目录表
正如下面概述的那样,我们正在推进新型放射免疫疗法和基于抗体的候选疗法的重点临床流水线的开发,旨在改善各种癌症患者的生活:
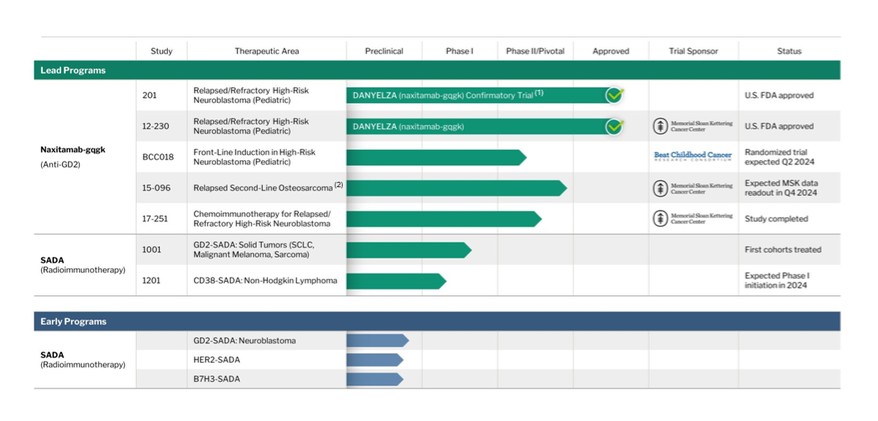
(1) | DANYELZA于2020年11月获得FDA的加速批准。支持BLA提交的关键注册研究,包括衡量药代动力学、毒性和疗效的研究12-230,以及另一项关键的第二阶段多中心研究,研究201,旨在证明使用当前良好制造实践或cGMP商业制造商的研究地点之间的可比性。研究201也是为了满足FDA的验证性研究和上市后要求而设计的。 |
(2) | 最初的研究代表了年龄从1岁到40岁的儿科和青年患者。 |
我们的使命是成为开发更好、更安全的放射免疫疗法和基于抗体的肿瘤疗法的全球领导者,以满足明显的未得到满足的医疗需求,并因此对患者的生活产生革命性影响。我们打算独立或与潜在合作伙伴合作,将我们的候选治疗方案推进和扩大到某些成人癌症适应症。
丹尼尔扎
DANYELZA是美国食品和药物管理局批准的第一个产品,是一种针对神经节苷脂GD2的重组人源化免疫球蛋白G,1K亚型,或IgG1mAb,针对神经节苷脂GD2,它在各种神经外胚层来源的肿瘤和肉瘤中高度表达。DANYELZA于2020年11月获得FDA的加速批准,用于与GM CSF联合治疗1岁及以上的儿童患者和患有R/R高危NB的成人患者,这些患者对以前的治疗表现出部分反应、轻微反应或病情稳定。我们正在将DANYELZA在美国商业化,并于2021年2月开始发货。截至2023年12月31日和2022年12月31日的年度,DANYELZA的销售额分别为8430万美元和4930万美元。2023年,SciClone制药国际有限公司在中国推出了治疗R/R高危NB患者的DANYELZA,并在以色列推出了武田以色列DANYELZA。此外,我们在2023年期间在巴西和墨西哥获得了DANYELZA的监管批准。
DANYELZA的加速批准受到某些上市后要求和承诺的约束,包括必须完成的临床益处验证性上市后试验,以便将生物制品许可证申请(BLA)转换为完全批准并防止FDA撤回许可证。FDA要求进行的验证性上市后临床试验是我们正在进行的201项研究,旨在招募至少80名可评估的患者,并报告总体应答率(ORR)、反应持续时间(DOR)、无进展生存期(PFS)和总生存期(OS)。ORR是主要的
7