目录表
正如2023年12月宣布的那样,我们正在为GM1神经节苷脂沉积症(GM1)、克拉贝病和异色性脑白质营养不良(MLD)的临床阶段儿科项目寻求潜在的外包许可机会。
我们有一条基因治疗管道,有可能解决多种神经退行性疾病。我们的发展计划包括:
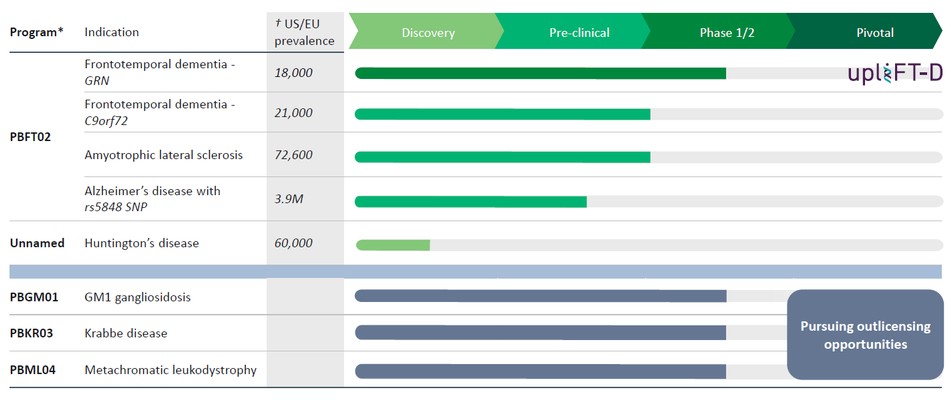
*仍有8个额外的CNS管道许可证选项;之前行使了3个许可证选项,权利随后归还给宾夕法尼亚大学。
第三方来源的†美国/欧盟流行率
PBFT02治疗FTD-GRN
我们目前正在开发PBFT02,它利用AAV1衣壳来提供GRNPGRN编码,用于治疗FTD-GRN。FTD-GRN是一种可遗传的FTD形式,由PGRN产量减少引起,原因是GRN吉恩。PGRN是一种复杂的高度保守的蛋白质,在细胞内稳态、神经发育和炎症中具有多种作用。在FTD中-GRN,PGRN缺乏会导致溶酶体功能障碍、神经炎症和神经变性。
目前,还没有被批准用于治疗FTD的疾病修改疗法-GRN。根据临床前研究的结果,我们认为PBFT02可能提供FTD-GRN预后显著改善的患者。我们选择AAV1衣壳和ICM给药作为PBFT02,是因为这种方法导致了广泛和强大的媒介在非人类灵长类动物(NHP)的大脑和脊髓中的传递,并且由于与其他被测试的血清型相比,AAV1在脑脊液(CSF)中获得了更高的PGRN水平。与健康受试者的脑脊液水平相比,ICM将AAV1注射到NHP可导致脑脊液中人PGRN水平的升高,并且超过了AAVhu68或AAV5的NHP的水平。我们拥有来自美国食品和药物管理局(FDA)的积极研究新药申请(IND),并在多个国家/地区批准了PBFT02的临床试验授权(CTA)。我们正在进行我们的Uplift-D试验,这是一项针对确诊为有症状的FTD患者的PBFT02的国际、多中心、开放标签、单臂1/2期临床试验。GRN.
我们报告了2023年12月和2024年5月在我们的Uplit-D试验的队列1中的三名患者的初步安全性和生物标志物数据。在这项试验中,在接受增强免疫抑制疗法的研究参与者中,剂量1的PBFT02治疗总体上耐受性良好。PBFT02剂量1导致脑脊液中PGRN浓度持续升高,治疗后30天(n=3)和6个月(n=2)的浓度分别为10.7~17.3 ng/m L和21.7~27.3 ng/m L,高于健康成人对照组的3.3~8.2 ng/m L(平均=4.8 ng/m L;n=61)。相比之下,在PBFT02治疗后,所有三名患者的血浆PGRN水平没有改变,在整个可用随访期内保持与基线浓度相似,低于健康成人对照组的水平。
25