アメリカ証券取引委員会
ワシントンD.C.,20549
表格10-K
X 1934年証券取引法第13又は15(D)節に提出された年次報告書
2019年12月31日までの財政年度
あるいは…。
O 1934年証券取引法第13条又は15(D)条に基づく移行報告書
そこからの過渡期について
委員会ファイル第001-36289号

Genocea生物科学社は
(登録者の正確な氏名はその定款に記載)
デラウェア州 | 51-0596811 | |
(登録設立又は組織の国又はその他の管轄区域) | (国際税務局雇用主身分証明書番号) | |
マサチューセッツ州ケンブリッジ市オークパーク通り100番地 | 02140 | |
(主にオフィスアドレスを実行) | (郵便番号) | |
(617) 876-8191
(登録者の電話番号、市外局番を含む)
同法第12条(B)に基づいて登録された証券:
クラスごとのタイトル | 取引記号 | 登録された各取引所の名称 | ||
普通株、額面0.001ドル | GNCA | ナスダック資本市場 | ||
同法第12条(G)により登録された証券:なし
登録者が証券法規則405で定義されている経験豊富な発行者である場合は、再選択マークで示してください。Oはx No
登録者がこの法第13節または第15節(D)節に基づいて報告を提出する必要がないかどうかを再選択マークで示す。Oはx No
再選択マークは、登録者が(1)過去12ヶ月以内(または登録者がそのような報告の提出を要求されたより短い期間)に、1934年の証券取引法第13条または15(D)節に提出されたすべての報告書を提出したかどうか、および(2)過去90日以内にそのような提出要件に適合しているかどうかを示す。XはO No
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−T規則405条(本章232.405節)に従って提出を要求した各相互作用データファイルを電子的に提出したか否かを示す。XはO No
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな報告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
大型加速ファイルサーバ | o | ファイルマネージャを加速する | x | |
非加速ファイルサーバ | o | 規模の小さい報告会社 | x | |
新興成長型会社 | o | |||
新興成長型企業であれば、登録者が延長された移行期間を使用しないことを選択したか否かを再選択マークで示し、取引所法第13(A)節に提供された任意の新たまたは改正された財務会計基準を遵守する。O
登録者が空殻会社であるか否かをチェックマークで示す(取引法第12 b-2条で定義されている)。Oはx No
ナスダック資本市場2019年6月30日(登録者が最近完了した第2四半期の最終営業日)に報告されたこのような株の終値によると、登録者の非関連会社が保有する投票権および無投票権を有する普通株の総時価は、73,983,101ドルである。
2020年2月11日現在、登録者普通株の流通株数は27,643,773株である。
引用で編入された書類
登録者の最終委託書のうち、その後提出される2020年株主総会に関連する部分は、参照によって本報告の第3の部分に組み込まれる。
カタログ
第1部 | ||
第1項。 | 業務.業務 | 4 |
第1 A項。 | リスク要因 | 16 |
項目1 B。 | 未解決従業員意見 | 41 |
第二項です。 | 属性 | 41 |
第三項です。 | 法律訴訟 | 41 |
第四項です。 | 炭鉱安全情報開示 | 41 |
第II部 | ||
五番目です。 | 登録者普通株市場、関連株主事項及び発行者による株式証券の購入 | 42 |
第六項です。 | 選定された財務データ | 43 |
第七項。 | 経営陣の財務状況と経営成果の検討と分析 | 43 |
第七A項。 | 市場リスクの定量的·定性的開示について | 49 |
第八項です。 | 財務諸表と補足データ | 49 |
第九項です。 | 会計と財務情報開示の変更と相違 | 49 |
第9条。 | 制御とプログラム | 49 |
プロジェクト9 B。 | その他の情報 | 50 |
第三部 | ||
第10項。 | 役員·幹部と会社の管理 | 52 |
第十一項。 | 役員と役員の報酬 | 52 |
第十二項。 | 特定の実益所有者の担保所有権及び経営陣及び関連株主の事項 | 52 |
十三項。 | 特定の関係や関連取引、取締役の独立性 | 52 |
14項です。 | チーフ会計士費用とサービス | 52 |
第4部 | ||
第十五項。 | 展示品と財務スケジュール | 53 |
第十六項。 | 表格10-Kの概要 | 53 |
2
前向きに陳述する
このForm 10-K年次報告書には、重大なリスクと不確実性に関する前向きな陳述が含まれている。前向きな陳述は歴史的事実でもなく、未来の業績の保証でもない。逆に、それらは私たちの現在の私たちの業務の未来、未来の計画と戦略、私たちの臨床結果と他の未来の状況に対する信念、期待、仮説に基づいている。“予想”、“信じる”、“考慮”、“継続”、“可能”、“推定”、“予想”、“予測”、“目標”、“計画”、“可能”、“計画”、“潜在”、“予測”、“プロジェクト”、“すべき”、“目標”、“将”、“将”、またはこれらの用語の否定または他の同様の表現は、すべての前向き表現がこれらの識別語を含むわけではないが、前向き表現を識別することを意図している。
本年度報告におけるForm 10−Kに関する前向きな陳述には、以下についての記述が含まれている
• | 我々はGen-009の臨床試験を行い、前臨床研究を継続し、Gen-011のための試験新薬(“IND”)を提出し、私たちの他の候補製品のための臨床前研究を継続し、免疫腫瘍学への投資を継続するのに必要な資金の時間と金額の推定を行った |
• | 支出、将来の収入、資本需要、現在および予想される現金資源の十分性、および追加融資に対する私たちの需要の推定 |
• | 私たちの候補製品のために規制承認のタイミングと能力を獲得し、維持する |
• | 戦略的パートナーシップ協定の潜在的な利点と私たちの戦略的パートナーシップ計画を達成する能力 |
• | 知的財産権の地位は |
• | 承認された候補製品の市場受容度および臨床的実用性の速度および程度 |
• | 候補製品を迅速かつ効率的に識別し開発する能力と |
• | 私たちの商業化、マーケティング、そして製造能力、そして戦略。 |
私たちは私たちの展望声明で開示された計画、意図、または予想を実際に達成できないかもしれません。あなたは私たちの展望的声明に過度に依存してはいけません。実際の結果または事件は、私たちが前向きな陳述で開示した計画、意図、および予想とは大きく異なるかもしれない。私たちはこの10-K表の年次報告書の警告声明に重要な要素を含んでおり、特に“リスク要因”の部分では、これらの要素は実際の結果や事件をもたらす可能性があり、私たちが行った前向き声明とは大きく異なると考えられる。私たちの展望的な陳述は、私たちが行う可能性のある任意の未来の買収、合併、処置、合弁または投資、または私たちが達成可能な協力または戦略的パートナーシップの潜在的な影響を反映しない。
Form 10-K年次報告書と私たちがForm 10-K年度報告書として提出した文書を完全に読み、私たちの将来の実際の結果が私たちが予想していたものと大きく異なる可能性があることを理解しなければなりません。私たちは法律の要求がなければ、新しい情報、未来の事件、または他の理由でも、いかなる前向きな陳述も更新する義務はない。
3
第1部
プロジェクト1.ビジネス
文意に加えて,本年度報告表格10−Kで言及されている“Genocea”,“我々”,“我々”はGenocea Biosciences,Inc.を指す。
概要
私たちは生物製薬会社で私たちのATLASを利用して新しい癌免疫療法の発見と開発を求めていますTM独自の発見プラットフォーム。ATLASプラットフォームは各患者のCD 4を分析します+CD 8と+患者の腫瘍中の各潜在標的或いは“抗原”に対する細胞の免疫反応を測定した。この方法は,患者が反応可能な抗原を認識することにより,免疫療法(例えば癌ワクチンや細胞療法)の抗原選択を最適化していると考えられる。したがって,ATLASはより多くの免疫原性と有効な癌免疫治療を引き起こすと信じられている
我々の最先端プロジェクトはGen−009であり,個人化された新しい抗原癌ワクチンであり,1/2 a期の臨床試験を行っている。Gen−009は、各患者のGen−009ワクチンに含まれるために、ATLASを使用して、各患者特有の免疫原性腫瘍変異を認識することを計画している。我々はGEN−011も進めており,新たな抗原特異的養子T細胞治療計画であり,ATLASにも依存している。私たちは2020年第2四半期に011世代IND申請を提出する予定だ
アトラス·プラットフォーム
ますます多くの人は、免疫系のT細胞腕を利用して腫瘍細胞を殺すことは多くの癌の治療に潜在力があると考えている。この方法は血液系悪性腫瘍に有効であり、最近ある固形腫瘍に対して有効である。この方法を用いたワクチンまたは細胞療法は、遺伝子変異由来の抗原のような腫瘍に存在する正常組織の特定の違いを対象としなければならない。しかし,この免疫療法の最適抗原は特に挑戦的であり,原因は2つあることが分かった。まず,ヒトT細胞反応の遺伝的多様性は,有効抗原が人によって異なることを意味する。次に,候補抗原の数は非常に多い可能性があり,ある癌では各患者に数千個もの候補抗原がある。したがって、有効な抗原選択システムは、各患者の腫瘍とそれらのT細胞バンクとを同時に考慮しなければならない。
Atlasは、各患者のヒト免疫系からのT細胞アームのコンポーネントを使用することによって、効率的な抗原選択を実現する。ATLASを用いて、各患者のT細胞の全面的な候補新抗原、腫瘍関連抗原と腫瘍関連ウイルス抗原に対する反応を測定し、個人癌を殺す可能性のある抗腫瘍T細胞反応に関連する標的を選択できるようにした。ATLASは最も包括的かつ最も正確な抗原発現系を代表していると考えられる。また,ATLASは新たな候補抗原特徴である抑制性T細胞反応を決定していると考えられる。従来,すべての候補抗原は有効な抗腫瘍反応(刺激性)の標的と考えられていたか,無関係と考えられていた。しかし,ATLASを用いてInhibigensと呼ばれる抑制性抗原が認識されているTMこれらは臨床前研究で腫瘍の進展を促進することが証明された。また、1つの抗原は、1つの患者において刺激的であってもよく、別の患者において抑制されてもよく、これは、各患者の潜在的な免疫原性抗原を選択することの重要性を強化することを見出した。
ATLAS製品の組み合わせは3つの特許シリーズを含む。最初の2つの家族は、発行された米国特許、特許期間はそれぞれ少なくとも2031年および2030年まで、および付与された外国特許および係属中の米国および外国出願を含む。第3のファミリーは、ATLASによる癌診断、予後および患者選択方法、および関連成分に関する。この一連の特許は現在、11の外国司法管轄区域における係属中の出願と係属中の米国特許出願を含む。これらの出願から発行される特許は,少なくとも1つの特許期間が2038年3月まで予定されている。
私たちの免疫腫瘍学プロジェクトは
我々の癌免疫療法は、T細胞に特定の癌標的の認識および攻撃を教育することを目的としたワクチンと、これらの標的を攻撃するように訓練されたT細胞を導入することを目的とした細胞療法とを含む。新しい抗原ワクチンは既存の癌治療法と組み合わせて使用することができ,個体の癌に対するT細胞反応を潜在的に誘導·増強し,より良い臨床結果を潜在的に産生すると考えられる。また,過継細胞治療により特定の新規抗原に対するT細胞群を分離·拡大することは意義のある臨床的メリットを提供すると考えられる。
我々が開発している積極的な免疫腫瘍学的計画を紹介する
4
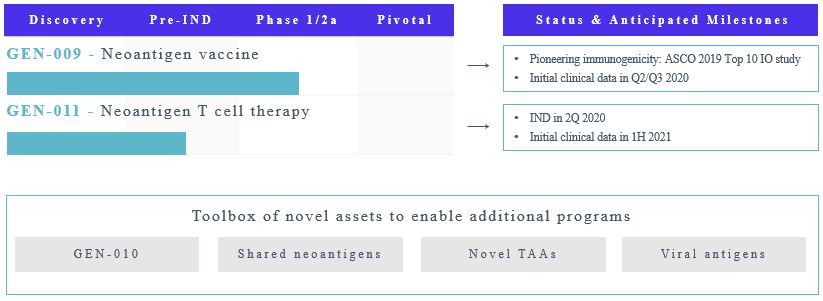
我々の主導プロジェクトGEN−009はアジュバントを有する新しい抗原ペプチド候補ワクチンである。ATLASを用いて特定の新しい抗原を認識し,ATLASが決定した患者の抗腫瘍免疫反応を刺激する新しい抗原のみを用いて,個々の患者に個性化ワクチンを作製した。著者らは現在一連の固形腫瘍タイプに対してGen-009の1/2 a期臨床試験を行っている
• | 試験のA部分は、ある疾患の証拠のない癌患者における単一療法としてのGen−009の安全性および免疫原性を評価することである |
• | 試験のB部分、著者らはすでに患者に投与し始め、Gen-009とICIの連合治療末期或いは転移性腫瘍患者の安全性、免疫原性と初歩的な抗腫瘍活性を評価することを目的とした。 |
2019年を通して臨床試験A部分のデータを示した。薬物服用患者8名からのデータでは,ATLASの潜在的抗原選択優位が確認されたと考えられる
• | 100%の患者が測定可能なCD 4を持っている+CD 8と+Gen-009ワクチンに対するT細胞の反応は |
• | ワクチン接種新抗原の99%(N=88接種抗原)の99%の応答が検出され,この応答率は以前に報告された新抗原候補ワクチンに対する応答を上回った |
• | Gen-009がCD 8を起こしました+T細胞体外反応、これはT細胞エフェクター機能の測定基準であり、41%のワクチン新抗原とCD 4に対して+T細胞は51%の新規抗原に反応した |
• | Gen−009は体外刺激試験を用いて広範な免疫反応を刺激し,中央記憶反応を測定する方法であり,新しい抗原の87%がCD 4を励起した+新規抗原の57%がCD 8応答を誘導+返事をする |
• | Gen−009耐性は良好であり、用量制限毒性は認められなかった |
• | 2020年1月31日まで、私たちはどんなワクチンを接種した患者がどんな病気の再発があるのか分からない |
任意のオープンラベル研究と同様に、私たちは、初期の臨床信号が見られることを確実にするために、より小さい患者グループを評価するために、登録を減速または一時停止することができる。著者らは2020年第2四半期あるいは第3四半期に著者らのGen-009部分B臨床試験にこれらの初歩的な臨床結果を報告する予定である。この評価に基づいて、私たちは研究を継続するのに適しているかどうかを考慮するつもりだ。
ATLASで決定された新しい抗原に対する養子T細胞療法であるGEN−011も進められている。腫瘍浸潤性リンパ球(TIL)治療などの養子T細胞療法は固形腫瘍に別の治療方法を提供した。TIL療法は、各患者の固形腫瘍からTILを抽出し、それらをインビトロで非特異的に拡大し、癌患者に再注入することに依存する。中期臨床研究では、免疫チェックポイント阻害剤(ICI)治療に失敗した患者は、測定可能かつ持続可能な腫瘍縮小を示す。GEN-011は、彼らの腫瘍ではなく、各患者の末梢血からATLASによって認識された新しい抗原特異的T細胞を抽出し、特異的に増幅する(TIL産生に対して行われたように)。Gen−011は,TIL療法と比較して,効力,治療効果,製造しやすい利点を提供すると信じている。2020年第2四半期に米国食品医薬品局(FDA)にGen−011に対するIND申請を提出する予定であり,2021年上半期に初歩的な臨床結果が予想される。
5
私たちはまたもっと多くのプロジェクトの機会を探索し続けるつもりだ。我々は、次世代抗原送達技術を採用した候補ワクチンであるGen−010を評価し続け、より良い免疫原性および/またはワクチン生産効率を提供する機会を提供するために、この技術を推進することができる。また,ATLASを用いて非個性化癌免疫治療のための新たな候補抗原を探している。これらの計画は、Epstein-Barrウイルス感染やInhibigensによって駆動される癌のような、共有された新しい抗原、非変異腫瘍関連抗原、ウイルス起源癌を対象とすることができるTM.
競争
バイオテクノロジーと製薬業界の特徴は,新技術と独自製品の開発競争が激しく迅速に変化することである。私たちは私たちの特許の組み合わせとT細胞ワクチンと細胞治療の専門知識が私たちに競争優位を提供すると信じているが、私たちはもっと規模が大きく、資金が豊富な製薬会社を含む多くの異なる源からの潜在的な競争に直面している。私たちは他の免疫腫瘍学会社と競争しなければならないだけでなく、私たちが商業化する可能性のあるどの製品も既存の治療法や将来出現する可能性のある新しい療法と競争しなければならない。
BioNTech SE,AureVac AG,Genentech,Inc.,Gritstone Oncology Inc.,メルク社,Moderna,Nouscom,Vaccibody ASなど,いくつかの会社が新たな抗原癌ワクチンの開発を試みている。ATLASプラットフォームの潜在的な能力に基づいて、Gen−009は、これらの候補製品のそれぞれに対して優れている、すなわち、各癌患者のために、その以前に存在する免疫反応に対する患者の新しい抗原を全面的に識別することに優れていると考えられる。ATLASを用いて個人癌ワクチンのための新しい抗原を選択することは,より有効なワクチンをもたらすと信じている。しかしながら、これらの会社のうちの1つまたは複数の企業が将来的にGen−009と類似またはより良い臨床結果を得ることは保証されず、私たちの将来の臨床試験が成功することも保証されない。
同様に,腫瘍抗原を認識するT細胞受容体(TCR)を転移させることにより形質導入されたT細胞,腫瘍から非特異的に濃縮されたT細胞(腫瘍浸潤性リンパ球)や,複数の腫瘍特異的抗原により増幅された末梢血液中のT細胞の新規抗原に対する細胞療法の開発が試みられている。これらの会社には、アキレス治療株式会社、適応バイオテクノロジー社、BioNTech SE社、細胞生物医学グループ会社、Eutilex社、GIlead Science社、Iovance BioTreateutics社、Marker治療会社、腫瘍治療科学会社、PACT製薬会社、ZiopHarm Oncology社が含まれています。GenoteaのATLAS真腫瘍抗原選択は、より的確で効果的な細胞治療をもたらすと信じていますが、これらの会社のうちの1つ以上または他の会社が将来Gen-011と類似または優れた臨床結果を得ることはできませんし、将来の臨床試験が成功する保証はありません。
私たちの多くの競争相手は、単独でも彼らとの戦略的パートナーでも、私たちよりも多くの財力、技術、人的資源を持っており、候補製品の発見と開発、FDAとワクチンの他の規制承認、およびこれらのワクチンまたは細胞療法の商業化の面でより多くの経験を持っている。したがって、私たちの競争相手は私たちよりもワクチンや細胞療法の承認を得ることに成功し、広範な市場で受け入れられるかもしれない。私たちの競争相手のワクチンまたは細胞療法は、私たちが商業化する可能性のある任意のワクチンまたは細胞療法よりも有効であるか、またはより効率的にマーケティングおよび販売され、私たちの製品を時代遅れにしたり、競争力を欠いたりする可能性がある。
バイオテクノロジーと製薬業界の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場と臨床試験の患者登録を確立し、そして私たちの計画と相補的或いは必要な技術を獲得する上で私たちと競争している。規模が小さいかスタートアップ段階にある会社も重要な競争相手になる可能性があり、特に大手や成熟会社との協力で手配する。
新薬の市場進出と先進技術の出現に伴い、私たちは激しい競争と日々の競争に直面すると予想している。我々が開発·商業化したいずれのワクチンや細胞療法も,有効性,安全性,管理·交付の利便性,価格,後発薬競争レベル,政府と他の第三者支払者の精算状況などに基づいて競争することが予想される。
もし私たちの競争相手が私たちが開発する可能性のあるどの製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会は減少または消失するかもしれない。私たちの競争相手も私たちよりも早くFDAや他の規制機関のその製品の承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立することをもたらすかもしれない。また、多くの場合、私たちの競争能力は、後発薬の使用を奨励することを求める保険会社または他の第三者支払者の影響を受ける可能性がある。
知的財産権
6
私たちは、内部開発でも第三者から許可を得ても、特許権を求め、維持し、擁護することを含む、当社の業務に重要なビジネス的意義を有するノウハウ、発明、改善を保護し、強化するために努力しています。私たちはまた、私たちの独自技術プラットフォームとノウハウに関連するビジネス秘密、持続的な技術革新、許可内の機会に依存して、ワクチンおよび細胞治療分野における私たちの独自の地位を強化し、維持しています。また,データ独占性,市場独占性,特許期間延長(可能であれば)による規制保護にも依存している。また、当社の会社名に対して商標保護を使用し、製品および/またはサービスが発売された場合に商標保護を行うことを期待しています。
私たちのビジネス成功は、私たちの業務に関連する重要な商業技術、発明および独自技術の特許および他の独自保護を取得および維持する能力があるかどうかにある程度依存するかもしれない;私たちの特許を擁護し実行すること、私たちの商業秘密を秘密にすること、および第三者が効果的に特許および独自の権利を実行することを侵害することなく運営することができる。私たちが第三者が私たちの製品を製造、使用、販売、提供、または輸入することを阻止する能力は、これらの活動をカバーする効果的かつ強制的に実行可能な特許または商業秘密に基づいて私たちが権利を持っている程度に依存するかもしれない。ライセンスおよび会社のすべての知的財産権について、私たちは、私たちの任意の未解決特許出願または将来提出される任意の特許出願が特許を得ることを保証することはできません。私たちはまた、私たちの任意の既存特許または将来私たちに付与される可能性のある任意の特許が、私たちの商業製品を保護し、これらの製品を製造する方法において商業的用途を有することを保証することはできません。
我々は、大量の特許および特許出願を開発または許可し、ワクチンおよび細胞治療製品の開発および商業化に関連する大量の技術的ノウハウおよび商業的秘密を有している。個別特許の期限は特許を取得した国の法的期限に依存する。私たちが出願を提出したほとんどの国では,特許期間は非仮出願を提出した日から20年である。米国では、特許期限調整(“特許期限調整”)によって特許の期限を延長することができ、この調整は、特許付与時の特許付与時の特許権者の損失を補償することができ、または、以前に提出された特許によって特許権者が最終的に放棄された場合には、特許期間を短縮することができる。
FDAが承認した薬物をカバーする特許期間も延長する資格があり、これは、FDA規制審査過程で失われた特許期間の補償として、米国特許の特許期間の回復を可能にする。ハッジ-ワックスマン法は特許期間を特許満了後最大5年間延長することを許可している。特許期間の延長の長さは,薬物が規制審査を受ける時間の長さと関係がある。1つの特許の残り期間を延長することは、製品承認日から合計14年間を超えることができず、承認された薬物に適用される特許を延長することしかできない。また、1つの特許は1回しか展示期間がないので、1つの特許が複数の製品に適用される場合は、1つの製品展示期間に基づくしかない。欧州や他の外国司法管区にも同様の規定があり、承認された薬物をカバーする特許の期限を延長する。可能な場合には,臨床試験の時間の長さや生物製品ライセンス申請(“BLA”)の提出に関わる他の要因に基づいて,我々の候補製品とその使用方法をカバーする特許出願のための特許期間を延長する予定である。
本年度報告Form 10-Kの日付まで、私たちの特許組み合わせは、以下の通りです
地図集
私たちの発見プラットフォーム特許の組み合わせは3つの特許シリーズを含み、現在発行された5つの米国特許を含む。我々は、ATLAS発見プラットフォームに関連する方法をカバーし、腫瘍細胞に発現される抗原を発見することを含む、総裁およびハーバード大学(“ハーバード”)研究員から得られた最初の特許ファミリーの独占的許可を持っている。この第1の特許シリーズは、米国特許9,051,564および9,920,314と、許可された米国特許と、ヨーロッパ、カナダ、およびオーストラリアで付与された特許とを含む。この一連の許可された外国特許は2027年に満期になる予定だ。米国特許9,051,564および9,920,314は、特許期限調整を含み、それぞれ2031年12月および2028年6月に延長される。私たちは、癌または腫瘍関連抗原を発見するための、我々が使用しているATLASプラットフォームに特化した第2の特許シリーズを完全に有する。第2の特許シリーズは、米国特許8,313,894,9,045,791および9,873,870、許可された米国特許出願、係属中の米国特許出願、欧州、カナダ、およびオーストラリアで発行された特許、および欧州における係属中の出願を含む。この家族に付与された外国特許の特許期間は2029年7月までである。米国特許8,313,894および9,045,791の条項は、それぞれ2030年8月および2029年8月に延長された特許期限調整を含む。米国特許9873870の有効期限は2029年7月である。私たちは、癌診断、予後、および患者のために選択された方法および関連成分を有する第3の特許ファミリーを完全に有する。この3番目の家族は現在、11の外国司法管轄区域で審査される申請と1つの被審アメリカ申請を含んでいる。私たちは他の3つの特許家族を完全に所有しており、各家族は未解決のPCT出願を含む, 2018年末に提出された臨時申請を優先すると主張した。これらのPCTの使用は、癌または腫瘍に関連する抗原をさらに選択し、免疫反応を再配向し、T細胞を再培養するためのATLASに基づく方法を対象としている。
7
許可協定
ハーバード大学
私たちはハーバード大学(“ハーバード”)と独占許可協定があり、3つの特許シリーズの独占的、世界的な、印税あり、再許可可能な許可、開発、製造、製造、使用、マーケティング、販売、輸入許可製品の提供、ATLAS発見プラットフォームに関する許可サービスを提供してくれます。私たちはまた、ある開発と規制のマイルストーンを達成する際に、ハーバードマイルストーンに合計160万ドルに上るお金を支払う義務がある。2019年12月31日現在、私たちは合計30万ドルのマイルストーン支払いを支払いました。本ライセンス契約により、合意された開発計画に従って、ビジネス上の合理的な努力を用いてライセンス製品を開発、マーケティング、販売することが義務付けられています。さらに、特定の開発マイルストーンを実現する義務があり、任意のタイプの製品やサービスの開発マイルストーンを満たすことができず、合理的な拡張や改訂提案がなければ、ハーバードは、製品またはサービスのタイプに応じてそのような製品に関する本プロトコルを終了する権利があり、またはそのような製品およびサービスに関する非独占的で再許可不可能な許可に許可を変換する権利がある。
特許権がカバーする製品や使用許可方法を許可して発見された製品を商業化した後、私たち、私たちの付属会社、私たちの許可者が販売しているこのような製品やサービスの純売上高にハーバード版税を支払う義務があります。製品やサービスの種類によって印税は違いますが、いずれも低い1桁になっています。製品の種類によっては、私たちの分ライセンシーが支払うべき販売ベースの印税は、適用される印税料率、またはその分のライセンシーから受け取った印税のより高い桁または低い2桁の割合のうちの大きい者である。使用料は、商業化された製品またはサービスの種類に応じて、ライセンス特許権に基づいて提出された最後の有効な権利要件が満了するまで、またはそのような製品またはサービスが初めて商業販売されてから10年以内に支払われなければならない。支払いが必要な任意の第三者支払いについては、ハーバード大学に支払うべき印税が減少する可能性があり、上限は特定の割合である。印税支払いに加えて、任意の再許可の下で任意の追加収入(現金または非現金)を取得した場合、特定のカテゴリの支払いを含まず、低いビット数から最高の下位2桁まで、特に再許可を含む許可範囲に応じて、ハーバードにそのような収入の一定の割合を支払わなければならない。
ハーバード大学とのこのライセンス契約は、ライセンス特許権に基づいて提出された最後の満期の有効権利要件が満了するまで、製品またはサービス、サービス、および国/地域に基づいて満期となる。私たちはいつでも事前にハーバード大学に合意を終了するように書面で通知することができる。もし私たちの重大な違約行為がまだ治癒されていない場合、もし私たちが借金を返済できない場合、破産や同様の状況、または私たちが私たちに付与された任意の特許の有効性に疑問を提起すれば、ハーバードも合意を終了することができる。
Oncovir許可と供給協定
2018年1月、私たちはOncovir,Inc.(“Oncovir”)と許可と供給協定を締結した。このプロトコルは、ヒルトノを我々の技術と組み合わせた製品(“組合せ製品”)の研究、開発、使用、販売、製造、商業化およびマーケティングのために、免疫調節剤およびワクチンアジュバントアジュバントヒルトノ(ポリILC)(“ヒルトノ”)を製造および供給するためのアンコヴィの条項および条件を規定する。ヒルトノは,我々独自のATLASプラットフォームを用いて認識された合成長ペプチドまたは新規抗原からなり,ヒルトノとともに調製されるGen−009のアジュバント成分である
Oncovirは、Hiltonolの使用を含むOncovirと組合せ製品の研究、開発、または商業化に関連するいくつかの知的財産権に再許可を付与する権利があるが、Hiltonolを製造または使用または販売するためにHiltonolを使用することはできない、非独占的で譲渡可能な印税付きグローバルライセンスを私たちに付与してくれた。ライセンスは、2028年1月25日遅く、または合意に従って私たちに許可された任意の特許の最後の有効な権利要件が満了した日に永久、全額支払い、および印税免除となります
この協定によると、各組合せ製品がある臨床試験マイルストーンを達成し、各組合せ製品がある地域で初めて発売許可を得た場合には、Oncovirに中の6桁低いマイルストーン支払いを支払い、組合せ製品の純売上高に応じて、製品ごとに1桁以下の等級別特許権使用料を支払う義務がある。
共同製品の開発を停止することを決定した場合、または適用可能な規制機関がヒルトンまたは連合製品が臨床的に安全でないか、または有効でないと判断した場合にプロトコルを終了することができます。合意のいずれか一方は、他方の重大な違約行為や、他方の破産、債務返済や解散のために合意を終了することもできる。
商業秘密
8
場合によっては、私たちは商業秘密に依存して私たちの技術を保護するかもしれない。しかし、商業秘密は保護するのが難しいかもしれない。私たちは、従業員、コンサルタント、科学コンサルタント、請負業者と秘密保持協定を締結することで、私たちのノウハウやプロセスを保護することを求めています。また、私たちは私たちのビルの実体安全と、私たちの情報科学技術システムの実体と電子安全を維持して、私たちの資料と商業秘密の完全性とセキュリティを維持することに力を入れています。私たちはこのような個人、組織、そしてシステムに自信がありますが、合意や安全措置は違反される可能性があり、私たちはどんな違反にも対応する十分な救済措置がないかもしれません。しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。私たちのコンサルタント、請負業者、または協力者が、私たちのために働いているときに他人が所有している知的財産権を使用する場合、関連するまたはそれによって生じるノウハウおよび発明の権利について論争が生じる可能性がある。
政府の監督管理
ワクチンや養子細胞療法などの生物製品は連邦食品、薬物と化粧品法案(FD&C法案)と公衆衛生サービス法案(PHS法案)及びその他の連邦、州、地方と外国法規の規制を受けている。FD&C法とPHS法及びその対応する法規は、他を除いて、生物製品の測定、製造、安全、効果、ラベル、包装、貯蔵、記録保存、流通、報告、広告とその他の宣伝方法を管理する。生物製品の臨床試験は開始前にFDAの審査を経なければならない。しかも、生物製品が発売される前にFDAの承認を受けなければならない。規制審査と承認を得る過程と、その後適切な連邦、州、地方、外国の法律·法規を遵守する過程には多大な時間と財力がかかり、必要な規制承認を得ることができない可能性がある。
アメリカの生物製品開発プロセス
米国食品医薬品局が生物製品が米国市場に参入する前に要求されるプログラムは、通常、以下の手順を含む
• | 良好な実験室操作規範(“GLP”)および適用された実験動物の人道使用要件または他の適用法規に基づいて非臨床実験室テストおよび動物研究を完成させる |
• | ヒト臨床試験の開始前に有効でなければならないIND出願をFDAに提出する |
• | FDAの一般的に良好な臨床実践(GCP)と呼ばれる法規および人体研究対象およびその健康情報を保護する任意の追加の要求に基づいて、各臨床試験を開始する前に、各臨床場所を代表する独立機関審査委員会(IRB)の承認を得ることを含む、提案された生物製品の期待される用途に対する提案生物製品の安全性および有効性を決定するために、十分かつ制御されたヒト臨床試験が行われる |
• | 非臨床試験および臨床試験結果からの安全性、純度および有効性の実質的な証拠を含む上場承認のためのBLAをFDAに提出する |
• | GMPに適合する状況を評価して、施設、方法、および生物製品の特性、強度、品質および純度を維持するのに十分な制御を保証し、適用可能であれば、FDAが現在ヒト細胞および組織製品を使用している良好な組織規範(GTP)を維持するために、バイオ製品を製造するためのFDAの1つまたは複数の製造施設の検査を満足的に完了させる |
• | FDAは、BLAを支持するデータを生成する非臨床試験場所および臨床試験場所を監査することができる |
• | FDAによるBLAの審査および承認、またはライセンス。 |
人体で任意の候補生物製品をテストする前に、この候補製品は臨床前研究段階に入る。臨床前研究は、非臨床研究とも呼ばれ、製品の化学、毒性と調合に対する実験室評価、及び候補製品の潜在的安全性と活性を評価する動物研究を含む。臨床前研究の進行はGLPを含む連邦法規と要求に適合しなければならない。
臨床試験スポンサーは臨床前試験の結果を生産情報、分析データ、任意の利用可能な臨床データ或いは文献、提案された臨床方案と共にFDAに提出し、INDの一部としなければならない。IND提出後も,いくつかの臨床前研究は継続する可能性がある。INDはFDAが30日以内に臨床試験を保留しない限り、FDAが受領してから30日後に自動的に発効する。この場合,INDスポンサーやFDAは臨床試験開始前に未解決の問題を解決しなければならない。FDAは臨床を強制する可能性もあります
9
臨床試験の前または間に、安全考慮または規定に適合しない理由で、いつでも生物製品候補製品を持っている。FDAが臨床的一時停止を強制する場合、研究はFDA許可なしに再開されず、その後、FDA許可の条件下でのみ再開される可能性がある。したがって,INDの提出によりFDAが臨床試験の開始を許可するか,あるいは開始すると,このような研究を一時停止または終了するという問題はないとは判断できない。
臨床試験は,合格した調査者の監督の下で健康なボランティアや患者に候補生物製品を服用することに関連し,これらの調査者は通常,試験スポンサーに雇用されたりコントロールされていない医師である。臨床試験は,臨床試験の目標,投与手順,被験者の選択と排除基準,および被験者の安全性を監視するためのパラメータを詳細に説明するプロトコルの下で行われ,何らかの有害事象(“副作用”)が発生した場合に臨床試験を停止する停止ルールを確保することを含む。各スキームおよびスキームの任意の修正は、INDの一部としてFDAに提出されなければならない。臨床試験は,すべての研究対象にインフォームドコンセントを要求することを含むFDAのGCP要求を含む規定に基づいて行われなければならない。さらに、各臨床試験は、IRBによって審査および承認されなければならないか、または臨床試験を行う各機関にサービスを提供しなければならない。IRBは試験参加者の福祉や権利の保障を担当し,臨床研究に参加する個人のリスクが最低に低下するかどうか,期待利益と比較して合理的かどうかなどの項目を考慮している。IRBはまた、各臨床試験対象またはその法律代表によって署名されなければならないインフォームドコンセントの形態および内容を承認し、完成まで臨床試験を監視しなければならない。
人体臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重複或いは合併する可能性がある
• | 第一段階:まず生物製品を健康な人体に導入し、その安全性、用量耐性、吸収、代謝、分布と排泄をテストする。深刻または生命を脅かす疾患のためのいくつかの製品の場合、特に製品がその固有の毒性のために健康ボランティアに道徳的に服用できない可能性がある場合、最初の人体テストは通常患者に行われる。 |
• | 第二段階:限られた患者集団において生物製品を評価し、可能な副作用と安全リスクを決定し、特定の目標疾患に対するこの製品の治療効果を初歩的に評価し、用量耐性、最適用量と用量計画を決定する。 |
• | 第三段階:地理的に分散した臨床試験地点で臨床研究を行い、拡大した患者群における生物製品の安全性、純度と潜在力を更に評価する。これらの臨床研究は製品の全体的なリスク/収益比を確定し、製品の承認と製品ラベルに十分な基礎を提供することを目的としている。 |
承認後の臨床研究は,4期臨床研究と呼ばれることがあり,最初の上場承認後に行うことができる。これらの臨床研究は,治療適応が予想される患者の治療から追加的な経験を得るためのものであり,特に長期安全なフォローアップのためである。
臨床開発のすべての段階において、監督管理機関はすべての臨床活動、臨床データと臨床試験調査人員に対して広範なモニタリングと監査を行うことを要求している。臨床研究結果を詳細に説明する年次進展報告はFDAに提出しなければならない。深刻かつ意外な副作用に対しては、直ちにFDAと調査者に書面のIND安全報告を提出しなければならず、任意の他の研究からの発見、実験室動物試験或いは体外試験は人類被験者に重大なリスクがあることを表明し、或いは任意の臨床上の深刻な疑わしい副作用の発生率は方案或いは研究者マニュアルに記載されているより増加する。スポンサーは15日以内にINDセキュリティ報告書を提出し,スポンサーがその情報有資格報告を確定した後でなければならない。スポンサーはまた、スポンサーが初めて情報を受け取ってから7日以内に、任意の意外、致命的、あるいは生命に危害を及ぼす疑いのある副作用をFDAに通知しなければならない。第1段階、第2段階、および第3段階の臨床研究は、もしあれば、任意の指定された時間内に成功しないかもしれない。FDAまたはスポンサーまたはそのデータ安全監視委員会は、研究対象または患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由で臨床試験を一時停止または終了することができる。同様に,臨床試験がIRBの要求に沿って行われていない場合,あるいは生物製品が患者に予期せぬ深刻な被害を受けた場合,IRBはその機関の臨床試験の承認を一時停止または終了することができる。すべての制御された臨床試験の発起人は、第一段階試験以外に、アメリカ国立衛生研究院によって維持されている公共臨床試験登録と結果データベースに格納するために、ある臨床試験情報を提出しなければならない, これらの報告は、http://Clinicaltrials.gov上で公開して得ることができる。
臨床研究と同時に、会社は通常追加の動物研究を完成し、生物製品の物理的特徴に関する追加情報を開発し、GMP要求に基づいて最終的に商業大量生産製品の技術を決定しなければならない。PHS法案は,生物製品を用いた外来製剤導入のリスク低減を支援するために,以下の属性を持つ製品の製造制御の重要性を強調している
10
正確には定義できません。製造過程は一貫して高品質の候補製品ロットを生産することができなければならず、他の以外に、スポンサーは最終生物製品の特性、強度、品質、効力と純度をテストする方法を開発しなければならない。また,適切な包装を選択·試験し,候補生物製品が賞味期限内に受け入れられない変質が生じないことを証明するために安定性研究を行わなければならない。
アメリカの審査と承認の流れ
生物製品の臨床試験が完了した後,生物製品の商業販売の前に,FDAによるBLAの承認を得なければならない。BLAは製品開発、実験室と動物研究、人体研究、製品製造と成分の情報、提案のラベル、その他の関連情報を含まなければならない。さらに、“小児科研究平等法”によれば、BLAまたはBLAの補充は、すべての関連する小児科亜群において生物製品が主張する適応の安全性および有効性を評価し、安全かつ有効な各小児科亜群に対する投与および投与を支持するためのデータを含まなければならない。FDAはデータの提出を延期することを許可するか、またはすべてまたは部分的な免除を与えることができる。検査や承認には多大な時間と労力が必要であり,FDAがBLAの届出を受ける保証はなく,届出しても,どの承認がタイムリーに承認されるか(あれば),どのような適応が承認されるか(あれば)は保証されない.
“処方薬使用料法案”(PDUFA)によると、2017年に再認可されてさらに5年間延長され、BLAごとに相当な使用料を伴わなければならない。PDUFAはまた,承認された生物プロジェクトごとに年間費用を徴収する。場合によっては、小企業が初めて出願した出願料を免除することを含む、費用を免除または減免することができる。
出願が提出されてから60日以内に、FDAは、機関が提出を受け入れる前に実質的に完了したかどうかを決定するためにBLAを審査する。FDAは、それが不完全であるか、または提出時に適切に審査できないと考えられる任意のBLAの提出を拒否することができ、より多くの情報の提供を要求することができる。この場合,BLAおよび付加情報を再提出しなければならない.再提出された申請はFDAが届出を受ける前にも審査を行わなければならない。提出された申請が受け入れられると,FDAはBLAの深い実質的な審査を開始する。FDAは、提案された製品がその予期される用途に対して安全かつ有効であるかどうか、許容可能な純度プロファイルを有するかどうか、および製品がGMP法規に従って生産されるかどうかを決定して、製品の識別、安全、強度、品質、効力、および純度を確保および保存するためにBLAを審査する。FDAは、新規な生物製品または安全性または有効性の問題を提起する生物製品の申請を諮問委員会に提出することができ、一般に、申請を承認すべきかどうか、およびどのような条件下で承認すべきかを審査、評価および提案するための臨床医および他の専門家を含むグループである。FDAは諮問委員会の提案に制限されていないが、それは決定を下す時にこれらの提案を慎重に考慮するだろう。生物製品の承認過程において、FDAはまた、生物製品の安全な使用を確保するために、リスク評価および緩和戦略(“REMS”)が必要であるかどうかを決定する。FDAがREMSが必要であると結論した場合,BLAのスポンサーは提案したREMSを提出しなければならず,必要であればFDAはREMSのないBLAを承認しないであろう。
BLAを承認する前に、FDAはこの製品を生産する施設を検査する。FDAは、製造プロセスおよび施設がGMP要件に適合していると判断しなければ、製品が要求された仕様内で一貫して生産されることを保証するのに十分であることを決定しない限り、この製品を承認しない。さらに、BLAを承認する前に、FDAは通常、IND研究要求およびGCP要求に従って臨床試験が行われることを確実にするために、1つまたは複数の臨床場所を検査する。GMP,GTP,GCPに適合することを確保するためには,申請者は訓練,記録保存,生産,品質管理に多大な時間,お金,労力をかけなければならない。
関連データおよび情報が提出されたにもかかわらず、FDAは、BLAがその承認の規制基準を満たしていないことを最終的に決定し、承認を拒否する可能性がある。臨床試験から得られたデータはつねに決定的ではなく,FDAのデータ解釈は我々の同じデータに対する解釈とは異なる可能性がある。機関が現在の形態のBLAを承認しないことを決定した場合、FDAは、FDAによって決定されたBLA内のすべての特定の欠陥を一般的に記載する完全な返信を発行するであろう。決定された欠陥は微小である可能性があり、例えば、ラベル変更が必要であるか、または重大であり、例えば、追加の臨床試験が必要である。さらに、完全な返信状は、出願人がとり得る、申請を承認条件に置くための提案行動を含むことができる。完全な返信が発行された場合、出願人は、BLAを再提出し、手紙で決定されたすべての不足点を解決するか、または出願を撤回することができる。
1つの製品が規制部門の承認を得た場合、この承認は、特定の疾患および用量に明らかに限定される可能性があり、または使用の適応が制限される可能性があり、これは、製品の商業的価値を制限する可能性がある。さらに、FDAは、いくつかの禁忌症、警告、または予防措置を製品ラベルに含めることを要求する可能性がある。FDAは、REMSの形態で製品の流通、処方または調剤に制限および条件を適用することができ、または他の方法で任意の承認範囲を制限することができる。そのほか、FDAは発売後の臨床試験の設計を要求する可能性があり、4期臨床試験と呼ばれることがある
11
生物製品の安全性と有効性をさらに評価し、商業化された承認された製品の安全性を監視する試験および監視計画をさらに評価する
米国の詐欺と乱用、透明性、プライバシー法
米国では、私たちの商業活動は、詐欺および乱用を防止すること、医療業界の他の人との相互作用の透明性を促進すること、個人情報のプライバシーを保護すること、研究の完全性を確保すること、または研究に参加する人間の被験者を保護することを目的とした多くの他の連邦、州、および地方法律によって制約されている。これらの法律は、米国司法省および司法省内の個別連邦検事室、米国衛生公衆サービス部(HHS)、HHSの各部門を含むが、これらに限定されないが、医療保険および医療補助サービスセンター(CMS)、監察長事務室、人間研究保護事務室、研究誠実事務室、その他の州および地方政府機関を含むが、異なる連邦および州法執行機関によって実行される。
現在、商業販売が許可されている製品はありませんが、将来規制やマーケティングの承認を受ける可能性のある任意の候補製品の将来の販売に関する活動によって、リベート法や虚偽申告法を含む医療保健の“詐欺や乱用”に関する様々な連邦·州法の制約を受ける可能性があります。バックオフ法は、一般に、購入、処方、または特定の薬剤の使用を含む業務を生成するために、製薬業者が任意の報酬を請求、提供、受け入れ、または支払うことを禁止する。虚偽精算法は、一般に、任意の虚偽または詐欺的な精算薬品またはサービス支払い申請の提出または提出をもたらす第三者支払者(連邦医療保険および医療補助を含む)に、誰もが知らず、自発的に第三者支払者に提出することを禁止する。これらの法律の具体的な規定はそれぞれ異なるにもかかわらず、それらの範囲は通常広く、これらの法律は特定の業界で実践されている法規、指導、または裁判所判決に適用されていないかもしれない。したがって、私たちの接近はこのような法律の挑戦を受ける可能性がある。
連邦政府と各州はすでに法律と法規を公布し、製品を販売する薬品メーカーの販売とマーケティングのやり方を規範化した。法律および法規は、製造業者と医療提供者との間の財務的相互作用を一般的に制限する;製造業者には、いくつかのコンプライアンス基準を採用することを要求し、および/または、政府および公衆にそのような相互作用を開示することを要求する。その中の多くの法律法規の要求が曖昧で、あるいは行政指導が必要である。法律およびその実施の曖昧性を考慮して、将来の任意の活動(私たちが連邦医療計画の候補製品の承認および/または補償を得たら)が挑戦される可能性がある。
私たちが個人の身分情報を運営、取得、保存している各管轄区では、プライバシーとセキュリティ法律の制約を受ける可能性があります。多くのアメリカ連邦と州法律は個人情報の収集、使用、開示と保存を規範化している。複数の外国の国でも、個人情報の収集、使用、開示、保存を管理する法律が制定されている。世界的に、私たちの業務に影響を及ぼす可能性のあるプライバシーやデータ保護問題がますます注目されている。“リスク要因-私たちの商業と産業に関するリスク”を見てください。
私たちの業務が上記の任意の衛生規制法律または私たちに適用される任意の他の法律に違反していることが発見された場合、私たちは民事、刑事および行政処罰、損害賠償、罰金、返還、Medicare、Medicaidおよび他の連邦医療計画への参加から除外される可能性があり、契約損害、名声損害、利益および将来の収益の減少、および私たちの業務を削減または再構築することを含む処罰を受ける可能性があります。
精算する
国内外の市場では、いかなる承認された製品の商業成功はある程度第三者支払者のこのような製品に対する保証範囲と十分な補償、例えば政府医療保健計画、個人健康保険会社と管理医療組織に依存する。ワクチン接種を提供する患者およびワクチン接種を提供する提供者は、通常、第三者支払者に依存して、関連する医療費の全部または一部を精算する。したがって、任意の承認されたワクチンの販売は、国内でも海外でも、私たちが承認したワクチンのコストがどの程度第三者支払者によって支払われるかに大きく依存するであろう。これらの第三者決済者は、医療製品やサービスの価格に挑戦し、コストを管理するための制御を実施することが増えている。医療費の抑制は連邦と州政府の優先事項となっており、薬品価格はこの努力の重点である。各国政府はコスト制御計画の実施に大きな興味を示し、価格制御、精算制限と代替後発薬の要求を含む。価格制御とコスト制御措置、および既存の制御·措置を講じている司法管轄区域でより制限的な政策をとることで、我々の純収入と業績をさらに制限することができる。第三者支払者は、承認されたリストまたは処方内の特定の製品に保証範囲を制限することができ、これは、FDAによって承認された特定の適応のすべての製品を含まない可能性がある。また、新たに承認された保健製品の精算状況には大きな不確実性がある。第三者支払人はますます医療製品の医療必要性と費用効果を検査するようになっている
12
安全性と有効性があります私たちは私たちの製品の費用効果を証明するために高価な薬物経済学的研究を行う必要があるかもしれない。第三者支払者が私たちの製品が他の療法と比較して費用効果があると思わない場合、支払人は承認後に私たちの製品をその計画下の福祉としてカバーしないかもしれません。あるいは、もし彼らがそう思う場合、支払いレベルは利益に基づいて私たちの製品を販売するのに十分ではないかもしれません。
アメリカ国内で、もし私たちが未来に適切な承認を得て、私たちの現在の任意の候補製品をマーケティングすれば、私たちはMedicaid、Medicare、340 B薬品定価計画に基づいてこれらの製品のために保険を求めるかもしれません。これらの計画は,複数の連邦や州機関によって管理され,65歳以上,低収入または障害のある個人に処方薬福祉を提供したり,弱者にサービスを提供する医療提供者が割引価格で処方薬を購入することを許可したりする。将来、私たちはまた承認された候補製品を政府調達業者に売却することを求めるかもしれない。政府福祉計画の下で私たちの製品に保険を提供したり、政府調達者に製品を販売するために、私たちの製品の価格を追跡して報告したり、特定の調達者に割引を提供したり、いくつかの使用にリベートを支払うことが要求される可能性があります
米国では,連邦政府と州政府は,医療コストを低減する取り組みを含む医療提供や支払いを改革するための立法を提案し,採択し続けている。例えば、2010年3月、米国議会は“患者保護·平価医療法案”と“医療·教育和解法案”(“医療改革法案”)を公布し、医療補助の拡大と個人医療保険の実施により医療保険のカバー範囲を拡大し、政府のヘルスケア計画下の薬品のカバー範囲の変更や精算範囲を含む医療カバー範囲を拡大した。トランプ政権政権中、医療改革法案の条項の全部または一部を改正または廃止しようと努力してきた。例えば、2017年末に税改正立法が公布され、2019年から十分な医療保険のカバー範囲を維持していない個人に対する税収処罰(いわゆる“個人強制令”)が廃止された。2018年5月の報告書では、国会予算事務所は、2018年に比べて2019年には未加入者数が300万人増加し、2028年には600万人増加すると推定しており、一部の原因は個人強制令を廃止したためだ。“医療改革法案”も司法的挑戦を受けた。2018年12月、複数の州総検事長が提起した挑戦の中で、ある連邦地方裁判所裁判官は、国会が個人強制条項を廃止すると、国会の税収権力に依存してこの法律の公布を支持していないため、“医療改革法案”全体が違憲と判断した。2019年12月連邦控訴裁判所は個人権限条項の違憲に同意しました, しかし,事件を地域裁判所に返送し,医療改革法案にどのような条項が分割可能であるか,継続可能かどうかをより詳細に評価する。地域裁判所が行動し、何らかの時間を要する可能性のある控訴を解決する前に、“医療改革法”は様々な面で機能している。
トランプ政権はまた、薬品の価格設定に重点を置いた取り組みを含む他の改革措置を打ち出している。例えば、2018年5月、トランプ総裁と衛生·公衆サービス部長官は、処方薬の価格と自己負担コストを低減する“青写真”を発表した。青写真中のいくつかの提案、及び青写真以来提出された関連薬品の価格設定措置は、製薬業の運営と精算に重大な変化を招く可能性がある。もう一つの例は、2019年に採択された立法で、メーカーが医療補助薬品リベート計画に基づいて報告したある価格の計算方式を改正し、国会予算事務所は、この改正は今後10年間で連邦政府のために約30億ドル節約されると推定している
政府関係者や立法者はまた、医薬品の輸入禁止を立法するなど、薬品の価格や支払いを規制する措置を実施するための他の努力を行っている。最近、公衆と政府は薬品定価に対してかなりの審査を行い、人々が考えている薬品コストが高すぎることを解決する提案を提出した。最近いくつかの州の立法は薬品コスト問題を解決するために努力しており、これらの努力は通常薬品コストの透明性の向上或いは薬品価格の制限に重点を置いている。
連邦や州レベルで新しい立法を採用することは、販売が承認されれば、私たちの候補製品に対する需要や定価に影響を与える可能性がある。しかし、“医療改革法案”や他の連邦や州改革努力の任意の変化の最終的な内容、時間、影響を予測することはできない。連邦や州医療改革が私たちの将来の業務や財務業績に悪影響を与えないことは保証されない。
アメリカ以外では、私たちの製品に十分なカバーと支払いを提供することを確保することは挑戦に直面するだろう。国際市場では,精算や医療保険支払い制度は国によって大きく異なり,多くの国で特定製品や療法に価格上限が設定されている。多くの国で、処方薬の価格設定は政府によって規制されている。政府当局との価格交渉は製品の監督マーケティング許可を受ける範囲をはるかに超えている可能性があり、私たちに臨床試験を要求し、私たちの候補製品或いは製品のコスト効果を他の利用可能な治療法と比較することができるかもしれない。このような臨床試験を行うことはコストが高く、私たちの商業化努力の遅延を招く可能性がある。第三者決済者は医療製品やサービスの価格に挑戦しており、多くの第三者支払者が新たに承認された医療製品の精算を制限している。多くのEU諸国の最近の予算圧力はまた各国政府に各種のコスト制御措置を考慮あるいは実施させ、例えば価格を凍結し、値下げ力を強化する
13
リベート。予算圧力が持続すれば、各国政府は追加的なコスト制御措置を実施するかもしれない。コストコントロールは、私たちが開発または販売する可能性のある製品のために制定された価格を下げるかもしれません。これは、製品収入や私たちに支払うべき印税を減少させることになります。薬品に対して価格制御や精算制限を実行する国が私たちのいかなる製品にも有利な精算と定価手配を許可することは保証されません。
外国監督管理
アメリカの法規以外に、私たちは各種の外国法規の制約を受けて、これらの法規は私たちの候補製品の臨床試験と商業販売と流通を管理しています。私たちがFDAの候補製品の承認を得るかどうかにかかわらず、私たちはこれらの国や地域で臨床試験や製品の販売を開始するために、外国や経済地域(例えばEU)の比較可能な規制機関の承認を得なければならない。臨床試験、製品許可、定価と精算を管理する審査手続きと要求は地によって異なり、時間はFDA承認の時間より長い或いは短い可能性がある。
米国以外のある国には,ヒト臨床試験開始前に臨床試験申請(“CTA”)の提出を要求する流れがあり,INDと類似している。例えば,ヨーロッパではCTAは主管する国家衛生当局や各社が臨床試験を行おうとしている国の独立倫理委員会に提出しなければならない。CTAが一国の要求に応じて承認されると,臨床試験開発はその国で行うことができる。すべての場合,臨床試験はGCPや他の適用法規の要求に基づいて行われなければならない。
EU規制制度の下で、会社は集中的または分散されたプログラムを通じてマーケティング許可申請を提出することができる。バイオテクノロジーで生産された薬品やエイズ,癌,神経変性疾患,糖尿病,ウイルス性疾患や孤児薬物などの特定の適応を指定する新たな活性物質を含む薬品では,集中手順は強制的であり,他の高度に革新的な薬物には選択可能である。集中手続きの下で、マーケティング申請はヨーロッパ薬品管理局に提出され、人が薬品委員会でそれを評価し、評価は通常、欧州委員会が意見を受けてから67日以内にすべてのEU加盟国に有効な単一マーケティング許可を授与する。最初のマーケティング許可の有効期限は5年ですが、一度更新すると、通常有効期限は制限されません。分権手続きは、1つの加盟国(“参照”加盟国と呼ばれる)に対して提出された申請に基づいて、1つまたは複数の“関連”加盟国によって承認されることを規定する。分散承認手続きによれば、出願人は、参照加盟国及び関連加盟国に申請又はファイル及び関連資料を提出する。会員国たちが有効な申請を受けてから120日以内に評価草案と関連材料草案を作成することを参照する。各関係加盟国は、加盟国の評価報告を参考にして90日以内に評価報告および関連材料を承認するかどうかを決定しなければならない。もし会員国がマーケティング許可を認めなければ、論争点は最終的に欧州委員会に提出されるだろう, その決定はすべての会員国に拘束力がある。英国が2016年にEU離脱、いわゆる英国離脱国民投票を投票したことを受けて、英国とEUがEU離脱条項について交渉するにつれ、集中承認手続きの範囲が変化する可能性がある。
製造業
私たちには何の生産施設もありません。私たちは現在依存しており、引き続き第三者に依存して私たちの候補製品を生産し、非臨床研究と臨床試験のために使用し、私たちの候補製品が市場の承認を得たら商業生産に使用されると予想される
私たちの執行官に関する情報は
次の表に2020年2月13日までの私たちの幹部1人あたりの名前、年齢、ポストを示します。
名前.名前 | 年ごろ | ポスト | ||
ウィリアム·クラーク | 51 | 社長と最高経営責任者 | ||
Girish Aakalu、博士。 | 45 | 首席商務官 | ||
トーマス·デイビス医学博士 | 56 | 首席医療官 | ||
ダイアナ·デュヴァル | 48 | 首席財務官 | ||
ジェシカ·ベック·フリークターナー博士 | 48 | 首席科学官 | ||
ナリンド·シンガー | 48 | 上級副社長製薬科学と製造 | ||
14
ウィリアム·キップ·クラークですChIPは2010年8月から2011年2月まで私たちのCEOを務めた後、2011年2月からCEO兼CEO総裁を務めています。2011年2月以来、キップはまだ私たちの取締役会に勤めている。Genoceaに入社する前は、万達製薬会社で首席商務官を務めていたが、2004年に人と共同で設立したバイオ製薬会社である。万達在任中、彼は会社の戦略と業務発展活動を指導し、業務発展取引と株式融資を通じて4億ドルを超える資金を調達し、核心的な役割を果たした。万達に入社する前、キップはバイオ製薬会社に投資するベンチャー企業Care Capitalの責任者で、これまでSmithKline Beecham(現グラクソ·スミスクライン)で様々なビジネスや戦略職を務めてきた。キップはハーバード大学の学士号とペンシルバニア大学ウォートンビジネススクールの工商管理修士号を持っています。
Girish Aakalu博士は2018年12月にGenoceaに加入して首席商務官を務めた。この職で、彼はGenoceaの事業発展努力をリードした。彼の広範な技能は業務開発、会社と研究開発戦略、製品組合せ管理、商業計画と連盟管理をカバーしている。Genoceaに加入する前に、Girishは2015年5月から2018年12月まで益プソングループに雇われ、2007年10月から2015年5月までファイザー会社で副総裁を務めた:全世界外部革新主管、そして退職前に取締役を務めた:ファイザー外部研究開発革新チーム戦略、革新と運営主管。彼のこれまでの職務には,Genentech,Inc.で業務開発や腫瘍学パイプライン市場計画職を務め,L.E.Kコンサルティング会社で生命科学コンサルティング経験を担当していた。彼はジョン·ホプキンス大学生物物理学学士号を取得し、普通と学部級の栄誉で卒業した;カリフォルニア工科大学で生物学修士号を取得した後、また細胞と分子神経生物学博士号を取得し、西北大学-ケロッグ管理学院で会社管理方面の幹部教育を完成した。
Thomas Davis,M.D.Tomは2018年10月にGenoceaに加入し,首席医療官を務め,免疫腫瘍学と抗癌剤開発において20年以上の学術と業界経験を有している。最近、2017年10月から2018年4月までの間にオランダの細胞治療会社Gadeta B.V.の首席医療官を務め、新たな癌標的を探すことに取り組んでいたが、そこで新たな細胞治療技術を一流のヒト臨床研究に持ち込んだ。Gadeta B.V.に加入する前に、2006年から2017年までCelldexの首席医療官を務め、戦略、戦術、実行を含む臨床と監督発展のあらゆる面を指導した。Celldex在任中、Tomは積極的に臨床科学、医療事務、安全、臨床運営、統計、監督事務とプロジェクト管理を確立し、監督し、大型グローバル製薬パートナーとの協力を管理し、投資家関係活動に参加した。Tomはこの業界に加入する前に、アメリカ国立癌研究所癌治療評価プロジェクトで臨床を指導し、スタンフォード大学でリツキシマブと独自型ワクチンの開発に従事していた。デイビス博士はジョンズ·ホプキンス大学で生物物理学学士号を取得し,ジョージタウン大学で生理学修士と医学博士号を取得し,スタンフォード大学で腫瘍学研究を完了した。
ダイアナ·デュヴァル。Dianthaは2019年3月にGenoceaに加入し、首席財務官を務めた。Genoceaに任命される前に、Duvallさんは2017年2月から2019年1月までBioverativ,Inc.の副主計長兼首席会計官総裁を務めた。これまでBiogen Inc.で働き、2016年2月から2017年1月までグローバルビジネスディレクター、2015年2月から2016年1月まで米国のビジネスディレクターを務めていた。2009年5月から2015年1月まで、彼女はメルク社で複数のポストを務めていた。彼女のメルクでの経験はベンチャー投資、業務開発、合弁企業と連合、そして運営制御と技術会計に関連している。彼女はまた、米国証券取引委員会報告、サバンズ-オクスリコンプライアンス、取引支援、リスク管理の面で豊富な経験を持ち、1996年から2009年まで普華永道で複数の健康業界の職を務めた。デュワールさんは東北大学の会計学修士と工商管理修士号、コルビー学院の文学学士号を持っています。
ジェシカ·ベック·フリークターナー博士は2007年にGenocea,すなわち同社設立直後にGenoceaに入社して以来,複数の科学職を務めてきた。彼女は2016年2月から私たちの首席科学官を務め、2014年2月から2016年1月まで研究部上級副総裁、2010年1月から2014年2月まで研究部総裁副主任を務めた。2007年から2014年2月まで、彼女はGenoceaで様々なポストを担当し、年功序列が高まっている。Genoceaに加入する前に、Flechtner博士は2006年6月から2007年3月までBioVest International,Inc.で免疫学顧問を務め、そこで彼女は検査方法の開発を指導し、同社の自己濾胞(非ホジキンリンパ腫)ワクチンの患者における成功を評価した。2001年から2005年まで,Mojave治療会社とMojave知的財産権を獲得したAntigenics Inc.(現在Agenus)の研究員として,Flechtner博士はタンパク質とポリペプチドに基づくワクチンと免疫療法を開発し,癌,感染症,自己免疫と過敏症に用いられている。彼女は出願中または発行された様々な特許の発明者であり、複数の同業者評議の科学出版物を持っている。フリークターナー博士はダナ·ファーバー癌研究所とハーバード医学院ハーヴィー·カントー博士の実験室で博士後の仕事を終え、コーネル大学で細胞免疫学博士号と動物科学学士号を取得した。彼女はアメリカ免疫学者協会、アメリカ癌研究協会、癌免疫治療学会、総裁コーネル女性委員会と生物女性協会のメンバーである。
ナリンド·シンガーですナリンドは2018年3月にGenoceaに入社し、製薬科学·製造部門の上級副社長を務めた。このポストでは,NarinderはGenocea製品の製造プロセス開発と製造を管理している。ナリンドは
15
生物製薬の技術開発、拡大、技術運営と製造サプライチェーンの面で豊富な経験を持っている。Genoceaに加入する前に、Narinderは2015年7月から2018年3月までMomenta製薬会社で薬品製品開発·製造副総裁を務め、Momenta生体模倣薬と新製品の組み合わせのプロセス開発と医薬製品製造を担当した。Momentaに加入する前に、ナリンドは2007年6月から2015年7月まで安進で取締役薬物製品技術部を務め、安進が生物製剤の製品組み合わせに基づく薬品開発のプロセス開発、商業化、製造と新技術開発を担当した。1997年から安進会社で様々な初級技術職を務め、キャリアを開始した。Narinderは統合したB.Tech/M.Techを獲得した。1995年にデリーインド工科大学生化学工学とバイオテクノロジー学士号、ヒューストン大学化学工学修士号、カリフォルニア大学ロサンゼルス校アンダーソン管理学院工商管理修士号を取得した。
従業員
2019年12月31日現在、私たちは59人の常勤従業員があり、そのうち46人が研究開発に従事し、13人が財務、法律、業務発展、人的資源、施設、情報技術、またはその他の一般的かつ行政的機能に従事している。私たちの従業員の中の一人も労働組合代表でもなく、集団交渉協定のカバー範囲もなく、私たちは何の停止も経験したことがない。私たちは私たちが従業員と仲がいいと思う。
企業情報
私たちは2006年8月にデラウェア州の法律に基づいて登録された。私たちの主な実行事務室はマサチューセッツ州ケンブリッジ市オーク公園通り100号5階にあります。郵便番号:02140、電話番号は(6178768191)。GenoceaとGenotea標識は登録商標です。
利用可能な情報
インターネットサイトhttp://www.genOcean a.comを設置しています。このサイトでは、私たちの10-Kフォームの年次報告、10-Qフォームの四半期報告、8-Kフォームの現在の報告および他のファイル、およびこのような報告とファイルのすべての修正を無料で閲覧し、アメリカ証券取引委員会(“アメリカ証券取引委員会”)に提出された時間後、合理的で実行可能な範囲内でこれらの報告とファイルをできるだけ早く無料で提供することができます。米国証券取引委員会はまた、米国証券取引委員会に電子的に提出された報告書、依頼書、および情報声明、および発行者(当社を含む)に関する他の情報を含む相互接続サイトを維持する。大衆は私たちがアメリカ証券取引委員会に提出した任意の文書をインターネットで得ることができる。我々のサイトアドレスへの参照は,サイト上の情報を参照することで組み込むものではなく,サイト上の情報も本ファイルの一部ではない.
第1 A項。リスク要因
私たちの財務状況と追加資本需要に関連するリスク
私たちは私たちの運営計画を実行し、持続的に経営する企業として運営を続けるために追加の資金が必要だ。
2019年12月31日までの年度監査財務諸表の作成仮説は、継続的な経営企業として運営していくとしていますが、継続的な運営赤字は、継続的な経営企業としての能力に大きな疑いを与えていると考えられます。私たちは引き続き公開または私募株式発行、戦略取引、私たちの市場持分発行計画に基づいて私たちの普通株を売却する収益、私たちの株式信用限度額、あるいは他の方法で私たちの運営に資金を提供する予定です。しかし、受け入れ可能な条件で、私たちは十分な追加融資を受けることができないかもしれないし、根本的にできないかもしれない。もし私たちが必要な時や魅力的な条件下で資金を調達できない場合、私たちはGen-009、Gen-011、および/または他の候補製品および他の会社の活動の開発を停止するなど、さらなるコスト削減戦略を実施することを余儀なくされるかもしれない
私たちは2006年に設立されてからすでに重大な損失が発生しており、予測可能な未来には引き続き重大な損失を受け、永遠に利益を達成したり維持したりする可能性があると予想している。
私たちは臨床段階のバイオテクノロジー会社で、私たちはまだ著しい収入を得ていない。設立以来、我々は毎年純損失を出しており、このうち2019年12月31日と2018年12月31日までの年間純損失はそれぞれ3900万ドルと2780万ドルだった。2019年12月31日現在、我々の累計赤字は約3.31億ドルです。私たちは今までどんな製品も商業化していませんし、販売製品から何の収入も得ていませんし、いつ製品収入が発生したり、利益を達成したりするかどうかもわかりません。これまで、私たちは主に何度も株式を公開し、私たちの普通株と優先株、債務手配を公募することで、私たちの業務に融資してきました
著者らは大部分の財力を研究開発に投入し、著者らの臨床と非臨床技術開発と開発活動を含む。私たちの将来の純損失額はある程度私たちのものにかかっています
16
将来の支出と私たちが株式発行や戦略取引を通じて資金を得る能力。私たちはまだ候補製品の重要な臨床研究を完成していません。もしあれば、商業化された候補製品を準備するのに数年かかります。私たちが規制部門の承認を得て候補製品を販売しても、私たちの将来の収入は、私たちの候補製品が承認された任意の市場の規模、私たちが十分な市場受け入れ能力、第三者支払者の精算、その他の要素に依存するだろう。
予測可能な未来には、巨額の費用と増加する運営損失が引き続き発生する見通しだ。私たちは次のような状況で、私たちの支出が大幅に増加すると予想する
• | 最先端の候補製品Gen-009の臨床試験を続けています |
• | Gen-011および私たちの他の候補製品のための非臨床、臨床、または他の研究を開始した |
• | 臨床試験や商業販売のための材料を製造し |
• | 臨床試験に成功した候補製品のために監督部門の承認を求める |
• | 販売、マーケティング、流通インフラを構築し、上場承認を得る可能性のある任意の製品を商業化する |
• | より多くの候補品を発見し開発することです |
• | 他の候補製品や技術を買収したり許可したり |
• | ライセンス契約に基づいて特許使用料、マイルストーン、または他の支払いを支払うこと |
• | 知的財産権の組み合わせを維持し、保護し、拡大する |
• | 技術人材を引きつけて引き留めること |
• | 上場企業としての私たちの運営と、私たちの製品開発·計画の将来の商業化努力を支援するために、追加のインフラを作成します。 |
我々の純損失は四半期ごとと毎年大きく変動する可能性があるため,我々の運営結果を経時的に比較することは将来の業績の良い指示ではない可能性がある。いずれの特定の四半期においても、私たちの経営業績は証券アナリストや投資家の予想を下回る可能性があり、これは私たちの株価を下落させる可能性がある。
利益を実現し、維持するためには、大量の収入を生む製品を開発し、最終的に商業化しなければならない。これは、私たちの候補製品の非臨床研究と臨床試験を完成させ、より多くの候補製品を発見し、これらの候補製品の監督管理許可を得ること、および私たちが規制承認を得る可能性のある任意の製品を含む、一連の挑戦的な活動で成功することを要求するだろう。私たちはただこのような活動の大多数の初期段階にいるだけだ。私たちはこのような活動で決して成功しないかもしれないし、たとえ私たちが成功しても、利益を達成するのに十分な収入が生まれないかもしれない。
医薬品開発に関連する多くのリスクや不確実性のため、費用を増加させる時間や金額、あるいはいつ、または利益を達成できるかどうかを正確に予測することはできない。FDAや欧州医薬品局が現在の予想外での研究を要求している場合、あるいは私たちの臨床試験や任意の候補製品の開発に遅延が生じた場合、私たちの費用が増加する可能性があります。
たとえ私たちが確実に利益を達成したとしても、私たちは四半期や年度の収益性を維持したり向上させることができないかもしれない。もし私たちが実現して利益を維持できなければ、会社の価値を下げ、資金を調達し、業務を拡大し、研究開発努力を維持し、製品を多様化し、さらには運営を継続する能力を弱める可能性がある。わが社の価値の低下はあなたの投資損失の全部または一部を招く可能性もあります。
私たちは私たちの目標を達成するために大量の追加資金を必要とし、必要な時に必要な資本を得ることができなければ、私たちの製品開発や商業化努力を延期、制限、減少、または終了させることを迫るだろう。
2019年12月31日現在、私たちの現金と現金等価物は4010万ドルです。私たちは予測可能な未来に、私たちはGen-009、Gen-011、および任意の他の新しい抗原癌ワクチン候補製品を開発するために大量の資源を投入し続けると信じている。これらの支出には、研究開発、新技術の獲得が可能であること、規制された承認および製品の製造、および販売が許可された製品(あれば)のマーケティングおよび販売に関連するコストが含まれる。さらに、他の予期しない費用も発生する可能性がある。私たちの計画と期待された臨床試験の結果は非常に不確定なため、私たちは合理的に成功を見積もることができません
17
私たちの候補製品の開発と商業化。また,臨床試験に関連する巨額の費用により,特定の候補製品のこのような試験を達成するのに十分な資本があるかどうかは確認できない
私たちの将来の資本需要は多くの要素に依存しています
• | 私たちが計画したGen-009とGen-011臨床試験の時間とコスト |
• | 計画された臨床試験のためのGen−009とGen−011の進捗、時間、およびコスト |
• | Gen-011のためのIND申請を含む規制承認の結果、時間、およびコストを求める; |
• | 私たちの他の候補製品と潜在候補製品の臨床前研究と臨床試験の開始、進捗、時間、コストと結果 |
• | 将来的には、協力、贈与、許可、相談、または私たちが確立する可能性のある他の予定の条項と時間; |
• | 任意の特許または他の知的財産権の許可、出願、起訴、弁護および実行に関連して、私たちが支払いを要求される可能性のある任意の支払いの金額および時間、または私たちが受け取る可能性のある任意の支払いの金額および時間は、私たちのライセンス契約に従ってマイルストーン支払い、使用料、および特許訴訟費用を支払う義務がある; |
• | 特許出願を準備、提出し、起訴し、私たちの知的財産権を維持し、保護し、知的財産権に関連するクレームを弁護するコスト; |
• | 私たちは他の製品や技術をどの程度許可したり取得したりします |
• | 上場承認文を受け取る |
• | 市場承認を得た場合、Gen−009およびGen−011および他の候補製品の商業化活動のコストは、製品販売、マーケティング、流通および製造能力を確立するコストおよび時間を含む |
• | 私たちの候補製品の商業販売から得られた収入。 |
私たちの現在の運営計画によると、私たちの既存の現金と現金等価物は、2021年第1四半期までの運営費用と資本支出需要をサポートするのに十分だと信じています
現在知られていない多くの要素のため、私たちの運営計画は変化する可能性があり、私たちは計画よりも早い追加資金が必要かもしれない。また、有利な市場条件や戦略的考慮により、現在または将来の運営計画のために十分な資金があると考えても、追加の資本を求めることができる。私たちが追加的な資金が必要な時、私たちは私たちが受け入れられる条項でこのような資金を得ることができないかもしれない。もし私たちが必要な時に利用できる十分な資金がない場合、私たちは、私たちの1つまたは複数の候補製品の非臨床研究、臨床試験または他の開発活動の延期、制限、減少または終了、または私たちの販売とマーケティング能力の確立、または他のおそらく私たちの候補製品を商業化するために必要な活動を延期、制限、または終了することを要求される。
私たちは、私たちが臨床開発を通じてGen-009やGen-011を成功的に推進し、任意の候補製品の規制承認を得るか、あるいは任意の候補製品または私たちの未来の任意の候補製品を商業化できるかどうかを決定することはできない。
現在、Gen-009は私たちの最先端の候補製品であり、私たちの将来の収入(もしあれば)はGen-009の成功した臨床進展、承認、商業化に大きく依存するだろう。Gen−009に加えて,Gen−011の臨床前作業を進めており,2020年第2四半期にINDをFDAに提出する予定である。私たちが商業化を開始することが許可される前に、Gen-009、Gen-011、および未来の候補製品は大量の臨床開発、テスト、および規制承認を必要とする。この過程には数年の時間がかかるかもしれないし、大量の資源の支出が必要になるかもしれないし、私たちは大量の追加資金を得る必要があると予想する。
私たちは現在Gen-009 B部分臨床試験を行っている。任意のオープンラベル研究と同様に、私たちは、初期の臨床信号が見られることを確実にするために、より小さい患者グループを評価するために、登録を減速または一時停止することができる。著者らは2020年第2四半期あるいは第3四半期に著者らのGen-009部分B臨床試験にこれらの初歩的な臨床結果を報告する予定である。この評価に基づいて、私たちは研究を継続するのに適しているかどうかを考慮するつもりだ。この研究を中止する決定は、Gen-009の臨床進展、承認、および商業化の遅延をもたらすだろう
18
追加資本の調達は、私たちの既存の株主を希釈し、私たちの運営を制限したり、不利な条項で私たちの技術または候補製品の権利を放棄することを要求する可能性があります。
これまで、相当な製品収入を生み出すことができれば、株式発行と戦略取引を組み合わせた方法で現金需要に融資する予定です。2018年1月には,普通株と普通株と優先株を同時に発行可能な引受権証および普通株を行使可能な引受権証(“同時発売”)により,約5,170万ドルの純収益を追加調達した。2019年2月、私たちは私募で約1,380万ドルの追加純収益を集めた。2019年6月、我々は普通株式と普通株が行使可能な引受権証を公開発行することにより、約3840万ドルの純収益を追加調達した。2019年10月、リンカーンパーク資本(“LPC”)と合意(“購入契約”)を締結し、250万ドルの追加純利益を集め、販売ごとに私たちの普通株の現行市場価格に基づいて、2750万ドルの普通株を単独で追加売却する権利があります。購入プロトコルは,我々がLPCに売却する普通株を5,227,323株普通株に制限し,購入合意日に発行された普通株の19.99%に相当する。購入契約も、LPCに任意の普通株の購入を指示することを禁止しており、もしその株式がLPCおよびその連合会社が当時実益を所有していた私たち普通株の他のすべての株式と合併すれば、LPCとその連合会社が任意の単一時点で実益が当時発行された普通株式総数の9.99%を超えることになる。Cowen and Companyとの市場持分発行計画に基づいて定期的に株を売却します, LLC(ATM)。私たちが株式または転換可能な債務証券を売却することによって追加資本を調達する場合、あなたの所有権資本は希釈され、条項は清算または株主としての権利に悪影響を及ぼす他の特典を含む可能性があります。債務融資が可能であれば、追加債務を招く、資本支出を行う、または配当を宣言するなど、私たちが何らかの行動をとる能力を制限または制限する契約を含むいくつかの合意が含まれるかもしれない。もし私たちが第三者との戦略的パートナーシップを通じてより多くの資金を調達するならば、私たちは私たちの技術または製品候補者、将来の収入フロー、研究計画、または製品候補者に対する貴重な権利を放棄しなければならないかもしれないし、私たちに不利な条項でライセンスを付与しなければならないかもしれない。もし私たちが必要な時に追加資金を集めることができない場合、私たちはGen-009、Gen-011、または私たちの他の候補製品に対する製品開発または商業化努力を延期、制限、減少または終了することを要求されるだろう。
もし私たちの優先株が普通株に変換されたり、発行された引受権証が行使された場合、私たちの株主は大量の追加希釈を受けるだろう。
2020年2月11日まで、私たちのAシリーズ転換可能優先株流通株は1,635株で、追加費用を支払うことなく204,375株の私たちの普通株に変換することができる。2020年2月11日まで、引受権証を行使する時に発行可能な普通株は5,122,183株であり、加重平均行権価格は1株7.66ドルである;発行済み株式オプションを行使する時に発行可能な普通株は1,310,927株であり、加重平均行権価格は1株11.72ドルである。我々Aシリーズの転換可能な優先株の流通株を普通株に変換したり、普通株の未償還オプションや株式権証を行使したりすることで、既存株主の権益を大幅に希釈する。どんな希釈や潜在的な希釈も、私たちの株主が彼らの株を売却することをもたらす可能性があり、これは私たちの普通株の株価を下方に移動させる可能性がある。
S-3表の棚登録声明によると、アメリカ証券取引委員会法規は、私たちが任意の12ヶ月の間に調達できる資金金額を制限します。
本年度報告をForm 10−K形式で提出してから60日間で,我々の公開流動資金は7500万ドル未満であった。したがって,Form S-3の一般的な指示I.B.6によれば,Form S-3登録声明を用いて任意の12カ月間に証券を初めて公開発行する(我々のATMを含む)ことで調達された資金は,我々の非関連会社が持つ投票権および無投票権のある普通株式の総時価の3分の1を超えてはならない.私たちは私たちの公開流通株が7500万ドルを超えるまでこの制限を受けている。もし私たちが別の形で新しい登録声明を提出することを要求されたら、私たちは追加費用が発生し、アメリカ証券取引委員会の審査によって遅延される可能性がある。
私たちの候補製品の臨床開発、規制審査、承認に関連するリスク
著者らはGen-009の臨床開発の成功に大きく依存し、Gen-009は現在活発な臨床試験にある唯一の候補製品である。Gen-009ワクチンの開発または商業化に成功しなかった場合、またはこの点でのいかなる重大な遅延も、私たちの業務、運営結果、および財務状況に重大な悪影響を及ぼすだろう
著者らは現在大量の努力と財政資源を投入してGen-009を開発しており、これは新しい抗原癌ワクチンであり、現在1/2 a期の臨床試験にある。私たちが製品収入を作る能力はGen-009臨床試験の成功とGen-009の成功開発と商業化に大きく依存する。Gen−009の成功した開発と商業化は以下のいくつかの要因に依存する
19
• | GEN-009に必要なすべての臨床試験を成功させた |
• | 監督部門のGen-009の上場承認を得た |
• | 私たちと第三者との間に製造と商業化の計画を作り |
• | 許容可能なGen−009のセキュリティおよび有効性の概要を確立すること; |
• | 医療支払者が患者に提供した009世代補償。 |
Gen−009の開発または商業化に成功しなかった場合、またはこの点でのいかなる重大な遅延も、私たちのビジネス、運営結果、および財務状態に重大な悪影響を及ぼすだろう。
私たちの活性候補製品は臨床開発の初期段階にあるため、失敗のリスクが高く、私たちは決して適切な製品の開発に成功したり、製品収入を生成したりすることはできないかもしれない。
われわれは現在Gen−009段階1/2 a臨床試験を行っている。我々は2020年第2四半期あるいは第3四半期に我々のGen-009第2部分B部分臨床試験の初歩的な臨床結果を報告する予定である。この評価に基づいて、私たちは研究を継続するのに適しているかどうかを考慮するつもりだ。この研究を中止する決定は、Gen-009の臨床進展、承認、および商業化の遅延をもたらすだろう。結果が成功しても,このような結果は後のより大規模な臨床試験では複製されない可能性がある。早期の小さな臨床試験が失敗する可能性がある他の原因の1つは,第3段階の開発や商業化に備え,生産規模を拡大する必要があることである。私たちの候補製品は複雑な製造技術を必要とする可能性があり、これらの技術を拡大することは製品の変化を招く可能性があり、製品が第三段階の試験期間中に更なるテストを行う時に現れるかもしれない
もし私たちの未来の臨床試験結果が私たちの候補製品に対する治療効果が定説がない場合、あるいは統計学的に意義のある臨床終点に達していない場合、あるいは私たちの候補製品に安全問題や副作用が存在すれば、候補製品の発売承認を阻止または延期される可能性がある。代替的に、私たちが規制部門の承認を得ても、この承認は、予期されたまたは期待されているような広範な適応や患者集団に適用されないか、または重大な使用または流通制限または安全警告を含むラベルが必要とされる可能性がある。私たちはまた、承認を得るために追加的または予期しない臨床試験を行うことを要求されるか、または規制部門の承認を維持するために追加の上場後試験要求を受ける可能性がある。また、規制当局は、その製品の承認を撤回したり、修正されたリスク評価および緩和戦略の形でその流通に制限を加えたりすることができる。
もし私たちの現在と未来の候補製品が規制部門の承認を得なければ、私たちの業務は悪影響を受けるだろう。
私たちの候補製品は研究、臨床試験、製造、輸入、輸出と商業化などの面で広範な政府法規の制約を受けている。任意の候補製品の商業販売の規制承認を得るためには、各目標適応における候補製品の使用が安全かつ有効であることを、広範な非臨床研究および臨床試験によって証明しなければならない。臨床試験は高価で時間がかかり、しかも結果は確定していない。私たちは、Gen-009、Gen-011、または私たちの現在または潜在的な他の臨床および非臨床候補製品に対する規制部門の承認を得ることができるかもしれないが、これらの製品は、いくつかの利用可能な地域または一部ではないが、すべての目標適応ではないが、すべての目標適応ではなく、規制部門の承認を得ることができ、それによって、私たちの候補製品のビジネス機会が限られているか、またはいかなる司法管轄区域でもこれらの候補製品のための規制部門の承認を得ないかもしれない
私たちの臨床試験に参加する患者を募集することは難しいかもしれませんが、候補製品の臨床試験を延期または阻止する可能性があります。
患者が著者らの候補製品の臨床試験に参加する資格を確定し、参加させることは著者らの成功に重要である。私たちの臨床試験の時間は私たちが患者を募集して私たちの候補製品のテストに参加する速度に依存する。もし患者が生物技術業界の不良事件の負の宣伝または他の原因(類似患者集団の競争的臨床試験を含む)のために著者らの研究に参加したくない場合、患者を募集し、研究を行い、監督機関の潜在的製品に対する承認を得る時間が延期または阻止される可能性がある。これらの遅延は,コスト増加,わが製品開発の遅延,我々の技術有効性試験の遅延,あるいは臨床試験の完全終了を招く可能性がある。
十分な数の患者を識別、募集、募集することができない場合があり、または研究において多様性を達成するために必要または所望の特徴を有する患者は、直ちに臨床試験を完了することができる。患者登録は以下の要素の影響を受ける
20
• | 調査中の病気の重症度は |
• | 学習プログラムの設計 |
• | 患者集団の規模は |
• | 試験に関する資格基準 |
• | 研究を受けた製品候補製品のリスクと収益を感知する |
• | 潜在患者に臨床試験場所の近似性と可用性を提供する |
• | 競争療法と臨床試験の有用性 |
• | 臨床試験への参加を促進するために努力しています |
• | 医師の患者への転職方法と |
• | 治療期間と治療後に患者を十分にモニタリングする能力がある。 |
十分な数の条件を満たす患者を募集できなければ、監督機関が要求する臨床試験に参加できなければ、臨床試験を開始したり、継続することができないかもしれない。計画通りの臨床試験を行うのに十分な数の患者を募集することが困難であれば、進行中または計画中の臨床試験を延期、制限または終了する必要があるかもしれず、いずれも私たちの業務に悪影響を及ぼす。
アメリカ国外で裁判を行う時、私たちは外国の管轄区域の要求を守ることができないかもしれない。
これまで米国以外では何の臨床試験も行われていない。もし私たちがどの国/地域で臨床試験の開始、登録、完成に成功しようとすれば、私たちが外国で業務を展開することに特有の多くのリスクは影響を受ける
• | 契約研究機関(“CRO”)と医師との関係を構築または管理することは困難である |
• | 臨床試験を行う異なる基準; |
• | 地元のコンサルタントや医師やパートナーを見つけることはできません |
• | 薬品と生物技術製品と治療の監督管理を含む各種の外国の法律、医療標準と監督管理要求の潜在的な負担を遵守する |
• | 米国国外で行われたBLAを支援する研究で得られたデータに対するFDAの受容可能性。 |
もし私たちがアメリカ国外での臨床試験の要求を満たすことに成功しなかったら、私たちは規制部門の候補製品に対する承認を得ることが遅れたり、得られなかったりする可能性がある。
著者らの臨床試験は重大な遅延に遭遇する可能性があり、あるいは著者らは安全性と有効性を証明できず、適用する監督管理機関を満足させることができないかもしれない。
規制部門から私たちの候補製品を販売する市場承認を得る前に、期待適応の候補製品の安全性と有効性を証明するために、広範な臨床試験を行わなければならない。臨床テストは高価で時間がかかり、しかも結果は確定していない。臨床試験が予定通りあるいは予定通りに完了することは保証されず,もしあれば。1つまたは複数の臨床試験の失敗は、試験の任意の段階で発生する可能性がある。成功を妨げたり、臨床開発をタイムリーに完成させたりする可能性のある事件は、
• | Gen-009臨床試験を行ったときの私たちまたは第三者の遅延は |
• | Gen-011のINDを含む規制機関との試験設計についての合意形成の遅延 |
• | 遅延は予期されるCROおよび臨床試験場所と許容可能な条項について合意した |
• | 各臨床試験地点で必要な機関審査委員会(“IRB”)の承認を得ることが遅延された |
• | 規制機関またはIRBは、ワクチンのような他の臨床試験によって提出された安全懸念を含む任意の理由で臨床休止を実施し、Gen-009またはGen-011の許容できないリスクを反映する可能性があり、または臨床操作または試験場所を検査した後; |
21
• | FDAの良好な臨床実践(“GCP”)または他の国/地域に適用される規制ガイドラインに従って操作できなかった |
• | 試験、検証、製造、臨床現場への候補製品の配送に遅延が発生した |
• | 患者が試験を完成していない或いは治療後のフォローアップに戻っていないことによる遅延 |
• | 臨床試験場所または患者が試験を終了したか、または投与を完了できなかった |
• | 臨床試験では、候補製品に関連する深刻な副作用が出現し、これらの候補製品は潜在的な利益を超えると考えられる |
• | 新しい臨床案の法規要件とガイドラインの変化を修正または提出する必要がある。 |
遅延は、上述の要素による遅延を含み、コストが高い可能性があり、そして著者らの臨床試験を完成する能力に負の影響を与える可能性がある。私たちは私たちが上述した要素や他のどんな要素によるどんな遅延もタイムリーにまたは完全に解決できるということを保証できない。その後の臨床試験の開始と完成に成功できなければ、規制部門の承認を得ることができず、候補製品を商業化することもできなくなるだろう。
我々の活性候補製品Gen−009、Gen−011、および我々の免疫腫瘍学計画によって産生される将来の潜在的製品候補は、ワクチン、免疫療法および医療治療の新しい方法であるT細胞に基づいて活性化されているか、またはT細胞に基づいて活性化されるであろう。
私たちはT細胞ワクチンと免疫療法技術の開発に集中しています。これはワクチン、免疫療法、医療の新しい方法であり、私たちの将来の成功はT細胞免疫療法の成功開発、特に私たちが積極的に開発した製品と現在と未来の候補製品に強く依存しています。したがって、私たちは製品開発の時間とコストを予測することが難しいかもしれない。ワクチンおよび免疫療法のためのT細胞方法の予見不可能な問題は、現在および将来の候補製品のさらなる開発または承認を阻害する可能性がある。我々や他のT細胞ワクチンや免疫療法を研究している人が将来遭遇する可能性のあるいかなる開発問題も重大な遅延や予期しないコストを招くことは保証されず,これらの開発問題が解決される保証はない。この方法の新規性により,我々が開発したワクチンや免疫療法には未知の安全リスクが存在する可能性がある。FDAなどの規制機関は,ワクチンや免疫療法によるまれかつ重篤な急性脳炎のリスクが低いことを証明するために,承認前に広範な安全試験を行うことを要求する可能性がある。承認されれば、ワクチンの新しい作用機序は医師と患者が私たちの製品の感知と吸収に不利な影響を与える可能性がある。
私たちの候補製品はすべての患者のために独特に製造されていて、私たちは生産中に困難に直面する可能性があり、特に私たちの製造能力を拡大する上で。もし私たちまたは私たちと契約した任意の第三者製造業者がこれらのタイプの困難に遭遇した場合、私たちは臨床試験に候補製品を提供したり、患者に私たちの製品を提供する能力(承認された場合)が延期または停止される可能性があり、あるいは商業的に実行可能なコスト構造を維持できないかもしれない。私たちのいくつかの第三者メーカーはアメリカ以外に位置しており、物流の原因による臨床材料の供給中断や、現地の監督監督による不利な規制行動をとるリスクに遭遇する可能性がある
私たちは私たちの候補製品をカスタマイズして設計して製造します。以下のような問題のため、このような候補製品を大量に製造することは、製品損失や故障を招きやすい
• | 患者の腫瘍や血液を採取することに関連する物流 |
• | 各患者の特定のバッチの独自性によって生じる可能性のある予測されていない特定のバッチ製造故障または問題; |
• | 品質管理検査に合格しなかった |
• | 安定性に置かれたロットは意外に失敗した |
• | 新しい分析方法や細胞選択や私たちの製造過程の他の構成要素 |
• | 個人化された製造に関連する巨大なコストは、私たちが発展し続ける能力に悪影響を及ぼすかもしれない |
• | 患者のための特定のバッチを成功的かつタイムリーに生産し、配布する |
• | ロットを患者の看護現場に搬送する過程で遭遇する積み込み問題; |
• | 私たちは単一供給者たちに依存している。 |
22
私たちのいくつかの候補製品は各患者のために生産されているので、私たちは、各患者のサンプル、これらのサンプルから得られた配列データ、これらの患者の免疫学的特徴を分析した結果、および各患者のためにカスタマイズされた製品に関連する身分チェーンの維持を要求されるであろう。このようなアイデンティティチェーンを維持することは困難で複雑であり、そうしないと、市場から任意の承認された製品を撤回することを含む、製品の混乱、不利な患者結果、製品損失、または規制行動をもたらす可能性がある。また、著者らの候補製品が早期臨床研究から後期臨床試験に発展し、承認と商業化に向かうに伴い、著者らは複雑な収集、分析、製造と交付過程の多くの方面で修正を行い、過程と結果の最適化に努力する予定である。これらの変化は予想された目標に達しない可能性があり、これらのいかなる変化も著者らの候補製品の表現が著者らの予想と異なることを招く可能性があり、それによって潜在的に臨床試験の結果に影響を与える。
我々のGen−009候補製品中のアジュバントを含む新規ワクチンアジュバントは、患者の安全リスクを増加させる可能性がある。
アジュバントは、ワクチン抗原に添加された化合物であり、免疫系の活性化を増強し、ワクチンの免疫反応及び効力を向上させる。新しいアジュバントを有するワクチンの開発は、治療薬の典型的な場合と比較して、承認前により多くの患者で評価する必要がある。FDAおよび他の規制機関および専門家委員会は、新規アジュバントを有するワクチンを評価するガイドラインを作成している。Gen−009を含む候補製品は、1つまたは複数の新しいアジュバントを含むことができる。いずれの新規抗原癌ワクチンも,アジュバントの存在により副作用がある可能性があり,患者にとって大きなリスクと考えられ,ワクチンの承認は保証されない。
もし私たちがアメリカ以外の管轄区で規制の承認を得られなければ、私たちはこれらの管轄区で私たちの製品を販売することができないだろう。
もし承認されたら、私たちは私たちの候補製品を国際市場に投入するつもりだ。このようなマーケティングは、各市場で個別の規制承認を得、多くの異なる規制要求を遵守する必要があるだろう。承認手続きは国によって異なり、追加テストの要求に関連する可能性があり、承認を得るのに要する時間は、FDAの承認を得るのに要する時間とは異なる可能性がある。また,米国以外の多くの国では,ワクチンはその国で販売が許可されるために承認されなければならない。場合によっては、私たちが私たちのワクチンのために受け取る予定の価格もまた承認されなければならない。FDAの承認は、他国又は管轄地域の規制機関の承認を確保するものではなく、外国規制機関の承認も他国の規制機関又はFDAの承認を確保することができない。外国規制機関の承認過程には、FDA承認の取得に関連するすべてのリスクが含まれる可能性がある。もしあれば、私たちは外国の規制部門の承認をタイムリーに得られないかもしれない。私たちは規制承認を申請できないかもしれないし、どの市場でも私たちのワクチンを商業化するために必要な承認を得ることができないかもしれない。
私たちの候補製品が規制部門の承認を得ても、これらの免疫療法は持続的な規制審査を受けることになり、多くの追加費用を招く可能性がある。さらに、私たちが開発している製品Gen-009、Gen-011、および任意の将来可能な免疫療法候補製品を含む私たちの候補製品は、承認されれば、ラベルや他の制限を受けるかもしれません。規制要件を遵守できなかったり、私たちの製品が予期しない問題に遭遇したら、処罰されるかもしれません。
私たちの候補製品のために得られた任意の規制承認は、その製品が発売される可能性のある承認適応の制限または承認条件によって制限される可能性があり、または第4段階の臨床試験を含む可能性の高い発売後試験要件を含むことができ、ワクチンまたは免疫療法の安全性および有効性を監視することが数年以内に行われる可能性がある。さらに、FDAが私たちの任意の候補製品を承認した場合、その製品の製造プロセス、ラベル、包装、流通、AE報告、貯蔵、広告、販売促進および記録は、広範かつ持続的な規制要件によって制限されるであろう。これらの要求には,安全や他の上場後の情報や報告,登録の提出,および我々が承認後に行った任意の臨床試験について,現在の良好な製造規範(CGMP)やGCPを継続して遵守することが含まれている。
その後、予期されていない重症度または頻度のAE、または製造操作またはプロセスを含む、以前に未知の承認された製品の問題が発見され、または法規要件を遵守できなかったことは、他を除いて、結果をもたらす可能性がある
• | 製品の販売または製造を制限し、製品を市場からリコールするか、または自発的または強制的に製品をリコールすること |
• | 罰金や警告状や臨床試験を一時停止した者は |
• | FDAは、我々が提出した係属中の出願または承認された出願の補充出願の承認を拒否するか、または製品ライセンスの承認を一時停止または撤回する |
23
• | 製品を差し押さえたり差し押さえたり、製品の輸入または輸出を許可することを拒否したり; |
• | 禁止または民事、刑事および/または行政処罰、損害賠償、罰金、返還、連邦医療保険、医療補助および他の連邦医療計画から除外され、私たちの業務を削減または再編する。 |
FDAの政策は変わる可能性があり、追加の政府法規が公布される可能性があり、これは私たちが獲得した候補製品に対する規制承認に影響を与える可能性がある。米国や海外の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが既存の要求の変化に適応できなかったり、新しい要求や政策を採用したり、あるいは規制適合性を維持できない場合、私たちは得られる可能性のあるいかなるマーケティング承認も失う可能性があり、私たちは利益を達成したり維持することができない可能性があり、これは私たちの業務に悪影響を及ぼすだろう。
私たちの第三者への依存に関するリスクは
私たちは第三者に依存して私たちの候補製品の技術開発、非臨床研究と臨床試験を行い、私たちの現役臨床開発製品Gen-009、Gen-011、および任意の他の未来の候補製品を含み、もし彼らが私たちに対する義務を正確かつ成功的に履行しなければ、私たちは監督部門の私たちの候補製品に対する承認を得ることができないかもしれない。
我々は、管理、監視、および他の方法でのGen−009およびGen−011臨床試験の実行を支援するために、第三者CROおよび他の第三者に依存し続けることを意図している。CRO,臨床データ管理組織,医療機関,臨床研究者など第三者に依存して臨床試験を行う予定である。私たちは他の多くの会社とこのような第三者の資源を争っている。私たちが通常依存している第三者はいつでも彼らの契約を終了することができ、代替手配を達成しなければならないことは、候補製品の開発と商業化を延期します。
私たちのこれらの第三者の研究開発活動への依存は、これらの活動に対する私たちの統制を減少させるだろうが、私たちの責任を軽減することはない。例えば、FDAおよび外国規制機関は、データおよび結果が信頼性および正確であることを保証し、試験参加者の権利、完全性、および機密性を保護するために、臨床試験結果を設計、実施、監視、記録、分析および報告する際に、GCPを含む法規および基準を遵守することを必要とする。われわれは第三者に依存して臨床試験を行っているが、各臨床試験がその全体的な研究計画と方案に従って行われることを確保する責任がある。
さらに、これらの第三者は、他のエンティティと関係がある可能性もあり、その中のいくつかは私たちの競争相手である可能性がある。もしこれらの第三者がそのプロトコル下の職責を成功裏に履行できなかった場合、もし彼らが獲得したデータの品質または正確性が彼らが臨床試験規程または規制要求を遵守できなかったことによって影響を受けた場合、あるいは彼らが臨床試験規程を遵守できなかった場合、あるいは予想された期限内に完成できなかった場合、私たちの候補製品の臨床試験は規制要求に適合しない可能性がある。もし臨床試験が監督管理の要求を満たしていない場合、あるいはこれらの第三者を交換する必要がある場合、非臨床開発活動或いは臨床試験は延長、延期、一時停止或いは終了される可能性がある。上記のいずれかの事件が発生した場合、私たちは規制部門の候補製品に対する承認をタイムリーにあるいは根本的に得ることができないかもしれない。
私たちはまた他の第三者に依存して私たちの臨床試験のための薬品の貯蔵と配布を望んでいる。私たちのディーラーのどんな業績ミスも、私たちの候補製品の臨床開発やマーケティング承認、あるいは私たちの製品の商業化を延期し、追加の損失を生じ、私たちの潜在的な製品収入を奪う可能性があります。
私たちは第三者に依存して製品製造のいくつかまたはすべての側面を行っているが、これらの第三者の表現は満足できないかもしれない。
私たちには生産施設や人員は何もありません。私たちは私たちの製品製造のすべての側面を独立して行うことを望んでいない。私たちはGen-009とGen-011を作ることで第三者に依存するつもりだ。私たちはまた第三者サプライヤーと製造業者に依存して他の臨床試験のためのワクチンを製造し、供給する。このような第三者への依存は、許容可能なコストまたは品質で十分な数の候補製品または製品またはそのような数を得ることができないリスクを増加させ、これは、私たちの開発または商業化努力を延期、阻止、または損害する可能性がある。
これらの第三者のいずれかはいつでも私たちとの契約を終了することができる。もし私たちが代替計画を達成する必要があれば、私たちの製品開発活動を延期するかもしれない。これらの第三者製造活動への依存は、これらの活動に対する私たちの制御を減少させるが、製造に関するすべての規定を遵守することを保証する責任は免除されない。
24
第三者製造商会に依存してリスクをもたらし、もし私たちが自分で候補製品を製造すれば、私たちはこれらのリスクの影響を受けません
• | ビジネス上合理的な条件で第三者と製造協定を交渉することはできない |
• | 規制コンプライアンスおよび品質保証を含む製造活動のすべての態様にサードパーティ製造業者が使用されるので、制御が減少する |
• | 私たちに代価または損害を与える方法で、または時間的に第三者との製造プロトコルを終了または更新しない; |
• | 私たちの臨床用品を生産する能力があるか、または生産する能力のある製造業者を得ることができず、遅延または追加の製造コストをもたらす |
• | 当社の契約製造業者は、当社のビジネス秘密および独自技術を含む、または第三者知的財産権の侵害を含む当社の固有情報を盗用する可能性があります |
• | 私たちの業務または運営とは無関係な条件による第三者製造業者またはサプライヤーの運営中断は、製造業者またはサプライヤーの倒産を含む。 |
これらの事件のいずれも、臨床試験の遅延や規制部門の承認を得られなかったり、将来の製品を商業化することに成功した能力に影響を与える可能性がある。いくつかの事件は、禁止、リコール、差し押さえ、または生産の完全な一時停止、または部分的な一時停止を含むFDAの行動の基礎となる可能性がある。
第三者メーカーは、米国以外のcGMP法規や同様の規制要件を遵守できない可能性がある。私たちまたは私たちの第三者製造業者が適用された法規を遵守できなかったことは、臨床封印、罰金、禁止、民事処罰、遅延、許可の一時停止または撤回、許可証の取り消し、候補製品または製品の差し押さえまたはリコール、運営制限、刑事起訴を含む制裁を実施する可能性があり、いずれも私たちの製品供給に重大な悪影響を及ぼす可能性があります。
私たちの候補製品と私たちが開発する可能性のあるどの製品も他の候補製品や製品と製造施設を競争する可能性があります。CGMP法規の下で運営されているメーカーの数は限られており、私たちのために製品を製造する能力があるかもしれない。
私たちの既存または未来のメーカーのどんな業績失敗も、臨床開発やマーケティング承認を延期する可能性がある。私たちは現在、Gen-009とGen-011に余分な供給源または第2の供給源を提供するように手配されていない。もし私たちの現在の契約製造業者が合意通りに履行できなければ、私たちはこれらのメーカーの交換を要求されるかもしれない。いくつかの潜在的な代替メーカーが私たちの候補製品を生産できると信じていますが、そのような代替製品を決定して同定する際に、追加のコストと遅延が生じる可能性があります。
私たちの現在と未来の他人が私たちの候補製品や製品を生産することへの依存は、私たちの将来の利益率と、マーケティングの承認をタイムリーかつ競争力的に獲得する製品の商業化能力に悪影響を及ぼす可能性があると予想されています。
もし私たちが私たちの製品を十分な数量や生産量で生産できない場合、あるいは規制機関の私たちの製品の製造施設の承認を得ることができなければ、製品開発、臨床試験、監督管理の承認、商業流通の面で遅延に遭遇する可能性があります。
私たちの臨床試験と私たちの候補製品の商業化を完成するには、十分な生産量と商業規模で私たちの候補製品を製造するために、施設を獲得または開発する必要があります。私たちは第三者が私たちの候補製品を製造したり管理したりした経験がありません。これらの製品は大規模な臨床試験や商業販売を支援するために必要です。これらの能力を確立する努力は、進度、拡大規模、再現性、生産量、純度、コスト、効力或いは品質など、最初の予想に達しない可能性がある。
私たちは第三者が私たちの候補製品を生産する臨床的かつ商業的なロット(必要であれば)に依存すると予想される。これらの第三者メーカーはまた、臨床材料や商業製品を生産するために、FDAの承認を得なければならない。私たちの製品は他の製品とこれらの施設の使用権を競争するかもしれません。第三者が他の製品にもっと高い優先権を与えれば、私たちの製品は製造が遅れる可能性があります。私たちは許容可能な条項やタイムリーに必要な第三者製造計画を達成できないかもしれない。さらに技術交渉の段階に入らなければならないかもしれません
25
契約を譲渡し、第三者メーカーと技術ノウハウを共有することは、時間がかかり、遅延を招く可能性があります。
私たちの契約製造業者への依存は、私たちの運営に悪影響を及ぼすかもしれないし、予見できない遅延や他の私たちがコントロールできない問題を招くかもしれない。契約の制限と専門知識を持つ第三者メーカーの数が限られているため、私たちの大口ワクチンを大規模に生産するためには、監督部門の許可と施設を得る必要があり、メーカーの交換は高価で時間がかかり、私たちのワクチン生産の中断を招く可能性がある。第三者製造業者たちはまた生産で困難に直面する可能性がある。これらの問題には
• | 生産コスト、規模拡大、生産量の困難 |
• | 原材料や供給品を得ることができません |
• | 品質管理と保証が不十分である |
• | 人材が足りない |
• | 厳格に施行された連邦、州、外国の法規を遵守できなかった。これらの法規は、製品を販売する可能性のある国によって異なる |
• | 資本資金が不足している。 |
したがって、どんな遅延または中断も、私たちの業務、財務状況、運営結果、およびキャッシュフローに実質的な悪影響を及ぼす可能性がある。
私たちは戦略的パートナーシップの構築と維持に成功できないかもしれませんが、これは私たちが製品を開発し、商業化する能力に悪影響を及ぼすかもしれません。
私たちの戦略の一部は評価であり、適切と考えられる場合には、主要なバイオテクノロジーや製薬会社とパートナーシップを構築することを含む、将来的に戦略的魅力があるときにパートナーシップを構築する。私たちは私たちの候補製品のために適切なパートナーを探す上で激しい競争に直面しており、交渉過程は時間がかかり複雑である。私たちが候補製品との協力を成功させるためには、潜在的なパートナーは、私たちが求めている条項や他の会社が許可することができる製品に基づいて、これらの候補製品が魅力的だと思う市場で経済的価値があると考えなければならない。たとえ私たちが戦略的パートナーシップの構築に成功したとしても、私たちが合意した条項は私たちに不利になる可能性があり、例えば、製品の開発や承認が延期または承認された製品販売に失望した場合、私たちはこのような戦略的パートナーシップを維持できないかもしれない。私たちの候補製品に関連する戦略的パートナー協定のいかなる遅延も、私たちの候補製品の開発と商業化を延期し、市場に進出してもそれらの競争力を低下させる可能性がある。
しかも、私たちの戦略的パートナーは彼らと私たちの合意に違反するかもしれないし、私たちはこのような合意の下で私たちの権利を十分に保護できないかもしれない。また、承認されれば、私たちの戦略パートナーは、私たちの候補製品の開発と商業化決定を制御するいくつかの権利について交渉することができ、私たちと同じ方法でこれらの活動を行わないかもしれない。
もし私たちが私たちの候補製品に関する戦略的パートナー関係を構築し、維持できなければ、私たちはこのような候補製品の開発に関連するすべてのリスクとコストを負担し、追加の資金を求め、より多くの従業員を雇用し、他の方法で私たちが予算を持っていない専門知識を開発する必要があるかもしれない。これは協力していない候補製品の開発に否定的な影響を及ぼすかもしれない。
また,我々は現在,我々の新しい抗原癌ワクチン候補のアジュバントや送達技術を持つ会社と戦略的パートナーシップを構築することを求めている。これらのパートナーシップの構築に成功できなければ,新たな抗原癌候補ワクチンを開発する能力は悪影響を受ける可能性がある
私たちの知的財産権に関するリスクは
もし私たちが私たちの候補製品に関連する知的財産権を獲得したり保護したりできなければ、私たちは私たちの市場で効果的に競争することができないかもしれない。
私たちは、特許、特許出願、ノウハウ、およびセキュリティプロトコルの組み合わせによって、私たちのプラットフォーム技術および候補製品に関連する知的財産権を保護します。バイオテクノロジー会社の特許地位は一般的に不確実であるが、それは複雑な法律と事実考慮に関連しているからだ。アメリカが採用している基準は
26
米国特許商標局(“米国特許商標局”)および外国特許庁は,特許を付与する際に常に統一的または予測可能に適用されるわけではない。例えば、バイオテクノロジー特許において許容される特許標的または特許請求の範囲については、世界的に統一された政策はない。私たちが所有しているまたは許可中の特許出願は、発行された特許を生成できない可能性があり、その声明は、米国または他の国/地域における私たちの発見プラットフォームまたは候補製品をカバーする。私たちの特許および特許出願または私たちの許可者の特許および出願に関連するすべての潜在的以前の技術が発見されたことは保証されず、私たちが開示していない以前の技術は、特許を無効にするために、または係属中の特許出願から特許が発行されることを阻止するために第三者によって使用される可能性がある。特許が確実に発行されても、これらの特許が私たちの発見プラットフォームまたは候補製品をカバーしていても、第三者はその有効性、実行可能性、または範囲に疑問を提起する可能性があり、これは、このような特許が縮小または無効になる可能性がある。また,我々の特許や特許出願が挑戦されていなくても,我々の特許や特許出願,あるいは我々の許可側の特許は,我々のプラットフォーム技術を十分に保護することができず,我々の候補製品に排他性を提供し,他の人が類似製品を用いて我々の特許を迂回して設計することを阻止したり,特許保護を求めていない司法管轄区域内で他の人が運営することを阻止したりする可能性がある.これらの結果のいずれも、第三者の競争を阻止する能力を弱める可能性があり、第三者競争は私たちの業務に悪影響を及ぼす可能性がある。
もし私たちが私たちのプラットフォームまたは候補製品の保有または許可された特許出願について発表できなかった場合、それらの保護の広さや強度が脅かされている場合、またはそれらが私たちの候補製品やATLAS発見プラットフォームに意味のある排他性を提供できなかった場合、それは会社が私たちとの協力を阻止し、私たちの1つまたは複数の製品、さらには任意の製品を開発または商業化する能力を制限または破壊する可能性がある。私たちまたは私たちの許可者たちは私たちの候補製品の様々な側面に関連して複数の特許出願を提出した。私たちは、どのような特許が発行されるか(あれば)、どのような特許の広さ、または発行された任意の特許が無効であるか、強制的に実行できないか、または第三者の挑戦を受けるかどうかを保証することはできない。これらの特許出願またはそれらが発行する可能性のある特許への成功反対、または私たちが所有または許可してくれた任意の他の特許出願または特許への成功反対は、私たちが開発する可能性のある任意の候補製品の商業化に必要な権利を奪う可能性がある。米国およびほとんどの他の国の特許出願は提出後しばらくは秘密であり、一部の特許出願は発行前に依然として秘密であるため、候補製品の任意の特定の態様に関連する特許出願を最初に提出した会社であるか、私たちまたは私たちの許可者が初めてであることを確認することはできない。
米国では,2013年3月16日までに提出された特許出願については,他の特許性要件を満たし,最初に発明した者が特許を有していると仮定しているが,米国以外では,まず特許出願を提出した者が特許を有している。2013年3月16日、米国は、初めて特許出願を提出した発明者が特許を取得する権利がある世界の他の場所のような“第1次出願”制度に移行した。既存の制度または現行制度の下で、第三者が特許発行の前に先行技術を提出することを許可する。さらに、米国および外国の特許制度は、第三者を許可するか、または場合によっては特許当局自身が訴訟を提起することを許可し、例えば、異議、派生、再審、当事者間の審査または妨害訴訟を含む、発行された特許の範囲および/または有効性に疑問を提起することを可能にする。このような提出、訴訟または訴訟において、当方または当方の許可側の特許権に不利な裁決を下すことは、当方の特許権の範囲を縮小したり、当方の特許権を無効にしたりする可能性があり、第三者に対する当方の競争地位に悪影響を及ぼす可能性があります。
しかも、特許の寿命は限られている。米国を含む多くの国では,特許の自然失効期間は出願日から20年である。ある国では異なる特許期間が延長される可能性があるが,すべての場合,特許の有効期限とその提供される保護は限られている。もし私たちが規制部門の承認を得るのに遅延があれば、私たちが特許保護された製品を販売する時間が短縮されるかもしれない。私たちは私たちが特許を起訴したどの国/地域でも特許期間の延長を受けることを望んでいる。このような可能な延長には、FDAによって承認された製品をカバーするために、特許期間を最大5年間延長することを可能にする米国の1984年の“医薬品価格競争および特許期限回復法”が許可された延長が含まれる。しかし、適用当局は、米国のFDAと他の国/地域のいずれの同等の規制機関も含めて、このような延期が利用可能かどうかの評価に同意しない可能性があり、私たちの特許延期の承認を拒否するか、または私たちが要求したよりも限られた延期を承認する可能性があるかもしれない。このような状況が発生すれば、私たちの競争相手は私たちの臨床と非臨床データを参考にすることで、私たちの開発と臨床試験への投資を利用することができ、その後、他の場合よりも早く彼らの製品を発売することができるかもしれない。
私たちのプラットフォームや候補製品では、世界各地のすべての国で特許の申請、起訴、強制執行は目を引くほど高価であり、米国以外のいくつかの国での知的財産権は米国ほど広くないかもしれない。また、いくつかの外国の法律は知的財産権の保護の程度はアメリカの連邦や州法律に及ばない。したがって、私たちは、第三者がアメリカ以外のすべての国で私たちの特許を侵害したり、アメリカや他の管轄区域で私たちの特許を侵害している製品を販売したり、輸入したりすることを防ぐことができないかもしれない。競争相手は私たちが特許保護を受けていない司法管轄区域で私たちの技術を使用して自分の製品を開発することができ、また、他の侵害製品を私たちが特許保護を持っている地域に輸出することもできるが、法執行力はアメリカに及ばない。これらの製品は私たちの製品と競争するかもしれませんが、私たちの特許や他の知的財産権は彼らの競争を効果的にまたは阻止するのに十分ではないかもしれません。
27
特許保護を失ったり、獲得できなかったりすることは、私たちの業務に実質的な悪影響を及ぼす可能性がある。私たちは競争相手が私たちの製品と似ているか同じ製品で市場に入るのを防ぐことができないかもしれない。
私たちは私たちの知的財産権を擁護したり実行したりする訴訟に巻き込まれるかもしれないが、これは高価で時間がかかり、成功しないかもしれない。
競争相手は私たちの特許を侵害したり、流用したり、他の方法で私たちの知的財産権を侵害したりする可能性があり、競争相手または他の第三者は、これらの権利の有効性または実行可能性を疑問視する可能性がある。侵害または無許可の使用に反撃するためには、または他の挑戦に対応するために、私たちの知的財産権を強制的に実行または擁護し、私たちの商業秘密を保護し、および/または私たち自身の知的財産権または他の人の固有の権利の有効性および範囲を決定するために訴訟を提起する必要があるかもしれない。そのような訴訟は高くて時間がかかるかもしれない。私たちは現在、潜在的な多くの競争相手と私たちよりも多くの資源を投入して知的財産権を起訴する能力を持っている。したがって、私たちは努力したにもかかわらず、私たちは第三者が私たちの知的財産権を侵害したり流用したりすることを防ぐことができないかもしれない。訴訟は巨額のコストと管理資源の移転を招く可能性があり、これは私たちの業務や財務業績を損なう可能性がある。さらに、論争のある訴訟では、裁判所または機関は、私たちが所有または許可している特許が無効または強制的に実行できないと判断することができ、または、私たちの特許が論争のある技術をカバーしていないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。どんな訴訟手続きの不利な結果も、私たちの1つ以上の特許を無効が宣言されるか、強制的に実行できないか、または狭い解釈される危険性に直面させる可能性がある。さらに、知的財産権訴訟は大量の開示を必要とするため、このような訴訟の間、私たちのいくつかの機密情報は開示によって漏洩される可能性がある。
第三者による知的財産権侵害や流用のクレームは、我々の開発と商業化努力を阻害または延期する可能性がある。
私たちのビジネス成功は、私たちの候補製品を開発、製造、マーケティング、および販売する能力、および第三者特許および独自の権利を侵害することなく、私たちまたは私たちのライセンシーの独自技術を使用する能力にある程度依存する。米国国内外では、特許侵害訴訟、介入、異議、再審及び米国特許庁と対応する外国特許庁に提起された各当事者間審査プログラムを含む生物技術と製薬業界特許及びその他の知的財産権に関する訴訟が大量にある。我々が開発している分野には,米国や外国から発行された特許や第三者が所有する未解決特許出願が多く存在し,我々の候補製品を開発することが可能である.バイオテクノロジーや製薬業界の拡張や特許の発行に伴い、我々の候補製品が第三者特許権侵害の告発を受けるリスクが高まる可能性がある。
第三者は私たちが許可されていない状況で彼らのノウハウを使用していると主張するかもしれない。材料、処方、製造方法、分析方法および/または治療方法のような、我々の製品または候補製品の使用または製造に関連する第三者特許または特許出願が存在する可能性がある。場合によっては、私たちはそのような関連する第三者特許または特許出願を識別できないかもしれない。例えば、2000年11月29日までに提出された出願およびその日後に提出されたいくつかの出願は、特許発行前に米国国外では提出されないが、依然として秘密にされるであろう。上記の例外を除いて、米国および他の地方の特許出願は、通常、最初に提出されてから約18ヶ月の待機期間後にのみ公表される。したがって、私たちのプラットフォーム技術または私たちの製品または候補製品に関する特許出願は、私たちが知らずに他の人によって提出されたかもしれない。さらに、いくつかの制限された場合、公表された保留特許出願は、我々のプラットフォーム技術、私たちの製品または候補製品、および/または私たちの候補製品の使用、分析および/または製造をカバーするために、後で修正することができる。
管轄権のある裁判所が任意の第三者特許を保有している場合、私たちの材料、調製、製造方法、分析方法および/または治療方法などをカバーする場合、そのような特許の所有者は、その特許が満了するまで、または私たちが許可を得ない限り、私たちが適用可能な候補製品を開発および商業化する能力を阻止することができるだろう。このようなライセンスは、もしあれば、受け入れ可能な条項で提供されないかもしれない。たとえ私たちが許可を得ることができても、これらの権利は非排他的である可能性があり、これは私たちの競争相手が同じ知的財産権を獲得することをもたらす可能性がある。最終的に、実際または脅威の特許侵害クレームにより、許容可能な条項で許可を得ることができない場合、製品の商業化を阻止されたり、何らかの態様の業務運営を停止させられたりする可能性がある。
我々にクレームをつけた当事者は、禁止または他の公平な救済を受ける可能性があり、これは、私たちの1つまたは複数の候補製品のさらなる開発と商業化を効果的に阻止することができる。結果にかかわらず、特許侵害や商業秘密の流用疑惑を弁護するのは費用が高く、時間がかかる可能性がある。したがって、私たちが最終的に勝訴したり、早い段階で和解が成立したりしても、このような訴訟は私たちに予期せぬ費用をもたらす可能性がある。また,訴訟や脅威訴訟は,我々の管理チームの時間や注意力に大きな要求を与え,会社の他の業務への追求を分散させる可能性がある.もし私たちが提出した権利侵害クレームが成功すれば、巨額の費用を支払う必要があるかもしれません
28
故意の侵害の3倍の損害賠償と弁護士費、印税の支払い、私たちの侵害製品の再設計、または第三者から1つ以上の許可を得ることは不可能かもしれないし、多くの時間とお金の支出を必要とする可能性がある損害賠償。
第三者が第三者がこの第三者のビジネス秘密を不正に入手して使用していると考えていれば、公金を流用した疑いに直面する可能性がある。第三者のビジネス秘密の盗用が発見された場合、このようなビジネス秘密のさらなる使用を阻止され、候補製品を開発する能力を制限され、損害賠償金の支払いが要求される可能性があります。任意の特許または他の知的財産権訴訟において、聴聞結果、動議裁決、および訴訟における他の一時的手続きが公表される可能性がある。もし証券アナリストや投資家がこれらの声明が否定的だと思うなら、私たちの製品、手続き、または知的財産権の知覚価値は低下するかもしれない。したがって、私たちの普通株の市場価格は下がるかもしれない。
私たちは私たちの知的財産権の一部を許可しました。もし私たちがこれらの手配の下で私たちの義務を履行できなかった場合、あるいは私たちのライセンシーが知的財産権を獲得して維持できなかった場合、私たちはこのような知的財産権を失ったり、そのような知的財産権の許可者に損害賠償金を支払うことができます。
私たちは私たちの業務に非常に重要な許可と協力協定に多くの署名をして、将来私たちは他の許可や協力協定を締結するかもしれません。例えば、私たちの発見プラットフォームは、学術や研究機関が独占的に許可した特許をめぐってある程度確立されている。ハーバード大学とOncovirと締結したライセンス内契約の説明については、“ビジネスライセンス契約”を参照されたい。これらの許可および他の許可は、すべての関連する使用分野および将来的に私たちの技術および製品の開発、またはそれを商業化するすべての地域でそのような知的財産権および技術を使用することを望む独占的な権利を提供しない可能性がある。私たちは合理的な費用や合理的な条項で追加的なライセンスを得ることができないかもしれない。したがって、私たちは競争相手が私たちのすべてのライセンスに含まれる地域で競争製品を開発し、商業化することを阻止できないかもしれない。この場合、私たちは、私たちの候補製品を再設計するために多くの時間と資源を必要とするか、または代替技術を開発または許可する必要があるかもしれません。これらは、技術的にも商業的にも不可能かもしれません。もし私たちがそれができなければ、影響を受けた候補製品を開発したり商業化することができないかもしれません。これは私たちの業務を深刻に損なう可能性があります。
私たちの既存の許可協定は、私たちが未来の許可協定が私たちに様々な勤勉さ、マイルストーン支払い、特許権使用料、その他の義務を負担することを要求すると予想している。例えば、我々の既存のライセンス契約では、将来の合意では、当社がライセンスしている技術の特許訴訟はライセンス側によって制御される可能性があり、ライセンス側の特許訴訟費用の償還を要求される可能性があると予想されています。もし私たちの許可者が彼らから私たちが許可した独自知的財産権の特許や他の保護を獲得し、維持できなかった場合、私たちは知的財産権に対する私たちの権利やこれらの権利に関する排他性を失う可能性があり、私たちの競争相手は知的財産権がカバーする競争製品を販売するかもしれない。さらに、私たちのライセンス契約では、私たちが許可した特許を侵害した第三者に対して訴訟を提起する責任があるかもしれません。私たちのライセンスパートナーとライセンス契約下の権利または義務の面で紛争、論争、分岐または不履行の問題があれば、私たちがそのような合意での支払い義務を履行できなかったことによる任意のこのような紛争、論争、または分岐が損害を受ける可能性があり、私たちの許可者は、影響を受けた許可を終了する権利がある可能性があり、薬物発見および開発作業において影響を受けた知的財産権を使用する能力、および影響を受けた候補製品のための協力またはマーケティング合意を達成する能力が悪影響を受ける可能性がある。例えば、知的財産権に関する紛争が発生する可能性があるが、許可協定を遵守しなければならない, ライセンス契約に従って付与された権利の範囲および他の解釈に関連する問題を含む;私たちの技術は、許可者が許可協定によって拘束されない知的財産権をどの程度侵害しているか、任意の協力開発関係下での特許および他の権利の再許可、許可協定下での職務義務およびどのような活動がこれらの職務義務を満たしているか、私たちの許可者および私たちおよびそのパートナーが知的財産権を共同で創造または使用することによって生じる発明および独自技術の発明または所有権、ならびに特許技術の発明優先である。私たちが許可している知的財産権をめぐる紛争が許容可能な条項で現在の許可手配の能力を維持していることを阻害したり、損害を与えたりすれば、影響を受けた候補製品の開発に成功し、商業化することができない可能性がある。
従業員および第三者と締結された秘密保護協定は、不正な独自情報の漏洩を防ぐことができない可能性がある。
特許提供の保護に加えて、私たちは、特許を申請できない可能性がある、または特許を出願しない可能性のある独自技術、特許を強制的に実行することが困難なプロセス、および私たちのプラットフォーム技術の任意の他の要素、ならびに特許がカバーされていないノウハウ、情報または技術に関する発見および開発プロセスをセキュリティプロトコルによって保護する。私たちは、従業員、コンサルタント、外部科学コンサルタント、請負業者、協力者と秘密保護協定を締結することで、私たちのノウハウやプロセスを保護することを求めています。私たちは合理的な措置を取って私たちのノウハウを保護しているにもかかわらず、私たちの従業員、コンサルタント、請負業者、または外部科学コンサルタントは、私たちのノウハウ情報を競争相手に意図的にまたは意図的に漏らしてしまう可能性がある。さらに、競合他社は、我々のノウハウを他の方法で取得したり、実質的に同じ情報および技術を独立して開発したりする可能性がある。
29
第三者が私たちのどんな独自技術を不法に取得して使用することを強制するのは高価で時間がかかり、結果は予測できない。また、米国以外の裁判所は米国裁判所のようにノウハウを保護しようとしない場合がある。私たちのノウハウを流用または不正に開示することは、私たちの競争的地位を損なう可能性があり、私たちの業務に実質的な悪影響を及ぼす可能性がある。
私たちの候補製品の商業化に関するリスクは
私たちの将来の商業成功は私たちの候補製品が承認されれば、医者、患者、第三者支払人と医学界の他の人の中で著しい市場受容度を得ることにかかっている。
私たちがGen-009、Gen-011、または私たちが将来開発または買収する可能性のある任意の他の製品のマーケティング承認を得たとしても、この製品は医師、第三者支払人、患者、および医学界の他の人の市場受け入れを得ることができないかもしれない。さらに、任意の承認された製品に対する市場の受け入れ度は、多くの他の要因に依存する
• | 臨床試験で証明されたこの製品の有効性と安全性 |
• | 製品の臨床適応を承認し、ラベル上で要求される可能性のある任意の警告を含む、規制部門によって製品と共に使用されることを許可するラベル; |
• | 医師や患者がこの製品を安全かつ有効な治療法として受け入れ,目標患者群が新たな治療法を試みる意欲と医師が新たな治療法を開発する意欲; |
• | 代替療法と比較して、治療のコスト、安全性、有効性 |
• | 第三者支払者と政府当局は十分な保険と補償を提供する |
• | 相手が便利で管理しやすい |
• | 副作用の発生率と重症度 |
• | 私たちの販売と市場普及の効果は |
• | 私たちの製品を他の薬(あれば)と一緒に使用することを制限する。 |
市場受容度は私たちが相当な収入を作る能力に必須的だ。任意の候補製品は,承認されて商業化されていれば,限られた能力のみを受け入れることができるか,あるいは全く受け入れないことができる。もしどんな承認された製品も市場に受け入れられなければ、私たちは相当な収入を生むことができないかもしれません。私たちの業務は影響を受けるでしょう。
もし私たちが販売、マーケティング、流通能力を確立できなければ、私たちの候補製品が承認されれば、私たちはそれを商業化することに成功できないかもしれない。
私たちは医薬製品を販売したり、マーケティングしたり、流通した経験もありません。私たちが発売許可を得た任意の製品をビジネスに成功させるためには、販売とマーケティング組織を構築する必要がある。
将来的には、いくつかの候補製品が承認された後、米国でマーケティングや共同普及を行うために、重要な販売·マーケティングインフラを構築したいと考えています。私たち自身の販売、マーケティング、そして流通能力を確立することはリスクに関するものだ。例えば、販売チームの採用と訓練は高価で時間がかかり、どんな製品の発表も延期される可能性がある。販売チームを募集し、マーケティング能力を確立する候補製品の商業発表が何らかの理由で延期または発生していない場合、これらの商業化費用を早期または不必要に発生させる。これは費用が高いかもしれません。もし私たちが私たちの販売とマーケティング担当者を維持したり再配置できなければ、私たちの投資は損失します。
私たち自身が製品を商業化するのを阻害するかもしれません
• | 十分な数の効果的な販売とマーケティング担当者を募集し、訓練し、維持することはできません |
• | 販売員は医者に触れることができない |
• | 未来の製品を処方するのに十分な数の医師が足りません |
• | 販売者が提供するセット製品の不足は、より広い製品ラインを持つ会社と比較して競争劣勢になる可能性がある |
30
• | 独立した販売およびマーケティング組織の作成に関連する予測不可能なコストおよび費用。 |
もし私たちが自分の販売、マーケティング、流通能力を確立することができなければ、第三者と合意してこれらのサービスを提供することができなければ、私たちの製品の収入と収益力(できれば)は、私たちが自分で開発したどの製品もマーケティング、販売、流通よりも低くなるかもしれません。さらに、私たちは第三者との販売、マーケティング、流通に成功できないかもしれません。あるいは私たちに有利な条項でそうすることができないかもしれません。私たちはこのような第三者に対して支配権がほとんどないかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが販売、マーケティング、流通能力を成功的に確立できなければ、私たち自身も第三者と協力しても、私たちの候補製品を商業化することに成功しないだろう。
私たちの候補製品はいくつかの細分化された市場のカバー範囲と精算範囲が限られているか、利用できないかもしれません。これは私たちが利益を得て製品を販売することを難しくするかもしれません。
任意の承認された製品の市場受容度および販売は、第三者支払者が十分な保険および補償を提供するか否かに大きく依存し、既存および将来の医療改革措置の影響を受ける可能性がある。政府医療計画、個人健康保険会社、管理医療組織のような第三者支払人は、どの薬に保険を提供し、精算レベルを確立するかを決定する。第三者支払者の製品使用状況の決定を含む第三者支払人の保証範囲と精算決定は多くの要素に依存する可能性がある
• | 健康計画の下で保障された福祉 |
• | 安全で効果的で医学的に必要なものです |
• | 特定の患者に適しています |
• | 費用対効果があります |
• | 実験的でも調査的でもない。 |
第三者支払者は,海外でも国内でも,政府のものでも商業的でも,ますます複雑な方法を開発して医療コストを抑えている。保証範囲と精算範囲は支払人によって違います。そのため、各政府や他の第三者支払人から製品の保証と精算承認を得るには、私たちの製品を使用する支持的な科学的、臨床的、費用対効果的なデータを個別に各支払人に提供する必要があるかもしれませんが、保証や精算の承認を得るのに十分なデータを提供できる保証はありません。私たちは私たちのどんな候補製品も保険や十分な精算を受けることができることを確実にすることができない。また、保険範囲の決定や補償金額が私たちの製品に対する需要を減少させないことを保証したり、製品価格を下げたり、割引を提供したりすることを要求することはできません。もし精算が得られない場合や限定精算だけでは、私たちは特定の製品を商業化できないかもしれない。また,米国では,第三者決済者が新薬のカバー範囲や精算レベルを制限することで医療コストを抑制しようとするようになってきている。したがって,第三者支払者が患者に新たに承認された薬物を多少補償するかどうかには大きな不確実性があり,逆に薬品定価に圧力を与える。
価格規制が施行されるかもしれないが、これは私たちの未来の収益性に悪影響を及ぼすかもしれない。
国際市場では,精算や医療保険支払い制度は国によって大きく異なり,多くの国で特定製品や療法に価格上限が設定されている。いくつかの国、特にEU加盟国では、処方薬の価格設定は政府によって統制されている。これらの国では、製品の発売許可を受けた後、政府当局との定価交渉にかなりの時間がかかる可能性がある。また,費用抑制措置の一部として,政府や他の利害関係者は保険,価格,補償レベルにかなりの圧力をかける可能性がある。政治、経済、規制の発展は定価交渉をさらに複雑化させる可能性があり、保険や補償を受けた後、定価交渉は継続される可能性がある。EU加盟国が使用する参考価格と平行分配、あるいは低価格と高価な会員国の間の裁定は、価格をさらに下げる可能性がある。いくつかの国では、私たちは臨床試験または他の研究を要求され、私たちの候補製品の費用効果を他の利用可能なワクチンと比較して、カバー範囲、精算または定価承認を得たり維持したりすることが要求されるかもしれない。第三者支払者や主管当局が割引を公表することは、公布国や他の国の価格や補償レベルにさらなる圧力を与える可能性がある。私たちの候補ワクチンが第三者支払者によって費用効果があるとみなされることは保証できません。十分な精算レベルが保証されていませんし、第三者支払者の精算政策が私たちの販売製品の収益性に悪影響を与えない保証もありません。もし私たちの製品が精算を受けられない場合、あるいは範囲や金額が制限されている場合、あるいは定価のレベルが満足できない場合, 私たちの業務は不利な影響を受けるかもしれない。
31
医療改革立法や医療業界や医療支出の他の変化がわれわれに与える影響は不明であり,われわれのビジネスモデルに悪影響を及ぼす可能性がある。
アメリカと一部の外国司法管轄区域では、立法構造が変化し続けている。私たちの収入の見通しはアメリカと海外の医療支出と政策の変化の影響を受けるかもしれない。私たちは、高度に規制された業界で運営されており、ヘルスケア獲得性、医療製品およびサービスの交付または支払い方法に関連する新しい法律または司法判断、または既存の法律または決定に対する新しい解釈は、私たちの業務、運営、および財務状況に悪影響を及ぼす可能性がある。米国では“医療改革法”が公布され,現在“医療改革法”の廃止や大幅な改正に努めており,医療改革の促進に非常に興味を持っていることが証明された。例えば、最近の税制改正立法では、十分な医療保険のカバー範囲を保っていない個人への税収処罰が廃止されている。“企業-政府規制-清算”を参照。米国内の連邦や州立法機関および外国政府は既存の医療保健立法の改正を考慮し続けている可能性が高い。
“医療改革法案”や米国国内や国外の他の連邦や州改革努力の最終的な内容、時間、影響を予測することはできない。医療改革が我々の業務や財務業績に悪影響を与えないことは保証されず,将来的に医療改革に関連する立法,司法あるいは行政改革が我々の業務にどのように影響するかを予測することもできない。
政府、保険会社、医療組織を管理し、他の医療サービス支払者が医療コストのコントロールあるいは低減に努力し続けることは、以下の点に悪影響を及ぼす可能性がある
• | 規制が承認される可能性のある薬品の需要 |
• | 私たちは私たちの製品に公平だと思う価格を設定することができます |
• | 私たちは製品の保証と清算の能力を得ることができます |
• | 私たちは収入を作り利益を達成したり収益性を維持したり |
• | 私たちは支払いの税金水準を要求された。 |
さらに、他のより広範な立法変化も採択され、これらの変化は、私たちの製品または候補製品の商業成功に悪影響を与え、それらの商業成功を阻止する可能性がある。改正2011年予算制御法または予算制御法には、2029年までに提供者に支払う医療保険を減らすことを含む連邦赤字削減のための条項が含まれている。Medicare、Medicaid、または他の公共援助または補助医療計画に影響を与える重大な支出削減、または任意のより広範な赤字削減努力または“予算制御法案”の立法代替措置として徴収される任意の重大な税金または費用、または他の態様は、私たちが予想する製品収入に悪影響を及ぼす可能性がある。
私たちは激しい競争に直面しており、これは他の人たちが私たちよりも早く、あるいは成功的に製品を発見、開発、商業化することにつながるかもしれない。
新薬製品の開発と商業化競争は激しい。私たちの将来の成功は候補製品の設計、開発、商業化の面で競争優位性を展示し、維持する能力にかかっている。優れた治療効果,利便性,耐性,安全性を有する新製品の設計,開発,商業化を目指している。多くの場合、私たちの商業化された製品は既存の市場をリードする製品と競争するだろう。
他に予測ツールを用いてワクチンやT細胞受容体療法を開発するための抗原を認識することを求めている会社としては,アキレス治療株式会社,適応バイオテクノロジー社,BioNTech SE,細胞生物医学グループ社,Eutilex社,遺伝子技術社,ギリッド科学社,Gritstone Oncology Inc.,Iovance BioTreateutics Inc.,Marker Treateutics Inc.,メルク社,Moderna,OncoTreatment Science Inc.,PACT Pharma Inc.,Vaccibody asおよびZiopharm Oncoly Inc.がある
私たちの多くの潜在的な競争相手は私たちよりも多くの財務、製造、マーケティング、薬物開発、技術、人的資源を持っている。特に大手製薬会社は、臨床テストの面で豊富な経験を持っており、患者の募集、監督管理の承認、患者の募集と医薬製品の製造を含む。特に、これらの会社は政府契約と贈与を獲得して、その研究開発を支援し、テストと臨床試験を行い、監督機関の製品の販売許可を獲得し、このような製品を大規模に製造し、マーケティングが許可された製品を獲得する上で、より多くの経験と専門知識を持っている。これらの企業の研究やマーケティング能力も私たちよりはるかに優れており、承認されたり、開発後期の段階にある製品を持っており、ターゲット市場で有力企業や研究機関と協力している可能性もあります
32
古い製薬会社も、新しい化合物の発見と開発を加速させたり、私たちが開発した製品を時代遅れにする可能性のある新しい化合物にライセンスを付与したりするために投資を大挙する可能性がある。これらのすべての要因のため、私たちの競争相手は、私たちの前に特許保護および/またはFDA承認、または製品の発見、開発、および商業化に成功するかもしれない。また、承認製品と競争するいかなる新製品も、価格競争を克服し、商業的に成功するために、効果、利便性、耐性、安全性の面で納得できる優位性を示さなければならない。私たちが潜在的な競争相手と効果的に競争できなければ、私たちの業務は増加せず、私たちの財務状況と運営は影響を受けるだろう。
私たちの製品は不良な副作用を引き起こす可能性があり、あるいは他の特性を持って、その規制承認を延期または阻止し、その商業潜在力を制限する可能性がある。
私たちの製品または開発中の競争製品が共通の行動メカニズムを使用することによる副作用は、私たちまたは規制機関の臨床試験の中断、延期、または停止を招く可能性があり、FDAまたは他の規制機関が規制承認を拒否し、製品責任クレームを招く可能性がある。私たちの候補製品による深刻な不良事件は私たちの候補製品の発展と私たちの全体業務に大きな影響を与える可能性があります。私たちはまだGen-009がAEsや深刻なAEsを引き起こすかどうかに関するいかなる情報も持っていない
もし私たちまたは他の人が上場承認を受ける前または後に、私たちの任意の候補製品による不良副作用を発見すれば、多くの潜在的な重大な負の結果を招く可能性がある
• | 臨床試験は放置されるかもしれません |
• | 私たちは規制部門の候補ワクチンの承認を得ることができないかもしれない |
• | 規制部門は私たちのワクチンの承認を撤回するかもしれない |
• | 規制部門はラベルに警告を追加することを要求するかもしれない |
• | 患者に配布するために、このような副作用のリスクを概説するための薬物ガイドラインが必要であるかもしれない |
• | 私たちは起訴され、患者への傷害に責任を負うかもしれない |
• | 私たちの名声は損なわれるかもしれない。 |
これらの事件のいずれも、私たちの製品に対する市場の受容度を達成または維持することを阻止し、商業化コストを大幅に増加させる可能性がある。
私たちの負債に関するリスクは
私たちの債務レベルと債務超過義務は私たちの財務状況に悪影響を及ぼす可能性があり、私たちの業務に資金を提供することを難しくするかもしれない。
2018年4月、Hercules Capital,Inc.(F/k/a Hercules Technology Growth Capital,Inc.)と改訂および再記述された融資および保証協定を締結した。(“Hercules”)は、後に2019年11月に改訂されます(改訂後は“2018年定期融資”)。2018年の定期融資は1,400万ドルまでの債務融資を提供した。2018年の定期ローンは2020年12月31日までに利息のみを支払います。その後、2021年5月満期日まで、元本や利息を含む分割払いを支払う義務があります。2019年12月31日現在、2018年の定期融資(改訂済み)での未返済額は1,340万ドル
2018年の定期融資下のすべての債務は、私たちの知的財産や許可内技術を含まず、私たちの既存の財産と資産のほとんどを担保にしています。このような債務は私たちに追加的な融資リスクをもたらす可能性があり、特に私たちの業務や現在の金融市場状況が満期時に私たちの未返済債務を返済または再融資するのに不利な場合がある。この債務はまた次のような事実を含む重要な否定的な結果をもたらすかもしれない
• | 私たちは利息と元金を支払うことで債務を返済する必要があり、これは私たちの運営、私たちの研究と開発、その他の一般企業活動を支援するための資金を減らすだろう |
• | 我々が2018年の定期融資における制限的な契約を遵守できなかったことは違約事件を招く可能性があり、違約を治癒または免除しなければ、この債務の返済義務を加速させ、Herculesはこのような債務を保証する資産における担保権益の強制執行を求める可能性がある。 |
33
もし私たちの現在の債務水準で追加的な債務を増加させれば、上記のリスクは増加するかもしれない。
私たちは満期債務の利息や元金を支払うのに十分な現金を持っていないかもしれない。もし私たちが満期になった時に計画通りに支払いをしなかった場合、あるいは他の方法で深刻に違約したり、2018年の定期融資違約事件を経験した場合、Herculesは私たちの総融資義務を加速させたり、私たちにその保証権益を強制的に執行したりするかもしれない。
2018年の定期融資項目での私たちの現在と未来の債務義務を履行できなかったことは、違約事件を招く可能性があります。また、私たちの業務で重大な不利な事件が発生し、違約事件が発生する可能性があるなど、私たちが完全にコントロールできないいくつかの事件も含まれています。契約違反事件の発生により、力神はすべての満期金額の返済を加速させる可能性がある。2018年の定期融資の満期額が加速すれば、私たちは私たちの債務を返済するために十分な資金がないか、追加の融資を手配できないかもしれない。さらに、Herculesはそのような債務を保証する資産における保証権益を強化することを求めるかもしれない。もし私たちが2018年の定期融資が加速した時にHerculesに満期金額を支払うことができない場合、またはHerculesが私たちの資産に対してその保証権益を強制的に実行して、Herculesに対する私たちの債務を保障することができなければ、私たちが業務を継続する能力が脅かされるかもしれない。
私たちは、これらの条項に違反すれば、2018年の定期融資項目の下で債務を加速させ、私たちの業務や将来性に実質的な悪影響を及ぼす可能性がある制限条項の制約を受けています。
2018年の定期融資は私たちに運営と他の制限を加えた。これらの制限は影響を与え、多くの点で、私たちの能力と任意の未来の子会社の能力を制限または禁止する
• | 特定の資産を処分する |
• | 私たちの業務範囲を変えて |
• | 合併や合併に従事する |
• | 追加的な債務を招く |
• | 資産留置権を設立する |
• | 配当金を支払って私たちの株を分配または買い戻すこと |
• | 付属会社と何らかの取引を行う。 |
このような制限的な協約は私たちが私たちの業務の最高の利益に合致すると思う行動を取ることを阻止するかもしれない。さらに、もし私たちがこのような制限的な条約に違反した場合、Herculesは2018年の定期融資下での私たちの債務を加速させるか、または私たちの資産を担保にしてその保証権益を強制的に実行するかもしれないが、いずれも私たちが事業を運営し続ける能力に大きな悪影響を及ぼすだろう。
私たちのビジネスや産業に関するリスクは
もし私たちが高級管理者と肝心な科学者を引き付けることができなければ、私たちは私たちの製品の開発に成功し、私たちの臨床試験を行い、そして私たちの候補製品を商業化することができないかもしれない。
私たちは社長と最高経営責任者Girish Aakalu博士、最高業務官Tom Davis医学博士、最高財務官Diantha Duvall、首席科学官Jessica Flechtner博士、製薬科学と製造部門の上級副総裁を含む、私たちの上級管理職メンバーに高く依存している。これらの人たちの誰かを失ったサービスは、研究、開発、商業化目標の達成を阻害する可能性がある。私たちはこのような高級管理者たちのすべてと雇用協定を持っている
合格した科学、臨床、製造、販売とマーケティング人員を募集し、維持することも著者らの成功の鍵となる。幹部や他の重要な従業員を失ったサービスは、私たちの研究開発と商業化目標の実現を阻害し、業務戦略を成功させる能力を深刻に損なう可能性がある。また、幹部やキーパーソンを交換することは困難かもしれませんし、私たちの業界では開発に成功し、規制承認や製品商業化に必要なスキルや経験を持つ個人の数が限られているので、長い時間がかかるかもしれません。この限られた人材バンクから募集する競争は非常に激しく、著者らの臨床開発計画の状況及び多くの製薬と生物技術会社の類似人員に対する競争によって、著者らは受け入れ可能な条件でこれらの肝心な人員を募集、訓練、維持或いは激励することができないかもしれない。私たちはまた、大学や研究機関から科学や臨床人を募集する競争に直面している。はい
34
また、私たちは、科学と臨床コンサルタントを含むコンサルタントとコンサルタントに依存して、私たちの研究開発と商業化戦略の制定を助けてくれます。私たちのコンサルタントやコンサルタントは、私たち以外の雇用主に雇われる可能性があり、他のエンティティと締結された相談または相談契約に基づいて約束することができ、これは、私たちが彼らを得る機会を制限するかもしれない。もし私たちが引き続き高い素質の人材を誘致し、維持することができなければ、私たちが成長戦略を推進する能力は制限されるだろう。
私たちの従業員、独立請負業者、主要な調査者、コンサルタント、ビジネスパートナー、サプライヤーは、規制基準と要求を遵守しないこと、およびインサイダー取引を含む不当な行為またはその他の不適切な活動に従事する可能性があります。
私たちは、従業員、独立請負業者、主要調査者、コンサルタント、ビジネスパートナー、サプライヤーが詐欺やその他の不正活動を行うリスクに直面しています。これらの当事者の不正行為は、FDAおよび外国規制機関のような法律に準拠できなかった故意、無謀、および/または職務怠慢行為を含む可能性がある;FDAおよび類似外国規制機関に真実、完全かつ正確な情報を提供すること、私たちが制定した製造基準を遵守すること、連邦、州および外国医療保健詐欺および法律法規を遵守すること、財務情報またはデータを正確に報告すること、または許可されていない活動を開示すること。特に、保健製品とサービスの普及、販売とマーケティング、および保健業界のいくつかの商業配置は、詐欺、リベート、自己取引、その他の乱用行為を含む不正行為を防止するための広範な法律と法規の制約を受けている。これらの法律および法規は、広範な価格設定、割引、マーケティング、構造および手数料、特定の顧客インセンティブ計画、および他のビジネススケジュールを制限または禁止する可能性があります。これらの法的拘束を受けた活動は,患者を募集して臨床試験を行う過程で得られた情報の不正使用に関するものである。このような不正行為を常に識別し、阻止できるわけではなく、私たちがそのような活動を発見し、防止するための予防措置は、未知または未管理のリスクまたは損失を効果的に制御することができないか、またはそのような法律や法規に準拠していないことによる政府の調査または他の行動または訴訟から私たちを保護することができないかもしれない。もし私たちにこのような訴訟を提起して、私たちが自分の権利を弁護したり、維持することに成功できなかったら、これらの行動は私たちの業務に大きな影響を与えるかもしれません, 民事、刑事および行政処罰の適用、損害賠償、罰金、返還、連邦医療保険、医療補助および他の連邦医療保健計画から除外される可能性があること、契約損害、名声損害、利益および将来の収益の減少、および私たちの業務を削減または再構築することが含まれており、これらはいずれも私たちの業務運営能力および私たちの運営結果に悪影響を及ぼす可能性がある。
私たちは私たちの成長を管理し、私たちの業務の成功を拡大する困難に直面するかもしれない。
私たちが臨床試験と商業化を通じて私たちの候補製品を推進することを求めるにつれて、私たちは私たちの開発、監督、製造、マーケティング、販売能力を拡大し、あるいは第三者と契約を締結し、これらの能力を提供する必要があるだろう。私たちの業務の拡大に伴い、様々な戦略パートナー、サプライヤー、他の第三者とのより多くの関係を管理する必要があると予想されます。未来の成長は経営陣の会員たちにもっと多くの重大な責任を負わせるだろう。私たちの将来の財務業績と私たちが候補製品を商業化し、効果的に競争する能力は、私たちが未来の成長を効果的に管理する能力にある程度依存するだろう。そのためには、私たちの開発作業や臨床試験を効率的に管理し、より多くの管理、行政を採用、訓練、統合することができなければなりません。必要であれば、販売やマーケティング担当者も含めなければなりません。私たちはこれらの任務を達成できないかもしれません。もし私たちがそのいずれかの任務を達成しなければ、私たちの会社の発展に成功するのを阻害するかもしれません。
私たちに製品責任訴訟を提起すれば、私たちは多くの責任を負い、候補製品の商業化を制限することを要求されるかもしれません。
私たちの候補製品の臨床テストのため、私たちは固有の製品責任リスクに直面しています。もし私たちがどんな製品を商業化すれば、私たちはもっと大きなリスクに直面します。例えば、私たちが開発したすべての製品が製品テスト、製造、マーケティング、または販売中にダメージを与えたり、不適切なことが発見されたと言われた場合、私たちは起訴されるかもしれない。このような製品責任クレームは、製造欠陥、設計欠陥、製品固有の危険について警告、不注意、厳格な責任と保証違反の告発を含む可能性がある。州消費者保護法によると、クレームも主張することができる。もし私たちが製品責任クレームで自分自身を弁護することに成功できなければ、私たちは重大な責任を招いたり、私たちの候補製品の商業化を制限することを要求されるかもしれません。成功的な防御であっても、多くの財政的で管理的な資源が必要だ。事件がどうであっても最終的な結果がどうであろうと、賠償責任は次のようになるかもしれない
• | すべての候補製品や私たちが開発する可能性のある製品の需要が減少した |
• | 私たちの名声とメディアの深刻な否定的な関心を損なう |
• | 臨床試験参加者の脱退 |
• | 関連訴訟の弁護費用が高い |
35
• | 経営陣の時間と資源を移転する |
• | 実験参加者や患者に多額の報酬を与え |
• | 製品のリコール、撤回またはラベル、マーケティング、または販売促進制限; |
• | 収入損失 |
• | 私たちが開発する可能性のある候補品を商業化することはできません |
• | 私たちの株価は下落しています。 |
潜在的な製品責任クレームを防止するために、許容可能なコストで十分な製品責任保険を得ることができず、我々が開発した製品の商業化を阻止または阻害する可能性がある。私たちは現在私たちの臨床試験に製品責任保険を提供して、総金額は500万ドルです。私たちは製品責任保険を引き受けますが、私たちに対するいかなるクレームも、裁判所の判決または和解の金額の全部または一部が私たちの保険範囲内にない、あるいは私たちの保険範囲を超えてしまう可能性があります。私たちの保険証書にも様々な例外があります。私たちは製品責任クレームの影響を受けるかもしれません。私たちは保険範囲を持っていません。私たちは裁判所によって裁決されたり、和解合意で交渉された私たちの保険範囲の制限を超えたり、私たちの保険カバー範囲内にない金額を支払わなければなりません。私たちはこれらの金額を支払うために十分な資本を得ることができないか、または十分な資本を得ることができないかもしれません。
私たちはこのような法律法規を守らずに環境法規を守らなければならないし、私たちに重大な責任を負わせるかもしれない。
私たちは業務のいくつかの面で危険化学品および放射性および生物材料を使用し、これらの材料の使用、生成、製造、配布、貯蔵、運搬、処理および処理を管理する様々な連邦、州、地方の法律と法規を遵守している。使用,製造,分配,貯蔵,運搬,処理,危険材料の処理による傷害や汚染のリスクを解消することはできない。汚染や傷害が発生したり、環境、職業健康および安全、ならびに輸出規制の法律および法規を遵守できなかった場合、私たちはそれによるいかなる損害にも責任を負う可能性があり、そのような責任は私たちの資産や資源を超える可能性がある。私たちは第三者汚染被害に保険をかけなかった。
私たちがデータ保護の法律および法規を遵守しないことは、政府の法執行行動、個人訴訟、および/またはマイナスの宣伝を招き、私たちの経営業績および業務に負の影響を与える可能性がある。
私たちはプライバシーとデータセキュリティに関するデータ保護法律と規制によって制限されている。データ保護の立法と監督管理構造は引き続き発展し、近年、プライバシーとデータ安全問題への関心はますます多くなっている。アメリカでは、多くの連邦と州法律法規は、州データ漏洩通知法、州健康情報プライバシー法及び連邦と州消費者保護法を含み、健康に関連する個人情報とその他の個人情報の収集、使用、開示と保護を管理している。データ保護の法律および法規を遵守しないことは、民事または刑事罰、個人訴訟および/または負の宣伝を含む可能性があり、私たちの経営業績および業務に負の影響を与える可能性がある政府の法執行行動をもたらす可能性がある。さらに、1996年の“健康保険携帯性·責任法案”(“健康情報技術促進経済·臨床健康法案”(総称して“HIPAA”と呼ぶ)改正)のプライバシーおよび安全要求に制約された第三者(例えば、私たちの製品を発行した医療提供者)から健康情報を取得することができる。私たちは、HIPAAによって直接規制されている“カバーエンティティ”または“ビジネス連絡先”ではないが、場合によっては、HIPAAの刑事条項は、“エンティティをカバーする”または“ビジネス連絡先”以外のエンティティに適用することができる。それに応じて。もし私たちが故意に不正または許可されていない方法でHIPAAによってカバーされたエンティティから個人識別可能な健康情報を取得または開示した場合、私たちは刑事罰を受けるかもしれない。
EUでは,個人健康データの収集と使用は,2018年5月に施行された“一般データ保護条例”(GDPR)の条項によって管轄されている。この条例は,個人データに関する個人の同意,個人への情報提供,主管国家データ保護機関へのデータ処理義務の通知,個人データの安全と秘密などについていくつかの要求を出している。GDPRはまた,個人データをEUから米国に移行するための厳しいルールを実施している。GDPRの要求や欧州連合加盟国の関連国家データ保護法を守らなければ、巨額の罰金やその他の行政処罰が科される可能性がある。米国では、いくつかの州の立法機関が新たなデータプライバシー立法の制定を検討している。すでに採択されたこのような立法の一例は、2020年1月1日に施行されるカリフォルニア消費者プライバシー法(CCPA)である。CCPAは、カバー会社が彼らのデータを処理するいくつかの詳細に関する権利を取得する権利と、彼らのデータを削除することを要求する権利と、彼らのデータを販売しないことを選択する権利とを含む、カリフォルニア消費者に(すべてのカリフォルニア住民を含むと定義される)いくつかの権利を与える。CCPAはまた、カリフォルニアの消費者にそのデータ処理活動に関する通知を提供することを、カバーする会社にいくつかの義務を課している。♪the the the
36
CCPAは,法律に違反した会社に巨額の罰金を科し,データ主体にその個人情報漏洩に対して法定または実際の損害賠償を求める私的訴訟権利を与えることを規定している。
情報技術システムの深刻な中断やセキュリティホールは、私たちの業務に悪影響を及ぼす可能性があります。
私たちはますます情報技術システム、インフラ、データに依存して私たちの業務を運営している。通常のビジネスプロセスでは、商業秘密または他の知的財産権、独自の商業情報、および個人情報を含む大量の機密情報を収集、格納、および送信する。重要なのは、私たちはこのような機密情報の機密性と完全性を維持するために、安全な方法でそうしなければならないということだ。私たちはまた私たちの運営要素を第三者にアウトソーシングしているので、私たちは多くの第三者サプライヤーを管理しています。彼らは私たちの機密情報にアクセスするかもしれません。我々の情報技術システムおよび我々と契約した第三者プロバイダのシステムの規模および複雑さ、ならびにこれらのシステム上に格納された大量の機密情報は、これらのシステムが、当社の従業員、コンサルタント、第三者プロバイダおよび/またはビジネスパートナーの不注意または意図的な行動、または悪意のある第三者からのネットワーク攻撃からのサービス中断またはセキュリティホールの影響を受けやすいようにする。ネットワーク攻撃の頻度,複雑さ,強度が増加しており,発見されにくくなってきている.ネットワーク攻撃には、サービスの信頼性に影響を与え、情報の機密性、完全性、および利用可能性を脅かすために、有害なマルウェア、恐喝ソフトウェア、サービス拒否攻撃、社会工学、および他の手段の配備が含まれる可能性がある。ネットワーク攻撃はまた、支払いまたは情報が予期されない受信者に送信されることをもたらすために、ネットワーク釣りまたは電子メール詐欺を含む可能性がある。
当社の情報技術システムまたは第三者サプライヤーの情報技術システムの重大な中断またはセキュリティホールは、当社の業務運営に悪影響を及ぼす可能性があり、および/または機密情報の損失、流用、および/または許可されていないアクセス、使用または開示をもたらし、または商業秘密または他の知的財産権、独自の商業情報および個人情報を含むが、これらに限定されない機密情報へのアクセスを阻止し、当社の財務、法律、商業および名声の損害をもたらす可能性があります。例えば、私たちの患者または従業員に関する個人情報を含む不正アクセス、使用、または漏洩をもたらすこのようなイベントは、連邦および/または州の違反通知法および外国と同等の法律を遵守することを要求する可能性があり、そうでなければ、個人情報のプライバシーおよび安全を保護する法律および法規に従って責任を負うことになる。セキュリティホールおよび他の不適切なアクセスを検出することは困難である可能性があり、それらを識別するための任意の遅延は、上述したタイプの被害を増加させる可能性がある。我々は,我々の情報技術システムやインフラを保護するためのセキュリティ対策を実施しているが,これらの措置が我々の業務に悪影響を及ぼす可能性のあるサービス中断やセキュリティホールを防止する保証はない.
私たちの普通株に関するリスクは
私たちの最大株主であるNew Enterprise Associates(“NEA”)は、私たちに大きな影響を与える可能性があり、制御権のいかなる変化も含めて、重要な取引結果に影響を与える能力を制限する可能性があります。
2020年1月31日現在、私たち最大の株主である恩賞投資実益が所有する株式総数は、私たちが発行した普通株の約30%を占めています。しかも、私たちの取締役会のメンバーの一人はNEAと関連がある。したがって,NEAは我々の業務に大きな影響を与えることが予想される.NEAはあなたの利益とは違う利益を持っているかもしれませんが、それはあなたが同意しない方法で投票するかもしれません。これはあなたの利益に不利になるかもしれません。私たちの株式の集中所有権は会社の支配権の変更を遅延、防止、または阻止する可能性があり、私たちの株主が会社を売却する時に普通株のプレミアムを得る機会を奪う可能性があり、私たちの普通株の市場価格に悪影響を及ぼす可能性があります。
私たちは私たちの普通株の市場価格を予測できないので、あなたが持っている私たちの普通株の株式を売ることは難しいかもしれません。
活発でない市場は、普通株を売却することで資金を調達する能力を弱める可能性があり、戦略的パートナー関係を達成したり、私たちの普通株を対価格で会社や製品を買収する能力を弱める可能性がある。私たちは私たちの普通株の取引価格を予測できない。今後の1つまたは複数の時期に、我々の経営結果は公開市場アナリストや投資家の予想を下回る可能性があり、これらや他の要因により、私たちの普通株の価格は低下する可能性がある。
私たちの株価が変動すれば、私たちの株主は大きな損失を受ける可能性があり、私たちは証券集団訴訟を含む証券関連訴訟に巻き込まれる可能性があり、これは経営陣の注意をそらし、私たちの業務を損害し、大きな責任を負わせる可能性があります。
私たちの株価は変動するかもしれません。一般的な株式市場,特にバイオ製薬会社の市場は極端な変動を経験しており,この変動は特定の会社の経営業績とは無関係であることが多い
37
このような変動のため、私たちの株主は大きな損失を受けるかもしれない。私たちの普通株の市場価格は多くの要素の影響を受けるかもしれません
• | 競争力のある製品や技術の成功 |
• | 私たちの候補製品の臨床試験結果は |
• | 臨床試験の結果が発表された時間は |
• | 競争相手製品の臨床試験の結果 |
• | 私たちの製品や競争相手の製品に対する規制行動や法律の発展 |
• | 特許出願、発行された特許または他の固有の権利に関連する発展または紛争; |
• | 私たちは他の候補製品や製品の結果を発見し、開発し、得るために努力している |
• | 財務状況と経営業績の実際または予想変動 |
• | 証券アナリストは私たちまたは私たちの競争相手や私たちの業界に関する研究報告を発表した |
• | 私たちまたは私たちの競争相手が私たちまたは私たちの競争相手が市場のアナリストに与える予測や導きに到達できなかった |
• | キーパーソンの増減 |
• | 買収、剥離、剥離、合弁企業、戦略投資、または業務戦略の変化など、私たちの競争相手の戦略決定 |
• | 私たちやこの業界の立法や他の規制発展に影響を与えることで; |
• | 投資家は会社の評価の変動は私たちと互角だと思っています |
• | 私たち、私たちの内部人、または他の株主は私たちの普通株を売却します |
• | ジャーナリズムや投資界の投機行為 |
• | 追加的な資金調達努力が発表されるか、または予想される |
• | 会計原則の変化 |
• | テロ行為、戦争行為、または広範囲の混乱期 |
• | 自然災害や他の災害 |
• | バイオ製薬株の市場状況の変化 |
• | 全体的な市場と経済条件の変化。 |
また、株式市場は最近、大幅な変動、特に製薬、バイオテクノロジー、その他の生命科学会社の株を経験している。製薬、バイオテクノロジー、その他の生命科学会社株の変動性は、その株に代表される会社の経営業績とは無関係であることが多い。私たちは単一の業界を経営しているので、私たちは特にこれらの要素の影響を受けやすいです。これらの要素は私たちの業界や私たちの製品に影響を与え、あるいは私たちの市場に小さい程度影響を与えます
また、将来のいかなる訴訟や訴訟も巨額の費用を招く可能性があり、私たちの経営陣の注意と資源を移し、判決を履行したり、訴訟を終わらせたりするために大量のお金を支払うことを要求する可能性もあります
ナスダック資本市場の継続的な上場要求を守れなかったことは私たちの普通株がナスダック資本市場から撤退する可能性があります。
もし私たちの株価が1株1.00ドルを割ったら、私たちはナスダック資本市場やナスダック世界市場で引き続き上場する資格がないかもしれない。上場を維持するためには、他の事項を除いて、1株1.00ドルの最低終値を維持しなければならない。もし私たちの普通株の終値が30営業日連続して1株1.00ドルを下回ったら、ナスダックの不足通知を受けます。ナスダックはもっと時間がかかるかもしれませんが、通常は180日、少なくとも10営業日連続で少なくとも1.00ドルの最低終値を維持することでコンプライアンスを回復します。
38
2018年6月15日、吾らはナスダック上場資産部から書面で通知を受け、吾らはナスダック上場規則第5450(A)(1)条を遵守できなかったと発表した。この日まで30営業日連続で、私たちの普通株の購入価格は上場を続ける1株1.00ドルの最低要求を下回ったからである。ナスダック上場規則第5810(C)(3)(A)条によれば、180暦の予備期限を取得し、又は2018年12月12日まで、遵守規則第5450(A)(1)条を回復する。我々は、2018年12月12日までにルール5450(A)(1)を遵守しないことを決定し、2018年11月19日に我々の普通株をナスダック世界市場への上場からナスダック資本市場への移行申請を提出した。このようにして、ナスダック資本市場に上場する企業に提供される追加180日間のコンプライアンス期間を取得する資格があり、公開保有株の時価継続上場の要求とナスダック資本市場の他のすべての初期上場基準を満たすことを前提としており、最低入札価格要件は除外し、第2コンプライアンス期間内に株式逆分割を行うことで不足点を補う予定であることを書面で通知する。最初の通知によると、吾らは譲渡申請において、ナスダック資本市場の他のすべての継続上場要求(入札価格要求を除く)を満たし、第2のコンプライアンス期間内に逆株式分割を行うことで不足点を補う予定であることを書面で通知した。2018年12月13日、私たちは2019年6月11日まで180個のカレンダーを追加したというナスダックから通知を受けました, ナスダック上場規則のうち1株1ドルの最低入札要求を再遵守する。これにより、2018年12月17日の寄り付き時には、我々の普通株の株式上場がナスダックグローバル市場からナスダック資本市場に移行した。私たちの普通株は“GNCA”のコードで取引を続けている
2019年5月22日、発行された普通株と発行された普通株を逆株式分割したところ、額面は0.001ドル、比率は8:1だった。そこで、2019年6月10日までに、我々普通株の入札価格は少なくとも10営業日連続で1株1.00ドル以上になり、ナスダックはナスダック上場規則の遵守を実現したことを示す書面通知を提供した。2019年6月10日までに1株1.00ドルの最低終値を再獲得したにもかかわらず、今後も遵守する保証はありません。私たちの普通株の撤退は投資家が私たちの普通株を取引する能力に深刻な影響を与え、私たちの普通株の流動性と価格にマイナスの影響を与える。また、私たちの普通株の退市は、受け入れ可能な条件で資金を調達する能力に実質的な悪影響を及ぼす可能性がある。ナスダックからの退市はまた、我々の既存または潜在的な第三者プロバイダおよびパートナーが自信を失う可能性があり、機関投資家が興味を失い、許可および協力の機会が減少する可能性があることを含む他の負の結果をもたらす可能性がある。
私たちは財務報告を実施し、有効な内部統制を維持することができず、私たちの財務諸表に重大な誤報を招く可能性があり、これは財務諸表を再記述する必要があり、投資家が私たちの報告書の財務情報に自信を失い、私たちの株価にマイナスの影響を与える可能性がある。
私たちはあなたに、私たちが財務報告書の内部統制のいかなる重大な弱点や重大な欠陥も未来に発見されないということを保証することはできません。必要な新しいまたは改善された制御を維持または実施できなかった場合、またはこれらの制御を実施する際に私たちが遭遇したいかなる困難も、追加の重大な弱点または重大な欠陥を招き、私たちが定期的な報告義務を履行できなかったこと、または私たちの財務諸表に重大な誤報を招く可能性がある。このような失敗は、財務報告書の内部統制の有効性を定期的に管理評価した結果にも悪影響を及ぼす可能性がある。重大な欠陥や重大な欠陥の存在は私たちの財務諸表にミスを招く可能性があり、財務諸表の再記述を招き、私たちは報告義務を履行できなくなり、投資家が私たちが報告した財務情報に自信を失い、私たちの株価の下落を招く可能性がある。
上場企業として大きなコストが発生しており、我々の経営陣は上場企業コンプライアンス計画に大量の時間を投入したいと考えている。
上場企業として、巨額の法律、保険、会計、その他の費用を招いた。しかも、私たちの行政者たちは追加的な任務を遂行しなければならない。私たちは発展する法律、法規、基準に適合するように資源を投入し、このような投資は一般的かつ行政費用の増加を招く可能性があり、製品開発活動から管理層の時間と注意を移す可能性がある。もし私たちが新しい法律、法規、標準を遵守する努力が規制機関や管理機関の予想活動と実践に関連する曖昧さによって異なる場合、規制機関は私たちに法的訴訟を提起する可能性があり、私たちの業務は損害を受ける可能性がある。最近の株主集団訴訟構造の変化により、役員や役員責任保険の価格がより高くなっている。この傾向が続くと、私たちは減少した保険範囲を受け入れることを要求されるか、あるいは保険を受けるためにより高い費用が発生するかもしれない。このような要素はまた、私たちが特に私たちの監査委員会と給与委員会に勤めていること、および合格した役員を引き付けて維持することをより難しくするかもしれない。
サバンズ-オキシリー法案は私たちに財務報告書に対する効果的な開示統制と手続きと内部統制を維持することを要求する。有効な制御措置を策定または維持できなかったやり方は、定期管理評価の結果に悪影響を及ぼす可能性がある。サバンズ·オクスリー法を遵守していることを証明できなければ内部統制は
39
もし私たちが過度な財務報告書が不十分だと思ったり、適時あるいは正確に財務諸表を作成できない場合、投資家は私たちの経営業績に自信を失う可能性があり、私たちの普通株価格は下落する可能性があります。しかも、もし私たちがこのような要求を満たし続けることができなければ、私たちはナスダックで上場し続けることができないかもしれない。
我々は、米国証券取引委員会がサバンズ-オキシリー法案第404条を実施するいくつかの規則を遵守しなければならない。この規則は、経営陣が私たちの四半期および年間報告書で財務および他の情報を認証し、私たちの第2の年間報告書から財務報告の内部統制の有効性に関する年間管理報告書を提供することを要求しなければならない。この評価には、経営陣または独立公認会計士事務所によって発見された財務報告内部統制の開示における任意の重大な弱点が含まれなければならない。規定された期間内に第404条の遵守を達成するために、コストが高く、挑戦的であることを記録し、評価するための財務報告書の内部統制の過程に参加する。この点では、外部コンサルタントを招聘することが可能な内部資源を継続的に提供する必要があり、詳細な作業計画により、財務報告内部制御の十分性を評価して記録し、適宜ステップ改善制御プログラムを採用し、制御措置がファイルのように機能しているかどうかをテストすることにより、財務報告内部制御の継続報告及び改善手順を実施する。私たちは努力したにもかかわらず、私たちは規定された時間枠内で結論を出すことができないかもしれない、すなわち私たちは財務報告書の内部統制に有効であり、404条の要求に適合する可能性がある。1つ以上の重大な弱点を発見すれば、財務諸表の信頼性に自信を失っているため、金融市場の不利な反応を招く可能性がある。
私たちの定款書類やデラウェア州法律の条項は逆買収の効力があり、買収が株主に有利であっても、他の会社が私たちを買収することを阻止し、私たちの株主が現在の経営陣を交換または更迭しようとすることを防止するかもしれません。
当社の改訂及び再記載された会社登録証明書及び改訂及び重述された付例に記載されている条項は、当社の制御権の変更又は当社経営陣の変更を阻止、遅延又は阻止する可能性があります。これらの条項はまた、投資家が将来私たちの普通株に支払いたいかもしれない価格を制限し、それによって私たちの普通株の市場価格を下げる可能性がある。また、我々の取締役会が責任を持って我々の管理チームのメンバーに命じているため、これらの規定は、株主が取締役会のメンバーを交換する難しさを増やすことで、現在の経営陣の任意の試みを交換または罷免することを阻害または阻止する可能性がある。他にもこれらの条項には
• | “空白小切手”優先株を許可し、株主の承認なしに私たちの取締役会から発行することができ、投票権、清算、配当、その他の私たちの普通株より優れた権利が含まれている可能性がある |
• | メンバーが3年間交互に勤務している分類された取締役会を作成した |
• | 私たちの株主特別会議は私たちの取締役会、私たちの会長、私たちのCEO、あるいは私たちの総裁だけが招集することができることを明らかにしてください |
• | 株主が書面の同意の下で行動することを禁止する |
• | 株主の年次株主総会での承認のための事前通知プログラムを作成し、指名された取締役会メンバーの人選を提案することを含む |
• | 私たちの役員は理由がある場合にのみ免職されることになっています |
• | 私たちの取締役会の空きは、当時在任していた取締役の大多数が埋めることしかできないことになっていた |
• | いかなる株主も取締役選挙で投票権を累積してはならないことを明確に規定している |
• | 当社の取締役会が当社の定款を修正、変更または廃止することを明確に許可します |
• | 私たちの定款の特定の条項を修正するためには、私たちの普通株式保有者の絶対多数票が必要だ。 |
これらの条項は単独でまたは一緒に敵意の買収、統制権の変更、または私たちの経営陣の変動を延期または阻止する可能性がある。
また、私たちはデラウェア州に登録して設立されたので、私たちはデラウェア州会社法第203条の規定によって管轄されています。この条項は、規定された方法で合併または合併が承認されない限り、私たちが発行した議決権のある株を15%以上保有している人が取引日後3年以内に私たちと合併または合併することを禁止しています。
40
私たちが改訂して再説明した会社登録証明書、私たちが改訂して再記述した定款、あるいはデラウェア州法律のいかなる遅延または制御権の変更を阻止する条項は、私たちの株主が彼らが持っている普通株から割増の機会を得ることを制限することができ、また一部の投資家が私たちの普通株のために支払う価格に影響を与える可能性があります。
私たちは純営業損失を利用して将来の課税収入を相殺する能力が制限されるかもしれません。
一般に、改正された1986年の“国内税法”(以下“税法”)第382条によると、会社が“所有権変更”後、その利用変更前の純営業損失(“NOL”)を利用して将来の課税所得額を相殺する能力が制限される。我々の既存のNOLは以前の所有権変更による制限を受けており,普通株や優先株のいずれかの後続発行により所有権変更が発生した場合,NOLを使用する能力は規則382節のさらなる制限を受ける可能性がある.州法によると、私たちのNOLもまた被害を受ける可能性がある。したがって、私たちは私たちのNOLの実質的な部分を利用できないかもしれない。また、私たちがNOLを利用する能力は私たちの収益性とアメリカ連邦課税収入にかかっている。私たちが設立されて以来、私たちは純損失が発生し、予測可能な未来に、私たちは引き続き重大な損失を受けると予想されている;したがって、私たちは私たちのNOLを使用するために必要なアメリカ連邦課税収入が発生するかどうか分からない。
我々が改訂して再記述した会社登録証明書は、デラウェア州に位置する州または連邦裁判所が私たちの株主のために開始する可能性のあるいくつかのタイプの訴訟および訴訟の唯一および独占フォーラムを指定し、これは、私たちまたは私たちの役員、役員、または従業員との紛争において有利な司法フォーラムを得ることを制限することができる。
限られた例外を除いて、デラウェア州に位置する州と連邦裁判所は、(1)私たちを代表して提起された任意の派生訴訟または訴訟、(2)私たちのいかなる取締役、上級管理者、または他の従業員の私たちまたは私たちの株主に対する受託責任に違反していると主張する任意の訴訟、(3)デラウェア州会社法の任意の規定に基づいて、私たちにクレームを提起する任意の訴訟であることを規定し、改訂し、再記載する。当社の改訂及び再記載された会社登録証明書又は改訂及び再記載された会社定款又は(4)内務原則に管轄されている請求の行動をわれわれに提出する他の任意のもの。任意の者又は実体が自社株の株式を購入又はその他の方法で取得する任意の権益は、当社の上述した改正及び再記載された会社登録証明書の規定に了承され、同意されたものとみなされる。このような裁判所条項の選択は、株主が司法裁判所において、私たちまたは私たちの役員、上級管理者、または他の従業員と紛争することに有利であると考えるクレームを提出する能力を制限する可能性があり、これは、私たちと私たちの役員、上級管理者、および従業員に対するこのような訴訟を阻止する可能性がある。あるいは、裁判所が、私たちが改訂および再記載した会社登録証明書のこれらの条項が、1つまたは複数の指定されたタイプの訴訟または法的手続きに適用されないことを発見した場合、またはそれを実行できない場合、私たちは、他の管轄区域でそのような問題の解決に関連する追加費用を発生させる可能性があり、これは、私たちの業務および財務状況に悪影響を及ぼす可能性がある。
私たちは予測可能な未来に私たちの株に何の現金配当も支払わないと予想されるので、資本付加価値(あれば)は私たちの株主の収益源になります。
あなたは私たちの普通株への投資に依存して配当収入を提供してはいけない。私たちは予測可能な未来に、私たちは普通株式保有者に現金配当金を支払わないと予想している。代わりに、私たちは私たちの業務を維持して拡大するためにすべての収益を維持する予定だ。さらに、私たちが現金配当金を支払う能力は現在、私たちの債務融資計画の条項によって禁止されており、将来の任意の債務融資計画には、私たちの普通株が発表または支払い可能な配当金の金額を禁止または制限する条項が含まれている可能性がある。そのため、投資家は、投資リターンを実現する唯一の方法として、価格上昇後に普通株を売ることに依存しなければならない。したがって、現金配当金を求める投資家は私たちの普通株を購入してはいけない。
項目1 B。未解決従業員意見
ない。
項目2.財産
私たちの主な実行事務室はマサチューセッツ州ケンブリッジ市オーク公園大通り100号5階にあります。郵便番号:02140です。私たちはこの住所に2つのレンタル契約があり、全部で約34,200平方フィートの実験室とオフィススペースを占有しています。2020年3月までに22,440平方フィートの実験室とオフィススペースを追加的に占有します。オフィススペースのみのレンタルは2020年2月に満了するが、実験室とオフィススペースのレンタルは2025年2月に満期となる。私たちの既存の施設は私たちの現在の運営を満たすのに十分だと信じていますが、近い将来、私たちの未来の実験室運営の需要を満たすために、既存施設の空間を拡大する必要があります。そうでなければ、新しい施設に搬入しなければなりません。
項目3.法的訴訟
通常の業務過程で、私たちは時々知的財産権、商業手配、その他の事項に関連する訴訟、クレーム、調査、法律手続き、訴訟脅威に関連する。吾らは現在、吾らが未解決の法律訴訟、仲裁手続、政府訴訟に関与していないと信じておらず、その結果、吾等に不利であれば、個別又は全体的に合理的に吾等の業務や経営業績に重大な悪影響を与えることが予想される。我々はいかなる重大な訴訟にも関与しないが、当該訴訟において、取締役、吾等の高級管理者又は共同経営会社のいずれか一方は、吾等又は吾等の付属会社に不利であるか、又は利吾等又は吾等の付属会社の重大な利益を有している。
プロジェクト4.鉱山安全情報開示
適用されません。
41
第II部
項目5.登録者普通株式市場、関連株主事項、発行者による株式証券の購入
市場情報
我々の普通株は2018年12月17日からナスダック資本市場で公開取引され、取引コードはGNCAである。これまで、私たちの普通株は2014年2月5日からナスダック世界市場で公開取引されていた
所持者
2020年2月11日現在、私たちの普通株には約14人の登録保有者がいます。この数字には,その株式が街中の被指名者が所有する実益所有者は含まれていない.
配当をする
私たちは普通株の現金配当金を発表したり支払ったりしたことがなく、私たちは予測可能な未来にも私たちの普通株に現金配当金を支払わないと予想している。
株式証券を購入する
本年度報告(Form 10−K)がカバーしている期間内に、吾等は登録株式証券を何も購入していない。
株式補償計画に基づいて発行された証券
次の表には、2019年12月31日現在の当社の株式給与計画の情報が含まれています
計画種別 | 未償還株式オプション及び株式承認証を行使する際に発行しなければならない証券数 | 未償還オプションと権利証の加重平均行権価格 | 持分補償計画に基づいて将来発行可能な証券の数 | ||||||||
証券保有者が承認した持分補償計画 (1) | 6,445,288 | $ | 8.48 | 245,430 | (2) | ||||||
(1)私たちが改訂し再編成した2014年の株式インセンティブ計画に関する情報が含まれている
(2)2020年1月1日に常緑樹条項に基づいて改正·再起動された2014年の株式激励計画で増加した1,098,116株は含まれていない。
42
項目6.選定された財務データ
適用されません。
プロジェクト7.経営陣の財務状況と経営成果の検討と分析
当社の財務状況と経営業績の検討と分析、および当社の財務諸表と関連付記をお読みください。これらはForm 10-K形式で今年度の報告書に登場します。本議論および分析に含まれるまたは本Form 10-K年次報告の他の部分に記載されている情報は、リスクおよび不確定要因に関する前向きな陳述を含む、我々の業務および関連融資の計画および戦略に関する情報を含む。本年度報告10-K表の“リスク要因”の部分に列挙された要素を含む多くの要因の影響により、我々の実際の結果は、以下の議論および分析に含まれる前向き陳述に記載または示唆された結果とは大きく異なる可能性がある。
概要
私たちは生物製薬会社で私たちのATLASを利用して新しい癌免疫療法の発見と開発を求めていますTM独自の発見プラットフォーム。ATLASプラットフォームは各患者のCD 4を分析します+CD 8と+患者の腫瘍中の各潜在標的或いは“抗原”に対する細胞の免疫反応を測定した。この方法は,患者が反応可能な抗原を認識することにより,免疫療法(例えば癌ワクチンや細胞療法)の抗原選択を最適化していると考えられる。したがって,ATLASはより多くの免疫原性と有効な癌免疫治療を引き起こすと信じられている
我々の最先端プロジェクトはGen−009であり,個人化された新しい抗原癌ワクチンであり,1/2 a期の臨床試験を行っている。Gen−009は、各患者のGen−009ワクチンに含まれるために、ATLASを使用して、各患者特有の免疫原性腫瘍変異を認識することを計画している。我々はGEN−011も進めており,新たな抗原特異的養子T細胞治療計画であり,ATLASにも依存している。私たちは2020年第2四半期に011世代IND申請を提出する予定だ
融資と業務運営
私たちは2006年8月に業務運営を開始した。これまで,我々の業務は組織と我々を備えた会社員に限られており,我々のATLASノウハウを買収·開発し,潜在的な候補製品を決定し,我々の候補製品のための臨床前研究や臨床試験を行ってきた。私たちはまだどんな製品収入も生まれておらず、予測可能な未来にもそのような収入は発生しないだろう。私たちは主に株式証券の発行、債務融資、贈与によって得られた金額を通じて私たちの業務に資金を提供します。2019年12月31日現在、3億993億ドルの株式証券発行総収益と債務融資の総収益、790万ドルの贈与を受けました。2019年12月31日現在、私たちの現金と現金等価物は4010万ドルです。
設立以来、私たちは重大な運営損失が発生した。2019年12月31日と2018年12月31日までの年度の純損失はそれぞれ3900万ドルと2780万ドルで、2019年12月31日までの累計赤字は3.31億ドルでした。私たちは予測可能な未来に巨額の費用と増加する運営損失が生じると予想している。私たちの純損失は四半期間と年度間に大きく変動する可能性があります。私たちは利益を達成するために相当な収入を作らなければならないだろうし、私たちは決してこれをしないかもしれない。
2019年10月、LPCと買収契約を締結し、協定によると、LPCは1株2.587ドルの買い取り価格で250万ドルの普通株を買収した。また、30ヶ月以内に、私たちは、販売ごとに私たちの普通株の現行市場価格に基づいて、最大2750万ドルの普通株を自ら決定する権利があります(ある所有権制限によって制限されています)。購入契約を締結する対価格として,承認料としてLPCに289,966株の普通株を発行した。
2019年6月、私たちは引受の公開発行を完了し、1株3.50ドルで1050万株の普通株を売却し、総収益は約3680万ドルだった。今回の引受の公開発行には引受業者160万株の超過配給選択権も含まれており、引受業者は2019年6月26日にこの選択権を全面的に行使した。これは550万ドルの追加毛収入を生み出した。約390万ドルの発売関連費用が発生し、純収益総額は3840万ドルとなった
2019年2月、私たちは普通株株式、普通株株式を購入する事前融資権証、および普通株株式を購入する引受権証を発行し、現金収益総額は約1,500万ドルである私募融資取引を完了した。私たちは120万ドルの発売関連費用が発生し、総純収益は1380万ドルになった
43
2018年1月、私たちは2回の引受公開を完了し、普通株、株式承認証、優先株を発行し、純収益は約5170万ドルだった。
私たちの連結財務諸表に示すように、私たちは現金を使って2019年12月31日までの年度の運営活動に3770万ドルの資金を提供し、2019年12月31日に4010万ドルの現金と現金等価物を持っています。また,我々は3.31億ドルの累積赤字を抱えており,予見可能な未来には,我々の候補製品の開発を継続するにつれて,重大な運営損失を招き続けると予想される。これまで、もしあれば、私たちが相当な製品収入を創出し、利益を実現しようとした場合、私たちは株式発行、戦略取引、私たちの市場株式発行計画に基づいて私たちの普通株の収益、その他の資金源を売却することで、私たちの現金需要に融資する予定です。もし私たちが必要な時にもっと多くの資金を集めることができなければ、Gen-009、Gen-011、その他の会社の計画や活動の開発を停止するなど、さらなるコスト削減戦略を実施する必要があるかもしれない。これらの要因に加え,これらの総合財務諸表が発表された日から少なくとも1年以内に運営に資金を提供するために必要な現金の予測に加え,我々が経営を継続している企業として経営を継続する能力が大きく疑われている。
2019年12月31日までの現金および現金等価物は、2021年第1四半期までの運営費および資本支出需要をサポートするのに十分であると信じています。
臨床試験に関連するコストは予測不可能である可能性があり,現在の現金や現金等価物残高に他の源から受け取った収益を加えることは保証されておらず,その間の試験や運営を支援するのに十分である。これらの資金は、Gen−009、Gen−011、または任意の他の候補製品に対して重要な臨床試験を行い、市場承認を求めるか、または商業的に発売することができるようにするのに十分ではないだろう。したがって、私たちは公開または私募株式発行、協力と許可手配、または他の出所を通じてより多くの資金を得ることを要求されるだろう。私たちは許容可能な条項で十分な追加資金を得ることができないかもしれないし、十分な追加資金を得ることができないかもしれません。これは、私たちの1つ以上の候補製品の臨床試験の開発または推進を一時停止または延期することを決定する可能性があります。同様に、我々の1つまたは複数の候補製品の臨床試験の開発または推進が軽率または非現実的であると考えられる場合、私たちは、そのような開発または進展を一時停止または延期することを決定するかもしれない。
財務概要
研究開発費
研究開発費には主に臨床前と臨床候補の推進によるコストが含まれています
• | 給料と関連費用 |
• | 我々の臨床試験と臨床前活動を行う契約研究組織(CRO)、契約製造組織(CMO)、コンサルタントおよび他のサプライヤーとの合意に基づいて発生する費用 |
• | 臨床試験材料および実験室用品の取得、開発、製造のコスト; |
• | 施設コスト、減価償却、その他の費用は、施設賃貸料とメンテナンス、保険その他の用品の直接費用と分担費用を含む。 |
我々は内部研究開発コストを発生した金額に基づいて運営費用に計上している。将来の研究および開発活動のための貨物およびサービスの払い戻し不可能な前金は、支払い時に支出するのではなく、活動が行われたときまたは貨物を受信したときに支出される
以下の表に我々の候補製品の研究開発費を以下のように示す(千単位)
十二月三十一日までの年度 | 増す | |||||||||||
2019 | 2018 | (減少) | ||||||||||
第1/2 aフェーズ計画 | $ | 16,462 | $ | 6,234 | $ | 10,228 | ||||||
発見と前編訳 | 7,141 | 14,888 | (7,747 | ) | ||||||||
他の研究と開発 | 3,349 | 4,087 | (738 | ) | ||||||||
総研究開発 | $ | 26,952 | $ | 25,209 | $ | 1,743 | ||||||
ステップ1/2 a計画は,ステップ1またはステップ2の開発活動である.DiscoveryとPre−INDには,第1段階開発を開始する前に我々の発見研究や翻訳科学を支援するためのコストが含まれている。他の研究と
44
開発には,施設コスト,減価償却費用,その他のコストを含む現役候補製品に特化して割り当てられていないコストが含まれている。
一般と行政費用
一般と行政費用は主に行政と他の行政機能者の賃金と関連費用を含む。その他の一般および行政費用には、施設費用、コンサルティング、会社や知的財産権の法律費用、会計サービスに関する専門費用が含まれています。
その他の収入(費用)
その他の収入(費用)には、権証負債の変化、利息費用、利子収入を差し引いた純額及び取引費用等の雑項目の他の費用が含まれる
重要な会計政策と重大な判断と見積もり
我々の経営陣の財務状況と経営結果の検討と分析は、米国公認会計原則(“GAAP”)に基づいて作成された我々の総合財務諸表に基づいている。公認会計原則に基づいて連結財務諸表を作成する際には、連結財務諸表と付記報告書に報告されている資産、負債、費用の金額に影響を与えるための推定と判断を行う必要がある。継続的に基づいて、以下に説明するものを含む、これらの推定および判断を評価する。私たちの見積もりは歴史的経験と当時の状況で合理的だと思う他の特定の市場や他の関連仮定に基づいています。これらの見積りと仮定は資産や負債の帳簿価値を判断する基礎を構成しており,これらの資産や負債の帳簿価値は他のソースからは明らかに見えない.実際の結果は,これらの推定や仮定とは大きく異なる可能性がある.
我々の重要会計政策は、10-K表年次報告第8項の付記2.重要会計政策要約により詳細に記載されているが、以下の会計政策は、私たちの報告書の財務結果を十分に理解し、評価するために最も重要であり、総合財務諸表を作成する際に使用するより重要な判断と推定に影響を与えると考えられる。
研究と開発費
連結財務諸表作成過程の一部として、私たちの研究と開発費用を見積もる必要があります。このプロセスは、未締結契約および調達注文を徹底的に検討し、内部スタッフによって評価され、提供されたサービスを決定することと、実際のコストを請求書または他の方法で通知されていないサービスに関するコストを推定することとを含む。私たちのほとんどのサービスプロバイダは毎月私たちが提供してくれるサービスや契約マイルストーンに達した時に私たちに借金の領収書を発行してくれます。我々は、当時知られていた事実と状況に基づいて、連結財務諸表において、各貸借対照表の日付までの計算すべき研究·開発費用を推定する。私たちは定期的にサービスプロバイダと私たちの推定の正確性を確認し、必要に応じて調整します。検討·開発費用を算定すべき例としては,臨床試験に関連するCROへの費用,臨床前·臨床材料や中間体に関連するCMOへの費用,臨床前開発活動に関連するサプライヤーへの費用が挙げられる。
われわれを代表して臨床試験を行っている臨床サイトとの契約による提供サービスの見積もりに基づいて臨床試験に関する費用を計算した。これらの合意の財務条項は交渉が必要であり、契約によって異なり、支払いの不均衡を招く可能性がある。その中のいくつかの契約下での支払いは、患者の成功登録および完了に必要なデータ提出のようないくつかの要素に依存する。サービス料金を記録する際には、各期間内に登録された患者数、サイト数、または提供されるサービス範囲など、サービスを提供する期間または契約において定義される他の観察可能および測定可能な進歩点に基づいて推定される。計算されたサービス料支出金額は、それによって生成された前払いまたは計上すべき金額を決定するために、契約課金スケジュールに従って支払われた実際の支払いと比較される。提供されたサービスの状態および時間の推定が、提供されたサービスの実際の状態および時間と異なる場合、任意の特定の期間内に高すぎるまたは低すぎる金額を報告する可能性がある。今まで、私たちの推定は発生した金額と実質的な差はなかった
償還可能証券を購入する引受権証
償還可能証券を購入するために2018年の公開発売時に発行された引受権証(“2018年公開株式公開証”)を貸借対照表上の負債として記録しました。2018年に株式証属負債分類を公開発売したため、著者らは報告日ごとに2018年に公開発売株式証の公正価値を再計量した。2018年の国民の推定公正価値を計算しました
45
モンテカルロシミュレーションを用いて権利証を発行する.モンテカルロシミュレーションは、我々の株価、株価の変動性、残存年数、期待配当収益率、および無リスク金利を含む想定された入力を必要とする。また、推定モデルは、2018年の株式公開発売承認期間内の毎年度期間に買収される可能性を考慮しており、買収事件が2018年の公開株式証の決算に影響を与える可能性があるためである。2018年に株式公開承認価値を公開発売する際に用いる仮定の変化は、2018年に株式公開承認証を公開発売する公正価値に重大な差が生じる可能性がある。
経営成果
2019年12月31日までと2018年12月31日までの年度比較
十二月三十一日までの年度 | 増す | |||||||||||
(単位:千) | 2019 | 2018 | (減少) | |||||||||
運営費用: | ||||||||||||
研究開発 | $ | 26,952 | $ | 25,209 | $ | 1,743 | ||||||
一般と行政 | 12,037 | 14,309 | (2,272 | ) | ||||||||
総運営費 | 38,989 | 39,518 | (529 | ) | ||||||||
運営損失 | (38,989 | ) | (39,518 | ) | (529 | ) | ||||||
その他の収入(支出): | ||||||||||||
株式許可証は価値変動を公正に許可する | 986 | 14,757 | (13,771 | ) | ||||||||
利子支出,純額 | (946 | ) | (1,021 | ) | (75 | ) | ||||||
その他の費用 | (1 | ) | (2,029 | ) | (2,028 | ) | ||||||
その他収入合計 | 39 | 11,707 | (11,668 | ) | ||||||||
純損失 | $ | (38,950 | ) | $ | (27,811 | ) | $ | 11,139 | ||||
研究開発費
2019年12月31日までの年度、研究開発費は2018年12月31日現在の2520万ドルから2700万ドルに増加し、170万ドルに増加した。この増加は主に外部製造コストが約220万ドル増加し,従業員数に関するコストが約60万ドル増加し,臨床コストが約30万ドル増加したが,コンサルティングや専門サービスコストは約130万ドル減少したためである
私たちは、私たちの臨床業務とGen-009計画に提供するサプライチェーン能力を発展させ続けるため、私たちの全体的な研究開発費用が増加すると予想しています。研究用新薬(“IND”)の準備·提出申請とその後の臨床試験開始に伴うGen−011関連費用の発生も予想される。
一般と行政費用
2019年12月31日までの1年間、一般·行政費は2018年12月31日現在の1,430万ドルから1,200万ドルに減少し、減少幅は約230万ドル。減少の主な原因は法的費用が約220万ドル減少したことだ。2018年、会社は株主訴訟に関する重大な法的費用が発生し、2019年に和解が成立した
私たちは今後、私たちの業務の期待成長を支援し、私たちの運営と組織能力を拡大するために、私たちの一般的かつ行政的費用が増加することを予想しています。また、私たちの最初の候補品が規制部門の承認を得る可能性があると考えるなら、コストを増加させ、商業発売に備えていくと予想されます。
株式許可証は価値変動を公正に許可する
株式承認証の公正価値変動は2018年に公開発売株式証の公正価値の非現金変動を反映し、このなどの権利証は発行当日にその公正価値によって入金され、そして任意の株式承認証の行使日及び各報告期間が終了した時に再計量される。収入の減少は私たちの株価が前年より減少したことによるものです。
利子支出,純額
46
利息支出純額は主に私たちの長期債務融資の利息支出を含み、私たちの現金等価物が稼いだ利息によって相殺されます。
その他の収入(費用)
2018年12月31日までの年度と比較して、2019年12月31日までの年度別収入(支出)は200万ドル減少した。2018年12月31日現在の年度別支出には、2018年の株式公開発売に関する権利証に割り当てられた220万ドルの取引コストが含まれています。
流動性と資本資源
概要
2006年の設立以来、私たちは主に普通株の公開発行、私たちの長期債務、私募を通じて私たちの普通株を運営に資金を提供してきた
2019年12月31日現在、私たちは約4010万ドルの現金と現金等価物を持っている。
2018年4月、私たちはHercules Capital,Inc.(“Hercules”)と改正され、再記述された融資および保証協定(“Hercules”)を締結し、その後、2019年11月に改正された(“2018年定期融資”)。2018年の定期融資は1,400万ドルの定期融資を提供します。2018年の定期融資は2021年5月1日に満期となり、変動金利で計算され、年利率は(I)8.00%または(Ii)3.00%プラス最優遇金利のうち大きい者になります。2018年の定期ローンは2021年1月1日までに利息のみを支払うことになっています。その後、支払いには満期の均等元金と利息分割払いが含まれます。2018年の定期ローンは前払いできますが、前払い料金がかかります。期限が切れた時、私たちは100万ドルの期末費用を支払う義務がある。2019年12月31日現在、同社の未返済借款は1340万ドル。
2019年10月、LPCと買収契約を締結し、協定によると、LPCは1株2.587ドルの買い取り価格で250万ドルの普通株を買収した。また、30ヶ月の間、私たちは販売するたびに私たちの普通株の現行市場価格に基づいて、私たちの普通株の現行市場価格に基づいて、最大2750万ドルの普通株を単独で売却する権利があります。購入契約を締結する対価格として,承認料としてLPCに289,966株の普通株を発行した。購入プロトコルは,我々がLPCに売却する普通株を5,227,323株普通株に制限し,購入合意日に発行された普通株の19.99%に相当する。購入契約はまた、LPCに任意の普通株の購入を指示することを禁止しており、これらの株式が当時LPCおよびその関連会社が実益所有していた私たちの普通株の他のすべての株式と合計した場合、LPCおよびその関連会社は任意の時点で当時の私たちの普通株式の総流通株の9.99%を超える実益所有権を持つことになる
2019年6月、私たちは10,500,000株の私たちの普通株の公開発行に関する引受契約を締結しました。1株当たり0.001ドルの価値があり、公開価格は1株3.5ドル、総収益は約3,680万ドルです(“2019年公開”)我々はまた、引受業者に最大1,575,000株の普通株を追加購入する選択権(“超過配給選択権”)を付与した。2019年6月、引受業者はこの選択権を全面的に行使した。私たちは引受業者から超過配給選択権を行使して約550万ドルの毛収入を得た。2019年の公開発売については、超過配給オプションを含め、約390万ドルの発売関連費用が発生し、総純収益は3840万ドルとなった。
2019年2月、私たちは指向性増発融資取引(“方向性増発”)を完了した。我々は,3,199,998株普通株(“株式”)、531,250株普通株を購入する予融資権証(“予融資権証”)と,最大932,812株普通株を購入する引受権証(“株式承認証”)を発行した。当該等株式、予融資権証及び私募株式承認証(総称して“単位”と呼ぶ)は単位当たり4.02元の購入価格で販売されている。私たちは約1380万ドルの現金純収益を受け取り、株の購入、事前出資の引受権証株、株式承認証株のために使用した。
キャッシュフロー
次の表は、2019年12月31日と2018年12月31日までの年間の私たちの現金源と使用状況(単位:千):をまとめています
47
十二月三十一日までの年度 | 増す | |||||||||||
2019 | 2018 | (減少) | ||||||||||
経営活動のための現金純額 | $ | (37,734 | ) | $ | (41,235 | ) | $ | (3,501 | ) | |||
投資活動のための現金純額 | (1,087 | ) | (131 | ) | 956 | |||||||
融資活動が提供する現金純額 | 52,901 | 55,455 | (2,554 | ) | ||||||||
現金と現金等価物の純増加 | $ | 14,080 | $ | 14,089 | $ | (9 | ) | |||||
経営活動
2019年12月31日までの1年間、運営中に使用された純現金は2018年12月31日現在の4120万ドルから3770万ドルに減少し、350万ドル減少した。使用した現金の純額が減少した原因は,売掛金と売掛金が運転資金面で有利に変化したためである
投資活動
2019年12月31日現在の年度では、投資活動用の現金純額が100万ドル増加し110万ドルに達しているが、2018年12月31日現在の年度では、投資活動用現金純額は10万ドルである。投資活動のための現金が増加したのは、購入した財産や設備が増加したためだ。
融資活動
2019年12月31日現在の年度、融資活動が提供する純現金は260万ドルから5290万ドル減少したが、2018年12月31日までの年度、融資活動が提供した現金純額は5550万ドルであった。2018年には,普通株と引受権証の公開により5,170万ドルの純収益が発生し,我々のATM施設によって発行された普通株の売却により290万ドルの純収益が生じたが,2019年には私募で1380万ドルの純収益,2019年の公募では3840万ドルの純収益,LPCとの購入合意による取引により250万ドルの純収益が生じた。2019年12月31日と2018年12月31日までの年間で、190万ドルと50万ドルの長期債務を支払いました。
運営資本要求
私たちの資本は主に補償と関連費用、臨床前と臨床材料の製造コスト、第三者臨床試験サービス、実験室と関連用品、法律とその他の監督管理費用及び一般管理費用に用いられる。私たちは近い将来、このような費用が依然として主要な運営資本要求になると予想している。
私たちは私たちの既存の現金と現金等価物が2021年第1四半期までの運営をサポートするのに十分だと予想しています。連結財務諸表に示すように、2019年12月31日現在、利用可能な現金と現金等価物4,010万ドルを持っています。また、2019年12月31日までの1年間、私たちが運営活動で使用した現金は3770万ドルでした。これらの要因に加え,これらの財務諸表が発表された日から少なくとも1年以内に運営に資金を提供するために必要な現金の予測に加え,我々が経営を継続している企業として経営を継続する能力が大きく疑われている。私たちの運営資本需要の予測は、正しくないことが証明される可能性があるという仮定に基づいており、私たちは予想よりも早くすべての利用可能な資本資源を使用するかもしれない。医薬製品の研究、開発と商業化に関連する多くのリスクと不確定性のため、私たちの運営資本需要の正確な金額を見積もることができない。私たちの未来の資金需要は多くの要素に依存するだろうが、これらに限定されない
• | Gen-009で行われた臨床試験の時間とコストを計画しています |
• | 計画中の臨床試験のためのGEN−009の製造の進捗、時間、およびコスト |
• | Gen-011のためのIND申請を含む規制承認の結果、時間、およびコストを求める; |
• | 私たちの他の候補製品と潜在候補製品の臨床前研究と臨床試験の開始、進捗、時間、コストと結果 |
• | 将来的には、協力、贈与、許可、相談、または私たちが確立する可能性のある他の予定の条項と時間; |
• | 任意の特許または他の知的財産権の許可、提出、起訴、抗弁および実行に関連する私たちは、支払いを要求される可能性があり、または私たちが受け取る可能性のある任意の支払いの金額および時間を含む |
48
私たちのライセンス契約によると、マイルストーン支払い、特許権使用料、特許訴訟費用を支払う義務があります
• | 特許出願を準備、提出し、起訴し、私たちの知的財産権を維持し、保護し、知的財産権に関連するクレームを弁護するコスト; |
• | 私たちは他の製品や技術をどの程度許可したり取得したりします |
• | 上場承認文を受け取る |
• | マーケティング承認を得た場合、Gen−009および他の候補製品の商業化活動のコストは、製品販売、マーケティング、流通、および製造能力を確立するコストおよび時間を含む |
• | 私たちの候補製品の商業販売から得られた収入。 |
私たちは臨床試験を完成するために大量の追加資金を得る必要があり、監督部門のGen-009、Gen-011と私たちの他の候補製品の承認を得る必要がある。私たちが普通株、転換可能証券、または他の株式証券を売却することで追加資本を調達する場合、私たちの既存株主の所有権権益は大幅に希釈される可能性があり、これらの証券の条項は、清算または既存株主の権利に悪影響を及ぼす可能性のある他の特典を含む可能性がある。さらに、債務融資(実行可能であれば)は、固定支払い義務の増加を招き、追加債務を招く、資本支出を招く、または配当を宣言するなど、私たちが具体的な行動をとる能力を制限する制限的な契約を含むプロトコルに関連する可能性があり、これらは、私たちの業務を展開する能力に悪影響を及ぼす可能性がある。もし私たちが必要な時や魅力的な条項で資金を調達できない場合、私たちはGen-009、Gen-011、または私たちの他の候補製品の開発を大幅に延期、削減または停止させることを余儀なくされる可能性があり、他の状況よりも望ましいより早い段階でパートナーを探すか、または他の条件よりも不利な条項でパートナーを探し、放棄することができるかもしれませんが、Gen-009、Gen-011、または私たちの他の候補製品に対する権利は、そうでなければ、私たちは開発または商業化自身を求めるだろう。
表外手配
私たちは何の表外の予定もありません。
第七A項。市場リスクに関する定性的と定量的開示
2019年12月31日現在、私たちは約4010万ドルの現金と現金等価物を持っている。私たちの投資活動の主な目標は、リスクを著しく増加させることなく元本を保存し、流動性を提供し、収入を最大化することです。私たちの市場リスクに対する主な開放は金利変動と関係があり、金利変動はアメリカの金利全体の水準変化の影響を受けている。私たちの現金と現金等価物の短期的な性質を考慮して、市場金利の突然の変化は私たちの財務状況および/または経営業績に実質的な影響を与えないと信じています。私たちはどんな派生金融商品も持っていない。
私たちは私たちの現金、現金等価物、有価証券に重大な違約リスクや流動性不足があるとは思わない。私たちの現金と現金等価物は過度のリスクを含まないと信じていますが、私たちの投資が将来市場価値の不利な変化の影響を受けないことを絶対に保証することはできません。さらに、私たちは1つ以上の金融機関で連邦保険限度額を超える大量の現金、現金等価物、および有価証券を持っている。
インフレは一般的に労働コストと臨床試験費用を増加させることで私たちに影響を及ぼす。2019年12月31日までの年度内に、インフレは我々の経営業績に実質的な影響を与えないと考えられる。
項目8.財務諸表と補足データ
我々の総合財務諸表および我々の独立公認会計士事務所の報告は本年度報告のF−1ページから始まり,フォーマットはForm 10−Kである。
項目9.会計·財務開示面の変更と会計士との相違
適用されません。
第9条。制御とプログラム
情報開示制御とプログラムの評価
我々は、1934年の証券取引法(以下、“取引法”という。)に基づいて提出または提出された報告書で開示を要求する情報を確保するために開示制御及び手続きを維持する(1)米国証券取引委員会規則及び表に指定された期間内に記録、処理、まとめ及び報告を行うこと、(2)必要な開示に関する決定を速やかに行うために、我々の経営陣に蓄積して伝達する。
我々の経営陣は、最高経営責任者とCEOの参加の下、2019年12月31日までの開示制御及び手続の有効性(取引所法案下の規則13 a−15(E)及び15 d−15(E)で定義されるような)を評価した。我々の経営陣は,どのような制御やプログラムも,どんなに設計や操作が良好であっても,その目標を実現するために合理的な保証を提供することしかできず,管理層は,可能な制御とプログラムのコスト-収益関係を評価する際にその判断を適用しなければならないことを認識している.我々の最高経営責任者および最高財務責任者は、上記の評価に基づいて、2019年12月31日現在、我々の開示制御および手続きは合理的な保証レベルで有効であると結論した。
49
財務報告の内部統制に関する経営陣の年次報告
私たちの経営陣は私たちの財務報告書に対する十分な内部統制を確立して維持する責任がある。財務報告の内部統制は、取引法規則13 a-15(F)および15 d-15(F)において、我々のCEOおよび私たちの最高財務責任者が設計または監督し、我々の取締役会、管理層、および他の人員によって実施されるプログラムとして定義され、私たちの財務報告の信頼性および公認会計原則(“GAAP”)に従って外部目的で作成された財務諸表に関する合理的な保証を提供し、以下の政策および手順を含む:
(1) | 資産取引および処置を合理的、詳細、正確かつ公平に反映する記録の保存に関連する |
(2) | 公認会計基準に基づいて財務諸表を作成し、収入および支出が管理層および取締役の許可のみで行われることを確保するために、必要に応じて取引を記録することを確保する合理的な保証を提供する |
(3) | 当社の財務諸表に重大な影響を及ぼす可能性のある不正資産の取得、使用、または処置を防止またはタイムリーに発見する合理的な保証を提供する。 |
最高経営責任者や最高財務責任者を含む経営陣の監督と参加の下、トレデビル委員会後援組織委員会が発表した“内部統制-総合枠組み(2013)”で提供された枠組みに基づいて、財務報告の内部統制の有効性を評価した。この評価によると、我々の経営陣は、財務報告書に対する内部統制が2019年12月31日から有効であると結論した。
財務報告の内部統制の変化
2019年12月31日までの四半期内に、取引法に基づく規則13 a-15(F)および15(D)-15(F)で定義されている財務報告の内部統制に変化はありません。これらの変化は、私たちの財務報告の内部統制に大きな影響を与えたり、合理的に大きな影響を与えたりする可能性があります。
プロジェクト9 B。その他の情報
2020年2月12日,我々の取締役会(“取締役会”)はGisela M.Schwab,M.D.,63歳を選挙し,独立した取締役会社として取締役会に入り,2020年2月14日から発効した。シュワブさんは会社の第2種役員メンバーになり、2022年に開催される株主総会で再任される
シュワブ博士は2016年2月からExelixis,Inc.(以下Exelixis)製品開発と医療事務総監兼首席医療官総裁を務めてきた。これまで,2008年1月から2016年2月までExelixis執行副総裁兼首席医療官を務め,2006年9月から2008年1月までExelixis高級副総裁兼首席医療官を務めていた。2002年から2006年まで、彼女はヒト抗体を基礎とする薬物開発会社Abgenix、Inc.(Abgenixと略称する)で高級副総裁兼首席医療官を務めた。1999年から2001年まで、彼女はAbgenixで臨床開発部副主任総裁も務めた。1992年から1999年までAbgenixで働く前に、彼女は安進会社でますます責任の大きい職を務め、最近担当した職務は取締役臨床研究と血液学/腫瘍学治療領域のチーム責任者である。2011年8月から2014年7月まで、シュワブ博士は上場バイオ製薬会社TopTarget A/Sの取締役会メンバーを務めた。2012年6月以来、私営生物製薬会社Cellerant Treeutics,Inc.の取締役会メンバーを務めており、2015年3月以来、ノルウェーバイオテクノロジー会社Nordic Nanovector A.S.の取締役会メンバーを務めてきた。彼女はハイデルベルク大学で医学博士号を取得し、アーランゲン-ニュルンベルク大学と国家癌研究所で訓練を受け、そして内科、血液学と腫瘍学取締役会の認証を受けた。私たちは、シュワブ博士の科学的専門家は、候補製品の開発と規制承認から商業発売までを推進し、彼女が私たちの取締役会のメンバーになる資格を持たせることを含むと信じている。
シュワブ博士は、年間35,000ドルの取締役費用を含む、我々の非従業員役員補償政策に基づいて取締役サービスの補償を受ける。我々の非従業員役員報酬政策と、私たちが改訂·再改訂した2014年株式インセンティブ計画と非限定株式オプション奨励協定によると、シュワブ博士は2020年2月14日に我々の普通株9,375株を購入する株式オプション奨励を受ける。
私たちの慣例によると、私たちはシュワブ博士と賠償協定を締結しました。この協定は、彼女の役員身分やサービスによって生じる可能性のある責任について賠償を要求しています。賠償協定はまた、シュワブ博士の任意の訴訟に関連する費用を事前に支払うことを規定している
50
彼女の重役の身分と関係があります。上記の説明は、我々のS−1表登録声明(登録番号333−197247)の添付ファイル10.1として米国証券取引委員会に提出され、参照によって本明細書に組み込まれる賠償協定の全文によって限定される。
シュワブ博士と他の誰との間には何の手配も了解もなく、シュワブ博士は取締役医師に選ばれた。上述したことに加えて、シュワブ博士の取引は、米国証券取引委員会S−K規則404(A)項に従って開示されなければならない
51
第三部
プロジェクト10.取締役、上級管理者、および企業管理
本報告第I部“業務−当社幹部に関する情報”というタイトルで提供されている当社幹部に関する情報のほか、本プロジェクトの要求に応じて提供される情報は、2020年株主総会のために作成された最終委託書を参考にして本明細書に組み込まれる。
項目11.役員および役員報酬
本第11項に要求する資料は、当社が2020年株主総会のために作成した最終委託書に参考方式で組み込まれている。
プロジェクト12.特定の実益所有者の保証所有権及び管理職及び株主に関する事項
本第12項に要求する資料は、当社が2020年株主総会のために作成した最終委託書に参考方式で組み込まれている。
第十三項特定関係及び関係者取引と取締役独立性
本第13項で要求した資料は、当社が2020年株主総会のために作成した最終委託書に参考方式で組み込まれている。
プロジェクト14.チーフ会計士費用とサービス
本14項で求めた資料は,当社が2020年株主総会のために作成した最終依頼書を参考に入れている。
52
第4部
プロジェクト15.証拠品および財務諸表の添付表
財務諸表
以下の財務諸表と補足データは本年度報告の一部としてForm 10−K形式で提出される。
独立公認会計士事務所報告
2019年12月31日と2018年12月31日現在の連結貸借対照表
2019年12月31日まで及び2018年12月31日までの総合経営及び全面赤字報告書
2019年12月31日までと2018年12月31日まで年度株主権益総合レポート
2019年12月31日と2018年12月31日までの各年度の連結現金フロー表
連結財務諸表付記
項目16.表格10-Kの概要
ない。
財務諸表明細書
これらは適用されない、または必要な資料が財務諸表または付記に含まれているので、すべての財務諸表の添付表は省略されている。
陳列品
法規S-K第601項の要求に従って提出された展示品は、参照によって本明細書に組み込まれるこの展示品の直前の展示品インデックスに列挙される。
53
Genocea生物科学社は
財務諸表索引
ページのページ | |
独立公認会計士事務所報告 | F-2 |
2019年12月31日と2018年12月31日現在の連結貸借対照表 | F-3 |
2019年12月31日まで及び2018年12月31日までの総合経営及び全面赤字報告書 | F-4 |
2019年12月31日までと2018年12月31日まで年度株主権益総合レポート | F-5 |
2019年12月31日と2018年12月31日までの各年度の連結現金フロー表 | F-6 |
連結財務諸表付記 | F-7 |
F-1
独立公認会計士事務所報告
会社の取締役会と株主
Genocea生物科学社は
財務諸表のいくつかの見方
当社は、添付Genocea Biosciences,Inc.(当社)2019年12月31日及び2018年12月31日までの総合貸借対照表、2019年12月31日及び2018年12月31日までの関連総合経営及び全面赤字報告書、株主権益(損失)及びキャッシュフロー、及び関連付記(総称して“財務諸表”と呼ぶ)を監査した。この等財務諸表は,当社の2019年12月31日および2018年12月31日の総合財務状況,および2019年12月31日および2018年12月31日までの総合運営結果およびキャッシュフローを各重大な面で公平に反映しており,米国公認会計原則に適合していると考えられる。
新しい会計基準を採用する
総合財務諸表付記2で述べたように、2016−02号会計基準更新(ASU)第2016−02号リース(テーマ842)及び2019年1月1日から施行される関連改正により、当社はリース会計方法を変更した。
会社が経営を続ける企業として経営を続ける能力
添付されている総合財務諸表の作成仮説会社は引き続き経営を継続する企業となる。財務諸表付記1で述べたように、当社は経営により経常赤字、運営資金が不足しており、当社の持続経営企業としての持続経営能力に大きな疑問を持っていることを示している。付記1はまた、これらの事項に関する経営陣のイベントや条件の評価、経営陣の計画について説明しています。合併財務諸表には、このような不確実性の結果生じる可能性のある調整は含まれていません。
意見の基礎
これらの財務諸表は会社の経営陣が責任を負う。私たちの責任は私たちの監査に基づいて会社の財務諸表に意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社と独立しなければならない。
私たちはPCAOBの基準に従って監査を行っている。これらの基準は、財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、エラーによるものであっても不正であっても、監査を計画し、実行することを要求する。当社はその財務報告の内部統制を監査することを求められておらず、私たちも招聘されて監査を行っていません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性に対する意見を表明するためではありません。したがって、私たちはそのような意見を表現しない
我々の監査には、財務諸表の重大な誤報リスクを評価するプログラム、エラーによるものであれ詐欺であっても、これらのリスクに対応するプログラムを実行することが含まれています。これらの手続きは、財務諸表中の金額および開示に関連する証拠をテストに基づいて検討することを含む。我々の監査には、経営陣が使用する会計原則の評価と重大な見積もり、財務諸表の全体列報を評価することも含まれています。私たちは私たちの監査が私たちの観点に合理的な基礎を提供すると信じている。
/S/安永法律事務所
私たちは2009年以来当社の監査役を務めてきた
ボストン、マサチューセッツ州
2020年2月13日
F-2
Genocea生物科学社は
合併貸借対照表
(単位は千、1株当たりのデータは除く)
十二月三十一日 2019 | 十二月三十一日 2018 | ||||||
資産 | |||||||
流動資産: | |||||||
現金と現金等価物 | $ | 40,127 | $ | 26,361 | |||
前払い費用と他の流動資産 | 1,457 | 696 | |||||
流動資産総額 | 41,584 | 27,057 | |||||
財産と設備、純額 | 2,617 | 2,582 | |||||
使用権資産 | 6,306 | — | |||||
制限現金 | 631 | 316 | |||||
他の非流動資産 | 1,473 | 1,160 | |||||
総資産 | $ | 52,611 | $ | 31,115 | |||
負債と株主権益 | |||||||
流動負債: | |||||||
売掛金 | $ | 553 | $ | 1,659 | |||
費用とその他の流動負債を計算しなければならない | 4,611 | 3,816 | |||||
賃貸負債 | 1,117 | — | |||||
長期債務の当期部分 | — | 5,257 | |||||
流動負債総額 | 6,281 | 10,732 | |||||
非流動負債: | |||||||
長期債務,当期分を差し引く | 13,407 | 9,565 | |||||
株式証法的責任 | 2,486 | 3,472 | |||||
賃貸負債、当期分を差し引く | 5,395 | — | |||||
他の非流動負債 | — | 11 | |||||
総負債 | 27,569 | 23,780 | |||||
引受金及び又は有事項(付記6) | |||||||
株主権益: | |||||||
優先株、額面0.001ドル;(2019年12月31日と2018年12月31日認可株式25,000,000株;2019年12月31日と2018年12月31日発行1,635株) | 701 | 701 | |||||
普通株は、額面0.001ドル;(2019年12月31日と2018年12月31日に発行された株式はそれぞれ85,000,000株と250,000,000株、2019年12月31日に発行·発行された株式は27,452,900株、2018年12月31日に発行·発行された株式は10,846,397株) | 27 | 11 | |||||
追加実収資本 | 355,268 | 298,627 | |||||
赤字を累計する | (330,954 | ) | (292,004 | ) | |||
株主権益総額 | 25,042 | 7,335 | |||||
総負債と株主権益 | $ | 52,611 | $ | 31,115 | |||
連結財務諸表の付記を参照。
F-3
Genocea生物科学社は
合併経営報告書と全面赤字
(単位は千、1株当たりのデータは除く)
十二月三十一日までの年度 | |||||||
2019 | 2018 | ||||||
運営費用: | |||||||
研究開発 | $ | 26,952 | $ | 25,209 | |||
一般と行政 | 12,037 | 14,309 | |||||
総運営費 | 38,989 | 39,518 | |||||
運営損失 | (38,989 | ) | (39,518 | ) | |||
その他の収入(支出): | |||||||
株式許可証は価値変動を公正に許可する | 986 | 14,757 | |||||
利子支出,純額 | (946 | ) | (1,021 | ) | |||
その他の費用 | (1 | ) | (2,029 | ) | |||
その他収入合計 | 39 | 11,707 | |||||
純損失 | $ | (38,950 | ) | $ | (27,811 | ) | |
総合損失 | $ | (38,950 | ) | $ | (27,811 | ) | |
1株当たり純損失--基本損失と赤字 | $ | (1.89 | ) | $ | (2.69 | ) | |
加重-1株当たり純損失を計算するための普通株式平均 | 20,644 | 10,321 | |||||
連結財務諸表の付記を参照。
F-4
Genocea生物科学社は
合併株主権益報告書(損失)
(単位:千)
優先して優先する | その他の内容 | 合計する | ||||||||||||||||||||
普通株 | 株 | 支払い済み | 積算 | 株主の | ||||||||||||||||||
株 | 金額 | 金額 | 資本 | 赤字.赤字 | 権益(赤字) | |||||||||||||||||
2017年12月31日残高 | 3,592 | $ | 3 | $ | — | $ | 258,140 | $ | (264,193 | ) | $ | (6,050 | ) | |||||||||
普通株発行,純額 | 7,230 | 8 | 701 | 38,077 | — | 38,786 | ||||||||||||||||
普通株発行 | 25 | — | — | 67 | — | 67 | ||||||||||||||||
株に基づく報酬費用 | — | — | — | 2,153 | — | 2,153 | ||||||||||||||||
債務改正に関する権証を発行する | — | — | — | 190 | — | 190 | ||||||||||||||||
純損失 | — | — | — | — | (27,811 | ) | (27,811 | ) | ||||||||||||||
2018年12月31日の残高 | 10,847 | $ | 11 | $ | 701 | $ | 298,627 | $ | (292,004 | ) | $ | 7,335 | ||||||||||
普通株発行,純額 | 16,530 | 16 | — | 54,653 | — | 54,669 | ||||||||||||||||
普通株発行 | 71 | — | — | 130 | — | 130 | ||||||||||||||||
株式オプションの行使 | 5 | — | — | 21 | — | 21 | ||||||||||||||||
株に基づく報酬費用 | — | — | — | 1,837 | — | 1,837 | ||||||||||||||||
純損失 | — | — | — | — | (38,950 | ) | (38,950 | ) | ||||||||||||||
2019年12月31日の残高 | 27,453 | $ | 27 | $ | 701 | $ | 355,268 | $ | (330,954 | ) | $ | 25,042 | ||||||||||
連結財務諸表の付記を参照。
F-5
Genocea生物科学社は
統合現金フロー表
(単位:千)
十二月三十一日までの年度 | |||||||
2019 | 2018 | ||||||
経営活動 | |||||||
純損失 | $ | (38,950 | ) | $ | (27,811 | ) | |
純損失と経営活動で使用した現金純額の調整 | |||||||
減価償却および償却 | 1,097 | 1,088 | |||||
株に基づく報酬 | 1,837 | 2,153 | |||||
収益を取引費用に分配する | — | 2,115 | |||||
株式証負債の公正価値変動を認める | (986 | ) | (14,757 | ) | |||
設備の売却益 | (29 | ) | (78 | ) | |||
繰延融資費用の核販売 | 110 | 355 | |||||
非現金利子支出 | 504 | 643 | |||||
経営性資産と負債の変動 | |||||||
前払い費用と他の流動資産 | (803 | ) | 53 | ||||
使用権資産賃貸負債を差し引いた純額 | 206 | — | |||||
他の非流動資産 | (423 | ) | (989 | ) | |||
売掛金 | (1,106 | ) | (2,103 | ) | |||
費用とその他の負債を計算すべきである | 809 | (1,904 | ) | ||||
経営活動のための現金純額 | (37,734 | ) | (41,235 | ) | |||
投資活動 | |||||||
財産と設備を購入する | (1,135 | ) | (241 | ) | |||
設備を売却して得た収益 | 48 | 110 | |||||
投資活動のための現金純額 | (1,087 | ) | (131 | ) | |||
融資活動 | |||||||
株式発行収益、発行コストを差し引く | 54,669 | 55,458 | |||||
繰延融資コストを支払う | — | (127 | ) | ||||
長期債務収益 | — | 592 | |||||
長期債務を償還する | (1,919 | ) | (535 | ) | |||
株式オプションを行使して得られる収益 | 21 | — | |||||
ESPPにより普通株式を発行して得た金 | 130 | 67 | |||||
融資活動が提供する現金純額 | 52,901 | 55,455 | |||||
現金、現金等価物、および限定的な現金の純増加 | $ | 14,080 | $ | 14,089 | |||
期初現金、現金等価物、および限定現金 | 26,678 | 12,589 | |||||
期末現金、現金等価物、および制限現金 | $ | 40,758 | $ | 26,678 | |||
非現金融資活動とキャッシュフロー情報の補充 | |||||||
レンタル負債に関連した支払済み現金 | $ | 1,637 | $ | — | |||
利子を支払う現金 | $ | 1,103 | $ | 1,074 | |||
債務改正で発行された引受権証 | $ | — | $ | 190 | |||
連結財務諸表の付記を参照。
F-6
Genocea生物科学社は
連結財務諸表付記
1.組織と運用
会社(The Company)
Genocea Biosciences,Inc.(以下は“会社”と略称する)は生物製薬会社であり、2006年8月16日にデラウェア州に登録して設立され、主要な営業場所はマサチューセッツ州ケンブリッジ市に位置する。同社はそのATLASを用いた新たな癌免疫療法の発見と開発を求めているTM独自の発見プラットフォーム。ATLASプラットフォームは各患者のCD 4を分析します+CD 8と+患者の腫瘍中の各潜在標的或いは“抗原”に対する細胞の免疫反応を測定した。同社は,この方法は免疫療法(例えば癌ワクチンや細胞療法)の抗原選択を最適化していると考えている。したがって,同社はATLASがより多くの免疫原性とより有効な癌免疫療法をもたらすと信じている。
同社の最先端の計画はGen−009であり,個性化された新しい抗原癌ワクチンであり,同社は1/2 a期の臨床試験を行っている。Gen−009は、各患者のGen−009ワクチンに含まれるために、ATLASを使用して、各患者特有の免疫原性腫瘍変異を認識することを計画している。同社はまたGen−011を進めており,同様にATLASに依存した新しい抗原特異的なT細胞治療計画であり,2020年第2四半期にGen−011にIND申請を提出することを目標としている。
同社はそのほとんどの努力を製品研究と開発、初歩的な市場開発、資金調達に投入している。現在まで、同社は、その主要な業務目的に関連するいかなる製品収入も生じておらず、バイオテクノロジーおよび製薬業界会社によく見られるいくつかのリスクおよび不確定要素の影響を受けているが、これらに限定されない:その臨床前および臨床試験成功の不確実性に関連するリスク、規制機関の候補製品の承認獲得に関する挑戦、薬品商業化に関連するリスク(マーケティングと販売が許可された場合)、第三者が会社の製品と競争する可能性のある新技術革新の潜在力を開発すること、キーパーソンへの依存、ノウハウ保護の挑戦、政府法規の遵守の必要性。薬物開発の高いコスト;政府法規の遵守;より多くの財政、技術、および他の資源を持つ会社からの競争、および必要に応じて業務の不確実性を支援するための追加資金を得ることができる
会計基準更新(“ASU”)、2014-15年度、財務諸表列紙-持続経営(サブテーマ205-40)、会計基準コード(“ASC”)205-40(“ASC 205-40”)とも呼ばれ、これらの義務が財務諸表発行後1年以内に満了するため、企業に条件および/またはイベントが将来の財務義務を履行する能力に重大な疑いを与えることが要求される。同社は2019年12月31日現在、累計3.31億ドルの赤字を計上しており、予想される将来、同社がその候補製品の開発を継続するにつれ、同社は引き続き重大な運営赤字となる見通しだ。これまで、企業が相当な製品収入を生み出して利益を得ることができれば、会社は株式発行、戦略取引、その他の資金源の組み合わせで現金需要に融資する予定である。もし会社が必要な時により多くの資金を調達できない場合、会社はGen-009、Gen-011、他社の計画や活動の開発を停止するなど、さらなるコスト削減戦略を実施することを要求される可能性がある
総合財務諸表に示すように、2019年12月31日に、会社は利用可能な現金および現金等価物4,010万ドルを持っています。また、2019年12月31日までの年間で、会社が経営活動で使用している現金は3770万ドル。これらの要因に加え、これらの総合財務諸表が発表された日から少なくとも1年以内に運営に資金を提供するために必要な現金の予測に加え、会社の継続経営企業としての能力が大きく疑われている。
付随する総合財務諸表は持続経営を基礎として作成され、正常業務過程で資産を現金化し、負債を返済することを考慮する。連結財務諸表は、記録された資産額の回収可能性および分類に関するいかなる調整も含まず、このような不確実性に起因する可能性のある負債額および分類のいかなる調整も含まない。
2019年5月22日から、当社は発行済み株式と発行済み普通株を逆株式分割し、額面は0.001ドル、割合は8:1であり、普通株の法定株式数を2.5億株から8.5億株に減少させた。これらの財務諸表と関連付記で提供された1株および1株当たりの情報は、8株交換株の逆株分割を反映するために遡及調整されている。
F-7
2.主な会計政策の概要
以下は,これらの財務諸表を作成する際に従う主な会計政策の概要である
列報根拠と合併原則
添付されている連結財務諸表には、すべての会社間口座および取引を解約した後の当社と完全子会社の口座が含まれています。同社は一つの部門として運営し,新しい癌免疫療法を発見·研究·開発し,商業化している
予算の使用
米国公認会計原則に従って財務諸表を作成することは、財務諸表と付記中の報告書の金額に影響を与えるために、管理層に推定と仮定を要求する。会社管理層は、臨床試験に計上されるべき費用に関する推定、前払いおよび計算すべき研究開発費に関する推定、株式に基づく補償費用、および償還可能な証券を購入する引受権証を含むが、これらに限定されない推定値を継続的に評価する。当社は過去の経験とその時点で部下が合理的と考えている他の特定の市場またはその他の関連仮定に基づいて推定しています。実際の結果は,これらの推定や仮定とは異なる可能性がある
現金と現金等価物
当社は流動性が高く、随時現金に変換でき、購入日から3ヶ月以内に満期になった投資のみを現金等価物としています。貨幣市場基金の期限が短いため、その帳簿価値は公正価値に近い。
前払い研究開発
当社が臨床前または臨床前材料および臨床前および臨床試験サービスを受け取る前に支払う現金前払いは前払い研究と開発コストとして記録されている。前払いは未来の研究開発費用に使用されるだろう。同社は研究開発コストを前払いする帳簿価値が完全に実現されると予想している。
財産と設備
財産と設備はコストから減価償却累計を引いて申告する.関連資産寿命の改善や延長ができないメンテナンスやメンテナンスは発生時に運営費用を計上し,主な増築と改造のコストは資本化に計上する。売却時には、関連コストや減価償却が勘定から差し引かれ、それによって生じる損益はいずれも経営報告書と総合損失に計上される。減価償却は直線法を用いてそれぞれの資産の推定耐用年数を記録し、具体的には以下の通りである
資産 | 使用寿命を見込む | |
実験室装置 | 5 | |
家具と事務設備 | 5 | |
コンピュータハードウェアとソフトウェア | 3-5年 | |
賃借権改善 | 耐用年数または残存賃貸期間が短い | |
内部で使用するソフトウェアを開発する
当社は、ASC 350-40“内部使用ソフトウェア”(以下、“ASC 350-40”と略す)の規定に従い、内部使用のために開発または取得したソフトウェアのコストを計算します。従業員が内部ソフトウェアを開発する際に発生する材料コスト、コンサルタントコスト、給与コスト、給与に関するコストは、発生時に資本化される。これらの費用は連結貸借対照表の財産と設備純額に計上される。プロジェクトの初期段階と実施後の段階で発生した費用は費用に計上される。償却は直線法を採用し、相応の資産の推定使用年数、すなわち3年から5年で記録する
長期資産減価準備
F-8
事件や環境変化が資産の帳簿価値を回収できない可能性があることを示した場合、当社は長期資産の潜在的減値を評価する。回収可能性の決定は、資産使用およびその最終処分によって生じる未割引将来のキャッシュフローの推定に基づく。このようなキャッシュフローが資産の帳簿額面を回収するのに不十分であることが予想される場合、その等資産はその推定公正価値に減記される。処分される長期資産は、帳簿または公正価値から売却コストのうち低いものを引いたものを基準とする。当社は2019年12月31日現在および2018年12月31日までに資産減価損失は確認されていません。
繰延融資コスト
発行コストは主に債務と株式融資に関する直接と増額費用を含み、ASU 2015-03、利息分配(特別テーマ835-30):債務発行コストを簡略化する列報(“ASU 2015-03”)による。当社は貸借対照表に債務負債に関する債務発行コストを列記し、債務負債の帳簿価値の直接控除として、ASU 2015-03の債務割引に対する会計処理に適合している。繰延債務融資コストの償却は有効金利法に従っている。
金融商品の公正価値
当社には公正価値記録された金融資産および負債がいくつかあり、公正価値計量会計基準に記載されている公正価値階層では1、2または3級に分類されている。
• | 第1レベル-公正価値は、アクティブ市場で企業が取得する能力がある同じ資産または負債の見積もり(未調整)を利用することによって決定される |
• | 第2レベル--公正価値は、金利、収益率曲線、および外貨スポット為替レートのようなアクティブ市場における類似資産および負債のオファーまたは他の市場で観察可能な投入を利用することによって決定される |
• | 第3級--価格或いは推定値は公正価値計量に重大な意義があり、観察できない投入が必要である。 |
会社の金融資産は現金等価物からなり、会社の金融負債は株式証負債からなる
当社の現金等価物の公正価値はアクティブ市場のオファーに基づいて決定されます。同社の現金等価物には、レベル1に分類された通貨市場基金が含まれている
当社の株式証負債の公正価値はモンテカルロシミュレーションを用いて確定した。付記9.公正価値を推定する際に用いられる仮説と方法の権証を参照。同社の株式引受証債務は3級に分類されている。
賃貸借証書
契約開始時に、当社は1つの手配がレンタルであるかどうかを決定し、レンタル期間は12ヶ月を超えた。賃貸開始時に次のいずれかの基準を満たす場合、賃貸は融資リースの条件を満たす:(I)リース資産の所有権はリース期間終了時に会社に移転し、(Ii)会社は賃貸資産購入の選択権を有し、このオプションは合理的に行使されることを決定し、(Iii)賃貸期間は賃貸資産の残存経済寿命の主要部分であり、(Iv)賃貸支払い総額の現在値がリース資産の全公正価値に等しいか、またはそれを超える。または(V)リース資産の性質は専用であり、レンタル期間の終了時にレンタル者に代替用途を提供しないことが予想される。他のすべての賃貸契約は経営賃貸契約と記載されています。ASU 2018-11レンタル(テーマ842)に準拠したすべてのレンタル:総合貸借対照表に計上されたリース使用権資産と賃貸負債を的確に改善します
ROU資産は会社がリース期間内に対象資産を使用する権利を代表し,リース負債は会社を代表してリースによるリース金の支払いを義務付けている。リース収益率資産および負債は、レンタル開始日にレンタル期間内にレンタル支払いの現在値を確認する。当社の大部分の借款は暗黙的な金利を提供していないため、当社はレンタル開始日に得られる情報に基づいて借入金金利を増加させて推定し、賃貸支払いの現在値を決定している。当社は確定しやすい状況で隠れた金利を使用しています。経営リースROU資産は、繰延賃貸支払いと未償却リースインセンティブにより減少した。当社の賃貸条項には、当社がその選択権を行使することを合理的に決定した場合に賃貸借契約を延長または終了する選択権が含まれています。経営的リースの固定リース支払いのリース料金は直線的に予想期間内に確認し,融資リースの固定リース支払いのレンタル料金は実際の利息を用いて確認する
F-9
方法はレンタル期間を超えます。当社はレンタルと非レンタル部分と賃貸契約を締結しており、これらの部分は通常単独で入金されています。非レンタル部分は、通常、発生時に料金を計上する公共エリアメンテナンスを含む。
研究開発費
研究·開発コストは発生時に費用を計上する。会社は科学研究機関や他社と様々な研究開発契約を結んでいます。将来の研究開発活動のための貨物またはサービスの払戻不可能な前払いは延期され資本化される。資本化金額は、支払時に支出するのではなく、貨物の交付またはサービスの提供に関する場合、または貨物を受信したときに支出する。関連する計算すべき負債の十分性を評価する際に、会社は、イベントの段階または完了状況、受信された請求書および契約コストを含む研究および/またはサービスの進捗状況を分析する
株に基づく報酬費用
同社は、ASC 718“補償-株式補償”に基づいて従業員の株式報酬を会計処理し、実際の没収状況を反映するように期間ごとに記録された金額を調整する。
2019年1月1日から、会社は、付与された日の推定公正価値に基づいて、必要なサービス期間内に非従業員コンサルタントを付与する株式オプション及び制限株式を含む株式の奨励に基づく株式ベースの報酬支出を確認する。2018年12月31日現在、会社は、奨励帰属日の公正価値に基づいて、非従業員コンサルタントに付与された株式報酬の株式報酬支出を確認する。株式ベースの報酬支出は、帰属期間中に確認され、その間にこれらの非従業員コンサルタントがサービスを提供することを前提としている。各財務報告期間が終了したとき、オプションに帰属していない公正価値は、当時の会社普通株の当時の公正価値とBlack-Scholesオプション定価モデルにおける最新の仮定を使用して再計量される
業績条件が達成された後に付与または開始された報酬については、隠れたサービス期間内に加速帰原因モデルを用いて業績条件を実現する可能性があると考えられた場合、会社は株式に基づく報酬支出を確認する。企業のいくつかの業績条件を含む報酬は、企業に付与される奨励数を推定することも要求され、業績条件が実現可能であると考えられた場合には、企業が獲得する奨励数を推定する
所得税
当社は貸借対照法で所得税を計算します。この方法によれば、繰延税金資産および負債の既存の資産および負債の財務諸表帳簿価額とそれぞれの課税ベースとの間の差額は、推定された将来の税金結果に起因して確認されることができる。繰延税金資産及び負債は、当該等の一時的な差額の年間の現行税率を予想どおり回収又は決済する。既存の証拠の重みに基づいて、繰延税金資産の一部または全部が現金化できない可能性が高い場合には、推定準備を計上する。
1株当たりの基本と償却純損失
1株当たり基本純損失の計算方法は,純損失を当期発行普通株の加重平均で割ったものである。会社が純損失を報告している期間については,1株当たり純損失を希釈することは1株当たりほぼ純損失と同様であり,希釈性普通株の効果が逆希釈であれば発行されていると仮定しないからである。同社は2019年12月31日と2018年12月31日までの年度純損失を報告した。
新会計公告
当社は2019年1月1日に以下の新たな会計公告を採用しました
FASBは2016年2月、ASC 840“レンタル”の既存指導意見の代わりにASU第2016-02号“レンタル”を発表し、2018年7月にASU第2018-11号“レンタル(テーマ842):的確な改善”を発表した。新基準は、テナントに貸借対照表上で12ヶ月を超える期限を超えるすべてのレンタルROU資産とリース負債を確認することを要求するROUを確立した。リースは融資型や経営型に分類され,その分類は損益表における費用確認のモデルと分類に影響を与える。ASC 842を採用すると、会社は2019年1月1日現在の総合貸借対照表でそれぞれ170万ドルのROU資産と180万ドルの関連経営賃貸負債を確認した。当社は改正された遡及採用方法を採用し、2019年1月1日を初適用の発効日としています
F-10
同社は条件に合ったすべての賃貸契約のために短期賃貸契約確認免除を選択した。当社はまた、2019年1月1日までに開始される賃貸契約の一括実際の便宜策を選択し、(I)いかなる満期または既存契約にテナントが含まれているかどうかを再評価しないこと、(Ii)任意の満期または既存賃貸契約の賃貸契約分類、および(Iii)任意の既存賃貸契約の初期間接コストを再評価することを可能にする。
2018年6月、FASBはASU番号2018-07を発表し、非従業員株式支払い会計を改善した。新基準は、ASC 718の範囲を非従業員に適用される株式ベースの取引に拡大することにより、取引が実際に融資形態でない限り、従業員および非従業員に支給される株式ベースの報酬の会計処理を大きく統一している。当社は2019年1月1日から発効するASU 2018−17号規定を採用しており、当社の2019年12月31日までの年度の総合財務諸表に大きな影響はありません。
以下の新しい会計公告が発表されましたが、2019年12月31日まで施行されていません
2016年6月、FASBは、実体に予想損失に基づく前向きな方法を使用して、貿易売掛金および販売可能な債務証券を含むいくつかのタイプの金融商品の信用損失を推定することを要求するASU 2016-13、金融商品-信用損失(特別テーマ326)を発表し、減値モデルを修正した。新しい基準は2020年1月1日から施行される。会社ポートフォリオの構成に基づいて、その中には通貨市場基金、及び会社の他の金融資産、現在の市場状況と歴史的信用損失活動の重要性のみが含まれており、会社はこの基準を採用することは総合財務状況、経営業績と関連開示に実質的な影響を与えないと予想される。
2018年8月、FASBはASU 2018-13、公正価値計量(テーマ820):開示フレームワーク-公正価値計量開示要求の変化を発表した。新しい基準は公共エンティティにいくつかの新しい情報を開示することを要求し、いくつかの開示要求を修正した。新しい基準は2020年1月1日から施行される。同社は、この基準を採用することがその開示に実質的な影響を与えないと予想している。
2018年8月、FASBは、ASU 2018-15、無形資産-営業権およびその他-内部使用ソフトウェア(サブトピック350-40):顧客がサービス契約であるクラウドコンピューティング手配で発生した実施コストの会計処理を発表した。ASU 2018−15は、どの実施コストを遅延させ、資産として確認するかを決定するために、サービス契約としてのクラウドスケジュール内のクライアントが、会計基準アセンブリ350~40内の内部使用ソフトウェアガイドに従うことを必要とする。新しい基準は2020年1月1日から施行される。当社は、この基準を採用することは、その総合財務状況、経営業績、関連開示に実質的な影響を与えないと予想している。
3.金融商品の公正価値
以下の表に、2019年12月31日現在と2018年12月31日までの公正価値で恒常的に計量されている資産と負債(単位:千)を示す
合計する | 市場オファーを活発にする (レベル1) | 大切な他の人 観測可能入力 (レベル2) | 意味が重大である 観測不可能な入力 (レベル3) | ||||||||||||
2019年12月31日 | |||||||||||||||
資産 | |||||||||||||||
現金等価物 | 39,971 | 39,971 | — | — | |||||||||||
総資産 | $ | 39,971 | $ | 39,971 | $ | — | $ | — | |||||||
負債.負債 | |||||||||||||||
株式証法的責任 | 2,486 | — | — | 2,486 | |||||||||||
総負債 | $ | 2,486 | $ | — | $ | — | $ | 2,486 | |||||||
2018年12月31日 | |||||||||||||||
資産 | |||||||||||||||
現金等価物 | 24,651 | 24,651 | — | — | |||||||||||
総資産 | $ | 24,651 | $ | 24,651 | $ | — | $ | — | |||||||
負債.負債 | |||||||||||||||
株式証法的責任 | 3,472 | — | — | 3,472 | |||||||||||
総負債 | $ | 3,472 | $ | — | $ | — | $ | 3,472 | |||||||
F-11
次の表は、会社3級株式証負債の変化を反映しています(単位:千):
株式証法的責任 | ||||
発行残高(2018年1月) | $ | 18,231 | ||
価値変動を公平に承諾する | (14,757 | ) | ||
引受権証を行使した | (2 | ) | ||
2018年12月31日の残高 | $ | 3,472 | ||
価値変動を公平に承諾する | (986 | ) | ||
2019年12月31日の残高 | $ | 2,486 | ||
4.財産と設備、純額
財産と設備の純額には、以下の項目が含まれる(千計)
十二月三十一日 | |||||||
2019 | 2018 | ||||||
実験室装置 | $ | 4,125 | $ | 3,761 | |||
内部開発のソフトウェア | 2,547 | 1,970 | |||||
賃借権改善 | 1,524 | 1,524 | |||||
家具と事務設備 | 456 | 447 | |||||
コンピュータハードウェア | 338 | 338 | |||||
内部開発のソフトウェアが開発中です | 97 | — | |||||
総資産と設備 | 9,087 | 8,040 | |||||
減価償却累計と償却 | (6,470 | ) | (5,458 | ) | |||
財産と設備、純額 | $ | 2,617 | $ | 2,582 | |||
2019年12月31日と2018年12月31日までの両年度の減価償却費用はそれぞれ70万ドル。2019年12月31日と2018年12月31日までの2年間で、社内で開発されたソフトウェアに関する償却はそれぞれ40万ドル。
F-12
5.課税費用およびその他の流動負債
計算すべき費用および他の流動負債には、以下の項目が含まれる(千で計算)
十二月三十一日 | |||||||
2019 | 2018 | ||||||
賃金総額と従業員に関するコスト | $ | 2,245 | $ | 2,147 | |||
研究開発コスト | 1,607 | 759 | |||||
その他流動負債 | 759 | 910 | |||||
合計する | $ | 4,611 | $ | 3,816 | |||
6.支払いの引受およびまたは事項
賃貸借契約を経営する
2019年1月1日現在、マサチューセッツ州ケンブリッジ市のマルチテナント建築の2階の実験室とオフィススペースについて2つの賃貸契約が締結されている
2019年3月に、当社は2020年2月までの一部オフィスビル賃貸について分譲契約を締結しました。当社は転貸での責任を保留しているため、賃貸負債を調整していませんが、転貸はレンタル費用の減少に反映されています。
2019年5月、同社はオフィスと実験室スペースの賃貸契約を締結し、2025年2月に延長した。レンタル期間の延長により、会社は純収益資産が540万ドル増加し、関連賃貸負債が530万ドル増加したことを確認した。延期された関連リース義務は、2019年12月31日現在の会社のROU資産と関連リース負債に含まれています。当社はリース期間をさらに5年間延長する権利があり、2019年12月31日現在、当社のROU資産や関連賃貸負債には含まれていません。
2019年7月、会社は2020年3月から2025年2月までの間にオフィスと実験室空間を増やす選択権を行使した。会社はオフィススペースを使用したりコントロールしたりする権利がないため、2019年12月31日現在、会社はROU資産と関連賃貸負債を決定する際に関連賃貸義務を計上していない。同社は増加した実験室やオフィススペースに関する賃貸義務を720万ドルとし、2020年3月にオフィス空間使用権を取得した後を賃貸負債に反映させている
2019年12月31日までの年度、転貸収入を差し引いたレンタル費用は150万ドルです
当社が経営している賃貸の加重平均残存期間と加重平均割引率は以下の通りです
2019年12月31日 | |||
加重平均残存賃貸年限(年) | 5.12 | ||
加重平均割引率 | 8.27 | % | |
融資リース
2019年12月、当社は15ヶ月間のラボ設備レンタル契約を締結しました。当社は、当社が購入資産選択権を持つ基準に基づき、賃貸期間終了時にこの合意を行使することを合理的に決定し、融資リースの条件を満たしていることを決定した。ROU資産と賃貸負債は7.95%の増量借入金金利を用いて計算される。本賃貸契約のレンタル料は2020年1月から支払いが開始されます。
F-13
以下の表は、会社総合貸借対照表における列報状況をまとめたものである
(千単位で)借りる | 分類する | 2019年12月31日 | ||||
資産 | ||||||
運営中です | 賃貸使用権資産 | $ | 6,156 | |||
金融 | 賃貸使用権資産 | 150 | ||||
リース資産総額 | $ | 6,306 | ||||
負債.負債 | ||||||
現在のところ | ||||||
運営中です | 賃貸負債 | $ | 990 | |||
金融 | 賃貸負債 | 127 | ||||
当面ではない | ||||||
運営中です | 賃貸負債、当期分を差し引く | 5,373 | ||||
金融 | 賃貸負債、当期分を差し引く | 22 | ||||
リース総負債 | $ | 6,512 | ||||
2019年12月31日現在、ASC 842規定によると、会社経営賃貸に関する最低賃貸支払いは以下の通り(千単位)
賃貸借契約を経営する | 融資リース | 合計する | |||||||||
2020 | $ | 1,477 | $ | 134 | $ | 1,611 | |||||
2021 | 1,473 | 23 | 1,496 | ||||||||
2022 | 1,511 | — | 1,511 | ||||||||
2023 | 1,548 | — | 1,548 | ||||||||
2024年とその後 | 1,853 | — | 1,853 | ||||||||
賃貸支払総額 | $ | 7,862 | $ | 157 | $ | 8,019 | |||||
計上された利息を差し引く | (1,500 | ) | (7 | ) | (1,507 | ) | |||||
合計する | $ | 6,362 | $ | 150 | $ | 6,512 | |||||
以下の情報はASC 840に基づいて開示されており、2019年1月1日からASC 842に適用されている。2018年12月31日現在、会社の初期期限が1年を超える経営賃貸契約における将来の最低約束は以下の通り(千単位)
賃貸承諾額総額 | ||||
2019 | $ | 1,637 | ||
2020 | 274 | |||
合計する | $ | 1,911 | ||
2018年12月31日までの年度には,ASC 840により,会社は150万ドルのレンタル料を支払った
2019年12月31日と2018年12月31日現在、会社と金融機関の未返済信用状はそれぞれ60万ドルと30万ドルで、オフィスや実験室スペースレンタルの保証金と関連している。この金額は預金現金で保証され、2025年2月28日に満期になる
契約義務
当社は、複数の契約研究機関(“CRO”)や契約製造機関(“CMO”)といくつかの合意を締結しており、その中には一般にキャンセル条項が含まれている
F-14
ハーバード大学許可協定
同社はハーバード大学(“ハーバード”)と独占ライセンス契約を締結し、3つの特許シリーズの独占、世界範囲内の、印税を負担する再許可可能な許可を会社に付与し、開発、製造、製造、使用、マーケティング、販売、輸入許可製品の提供、ATLAS発見プラットフォームに関する許可サービスを提供する。同社には、ある開発と規制のマイルストーンを実現する際に、ハーバードマイルストーンに合計160万ドルにのぼる金を支払う義務がある。同社は2019年12月31日現在、合計30万ドルのマイルストーン支払いを行っている。本ライセンス契約によると、当社は合意した開発計画に従って、ビジネス上の合理的な努力を用いてライセンス製品を開発、マーケティング、販売することが義務付けられています。さらに、会社は、指定された開発マイルストーンを達成する義務があり、会社が任意のタイプの製品またはサービスの開発マイルストーンに到達できず、合理的な拡張や改訂提案がない場合、ハーバードは、製品またはサービスのタイプに応じて、そのような製品に関する本合意を終了するか、またはそのような製品およびサービスに関する非独占的、再許可不可能な許可に変換する権利がある。
ライセンス特許権がカバーする製品または使用許可方法で発見された製品を商業化した後,会社は,会社,会社付属会社,会社の再被許可者が販売しているこのような製品やサービスの純売上高についてハーバード特許権使用料を支払うことが義務付けられている。製品やサービスの種類によって印税は違いますが、いずれも低い1桁になっています。製品タイプによっては、当社の分被許可者が支払うべき販売ベースの特許権使用料は、適用される特許権使用料率又は当社が当該分被許可者から取得した特許権使用料の高い桁又は低い2桁の割合のうちの大きい者をとる。使用料は、商業化された製品またはサービスの種類に応じて、ライセンス特許権に基づいて提出された最後の有効な権利要件が満了するまで、またはそのような製品またはサービスが初めて商業販売されてから10年以内に支払われなければならない。支払いが必要な任意の第三者支払いについては、ハーバード大学に支払うべき印税が減少する可能性があり、上限は特定の割合である。特許使用料の支払いに加えて、会社が任意の分割許可の下で任意の追加収入(現金または非現金)を取得した場合、会社はハーバード大学にそのような収入の一定の割合を支払わなければならず、特定のカテゴリの支払いは含まれておらず、分許可を含む許可範囲に応じて、より低いビット数から最高の低い2桁まで様々である。
ハーバード大学とのこのライセンス契約は、ライセンス特許権に基づいて提出された最後の満期の有効権利要件が満了するまで、製品またはサービス、サービス、および国/地域に基づいて満期となる。当社は随時書面でハーバード大学に本契約の終了を通知することができます。もし会社の重大な違約行為がまだ治癒されていない場合、もし私たちの破産、破産、または同様の場合、または会社が私たちに付与された任意の特許の有効性に疑問を提起した場合、ハーバードも合意を終了することができる。
Oncovir許可と供給協定
2018年1月、当社はOncovir,Inc.(“Oncovir”)とライセンスと供給契約を締結しました。このプロトコルは、アウンコビル社が会社の技術と組み合わせた製品(“組合せ製品”)の研究、開発、使用、販売、製造、商業化およびマーケティングのために、免疫調節剤およびワクチンアジュバントアジュバントヒルトノ(“ヒルトノ”)を製造および供給する条項および条件を規定する。ヒルトノは,同社独自のATLASプラットフォームを用いて識別された合成長ペプチドまたは新規抗原からなり,ヒルトノとともに調製されるGen−009のアジュバント成分である
Oncovirは、Hiltonolの使用を含むOncovirの研究、開発、または商業化組合せ製品に関する特定の知的財産権にHiltonolを使用することを含む、排他的で譲渡可能であり、特許使用料を負担するグローバルライセンスを同社に付与する権利があるが、Hiltonolを単独で製造または使用または販売するために使用することはできない。このライセンスは、2028年1月25日遅く、または合意に従って会社に付与された任意の特許の最後の有効な権利要件が満了した日に、永久、全額支払い、および印税免除となる。
この合意によると、同社は、各組み合わせ製品がある臨床試験のマイルストーンに達し、ある地域で初めて各組合せ製品を承認した場合、Oncovirに中の6桁低いマイルストーン支払いを支払い、組合せ製品の純売上に応じて、製品ごとに1桁から1桁の等級別特許権使用料を支払う義務がある。
当社は、共同製品の開発を停止することを決定した場合、または会社または適用される規制機関がヒルトンまたは共同製品が臨床的に安全でないか、または有効でないと判断した場合に合意を終了することができる。合意のいずれか一方は、他方の重大な違約行為や、他方の破産、債務返済や解散のために合意を終了することもできる。
F-15
7.長期債務
2018年4月、当社はHercules Capital,Inc.(“Hercules”)と改訂および再記述された融資および保証協定(“Hercules”)を締結し、その後2019年11月に改訂された(“2018年定期融資”)。2018年の定期融資は1,400万ドルの定期融資を提供します。2018年の定期ローンは2021年5月1日に満期になり、利息で計算すると、年利は(I)8.00%または(Ii)3.00%プラス最優遇金利の和に等しい。2018年の定期ローンは2021年1月1日までに利息のみを支払うことになっています。その後、支払いには満期の均等元金と利息分割払いが含まれます。2018年の定期ローンは前払いできますが、前払い料金がかかります。会社は満期時に100万ドルの期末費用を支払う義務がある。当社は2018年の定期融資の2019年11月改正案を評価し、ASC 470-50債務(主題470)による改正案であると結論した
2018年の定期融資は、会社のほとんどの資産(知的財産権を除く)への留置権を担保としている。Herculesは特定の現金、現金同等物、および投資口座で完全な優先保証権益を持っている。2018年の定期融資には、非金融契約、陳述、および重大な悪影響条項が含まれています。金融契約はありません。“重大な悪影響”とは、(I)会社の業務、運営、財産、資産または状況(財務または他の態様)、(Ii)会社が融資文書の条項に従って担保債務を履行する能力、または代理人または貸金人が担保債務に関する任意の権利または救済措置を実行する能力、または(Iii)担保品または代理人の担保権またはその等の留置権に対する優先権を意味する。ローン協定によると、重大な悪影響を与えるか、または合理的に予想される重大な悪影響を与える事件はすべて違約事件であり、Herculesは違約事件と同じ条項に従って融資協定項の下で満期になった金の返済を加速することができる。2019年12月31日現在、当社は2018年定期融資のすべての契約を遵守しています。2018年の定期融資は、コントロール権が変化した場合に自動的に償還することができます。当社は、ローン返済を加速させる項目での未返済金額は手が届かないと考えているため、債務残高は2019年12月31日の契約支払条項に基づいて分類されています。
以前2014年に発行された定期融資について、会社はHerculesに普通株式承認証を発行したが、この株式承認証は2019年11月に満期となり、行使されなかった。2018年の定期融資について、当社はHerculesに普通株式承認証(“Hercules株式承認証”)を発行します。付記9.手令
2019年12月31日と2018年12月31日現在、会社の未返済借入金はそれぞれ1,340万ドルと1,480万ドルです。2019年12月31日と2018年12月31日までの年間で、会社は190万ドルと50万ドルの長期債務を支払った。2019年12月31日と2018年12月31日までの年度の利息支出はそれぞれ160万ドルと170万ドル。
将来の元金支払いは、期末費用を含めて、以下のようになります(千で計算)
十二月三十一日 | |||
2019 | |||
2020 | $ | — | |
2021 | 13,960 | ||
合計する | $ | 13,960 | |
8.株主権益
2019年公募株
2019年6月、当社は10,500,000株の自社普通株について公開販売契約を締結し、1株当たり額面0.001ドル、1株当たり3.5ドル、総収益約3,680万ドル(“2019年公開発売”)とした。会社はまた、引受業者に最大1,575,000株の普通株を購入する選択権(“超過配給選択権”)を付与した。2019年6月26日、引受業者はこの選択権を全面的に行使した。同社は引受業者から超過配給選択権を行使して約550万ドルの毛収入を得た。2019年の公開発売については、超過配給選択権を含め、会社は純収益3840万ドルを受け取った。
私募する
2019年2月、当社は指向性増発融資取引(略称“方向性増発”)を完了した。当社は3,199,998株の普通株、531,250株の普通株を購入可能な予備資本権証(“資本金権証”)及び最大932,812株の普通株(“株式承認証”)を購入可能な引受権証(“私募株式承認証”)を発行する。当該等株式、予融資権証及び私募株式承認証(総称して“単位”と呼ぶ)は単位当たり4.02元の購入価格で販売されている。同社は約1380万ドルの現金純収益を獲得し、株の購入、予め出資した引受権証株、株式承認証株に用いられている。付記9.手令
F-16
同社は2回目の終値(“2回目の終値”)時に普通株を増発する権利があり、総収益は最高2420万ドルに達する。第二次閉鎖の発生は,Gen−009に対する会社の1/2 a期臨床試験A部分の主な結果および会社取締役会が第二次閉鎖を継続することを決定したことを条件とした。2019年6月、同社は今回の試験の主な結果を発表したが、2回目の結審を行わないことを選択した。2回目の終値に代わり、同社は2019年の公募株を継続している。
2018年公募株
2018年1月、当社は2つの引受契約を締結し、1部目は6,670,625株当社普通株の公開発売について、1株当たり額面0.001ドル、および株式承認証付きで、1株8ドルの合併価格で最大3,353,313株普通株(“2018年公開株式公開発売”)を購入し、総収益は約5,340万ドル(“2018年普通株発行”)、2部目は公開発売1,635株当社Aシリーズについて優先株(“優先株”)を転換でき、1株当たり額面0.001ドル。204,375株普通株、および対応する株式承認証に変換することができ、総収益約160万ドルで最大102,188株の普通株を購入することができる(“優先株発行”、2018年普通株発行とともに“2018年1月融資”と呼ぶ)。同社は約100万ドルの毛収入を得て、119,718株の普通株と引受権証を発行し、引受業者が超過配給選択権を行使した中から最大179,757株の普通株を購入した。
優先株
1株の優先株は125株の普通株に変換することができるが、株式配当と株式分割によっていくつかの調整を行う必要がある。優先株保有者は、普通株が実際に支払った配当と同じ配当を得る権利があり、取締役会が普通株に転換した上で普通株に配当金を支払うことを宣言していれば。
発行コスト
2018年1月の融資に関連して、当社は約400万ドルの発行コストを発生させた。当社は約260万ドルの発行コストを追加実収資本内の普通株と優先株に割り当て、2018年に株式公開承認証を公開発売した負債に分類された約140万ドルの発行コストを直ちに支出した
株式承認証
付記9.手令
ヘラクレス
Herculesとの2018年融資協定については、当社は2014年11月の株式函合意(“改訂株式箱合意”)についても改訂した。Herculesは任意のまたは複数の後続私募株式融資に参加する権利があり、金額は最大200万ドルに達し、その条項と条件は、その後の各株式融資で他の投資家が購入した条項と条件と同じだ。改訂株式通信契約は、(1)Herculesが合計2,000,000ドルの後続持分融資証券を購入した場合に終了し、(2)(A)2018年の定期融資項目の下のすべての債務を返済するか、または(B)Herculesが株式証発行期間の満了または終了を承認したときに終了する。付記9.手令
リンカーンパークキャピタルと合意しました
2019年10月、当社はリンカーンパーク資本会社(“リンカーンパーク資本”)と購入契約を締結し、合意に基づき、リンカーンパーク資本は1株2.587ドルの購入価格で当社の普通株株式を購入し、約2,500,000ドルの純収益を得た。また、30ヶ月以内に、会社は売却ごとに普通株の現行市場価格に基づいて、最大2750万ドルの会社普通株を自己決定する権利がある。購入契約を締結する代償として,会社は承認費として289,966株の普通株をLPCに発行した。購入プロトコルは,我々がLPCに売却する普通株を5,227,323株普通株に制限し,購入合意日に発行された普通株の19.99%に相当する。購入契約はまた、LPCに任意の普通株の購入を指示することを禁止しており、これらの株式が当時LPCおよびその関連会社が実益所有していた私たちの普通株の他のすべての株式と合計した場合、LPCおよびその関連会社は任意の時点で当時の私たちの普通株式の総流通株の9.99%を超える実益所有権を持つことになる。会社は、追加株式を売却する権利は、基準を満たす独立株オプションを代表することを決定した
F-17
ASC 815派生ツールおよびヘッジ保証によれば、派生ツールの公正価値はゼロであるので、追加の会計処理を必要としない
場内株式発行計画
2015年、当社はCowen and Company,LLCと市場に設立された株式発行計画(“ATM”)を締結し、この計画により、当社は時々現行の市場価格で普通株を発行·販売することができる。同社は2019年12月31日現在、現金自動支払機で合計463,887株を売却し、約400万ドルの純収益を得ている。
9.手令
2019年12月31日現在、会社は株式承認証を行使していない以下の潜在的に普通株式を発行できる
株 | 行権価格 | 期日まで | 分類する | ||||||||
力神令状 | 41,177 | $ | 6.80 | Q2 2023 | 権益 | ||||||
2018年公募株式証明書 | 3,616,944 | $ | 9.60 | Q1 2023 | 負債.負債 | ||||||
私募株式証明書 | 932,812 | $ | 4.52 | Q1 2024 | 権益 | ||||||
あらかじめ出資して株式証明書を発行する | 531,250 | $ | 0.08 | Q1 2039 | 権益 | ||||||
5,122,183 | |||||||||||
力神令状
行権価格および株式数は、合併イベント、普通株式再分類、普通株式分割または合併、またはいくつかの配当金支払い時に調整することができる。当社は、Hercules承認株式証はASC 480に基づいて権益分類を行い、すべての届出期間の負債と権益(“ASC 480”)を区別すべきであることを決定した
2018年公募株式証明書
行権価格および株式数は、合併イベント、普通株式再分類、普通株式分割または合併、またはいくつかの配当金支払い時に調整することができる。“買収”が発生した場合、一般的に合併または合併を含むと定義され、当社の50%以上の投票権を有する証券の売却、自社の全資産または実質的な全資産または投票権を有する証券の売却、または2018年に株式承認証が定義された他の支配権変更取引を公開発売することにより、当社は2018年度に公開発売承認持分証保有者が既存または買収エンティティ(“買収者”)から新たな引受権証を取得することを確保するために最大の努力を尽くす責任がある。買収側株式を購入する新株式証の満期日は2018年公開株式公開承認証と同様であり、執行価格は買収側株式価値と当社普通株の割合に基づくべきである。もし会社が最善を尽くしたにもかかわらず、買収側が上述したように買収中に新たな引受証を発行することを促すことができなかった場合、会社の株主が買収で現金を獲得した場合、会社は現金決済で2018年に株式承認証を公開発売し、会社の株主が買収で株を獲得する場合、会社は株式承認証所持者1人当たり普通株を発行する。
当社は、2018年に株式公開発行承認証をASC 480に基づいて責任種別に分類すべきであることを決定した。2018年に株式証属負債分類を公開発売したため、当社は報告日ごとに公正価値を再計量した。当社は2018年に株式証明書を公開発売したことを初歩的に記録し、公正価値は約1,820万ドルと推定されている。当社が公正価値に応じて2018年に株式承認証を公開発売したことについて、当社は2019年12月31日および2018年12月31日までにそれぞれ100万ドルおよび1,480万ドルの収入を記録しました。2019年12月31日と2018年12月31日現在、権利証負債の公正価値はそれぞれ約250万ドルと350万ドルである
F-18
次の表は、モンテカルロシミュレーションモデルにおける株式証負債の2019年12月31日と2018年12月31日までの公正価値を推定するための仮定を詳細に説明する
2019年12月31日 | 2018年12月31日 | |||||||
株価.株価 | $ | 2.07 | $ | 2.32 | ||||
波動率 | 50.0% - 116.6% | 50.0% - 111.3% | ||||||
残り期限(年) | 3.1 | 4.1 | ||||||
期待配当収益率 | — | % | — | % | ||||
無リスク金利 | 1.6 | % | 2.4% - 2.5% | |||||
年間捕獲イベント確率範囲 | 20.0 | % | 0.0% - 30.0% | |||||
私募及び前払い資金株式承認証
株式配当、分割、株式合併、再分類、再編或いは制御権変更が私たちの普通株に影響を与える場合、株式承認証の行権価格は適切に調整される。当社は、私募株式証及び事前資本権証は、2019年12月31日までの間にASC 480に基づいて権益分類を行うことを決定した。同社はまた、ASC 260の1株当たり収益の決定に基づいて、事前資金権証は基本1株当たり収益の決定に含まれるべきであることを決定した。
10.株式および従業員福祉計画
会社はサービス条件付き株式オプションを発行し、これらの条件は通常報酬の帰属である。同社は株式オプションも発行し、ある業績条件の満足に応じて付与している
二零一四年株式激励計画(“二零一四年株式激励計画”)はその後、二零一八年六月に改訂及び再記述し、当社の主要従業員及び取締役、顧問及び顧問に奨励性株式オプション、非制限株式オプション、制限性株式及びその他の奨励を付与することを規定している。改正された2014年の株式計画では、発行可能な株式数は毎年1月1日に自動的に増加し、発行金額は、会社が前年12月31日までに終値した時点で発行された普通株の4.0%または会社取締役会が決定した株式数に等しく、両者の間の小さい者となる。2020年1月1日、改訂された2014年の株式計画に基づいて発行可能な株式総数。この規定により1,098,116株増加した。
2019年12月31日現在、当社の株式計画に基づいて発行するために1,568,535件のオプション奨励が予約されており、残りの245,430件の奨励は将来の授与が可能です
株式オプション公正価値の決定
同社は授与日にブラック·スコアーズオプション定価モデルを採用し、サービスと業績に基づく授与基準に従って、従業員、コンサルタント、取締役に株式オプションの公正価値を計量した。
当社には、その履歴データのみを用いて変動率と予想期間を計算するのに十分な履歴データがありません。そこで,当社は加重平均変動率を採用し,当社の初公募以来の変動性およびいくつかの基準会社の変動性を考慮した。類似した実体を識別するために,同社は業界,取引履歴長,ライフサイクル段階などの特徴を考慮している。仮定された配当率は、予測可能な未来に配当金を派遣しない会社の予想に基づいている。平均予想寿命は、SAB 110に記載された“簡略化方法”に従って決定される、すなわち、ホーム日と契約期間終了との間の中間点である。無リスク金利は米国債の暗黙的な収益を参考にして決定され、残り期限は付与された日に仮定した期待寿命に等しい
ブラック·スコアーズオプション定価モデルで使用される加重平均は以下のように仮定される
十二月三十一日までの年度 | ||||
2019 | 2018 | |||
予想変動率 | 79.7% | 78.6% | ||
無リスク金利 | 2.3% | 2.8% | ||
予想期限(年単位) | 6.0 | 6.0 | ||
期待配当収益率 | 0% | 0% | ||
F-19
下表は、従業員と非従業員の株式オプション活動(千株単位)をまとめた
株 | 加重平均 行権価格 | 加重平均 残り 契約書 期限(年) | 骨材 内在的価値 | ||||||||||
2018年12月31日現在返済しておりません | 893 | $ | 18.79 | ||||||||||
授与する | 679 | $ | 4.36 | ||||||||||
鍛えられた | (5 | ) | $ | 4.32 | |||||||||
キャンセルします | (244 | ) | $ | 17.64 | |||||||||
2019年12月31日現在返済しておりません | 1,323 | $ | 11.65 | 8.0 | $ | — | |||||||
2019年12月31日に行使可能 | 522 | $ | 20.09 | 6.6 | $ | — | |||||||
2019年12月31日および2018年12月31日までに、当社はそれぞれ678,710株および657,375株の普通株を購入するために株式オプションを付与し、加重平均授受日の公正価値はそれぞれ4.36ドルおよび6.96ドルであった。
2019年12月31日現在、会社の株式計画により付与された従業員と非従業員の株式オプションに関する未確認報酬コスト総額は300万ドルである。同社は2.65年の残り加重平均期間内にこのコストを確認する予定だ。
株に基づく報酬費用
株式に基づく報酬支出総額は、従業員および非従業員に付与された株式オプションおよび制限株であることが確認され、会社の総合経営報告書で以下のように報告されている(千計)
十二月三十一日までの年度 | ||||||||
2019 | 2018 | |||||||
研究開発 | $ | 725 | $ | 620 | ||||
一般と行政 | 1,112 | 1,533 | ||||||
合計する | $ | 1,837 | $ | 2,153 | ||||
従業員株購入計画
2014年2月、会社取締役会は2014年度従業員株購入計画(“2014 ESPP”)を採択し、その後2018年6月に改訂を行った。改訂された2014年従業員持株計画は、計画に参加した合格社員に最大337,597株の普通株を発行することを許可した。改訂された2014年ESPPは、例年の1月1日から6月30日まで、7月1日から12月31日までの6カ月のオプション期間を規定している。当社は2019年12月31日まで及び2018年12月31日までにそれぞれ改訂された2014年度株主権益計画に基づいて71,118株及び23,417株の株式を発行する。2019年12月31日現在、同計画によると、今後も217,985株が発行可能となっている。
401(K)貯蓄計画
2007年、会社は国税法第401(K)節(“401(K)計画”)に基づいて固定払込貯蓄計画を構築した。401(K)計画は、規定された最低年齢およびサービス要件に適合するすべての従業員をカバーし、参加者が税引前に年間給与の一部の支払いを延期することを可能にする。2015年1月1日から、会社は同計画の参加者に相応の寄付を提供するようになった。2019年12月31日と2018年12月31日までの年間で、当社は同計画の参加者に相応の寄付金を提供し、総額はそれぞれ20万ドルと10万ドルだった。
F-20
11.1株当たり純損失
2019年12月31日と2018年12月31日までの年度の1株当たり基本と希釈後の純損失は以下のように計算される
十二月三十一日までの年度 | ||||||||
2019 | 2018 | |||||||
1株当たりの純損失は | ||||||||
分子: | ||||||||
純損失 | $ | (38,950 | ) | $ | (27,811 | ) | ||
分母: | ||||||||
加重平均発行済み普通株式-基本(単位 数千人) | 20,644 | 10,321 | ||||||
普通株等価物株式の希釈効果 普通株式オプションと制限株から発生します 職場.職場 | — | — | ||||||
加重平均発行済み普通株式-希薄化 | 20,644 | 10,321 | ||||||
1株当たり純損失--基本損失と赤字 | $ | (1.89 | ) | $ | (2.69 | ) | ||
以下、2019年12月31日と2018年12月31日までの発行済み普通株等価物を換算ベースで列記し、その逆希釈効果(千単位)のため、列報期間中の1株当たり純損失計算には含まれていない
十二月三十一日までの年度 | ||||||
2019 | 2018 | |||||
株式オプション | 1,323 | 893 | ||||
株式承認証 | 4,591 | 3,668 | ||||
合計する | 5,914 | 4,561 | ||||
未償還株式オプションには、発行された普通株式等価物を計算する際に業績条件を満たしていない業績に基づく帰属基準は含まれていない
12.所得税
2019年12月31日および2018年12月31日まで、当社は当期または繰延所得税の支出や利益を記録していません。同社の所得税前損失には国内損失のみが含まれています。同社の繰延税金資産の重要な構成要素は、以下の通りである
F-21
十二月三十一日 | |||||||
2019 | 2018 | ||||||
繰延税金資産: | |||||||
アメリカと各州の純営業損失が繰り越す | $ | 56,906 | $ | 49,614 | |||
資本化R&D | 28,427 | 25,366 | |||||
研究開発単位 | 11,717 | 10,445 | |||||
リース責任 | 1,779 | — | |||||
株に基づく報酬 | 1,053 | 1,989 | |||||
減価償却および償却 | 545 | 640 | |||||
費用を計算する | 507 | 450 | |||||
他の一時的な違い | 38 | 85 | |||||
繰延税金資産総額 | 100,972 | 88,589 | |||||
推定免税額を差し引く | (99,249 | ) | (88,589 | ) | |||
繰延税金資産から推定免税額を差し引く | $ | 1,723 | $ | — | |||
繰延税金負債: | |||||||
ROU資産 | $ | (1,723 | ) | $ | — | ||
繰延税金負債総額 | (1,723 | ) | — | ||||
繰延税金項目純資産(繰延税金項目負債) | $ | — | $ | — | |||
当社はすでにその繰延税金資産の現金化に影響するプラスと負の証拠を評価した。同社の経営赤字の歴史によると、同社は繰延税金資産の利益が実現できない可能性があると結論した。これにより、当社は2019年12月31日および2018年12月31日までの繰延税金資産について全額評価を提供する準備をしています。2019年12月31日までの年間推計額は約1,070万ドル増加しており、純運営損失が生じていることが要因となっている
法定連邦所得税税率で計算される所得税費用と連結財務諸表に反映される所得税の入金は以下のとおりである
十二月三十一日までの年度 | ||||||
2019 | 2018 | |||||
法定税率で計算される連邦所得税支出 | 21.0 | % | 21.0 | % | ||
州所得税、連邦福祉を差し引いた純額 | 6.3 | % | 8.9 | % | ||
恒久的差異 | 0.0 | % | 9.0 | % | ||
信用を研究開発する | 3.3 | % | 6.7 | % | ||
評価免除額を変更する | (27.4 | )% | (43.1 | )% | ||
その他、純額 | (3.2 | )% | (2.5 | )% | ||
実際の税率 | 0.0 | % | 0.0 | % | ||
2019年12月31日と2018年12月31日現在、当社の米国連邦純営業損失はそれぞれ約2.115億ドルと1.848億ドルで、将来の所得税負債を相殺し、2038年前の異なる日に満期にすることができます。2017年12月31日以降の納税年度に発生した連邦純営業損失は無期限に繰り越すことができます。2019年12月31日と2018年12月31日現在、米国各州における会社の純営業損失はそれぞれ約1兆977億ドルと1兆711億ドルであり、将来の所得税負債を相殺し、2039年までの異なる期日で満期にすることができる
2019年12月31日および2018年12月31日まで、当社はそれぞれ約890万ドルおよび780万ドルの連邦研究開発税収控除繰越を持ち、異なる期日で2039年まで満期になる将来の税務負担を減らすために使用することができます。2019年12月31日および2018年12月31日まで、当社はそれぞれ約350万ドルおよび320万ドルの国家研究開発税収控除繰越を持ち、異なる日に満期から2034年までの将来の税務負担を減らす。
1986年に改正された“国税法”(以下、“国税法”)の規定によると、純営業損失と税収控除の繰越は国税局と国家税務機関が審査と可能な調整を行わなければならない。ネットワークがあります
F-22
大株主の所有権権益が3年間の累計変動が50%を超える(それぞれ規則382及び383節及び類似の国家条文によって定義される)場合、運営損失及び税額控除繰越は年次制限を受ける可能性がある。これは、将来の課税収入または納税義務を相殺するために毎年使用できる税金属性の数を制限することができるかもしれない。年間限度額の金額は、所有権変更直前の会社価値に基づいて決定される。その後の所有権変更は今後数年間の制限にさらに影響を及ぼすかもしれない。当社は設立以来、いくつかの融資を完了しており、これらの融資は、規則382及び383条で定義された制御権の変更を招いた可能性があり、あるいは後日の制御権の変更を招く可能性がある。
当社は所得税支出における不確定な税収状況に関する利息と罰金を確認します。2019年12月31日および2018年12月31日まで、当社は不確定税務状況に関する課税利息や罰金はなく、当社の経営報告書および全面赤字では何の金額も確認していません。
2019年12月31日までの全年度で、同社は研究単位を発生させたが、合格した活動を記録するための研究は行われていない。この研究は会社の研究開発信用の転換の調整につながるかもしれない。しかし、研究が完了し、いかなる調整も理解されるまで、ここ数年は不確定な税収状況として何の金額も報告されていない。すでに当社の研究開発信用について全額評価を準備し、もし調整が必要であれば、この調整は研究開発信用の繰越のために設立された繰延税金資産の調整と推定値から相殺される。
その会社はアメリカとマサチューセッツ州連邦に所得税申告書を提出した。連邦と州所得税申告書は通常、2015年12月31日から2019年12月31日までの納税年度の税務審査を受けなければならない。会社が税務属性の繰り越しを持っている場合、その属性を生成した納税年度は、国税局、州または外国の税務機関が審査した後も将来の期間に使用される程度に調整することができる。
13.四半期財務情報(監査されておらず、単位は千、1株当たりのデータは除く)
3ヶ月が経ちました | ||||||||||||||||
March 31, 2019 | June 30, 2019 | 2019年9月30日 | 2019年12月31日 | |||||||||||||
運営費 | $ | 9,477 | 10,066 | 9,584 | 9,862 | |||||||||||
純収益(赤字) | (15,567 | ) | (6,495 | ) | (7,532 | ) | (9,356 | ) | ||||||||
1株当たり純損失--基本損失と赤字 | $ | (1.22 | ) | $ | (0.42 | ) | $ | (0.28 | ) | $ | (0.34 | ) | ||||
加重-1株当たり純損失を計算するための普通株平均-基本的かつ希薄化 | 12,713 | 15,344 | 26,681 | 27,620 | ||||||||||||
3ヶ月が経ちました | ||||||||||||||||
March 31, 2018 | June 30, 2018 | 2018年9月30日 | 2018年12月31日 | |||||||||||||
運営費 | $ | 10,384 | 9,788 | 10,460 | $ | 8,886 | ||||||||||
純損失 | (15,890 | ) | (4,438 | ) | (7,833 | ) | 350 | |||||||||
1株当たり純損失--基本損失と赤字 | $ | (1.78 | ) | $ | (0.42 | ) | $ | (0.72 | ) | $ | 0.03 | |||||
加重-1株当たり純損失を計算するための普通株平均-基本的かつ希薄化 | 8,905 | 10,693 | 10,829 | 10,847 | ||||||||||||
F-23
サイン
1934年“証券取引法”第13又は15(D)節の要求に基づき、登録者は2020年2月13日に以下の署名者がその代表を代表して本報告に署名することを正式に許可した。
Genocea生物科学社は | ||
差出人: | /s/ウィリアム·クラーク | |
ウィリアム·クラーク | ||
社長と最高経営責任者 | ||
本報告は、1934年の証券法の要求に基づき、以下の者によって登録者として指定日に署名された。
サイン | タイトル | 日取り | ||
/s/ウィリアム·クラーク | 社長と取締役CEO | |||
ウィリアム·クラーク | (首席行政主任) | 2020年2月13日 | ||
/s/Diantha Duvall | 首席財務官 | |||
ダイアナ·デュヴァル | (首席財務官と首席会計官) | 2020年2月13日 | ||
/s/Kenneth Bate | ||||
ケネス·ベター | 役員.取締役 | 2020年2月13日 | ||
/s/アリ·ベヘバハニ | ||||
Ali·ベヘバハニ | 役員.取締役 | 2020年2月13日 | ||
/s/カトリーヌ·ボスリー | ||||
カトリーヌ·ボスリー | 役員.取締役 | 2020年2月13日 | ||
/s/ロナルド·クーパー | ||||
ロナルド·クーパー | 役員.取締役 | 2020年2月13日 | ||
/s/Michael Higgins | ||||
マイケル·ヒギンズ | 役員.取締役 | 2020年2月13日 | ||
/s/ハワード·マイヤー | ||||
ハワード·メイヤー医学博士 | 役員.取締役 | 2020年2月13日 | ||
/s/George Siber | ||||
ジョージ·シーバー医学博士 | 役員.取締役 | 2020年2月13日 | ||
展示品 番号をつける | 展示品 | |
3.1 | 第5回改訂及び再署名された会社登録証明書(2014年2月12日に提出された会社現在8-Kレポート第001-36289号ファイルの添付ファイル3.1編入を参照) | |
3.2 | 改訂された会社登録証明書の改訂証明書(2018年6月25日に提出された会社現在の8-K表第001-36289号ファイルの添付ファイル3.1を参照して編入) | |
3.3 | 改訂された会社登録証明書の改訂証明書(当社が2019年5月21日に提出した8-K表第001-36289号ファイルの添付ファイル3.1を参照して編入) | |
3.4 | 改訂·再改訂された定款(2014年2月12日に提出された会社現在8-Kレポート第001-36289号ファイルの添付ファイル3.2を参照して編入) | |
4.1 | 普通株式証明書フォーマット(2013年12月23日に提出されたS-1表第333-193043号ファイルの会社登録説明書添付ファイル4.1参照) | |
4.2 | 4回目の改訂及び再署名された登録権協定(会社が2013年12月23日に提出したS−1表を参照することにより、文書番号333−193043の登録説明書添付ファイル4.5を編入) | |
4.3 | Aシリーズ転換可能な優先株の優先株、権利、および制限指定証明書(2018年1月19日に提出された会社の現在の8-Kレポート第001-36289号ファイルの添付ファイル4.1を参照して編入) | |
4.4 | Genocea Biosciences,Inc.普通株を購入するA類株式証フォーマット(2018年2月16日に提出された会社8-K年報第001-36289号ファイルの添付ファイル4.5を参照して編入) | |
4.5 | 会社とHercules Capital,Inc.が2018年4月24日に締結した引受権証明書契約(2018年4月30日に提出された会社の現在の8-Kレポート第001-36289号ファイルの添付ファイル4.1を参照して編入) | |
4.6 | Genocea Biosciences,Inc.普通株の事前出資株式承認証表を購入する(2019年2月12日に提出された会社8-K表第001-36289号書類の添付ファイル4.1を参照して編入) | |
4.7 | Genocea Biosciences,Inc.普通株を購入するB類株式証明書表(会社が2019年2月28日に提出したForm 10-K年報第001-36289号の添付ファイル4.8を参照して編入) | |
4.8* | 1934年“証券取引法”第12条に基づいて登録された登録者証券の説明。 | |
10.1 | 取締役賠償協議表(2013年12月23日に提出された会社レジストリS-1第333-193043号添付ファイル10.1参照) | |
10.2++* | 2012年11月19日にGenocea Biosciences,Inc.と総裁とハーバード大学研究員との間の許可協定が改訂され再署名された | |
10.3 | 会社とGenocea Biosciences,Inc.との間のリース期間は2012年7月3日である(2013年12月23日会社S-1表登録説明書の添付ファイル10.8,ファイル番号333-193043参照) | |
10.4 | 転貸に関する合意は,2012年7月9日にTBCI,LLC,FoldRx PharmPharmticals,Inc.,Pfizer Inc.とGenocea Biosciences,Inc.が締結された(2013年12月23日に提出された会社S-1表登録声明の添付ファイル10.9参照により統合,文書番号333-193043) | |
10.5† | Genocea Biosciences,Inc.は2013年6月24日に改訂された2007年株式インセンティブ計画を改訂し再起動した(2013年12月23日に提出されたS-1表登録説明書第333-193043号添付ファイル10.10を参照して編入) | |
10.6† | ウィリアム·クラークとGenocea Biosciences,Inc.が2014年1月16日に改訂·再署名した招聘状協定(2014年1月23日に改訂された会社S-1表登録声明の添付ファイル10.12を参照して書類第333-193043号に編入) | |
展示品 番号をつける | 展示品 | |
10.7† | Genocea Biosciences,Inc.は2014年株式インセンティブ計画を改訂·再起動した(添付ファイル10.1を参照して2018年6月25日に提出された現在の報告Form 8-K,文書番号001-36289) | |
10.8† | Genocea Biosciences,Inc.現金報酬計画(添付ファイル10.16を参照して2014年1月13日に改訂されたS-1表、ファイル番号333-193043の登録声明に組み込まれている) | |
10.9† | Genocea Biosciences,Inc.改正および再制定された2007年株式インセンティブ計画に基づいて付与された非法定株式オプション表(添付ファイル10.20を参照して2013年12月23日に提出されたS-1表、文書番号333-193043の登録声明に組み込まれている) | |
10.10† | Genocea Biosciences,Inc.改正および再制定された2007年持分インセンティブ計画に基づいて付与されたインセンティブ株式オプション表(添付ファイル10.21を参照して2013年12月23日に提出されたS-1表、文書番号333-193043の登録声明に組み込まれている) | |
10.11† | Genocea Biosciences,Inc.2014年持分インセンティブ計画下のインセンティブ株式オプション表(2014年1月13日に改訂された会社S-1表登録説明書第333-193043号添付ファイル10.22を参照して編入) | |
10.12† | Genocea Biosciences,Inc.2014年株式インセンティブ計画下の非法定株式オプション表(会社が2014年1月13日に改訂したS-1表登録説明書添付ファイル10.23,文書333-193043号参照) | |
10.13† | 改訂されたGenocea Biosciences,Inc.2014年従業員株式購入計画(2018年6月25日に提出された会社現在8-Kレポート第001-36289号ファイルの添付ファイル10.2を参照して編入) | |
10.14† | Genocea Biosciencesによると、Inc.が2013年5月13日に改訂され、再編成された2007年株式インセンティブ計画は、カトリーヌ·ボスリーの非法定株式オプションを付与する(2014年1月13日に改訂された会社S-1表登録説明書第333-193043号ファイル添付ファイル10.27を参照して編入) | |
10.15† | Genocea Biosciencesによると、Inc.が2013年11月5日に改訂され、再編成された2007年株式インセンティブ計画は、カトリーヌ·ボスリーの非法定株式オプションを付与する(2014年1月13日に改訂された会社S-1表登録説明書第333-193043号ファイル添付ファイル10.28を参照して編入) | |
10.16 | 会社とHercules Technology Growth Capital,Inc.が2014年11月20日に締結した株式書簡協定(添付ファイル10.2を参照して2014年11月21日に提出された8-K表第001-36289号文書の現在の報告に組み込まれている) | |
10.17 | 会社とスミソニアン学会の転貸協定は、2015年6月15日(2015年6月19日に提出された会社現在8-Kレポート第001-36289号ファイルの添付ファイル10.2参照) | |
10.18 | リース第一修正案は、2016年5月16日、デラウェア州の有限責任会社100 Discovery Park DE,LLC(100 Discovery Park Realty Trust受託者の権益相続人として)とGenocea Biosciences,Inc.との間(2016年8月5日に提出された会社10-Qテーブルの添付ファイル10.30を参照して統合され、ファイル番号001-36289) | |
10.19+ | 会社がOncovir,Inc.と2018年1月26日に締結したライセンスおよび供給契約(2018年2月16日に提出された会社10-K年間報告書第001-36289号ファイルの添付ファイル10.32を参照して編入)。 | |
10.20+ | 会社といくつかの銀行および他の金融機関または実体との間で2018年4月24日にHercules Capital,Inc.と締結された融資および保証協定に改訂および再署名された(2018年8月3日に提出された会社10-Q四半期報告書第001-36289号文書の添付ファイル10.4を参照して組み込まれる) | |
10.21 | 2018年4月24日Hercules Capital,Inc.(Hercules Technology Growth Capital,Inc.)社(F/k/a Hercules Technology Growth Capital,Inc.)の株式書簡プロトコル修正案(2018年4月30日に提出された会社の現在8-K表第001-36289号文書の添付ファイル10.1を参照して組み込む) | |
展示品 番号をつける | 展示品 | |
10.22 | 当社、いくつかの銀行および他の金融機関または実体がHercules Capital,Inc.が2019年11月14日に締結した改正および再署名された融資および保証協定の第1の修正案(2019年11月19日に提出された会社の現在の8-Kレポート第001-36289号文書の添付ファイル10.1を参照して編入する) | |
10.23* | 2019年5月1日,デラウェア州有限責任会社100 Discovery Park DE,LLC(100 Discovery Park Realty Trustの受託者としてTBCI,LLCの権益相続人として)とGenocea Biosciences,Inc.との間の賃借第2改正案 | |
21.1 | 会社子会社リスト(2016年2月17日に提出された会社10-K表添付ファイル21.1,001.36289号書類合併参照) | |
23.1* | 安永法律事務所が同意した | |
31.1* | 最高経営責任者は2002年サバンズ·オキシリー法案第302条の規定による認証 | |
31.2* | 2002年サバンズ·オキシリー法第302条の規定による首席財務官の証明 | |
32.1** | CEOは2002年サバンズ·オキシリー法案第906条による定期財務報告の証明 | |
32.2** | 2002年サバンズ·オキシリー法第906条による定期財務報告の証明 | |
101. INS* | XBRLインスタンスドキュメント | |
101. SCH* | XBRL分類拡張アーキテクチャ | |
101. CAL* | XBRL分類はリンクライブラリをトポロジ計算できる | |
101. DEF* | XBRL分類拡張定義リンクライブラリ | |
101. LAB* | XBRL分類拡張タブリンクライブラリ | |
101. PRE* | XBRL分類拡張プレゼンテーションリンクライブラリ | |
_________________________
* | 本局に提出します。 |
** | 手紙で提供する。 |
† | 契約または補償計画を管理すること。 |
+ | 機密処理の要求により,本展示品の内容の一部(星番号で示す)は省略されており,本展示品は単独で米国証券取引委員会に提出されている. |
++ | 本展示品のいくつかの部分(星番号で表される)は省略されている。登録者は、それらが実質的ではないと判断しているので、開示されている場合、登録者に競争損害を与える可能性がある。 |