根据规则
424(B)(4)提交
注册号码333-276523
招股说明书
14,500,000股

Kyverna治疗公司
普通股
这是Kyverna治疗公司普通股的首次公开发行。我们将发行14,500,000股我们的普通股。
在此次发行之前,我们的普通股还没有公开市场。首次公开募股价格为每股22.00美元。我们的普通股已获准在纳斯达克全球精选市场上市,交易代码为KYTX。
我们是一家新兴成长型公司和一家较小的报告公司,每个公司都符合联邦证券法的定义,因此,我们已选择遵守本招股说明书的某些降低的报告要求,并可能在未来的备案文件中选择这样做。见招股说明书摘要和作为一家新兴成长型公司和一家较小的报告公司的影响。
投资我们的普通股涉及很高的风险。请参阅第16页开始的本招股说明书中题为风险因素的部分,以了解您在购买我们普通股股票之前应考虑的因素。
美国证券交易委员会或任何其他监管机构都没有批准或不批准这些证券,也没有对本招股说明书的准确性或充分性进行评估。任何相反的陈述都是刑事犯罪。
| 人均分享 | 总计 | |||||||
| 首次公开募股价格 |
$ | 22.00 | $ | 319,000,000 | ||||
| 承保折扣和佣金 (1) |
$ | 1.54 | $ | 22,330,000 | ||||
| 扣除费用前的收益将捐给Kyverna治疗公司。 |
$ | 20.46 | $ | 296,670,000 | ||||
| (1) | 有关向承销商支付的赔偿的说明,请参阅本招股说明书中题为承销的部分。 |
我们已授予承销商以首次公开发行价格减去承销折扣和佣金从我们手中购买最多2,175,000股额外股票的选择权 。
承销商预计在2024年2月12日左右向买家交付股票 。
| 摩根大通 | 摩根士丹利 | Leerink合作伙伴 | 富国银行证券 |
招股说明书日期为2024年2月7日。
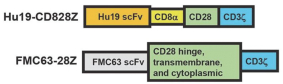
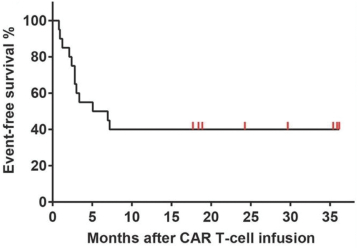
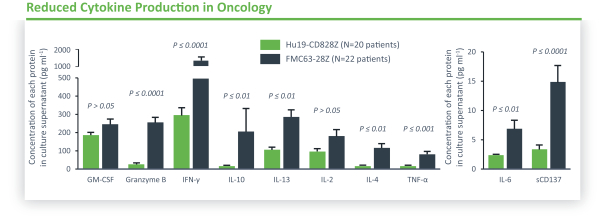
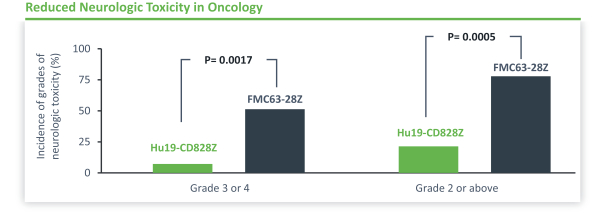
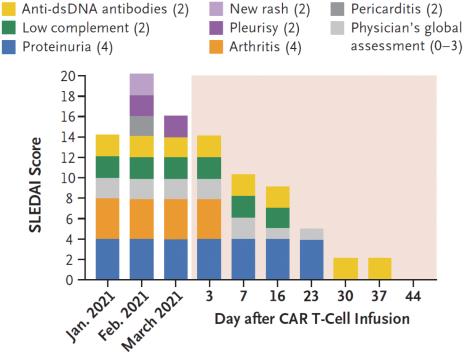
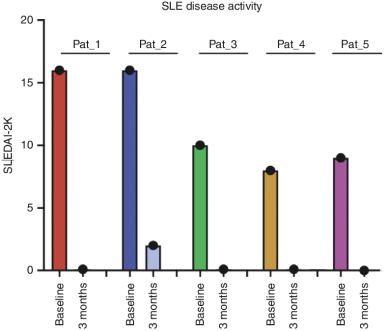
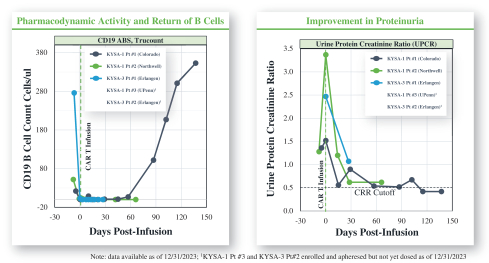
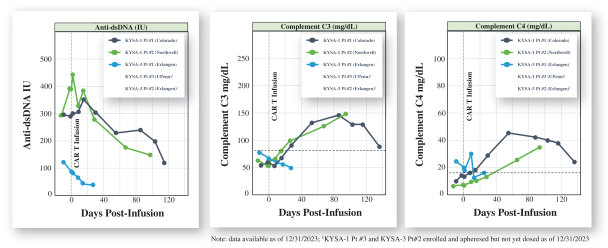
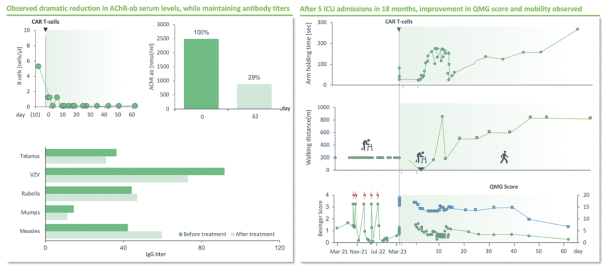
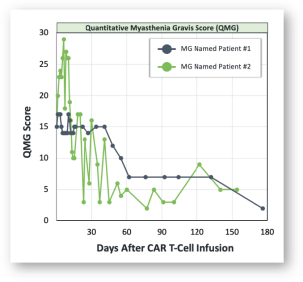
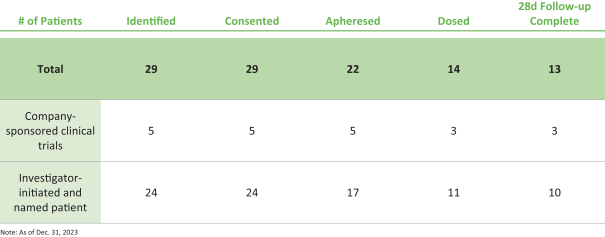
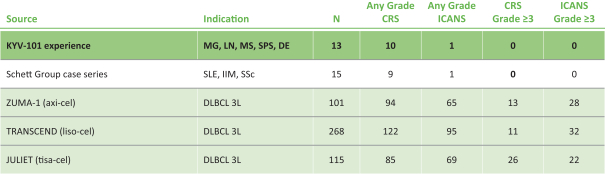
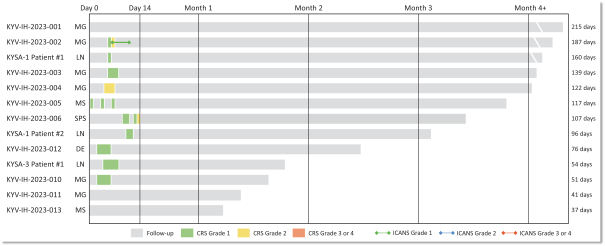
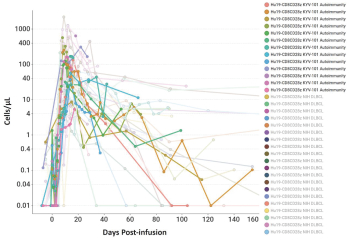
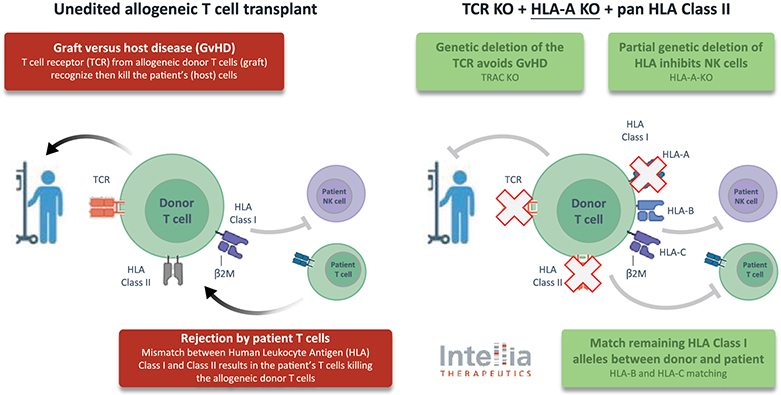
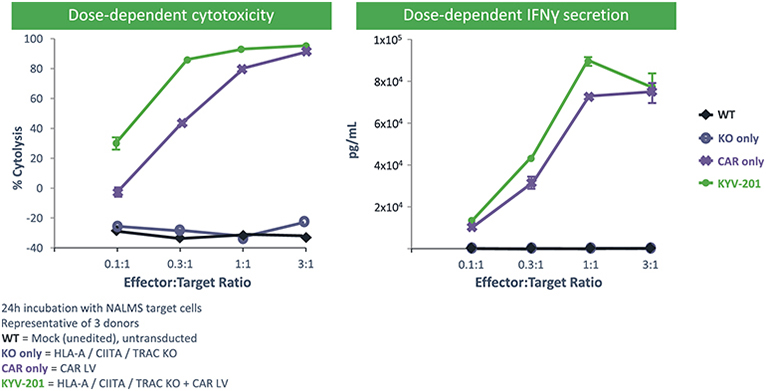
 ).
).