目录表
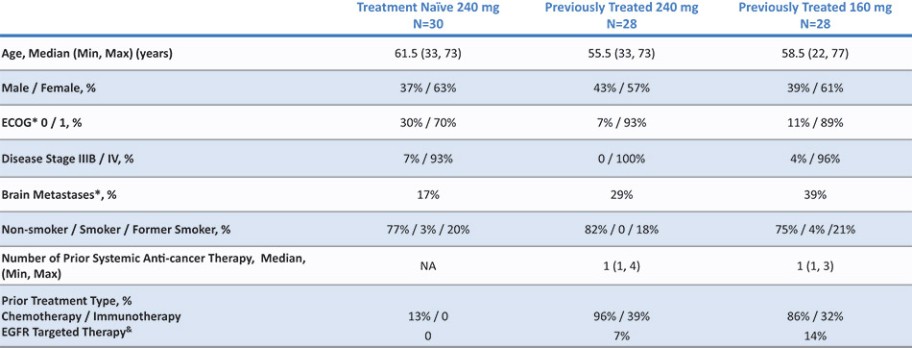
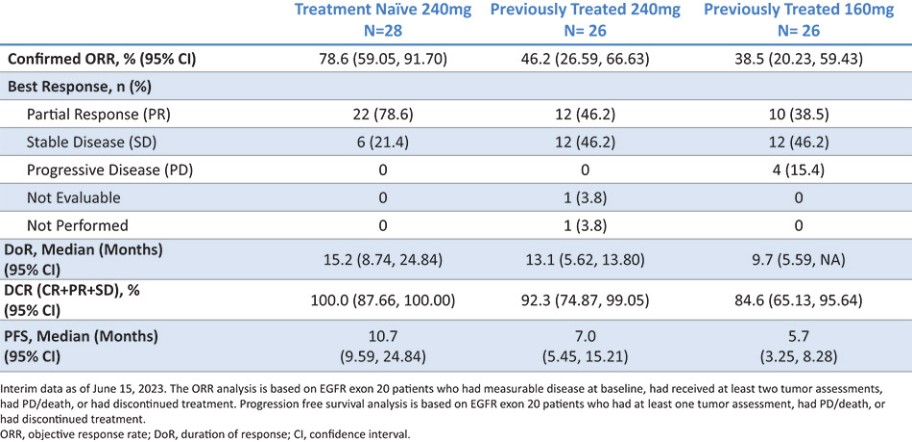
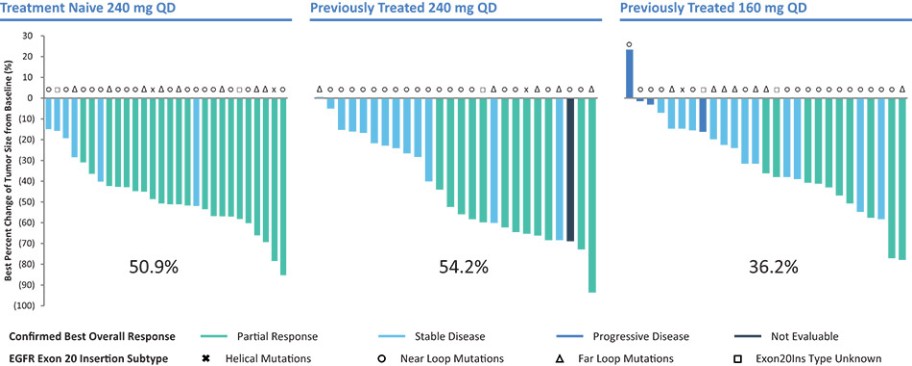
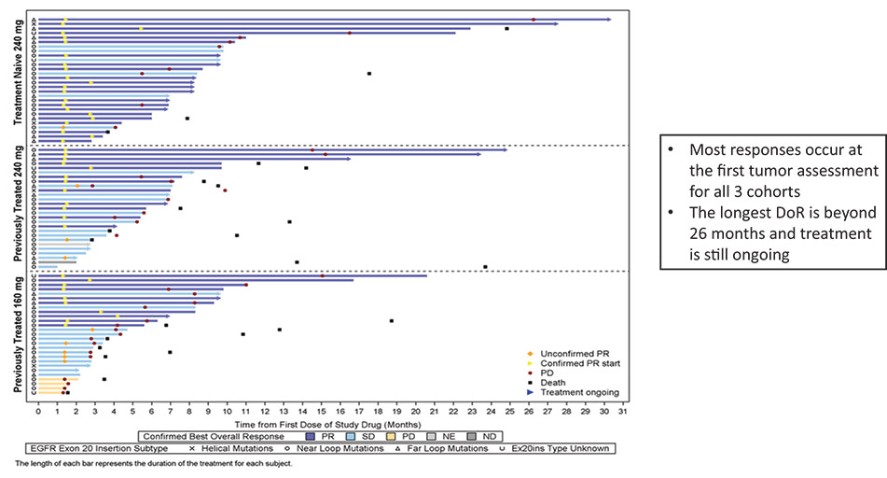
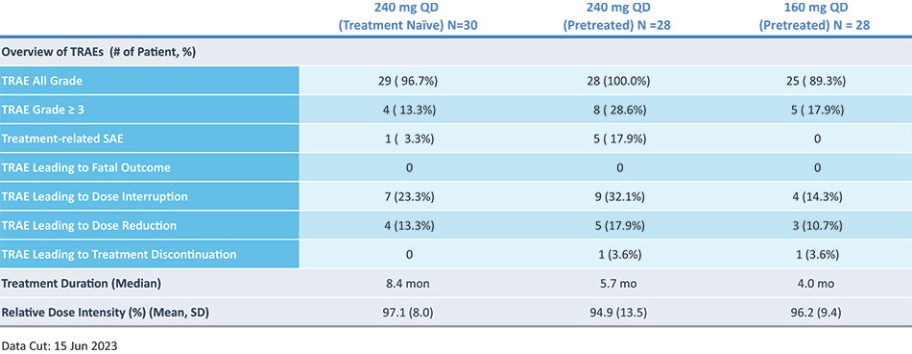
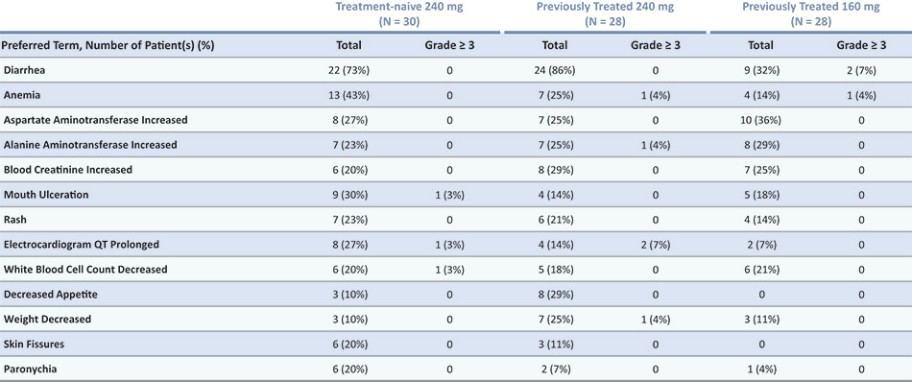
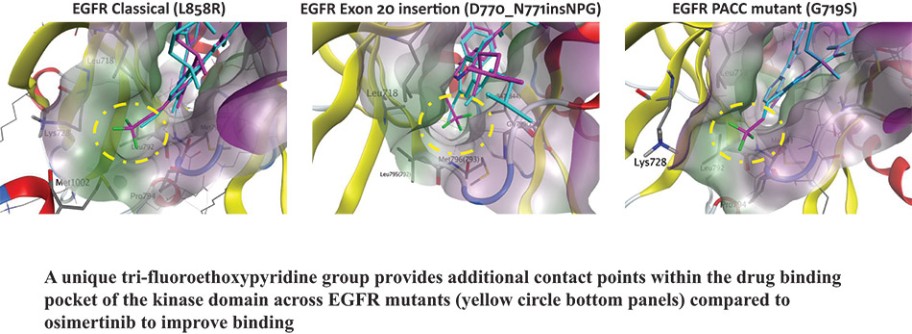
结果可能不代表最终结果;然而,我们相信这些中期临床结果强调了呋喃莫替尼在其肿瘤中含有不常见的EGFRm的患者中的潜力。
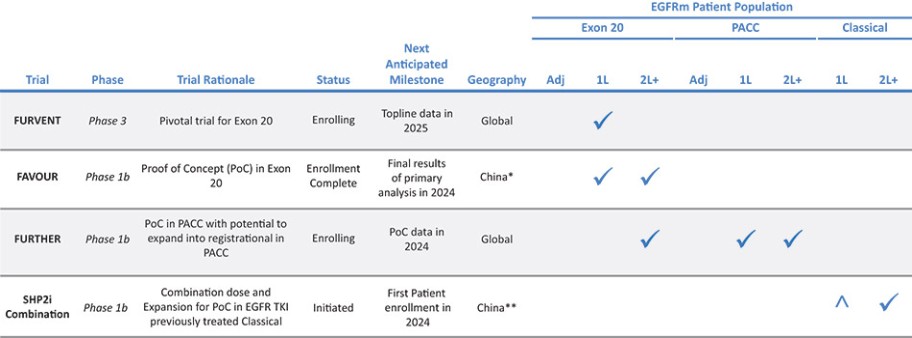
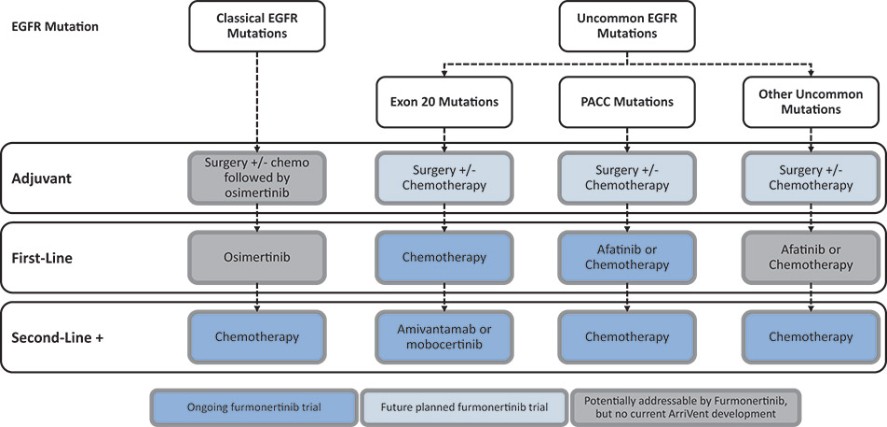
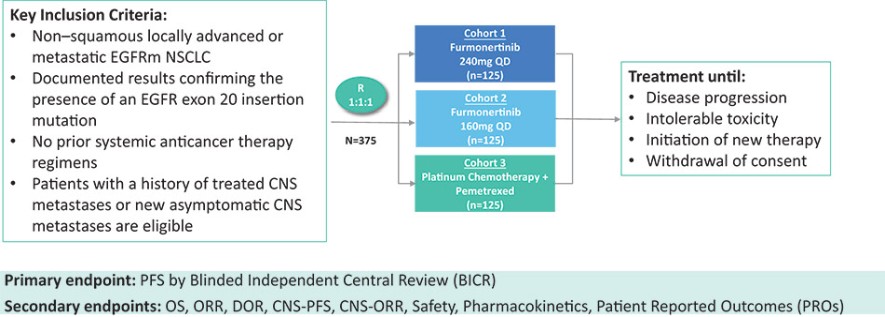
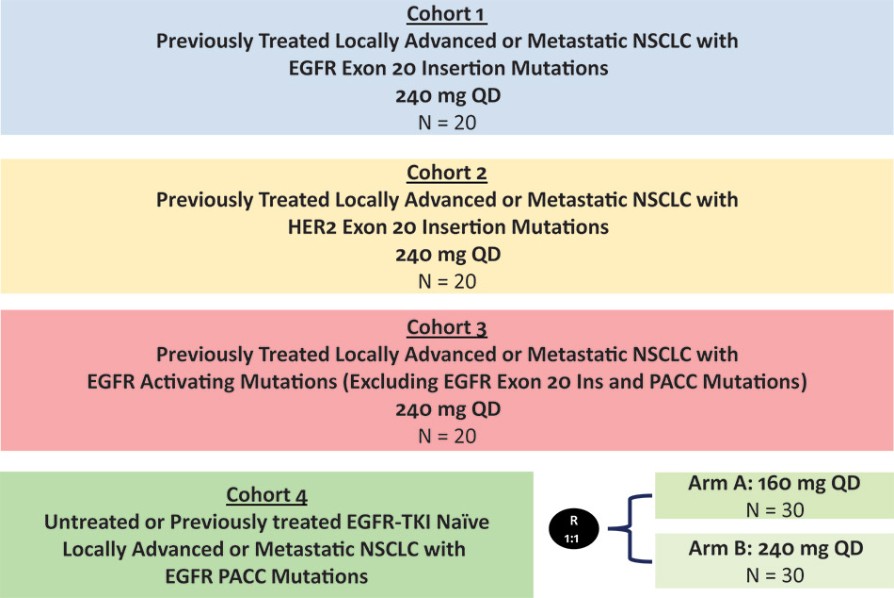
我们目前正在进行一项关键的第三阶段临床试验,即FURVENT临床试验,对象是具有外显子20插入突变的一线局部晚期或转移性非鳞状细胞癌EGFRm NSCLC患者,我们预计2025年该试验将提供TOPLINE数据。此外,我们正在对激活了EGFRm的非小细胞肺癌患者进行1b期临床试验,这是进一步的临床试验,包括一个具有PACC突变的队列已经完全登记,我们预计这个队列的概念数据将在2024年得到证明。根据数据审查和监管反馈,我们打算将这一1b期临床试验扩大为针对PACC突变的EGFRm非小细胞肺癌患者的潜在注册临床试验。
自2021年成立以来,我们利用我们的全球网络和我们在业务发展交易方面的经验,建立了强大的肿瘤学渠道。2021年,我们从Allist获得了在全球范围内开发和商业化呋莫替尼的权利,但包括内地中国、香港、澳门和台湾在内的大中国除外。我们计划继续从事业务开发活动,在目前未得到批准的疗法服务的地区寻找更多的创新疗法。这一更广泛的战略包括评估旨在扩大我们差异化和下一代肿瘤学资产开发渠道的研究合作、伙伴关系和许可安排。下表汇总了我们目前正在开发的最高级阶段:
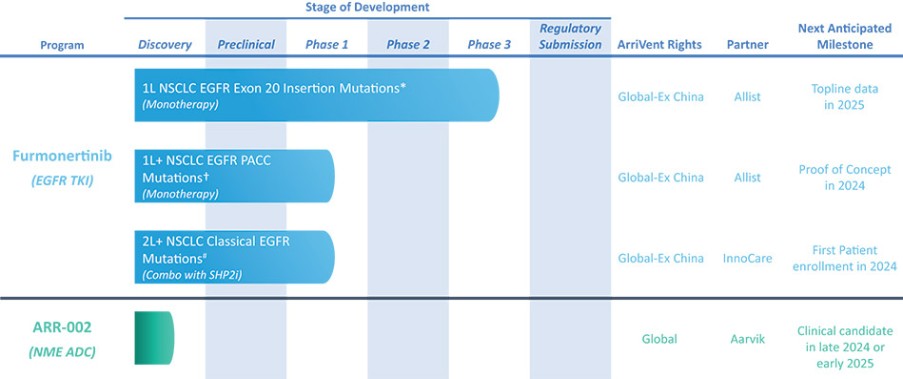
NSCLC:非小细胞肺癌;EGFR:表皮生长因子受体;PACC:P-loopα-c螺旋压缩
名单:上海艾力士制药有限公司;InnoCare:北京Innocare医药科技有限公司;Aarvik:Aarvik治疗公司;1L:一线治疗;1L+:治疗单纯且以前接受非TKI治疗;2L+:二线或更大治疗;SHP2i:Shp2抑制剂。
* | 针对非小细胞肺癌EGFR外显子20插入突变的一线治疗药物Furmonertinib的研究是基于Allist公司正在进行的1b阶段研究和正在进行的FURVENT第三阶段研究。这些研究尚未完成,也没有针对这一适应症进行第二阶段研究。 |
† | 正在进行的关于Furmonertinib治疗EGFRm NSCLC的进一步1b阶段研究包括PACC突变(一线或更高)和外显子20插入突变(二线或更高)的队列。 |
# | 对Furmonertinib与SHP2i联合二线或更大剂量治疗EGFRm NSCLC的评估是基于与InnoCare合作进行的1b期研究。 |
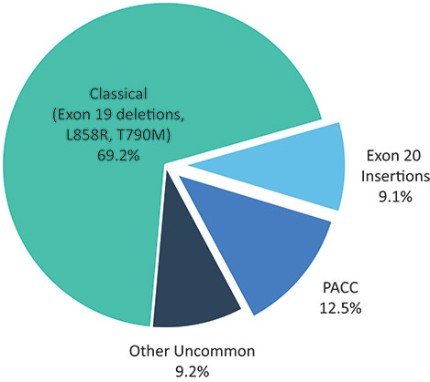
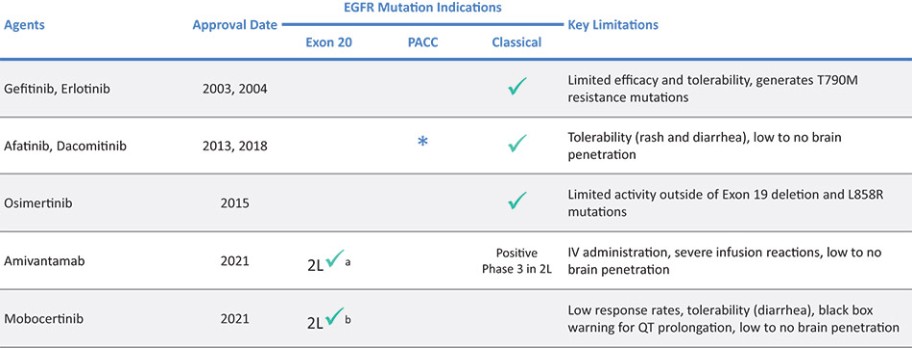
目前批准的EGFR TKIs已取得相当大的商业成功,并已成为典型EGFRm非小细胞肺癌患者的标准治疗方案,约占所有EGFRm非小细胞肺癌患者的69%。然而,这些疗法中的许多,包括阿斯利康(阿斯利康)的第三代EGFR TKI,奥西莫替尼(Tagrisso)®),对相当一部分患有罕见的EGFRm的非小细胞肺癌患者最低限度地有效或未被批准使用。这种未得到满足的需求使许多EGFRm非小细胞肺癌患者几乎没有有效的治疗选择。呋莫那替尼目前由中国的Allist批准并商业化分配,作为治疗
6