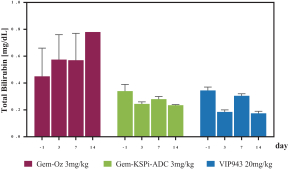
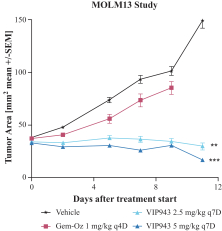
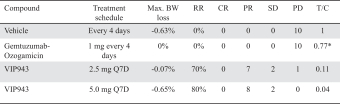
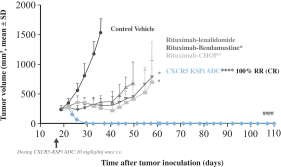
| • | 有效载荷设计用于: |
| • | 使用我们的蜂窝捕捉器降低渗透性™确保癌细胞蓄积的技术 |
| • | 以较低的流出量增加渗透性,以便在TME中释放。 |
VersAptx平台使我们能够针对特定目标优化这些技术,并开发旨在解决第一代生物结合物的安全性和有效性挑战的生物结合物。
下图总结了我们多样化的渠道:

我们卓有成效的管理团队在肿瘤学方面拥有超过120年的集体经验,在成功的药物开发、审批和价值创造方面有着久经考验的记录。他们从Pharmacclics LLC、Acerta Pharma LLC、阿斯利康、拜耳股份公司、安进、基因泰克公司、礼来公司和强生公司等领先公司吸引人才,为多项药物审批和重磅退出做出了重大贡献,包括以70亿美元收购Acerta Pharma的Calquence®阿斯利康和价值9.75亿美元的Imbrovica®Janssen PharmPharmticals(现为强生创新医药)和Pharmaccle ics之间的合作伙伴关系。
我们相信,我们多样化的渠道,包括我们的下一代VersAptx平台和我们的管理团队,使我们能够成功地为患者开发范式转换疗法。
战略
我们的目标是通过开发安全、有效和耐受性良好的差异化和新疗法,成为领先的生物制药公司。我们力求通过实施以下战略来实现这一目标:
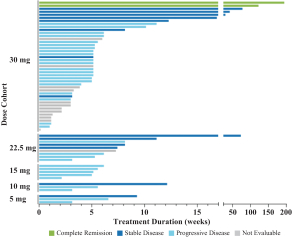
| • | VIP943、VIP236和Enitociclib在临床开发和治疗中的进展证明-概念 |
| • | 在2025年底或2026年初将VIP924进展为IND应用程序 |
| • | 利用我们的下一代VersAptx平台扩展我们的渠道,并与其他公司合作开发独特、安全和有效的ADC和SMDC |
| • | 形成战略合作伙伴关系/协作,最大限度地发挥我们的渠道和VersAptx平台的潜力 |
7