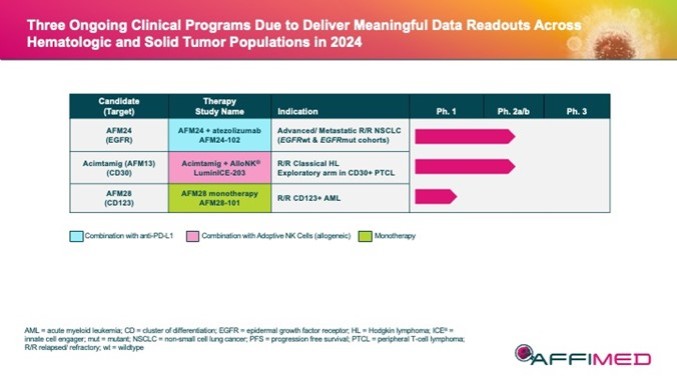
本附注所载的股份及每股资料追溯反映于2024年3月8日生效的反向股票分拆的影响(见附注12)。
带服务条件的股份支付
大多数奖项都是以分期付款的形式发放的三年并且可以被行使到10年在授予之日之后。在2023年和2022年,集团授予822,175和491,560分别奖励员工、管理委员会成员和监事会成员。这些奖励在2023年授予日的公允价值为欧元。6.0百万(美元)6.4百万)。
在2023年,167,260由于与非雇员的雇佣关系终止或咨询协议终止,员工持股2014年的奖励被取消或没收(2022:27,743),以及不是期权已被行使(2022年: 4,344期权的行使加权平均行权价为#美元。25.2).
截至2023年12月31日,2,181,8882014年ESOP奖项杰出(2022年12月31日: 1,526,973), 1,240,852奖项(2022年12月31日: 851,086)被授予。于2023年12月31日尚未行使的期权的行使价格在美元范围内3.5至$134.7 (2022: $13.0至$134.7),加权平均剩余合同期限为 7.3年份(2022年:7.4年),加权平均行使价为美元35.7 (2022: $49.1). 2023年和2022年,本集团预计年度没收率约为 4未归属期权的%。
市场状况下的股份支付
在2022年期间,公司发布了282,500向管理委员会成员和员工提供基于市场的绩效条件的期权。每笔赠款包括 三份额,由此 一- 当成交量加权平均股价高于之前时,授予总额的三分之一将归属 三十交易天数达到美元120, $150、和$180,分别。除控制权变更外,该等期权于授予日期一周年时归属。截至2023年12月31日, 20,000期权被取消。2022年授予日奖励的公允价值为欧元2.9百万(美元)3.2百万),期权的合同期限为 两年.授予日期后两年内任何未授予的奖励将失效。
基于股份的支付费用
2023年,费用为欧元10,714被认为影响研究和开发费用(欧元6,014)以及一般和行政费用(欧元4,700). 2022年,费用为欧元19,110被认为影响研究和开发费用(欧元10,351)以及一般和行政费用(欧元8,759). 2021年,费用为欧元11,820被认为影响研究和开发费用(欧元5,892)以及一般和行政费用(欧元5,928).
公允价值计量
Affimed N.V.发行的带有服务条件的股票期权的公允价值是使用Black-Scholes-Merton公式估计的。该公式根据标的工具的价值、行权价格、股价回报的预期波动率、股息、无风险利率和期权到期时间等输入参数来确定期权的价值。
在市场条件下,股票期权的公允价值是通过使用蒙特卡罗模拟法确定的,该模拟法将需要达到的障碍(或障碍)作为额外的输入参数。股份权益结算补偿计划的公允价值于授出日计量,补偿成本于归属期间确认,并相应增加权益。预计将授予的股票期权数量是在每个衡量日期估计的。