分野の各候補製品のために、治療方法特許を含む物質成分および/または関連方法特許を求めている。米国特許制度は一時的および非臨時特許出願の提出を許可する。臨時特許出願は、米国特許商標局(USPTO)によって特許出願が可能かどうかを審査し、提出日12ヶ月後に自動的に失効することはない。したがって、臨時特許出願は発行された特許に成熟することはできない。他に加えて、一時特許出願は、一般に、後に提出される非一時的特許出願のためのより早い有効な提出日を確立するために使用される。非仮特許出願は米国特許商標局によって審査され,米国特許商標局が要求する発明が特許性基準に適合すると判断すると,非仮特許出願は特許に成熟することができる。
17
個別特許の展示期間は、特許出願の提出日、要求される優先権日、及びこれらの特許を取得した国の適用法により決定された特許の法的期限に依存する。一般に,米国で提出された出願から発行される特許の有効期間は20年であり,最も早い非仮出願日から計算される。さらに、場合によっては、特許期間は、FDA規制審査期間によって実際に失われた部分期間を再取得するために延長することができるが、回復期間は5年を超えることはできず、回復期間を含む総特許期間は、FDA承認後14年を超えてはならない。さらに、特許期限の調整は、米国特許商標局がオフィスに訴訟を提起している間のいくつかの遅延を説明するために、期限を延長することができる。非米国特許の有効期限は,適用される現地法の規定によって異なるが,通常,非米国特許の有効期限は最も早い国際出願日から20年であり,利用可能な特許期間の延長は含まれていない。特許提供の実際の保護は製品によって異なり,特許のタイプ,そのカバー範囲,特許期間延長の可能性,特定国の法的救済措置,特許の有効性と実行可能性を含む多くの要因に依存する。
我々の製品などタンパク質に基づく生物製品に関する国内や国際特許法は未解決のままである。これまで、米国、ヨーロッパ、または他の国は、特許資格またはその分野の特許によって許容される権利要件の広さについて一致した政策を策定していない。米国または他の国の特許法または特許法解釈の変化は、私たちの発明を保護し、私たちの知的財産権を実行する能力を弱める可能性がある。したがって、私たちは、私たちの特許または第三者特許に付与される可能性のある特許の広さまたは実行可能性を予測することができない。バイオテクノロジーと製薬産業の特徴は広範な知的財産権訴訟だ。私たちが私たちの候補製品と技術の特許地位を維持して強化する能力があるかどうかは、私たちが私たちの特許のために有効な特許請求を獲得することに成功し、特許が付与された後にこれらの権利要件を強制的に実行できるかどうかにかかっている。私たちは私たちのどんな特許出願がどんな特許の発行につながるのか分からない。私たちが発表した特許は挑戦、無効、または回避される可能性があり、任意の発行された特許によって付与された権利は、同様の技術を持つ競争相手に対抗するために十分な保護または競争優位性を提供できない可能性がある。さらに、私たちの競争相手は、私たちの特許を侵害することなく、類似した薬物を独立して開発し、商業化するか、または私たちの技術、ビジネスモデル、または戦略を複製することができる。私たちの候補製品から開発された任意の薬物の臨床開発と規制審査には多くの時間を要する可能性があるため、私たちの任意の薬物が商業化される前に、任意の関連特許は商業化後に短期的に満了または有効に維持され、そのような特許の任意の利点を弱める可能性がある。
私たちの世界特許権は、独自資本および認可中の特許および特許出願、治療方法を含む物質に関する組成物および/または関連方法を含み、33個の特許系列からなり、合計330件以上の発行された特許または特許出願がある。我々の特許及び特許出願(発行された場合)は、2023年から2042年の間に満了する予定であるが、関連する司法管轄区域で提供可能ないかなる特許期間の延長も含まれていない。
商業秘密
特許に加えて、私たちは非特許の商業秘密とノウハウ、そして持続的な技術革新に依存して、私たちの競争地位を発展させ、維持しています。私たちは、私たちのビジネスパートナー、協力者、従業員、コンサルタントとの秘密協定、および私たち従業員との発明譲渡協定の一部を使用して、私たちの独自の情報を保護することを求めています。これらの協定は、当社の独自の情報を保護し、発明譲渡協定の場合、従業員が開発した技術の所有権を付与することを目的としています。私たちはこのような合意に違反するかもしれないし、私たちはそのような違反に対応するための十分な救済策を持っていないかもしれないし、私たちの固有の情報を不正に開示するのに十分な救済策がないかもしれない。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。もし、私たちのビジネスパートナー、協力者、従業員、およびコンサルタントが、私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連するまたはそれによって生じるノウハウおよび発明の権利について論争が生じる可能性がある。
商標
私たちはまた商標によって私たちの競争地位を発展させ、維持し、私たちは私たちの業務に関連する商標権を追求し、獲得し続けている。私たちは、私たちの商標の価値を維持し、強化し、これらの商標の不正使用を防止するための強力な世界的商標登録·法執行計画を持っている。私たちの世界的な商標の組み合わせは、178件以上の登録および処理される出願を含む16の異なる商標シリーズを含む。
政府規制と製品審査
私たちはアメリカで運営されているバイオ製薬会社として広く規制されている。われわれのT細胞免疫療法は,承認されれば生物製品や生物製品として規制される。この分類により,我々の製品の商業生産は登録された施設で行う必要があり,現在の生物製剤の良好な製造規範(CGMP)に適合している。FDAは人体細胞或いは組織に基づく製品を最小操作或いは最小操作を超える製品に分類し、最小操作を超える製品は製品の安全性を証明するために臨床試験を必要とすることを確定した
18
有効性と販売許可を提出したBLA。我々の候補製品は最小限の操作だけではないと考えられており,発売される前に臨床試験で評価し,BLAを提出·承認する必要がある。
米国(連邦、州と地方各レベル)とその他の国の政府当局は、私たちが開発している生物製薬製品の研究、開発、テスト、製造、品質管理、承認、ラベル、包装、貯蔵、記録保存、販売促進、広告、流通、追跡と追跡、承認後のモニタリングと報告、マーケティングと輸出入などの方面に対して広範な監督管理を行っている。私たちの候補製品はFDAの承認を得なければ、アメリカで合法的に発売され、適切な外国監督管理機関の許可を得て、国外で合法的に発売することができる。一般的に、私たちの他の国での活動は、重要な違いがあるかもしれないにもかかわらず、米国と類似した性質と範囲の規制を受けるだろう。また、欧州規制のいくつかの重要な側面は集中的に処理されているが、具体的な国の規制は多くの点で不可欠である。規制マーケティングの承認を得る過程とその後、適切な連邦、州、地方、外国の法律法規を遵守する過程には、多くの時間と財力が必要だ。
アメリカ製品開発プロセス
米国では、FDAは“連邦食品、薬物と化粧品法”(FDCA)、“公衆衛生サービス法”(PHSA)及びその実施条例に基づいて薬品と生物製品を規制している。規制の承認を得て、その後、適切な連邦、州、地方、外国の法規と条例を遵守する過程には、多くの時間と財政資源が必要だ。製品開発過程,承認過程又は承認後のいずれかの場合,出願人が適用される米国の要求を遵守できない場合には,行政又は司法制裁を受ける可能性がある。FDAの制裁は、承認保留申請の拒否、承認の撤回、臨床一時停止、警告または他の実行手紙、製品のリコールまたは市場からの撤回、製品の差し押さえ、生産または流通禁止の完全または部分的な一時停止、罰金、政府契約の拒否、原状回復、返還、返還、または民事または刑事罰を含むことができる。どんな機関や司法法執行行動も私たちに実質的な悪影響を及ぼすかもしれない。米国食品医薬品局が生物製品が米国市場に参入する前に必要なプログラムには、通常、以下のような態様が含まれる
人体で任意の生物候補製品をテストし、私たちの候補製品を含む前に、候補製品は臨床前試験段階に入る。臨床前試験は、非臨床研究とも呼ばれ、製品の化学、毒性と調合に対する実験室評価、及び候補製品の潜在的安全性と活性を評価する動物研究を含む。臨床前試験の実施は適用されるGLPを含む連邦法規と要求に適合しなければならない。臨床試験スポンサーは臨床前試験の結果を生産情報、分析データ、任意の利用可能な臨床データ或いは文献、提案された臨床試験方案と共にINDの一部としてFDAに提出しなければならない。IND提出後も,いくつかの臨床前試験が継続される可能性がある。INDはFDAが提案された臨床試験に対して懸念または問題を提起し、30日以内に臨床試験を一時停止しない限り、FDAが受信後30日以内に自動的に発効する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。FDAは強制的に
19
臨床試験対象は受け入れられない重大なリスクに直面している或いはFDAの要求に符合しないため、臨床試験前或いは臨床試験期間中のいつでも候補生物製品を持っている。FDAが臨床一時停止を強制した場合、FDA許可なしに、試験は米国で継続または再開されず、その後、FDA認可の条項の下でしか行われない可能性がある。したがって,INDの提出によりFDAが臨床試験の開始を許可するか,あるいは開始すると米国でのこのような試験を一時停止または終了するという問題はないとは考えられない。
臨床試験は合格した調査者の監督の下で患者にバイオ製品候補薬を投与することに関連し、通常は試験スポンサーに雇われたりコントロールされたりしない医師である。臨床試験は,いくつかの有害事象発生時に臨床試験が停止されることを確保する停止規則を含む,臨床試験の目標,投与手順,被験者の組み入れと排除基準,および被験者の安全性を監視するためのパラメータを詳細に説明したプロトコルで行われる。各スキームおよびスキームの任意の修正は、INDの一部としてFDAに提出されなければならない。臨床試験はFDAがGCP要求を含む規定に基づいて行わなければならず、すべての研究患者にインフォームドコンセントを提供することを含む。さらに、各臨床試験は、臨床試験を行う各機関の独立したIRBによって審査および承認されるか、またはサービスを提供しなければならない。IRBは試験参加者の福祉や権利の保障を担当し,臨床試験に参加する個人のリスクが最低に低下するかどうか,期待利益と比較して合理的かどうかなどの項目を考慮している。IRBはまた、各臨床試験対象またはその法律代表によって署名されなければならないインフォームドコンセント文書の形態および内容を承認し、完成まで臨床試験を監視しなければならない。いくつかの研究はまた、データ安全監視委員会と呼ばれる臨床研究スポンサーによって組織された合格専門家からなる独立したグループの監督を含む, これは、研究からのいくつかのデータへのアクセスに基づいて、研究が指定されたチェックポイントで進むことができるかどうかを許可し、対象に対して許容できない安全リスクまたは他の理由があると判断した場合、例えば有効性を示さない場合、臨床試験を停止することができる。現在行われている臨床研究や臨床研究結果を公的登録機関に報告することに関する要求もある。
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複或いは合併する可能性がある
承認後の臨床試験は,4期臨床試験と呼ばれることがあり,最初の上場承認後に行うことができ,FDAが要求する場合もある。これらの臨床試験は,治療適応が予想される患者の治療から追加的な経験を得るためのものであり,特に長期安全なフォローアップのためである。臨床開発のすべての段階において、監督管理機関はすべての臨床活動、臨床データと臨床試験調査人員に対して広範なモニタリングと監査を行うことを要求している。臨床試験結果を詳細に説明する年次進展報告はFDAに提出しなければならない。深刻かつ予期しない不良事件に対して、迅速にFDAと調査人員にIND安全報告を提出しなければならず、その他の研究結果はこの薬物に暴露された人類は重大なリスクが存在することを表明し、実験室動物試験或いは体外試験は人類患者に対して重大なリスクがあることを表明し、或いは任意の臨床上の深刻な疑わしい副作用の発生率は方案或いは研究者マニュアルに列挙された比率より高い臨床重要な増加である。スポンサーは15日以内にINDセキュリティ報告書を提出し,スポンサーがその情報有資格報告を確定した後でなければならない。スポンサーはまた、スポンサーが初めて情報を受け取ってから7日以内に、任意の意外、致命的、あるいは生命に危害を及ぼす疑いのある副作用をFDAに通知しなければならない。第1段階、第2段階、および第3段階の臨床試験は、もしあれば、任意の指定された時間で成功しない可能性がある。FDAまたはスポンサーまたはそのデータ安全監視委員会は、他の無関係な免疫療法試験から推定されるリスクを含む、研究患者のリスクへの曝露が許容できない健康リスクを構成することを発見することを含む、随時様々な理由で臨床試験を一時停止または終了することができる。類似, もし臨床試験がIRBの要求に従って行われない場合、あるいは生物製品が患者が予期しない深刻な損傷を受けることに関連している場合、IRBはその機関の臨床試験の承認を一時停止または終了することができる。
20
臨床試験と同時に、会社は通常追加の研究を完成しなければならず、生物製品の物理的特徴に関する追加情報を開発し、cGMP要求に基づいて商業大量生産製品のプロセスを最終的に決定しなければならない。生物製品を用いた外来製剤導入のリスク低減を支援するために,FDCA,PHSA,FDAの法規は,属性が正確に定義できない製品の生産制御の重要性を強調している。製造過程は一貫して高品質の候補製品ロットを生産することができなければならず、他の以外に、スポンサーは最終生物製品の特性、強度、品質、効力と純度をテストする方法を開発しなければならない。また,適切な包装を選択·試験し,候補生物製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
アメリカBLAの審査と承認プロセス
生物製品の臨床試験が完了した後,生物製品の商業マーケティングの前に,創新者生物製品のBLAに対するFDAの承認を得なければならない。提出されたBLAには、製品開発、実験室と動物研究、人体試験、製品製造と成分の情報、提案されたラベル、その他の関連情報が含まれなければならない。テストや承認過程には多大な時間と労力が必要であり,FDAがBLAの届出を受ける保証はなく,届出してもどの承認もタイムリーに承認される保証はない.
改正された“処方薬使用料法案”(PDUFA)によると、各BLAは相当な使用料を伴わなければならない。FDAは毎年イノベーティブ生物製品を許可するPDUFA使用料を調整している。PDUFAは革新者生物製品に年間計画費を徴収している。場合によっては、小企業が初めて出願した出願料を免除することを含む、費用を免除または減免することができる。また,孤児薬の創新者生物製品に指定されているBLASは使用料を評価しておらず,この製品に孤児適応も含まれていない限りである。
出願が提出されてから60日以内に、FDAは、BLAが提出された文書を審査して、機関が提出を受ける前に実質的に完了したかどうかを決定する。FDAは、それが不完全であるか、または提出時に適切に審査できないと考えられる任意のBLAの提出を拒否することができ、より多くの情報の提供を要求することができる。この場合,BLAおよび付加情報を再提出しなければならない.再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると,FDAはBLAの深い実質的な審査を開始する。FDAは、提案された製品が安全であるかどうか、有効であるかどうか、および/またはその予期される用途に有効であるかどうか、許容可能な純度プロファイルを有するかどうか、および製品がcGMPに従って製造されているかどうかを決定して、製品の特性、安全性、強度、品質、効力および純度を確保および保存するためにBLAを審査する。FDAは、新規な生物製品または安全性または有効性の問題を提起する生物製品の申請を諮問委員会に提出することができ、一般に、申請を承認すべきかどうか、およびどのような条件下で承認すべきかを審査、評価および提案するための臨床医および他の専門家を含むグループである。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。生物製品の審査過程において、FDAはまた、生物製品の安全な使用を確保するためにリスク評価と緩和策(REMS)が必要かどうかを確定した。REMSは、既知または潜在的に薬物に関連する深刻なリスクを管理し、これらの薬剤の安全な使用を管理することによって、薬物ガイドライン、医師のコミュニケーション計画、または安全な使用を確保する要素を含む可能性があるこれらの薬剤を継続的に得ることができる安全戦略である, 例えば、制限された配信方法、患者登録表、および他のリスク最小化ツール。FDAがREMSが必要であると結論した場合,BLAのスポンサーは提案したREMSの承認を提出しなければならない。必要であれば、FDAはREMSのないBLAを承認しないだろう。
BLAを承認する前に、FDAはこの製品を生産する施設を検査する。FDAは、製造プロセスおよび施設がcGMP要件に適合していると判断しなければ、製品が要求された仕様内で一貫して生産されることを保証するのに十分であることを決定しない限り、この製品を承認しないであろう。免疫療法製品については,メーカーが適用範囲内でGTPSに適合していなければ,FDAもこの製品を承認しない。GTPは、ヒト細胞、組織、および細胞および組織ベースの製品(HCT/Ps)を製造するための方法および施設および制御を管理するFDAの法規制および指導文書であり、これらの製品は、ヒトレシピエント内に移植、移植、注入または転移するためのヒト細胞または組織である。GTP要求の主な目的は,細胞や組織に基づく製品の製造方式の確保であり,感染症の導入,伝播,伝播を防止することを目的としている。FDAの規定はまた、組織機関がFDAに彼らのHCT/Pを登録し、適用した場合にスクリーニングとテストを通じてドナーを評価することを要求する。さらに、BLAを承認する前に、FDAは通常、IND試験要求およびGCP要求に従って臨床試験が行われることを確実にするために、1つまたは複数の臨床場所を検査する。CGMP,GTP,GCPに適合することを確保するためには,申請者は訓練,記録保存,生産,品質管理に多大な時間,お金,労力をかけなければならない。
21
関連データおよび情報が提出されたにもかかわらず、FDAは、BLAがその承認の規制基準を満たしていないことを最終的に決定し、承認を拒否する可能性がある。臨床試験から得られたデータはつねに決定的ではなく,FDAのデータ解釈は我々の同じデータに対する解釈とは異なる可能性がある。FDAが現在の形態のBLAを承認しないと決定した場合、FDAは、FDAによって決定されたBLA内のすべての特定の欠陥を記述する完全な返信を発行するであろう。決定された欠陥は微小である可能性があり、例えば、ラベル変更が必要であるか、または重大であり、例えば、追加の臨床試験が必要である。さらに、完全な返信状は、出願人がとり得る、申請を承認条件に置くための提案行動を含むことができる。完全な返信が発行された場合、出願人は、BLAを再提出し、手紙で決定されたすべての不足点を解決するか、または出願を撤回することができる。
製品が規制部門の承認を得た場合、承認は特定の疾患および用量に限定される可能性があり、または使用適応が制限される可能性があり、これは製品の商業的価値を制限する可能性がある。さらに、FDAは、いくつかの禁忌症、警告、または予防措置を製品ラベルに含めることを要求する可能性がある。FDAは、リスク管理計画またはREMSの形態で、製品流通、処方または調剤に制限および条件を適用することができ、または他の方法で任意の承認範囲を制限することができる。そのほか、FDAは発売後の臨床試験を要求する可能性があり、時々第四段階の臨床試験と呼ばれ、生物製品の安全性と有効性を更に評価することを目的とし、そして商業化された承認製品の安全性を監督するためにテストと監督計画を要求する可能性がある。
そのほか、“小児科研究公平法”(PREA)は申請者に関連小児科群中のいくつかの薬物と生物製品を研究することを要求し、もしこの薬物が児童に対して安全かつ有効であることを発見すれば、その製品の小児科ラベルを獲得する可能性がある。2012年の食品と薬物管理局の安全と革新法案(FDASIA)の公布に伴い、スポンサーはBLAに初歩的な小児科研究計画を提出しなければならない。小児科研究計画は、提案された1つまたは複数の小児科研究の概要、出願人が計画した研究、研究目標および設計、任意の延期または免除請求、および法規要件の他の情報を含み、FDAの同意を得なければならない。FDAまたは出願人はいつでも計画の修正を要求することができる。
FDAは、データの提出を延期することを許可するか、または小児科研究にすべてのまたは部分的な免除を与えることができ、その能動的または申請者の要求に応じてすべての小児科患者または亜群の研究を含むことができる。延期要求および延期要求に関する他の要求および手続きは、“連邦延期審査法”に記載されている。法規が別途要求されない限り、小児科データ要求は孤児の称号を有する製品には適用されない。小児科専門権は、米国では非特許マーケティング専門権であり、承認された場合、任意の既存の規制専有権の条項に追加の6ヶ月間の市場保護を追加することができる。BLAスポンサーから提出された小児科データがこのようなデータに対するFDAの書面要求に公平に応答すれば,この6カ月の排他性を与えることができる。これらのデータは,この製品が研究されている小児科群で有効であることを証明する必要はなく,逆に臨床試験がFDAの要求に公平に応答していると考えられれば,追加的な保護が得られる。要求された小児科研究報告が法定期限内にFDAに提出され、FDAによって受け入れられた場合、製品の法定または規制排他性または特許保護期間にかかわらず6ヶ月間延長される。これは特許期間の延長ではないが、FDAが別の出願を承認できない規制期間を効果的に延長する。
アメリカの孤児薬物の称号
孤児医薬品法によれば、FDAは、米国では通常20万人未満に影響を与え、米国では20万人を超える影響を与え、米国ではそのような疾患または疾患を治療する薬剤または生物薬剤を開発および提供することが合理的に期待されていない孤児の称号を付与することができ、このような疾患または疾患を治療する薬剤または生物学的薬剤を米国で開発および提供するコストは、米国での薬剤または生物学的薬剤または生物学的薬剤の販売から回収される。BLAを提出する前に,指定孤児薬を申請しなければならない。FDAが孤児薬物指定を承認した後、FDAは、治療薬の模倣薬識別情報およびその潜在的孤児の使用を開示する。孤児薬物の指定は、規制審査または承認過程においていかなる利点も伝達されず、規制審査または承認過程の持続時間を短縮することもない。
孤児薬物指定を有する製品がその後、このような指定された疾患を有するFDAの最初の承認を得た場合、この製品は、孤児製品の排他性を得る権利があり、これは、FDAが完全なBLAを含む他の出願を承認しない可能性があり、限定された場合、例えば孤児薬物に対して排他的な製品に対する臨床的利点を示さない限り、7年以内に同じ生物適応を販売することを意味する。孤児薬物の排他性は、FDAが同じ疾患または条件のために異なる薬剤または生物学的薬剤を承認することを阻止しないか、または異なる疾患または条件のための同じ薬剤または生物学的薬剤を使用することを阻止しない。孤児薬を指定する他の利点は、いくつかの研究の税金控除およびBLA申請使用料の免除を含む。
指定された孤児薬物が孤児が指定された適応よりも広い用途で承認された場合,孤児薬物の排他性を得ることはできない。また、FDAが後で決定すれば、米国での独占営業権を失う可能性がある
22
指定された要求に重大な欠陥がある場合、または製造業者は、このようなまれな疾患または疾患を有する患者の需要を満たすのに十分な数の製品を保証することができない。
アメリカの急速な開発と審査計画
FDAは、いくつかの基準に適合した新製品の審査プロセスを加速または促進するための迅速なチャネル計画を持っている。具体的には、新製品が深刻または生命に危険な疾患や状況を治療し、その疾患または状況が満たされていない医療需要を満たす潜在力を示す場合、迅速なチャネル指定を受ける資格がある。高速チャネル指定は,製品と研究中の特定の適応の組合せに適している.高速チャネル指定を取得した製品の場合、FDAは、完全な出願を提出する前に、BLAの部分を検討することをスクロールする可能性がある。スクロール提出を求めるスポンサーは、BLAの各部分の提出スケジュールを提供しなければならず、FDAはスクロール提出に同意しなければならず、スケジュールは許容可能である。また、スポンサーは、BLA第1部を提出する際に必要な使用料を任意に支払わなければならない。
迅速なチャネル指定を有する製品を含む任意の製品は、優先審査および承認の加速など、FDAの開発および審査を加速させるための他のタイプの計画に参加する資格がある可能性がある。重篤な疾患を治療するための製品であり、満足できる代替療法なしに安全で有効な治療を提供する可能性がある場合、または市販されている製品と比較して、疾患の治療、診断または予防において有意に改善されている場合、優先審査を受ける資格がある。FDAは、優先審査として指定された新製品の申請を評価するために追加のリソースを使用することを試み、審査を促進するために努力する。FDAは,出願提出日から6カ月以内に優先出願に行動する予定であるが,通常出願の優先出願日から10カ月間である。
しかも、製品は加速された承認を受ける資格があるかもしれない。深刻なまたは生命を脅かす疾患または状態の治療における研究された製品の安全性および有効性が、臨床的利益を合理的に予測する可能性のある代替終点に有効であるか、または不可逆的な発病率または死亡率よりも早く測定することができる臨床終点に有効であると判定された場合、病状の重症度、希少性または流行率、および代替治療を利用可能または不足し、不可逆的な発症率または死亡率または他の臨床的利益への影響を合理的に予測することが可能であることを考慮すると、加速承認を得ることができる。承認の一つの条件として、FDAは加速承認を得た薬物或いは生物製品のスポンサーに十分かつ良好に制御された発売後の臨床研究を要求し、臨床利益を証明する可能性がある。また、FDAは現在、承認を加速させる条件として宣伝材料を事前承認することを求めており、製品商業発売の時期に悪影響を及ぼす可能性がある。
再生医学高度療法(RMAT)はFDAが2017年に設立したものであり、目的は以下の基準に適合する任意の薬物の有効な開発計画を促進し、それの審査を加速することである:(1)細胞療法、治療用組織工学製品、ヒト細胞と組織製品、またはこのような治療法または製品を使用する任意の組み合わせ製品として定義されているRMATの資格に適合するが、限られた例外である;(2)深刻に生命に危険な疾患または状態を治療、修正、逆転または治癒することを目的としている;および(3)このような疾患または状態の未満足な医療需要を満たす可能性があることを初歩的な臨床証拠は示す。RMATの指定は、FDAとのより頻繁な会議の開催、候補製品の開発計画の検討、審査および優先審査の資格のスクロールなど、潜在的な利点を提供する。RMAT認証が付与された製品も、長期的な臨床的利益を合理的に予測する可能性のある代替物または中間終点に基づいて、またはより多くの場所に拡張することによって加速承認を得ることを含む、大量の場所から取得されたデータに依存する資格がある。承認が得られると、適切なとき、FDAは、臨床証拠、臨床研究、患者登録、または電子健康記録のような他の真の証拠源を提出することによって、より大きな検証データセットを収集することによって、または承認前にすべての治療を受けた患者を承認後に監視することによって、加速承認の下で承認後の要求を満たすことを可能にすることができる。
画期的な治療指定は、深刻または生命に危険な疾患を治療する製品の開発と審査を加速することも目的である。FDAの指定は、1つの候補製品が単独で、または他の薬物および生物製品と組み合わせて使用されることを証明する予備的な臨床証拠を必要とし、1つまたは複数の臨床的重要終点において、臨床開発早期に観察される実質的な治療効果など、現在利用可能な治療法よりも実質的な改善を示す。画期的な治療指定は、指定のすべての利点を迅速に追跡することを伴うが、これは、いくつかの条件が満たされる場合、スポンサーが、一部の申請を提出する提案スケジュールについてFDAと合意すること、およびFDAが審査を開始する前に適用される使用料を支払うことを含む、BLAの提出部分をスクロールして審査することができることを意味する。
迅速チャネル指定、優先審査、RMAT、および画期的な治療指定は、承認の基準を変更することはありませんが、開発または承認プロセスを加速させる可能性があります。もし製品がこれ以上適用された基準を満たしていなければ、FDAはこのような指定を取り消すことができる。
23
アメリカの承認後に要求する
我々がFDAの承認を得たどの製品も、記録保存要件、製品副作用の報告、FDAへの最新の安全および治療効果情報の提供、製品サンプルおよび流通要件、およびFDAの宣伝および広告要件を遵守することを含むFDAの持続的な規制を受けなければならない。その中には、他に加えて、直接消費者向けの広告基準、製品承認用途に記載されていない製品(“非ラベル使用”と呼ばれる)のための製品の普及を制限するか、業界支援の科学的および教育活動の制限、およびインターネットに関連する販売促進活動の要求を含む。医師は合法的な製品を非ラベル用途のために処方することができるが、医師がこのような製品が彼/彼女の専門的な医学判断において適切であると思っている場合、メーカーはラベル外用途を販売または普及させてはならない。しかしながら、場合によっては、製品承認されたラベルと一致する真で誤解されない情報の共有を可能にする。
そのほか、処方薬製品の流通は、多くの処方を必要とする生物製品を含み、“処方薬販売法”(PDMA)の制約を受け、この法案は連邦レベルの薬品サンプルの分配を規定し、各州の薬品流通業者の登録と監督管理に最低基準を設定した。PDMA,州法ともに処方薬製品サンプルの分配を制限し,分配における責任の確保が求められている。
また、品質管理及び製造プロセスは、製品の長期安定性を確保するために、承認された後も適用される製造要件に適合し続けなければならない。CGMP条例では,他の事項のほかに,品質管理と品質保証およびそれに応じた記録やファイルの保守が要求され,cGMPから外れた状況を調査·是正する義務がある.承認製品の製造·流通に参加するメーカーおよび他のエンティティは、FDAおよびある州機関にその工場を登録し、cGMPおよび他の法律を遵守することを確実にするために、FDAおよび特定の州機関の定期的な抜き打ち検査を受けなければならない。そのため、メーカーはcGMPコンプライアンスを維持するために、生産と品質管理に時間、お金、労力をかけ続けなければならない。承認後に製品が発見された問題は、製品のリコールまたは市場からの撤回を含む製品、製造業者、または承認されたBLA所有者の制限をもたらす可能性がある。また,製造プロセスの変更は厳しく規制されており,変更の重要性に応じてFDAの承認を得て実施する必要がある可能性がある。新たな適応や声明を増やすなど、製品の他のタイプの変化を承認するには、FDAのさらなる審査と承認も必要である。
FDAはまた、上場後テスト、いわゆる第4段階テストを要求し、許可された製品の効果を監視するために監視を行う可能性がある。製品が以前に未知の問題を発見し、あるいは適用されたFDA要求を遵守できなかったことは、負の結果をもたらす可能性があり、負の宣伝、司法或いは行政法執行、FDAの警告状、強制要求の是正広告或いは医師とのコミュニケーション、民事或いは刑事罰などを含む。新たに発見または開発された安全性または有効性データは、新たな警告や禁忌症を増加させることを含む製品承認のラベルを変更する必要がある場合があり、他のリスク管理措置を実施する必要がある可能性もある。また、新しい立法による要求、またはFDAの政策が変更される可能性がある新しい政府要求を確立することが可能であり、規制部門が私たちが開発している製品を承認することを延期または阻止する可能性がある。
アメリカマーケティング排他性
生物製品価格競争と革新法(BPCIA)はPHSAを改正し、FDAが類似バージョンの革新生物製品を許可することを許可し、通常は生物模倣薬と呼ばれる。生物類似物の承認を求める競争相手は申請を提出しなければならず、その分子が承認された革新者の生物と高度に類似していることを証明しなければならない。臨床上の不活性成分に微小な差があるにもかかわらず、生物製品と参考製品は安全性、純度と効力の面で臨床的に意義のある差がなく、これは通常分析研究、動物研究と臨床研究によって証明できる。さらに、参照製品と生物学的に類似した製品は交換可能であると考えられ、製品が任意の所与の患者において参照製品と同じ臨床結果を生成することが予想されることが証明され、複数回投与された製品の場合、交換可能な生物学的類似製品および参照生物製品は、参照生物製品の独占的使用と比較して安全リスクを増加させることなく、または治療効果を低下させるリスクを増加させることなく、以前の投与後に交互または交換することができる。FDA承認された参照生物と類似しているか、または交換可能であることが証明された製品であって、承認された製品を発売するのに要するコストおよび時間を減少させる可能性がある製品。生物製品のより大きく、しばしばより複雑な構造に関する複雑さ、及びこのような製品を製造するプロセスは、重大な障害を構成し、FDAのBPCIAの実施を緩和した。
しかし,BPCIAは生物模倣薬のBLASの承認申請を生物の許可日を参考にして4年後まで提出することを禁止している。また、革新者生物製品が初歩的な市場承認を得てから12年以内に、FDAは生物類似申請を許可しない可能性がある。この12年間の参照製品独占期間内に、FDAが競合製品の完全なBLAを承認した場合、出願人自身の臨床前データおよび十分かつ良好に制御された臨床試験からのデータを含み、別の会社はFDA許可を得て参照製品の競合バージョンを販売することができる
24
その製品の安全性、純度、力価。BPCIAはまた、交換可能な製品として承認された生物模倣薬のためのいくつかの排他的期限を設けている。この節では,FDAが“交換可能”と考えている製品が本当に州薬剤法に管轄されている薬局に取って代わられるかどうかは不明である。FDAが要求すれば、この12年間のデータ独占期間は6ケ月延長でき、合計12.5年であり、革新者会社はこの製品の小児科臨床研究を完成した。
生体模倣薬の開発·販売(承認された場合)は、2022年の生体類似ユーザ料金改正案(BsUFA)に基づいて使用料を徴収し、この改正案は現在2027年9月に適用されており、その後更新または改正される可能性がある。スポンサーは、IND提出前またはFDAが第1回BPD会議を承認した後7つのカレンダー日以内に初期生物類似生物製品開発(BPD)費用を提出しなければならず、その後、スポンサーが提出を受け入れられたBPDを提出するまで、またはスポンサーがBPD計画への参加を終了するまでBPDを毎年提出しなければならない。スポンサーが2会計年度連続でBPD年度費用を支払わなければ、FDAはBPD計画からスポンサーを除去することもできる。BPD計画への参加を停止したが,FDAと再協力して製品開発を行いたいスポンサーは,これまでに評価されたBPD費用と再活性化費用をすべて支払わなければならず,BPD年度費用の影響を受ける。スポンサーが生物類似物のBLAを提出すると、彼らは申請料を払わなければならない。また,生物的に類似したBLAが承認されると,スポンサーは年間計画費用を支払う必要がある。FDAはBsUFA項の具体的な費用金額を毎年修正している。
BPCIAは複雑であり、FDAによって解釈され、実施され続けている。また、国会が12年間の参考製品専門期間を短縮すべきかどうかも検討した。BPCIAの他の面では,そのいくつかがBPCIAの排他的条項に影響を与える可能性があり,最近の訴訟のテーマでもある.したがって、生物多様性条約の最終実行には大きな不確実性がある。
FDAが私たちの候補製品の使用時間、期限、詳細を承認することによると、私たちのいくつかの米国特許が承認されれば、1984年の“薬品価格競争および特許期限回復法”(一般に“ハッジ-ワックスマン法案”と呼ばれる)によって限られた特許期間の延長を受ける資格がある可能性がある。ハッジ·ワックスマン法案は、製品開発およびFDA規制審査中に失われた特許期間の補償として、最長5年間の特許回復期限を許可する。しかし,特許期限の回復は特許の残存期間を延長することはできず,製品承認日から合計14年を超えることはできない。特許期間回復期間は、一般に、INDの発効日とBLA提出日との間の時間の半分に、BLA提出日とその出願が承認された日との間の時間を加える。承認された製品に適用される特許は1つのみ延期する資格があり,延期出願は特許が満了する前に提出されなければならない。米国特許商標局はFDAと協議し,任意の特許期限の延長または回復の出願を審査·承認する。将来、私たちは、臨床試験の期待長と関連BLAの提出に関連する他の要因に依存して、現在の満期日以降の特許寿命を延長するために、現在所有または許可されている特許出願のうちの1つの特許期間を回復することを意図しているかもしれない。
アメリカで承認された製品を精算します。
国内外の市場では、任意の承認された製品の販売と精算は、政府の医療計画、商業保険、管理の医療機関のような第三者支払人がこのような製品コストを支払う程度にある程度依存する。第三者支払者は彼らがどの薬を保証し、精算金額を確定するかを決定する。新たに承認された薬物については,保険取得や精算に大きな遅延がある可能性があり,保険範囲はFDAや同様の外国規制機関が承認する目的よりも限られている可能性がある。適用すれば、新薬の一時精算金額も私たちのコストを支払うのに十分ではないかもしれませんし、一時的なものかもしれません。販売率は薬の使用や使用の臨床環境によって異なる可能性があります, 低コストの薬品のために設定された精算レベルに基づいて、他のサービスの既存の支払いに組み込むことが可能である。政府の医療保健計画や個人支払者が要求する強制的な割引やリベート、および将来、米国より低い価格で販売される可能性のある国からの輸入薬品の法律が緩和されることで、薬品の純価格が低下する可能性がある。薬品の保証と精算政策は支払人によって異なる可能性がある。米国の第三者支払者は薬品の保証と精算に統一的な政策がないからである。米国の第三者支払者は、自分の精算政策を制定する際に、通常連邦医療保険保険政策と支払い制限に依存している。保険の獲得と精算に重大な遅延が生じる可能性があります。保険と精算を確定する過程は往々にして時間も高く、これは私たちの製品を使用するために単独で各支払人に科学的と臨床支援を提供することを要求します。保険或いは十分な精算を受けることを保証することはできません。政府当局と第三者支払人が私たちの薬品の保険や精算についてどのように決定するか予測するのは難しい。
これらの第三者決済者は、医療製品やサービスの価格に挑戦し、コストを管理するための制御を実施することが増えている。医療費の抑制は連邦と州政府の優先事項となっており、薬品価格はこの努力の重点である。各国政府はコスト制御計画の実施に大きな興味を示し、価格制御、精算制限と代替後発薬の要求を含む。アメリカでは
25
すでに数回の国会調査を行い、そして連邦と州立法を提出し、公布し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、連邦医療保険下の薬品コストを下げ、そして政府計画の薬品精算方法を改革することを目的とした。例えば、“2021年総合支出法案”には、いくつかの薬品価格報告および透明性措置が含まれており、例えば、いくつかの連邦医療保険計画開発ツールが、連邦医療保険D部分の処方薬福祉情報をリアルタイムで表示することを要求し、団体および医療保険発行者が衛生および公共サービス部、労働部、および財務省の秘書に薬局福祉および薬品コストの情報を報告することを要求する。また、2021年7月9日、バイデン総裁は、処方薬問題を解決するいくつかの取り組みを含む米国経済競争を促進する行政命令を発表した。他の条項では、行政命令は、バイデン政府は“医療保険の薬品価格交渉の許可、インフレ上限の実施、その他の関連改革による処方薬の低減を含む積極的な立法改革を支持する”と規定している。行政命令への応答として、2021年9月9日、衛生·公衆サービス部は高い薬価に対応する総合計画を発表し、その中で国会とこの機関が取ることができる潜在的な立法政策と行政ツールを決定し、薬品価格をより負担と公平にし、処方薬業界全体の競争を改善と促進し、科学革新を促進した。最近では2022年8月に総裁·バイデンが“2022年インフレ率低減法”に署名し,医療保険計画の重大な改革を行った, 薬品定価改革と連邦医療保険D部分の福祉設計の変更を含む。その他の改革では,2022年の“インフレ率低減法案”は,連邦医療保険B部分とD部分で精算された製品に対してインフレリベートを実施し,これらの製品の価格増加がインフレよりも速いことを前提としており,連邦医療保険D部分福祉を改正し,2025年から福祉年度自己支出上限を2,000ドルとするとともに,製薬業者に新たな割引義務を課し,2026年から連邦医療保険·医療補助サービスセンターと価格交渉を行った後,連邦医療保険B部とD部分がカバーする固定数の薬品やバイオ製品のための“最高公平価格”を構築している。州レベルでは、立法機関は、価格や患者の精算制限、割引、特定の製品への参入の制限、マーケティングコストの開示と透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入と大量購入も奨励されている。
アメリカ国内で、もし私たちが将来適切な承認を得て私たちの任意の候補製品をマーケティングすれば、私たちはMedicaid、Medicare、公衆衛生サービス(PHS)薬品定価計画に基づいて、これらの製品の承認とカバーを求め、製品を連邦機関に販売することを求めるかもしれない。
医療補助は連邦と州の共同計画であり、各州が管理し、低収入と障害受益者に向けている。医療補助薬品リベート計画によると、メーカーは州医療補助計画で精算された保険外来薬の単位ごとにリベートを支払わなければならない。各製品の税金還付金額は法律で規定されており、ある価格の上昇幅がインフレを超えた場合、追加の割引を受ける可能性がある。
連邦医療保険は連邦政府が管理する連邦計画であり、65歳以上の個人及びある障害者をカバーする。連邦医療保険D部分は、登録された連邦医療保険患者に自己管理された外来薬(すなわち、通常薬局によって配布され、医師によって管理される必要がない薬剤)の保険を提供する。Medicare Part Dは米国政府によって承認された個人処方薬計画によって管理され、各薬物計画はCMS規則と要求に基づいて独自のMedicare Part D処方薬の保証範囲と定価を確立し、薬物計画は時々これらの規則と要求を修正する可能性がある。
連邦医療保険B部は,入院環境で使用されている注射薬の多くと,病院外来部や医師室の免許を有する医療提供者が管理する薬剤をカバーしている。連邦医療保険B部分は連邦医療保険行政請負業者が管理しており,彼らは通常CMS規則と要求に基づいて保険決定を行うことを担当している。いくつかの支払い調整および制限に基づいて、Medicareは、一般に、製造業者によって報告された平均販売価格のパーセンテージに基づいて、B部分保証薬の費用を支払う。
連邦機関が連邦供給スケジュール(FSS)を通じて薬品を購入する場合、薬品は割引定価の影響を受ける。ある連邦機関が保証と支払いを受ける薬品、及びMedicaid、Medicare Part BとPHS薬品定価計画(通常340 B薬品定価計画と呼ばれる)下の保険には、FSSが必要である。FSS定価は定期的に退役軍人事務部と交渉している。FSS定価は、メーカーがその最恵国非連邦顧客から受け取った製品価格を超えないことを目的としている。また,退役軍人管理局,国防省(軍人や家族がTRICARE小売薬局で計画して購入した薬品を含む),沿岸警備隊やPHSで購入した薬品の価格は定価上限(“連邦最高価格”と呼ぶ)に制限されており,価格上昇幅がインフレを超えると追加割引の影響を受ける可能性がある。
医療補助薬品リベート計画下の薬品カバー範囲を維持するために,メーカーはPHS薬品定価計画に基づいてある購入者に割引を提供することを要求されている。割引を受ける資格のある購入者は提供が含まれています
26
不比例の経済困難患者,コミュニティ衛生診療所,他の実体はPHSの衛生サービス贈与を受けている。
2010年3月、米国議会は、政府医療計画下の薬品のカバー範囲および支払い方法の改正を含む“医療·教育調整法案”(総称して“平価医療法案”と総称する)によって改正された“患者保護·平価医療法案”を公布した。公布以来、“平価医療法案”の多くの内容が司法や国会から疑問視されており、連邦政府の行政·立法部門は“平価医療法案”のいくつかの側面の廃止または代替に努めている。例えば、国会はまだ全面的な廃止立法を通過していないが、“平価医療法案”の一部の条項を改正するための法律が制定されている。例えば、“平価医療法案”の医療保険購入の個人権限を遵守しないため、2019年1月1日から処罰が廃止された。ACAのいくつかの強制費用の実施を延期し、連邦医療保険D部に加入している製薬業者が不足している販売時点保険ギャップ割引を増加させるためである。2021年6月、米国最高裁はACAのいくつかの合憲性を疑問視する訴訟を却下したが、合憲性議論の是非について判断しなかった。さらに,2021年米国救援計画,Pub.2021年3月11日に公布された法律第117-2号は、2021年と2022年に補助金を受ける資格のある人に対する保険税控除援助を一時的に増加させ、400%の連邦貧困レベル制限を撤廃し、そうでなければ、保険税控除を受ける資格がある目的に適用される。最近、2022年のインフレ削減法案は、増加した税収控除援助と400%の連邦貧困上限の廃止を2025年に延長する。バイデン政府の医療改革措置や今後のいかなる訴訟が、平価医療法案と我々の業務にどのように影響するかは不明である。
アメリカ医療保険法
医療提供者と第三者支払者は、私たちが規制部門の承認を得た任意の候補製品の推薦と処方において主な役割を果たすだろう。私たちの現在と未来の第三者支払者と顧客との手配は、広範に適用される詐欺や乱用、その他の医療法律や法規に直面する可能性があり、これらの法律と法規は、私たちの研究やマーケティング、販売、流通、私たちの製品の業務または財務配置と関係を制限するかもしれません。バイオ製薬会社として、医療サービスの転転を制御したり、連邦医療保険、医療補助または他の第三者支払者に請求書を発行したりすることはありませんが、詐欺や乱用、および患者の権利に関する連邦および州医療法律法規は現在も将来も私たちの業務に適用されています。適用される連邦と州医療法律と法規によると、私たちの運営能力に影響を与える可能性のある制限は、
27
私たちが第三者の業務手配と適用される医療法律や法規に適合していることを確保するためには、大量のコストがかかります。政府当局は、我々の業務やり方は、現在または将来、医療法律や法規の適用に関連する法規、法規または判例法に適合していないと結論するかもしれない。もし私たちの運営がこれらの法律または任意の他の政府法規に違反していることが発見された場合、私たちは重大な民事、刑事と行政処罰、損害賠償、罰金、監禁、連邦医療保険と医療補助、返却、追加の報告要件または監督(私たちが会社の誠実な合意または同様の合意の制約を受けている場合)、および私たちの業務の削減または再編のような政府援助の医療計画から除外される可能性がある。
外国監督管理
アメリカの法規以外に、私たちは各種の外国法規の制約を受けて、これらの法規は私たちの候補製品の臨床研究、商業販売と流通、及び医療保健専門家との相互作用を管理している。私たちがFDAの候補製品の承認を得るかどうかにかかわらず、私たちはこれらの国や地域で臨床研究や製品の販売を開始するために、外国や経済地域(例えばEU)の比較可能な規制機関の承認を得なければならない。臨床研究、製品許可、定価と精算を管理する審査手続きと要求は地によって異なり、時間はFDA承認の時間より長い或いは短い可能性がある。
米国以外のある国には,ヒト臨床研究を開始する前に臨床試験申請(CTA)の提出を要求するプログラムがあり,米国のINDに似ている。例えば,EUでは,EU臨床試験条例536/2014(CTR)の要求に基づき,CTAは主管する国家衛生当局に提出しなければならないか,EU中央ポータルサイト,臨床試験情報システム(CTIS)および会社が臨床研究を展開しようとしている国ごとの独立倫理委員会に単一申請を提出しなければならない。CTAが一国の要求に応じて承認されると,臨床研究開発はその国で行うことができる。すべての場合,臨床研究はGCPや他の適用法規の要求に基づいて行われなければならない。出願手続の一部として、スポンサーは、申請の検証及び評価を調整するために、報告書を提出する加盟国を提案しなければならない。報告書を提出した会員国は他の関連会員国と協議して調整しなければならない。もし承認されれば、スポンサーはすべての関連会員国で臨床試験を開始することができる。しかし、関連会員国は限られた状況で“選択脱退”原子力規制書を発表することができる。このような場合、臨床試験はこの会員国で行われてはいけない。CTRはまた、安全報告に関する規則を簡素化および簡略化し、EUデータベースに臨床試験結果要約を強制的に提出するなど、透明性要件を強化することを目的としている。CTRによると,2022年1月31日から臨床試験スポンサーはCTIを使用することができたが,CTIの使用義務はなかった。2023年1月31日から、臨床試験スポンサーはCTISを使用してEUや欧州経済圏での新たな臨床試験の開始を申請しなければならない, しかし,これまでの法律で承認された臨床試験によると,臨床試験指令(CTD)は2025年1月31日までCTDで動作し続けることができ,スポンサーはCTRを遵守してCTIにこれらの研究の情報を記録しなければならない。2022年1月31日から、EU加盟国と欧州経済圏諸国の国家規制機関は、その法的責任を履行し、CTI使用の臨床試験を評価·監督し始めた。
EU規制制度の下で、会社は国家、集中または分散、または相互承認の手続きの下でマーケティング許可申請を提出することができる。私たちは集中的な手続きを使いたいと思っていますこれは医療上の義務的な手続きです
28
エイズ、癌、神経変性疾患、糖尿病、ウイルス性疾患および指定された孤児薬物などの特定の適応、ならびに任意の他の高度に革新的な薬剤の治療のための、バイオテクノロジーによって生産された製品または新規活性物質を含む医薬製品。中央プログラムにより,マーケティング申請はEMAに提出され,人間が薬品委員会で評価した。もし委員会が有利な意見を発表した場合、これは一般に、欧州委員会が意見を受け取ってから67日以内にすべてのEU加盟国に有効な単一のマーケティング許可を付与することにつながる。最初のマーケティング許可の有効期限は5年ですが、一度更新すると、通常有効期限は制限されません。
(1)製品のリスク·利益バランスが正である場合、(2)申請者が必要な全面的な臨床研究データを提供することができる可能性が高い、(3)満たされていない医療需要が満たされる、および(4)医薬品の即時発売による公共健康へのメリットが依然として追加データが必要であるという事実に固有のリスクを超える場合、不完全な臨床データに基づいて、限られた数の人用医薬品(EU法律に従って孤児医薬品として指定された製品を含む)に対するEUでの限定された数の人用医薬品の条件付きマーケティング許可を可能にする。具体的義務には,進行中または新たな研究の完了に関する義務と,薬物警戒データの収集に関する義務が含まれており,条件付きマーケティング認可に具体的に規定することができる。条件付きマーケティング許可の有効期間は1年であり、リスク-収益バランスが正のままであり、条件の追加または修正の必要性を評価した後、毎年更新することができる。スポンサーはまた、“特別な状況”と呼ばれる異なるマーケティング許可経路を使用することができ、この場合、許可後であっても全面的なデータを得ることができない場合(例えば、まれな疾患または疾患の場合)、欧州委員会は、特定の疾患または疾患について製品のマーケティング許可を与える。特殊な場合には薬品の上場許可を得たスポンサーには継続的な発売後の義務があり,その製品のメリットを確認し続けなければならない。“特殊状況”の監督管理ルートによって付与された上場許可が引き続き存在するかどうかは、毎年再評価を行う必要がある。年間再評価は、営業許可を維持、変更、または一時停止すべきかどうかを決定します, スポンサーがその上場後の義務と製品のリスク·収益の概要を履行することに基づく。
アメリカのように、私たちはMAAを申請する前に、EUで特定の適応を治療する孤児薬として製品を指定することを申請することができる。ヨーロッパの孤児薬物は、承認された適応が最長11年の孤児市場排他性を含む経済とマーケティング利益を有しており、他の申請者がその製品が孤児指定製品よりも安全で、有効であることを証明できない限り、または他の点で孤児指定製品よりも優れていることを証明することができる。良質な計画はヨーロッパ薬品管理局が設立したものであり、目的はヨーロッパ連合の新薬の開発促進と促進を助けることであり、これらの新薬は満たされていない医療需要領域で重大な治療優勢を持つ潜在力を示した。Prime認証のメリットは加速評価の潜在力を早期に確認し、早期に関連監督委員会と対話と相互作用を強化し、発展選択を討論し、肝心な発展マイルストーンで科学提案を提供し、及びEMAからの積極的な監督管理支持を提供し、製品が更に速いMAAを獲得させることを含む。
EUでは,新医薬製品を開発する会社はEMAの小児科調査計画(PIP)に同意し,このPIPに基づいて小児科臨床試験を行わなければならない。製品のMAAは,免除が適用されない限り,PIPによる小児科臨床試験の結果を含まなければならず,この場合,小児の症例研究(例えば,疾患や状況が成人でのみ発生する場合)を必要としない。あるいは延期が許可されており、この場合、小児科臨床試験は遅く完了しなければならない。PIPによる小児科臨床試験により発売許可を得た製品には,補充保護証明書(承認時に有効であれば)によって6カ月の保護延長を得る資格があり,あるいは孤児医薬製品に対しては,孤児市場を2年間排他的に延長する資格がある。この小児科奨励は特定の条件の制約を受け,PIPに適合したデータを開発·提出する際に自動的に獲得されない。
アメリカ以外にも、私たちの製品が十分なカバー範囲と支払いを確保する上で他の挑戦があります。多くの国で、処方薬の価格設定は政府によって規制されている。政府当局との価格交渉は製品の監督管理の許可を受ける範囲をはるかに超えている可能性があり、そして私たちに臨床研究を要求し、私たちの候補製品或いは製品のコスト効果を他の利用可能な治療法と比較することができるかもしれない。このようなタイプの臨床研究を行うことはコストが高く、私たちまたは私たちの商業化パートナーの商業化努力の遅延を招く可能性がある。第三者決済者は医療製品やサービスの価格に挑戦しており、多くの第三者支払者が新たに承認された医療製品の精算を制限している。多くのEU諸国の予算圧力もまた、価格の凍結、値下げの増加、リベートなど、各国政府に様々なコスト制御措置を考慮または実施させている。予算圧力が持続すれば、各国政府は追加的なコスト制御措置を実施するかもしれない。コストコントロールは、私たちが開発または販売する可能性のある製品のために制定された価格を下げるかもしれません。これは、製品収入や私たちに支払うべき印税を減少させることになります。薬品に対して価格制御や精算制限を実行する国が私たちのいかなる製品にも有利な精算と定価手配を許可することは保証されません。
ヨーロッパ共同体は現在薬品の法律枠組みに対して大規模な審査を行っており、その中には医薬製品に提供する監督管理保護、例えばデータ独占性、マーケティング保護、孤児適応の市場独占性と小児科普及が含まれている。EUが現在提供している保護は今後数年で減少すると予想され、新しいEU
29
立法提案はこのスケジュールがさらに延長される可能性があるにもかかわらず、2023年第2四半期に欧州共同体によって発表される予定だ。
イギリスの離脱とイギリスの規制枠組み
2016年の国民投票結果に続き、英国は2020年1月31日にEU離脱、通称離脱となった。英国とEUが合意した正式な離脱手配によると、英国には2020年12月31日(移行期間)まで移行期間があり、その間EUルールは適用され続けている。2020年12月には、英国とEU間の将来の貿易関係を概説し、各EU加盟国とイギリスの承認を得た英国-EU貿易·協力協定(同協定)が達成された。
イギリスが私たちの業務と私たちの候補製品に適用される規制枠組みの大部分はEU指令と法規から来ているため、イギリス立法は既存のEU法律を維持している。しかし、イギリスは新しい立法を起草しているが、イギリスはCTRのような新しいEU法律を施行していない;したがって、イギリスは離脱し、イギリスとEUの候補製品の開発、製造、輸入、承認、商業化に関する私たちの規制制度に実質的な影響を与え続けるだろう。大ブリテン(イングランド、スコットランド、ウェールズからなる)は、欧州経済地域からマーケティング権限を付与された手続きの管轄を受けなくなった(北アイルランドは集中権限手続きの管轄を受け、北アイルランド議定書に基づいてEUの法的枠組みが北アイルランドに適用され続けるので、分権または相互承認手続きの管轄を受けることができる)。イギリスで薬品を販売するには単独のマーケティング許可が必要だ。イギリスのヘルスケア製品規制機関(MHRA)が、それが受け取る可能性のあるますます多くのマーケティング許可申請を処理するのに十分な準備があるかどうかは不明である。いかなるマーケティング承認を得ることもできない点でのいかなる遅延も、私たちの候補製品がイギリスやEUで商業化されることを延期または阻止し、私たちの収入の創出、利益を達成、維持する能力を制限するだろう。
この協定は連合王国とEU間の医薬製品ゼロ関税貿易を規定しているが、このような貿易は過渡期終了前には存在しない追加の非関税コストを生じる可能性がある。また、連合王国が医薬製品に関する規制の観点でEUに逆行すれば、将来的に関税が徴収される可能性がある。したがって、私たちは現在も未来も、事業を運営する重大な追加費用(過渡期が終了する前の場合と比較して)に直面する可能性があり、これは、私たちの収入を創出したり、業務の収益性を達成したりする能力を著しく損害したり、延期したりする可能性がある。イギリスの離脱やその他の理由で、国際貿易、関税、輸出入法規の任意のさらなる変化は、予期しない関税コストまたは他の非関税障壁をもたらすかもしれない。これらの事態が発展したり、そのいずれかが起こりうるとの見方は、世界貿易、特に影響を受けている国と連合王国との貿易を大幅に減少させる可能性がある。イギリスの離脱は、特にEUから来た従業員たちのために、私たちが従業員を引き付け、維持する能力にマイナスの影響を与える可能性もある。
英国の離脱後,英国が孤児に指定された基礎はEUとほぼ同じであるが,このような状況によるイギリスでの一般的な程度である。したがって,現在イギリスで孤児条件に指定されている条件は孤児条件として指定されなくなり,EUでは現在孤児条件に指定されていない条件はイギリスで孤児条件として指定される。
法規を付加する
バイオ製薬会社として、医療サービスの転転を制御したり、連邦医療保険、医療補助または他の第三者支払者に請求書を発行したりすることはありませんが、詐欺や乱用、および患者の権利に関する連邦および州医療法律法規は現在も将来も私たちの業務に適用されています。私たちはまた、“職業安全と健康法”、“環境保護法”、“有毒物質制御法”、“資源保護と回収法”、その他の既存と潜在的な連邦、州あるいは地方法規の規制を受けている。これらの法律と他の法律は私たちの様々な生物と化学物質の使用、処理、処分、これらの物質と化学物質は私たちの業務、そして私たちの業務から発生する廃棄物を規範化している。私たちの研究と開発は危険材料、化学品、ウイルスの制御された使用に関するものだ。このような材料を処理·処分するセキュリティプログラムは州や連邦法規で規定されている基準に適合していると信じているが,これらの材料による意外な汚染や傷害のリスクは完全には解消できない。もしこのような事故が発生した場合、私たちはそれによるいかなる損害に対しても責任を負う可能性があり、このような責任は私たちの資源範囲を超える可能性がある。
製造業
2022年4月、カリフォルニア州千オーク市に位置するAtara T細胞運営および製造施設(ATOM施設)に関連する特定の資産のすべての権利、所有権、および権益をFDBに売却する。私たちは教師級のサービスや
30
FDB(Fujifilm MSA)との供給協定は2022年4月に発効し、最長10年間延長できる。富士フイルムMSAによると、FDBはcGMP規格に基づいて特定の数量の製品と候補製品を提供する。富士フイルムMSAはFDBから私たちの製品と候補製品だけを購入することを要求していません。患者に対する私たちの期待とEUの製品に対する需要によると、私たちの現在のEbvallo在庫は2023年末までのEUの商業需要を満たすのに十分だと信じている。
著者らは引き続き著者らのEBV T細胞製造プラットフォームを拡大し、単一ドナーの白血球分離採集の製品生産量を向上させ、そしてすでに生成したデータは攪拌槽バイオリアクターを使用して生産量と細胞成長生産性を高めることを実証した。私たちの拡張可能な技術は、生物コストのような製品製造を提供する重要な推進要素になり、私たちのCAR T計画を含めて、私たちの製品の組み合わせで利用できると信じています。我々は,新冠肺炎の大流行に関連する臨床前と臨床細胞療法に用いた原材料と消耗材の供給が一時的に中断し,我々の候補製品に原材料を提供する白血球分離採取を含む一時的な中断を開発·製造した。これらの原材料や他の必要な原材料をタイムリーに得ることができなければ、私たちの業務運営や製造能力は悪影響を受ける可能性があります。
FDBに加えて,2019年12月に締結されたビジネス製造サービスプロトコル(CRL MSA)によりCharles River実験室社(CRL)と連携する.CRL MSAにより、CRLは私たちの製品といくつかの候補製品に製造サービスを提供します。2023年2月、CRL MSAを修正し、期限を2023年9月30日の早い時期に延長するか、またはいくつかのロットの製品および候補製品を受信します。
私たちの現在の製造戦略は、各候補製品を評価し、私たちの製造ネットワークのどの場所が適切な段階の技術、品質、法規遵守要求を提供するかを決定することである。また、私たちは、ネットワーク全体でそれに応じて製造能力と能力を計画することを確実にするために、候補製品の長期供給需要を定期的に評価する。我々の製造ネットワークは,我々自身の工場と我々のパートナーの製造能力からなり,QIMR Berghoferの付属会社MSKとQ−Gen Cell Treeutics,SAFC Carlsad,Inc.,FDBとCRLを含む契約製造組織(CMO)である。この戦略的方法は、私たちの臨床的および商業的生産需要を支援し、時間または能力制限を解決し、適切な場合に供給冗長性を提供するための柔軟性を提供してくれる。
我々のT細胞候補製品は,FDAとEMA互換性の採取センターを介して健康で同意した第三者ドナーから得られた血液由来の出発材料を必要とする。我々の生産操作は連邦法規の良好な製造規範(GMP)及び良好なティッシュ規範(GTP)に従っている。GTPはFDAの法規制およびガイドラインであり,HCT/Psを製造するための方法およびHCT/Psを製造するための施設および制御を管理し,HCT/Psはヒトレシピエントに移植,移植,注入または転移するためのヒト細胞または組織である。GTP要求の主な目的は,細胞や組織に基づく製品の製造方式の確保であり,感染症の導入,伝播,伝播を防止することを目的としている。FDAの規定はまた、組織機関がFDAに彼らのHCT/Pを登録し、適用した場合にスクリーニングとテストを通じてドナーを評価することを要求する。
我々のパートナーと合意することにより,生産臨床研究に関連する薬物供給に関連するいくつかの製造プロセス技術を使用する権利を獲得した。これらの材料は、重要な出発材料および中間体、ならびに既存の臨床研究材料の在庫を含む臨床研究材料の製造を支持する材料を含む。私たちは第三者から供給を受けて、私たちが健康で同意した第三者ドナーから寄付するために必要な出発材料を持っていることを確実にすることができる。
人的資本管理
2022年12月31日まで、私たちは334人の従業員を持っている。私たちは、私たちの業務の成功は私たちが合格した人材を誘致し、維持する能力にある程度依存すると信じている。私たちの人的資本戦略は、協力と革新の文化を育成し、多様性と包容性を抱きながら、私たちの業務目標を成功させることを目的としている。著者らは従業員の尊敬度、空いている率、求人時間、昇進率、業績格付け、後任の深さ、留任、平等な雇用機会のコンプライアンス、給与公平性と多様性代表などの人的資本指標の洞察を通じて、私たちの成功を監視した。我々の報酬政策と持分インセンティブ計画の主な目的は、株式ベースの報酬奨励と現金ベースの業績ボーナスを付与することで業績報酬を支払い、従業員と役員を吸引、維持、激励することである。私たちの従業員のうち一人も労働組合代表でもなく、集団交渉合意の一方でもなく、私たちは従業員との関係が良いと思います。
新冠肺炎商業動態
著者らは引き続き新冠肺炎疫病が著者らの業務と運営に与える影響を監視し、そしてすでにこのような影響を最小限に下げ、業務の連続性を維持する措置を取った。私たちは一部の従業員を遠隔地に移行し
31
在宅勤務モデルは、必要な対面実験室機能を維持しながら、肝心な研究、開発と製造優先事項を推進する。私たちは私たちの現場職員たちを支援するために安全協定と手続きを施行した。
著者らの臨床研究と運営チームは臨床サイトと協力し、新冠肺炎の大流行の影響を最小限に下げた。必要な場所では,遠隔研究アクセス,遠隔医療,在宅保健,その他の方法を利用して,キー終点データを保持しながら患者に連続的な看護を提供することを確保している。
今まで、新冠肺炎疫病はまだ著者ら或いは著者らのパートナーの臨床、研究開発、監督と製造運営或いはスケジュールに実質的な影響を与えていない。しかし,大流行開始時には,再経験する可能性があり,臨床研究操作の一時的な遅延,結果として新冠肺炎を経験した。
新冠肺炎疫病は私たちの業務と運営にどの程度の影響を与える可能性があり、未来の事態の発展に依存し、これらの事態の発展は不確定であり、予測も困難である。
新冠肺炎の流行が我々の業務、財務状況、運営のリスクおよび不確実性に影響を及ぼす可能性のあるより多くの情報については、“1 A”というタイトルの部分を参照されたい。本年度報告表格10−K第I部第1 A項下の“リスク要因”。
企業情報
私たちは2012年にデラウェア州に登録して設立した。当社の主要会社事務所はカリフォルニア州91320千オーク市Conejo Spectrum St.2380 Suite 200、郵便番号:(8056234211)。私たちのサイトの住所はwww.atarabio.comです。
利用可能な情報
我々は,Form 10−K年次報告,Form 10−Q四半期報告,Form 8−K現在報告,依頼書,その他の資料を米国証券取引委員会(米国証券取引委員会)に提出した。私たちが電子的にアメリカ証券取引委員会にこのような報告書を提出または提供した後、私たちは合理的で実行可能な範囲でできるだけ早く私たちのサイトを通じてこれらの報告書を無料で提供します。当社のウェブサイトに含まれている、または当社のウェブサイトを介してアクセス可能な情報は、本10-Kフォーム年次報告書または米国証券取引委員会に提出された任意の他の文書の一部でもなく、引用によって本報告に組み込まれているわけでもありません。
米国証券取引委員会には、米国証券取引委員会に電子的に提出された報告書、依頼書、情報声明、その他の発行者に関する情報が含まれており、サイトはwww.sec.govである。
32
第1 A項。国際ロータリーSK因子
私たちの普通株に投資することは高い危険と関連がある。投資家は、我々の普通株に投資する前に、本10-K表年次報告に含まれる他の情報に加えて、本報告書の“経営層の財務状況および経営結果の議論および分析”と題する部分、および我々の連結財務諸表および関連説明を含む以下に説明するすべてのリスク要因および不確定要因を慎重に考慮すべきである。
以下に述べるリスクはわが社に関連する唯一のリスクではないかもしれませんが、現在重要でない他のリスクも私たちに影響を与えている可能性があります。これらのリスクのいずれかが現実になった場合、以下に説明するリスクを含む場合、私たちの業務、競争地位、名声、財務状況、運営結果、キャッシュフロー、および将来の見通しは深刻な損害を受ける可能性がある。この場合、我々証券の市場価格は下落する可能性があり、投資家は投資の全部または一部を損失する可能性がある。
私たちの財務業績と資本需要に関連するリスク
私たちは設立以来大きな損失を被っており、予測可能な未来にも大きな損失を被ることが予想される。
私たちは臨床段階の生物製薬会社です。生物製薬製品開発への投資は非常に高い投機性があり、それは大量の前期資本支出を必要とし、及び候補製品は有効であることを証明できず、監督管理の許可を得られない或いは商業上実行可能な重大なリスクがないからである。私たちは、EUで承認され、商業製品販売から何の収入も生じず、私たちの持続的な運営に関連する大量の研究開発や他の費用が発生し、このような費用が引き続き発生すると予想される製品、Ebvalloを持っている。したがって、私たちが設立されて以来、私たちは年度報告期間ごとに重大な運営損失を発生させた。2022年12月31日までの1年間に、純損失2億283億ドルを報告します。
今まで、私たちは商業製品販売から何の収入も得ていない。私たちはいつ、あるいは商業製品の販売から十分な収入が発生して、私たちの運営費用を相殺するかどうかわかりません。私たちは、予測可能な未来に、私たちが引き続き研究、開発、規制機関が私たちの候補製品、および私たちが買収、許可、あるいは開発する可能性のある他の候補製品の承認を求めるにつれて、巨額の費用と運営損失を招き続けると予想している。私たちは予測できない費用、困難、合併症、遅延、および私たちの業務に悪影響を及ぼす可能性のある他の未知の要素に直面するかもしれない。私たちの未来の純損失の規模は私たちの支出の変化率と私たちが収入を作る能力にある程度依存するだろう。もし私たちの候補製品が臨床研究で失敗した場合、あるいは規制部門の承認を得なかった場合、あるいは承認され、市場の承認を得られなかった場合、私たちは決して利益を上げないかもしれない。私たちが未来に利益を達成しても、私たちはその後の時期に利益を維持できないかもしれない。私たちが既存の候補製品の研究と開発に投資し続け、調査して新しい候補製品を獲得する可能性があることに伴い、将来私たちの費用が増加する可能性がある。
私たちの経営の歴史は限られていて、これは私たちの業務のこれまでの成功度を評価することを難しくし、私たちの未来の生存能力を評価することも難しいかもしれません。
これまで、当社の業務は、当社の組織と配備に限られており、製品や技術的権利を獲得し、候補製品のための製品開発活動を行ってきました。私たちは、いかなる第三段階の臨床研究を成功させ、アメリカの監督管理機関の許可を得て、商業規模の製品を一貫して生産したり、第三者代表を手配して、あるいは私たちの任意の候補製品の商業化に必要な販売とマーケティング活動を行うことができるか、あるいは第三者代表を手配して私たちにそうすることができることを証明していません。また,我々の候補T細胞製品の背後にある養子免疫治療技術は,我々の次世代CAR T計画を含め,新たであり,ほとんど検証されていない。我々の将来の成功,発現あるいは生存能力に関する予測,特に急速な発展を考慮した免疫治療分野は,不正確であることが証明されている可能性がある。
また,若い企業としては,予見できない費用,困難,合併症,遅延などの既知や未知の要因に遭遇する可能性がある。様々な要因により、私たちの財務状況と経営業績は四半期ごとと毎年大幅に変動し続け、その多くの要素はコントロールできないと予想しています。したがって、私たちのどの四半期や年間業績も未来の経営業績を予測することはできません。
33
私たちは商業製品の収入がありません。私たちは商業製品の販売から収入を得ないかもしれないし、利益を達成しないかもしれない。
今まで、私たちは商業製品販売から何の収入も得ていない。私たちは連合でEbvalloという製品の規制承認を得た。私たちは、Pierre Fabre商業化プロトコルに従って、EbvalloのEUでの商業化権利をPierre Fabreにアウトソーシングし、HCRxプロトコルに従ってPierre Fabre商業化プロトコル下のいくつかの特許使用料とマイルストーン権利をHCRxに販売したが、特定の上限によって制限されている。私たちが製品販売から収入を創出し、利益を達成する能力は、Pierre Fabre商業化プロトコルおよびHCRxプロトコルに支配され、私たちの商業化パートナーが製品の商業化に成功する能力に応じて、私たちの現在の任意の製品および製品候補、および私たちが将来開発、許可または買収する可能性のある他の候補製品を含む。私たちが製品販売から収入を得て利益を達成する能力はまた、私たちが能力があるかどうかを含む多くの他の要素に依存するだろう
私たちがEbvalloまたは規制の承認を受けた任意の候補製品から得られる収入は、私たちが規制の承認を得た地域の市場規模、製品の受け入れ可能な価格、任意の価格で補償を受ける能力、およびパートナーとその地域について達成された商業化合意の条項と条件に部分的に依存するだろう。Ebvalloマーケティング許可の承認と譲渡に関連して私たちに支払われるいくつかのマイルストーン支払いに加えて、HCRx合意の下で適用される特許権使用料の上限に達するまで、Pierre Fabreから意味のあるマイルストーンや特許使用料支払いは何年もかかるかもしれません。もし私たちの解決可能な疾患患者の数が私たちが推定したほど多くなければ、規制部門が承認した適応は私たちが予想していたより狭い、あるいは競争、医師の選択、治療ガイドライン、または解決可能な疾病の発病率の低下により、合理的に治療を受ける人たちを縮小し、承認されても、私たちのパートナーは私たちの製品を商業化することに成功できないかもしれない。私たちがパートナーから受け取る可能性のある任意のマイルストーンと特許権使用料の支払いの時間と金額、そして私たちの製品の商業的成功は、私たちのパートナーの努力、資源分配、定価と清算交渉、そして私たちの製品の成功した商業化にかかっています。したがって、私たちが製品収入を生み出しても、私たちは利益を得ることができず、運営を継続するために追加資金を得る必要があるかもしれない。もし私たちが利益を上げることができない場合、あるいは持続的に利益を上げることができなければ、計画通りに運営を続けることができず、運営を減らすことを余儀なくされる可能性がある。
私たちは私たちの目標を達成するために大量の追加資金を必要とし、必要な時に必要な資本を得ることができなければ、私たちの製品開発や製造努力を延期、制限、減少、または中止させることができるかもしれない。
著者らは予測可能な未来に大量の資源を投入し、著者らのT細胞免疫治療候補製品の臨床開発と生産を継続し、そして著者らの臨床前研究導管を推進と拡大することを望んでいる。我々はまた,我々の製品や候補製品の開発と製造,パートナーから許可や独占許可を得る技術の開発や製造に資源を投入していく予定である.これらの支出には、研究および開発、可能性のある新たな候補製品または技術の獲得、臨床前および臨床研究、および規制承認および製品の製造に関連するコストが含まれる。すべてのライセンス内のパートナーと締結したライセンス契約条項によると、私たちはいくつかの開発、規制、
34
ビジネスのマイルストーンです。さらに、他の予期しない費用も発生する可能性がある。私たちが行っている、計画的、予想された臨床研究の設計と結果は高度な不確実性があるため、私たちは私たちの製品と候補製品の開発と商業化に成功するために必要な実際の数量を合理的に見積もることができない。
私たちの将来の資本需要は多くの要素に依存しています
2022年12月31日までの既存の現金、現金等価物、短期投資に加え、2023年1月にピエール·ファブレ商業化協定下のいくつかのマイルストーンを実現するために受け取った4000万ドルが、2024年第2四半期まで私たちの計画中の運営に資金を提供するのに十分になると予想される。2022年12月31日現在、私たちの現金、現金等価物、短期投資総額は2兆428億ドルです。しかし、現在知られていない多くの要素のため、私たちの運営計画は変化する可能性があり、私たちは計画よりも早い追加資金が必要かもしれない。
マイルストーンと特許使用料を除いて、私たちはPierre Fabre商業化協定に基づいて、HCRx協定の条項に従って他の資金を受け取ることができる約束された外部資金源を持っていない。Ebvalloマーケティング許可の承認と譲渡に関連して私たちに支払われるいくつかのマイルストーン支払いに加えて、HCRxプロトコルで適用される特許権使用料の上限(あれば)に達するまで、Pierre Fabreから意味のあるマイルストーン支払いや特許権使用料支払いは受けられません。追加的な公共または私募株式発行または債務融資を通じて、潜在的な協力、協力または他の戦略的手配、または上記の各項目の組み合わせによって、追加資金の獲得を日和見的に求めることが予想されるが、追加資金が必要な場合、私たちは受け入れられる条項で追加資金を得ることができないかもしれない、または全くないかもしれない。もし私たちが適時に十分な資金を得ることができなければ、私たちは私たちの運営に資金を提供するのに十分な運営資金がないかもしれないし、経営を続けることができない場合、私たちは私たちの1つまたは複数の候補製品の臨床前研究、臨床研究、または他の開発活動を延期、制限、減少、または中止することを要求されるかもしれない。
追加資本を調達することは、私たちの既存の株主を希釈し、私たちの運営を制限したり、私たちに不利な条項で私たちの候補製品に対する権利を放棄することを要求するかもしれません。
私たちは私募と公開発行および債務融資を含む様々な方法で必要な追加資本を求めるかもしれない。例えば、2022年12月、私たちはピエール·ファブル商業化協定に従って、私たちの特定の特許使用料とマイルストーン権利をHCRxに売却しましたが、指定された上限があります。私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、または既存の株式証所有者が普通株を購入する権利を行使する場合、既存の株主の所有権権益は希釈され、条項は清算または他の株主権利に悪影響を及ぼす特典を含む可能性がある。もし株式推定値が、私たちの普通株の取引価格を含む場合、経済中断や他の不確実性によって低くされ、例えば、新冠肺炎の流行、上昇するインフレ圧力、ウクライナへの持続的なロシアの侵入、あるいは他の要素により、このような希釈の潜在的な幅は増加するだろう。債務融資が可能であれば、関連する可能性のある合意は、追加債務を招くこと、資本支出を行うこと、許可手配を達成すること、または配当を宣言することを含む、いくつかの行動をとる能力を制限または制限する契約を含む。もし私たちが第三者からより多くの資金を調達した場合、私たちは私たちの技術または製品候補に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利な条項でライセンスまたは他の権利を付与しなければならないかもしれない。もし私たちが必要な時に株式や債務融資を通じて追加資金を調達できない場合、私たちは候補製品に対する私たちの製品開発を延期、制限、減少、または終了し、他の人に候補製品の開発とマーケティングの権利を与えることを要求されるかもしれません。そうでなければ、私たちは自分を発展させたり、私たちの業務に不利な他の行動を取りたいです。
35
私たちが発表したリストラは予想された節約をもたらさない可能性があり、総コストと支出が予想以上に増加し、私たちの業務を混乱させる可能性がある。
2022年8月、経営陣メンバーを含む会社の全部門で約20%の人員削減を発表した。人員の減少は,キー研究開発計画をめぐる優先順位と我々の費用状況の減少を反映している。予見できない困難、遅延或いは意外なコストのため、私たちは私たちの再構成努力による期待収益、節約とコスト構造の改善を完全に或いは部分的に実現できないかもしれない。再編から予想される運営効率とコスト節約を実現できなければ、私たちの運営業績や財務状況は不利な影響を受けるだろう。私たちはまた私たちが今後もっと多くのリストラや再編活動をする必要がないということを確信できない。また、私たちのコスト節約計画は私たちの運営を混乱させるかもしれません。これは私たちの製品収入を作る能力に影響を与えるかもしれません。また、私たちのリストラは、計画されたリストラを超えた自然減員や、私たちの日常運営の中断など、予期せぬ結果をもたらす可能性がある。私たちのリストラはまた、私たちの業務に重要な合格管理、科学、臨床、製造人員を吸引し、維持する能力を損なう可能性がある。合格者を誘致または維持できなかった場合は、将来的にEUまたは私たちの候補製品のEbvalloの開発と商業化を阻害する可能性がある。
富士フィルム取引のすべての予想されるメリットを実現する保証はありません。私たちは思わぬ挑戦に直面するかもしれません。
私たちは富士フィルム取引の予想収益の一部または全部を実現できないかもしれません。私たちは(I)私たちがFDBに提供するいくつかの移行サービスの提供と(Ii)FDBが富士フィルムMSAによるサービスを提供することに関連するリスクを含む取引完了後のリスクに遭遇する可能性があります。Atom施設の従業員が富士フイルム取引によってFDBに移転されたため、より多くの困難や機関知識の損失に遭遇する可能性があり、これは私たちの業務を損なう可能性があります。過渡期には、富士フイルムの取引には多大な時間と資源が必要となり、これは私たちの業務を混乱させ、経営陣の他の責任への注意を分散させる可能性があり、賠償や他の財務計画を含むATOM施設の財務活動への損失や継続を招く可能性があり、これは私たちの財務業績に悪影響を及ぼす可能性がある。
私たちの製品や候補製品開発に関するリスク
私たちの開発は全体的にまだ初期段階であり、臨床開発では一部の候補製品しかない。私たちの他のすべての候補製品はまだ臨床前開発段階にある。もし私たちまたは私たちの協力者が私たちの製品や候補製品の開発、製造、商業化に成功できなかった場合、あるいはそうする過程で重大な遅延に遭遇すれば、私たちの業務は深刻な損害を受ける可能性がある。
私たちの開発は全体的にまだ初期段階にあり、一部の候補製品だけが臨床開発段階にある。私たちの候補製品のほとんどは現在臨床前開発段階にあります。私たちは大量の資源を投入して潜在的な候補製品を決定し、開発し、臨床前と臨床研究、製造活動を行い、私たちの製品と候補製品の商業発表に準備した。承認されれば、私たちが私たちの製品や候補製品を販売することから収入を得る能力は、私たちの製品と候補製品の成功した開発、製造、私たちのパートナーの最終的な商業化に大きく依存するだろう。
私たちの製品と候補製品の成功は以下の要素を含む多くの要素に依存するだろう
36
もし私たちがこれらの要素のうちの1つまたは複数をタイムリーにまたは根本的に達成できなければ、私たちは重大な遅延に遭遇したり、私たちの製品や候補製品を開発して商業化することができなくなり、これは私たちの業務に実質的な損害を与える可能性がある。
私たちの業務と運営は、新冠肺炎の大流行の変化と持続的な影響、特にこの流行病に関する任意の新しい変種あるいは灰再発を含む衛生流行病と流行病の影響を受け、将来的に影響を受ける可能性がある。新冠肺炎疫病は引き続き私たちの業務と運営に影響を与え、私たちの将来の業務と運営、そして私たちが依存している第三者の業務と運営に実質的な悪影響を及ぼす可能性がある。
私たちの業務は、進行中の新冠肺炎の大流行を含む衛生流行病と流行病の悪影響を受ける可能性があり、それは世界各地で公衆衛生と経済に大きな挑戦をもたらし、すでに私たちの従業員、患者、コミュニティ、業務運営、そしてアメリカ経済と金融市場に影響を与え続けている。持続的な新冠肺炎の流行により、私たちは大部分の従業員を在宅勤務モードに転換した。我々は引き続き必要な対面製造と実験室機能を維持し、肝心な研究、開発、製造優先事項を推進する。これらの措置について、私たちは、私たちが法律を適用して許可された場合に、私たちのオフィスや施設を運営し続ける可能性のある任意の決定を含む、新しい冠肺炎および私たちの行動および対応に関連するクレームに基づいて、または制限される可能性がある。将来可能な州行政命令、地方亡命命令、政府の強制隔離、私たちの在宅勤務政策と他の類似行動の影響は生産性に負の影響を与え、私たちの業務を混乱させ、私たちの臨床計画とスケジュールを延期し、その程度は制限の長さと深刻さ、そして私たちの正常な業務展開能力に対する他の制限に部分的に依存するかもしれない。
外国、連邦、州、地方政府が講じたさらなる隔離、現地または同様の制限および他の行動、またはそのような命令、閉鎖または他の業務運営の制限が回復する可能性があると考える考え、特に持続的な新冠肺炎の大流行または他の感染症に関連する問題は、米国および他の国における私たちの製造能力および第三者製造施設、または材料の利用可能性またはコストに影響を与え、私たちのサプライチェーンを乱す可能性がある。特に、標準輸送ルートは影響を受け、私たちと他の製造、テスト、製品処分、CMOと外部テスト実験室は強化されたリスク評価と緩和措置の影響を受けた。また,白血球分離収集の供給が中断され,継続的に中断される可能性があり,これらの収集は我々の製品に用いた原材料を提供する。
著者らの臨床試験も衛生流行病の影響を受ける可能性があり、すでに進行と変化している新冠肺炎の大流行の影響を受けている。新冠肺炎疫病のため、臨床サイトの起動と患者登録はすべて遅延が出現し、これは病院資源が臨床試験ではなく、新冠肺炎に優先的に応用されているため、或いは著者らの試験に関連する疾病に影響する執業モードを変更したためである。もし隔離が患者の流動を阻害したり、医療サービスを中断したり、或いは患者が新冠肺炎に感染したり、隔離を余儀なくされた場合、一部の患者は臨床試験方案を遵守できない可能性がある。例えば、著者らが研究している多くの臨床試験サイトは、EBウイルス+PTLD患者に対するTabcelの第三段階臨床試験を含み、依然として患者に開放されているが、いくつかのサイトは新冠肺炎に関連する政府命令または新冠肺炎感染に対する恐怖により、患者の臨床サイトへのアクセス能力を制限し、患者の臨床サイトへのアクセス能力を制限し続ける可能性がある。新冠肺炎に関連する旅行制限も重要な臨床試験活動を中断する可能性があり、例えば臨床試験現場のデータモニタリングと有効性、安全性と転換性データの収集、処理と分析。新冠肺炎が大流行した当初は
37
幹細胞と固体臓器移植数の一時的な低下は,TAABCELの第三段階研究に適合する患者数を減少させた可能性がある。2020年4月、著者らは経前症候群患者ATA 188のEMBOLD研究のスクリーニングと登録作業の一時停止を開始した。EMBOLD研究中の患者のスクリーニングと登録を回復し、2020年6月に本研究の第一名の患者を募集することができるが、行われている新冠肺炎の大流行は臨床研究中の患者のスクリーニングと登録を一時停止する必要があるかもしれない。同様に,主な調査者や現場スタッフの能力が悪影響を受ける可能性があり,医療提供者として新冠肺炎への開放が増加している可能性がある。
また、持続的かつ変化し続ける新冠肺炎の大流行の進行的な影響が私たちの業務や運営結果に悪影響を与える場合、“リスク要因”の一部の他の場所に記載されている多くの他のリスクや不確実性をシナリオする効果もある可能性がある。
私たちの未来の成功は私たちの製品候補製品の規制承認にかかっている。
私たちはただ一つの製品EbvalloだけがEU規制部門の承認を受けた。現在、私たちが優先している臨床段階の候補製品には、米国のATA 188とTAB-CELが含まれています。私たちの業務は、規制部門の承認を得ることができるかどうか、承認されれば、私たちの候補製品をタイムリーに商業化することに成功したパートナーを見つけることができるかどうかに大きく依存しています。
FDAの候補製品に対する規制承認が得られないまで、私たちも私たちのパートナーもアメリカで候補製品を商業化することはできません;同様に、比較可能な外国規制機関の規制承認を得なければ、私たちも私たちのパートナーもアメリカ以外で候補製品を商業化することはできません。目標適応のための任意の候補製品の商業販売の規制承認を得る前に、私たちは、候補製品が目標適応に使用されることが安全かつ有効であることを証明するために、臨床前および臨床研究で収集された大量の証拠を利用しなければならず、候補製品の製造施設、プロセス、および制御は、安定性、安全性、純度および効力を保証するために十分である。
FDAと類似の外国監督管理機関の承認を得るのに要する時間は予測できないが、通常は臨床前と臨床研究開始後数年後に必要であり、そして多くの要素に依存し、監督機関のかなり大きな適宜決定権を含む。私たちの候補製品の新規性は規制部門の承認を得る上でさらなる挑戦をもたらすかもしれない。例えば、FDAと類似の外国の監督管理機関は、T細胞免疫療法の開発と商業化を監督する上で経験が限られており、特に同種異体T細胞候補製品とCAR T療法は、これらの候補製品の異なるバージョンの比較可能性を評価することを含む。また、承認政策、法規、監督の立場、或いは承認を得るために必要な臨床と他のデータのタイプと数量は候補製品の臨床開発過程中と全体の監督との相互作用過程で変化する可能性があり、そして司法管轄区域によって異なる可能性があり、特に新しい治療法に対して。ECはEBV+PTLD患者の単一療法としてEbvalloのマーケティング許可申請を承認しており,これらの患者は“特殊な場合”で少なくとも1回の治療を受けており,ECが“許可後も全面的なデータを得ることができない”場合にマーケティング許可を承認する方法である。マーケティング許可の特別な場合、私たちの商業パートナーピエール·ファブレは、Ebvalloの利点を確認し続け、もし私たちの他の候補製品がこの方法で承認された場合、継続的な発売後の義務があります, 私たちまたは私たちの未来の商業パートナーはこの義務によって制限されるだろう。Ebvalloマーケティング許可の継続は毎年再評価されなければならない。年次見直しは,Pierre Fabreが上場後の義務を履行している状況とEbvalloのリスク/収益状況に基づいて,Ebvalloのマーケティング許可を維持,変更または一時停止すべきかどうかを決定する。もし私たちまたは私たちの商業パートナーが進行中の上場後の義務を履行しない場合、または欧州委員会がEbvalloのリスク/収益状況が受け入れられないと判断した場合、欧州委員会はEbvalloの上場承認を変更または一時停止する可能性がある。私たちはまだ他の候補製品の規制承認を得ていません。私たちの既存の任意の候補製品または任意の未来の候補製品は決して規制承認を受けないかもしれません。
私たちの候補製品はFDAや同様の外国規制機関の規制承認を得ることができないかもしれません
38
FDAまたは同様の外国の規制機関は、承認を支援するための追加のCMC情報、臨床前または臨床データを含むより多くの情報を必要とする可能性があり、これは、承認および私たちの商業化計画を延期または阻止するか、または開発計画を放棄することを決定する可能性がある。例えば、2022年2月のB型会議では、キー対立遺伝子研究で使用されているラベル製品バージョンと予想される商業製品との比較性についてFDAと一致することはできなかった。FDAは最初に著者らがこの商業製品に対して新しい臨床試験を行うことを提案し、比較性の不足の問題を解決し、そして期待商業製品とのより多くの臨床経験を獲得した。2023年2月にFDAと臨床上の問題について会議を行い,BLAをTABCELに提出する可能性がある。我々は,異なるプロセスバージョンを集約することが可能な臨床データの比較可能性を含めて,CMCが潜在的なBLA提出に関連する事項をさらに検討する近未来会議を開催したい。新たな臨床試験を必要とせずにタグ細胞にBLAを提出できるようにFDAとこのような潜在的な経路を検討し続けているが,最終的には現在の臨床データセットをBLAに提出する経路についてFDAと合意できない可能性がある。この場合、TAB−CELのBLAをサポートするための追加の1つまたは複数の臨床試験が必要とされる可能性があり、これは、BLA提出のかなりの遅延をもたらすか、またはBLAの提出を継続しないことをもたらす可能性がある。臨床試験を行うことは困難すぎる或いは高すぎることが証明される可能性があり、この方案に基づいて臨床試験を設計し、十分な患者を募集し、治療とデータ収集を完成する過程は大量の時間、精力と資源を必要とする可能性がある。臨床試験を終えても, この研究はあらかじめ指定された終点に適合していない可能性があり,予定の終点に達してもFDAはラベルのためのBLAの提出·承認に十分な臨床試験に同意していない可能性があり,あるいはデータはBLAを提出するのに十分であるが,我々が最初に申請したよりも限られた適応しかサポートできないと考えられる可能性がある。
様々な要因により、我々パートナーとの開発活動および/または商業化計画は、政府または規制部門の遅延によって損害または遅延を受ける可能性がある。これらの要因には、政府および規制当局者が規制文書を審査するか、または私たちと接触する能力が制限されている(持続的かつ進化し続ける新冠肺炎の大流行を含む世界的な健康懸念や他の原因によって引き起こされる);政府規制要件、政策、ガイドラインまたは優先順位、政府資源の再分配または利用可能性の変化、または他の理由では、FDAまたは他の規制機関が私たちが提出した任意の申請を審査して処理する能力を著しく遅らせる可能性があり、または他の規制遅延を提出または引き起こす可能性がある。世界的な健康懸念がFDAまたは他の規制機関の定期的な検査、影響審査または他の規制活動を阻害し続ける場合、FDAまたは他の規制機関が私たちが提出した規制文書を適時に審査して処理する能力に深刻な影響を与える可能性があり、これは私たちの業務に実質的な悪影響を及ぼす可能性がある。例えば、新冠肺炎の流行に対応するため、米国食品薬品監督管理局は2020年3月10日、外国製造施設の大部分の検査と国内製造施設の製品検査を2020年4月に延期する意向を発表した。FDAは2020年3月18日,国内製造施設の通常監督検査を一時的に延期し,臨床試験の指導を行う予定であると発表した。FDAは2020年7月10日、“重要な任務”とされる現場検査の再起動目標の実現に努めていると発表した。2021年5月,食品医薬品局は2020年8月に初めて発表された指導意見を更新し,新冠肺炎流行中にどのように検査を行う予定であるかを明らかにした, それがどのような検査が“重要な任務”であるかをどのように決定するかを含む。さらに、FDAは、2021年4月14日に、ある薬物製造施設および臨床研究場所の自発的な遠隔インタラクション評価を行う計画を記述した指導文書を発表した。ガイドラインによると,FDAは対面検査が優先されず,キータスクとされている場合や,直接検査が旅行制限を受けている場合であるが,FDAが遠隔評価が適切であると判断した場合には,このような遠隔インタラクション評価を要求する予定である.FDAは、このリスクに基づく評価システムを使用して、特定の地理的地域内で発生する可能性のある規制活動種別を決定し、重要な任務検査からすべての規制活動を回復することを意図している。その後,米国食品薬品監督管理局は進行中の新冠肺炎の大流行に対応するために検査活動を調整した。FDAは2021年12月29日にその検査活動を一時的に変更し、従業員と
39
規制されている会社。FDAは2022年2月2日、全製品分野の国内監督検査を2022年2月7日に再開すると発表した。FDAは2022年7月、遠隔規制評価に関する政策について概説した指導文書草案を発表した。FDAがいつ新冠肺炎のせいで検査の一時停止または回復を決定するかどうかを予測することはできない。新冠肺炎疫病に対して、アメリカ以外の規制機関は類似した制限或いは他の政策措置をとる可能性がある。FDAや他の衛生機関の政策や指導が臨床研究なしにわれわれの施設や臨床試験地点で行ったいかなる検査にどのように影響するかは不明である。
もし私たちが確かに承認された場合、規制機関は私たちの任意の候補製品の適応が私たちが要求しているものよりも少ないか限られているかもしれない(最も商業的な見通しのある適応を承認できないことを含む)、高価な発売後の臨床研究の表現によって承認されるかもしれない、またはそのラベルが候補製品の商業化に成功するために必要または必要なラベル宣言を含まないことを承認する可能性がある。さらに、FDA、EMAおよび他の規制機関の臨床研究要求およびこれらの規制機関が候補製品の安全性と有効性を決定するための基準は、潜在的製品のタイプ、複雑性、新規性および予想用途および市場に基づいて決定される。新しい候補製品、例えば我々の新型T細胞候補製品および次世代CAR T計画の場合、規制承認プロセスは、他のより有名または広く研究された候補医薬品または他の製品よりも複雑になる可能性があるため、コストが高く、時間がかかる。ノワールのKymriahのような既存の自己CAR T療法に対する欧州委員会とFDAの承認®ギレドのイエスカタと®多分、このような規制機関が私たちの治療法を承認する必要があるかもしれないということを見せてくれないかもしれない。私たちは現在私たちの候補製品に対して多くの臨床試験を行っている。もし私たちの1つまたは複数の臨床試験において不良安全性問題、臨床保留または他の不良発見が出現した場合、このような事件は私たちの他の同じ或いは関連する候補製品の臨床試験に不利な影響を与える可能性がある。さらに、我々の候補製品は、臨床研究において不良である可能性があり、または既存の自己CAR T療法のような以前に承認された製品と区別される有害事象に関連する可能性がある。例えば、同種候補製品は、自己製品が経験したことのない有害事象をもたらす可能性がある。候補製品がFDAおよび同様の外国規制機関の承認を得ることに成功したとしても、どの承認も、特定の年齢層の使用制限、警告、予防措置または禁忌症に関連する重大な制限を含むことができ、または重い承認後の研究またはリスク管理要件の影響を受ける可能性がある。私たちが1つ以上の司法管轄区域で私たちの候補製品のうちの1つに対する規制承認を得ることができない場合、または任意の承認に重大な制限が含まれている場合、私たちは製品を開発し続けたり、候補製品に起因することができる収入を生成するのに十分な資金を得ることができないかもしれない。さらに、現在または将来の候補製品に対する私たちの任意の規制承認は、一旦取得されると、対応する規制機関が1つの地域または国/地域で撤回される可能性がある。
私たちのT細胞免疫療法製品と候補製品と私たちの次世代CAR T計画は新しい治療方法を代表しており、これらの方法はより厳格な監督審査、臨床開発の遅延、或いは私たちは監督許可、製品候補の商業化或いは支払人カバーを実現できないかもしれない。
我々の将来の成功は,T細胞免疫療法の成功と商業化,および我々の次世代CAR T計画,特に我々の開発製品候補に依存する。これらの計画、特に私たちの同種異体T細胞製品とドナーからの生物工学候補製品のパイプラインは、癌や他の疾患を治療するための免疫療法の新しい方法を表しているため、私たちの候補製品の開発と商業化は、私たちを多くの挑戦に直面させている
40
私たちは、私たちのT細胞免疫療法製品および候補製品に関連する製造プロセスが十分な満足できる製品供給を生成することを保証することはできず、これらの製品は安定、安全、純粋かつ有効であり、または私たちのパートナーの歴史上生産されたT細胞に匹敵することができ、拡張可能または利益になる。
さらに、実際または知覚される安全性の問題は、新しい治療方法または新しい治療方法を採用することを含み、被験者の臨床研究への参加意欲に悪影響を及ぼす可能性があり、または、私たちの候補製品が関連する規制機関の承認を得た場合、医師が新しい治療機序を購読する可能性がある場合、または患者が規制部門がこのメカニズムを承認したにもかかわらず、新しい治療を受けることに同意する可能性がある。FDAまたは他の適用可能な規制機関は、我々の製品のメリットまたはリスクを伝達するために、特定の上場後研究または他の情報を要求することができる。新しいデータは規制承認の前または後の任意の時間に私たちの候補製品の新しいリスクを明らかにするかもしれない。
医師、病院と第三者支払人は新製品、技術と治療実践の使用においてよく行動が遅く、これらの製品、技術と治療実践は追加の前期コストと訓練を必要とする。医師はこのような新しい療法に関する研修を受けたくない可能性があり,複雑すぎて適切な訓練なしには採用できない,あるいは費用対効果に適合していないと考え,施行しないことを選択する可能性がある。これらや他の要因から,病院や支払者はこのような新しい療法の利点を決定する可能性があり,そのコストを超えないか,あるいはそれを超えない可能性がある。
臨床前研究或いは早期臨床研究の結果は必ずしも未来の結果を予測できるとは限らない。我々の既存の臨床研究候補製品、及び著者らが臨床研究に入った任意の他の候補製品は、今後の臨床研究において有利な結果が得られない可能性があり、監督部門の承認を得られない可能性がある。
臨床前研究と早期臨床研究の成功は今後の臨床研究が十分なデータを産生して研究薬物の有効性と安全性を証明することを確保しない。同様に,製薬やバイオテクノロジー業界のいくつかの会社は,我々よりも多くの資源や経験を持つ会社を含め,臨床研究においても大きな挫折を経験しており,早期の臨床前研究や臨床研究でも有望な結果が見られている。著者らの候補製品の早期臨床前研究或いは臨床研究は結果を報告したが、多種の要素のため、同一候補製品の異なる臨床試験間の安全性と有効性結果は有意差が存在する可能性がある。私たちが行う可能性のある臨床研究あるいは行われている臨床研究が十分な有効性と安全性を証明するかどうかは、規制機関が任意の特定の司法管轄区で任意の候補製品を販売することを許可することを招く。
41
TAABCELは主にMSKの研究者が後援した研究性新薬(INDS)申請下の単中心研究で評価を行い,われわれが拡大した参入計画で評価を行い,われわれが今後の臨床研究ですでにあるいは使用可能な異なる反応基準と終点を利用している。早期研究の結果は我々が行った後期研究では再現できないかもしれない。例えば,我々が現在EBV+PTLDで行っている対立遺伝子研究は,ゼロ仮説として20%のOORを排除するためのものである。このことは,研究終了時に少なくとも1剤のTabcel治療を受けた患者において,OORの95%信頼区間下限が20%を超えていれば,PTLD治療の主な終点に達することが期待できることを意味している。たとえば,対立遺伝子キューに33名の患者が登録されていると仮定すると,観察されたOORは約37%以上でそのキューの主要終点を満たすことが期待できる.また、著者らが改訂した対立遺伝子研究方案は中期分析と最終研究分析を含む。我々は以前FDAからフィードバックを受けており,対立遺伝子研究の中期解析はBLAの承認を支持するには不十分である可能性が考えられた。また、対立遺伝子研究に使用されている異なる技術バージョンの比較可能性に関するFDAの結論によると、対立遺伝子研究の総サンプル量と統計方法を修正する必要があるかもしれない。Ebvalloは特別な状況で承認されているので、Ebvalloマーケティング許可の継続は毎年再評価される必要がある。年次見直しは,Pierre Fabreが上場後の義務を履行している状況とEbvalloのリスク/収益状況に基づいて,Ebvalloのマーケティング許可を維持,変更または一時停止すべきかどうかを決定する。
TAABCELの規制承認については,独立した放射線科医および/または腫瘍専門家を用いて反応を評価する予定であり,これらの評価は研究者が報告した評価とは関連しない可能性がある。そのほか、TABCELの第二段階の臨床研究は1組の異なるタイプのEBV駆動悪性腫瘍患者、HCT後のEBV+PTLDとSOT後のEBV+PTLDを含む。これらの2期研究は単一疾患状態におけるTAABCELの治療効果を評価するために設計されたものではなく,今後承認が求められる可能性がある。
また,最終的な研究結果は中期研究結果と一致しない可能性がある。展望性設計研究からの治療効果データは遡及性サブグループ分析からのデータと明らかに異なる可能性がある。さらに、同種候補製品の臨床研究から得られた臨床データは、自己候補製品と比較して同じまたはそれ以上の結果を生成しない可能性がある。もし後期臨床研究に有利な結果が生じなければ、私たちがどの候補製品のために規制承認を得る能力も不利な影響を受ける可能性がある。私たちが規制承認申請が私たちの候補製品を市場に出すのに十分なデータがあると思っても、FDAや他の規制機関は同意しない可能性があり、追加の臨床研究を要求するかもしれません。
私たちまたは私たちのパートナーは、監督機関と共有されている臨床研究の一時的な“トップライン”および予備データが、より多くの患者データの取得に伴って変化する可能性があり、監査および検証手続きによって制限される可能性があり、最終データに大きな変化をもたらす可能性がある。
私たちまたは私たちのパートナーは、臨床研究からの一時的な“トップライン”または予備データを時々発表したり、監督当局と共有したりするかもしれない。臨床研究の中期データは患者登録の継続とより多くの患者データの獲得に伴い、1つ或いは複数の臨床結果が実質的に変化する可能性があるというリスクに直面している。予備データまたは“トップライン”データは依然として監査とチェック手続きを受けなければならず、これは最終データが以前に発表された予備データと大きく異なる可能性がある。したがって、最終データを得る前に、中期と予備データを慎重に見なければならない。初期または中期データと最終データとの間の不利な差は、適用データの影響を受ける任意の候補製品に対する監督管理部門の承認に影響を与え、その将来性に重大な損害を与える可能性がある。
臨床薬物開発は長くて高価な過程に関連し、結果は不確定である。
臨床試験費用は高価であり,完成まで数年かかる可能性があり,その結果自体も確定していない。臨床研究過程では、いつでも失敗する可能性がある。臨床前と臨床研究で進展が得られたにもかかわらず、臨床研究後期段階の候補製品は必要な安全性と有効性特徴を示すことができないかもしれない。
進行中や将来の臨床研究に遅延が生じる可能性があり,臨床研究が時間どおりに開始されるかどうか,被験者を募集するかどうか,再設計や予定通りに完成する必要があるかどうかは分からない。FDAや同様の外国の規制機関が将来的に私たちの候補製品の臨床研究を中断しないことは保証されない。臨床研究は、例えば、様々な原因で遅延、一時停止または早期終了する可能性がある
42
患者の入選は臨床研究時間の重要な要素であり、それは多くの要素の影響を受けている
43
例えば、2018年の間、私たちはTab-celの対立遺伝子研究のためにより多くの臨床サイトを活性化し、その間にヒト白血球抗原の被覆率を増加させた。したがって,われわれが検討した入学者数は2018年初めに制限され,臨床地点やヒト白血球抗原カバー範囲の増加に伴い,入学者数は1年間増加した。しかし、2019年5月、我々はEBV+PTLD患者に対するTAABCELの第三段階研究の登録進展が予想より遅いことを発表した。われわれの多くの候補製品はまれな疾患を治療するために設計されているため,特定の疾患に対する潜在的な患者池は小さい。FDAまたは他の規制機関の要求に基づいて、これらの研究に参加するのに十分な数の合格参加者を見つけることができない場合、TABCEL、ATA 188、または任意の他の候補製品の臨床研究を開始または継続することができない可能性がある。持続的な新冠肺炎の大流行の絶えず変化の影響により、著者らはある臨床試験(著者らの対立遺伝子研究を含む)の臨床試験サイトの起動と患者募集においていくつかの一時的な遅延を経験した。私たちの臨床研究で十分な数の患者を募集することができても、登録速度が予想より遅くなれば、候補製品の開発コストが増加する可能性があり、私たちの研究は完成を遅らせるかもしれないし、私たちの研究は高すぎて完成できないかもしれない。
著者らはCRO、他のサプライヤーと臨床研究サイトに依存して、著者らの臨床研究が正確かつ適時に行われることを確保し、著者らは彼らが約束した活動に対して合意があるが、私たちは彼らの実際の表現に対する影響は限られている。さらに、これらの第三者は、他のエンティティと関係がある可能性もあり、その中のいくつかは私たちの競争相手である可能性がある。CRO依存はリスクをもたらし,我々自身が臨床研究を行えば,CROによる臨床サイト起動と監視,CROが財務資源を維持して我々の合意下の義務を履行できないこと,CROが制御できない要因によりこれらの合意に違反する可能性,これらの合意下の義務を正確に履行できなかったこと,CROがそれ自身の業務優先事項に基づいて合意を終了または更新しなかった可能性を含むこれらのリスクの影響を受けない。これらの第三者の研究開発活動への依存は、これらの活動に対する私たちの統制を減少させるだろうが、私たちの規制責任は免除されないだろう。例えば、私たちのすべての臨床試験が全体調査計画、試験の研究案、統計分析計画、および他の研究特定文書(例えば、監視および盲法計画)に従って行われることを確実にする責任を引き続き担当します。また、FDAは、一般に良好な臨床実践(GCP)と呼ばれる基準、国際人用医薬品技術要求調整理事会(ICH)のガイドライン、ならびにインフォームドコンセント過程、安全報告要件、データ収集ガイドライン、およびその他の進行を遵守することを要求している, データおよび報告の結果が信頼性かつ正確であることを保証し、試験参加者の権利、完全性、および機密性を保護するために、臨床試験の結果を記録し、報告する。EMAはまた私たちに似たような基準を遵守することを要求する。規制機関は,試験スポンサー,主要調査者,試験地点を定期的に検査することでこれらのGCP要求を実行する。もし私たちまたは私たちの任意のCROが適用されたGCP要件を遵守できなかった場合、私たちの臨床試験で生成された臨床データは信頼できないと考えられるかもしれません。FDA、EMAまたは同様の外国の規制機関は、私たちの上場申請を承認する前に追加の臨床試験を行うことを要求するかもしれません。特定の規制機関が検査を行った後、この規制機関は、私たちの任意の臨床試験がGCPおよび他の適用法規に適合しているかどうかを確認することは保証されない。また,われわれの臨床試験では,適用された現行良好生産規範(CGMP)と現行良好組織規範(CGTP)法規に基づいて生産された製品を用いなければならない。もし私たちがこれらの規定を守らなければ、私たちは新しい臨床試験を行う必要があるかもしれません。これは上場承認過程を延期します。いくつかの行われている臨床試験を一定の時間範囲で登録し、政府が援助したデータベースClinicalTrials.gov上でいくつかの完了した臨床試験の結果を公表することも要求されている。そうしないと罰金、否定的な宣伝、そして民事と刑事制裁につながるかもしれない。
44
もし私たちの候補製品に対して任意の臨床研究を行い、完成または終了する時に遅延または品質の問題に遭遇した場合、その候補製品の承認と商業見通しは損なわれ、候補製品から製品収入を得る能力は延期されるだろう。そのほか、臨床研究を完成するいかなる遅延も著者らのコストを増加し、著者らの候補製品の開発と審査過程を緩和し、そして著者らの創立能力を脅かす。私たちの候補製品の臨床研究を完成する上でのいかなる遅延も商業独占期間を短縮する可能性がある。さらに、臨床研究の開始や完成を遅延させる可能性のある多くの要素は、最終的に私たちの候補製品が規制部門の承認を得ることを拒否される可能性もある。
我々の製品および候補製品、それらを伝達するための方法、またはその用量レベルは、副作用をもたらす可能性があり、またはその規制承認を遅延または阻止し、承認ラベルを制限するための他の商業イメージ、または任意の規制承認後に重大な負の結果をもたらす可能性がある特性を有する。
私たちの製品および候補製品によって引き起こされる副作用、その投与方法または用量レベルは、私たちまたは規制機関の臨床研究の中断、延期、または一時停止を招く可能性があり、より厳しいラベルまたはFDAまたは他の同様の外国規制機関の規制承認遅延または拒否を引き起こす可能性がある。私たちまたは私たちのパートナーが臨床研究で遭遇する可能性のある安全または毒性の問題のために、私たちまたは私たちのパートナーは、マーケティングの任意の候補製品の承認を得ることができない可能性があり、これは、そのような候補製品のための製品または特許使用料収入を創出したり、利益を達成することを阻止するかもしれません。私たちの研究結果は、受け入れられない重症度と副作用の発生率、あるいはリスクが私たちの製品や候補製品を超える利点を示しているかもしれない。この場合、私たちの研究は一時停止または終了される可能性があり、FDAまたは同様の外国規制機関は、私たちの任意またはすべての目標適応の候補製品の開発を停止または拒否するように命令することができる。薬物に関連する副作用は、患者の募集に影響を与える可能性があり、または対象が研究を完了する能力、または潜在的な製品責任クレームを引き起こす可能性がある。
また、もし私たちの候補製品が監督部門の許可を得て、私たちまたは他の人が後にその製品が不良副作用をもたらしていることを発見したら、多くの潜在的な負の結果を招く可能性があります
これらの事件のいずれも、我々の製品および候補製品の使用を減少させるか、または他の方法でその商業成功を制限し、影響を受けた候補製品に対する市場の受容度を達成または維持することを阻止または維持する可能性がある(適用される規制機関の承認が得られた場合)。
私たちの製品および候補製品の市場機会は、以前の治療に適合していないか、または通過できなかった患者に限定される可能性があり、小さい可能性がある。
FDAは通常新たな癌療法のみを承認し,最初は再発や難治性転移疾患の患者にのみ適用される。このような状況下でTabCELとわれわれの他の腫瘍学候補製品の初歩的な承認を求めることを希望する。その後,十分な利益が証明された製品については,あれば早期の治療経路の承認を求める可能性があり,第一線の治療となる可能性があるが,我々の製品や候補製品が承認されても早期の治療経路のために承認される保証はなく,いずれかの承認を得る前に追加の臨床試験を行わなければならない。
私たちの目標疾患を患っている人の数の予測とこれらの疾患患者のサブセットの予測は二線以上の治療を受けることができ、私たちの製品治療から利益を得る可能性があります
45
候補品は私たちの現在の信念と推定に基づいていますこれらの推定は、科学文献、診療所調査、患者基金会あるいは私たち自身の市場研究を含む様々な源から来ており、正しくないことが証明されているかもしれない。そのほか、新しい研究、製品承認或いは市場研究はこれらの疾病の推定発病率或いは流行率を変える可能性があり、患者数は予想より低いかもしれない。さらに、私たちの候補製品の潜在的にアドレス指定可能な患者集団は限られているかもしれないし、または私たちの候補製品の治療を受け入れられないかもしれない。例えば、我々の製品TAB−CELは、最初に侵襲性EBV+PTLDを有し、リツキシマブまたはリツキシマブプラス化学療法を通過できなかった患者集団を対象としていることが望ましい。私たちのビジネスパートナーは私たちの製品や候補製品の市場機会を違う推定をするかもしれない。新冠肺炎の大流行開始時には,最初に幹細胞と実体臓器移植数の一時的な鈍化が観察された。これらの減少は一時的であるが,この数が減少するとPTLD発生率が低下し,タバコ需要が減少する可能性がある。私たちの製品や候補製品がかなりの市場シェアを獲得していても、潜在的なターゲット層が少ないため、規制部門のより多くの適応の承認を得なければ、決して利益を達成しないかもしれない。
私たちは私たちの候補製品の孤立した薬物独占経営権を獲得したり維持できないかもしれない。
一部の司法管轄区域の監督当局は、アメリカ、EU、イギリス(UK)を含み、患者数が相対的に少ない薬物を孤児薬物に指定する可能性がある。孤児医薬品法によれば、1つの製品がまれな疾患または疾患を治療するための薬剤である場合、FDAはそれを孤児薬として指定することができる。米国では、まれな疾患または疾患は、通常、年間20万人未満の患者数として定義されている。FDAおよびEMAは、HCTまたはSOTの後にEBV+PTLDのために使用される薬物孤児薬物名を承認している。
一般に、孤児薬物名を有する製品が、その後、そのような名称を有する適応の最初の規制承認を得た場合、製品は、一定期間内に市場排他期間を得る権利があり、これは、EMAまたはFDAがこの期間内に同じ生物の同じ適応を承認する別のマーケティング申請を阻止するであろう。適用期間は米国では7年,ヨーロッパでは10年である。薬物が孤児薬物指定の基準に適合しなくなった場合、またはその薬剤の利益が十分に高く、市場排他性を得る理由がなくなった場合、ヨーロッパの排他的期間は6年に短縮されることができる。また、欧州委員会は2023年第2四半期に新たな適用法枠組みを発表する予定で、これらの期間はEUで短縮される見通しだ。FDAまたはEMAが、指定された要求に重大な欠陥があると判断した場合、または製造業者が稀な疾患または疾患を有する患者の需要を満たすのに十分な数の薬剤を保証できない場合、孤児薬の排他性を失う可能性がある。米国では、FDAは、孤児薬物に対して排他的な製品に対する臨床的優位性を示すなど、持続的な孤児薬物排他期によって阻害された上場申請を限られた状況で承認することができ、またはFDAが孤児薬物排他性保持者がそれが承認された疾患または状態に罹患している患者の需要を満たすために十分な数の孤児薬物の供給を確保できることを証明していないことを証明することができる。したがって、私たちの候補製品が孤児の独占特許を取得しても、FDAは、同じ適応または疾患の治療に使用するために、異なる有効成分を有する他の医薬または生物学的製品を承認することができるかもしれない。
また,国会は最近の11に対応するためにFDCAの孤児薬物条項の更新を検討しているこれは…。巡回判決。孤児薬物条項のいかなる変化も私たちが孤児薬物の独占経営権を獲得する機会或いは成功の可能性を変える可能性があり、そして私たちの業務、財務状況、運営業績、キャッシュフローと将来性に重大な不利な影響を与える可能性がある。
私たちが製品の孤児薬物排他性を獲得しても、この排他性は、異なる薬物が同じ条件のために承認される可能性があるので、その製品を競争から維持または効果的に保護することができない可能性がある。
FDAのBTDとEMAのPrime指定は、より速い開発や規制審査や承認過程を招くことがなく、我々の候補製品が上場承認される可能性も増加しない可能性がある。
われわれはすでに米国でTAABCELのBTDを獲得し,リツキシマブを通過できなかったEBV+PTLD患者の治療に用いられているにもかかわらず,これらの指定はより速い開発や規制審査を招くことはなく,われわれの成功の可能性も増加しない可能性がある。画期的な治療法は、1つまたは複数の他の薬剤または生物学的製品と単独でまたは1つまたは複数の他の薬剤または生物学的製品と組み合わせて重篤または生命に危険な疾患または状態を治療することを目的とした医薬または生物学的製剤として定義され、予備的な臨床的証拠は、1つまたは複数の臨床的に重要な終点において、例えば臨床開発早期に観察される実質的な治療効果のような既存の治療法よりも有意な改善を示す可能性があることを示す。画期的な治療法に指定された候補製品に対して、FDAと研究スポンサーとの間の相互作用とコミュニケーションは、臨床開発の最も有効な方法を決定するのを助けることができ、同時に無効な制御方案中の患者数を最小限に下げることができる。FDAによって画期的な治療法に指定されている生物製品も,優先審査のような他の迅速審査計画を受ける資格がある可能性がある。我々のBTDによれば、米国でEBV+PTLDのTAB-CELのためのBLAに対してスクロール提出戦略をとる可能性があります。スクロール審査過程はFDAとの継続的なコミュニケーションやフィードバックの機会を提供する可能性がありますが、これは上場承認のより速い時間を招くことはなく、TABCELが最終的に承認されるかどうかにも関係ありません。FDAは私たちに問題と問題を提起するかもしれません。これらの問題は私たちのBLA提出の開始と完成を延期し、完全なBLAを受け入れるかもしれません
46
BLAの届出と承認。私たちはFDAの質問に満足またはタイムリーな応答を提供できないかもしれないし、必要なデータを収集して、計画的に私たちのBLA提出を準備することができないかもしれない。もし私たちがFDAが提起する可能性のあるすべての問題や懸念を解決できない場合、あるいはBLAを準備するために必要なデータをタイムリーに得ることができなければ、私たちはBLAをタイムリーに起動して完成させ、最終的にFDAの承認を得ることができないかもしれない。また,スクロール審査中に我々のBLAを提出しても,FDAは提出が完了すると考えられるまでスクロール審査中に我々のBLAの内容の一部を審査しないことを決定する可能性がある.
PRIME指定は、満足されていない医療ニーズを有する患者を治療するために、EMAの開発と見直しを支援する新しい療法を加速する。この指定と加速評価に関する機会にもかかわらず,環境評価局はMAA審査を行うのに余分な時間を要することを決定し,MAAを標準審査スケジュールに変換することができる。
画期的な治療法への指定はFDAが適宜決定し,Primeを用いてEMAが適宜決定する。候補製品のBTDまたはPrime称号を受信することは、非迅速FDAまたはEMA審査手順に従って承認を考慮する薬剤と比較して、より速い開発プロセス、審査または承認をもたらすことができず、FDAまたはEMAの最終承認を保証することもできないかもしれない。さらに、FDAまたはEMAは、その後、製品がもはや資格条件に適合しないことをそれぞれ決定し、BTDまたはPrime称号を撤回するか、またはFDAまたはEMAがそれぞれ審査または承認される期間を短縮しないことを決定することができる。例えば、2022年6月、FDAは、指定された要求に適合しなくなった製品のBTDをキャンセルする際のFDAの考慮要因について概説するガイドライン草案を発表した。
FDAの高速チャネル指定は、他の現在または将来の候補製品を付与しても、より速い開発や規制審査、許可プロセスを招くことなく、私たちの候補製品がマーケティング許可を得る可能性を増加させることはない。
私たちは私たちの未来の1つ以上の候補製品のための迅速なチャネル認証を求めるかもしれない。2021年12月、ATA 188は、PPMSおよびSPMS患者を治療する迅速チャネル指定を取得した。1つの医薬または生物学的製品が、深刻なまたは生命に危険な疾患または状態を治療することが意図されており、そのような疾患または状態を解決するための満たされていない医療需要の潜在力を示す場合、医薬品スポンサーは、特定の適応に対するFDAの迅速なチャネル指定を申請することができる。私たちは私たちの候補製品のための高速チャネル指定を求めるかもしれませんが、FDAがこの指定を私たちが提案した任意の候補製品に与える保証はありません。FDAが提供する政策およびプログラムによれば、迅速チャネル開発製品のスポンサーが提出したマーケティング申請は、優先審査を受ける資格がある可能性があるが、高速チャネルの指定は、FDAのどのような資格または最終的なマーケティング許可も保証しない。FDAは広範な裁量権を持ち,高速チャネル認証を付与するかどうかを持っているため,特定の候補製品がこのような認証を受ける資格があると考えても,FDAが付与を決定する保証はない。高速チャネル認証を確実に取得したとしても、従来のFDAプログラムや経路と比較して、より速い開発過程、審査または許可を経験することはなく、高速チャネル認証を得ることは、最終的にFDA許可を得ることを保証することはできない。また,FDAはその指定が我々の臨床開発計画データの支持を得なくなったと考える場合を含む迅速チャネル指定をいつでも撤回する可能性がある。
国際司法管轄区で規制機関や支払人の承認を得ることができなければ、私たちの候補製品は海外で販売できないだろう。
米国の法規を除いて、EU、イギリス、多くのアジア諸国、および他の司法管轄区域で私たちの製品をマーケティングして販売するためには、私たちまたは私たちの現在または未来の商業化パートナーは単独の規制承認を得なければならず、臨床と製造の観点から多くの異なる規制要求を遵守しなければならない。承認手続きは国によって異なり、追加的なテストが含まれるかもしれない。承認を得るのに要する時間は、FDA承認を得る時間とは大きく異なる可能性がある。米国以外の規制承認プロセスには、通常、FDA承認の取得に関するすべてのリスクが含まれており、追加のリスクも含まれている可能性がある。ある国で受け入れられた臨床研究は他の国の規制機関に受け入れられないかもしれない。また、米国以外の多くの国は、製品がその国での販売を許可される前に、精算承認を受けなければならないことを要求している。特定の国/地域で販売が許可された候補製品は、その国/地域で精算承認を受けない可能性がある。私たちはもしあれば、アメリカ以外の規制機関や支払人当局の承認をタイムリーに得ることができないかもしれない。監督機関または支払人の承認は、他の国または司法管轄区域の任意の他の規制機関または支払人当局の承認を確保しない。私たちは規制承認を申請できないかもしれないし、どの市場でも私たちの製品を商業化するために必要な承認を得ることができないかもしれない。米国、EU、イギリス、アジア、または他の地域の規制機関または支払人当局による私たちの任意の候補製品の承認を得ることができなければ、その候補製品のビジネス見通しは著しく低下する可能性がある。
私たちの製品候補が規制部門の承認を得ても、彼らは将来の発展と規制面の困難に直面する可能性がある。
私たちまたは私たちのパートナーが候補製品の規制承認を得ても、FDAと同様の外国の監督管理機関によって製造、品質管理、さらなる開発、ラベル、
47
包装、貯蔵、流通、不良事件報告、安全監督、輸出入、広告、普及、記録と報告安全及びその他の発売後の情報。これらの要求は、安全と他の上場後の情報と報告、登録の提出、および私たちおよび/または私たちのCMOとCROが私たちが行った任意の承認後の臨床研究の持続的なコンプライアンスを含む。それらはまた、FDAまたは同様の外国規制機関によって課せられた任意の承認後に承認条件として要求または承諾されたか、または適用される場合、任意のリスク評価または緩和戦略(REMS)を含む。どの製品の安全状況も承認された後、FDAや同様の外国規制機関の密接な監視を受け続ける。FDAまたは同様の外国規制機関が、我々の任意の候補製品が承認された後に新しいセキュリティ情報を認識した場合、彼らは、ラベルの変更またはリスク評価および緩和策の確立、製品の指示用途またはマーケティングに重大な制限を加えるか、または可能性の高い承認後の研究または発売後の監督に持続的な要求を加えることができるかもしれない。
また、薬品メーカー及びその施設はFDAと他の監督機関の初歩的かつ持続的な審査と定期検査を受けて、現在のcGMP、現在のGCP、現在のCGTPとその他の法規に適合することを保証しなければならない。もし私たちまたは監督管理機関が、ある製品に以前に未知の問題、例えば意外な深刻性や頻度の不良事件、またはその製品の製造施設に問題があることを発見した場合、監督管理機関は、製品のリコールを要求するか、市場から製品を撤回するか、または生産を一時停止することを含む、製品、製造施設、または私たちに制限を加える可能性がある。もし私たち、私たちの候補製品、あるいは私たちの候補製品の製造施設が適用される規制要求を守らなければ、規制機関はできます
上述した任意の事件や処罰の発生は、負の宣伝または製品の商業化に成功する能力を生み出したり、抑制したりする可能性もある。
米国で承認された候補製品の広告および普及は、FDA、司法省(DoJ)、衛生·公衆サービス部(HHS)監察長室、州総検事長、米国議会議員、公衆の厳しい審査を受ける。さらに、米国国外で承認された候補製品の広告および普及は、比較可能な外国の実体および利害関係者の厳しい審査を受ける。例えば、ある会社はその薬品の“ラベル外”用途を普及させないかもしれない。ラベル外使用とは、米国FDAによって承認されたラベルに記載されていない製品の適応に製品を使用するか、または他の管轄地域で適用規制機関の承認とは異なる使用を意味する。一方,医師は非ラベル用途のための製品を開発する可能性がある。FDAおよび他の規制機関は、医師が独立した医療判断において行う薬物治療選択を規範化していないが、彼らは、マーケティング許可が発行されていない製品のラベル外用途に関連する会社またはその販売者からの販売促進情報を制限している。しかしながら、会社は、FDAによって承認された製品ラベルと一致する真で誤解されない情報を共有するかもしれない。FDAまたは同様の外国機関の実行書、調査および調査、ならびに民事および刑事制裁を受けるための、許可されていないまたはラベル外での私たちの製品の宣伝のための実際または宣伝されていると言われている違反を含む。ラベルや宣伝要求を守らないと実際または言われている行為は、罰金、警告状、医療従事者への強制的な訂正情報の提供、禁止または民事または刑事罰を招く可能性がある。
様々な政府機関と組織が発表した法規、ガイドライン、提案は私たちの候補製品の使用に影響を与える可能性があります。
われわれの治療の適応に対して代替療法を提唱する法規,アドバイスや他のガイドラインの変化は,われわれの製品の使用を減少させる可能性がある。例えば,国家総合癌ネットワークガイドラインでは,EBV特異的T細胞を用いた治療は持続的あるいは進行性EBV+PTLDの推奨治療法と考えられているが,将来的には提案されている
48
政府機関、専門協会、実践管理団体、個人健康/科学財団、その他の組織のガイドラインは、私たちの候補製品を開発する能力の低下を招く可能性があり、あるいは適用される規制機関の承認を得ると、私たちの製品の使用を減少させる可能性がある。
私たちは新しい潜在的な候補製品を識別、取得、開発、または商業化することに成功できないかもしれない。
私たちの業務戦略の一部は新しい候補製品を識別して検証することで、私たちの候補製品ルートを拡大することで、私たちは自分で開発、許可したり、他の方法でこれらの候補製品を獲得することができます。また、既存の候補製品が規制部門の承認を得ていない場合や商業化に成功していない場合、私たちの業務の成功は、内部開発、許可、または他の買収を通じて私たちの製品ラインを拡大する能力にかかっています。私たちは関連した候補製品を決定できないかもしれない。もし私たちがこのような候補製品を決定したら、私たちはそれから許可を得たり、それらを取得したいいかなる第三者とも受け入れ可能な条項を達成できないかもしれない。私たちが確定、取得、許可、または開発した任意の候補製品は、その目標疾患に対して安全でないか、または有効ではないかもしれないし、適時にマーケティング許可を得ることができない可能性もあり、甚だしきに至っては全くできないかもしれない。
製造業に関するリスク
私たちは大量の製造リスクに直面していますが、いずれも私たちのコストを大幅に増加させ、私たちの製品と候補製品の供給を制限する可能性があります。
既存製品および候補製品の許可を得るとともに、パートナーから製造プロセス技術を取得し、場合によってはプロセス中間体および臨床材料の在庫を得る。製造プロセス、テスト、および関連する技術ノウハウを移行することは複雑であり、時間経過とともに変化する可能性のある記録ありおよび無記録プロセスの審査および組み入れに関する。さらに、生産を異なる施設に移転することは、特定の施設の特定の要件を満たすために、新しいまたは異なるプロセスおよび/または装置を利用する必要がある可能性がある。すべての段階は遡及されて適切な規制に適合しているかどうかを同時に確認する。したがって,我々のパートナーがすべての関連技術を十分に譲渡していない,あるいはこれまでの実行が適用されなかったリスクがある.
また、私たちは重大な開発と拡大を行い、これらの技術を移転し、各種の研究、臨床研究、商業投入のために私たちのすべての製品と候補製品を生産する準備をする必要がある。もし私たちが第三者CMOネットワーク内で生産を移転することを選択した場合、新しいまたは“受信”施設で生産された製品が元のまたは“送信”施設で生産された製品に相当することを証明しなければならない。適用される規制機関ごとに類似した薬物製品が生産されていることを証明することはできず,候補製品の開発が遅れる可能性がある。
私たちのいくつかの製品と候補製品の製造プロセスは最初に私たちのパートナーによって臨床目的で開発されました。我々は,先進的な臨床研究や商業化要求を支援するために,我々のパートナー開発の流れと我々開発の流れを発展させる予定である。私たちはまた、先進的な臨床研究と商業化要求を支援するために、Ataraから始まった技術を発展させるつもりだ。商業上実行可能な製造技術の開発は困難と不確定な任務であり、しかも高級臨床研究或いは商業化に必要なレベルへの拡張に関連するリスクはコスト超過、技術拡大の潜在問題、技術再現性、比較可能性問題、安定性、安全性、純度と効力問題、試薬或いは原材料の一致性と即時性を含む。我々は生産製品と候補製品の製造施設は持続的な新冠肺炎疫病、地震とその他の自然或いは人為的災害、設備故障、労働力不足、電力故障と多くの他の要素の不利な影響を受ける可能性がある。そのほか、新冠肺炎疫病或いはその他の全世界圧力による原材料不足により、著者らの臨床前と臨床細胞療法の開発と製造に使用された原材料と消耗材の供給はすでに一時的な中断が続いている可能性があり、その中には私たちの製品と候補製品で使用されている出発材料を供給する白血球分離器の収集、細胞治療製造の専用原材料と消耗材を含む。これらの原材料や他の必要な原材料をタイムリーに得ることができなければ、私たちの業務運営や製造能力は悪影響を受ける可能性があります。
細胞療法の製造過程は汚染、設備故障或いは設備の設置或いは操作が不適切、サプライヤー或いはオペレータのミスにより製品損失を招きやすい。私たちのどの製品と候補製品に対しても、正常な製造と流通プロセスとの微小な偏差が生産良率の低下、肝心な製品の品質属性への影響、その他の供給中断を招く可能性がある。製品の欠陥も意外に起こる可能性がある。試薬中または私たちの製品および候補製品において微生物、ウイルスまたは他の汚染が発見された場合、または私たちの製品および候補製品を製造する製造施設において微生物、ウイルスまたは他の汚染が発見された場合、これらの製造施設は、汚染を調査および修復することができるように、長い間閉鎖する必要があるかもしれない。我々のT細胞免疫治療製品および候補製品は第三者ドナーの血液から収集した細胞から製造されているため,製造過程は第三者ドナー材料の有用性の影響を受ける。商業化できる製品を開発する過程は特に挑戦的かもしれません
49
そうでなければ、安全で効果的であることが証明される。このような製品と候補製品の製造は複雑な過程と関連がある。その中のいくつかの過程は専門的な設備と高い技術と訓練された人員を必要とする。これらの製品や候補製品の製造過程は、製造過程全体で無菌条件を維持する必要性を考慮すると、追加リスクの影響を受けやすい。ドナー材料であっても、製造中に使用される材料中のウイルスまたは他の病原体の汚染であっても、または微生物材料がそのプロセス中の任意の点に入ることは、汚染または使用できない製品をもたらす可能性がある。製品および候補製品を製造するためのウイルス試薬を製造するための組換えウイルス試薬製造システムにおいてもウイルス汚染物質が発生する可能性がある。これらのタイプの汚染は製品製造の遅延を招く可能性があり、候補製品の開発遅延を招く可能性がある。このような汚染物質はまた副作用の危険を増加させる可能性がある。さらに、我々の同種異体製品は最終的に多くの単独の細胞中間体または細胞製品ロットから構成され、各ロットは異なるヒト白血球抗原特徴を有する。そのため、患者治療のための適切な細胞製品ロットの選択と分配は臨床操作、サプライチェーンと品質保証人員の間の密接な協調が必要である。
私たちまたは私たちのCMOが私たちの製品および候補製品の製造運営に悪影響を及ぼすいかなる発展も、ロット故障、在庫不足、出荷遅延、製品撤回またはリコール、または私たちの薬品供給の他の中断を招く可能性があり、これは私たちの候補製品の開発を遅延させる可能性があります。私たちはまた、規格に適合しない薬品の供給によって他の費用と支出が発生し、コストの高い救済措置を取らなければならないかもしれないし、より高価な製造代替案を求めなければならないかもしれない。医師、医療支払者、患者、または私たちの製品開発努力を支持する医療コミュニティ(病院および外来診療所を含む)における私たちの製品および候補製品の需要を満たすことができないことは、私たちの名声と私たちの製品の名声を損なう可能性があります。
私たちのCMO工場で生産された候補製品の遅延規制承認は、私たちの開発計画を延期し、収入を創出する能力を制限する可能性があります。
ARC内部の研究開発,プロセスと分析開発実験室およびコロラド州オーロラにおける我々の施設は現在,われわれの臨床前·中期/後期開発活動を支援している。臨床開発を支援する特定製品資格認証が完了しており,我々CMOの施設では商業生産資格認証活動を行っている。もし私たちのCMO工場の候補製造製品に対する適切な規制承認が延期されたら、私たちは十分な数の候補製品を生産できないかもしれません。これは私たちの開発活動と成長機会を制限します。
我々の施設およびCMOの施設は、“第三者への依存に関するリスク”に記載されているような製造リスクに加えて、cGMPおよびGTPに適合することを確実にするために、FDA、EMAまたは他の同様の規制機関の持続的な定期検査を受ける。私たちまたは私たちのパートナーがこれらの法規または他の法規要件を遵守し、記録することができない場合、臨床または将来の商業用途のための製品供給に重大な遅延をもたらす可能性があり、臨床研究の終了または棚上げを招く可能性があり、または候補製品の商業マーケティング申請の提出または承認を遅延または阻止する可能性がある。私たちはまた次のような点で問題に直面するかもしれない
適用される法規を遵守しないことは、罰金、禁止、民事処罰、私たちの1つ以上の臨床研究の一時停止または棚上げを要求すること、規制機関が私たちの候補製品の上場を承認できなかったこと、遅延、一時停止または承認の撤回、許可証の取り消し、候補製品の差し押さえまたはリコール、運営制限、刑事起訴を含む私たちに制裁を加える可能性があり、これらはすべて私たちの業務を損なう可能性がある。
先進的な製造技術やプロセス制御を開発することは高価で時間がかかり,我々や我々のCMOの施設を活用する必要がある。製造技術やプロセス制御を進められなかったことは私たちの候補製品の承認を遅らせる可能性があります。これ以上の投資がなければ、製造技術の進歩は私たちの候補製品を製造する施設や設備が不足したり時代遅れになったりする可能性がある。
私たちの製品グループの多くの候補製品は、適用される規制機関の承認を得たら、市場ニーズを満たすために大量の商業供給が必要になるかもしれない。これらの場合、私たちは最初の生産レベルでの生産過程の重要な要素を増加または“拡大”する必要があるかもしれない。もし私たちがそれができない場合、あるいは規模を拡大するコストが私たちにとって経済的に不可能である場合、あるいは第三者サプライヤーが見つからなければ、未来の需要を満たすのに十分な数の候補製品を生産できないかもしれない。
50
もし私たちのCMOの1つ以上の施設が破損したり破壊されたり、あるいはこれらの施設の生産が他の理由で中断された場合、私たちの業務は負の影響を受けるだろう。
もし私たちのCMOの任意の製造施設または任意のそのような施設の設備が破損または破壊された場合、私たちはそのような製造能力を迅速または安価に交換できないか、または全く交換できない可能性がある。一時的または長期的な運営中断または施設またはその設備の損失がある場合、供給を維持するのに必要な時間内に生産を他方の第三者に移転することができない可能性がある。生産を他方に移すことができても、移転は高価で時間がかかる可能性があり、特に新工場は必要な規制要求を遵守する必要があるため、またはその工場で生産された任意の製品を販売する前に規制部門の承認が必要となる可能性がある。そのような事件は私たちの臨床研究を延期したり、私たちの商業製品の収入を減少させるかもしれない。
現在、私たちは私たちの財産損失に保険を提供し、業務中断と研究開発回復費用を支払います。しかし、私たちの保険カバー範囲は私たちを清算しないかもしれないし、私たちが受ける可能性のある費用や損失を清算するのに十分ではないかもしれない。もし私たちの現在の製造施設やプロセスに悲劇的な事件や故障が発生したら、私たちは候補製品に対する要求を満たすことができないかもしれません。
私たちの第三者への依存に関するリスク
臨床およびビジネススケジュールの維持は、製造を支援するために当社のエンドツーエンド·サプライチェーンネットワークに依存し、第三者サプライヤーやCMOの問題に遭遇した場合、私たちの製品および候補製品の開発および/または商業化を延期する可能性があります。
私たちは、私たちのCMOまたは私たちのパートナーに依存して、現在私たちの製品および候補製品を生産し、私たちの製品および候補製品の製造またはテストに組み込まれたり使用されたりする材料を取得します。私たちのCMOやパートナーは私たちの従業員ではなく、CMOやパートナーとの合意に基づいて私たちに提供された救済措置を除いて、私たちが行っている臨床、非臨床、および臨床前プロジェクトの供給を作るのに十分な時間と資源(経験豊富な従業員を含む)が投入されているかどうかを直接制御することはできない。私たちの製品と候補製品のCMOは、私たちの規制届出に関する承認前検査を受ける準備ができている必要があり、私たちはこのような検査を通じて彼らに十分な支援を提供できるかどうかを確認することができず、彼らがこのような検査を通過することに成功するかどうかを確認することもできません。
私たちの活動を支援するために、臨床および商業材料に対する予想される供給需要を満たすために、規制部門の承認とTAB-CEL、ATA 188、私たちの次世代CAR T計画によって生成された任意の候補製品または任意の他の候補製品の商業製造を通じて、これらの材料の製造をCMOに移す必要がある。どこで生産しても,キー原材料や試薬のサプライヤーと関係を発展させ,生産規模を拡大し,これらの施設で生産された材料が従来生産されていた材料と比較可能であることを証明する必要がある。移転製造プロセス、分析方法と専門技術は複雑であり、時間の経過とともに変化する可能性のある文書記録と無文書記録の審査と組み入れの過程に関する。
さらに、生産を異なる施設に移転するためには、特定の施設の具体的な要件を満たすために、新しいプロセスまたは異なるプロセスを利用する必要がある場合がある。いくつかの製造プロセスとプロセス改善の移転を支援するために、より多くの比較可能な作業を行う必要があると予想される。以前に生産された材料と我々または我々のCMOが生成した材料との比較性を証明するための研究(および関連評価)が完了するまで、すべての関連する技術的ノウハウおよびデータが製造プロセスに十分に組み込まれていることを決定することはできない。
もし私たちまたは私たちのCMOが比較可能な製品と候補製品の移転と生産に成功できなければ、私たちの製品と候補製品をさらに開発して製造する能力は負の影響を受ける可能性がある。
私たちはまだ私たちのいくつかの製品と候補製品のために追加のCMOを探して生産供給を続ける必要があるかもしれない。我々の製造プロセスの性質から,我々の候補T細胞免疫療法製品を生産するために必要な技能と能力を有するCMOおよびそのような製品を製造するための重要な中間体や試薬の数は限られている。もし私たちが現在使用しているCMOが大規模化生産できない場合、あるいは他の方面で何か問題があったら、私たちはまだ代替サプライヤーを確定していません。
私たちは私たちのCMOと製造ネットワークに頼って私たちの製品と候補製品を生産します。私たちのこれらの製品と候補製品の供給はこれらの施設の絶え間ないかつ効率的な運行に依存しており、これらの施設は設備故障、労働力或いは原材料不足、公衆衛生流行病、自然災害、電力故障、ネットワーク攻撃、および多くの他の要素の不利な影響を受ける可能性がある。もし私たちがどんな製造やサプライチェーンの困難に直面したら、私たちは私たちの製品と製品候補に対する需要を満たすことができないかもしれない。
51
細胞療法の製造はFDAと世界各地の類似の監督管理機関によって複雑かつ厳格な監督管理を行い、必要な製造と監督管理の専門知識と施設を持つ代替第三者サプライヤーが存在するにもかかわらず、代替サプライヤーを手配し、製造手順と分析方法をこれらの代替サプライヤーに移し、これらの新しいサプライヤーが生産した材料が比較可能性が高い可能性があることを証明し、大量の時間を必要とする。どんな製品、候補製品、または中間体の新しい製造業者も、適用される規制要件に適合することが要求されるだろう。これらのメーカーは、コストや十分な量で私たちの製品や候補製品を製造できないかもしれませんし、私たちの候補製品の開発や商業的に成功した製品をタイムリーに完成させることができないかもしれません。もし私たちが代替の第三者製造源を手配できない場合、あるいは商業的に合理的な条項や適時にそうすることができなければ、私たちは候補製品の開発を完成させたり、マーケティングや流通を行うことができないかもしれません。さらに、FDAまたは同様の規制機関がこれらの材料の候補製品仕様および比較可能な方法または評価に同意しない場合、規制当局は、接続可能性試験を含む追加的な研究を要求する可能性があり、私たちの候補製品のさらなる臨床開発または商業発売は大幅に遅れる可能性がある。
第三者製造商会に依存してリスクをもたらし、もし私たちが自分で製品や候補製品を製造すれば、私たちは第三者の法規遵守性と品質保証に依存することを含むこれらのリスクの影響を受けず、第三者メーカーは製造協定の規定された義務を履行するための財政資源を維持していない可能性があり、第三者は私たちの制御できない要素によって製造合意に違反する可能性があり、私たちの規格に従って私たちの候補製品または私たちまたは私たちのパートナーが最終的に商業化される可能性のあるいかなる製品を製造できなかったか、私たちのビジネス秘密やノウハウを含む、私たちの独自の情報を流用し、第三者が私たちの候補製品に十分な時間または資源を投入する可能性がない、または私たちまたは私たちのパートナーが最終的にその業務優先度に基づいてそれを商業化する可能性のある任意の製品、第三者が他方によって買収され、その業務優先度を変更する可能性、および第三者がそれ自身の業務優先度に基づいて、私たちにコストまたは損害を与えた時間に合意を終了または更新しない可能性がある。富士フィルムが富士フイルムMSAでの義務を十分に履行していない場合、あるいは私たちの製品や候補製品に十分な時間や資源を投入していなければ、私たちの製品の商業化を含め、私たちの運営は不利な影響を受ける可能性がある。同様に,CRLがCRL MSAが規定する義務を十分に履行していない場合や,我々の製品や候補製品に十分な時間や資源が投入されていなければ,我々の製品の商業化を含めて我々の運営は悪影響を受ける可能性がある.私たちはまたCMOとのいくつかの合意で製品とサービスに対してキャンセルできない最低購入約束をした, もし私たちがこのような最低購入約束を履行しなければ、私たちは適用された最低購入約束と私たちが所与の期間内に実際に購入した製品とサービスとの間の差額をこれらのCMOに支払う必要があるだろう。さらに、FDAおよび他の規制機関は、私たちの候補製品および私たちまたは私たちのパートナーが最終的に商業化される可能性のあるどの製品も、cGMP、CGTP、および同様の規制管轄基準で生産されなければならないことを要求する。これらの要求は、他にも、品質管理、品質保証、および記録およびファイルの維持を含む。FDAまたは同様の外国規制機関は、新しい基準を随時実施したり、製品製造、包装、またはテストの既存の基準の解釈および実行を変更したりすることもできる。私たちは私たちの製造業者を監視しているにもかかわらず、私たちは彼らに誠実で正確な情報を提供することに依存しているが、私たちは製造業者にこれらの法規と標準を遵守する制御が限られている。当社の第三者製造業者は、十分な数の候補製品をタイムリーに配送できなかったことを含む、cGMPまたはCGTPに準拠できなかったか、または規制部門の承認を得ることができなかった候補製品のいずれかを遅延させる可能性があります。さらに、このような失敗は、FDAが警告状を発行すること、以前に私たちに付与された候補製品の承認を撤回すること、または候補製品の外部供給のリコールまたは差し押さえ、生産の完全または部分的な一時停止、進行中の臨床研究の一時停止、承認保留申請または補充申請の拒否、拘束または製品の承認の拒否、製品の輸出入の許可の拒否、禁止または民事および刑事罰の適用を含む他の規制または法的行動の根拠となる可能性がある。
私たちは第三者サプライヤーとテスト実験室に依存して、私たちの製品と候補製品を生産またはテストするための重要な材料を提供します。供給者関係のどんな重大な中断も私たちの業務を損なう可能性がある。進行中の臨床研究に候補製品方面のいかなる重大な遅延を提供することは、著者らの臨床研究、製品テストの起動或いは完成、及び著者らの候補製品の潜在的な監督管理許可を大幅に延期する可能性がある。原材料やコンポーネントを購入できない場合や承認された仕様を満たすことができない場合、私たちの製品や候補製品の商業発表が遅れる可能性があり、あるいは供給不足が発生する可能性があり、私たちの製品や候補製品を販売することから収入を得る能力を弱める可能性があります。
私たちはEUとアメリカ以外のいくつかの国でEbvalloを商業化することにピエール·ファブレに依存している。Pierre Fabreがその契約、規制、または他の義務を履行できなかったことは、HCRx協定の下での私たちの業務と私たちの義務に悪影響を及ぼすかもしれない。
我々はすでにヨーロッパでEbvalloに対するPierre Fabre商業化協定を締結し,EBV陽性癌の治療に中東,アフリカ,東欧,中央アジア(地域)の新興市場を選択した。したがって、私たちは領土でEbvalloのマーケティングと商業化、交渉定価と補償を含むピエール·ファブルに完全に依存している。Ebvalloマーケティングの承認と譲渡に関するいくつかのマイルストーン支払いを除いて
52
許可された後、私たちはHCRx協定で適用される特許使用料の上限(あれば)に達するまで、Pierre Fabreから意味のあるマイルストーンや特許使用料支払いを受けないだろう。しかも、私たちが受け取ることができる任意のマイルストーンと特許税の支払いの時間と金額 ピエール·ファブル商業化協定およびEbvalloのこの領土での商業成功は、他を除いて、Ebvalloに対するピエール·ファブレの領土での努力、資源分配、価格設定、補償交渉、および商業化の成功に依存するであろう。
Pierre Fabre商業化協定の条項によると、EUがEBV+PTLD患者にEbvalloを使用するマーケティング許可を得た場合、マーケティング許可をPierre Fabreに譲渡しなければならない。Pierre Fabreは領土内のすべての他の規制承認を獲得し、領土内のすべての規制承認を維持する責任があるだろう。私たちはPierre Fabreが多くと異なる法規要求を遵守することに依存し、適用されれば、製造、品質管理、さらなる開発、ラベル、包装、貯蔵、流通、不良事件報告、安全監視、輸出入、広告、販売促進、記録保存とその他の発売後の情報を記録し、報告する。ピエール·ファブレの個人的な努力をコントロールすることはできませんが、ピエール·ファブルのパフォーマンスが予想以上であれば、私たちはピエール·ファブレの商業化協定を中止する能力が限られています。Pierre Fabreが規制要件を遵守し、領土内でEUのマーケティング許可および他の規制承認および/またはそれの私たちに対する義務を維持するのに十分な時間とエネルギーを投入できなかった場合、私たちの財務業績と運営およびHCRx協定下での私たちの義務に悪影響を及ぼすかもしれない。
私たちはまた、この領土におけるEbvalloの商業化に関するすべての適用法を遵守するピエール·ファブレに依存している。Pierre FabreはEbvalloを商業化するのに十分な時間とエネルギーを投入できなかった;将来の特許権使用料とマイルストーン支払いを含む彼らの義務を履行できなかった;危機が発生した時に業務連続計画を十分に配置できなかった;および/または私たちとの重大な相違を満足に解決できなかった、または他の要素を処理することができなかったことは、私たちの財務業績と運営、HCRx合意下での私たちの義務に悪影響を及ぼす可能性がある。さらに、ピエール·ファブレが私たちへの義務を履行している間に任意の法律または法規に違反または非難された場合、可能な法的結果を含む財務的名声被害や他の否定的な結果を受ける可能性がある。
ピエール·ファブレ商業化協定または付属協定の任意の終了、違約、または満了は、締結されると、費用、マイルストーン、および特許権使用料を得る私たちの権利を減少またはキャンセルする可能性があり、それによって、私たちの財務状況およびHCRx協定下での私たちの義務に大きな悪影響を及ぼす可能性がある。この場合、私たちはより多くの努力をし、Ebvalloの領土内での規制承認および商業化に関する追加費用を生成する必要があるかもしれない。あるいは、私たちは新しい商業化パートナーを探して取引をしようと試みるかもしれないが、適切なパートナーを見つけることができるか、またはPierre Fabre商業化協定のような条項や私たちに有利な条項に従って取引できる保証はない。
私たちは、私たちが将来形成する可能性のある戦略連合のメリット、あるいは将来の潜在的な製品買収や許可のメリットを意識していないかもしれない。
我々は、より多くの戦略的同盟、商業的パートナーシップの構築、合弁企業または協力の作成、第三者とのライセンス契約、製品または事業の買収を希望するかもしれませんが、いずれの場合も、既存の業務を補完または拡大すると考えられています。これらの関係や取引、または同様に、非日常的な費用や他の費用を発生させ、短期的および長期的な支出を増加させ、既存の株主を希釈した証券を発行し、関係の対象となる製品の潜在的な収益性を低下させたり、私たちの管理や業務を混乱させたりする必要があるかもしれない。また、適切な戦略連合や取引を求める上で、私たちは激しい競争に直面しており、交渉過程は時間も複雑であり、そうしたいとしても、いずれの取引も達成できる保証はない。また、将来の候補製品および計画のための戦略同盟または他の代替手配を確立する努力は成功しない可能性があり、私たちの研究開発ルートが不足している可能性があるため、私たちの候補製品および計画は協力努力の早期開発段階にあると考えられる可能性があり、第三者は私たちの候補製品および計画が積極的な収益/リスクプロファイルを示すために必要な潜在力を持っているとは思わないかもしれない。私たちの候補製品に関連する新しい戦略連合協定のいかなる遅延も、私たちの候補製品の開発と商業化を延期し、市場に進出してもそれらの競争力を低下させる可能性がある。また、構築された戦略連合協定の終了は、関連合意によって得られる可能性のある任意の将来の資金を終了し、新たなパートナーを探して開発または商業化しなければならないだろう, このような開発や商業化を減少または放棄したり、製品の開発および商業化に関連して資金を提供したりする。もし私たちが新しいパートナーを探しているが、受け入れ可能な条件でそうすることができない場合、あるいは自分で製品の開発や商業化を行うのに十分な資金がない場合、私たちの業務を損なう可能性のある開発や商業化を減少または放棄することを含む他の戦略的選択を模索しなければならない。例えば、2022年7月31日から、私たちはベイヤ終了プロトコルに従ってベイヤプロトコルを終了した。したがって、我々は、ATA 2271およびATA 3271のさらなる開発および製品商業化の責任を負い、承認されれば、ATA 2271および/またはATA 3271の新しいパートナーと新たな戦略的協力を達成するまでである。
53
もし私たちが製品や買収業務を許可すれば、これらの取引を私たちの既存の運営や会社文化と組み合わせることに成功できなければ、これらの取引のメリットを実現できないかもしれません。買収が可能かもしれない後、私たちがこの取引の合理的な財務や戦略的結果を証明することが達成されるかどうかは確信できない。
私たちの知的財産権に関するリスクは
もし私たちが候補製品のために十分な知的財産権保護を得ることができなければ、あるいは知的財産権保護の範囲が十分でなければ、私たちの候補製品の商業化と効果的な競争に成功する能力は不利な影響を受ける可能性がある。
私たちは、特許、商標、商業秘密、および秘密協定の組み合わせによって、私たちの技術、製品、および候補製品に関連する知的財産権を保護し、これらの特許、商標、商業秘密および秘密協定は、私たちが所有しているか、または私たちのパートナーによって所有されているか、または所有している。私たちが私たちの技術、発明、特許、特許出願または他の知的財産権に言及すると、私たちは私たちの知的財産権保護と私たちの業務に重要な私たちの知的財産権保護および私たちの業務に重要な私たちの所有または所有する権利と私たちの許可の権利を意味する。例えば、私たちがパートナーから許可を得た製品、候補製品、およびプラットフォーム技術は、主に、私たちが許可したパートナーの特許または特許出願、ならびに秘密技術および商業秘密によって保護される。もし私たちが依存している知的財産権が十分に保護されていなければ、競争相手は私たちの技術を使用して、私たちが持つ可能性のあるいかなる競争優位性を侵食または否定するかもしれない。
発明の特許性およびバイオテクノロジー分野特許の有効性,実行可能性,範囲は不確定であり,複雑な法律,科学的,事実的考慮に関連しており,近年重大な訴訟のテーマとなってきた。また、米国特許商標局(USPTO)および非米国特許庁が特許を付与する際に適用される基準は、常に統一的または予測可能ではない。例えば、バイオテクノロジー特許において許容される特許標的または特許請求の範囲については、世界的に統一された政策はない。
私たちの特許および特許出願に関連するすべての潜在的に関連する以前の技術が私たちに知られているか、または検索の場合に発見されたことを保証することはできない。私たちは、既存技術が、発行された特許を無効にするために使用されるか、または係属中の特許出願を特許発行として阻止するために使用されることができることを知らないかもしれない。私たちが知っているかもしれませんが、私たちは、私たちの特許または特許出願の特許請求の有効性または実行可能性に影響を与えるとは考えられませんが、最終的には、特許請求の有効性または実行可能性に影響を及ぼすことが発見される可能性があります。これらおよび他の要因のため、私たちの特許出願は、発行された特許が、私たちの製品および米国または他の国/地域の候補製品をカバーすることを招くことができないかもしれない。
特許が特許出願から成功裏に発表されたとしても、これらの特許が我々の製品および候補製品をカバーしていても、第三者はその有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、これらの特許が縮小され、無効であるか、または実行不可能であると認定される可能性がある。もし挑戦されたら、私たちの特許は裁判所によって効果的または強制的に執行可能だと宣言される保証はない。
挑戦されていなくても、私たちの特許や特許出願や他の知的財産権は、私たちの知的財産権を十分に保護できず、私たちの製品や候補製品に排他性を提供したり、他の人が私たちの声明を中心に設計することを阻止したりする可能性があります。他の人は、私たちの製品および候補製品と同じ効果を有し、私たちの特許または他の知的財産権を侵害しない製品を独立して開発するか、または他の人は、私たちが発表した私たちの製品および候補製品をカバーする特許の権利要件を中心に設計される可能性がある。もし私たちの特許と特許出願が私たちの製品と候補製品に提供する保護の広さや強度が脅かされたら、私たちの製品と候補製品を商業化する能力を危険にさらし、会社が私たちと協力することを阻止するかもしれません。
私たちはまた、知的財産権を持つ第三者に許可を求めることを望むことができ、この許可は、私たちの製品および候補製品の排他性を提供すること、または制限されない方法で候補製品を開発し、それを商業化する能力を提供することに役立つ可能性がある。私たちが商業的に合理的な条項でそのような第三者から許可を得ることができるか、あるいは全く保証されない保証はない。
さらに、米国特許商標局および各種外国政府特許機関は、特許出願中にいくつかのプログラム、文書、費用支払い、および他の同様の規定を遵守することを要求する。多くの場合、適用規則に従って滞納金を支払うか、または他の方法で不注意を是正することによって失効を是正することができるが、場合によっては、規定を遵守しないことは、特許または特許出願が放棄または失効され、関連法ドメインの特許権の一部または全部の喪失をもたらす可能性がある。
私たちと私たちのパートナーは、私たちの製品および候補製品をカバーするか、またはこれらの候補製品を使用または製造する方法を含む複数の特許出願を提出した。私たちは何の保証も提供できません。もしあれば、これらの特許に関連するものを発行します
54
未完了の特許出願、最終的に発行される任意のそのような特許の広さ、または発行された任意の特許が無効であることが発見されるかどうか、強制的に実行できないか、または第三者によって脅かされるであろうか。米国およびほとんどの他の国の特許出願は出願後しばらくは秘密であり、一部の特許出願は発行前に依然として秘密であるため、我々または我々のパートナーが製品または候補製品に関連する特許出願を最初に提出した会社であることを確認することはできない。私たちまたは私たちのパートナーはまた、特許侵害訴訟、妨害または派生訴訟、反対、および私たちの特許関連訴訟に巻き込まれる可能性がある各方面間米国特許商標局,欧州特許庁及びその他の非米国特許庁に提出された認可後審査手続。
許可を得ても、特許の寿命は限られている。アメリカでは、特許の自然消滅は一般的に出願20年後に発生する。いくつかの条件が満たされれば、様々な延期がある可能性があるにもかかわらず、特許の有効期間およびその提供される保護は限られている。もし私たちが臨床研究中や規制承認を得る上で遅延に遭遇した場合、私たちの任意の製品と特許保護された製品を独占的に販売することができ、承認されれば、短縮される可能性がある。私たちの製品と候補製品をカバーする特許を取得しても、製品の特許有効期限が切れると、私たちは競争相手が私たちの候補製品と似ているか同じ製品で市場に参入することを阻止できないかもしれないので、生物類似製品からの競争を受けやすいかもしれません。
また、私たちのいくつかの特許権と技術の研究はアメリカ政府によって資金援助されている。したがって、政府はこれらの特許権と技術に対して一定の権利を持っている。政府の援助の下で新しい技術を開発する場合、政府は通常、政府が米国または米国に代わって発明を実施するための非独占的許可を許可することを含む、生成された任意の特許のいくつかの権利を得る。これらの権利は、政府が私たちが依存している機密情報を第三者に開示し、第三者が私たちが許可した技術を使用または使用することを許可する先行権を行使することを可能にするかもしれない。政府が政府援助の技術の実用化を実現できなかったため、健康や安全需要を緩和し、連邦法規の要求を満たすために行動する必要があると判断した場合、または米国工業を優先することができれば、政府はそのデモ権利を行使することができる。さらに、政府によって援助された研究によって生成された任意の発明の権利は、これらの発明を体現する製品を米国で製造するいくつかの要件によって制約される可能性がある。
もし私たちが第三者の知的財産権侵害で起訴されれば、それによって生じる訴訟は費用が高く時間がかかる可能性があり、私たちまたはパートナーの開発と商業化努力を阻害または延期する可能性がある。
私たちのビジネスの成功は、私たちと私たちのパートナーが第三者の特許と独自の権利を侵害しないことにある程度かかっている。アメリカ国内外で、大量の訴訟とその他の対抗性訴訟があり、生物技術と製薬業界の特許とその他の知的財産権に関連し、特許侵害訴訟、妨害或いは派生訴訟、反対と各方面間米国特許商標局と非米国特許庁で行われた認可後審査手続。我々が開発している分野には,米国や非米国から発行された特許および第三者が所有する未解決特許出願が多く存在し,我々の製品や候補製品を開発することが可能である.バイオテクノロジーや製薬業界の拡張や特許の発行に伴い、我々の製品や候補製品が第三者特許権侵害のクレームを受けるリスクが増加する可能性がある。なぜなら、私たちを含む業界参加者は、どの特許が様々なタイプの製品または使用方法をカバーしているかを常に明らかにしていない可能性があるからである。特許のカバー面は裁判所の解釈に依存し、解釈は常に統一的または予測可能ではない。
第三者は、既存または未来の知的財産権に基づいて、私たちが彼らの独自技術を不正に使用していると主張する権利侵害を私たちに提起するかもしれない。材料、処方、製造方法、または治療方法に要求される当社の製品または候補製品の使用または製造に関連する第三者特許または特許出願が存在する可能性がある。例えば、これらの出願は、通常、提出日後の一定期間秘密に維持されているので、私たちの製品および候補製品に関する特許出願は、私たちが知らずに他の人によって提出される可能性がある。すでに公表されている係属中の特許出願であっても、私たちが知っているいくつかの特許出願を含めて、私たちの製品および候補製品、またはそれらの使用または製造をカバーするために、後で修正されることができる。さらに、私たちは、私たちの活動に関連すると考えられる第三者特許または特許出願を分析し、私たちの任意の製品および候補製品に対して自由に運営できると考えているかもしれませんが、私たちの競争相手は、私たちが関係ないと思う特許を含む発表されたクレームを得るかもしれません。これは、私たちの努力を阻害したり、私たちの任意の製品、候補製品、または私たちの活動が彼らのクレームを侵害する可能性があります。
もし私たちまたは私たちのパートナーが特許侵害を起訴された場合、私たちは私たちの候補製品、製品、および方法が関連特許を侵害していないか、特許主張が無効であるかを証明する必要があり、私たちはそれができないかもしれない。特許が無効であることを証明することは困難であり,関連訴訟で勝訴しても,我々は巨額のコストを招く可能性があり,我々管理者や科学者の時間と注意力が他の活動から移行する可能性がある。管轄権のある裁判所が発行された任意の第三者特許を持っている場合、私たちの材料、処方、製造方法、または治療方法の様々な態様をカバーしている場合、関連特許が満了するまで、関連製品または候補製品の開発、製造、または商業化を停止、製造、または商業化することを余儀なくされる可能性がある。あるいは許可証の取得を希望したり要求されたりするかもしれません
55
権利侵害技術を使用し、侵害製品または候補製品の開発、製造または販売を継続する。しかし、私たちは商業的に合理的な条項で必要な許可証を得ることができないかもしれないし、全く得られないかもしれない。たとえ我々が許可を得ることができても,これらの権利は非排他的である可能性があり,これは我々の競争相手が許可を得た同じ知的財産権へのアクセス権をもたらす可能性がある.
私たちは第三者の機密情報や商業秘密を流用する疑いに直面するかもしれない。もし私たちが第三者の商業秘密を盗用したことが発見されたら、私たちはこれらの商業秘密のさらなる使用を阻止されるかもしれません。これは私たちの候補製品を開発する能力を制限するかもしれません。
結果にかかわらず、知的財産権クレームの弁護は高価で時間がかかるかもしれない。したがって、私たちが最終的に勝訴したり、最終判決の前に和解が成立したりしても、どの訴訟も思わぬ費用を私たちにもたらす可能性がある。また,訴訟や脅威訴訟は,我々の管理チームの時間や注意力に大きな要求を与え,会社の他の業務への追求を分散させる可能性がある.任意の知的財産権訴訟において、聴聞結果、動議裁決、および訴訟における他の一時的手続きが公開される可能性があり、これらの発表は、私たちの製品、候補製品、プログラム、または知的財産権の知覚的価値に悪影響を及ぼす可能性がある。もし私たちの知的財産権に対するクレームが成功すれば、私たちは3倍の損害賠償と弁護士費を含めて巨額の損害賠償を支払わなければならないかもしれない(もし私たちが故意に特許侵害が発見された場合)、あるいは私たちの侵害製品や候補製品を再設計することは不可能かもしれないし、多くの時間とお金の支出が必要かもしれない。金銭賠償を支払う以外に、私たちは貴重な知的財産権や人員を失う可能性があり、私たちにクレームをつけた当事者は禁止令や他の衡平法の救済を受ける可能性があり、これは私たちの業務行為に制限を加えるかもしれない。私たちはまた、訴訟前に特許侵害クレームを解決するためにライセンス契約を締結することを選択することができ、これらのライセンス契約のいずれも、印税および他の可能性のある高額な費用の支払いを要求する可能性がある。上記のすべての状況のため、任意の実際または脅威の知的財産権クレームは、私たちまたは私たちのパートナーが製品または候補製品を開発または商業化することを阻止し、またはいくつかの態様のビジネス運営を停止させる可能性がある。
私たちは世界各地で私たちの知的財産権を保護できないかもしれない。
世界のすべての国で私たちのすべての製品と候補製品の申請、起訴、強制執行、特許保護の費用は目を引くほど高いだろう。私たちのアメリカ以外のある国の知的財産権は、アメリカの知的財産権が広くないかもしれません。また、ある外国の法律の知的財産権の保護の程度はアメリカの法律とは違います。したがって、私たちと私たちのパートナーは、米国以外の国で私たちの発明を実施したり、アメリカや他の司法管轄区域で私たちの発明を使用して製造した侵害製品を販売または輸入することを阻止することができないかもしれません。競争相手は、私たちがまだ特許保護を受けていないか、または関連特許の下で独占的な権利を持っていない司法管轄区で私たちの技術を使用して自分の製品を開発することができ、私たちと私たちのパートナーに特許保護を持っているかもしれませんが、法執行力はアメリカの地域に比べて他の侵害製品を輸出することができません。これらの侵害製品は、私たちまたは私たちのパートナーが特許を発行していないか、または関連特許の下で独占的な権利を持っていない私たちの司法管轄区域で私たちの製品や候補製品と競争しているか、あるいは私たちの特許主張と他の知的財産権が彼らの競争を効果的に阻止できないか、または彼らを阻止するのに十分ではないかもしれません。
多くの会社は外国の管轄区域の知的財産権の保護と保護に重大な問題に直面している。特定の国の法律制度、特に特定の発展途上国の法律制度は、特許や他の知的財産権保護の実行、特に生物製薬に関連する特許保護を支持しておらず、これは、私たちと私たちのパートナーが、私たちの特許の侵害を阻止したり、私たちの知的財産権に違反する競争製品を販売したりすることを困難にする可能性がある。外国の管轄区域で私たちの特許権を強制的に執行する訴訟手続きは巨額のコストを招く可能性があり、私たちの業務の他の方面への注意を移すことは、私たちの特許が無効または偏狭に解釈されるリスクに直面する可能性があり、私たちの特許出願を発表できないリスクに直面させ、第三者が私たちまたは私たちのパートナーにクレームを提起する可能性がある。私たちまたは私たちのパートナーは、私たちまたは私たちの許可者が起こしたいかなる訴訟でも勝利しないかもしれません。私たちまたは私たちのライセンシーが勝訴しても、判決された損害賠償または他の救済措置(もしあれば)は商業的な意味がないかもしれません。
私たちは私たちのパートナーから知的財産権のかなりの部分の許可を得た。もし私たちがこれらのパートナーとの任意の許可協定に違反すれば、私たちは私たちの1つまたは複数の製品および候補製品を開発し、潜在的に商業化し続ける能力を失うかもしれない。
我々は,MSK,QIMR Berghofer,Moffittを含むパートナーと,ライセンスプロトコルにより我々の業務に非常に重要な権利を持っている.我々の発見と開発プラットフォームは,我々のパートナーから得られた特許権を中心にある程度構築されている.私たちの既存のライセンス契約によると、開発や商業化活動に関する職務義務、あるマイルストーンを実現する際の支払い義務、製品販売の特許使用料など、様々な義務があります。もし私たちと私たちの取引相手との間に、これらの許可協定の下での私たちの権利または義務について、私たちが勤勉または支払いを満たすことができなかったことによるいかなる衝突、論争、または相違を含む、紛争、論争、分岐、または不履行の問題が存在すれば、
56
義務、私たちは損害賠償を支払う責任があるかもしれません。私たちの取引相手は影響を受けた許可証を終了する権利があるかもしれません。私たちのパートナーのうちの1つとの任意の許可合意を終了することは、薬物発見および開発作業において、ライセンス契約によって拘束された知的財産権を利用する能力、影響を受けた1つまたは複数の製品および候補製品に対して将来の協力、許可および/またはマーケティング協定を達成する能力、および私たちまたは私たちのパートナーが影響を受ける製品および候補製品の商業化の能力に大きな悪影響を与えるであろう。また、これらの許可協定のいずれかの分岐は、私たちとパートナーとの関係を損なう可能性があり、これは、私たちの業務の他の側面に悪影響を及ぼす可能性があります。
私たちは私たちの知的財産権を保護または実行する訴訟に巻き込まれる可能性があり、これは高価で、時間がかかり、成功しない可能性があり、私たちの業務の成功に実質的な悪影響を及ぼすかもしれない。
第三者は私たちの特許を侵害したり、流用したり、他の方法で私たちの知的財産権を侵害する可能性がある。我々の特許出願は、特許が出願から発行されるまで、および発行されるまで、発行された特許要件が技術の範囲をカバーする限り、これらの出願に要求される技術を実施する第三者に対して実行することはできない。将来的に、私たちまたは私たちのパートナーは、私たちまたは私たちのパートナーの知的財産権を強制または保護し、私たちまたは私たちのパートナーの商業秘密を保護し、または私たちの知的財産権の有効性または範囲を決定するために、法的手続きを開始することを選択することができる。私たちまたは私たちのパートナーは、侵害者が提起したと思われる任意のクレームに対して、これらの当事者が私たちまたは私たちのパートナーに反訴し、私たちまたは私たちのパートナーが彼らの知的財産権を侵害したり、私たちの知的財産権を無効にしたと主張することを促進するかもしれません。
第三者によって引き起こされる、我々または我々のパートナーによって提起される、またはUSPTOまたは任意の非米国特許機関によって提起される干渉または派生プログラムは、我々の特許または特許出願に関連する発明または発明権事項の優先権を決定するために必要である可能性がある。私たちや私たちのパートナーも再審や異議手続きのような他の手続きに参加することができます各方面間米国特許商標局またはその外国同業者における、私たちの知的財産権または他の人の知的財産権に関連する審査、認可後の審査、または他の認可前または認可後の手続き。これらの訴訟のいずれの不利な結果も、私たちまたは私たちのパートナーに関連技術の使用を停止し、私たちの製品および候補製品を商業化することを要求するか、または勝利者から許可権利を得ようとするかもしれない。もし勝利者が商業的に合理的な条項で私たちまたは私たちのパートナーに許可を提供しなければ(何かの許可を提供すれば)、私たちの業務は損害を受ける可能性がある。たとえ我々または我々の許可者が許可を得ても,我々の競争相手が許可または我々の許可側の同じ技術にアクセスできるように非排他的である可能性がある.さらに、私たちの特許および特許出願によって提供される保護の広さまたは強度が脅かされている場合、会社が私たちと協力し、現在または将来の製品および候補製品を許可、開発、または商業化することを阻止するかもしれない。
どんな知的財産権訴訟も費用が高くて時間がかかるかもしれない。私たちまたは私たちのパートナーと比較して、私たちまたは私たちのパートナーは、このような訴訟における相手が、このような法的行動を起訴するためにより多くの資源を投入する能力があるかもしれない。したがって、私たちまたは私たちのパートナーは努力しているにもかかわらず、私たちまたは私たちのパートナーは、第三者が私たちの知的財産権を侵害したり、流用したりすることを阻止できないかもしれません。特に法律ではアメリカのように私たちの権利を十分に保護する国がないかもしれません。関連訴訟で勝訴しても、私たちは巨額のコストを招く可能性があり、私たちの管理者や科学者の時間と注意は他の活動に移される可能性があります。もし私たちが故意に特許を侵害したことが発見されたら、私たちは3倍の損害賠償と弁護士費を含む金銭損害賠償責任を負われるかもしれない。さらに、侵害訴訟では、裁判所は、私たちの1つまたは複数の特許の全部または一部が無効または強制的に実行できないと判断するか、または私たちの特許が関連技術をカバーしないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。どんな訴訟手続きの不利な結果も、私たちの1つ以上の特許を無効が宣言されるか、強制的に実行できないか、または狭い解釈される危険性に直面させる可能性がある。
さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。さらに、公聴会、動議、または他の一時的な手続き、または事態の発展の結果が発表される可能性がある。
もし私たちが私たちの商業秘密と他の固有情報の機密性を保護できなければ、私たちの技術的価値は実質的な悪影響を受ける可能性があり、私たちの業務は損なわれるかもしれない。
特許提供の保護を求めることに加えて、私たちは、特許を出願できない独自技術、特許を実施しにくいプロセス、および私たちの技術、発見および開発中に特許に含まれないノウハウ、情報または技術に関する他の要素を保護するために、商業秘密保護および秘密保護プロトコルに依存して保護する。我々がパートナーから許可を得たT細胞免疫療法候補製品やプラットフォーム技術は,主に機密技術や商業秘密として保護されている。第三者による私たちの機密固有情報の開示または流用は、我々の候補製品と実質的に類似したまたは競争力のある製品を開発し、それを商業化し、市場における私たちの競争的地位を侵食することを含む、競争相手に当社の技術成果を迅速に複製または超えることを可能にする可能性がある。
57
商業秘密は保護するのが難しいかもしれない。私たちは、私たちの従業員、コンサルタント、CMO、および外部科学コンサルタント、請負業者、協力者と秘密協定および発明譲渡協定を締結することによって、私たちのノウハウおよびプロセスを保護することを求めています。このような協定は私たちの固有の情報を保護することを目的としている。私たちは合理的な措置を取って私たちの商業秘密を保護しているにもかかわらず、私たちの従業員、コンサルタント、請負業者、または外部科学コンサルタントは、私たちの商業秘密または機密、独自の情報を競争相手に意図的にまたは意図的に漏らしてしまう可能性がある。また,競争相手は我々のビジネス秘密を他の方法で取得したり,基本的に同じ情報や技術を独立して開発したりする可能性がある.もし私たちの任意の機密固有情報が競争相手によって合法的に取得または独立して開発された場合、私たちはその競争相手がその技術または情報を使用して私たちと競争することを阻止する権利がなく、これは私たちの競争地位を損なう可能性がある。
第三者が私たちのいかなる商業秘密を不法に取得して使用することを強要するかは高価で時間がかかり、結果は予測できない。さらに、特定の国/地域の法律は、商業秘密などの独自の権利の保護の程度や方法が米国の法律とは異なる。当社の商業秘密を第三者に流用または不正に開示することは、市場での競争優位性を損なう可能性があり、私たちの業務、運営結果、および財務状況に実質的な悪影響を及ぼす可能性がある。
私たちの製品や候補製品の商業化に関連するリスク
私たちのビジネス成功は、医師、患者、医療支払者、医療コミュニティ(病院および外来診療所を含む)における幅広い市場受容度を得るために、私たちの製品および候補製品に依存する。
私たちが将来開発または買収する可能性のある任意の候補製品が規制部門の承認を得ても、この製品は医師、医療支払者、患者、あるいは私たちの製品開発努力を支援する医療コミュニティ(病院や外来診療所を含む)の市場受け入れを得ることができない可能性がある。私たちのすべての製品と私たちが承認された候補製品に対する市場の受け入れの程度は、多くの要素に依存します
58
私たちの製品や候補製品を商業化することができても、パートナーが私たちの製品を商業化することを求めているアメリカや他の国/地域では、製品は第三者支払者から保険と十分な補償を受けることができない可能性があり、これは私たちの業務を損なう可能性があります。
私たちがいかなる製品を商業化することに成功した能力は、政府衛生行政当局、個人健康保険会社、その他の組織がこれらの製品と関連治療に保険と十分な補償を提供する程度にある程度依存する。
政府当局と第三者支払者、例えば個人健康保険会社や健康維持組織は、どの薬をカバーし、精算レベルを確立するかを決定する。医療産業の主な傾向は費用を抑えることだ。政府当局と第三者支払者は,特定の薬物のカバー範囲や精算金額を制限することでコストを抑制しようとしている。ますます多くの第三者支払人は製薬会社に価格に基づいて所定の割引を提供し、医療製品の定価に挑戦することを要求している。監督管理の承認に必要なデータを得る以外に、第三者支払人は追加の臨床証拠を求め、特定の患者集団のために著者らの製品をカバーする前に、これらの患者の臨床利益と価値を証明する可能性がある。私たちのパートナーが商業化したどの製品にもカバー範囲と十分な精算があることを確実にすることはできません。もし精算があれば、清算レベルはいくらですか。米国などの一部の国では,より多くのコストが支払者から患者に移行する傾向にあり,より高い患者自己負担や他の行政負担が患者や医療専門家の需要を減少させる可能性がある。挑戦的な経済環境の下で、状況は特にそうかもしれない。保証範囲と精算は、私たちが規制承認を受けた任意の製品または候補製品の需要または価格に影響を与える可能性があり、最終的には、私たちのパートナーが規制承認を得た任意の製品または候補製品を商業化する能力に影響を与える可能性がある。
新たに承認された薬物については,保険取得や精算に大きな遅延がある可能性があり,保険範囲はFDAや同様の外国規制機関が承認する目的よりも限られている可能性がある。さらに、保険や精算を受ける資格があることは、すべての場合にどんな薬物の費用を支払うか、研究、開発、製造、販売、流通を含む私たちのコストを支払うことを意味するわけではない。適用すれば、新薬の一時精算レベルも私たちのコストを支払うのに十分ではないかもしれません。一時的なものかもしれません。販売率は薬物の使用や臨床環境によって異なる可能性があり,すでに低コスト薬物のために設定された精算レベルに基づいている可能性があり,他のサービスの既存支払いにも組み込まれている可能性がある。政府の医療保健計画や個人支払者が要求する強制的な割引やリベート、および将来、米国より低い価格で販売される可能性のある国からの輸入薬品の法律が緩和されることで、薬品の純価格が低下する可能性がある。薬品の保証と精算政策は支払人によって異なる可能性がある。米国の第三者支払者は薬品の保証と精算に統一的な政策がないからである。米国の第三者支払者は、自分の精算政策を制定する際に、通常連邦医療保険保険政策と支払い制限に依存している。保険の獲得と精算の面で重大な遅延が発生する可能性があります。保険と精算を確定する過程はよく時間もかかりますし、費用もかかりますので、各支払人にそれぞれ私たちの製品を使用する科学的と臨床支援を提供することが要求されます, 保険を受けたり、十分な補償を受けることは保証されない。政府当局と第三者支払人が私たちの薬品の保険や精算についてどのように決定するか予測するのは難しい。私たちのパートナーは、政府援助および個人支払者から、私たちが開発した任意の承認された製品の保証範囲と利益の収益率を迅速に得ることができず、これは、私たちの経営業績、私たちが必要な資本を調達する能力、および私たちの全体的な財務状況に実質的な悪影響を及ぼす可能性がある。
現在と将来の立法は、不利になる可能性のある価格設定法規や他の医療改革措置を含み、規制部門の私たちの候補製品に対する承認を得る難しさとコストを増加させ、私たちの製品や候補製品の価格に影響を与える可能性がある。
新薬製品を管理する監督管理審査、カバー範囲、定価と精算などの方面の法規は国によって異なる。米国や一部の外国司法管轄地域では、医療システムに関する立法や規制改革、提案された改革は、候補製品の規制承認を阻止または延期し、承認後の活動を制限または規制し、規制承認された製品や候補製品の販売に成功する能力に影響を与える可能性がある。特に,2010年3月に“医療·教育協調法案”により改正された“患者保護·平価医療法案”(総称して“平価医療法案”)が公布され,医療保健が政府や民間保険会社から資金を提供する方式が大きく変更され,米国製薬業に大きな影響を与え続けている。他の事項を除いて、“平価医療法案”及びその実施条例は、ある薬品と生物製品に対して、私たちの候補製品を含めて、メーカーが医療補助薬品還付計画の下で不足している税金還付を計算し、医療補助薬品還付計画の下でメーカーが不足している最低医療補助税金還付を増加させ、医療補助薬品還付計画を医療補助管理の看護組織に登録された個人の処方に拡大し、メーカーにあるブランドの処方薬に対して新しい年会費と税を徴収することを要求する新しい方法を解決した
59
連邦政府の比較有効性研究の項目を増加させる激励措置に対して、新しい連邦医療保険D部分カバーギャップ割引計画を構築した。
“平価医療法案”が公布されて以来、米国は他の立法改正も提出し、可決した。2011年8月、“2011年予算抑制法案”などの法案は米議会のための支出削減措置を制定した。赤字削減合同特別委員会の任務は、2013年から2021年までの間に少なくとも1.2兆ドルの赤字削減を提案することであるが、同委員会は必要な目標を達成できず、立法をいくつかの政府プロジェクトに自動的に削減することを触発した。これには、2013年4月に施行された各年度に提供者に支払われる連邦医療保険総額が2%減少することが含まれており、その後の立法改正により、国会が追加行動(2022年7月1日に満了した一時停止を除く)をとらない限り、この削減は2031年まで有効となる。新冠肺炎の流行期間の一時停止を補うために、2030年、連邦医療保険の支払いは上半期に2.25%、下半期に3%減少する。2013年1月、2012年に“米国納税者救済法”が公布され、それ以外にも、病院や外来診療所を含むいくつかの医療サービス提供者に支払われる医療保険がさらに減少し、政府が提供者に多額を取り戻す訴訟時効期間を3年から5年に延長した。
司法や国会は“平価医療法案”の多くの内容を疑問視しており、連邦政府の行政や立法部門は“平価医療法案”のいくつかの側面を廃止または代替しようと努力している。また、米国議会は“平価医療法案”の全部または一部を廃止または廃止し、代替する立法も検討している。米国議会はまだ全面的な廃止立法を通過していないが、“平価医療法案”の一部の条項を改正し、例えば2019年1月1日から処罰を廃止する法律を制定している。その理由は、“平価医療法案”を遵守しない個人権限による医療保険の購入、特定の強制費用の実施の廃止、連邦医療保険D部に加入する製薬業者の不足している販売時点カバーギャップ割引を増加させることである。2021年6月17日、米国最高裁は、個人権限なしにこの法案に対するいくつかの州の法的挑戦を却下した。“平価医療法案”全体は違憲だ。最高裁はこの訴訟を却下したが、各州の合憲性論点の是非については裁かなかった。“平価医療法案”への法的挑戦が未解決の際,2021年1月28日に総裁·バイデンが行政命令を発表し,2021年2月15日から2021年8月15日までの特別保険期間を開始し,“平価医療法案”市場で医療保険を獲得することを目的とした。行政命令はまた、仕事の要求を含む医療補助モデルプロジェクトおよび免除計画の再検討を含む、特定の政府機関に、医療保健の取得を制限する既存の政策および規則を検討するように指示した, 医療補助や“平価医療法案”による医療保険カバー範囲の獲得に不必要な障害を与える政策もある。最近,“インフレ率低減法”は“平価医療法”により医療保険を購入した個人により多くの補助金を提供する規定を延長した。増強後の補助金は当初米国救援計画の一部として採択され、計画は2022年に満期になり、現在は2025年に延長されている。将来、“平価医療法案”により多くの挑戦および/または修正案が提起されるかもしれない。米国最高裁の裁決,その他のこのような訴訟,バイデン政府の医療形態措置が“平価医療法案”や我々の業務にどのように影響するかは不明である。
外国、連邦と州の各レベルはすでに立法と監督管理提案を引き続き提出することが可能であり、医療保健の獲得性を拡大し、医療保健コストをコントロール或いは低減することを目的としている。私たちは未来に取られる可能性のある計画を予測できない。政府、保険会社、医療組織、医療サービスを管理する他の支払者は、価格制御を実施することを含む医療コストの制御または低減に努力し続けており、私たちの製品および規制の承認を得た候補製品の需要および私たちの製品のために公平と思われる価格を設定する能力に悪影響を及ぼす可能性がある。連邦医療保険や他の政府が計画している精算のいかなる減少も、個人支払者の支払いの同様の減少を招く可能性がある。
承認後の要求を拡大し、医薬品の販売や販売促進活動を制限するための立法·規制提案がなされている。私たちは、より多くの法律変更が公布されるかどうか、あるいはアメリカや外国の法規、指導、解釈が変わるかどうか、あるいはこれらの変化が私たちの製品や候補製品の規制承認にどのような影響を与える可能性があるかどうかを判断することはできません。アメリカ、EU、その他の潜在的な重要な市場では、私たちの製品と候補製品、政府当局と第三者支払人はますます多くの医療製品とサービスの価格、特に新しい革新的な製品と療法を制限または規制しようとしており、これはいくつかの市場でのいくつかの製品の平均販売価格を低くしている。アメリカでは、議会は数回の調査を行い、連邦と州立法を提出し、公布し、薬品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、政府計画の薬品精算方法を改革することを目的とした。例えば、2021年の総合支出法案は、連邦医療保険D部分の処方薬福祉情報の新しい要求をリアルタイムで表示するためのいくつかの連邦医療保険計画開発ツールのようないくつかの薬品価格報告および透明性措置を含み、団体および医療保険発行者に衛生および公共サービス部部長、労働大臣および財務大臣に薬局福祉および薬品コストの情報を報告することを要求する。最近は2022年8月16日に総裁·バイデンが2022年のインフレ率低減法案に署名し、医療保険の許容を含む2026年から, 連邦医療保険B部分とD部分で覆われたいくつかの薬品と生物製品のために“最高公平価格”を設定した;2023年から罰が向上した
60
連邦医療保険B部分とD部分で覆われた製品の価格はインフレよりも速く,2025年から連邦医療保険D部分でカバーされている製品の製薬やバイオメーカーに新たな割引義務が課せられている。
米国でも薬品価格決定に関する行政事態が発生している。例えば、総裁·バイデンが2021年7月9日に発表した行政命令に応えるために、いくつかの処方薬の措置を含むため、衛生·公衆サービス部は2021年9月9日に高い薬価に対応する総合計画を発表し、その中で国会とこの機関が取ることができる潜在的な立法政策と行政ツールを決定し、薬品価格をより負担し、より公平にし、処方薬業界全体の競争を改善し、促進し、科学的な革新を促進する。また、2022年2月2日、バイデン政府は2016年に最初にスタートしたがん月面着陸計画に引き続き取り組むと表明した。政府は声明で、このイニシアティブの新しい目標は、より広範な先端癌療法の獲得を確保し、強力な新しい治療ルートに投資するための不平等問題の解決を含むと指摘した。2022年9月12日、総裁·バイデンは行政命令を発表し、バイオテクノロジーと生物製造革新を推進した。この命令は、バイデン政府はいくつかの方法を通じて医療保健領域の生物技術と生物製造の進歩を支持し、衛生と公衆サービス部に命令発表後180日以内に報告書を提出し、どのように生物技術と生物製造を利用して医学突破を実現し、全体の疾病負担を軽減し、そして健康結果を改善することを評価することを指示した。最近、総裁·バイデンは2022年10月14日に米国の処方薬コストの削減に関する行政命令を発表した, これは、CMS革新センターによってテストされた新しい医療支払いと交付モードを選択するかどうかを考慮するかどうかを考慮することを衛生公衆サービス部に指示し、これらのモードは薬品コストを低減し、連邦医療保険と医療補助計画の受益者が革新的な薬物療法を獲得することを促進する。この行政命令はまた、衛生·公衆サービス部秘書に、この行政命令が発行された日から90日以内に報告書を提出するように指示し、常用薬物費用の分担の低減と質の高い看護を促進する価値別支払いのモードを支持する可能性があることを説明した。州レベルでは、立法機関は、価格や患者の精算制限、割引、特定の製品への参入の制限、マーケティングコストの開示と透明性措置を含む薬品および生物製品の価格設定を制御するための法規を立法および実施することが増えており、場合によっては、他の国からの輸入と大量購入も奨励されている。また、米国の管理型医療保健に対する日々の重視、及びEUの国家と地域の定価及び精算制御に対する日々の重視は、製品の定価、精算と使用に追加的な圧力をもたらし、これは私たちの未来の製品販売に不利な影響を与える可能性がある。これらの圧力は管理型医療グループ、他の保険会社の規則とやり方、司法裁決及び連邦医療保険、医療補助と医療改革、薬品精算政策と一般定価に関する政府法律法規から来る可能性がある。
また、新たに承認された保健製品の清算状態には重大な不確実性がある。私たちは私たちの製品と候補製品の費用効果を証明するために高価な薬物経済学的研究を行う必要があるかもしれない。第三者支払者が、私たちの製品および候補製品が他の療法と比較して費用対効果があると思わない場合、支払者は、承認後に私たちの製品および候補製品をその計画の下の福祉としてカバーしないかもしれない、または、支払いレベルが利益に基づいて私たちの製品を販売するのに十分ではないと思う場合がある。
海外市場では価格規制が行われる可能性があり、これは私たちの将来の収益性に悪影響を及ぼすかもしれない。
いくつかの国、特にEU加盟国とイギリスでは、処方薬の価格設定は政府によって統制されている。これらの国では、製品の規制承認を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。また、費用抑制措置の一部として、各国政府や他の利害関係者は価格や補償レベルにかなりの圧力をかける可能性がある。政治、経済、規制面の事態は定価交渉をさらに複雑化させる可能性があり、補償を受けた後、定価交渉が継続される可能性がある。EUの各加盟国が使用する参考価格と平行分配、あるいは低価格と高価な会員国の間の裁定は、価格をさらに下げることができる。いくつかの国では、私たちまたは私たちの協力者は、私たちの製品および候補製品の費用効果を他の利用可能な療法と比較して、精算または価格設定の承認を得たり維持したりするために、臨床研究または他の研究を要求される可能性がある。第三者支払者や主管当局が割引を公表することは、公布国や他の国の価格や補償レベルにさらなる圧力を与える可能性がある。もし私たちの製品が精算を受けられない場合、あるいは範囲や金額が制限されている場合、あるいは定価レベルが満足できなければ、私たちの業務は不利な影響を受ける可能性があります。
61
私たちは激しい競争に直面しており、これは他の人たちが私たちよりも製品の発見、開発、商業化に成功する可能性がある。
私たちは多くの製薬やバイオテクノロジー企業、学術機関、政府機関、民間、公共研究機関からの現在の製品や候補製品に対する競争に直面している。もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少ない、またはより安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消滅するだろう。さらに、新しい上流製品または治療レジメンの変化が、現在または将来の目標疾患の全体的な発病率または流行率を低下させる場合、私たちのビジネス機会は減少または消失するであろう。関連する規制機関の承認を得られれば、競争は私たちの製品と候補製品の販売と価格設定圧力を減少させる可能性がある。また、私たちの候補製品開発の重大な遅延は、私たちの競争相手が私たちより先に製品を市場に出し、私たちの製品と候補製品のいかなる商業化能力を弱めるかもしれません。
FDAが承認したEBV+PTLD治療の製品は現在のところなく,EbvalloのほかにECが承認したこの適応の製品もない。しかし、いくつかの市場の製品および治療法は、いくつかの医療専門家および機関によってラベル外でEBV+PTLDの治療、例えばリツキシマブおよび連合化学療法レジメンに使用されていることを知っている。そのほか、いくつかの会社と学術機構はEBV+PTLDと他のEBV駆動の疾病のために候補製品を開発しており、Viracta Treateutics,Inc.を含み、それは重要な第二段階の臨床研究を行っており、Nanatinostat(以前はTractinostat、あるいはVRX-3996と呼ばれていた)を抗ウィルス薬物valganciclovirと併用して再発/難治性EBV+リンパ腫を治療している。アロビル社(前身はウイルス細胞)は、難治性感染を有する異遺伝子造血幹細胞移植レシピエントの6種類のウイルスに対して、ウイルスに関連する出血性膀胱炎の2つの第3段階試験を行い、同種造血幹細胞移植後の患者とターザ治療個人有限会社のBKV、サイトメガロウイルス、アデノウイルス、EBウイルス、HHV 06とJCVを予防する第3段階試験を開始した異遺伝子多ウイルスT細胞製品である同種多ウイルス卵巣移植患者の第2段階臨床研究を完了した。
多発性硬化症は市場競争が激しく、米国およびEUは、臨床隔離症候群、再発緩和多発性硬化症(RRMS)、二次性進展性多発性硬化症(SPMS)、および原発進行性多発性硬化症(PPMS)を含む4つの模倣薬または生物学的等価物を含む少なくとも20の療法を承認した。MS市場には主要な国際的に完全に統合された製薬会社と老舗のバイオテクノロジー会社を含む多くの競争相手がいる。最近,TG Treeuticsで販売されているBriumvi(Ublituximab),ジョンソンから販売されているPonvory(S 1 P調節剤)とKesimpta®米国および/またはEUで再発形態のMSの治療のために許可されたノ華社によって販売されている(抗CD 20モノクロナル抗体)。
第3段階研究では、再発および/または進行型多発性硬化症に対する多くの候補開発薬があり、将来的には、KGaAのBruton‘sチロシンキナーゼ(BTK)阻害剤evobrutinib、羅氏のBTK阻害剤evobrutinib、サイノフェナントレンのBTK阻害剤fenebrutinib、tlebrutinibおよびAB Scienceのチロシンキナーゼ阻害剤Masitinibを含む2つまたは2つの適応のうちの1つまたは2つでより多くの新薬が承認される可能性がある。MediciNovaは、不活性SPMSにおけるPDE阻害剤イソブチスト(MN 166)の第3段階研究を開始することを計画している。
現在6つの自己CAR T療法がアメリカおよび/またはEUで承認されていますノワールのKymriah®(Tisagenlecleucel)、Gilead/KiteのYescarta®(Axicabagene Cilolucel)およびTecartusTM百時美施貴宝のBrexucabagene autolucel)と®(リソカタエン)およびアベマ(アダカタエン白血球)、ならびにジョンソンおよびレジェンド生物のカリクティ(シラカタエン自生白血球)。多くのCARを介した細胞療法が開発されており,多くは自己であるにもかかわらず,同種異体や既製細胞療法も含まれている。現在、複数の異遺伝子CARプラットフォームが開発されており、方法が異なり、ドナー細胞が患者の身体を異体あるいは患者の身体でドナー細胞を拒絶することを最大限に減少させる。これらの方法は、遺伝子編集を使用してTCRを除去または阻害することと、TCRを含まない細胞タイプを使用することとを含む。多くの臨床段階の同種異体CAR計画は細胞型としてα−βT細胞を用い,T細胞受容体やヒト白血球抗原の遺伝子編集を第一選択の技術的方法としているが,他の戦略も開発されている。これらの他の方法のいくつかは、私たちが使用する方法よりも有利な特徴を有する可能性があり、これは、我々の製品よりも潜在的なパートナーまたは顧客に好まれることをもたらすであろう。我々の将来の疾患によれば,関心のある適応において,自己および異体CAR療法および他の方法(例えば,小分子,抗体,二重特異性)からの競争に直面する可能性がある。
62
著者らの現在或いは未来の目標疾患に対する多くの承認或いは常用の薬物と治療法は、EBV+PTLDとMSを含み、すべて公認され、そして医者、患者と第三者支払人に広く受け入れられている。その中のいくつかの薬物はブランド薬物であり、特許によって保護され、他の薬物および栄養補助剤は模倣薬に基づいて提供される。保険会社および他の第三者支払者は、非特許製品または特定のブランド製品の使用を奨励することができる。もし私たちの製品と候補製品が承認されれば、それらの価格は競争相手の模造薬より著しく高いと予想しています。差別化され、納得できる臨床的証拠が不足している場合、価格割増は、現在承認されているまたは一般的な治療法よりも当社の製品のより良い採用を阻害する可能性があり、これは私たちの業務に悪影響を及ぼす可能性がある。また,多くの会社が新たな治療法を開発しており,我々の製品が臨床開発を継続していることに伴い,看護基準がなぜなるかは予測できない。
私たちの多くの競争相手あるいは潜在的な競争相手は研究開発、製造、臨床前テスト、臨床研究を行い、監督管理の許可とマーケティング許可を得た製品の面で私たちよりずっと大きい市場地位、財力と専門知識を持っているため、私たちより競争優位を持っているかもしれません。規模の小さい企業や初期段階にある企業も、大型·成熟企業との連携による手配、または大きな会社に買収された場合など、重要な競争相手であることが証明される可能性がある。これらの第三者は合格した科学と管理者を募集と維持し、臨床研究サイトと臨床研究患者登録を確立し、著者らの計画と相補的あるいは私たちの業務に有利な技術と技術許可証を獲得する面で私たちと競争している。
これらの要因により、これらの競争相手は、特許保護や他の知的財産権を取得する前に、規制機関の製品の承認を得る可能性があり、これは、私たちの候補製品を開発または商業化する能力を制限するだろう。私たちの競争相手はまた私たちより安全で、より効果的で、より広く使用され、より安い薬物を開発するかもしれませんし、彼らの製品を製造してマーケティングする上で、私たちよりも成功するかもしれません。私たちが開発と他の費用を回収する前に、これらの明らかな利点は私たちの候補製品を時代遅れにしたり、競争力を失ったりするかもしれない。
Healthcare Royalty Partnersと合意した特許権使用料融資合意によると,我々は何らかの契約義務の制約を受けており,これらの義務を履行できなければ,損害クレームの制約を受ける可能性がある.
2022年12月、我々はHCR Molag Fund、L.P.(HCRx)と購入契約(HCRxプロトコル)を締結した。HCRx協定の条項によると、私たちはPierre Fabreによる商業化協定として3,100万ドルの現金を受け取り、私たちはPierre FabreからEU Ebvalloの未来の特許使用料の一部と特定のマイルストーンの権利を得る権利がある。HCRx協定には、当事者に有利な陳述と保証、契約および賠償義務を含むいくつかの慣例条項および条件が含まれている。このような条項の中には、私たちがPierre Fabre商業化協定を遵守することに関するいくつかの条項がある。Pierre Fabre商業化プロトコルやHCRxプロトコルに違反していると実際にまたは言われている場合、私たちはHCRxの損害クレームを受け、費用の高い訴訟に直面する可能性がある。
我々が開発した候補製品は生物製品(生物製品)として規制されることが予想されるため,予想よりも早く競争される可能性がある。
2009年に生物製品価格競争と革新法(BPCIA)は“平価医療法”の一部として公布され、生物類似と交換可能な生物製品を承認するための簡略化された方法を確立することを目的とした。規制経路はFDAのために生物類似生物製品を審査および承認する法律的権威を確立し、生物類似体と承認された生物製品との類似性に基づいて、それを“交換可能な”生物製剤として指定することを含む。BPCIAによると、バイオ類似製品の申請は、BLAによる参考製品の承認12年後にのみFDAの承認を得ることができる。この12年間の独占期間内に、FDAが競合製品の完全なBLAを承認した場合、スポンサー自身の臨床前データおよび十分かつ良好に制御された臨床試験からのデータを含み、その製品の安全性、純度および有効性を証明するために、別の会社は依然としてこの参照製品の競合バージョンを販売する可能性がある。この法律は複雑であり、FDAはまだ説明して施行している。したがって、その最終的な影響、実施、そして意味には不確実性がある。FDAがBPCIAを実施するためのプロセスをいつ完全に採用できるかは不明であるが,これらのプロセスのいずれも我々の生物製品の将来のビジネス見通しに重大な悪影響を及ぼす可能性がある。
我々の製品と我々が開発したBLAによって米国で生物製品として承認された候補製品はいずれも12年の排他期を得る資格があるべきであると考えられる。しかしながら、国会の行動または他の理由により、この排他性は短縮される可能性があり、またはFDAは候補製品を競合製品の参考製品とみなさず、予想よりも早く後発薬競争の機会を創出する可能性がある。さらに、承認されると、生物学的類似体が、どの程度、非生物製品に類似した伝統的な模倣薬代替の方法で任意の参照製品を置換するかは、まだ発展中のいくつかの市場および規制要因に依存するであろう。
63
さらに、私たちの製品生物に似た生物製品を承認することは、市場投入コストを著しく低下させる可能性があり、価格が私たちの製品よりも著しく低い可能性があるので、私たちの業務に実質的な悪影響を及ぼす可能性があります。
もし私たちが第三者と合意して私たちの製品と候補製品をマーケティングして販売することができなければ、私たちは私たちの製品を販売することから何の収入も得られないかもしれません。
FDAや同様の外国規制機関によって承認される可能性のあるすべての製品をマーケティングするためには、第三者と協定を締結して、私たちの製品をマーケティングして販売しなければなりません。私たちが第三者とこのような合意に到達できるか、または商業的に合理的な条件で、またはタイムリーにそうすることができる保証はない。我々の製品をマーケティングおよび販売するために第三者と合意に達したり遅延したりすることができなかった場合、これらの製品の商業化に悪影響を及ぼすであろう。私たちは私たちの製品をマーケティングして販売するための適切な第三者を見つけることができるか、または第三者と私たちに有利または許容可能な条項を合意することができるか、または根本的にできないという保証はない。もし私たちが第三者と合意して私たちの製品をマーケティングして商業化することができなければ、私たちは自分で製品を商業化することを含め、他の戦略的選択を模索する必要があり、私たち自身が製品を商業化することに成功できる保証はないかもしれない。私たちは多くの会社と競争しているかもしれません。これらの会社は現在広範で資金的な販売とマーケティング業務を持っています。販売やマーケティング機能を実行するのに十分な規模、適切なタイミング、訓練された第三者がなければ、私たちはこれらのより成熟した会社と成功的に競争することができないかもしれない。
私たちは従業員基盤の側面と、私たちの運営を成功的に管理することを含めて、私たちの成長を管理する上で困難に直面するかもしれない。
2022年12月31日現在、2022年10月に2022年8月に発表されたリストラ計画の一部として解雇された従業員は含まれていない334人の従業員がいる。私たちは私たちの持続的な開発活動を支援するために私たちの業務規模を管理し、私たちのパートナーが私たちの製品や候補製品の潜在的な商業化を支援することに困難に直面するかもしれません。私たちの発展と商業化計画や戦略の発展に伴い、あるいは将来の任意の買収により、私たちの業務規模を管理するために、私たちの管理、運営、財務、その他の手続きやプロセスを改善し続けなければならない。私たちの既存の管理、人員、そしてシステムは未来のどんな成長も支援するのに十分ではないかもしれない。今後の成長は、経営陣のメンバーにより多くの重大な責任を負わせるだろう
私たちの業務の拡大に伴い、様々な戦略パートナー、サプライヤー、他の第三者とのより多くの関係を管理する必要があります。私たちの未来の財務パフォーマンスと効果的な競争の能力は、私たちが未来のどんな成長の能力を効果的に管理するかにある程度依存するだろう。そのため、著者らは著者らの開発仕事及び臨床前と臨床研究を有効に管理し、更に多くの管理、研究開発、製造と行政人員を募集、訓練と統合することができなければならない。私たちはこのような任務のいずれも達成できず、私たちの会社の発展に成功することを阻害するかもしれない。
私たちの普通株式所有権に関連するリスク
私たちの経営業績にかかわらず、私たちの株価はずっと変動し続け、下落する可能性があります。
私たちの株価は過去に変動しており、将来も変動することが予想される。2020年1月1日から2022年12月31日まで、私たちの普通株の報告販売価格は1株2.83ドルから28.20ドルの間で変動します。一般的な株式市場、特にバイオテクノロジー会社の市場は極端な変動を経験しており、この変動は往々にしてある会社の経営業績とは無関係である。このような変動により、投資家は私たちの普通株に投資する時に損失を受けるかもしれない。私たちの普通株の市場価格は以下の要素を含む多くの要素の影響を受ける可能性がある
64
また、株式市場全体、特にバイオテクノロジーや製薬株式市場は、ある会社の経営業績とは無関係な大幅な変動を経験しており、多くの会社の株価下落を招いている。例えば、薬品定価や製薬会社の値上げに関する否定的な宣伝は、バイオテクノロジーや製薬株式市場に負の影響を与え続ける可能性がある。同様に、米国の社会、政治、規制および経済条件の重大な変化、または対外貿易および医療支出および交付を管理する法律および政策の重大な変化により、“平価医療法案”の全部または一部を廃止および/または置換する可能性があること、または米国と外国政府の政策やその他の原因による関税や他の自由貿易制限の変化により、金融市場は大きな変動を経験する可能性があり、これはバイオテクノロジーや製薬株の市場に負の影響を与える可能性もある。このような市場変動は私たちの普通株の取引価格に悪影響を及ぼすかもしれない。
過去には,それらの証券が市場価格変動時期を経験した会社に対して,集団訴訟が提起されることが多かった。私たちに提起されたどのような訴訟も巨額のコストを招き、経営陣の注意と資源を移転させる可能性があり、これは私たちの臨床研究やパートナーの商業化努力の遅延を招く可能性がある。
私たちの主要株主は私たちのかなりの割合の株を持っていて、株主の承認が必要な事項に大きな制御を加えることができるだろう。
私たちの主要株主は私たちが発行した普通株式の大きな部分を持っている。これらの株主は株主の承認を必要とするすべての事項の結果を決定することができるかもしれない。例えば、これらの株主は、取締役選挙を制御し、私たちの組織文書を修正し、任意の合併、資産の売却、または他の重大な会社取引を承認することができるかもしれない。これは私たちの普通株に対する自発的な買収提案や要約を阻止したり阻止したりするかもしれない。私たちの主要株主の利益は常に他の株主の利益と一致しているわけではなく、彼らの行動は彼らの最適な利益を促進する可能性があり、必ずしも他の株主の利益ではなく、彼らの普通株のためのプレミアムを求め、私たちの普通株の市場価格に影響を与える可能性がある。
65
公開市場で私たちの大量の普通株を売ることは私たちの株価を下落させるかもしれない。
公開市場で私たちの大量の普通株を売ることはいつでも起こる可能性がある。これらの売却、あるいは市場で大量の株式保有者が株を売却しようとしているとの見方は、我々の普通株の市場価格を低下させる可能性がある。さらに、いくつかの条件の制限の下で、私たち普通株のいくつかの保有者は、彼らの株式に関する登録声明を提出することを要求する権利があり、または彼らの株を私たち自身または他の株主のために提出する可能性のある登録声明に含める権利があるだろう。私たちはすでに登録して、私たちが株式補償計画に従って発行可能なすべての普通株を登録し続けるつもりです。これらの株を登録すると、発行時に公開市場で自由に売ることができますが、付属会社に適用される数量制限を受けています。
上場企業としては、コストを増やし続けており、当社の経営陣は上場企業コンプライアンス計画に多くの時間を費やすことが予想されています。
上場企業として、私たちはすでに巨額の法律、会計、その他の費用を負担し続けている。私たちは、我々の業務および財務状況に関する年間、四半期、および現在の報告書を米国証券取引委員会に提出することを要求する取引法の報告要件を遵守しなければならない。また、“サバンズ-オクスリ法案”および米国証券取引委員会とナスダックが後に“サバンズ-オクスリ法案”を実施するために採択された規則は、効率的な情報開示と財務制御の確立と維持を要求し、コーポレートガバナンスのやり方を変更することを含む上場企業に重大な要求を提出した。また、2010年のドッド·フランクウォール街改革と消費者保護法によると、米国証券取引委員会は、現在私たちの強制的な“報酬発言権”投票要件に適用されるような他の規則や規定を採択している。株主急進主義、現在の政治環境、将来の規制改革の可能性は、多くの新たな規制·開示義務を招く可能性があり、これは追加のコンプライアンスコストを招き、現在予見できない方法で業務を運営する方法に影響を与える可能性がある。
上場企業に適用される規則と法規は、私たちの法律と財務コンプライアンスコストを大幅に増加させ、いくつかの活動をより時間と高価にした。これらの要求が私たちの経営陣と従業員の注意を他の業務から移すと、それらは私たちの業務、財務状況、運営結果に実質的な悪影響を及ぼす可能性がある。増加したコストは私たちの純収益を減少させ、あるいは私たちの純損失を増加させ、他の業務分野のコストを下げたり、私たちの製品やサービスの価格を向上させることを要求するかもしれません。
私たちは予測可能な未来に私たちの株にいかなる現金配当金も支払わないと予想されるので、資本付加価値(あれば)は私たちの株主の潜在的な収益の唯一の源になるだろう。
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、私たちの未来のすべての収益を維持するつもりで、もしあれば、私たちの業務の成長と発展に資金を提供します。しかも、未来のどんな債務協定の条項も私たちが配当金を支払うことを阻止するかもしれない。したがって、予測可能な未来には、私たち普通株の資本付加価値(あれば)が私たちの株主の唯一の収益源になるだろう。
将来的に私たちの株主の所有率をさらに希釈し、私たちの株式インセンティブ計画に基づいて、私たちの株主の所有率をさらに希釈し、株価を下落させる可能性があることを含む、私たちの普通株を売却して発行する権利、または普通株を購入する権利。
私たちは未来に私たちが計画した業務を継続するために多くの追加資本が必要になると予想する。資本を調達するために、私たちは、1回または複数回の取引において、時々決定された価格および方法で、大量の普通株または普通株または普通株に交換可能な証券を販売することができる。これらの将来発行される普通株または普通株関連証券は、未償還オプションまたは株式承認証の行使、および買収または許可証内の発行に関連する任意の追加株式(ある場合)に加えて、私たちの投資家に重大な希釈をもたらす可能性がある。このような売却はまた、私たちの既存株主の実質的な希釈をもたらす可能性があり、新しい投資家は私たちの普通株式保有者よりも優先的な権利、優遇、特権を得ることができる。株式推定値が、我々普通株の取引価格を含めると、経済中断やその他の要因で押し下げられ、このような希釈の潜在幅が増加する。私たちの持分インセンティブ計画によると、私たちの報酬委員会は、私たちの従業員、非従業員取締役、コンサルタントに株式に基づくインセンティブ奨励を付与する権利がある。私たちの株式激励計画によると、将来的にRSU、オプションおよび他の株式奨励および普通株の発行は希釈を招き、私たちの普通株の市場価格に悪影響を及ぼす可能性がある。
66
私たちの定款文書やデラウェア州法律のいくつかの条項は反買収効果があるかもしれませんが、買収が株主に有利であっても、他の会社が私たちを買収することを阻止し、私たちの株主が現在の経営陣を交換または更迭しようとすることを阻止するかもしれません。
当社の会社登録証明書(会社登録証明書)の改訂及び再記載の定款(付則)及びデラウェア州の法律は、第三者が我々を買収したり、買収コストを増加させたりすることを困難にする可能性があり、そうしても株主に利益を与えたり、現在の経営陣を罷免したりすることができる。これらの条項には
上記のいずれの要因も、株主が管理職メンバーを任命する取締役会メンバーを交代させることを困難にし、それによって、株主が現在の管理職を交代または更迭するいかなる試みを挫折または阻止する可能性がある。
また、私たちはデラウェア州で登録されているので、私たちはデラウェア州会社法第203条の管轄を受けています。この条項は、これが私たちの株主の意志に合っているかどうか、あるいは私たちに有利であるかどうかにかかわらず、誰かが私たちを買収したり、私たちと合併したりすることを阻止、遅延または阻止するかもしれません。デラウェア州の法律によると、一般的に、会社は、その株を保有する任意の15%以上の株主と商業合併を行ってはならない。当該株を保有している株主がその株を3年間保有しているか、または他の事項を除いて、取締役会はその取引を承認している。私たちの会社の登録証明書や定款やデラウェア州法律の遅延または制御権の変更を阻止する条項は、私たちの株主が彼らが持っている普通株からプレミアムを得る機会を制限する可能性があり、また一部の投資家が私たちの普通株に支払いたい価格に影響を与える可能性があります。
私たちの規約は、デラウェア州に位置する州または連邦裁判所を、私たちと株主とのほとんどの紛争の唯一のおよび独占的なフォーラムとして指定しており、これは、私たちの株主が私たちまたは私たちの現職または元役員、役員、株主、または他の従業員との紛争で有利な司法フォーラムを得る能力を制限するかもしれない。
私たちの定款規定は、私たちが書面で代替裁判所を選択することに同意しない限り、デラウェア州衡平裁判所は、以下の唯一かつ独占裁判所でなければならない:(I)デラウェア州法律に基づいて私たちを代表して提起された任意の派生訴訟または訴訟、(Ii)会社の現職または前取締役役員または他の従業員が私たちまたは私たちの株主に対する信頼責任に違反していると主張するいかなる訴訟も、(Iii)私たちまたは私たちの任意の取締役、役員、(Iv)内務原則に基づいて吾等にクレームを提起する任意の訴訟、又は(V)DGCL第115条の定義に基づいて“内部会社クレーム”を提起する任意の訴訟、又は当社登録証明書又は付例(両方とも随時改訂することができる)の任意の条文に基づいて生成される任意の訴訟。上記の規定は、証券法に基づいて提起されたいかなるクレームにも適用されず、私たちが書面で代替裁判所を選択することに同意しない限り、米国連邦地域裁判所は、証券法に基づくクレームを解決するための任意の訴訟を解決する唯一かつ独占的な裁判所となる。
これらの裁判所条項の選択は、株主が司法裁判所において、そのようなクレームに関連する訴訟を阻害する可能性がある私たちまたは任意の現職または前任取締役、上級管理職、または他の従業員と紛争することに有利であると考えられるクレームを司法裁判所で提出する能力を制限する可能性がある。裁判所がこれらの規定を強制執行するかどうかには不確実性があり,他の定款文書では選択のような裁判所条項の実行可能性が法的手続きで疑問視されている。裁判所が認定する可能性がある
67
これらのタイプの条項は適用されないか、または実行不可能であり、もし裁判所が選択された裁判所条項が訴訟で適用されないか、または実行できないことを発見した場合、私たちは、他の管轄区域でそのような訴訟を解決することに関連する追加コストを生じる可能性があり、これは、私たちの業務、運営結果、および財務状態を損なう可能性がある
証券や業界アナリストが我々の業務に関する研究報告を発表しない場合、あるいは不正確または不利な研究報告を発表しなければ、私たちの株価や取引量は低下する可能性がある。
私たちの普通株の取引市場は、証券や業界アナリストが発表した私たちと私たちの業務に関する研究と報告にある程度依存するだろう。もし私たちの証券や業界アナリストが私たちの株式格付けを引き下げたことを報道したり、私たちまたは私たちの業務に不利な研究報告を発表したりすれば、私たちの株価は下落する可能性がある。これらのアナリストのうちの1人以上がわが社への報道を停止したり、私たちに関する報告書を定期的に発表できなかったりすれば、私たちの株に対する需要が減少する可能性があり、これは私たちの株価や取引量を低下させる可能性がある。
一般リスク因子
私たちの未来の成功は私たちの行政人員を維持できるかどうか、そして合格した人員を吸引、維持、激励できるかどうかにかかっている。
私たちは私たちの幹部と他の重要な従業員に高度に依存して、私たちの任意の幹部や他の重要な従業員のサービスを失って、科学、技術、あるいは管理者を含めて、私たちの会社の目標の達成を阻害するかもしれません。2022年8月、経営陣メンバーを含む会社の全部門で約20%の人員削減を発表した。経営陣のメンバーと他のキーパーソンの流失は、職責と任務を調整できなかったこと、新しい管理システムとプロセスを作成する必要性、企業文化への影響及び歴史知識の保留を含む多くのリスクに直面させた。
私たちの成功は私たちが募集し、維持し、管理し、奨励する能力にかかっている。私たちは私たちの従業員と雇用協定や招聘書を締結しているにもかかわらず、これらの文書は“勝手”雇用を規定しており、これは私たちのどの従業員も通知するかどうかにかかわらず、いつでも私たちの仕事を離れることができることを意味する。私たちの産業や地理的地域では、技術者に対する競争が激しく、これは私たちが受け入れ可能な条件で合格者を採用し、維持する能力を制限するかもしれない。価値のある従業員をわが社に引き付けるために、給料や現金インセンティブのほか、時間の経過とともに付与された持分奨励を提供しています。株式奨励の従業員に対する価値は、私たちの株価変動の大きな影響を受ける可能性があり、これらの変動は私たちの制御範囲を超えており、いつでも他社が提出したより利益のある見積もりを相殺するのに十分ではないかもしれない。
顧客と第三者支払者との関係は、刑事制裁、民事処罰、契約損害、名声損害、利益および将来の収益の減少に直面する可能性がある反リベート、詐欺および乱用、プライバシーおよび他の法律法規の制約を受けることになります。
医療提供者は,医師や第三者支払者を含め,我々の製品を推薦·処方し,規制部門の承認を得た任意の候補製品を推薦する際に主な役割を果たすであろう。私たちの現在と未来の第三者支払者と顧客との手配は、広範に適用される詐欺や乱用、その他の医療法律や法規に直面する可能性があり、これらの法律と法規は、私たちの研究やマーケティング、販売、流通、私たちの製品の業務または財務配置と関係を制限するかもしれません。バイオ製薬会社として、医療サービスの転転を制御したり、連邦医療保険、医療補助または他の第三者支払者に請求書を発行したりすることはありませんが、詐欺や乱用、および患者の権利に関する連邦および州医療法律法規は現在も将来も私たちの業務に適用されています。もし私たちの候補製品がFDAの承認を得て、私たちのパートナーがアメリカでこれらの製品を商業化し始めたら、私たちのこのような法律の下での潜在的なリスクは著しく増加し、私たちはこのような法律を遵守することに関連するコストも増加する可能性がある。他の事項以外に、これらの法律は、現在の主要な研究者と研究患者との活動、提案と将来の販売、マーケティングと教育計画、流通協定、割引、手数料補償、いくつかの患者支援サービス、および一般的な他の業務手配に影響を与える可能性がある。また,我々の製品や米国以外の任意の候補製品の承認や商業化も,ここで述べた医療保健法や他の外国法の外国等価物の制約を受ける可能性がある。適用される連邦と州医療保健法律と法規によると、いくつかの業務手配と私たちの運営能力に影響を与える可能性のある制限は含まれていますが、これらに限定されません, 以下の内容:
68
私たちが第三者の業務手配と適用される医療法律や法規に適合していることを確保するためには、大量のコストがかかります。政府当局は、我々の業務やり方は、現在または将来、医療法律や法規の適用に関連する法規、法規または判例法に適合していないと結論するかもしれない。もし私たちの運営がこれらの法律または任意の他の政府法規に違反していることが発見された場合、私たちは重大な民事、刑事と行政処罰、損害賠償、罰金、監禁、政府援助の医療計画から除外された(例えばMedicareおよびMedicaid)、返還、追加の報告要求または監督(私たちが会社の誠実な合意または同様の合意の制約を受けている場合)、および私たちの業務の削減または再編、名声損害、契約損害、利益および将来の収益の減少、これらはいずれも私たちの業務運営能力と私たちの運営結果に悪影響を及ぼす可能性がある。もし私たちがそれと業務往来があると予想される任意の医師や他の医療提供者や実体が適用法を遵守していないことが発見された場合、彼らは政府の援助された医療計画から除外されることを含む重大な刑事、民事または行政制裁を受ける可能性がある。
私たちの従業員は規制基準や要求を守らないことを含む不正行為や他の不正活動に従事する可能性があり、これは私たちに重大な責任を与え、私たちの名声を損なう可能性がある。
我々は、fdaの規定や同様の外国規制機関のような規定を故意に遵守しないこと、fdaまたは同様の外国規制機関に正確な情報を提供すること、私たちが制定した製造基準を遵守すること、連邦および州医療詐欺および乱用法律法規、および比外国規制機関によって制定および実行可能な類似の法律および法規を遵守することを含む、従業員詐欺または他の不正行為のリスクに直面している
69
規制当局は、財務情報やデータを正確に報告したり、許可されていない活動を開示したりする。従業員の不当行為はまた臨床研究過程で得られた情報を不当に使用する可能性があり、これは規制制裁と著者らの名声に深刻な損害を与える可能性がある。従業員の不正行為を常に識別し、阻止することができるわけではなく、このような行為を発見し、防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御することができないか、またはそのような法律や法規を遵守しないことによる政府の調査または他の行動または訴訟から私たちを保護することができない可能性がある。もし私たちにこのような行動を取って、私たちが自分の権利を弁護したり、維持したりすることに成功しなかったら、これらの行動は、巨額の罰金や他の制裁を加えることを含む、私たちの業務および業務結果に大きな影響を及ぼすかもしれない。
私たちに対する製品責任訴訟は、私たちが重大な責任を負い、私たちが開発する可能性のある任意の製品の商業化を制限する可能性があります。
私たちは人体臨床研究で私たちの候補製品をテストすることに関連する固有の製品責任リスクに直面しています。もし私たちが開発する可能性のある任意の製品を商業的に販売すれば、私たちはより大きなリスクに直面します。私たちの臨床研究に参加した被験者、患者、医療提供者、あるいは他の使用、管理、または販売した人は、私たちに製品責任クレームを出すかもしれません。もし私たちが私たちの候補製品や製品に被害を与えるクレームを自己弁護することに成功できなければ、私たちは重大な責任を招くかもしれない。是非曲直や最終結果にかかわらず、賠償責任は
私たちは現在、製品責任保険のレベルを持っていて、これは似たような場合の会社の慣例であり、予測可能なリスクを提供するのに十分な保険だと思いますが、私たちが生じる可能性のあるすべての責任をカバーするのに十分ではないかもしれません。私たちは可能などんな責任にも対応するために、合理的な費用や十分な金額で保険範囲を維持することができないかもしれない。必要であれば、私たちが開発している候補商品に対する規制機関の承認を得たら、私たちの製品保険範囲を拡大し、商業製品の販売を含めることができるかもしれませんが、規制機関の承認を得た製品のために商業的に合理的な商品責任保険を得ることができないかもしれません。予期せぬ副作用を有する薬物に基づく集団訴訟では,すでに多くの判決が下されている。成功した製品責任クレームまたは一連の私たちに対するクレーム、特に判決が私たちの保険範囲を超えた場合、私たちの現金を減少させ、私たちの業務に悪影響を及ぼす可能性があります。
もし私たちと私たちの第三者製造業者が環境、健康、安全法律法規を遵守できなかったら、私たちは罰金や処罰を受けたり、コストを発生したりする可能性があり、これは私たちの業務成功に実質的な悪影響を及ぼすかもしれない。
私たちと私たちの第三者製造業者は、実験室プログラムおよび危険材料と廃棄物の処理、使用、貯蔵、処理および処理を管理する法律法規を含む、多くの環境、健康および安全法律法規の制約を受けている。私たちの行動は化学物質と生物学的材料を含む危険で燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物製品を発生させるだろう。私たちは一般的に第三者と契約を結び、このような材料と廃棄物を処理する。私たちはこのような材料が汚染や傷害をもたらす危険を除去することができない。もし私たちまたは私たちの第三者製造業者が危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによるいかなる損害に責任を負うかもしれません。どんな責任も私たちの資源範囲を超える可能性があります。私たちはまた民事や刑事罰金と処罰に関連した巨額の費用を発生させるかもしれない。
70
危険材料の使用による従業員の負傷により発生する可能性のあるコストや費用を支払うために労働者補償保険を維持しているが、保険限度額は、同様の状況の会社にとっては、予見可能なリスクを提供するのに十分な保険であると考えられるが、潜在的な責任に十分な保険を提供できない可能性がある。私たちは私たちが生物や危険材料を貯蔵したり処分したりすることで、私たちが提起した環境責任や有毒侵害請求に保険を提供することはできません。
また、現在または未来の環境、健康、安全法律法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。これらの法律法規を遵守しないことは、巨額の罰金、処罰、または他の制裁を招く可能性もあり、これは私たちの業務、財務状況、運営結果、および将来性に悪影響を及ぼす可能性がある。
私たち、私たちの顧客またはサプライヤーは、プライバシー、データ保護、データセキュリティに関連するますます厳格な法律、法規、契約義務を遵守できなかったと実際にまたは考えており、私たちの名声を損なう可能性があり、巨額の罰金と責任を直面させます。
私たちはプライバシー、データ保護、データセキュリティに関する多くの国内と海外の法律法規に支配されているか、その範囲は変化しており、異なる応用と解釈の影響を受け、異なる国の間で一致しない、あるいは他の規則と衝突している可能性がある。プライバシー、データ保護、データセキュリティに関する顧客および第三者への契約義務の条項も遵守しなければなりません。私たち、私たちの顧客、私たちのサプライヤー、あるいは他の関連第三者は、これらの法律、法規と義務を実際に処理または遵守できなかったと考えて、私たちのコンプライアンスと運営コストを増加させ、私たちを規制機関の審査、行動、罰金、処罰に直面させ、規制機関が私たちの臨床試験活動を拒否、制限、あるいは混乱させ、名声損害を招き、顧客の流失を招き、私たちの製品の使用を減少させ、訴訟と責任を招き、そうでなければ、私たちの業務、財務状況、運営結果に実質的な悪影響を与える可能性がある。
例えば,EUは2018年5月に“一般データ保護条例(EU)2016/679(EU GDPR)”を採択し,個人(あるいはデータ主体)の個人情報を扱うことに厳しい要求を出している.EU GDPRは、個人情報の収集、使用、開示、移転および他の処理を管理し、すべてのEU加盟国が直接効力を有し、欧州経済地域(EEA)以外の組織が欧州経済地域内の個人がこれらの個人に商品またはサービス(“方向性テスト”)を提供するか、またはその行動(“監視テスト”)の個人情報を監視する際に治外法権効果を有する。したがって、EU GDPRは私たちに適用され、私たちがEU加盟国に設立されている限り、私たちはEU加盟国の機関を背景に個人情報を処理しているか、または私たちは目標テストまたは監視テストの要求に適合している。
EU GDPRは、管理者および処理者に対して、処理された個人情報を保護するための適切な技術および組織措置を要求することを含む契約プライバシー、データ保護、およびデータセキュリティ約束、(2)個人がそのデータ保護権を行使する手段(例えば、個人情報を削除する権利)を決定する手段(例えば、個人情報の権利を削除する)、(3)保持および処理された個人情報量の制限、(4)敏感な情報(例えば、健康データ)に関する追加的な要求、を含む、深刻かつ包括的なプライバシー、データ保護、およびデータセキュリティ義務を規定する。(V)規制当局および/または関係者に、遅延すべきでない(および可能な場合には72時間以下)資料漏洩通知規定を行う。(Vi)資料当事者の有効な同意を得る規定を強化する。(Vii)任意の新製品またはサービスを開発する際には、保障資料の責任を考慮しなければならない。(Viii)臨床試験対象者および調査者により詳細な私的通知を提供する。EU GDPRはまた、EU加盟国は、遺伝、生体認証、または健康データの処理を制限するさらなる法律および法規を導入することができ、これは、EUの個人情報を収集、使用、共有する能力を制限し、コンプライアンスコストを増加させ、私たちの業務に悪影響を与え、財務状況を損なうことを要求する可能性がある。
EU GDPRはまた、個人情報を欧州経済圏から米国や他の国に移転することを制限しており、移転当事者が移転した個人情報を保護する具体的な保障措置を実施している限り、欧州委員会はこれらの国が“十分な”データ保護法を持っていることを認めない。イギリス(以下に述べる)とスイスのデータ保護法にも同様の制限がある。米国会社がEUとスイスから個人情報を輸入することを許可する主要な保障措置の一つであり、歴史的にはEU-米国プライバシー盾フレームの認証であり、この枠組みはそれぞれ米国商務省とスイス-米国プライバシー盾フレームによって管理されている。しかし、欧州連合裁判所(CJEU)は2020年7月の“Schrems II”裁決で、EU-米国プライバシー保護枠組みを国際移転合法化のメカニズムとして無効と発表した。同様に、スイス連邦データ保護と情報専門家はSchrems IIの決定に基づいてスイス-米国プライバシー盾の枠組みが不十分であることを宣言した。また、欧州委員会の新しいバージョンの標準契約条項(新しいEU SCC)は2021年6月に採択され、この条項は現在、個人情報をEUから米国に合法的に移転する主要な保障措置であり、締結当事者に重い義務を課している。これらの新しいEU SCCは、将来のすべての新しい契約(個人情報の制限移転がある)のために使用されなければならず、2021年9月27日までに締結された既存の契約は、2022年12月27日までに更新されることが要求される。だから、
71
私たちまたは私たちのサプライヤーがEUから行った任意の個人情報移転は、欧州データ保護法に適合していない可能性があり、EU GDPRが国境を越えたデータ転送制限に違反したための厳しい制裁に直面し、欧州データ保護法に制約されている会社の需要を減少させる可能性があります。
お客様、パートナー、サプライヤーに協力してEU GDPRを遵守したり、EU GDPRを自分で守ったりすることで、巨額の運営コストが発生したり、ビジネス慣行の変更が要求されたりする可能性があります。正確な実施が得られない場合や企業内の人員が必要なプログラムを完全に遵守できないリスクもある可能性がある。
2022年10月7日、米国の総裁は行政命令を発表し、新しい大西洋横断データプライバシーの枠組みに便宜を提供した。これはプライバシーの盾に続く新たなEU-米国の十分なメカニズムである。2022年12月13日、欧州委員会はまた、Schrems IIで提案された懸念を解決することができる新しい行政命令および大西洋横断データプライバシーの枠組みが指摘された十分性決定草案を公表した。十分性決定草案がEU委員会の承認を得て実施されれば、この協定は個人情報の大西洋横断的な流れを促進し、EUから米国への個人情報のデータ転送メカニズム(EU SCCおよび拘束力のある会社ルールを含む)に追加的な保障を提供する。締約国が新たな大西洋横断データプライバシー枠組みに依存する前に、EUと米国は立法と規制の手順を取らなければならない。Schrems IIの決定は、企業にプライバシー影響評価を要求することを招き、他に加えて、被援助国の個人情報取得に関する法律を評価し、追加措置を実施する必要があるかどうかを考慮し、データ保護レベルがEUと“ほぼ同じ”ことを確保するために、SCCの下で追加のプライバシー保護を提供する。したがって、現在の新しいEU SCCは依然として個人情報がEUから米国に送信される主な保障である。そのため、現在の法的地位は私たちの国境を越えたデータの流れに影響を与え、コンプライアンスコストを招く可能性がある。
EU GDPRを遵守することは、厳格で時間のかかるプロセスに関連しており、これにより、いくつかの運用コストが生じ、および/または、私たちのビジネス慣行を変更することが要求される可能性があります。正確な実施が得られない場合や企業内の人員が必要なプログラムを完全に遵守できないリスクもある可能性がある。
EU GDPRを遵守しなければならない企業は、より強力なデータ保護要求の規制実行を含むより多くのコンプライアンス義務とリスクに直面し、遵守しないと2000万ユーロまたは世界総合売上高4%までの巨額の罰金、およびデータ処理の制限または禁止に処せられる可能性がある。EU GDPRはまた、データ主体と消費者協会に対して個人訴訟を提起する権利を付与し、監督管理当局に苦情を提出し、司法救済を求め、EU GDPR違反による損害について賠償を受けることができる。
また、連合王国のEU離脱後、2018年の“欧州連合(離脱)法”第3条に基づき、EU GDPRの義務はほぼ変わらない形で英国に適用され続けている。英国GDPRは2018年の英国データ保護法と併存しており、英国GDPRのいくつかの減税を英国法に盛り込んでいる。イギリスGDPRによると、連合王国に設立された会社とイギリスに設立されていない会社がイギリス個人への商品やサービスの提供に関する個人情報を処理したり、彼らの行動を監督する会社は、イギリスGDPRの制約を受ける--その要求はEU GDPRの要求とほぼ一致するため、類似のコンプライアンスおよび運営コストを招く可能性があり、潜在的罰金は最高1750万GBまたは世界総合売上高の4%に達する。したがって、私たちは2つの平行したデータ保護制度に直面する可能性があり、すべての制度は罰金を許可し、異なる法執行行動を取ることができる。また,イギリス情報コミッショナー事務室(ICO)は,イギリス国際データ転送プロトコル,および新しいEU SCCSの国際データ転送付録と呼ばれる独自形式のEU SCCSを公表していることを指摘すべきである。ICOはまた,各エンティティがEUやイギリス風の譲渡影響評価を選択できるにもかかわらず,そのバージョンの譲渡影響評価と国際譲渡に関する情報ガイドラインを発表した.英国と米国間の国際データ伝送では、英国と米国が十分な合意について交渉しているという。
欧州やイギリス以外の他の国は、同様の国境を越えたデータ伝送制限やローカルデータ駐留を要求する法律を制定または検討しており、サービスや運営業務を提供するコストや複雑さを増加させる可能性がある。例えば、ブラジルでは最近、一般データ保護法(Lei Geral de Prote≡o de Dados PessoaisまたはLGPD)(第13,709/2018法律)が公布され、個人情報の処理が広く規制され、EU GDPRやイギリスGDPRに相当するコンプライアンス義務と処罰が加えられている。
米国でもプライバシー、データ保護、データセキュリティに対する規制が厳しくなっている。例えば、2020年1月1日に施行されたカリフォルニア消費者プライバシー法(CCPA)は、カリフォルニア住民により大きな権利を与え、彼らの個人情報にアクセスして削除し、特定の個人情報を共有しないことを選択し、彼らの個人情報がどのように使用されるかに関する詳細な情報を得ることができる。CCPAは違反行為に対する民事処罰と,データ漏洩に対する個人訴権を規定しており,データ漏洩訴訟が増加すると予想される。CCPAは私たちのコンプライアンスコストと潜在的な責任を増加させるかもしれない。“包括的平和協定”
72
2023年1月1日、CCPAに対するカリフォルニアプライバシー権法案(CPRA)の改正案が全面的に発効すると、CCPAは大幅に拡大した。他の事項以外に、CPRA修正案はカリフォルニア住民にある敏感な個人情報の使用を制限する能力を与え、更に文脈を越えた広告の使用を制限し、個人情報を保留する制限を確立し、CCPA個人訴訟権利制約を受けたデータ漏洩タイプを拡大し、16歳以下のカリフォルニア住民に関連するCCPA違反行為に対する処罰を加重することを規定し、新しいカリフォルニアプライバシー保護局を設立して新しい法律を実施し、実行する。コロラド州、コネチカット州、ユタ州、バージニア州のような他の州では、同様の全面的なプライバシー法が採択され、2023年に施行され、いくつかの州が独自バージョンのプライバシー法を検討しており、米国がより厳しい州プライバシー、データ保護、データセキュリティ立法に傾いている傾向を示しており、これは私たちの潜在的な責任を増加させ、私たちの業務に悪影響を及ぼす可能性がある。
適用される米国や外国のプライバシー、データ保護、データセキュリティの法律法規を遵守することは、政府の調査を招いたり、巨額のコストを招いたり、私たちの業務に不利な方法で私たちの業務のやり方やコンプライアンスを変更することを要求する可能性があります。さらに、これらの異なる法律を遵守することは、私たちがデータを収集、使用、開示する能力を制限し、または場合によっては、いくつかの司法管轄区域で私たちが運営する能力に影響を与える契約でより重い義務を負うことを要求するかもしれない。米国および外国のプライバシー、データ保護、およびデータセキュリティ法律法規を遵守しないことは、政府の調査または法執行行動(民事または刑事罰を含む可能性がある)、私たちに対する個人訴訟、クレームまたは公開声明および/または負の宣伝を招き、私たちの経営業績および業務に負の影響を与える可能性がある。私たちがプライバシー権を侵害し、プライバシー、データ保護、データセキュリティ法律を遵守できなかったと主張したり、私たちの契約義務に違反したりして、私たちが責任を負わなくても、弁護の費用が高く、時間がかかる可能性があり、負の宣伝を招く可能性があり、私たちの業務、名声、財務業績、業務、および運営に実質的な悪影響を及ぼす可能性がある。また、当社の顧客業務に適用される法律、法規、政策を遵守するコストやその他の負担は、私たちの製品やサービスの採用と使用を制限し、私たちの製品やサービスに対する全体的な需要を減少させる可能性があります。
もし私たちのセキュリティ措置が破壊された場合、あるいは私たちの情報技術システムやサプライヤーの情報技術システムと他の関連第三者が故障したり、セキュリティホール、データ損失や漏洩、その他の中断を受けたりすることは、私たちのサービスの深刻な中断を招き、私たちの業務に関連する敏感な情報を危険にさらし、私たちの名声を傷つけ、私たちの違反通知義務をトリガし、私たちの重要な情報へのアクセスを阻止し、私たちを責任または他の私たちの業務への悪影響に直面させる可能性があります。
私たちの通常のビジネスプロセスでは、当社または他の当事者によって所有または制御される個人情報(健康情報を含む)、知的財産権、ビジネス秘密、および独自の商業情報を含む独自、機密および敏感な情報を収集、処理、格納することができます。私たちはこのような情報の機密性、完全性、そして利用可能性を維持するために安全な方法でそうしなければならない。私たちは、アクセス権限を失うリスク、不適切な使用または開示、不適切な修正、および私たちがキー情報の制御を十分に監視、監査、修正できないリスクを含む、これらの重要な情報の保護に関連するいくつかのリスクに直面している。このリスクは私たちの業務要素を扱う第三者サービスプロバイダに延長される。
私たち、私たちのパートナー、私たちのCRO、私たちのCMO、および私たちは、人的資源、財務報告および制御、規制コンプライアンス、および他のインフラ運営を処理するシステムなど、情報技術および電気通信システムに依存して重要に運営されている他のビジネスサプライヤーに依存しています。セキュリティ対策が実施されているにもかかわらず、我々の情報技術システムおよび我々の第三者サプライヤーおよび他の請負業者およびコンサルタントのシステムの規模および複雑さ、ならびに彼らが維持する独自、機密および敏感な情報の数が増加していることを考慮すると、このような情報技術システムは、故障、サービス中断、システム障害、自然災害、テロ、戦争、電気通信および電気故障、ならびに私たちの人員、第三者サプライヤー、請負業者、コンサルタント、業務パートナーおよび/または他の第三者の不注意、または意図的な行為によるセキュリティホールが発生しやすい可能性がある。または悪意のある第三者のネットワーク攻撃(有害なマルウェアの配備、恐喝ソフトウェア、サービス拒否攻撃、社会工学、および他の手段を含み、サービスの信頼性に影響を与え、情報の機密性、完全性および利用可能性を脅かす)、これは、私たちのシステムインフラ、または私たちの第三者サプライヤーおよび他の請負業者およびコンサルタントのシステムインフラに危害を及ぼす可能性があり、またはデータ漏洩を引き起こす可能性がある。世界各地からの未遂攻撃と侵入の数、強度と複雑性の増加に伴い、セキュリティホールや破壊のリスクは普遍的に増加し、特に信頼できる内部者の意外な行為や不注意、ネットワーク攻撃またはネットワーク侵入により、コンピュータハッカー、ウイルス、外国政府とネットワークテロリストを含む。また…, 新冠肺炎疫病に関連する現地亡命或いは類似の制限により、ホームネットワーク上で操作するコンピュータの使用率が増加し、我々のシステムがセキュリティホールの影響を受けやすくなる可能性がある。たとえば,MSKは2021年3月に,MSKはAccellion,Inc.データ漏洩の影響を受ける多くのクライアントの1つであり,Accellion,Inc.は文書共有システムを提供することを通知した.MSKはその後,我々の生産停止計画に関連するいくつかの文書が侵入の影響を受けていることを通知し,この妥協は重要ではないと考えている.私たちは不正アクセス、使用または漏洩から敏感なデータを保護する措置を取っているが、私たちと私たちの第三者サービスプロバイダはしばしばネットワーク攻撃を防御し、対応しており、私たちの情報技術およびインフラはハッカーやウイルスの攻撃を受けやすいか、または人員ミス、汚職、または他の悪意または意図しない中断によって破壊されやすいかもしれない。このような侵入または中断は、私たちのネットワークを危険にさらす可能性があり、そこに格納されている情報は、許可されていない当事者によってアクセス、操作、開示、紛失、または盗まれる可能性がある。
73
私たちの情報技術や電気通信システム、あるいは私たちの第三者サービスプロバイダが使用するシステムの故障や深刻な停止は、私たちのテストや研究開発活動を阻止し、私たちの管理業務の行政面を阻止することを含む、私たちの運営を深刻に中断させる可能性があります。例えば、完了した、行われている、または計画されている臨床研究データの損失は、私たちの規制承認作業を遅延させ、データを回復または複製するコストを著しく増加させる可能性がある。さらに、我々が第三者から調達した複雑なオペレーティングシステムソフトウェアおよびアプリケーションは、脆弱性、“エラー”および他の問題を含む設計または製造上の欠陥を含む可能性があり、これらの問題は、意外にも私たちのネットワーク、システムの動作、または個人情報または他のデータの処理を妨害する可能性がある。もしどんな中断やセキュリティホールが私たちのデータやアプリケーションを紛失したり破損したり、あるいは機密または独自の情報を適切に開示しない場合、私たちは責任を招く可能性があり、私たちの候補製品のさらなる開発は延期される可能性があり、私たちの業務は他の不利な影響を受ける可能性があります。
私たちはすべてのタイプのセキュリティ脅威を予見できないかもしれないし、このようなすべてのセキュリティ脅威に対して有効な予防措置を実施できないかもしれない。私たちはまた攻撃の危険に効果的に対応、抑制、または軽減できないかもしれない。サイバー犯罪者が使用する技術はしばしば変化し,起動まで識別されない可能性があり,外部サービスプロバイダ,組織犯罪分岐機関,テロ組織,敵対する外国政府や機関やネットワークセキュリティ研究者などの外部団体から様々なソースから来る可能性がある.任意の中断またはセキュリティホールが、私たちのデータまたはアプリケーション、または当社の第三者サプライヤー、他の請負業者およびコンサルタントのデータまたはアプリケーションを紛失または破損させたり、機密または独自の情報を適切に開示しない場合、私たちの製品およびサービスのさらなる開発および商業化は遅延する可能性があります。
重大なセキュリティホールや中断に関連するコストは巨大である可能性があり、そのようなリスクに対して提供されるネットワークセキュリティ保険の限度額を超える可能性がある。もし私たちの第三者サプライヤー、他の請負業者、およびコンサルタントの情報技術システムが中断またはセキュリティホールの影響を受ける場合、私たちはこのような第三者に対抗するための十分な追跡権がない可能性があり、このようなイベントの影響を軽減するために大量の資源を費やし、将来のこのような事件の発生を防止するために保護措置を制定し、実施しなければならないかもしれない。
私たちのデータ保護努力と情報技術への私たちの投資は、私たちのシステムまたは私たちの第三者サプライヤー、他の請負業者、コンサルタントのシステムに重大な障害、データ漏洩、侵入、または私たちの名声、業務、運営、または財務状況に重大な悪影響を及ぼす可能性のあるネットワークイベントを防止することを保証することはできません。例えば、このような事件が発生して私たちの運営が中断されたり、私たちの第三者サプライヤーや他の請負業者やコンサルタントの運営が中断されたりすると、私たちの計画が実質的に中断される可能性があり、私たちのサービスや技術の開発が延期される可能性があります。さらに、我々の内部情報技術システムまたは第三者サプライヤー、他の請負業者およびコンサルタントシステムの深刻な中断またはセキュリティホールは、機密情報(商業秘密または他の知的財産権、独自業務情報および個人情報を含む)の損失、流用および/または不正アクセス、使用または開示、またはアクセスを阻止する可能性があり、これは、財務、法律、商業および名声を損なう可能性がある。例えば、不正アクセス、使用または個人情報(私たちの顧客または従業員に関する個人情報を含む)をもたらすこのような事件は、私たちの名声を直接損なう可能性があり、連邦および/または州の違反通知法および外国等価法律に遵守させ、是正措置を強制することができます。そうでなければ、個人情報のプライバシーや安全を保護する法律および法規に基づいて責任を負うことになり、重大な法律や財務リスクおよび名声被害を招き、私たちの業務に悪影響を及ぼす可能性があります。
私たちは、不正アクセス、使用、または漏洩から敏感なデータを保護する措置を取っているが、私たちの情報技術およびインフラは、ハッカーやウイルスの攻撃を受けやすいか、または人員ミス、汚職、または他の悪意または意図しない中断によって破壊される可能性がある。このような侵入または中断は、私たちのネットワークを危険にさらす可能性があり、そこに格納されている情報は、許可されていない当事者によってアクセス、操作、開示、紛失、または盗まれる可能性がある。
このような任意の情報のアクセス、漏洩、または他の損失は、法的クレームまたは訴訟、国内または海外のプライバシー、データ保護およびデータセキュリティ法律(例えば、HIPAAおよびHITECH)に基づく責任、および処罰をもたらす可能性がある。いくつかのセキュリティホールは、影響を受けた個人、HHS部長に通知しなければならないが、広範な違反に対しては、メディアまたは州総検察長に通知する必要があるかもしれない。そのような通知は私たちの名声と競争能力を損なうかもしれない。我々はセキュリティ対策を実施しているが,これらのデータは現在様々なチャネルでアクセス可能であり,攻撃からデータを保護できる保証はない.許可されていないアクセス、紛失、または伝播は、私たちの分析、研究開発活動、会社の財務情報の収集、処理、準備、および私たちの業務を管理する行政面の能力を含む、私たちの名声を傷つけたり、私たちの運営を混乱させたりする可能性があります。
このような法律違反に対する処罰はそれぞれ違う。例えば、HIPAAとHITECH要求を守らない処罰は、重大な民事罰金を含めて大きく異なり、場合によっては、刑事罰、毎回の違反および/または監禁に最高25万ドルの罰金が科せられる。HIPAAに違反し、個人識別可能な健康情報を故意に取得または開示する人は、最高50,000ドルの刑事罰および最高1年の禁固に直面する可能性がある。犯人は…
74
もし不法行為が偽りの口実或いは意図が商業利益、個人利益或いは悪意をもって売却、譲渡或いは識別可能な健康情報を使用することに関連する場合、処罰を強化する。
また、カリフォルニア州やマサチューセッツ州などでも同様のプライバシー法律や法規が施行されており、“カリフォルニア医療情報秘密法”のような、健康情報や他の個人識別情報の使用や開示に制限的な要求が加えられている。HIPAAは必ずしも先制されるとは限らず、特に国がHIPAAよりも個人に大きな保護を与えた場合。州の法律がもっと保護されているところで、私たちはもっと厳格な規定を守らなければならない。違反者に罰金と罰を加えるほか、いくつかの州法は、自分の個人情報が乱用されたと思う個人に個人訴訟権利を提供している。例えば、カリフォルニアの患者プライバシー法は最高25万ドルの罰金を規定し、被害者側が損害賠償を要求することを許可する。同様に、CCPAは、ある個人情報が、企業が合理的なセキュリティプログラムを実施および維持できなかったために、不正なアクセスおよび流出、盗難、または開示を受けた場合、消費者に個人訴訟を提起することを可能にする。連邦と州の法律の相互作用は裁判所と政府機関の異なる解釈を受ける可能性があり、私たちおよび私たちが受け入れ、使用し、共有するデータに複雑なコンプライアンス問題をもたらし、私たちを追加的な費用、不利な宣伝と責任に直面させるかもしれない。また,規制機関のプライバシー問題への関心が高まっていることや,個人情報保護に関する法律法規が拡大し複雑になっていることにより,我々の業務に対する潜在的なリスクが悪化する可能性がある.特定のタイプの敏感なデータの保護の強化、遺伝データの処理に関する法律または法規の変化、およびデータセキュリティインフラの強化に対する顧客の需要の増加, 私たちが製品を提供するコストを大幅に増加させ、私たちの製品に対する需要を減らし、私たちの収入を減少させ、および/または追加の債務を負担させるかもしれない。
私たちまたは私たちの顧客に不利な税金法律または法規の変化は、私たちの業務、キャッシュフロー、財務状況、または経営結果に悪影響を及ぼす可能性があります。
私たちはアメリカとアメリカ以外の異なる司法管轄区で所得税と非所得税を納めています。私たちの業務と財務状況は連邦、州、現地あるいは国際税法の変化、税収管轄区域の行政解釈、決定、政策と立場の変化、会計原則の変化、源泉徴収税の適用性、そして私たちの業務運営の変化の悪影響を受ける可能性があります。例えば、“税法”、“コロナウイルス援助、救済·経済安全法”(CARE法案)、米国救助法などの米国の法律は、会社の税率、私たちの運営に関連する繰延純資産の潜在的な現金化、外国収益の課税と費用の控除に大きな変化をもたらし、私たちの財務状況や運営結果に大きな影響を与える可能性がある。
私たちは純営業損失の繰越とある税務資産を使って将来の課税収入や税金を相殺する能力が一定の制限を受ける可能性があります。
私たちは連邦と州の純営業損失(NOL)といくつかの他の税金属性を使用して、潜在的な将来の課税所得額と関連する所得税を相殺する能力は私たちの未来の課税所得額の発生に依存して、私たちは私たちがいつ、あるいは私たちのすべてのNOLまたは他の税金属性を使用するのに十分な課税収入が発生するかどうかを予測することができない。
2022年12月31日まで、前期損失のため、大量のアメリカ連邦と州NOLがあります。減税·雇用法案(Tax Act)によると、2018年1月1日以降の納税年度に発生した連邦NOLは無期限に繰り越すことができるが、このような連邦NOLの使用は今年度の課税所得額の80%を超えてはならない。CARE法案はこの80%の課税収入制限を一時停止し、NOL繰越が2021年1月1日までの納税年度に課税収入を完全に相殺することを許可しており、これは我々の財務諸表に影響を与えない。各州が税法やCARE法案をどの程度遵守しているかは不明である。税法もCARE法案も私たちの財務諸表に実質的な影響を与えなかった。
また、改正された1986年の国内税法(以下、“税法”と略す)第382条の規定によると、所有権変更を経験した場合、任意の課税年度にこれらのNOLや他の税属性(例えば連邦税収控除)を利用する能力が制限される可能性がある。一般に、1人または複数の株主または1社の株式の少なくとも5%を保有するグループ株主が3年間のテスト期間内に、その所有権がその最低持株割合より50ポイント以上増加すると、第382条に規定する所有権変更が発生する。似たような規則は州税法に適用されるかもしれない。2022年12月31日までの株式取引について第382節の分析を行い,成立以来所有権変更を経験しており,準則第382節の規定により,何らかの変更前のNOLと信用を使用する能力が制限されると結論した。また、将来の株式発行や株式所有権の他の変化により、私たちは後続の所有権変化を経験する可能性があり、その中のいくつかは私たちがコントロールできない。したがって、我々の財務諸表に記載されているNOLと税収控除の金額は限られている可能性があり、2018年1月1日までに発生したNOLであれば、満期になって使用されない可能性があります。私たちのNOLのこのような実質的な制限や満期は、私たちの未来の納税義務を効果的に増加させ、私たちの未来の経営業績を損なうかもしれません。州税法の類似規定は、累積された州税収属性の使用を制限するのにも適用可能である。NOLの使用を一時停止するような規制の変化
75
あるいは他の予見できない理由は、私たちの既存の税務属性の失効、価値の低下、または将来の所得税負債を相殺できない可能性がある。
業務中断は私たちの将来の収入と財務状況を深刻に損害し、私たちのコストと支出を増加させるかもしれない。
私たちの業務は地震、電力不足、電気通信故障、水不足、洪水、ハリケーン、台風、火災、極端な天気条件、医療流行病、その他の自然或いは人為的災害或いは業務中断の影響を受ける可能性があり、私たちは主に自己保険です。私たちの2つの会社の場所はカリフォルニア州千樫市にあり、地震と火災が多発する地域です。このような業務中断の発生は、私たちの運営と財務状況を深刻に損害し、私たちのコストと支出を増加させる可能性がある。私たちは第三者製造業者に依存して私たちの製品と候補製品を生産する。もしこれらのサプライヤーの運営が人為的あるいは自然災害あるいは他の業務中断の影響を受け、進行中の新冠肺炎疫病を含む場合、私たちが製品と候補製品の臨床供給を獲得する能力は妨害される可能性がある。
76
情報技術EM 1 B.未解決従業員意見
ない。
I項目2.特性
当社の本社はカリフォルニア州千樫市にあり、2026年2月に期限が切れる賃貸契約によると、同社は約51,160平方フィートのオフィススペースを持っています。
2021年3月、カリフォルニア州千オーク市で約33,659平方フィートのオフィス、実験室、倉庫スペースを購入する新しいレンタル契約を締結しました。本借約の最初の10.5年レンタル期間は2021年8月に開始されました。最初のレンタル期間の後、私たちはレンタル期間をさらに2つ5年間延長することを選択することができます。また、2021年には、コロラド州オーロラのオフィスと実験室のために改訂されたレンタル契約を締結し、追加の実験室空間を増加させました。
2022年4月,富士フイルム取引の一部として,カリフォルニア州千樫市の約90,580平方フィートのオフィス,実験室,細胞治療製造空間のリースをFDBに譲渡し,リースは2033年4月に満期となる。私たちはまだ賃貸譲渡に関する義務について連帯責任を持っている。
2025年5月までのレンタル不可契約によると、カリフォルニア州サンフランシスコ南部でオフィススペースをレンタルしました。2022年10月、私たちは第三者とこのオフィス空間の分譲協定を締結した。分譲期間は2022年11月に開始され、2025年5月に満了し、分譲期間を延長する選択権はない。
私たちは私たちがカリフォルニア州千樫市とコロラド州オロラ市で借りた空間を使って、私たちの翻訳と臨床前科学、分析開発と過程科学機能に使用した。これらの施設は私たちの製品パイプラインとプロセス開発を支持し、私たちの異遺伝子細胞治療プラットフォームを利用して革新を推進する。私たちは私たちの既存の施設の運営状況が良好で、業務を展開するのに適していると信じている。
情報技術EM 3.法的訴訟
ない。
情報技術EM 4.炭鉱安全情報開示
適用されません。
77
P芸術二
情報技術EM 5.登録者普通株、関連株主事項、発行者が株式証券を購入する市場
市場情報
私たちの普通株は2014年10月16日からナスダック世界ベスト市場に上場し、取引コードはATRAである。その前に、私たちの普通株は市場を公開しなかった。
2023年1月31日、私たちの普通株には登録されている株主が5人います。私たちの多くの株は仲介人や他の機関が私たちの株主を代表して保有しているので、これらの記録保有者が代表する株主の総数を見積もることはできません。
配当政策
私たちは私たちの株の現金配当金を発表したり支払ったりしたことがない。私たちは現在、私たちの未来のすべての収益(もしあれば)を保留し、私たちの業務の成長と発展に資金を提供し、予測可能な未来に現金配当金を発表または支払うつもりはありません。配当金をさらに決定するいかなる決定も、私たちの取締役会が適宜決定し、適用法律の制約を受け、私たちの財務状況、経営結果、資本要求、一般業務状況及び私たちの取締役会が関連すると考えている他の要素に依存する。
株式補償計画に基づいて発行された証券
当社の株式報酬計画に関する情報は、ここに組み込まれ、10-K表形式の本年度報告第3部第12項を参考にします。
株式表現グラフ
次の図は,2022年12月31日までの5年間に会社普通株ナスダック総合指数とナスダックバイオテクノロジー指数に対する100ドル投資の指数累積総リターンを比較したものである。
取引法第18節の規定によれば、本業績グラフは、“募集材料”または米国証券取引委員会によって“保存”されてはならず、参照によってAtara BioTreateutics,Inc.特に参照によってそのような文書に組み込まれない限り、証券法または取引法に基づいて提出された任意の文書とみなされてはならない。私たちの普通株の過去の表現は未来の表現を暗示することはできない。
78
5年間の累計総収益比較

12月31日まで |
|
Atara生物治療会社 |
|
|
ナスダック複合体 |
|
|
ナスダック生物技術は |
|
|||
2017 |
|
|
100.00 |
|
|
|
100.00 |
|
|
|
100.00 |
|
2018 |
|
|
191.93 |
|
|
|
96.12 |
|
|
|
90.68 |
|
2019 |
|
|
90.99 |
|
|
|
129.97 |
|
|
|
112.81 |
|
2020 |
|
|
108.45 |
|
|
|
186.69 |
|
|
|
141.78 |
|
2021 |
|
|
87.07 |
|
|
|
226.63 |
|
|
|
140.88 |
|
2022 |
|
|
18.12 |
|
|
|
151.61 |
|
|
|
125.52 |
|
第六項です[R保存された]
必要ではありません。
79
情報技術EM 7.経営陣の財務状況と経営結果の検討と分析
あなたは私たちの財務状況と経営結果の以下の議論と分析、ならびに私たちの監査された総合財務諸表と本年度報告書の他の場所に関する付記を読むべきです。本議論および本年度報告の他の部分は、私たちの計画、目標、期待、および意図の陳述のようなリスクおよび不確実性に関する前向きな陳述を含む。本年度報告の“リスク要因”の一部に列挙された要素を含む多くの要素の影響により、我々の実際の結果は、以下の議論と分析に含まれる前向き陳述に記述または示唆された結果とは大きく異なる可能性がある。
概要
Atara BioTreateuticsはT細胞免疫療法の先駆者であり、その新型同種異体EBウイルス(EBV)T細胞プラットフォームを用いて癌と自己免疫疾患患者のための変革性療法を開発した。Tabelecleucelは私たちがアメリカで第三段階の臨床開発の主要なプロジェクトであり、すでに欧州委員会(EC)のマーケティング許可(MAA)を獲得し、独自名のEbvalloで欧州連合(EU)で商業販売を行っている。われわれは最先端の同種異体T細胞免疫治療会社であり,高度に満たされていない医療ニーズの高い患者に迅速に既製の治療を提供する予定である。著者らのプラットフォームはEBV T細胞の独特な生物学を利用し、そして統合工学キメラ抗原受容体(CARS)或いはT細胞受容体(TCR)を通じて広範なEBV駆動疾患或いはその他の深刻な疾患を治療する能力がある。Ataraは、強力な導管を作成するためにTCRまたはHL A遺伝子編集を必要としないこのプラットフォームを適用している。私たちの戦略的なポイントは
上述の戦略重点以外に、著者らは多くの臨床と臨床前プロジェクトがあり、ATA 2271を含み、腫瘍抗原間硫黄蛋白を発現する固形腫瘍に対する自己CAR T免疫療法は、現在第一段階に開発されている;及びATA 3271は、間硫黄蛋白に対する同種異体CAR T免疫療法であり、現在臨床前開発段階にある。
我々のT細胞免疫治療プラットフォームは同種異体と自己プログラムを行う能力を含み、広範な標的と疾患に適用可能である。我々の既存の同種異体T細胞プラットフォームは、患者が必要とする前に製造され、在庫に貯蔵されたT細胞免疫治療製品を迅速に提供することができ、各製造されたバッチ細胞は無数の潜在的な患者に治療を提供することができる。これは自己治療とは異なり,自己治療では,個々の患者自身の細胞が抽出されなければならず,体外で遺伝子改変を行い,患者に返送するには複雑な物流ネットワークが必要である。われわれの同種移植計画では,患者独自の免疫状況に応じて適切な細胞集を選択して使用した。私たちの契約製造組織(CMO)はTABCELの商業生産鑑定活動を完了しており、私たちの他の契約製造組織は現在TABCELの商業生産鑑定活動を完了しており、同時に私たちは私たちの商業製品供給戦略に基づいて在庫を構築している。
2021年10月、我々はPierre Fabreと商業化協定(Pierre Fabre商業化協定)を締結し、これにより、Pierre Fabreに独占的、限られた分野の許可を付与し、規制承認後にヨーロッパでEbvalloを商業化·流通し、中東、アフリカ、東欧、中央アジアで新興市場を厳選した。私たちは北米、アジア太平洋地域、ラテンアメリカのTabcelを含む他の主要市場に完全な権利を保持している。Pierre Fabre商業化プロトコルの想定によると,Pierre Fabreと(I)製造·供給プロトコル(Ii),薬物警戒プロトコル(Iii),品質プロトコルをそれぞれ締結し,Pierre Fabreとの協力関係をさらに進める。2022年9月、欧州委員会(EC)がEBV+PTLDのEbvalloの使用を許可し、その後、Pierre FabreにMAA移転申請を提出した後、我々は、(I)この地域におけるEbvalloの純売上高の割合を占めるEbvalloの資格がある特許権使用料の割合と、(Ii)Pierre FabreのEbvalloの購入価格の値上げと引き換えに、Pierre Fabreから追加の3,000万ドルのマイルストーン支払いを取得した。また,Pierre Fabre商業化プロトコルに従ってPierre Fabreに何らかのサービスを提供する期限を延長することにも同意した.2022年12月、ピエールのもとで、エブワロで特許使用料と特定のマイルストーンを獲得する権利の一部を売却しました
80
HCR Molag Fund L.P(HCRx)とFabre商業化協定を締結し,総投資額は3,100万ドル,上限はHCRx総投資額の185%から250%であった。
2020年12月に,吾らはバイエルと研究,開発および許可協定(バイエル許可協定)を締結し,これにより,吾らは当社とその連属会社が所有または制御しているATA 2271およびATA 3271またはATA 3271に関する適用特許およびノウハウに基づいて,バイエルに独占的な限られた分野許可を付与した。バイエル許可協定によると、私は2021年3月にバイエルと(I)製造および供給協定(バイエル製造協定)、(Ii)薬物警戒協定;(Iii)品質協定;および(Iv)バイエルと技術移転協定を締結し、私たちとバイエルとの協力を更に推進する。バイエル許可プロトコル,製造とプロビジョニングプロトコルおよび技術移転プロトコルを総称してバイエルプロトコルと呼ぶ.2022年5月、バイエルはベイヤ協定の終了決定を通知し、2022年8月2日にバイエルと終了、改訂、計画譲渡協定(バイエル終了協定)を締結し、ベイヤ協定を終了し、ATA 2271とATA 3271の全製品開発権をAtaraに返還し、2022年7月31日から発効した。
著者らはまた、スローン-キャトリン癌センター(MSK)、クイーンズランド医学研究所理事会(QIMR Berghofer)とH.Lee Moffitt癌センターと研究所(Moffitt)を記念するなど、有力な学術機関と研究協力を行い、これにより、斬新かつ独自の技術とプロジェクトの権利を獲得した。
我々のカリフォルニア州千樫市(ARC)とコロラド州オロラ市の研究機関には,我々の翻訳と臨床前科学,分析開発と過程科学機能が含まれている。これらの施設は私たちの製品ライン、技術開発を支持し、私たちの同種異体細胞治療プラットフォームを利用して革新を推進する。
2022年1月、富士フイルムカリフォルニアバイオテクノロジー社(FDB)および富士フイルムホールディングス米国(Fujifilm Holdings America Corporation)と資産購入協定を締結し、カリフォルニア州千どん市にあるAtara T細胞運営·製造施設(ATOM施設)関連資産のすべての権利、所有権、権益を1億ドルの現金で売却し、資産購入協定(富士フイルム取引)に基づいて可能な取引後調整を行った。富士フイルム取引は2022年4月4日に完了し,ATOM施設のリースをFDBに譲渡したことが富士フイルム取引の完了につながっていた。また、取引終了時に発効し、最長10年間延長できるFDB(Fujifilm MSA)とプライマリサービスとプロビジョニング協定を締結しました。富士フイルムMSAによれば、FDBはcGMP規格に従って特定の数の細胞治療製品(承認された場合)および候補製品を提供する。富士フイルムMSAはFDBから製品と候補製品だけを購入することを要求していません。患者に対する私たちの期待とEUの製品に対する需要によると、私たちの現在のEbvallo在庫は2023年末までのEUの商業需要を満たすのに十分だと信じている。
我々はまた,2019年12月に締結したビジネス製造サービスプロトコル(CRL MSA)に基づいてCharles River実験室(CRL)と連携している.CRL MSAにより、CRLは私たちの製品といくつかの候補製品に製造サービスを提供します。2023年2月、CRL MSAを修正し、期限を2023年9月30日の早い時期に延長するか、またはいくつかのロットの製品および候補製品を受信します。
我々は製品やサービスに対してキャンセル不可能な最低約束があるが,臨床研究組織やCMOと1年以上の契約を締結しなければならない。
2022年8月、私たちの活動を研究開発を中心としたより簡素化された組織として、私たちの革新的なパイプをさらに推進し、現金消費を削減するために、約20%のリストラを発表しました。リストラには約600万ドルの総再編費用が含まれる予定で、主に解散費、カリフォルニア労働者調整·再訓練通知法に基づく60日間の通知期間の賃金、退職後しばらくの持続医療保険が含まれる。ほとんどの場合、解散費は2022年10月に一度に支払われる。通知された従業員の中には、昇給の形で離職福祉を提供することが規定されている雇用協定があり、これらの福祉は2022年10月から2023年11月までの間に支払われる。すべての解散費は現金支出です。リストラでは、2023年度までに、報酬に関するコスト低下により、年化運営費が約2500万ドル削減されると予想している。
2022年12月、我々はデラウェア州の有限組合企業HCR Molag Fund,L.P.(HCRx)と購入契約(HCRx協定)を締結した。HCRx協定の条項によると、HCRxがEbvalloの部分階層、販売に基づく特許権使用料、金額の中央値の1桁から重要な2桁まで、いくつかの記念碑的な支払いと引き換えに、総投資額3,100万ドルを受け取り、両者ともPierre Fabre商業化協定に従ってPierre Fabreによって支払われている。HCRx協定によると、HCRxに支払われる特許使用料およびマイルストーン総額の上限は、HCRx総投資額の185%~250%であり、これは、このような特許使用料およびマイルストーンのスケジュールに依存する。
81
財務概要
私たちの運営の歴史は限られている。2012年の設立以来、著者らは臨床前と臨床研究を行い、臨床研究のための材料を獲得或いは製造し、これらの操作に一般的かつ行政的な支持を提供することを含む、著者らの候補製品を決定、獲得、開発するためにほとんどの資源を投入した。
2022年、2021年、2020年12月31日までの年度の純損失はそれぞれ2.283億ドル、3.401億ドル、3.066億ドルだった。2022年12月31日現在、私たちの累計赤字は17億ドルです。我々のほとんどの純損失は,我々の研究や開発計画に関するコストと我々の運営に関する一般的かつ行政費用によるものである。2022年12月31日現在、私たちの現金、現金等価物、短期投資総額は2兆428億ドルで、私たちはこれらの資金で私たちの運営に資金を提供するつもりです。
収入.収入
私たちは製品の商業販売から収入を得たことがなく、設立以来赤字になっています。これまで,我々の収入は完全にバイエルやピエール·ファブレとの合意に由来しており,主に前払い許可費,研究,プロセス開発と翻訳活動の費用および技術移転費用に関連している。
私たちは、Pierre Fabre商業化協定と未来の協力、研究、許可パートナーから生成されたどんな収入も、マイルストーンと他の支払いの時間と数量によって毎年変動すると予想している。
研究と開発費
設立以来、私たちの総運営費用の最大の部分は、私たちの候補製品の臨床前と臨床開発を含む研究と開発活動への投資である。研究開発費には、主に、株式ベースの報酬、臨床前および臨床研究を行う契約研究組織および調査場所との合意に基づいて生じる費用、CMOとの合意に基づいて発生する費用、許可および研究開発協定に基づいて支払う費用、他の外部サービスおよび相談費用、施設、情報技術および管理費用を含む研究開発および規制支援従業員の報酬および福祉が含まれる。研究·開発コストは発生時に費用を計上する。
私たちは私たちの候補製品の開発に引き続き投資する予定です。私たちが現在計画している研究と開発活動は
また,我々の候補製品パイプラインと我々の業務の価値を確立し続けるために,新候補製品の開発に投資することが重要であると考えられる.我々は,われわれの最も有望な早期候補製品を臨床前開発に進めていく予定であり,今後数年でこれらの早期計画をヒト臨床研究に進めることを目標としている。
82
現在と未来の臨床前と臨床開発計画への支出は時間と完成コストの多くの不確実性の影響を受ける。私たちの候補製品の臨床研究と開発の持続時間、コスト、時間は様々な要素に依存します
必要な臨床研究を行い、FDAと他の監督管理機関の承認を得る過程は高価で時間がかかり、著者らの候補製品の開発成功も非常に不確定である。我々の研究·開発プロジェクトに関するリスク·不確定要因は,本報告の“1 A”と題する章でより包括的に議論されている。リスク要因ですこれらのリスクと不確実性のため、私たちはどの程度の確実性で私たちの研究開発プロジェクトの持続時間と完成コストを決定することができませんか、あるいは私たちがいつ、あるいはどの程度規制部門の承認を得た候補製品の商業化および販売から収入を得るかどうかを決定することはできません。
一般と行政費用
一般および行政費用は、主に、株式ベースの報酬を含む法律、人的資源、財務、商業および他の一般および行政従業員の報酬および福祉、法律、特許、人的資源、監査および会計サービスを含む専門サービス費用、他の外部サービスおよびコンサルティング費用、ならびに情報技術および間接費用を含む。
ATOM設備の売却益
ATOMローンを売却する収益には,開発銀行から受け取った対価格,取引コストの減算,資産売却の帳簿価値がある.
利息とその他の収入,純額
利息とその他の収入(費用)は、純額は主に私たちの現金、現金等価物、短期投資で稼いだ利息からなる。
所得税支給
所得税の支出は主にアメリカ各州と外国司法管轄区の所得税を含む。2022年、2021年、2020年12月31日までの年間有効税率は0%です。
重要な会計政策と重大な見積もり
私たちの経営陣は、私たちの財務状況と経営結果の討論と分析は、私たちの総合財務諸表に基づいています。これらの総合財務諸表は、アメリカで公認されている会計原則に基づいて作成されています。これらの財務諸表の作成は、報告書に影響を与える資産、負債、費用金額の推定と仮定を要求しています。持続的な基礎の上で、私たちは私たちの重要な会計政策と推定を評価する。我々の推定は,歴史的経験と当時の状況で合理的であると考えられる様々な他の仮定に基づいており,その結果形成される
83
資産や負債の帳簿価値を判断する基礎は、これらの資産や負債の帳簿価値は他の出所からは明らかではないように見える。異なる仮定や条件では、実際の結果はこれらの推定値とは異なる可能性がある。私たちの重要な判断と推定詳細は以下のとおりであり、私たちの重要な会計政策は添付されている総合財務諸表付記2により全面的に記述されている。
収入確認
非ライセンス契約の収入は、義務を履行していることが確認され、顧客が約束された商品またはサービスの制御権を取得した場合、その金額は、これらの商品やサービスと交換することが予想される対価格を反映している。私たちの外部許可協定による収入は返済する必要がありません。通常、前払い費用、開発、監督と商業マイルストーン支払い、被許可者の未来の製品販売の印税が含まれています。
私たちの外部許可協定には、許可の形で知的財産権を譲渡し、研究開発サービスを提供することを約束し、協力者と共にいくつかの開発委員会に参加することを約束することが含まれる可能性がある。私たちはこのような合意の約束が異なる履行義務とみなされているかどうかを評価し、単独で計算しなければならない。このような約束が違うかどうかを確認するために判断が必要だ。
各プロトコルにおける取引価格は,契約義務ごとの独立販売価格(SSP)に応じて決定された履行義務に割り当てられる.我々のライセンス技術は初期段階にあるため、このような技術の許可は、一般に、これらのプロトコルにおける追加的な約束と組み合わされて、総合的な義務として履行される。
払戻不可能な前払い許可料に関する収入は,許可料や他の承諾が単独の履行義務として入金できない場合には,履行義務の性質に基づく適切な確認方法を用いて期待履行期間中に収入として確認する。私たちは判断を利用して義務履行の交付モデルを評価する。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。予測コストや患者需要の範囲やタイミングなど、予測コストや推定の重大な変化は、将来期間に確認された収入の時間や金額、または変化期間確認の累積収入の調整に大きな影響を与える可能性がある。
開発、規制、または商業マイルストーン支払いを含む各合意の開始時に、マイルストーンが実現可能であると考えられているかどうかを評価し、可能な金額法を用いて取引価格に含まれる金額を推定する。大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。取引価格は関連するSSPによってプロトコル中の履行義務ごとに割り当てられる.私たちは通常コストプラス利益率モデルを使用してSSPを決定します。規制承認のような我々または被許可者の制御範囲内でのマイルストーン支払いではなく、これらの承認を受ける前に実現可能であるとは考えられない。各報告期間の終了時に、このような各マイルストーンの実現確率および任意の関連制限を再評価し、必要に応じて全体の取引価格の推定値を調整します。いずれの調整も累積追跡をもとに記録されており,調整期間中の収入や収益に影響を与える.
特定の判断は私たちの収入確認政策の適用に影響を及ぼすだろう。例えば、短期および長期繰延収入は、収入確認時間の最適な推定値に基づいて記録される。短期繰延収入は今後12カ月で収入として確認される予定の金額からなり、長期繰延収入は今後12カ月で確認されないと予想される金額からなる。この推定は,我々が予測した患者需要と現在の運営計画に基づいており,患者需要や我々の運営計画が将来的に変化すれば,今後12カ月以内に異なる額の繰延収入を確認する可能性がある。
研究と開発費用を計算すべきである
財務諸表作成過程の一部として、臨床研究や臨床候補製品製造に関連する費用を含む研究開発費用に関連する費用の見積もりと計上が必要である。この過程には、契約および調達注文の審査、私たちを代表するサービスの決定と評価、および実際に発生した費用に関する費用が領収書を発行されていないか、または他の方法で通知されていないと推定されることが含まれています。
臨床前研究、臨床研究と製造活動のコストは著者らのサプライヤーが特定の任務を達成する方面の進展を評価した上で確認したものであり、使用したデータは患者登録、臨床サイト活性化或いはサプライヤーが私たちに提供したその実際のコストに関する情報を含む。これらの活動の支払いは個別契約の条項に基づいており,支払い時間はサービスを提供する期間と大きく異なる可能性がある.カウントを計算すべきだと確信しています
84
学習進行または完了状態または交付された貨物およびサービスについて、適用者および外部サービスプロバイダを介して報告書を提出し、議論する。私たちの各貸借対照表の日付までの課税費用の推定は、当時既知の事実と状況に基づいている。履行前に支払われた費用を前払い料金として繰延し、サービス提供時にサービス期間内に償却する。
2022年と2021年12月31日までの年次では,我々の研究開発費に対する見積もりには実質的な変化はなかった。将来的に計算すべき研究や開発費用の見積もりに合理的な可能性は大きく変化していないと考えられる。しかし,実際の結果が我々の予想と一致しなければ,計算すべき研究開発費の変化の影響を受ける可能性があり,これらの変化は重大である可能性があり,あるいは我々の財務諸表で報告されている計算すべき研究開発費は,計算すべき研究開発の実際の経済コストを代表できない可能性がある。
株に基づく報酬
限定株式単位(RSU)、株式オプション、および従業員株式購入計画を含む株式ベースの報酬計画がある。株式ベースの報酬計画の完全な議論については、本報告の第8項.財務諸表および補足データに含まれる合併財務諸表付記の2-“重要会計政策概要”および付記11-“株主権益”を参照されたい。我々は、発行された株式ツールの公正価値に基づいて、RSUと普通株式で決済可能な株式オプションを付与する支出を含む株式ベースの報酬支出を会計処理する。公正価値は計量日付によって決定され、この日付は一般的に授与日である。私たちのRSUの公正価値は私たちの普通株の計量日の市場価格によって計量されます。私たちの株式オプション報酬の公正価値は授与日にブラック·スコアーズ推定値モデルを用いて決定された。
ブラック·スコアーズの評価モデルが従業員の株式報酬に用いられた仮説には
業績帰属基準を採用した奨励については、各報告期間終了時に業績条件に達する可能性を評価し、業績条件を満たす可能性が高い場合に株式に基づく報酬コストの確認を開始した。サービスと業績条件の制限を同時に受けた奨励については、業績条件を満たす可能性が高いまで、何の費用も確認されない。私たちは、株式に基づく報酬支出の将来推定や仮定が合理的でない可能性が大きく変化することを決定するために使用されると考えられる。しかしながら、実際の結果が私たちの推定や仮定と一致しない場合、株式ベースの報酬支出の変化に直面する可能性があり、これらの変化は重大である可能性があり、または私たちの財務諸表で報告されている株式ベースの報酬支出は、株式ベースの報酬の実際の経済コストを表すことができない可能性がある。
所得税会計
Atara所得税支出の構成要素および2022年12月31日現在の一時的な差の完全な議論については、本報告の財務諸表および補足データを含む合併財務諸表付記12“所得税”を参照されたい。
私たちの総合的で有効な所得税率は、私たちが業務を展開している各司法管轄区で提供されている税務計画の機会の影響を受けています。私たちの税務状況を評価する際には、不確実な可能性がある場合も含めて、重大な判断が必要だ。Ataraはまた私たちの繰延純税資産の現金化を判断することを要求された。経営陣はすべての積極的で消極的な証拠を評価し、過去と未来の事件を判断して、より可能性があるかどうかを判断する
85
繰延税金資産の全部または一部は現金化できないかもしれない。適用されれば、繰延税金資産に推定値を計上して、将来実現できない可能性のある税金優遇を相殺することができる。
私たちは私たちが不確定な所得税の頭寸や私たちの有効な所得税率の負債に大きな変化がある合理的な可能性があるとは思わない。しかし、実際の結果が私たちの推定や仮定と一致しなければ、私たちは大きな損失に直面するかもしれない。2022年12月31日まで、Ataraが記録した評価額は約4.225億ドルに割り当てられ、主に純運営損失の繰越、資本化研究費用、税収控除、株式に基づく報酬に用いられている。
経営成果
2022年、2021年と2020年12月31日終了年度比較
許可と協力収入
示した期間の許可と連携収入は以下のとおりである
|
|
十二月三十一日までの年度 |
|
|
(減少を)増やす |
|
||||||||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|
2022年は2021年と比較して |
|
|
2021年は2020年と比較して |
|
|||||
|
|
(単位:千) |
|
|||||||||||||||||
許可と協力収入 |
|
$ |
63,573 |
|
|
$ |
20,340 |
|
|
$ |
— |
|
|
$ |
43,233 |
|
|
$ |
20,340 |
|
2022年には許可·協力収入は6,360万ドルだったが、2021年には2,030万ドル、2020年にはゼロ収入となった。2022年の成長は主にベイヤ協定の終了により、ベイヤ協定に関連する残りの繰延収入が2022年第2四半期に確認された。ベイヤ協定の終了により、今後数四半期の許可と協力収入が大幅に低下することが予想される。
研究開発費
本報告に記載されている間、研究および開発費用は、計画的に区分された以下の費用を含む
|
|
十二月三十一日までの年度 |
|
|
(減少を)増やす |
|
||||||||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|
2022年は2021年と比較して |
|
|
2021年は2020年と比較して |
|
|||||
|
|
(単位:千) |
|
|||||||||||||||||
付箋式費用 |
|
$ |
40,597 |
|
|
$ |
50,086 |
|
|
$ |
61,196 |
|
|
$ |
(9,489 |
) |
|
$ |
(11,110 |
) |
ATA 188、CAR Tなどの番組費 |
|
|
45,597 |
|
|
|
36,424 |
|
|
|
25,124 |
|
|
|
9,173 |
|
|
|
11,300 |
|
従業員と管理費用 |
|
|
186,339 |
|
|
|
195,491 |
|
|
|
158,330 |
|
|
|
(9,152 |
) |
|
|
37,161 |
|
研究開発費総額 |
|
$ |
272,533 |
|
|
$ |
282,001 |
|
|
$ |
244,650 |
|
|
$ |
(9,468 |
) |
|
$ |
37,351 |
|
2022年、Tabcel支出は4060万ドルだったが、2021年と2020年はそれぞれ5010万ドルと6120万ドルだった。2022年の減少は、臨床試験とEUラベル申請と承認活動の減少によるものである。2021年の低下は,主に2020年に我々の製造工場のラベルやプロセス性能同定活動の確立に関する生産活動の増加によるものである。
2022年のATA 188、CAR T、その他のプロジェクト支出は4560万ドルだが、2021年と2020年はそれぞれ3640万ドルと2510万ドルである。2022年の増加は,ATA 188臨床試験と臨床供給コストの上昇およびIND届出のATA 3219製造活動,2021年期間に記録されたQIMRのATA 188第2段階起動マイルストーンとMSKのCAR T関連許可費および計画休止後のATA 3271によるIND有効化活動の減少によって相殺された。2021年の増加は主に研究、開発と臨床試験コストと関係があり、これらのコストは著者らのATA 188第二段階EMBOLD研究の登録と更なる推進と関係がある。
2022年の従業員と管理費は1兆863億ドルだったが、2021年と2020年はそれぞれ1兆955億ドル、1億583億ドルだった。2022年の減少は主にATOM施設の売却による補償関連コストと施設関連コストの低減であり,FDBの製造サービスコストの一部によって相殺されている。2022年は2021年に比べて賃金および関連費用が930万ドル減少し,施設関連費用が360万ドル減少し,外部サービス費用が370万ドル増加した。2021年の増加は,主に従業員数の増加による報酬に関するコスト増加と,研究開発活動の拡大を支援するための施設関連コストの増加である。2020年に比べて2021年の賃金および関連費用は1930万ドル増加し,施設関連費用は1170万ドル増加し,外部サービス費用は620万ドル増加した。
86
本報告で述べたすべての時期の研究·開発費用総額は,新冠肺炎の大流行により大きな影響を受けていない。
一般と行政費用
上記期間の一般料金および行政費用は以下のとおりである
|
|
十二月三十一日までの年度 |
|
|
(減少が)増加する |
|
||||||||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|
2022年は2021年と比較して |
|
|
2021年は2020年と比較して |
|
|||||
|
|
(単位:千) |
|
|||||||||||||||||
一般と行政費用 |
|
$ |
71,553 |
|
|
$ |
78,801 |
|
|
$ |
64,402 |
|
|
$ |
(7,248 |
) |
|
$ |
14,399 |
|
2022年の一般と行政費用は7160万ドルだが、2021年と2020年はそれぞれ7880万ドルと6440万ドルだ。2022年の低下は主に米国の売店商業活動の減少によって推進された。2021年の増加は、主にEUで予想されるEbvalloおよびTabcelの米国での商業化を支援するために、従業員数や活動を増加させ、報酬に関するコストを増加させたためである。
すべての期間の一般と行政費用総額は新冠肺炎の大流行による大きな影響を受けなかった。
流動性と資本資源
流動資金源
2012年の設立以来、私たちは主に普通株と優先株の発行、普通株を購入するための事前融資権証、バイエル許可協定、Pierre Fabre商業化協定の前払い費用とマイルストーン支払い、私たちのATOM施設の売却を通じて私たちの運営に資金を提供してきました。
過去3年間、コーエン社(Cowen)と2つの独立した販売協定を締結した:2020年2月(2020年ATM施設)と2021年11月(2021年ATM施設)。各ATM機は、総発行価格1.00億ドルの普通株を販売代理として提供または提供してくれます。我々は、ATM施設に基づいてこれらの株式を発行·売却することは、改正された1933年証券法(証券法)第415条に規定する“市場で”発行され、証券法に基づいて登録されているとみなされる。各ATM施設で販売されている普通株の総販売収入のうち、最高3.0%の手数料を支払うことができる。
2022年12月31日までの1年間に、2021年のATMメカニズムにより合計1,618,672株の普通株を売却し、平均価格は1株13.84ドルで、手数料と私たちが支払うべき他の発売費用を差し引いた総収益は2,240万ドル、純収益は2,200万ドルだった。
2022年12月31日まで、私たちは2020年のATM施設を十分に利用して、5590万ドルの普通株式の残りがあり、2021年にATM施設の下で販売することができます。
2020年12月、私たちは5,102,041株の普通株の公開発行を完成し、公開発行価格は1株24.5ドル、及び予融資権証は1株承認株式証24.4999ドルの公開発行価格で2,040,816株普通株を購入した。引受割引と手数料および支払うべき発売費用を差し引いた後、約1億643億ドルの純収益を得た。
二零二年第二四半期に、引受業者に付与された全面引受権を行使し、1株11.32ドルで普通株を公開発売することと、株式承認証1部当たり11.3199ドル公開発売価格で2,866,961株普通株を購入する予定資本権証を含む14,958,039株の公開発売を完了した。保証割引と手数料と私たちが支払うべき発売費用を差し引いた後、約1.893億ドルの純収益を得ました。
設立以来、私たちの毎年の運営は赤字と負のキャッシュフローを出しており、2022年12月にEUがEbvalloから商業化収入を生み出すことはまだ始まっていない。私たちは継続して私たちの持続的な運営に関連した巨額の研究開発やその他の費用を発生させ、予測可能な未来に損失が出ることを予想している。したがって、私たちは私たちの運営を支援するための追加の資本が必要になり、私たちは株式発行、債務融資、他の第三者融資、マーケティング、流通手配、その他の協力、戦略連合、許可手配を通じて資金を調達することができる。制限契約を含む可能性のある条件で資金を借り入れることができます
87
我々の業務、資産への留置権、高有効金利、および現金資源の削減と将来の資本市場への参入を制限する償還条項。さらに、2021年のATM施設を利用して、潜在的な商業化、協力、協力または他の戦略的配置、または上記の各項目の組み合わせによって、追加資金を得る機会を求めることを含む、追加の公共または私募株式発行または債務融資を継続する予定だ。ある程度、私たちは株式証券を発行することで追加資本を調達し、私たちの株主は深刻な希釈を経験するかもしれない。私たちが商業化、協力、または協力を通じて追加資金を調達する場合、私たちは、私たちの不利な条項にライセンスまたは他の権利を付与するために、または私たちの株主に重大な希釈をもたらす可能性のある株式を発行するために、いくつかの技術的権利またはいくつかの地域で私たちの製品をマーケティングおよび販売する権利を放棄することを要求される可能性がある。
即時需要を超えた現金は、主に流動性と保証のために、私たちの投資政策に基づいて投資されるだろう。現在、私たちの現金、現金等価物および短期投資は、通貨市場基金、米国財務省、政府機関および会社の債務、商業手形、および資産保証証券を含む銀行および信託口座に保管されている。
示された日付まで、私たちの現金、現金等価物、および短期投資残高は以下の通りです
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
現金と現金等価物 |
|
$ |
92,942 |
|
|
$ |
106,084 |
|
短期投資 |
|
|
149,877 |
|
|
|
264,984 |
|
現金、現金等価物、短期投資総額 |
|
$ |
242,819 |
|
|
$ |
371,068 |
|
契約義務と約束
キャンセルできない賃貸契約によると、カリフォルニア州千樫市にある会社本部、約51,160平方フィートのオフィススペースをレンタルした。本契約の初期期限は2026年2月に満了します。最初の期限内の契約債務総額は850万ドルだった。私たちは最初のレンタル期間の後に5年間延長することを選択することができる。
キャンセルできないレンタル契約によると、カリフォルニア州サンフランシスコ南部でオフィススペースを借りました。2021年12月に第2次改正を行い,レンタル期間を2025年5月に延長した。修正されたレンタル契約には、レンタル期間の延長のオプションは含まれていません。2022年10月、私たちは第三者とこのオフィス空間の分譲協定を締結した。分譲期間は2022年11月に開始され、2025年5月に満了し、分譲期間を延長する選択権はない。その後、私たちは会社本社をカリフォルニア州千樫市のオフィスに移した。
2019年5月、コロラド州オロラ市で約8,800平方フィートのオフィスと実験室空間レンタル契約を締結しました。本契約の初期期限は2024年4月に満了します。2021年2月、私たちはさらにこの賃貸契約を修正し、2861平方フィートの実験室空間を増加させた。レンタル期間内の契約義務は実質的ではありません。最初のレンタル期間の後、私たちはレンタル期間をさらに2つ5年間延長することを選択することができます。
2021年3月、カリフォルニア州千オーク市で約33,659平方フィートのオフィス、実験室、倉庫空間の賃貸契約を締結しました。本借約の最初の10.5年レンタル期間は2021年8月に開始され、最初のレンタル期間内の契約債務総額は2,100万ドルである。最初のレンタル期間の後、私たちはレンタル期間をさらに2つ5年間延長することを選択することができます。
2017年2月、カリフォルニア州千樫市で約90,580平方フィートのオフィス、実験室、細胞治療製造スペースを購入した賃貸契約(ATOM Lease)を締結した。本賃貸借契約の最初の15年リース期間は2018年2月に開始され、最初のレンタル期間内の契約債務総額は1,640万ドルであった。私たちは最初のレンタル期間の後に2つのレンタル期間を延長することができて、それぞれ10年と9年です。2022年4月,ATOM施設をFDBに売却する取引が完了した後,ATOMをFDBにリース譲渡した。私たちはまだATOMレンタルに関する義務に対して連帯責任を負っている。賃貸責任の更なる資料は、本報告第8項に記載されている総合財務諸表付記8-“リース”を参照されたい。財務諸表及び補足資料。
正常な業務過程において、私たちは臨床研究機関と臨床研究契約を締結し、CMOと臨床と商業材料契約を締結し、他のサプライヤーと臨床前研究と用品契約、及び運営目的のための他のサービスと製品契約を締結した。これらの契約は一般に通知期間後に便宜上契約を終了することに規定されている.我々は製品やサービスに対してキャンセル不可能な最低約束があるが,臨床研究機関やCMOと1年以上の契約を締結しなければならない。総合財務付記における付記10を参照--“負担及び又は事項”
88
プロジェクト8.本報告書の財務諸表および補足データは、私たちの契約義務および約束をさらに理解するために使用されます。
キャッシュフロー
次の表は、以下の各期間の主要な現金源と用途を詳細に説明する
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
提供された現金純額(使用): |
|
|
|
|
|
|
|
|
|
|||
経営活動 |
|
$ |
(270,430 |
) |
|
$ |
(220,522 |
) |
|
$ |
(180,759 |
) |
投資活動 |
|
|
202,956 |
|
|
|
22,258 |
|
|
|
(120,728 |
) |
融資活動 |
|
|
53,084 |
|
|
|
103,944 |
|
|
|
427,574 |
|
現金、現金等価物および制限現金純増加(マイナス) |
|
$ |
(14,390 |
) |
|
$ |
(94,320 |
) |
|
$ |
126,087 |
|
経営活動
2022年の経営活動のための現金純額は2兆704億ドルだったが、2021年は2億205億ドルだった。4,990万ドル増加したのは,主に2021年にPierre Fabreから4500万ドルの前払い費用が受信され,2022年には類似したキャッシュフローが受信されなかったためであり,残りの差は純運営資本の減少によるものである。
2021年の経営活動のための現金純額は2億205億ドルだったが、2020年は1兆808億ドルだった。3,970万ドル増加は主に純損失が3,350万ドル増加および運営資金純額が増加したが,Pierre Fabre商業化合意により受信された4,500万ドル分が相殺された。
投資活動
2022年の投資活動によって提供される現金純額には、満期および売却可能な証券が受信した2.93億ドルと、ATOM融資機構が受信した純利益9480万ドルが含まれるが、売却可能な証券の購入に使用される1.806億ドルと、財産および設備を購入する420万ドルが部分的に相殺される。
2021年の投資活動によって提供される現金純額は、主に、売却可能な証券の満期および販売から受け取った3.34億ドルを含み、売却可能な証券の購入に使用される3.011億ドルと、財産および設備を購入するための1060万ドルが部分的に相殺される。
2020年の投資活動のための現金純額は、主に売却可能な証券を購入するための4.259億ドルと財産や設備を購入するための450万ドルが含まれるが、売却可能な証券の満期と販売から受け取った3.097億ドルはこの数字を部分的に相殺した。
融資活動
2022年の融資活動で提供される現金純額には、主にATM施設の純収益2190万ドル、将来の特許使用料を売却する純収益3060万ドル、従業員株奨励取引の純収益190万ドルが含まれる。
2021年の融資活動で提供される現金純額には、主にATM施設からの純収益9870万ドルと従業員株奨励取引からの純収益680万ドルが含まれ、一部はRSU株の純決済に関する120万ドルの税金によって相殺される。
2020年の融資活動で提供される現金純額は、主に普通株と事前融資権証の2回の引受公開発売で得られた純収益の合計3.538億ドル、ATM施設からの純収益6920万ドル、および従業員株奨励取引の純収益670万ドルであり、一部はRSU株の純決済に関する150万ドルの税金で相殺されている。
89
運営資本要求及び運営計画
今まで、私たちは商業製品販売から何の収入も得ていない。私たちはいつ、あるいは商業製品の販売から十分な収入が発生して、私たちの運営費用を相殺するかどうかわかりません。予測可能な未来には、引き続き赤字が発生すると予想され、引き続き私たちの候補製品を開発し、規制部門の承認を求め、任意の承認された製品を商業化するようになり、累積赤字が増加することが予想される。私たちは新製品の開発に固有のすべてのリスクを受けて、私たちは予測できない費用、困難、複雑な状況、遅延、その他私たちの業務に悪影響を及ぼす可能性のある未知の要素に直面する可能性があります。私たちは私たちが計画した行動に資金を提供するために多くの追加資金を調達する必要があると予想する。
2022年12月31日までの既存の現金、現金等価物、短期投資に加え、2023年1月にピエール·ファブレ商業化協定下のいくつかのマイルストーンを実現するために受け取った4000万ドルが、2024年第2四半期まで私たちの計画中の運営に資金を提供するのに十分になると予想される。私たちのすべての候補製品が規制部門の承認を得る過程を完成させるために、私たちは多くの追加資金が必要になるだろう。さらに、追加的な公共または私募株式発行または債務融資を通じて、潜在的な商業化、協力、協力、または他の戦略的配置、またはこれらの組み合わせによって、日和見主義的に追加資金の獲得を求め続ける予定だ。
私たちの運営資本需要の予測は、正しくないことが証明される可能性があるという仮定に基づいており、私たちは予想よりも早くすべての利用可能な資本資源を使用するかもしれない。医薬製品の研究、開発と商業化に関連する多くのリスクと不確定性のため、私たちの運営資本需要の正確な金額を見積もることができない。私たちの未来の資金需要は多くの要素に依存するだろうが、これらに限定されない
私たちが運営から十分な現金純流入を生み出すことができるまで(私たちは決してそれをしないかもしれない)前に、私たちの長期資本需要を満たすことは、公共および私募株式および債務資本市場への参入に大きく依存し、運営によって発生した現金と私たちの現金残高が投資して稼いだ利息収入に大きく依存する。私たちは引き続き株式と債務資本市場への進出を求めて、私たちの発展努力と業務を支援する予定です。ある程度、私たちは株式証券を発行することで追加資本を調達し、私たちの株主は深刻な希釈を経験するかもしれない。私たちが商業化、協力、または協力を通じて追加資金を調達する場合、私たちは、私たちの不利な条項にライセンスまたは他の権利を付与するために、または私たちの株主に重大な希釈をもたらす可能性のある株式を発行するために、いくつかの技術的権利またはいくつかの地域で私たちの製品をマーケティングおよび販売する権利を放棄することを要求される可能性がある。
経済状況、普遍的な世界経済の不確実性、政治的変化、その他の要因により、必要に応じて追加資本を獲得するかどうか、あるいはあれば、合理的な条件で追加資本を得ることができるだろう。不安定な世界金融市場、普遍的な経済不確実性、または他の要因により、私たちはより多くの資金を集めることができなければ、私たちは私たちの1つまたは複数の候補製品の臨床前研究、臨床研究、または他の開発活動を延期、制限、減少、または中止させることを余儀なくされるだろう。
90
情報技術EM 7 Aです。市場リスクの定量的·定性的開示について
金利市場リスク
私たちは金利の変化と関連した市場リスクに直面している。2022年12月31日現在、私たちの現金、現金等価物、短期投資総額は2兆428億ドルです。私たちの市場リスクに対する主な開口は金利感受性であり、金利感受性はアメリカの金利全体の水準変化の影響を受けており、特に私たちの投資は短期証券だからです。私たちの供給可能な証券は金利リスクの影響を受け、市場金利が上昇すれば、私たちの売却可能な証券の価値は低下し、もし私たちが投資満期前に売却を余儀なくされれば、損失を招く可能性がある。私たちは現在金利リスクの開放をヘッジしていない。私たちポートフォリオの短期的な存続期間と私たちの投資の低リスク状況により、100ベーシスポイントの金利が直ちに変化することは、私たちのポートフォリオの公平な市場価値に実質的な変化をもたらすことはありません。
私たちの投資活動の主な目標は、リスクを著しく増加させることなく、投資から得られる収入を最大化することであり、保証と流動性である。この目標を達成するために、通貨市場基金、米国財務省、政府機関および会社債務債券、商業手形、および資産保証証券を含む、我々の現金等価物および様々な証券に対する短期および長期ポートフォリオを維持する。これらの証券はいずれも売却可能な証券に分類されているため、公正価値で貸借対照表に計上され、未実現収益または損失は他の全面収益(損失)を累積する単独構成要素として報告されている。米国財務省、米財務省保証証券または通貨市場基金の債務を除いて、いずれの発行者に対する証券保有量も私たちポートフォリオの5%を超えない。
91
項目8.財務状況TSと補足データ
連結財務諸表索引
|
|
ページ |
独立公認会計士事務所報告 (PCAOB ID |
|
93 |
合併貸借対照表 |
|
96 |
合併経営報告書と全面赤字 |
|
97 |
株主権益合併報告書 |
|
98 |
統合現金フロー表 |
|
99 |
連結財務諸表付記 |
|
100 |
92
“独立公認会計士報告書”アイレード会計士事務所
Atara BioTreateutics,Inc.の株主と取締役会へ
財務諸表のいくつかの見方
Atara BioTreateutics,Inc.およびその子会社(“当社”または“Atara”)の2022年12月31日と2021年12月31日までの総合貸借対照表、2022年12月31日までの3年度の関連総合経営報告書と全面赤字、株主権益と現金流量、および関連付記(総称して“財務諸表”と呼ぶ)を監査した。財務諸表は,すべての重要な面で,会社の2022年12月31日と2021年12月31日までの財務状況,および2022年12月31日までの3年度の経営結果とキャッシュフローを公平に反映しており,米国公認の会計原則に適合していると考えられる。
我々はまた、米国上場会社会計監督委員会(PCAOB)の基準に従って、会社が2022年12月31日までの財務報告の内部統制を監査し、根拠を監査した内部統制--統合フレームワーク(2013)テレデビル委員会は、組織委員会が発表した報告書と2023年2月8日の報告書を後援し、会社の財務報告書の内部統制について保留のない意見を表明した。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
重要な監査事項
以下に述べる重要な監査事項とは、財務諸表を当期監査する際に生じる、監査委員会に伝達または要求された事項であり、これらの事項は、(1)財務諸表に対して大きな意味を有する勘定または開示に関し、(2)私たちが特に挑戦的で主観的または複雑な判断に関するものである。重要監査事項の伝達は、財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、次の重要な監査事項を伝達することによって、重要な監査事項又はそれに関連する勘定又は開示について個別の意見を提供することもない。
ライセンスと連携収益と繰延収入−外部ライセンスプロトコルの会計処理−財務諸表付記2および5を参照s
重要な監査事項の説明
同社はバイエルとピエール·ファブレといくつかの外部許可協定を締結した。
2020年から2021年の間に、会社はバイエル株式会社(“バイエル”)と研究、開発と許可協定、技術移転協定、および製造と供給協定を締結した。
2022年に、当社はバイエルと終了合意を締結し、これによりすべての既存の合意を終了する。本合意の条項によれば、ATA 2271およびATA 3271に関連するすべての製品開発および商業化権利はAtaraに回復され、バイエルは、会社が有効日を終了する前に行ったいくつかの活動についてAtaraに追加料金を支払う。
93
また,2021年に当社はピエール·ファブレ医薬品社(“ピエール·ファブレ”)と商業化協定を締結した。合意条項によると,同社はPierre Fabreの商業化および流通治療のライセンスを付与し,治療の製造およびPierre Fabreへの治療の供給,および関連する細胞選択サービスを担当する。
当社は2022年にPierre Fabre商業化協定改訂協定(“PF改訂”)を締結した。PF修正案の条項によると、Ataraは、(I)Ataraが取得する資格のある特許使用料、および(Ii)Pierre Fabre購入の供給の値上げと引き換えに、追加のマイルストーン支払いを受ける権利がある。また、Ataraは、Pierre Fabre商業化プロトコルに従ってPierre Fabreにいくつかのサービスを提供する期限を延長することに同意した。
会社は、顧客が承諾した商品またはサービスの統制権を取得したとき、彼らの履行義務を満たしているため、非許可契約の収入を確認する。2022年12月31日現在、会社は対外許可協定に基づいて6,360万ドルの収入を確認し、繰延収入は8,500万ドルであり、そのうち800万ドルは流動負債に含まれ、7,700万ドルは長期負債に含まれている。
外部ライセンス契約、確認された収入、および収入として確認されるべき推定繰延収入の会計確認を重要な監査事項とします。許可外プロトコルに適用される会計文書を決定するために必要な判断、履行義務の完了の進捗を推定·測定する方法、義務履行を完了する推定契約期限を考慮すると、このような判断や推定には、許可外プロトコルの複雑さや、監査プログラムの実行やそれなどのプログラムの結果を評価する際に適用される監査者の判断力が高いため、監査には広範な監査作業が必要である。
監査で重要な監査事項をどのように処理するか
我々の監査手続きは、合意に適した会計文書を決定し、管理層の進捗を評価する方法に関するものであり、
94
計算すべき研究と開発費用および前払い研究と開発費用−財務諸表付記2参照−
重要な監査事項の説明
同社は,サプライヤーが特定のタスクを達成する進捗の評価に基づき,研究や開発費用によるコストを確認している。支払い時間はコストが料金として確認されている期間とは大きく異なるかもしれません。あらかじめ支払われた費用を前払い費用として繰延し、サービス提供時にサービス期間内に償却する。領収書が発行されていないか、または支払われていないサービス費用は、計算すべき費用であることが確認されました。
特定のタスクを達成するためのサプライヤーの進展を推定する際に、同社は、患者登録、臨床サイト活性化、またはサプライヤー情報などの実際に発生するコストのようなデータを使用する。これらのデータは,学習進捗や完了状態やサービス完了状況に関する会社員や外部サービスプロバイダの報告やその議論によって得られる.
現在行われている研究·開発活動の数および推定関連すべき計や前払い費用に係る主観性に鑑み,監査に係るべき計と前払い研究·開発費用は特に主観的判断である。
監査で重要な監査事項をどのように処理するか
私たちの計算と前払い研究開発費に関する監査プログラムには以下のものが含まれています
/s/
2023年2月8日
2013年以来、当社の監査役を務めてきました。
95
Atara生物治療会社
合併B割当書
(千単位で、1株当たりを除く)
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2022 |
|
|
2021 |
|
||
資産 |
|
|
|
|
|
|
||
流動資産: |
|
|
|
|
|
|
||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
短期投資 |
|
|
|
|
|
|
||
制限現金 |
|
|
|
|
|
|
||
売掛金 |
|
|
|
|
|
|
||
棚卸しをする |
|
|
|
|
|
|
||
その他流動資産 |
|
|
|
|
|
|
||
流動資産総額 |
|
|
|
|
|
|
||
財産と設備、純額 |
|
|
|
|
|
|
||
経営的リース資産 |
|
|
|
|
|
|
||
制限された現金-長期 |
|
|
|
|
|
|
||
その他の資産 |
|
|
|
|
|
|
||
総資産 |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
負債と株主権益 |
|
|
|
|
|
|
||
流動負債: |
|
|
|
|
|
|
||
売掛金 |
|
$ |
|
|
$ |
|
||
補償すべきである |
|
|
|
|
|
|
||
研究と開発費用を計算すべきである |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
||
その他流動負債 |
|
|
|
|
|
|
||
流動負債総額 |
|
|
|
|
|
|
||
繰延収入-長期 |
|
|
|
|
|
|
||
賃貸負債を経営しています--長期 |
|
|
|
|
|
|
||
将来の収入の販売に関する負債-長期 |
|
|
|
|
|
|
||
その他長期負債 |
|
|
|
|
|
|
||
総負債 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
||
株主権益: |
|
|
|
|
|
|
||
普通株式--$ |
|
|
|
|
|
|
||
追加実収資本 |
|
|
|
|
|
|
||
その他の総合収入を累計する |
|
|
( |
) |
|
|
( |
) |
赤字を累計する |
|
|
( |
) |
|
|
( |
) |
株主権益総額 |
|
|
|
|
|
|
||
総負債と株主権益 |
|
$ |
|
|
$ |
|
||
連結財務諸表の付記を参照。
96
Atara生物治療会社
合併報告書運営と全面赤字
(千単位で、1株当たりを除く)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
許可と協力収入 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
運営費用: |
|
|
|
|
|
|
|
|
|
|||
研究開発 |
|
|
|
|
|
|
|
|
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
総運営費 |
|
|
|
|
|
|
|
|
|
|||
運営損失 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他の収入(費用)、純額: |
|
|
|
|
|
|
|
|
|
|||
ATOM売却融資の収益(付記7参照) |
|
|
|
|
|
|
|
|
|
|||
利息とその他の収入,純額 |
|
|
|
|
|
|
|
|
|
|||
その他の収入を合計して純額 |
|
|
|
|
|
|
|
|
|
|||
所得税準備前の損失を差し引く |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
所得税支給 |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
その他総合(赤字)収益: |
|
|
|
|
|
|
|
|
|
|||
証券売却可能な未実現収益 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
総合損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
普通株は基本と希釈して純損失 |
|
$ |
( |
) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|
|||
基本と希釈加重平均流通株 |
|
|
|
|
|
|
|
|
|
|||
連結財務諸表の付記を参照。
97
Atara生物治療会社
年合併変動表株主権益
(単位:千)
|
|
|
|
|
|
|
|
|
|
|
積算 |
|
|
|
|
|
|
|
||||||
|
|
ごく普通である |
|
|
その他の内容 |
|
|
他にも |
|
|
|
|
|
合計する |
|
|||||||||
|
|
在庫品 |
|
|
支払い済み |
|
|
全面的に |
|
|
積算 |
|
|
株主の |
|
|||||||||
|
|
株 |
|
|
金額 |
|
|
資本 |
|
|
収入を損ねる |
|
|
赤字.赤字 |
|
|
権益 |
|
||||||
2020年1月1日の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||||
普通株式と事前融資権証は以下のように発行します |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
ATM施設で普通株を発行し,純額 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
事前出資の引受権証を行使する |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
RSU和解、株式差し押さえ後の純額 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
社員株奨励に応じて普通株を発行する |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
証券売却可能な未実現収益 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
2020年12月31日の残高 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||||
ATM施設で普通株を発行し,純額 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
RSU和解、株式差し押さえ後の純額 |
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
社員株奨励に応じて普通株を発行する |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
証券売却可能な未実現損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
2021年12月31日現在の残高 |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
|
||||
ATM施設で普通株を発行し,純額 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
RSU和解、株式差し押さえ後の純額 |
|
|
|
|
|
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
||
社員株奨励に応じて普通株を発行する |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
株に基づく報酬費用 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
||
純損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
証券売却可能な未実現損失 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
2022年12月31日現在の残高 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
||||
連結財務諸表の付記を参照。
98
Atara生物治療会社
合併状態キャッシュフロープロジェクト
(単位:千)
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
経営活動 |
|
|
|
|
|
|
|
|
|
|||
純損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
純損失と経営活動で使用される現金純額の調整: |
|
|
|
|
|
|
|
|
|
|||
ATOM設備の売却益 |
|
|
( |
) |
|
|
|
|
|
|
||
株に基づく報酬費用 |
|
|
|
|
|
|
|
|
|
|||
減価償却および償却費用 |
|
|
|
|
|
|
|
|
|
|||
非現金でレンタル料金を扱っております |
|
|
|
|
|
|
|
|
|
|||
投資割増償却 |
|
|
|
|
|
|
|
|
|
|||
他の非現金プロジェクト、純額 |
|
|
|
|
|
|
|
|
|
|||
経営性資産と負債変動状況: |
|
|
|
|
|
|
|
|
|
|||
売掛金 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
棚卸しをする |
|
|
( |
) |
|
|
|
|
|
|
||
その他流動資産 |
|
|
|
|
|
|
|
|
( |
) |
||
経営的リース資産 |
|
|
|
|
|
|
|
|
|
|||
その他の資産 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
売掛金 |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
補償すべきである |
|
|
( |
) |
|
|
|
|
|
|
||
研究と開発費用を計算すべきである |
|
|
|
|
|
( |
) |
|
|
|
||
その他流動負債 |
|
|
|
|
|
|
|
|
( |
) |
||
収入を繰り越す |
|
|
( |
) |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
その他長期負債 |
|
|
|
|
|
( |
) |
|
|
|
||
経営活動のための現金純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
投資活動 |
|
|
|
|
|
|
|
|
|
|||
短期投資を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
短期投資満期と売却による収益 |
|
|
|
|
|
|
|
|
|
|||
財産と設備を購入する |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
ATOM施設を売却する純収益 |
|
|
|
|
|
|
|
|
|
|||
投資活動提供の現金純額 |
|
|
|
|
|
|
|
|
( |
) |
||
融資活動 |
|
|
|
|
|
|
|
|
|
|||
普通株の年間売却と事前出資株式証明書による収益 |
|
|
|
|
|
|
|
|
|
|||
ATM施設で普通株の収益,純額を発行する |
|
|
|
|
|
|
|
|
|
|||
従業員株奨励の収益 |
|
|
|
|
|
|
|
|
|
|||
将来の収入の収益を売却し,純額 |
|
|
|
|
|
|
|
|
|
|||
限定株単位株式純決算に関する税金 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
融資リース債務の元金支払い |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
他の資金調達活動、純額 |
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
融資活動が提供する現金純額 |
|
|
|
|
|
|
|
|
|
|||
現金、現金等価物、および制限現金の増加(減少) |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
期初現金、現金等価物、および限定現金 |
|
|
|
|
|
|
|
|
|
|||
期末現金、現金等価物、および制限現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
非現金投融資活動 |
|
|
|
|
|
|
|
|
|
|||
支払すべき帳簿その他の金に含まれる財産及び設備購入 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
引受の公開発行に関する課税コスト |
|
$ |
|
|
$ |
|
|
$ |
|
|||
ATM施設に関する課税コスト |
|
$ |
|
|
$ |
|
|
$ |
|
|||
販売将来の収入に関する課税取引コスト |
|
$ |
|
|
$ |
|
|
$ |
|
|||
補充キャッシュフロー開示 |
|
|
|
|
|
|
|
|
|
|||
利子を支払う現金 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
所得税の現金を納める |
|
$ |
|
|
$ |
|
|
$ |
|
|||
連結財務諸表の付記を参照。
99
Atara生物治療会社
総合備考財務諸表
Atara BioTreateutics,Inc.(“Atara”,“We”,“Our”または“The Company”)が成立したはい
われわれはいくつかのT細胞免疫療法が臨床開発されており,次世代同種異体キメラ抗原受容体T細胞(CAR T)計画が複数行われている。私たちの最先端のT細胞免疫治療計画Tabcel®欧州委員会(EC)のマーケティング許可を得て、欧州連合(EU)で商業販売と使用が可能であり、現在米国では第3段階で開発されている。2021年10月、吾らはPierre Fabre Medicant(“Pierre Fabre”)と商業化協定(“Pierre Fabre商業化協定”)を締結し(2022年9月に改正された)、これにより、私は規制承認後、Pierre Fabreに独占的な有限領域許可を付与し、ヨーロッパおよび選定された中東、アフリカ、東欧および中央アジア(“地域”)新興市場での商業化および流通Tabcelを付与した。私たちは北米、アジア太平洋地域、ラテンアメリカのTabcelを含む他の主要市場に完全な権利を保持している。詳細は注5を参照されたい。
2020年12月、我々はバイエル株式会社(“バイエル”)と研究、開発、許可協定(“バイエル許可協定”)を締結し、この協定に基づいて、当社およびその関連会社が所有または制御するATA 2271およびATA 3271またはATA 3271に関連する適用特許および独自技術に関連する独占的、限られた分野の許可をバイエルに付与した。2021年3月に、バイエル許可協定が期待し、さらに協力を推進するために、吾らは(I)製造及び供給協定;(Ii)薬物警戒協定;(Iii)品質協定;及び(Iv)技術移転協定(総称してバイエル許可協定、製造及び供給協定及び技術移転協定と総称し、総称して“バイエル協定”と呼ぶ)を締結した。2022年5月、バイエルはベイヤ協定の終了決定を通知し、2022年8月2日にバイエルと終了、改訂、計画移転協定(“バイエル終了協定”)を締結し、ATA 2271とATA 3271に関連するすべての製品開発と商業化権利を返還し、2022年7月31日から発効した。詳細は注5を参照されたい。
我々はスローン·キャトリン癌センター(“MSK”)を記念するT−cell候補製品からの許可権,MSKおよびH.Lee Moffitt癌センター(“Moffitt”)からの我々の次世代CAR Tプロジェクトに関する権利,およびクイーンズランド医学研究所理事会(“QIMR Berghofer”)からのノウハウおよび技術の権利を有している。詳細は付記10を参照されたい。
2022年1月、我々は富士フイルムカリフォルニア生物科学技術会社(“FDB”)および(ある限られた目的について)富士フイルムホールディングス米国社と資産購入協定を締結し、カリフォルニア州千どん市に位置するAtara T細胞運営および製造施設(“ATOM施設”)の当社のすべての権利、所有権および権益を$で売却した
2022年8月、私たちは人員削減計画を発表して、私たちの従業員数を約半分に減らしました
前年度のいくつかの非実質的な金額は、本年度の列報に適合するように、合併キャッシュフロー表と連結財務諸表付記で再分類されている。
合併原則
連結財務諸表はAtaraと私たちの完全子会社の勘定を含む。すべての会社間残高と取引は合併で流された。
100
市場と地理情報を細分化する
私たちの業務を経営し管理するのは
ドルの中で
予算の使用
当社は米国公認会計原則(“米国公認会計原則”)に基づいて総合財務諸表を作成し、この基準は総合財務諸表及び付記された金額に影響を与える推定及び仮定を行うことを要求している。基本取引が完了するまで,推定と仮定の不確実性の程度は時間の長さとともに増加する.これらの財務諸表を作成する際に依存する重大な推定および仮定には、収入確認、計算すべき研究および開発費用、株式ベースの報酬費用および所得税に関する推定および仮定が含まれる。しかも、私たちは既存の市場情報を使用して私たちの短期投資の公正な価値を評価する。実際の結果はこのような推定とは大きく異なるかもしれない。実際の金額が見積もりと異なる場合は、実金額既知期間の総合経営実績に最新の状況を計上します。歴史的に見て、私たちのどの年の見積もりと実際の金額との総差額(あれば)は、私たちの連結財務諸表に実質的な影響を与えていません。
流動性リスク
設立以来、私たちは主に公共と私募株式融資および商業化、許可、協力協定の収入に依存して、私たちの運営に資金を提供する重大な運営損失が発生した。私たちが赤字を続けているため、私たちの利益への移行は、候補製品の開発成功、承認、商業化、そして私たちのコスト構造を支援するのに十分な収入を達成することにかかっている。私たちは持続的な運営現金の流入や利益を達成しないかもしれない。2022年12月31日までに、既存の現金、現金等価物、短期投資を予定しています$ごとに
信用リスクと他の不確実性の集中
現金と現金等価物を管理層に預けて高信用品質を有すると考えられる金融機関に預け、これらの機関の金額が連邦預金保険会社の保険金額を超える場合がある。我々はまた、通貨市場基金、米国財務省、政府機関、会社債務債券、商業手形、預金証書、資産保証証券に短期投資を行い、これらの証券は一定の信用リスクを受ける可能性がある。しかし、私たちはハイレベルのツールに投資し、どの発行者への開放を制限し、金融機関と発行者の持続的な信用を監視することで、リスクを緩和します。
貨幣換算
外貨建てでの取引と貨幣資産負債はそれぞれ取引日と資産負債表日ごとに現在の為替レートでドルに換算され、外貨変動の収益または損失は利息とその他の収入(費用)、総合経営報告書と全面赤字純額で確認される。2022年12月31日現在の外貨建て貨幣資産と負債実質的なものではありません
現金、現金等価物、短期投資
現金と現金等価物は高流動性投資と定義され、元の満期日は
101
原始期限よりも
私たちのポートフォリオ全体が現在の業務に利用可能であると考えられているので、私たちは、その期限が現在の貸借対照表の日付から1年を超えても、すべての投資を売却および流動資産に分類する。売却可能な証券は公正価値に従って勘定し、収益と損失を実現していないことは累計他の総合損失の中に列報し、これは合併貸借対照表中の株主権益の単独構成部分である。
証券の償却コストは割増償却と満期割引の増加に応じて調整され、いずれも利息とその他の収入(費用)に計上され、総合経営報告書と全面赤字で純額となっている。
公正価値計量
当社のいくつかの金融商品には、現金等価物、売掛金、その他の流動資産、売掛金および売掛金の帳簿価値があり、満期日が短いため公正価値に近い。短期投資は売却可能な証券からなり,公正価値に基づいて帳簿に記入される.
金融商品
適用される公認会計原則に基づいて、私たちの金融資産は公正価値によって経常的な基礎に従って計量し、以下の階層構造を用いて評価投入の優先順位を決定する
レベル1:アクティブ市場における同じ資産または負債のオファーにアクセスすることができます
第2段階:観察可能な市場の投入または観察不可能な投入に基づいて、見積もり、金利、収益率曲線などの市場データの実証を得る
レベル3:市場データで確認されていない観察不可能なデータ点の入力
私たちは四半期ごとに価値階層分類を公正に許可する。評価投入の能力の変化を観察することは公正価値階層構造中のある証券のレベルの再分類を招く可能性がある。移転による実イベントや環境変化が発生している間,流入と流出公正価値レベル内のレベルの遷移を確認した.表示されたいずれの期間においても、レベル1、レベル2、レベル3の間で遷移は発生しない。
金融資産および負債の公正な価値が、市場で観察可能な投入を使用して決定される場合、または主に観察可能な市場データから導出または観察可能な市場データの確認を得ることができ、例えば、類似した証券の定価、最近実行された取引、収益率曲線を有するキャッシュフローモデルおよび基準証券のような場合、金融資産および負債は第2のレベルとみなされる。また,二次金融商品の価値は同種の金融商品やモデルとの比較によって行われ,これらの金融商品やモデルは観察しやすい市場データを基礎としている.米国財務省、政府機関と会社の債務債券、商業手形と資産支援証券の推定値は、主に比較可能な証券の市場価格、売買オファー、金利収益率、早期返済利差を使用し、第2段階に含まれる。
金融資産および負債の公正価値が、定価モデル、キャッシュフロー方法、または同様の技術を使用して決定され、少なくとも1つの重要なモデルの仮定または投入が観察不可能である場合、レベル3とみなされる。私たちはレベル3の金融資産や負債を持っていない。
102
売掛金純額
売掛金は、作成された準備金の可変対価格推定を差し引いて記録されており、これらの可変対価格は、米国の限られた数の専門薬局と専門販売店との契約で提供される割引と、記憶容量別の使用課金の結果である。この準備金は売掛金の減少に分類される。
著者らは現在予想されている信用損失モデル或いはCECLモデルを用いて不良債権準備を推定する。CECLモードでは,不良債権準備は売掛金から予想される純額を反映している。我々は,資産の剰余コスト,リスクが小さくても損失リスク,資産契約期間内の損失,および我々が得ることができる他の関連情報からこれらのキャッシュフローの回収可能性を評価する.売掛金が回収されない可能性が高い場合には、売掛金残高と引当金が相殺される。私たちの売掛金の性質と歴史を考慮して、本報告で述べた期間まで、不良債権準備をする必要はないことを確認した。
棚卸しをする
在庫は、コストまたは推定可変動純値のうち低いものを基準に、特定確認基準に従って申告します。私たちは実際のコストを使って在庫のコストベースを決定します。在庫には原材料、製品、生産品が含まれている。1年間の予測売上高を超える製品バンク存在合併貸借対照表では非流動“他の資産”に分類される
候補品が規制承認され、このような在庫を生産する製造施設が関連規制機関の資格を得ると、コストを在庫に資本化するようになった。規制部門の承認と資格を得る前に、候補製品に関する生産コストを研究開発費として記録する。商業販売に利用可能な製品はすべて在庫に記録され、その後臨床研究に使用される場合、このような在庫コストは研究および開発費用に記録される。
私たちは定期的に在庫の回収可能性を評価し、プロジェクトが時代遅れで、欠陥があるか、あるいは予想される販売需要を超えると確定する時に在庫の帳簿価値を低下させます。超過、不良品、時代遅れの在庫品の在庫減記は販売コストとして記録されている。本報告書で述べている間、私たちの在庫は減記されていません。
財産と設備、純額
財産及び設備をコスト別に列記し、資産の推定耐用年数内に直線法で減価償却し、範囲を至れり尽くせり
長寿資産
事件や環境変化が私たちの長期資産が回収できない可能性があることを示した場合、長期資産の帳簿価値を評価します。資産の使用とその最終処分によって予想される将来のキャッシュフローが資産の帳簿価値よりも少ないと推定された場合には、減値損失が確認される。これまでに
資産廃棄債務(“ARO”)
AROはリース改善に関する長期資産廃棄に関する法的義務である。これらの負債は最初に公正価値で入金され,関連資産廃棄コストは関連資産の帳簿価値を負債と同じ金額に増加させることで資本化される。資産廃棄コストはその後、関連資産の使用年数内に減価償却される。初歩的な確認の後、同社は、時間の推移および未割引キャッシュフローの元の推定時間または金額の改訂によるARO負債の期間変化を記録した。関連債務が返済された後、会社はARO負債を再確認しないだろう。
103
賃貸借証書
私たちは契約の開始時に契約がレンタルであるかどうか、あるいはレンタルを含むかどうかを確認します。経営リース総合貸借対照表に計上されている経営リース資産、その他の流動負債および経営賃貸負債。私たちの政策はレンタル期間が12ヶ月以下の短期経営賃貸の使用権資産と賃貸負債を確認しないことであり、私たちは直線原則に従ってレンタル期間内にこれらのレンタルの短期賃貸費用を確認します。長期経営リースROU資産と長期経営リース負債を分けて示し、今後12ヶ月間対応する経営リース負債を他の流動負債に計上する。融資リースROU資産を他の資産に計上し、関連融資リース負債を他の流動負債及び他の長期負債に計上する。
リース資産およびリース負債は、発効日にレンタル期間内に将来最低賃貸支払いの現在値を確認します。レンタル期間には、開始日に行使する継続選択権を合理的に決定することが含まれています。開始日に将来の最低レンタル支払いを計算するためのレンタル条項には、継続オプションは含まれていません。私たちのほとんどのレンタルは暗黙的な金利を提供していないので、私たちは開始日に利用可能な情報に基づく増分借入金金利を使用して未来の支払いの現在値を決定します。私たちの賃貸の逓増借款金利は、賃貸期限と賃貸支払いの通貨に基づいて決定され、担保の影響に応じて調整された。レンタル資産には、レンタル報酬および生成された初期直接コストは含まれていない支払いされた任意のレンタル金額も含まれています。最低賃貸支払いの経営リース料金はレンタル期間内に直線原則で確認されます。融資リース資産は、リース期間または資産の推定耐用年数の中で短い時間で償却される。
私たちの施設と設備経営リースはレンタルと非レンタル構成要素を含み、私たちはすでに政策選択をして、レンタルと非レンタル構成要素を単一のレンタル構成要素として計算します。
吾らは他方と分譲契約を締結したり、レンタル契約を他方に譲渡したりしており、吾らはいくつかの借款の分譲者とみなされている。賃貸料収入はどの列報期間にも重要ではなく、賃貸料収入を賃貸料支出から運営費を引いたものと記す。
研究と開発コストの応計
著者らはサプライヤーが特定の任務を達成する進捗の評価に基づいて、患者登録、臨床サイト活性化などのデータ或いはサプライヤーが私たちに提供したその実際のコストに関する情報を用いて、研究開発コストを推定する計算すべき項目を記録した。これらの活動の支払いは個別契約の条項に基づいており,支払い時間はサービスを提供する期間と大きく異なる可能性がある.我々は、研究の進行または完了状態または完了したサービスに関する内部人員および外部サービスプロバイダの報告および議論によって、評価すべきカウントを決定する。私たちの各貸借対照表の日付までの課税費用の推定は、当時既知の事実と状況に基づいている。履行前に支払われた費用を前払い料金として繰延し、サービス提供中に償却する。
将来の収入を売る
ASC 470に従って将来の収入を債務売却として会計処理する範囲で、実際の金利法を用いて関連プロトコルの推定寿命内に負債を償却し、将来の収入の売却に関連する利息支出を確認する。負債および関連利息支出は、現在の合意有効期間内の予想される将来の支払いに対する私たちの推定に基づいています。内部予測と外部資源からの予測に合わせて、報告期間ごとの予想支払金額と時間を再評価し、予想される実際の金利で負債帳簿価値の利息支出を記録する。
収入確認
会計基準符号化主題606(会計基準更新(ASU)2014−09)の範囲内で決定された契約については、取引先と契約した収入そして、すべての後続の改訂(総称して“ASC 606”と呼ばれる)と、収入は、契約履行義務を履行し、顧客が約束された商品またはサービスの制御権を取得したときに確認される。確認された収入額は、これらの商品とサービスと交換するために、私たちが獲得する権利があると予想された対価格を反映している。この核心原則を実現するために,以下の5つのステップ(I)顧客との契約の決定,(Ii)契約における履行義務の決定,(Iii)取引価格の決定,(Iv)契約に取引価格を割り当てる履行義務,および(V)契約履行義務を履行する際に収入を確認する5つのステップを採用する.顧客の意図と支払い承諾に応じた対価格の能力を決定し、譲渡された商品とサービスのほぼすべての対価格を実質的に受け取ることができる場合にのみ、5段階モデルを契約に適用する。
104
契約で約束された履行義務は,顧客に移行する貨物やサービスによって決定され,これらの貨物とサービスは区別できるとともに,契約範囲内で異なる.1つの契約に複数の約束された商品およびサービスが含まれている場合、約束された商品およびサービスが契約の文脈で区別可能であるかどうかを決定するために、判断を運用する。これらの基準を満たしていなければ、約束された貨物とサービスは総合履行義務として入金されるだろう。
取引価格は、商品とサービスを顧客に移転するために、私たちが獲得する権利のある対価格によって決定されます。取引価格に可変対価格が含まれている場合には、可変対価格の性質に基づいて、期待値方法または最も可能な金額方法を用いて、取引価格に含まれるべき可変対価格金額を推定する。私たちの判断によると、契約項での累積収入が将来的に大きな逆転が起こらない可能性が高い場合、可変対価格は取引価格に含まれる。可変考慮に対する制約の影響を含む任意の推定は、どのような変化があるかどうかを決定するために、各報告期間において評価される。取引価格を決定するには重要な判断が必要であり、私たちの外許可プロトコルは付記5でより詳細な議論があります。私たちの外許可プロトコルには重要な融資部分は含まれていません。
契約が単一履行義務を含む場合、取引価格全体をその単一履行義務に割り当てる。複数の履行義務を含む契約要求は、取引価格が可変でない限り、相対的に独立した販売価格に基づいて取引価格を個々の履行義務に割り当て、かつ、履行義務または単一履行義務の一部を構成する異なるサービスに完全に割り当てられる基準に適合する。受信した対価格を相対的に独立した販売価格に応じて単独の履行義務の間に割り当てる.私たちは通常予想コストと利益率方法を使って独立販売価格を決定します。
時間が経つにつれても、ある時点でも、私たちは義務を履行するつもりだ。(I)顧客が我々の業績が提供する利益を同時に受け取り、消費する場合、(Ii)私たちの業績は、資産作成または増強時に顧客が制御する資産を創造または増強するか、または(Iii)私たちの業績は、代替エンティティが使用可能な資産を作成しておらず、これまでに完了した業績支払いを強制的に実行する権利がある場合、収入は時間の経過とともに確認される。私たちは報告期間ごとに進捗指標を評価し、必要に応じて業績指標と関連収入確認を調整する。私たちが一定期間義務を履行していない場合、ある時点で約束された貨物またはサービスの制御権を顧客に移すことによって、関連する履行義務を履行する。
2022年12月31日現在、私たちの繰延収入は、ASC 606の範囲に属するピエール·ファブレ商業化協定に関連している。付記5でさらに詳細に議論されているように、これらの手配の条項には、払い戻し不可能な前払い費用、開発、規制、および商業マイルストーン支払い、研究開発資金支払い、およびライセンス製品の純売上の印税の一部または全部について支払われる可能性がある金が含まれている。これらの支払いは約束された貨物やサービスと関連があり、私たちは基本的な履行義務を履行した後にその収入を確認するつもりだ。
知的財産権許可証:私たちの知的財産権許可が手配で決定された他の履行義務とは異なると判断された場合、許可が顧客に譲渡され、顧客が許可を使用して利益を得ることができる場合、許可に割り当てられた対価格収入を確認します。他の承諾と組み合わせたライセンスについては、合併履行義務が時間の経過とともに履行されるか、ある時点で履行されるかを決定するために、合併履行義務の性質を判断するために利用され、時間の経過とともに収入を確認するための進展を測定するための適切な方法が決定される。
前払金:前払いおよび費用は、受領または満了時に繰延収入として記録され、これらの手配の義務が履行されるまで、将来の期間に収入を確認する必要がある場合があります。
一里塚払い:開発マイルストーンの支払いを含む各手配の開始時に、マイルストーンに達する可能性を評価し、可能な金額法を用いて取引価格に含まれる金額を推定します。将来大きな収入逆転が起こらない可能性が高い場合、関連するマイルストーン価値は取引価格に含まれるだろう。そして,取引価格は相対的に独立した販売価格で個々の履行義務に割り当てられ,そのため,収入を契約下の履行義務が履行された場合に確認する.その後の報告期間ごとに終了した場合には,このような発展マイルストーンを達成する可能性や任意の制限を再評価し,必要に応じて全体の取引価格の見積りを調整する.いずれの調整も累積追跡原則で入金されており、これは許可証と連携収入および総合業務報告書と調整期間内の全面的な損失に影響を与える。
105
印税:販売ベースの特許使用料を含む手配については、販売レベルに基づくマイルストーン支払いを含み、許可が特許使用料に関連する主要項目とみなされる場合、(I)関連販売が発生した場合、または(Ii)特許権使用料の一部または全部が割り当てられた履行義務が履行されたか、または部分的に履行されたときに収入を確認する。今まで、私たちは私たちの対外許可協定によって生成されたいかなる特許権使用料収入も確認していない。
特定の判断は私たちの収入確認政策の適用に影響を及ぼすだろう。例えば、短期および長期繰延収入は、収入確認時間の最適な推定値に基づいて記録される。短期繰延収入は今後12カ月で収入として確認される予定の金額からなり、長期繰延収入は今後12カ月で確認されないと予想される金額からなる。この推定は,我々が予測した患者需要と現在の運営計画に基づいており,患者需要や我々の運営計画が将来的に変化すれば,今後12カ月以内に異なる額の繰延収入を確認する可能性がある。
研究開発費
研究開発費用には、研究開発活動を行うことによって生じるコスト、研究開発従業員の報酬および福祉、臨床および臨床前研究を行う契約研究組織および研究場所との合意に基づいて生じる費用、契約製造組織の合意に基づいて生成される臨床研究材料および他の候補製品の生産を支援する物品の取得および製造に関する費用、許可および研究開発協定に基づいて支払われる費用、他の外部サービスおよびコンサルティングコスト、ならびに施設、情報技術および管理費用が含まれる。研究·開発コストは発生時に費用を計上する。
株に基づく報酬費用
我々は、発行された株式ツールの公正価値に基づいて、制限的普通株奨励(“RSA”)、制限株式単位(“RSU”)を付与する支出、および普通株式で決済可能な株式オプションを含む株式ベースの報酬支出を計上する。公正価値は計量日付によって決定され、この日付は一般的に授与日である。私たちのRSUの公正価値は私たちの普通株の計量日の終値によって計量されます。私たちの株式オプション報酬の公正価値は授与日にブラック·スコアーズ推定値モデルを用いて決定された。
付与された株式オプションの公正価値を決定する際には、ブラック·スコアーズ推定値モデルを用いて、
所期期限我々は、過去の情報が限られているので、将来の行使パターンおよび帰属後の雇用終了行動の期待を発展させることができないので、“簡略化”方法を使用して予想期限(期待期限がオプションの帰属時間および契約期間の平均値として決定される)を得る。
予想変動率2021年までに、予想変動率は、同様の条項の比較可能な上場企業変動率を使用して推定される。変動性は2021年からAtaraの履歴変動率と類似条項で上場企業の変動率の平均値を用いて推定される。
配当を期待する·我々は従来、株主に配当金を発表または支払いする計画もなく、配当金を支払う計画もなかった。したがって、予想配当収益率は
無リスク金利-リスクなし金利は、米国債収益率および関連報酬の期待期間に基づいています。
業績帰属基準を採用した奨励については、各報告期間終了時に業績条件に達する可能性を評価し、業績条件を満たす可能性が高い場合に株式に基づく報酬コストの確認を開始した。サービスと業績条件の制限を同時に受けた奨励については、業績条件を満たす可能性が高いまで、何の費用も確認されない。時間帰属基準を有する報酬の株式補償費用は、必要なサービス期間内に直線ベースの費用であることが確認される。業績および他の帰属基準の報酬を有する株式ベースの報酬費用は、加速階層帰属モデルの下で費用として確認される。株式奨励金が発生した時、私たちはそれらの没収を考慮するつもりだ。
106
支払い計画を確定する
私たちは合格したのがある
所得税
私たちは貸借対照法を使用して所得税を計算する。財務諸表の帳簿金額と資産および負債の課税基準との間の一時的な差によって生じる予想される将来の税務結果について、私たちは、差が逆転すると予想される制定された税率を使用して、繰延税金資産と負債を記録する。繰延税項目の純資産を最も顕在化する可能性のある額に減らす必要があれば、推定免税額を提供する。既存の証拠によると、私たちは現在、私たちの繰延税金資産が未来に利用される可能性が高いという決定を支持できない。そこで,2022年12月31日と2021年12月31日までの全額推定手当を記録した。私たちはその逆転を支持する十分な証拠があるまで、推定免税額を維持するつもりだ。
税務状況が監査期間中により持続する可能性がある場合には、不確定な税収状況に関連する税収割引が確認される。未確認の税金優遇に関する利息と罰金は所得税支給に含まれている。
総合収益(赤字)
総合収益(損失)は、企業が一定期間内に非所有者由来の取引によって発生する権益変化と定義される。我々のその他の総合収益(損失)は完全に売却可能な証券の未実現収益(損失)からなり、税額を差し引いて列記する。列報のいずれの期間においても、他の全面収益(損失)から純損失までの重大な再分類はなかった。
最近の会計公告
財務会計基準委員会(“FASB”)が最近発表した任意のASUの適用性と影響を考慮する。私たちの評価によると、ASUSは私たちの総合財務諸表への影響が最も少ないと判断されました。
1株当たりの基本純損失の計算方法は、純損失を期間中に発行された普通株と予融資権証の加重平均株式数で割ったものであり、普通株等価物は考慮しない。1株当たりの普通株式償却純損失の計算方法は、純損失を当期に発行された普通株、予備融資承認株式証と普通株等価物の加重平均で割る。行使価格は無視でき、しかも事前資本権証は完全に帰属し、行使可能であるため、予備資本権証は1株当たりの普通株の基本及び償却純損失の計算に計上される。普通株等価物は、その影響が希薄化である場合にのみ普通株1株当たりの純損失の計算に計上される。
非帰属RSU、非帰属実績ベースRSU、および業績に基づく普通株購入オプション(それぞれの期末までに既定の業績基準に達している)、普通株購入のための既存および非帰属オプション、および我々の従業員による株式購入計画(“ESPP”)に従って発行される株を含む潜在的希薄化証券は、その影響が逆薄であるため、1株当たり純損失を希釈する計算から除外されている。したがって,基本1株当たり普通株と希釈後の1株当たり純損失を計算するための分母は列報のすべての期間で同じである。
次の表は、上場日までに発行された証券に基づいて発行された潜在的な普通株を表しており、これらの株は普通株希釈純損失の計算には含まれておらず、これらの株を含めると逆希釈効果が生じるからである
|
12月31日まで |
|
|||||||||
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
帰属しないRSU |
|
|
|
|
|
|
|
|
|||
既得オプションと非既得オプション |
|
|
|
|
|
|
|
|
|||
ESPP株式購入権 |
|
|
|
|
|
|
|
|
|||
合計する |
|
|
|
|
|
|
|
|
|||
107
次の表は、各期間末までに販売可能な証券の推定公正価値と関連推定値の投入階層構造をまとめた
|
|
|
|
合計する |
|
|
合計する |
|
|
合計する |
|
|
合計する |
|
||||
|
|
|
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
推定数 |
|
||||
2022年12月31日まで: |
|
入力レベル |
|
コスト |
|
|
利得 |
|
|
損 |
|
|
公正価値 |
|
||||
|
|
|
|
(単位:千) |
|
|||||||||||||
貨幣市場基金 |
|
レベル1 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
アメリカ財務省債務 |
|
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
政府機関義務 |
|
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
会社債務義務 |
|
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
商業手形 |
|
レベル2 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
資産支援証券 |
|
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
売却可能証券総額 |
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
差し引く:現金等価物に分類される金額 |
|
|
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
( |
) |
|
短期投資の金額に分類する |
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
合計する |
|
|
合計する |
|
|
合計する |
|
|
合計する |
|
||||
|
|
|
|
償却する |
|
|
実現していない |
|
|
実現していない |
|
|
推定数 |
|
||||
2021年12月31日まで: |
|
入力レベル |
|
コスト |
|
|
利得 |
|
|
損 |
|
|
公正価値 |
|
||||
|
|
|
|
(単位:千) |
|
|||||||||||||
貨幣市場基金 |
|
レベル1 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
アメリカ財務省債務 |
|
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
政府機関義務 |
|
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
会社債務義務 |
|
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
商業手形 |
|
レベル2 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
資産支援証券 |
|
レベル2 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
売却可能証券総額 |
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
差し引く:現金等価物に分類される金額 |
|
|
|
|
( |
) |
|
|
|
|
|
|
|
|
( |
) |
||
短期投資の金額に分類する |
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
契約期日によって計算すると、私たちが証券を売ることができる分担コストと公正価値は以下の通りである
|
2022年12月31日まで |
|
|
2021年12月31日まで |
|
||||||||||
|
償却する |
|
|
推定数 |
|
|
償却する |
|
|
推定数 |
|
||||
|
コスト |
|
|
公正価値 |
|
|
コスト |
|
|
公正価値 |
|
||||
|
(単位:千) |
|
|
(単位:千) |
|
||||||||||
1年以内に満期になる |
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
1年から5年で成熟します |
|
|
|
|
|
|
|
|
|
|
|
||||
売却可能証券総額 |
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
私たちは現在と予想されている未来の世界経済と市場状況を考慮して、新冠肺炎疫病とウクライナ戦争を含み、私たちの投資が大きな影響を受けていないことを確認した。2022年12月31日現在、私たちが保有している売却可能な証券の発行者の信用状況が悪化しているという重大な事実や状況はなく、これらの証券が満期になったり、その償却コストに基づいてこれらの証券を回収したりすることを要求したり、売却しようとしていません。公正価値がその余剰コスト基礎より低いすべての証券に対して、著者らは公正価値が余剰コスト基礎より低い低下は信用と関係がなく、損失準備も記録されていないことを確定した。2022年、2022年、2021年、2020年12月31日までの年間で、私たちが投資した減価損失は確認されていません。
108
減価を確認·計量するために,実際の方便を選択し,適用すべき利息を我々の売却可能証券の公正価値と余剰コストベースから除外した。私たちは私たちの売却可能な証券に関連する計算すべき利息を他の流動資産に計上し、私たちの総合貸借対照表の短期投資とは独立している。2022年12月31日と2021年12月31日まで利息は#ドルと計算されなければなりません
また、貨幣市場基金担保の制限的な現金は公正な価値で計量された金融資産であり、公正価値レベルの下の一級金融商品である。
以下の表は、連結貸借対照表内の現金、現金等価物、および制限現金を照合し、これらの現金の合計は、合併キャッシュフロー表の同じ額の総額である
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
現金と現金等価物 |
|
$ |
|
|
$ |
|
||
制限された現金--短期 |
|
|
|
|
|
|
||
制限された現金-長期 |
|
|
|
|
|
|
||
現金総額、現金等価物、および限定現金 |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
ピエール·ファブレ商業化協定
2021年10月、私たちはPierre Fabre商業化協定を締結し、この協定に基づいて、私たちはPierre Fabreに独占的で場所の限られた許可を与え、ヨーロッパでのEbvalloの商業化と流通を許可し、規制の承認を得た後に領土内の新興市場を選択することを許可した。Ataraは北米、アジア太平洋地域、ラテンアメリカを含む他の主要市場におけるEbvalloのすべての権利を保持している。2022年9月、Pierre Fabre商業化協定の第1号改正案(“PF改正案”)を締結した。PF修正案の条項によると、欧州委員会がEBV+PTLDのためのEbvalloの使用を許可し、その後、Pierre Fabreにマーケティング許可申請(MAA)を提出した後、私たちは追加の$を得る権利がある
著者らは進行中の3期対立遺伝子臨床研究と2期多列臨床研究の結論に責任を持っている。また,ピエール·ファブレ欧州商業化協定の条項に基づき,規制機関の承認を得るためにEBV陽性リンパ増殖性疾患の治療のための何らかの他の活動の費用を担当する。Pierre Fabreは他のすべての規制承認を獲得して維持する責任があり、Ebvalloのこの領土での商業化と流通に責任があるだろう。私たちはこの協定に従って私たちが単独で開発した任意の知的財産権を持つつもりだ。
ピエール·ファブレは私たちに#ドルの現金を前払いした
私たちはピエール·ファブレと単独の製造と供給協定を締結し、2023年12月31日までの固定価格と2024年1月1日以降のコストと保証金に基づいて、私たちはピエール·ファブルのためにEbvalloを製造し、領土で使用する。私たちはピエール·ファブルのEbvalloの製造と供給を担当していますピエール·ファブルの最低コストで領土で商業化しています
109
技術ピエール·ファブレも製造責任を直接担うことを選択し、関連する製造技術を得ることができる。
双方がこの日までに関連するセル選択技術をPierre Fabreに譲渡することに同意しない限り、私たちはまた、一定期間内にセル選択サービスを自費で提供する責任がある。この期間の後、私たちがセル選択サービスを継続することに同意すれば、費用はピエール·ファブレによって完全に負担されるだろう。
私たちはPierre Fabreと共同指導委員会を設立し、合意に含まれた商業化活動について監督、意思決定、実施指導を提供した。
我々は、ASC 606に基づいてこのスケジュールを評価し、Pierre Fabre商業化プロトコルにおける約束が顧客との取引を表すと結論した。私たちの結論は、Pierre Fabre商業化協定は、ライセンスの形で知的財産権の譲渡、Ebvalloの製造と供給の潜在力を含む 少なくとも7年および技術移転の前に、少なくとも3年および技術移転の前に細胞選択サービスを提供し、合同委員会に参加する義務がある可能性がある。他のサービスがなければ、ピエール·ファブレはライセンスから利益を得ることができず、その逆も同様であるから、私たちはこのような約束は明らかではないと結論した。したがって、許可、製造と供給、電池選択、およびJSCへの参加は単一の履行義務である。
Pierre Fabre商業化協定によると
私たちは、マイルストーン金額が完全に実現の可能性に基づいているか、またはまだ稼いでいないため、残りの潜在開発および商業マイルストーン支払いを取引価格から除外する資格がある。将来の特許権使用料および販売ベースのマイルストーン支払いは、潜在的な支払いが販売ベースの対価格を表すので、取引価格には含まれない。各報告期間の終了時および不確定イベントが解決された場合や他の状況が変化した場合に取引価格を再評価し、必要に応じて取引価格の推定値を調整する。
バイエル協定
研究·開発·許可協定
2020年12月,我々は固形腫瘍を治療するための間コルチゾールガイドのCAR T細胞療法を開発するためのバイエル許可協定を締結し,これにより,我々およびその関連会社が所有または制御しているカバーまたはATA 2271およびATA 3271(“ライセンス製品”)に関する適用特許とノウハウを含む独自の領域限定許可をバイエルに付与した。
バイエル許可協定の条項によると、著者らはMSKと協力して、ATA 2271のすべての共通合意した臨床前と臨床活動を担当し、人類の第一段階の臨床研究の最初まで、それからバイエルはATA 2271の更なる開発を担当し、費用は自費である。バイエルはATA 3271の開発を担当しているが、著者らが実行するATA 3271に関連するいくつかの双方が合意した臨床前、翻訳、製造、サプライチェーン活動を除いて、各活動の費用はベイヤが負担する。バイエルは独自にライセンス製品を商業化し、費用はバイエルが負担する。
2020年12月に現金前払いを受け取りました
最初の取引価格は1ドルが含まれています
110
“技術移転協定”
2021年3月,吾らはバイエルと技術移転プロトコル(“バイエル技術譲渡プロトコル”)を締結し,ベイヤライセンスプロトコルにおけるCMCサービスの一部として開発されたATA 3271製造プロセスをバイエルに譲渡した.契約を結ぶと,われわれはバイエルに領収書を発行した
我々は、ASC 606に基づいてこのスケジュールを評価し、バイエル技術移転プロトコルにおける約束は、顧客との取引を表すと結論した。我々の結論は,バイエル技術移転プロトコルはバイエル許可プロトコルと合併し,このプロトコルの修正として計算すべきであり,バイエル技術移転プロトコルは以下の約束を含む:(I)技術移転サービスと(Ii)技術移転サービスを提供するために必要な材料を含む.ASC 606によれば、技術移転サービスおよび技術移転サービスに必要な材料供給は、互いに高度に依存しているので、互いに区別されていないと判断される。また,技術移転サービスに必要な技術移転サービスと材料供給は,バイエル許可プロトコルで決定された許可,早期開発,CMCサービスに高度に依存していると結論した.したがって、私たちはこのような約束を単一の履行義務に統合することにした。
バイエル技術移転協定によると、適切な取引価格を評価するために、私たちは確定します
我々は,コストに基づく入力法を採用し,実際に発生したコストが合併履行義務に期待される予算コスト総額に対して収入を確認する.
製造と供給協定
2021年3月、私たちはベイヤと製造と供給協定(“ベイヤ製造協定”)を締結し、この協定はベイヤ許可協定の一部とみなされ、第一段階と第二段階の間皮間甲素ガイドの異体CAR T細胞療法を生産し、ベイヤが臨床試験で使用するために、価格は私たちのコストに基づいて合理的な利益率を加え、これは私たちの独立販売価格と一致する。ベイヤ製造プロトコルによると、私たちはまた、これらのサービスに対する私たちの独立販売価格と一致する価格でベイヤにストレージと流通サービスを提供します。
ベイヤ製造協定を締結した後、バイエルは拘束力のある製造サービスと倉庫サービス調達注文を提出し、私たちは承認した。製造サービスの任意の費用は以下のように領収書を発行する:(I)
2021年3月、バイエルに領収書を発行しました
我々は、ASC 606に基づいてこのスケジュールを評価し、製造および供給プロトコルにおける約束が顧客との取引を表すと結論した。私たちの結論は、バイエル製造協定は、(I)製造サービス、(Ii)月ごとに提供される倉庫サービス、および(Iii)流通サービスを含む。ASC 606,吾らによれば、技術移転完了前に提供されることが予想される6つの初期調達注文の製造サービスは、ベイヤ許可プロトコルおよびベイヤ技術譲渡プロトコルの下で開発および譲渡されている製造プロセスに高度に依存しているので、ASC 606,吾らは決定している。したがって、私たちはこのような約束を単一の履行義務に統合することにした。私たちはまた他のすべてのサービスが違うし、個別的に義務を履行していると確信する。私たちは、6ロットの貨物を製造と供給する初期拘束性注文はベイヤ許可協定と合併し、バイエル技術移転協定と共にこの合意の修正を計上すべきであることを決定した。また,バイエルからの拘束力のある調達注文はバイエル製造プロトコルとともに製造サービスと倉庫サービスの契約を構成し,バイエルからの出荷注文は流通サービスの契約を構成していると結論した.また、月ごとに提供されるストレージサービスと配信サービスが異なることと、個別の履行義務があることを確認します。上記のすべての履行義務はその独立価格に基づいて価格設定される。
ベイヤ製造協定によると、適切な取引価格を評価するために、私たちは確定します
111
性能バイエル許可協定によって決定された義務。最初の6ロットの製造サービスの収入は、実際に発生したコストが合併履行義務に対して予想される予算コスト総額に基づいて確認される。これらのサービスの提供にともない,ストレージサービスの収入は時間の経過とともに確認される.流通サービスの収入は,バイエルが指定した臨床地点に製品が渡されたときに確認される。
ベイヤ協定終了と収入確認
2022年5月、バイエルはバイエル協定の終了決定を通知し、2022年8月2日、私たちはバイエル終了協定を締結し、発効日は2022年7月31日です。終了発効日からATA 2271とATA 3271に関連する全製品開発権がAtaraに返還される。有効日を終了する前にAtaraが行ったいくつかの活動への見返りとして、バイエルはAtaraに#ドルを支払った
2022年12月、我々はデラウェア州有限組合企業HCR Molag Fund,L.P.(“HCRx”)と購入契約(“HCRx協定”)を締結した。#ドルの支払いで
HCRxプロトコルによると,HCRxはEbvalloの領土内での純売上(Pierre Fabre商業化プロトコルの定義)について階層的特許権使用料を獲得する権利があり,金額は1桁の中央桁から2桁まで様々である年間純売上高をベースにしています。HCRxはまたPierre FabreからAtaraにいくつかの記念碑的な支払いを支払う権利がある。HCRxへの特許使用料とマイルストーン総額の上限は
投資額$の総収益
記録された負債の償却を決定するためには、HCRxが受け取る将来の支払い総額を推定する必要がある。これらの金額の合計から$を差し引く
次の表は、2022年12月31日までの年間販売“HCRx協定”における将来の収入に関する負債の変化を示しています
|
|
12月31日までの年度 |
|
|
|
|
2022 |
|
|
|
|
(単位:千) |
|
|
将来の収入·期初残高の売却に関する負債 |
|
$ |
|
|
将来の収入の収益を売却し,純額 |
|
|
|
|
減算:債務発行コスト |
|
|
( |
) |
将来の収入·期末残高に関する負債の売却 |
|
$ |
|
|
112
2022年4月4日、ATOM施設のFDBへの売却取引が完了し、純収益は#ドルです
(単位:千) |
|
|
|
|
ATOM施設を売却する純収益 |
|
$ |
|
|
|
|
|
|
|
売却された資産: |
|
|
|
|
その他流動資産 |
|
$ |
|
|
財産と設備、純額 |
|
|
|
|
その他の資産 |
|
|
|
|
差し引く:売却資産 |
|
|
|
|
ATOM設備の売却益 |
|
$ |
|
|
販売に関連して、FDBと移行サービス協定(“TSA”)を締結し、この合意に基づいて、情報技術、金融、技術運営を含むが、これらに限定されないいくつかの機能の移行に協力している。FDBは,我々のTSAに関するすべての第三者費用と,我々の従業員がTSAが規定する要求を満たすためにかかる時間をコストごとに補償する.精算は関連運営費の減少額と記載されており、従業員時間精算に関する金額は2022年12月31日までの年度内では重要ではない。FDBは2022年12月31日現在、TSAに基づいて当社に借りているいかなる金額も他の流動資産に含まれています。
キャンセルできないレンタル契約によると、カリフォルニア州サンフランシスコ南部でオフィススペースを借りました。2021年12月、私たちは大家さんと2つ目の修正案を締結し、レンタル期間を延長しましたそれは.修正されたレンタル契約には、レンタル期間の延長のオプションは含まれていません。修正された賃貸契約について、私たちは$の金額の信用状を保留する必要があります
2021年3月に新しい賃貸契約を締結しました
二零一七年二月、私たちは賃貸契約(“ATOMレンタル”)を予約して、レンタル期間は約
私たちは、組み込み賃貸を決定するためのサプライヤー契約を評価し、Ataraが資産使用の実質的にすべての経済的利益を得る権利があり、資産の使用を指導することができるので、Fujifilm MSAがASC 842リースを構成するプロジェクトを含むと判断した。私たちの結論は、Fujifilm MSAは、Atara製品を製造するためのいくつかの専用加工チャンバの組み込み運営リースと、Ataraの使用専用の特定のアイスボックスの組み込み融資リースとを含む。Fujifilm MSAには加工室と冷蔵室を支払い支払いする契約義務が含まれており、各期間は#年である
113
12月31日現在、私たちの経営リースと融資リース項目の賃貸負債の満期日2022年の数字は以下の通り
|
賃貸借契約を経営する |
|
融資リース |
|
||||
12月31日までの年度 |
|
(単位:千) |
|
|||||
2023 |
|
$ |
|
|
$ |
|
||
2024 |
|
|
|
|
|
|
||
2025 |
|
|
|
|
|
|
||
2026 |
|
|
|
|
|
|
||
2027 |
|
|
|
|
|
|
||
その後… |
|
|
|
|
|
|
||
賃貸支払総額 |
|
$ |
|
|
$ |
|
||
差し引く:利息を表す額 |
|
|
( |
) |
|
|
( |
) |
賃貸負債現在価値 |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
2022年12月31日現在の残高 |
|
|
|
|
|
|
||
その他流動負債 |
|
$ |
|
|
$ |
|
||
賃貸負債を経営しています--長期 |
|
|
|
|
|
— |
|
|
その他長期負債 |
|
|
— |
|
|
|
|
|
合計する |
|
$ |
|
|
$ |
|
||
レンタル料の構成は以下のとおりである
|
|
現在までの年度 |
|
|
現在までの年度 |
|
|
現在までの年度 |
|
|||
|
|
2022年12月31日 |
|
|
2021年12月31日 |
|
|
2020年12月31日 |
|
|||
|
|
(単位:千) |
|
|||||||||
経営リースコスト: |
|
|
|
|
|
|
|
|
|
|||
リースコストを経営する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
短期賃貸コスト |
|
|
|
|
|
|
|
|
|
|||
リース総コストを経営する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
融資リースコスト: |
|
|
|
|
|
|
|
|
|
|||
費用を償却する |
|
$ |
|
|
$ |
|
|
$ |
|
|||
賃貸負債利息 |
|
|
|
|
|
|
|
|
|
|||
融資リース総コスト |
|
$ |
|
|
$ |
|
|
$ |
|
|||
賃貸契約に関するその他の資料は以下の通り
|
|
現在までの年度 |
|
|
現在までの年度 |
|
|
現在までの年度 |
|
|||
|
|
2022年12月31日 |
|
|
2021年12月31日 |
|
|
2020年12月31日 |
|
|||
|
|
(単位:千、レンタル期間と割引率を除く) |
|
|||||||||
キャッシュフロー情報を補完する |
|
|
|
|
|
|
|
|
|
|||
計量に含まれる金額のための現金 |
|
|
|
|
|
|
|
|
|
|||
レンタル経営キャッシュフロー |
|
$ |
|
|
$ |
|
|
$ |
|
|||
融資リースの運営キャッシュフロー |
|
|
|
|
|
|
|
|
|
|||
融資リースのキャッシュフロー融資 |
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||
レンタル義務と引き換えに経営的リース資産: |
|
$ |
|
|
$ |
|
|
$ |
|
|||
賃貸義務と引き換えの融資リース資産: |
|
|
|
|
|
|
|
|
|
|||
賃貸資産を経営する非現金が増加した理由は |
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||
加重平均残余レンタル期間 |
|
|
|
|
|
|
|
|
|
|||
賃貸借契約を経営する |
|
|
|
|
|
|
||||||
融資リース |
|
|
|
|
|
|
||||||
加重平均割引率 |
|
|
|
|
|
|
|
|
|
|||
賃貸借契約を経営する |
|
|
% |
|
|
% |
|
|
% |
|||
融資リース |
|
|
% |
|
|
% |
|
|
% |
|||
114
資産廃棄義務
我々の資産廃棄義務(“ARO”)には,カリフォルニア州千樫市にあるATOM施設のテナント改善施設を撤去し,施設を賃貸契約に規定されている条件に戻す契約要件が含まれている。我々は2022年4月にATOM施設をFDBに売却する取引が完了した後,ATOMをFDBにリース譲渡したが,大家から更新を受けていない。そのため,ATOM施設に関連するAROは我々の貸借対照表に残っている.ARO負債の公正価値を他の長期負債に計上し、ARO資産を発生期間の長期資産に計上する。ARO資産の公正な価値は賃貸期間内に償却される。我々のAROの公正価値は,我々の信用調整無リスク金利を用いて関連資産の推定寿命内の予想キャッシュフローを割引することで推定される.AROの資産と負債は2022年12月31日と2021年12月31日まで実質的ではない。
2022年8月8日に戦略的リストラ計画を発表しました
私たちは部隊の削減に関する以下の再構成費用を記録した
|
|
十二月三十一日までの年度 |
|
|
|
|
2022 |
|
|
|
|
(単位:千) |
|
|
|
|
|
|
|
研究開発費 |
|
$ |
|
|
一般と行政費用 |
|
|
|
|
再編成費用総額 |
|
$ |
|
|
以下の再編負債活動は、2022年12月31日終了年度の有効債務減少に関連して、すべてのドル
|
|
再編成費用総額 |
|
||
|
|
(単位:千) |
|
||
負債残高、2022年1月1日 |
|
$ |
|
|
|
料金を取る |
|
|
|
|
|
現金払い |
|
|
|
( |
) |
負債残高、2022年12月31日 |
|
$ |
|
|
|
MSKプロトコル
2015年6月,MSKと3種類の臨床段階T細胞療法の独占ライセンス契約を締結した。私たちは、特定の規制と販売関連マイルストーンの完成状況に応じてMSKに支払い、将来の開発許可製品候補製品(ある場合)の製品販売状況に応じてMSKに1桁の中央値パーセントの階層印税を支払うことを要求された。また、場合によっては、MSKに一定の最低年間印税を支払うことを要求され、これらの費用は、同じ年の期間に支払われた当然の印税を免除することができる。私たちはまた、再許可によって許可された権利のために私たちが受け取った任意の代価の低い2人の数百分を支払うことを要求された。ライセンス契約は、製品及び国/地域の最終期限で満了する:(I)各ライセンス製品に関連する最終ライセンス特許権の満了、(Ii)ライセンス製品毎に法的に付与された任意の市場排他期間の満了、及び(Iii)各国·地域における初の商業販売ライセンス製品後の指定年数。許可協定が満了した後、Ataraは許可製品に対する非独占的な権利を保持するだろう。
2018年5月と12月、私たちはMSKから追加の技術許可を得た。特定の開発、法規、販売に関するマイルストーンの達成に応じて追加のマイルストーン支払いと、将来の開発許可製品候補製品による製品販売(ある場合)に応じて中間桁数百点の等級別特許使用料を支払う義務があります。
115
2021年3月、MSKとのライセンス契約を修正し、再確認し、いくつかの権利の許可を終了し、既存のプロトコルではカバーされていない他の独自技術的権利を許可しました。
QIMR Berghoferプロトコル
2015年10月、QIMR Berghoferと独占ライセンス契約と研究開発協力協定を締結した。ライセンス契約の条項により,我々は独自の世界的許可を得て,QIMR Berghoferが開発した技術とノウハウを用いて同種異体T細胞治療プロジェクトを開発·商業化した。2016年9月19日、独占ライセンス協定と研究開発協力協定の改正と再記述が行われた。改訂·再記述されたプロトコルによると、我々は、追加T−cell計画を開発·商業化する独自のグローバルライセンスと、2018年6月に行使したライセンス追加技術の選択権とを取得した。2019年8月、私たちは特定の権利に対する私たちの許可を終了するために、QIMR Berghoferとの許可協定と研究開発協力協定をさらに修正して再確認しました。我々の現在のライセンス契約では,将来の製品販売(あれば)に応じてQIMR Berghoferに様々なマイルストーンや特許権使用料を支払うことも規定されている。我々の現在の研究開発協力協定の条項によると,協力して開発したプロジェクトに関する合意開発活動のコストを返済する必要がある.これらの支払いは関連開発期間中に直線的に支出される。この協定はまた、ある発展と規制マイルストーンの成果に基づいて、QIMR Berghoferに様々な記念碑的なお金を支払うことを規定している。
その他ライセンス内プロトコルと連携プロトコル
私たちは時々他の当事者たちと他の許可と協力協定を締結する。例えば,我々は2018年8月にモフェットがんセンターから次世代CAR Tプロジェクトに関する権利を獲得し,これらの許可に関する協賛研究による協力に同意した。2018年12月には,米国国立衛生研究院からMSKと協力した次世代CAR Tプロジェクトに関する権利も獲得した。
上記の各合意下のマイルストーンと特許権使用料は、将来の事件に応じて決定され、基本的なマイルストーンを実現したり、特許使用料を稼いだりする際に費用と表記されます。2022年と2021年12月31日までに
CRL製造プロトコル
2019年12月、2021年3月にCharles River実験室社(“CRL”)に買収されたCognate BioServices,Inc.と商業製造サービス契約(“CRL MSA”)を締結した。
CRL MSAにより、CRLは私たちの製品といくつかの候補製品に製造サービスを提供します。2023年2月、CRL MSAを修正し、期限を2023年9月30日の早い時期に延長するか、またはいくつかのロットの製品および候補製品を受信します。
富士フィルムメインサービスと供給契約
2022年1月に富士フイルムMSAに加入し、2022年4月4日にATOM施設の売却完了後に発効し、延長可能となった
他の研究、開発、製造協定
著者らは正常な業務過程中に臨床研究機関と臨床試験契約を締結し、CMOと臨床用品契約を締結し、そして他のサプライヤーと臨床前研究、用品とその他のサービス契約を締結し、著者らの運営目的を満たす可能性がある。これらの契約は一般的に通知後に契約を終了することに規定されている。2022年12月31日と2021年12月31日までに
116
最低の約束
2022年12月31日まで、臨床研究組織とCMOと締結された期限が1年を超える協定を除いて、製品とサービスのキャンセル不可の最低約束は以下の通りである
例年 |
|
2022年12月31日までの残り最低約束 |
|
|
|
|
(単位:千) |
|
|
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
2027 |
|
|
|
|
合計する |
|
$ |
|
|
私たちはすでに$を使いました
2022年12月31日までに
賠償協定
正常な業務過程において、私たちは様々な陳述と保証が含まれている契約と協定を締結し、特定の責任を賠償することを規定している。これらの合意の下でのリスクは未知であり、未来に私たちに提起される可能性があるが、まだ提起されていないクレームに関連しているからだ。今まで、私たちは何のクレームも支払わなかったし、私たちの賠償義務に関連したいかなる訴訟も弁護されなかった。しかし、このような賠償義務のため、私たちは未来に費用を記録するかもしれない。私たちの役員や幹部が私たちの要求に応じてこのような身分でサービスする場合、特定の事件や事件について賠償義務を提供する義務もありますが、一定の制限があります。今まで、誰もクレームを出していません。私たちはこのような賠償協定の公平な価値が最も低いと思います。そこで私たちは
事件があったり
私たちは時々法的手続きと、私たちの正常な業務過程または他の側面で発生した要求、クレーム、そして脅威訴訟に巻き込まれるかもしれない。いかなる訴訟の最終結果も不確定であり、不利な結果は私たちの運営結果や財務状況に悪影響を及ぼす可能性がある。結果にかかわらず,弁護コスト,管理資源分流などの要因により,訴訟は我々に悪影響を与える可能性がある.私たちは現在、実質的な法的手続きに参加していない。
私たちの法定株式は
株式発行
2019年7月に販売を受けた公開発行の一部として、私たちは購入のために事前融資権証を発行して販売しました
すべての前払い援助権証は所有者に購入権を持たせます
117
2020年第2四半期に発行され販売されました
2020年12月に発行され販売されました
2020年に公開発行される一部の発行·売却の予融資権証の条項は、2019年の発行·販売と類似している。2022年12月31日現在、2020年の引受販売として一部発行·販売されているすべての予融資権証は返済されていない。
ATM施設
過去3年間、コーエン社(Cowen)と2つの独立した販売協定を締結した:2020年2月(“2020年ATM施設”)、2021年11月(“2021年ATM施設”)。各ATMサービスは、当社の普通株を販売する株式を提供または提供し、その総発行価格は最高$に達する
2021年12月31日までの事業年度では、全部で販売しております
2022年12月31日までの年間で、全部で販売しました
2022年12月31日まで2020年のATM施設を活用して$を持っています
持分激励計画
2014年3月、2014年10月15日に初公募株(IPO)定価に基づいて改訂·再記述された2014年株式インセンティブ計画(“2014 EIP”)を採択した。
2014年EIPでは、2015年から2024年までの財政年度ごとの最初の営業日に、発行可能株式数が毎年増加し、相当すると規定されています
2014年のEIPの条項によると、従業員、取締役、コンサルタント、および他のサービスプロバイダに株式オプション、RSA、およびRSUを付与することができます。RSUは通常付与されます
2018年2月には、新入社員にオプション、株式付加価値権、RSA、RSUを付与することができる2018年インセンティブ計画(“インセンティブ計画”)を採択しました。2020年11月、2021年9月、2022年6月、私たちはインセンティブ計画を修正し、追加的な
118
2020年、私たちは私たちのある従業員に業績に基づく奨励を授与し、私たちの臨床計画に関連する特定の会社の業績基準を達成すれば、普通株を発行できることを規定した。最終的に授与された業績に基づく奨励の数量は、いつ、どのような業績基準を達成したか、及び従業員が2014年の企業資源計画で定義した持続サービス年限に依存し、授与日までである。ライセンス契約の規定の期限までに達成された業績基準は一つもなく、これらの基準はその後没収される。
株式オプションを付与した場合,行権価格は下回らない
2022年には、私たちの従業員の一部に業績に基づく株式オプションを付与し、業務発展計画に関する特定の会社の業績基準を達成すれば、株式オプションを発行して普通株を購入できることを規定しています。最終的に付与された業績ベースの奨励の数は、業績基準が指定されたスケジュール内で達成されたかどうか、および従業員が2014年の企業資源計画で定義された継続サービス時間が授与される日までに決定される。2022年12月31日現在、業績基準が達成されていないことは一つもなく、奨励されていない金額は重要ではない。
2022年12月31日までに
限定株単位
以下は、2014年のEIPと誘導計画におけるRSU活動の概要である
|
|
RSU |
|
|||||
|
|
株 |
|
|
重みをつける |
|
||
2021年12月31日現在の残高 |
|
|
|
|
$ |
|
||
授与する |
|
|
|
|
$ |
|
||
没収される |
|
|
( |
) |
|
$ |
|
|
既得 |
|
|
( |
) |
|
$ |
|
|
2022年12月31日現在の残高 |
|
|
|
|
$ |
|
||
2022年,2021年および2020年12月31日までに年度内にロットされた買い戻し単位の加重平均払出日の公正価値は
われわれのRSU和解手続きによると、従業員に付与されたいくつかのRSUについては、和解時に株式を差し押さえて、推定された賃金源泉徴収義務を支払う。2022年の間和解しました
119
株式オプション
以下は、2014年のEIPとインセンティブ計画における株式オプション活動の概要です
|
|
株 |
|
|
重みをつける 平均行権価格 |
|
|
加重平均 |
|
|
骨材 |
|
||||
2021年12月31日現在の残高 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
授与する |
|
|
|
|
|
|
|
|
|
|
|
|
||||
鍛えられた |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
没収または期限切れ |
|
|
( |
) |
|
|
|
|
|
|
|
|
|
|||
2022年12月31日現在の残高 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
帰属しており予想されています |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
2022年12月31日から行使可能 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
総内在価値は、2022年12月31日の終値と発行された現金オプションの行権価格との差額を表しています。2022年12月31日まで一ドルあります
オプション:
発行済みオプションごとの公正価値は,付与された日にBlack-Scholes推定モデルを用いて推定される.次の表は、Black-Scholesモデルの入力として使用される加重平均仮定と、それによって生成された加重平均付与日が示された期間に付与された株式オプションの公正価値とをまとめる
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
仮定: |
|
|
|
|
|
|
|
|
|
|||
所期期間(年) |
|
|
|
|
|
|
|
|
|
|||
予想変動率 |
|
|
% |
|
|
% |
|
|
% |
|||
無リスク金利 |
|
|
% |
|
|
% |
|
|
% |
|||
期待配当収益率 |
|
|
% |
|
|
% |
|
|
% |
|||
公正価値: |
|
|
|
|
|
|
|
|
|
|||
加重平均推定授出日1株当たり公正価値 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
付与したオプション |
|
|
|
|
|
|
|
|
|
|||
総推定付与日公正価値 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
2022年,2021年および2020年12月31日まで年度内に帰属する株式オプションの推定公正価値はい$です
従業員株購入計画
2014年5月、2014年10月15日にIPO定価で発効する2014年従業員株購入計画(“2014 ESPP”)を採択した。2014年にESPPは、条件を満たす従業員が所定の提供期間中に賃金控除により普通株を割引で購入することを許可した。条件に合った従業員は以下のように会社普通株を購入することができます
2022年12月31日まで一ドルあります
120
2014年ESPPは、2015年から2024年までの財政年度ごとの第1営業日は、この計画に基づいて発行可能な株式数が毎年増加し、(I)に相当すると規定している
保留株
12月31日まで、私たちの株式激励計画によると、以下の普通株は未来の発行のために予約します2022:
|
|
保留株式総数 |
|
|
2014持分インセンティブ計画 |
|
|
|
|
2018年インセンティブ計画 |
|
|
|
|
2014年度従業員株購入計画 |
|
|
|
|
普通株予約株式合計 |
|
|
|
|
株に基づく報酬費用
すべての株式奨励に関する株式報酬支出総額は以下のとおりである
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
研究開発 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
一般と行政 |
|
|
|
|
|
|
|
|
|
|||
株式に基づく報酬総支出 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
列報の期間ごとに、所得税準備前の損失を差し引くと以下のようになる
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
アメリカです |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
外国.外国 |
|
|
|
|
|
|
|
|
|
|||
所得税未払い準備前の総損失 |
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
( |
) |
各期間の所得税準備金は以下のように構成される
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
|
|
(単位:千) |
|
|||||||||
現行の所得税引当金: |
|
|
|
|
|
|
|
|
|
|||
状態.状態 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
外国.外国 |
|
|
|
|
|
|
|
|
|
|||
所得税当期準備金総額 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
法定税率と実際の税率の入金状況は以下のとおりである
|
|
十二月三十一日までの年度 |
|
|||||||||
|
|
2022 |
|
|
2021 |
|
|
2020 |
|
|||
法定税率で徴収される連邦所得税 |
|
|
% |
|
|
% |
|
|
% |
|||
税収控除を検討する |
|
|
% |
|
|
|
|
|
|
|||
株に基づく報酬 |
|
|
( |
%) |
|
|
( |
%) |
|
|
( |
%) |
他にも |
|
|
( |
%) |
|
|
( |
%) |
|
|
% |
|
評価免除額を変更する |
|
|
( |
%) |
|
|
( |
%) |
|
|
( |
%) |
実際の税率 |
|
|
% |
|
|
% |
|
|
% |
|||
121
繰延税項資産及び負債は、(A)財務報告用途の資産及び負債額面と所得税用途に用いられる金額との間の一時的な差額及び(B)営業損失及び税項相殺繰越による税項純影響を反映する
|
|
12月31日まで |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
繰延税金資産: |
|
|
|
|
|
|
||
純営業損失が繰り越す |
|
$ |
|
|
$ |
|
||
資本化研究費 |
|
|
|
|
|
|
||
税金の繰り越しを免除する |
|
|
|
|
|
|
||
株に基づく報酬 |
|
|
|
|
|
|
||
収入を繰り越す |
|
|
|
|
|
|
||
リース負債を経営する |
|
|
|
|
|
|
||
許可証料 |
|
|
|
|
|
|
||
他にも |
|
|
|
|
|
|
||
繰延税金資産総額 |
|
|
|
|
|
|
||
推定免税額 |
|
|
( |
) |
|
|
( |
) |
繰延税金資産総額 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
繰延税金負債: |
|
|
|
|
|
|
||
経営的リース資産 |
|
|
( |
) |
|
|
( |
) |
繰延税金負債総額 |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
||
繰延税項目純資産(負債) |
|
$ |
|
|
$ |
|
||
2022年1月1日から、“減税·雇用法案”(以下、“税法”と略す)は、今年度の研究開発支出を差し引く選択肢を廃止し、納税者に米国国税法(IRC)第174条の規定に基づいてこれらの支出を資本化するよう要求した。資本化された支出は1年以内に償却される
私たちの税金の繰越は増加しました
繰延税金資産の現金化能力を決定する際には、定期的にプラスとマイナスの証拠を評価する。既存の証拠の重みによると、我々の歴史的経営業績と成立以来報告された累積純損失を含めて、2022年12月31日と2021年12月31日までの繰延税項純資産に対して全額推定値を維持している。私たちは繰延税金資産の全額評価支出を維持し、評価取り消しを支持する十分な肯定的な証拠があるまで維持するつもりだ。推定免税額は#ドル増加した
2022年8月には、“チップと科学法案”(“チップ法案”と略す)と“インフレ低減法案”(略称“インフレ低減法案”)が可決され、どちらの法案も我々の財務諸表に実質的な影響を与えないと予想される。また、2022年1月1日から、カリフォルニア州の純営業損失控除と商業信用の臨時限度額が回復した。
“米国救援計画法案”は2021年3月11日に署名されて法律となった。ARAは我々の財務諸表に実質的な影響を与えないと予想されるが,ARAが2027年に発効したIRC第162(M)条に変化をもたらす可能性があることを考慮して,監視·評価を継続する。
税法によると、2018年1月1日以降に開始された納税年度から発生する連邦純営業損失は無期限に繰り越すことができるが、このような連邦純営業損失の利用は今後数年間の課税所得額の80%を超えてはならない。公布以来、米国国税局と財務省は、税法に関連するいくつかのテーマを明確にする指導意見を含む最終的かつ提案された法規を発表してきた。すべての州が税法に適合しているわけではないし、他の州の税法に対するコンプライアンスがそれぞれ異なるわけではない。
122
2022年12月31日現在、連邦所得税の目的で、我々の純営業損失は約$に繰り越されています
改正されたIRC第382条によると、会社に“所有権変更”が発生した場合、純営業損失や税収控除繰越の使用には大きな制限がある。一般に、第382条“所有権変更”とは、所定のテスト期間内に、1つまたは複数の株主またはグループ株主が1社の少なくとも5%の株式を保有していれば、その所有権がその最低持株割合より50ポイント以上増加すると、所有権変更が発生することを意味する。似たような規則は州税法に適用されるかもしれない。したがって,純営業損失や税収控除を利用する能力は,このような所有権の変化によって制限される可能性があり,この制限により繰越が使用前に満期になる可能性がある。
我々は2022年12月31日までの株式取引の第382節の研究を完了した。この研究の結論は,成立以来所有権変更を経験しており,繰越営業損失純額の利用が年間制限されることである。しかし、年間制限は、使用前に税務属性の繰越を招くことはないと予想される。
12月31日終了年度に税収優遇総額(利息や罰金を含まない)残高の変化が確認されなかった2020年、2021年、2022年は以下の通り
|
(単位:千) |
|
|
2020年1月1日の残高 |
$ |
|
|
今年度に関連する税務職毛数が増加する |
|
|
|
前年度に関連する税務職毛数が増加した |
|
|
|
前年度に関連した税務職毛数の減少 |
|
( |
) |
2020年12月31日の残高 |
|
|
|
今年度に関連する税務職毛数が増加する |
|
|
|
前年度に関連する税務職毛数が増加した |
|
|
|
前年度に関連した税務職毛数の減少 |
|
|
|
2021年12月31日現在の残高 |
|
|
|
今年度に関連する税務職毛数が増加する |
|
|
|
前年度に関連する税務職毛数が増加した |
|
|
|
前年度に関連した税務職毛数の減少 |
|
( |
) |
2022年12月31日現在の残高 |
$ |
|
|
私たちは現在、米国の繰延税純資産に全額推定手当を持っており、今後いかなる不確定な税収状況が有利に解決されれば、有効税率優遇の時間に影響を与えるだろう。未確認の税金優遇を廃止することは私たちの実際の税率に影響を与えません。私たちが引き続き私たちの繰延税金資産に対して全額評価準備を維持すれば。
私たちの政策は、不確定な税金状況に関連する利息と罰金を所得税条項の構成要素とすることである。私たちは
私たちの重要な管轄区域はアメリカ連邦管轄区とカリフォルニア州管轄区です。私たちのすべての納税年度はまだアメリカ連邦とカリフォルニア税務当局の審査を受けることができます。私たちはまた私たちが経営している他の州、地方、外国司法管轄区に書類を提出して、このような納税年度は依然として審査することができます。
2022年12月31日現在、私たちはその海外収益について恒久的な再投資を行っておらず、繰延所得税や源泉徴収税も記録されていません。これらの税収は財務諸表にとって重要ではないからです。
123
棚卸しをする
在庫には以下の内容が含まれている
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
原材料.原材料 |
|
$ |
|
|
$ |
|
||
製品の中で |
|
|
|
|
|
|
||
総在庫 |
|
$ |
|
|
$ |
|
||
財産と設備、純額
各期間が終了するまで、財産および装置は、以下のものを含む
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
賃借権改善 |
|
$ |
|
|
$ |
|
||
実験室装置 |
|
|
|
|
|
|
||
機械と設備 |
|
|
|
|
|
|
||
コンピュータ装置及びソフトウェア |
|
|
|
|
|
|
||
家具と固定装置 |
|
|
|
|
|
|
||
建設中の工事 |
|
|
|
|
|
|
||
財産と設備、毛額 |
|
|
|
|
|
|
||
減算:減価償却累計と償却 |
|
|
( |
) |
|
|
( |
) |
財産と設備、純額 |
|
$ |
|
|
$ |
|
||
減価償却と償却費用は#ドルです
その他流動負債
各期間が終了するまで、他の流動負債は以下のものを含む
|
|
十二月三十一日 |
|
|
十二月三十一日 |
|
||
|
|
2022 |
|
|
2021 |
|
||
|
|
(単位:千) |
|
|||||
営業費用を計算する |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|||
|
|
|
|
|
|
|||
その他負債を計算すべき |
|
|
|
|
|
|
||
その他流動負債総額 |
|
$ |
|
|
$ |
|
||
124
ITEM 9.会計·財務開示における変化と会計士との相違
ない。
情報技術EM 9 A。制御とプログラム
情報開示制御とプログラムの評価
最高経営責任者と最高財務責任者の監督の下で、2022年12月31日までの開示制御および手順(取引所法案規則13 a-15(E)および15 d-15(E)で定義されている)の有効性を評価した。この評価に基づいて 我々の最高経営責任者および最高財務官は、取引所法案に基づいて提出または提出された報告書で開示を要求する情報が、米国証券取引委員会規則および表で指定された期間内に記録、処理、集計および報告され、これらの情報が蓄積され、我々のCEOおよび最高財務官を含めて、開示すべき内容をタイムリーに検討するために、取引所法案に基づいて提出または提出された報告書に開示されることを保証するために、我々の開示制御および手続きが2022年12月31日から有効であると結論した。我々の開示制御およびプログラムを設計·評価する際に、管理層は、任意の開示制御およびプログラムは、設計および動作がどんなに良好であっても、予想される制御目標を達成するために合理的な保証を提供することしかできないことを認識している。また、開示制御およびプログラムの設計は、管理層に、そのコストに対する可能な制御およびプログラムの利益を評価する際に判断することが要求されるリソース制限が存在するという事実を反映しなければならない。
財務報告の内部統制に関する経営陣の報告
我々の経営陣は、“取引法”ルール13 a-15(F)および15 d-15(F)に定義されている財務報告の十分な内部統制の確立と維持を担当している。我々の経営陣は,2022年12月31日までの財務報告内部統制の有効性を以下の基準に基づいて評価した内部制御--統合フレームワーク(2013)テレデビル委員会が組織委員会の発表を後援します。
評価結果によると、経営陣は、財務報告に対する内部統制は2022年12月31日から有効であると結論した。2022年12月31日現在、財務報告の内部統制の有効性は、本年度報告第10-K表の第9 A項に含まれる独立公認会計士事務所徳勤会計士事務所が監査している。
制御やプログラムへの固有の制限
私たちの経営陣は、CEOやCEOを含め、私たちの開示制御や手続き、および私たちの内部統制がすべてのミスやすべての詐欺を防ぐことができることを期待していません。制御システムの設計や動作がどんなに良くても,合理的な保証を提供し,制御システムの目標を実現することしかできない.制御システムの設計は資源の制限を反映しており,制御の利点はそのコストに対して考慮しなければならない.すべての制御システムには固有の限界があるため、どの制御評価も、社内のすべての制御問題や不正イベントが検出されたか、または検出されることを絶対に保証することはできない。これらの固有の制限は財務報告手続きの既知の特徴であるため、これらのリスクを除去するのではないにもかかわらず、保護措置をプログラム中に設計することが可能である。これらの固有の限界には,意思決定における判断が誤りである可能性があり,故障発生は簡単な誤りや誤りによる現実がある.ある人の個人的な行動、2人以上の結託、または制御の管理によって制御を凌駕することによって制御を回避することができる。いずれの制御システムの設計も,将来のイベントの可能性に対する何らかの仮定にある程度基づいている.私たちの開示制御およびプログラムは、その目標を達成するための合理的な保証を提供することを目的としているが、どの設計もすべての未来の条件でその目標を達成することに成功する保証はない。時間の経過とともに,条件の変化や政策やプログラムに対する遵守度の悪化により,制御が不十分になる可能性がある.費用対効果を有する制御システムの固有の制限により、エラーまたは詐欺によるエラー陳述が発生し、発見されない可能性がある。
私たちは、私たちの開示制御とプログラムの設計と有効性を継続的に検討し、評価し、時間の経過とともに私たちの制御とプログラムを改善し、私たちが将来発見する可能性のある任意の欠陥を修正するつもりです。我々の最高経営責任者および最高財務責任者は、2022年12月31日現在、“取引法”規則13 a-15(E)で定義されているように、開示制御およびプログラムの設計が有効であると結論しているが、将来的に私たちの業務に影響を与えるイベントは、私たちの開示制御およびプログラムを大幅に修正する可能性がある。
125
財務報告の内部統制の変化
12月31日までの3ヶ月以内に、取引法第13 a-15(D)および15 d-15(D)条に要求される我々の評価に基づいて決定された財務報告の内部統制には何の変動もなく、これらの変動は、財務報告の内部統制に大きな影響を与えているか、または財務報告の内部統制に大きな影響を与える可能性がある。新冠肺炎の流行により、私たちの多くの従業員は遠隔で働いているにもかかわらず、私たちの財務報告の内部統制は何の実質的な影響を受けていない。我々は,我々の内部制御設計と運営有効性への影響を最小限に抑えるために,新冠肺炎の状況を継続的にモニタリング·評価している。
独立公認会計士事務所報告
この10-K表年次報告書は、私たちの独立公認会計士事務所が発行した認証報告書を含む。
126
独立公認会計士事務所報告
Atara BioTreateutics,Inc.の株主と取締役会へ
財務報告の内部統制については
Atara BioTreateutics,Inc.及びその子会社(“当社”)の2022年12月31日までの財務報告内部統制を監査し、根拠内部統制--統合フレームワーク(2013)テレデビル委員会(COSO)が主催して組織委員会が発表した。2022年12月31日現在、当社はすべての重要な面で財務報告に対する有効な内部統制を維持しており、その根拠は内部統制--統合フレームワーク(2013)COSOから発表されます。
我々はまた、米国上場企業会計監督委員会(PCAOB)の基準に従って、当社の2022年12月31日現在と2022年12月31日までの年度の総合貸借対照表と関連する総合経営報告書と全面赤字、株主権益と現金流量、および2023年2月8日までの報告を監査し、これらの財務諸表に対して保留のない意見を表明した。
意見の基礎
会社経営陣は、財務報告を効果的に内部統制し、添付されている“経営陣財務報告内部統制報告”に含まれる財務報告の内部統制の有効性を評価する責任がある。私たちの責任は私たちの監査に基づいて、会社の財務報告書の内部統制に意見を述べることです。私たちはPCAOBに登録されている公共会計士事務所で、アメリカ連邦証券法およびアメリカ証券取引委員会とPCAOBの適用規則と法規に基づいて、私たちは会社と独立しなければなりません。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務報告の有効な内部統制がすべての重要な面で維持されているかどうかを決定するために、合理的な保証を得るために監査を計画し、実行することを要求する。我々の監査には,財務報告の内部統制を理解すること,重大な弱点があるリスクを評価すること,評価されたリスクテストに基づいて内部統制の設計·運用有効性を評価すること,および状況下で必要と考えられる他のプロセスを実行することが含まれる。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
財務報告の内部統制の定義と限界
会社の財務報告に対する内部統制は、公認された会計原則に基づいて、財務報告の信頼性と外部目的の財務諸表の作成に合理的な保証を提供することを目的としたプログラムである。会社の財務報告に対する内部統制には、(1)合理的で詳細かつ正確かつ公平に会社の資産を反映した取引および処分に関する記録の保存、(2)公認された会計原則に従って財務諸表を作成するために必要な取引が記録されている合理的な保証を提供し、会社の収入および支出は会社の経営陣と取締役の許可のみに基づいて行われる。(三)財務諸表に重大な影響を及ぼす可能性のある不正買収、使用、または処分会社の資産を防止またはタイムリーに発見する合理的な保証を提供する。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的にどのような有効性評価を行うかの予測は,条件の変化により制御不足のリスクが生じる可能性があり,あるいは政策やプログラムの遵守度が悪化する可能性がある.
/s/徳勤法律事務所
カリフォルニア州サンフランシスコ
2023年2月8日
127
イットM 9 Bです。その他の情報
ない。
プロジェクト9 Cです。外国の開示について検査妨害不公正裁決
適用されません。
128
パ.パRT III
改正された1934年証券取引法第14 A条に基づき,2022年12月31日より遅くない120日後に2023年度株主総会の最終依頼書(最終依頼書)を提出する予定であるため,本年度報告10−K表では第III部に要求されるいくつかの情報を省略し,最終依頼書に含まれるいくつかの情報を引用して本明細書に組み込む。
Iプロジェクト10.役員、役員、および企業管理
この条項が要求する情報はここで私たちの最終依頼書を引用する。
イットM 11.役員報酬
この条項が要求する情報はここで私たちの最終依頼書を引用する。
Iプロジェクト12.特定の実益所有者の保証所有権と管理職および関連株主事項
この条項が要求する情報はここで私たちの最終依頼書を引用する。
情報技術EM 13.いくつかの関係や関連取引、および取締役独立性
この条項が要求する情報はここで私たちの最終依頼書を引用する。
情報技術EM 14.チーフ会計士費用とサービス
この条項が要求する情報はここで私たちの最終依頼書を引用する。
129
P第四条
プロジェクト15.展示品と展示品社会決算表
(A)(1)財務諸表。
項目15の部分に対する回答は上記項目8に記載されている.
(A)(2)財務諸表付表。
これらは必要ではないので、または上記第8項の財務諸表または付記に必要な資料が提供されているので、すべての付表は省略される。
(A)(3)展示品。
130
展示品索引.索引
展示品 |
|
|
|
引用で編入する |
|
保存済み |
|||||
番号をつける |
|
展示品説明 |
|
表 |
|
|
展示品 |
|
提出日 |
|
ここから声明する |
3.1 |
|
Atara BioTreateutics,Inc.社登録証明書の改訂と再発行 |
|
S-1 |
|
|
3.2 |
|
06/20/2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3.2 |
|
二番目の改正とAtara BioTreateutics,Inc.の付則を再修正する。 |
|
8-K |
|
|
3.1 |
|
09/27/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.1 |
|
普通株の書式 |
|
S-1/A |
|
|
4.1 |
|
07/10/2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.2 |
|
2019年前払い資金株式証明書表 |
|
8-K |
|
|
4.1 |
|
07/22/2019 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.3 |
|
2020年5月事前出資株式証表 |
|
8-K |
|
|
4.1 |
|
05/28/2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.4 |
|
2020年12月事前出資株式証表 |
|
8-K |
|
|
4.1 |
|
12/09/2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.5 |
|
証券説明書 |
|
10-K |
|
|
4.4 |
|
02/27/2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1* |
|
2014年の株式インセンティブ計画を改訂して再確認します |
|
10-Q |
|
|
10.2 |
|
08/08/2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.2* |
|
2014年株式インセンティブ計画下のオプション協定とオプション付与通知のフォーマット |
|
S-1 |
|
|
10.2 |
|
06/20/2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.3* |
|
2014年持分インセンティブ計画の下で制限株式単位協定及び制限株式単位付与通知のフォーマット |
|
10-Q |
|
|
10.1 |
|
11/07/2019 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.4* |
|
2014年度従業員株購入計画 |
|
S-1/A |
|
|
10.8 |
|
07/10/2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.5* |
|
Atara BioTreateutics,Inc.2018年誘導計画の第3回改訂と再改訂 |
|
S-8 |
|
|
4.3 |
|
07/22/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.6* |
|
インセンティブ計画下限売却契約と限定株授権書のフォーマット |
|
10-Q |
|
|
10.2 |
|
11/07/2019 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.7* |
|
株式オプション協定フォーマット及びインセンティブ計画下の株式オプション付与通知 |
|
10-Q |
|
|
10.3 |
|
05/08/2018 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.8* |
|
割引承認通知書及び優遇承認契約のフォーマット |
|
10-Q |
|
|
10.3 |
|
08/07/2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.9* |
|
Atara BioTreateutics,Inc.とその各役員と役員との間で締結された賠償協定のフォーマット |
|
S-1 |
|
|
10.9 |
|
06/20/2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.10* |
|
Atara BioTreateutics,Inc.とその幹部間の雇用合意フォーマット。 |
|
10-Q |
|
|
10.4 |
|
08/01/2018 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.11* |
|
行政人員採用協議形式 |
|
10-Q |
|
|
10.2 |
|
08/08/2019 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.12* |
|
役員採用協定は,2019年5月23日,Pascal TouchonとAtara BioTreateutics,Inc.によるものである. |
|
8-K |
|
|
10.1 |
|
05/28/2019 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.13 |
|
Atara BioTreateutics,Inc.とスローン·キャトリンがんセンターが署名した独占許可協定は2015年6月12日 |
|
S-1 |
|
|
10.30 |
|
06/29/2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.14 |
|
Atara BioTreateutics,Inc.スローン·キャトリンがんセンターとの間の独占許可協定の改正案第1号改正案は,2018年8月30日となっている |
|
10-K |
|
|
10.14 |
|
02/26/2019 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.15 |
|
Atara BioTreateutics,Inc.とクイーンズランド医学研究所理事会との間の独占的許可協定の改正と再署名は、2016年9月23日、改訂された |
|
10-Q |
|
|
10.1 |
|
08/01/2018 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.16 |
|
Atara BioTreateutics,Inc.とクイーンズランド医学研究所理事会が2016年9月23日に改訂し、再署名した“研究と開発協力協定”は、改訂された |
|
10-Q |
|
|
10.2 |
|
08/01/2018 |
|
|
131
展示品 |
|
|
|
引用で編入する |
|
保存済み |
|||||
番号をつける |
|
展示品説明 |
|
表 |
|
|
展示品 |
|
提出日 |
|
ここから声明する |
|
|
|
|
|
|
|
|
|
|
|
|
10.17+ |
|
Atara BioTreateutics,Inc.とクイーンズランド医学研究所弁護士との間の第2回改訂と再改訂の研究·開発協力協定は、2019年8月28日となっている |
|
10-Q |
|
|
10.3 |
|
11/07/2019 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.18+ |
|
Atara BioTreateutics,Inc.とクイーンズランド医学研究所弁護士との間の第2回改正と再署名の独占ライセンス契約は、2019年8月28日となっています |
|
10-Q |
|
|
10.4 |
|
11/07/2019 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.19+ |
|
Atara BioTreateutics,Inc.とクイーンズランド医学研究所弁護士との間の第3回改正と再署名の独占許可協定は,2020年8月26日である |
|
10-Q |
|
|
10.1 |
|
11/09/2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.20+ |
|
Atara BioTreateutics,Inc.とクイーンズランド医学研究所弁護士との間の第3回改訂·再改訂の研究·開発協力協定は、2020年8月26日に日が定められている |
|
10-Q |
|
|
10.2 |
|
11/09/2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.21 |
|
Atara BioTreateutics,Inc.とCognate BioServices,Inc.の間の開発と製造サービス協定は、2015年8月10日に改訂された |
|
10-Q |
|
|
10.3 |
|
08/01/2018 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.22+ |
|
Atara BioTreateutics,Inc.及びCognate BioServices,Inc.が2018年11月4日に改正·再署名した“開発·製造サービス協定”の改正案第2号 |
|
10-Q |
|
|
10.5 |
|
11/07/2019 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.23+ |
|
Atara BioTreateutics,Inc.とCogate BioServices,Inc.との間の開発·製造サービス協定の修正案第3号改正案は,2019年6月28日とした |
|
10-Q |
|
|
10.6 |
|
11/07/2019 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.24+ |
|
Atara BioTreateutics,Inc.とCogate BioServices,Inc.の間の開発·製造サービス協定第4号修正案は,2019年11月4日とした |
|
10-K |
|
|
10.22 |
|
02/27/2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.25+ |
|
Atara BioTreateutics,Inc.とCogate BioServices,Inc.との間の開発·製造サービス協定の修正案第5号改正案は,2019年11月27日とした |
|
10-K |
|
|
10.23 |
|
02/27/2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.26+ |
|
Atara BioTreateutics,Inc.とCognate BioServices,Inc.の間の商業製造サービス協定は,2020年1月1日である |
|
10-K |
|
|
10.24 |
|
02/27/2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.27+ |
|
Atara BioTreateutics,Inc.とCognate BioServices,Inc.の間の商業製造サービス協定の修正案第1号修正案は,2021年9月1日である |
|
10-Q |
|
|
10.1 |
|
11/04/2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.28 |
|
BXP 611 Gateway Center LPとAtara BioTreateutics,Inc.の間のオフィスレンタルは、2015年12月9日 |
|
10-K |
|
|
10.29 |
|
03/04/2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.29 |
|
BXP 611 Gateway Center LPとAtara BioTreateutics,Inc.の間のレンタル第1修正案は、2020年10月21日 |
|
10-Q |
|
|
10.4 |
|
11/09/2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.30 |
|
千オーク工業ポートフォリオ有限責任会社とAtara BioTreateutics,Inc.の間の標準工業リースは、2017年2月6日です |
|
10-Q |
|
|
10.1 |
|
05/04/2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.31+ |
|
Atara BioTreateutics,Inc.とバイエル株式会社との間の研究,開発と許可協定は,2020年12月4日である |
|
10-Q |
|
|
10.1 |
|
05/04/2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.32 |
|
ロサンゼルス地区第2号有限責任会社がAtara BioTreateutics,Inc.と締結した賃貸契約日は2021年3月17日 |
|
10-Q |
|
|
10.2 |
|
05/04/2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.33+ |
|
Atara BioTreateutics社とスローン·キャトリンがんセンターが初めて改正·再署名した独占許可協定は、2021年3月22日に日となった |
|
10-Q |
|
|
10.3 |
|
05/04/2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.34+ |
|
2021年4月21日の第3回改正と再署名の許可協定 |
|
10-Q |
|
|
10.1 |
|
08/09/2021 |
|
|
132
展示品 |
|
|
|
引用で編入する |
|
保存済み |
|||||
番号をつける |
|
展示品説明 |
|
表 |
|
|
展示品 |
|
提出日 |
|
ここから声明する |
|
|
|
|
|
|
|
|
|
|
|
|
10.35+ |
|
Atara BioTreateutics,Inc.とPierre Fabre Medicant間の商業化協定は,2021年10月2日である |
|
10-K |
|
|
10.35 |
|
02/28/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.36 |
|
Atara BioTreateutics,Inc.と611 Gateway Center LP,LLC間のレンタルに関する第2の修正案は,2021年12月9日である |
|
10-K |
|
|
10.36 |
|
02/28/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.37+ |
|
Atara BioTreateutics,Inc.クイーンズランド医学研究所弁護士と2021年12月17日に調印された第4回改訂と再改訂の研究·開発協力協定 |
|
10-K |
|
|
10.37 |
|
02/28/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.38+ |
|
Atara BioTreateutics,Inc.とクイーンズランド医学研究所弁護士との間の第4回改正と再署名の独占許可協定は,2021年12月17日である |
|
10-K |
|
|
10.38 |
|
02/28/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.39* |
|
Atara BioTreateutics,Inc.役員採用契約のフォーマット |
|
10-K |
|
|
10.39 |
|
02/28/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.40+ |
|
資産購入契約は,2022年1月26日,Atara BioTreateutics,Inc.,Fujifilm DiSynth BioTechnologies California,Inc.とある限られた目的,Fujifilm Holdings America Corporationによって締結された |
|
8-K |
|
|
2.1 |
|
04/04/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.41 |
|
2022年1月26日現在、Atara BioTreateutics,Inc.およびFujifilm DiSynth BioTechnologies California,Inc.が署名したプライマリサービスおよび供給プロトコル。 |
|
10-Q |
|
|
10.1 |
|
05/05/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.42 |
|
Atara BioTreateutics,Inc.とCharles River実験室会社の間で2022年5月31日に署名された商業製造サービス協定の修正案第2号。 |
|
10-Q |
|
|
10.1 |
|
08/08/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.43+ |
|
Atara BioTreateutics,Inc.とCharles River実験室社の間で2022年8月1日に署名された商業製造サービス協定の修正案第3号。 |
|
10-Q |
|
|
10.1 |
|
11/08/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.44 |
|
Atara BioTreateutics,Inc.とバイエル株式会社の間で2022年8月2日に署名された終了、改訂、および計画移転協定 |
|
10-Q |
|
|
10.2 |
|
11/08/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.45+ |
|
Atara BioTreateutics,Inc.及びPierre Fabre Medicantが2022年9月28日に署名した商業化協定の改正案第1号 |
|
10-Q |
|
|
10.3 |
|
11/08/2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.46+ |
|
Atara BioTreateutics,Inc.とHCR Molag Fund,L.P.との間の売買合意は,2023年12月20日である |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
21.1 |
|
付属会社名簿 |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
23.1 |
|
独立公認会計士事務所の同意 |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
24.1 |
|
授権書(署名ページに含まれる) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.1 |
|
2002年サバンズ-オキシリー法第302条に基づく“1934年証券取引法”第13 A-14 A条及び第15 D-14 A条による最高経営責任者の証明 |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
133
展示品 |
|
|
|
引用で編入する |
|
保存済み |
|||||
番号をつける |
|
展示品説明 |
|
表 |
|
|
展示品 |
|
提出日 |
|
ここから声明する |
31.2 |
|
2002年サバンズ-オキシリー法第302条に基づく“1934年証券取引法”第13 A-14 A条及び第15 D-14 A条による首席財務官の証明 |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
32.1(1) |
|
米国法第18編第1350条に基づいて発行された最高経営責任者·最高財務責任者証明書によると、この条項は“2002年サバンズ·オックススリー法案”第906条に基づいて可決された |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
101.INS |
|
連結されたXBRLインスタンス文書-インスタンス文書は、そのXBRLタグがイントラネットXBRL文書に埋め込まれているので、対話データファイルには表示されない |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
101.書院 |
|
イントラネットXBRL分類拡張アーキテクチャ文書 |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
101.カール |
|
インラインXBRL分類拡張計算リンクライブラリ文書 |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
101.def |
|
インラインXBRL分類拡張Linkbase文書を定義する |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
101.介護会 |
|
XBRL分類拡張ラベルLinkbase文書を連結する |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
101.Pre |
|
インラインXBRL分類拡張プレゼンテーションLinkbaseドキュメント |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
104 |
|
表紙インタラクションデータファイル(イントラネットXBRL文書に埋め込む) |
|
|
|
|
|
|
|
|
X |
その展示品の一部は秘密待遇を受けた。
+本展示品の部分は省略されており、(I)実質的ではなく、(Ii)が公開されていれば、競争被害をもたらす可能性があるからである。
*管理契約または補償計画またはスケジュールを示します。
項目16.表格10-Kの概要
ない。
134
標札すきま
改正された1934年証券取引法第13又は15(D)節の要求に基づいて、登録者は8月8日にカリフォルニア州千樫市で次の署名者によって正式に本報告書に署名することを許可されたこれは…。2023年2月1日。
|
Atara生物治療会社 |
||
|
|
|
|
|
差出人: |
|
/s/Pascal Touchon |
|
|
|
パスカル·トゥーチン |
|
|
|
社長と最高経営責任者 |
このような陳述により、以下の署名のすべての人が、Pascal TouchonおよびUtpal Koppikarを構成して任命し、彼らの各々が、彼または彼女の真および合法的な事実上の代理人および代理人として、誰もが彼または彼女の名義、場所または代替、任意およびすべての身分で、本Form 10-K年次報告の任意およびすべての修正案(発効後の修正案を含む)に署名し、その証拠物およびそれに関連する他の文書とともに、証券取引委員会に提出し、上記の事実上の代理人および代理人を付与することを知っている。そして、彼らの各々は、その場所およびその周囲で必要かつ行わなければならないすべてのことを行い、実行する権利が完全にあり、可能であるか、または自ら行うことができるすべての意図および目的を尽くし、ここで上記のすべての事実弁護士および代理人を承認し、確認することができ、またはそれらの1人または複数の代理人は、本条例によってなされたすべてのことおよび事柄として合法的にまたは手配することができる。
本報告は、1934年の証券取引法の要求に基づき、以下の者によって登録者として指定日に署名された。
サイン |
|
タイトル |
|
日取り |
|
|
|
|
|
/s/Pascal Touchon |
|
|
|
|
パスカル·トゥーチン |
|
取締役最高経営責任者総裁 |
|
2023年2月8日 |
/s/Utpal Koppikar |
|
|
|
|
Utpal Koppikar
|
|
首席財務官 |
|
2023年2月8日 |
|
|
|
|
|
/キャロル·G·ガラゲル |
|
|
|
|
キャロル·G·ガラゲル製薬会社ですD。 |
|
役員、議長 |
|
2023年2月8日 |
|
|
|
|
|
/s/Eric L.Dobmeier |
|
|
|
|
エリック·L·ドブマイヤー |
|
役員.取締役 |
|
2023年2月8日 |
|
|
|
|
|
/s/Matthew K.Fust |
|
|
|
|
マシュー·K·フォスター |
|
役員.取締役 |
|
2023年2月8日 |
|
|
|
|
|
/s/ウィリアム·K·ヘイデン |
|
|
|
|
ウィリアム·Kハイデン |
|
役員.取締役 |
|
2023年2月8日 |
|
|
|
|
|
/s/アミート·マリク |
|
|
|
|
アミート·マリク |
|
役員.取締役 |
|
2023年2月8日 |
|
|
|
|
|
マリア·グラツィア·ロンカロロ |
|
|
|
|
マリア·グラツィア·ロンカロロ医学博士 |
|
役員.取締役 |
|
2023年2月8日 |
|
|
|
|
|
ベス·セデンバーグ |
|
|
|
|
ベス·セデンバーグ医学博士 |
|
役員.取締役 |
|
2023年2月8日 |
135