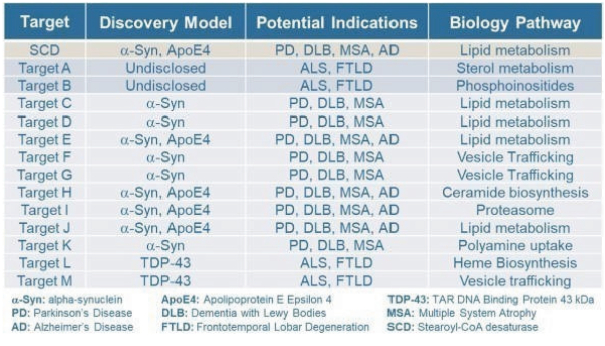
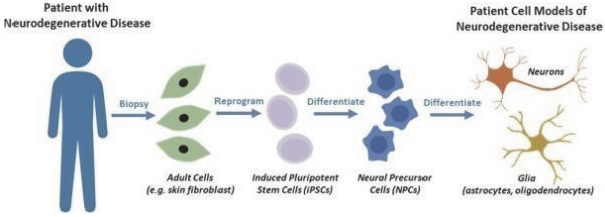
高スループット技術の化学ライブラリーを用いて,誤ったフォールディングから細胞を保護するヒト疾患関連タンパク質による毒性の化学物質を選択した。そして、一連の化学遺伝技術を用いて、これらの保護分子の生物標的と経路を発見した。我々の技術は単一遺伝子欠損酵母集合のスクリーニングも許可しているため、救助が観察された場合、この酵母菌株中の欠損遺伝子は誤って折り畳まれたヒト疾患関連蛋白の毒性改善に関与していると推定される。原始酵母系の唯一の修正は張本人の誤ったフォールディング蛋白を導入することであるため、それによって産生される毒性の分子や遺伝子欠損から細胞を保護することは意義がある。保護性分子や生物標的の発現,特に従来未知の場合には,新たなあるいは未開発の研究分野を明らかにすることができる。小分子と遺伝子救援スクリーニングとの間の相補と重複は、相互に関連する生物過程の強力なネットワークを作成する可能性があり、以前未知の治療標的をさらに識別することができると信じている。我々は,情報学,先端幹細胞,IPSC実験技術を用いて,酵母系からヒト疾患関連細胞までのこれらの細胞保護発現を探索した。発見エンジンは最終的に新たなプログラムを出力するように設計されている:新しい生物標的を持つ分子は,標準的な臨床前薬物開発過程で進展することができる
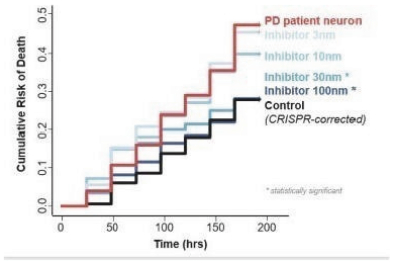
私たちの独自の発見エンジンは、神経変性をめぐる複雑な生物学の知識を大きく拡張する可能性があり、 は、現在存在する伝統的で限られた仮説のセットではなく、発見計画を開始することをさらに可能にすると信じている。生酵母系でヒットをスクリーニングすることは、非生試験管系で開始されるいくつかの典型的な方法よりも速く生物関連読み取りを提供するので、時間を節約することができる。さらに、酵母細胞膜と血液脳関門との間の共通の特徴、例えば、同様の膜透過性、極性、および非天然化合物を除去するための薬物ポンプは、細胞内救助を達成するために酵母細胞に浸透することができる分子も血液脳関門を貫通する可能性があることを意味する。また,我々の分子brは罹患したヒト細胞でテストされている体外で臨床前の発展の始まりは終わりではなく.人体組織がどのような実際のテストを行う前に多輪動物研究を行うより伝統的なパラダイムに比べて,このような環境での成功はプロジェクトに対する信頼を高めることができると考えられる
Discovery Engineの主な利点は
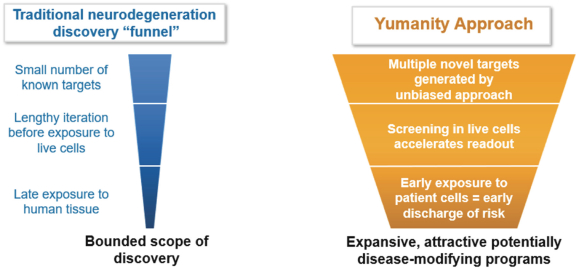
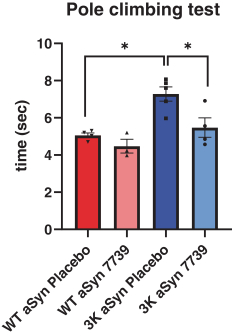
我々は,発見エンジンの強力な機能を利用して,一連の有望な新薬標的と分子の強力な組合せを生成する。そして,最も有望な目標を優先的に選択し,薬物発見計画を加速し,化合物を臨床前と最終的な臨床開発に進める。これまでに,この方法は を発見している
4