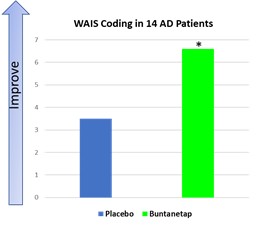
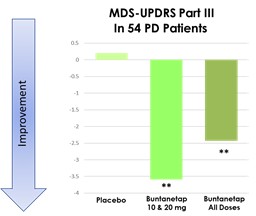
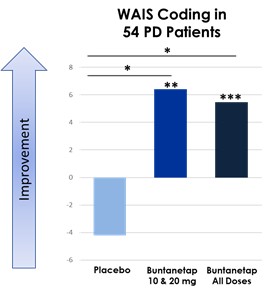
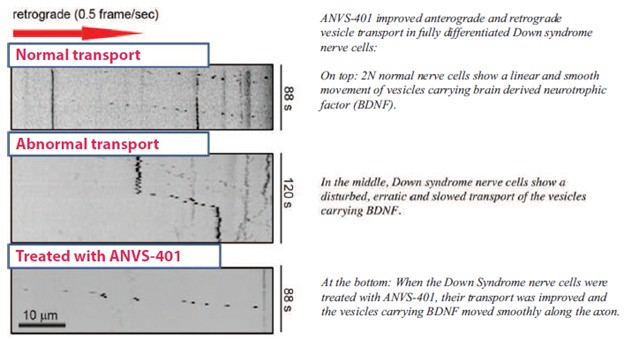
美国
美国证券交易委员会
华盛顿特区,20549
表格:10-K
☒ | 根据1934年《证券交易法》第13或15(D)款提交的年度报告 |
| |
截至本财年的12月31日, 2023 |
|
☐ | 根据1934年《证券交易法》第13或15(D)款提交的过渡报告 |
| |
在过渡时期, 到 |
委员会文件编号:001-39202
Annovis Bio,Inc.
(注册人的确切姓名载于其章程)
| |
特拉华州(述明或其他司法管辖权
公司或组织) | 26-2540421(税务局雇主
识别号码) |
101 Lindenwood Drive,Suite 225
马尔文, 帕19355
(主要执行机构地址,包括邮政编码)
(484) 875-3192
(注册人的电话号码,包括区号)
根据该法第12(B)款登记的证券:
| | | | |
每个班级的标题 | | 交易符号 | | 注册的每个交易所的名称 |
普通股,每股票面价值0.0001美元 | | anvs | | 纽约证券交易所 |
根据该法第12(G)款登记的证券:无。
用复选标记表示注册人是否为证券法规则第405条所定义的知名经验丰富的发行人。是☐ 不是 ☒
如果注册人不需要根据交易法第13条或第15条(d)款提交报告,则用复选标记进行标记。是的 ☐ 不是 ☒
用复选标记表示注册人(1)是否在过去12个月内(或注册人被要求提交此类报告的较短期限内)提交了1934年《证券交易法》第13节或15(D)节要求提交的所有报告,以及(2)在过去90天内是否符合此类提交要求。是 ☒*不是。☐
用复选标记表示注册人是否已在过去12个月内(或在注册人被要求提交此类文件的较短时间内)以电子方式提交了根据S-T规则第405条(本章232.405节)要求提交的每个交互数据文件。是 ☒*不是。☐
用复选标记表示注册人是大型加速申报公司、加速申报公司、非加速申报公司、较小的报告公司或新兴成长型公司。请参阅《交易法》第12b-2条规则中“大型加速申报公司”、“加速申报公司”、“较小申报公司”和“新兴成长型公司”的定义。
| | | |
大型数据库加速了文件管理器的使用☐ | 加速的文件管理器☐ | 非加速文件服务器 ☒ | 规模较小的报告公司。☒ |
新兴成长型公司☒ | | | |
如果是一家新兴的成长型公司,用复选标记表示注册人是否已选择不使用延长的过渡期来遵守根据交易所法案第13(A)节提供的任何新的或修订的财务会计准则。☒
用复选标记表示注册人是否提交了一份报告,证明其管理层根据《萨班斯-奥克斯利法案》(《美国联邦法典》第15编,第7262(B)节)第404(B)条对其财务报告的内部控制的有效性进行了评估,该评估是由编制或发布其审计报告的注册会计师事务所进行的。☐
如果证券是根据该法第12(B)条登记的,应用勾号表示登记人在备案中的财务报表是否反映了对以前发布的财务报表的错误更正。☐
检查是否有任何错误更正是重复的,需要根据第240.10D—1(b)节对注册人的执行官在相关恢复期内收到的基于激励的补偿进行恢复分析。 ☐
检查注册人是否为空壳公司(如交易法第12b—2条所定义)。是的 ☐不是的。☒
截至2023年6月30日,也就是注册人最近完成的第二财季的最后一个营业日,注册人的非关联公司持有的注册人普通股的总市值为$91,527,093根据纽约证券交易所公布的收盘价
截至2024年3月26日,发行人普通股的流通股数量为 11,011,299.
引用成立为法团的文件
如本报告中明确描述的,注册人2024年股东周年大会委托书的某些部分通过引用纳入本年度报告表格10—K的第三部分。