アメリカ アメリカ
証券取引委員会
ワシントンD.C.,20549
表
(タグ 一)
| 1934年証券取引法第13又は15(D)節に基づいて提出された年次報告 |
締め切りの財政年度について
あるいは…。
| 1934年証券取引法第13条又は15条に基づいて提出された移行報告 |
への過渡期について
手数料ファイル番号
(登録者がその定款に明記されている氏名)
(State or Other Jurisdiction of 会社(br}や組織) |
(I.R.S. Employer 標識 番号) |
| (主に実行オフィスアドレス ) | (Zip コード) |
登録者の電話番号は市外局番を含んでいます
同法第12条(B)に基づいて登録された証券:
| クラスごとのタイトル | 取引 個の記号 | 登録された各取引所の名称 | ||
|
|
同法第12条(G)により登録された証券:なし
登録者が証券法ルール405で定義されている有名な経験豊富な発行者であるかどうかをチェックする.はい。☐
登録者が当該法第13条又は第15条に基づいて報告書を提出する必要がない場合は,フックで
を示してください。はい。☐
再選択マークは、登録者が、(1)過去12ヶ月以内(または登録者がそのような報告を提出する必要があるより短い期間)に、1934年の証券取引法第13または15(D)節に提出されたすべての報告書を提出したかどうか、および(2)
が過去90日以内にそのような提出要件に適合しているかどうかを示す
再選択マークは、登録者が過去12ヶ月以内(または登録者がそのような文書の提出を要求されたより短い時間以内)に、S−Tルール(本章232.405節)405条に従って提出を要求した各対話データファイルを電子的に提出したか否かを示す
登録者が大型加速申告会社,加速申告会社,非加速申告会社,小さな申告会社,あるいは新興成長型会社であることを再選択マークで示す。取引法第12 b-2条の規則における“大型加速申告会社”、“加速申告会社”、“小申告会社”、“新興成長型会社”の定義を参照されたい。
| 大型 加速ファイルサーバ | ☐ | 加速した ファイルマネージャ | ☐ | |
| ☒ | 小さな報告会社 | |||
| 新興成長型会社 |
もしbrが新興成長型会社である場合、登録者が延長された移行期間を使用しないことを選択したかどうかを再選択マークで示して、取引法第13(A)節に従って提供された任意の新しいまたは改正された財務会計基準を遵守してください
登録者が報告書を提出したか否かを再選択マークで示し、その経営陣が“サバンズ-オキシリー法案”(“米国法典”第15編7262(B)節)第404(B)条に基づいてその財務報告の内部統制の有効性を評価したことを証明する
登録者が空殻会社である場合(同法第12 b-2条で定義されているように)、フックで表される。はい。☐
登録者の非関連会社が保有する投票権と無投票権普通株の総時価は#ドルである
2022年12月14日までの登録者の普通株当たりの流通株数を示す
| クラス | 株式数: | |
| 普通株、額面0.0001ドル |
参照により組み込まれた文書
カタログ表
| 第 部分I | 1. | 業務.業務 | 4 |
| 1A. | リスク要因 | 32 | |
| 1B. | 未解決 従業員意見 | 74 | |
| 2. | 属性 | 74 | |
| 3. | 法的訴訟 | 74 | |
| 4. | 鉱山安全開示 | 74 | |
| 第 第2部分 | 5. | 登録者普通株,関連株主事項及び発行者が株式証券を購入する市場 | 75 |
| 7. | 経営陣の財務状況と経営結果の検討と分析 | 75 | |
| 7A. | 市場リスクの定量的·定性的開示について | 85 | |
| 8. | 財務諸表と補足データ | 86 | |
| 9. | 会計·財務開示面の変更と会計士との相違 | 109 | |
| 9A. | 制御 とプログラム | 109 | |
| 9B. | その他 情報 | 109 | |
| 第 第3部分 | 10. | 役員·役員·会社管理 | 110 |
| 11. | 役員報酬 | 114 | |
| 12. | 安全な利益所有者と経営陣の所有権および関連する株主事項 | 118 | |
| 13. | いくつかの関係や関連取引、および取締役の独立性 | 119 | |
| 14. | 依頼者 は課金とサービス | 120 | |
| 第4部 | 15. | グラフ、財務諸表明細書 | 121 |
| 16. | 表 10-K要約 | 121 |
| -2- |
前向き陳述に関する特別説明
本報告書には,1995年の民間証券訴訟改革法で指摘された前向きな陳述が含まれている。前向き 陳述はすべての非歴史的事実の陳述を含む。場合によっては、用語 を使用して、“可能”、“将”、“すべき”、“可能”、“将”、“予想”、“計画”、“予想”、“予想”、“信じ”、“推定”、“プロジェクト”、“予測”、“潜在”またはこれらの用語の負の意味、ならびに前向き陳述を識別することを目的とする同様の表現および同様の用語を識別するために前向き陳述を識別することができる。これらの陳述は、会社の未来の事件に対する現在の見方を反映し、仮説とリスクと不確定要素の影響を受けることに基づいており、これらのリスクと不確定要素は、本10-K年度報告書の第I部分第1 A項“リスク要素”で述べたリスクと不確定要素を含む。このような不確実性を考慮して、あなたはこのような前向きな陳述に過度に依存してはいけない。これらの前向き は、会社が本10-K年度報告の日までの推定および仮定のみを代表しており、法律で規定されている を除いて、会社は、本年度報告が10-K形式で発表された後の新しい情報、未来のイベント、または他の理由のために、任意の前向き陳述を公開更新または審査する義務がない。このForm 10-K年次報告書とこのForm 10-K年次報告書で参照され、添付ファイルとしてアーカイブされたファイルを読み、会社の将来の実際の結果が会社が予想しているものと大きく異なる可能性があることを理解しなければなりません。当社はこれらの警告的声明によりそのすべての 前向き声明を限定している。このような陳述は、以下の に関する以下の陳述を含むことができるが、これらに限定されない
私たちは運営履歴と運営損失の歴史が足りません
私たちの現在と未来の資本需要と私たちの資本需要を満たす能力
私たちは必要な製品の臨床試験を完成させ、食品と薬物管理局あるいは異なる司法管轄区の他の規制機関の許可を得ることができる
最近の新冠肺炎の大流行は著者らの臨床開発計画とスケジュールへの影響を含むわが業務に対する潜在的な影響を含む
私たちの特許と他の知的財産権の有効性を維持または保護する能力;
私たちは主な実行メンバーの能力を維持します
私たちは内部で新しい発明と知的財産権を開発する能力;
現行法の解釈と将来の法律の段落;
投資家は私たちのビジネスモデルを受け入れます
私たちの費用と資本需要の推定の正確さ
私たちは成長能力を十分に支持している。
| -3- |
第 部分I
第 項1.業務
概要
十四行詩 BioTreateutics Holdings,Inc.は臨床段階、腫瘍学に集中する生物技術会社であり、革新 単特効或いは二特効生物薬物の特許プラットフォームを持っている。Fと呼ぶHAB(完全ヒトアルブミン結合)は,ヒト血清アルブミンに結合してその上に“便乗”する完全ヒト単鎖抗体断片を用いて標的組織 に輸送される。Fを設計しましたHABは癌腫瘍への薬物の蓄積を改善し、体内での活動時間を延長するように構築された。F.FHAB発育候補は哺乳動物細胞培養において産生され、これによりグリコシル化および天然サイトカインに類似した生物構造が生じる体内にあるそれは.私たちのFはHAB技術は著者らの生物製薬プラットフォームの1つの顕著な特徴であり、未来の一連の人類疾病領域における薬物開発に非常に適しており、腫瘍学、自己免疫、病原性、炎症性と血液学疾患を含む。
我々の現在の内部パイプライン開発活動はサイトカインに集中しており,これは1種類の細胞シグナルペプチドであり,他の重要な機能において有効な免疫調節剤である。独立および相乗作用の下で、特定のサイトカインは、癌および病原体に対して免疫細胞の活性化および成熟を調節する能力を示すようになっている。しかし、サイトカイン自体は特定の組織に優先的に蓄積することはなく、そして迅速に体内から除去され、伝統的なサイトカイン治療方法は通常高用量と頻繁な投与量が治療効果を達成するために必要である。これは治療効果の低下を招き,潜在的な全身毒性を伴う可能性があり,これらの薬物の治療応用に挑戦している。
十四行詩 はサプライヤーネットワークを含む効率的な研究開発プラットフォームを構築し、救済費用を助け、 の実行時間を短縮する。多くのサプライヤーは戦略パートナーであり、会社に優先順位を提供し、 コストを協議する。この方法の主な利点は、プロジェクトの最適化への直接投資を含み、その費用はプロジェクト数に応じて迅速に増加または減少することができる。Sonnetプラットフォームのコストメリットはサプライヤーネットワーク 選択プロセスから始まり、CMCは最初の薬物開発ステップの中で最も高価なコンポーネントの一つである。十四行詩は、インドでCMC戦略パートナーを選択し、交渉によって、同様の米国やヨーロッパのサプライヤーから発生した費用を大幅に下回るコストを達成した。十四行詩は,同社が行っている3つの臨床試験のうち2つをオーストラリアで行っており,オーストラリア政府の開発信用計画により,米国の試験に比べてコストが大幅に低下している。十四行詩はまた、米国、イギリス、ドイツ、スイスからのトップクラスの研究開発サプライヤーとインドとオーストラリアの実行を調整し、会社の運営費用の大部分をその薬物開発パイプラインに利用することを目標としている。
パイプ
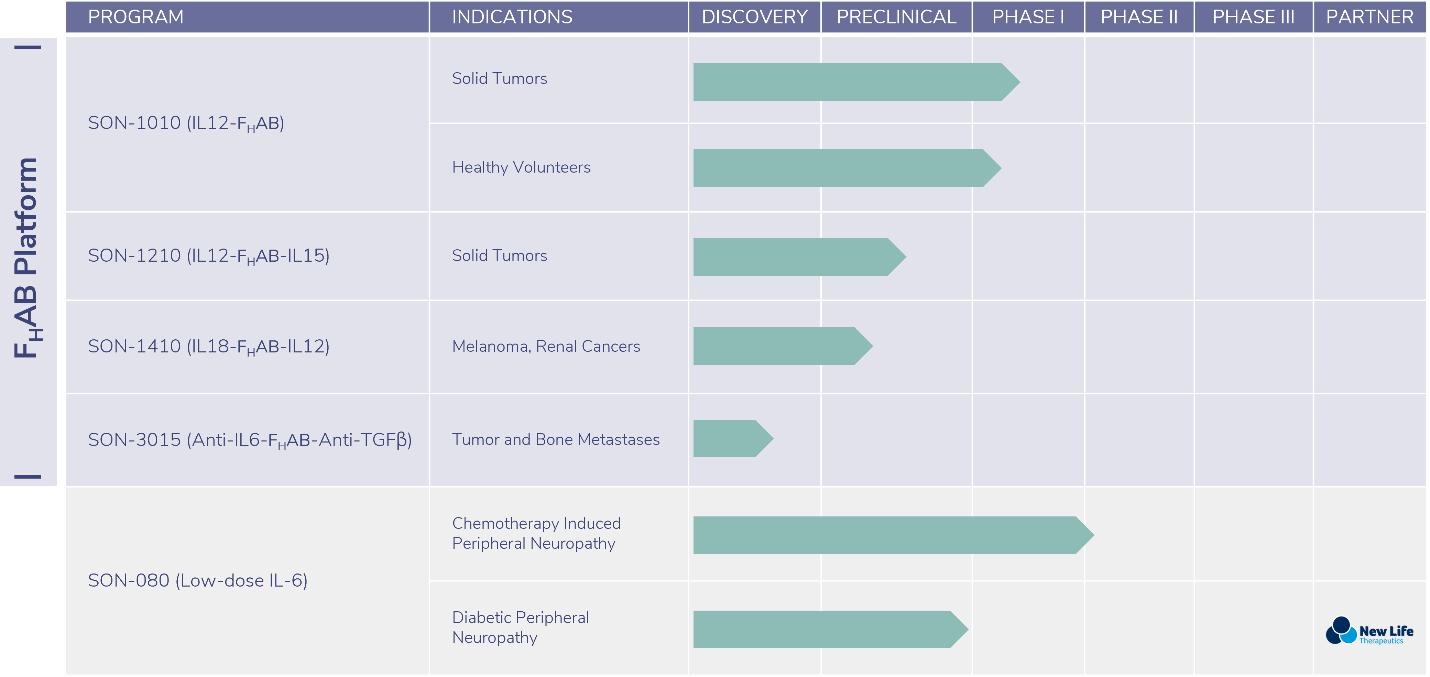
われわれには一連の治療化合物があり,主に高度に満たされていない医療ニーズの腫瘍学的適応に集中している。
| ● | 当社の先行する独自資産SON-1010は、Fに共有結合されたインターロイキン12(IL-12)の全ヒト一本鎖バージョンですHAB機構,そのために固形腫瘍の臨床開発を行っている。十四行詩は、非ヒト霊長類動物(“NHP”)のGLP毒性研究を完了し、臨床のための液体および凍結乾燥形態のこの医薬製品の製造に成功した。2022年上半期、私たちはアメリカで癌患者の第一段階臨床試験を開始し、健康ボランティアでオーストラリアの臨床試験を開始し、この化合物の薬物動態(PK)と薬効学(PD)を研究した。この2つの臨床研究の中期安全性データはいずれも2022年第4四半期に公表された。これらのデータセット は次の段階の臨床開発の段階であり、検査点阻害剤との併用治療を探索することを含む。 の2つの試験の初歩的な分析データは2023年の第2のカレンダー四半期に発表される予定である。 | |
| ● | 同社は2020年4月にインターロイキン6(“IL-6”)の全人バージョンのグローバル開発権を獲得した。この候補薬をSON−080と呼び,その目標適応は化学療法による末梢神経病変(CIPN) と糖尿病末梢神経病変(DPN)であるからである。十四行詩のCIPN 1 B期/IIA臨床試験はオーストラリアで行われている。この研究は2023年上半期に初歩的なトップクラスの臨床安全性データを生成する可能性がある。会社が2021年5月にシンガポール新生命治療プライベート有限会社(“新生命”)と達成した許可合意によると、十四行詩と新生命は共同でDPNの中でSON-080の開発を担当し、2023年下半期に前アメリカの先導的治療効果研究を開始することを目標としている。 | |
| ● | SON-1210 (IL12-FHAB-IL 15)はFに結合した鉛二重特異性化合物ですHABは全ヒトIL−12と全ヒトIL−15(IL−15)を用いて構築した。この化合物は結腸直腸癌を含む固形腫瘍への適応が開発されている。非ヒト霊長類(NHP)研究の生命部分は2022年第4四半期に完成する見込みであり,規制認可手続きは2023年上半期に開始される。2023年第1四半期に凍結乾燥薬物製剤“br”を調製することにより、生産と薬品供給を確保し、この製剤は初めての人体臨床試験に適用されることが予想される |
私たちの発見ルートでは調査しています
| ● | SON-1410 (IL18-FH黒色腫および腎癌を治療するインターロイキン18(IL-18)およびインターロイキン12(IL-12)の二重特異的組み合わせであるAB-IL 12)。CELL シリーズ開発とプロセス開発が行われており,早期開発概念検証に少量のほか,処方や分析方法開発活動に適した早期実験的薬剤を提供している体外培養研究。brプロセス開発活動は2023年まで続き、初歩的な応用に適した薬物が生産される可能性がある体内で 2023年に暦年が終わる前にマウス研究を行った。 |
| ● | SON−3015 (抗IL 6−FHAB-抗形質転換増殖因子β)、抗IL-6と抗腫瘍増殖因子βの二重特異的組み合わせは、腫瘍および骨転移に用いられる。初期の二重特異性薬が生産され将来のために保存されています体内にあるネズミ研究。十四行詩は、費用を下げるために、SON-3015開発計画を保留することを選択した。 |
| -4- |
私たちの治療化合物の開発と商業化の面で、私たちは多くの挑戦と不確実性に直面していますHエービー技術です。本募集説明書の他の部分に含まれる“リスク要因”と、本募集説明書の文書に引用して“リスク要因”と題する章を参照してください。
戦略.戦略
私たちの目標はルートを迅速に推進し治療機能を利用することですHABプラットフォームは生物薬物の発見、開発と商業化の先頭になる。Sonnetは設立以来,診療所候補プロセスの迅速な推進に専念するとともに,適切なパートナーとの連携関係の構築にも取り組んできた。協調関係対話の発展にともない,Sonnet計画 は最大の戦略的利益を持つ資産に費用を優先的に割り当てる.そのため,Sonnetは運営費を約30%削減しようと努力しており,将来のパイプライン拡張に資金を提供するための許可協定を交渉しようとしている.早期発表の一例として、Janssenは、我々の3つのパイプライン化合物SON−1010、SON−1210およびSON−1410とその細胞治療製品との組み合わせ を評価している。
FHAB 計画進展:2021年下半期にSON−1010のINDをFDAに提出し,2022年初めにこの機関に対するすべての回答 を完了した。私たちの最初のヒト臨床研究は2022年の第1四半期に開始されましたわれわれの最初の二重特異性候補薬SON−1210については,GMP原料薬が生産されており,2023年上半期に臨床応用される予定である。
SON-080は次の臨床開発段階に入る:SON-080は研究中の低用量インターロイキン6(IL-6)の全ヒトバージョンであり、化学療法による末梢神経病変(CIPN)の治療に用いられる。SON-080はすでに癌患者のI/II期臨床試験 を完成することに成功し、著者らは2022年下半期にCIPN患者に対するIb/IIa期の治療効果の試験研究を開始した。
製造プラットフォーム:十四行詩化合物は業界標準哺乳動物細胞(中国ハムスター卵巣/CHO)宿主細胞系を用いて生産され、この宿主細胞系は最先端の製造技術と技術を用いて迅速な拡大と商業製造を許可する。哺乳動物の細胞培養系はグリコシル化作用と天然サイトカインに類似した生物構造を体内にあるそれは.臨床応用サイトカインの製造、即ちそれらの生産と精製は、明らかな技術挑戦をもたらした。そのため、十四行詩 は1種の連続強化灌流技術を開発して商業生産量を製造し、著者らはこれらの製造と下流技術開発ステップ中のいくつかのステップのために知的財産権保護 を求めている。
規制戦略:Sonnetの候補薬物は既存療法と大きく異なり、生物製薬薬物開発の潜在的な突破を代表していると考えられる。規制当局と画期的な治療指定を求める努力をし,臨床開発スケジュールを加速させる可能性がある。
パイプラインライセンス機会:リーディングバイオ製薬会社との協力機会を求めて、パイプライン資産を開発して商業化しています。
FHAB 技術拡張:十四行詩はFを探索しているHAB技術はその治療展開を意図的に拡大した外部パートナーと許可され、これはワクチン、抗体薬物 結合体などの他の分野でのこのプラットフォームの応用をもたらし、キメラ抗原受容体(CAR)T細胞技術の補充となる可能性があると信じている。Fとの排他性{brを確保するために仮特許が提出されたHこの田野にあります。
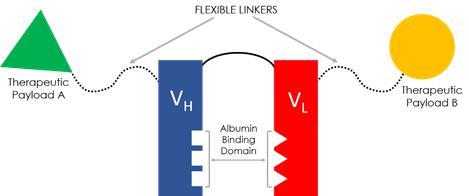
The FHAB技術
私たち 独自FHAB技術は生物製薬薬物開発の現有方法のいくつかの重要な欠陥を解決することを目的としている。Fを設計しましたHABはプラグアンドプレイ用のモジュール化構造として、異なる治療ペイロードの再配置を必要とすることなく、新しい化学実体を革新するために使用される。すべての生物薬物の場合のように,用量レベルと投与頻度 はキー変数であり,開発過程の障害となることが多い。注射後,ポリペプチド,タンパク質,融合蛋白,抗体などを含む巨大分子療法は完全に保たなければならず,特定の毒性閾値を超えることなく体内指定のbr標的を達成することができる。最後に、それらはまた商業的に魅力的な方法で生産されなければならない。
十四行詩のプラットフォーム技術はヒト血清アルブミン(HSA)を治療用シャトル分子として利用することを目的としている。ヒト血清アルブミンは自然に血液と血漿中の主要なタンパク質中に存在する。アルブミンは炎症、高代謝組織のエネルギー源であり、腫瘍を含む。栄養に対する積極的な需要により、癌細胞はSPARC(分泌蛋白酸性、システイン豊富)とgp 60(Albondin糖タンパク質)のようなアルブミン結合蛋白を過剰に発現する。
| -5- |
XOMA(US)LLC(“XOMA”)は、2012年7月23日の発見協力プロトコルと2019年5月7日の発見協力プロトコル修正案 (総称して“連携プロトコル”と呼ぶ)に基づいて、非排他的で譲渡不可能なbrライセンスおよび/または抗体および関連タンパク質の発見、最適化および開発に関連するいくつかの材料、技術および情報を使用する権利をSonnetに付与し、それに基づいて製品(各製品を“製品”)を開発および商業化する。協力プロトコルは、生物学的配列を探すための完全なヒト単鎖抗体br断片(‘scFv’)を産生することを意図した全ヒトファージライブラリーを使用するライセンスを含む。厳格な基準を用いて、十四行詩は数百万の単鎖抗体をヒト血清アルブミンに結合して十四行詩のFを生成するHAb, はヒト血清アルブミンに結合しており,3つの主要ドメインを有する球状タンパク質である。アルブミンドメイン1と3がFcRNへの結合に関与していることが知られている。これにより十四行詩はドメイン2に特定された単鎖抗体結合子を選択して表現することができ、これは十四行詩Fの基礎であるHAB プラットフォーム。
十四行詩(Br)は、製品に関するいくつかの開発と承認マイルストーンを実現する際に、製品ごとにXOMAに合計375万ドルを支払うか、マイルストーンで支払う義務があります。十四行詩はまた、十四行詩販売製品の純売上高のためにXOMAに低い一桁版税を支払うことに同意した。各製品の使用料は、(I)初の商業販売(提携協定の定義参照)後12(Br)年(12)および(Ii)協力協定によってカバーされる発行された特許の最後の有効な クレームが満了する日(遅い者を基準とする)まで国/地域で支払われる。また,十四行詩は,XOMAに指定された金額を支払うことで,1製品あたりの印税料率を下げる権利がある.協力協定は、いずれか一方によって を終了することができ、通常の賠償条項を含むことができる。
14行詩のFHABは種(ヒト,マウス,カニクイザル)にまたがる血清アルブミンとの高い結合親和性を示しており,免疫原性がほとんどなく,新生児FcRNを介したアルブミン循環の利点を保持しており,血清半減期を4週間延長することができる。モノクロナル抗体(MAbs)とは異なり,この結合は抗体依存性細胞毒性(ADCC)や補体依存性細胞毒性(CDC)を引き起こさない。“F”HAB構築はイオン疎水機構により血清アルブミンを物理的に結合しており,化学,共有結合に依存する技術よりも明らかな優位性を提供していると考えられる。共有鍵が切れたら,これ以上変化することはできないが,十四行詩のFHABはアルブミンに結合し,結合を解除し,再結合する能力を有する。アルブミン受容体gp 60とSPARC,Fを探すHABは固有の生物機序を利用して治療ペイロードを腫瘍微小環境に配向輸送する。
十四行詩のもう一つの独特な優位性Hエービーはそのリンカのデザインです。1つまたは2つの巨大分子を連結するための治療ペイロード、 は単一または二重特異性活性のために使用され、私たちのG 4 S(グリシン、セリン)ポリペプチドリンカーは柔軟であり、同時に長さは十分に長く、空間障害 を防止することができ、緊密な組織基質への浸透を増強するために棒状構造を呈することができる。治療機能ドメイン間の距離 を保つことを除いて,十四行詩リンカーは完全にヒトであり,リンカー構造的にはペイロード結合領域を含む非免疫原性である。二重特異性構造において、治療ペイロードの方向を操作することができ、潜在的な治療効果を改善することができる。
| -6- |
最終的なキーデザインコンポーネントとしてFHABは哺乳動物細胞培養中に産生され、特に中国ハムスター卵巣(CHO)細胞であり、グリコシル化作用を低下させ、あるいは潜在的に免疫原性を除去する。CHOを用いて、著者らは様々な低分子治療性タンパク質(例えば、IL-12、IL-15、IL-18、抗IL-6および抗形質転換増殖因子βなどの組換えサイトカイン)とのいくつかの異なる融合構築物を作成した。細胞因子を含む組換え治療性蛋白は、すでに巨大な治療潜在力を示したが、組織特異性が乏しい可能性があり、これは毒性を招く可能性がある。それらが小さい(HAB由来化合物)ため,それぞれの露出組換えサイトカインと比較して有意に長い血清半減期,改善した組織蓄積,有意な腫瘍抑制活性を示した。
要約では私たちのFはHAB技術はモジュール化、多機能ステントを支持し、このステントは広範な多標的候補治療 を産生するようにカスタマイズすることができる。既存のアルブミン結合技術と比較してFHABの違いは、標的組織をよりよく貫通することを目的とした線形棒状形状を有し、完全に人間的に設計され、免疫原性を低下させることができ、哺乳動物グリコシル化とFcRN結合が血清半減期を延長することができることである。重要なのはFHAB誘導療法は、標的送達、毒性の減少、およびより広い治療窓の潜在力を有し、カスタマイズされた単一または二重特異性作用機序を利用する追加の利点を有する。
Fの応用を拡張した HABテクノロジー:
免疫療法: 私たちはFを信じているHABプラットフォームは特定の組織に対する生物薬物を革新することができ、同時に治療半減期を増加させることができる。FとしてHAB構築の目的は2種類の協同免疫治療化合物の同時展開を実現することであり,従来開発されていなかった免疫治療の進展への道を想定している。
薬物br結合:FとHAB技術、様々な医薬化合物がFに結合することができるHABステントの組み合わせ はわれわれの第一波サイトカイン管の外に伸びており,無数の疾患領域の発展に機会を提供している。
ワクチン: ワクチン開発者は,ワクチンとアルブミンなどの天然担体を結合することでワクチン効率を向上させることを求めている。Fを信じていますHABプラットフォームはモジュール化されたステント構造を有し、有効な担体としてワクチンをリンパ節に輸送し、浸透を改善し、提示し、半減期を延長することができる。
CAR T細胞療法:CAR T細胞療法は患者自身のT細胞を遺伝子修飾して癌細胞を認識し、腫瘍をより効率的に標的と殺傷することに関連する。著者らは、インターロイキン標的を利用して構築した十四行詩は系統的に連合応用し、CAR T細胞の治療効果を増強できると信じている。
パイプ の概要
次の表は、特定の目標適応症を開示したパイプライン計画に関する情報をまとめています
| -7- |
SON-1010
インターロイキン12(IL-12)は循環サイトカインであり、すでに先天性免疫と獲得性免疫に対して多種の作用を発揮することが証明された。これらの免疫機能は癌細胞と病原体を攻撃する上で重要である。IL-12は樹状細胞、単球とマクロファージから産生されるヘテロ二量体サイトカインであり、抗原提示細胞(APC)とも呼ばれる。IL-12はT細胞とナチュラルキラー細胞(NK細胞)のインターフェロン-オスミウムの分泌を誘導し、活性化されたT細胞とNK細胞の増幅と生存を促進し、細胞毒T細胞の殺傷活性を増強し、Th 1補助エフェクター細胞の分化を支持し、抗体依存の細胞毒作用を増強する体外培養癌患者の末梢血リンパ球の抗腫瘍活性体内にある[br]マウス黒色腫、結腸癌、乳癌および肉腫モデルの抗腫瘍活性。
2021年5月、我々は、非ヒト霊長類動物(NHP)においてSON-1010のGLP反復用量研究に成功したことを発表した。この研究の目的はNHPにおけるSON-1010の毒性を評価し、3種類の異なる用量レベルの反復皮下投与方案 を用いて未治療対照群と比較し、そして任意の不良結果の潜在的可逆性を評価することである。研究結果には
| ● | 重複投与後に観察されなかった有害事象レベル(NOAEL)はNHP予想等量ヒト臨床投与量の50倍以上であり,サイトカイン放出症候群の証拠はない。 | |
| ● | 血清試料の薬物動態(PK)分析により,IL 12−FHABは組換えヒトIL−12よりも強い特性を有し,NHPにおける半減期は約40 br}時間であることが確認された。 | |
| ● | インターフェロン-γは抗腫瘍機序に関連する重要な多効性サイトカインであり、IL 12-FHAB投与後に明らかに増加する。 | |
| ● | SON-1010は臨床観察、体重、臨床病理、サイトカインと免疫表現型に相関変化が発生し、これらのすべての変化は以前非ヒト霊長類動物で観察された標的効果と一致した。 | |
| ● | 38日目には,すべての対象者がベースライン(研究前)値に回復した。 | |
| ● | 検査のすべての用量レベルでは,反復投与は耐性であった。 |
2021年2月、我々は、GLP毒性研究の設計に情報を提供し、IND提出に準備するために使用されるSON-1010のNHP非GLP反復用量毒理学研究の完成に成功したことを発表した。この研究の目的は2種類の異なる用量の繰り返し投与方案におけるSON-1010の毒性を評価し、そして更なるIND安全性と毒性研究を設計するために肝心なデータを収集することである。この研究には静脈と皮下投与経路を含め,計2回投与し,14日間隔で投与した。この研究で用いられた高用量率は,患者が予想していた臨床曝露レベルの50倍を超えていた。研究結果には
| ● | 検査された2つの用量レベルでは,静脈および皮下経路による反復投与は許容可能である。通常観察されているように,IL−12を用いると白血球数が低下し,肝酵素(ALTとAST)が上昇する。これらは一過性の影響であり,2回目の注射後7日でベースラインレベルに回復した。 | |
| ● | SON-1010は生理観察、体重、病理、サイトカインと免疫表現型に関連する変化が発生し、これらのすべての変化は以前単剤研究で観察された標的効果と一致した。 | |
| ● | 1回目のSON−1010投与後,インターフェロン−γレベルは有意に上昇したが,2回目の投与後,インターフェロン−γレベルは低下した。この傾向は,ヒトおよびNHPにおけるIL−12の他の研究について発表されたデータに従っている。 | |
| ● | 薬物動態分析により、SON-1010を皮下注射した動物の平均血清半減期は約40時間であることが示された。これは以前に行われた用量増加段階の研究データと一致し、この段階の半減期は裸組換えヒトIL-12 13-19時間の半減期と比較して有意に改善した。 |
また、臨床前研究により、Fを含まないIL-12と比べ、SON-1010は腫瘍成長に対して更に大きな抑制作用があることが示唆されたH黒色腫マウスモデルにおけるAB(露出/独立IL-12)の役割。以下の図2は,このマウス黒色腫研究から,SON−1010が独立したIL−12 WT(野生型)と比較して腫瘍減少において30~50倍増加していることを示している。
また,同一モデルではSON−1010は腫瘍中の蓄積濃度が高く,血清,脾,腫瘍に滞留する時間はFのないIL−12 WTより有意に長かったHABは,投与頻度を低くし,投与量を低くする可能性がある。
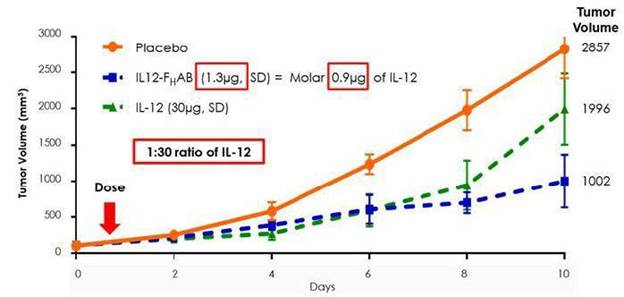
図 2:IL-12(1μg)とIL 12-FHAB(1.3マイクログラム)はインビトロでモルに相当し、類似した生物活性を有するが、インビボでIL 12-FはHABの効力はIL−12の約30倍である(10日目には1.3μg IL−12−FHAB > IL-12 30µg).
B 16 G腫瘍モデルを使用した別の臨床前研究では、SON-1010は、IL-12 WT単独よりも良い用量反応を示し、より長い生存期間を示した(図3および図4)。
この研究の結果により、SON-1010は腫瘍体積の縮小と生存期間の延長においてIL-12単独よりも大きな作用がある可能性が示唆された。
| -8- |
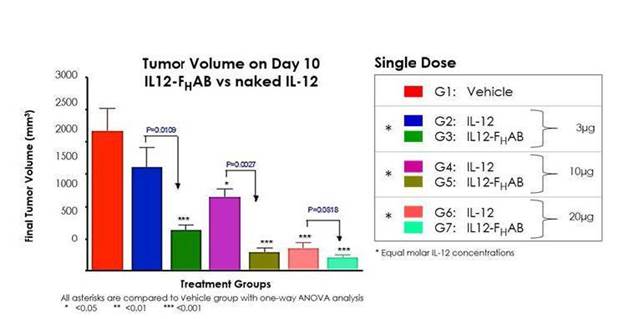
図3:腫瘍体積分析によりIL-12 WTとIL 12-Fの腫瘍は用量依存性の減少を示したHAB処理マウスは,賦形剤対照群と比較した。IL 12-FH等モル用量のIL−12 WT処理マウスと比較して,AB処理マウスの腫瘍体積は統計的に有意に減少した。その結果,IL−12の抗腫瘍活性は血清半減期の延長に伴い潜在的に増強することが分かったHエービー連鎖です。
以下の図4では,SON−1010またはIL−12 WT治療を受けた動物の生存率を比較するためにKaplan−Meier分析を行い,腫瘍成長百分率低下(図3)と生存時間増加 (図4)との相関を示している。この研究では,SON−1010で治療された動物の腫瘍成長は,露出IL−12 WT治療で観察されたより速い腫瘍成長と比較して,より長い生存時間と相関していた。
最低用量のSON−1010(3μg)の生存率は,最高用量のIL−12 WT(30μg)に相当する。全用量のSON−1010は、14日目および17.5日目の生存期間が賦形剤よりも50%増加した。
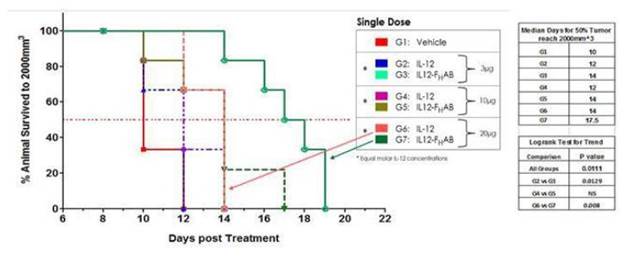
図4:マウスB 16 F腫瘍生存率に対するKaplan−Meierの評価により,IL 12−Fを用いて生存率が向上することが示されたHエービー治療です。投与量10マイクログラムと20μgの独立IL−12 WTの2日目と4日目の生存率はそれぞれ担体対照群(10日)より50%高かった。すべての用量のIL 12-FHABの14日目と17.5日目の生存率はそれぞれ50%であった。最低用量でのIL 12−Fの生存率HAB は最高用量のみのIL−12に相当する。
| -9- |
私たちは完成しました体外培養親和性と結合動力学の薬理学研究により、SON-1010は血清アルブミン中でハムスター、ラット、カニクイザルとヒトに対して種交差反応能力を有することが分かった。その結果、SON-1010はカニクイザルとヒトに対して種特異性を有し、更なる臨床前毒理仕事に種選択の指導を提供した。PK/PDおよび用量反応を評価することを目的としたヒト化マウスモデル(SCID) の研究が完了した。この仕事は非ヒト霊長類(NHP)研究における投与決定に根拠を提供した。
このNHP用量範囲研究の目的は、SON-1010が組換えヒトIL-12(以前にヒト化マウスモデルで示されていた)と比較した増強PKスペクトルを確認すること、2つはSON-1010 INDを設計するために必要な後続のNHP研究を通知し、そのリスクを低下させるために用量増加を実行することである。この研究のデータは,健康なカニクイザルでは,男女問わず単剤SON−1010の用量レベルがヒト臨床試験で予想される曝露量の50倍を超え,耐性が良好であることを示している。また,インターフェロン−γ(インターフェロン−γ)の検出により,SON−1010は持続的かつ有効な標的PD効果を産生し,インターフェロン−DNAは抗腫瘍活性の重要なバイオマーカーである。臨床化学と病理パラメータで標的と一過性の変化が認められたが,投与後14日から21日で完全に消失した。研究で試験されたすべての用量レベルにおいて、腫瘍壊死因子-α、IL-1βおよびIL-6を含むサイトカインの不均衡の兆候、または炎症性サイトカインの制御されない増加は明らかになかった。研究動物からの血清試料の薬物動態分析では,皮下投与の平均半減期は40.0(±6.9)時間,静脈投与の平均半減期は27.45(±2.8)時間であった。これらの結果はB 16 F 10黒色腫マウスモデル研究に基づいて、マウスIL-12と比べ、マウスバージョンのSON-1010は類似治療効果を達成するのに必要な投与量は20倍減少した。以上より,観察された延長的半減期,改善された治療窓と減少した用量需要, は十四行詩のFをHAB技術は、潜在的な免疫腫瘍治療の重要な優勢としてSON-1010を代表した。
薬物製剤(液体と凍結乾燥)以外に、SON-1010を発現する主細胞バンクの仕事、調合開発と技術開発活動はすでに完成した。プロセス移行とcGMP製品製造が完了した。GLPは,INDを有効にするbr毒理学研究が完了した。私たちは2021年下半期にINDをFDAに提出した。
FDAの承認後、著者らは2022年4月にアメリカで固形腫瘍患者に対する臨床試験を開始した。著者らはまた2022年7月に健康ボランティアの中でオーストラリアの単回漸増用量(SAD)の臨床研究をスタートし、PKとPDを詳細に研究し、潜在的なbr}共同研究に準備した。SON−1010のいくつかのキューにおける安全性はすでに2段階の臨床試験で正式に審査されており、早期データの獲得に伴い、用量は増加し続けている。これまで,この新しい方法を用いてサイトカインによる免疫療法の安全性を向上させ,用量制限毒性は発生していない。臨床用量増加戦略は健康ボランティアの研究においてこのような非細胞毒性薬物を使用する能力に基づいて開発され、この研究は以前の癌治療効果の解釈挑戦を受けることなく、迅速にきれいなPK/PDデータを提供することができる。最初の安全性およびPDデータは、SON-1010の免疫反応を評価し、さらなる用量増加の影響を予測するために使用されている。IL−12はインターフェロン−γの産生を刺激することが証明されており,その抗腫瘍作用に必要である。十四行詩は既知のIL-12用量時間測定の保護作用を利用して、毒性を最低に低下させ、最大耐容量(MTD)を延長した。
SON−080 (化学療法による末梢神経障害)
我々の管路拡張により,IL−6(IL−6)が独立分子として送達する際に重要な生物学的特性 を有することが確認された。著者らの先行臨床段階資産SON-080は中国ハムスター卵巣(CHO)細胞で製造された完全ヒトバージョンのIL-6である。SON−080は血小板減少を有する癌患者と健常ボランティアに対するI/II期臨床試験を完了し,化学療法による末梢神経病変(CIPN)の次の開発段階に入り,CIPNは癌抗腫瘍薬治療のよく見られる副作用である。CIPNは衰弱した疾患であり、四肢の痛み、麻痺、痛みを示す。brは、特定の癌プランを受けた患者のうち、70%もの患者がCIPNを患っており、患者が早期に化学療法を放棄する主な原因であると報告されている。CIPNの臨床症状を複製するための動物実験では、SON-080は損傷神経を修復する潜在力を含む疾患の特性を修正することを示した。
臨床前の仕事に基づいて、SON-080は損傷した神経を再生する可能性があると信じており、それによって痛みに関連する症状を解決できるだけでなく、CIPN患者がよく経験する深刻な不快感と運動障害を解決することができる。神経系において、IL-6は潜在的な神経栄養様活性を有し、抗アポトーシス遺伝子発現を誘導し、神経細胞を毒性損傷から保護し、神経再生と髄鞘再生を促進する。SON−080は,シスプラチン,パクリタキセルあるいはビンクリスチン誘導CIPNの様々な臨床前モデルにおいて,神経再生を誘導し正常神経機能(図5)と感覚(図6)を再構築する潜在力を有することが証明されている。br}は2型糖尿病神経病変や他の神経系や他の臓器に影響する疾患の臨床前モデルにおいてもSON−080の活性が観察されている。この広範な活性はSON-080の作用機序が所与の化学療法薬物クラスに限定されない可能性があり、そして普遍的な神経保護-神経修復反応を引き起こす可能性があることを表明した。また,臨床前データ はSON−080が潜在的な神経疾患予防と治療活性を有することを指摘している(図6)。これは依然として神経障害を有する癌生存者を治療する可能性をもたらし、米国の1400万癌生存者のうち、この集団は10%~60%を占めている。
| -10- |
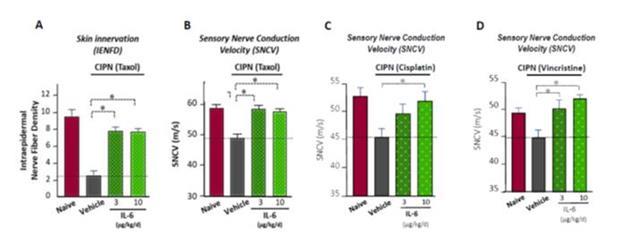
図5:パクリタキセルまたはシスプラチン誘導ラット神経病変に対するSON−080(IL−6)の組織学的(IENFD)または生理(SNCV)レベルの活性測定

図6:予防と治療活動が正常感受性の回復を促進していることを示している(ここでは,シスプラチン誘導末梢神経病変に熱刺激に対する行動反応を用いている)。
SON−080 は化学療法による血小板減少を有する200名以上の癌患者のI/II期研究を完了している。試験参加者は毎日0.25~32マイクログラム/kgの投与量、あるいは週3回皮下注射した。これらの試験では,75%を超える治療を受けた患者に固形腫瘍が存在し,IL−6の累積用量は平均8000μg範囲(122−54880μg),平均治療持続期間は28日であった。1つの試験には6つの化学療法周期があり、IL-6治療期間は203日と長い。これらの試験では癌や神経病変の悪化は認められなかった。
SON-080の最大耐容量(MTD)は4つの研究で確定され、方法はすでに確立された共通毒性標準を採用し、逐次投与量群の待ち行列投与量の増加によって決定される。1日投与時,皮下注射後のMTD値は3~8μg/kgと決定された。週3回投与し、MTDは>10μg/kgと推定された。これらの研究の中で、治療制限用量を確定する最も臨床相関性のある明らかな毒性はインフルエンザ様症状と神経皮質毒性であり、傾眠、騒動、困惑、幻覚と方向性障害として表現される。使用量はMTDより50倍少ない と予想され,将来的にはより良い有害事象プロファイルが出現することが予想される。
以下の図7にI/II期臨床研究報告の有害事象(AE)と重篤な有害事象(SAE)をまとめ,これらの有害事象はIL−6治療によるものと考えられる。
| -11- |
| 合計 名患者(n=214) | No. of AEs in at least 10% of patients treated with IL-6 | No. of SAEs in at least 2% of patients treated with IL-6 | ||||||
| 高熱が高い | 151 (70.6%) | 19 (8.9%) | ||||||
| 過酷である | 120 (56.1%) | - | ||||||
| 好中球減少症 | 31 (14.5%) | 15 (7.0%) | ||||||
| 血小板減少症 | 48 (22.4%) | 15 (7.0%) | ||||||
| 貧血 | 64 (29.9%) | 13 (6.1%) | ||||||
| 吐く | 88 (41.1%) | 10 (4.7%) | ||||||
| 吐き気がする | 106 (49.5%) | 8 (3.7%) | ||||||
| 疲れている | 82 (38.3%) | - | ||||||
| 脱水する | 7 (3.3%) | |||||||
| 呼吸が苦しい | 37 (17.3%) | 7 (3.3%) | ||||||
| 腹痛 | 27 (12.6%) | 6 (2.8%) | ||||||
| めまいがする | 41 (19.2%) | 5 (2.3%) | ||||||
| 頭が痛い | 68 (31.8%) | 5 (2.3%) | ||||||
| 便秘する | 51 (23.8%) | - | ||||||
| 腹をくだす | 50 (23.4%) | - | ||||||
| 注射用br部位紅斑 | 46 (21.5%) | - | ||||||
| フィブリノーゲンが増加する | 45 (21.0%) | - | ||||||
| 拒食症 | 45 (21.0%) | - | ||||||
| 多汗症 | 41 (19.2%) | - | ||||||
| すっかり元気がない | 40 (18.7%) | - | ||||||
| せきが出る | 39 (18.2%) | - | ||||||
| 眠れない | 35 (16.4%) | - | ||||||
| 虚弱である | 34 (15.9%) | - | ||||||
| 血液中アルカリホスファターゼ上昇 | 33 (15.4%) | - | ||||||
| インフルエンザ様症状 | 28 (13.1%) | - | ||||||
| 抜け毛 | 28 (13.1%) | - | ||||||
| 粘膜炎症 | 27 (12.6%) | - | ||||||
| 背中が痛い | 26 (12.1%) | - | ||||||
| 眠くなりそうだ | 26 (12.1%) | - | ||||||
| 痛みがある | 24 (11.2%) | - | ||||||
| 食欲低下 | 24 (11.2%) | - | ||||||
| ビリルビン 増加 | 23 (10.7%) | - | ||||||
| 関節が痛い | 23 (10.7%) | - | ||||||
| 周囲性水腫症 | 22 (10.3%) | - | ||||||
| 血小板数が減少する | 22 (10.3%) | - | ||||||
| 血尿 | 22 (10.3%) | - | ||||||
| 静脈閉塞型肝疾患 | - | 5 (2.3%) |
図7:同時にまたは化学療法後にIL−6治療を受けた癌患者の副作用と副作用。試験用量は0.25~26μg/kgの範囲を含み、総薬物曝露は1~54,880 mgである。
これらのbrデータは我々がCIPNで行う臨床試験の基礎を構成しており,われわれの臨床前研究所が支持しているように,投与量はMTDを大きく下回ることが予想される。比較として、我々の目標用量は、類似時期用量が達成した平均累積用量 よりも25倍低い累積用量を提供する。SON−080は糖尿病末梢神経病変(DPN)や潜在的な他の神経系疾患を含む他の神経疾患を治療する大きな潜在力があると信じており,現在これらの機会の将来の開発経路を評価している。十四行詩は2022年7月にCIPNでSON-080を使用して前アメリカ段階1 b/2 a試験規模の治療効果研究を開始した。この研究は2023年上半期に初歩的なトップクラスの臨床安全性データを生成する可能性がある。
| -12- |
SON−080 (糖尿病末梢神経病変)
われわれとSON−080のCIPN計画に加えて,われわれのDPN計画は,われわれがSON−080と行うCIPN研究で収集したデータに基づいて,糖尿病末梢神経病変(DPN)へのIL−6の臨床応用を探索する可能性がある。現在50%-80%の糖尿病患者はDPNと診断されている。世界保健機関(WHO)の予測によると、2030年までに、糖尿病罹患率は3億5千万人を超えると推定されている。神経病変は進行性であり,糖尿病の連続過程で進行する。この場合には,明らかな原因のない難治性疼痛や,バランスの崩れ,感覚不足,自律神経機能障害など,痛みとは無関係な症状がある。このような赤字は生活の質を悪化させ、期待寿命を短縮させる。糖尿病性足潰瘍は糖尿病医療看護に関連する主要なコストであり、DPNの発展と直接関連している。
重篤であるにもかかわらず,現在の治療はDPNの疼痛部分のみであり,疾患進展や痛みに関係のないbr症状は解決されていない。そのほか、現在鎮痛に応用されている少数の薬物(即ち欣百達、Lyrica、カンナビノイド、オピオイド)は部分的に有効であり、しかも主要な副作用と関連しており、これは通常それらの患者への看護を延期する。そのため、DPNは依然として大きな商業市場の潜在力を持つ満足されていない医療需要である。
長い間、トレーニングはずっとWHOと看護人員によって糖尿病を治療と潜在的に予防する有効な手段とされ、いくつかの試験研究はすでに証拠を提供して糖尿病の栄養改善における作用を支持した。しかし,多くの糖尿病患者は身体的にトレーニングを行うことができない。定期トレーニングは糖尿病関連マーカー(HbA 1 c,グルコース恒常性)を改善し,心拍変動性を改善し,神経機能や血流の回復を刺激することが知られている。最近の証拠により、IL-6は運動過程中に放出され、体力活動のいくつかの有益な効果を媒介している。十四行詩はすでにDPN動物モデルの臨床前仕事を完成し、この動物モデルにおいて、外因性IL-6の投与は表皮神経密度、神経機能、血流と疼痛或いは妨害刺激に対する反応において回復活性を示した。この場合,IL−6は将来のDPN治療の重要な疾患修正療法となる可能性がある。
試験管の中で低突起グリア細胞或いは器官培養に関するデータにより、IL-6はシュワン細胞或いは少突起グリア細胞を通じて髄鞘遺伝子の発現を誘導する可能性があることが示唆された(図8)。
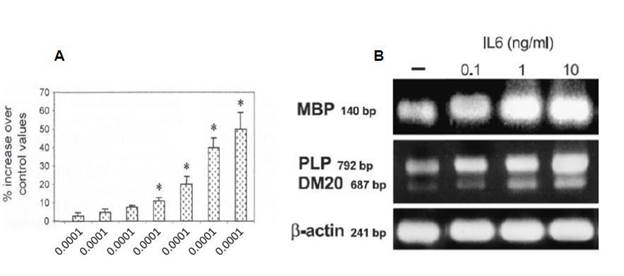
図8:ミエリン塩基性蛋白(MBP)、蛋白リポ蛋白(PLP)及びそのスプライシング変異体発現(B)は少突起グリア細胞の生存と分化を評価する。
Valerio ら,Mol Cell Neurosci 21(2002)602-615である.
Pizzi らは、Mol Cell Neurosci 25(2004)301−311である。
| -13- |
IL-6の神経保護活性はすでに興奮性毒性を含む多種の模式を用いて評価されている。IL-6は、ニューロン保護に加えて、軸索再生および機能的シナプスの回復を促進する可能性がある(図9)。
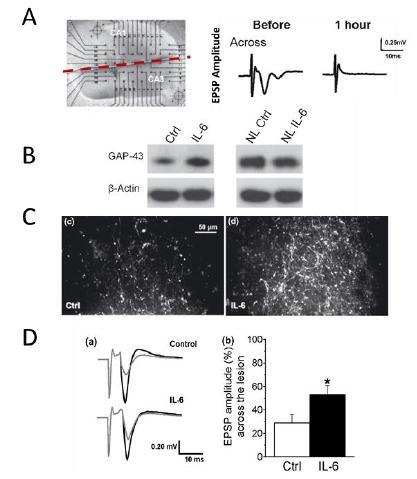
図9:海馬半切切片の軸索再生活性(A)、損傷切片中の成長関連蛋白43(GAP 43)の発現は増加したが、正常切片(NL)には発現(B)が見られなかった。損傷(C)の軸索再生活性と(A)興奮性シナプス後電位(EPSP)が抑制される機能回復(D)。
Hakkoum ら、神経化学雑誌100(2007)747-757。
DPN臨床前モデルにおけるIL-6の活性はすでに3つの独立した実験室によって評価された。この仕事により、IL-6は神経病変において用量依存的に 陽性活性を示し、神経病変樹立後(即ち糖尿病と続発性神経病変誘導後4週間)の正常な生理パラメータの回復にも役立つ可能性がある。運動(図10−A)および感覚(図10−B)の神経機能(伝達速度)上で有益な活動が観察され,熱(図10−C)および触覚(図10−D)知覚を測定することで行動が観察される。先に体外で観察された髄鞘や軸索への直接影響に加え,IL−6は神経中の微小血管血流の回復に活性が認められ(図10−E),糖尿病神経病変の主要な駆動因子である。神経障害の発展過程においてIL-6の予防的治療を受けた動物の神経の組織学的分析により、IL-6は髄鞘に対して保護作用があり、神経繊維の完全性、神経伝達速度と感覚を保護する上で作用する可能性があることを表明した。
| -14- |
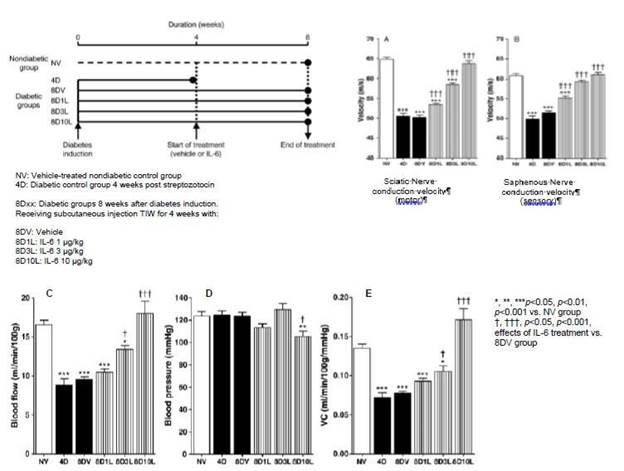
図10:IL-6のストレプトゾシンによる糖尿病神経病変ラットに対する治療作用。
キャメロンら、Exp Neurol 207(2007)23-29。
腫瘍学を除く15項目の先導的研究は,十四行詩に属さない独立した学術団体によって行われ,合計167名の被験者が,運動および代謝におけるIL−6の役割を評価するために27名の2型糖尿病患者を含む。同業者評議の結果により、低用量のIL-6は運動のいくつかの有益な方面をシミュレーションし、抗炎症分子の発現、脂肪代謝の増加、インシュリン分泌の減少と筋肉中のSTAT 3シグナル経路の活性化を含む。
これらのデータは,DPNにおけるIL−6の臨床開発に強力な支援を提供していると信じている。その作用機序と潜在的な疾病改善活性を通じて、低用量のIL-6は糖尿病患者の神経病変症状と心臓自律神経病変(CAN)に治療方案を提供する可能性がある。我々は,SON-080に対して我々が行ったCIPN研究から収集したデータを用いて,SON-080がDPNにおいて可能な次の開発ステップを決定するために我々の意思決定に情報を提供する予定である.会社が2021年5月にシンガポール新生命治療有限会社と締結した許可協定によると、私たちは新生命と共同でDPNの中でSON-080の開発を担当し、2023年下半期に前アメリカの先導的治療効果研究を開始することを目標としている。
SON-080: 新生命治療プロトコル
2021年5月、私たちは以下のように詳細に説明する許可協定に署名することを発表しました(“新生命協定”)。br}は私たちのIL-6(SON-080)資産をシンガポール新生命治療プライベート有限会社(“新生命”)に許可することになりました。許可された領土には、シンガポール、マレーシア、インドネシア、タイ、フィリピン、カンボジア、ブルネイ、ベトナム、ミャンマー、ラオス人民民主共和国の10カ国が含まれる。2021年6月と7月に、Sonnet BioTreateutics CH SA(Sonnet BioTreateutics,Inc.)を修正しました。新生命協定(第1修正案)の一方と我々はそれぞれSonnet BioTreeutics,Inc.を新生命協定(第2修正案)での履行保証人とした。Sonnet が2020年8月に意向書に署名した際に受け取った最初の500,000ドルのほか、Sonnetは新生活協定に署名した際に他の500,000ドルの払い戻し不可の前金を受け取りました。 新生活協定の条項によると、十四行詩は早期商業販売マイルストーンに達してから30日以内に100万ドルの繰延許可料を受け取ることができ、合計1,900万ドルに達するマイルストーン支払いと商業販売の12%から30%までの等級別特許権使用料 を受け取ることができる。
十四行詩と新生計画は2023年下半期にDPNで前アメリカ試験規模の治療効果研究を開始する。
| -15- |
SON-1210
SON−1210, 我々の主導的二重特異性構築物は,Fに結合したIL−12とIL−15を結合したHアブ。これらのサイトカインは協同生物活性に基づいて選択される。
IL−15はその特異的受容体IL 15 Rαを介して作用し,抗原提示樹状細胞,単球,マクロファージに発現する。 は上記IL−12の潜在的抗腫瘍特性に加えて,IL−15は以下の相補的なbr活性を増加させる可能性があると考えられる
| ● | T、Bとナチュラルキラー(NK)細胞の分化と増殖を誘導する | |
| ● | CD 8+T細胞の殺傷活性を増強する | |
| ● | 長時間作用CD 8+記憶性T細胞を誘導し,癌に対する免疫監視を増強し,数ヶ月/数年持続する | |
| ● | B細胞分化と免疫グロブリン合成を刺激する | |
| ● | 樹状細胞の成熟を誘導する | |
| ● | Up IL−12 b 1受容体の発現調節 |
インターロイキン12および15の相互生物活性の概要:
| ● | IL-12:IL-15 Rα受容体、インターフェロン、NK/T細胞、TH 1(腫瘍殺傷)を増加させ、Tregを低下させる | |
| ● | IL-15:IL-12β-1受容体、NK細胞、CD 8記憶力を増加させ、アポトーシスを減少させる |
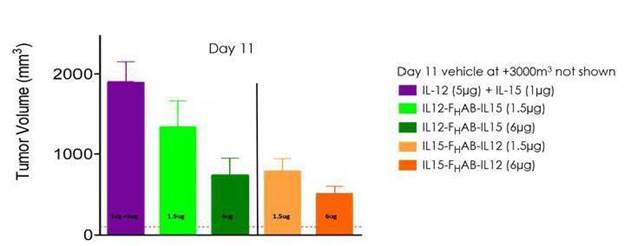
図11:これらのデータは、黒色腫マウスモデルにおいて、露出したIL−12およびIL−15を併用するよりも、SON−1210の方が腫瘍成長抑制作用が強いことを示している。
図 12:IL-12とIL-15とFの組み合わせHABはIL 12−Fと比較して協同活性を示し,腫瘍体積の縮小をもたらしたH黒色腫マウスモデルにAB単独を用いた
非ヒト霊長類(NHP)非GLP投与量増加研究は2022年9月に完成し、GLP繰り返し用量NHP研究も2022年第4四半期に完成した。原料薬cGMPの生産はすでに完成し、薬物の凍結乾燥製剤は2023年初めに生産され、初のヒト(FIH)の臨床研究を支持する。最初の毒性材料は非GLP研究を支持していたが,GLP研究はFIH臨床研究と同じロットのGMP薬を行っている。SON−1210の規制 許可プロセスは2023年上半期に開始される予定である。
非ヒト霊長類(NHP)研究の生命部分は2022年第4四半期に完成する見込みであり,規制認可手続きは2023年上半期に開始される。2023年第1四半期に凍結乾燥薬物製剤“br”を調製することにより、生産と薬品供給を確保し、この製剤は初めての人体臨床試験に適用されることが予想される。
| -16- |
発見 資産:SON-1410(IL 18-FHAB−IL 12)及びSON−3015(抗IL 6−FHAB−抗形質転換成長因子β)
2021年8月、我々はマウス黒色腫モデルの比較研究を完了した後、新たな開発候補化合物を選択したことを発表した。この候補化合物はSonnetの第2の二機能性化合物を代表し、インターロイキン12(IL-12)を同社の完全ヒトアルブミン結合(F)に結合させたHAB)プラットフォーム。SON−1410の標的適応は、黒色腫および腎癌であろう。
IL 18-FHマウス黒色腫研究では、AB−IL 12 は、プラセボと比較して統計的に有意な腫瘍縮小を示し、用量反応を示した
| 化合物 | Day 0, Single Dose Tumor @ 100 mm3 | 8日目腫瘍体積(mm3 +/- SEM), N=8 | 8日目腫瘍は8%縮小 | |||||
| プラセボ | 北米.北米 | 1747 +/- 301 | - | |||||
| IL 18-FHAB-IL 12 | 1 µg | 918 +/- 130 | 47 | % | ||||
| IL 18-FHAB-IL 12 | 5 µg | 619 +/- 141 | 65 | % | ||||
単独のマウス研究も行い,選択したIL 18−Fバージョンを比較したHGMCSF-FはAB-IL 12と他の2つの候補ですHAB−IL 18及びGMCSF−FHAB-IL 12です。比較データによると,IL 18−Fを用いると腫瘍体積は有意に縮小し,インターフェロンレベルと免疫細胞反応(NK,NKT,Th 1と細胞毒性CD 8 T細胞)レベルの方が高かったHAB−IL 12とGMCSF−Fの比較HAB−IL 12 またはGMCSF−FHAB-IL 18です。
SON−1410のセルライン開発活動が開始されており,その後プロセス開発,製剤開発,拡大製造が行われている。この化合物の開発活動が行われており,最初の生産に適した薬剤が生産されている可能性がある体内にあるネズミ研究は2023年まで例年終了した。
転化成長因子-β-1/IL-6の生物学的特性は腫瘍の総生存期間を予測する重要な指標であり、SON-3015連合標的IL-6と転化成長因子-β1シグナル経路は腫瘍と骨転移を治療する将来性のある策略である可能性がある。形質転換成長因子βは分解した骨から放出され,IL−6の産生を促進し,骨転移の悪循環を招く。骨環境におけるFcRNの高発現は抗IL−6−F二重構造蛋白の骨への蓄積を招くHAB-抗形質転換成長因子βは、骨転移を潜在的に抑制または遮断するために選択された。[br}十四行詩は、費用を低減するためにSON-3015開発計画を保留することを選択した。
| -17- |
私たちの治療化合物の開発と商業化の面で、私たちは多くの挑戦と不確実性に直面していますHエービー技術です。本募集説明書の他の部分に含まれる“リスク要因”と、本募集説明書の文書に引用して“リスク要因”と題する章を参照してください。
競争
Br製薬と生物技術業界の特徴は技術の迅速な進歩、競争の激しい及び専有製品に対する高度な重視である。著者らは私たちの技術、開発経験と科学知識が私たちに競争優位を提供していると信じているが、著者らは大型製薬と生物技術会社、学術機関、政府機関とその他の研究、特許保護を求め、癌免疫療法の研究、開発、製造と商業化のための協力手配の公共と個人研究組織を含む多くの異なる源からの潜在的な競争に直面している。私たちが開発と商業化に成功した任意の候補製品は、将来発売される可能性のある新しい免疫療法と競争するだろう。
われわれは製薬,バイオテクノロジー,その他の免疫腫瘍治療の開発に関する市場分野で競争を展開している。他の多くの会社はすでに商業化および/または癌免疫腫瘍学治療方法を開発しており、大手製薬会社と生物技術会社、例えば安進、アスリコン/医学免疫会社、百時美施貴宝、メルク、ノワール、ファイザーと羅氏/遺伝子テークなどを含む。
私たちは製薬やバイオテクノロジー会社からの激しい競争に直面しており,これらの会社は癌環境において特定のサイトカインや他の巨大分子を免疫調節療法として使用することを目指している。これらは、一般に、単特異性または二重特異性抗体、融合タンパク質、抗体薬物結合体、および標的ワクチンを含む。
我々の主要候補製品SON−080については,Apexian PharmPharmticals,Inc.,Aphios Corporation,Asahi Kasei Corporation,MundiPharma EDOおよびRegenacy PharmPharmticals, Inc.を含むCIPN治療製品を開発している他の会社も知られているが,我々は疾患修正サイトカインを用いてCIPNを治療する唯一の会社であると信じている。
最初のFではHABから派生する候補案SON−1010に加えて,Celsion Corporation,Eli Lilly,Inovio PharmPharmticals,Inc.,Inc.,Intrexon Corporation,Codiak Biosciences,Xolio Treeutics,Werewolf Treeutics,トンボTreeutics,OncoSec Medicalが開発されている計画を含むが,他に競合他社のIL−12計画があることが知られている。私たちのFはH集積したIL−12は腫瘍を標的とした強化されたPKプロファイルを持ち,競争相手とは異なるようにした。
初期のパイプラインFについてHAB製品候補製品SON−1210、SON−2014、およびSON−3105は、これらの特定のデュアル特定計画上で動作する他の他の競合会社があることを知らない。
私たちと比較して、私たちは競争あるいは未来に競争する可能性のある多くの会社は研究開発、製造、臨床前テスト、臨床試験を行い、監督管理許可とマーケティング承認薬物を獲得する方面でより多くの財務資源と専門知識を持っている。製薬、バイオテクノロジー、診断業界の合併と買収は、私たちの数の少ない競争相手により多くの資源を集中させる可能性がある。規模が小さいか早い段階にある会社も重要な競争相手になる可能性があり、特に大企業や成熟会社との協力で手配する。これらの競争相手はまた、合格した科学と管理者を募集と維持し、臨床試験場を構築し、私たちの臨床試験のために被験者を募集し、私たちの計画と相補的あるいは必要な技術を獲得する面で私たちと競争している。
もし私たちの競争相手が私たちまたは私たちの協力者が開発する可能性のある任意の製品よりも安全で、より効果的で、副作用が少なく、より便利で、より安い製品を開発し、商業化すれば、私たちのビジネス機会が減少または消失することを見ることができるかもしれない。私たちの競争相手も私たちよりも早くFDAや外国の規制機関のその製品の承認を得ることができます。これは、私たちの競争相手が私たちまたは私たちの協力者が市場に入る前に強力な市場地位を確立することをもたらすかもしれません。承認されれば、私たちのすべての候補製品の成功に影響を与える重要な競争要素は、それらの治療効果、安全性、利便性、価格、セット診断の有効性(必要であれば)、生物類似或いは模倣薬の競争レベル及びbr}政府と他の第三者支払人が精算できるかどうかである可能性がある。
| -18- |
製造業
著者らは契約開発と生産組織(CDMO)に依存してFDAの現在の良好な生産規範(CGMP)に基づいて著者らの候補薬物を生産し、著者らの臨床試験に用いた。生物薬品の生産は広範なcGMP法規の制約を受け、これらの法規は各種のプログラムとファイルの要求を規定し、そして記録保存、生産技術と制御、人員と品質管理のすべての領域を管理する。著者らのパイプライン分子は標準的な工業中国ハムスター卵巣(CHO)プラットフォームを用いて、既製の原材料を用いてよく見られる生化学工程を行った。
規制承認と商業製造を通じて私たちの臨床用品に対する予想需要を満たし、私たちの活動を支援するために、現在協力しているCDMOは生産規模を拡大する必要があり、そうでなければ代替サプライヤーを探す必要がある。我々の現在のCDMOは万能と興味を持ち,必要に応じて容量を増加させることができると信じている.CDMOの見通しは強力であり、多様な潜在的な代行源がある。我々の現在のCDMO が大規模化生産できなければ,代替サプライヤーと交渉していないが,我々は既存のCDMOと拡張対話を行っている.我々とCDMOとの関係 は薬品開発と製造において豊富な経験を持つ内部者によって管理されている。
もし私たちが十分な数の候補薬を得ることができない場合、あるいは直ちに原材料を得ることができなければ、進行中の臨床試験を延期し、代替メーカーを探す必要があるかもしれないが、これは高価で時間がかかる。
ライセンス と他のビジネススケジュール
楊森製薬会社(ジョンソン)
2022年10月、十四行詩は、ジョンソン傘下のヤンソン製薬会社の一つであるヤンソンバイオテクノロジー社(Janssen Biotech,Inc.)との協力合意を発表した体外培養そして体内の薬効SON−1010(IL 12−F)HAB), SON-1210 (IL12-FHAB-IL 15) とSON-1410(IL 18-FHAB-IL 12)は、いくつかのJanssen特許細胞治療資産と共に評価される。合意 はジョンソン·イノベーションによって促進される.合意条項によると,十四行詩は対面コミュニケーションのために3種類の参考化合物 を提供しなければならないはい体外培養と体内にある効能研究。合意条項に成功して遵守すれば, Janssenはその選択権を行使することができ,Sonnetは協力の拡大を求めることができる.
新生活
当社は2021年5月に、新生命治療プライベート株式会社(“新生命”)とライセンス契約(“新生命協定”)を締結した。新生命協定によると、当社はマレーシア、シンガポール、インドネシア、タイ、フィリピン、ベトナム、ブルネイ、ミャンマー、ラオス人民民主共和国及びカンボジア (“専属地域”)で特定組換えヒトインターロイキン6、SON−080(“化合物”)を含む医薬製剤(この等の製剤及び“製品”)を開発及び商業化し、ヒト糖尿病末梢神経病変 (“DPN領域”)を予防、治療又は緩和する。新しい生命は、(1)独占許可証の範囲を拡大して、ヒト化学療法による末梢神経障害(“CIPN領域”)を予防、治療または緩和するための拡大オプションを行使することができ、このbrオプションは非排他的であり、2021年12月31日に満了する;および/または(2)許可証の地域範囲は、人民のbr}Republic of China、香港および/またはインドを含み、このオプションは独占的であり、2021年12月31日にも満了する。これらの選択肢を行使すれば、CIPN油田と領土拡張の条項は双方が協議する。2021年6月と7月に、Sonnet BioTreateutics CH SA(Sonnet BioTreateutics,Inc.)となるように“新生プロトコル”を修正した。新生命協定(第1修正案)の当事者と我々はそれぞれSonnet BioTreateutics,Inc.を新生命協定(第2修正案)での履行保証人, とした。
その会社は世界のどこでも化合物と製品を生産するすべての権利を維持するだろう。当社は新生活と後続供給協定を締結し、この合意に基づき、当社は双方が協議した条項に従って、専属地域のDPN油田(及びCIPN油田、適用すれば)に新生活製品を供給して開発及び商業化に供する。会社のbrはまた、新生活 が許可証から利益を得ることに役立ついくつかの臨床前と臨床開発技術の譲渡に協力する。
| -19- |
New Lifeは,専属地域での臨床研究と追加の非臨床研究,およびDPN領域(とCIPN分野,適用すれば)製品の他の開発と規制活動および製品商業化の費用 を負担して担当する。
New Lifeは2020年8月に50万ドルの払い戻し不可能な前払い現金を会社に支払い,2021年6月にNew Lifeプロトコルを実行して50万ドルの払い戻しができない前払い現金 を会社に支払った。新しい生命には、いくつかのマイルストーンを満たしたときに100万ドルの払戻不可能な延期許可料の追加支払いと、いくつかの開発および商業化マイルストーンの実現に応じて会社に支払う可能性のある1,900万ドルまでの追加マイルストーンの支払いが義務付けられている。また、特許権使用料期間内(以下のように定義する)では、新生命には、製品の専属地域での年間純売上高に応じて12%から30%の等級2桁の特許権使用料を会社に支払う義務がある。 “特許権使用料条項”とは、専属地域内で製品や国/地域に基づいて、そのような製品が専属地域で初めて商業販売された日(ある条件に応じて)から から新生命がDPN分野(またはCIPN分野)で商業化されるまでのことである。(適用される場合)。
新生命協定は、製品ごとに国/地域ごとに有効になり、最終満期国/地域の最終満期製品の特許権使用料の期限が満了したときに終了するが、以下の条件によって制限される:(I)当事者の早期解約権 は、他方の重大な違約または破産または倒産により早期に終了する権利と、(Ii)自社の買い戻し権と新生命の復帰権(以下、定義とする)を含む。
また,New Lifeは当社がNew Lifeを買い戻す権利の独占的選択権を付与し,会社 はNew Life権利を規定すべき条項に従って専属地域の1つまたは複数の国または地域でDPN現場および/またはCIPN現場(例えば適用)に関する製品を買い戻す権利を付与し,このような選択権は適用製品の第3段階試験開始時に終了する。
Xoma
十四行詩 (Oncobiologics,Inc.(“Oncobiologics”)の利益相続人として,Oncobiologicsが2015年4月6日にある資産をSoncobiologics に剥離しながらOncoBioicsの株主にSonnetの全株式を比例的に割り当てた後, とXOMA(US)LLC(“XOMA”)は,2012年7月23日の発見協力プロトコルと2019年5月7日の発見br}連携プロトコル修正案(総称して“提携プロトコル”と呼ぶ)の一方であり,このプロトコルによりXOMAはSonnet に非協力的なプロトコルを付与した.譲渡不可能なライセンスおよび/または抗体および関連タンパク質に関連するいくつかの材料、技術および関連情報を発見、最適化および開発する権利を使用し、それに基づいて製品(各製品)を開発および商業化する。br}十四行詩は、製品に関連するいくつかの開発および承認マイルストーンを実現する際に、製品ごとに合計375万ドルまたはマイルストーン支払いをXOMAに支払う義務がある。十四行詩はまた、十四行詩販売製品の純売上高のためにXOMAに低い一桁版税を支払うことに同意した。各製品の使用料は、(I)最初の商業販売(連携協定に定義されているように)後に指定されたbr期間および(Ii)連携プロトコルによってカバーされる発行特許の最後の有効クレームの最後の 満了日まで国/地域で支払われる(遅い者を基準とする)。また,十四行詩はXOMAに指定金額を支払うことにより, は製品をもとに印税料率を下げる権利がある.協力協定は、いずれか一方によって を終了することができ、通常の賠償条項を含むことができる。
アリス
2015年8月28日、現在Sonnetの完全子会社である救済は、メルクKGaA(“ARES”)の完全子会社Ares Tradingとライセンス契約(“ARESライセンス契約”) に署名した。ARES許可協定の条項によると、ARESは自社に再許可可能な独占的、世界的に印税負担のある特許許可を付与し、アトクシャキンアルファ(“アトサキン”)の製品(各製品は1つの“製品”)を研究、開発、使用、商業化し、アトクシャキンは末梢神経疾患および血管合併症を治療する低用量ヒトインターロイキン6製剤である。ARESライセンスプロトコルには,Atex akinを用いたi)糖尿病神経病変の治療,ii)化学療法による末梢神経病変,iii)血管合併症の保護の3つの特許が含まれている。
| -20- |
ARESライセンス契約により、当社が販売している製品の純売上高に応じてARESに1桁の高額印税を支払います。印税は、(I)その国/地域で最初の商業販売(ARES許可協定に定義されているように)が行われた後の指定された時間帯まで、(I)その国/地域における製品の有効なクレームがカバーされる最後の日 まで製品および国/地域によって支払われる。製品が国/地域の有効クレームの範囲内にない場合、またはその製品が国/地域で初めて商業販売された日から12(12)周年前に、有効クレームが期限切れまたは失効した場合、印税税率は50%(50%)減少する。また,再許可 イベント(“再許可受領書”)から受け取った収益の割合である再許可費用をARESに支払い,浮動比例表(再許可イベントが発生した臨床開発の後期段階ではこの割合が低下する)を用いて,低い2桁から高いビット数に減少させる。便宜上、会社はARES許可プロトコルを随時終了したり、いずれか一方が合意に違反した場合には他方が終了したりすることができる。ライセンス 協定は慣例的な賠償条項を含む。
Aresライセンス協定は、再ライセンスに関連する一部の条項および条件の適用範囲を明確にするために、2021年11月1日に改正された。特に:
| ● | 十四行詩(br}は現在、ARESの事前書面同意なしに第三者に再許可を付与することが許可されており、任意のこのような再許可の財務状況 は十四行詩の善意で確定された公平な市場価値を反映すべきであることを前提としている。 | |
| ● | 十四行詩が再許可受領書からAREに報酬を支払う初期条件は不明であるため,ARES許可プロトコル を明らかにし,第1段階臨床試験完了前または後に関連する再許可 協定に署名すれば,十四行詩はARESにすべての再許可受領書の一定割合を支払わなければならない(最初のARES許可プロトコルにのみ設定された第1段階臨床試験完了後に関連する 再許可合意に署名した場合ではない)と規定されている。 | |
| ● | 双方は、上記の明確化は、将来の再許可協定にのみ適用され、新しい生命協定によって生成される可能性のある特許権使用料 (ただし、マイルストーン支払いには適用されない)に適用されることに同意する。 |
知的財産権
私たちの特許の組み合わせでは、私たちは2つの発行された特許(アメリカと日本)があり、完全ヒトアルブミン結合ドメイン(Fを含む)の多くのbr融合タンパク質に対する特許出願を提出しましたHAB)です。承認された場合、それによって生成された特許は、2038年から2041年の間に満了するが、場合によっては特許期間が延長される可能性がある。特許出願書類は、以下のものを含む
WO/2018年/151868に対応する国家届出書類-この出願は、完全ヒトのアルブミン結合ドメイン(F)を対象としているH単鎖抗体を含むAb) 融合タンパク質(例えば抗形質転換増殖因子β、PD-L 1、腫瘍壊死因子、インターロイキン1、インターロイキン6、インターロイキン8など)、蛋白とサイトカインを融合する(例えばIL 2-FHAB、IL 12-FHAB、IL 15-FHAB,IL 7-FHABなど)IL 12-Fのような2つのサイトカインの組み合わせですHAB-IL 15、GMcSF-FHAB−IL 18及びIL 18−FHAB-IL 12;およびこのFを用いた処理方法Hエービー融合タンパク質です。特許は2021年6月8日に米国で発行され,名称は米国特許番号11,028,166である。また、日本国家段階申請で付与の決定を受けた。米国の特許11,028,166号特許は現在2039年3月26日に満了する予定であるが,日本の特許出願が承認されれば,現在2038年2月20日に満了する予定である。オーストラリア、ブラジル、カナダ、中国、EU、香港、インド、ニュージーランド、ロシアも国家段階の申請を待っている。
| -21- |
◌ 米国特許出願(U.S.15/932,387)およびPCT特許出願(PCT/US 2018/00085)が最初に受け取った出願日は、2018年2月20日であり、米国仮特許出願U.S.62/459,975およびU.S.62/459,981が優先的利益を要求する1周年記念日 の4日後である。米国特許出願およびPCT特許出願については、米国仮特許出願US 62/459,975およびUS 62/459,981に優先権を回復する出願が承認された。その後、PCT特許出願は、オーストラリア、ブラジル、カナダ、中国、ヨーロッパ、インド、日本、ニュージーランド、ロシアにおいて国家段階特許出願を提出した。しかし,これらの管轄区域の特許法が異なるため,これまで米国62/459,975 と米国62/459,981に対する優先権要求はオーストラリア,ヨーロッパ,インド,日本,ニュージーランド,ロシアでのみ受け入れられてきた。
米国における抗IL−6−Fに対する仮出願H抗IL−6−Fを含むAB融合タンパク質HAB、抗IL-6-FH抗形質転換成長因子β及び抗IL−6−FHAB−抗−IL 8融合蛋白;およびこの融合蛋白を用いた治療法は2021年9月22日にUS 63/245,702 に再提出された。科学的な挑戦により,仮特許出願後1年以内に支援データが得られなかったことが大きいため,この特許は放棄された。しかし、最近の技術的ブレークスルーは、この廃棄された 特許を2023年に再提出することを可能にする。
米国の一時的出願は、抗原/アルブミン結合ドメイン結合体を含み、このような結合体を用いた治療方法は、2021年5月11日にUS 63/187,278に再提出される。一時的な特許請求の範囲をサポートするデータは生成されておらず、したがって、特許 は放棄される。
米国仮出願は、低用量IL 6を用いて年齢に関連する虚弱を治療する方法を指し、出願番号63/197,097として2021年6月に提出され、2022年6月にPCT特許に変換される。
米国仮出願は,アルブミン結合ドメインを有する新規抗体薬物結合体(ADCPlatform)を指し,2021年12月7日に提出され,出願番号は63/286,996であった。
2022年5月27日に出願番号63/346,368として出願されたIL−12−アルブミン結合ドメイン融合タンパク質製剤及びその使用方法に対する米国仮出願。
米国仮出願は,組換えIL−12アルブミン結合ドメイン融合蛋白を用いた癌治療方法について,2022年11月2日に提出され,出願番号は63/421,846であった。
私たちの商標の組み合わせについて、私たちは世界知的所有権局(WIPO)のSonnet BioTreateuticsとFの国際登録許可を得ましたHABタグは,各タグの発効日は9月1日である.17、2020年。また、これら2つの商標はいずれもEU知的財産権局(EUIPO)によって発表され、発効日はそれぞれ2020年11月30日と2020年12月6日。2021年、米国特許商標局は、両方の出願が反対期間の完了に成功し、受け入れ可能な使用声明を提出して登録するまで成熟したことを示す2つの商標の補助金通知を発表した。そこで,USPTOはSonnet BioTreateuticsとFの使用宣言ごとの手当通知を発表したHAB申請は,まもなく登録証明書 を受け取る予定である.
スイス商標局は9月9日にSonnet BioTreeuticsおよびFHAB商標保護を付与した。それぞれ2021年10月14日と2021年10月26日 で、国際商標登録番号によって保護されている。1558330および1558471。
カナダ知的財産権局は2022年6月8日にSonnet BioTreateutics商標保護権を付与し,国際商標登録番号1558330によって保護されているが,FHAB商標は国際商標登録番号15584471によって保護されており,反対期間は2022年11月16日から18カ月間である。
| -22- |
Sonnet BioTreeutics商標は,スイスとカナダのほかに,オーストラリア,EU,日本,メキシコ,韓国,イギリスで保護されており,各国に登録番号がある。有効登録日は1558330年9月17日、更新日は9月17日です。17年2030年です同様に、FHAB商標はオーストラリア、中国、EU、日本、メキシコ、韓国、イギリスで保護され、各ケースに登録番号がある。1558471、保護日は9月1日です。2020年7月17日、更新日は9月9日です17年2030年です
十四行詩生物治療商標は、ある競争相手のbr社に対する潜在的な非使用クレームが原因で最初に中国で却下されたが、私たちの法律事務所は、最初の42種類の却下が成功したため、同じ商標の2つの新しい商標申請も2021年に登録および/または発表できるという自信を持っているが、2025年までにこれらの商標に対してbr非使用抹消申請を開始することができ、これらの未解決の42種類の出願 が中国に登録される可能性が高いと予想される時間枠である。
従業員
2022年9月30日現在、私たちは12人のフルタイム従業員がいます。私たちの従業員は労働組合代表もなく、集団交渉合意のカバー範囲もなく、私たちは従業員との関係が良いと信じている。また,独立請負業者や 他の第三者を用いてその業務の様々な側面に協力している.
政府の法規
薬品(生物製品を含む)の研究、開発、テスト、製造、品質管理、承認、包装、貯蔵、記録保存、ラベル、広告、販売促進、流通、マーケティング、承認後の監視と報告及び輸出入はすべてアメリカ連邦、州と地方政府当局及びその他の国家と司法管轄区域の広範な監督管理を受けている。いくつかの司法管轄区域はまた薬品の価格設定を規制している。アメリカ及びその他の国と司法管轄区でマーケティング許可を得る流れ、その後の適用法規と法規及びその他の監督管理機関の遵守には、大量の時間と財力が必要である。
アメリカの生物製品許可証と規制
アメリカでは、生物製品或いは生物製品は“公衆衛生サービス法”(PHSA)と“連邦食品、薬品と化粧品法”(FDCA)及びその実施条例の規制を受けている。製品開発過程のいつでも適用される要求 を遵守できない場合、出願人は、研究、監督審査および承認、 および/または行政または司法処罰を行う上で遅延を受ける可能性がある。これらの制裁は、FDAが許可を拒否することに限定されないが、申請者が臨床試験を継続することを許可すること、係属中の申請の承認の拒否、免許の取り消しまたは免許の取り消し、承認の撤回、製品のリコール、製品の差し押さえ、生産または流通の一時停止、禁止、罰金、調査、および民事および刑事罰を含むことができる。生物製品の候補製品はFDAが発行した生物許可証を取得しなければ、アメリカで合法的に発売されることができない。
| -23- |
FDAが米国で生物学的ライセンスを取得するために必要なbrの流れは、一般に以下に関連する
広範な非臨床或いは臨床前実験室試験と臨床前動物試験を完成し、良好な実験室実践、或いはGLPを含む適用法規による実験動物の人道使用と調合研究への適用要求;
Brは、任意のヒト臨床試験を開始する前に、研究中の新薬またはIND申請を米国食品·薬物管理局に提出する。このような裁判が始まる前に継続される許可を得なければならない
Brは、FDAの法規(一般に良好な臨床実践と呼ばれる)またはGCPおよび人体研究対象およびその健康情報を保護する任意の追加要求に基づいて、各提案された適応について候補製品の安全性、効力および純度を決定し、提案された生物製品の期待用途に対する安全性および有効性を決定するために、十分かつ良好な人体臨床試験を制御する。FDAはまた、著者らの臨床試験の前または期間の任意の時間に、安全考慮または規定に適合しない理由で、生物製品候補製品 に対して臨床保留を実施する可能性がある。FDAが臨床保留を強制的に実施した場合、試験 はFDA許可なしに再開することができず、その後、FDA認可の条項に従ってしか行うことができない
Brは、臨床開発および推奨ラベルの製造および成分に関する詳細な情報を提出することを含む、1つまたは複数の推奨適応をマーケティングすることを要求する生物学的製品のために準備され、食品および医薬管理局に生物学的ライセンス申請を提出する
食品医薬品局審査部門によって決定された食品·薬物管理局顧問委員会による製品の審査
現在の良好な製造仕様またはcGMP要件に適合する場合を評価し、施設、方法、および製品の特性、強度、品質、および純度を維持するのに十分な施設、方法、および制御が、現在の良好な製造仕様またはcGMP要件に適合することを評価するために、FDAによる1つまたは複数の製造施設(サードパーティを含む)の1回または複数回の検査を円満に完了する
GCPに適合し、BLAをサポートする臨床データの完全性を保証するために、1つまたは複数の臨床研究会場のFDA審査を満足的に完了した
使用料を支払い、FDAがBLAおよび新生物製品の許可を承認することを確実にする
は、リスク評価の実施と緩和戦略の潜在的な要求、 またはREMS、およびFDA要求の任意の承認後研究を含む任意の承認後要求を遵守する。
非臨床研究と探索的新薬応用
各候補製品は人体試験を行う前に非臨床試験を受けなければならない。これらのテストは製品化学、調合と安定性の実験室評価、及び活性と毒性潜在力を評価する動物研究を含み、適用する法規を遵守して行わなければならない。非臨床試験の結果は,生産情報や分析データとともにIND申請の一部としてFDAに提出された。INDはFDAが受領した30日後に自動的に発効し、それ以前にFDAが提案された臨床試験の製品または懸念または問題を提起しない限り、ヒト研究対象が不合理な健康リスクにさらされることを懸念することを含む。この場合,INDスポンサーやFDAは臨床試験開始前にFDAの未解決の問題を解決しなければならない。
INDの提出は、FDAが試験開始を許可しないか、または試験を発起人が最初にINDに指定された条項で行うことを許可しない可能性がある。FDAが懸念または問題を提起した場合、それは安全問題または規定に適合していないため、生物製品候補製品に対して臨床保留を実施することを選択することができる。FDAが臨床保留を強制する場合、試験はFDA許可なしに を再開してはならず、FDA許可の条項の下でのみ再開することができる。
| -24- |
ヒトbr支持血中乳酸の臨床試験
臨床 試験はGCP要求に基づいて、合格した首席研究者の監督の下で、研究製品を健康ボランティア或いは治療を受ける疾患患者に候補することに関連している。INDの一部として,各臨床試験のレジメンおよび任意の後続レジメン修正をFDAに提出しなければならない。米国国外で臨床試験を行うスポンサーはFDAの認可を得ることができるが,INDによる臨床試験を希望している。外国の臨床試験がINDで行われていない場合,スポンサーはBLAを支援するために臨床試験のデータをFDAに提出することができ,臨床試験が良好でGCPに適合していれば,独立した道徳 委員会で審査·承認され,FDAは現場検査(必要に応じて)により研究データを検証することができる。
また,各臨床試験は機関審査委員会またはIRBが審査·承認しなければならず,審査委員会またはIRBは臨床試験を行う各機関で集中的または単独で行われ,米国国外で行われた試験については,上記の独立倫理委員会が審査·承認しなければならない。IRBは臨床試験設計、患者インフォームドコンセント、倫理要素と人類被験者の安全などの要素を考慮する。IRBの運営はFDAの規定に適合しなければならない。FDA、IRBまたは臨床試験スポンサーは、臨床試験がFDAの要求に従って行われていないこと、または被験者または患者が受け入れられない健康リスクに直面していることを発見することを含む、様々な理由で臨床試験を随時一時停止または中止することができる。臨床検査はまた広範なGCP規則とインフォームドコンセントの要求を満たさなければならない。そのほか、いくつかの臨床試験は臨床試験スポンサーが組織した独立した合格専門家グループによって監督され、データ安全監視委員会或いは委員会と呼ばれる。 このグループは研究中のあるデータへのアクセスに基づいて、計画に従って研究を継続し、研究方法を変更したり、指定されたチェックポイントで研究を停止することを提案することができる。
臨床試験は通常3つの連続段階に分けて行われ、これらの段階は重なる可能性があり、合併する可能性もある。承認された後に追加的な研究が必要かもしれない。
第1段階:この生物製品は,まず健康なヒトボランティアに導入され,安全性試験が行われる。いくつかの深刻または生命に危険な疾患の製品の場合、特に製品自体の毒性が高く、健康ボランティアに道徳的に使用できない可能性がある場合、最初の人体テストは、通常、癌患者のような患者において行われる。
第二段階:限られた患者集団において候補生物製品を評価して、可能な副作用と安全リスクを決定し、特定の標的疾患に対するこの製品の治療効果を初歩的に評価し、用量耐性、最適用量および用量計画を決定する。
第三段階:拡大した患者群と地理的に分散した臨床研究地点で臨床試験を行い、投与量、臨床治療効果、効力と安全性を更に評価する。これらの試験は製品の全体的なリスク/収益比率を確定し、製品ラベルに十分な根拠を提供することを目的としている。
第四段階:承認後の臨床試験或いは第四段階の臨床試験は初歩的な発売許可後に行うことができる。それらは、予期される治療適応患者の治療にbrの追加の経験、特に長期安全追跡を提供する。FDAが製品を承認した場合、ある会社は承認を必要としない臨床試験を行っており、会社はこれらの臨床試験のデータを使用して、任意の第4段階の臨床試験のすべてまたは一部の要求を満たすことができ、または製品ラベルの変更を要求することができる。必要な4期臨床試験を行う上で職務調査を行うことができなければ,製品の承認撤回につながる可能性がある。
CGMP要求 に該当する
BLAを承認する前に、FDAは通常、製造プロセスおよび工場がcGMP要求に完全に適合し、必要な仕様と一致することを保証するために、この製品を生産する工場を検査する。製造業者や製品製造·流通に関連する他の企業もFDAやある州の機関に登録しなければならない。国内と海外の製造企業は初めて生産過程に参加する際にFDAに登録して付加情報を提供しなければならない。未登録の工場で製造または輸入されたいずれの製品もFDCA下の誤ったブランドとみなされる。機関は政府当局の定期的な抜き打ち検査を受けるかもしれない。メーカー はその工場に関する記録を提供する必要があるかもしれない。
| -25- |
BLA の審査と承認
製品候補開発,非臨床試験,臨床試験の結果はBLAの一部としてFDAに提出され,その製品の販売許可証 を申請する。BLAには,製品製造や組成に関する広く詳細な情報と,提案されたラベルやユーザ料金の支払いが含まれていなければならない.FDAは、BLAが申請を受け入れるのに十分であるかどうかを決定するために、出願を提出した後60日の間予備審査を行う。提出された申請が受け入れられると、FDAは深い検討を始めた。FDAは、標準出願の予備審査(優先審査の場合、6ヶ月の期間がある)を完了し、出願人に応答する12ヶ月の期間がある。FDAは常にその目標日を達成するわけではなく、審査プロセスは、FDAがより多くの情報を提供することまたは明確化を要求することによって、 を著しく延長する可能性がある。FDAが要求する場合、または出願人が目標日より前の最後の3ヶ月以内に提出中に提供された情報に関する他の情報を提供または明確にした場合、審査プロセスおよび目標日は3ヶ月延長することができる。
FDAによる出願の評価および関連情報によれば、FDAは、承認書、拒否書、または完全な返信を発行することができる。この製品のビジネスマーケティングを承認し、特定の適応の具体的な処方情報 を提供する。PHSAによれば、FDAが製品が安全で純粋で有効であると判断し、製品を製造する施設が、その継続安全、純粋および有効性を保証するための基準に適合する場合、FDAはBLAを承認することができる。申請が承認されていない場合、FDAは、申請が最終的に承認されることを確実にするために満たされなければならない条件が含まれ、可能な場合には、申請の承認を得るためにスポンサーがとりうる提案行動を列挙する完全な返信を発行することができる。完全な返信を受信したスポンサーは、FDAによって決定された問題に対する完全な応答を表す情報をFDAに提出することができる。“処方薬使用料法案”(PDUFA)によると,このような再提出は,申請者が行動書簡を返信する際に提出される情報に基づいて, 1つまたは2つに分類される.FDAがPDUFA同意の目標と政策に基づいて、FDAは1種類の再提出された申請を2ヶ月間審査し、6ヶ月間に2種類の再提出された申請を審査する。 FDAは完全な返信で決定された問題が解決されるまで申請を承認しない。FDAが機関または製品が機関の要求に適合していないと判断した場合、否定文を発行する。
FDAはまた、申請が承認されるべきかどうかを決定するために、審査、評価、および拘束力のない提案を諮問委員会に提出することができる。特に,FDAは新たな生物製品や安全性や有効性の問題を提起した生物製品の申請を諮問委員会に提出する可能性がある。
FDAが新製品を承認する場合、FDAは、その承認された適応使用を制限することができ、禁忌症、警告、または予防措置を製品ラベルに含めることを要求することができる。また、FDAは、承認後の製品の安全性をさらに評価するための4期の臨床試験を含む承認後の研究を要求する可能性がある。FDAはまた、製品が商業化された後にそれを監視するためにテストおよび監視計画を要求するか、またはREMSを含む流通制限または他のリスク管理機構を含む他の条件を適用して、製品の利益が潜在的リスクよりも大きいことを保証することを容易にすることができる。FDAは発売後の研究やモニタリング計画の結果に基づいて、製品のさらなるマーケティングを阻止または制限する可能性がある。承認された後、新しい適応の増加、製造変更、および追加のラベル宣言など、承認された製品の多くのタイプの変更は、さらなるテスト要件およびFDAの審査および承認を受けなければならない。
迅速な追跡、画期的な治療、優先的な指定の検討
FDAはある製品を指定して迅速な審査を行う権利があり、もしこれらの製品が深刻な或いは生命に危害を及ぼす疾病或いは状況を治療する時に満足されていない医療需要を満たすことを目的としている場合。これらの計画は,(I)高速チャネル指定,(Ii)画期的治療指定,(Iii)優先審査指定と呼ばれる。
| -26- |
迅速審査:1つの製品が(単独でまたは1つまたは複数の他の製品と組み合わせて使用されることが意図されている)深刻または生命に危険な疾患または状態を治療するために使用されることが意図されており、そのような疾患または状態が満たされていない医療需要を解決する可能性を示す場合、FDAは、製品を迅速軌道審査に指定することができる。スポンサーはFDAとより多くのインタラクションを行う可能性があり、FDAは申請が完了する前にFast Track製品申請の部分を審査する可能性がある。スポンサーはまた、残りの情報を提出するスケジュールを提供しなければならず、FDAの承認を得なければならず、スポンサーは適用された使用料を支払わなければならない。しかし、FDAが高速チャネル申請を審査する期間目標は、申請の最後の部分が提出されてから開始されます。 FDAは高速チャネル指定を撤回する可能性があります。
突破的療法:製品は、重症または生命に危険な疾患または状態の治療のために単独または1つまたは複数の他の製品と組み合わせて使用されることが意図されている画期的療法として指定することができ、予備臨床証拠は、製品が1つまたは複数の臨床的に重要な終点(例えば、臨床開発早期に観察された重大な治療効果)において有意な改善を示す可能性があることを示す場合、画期的な治療法として指定される可能性があり、迅速な審査を受ける資格がある可能性がある。FDAは画期的な治療法の面で何らかの行動をとるかもしれない。
優先審査:1つの製品が深刻な場合、FDAは、承認された場合、他の利用可能な治療法と比較して、安全性または有効性の面で有意な改善を提供するように、製品を優先的に審査することができる。この評価はFDAによって具体的な状況に基づいて行われる。このような申請の評価に全体的な関心およびリソースを誘導することを目的とした優先指定を優先的に指定し、FDAがマーケティング申請に行動する目標を10ヶ月から6ヶ月に短縮する。
Br承認経路を加速した
FDAは患者に既存の治療よりも意義のある治療利点を提供し、その基礎はこの製品が代替終点に影響を与えることを決定することであり、この代替終点は臨床利益を予測する可能性が高い、深刻または生命に危険な疾患の製品の承認を加速することができる。製品が中間臨床終点に影響を与え、不可逆的発病率または死亡率またはIMMへの影響よりも早く測定することができ、疾患の重症度、希少性または流行率、および代替治療の利用可能または不足を考慮すると、IMMまたは他の臨床的利益に対する影響を合理的に予測する可能性があり、FDAもこのような疾患の加速承認を許可することができる。加速された承認を得た製品は、従来承認された製品と同じ安全かつ有効性法定 基準に適合しなければならない。
承認の加速について言えば、代替終点は1つの標識であり、例えば実験室測定、放射画像、バイタルサイン或いは他の臨床利益を予測できると思われるが、それ自体は臨床利益の測定基準ではない。エージェント端末 は、通常、臨床端末よりも容易または迅速に測定を行う。中間臨床終点は治療効果の測定であり、それは製品の臨床利益、例えばIMMに対する効果を合理的に予測することが可能であると考えられる。加速承認経路は病気経過が長く、製品の期待される臨床利益を測定するために時間を延長する必要がある環境に最もよく用いられ、代用或いは中間臨床終点への影響は非常に速く発生した。そのため、加速承認は様々な癌を治療するための製品の開発と承認に広く使用されており、その中で治療の目標は通常生存率を向上させたり、発病率を低下させたりすることであり、典型的な病気経過の持続時間は長いbr}を必要とし、臨床あるいは生存利益を証明するための大型試験が必要な場合もある。
加速承認経路は、一般に、製品の臨床的利益を検証し、説明するために、スポンサーが勤勉な方法で承認後に追加の検証性研究を行うことに同意することに依存する。そのため、この基礎の上で承認された候補製品 は厳格な発売後のコンプライアンス要求を守らなければならず、4期或いは承認後の臨床試験を完成して臨床終点への影響を確認することを含む。必要な承認後研究を行わない場合、あるいは発売後の研究期間中に臨床利益が確認できなければ、FDAがこの製品の市場からのリコールを加速することを許可する。加速法規により承認された候補製品の販売促進材料 はすべてFDAの事前審査を経なければならない。
| -27- |
承認後条例
監督管理部門の許可を得ても、上場製品は不良事件報告、記録保存、マーケティングとcGMPコンプライアンスの要求と制限を含む連邦、州と外国の法律法規の持続的な全面的な要求を守らなければならない。薬物承認後に報告された不良事件は上場製品の使用に追加的な制限を加えることを招く可能性があり、或いは追加の発売後の研究或いは臨床試験を要求する可能性がある。
適用される連邦、州、地方の法律法規を基本的に遵守するには、多くの時間と財力が必要だ。FDAの生物製品に対する厳格かつ広範な監督管理は承認された後も継続しており、特にcGMP要求の面である。生物製品製造業者および他の承認された生物製品の製造および流通に関連するエンティティは、FDAおよび特定の州機関にその機関を登録し、cGMP要求および他の法律を遵守することを確実にするために、FDAおよび特定の州機関の定期的な抜き打ち検査を受けなければならない。私たちは依存し、私たちが商業化する可能性のある任意の製品の臨床的および商業的ロットを生産するために第三者に依存し続けることが予想される。私たちの製品のメーカー は、品質管理と品質保証 および記録と文書のメンテナンスを含むcGMP法規に適用される要求を守らなければなりません。他の生物製品に適用される承認後の要求は記録保存要求、副作用報告と報告最新の安全と治療効果情報を含む。
もし 以前に未知の問題が発見された場合、あるいは製品を承認したメーカーや販売促進に関する適用法規要求 を遵守できなかった場合、その製品の販売を制限したり、その製品を市場からのリコールや重大な行政、民事または刑事制裁を招く可能性がある。
孤児薬名
米国の孤児薬物指定は、スポンサーが稀な疾患または疾患のための製品を開発することを奨励することを目的としている。米国では、法律は、まれな疾患または疾患を、米国で200,000人未満の疾患または疾患に影響を与えるか、または米国で200,000人を超える疾患に影響を与えると定義し、この疾患または疾患に対する製品の開発および提供コストを米国での製品の販売から回収することを合理的に予想することができない。
孤児 がFDAの承認を得た場合、製品発売承認日から7年以内に、会社は税収控除と市場独占経営権を獲得する資格がある。孤児製品に指定された申請は、製品上場承認申請が提出される前のいつでも提出することができる。受け入れ可能な申請により,FDA孤児製品開発オフィス(OOPD)は製品を孤児薬に指定する可能性がある。そして、この製品は任意の他の製品のように審査と承認の流れを通過しなければなりません。 疾患発症率の変化により,孤児薬の指定が撤回される可能性がある。
スポンサーは、以前承認されていなかった製品のために孤児薬物指定を申請することができ、または発売された製品のために新しい孤児薬物指定を申請することができる。また、スポンサーがそのbr製品が第1の薬剤よりも臨床的に優れている可能性があるという合理的な仮定を提出することができる場合、承認された孤児薬と同じ製品のスポンサーは、後続製品の申請および同じ珍しい疾患または疾患を取得する孤児薬物のために指定することができる。複数のスポンサーは、同じ製品の孤児薬物指定brを同じ稀な疾患または疾患のために得ることができるが、孤児薬物指定を求める各スポンサーは、完全な指定申請を提出しなければならない。
専門期間はFDAが上場申請を承認した日から,この製品が指定されている 適応のみに適用される。FDAは、同じ製品の第2の出願を異なる使用のために承認することができ、または同じ使用のために製品の臨床的により優れたバージョンを申請することができる。しかしながら、FDAは、スポンサーの同意またはbrスポンサーが十分な数を提供できない限り、他の製造業者によって生産された同じ製品を同じ適応のために市場独占期間内に承認することはできない。
| -28- |
小児科研究
“小児科研究公平法”によると、いくつかの承認申請は、関連する小児科群における試験薬物の安全性と有効性の評価を通常臨床研究データに基づいて含まなければならない。FDAは会社の要求またはFDAの計画に応じて、小児科評価の要求を免除または延期することができる。FDAは、リスク評価と緩和策が新製品の利益がリスクよりも大きいことを確保するために必要であると認定するかもしれない。REMは、薬物の安全な使用に必要であると考えられるFDAが薬物を安全に使用するために必要であることに依存して、薬物ガイドラインまたは患者パッケージから誰が薬物を処方または分配できるかの制限まで様々な要素を含むことができる。スポンサーはFDAとの第2段階会議終了後にIND に予備小児科研究計画を提出する必要がある
EU医薬品承認条例と手続き
米国以外でいかなる製品を販売するためにも、会社は他の国·地域や司法管轄区の多くの法規要求を守らなければならない。製品がFDAの承認を得ているか否かにかかわらず、申請者は外国の監督管理機関よりも必要な許可を得る必要があり、その後、これらの国または司法管轄区でこの製品の臨床試験またはマーケティングを開始することができる。
臨床試験承認br
現在適用されている臨床試験指令2001/20/ECとGCPに関する指令2005/28/ECに基づいて、EUはすでに加盟国の国家立法を通じて臨床試験審査制度を実施した。この制度によれば,出願人はその中で臨床試験を行うEU加盟国の主管国当局の承認を得なければならず,複数の加盟国で臨床試験を行う場合には,複数の加盟国で承認されなければならない。また,独立した倫理委員会が賛成の意見を発表した後のみ,申請者は特定の研究地点で臨床試験を開始することができる。臨床試験申請或いはCTAは2001/20/EC号指令と2005/28/EC号指令及び加盟国の対応国家法律で規定された支持情報を含む研究用薬品ファイルを添付しなければならず、適用されたガイドライン文書にさらに詳細に説明されている。
2014年4月,EUは現在の臨床試験指令2001/20/ECに代わる新たな臨床試験条例(EU)第536/2014号を採択した。新しい臨床試験条例は2019年または2020年に適用される予定だ。それはEUの現在の臨床試験承認制度を徹底的に改革するだろう。具体的には、すべての加盟国に直接適用されるこの新しい規定は、EUの臨床試験の承認手続きを簡略化することを目的としている。例えば,新たなbr臨床試験条例では簡略化された申請手順が規定されており,単一入口点と厳密に定義された締め切りを用いて臨床試験申請の評価を行っている。
マーケティング ライセンス
EU規制システムに基づいて製品のマーケティング許可を得るためには、申請者は、EU規制機関が管理する中央手続きまたはEU加盟国主管当局が管理するプログラムのうちの1つ(分散プログラム、国家プログラムまたは相互承認プログラム)を提出しなければならない。連合に設立された申請者だけがマーケティング許可を得ることができる。出願人は、EMAが特定の製品の免除、カテゴリ免除、またはPIPに含まれる1つまたは複数の措置を許可しない限り、EMAによって承認された小児科集団のすべてのサブセットをカバーする小児科調査計画(PIP)に含まれるすべての措置を証明しなければならない。
集中化手続きは、すべての欧州連合加盟国に対して有効な単一マーケティング許可を欧州委員会によって付与することを規定している。特定の製品には、特定のバイオテクノロジープロセスによって製造された医薬品、孤児医薬品として指定された製品、高度な治療製品、および癌の治療のための製品を含む特定の疾患を治療するための新しい活性物質を含む製品が強制的である。他の疾患を治療するための新しい活性物質を含む製品、および高度に革新的な製品または患者の利益に適合する集中プロセスについては、 集中プロセスは任意である可能性がある。
| -29- |
集中化プログラムによれば、EMAに設立された人は、そのリスク/利益プロファイルを決定するために、医薬品委員会またはCHMPを用いて製品の評価を担当する。中央手続きの下で、重大な影響評価を評価する最長期限は210日であり、申請者がCHMP質問に回答する際に補足情報または書面または口頭解釈を提供する時間は含まれていない。特殊な場合、公衆衛生の観点、特に治療革新の観点から見れば、1種の医薬製品は重大な価値があり、CHMPは加速評価を承認する可能性がある。
ライセンスと更新期間
マーケティング許可は原則として5年有効であり、5年後にはEMAまたはライセンス加盟国の主管当局によるリスク収益残高の再評価によって更新することができる。更新後、マーケティング許可の有効期限は無期限であり、欧州委員会または主管当局がbr薬物警戒に関する正当な理由に基づいて、5年間の継続を継続することを決定した。認可が無効になってから3年以内にEU市場(集中手続きであれば)または認可加盟国の市場に薬品を投入しないいかなる許可もない。
営業許可後の規制要件
Brの承認後、マーケティング許可の保持者は、医薬製品の製造、マーケティング、普及、販売に適用される一連の要求を遵守しなければならない。これらの措置には,EUの厳格な薬物警戒や安全報告規則の遵守が含まれており,これらの規則により,認可後の研究や追加的なモニタリング義務を実施することができる。また、許可製品の製造も、欧州薬品管理局のGMP要求とEUの他の監督機関の同様の要求を厳格に遵守して、薬品の安全と身分を確保しなければならないが、許可製品の製造、加工、包装も欧州薬品管理局のGMP要求と同様の要求を厳格に遵守しなければならない。最後に、ライセンス製品のマーケティングと普及は、業界後援の継続医学教育と薬品処方者および/または公衆向けの広告を含み、EU 2001/83 EC指令の下で厳格に規制されている。改訂された
孤児薬品名と排他性
第(Br)(EC)第141/2000号法規及び第847/2000号法規によると、製品は欧州委員会により孤児薬物として指定されることができ、そのスポンサーが、(1)申請時にEUの万分の5以下の人に影響を与える生命又は慢性衰弱に危害を及ぼす疾患、又は(2) 生命に危険な疾患、又は(2)生命にかかわる疾患を診断、予防又は治療することができることを証明することができる。インセンティブがなければ、その薬剤の欧州連合での販売は、必要な投資が合理的であることを証明するのに十分な見返りをもたらす可能性が低い。上記2つの場合のいずれについても、出願人は、EU許可された関連疾患の診断、予防または治療の好ましい方法が存在しないことを証明しなければならないか、またはそのような方法が存在する場合、薬剤は、疾患に適した製品と比較して有意な利点を有しなければならない。孤児薬物の指定は費用減免、監督管理援助、EU集中販売許可の申請などのメリットを提供することができる。孤立した薬物の販売許可は10年間の市場独占期間をもたらすだろう。しかし,5年目の終了時にその製品が指定孤児薬の基準を満たしていないと判定されれば,市場専門期間は6年に短縮できる。
アメリカの組み合わせ製品
いくつかのbr製品、すなわち組み合わせ製品は、通常、異なるタイプの監督管理機関によって規制される成分から構成される可能性があり、通常はFDAの異なる中心によって規制される。組合せ製品は、(I)単一の実体として製造される物理的、化学的または他の方法で組み合わせまたは混合された2つ以上の規制されたbr成分からなる製品であってもよく、(Ii)2つ以上の別個の製品は、1つの別個のパッケージに包装されているか、または1つのユニットとして、医薬品および器具製品、器具および生物学的製品または生物学的および医薬品製品からなる。(3)個別に包装された医薬品、器具または生物製品は、その研究計画または提案されたラベルに従って、承認された個別に指定された医薬品、器具または生物物品と共にのみ使用され、両方とも所望の用途、適応または効果を達成する必要があり、提案された製品が承認された後、例えば、予期される用途、剤形、濃度、投与経路の変化、brまたは用量の大きな変化を反映するために、製品のラベルを変更する必要がある。または(Iv)個別に包装された任意の研究用医薬、装置または生物製品であって、その提案されたラベルは、別の個別に指定された研究用医薬、装置または生物学的製品にのみ使用され、これら2つの医薬、装置または生物学的製品は、所望の使用、適応または効果を達成するためにbrを必要とする。FDAは,主要な管轄権を持つセンターや先頭センターを指定して組合せ製品を審査することを担当しており,この決定は組合せ製品の“主な行動パターン”に基づいている。スポンサーは、組合医薬品事務室に指定された要求を提出することによって、管轄権決定を要求することができる。
| -30- |
アンティクリールと合併してRelipmentを買収する
本10-K表年次報告は、Sonnet BioTreateutics Holdings,Inc.(“Sonnet Holdings”,“WE”,“Us”,“br}”Our“または”Company“)から提出され,前身は強アンティクリールホールディングスである。2020年3月31日まで,国内と国際的に所有,経営,特許経営の急速なレジャー飲食概念を有している。先に開示したように、当社は2020年4月1日に十四行詩生物治療会社(“十四行詩”)との合併取引を完了し、十四行詩は当社の全額付属会社(“合併”)となった。2020年4月1日,合併に関連して会社は“Sonnet BioTreateutics Holdings,Inc.”と改称した。十四行詩は2015年4月6日にニュージャージー州社として登録設立された。
合併は当社から逆合併とみなされ、米国公認会計原則(“米国公認会計原則”)に基づいて逆資本再編成として入金される。会計目的については、十四行詩は同社を買収したとみなされている。
合併後及び合併前に、当社は、当社の飲食業務に関するすべての資産及び負債を、新たに設立された当社全資付属会社amergent Hotel Group,Inc.(“amergent”)に譲渡します。この配当金は、上記のような会社の飲食業務への貢献と譲渡とともに、“分割”と呼ばれる。剥離するまで、アメフトは何の業務も運営していなかった。
分割·合併の結果、2020年4月1日以来、会社は十四行詩とその直接·間接子会社を通じて運営されており、会社の継続業務は十四行詩業務である。
また、2020年4月1日、合併に関連し、合併前に、十四行詩は救済治療持株会社(“救済 ホールディングス”)からアトサキンα(低用量インターロイキン6、IL-6、現在“SON-080”)の世界開発権の買収を完了し、救済ホールディングスの全額付属会社救済治療会社(“救済”)を買収し、合併中に救済持株会社に合計757,933株会社普通株のSonnet普通株を発行することに転換した。
会社 と既存の情報
デラウェア州の法律によると,同社は1999年10月21日に設立され,本名はTulvine Systems,Inc.である。2005年4月25日、Tulvine Systems,Inc.は完全子会社Chonancleer Holdings,Inc.を設立し、2005年5月2日、Tulvine Systems,Inc.はChancleer Holdings,Inc.と合併してChancleer Holdings,Inc.と改称した。2020年4月1日、当社、SonnetとBiosub Inc.との間の2019年10月10日の合併協定及び計画の条項により、当社はSonnet BioTreateutics,Inc.(以下Sonnet)との業務統合を完了した。当社の全資附属会社(“合併附属会社”)(“合併協議”)は、同協定によると、合併附属会社は十四行詩と合併して十四行詩に編入され、十四行詩は当社の全額付属会社(“合併”)として存続する(“合併”)。合併協議の条項によると、当社は合併直前に十四行詩株主に普通株 を発行し、為替レートは一株当たり十四行詩普通株を発行した0.106572株の発行済み普通株 である。合併に関連して,同社はその名称を“強アンティクリールホールディングス”から“Sonnet BioTreateutics Holdings,Inc.”に変更し,会社が経営している業務はSonnetが展開する業務となった。
私たちの主な実行事務室はニュージャージー州プリンストン08540号102号室から中心100号を見下ろすところにあります。私たちの電話番号は(609) 375-2227、会社サイトはhttps://www.sonnetBio.com/です。本年度報告の表 10-Kにサイトアドレスを含め,非アクティブテキストとしてのみ参照し,我々のサイトへのアクティブリンクとする予定はない.ウェブサイト上の情報は、本10-Kフォーム年次報告に参照して組み込まれていない。
本報告書Form 10-K、四半期報告Form 10-Q、現在の報告Form 8-K、およびこれらの報告のすべての修正、および私たちが米国証券取引委員会(“米国証券取引委員会”)に提出した他の文書は、米国証券取引委員会に電子的に提出された後、または米国証券取引委員会に電子的に提出された後、合理的で実行可能な範囲内でできるだけ早く私たちのサイトの投資家によって部分的に無料で取得することができる。私たちがアメリカ証券取引委員会に記録した書類を得ることができますWwwv.sec.gov.
| -31- |
1 a項目.リスク要因です
私たちの普通株に投資することは、あなたのすべての投資損失のリスクを含む高いリスクと関連がある。あなたは、以下に説明するリスクおよび不確実性、ならびに本報告書および米国証券取引委員会に提出された他の報告書に含まれる他の情報を慎重に考慮しなければならない。次に列挙された危険は私たちが直面している唯一の危険ではない。他のリスクおよび不確実性要因が存在する可能性があり、これらのリスクおよび不確実性は、私たちの業務、運営、および財務状況にも悪影響を及ぼす可能性がある。実際に以下のいずれかのリスクが発生した場合、私たちの業務、財務状況および/または運営は影響を受ける可能性があります。この場合、私たちの普通株の価値は下がるかもしれません。あなたはすべてのbrを失ったり、あなたが私たちの普通株に支払ってくれたお金の大部分を失うかもしれません。
リスクファクターの概要
| ● | 私たち は重大な運営損失の歴史があり、予測可能な未来に重大かつ増加する損失が予想され、 私たちは永遠に利益を実現したり維持したりすることはできないかもしれない。 | |
| ● | 私たちの運営中の度重なる赤字は、私たちの持続経営企業としての持続的な経営能力を大きく疑っています。 | |
| ● | 私たちは大量の追加資金が必要になります。もし私たちが必要な時に資金を集めることができなければ、私たちは私たちの製品発見と開発計画や商業化努力を延期、減少、またはキャンセルさせられるかもしれません。 | |
| ● | 新冠肺炎コロナウイルスの大流行或いは任意の他の伝染病の広範な爆発は著者らの業務、財務状況と運営業績に実質的な不利な影響を与える可能性がある。 | |
| ● | 私たち は私たちの内部開発計画の成功に大きく依存して、私たちの候補製品は臨床試験に成功し、監督管理の許可を得る或いは商業化に成功できないかもしれない。 | |
| ● | 私たちのbrは開発作業の非常に早い段階にあり、私たちの候補製品は新しい薬物カテゴリーを代表しており、それらが治療法として確立されるまで、より厳しい規制審査を受ける可能性がある。 | |
| ● | 私たちが必要な臨床前研究と臨床試験を完成しても、マーケティング承認の流れは非常に高価で、時間がかかり、確定しておらず、私たちあるいは任意のパートナーが一部またはすべての候補製品の商業化の承認を得ることを阻止する可能性があります。 したがって、私たちまたは任意のパートナーがいつ、どの地域で候補製品を商業化するマーケティング承認を得るかを予測することはできません。 | |
| ● | 私たちは激しい競争に直面しています。もし私たちの競争相手が私たちが開発した候補製品よりも効果的で、より安全で、あるいは安い製品を開発·販売すれば、私たちのビジネス機会はマイナスの影響を受けるでしょう。 | |
| ● | 任意の現在または未来の候補製品の商業成功は医師、患者、支払人、および医学界の他の人の市場に対する受け入れの程度に依存する。 | |
| ● | いくつかの候補製品については、私たちは開発および商業化パートナーに依存して臨床試験 を開発し、規制部門の候補製品の承認を得、承認された場合、候補製品をマーケティングおよび販売する可能性がある。もしこのような協力者が期待通りに実行できなかった場合、私たちがこのような候補製品から将来の収入を得る潜在力は著しく低下し、私たちの業務は損害を受けるだろう。 | |
| ● | 我々 は,独立した臨床研究者とCROを含む第三者に依存して,我々の候補製品のいくつかの臨床試験 を行って支援する。第三者が候補製品の臨床開発における義務を履行できなかったことは、候補製品が規制部門の承認を得る能力を延期または弱める可能性がある。 | |
| ● | もし私たちの製品や候補製品のために特許や他の知的財産権保護を獲得して維持することができない場合、あるいは取得した特許や他の知的財産権保護の範囲が十分に広くなければ、私たちの競争相手は私たちと似ているまたは同じ製品を開発して商業化する可能性があり、私たちの製品と候補製品を商業化することに成功した能力は悪影響を受ける可能性がある。 | |
| ● | 我々 は我々の組織を拡大することを望んでいるため,我々は我々の成長を管理する際に困難に遭遇する可能性があり,これは我々の 運営を中断する可能性がある. | |
| ● | 私たち は予測可能な未来に現金配当金を支払わないと予想しているので、投資家は彼らのbr投資が現金配当金を得ることを期待すべきではない。 |
| -32- |
私たちの財務状況と追加資本需要に関するリスク
私たち は重大な運営損失の歴史があり、予測可能な未来に重大かつ増加する損失が予想され、 私たちは永遠に利益を実現したり維持したりすることはできないかもしれない。
私たちは短期的には私たちの運営融資に必要な収入や収益力が生じないと予想しています。2022年と2021年9月30日までの年度までの純損失はそれぞれ2,970万ドルと2,500万ドルです。2022年9月30日現在、私たちの累計赤字は9140万ドルです。
これまで、私たちは何の製品も商業化していませんし、製品販売から何の収入も得ていません。十分な製品販売収入が実現されていなければ、私たちは将来決して利益を達成しないかもしれません。我々はほとんどの財政資源と努力を臨床前研究と臨床試験を含む研究と開発に投入した。私たちの純損失は四半期と年度によって大きく変動する可能性があります。純損失と負のキャッシュフローはすでにわれわれの株主(赤字)権益や運営資本に悪影響を与え続けている。
私たちは予想しています
私たちの主要候補製品SON-080および他の候補製品の開発と臨床試験を継続した
Brは任意の未来の候補製品のために起動し、研究、臨床前と臨床開発を継続する
より多くの候補製品の発見と開発を求め、私たちの臨床製品ラインをさらに拡大した
臨床試験に成功した任意の候補製品のためにマーケティングと監督管理の承認を求める
臨床開発と潜在的な商業化のためにより大量の候補製品を生産する必要がある
私たちの知的財産権の組み合わせを維持し、拡大し、保護する
臨床、品質管理、科学者のような多くの人員の募集と保留を含む、私たちの研究開発インフラを拡張します
市場で承認された製品を商業化するために、将来的に販売、マーケティング、流通、および他の商業インフラを確立する
私たちの製品開発と商業化を支援し、上場企業の義務を履行するのを助けることを含む、運営、財務、管理情報システムと人員を増加させる
デバイスと物理インフラを追加して、私たちの研究開発を支援します。
私たちが利益を達成して利益を維持する能力は私たちが製品を許可して収入を創出する能力にかかっている。製品収入の発生は、私たちの1つまたは複数の候補製品の市場承認を得て、商業化に成功する能力があるかどうかにかかっている。
| -33- |
成功したbr商業化は、私たちの候補製品の臨床試験を完成させ、これらの候補製品のマーケティング承認を獲得し、私たちまたは任意のパートナーがマーケティング許可を得ることができる製品を製造、マーケティング、販売することを含む重要なマイルストーンを実現する必要があり、任意の発売後の要求を満たし、個人保険または政府支払者から私たちの製品の精算を得ることができる。これらの活動に関連する不確実性とリスクのため、私たちは収入の時間と金額、そして私たちがいつ利益を達成できるかどうかを正確に予測することができない。私たちとどの協力者もこのような活動で決して成功しないかもしれません。たとえ私たちまたはどんな協力者が成功しても、私たちは利益を達成するために十分な収入を生成しないかもしれません。たとえ私たちが利益を達成したとしても、私たちは四半期や年間収益性を維持したり向上させることができないかもしれない。
私たちのbrは利益を上げて利益を維持できなければ、私たちの普通株の市場価格を下げ、資金を集め、業務を拡大し、製品を多様化したり、運営を継続する能力を弱める可能性があります。もし私たちが引き続き損失を被ったら、投資家は彼らの投資から何の見返りも得られず、彼らのすべての投資を失うかもしれない。
私たちの限られた運営の歴史は、私たちの業務のこれまでの成功度を評価することが難しくなり、私たちの将来の生存能力を評価することも困難になるかもしれません。
私たちの業務は2015年に運営を開始しました。これまで,我々の業務は我々の会社に資金と人員を提供し,我々のbr技術を開発し,我々の候補製品のための臨床前研究と早期臨床試験を行い,戦略的協力を求めて候補製品を進めてきた。私たちはまだ後期臨床試験に成功し、brの上場許可を得て、商業規模の製品を製造したり、第三者代表が私たちを手配したり、成功した製品の商業化に必要な販売とマーケティング活動を行うことができないことを証明していません。したがって、あなたは会社が開発初期段階でよく遭遇するコスト、不確実性、遅延、困難に基づいて、私たちの将来性、特に私たちのような臨床段階の生物製薬会社を考慮すべきです。もし私たちがより長い運営歴史あるいは医薬製品の開発と商業化に成功した歴史を持っていれば、あなたは私たちの未来の成功あるいは生存能力に対するいかなる予測も のように正確ではないかもしれない。
私たちのビジネス目標を達成する際には、予見できない費用、困難、複雑な状況、遅延、他の既知または未知の要素に遭遇する可能性があります。私たちは最終的には、発展に集中している会社からビジネス活動を支援できる会社に移行する必要があります。私たちはこのような移行では成功しないかもしれません。
様々な要因により、私たちの財務状況と経営業績は四半期ごとと毎年大幅に変動し続けると予想されていますが、その多くの要素は制御できません。したがって、あなたは将来の運営業績の指標として、どんな四半期や年間業績にも依存してはいけません。
私たちの運営中の度重なる赤字は、私たちの持続経営企業としての持続的な経営能力を大きく疑っています。
我々のbrは設立以来運営活動の中で経常的な損失と負のキャッシュフローが発生しており,我々は予見可能な未来には,主に我々の潜在製品の研究開発コストにより,運営活動に損失と負のキャッシュフローが生じることが予想される。2022年9月30日現在、私たちは310万ドルの現金を持っており、株主赤字は250万ドルだ。私たちは、2022年9月30日の現金に、2022年9月30日以降と本報告日までにBTIG,LLCとの市場発売で調達した460万ドルの純収益を加えて、2023年3月までの運営に資金を提供すると信じています。私たちは私たちの運営を支援するために多くの追加資金が必要になるだろう。このような要素は私たちの持続的な経営能力に大きな疑いを抱かせる。付随する総合財務諸表は持続経営を基礎として作成され、正常業務過程で資産を現金化し、負債を返済することを考慮する。合併財務諸表には、この不確実性の結果に起因する可能性のあるいかなる調整も含まれていない。
私たちは将来的に株式や債務融資、パートナー関係、協力、または他のソースを通じてより多くの資金を必要とし、私たちが計画している開発活動を展開する。必要に応じて追加資金を得ることができない場合、私たちはそのような資金を受け取るまで運営 を延期または削減する必要があるかもしれない。様々な内部および外部要因は、私たちの候補製品がいつマーケティングおよび商業化に成功したかどうかの承認に影響を与えるだろう。私たちの候補製品の規制承認と市場受容度、これらの候補製品の開発と商業化の時間長 およびこれらの候補製品の開発と商業化コストおよび/またはそれらの承認過程のどの段階での失敗も、私たちの財務状況と将来の運営に大きな影響を与えるだろう。
| -34- |
設立以来,運営 は主に組織我々,融資獲得,研究と開発による技術開発,臨床前研究を行ってきた。私たちはその製品が開発中の会社に関連するリスクに直面している。これらのbrリスクには、その研究開発を達成し、その研究開発目標を実現するために追加の資金が必要であり、その知的財産権の保護、技術者の採用と維持及び肝心な管理層メンバーへの依存が含まれている。
私たちの持続的な経営企業としての持続的な経営能力は、追加株式や債務資本を調達したり、非コア資産を剥離して余分な現金を調達する能力にかかっています。もし私たちが十分な追加資金を集めることができない場合、私たちはいくつかの臨床活動を延期または停止することを含むコスト削減措置をとる必要があるかもしれない。
未来のいかなる融資の源、時間と可用性は主に市場状況に依存し、もっと具体的には、著者らの臨床開発計画の進展に依存する。必要な時には、資金が全くないか、あるいは私たちが受け入れられる条件の下にあるかもしれない。必要な資金が不足しているため、計画の一部または全部の臨床試験を延期、削減、または廃止する必要があるかもしれない。他の要因を除いて、これらのbr要因は、私たちの持続的な経営企業としての持続的な経営能力を大きく疑わせた。
以下でさらに議論する新冠肺炎による潜在的な経済影響と持続時間は評価や予測が困難である可能性があるが、広範囲の流行病は世界金融市場の深刻な混乱を招き、私たちの資本獲得能力を低下させる可能性があり、このbr}は将来私たちの流動性に負の影響を与える可能性がある。また、新冠肺炎の伝播による景気後退や市場回復は、私たちの業務と私たちの普通株の価値に実質的な影響を与える可能性がある。
私たちは大量の追加資金が必要になります。もし私たちが必要な時に資金を集めることができなければ、私たちは私たちの製品発見と開発計画や商業化努力の延期、減少、またはキャンセルを余儀なくされるかもしれません。
臨床前研究と臨床試験を含む薬物製品を開発することは、非常に時間がかかり、高価かつ不確定な過程であり、完成するのに数年かかる。例えば,2022年と2021年9月30日までの年度では,それぞれ2,770万ドルと2,260万ドルの純現金を我々の経営活動に使用しており,基本的にはこれらの活動が研究開発活動に関連している。私たち は、私たちが行っている活動に関連する費用が増加することを予想して、特に私たちが新しい臨床試験を開始した時、私たちの現在の候補製品または任意の未来の候補製品のために新しい研究と臨床前開発作業を開始し、マーケティング承認を求める。さらに、私たちの任意の候補製品がマーケティング承認を受けた場合、このような販売、マーケティング、製造、および流通はパートナーの責任ではないので、製品販売、マーケティング、製造および流通に関連する巨額の商業化費用 が生じる可能性がある。また、合併により、上場企業の運営に関連した巨額のコストが発生し続ける。したがって、私たちは私たちの持続的な運営に関連した多くの追加資金を得る必要があるだろう。もし私たちが必要な時や魅力的な条件下で資金を集めることができなければ、私たちは私たちの研究開発計画や将来の商業化努力を延期、減少、または廃止することを余儀なくされるかもしれない。
我々 は,我々が開発している候補製品や,我々が開発する可能性のある他の候補製品の開発を進めるために多くの資金が必要となる.さらに、私たちは私たちの候補製品の将来の開発のために1つまたは複数のパートナーを探すかもしれませんが、適切なbr条項でタイムリーに、または私たちのどの候補製品とも協力できないかもしれません。いずれにしても、私たちの既存の現金は、私たちが計画しているすべての仕事に資金を提供するのに不足し、私たちのどの候補製品の開発完了にも資金を提供するのに十分ではないだろう。したがって、私たちは公開または私募株式発行、債務融資、協力、許可手配、または他の出所を通じてより多くのbr資金を得ることを要求されるだろう。私たちは約束された外部資金源を持っていない。私たちは許容可能な条項や全部で十分な追加融資を受けることができないかもしれない。私たちが必要な時に資金を調達できなかったことは、私たちの財務状況と私たちの業務戦略を実施する能力にマイナス影響を与えるだろう。
| -35- |
私たちのbr}推定は間違っていることが証明される可能性があり、私たちは現在予想されているよりも早く利用可能な資本資源を使用することができる。さらに、変化している場合、その中のいくつかは私たちの制御を超えている可能性があり、私たちの資本消費速度が私たちの現在の予想よりも大きく速くなる可能性があり、私たちは計画よりも早く追加資金を求める必要があるかもしれません。私たちの将来の資金需要は、短期的にも長期的にも、多くの要素に依存するだろう
私たちの現在と未来の候補製品の臨床試験の範囲、進捗、時間、コストと結果、ならびに研究と臨床前開発の仕事
私たちは任意の協力、許可、または他の手配の条項と時間を達成することができるかどうか
私たちのパイプラインのための1つまたは複数の未来の候補製品を決定することができます
私たちが求めている未来の候補製品の数とその開発需要
規制承認の結果、時間、コストを求める
市場の承認を得た私たちの任意の候補製品の商業化活動コスト は、製品販売、マーケティング、流通、および製造能力を確立するコストおよび時間を含む協力者の責任ではない
私たちの現在と未来の候補製品の商業販売収入(あれば)と販売許可の領収書;
私たちの研究開発とビジネスインフラの拡大に伴い、私たちの従業員の増加と関連コスト
特許出願の準備、提出、起訴、知的財産権の維持および保護のコストは、知的財産権に関連するクレームを実行し、保護することを含む
上場企業の運営コスト。
追加資本を調達することは、私たちの既存の株主を希釈し、私たちの運営を制限したり、私たちが貴重な権利を放棄したりする可能性がある。
私たちのbrは、公共と私募株式発行、債務融資、戦略的パートナーシップと 連盟、許可手配または貨幣化取引を組み合わせた方法で追加資本を求めることができる。もし私たちが株式、転換可能な債務証券、または他の株式ベースの派生証券を売却することによって追加資本を調達する場合、あなたの所有権権益は希釈され、条項は清算または株主としての他の権利に悪影響を及ぼす特典を含む可能性がある。私たちが発生したどんな債務も、固定支払義務の増加を招き、制限契約、例えば、私たちが追加債務を発生させる能力の制限、私たちが知的財産権を得ることができる能力の制限、および私たちの業務を展開する能力に悪影響を及ぼす可能性のある他の運営制限に関連する可能性があります。さらに、私たちは、株式でも債務でも、そのような発行の可能性にかかわらず、私たちの普通株の市場価格を下落させる可能性があり、既存の株主は、私たちの融資計画やそのような融資の条項に同意しない可能性があります。もし私たちが戦略的パートナーシップと連合、許可手配、または第三者との金銭化取引を通じてより多くの資金を調達するならば、私たちは私たちの技術または候補製品に対する貴重な権利を放棄しなければならないかもしれないし、または私たちに不利な条項でライセンスを付与しなければならないかもしれない。私たちは許容可能な条項で十分な追加融資を得ることができないかもしれないし、十分な追加融資を得ることができないかもしれない。もし私たちが必要な時に追加資金を集めることができない場合、私たちは私たちの製品開発や将来の商業化努力を延期、制限、減少、または中止する必要があるかもしれないし、あるいは私たちが開発とマーケティングをより望んでいた候補製品を開発し、マーケティングする権利を与える必要があるかもしれない。
| -36- |
我々の候補製品の発見,開発と規制承認に関するリスク
新冠肺炎コロナウイルスの大流行或いはいかなる他の伝染病の広範な爆発はすべて著者らの業務、財務状況と運営業績に実質的な不利な影響を与える可能性がある。
著者らのbrは衛生流行病或いは伝染病の爆発に関連するリスクに直面し、例えば最近世界各地で発生した高伝染性と病原性コロナウイルス新冠肺炎である。このような伝染病の爆発は広範な健康危機を招く可能性があり、これは多くの国の一般的な商業活動や経済·金融市場に悪影響を及ぼす可能性がある。
2019年12月、新型コロナウイルス新冠肺炎が武漢中国で出現し、2020年3月11日に世界保健機関によって大流行と発表された。新冠肺炎はどの程度著者らの臨床前と臨床試験運営に影響する可能性があり、brは未来の事態の発展に依存し、これらの事態の発展は非常に高い不確定性を持っており、例えば疫病の持続時間と地理範囲、新冠肺炎の重症度及び新冠肺炎の行動の制御と治療の有効性を自信的に予測できない。
現在まで、世界の多くの国は旅行と大衆集会に対して隔離と制限を実施し、新冠肺炎の伝播を緩和し、不要な業務を閉鎖した。国や州や地方司法管轄区域の継続制限に伴い,我々が業務を継続する能力も制限される可能性がある。このようなイベントは、業務、供給、および薬品生産の中断を招き、運営の減少を招く可能性があり、そのいずれも、私たちの業務、財務状況、および運営結果に重大な影響を与える可能性がある。
このような大流行または爆発は、臨床試験地点、CROおよび/または試験モニタ、および試験を支持する他の重要なサプライヤーおよびコンサルタントの安全を確保することを困難にする可能性がある。また、疫病或いは臨床試験地点付近の疫病は私たちの患者募集能力に影響する可能性がある。これらの状況や新冠肺炎に関連する他の状況は、我々の臨床試験計画の遅延を招く可能性があり、予想コストを増加させる可能性があり、これらはすべて私たちの業務およびその財務状況に実質的な悪影響を及ぼす可能性がある。
具体的には,我々のパイプライン製品(SON−1010型を除く)の生産は新冠肺炎に関するサプライチェーン問題の遅延を受け,特に原材料の供給は,媒体,樹脂,分析キットの供給に加え,国際輸送遅延を含む。
新冠肺炎による潜在的な経済影響及びその持続時間は評価或いは予測が困難である可能性があるが、広い範囲の疫病は全世界の金融市場の深刻な混乱を招き、私たちの資本獲得能力を低下させる可能性があり、これは未来に私たちの流動性に負の影響を与える可能性がある。また、新冠肺炎の伝播による景気後退や市場回復は、私たちの業務と私たちの普通株の価値に大きな影響を与える可能性がある。
Br新冠肺炎疫病はまた著者らの従業員と協力各方面が著者らの非臨床、臨床とbr薬物生産活動を展開する能力に影響する可能性がある。私たちは臨床サイト、研究者および他の研究者、コンサルタント、独立請負業者、契約研究組織および他の第三者サービスプロバイダに依存して、私たちの管理、監視、および他の方法で私たちの非臨床研究と臨床試験を実行するのを助けるかもしれない。私たちはまた、コンサルタント、独立請負業者、契約製造組織、および他の第三者サービスプロバイダに依存して、私たちの管理、監視、および他の方法で私たちの原料薬の生産、調合、および薬品製造活動を実行することを支援することに依存するかもしれない。新冠肺炎は、これらの外部の個人、組織、または会社が私たちのプロジェクトに十分な時間と資源を投入したり、出張のために仕事を実行したりする能力に影響を与える可能性がある。
新冠肺炎の発生が現在或いは未来の臨床研究に対する潜在的なマイナス影響は監督管理機関からのフィードバックの遅延、新しい臨床研究の開始及び被験者を登録している研究に参加することを含む。潜在的な負の影響 はまた、研究サイトで研究アクセスを行うことができないこと、安全性と有効性データの収集が不完全であること、進行中の研究中の被験者の中退率が高いこと、研究データがデータベースサイトに入る遅延、研究データの監視が研究サイトの物理的アクセス制限による遅延、クエリに対するサイトの応答遅延、データベースロックの遅延、データ分析の遅延、トップラインデータのタイムリーな遅延、および研究報告の完了遅延を含む。新しい或いは絶えず悪化する新冠肺炎の中断或いは制限は著者らの非臨床研究、臨床試験と薬物製造活動に更なる負の影響を与える可能性がある。
| -37- |
私たち は私たちの内部開発計画の成功に大きく依存して、私たちの候補製品は臨床試験に成功し、監督管理の許可を得る或いは商業化に成功できないかもしれない。
私たちの将来の成功は私たちの内部開発計画と私たちのチャネル計画候補製品の成功に大きく依存するだろう。
私たちのルートと他の候補製品を商業化することに成功する能力は、私たちの能力にかかっています
臨床前研究と臨床試験に成功した
Brはアメリカ食品医薬品局、ヨーロッパ医薬品局、その他の類似した監督管理機関の監督許可を得た
当社の候補製品を開発および/または商業化するために、第三者との協力を確立し、維持するために、または強力な開発、販売、流通、およびマーケティング能力を確立し、維持し、製品を開発し、任意の承認された製品の商業販売を開始するのに十分である
政府医療システムや保険会社などの支払者から保険と十分な補償を受け、商業的に魅力的な価格設定レベルを実現する
医師、医療支払者、患者、医学界が私たちの製品候補を受け入れることを確保します
検証の流れにより、商業化に成功するために、監督管理機関(食品医薬品局を含む)によって検査および承認された製造施設で十分な数の候補製品が生産される
臨床試験および商業化により費用が増加した場合に、私たちの支出を管理します
は、任意の承認された製品および候補製品のために十分な知的財産権を取得し、強制的に実行する。
製薬業界で大量に開発されている薬物のうち,一部の薬物のみがFDAに新薬申請(NDA)や生物製品許可申請(BLA)を提出しており,より少ない薬物が商業化承認されている。さらに、私たちが私たちの候補製品をマーケティングする規制の承認を得たとしても、このような承認は、製品をマーケティングする可能性のある指定された用途や患者集団によって制限される可能性があります。したがって、私たちが開発計画に資金を提供し続けるために必要な資金を得ることができても、私たちの候補製品が開発または商業化に成功することを保証することはできません。もし私たちが開発や規制部門の承認を得ることができなければ、あるいは私たちの候補製品が商業化に成功できなければ、私たちは私たちの業務を継続するのに十分な収入を生むことができないかもしれません。
私たちのbrは開発作業の非常に早い段階にあり、私たちの候補製品は新しい薬物カテゴリーを代表しており、それらが治療法として確立されるまで、より厳しい規制審査を受ける可能性がある。
我々のbr候補パイプライン製品は、完全なヒトアルブミン結合ドメインを使用してbr治療製品を提供することを含む新しい治療方法を代表する。私たちの候補製品は、それらのbrが有する可能性があると考えられる任意のまたは全ての薬理学的利益を患者に示すことができないかもしれない。私たちはまだ、これらまたは任意の他の候補製品の有効性および安全性 を臨床試験中または後に市場承認を得ることに成功しないかもしれない。
規制機関は我々の候補製品に経験がなく,我々の開発計画に含まれる安全性と有効性を超える証拠の提供を要求する可能性がある.この場合、私たちの候補製品の開発は予想されるコストや時間よりも高い可能性があり、私たちの候補製品は不可能であることが証明される可能性がある。
| -38- |
我々の開発努力が成功しなければ,候補製品の開発を進め,br製品を商業化し,資金を調達し,業務を拡大したり,運営を継続することができない可能性がある。
我々の候補製品とどのパートナーの製品も時間がかかり高価な臨床前と臨床試験が必要となり,その結果は予測できず,失敗のリスクが高い。私たちまたは彼らの候補製品の臨床前または臨床試験がFDA、EMAおよび任意の他の同様の規制機関に安全性および有効性を満足に証明できなかった場合、追加のコストまたは遅延完了が生じる可能性があり、これらの候補製品の開発または彼らの開発は放棄される可能性がある。
米国のFDA、EU、欧州経済区のEMAおよび他の司法管轄区の他の同様の規制機関は、新しい候補製品を承認しなければならず、その後、これらの地域でマーケティング、普及、または販売を行うことができる。我々はこれまでINDやBLAをFDAに提出したこともなく,類似した外国規制機関に類似した薬品承認文書を提出したこともなかった。私たちは、私たちの候補製品が特定の適応に対して安全かつ有効であることを証明するために、前臨床研究および臨床試験からのデータをこれらの規制機関に提供しなければならない。そして、商業販売のために許可されることができる。私たちの候補製品の臨床試験が成功するかどうか、または私たちの任意の候補製品がFDA、EMA、または任意の他の類似した規制機関の承認を得ることができない。
臨床前研究と臨床試験は長く、高価で予測できない過程であり、広範な遅延の影響を受ける可能性がある。いずれの臨床試験も計画的あるいは計画的に完了することは保証されない(あれば)。候補製品の商業化を達成するために必要な臨床前研究および臨床試験は数年かかり、大量のbr支出を必要とする可能性があるが、遅延または失敗は本質的に予測不可能であり、任意の段階で発生する可能性がある。私たちが予想している試験および試験に加えて、私たちの候補製品に対して追加の臨床試験または他の試験 を行う必要があるかもしれません。これは、追加の計画外コスト を生成したり、臨床開発の遅延をもたらす可能性があります。さらに、進行中または計画中の臨床試験に関連する計画を再設計または他の方法で修正する必要があるかもしれないが、臨床試験の設計を変更するには高価で時間がかかる可能性がある。1つまたは複数の実験で不利な結果が出たことは、私たちの候補製品と私たちの大きな挫折になるだろう。1つまたは複数の試験における不利な結果 は、私たちの業務、財務状態、運営結果、および将来の成長見通しに実質的な悪影響を及ぼす可能性がある1つまたは複数の製品開発計画を延期、縮小またはキャンセルする必要があるかもしれない。
臨床試験の開始遅延または完成をもたらす多くの要素は、最終的に私たちの候補製品が上場承認を拒否される可能性もある。FDA、EMA、あるいは他の類似した規制機関は、私たちの臨床試験設計と臨床試験データの解釈に同意しないかもしれない、あるいはbrを審査し、私たちの臨床試験設計をレビューした後であっても、承認要求を変更する可能性がある。
私たちの候補製品の臨床試験で、私たちは以下のリスクを含む多くのリスクに直面している
同じ適応に対して、候補製品は、既存の承認された製品よりも無効または劣る
候補製品は、許容できない毒性または許容できない副作用に関連するか、またはそれに関連する
Br患者は死亡または副作用を受ける可能性があり、その原因はテスト中の候補製品に関連している可能性があり、それとは無関係である可能性がある
この結果は早期試験の陽性結果を確認できない可能性がある
Br結果は、継続試験または発売承認のために、我々の候補製品の安全性と有効性を決定するために、食品·薬物管理局、食品薬品監督管理局または他の関連監督機関が要求する統計的有意性レベルに適合しない可能性がある
私たちの協力者はその契約を履行できないか、または履行したくないかもしれない。
| -39- |
また, は計画目的で,様々な科学,臨床,法規,他の製品開発目標を達成する時間を見積もることがある. これらのマイルストーンには、科学研究、臨床試験の開始または完成、監督文書の提出、あるいは商業化目標に対する私たちの期待が含まれるかもしれない。私たちは、進行中の臨床試験の完了、他の臨床計画の開始、市場承認または製品の商業発表を受けるなど、いくつかのマイルストーンの予想される時間を時々公開するかもしれない。その中の多くのマイルストーンの達成は私たちの統制範囲を超えているかもしれない。このようなすべてのマイルストーンは様々な仮定に基づいており、これはマイルストーンの実現時間が私たちの推定と大きく異なることをもたらすかもしれない。もし私たちが予想された時間範囲でマイルストーンを達成できなかったら、私たちの候補製品の商業化は延期される可能性があり、私たちはいくつかの契約支払いを得る資格がないかもしれません。これは、私たちの業務、財務状況、運営結果、および将来の成長見通しに実質的な悪影響を及ぼす可能性があります。
われわれは患者をわれわれの臨床試験に組み込むことが困難であることが発見される可能性があり,候補製品の臨床試験の継続を延期または阻止する可能性がある。
Brを確定し、患者に著者らの候補製品に参加する資格を持たせる臨床試験は著者らの成功に重要である。われわれの臨床試験の時間は,患者の参加を募集する能力と必要なフォローアップ期間の完了状況に依存する。新しい治療方法に関連する不良事件の負の宣伝、類似患者群の競争的臨床試験、現在の治療方法の存在或いはその他の原因により、患者は著者らの臨床試験に参加したくないかもしれない。まれな疾患である可能性のある1つまたは複数の候補製品に対するいくつかの兆候については、登録リスクが増加し、これは、私たちが計画している臨床試験において登録可能な患者プールを制限する可能性がある。患者を募集し、br試験を行い、私たちの候補製品の規制承認を得るスケジュールが遅延する可能性があり、これはコスト増加、私たちの候補製品の推進の遅延、試験候補製品の有効性の遅延、あるいは臨床試験の完全な終了を招く可能性がある。
十分な数の患者または必要なまたは必要な特徴を有する患者を識別、募集、募集することができない可能性があり、br}は直ちに我々の臨床試験を完了することができない。例えば,我々が最初に対象とした適応の性質により,末期疾患が進行した患者はわれわれの候補製品を用いた治療に適していない可能性があり,われわれの臨床試験に参加する資格がない可能性がある。したがって、私たちの目標疾患患者の早期診断は私たちの成功に必須的だ。患者の登録と試験完了は以下の要素の影響を受ける
Br患者集団の規模と被験者を識別する流れ;
試験シナリオ設計;
資格と排除基準;
これまで研究されてきた候補製品の安全概要;
研究された製品候補製品の知覚リスクおよび収益;
私たちの病気を治療する方法のリスクと利益
競争療法と臨床試験の有用性;
調査中の疾患の重症度;
Br登録時の被験者の疾患進行度;
潜在的被験者の臨床試験地点の近似性および有用性;
当事者の同意を得て維持する能力;
試験終了前に登録された被験者が退出するリスク;
| -40- |
Br先生の患者紹介方法;
Brは、治療中および治療後に被験者の能力を十分に監視する。
また、SON-080のパイロット規模フィージビリティスタディの臨床開発は現在、アメリカ以外で行われる予定です。私たちがどの外国でも臨床試験の開始、登録、完了に成功する能力は、外国で業務を展開するbr独自の多くのリスクの影響を受けています
学術パートナーまたは契約研究組織またはCROと医師との関係を構築または管理することは困難である
臨床試験を行う異なる基準;
いくつかの国/地域では、私たちの新しい方法に関連する合意を検討するのに十分な規制専門知識を有する成熟したグループが不足している
私たちは現地で合格したコンサルタント、医者、パートナーを見つけることができません
様々な外国の法律、医療標準と法規要求の潜在的負担を遵守し、薬品と生物技術製品と治療の法規 を含む。
計画通りの臨床試験を行うのに十分な数の患者を募集することが困難な場合、進行中または計画中の臨床試験の終了を延期、制限、または中止する必要があるかもしれませんが、いずれも私たちの業務、財務状況、運営結果、および将来性に悪影響を与えます。
臨床前研究と早期臨床試験の結果は未来の臨床試験の結果を予測できないかもしれない。
臨床前研究と早期臨床試験の結果は後続の臨床試験の成功を予測できない可能性があり、臨床試験の中期結果も完成した臨床試験の結果を予測できるとは限らない。製薬やバイオテクノロジー業界の多くの会社が早期開発に積極的な成果をあげた後,後期臨床試験で大きな挫折を経験し,類似した挫折に直面する可能性がある。例えば,SON−080のIIa期試験は米国国外で行われ,われわれやわれわれの協力者による後期臨床試験では,これらの発見は将来の世界的な臨床試験地点の試験で複製されない可能性がある。臨床試験の設計はその結果が製品の承認を支持するかどうかを確定することができるが、臨床試験設計中の欠陥は臨床試験の進展が良好になるまで明らかにならない可能性がある。私たちは市場承認を支援するために、br臨床試験を設計し、実行することができないかもしれない。
臨床前データと臨床データはよく異なる解釈と分析の影響を受けやすい。多くの会社は彼らの候補製品 は臨床前研究と臨床試験で満足できると思っているが、まだ候補製品の市場許可を得られなかった。私たちまたは任意のパートナーが私たちの候補製品の臨床試験結果が発売される価値があると思っていても、FDAや同様の外国の規制機関は同意しない可能性があり、私たちの候補製品の発売を承認しないかもしれない。
ある場合、多種の要素のため、同一候補製品の異なる臨床試験の安全性或いは有効性結果は有意差が存在する可能性があり、方案中に規定された試験プログラムの変化、患者群の大きさとタイプの差異、投与方案と他の臨床試験方案の変化と遵守状況、及び臨床試験参加者の退学率を含む。もし私たちが候補製品の臨床試験で積極的な結果を得なければ、私たちの最先端の候補製品の開発スケジュール および規制承認と商業化の見通し、そしてそれに応じた私たちの業務と財務の見通しは負の影響を受けるだろう。
| -41- |
現在或いは未来の候補製品の単独使用或いは他の承認された製品或いは研究新薬と一緒に使用する場合、不良副作用或いは他の特性を招く可能性があり、その臨床開発を停止し、その上場承認を阻止し、その商業潜在力を制限し、或いは深刻な負の結果を招く可能性がある。
不良なbr或いは臨床上コントロールできない副作用が発生する可能性があり、そして私たち或いは監督機関の臨床試験の中断、延期或いは一時停止を招き、更に厳格なラベル或いはFDA或いは類似の外国の監督管理機関が上場承認を遅延或いは拒否する可能性がある。私たちの実験結果は、副作用または予期しない特徴の高さと受け入れられない深刻さおよび流行率を示すかもしれない。
もし私たちの候補製品が開発中に受け入れられない副作用が発生した場合、私たち、FDAまたは同様の外国の規制機関、機関審査委員会またはIRBsまたは私たちが研究を行っている機関の独立道徳委員会、またはbr}データ安全監視委員会またはDSMBは、私たちの臨床試験を一時停止または終了することができ、またはFDAまたは同様の外国規制機関は、臨床試験を停止するか、または私たちの製品候補製品の任意またはすべての目標適応を承認することを拒否するように命令することができる。治療に関連する副作用は、患者募集または被験者が試験を完了する能力に影響を与える可能性があり、または潜在的な製品責任クレームをもたらす可能性がある。また、治療医療従事者は、これらの副作用を正確に識別または管理できない可能性がある。私たちの候補製品を使用する医療従事者を訓練して、私たちの臨床試験および任意の候補製品が商業化された後の副作用の概要を理解する必要があるかもしれない。私たちの候補製品の潜在的な副作用を識別または管理する上での訓練不足は、患者の負傷または死亡を招く可能性がある。これらの状況のいずれも、影響を受けた候補製品に対する市場の受容度を達成または維持することを阻止し、私たちの業務、財務状況、および将来性を深刻に損なう可能性がある。
また,われわれの候補製品の臨床試験は丁寧に定義された患者群で行われており,これらの患者は臨床試験に入ることに同意している。したがって,われわれの臨床試験では,候補製品の有意なプラス効果が実際のプラス効果(あれば)よりも大きいか,あるいは不良副作用を認識できない可能性がある。もし候補製品が承認された後、私たちまたは他の人がその製品の効果が以前に考えられていたほど有効ではないことを発見した場合、あるいは以前に確定されていなかった不良副作用 をもたらした場合、以下のいずれかの結果が発生する可能性がある
監督管理部門は、製品の承認を取り消すことができ、または製品を差し押さえることができる
私たちまたは任意の協力者は、製品をリコールする必要があるか、または製品の投与方法の変更を要求されるか、または追加のbr臨床試験を行う必要がある場合があります
特定の製品のマーケティングまたは製造プロセスに追加の制限を適用することができる
私たちは罰金、禁止、あるいは民事または刑事罰を受けるかもしれない
Br規制機関は、ブロック警告または禁忌症のようなラベル宣言の追加を要求する可能性がある
私たちまたは任意の協力者は、患者に配布するために、以前に決定されていなかった副作用のリスクを概説するための薬物ガイドラインの作成を要求される可能性がある
私たちまたは任意の協力者は起訴され、患者に与える傷害に責任を負うかもしれない
製品はそんなに競争力がなくなる可能性があります;
私たちの名声は影響を受けるかもしれない。
もし私たちが現在または未来の任意の候補製品が臨床試験で安全性と有効性を証明できない、あるいは市場の承認を得られなければ、私たちは収入を生むことができなくなり、私たちの業務は損害を受けるだろう。これらの事件のいずれも私たちの業務と運営を損なう可能性があり、 は私たちの普通株の価格に悪影響を及ぼす可能性がある。
| -42- |
私たち は他の候補製品を決定または発見することで成功しないかもしれない。
我々が現在開発している候補製品以外にも,他の治療機会を探る予定であるにもかかわらず,様々な理由で臨床開発のための他の候補製品を決定できない可能性がある。例えば、私たちの研究方法は、潜在的な候補製品の識別に成功しない可能性があり、または私たちが発見した製品が、規制部門の承認を得ることができないか、または不可能にするために、有害な副作用または他の特徴を有する可能性がある。商業販売の前に、他の候補製品は臨床前研究、臨床試験とFDAおよび/または適用される外国の監督管理機関の承認を含む追加的で時間のかかるbr開発作業が必要となる。すべての候補製品は薬品開発に固有の失敗リスク が出現しやすい。もし私たちがもっと多くの潜在的な候補製品を発見して開発できなければ、私たちは私たちの業務を発展させることができないかもしれません。私たちの運営結果は実質的な損害を受けるかもしれません。
我々 は、特定の候補製品または指示を追求するために限られたリソースを費やす可能性があり、より利益またはより成功する可能性の高い候補製品または指示 を利用することができない。
我々は財務や管理資源が限られているため, が最も成功する可能性があると考えられる特定の適応に焦点を当てて候補製品を開発する予定であり,市場承認においても商業化においても。したがって、私たち は、他の候補製品を求めることを放棄または延期するか、またはより大きな商業的潜在力を有することが証明される可能性のある他の指示 を放棄または延期する可能性がある。
私たちの資源配分決定は私たちが実行可能な商業製品や利益のある市場機会を利用できないかもしれない。現在と未来の研究開発計画および特定の適応候補製品への支出は、いかなる商業的に実行可能な候補製品も生じないかもしれない。もし私たちが特定の候補製品の商業的潜在力や目標市場を正確に評価できなければ、候補製品の独占的な開発と商業化の権利を保持することが私たちにより有利な場合、私たちは協力、許可、または他の印税配置を通じて候補製品の貴重な権利を放棄するかもしれない。
私たちは潜在的な製品責任に直面していますが、私たちに対するクレームが成功すれば、大量の責任とコストを招く可能性があります。もし私たちの候補製品を使用して患者を傷つけた場合、あるいはこのようなダメージが私たちの候補製品と関係がなくても、患者を傷つけたと考えられ、私たちの規制承認が撤回されるか、または他の方法で否定的な影響を受ける可能性があり、私たちはコストが高く破壊的な製品責任クレームを受ける可能性がある。
臨床試験で私たちの候補製品を使用し、市場の承認を得たいかなる製品を販売することは、私たちを製品責任クレームのリスクに直面させます。患者、医療提供者、製薬会社、または販売、または他の方法で私たちの製品に接触した他の人は、私たちに製品責任クレームを提起するかもしれません。私たちの候補製品は不良事件を引き起こす可能性があります。もし私たちが製品責任クレームに成功的に対抗できなければ、私たちは大量の責任とコストを招くかもしれない。さらに、利点または最終結果にかかわらず、製品責任クレームは、以下のような結果をもたらす可能性がある
| ● | 私たちのビジネス的名声は損なわれています | |
| ● | 臨床試験参加者の脱退 | |
| ● | 患者や他のクレーム者に巨額のお金の報酬を与えます | |
| ● | 関連訴訟による費用 ; | |
| ● | 経営陣の関心を主要業務から分散させる | |
| ● | 候補品を商業化することはできません | |
| ● | 私たちの候補製品に対する需要が減少した(商業販売のための承認が得られれば)。 |
| -43- |
私たちの現在の臨床計画を考慮して、私たちは製品責任保険を購入するつもりですが、私たちは責任による損失から私たちを守るために、合理的なコストあるいは十分な金額でbr保険範囲を維持することができないかもしれません。私たちは新製品を商業化するたびに私たちの保険範囲を拡大するつもりですが、私たちは商業的に合理的な条項や十分な金額で製品責任保険を受けることができないかもしれません。予期せぬ悪影響を及ぼす薬物や治療に基づく集団訴訟では,多額の判決が下されることがある。成功した製品責任クレームあるいは私たちに対する一連のクレーム は私たちの株価下落を招く可能性があり、もし判決が私たちの保険範囲を超えたら、私たちの運営と業務業績に不利な影響を与えるかもしれない。
私たちのいくつかの候補製品が対象とする疾患を有する患者brは、通常、深刻なbrおよび末期疾患にあり、既知かつ未知の重大な事前存在および潜在的に生命を脅かす健康リスクを有する。治療中、患者は、私たちの候補製品に関連する可能性のある原因によって、死を含む有害事象を受ける可能性がある。このような事件は、私たちの製品をマーケティングする機会を得るために、怪我をした患者に巨額の費用を支払うこと、遅延、負の影響を与えるか、または維持することを要求するコストの高い訴訟に直面する可能性がある。私たちの商業化努力を一時停止したり放棄したりすることを要求したりする。有害事象が我々の製品に関連しているとは考えられない場合でも,この場合の 調査は時間や不確実性が高い可能性がある.これらの調査は、私たちの販売作業を中断し、私たちの規制承認の流れを遅らせるか、あるいは私たちの製品候補の受け入れや維持の規制承認タイプに影響し、制限する可能性があります。これらの要因により、製品責任クレームは弁護に成功しても、私たちの業務、財務状況、あるいは運営結果に重大な悪影響を及ぼす可能性があります。
私たち は、より速い開発過程やより速い規制経路などの利点を提供することを目的としたFDAおよび他の同様の規制機関に認証を求めることができるが、このような認証を成功させることは保証されない。さらに、私たちの1つまたは複数の候補製品がそのような指定を取得しても、そのような指定の予期される利点を達成することができない可能性がある。
FDAと他の類似した監督管理機関は候補製品に特定の名称を提供し、重大な満足されていない医療需要の状況に対する薬物製品の研究開発を奨励することを目的としている。これらの指定は、規制機関との追加的な相互作用、加速可能な規制経路、および優先審査のような利点をもたらす可能性がある。私たちは私たちの他の候補物がそのような認証に成功することを保証できない。また,このような指定は開発や承認の流れを加速させることができるが,通常承認基準を変更することはない. 1つ以上の候補製品のこのような指定を得ても,期待される の利点を実現する保証はない.
例えば、私たちは私たちの1つまたは複数の候補製品のための画期的な治療認証を求めるかもしれない。画期的な治療法は、初歩的な臨床証拠が、1つまたは複数の臨床的に重要な終点(例えば、臨床開発早期に観察される実質的な治療効果)において既存の療法よりも顕著に改善されたことを示す場合、単独でまたは1つまたは複数の他の療法と組み合わせて、深刻または生命に危険な疾患または状態を治療することを意図していると定義される。画期的な治療法に指定されている療法では,FDAと試験スポンサーとのインタラクションやコミュニケーションは,臨床開発の最も有効な経路を決定するのに役立ち,無効対照案の患者数を最小限に抑えることができる。brはFDAによって画期的な療法に指定された療法も加速承認を得る資格がある。画期的療法 に指定されたのはFDAの裁量権である。したがって,我々の候補製品の1つが画期的療法として指定された基準に適合していると考えても,FDAは同意せず,このような指定を行わないことにした可能性がある。いずれの場合も,FDAの従来の手順による承認を考慮した療法と比較して,候補製品に対する画期的な治療指定を受けることは,より速い開発過程,審査または承認を招くことはなく,FDAの最終承認を確保することもできない可能性がある。また,我々の1つまたは複数の候補製品が画期的療法の条件を満たしていても,FDAは今後これらの候補品が資格条件を満たしていないことを決定する可能性がある。
私たちはまた私たちのいくつかの候補製品のために高速チャネル認証を求めるかもしれません。治療法が重篤または生命に危険な疾患を治療することを意図しており、そのような疾患が満たされていない医療需要を解決する可能性を示す場合、治療スポンサーは、br}迅速チャネル認証を申請することができる。FDAはその称号を付与するか否かを決定する幅広い裁量権を持っているため,特定の候補製品がその称号を獲得する資格があると考えても,FDAがその称号を付与することを決定する保証はない。我々が高速チャネル認証を確実に取得しても,従来のFDAプログラムと比較して,より速い開発プロセス,審査または承認を経験しない可能性があり, かつ高速チャネル認証を得ることはFDAの最終承認を保証することはできない.FDAが我々の臨床開発計画のデータがこの指定を支持しなくなったと考えた場合,その指定を撤回する可能性がある。
| -44- |
私たち は私たちの1つ以上の候補製品のための優先審査指定を求めるかもしれませんが、私たちはそのような指定を受けないかもしれません。たとえそうしても、そのような指定はより速い規制審査や承認過程をもたらすことができないかもしれません。
FDAが候補製品が重篤な疾患を治療する方法を提供すると判断し、承認された場合、製品が安全性または有効性の面で有意な改善を提供する場合、FDAは、候補製品を優先的に検討するように指定することができる。優先審査を指定することは、FDA審査申請の目標が標準的な10ヶ月の審査期間ではなく、6ヶ月であることを意味します。 私たちは私たちの候補製品の優先審査を要求するかもしれません。FDAは候補製品に優先審査地位を付与するか否かについて広範な裁量権を有しているため,特定の候補製品にこのような資格や地位を得る資格があると考えても,特にその候補品が突破療法指定を受けていれば,FDAはその資格を付与しないことを決定する可能性がある。また,FDAの従来のプログラムと比較して,優先審査指定が必ずしも開発を加速するとは限らず,規制審査や承認過程を加速させるとは限らず,必ずしも承認面の優位性をもたらすとは限らない.FDAの優先審査を受けることは、6ヶ月の審査期間内にまたは全く承認されないことを保証しない。
1つの管轄区域で現在および将来の候補製品のマーケティング承認を取得し、維持することは、他の管轄区域で現在および将来の候補製品のマーケティング承認を得ることに成功することを意味するわけではない。
1つの管轄地域で現在および将来の候補製品のマーケティング承認を取得し、維持することは、他の任意の管轄エリアでマーケティング承認を得ることができるか、または維持することができる保証はなく、1つの管轄エリアでマーケティング承認を得ることができないか、または遅延することは、他の管轄エリアのマーケティング承認プロセスに悪影響を及ぼす可能性がある。例えば、FDAが候補製品の販売を許可しても、外国司法管轄区の比較可能な規制機関は、これらの国/地域における候補製品の製造、マーケティング、および普及を承認しなければならない。審査手続きは司法管轄区域によって異なり、1つの司法管轄区で行われた臨床研究は他の管轄区の監督機関に受け入れられない可能性があるため、米国と異なるか、あるいはアメリカより大きい要求と行政審査期限に関連する可能性がある。米国以外の多くの司法管轄区では、候補製品は先に精算許可を得なければならず、その後、この管轄区で販売することができる。場合によっては、私たちが私たちの製品のために受け取る価格もまた承認されなければならない。私たちは国際市場で精算や価格決定の承認を受けた経験がない。
市場の承認を得て監督管理要求を遵守することは私たちに重大な遅延、困難とコストをもたらす可能性があり、brは私たちの製品がアメリカ以外のある国/地域で発売されることを延期または阻止する可能性がある。もし私たちが国際市場の規制要求を遵守せず、および/または相応のマーケティング承認を得なければ、私たちの目標市場は を減少させ、候補製品市場の潜在力を十分に発揮する能力は損なわれるだろう。
私たちの候補製品の商業化に関するリスクやその他のコンプライアンス問題
必要な臨床前研究と臨床試験を完了しても、マーケティング承認の流れは高価で、時間がかかり、不確定であり、私たちまたは任意の協力者が候補製品の一部または全部を商業化する承認を得ることを阻止する可能性がある。したがって、私たちまたはどの協力者がいつ、どの地域で候補製品を商業化するマーケティング承認を得るかを予測することはできない。
アメリカと海外でマーケティング承認を得る過程は長く、高価で不確実である。最終的に承認された場合、br年が必要となる可能性があり、関連する候補製品のタイプ、複雑性、および新規性を含む様々な要因によって大きく変化する可能性がある。上場承認を得るためには、各治療適応に対して監督管理機関に広範な臨床前と臨床 データと支持情報を提出し、候補製品の安全性と有効性を確定する必要がある。市場承認を得るためには,製品製造過程に関する情報を規制機関に提出し,規制機関が製造施設を検査する必要がある。FDAまたは他の規制機関は、私たちの候補製品が安全ではなく、中程度の効果しかない、あるいは不良または意外な副作用、毒性、または他の製品が上場承認を得ることができない、または商業使用を阻止または制限する特徴を認定する可能性がある。私たちの最終的に得られたどのマーケティング承認も制限されたり、制限されたり、承認された後の約束が制限されたりして、承認された製品が商業的に生存できないようにする可能性がある。
| -45- |
さらに、開発中の市場承認政策の変更、追加法規、法規またはガイドラインの制定または公布の変更、または各提出された製品申請に対する規制審査の変更は、申請承認の遅延 または拒否をもたらす可能性がある。監督管理機関は承認過程においてかなりの自由裁量権を持っており、任意のbrの申請を拒否する可能性があり、あるいは私たちのデータが承認を得るのに十分ではないと決定する可能性があり、追加の臨床前、臨床或いは他の研究を行う必要がある。臨床前と臨床テストから得られたデータの異なる解釈は候補製品の上場承認を延期、制限、或いは阻止する可能性がある。適切な規制機関が候補製品を審査して承認するまで、私たちは製品を商業化することができない。私たちの候補製品が臨床試験で安全性と有効性を示しても、規制機関はその審査プロセスを適時に完成できない可能性があり、あるいは監督部門の承認を得ることができない可能性がある。FDA諮問委員会または他の規制機関が承認または制限を提案しない場合、追加の遅延を招く可能性がある。また、製品開発、臨床試験、審査過程において、将来の立法または行政行動における追加の政府規制または規制機関の政策の変化によって遅延や拒否が生じる可能性がある。私たちが最終的に得たどんなマーケティング承認も制限されたり、制限されたり、承認された後の約束が制限されたりして、承認された製品が商業的に不可能になる可能性があります。
また,われわれの臨床試験の首席研究員は時々私たちの科学コンサルタントやコンサルタントを務め,このようなサービスに関する報酬 を得る可能性がある。場合によっては、私たちはFDAまたは他の規制機関にいくつかの関係を報告する必要があるかもしれない。FDAや他の規制機関は、私たちと主要調査員との財務関係が利益の衝突をもたらしたり、他の方法で研究の解釈に影響を与えたりすると結論するかもしれない。したがって,FDAや他の規制機関は,適用された臨床試験現場で生成されたデータの完全性を疑問視する可能性があり,臨床試験自体の効用が脅かされる可能性がある。これは、FDAまたは他の規制機関が私たちの上場申請の承認を遅延または拒否することを招き、最終的に私たちの1つまたは複数の候補製品の上場承認が拒否される可能性がある。
さらに、規制当局は、私たちの候補製品の商業化に必要または必要なラベル宣言 を承認しないかもしれない。例えば、規制機関が承認する可能性のある候補製品の適応は要求よりも少ないか少ないか、上場後の研究の表現に基づいて承認される可能性もある。監督管理機関は患者数が少なく、異なる薬物調合或いは異なる製造技術に適した候補製品を承認する可能性があり、私たちが求めている製品ではない。もし私たちが必要な規制承認を得ることができない場合、あるいは規制承認が私たちが予想しているよりも限られていれば、私たちの業務、見通し、財務状況、運営結果は影響を受ける可能性がある。
任意の遅延が必要な承認を得たか、または得られなかったbrは、特定の候補製品から収入を得る能力に悪影響を及ぼす可能性があり、これは私たちの財務状況に重大な損害を与え、私たちの普通株価格に悪影響を及ぼす可能性がある。
私たち は現在、私たちの候補製品に関するマーケティング、販売、流通インフラを持っていません。もし私たちが自分自身やマーケティングパートナーとの協力を通じて私たちの販売、マーケティング、流通能力を発展させることができなければ、私たちの候補製品を商業化することに成功しないだろう。
私たち は現在、マーケティング、販売、または流通能力がなく、私たちの組織内での販売またはマーケティング経験は限られています。 もし私たちの1つまたは複数の候補製品が承認された場合、私たちは、その候補製品を商業化するため、またはこの機能を第三者 にアウトソーシングするために、技術的な専門知識と流通能力を支援する販売およびマーケティング組織を構築するつもりです。私たち自身の販売とマーケティング能力を確立し、 第三者と合意してこれらのサービスを実行することにはリスクがあります。
| -46- |
採用と訓練内部の商業組織は高価で時間がかかり、どんな製品の発表も延期される可能性がある。これらの費用の一部または全部は、私たちの任意の候補製品が承認される前に起こるかもしれない。販売チームを募集し、マーケティング能力を確立する候補製品のビジネス発表が何らかの理由で延期または発生していない場合、これらの商業化費用を早期にまたは不必要に発生させる。もし私たちが私たちの販売とマーケティング担当者を維持したり再配置することができなければ、コストが高いかもしれませんし、私たちの投資は損失します。さらに、アメリカや私たちが狙う医療市場で十分な規模や十分な専門知識を持つ他のターゲット市場で販売員を募集することはできないかもしれません。
候補製品の商業化を阻害する可能性がある要因は、
十分な数の効果的な販売およびマーケティング担当者を募集、訓練、維持することができない
販売員は、医師に接触することができないか、または私たちが開発する可能性のある未来の製品の処方を出すのに十分な数の医師を説得することができない
販売員が提供する補完的な治療不足は、より広い製品ラインを持つ会社に対して競争上不利になる可能性があります
独立した販売およびマーケティング組織の作成に関連する予期しないコストおよび費用。
もし私たちが第三者と合意して販売、マーケティング、流通サービスを実行すれば、私たちの製品の収入またはこれらの収入の流れから私たちにもたらす収益力は、私たちが自分たちが開発した任意の候補製品をマーケティングして販売する場合よりも低いかもしれません。また、私たちは第三者と合意して私たちの候補製品を販売し、販売することに成功できないかもしれません。あるいは は私たちに有利な条項でそうすることができないかもしれません。私たちはこれらの第三者に対してほとんどコントロール権がないかもしれません。彼らのいずれも必要な資源と注意力を投入して、私たちの候補製品を効果的に販売し、マーケティングすることができないかもしれません。もし私たちが販売とマーケティング能力を確立することに成功できなければ、私たち自身も第三者と協力しても、私たちの候補製品を商業化することに成功できないかもしれません。
承認されれば、我々が開発した現在または将来の候補製品のいずれかのbr市場機会は、既存の療法または以前の療法を受ける資格がない患者に限定される可能性があり、したがって、小さい可能性がある。
癌療法は1線、2線、または3線と記述されることがあり、FDAは通常、最初に三線使用の新しい療法のみを承認する。癌が十分に早く発見されたとき、第一線の治療は、通常、化学療法、ホルモン治療、手術、放射線治療、免疫治療、またはこれらの療法の組み合わせであり、治癒を必要とすることなく、癌を治癒または延長するのに十分である場合がある。以前の治療が無効であった場合、患者に二線と三線治療を実施する。私たちは最初に、1つまたは複数の以前の治療を受けた患者の治療として、SON−080およびbrが開発した任意の他の候補製品の承認を求めることができる。その後,十分なメリットが証明された製品(あれば)については,第一線療法としての承認を求める可能性が予想されるが,我々が開発した候補製品が承認されても一次療法として承認されることは保証されず,いずれかの承認を得る前に追加的な臨床試験を行わなければならない可能性がある。
私たちが対象としている癌患者の数は予想を下回るかもしれない。さらに、承認された場合、私たちの現在の計画または将来の候補製品の潜在的にアドレス指定可能な患者の数は制限されるかもしれない。私たちが任意の候補製品のためにかなりの市場シェアを得ていても、承認された場合、潜在的なターゲット層が少ない場合、私たちは、第一線または二次治療として使用することを含む追加の適応のマーケティング承認を得られなければ、決して利益を達成できない可能性がある。
| -47- |
候補製品の上場承認を得ても、継続的な規制義務と持続的な規制審査を受けることになり、これは多くの追加費用を招く可能性があり、規制要件を遵守できなかったり、意外な製品問題に遭遇したりすれば、処罰される可能性があります。
現在または将来の任意の候補製品について、私たちが受信した任意の マーケティング承認は、製品上場の承認指示用途または承認条件によって制限される可能性があり、または、候補製品の安全性および有効性を監視するために、コストが高い可能性のある上場後のテストおよび監視要件を含む可能性がある。FDAはまた、任意の候補製品を承認する条件としてリスク評価および緩和戦略またはREMSを要求することができ、薬物ガイドライン、医師コミュニケーション計画、または安全使用を保証する他の要素の要件、例えば、制限分配方法、患者登録 および他のリスク最小化ツールを含むことができる。FDAまたは同様の外国規制機関が候補製品を承認する場合、候補製品の製造、ラベル、包装、流通、不良事件報告、貯蔵、広告、販売促進、輸出入と記録は広範かつ持続的な監督管理要求を受ける。これらの要件は、製品承認のラベルに含まれていない使用のための承認された製品の普及を禁止するための使用、安全および他の発売後の情報および報告書の提出、登録、および承認後に我々が行った任意の臨床試験について、現在の良好な製造実践(CGMP)および良好な臨床実践(GCP)を継続的に遵守することを含む。その後、任意の承認された候補者には、意外な重大性または頻度の有害事象、または私たちの第三者製造業者または製造プロセス、または規制要件を遵守できなかったことを含む以前に未知の問題が存在することが発見され、以下のことが含まれる可能性がある
製品のラベル、流通、マーケティングまたは製造を制限し、製品を市場からリコールするか、またはリコールする
タイトルおよび警告状はなく、または臨床試験を一時停止した
食品および医薬品局は、係属中の出願または私たちが提出した承認された申請の追加の承認を拒否するか、または許可証の一時停止または取り消し;
発売後の研究或いは臨床試験の要求;
第三者支払人に対する保証の制限;
罰金、返却または利益または収入の返還;
上場承認の一時停止または撤回;
製品が差し押さえられたり、差し押さえられたり、製品の輸入または輸出を許可することを拒否したり、
民事または刑事罰の命令または適用を禁止する.
FDAや他の規制機関の政策は変わる可能性があり、製品の上場承認を阻止、制限、または延期するための追加の政府法規が公布される可能性がある。米国や海外の将来の立法や行政行動によって生じる可能性のある政府規制の可能性、性質、程度を予測することはできない。もし私たちが既存の要求の変化にゆっくりまたは適応できない場合、あるいは新しい要求や政策を採用することができない場合、または私たちがコンプライアンスを維持できない場合、私たちは私たちが獲得する可能性のあるいかなるマーケティング承認も失う可能性があり、私たちは利益を達成したり維持することができないかもしれない。
私たちの競争は激しい競争に直面しています。もし私たちの競争相手が私たちが開発した候補製品よりも効果的で、より安全で、あるいはもっと安い製品を開発·販売すれば、私たちのビジネス機会はマイナスの影響を受けます。
生命科学業界は競争が激しい。我々が現在開発している療法が承認されれば,他の製品や現在存在する,開発中あるいは将来開発される療法 と競合し,その中のいくつかは現在知られていないかもしれない。
| -48- |
私たちはアメリカと国際に競争相手がいて、大型多国籍製薬会社、老舗バイオテクノロジー会社、専門製薬会社、大学、その他の研究機関を含みます。私たちと比較して、私たちの多くの競争相手はより多くの財務、製造、マーケティング、製品開発、技術、人的資源を持っている。大手製薬会社、特に大手製薬会社は、臨床テスト、市場承認、患者募集と薬品 製品の製造において豊富な経験を持っている。これらの会社の研究やマーケティング能力も私たちよりはるかに優れており、承認されたり、開発後期の段階にある製品や、私たちのターゲット市場でリーディングカンパニーや研究機関との協力手配も持っているかもしれません。古い製薬会社はまた、新しい化合物の発見と開発を加速させたり、私たちが開発した候補製品を時代遅れにする可能性のある新しい化合物の使用を許可したりするために投資する可能性がある。製薬とバイオテクノロジー業界の合併と買収は、より多くの資源を私たちの数の少ない競争相手 に集中させる可能性がある。これらのすべての要因により、私たちの競争相手は、私たちの前に特許保護および/またはマーケティング承認を得ることに成功するか、または私たちの前に私たちの分野の製品を発見、開発、商業化するかもしれない。
多くの主要な製薬会社とバイオテクノロジー会社を含む多くの会社が癌治療法を開発またはマーケティングしている。これらの治療方法は伝統的な化学療法などの小分子薬物製品も含まれており、新しい免疫療法も含まれている。例えば,多くの多国籍企業や大手バイオテクノロジー会社は,Astellas Pharma Inc.,Seattle Genetics, Inc.,アスリコンとグラクソ·スミスクラインを含み,我々のパイプラインプロジェクトのための目標開発プロジェクトを探索している。
もし私たちの競争相手が私たちが開発する可能性のあるどんな製品よりも安全で、より効果的で、影響が少ない、またはより深刻ではなく、より便利で、より広いラベル、より効果的なマーケティング、精算またはより安い製品を得ることができれば、私たちのビジネス機会は減少または消滅するかもしれない。私たちの競争相手も私たちよりも早くFDA、EMA、または他の製品の市場承認を得ることができ、これは私たちの競争相手が私たちが市場に入る前に強力な市場地位を確立できるようにするかもしれない。我々が開発した候補製品が市場承認を得ても,それらの価格は競合製品よりも顕著な割増(その時点で承認された場合)が高く,競争力の低下を招く可能性がある.
規模の小さい や他のスタートアップ会社も重要な競争相手であることが証明されている可能性がある.これらの第三者は合格した科学と管理人員を募集と維持し、臨床試験場と臨床試験患者登録を確立する方面及び著者らの計画と相補的或いは必要な技術を獲得する面で著者らと競争を展開している。しかも、生物製薬産業の特徴は技術の変化が迅速だということだ。もし私たちが技術変革の先端を維持できなければ、私たちは効果的に競争できないかもしれません。技術進歩や競争相手が開発した製品は、私たちの候補製品を時代遅れにしたり、競争力を低下させたり、経済的ではないかもしれません。
任意の現在または未来の候補製品の商業成功は医師、患者、支払人、および医学界の他の人の市場に対する受け入れの程度に依存する。
私たちは製品を商業化したことがありません。私たちの候補製品のためにいかなる規制許可を得ても、私たちの候補製品の商業成功は医療界、患者と支払人が私たちの候補製品を受け入れるかどうかにある程度依存し、brは安全で経済的に効率的です。私たちが市場に発売したどの製品も医者、患者、支払人、医学界の他の人の認可を得られないかもしれない。医師は通常,患者を既存の治療法から転換することを望まず,新たな,より有効あるいはより便利な治療法が市場に参入した場合でも同様である。さらに、患者は通常、彼らが現在受けている治療brに適応し、医師が製品の交換を提案しているか、あるいは既存の治療がbrの精算が不足しているため、治療の交換を要求しない限り、患者は交換したくない。
これらの候補製品が商業販売のために許可された場合、その市場受容度は、多くの要因に依存するであろう
| ● | 代替療法と比較した潜在的な治療効果と潜在的優位性 | |
| ● | 製品承認ラベルに含まれる任意の制限または警告を含む任意の副作用の頻度および重症度; | |
| ● | 私たちの候補製品を管理する後続の要求は、任意の副作用の頻度および重症度をもたらす |
| -49- |
| ● | 相手が便利で管理しやすい | |
| ● | 対象患者群が新たな療法を試みる意欲と,医師がこれらの治療法を処方する意欲 | |
| ● | マーケティングと流通支援の実力と競争製品が市場に参入するタイミング; | |
| ● | 私たちの製品や競争相手の製品や治療法を宣伝し | |
| ● | 十分な第三者保険カバー範囲と十分な精算。 |
もし候補製品が臨床前研究と臨床試験において良好な治療効果と安全性を示していても、この製品が商業販売に使用されることが許可されていれば、市場受け入れ程度はこの製品が発売されてから知ることができる。医療界や支払者に私たちの候補製品のメリットを知ってもらうために努力しており,これには大量の資源が必要であり,決して成功しないかもしれない。このような教育市場の努力は、私たちの競争相手が販売する従来の技術よりも多くの資源を必要とするかもしれません。特に私たちの14行の詩近づいています。もしこれらの製品が十分な受容度に達していなければ、私たちは著しい製品収入が生じないかもしれないし、利益を上げることができないかもしれない。
もし私たちの候補製品の市場機会が私たちが思っているより小さいなら、私たちの製品収入は不利な影響を受ける可能性があり、私たちの業務は影響を受ける可能性があります。
私たちは現在私たちの研究と製品開発を腫瘍適応の治療と私たちの製品Fに重点を置いていますHAB候補 は固形腫瘍に対するものである。これらの疾患を有する人の数と、私たちの候補製品治療から利益を得る可能性があるこれらの疾患患者のサブセットとの理解は、推定に基づいている。これらの推定は不正確であることが証明される可能性があり,新しい研究はこれらの疾患の推定発症率や流行率を低下させる可能性がある。患者識別brは患者集団を解決する能力にも影響する。患者識別における努力が成功しなかったり,効果が期待されていなかったりすると,我々が探している機会全体を解決できない可能性がある。
新承認製品の保険カバー範囲と精算状態は確定していません。もし私たちの任意の候補製品が十分な保証範囲と精算を得ることができなかった場合、承認されれば、これらの製品を販売する能力を制限し、収入を創出する能力を低下させる可能性があります。
私たちの候補製品が市場で承認された時、彼らのコストは高くなると予想されています。政府と個人支払者の獲得性と精算範囲は大多数の患者が高価な治療費用を負担できる鍵である。私たちの候補製品の販売は、私たちの候補製品の国内および海外での販売状況に大きく依存し、私たちの候補製品の費用は、個人健康保険会社、健康維持、管理医療、薬局福祉および同様の医療保健管理組織のような個人支払者によってどの程度支払われるか、またはMedicareおよびMedicaidのような政府医療計画によって精算される。私たちは保証と精算の承認を得るのに十分なデータを提供できないかもしれない。精算が得られない場合や数量限定で精算を受けることができなければ、承認されても候補製品を商業化することに成功しない可能性がある。保険を提供しても、承認された精算金額は、十分な投資リターンを達成するために十分な価格を確立したり維持したりするのに十分ではないかもしれません。
新承認製品の保険カバー範囲や精算に関する重大な不確実性。アメリカでは、新薬の保証と精算に関する主な決定は通常医療保険と医療補助サービスセンター(CMS)によって行われ、CMSはアメリカの衛生と公衆サービス部の一つの機関であり、CMSは新薬がMedicareの下で保険と精算を受けるかどうか、及びどの程度精算するかを決定する。個人支払者はCMSに大きく従う傾向がある。CMSが我々のような新製品の保証範囲と精算がどのような決定を下すかを予測することは難しい。これらの新製品は既定のbr実践と前例がないからである。第三者支払人の保険範囲と精算は多くの要素に依存する可能性があり、 は第三者支払人の製品使用の決定を含む:(1)その医療計画下の保険福祉、(2)安全、br}は有効で医療必要性がある、(3)特定の患者への適用、(4)費用効果、および(5)試験的でも試験的でもない。米国では、第三者支払人の間に統一された製品保証と精算政策はない。そのため、保険範囲の確定過程は通常時間がかかり、高価な過程であり、各支払人にそれぞれ私たちの製品を使用する科学と臨床支持を提供する必要があるが、保険範囲と十分な精算が一貫してbrを応用するか、あるいは最初に得られることを保証することはできない。特定の製品の保険を受けても,それによる精算支払率は収益性を実現したり維持したりするのに不十分である可能性があり,あるいは患者が受け入れられないと考えられる高額な自己負担費用が必要となる可能性がある。第三者支払者は保証範囲を承認リストの特定の薬品に制限する可能性がある, 処方集とも呼ばれ,特定の適応のすべての承認されたbr薬は含まれていない可能性がある。
| -50- |
また, 第三者支払者は,候補製品を使用した後に必要な長期的な後続評価をカバーしない可能性があり,十分な補償を提供しない可能性がある.患者が私たちの候補製品を使用する可能性はあまりありません。保険を提供し、私たちの候補製品の大きなコストを支払うのに十分な費用を精算しなければなりません。私たちの候補製品は従来療法の商品コストよりも高い可能性があり、長期的な後続評価が必要かもしれないため、保険と販売率は利益を達成するのに十分ではない可能性が高い可能性がある。新たに承認された製品の保険範囲や精算に関する不確実性が大きい。第三者決済者が私たちの候補製品の保証範囲と精算についてどのように決定するか予測するのは難しいです。
また、アメリカと海外の政府と第三者支払人が制限を強化したり、医療コストを下げたりすると、brのような組織が新承認製品の保証範囲と精算レベルを制限する可能性があるため、brを保証したり、候補製品に十分な支払いを提供できない可能性があります。特殊薬品の価格設定実践におけるアメリカの立法と法執行の興味はずっと増加している。具体的には、アメリカ議会は最近いくつかの調査を行い、連邦と州立法を提出し、薬品定価の透明性を高め、連邦医療保険制度下の処方薬のコストを下げ、定価とメーカー患者計画との関係を審査し、政府計画薬品精算方法を改革することを提案した。管理型ヘルスケアの傾向、ヘルスケア組織のますます増加する影響力、コスト制御措置、追加の法的変化により、私たちのどの候補製品の販売も定価圧力に直面することが予想される。
アメリカ以外では、ある国/地域、EUを含むいくつかの加盟国は、薬品br製品のために価格と精算、つまりEUで通常言われている医薬製品を制定している。これらの国/地域は価格を制定する上で広範な自由裁量権を持っており、このような価格や精算が私たちまたはパートナーに受け入れられるかどうかは確認できません。もしこれらの管轄区域の規制機関が設定した価格や精算レベルが私たちまたは私たちの協力者に商業的な魅力がなければ、私たちまたは私たちの協力者のこれらの国/地域での販売収入と私たちの薬品の潜在的な収益力は負の影響を受ける。brはますます多くの国が積極的に行動しており、コスト削減の努力をその国営医療システムの薬品に集中させることで、巨額の赤字予算を削減しようとしている。これらの国際価格制御努力は世界のすべての地域に影響を与えているが、EUの影響が最も大きい。また、製品の販売価格が承認されてから合法的に販売することを要求する国·地域もある。多くの国/地域では、定価審査期間は、マーケティングまたは製品許可を得た後に開始される。ある国/地域で精算または定価の承認を得るためには、私たちまたは任意の協力者が臨床試験を行う必要があるかもしれません。私たちの製品のコスト効果を他の利用可能な療法と比較します。そのため、特定の国/地域での製品のマーケティング承認を得ることができますが、その後、製品の精算承認が遅れたり、価格規定の制約を受けたりする可能性があり、これは私たちの製品の商業発表を延期する可能性があり、長い時間があるかもしれません, これは、私たちがこの特定の国/地域で製品を販売することによるbr}収入に悪影響を及ぼす可能性がある。
また、米国や海外の政府や支払人が医療コストを制限または低減するための努力は、このような組織が許可された新製品の精算範囲や精算レベルを制限する可能性があるため、私たちの候補製品に十分な精算を提供できない可能性がある。専門的な薬物実践における米国の立法と法執行の関心は増加している。具体的には、米国議会は最近数回の調査を行い、薬品定価の透明性を高め、連邦医療保険下の処方薬のコストを下げ、価格設定とメーカー患者計画との関係を審査し、薬品の政府計画精算方法を改革することを提案した。br}は医療管理の傾向により、ヘルスケア組織の影響力がますます大きくなり、追加の立法が変化し、私たちの任意の候補製品を販売する際に価格設定圧力に直面することが予想される。全体的には,医療費,特に処方薬や他の治療費の下り圧力が非常に大きくなっている。そのため、新製品の参入にはますます高い壁が設けられている。もし私たちの製品が精算を受けられない場合、あるいは範囲や金額が制限されている場合、あるいは定価レベルが満足できなければ、私たちの業務は損害を受ける可能性があります。
| -51- |
FDAまたは同様の外国規制機関が、私たちの任意の候補製品の模造バージョンが発売承認 を得ることを許可する場合、またはそのような機関が、そのような製品の模倣バージョン を承認する前に、そのような製品に適切なデータ独占期間を付与しない場合、そのような製品の販売は悪影響を受ける可能性がある。
米国では、製造業者は、簡略化された生物学的ライセンス申請またはABLASを提出することによって、BLAに従ってFDAによって承認された生物学的類似バージョンの承認を求めることができる。ABLAを支援するためには,生物類似メーカーは通常,その製品が原始生物製品と類似していることを証明しなければならない。生物類似製品を市場に出すコストは、生生物製品brよりも低い可能性があり、生物類似製品を生産する会社は、これらの製品をより低い価格で提供することができる場合がある。そのため,生物類似製品を発売した後,生生物製品の売上の大部分が生物類似製品に奪われる可能性があり,生生物製品の価格が低下する可能性がある。
FDAは、生生物の任意の適用可能な非特許特定期間が満了する前に、生物類似製品のABLAの審査または承認を受け入れるか、または承認することができない。公衆衛生サービス(PHS)法案はBLAによって承認された生物に12年間の非特許専有期間を提供している。
私たちの製品は生物類似バージョンの製品からの競争に直面する可能性があり、私たちの将来の収入、収益力、キャッシュフローにマイナスの影響を与える可能性があり、これらの候補製品から投資収益を得る能力を大きく制限するかもしれない。
私たちbrは連邦と州の医療詐欺と乱用法律、虚偽声明法律、健康情報プライバシーと安全法律、その他の医療法律と法規の制約を直接または間接的に受ける可能性がある。もし私たちがこのような法律を遵守できないか、または完全に遵守できなければ、私たちは巨額の処罰に直面するかもしれない。
もし私たちの候補製品がFDAの承認を得て、アメリカでこれらの製品を商業化し始めた場合、私たちの運営は直接または間接的に私たちの当事者、顧客、および購入者によって様々な連邦と州詐欺と乱用法律法規を遵守し、連邦医療保健計画反リベート法規、br連邦民事と刑事虚偽請求法案、および医師支払い日光法案と法規を含むが、これらに限定されない。このような法律は私たちが提案した販売、マーケティング、そして教育計画などに影響を及ぼすだろう。また、私たちは連邦政府と私たちが業務を展開している州の患者プライバシー法の制約を受けるかもしれない。私たちの運営に影響を与える法律は含まれているが、これらに限定されない
| ● | リベート条例は、他の事項に加えて、個人または実体が直接または間接的に、現金または実物の形態で直接または間接的に、公開的または間接的に、公開的または隠蔽的に要求、受信、提供、提供または支払いを禁止し、個人の推薦、または購入、レンタル、注文、手配、または連邦医療計画に従って支払い可能な任意の商品、施設、物品、またはサービスを誘引または応答するために、任意の報酬(任意のリベート、賄賂またはリベートを含む)を支払うことを禁止する。例えば連邦医療保険と医療補助プログラム。“報酬”という言葉は価値のあるものを含むと広く解釈されている。個人またはエンティティは、“反リベート法規”またはその法規に違反する特定の意図を実際に知る必要がなく、 違反を実施することができる。さらに、政府は、連邦虚偽請求法案または連邦民事罰金の目的に基づいて、反リベート法規違反によって生じる物品またはサービスのクレームが虚偽または詐欺的クレームを構成することを含むと断言することができる。逆リベート法規は、医薬品製造業者とbrの処方者、購入者、および処方マネージャーとの間の配置に適用されると解釈されている。いくつかの法定例外と規制避難港はいくつかの一般的な活動を起訴から保護することができる |
| -52- |
| ● | FCAを含む連邦民事および刑事虚偽申告法および民事金銭罰法は、個人または実体に刑事および民事罰を科すことを含む。理由は、故意に連邦政府に虚偽または詐欺的な支払い請求を提出するか、または連邦政府に虚偽または詐欺的な支払い請求を提出または誘導すること、金銭または財産を連邦政府に支払うかまたは移転する虚偽または詐欺的クレームまたは義務に対して、記録材料の作成または使用を引き起こす虚偽陳述br}を含む。メーカー は、政府支払人に直接クレームを提出していなくても、FCAによって責任を負うことができ、虚偽または詐欺的クレームの提出を“招く”と考えられている場合。FCAはまた、FCA違反行為を告発し、いかなる金銭的回収も共有する“密告者”としての個人代表が連邦政府を代表して訴訟を起こすことを許可した | |
| ● | 議定書の受益者誘因条項は、他の事項に加えて、無料または公平な市場価値よりも低い任意の物品またはサービスの譲渡(限られた例外)を含むが、受益者が連邦または州政府によって計算可能な物品またはサービスを選択する特定のサプライヤーを選択することに影響を与える可能性があることを知っているか、または受益者に影響を与える可能性があることを知っているべきである | |
| ● | 1996年に連邦健康保険携帯および責任法案、またはHIPAAは、任意の医療福祉計画を詐欺の任意の医療福祉計画を意図的かつ意図的に実行または実行しようとするいかなる人も意図的かつ意図的に実行または実行しようとすることを禁止する新しい連邦刑法を作成し、または虚偽または詐欺的な言い訳、陳述または約束によって、支払者(例えば、公共または個人)にかかわらず、故意および故意に偽造することなく、任意の医療福祉計画が所有または管理または制御された任意の金銭または財産を得ることを禁止する。任意のトリックまたは手段で重大な事実を隠蔽または隠蔽するか、または医療事項に関連する医療福祉、プロジェクトまたはサービスの交付または支払いに関する任意の重大な虚偽、架空または詐欺的な陳述または陳述を行う。“反リベート法規”と同様に、個人またはエンティティは、法規または法規違反の具体的な意図を実際に知る必要がなく、違反を実施することができる | |
| ● | HIPAAは、2009年に“衛生情報技術促進経済·臨床健康法”及びそのそれぞれの実施条例改正された“HIPAA”、“br}は、特定の医療保健提供者、健康計画及び医療情報交換所(カバー実体と呼ぶ)及びそのそれぞれの商業パートナー、それを代表してサービスを提供する個人及び実体に対して要求を提出し、個人が健康情報を識別できるプライバシー、安全及び伝送に関する個人識別可能な健康情報の使用又は開示に関するものである | |
| ● | “医療·教育調整法”によって改正された“患者保護および平価医療法案”(または総称してACAと呼ばれる)によって規定される米国連邦透明性要件は、一般に“医師支払い日光法案”と呼ばれる条項を含み、この条項は、適用される医薬品、設備、バイオ製品および医療用品製造業者がMedicareに従って支払い、医療補助または児童健康保険計画(いくつかの例外を除く)に毎年CMSに報告および医師(医師、歯科医を含むと定義され、視光師,足科医と脊椎マッサージ師) と教育病院,および上記医師とその直系親族が持つ所有権と投資権益。2022年から適用されるメーカーは,医師アシスタント,看護師従事者,臨床看護師専門家,登録看護師麻酔科医,登録看護師助産師への支払いと移転価値の情報を報告することを要求される | |
| ● | 連邦政府価格報告法は、複雑な定価指標を正確かつ適時に計算し、政府プロジェクトに報告することを要求している | |
| ● | 連邦 消費者保護と不正競争法は,市場活動と消費者を損なう可能性のある活動を広く規制している。 |
| -53- |
さらに、我々は、上記の各医療法律法規の州および海外等価物に制限されており、いくつかの可能な範囲が広く、任意の支払人に適用可能なbr}が含まれている。米国の多くの州では、研究、流通、販売またはマーケティング、および非政府支払人(個人保険会社を含む)によって精算された医療項目やサービスに関するクレームを含むが、これらに限定されないが、研究、流通、販売またはマーケティング手配、および非政府支払人(個人保険会社を含む)によって精算された医療項目やサービスに関連するクレームを含む“反リベート法規および虚偽請求法案”のような法律が可決されている。また、いくつかの州は、2003年4月の総監察長事務室の薬品メーカーコンプライアンス計画ガイドラインおよび/またはアメリカの薬品研究とメーカーが医療保健専門家と相互作用するbr守則を遵守するように製薬会社に要求する法律を採択している。いくつかの州では、他のマーケティング制限を実施したり、製薬会社に州政府へのマーケティングや価格開示を要求したりしている。これらの州を遵守する必要がある要求には曖昧な があり,適用される州の法律要求を守らなければ処罰される可能性がある.最後に,健康情報のプライバシーや安全を管理する国家法や外国法もあり,その多くの法律は互いに大きく異なり,HIPAAは先制されず,コンプライアンス作業を複雑化することが多い。
これらの法律の広さと、利用可能な法定例外と安全港の規制の狭さにより、私たちのいくつかの業務活動は、1つ以上のこのような法律の挑戦を受ける可能性がある。法執行部門はますます詐欺と法の乱用に集中しており、私たちのいくつかの接近はこのような法律の挑戦を受けるかもしれない。私たちの現在と将来の第三者との業務配置と、私たちの業務が全体的に適用される医療法律法規に適合することを確保する努力 は大量のコストに関連する。もし私たちの業務が、医師や他の医療提供者との手配を含む場合、その中のいくつかは、サービスを提供するための補償として株式オプションを取得し、私たちに適用される任意のこのような法律または任意の他の政府法規に違反していることが発見された場合、私たちは、行政、民事および刑事罰、損害賠償、罰金、返還、契約損害、名声損害、利益減少および将来の収益を含むが、我々の業務の削減または再編を含む罰を受ける可能性があります。連邦および州医療計画(例えば、MedicareおよびMedicaid)、追加報告要件および/または監督(これらの法律を遵守しない疑いを解決するために企業誠実協定または同様のbr協定の制約を受けている場合)、および個人監禁を排除し、これらのいずれも、私たちの業務運営能力および財務業績に悪影響を及ぼす可能性がある。これらの法律に違反した行為は,たとえ弁護に成功しても, Br}は製薬業者が巨額の法的費用を招き,経営陣の業務運営への注意をそらす可能性がある。今後発売される製品の販売を禁止または制限または撤回することは、不利な方法で業務に大きな影響を与える可能性がある。
ヘルスケア立法改革措置や国家予算社会保障制度への制限は,我々の業務や運営結果に実質的な悪影響を及ぼす可能性がある。
支払先は,国内でも海外でも,政府や個人でも,医療コスト を制御するためのますます複雑な方法が開発されており,必ずしも我々が開発している新しい技術に特化しているわけではない。米国や一部の外国司法管轄地域では,医療システムの立法や規制に多くの変化が生じており,brは我々の製品販売収益力に影響を与える可能性がある。特にアメリカでは、ACAは2010年に公布され、他の事項以外に、それは生物製品を低コストの生物模倣薬の潜在的な競争を受けさせた;新しい方法を解決し、この方法を通じて、吸入、輸液、点滴、移植或いは注射の薬物に対して、計算メーカーがMedicaid薬品リベート計画の下で不足したリベートを増加した;大多数のメーカーがMedicaid薬品リベート計画の下で不足していた最低Medicaidリベートを増加した;Medicaid 薬品リベート計画をMedicaid管理の看護組織に登録された個人処方に拡大した;メーカー にあるブランド処方薬に新しい年費と税金を徴収することを要求した;連邦政府の比較有効性研究のプロジェクトを増加させるためにインセンティブを提供する。
公布以来、ACAのいくつかの側面は司法と国会の挑戦を受けてきたし、ACAのいくつかの側面を廃止または置換するための今回の政府の努力は最近行われてきた。また、トランプ総裁は2017年1月以降、ACAのいくつかの条項の実施を延期したり、ACAが許可しているいくつかの医療保険要求を回避したりするための2つの行政命令と他の指示に署名した。また,CMSは最近,2020年から各州により大きな柔軟性を与え,個人や小団体市場の保険会社に基準を設定する上で,ACAがこのような市場で販売されている保険計画に要求される基本的な健康福祉を緩和する可能性がある最終規則を発表した。
| -54- |
また,国会ではACAの全部または一部を廃止·置換する立法が審議されている。国会ではまだ全面的な廃止立法は成立していないが、ACAの下にある税収実施に影響を与える2つの法案が署名されて法律となっている。2017年減税·雇用法案(TCJA)には、ACAが1年の全部または一部で合格医療保険を維持できなかった個人に対して実施された税収に基づく分担責任支払いを2019年1月1日から廃止する条項が含まれており、一般に“個人強制医療保険”と呼ばれている。また、2018年1月22日、トランプ総裁は、ACAが規定する特定の費用の実施を延期し、ある高コスト雇用主が後援する保険計画に対していわゆる“キャデラック”税“br”、ある高コスト雇用主が後援する保険計画に年会費を徴収し、市場シェアに応じてある医療保険提供者に年会費を徴収すること、非免除の医療機器に対して医療機器税を徴収することを含む2018年度の支出に関する持続的な決議に署名した。また,2018年に両党予算法案(BBA)などがACAを改正し,2019年1月1日から施行され,Medicare D部分に参加する製薬メーカーが不足している販売時点割引を50%から70%に引き上げ,Medicare薬物計画の多くのカバーギャップを縮小し,通常は“ドーナツ穴”と呼ばれている。最近、 2018年7月, CMSは、ACAリスク調整計画に基づいて、あるACA合格健康計画と健康保険発行者に対してさらなる入金と支払いを可能にし、CMSがこのリスク調整を決定する方法に関する連邦地域裁判所の訴訟結果に応答する最終規則を発表した。議会はまたACAの他の要素を廃止または代替するための追加的な立法を考慮することができる。したがって、ACAのすべての影響、その内容を廃止または置換する任意の法律、および私たちの業務をめぐる任意の廃止または代替立法の政治的不確実性はまだ不明である。
また、ACAが公布されて以来、米国は他の立法改正を提案し、採択した。2011年8月には、他の事項を除いて、“2011年予算制御法案”が国会支出削減措置を制定した。赤字削減合同特別委員会の任務は,2013年から2021年までに少なくとも1.5兆ドルの赤字削減を提案することであるが,同委員会は必要な目標を達成できず,いくつかの政府計画の自動削減を触発した。これには、2013年4月に施行された各年度に提供者に支払われる医療保険総金額が2%減少することが含まれており、その後の立法改正により、BBAが含まれており、追加の国会行動がとられない限り、2027年まで有効となる。2013年1月、2012年に“米国納税者救済法”が署名されて法律となり、他の事項に加えて、病院やがん治療センターを含むいくつかの医療サービス提供者への医療保険支払いがさらに減少し、政府が医療サービス提供者に多額の支払いを取り戻す訴訟時効が3年から5年に延長された。
また、br政府は最近、薬品メーカーがその上場製品の価格設定方式の審査を強化し、これはいくつかの国会調査を招き、連邦と州立法を提出し、公布し、製品定価の透明性を高め、定価とメーカー患者計画との関係を審査し、そして 政府計画薬品精算方法を改革することを目的とした。例えば、今回の政府は薬品価格を低減し、薬品の自己負担コストを低減するための“青写真” を発表し、その中にはメーカーの競争を増加させる追加の提案が含まれており、一部の連邦医療保健計画の交渉力を増加させ、製造業者にその製品の価格 を下げるよう激励し、消費者が支払う薬品の自己負担コストを減少させる。例えば、2018年11月、CMSは、6つの保護された処方カテゴリにおける薬剤の割引を交渉し、場合によってはこれらの薬剤を排除し、Medicare Advantageが医師が管理する薬剤の階段療法のような特定の薬剤管理ツールを使用することを可能にするために、D部分下のMedicare処方薬計画により大きな価格設定透明性およびより大きなbr柔軟性を提供するための提案ルールを発表する。その多くの措置および他の提案措置は、追加の立法許可によって発効する必要があるにもかかわらず、国会とトランプ政権は、br薬品コストを制御するために新たな立法および/または行政措置を求め続けると表明している。
外国、連邦、州の各レベルはすでに立法と監督管理提案を引き続き提出することが可能であり、医療保健の獲得性を拡大し、医療保健コストを制御或いは低減することを目的としている。私たちは未来に採用される可能性のある計画を予測できない。これらの政府および他の支払者は、医療コストの制御または低減、および/または価格規制の実施に努力し続けており、以下の点に悪影響を及ぼす可能性がある
| ● | もし私たちが規制部門の承認を得て、私たちの候補製品の需要を得たら | |
| ● | 私たちは私たちの製品に公平だと思う価格を設定することができます | |
| ● | 私たちは収入を創出し、利益を達成するか、または収益性を維持する | |
| ● | 税金を払う必要があります | |
| ● | 資金の入手可能性。 |
| -55- |
連邦医療保険または他の政府計画の支払いを拒否または減少させるいかなる場合も、同様の民間支払者の支払いを拒否または減少させる可能性があり、これは、私たちの将来の収益性に悪影響を及ぼす可能性がある。
我々の は,米国で1977年に改正された“反海外腐敗法”や“反海外腐敗法”やその他の反腐敗法および輸出規制法,輸入·税関法,貿易·経済制裁法,その他我々の業務を管理する法律の制約を受けている。
我々の運営は反腐敗法に拘束されており,“海外腐敗防止法”,米国“反海外腐敗法”第18章201節に含まれる米国国内賄賂法規,“米国旅行法”および我々が業務を展開している国/地域に適用される他の反腐敗法律を含む。“贈収賄法”、“海外腐敗防止法”および他のbr法は、一般に、業務または他の業務優位性を得るために、私たちおよび私たちの従業員および仲介機関が直接または間接的に許可、承諾、不正または禁止されたお金または任意の他の価値のあるものを提供または提供することを禁止している。反収賄法によると、私たちはまた、私たちと関連のある人が賄賂犯罪を実施することを阻止できなかったために責任を負う可能性があります。私たちと私たちのビジネスパートナーは複数の司法管轄区で業務を展開しており、これらの司法管轄区域は“収賄法”や“海外腐敗防止法”に違反するリスクがある可能性があり、私たちは第三者との協力や関係に参加しており、これらの第三者の腐敗や不正活動は、たとえ私たちが明確に許可していなくても、このような活動を明確に許可していなくても、“収賄法”、“反海外腐敗法”または現地反腐敗法によって責任を負う可能性がある。さらに、将来の規制要求の性質、範囲、または効果を予測することはできません。私たちの国際業務は、これらの規制要求に支配される可能性があり、既存の法律が管理または解釈される可能性がある方式も予測できません。
私たちの国際業務を管理する他の法律と法規はまた、イギリスとアメリカ政府及びEU当局が管理する法規を含み、適用される輸出規制法規、ある国と人員に対する経済制裁と禁輸、反マネーロンダリング法、輸入と税関要求及び貨幣両替法規を含み、総称して貿易規制法と呼ばれる。
私たちが貿易統制法を含むすべての適用された反腐敗法律を遵守することを完全に効果的に確保することは保証されない。もし私たちが“反収賄法”、“海外腐敗防止法”および他の腐敗防止法または貿易統制法を遵守しなければ、私たちは刑事と民事処罰、返還と他のbr制裁と救済措置、そして法的費用を受ける可能性があり、これは私たちの業務、財務状況、運営結果、流動性に悪影響を及ぼす可能性がある。同様に、イギリス、アメリカ、または他の当局が“収賄法”、“海外腐敗防止法”、他の反腐敗法律または貿易規制法に違反する可能性があるいかなる調査も、私たちの名声、私たちの業務、経営結果、および財務状況に悪影響を及ぼす可能性がある。
もし私たちが環境、健康、安全法律法規を遵守できなかった場合、私たちは罰金や処罰を受けたり、brコストが発生したりする可能性があり、これは私たちの業務成功に重大な悪影響を及ぼす可能性がある。
私たちbrは多くの環境、健康と安全法律法規の制約を受けて、実験室の手続き及び危険材料と廃棄物の処理、使用、貯蔵、処理と処理を管理する法律法規を含む。私たちの業務は化学品と生物学的材料を含む危険で燃えやすい材料の使用に関するものだ。私たちの業務はまた危険な廃棄物製品を発生させるだろう。私たちは一般的に第三者とこのような材料と廃棄物を処理する契約を締結する。私たちはこのような材料が汚染や傷害をもたらす危険をなくすことができない。もし私たちが危険な材料を使用して汚染や損傷をもたらしたら、私たちはそれによって発生したいかなる損害にも責任を負わなければならず、どんな責任も私たちの資源の範囲を超える可能性がある。私たちはまた民事または刑事罰金に関連した巨額のコスト と処罰を生じる可能性がある。また、環境法律や法規は複雑で、変化が頻繁で、より厳しくなる傾向にあります。 私たちはこのような変化の影響を予測することができず、私たちの将来のコンプライアンスを決定することもできません。また,現在または将来の環境,健康,安全法律法規を遵守するためには,巨額のコスト が生じる可能性がある。これらの現在または未来の法律法規は、私たちの研究、開発、または生産努力を損なう可能性がある。このような法律法規を遵守しないことはまた巨額の罰金、処罰、または他の制裁につながる可能性がある。
| -56- |
我々は、危険材料の使用や他の仕事に関連する傷害による従業員への傷害によるコストおよび支出を支払うために労働者補償保険を維持しているが、この保険は潜在的なbr責任に十分な保険を提供できない可能性がある。また、現在または未来の環境、健康、安全に関する法律や法規を遵守するためには、巨額のコストが生じる可能性がある。このような現行または未来の法律法規は私たちの研究、開発、または生産努力を損なうかもしれない。これらの法律法規を遵守しないことはまた、巨額の罰金、処罰、または他の制裁または責任をもたらす可能性があり、これは、私たちの業務、財務状況、運営結果、および将来性に重大な悪影響を及ぼす可能性がある。
私たちの国際業務に関するリスク
私たちの子会社救済はアメリカ国外にあるため、私たちは国際業務に関連する経済、政治、規制、その他のリスクに直面している。
Relipmentの本社はスイスにあるため、私たちの業務はアメリカ以外の地域で業務を展開することはリスクの影響を受けます。私たちの多くのサプライヤーと臨床試験関係はアメリカ以外にあります。したがって、私たちの将来の業績は様々な要素の影響を受けるかもしれない
| ● | インフレ、特に非アメリカ経済と市場の政治的不安定を含む経済的疲弊 | |
| ● | 異なるbrと変化する製品承認法規の要求; | |
| ● | 異なる法ドメインは、そのようなドメインで動作する自由を確保、維持、または獲得する上で異なる問題が生じる可能性がある | |
| ● | 知的財産権の保護を減らすかもしれません | |
| ● | 複数の司法管轄区域の異なる、複雑かつ絶えず変化する法律、法規と裁判所システムの遵守及び各種の外国の法律、条約と法規の遵守に困難がある | |
| ● | 米国以外の規制と関税、関税、貿易障壁を変える | |
| ● | ポンド、ドル、ユーロ、通貨規制の非米国通貨レートの変化 ; | |
| ● | 貿易各国政府の保護措置、輸出入許可要求またはその他の制限行動 | |
| ● | ある非アメリカ市場では異なる精算制度と価格規制が行われています | |
| ● | 税法変更による否定的な結果 | |
| ● | 海外居住または旅行従業員の税法、就業法、移民法と労働法を遵守し、異なる司法管轄区で私たちの株式オプション計画または株式激励計画によって付与されたオプションに対する可変税収 待遇を含む | |
| ● | 労働力の労働騒乱はアメリカよりも一般的な国の不確実性である | |
| ● | 解雇ミス、差別、誤分類、または他の労働法違反のクレームまたは他の告発された行為を含む、現職または前任者またはコンサルタント単独または集団訴訟の一部として私たちに提起された行政訴訟 | |
| ● | 異なる労使関係を含む人員配置と国際業務の管理に関する困難 | |
| ● | 海外の原材料供給や製造能力に影響を与えるいかなる事件による不足 | |
| ● | 業務brは、地政学的行動(戦争やテロを含む)や自然災害(地震、台風、洪水、火災を含む)による中断。 |
| -57- |
ヨーロッパ データ収集は,個人情報の使用,処理,国境を越えた転送に関する制限的法規によって管轄されている.
EU情報システムにおける個人健康データの収集と使用はデータ保護指令の規定に管轄されており,2018年5月25日からGDPRに置き換えられている。これらの指令は,個人データに係る個人の同意,個人への情報提供,主管国家データ保護機関へのデータ処理義務の通知,個人データのセキュリティと秘密についていくつかの要求を行っている.データ保護指令とGDPRはまた,個人データをEUから米国に移行するための厳しいルールを実施している。データ保護指令,GDPRおよびEU加盟国の関連国家データ保護法の要求を遵守できなければ,罰金やその他の行政処罰を受ける可能性がある。データ保護指令はEU以外に本部を置く組織には適用できないが,GDPRはそのカバー範囲をEU住民に商品やサービスを提供するどの企業にも拡大しており,その位置にかかわらず である。この拡張はEU加盟国の任意の潜在的な臨床試験活動に統合されるだろう。GDPRは,EUに住むデータオブジェクトの健康や遺伝情報を含む“敏感な情報” の特殊な保護を含む個人データのコントローラやプロセッサに厳しい要求を加えている.GDPRは,個人がその個人情報の処理に反対する機会があることを許可し,個人情報の削除を要求する場合があり, は個人に明確な権利を提供し,個人がその権利が侵害されていると考えた場合に法的救済を求める.さらに進む, GDPRは,個人データをEUから米国や他の“十分な”プライバシー保護を提供していないと考えられる地域に移行するための厳しいルールを実施している.GDPRやEU加盟国の関連国家データ保護法の要求を守らないことは、GDPRと少しずれている可能性があり、 は世界の収入の4%までの罰金、または20,000,000,000ユーロを招く可能性があり、金額が大きい者を基準とする。GDPRを実施した結果,新たなデータ保護ルールの遵守を確保するために追加的なメカニズムを構築する必要があるかもしれない.
我々の第三者依存に関するリスク
いくつかの候補製品については、私たちは開発および商業化パートナーに依存して臨床試験を開発および行うことができ、 は規制部門の候補製品の承認を得、承認された場合、候補製品をマーケティングおよび販売する。もしこのような協力者の表現が期待に達しなければ、私たちがこれらの候補製品から将来の収入を得る潜在力は著しく低下し、私たちの業務は損害を受けるだろう。
いくつかの候補製品については,我々の開発とビジネスパートナーに依存して候補製品の臨床 試験を開発,行い,承認された後に商業化する。
私たちの現在の協力と私たちが未来に参加するどんな協力も多くのリスクに直面するだろう
| ● | 協力者(Br)は、彼らが協力の仕事や資源に適用されることを決定する上で大きな裁量権を持っている | |
| ● | 協力者 は予想通りに義務を履行していないか、または適時に責任を履行できなかったり、責任を完全に履行していない可能性がある |
| -58- |
| ● | 協力者 は、監督部門の許可を得た任意の候補製品を開発および商業化してはならない、または臨床前研究または臨床試験結果、協力者の戦略重点の変化または利用可能な資金または外部要素(例えば、買収)に基づいて、継続しないか、または開発または商業化計画を継続しないことを選択することができる | |
| ● | 協力者は臨床前研究或いは臨床試験を延期し、臨床試験に資金不足を提供し、臨床前研究或いは臨床試験を停止し、或いは候補製品を放棄し、新しい臨床試験を繰り返し或いは行うことができ、或いは新しい調合の候補製品に臨床試験を行うことを要求することができる | |
| ● | 私たち は、協力の下で開発または商業化されている候補製品に関するいくつかの情報をアクセスできないか、または開示することが制限されている可能性があり、したがって、そのような候補製品の状態を株主に通知する能力は限られている可能性がある | |
| ● | 協力者 は、協力者が競争力のある製品の方が開発に成功する可能性があると考えているか、または私たちよりも経済的に魅力的な条項で商業化できることを前提として、第三者開発と直接または間接的に我々の候補製品と競合する製品 を独立して開発または間接的に開発することができる | |
| ● | Br協力は候補製品開発および/または臨床前研究または臨床試験をbr協力の一部として行わない可能性があり、協力は成功しないかもしれない | |
| ● | 協力者と共同開発された候補製品は、私たちの協力者によって彼ら自身の候補製品や製品と競争するとみなされる可能性があり、これは、協力者が私たちの候補製品の商業化を停止することを招く可能性がある | |
| ● | 規制の承認を得て、私たちの1つまたは複数の候補製品に対してマーケティングおよび流通権限を有するパートナーは、そのような候補製品をマーケティングおよび流通するのに十分なリソースを投入しない可能性がある | |
| ● | 協力者 は、私たちの知的財産権を正確に維持または擁護できないかもしれない、または何らかの方法で私たちの固有の情報を使用して、私たちの知的財産権または固有の情報を危険にさらすか、または私たちを潜在的な訴訟に直面させる可能性がある。 |
さらに、いくつかの協力および商業化プロトコルは、このようなプロトコルを終了する権利を私たちの協力者に提供し、これらの権利は条件によって制限される可能性があり、条件によって制限されない可能性もあり、これらの権利を行使すれば、私たちの製品開発作業に悪影響を与え、新しい協力者を引き付けることを困難にする可能性がある。この場合、私たちはこのような候補製品または製品の開発および商業化の規模および努力範囲を制限する必要があるかもしれない;私たちはさらなる開発または代替戦略協力の決定に資金を提供するために追加融資を求める必要があるかもしれない;私たちはそのような候補製品または製品の印税およびマイルストーン支払いから将来の収入を得る潜在力は大幅に減少、延期または廃止されるだろうし、 これは私たちの業務および将来の成長見通しに悪影響を及ぼす可能性がある。有形および無形資産の権利 を回収し、協力終了後に候補製品や製品を推進するために必要な知的財産権は契約によって制限される可能性があり、終了後に計画を進めることができない可能性があります。
もし私たちの開発や商業化パートナーと衝突すれば、彼らは自分の利益のために行動するかもしれません。これはわが社の利益に反するかもしれません。
私たち は将来私たちの開発や商業化協力者と食い違うかもしれない。以下の1つまたは複数の理由により、第三者との連携および許可スケジュールが衝突する可能性があります
| ● | マイルストーン、特許権使用料、適用協定によって支払われるべき他のお金に関する紛争 ; | |
| ● | 知的財産権の所有権または許可範囲に分岐 ; |
| -59- |
| ● | いずれの報告義務の範囲についても分岐 ; | |
| ● | 協力者は、その開発および商業化活動の進捗状況を私たちに知らせることを望まない、またはこれらの活動の公開開示を許可すること;および | |
| ● | パートナーに関する議論、または我々の製品および候補製品の開発または商業化努力に関する議論。 |
私たちの開発と商業化パートナーとの衝突は、私たちの業務、財務状況、または運営結果、および将来の成長見通しに重大な悪影響を及ぼすかもしれません。
我々 は,独立した臨床研究者とCROを含む第三者に依存して,我々の候補製品のいくつかの臨床試験 を行って支援する。第三者が候補製品の臨床開発における義務を履行できなかったことは、候補製品が規制部門の承認を得る能力を延期または弱める可能性がある。
著者らのbrは独立した臨床研究者、学術パートナー、監督事務顧問と第三者CROを含む第三者に依存し、引き続き依存することを計画し、いくつかの場合にはこのような臨床試験を賛助し、そして監督機関と接触し、著者らが行っている臨床前と臨床プロジェクトのデータを監視し、管理することを含む。私たちの候補製品は実用価値のある臨床治療領域の広さを持っている可能性があると考えていることから、内部規制の専門知識を確立するのではなく、外部サービスプロバイダに依存し続けるつもりである。
場合によっては、このような第三者のいずれかは私たちとの契約を終了することができる。私たちは代替のbr計画を達成できないかもしれないし、商業的に合理的な条項でそうすることができないかもしれない。また、新しい契約研究機関が仕事を始めた時、自然な過渡期があった。そのため、遅延が生じる可能性があり、これは予想される臨床開発スケジュールを満たす能力に負の影響を与え、私たちの業務、財務状況、将来性を損なう可能性がある。
私たちのbrはまだ私たちのすべての臨床前研究と臨床試験が適用された 方案及び法律、法規と科学標準に従って行われることを保証する責任があり、私たちのこれらの第三者への依存は私たちの監督責任を解除しない。我々と我々の第三者請負業者およびCROは、FDA、欧州経済地域(EEA)加盟国の主管当局、および同様のbr外国規制機関によって臨床開発において実行される法規およびガイドラインであるGCP要件の遵守を要求されている。規制機関は,試験スポンサー,主要調査員,試験場を定期的に検査することでこれらのGCP要求を実行している。もし私たちが私たちの任意の学術パートナーまたはCROを十分に監視できなかった場合、または私たちまたは私たちの任意の学術パートナーまたはCROがその契約義務または義務を成功裏に履行できず、予期された最終期限内に達成できなかった場合、または彼らが得た臨床データの品質または正確性が、私たちの臨床方案または法規の要件または任意の他の理由に従わずに損害を受けた場合、私たちの臨床試験で生成された臨床データは信頼できないとみなされる可能性がある、FDA。EMAや同様の外国の規制機関は、追加の臨床試験を要求し、その後、私たちのマーケティング申請を承認することができるかもしれません。私たち、私たちの学術パートナー、あるいは私たちのCRO、あるいは私たちの臨床試験に関連するサービスを提供する第三者を監督検査した後、このような監督機関は、私たちの任意の臨床試験がGCP規定に適合していると判断することを保証することはできません。また、, われわれの臨床試験は適用されたCGMP法規により生産された製品を用いて行わなければならない。もし私たちがこれらの規定を守らなければ、私たちは臨床試験を繰り返す必要があるかもしれません。これは規制部門の承認過程を延期します。
また、brは、私たちが臨床試験を行っている第三者が私たちの従業員ではなく、私たちとこのような請負業者との合意によって私たちに提供されている救済措置に基づいて、私たちが行っている開発プロジェクトに十分な時間、スキル、資源を投入するかどうかを制御することができません。brこれらの請負業者はまた、私たちの競争相手を含む他のビジネスエンティティと関係がある可能性があり、彼らはこれらのエンティティのための臨床試験や他の薬物開発活動を行う可能性があり、これは、彼らが適切な時間を私たちの臨床プロジェクトに投入する能力を阻害する可能性があります。もしこれらの第三者が、臨床研究者を含めて、契約の履行に成功していない場合、もし私たちが予想された期限内にbrを完成したり、法規の要求や私たちが規定した方案に基づいて臨床試験を行うことができなければ、私たちは私たちの候補製品の上場承認を得ることができないかもしれません。もしこのような状況が発生したら、私たちは私たちの候補製品を商業化する努力を成功させる努力を延期することができないか、または延期するかもしれない。
| -60- |
さらに、可能な研究者が支援する試験については、これらの試験の設計または実施を制御することはなく、FDAまたはEMAは、これらの研究者が後援する試験が、試験の設計要素またはbr}の実行または安全問題または他の試験結果を含む、我々によっても第三者によって制御されても、将来の臨床試験または市場承認に十分な支援を提供しているとは思わないかもしれない。私たちは、私たち自身が調査員支援実験から提出した規制文書を含む、調査員支援実験に関するいくつかの情報br権利を提供してくれることを願っています。しかし,研究者が試験のデータを賛助する時間や報告を制御することはなく,研究者が試験を支援するデータを持つこともない。われわれbr}が研究者が後援した試験結果を確認あるいは複製できなければ,あるいは陰性結果が得られれば,さらなる臨床開発をさらに延期または阻止する可能性が高い。また,研究者や機関が候補製品の臨床開発における義務に違反している場合や,データが我々が獲得可能な第1の知識と比較して不十分であることが証明されていれば,任意の将来の臨床試験を自ら設計·実施する能力が悪影響を受ける可能性がある。さらに、FDAまたはEMAは、これらの研究者が後援する試験によって生じる臨床前、生産または臨床データの参照権の十分性、または臨床前の私たちの解釈に同意しない可能性がある, これらの研究者が協賛した試験の製造や臨床データ。そうであれば、FDAまたはEMAは、追加の臨床前、生産、または臨床データを取得して提出することを要求するかもしれない。
私たちの は、第三者に候補製品を生産することに依存しようとしており、これは、許容可能なコストで十分な数のそのような候補製品またはそのような数を得ることができないリスクを増加させ、これは、私たちの開発 または商業化努力を延期、阻害、または損害する可能性がある。
我々のbrは,我々が開発しているか,あるいは我々の開発計画で評価している候補製品を生産するための臨床や商業用品の製造施設を所有または運営していない。私たちの薬物製造における経験は限られており、資源と能力が不足しており、臨床或いは商業規模で私たちの任意の候補製品を生産することができない。私たちは第三者 に依存して私たちの候補製品を供給し、私たちの戦略は私たちの候補製品と製品のすべての製造を第三者 にアウトソーシングすることです。
候補製品の臨床試験を行うためには、これらの製品を大量に生産する必要があるかもしれない。私たちの第三者製造業者は、タイムリーに、または費用効果のある方法で、または私たちの任意の候補製品の生産能力を向上させることに成功できないかもしれません。さらに、拡張活動中および他の任意の時間に品質の問題が発生する可能性がある。例えば、私たちの候補製品の安定性に関する持続データは私たちの候補製品の有効期限を短縮し、臨床試験材料の供給不足を招き、臨床試験の遅延を招く可能性がある。これらの第三者製造業者が私たちの候補製品の生産 を十分な品質および数量で成功的に拡大することができない場合、候補製品の開発、試験、および臨床試験は延期または不可能になる可能性があり、候補製品の規制承認または商業発表が遅延または入手できない可能性があり、これは私たちの業務を深刻に損なう可能性がある。
我々は新たな第三者製造商会を用いて生産遅延や候補製品供給不足のリスクを増加させているが,我々はこれらのメーカーに製造技術を譲渡し,彼らが我々の候補製品を製造した経験を得ているからである.たとえ 第三者メーカーが私たちの候補製品を製造する上で豊富な経験を得ても、あるいは製造プロセスの最適化に成功したと考えても、そのメーカーが私たちの候補製品をタイムリーまたは時間とともに連続的に生産する保証はない、あるいは全く保証されない。
もし私たちが第三者が使用する製造プロセスを変更する必要があれば、私たち は遅延するかもしれない。また、承認された製造プロセス を変更する場合、FDAや同様の外国機関が新しい製造プロセスが使用できる前に検討する必要がある場合、延期される可能性があります。
| -61- |
我々 はアウトソーシングモデルを用いて我々の候補製品を生産し,許可された薬品契約開発·製造組織と良好な製造規範やGMPを締結した。臨床用品や非臨床用品および充填剤サービスを提供するためにいくつかの第三者サプライヤーを招聘しているが,現在第三者メーカーと長期商業用品の合意は締結されていない。将来、私たちは第三者製造業者と私たちが開発した任意の候補製品の商業供給について合意できないかもしれないし、受け入れ可能な条項でそうすることができないかもしれない。たとえ第三者製造業者と 手配を確立し、維持することができても、第三者メーカーに依存することはリスクをもたらす
| ● | 第三者に依存して製造プロセス開発、コンプライアンス、品質保証を行う | |
| ● | サードパーティの能力および進捗制限による供給可用性制限 ; | |
| ● | 私たちがコントロールできない要素のため、第三者は製造協定に違反する可能性がある | |
| ● | 私たちにとってコストが高い場合、または不便な場合、 第三者は製造プロトコルを終了または更新しない可能性がある。 |
第三者 メーカーは、cGMP要求や米国以外の同様の規制要求を遵守できない可能性がある。私たちが適用要件を遵守できなかったか、または第三者メーカーが関連要求を遵守できなかったことは、罰金、禁止、民事処罰、遅延、承認の一時停止または撤回、許可証の取り消し、候補製品または製品の差し押さえまたはリコール、運営制限、および/または刑事起訴を引き起こす可能性があり、いずれも私たちの候補製品の供給に重大かつ不利な影響を与える可能性があります。さらに、SON-080を含むいくつかの候補製品を開発する予定であり、毒素 または特定の許可および許可を有する専門施設でしか生産できない他の物質を使用して、私たちまたは私たちの製造業者がこのような許可および許可を維持することを保証することはできない。これらの専門化要件はまた、私たちが招聘することができる潜在的な製造業者の数を制限して、私たちの候補製品を生産し、代替メーカーに移行する任意の努力を破壊することができます。
私たちの将来の候補製品と私たちが開発する可能性のあるどの製品も他の候補製品や製品と生産施設を競争する可能性があります。CGMPの要求に応じて運営し、私たちのために製品を生産することができるメーカーの数は限られています。
もし、私たちの臨床前試験および臨床試験に任意の材料または製品を提供する第三者が何らかの理由で任意の材料または製品の提供を停止した場合、交換サプライヤーまたは製造業者の資格を決定および決定する際に、これらの試験および試験の推進を遅延させる可能性が高く、私たちに有利な条項で代替供給を得ることができない可能性がある。また、十分な候補製品やこれらの候補製品を製造するための物質を得ることができなければ、候補製品を開発し、効率的に競争することはより難しくなる。
私たちの現在と予想されている未来の他の候補製品生産への依存は、私たちの将来の利益率と、私たちが候補製品を開発し、市場の承認を得た任意の製品を商業化する能力に悪影響を及ぼすかもしれない。
私たちの第三者への依存は、私たちのビジネス秘密を共有することを要求し、これは、競争相手がこれらの秘密を発見したり、私たちのビジネス秘密が盗用されたり、漏洩したりする可能性を増加させる。
私たちは第三者に依存して私たちの候補製品を生産し、様々な組織や学術機関と協力して私たちの候補製品を開発しているので、私たちは時々彼らと商業機密を共有しなければならない。私たちは、独自の情報の研究または開示を開始する前に、私たちの協力者、コンサルタント、従業員、およびコンサルタントと秘密保護協定、材料譲渡協定、共同研究協定、コンサルティングプロトコル、または他の同様の合意を締結することによって、私たちのノウハウを保護することを求めています。これらの合意は、一般に、第三者が商業秘密のような私たちの機密情報を使用または開示する権利を制限する。
| -62- |
第三者と協力する際に契約条項が採用されているにもかかわらず、商業秘密および他の機密情報を共有する必要があるbrは、そのような商業秘密が我々の競争相手に知られ、意図せずに他の人の技術に組み込まれるか、またはこれらの合意に違反した場合に開示または使用されるリスクを増加させる。私たちの独自の地位が私たちのノウハウおよび商業秘密にある程度基づいていることを考慮すると、競争相手は、私たちの商業秘密または他の許可されていない使用または開示が私たちの競争地位を損なうことを発見し、私たちのビジネスに実質的な悪影響を及ぼす可能性がある。
さらに、これらの合意は、通常、私たちの協力者、コンサルタント、従業員、およびコンサルタントが、私たちのビジネス秘密に関連する可能性のあるデータを発表することを制限します。私たちの学術協力者は通常、事前に私たちに通知して、指定された時間を延期して、協力によって生じる知的財産権を保護するために、データを発表する権利があります。他の場合、配布権は私たちが独占的に制御し、場合によっては他の当事者とこれらの権利を共有する可能性があります。 私たちは私たちのビジネス秘密を保護しようと努力していますが、私たちの競争相手はこれらの合意に違反して、独立して情報を開発したり、発表したり(私たちの商業秘密を含む)ことによって、私たちのビジネス秘密を発見することができます。公開時には独占権や他の保護された権利はありません。競争相手は私たちのビジネス機密が私たちの競争地位 を壊し、私たちの業務に悪影響を及ぼすことを発見した。
私たちの知的財産権に関するリスク
もし私たちが私たちの製品や候補製品のために特許や他の知的財産権保護を獲得して維持することができない場合、あるいは が獲得した特許や他の知的財産権保護の範囲が広くなければ、私たちの競争相手は私たちと似ているまたは同じ製品を開発して商業化する可能性があり、私たちの製品と候補製品を商業化することに成功する能力は不利な影響を受ける可能性がある。
私たちの効果的な競争の能力は、技術と製造プロセスの独自の性質を維持する能力にある程度依存するだろう。私たちは研究、製造、その他の技術、特許、商業秘密、許可協定、契約条項によって私たちの知的財産権を確立し、私たちの製品と候補製品を保護します。しかし、このような法的手段は限られた保護しか提供できず、私たちの権利を十分に保護できないかもしれない。2022年12月14日現在、我々の知的財産権の組み合わせは、米国での発行済み特許1件、日本でのライセンス決定1件、および10件の5007特許シリーズ中のPCT出願 を含む14件の処理すべき出願および特許を含み、配合、製造プロセス、および使用方法 に関する4つの係属中の仮出願が含まれている。
場合によっては、適切と考えられる場合には、現在および将来の製品および私たちの業務に重要な候補製品に関連する特許出願を、少なくとも場合によっては、米国以外の1つまたは複数の国/地域に提出することによって、米国に特許出願を提出することによって、私たちの独自の地位を保護することを継続して求めるつもりである。しかしながら、現在出願されている特許 が特許として発行されるかどうか、またはそれによって生成された任意の特許の権利要件が競争優位性を提供するかどうか、または現在または未来の製品および候補製品に関連する特許出願 を将来的に成功的に出願できるかどうかを予測することはできない。しかも、特許出願と承認過程は高価で時間がかかる。私たちはすべての必要または望ましい特許出願を合理的な費用やタイムリーに提出して起訴することができないかもしれない。さらに、私たちまたは任意の未来のパートナー、協力者、または許可者は、特許保護を得る前に、開発および商業化活動における発明の出願可能な特許の態様を決定することができない可能性がある。したがって、私たちは追加の特許保護を求める予想される潜在的な機会 を逃すかもしれない。特許出願の準備または提出中には、 または将来的に生じる可能性があり、例えば、適切な優先権請求、リスト、特許請求の範囲、または特許期限調整要求に関して、形態的な欠陥が存在する可能性がある。もし私たちがそのような特許および他の知的財産権を確立、維持、または保護できなかった場合、そのような権利は減少またはキャンセルされる可能性がある。私たちの特許または特許出願が形式、準備、起訴または実行において重大な欠陥がある場合、 そのような特許は無効および/または実行不可能である可能性があり、そのような出願は決して有効ではないかもしれない, 実行可能な特許。
私たちの特許および特許出願が疑問視されなくても、もしそれらが発行された場合、私たちの特許および特許出願は私たちに意味のある保護を提供しないかもしれないし、または非侵害的に類似または代替技術または療法を開発することによって、競争相手が私たちの特許主張を迂回することによって設計されることを阻止するかもしれない。例えば、第三者は、私たちの1つまたは複数の候補製品と同様の利益を提供する競争療法を開発することができるが、これは私たちの特許保護範囲を超えている。候補製品の保有または出願の特許および特許出願に提供される特許保護が十分に広くなく、このような競争を阻害するのに不十分であれば、候補製品の商業化に成功する能力は負の影響を受ける可能性がある。
| -63- |
“業務”というタイトルで議論されているように、我々の国際特許出願番号 PCT/US 2018/00085のPCT特許出願が受信した出願出願日は2018年2月20日であり、これは、米国仮特許出願米国62/459,975号及び米国62/459,981号の提出から1周年後の4日間であり、PCT受信局のコンピュータ問題により、PCT特許出願は優先的な利益を要求する。PCTは、米国 仮特許出願(U.S.62/459,975およびU.S.62/459,981)の出願日に優先権を回復したが、本PCT特許出願から提出された国家段階特許出願 のいくつかの国は、カナダおよび中国を含むこの回復を受け入れず、回復手順 はブラジルで待機している。優先度が回復していない場合、従来技術は、これらの特許出願に適用される可能性があり、そうでなければ、これらの特許出願は、PCT/US 2018/00085出願の他の特許出願に適用されない可能性がある。これは、ブラジル、カナダ、中国で出願された特許出願の範囲または広さに影響を与える可能性があり、またはこれらの国/地域で特許を取得できない可能性がある。
他の 側は、我々の方法に関連しているか、または競争力を有する可能性のある技術を開発または開発しており、特許出願 が提出されているか、または提出されている可能性があり、我々の特許出願と重複または衝突する権利要件を取得しているか、または衝突している可能性があり、彼らの多くは、より多くのリソースを有し、競合技術に多くの投資を行っており、彼らは、 と同じ成分、配合または方法、または私たちの特許地位を支配する可能性のある主題を要求している。しかも、外国の法律はアメリカの法律のように私たちの権利を保護しないかもしれない。したがって、私たち が将来獲得する可能性のあるいかなる特許も、他の人が私たちの製品や候補製品と類似した製品 を商業化することを阻止するのに十分ではなく、十分かつ持続的な特許保護を提供してくれないかもしれない。
バイオテクノロジーと製薬会社の特許地位は一般的に非常に不確実だ。これまで、米国または多くの外国司法管轄区域では、バイオテクノロジーおよび薬物特許で許容される権利要件の広さに関する一致した政策は出現していない。また、薬物化合物特許権の決定は、一般に複雑な法律および事実の問題に関連しており、これはここ数年、多くの訴訟のテーマとなっている。そのため,我々の特許権の発行,範囲,有効性,実行可能性,商業価値は大きな不確実性を持っている.私たちの競争相手はまた彼ら自身の製品を販売することを許可して、私たちの製品と似ているか、あるいは他の面で私たちの製品と競争することを求めるかもしれません。代替的に、私たちの競争相手は、FDAにANDAを提出することによって、私たちの特許が無効であるか、強制的に実行できないか、または侵害されていないと主張して、任意の承認された製品の模造バージョンの販売を求めることができる。この場合、私たちは、訴訟を提起することによって特許侵害を告発することを含む、私たちの特許を擁護または維持する必要があるかもしれない。上記の任意のタイプの訴訟では、裁判所または他の管轄権を有する機関は、私たちの特許が無効であるか、または強制的に実行できないことを発見するか、または私たちの競争相手が非侵害的に競争していることを発見する可能性がある。したがって、効果的かつ強制的に実行可能な特許を有していても、これらの特許は、私たちのビジネス目標を達成するのに十分な競合製品またはプロセスを保護することができない可能性がある。
将来的には、私たちの1つまたは複数の製品および候補製品は、第三者から内部許可を得ることができる。したがって、場合によっては、潜在的な特許保護の利用可能性および範囲は、特許出願がいつ提出されるか、または特許出願を提出するかどうかに関する決定のような、我々の許可者または発明者の前の決定に基づいて制限される。私たちはいかなる理由でもこのような知的財産権を獲得、維持、強制または保護することができず、第三者、特に開発、製造、流通能力が確立された成熟し、資金がより十分な競争相手、それと競争する製品を生産すること、または私たちが商業的に実行可能な基礎の上で私たちの製品および候補製品を開発、製造、マーケティングする能力に影響を与える可能性があり、これは私たちの業務に重大な悪影響を及ぼす可能性がある。
特許保護に加えて、商業秘密、ノウハウ、および他の保護が困難な非特許技術にも大きく依存することが予想される。私たちは、サプライヤー、従業員、コンサルタント、および他の固有情報にアクセスできる可能性のある人と秘密保護協定を締結することによって、このような保護を求めることができるが、これらの合意が違反されないこと、任意の違反救済措置があること、または私たちのビジネス秘密、ノウハウ、および他の非特許ノウハウが、私たちの競争相手に知られているか、または独立して開発されないことを保証することはできない。もし私たちが知的財産権の保護に成功しなければ、私たちの製品の販売は影響を受ける可能性があり、私たちの創造能力は深刻な影響を受ける可能性がある。
| -64- |
私たちの製品および候補製品をカバーする発行された特許が法廷または行政手続きで疑問視されれば、無効または実行不可能と認定される可能性がある。私たちは法廷で私たちの商業秘密を保護できないかもしれない。
もし私たちが第三者に対して私たちの製品または候補製品をカバーする特許を強制的に執行するために第三者に対して法的訴訟を起こした場合、 にこのような特許問題が発生した場合、被告は私たちの製品または候補製品をカバーする特許を無効または強制的に実行できないと反訴することができる。 米国の特許訴訟では、被告が無効または強制執行できないと主張する反訴が一般的である。有効性を疑問視する理由は、新規性の欠如、明らかな、書面での記述、または有効化されていないことを含む、いくつかの法定要件のいずれかを満たしていないと言われている可能性がある。主張を実行できない理由は,特許起訴に関係する人が起訴期間中に米国特許商標局に特許の特許性に関する情報を隠したり,誤った声明をしたりしたからかもしれない. 第三者は米国や海外の行政機関に類似したクレームを出すことができ, 訴訟の背景以外でもそうである.このようなメカニズムには、再審査、認可後の審査、当事者間審査、および外国の管轄区域で行われる同等の手続きが含まれています。上記のいずれの手続きでも不利な裁決を下すことは、私たちの製品または候補製品をカバーしないように、私たちの特許が撤回、キャンセル、または修正される可能性があります。法的に無効と強制執行できないと断言した後の結果は予測できない。例えば、有効性の問題については、私たちは特許審査員と私たちが起訴中に知らなかった既存技術を無効にしていないと判断できない。もし被告または第三者が法的に無効または強制執行できないと主張すれば、私たちは少なくとも部分的、さらにはすべてを失うかもしれない, 私たちの1つ以上の製品と候補製品の特許保護。このような特許保護の喪失は私たちの業務に実質的な悪影響を及ぼすかもしれない。
しかも、私たちのビジネス秘密は競争相手に知られたり独立して発見されるかもしれない。競争相手や他の第三者 は、私たちの製品と候補製品を購入する可能性があり、私たちの開発作業から得られた競争優位性の一部または全部を複製し、故意に私たちの知的財産権を侵害、あるいは他の方法で私たちの知的財産権を侵害し、私たちの保護された技術をめぐって設計したり、彼ら自身の私たちの知的財産権に属さない競争技術を開発しようとしています。もし私たちの任意の商業秘密が競争相手または他の第三者によって合法的に取得または独立して開発された場合、私たちは彼らまたは彼らが情報を伝達する人がその技術や情報を使用して私たちと競争することを阻止する権利がないだろう。もし私たちのビジネス秘密が十分に保護されていない場合、または競争相手に対する優位性を提供するのに十分でなければ、私たちの競争地位は不利な影響を受ける可能性があり、私たちの業務も影響を受ける可能性がある。また,我々のビジネス秘密を保護するための措置が不十分であると考えられれば,第三者に対して我々のビジネス秘密を盗用する十分な追跡権がない可能性がある.
私たち は特許や他の知的財産権の発明権または所有権のクレームを受ける可能性がある。
私たちのbrは、私たちが所有または将来所有または許可する可能性のある特許および知的財産権の所有権に対する元従業員、協力者、または他の第三者からクレームを受ける可能性がある。私たちの政策は、私たちが知的財産権開発に参加する可能性のある従業員と請負業者に、このような知的財産権を譲渡する合意を実行することを要求することであるが、実際に私たちが自分の知的財産権を開発しているすべての側とそのような合意を実行できない可能性があり、または このような譲渡は自動的に実行されないか、または違反される可能性がある。例えば、私たちの製品や候補製品の開発に参加している従業員、コンサルタント、あるいは他の人の義務衝突により、私たちは所有権紛争の影響を受ける可能性があります。在庫または所有権を疑問視する任意のクレームを弁護する必要があるかもしれない。もし私たちがこのようなクレームを弁護できなかった場合、私たちはお金の損害賠償を支払う必要があり、知的財産権の独占的な所有権や使用権のような貴重な知的財産権を失う可能性があり、これは私たちの業務、運営結果、および財務状況に悪影響を及ぼす可能性がある。
特許保護の獲得と維持は、政府特許機関が提出した様々なプログラム、書類提出、費用支払い、および他の要求 を遵守することに依存し、これらの要求に適合しなければ、私たちの特許保護は減少またはキャンセルされる可能性がある。
定期 特許と出願の維持費,継続費,年会費,各種他の政府費用は,特許や出願の有効期間内にいくつかの段階でUSPTOや米国以外の各種政府特許機関に支払う必要がある。米国特許商標局および各種非政府特許機関は、特許出願中および特許発行後に、いくつかのプログラム、文書、費用支払い、および他の同様の条項を遵守することを要求する。Brの場合、規定を遵守しないことにより、特許または特許出願が放棄または失効され、関連する管轄区域の特許権の一部または全部が失われる可能性がある。私たちが将来締結した1つまたは複数のライセンスの条項は、製品の組み合わせにおける特許を維持または起訴する能力を提供してくれないかもしれませんので、メンテナンスまたは起訴するために第三者に依存しなければなりません。
| -65- |
もし私たちが私たちの製品や候補製品のために特許期間の延長とデータ独占権を得なければ、私たちの業務は深刻な損害を受ける可能性がある。
特許 の寿命は限られている.米国では、すべての維持費が適時に支払われる場合、特許の自然有効期限は、通常、米国で最初の非臨時出願日から20年である。様々な延期があるかもしれないが、特許の有効期限とその提供される保護は限られている。私たちの候補製品をカバーする特許を取得しても、候補製品の特許有効期限が切れたら、競争製品からの競争を受け入れるかもしれません。新製品候補製品の開発、テスト、および規制審査に要する時間を考慮すると、これらの候補製品を保護する特許は、これらの候補製品の商業化前または直後に満了する可能性がある。したがって、私たちの特許の組み合わせは、他の会社が私たちと似ているか同じ製品を商業化することを排除するのに十分な権利を提供できないかもしれない。
将来的に、私たちが現在または未来の候補製品のうちの1つをカバーする発行された特許を取得した場合、そのような候補製品の任意の上場承認の時間、持続時間、およびFDAによる詳細に応じて、この特許は、1984年の“医薬品価格競争および特許期限回復法”または“ハッジ·ワックスマン修正案”に従って限られた特許期間延長を受ける資格がある可能性がある。“ハッジ·ワックスマン修正案”は、FDA規制審査中に失われた特許期間の補償として、特許延長期間を最大5年とすることを許可する。特許期間の延長 1つの特許の残り期間は、製品が承認された日から14年を超えてはならず、1つの特許 しか延長できず、承認された薬物、その使用方法、または製造方法に関する権利要件しか延長できない。 1つの特許は1回しか延長できず、単一の承認に基づく製品のみである。しかし、例えば、製品候補製品の承認前に付与された特許を得ることができなかったこと、br}テスト段階または監督審査中に職務調査を行うことができなかったこと、適用された最終期限内に出願を行うことができなかったこと、関連特許が満了する前に出願することができなかったこと、または適用要件を満たすことができなかったため、延期を得ることができない可能性がある。第三者によって付与された特許は、 特許期間の延長に適用されない可能性がある。また、特許保護の適用期間や範囲は、私たちが要求しているものよりも短い可能性があります。 もし私たちが特許期限の延長を得ることができない場合、あるいはこのような延長の期限が私たちが要求しているものよりも短い場合、私たちの競争相手は私たちの特許が満期になった後に競争製品のbrの承認を受ける可能性があり、私たちの収入は大幅に減少する可能性があります。
米国や他の管轄区特許法の変更 は特許の全体的な価値を低下させ,製品や候補製品を保護する能力を弱める可能性がある。
米国または他の管轄区域の特許法または特許法解釈の変更 は、特許出願をめぐる起訴および発行された特許の実施または保護の不確実性 およびコストを増加させる可能性がある。2011年9月16日、“ライシー·スミス米国発明法”または“ライシー·スミス法案”が法律に署名された。実施時、“ライシー·スミス法案”には、米国特許法のいくつかの重大な改正が含まれており、これらの改正は、特許権がどのように起訴され、強制執行され、保護されるかに影響を与える。特に、“Leahy-Smith Act”は、米国を“先行発明”制度から“先行出願”制度に変更する条項をさらに含み、第三者が特許訴訟中に米国特許商標局に以前の技術を提出することを許可し、米国特許商標局によって管理されているライセンス後訴訟手続が特許有効性を攻撃することを規定する追加プログラムを含む。先行出願制度の下で、特許可能性の他の要件 を満たすと仮定すると、最初に特許出願を提出した発明者は、通常、1つの発明の特許を取得する権利があり、それ以前に別の発明者がなされたか否かにかかわらず、別の発明者が発明を行う権利がある。米国特許商標局は、“ライシー·スミス法案”の管理を管理する新しい法規やプログラムを制定し、“ライシー·スミス法案”に関連する特許法の多くの実質的な改正、特に最初の提出条項の改正は、2013年3月16日に施行された。Leahy-Smith法案が我々の業務運営にどのような影響を与えるかは不明である(もしあれば)。しかし、Leahy-Smith法案およびその実施は、私たちの特許出願をめぐる起訴および私たちが発行した特許の実施または保護の不確実性およびコストを増加させる可能性があり、これらのすべては、私たちの業務に実質的な悪影響を及ぼす可能性がある。
また,生物製品や薬品の開発と商業化における会社の特許地位は特に は不確定である。米国連邦巡回控訴裁判所および米国最高裁判所の最近の裁決は、場合によっては入手可能な特許保護範囲を縮小し、場合によっては特許所有者の権利を弱める。この一連の事件は特許取得後の有効性と実行可能性の面で不確実性をもたらしている。米国議会、連邦裁判所、USPTOの将来の行動 によると、特許を管理する法律や法規は予測不可能な方法で変化する可能性があり、これは、私たちの既存の特許の組み合わせや、私たちの将来の知的財産権の保護と実行能力に重大な悪影響を及ぼす可能性がある。
| -66- |
私たちの1つ以上の製品と候補製品のために特許保護を求める努力は、上記の決定、他の案件の裁決、またはUSPTOが発表した指導やプログラム変更の負の影響を受けないことを保証することはできません。歴史と未来の事件における裁判所の裁決が生命科学会社がその製品に関連する特許を将来獲得または実行する能力にどのような影響を与える可能性があるかを完全に予測することはできない。これらの裁決,USPTOが発表したガイドラインや他の案件の裁決,あるいはUSPTO指導意見やプログラムの変化は,我々の既存の特許権および我々の将来の知的財産権を保護·実行する能力に重大な悪影響を及ぼす可能性がある.
私たち は世界的に私たちの知的財産権を保護できないかもしれない。
世界のすべての国/地域で製品や候補製品の申請、起訴、維持、弁護、強制特許の執行費用は目を引くほど高く、米国以外のいくつかの国/地域での知的財産権は米国の知的財産権ほど広くないかもしれない。特許性に対する要求はある国で異なる可能性があり,特に発展途上国では, したがって,我々が確かに特許保護を求めている国であっても,どの特許も我々の製品をカバーする声明を発表する保証はない.将来のいかなるライセンス契約に基づいてアメリカ国内または海外で特許権を取得または維持することは保証されません。また、一部の国の法律は知的財産権の保護の程度はアメリカの連邦法律や州法律に及ばない。したがって,米国以外のすべての国/地域で第三者が我々の発明を実施することを阻止することはできない可能性があり,特許保護を求めている司法管轄区においても,第三者が米国や他の管轄区で我々の発明を用いて製造した製品を販売または輸入することを阻止することはできない.競争相手は私たちが特許保護を申請していない管轄区域で私たちの技術を使用して彼ら自身の製品を開発し、さらに他の侵害製品を私たちが特許保護を持っている地域に輸出することができるが、法執行力はアメリカに及ばない。これらの製品 は私たちの製品および候補製品と競争する可能性があり、私たちの特許または他の知的財産権はそれらの競争を効果的にまたは阻止するのに十分ではないかもしれない。
多くの会社は外国の管轄区域の知的財産権の保護と保護に深刻な問題に直面している。特定の国の法律制度、特にある発展途上国の法律制度は、特許、商業秘密、および他の知的財産権保護の実施、特にバイオテクノロジーおよび医薬製品に関する保護を支持しておらず、これは、私たちの特許またはマーケティング競争相手製品が私たちの独占権を侵害することを全体的に阻止することを困難にする可能性がある。例えば,多くの外国国に強制許可法があり,これらの法律により,特許所有者は第三者 に許可を付与しなければならない。外国の管轄区域で私たちの特許権の訴訟を実行することは、取得しても、巨額のコストを招き、私たちの努力と注意を私たちの業務の他の側面から移転させる可能性があり、私たちの特許は無効または狭義に解釈されるリスクに直面する可能性があり、私たちの特許出願は発表できない可能性があり、第三者が私たちにクレームを提起する可能性がある。私たちは私たちが始めたどんな訴訟でも勝てないかもしれないし、損害賠償や他の救済措置があれば、商業的な意味がないかもしれない。私たちは主要市場における私たちの製品の知的財産権を保護するつもりですが、私たちが製品を販売したいすべての司法管轄区域で同様の努力を展開したり維持したりできることを保証することはできません。したがって、私たちが世界的に知的財産権を実行する努力は、私たちが開発した知的財産権から顕著なビジネスメリットを得るのに十分ではないかもしれません。
もし私たちが第三者の知的財産権侵害で起訴された場合、このような訴訟は費用が高く時間がかかる可能性があり、brは私たちの候補製品の開発または商業化を阻止または延期する可能性がある。
当社のビジネス成功は、第三者の知的財産権および他の独自の権利を侵害することなく、当社の候補製品を開発、製造、マーケティング、販売する能力にある程度依存しています。第三者は、米国および非米国から発行された特許、ならびに化合物、化合物の製造方法および/または我々が開発している候補製品の疾患適応を治療するための使用方法に関する係属中の特許出願を有する可能性がある。もし任意の第三者特許または特許出願が私たちの候補製品またはその使用または製造方法をカバーしていることが発見された場合、私たちと私たちの協力者または分が許可されていない場合、計画通りに私たちの候補製品を自由に生産または販売することができない可能性があり、許可は商業的に合理的な条項で獲得できない可能性があり、 または全く得られないかもしれない。この場合、私たちはまた私たちの協力者または分譲許可者に賠償を要求されるかもしれない。
| -67- |
バイオテクノロジーや製薬業界には大量の知的財産権訴訟があり、私たちは、私たちの製品知的財産権に関連する訴訟や他の対抗訴訟の当事者になったり、妨害や米国特許商標局の許可後訴訟を含むbr候補製品を脅かされたりする可能性がある。我々の候補製品の組成、使用または製造に関連する材料、調製、製造方法または処理方法の第三者特許または特許出願 が存在する可能性がある。私たちは、関連特許の識別、特許請求の範囲、または関連特許の満了を含むが、関連特許の検索または分析を含む保証はなく、完全または完全であり、私たちの候補製品とのいかなる司法管轄区域での商業化または必要なbrの米国および海外でのすべての特許および保留出願を識別したかを決定することもできない。特許出願が発行されるまでに数年かかる可能性があるため、現在処理されている特許出願が存在する可能性があり、これらの出願は、私たちの候補製品が発行された特許を侵害していると告発される可能性がある。さらに、第三者は将来的に特許を取得し、私たちの技術を使用してこれらの特許を侵害したと主張する可能性がある。したがって, 第三者は現在存在または将来出現する知的財産権に基づいて侵害請求を行う可能性がある.知的財産権訴訟の結果は不確定要因の影響を受け,これらの不確定要因は事前に十分に定量化できない。製薬とバイオテクノロジー産業は多くの特許を生み出しており、私たちを含む産業参加者はいつもこれを知っているわけではないかもしれない, これらの特許は様々な種類の製品または使用または製造方法をカバーする。特許提供の保護範囲は裁判所によって解釈され、解釈はいつも一致しているわけではない。もし私たちが特許侵害で起訴された場合、私たちの候補製品、製品、または方法が関連特許を侵害していない特許主張{br]、または特許主張が無効または実行不可能であることを証明する必要があり、私たちはそれをできないかもしれない。無効性を証明することは難しい。例えば,米国では が無効性を証明するためには,発行された特許が有する有効性の推定を覆すために,明確かつ納得できる証拠を提示する必要がある.私たちがこれらの訴訟で勝訴しても、私たちは巨額のコストを生む可能性があり、私たちの経営陣と科学者の時間と注意力はこれらの訴訟に移される可能性があり、これは私たちの業務と経営業績を深刻に損なう可能性があります。しかも、私たちはこのような行動を円満に達成するための十分な資源がないかもしれない。
もし私たちが第三者の知的財産権を侵害していることが発見された場合、裁判所の命令を含む開発、製造、または商業化侵害候補製品または製品の開発停止、製造または商業化を余儀なくされる可能性がある。代替的に、権利侵害技術を使用して、権利侵害製品の候補製品または製品の開発、製造、または販売を継続するために、このような第三者の許可を得る必要があるかもしれない。
しかし、私たちは商業的に合理的な条項や必要なライセンスを得ることができないかもしれません。たとえ私たちが許可を得ることができても、それは非排他的で、私たちの競争相手が私たちに許可された同じ技術にアクセスできるようにすることができ、しかも、それは商業市場での私たちの成功した競争の能力を阻害または破壊する条項を含むことができる。さらに、もし私たちが故意に特許を侵害したことが発見された場合、私たちは3倍の損害賠償と弁護士費を含む金銭損害賠償責任を負うことになるかもしれない。権利侵害発見は、候補製品を商業化したり、一部の業務を停止させたりすることを阻止する可能性があり、これは私たちの業務を損なう可能性があります。第三者の機密情報や商業秘密を盗用した疑いは、私たちの業務に類似した負の影響を与える可能性があります。
私たち は第三者からクレームを受ける可能性があり、私たちの従業員や私たちが彼らの知的財産権を流用していると主張したり、私たち自身の知的財産権を持っていることを要求したりします。
私たちの多くの現職および元従業員は、私たちの上級管理職を含み、以前は競争相手または潜在的な競争相手である可能性のある会社を含む大学や他のバイオテクノロジーや製薬会社に雇われていた。一部の従業員は、このような以前の仕事に関連する独自の権利、秘密およびスポーツ禁止協定、または同様の合意に支配されている可能性がある。私たちの従業員が私たちのために働いているときに他人の固有の情報またはノウハウを使用しないことを保証するために努力しているが、私たちの従業員は、商業秘密または他の固有の情報を含む任意の第三者の知的財産権を使用または開示しているという疑惑を受ける可能性がある。このようなクレームを弁護するために訴訟を提起する必要があるかもしれない。もし私たちがこのようなクレームを弁護できなかった場合、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権や人員を失ったり、損害を受けたりする可能性がある。このような知的財産権 は第三者に付与することができ、私たちの技術または製品を商業化するためには、第三者から許可を得る必要があるかもしれません。そのような許可は商業的に合理的な条項や根本的に存在しないかもしれない。私たちがこのようなクレームを弁護することに成功しても、訴訟は巨額のコストを招き、経営陣の注意を分散させる可能性がある。
| -68- |
また、私たちは通常、知的財産権開発に参加する可能性のある従業員、コンサルタント、および請負業者に、このような知的財産権を譲渡する合意を実行することを要求しているが、私たちは、私たちがこのような知的財産権を実際に開発することに成功しておらず、私たち自身の知的財産権のそれぞれとこのような合意を実行することに成功することができず、このような知的財産権の所有権に関連するクレームを私たちにまたは提示する可能性がある。もし私たちがこのようなクレームを起訴したり、弁護することができなければ、金銭的損害賠償を支払う以外に、私たちは貴重な知的財産権を失う可能性がある。たとえ私たちがこのようなクレームを起訴または抗弁することに成功しても、br訴訟は巨額のコストを招き、私たちの上級管理者と科学者の注意を分散させる可能性がある。
私たち は、私たちの特許や他の知的財産権を保護または強制する訴訟に巻き込まれる可能性があり、これは高価で時間のかかるbrであり、成功しないかもしれない。
競争相手は私たちの特許、商標、著作権、または他の知的財産権を侵害する可能性がある。権利侵害や不正使用に反撃するために、私たちは権利侵害請求を要求されるかもしれません。これは高価で時間がかかり、私たちの管理者と科学者の時間と注意力を分散させているかもしれません。さらに、私たちの特許は発明権、優先権、または有効性紛争に巻き込まれる可能性がある。このようなクレームに反撃するか、またはbrを弁護することは、高価で時間がかかる可能性があり、私たちの相手は、これらの法的行動を起訴するために、私たちよりも多くの資源を投入する能力があるかもしれない。私たちが認定された侵害者に対するいかなるクレームも、これらの当事者が私たちに反クレームを提起することを促す可能性があり、私たちは彼らの特許を侵害したと主張し、また、私たちの特許は無効であるか、強制的に執行できないか、または両方を持っていると主張する。
侵害訴訟では、裁判所は、ある特許が無効または強制執行できないと判断するか、または私たちの特許が関連技術をカバーしていないことを理由に、他方の関連技術の使用を阻止することを拒否することができる。したがって、私たちは努力したにもかかわらず、私たちは第三者が私たちが所有または制御している知的財産権を侵害または流用することを阻止できないかもしれない。 任意の訴訟手続きにおける不利な結果は、私たちが所有または許可している1つまたは複数の特許が無効または狭義に解釈されるリスクに直面する可能性がある。また,知的財産権訴訟に関する大量の発見により,このような訴訟中に,我々のいくつかの機密情報が開示によって漏洩する可能性がある.
たとえ裁決が我々に有利であっても,裁判所はさらなる侵害活動を禁止する禁止令を付与せず,金銭的損害賠償のみを決定する可能性があり,十分な救済措置ではない可能性がある。知的財産権請求に関する訴訟や他の法的手続きは、私たちに巨額の費用を発生させ、私たちの人員の正常な責任を分散させる可能性があります。また、 は、公聴会、動議、または他の一時的な手続きまたは事態の発展の結果を公表する可能性があり、証券アナリストや投資家がこれらの結果がマイナスであると考える場合、私たちの普通株の価格に大きな悪影響を及ぼす可能性がある。このような訴訟または訴訟は、私たちの運営損失を大幅に増加させ、開発活動または任意の将来の販売、マーケティング、または流通活動に使用することができるリソースを減少させる可能性がある。
私たちのbrは、このような訴訟または訴訟手続きを十分に展開するために十分な財政的または他の資源がないかもしれない。私たちのいくつかの競争相手は、より多くの財力とより成熟して発展した知的財産の組み合わせを持っているので、私たちよりも効率的にこのような訴訟または訴訟の費用を負担することができるかもしれない。特許訴訟または他の訴訟の開始および継続によって生じる不確実性は、市場における我々の競争能力に重大な悪影響を及ぼす可能性がある。
もし我々が将来的に第三者とのいかなる知的財産権許可義務も履行できなければ,我々の業務に非常に重要な許可権 を失う可能性がある.
私たちが私たちの候補製品チャンネルを作るために努力していることを考慮して、私たちは未来に許可協定を締結するかもしれない。私たちはこのような許可協定が私たちに様々な職務調査、マイルストーン支払い、特許権使用料、保険、その他の義務を課すことを予想しています。もし私たちがこれらの許可下の義務を守ることができなければ、私たちの許可者はこれらの許可協定を終了する権利があるかもしれません。この場合、私たちはこれらの合意がカバーするいかなる製品も販売できないかもしれません。あるいは私たちの許可者は許可を非独占許可に変換する可能性があり、これは許可プロトコルによって開発された候補製品の価値に悪影響を及ぼすかもしれません。これらの許可プロトコルを終了するか、または私たちの許可権利を減少またはキャンセルすることは、私たちが条項の悪い新しい許可を協議したり、許可を回復しなければならない可能性があります。
| -69- |
もし 我々の商標や商号が十分に保護されていなければ,私たちの興味のあるタグに名前認知度 を作ることができない可能性があり,我々の業務は悪影響を受ける可能性がある.
私たちの商標または商号は、疑問、侵害、回避、または汎用商標として発表されるか、または他の商標が侵害されたと認定される可能性があります。私たちの商標は登録および一般法の保護に依存しています。私たちはこれらの商標や商品名に対する私たちの権利を保護できないかもしれないし、これらの名前の使用を停止させることができないかもしれません。私たちは関心のある市場で潜在的なパートナーや顧客の名前の承認を得るために必要です。商標登録過程で、私たちは拒否を受けるかもしれない。私たちはこのような拒否に答える機会があるにもかかわらず、私たちはこのような拒否を克服できないかもしれない。また,米国特許商標局や多くの外国司法管轄区の類似機関では,第三者は係属中の商標出願に反対し,登録商標の抹消を求める機会がある。私たちの商標に反対またはキャンセル訴訟を提起するかもしれません。私たちの商標は存在し続けることができないかもしれません。 私たちの商標と商号に基づいて名称を確立することができなければ、私たちは効果的に競争できないかもしれません。 私たちの業務は悪影響を受ける可能性があります。
従業員事務と管理成長に関するリスク
私たちの業務は限られた従業員だけが私たちの業務を管理して運営しています。
2022年9月30日までに、約12人のフルタイム従業員がいます。また,独立請負業者や他の第三者 を用いて我々の業務の様々な側面に協力している.候補製品開発への関心は、現金使用 を最適化し、効率的な方法で私たちの業務を管理·運営することを求めています。私たちが私たちの候補製品を開発したり、私たちの業務を運営したり、私たちが本来達成を求めていたすべての目標を達成するために、私たちが十分な人員レベルを雇用または維持できることを保証することはできません。
ネットワーク攻撃brまたは電気通信または情報技術システムにおける他の障害は、情報の盗難、データ破損、および深刻なトラフィック運用中断をもたらす可能性がある。
我々は,情報技術やIT,システム,ネットワークを利用して,我々の業務活動に関連する電子情報を処理,転送,蓄積する.デジタル技術の使用がますます多くなるにつれて、意図的な攻撃および不正なコンピュータシステムおよびネットワークへの不正アクセスを試みることを含むネットワークイベントも、頻度および複雑性を増加させる。これらの脅威は,我々のシステムやネットワークのセキュリティ,データのセキュリティ,および可用性と完全性にリスクを与える.
私たちの未来の成功は私たちが肝心な従業員、顧問と顧問、及び合格したbr人員を引きつけ、維持し、激励できるかどうかにかかっている。
我々 は役員チームの主要メンバーやキースタッフに強く依存しており,彼らのサービスを失うことは我々の目標の実現に悪影響を与える可能性がある.私たちは私たちのいくつかの幹部と雇用協定を締結したが、彼らの誰でもいつでも私たちの職場から離れることができる。私たちはこのような個人の生命や私たちの他の従業員の生命のために“キーパーソン”保険証書を維持しない。1人以上の既存従業員を失ったサービスは、私たちの研究開発と商業化目標の達成を阻害するかもしれない。また、幹部や他の重要な従業員を交換することは困難かもしれませんし、私たちの業界では開発に成功し、マーケティング承認や製品商業化に必要なスキルや経験を持っている人の数が限られているので、時間がかかるかもしれません。
私たちの業務のために他の合格した従業員、コンサルタント、コンサルタントを募集し、維持し、科学と技術者を含むことも、私たちの成功の鍵となるだろう。現在、私たちの産業にはスキルのある幹部が不足しており、このような状況は続く可能性が高い。そのため,技能人材に対する競争は非常に激しく,流出率が高い可能性がある。多くの製薬会社とバイオテクノロジー会社が類似したスキルを持つ人員を争っているため、受け入れ可能な条件で人材を誘致し、維持することはできないかもしれない。そのほか、臨床前或いは臨床試験で成功できなかったことは、合格者の採用と維持を更に挑戦的になる可能性がある。
| -70- |
また、私たちは、科学や臨床コンサルタントを含むコンサルタントやコンサルタントに依存して、私たちの研究、開発、商業化戦略の策定を支援してくれます。私たちのコンサルタントとコンサルタントは他のエンティティに雇われる可能性があり、これらのエンティティとの相談やコンサルティング契約に基づいて約束することができ、これは彼らの私たちへの利用可能性を制限するかもしれない。もし私たちが引き続き高い素質の人材を誘致し、維持することができなければ、候補製品を開発し、商業化する能力は制限されるだろう。
役員、重要な従業員、コンサルタント、コンサルタントを募集したり失うことができないサービスは、私たちの研究開発と商業化目標の進展を阻害する可能性があります。
私たちのbr従業員、独立請負業者、顧問、協力者と契約研究組織は不正行為或いは他の不当な活動に従事する可能性があり、規制基準と要求を守らないことを含み、これは私たちに重大な責任をもたらし、私たちの名声を損なう可能性がある。
私たちbrは、従業員、独立請負業者、コンサルタント、協力者、契約研究組織が詐欺行為や他の不正活動に従事する可能性があるリスクに直面しています。これらの当事者の不正行為には、(1)FDA法規または同様の非米国規制機関の類似法規、そのような規制機関に真実で完全かつ正確な情報を報告することを要求する法律、 (2)製造基準、(3)連邦および州医療詐欺および法律法規の乱用、および同様の非米国規制機関によって制定され実行される類似の法律を含む、意図的、無謀、および/または不注意が、私たちに以下の規定に違反する不正活動を行うか、または開示することができる。(4)財務情報又はデータを正確に報告することを要求する法律。特に、医療業界の販売、マーケティング、業務配置は、詐欺、不正行為、リベート、自己取引、賄賂、その他の乱用行為を防止するための広範な法律法規の制約を受けている。これらの法律法規は、広範な価格設定、割引、マーケティングおよび販売促進、販売手数料、顧客インセンティブ計画、および他のビジネススケジュールを制限または禁止しています。従業員や協力者の不適切な行為はまた、臨床試験中に得られた不適切な使用(取引を含む)によって得られた情報に関連する可能性があり、これは規制制裁を招き、私たちの名声を深刻に損なう可能性がある。私たちは行為と商業道徳基準を持っているが、常に不当な行為を識別し、阻止することができるわけではなく、私たちがこのような活動を発見し、防止するための予防措置は、未知または未管理のリスクや損失を効果的に制御できないかもしれないし、政府の調査またはそのような法律を遵守できないことによる他の行動や訴訟から私たちを保護することができないかもしれない, 基準や法規ですもし私たちにこのような訴訟を提起し、私たちが自分の権利を弁護したり、維持することに成功しなかった場合、これらの訴訟は、民事、刑事、行政処罰の適用、br損害賠償、罰金、連邦医療保険、医療補助および他の連邦医療保健計画から除外される可能性があり、もし私たちが会社の誠実な合意または同様の合意の制約を受けて、これらの法律違反の告発、監禁、契約損害、名声損害などの追加的な報告要件および/または監督を解決するために、私たちの業務および運営結果に重大な影響を与える可能性があります。利益と将来の収益の減少、そして私たちの業務の削減、これらはいずれも私たちの業務運営能力と私たちの運営業績に大きな悪影響を及ぼす可能性があります。
私たちは私たちの組織を拡大することを望んでいるので、私たちは私たちの成長を管理する際に困難に直面する可能性があり、これは私たちの運営を中断する可能性があります。
私たちの従業員の数と業務範囲は大幅に増加することを予想しています。特に薬品製造、規制事務と販売、マーケティングと流通の分野で、私たちの上場企業の運営を支持します。これらの成長活動を管理するためには、私たちの管理、運営、財務システムを継続して実施し、改善し、私たちの施設を拡大し、より多くの合格者を募集し、訓練し続けなければならない。私たちの経営陣はこのような成長活動を管理するために多くのエネルギーを投入しなければならないかもしれない。しかも、私たちの予想成長は私たちの歴史上の地域以外の地理的地域に移転する必要があるかもしれない。例えば、私たちはニュージャージー州プリンストンに事務所を設置して、私たちの多くの財務、管理、行政者はそこで働いています。私たちの財務資源が限られているため、私たちの管理チームはこのような成長を期待している会社を管理する上での経験も限られているため、私たちは私たちの業務の拡張や移転を効果的に管理することができず、重要な従業員を維持したり、より多くの合格者を確定、採用、訓練することができないかもしれない。私たちの業務の拡張や移転を効果的に管理できないことは、私たちのインフラが弱くなり、操作ミス、ビジネスチャンスの喪失、従業員の流失、余剰従業員の生産性の低下を招く可能性があります。私たちの予想成長はまた、大量の資本支出 を必要とする可能性があり、より多くの候補製品を開発するような他のプロジェクトから財務資源を移転するかもしれない。もし私たちが期待成長を効果的に管理できなければ、私たちの支出は予想よりも増加するかもしれません。私たちの創造能力は低下するかもしれません。私たちは私たちの業務戦略を実施できないかもしれません, 私たちの候補製品の成功的な商業化を含む。
| -71- |
私たちの普通株に関するリスク
私たちの普通株の市場価格は大きく変動するかもしれない。
我々の普通株の市場価格は変動が大きい可能性があり、以下の要素の影響を受ける可能性がある
| ● | 当社の四半期または年度の経営業績の実際または予想変動 | |
| ● | 財務またはトラフィック推定または予測の変化 ; | |
| ● | 一般的な市場状況 | |
| ● | 私たちと類似した会社の経済表現や市場評価の変化 ; | |
| ● | アメリカや他の地域の一般的な経済や政治状況。 |
特に,我々のようなバイオテクノロジー会社の市場価格は非常に不安定であり,原因としては :
| ● | 私たちの製品の臨床試験を遅延したり、FDAおよび他の規制機関の承認を得ることができませんでした | |
| ● | 会社の知的財産権に関する事態や紛争 | |
| ● | このような会社や競争相手の技術革新は | |
| ● | 会社のような市場推定値変化 ; | |
| ● | このような会社またはその競合他社は、重大な契約、買収、戦略的パートナーシップ、合弁企業、資本約束、新技術または特許を発表する | |
| ● | 重大な取引を完了できなかったか、またはサプライヤーと協力して製品を生産することができなかった。 |
証券市場は特定会社の経営業績に関係なく重大な価格や出来高変動を時々経験している。このような市場変動はまた私たちの普通株の市場価格に実質的な悪影響を及ぼす可能性がある。
私たちは予測可能な未来に現金配当金を支払わないと予想しているので、投資家は彼らの投資に現金配当金があることを期待してはいけない。
私たちの取締役会は予測可能な未来に現金配当金を支払うつもりではなく、すべての収益とすべての収益を保留して業務成長に資金を提供するつもりです。今まで、私たちは現金配当金を支払わなかったし、私たちの普通株が永遠に現金配当金を支払うことを保証することもできない。
上場企業として,我々 は巨大なコストを発生させ,大量の管理時間を投入し,これらのコスト が増加することが予想される.
上場企業として、私たちは多くの法律、会計、その他の費用を負担した。例えば、私たちは、“サバンズ-オキシリー法案”および“ドッド·フランクウォール街改革および消費者保護法案”のいくつかのbr要求、および効果的な情報開示および財務制御の確立と維持、およびコーポレートガバナンス実践における変化を含む、米国証券取引委員会がその後に実施するルールおよび条例 を遵守することを要求される。私たちは、このような要求を遵守することが、私たちの法律と財務コンプライアンスコスト を増加させ、いくつかの活動をより時間とコストを高くすると予想する。また、私たちの経営陣や他の人たちは、これらの上場企業要求に多くの時間を使用するために、運営や他の業務から注意をそらす必要があると予想されています。特に、私たちは、“サバンズ-オクスリー法案”404条の要件を遵守することを確保するために、巨額の費用を発生させ、大量の管理業務を投入する予定です。私たちには現在内部監査機能がなく、適切な上場企業の経験と技術会計知識を持つ追加会計·財務者を募集または契約して招聘する必要があるかもしれない。
| -72- |
私たちの内部統制の有効性には制限がある可能性があります。もし私たちの制御システムがミスや詐欺を防ぐことができなければ、私たちの会社に大きな被害を与える可能性があります。
サバンズ·オキシリー法404条によると、私たちは財務報告書の内部統制の有効性に関する報告書を私たちの経営陣が提出しなければならない。この評価には、財務報告書の内部統制で発見された私たちの経営陣の重大な弱点を開示する必要があるだろう。重大な欠陥とは、財務報告の内部統制に欠陥或いは欠陥の組み合わせが存在し、年度或いは中期合併財務諸表の重大な誤報が合理的な可能性があり、適時に防止或いは発見できないようにすることである。
効率的な財務報告内部統制は、信頼性とタイムリーな財務報告を提供するために必要であり、適切なbr開示制御およびプログラムと共に詐欺を合理的に発見し、防止することを目的としている。必要な新しいまたは改善された制御措置を実施できなかったか、または実施中に遭遇した困難は、私たちの報告義務を履行できない可能性がある。未発見の材料 財務報告の内部統制に弱点があり、財務諸表の再記述を招く可能性があり、救済費用 が要求されています。
さらに、私たちは財務報告書の開示統制や内部統制がすべてのミスとすべての詐欺を阻止しないと予想している。制御システムは、設計や動作がどんなに良くても、絶対的な保証ではなく、合理的な保証しか提供できず、制御システムの目標が実現できることを確保する。また,制御システムの設計は,資源制約が存在し,そのコストに対する制御の利点を考慮しなければならないという事実を反映しなければならない.すべての制御システムの固有の限界により、すべての制御問題および不正事例が検出されたことを絶対的に保証することはできない。もし私たちの制御システムがエラーや詐欺を検出したり防止しなかったら、私たちに深刻な否定的な影響を及ぼすかもしれない。
このような状況が発生すれば、大衆のわが社に対する見方にマイナス影響を与える可能性があり、これは私たちの株価にマイナス影響を与える可能性がある。
私たち は財務報告の内部制御の分析を適時に完了できない可能性があり、あるいはこれらの内部制御 は有効であると確定できない可能性があり、これは投資家がわが社の自信に悪影響を与え、私たちの 普通株の価値に影響を与える可能性がある。
私たち は財務報告書の内部統制の評価とテストを完了できないかもしれない。評価およびテスト中に財務報告の内部統制に1つまたは複数の重大な弱点が発見された場合、私たちの内部統制が有効であるとは断言できないだろう。
もし私たちが財務報告の内部統制に有効であると断言できない場合、あるいは適用されれば、私たちの独立公認会計士事務所は私たちの内部統制の有効性に意見を述べることができず、私たちの財務報告の正確性と完全性に対する投資家の信頼を失う可能性があり、これは私たちの普通株式価格の下落を招き、私たちはアメリカ証券取引委員会の調査または処罰を受ける可能性がある。私たちはまた四半期ごとに内部統制と手続きの側面で行われた変更を開示することを要求されるだろう。
デラウェア州法の反買収条項は、合併後の会社を買収することをより困難にする可能性があり、合併後の会社株主が合併後の会社経営陣を交換または更迭しようとすることを阻止する可能性がある。
合併後の会社はデラウェア州に登録して設立されるため、デラウェア州汎用会社法第203節またはDGCLの条項によって管轄され、発行された合併会社の投票権株の15%を超える株主合併 または合併後の会社との合併を禁止する。これらの条項は、潜在的な買収者に合併後の会社取締役会との交渉を要求することで、より高い入札を得る機会を提供すると信じているが、ある株主が要約が有益であると考える可能性があれば、これらの条項はさらに適用可能である。また、これらの規定は、株主が管理職メンバーを任命する取締役会メンバーを変更することを困難にし、それによって、合併後の会社株主の既存の管理層の交換または更迭を挫折または阻止する可能性がある。
| -73- |
役員brと高級乗組員の責任は限られている。
デラウェア州の法律で許可されているため、我々の定款は取締役が取締役の受託責任に違反して負う金銭損害賠償責任を制限していますが、場合によっては責任は除外されています。私たちの定款条項とデラウェア州法律のため、株主は限られた権利 だけが受託責任に違反した取締役に賠償する可能性があります。
一般リスク因子
ネットワーク攻撃brまたは電気通信または情報技術システムにおける他の障害は、情報の盗難、データ破損、および深刻なトラフィック運用中断をもたらす可能性がある。
我々は,情報技術やIT,システム,ネットワークを利用して,我々の業務活動に関連する電子情報を処理,転送,蓄積する.デジタル技術の使用がますます多くなるにつれて、意図的な攻撃および不正なコンピュータシステムおよびネットワークへの不正アクセスを試みることを含むネットワークイベントも、頻度および複雑性を増加させる。これらの脅威は,我々のシステムやネットワークのセキュリティ,データのセキュリティ,および可用性と完全性にリスクを与える.
普通株、転換可能証券、引受権証またはオプションを増発するため、私たちの普通株はさらに希釈される可能性がある。
従来、資金調達のために、普通株式、転換可能証券(例えば、転換手形)、権利証が発行されていた。私たちはまた、私たちの従業員、役員、およびあるサプライヤーへのサービス補償と奨励的補償として普通株式を発行しました。私たちは、その中のいくつかの証券を行使する際に発行し、将来的にこれらの目的のために保留されている株式を増加させる可能性があるために普通株式を保持しています。私たちが普通株式、転換可能証券、オプション、および引受権証を発行することは、私たちの株主の権利に影響を与える可能性があり、私たちの普通株の市場価格を低下させるか、あるいは発行された株式証明書の発行された株式価格の調整を招く可能性があります(これらの証券は、より多くの普通株br株を行使することができます)、またはある株主に追加の普通株を発行させる可能性があります。
将来売る資格のある株 は市場に悪影響を及ぼす可能性がある。
証券法第144条規則によると、私たちの一部の株主は時々通常ブローカー取引を通じて公開市場でその普通株式の全部または一部を販売する資格があるかもしれないが、いくつかの制限を受けている。全体的には、第144条によれば、最初の3ヶ月は非関連会社の株主であり、6ヶ月後に私たちの普通株の株式を自由に売却することができるが、現在の公開情報要求を遵守しなければならない。関連会社は、6ヶ月後に規則144の数量、販売方法、最新の公開情報、および通知に従って、私たちの普通株の株の売却を要求することができます。規則144に基づいて私たちの普通株を売却することは、私たちの普通株の市場価格に重大な悪影響を及ぼす可能性があります。
項目 1 B.未解決の従業員のコメント。
は適用されない.
第 項2.属性.
同社は短期勤務使用契約に依存して事務や会議空間を調達している。
第3項:法的訴訟。
私たち は現在どんな重大な法的手続きの影響を受けていない。しかし、私たちは時々私たちの正常な業務過程で現れる様々な法的手続きの側になるかもしれない。
第br項4.鉱山安全情報開示
は適用されない.
| -74- |
第 第2部分
第 項5.登録者普通株市場、関連株主事項、発行者が株式証券を購入する。
市場情報
同社の普通株はナスダック資本市場で取引されており、コードは“SONN”である
所持者
2022年12月14日までに、私たちは132人の普通株式保有者がいます。記録保持者の数は譲渡エージェントの記録によって決定され,普通株の受益者を含まず,その株は各種証券仲介人,取引業者,登録決済機関の名義で保有される.我々の普通株の譲渡代理は証券譲渡会社,ダラス公園路北2901号,Suite 380,Plano,TX 75093である.
配当をする
私たちは普通株の現金配当金を発表したり、支払ったことがありません。予測可能な未来では、私たちは普通株の現金配当金を発表または支払うつもりはありませんが、現在は任意の未来の収益を維持し、私たちの業務の発展と成長に資金を提供するつもりです。普通株が現金配当金(あれば)を支払うことは完全に私たちの取締役会が適宜決定し、私たちの収益、資本要求、財務状況、その他の関連要素に依存します。
第 項6[保留されている]
第br項7.経営陣の財務状況と経営成果の検討と分析
以下のbr}経営陣の財務状況と経営成果の検討と分析(“MD&A”)は、当社が本報告で述べた期間の財務状況とその歴史的経営業績の理解を支援することを目的としている。本MD&Aは,本年度報告(Form 10−K)に含まれる監査された合併財務諸表とその付記とともに読まなければならない。本MD&Aは、リスクおよび不確定要因に関する前向きな陳述を含む可能性がある。前向きな説明に関する議論は、参照によって本明細書に組み込まれる上記の“前向きな陳述に関する特別な説明”のタイトルの下の情報を参照されたい。
概要
十四行詩 BioTreateutics Holdings,Inc.(“十四行詩”、“私たち”あるいは“会社”)、 は臨床段階で腫瘍学に集中する生物技術会社であり、単一或いは双特定の作用を革新する生物薬物の独自のプラットフォームを持っている。Fと呼ぶHAB(完全ヒトアルブミン結合)は,完全ヒト単鎖抗体断片を用いてヒト血清アルブミンに結合し,その上に“便乗”し,標的組織に輸送する。特定の組織における薬物の蓄積を改善し,体内での活動時間を延長するためにbr構造を設計した。F.FHAB は哺乳動物細胞培養において発育候補遺伝子を産生し、グリコシル化を実現し、免疫原性のリスクを低下させる。私たちはFを信じているH著者らは2021年6月にアメリカ特許を獲得したAB技術は著者らの生物製薬brプラットフォームの1つの顕著な特徴であり、未来の一連の人類疾病領域における薬物開発に非常に適しており、腫瘍学、自己免疫、病原性、炎症と血液学疾患を含む。
我々の現在の内部パイプライン開発活動はサイトカインに集中しており,これは1種類の細胞シグナルペプチドであり,他の重要な機能において有効な免疫調節剤である。独立および相乗作用の下で、特定のサイトカインは、癌および病原体に対して免疫細胞の活性化および成熟を調節する能力を示すようになっている。しかし、それらは特定の組織に優先的に蓄積し、迅速に体内から除去することはないため、サイトカイン治療を用いて治療効果を達成する伝統的な方法は通常高用量と頻繁な用量を使用する必要がある。これは治療効果の低下を招き,潜在的な全身毒性を伴う可能性があり,これらの薬物の治療応用に挑戦している。
| -75- |
我々の先行特許資産SON-1010は、Fに共有結合されたインターロイキン12(IL-12)の完全人間バージョンであるHAB 構築は,非小細胞肺癌や頭頸部癌を含む固形腫瘍適応の臨床開発を行う予定である。我々はすでに非ヒト霊長類(NHP)GLP毒性研究を完成し、臨床で使用されている医薬製品の生産に成功した。2022年3月、FDAは我々のSON-1010研究新薬(IND)申請を許可した。このbrは2022年の第2のカレンダー四半期に固形腫瘍腫瘍患者に対するアメリカ臨床試験(SB 101)を開始することができるようにした。 2021年9月、著者らは完全所有のオーストラリア子会社SonnetBio Pty Ltdを設立し、ある臨床試験を行った。著者らは承認を得て、2022年第3四半期に健康ボランティアに対するSON-1010オーストラリア臨床研究(SB 102)を開始した。SB 101とSB 102の研究の初歩的な安全性と耐性データは2022年第4四半期に公表され、2つの研究の初歩的な薬物動態学と薬効学データは2023年第2四半期に発表される予定である。
我々 は2020年4月にRelipment Treeutics SAの流通株を買収することにより,我々の最先端化合物SON−080の世界開発権を獲得し,SON−080は完全人間バージョンのインターロイキン6(IL−6)である。SON−080治療化学療法による末梢神経病変(“CIPN”)と糖尿病末梢神経病変(“DPN”)の目標適応を進めている。我々は の承認を得ており,CIPNではSON−080を用いて前米国段階1 b/2 a研究を開始することができる。この研究は2023年上半期に初歩的な臨床安全性データを生成する可能性がある。我々が2021年5月にシンガポールNew Life Treeutics Pte,Ltd.(“New Life”)と締結した許可協定 に基づき,New Lifeと共同でDPNにおけるSON−080の開発を担当する。CIPNセキュリティデータが利用可能になると,2023年下半期に第2段階研究 を開始することを目標としている。
SON-1210 (“IL12-FHAB-IL 15“),我々の主要二重特異性構築物はFに結合しているH完全なヒトIL-12と完全なヒトIL-15(“IL-15”)を用いる。この化合物は結腸直腸癌を含む固形腫瘍適応のために開発されている。NHP研究は2022年第4四半期に完成する見込みであり、2023年上半期に規制認可プログラム を開始する予定である。2023年第1四半期に凍結乾燥薬物製剤を調製することにより、生産と薬品供給を確保し、この製剤は初の人体臨床試験に適用されると予想される。
SON−1410(IL 18−F)のユニット系開発とプロセス開発H現在インターロイキン18(“IL−18”)とIL−12の二重特異性結合が行われており,早期開発のための体外概念検証研究の少量に加え,製剤や分析法開発活動に適した早期実験的薬剤を提供する。プロセス開発活動は2023年まで続き,2023年末までに初歩的な体内マウス研究に適した薬物を生産する可能性がある。
SON−3015(抗IL 6−F)の配列確認が完了しましたHAB−抗形質転換成長因子β)。初期の二重特効薬が生成され、将来の使用のために保存されています体内にあるネズミの研究です
我々 は設立以来,経常的な運営損失と運営キャッシュフローは負である。私たちが利益を達成するのに十分な製品を生産できるかどうかは、1つまたは複数の現在または未来の候補製品の成功した開発および最終商業化に大きく依存するかもしれない。2022年と2021年9月30日までの年間純損失はそれぞれ2,970万ドルと2,500万ドルです。2022年9月30日まで、私たちは310万ドルの現金を持っている。少なくとも今後数年以内には,巨額の費用 が発生し,運営損失が増加することが予想される。私たちは私たちが行っている活動によって私たちの費用と資本需要が大幅に増加すると予想している
| ● | 候補製品のための追加の臨床試験を行った | |
| ● | 他の候補製品を発見し開発するために を続ける; | |
| ● | または許可を得る他の候補製品および技術; | |
| ● | 私たちの知的財産権の組み合わせを維持し、拡大し、保護する |
| -76- |
| ● | 追加の臨床、科学、商業者を雇う | |
| ● | 我々が規制承認を得ることが可能な任意の候補製品の商業数量を提供するのに十分な商業製造源およびセキュリティサプライチェーン能力を確立する | |
| ● | 臨床試験を成功させた候補製品のために監督管理の承認を求める | |
| ● | 販売、マーケティング、流通インフラを構築し、規制の承認を受ける可能性のある任意の製品を商業化する | |
| ● | 私たちの製品開発と計画の将来の商業化努力を支援する人員 を含む、brの運営、財務、管理情報システムと人員を増加させ、公共報告会社としての私たちの運営を支援します。 |
私たち は、許可収入を受けて、および/または臨床 開発に成功し、規制部門から候補製品の承認を得ない限り、製品販売から収入を得ません(もしあれば)。もし私たちが規制部門から任意の候補製品の承認を得て、商業化パートナー関係に加入していなければ、製品販売、マーケティング、流通を支援するための内部商業化能力の発展に関連した巨額の費用が発生すると予想される。上場企業として、運営に関わる巨額のコストが発生し続ける。
したがって、私たちは私たちの持続的な運営を支援し、私たちの成長戦略を実施するために多くの追加資金を必要とするだろう。製品販売から相当な収入を得ることができる前に、株式、債務融資、または他の資本源(他社との協力や他の戦略取引を含む場合があります)を売却することで、当社の運営に資金を提供する予定です。私たちは必要な時に優遇条項で追加資金を調達したり、他の合意や手配を達成することができないかもしれません。もし私たちが資金を調達できない場合、あるいは必要な時にそのような合意に到達できない場合、私たちは私たちの1つ以上の候補製品の開発と商業化を大幅に延期、減少、またはキャンセルしなければならないかもしれません。または潜在的なライセンス内または買収を延期しなければなりません。
製品開発に関連する多くのリスクや不確実性のため、費用が増加する時間や金額を予測することもできず、いつ実現したり、利益を維持したりすることができるかどうかも予測できない。たとえ私たちが製品販売を作ることができても、私たちは利益を上げることができないかもしれない。もし私たちが利益を上げることができない場合、または持続的に利益を上げることができない場合、私たちは計画通りに運営を継続することができず、運営を減少または終了させることができないかもしれない。
我々は2015年に設立されて以来、ほとんどの努力と財務資源を投入して、会社員、br}業務計画、資金調達、候補製品の買収または発見、関連知的財産権の保護、および候補製品のための発見、研究、開発活動を展開してきた。私たちは販売を許可された製品は何もなく、製品販売から何の収入も得ていません。これまで、私たちの運営資金は主に普通株の売却、株式承認証、転換可能な債券の発行の収益から来ています。
新冠肺炎大流行
2019年12月、新型コロナウイルス新冠肺炎が武漢中国で出現し、2020年3月11日に世界保健機関によって大流行と発表された。世界の多くの国は旅行や大規模な集会に対して隔離と制限を実施し、新冠肺炎の伝播を緩和し、不要な企業を閉鎖している。国や州や地方司法管区が規制を継続することに伴い、私たちが業務を継続する能力も制限される可能性がある。このような事件は、業務、供給、および薬品生産の中断を招き、運営の減少を招く可能性があり、そのいずれもが、私たちの業務、財務状況、および運営結果に大きな影響を与える可能性がある。
このような大流行または爆発は、臨床試験地点、CROおよび/または試験モニタ、および試験を支持する他の重要なサプライヤーおよびコンサルタントの安全を確保することを困難にする可能性がある。また、疫病或いは臨床試験地点付近の疫病は私たちの患者募集能力に影響する可能性がある。これらの状況や新冠肺炎に関連する他の状況は、我々の臨床試験計画の遅延を招く可能性があり、予想コストを増加させる可能性があり、これらはすべて私たちの業務およびその財務状況に実質的な悪影響を及ぼす可能性がある。
| -77- |
具体的には,我々のパイプライン製品(SON−1010型を除く)の生産は新冠肺炎に関するサプライチェーン問題の遅延を受け,特に原材料の供給は,媒体,樹脂,分析キットの供給に加え,国際輸送遅延を含む。
新冠肺炎による潜在的な経済影響及びその持続時間は評価或いは予測が困難である可能性があるが、広い範囲の疫病は全世界の金融市場の深刻な混乱を招き、私たちの資本獲得能力を低下させる可能性があり、これは未来に私たちの流動性に負の影響を与える可能性がある。また、新冠肺炎の伝播による景気後退や市場回復は、私たちの業務と私たちの普通株の価値に大きな影響を与える可能性がある。
Br新冠肺炎疫病はまた著者らの従業員と協力各方面が著者らの非臨床、臨床とbr薬物生産活動を展開する能力に影響する可能性がある。私たちは臨床サイト、研究者および他の研究者、コンサルタント、独立請負業者、契約研究組織および他の第三者サービスプロバイダに依存して、私たちの管理、監視、および他の方法で私たちの非臨床研究と臨床試験を実行するのを助けるかもしれない。私たちはまた、コンサルタント、独立請負業者、契約製造組織、および他の第三者サービスプロバイダに依存して、私たちの管理、監視、および他の方法で私たちの原料薬の生産、調合、および薬品製造活動を実行することを支援することに依存するかもしれない。新冠肺炎は、これらの外部の個人、組織、または会社が私たちのプロジェクトに十分な時間と資源を投入したり、出張のために仕事を実行したりする能力に影響を与える可能性がある。
新冠肺炎の発生が現在或いは未来の臨床研究に対する潜在的なマイナス影響は監督管理機関からのフィードバックの遅延、新しい臨床研究の開始及び被験者を登録している研究に参加することを含む。潜在的な負の影響 はまた、研究サイトで研究アクセスを行うことができないこと、安全性と有効性データの収集が不完全であること、進行中の研究中の被験者の中退率が高いこと、研究データがデータベースサイトに入る遅延、研究データの監視が研究サイトの物理的アクセス制限による遅延、クエリに対するサイトの応答遅延、データベースロックの遅延、データ分析の遅延、トップラインデータのタイムリーな遅延、および研究報告の完了遅延を含む。新しい或いは絶えず悪化する新冠肺炎の中断或いは制限は著者らの非臨床研究、臨床試験と薬物製造活動に更なる負の影響を与える可能性がある。現在、私たちはこの大流行が未来の行動に及ぼす潜在的な影響を定量化することができない。
運営結果の構成要素
協力 収入
協力 収入は現在New Lifeと2021年5月に締結した許可手配(“New Lifeプロトコル”)から来ており、この協定 はNew Lifeに独占許可(再許可の権利がある)を付与し、特定の組換えヒトインターロイキン6、SON-080(“化合物”)を含む医薬製剤 (このなどの製剤、“製品”) を開発、商業化するために、ヒト糖尿病末梢神経病変(“DPN領域”)の専属区域 の予防、治療或いは緩和に用いられている。我々は,この手配に基づいて,(I)排他的地域内での開発,マーケティング,輸入,使用および商業化製品のライセンス(“ライセンス”),および(Ii)技術移転と臨床開発 と規制活動(“研究開発活動”)を決定した。私たちはライセンスと研究開発活動がお互いに区別されていないことを確認したので、これらの重大な約束を履行義務に統合した。この協定によると、私たちは合計100万ドルの前払い現金支払いを受け取り、これらの現金は全額単一業績義務に割り当てられ、研究開発サービスの見積もり業績期間中にbr}を確認する。
| -78- |
運営費用
研究と開発費
研究と開発費用には、主に私たちの候補製品の発見と開発に関するコストが含まれています。私たちは発生した状況に応じて研究と開発コストを計上しています
| ● | 研究開発に従事している従業員の給料、株式給与、関連福祉を含む従業員に関する支出 | |
| ● | コンサルタントや臨床研究機関などの第三者との合意を含む、我々の候補製品の臨床前および臨床開発に関連する費用br; | |
| ● | コンサルタントや契約製造組織などの第三者との合意を含む、我々の臨床前研究および臨床試験のための医薬製品を製造するコスト | |
| ● | 施設、減価償却およびその他の費用、賃貸料および施設維持および保険の直接費用または分担費用を含む; | |
| ● | 法規遵守要求に関するコスト ; | |
| ● | 第三者許可プロトコルにより支払い を行う. |
我々 は,サービスプロバイダが提供する情報に基づいて特定のタスクを達成する進捗を評価し,外部開発コストを確認する.このプロセスは、未完了契約および購入注文を審査し、担当者とコミュニケーションして、私たちの代わりに実行されたサービスを決定することと、請求書を受信していない場合、または他の方法で実際のコストを通知する場合に、実行されたサービスレベルおよびそのサービスのために生じる関連コストを推定することとを含む。将来的に受け取った研究開発活動のための貨物またはサービスの前払いは、前払い費用として払い戻しできません。商品の配送またはサービスが完了した場合、このような金額は費用として確認されます。
私たちの直接研究開発費用は主に外部コスト、例えば外部コンサルタント、CRO、CMO、研究実験室に支払う費用を含み、これらの費用は臨床前開発、プロセス開発、製造、臨床開発活動と関係がある。私たちの直接研究開発費用には、第三者許可プロトコルによる費用も含まれている。従業員コストや発見作業、実験室用品、施設に関するコスト(減価償却や他の間接コストを含む)を特定の候補製品に割り当てることはありませんが、これらのコストは複数の計画に配置されているため、単独では分類されていません。私たちは主に内部資源を用いて研究と発見を行い、臨床前開発、プロセス開発、製造、臨床開発活動を管理しています。これらの従業員は複数の計画で働いているため,候補製品に応じてコスト を追跡しない.
我々 は,予見可能な未来に,我々の候補製品の開発を進めていくことにより,我々の研究開発費が増加することを予想している.私たちの候補製品が開発に成功するかどうかは大きな不確実性を持っている。現在、臨床開発に関連する多くのリスクと不確定性のため、以下の方面のリスクと不確定性を含むため、私たちは現在のパイプの余剰開発または私たちが開発する可能性のある未来の候補製品を完成させるために必要な仕事の性質、時間、コストを合理的に見積もることができない、あるいは 私たちの現在のパイプの余剰開発または私たちが開発する可能性のある未来の候補製品を完成するために必要な仕事の性質、時間、コストを知ることができない
| ● | 臨床前と臨床開発活動の時間と進捗; | |
| ● | 私たちは臨床前と臨床プロジェクトの数と範囲を決定しました | |
| ● | 私たちは現在の研究開発計画を維持し、新しい計画を立てることができます | |
| ● | 性新薬を研究することによって、適切な安全概況を研究できるようにした | |
| ● | 成功したbr患者の登録を行い、臨床試験を開始し、完成した | |
| ● | 臨床試験に成功し、その安全性、耐性と治療効果状況はFDA或いは任意の類似した外国の監督管理機関を満足させた | |
| ● | 適用規制機関の規制承認を受けた; |
| -79- |
| ● | 適用規制機関からの任意の上場承認の時間、受信、および条項 | |
| ● | 私たちは新しい許可や協力計画を確立することができます | |
| ● | 第三者製造業者とbr協定を確立し、私たちの臨床試験および商業生産(私たちの任意の候補製品が承認されたら)に臨床供給を提供する | |
| ● | 臨床試験と商業投与に使用できる臨床レベルと商業レベルの薬物処方を開発し、適時に提供した | |
| ● | 特許請求の範囲および他の知的財産権の取得、維持、擁護、および実行; | |
| ● | 候補製品の商業販売を開始し(承認されれば)、単独でも他との協力でも ; | |
| ● | 承認後、候補製品の持続的に許容可能なセキュリティプロファイルを維持する;および | |
| ● | 新冠肺炎が運営に及ぼす潜在的な影響は,臨床試験の時間,原材料の可用性,検出施設へのアクセスや保護能力などに影響する可能性がある。 |
我々の候補製品の開発については、これらの変数のいずれかの結果が変化しても、候補製品の開発に関連するコストおよびスケジュールを著しく変更することが可能である。私たちのどんな候補製品も規制部門の承認を得ることができないかもしれない。
一般料金 と管理費用
一般費用と行政費用には、株式で計算された報酬を含む行政、財務、行政機能者の賃金と関連費用が主に含まれる。一般および行政費用には、直接分担された施設関連コスト、例えば、法律、特許、コンサルティング、会計、監査サービスの専門費用も含まれる。
継続的な研究活動や候補製品の開発を支援するために従業員数を増やすことにより,我々の一般的かつ管理費は将来的に増加するであろう。上場企業に関連する会計、監査、法律、監督、コンプライアンスと取締役と役員の保険コスト、投資家と広報費用を引き続き発生させていきます。
外国為替損失
外国為替損失にはドル以外の通貨での取引の為替変動が含まれる。
その他 収入
2021年にPPPローン免除に関する他の収入を確認した。
| -80- |
運営結果
2022年9月30日と2021年9月30日までの年次比較
次の表は、2022年9月30日と2021年9月30日までの年間運営結果をまとめています
| 9月30日までの年度は | ||||||||||||
| 2022 | 2021 | 変わる | ||||||||||
| 協力 収入 | $ | 349,943 | $ | 483,626 | $ | (133,683 | ) | |||||
| 運営費用 | ||||||||||||
| 研究開発 | 21,444,019 | 16,634,553 | 4,809,466 | |||||||||
| 通常 と管理 | 8,575,283 | 8,936,509 | (361,226 | ) | ||||||||
| 運営費総額 | 30,019,302 | 25,571,062 | 4,448,240 | |||||||||
| 運営損失 | (29,669,359 | ) | (25,087,436 | ) | 4,314,557 | |||||||
| 利息収入 | — | 15 | (15 | ) | ||||||||
| 為替為替損失 | (52,482 | ) | (22,041 | ) | (30,441 | ) | ||||||
| その他 収入 | — | 125,501 | (125,501 | ) | ||||||||
| 純損失 | $ | (29,721,841 | ) | $ | (24,983,961 | ) | $ | (4,737,880 | ) | |||
協力 収入
2022年9月30日までの年間で30万ドルの新生活協定に関する収入が確認されたが,2021年9月30日までの年間で確認された収入は50万ドル であった。
研究と開発費
2022年9月30日までの年度の研究·開発費は2,140万ドルであるのに対し,2021年9月30日までの年度は1,660万ドルである。480万ドル増加の要因はIL 12−F細胞系の開発支出の増加であるHAB, IL 12-FHAB-IL 15およびSON-080は、SON-1010の第1段階臨床試験を開始し、CIPNのSON-080と1 b/2 a段階の試験的治療効果研究を準備する;買収が行われている研究開発;様々な許可および研究開発協定(XOMA協力協定を含む)に関連するマイルストーン支払い ;および私たちが私たちの候補製品を開発し続けるにつれて、賃金支出が増加する。
一般料金 と管理費用
2022年9月30日までの年度の一般·行政費は860万ドルであるのに対し,2021年9月30日までの年度は890万ドルである。40万ドル減少の主な原因は賃金と株式ベースの給与支出の減少だ。
その他 収入
2021年6月の購買力平価ローン免除に関する他の収入は10万ドル。
流動性 と資本資源
これまで、私たちは主に普通株の売却、株式承認証、転換可能な債券の発行収益を通じて運営に資金を提供してきた。私たちは2023年度にもっと多くの証券を販売する必要があるだろう。将来株式や債務融資があるかどうかは特定できないし、受け入れ可能な条項で融資されるかどうかも確定できず、現在のところこれらの 事項の結果を予測することはできない。
2022年9月30日と2021年9月30日までの年間で、私たちはそれぞれ2,970万ドルと2,500万ドルの重大な純損失が発生しました。今後12ヶ月以降、私たちは引き続き重大な運営費用と純損失を発生させる予定です。私たちの純損失は、各四半期と毎年大きく変動する可能性があります。これは、私たちの研究開発の段階と複雑さ、関連するbr支出、私たちの技術許可の追加支払い(ある場合)、および私たちの現在または未来に達成可能な任意の協力によって受信された支払いに依存します。
我々 は,いくつかの条件やイベントが存在するかどうか(総合的に考える)ことを評価しており,我々の経営継続能力 を大きく疑っている.私たちは、2022年9月30日の現金310万ドルに、2022年9月30日から2022年12月15日まで2022年販売協定(以下、定義)に基づいて普通株を売却して得られた460万ドルの純収益を加え、2023年3月まで予定されている運営に資金を提供すると信じている。私たちは私たちの運営に資金を提供するために多くの追加資金が必要になるだろう。これらの要素は私たちの持続的な経営企業としての能力を大きく疑っています。
| -81- |
次の表は私たちの年間の現金源と使用状況をまとめています
| 9月30日までの年度は | ||||||||
| 2022 | 2021 | |||||||
| 純額 経営活動で使用した現金 | $ | (27,687,278 | ) | $ | (22,552,245 | ) | ||
| 投資活動用現金純額 | (896,477 | ) | (3,623 | ) | ||||
| 純融資活動から提供された現金 | 4,014,567 | 42,828,032 | ||||||
| 現金純増加 (減少) | $ | (24,569,188 | ) | $ | 20,272,164 | |||
操作 活動
2022年9月30日までの年間で、私たちは経営活動に2,770万ドルの現金を使用しましたが、これは主に私たちの純損失2,970万ドルによるものです。この額は90万ドルの株式ベースの報酬支出と100万ドルの買収における研究開発および運営資産と負債の純減少10万ドルによって相殺された。
2021年9月30日までの年間で、私たちは経営活動に2,260万ドルの現金を使用しましたが、これは主に私たちの純損失2,500万ドルによるものです。この額は、株式ベースの報酬支出140万ドルと運営資産と負債の純減少100万ドルで相殺された。
投資 活動
2022年9月30日までの年間で、購入買収のために90万ドルを使用している研究開発を行っている。
2021年9月30日までの年間で、3,623ドルのオフィス家具とコンピュータ機器を購入しました。
活動に資金を提供する
融資活動が提供する現金純額は2022年9月30日までの年間400万ドルで、主に優先株売却と引受権証の純収益および普通株売却の純収益を含む。
2021年9月30日までの年間で,融資活動が提供する現金純額は4,280万ドルであり,主に普通株の売却と引受権証の純収益 を含む。
資金需要
われわれの持続活動に関する費用は大幅に増加することが予想され,特に臨床前活動や開発中の候補製品の臨床試験を進めている場合である。また,上場企業運営 に関する余分なコストが発生することが予想される.私たちの運営費の時間と金額は主に見られるだろう
| ● | 現在または未来の候補製品のbr範囲、数量、起動、進捗、時間、コスト、設計、持続時間、任意の潜在的遅延、br}臨床試験および非臨床研究の結果 ; | |
| ● | 私たちはこれらの候補製品のための臨床開発計画を立てた | |
| ● | 私たちは許可を得ることができる候補製品とプログラムの数量と特徴を開発または獲得する |
| -82- |
| ● | FDAや同様の外国規制機関が設立した規制要件を満たすために、審査、承認、または他の行動の結果、時間、コストを規制する。Brを含むFDAまたは同様の外国規制機関は、私たちの候補製品について、現在予想されているよりも多くの研究を行うことを要求するかもしれない | |
| ● | 私たちが候補製品の発売承認を得る能力; | |
| ● | 特許主張を出願、起訴、弁護および実行し、私たちの候補製品をカバーする他の知的財産権のbrコスト; | |
| ● | 私たちの知的財産権の組み合わせ範囲を維持、拡張、保護する能力は、第三者が私たちまたは私たちの候補製品に対して提起した特許侵害訴訟を含む知的財産紛争を弁護するコストを含む | |
| ● | 候補製品に関する商業規模アウトソーシング製造活動のコストと完了時間; | |
| ● | 私たちのbrは、特典条項で許可、連携または同様の手配を確立および維持する能力、および任意の新しい許可、連携、または同様の手配に基づいて開発または商業化責任をどの程度保留するかどうか、および任意の新しい許可、連携または同様の手配に基づいて開発または商業化責任をどの程度保留するかどうかを決定する能力である | |
| ● | 私たちが自ら商業化製品を選択した地域で規制許可を得た任意の候補製品のための販売、マーケティング、および流通能力を確立するためのコスト ; | |
| ● | 私たちが買収したり投資したりした他の事業、製品、技術の成功 | |
| ● | 企業、候補製品、および技術を買収、許可または投資するコスト; | |
| ● | 私たちはより多くの管理者や科学と医療者を雇うことが必要です | |
| ● | 米国で上場企業として運営されているbrコストには、我々の業務のための追加の財務·報告システムおよび他の内部システムやインフラの需要が含まれている | |
| ● | 市場はすべての製品が商業販売のために承認される限り、私たちの候補製品を受け入れます | |
| ● | 競争の技術と市場発展の影響 | |
| ● | 新冠肺炎疫病は著者らの臨床試験と運営に潜在的な影響を与える。 |
私たちが相当な製品収入を生成することができる前に、私たちは株式発行、債務融資、協力、戦略連合、および第三者のマーケティング、流通または許可手配との組み合わせによって私たちの現金需要を満たすことが予想される。 もし私たちが株式または転換可能な債務証券を売却することで追加資本を調達すれば、私たちの所有権権益は大幅に希釈される可能性があり、このような証券の条項には清算または他の私たちの株主権利に悪影響を与える特典が含まれている可能性がある。債務融資および優先持分融資(利用可能な場合)は、追加債務を招くこと、資本支出を行うこと、または配当を宣言することなど、特定の行動をとる能力を制限する制限br契約を含むプロトコルに関連する可能性がある。もし私たちが第三者との協力、戦略連合またはマーケティング、流通または許可手配によって資金を調達する場合、私たちは技術、将来の収入フロー、研究計画、または候補製品の貴重な権利を放棄しなければならないかもしれません。もし私たちが必要な時に株式や債務融資または他の手配を通じてより多くの資金を調達することができない場合、私たちは製品開発または将来の商業化努力を延期、減少、停止またはキャンセルし、資産を売却したり、開発とマーケティングを希望する候補製品の権利を付与したり、運営を停止したりする必要があるかもしれない。
| -83- |
個人配給優先株
2022年8月、著者らは数名の認可投資家と証券購入協定(“優先株合意”)を締結し、(I)を合わせて22,275株の著者らの第3シリーズ転換可能優先株、1株100ドル、(Ii)225株私たちの第4シリーズ転換可能優先株、1株100ドル、及び(Iii)第3シリーズ株式承認証で、私募方式で最大276,140株の普通株を購入し、総収益は230万ドルであった。10万ドル発行され、純収益は210万ドル。シリーズ3転換可能優先株株は合計546,760株我々の普通株に変換でき、シリーズ4の転換可能優先株株は合計5,523株普通株 に変換でき、それぞれの場合の転換価格は1株4.074ドルである。第三シリーズ株式証の行使価格は1株4.074ドルで、発行後6ヶ月で行使でき、発行日から5年で満期になる。2022年9月には、優先SPAに関連して発行されたすべての優先株が普通株に変換される。
市場で普通株式を発行する
我々は2022年8月15日にBTIG,LLC(“BTIG”)と市販協定(“2022年販売協定”)を締結した。“2022年販売契約”によると、吾らは時々販売代理および/または依頼人として、BTIGを通じて当社の普通株株式を発売·販売することができ、総発行価格は最高2,500万ドルに達するが、“2022年販売協定”は吾などの普通株の発売および売却可能金額に対する若干の 制限を受けなければならない。“一般的な説明I.B.6”で提供されているのは我々の制限に適しているからである.2022年販売協定の条項によると、2022年8月15日に提出された目論見書付録に基づいて、総販売価格610万ドルに達する株を発売することができる。2022年11月9日現在、合計2,477,287株の普通株を売却し、総収益は610万ドルであり、2022年11月9日付の目論見書補足書類を提出し、2022年の販売契約に基づいて、合計170万ドルの総発行価格で私たちの普通株を売却する場合を登録しました。2022年12月14日現在、2022年11月9日の目論見書付録によると、合計579,239株の普通株が売却され、総収益は70万ドルである。“2022年販売協定”によると、株式売却を行う義務はありません。私たちが下した任意の決定は、市場状況や融資ニーズなどに依存します。
契約義務と約束
下表は、同社の2022年9月30日までの契約義務とその義務が将来期間の流動性とキャッシュフローに及ぼす影響をまとめた
| 1年未満 | 1 から3年 | 4 から5年 | 5年以上 | 合計する | ||||||||||||||||
| 運営 レンタル(1) | $ | 79,259 | $ | 189,101 | $ | 48,216 | $ | — | $ | 316,576 | ||||||||||
| 債務 債務(2) | 748 | — | — | — | 748 | |||||||||||||||
| 合計する | $ | 80,007 | $ | 189,101 | $ | 48,216 | $ | — | $ | 317,324 | ||||||||||
(1) は、当社がニュージャージー州プリンストンに位置するオフィスビル賃貸契約に基づいて負う義務を反映している。
(2) ある関連先に発行された無担保対応チケットを反映する.
私たちが上の表に反映した支払い承諾を持つ契約以外に、私たちはbr}正常な業務過程であるCRO、CMOと他の第三者と臨床前研究とテスト、臨床 試験と製造サービスについて他の契約を締結した。これらの契約には最低調達承諾は含まれておらず,事前通知後に をキャンセルすることができるため,上記の契約義務や承諾表には含まれていない.キャンセル時に支払われるべき金額は、キャンセルの日まで、当社のサービスプロバイダに対するキャンセル不可債務を含む、提供されるサービスの支払いおよび発生した費用のみを含む。
| -84- |
重要な会計政策と試算
我々の経営陣の財務状況と運営結果の検討と分析は、米国公認会計基準に基づいて作成された我々の合併財務諸表に基づいている。これらの財務諸表を作成するには、財務諸表に報告されている資産、負債および費用金額、または資産および負債の開示に影響を与えるために、推定および判断を行う必要があります。私たちは私たちの推定と判断を継続的に評価し、 研究開発費の計算に関連する見積もりと判断を含む。我々の見積りは,歴史的経験,既知の傾向や事件,および当時の状況では合理的と考えられていた様々な他の要因に基づいており,これらの要因の結果は資産や負債の帳簿価値を判断する基礎を構成しているが,これらの価値は他のソースからは明らかに見えない.異なる仮定または条件では、実際の結果は、これらの推定値とは異なる可能性がある。
本10−K表の他の部分に含まれる総合財務諸表付記 は、当社の主要会計政策をより詳細に紹介しているが、以下の会計政策が総合財務諸表作成に用いる判断と見積もりが最も重要であると考えられる。
研究開発費
研究·開発費用には、我々のバイオ製薬製品開発に関するすべての直接·間接コストが含まれています。これらの費用には、人員コスト、相談費、第三者に支払われる研究、開発、製造サービス費用が含まれています。これらのコストは発生時に費用を計上しています。
各報告期間終了時に、契約で定義された進捗評価基準に基づいて、第三者サービスプロバイダに支払われる金額と、関連項目が完了した推定進捗とを比較する。我々が評価を作成する際に考慮する要因は, サービスプロバイダによるコスト,実現のマイルストーン,および我々のサービスプロバイダの努力に関する他の基準である.より多くの情報が得られるにつれて,このような 推定値が変化する可能性がある.第三者サービスプロバイダへの支払い時間 および提供されたサービスの進展を推定することに基づいて、これらのコストに関連する前払い費用または負債を記録する。または開発または規制マイルストーン支払いは、そのような事項または関連する解決策 で確認されている。2022年9月30日現在、私たちは計算研究と開発費用の以前の見積もりに何の実質的な調整も行っていません。
最近会計公告が発表された
最近発表された我々の財務状況や経営結果に影響を与える可能性のある会計声明の説明 は、本リスト10-Kの他の部分に含まれる連結財務諸表の付記2に開示されている。
第 7 A項。市場リスクに関する定性的で定量的な開示。
は適用されない.
| -85- |
第br項財務諸表および補足データ
十四行詩生物治療持株会社
連結財務諸表インデックス
| ページ | |
| 独立公認会計士事務所報告
ペンシルバニア州フィラデルフィア監査役事務所ID: |
87 |
| 合併貸借対照表 | 89 |
| 統合の作業報告書 | 90 |
| 合併 株主権益変動表(損失) | 91 |
| 統合されたキャッシュフロー表 | 92 |
| 連結財務諸表付記 | 93 |
| -86- |
独立公認会計士事務所報告
株主と取締役会へ
十四行詩生物治療ホールディングス:
連結財務諸表に関する意見
我々は,Sonnet BioTreateutics Holdings,Inc.とその子会社(当社)が2022年9月30日と2021年9月30日までの連結貸借対照表,同年度までの関連総合経営報告書,株主権益(損失)の変化とbr}現金流量,および関連付記(総称して総合財務諸表と呼ぶ)を監査した。連結財務諸表は,すべての重要な面で,会社の2022年9月30日,2022年と2021年までの財務状況,およびそれまでの年度までの経営成果とキャッシュフローを公平に反映しており,米国公認の会計原則に適合していると考えられる。
注目を行っている
添付されている総合財務諸表 は、当社が経営を継続すると仮定して作成されています。総合財務諸表付記1で述べたように、当社は設立以来経常赤字や運営キャッシュフローが負の場合 であり、その研究開発活動に資金を提供し続けるために大量の追加融資が必要となり、その継続経営能力に大きな疑いを抱かせる。経営陣は、これらの事項の計画についても付記1で説明しています。連結財務諸表には、このような不確実性の結果による可能性のあるいかなる調整も含まれていません。
意見を求める根拠
これらの合併財務諸表は会社経営陣が担当しています。私たちの責任は、私たちの監査に基づいてこれらの連結財務諸表に対して意見を発表することです。私たちは米国上場企業会計監督委員会(PCAOB)に登録されている公共会計士事務所であり、米国連邦証券法および米国証券取引委員会とPCAOBの適用規則と法規に基づいて、会社を独立させなければならない。
私たち はPCAOBの基準に従って監査を行います。これらの基準は、合併財務諸表に重大な誤報がないかどうかに関する合理的な保証を得るために、監査を計画し、実行することが要求されます。当社はその財務報告書の内部統制を監査する必要もありません。私たちの監査の一部として、財務報告の内部統制を理解することが求められていますが、社内財務報告の内部統制の有効性に対する意見を表明するためではありません。したがって、私たちはそのような意見を表現しない。
我々のbr監査には、連結財務諸表の重大な誤報リスクを評価するプログラム(エラーによるものであっても詐欺によるものであっても)、およびこれらのリスクに対応するプログラムを実行することが含まれる。これらの手続きは、連結財務諸表中の金額および開示に関する証拠をテストに基づいて検討することを含む。私たちの監査には、評価に使用される会計原則と経営陣による重大な推定と、連結財務諸表の全体的な報告書を評価することも含まれています。私たちの監査は私たちの意見に合理的な基礎を提供していると信じています。
| -87- |
重大監査事項
以下に述べる重要な監査事項とは、合併財務諸表を当期監査する際に生じる事項であり、 は監査委員会に伝達または要求され、(1)総合財務諸表に対して重大な意義を有する勘定または開示に関するものであり、(2)私たちが特に挑戦的で主観的または複雑な判断に関するものである。重要な監査事項のコミュニケーション は、合併財務諸表に対する私たちの全体的な意見をいかなる方法でも変えることはなく、私たち は次の重要な監査事項を伝達することによって、重要な監査事項或いはそれに関連する勘定或いは開示について単独の意見を提供することはない。
研究開発費の前払いと研究開発費の計上
総合財務諸表付記2及び付記3に記載されているように、研究及び発展コストは発生した時点で提案され、その中には、第三者に研究、発展及び製造サービスのために支払うべき金が含まれている。各報告期間終了時に,会社 は契約で定義された進捗評価基準に基づいて,第三者サービスプロバイダに支払われた金と関連項目完了の推定進捗を比較する.会社が試算を作成する際に考慮する要因には,サービスプロバイダによるコスト,実現のマイルストーン,そのサービスプロバイダの努力に関する他の基準がある。第三者サービスプロバイダへの支払い時間 および会社が提供するサービスの進展を推定することに基づいて、会社は、これらのコストに関連する前払い費用または負債を記録するであろう。2022年9月30日現在,同社が報告した前払い費用やその他の流動資産は240万ドルであり,その一部はこれらのコストに関係しており,計算すべき研究開発費は160万ドルである。
我々 は,第三者サービスプロバイダのいくつかの前払いと計算すべき研究開発費を重要な 監査事項として決定する.完成研究と開発プロジェクトの推定進捗を評価するには、上記の要素を含み、特に監査員の主観的な判断が必要である。
以下は私たちがこの重要な監査問題を解決するために実行した主な手続きだ。当社の2022年9月30日までに発生した前払いと対応研究開発費の見積もりを評価するために,(1)第三者サービスプロバイダから受信したプロジェクト状態に関する契約,領収書,通信におけるbr条項,(2) が第三者サービスプロバイダに確認書を送信したこと,および(3)研究開発活動状況の監視と追跡を担当する者に聞いた。
/s/
私たち は%sがある2015年から当社の核数師を務めています。
2022年12月15日
| -88- |
十四行詩生物治療持株会社
合併貸借対照表
| 9月30日 | ||||||||
| 2022 | 2021 | |||||||
| 資産 | ||||||||
| 現在の 資産: | ||||||||
| 現金 | $ | $ | ||||||
| 前払い料金と他の流動資産 | ||||||||
| 流動資産合計 | ||||||||
| 財産と設備、純額 | ||||||||
| 運営 レンタル使用権資産 | ||||||||
| 延期された 製品コスト | ||||||||
| 総資産 | $ | $ | ||||||
| 負債 と株主(損失)権益 | ||||||||
| 流動負債 : | ||||||||
| 係り先 備考 | $ | $ | ||||||
| 売掛金 | ||||||||
| 課税費用 | ||||||||
| 運営 賃貸負債 | ||||||||
| 繰延収入 | ||||||||
| 流動負債合計 | ||||||||
| 運営 賃貸負債 | ||||||||
| 総負債 | ||||||||
| 引受 と事項があります(注5) | ||||||||
| 株主 (赤字)権益: | ||||||||
| 優先株 ;$額面:株式を授権する発行済みまたは発行済み株式 | ||||||||
| 普通株額面:ライセンス株;そして2022年9月30日と2021年9月30日にそれぞれ発行·発行された株 | ||||||||
| 追加実収資本 | ||||||||
| 累積赤字 | ( | ) | ( | ) | ||||
| 株主権益合計 | ( | ) | ||||||
| 負債と株主権益の合計 | $ | $ | ||||||
合併財務諸表の付記を参照
| -89- |
十四行詩生物治療持株会社
統合の作業報告書
| 9月30日までの年度は | ||||||||
| 2022 | 2021 | |||||||
| 協力 収入 | $ | $ | ||||||
| 運営費用 : | ||||||||
| 研究開発 | ||||||||
| 通常 と管理 | ||||||||
| 運営費総額 | ||||||||
| 運営損失 | ( | ) | ( | ) | ||||
| 利息収入 | ||||||||
| 為替為替損失 | ( | ) | ( | ) | ||||
| その他 収入 | ||||||||
| 純損失 | $ | ( | ) | $ | ( | ) | ||
| 各 は情報を共有する: | ||||||||
| 1株当たり基本と希釈後の純損失 | $ | ( | ) | $ | ( | ) | ||
| 加重平均流通株、基本株、希釈株 | ||||||||
合併財務諸表の付記を参照
| -90- |
十四行詩生物治療持株会社
合併 株主権益変動表(損失)
| 優先株 | 普通株 株 | 追加の 個の実収 | 積算 | |||||||||||||||||||||||||
| 株 | 金額 | 株 | 金額 | 資本 | 赤字.赤字 | 合計する | ||||||||||||||||||||||
| 2020年10月1日の残高 | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||||||
| 普通株と引受権証を売却して発行コストを差し引く | — | |||||||||||||||||||||||||||
| 制限株式単位が帰属する場合に普通株式 を発行する | — | ( | ) | |||||||||||||||||||||||||
| 株式引受証純額株式決済 | — | ( | ) | |||||||||||||||||||||||||
| 許可演習 | — | |||||||||||||||||||||||||||
| 株式ベースの報酬 | — | — | ||||||||||||||||||||||||||
| 純損失 | — | — | ( | ) | ( | ) | ||||||||||||||||||||||
| 2021年9月30日の残高 | ( | ) | ||||||||||||||||||||||||||
| 優先株と普通株の株式承認証を売却し,発行コストを差し引く | — | |||||||||||||||||||||||||||
| 優先株を普通株に転換する | ( | ) | ( | ) | ||||||||||||||||||||||||
| 普通株を売却し,発行コストを差し引く | — | |||||||||||||||||||||||||||
| 制限株式単位が帰属する場合に普通株式 を発行する | — | ( | ) | |||||||||||||||||||||||||
| 株式ベースの報酬 | — | — | ||||||||||||||||||||||||||
| 純損失 | — | — | ( | ) | ( | ) | ||||||||||||||||||||||
| 2022年9月30日の残高 | $ | $ | $ | $ | ( | ) | $ | ( | ) | |||||||||||||||||||
合併財務諸表の付記を参照
| -91- |
十四行詩生物治療持株会社
統合されたキャッシュフロー表
| 9月30日までの年度は | ||||||||
| 2022 | 2021 | |||||||
| 経営活動からのキャッシュフロー: | ||||||||
| 純損失 | $ | ( | ) | $ | ( | ) | ||
| を調整し、純損失と経営活動で使用されている現金純額を照合する: | ||||||||
| 進行中の研究開発を買収しました | ||||||||
| 減価償却 | ||||||||
| レンタル使用権資産の償却を経営する | ||||||||
| 株式ベースの報酬 | ||||||||
| 非現金支払利息 | ||||||||
| 手形対応への許し | ( | ) | ||||||
| 経営資産と負債の変化 : | ||||||||
| 前払い料金と他の流動資産 | ( | ) | ( | ) | ||||
| その他 資産 | ||||||||
| 売掛金 | ||||||||
| 課税費用 | ||||||||
| 運営 賃貸負債 | ( | ) | ( | ) | ||||
| 繰延収入 | ( | ) | ||||||
| 純額 経営活動で使用した現金 | ( | ) | ( | ) | ||||
| 投資活動によるキャッシュフロー: | ||||||||
| 購入 が行っている研究と開発 | ( | ) | ||||||
| 財産と設備を購入する | ( | ) | ||||||
| 投資活動用現金純額 | ( | ) | ( | ) | ||||
| 資金調達活動によるキャッシュフロー: | ||||||||
| 優先株と普通株の株式承認証を売却して得られる収益は発行コストを差し引く | ||||||||
| 普通株売却で得られる収益(発行コストを差し引く) | ||||||||
| 株式承認証を行使して得られた収益は発行コストを差し引く | ||||||||
| 関連側手形の償還 | ( | ) | ||||||
| 純融資活動から提供された現金 | ||||||||
| 現金純増加 (減少) | ( | ) | ||||||
| 現金、 年明け | ||||||||
| 現金、年末 | $ | $ | ||||||
| 非現金経営、投資、融資活動の追加: | ||||||||
| レンタル修正のため経営リース使用権資産と負債を変更 | $ | $ | ||||||
| 株式承認純額(Br)決済 | $ | $ | ||||||
| 制限株式単位が帰属する場合に普通株式 を発行する | $ | $ | ||||||
| 計上すべき費用における進行中開発 | $ | $ | ||||||
| 計上すべき費用のうち普通 株式発行コスト | $ | $ | ||||||
合併財務諸表の付記を参照
| -92- |
十四行詩生物治療持株会社
連結財務諸表付記
1. 業務の組織と記述
業務説明
十四行詩br生物治療会社(“前の十四行詩”)は2015年4月6日にニュージャージー州の会社に登録された。Pre Sonnetは2020年4月1日に公開持ち株の強泣きクリールホールディングス(“最強クリール”)との合併を完了した。合併後、強音クリーアはSonnet BioTreateutics Holdings,Inc.(“Sonnet”または“会社”)と改名した。十四行詩は臨床段階、腫瘍学に集中する生物技術会社であり、革新単一或いは双特定作用生物薬物の独自のプラットフォームを持っている。Fと呼ぶHAB (完全ヒトアルブミン結合)は,完全ヒト単鎖抗体断片(ScFv)を用いてヒト血清アルブミン(“hSA”)に結合し,その上にヒッチハイク を用いて標的組織に輸送する。十四行詩の主要な専有資産SON-1010はインターロイキン12(IL-12)の完全人間バージョンであり、Fと共有結合しているHAB構造,Sonnetは非小細胞肺癌や頭頸部癌を含む固形腫瘍適応の臨床開発を行う予定である。十四行詩は、非ヒト霊長類(NHP)GLP毒性研究を完成させ、臨床で使用される医薬製品の製造に成功した。FDAは2022年3月にSonnetのSON−1010研究新薬(“IND”)申請を承認した。これにより、同社は2022年第2四半期に固形腫瘍患者に対する米国臨床試験(SB 101)を開始することができる。2021年9月、同社はオーストラリアに完全子会社SonnetBio Pty Ltdを設立し、いくつかの臨床試験を行った。十四行詩 は承認を得て、2022年第3四半期に健康ボランティアの中でSON-1010に関するオーストラリア臨床研究(SB 102) を開始した。SB 101とSB 102の研究の初歩的な安全性と耐性データは2022年の第4のカレンダー四半期に発表され、2つの研究の初歩的な薬物動態学と薬効学データは2023年の第2のカレンダー四半期に発表される予定である。
同社は2020年4月にRelipment Treeutics SAの流通株を買収することにより,その最先端化合物SON−080の世界開発権を獲得し,SON−080は完全ヒトバージョンのインターロイキン6(IL−6)である。十四行詩はSON−080治療化学療法による末梢神経病変(CIPN)と糖尿病末梢神経病変(DPN)の目標適応を進めている。十四行詩は、CIPNにおいてSON−080を使用して前の米国段階1 b/2 a研究を開始することができることが承認された。この研究は2023年上半期に初歩的な臨床安全性データを生成する可能性がある。会社が2021年5月にシンガポール新生命治療プライベート有限会社(“新生命”) と締結した許可協定によると、十四行詩と新生命は共同でDPNにおけるSON-080の開発を担当する。CIPNセキュリティデータが利用可能になると、2023年下半期に第2段階研究を開始することを目標としている。
SON−1210 (IL 12−FHAB-IL 15),十四行詩の主要な双特定構築体は,Fを結合しているHABは全ヒトIL−12と全ヒトインターロイキン15(“IL−15”)を含む。この化合物は結腸直腸癌を含む固形腫瘍への適応が開発されている。非ヒト霊長類(NHP)の研究は2022年第4四半期に完成する見込みで、Sonnetは2023年上半期に規制認可手続きを開始する予定だ。2023年の第1四半期に凍結乾燥薬物製品の調合を調製することによって、生産と薬品の供給を確保し、この製剤は初めての人体臨床試験に適用されると予想される。
SON−1410(IL 18−F)のユニット系開発とプロセス開発H現在インターロイキン18(“IL−18”)とIL−12の二重特異性結合が行われており,早期開発のための体外概念検証研究の少量に加え,製剤や分析法開発活動に適した早期実験的薬剤を提供する。プロセス開発活動は2023年まで続き,2023年末までに初歩的な体内マウス研究に適した薬物を生産する可能性がある。
Br社はSON-3015(抗IL 6-F)配列確認を完了しましたHAB−抗形質転換成長因子β)。すでに早期の二重特異性薬物が産生され,保存されており,将来生体マウス研究に使用されている。
| -93- |
十四行詩生物治療持株会社
連結財務諸表付記
全世界大流行-新冠肺炎
2020年3月10日、世界保健機関は新型新冠肺炎ウイルスを全世界大流行と同定した。このような疾患の可能な影響には重大な不確実性 が存在し、その中で会社計画の臨床試験に重大な影響を与える可能性がある。 のような大流行や爆発は、臨床試験場所、臨床研究組織(CRO)、 および/または試験モニター、および試験を支持する他の重要なサプライヤーやコンサルタントの安全を確保することが困難になる可能性がある。そのほか、疫病或いは臨床試験地点付近の疫病は会社が患者を募集する能力に影響する可能性がある。これらの状況や他の新冠肺炎に関連する状況は、会社の臨床試験計画の遅延を招く可能性があり、予想コストを増加させる可能性があり、これらのすべての は会社の業務とその財務状況に重大な悪影響を与える可能性がある。
流動性
設立以来,同社は運営により経常赤字と負キャッシュフローが出現し,予見可能な未来には,主にその潜在製品候補製品の研究開発コストにより,運営に損失が生じることが予想される。
会社はその現金を$としている
当社は2022年8月15日にBTIG,LLCと市販協定(“BTIG”)(“2022年販売協定”)を締結した。2022年の販売契約によると、会社は時々BTIGを介して販売代理および/または依頼者として普通株式を発売することができ、総発行価格は最高$に達する
Br社は、将来的に株式または債務融資、パートナー関係、協力または他のソースを通じて追加資本 を獲得して、会社計画の開発活動を展開する予定です。必要に応じて追加資金を得ることができない場合、会社はこのような資金を受け取るまで、運営を延期、削減、または停止する必要があるかもしれない。各種の内部と外部要素は会社の候補製品がいつ発売されるか、商業化に成功するかどうかに影響を与える。監督管理部門の会社候補製品の承認と市場受容度、これらの製品の開発と商業化の時間長とコスト、および/またはこれらの候補製品の承認過程のどの段階での失敗は、会社の財務状況と将来の運営に重大な影響を与える。
| -94- |
十四行詩生物治療持株会社
連結財務諸表付記
設立以来,運営には主に組織会社,融資獲得,研究と開発による技術開発,臨床前研究がある。当社はその製品が開発段階にある会社に関するリスクに直面しています。これらのリスクは大量の追加資金を必要とし、その研究開発を完成し、その研究開発目標を実現し、その知的財産権を保護し、技術者を採用と維持し、肝心な管理職メンバーへの依存を含む。
2. 重要会計政策の概要
a. 陳述の基礎
添付されている連結財務諸表は、米国公認会計原則(“米国公認会計原則”)に従って作成されている。本付記の適用指針に対するいかなる言及も財務会計基準委員会(FASB)が公布した会計基準編纂(ASC)と会計基準更新(ASU)に掲載されているアメリカ公認会計原則を指す。
b. 整固する
連結財務諸表には、当社及びその完全子会社の勘定が含まれている。すべての会社間口座とbr取引は合併中にキャンセルされました。
c. 予算の使用
アメリカ公認会計原則に基づいて総合財務諸表を作成するには、管理層が総合財務諸表及び付記した金額に影響を与える推定及び仮定 を行う必要がある。これらの連結財務諸表に反映される重大な見積もりおよび仮定 には、研究開発費の計上費用が含まれている。状況,事実,経験の変化に応じて,見積りと仮説 を定期的に審査する.推定された変化は,それらが知られている時間帯 に記録される.実際の結果は経営陣の推定とは異なるかもしれない。
d. 市場情報を細分化する
運営部門は企業の構成要素として定義され、その独立した離散情報は、首席運営意思決定者または意思決定グループが資源をどのように割り当てるかを決定し、業績を評価する際に評価することができる。会社は細分化された市場でbrを運営し、その業務を管理している。
e. 金融商品の公正価値
経営陣 は,このような金融商品の短期的な性質により,当社の金融商品(売掛金を含む)の帳簿価値はおおむね公正価値であると考えている.
| -95- |
十四行詩生物治療持株会社
連結財務諸表付記
f. 財産と設備
財産や設備はコストに応じて入金され、資産の推定耐用年数内に直線法で減価償却される。未使用寿命の延長や資産改善のためのメンテナンス·メンテナンス支出 は,発生時に費用を計上する。廃棄または販売時には、資産を処分するコスト及び関連減価償却及び償却が勘定から差し引かれ、それによって生じるいかなる損益も総合経営報告書に計上される。
g. 長期資産減価準備
事件や環境変化が資産の帳簿価値が回収できない可能性があることを示す場合、会社は財産や設備の減価などの長期資産を審査する。保有·使用する資産の回収可能性は,資産の帳簿価値とその資産が予想される未割引将来のキャッシュフローとを比較することで測定した。1つの資産の帳簿金額 がその推定された未割引将来のキャッシュフローを超える場合、その資産の帳簿価値がその資産の推定公正価値を超える金額について減価費用を確認する。2022年9月30日と2021年9月30日までの財政年度では、減価費用は記録されていない。
H. 発売延期コスト
持分発行に関する法的コスト及びその他のコストは、受領時に繰延発行コストに計上され、持分発行の収益
から差し引かれる。もし融資を放棄すれば、繰延発行費用は費用に計上されるだろう。2022年9月30日現在、同社は$を所有している
i. 協力収入
協働 配置は、(I)ライセンス、(Ii)研究開発活動、および(Iii)いくつかの材料の製造および供給を含む可能性がある複数の構成要素を含むことができる。これらの手配に基づいて支払われるお金には、払戻不可能な支払い、前金、重大な規制および開発活動を完了する際のマイルストーン支払い、販売マイルストーン、製品販売の印税が含まれる可能性があります。収益が今後一定期間にわたって大きな逆転リスクが存在しない可能性が高いまで、可変対価格金額が制限されています。
会社が協力手配義務を履行する際に確認すべき適切な収入金額を決定する際に、会社は、(I)契約で約束された貨物またはサービスを決定するステップと、(Ii)区別できるかどうかを含む契約義務またはサービスが履行義務であるかどうかを決定するステップと、(Iii)可変対価格の制限を含む取引価格の計量と、(Iv)取引価格を履行義務 に割り当てるステップと、(V)収入を会社が各履行義務を履行することを確認するステップとを実行する。
契約義務が異なる履行義務を代表するかどうかを評価し、取引価格を契約内に割り当てる履行義務、いつ履行義務を履行したかの決定、および 可変対価格の確認を評価する際に、会社は重大な判断を適用する。会社が契約条項に基づいてその履行義務を履行する前に対価格を受けた場合、契約負債は繰延収入と表記される。貸借対照表後12カ月以内に収入と確認された繰延収入 は流動負債に分類される予定である。当社は2021年5月に新生活とライセンス契約(“新生活協定”)を締結した。新生命協定のさらなる検討については,付記6を参照されたい。
j. 研究開発費
研究と開発費用には,会社バイオ製薬br製品開発に関するすべての直接·間接コストが含まれている。これらの費用には、人員コスト、相談費、第三者に支払う研究開発と製造サービス費用が含まれています。 これらのコストは発生時に費用を計上する。
報告期間終了時に、会社は契約で定義された進捗評価基準に基づいて、第三者サービスプロバイダに支払われた金額と関連項目が完了した推定進捗を比較する。会社が準備時に考慮した要因 は,サービスプロバイダによるコスト,実現のマイルストーン,そのサービスプロバイダの努力に関する他の基準を含むと推定される.より多くの情報を得るにつれて、これらの推定値は変化するかもしれない。サービス提供者への支払い時間および会社が提供するサービスの進展を推定することに基づいて、会社は、これらのコストに関連する前払い費用または負債を記録するであろう。代表会社が研究·開発サービスを実行する第三者に支払うマイルストーン前金は、サービスを提供する際に費用を計上する。または開発または規制マイルストーン(Br)支払いは、関連する事項が解決された後に確認される。
| -96- |
十四行詩生物治療持株会社
連結財務諸表付記
k. 外貨?外貨
取引 ドル以外の通貨での取引為替変化による損益を取引発生期間の経営活動に計上し、連結経営報告書の為替損失項目で報告する。
当社は、付与日の推定公正価値に基づいて、従業員と非従業員の株式分類株式奨励を計量し、必要なサービス期間(すなわち、対応する奨励の帰属期間)内の補償費用を確認する。当社は発生した没収行為を計算します。サービスの帰属条件に基づく株式奨励について、 社は、サービス期間内に報酬支出を直線的に確認する。当社はその総合経営報告書において株式ベースの報酬 費用を分類し,受賞者の賃金コストを分類する方式や受賞者のサービス支払いを分類する方式と同様である。
m. その他の収入
2020年3月、米国連邦政府はPaycheck保護計画(PPP)の条項を含む“コロナウイルス援助、救済、経済安全法案”(略称“CARE法案”)を公布した。2020年5月、当社は10万ドルのPPPローンを取得しました。 CARE法案の条項によると、当社のPPPローンと関連利息は2021年6月に全額免除されます。購買力平価融資の免損額 は2021年9月30日までの年度総合経営報告書における他の収入に計上されている。
n. 所得税
会社は貸借対照法を用いて所得税を計算する。繰延税項資産及び負債は、既存資産及び負債の帳簿金額とそのそれぞれの課税基礎との間の差額、及び営業損失及び信用繰越間の差額による推定将来の税項結果によって確認される。税金資産及び負債を繰延して税率計量を策定し、その等の一時的な差額を回収又は決済する予定の年度の課税収入に適用されることが予想される。繰延税金資産の一部または全部が現金化できない可能性が高い場合には、推定準備を提供することができる。当社はその所得税申告書に採用されているか予想されている不確定税収状況のメリットを確認しております このような状況がさらに続く可能性があれば。
o. 逆株分割
2022年9月16日、同社はデラウェア州国務長官に改訂された会社登録証明書修正証明書を提出し、この証明書は実現した
| -97- |
十四行詩生物治療持株会社
連結財務諸表付記
基本 1株当たりの純損失の計算方法は,純損失を 期間ごとに発行された普通株の加重平均株式数で割る(および対価格行使を少なくまたは必要としない潜在的な普通株)である。2022年9月30日と2021年9月30日までの年間で、基本加重平均発行された普通株式数にはB系列権証が含まれており、行権価格は$である。 一株ずつです。
希釈後の1株当たり損失には、普通株式承認株式証や株式オプションなどの証券による影響(ある場合)を行使または転換する可能性があり、このような証券は普通株の増資株式を発行することを招く。1株当たり純損失を希釈する場合,普通株の加重平均株数は1株当たりほぼ純損失と同じであるが,これは純損失が存在する場合,希釈証券 は計算に含まれず,影響が逆希釈であるためである。
| 9月30日 | ||||||||
| 2022 | 2021 | |||||||
| 普通株式証明書 | ||||||||
| 引受業者は持分証を承認する | ||||||||
| プライベート株式証明書 | ||||||||
| ジャンティクリール株式承認証 | ||||||||
| Cシリーズ株式承認証 | ||||||||
| シリーズ 3株式承認証 | ||||||||
| 無許可限定株式単位 | ||||||||
q. 最近の会計声明
最近 が発表した
2021年5月、FASBはASU 2021-04を発表した1株当たり収益(主題260)、債務修正および補償(主題470-50)、補償-株式補償(主題718)、ならびに派生ツールおよびヘッジ-エンティティ自己持分契約(主題815-40):独立株式のいくつかの修正または交換に対する発行者の会計 -分類書面コールオプションそれは.ASU 2021-04の改訂は、修正または交換後も持分分類を維持する発行者による独立株式分類 書面コールオプション(例えば、権利証)の会計処理の多様性を明確にし、低減するための指導を提供する。このガイドラインは、2021年12月15日以降の会計年度(移行期間を含む)に有効であり、早期採用が許可されている。当社は現在、本公告を採用した総合財務諸表への影響を評価しています。
最近 を採用しました
2019年12月、FASBはASU 2019-12を発表した所得税主題740−所得税の会計処理の簡略化その目的は所得税会計に関連する様々な側面を単純化することだ。ASU 2019−12は、主題740における一般原則のいくつかの例外 を削除し、主題740の一貫した適用を改善するために、既存のガイドを明確におよび修正した。2021年10月1日にASU 2019-12 を採用することは、連結財務諸表に何の影響もありません。
2020年8月、FASBはASU 2020-06を発表債務--転換可能な債務およびその他の選択(主題470-20)および派生ツールおよびヘッジ--エンティティ自己資本の契約(主題815-40)それは.ASU 2020−06は、埋め込まれた変換機能を個別に計算する必要があるASC 470−20の収益変換および現金変換会計モデルをキャンセルし、契約が株式分類を行う資格があるかどうかを決定するために決済評価を簡略化する。また、新しい案内要求 エンティティは、IF変換方法を使用して、すべての変換可能なツールの1株当たりの収益を計算し、現金または株式で決済可能なツールの株式決済の影響を含む。当社は2021年10月1日にASU 2020-06改正トレーサビリティ法を採用し、2021年初めまでのすべての未返済金融商品に指針を適用します。ASU 2020-06年度を採用した後、利益剰余金期初め残高はASU 2020-06年度の採用による累積影響調整はありません。
| -98- |
十四行詩生物治療持株会社
連結財務諸表付記
3. 費用を計算する
計算すべき費用 には:
| 9月30日 | ||||||||
| 2022 | 2021 | |||||||
| 専門費用 | $ | $ | ||||||
| 報酬 と福祉 | ||||||||
| 研究開発 | ||||||||
| 他にも | ||||||||
| $ | $ | |||||||
4. 賃貸借証書
当社は2019年12月に36カ月間のニュージャージー州プリンストンオフィスビル賃貸契約を締結し、レンタル期間は2020年2月1日から開始した。2022年5月、当社は既存の賃貸契約を改訂し、レンタル期間を約3年間延長し、レンタル修正に計上しました。経営リース使用権資産と負債は修正日に再計量され、両者とも#ドル増加した
2022年9月30日と2021年9月30日までの年間レンタル費用 は以下のように構成されています
| レンタル料金 | 2022 | 2021 | ||||||
| 運営 レンタル料 | $ | $ | ||||||
| 短期レンタル料金 | ||||||||
| レンタル総コスト | $ | $ | ||||||
2022年9月30日、加重平均残存賃貸期間は
2022年9月30日と2021年9月30日までの年度経営リースに関するキャッシュフロー情報は以下の通り
| 2022 | 2021 | |||||||
| 賃貸負債に含まれる金額を計量するために支払う現金: | ||||||||
| 運営 はレンタルを運営するキャッシュフローから | $ | $ | ||||||
| -99- |
十四行詩生物治療持株会社
連結財務諸表付記
2022年9月30日まで、レンタルをキャンセルできない未来の最低レンタル支払いは以下の通りです
| 会計年度 年 | ||||
| 2023 | $ | |||
| 2024 | ||||
| 2025 | ||||
| 2026 | ||||
| 未割引のレンタル支払い合計 | ||||
| 差し引く: 利子を計上する | ( | ) | ||
| 賃貸負債合計 | $ | |||
5. 引受金とその他の事項
法的手続き
当社はその正常な業務過程で発生した様々な訴訟、クレーム、その他の法律手続きに時々参加しています。これらの問題の結果は不確定であるが,経営陣はこれらの問題を解決する最終コストが会社の総合財務状況,経営業績やキャッシュフローに大きな悪影響を与えないと予想している。
ライセンス プロトコル
2012年7月、当社はXOMA(US)LLC
(“XOMA”)と発見協力協定(“協力合意”)を締結し、これにより、XOMAは当社に非排他性、譲渡不可許可及び/又は権利を付与し、抗体及び関連タンパク質の発見、最適化及び開発に関連するいくつかの
材料、技術及び関連資料を使用し、それに基づいて製品を開発及び商業化する。会社はXOMAへの支払いまたはマイルストーン支払いが義務付けられており,総額は
$である
当社は2015年8月、メルクKGaA(“ARES”)の完全子会社Ares Tradingとライセンス契約(“ARESライセンス契約”)を締結した。ARESライセンス契約の条項によると、ARESは、アトクシャキン(“アトシャキン”)を研究、開発、使用、商業化するための再許可可能なグローバル独占特許使用許可を同社に付与しており、アトクシャーキンは末梢神経疾患および血管合併症を治療する低用量ヒトIL-6製剤である。ARESライセンス契約によると、会社はその販売製品の純売上高に応じてARESに1桁の高額特許権使用料を支払う。印税 は、(I)その国/地域で初めて商業販売された指定された時間帯および(Ii)その製品がその国/地域で有効にクレームされた最終日(遅い者を基準とする)まで製品および国/地域によって支払われる。
| -100- |
十四行詩生物治療持株会社
連結財務諸表付記
2019年1月、当社はSartorius Stedim
Cellca GmbH(“Cellca”)とフレームワークサービス及び許可プロトコル(“Cellcaプロトコル”)を締結し、このプロトコルに基づいて、Cellcaは当社にグローバル、非排他性、永久性及び譲渡不可能なbr}ライセンスを付与し、開発、製造又は製造、使用、販売、輸入、輸出及び/又はCellca生産指定遺伝子組換え細胞系を他の方法で商業化し、このような細胞系のための上流生産工程の製品を開発、製造又は製造し、販売、輸入、輸出及び/又は他の方法でCellca生産指定遺伝子組換え細胞系を商業化し、当該等の細胞系のために上流生産工程の製品を開発、製造又は製造する。Cellcaプロトコル
は、どちらか一方が6ヶ月の通知を出して終了しない限り、または正当な理由で終了した場合、14日間通知を与えます。
2021年10月に、当社はBrink Biologics Inc.
(“Brink”)と非独占許可プロトコル(“Brinkプロトコル”)を締結し、これによりBrinkは当社の非独占、譲渡不可能許可及び有限権利
再許可いくつかの材料及び関連資料を授与し、細胞に基づく分析を発展させ、製品生産及び商業化に必要なロット、品質制御、安定性、効力、効力或いは任意の他のタイプの分析を行う。Brinkプロトコルは2020年11月1日に発効し、
の初期期限は3年であり、いずれか一方が書面で
を終了しない限り、または会社が許可財産を製品開発段階に使用することを停止しない限り、自動的に1年更新される。製品開発段階では,会社
は毎年約$を支払う義務がある
当社は2022年2月にInvivoGen
SAS(“InvivoGen”)と生体材料許可プロトコル(“InvivoGenプロトコル”)を締結し,これによりInvivoGenは当社に世界的な非独占許可を付与し,当社が何らかの
電池を用いて研究,開発および/または品質管理を報告することを許可した。InvivoGenプロトコルの初期期限は3年
であり,会社が書面で通知して約ユーロを支払った場合,さらに2回3年間延長することができる
2022年3月,当社はProteoNic
B.V.(“ProteoNic”)と材料譲渡および許可プロトコル(“ProteoNicプロトコル”)を締結し,これによりProteoNicは自社に非排他性,譲渡不可能,再許可不可能なbr}ライセンス(ProteoNicプロトコル別規定者を除く)を付与し,自社細胞系用ベクターを産生するためのプラスミドおよびDNA配列を含み,当社の製品の研究,開発および商業化に供されている。br}ライセンスはいずれも終了し続ける。その会社はProteoNicに支払う義務があります。マイルストーンの支払いがあります。総額は最大でユーロに達します
| -101- |
十四行詩生物治療持株会社
連結財務諸表付記
研究と開発協定
2021年12月に、当社はNavigo Proteins GmbH(“Navigo”)と研究開発プロトコル(“Navigoプロトコル”)を締結し、このプロトコルに基づいて、Navigoは特定の評価および開発プログラムを実行して、いくつかのbr}材料を評価してその商業潜在力を決定する。Navigo協定の条項によると、会社は、Navigoにいくつかの技術を使用して評価および開発活動を行う免印税、 非独占、世界、再許可不可能、譲渡不可能な権利および許可、およびNavigoが会社に(I)研究、開発、使用、販売、流通、輸入または他の方法である材料を商業的に利用する独占、世界、永久、撤回不可能、再許可可能、譲渡可能、印税免除の権利および許可、および(Ii)非独占的、世界的範囲、永久的、再許可可能、および(Ii)非独占的、世界的範囲、永久的、再許可可能、このような材料を製造または製造した権利およびライセンスは、br}を譲渡することができない。その会社は$を支払った2022年9月30日までの年度の総合経営報告書には、買収が行われている研究開発費および研究開発費に計上された百万元の技術アクセス費と記されている。会社はNavigoに合計$までの支払いまたはマイルストーンの支払いを義務付けています“ナビゲーションプロトコル”で概説されたいくつかの評価と発展マイルストーンを実現した上で、100万ユーロに達した。
雇用契約
Br社はすでにその高級管理者とある従業員と雇用契約を締結しており、会社が無断で雇用を終了したり、従業員が十分な理由で雇用を中止したりした場合、解散費と継続福祉を提供することが規定されており、両者は契約中の定義 を満たしている。また,制御権変更後に雇用関係を終了すると,会社 が無断で終了しても,従業員が正当な理由で終了しても,従業員の初期株式オプション付与されていない任意の未付与部分はただちに に帰属する.
6. 協力協定
新生命協定によると,当社はマレーシア,シンガポール,インドネシア,タイ,フィリピン,ベトナム,ブルネイ,ミャンマー,マレーシア,シンガポール,インドネシア,タイ,フィリピン,ベトナム,ブルネイ,ミャンマーで特定組換えヒトIL−6,SON−080(“化合物”)を含む医薬製剤(これらの製剤を“製品”)を開発·販売し,ヒト糖尿病末梢神経病変を予防,治療または緩和する(“DPN領域”)。ラオス人民民主共和国とカンボジア(“排他的領土”)。br}新生活は、(1)専属許可証の範囲を拡大する権利があり、2021年12月31日に満了する化学療法によるヒト末梢神経障害(“CIPN領域”)を予防、治療または緩和することを含む。人民Republic of China、香港および/またはインドを含む、および/またはbr}(2)ライセンスの地域範囲は、2021年12月31日に満了する独占的なbrである。
その会社は世界のどこでも化合物と製品を生産するすべての権利を維持するだろう。当社は新生活と後続供給協定を締結し,この合意に基づき,当社は排他的地域のDPN油田での開発および商業化のために双方が協議した条項で新生活に製品を供給する。同社はまた、新生命がライセンスから利益を得ることができるいくつかの臨床前と臨床開発技術ノウハウの譲渡に協力する。
新人寿は、専属区域内のDPN領域での臨床研究と他の非臨床研究及びその他の開発と監督活動及び製品商業化の費用と責任を負担し、責任を負う。
ニューライフは同社に$を支払いました
新生命協定は、製品ごとに国/地域ごとに有効になり、最終満期国/地域の最終満期製品の特許権使用料の期限が満了したときに終了するが、以下の条件によって制限される:(I)当事者の早期解約権 は、他方の重大な違約または破産または倒産により早期に終了する権利と、(Ii)自社の買い戻し権と新生命の復帰権(以下、定義とする)を含む。
また,New Lifeは当社にNew Lifeの権利(“買い戻し権”)の独占選択権を付与し,New Life権利は合意すべき条項に従って専属地域の1つまたは複数の国のDPN現場製品を買い戻す権利(“買い戻し権”)を付与し,このような選択権は製品を適用する第3段階の実験開始時に終了する.
| -102- |
十四行詩生物治療持株会社
連結財務諸表付記
収入 確認
同社はまずASC 808に基づいて新生命協定を評価した協力手配新しい生命プロトコルまたは新しい生命プロトコル内のアカウント単位が、双方のリスク、リターン、および活動に基づく協力スケジュール を表すかどうかを決定するために使用される。会社は新生命が顧客を代表すると判断し、ASC 606の関連指導を応用した顧客と契約を結んだ収入 新しい生命協定の下で適切な会計を評価するために。本ガイドラインによると、会社はこの手配に基づいて、(I)専属区域内での現場開発、マーケティング、輸入、使用および商業化製品の許可証(“ライセンス”)、および(Ii)技術移転と臨床開発 と監督活動(“研究開発活動”)を決定した。CIPN領域および地域を拡張するオプションおよび将来の供給プロトコルは、材料権利をbrクライアントに譲渡しない限り、これらの調達は別個の契約として入金されるオプション調達である。その会社はこのような単独の契約を評価し、何の実質的な権利が存在するかを決定しなかった。会社はライセンスと研究開発サービスが互いに区別されていないことを確定したため、これらの重大な約束を履行義務 に統合した。
会社は単一履行義務の初期取引価格を$と決定した
連携
が単一業績義務から得られた収入は、研究開発サービスの見積もり業績で確認される。会社
確認しました$
7. 株主権益
私募優先株
当社は2022年8月に、数名の認可投資家と証券購入協定(“優先SPA”)を締結し、(I)の発行及び販売を合算したその第3シリーズは優先株株に転換し、価値を述べる$1株、
(Ii)その第4シリーズ転換可能優先株株、陳述価値$1株当たり、および(Iii)シリーズ3株式権証、最大購入
| -103- |
十四行詩生物治療持株会社
連結財務諸表付記
系列3と系列4は優先株に変換可能な
株に投票権はないが,単一種別の普通株株主として改訂された会社登録証明書について投票する権利があり,普通株流通株の逆分割
を実現する.シリーズ3転換可能な優先株の1株当たり換算後に投票する権利があり、
私募は2022年8月15日に終了し、株主は2022年9月15日投票で逆株式分割を承認した。
シリーズ3とシリーズ4は優先株に変換可能
普通株 株
同社は2022年9月30日までに販売したBTIGと締結した2022年販売契約に基づき、普通株を発行し、総収益は$
また、2022年9月30日までの年度内に、当社は発行します制限株式単位が帰属する場合の普通株。
2021年8月,同社は一次引受の公募株を完成させ,約$を獲得した
今回の発行については,会社は引受業者に30日間の超過配給選択権を付与して,最も多く購入する
また
では、株式購入承認証
当社は2021年2月にBTIGと市場販売協定(“2021年販売協定”)を締結した。“2021年販売契約”によると、当社はBTIGを介して時々販売代理および/または依頼者として普通株br株を発売する能力があり、総発行価格は最高$に達する
また、2021年9月30日までの年度内に、当社は発行します制限株式単位が帰属する場合の普通株。
| -104- |
十四行詩生物治療持株会社
連結財務諸表付記
修正と練習を許可する
B系列権証保有者は、2021年9月30日までの年度内に行使される株式証明書で得られた金は$である
2021年9月30日までの年間で、強アンティクリール株式証購入
2021年9月30日までの年間で2021年の公開発行と同時に販売される予融資権証の純シェア決済 は、発行につながる普通株です。
普通株式証明書
2022年9月30日まで、以下の株式分類株式証と関連条項はまだ期限が切れていない
株式承認証 卓越した | 演習 価格 | 期限切れ日 | ||||||||
| 普通株式証明書 | $ | |||||||||
| 引受業者は持分証を承認する | $ | |||||||||
| プライベート株式証明書 | $ | |||||||||
| ジャンティクリール株式承認証 | $ - $ | |||||||||
| Bシリーズ株式承認証 | $ | |||||||||
| Cシリーズ株式承認証 | $ | |||||||||
| シリーズ 3株式承認証 | $ | |||||||||
上述したように、第3シリーズ株式承認証は2022年に発行され、私募優先株と関係がある。私募で得られた純額
は発行日の相対公正価値に応じて系列3及びシリーズ4に優先株及び系列3株式承認証を転換することができ,分配金額は$である
| 行権 価格 | $ | |||
| 用語.用語 | ||||
| 期待変動 | % | |||
| 配当金 収益率 | % | |||
| 無リスク金利 | % |
2020年4月、当社は“2020年総合持分インセンティブ計画”(以下、“計画”)を採択した。2022年9月30日現在、この計画により発行可能な普通株式総数はそれは.この計画に基づいて発行可能な株式数を毎年1月1日に普通株式流通株の4%増加させる。本計画は、株式オプション、制限株式単位および奨励、株式付加権、および適切と考えられる他の種類の奨励を含む、本計画の条項に基づいて株式ベースの報酬を付与することを可能にする。奨励条項は会社の取締役会によって決定される。
| -105- |
十四行詩生物治療持株会社
連結財務諸表付記
制限された 在庫単位
付与されていないRSUは、サービス終了時に没収される。RSUの公正価値は,日会社普通株に付与された公正時価 に等しい.RSU費用は授権期間内に直線的に償却される。
| 9月30日までの1年間 | ||||||||
| 2022 | 2021 | |||||||
| 研究開発 | $ | $ | ||||||
| 通常 と管理 | ||||||||
| $ | $ | |||||||
この計画におけるRSU活動を以下の表にまとめる
| RSU | 重みをつける Average Grant Date Fair Value | |||||||
| 2020年10月1日未帰属残高 | $ | |||||||
| 授与する | $ | |||||||
| 既得 | ( | ) | $ | |||||
| 没収される | ( | ) | $ | |||||
| 2021年9月30日現在の未帰属残高 | $ | |||||||
| 授与する | $ | |||||||
| 既得 | ( | ) | $ | |||||
| 没収される | ( | ) | $ | |||||
| 2022年9月30日の未帰属残高 | $ | |||||||
2022年9月30日現在,無帰属RSUに関する未確認補償支出総額は$である百万, は以下の重み付き平均期間で確認されると予想される.
| -106- |
十四行詩生物治療持株会社
連結財務諸表付記
9. 所得税
2022年9月30日現在、同社は$を所有している
国税法の所有権条項の変更により、当社の純営業損失繰越の可獲得性 は将来の期間の課税収入の年間制限を受ける可能性があり、このような繰越の最終使用 を大きく制限する可能性がある。当社はその持分融資が実益所有権に及ぼす歴史や潜在的な影響を分析していないため、繰り越しの純営業損失が“国内収入法典”第382節の制限を受けるかどうかは未定である。制限のある範囲では繰延税項資産は減少するが,推定免税額は相殺的に減少する。
不確定な税収頭寸が存在する場合、当社は、 を実現することを可能にするために、税収頭寸の税収割引を確認する。税務優遇がより実現可能かどうかに関する決定は、税務状況の技術的利点および入手可能な事実と状況の考慮に基づいている。当社はその総合経営報告書の所得税引当金内で、未確認の税収割引のために計上すべき利息と罰金を確認します。未確認のbr税金割引が記録されていません。
| -107- |
十四行詩生物治療持株会社
連結財務諸表付記
繰延税金の一時的な差をもたらす税収の影響は以下のとおりである
| 9月30日 | ||||||||
| 2022 | 2021 | |||||||
| 繰延納税資産: | ||||||||
| 純営業損失繰越 | $ | $ | ||||||
| 研究と開発信用繰り越し | ||||||||
| 償却する | ||||||||
| 株式ベースの報酬 | ||||||||
| 運営 賃貸負債 | ||||||||
| 費用とその他を計算すべきである | ||||||||
| 財産 と設備 | ||||||||
| 繰延税金資産総額 | ||||||||
| 減算: 推定免税額 | ( | ) | ( | ) | ||||
| 繰延納税義務: | ||||||||
| 運営 レンタル使用権資産 | ( | ) | ( | ) | ||||
| 純繰延税金資産 | $ | $ | ||||||
会社は2022年9月30日と2021年9月30日までの年間で、いかなる所得税支出や福祉も記録していない。連結財務諸表に反映される法定連邦所得税率で計算される所得税(費用) 福祉と所得税の入金は以下のとおりである
| 9月30日までの1年間 | ||||||||
| 2022 | 2021 | |||||||
| アメリカ連邦法定金利 | ( | )% | ( | )% | ||||
| 州税、連邦福祉の純額を差し引く | ( | ) | ( | ) | ||||
| 評価免税額の変更 | ||||||||
| 信用を研究·開発する | ( | ) | ||||||
| 恒久的差異 | ( | ) | ||||||
| 海外の税率の違い | ||||||||
| 他にも | ||||||||
| 実際の所得税率 | % | % | ||||||
2022年8月、米国は“2022年インフレ削減法案”(IRA)を公布した。アイルランド共和軍には会社が最低税額を代替することを含む税金に関する規定が含まれています
10. 後続事件
会社は貸借対照表の日付から2022年12月15日までの後続事件を評価し、2022年12月15日は連結財務諸表を発表できる日である。
前に発表したように、当社はナスダック上場規則第5550(B)(1)条(以下、規則と略す)に適合しておらず、この規則はナスダック資本市場に上場する会社
が少なくとも$を維持しなければならないことを要求している
2022年11月9日、同社は普通株の要約と販売を登録し、総発行価格は最高$に達する目論見補充書類を提出した
その後
から2022年9月30日から本出願の日まで、会社は販売しました普通株で総収益
$と交換する
| -108- |
第 項9.会計と財務開示に関する変更と分岐。
ない。
第 9 A項。制御とプログラムです
私たちの情報開示制御評価に関する結論
我々のbrは、1934年の証券取引法(改正証券取引法)に基づいて提出された定期報告で開示された重大な情報が、米国証券取引委員会規則および表に指定された期間内に記録、処理、まとめ、報告され、適切な保証を提供し、必要な開示について決定するために、適切な保証を提供するために、開示制御および手続きを維持することを目的としている。私たちは、経営陣(私たちの主要幹部や首席財務官を含む)の監督と参加の下で、取引法規則13(A)-15(E)の定義に適合する経営陣(私たちの主要幹部および最高財務官を含む)の監督と参加の下で、開示制御とプログラムの設計と操作の有効性を評価した。この評価によると、我々の最高経営責任者および最高財務官は、2022年9月30日までに、我々の開示制御プログラムおよびプログラムが有効であると結論した。
経営陣財務報告内部統制年次報告書
我々の経営陣は、取引法規則13 a-15(F) および15 d-15(F)の規定に従って、財務報告に対する十分な内部統制の確立と維持を担当している。財務報告の内部統制はアメリカ公認会計原則に基づいて、財務報告の信頼性と外部報告目的の財務諸表の作成に合理的な保証を提供する過程である。
財務報告に対する内部統制は、(1)取引および資産処置を合理的、詳細、正確かつ公平に反映する記録の維持に関連する政策およびプログラム、(2)米国公認会計原則に基づいて財務諸表を作成し、管理層および取締役の許可のみに基づいて収支を行うために必要な記録を確保するための合理的な保証を提供する。(3) は、TIS財務諸表に重大な影響を与える可能性のある不正取得、使用、または処理を防止またはタイムリーに発見することについて、我々の資産を合理的に保証する。
我々の経営陣(CEOやCEOを含む)の監督と参加の下で,我々はbrの枠組みで決定された基準に基づいて財務報告の内部統制の有効性を評価した内部統制--統合フレームワーク(2013)トレデビル委員会が後援して組織委員会が発表した。この評価によると、我々の経営陣は、財務報告に対する内部統制が2022年9月30日から有効であると結論した。
その固有の限界のため、財務報告書の内部統制は誤った陳述を防止したり発見できない可能性がある。また,将来的に任意の有効性評価を行う予測は, 条件の変化により制御が不十分になる可能性がある,あるいは政策やプログラムの遵守度が悪化する可能性があるというリスクの影響を受ける可能性がある.
本 年次報告書には,我々の独立公認会計士事務所の認証報告は含まれておらず,我々は“非加速申請者”であるため,加速申請者に適した上場企業の各種報告要求の何らかの免除を利用する可能性があり,サバンズ−オクスリー法案 第404(B)節の監査師認証要求を遵守する必要はないがこれらに限定されない。
財務報告内部統制変更
2022年9月30日までの第4四半期において、財務報告の内部統制(“取引法”第13 a-15(F)および15 d-15(F)条の定義による)には何の変化もなく、財務報告の内部統制に大きな影響を与えたり、それに大きな影響を与えたりする可能性が高い。
第 9 B項。他の情報。
ない。
第 9 C項.検査を妨害する外国司法管轄区域を開示する。
は適用されない.
| -109- |
第三部
プロジェクト 10.取締役、行政者、会社ガバナンス
役員.取締役
次の表に当社の現取締役に関するいくつかの情報を示します。取締役の任期は次年度の株主総会および後継者が選出され資格を得るまでです。
| 役員.取締役 | 年ごろ | Year First 取締役になりました | ||
| パンカジ·モーハン博士 | 58 | 2020 | ||
| Nailesh バート | 50 | 2020 | ||
| アルバート·デルネス | 60 | 2020 | ||
| ドナルド·グリフィス | 74 | 2020 | ||
| ラグ ラオ | 60 | 2020 | ||
| ローリーマクニール | 50 | 2022 |
以下は、現在当社取締役を務めている個人の簡単な履歴書であり、その根拠は、同社等が当社に提供している資料である。
パンカj Mohan博士
Pankaj Mohan博士は2015年に十四行詩を創立し、その後も取締役会のメンバーを務め、合併終了時に私たちの取締役会(“取締役会”)の議長に任命された。モハン博士は2018年6月に十四行詩主席に就任し、2019年1月に十四行詩最高経営責任者に就任し、合併完了時に総裁兼当社の最高経営責任者に任命された。2011年1月から2018年6月まで、2011年に設立された腫瘍生物製剤会社(現在はOutlook治療会社;ナスダック:OTLK)の最高経営責任者兼会長を務めている。これまで、モハン博士は百時美施貴宝社で生物工芸と製品開発の業務運営とポートフォリオ管理担当を務め、遺伝子テーク社で生物工芸工学部門の役員を務めていた。これまでモハン博士は礼来会社で高級マネージャーを務めていた。1993年5月から1996年4月まで、モハン博士はイギリスロンドン大学学院生物化学工学高級センターでアシスタント教授(講師/研究員)を務めた。Mohan博士は,英バーミンガム大学バーミンガム大学化学工学学院の生化学工学博士号,英国ロンドンミドルサックス大学商学院の財務管理修士号,デューク大学福庫商学院の幹部管理課程(AMP)およびインドロッキーインド工科大学の化学工学学士号を有している。彼は生物過程操作に関する業界参考書(McGraw Hill)の著者でもある。会社はモハン博士が取締役会に貴重な貢献をすることができると信じている。生物製薬業界を広く理解しており、以前幹部を務めた経験があるからである。
Nailesh バート
Nailesh Bhattは2018年7月からSonnet取締役会に勤務しており、合併終了時に私たちの取締役会メンバーに任命されています。バートさんは2018年1月以降、VGYAAN PharmPharmticals LLCのCEO兼取締役会メンバーを務めており、VGYAAN PharmPharmticals LLCは臨床的に重要な薬剤の開発に注力し、商業化している会社です。これまで、バートさんは2001年11月にProximare社を創業し、取締役社長を務めています。 Proximareは製薬業界に専念する戦略コンサルティング会社です。バートさんは2018年4月以降、Azity製薬会社の取締役会メンバーも務めています。2015年6月、バートさんはProximare生命科学基金を設立しました。バートさんはボストン大学で文学の学士号を取得し、生物学を専攻しています。会社は、製薬業界でフォーチュン500社のスタートアップ企業と長年協力してきた経験から、取締役会に貴重な貢献をすることができると信じています。
| -110- |
アルバート·デルネス
アルバート·デルネスさんは、2019年10月からSonnetの取締役会に在籍しており、合併完了時に取締役会メンバーに任命されています。 デルネスさんは、バイオ製薬業界で公認されているバイオテクノロジー工学の専門家であり、上流、下流、灌流/完了プロセスに関して専門的な知識を有しています。ダイネスは2019年7月以来、利邦コンサルティング会社傘下の取締役会社Coment Engineering Services,Inc.の取締役社長を務めており、同社は利邦の生命科学部門である。1988年、Dyrnessさんが他の人と共同でComent Engineering Services,Inc.を設立したのは、エネルギーと生命科学産業にサービスを提供するエンジニアリングコンサルティング会社である。Comentは最初に旧金山湾区に4人の従業員しかいなかったが,現在130人以上のエンジニアに発展し,カナダトロント,シンガポール,ノースカロライナ州ローリー,オレゴン州ポートランド,ボストン,マサチューセッツ州オーウェン,カリフォルニア州サンラモンに事務所を設置している。2016年、デルネスは社長兼Comentの首席技術官となり、2017年に700人の従業員を持つエンジニアリングコンサルティング会社リバーンコンサルティングと合併した。2015年12月から2017年9月まで、腫瘍学会社(現在Outlook Treateutics,Inc.;ナスダックコード:OTLK)取締役会のメンバーも務めた。 1986年、Dyrnessさんはマサチューセッツ工科大学を卒業し、そこで彼は機械工学と創業精神を学んだ。 会社はDyrnessさんがNisダック上場企業に長年の経験を持っているため、Dyrnessさんが生物製薬業界に含まれる長年の創業経験を加えたので、貴重な貢献をすることができると信じている。
公認会計士ドナルド·グリフィス
2015年4月の設立以来、公認会計士Donald Griffithは十四行詩取締役会のメンバーを務め、2015年4月から2018年6月まで十四行詩取締役会議長を務め、合併終了時に私たちの取締役会メンバーに任命された。グリフィスさんは、Sonnetの財務ディレクターを2019年1月1日から務めており、統合以来、当社の財務ディレクターを務めています。財務総監を務める前、2015年4月から2016年12月までの間にSonnetの最高経営責任者と財務責任者を務めた。以前、グリフィスさんは、2011年から2018年までの間、会社の最高財務責任者兼腫瘍生物製剤会社(現Outlook Treateutics;ナスダック:OTLK)の役員を務めていました。グリフィスさんは40年以上の財務および会計経験を有しており、LLCの創始者でありパートナーであるニュージャージー州の会計士Stolz&Griffithです。当社は、グリフィスさんが長年の財務および製薬業界の経験によって、取締役会に貴重な貢献をすることができると信じています。
ラグ ラオ
ラグは、2019年11月からSonnetの取締役会に勤めており、合併完了時には当社の取締役会メンバーに任命されています。 ラオさんは、連続創業者、戦略的ビジネスコンサルタント、エンジェル投資家です。ラオはいくつかのハイテク会社を設立し、拡張し、成功させた。彼の33年間のキャリアの中で、Raoさんは、USA.gov、TSA Screen Gateway、Cancer.gov、その他の電子政府の行動など、顧客に戦略的助言と注目のプロジェクトを提供してきました。Vistageプリンストン大学の会長として、2012年7月から2017年3月までの間に、Raoさんは、総収入が20億ドルを超える企業のミドルエンド市場CEOとビジネスリーダーの3つの実績の高い同業者諮問委員会を経営しています。1995年8月から2008年7月まで、InfoZenの会長と社長として、Raoさんは10億ドル以上の米連邦政府契約を管理していた。ラオさんはインダス企業家協会(tie.org)20年の特許会員であり、インディアン協会の5年間のスポンサーでもあります。彼はCellix BioSciences(2016年1月から2017年1月)、br}Paper Battery Company(2009年1月から2018年12月)、Kovid Group(2016年2月から2017年10月)、WizNucleus(2010年6月から現在)、InfoZen(1995年8月 から2008年7月)を含むいくつかの会社で取締役会を務めている。ラオさんは、社会的起業とコミュニティサービスに積極的に参加しています。彼は2012年12月に他人と共同でヒンドゥー教ユダヤ人連盟を創設し、2007年3月に世界各地の宗教多様性を維持するために宗教自由フォーラムの創設に参加した。彼は無限基金(ニュージャージー州)、アザ·ヴィディア·グルクーラム(ペンシルベニア州)、家庭サービス機関(メリーランド州)で非営利取締役会の職を務めていた。Raoさんは、ジョージワシントン大学金融MBA号(1991年12月)、バージニア工科大学コンピュータ科学修士号(1986年12月)を所有している, インド工科大学マドラス工科大学電気工学学士号(1984年6月)当社は、Raoさんが、生物科学技術分野での を含む25年間の創業経験と、15年間の幹部経験を持っているため、取締役会に貴重な貢献をすることができると信じています。
| -111- |
ローリーマクニール
Lori McNeillは2022年9月から私たちの取締役会に勤めており、2019年9月以来私たちのビジネス諮問委員会の議長を務めています。McNeillさんは2016年からずっとMcNeill Consulting,LLCの創業者兼最高経営責任者であり、同社は管理コンサルティング会社であり、より有効なリーダーの育成に専念し、変革管理措置のシームレスな進行を確保している。McNeillさんは医療保健業界で20年以上のbr経験を持ち、その中で13年にファイザーで働き、その中に総合健康業務部門で全世界運営事務室主任を務めることを含む。2020年から2021年まで、McNeillさんはGlobal PPE、Inc.の首席運営官兼取締役会議長を務め、同社は医療保健と政府実体の新冠肺炎疫病に対抗する全世界の個人保護設備と安全用品サプライヤーである。彼女は複数の機関から認められた:世界100強女性リーダーシップ -グローバル国際貿易促進委員会、2021年、Changemakerサミット賞受賞者、2021年、女性リーダーシップ状況 -HR.com封面記事、2020;およびファイザー国際革新卓越賞、2011、現在女性経済学グローバル会長 。当社はMcNeillさんが役員会に貴重な貢献をする能力があると信じている。彼女は医療業界で20年以上の経験を持っており、高級指導職を含むからである。
執行官
次の表に会社の現在の幹部に関するいくつかの情報を示します
| 執行官 | 年ごろ | ポスト とオフィス | ||
| パンカジ·モーハン博士 | 58 | 社長 とCEO | ||
| ジェイ·クロス | 52 | 最高財務官 | ||
| ジョン·K·シーニー博士 | 70 | 首席科学官 | ||
| スーザン·デクスター | 67 | 首席技術官 | ||
| リチャード·ケニー医学博士です | 64 | 首席医療官 |
以下は、現在当社の役員を務めている個人の簡単な履歴書であり、その根拠は、このような人が当社に提供している情報である。
パンカj Mohan博士
ガイド下の 説明を参照してください。
ジェイ·クロス
Jay クロスは2019年5月に十四行詩に加入し、その後同社の首席財務官と首席業務官を務め、合併完了時に会社の首席財務官に任命された。十四行の詩に参加する前に、クロスさんは、2015年11月~2019年3月にチャールダン資本の医療投資銀行のチームで取締役社長を務めている間、バイオ製薬に専念しました。これまで、クロスさんは2014年5月から2015年6月まで、Alere Financial Partnersで取締役を務め、2011年5月から2013年10月までBalyasny Asset Managementがシニアアナリスト を担当しました。1999年にHambrecht&Quistのヘルスケア株研究チームでバイオテクノロジーを担当するアシスタントアナリストを務め、金融キャリアを開始した。クロスさんはエール大学医学部で修士号、ワシントン大学で心理学の学士号を取得している。
ジョン·K·シーニー博士
John K.Cini博士は2015年に共同で十四行詩を創立し、その後同社の首席科学官を務め、合併終了時に同社の首席科学官に任命され、同社の発見と開発計画の監督と指導を担当した。彼の職責は癌と免疫腫瘍学標的の選択過程及び概念検証テストを監督することである。十四行詩に加入する前に、2011年1月から2015年4月まで腫瘍学会社で発見と発展科学部総裁副主任を務めた。Cini博士はすでに20種類以上の新型モノクロナル抗体製品を発見段階からIND段階に推進することに成功した。彼は多くの新製品と調合特許と応用を持っている。また、早期発見、研究開発と生産、臨床試験と商業化を通じて、いくつかの成功した新型生物製品に直接参与した。これまでのポストには,美徳士(2010年に百時美施貴宝に買収された)で取締役幹部を務め,ジョンソン(生物科学技術整形外科や薬物研究)で発見科学指導職を務め,バイエルを務めていた。シーニー博士の系統生物学方面の専門治療領域は腫瘍学、免疫腫瘍学、炎症、骨粗鬆症、傷口癒合、外科癒着と細胞老化を含む。シーニー博士はノーステキサス大学の生化学博士号を持っています。
| -112- |
スーザン·デクスター
スーザン·デクスターは2019年5月から十四行詩の契約顧問を務め、首席技術官を務め、合併完了時に会社の常勤首席技術官に任命された。彼女は30年以上のバイオテクノロジー科学、製造と業務開発経験で十四行詩に来て、3つのスタートアップ会社に直接参加し、多くのM&A活動に参加した。彼女のCMC生物製薬プロセス開発における専門知識の範囲は細胞系開発からプロセス開発から商業製造までである。2008年9月から合併完了まで、デクスターさんはレザム生物製薬グループで取締役マネージャーを務め、製品開発サービスを担当し、動物毒理学前、臨床前毒素研究とCMC関連活動に関連する活動と規律を管理し、IND届出、cGMP活動の品質監督とその他のCMCサプライチェーン関連活動 を含む。彼女はXcellerex,Inc.からLBG,Xcellerex,Inc.に加盟し,CDMOと生物処理一次使用技術の開発者である。2004年4月から2008年9月まで、彼女はXcellerexの首席業務官を務めた。Xcellerexに加入する前に,1998年7月から2004年4月まで陶氏化学社のCDMOで業務発展副総裁を務め,デクスターさんの働きかけで2000年にCollaborative BioAllianceを買収した取引であり,br取締役はCelltech Biologicsの業務開発副社長であり,生物製剤CDMOに買収された。1986年から1998年7月まで、彼女はCelltech/Lonzaで働いていた。デクスターさんはワシントン大学の免疫学とマーケティング学の栄誉学位と、ハーバード大学から発行された“弁護士交渉”と“非金融マネージャー金融”証明書を持っている。彼女はロンドン大学生物工学学部名誉退職教授でもある, 1999年から2006年までの間に,“生物製剤施設プロジェクト管理”の授業とシンポジウムを大学院生,博士,大学院生に教授した。
リチャード·ケニー医学博士です
リチャード·ケニー医学博士は2021年4月以来会社の首席医療官を務めてきた。Kenney博士は生物製剤の翻訳段階開発および臨床前,臨床段階と商業化ワクチンと免疫療法の商業化戦略と企業管理において20年以上の経験を持っている。Clinreg Biologicsの総裁として,臨床や生物製品規制事務や医療モニタリングや薬物警戒などに戦略的相談を提供してきた。Kenney博士は最近X−VAX技術会社で首席開発官を務め,これまで免疫設計とCrucell Hollandで首席医療官を務め,そこで臨床開発と監督事務チームを指導していた。ケニー博士はFDAで研究員/審査員を6年以上務め,デューク大学と米国国立衛生研究院で大学院生研修を受けている。ケニー博士はジョージ·ワシントン大学の化学学士号とハーバード医学院の医学博士号を取得した。
第十六条第十四節実益所有権報告適合性
改正された1934年“証券取引法”第(br}16(A)節は、当社の役員と役員、高級管理者及び当社の登録種別株式証券の10%を超える実益所有者 を有する米国証券取引委員会に所有権及び所有権変更報告を提出することを要求する。米国証券取引委員会法規は、これらの者に、彼らが提出したすべての第16(A)節の表のコピーを会社に提供することを要求する。
当社が当社に提供した表3、4、5のコピーのみを審査し、当社は、2022年9月30日までの財政年度中に、会社のすべての役員、役員、その他の適用株主が取引所法案第16(A)節に要求したすべての報告をタイムリーに提出したと考えており、以下の場合を除く:Don Griffith、Nailesh Bhatt、Susan Dexter、Pankaj Mohan、John Cini、Raghu Rao、Jay Cross、Albert Dyrnessの表4 Sは202年4月8日に提出された。
ビジネス行為と道徳的基準
会社はその役員、上級管理者、従業員に適用される“ビジネス行為と道徳規範”を採択した。商業行為と道徳規則の目的は不当な行為を阻止し、そして会社の役員、高級管理者と従業員に指導を提供し、彼らが道徳問題を認識と処理し、不道徳或いは不法行為を通報するメカニズムを提供し、そして会社の誠実と問責文化に積極的に貢献することである。会社のビジネス行為や道徳基準は会社のサイトで公開して取得できます。サイトはhttps://www.sonnetBio.com/です。当社が“商業行為および道徳的規則”を実質的に修正または任意の免除を付与する場合、その取締役または幹部の“商業行為および道徳的規則”条項の任意の黙示放棄を含む場合、当社は、そのウェブサイト上または現在のForm 8-K報告書において、そのような修正または放棄の性質を開示する。
| -113- |
監査委員会
取締役会は監査委員会を設立し、現在はバートさん(会長)、デネスさん、ラオさんから構成されています。監査委員会の主な機能は、会社の総合財務諸表と会社が提供する他の財務情報の完全性、会社が法律と法規の要求を遵守する状況、会社の内部会計と財務制御制度、独立監査師の採用、資格、業績、報酬と独立性、関連側取引及び会社の商業行為と道徳基準の遵守状況である。
監査委員会の各メンバーは、米国証券取引委員会の適用規則とナスダック株式市場の適用規則に基づいて定義されているので、“独立”である。取締役会は、各監査委員会のメンバーが財務と監査事務の面で十分な知識を持っており、監査委員会に在任できることを確定した。取締役会は、Raoさんを“米国証券取引委員会適用規則およびナスダック証券市場適用規則によって定義された監査委員会財務の専門家”と認定しました。当社の取締役会は監査委員会の定款を通過しており、以下のサイトで調べることができます:https://www.sonnetBio.com/。
プロジェクト 11.役員報酬
集計表 給与表
次の表は、2022年度に当社の最高経営責任者を務める各役員、2022年9月30日までに当社役員を務める2人の最高報酬の役員、および最大で開示情報を得るべき他の2人の個人が2022年度に獲得した報酬 を示しています。以下の表に示す人員を本稿では“指定された実行官”と呼ぶ
集計表 給与表
| 氏名と主要職務 | 年.年 | 賃金.賃金 ($) | ボーナス.ボーナス ($) | 在庫品 賞.賞 ($)(1) | 選択権 賞.賞 ($)(1) | All other 補償する ($) | 合計 ($) | |||||||||||||||||||||
| パンカj·モハン博士 | 2022 | 559,729 | 218,680 | 144,061 | - | 82,923 | 1,005,393 | |||||||||||||||||||||
| 社長 とCEO | 2021 | 512,236 | 279,930 | - | - | - | 792,165 | |||||||||||||||||||||
| ジョン·シーニー博士です | 2022 | 413,048 | 113,899 | 36,015 | - | 52,014 | 614,976 | |||||||||||||||||||||
| 首席科学官 | 2021 | 382,594 | 142,505 | - | - | - | 525,099 | |||||||||||||||||||||
| ジェイ·クロス | 2022 | 403,676 | 127,649 | 30,128 | - | 43,358 | 604,811 | |||||||||||||||||||||
| 最高財務官 | 2021 | 375,767 | 156,878 | - | - | - | 532,645 | |||||||||||||||||||||
| (1) | 代表は、財務会計基準委員会(“FASB”)会計基準編纂(“ASC”)テーマ718に基づいて計算された2022年と2021年の贈与の総贈与日公正価値を代表する。この計算は、サービスベースの帰属に関連する没収に対していかなる 推定も行わないが、実行者が必要なサービスの実行を全額帰属する 報酬を与えると仮定する。 |
| -114- |
Narrative 報酬集計表開示
雇用契約
指名された執行幹事ごとの雇用協定や手配の具体的な条項は以下のとおりである。
十四行詩(Br)は、2018年12月31日にMohan博士と改訂された雇用協定(“Mohan合意”)を締結し、CEOを務める雇用条項を列挙し、合併完了時に当社が負担する。雇用協定によると、Mohan博士は他の事項を除いて、(I)490,000ドルの年間総基本給を得る権利があり、(Ii)自社が戦略取引から得た毛収入の5.4%に相当するボーナスを得る資格があり、(Iii)前条項のボーナスが基本給の50%を下回るいずれの年度についても、業績に基づく現金ボーナスを追加発行し、その年度の現金ボーナス総額が基本給の50%に達するように取締役会が決定する。雇用協定はその条項によって終了されなければならない。モハン博士の雇用契約によると、“支配権変更”の前または後の2ヶ月以内またはその後の12ヶ月以内に無断解雇や“正当な理由”で解雇された場合、(I)18ヶ月の基本給を得る権利があり、(Ii)ボーナスは発生当時の業績ボーナスを終了し、12で割ったものに等しく、その後 に18を乗じ、(Iii)COBRAに従って保険を継続すれば、継続保証に必要なコブラ保険料を支払うのは、br(A)終了日から18ヶ月後、(B)新就職や自己雇用に関する実質的に同値な健康保険を受ける資格がある日、または(C)コブラ継続保険を受ける資格がない日。モハン博士が無断で終了したり、コントロール変更に合わない“十分な理由”で解雇された場合、(I)18ヶ月の基本給を得る権利があり、(Ii)彼が終了した業績年度のいかなる業績ボーナスも, と(Iii)もし彼がCOBRAに従って保険を継続し、継続保険に必要なCOBRA保険料を支払うと、(Br)終了日から18ヶ月後まで、(B)新規雇用や自己雇用に関連する実質と同等の医療保険を取得する資格がある日、または(C)COBRA継続保険資格を満たしていない日。
十四行詩(Br)は2020年1月10日にCini博士と改訂された雇用協定(“Cini合意”)を締結し、首席科学官を務める雇用条項を列挙し、この協定は当社が合併完了時に負担する。雇用協定によると、Cini博士は、(I)年間37万ドルの総基本給、(Ii)自社が戦略取引から得た総収入1.1%に相当するボーナスを得る資格があること、および(Iii)前の条項のボーナスが基本給の35%を下回るいずれの年度についても、業績に基づく現金ボーナスを追加発行し、その年度の現金ボーナス総額を基本給の35%に達するように取締役会が決定する権利がある。雇用協定はその条項によって終了されなければならない。Cini博士の雇用契約によると、“支配権変更”の前または後の2ヶ月以内またはその後の12ヶ月以内に“原因”または“正当な理由”で解雇された場合、(I)12ヶ月の基本給と(Ii)COBRAに従って保険を継続し、継続保証に必要なCOBRA保険料brを支払う権利があり、(A)終了日後18ヶ月まで、(B)新規雇用や自己雇用に関連する基本的に同等の健康保険を取得する資格がある。あるいは(C)彼がCOBRA更新資格を満たしていない日 である.Cini博士が“原因”や“制御変更”に該当しない“十分な理由”で解雇された場合、(I)9ヶ月の基本給と(Ii)COBRAに従って保証を継続すれば、(A)終了日後12ヶ月の中で最も早い1ヶ月までのCOBRA保険料を支払う権利がある, (B)新規雇用または自己雇用に関連する基本的に同等の医療保険を取得する資格がある日、または(C)コブラ継続保険を受ける資格がない日。
二零二零年一月十日にクロスさんと雇用契約(“クロス合意”)を締結した二零四行詩において、合併完了時に当社が負担する首席財務官の採用条項を列挙する。雇用契約によれば、クロスさんは、(I)年間基本給総額365,000ドル、および(Ii)取締役会で決定された最高40%までの基本給の業績現金配当金を取得する権利を有しています。雇用協定はその条項によって を終了する。クロスさんの雇用契約によれば、“支配権変更”の前後2ヶ月以内又は12ヶ月以内に“正当な理由”により解雇された場合には、(I)12ヶ月分の基本給を得る権利があり、(Ii)解雇が発生した実績年度の業績ボーナス、並びに(Iii) COBRAに従って保険加入を継続すれば、(A)終了日後18ヶ月まで継続加入に必要なCOBRA保険料を支払うことができ、(B)新規雇用または自己雇用に関連する基本的に同値な健康保険を取得する資格がある日、または(C)コブラ継続保険を受ける資格がない日。クロスさんが“理由”または“統制権変更”の“十分理由”を満たさずに解雇された場合、彼は (I)9ヶ月の基本給を得る権利があり、(Ii)業績年間のボーナスの中で任意の業績ボーナスを解雇し、(Iii)COBRAの下で保険加入を継続した場合、(A)終了日以降12ヶ月以内に最も早い までの継続加入に必要なCOBRA保険料を支払う。(B)彼は、新規雇用または自己雇用に関する基本的に同値な健康保険を取得する資格がある日 , あるいは(C)彼は“コブラ”の更新資格の日付を満たしていない。
| -115- |
その他 プロトコル
当社は2020年4月1日にDexterさんと雇用協定(“Dexter合意”)を締結し、首席技術官を務める雇用条項を明らかにした。雇用協定によると、Dexterさんは、(I)310,000ドルの年間基本給総額と、(Ii)取締役会によって決定された最高35%の基本給の業績現金ボーナスを含む権利がある。雇用協定はその条項によって終了されなければならない。デクスターさんの雇用契約によると、“支配権変更”の前または後の2ヶ月以内に“原因”や“正当な理由”で解雇された場合、(I)12ヶ月の基本給を得る権利があり、(Ii)解雇された業績年度のいかなる業績ボーナスも、(Iii)COBRAによって速やかに保証を継続した場合、(A)終了日後18ヶ月まで、継続保証に必要なCOBRA保険料brを支払う権利がある。(B)新規雇用または自己雇用に関連する基本的に同等の医療保険を取得する資格がある日、または(C)COBRA継続保険資格を満たしていない日。Dexterさんが“原因”や“統制権変更”に該当しない“十分な理由”がない場合に解雇された場合、(I)9ヶ月の基本給を得る権利があり、(Ii)解雇された業績年間のいかなる業績ボーナスも、(Iii)COBRAに従って保険を継続し、COBRA保険料brを支払い、(A)終了日から12ヶ月後まで、(B)新規雇用や自己雇用に関連する基本的に同等の医療保険を受ける資格がある日まで保証を継続する権利がある, または(C)彼女がCOBRA継続保証資格を満たしていない日。
財政年度末未返済の持分奨励
以下の表は、普通株を購入する未行使オプション、指定役員毎に付与されておらず、2022年9月30日までに発行された普通株および普通株の制限株に関するいくつかの情報を1つずつ示している。
2022年度末未償還株式奨励
| 株 奨励 | ||||||||
| 名前.名前 | 持分インセンティブ計画奨励:まだ付与されていない未獲得株式、単位または他の権利の数(#) | 持分インセンティブ計画奨励:まだ付与されていない未獲得株式、単位または他の権利の市場または配当価値($) | ||||||
| パンカj·モハン博士 | 22,866 | (1) | 33,613 | |||||
| ジョン·シーニー博士です | 5,716 | (1) | 8,403 | |||||
| ジェイ·クロス | 4,782 | (1) | 7,030 | |||||
| (1) | 各 制限株式単位(“RSU”)賞は2023年1月1日に100%授与される。 |
役員報酬
非従業員役員報酬政策
合併に関連して、取締役会は取締役非従業員取締役の新しい報酬政策を承認した。取締役会と委員会会議に出席する合理的な費用を精算するほか、本政策では以下の現金補償を規定している
各非従業員取締役は私たちから35,000ドルの年会費を得る権利があります
私たちの監査委員会の議長は私たちから15,000ドルの年会費を得るだろう
| -116- |
私たちの給与委員会の議長は私たちから10,000ドルの年会費を得るだろう
私たちの指名と会社管理委員会の議長は私たちから8,000ドルの年会費を得るだろう
監査委員会、報酬委員会、および指名および会社統治委員会の非議長メンバー1人は、それぞれ7,500ドル、5,000ドル、4,000ドルの年会費を得る。
取締役会に参加した非従業員取締役の各々は、合併完了時に会社が完全に希釈した発行された普通株式の0.080%を購入することができ、これは3年以内に毎年33%付与され、最初の帰属日は授与日の1年記念日に発生する。非従業員取締役1人はまた、年次オプション付与を受け、合併完了時に会社の0.040%の完全希釈後に普通株式を発行し、授与日の1周年または次年度株主総会の早い日に100%を付与する。支配権が変更されると、当社の持分インセンティブ計画によって定義されるように、これらのオプション関連株式の100%は、制御権変更前に帰属され、行使可能となる。
次の表に記載されているbrを除いて、非従業員取締役は2022年度に現金または持分報酬を受信していない
役員報酬
| 名前.名前 | Fees Earned or Paid in Cash ($) | 在庫品 賞.賞 ($)(1) | 選択権 賞.賞 ($)(1) | All Other 補償する ($) | 合計 ($) | |||||||||||||||
| ネレッシュ·バート(2) | 54,000 | 6,368 | - | - | 60,368 | |||||||||||||||
| アルバート·デネス(3) | 55,500 | 6,368 | - | - | 61,868 | |||||||||||||||
| ドナルド·グリフィス(4) | - | 6,750 | - | 134,634 | 141,384 | |||||||||||||||
| ラグ·ラオ(5) | 56,500 | 6,368 | - | - | 62,868 | |||||||||||||||
| ローリー·マクニール(6) | - | - | - | - | - | |||||||||||||||
| (1) | 代表 FASB ASCテーマ718によって計算された2022年贈与の総支出日公正価値。この計算 は、サービスベースの帰属に関連するいかなる没収推定値にも影響を与えないが、行政者が全額帰属を奨励するために必要なbr}サービスを履行すると仮定する。 | |
| (2) | バートさんは2022年9月30日までに、1,010個の限定株式単位を合計保有している。 | |
| (3) | Dyrnessさんは2022年9月30日までに1,010個の制限株式単位を合計保有している。 | |
| (4) | グリフィスさんは、2019年1月1日から十四行の財務総監を務め、合併後は当社の財務総監を務めます。“その他のすべての報酬”の額は、グリフィスさんが2022年度に受け取る給与とボーナスを表します。グリフィスさんとの雇用契約について以下の説明を参照。 | |
| (5) | 饒さんは2022年9月30日までに限定株1,010株を保有している。 | |
| (6) | マクニールさんは2022年9月25日に取締役会に参加した。 |
取締役とのその他の合意
十四行詩(Br)は、2019年1月1日にGriffithさんと財務総監を務める雇用条項を締結する。
| -117- |
第br項12.ある実益所有者及び管理層の保証所有権及び関連株主事項。
安全な利益を得る所有者と経営陣の所有権
次の表は、2022年12月14日までの当社の普通株実益所有権のある情報を示しており、具体的には、(I)当社の現職取締役一人ひとり、(Ii)指名された役員一人ひとり、(Iii)すべての現役員と取締役を全体として、および(Iv)当社が知っている実益ごとに5%(5%)を超える自社普通株流通株を持っている人である。
次の表について言えば、実益所有権は適用されたアメリカ証券取引委員会規則によって決定され、情報は必ずしも実益所有権が他の目的に使用されていることを表すとは限らない。表内の付記に別途明記されているほか、当社は、表に記載されている各個人又は実体が、その個人又は実体実益に対して所有するすべての当社普通株株式に対して独占投票権及び投資権を有する(又はその配偶者とそれ等の権力を共有する)と信じている。米国証券取引委員会の規則によれば、2022年12月14日以降60日以内に行使可能なオプション(“現在行使可能なオプション”)に基づいて発行可能な当社普通株は、発行済み株式とみなされるので、表で指名された個人または実体が実益所有として報告された株式数に含まれ、その個人または実体実益が所有する普通株式のパーセンテージ を計算するために使用される。しかしながら、他の任意の個人または実体実益が所有する普通株のパーセンテージを計算する際に、これらの株は流通株とはみなされない。
次の表に示す各個人または実体実益が所有する普通株式の割合は、2022年12月14日までに発行および発行された7,934,156株の普通株式に基づいており、その個人または実体が保有する任意の現在行使可能なオプションを行使するために発行された株式に基づいている。
Title of クラス | 受益者の名前または名称および住所* | 所有権の金額と性質から利益を得る | クラスパーセント | |||||||
| 任命された役員と役員: | ||||||||||
| パンカj·モハン博士 | 115,524 | (1) | 1.5 | % | ||||||
| ナイレッシュ·バート | 1,962 | (2) | ** | |||||||
| アルバート·デルネス | 2,027 | (3) | ** | |||||||
| ドナルド·グリフィス | 7,584 | (4) | ** | |||||||
| ラグ·ラオ | 1,604 | (5) | ** | |||||||
| ローリー·マクニール | - | - | ||||||||
| ジョン。K.Cini博士。 | 21,720 | (6) | ** | |||||||
| ジェイ·クロス | 10,368 | (7) | ** | |||||||
| すべての現執行幹事と取締役 を全体(10人)とする | 172,507 | 2.2 | % | |||||||
| 5%保有者 | ||||||||||
| ありません | ||||||||||
| (*) | 別の説明がない限り、住所はc/o Sonnet BioTreateutics,Inc.,100は中心、102号室、プリンストン、ニュージャージー州、08540を見下ろす。 | |
| (**) | は1%未満である. | |
| (1) | (Br)(1)モハン家のオフィスが保有している66,478株の普通株、モハン博士は配偶者のスワティ·モハンと投票権と処分権を共有し、(2)モハン博士の子供のパンクリ·モハンは個人が保有している570株の普通株、モハン博士はパンキュリー·モハンと投票権と処分権を共有している。および(Iii)は、2022年12月14日から60日以内にモハン家族事務室が保有する引受権証によって発行された304株の普通株を行使することができ、モハン博士とスワティ·モハンが共同で投票権と処分権を行使することができる。22,866個の限定株式単位を含め、2022年12月14日から60日間、普通株と帰属株で決済される。 |
| -118- |
| (2) | 1,010個の限定株式単位を含み、これらの単位は2022年12月14日から60日以内に普通株と帰属株の形で決済される。 | |
| (3) | 2022年12月14日から60日以内に行使可能な引受権証が発行可能な普通株式91株を含む。1,010個の限定株式単位を含み、これらの単位は2022年12月14日から60日以内に普通株と帰属株の形で決済される。 | |
| (4) | 1,071個の限定株式単位を含み、これらの単位は2022年12月14日から60日間、普通株と帰属株の形で決済される。 | |
| (5) | 2022年12月14日から60日以内に株式承認証を行使して発行可能な7株普通株を含む。1,010個の制限株式単位を含み、これらの単位は2022年12月14日から60日以内に普通株と帰属株の形で決済される。 | |
| (6) | 5,716個の限定株式単位を含め、2022年12月14日から60日間、普通株と帰属株の形で決済される。 | |
| (7) | 4,782個の限定株式単位は含まれておらず、これらの単位は2022年12月14日から60日以内に普通株で決済および帰属される。 |
資本報酬計画情報
次の表は、2020年総合持分インセンティブ計画(“2020計画”)が発行可能な会社普通株を含む、2022年9月30日までに会社の既存株式補償計画に基づく情報を提供します。
| 資本報酬計画情報 | ||||||||||||
| 計画種別 | (a) Number of securities to be issued upon exercise of outstanding options, warrants and rights | (b) Weighted average exercise price of outstanding options, warrants and rights | (c) Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | |||||||||
| 証券保有者が承認した持分補償計画(1) | 47,798 | 適用されない | - | |||||||||
| 株式補償計画は証券保有者の許可を得ていない | 適用されない | 適用されない | 適用されない | |||||||||
| 合計する | 47,798 | - | - | |||||||||
| (1) | RSUは行権価格を持たないため,重み付き平均行権価格はRSU決済に関する発行する株式を反映しない.2022年9月30日まで、RSUを除いて、私たちの持分補償計画には未返済のオプション、株式承認証あるいは権利はありません。 |
第 項13.ある関係と関連取引、および取締役独立性。
当社は、役員や役員の報酬スケジュールを指定するほか、2021年の財政年度開始以来、当社が参加または参加する各取引と一連の類似取引を紹介している
| ● | 過去の2つの完全財政年度末の総資産平均の1%を超えるか、または12万ドルを超えるか、またはそれを超えるか、またはそれを超えるか、またはより小さい者を基準とする |
| -119- |
| ● | 当社の任意の取締役、取締役の被著名人、幹部、または当社の普通株を5%以上保有するbrの任意のメンバーまたは上記の人の任意の直系親族は、かつてまたは直接的または間接的な重大な利益を持つことになる。 |
給与 当社が指定した役員および役員の報酬スケジュールは“役員報酬”の一節に掲載されています。
賠償協定
会社はすべての現職役員と役員と賠償協定を締結した。これらの合意は、デラウェア州の法律で許可されている最大範囲で、これらの個人が会社にサービスを提供することによって生じる可能性のある責任を賠償することを会社に要求し、彼らが賠償を受ける可能性があるため、彼らを起訴することによって発生した費用を前借りする。同社は将来の役員や役員と賠償協定も締結しようとしている。
取締役 独立
Br社は現在6人の取締役会で管理されている。当社は、ナスダックの株式市場規則に基づいて定義されている、バート·さん、デニス·さん、ラオ·さん、およびマクニール夫人を“独立”と決定しました。
第br項14.総会計士料金とサービス
以下の表は、ピマウェイ会計士事務所、当社の独立公認会計士事務所(および合併前の十四行詩)が過去2会計年度にそれぞれ毎年提供している専門サービス費用をまとめています
| 費用別 | 2022 | 2021 | ||||||
| 料金を審査する | $ | 435,000 | $ | 509,500 | ||||
| 監査関連費用 | - | - | ||||||
| 税金.税金 | 33,500 | 34,000 | ||||||
| 他のすべての費用 | - | - | ||||||
| 総費用 | $ | 468,500 | $ | 543,500 | ||||
監査費用
監査法人年度連結財務諸表及び審査会社四半期中期連結財務諸表に関する専門サービス費用、及び登録声明、慰問状及び同意書に関する費用をいう。
監査に関する費用
2022年または2021年には、私たちの独立監査人は監査に関連した費用を発生させなかった。
税 手数料
税務 費用は税務コンプライアンス、税務相談、税務計画と税務準備サービスと関係があります。
すべての その他の費用
総会計士が提供する製品やサービスに関する費用は、上記各節で報告したサービスは含まれていない。
| -120- |
監査委員会は給与を任命し、決定し、独立監査役を監督する仕事を担当する。監査委員会 は、その定款で概説された任意の提案された監査および非監査サービスを実行するために、独立監査師の提案を審査し、承認する必要がある。監査委員会は、その定款の外で監査及び非監査関連サービスを事前に承認するための政策及び手続を策定していない。取引法第10 A条の要求に基づき、我々の監査委員会は、2022年から2021年までの間に提供されるすべての監査および非監査サービスおよびこれらのサービスに支払われる費用をピマウェイ有限責任会社に許可した。しかし,“取引法”第10 A(I)(1)(B)節の“最低限”条項を満たしていれば,会社に非監査サービスを提供する事前承認要求 を免除することができる.
監査委員会は、上記の監査に関連する費用、税費、その他のすべての費用の提供が、ピマウェイ会計士事務所の独立性を維持することに適合しているかどうかを考慮し、2022年度および2021年度のこのようなサービスが互換性があることを決定した。br}のすべてのサービスは、監査委員会によって取引所法案S-Xルール2-01によって承認されたが、brルールは適用される。
監査委員会は、経営陣と監査された連結財務諸表の審査及び検討を担当し、独立公認会計士と上場企業会計監督委員会第1301号監査基準に要求される事項を検討する監査委員会とのコミュニケーション上場企業会計監督委員会の適用要求に基づき、独立公認会計士と監査委員会が独立性についてコミュニケーションを行うことに関する独立公認会計士の書面開示を受け、独立公認会計士とその独立性を検討し、監査された合併財務諸表を当社の10-K表年次報告書に組み入れることを提案する。
第4部
第 項15.物証、財務諸表付表。
(a)(1) 財務諸表それは.本報告の一部として提出された財務諸表は総合財務諸表インデックス に列挙されている。
(a)(2) 財務諸表明細書それは.表が省略されているのは、適用されないため、または必要な情報が連結財務諸表または付記に表示されているためである。
(a)(3) 展示品です。“展示品索引”を参考にしてください。本年度報告は,表格10−Kの形で登録または引用により本年度報告に組み込まれ,S−K規則601項に従って番号付けされる。
第 項16.表格10-K要約
ない。
| -121- |
サイン
1934年の証券取引法第13または15(D)節の要求によると、登録者は、本報告が正式に許可された署名者によってその署名を代表するように正式に促進された。
Sonnet BioTherapeutics, Inc. (登録者) | ||
| 日付: 2022年12月15日 | /s/ Pankaj Mohan博士 | |
| パンカジ·モーハン博士 | ||
社長 とCEO (CEOと 正式なサイン人) | ||
| 日付: 2022年12月15日 | /s/ ジェイ·クロス | |
| ジェイ·クロス | ||
Chief Financial Officer (最高財務会計官 ) |
署名 と依頼書
以下の署名のすべての人がここでPankaj Mohan、Ph.DおよびJay Crossを構成して任命し、彼らのすべての人は、彼の真の合法的な代理人、代理人、事実代理人であり、十分な代替と再代理の権力を持っており、br}は彼の名義、位置、およびすべての身分の代わりに、(I)本10-K表年次報告の任意およびすべての修正案およびその中のすべての付表と証拠に対して行動し、米国証券取引委員会(SEC)に署名とアーカイブを行い、(Ii)が行動することに注意してください。これに関連する必要または適切な証明書、文書、プロトコル、および他の文書に署名およびアーカイブし、(Iii) は、その可能性または自ら取ることができるすべての意図および目的を尽くし、ここで、代理人、代理人およびエージェント、またはそれらの任意の代替者が合法的にまたはそれに至ることができるすべてのことを承認、承認および確認することができる。
本報告は、1934年の証券取引法の要求に基づき、登録者として指定日に登録者の名義で以下の者によって署名された。
| サイン | タイトル | 日取り | ||
| /s/ Pankaj Mohan博士 | 社長、CEO兼会長 | 2022年12月15日 | ||
| パンカジ·モーハン博士 | (CEO ) | |||
| /s/ ジェイ·クロス | 最高財務官 | 2022年12月15日 | ||
| ジェイ·クロス | (最高財務会計官 ) | |||
| /s/ アルバート·デルニス | 役員.取締役 | 2022年12月15日 | ||
| アルバート·デルネス | ||||
| /s/ ナエレシュ·バート | 役員.取締役 | 2022年12月15日 | ||
| Nailesh バート | ||||
| /s/ ラグラオ | 役員.取締役 | 2022年12月15日 | ||
| ラグ ラオ | ||||
| /s/ ドナルド·グリフィス | 役員.取締役 | 2022年12月15日 | ||
| ドナルド·グリフィス | ||||
| /s/ ローリー·マクニール | 役員.取締役 | 2022年12月15日 | ||
| ローリーマクニール | ||||
| -122- |
展示品インデックス
展示品 違います。 |
説明する | |
| 2.1 | 合意 と合併計画は、日付は2019年10月10日で、会社と会社の間、十四行詩子会社である。合併子会社(2019年10月11日に提出された表格8-K現在の報告書の添付ファイル2.1として提出され、参照によって本明細書に組み込まれます) | |
| 2.2 | 当社、十四行子会社と連結子会社との間で2020年2月7日に合併協定及び計画の第1号改正案が提出された(2020年2月7日に提出された会社の現在の8-Kレポートの添付ファイル2.1として提出され、参照により本明細書に組み込まれる)。 | |
| 2.3 | Sonnet BioTreateutics,Inc.とRelipment Treateutics Holding SAとの間の株式交換協定は、2019年8月9日である(2019年11月27日に米国証券取引委員会に提出された会社S-4表登録声明の添付ファイル2.10を参照することにより) | |
| 3.1 | 改訂されたSonnet BioTreateutics Holdings,Inc.の登録証明書(会社が2020年12月17日に米国証券取引委員会に提出したForm 10−K年次報告書の添付ファイル3.1を引用して合併した)。 | |
| 3.2 | シリーズ3優先株の優先株、権利、および制限指定証明書(添付ファイル3.1を参照して、2022年8月15日に米国証券取引委員会に提出された現在の8-K表報告書に組み込まれています)。 | |
| 3.3 | 第4シリーズ優先株の優先株、権利、および制限指定証明書(添付ファイル3.2を参照して、2022年8月15日に米国証券取引委員会に提出された現在の8-K表報告書に組み込まれています)。 | |
| 3.4 | 日付は2022年9月16日のSonnet BioTreateutics Holdings,Inc.の修正された登録証明書修正証明書である(添付ファイル3.1を参照して2022年9月19日に米国証券取引委員会に提出された現在の8-K表報告書に組み込まれている)。 | |
| 3.2 | Sonnet BioTreateutics Holdings,Inc.の規約を改訂·再改訂した(添付ファイル3.3を参照して2022年8月15日に米国証券取引委員会に提出されたbr}Form 8−Kの現在の報告書に組み込まれている)。 | |
| 4.1 | 普通株式証明書表 (2011年12月2日に米国証券取引委員会に提出されたS-1表登録説明書添付ファイル4.1(登録番号: 333-178307)を参照)。 | |
| 4.2 | 2017年5月4日の引受権証表 (添付ファイル4.2を参照して、2017年5月5日に米国証券取引委員会に提出された現在の8-K表報告書に組み込まれています)。 | |
| 4.3 | 剥離 実体株式証は、日付は2020年4月1日である(添付ファイル4.1を参照して会社に編入し、2020年4月3日にアメリカ証券取引委員会の現在の8-K表報告 )に提出する。 | |
| 4.4 | Sonnet BioTreateutics,Inc.変換されたForm (会社を参照して2020年8月14日に米国証券取引委員会に提出されたForm 10-Q四半期報告書の添付ファイル4.2によって組み込まれる)。 | |
| 4.5 | A/Bシリーズ株式証表 (会社が2020年2月7日に米国証券取引委員会に提出したS-4/A表登録説明書添付ファイル4.16を参照)。 | |
| 4.6 | Cシリーズ株式証明書の表 (2020年8月4日に米国証券取引委員会に提出された現在の8-K表報告書の添付ファイル4.1を参照して組み込む)。 | |
| 4.7 | 当社とその中に列挙されたいくつかの投資家との間で2020年2月7日に締結された権利協定を登録する(当社を参照して2020年2月7日に米国証券取引委員会に提出されたS-4/A表登録説明書添付ファイル4.17)。 | |
| 4.8 | 証券説明 * | |
| 4.9 | 日付は2021年8月24日の事前資本権証表 (2021年8月25日に米国証券取引委員会に提出された会社の現在の報告書の8-K表の添付ファイル4.2を参照して編入されます)。 | |
| 4.10 | 日付は2021年8月24日の引受業者株式証明書表 である(会社が2021年8月16日に米国証券取引委員会に提出したS-1/A表登録説明書の添付ファイル4.9を参照して組み込まれる)。 | |
| 4.11 | 日付は2021年8月24日の一般権証表 (当社の2021年8月25日に米国証券取引委員会に提出された8-K表の添付ファイル4.4を参照して編入されます)。 |
| -123- |
| 10.1 | GEM Global Year Fund LLC SCSとSonnet BioTreateutics,Inc.の共通の株式購入契約は、2019年8月6日である(2019年11月27日に米国証券取引委員会に提出された会社S-4表登録説明書添付ファイル10.54を参照することにより)。 | |
| 10.2 | GEM Global Year Fund LLC SCSとSonnet BioTreateutics,Inc.が2019年9月25日に締結した普通株購入協定修正案 (2019年11月27日に米国証券取引委員会に提出された会社S-4表登録説明書添付ファイル10.55を参照することにより)。 | |
| 10.3 | GEM Global Year Fund LLC SCS,Sonnet BioTreateutics,Inc.とChancleer Holdings,Inc.2020年2月7日に締結された普通株購入協定の書簡と第2号修正案(合併日は2020年2月7日の会社S-4表登録声明の添付ファイル10.60)。 | |
| 10.4 | Pankaj MohanとSonnet BioTreateutics,Inc.が2018年12月31日に締結した雇用契約(添付ファイル10.56 を参照して会社に組み込まれ、2020年2月7日に米国証券取引委員会に提出されたS-4表登録声明に提出される)。ガンギエイ | |
| 10.5 | John CiniとSonnet BioTreateutics,Inc.が2020年1月10日に締結した雇用協定(添付ファイル10.58 を参照することにより、2020年2月7日に米国証券取引委員会に提出されたS-4表登録声明に組み込まれる)。ガンギエイ | |
| 10.6 | Jay CrossとSonnet BioTreateutics,Inc.との間の雇用契約は,2020年1月10日(添付ファイル10.57 を参照して会社に編入され,2020年2月7日に米国証券取引委員会に提出されたS-4表登録声明に提出される).ガンギエイ | |
| 10.7 | スーザン·デクスターが会社と締結した雇用協定は、2020年4月1日である(添付ファイル10.7を参照して会社に組み込まれ、2020年4月3日に米国証券取引委員会の現在の8-K表報告書に提出される)。ガンギエイ | |
| 10.8 | Donald GriffithとSonnet BioTreateutics,Inc.の間の要約書簡は、2019年1月1日である(添付ファイル10.59 を参照して会社に組み込まれ、2020年2月7日に米国証券取引委員会のS-4表登録声明に提出される)。ガンギエイ | |
| 10.9 | 十四行詩生物治療持株会社2020年総合持分インセンティブ計画(会社が2020年5月20日に米国証券取引委員会に提出したS-8表登録声明の添付ファイル4.2を引用して組み込む)。ガンギエイ | |
| 10.10 | 限定株式単位賞表 (2020年7月9日に米国証券取引委員会に提出された8-K表(文書番号:001-35570)添付ファイル10.1を参照)。ガンギエイ | |
| 10.11 | ライセンス ARES Trading SAとRelipment Treeutics SAとの間の合意は、2015年8月28日である(添付ファイル10.51 を参照して会社に組み込まれ、2020年2月7日に米国証券取引委員会に提出されたS-4表登録声明に提出される) | |
| 10.12 | Discovery XOMA(US)LLCとOncobiologics,Inc.が2012年7月23日に署名した協力協定(添付ファイル10.52を参照することにより、同社が2020年2月7日に米国証券取引委員会に提出したS-4表登録声明に組み込まれる) | |
| 10.13 | XOMA(US)LLCとSonnet BioTreateutics,Inc.が2019年5月7日に締結したDiscovery協力協定修正案(2020年2月7日に米国証券取引委員会に提出された会社S-4表登録声明添付ファイル10.53に合併)。* | |
| 10.14 | 証券購入協定は,日付が2020年2月7日であり,強ザクリールホールディングス,Sonnet BioTreateutics,Inc.とその投資家 側が締結した(2020年2月7日に米国証券取引委員会に提出された会社S-4表登録声明の第10.64号添付ファイルを引用することにより). |
| -124- |
| 10.15 | Sonnet BioTreateutics Holdings, Inc.および所持者(添付ファイル10.1を参照することにより、2020年8月4日に米国証券取引委員会に提出された現在の8-Kフォーム(ファイル番号001-35570)に組み込まれ、2020年8月3日にSonnet BioTreateutics Holdingsと所有者の間で署名された引受権証行使と総合改訂プロトコル表 が組み込まれる。 | |
| 10.16 | Sonnet BioTreateutics Holdings,Inc.の譲渡と仮想雇用協定は,2020年4月1日に施行される(同社が2020年12月17日に米国証券取引委員会に提出したForm 10−K年次報告書の添付ファイル10.16を参照することにより) | |
| 10.17 | Pankaj Mohanと当社が2020年11月23日に締結した役員採用協定第1号改正案(引用により2020年12月17日に米国証券取引委員会に提出されたForm 10−K年度報告添付ファイル10.17) | |
| 10.18 | ジョン·シーニーと会社が2020年11月23日に締結した役員採用協定の第1号改正案(2020年12月17日に米国証券取引委員会に提出された会社10-K表年次報告書の添付ファイル10.18を引用することにより組み入れられる) | |
| 10.19 | 賠償協定表 (引用会社が2020年12月17日に米国証券取引委員会に提出した10−K表年次報告書の添付ファイル10.19を編入する) | |
| 10.20 | 当社と必和必拓が2020年2月5日に締結した場外販売協定(2021年2月5日に米国証券取引委員会に提出された8-K表(ファイル番号001-35570)は添付ファイル1.1を参照して組み込む)。 | |
| 10.21 | ライセンス契約は、2021年5月2日に、Sonnet BioTreateutics,Inc.およびNew Life Treateutics Pte,Ltd.(参照合併により2021年5月17日に米国証券取引委員会に提出されたForm 10-Q四半期報告添付ファイル10.1)である。 | |
| 10.22 | Sonnet BioTreateutics,Inc.およびNew Life Treateutics Pte,Ltd.は、2021年6月11日に最初の許可協定修正案に署名した(2021年12月17日に米国証券取引委員会に提出されたForm 10-K年度報告書の添付ファイル10.22を参照することによって組み込まれる)。 | |
| 10.23 | 第2のライセンス契約修正案は、2021年7月7日に、Sonnet BioTreateutics CH SA、Sonnet BioTreateutics,Inc.およびNew Life Treateutics Pte,Ltd.(2021年12月17日に米国証券取引委員会に提出されたForm 10-K年度報告添付ファイル10.23を参照して統合された)である。 | |
10.24
|
ARES Trading SAおよびSonnet BioTreateutics CH SAが2021年11月1日に署名したライセンス契約および和解協定を改訂する(2021年12月17日に米国証券取引委員会に提出された10-K表年次報告書添付ファイル10.24を参照して組み込まれる)。 | |
| 10.25 | 会社と必和必拓が2022年8月15日に締結した場外販売協定(2022年8月15日に米国証券取引委員会に提出された8-K表の添付ファイル1.1)に統合された。 | |
| 21.1 | 当社の付属会社です。* | |
| 23.1 | ピマウェイ有限責任会社の同意。* | |
| 24.1 | 授権書 (署名ページに含まれる)* | |
| 31.1 | 改正証券取引法1934年公布の第13 a-14(A)及び15 d-14(A)条に基づいて発行された最高経営責任者証明書 | |
| 31.2 | 改正された1934年の証券取引法第13 a-14(A)及び15 d-14(A)条に基づいて発行された首席財務官証明書 |
| -125- |
| 32.1 | 2002年にサバンズ·オクスリ法案第906節で可決された“米国法典”第18編1350条に基づく最高経営責任者証明書。** | |
| 32.2 | 2002年にサバンズ·オクスリ法案第906条で可決された“米国法典”第18編第1350条に規定されている首席財務官証明書。** | |
| 101.INS | 相互接続 XBRLインスタンス文書-このインスタンス文書は、そのXBRLタグがイントラネットXBRL文書に埋め込まれているので、対話データファイルには表示されない。* | |
| 101.書院 | 連結 XBRL分類拡張アーキテクチャ文書。* | |
| 101.カール | 連結 XBRL分類拡張はLinkbase文書を計算する.* | |
| 101.def | 連結 XBRL分類拡張Linkbase文書を定義します* | |
| 101.介護会 | XBRL分類拡張ラベルLinkbase文書を連結する.* | |
| 101.Pre | インライン展開XBRL分類拡張プレゼンテーションLinkbaseドキュメント* | |
| 104 | 表紙 ページ相互データファイル(添付ファイル101に含まれるイントラネットXBRL形式)。* |
| * | 同封してアーカイブする。 |
| ** | 同封で提供します。 |
| *** | アーカイブ;S-K条例第601(B)(10)項の規定により、一部の証拠品は省略されている。任意の漏れ部分のコピー は、要求に応じて証券取引委員会に提供される。 |
| # | S−K条例第601(B)(2)項によれば、本プロトコルの付表および証拠品は省略されている。何か漏れたスケジュールおよび/または証拠品があれば、米国証券取引委員会に提供することを要求しなければならない。 |
| † | 管理契約または報酬計画、契約または手配を表します。 |
| -126- |