目录表
我们保留Nefecon在全球范围内的权利,但不包括在我们已建立战略合作的地区。2019年,我们签订了一项协议,根据该协议,我们授予珠峰药物二期有限公司(即珠峰)独家许可,在中国大区和新加坡开发和商业化治疗IgAN的奈非康。2022年3月,我们扩大了协议覆盖的地区,将韩国包括在内。2021年7月,我们与Stada签订了一项许可协议,将Nefecon在欧洲经济区、英国以及瑞士(如果获得批准)用于治疗IgAN的药物商业化。2022年12月,我们与Viatris Inc.或Viatris的子公司Viatris PharmPharmticals Japan Inc.签订了一项独家许可协议,在日本注册治疗IgAN的Nefecon并将其商业化。
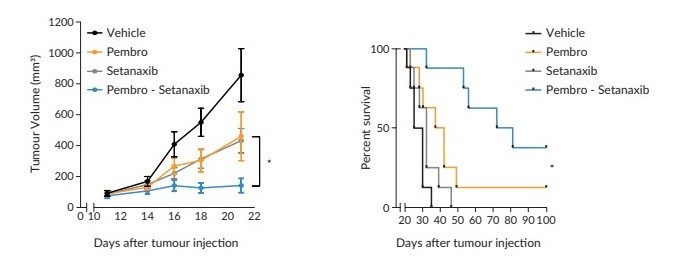
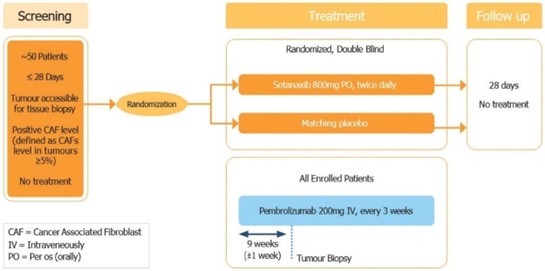
我们还在开发一种新的烟酰胺腺嘌呤二核苷酸磷酸,或NADPH,氧化酶,或NOX抑制剂,我们打算主要开发用于具有纤维化病理的孤儿疾病,主要关注肾脏和肝脏疾病。在这个平台上,我们正在开发一种氮氧化物抑制剂setanaxib,用于治疗原发性胆管炎(PBC)。我们目前正在评估Transform研究中的setanaxib,这是一项2b期临床试验,在该试验中,我们于2022年2月随机选择了第一名患者。Setanaxib将用于大约70-80名PBC和肝脏僵硬程度升高以及对熊去氧胆酸不耐受或反应不足的患者。熊去氧胆酸是一种仿制药,也被称为熊去氧胆酸或UDCA,在北美、欧洲、以色列、澳大利亚和新西兰的80-130个研究中心进行的全球试验中进行。主要终点是碱性磷酸酶(ALP)的降低,关键的次要终点包括肝脏僵硬的变化以及对疲劳和瘙痒(瘙痒)的影响。根据第一阶段研究的有利安全性数据,这项试验将评估两种剂量方案:每天1200毫克和每天1600毫克。我们预计数据将在2024年第三季度读出,这一分析将决定未来潜在的3期研究将使用何种剂量的setanaxib。Setanaxib于2021年8月被FDA授予快车道称号。我们还在进行一项概念验证,即2期临床试验,将setanaxib与检查点抑制剂pembrolizumab联合应用于头颈部鳞状细胞癌(SCCHN),以探索setanaxib作为治疗肿瘤相关成纤维细胞(CAF)水平高的癌症的方法。我们目前还在进行setanaxib治疗Alport综合征的第二阶段临床试验,该试验于2023年11月启动。
我们的管道
下表汇总了我们的主要候选产品组合的开发阶段和状态:

78