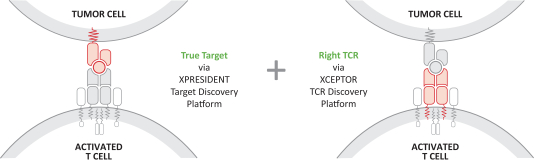
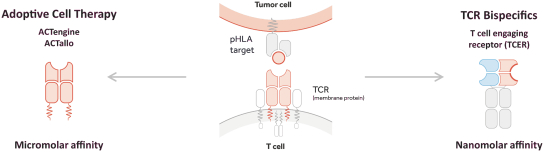
(“ACT”)和抗体样双特异性,也称为T细胞接合受体(“TCER”)。每种疗法都设计有不同的属性和作用机制,以产生目标癌症患者群体所需的治疗效果。我们目前的渠道如下所示,包括几个专有的和合作的基于TCR的临床和临床前开发的候选产品。
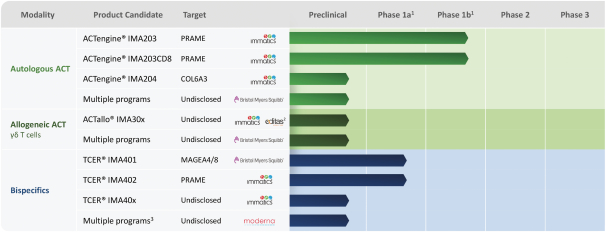
1阶段1a:剂量递增,阶段1b:剂量扩大;2Imatics的专有ACTallo平台,利用Editas的CRISPR基因编辑技术;3启用了信使核糖核酸体内表达的TCER分子;IMA203队列b(IMA203结合免疫检查点抑制剂)先前已被剥夺
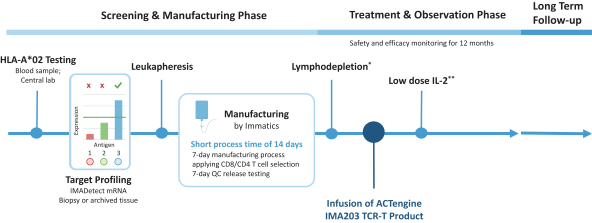
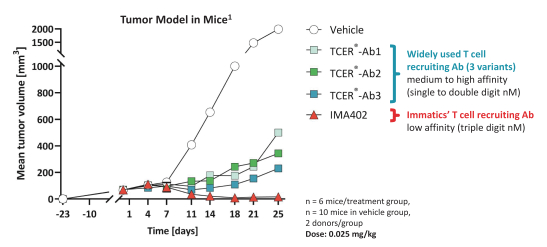
自体ACTEngineTCR-设计T细胞(“TCR-T”)在肿瘤负担较高的患者中已显示出较强的临床抗肿瘤活性。它们被设计为单次输液治疗。(“一劳永逸”)在专门的医疗中心进行管理,并需要个性化的自体细胞供应链。除了我们的自体ACTEngine候选产品外,我们还在建造一种同种异体,现成的,平台(ACTallo)基于同种异体(即第三方捐赠者衍生的)伽马三角洲t细胞。我们正在筹备的最先进的细胞治疗计划是两个ACTEngine计划,IMA203(第一代)和IMA203CD8(第二代),这两个计划都针对HLA-A*02-已呈现源自PRAME的多肽,在大量实体癌症中高表达。这两个项目都处于临床10期亿开发阶段。
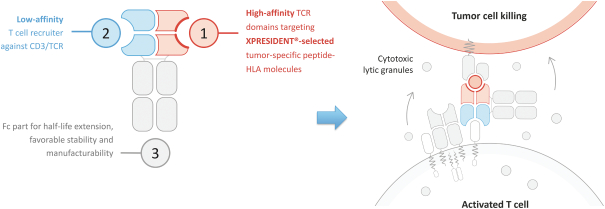
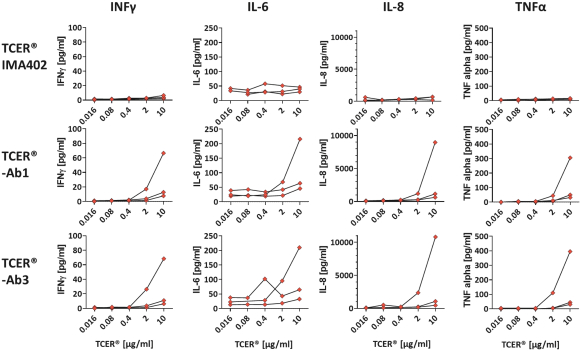
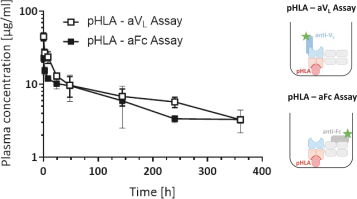
TCER旨在提供现成的并使用用于其他生物制品的标准药品供应链渠道进行部署。我们相信,TCERs可以在不需要专门医疗中心的情况下,对更广泛的患者群体进行治疗,因此可以提供有利的商业特征。尽管TCER将需要多轮剂量,但它们旨在用于门诊设置。我们最先进的TCER计划是IMA401,目标是HLA-A*02-已呈现来源于MAGEA4和/或MAGEA8的多肽,以及靶向An的IMA402HLA-A*02-已呈现来自PRAME的多肽。IMA401和IMA402都针对广泛的实体癌症,并处于临床第一阶段剂量递增。
此外,我们认为我们的两种治疗方式似乎都适合不同的靶点。而当TCR—T对于具有严格的肿瘤选择性和每细胞低拷贝数的靶标,TCER分子可能需要每细胞拷贝数较高的靶标,并可用于具有
61