目录
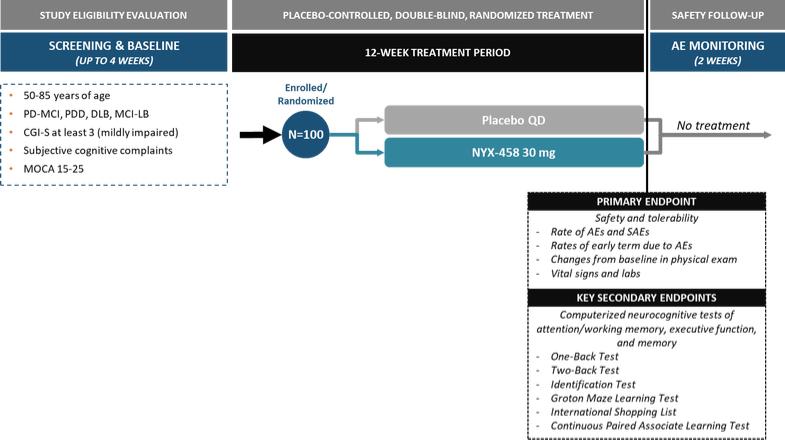
| NYX-458的第二阶段研究目前正在进行中,这将为NYX-458的未来发展提供信息。基于相关的、可翻译的、非人类灵长类动物模型中令人信服的数据,我们相信NYX-458如果获得批准,可能会对与路易体痴呆相关的认知障碍的现有治疗方法提供实质性的改进。 |
| ● | 通过利用我们的NMDAR调制器发现平台,在NMDAR生物学领域建立和扩大我们的领先地位,继续扩大我们的渠道。我们打算利用我们的发现平台,开发一系列大脑和神经系统疾病的广泛管道和产品组合。我们的研发流程由我们的1,000多种独特的、合成的小分子NMDAR调节剂库提供支持,这些调节剂来自我们广泛的原创性研究,并发现了一种新的结合结构域,我们相信这种结合结构域可以安全有效地增强突触的可塑性。所有这些化合物的设计都符合良好的中枢神经系统(CNS)、安全性和药代动力学(PK)标准。我们还计划继续寻找依赖NMDAR的生物标志物,这可能有助于我们未来候选产品的开发。 |
| ● | 优化我们候选产品的开发和商业潜力。在我们选定的适应症中,我们拥有NYX-2925、NYX-783和NYX-458的全球商业权。我们的主要战略是独立追求我们候选产品的开发和商业化。我们组建了一支经验丰富的管理团队,能够沿着药物开发和商业化的整个价值链执行。随着我们继续建立和发展我们的产品组合,我们可能会机会主义地寻求战略合作伙伴关系,使我们的管道价值最大化。 |
来自我们的Discovery平台的候选产品
下表汇总了我们从Discovery平台生成的全资候选产品管道的当前开发状况:

虽然我们所有的候选产品和我们发现平台中的其他分子都调节NMDAR,但它们是不同的化学实体,具有不同的药理特性。我们的每个分子都在结合域内唯一地结合,导致不同的活性、效力和NMDAR亚型选择性特征。我们通过在大脑和神经系统疾病的不同临床前模型中询问我们的分子来评估这些变异的治疗意义。我们从这些临床前研究中收集的数据表明,哪些分子更适合不同的适应症,并相应地为我们的开发决策提供信息。
7