技术平台,我们称之为Viaskin,是一种潜在治疗免疫疾病的创新方法,主要关注食物过敏。在EPIT中,完整的皮肤通过Viaskin技术使用含有微克量食物蛋白质的贴片暴露在过敏原中。通过EPIT施加的过敏原被特殊的抗原提呈细胞(表皮内的朗格汉斯细胞)以及真皮树突状细胞捕获在皮肤浅层,从而限制接触血液。在实验模型中,EPIT诱导了一群具有特定特性的调节性T细胞,即Tregs,从而抑制过敏反应。症状和防止进一步敏感化的保护。EPIT诱导的表观遗传修饰有利于Treg介导的免疫反应和下调的Th2反应,并可能在效应的可持续性中发挥作用。根据我们的试验和研究,我们相信EPIT有可能提供所有预期的过敏疾病修改治疗的好处,同时避免严重或危及生命的过敏反应。
Viaskin补丁操作机制的关键元素如下所示:
| • | 该贴片的中心含有一层干燥的过敏原,无需事先准备,即可放置在完整的皮肤上。 |
| • | 在皮肤和贴片中心之间形成的冷凝室会使皮肤过度水合,并积累水分。 |
| • | 水的积累溶解了过敏原。由于这个冷凝室,表皮变得更具渗透性,允许过敏原进入表皮。 |

一旦进入表皮,过敏原就会被一群高度专业化的细胞捕获:朗格汉斯细胞。这些细胞可以捕获皮肤表面的蛋白质,对其进行处理,并将其表位呈现给淋巴结中的T淋巴细胞。
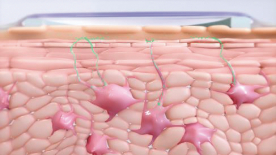
在涂抹Viaskin贴片后,表皮中的朗格汉斯细胞捕获角质层(皮肤最外层)内的花生过敏原(以绿色表示),使过敏原溶解并渗透到皮肤中。
3