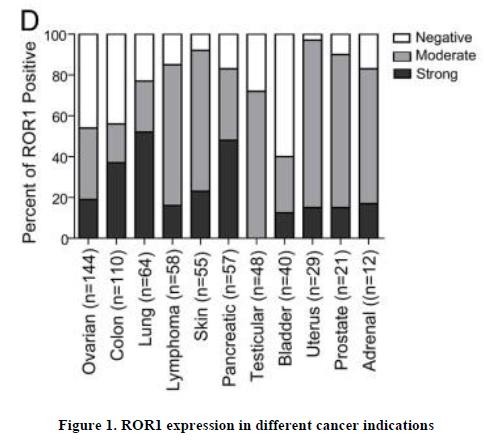
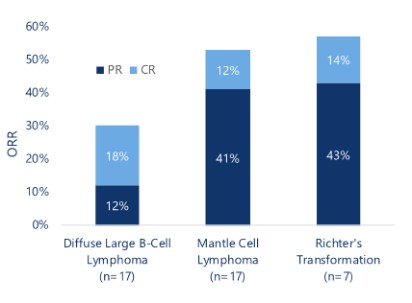
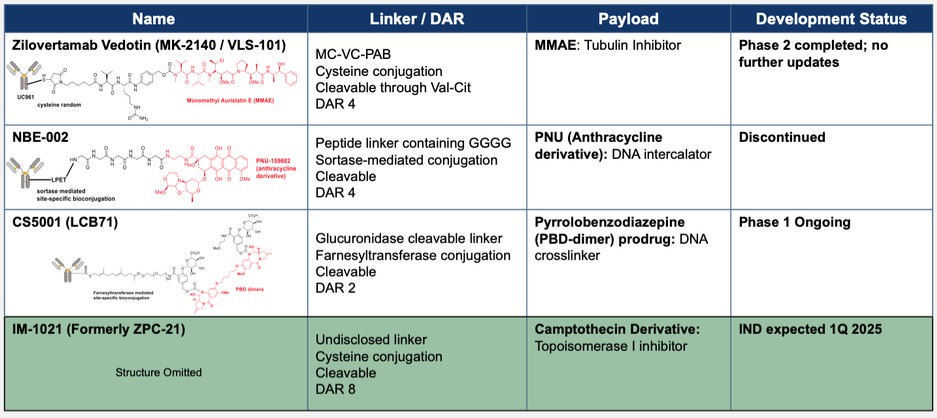
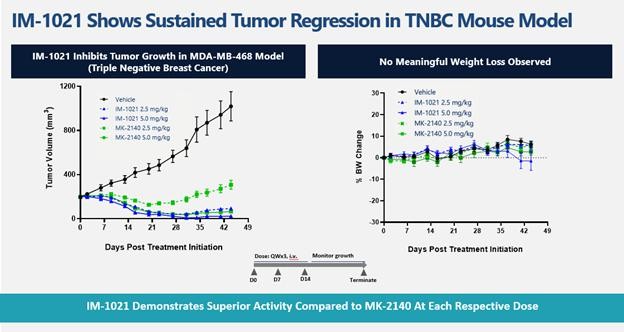
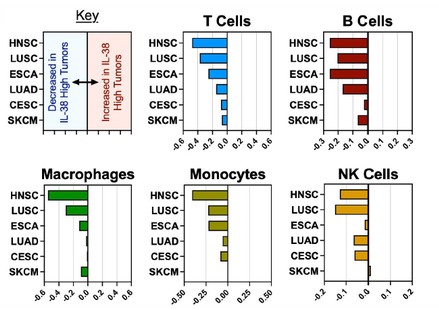
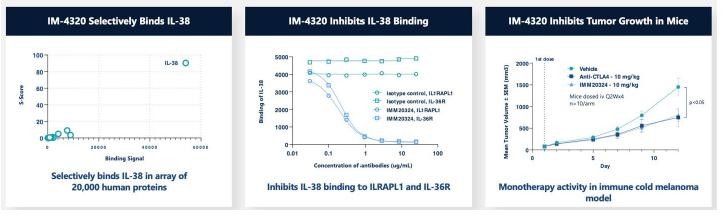
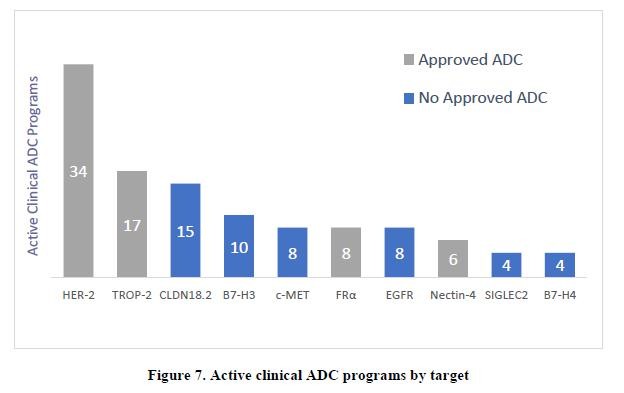
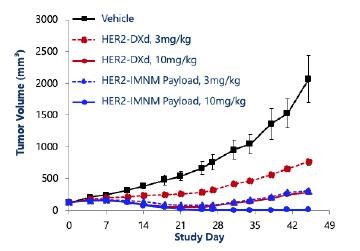
目录表
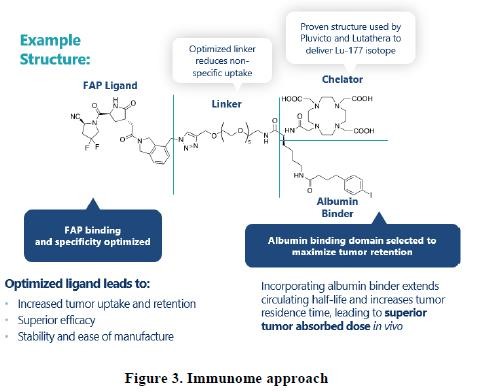
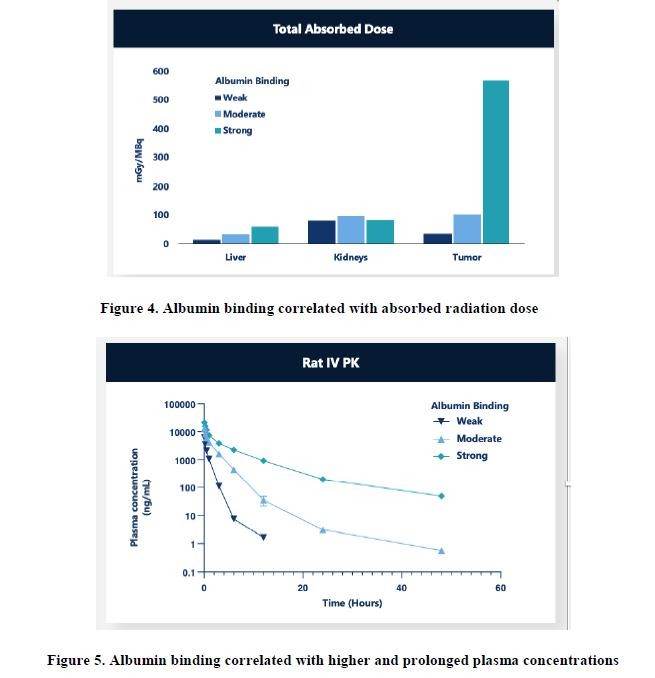
免疫组管路
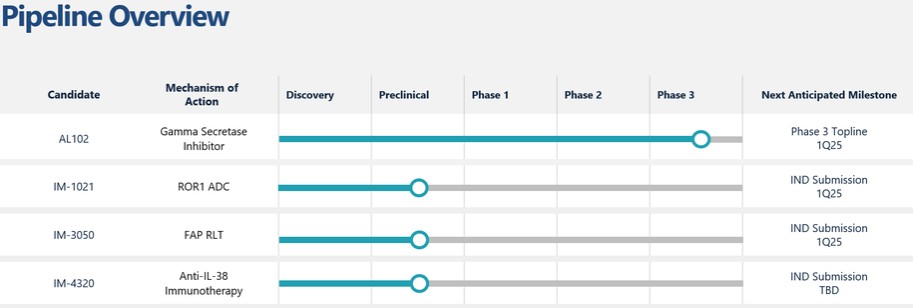
AL102(伽玛分泌酶抑制剂)
2024年2月5日,我们与Ayala签订了Ayala购买协议,根据协议,我们获得了包括AL102权利在内的资产,AL102是一种GSI,目前正在进行治疗硬纤维瘤的第三阶段试验评估。我们于2024年3月25日完成了阿亚拉购买协议预期的交易,包括购买AL102。
我们对AL102的兴趣在很大程度上是对第二阶段数据的回应,2023年10月,Ayala在欧洲医学肿瘤学学会大会(ESMO)上分享了该数据的初步版本。Ayala的数据显示,在有意治疗的人群中,客观应答率(ORR)为64%,在可评估的患者中为75%。其他反应指标,包括MRI测量的肿瘤体积和T2成像估计的细胞密度,也显示出深度反应。
疾病背景
硬纤维瘤,也称为侵袭性纤维瘤病或硬纤维样型纤维瘤病,是一种衰弱、疼痛和侵袭性的非转移性软组织肿瘤,容易复发。硬纤维瘤根据发生在体内的位置而定,可能会导致虚弱的疼痛、畸形,在某些情况下,还会导致危及生命的器官损伤。这些肿瘤很罕见,在美国每年大约有1,000-1,650名患者被诊断出来,大约5,500-7,000名患者得到积极治疗。它们通常发生在15岁到60岁之间的人群中,最常见的是青春期早期,高峰在30岁左右,女性比男性更常见。
硬纤维样瘤发生在结缔组织中,可以发生在身体任何有结缔组织的地方。这些肿瘤具有局部侵袭性,这意味着虽然它们不会转移到身体的其他部位,但它们可以生长到周围或邻近的组织中。虽然一些硬纤维瘤患者没有症状,但其他人可能会出现疼痛、肿胀、睡眠困难和行动不便。症状在很大程度上取决于肿瘤的位置和周围组织的侵犯或压迫程度。与硬纤维瘤相关的疼痛和身体限制导致高临床负担和生活质量下降。此外,在丹麦进行的一项研究发现,在确诊后1年和3年,与匹配的患者相比,硬纤维瘤患者的医疗资源利用率显著更高,包括住院和门诊就诊以及住院天数的增加。
绝大多数(85%-90%)的硬纤维样瘤是散发性的,由编码β-连环蛋白的CTNNB1基因的体细胞突变引起。硬纤维样瘤也可能是由于APC基因的胚系突变引起的,这也与家族性腺瘤性息肉病有关,导致β-连环蛋白在细胞核内积聚,并驱动其靶基因的过度表达。具有这些突变的患者发生硬纤维瘤的危险因素包括创伤(特别是腹部手术)、雌激素和怀孕。
7