目录表
分子的ETB平台技术
虽然化疗仍然是大多数癌症治疗的基石,但新的和有针对性的治疗类别的出现极大地改变了疾病治疗的结果。单抗、信号转导抑制剂和最近的免疫肿瘤学的出现在复发和难治性环境中都提供了实质性的临床益处,当联合使用时,在早期的治疗路线中。分子认为,ETB代表了一类具有不同生物学特性的新型靶向制剂,它们非常适合改善癌症患者的预后。
ETB似乎能诱导非或不良内化靶标的内化,具有不同的作用机制(酶促和不可逆转的核糖体失活),具有相对可预测的PK谱,并且可以很容易地按照cGMP标准制造。从抗体样靶向结构域的库中,分子的研究和设计平台允许全面的体外培养根据亲和力和特异性、效力和表达来选择针对给定靶标的前导ETB。铅的选择是通过使用动物模型来证实的,以验证PK、吸收、分布、代谢和排泄以及效力。ETB通过一种分化的作用机制具有强大的直接细胞杀伤作用,可以迫使受体内化,并可用于将外源I类抗原等有效载荷输送到胞浆。
在所有临床阶段的ETB中,分子公司使用一种高度有效和适当去免疫的SLTA支架,临床前和动物研究表明,该支架可显著降低先天和获得性免疫原性反应。对于肿瘤已被证明对T细胞参与敏感的适应症,分子公司开发了ETBS,它可以递送外来的I类病毒抗原,呈现在肿瘤表面:分子的抗原种子技术,这是一种免疫肿瘤学的差异化方法。分子已经将其抗原种子技术集成到针对ETB的PD-L1中,MT-6402,并继续建立动物模型,以进一步验证和筛选更多的ETB候选对象来支持这一方法。
分子公司认为,其专有的ETB技术平台代表了肿瘤学领域的一种差异化方法。EBCs具有基于抗体的治疗方法的靶向特异性,但提供高度有效的有效载荷,以一种不受传统化疗耐药机制或靶点内化限制的方式扰乱蛋白质合成,而不是ADC。分子还在寻求扩大药物治疗的潜在靶标的宇宙,方法是利用ETB的能力,针对通常不内化的受体强制内化。
肿瘤学治疗需要新的作用机制,分子公司认为,其ETB平台技术的不同作用机制可能提供超过现有治疗模式的独特好处。
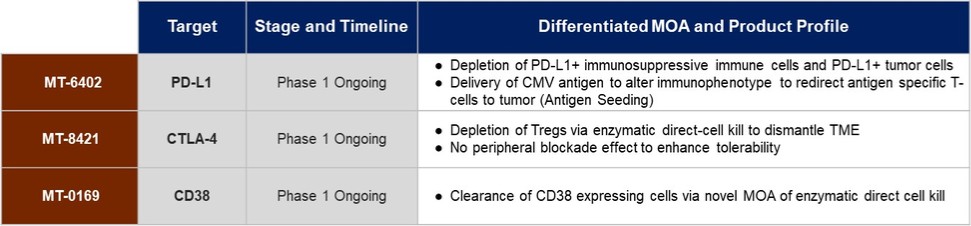
ETB产品线
分子公司正在开发一条ETB管道,分子公司相信这将有能力为癌症患者提供有意义和长期的好处。分子计划将每一种药物作为单一药物和/或与其他疗法联合开发,如果适用的话。下表描述了分子公司目前的流水线:
9