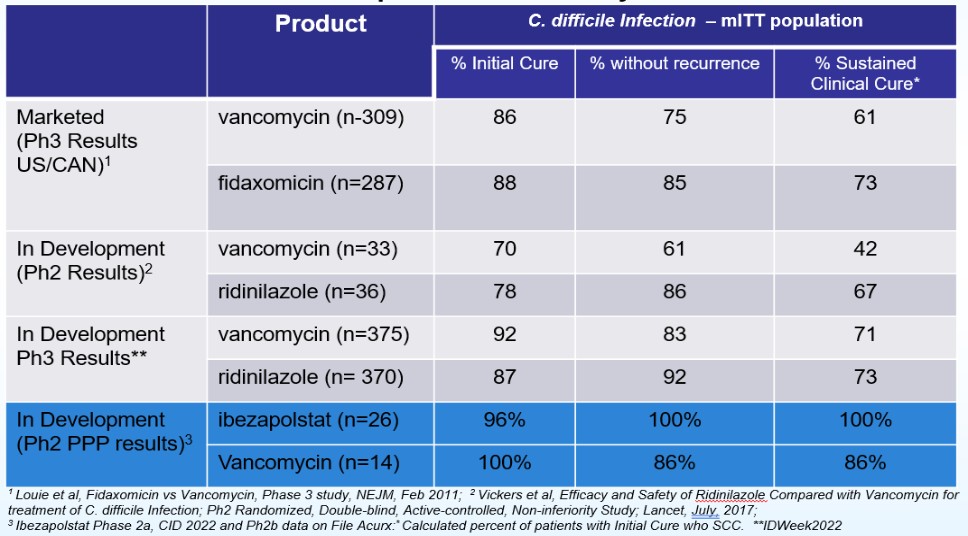
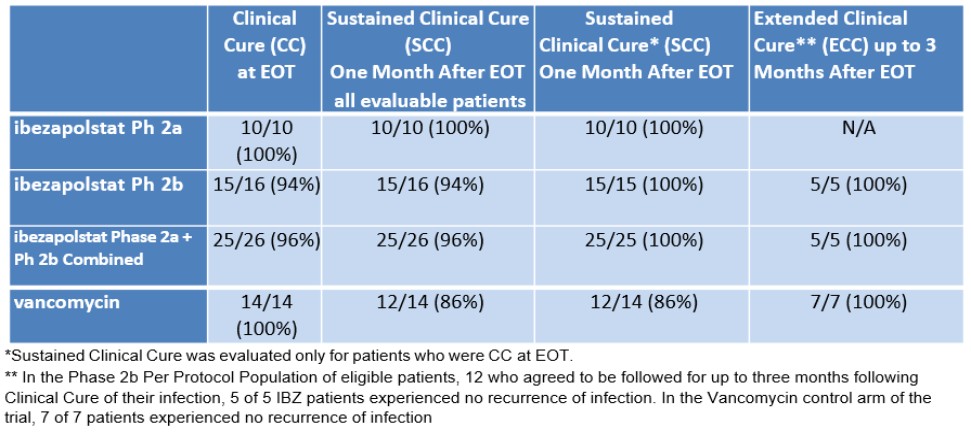
附注4-发行股权
2021年6月23日,Acurx Pharmaceuticals,LLC更名为公司,并更名为Acurx Pharmaceuticals,Inc。公司的注册证书授权 200,000,000普通股,其中14,468,229是已发布且截至2023年12月31日尚未偿还。
2021年6月29日,本公司完成IPO发行2,875,000普通股,价格为$6.00每股,产生净现金收益约为$14.8100万美元,现金发行成本约为$2.4万未发行的A类和B类会员权益根据转换率转换为普通股股份 一- 两名会员权益未解决,导致转换 14,082,318A类和B类会员权益7,041,208普通股股份。购买A类会员权益的凭证同时转换为购买普通股的凭证 一- 对于两个转换比,导致 1,437,577认股权证以加权平均行权价$购买普通股2.88.
与IPO相关,公司发行了 150,000向承销商发出的认股权证。每份认股权证可行使4.5自2021年12月21日起,行使价为美元7.50每股。该公司采用布莱克-斯科尔斯模型计算权证的价值,估计公允价值为#美元。618,000。计算中使用的投入如下:四还有半年的期限,0.79%无风险利率,授权日股价为$6.26,和一个94利用可比公司的%波动率。这一数额既记录为额外实收资本的增加,也记录为发行的非现金发行成本。
2022年7月25日,公司与公司两名高管和公司一名董事会成员(统称为“关联投资者”)以及一名美国机构投资者(“投资者”)签订了证券购买协议(“购买协议”),根据该协议,公司在登记直接发行中发行并出售了总计 1,159,211普通股,面值$0.001每股及预先出资认股权证购买合共130,769普通股。关联投资者总共购买了59,211普通股,收购价为$3.80每股。投资者购买了总计1,100,000普通股,收购价为$3.25每股,以及总计130,769预融资权证,收购价为$3.2499根据预付资金的授权书。出售给投资者的预融资权证的行使价为$。0.0001,可以立即进行练习。 截至2022年12月31日,所有预筹资金认购权均已行使。 该公司还向联属投资者和投资者发行了A系列购买期权 1,289,980购买普通股及B系列认股权证的股份1,289,980普通股,所有这些股票都被认为是权益类的。这些认股权证包括59,211A系列权证和一系列59,211B系列向关联投资者发行认股权证,行使价为每股$。3.55和一组1,230,769A系列权证和一系列1,230,769B系列认股权证向投资者发行,行权价为每股$3.25. A系列认购证可于2023年1月27日开始行使,投资者将于2029年5月18日到期,关联公司将于2028年1月27日到期。B系列配股可于2023年1月27日开始行使,投资者将于2029年5月18日到期,关联公司将于2024年1月27日到期。登记直接发行于2022年7月27日结束。 截至2023年12月31日, 682,769已行使B系列认购权,公司收到约美元2.2这些搜查令的收益为数百万美元。
本公司从登记直接发售中所得的总收益为$4.2扣除配售代理费及公司应付的其他发售费用后的百万元及所得款项净额约为$3.7百万美元。
于2022年7月25日,本公司与两名配售代理订立与登记直接发售有关的配售代理协议(“配售代理协议”),根据该协议,本公司向配售代理支付现金费用$287,874并向各安置代理发出一份63,018购买普通股的认股权证。认股权证的行使价为$3.60每股(代表 110%登记直接向投资者和关联投资者出售的普通股总数的加权平均公开发行价),并于2027年7月27日到期。该公司采用布莱克-斯科尔斯模型计算权证的价值,估计公允价值为$171,409。计算中使用的投入如下:五-一年任期,2.82%无风险利率,授权日股价为$3.70以及一个95波动率%