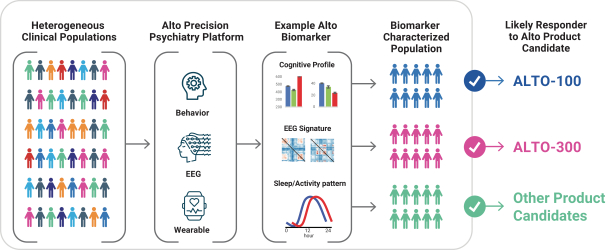
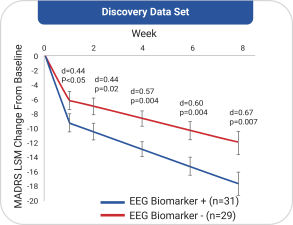
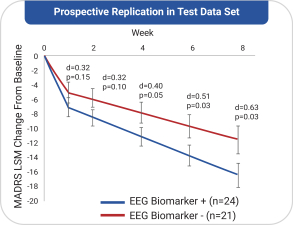
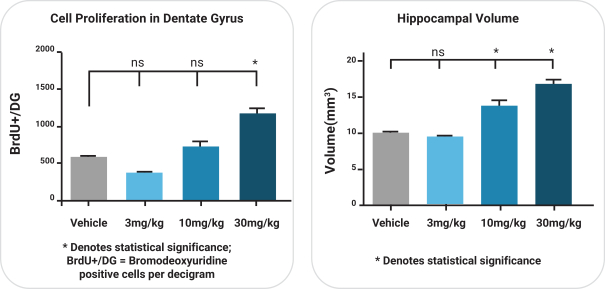
依据第424(B)(4)条提交
注册号码333-276495及333-276828
8040,000股

普通股
我们将提供8,040,000股普通股。这是我们的首次公开募股,在此之前,我们的普通股还没有公开市场。首次公开募股价格为每股16.00美元。我们的普通股已被批准在纽约证券交易所上市,股票代码为ANRO。
我们是一家新兴的成长型公司和一家规模较小的报告公司,根据美国联邦证券法的定义,因此,我们已选择遵守本招股说明书的某些减少的报告要求,并可能选择在本次发行结束后的未来报告中这样做。见 题为招股说明书摘要和作为一家新兴成长型公司和一家较小的报告公司的影响的章节。
投资我们的普通股是有风险的。请参阅本招股说明书第17页开始的题为风险因素的部分,了解您在购买我们普通股股票之前应考虑的因素。
美国证券交易委员会或任何其他监管机构都没有批准或不批准这些证券,也没有对本招股说明书的准确性或充分性做出判断。任何相反的陈述都是刑事犯罪。
| 每股 | 共计 | |||||||
| 首次公开募股价格 |
$ | 16.00 | $ | 128,640,000 | ||||
| 承保折扣及佣金(1) |
$ | 1.12 | $ | 9,004,800 | ||||
| 扣除费用前的收益将捐给Alto神经科学公司。 |
$ | 14.88 | $ | 119,635,200 | ||||
| (1) | 有关承保补偿的其他信息,请参阅标题为承保的章节。 |
应我们的要求,承销商已预留本招股说明书提供的高达5%的股份,以通过定向股票计划以首次公开募股价格出售给某些个人,包括我们的董事、高级管理人员、员工和管理层确定的其他个人。请参阅标题为承销定向股票计划的部分。
普通股预计将于2024年2月6日左右交割。
我们已授予承销商自本招股说明书之日起为期30天的选择权,可额外购买最多1,206,000股普通股。如果承销商全面行使选择权,我们应支付的承保折扣和佣金总额为10,355,520美元,扣除费用前,我们获得的总收益将为137,580,480美元。
| 杰富瑞 | TD Cowen | Stifel | 威廉·布莱尔 |
|
贝尔德 | ||||
招股说明书日期:2024年2月1日