目录表
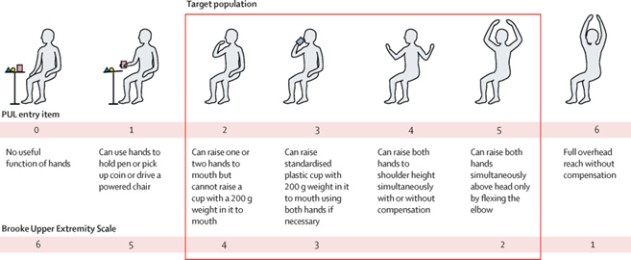
上肢性能(PUL条目) (1)
(CAP—1002当前DMD目标人群)
| (1) | 图片来源:HOPE—2 柳叶刀 出版物(2022年3月) |
队列B的入组正在进行中,该队列旨在入组约44名受试者,以1:1的比例随机分配至CAP—1002或安慰剂组。在4次CAP—1002或安慰剂给药后,将在第12个月对每个个体队列进行主要疗效和安全性分析。我们计划于2024年第二季度完成队列B的入组。队列B使用在我们圣地亚哥工厂生产的产品。
HOPE—3研究的主要结局指标将是上肢功能("PUL")v2.0,这是一种专门设计用于评估高位(肩)、中段(肘)和远端(腕和手)功能的经验证的工具,其概念框架反映上肢功能的无力进展。HOPE—3还将测量各种次要终点,包括心功能评估。
根据我们的RMAT指定,在2023年第三季度,我们与FDA举行了一次B类会议,讨论了我们的生产计划,预计可能提交BLA申请。在这次会议上,我们确认了我们的第三阶段,希望—3计划的一致性。此外,我们还讨论了商业生产活动的计划,包括效价测定和其他产品放行标准,以支持商业化。我们计划在2024年第一季度与FDA会面,继续讨论我们的BLA途径。在即将举行的B类会议中,我们打算讨论我们进一步的CMC商业发布计划(如果获得批准),以加快我们的BLA提交途径。我们的最终目标是提交BLA,允许使用我们圣地亚哥工厂生产的CAP—1002商业产品。
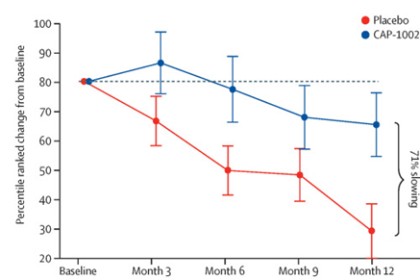
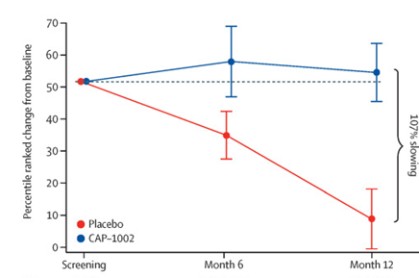
II期HOPE—2临床试验: HOPE—2是一项随机、双盲、安慰剂对照临床试验,在美国多个研究中心进行,并于2021年完成。该临床试验旨在评价CAP—1002重复静脉给药的安全性和有效性,用于有骨骼肌损害证据的男孩和年轻男性,无论活动状态如何。研究中约90%的患者为非活动性患者,所有患者均接受稳定的类固醇治疗。两个治疗组的人口统计学和基线特征相似。希望2的最终一年结果发表在 《柳叶刀》2022年3月,表明该试验达到了PUL v1.2的中级维度的主要疗效终点(p = 0.01)和完整PUL v2.0的其他阳性终点(p = 0.04)。虽然中级PUL v1.2是试验确定的主要终点,但我们也使用PUL v2.0进行了分析,因为FDA建议使用更新后的PUL v2.0作为主要疗效终点以支持BLA。左心室射血分数(LVEF)(心脏泵功能的总体指标)在安慰剂组随时间推移降低,但CAP—1002组改善,显示心脏病进展减慢107%(p = 0.002)。此外,数据表明心脏功能的整体改善,如指数容积(LVESV,LVEDV)测量。这些是心脏功能的替代指标,被认为与长期结局相关。此外,数据显示生物标志物CK—MB减少,CK—MB是一种只有在心肌细胞损伤时才会释放的酶。在正常人受试者中,血液中通常没有可测量的CK—MB。众所周知,DMD中的持续性肌细胞损伤导致与心肌细胞损失相关的病理性高酶水平。据我们所知,这是DMD中第一个将心脏功能稳定与细胞损伤生物标志物减少相关的临床研究。 除了类固醇,DMD的功能保留是罕见的。安慰剂组患者的结果与自然病史一致,但在治疗组中,大多数患者在一年治疗期间这些终点稳定或改善。CAP—1002在整个研究期间通常安全且耐受良好。除了过敏症
7