
美国 美国
证券交易委员会
华盛顿特区,20549
表格
对于
截止的财政年度
或
对于 ,过渡期从_
佣金
文件编号:
(注册人的确切名称与其章程中规定的名称相同)
| (州或 公司或组织的其他管辖权) | (I.R.S.雇主 识别码) |
(主要执行办公室地址 )
(注册人的电话号码,包括区号)
根据该法第12(B)条登记的证券:
| 每个班级的标题 | 交易 个符号 | 注册的每个交易所的名称 | ||
根据该法第12(G)条登记的证券:
无
如果注册人是证券法规则405中定义的知名经验丰富的发行人,请用复选标记表示
。是的☐
如果注册人根据《交易法》第13或15(d)条无需提交报告,请使用复选标记进行标记。是的,
用复选标记检查
注册人是否(1)在过去12个月内(或注册人被要求提交此类报告的较短时间内)提交了1934年证券交易法
第13条或第15(d)条要求提交的所有报告,以及(2)在过去90天内是否
遵守此类提交要求。
用复选标记表示注册人是否在过去12个月内(或在注册人被要求提交和张贴此类文件的较短时间内)以电子方式提交了根据S-T规则(本章232.405节)第405条要求提交的每个交互数据文件。
用复选标记表示注册人是大型加速申报公司、加速申报公司、非加速申报公司、较小的申报公司或新兴成长型公司。请参阅《交易法》第12b-2条规则中“大型加速申报公司”、“加速申报公司”、“较小报告公司”和“新兴成长型公司”的定义。
| 大型 加速文件服务器 | ☐ | 加速的 文件管理器 | ☐ |
| ☒ | 较小的报告公司 | ||
| 新兴的 成长型公司 |
如果
是一家新兴成长型公司,请用复选标记表示注册人是否已选择不使用延长的过渡期来遵守根据《交易法》第13(A)节提供的任何新的或修订的财务会计准则。
用复选标记表示注册人是否提交了一份报告,并证明其管理层根据《萨班斯-奥克斯利法案》(《美国法典》第15编第7262(B)节)第404(B)条对其财务报告内部控制的有效性进行了评估
编制或发布其审计报告的注册会计师事务所。
如果证券是根据该法第12(B)条登记的,请用复选标记表示备案文件中包括的注册人的财务报表是否反映了对以前发布的财务报表的错误更正。
用复选标记表示这些错误更正中是否有任何重述需要根据§240.10D-1(B)对注册人的任何高管在相关恢复期间收到的基于激励的薪酬进行恢复分析。☐
用复选标记表示注册人是否是空壳公司(如该法第12b-2条所界定)。是,☐不是
说明
非关联公司持有的有表决权和无表决权普通股的总市值,该市值参考
普通股最后一次出售的价格,或该普通股的平均出价和要价,截至注册人
最近完成的第二财政季度的最后一个营业日:$
截至2024年3月15日,注册人普通股的流通股数量为 .
通过引用并入的文档
目录表
| 第一部分 | 1 | |
| 第 项1. | 业务 | 1 |
| 第 1a项。 | 风险因素 | 38 |
| 项目 1B。 | 未解决的员工意见 | 62 |
| 项目 1C。 | 网络安全 | 62 |
| 第 项2. | 属性 | 63 |
| 第 项3. | 法律诉讼 | 63 |
| 第 项。 | 煤矿安全信息披露 | 63 |
| 第II部 | 64 | |
| 第 项5. | 注册人普通股市场、相关股东事项与股权证券发行人回购 | 64 |
| 第 项6. | 已保留 | 65 |
| 第 项7. | 管理层对财务状况和经营成果的探讨与分析 | 66 |
| 第 7A项。 | 关于市场风险的定量和定性披露 | 75 |
| 第 项8. | 财务报表和补充数据 | 76 |
| 第 项9. | 会计与财务信息披露的变更与分歧 | 77 |
| 第 9A项。 | 控制和程序 | 77 |
| 第 9B项。 | 其他信息 | 77 |
| 第 9C项。 | 关于妨碍检查的外国司法管辖区的披露 | 77 |
| 第三部分 | 78 | |
| 第 项10. | 董事、高管与公司治理 | 78 |
| 第 项11. | 高管薪酬 | 78 |
| 第 项12. | 某些实益拥有人的担保所有权以及管理层和相关股东的事项 | 78 |
| 第 项13. | 某些关系和相关交易,以及董事的独立性 | 78 |
| 第 项14. | 首席会计师费用及服务 | 78 |
| 第四部分 | 78 | |
| 第 项15. | 展品和财务报表附表 | 78 |
| 第 项16. | 表格10-K摘要 | 79 |
| 签名 | 80 | |
| i |
注意事项 注意事项
本 表格10—K的年度报告包含1933年《证券法》第27A条(经修订)和1934年《证券交易法》第21E条(经修订)定义的前瞻性陈述。这些前瞻性陈述包括我们对未来的期望、信念、意图和策略。
These and other factors that may affect our financial results are discussed more fully in “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in this report. Moreover, we operate in a very competitive and rapidly changing environment, and new risks emerge from time to time. It is not possible for us to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this report may not occur and actual results could differ materially and adversely from those anticipated or implied in our forward-looking statements. Although we believe that the expectations reflected in our forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances described in the forward-looking statements will be achieved or occur. Moreover, neither we nor any other person assumes responsibility for the accuracy and completeness of the forward-looking statements. We caution readers not to place undue reliance on any forward-looking statements. We do not undertake, and specifically disclaim any obligation, to update or revise such statements to reflect new circumstances or unanticipated events as they occur, and we urge readers to review and consider disclosures we make in this and other reports that discuss factors germane to our business. See in particular our reports on Forms 10-K, 10-Q, and 8-K subsequently filed from time to time with the Securities and Exchange Commission.
除 另有说明外,本报告中的所有股份和股价均适用于2023年5月17日以 一比一的比率实施的远期股票分割。
行业和市场数据
本 报告,特别是“业务”部分,包含基于独立行业、政府和非政府组织出版物或其他公开可用信息的观察、统计数据、估计和预测,以及 基于我们内部来源的其他信息。虽然我们认为本报告中提到的第三方来源是可靠的,但 与预测有关的估计涉及许多假设,受到风险和不确定性的影响,并且 可能会根据各种因素(包括在标题为“”的章节中讨论的因素)而发生变化风险因素"以及本报告的其他地方。这些和其他因素可能导致结果与独立 方和我们所作估计中所表达的结果有重大差异。
本报告文本中的某些 信息载于独立行业政府和非政府组织出版物中。 这些出版物的来源如下:
| ● | Stacy 和Belkaid研究, 阿波罗·斯泰西和亚斯明Belkaid,皮肤健康的微生物守护者。 Science,2019年1月18日;363(6424):227—228。Doi:10.1126/science.aat4326。PMID:30655428 | |
| ● | 哦 研究,Zhou W,Spoto M,Hardy R,Guan C,Fleming E,Larson PJ,Brown JS,Oh J.主机特定 进化和传播动力学塑造葡萄球菌的功能多样性 表皮在人体皮肤中。cell. 2020年2月6日;180(3):454—470.e18. doi:10.1016/j.cell.2020.01.006。 Epub 2020 Jan 30. PMID:32004459;PMCID | |
| ● | 佐藤 研究,Satoh TK,Mellett M,Meier—Schiesser B,Fenini G,Otsuka A,Beer HD,Rordorf T,Maul JT,Hafner J,Navarini AA,Contassot E,French LE. IL—36 γ引起皮肤毒性 EGFR/MEK抑制作用和痤疮角质杆菌。J临床投资2020年3月2日;130(3):1417—1430. doi:10.1172/JCI128678。PMID:31805013;PMCID:PMC7269569 | |
| ● | Barbati 研究,儿童内瑟顿综合征:管理和未来展望,Federica Barbati, Mattia Giovannini Teresa Oranges,Lorenzo Lodi,Simona Barni,Elio Novembre,Ermanno Baldo, Mario Cristofolini、Stefano Stagi、Silvia Ricci、Francesca Mori、Cesare Filippeschi、Chiara Azzari和Giuseppe Indol;儿科前沿,2021年5月 | |
| ● | 日 Netherton综合征研究:病例报告和文献回顾,Joannie D. Sun,MD, Kenneth G. Linden,博士,医学博士,国际皮肤病学杂志2006 | |
| ● | 孤儿网, Netherton综合征,Orphanet:Netherton综合征 |
| II |
风险 因素汇总
我们的 业务受到许多风险和不确定性的影响,包括本年度报告 表格10—K中的“风险因素”中所述的风险和不确定性。这些风险包括但不限于以下:
| ● | 我们 是一家经营历史有限的早期临床生物制药公司; | |
| ● | 我们 有重大经营亏损的历史,并预计将持续经营亏损 在可预见的将来; | |
| ● | 我们 预计我们将需要额外的资金来执行我们的业务计划和资金运营, 可能无法以合理条件或根本无法获得额外融资; | |
| ● | 我们的微生物文库和基因工程平台的临床和商业用途 是不确定的,可能永远不会实现; | |
| ● | 我们的 候选产品处于早期开发阶段,因此需要大量 额外的临床前和临床试验; | |
| ● | 我们 将需要扩大我们的组织规模,并且我们可能会在管理方面遇到困难 这种增长; | |
| ● | 我们 目前没有销售和营销机构; | |
| ● | 我们 在可预见的未来,将完全依赖第三方来制造我们的 用于商业销售的候选产品; | |
| ● | 我们的 商业模式包括我们专有微生物菌株的潜在外授权 库或我们的候选产品到其他制药公司;但是,技术 制药行业的许可证是一个漫长的过程,并存在多种风险 以及我们无法控制的因素; | |
| ● | 我们的 企业可能因失去关键人员而受到影响; | |
| ● | 如果 产品责任诉讼针对我们,我们可能会承担重大责任, 可能要求限制我们候选产品的商业化; | |
| ● | 我们的 如果信息技术系统出现故障,业务运营可能会受到影响 或安全漏洞; | |
| ● | 我们 面临来自其他生物技术和制药公司的激烈竞争,目标是 医学皮肤病学指征; | |
| ● | 我们的 成功完全取决于我们获得产品市场批准的能力 FDA和外国司法管辖区的监管机构的候选人,我们 打算推销我们的候选产品,但不能保证; | |
| ● | 我们的 临床试验可能无法证明安全性和有效性的实质性证据 我们的候选产品或任何未来的候选产品; | |
| ● | 结果 我们候选产品的临床前研究可能无法预测 未来的临床前研究或临床试验; | |
| ● | 甚至 如果我们的任何候选产品获得了监管部门的批准,我们可能无法 成功地将产品和我们从其销售中获得的收入商业化,如果 任何,可能是有限的; | |
| ● | 当前 未来的立法可能会增加我们获得上市许可的难度和成本 我们的候选产品并将其商业化,并影响我们可能获得的价格; | |
| ● | 它 保护我们的知识产权既困难又昂贵,而且我们无法确保 保护这些权利; | |
| ● | 我们的 候选产品可能会侵犯他人的知识产权,这可能会增加 我们的成本和延迟或阻碍我们的开发和商业化努力; | |
| ● | 安 我们的股票可能无法发展活跃,流动性和有序的交易市场; | |
| ● | 未来 融资可能会稀释您的所有权,并对我们的运营造成其他不利影响; | |
| ● | 本公司股份的市价可能会波动及波动; | |
| ● | 我们的 未能满足纽约美国证券交易所的持续上市要求可能导致 我们的普通股退市; | |
| ● | 如果 我们未能维持一个有效的财务报告内部控制系统,我们 可能无法准确报告我们的财务业绩或防止欺诈; | |
| ● | 我们 根据特拉华州总公司法第204条批准了某些公司行动 法律,或DGCL;但是,不能保证不会提出质疑的索赔 批准书或相关公司行动的有效性;以及 | |
| ● | 我们的章程文件和特拉华州法律可能会阻止股东认为有利的收购。 |
| 三、 |
第 部分I
第 项1.业务
背景
Azitra, Inc.成立于2014年1月2日,是一家特拉华州公司,目的是使用工程蛋白质和局部活性生物制剂产品开发精准皮肤病学的创新疗法 。自我们成立以来,我们已经建立了一个专有平台,其中包括 一个微生物库,由大约1500种独特的细菌菌株组成,可以筛选独特的治疗特性。 该平台由人工智能和机器学习技术增强,可分析、预测并帮助筛选我们的 菌株库中的药物类分子。该平台还利用了一种获得许可的基因工程技术,能够 转化以前遗传上难以处理的菌株。我们尚未开始商业运营。除非另有说明, 术语“Azitra”、“公司”、“我们”和“我们的”指Azitra,Inc.及其全资子公司。
概述
我们 是一家早期临床生物制药公司,专注于使用工程 蛋白质和局部活性生物治疗产品开发精准皮肤病的创新疗法。我们已经建立了一个专利平台,其中包括一个微生物文库,该文库由大约1,500个独特的细菌菌株组成,可以筛选出独特的治疗特征。该平台得到了 人工智能和机器学习技术的增强,该技术可以分析、预测和帮助筛选我们的菌株库中类似药物的 分子。该平台还利用了一项获得许可的基因工程技术,可以转化以前在基因上难以处理的菌株。我们最初的重点是开发基因工程菌株。表皮葡萄球菌, 或 表皮葡萄球菌,我们认为这是皮肤病治疗工程的最佳治疗候选种。 特定物种在皮肤中表现出许多描述良好的特性。截至本报告的日期,我们已经在微生物库中确定了超过60种不同的细菌物种,我们认为这些细菌能够被改造以创造具有显著治疗效果的活的生物体 或工程蛋白质。
我们是基因工程细菌用于皮肤病治疗的先驱。我们的目标是利用我们的平台和内部微生物库细菌菌株来创造新的疗法,这些疗法要么是工程生物,要么是工程蛋白质或多肽,用于治疗皮肤病。我们最初的重点是开发我们目前的候选产品,包括:
| ● | ATR-12, 一个转基因菌株表皮葡萄球菌为了治疗孤儿病,Netherton 综合征,一种慢性且有时致命的皮肤疾病,估计影响大约 每100,000人中有1至9人,但其患病率可能因误诊而被低估 与其他皮肤病的相似性引起的我们获得了儿科罕见病称号 美国食品药品监督管理局(FDA)于2019年发布了ATR—12。12月 2022年,我们提交了一份1b期临床试验新药申请(IND) 在Netherton综合征患者中进行ATR—12试验,并于2023年1月27日收到通知 FDA关于拟定阶段的"研究可以继续进行" 1b临床试验提交IND后生产报告后,我们已开始运行 2023年12月,我们的1b期临床试验。我们希望报告初始 2024年下半年的安全性。 |
| ● | ATR-04, 一株转基因菌株表皮葡萄球菌用于治疗接受表皮生长因子受体抑制剂或EGFRi靶向治疗的癌症患者出现的丘疹丘疹。我们打算在2024年年中之前将IND提交给正在接受EGFRi靶向治疗的某些癌症患者进行1b期临床试验。如果FDA批准我们的IND,我们预计将于2024年第四季度开始我们的1b期临床试验。 |
| ● | ATR-01,一种用于治疗寻常型鱼鳞病的工程重组人细丝蛋白,这是一种慢性、干燥(异常干燥)、鳞片状皮肤病,估计在250人中的发病率和患病率为1。这表明美国的患者总数为130万。 我们计划在2024年完成Lead优化和IND启用研究,以支持计划于2025年下半年提交的IND申请。 |
| 1 |
| ● | 我们和拜耳的消费品部门拜耳或国际生命科学公司拜耳正在研究和开发两种不同的细菌微生物菌株。我们于2019年12月与拜耳签订了联合开发协议(JDA)。根据JDA的条款,我们负责测试我们的细菌菌株及其天然产物库中的关键临床前特性。在从数百个菌株中进行筛选后,我们和拜耳选择了两个特定的菌株进行进一步的开发。拜耳拥有授予这些菌株专利权的独家选择权。2020年12月,拜耳购买了800万美元的B系列优先股, 这些优先股转换为我们普通股的1,449,743股,约占我们普通股流通股的12.0% 。 |
我们 还与卡内基梅隆大学和弗雷德·哈钦森癌症中心或弗雷德·哈奇的团队建立了合作伙伴关系,这两个中心是美国两个主要的学术中心。我们与卡内基梅隆大学团队的合作利用了全基因组测序的力量。这一合作伙伴关系正在挖掘我们专有的细菌菌株库,以寻找新的、类似药物的多肽和蛋白质。该团队开发的人工智能/机器学习技术根据微生物的基因序列预测微生物制造的分子。然后,该系统将预测结果与通过串联质谱学和/或核磁共振成像实际做出的产品进行比较,以完善未来的预测。这些预测可以与公开可用的2D和3D蛋白质数据库进行比较,以选择类似药物的结构。
我们 持有Fred Hutch的全球独家许可,允许将其获得专利的SyMPL技术用于基因工程的所有领域,包括发现、开发和商业化皮肤疾病的工程微生物疗法和微生物衍生的多肽和蛋白质 。我们正在利用我们许可的专利权来构建质粒,以便进行以前从未实现过的基因转化。我们与Fred Hutch的合作是由微生物工程专家Christopher Johnston博士领导的,他也是SyMPL技术的创新者。
除了我们的三个主要候选产品和与拜耳的合作之外,我们的目标是开发广泛的候选产品组合, 专注于扩展我们的精确皮肤病平台的应用。我们相信,我们已经在推动精准皮肤病生物制剂的开发方面确立了独特的地位。

| 2 |
我们的 业务战略
我们 打算通过开发从我们的专利微生物库中挑选的约1,500种独特细菌菌株中挑选出来的基因工程 蛋白质,为精密皮肤病创造广泛的候选产品组合。我们的策略如下:
| ● | 打造一家可持续发展的精准皮肤病公司。我们的目标是打造一家领先的精准皮肤病公司,拥有可持续的候选产品流水线。为此,我们 专注于快速推进我们目前的生物治疗候选产品线,同时 积极开发其他候选产品。我们目前的每个候选产品 都是专有产品,正在申请专利。我们预计,我们开发的大多数(如果不是全部)基因工程产品候选产品将有资格获得专利保护。 |
| ● | 推动我们的主要候选产品ATR-12和ATR-04通过临床试验。我们预计 将在2024年下半年报告我们的ATR-12在Netherton综合征患者中的1b期临床试验的初步安全性结果,目前正计划在某些情况下开始我们的ATR-04的1b期试验2024年第四季度接受EGFRi治疗的癌症患者。我们已经批准了ATR-12的IND,预计将在2024年年中提交ATR-04的IND。 |
| ● | 通过有选择地探索战略合作伙伴关系,最大限度地发挥我们精准皮肤科项目的潜力, 拓宽我们的平台。我们打算保留对我们所有核心技术和候选产品的重要权利。但是,我们将继续评估 战略合作伙伴可以帮助我们加快技术和候选产品开发的合作机会,提供获得协同组合的途径,或者 提供专业知识,使我们能够扩展到不同类型皮肤病的治疗 。我们还可以通过选择性地许可技术或候选产品来扩大我们平台的覆盖范围。此外,我们还将考虑将我们的某些专有技术授权给我们自己并不追求的适应症和行业 。我们相信,我们的基因工程技术和技术在医药领域之外具有适用性,包括化妆品,以及清洁燃料的生成和生物修复。 |
| ● | 利用我们的学术合作伙伴关系。我们目前与弗雷德·哈钦森癌症中心、耶鲁大学、杰克逊基因组医学实验室和卡内基梅隆大学的研究人员建立了合作伙伴关系。我们拥有弗雷德·哈钦森癌症中心的独家许可证,其中包括DNA技术,这些技术可以对以前在遗传上难以解决的菌株进行基因转化。我们与卡内基·梅隆大学的研究人员合作,建立在人工智能和机器学习技术的基础上, 预测由我们图书馆的微生物制造的类似药物的分子。我们与杰克逊实验室的朱莉娅·吴博士签订了持续的科学顾问委员会合同,并通过赞助小鼠实验的研究协议与杰克逊实验室进行了合作。 我们希望利用这些合作伙伴关系,并有可能扩大它们或形成其他形式学术合作伙伴关系,以支持我们的工程平台并扩展我们的研发管道 。 |
| ● | 经验丰富的管理团队和董事会. We are led by Francisco D. Salva, our chief executive officer, and Travis Whitfill, our co-founder and chief operating officer, who have more than 35 years of combined experience in the management of biotechnology companies and healthcare investing. Mr. Salva was previously a co-founder of Acerta Pharma, which was sold to AstraZeneca for approximately $6.3 billion in a staged acquisition beginning in 2016. He also worked on the turnaround of Pharmacyclics, which subsequently sold to Abbvie for approximately $21 billion in 2015. Before that, Mr. Salva spent almost a decade in life sciences venture capital. Mr. Whitfill served as associate research scientist and currently serves as assistant professor adjunct at Yale University with appointments in the Departments of Pediatrics and Emergency Medicine. He spent nearly a decade in venture capital as a partner in a biotech-focused venture capital fund, Bios Partners. He has led numerous grant-funded projects, holds nearly a dozen patents and has co-authored over 60 publications. Our board of directors, or Board, is comprised of renowned group of senior executives, scientists and investors in the biotechnology industry. |
| 3 |
我们的微生物文库和微生物药物传递平台
共生微生物存在于人体表面或粘膜中,不会损害人类健康。它们作用于宿主的免疫系统,以诱导保护性反应,防止感染病原体的侵袭和侵袭,从而在维持人类许多器官系统的健康方面发挥关键作用,特别是在皮肤中。皮肤上分布着各种各样的微生物群落,一平方厘米可以容纳多达十亿个微生物。这些不同的细菌、真菌、螨虫和病毒群落可以提供对疾病的保护,并在皮肤上形成动态而独特的生态位。它们共同构成了皮肤微生物群。
许多由基因驱动的人类疾病在系统上或部分上与特定蛋白质的功能障碍有关,这些蛋白质因突变而缺失或功能惰性。自大约1982年以来,生物制药行业一直在对细菌微生物中的重组蛋白进行基因工程 ,目的是提供模仿或支持人体正常功能的蛋白质和多肽的疗法。几十年来,绝大多数基因工程一直局限于初级大肠杆菌以及少数其他细菌 物种,其中许多可以致病,导致感染。相比之下,我们选择将重点放在表皮葡萄球菌因为它作为一种在皮肤上自然存在的共生微生物具有有益的作用。我们的目标是利用我们的平台和60多种细菌的内部微生物库来设计并通过皮肤角质层将共生皮肤细菌直接输送到目标皮肤。在皮肤的这些更深层次,工程微生物可以产生缺失或惰性的蛋白质,从而解决潜在的疾病原因。
S.和我们的专有微生物文库
表皮葡萄球菌是一种强有力的治疗候选物种,因为皮肤中有许多描述良好的特性。表皮葡萄球菌 是一种革兰氏阳性细菌,普遍存在于人类皮肤和粘膜菌群中。作为最早的皮肤殖民者之一, 表皮葡萄球菌在皮肤免疫和维持微生物群落动态平衡方面起着重要作用。表皮葡萄球菌 已知与宿主有一种有益的皮肤共生关系。该物种对致病菌株 显示出抑制作用,金黄色葡萄球菌,或金黄色葡萄球菌,以及压力痤疮丙酸杆菌,或痤疮假单胞菌. 表皮葡萄球菌 诱导角质形成细胞产生抗菌肽,并通过免疫细胞信号产生CD4+和CD8+T细胞的非炎性T细胞积聚。T细胞反应诱导损伤后皮肤重新上皮化,加速修复和伤口闭合。基于这些原因,我们认为表皮葡萄球菌作为局部递送治疗性蛋白质的载体提供了几个优点。
在他们2019年的研究中,世界领先的皮肤微生物组专家Stacy和Belkaid描述了表皮葡萄球菌作为“皮肤微生物区系的‘海报孩子’,它展示了微生物对皮肤生理和健康所能起到的不同功能。”表皮葡萄球菌具有巨大的菌株多样性,可用于治疗目的。在2020年的ONG研究中,朱莉娅·吴的实验室报告称,1,482株独特的表皮葡萄球菌只出现在五个个体身上。这些菌株不仅具有显著的遗传多样性,而且具有较大的表型多样性。我们相信这种巨大的菌株间差异表皮葡萄球菌可以被利用。为此,我们收集了健康志愿者的样本,以开发和鉴定我们自己的菌株 库表皮葡萄球菌其中包括900多个独特的表皮葡萄球菌具有治疗用途潜力的菌株。我们已经使用这个微生物文库对选定的特性进行了筛选,包括抗菌肽分泌,金黄色葡萄球菌杀伤力、抗生素敏感性和其他与治疗相关的特征。我们还收集了图书馆中的其他物种,其中包括大约60种不同的皮肤共生物种,也可以出于治疗目的进行筛选。
| 4 |
图1.Azitra微生物文库中的代表性物种

我们的微生物文库的预测性分析
生物制药行业在鉴定和分离数千种细菌方面取得了成功。然而,只有相对较少的几个这样的物种,据信不到20个,已经被改造成生产具有治疗潜力的蛋白质或多肽。我们已经与卡内基梅隆大学的研究和开发集团Chia Biosciences,Inc.建立了合作伙伴关系。通过我们与中国生物科学公司的合作,我们能够使用他们专有的基因组和多肽人工智能和机器学习系统NRPMiner, 来开发和确认我们专有的 菌库产生的蛋白质、多肽和小分子的天然产物预测。这些预测通过串联质谱学或核磁共振得到证实。然后将信息反馈给机器学习算法以改进预测。它还可以与现有的2D和3D蛋白质数据库进行比较 ,以查找我们的产品与现有蛋白质和多肽药物的结构同源性。我们相信,我们与卡内基 梅隆大学团队的合作为我们提供了一种可扩展和容忍修改的方法,以加速我们微生物 库中的治疗发现。
我们微生物生产的药物的交付
将基因工程蛋白运送到皮下靶点的过程受到天然屏障和角质层防御的阻碍。这是皮肤最外面的一层,它起到了屏障的作用,防止不需要的物质进入身体。为了应对这一挑战,我们开发了一种专利工艺,能够以一种绕过通常无法穿透的角质层的方式促进蛋白质的输送。该策略利用特定微生物渗透到皮肤更深层的能力。在那里,转基因微生物充当微型工厂,在需要的地方生产治疗性蛋白质或分子。
我们治疗皮肤病的蛋白质输送能力是基于工程技术表皮葡萄球菌和其他微生物分泌蛋白质,将药物输送到皮肤。我们相信,任何数量的蛋白质都可以由我们的细菌进行工程和编码,产生 并输送到皮肤上,以治疗各种皮肤疾病。我们还在其平台中添加了关键的专有功能,以促进蛋白质的交付。该系统的一个关键特征是它绕过了通常无法穿透的皮肤屏障,这是一个局部蛋白质输送的问题。皮肤屏障由角质层组成,由去核的角质形成细胞封闭,并由许多结构、物理和生化特性形成。其他经皮给药挑战是由于蛋白质对酶消化的敏感性,以及疏水表面和组成角质层的相连角质细胞层造成的溶解和扩散障碍。我们通过利用自然归位的表皮葡萄球菌角质层以下的层。 在临床前研究中,我们表明表皮葡萄球菌栖息在角质层下面的几层,将蛋白质输送到更深的表皮。
为了扩展我们的重组蛋白质构建能力,我们获得了专有技术的独家许可,该技术可以伪装我们的基因工程DNA序列,使其能够在以前难以处理的细菌物种中生产蛋白质。弗雷德·哈钦森癌症中心或弗雷德·哈奇的这项技术扩大了可以转基因的细菌物种的范围。它 基于限制修改系统-静默SyMPL工具集。SyMPL技术平台使人造DNA对细菌的防御系统不可见。从理论上讲,这种方法可以应用于任何类型的细菌。我们当前的候选产品未采用SyMPL技术平台,但我们预计未来的部分或全部候选产品将采用此技术平台。
| 5 |
事实上,所有自然产生的细菌菌株都有称为限制修饰系统的防御机制。四种类型的限制修饰系统识别和防御用于编码重组蛋白质的外来DNA的插入。S的功能基因工程。表皮炎(以及金黄色葡萄球菌)以前受到限制,因为在这些细菌物种的几乎所有菌株中都存在I型和IV型限制系统 。这些限制系统识别来自标准克隆扩增系统(例如(E.Coli)并阻碍外来DNA在微生物中的结合。表皮葡萄球菌由于其遗传上的顽固性,曾被认为是一种“不可转化”的菌株。然而,我们已经能够克服S. 表皮‘防御。
目前的基因工程流程添加了特定的修饰来伪装人造DNA,以欺骗细菌认为入侵者 是其自身DNA的一部分。此方法通常需要花费相当多的时间和资源来尝试将正确的伪装与每个特定的识别主题相匹配。相比之下,Fred Hutch的SyMPL技术平台是一种系统化的“工程隐身”方法 ,以克服限制修改防御系统。这些限制修饰防御系统保护微生物免受外来DNA的影响,并阻碍绝大多数基因工程方法。SyMPL技术平台基于构建微环DNA质粒的能力,这些DNA质粒缺乏任何目标识别基序,供微生物的防御系统识别。该技术使用目标细菌基因组序列中的基因组和甲基组来识别限制酶修饰的目标基序。然后将它们从遗传工具的核苷酸序列中消除硅片。生成的序列用于构建限制 修改、SyMPL工具。这些基因被繁殖,然后用于基因转化。“通过工程进行隐形” 方法不仅能够在遗传上难以处理的细菌菌株中进行转化,而且它还被证明能够显著提高转化效率。原则性实验证明,转基因菌落的产量提高了10,000倍以上。
2022年1月,Fred Hutch向我们授予了SyMPL技术平台在所有使用领域的全球独家专利权和非独家全球专利权使用费许可。有关根据弗雷德·哈奇许可协议获得的知识产权的更多信息,请参阅标题为“营业执照和知识产权.”
我们的 候选产品
ATR-12治疗Netherton综合征
ATR-12是我们的专利和正在申请专利的候选药物,它包含一种新的菌株表皮葡萄球菌该基因经过基因修饰,可以表达和分泌全长蛋白质的活性片段,称为淋巴上皮型Kazal相关抑制物,或Lekti。它还被设计成营养缺陷型,这意味着它需要配方中的D-丙氨酸营养才能存活和繁殖。这提供了额外的安全级别,防止潜在的系统性感染。ATR-12是一种局部应用,旨在通过用人重组lekti的活性片段 或rhLEKTI-D6替换缺陷的lekti来解决Netherton综合征的根本原因,以对抗在Netherton综合征患者中观察到的皮肤丝氨酸蛋白酶活性失调。丝氨酸蛋白酶活性失控导致严重的皮肤屏障缺陷,并导致角质形成细胞和免疫细胞释放促炎和促过敏介质。截至本报告发表之日,还没有治愈或有效治疗Netherton综合征的已知疗法。 我们相信ATR-12有可能成为有效治疗这种皮肤病的第一种疗法。根据Barbati和Sun的研究,我们认为ATR-12代表着到2030年年中潜在的2.5亿美元的全球销售机会。
Netherton 综合征概述
Netherton综合征是一种罕见的常染色体隐性遗传病,估计每200,000人中就有一人患病,但由于误诊,其患病率可能被低估了。这是一种慢性皮肤疾病,以严重的炎症、瘙痒、鳞屑、红肿和脱水为特征。出生时患有Netherton综合征的婴儿可能会遭受发育不良的痛苦,据报道,大约十分之一的Netherton综合征婴儿在出生后的第一年死亡。那些幸存下来的人一生都面临着皮肤病的挑战,包括红色、鳞片状皮肤、头发缺陷以及持续高于正常水平的感染和过敏风险。
| 6 |
Netherton综合征是由基因突变引起的Spink5基因,它编码淋巴上皮型Kazal相关抑制物,或lekti。LEKTI的功能是抑制表皮中的酶,如激肽释放酶5、7和14,或KLK5、KLK7和KLK14,它们促进皮肤细胞在一个称为脱皮的过程中脱落。当Lekti缺失或活动减少时,会导致皮肤过度脱皮,皮肤敏感、开放,并出现红色和鳞片。伴随而来的是角质层的脱离,导致严重的屏障功能障碍、脱水和潜在的环境因素,如化学物质。与健康志愿者相比,Netherton综合征患者的皮肤组织病理学和免疫荧光染色显示皮肤中没有Lekti和异常,如角化过度、表皮增厚和嗜碱性角化透明质颗粒减少。

图2:Netherton综合征的病理生理学和LETKI缺乏症
Netherton综合症的严重程度从轻微的皮肤红斑到危及生命不等。疾病的严重程度与皮肤上LEKTI功能丧失的程度直接相关。Netherton综合征在出生后不久出现,在婴儿出生的第一年最为严重。在大多数情况下,第一年以上的存活是常见的,但这种疾病的影响是一个终生的挑战。
截至本报告之日 ,内瑟顿综合征尚无已知的治愈方法,治疗选择有限。治疗Netherton综合征严重皮肤表现的皮肤病学干预措施 包括保湿剂、局部皮质类固醇和钙调磷酸酶抑制剂, 所有这些措施都受到限制,因为它们不能提供持续的治疗。考虑到新生儿阶段疾病的严重程度,除了治疗这些患者经常出现的感染外,还需要体液/电解质 和饮食支持。虽然免疫球蛋白治疗治疗与Netherton综合征相关的免疫缺陷的成功率有限,但目前无法持续修复由LEKTI失调引起的皮肤屏障缺陷。
我们的解决方案-ATR-12用于治疗Netherton综合征
ATR-12是一种外用软膏,含有表皮葡萄球菌菌株SE351,已经经过基因改造,表达了来自 染色体的lekti。SE351菌株还被设计成对D-丙氨酸具有营养缺陷性,这意味着如果没有配方中提供的外源D-丙氨酸营养,它就不能生存。ATR-12旨在通过将缺乏/功能失调的lekti替换为具有活性的重组人全长蛋白片段rhLEKTI-D6来解决Netherton综合征的根本原因。治疗包括将ATR-12应用于受影响的地区。由SE351生产的rhLEKTI-D6将对抗Netherton综合征患者观察到的皮肤丝氨酸蛋白酶活性失调,恢复皮肤屏障功能,减少炎症。我们认为,这种方法的重要优点之一是,随着时间的推移,有可能将rhLEKTI-D6输送到角质层和表皮的下层,这是Netherton综合征患者调节失调的主要部位。
| 7 |
表皮葡萄球菌SE351是从我们的专利菌株库中挑选出来的将重组人LEKTI-D6运送到皮肤上的菌株。 该菌株的特点是毒力低,是一种不形成生物膜的宿主菌株。为了进一步提高ATR-12的安全性,我们已经对D-丙氨酸的微生物进行了改造,使其具有营养缺陷性。工程营养缺陷症的关键优势是能够控制生长和阻止潜在的感染。全长人类LETKI是一种15个结构域的蛋白质(145 KDa),太大了,无法可靠地在细菌中表达和分泌。有证据表明,全长蛋白片段足以对抗Netherton综合征患者观察到的失调的皮肤丝氨酸蛋白酶活性,我们选择D6在表皮葡萄球菌.
在2020年5月,我们从FDA获得了ATR-12的罕见儿科疾病称号。因此,如果我们能够在儿科方面获得FDA对ATR-12的批准,我们将有资格收到优先审查凭证,我们可以使用该凭证在六个月的加速期内获得FDA对此或其他候选药物的新药申请或生物制品许可证申请的审查 。这些 代金券通常可以转让,有些代金券的售价已超过1亿美元。
ATR-12的临床前 数据
截至本报告之日 ,我们已经进行了几次 体内和离体这些实验共同支持ATR—12作为内瑟顿综合征患者的疾病改善疗法的潜在疗效。转基因菌株S.配制的ATR—12制剂中使用的表皮被称为SE351。于二零二一年,我们进行了 体外培养评估外源应用SE351在无菌重建人类表皮中的能力的研究。SE351成功地在重建的人类表皮上殖民化, 此外, 表皮葡萄球菌在没有D—丙氨酸存在的情况下发生了定殖,证实了必须为 SE351在皮肤上生长提供D—丙氨酸。这些数据表明,SE351能够在人体皮肤上定殖,并且可以通过补充 D—丙氨酸来控制定殖。
此外, 体外培养使用添加了KLK5的健康志愿者的胶带剥离皮肤来模拟Netherton综合征的研究表明,稀释的SE351培养上清液可剂量依赖性地抑制胰蛋白酶样活性(KLK5活性)。当加入含有≥0.5%的SE351培养上清液时,Netherton综合征代谢物中的胰酶样活性恢复到正常健康水平。

图 3:离体人皮带提取液辅以病级KLK5活性建立Netherton综合征模型
在 此外,来自离体猪皮肤模型显示,单次局部注射3个剂量水平的ATR-12可导致活性rhLEKTI-D6分泌 。最后,来自离体健康人体皮肤模型显示,单次局部剂量的ATR-12最大预期剂量为109Cfu/g可将足够活性的rhLEKTI-D6送入角质层的下层,有效地抑制激肽释放酶5(KLK5),达到Netherton综合征患者的典型水平。
| 8 |
具体地说,来自离体健康的人体皮肤模型显示,单次局部注射ATR-12的最大预期剂量为109每克集落形成单位(CFU/g)可将足够活性的rhLEKTI-D6送入角质层的下层 ,有效抑制KLK5,达到Netherton综合征患者的典型水平。提取层中Lekti 活度的量来自于离体用安慰剂和ATR—12处理的人皮肤。采集 在皮肤贴敷后立即进行(T = 0小时,白色条)或在30 ℃下孵育8小时后(T = 8小时,黑色条)。通过将pmol量加入安慰剂(灰色条)或ATR—12(黑色条)样品的第1—30层,获得总 LEKTI活性水平。 数据为3个独立样本(N = 3)的平均值±标准差(SD)。使用 双因素方差分析进行统计分析,**代表p

图 4:安慰剂和ATR-12处理的皮肤样品在孵育0和8小时后的LETKI活性
此外,单次治疗剂量的ATR-12在24小时的孵育中产生的LEKTI活性是8小时孵育的~2倍。 这表明随着时间的推移,ATR-12持续产生功能性的重组人LEKTI-D6。
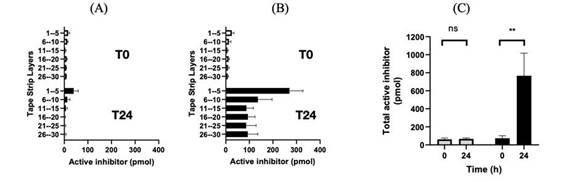
图5:24小时孵育后安慰剂和ATR-12处理的皮肤样本中的LETKI活性
在试管中Azitra进行的化学计量学研究表明,KLK5需要在rhLEKTI-D6蛋白上有2个摩尔当量的抑制 (IC测量50)。历史研究表明,Netherton综合征患者的KLK5含量是正常皮肤的~6倍。这相当于每一特定区域60 pmoL的KLK5。以上研究表明,SE351在8小时内释放了350 pmol的重组人LEKTI-D6,在24小时内释放了700 pmol的重组人LEKTI-D6。这比预计的活动所需金额高出5至11倍。
| 9 |

图 6:离体抑制KLK5的LEKTI-D6的化学计量比
在2022年,我们获得了与FDA的IND前通信,目的是讨论我们建议的ATR-12监管途径,并从FDA获得导致提交和接受ATR-12的IND申请的临床前计划指导。2022年12月,我们提交了一份IND申请,对Netherton综合征患者进行ATR-12的首次人体试验。我们IND建议对Netherton综合征患者的ATR-12进行1b期多中心、随机、双盲、单剂量水平的安慰剂对照临床研究。主要终点 是安全性,次要终点将包括疗效和药代动力学信号。探索性终点包括免疫和炎症机制生物标记物。2023年1月27日,我们收到FDA的通知,关于拟议的1b期临床试验的研究可能会继续进行,初步安全结果预计在2024年下半年。
ATR-04用于治疗EGFRi相关性皮疹
ATR-04是我们的专利和正在申请专利的候选药物,它包含一种新的菌株表皮葡萄球菌,SE484,经基因改造为营养缺陷型D-丙氨酸。ATR-04是一种局部应用,旨在解决接受表皮生长因子受体抑制剂(EGFRi)靶向治疗的癌症患者所经历的丘疹/丘疹。我们相信,这款候选产品 代表着2030年前10亿美元的潜在全球销售机会。
EGFRi-关联的 皮疹概述
靶向癌症疗法为诊断为各种肿瘤类型的患者带来了显著的治疗进展,但它们也与独特的皮肤病毒性有关,这些毒性可能会阻碍治疗努力,并给患者造成严重的身体和心理不适 。预防和处理这些毒性可能会使患者更好地耐受治疗,延长治疗时间,从而有可能从药物中获得最大的临床益处。其中一类靶向癌症治疗包括EGFR抑制剂。EGFR是细胞表面的一种蛋白质,有助于细胞生长和分裂。它也是某些恶性肿瘤的关键因素,它的活性促进了肿瘤的生长、侵袭和转移。全身暴露于EGFRi制剂会抑制靶肿瘤部位的EGFR,同时也会抑制全身的EGFR。在皮肤中,EGFR调节多种角质形成细胞的功能,包括增殖、黏附、迁移、存活和分化。因此,抑制皮肤中的EGFR会导致不良皮肤反应,这使得患者很难坚持这些有效的治疗方法。
皮肤病毒性是EGFRi靶向治疗中最常见的副作用之一。丘疹是EGFRi治疗中最早也是最常见的皮肤科不良事件,通常发生在50%-80%的患者中,具体取决于药物、正在治疗的癌症和治疗方案。丘疹皮疹的出现是一种剂量依赖的皮肤药物反应,通常在最初的一到两周内出现,在治疗后三到四周达到高峰。皮疹的强度可能在两周后开始减轻,但可以在EGFRi治疗的整个过程中持续。皮疹的临床特征是细嫩的红斑丘疹,几天后演变为脓疱疹,然后在面部、头皮、胸部和上背部形成结痂。皮疹通常伴有严重的干燥症,有时还会出现严重的皮肤细菌感染,主要是金黄色葡萄球菌。虽然大多数皮疹被认为是轻度到中度,但也有一些是严重的。在许多情况下,皮疹会导致严重的生活质量问题,甚至可能导致EGFRi治疗中断或停止.
| 10 |
目前接受EGFRi治疗的患者的皮疹治疗标准因皮疹的严重程度而异。通常,皮肤保湿剂、局部类固醇和多西环素从EGFRi治疗开始就预防性使用,并在整个治疗期内持续使用。如果皮疹继续发展,就需要口服类固醇和/或抗生素。然而,已知有与这些辅助疗法相关的全身不良反应,我们认为医生和患者试图 限制其使用。此外,研究表明,口服抗生素会导致肠道微生物群的破坏,进而导致靶向治疗的有效性降低,包括EGFRi。鉴于这些患者持续出现皮疹的高发病率,以及与抗生素对这些治疗的潜在影响相关的担忧,我们认为显然存在着对额外安全有效的辅助治疗的医疗需求,以解决丘疹-丘疹皮疹。
基于佐藤和利希滕贝格进行的研究,细胞因子,白介素36伽马,或白介素36γ,以及金黄色葡萄球菌与接受EGFR治疗的患者所经历的皮疹相关,并在其中发挥重要作用。IL-36γ在接受EGFR i治疗的患者皮肤中升高。2020年,Satoh使用基因表达谱确定IL-36γ是EGFRi/MEKi皮肤毒性的候选驱动因素。它由EGFR抑制诱导, 痤疮皮肤杆菌协同诱导皮肤中的IL-36γ,随后诱导IL-8和NF-κB,导致皮肤嗜中性粒细胞。IL-36γ可能是治疗EGFRi诱导的皮疹的关键治疗靶点。2013年,Lichtenberger注意到EGFRi治疗患者(n=107)的细菌感染率较高(70%),并提出了EGFR消融导致 金黄色葡萄球菌-在小鼠中诱导感染。该研究指出,大多数患者的 金黄色葡萄球菌(54%).从机制上讲,作者注意到EGFRi治疗损害宿主防御:抗微生物肽的表达受损,特别是抗 金黄色葡萄球菌以及紧密连接的表达降低。此外,该研究显示EGFR消融导致皮肤屏障缺陷以及受损的皮肤免疫应答和细胞因子表达。
我们的解决方案-ATR-04用于治疗EGFRi相关性皮疹
ATR-04是我们用于治疗EGFRi相关皮疹的配方候选药物产品。它包括一种新的营养缺陷型菌株表皮葡萄球菌从我们的微生物菌种库中选择的菌株,基于所需的IL-36γ还原特性和抑制金黄色葡萄球菌以及它的生物膜。目前的铅菌株被称为SE484。然后,我们将SE484进行基因工程,使其成为营养缺乏型D-丙氨酸,并创造出我们的候选药物产品ATR-04。
从我们的微生物文库中选择SE484是基于关键特性,如对IL-36γ的抑制以及对S 金色。总之,我们预计这些作用机制将显著降低接受EGFRi治疗的患者的皮疹严重程度。
我们 相信ATR-04有可能解决目前治疗EGFRi相关皮疹的局限性:
| ● | 减少了抗生素的使用。从我们对临床医生和主要意见领袖的调查来看,从业者不愿给接受EGFRi治疗的患者开全身抗生素。这些 患者通常可以服用抗生素超过12个月,并遭受与抗生素相关的不良事件。我们相信ATR-04将减少这些患者对抗生素的需求,并导致较少的不良事件,因为EGFRi和抗生素的使用。 |
| ● | 改善了 EGFRi合规性。高达20%的接受EGFRi治疗的患者因不良事件(主要是皮疹)而停止治疗。我们相信,我们可以降低接受EGFRi治疗的患者的停用率,从而提高依从性。 |
| ● | 更高的生活质量。许多接受EGFRi治疗的患者报告说,由于不良事件和丘疹结节皮疹,生活质量较差。目前的治疗方案不能充分减少这些不良事件。我们相信,在接受EGFRi治疗的患者中使用ATR-04将会减轻皮疹的严重程度,从而提高生活质量。 |
| 11 |
ATR-04的临床前数据
我们根据安全性(例如,缺乏抗生素耐药性)和生物活性(例如,IL-36γ抑制和活性与金黄色葡萄球菌),并指定SE484为我们的主要候选菌株。在将该菌株设计为D-丙氨酸的营养缺陷型菌株后,我们提名该候选菌株作为ATR-04药物产品配方中的活性微生物。
EGFR相关皮疹是一种以皮肤发红、瘙痒和刺激为特征的疾病,由某些癌症治疗引起。 基因表达谱(Satoh Et Al 2020)显示,与健康捐赠者的皮肤相比,EGFR相关皮疹患者的皮肤活检样本中细胞因子IL-36γ(IL-36γ)和IL-8水平升高。这些是促炎细胞因子,是免疫系统的信号分子,可增加免疫反应的强度,并可能导致组织损伤。 除了细胞因子水平升高外,接受EGFRi治疗的患者还会损害皮肤屏障功能。感染致病菌株 金黄色葡萄球菌加重EGFRi诱导的皮肤病。
我们的工作重点是确定一个表皮葡萄球菌菌株,一种皮肤共生体,可降低IL-36γ水平,从而减少与EGFRi相关的皮疹。我们推测,许多生活在人类皮肤上的细菌物种可能在那里生存下来,因为它们已经进化出减少人类免疫系统对它们存在的反应的方法,我们或许能够识别出一种存活下来的人类皮肤共生细菌,从而特别地降低IL-36γ的活性。
要 确定这样的表皮葡萄球菌菌株,我们开发了一种体外培养检测培养的人皮肤细胞产生IL-36、γ和IL-8的水平。我们使用的细胞系被称为HaCaT,它来自人类角质形成细胞,角质形成细胞是表皮中的一种细胞类型。为了模拟表皮生长因子受体相关疾病皮肤的炎症表型,用免疫刺激剂多肌苷:多胞苷或多聚I:C刺激HaCaT细胞,使其分泌高水平的IL-36γ和IL-8。本试验用来鉴定和评价不同菌株的能力。表皮葡萄球菌可降低IL-36γ和IL-8水平。
我们根据安全性(例如,缺乏抗生素耐药性)和生物活性(IL-36γ抑制和抗病毒活性)筛选了100多个菌株金黄色葡萄球菌),并指定SE484为我们的主要候选菌株。在将该菌株改造成对D-丙氨酸具有营养缺陷型 ,以便只有在提供D-丙氨酸的情况下才能生长之后,我们还消除了一个抗生物质抗性基因。然后,我们提名该候选微生物作为ATR-04药品配方中的活性微生物。
为了在类皮肤模型上测试SE484降低IL-36γ的能力,使用厄洛替尼诱导重建的人表皮分泌IL-36γ。同时应用SE484和厄洛替尼可将IL-36γ降低到与未用厄洛替尼治疗的相似的水平,这表明SE484作用于皮肤模型以减少这种促炎细胞因子。图7显示了两个测试SE484降低IL-36γ的实验结果。在图7A中,将来自SE484培养物的无细胞上清液(CFs)应用于RHE,而在图7B中,将SE484的活细胞(1x108CFU或1x109CFU),检测SE484降低IL-36γ的能力。在这两种情况下,无细胞培养上清液或SE484细胞,厄洛替尼诱导的IL-36γ 水平均降低。
| 12 |
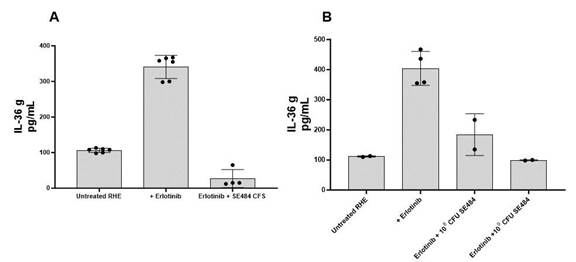
图7.SE484对RHE的抗IL-36G活性将重建的人表皮或RHE单独用1 mM厄洛替尼处理72小时,或用来自SE484培养(A)的无细胞培养上清(CFs)处理,或用约10 mM厄洛替尼处理72小时。8或109SE484(B)的CFU。用酶联免疫吸附试验检测培养上清液中IL-36γ水平。

图8.Poly I:C诱导的IL-8被SE484的CS降低。来自SE484的培养基可阻止聚I:C(红色箭头)对IL-8的刺激和释放。使用Poly I:C的抑制剂作为对照(蓝色箭头)。数据代表两个独立的 实验。CS=培养上清液。
图8显示了SE484培养上清对PolyI:C诱导的IL-8的抑制作用,与IL-36γ相似,当PolyI:C 加入HaCaT细胞时,IL-8也被分泌,比本底高出数倍(如未处理的HaCaT和经SE484处理的HaCaT所见)。然而,在SE484存在的情况下,检测到较低水平的IL-8,从而进一步证明了SE484抑制参与EGFRi相关皮疹的促炎 途径的有效性。
| 13 |
我们的 结果显示表皮葡萄球菌菌株从健康人志愿者中分离得到的SE484可以降低HaCaT细胞产生的IL-36γ和IL-8的水平(分别见图7和图8),从而有助于治疗EGFRi-相关皮疹。除了抗IL-36γ特性外,SE484对不同的甲氧西林耐药株也具有广泛的活性S 金色,或MRSA,菌株类型以及对甲氧西林敏感金黄色葡萄球菌,或MSSA。SE484降低IL-36γ/IL-8水平及其抗肿瘤活性的研究金黄色葡萄球菌经过改造的D-丙氨酸营养缺陷症使我们能够提名菌株SE484作为ATR-04药物产品配方中的活性微生物,以形成治疗的基础并降低EGFRi皮疹的严重程度。
我们 还表明,SE484可帮助体外培养抑制对甲氧西林耐药的已知毒力菌株USA300和对甲氧西林敏感的MSSA。以下数据表明,ATR-04降低了致病能力金黄色葡萄球菌细菌 种,用于生长和引发EGFRi皮疹患者的感染。

图9.表达Epidermin的SE484杀死金黄色葡萄球菌在体外琼脂平板上具有与莫匹罗星相似的活性。
我们 正在提议在患者中对ATR—04制剂中的SE 484进行初步研究。预期这是一项在开始EGFRi治疗的结直肠癌或头颈癌患者中进行的Ib期多中心、随机、双盲、单剂量、安慰剂对照试验。 主要终点是安全性,次要终点将包括疗效和生活质量或QoL我们计划在2024年年中提交 IND。待FDA批准IND后,我们预计将于2024年第四季度开始1b期临床试验。
ATR-01治疗寻常型鱼鳞病
ATR-01是我们用于治疗寻常型鱼鳞病的候选药物。该项目目前正在研究人类微丝蛋白的专利和正在申请专利的新型工程片段。ATR-01正在开发为一种局部应用,旨在治疗寻常型鱼鳞病,这是一种慢性鳞状皮肤病,估计发病率和患病率为1/250,使美国的患者总数达到130万人。寻常型鱼鳞病是由编码微丝蛋白的基因功能缺失突变引起的。利用蛋白质工程的合成生物学工具,我们在微丝蛋白上附加了一种细胞穿透肽,这有助于更深层次地将微丝蛋白输送到皮肤。这是为了克服皮肤屏障的不可穿透性,否则会限制局部蛋白质的传递。
寻常型鱼鳞病概述
寻常型鱼鳞病,或IV,是一种慢性、干燥性、鳞片状皮肤病,估计发病率和患病率为1/250,这使得美国的患者总数达到130万人。IV的临床特征通常出现在2个月左右,包括全身干燥和腹部、胸部和四肢伸肌表面突出的细小、白色到灰色的鳞片。 虽然罕见,但一些IV患者也会出现汗量减少和热耐受。长期以来,IV的发病机制一直被认为是表皮角质透明质颗粒的减少或数量减少,甚至完全不存在。此外,IV患者患特应性皮炎、哮喘和过敏的风险增加。
| 14 |
寻常型鱼鳞病是一种常染色体半显性遗传病,由编码微丝蛋白的基因功能缺失突变引起。微丝蛋白是一种必需的结构蛋白,来源于蛋白原蛋白,它在角质层分解成单独的微丝蛋白单位。这些 通过与角质形成细胞细胞骨架中的角蛋白和其他中间丝蛋白结合来增强皮肤屏障。许多研究 已经发现了功能丧失突变外挂在IV患者中,这些突变与角蛋白细丝紊乱、皮肤屏障缺陷和角质层微骨折相关,从而导致经皮变应原增敏。此外,丝状蛋白及其分解产物在皮肤中还具有显著的附加功能,包括滋润皮肤(通过吸湿性氨基酸或“天然保湿因子”),影响抗菌分子的产生(特别是针对S 金色)和保持有益的脂类分布和皮肤的PH值。
治疗IV几乎没有有效的疗法。目前IV的治疗选择主要包括局部水分蒸发抑制剂 (例如氯化钠、尿素、乳酸、水杨酸),以及少量的保湿剂(例如甘油、丙二醇)。也可以使用局部维甲酸来减缓人体皮肤细胞的产生。然而,长期使用维甲酸并不理想。特别令人担忧的是所有维甲酸的致畸作用,这限制了它们在有生育潜力的妇女中的使用。长期使用维甲酸治疗的慢性毒性可能会导致骨骼异常。此外,儿童长期使用维甲酸可能会抑制他们的生长。值得注意的是,许多IV患者由于自我意识和社交尴尬而显著降低了生活质量,并对家庭生活、教育/职业生活甚至休闲/体育活动产生了负面影响。
我们治疗寻常型鱼鳞病的解决方案-ATR-01
现已知,IV是由编码微丝蛋白的基因功能丧失突变引起的,导致角蛋白细丝组织紊乱、皮肤屏障缺陷和角质层微骨折,以及细菌和病毒皮肤感染导致经皮变应原增敏。我们正在开发ATR-01作为IV的一种新的治疗方式,直接针对这种疾病的病理生理学。ATR-01由人Flg蛋白的Flg9-10功能单位和一个附着的细胞穿透肽组成。目标 是通过局部涂抹和通过细胞穿透肽更深入地渗透皮肤来补充hFlg的稳定释放。
ATR-01的临床前 数据
人类Flg单位(结构域9-10)在人体皮肤外植体(来自整形手术)上进行了评估离体。外植体的皮肤屏障被反复剥离胶带破坏,导致经皮水分损失或TEWL值较正常皮肤显著增加。如下例所示,每天局部应用含有细胞穿透肽的人微丝蛋白单元5天后,TEWL迅速改善,呈剂量依赖性(未显示),提示皮肤屏障得到改善。因此,局部注射重组hFLG单位和细胞穿透肽可以改善/加速受损的人皮肤屏障的修复。

| 15 |
图 10:剥离的磁带上的局部微丝胶合应用离体使用人类微丝凝聚蛋白后的人体皮肤。
最后, 我们已经证明,外用微丝蛋白可以改善微丝蛋白缺陷小鼠模型的皮肤屏障缺陷。将重组小鼠微丝蛋白,或mFlg,应用于片状尾巴或FT小鼠的尾部(一种在细丝研磨基因),每天一次,持续2周(50微克总蛋白质/尾部或15.2微克总蛋白质/厘米2)。与赋形剂(“基线”组)相比,每日服用mFlg显著改善了FT小鼠在治疗时的经皮水分损失(FT+Flg组)。X轴上的第三个 组是正常、对照、野生型组(WT),它没有细丝研磨吉恩被击倒了。重组mFlg联合细胞穿透肽处理受损小鼠皮肤可改善受损小鼠皮肤屏障(见下图A)。此外,对小鼠尾部切片的表皮的组织学分析显示,使用mFlg处理有改善角质层厚度的趋势(见下图B)。在这张图中,Y轴表示角质层增厚,角质层在超过正常厚度后开始分裂或结垢。用mFlg治疗后,6个样本中有4个样本的厚度有所改善。

其他 个潜在候选产品
除了我们的三个主要候选产品外,我们的目标是开发广泛的候选产品组合,专注于扩大我们的精确皮肤病平台的应用 。我们拥有发现和开发精准皮肤病治疗产品的专有平台。我们的平台围绕由大约1,500个独特的细菌菌株组成的微生物文库构建,以筛选独特的治疗特性,并利用微生物基因技术来分析、预测和设计皮肤微生物产生的蛋白质、多肽和分子。通过SyMPL技术的独家许可,我们对难处理的微生物物种进行基因工程的能力得到了独一无二的利用。
拜耳 联合开发协议
2019年12月,我们与拜耳签订了联合开发协议(JDA),根据该协议,我们同意联合开发从我们的专有微生物文库中挑选出来的某些菌株。我们和拜耳已经同意在身份识别和在试管中和离体用于外用制剂的微生物菌株的特性。拜耳在签署联合开发协议时向我们一次性支付了150,000美元 ,并同意偿还我们的开发费用。2021年10月,拜耳扩大了期权协议 ,并支付了375,000美元用于额外的表征工作。我们已授予拜耳获得JDA项下开发活动的最多六个品种的独家版税许可的选择权,包括任何相关专利权的独家版税许可 。在对数百个菌株进行筛选后,我们和拜耳选择了两个特定的菌株进行进一步研究在试管中和离体表征,我们打算开发作为潜在的非处方化妆品产品。我们在2023年第四季度完成了 表征工作并将数据交付给拜耳,届时拜耳有12个月的时间来行使 其许可菌株和相关专利的选择权。表征研究结束并将数据 交付给拜耳后,JDA将结束,但拜耳可选择许可菌株和相关专利。截至本报告日期,我们 尚未与拜耳谈判商业许可协议,我们将不会这样做,直到拜耳行使其选择权 以获得独家附带使用费的许可。
| 16 |
2020年9月,拜耳的风险投资集团Leaps by Bayer购买了我们B系列优先股800万美元。
销售 和市场营销
鉴于我们所处的发展阶段,我们尚未建立商业组织或分销能力。我们计划在美国建立有重点的 能力,将我们的开发计划商业化,重点是用于皮肤病治疗的活生物治疗产品和重组蛋白 ,我们相信,在美国,患者群体和针对我们目标适应症的医学专家足够集中,使我们能够通过有针对性的销售团队有效地推广我们的产品,如果批准进行商业销售的话。在商业化可能对我们来说资本效率较低的其他市场,我们可能会有选择地寻求与第三方的战略合作 ,以最大限度地发挥我们候选产品的商业潜力。
制造业
我们 不拥有或运营用于生产当前候选产品的制造设施。我们目前依赖第三方合同制造商提供我们所需的所有原材料、制造设备和活性药物成分,以及我们的 临床前研究和临床试验。虽然我们能够在我们的康涅狄格州格罗顿工厂为我们的临床试验 生产成品,但我们将依赖第三方生产我们用于商业销售的成品。我们与这些第三方中的任何一方都没有长期的 协议。我们目前也没有关于制造3期临床试验或商业用品的任何合同关系。我们打算与第三方合同制造商和一个或多个备份制造商签订协议,以用于未来的生产。我们正在分析为未来的开发和我们开发的任何产品的商业 量建立制造能力的可行性。此类产品将需要在符合FDA和我们正在寻求批准的其他司法管辖区监管机构的要求的设施和工艺中进行生产。
竞争
生物制药行业的特点是技术快速进步、竞争激烈以及对专利药物的高度重视。虽然我们相信我们的知识、经验和科学资源为我们提供了竞争优势,但我们面临着来自许多不同来源的潜在竞争,包括其他生物制药公司、学术机构和政府机构 以及公共和私人研究机构。我们成功开发和商业化的任何候选药物都将与现有疗法和未来可能出现的新疗法展开竞争。
Netherton综合征
关于Netherton综合征,到目前为止还没有药物被FDA批准,特别是针对Netherton综合征。标准护理包括使用温和/柔软的非洗涤剂液体洁面油清洁皮肤,最好是酸性(5)。由于Netherton综合征患者的皮肤最常见的是干燥、鳞屑和脱皮,因此也经常使用润肤剂和保湿霜。水杨酸、尿素或α-羟基酸等角质溶解药物通常具有刺激性,Netherton综合征患者对其耐受性不佳。Netherton综合征患者的皮肤易受细菌感染。有限的感染用局部抗生素治疗,治疗时间很短,最长可达2周。口服抗生素也可以用来治疗病原体金黄色葡萄球菌和链球菌可以导致更极端感染的菌株。漂白剂也被推荐每周洗两到三次,因为它们有抗菌作用。外用皮质类固醇通常用于治疗与非感染性Netherton综合征皮损相关的炎症和过度增殖,但由于其不良反应,必须加以限制。这些不良事件包括氨基酸尿症、库欣综合征、皮肤萎缩、肾上腺功能不全、生长迟缓、高血压和虚弱。过度使用局部类固醇甚至会导致角质层的丧失,从而加剧皮肤屏障的缺陷。全身性维甲酸对Netherton综合征显示出不同程度的疗效,但也具有骨毒性和致畸作用。局部的钙调神经磷酸酶抑制剂已被用于减少红斑(红肿),但患者 已表现出快速反应,并随着治疗时间的延长而降低疗效。这些免疫调节剂还存在严重不良影响的风险,包括增加感染、肿胀、灼热感和刺痛感的风险。光疗法(窄带UVB(NB-UVB)和补骨脂素-UVA(PUVA))也已被研究用于Netherton综合征患者,但由于其可能导致红斑和皮肤癌风险增加而受到限制。
| 17 |
我们 还意识到Sixera Pharma于2021年12月在欧洲启动了一项临床试验,使用SXR—1096(一种局部小分子KLK抑制剂) 治疗Netherton综合征。此外,Quoin Pharmaceuticals于2022年12月和2023年3月启动了两项使用局部小分子广谱 丝氨酸蛋白酶抑制剂QRX 003的临床试验。勃林格殷格翰还在招募一项使用spesolimab(一种IL—36抑制剂)治疗Netherton综合征的II/III期临床试验。Krystal Biotech、MatriSys和BridgeBio报告说,他们正在开发处于临床前阶段的Netherton综合征项目。
如果我们的竞争对手开发和商业化比ATR-12或我们可能开发的任何其他药物更安全、更有效、副作用更少或更少、更方便或更便宜的药物,我们的商业机会可能会减少或消失。 我们的竞争对手也可能比我们更快地获得FDA或其他监管机构对其药物的批准, 这可能会使我们的竞争对手在我们能够进入市场之前建立强大的市场地位。与我们相比,我们正在竞争或未来可能竞争的许多公司在研发、制造、临床前试验、进行临床试验、获得监管批准和营销 药品方面拥有更多的财务资源和专业知识。制药和生物技术行业的合并和收购可能会导致更多的资源集中在我们数量较少的竞争对手中。规模较小或处于早期阶段的公司也可能成为重要的竞争对手,尤其是通过与大公司和成熟公司的合作安排。这些竞争对手还在招聘和留住合格的科学和管理人员、为临床 试验建立临床试验站点和受试者注册以及在获取与我们的计划互补或可能需要的技术方面与我们展开竞争。
EGFRi相关 皮疹
迄今为止,FDA尚未专门批准任何药物用于治疗EGFRi相关皮疹。大多数持续接受抗EGFR治疗的患者(估计高达90%)发生皮肤病不良事件,尤其是丘疹脓疱性皮疹、 瘙痒(瘙痒)、干燥症(干燥)和甲沟炎(指甲感染)。丘疹脓疱或痤疮样皮疹是EGFR在皮肤上最常见的不良 事件。这种皮疹在许多患者中对EGFRi治疗的依从性产生负面影响。严重病例需调整剂量或停止 治疗。由于循证对照试验仍然非常稀少,EGFRi皮肤毒性的治疗主要 依赖于医师经验和专家共识会议的建议。因此,在EGFRi皮疹的临床治疗中存在地理差异 甚至不一致。对于痤疮样皮疹,在欧洲避免使用局部皮质类固醇,但在美国经常使用。此外,局部治疗经常根据个体患者定制 ,并可能根据具体情况进行更改。没有任何局部治疗方案普遍适用于所有患者。
我们 了解到以下第二阶段计划正在开发针对EGFRi相关皮疹的候选研究药物。Lutris Pharma正在美国和以色列开发外用B-Raf抑制剂LUT014。大同药业正在韩国开发DWP708。
知识产权
概述
我们 积极寻求通过各种方式保护我们的专有技术、发明、发明改进和其他对我们的业务发展具有重要商业意义的知识产权,例如寻求、维护和捍卫专利权,无论是内部开发还是从第三方获得许可。我们还可以依靠与我们的专有技术平台相关的商业秘密和技术诀窍,依靠持续的技术创新和未来的许可机会来发展、加强和保持我们在基因治疗领域的地位,这可能对我们的业务发展至关重要。还可以通过数据独占性、市场独占性和专利期延长提供额外的监管保护 。
| 18 |
截至本报告之日,我们拥有或独家许可了3项已颁发的美国专利、12项未决的美国专利申请、3项未决的PCT申请以及57项对我们的业务发展至关重要的其他外国专利和专利申请。
我们的 政策是提交专利申请,以保护专有技术、发明和对发明的改进以及可能对我们业务发展具有重要商业意义的其他知识 产权。我们还打算寻求额外的专利保护或 依靠专有技术或商业秘密权利来保护可能用于生产和开发我们的活体生物 产品的其他技术。如下所述,我们是独家许可协议的一方,该协议授予我们在我们的 活体生物产品以及我们产品的制造和开发中使用特定技术的权利。
我们的 专利组合
我们的专利组合广泛涵盖治疗细菌的药物组合物,这些药物组合物含有用于治疗异常皮肤状况的治疗细菌,以及制造和使用这些重组细菌的方法。在我们最广泛的申请中,我们获得了一项美国专利 ,该专利保护了使用一种细菌菌株治疗异常皮肤状况的药物组合物,该细菌菌株表达了具有治疗效果的重组多肽。这项专利将于2035年5月到期。具体地说,本专利涉及含有一种或多种以下细菌菌株的药物组合物:双歧杆菌、短杆菌、丙酸杆菌、乳球菌、链球菌、葡萄球菌、乳杆菌、肠球菌、小球菌属、明串珠菌或葡萄球菌,其中该细菌菌株经过改造可生产用于治疗异常皮肤状况的治疗性多肽。我们相信,这项专利为我们使用重组细菌治疗皮肤病和疾病提供了广泛的保护。到2035年5月到期。
针对我们最先进项目的专利申请摘要如下。
ATR-12
我们的 ATR-12候选产品拥有3项已颁发的美国专利、7项未决的美国专利和31项未决的外国专利 和专利申请。这些专利和专利申请代表了十个权利要求家族,其中包括表皮葡萄球菌重组治疗性多肽的表达--营养缺陷型表皮葡萄球菌、 和重组表皮葡萄球菌表达治疗性lekti蛋白的菌株,以及ATR-12的配方。发布的其中一项美国专利涉及一种含有治疗异常皮肤疾病的治疗性多肽的重组细菌菌株,将于2035年到期。第二项颁发的美国专利涵盖了一种营养缺乏症表皮葡萄球菌2039年到期如果要 从未决专利申请中授予额外专利,它们将在2035年至2044年之间到期。
ATR-04
我们的 ATR-12候选产品受一项已颁发的美国专利、两项待决的美国专利申请和17项待决的外国申请的约束。 这些专利和专利申请代表了针对营养缺陷型细菌及其治疗疾病用途的两类权利要求。我们有一项已颁发的美国专利,涵盖ATR-04。如果授予更多专利,它们也将在2039年到期 。
| 19 |
专利期限和期限延长
个别专利有不同的期限,具体期限取决于专利申请的提交日期或专利颁发日期以及获得专利的国家/地区的专利的法律期限。通常,为在美国提交的申请颁发的实用新型专利的有效期为自非临时专利申请的最早有效申请之日起20年。此外,在 某些情况下,美国专利的有效期可以延长,以重新获得美国专利商标局或USPTO的一部分,延迟发布专利,以及因FDA监管审查期间而实际失去的一部分期限 。但是,对于FDA的组件,恢复期限不能超过5年,恢复期限不能从FDA批准之日起延长 14年以上。此外,只有一项适用于批准的药物的专利有资格延期, 并且只有那些涉及批准的药物、其使用方法或制造方法的权利要求才能延长。外国专利的有效期根据适用的当地法律规定有所不同,但通常也是自最早生效日期起计的20年。根据美国专利商标局和各个外国司法管辖区的要求,专利的所有税金、年金或维护费都必须及时支付,以便专利在这段时间内保持有效。
专利提供的实际保护可能因国家/地区的不同产品而有所不同,并可能取决于许多因素,包括专利的类型、其覆盖范围、与监管相关的延期的可用性、特定国家/地区的法律 补救措施的可用性以及专利的有效性和可执行性。
我们的专利和专利申请可能会受到其他人的程序或法律挑战。我们可能无法获得、维护和 保护开展业务所需的知识产权,并且我们可能会受到侵犯或以其他方式侵犯他人知识产权的索赔,这可能会对我们的业务造成实质性损害。有关更多信息,请参阅标题为 “风险因素-与我们的知识产权有关的风险.”
交易秘密和诀窍
我们 还可能依靠商业秘密、技术诀窍、持续的技术创新和机密信息来发展和维护我们的 专有地位,并保护我们业务中不受专利 保护或我们认为不适合专利保护的方面,包括我们生产活性生物治疗产品的专有工艺。我们寻求通过与我们的员工、顾问、科学顾问、承包商和其他可能可以访问专有信息的人签订保密协议和发明转让协议来保护我们的专有技术和流程,根据这些协议,他们有义务将他们在受雇期间或服务期限内做出的发明转让给我们。这些协议可能会被违反,我们可能没有足够的 任何违反的补救措施。此外,我们的商业秘密可能会被竞争对手知晓或独立发现。对于我们的承包商、商业合作伙伴、合作者、员工和顾问在为我们工作时使用他人拥有的知识产权的程度,可能会就相关或由此产生的专有技术和发明的权利产生争议。有关更多信息,请参阅 标题为“风险因素-与我们的知识产权有关的风险.”
我们 还试图通过维护我们场所的物理安全以及我们信息技术系统的物理和电子安全来保护我们的数据和商业秘密的完整性和机密性。
与弗雷德·哈奇森癌症中心签订独家许可协议
2022年1月,我们与弗雷德·哈钦森癌症中心或弗雷德·哈奇签订了独家许可协议。根据我们与Fred Hutch的协议,我们在由Fred Hutch开发和拥有的与SyMPL技术相关的某些专利下获得了全球独家许可,可以开发、制造、分销、分销、使用、研究、改进、进口、提供销售、销售和以其他方式商业化此类专利涵盖的产品。此类独家许可受Fred Hutch和美国政府保留的某些权利的约束。弗雷德·哈奇授予我们的专利权包括两个专利申请系列 ,这些专利申请针对的是绕过限制修饰系统的方法,以便更容易地将异种DNA引入工程微生物。 这些专利申请以及这些申请颁发的任何专利将使我们能够生产更多用于疾病治疗的改良微生物。我们当前的候选产品未采用SyMPL技术平台,但我们预计部分或全部未来候选产品将采用SyMPL技术平台。如果发行,这两个家族将分别于2037年和2040年到期。
考虑到根据Fred Hutch许可协议授予我们的许可,我们象征性地向Fred Hutch支付了预付款。此外, 我们需要向Fred Hutch支付某些开发和商业里程碑付款,并按许可产品的净销售额支付个位数版税 。弗雷德·哈奇协议还要求我们向弗雷德·哈奇报销许可专利的起诉和维护费用。
| 20 |
根据弗雷德·哈奇许可协议,我们必须以商业上合理的努力,通过一个积极而勤奋的计划,将许可产品推向市场,以利用许可的专利权。Fred Hutch许可协议的期限将持续 ,直至(I)许可专利到期或(Ii)自许可产品首次销售之日起十年内。如事先书面通知Fred Hutch,我们可随时终止Fred Hutch许可协议。如果我们严重违反协议并且未能在指定的治愈期限内纠正此类违规行为,或者如果 我们破产或资不抵债,则Fred Hutch有权终止该许可协议。有关根据弗雷德·哈奇许可协议获得的知识产权的更多信息,请参阅标题为“营业执照和知识产权.”
我们 还在美国和七个国家/地区拥有我们公司名称和设计的注册商标。
政府法规
制药公司受到外国、联邦、州和当地机构的广泛监管,如美国FDA,以及全球大多数国家和地区的各种类似机构。药品的研究、开发、测试、制造、分销、包装、标签、储存、记录保存、营销和销售在美国和其他国家均受政府监管。此外, 在美国,我们必须遵守FDA制定的规章制度,要求提交数据,表明我们的候选产品 是安全有效的,并根据cGMP规定生产。如果我们不遵守适用的要求, 我们可能会被罚款,政府可能会拒绝批准我们的营销申请,或者允许我们制造或营销我们的候选产品, 我们可能会受到刑事起诉。我们、我们的制造商和临床研究组织也可能受到其他外国、联邦、州和当地法律的监管,包括但不限于美国《职业安全与健康法》、《资源保护和回收法》、《清洁空气法》、进出口和海关法规以及其他 国家的法律法规。美国政府加强了针对国内和国际非法营销行为的执法活动。 因此,制药公司必须确保其遵守《反海外腐败法》和联邦医疗欺诈 以及包括《虚假索赔法》在内的滥用法律。
这些 监管要求会影响我们的运营,并且因国家/地区的不同而有所不同,因此确保一个国家/地区获得适用的监管批准并不意味着另一个国家/地区的批准。审批程序成本高,人力密集, 通常持续多年,需要高技能和专业资源。
FDA 上市审批流程
在美国,我们的候选产品根据联邦食品、药物和化妆品法案(FDCA)、公共卫生服务法案(PHSA)和FDA颁布的法规作为生物制品受到FDA的监管。在产品开发流程(包括临床前测试、临床测试、审批流程或审批后流程)期间的任何时间未能遵守适用要求 可能会使申请人在进行临床试验、监管审查和批准和/或行政或司法制裁方面受到延误。这些制裁可能包括但不限于FDA拒绝允许申请人继续进行临床测试、拒绝批准未决申请、吊销或吊销执照、撤回批准、警告信、不良宣传、客户通知、产品召回、产品扣押、拒绝批准出口或进口批准、完全或部分暂停生产或分销、同意法令、禁令、罚款,以及由FDA或美国司法部或其他政府实体提起的民事或刑事调查和处罚。
新生物在美国上市前通常需要采取的步骤通常包括:
| ● | 完成临床前实验室测试和根据FDA现行良好实验室操作规程进行的动物研究; |
| 21 |
| ● | 向FDA提交IND,该IND必须在临床试验开始之前生效,并且必须 每年更新或发生重大变化; |
| ● | 在试验开始前,每个治疗地点的独立机构审查委员会或伦理委员会批准 ; |
| ● | 执行充分和良好控制的人体临床试验,以确定拟议生物产品候选的安全性、纯度和效力,以确定其拟议的使用适应症; |
| ● | 提交支持安全性和有效性的数据,以及关于临床开发和拟议标签中产品的制造和成分的详细信息。 |
| ● | 在所有关键临床试验完成后,准备并向FDA提交BLA; |
| ● | 令人满意的 完成FDA咨询委员会的审查,如果适用; |
| ● | FDA在收到BLA后60天内作出的提交审查申请的决定; |
| ● | 令人满意的 完成FDA对生产建议产品的一个或多个制造设施的批准前检查,以评估是否符合cGMP标准,并确保这些设施:方法和控制足以保持生物制品的持续安全性、纯度和效力,以及选定的临床调查地点,以评估对良好临床实践或GCP的遵从性; |
| ● | 令人满意的 完成FDA对非临床和临床试验地点的任何审计,以确保符合CGP要求和支持BLA的临床数据的完整性; |
| ● | 支付使用费,并确保FDA批准拟议的使用适应症的BLA; |
| ● | FDA审查和批准BLA,以允许在美国使用的特定适应症的产品进行商业营销;以及 |
| ● | 遵守任何审批后要求,包括REMS和FDA要求的任何审批后研究 。 |
临床前研究和探索性新药应用
临床前试验包括对产品的化学成分、配方和稳定性进行实验室评估,以及对动物进行动物研究以评估其潜在的疗效和毒性。进行临床前试验和用于试验的化合物配方必须符合联邦法规和要求。临床前试验的结果连同生产信息和分析数据一起作为IND申请的一部分提交给FDA。即使在提交IND申请后,某些临床前试验仍可能继续。 IND在FDA收到后30天自动生效,除非在此之前FDA对拟议的临床试验的产品或进行提出担忧或问题,包括担心人类研究志愿者将面临不合理的健康风险 。在这种情况下,IND赞助商和FDA必须在临床试验开始之前解决FDA的任何悬而未决的问题。
因此,提交IND可能导致FDA不允许临床试验开始,或允许临床试验按发起人最初在IND中指定的条款开始。如果FDA在最初的30天期间或在IND过程中的任何时候提出担忧或问题,它可以选择实施部分或全部临床搁置。FDA发布的这一命令将推迟拟议的临床试验或导致正在进行的临床试验的一个阶段的启动延迟,直到所有悬而未决的问题得到充分解决,FDA已通知该公司可能会继续进行调查。这可能会导致在及时完成计划的临床试验方面出现重大延误或困难。
| 22 |
临床试验
临床 试验涉及根据良好临床实践或GCP要求,在合格的首席研究人员的监督下,将研究产品候选给健康志愿者或要接受治疗的疾病患者 。临床 研究是在详细说明研究目标、哪些类型的患者可以参加研究、测试和程序的时间表、药物、剂量和研究时间、以及用于监测安全性的参数和要评估的疗效标准的方案下进行的。作为IND过程的一部分,每项临床研究的方案和任何后续方案修改都必须提交给FDA。
希望在美国境外进行临床试验的赞助商可以(但不需要)获得FDA的授权,根据IND进行临床试验。如果在美国境外的临床试验不是根据IND进行的,只要临床试验的进行符合GCP的精神,并符合国际临床研究道德行为指南《赫尔辛基宣言》和/或进行临床试验的一个或多个国家的法律法规,赞助商可以将临床试验的数据提交给FDA以支持BLA,以为临床试验的参与者提供更大保护的 。
IRB还必须集中或单独审查将进行临床试验的每个机构的每项临床试验。IRB将考虑临床试验设计、患者知情同意、伦理因素、人类受试者的安全、该机构可能的责任,以及在适当情况下保护人类受试者的隐私。IRB必须按照FDA的规定进行操作。FDA、IRB或临床试验赞助商或首席研究人员可因各种原因在任何时间暂停或中止临床试验,包括发现临床试验未按照FDA要求进行,或受试者或患者面临不可接受的健康风险。临床测试还必须满足广泛的GCP规则和知情同意的要求。此外,一些临床研究由临床研究赞助商组织的一个独立的合格专家小组监督,称为数据安全监测委员会或委员会。根据对研究中某些数据的访问,该小组建议是否可以在指定的检查点进行试验。临床研究赞助商 还可以根据不断变化的业务目标和/或竞争环境暂停或终止临床试验。
临床试验通常分三个连续阶段进行,但这些阶段可能会重叠或合并。详细说明临床试验阶段结果的年度进度必须提交给FDA。
| ● | 阶段 1临床试验通常在健康志愿者的小组中进行,以评估 各种给药方案和药代动力学的安全性和耐受性。对于某些产品 用于孤儿、严重或危及生命的疾病,特别是当产品毒性过大时 为了给健康人施用,可以在个体中进行初始临床试验 患有特定疾病,该疾病指示使用测试产品。这些审判 患者通常被称为1b期试验。如果它们包含一个设计,以建立 在特定剂量下,它们通常被称为Ib/2a期临床试验。不过, 通常需要额外的II期(有时称为2b期)临床试验来完善 选择用于关键的III期临床试验的最终剂量。 |
| ● | 阶段 2临床试验通常在有限的患者人群中进行,以确定 可能的不良反应和安全风险,评估候选产品的有效性 针对特定的目标适应症,并确定耐受性和最佳剂量。多个 申办者可进行II期临床试验,以在开始前获取相关信息 更大、更昂贵的III期临床试验。 |
| ● | 阶段 3如果II期临床试验证明剂量范围 候选产品的潜在有效性和可接受的安全性特征。阶段 进行了3项临床试验,以进一步评估更多患者的剂量, 提供临床疗效的实质性证据,并进一步测试扩展的安全性 在多个地理上分散的临床试验中心和不同的患者人群。 可以设计一项控制良好、统计学稳健的III期试验来提供数据 监管机构将用来决定是否批准,如果批准, 如何适当地给药物贴标签:此类III期研究被称为“关键”。 |
| 23 |
如果FDA认为临床研究没有按照FDA的要求进行,或者参与者面临不可接受的健康风险,FDA可以在任何时候下令暂时或永久停止临床研究或施加其他制裁。在某些情况下,FDA可能会批准候选产品的BLA,但要求赞助商进行额外的临床试验,以在BLA批准后进一步评估该药物的安全性和有效性。此类批准后试验通常被称为4期临床试验。这些研究用于从预期治疗适应症患者的治疗中获得其他经验数据,并在根据加速审批法规批准的药物或生物制品的情况下记录临床益处。 如果FDA批准了产品,而公司正在进行不需要批准的临床试验,则公司可以 使用这些临床试验的数据来满足任何第四阶段临床试验的全部或部分要求,或请求更改 产品标签。未能对进行4期临床试验进行尽职调查可能会导致撤回对产品的批准 。
符合cGMP要求
当候选产品通过临床测试阶段时,生产流程将进一步定义、细化、控制和验证。 FDA要求的控制和验证级别随着临床研究的进展而提高。FDA通常不会批准BLA申请,除非它确定制造工艺和设施完全符合cGMP要求,并且 能够确保产品在所要求的规格内一致生产。PHSA强调了对属性无法精确定义的生物制品等产品进行生产控制的重要性。
我们 和我们赖以生产我们的候选产品及其各自成分(包括活性药物成分或原料药)的第三方制造商必须遵守药品的制造、包装和标签符合cGMP的要求。为了符合cGMP要求,制造商必须继续花费时间、金钱和精力来满足与人员、设施、设备、生产和工艺、标签和包装、质量控制、记录保存和其他要求有关的要求。
制造商和其他涉及产品制造和分销的企业也必须向FDA和某些州监管机构注册。美国和非美国制造企业在首次参与制造过程时,必须注册并向FDA提供附加信息。由未经注册的工厂制造或进口的任何产品,无论是在美国还是非美国,都被视为在FDCA下有错误的品牌。机构可能会受到政府当局的定期突击检查,以确保遵守cGMP和其他法律。检查必须遵循“基于风险的时间表” ,这可能会导致某些机构更频繁地被检查。制造商可能还必须应要求提供有关其工厂的电子或实物记录。推迟、拒绝、限制或拒绝FDA的检查可能会导致产品被视为掺假。
BLA 提交和审查
假设 根据所有适用的法规要求完成了所有要求的测试,则将以BLA的形式向FDA提交有关候选产品的详细信息,请求批准针对一个或多个适应症销售该产品,并支付 使用费,除非放弃。BLA包括从相关的非临床和临床研究中获得的所有相关数据,包括阴性或不明确的结果以及阳性结果,以及有关化学、制造、对照和建议的 标签等的详细信息。为支持上市审批,提交的数据必须在质量和数量上足以确定候选产品的安全性和有效性,使FDA满意。FDA还在批准BLA之前对制造商和实验室进行预批准 检查。
如果BLA提交的申请被接受,FDA将开始对BLA进行深入审查。根据《处方药使用费法案》(PDUFA),FDA的目标是在提交后十个月内完成初步审查并对申请人做出回应,除非申请 涉及未满足的医疗需求,或涉及严重或危及生命的适应症,在这种情况下,目标可能是在提交BLA后六个月内 。但是,PDUFA目标日期不是法定要求,FDA的回应通常比最初的PDUFA目标日期晚几个月。此外,如果FDA要求或BLA发起人以其他方式提供关于BLA中已经提供的信息的补充信息或澄清,则PDUFA项下的审查过程和目标答复日期可被延长。因此,BLA审查 过程可能非常漫长。在审查BLA期间,FDA可以将申请提交给咨询委员会进行审查、评估和建议,以确定是否应该批准申请。FDA不受咨询委员会的建议的约束,但它通常遵循这样的建议。来自临床研究的数据并不总是决定性的,FDA和/或其任命的任何咨询委员会可能会以不同于申请者的方式解释数据。
| 24 |
在 FDA评估BLA并检查将生产和测试药品和/或其原料药的制造设施后,它 将批准药物产品的商业销售,并提供特定适应症的处方信息,或发布完整的 回复信,表明申请尚未准备好批准,并说明为确保BLA获得批准必须满足的条件。如果完整的回复信需要额外的数据,而申请人随后提交了该数据,FDA仍可能最终判定BLA不符合其批准标准。FDA还可以通过风险评估和缓解策略或REMS计划来批准BLA以降低风险,其中可能包括药物指南、医生沟通计划或确保安全使用的要素,如受限分发方法、患者登记和其他风险最小化工具。 FDA还可能以更改建议的标签、制定适当的控制和规范或承诺进行上市后测试为条件进行批准。此类上市后测试可能包括第四阶段临床试验和监测,以便 在批准后进一步评估和监测产品的安全性和有效性。对严重或危及生命的适应症的产品的监管批准可能需要对临床研究的参与者进行长期跟踪,以确定该药物的总体生存益处 。
如果FDA批准了我们的候选产品之一,我们将被要求遵守批准后的一些监管要求。 我们将被要求向FDA报告某些不良反应和生产问题,提供最新的安全性和有效性信息,并遵守有关我们任何候选产品的广告和促销标签的要求。此外,质量控制和制造程序在批准后必须继续符合cGMP,FDA定期检查 制造设施以评估是否符合cGMP,这规定了广泛的程序性,实质性和记录保存要求。 如果我们寻求对批准的产品进行某些更改,例如某些制造更改,我们可能需要FDA审查和批准 才能实施更改。
虽然 医生可以将产品用于未经FDA批准的适应症,但我们不能将产品用于未经批准的适应症 。确保FDA批准新适应症类似于批准原始适应症的流程 ,除其他事项外,还需要提交充分且受控的研究数据,以证明该产品在新适应症中的安全性和有效性。即使进行了这样的研究,FDA也可能不会及时批准任何变化,或者根本不批准。
FDA还可能要求进行上市后测试或第四阶段测试,以及风险最小化行动计划和监督,以监控可能限制产品分销或使用的批准产品或场所条件或批准的影响。
快速跟踪、突破性治疗和优先审查指定
FDA有权指定某些产品进行快速审查,如果这些产品旨在满足在治疗严重或危及生命的疾病或状况或在紧急情况下未得到满足的医疗需求。这些计划是快速通道指定、突破性治疗指定和优先审查指定。
具体而言,如果一种产品是用于治疗严重或危及生命的疾病或病症,并且有可能满足此类疾病或病症的未得到满足的医疗需求,则FDA可指定该产品进行快速通道审查,无论该产品是单独使用还是与一种或多种其他产品联合使用。对于Fast Track产品,赞助商可能会与FDA有更多的互动,FDA可能会在申请完成之前对Fast Track产品申请的部分进行审查。如果FDA在对赞助商提交的临床数据进行初步评估后确定快速通道产品可能有效,则可以进行滚动审查。 赞助商还必须提供提交剩余信息的时间表,并且赞助商必须 支付适用的用户费用。然而,FDA审查快速通道申请的时间段目标直到申请的最后 部分提交后才开始。此外,如果FDA认为快速通道指定不再得到临床试验过程中出现的数据的支持,则FDA可能会撤回该指定。
| 25 |
此外,2012年,国会颁布了《食品和药物管理局安全与创新法案》,简称FDASIA。这项法律建立了一个新的监管方案,允许加快对被指定为“突破性疗法”的产品的审查。如果一种产品旨在单独或与一个或多个其他产品联合用于治疗严重或危及生命的疾病或状况,并且初步临床证据表明该产品可能在一个或多个临床重要终点显示出比现有 疗法显著改善的效果,例如在临床开发早期观察到显著的治疗效果,则该产品可被指定为 突破性疗法。 FDA可能会针对突破性疗法采取某些行动,包括在整个开发过程中与赞助商举行会议;及时向产品赞助商提供有关开发和批准的建议;让更多的高级人员参与评审流程;为评审团队指派一名跨学科的项目负责人;以及采取其他步骤以高效的方式设计临床试验。
此外,如果一种产品治疗严重疾病,并且如果获得批准,将在安全性或有效性方面有显著改善,则FDA可指定该产品进行优先审查。FDA根据具体情况确定,与其他可用的疗法相比,建议的产品是否有显著改善。显著的改善可能表现为:有证据表明在治疗某种疾病方面提高了 有效性,消除或大幅减少了限制治疗的产物反应,有记录的 患者依从性的提高可能导致严重结果的改善,以及新的 亚群的安全性和有效性的证据。优先指定的目的是将整体注意力和资源引导到对此类申请的评估上,并将FDA对营销申请采取行动的目标从10个月缩短到6个月。
此外,如果美国政府已指定实际或潜在的紧急情况,卫生与公众服务部部长可授权销售未经批准的药品和生物制品。在指定紧急情况后,FDA可以根据FDCA建立的标准,为特定产品的使用颁发紧急使用授权。EUA是特定于产品的,并受特定的 条件和限制。一旦作为EUA基础的紧急情况结束,那么EUA就会终止。
儿科 罕见病指定和优先审查凭证
根据修订后的FDCA,FDA鼓励开发符合“罕见儿科疾病”定义的药物和生物制品。“定义为一种严重的或危及生命的疾病,其严重的危及生命的表现主要影响从出生到18岁的个人,该疾病在美国影响不到200,000人,或者在美国影响到200,000人或更多人,并且没有合理的预期在美国开发和制造治疗此类疾病或疾病的药物或生物制剂的成本将从在美国销售此类药物或生物制剂中获得。罕见儿科疾病候选产品的 赞助商可能有资格获得优惠券,该优惠券可用于在罕见儿科疾病药物产品批准之日后获得后续人类药物或生物应用的优先审查 ,称为优先审查优惠券,或PRV。赞助商可以在提交其保密协议或BLA之前向FDA申请罕见儿科疾病的指定。一种罕见的儿科疾病指定并不保证赞助商在其NDA或BLA获得批准后会收到PRV。此外,选择不提交罕见儿科疾病指定请求的赞助商,如果他们请求原始凭证,则在其营销申请获得批准 后仍可收到PRV。市场推广申请,并满足所有资格条件 。如果收到PRV,它可能会被出售或转让不限次数。国会已将PRV计划延长至2024年9月30日,并有可能批准PRV至2026年。
审批后条例
一旦获得监管机构批准的产品上市或现有产品的新适应症,赞助商将被要求 遵守批准后的监管要求,包括FDA可能作为批准条件 强加的任何批准后要求。赞助商将被要求向FDA报告某些不良反应和生产问题,提供最新的安全性和有效性信息,并遵守有关广告和促销标签要求的要求。制造商及其某些分包商必须向FDA和某些州机构注册其机构,并接受FDA和某些州机构的定期突击检查,以了解是否符合持续的法规要求,包括对药品制造商施加某些程序和文件要求的cGMP法规。因此,赞助商及其第三方制造商必须继续在生产和质量控制方面投入时间、金钱和精力,以保持 遵守cGMP法规和其他法规要求。
| 26 |
产品也可能需要正式批次发布,这意味着制造商需要对 产品的每个批次进行某些测试,然后才能发布分销。如果产品需要正式发布,制造商必须向FDA提交每个批次的样品,以及显示该批次生产历史摘要和制造商对该批次进行的所有测试结果的发布协议。此外,FDA可能还会对一些 产品的批次进行某些验证性测试,然后再放行批次进行分销。最后,FDA将进行与药品的安全性、纯度、效力和有效性有关的实验室研究。
批准后,如果没有遵守监管要求和标准,或者产品上市后出现问题,FDA可能会撤回批准。如果后来发现产品存在以前未知的问题,包括未预料到的严重程度或频率的不良事件,或生产工艺,或未能遵守法规要求,可能会 导致修订批准的标签以添加新的安全信息;实施上市后研究或临床试验以评估新的安全风险;或根据REMS计划实施分销或其他限制。除其他事项外,其他潜在后果包括:
| ● | 限制 在产品的销售或生产过程中,完全从 市场或产品召回; |
| ● | 罚款, 批准后临床试验的警告信或搁置; |
| ● | 拒绝 FDA批准待决的BLA或已批准BLA的补充,或暂停或撤销 产品许可证批准; |
| ● | 产品 扣押、扣留或拒绝准许进口或出口产品;或 |
| ● | 禁令或施加民事或刑事处罚。 |
FDA对投放市场的产品的营销、标签、广告和促销进行严格管理。药品和生物制品 只能针对批准的适应症并根据批准的标签的规定进行推广。FDA和其他机构 积极执行禁止推广非标签用途的法律和法规,被发现以不正当方式推广非标签用途的公司可能会承担重大责任。
孤儿 药品名称
根据《孤儿药品法》,FDA可以授予一种药物孤儿称号,该药物旨在治疗在美国影响少于200,000人的罕见疾病或疾病,或在其他有限的情况下。孤儿药物指定(ODD)向拥有将特定产品推向市场的ODD的公司规定了七年的市场排他性,独立于专利保护。此外,开发孤儿药物的公司有资格享受某些激励措施,包括对合格的临床测试给予税收抵免。此外,已获得孤儿药物指定的产品的BLA 不受处方药使用费的限制,除非申请包括 药物指定用于治疗罕见疾病或病症以外的其他适应症。
| 27 |
为了获得排他性,如果具有孤儿药物指定的产品随后获得了FDA对其具有此类指定的疾病或情况的第一次批准 ,该产品有权获得孤儿药物排他性,这意味着FDA在七年内不得批准任何其他申请在同一适应症内销售相同的活性部分,除非在有限的情况下,例如 另一种药物显示出优于具有孤儿排他性的药物的临床优势。此外,医生可能会开出用于非标签用途的产品 ,从而破坏我们的排他性。如果竞争对手先于我们获得了对相同适应症的相同活性部分的批准,则孤立药物独占可能会在 七年内阻止我们的某个候选产品获得批准,除非我们能够证明我们的产品在临床上是优越的。
赞助商可以为以前未获批准的产品申请孤儿药物指定,或为已上市的产品申请新的孤儿药物指定。 此外,如果赞助商能够提出其 产品可能在临床上优于第一个批准的产品的合理假设,则该产品与已批准的孤儿药物相同的产品可以申请并获得针对相同罕见疾病或疾病的后续产品的孤儿药物指定。多个赞助商可以因相同的罕见疾病或疾病获得同一产品的孤儿药物指定,但每个寻求孤儿药物指定的赞助商必须提交完整的孤儿药物指定申请,并且只有第一个获得该药物的孤儿适应症批准的赞助商才能获得市场排他性, 有效地阻止了FDA批准同一药物和相同适应症的竞争对手正在开发的产品,除非 竞争对手能够证明正在开发的产品在临床上优于批准的产品,或者批准的 产品供应不足。为了允许FDA结束另一家制造商的孤儿排他期,FDA必须确定该制造商已经通过证明后一种药物更安全、更有效,或者 在其他方面对患者护理做出了重大贡献,从而证明了该制造商的临床优势。
专营期从FDA批准上市申请之日开始,仅适用于该产品已被指定的 适应症。FDA可能会批准同一产品用于不同用途的第二次申请,或随后针对同一适应症的不同药物的 申请。指定孤儿药物既不会缩短药物的开发时间或监管审查时间,也不会在监管审查或批准过程中给药物带来任何优势。
我们 可能计划在美国、欧盟和其他特定产品感兴趣的地区为我们的一些候选产品申请孤儿药物指定和独家经营权。我们不能保证我们在任何司法管辖区的任何产品都会获得孤儿药物称号 。即使我们能够获得产品的孤儿药物称号,我们也不能确保该产品将获得 批准,我们是否能够在获得批准后获得孤儿药物独家经营权(如果有的话),或者我们是否能够保持授予的任何独家专利权 。
生物仿制药和排他性
2010年3月23日签署成为法律的《患者保护和平价医疗法案》(ACA)包括一个副标题,名为《2009年价格竞争和创新法案》(BPCIA)。BPCIA建立了一个管理方案,授权FDA批准生物仿制药和可互换的生物仿制药。FDA已经发布了几份指导文件草案,概述了审查和批准生物仿制药的方法。与生物制品更大且往往更复杂的结构相关的复杂性,以及制造此类产品的工艺 ,对实施构成了重大障碍,FDA仍在制定这些障碍。
根据《生物产品检验法》,制造商可以提交一份生物制品的许可申请,该生物制品与先前批准的生物制品或参比产品具有生物相似性或互换性。为了让FDA批准生物相似产品,必须发现参考产品和建议的生物相似产品在安全性、纯度和效力方面没有临床上有意义的差异。为使FDA批准生物相似产品可与参考产品互换,该机构必须发现该生物相似产品可预期产生与参考产品相同的临床结果,并且(对于多次给药的产品)生物制剂和参考生物制剂可在先前给药后 在不增加安全风险或相对于单独使用参考生物制剂而降低疗效的情况下进行切换。
根据BPCIA,生物相似产品的申请必须在参考产品获得批准之日起4年后才能提交给FDA。FDA可能在参考产品获得批准之日起12年后才会批准生物相似产品。 即使一种产品被认为是有资格独家销售的参考产品,如果FDA批准该产品的完整BLA,该产品包含赞助商自己的临床前数据和来自充分的临床试验和良好控制的临床试验的数据,以证明其产品的安全性、纯度和有效性,则另一家公司可以销售该产品的竞争版本 。BPCIA还为被批准为可互换产品的生物仿制药设立了某些排他性期限。
| 28 |
美国以外的法规
为了在美国境外销售任何产品,公司还必须遵守其他国家/地区和司法管辖区关于质量、安全性和有效性的众多且各不相同的监管要求,其中包括临床试验、营销授权、商业销售和药品分销。无论产品是否获得FDA批准,该公司 都需要获得类似非美国监管机构的必要批准,然后才能在这些国家或司法管辖区开始临床试验或产品营销。审批流程最终因国家和司法管辖区而异 ,可能涉及额外的产品测试和额外的行政审查期。在其他国家和司法管辖区获得批准所需的时间可能与获得FDA批准所需的时间不同,甚至可能更长。在一个国家/地区或司法管辖区获得监管批准并不能确保在另一个国家/地区获得监管批准,但在一个国家/地区 或司法管辖区未能或延迟获得监管批准可能会对其他国家/地区的监管流程产生负面影响。
欧盟的法规和销售授权
欧洲药品管理局(简称EMA)是欧盟或欧盟的科学机构,负责协调对药品和生物制品等新的和批准的医药产品进行评估和监测。它负责对欧盟营销授权申请进行科学评估,并制定技术指导并向赞助商提供科学建议。
欧盟的医药产品审批流程与美国的流程大致相同,通常也包括圆满完成以下各项:
| ● | 临床前 实验室试验、动物研究和制剂研究均按照 适用的欧盟药物非临床研究质量管理规范法规; |
| ● | 提交 欧盟成员国的相关监管机构或国家监管机构,临床试验 每个临床试验的试验申请或CTA,必须在人类临床试验之前获得批准 审判可以开始; |
| ● | 性能 充分且控制良好的临床试验,以确定该药物的安全性和有效性 每个建议适应症的产品; |
| ● | 提交 上市许可申请或MAA的相关国家主管部门,其中 包括支持安全性和有效性的数据以及生产的详细信息 临床开发中产品的成分和拟定标签; |
| ● | 满意 相关国家主管部门完成对生产设施的检查 或设施,包括第三方的设施,在那里生产产品,以评估 符合cGMP; |
| ● | 潜力 对生成数据的非临床和临床试验中心进行稽查,以支持 MAA;及 |
| ● | 审核 并在进行任何商业营销之前获得MAA相关国家机构的批准, 产品的销售或发货。 |
| 29 |
临床前研究
临床前试验包括对产品化学成分、配方和稳定性的实验室评估,以及对动物潜在疗效和毒性的评估研究。进行临床前试验和用于试验的化合物配方必须符合欧盟相关法规和要求。临床前试验的结果,连同相关的生产信息和分析数据,在寻求批准开始临床试验时作为CTA的一部分提交,在寻求营销授权时与MAA一起提交。
临床试验批准
在欧盟进行临床试验(包括CGCP)的要求 在当前的临床试验指令2001/20/EC和 GCP指令2005/28/EC中执行。根据修订后的2001/20/EC号指令和2005/28/EC号指令,欧盟已通过成员国的国家立法实施了临床试验审批制度。根据这一制度,必须获得计划进行试验的国家主管部门的批准,如果要在多个成员国进行临床试验,则必须在多个成员国获得批准。为此,必须提交CTA,该文件必须有调查药品档案或IMPD,以及指令2001/20/EC和指令2005/28/EC以及其他适用的指南文件所规定的进一步支持信息。此外,只有在主管伦理委员会对该国的临床试验申请发表了积极的意见后,才能开始临床试验。
2022年1月31日,《临床试验条例》(EU)第536/2014号取代了现行的《临床试验指令2001/20/EC》。为确保临床试验规则在整个欧盟范围内保持一致,通过了《临床试验条例》(EU)第536/2014号,作为一项直接适用于所有欧盟成员国的条例。然而,《临床试验指令2001/20/EC》仍将在《临床试验条例》生效之日起三年内适用于(I)在申请前提交的临床试验申请,以及(Ii)如果发起人选择旧系统,在申请后一年内提交的临床试验申请。
第(Br)(EU)2014号法规旨在简化欧盟临床试验的审批流程。该条例的主要特点包括:
| ● | A 通过单个入口点简化申请程序,称为临床试验 信息系统; |
| ● | A 为申请准备和提交的一套文件以及简化文件 报告程序可避免赞助商提交大致相同的信息 分别向不同的国家主管部门通报; |
| ● | A 临床试验申请评估协调程序,分为 分两部分; |
| ● | 严格来说 确定临床试验申请评估的截止日期;以及 |
| ● | 伦理委员会根据国家《国家标准》参与评估程序 相关成员国的法律,但在法规规定的总体时间内 (EU)第536/2014号。 |
营销 授权
在欧盟成员国销售产品的授权是按照以下四种程序之一进行的:集中程序、相互承认程序、分散程序或国家程序。
| 30 |
集中式 流程
通过 集中程序,申请者可以根据单个 申请获得在所有欧盟成员国都有效的营销授权。某些医药产品,包括通过生物技术方法开发的产品,必须经过集中的上市授权程序,如果根据EMA的意见,如果得到欧盟委员会的批准,则 自动在所有欧盟成员国有效。赞助商可以选择通过其他类别的 产品的集中程序提交MAA。
集中程序对于某些类型的产品是强制性的,例如来自生物技术过程的药物,如基因工程 ,高级治疗药物,如基因疗法或组织工程药物,孤儿药物,以及含有用于治疗艾滋病毒、艾滋病、癌症、糖尿病、神经退行性疾病、自身免疫和其他免疫功能障碍以及病毒疾病的含有新活性物质的医药产品。
如果其他医药产品含有新的活性物质、申请人 证明相关医药产品构成了重大的治疗、科学或技术创新,或者批准的批准符合欧盟的公共利益,则可选择采用 集中授权程序。
管理 程序
根据集中程序,EMA的人类医药产品委员会,或CHMP作为科学委员会,代表EMA就人类使用的医药产品的安全性、有效性和质量提出意见。CHMP由每个成员国的国家药品主管部门提名的专家组成,其中一人被任命为评估协调工作的报告员,可能的话还会有另一名委员会成员担任联合报告员。批准后,报告员(S)继续对产品的整个生命周期进行监测。CHMP有210个有效天数来采纳关于是否应授予营销授权的意见 。在请求额外信息的情况下,该过程通常需要更长的时间,这会触发程序时间表中的时钟停止。这一过程很复杂,需要与成员国的监管当局和一些专家进行广泛的磋商。如果从公共卫生的角度,特别是从治疗创新的角度,就一种具有重大意义的药物提出上市许可申请,则申请人可根据第14(9)条(EC)第726/2004号条例,请求加快评估程序。如果CHMP接受这样的请求,210天的时限将减少到150天,但如果CHMP认为不再适合进行加速评估,也可以恢复到集中程序的标准时限 。一旦程序 完成,就会生成一份欧洲公共评估报告,简称EPAR。如果意见是否定的,则提供关于得出这一结论的理由的信息。CHMP意见通过后,关于MAA的决定必须由欧盟委员会在咨询欧盟成员国后 通过,总共可能需要60多天的时间。药品获得授权并投放市场后,必须对与其质量、安全性和有效性有关的所有方面进行 审查,这是维持上市授权的条件。
有条件的 审批
在特定情况下,欧盟立法(第14(7)条条例(EC)第726/2004号和第(EC)507/2006号条例)允许申请者在获得申请全面营销授权所需的全面临床数据之前获得有条件营销授权。如果 (1)产品的风险-效益平衡为正,(2)申请人很可能能够提供所需的全面临床试验数据,(3)产品满足未得到满足的医疗需求,以及(4)相关药物产品立即上市对公共健康的益处大于仍需要额外数据这一事实所固有的风险,则可对产品(包括被指定为孤儿药品的药品)授予此类有条件批准。有条件的营销授权 可能包含营销授权持有人必须履行的具体义务,包括完成正在进行的或新的研究的义务,以及收集药物警戒数据的义务。有条件营销授权的有效期为一年,如果风险-收益平衡保持为正,并且在评估是否需要额外的 或修改的条件和/或特定义务后,可以每年续签。上述集中程序的时间表也适用于CHMP对有条件营销授权申请的审查。
| 31 |
营销 特殊情况下的授权
根据第(Br)条第(8)款(EC)第726/2004号条例,在特殊情况下,申请人能够证明无法提供(由于立法中预见的特定原因)综合数据(符合经修订的2001/83/EC指令附件I中的要求)的产品可能有资格获得上市许可。此类授权每年都会进行审查 以重新评估风险与收益的平衡。在特殊情况下履行作为营销授权的一部分而施加的任何特定程序/义务旨在提供有关产品安全有效使用的信息,通常不会导致完成完整的档案/批准。
儿科研究
在欧盟获得营销授权之前,申请人必须证明符合EMA批准的涵盖所有儿科人群的儿科调查计划(PIP)中包括的所有措施,除非EMA已批准(1)特定产品的豁免、(2)类别豁免或(3)推迟PIP中包括的一项或多项措施。所有销售授权程序的各自要求在(EC)第1901/2006号条例,即所谓的儿科条例中规定。当公司希望为已获授权的药物添加新的适应症、药物形式或给药途径时,此要求 也适用。EMA的儿科委员会,或PDCO,可能会批准某些药物的延期,允许公司推迟儿童药物的开发,直到有足够的信息证明其对成人的有效性和安全性。当不需要或不适合在儿童中开发药物时,PDCO也可以批准豁免,例如只影响老年人口的疾病。
在提交营销授权申请或修改现有营销授权之前,EMA确定公司 确实遵守了每个相关PIP中列出的商定研究和措施。
授权和续订期限
营销授权原则上有效期为五年,营销授权可在五年后根据EMA或国家当局重新评估风险-收益平衡的基础续签。为此,营销授权书持有人必须在营销授权书失效前至少九个月向EMA或主管当局提供关于质量、安全性和有效性的文件的合并版本,包括自营销授权书颁发以来引入的所有变化。一旦续签,上市授权将在无限期内有效,除非欧盟委员会或 国家当局基于与药物警戒相关的正当理由决定继续进行一次为期五年的续签。 任何授权之后,如果没有在授权后三年内将药品实际投放到欧盟市场(如果是集中式程序)或授权成员国的 市场,则将不再有效,即所谓的“日落条款”。
孤儿 药品名称和排他性
欧盟委员会可以对赞助商可以确定用于诊断、预防或治疗 (1)危及生命或慢性衰弱的疾病,或(2)威胁生命、严重衰弱或严重慢性疾病的产品授予孤儿药品称号,如果没有激励措施,该药物在欧盟的销售不太可能产生足够的回报,以证明必要的投资是合理的。此外,赞助商必须证明欧盟没有批准的其他令人满意的诊断、预防或治疗这种疾病的方法,或者如果存在这样的方法,建议的孤儿药物将对患者有重大好处。
孤儿 药物指定提供许多好处,包括费用减免、监管协助和申请集中的欧盟营销授权(请参阅“-政府法规和产品审批-美国境外的监管-集中的授权程序”),以及在获得营销授权后10年的市场排他性。在此市场独占期内,EMA、欧盟委员会或成员国均不能接受申请或授予销售授权 含有与授权孤儿药品相似的一种或多种活性物质的药品,以及用于相同治疗适应症的药品。如果在第五年结束时确定不再符合孤儿药物指定标准,则授权治疗适应症的市场专营期可缩短至六年,包括证明该产品具有足够的利润而不足以证明维持市场专有性是合理的。此外,在市场独占期结束之前,竞争对手的类似药品可以获得授权,包括如果 被证明比已经批准的孤儿药物更安全、更有效或在临床上更好,或者如果已经批准的孤儿药物的上市授权持有人无法供应足够数量的产品。
| 32 |
如果被指定为孤儿药物的医药产品的MAA包括按照商定的PIP进行的所有研究的结果,并且随后在授予的上市授权中包含相应的声明,则十年的市场专营期将延长至十二年。
监管数据保护
欧盟立法还规定了监管数据和市场排他性制度。在获得营销授权后,基于完整独立数据包获得批准的新化学品 实体将受益于八年的数据独占权和额外的两年市场独占权。数据排他性使欧盟的监管机构无法参考创新者的数据来评估通用或生物相似(缩写)应用。在额外的两年市场独占期内,可以提交仿制药或生物相似药的上市授权,并可以参考创新者的数据,但在市场独家经营权到期之前,不能销售仿制药或生物相似药 。如果在这十年的前八年中,营销授权持有人或MAH获得了一个或多个新的治疗适应症的授权,那么整个十年的期限将延长到最多11年 ,在授权之前的科学评估中,这些适应症被认为与现有疗法相比可以带来显著的临床 益处。即使一种化合物被认为是一种新的化学实体,创新者能够 获得数据独占期,但另一家公司也可以销售该药物的另一版本,前提是该公司基于具有药物试验、临床前试验和临床试验的完整独立数据包的MAA获得 营销授权。但是,被指定为孤儿药品的产品在获得上市许可后,享有10年的孤儿市场专营权(另见“项目4.B--政府法规和产品审批--欧盟的法规和营销--孤儿药品的指定和专营权”)。根据欧盟营销授权流程的时间和持续时间,产品可能有资格获得最多五年的补充保护证书,或 SPC。这种SPC延长了该药物基本专利下的权利。
获得营销授权后的监管要求
如果 我们在欧盟获得医药产品的授权,我们将被要求遵守适用于医药产品的制造、营销、促销和销售的一系列要求:
药物警戒
例如,我们 将必须遵守欧盟严格的药物警戒或安全报告规则,根据这些规则,可以实施授权后研究和额外的监测义务。
其他 要求涉及,例如,按照良好制造规范标准制造产品和原料药。 欧盟监管机构可能会进行检查,以验证我们是否符合适用的要求,我们将不得不继续花费时间、金钱和精力来保持合规性。不遵守欧盟关于安全监测或药物警戒的要求,以及与为儿科人群开发产品相关的要求,也可能导致欧盟的重大经济处罚 。同样,不遵守欧盟关于保护个人数据的要求也可能导致重大处罚和制裁。如果我们不遵守当地适用的要求,个别欧盟成员国也可能实施各种制裁和处罚。
| 33 |
制造业
必须持有单独的制造商许可证的授权药品的生产必须遵守EMA的cGMP要求和其他国家主管部门的类似要求,这些要求规定了药品生产、加工和包装过程中使用的方法、设施和控制措施,以确保其安全性和身份。EMA通过强制登记设施和检查这些设施来执行其cGMP要求。EMA可能对这些检查起到协调作用 ,而执行这些检查的责任则由制造商所属的成员国主管当局负责。不遵守这些要求可能会中断供应并导致延误、意外成本和收入损失, 并可能使申请者面临潜在的法律或监管行动,包括但不限于警告信、暂停生产、扣押产品、禁令行动或可能的民事和刑事处罚。
营销 和促销
欧盟严格监管授权药品的营销和推广,包括行业赞助的继续医学教育和面向药品处方者和/或普通公众的广告。适用法规旨在确保营销授权持有者提供的有关其产品的信息 真实、平衡,并准确反映EMA或授权成员国的国家当局授权的安全性和功效声明。不遵守这些要求 可能会导致负面宣传、警告信、改正广告以及潜在的民事和刑事处罚。
其他美国医保法和合规性要求
对于在美国分销的产品,我们还将接受联邦 政府和我们开展业务的州的额外医疗法规和执法。适用的联邦和州医疗法律法规包括 以下内容:
| ● | 联邦医疗保健反回扣法规禁止,除其他外,个人故意 并故意直接或间接索取、提供、接受或提供报酬, 以现金或实物,诱导或奖励个人转介,或 购买、订购或推荐任何商品或服务,并可付款 根据联邦医疗保健计划,如医疗保险,医疗补助或其他政府计划。 个人或实体无需实际了解联邦反回扣法规 或有特定意图违反,即已违反;此外,物品或服务 因违反联邦反回扣法规而导致的,可能构成虚假或 为《虚假索赔法》的目的而进行的欺诈性索赔; |
| ● | 《患者转诊伦理法案》,通常称为《斯塔克法》及其相应的法规,禁止医生将根据Medicare或Medicaid计划报销的指定医疗服务(包括门诊药物)转介给医生或其直系亲属与其有经济关系或所有权利益的实体,但狭隘的监管例外情况除外,并禁止这些实体就向转介受益人提供的物品或服务付款向Medicare或Medicaid提出索赔 ; |
| ● | 联邦虚假索赔法案对故意提供或导致提交的个人或实体施加刑事和民事处罚,包括民事举报人或准诉讼,向联邦政府提出虚假或欺诈性的付款要求,或作出虚假陈述以逃避、减少或隐瞒向联邦政府付款的义务 ; |
| ● | 健康 《1996年保险流通和责任法》规定了刑事和民事责任 执行欺诈任何医疗福利计划的计划,并施加义务, 包括关于保护隐私、安全性的强制性合同条款 以及个人可识别健康信息的传输。该法规还禁止 故意伪造、隐瞒或掩盖重要事实或作出任何 与医疗福利的交付或支付有关的重大虚假陈述, 项目或服务; |
| 34 |
| ● | 联邦虚假陈述法规禁止故意伪造、隐瞒 或掩盖重要事实或作出任何重大虚假陈述 提供或支付医疗福利、物品或服务; |
| ● | 根据ACA制定的《医师支付阳光法案》及其实施条例, 要求指定的药品、器械、生物制品和医疗用品制造商 可根据Medicare、Medicaid或儿童健康保险支付 计划,除特定例外情况外,每年向医疗保险和医疗补助中心报告 服务或CMS,与付款或其他"价值转移"相关的信息 给医生做的。所有这类报告的资料均公开; |
| ● | 类似的州法律法规,如州反回扣和虚假索赔法,可能适用于涉及非政府第三方付款人(包括私营保险公司)报销的医疗项目或服务的销售或营销安排和索赔。一些州法律要求制药公司遵守制药行业的自愿合规指南和联邦政府颁布的相关合规指南。 |
由于这些法律的广度以及法定例外和可用避风港的狭隘,我们 未来的一些商业活动可能会受到一项或多项此类法律的挑战。努力确保我们与第三方的业务安排符合适用的法律和法规,这将涉及大量成本。政府 当局可能会得出结论,我们的业务实践可能不符合当前或未来涉及 适用的欺诈和滥用或其他医疗保健法律和法规的法律、法规或判例法。如果我们的运营被发现违反了这些 法律或任何其他可能适用于我们的政府法规,我们可能会受到重大的民事、刑事和行政 处罚、损害赔偿、罚款、被排除在政府资助的医疗保健计划(如Medicare和Medicaid)之外,以及削减或 重组我们的运营。如果我们预期与之开展业务的任何医生或其他提供者或实体被发现 不符合适用法律,他们可能会受到刑事、民事或行政制裁,包括将 排除在政府资助的医疗保健计划之外。
报销
我们候选产品在美国的销售额 可能在一定程度上取决于第三方付款人支付候选产品成本的程度,如政府医疗计划、商业保险和管理的医疗保健组织。这些第三方付款人对医疗产品和服务的收费提出了越来越多的挑战。此外,控制医疗成本已成为联邦和州政府的优先事项,药品价格一直是这一努力的重点。美国政府、州立法机构和外国政府对实施成本控制计划表现出了极大的兴趣,包括价格控制、限制报销和非专利产品的替代要求。采用 价格控制和成本控制措施,以及在现有控制和措施的司法管辖区采用更严格的政策, 可能会进一步限制我们的净收入和业绩。如果这些第三方付款人不认为我们的候选产品与其他可用的疗法相比具有成本效益 ,他们可能不会在批准后将我们的候选产品作为其计划下的一项福利覆盖,或者,如果他们这样认为,付款水平可能不足以让我们在有利可图的基础上销售我们的候选产品。
为了确保可能被批准销售的任何产品的承保范围和报销,我们可能需要进行昂贵的药物经济学研究,以证明该产品的医疗必要性和成本效益,以及获得FDA、EMA或其他类似监管批准所需的成本。我们的候选产品可能不被视为具有医疗必要性或成本效益。 付款人决定为药品提供保险并不意味着将批准适当的报销率。 第三方报销可能不足以使我们能够将价格保持在足够高的水平,以实现产品开发投资的适当回报。
| 35 |
定价 和报销方案因国家/地区而异。一些国家规定,只有在商定了报销价格之后,才能销售药品。一些国家可能要求完成额外的研究,将特定候选产品的成本效益与目前可用的疗法进行比较。进行此类研究的成本可能很高,并导致我们的商业化工作延迟 。欧盟为其成员国提供了多种选择,以限制其国家医疗保险制度提供报销的药品范围,并控制供人使用的医疗产品的价格。欧盟成员国 可以批准药品的具体价格,或者转而对将药品投放市场的公司的盈利能力采取直接或间接控制制度。其他成员国允许公司自行确定药品价格,但监督和控制公司利润。总体上,医疗成本,特别是处方药的下行压力变得非常大。因此,对新产品的进入设置了越来越高的壁垒。此外,在某些国家/地区,来自低价市场的跨境进口产品会带来竞争压力,可能会降低一国国内的定价水平。不能保证 任何对药品实行价格控制或报销限制的国家/地区都会允许对我们的任何产品进行优惠的报销和定价安排。
如果政府和第三方付款人未能提供足够的保险和报销,我们获得监管部门批准用于商业销售的任何产品的适销性可能会受到影响。此外,美国对管理式医疗的重视程度也有所提高 ,我们预计这将继续增加药品定价的压力。承保政策、第三方报销费率和药品定价规则可能随时更改。 即使我们获得监管部门批准的一个或多个产品获得了有利的承保范围和报销状态,未来也可能会实施不太有利的承保政策和报销费率。
医疗保健 改革
在美国,已经并将继续有许多重要的立法举措来控制医疗成本。ACA于2010年3月在美国颁布,其中包含的条款可能会降低药品的盈利能力,包括 例如,增加受Medicaid药品返点计划约束的药品的返点、将Medicaid返点扩大到Medicaid管理的护理计划、对某些Medicare Part D受益人强制折扣,以及根据制药公司在联邦医疗保健计划的销售中所占份额 计算的年费。
此外,自《ACA》颁布以来,还提出并通过了其他立法修改。这些变化包括从2013年4月1日起将向提供商支付的医疗保险总金额削减 每财年2%,由于随后对法规进行的立法修订,这些削减将持续到2027年,除非国会采取额外行动;然而,根据CARE法案和随后的 立法,由于新冠肺炎疫情,这些削减将从2020年5月1日起暂停至2022年3月31日。2013年1月,《2012年美国纳税人救济法》签署成为法律,其中包括减少了对几家医疗服务提供者的医疗保险支付, 并将政府向提供者追回多付款项的诉讼时效期限从三年延长至五年。这些 新法律可能会导致医疗保险和其他医疗保健资金的进一步减少,如果获得批准,这可能会对我们药品的 客户以及我们的财务运营产生实质性的不利影响。
此外, 最近,政府加强了对制造商为其商业产品定价的方式的审查。 美国国会最近进行了几次调查,提出并颁布了联邦和州立法,旨在提高药品定价的透明度,审查定价与制造商患者计划之间的关系,降低医疗保险下的药品成本,并改革政府计划的药品报销方法。FDA于2020年9月24日发布了最终规则,自2020年11月30日起生效,为各州制定和提交来自加拿大的药品进口计划提供了指导。此外,2020年11月20日,美国卫生与公众服务部(HHS)敲定了一项法规,将药品制造商对D部分下的计划赞助商的降价安全港保护 直接或通过药房福利经理删除,除非法律要求降价。该规则还为反映在销售点的降价创造了新的安全港,并为药房福利经理和制造商之间的某些固定费用安排创造了安全港。 在州一级,立法机构越来越多地通过立法并实施旨在控制药品和生物制品定价的法规,包括价格或患者报销限制、折扣、对某些产品准入的限制 以及营销成本披露和透明度措施,在某些情况下,旨在鼓励从其他国家进口和 批量购买。
| 36 |
最近,2022年8月16日,2022年《降低通货膨胀率法案》或《爱尔兰共和军》签署成为法律。除其他事项外,爱尔兰共和军指示 卫生与公众服务部或HHS协商医疗保险覆盖的某些高支出、单一来源的药物和生物制品的价格 。谈判价格将于2026年首次生效,将以法定最高价格为上限 ,这意味着相对于批发商和直接购买者的平均价格有很大折扣。从2023年开始,该法律还将惩罚那些以高于通货膨胀率的速度提高联邦医疗保险B部分和D部分药品价格的药品制造商。此外,从2025年开始,IRA通过显著降低受益人的最大自付成本并创建新的制造商折扣计划,消除了Medicare Part D计划下的“甜甜圈漏洞”。爱尔兰共和军允许卫生和公众服务部部长在最初几年通过指导而不是监管来实施许多这些规定。HHS已经并将继续在这些计划实施时发布和更新指导,尽管爱尔兰共和军可能会受到法律挑战。目前尚不清楚IRA 将如何实施,但可能会对制药行业产生重大影响此外,为了回应拜登政府2022年10月的行政命令,HHS于2023年2月14日发布了一份报告,概述了医疗保险和医疗补助创新中心 测试的三种新模型,这些模型将根据它们降低药品成本、促进可获得性和提高医疗质量的能力进行评估 。目前还不清楚这些模型是否会在未来的任何医疗改革措施中使用。
尽管其中许多措施以及其他拟议的措施可能需要通过额外的立法授权才能生效,但国会 已表示将继续寻求新的立法措施来控制药品成本。
CMS 发布了一项最终规则,于2019年7月9日生效,该规则要求处方药和生物 产品的直接面向消费者的广告(可通过或根据联邦医疗保险或医疗补助付款)在广告中包括该药物或生物制品的批发采购成本或标价,如果该药物或生物制品每月供应或通常疗程的成本等于或大于35美元。违反这些要求的处方药和生物制品将被列入公开名单。
任何 医疗改革措施如果获得批准,都可能减少对我们产品的最终需求,或给我们的产品定价带来压力。 美国各州也越来越积极地通过立法和实施旨在控制药品定价的法规,包括价格或患者报销限制、折扣、对某些产品的限制 准入和营销成本披露和透明度措施,在某些情况下,旨在鼓励从其他国家/地区进口和批量购买。此外,地区医疗当局和个别医院越来越多地使用招标程序 来确定哪些药品和供应商将被纳入其处方药和其他医疗保健计划。 我们预计未来将采取更多的州和联邦医疗改革措施。
我们 预计未来将采取更多的州和联邦医疗改革措施,以及外国政府的法律改革,其中任何一项都可能限制政府为医疗保健产品和服务支付的金额,这可能导致对我们候选产品的需求减少或额外的定价压力。
员工
截至本报告之日 ,我们有10名员工和全职顾问,包括我们的执行官,提供管理和 财务服务,并承担一般行政责任。我们相信,我们与 员工保持着令人满意的工作关系,我们没有遇到任何重大的劳资纠纷或在招聘员工方面遇到任何困难。 我们的员工都没有工会代表。
人力资源 资本资源
我们的人力资本资源目标包括识别、招聘、留住、激励和整合我们现有的 和新员工、顾问和顾问。我们的股权和现金激励计划的主要目的是通过授予基于股票和现金的薪酬奖励来吸引、留住和奖励人员,以通过激励这些人员尽其所能并实现我们的目标来增加股东价值和公司的成功。
| 37 |
可用信息
我们的 网站位于www.azitrainc.com。我们网站上的信息或通过我们网站访问的信息不属于本年度报告表格 10—K的一部分。本年度报告的表格10—K的副本位于美国证券交易委员会公共参考室,地址为100 F Street,NE,Washington,D.C. 20549。有关公共资料室运作的信息可致电SEC 1—800—SEC—0330获得。SEC还 在www.sec.gov上维护了一个网站,其中包含有关我们提交文件的报告和其他信息。
第 1a项。风险因素
投资我们的普通股涉及很高的风险。在购买我们的普通股之前,您应该仔细阅读和考虑以下风险因素以及本报告中包含的所有其他信息,包括我们的财务报表和相关说明。这些风险因素中的每一个,无论是单独存在还是合并在一起,都可能对我们的业务、经营业绩和财务状况产生不利影响, 也会对我们普通股的投资价值产生不利影响。可能还有我们目前不知道的或我们目前认为不重要的额外风险,这也可能损害我们的业务和财务状况。如果发生下面所述的任何事件,我们的财务状况、我们获得资本资源的能力、我们的经营业绩和/或我们未来的增长前景可能会受到重大和不利的影响 我们的普通股市场价格可能会下跌。因此,您可能会损失您对我们普通股的部分或全部投资。
与我们的业务相关的风险
我们是一家处于早期阶段的临床生物制药公司,运营历史有限.
我们 是一家成立于2014年1月2日的早期临床生物制药公司,运营历史有限。除了有限的赠款和服务收入外,我们尚未 开始创收业务。到目前为止,我们的业务包括开发我们的专有微生物文库,鉴定、表征、基因工程和某些细菌种类的测试 以提供治疗效果,以及我们最初的候选产品的开发。我们有限的运营历史使潜在投资者很难评估我们的技术或未来的运营。作为一家早期临床生物制药公司,我们 面临与新业务相关的组织、融资、支出、并发症和延误所固有的所有风险。因此,您应该根据 公司在发展早期经常遇到的成本、不确定性、延误和困难来考虑我们的前景,特别是像我们这样的早期临床生物制药公司。潜在投资者应仔细考虑一家经营历史有限的公司将面临的风险和不确定性。尤其是, 潜在投资者应考虑到我们可能无法:
| ● | 成功 实施或执行我们的业务计划,或我们的业务计划是健全的; | |
| ● | 成功 完成临床前和临床试验并获得监管部门的上市批准 我们的候选产品; | |
| ● | 成功地 展示了我们的精准皮肤病候选产品 与市场上当前的产品之间的良好差异化; | |
| ● | 成功 签约生产我们的临床药物产品并建立商业药物供应 ; | |
| ● | 确保我们的候选产品获得市场排他性或足够的知识产权保护; | |
| ● | 吸引并保留一支经验丰富的管理和咨询团队;以及 | |
| ● | 在资本市场筹集足够的资金来实施我们的业务计划,包括我们的候选产品的临床开发、监管批准和商业化。 |
投资者 应根据发展中公司在竞争环境中遇到的不确定性来评估对我们的投资。 不能保证我们的努力会成功,也不能保证我们最终能够实现盈利。如果我们不能成功 执行上述任何一项,我们的业务可能不会成功,您的投资将受到不利影响。您必须准备好 失去所有投资。
| 38 |
我们 有重大运营亏损的历史,并预计在可预见的未来将继续运营亏损.
截至2023年12月31日和2022年12月31日的财年,我们分别产生了1260万美元和1340万美元的净亏损。截至2023年12月31日,我们的累计赤字为4860万美元。我们预计,在没有任何有意义的 收入的情况下,我们将继续产生大量开支,除非我们能够获得监管部门的批准,并成功地将我们的至少一个候选产品商业化。 我们还认为,自本报告之日起,我们至少需要4到6年时间才能获得第一个候选药物的监管批准 ,前提是我们能够获得监管批准。即使我们能够将我们的候选产品商业化 ,也不能保证我们将产生可观的收入或实现盈利。
我们 预计在推进候选产品向商业化方向发展的过程中,将有大量的研究、监管和开发费用。 因此,我们预计在可预见的将来将产生重大损失,而且这些损失将不断增加。我们不确定 何时或是否能够实现或维持盈利能力。如果我们在未来实现盈利,我们可能无法维持 在以后的时期的盈利。未能实现并保持盈利可能会削弱我们维持运营的能力,并 对我们的业务和筹集资金的能力造成不利影响。如果我们无法在合理的时间内产生正现金流, 我们将无法进一步推进我们的业务计划或继续运营,在这种情况下,您可能会失去全部投资。
我们的独立注册会计师事务所 截至2023年12月31日止年度的报告指出,由于我们的累计赤字、 经常性和来自运营的负现金流,我们的持续经营能力存在重大疑问。
我们 预计我们将需要额外的资金来执行我们的业务计划和为运营提供资金,而这些额外的资金可能无法以合理的条款或根本无法获得。
截至2023年12月31日,我们的总资产为510万美元,营运资金为932,384美元。2024年2月16日,我们完成了16,667,000股普通股的公开发行,发行价为每股0.30美元,扣除承销商折扣和发行费用后,我们收到了约440万美元的净收益。我们认为,截至本报告日期,我们的手头现金 将不足以支付我们未来12个月的拟议运营计划。我们打算通过各种 融资来源寻求额外资金,包括出售我们的股权、技术的许可费以及与行业合作伙伴的合资企业。 此外,我们还将考虑现有业务计划的替代方案,以使我们能够以较少的资金实现创收业务和 有意义的商业成功。但是,无法保证此类资金 将以商业上合理的条款提供(如果有的话)。如果无法以令人满意的条件获得此类融资,我们可能无法进一步执行 我们的业务计划,我们可能无法继续运营,在这种情况下,您可能会失去全部投资。
我们的微生物文库和基因工程平台的临床和商业用途是不确定的,而且可能永远不会实现。
我们 构建了一个专有平台,其中包括一个微生物文库,该文库由大约1,500个独特的细菌菌株组成,可以 筛选独特的治疗特征。该平台通过人工智能、机器学习和遗传工程技术进行了扩展。到目前为止,我们的重点是开发基因工程菌株表皮葡萄球菌,我们认为它是皮肤病治疗工程的最佳治疗候选物种。然而,我们认为,基因工程表皮葡萄球菌是一种新的、未经证实的治疗模式我们最近启动了ATR—12的Ib期临床试验,预计在2024年上半年入组第一名患者,并预计在 2024年年中之前为ATR—04的Ib期临床试验提交IND。然而,截至本报告之日,我们已经检测和评估了我们的专利菌株, 表皮葡萄球菌在临床前 研究中,尚未进行任何旨在评估安全性、耐受性或疗效的临床试验。此外,早期临床试验的成功并不能保证大规模临床试验的成功,也不能预测最终结果。即使 我们拟定的1b期临床试验完成后,我们的初始候选产品也只能在少数患者中进行测试。 这些临床试验的结果可能不一定表明我们候选产品的安全性、耐受性或疗效 或我们扩展到更大的临床试验。在此之前(如果有的话),由于我们能够向FDA提供大量临床证据,以支持安全性、有效性、纯度和效价的声明,足以使FDA批准我们的专利候选产品 用于任何适应症,我们的专利微生物库和基因工程平台仍将未经证实。
| 39 |
我们的候选产品处于早期临床试验或临床前开发的早期阶段,因此它们将需要广泛的临床前和临床测试。临床前研究或早期临床试验的成功可能不代表未来临床试验的结果 ,我们不能向您保证任何正在进行的、计划中的或未来的临床试验将导致足够的结果 以获得必要的监管批准。
Because our product candidates are in early stages clinical trials or of preclinical development, they will require extensive preclinical and clinical testing. We recently initiated our Phase 1b clinical trial for ATR-12 and expect to enroll our first patient in the first half of 2024, and expect to file an IND for a Phase 1b clinical trial of ATR-04 by mid-2024. However, we have not conducted meaningful preclinical studies for any of our other product candidates. Success in preclinical testing and early-stage clinical trials does not ensure that later clinical trials will generate the same results or otherwise provide adequate data to demonstrate the efficacy and safety of a product candidate. Preclinical studies and Phase 1b clinical trials are primarily designed to test safety, to study pharmacokinetics and pharmacodynamics and to understand the side effects of product candidates at various doses and schedules. Phase 1b clinical trials also test how well a certain disease responds to a new treatment. Success in preclinical studies and earlier clinical trials does not ensure that later efficacy trials will be successful, nor does it predict final results. Our product candidates may fail to show the desired safety and efficacy in clinical development despite positive results in preclinical studies or even if they successfully advance through earlier clinical trials.
此外,临床试验的设计可以确定其结果是否支持产品的批准,而临床试验设计中的缺陷可能在临床试验进展良好之前不会变得明显。作为一个组织,我们在设计临床试验方面的经验有限,可能无法设计和执行临床试验来支持监管部门的批准。制药和生物技术行业的许多公司都遭遇了重大挫折,包括在临床前测试和早期临床试验取得了令人振奋的结果后,仍未能在后期临床试验中取得成功。从临床前和临床活动中获得的数据可能会受到不同的解释,这可能会推迟、限制或阻止监管部门的批准。
此外, 我们无法确定是否或何时可能为我们的任何候选产品提交生物制品许可证申请或BLA以供监管部门批准,或任何此类BLA是否会被FDA接受审查,或任何BLA是否会在 审查后获得批准。即使我们的临床试验按计划完成,我们也不能确定他们的结果是否支持我们建议的适应症。 临床前试验和早期临床试验的成功并不能确保以后的临床试验也会成功,我们也不能 确保以后的临床试验的结果将复制以前的临床试验和临床前试验的结果。临床 试验过程可能无法证明我们的候选产品对于其建议的用途是安全有效的。此失败可能会 导致我们放弃某个候选产品,并可能延迟其他候选产品的开发。我们临床试验的任何延迟或终止都将延迟并可能阻止向FDA提交任何BLAS,并最终影响我们将候选产品商业化并创造产品收入的能力。
我们 将需要扩大我们组织的规模,我们在管理这种增长时可能会遇到困难.
随着我们的开发和商业化计划和战略的发展,我们将需要扩大员工和顾问/承包商的规模 。未来的增长将使管理层成员承担更多的责任,包括需要确定、招聘、维持、激励和整合更多的员工。此外,我们的管理层可能不得不将不成比例的 注意力从我们的日常活动中转移出来,并投入大量时间来管理这些增长活动。我们未来的财务业绩、我们将我们的候选产品和任何其他候选产品商业化的能力以及我们有效竞争的能力 在一定程度上将取决于我们有效管理未来增长的能力。
| 40 |
如果我们不能成功地吸引和留住高素质的人才,我们就可能无法成功地实施我们的业务 战略。此外,失去高级管理层的服务将对我们的业务前景造成不利影响.
我们的管理团队在药物开发和商业化的许多不同方面拥有专业知识。然而,我们在竞争激烈的制药行业中的竞争能力在很大程度上取决于我们能否吸引和留住高素质的管理、科学和医疗人员。当我们进一步开发我们的候选产品时,我们将需要招聘更多的人员。我们市场对技术人员的竞争非常激烈,而对经验丰富的科学家的竞争可能会限制我们以可接受的条件聘用和留住高素质人员的能力。尽管我们努力留住有价值的员工,但我们的管理、科学和医疗团队的成员可能会在短时间内终止与我们的雇佣关系。失去任何首席执行官或其他关键员工的服务,或我们无法聘用目标高管,可能会潜在地损害我们的业务、经营业绩或财务状况。 我们尤其认为,失去首席执行官的服务将对我们的业务产生实质性的不利影响。
与我们竞争人才的其他生物制药公司比我们拥有更多的财务和其他资源,不同的风险 概况,以及更长的行业历史。它们还可以提供更多样化的机会和更好的职业晋升机会 。这些特点中的一些可能比我们所提供的更吸引高素质的应聘者。如果我们不能 继续吸引和留住高素质的人才,我们开发和商业化候选产品的速度和成功将受到限制。
我们 目前没有销售和营销组织。如果我们无法建立令人满意的销售和营销能力,或无法获得第三方销售和营销关系,我们可能无法成功地将我们的任何候选产品商业化.
目前,我们没有销售或营销人员。在我们的一个或多个药物产品初步获得必要的监管批准后,我们计划在美国建立专注于将我们的开发计划商业化的能力, 我们的开发计划侧重于用于治疗皮肤病的活性生物治疗产品和重组蛋白,我们相信,在那里,我们目标适应症的患者群体 和医学专家足够集中,使我们能够通过有针对性的销售团队有效地推广我们的产品 。在商业化可能对我们来说资本效率较低的其他市场,我们可能会有选择地寻求与第三方的战略合作,以最大限度地发挥我们候选产品的商业潜力。在某些情况下,我们 可能会寻求我们的微生物文库或专利权的许可,或者达成联合开发安排。如果我们在招聘销售和营销人员、建立销售和营销基础设施或与第三方达成适当的合作安排方面不成功 ,我们将很难成功地将我们的候选产品商业化,这将对我们的业务、运营业绩和财务状况产生不利影响。
即使我们签订了第三方营销和分销安排,我们对这些第三方的销售、营销和分销活动的控制也可能是有限的,甚至没有控制权。我们未来的收入可能在很大程度上取决于这些第三方努力的成功。 在建立销售和营销基础设施方面,我们将不得不与资金雄厚的老牌制药公司和生物技术公司竞争,以招聘、聘用、培训和留住销售和营销人员。可能阻碍我们 建立内部销售组织或与第三方达成协作安排的因素包括:
| ● | 我们无法招聘和留住足够数量的有效销售和营销人员; | |
| ● | 销售人员无法接触或说服足够数量的医生开出我们的任何候选产品; | |
| ● | 销售人员缺乏可供补充的产品,这可能使我们相对于拥有更广泛产品线的公司处于竞争劣势;以及 | |
| ● | 与创建内部销售和营销组织相关的不可预见的成本和费用。 |
| 41 |
在可预见的将来,我们 将完全依赖第三方生产我们的候选产品用于商业销售, 如果这些第三方无法获得FDA或类似的外国监管机构的生产批准、无法向我们提供足够数量的候选产品或无法以可接受的质量水平或价格进行商业化,我们候选产品的商业化可能会被暂停、推迟或利润下降.
我们 不拥有或运营用于商业生产当前候选产品的制造设施。我们目前依赖 第三方合同制造商提供我们临床前研究和临床试验所需的所有原材料、制造设备和活性药物成分 。尽管我们能够在我们的康涅狄格州格罗顿工厂为我们的临床试验 生产成品,但我们将依赖第三方生产我们用于商业销售的成品。我们没有与这些第三方中的任何一方签订长期协议。我们目前也没有任何商业用品的合同关系。 我们打算与第三方合同制造商和一个或多个备份制造商达成协议,以供未来生产。 我们正在分析为我们开发的任何产品的未来开发和商业批量建立制造能力的可行性 。此类产品将需要在符合FDA和我们正在寻求批准的其他司法管辖区监管机构的要求的设施和工艺中生产。同时,如果我们的任何候选产品被批准商业化,我们将有义务依赖合同制造商进行临床前研究、临床试验和商业生产。
The facilities used by us or any contract manufacturer to manufacture our raw materials, manufacturing devices, active pharmaceutical ingredients and finished products must be approved by the FDA or comparable foreign regulatory authorities. Such approvals are subject to inspections that will be conducted after we submit a BLA to the FDA or their equivalents to other relevant regulatory authorities. Until such time, if ever, as we establish our own manufacturing facilities, we will not control the manufacturing process of our product candidates and will be completely dependent on our contract manufacturing partners for compliance with Current Good Manufacturing Practices, or cGMPs, for manufacture of our raw materials, manufacturing devices, active pharmaceutical ingredients and finished products. These cGMP regulations cover all aspects of the manufacturing, testing, quality control, storage, distribution and record keeping relating to our product candidates. If our contract manufacturers do not successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or others, we will not be able to secure or maintain regulatory approval for product made at their manufacturing facilities. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which could significantly delay our clinical trials and impact our ability to develop, manufacture, obtain regulatory approval for or market our product candidates, if approved. Likewise, we could be negatively impacted if any of our contract manufacturers elect to discontinue their business relationship with us.
我们的合同制造商将接受FDA以及相应的州和外国机构的持续定期突击检查,以了解是否符合cGMP和类似的法规要求。我们无法控制我们的合同制造商遵守这些法规和标准的情况。如果我们的任何合同制造商未能遵守适用的法规,可能会导致对我们实施制裁,包括罚款、禁令、民事处罚、未能批准我们的任何产品上市 候选产品、延迟、暂停或撤回批准、无法提供产品、运营限制和刑事起诉, 任何这些都可能对我们的业务产生重大不利影响。此外,我们将无法控制我们的合同制造商保持足够的质量控制、质量保证和合格人员的能力。如果我们的合同制造商未能遵守或保持这些标准中的任何一项,可能会对我们开发、制造、获得监管机构批准或营销我们的任何候选产品的能力产生不利影响。
如果, 由于任何原因,这些第三方不能或不愿意履行我们的职责,我们可能无法找到替代制造商或配方商,也无法与他们达成有利的协议,我们不能确定任何此类第三方是否具有满足未来要求的制造能力 。如果这些制造商或任何成品药品替代制造商在各自的生产流程中遇到我们所需的原材料、制造设备、活性药物成分或成品的任何重大 困难,或者因任何原因而停止与我们的业务往来,我们的临床试验可能会出现重大延误 ,我们的任何候选产品的供应可能会出现重大中断,或者可能根本无法提供我们的候选产品 。
| 42 |
任何制造问题或失去一家代工制造商都可能对我们的运营造成中断,并导致开发和临床试验延迟和销售损失。此外,我们将依赖第三方提供生产我们的产品所需的原材料 候选产品。任何这种对供应商的依赖都可能涉及几个风险,包括可能无法获得关键材料,以及对生产成本、交付时间表、可靠性和质量的控制减少。供应商问题对我们的合同制造商之一的运营造成的任何意外中断都可能延迟我们的任何候选产品的发货,增加我们销售商品的成本,并导致临床试验延迟或销售损失。
我们的 业务模式包括可能将我们专有微生物文库或我们的候选产品的菌株授予其他 生物制药公司,然而生物制药行业的技术许可是一个漫长的过程,并受到几个我们无法控制的 风险和因素的影响,我们无法预测我们能否成功地超越我们的技术许可或建立新的许可关系所需的时间 。
我们的商业模式包括将我们的专有微生物文库或我们的候选产品 中的菌株潜在地外包或联合开发给其他生物制药公司。任何此类安排通常都会从我们的潜在合作伙伴或被许可方进行初步可行性测试和评估开始。假设可行性测试成功,我们将成功的测试转换为商业许可或联合开发协议的能力取决于许多风险和因素,其中许多风险和因素不在我们的控制范围之内,包括:
| ● | 制药行业成员普遍采用和采用新技术的速度; | |
| ● | 我们的 潜在被许可人对销售皮肤科产品的经济效益的内部评估 可能与目前正在开发或商用的其他产品竞争的产品 由我们的潜在合作伙伴或被许可人进行销售,而不管他们认为的好处或优势如何 我们的技术或产品; | |
| ● | 我们潜在合作伙伴/被许可方的内部预算和产品开发问题,包括他们将资本和人力资源投入到我们的技术或产品的开发和商业化的能力;以及 | |
| ● | 我们的潜在合作伙伴/被许可方是否愿意接受我们对预付费用和持续版税的要求。 |
此外,我们认为,在许多情况下,我们的潜在合作伙伴或被许可方可能会与我们一起进行早期可行性测试,作为他们评估多种药物和药物输送方案的一部分,并在做出任何决定或承诺开发新药产品之前 。因此,即使我们的平台在早期可行性研究中成功,我们的潜在合作伙伴/被许可人 可能会出于与我们的技术性能无关的原因而决定不与我们签订许可协议。因此,我们 无法预测我们提议的许可模式将在多大程度上成功。
如果对我们提起产品责任诉讼,我们可能会承担大量责任,并可能被要求限制我们候选产品的商业化 .
由于我们候选产品的临床测试,我们 将面临潜在的产品责任风险,如果我们将任何候选产品商业化,我们还将面临更大的此类责任风险。例如,如果我们开发的任何产品(包括我们的任何候选产品)或我们在候选产品中使用的任何材料涉嫌造成伤害或在产品测试、制造、营销或销售过程中被发现 不适合,我们可能会被起诉。任何此类产品责任索赔可能包括对制造缺陷、设计缺陷、未能就产品固有危险发出警告、疏忽、严格责任和违反保修的指控。在美国,根据州消费者保护法,也可以对我们提出索赔。如果我们不能成功地针对产品责任索赔进行辩护,我们可能会承担巨额责任或被要求限制我们的候选产品的商业化 。即使成功地对这些索赔进行辩护,也需要我们使用大量的财务和管理资源。 无论案情如何或最终结果如何,责任索赔可能会导致:
| ● | 减少了对我们的任何候选产品或我们可能开发的任何未来产品的需求; |
| 43 |
| ● | 损害我们的声誉。 | |
| ● | 未能获得监管部门对我们的候选产品的批准; | |
| ● | 退出我们临床试验的参与者 ; | |
| ● | 与我方相关诉讼辩护相关的费用 ; | |
| ● | 转移我们管理层的时间和资源; | |
| ● | 向试验参与者或患者发放巨额 金钱奖励; | |
| ● | 产品 召回、撤回或贴标签、营销或促销限制; | |
| ● | 无法将我们的部分或全部候选产品商业化;以及 | |
| ● | 我们股票的价值下跌了。 |
截至本报告发布之日,我们投保的产品责任保险足以满足我们目前的临床测试和开发水平 。但是,在我们开始商业销售我们的初始产品时,我们将需要额外的产品责任保险。 我们无法以可接受的成本获得并保留足够的产品责任保险,以防范潜在的产品责任索赔。 我们开发的产品可能会阻止或阻碍其商业化。尽管我们将努力获得并维持我们认为足够的此类保险金额,但任何针对我们提出的索赔都可能导致法院判决或和解的金额不在我们的保险范围内,或者超出我们的保险范围。 我们的保险单也将有各种排除,我们可能会受到我们没有保险范围的产品责任索赔的影响。 因此,我们可能需要支付超出我们的承保范围限制或 不在我们的保险范围内的任何由法院裁决或在和解协议中协商的任何金额,并且我们可能没有或能够获得足够的资本来支付这些金额。
我们的内部计算机系统,或我们的协作者或其他承包商或顾问的系统,可能会出现故障或遭遇安全漏洞,这 可能会导致我们的产品开发计划发生实质性中断。
我们的内部计算机系统以及我们当前和未来的任何合作者和其他承包商或顾问的计算机系统容易受到计算机病毒、未经授权的访问、自然灾害、恐怖主义、战争以及电信和电气故障的破坏。虽然我们迄今尚未经历任何此类重大系统故障、事故或安全漏洞,但如果发生此类事件并导致我们的运营中断,则可能导致我们的开发计划和业务运营中断,无论是由于我们的商业机密或其他专有信息的丢失,还是由于其他类似的中断。例如,临床试验数据的丢失可能会导致我们的监管审批工作延迟,并显著增加我们恢复或复制数据的成本。如果任何中断或安全漏洞 导致我们的数据或应用程序丢失或损坏,或不适当地披露机密或专有信息,我们可能会承担责任,我们的竞争地位可能会受到损害,我们候选产品的进一步开发和商业化可能会推迟 。
我们 可能会因公司和供应商的信息系统和网络中维护的信息(包括员工和研究对象的个人信息以及公司和供应商的机密数据)被盗用、误用、泄露、篡改或故意或意外泄露或丢失而面临风险。此外,外部各方可能试图侵入我们的系统或我们供应商的系统,或欺诈性地诱使我们的人员或我们供应商的人员披露敏感信息,以便 访问我们的数据和/或系统。我们的数据和系统可能会受到威胁,包括恶意代码和病毒、网络钓鱼和其他网络攻击。随着时间的推移,这些威胁的数量和复杂性不断增加。如果我们的信息技术系统或供应商的信息技术系统发生重大破坏或数据意外或故意丢失,可能会损害市场对我们安全措施有效性的看法 并可能损害我们的声誉和信誉。我们可能需要 花费大量资金和其他资源来修复或更换信息系统或网络。此外,我们可能会 受到个人和团体在涉及隐私问题的私人诉讼中采取的监管行动和/或索赔,这些诉讼涉及与数据收集和使用实践和其他数据隐私法律法规相关的隐私问题,包括对数据的滥用或不当披露的索赔,以及不公平或欺骗性的做法。
尽管 我们开发和维护旨在防止这些事件发生的系统和控制,并且我们有识别和缓解威胁的流程 ,但这些系统、控制和流程的开发和维护成本高昂,并且需要持续监控和更新 ,因为技术变化和克服安全措施的努力变得越来越复杂。此外,尽管我们做出了努力,但这些事件发生的可能性无法完全消除。随着我们将更多的信息系统外包给供应商,与付款人和患者进行更多的电子交易,以及更多地依赖基于云的信息系统,相关的安全风险 将会增加,我们将需要花费更多的资源来保护我们的技术和信息系统。此外, 不能保证我们的内部信息技术系统或我们第三方承包商的系统,或我们的顾问为实施足够的安全和控制措施所做的努力 是否足以在系统故障时保护我们免受故障、服务中断、数据恶化或丢失,或者在发生网络攻击、安全漏洞、工业间谍攻击或可能导致财务、法律、商业或声誉损害的内部威胁攻击时防止数据被盗或损坏。
| 44 |
我们 面临其他生物技术和制药公司针对医用皮肤病适应症的激烈竞争,如果我们不能有效竞争,我们的经营业绩将受到影响。
皮肤病治疗市场竞争激烈,并受到重大技术发展的引领。我们预计,如果我们 成功获得监管部门对我们的候选药物的批准,我们将面临来自我们行业将推出的其他批准疗法或 药物的激烈竞争。即使其他品牌、仿制药或非处方药的效果较差,但基于成本或便利性,它可能会比我们的产品更快地被医生和患者采用。
与产品监管相关的风险
我们的成功完全取决于我们能否获得FDA和外国司法管辖区监管机构对我们候选产品的上市批准,我们打算在这些司法管辖区营销我们的候选产品,这一点无法得到保证。
我们 在获得FDA批准 BLA之前,不允许在美国将候选产品作为处方药产品销售,也不允许在任何外国国家销售,直到我们获得这些国家的必要批准。在美国, FDA通常要求完成每种生物制品的临床试验,以确定其安全性和有效性,以及广泛的药物开发 ,以确保BLA获得批准之前的质量。在大量正在开发的生物制剂中,只有一小部分 导致向FDA提交BLA,最终获得商业化批准的产品甚至更少。截至本报告之日, 我们尚未向FDA提交BLA或向其他监管机构提交任何候选产品的类似申请。
我们的 成功取决于我们是否获得上述监管批准,而此类监管批准的发放是不确定的 ,并受到许多风险的影响,包括:
| ● | 此类 当局可能不同意我们的临床试验或任何合作者的临床试验的数量、设计、规模、实施或实施; | |
| ● | 此类 当局可能不同意我们对临床前研究或临床 试验数据的解释,或使用研究结果作为我们当前或未来候选产品的前兆 ; | |
| ● | 毒理学研究结果可能不支持为我们的候选产品提交研究新药申请、 或IND或BLA; | |
| ● | FDA或类似的外国监管机构或机构审查委员会 可能不同意我们临床试验的设计或实施; | |
| ● | 我们 可能无法为我们的候选产品的安全性和 有效性提供可接受的证据; | |
| ● | 我们临床试验的 结果可能不令人满意,或者可能不符合FDA、欧洲药品管理局或EMA或其他监管机构为我们的任何候选产品获得上市批准所要求的统计 水平或临床意义; | |
| ● | 我们的候选产品在特定临床试验中的剂量可能不是最佳的 水平; | |
| ● | 我们临床试验中的患者 可能会出现不良反应,原因可能与我们的候选产品有关,也可能不相关。 | |
| ● | 从临床试验收集的数据可能不足以支持提交BLA或其他提交或获得美国或其他地方的监管批准; | |
| ● | FDA可能要求制定风险评估和缓解策略,或REMS,作为批准的条件; |
| 45 |
| ● | FDA或类似的外国监管机构可能无法批准与我们签订临床和商业用品合同的第三方制造商的制造工艺或设施 ;以及 | |
| ● | FDA或类似的外国监管机构的批准政策或法规 可能会发生重大变化,导致我们的临床数据不足以批准我们的候选产品 。 |
获得监管部门批准的过程非常昂贵,通常需要多年时间(如果最终获得批准的话),而且可能会根据涉及的候选产品的类型、复杂性和新颖性、寻求监管部门批准的司法管辖区以及监管机构的重大自由裁量权等因素而有很大差异。开发期间监管审批政策的变化、附加法规或法规的变更或提交的产品申请的监管审查的变更 可能会导致申请审批或拒绝的延迟。在一个司法管辖区获得监管批准并不一定意味着候选产品将在我们可能寻求批准的所有司法管辖区获得监管批准,但在一个司法管辖区未能获得批准可能会对我们在不同司法管辖区寻求批准的能力产生负面影响。由于上述原因或任何其他原因,如果我们的候选产品未能获得监管部门的批准,我们将无法将我们的候选产品 商业化,我们的创收能力将受到严重影响。
此外,FDA、EMA或其他监管机构还可能批准的候选产品的适应症少于或超过我们的要求,可能会对某些年龄段的使用限制、警告、预防措施或禁忌症施加重大限制,或者可能会根据昂贵的上市后临床试验或风险缓解要求的表现而批准。FDA、EMA或其他监管机构也可能不接受我们认为对于我们的候选产品成功商业化是必要或可取的标签声明。
2022年12月,美国国会颁布了一项新法律,即《2022年化妆品现代化监管法案》,简称MOCRA。MOCRA将要求化妆品制造商或进口商:确保其手头有其产品和成分的安全性证明;满足 增加的注册、记录保存和报告要求;在其标签上包括香料和过敏原信息;并准备满足FDA将颁布的良好制造规范要求。这些额外要求可能会影响预算 和时间表。
我们的临床试验可能无法证明我们的候选产品或任何未来候选产品的安全性和有效性的实质性证据 ,这将阻止、推迟或限制监管批准和商业化的范围.
Our business model depends entirely on the successful development, regulatory approval and commercialization of our product candidates, which may never occur. We recently initiated our Phase 1b clinical trial for ATR-12 and expect to enroll our first patient in the first half of 2024 and expect to file an IND for a Phase 1b clinical trial of ATR-04 by mid-2024. However, all of our other product candidates are in the early stages of development and as of the date of this report we have not progressed any of our product candidates, other than ATR-12 and ATR-04, beyond performance characterization and animal testing We may not be successful in obtaining approval from the FDA or comparable foreign regulatory authorities to start clinical trials for ATR-04 or any of our other product candidates. If we do not obtain such approvals as presently planned, the time in which we expect to commence clinical programs for any product candidate will be extended and such extension will increase our expenses, delay our potential receipt of any revenues and increase our need for additional capital. Moreover, there is no guarantee that we will receive approval to commence human clinical trials or, if we do receive approval, that our clinical trials will be successful or that we will continue clinical development in support of an approval from the FDA or comparable foreign regulatory authorities for any indication. We note that most product candidates never reach the clinical development stage and even those that do commence clinical development have only a small chance of successfully completing clinical development and gaining regulatory approval. Success in early phases of preclinical and clinical trials does not ensure that later clinical trials will be successful, and interim results of a clinical trial do not necessarily predict final results. A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent our ability to receive regulatory approval or commercialize our product candidates. Therefore, our business currently depends entirely on the successful development, regulatory approval and commercialization of our product candidates, which may never occur.
| 46 |
我们候选产品的临床前研究的结果可能不能预测未来的临床前研究或临床试验的结果。
要获得上市和销售我们的任何候选产品所需的监管批准,我们必须通过广泛的临床前研究和临床试验证明我们的候选产品在人体内是安全、纯净和有效的。IND必须经过广泛的临床前研究(包括临床前实验室测试、动物实验和符合良好实验室规范的配方研究)才能提交FDA 并生效,这是在美国进行人体临床试验的先决条件。临床前研究的成功并不能确保以后的临床前研究或临床试验也会成功。生物技术和制药行业的一些公司在临床试验中遭遇了重大挫折,即使在早期的临床前研究中取得了积极的结果。这些挫折是由临床试验进行期间的临床前发现以及在临床试验中进行的安全性或有效性观察(包括以前未报告的不良事件)等原因造成的。临床试验的设计可以决定其结果是否支持产品的批准, 在临床试验进展良好之前,临床试验设计中的缺陷可能不会变得明显。此外,我们或我们的研究人员可能无法控制受试者是否遵守临床试验方案的重要方面。此外,临床前和临床数据往往容易受到不同解释和分析的影响。尽管在早期的研究中有任何潜在的有希望的结果,但我们不能确定我们不会面临类似的挫折。此外,我们的临床前动物研究结果可能不能预测后续人体临床试验的结果。临床试验中的候选产品可能无法显示出所需的药理特性或安全性和有效性特性,尽管已通过临床前研究取得进展。
如果我们在候选产品的临床前研究或临床试验中未能收到积极的结果,我们最先进的候选产品的开发时间表和监管批准以及商业化前景将相应地受到负面影响,我们的业务和 财务前景将受到负面影响。
对我们的任何候选产品进行任何针对任何适应症的必要研究的任何 终止或暂停,或开始或完成的任何延误都可能导致我们的成本增加,延迟或限制我们的创收能力,并对我们的商业前景产生不利影响 .
临床研究的开始和完成可能会因多种原因而推迟,包括与以下方面有关的延迟:
| ● | FDA或类似的外国监管机构未能批准继续进行, 搁置了临床研究; | |
| ● | 临床试验受试者 未能以我们预期的速度登记或继续登记参加我们的试验; | |
| ● | 生产我们的任何候选产品的工厂因违反cGMP要求或其他适用要求而被FDA或其他政府或监管机构责令暂时或永久关闭,或制造过程中候选产品的交叉污染 ; | |
| ● | 可能需要或希望对我们的制造流程进行的任何 更改; | |
| ● | 受试者 为我们正在开发的产品的适应症选择替代疗法 候选对象,或参与竞争性临床研究; | |
| ● | 出现严重或意想不到的药物不良反应的受试者; | |
| ● | 类似技术和产品的临床测试报告 引发了安全性和/或有效性问题 ; | |
| ● | 第三方 临床研究人员被吊销进行临床试验所需的执照或许可 ,未按预期时间表进行临床试验或采用符合临床试验规程、cGMP要求的方法,或者其他未及时、准确地进行数据收集和分析的第三方; |
| 47 |
| ● | FDA、类似的外国监管机构或IRBs对临床研究地点进行检查,发现 违反规定的行为,要求我们采取纠正措施,导致暂停或终止一个或多个地点,或对整个研究实施临床暂停, 或禁止我们使用部分或全部数据来支持我们的营销应用程序; | |
| ● | 第三方承包商因违反监管要求而被FDA或其他政府或监管机构禁止或暂停或以其他方式处罚,在这种情况下,我们可能需要 寻找替代承包商,我们可能无法使用此类承包商提供的部分或任何数据来支持我们的营销应用程序; | |
| ● | 一个或多个研究所拒绝批准、暂停或终止研究地点的研究,禁止招收更多的受试者,或撤回对试验的批准; 与潜在合同研究机构、或CRO和临床试验地点就可接受的条款达成协议,条款可进行广泛谈判 ,不同CRO和试验地点的条款可能存在较大差异; | |
| ● | 临床站点偏离试验方案或退出试验; | |
| ● | 增加 个新的临床试验地点; | |
| ● | CRO因任何原因不能进行任何临床试验;以及 | |
| ● | 政府 或监管延迟或需要暂停或终止试验的“临床搁置” 。 |
产品 如果我们在测试或审批方面有延误,或者如果我们需要执行比计划更多的 或更大的临床研究,我们任何候选产品的开发成本都将增加。此外,法规要求和政策可能会发生变化,我们可能需要修改 研究方案以反映这些变化。修正案可能要求我们向FDA、类似的外国监管机构和IRBs重新提交我们的研究方案进行重新审查,这可能会影响该研究的成本、时间或成功完成。如果我们遇到 延迟完成,或者如果我们、FDA或其他监管机构、IRB或其他审查实体,或我们的任何临床研究站点暂停或终止我们对任何候选产品的任何临床研究,其商业前景可能会受到实质性的 损害,我们创造产品收入的能力将被推迟。完成临床试验的任何延误都将增加我们的成本, 减慢我们的开发和审批流程,并危及我们开始产品销售和创造收入的能力。任何此类 事件都可能严重损害我们的业务、财务状况和前景。此外,许多导致或导致临床研究终止或暂停,或临床研究开始或完成延迟的因素,最终也可能导致我们的候选产品被监管部门拒绝批准。此外,如果一项或多项临床研究被推迟,我们的竞争对手可能会 先于我们将竞争产品推向市场,而我们任何受影响的候选产品的商业可行性可能会显著 降低。
即使 如果我们的任何候选产品获得监管部门的批准,我们也可能无法成功地将该产品商业化。 我们从其销售中获得的收入(如果有)可能是有限的。
如果 被批准上市,我们候选产品的商业成功将取决于医疗 社区(包括医生、患者和医疗保健付款人)对每种产品的接受程度。市场对我们的任何候选产品的接受程度将 取决于许多因素,包括:
| ● | 展示临床安全性和有效性; | |
| ● | 相对 方便、给药负担和易于管理; | |
| ● | 任何不良影响的流行率和严重程度; | |
| ● | 医生为我们的候选产品开处方的意愿,以及目标患者群体尝试新疗法的意愿。 | |
| ● | 我们的候选产品与竞争产品相比的功效 ; | |
| ● | 针对我们的候选产品可能获得批准的适应症 推出未来可能出现的任何新产品; | |
| ● | 新的 程序或疗法,可减少我们的候选产品可能显示实用的任何适应症的发生率; | |
| ● | 定价 和成本效益; | |
| ● | 在适用的治疗和疫苗指南中包括或省略我们的候选产品; | |
| ● | 我们自己或任何未来合作伙伴的销售和营销策略的有效性; | |
| ● | 监管部门批准的标签中包含的限制或警告; |
| 48 |
| ● | 我们 能够从政府医疗保健计划(包括Medicare和Medicaid)获得并维持足够的第三方保险或报销,私人健康保险公司和其他第三方付款人,或从管理治疗药物定价和使用的政府机构获得必要的定价批准;和 | |
| ● | 患者在没有第三方保险或报销或政府定价审批的情况下自付费用的意愿 。 |
如果我们的任何候选产品获得批准,但没有获得医生、医疗保健付款人和患者的足够程度的接受,我们可能无法产生足够的收入,我们可能无法实现或维持盈利。我们努力让医疗界和第三方付款人了解我们的候选产品的好处,这可能需要大量资源,而且可能永远不会成功。
此外,即使我们获得了监管部门的批准,任何批准的时间或范围也可能会禁止或降低我们将候选产品成功商业化的能力。 例如,如果审批过程太长,我们可能会错过预期的市场机会,并使其他 公司有能力开发竞争产品或建立市场主导地位。我们最终获得的任何监管批准都可能是有限的,或者受到限制或审批后承诺的限制,从而使我们的候选产品在商业上不可行。例如, 监管机构可能会批准我们的任何候选产品的适应症少于或超过我们要求的范围,可能不会批准我们打算对任何候选产品收取的价格,可能会批准取决于昂贵的上市后临床试验的表现,或者可能批准我们的任何候选产品的标签不包括该适应症成功商业化所必需或需要的标签声明 。此外,FDA或类似的外国监管机构可以在批准上附加条件,或要求风险管理计划或风险评估和缓解战略(REMS),以确保药物的安全使用。 此外,如果产品不符合监管标准或在产品首次上市后出现问题,可能会撤回产品批准。上述任何一种情况都可能对我们的候选产品的商业成功造成实质性损害。
即使 如果我们的任何候选产品获得了市场批准,我们也将受到持续的义务和持续的监管 审查,这可能会导致大量的额外费用。此外,我们的候选产品可能受到标签和其他 限制并退出市场,如果我们未能遵守监管要求或 我们的候选产品遇到意想不到的问题,我们可能会受到处罚。
即使 如果我们的任何候选产品获得了监管部门的批准,FDA或外国同等机构仍可能对其指示用途或营销或批准条件施加 重大限制,或对可能 昂贵且耗时的审批后研究(包括4期临床试验)和上市后监测以监测安全性和 疗效施加持续要求。我们的候选产品还将遵守有关制造、标签、包装、存储、分销、安全监控、广告、促销、记录和报告不良事件及其他上市后信息的持续法规要求 。这些要求包括向FDA注册,以及对于我们在批准后进行的任何临床试验,继续遵守当前的良好临床实践法规或CCP。此外,药品制造商及其设施 要接受FDA和其他监管机构的持续审查和定期检查,以确保符合当前的cGMP、与质量控制、质量保证和相应的记录和文件维护相关的要求。
FDA有权要求将REMS作为BLA的一部分或在批准后进行,这可能会对批准的药物的分发或使用施加进一步的要求或限制,例如将处方限制在经过 专门培训的特定医生或医疗中心,将治疗限制为满足某些安全使用标准的患者,或要求患者测试、监测和/或在登记中登记。
对于与我们的候选产品相关的销售和营销活动,广告和促销材料除了遵守美国其他适用的联邦、州和地方法律以及其他国家/地区的类似法律要求外,还必须符合FDA的 规则。 在美国,向医生分发产品样本必须符合美国处方药 营销法的要求。根据更改的性质,申请持有人必须获得FDA批准才能更改产品和制造。 我们还可能通过我们的客户和合作伙伴直接或间接地受到各种欺诈和滥用法律的约束,包括但不限于美国反回扣法规、美国虚假索赔法案和类似的州法律,这些法律除其他外,还会影响我们拟议的销售、营销和科学/教育资助计划。如果我们参加了美国医疗补助药品回扣计划、美国退伍军人事务部的联邦供应时间表或其他政府药品计划,我们将受到有关报告和付款义务的复杂法律和 法规的约束。所有这些活动还可能受到美国联邦和州 消费者保护和不正当竞争法律的约束。在其他国家的许多这样的领域也存在类似的要求。
| 49 |
此外,如果我们的任何候选产品被批准用于特定用途,我们的产品标签、广告和促销 将受到监管要求和持续的监管审查。FDA严格监管关于处方药的促销声明。特别是,产品不得用于未经FDA批准的用途,如产品批准的标签中所反映的那样。如果我们的候选产品获得了市场批准,医生仍然可以合法地 以与批准的标签不一致的方式向他们的患者开出我们的产品。如果我们被发现推广此类 标签外使用,我们可能会面临重大责任和政府罚款。FDA和其他机构积极执行禁止推广标签外使用的法律和法规,被发现不当推广标签外使用的公司可能会受到重大制裁。联邦政府已对涉嫌不当促销的公司处以巨额民事和刑事罚款,并禁止几家公司从事标签外促销。FDA还要求公司 签订永久禁令的同意法令,根据这些法令,特定的促销行为将被改变或减少。
如果我们或监管机构发现候选产品存在以前未知的问题,例如未预料到的严重程度或频率的不良事件、生产产品的设施存在问题,或者我们或我们的制造商未能遵守适用的 监管要求,我们可能会受到以下行政或司法制裁:
| ● | 限制产品的销售或制造、从市场上召回产品、或自愿或强制召回产品; | |
| ● | 发出警告信或无标题信; | |
| ● | 临床 坚持; | |
| ● | 禁制令或施加民事或刑事处罚或罚款; | |
| ● | 暂停或撤回监管审批; | |
| ● | 暂停任何正在进行的临床试验; | |
| ● | 拒绝批准我们提交的待决申请或已批准申请的补充申请,或暂停或撤销产品许可批准; | |
| ● | 暂停 或对运营施加限制,包括代价高昂的新制造要求; 或 | |
| ● | 产品 扣押、扣押或拒绝允许进口或出口产品。 |
发生上述任何事件或处罚可能会抑制我们将候选产品商业化并创造收入的能力。 无论是在审批前还是审批后,不利的监管行动也可能导致产品责任索赔并增加我们的产品责任风险。
我们 也无法预测美国或国外未来的立法或行政 行动可能产生的政府监管的可能性、性质或程度,遵守此类监管可能代价高昂,并消耗大量财务 和管理资源。如果我们或任何未来的营销协作者或合同制造商缓慢或无法适应现有要求的变化或新要求或政策的采用,或者无法保持合规性,则可能会 延迟或阻止我们产品的推广、营销或销售,这将对我们的业务和运营结果产生不利影响。
| 50 |
在一个司法管辖区获得并保持对我们候选产品的监管批准并不意味着我们将在其他司法管辖区成功获得 我们候选产品的监管批准。
在一个司法管辖区获得并保持对我们的候选产品的监管批准并不能保证我们能够在任何其他司法管辖区获得或 维持监管批准,但在一个司法管辖区未能或延迟获得监管批准可能会对其他司法管辖区的监管审批流程产生负面影响。例如,即使FDA批准了候选产品的上市 ,外国司法管辖区的可比监管机构也必须批准该候选产品在这些国家/地区的制造、营销和推广。审批程序因司法管辖区而异,可能涉及与美国不同的要求和行政审查期限,包括额外的临床前研究或临床试验,因为在一个司法管辖区进行的临床研究 可能不会被其他司法管辖区的监管机构接受。在美国以外的许多司法管辖区,候选产品必须先获得报销批准,然后才能在该司法管辖区批准销售。在某些情况下,我们打算对产品收取的价格也需要审批。
获得外国监管批准并遵守外国监管要求可能会给我们带来重大延误、困难和 成本,并可能推迟或阻止我们的候选产品在某些国家/地区推出。如果我们未能遵守国际市场的监管要求和/或获得相应的营销批准,我们的目标市场将会减少 ,我们充分发挥候选产品市场潜力的能力将受到损害。
即使 虽然我们可以为候选产品申请孤儿药物指定,但我们可能无法获得孤儿药物营销排他性。
我们 认为,在某些情况下,我们的候选产品可能有资格获得FDA的孤立药物地位。不能保证FDA将批准我们的任何候选产品未来的任何孤儿药物指定申请,这将使我们没有资格获得 孤儿药物指定的额外排他性和其他好处。
根据《孤儿药品法》,FDA可以将用于治疗罕见疾病或疾病的药物授予孤儿药物称号,这种疾病或疾病通常是指在美国影响不到200,000人的疾病或疾病,因此没有合理的预期 在美国开发和提供治疗此类疾病或疾病的药物的成本将从产品的销售中收回 。在提交BLA之前,必须申请指定孤儿药物。在FDA批准孤儿药物指定后, FDA将公开披露治疗剂的身份及其潜在的孤儿用途。孤立产品命名不会 在监管审批过程中传达任何优势或缩短持续时间。除了可能的独占期 外,孤儿指定还使公司有资格在四年内每年获得高达650,000美元的赠款资金,以支付临床试验费用、临床研究费用的税收抵免,以及可能免除FDA申请用户费用。
如果具有孤儿称号的产品随后获得FDA对其具有孤儿称号的疾病或病症的第一次批准,则该产品有权获得孤儿药品排他性,这意味着FDA在七年内不得批准同一药物在同一适应症下销售的任何其他申请,除非在有限情况下,例如:(I)该药物的孤儿称号被撤销;(Ii)其上市批准被撤回;(Iii)孤儿排他性持有者同意批准另一申请人的 产品;(Iv)孤儿排他性持有者不能保证获得足够数量的药物;或(V)竞争对手的产品显示出相对于具有孤儿排他性的产品的临床优势。如果被指定为孤儿产品的药物获得了 比指定范围更广的适应症的上市批准,则该药物可能没有资格获得孤儿药物独家经营权。如果我们选择寻求此类申请,不能保证我们的任何候选产品在我们认为 可能符合条件的适应症中将获得孤儿药物指定。
当前的 和未来的立法可能会增加我们获得候选产品的上市批准并将其商业化的难度和成本 ,并影响我们可能获得的价格。
在 美国和一些外国司法管辖区,有关医疗保健系统的立法和法规更改以及拟议的更改 可能会阻止或推迟我们候选产品的上市审批,限制或规范审批后活动 ,并影响我们销售候选产品的盈利能力。已经提出了立法和监管建议,以扩大批准后的要求,并限制药品的销售和促销活动。我们不知道是否会颁布额外的立法变更,或FDA的法规、指南或解释是否会更改,或此类变更对我们候选产品的上市审批(如果有)可能产生什么影响。此外,美国国会对FDA审批过程的更严格审查可能会显著推迟或阻止上市审批,并使我们受到更严格的产品标签和上市后测试及其他要求的约束。
| 51 |
在美国,《联邦医疗保险现代化法案》(Medicare Modinization Act,简称MMA)改变了联邦医疗保险覆盖和支付药品的方式。立法扩大了老年人购买药品的医疗保险覆盖范围,并引入了基于药品平均销售价格的新报销方法。此外,这项立法授权Medicare Part D处方药计划使用处方,在这些处方中, 可以限制任何治疗类别涵盖的药物数量。由于这项立法和联邦药品覆盖范围的扩大,我们预计将会有更多的压力来控制和降低成本。这些降低成本的举措 以及该立法的其他条款可能会降低我们为候选产品提供的覆盖范围和价格,并可能严重 损害我们的业务。虽然MMA仅适用于Medicare受益人的药品福利,但私人付款人在设置自己的报销费率时通常遵循Medicare承保政策和支付限制,而MMA导致的任何报销减少 可能会导致私人付款人的付款减少类似的情况。
《患者保护和平价医疗法案》经2010年《医疗保健和教育负担能力协调法案》(或统称为ACA)修订,是一部全面的法律,旨在扩大医疗保险的可及性,减少或限制医疗支出的增长,加强针对欺诈和滥用的补救措施,为医疗保健和医疗保险行业增加新的透明度要求, 对医疗行业征收新的税费,并实施额外的医疗政策改革。出于报告目的,ACA修改了“制造商平均价格”的定义,这可能会增加对各州的医疗补助药品退税金额。该法律还对生产或进口品牌处方药产品的公司征收了一笔可观的年费。此外,2022年8月16日,总裁·拜登签署了2022年通胀削减法案,将个人在ACA市场购买医疗保险的增强补贴延长至2025年。从2025年开始,IRA还通过显著降低受益人的最大自付成本和创建新的制造商折扣计划来消除Medicare Part D计划下的“甜甜圈洞” 。目前尚不清楚这些挑战以及拜登政府的医疗改革措施将如何影响ACA和我们的业务。
此外,自ACA颁布以来,美国还提出并通过了其他立法修改。2011年,美国 国会颁布了《2011年预算控制法》或《预算控制法》,其中包括旨在减少联邦赤字的条款。预算控制法案导致从2013年开始对医疗保险提供者的支付减少2%, 由于随后对该法规的立法修订,该法案将在没有国会额外行动的情况下一直有效到2027年。然而, 根据CARE法案和后续立法,由于新冠肺炎大流行,这些削减将从2020年5月1日至2022年3月31日暂停。2013年1月,2012年《美国纳税人救济法》签署成为法律,其中包括减少向多家医疗服务提供者支付的医疗保险费用,并将政府向服务提供者追回多付款项的诉讼时效期限从三年延长至五年。这些新法律可能会导致联邦医疗保险和其他医疗保健资金的进一步减少,如果获得批准,这可能会对我们药品的客户以及我们的财务运营产生实质性的不利影响。如果政府支出进一步削减,预计的预算缺口也可能影响FDA等相关机构继续以当前水平运作的能力,这可能会影响相关机构及时审查和批准研发、制造和营销活动的能力,这可能会推迟我们开发、营销和销售任何候选产品的能力。 此外,任何影响Medicare、Medicaid或其他可能实施的公共资助或补贴医疗计划的重大支出削减,或可能对我们征收的任何重大税收或费用,作为任何更广泛的赤字削减努力的一部分 或对《预算控制法案》的立法替代,可能会对我们预期的产品收入产生不利影响。
| 52 |
此外, 最近,政府加强了对制造商为其商业产品定价的方式的审查。 美国国会最近进行了几次调查,提出并颁布了联邦和州立法,旨在提高药品定价的透明度,审查定价与制造商患者计划之间的关系,降低医疗保险下的药品成本,并改革政府计划的药品报销方法。2020年9月24日,FDA 发布了一项最终规则,为各州制定和提交来自加拿大的药物进口计划提供指导。此外,2020年11月20日,美国卫生与公众服务部(HHS)敲定了一项法规,取消了药品制造商对D部分下计划赞助商降价的安全港保护 直接或通过药房福利经理,除非 法律要求降价。该规则还为反映在销售点的降价创造了新的安全港, 也为药房福利经理和制造商之间的某些固定费用安排创造了安全港。2022年8月16日, 国会颁布了《2022年通胀削减法案》,其中包含多项与处方药成本有关的条款,包括对联邦政府价格谈判的要求、返点要求,以及对Medicare Part D投保人的自付支出上限。 在州一级,立法机构越来越多地通过立法并实施旨在控制药品和生物制品定价的法规,包括价格或患者报销限制、折扣、对某些产品准入的限制 以及营销成本披露和透明度措施,在某些情况下,旨在鼓励从其他国家进口和 批量购买。
2020年11月20日,卫生和公众服务部监察长办公室敲定了对联邦反回扣法规的进一步修改。根据《最终规则》,卫生与公众服务部监察长办公室根据《反回扣法规》为临床医生、提供者和其他人之间的某些协调护理和基于价值的安排增加了安全港保护,但取消了 制药商直接或通过药房福利经理对D部分下的计划赞助商降价的安全港保护,除非法律要求降价 。该规则还为反映在销售点的降价创造了新的安全港,以及为药房福利经理和制造商之间的某些固定费用安排创造了安全港。此规则(有例外情况)于2021年1月19日生效。我们将继续评估这些规则将对我们的业务产生什么影响(如果有的话)。CMS于2019年7月9日发布了最终规则, 要求处方药和生物制品的直接面向消费者的广告(可通过或根据Medicare或Medicaid付款)在广告中包括该药物或生物制品的批发采购成本或标价,如果该药品或生物制品的每月供应或通常疗程的批发价等于或大于35美元。违反这些要求的处方药和生物制品将被列入公共名单。任何采用的医疗改革措施 如果获得批准,可能会降低对我们产品的最终需求,或者给我们的产品定价带来压力。美国各个州也越来越积极地通过立法和实施法规来控制药品定价,包括价格或患者报销限制、折扣、对某些产品准入和营销成本披露的限制 以及透明度措施,在某些情况下,旨在鼓励从其他国家进口和批量采购。此外,地区医疗当局和个别医院越来越多地使用招标程序来确定哪些药品 以及哪些供应商将包括在其处方药和其他医疗保健计划中。我们预计未来将采取更多的州和联邦医疗改革措施。
在欧洲联盟或欧盟提供医疗保健,包括医疗服务的建立和运营以及药品的定价和报销,几乎完全是国家法律和政策的问题,而不是欧盟的法律和政策。各国政府和卫生服务提供者在提供卫生保健以及产品定价和报销方面有不同的优先事项和方法 。然而,总的来说,大多数欧盟成员国的医疗预算限制导致相关医疗服务提供商对药品的定价和报销进行了限制。再加上希望开发和营销产品的欧盟和国家监管机构的负担不断增加,这可能会阻止或推迟我们的候选产品的上市审批,限制 或监管审批后的活动,并影响我们将任何获得上市审批的产品商业化的能力。在美国和欧盟,都提出了立法和监管建议,以扩大审批后的要求,并限制 药品的销售和促销活动。我们不知道是否会颁布额外的法律变更, 或法规、指南或解释是否会更改,或此类更改对我们的候选产品的上市审批 可能会产生什么影响。
| 53 |
第三方保险和报销、医疗成本控制计划和治疗指导方针可能会限制我们未来的收入。
我们 能否成功营销我们的候选产品,部分取决于政府卫生行政部门、私人健康保险公司和其他组织为我们候选产品和相关治疗提供的报销水平。 我们的任何候选产品经常通过国家医疗保险计划下的报销计划销售的国家/地区 要求药品制造商和销售商在初始价格和任何后续价格上涨时获得政府批准。在某些国家,包括美国,政府资助和私人医疗保健计划可能会对价格施加巨大的间接压力。如果未批准足够的价格或覆盖范围 且无法获得报销或报销范围有限,我们可能无法销售我们的候选产品以盈利。越来越多的第三方付款人试图通过 可能影响我们产品开发的方式来控制医疗成本,包括:
| ● | 未能批准或者对保健品价格提出异议的; | |
| ● | 从价格较低的司法管辖区引入 再进口计划; | |
| ● | 限制新治疗产品的覆盖范围和报销金额; | |
| ● | 拒绝 或限制监管机构批准但被第三方付款人视为试验性或调查性产品的承保范围;以及 | |
| ● | 当批准的产品用于未获得监管部门 市场批准的方式时,拒绝提供保险。 |
我们与客户和付款人的关系受到适用的反回扣、欺诈和滥用以及其他医疗法律法规的约束, 如果违反这些法律法规,我们可能面临刑事制裁、民事处罚、被排除在政府医疗保健计划之外、合同损害、声誉损害以及利润和未来收入减少。
医疗保健 提供者、医生和其他人将在推荐和处方我们获得市场批准的任何产品时发挥主要作用。我们未来与第三方付款人和客户的协议可能使我们面临广泛适用的欺诈和滥用 和其他医疗法律法规,主要是在美国,这可能会限制我们营销、销售和分销我们获得营销批准的产品的业务或财务安排 和关系。根据 适用的医疗法律法规的限制,包括:
| ● | 联邦反回扣法规禁止明知和故意提供、支付、招揽或收取任何形式的报酬,以换取或引诱 (I)转介某人,(Ii)提供或安排提供根据Medicare、Medicaid或其他政府计划可报销的物品或服务,或 (Iii)购买、租赁或订购或安排或推荐购买、租赁或订购根据Medicare可报销的任何物品或服务。医疗补助或其他政府计划。 个人或实体不需要实际了解法规或具体意图即可违反法规 即可实施违规。此外,政府可主张 根据《虚假索赔法》,包括因违反联邦《反回扣条例》而产生的物品或服务的索赔构成虚假或欺诈性索赔; | |
| ● | 联邦虚假索赔法案对故意向联邦政府提交或导致提交的个人或实体施加民事处罚,并规定民事举报人或准诉讼。虚假或欺诈性的付款要求,或者为逃避、减少或隐瞒向联邦政府付款的义务而做出虚假陈述的; | |
| ● | HIPAA 对执行诈骗任何医疗福利计划的计划或做出与医疗保健事项相关的虚假陈述的行为追究刑事和民事责任。与联邦《反回扣法规》类似,个人或实体不需要实际了解法规或违反法规的具体意图即可实施违规; | |
| ● | 联邦虚假陈述法禁止明知和故意伪造、隐瞒、掩盖重大事实或作出任何与医疗保健福利、项目或服务的交付或付款有关的重大虚假陈述。 | |
| ● | 根据ACA创建的《医生支付阳光法案》及其实施条例, 该法案要求药品、器械、生物制品和医疗用品的指定制造商 可根据联邦医疗保险获得付款,医疗补助或儿童健康保险计划,除特定的例外情况外,每年向CMS报告与支付 或向医生进行的其他“价值转移”有关的信息。所有此类报告信息 均可公开获取; |
| 54 |
| ● | 类似的州和非美国的法律法规,例如某些州的反回扣和虚假索赔 法律可能适用于任何第三方付款人报销的项目或服务,包括商业保险公司 ;州法律要求制药公司遵守 行业的自愿合规指南和联邦政府颁布的适用合规指南,或以其他方式限制可能向医疗保健提供者和其他潜在转介来源支付的款项;州法律,要求药品制造商报告与向医生和其他医疗保健提供者支付或以其他方式转移价值或营销支出有关的信息。以及在某些情况下管理健康信息隐私和安全的州法律,其中许多法律在许多方面存在重大差异,可能不会产生相同的效果,从而使合规工作复杂化;以及 | |
| ● | 由CMS监管和HHS监察长办公室或美国司法部执行。 |
由于这些法律的广度以及法定例外和可用避风港的狭隘,我们 未来的一些商业活动可能会受到一项或多项此类法律的挑战。
确保我们与第三方的业务安排符合适用的医疗法律法规的努力将涉及 巨额成本。政府当局可能会得出结论,我们的业务实践可能不符合当前或未来涉及适用欺诈和滥用或其他医疗保健法律和法规的法规、法规或判例法。如果我们的业务 被发现违反了任何这些法律或任何其他可能适用于我们的政府法规,我们可能会受到重大的民事、刑事和行政处罚、损害赔偿、罚款、被排除在美国政府资助的医疗保健计划之外,如Medicare 和Medicaid,以及削减或重组我们的业务。如果我们希望与之开展业务的任何医生或其他提供者或实体被发现不符合适用法律,他们可能会受到刑事、民事或行政 制裁,包括排除在政府资助的医疗保健计划之外。
与我们知识产权有关的风险
保护我们的知识产权是困难和昂贵的,我们无法确保这些权利的保护。
我们的商业成功将在一定程度上取决于我们是否有能力在必要时针对第三方 挑战起诉和捍卫我们的专利权,并成功地针对第三方竞争对手实施这些专利权。生物技术公司的专利地位可能高度不确定,涉及复杂的法律、科学和事实问题,其中重要的法律原则仍未解决。 专利法或专利法解释的变化可能会降低我们知识产权的价值。因此, 我们无法预测在我们或我们的专利权许可人提交的任何专利申请中可能允许或可强制执行的索赔的广度 。我们持有或许可的与我们的微生物平台和相关技术相关的专利和专利申请 可能会受到第三方的挑战、无效或规避,并且可能无法保护我们免受具有类似产品或技术的竞争对手的影响。
由我们持有或授权给我们的专利权提供的未来保护程度是不确定的,因为法律手段只能提供有限的 保护,而可能无法充分保护我们的权利,使我们能够获得或保持我们的竞争优势,或为我们提供任何竞争优势。我们不能确定第三方拥有的任何专利申请不会优先于我们提交或我们持有许可权的专利申请,也不能确定我们不会卷入美国或外国专利局的干扰、反对或无效诉讼 。
此外, 如果我们对第三方提起法律诉讼,以强制执行涵盖我们任何候选产品的专利,被告 可以反诉该专利无效和/或不可强制执行。在美国的专利诉讼中,被告声称无效和/或不可执行的反诉是司空见惯的。质疑有效性的理由包括据称未能满足 几项法定要求中的任何一项,包括缺乏新颖性、明显或无法实施。不可执行性断言的理由包括 与专利起诉有关的人在起诉期间向美国专利商标局或USPTO隐瞒相关信息或做出误导性陈述的指控。第三方也可以向美国或国外的行政机构提出类似的索赔,即使在诉讼范围之外也是如此。这类机制包括复审、赠款后审查和外国司法管辖区的同等程序,例如反对程序。此类诉讼可能导致撤销或修改由我们持有或授权给我们的专利,使其不再涵盖我们的候选产品或竞争产品。在法律上断言无效和不可执行之后的结果 是不可预测的。例如,关于有效性,我们不能确定 没有无效的先前技术,而我们、我们专利权的任何许可人或专利审查员在起诉期间并不知道这一点。 如果被告胜诉,无效和/或不可强制执行的法律主张成立,我们将失去对我们任何候选产品的至少部分,甚至全部 专利保护。这种专利保护的丧失将对我们的业务产生实质性的不利影响。
| 55 |
我们 依靠专有技术和商业秘密来保护技术,特别是在我们认为专利保护不合适或 可以获得的情况下。然而,专有技术和商业秘密很难保护。虽然我们要求员工、学术合作者、顾问和其他承包商签订保密协议,但我们可能无法充分保护我们的商业秘密或其他专有或许可信息。通常,研究合作者和科学顾问有权发布数据和信息,而我们可能有权发布这些数据和信息。强制执行第三方非法获取和使用我们的任何商业秘密的声明是昂贵和耗时的 ,结果不可预测。此外,与专利相比,法院有时更不愿意保护商业秘密。此外,我们的竞争对手可以自主开发同等的知识、方法和诀窍。
如果 我们无法为我们的候选产品或我们的技术获得或维护专利保护或商业秘密保护,第三方 可能会使用我们的专有信息,这可能会削弱我们在市场上的竞争能力,并对我们创造 收入和实现盈利的能力产生不利影响。
我们的 候选产品可能会侵犯他人的知识产权,这可能会增加我们的成本,延迟或阻碍我们的开发和商业化努力。
我们的成功在一定程度上取决于避免侵犯他人的专有技术。制药业的特点是涉及专利和其他知识产权的诉讼频繁。识别可能与我们的专有技术相关的第三方专利权是困难的,因为专利搜索由于 专利之间的术语差异、数据库不完整以及难以评估专利权利要求的含义而不完善。此外,由于专利申请 在申请发布之前一直保密,我们可能不知道我们的任何候选产品或任何未来候选产品的商业化 可能会侵犯第三方专利。可能会有某些已颁发的专利和专利申请声称 我们可能需要获得许可才能对我们的任何候选产品进行研究、开发或商业化,而我们 不知道这些专利和专利申请是否可以按商业合理的条款获得许可,或者根本不知道。第三方提出的任何专利侵权索赔都将非常耗时,并且可能:
| ● | 结果 诉讼费用高昂; | |
| ● | 转移我们技术人员和管理人员的时间和注意力; | |
| ● | 防止 我们将产品商业化,直到主张的专利到期或最终被法院裁定为无效或未被法院侵犯; | |
| ● | 要求我们停止或修改我们对该技术的使用和/或开发非侵权技术; 或 | |
| ● | 要求 我们签订版税或许可协议。 |
第三方 可能拥有可能阻止我们的任何候选产品上市的专有权利。任何针对我们要求损害赔偿并试图禁止与我们的任何候选产品或我们的工艺有关的商业活动的专利相关法律诉讼都可能 使我们承担潜在的损害赔偿责任,并要求我们获得许可证才能继续制造或销售我们的任何候选产品或任何未来的候选产品。我们无法预测我们是否会在任何此类诉讼中获胜,也无法预测这些专利所需的任何许可证是否会以商业上可接受的条款提供(如果有的话)。此外,如果有必要,我们不能确定我们 是否可以重新设计我们的候选产品或任何未来的候选产品或流程,以避免侵权。因此, 司法或行政诉讼中的不利裁决,或未能获得必要的许可证,可能会阻止我们 开发和商业化我们的任何候选产品或未来的候选产品,这可能会损害我们的业务、财务状况和经营业绩。
| 56 |
我们 预计还有其他公司,包括主要的生物制药公司,在与我们推荐的候选产品竞争的领域开展工作,这些领域已经或可能导致提交可能被认为与我们的活动相关的专利申请。 如果我们要在法庭上挑战这些或任何已发布的美国专利的有效性,我们将需要克服适用于每一项已发布的美国专利的法定有效性推定 。这意味着,为了胜诉,我们必须就专利权利要求的无效性提出明确而令人信服的证据。如果我们要在USPTO的专利审判和上诉委员会的行政审判中质疑这些或任何已颁发的美国专利的有效性,我们将必须证明 权利要求是不可专利的,因为证据占优势。不能保证陪审团和/或法院会在侵权、有效性或可执行性问题上做出有利于我们的裁决。即使我们成功了,诉讼也可能导致巨额成本,并 分散管理层的注意力。
我们 可能会受到以下指控:我们错误地从竞争对手那里雇佣了一名员工,或者我们或我们的员工错误地使用了 ,或者泄露了他们前雇主的所谓机密信息或商业秘密。
由于 在我们的行业中很常见,我们将聘用以前在其他生物制药公司工作过的人员,包括我们的竞争对手或潜在竞争对手。虽然目前没有针对我们的索赔待决,但我们未来可能会受到索赔 ,即我们的员工或潜在员工对其前雇主负有持续义务(例如竞业禁止义务或非征集义务),或者我们的员工或我们无意或以其他方式使用或泄露其前雇主的商业秘密或 其他专有信息。可能有必要提起诉讼来抗辩这些指控。即使我们成功地对这些索赔进行了辩护,诉讼也可能导致巨额成本,并分散管理层的注意力。
与拥有我们的普通股有关的风险
我们股票的市场价格可能会受到波动和波动的影响。你可能会失去全部或部分投资.
我们普通股的市场价格受各种因素的影响而波动很大,其中有些因素超出了我们的控制范围。 自2023年6月首次公开募股(IPO)以每股5美元的价格出售我们的普通股股份以来, 我们的普通股报告的 在2024年2月29日之前,我们的普通股的高、低销售价格范围从5. 18美元到0. 20美元不等。我们在纽交所美国股票的市场价格 可能会因多种因素而波动,其中一些因素超出了我们的控制范围,包括但不限于 :
| ● | 我们和我们的竞争对手的经营结果和财务状况的实际或预期变化 ; | |
| ● | 如果我们的股票在分析师的跟踪范围内,证券分析师的收益预期或推荐发生变化 ; | |
| ● | 市场对我们候选产品的接受度; | |
| ● | 他人开发技术创新或有竞争力的新产品; | |
| ● | 我们发布的技术创新或新产品公告 ; | |
| ● | 发布我们的候选产品的临床前或临床试验结果; | |
| ● | 我们未能实现公开宣布的里程碑; | |
| ● | 我们开发和营销新的或增强的产品的支出与这些产品产生的销售之间的延迟 ; | |
| ● | 有关知识产权的发展 ,包括参与由我们提起或针对我们提起的诉讼 ; | |
| ● | 监管事态发展和监管当局关于批准或拒绝新产品或改良产品的决定。 | |
| ● | 更改我们用于开发、获取或许可新产品、技术或业务的金额 ; | |
| ● | 更改我们用于推广候选产品的支出 ; | |
| ● | 我们的 出售或建议出售,或我们的主要股东在未来出售我们的股票或其他证券; |
| 57 |
| ● | 关键人员变动 ; | |
| ● | 我们或竞争对手的研发项目的成功或失败; | |
| ● | 我们股票的交易量;以及 | |
| ● | 一般 经济和市场状况以及其他因素,包括与我们的经营业绩无关的因素。 |
这些 因素和任何相应的价格波动可能会对我们股票的市场价格产生重大不利影响,并导致我们的投资者遭受重大损失。在过去,在市场波动之后,上市公司股东经常提起证券集体诉讼。如果我们卷入证券诉讼,可能会给我们带来巨额成本,并将我们管理层的资源和注意力从我们的业务上转移出去。
我们的 未能满足纽约证券交易所美国人的持续上市要求可能会导致我们的普通股退市。
如果 我们未能满足NYSE American的持续上市要求,例如公司治理要求或最低收盘价要求,NYSE American可能会采取措施将我们的普通股退市。这样的退市可能会对我们的普通股价格产生负面的 影响,并会削弱您在您愿意的时候出售或购买我们的普通股的能力。如果发生退市事件,我们不能保证我们为恢复遵守上市要求而采取的任何行动将 允许我们的普通股重新上市,稳定市场价格或提高我们普通股的流动性,防止我们的普通股 跌破NYSE American的最低出价要求,或防止未来不遵守NYSE American的 上市要求。
未来的增资可能会稀释您的所有权和/或对我们的运营产生其他不利影响.
如果 我们通过发行股权证券筹集额外资本,我们现有股东的所有权百分比将会减少,这些 股东可能会经历大幅稀释。如果我们通过发行债务证券筹集更多资金,这些债务证券将 拥有优先于我们普通股的权利,而发行的债务证券的条款可能会对我们的业务施加重大限制,包括对我们资产的留置权。如果我们通过协作和许可安排筹集更多资金,我们可能会被要求 放弃对我们的知识产权或候选产品的某些权利,或按对我们不利的条款授予许可。
有资格未来出售的股票 可能会对我们普通股的市场产生不利影响.
截至本报告日期,我们有28,804,643股已发行和流通的普通股,所有这些股票都有资格根据《证券法》颁布的规则144通过公开市场的普通经纪交易进行销售,但须遵守规则144下的某些 限制,约925,我们的管理人员和董事持有的774股普通股, 受IPO禁售,将于2024年8月到期。我们已授予可转换 优先股和可转换承兑票据的前持有人索购和附带登记权,据此,他们可要求登记转售最多9,542,519股普通股 。根据第144条或根据任何转售报告出售我们的普通股的任何实质性出售可能对我们的普通股的市场价格产生重大不利影响。
根据《就业法案》,我们 是一家“新兴成长型公司”,我们不能确定适用于新兴成长型公司的披露要求的降低是否会降低我们的普通股对投资者的吸引力.
根据《就业法案》的定义,我们 是“新兴成长型公司”,我们可以利用适用于非“新兴成长型公司”的其他上市公司的各种 报告要求的某些豁免,包括但不限于:
| ● | 未要求遵守《萨班斯-奥克斯利法案》第404节的审计师认证要求; |
| 58 |
| ● | 在我们的定期报告和代理声明中减少了关于高管薪酬的披露义务 ; | |
| ● | 免除 就高管薪酬进行不具约束力的咨询投票和 股东批准任何黄金降落伞付款的要求;以及 | |
| ● | 延长了 可用于遵守新的或修订的会计准则的过渡期。 |
我们 已选择利用《就业法案》提供的所有福利,包括上文讨论的豁免。我们无法预测投资者是否会因为我们可能依赖这些豁免而发现我们的普通股吸引力下降。如果一些投资者因此发现我们的普通股吸引力下降,我们的普通股可能会出现不那么活跃的交易市场,我们的股价可能会更加波动。
我们 将在五年内保持“新兴成长型公司”的地位,但如果我们的收入超过 12.35亿美元,如果我们在三年期内发行超过10亿美元的不可转换债券,或者如果非关联公司持有的普通股 市值在未来任何一年的6月30日超过7亿美元,我们将很快失去这一地位。
根据《就业法案》,我们作为“新兴成长型公司”的身份可能会使我们在需要时筹集资金变得更加困难 .
由于我们作为一家“新兴成长型公司”获得了各种报告要求的豁免,因此我们对投资者的吸引力可能会降低 ,因此我们可能很难在需要时筹集更多资本。如果投资者认为我们的报告不如行业中的其他公司透明,他们可能无法将我们的业务与行业内的其他公司进行比较。 如果我们无法在需要时筹集额外资本,我们的财务状况和运营结果可能会受到实质性的 和不利影响。
我们 过去没有为我们的普通股支付股息,目前也没有支付此类股息的计划.
我们 计划在我们有收益的范围内,将所有收益再投资,以支付运营成本,否则将成为并保持竞争力。 在可预见的将来,我们不打算就我们的普通股支付任何现金股息。我们不能向您保证,我们将在任何时候, 产生足够的盈余现金,将可用于分配给我们的普通股持有人作为股息。 因此,您不应期望从我们的普通股中获得现金股息。
如果股票研究分析师不发表关于我们业务的研究或报告,或者如果他们发表不利评论或下调我们的股票评级,我们的股票价格可能会下跌.
我们股票的交易市场将部分依赖股票研究分析师发布的关于我们和我们业务的研究和报告。 如果有的话。我们对这些分析师没有控制权,他们也没有承诺要写关于我们的研究报告。如果没有发布关于我们或我们业务的研究报告,或者如果一个或多个股票研究分析师 下调了我们的股票评级,或者如果这些分析师发布了其他不利的评论或停止发布关于我们或我们业务的报告,则我们的股票价格可能会下跌。
我们 可能面临更高的证券集体诉讼风险。
从历史上看, 证券集体诉讼通常是在证券市场价格下跌后针对公司提起的。 这种风险对我们尤其重要,因为生物技术和制药公司近年来经历了重大的股价波动 。如果我们被起诉,可能会导致大量成本,并转移管理层的注意力和资源, 这可能会损害我们的业务。我们为董事和高级管理人员购买了保险,我们认为该保险足以保护我们免受潜在的 索赔;但是,我们有责任满足保单规定的某些免赔额,并且在任何情况下,我们无法向您保证 保险范围将充分保护我们免受索赔。此外,保险成本可能会增加, 承保范围可能会减少。因此,我们可能无法以合理的成本维持目前的保险水平,或根本无法维持。
| 59 |
我们的章程文件和特拉华州法律可能会阻止股东认为有利的收购。
本公司经修订和重述的公司注册证书、经修订和重述的公司章程以及特拉华州法律的适用条款 可能会 延迟或阻碍涉及实际或潜在的控制权变更或管理层变更的交易,包括 股东可能会因其股份获得溢价的交易,或本公司股东可能会认为 符合其最佳利益的交易。我们修订和重述的公司注册证书和修订和重述的章程中的规定:
| ● | 限制可以召开股东大会的人; | |
| ● | 限制 我们的股东向我们的年度股东大会提出事项,或者如果没有遵循适当的程序,则禁止 在我们的年度股东大会上提名董事。 | |
| ● | 是否没有规定股东通过书面同意采取行动; | |
| ● | 是否没有规定累积投票权;以及 | |
| ● | 提供 所有董事会空缺可由当时在任的大多数董事投赞成票填补 ,即使不足法定人数。 |
DGCL第 203条可能会限制我们与实益拥有我们已发行 有表决权股票15%或更多的人进行任何业务合并的能力,除非满足某些条件。这一限制在股份收购后持续三年。 这些规定可能会加强我们的管理团队,并可能剥夺您以高于当前价格的溢价将您的股份出售给 潜在收购者的机会。这种可能无法获得控制权溢价的情况可能会降低我们普通股的价格 。
一般风险因素
会计准则的变化以及管理层与复杂会计事项相关的主观假设、估计和判断可能会对我们的财务状况和经营结果的报告产生重大影响。
会计 美国普遍接受的会计原则以及相关的会计声明、实施指南和解释 我们适用于与我们的业务相关的广泛事项,如长期资产减值会计和基于股份的薪酬,这些都是复杂的,涉及我们管理层的主观假设、估计和判断。这些规则或其解释的变化,或我们管理层对基本假设、估计或判断的变化,可能会显著改变我们的报告或预期财务业绩,或使其大幅波动。
如果未能根据《萨班斯-奥克斯利法案》第404条对财务报告进行有效的内部控制,可能会对我们的业务、财务状况和经营结果产生重大不利影响。
我们的管理层负责建立和维护对财务报告的充分内部控制。财务报告内部控制是一个旨在根据美国公认会计原则(GAAP)对财务报告的可靠性和财务报表的编制提供合理保证的过程。根据上市公司会计监督委员会(PCAOB)制定的标准,当控制的设计或操作不允许管理人员或人员在正常履行其指定职能的过程中防止或及时发现错误陈述时,财务报告的内部控制存在缺陷。PCAOB将重大缺陷定义为财务报告内部控制的缺陷或缺陷的组合,因此有合理的可能性年度或中期财务报表的重大错报将不会得到及时防止或发现和纠正。
| 60 |
根据萨班斯-奥克斯利法案第404条,我们 必须为我们向美国证券交易委员会提交的第二份10-K表格年度报告以及此后每年提交一份关于我们财务报告内部控制有效性的报告,其中包括管理层的报告。 我们的审计师还需要在我们是加速申报公司或大型加速申报公司而不再是新兴成长型公司或较小的报告公司时证明我们财务报告内部控制的有效性。如果我们无法 断言我们对财务报告的内部控制是有效的,或者在未来需要时,如果我们的独立注册会计师事务所无法对我们的财务报告内部控制的有效性发表无保留意见, 投资者可能会对我们财务报告的准确性和完整性失去信心,我们普通股的市场价格可能会受到不利影响,我们可能会成为我们普通股上市交易所、美国证券交易委员会或其他监管机构的诉讼或调查对象。这可能需要额外的财务和管理资源,并可能对我们的业务、财务状况和运营结果产生重大不利影响。
我们管理团队有限的上市公司经验可能会对我们遵守美国证券法的报告要求的能力产生不利影响 ,这可能会对我们的业务产生实质性的不利影响。
我们的 官员的上市公司经验有限,这可能会削弱我们遵守法律和监管要求的能力,例如 萨班斯-奥克斯利法案规定的要求。此类责任包括遵守联邦证券法并及时披露所需信息。 任何此类缺陷、弱点或不合规性都可能对我们遵守1934年《证券交易法》或《交易法》的报告要求的能力产生重大不利影响,《交易法》是维持我们的上市公司地位所必需的。如果我们未能履行这些义务,我们继续作为美国上市公司的能力将处于危险之中 ,在这种情况下,您可能会失去对我们公司的全部投资。
我们 发现了我们在财务报告内部控制方面的重大弱点,我们可能会在未来发现其他重大弱点,这些弱点可能会导致我们无法履行报告义务或导致我们的财务报表出现重大错报。 如果我们未能纠正任何重大弱点,或者如果我们未能建立和保持对财务报告的有效控制, 我们准确及时报告财务业绩的能力可能会受到不利影响。
有效的财务报告内部控制对于我们提供可靠的财务报告是必要的,再加上充分的披露 控制和程序旨在防止欺诈。任何未能实施所需的新的或改进的控制措施,或在实施过程中遇到困难,都可能导致我们无法履行我们的报告义务。此外,我们根据《萨班斯-奥克斯利法案》第404条进行的任何测试,或在需要时由我们的独立注册会计师事务所进行的后续测试, 可能会揭示我们在财务报告内部控制方面的缺陷,这些缺陷被认为是重大弱点,或可能需要对我们的财务报表进行 前瞻性或追溯性更改,或确定需要进一步关注或改进的其他领域。劣质的内部控制还可能导致投资者对我们报告的财务信息失去信心,这可能对我们普通股的交易价格产生负面影响 。还有一个风险是,我们和我们的独立注册公共会计师事务所(如果将来适用)都不能在规定的时间范围内得出结论,即财务报告的内部控制 按照第404条的要求有效。因此,投资者可能会对我们的财务和其他公开报告失去信心,这 将损害我们的业务和我们普通股的交易价格。
在编制本报告所包含的财务报表的过程中,我们和我们的独立注册会计师事务所发现了一个重大弱点,因为它与会计职能缺乏充分的分离有关。我们正在实施旨在改善财务报告内部控制并弥补这一重大弱点的措施 。我们打算在我们的会计基础设施内增加人员 ,以促进会计职能的适当分离。
我们 可能在财务报告的内部控制中发现未来的重大弱点,或无法满足上市公司对我们提出的要求,包括萨班斯-奥克斯利法案的要求,我们可能无法准确报告我们的财务 结果,或在法律或证券交易所法规要求的时间范围内报告这些结果。我们不能保证我们现有的材料缺陷将得到补救,或者不会存在或以其他方式发现其他材料缺陷,任何可能对我们的声誉、财务状况和运营结果产生不利影响的情况 。
| 61 |
作为一家向美国证券交易委员会报告的上市公司,我们 已经并将继续大幅增加成本,我们的管理层将被要求投入大量时间来履行合规义务.
作为一家向美国证券交易委员会报告的上市公司,我们将继续产生重大的法律、会计和其他费用,这是我们作为私营公司没有发生的 。我们受《交易所法案》的报告要求、《萨班斯-奥克斯利法案》和《多德-弗兰克华尔街改革与保护法》的报告和治理条款以及美国证券交易委员会随后实施的规则的约束,这些规则对上市公司提出了重大要求,包括要求建立和维持有效的 披露和财务控制,以及改变公司治理做法。这些法律中有重要的公司治理和报告 条款,这将增加我们的法律和财务合规成本,使一些活动更加困难、耗时 或成本高昂,还可能给我们的人员、系统和资源带来不必要的压力。我们的管理层和其他人员将需要投入大量时间来制定这些规定。此外,我们预计这些规章制度将使我们获得董事和高级职员责任保险变得更加困难和更加昂贵,我们可能被要求接受降低的保单限额和承保范围 或产生更高的费用才能获得相同或类似的承保范围。因此,我们可能更难吸引和留住合格的人员加入我们的董事会我们的董事会委员会或担任高管。
不利的 地缘政治和宏观经济发展可能对我们的业务、财务状况或经营业绩产生不利影响。
我们的业务可能会受到美国和全球经济、美国和全球金融市场状况以及 不利的地缘政治和宏观经济发展的不利影响,包括通货膨胀率上升、新冠肺炎疫情的持续不利影响、乌克兰/俄罗斯和以色列/巴勒斯坦冲突和相关制裁、银行倒闭以及与这些状况相关的经济不确定性。
例如,通货膨胀率,特别是美国的通货膨胀率最近上升到了多年未见的水平,通货膨胀的加剧可能会导致我们的运营成本增加(包括我们的劳动力成本),流动性减少,我们获得信贷或以其他可接受的条件筹集资本的能力受到限制 。为了应对不断上升的通胀,美国联邦储备委员会已经提高了利率,并可能再次提高利率,再加上政府支出的减少和金融市场的波动,可能会产生进一步增加经济不确定性和加剧这些风险的影响。
此外,在2022年2月俄罗斯入侵乌克兰和2023年10月以色列/巴勒斯坦冲突爆发后,全球金融市场经历了波动,包括对俄罗斯的经济制裁和出口管制以及俄罗斯采取的反措施。这些制裁和反措施的全面经济和社会影响,加上乌克兰和加沙正在发生的 军事冲突,可以想象,这种冲突可能会扩大,仍然不确定;然而,冲突和相关制裁 已经并可能继续导致欧洲和全球贸易、商业、价格稳定性、信贷可用性和/或供应链 连续性中断,并给全球市场带来了巨大的不确定性。虽然我们目前 不在俄罗斯、乌克兰或中东开展业务,但由于这些冲突的不利影响将继续影响我们的业务, 经营业绩可能会受到不利影响。
项目 1B。未解决的员工意见
不适用 。
项目 1C。网络安全
风险 管理和战略。我们采用流程来评估、识别和管理来自网络安全威胁的重大风险。 我们与EBM,Inc.合作,在终端采用一流的终端检测和响应功能,提供全天候SOC。我们还在可能的情况下使用MFA和地理位置强制执行。我们持续监控和维护我们的网络基础设施,包括一致的 漏洞监控和修复。我们还每季度开展网络钓鱼活动,并对我们的最终用户进行网络安全意识培训 ,以保持他们对威胁的警惕。
| 62 |
尽管我们开发和维护旨在防止网络安全威胁发生的系统和控制,并且我们有识别和缓解威胁的流程,但这些系统、控制和流程的开发和维护成本高昂,需要持续监控 ,并随着技术变化和克服安全措施的努力变得日益复杂而不断更新。此外,尽管我们做出了努力,但这些事件发生的可能性无法完全消除。随着我们将更多的信息系统外包给 供应商,与服务提供商和患者进行更多的电子交易,并更多地依赖基于云的信息系统, 相关的安全风险将会增加,我们将需要花费额外的资源来保护我们的技术和信息系统。 此外,不能保证我们的内部信息技术系统或我们的第三方承包商的系统,或者我们的 顾问为实施适当的安全和控制措施所做的努力,将足以保护我们免受系统故障、服务中断、数据恶化或丢失的影响,或防止数据在发生网络攻击、安全漏洞、工业间谍攻击或内部威胁攻击时被窃取或损坏,这些攻击可能导致财务、法律、商业或声誉损害。
截至本报告日期 ,我们不知道网络安全威胁的任何风险,包括之前的任何网络安全事件 已经或很可能对我们产生重大影响的任何风险,包括我们的业务战略、 运营结果或财务状况。
治理. 我们的高级管理团队定期评估和管理来自网络安全威胁的重大风险,包括与我们的IT团队和第三方服务提供商进行审查 。所有员工和顾问都应向我们的高级管理层报告任何 可能表明网络安全威胁或事件的不正常或可疑活动。我们董事会的审计委员会 评估我们的网络安全评估和管理政策,包括每季度与我们的高级管理人员和独立 注册会计师事务所面谈。
第 项2.属性
我们的行政办公室位于康涅狄格州06405号布兰福德商业园大道21号约12,030平方英尺的租赁办公和实验室空间内。租约将于2027年到期,取决于我们是否选择将租约再延长两次,为期五年。根据租约,我们目前每月支付14,035美元,到2024年将增加到14,385美元,外加我们按比例分摊的某些物业运营费用 。
我们 还租用了约1,093平方英尺的额外实验室空间,位于康涅狄格州格罗顿申内科塞特路93号 06340。租约将于2024年4月到期,视我们是否选择将租约再延长一年而定。根据租约,我们每月支付7,235美元,外加我们按比例分摊的某些物业运营费用。
我们 还租用了约1,868平方英尺的办公和实验室空间,位于加拿大魁北克省拉瓦尔的卡地亚大道500号。根据租约,我们每月支付6,515美元。租约将于2024年4月到期,取决于我们是否选择将租约再延长 一年。
我们 相信我们的设施足以满足我们目前的需求,并且可以根据需要以商业上合理的条款获得更多空间。
第 项3.法律诉讼
截至本报告日期 ,我们或我们的财产没有受到任何法律程序的影响。我们可能会不时地卷入在其正常业务过程中产生的法律程序和索赔。此类事项受许多不确定因素和结果的影响,不能有把握地预测。
第 项4.矿山安全信息披露
不适用 。
| 63 |
第 第二部分
第 项5.注册人普通股市场、相关股东事项和发行人回购股权证券
市场信息
我们的 普通股在纽约证券交易所的交易代码为"AZTR"。
记录持有者
截至2024年3月15日,共有34名普通股持有人。
分红政策
我们 从未宣布或支付过普通股的现金股息。我们目前打算保留收益,为我们业务的运营和扩张提供资金。
权益 薪酬计划信息
我们 采用了Azitra,Inc. 2016年股票激励计划,或2016年计划,规定授予非合格股票期权和奖励 股票期权以购买我们的普通股股份,并授予限制性和无限制性股票授予和限制性股票 单位。我们目前已根据2016年计划保留242,340股普通股。2016年计划的目的是为 符合条件的参与者提供获得本公司所有权权益的机会。我们公司的所有管理人员、董事、员工和顾问 都有资格参加2016年计划。2016年计划规定,购股权的行使价不得低于本公司普通股股份于授出日期的公平市价。
2023年3月,我们的董事会和股东批准并采纳了Azitra,Inc. 2023年股票激励计划,或2023年计划,规定授予 非合格股票期权和奖励股票期权以购买我们的普通股股份,并授予限制性的 和非限制性的股票授予和限制性的股票单位。我们目前已根据2023年计划预留2,000,000股普通股。2023年计划的目的是为合资格参与者提供收购本公司所有权权益的机会。 本公司的所有高级管理人员、董事、员工和顾问都有资格参加2023年计划。2023年计划规定 购股权不得以低于授出日期我们普通股股份的公平市值的行使价授出。
下表载列截至2023年12月31日有关我们的2016年计划和2023年计划的某些信息,根据该计划,我们的股本证券 获授权发行。
| 计划类别 | (a)行使未行使购股权时将予发行的证券数目 | (b) 加权的- 未行使期权的平均行使价 | (c) 根据股权补偿计划可供日后发行的证券数目(不包括(a)栏反映的证券) | |||||||||
| 证券持有人批准的股权补偿计划 | 1,288,255 | $ | 1.37 | 2,202,340 | ||||||||
| 未经证券持有人批准的股权补偿计划 | - | - | - | |||||||||
| 总计 | 1,288,255 | $ | 1.37 | 2,202,340 | ||||||||
| 64 |
未登记 股权证券的销售和收益的使用
未登记 股权证券销售
在2023年6月21日我们首次公开发行结束之际,我们发行了8,951,526股普通股,用于 优先股可换股票据的所有流通股的转换。我们认为,根据《证券法》第4(a)(2)条和《证券法》第506条,我们对上述证券的要约、销售和发行 作为不涉及公开发行的交易,豁免了《证券法》下的登记。所有投资者都是老练的、经认证的投资者,其定义见《证券法》第501条,并表示他们购买证券的意图仅用于投资,而不是考虑 或出售与其任何分销有关的证券,并且在这些交易中发行的股票证书、票据和 权证上放置了适当的图例。
IPO所得款项的用途
2023年6月15日,我们在表格S—1上的注册声明,经修订(文件编号333—269876)已被美国证券交易委员会宣布生效, 与我们的首次公开发行普通股有关,根据该计划,我们于2023年6月15日发行并出售了1,500股,000股普通股 ,以每股5美元的公开发行价,总收益为750万美元,净收益为600万美元,扣除承销折扣和发行费用后, 。IPO由ThinkEquity LLC承销
截至2023年12月31日 ,我们估计我们已将IPO所得款项净额中约4,661,707美元用于临床试验 和产品开发、研发、临床制造以及营运资金和其他一般企业用途 。绝大部分未动用的发行净所得款项都存放在计息账户中。根据《证券法》 第424(b)(4)条于2023年6月21日向SEC提交的最终招股说明书中所述,我们对IPO所得款项净额的使用没有重大 变化。除了在日常业务过程中支付高级职员薪酬以及支付给非雇员 董事作为董事会或董事会委员会服务的薪酬外,我们使用的首次公开募股所得款项净额均不直接或间接 支付给(i)我们的董事或高级职员或其联系人,(ii)拥有我们10%或以上普通股的人士,或 (iii)我们的关联公司。
第 项6.保留
| 65 |
项目 7.管理层对财务状况及经营业绩的讨论与分析
以下讨论和分析应与本报告其他部分所载的综合经审计财务报表及其相关附注一并阅读。本年度报告表格10—K中包含的信息并不完整描述 我们的业务或与我们普通股投资相关的风险。我们敦促您仔细审查和考虑我们在本报告和我们向美国证券交易委员会(SEC)提交的其他文件中所做的各种 披露。
在 本报告中,我们就我们的业务和前景作出并不时作出书面和口头陈述,例如 未来业绩预测、管理层计划和目标陈述、市场趋势预测,以及其他事项 1933年《证券法》第27A条和《证券法》第21E条所指的前瞻性陈述 1934年的交易法。包含"将可能导致"、"预计"、"将 继续"、"预计"、"估计"、"项目"、"相信"、"预期"、"预期"、"打算"、"目标"、"计划"、"目标"、"目标"、"计划"、"目标"、"应该"或类似表述的声明,即为前瞻性声明,这些声明可能出现在我们的文件、报告、向SEC提交 的文件、新闻稿、高级管理人员或其他代表向分析师、股东、 投资者、新闻机构和其他人所作的书面或口头报告,以及与管理层和我们的其他代表进行的讨论。
我们的 未来业绩(包括与前瞻性陈述相关的业绩)涉及许多风险和不确定性,包括本报告“风险因素”一节中包含的那些 风险。无法保证任何 前瞻性陈述中反映的结果将得以实现。任何前瞻性声明仅限于作出该声明之日。 我们的前瞻性声明基于假设,有时这些假设基于供应商、政府机构和其他来源的估计、数据、通信和其他信息 ,这些信息可能会被修改。除法律要求外,我们不承担 任何义务更新或保持最新(i)任何前瞻性声明,以反映 此类声明日期之后发生的事件或情况,或(ii)可能导致我们未来业绩与历史业绩 或趋势、我们预期或计划的业绩有重大差异的重要因素,或不时反映在任何前瞻性陈述中。
一般信息
我们 成立于2014年1月,是一家生物制药公司,专注于使用 工程蛋白质和活生物制剂产品开发精准皮肤病的创新疗法。我们是一家早期临床生物制药公司,尚未开始 商业运营。
迄今为止,我们主要通过一系列可转换优先股和可转换承兑票据的私人配售以及于2023年6月21日结束的首次公开发行(IPO)普通股,将我们的业务资本化。与我们的首次公开募股有关, 我们以每股5美元的公开发行价发行了150万股普通股。在IPO结束的同时,我们所有 的可转换优先股和可转换承兑票据已转换为总共8,951,526股普通股 。
2024年2月15日,我们完成了16,667,000股普通股的公开发行,发行价为每股0.30美元。扣除承销折扣 和发行相关费用后,我们 收到的总收益约为500万美元,净收益约为440万美元。我们已授予承销商超额配售权,以公开发行减去承销商折扣的方式购买额外2,500,000股我们普通股,为期45天,截至2024年3月29日。
除 另有说明外,本报告中的所有股份及股价金额均适用于2023年5月17日按7. 1比1的比率进行的远期股票拆分。
| 66 |
概述
我们 专注于开发使用工程蛋白质和局部活性生物制剂的精准皮肤科创新疗法。 我们已经建立了一个专有平台,其中包括一个微生物库,由大约1500种独特的细菌菌株组成, 可以对这些菌株进行独特的治疗特性筛选。该平台通过人工智能和机器学习技术进行了增强,该技术分析、预测并帮助筛选我们的菌株库中的药物类分子。该平台还利用了获得许可的 基因工程技术,可以转化以前遗传上难以处理的菌株。我们最初的重点是 开发基因工程菌株, 表皮葡萄球菌或S.表皮,我们认为是皮肤病治疗工程的最佳候选治疗物种。该特定物种在皮肤中表现出许多 描述良好的特性。截至本报告之日,我们已在微生物库中确定了超过60种不同的 细菌物种,我们认为这些细菌物种能够被改造以产生具有显著 治疗效果的活生物体或工程蛋白质。
我们 是皮肤科治疗用基因工程细菌的先驱。我们的目标是利用我们的平台和内部微生物库细菌菌株来创造新的疗法,这些疗法要么是工程生物,要么是工程蛋白质或多肽,用于治疗皮肤病。我们最初的重点是开发我们目前的候选产品,包括:
| ● | ATR—12, 一种转基因菌株。用于治疗孤儿病的表皮,Netherton 综合征,一种慢性且有时致命的皮肤疾病,估计影响大约 每100,000人中就有一人,但其患病率可能因误诊而被低估 与其他皮肤病相似2022年12月,我们提交了一份研究报告 ATR—12治疗Netherton综合征的Ib期临床试验的新药申请(IND) 2023年1月,我们收到FDA的通知,称"研究 “可以继续进行”关于拟议的1b期临床试验。提交 后 IND后生产报告,我们开始了1b期临床的运营活动 2023年12月审判。我们预计将在下半年报告初步安全性结果 2024. |
| ● | ATR—04, 一种转基因菌株。表皮用于治疗丘疹脓疱性皮疹 接受表皮生长因子受体抑制剂(或EGFRi)靶向治疗的癌症患者 疗法我们打算为某些癌症患者的1b期临床试验提交IND 2024年年中接受EGFRi靶向治疗。根据FDA对IND的批准,我们希望 我们将于2024年第四季度开始1b期临床试验。 |
| ● | ATR-01,一种用于治疗寻常型鱼鳞病的工程重组人细丝蛋白,这是一种慢性、干燥(异常干燥)、鳞片状皮肤病,估计在250人中的发病率和患病率为1。这表明美国的患者总数为130万。 我们计划在2024年完成Lead优化和IND启用研究,以支持计划于2025年下半年提交的IND申请。 |
| ● | 两个 我们和拜耳消费者正在研究和开发的单独的细菌微生物菌株 Care AG,拜耳公司的消费品部门,或拜耳,国际生命科学公司 公司我们于2019年12月与拜耳签订了联合开发协议(JDA)。 根据JDA的条款,我们负责测试我们的细菌菌株库 以及它们的天然产物的关键临床前特性。在筛选了数百个 我们和拜耳选择了两种特殊的菌株,以进一步推进 发展拜耳拥有这些菌株专利权的独家选择权。 2020年12月,拜耳购买了800万美元的B轮优先股,将 1,449,743股普通股,约占我们流通股的4.5% 截至本报告日期的普通股。 |
| 67 |

我们 还与卡内基梅隆大学和弗雷德哈钦森癌症中心(Fred Hutchson Cancer Center)的团队建立了合作关系,这两个 美国首屈一指的学术中心。我们与卡内基梅隆大学团队的合作也充分利用了全基因组测序的威力。这一合作关系正在挖掘我们专有的细菌菌株库,以获取新的药物,如肽 和蛋白质。该团队开发的人工智能/机器学习技术可以预测微生物 根据其基因序列制造的分子。然后,该系统将预测结果与通过串联质谱 和/或核磁共振成像实际得到的产物进行比较,以完善未来的预测。这些预测可以与公开可用的2D和3D蛋白质数据库进行比较,以选择类似药物的结构。
我们 拥有Fred Hutch在全球范围内的独家许可,将其专利的SyngenicDNA微环质粒(SyMPL)技术用于所有基因工程领域 ,包括发现、开发和商业化工程微生物疗法和用于皮肤病的微生物衍生肽和蛋白质。我们正在利用我们的专利权来构建质粒,以便进行以前从未实现的基因转化 。迄今为止,我们的团队已成功设计了不使用SyMPL 技术的主要治疗候选药物。然而,我们相信SyMPL将打开对扩大的微生物 物种进行基因转化的能力,我们预计我们未来的部分或所有候选产品将采用SyMPL技术。我们与 Fred Hutch的合作由微生物工程专家Christopher Johnston博士领导,也是SyMPL技术背后的创新者。
我们的 战略
除了我们的三个主要候选产品和与拜耳的合作之外,我们的目标是开发广泛的候选产品组合, 专注于扩展我们的精确皮肤病平台的应用。我们相信,我们已经在推动精准皮肤病生物制剂的开发方面确立了独特的地位。
我们 打算通过开发从我们的专利微生物库中挑选的约1,500种独特细菌菌株中挑选出来的基因工程 蛋白质,为精密皮肤病创造广泛的候选产品组合。我们的策略如下:
| ● | 生成 一家可持续的精密皮肤科公司.我们的目标是打造领先的精度 皮肤科公司拥有可持续的候选产品管道。为此,我们 专注于快速推进我们目前的活体生物多样性候选人管道,同时 积极开发新的产品。我们当前的每个候选产品 是所有的,并受待决专利申请的限制。我们预计,如果不是全部的话, 我们开发的基因工程候选产品将符合专利保护的资格。 |
| 68 |
| ● | 预付款 我们的主要候选产品ATR—12和ATR—04通过临床试验,.我们希望 报告在Netherton进行ATR—12的Ib期临床试验的初步安全性结果 综合征患者将于2024年下半年开始治疗,目前正计划开始一个阶段 1b在第二次接受EGFRi治疗的某些癌症患者中进行的ATR—04试验 2024年的一半。我们已批准ATR—12的IND,并预计在2024年年中提交ATR—04的IND。 |
| ● | 通过有选择地探索战略合作伙伴关系,最大限度地发挥我们精准皮肤科项目的潜力, 拓宽我们的平台。我们打算保留对我们所有核心技术和候选产品的重要权利。但是,我们将继续评估 战略合作伙伴可以帮助我们加快技术和候选产品开发的合作机会,提供获得协同组合的途径,或者 提供专业知识,使我们能够扩展到不同类型皮肤病的治疗 。我们还可以通过选择性地许可技术或候选产品来扩大我们平台的覆盖范围。此外,我们还将考虑将我们的某些专有技术授权给我们自己并不追求的适应症和行业 。我们相信,我们的基因工程技术和技术在医药领域之外具有适用性,包括化妆品,以及清洁燃料的生成和生物修复。 |
| ● | 利用我们的学术合作伙伴关系。我们目前与弗雷德·哈钦森癌症中心、耶鲁大学、杰克逊基因组医学实验室和卡内基梅隆大学的研究人员建立了合作伙伴关系。我们希望利用这些合作伙伴关系,并可能 扩大它们或形成其他学术合作伙伴关系,以支持我们的工程平台并 扩大我们的研发渠道。 |
| ● | 扩展 我们的其他潜在候选产品。除了我们的三个主要候选产品外, 我们的目标是开发广泛的候选产品组合,专注于扩大我们的精确皮肤病平台的应用 。我们拥有发现和开发精准皮肤病治疗产品的专有平台。我们的平台围绕由大约1,500个独特细菌菌株组成的微生物文库构建,以筛选独特的治疗特性,并利用微生物基因技术来分析、预测和设计皮肤微生物产生的蛋白质、多肽和分子。我们对难处理的微生物物种进行基因工程的能力由我们获得SyMPL技术的独家许可 独一无二地加以利用。 |
运营结果
我们 是一家早期临床生物制药公司,成立于2014年1月,运营历史有限。除了通过与拜耳的JDA获得的有限服务收入外,我们尚未开始 创收业务。根据JDA的条款,我们 负责测试我们的微生物菌株库及其天然产物的关键临床前特性,拜耳将 补偿我们的开发成本。迄今为止,我们的业务包括开发我们的专有微生物库,从我们的微生物库中鉴定、 表征和测试我们认为能够被改造 以提供显著治疗效果的某些细菌物种,以及开发我们的初始候选产品。
| 69 |
截至2023年12月31日的年度与截至2022年12月31日的年度比较
下表概述了我们在截至2023年12月31日和2022年12月31日的年度内与以下项目相关的运营结果以及这些项目的百分比变化。
| 截至十二月三十一日止的年度: | ||||||||||||||||
| 2023 | 2022 | $Change | 更改百分比 | |||||||||||||
| 服务收入关联方 | $ | 686,000 | $ | 284,000 | $ | 402,000 | 142 | % | ||||||||
| 总收入 | 686,000 | 284,000 | 402,000 | 142 | % | |||||||||||
| 运营费用: | ||||||||||||||||
| 一般和行政 | 4,493,332 | 3,639,666 | 853,666 | 23 | % | |||||||||||
| 研发 | 3,809,063 | 6,097,938 | (2,288,875 | ) | (38 | )% | ||||||||||
| 总运营费用 | 8,302,395 | 9,737,604 | (1,435,212 | ) | (15 | )% | ||||||||||
| 运营亏损 | (7,616,395 | ) | (9,453,604 | ) | 1,837,209 | (19 | )% | |||||||||
| 营业外收入(支出) | ||||||||||||||||
| 利息收入 | 1,577 | 4,818 | (3,241 | ) | (67 | )% | ||||||||||
| 利息支出 | (167,726 | ) | (251,891 | ) | 84,165 | (33 | )% | |||||||||
| 员工留任积分 | — | 229,813 | (229,813 | ) | (100 | )% | ||||||||||
| 其他收入 | — | 65,849 | (65,849 | ) | (100 | )% | ||||||||||
| 应付账款的宽免 | 56,285 | — | 56,285 | 100 | % | |||||||||||
| 可转换票据公允价值变动 | (3,630,100 | ) | (1,250,000 | ) | (2,380,100 | ) | 100 | % | ||||||||
| 其他收入(费用) | 89,886 | (25,351 | ) | 115,237 | (455 | )% | ||||||||||
| 营业外费用合计 | (3,650,078 | ) | (1,226,762 | ) | (2,423,316 | ) | 198 | % | ||||||||
| 所得税前亏损 | (11,266,473 | ) | (10,680,366 | ) | (586,107 | ) | 5 | % | ||||||||
| 所得税费用 | (17,308 | ) | — | (17,308 | ) | 100 | % | |||||||||
| 净亏损 | (11,283,781 | ) | (10,680,366 | ) | (603,415 | ) | 6 | % | ||||||||
| 优先股股息 | (1,355,347 | ) | (2,768,984 | ) | 1,413,637 | (51 | )% | |||||||||
| 普通股股东应占净亏损 | $ | (12,639,128 | ) | $ | (13,449,350 | ) | $ | 810,222 | (6 | )% | ||||||
服务 收入相关方
截至2023年12月31日止年度,我们 在拜耳JDA下产生了70万美元的服务收入,而截至2022年12月31日止年度,JDA下的服务收入为 30万美元。服务收入增加40万美元是由于 2023年可偿还开发费用增加。
| 70 |
常规 和管理
截至2023年12月31日止年度的一般 及行政成本较上年同期增加854,000美元,或23%,至450万美元。 这一增长主要是由于与上市公司的出现有关的费用增加了856,000美元,包括与会计和财务有关的额外行政费用216,000美元,增加的法律费用200,000美元, 纽约证券交易所美国证券交易所上市费用110,000美元,董事和高级管理人员保险费增加000美元,投资者和公共关系费用增加97,000美元。与去年同期相比,一般和行政费用的增加还包括知识产权法律费用354,000美元,这是由于某些专利族的减值和费用化,但由于某些职位 部分年份未填补,工资和相关费用减少378,000美元,以及其他间接费用净增加22,000美元。
研究和开发
研究和开发费用包括所有研究人员的工资和福利、支付给合同研究机构的费用、支付给研究顾问的费用和购买实验室用品的费用。这些支出被政府拨款支付的收入所抵消。 我们通过与各种联邦机构和非营利研究机构签订合同,为我们进行的一般研究创造拨款收入 。这些赠款安排也不符合收入确认的标准,根据这些赠款合同赚取的金额 被记录为负研究和开发费用。
截至2023年12月31日止年度,研发费用较上年同期减少230万美元,或37%,至380万美元。减少的主要原因是,由于我们的Netherton计划向诊所过渡, 研发相关成本减少了190万美元, 由于员工减少,工资和相关成本净减少了556 000美元,减少了183美元,由于顾问使用率下降,被由于推进CTAR计划而导致的研发费用增加 378,000美元以及其他费用净增加39,000美元所抵消。在2023或2022财年,我们分别收到了0美元和4,426美元的政府和非营利组织赠款收入。
我们 预计我们的研发费用将在未来大幅增加,主要原因是我们计划的临床试验活动 和持续开发候选产品。
营业外 收入(费用)
我们的 营业外收入(支出)包括可退还的研发信贷、认股权证估值、债务发行摊销 成本、免除应付账款、外币换算损失、雇员留用信贷、 可转换票据公允价值变动、利息收入和利息支出。截至2023年12月31日止年度,非营业收入(费用)较2022财年同期增加240万美元,或198%。增加的主要原因是 可换股票据公允价值变动导致的增加240万美元,以及员工留用信贷导致的收入减少20万美元。这一增加额被免除应付账款导致的收入增加10万美元和其他收入(支出)导致的净增加10万美元所部分抵消。
财务状况
As of December 31, 2023, we had total assets of approximately $5.1 million and working capital of approximately $0.9 million. As of December 31, 2023, our liquidity included approximately $1.8 million of cash and cash equivalents. On February 15, 2024, we completed a public offering of 16,667,000 shares of our common stock, at an offering price of $0.30 per share, in which we received net proceeds of approximately $4.4 million, after deducting underwriting discounts and offering-related expenses. After giving effect to the February 2024 public offering, we believe that our cash on hand as of the date of this report will not be sufficient to cover our proposed plan of operations over the next 12 months. We intend to seek additional funds through various financing sources, including the sale of our equity, licensing fees for our technology and joint ventures with industry partners. In addition, we will consider alternatives to our current business plan that may enable us to achieve revenue producing operations and meaningful commercial success with a smaller amount of capital. However, there can be no guarantees that such funds will be available on commercially reasonable terms, if at all. If such financing is not available on satisfactory terms, we may be unable to further pursue our business plan and we may be unable to continue operations, in which case you may lose your entire investment.
| 71 |
如果 我们通过出售股权或可转换债务证券筹集额外资本,我们普通股股东的 所有权权益将被稀释,这些证券的条款可能包括清算或其他对 普通股股东权利产生不利影响的优先权。债务融资和优先股权融资(如有)可能涉及协议,这些协议包括限制或限制我们采取特定行动的能力,例如产生额外债务、进行收购或资本支出 或宣布股息。如果我们通过与第三方的合作、战略联盟或营销、分销或许可安排筹集额外资金,我们可能不得不放弃对我们的技术、未来收入来源、研究项目 或候选产品的宝贵权利,或以可能不利于我们的条款授予许可。如果我们无法在需要时通过 股权或债务融资或其他安排筹集额外资金,我们可能会被要求延迟、限制、减少或终止我们的研究、产品开发或未来的商业化努力,或授予开发和销售我们本来希望 自己开发和销售的候选产品的权利。
截至本申请之日,管理层已确定,基于我们缺乏商业运营收入、重大亏损以及 需要筹集额外资金以支持持续运营, 对我们继续作为持续经营企业的能力存在重大疑问。
现金流
下表显示了我们在所示时期的现金流摘要:
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 用于经营活动的现金净额 | $ | (7,362,375 | ) | $ | (8,349,469 | ) | ||
| 用于投资活动的现金净额 | $ | (318,259 | ) | $ | (336,761 | ) | ||
| 融资活动提供的现金净额 | $ | 5,983,967 | $ | 4,134,624 | ||||
| 现金净减少 | $ | (1,696,667 | ) | $ | (4,551,606 | ) | ||
操作 活动
截至2023年12月31日止年度,经营活动使用了740万美元现金,主要是由于我们的净亏损1130万美元, 被390万美元的非现金项目抵消。在2022财年的可比期间,经营活动使用了830万美元的现金,主要原因是我们1070万美元的净亏损被240万美元的非现金项目抵消。
投资 活动
截至2023年12月31日止年度,投资活动使用了30万美元现金,主要由30万美元递延专利成本以及专利和商标成本驱动。在2022财年的可比期间,投资活动使用了30万美元的现金,主要 由30万美元的递延专利成本以及专利和商标成本驱动。
为 活动提供资金
在截至2023年12月31日的一年中,融资活动提供了600万美元的现金,主要由我们首次公开募股的收益推动。在2022财年的可比期间,融资活动提供了410万美元的现金,主要由可转换票据的收益推动。
| 72 |
关键的会计政策和重要的判断和估计
我们的 财务报表和相关公共财务信息基于美国公认的会计原则的应用 (“GAAP”)。GAAP要求使用对报告的资产、负债、收入和费用金额有影响的会计原则的估计、假设、判断和主观解释 。这些估计还可能影响我们的外部披露中包含的补充信息,包括有关意外情况、风险和财务状况的信息 。我们相信,我们对估计和基本会计假设的使用符合公认会计准则,并得到一致和保守的应用。我们的估计基于历史经验和我们认为在这种情况下合理的各种其他假设。 在不同的假设或条件下,实际结果可能与这些估计大不相同。我们将继续监测在编制财务报表过程中做出的重大估计。
我们的财务报表附注2总结了我们的重要会计政策。虽然所有这些重要的会计政策都会对我们的财务状况和运营结果产生影响,但我们认为其中某些政策至关重要。被确定为 关键的政策是那些对我们的财务报表有最重大影响并要求管理层使用更大程度的判断和估计的政策。实际结果可能与这些估计不同。我们的管理层认为,鉴于目前的事实和 情况,应用任何其他合理的判断或估计方法不太可能对本报告所述期间的运营业绩、财务状况或流动资金造成影响。
财务报告内部控制
财务报告内部控制是一个旨在为财务报告的可靠性和根据美国公认会计准则编制财务报表提供合理保证的过程。根据上市公司会计监督委员会(PCAOB)建立的标准,当控制的设计或操作 不允许管理层或人员在履行其指定职能的正常过程中及时防止或发现错误陈述 时,财务报告的内部控制存在缺陷。PCAOB将重大缺陷定义为财务报告内部控制的缺陷或缺陷的组合,使得年度或中期财务报表的重大错报有可能无法得到及时防止或发现和纠正。
在编制截至2023年12月31日和2022年12月31日的年度财务报表期间,我们和我们的独立注册会计师事务所 发现了一个重大弱点,因为它与会计职能缺乏充分分离有关。我们正在 实施旨在改善财务报告内部控制并弥补这一重大缺陷的措施。 我们打算在我们的会计基础设施中增加足够的人员,以促进会计职能的适当分离 并对我们内部编制的财务报表进行适当的审查。
收入 确认
正如本报告其他部分所载经审计财务报表附注2所述,根据会计准则编撰,或ASC,606,与客户签订合同的收入,实体在其客户获得对承诺的货物或服务的控制权时确认收入,其金额反映该实体预期从这些货物或服务交换中获得的对价。
为了确定确定在ASC606范围内的安排应确认的适当收入金额,我们执行以下五个步骤:(I)确定与客户的合同(S),(Ii)确定合同中承诺的货物或服务,并确定承诺的货物或服务是否为履约义务,(Iii)计量交易价格,(Iv)将交易价格分配给履约义务,以及(V)当(或作为)我们满足每项履约义务时确认收入。我们仅在实体可能收取其有权获得的对价以换取其转让给客户的商品或服务时,才将五步模型应用于合同。
当提供可选商品或服务时,我们评估选项以确定选项是否授予客户物质权利。此 确定包括选项的定价是否为客户在没有签订 合同的情况下不会收到的金额。如果我们得出结论,期权传达了一项实质性权利,它将作为一项单独的履行义务入账。在确定合同中的 履约义务时,我们确定了那些不同的承诺。当客户可以单独或与现成的资源一起从货物或服务中受益,并且货物或服务可与合同中的其他承诺分开识别时,承诺的货物或服务被视为不同的 。如果承诺不明确,则将其与合同中的其他承诺合并,直到合并后的承诺组能够区分。
| 73 |
我们 根据转让合同中承诺的货物或服务预期收到的对价金额来估计交易价格 。对价可以包括固定对价和变动对价。在每个包含可变对价的安排开始时,我们评估潜在付款的金额和收到付款的可能性。 如果很可能不会发生重大收入逆转,则可变对价包括在交易价格中。 对于包括基于许可化合物的基于销售的版税的合同,我们在相关销售发生的日期确认收入。 最后,我们通过分析相对于承诺商品和服务的独立销售价格的承诺对价,以及相对于承诺商品和服务转让的付款时间,来确定合同是否包含重要的融资部分。在每个报告日期,我们重新评估交易价格和履约义务实现的可能性以及对交易价格的相关限制。如有必要,我们会调整交易价格,根据之前受限制的金额的进度记录累计追赶进度。
收入 在(或作为)履行义务控制权转移到客户时确认。当综合履约义务 包含承诺的许可证和相关服务或其他承诺时,需要管理层判断以确定收入确认的适当时间 。为此,我们必须确定合同中的一个或多个主要承诺,以确定收入是在某个时间点确认还是在某个时间确认。如果随着时间的推移,我们必须确定适当的进展衡量标准。如果许可证被认为是履行义务中的主要承诺,我们必须确定许可证的性质,无论是功能性的还是象征性的 知识产权,以得出时间点或长期收入确认最合适的结论。确定功能性 或象征性知识产权需要评估客户是否能够在其当前状态下利用许可证并从中受益,或者许可证的效用是否依赖于我们正在进行的活动或受我们的影响或与我们相关联。
在每个报告日期,我们都会计算一段时间内转移的履约义务的进度。计算一般采用基于迄今发生的费用与估计总费用之比的投入计量,以完成履约义务的转移。
研究和开发费用
研究和开发费用主要包括与我们的候选产品开发相关的成本。我们会按实际发生的费用来支付研发费用。
我们 根据对已完成工作的比例的估计,为我们的供应商执行的临床前研究和临床试验活动计提费用。我们通过审查合同、供应商协议和采购订单,并通过与我们的内部临床人员和外部服务提供商讨论试验或服务的进度或完成阶段以及为此类服务支付的商定费用来确定估计数。然而,临床试验的实际成本和时间是高度不确定的,受到风险的影响,可能会根据包括我们的临床开发计划在内的许多因素而变化。
我们 根据当时已知的事实和情况,在财务报表中对截至每个资产负债表日期的预付/应计费用进行估计。如果服务执行的实际时间或工作水平与估计值不同,我们将相应地调整应计项目。不可退还的商品和服务预付款,包括将在未来研发活动中使用的临床试验费用、工艺开发费用或临床用品的制造和分销费用,将延期 并在相关商品或服务被消费或提供服务期间确认为费用。
| 74 |
基于股份的薪酬
我们 根据授予日基于股票的奖励的估计公允价值来衡量所有基于股票的奖励的薪酬支出。 我们使用Black-Scholes期权定价模型对基于股票的奖励进行估值。我们以直线方式确认所需服务期内的补偿费用 ,这通常是奖励的授权期。我们尚未根据市场或业绩条件颁发奖励,其归属取决于市场或业绩条件。
柏力克—舒尔斯期权定价模式要求使用主观假设,其中包括预期股价波动率和 相关普通股在授出日期的公允价值。请参阅 本报告其他部分的经审计财务报表附注10,了解我们在应用柏力克—舒尔斯期权定价模型时使用的某些特定假设 以确定我们授予的奖励的估计公允价值。
评估权证的公允价值
我们 采用布莱克-斯科尔斯方法在每个报告期对我们的未偿还认股权证进行估值,公允价值的变化在 经营报表中确认。认股权证负债的估计公允价值是使用第3级投入确定的。布莱克-斯科尔斯模型中固有的假设与预期股价波动、预期寿命、无风险利率和股息收益率有关。我们根据市场参与者的假设估计我们普通股的波动率,并与用于评估我们股票期权的波动率相匹配。 无风险利率基于授予日的美国财政部零息收益率曲线,到期日与认股权证的预期剩余期限相似。我们的未清偿认股权证的预期寿命假设与其剩余的合同期限相等。股息率基于历史利率,公司预计历史利率将保持在零。
最近 会计声明
请参阅 本报告其他地方的经审计财务报表附注2,了解适用于我们财务报表的最新会计声明的描述 。
第 7A项。关于市场风险的定量和定性披露
不适用 。
| 75 |
第 项财务报表和补充数据
合并财务报表索引
| 独立注册会计师事务所报告(PCAOB ID号 |
F-1 |
| 截至2023年12月31日和2022年12月31日的合并资产负债表 | F-2 |
| 截至2023年12月31日和2022年12月31日的合并业务报表 | F-3 |
| 截至2023年及2022年12月31日止年度的股东亏损综合变动表 | F-4 |
| 截至2023年12月31日和2022年12月31日的合并现金流量表 | F-5 |
| 合并财务报表附注 | F-6 |
| 76 |
独立注册会计师事务所报告{br
致 公司股东和董事会
Azitra, Inc.
Grassi & Co. CPA,P.C.
纽约杰里科
对财务报表的意见
我们 审计了随附的Azitra,Inc.的资产负债表。(the截至2023年12月31日及2022年12月31日止年度的相关 经营、股东权益和现金流量表以及相关附注(统称 财务报表)。我们认为,财务报表在所有重大方面公允列报了公司截至2023年和2022年12月31日的财务状况,以及截至该日止年度的经营成果和现金流量, 符合美利坚合众国公认的会计原则。
对公司作为持续经营企业的持续经营能力有很大的怀疑
随附财务报表的编制假设Azitra,Inc.,将继续经营下去如财务报表附注 1所述,公司的重大经营亏损对该实体的持续经营能力产生了重大疑问。管理层对事件和条件的评估,以及管理层关于 这些事项的计划,也在附注1中描述。财务报表不包括可能由这种不确定性的结果导致的任何调整 。我们的意见不会就此事作出修改。
征求意见的依据
这些 财务报表由公司管理层负责。我们的责任是根据我们的审计对公司的财务报表发表意见。我们是一家在美国上市公司会计监督委员会(PCAOB)注册的公共会计师事务所,根据美国联邦证券 法律以及美国证券交易委员会和PCAOB的适用规则和法规,我们必须与公司保持独立。
我们 按照PCAOB的标准进行审计。这些标准要求我们计划和执行审计,以获得关于财务报表是否没有重大错报的合理保证,无论是由于错误还是舞弊。公司 不需要对其财务报告的内部控制进行审计,也没有聘请我们进行审计。作为我们审计的一部分,我们需要了解财务报告的内部控制,但不是为了对公司财务报告内部控制的有效性发表意见。因此,我们不表达这样的意见。
我们的 审计包括执行程序,以评估财务报表重大错报的风险,无论是由于错误 还是欺诈,以及执行应对这些风险的程序。这些程序包括在测试的基础上审查与财务报表中的金额和披露有关的证据。我们的审计还包括评估管理层使用的会计原则和作出的重大估计,以及评估财务报表的整体列报。我们相信,我们的审计 为我们的观点提供了合理的基础。

我们 自2022年以来一直担任本公司的审计师。
2024年2月23日
| F-1 |
AZITRA, Inc.
合并资产负债表
| 2023年12月31日 | 2022年12月31日 | |||||||
| 资产 | ||||||||
| 流动资产: | ||||||||
| 现金和现金等价物 | $ | $ | ||||||
| 应收账款 | ||||||||
| 应收账款关联方 | ||||||||
| 应收税额抵免 | ||||||||
| 应收所得税 | ||||||||
| 递延发售成本 | ||||||||
| 预付费用 | ||||||||
| 流动资产总额 | ||||||||
| 财产和设备,净额 | ||||||||
| 融资租赁使用权资产 | ||||||||
| 经营性租赁使用权资产 | ||||||||
| 无形资产,净额 | ||||||||
| 递延专利成本 | ||||||||
| 其他资产 | ||||||||
| 总资产 | $ | $ | ||||||
| 负债、优先股和股东权益 | ||||||||
| 流动负债: | ||||||||
| 应付帐款 | $ | $ | ||||||
| 流动融资租赁负债 | ||||||||
| 当期经营租赁负债 | ||||||||
| 应计费用 | ||||||||
| 合同责任-关联方 | ||||||||
| 流动负债总额 | ||||||||
| 长期融资租赁负债 | ||||||||
| 长期经营租赁负债 | ||||||||
| 认股权证法律责任 | ||||||||
| 可转换应付票据,净额 | ||||||||
| 总负债 | ||||||||
| 承付款和或有事项(附注14) | ||||||||
| 优先股: | ||||||||
| A系列可转换优先股;美元票面价值;2023年12月31日和2022年12月31日授权的股票;和分别于2023年12月31日和2022年12月31日发行和发行的股份;清算价值为$ | ||||||||
| A系列-1可转换优先股;$票面价值;2023年12月31日和2022年12月31日授权的股票;和分别于2023年12月31日和2022年12月31日发行和发行的股份;清算价值为$ | ||||||||
| B系列可转换优先股;美元票面价值;2023年12月31日和2022年12月31日授权的股票;和分别于3023年12月31日及2022年12月31日发行及发行在外的股份;清盘价值为美元 | ||||||||
| 股东权益(亏损) | ||||||||
| 普通股;美元面值,2023年12月31日和2022年12月31日授权的股票,和于二零二三年及二零二二年十二月三十一日已发行及流通股 | ||||||||
| 额外实收资本 | ||||||||
| 累计赤字 | ( | ) | ( | ) | ||||
| 股东权益合计(亏损) | ( | ) | ||||||
| 负债、优先股和股东权益总额(赤字) | $ | $ | ||||||
附注是这些合并财务报表的组成部分。
| F-2 |
AZITRA, Inc.
合并的 运营报表
| 截至12月31日止年度, | ||||||||
| 2023 | 2022 | |||||||
| 服务收入关联方 | $ | $ | ||||||
| 总收入 | ||||||||
| 运营费用: | ||||||||
| 一般和行政 | ||||||||
| 研发 | ||||||||
| 总运营费用 | ||||||||
| 运营亏损 | ( | ) | ( | ) | ||||
| 营业外收入(费用) | ||||||||
| 利息收入 | ||||||||
| 利息支出 | ( | ) | ( | ) | ||||
| 员工留任积分 | ||||||||
| 其他收入 | ||||||||
| 应付账款的宽免 | ||||||||
| 可转换票据公允价值变动 | ( | ) | ( | ) | ||||
| 其他收入(费用) | ( | ) | ||||||
| 营业外费用合计 | ( | ) | ( | ) | ||||
| 所得税前亏损 | ( | ) | ( | ) | ||||
| 所得税费用 | ( | ) | ||||||
| 净亏损 | ( | ) | ( | ) | ||||
| 优先股股息 | ( | ) | ( | ) | ||||
| 普通股股东应占净亏损 | $ | ( | ) | $ | ( | ) | ||
| 每股基本和稀释后净亏损 | ) | ) | ||||||
| 加权平均已发行普通股、基本普通股和稀释普通股 | ||||||||
附注是这些合并财务报表的组成部分。
| F-3 |
AZITRA, Inc.
合并的股东亏损变动表
| A系列可转换优先股 | A-1系列可转换优先股 | B系列可转换优先股 | 普通股 | 额外的实收- | 累计 |
股东总数 股权 | ||||||||||||||||||||||||||||||||||||||
| 股票 | 金额 | 股票 | 金额 | 股票 | 金额 | 股票 | 金额 | 资本 | 赤字 | (赤字) | ||||||||||||||||||||||||||||||||||
| 余额-2021年12月31日 | $ | | $ | $ | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||||||||||||||||||||||
| 基于股票的薪酬 | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||
| 股票期权的行使 | — | — | — | |||||||||||||||||||||||||||||||||||||||||
| 净亏损 | — | — | — | — | ( | ) | ( | ) | ||||||||||||||||||||||||||||||||||||
| 余额-2022年12月31日 | $ | $ | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||||||||||||||||||||||||
| 发行B系列可转换优先股 | — | — | — | |||||||||||||||||||||||||||||||||||||||||
| 转换可转换应付票据 | — | — | — | |||||||||||||||||||||||||||||||||||||||||
| 转换优先股 | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||||||||||||||
| 首次公开发行,扣除发行成本 | — | — | — | |||||||||||||||||||||||||||||||||||||||||
| 基于股票的薪酬 | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||
| 净亏损 | — | — | — | — | ( | ) | ( | ) | ||||||||||||||||||||||||||||||||||||
| 余额-2023年12月31日 | $ | $ | $ | $ | $ | ( | ) | $ | ||||||||||||||||||||||||||||||||||||
附注是这些合并财务报表的组成部分。
| F-4 |
AZITRA, Inc.
合并现金流量表
| 截至12月31日止年度, | ||||||||
| 2023 | 2022 | |||||||
| 经营活动的现金流: | ||||||||
| 净亏损 | $ | ( | ) | $ | ( | ) | ||
| 对净亏损与经营活动中使用的现金净额进行的调整: | ||||||||
| 折旧及摊销 | ||||||||
| 债务贴现摊销 | ||||||||
| 使用权资产摊销 | ||||||||
| 可转换票据的应计利息 | ||||||||
| 基于股票的薪酬 | ||||||||
| 认股权证负债的公允价值变动 | ( | ) | ( | ) | ||||
| 可转换票据公允价值变动 | ||||||||
| 应付账款的宽免 | ( | ) | ||||||
| 财产和设备处置损失 | ||||||||
| 无形资产减值和递延专利成本 | ||||||||
| 经营性资产和负债变动情况: | ||||||||
| 应收账款 | ( | ) | ||||||
| 预付费用 | ( | ) | ( | ) | ||||
| 其他资产 | ( | ) | ||||||
| 应收税额抵免 | ( | ) | ||||||
| 应付账款和应计费用 | ( | ) | ||||||
| 应收所得税 | ( | ) | ||||||
| 经营租赁负债 | ( | ) | ( | ) | ||||
| 合同责任 | ( | ) | ||||||
| 用于经营活动的现金净额 | ( | ) | ( | ) | ||||
| 投资活动产生的现金流: | ||||||||
| 购置财产和设备 | ( | ) | ( | ) | ||||
| 出售财产和设备所得收益 | ||||||||
| 递延专利成本的资本化 | ( | ) | ( | ) | ||||
| 许可证的资本化 | ( | ) | ||||||
| 专利和商标费用的资本化 | ( | ) | ( | ) | ||||
| 用于投资活动的现金净额 | ( | ) | ( | ) | ||||
| 融资活动产生的现金流 | ||||||||
| 可换股票据所得款项(扣除发行成本) | ||||||||
| 支付递延发售费用 | ( | ) | ||||||
| 融资租赁本金支付 | ( | ) | ||||||
| 扣除交易成本后的首次公开募股所得款项 | ||||||||
| 行使股票期权所得收益 | ||||||||
| 融资活动提供的现金净额 | ||||||||
| 现金和现金等价物净变化 | ( | ) | ( | ) | ||||
| 期初现金及现金等价物 | ||||||||
| 期末现金及现金等价物 | $ | $ | ||||||
| 补充披露非现金投融资信息: | ||||||||
| 取得使用权资产以换取租赁负债 | $ | $ | ||||||
| 获得使用权资产以换取融资租赁负债 | $ | $ | ||||||
| 将票据转换为普通股 | $ | $ | ||||||
| 票据转换为B系列可转换优先股 | $ | $ | ||||||
| 将A、A—1和B系列可转换优先股转换为普通股 | $ | $ | ||||||
附注是这些合并财务报表的组成部分。
| F-5 |
AZITRA, Inc.
合并财务报表附注
截至二零二三年及二零二二年十二月三十一日止年度
1. 业务的组织和性质
Azitra, Inc.成立于2014年1月2日。这是一家合成生物学公司,专注于筛选和基因工程的 皮肤微生物。我们的使命是发现和开发新的疗法,创造治疗皮肤病的新范式。该公司的 发现平台筛选了具有有益效果的天然细菌细胞。然后对这些微生物进行基因组测序 并进行工程改造以制造细胞疗法、重组治疗蛋白质、肽和小分子,用于精确治疗 皮肤病。2023年5月17日,公司更名为“Azitra Inc”。
公司在加拿大蒙特利尔设有办事处,用于开展某些研究活动。在2022年和2023年期间,该地点和在那里进行的操作保持一致。本公司亦于二零二一年在康涅狄格州格罗顿开设制造及实验室。
远期 股票拆分、面值变动和首次公开发行
2023年6月,公司完成了首次公开募股(IPO),发行并出售 以
对公众的价格,每股该股于2023年6月16日开始在纽约美国证券交易所交易,代码为“AZTR”。
本公司从发售中收到的净所得款项为美元
紧接
公司注册声明生效前,公司实施了
去 关注事项
综合财务报表乃按持续经营基准编制,该基准假设本公司将于可预见将来继续经营
,并预期在正常业务过程中变现资产及清偿负债。
然而,管理层已确定以下条件和事件,这些条件和事件对公司
的持续经营能力造成了不确定性。截至2023年12月31日止年度,本公司累计亏损为美元。
管理层 计划继续通过股权和债务融资筹集资金,为运营和营运资金需求提供资金;然而,公司 将需要大量额外资金来完成其产品的开发,并为 公司预计在未来几年内产生的额外损失提供资金,因为公司尚未获得产品收入。无法保证 公司将成功获得额外融资以满足其运营需要。
这些 条件和事件对公司自财务报表可供发布之日起12个月内持续经营的能力造成了不确定性。财务报表不包括如果公司无法持续经营,则可能需要进行的任何调整 。
| F-6 |
AZITRA,INC.
合并财务报表附注
截至二零二三年及二零二二年十二月三十一日止年度
2. 重要会计政策摘要
会计基础
公司的财务报表是根据美国公认会计原则(“美国公认会计原则”)编制的。
使用预估的
按照美利坚合众国公认的会计原则编制财务报表需要 管理层作出影响资产负债表日期资产和负债报告金额的估计和假设。 这些估计和假设是基于当前事实、历史经验和在当时情况下被认为是合理的各种其他因素 ,这些结果构成了对资产和负债账面值作出判断的基础,以及 记录无法从其他来源显而易见的费用。虽然管理层认为估计和假设是适当的,但实际结果可能与这些估计存在重大不利差异。
现金 和现金等价物
就资产负债表及现金流量表而言,本公司将所有手头现金、活期存款及所有原始到期日为三个月或以下的高流动性投资视为现金等价物。
财产 和设备
财产和设备按成本入账。折旧是在估计使用年限内使用直线法计算的,其范围为
应收账款
公司的应收账款按成本减去预期信用损失准备金计提。本公司定期评估其应收账款,并根据过去的核销、催收和当前状况计提预期信贷损失准备。在2023年12月31日和2022年12月31日,没有信贷损失拨备。
延期的 产品成本
公司将递延发售成本资本化,这些成本主要包括与公司首次公开募股相关的直接、递增法律、专业、会计和其他 第三方费用。2023年6月,本公司完成首次公开募股,并在股东权益表中将该等金额与首次公开募股的总收益相抵销。在第四季度,该公司产生了67,859美元的与后续发行相关的成本。
租契
2016年2月,财务会计准则委员会(“FASB”)发布了ASU第2016-02号租赁(“主题842”)。ASU 2016-02要求承租人在资产负债表上列报所有租期超过12个月的使用权资产和租赁负债 。
在计算ASU 2016-02的影响时,公司选择了过渡方法,因此没有重述可比期间。本公司 选择将非租赁组成部分作为其相关租赁组成部分的一部分进行会计处理。租赁会计处理涉及重大 判断,包括对租赁期、租赁付款和贴现率作出估计。根据该指引, 本公司就所有租期超过12个月的租赁确认使用权资产及租赁负债。租赁根据协议的经济实质分类为 经营租赁或融资租赁。
| F-7 |
AZITRA,INC.
合并财务报表附注
截至二零二三年及二零二二年十二月三十一日止年度
2. 重要会计政策摘要(续)
公司拥有建筑物的运营租约。目前,该公司有3个经营性租赁,ROU资产和租赁负债总额为
$
截至2023年12月31日,公司拥有经营性使用权资产,净价值为$
于
2023年,本公司就若干设备的使用订立租赁,该租赁被分类为融资租赁。融资租赁的期限为
无形资产
无形资产包括商标和专利。与专利和商标申请的提交和起诉直接相关的所有成本都是资本化的。专利在正式批准后在其各自的剩余使用寿命内摊销。商标具有无限的生命期 。
公司按照ASC主题350对其他无限期无形资产进行核算,商誉及其他无形资产
(ASC 350)。ASC 350要求,具有无限使用寿命的无形资产必须至少每年进行一次减值测试
,或者当事件或情况变化表明账面值可能无法收回时。使用寿命有限的无形资产将继续在其使用寿命内摊销。与许可协议有关的减值损失为美元
延期的专利成本
递延
专利费用是指与提交待批准专利申请有关的法律和归档费用。
这些递延成本将在专利获得正式批准后在其估计使用寿命内开始摊销。如果专利
未被颁发,则与专利相关的费用将在专利被驳回的年份计入费用。截至2023年12月31日和2022年12月31日止年度,与递延专利成本相关的减值损失为美元
长期资产减值
根据ASC主题360-10《长期资产减值或处置会计》(ASC 360-10),本公司的政策 是在发生事件或环境变化表明资产的账面价值可能无法收回时,审查其长期资产的减值。为配合本次审核,本公司亦会重新评估该等资产的折旧期。 当资产的使用及最终处置所产生的未贴现预期未来现金流量总和少于其账面金额时,本公司确认减值亏损。如果一项资产被视为减值,应确认的减值按该资产的账面价值超过该资产的公允价值的金额计量,该金额是根据该资产产生的未来营运现金流量净值的现值 确定的。
| F-8 |
AZITRA,INC.
合并财务报表附注
截至二零二三年及二零二二年十二月三十一日止年度
2. 重要会计政策摘要(续)
可转换债务和权证会计
认股权证
根据对权证具体条款的评估和ASC 480中适用的权威指导,公司将权证作为股权分类或负债分类工具进行会计处理。区分负债与股权(“ASC 480”)和ASC 815,衍生工具和套期保值(“ASC 815”)。评估考虑认股权证是否为ASC 480所指的独立财务工具,是否符合ASC 480所指的负债定义,以及认股权证是否符合ASC 815中有关股权分类的所有要求,包括权证是否与本公司本身的普通股挂钩,以及其他股权分类条件。这项评估需要使用专业判断,在权证发行时以及在权证尚未结清的每个季度结束日进行。
对于 满足所有股权分类标准的已发行认股权证,认股权证必须在发行时记录为额外 实缴资本的一部分。对于不符合所有权益分类标准的已发行认股权证,认股权证 须按其于发行日期及其后各结算日的初始公允价值入账。认股权证的估计公允价值的变动在经营报表中确认为其他收入项下的非现金收益或亏损。
可转换债务
When the Company issues debt with a conversion feature, it first assesses whether the debt should be accounted for in accordance with ASC 480 – Distinguishing Liabilities from Equity. If the debt does not meet the criteria of an ASC 480 liability, the note’s conversion features require bifurcation in accordance with ASC 815 – Derivatives and Hedging. If the Company determines the embedded conversion feature requires bifurcation in accordance with ASC 815, the Company also considers if it can elect the fair value option. The Convertible Notes were converted into the Company’s common stock on the Closing Date of the Company’s IPO. Under 815-15-25, the election can be at the inception of a financial instrument to account for the instrument under the fair value option under ASC 825. The Company has made such election for the 2022 Convertible Notes. Using the fair value option, the convertible promissory note is to be recorded at its initial fair value on the date of issuance, and each balance sheet date thereafter. The Company evaluates the change based on the conversion price at the current market value. When recognized, changes in the estimated fair value of the notes are recognized as a non-cash gain or loss in Other Income (Expense) on the statements of operations. The Company elected the fair value option for the 2022 Convertible Notes and recorded the notes at their initial fair values with subsequent changes in fair value recorded in earnings.
可转换 优先股
由于 可转换优先股股东在发生被视为清盘事件时拥有清算权,而在某些情况下, 并非完全在本公司的控制范围内,需要赎回当时尚未发行的可转换优先股,因此,本公司将可转换优先股归入资产负债表夹层股权。
如附注8所述,于本公司首次公开招股截止日期,可换股优先股转换为本公司的普通股。
收入
公司遵循五根据ASC 606确认与客户的合同收入的步骤 (“ASC 606”),包括:
| ● | 步骤 1:确定与客户的合同(S) |
| F-9 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
2.主要会计政策摘要(续)
| ● | 步骤 2:确定合同中的履约义务 | |
| ● | 步骤 3:确定成交价格 | |
| ● | 步骤 4:将交易价格分摊到合同中的履约义务 | |
| ● | 步骤 5:在履行履约义务时(或作为)确认收入 |
该公司通过与一家研究合作伙伴签订的联合开发协议获得服务收入。随着合同工作的完成,公司使用输入法确认与协议研究和开发方面相关的收入。
公司还产生赠款收入,这是从与各联邦机构和非营利性研究机构签订的合同中收到的款项,用于公司为进一步产品开发而进行的一般研究,因此被视为对公司的贡献 。合同的期限一般为一年或更长时间,任何一方都可以取消。本公司的结论是,由于本公司 是该安排的唯一积极参与者,因此赠款安排不符合被视为FASB ASC主题808项下的合作安排的标准。赠款安排也不符合主题606下的收入确认标准,因为美国政府不符合客户的定义。
如果发生了符合条件的费用,并且可实现或已实现并赚取了支付权,则根据这些赠款合同赚取的金额 将作为研发费用的减少额入账。本公司相信这一政策与主题606一致,以确保 确认反映了向客户转让承诺的商品或服务的金额,反映了 公司希望有权获得的对价,以换取这些商品或服务,即使没有主题606中定义的交换。 此外,公司已确定,将收到的金额确认为已产生的成本和可实现的金额 类似于主题606下的随着时间推移转移服务控制权的概念。
预付补助金的收据 如果未按照赠款中与允许成本有关的条件使用,或在执行合同之前从研究合作伙伴收取与服务收入安排有关的资金时,应返还给供资机构,在随附的资产负债表中被归类为合同负债。
研究和开发
本公司按照会计准则编纂(ASC)小标题730-10核算研发费用,研究和开发
。因此,内部研究和开发成本在发生时计入费用。研发成本包括与人力、材料和用品相关的成本。将用于未来研发活动的货物和服务的预付款在活动完成或收到货物时计入费用。发生的研发成本
为$
截至2023年12月31日和2022年12月31日,该公司有一笔应收州税收抵免美元
公司根据ASC 718对股票薪酬进行核算,薪酬--股票薪酬(ASC 718)。ASC 718要求 员工的股票期权和根据股票参与计划购买股票的权利必须按公允价值入账。ASC 718要求 与以股份为基础的支付交易相关的补偿成本在财务报表中确认为运营费用。 根据该方法,所有授予或修改的奖励的补偿成本均按授予日的估计公允价值计量,并 计入员工提供服务以换取奖励的归属期间的补偿费用。对于绩效条件影响归属的奖励 ,公司在确定可能达到绩效条件时确认补偿费用。当没收发生时,本公司确认没收的影响。
| F-10 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
2.主要会计政策摘要(续)
该公司使用布莱克-斯科尔斯期权定价模型来确定其股票期权的公允价值。布莱克-斯科尔斯模型包括各种假设,包括标的普通股的价值、股票期权的预期寿命、预期波动率和预期无风险利率。这些假设反映了公司的最佳估计,但它们涉及基于一般不受公司控制的市场情况的固有不确定性 。因此,如果使用其他假设,基于股票的薪酬成本可能会受到重大影响。此外,如果公司对未来的赠款使用不同的假设,基于股票的薪酬成本可能会在未来期间受到重大影响。
公司按照会计准则更新(ASU)2018-07号更新的ASC 718的规定,对发行给非员工的股权工具进行会计核算。对非员工股份支付会计的改进,这将ASC 718的范围扩大到包括对非员工的基于股票的支付交易。
在对使用Black-Scholes期权定价模型发行的期权进行估值时,使用了以下假设:
预期波动 。本公司股票的预期波动率是根据同行公司的平均波动率估计的。
预期的 期限。期权的预期期限采用简化方法估计,该方法基于每个授予的归属期限和合同 期限,或者对于具有分级归属的奖励的每个归属部分。
基础 普通股价值。在公司首次公开募股之前,公司股票的基本普通股价值由第三方估值专家进行估计,当时公司利用其在授予日在纽约证券交易所美国证券交易所的交易价格。
无风险利率 。该公司的无风险利率以美国国库券的隐含收益为基础,其条款等于标的授权书的预期期限。
股息 收益率。布莱克-斯科尔斯估值模型要求以单一的预期股息收益率作为输入。本公司过去没有就普通股支付股息 ,近期也不会对普通股支付股息。因此,公司使用的股息 收益率百分比为零。
所得税 税
公司采用ASC 740中规定的所得税责任会计方法,所得税会计。根据此方法,递延税项资产及负债按资产及负债的账面值及计税基准之间的暂时性差异及经营亏损结转净额的预期未来税务后果确认,均按现行的税率计算。
管理层 已评估了与不确定所得税状况相关的ASC指引的影响,并得出结论认为,本公司于2023年12月31日和2022年12月31日没有重大 不确定所得税状况风险。截至2023年12月31日,税务机关尚未审查公司的所得税 纳税申报表。
| F-11 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
2.主要会计政策摘要(续)
公司使用本期发行在外的普通股加权平均股数 计算归属于普通股股东的每股加权平均股基本净亏损。本公司使用本期发行在外的普通股和潜在摊薄普通股的加权平均数计算归属于普通股股东的每股加权平均股的摊薄净亏损。 潜在发行在外的普通股包括股票期权、认股权证和优先股,如果其影响具有反摊薄作用,则不包括在内。 截至2023年及2022年12月31日止财政年度,摊薄加权平均普通股不包括任何增量股份。
某些 风险和不确定性
公司的活动受到重大风险和不确定性的影响,包括无法获得额外资金 以正确执行公司的业务计划的风险。本公司面临制药行业公司常见的风险 ,包括但不限于本公司或其竞争对手开发新技术创新、依赖 关键人员、依赖第三方制造商、保护专利技术以及遵守法规要求。
公允价值计量
公司按公允价值经常性地承担某些负债。使用由三个级别组成的公允价值层次结构来确定公允价值评估技术的输入的优先顺序:
| ● | 级别 1-投入基于在活跃市场交易的相同工具的可观察或报价 。 | |
| ● | 第2级-投入基于活跃市场中类似工具的报价, 非活跃市场中相同或类似工具的报价,以及基于模型的估值技术,其所有重要假设均可在市场上观察到,或可由资产或负债的基本完整期限的可观察市场数据来证实。 | |
| ● | 级别 3-投入通常是不可观察的,通常反映了管理层对市场参与者在为资产或负债定价时将使用的假设的估计。因此,公允价值是使用基于模型的技术来确定的,这些技术包括期权定价模型、贴现现金流模型和类似技术。 |
在确定公允价值时,本公司采用估值技术,最大限度地利用可观察到的投入和最大限度地减少使用不可观察到的投入,并在评估公允价值时考虑交易对手信用风险。
金融工具
公司的金融工具主要包括应收账款、应付账款、应计负债和长期债务。对于应收账款、应付账款和应计负债,由于该等票据的短期到期日,账面金额接近公允价值。本公司长期债务的估计公允价值接近账面价值。
信贷风险和资产负债表外风险集中
可能使公司面临集中信贷风险的金融工具主要包括现金和现金等价物。现金 等价物偶尔投资于存单。公司将其每项现金余额与高质量和 认可的金融机构保持,因此,此类资金不会面临超出与商业银行关系相关的正常信贷风险 之外的异常信贷风险。在金融机构的存款可能不时超过联邦保险限额。 公司未经历任何现金存款损失。本公司以货币市场账户的形式在管理层认为信誉良好的金融机构保存部分现金和现金等价物余额。
| F-12 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
2.主要会计政策摘要(续)
最近 会计声明
2019年12月,FASB发布了ASU第2019-12号,所得税(话题740):简化所得税的会计核算. 本标准通过删除以前规定的各种例外情况,以及提供 额外的所得税报告要求,简化了所得税的会计处理。会计准则于二零二二年一月一日对本公司生效。本公司已于2022年1月1日采用 该准则,该准则对财务报表没有重大影响。
2016年6月,FASB发布了ASU编号1026—13, 金融工具—信用损失(主题326):金融工具信用损失的度量(ASC 326)以预期损失方法取代已发生损失方法。此标准称为 当前预期信贷损失(“CECL”)方法。CECL本准则自2023年1月1日起对本公司生效。本公司已采纳该准则,自二零二三年一月一日起生效,对财务报表并无重大影响。
管理层 不认为最近发布但尚未生效的任何其他会计准则会对随附的财务报表产生实质性影响。随着新的会计公告的发布,公司将采用在这种情况下适用的会计公告。
3. 员工留任积分
CARES法案提供了员工留用抵免("CARES员工留用抵免"),这是一种针对
某些就业税的可退还税收抵免,最高可达$
4. 财产和设备
于2023年12月31日及2022年12月31日,物业 及设备包括以下各项:
| 2023年12月31日 | 2022年12月31日 | |||||||
| 实验室设备 | $ | $ | ||||||
| 计算机和办公设备 | ||||||||
| 家具和固定装置 | ||||||||
| 租赁权改进 | ||||||||
| 建筑设备 | ||||||||
| 总资产和设备 | ||||||||
| 减累计折旧和摊销 | ( | ) | ( | ) | ||||
| 财产、厂房和设备合计(净额) | $ | $ | ||||||
| F-13 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
4.物业及设备(续)
折旧
费用为$
截至2023年12月31日和2022年12月31日止年度,处置物业和设备的损失记录为美元,
5. 无形资产
无形资产 由以下资产组成:
2023年12月31日:
| 预计使用寿命 | 总金额 | 累计摊销 | 减损 | 净额 | ||||||||||||||
| 商标 | $ | $ | $ | $ | ||||||||||||||
| 专利 | ||||||||||||||||||
| 许可协议 | ||||||||||||||||||
| 无形资产 | $ | $ | $ | $ | ||||||||||||||
2022年12月31日 :
| 预计使用寿命 | 总金额 | 累计摊销 | 减损 | 净额 | ||||||||||||||
| 商标 | $ | $ | $ | $ | ||||||||||||||
| 专利 | ||||||||||||||||||
| 许可协议 | ||||||||||||||||||
| 无形资产 | $ | $ | $ | $ | ||||||||||||||
截至2023年12月31日和2022年12月31日止年度,与无形资产有关的摊销费用为美元,
6. 应计费用
应计费用 包括以下费用:
| 2023年12月31日 | 2022年12月31日 | |||||||
| 应计费用: | ||||||||
| 员工工资单和奖金 | $ | $ | ||||||
| 休假 | ||||||||
| 研究和开发项目 | ||||||||
| 利息 | ||||||||
| 专业费用 | ||||||||
| 其他 | ||||||||
| 应计费用总额 | $ | $ | ||||||
| F-14 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
6.应计费用(续)
公司根据对收到的服务的评估和根据合同安排的条款支出的工作,计提与第三方开展的开发活动有关的费用。根据其中一些合同支付的费用取决于研究和非临床试验里程碑。在某些情况下,向公司供应商支付的款项可能会超过所提供的服务水平,从而导致预付费用。在应计服务费时,公司估计将提供服务的时间段 以及每个时间段需要花费的工作量。如果实际执行服务的时间或努力程度与估计值不同,公司将相应调整应计或预付费用。本公司自成立以来并未 应计成本与实际成本之间出现任何重大差异。
7. 可转债
自2021年1月5日起,本公司订立票据购买协议,发行最多$
于
二零二二年九月,本公司订立可换股票据购买协议(该协议),以发行最多$
于
二零二三年一月,本公司选择转换二零二一年可换股票据,包括应计但尚未支付的利息,
于
二零二三年二月,二零二二年可换股票据经修订,将到期日延长至
| F-15 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
7.可换股债券(续)
于
二零二三年四月及六月,二零二二年可换股票据经进一步修订,将到期日延长至
2023年6月21日起,2022年可换股票据,包括应计利息$
公司评估了与2021年可转换票据相关的票据购买协议的条款和条件,以评估ASC 480-区分负债与股权以及ASC 815-衍生品和对冲项下的会计考虑事项。 公司认定可转换票据不符合根据ASC 480负债进行会计处理的任何标准。公司 还根据ASC 815中的指导对嵌入式功能进行了评估,并确定嵌入式功能不符合任何分支标准 。
可转换 应付票据包括以下内容:
| 2023年12月31日 | 2022年12月31日 | |||||||
| 2021年可转换票据 | $ | $ | ||||||
| 2022年可转换票据 | ||||||||
| 可转换票据总额 | $ | $ | ||||||
利息
费用为$
8. 股东权益
2023年5月17日,公司实施了一项
普通股 股票
2023年和2022年12月31日,根据公司修订和重述的公司注册证书,公司被授权 签发 $的股票面值普通股。
公司目前有 为未来发行而预留的普通股股份,用于潜在行使于2023年12月31日尚未行使的股票期权和认股权证 。
| F-16 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
8.股东权益(续)
优先股 股票
2023年12月31日和2022年12月31日,根据公司修订和重述的公司注册证书,公司已授权 股$面值优先股。
2023年1月,公司发布了其B系列优先股股份与转换2021年可换股票据有关,转换价为
系列A、系列A—1和系列B优先股具有以下权利、优先权和特权:
转换
优先股根据持有人的选择权根据预定义的公式转换为普通股。优先股持有人在公司首次公开募股时将这些股份转换为普通股。出于转换目的,初始转换价格
为$
在 公司于2023年6月首次公开募股时,所有未发行优先股转换为普通股,导致 , ,以及分别以普通股换取A系列、A—1系列和B系列优先股, 。转换时并无损益。
投票权 权利
A系列、A-1系列和B系列优先股的持有人有权就提交给公司股东以供其在公司的任何股东大会上采取行动或审议的任何事项投票(或经股东书面同意以代替会议),每名优先股流通股持有人有权投出的投票数等于该股东持有的优先股的全部普通股股数 截至确定有权就该事项投票的股东的记录日期为止。A系列和A-1系列优先股的持有者每人有权选择公司的一只董事。B系列股票的持有者有权选举两名董事会成员。在公司注册证书所述的特定情况下,每类优先股可将该等董事免职,并填补因该等董事辞职、死亡或免任而造成的任何空缺 。
分红
A系列优先股的持有者有权按以下比率获得股息
A-1系列股票的持有者有权按以下比率获得股息
| F-17 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
8.股东权益(续)
B系列股票的持有者有权按以下比率获得股息
清算
在本公司发生任何清算、解散或清盘的情况下,优先股持有人有权在普通股持有人之前并优先于普通股持有人获得相当于A系列、A系列1或B系列优先股的原始发行价的金额,外加已宣布和/或应计但未支付的股息。在发生任何此类清算事件时,在支付需要支付给优先股股东的所有优先金额后,公司可供分配给其股东的剩余资产应根据每个优先股和普通股持有人持有的股份数量按比例分配给优先股和普通股股东,为此将所有此类证券视为已根据紧接该清算事件之前的公司注册证书的条款转换为普通股。
9. 认股权证
该公司发行了认股权证以购买
该公司亦于2016及2019年发行认股权证,但该等认股权证不符合ASC 480分类为负债的标准,而是符合股权分类标准。这个2016年发行的权证的加权平均行使价格为1美元。和加权的 平均期限三年,于2023年6月首次公开招股时届满。
下表汇总了有关2023年12月31日未到期认股权证的信息:
| 未清偿认股权证 | 可撤销的令状 | |||||||||||||||||||||||||||||
| 授予的年份 | 行权价格 | 截至2023年12月31日的认股权证数目 | 加权平均剩余合同寿命 | 体重平均锻炼价格 | 截至2023年12月31日的认股权证数目 | 加权平均剩余合同寿命 | 加权平均行权价 | |||||||||||||||||||||||
| 2018 | $ | 年份 | $ | 年份 | $ | |||||||||||||||||||||||||
| 2019 | $ | 年份 | $ | 年份 | $ | |||||||||||||||||||||||||
| 2023 | $ | 年份 | $ | 年份 | $ | |||||||||||||||||||||||||
| $ | $ | |||||||||||||||||||||||||||||
| F-18 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
10. 股票期权
2023年3月,公司董事会和股东批准了2023年股票激励计划(简称2023年计划)。 2023年计划允许委员会授予以激励性和非法定股票期权、限制性股票奖励、限制性股票单位和其他基于股票的奖励形式向员工、董事和非员工发放的普通股股票。截至2023年12月31日,购买选项普通股已被授予,并根据2023年计划和根据该计划,普通股 可供授予。
2016年,公司制定了Azitra Inc.2016股票激励计划(以下简称计划),规定向公司员工、高级管理人员、董事、顾问和顾问授予股票期权和限制性股票。截至2023年12月31日,购买选项 普通股已被授予,并根据2016年计划和根据该计划,普通股 可供授予。
在截至2023年12月31日的年度内,公司确认股票薪酬支出为$
| 未完成的期权 | 可行使的期权 | |||||||||||||||||||||||||
| 行权价格 | 截至2023年12月31日的期权数量 | 加权平均剩余合同寿命 | 体重平均锻炼价格 | 截至2023年12月31日的期权数量 | 加权平均剩余合同寿命 | 加权平均行权价 | ||||||||||||||||||||
| $ | 年份 | $ | 年份 | $ | ||||||||||||||||||||||
| $ | 年份 | $ | 年份 | $ | ||||||||||||||||||||||
| $ | 年份 | $ | 年份 | $ | ||||||||||||||||||||||
| $ | 年份 | $ | 年份 | $ | ||||||||||||||||||||||
| 股票 | 加权平均行权价 | |||||||
| 截至2021年12月31日的未偿还债务 | $ | |||||||
| 已锻炼 | ( | ) | ||||||
| 被没收 | ( | ) | ||||||
| 在2022年12月31日未偿还 | $ | |||||||
| 授与 | ||||||||
| 已锻炼 | ||||||||
| 被没收 | ( | ) | ||||||
| 截至2023年12月31日的未偿还债务 | $ | |||||||
| F-19 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
10.股票期权(续)
有 2016年计划下可供未来授出的股份, 在2023年12月31日的2023年计划中。
| 2023年12月31日未偿还的期权 | 在2023年12月31日可行使的期权 | |||||||
| 2023年12月31日的股票价值 | $ | $ | ||||||
| 行权价格 | ||||||||
| 每股内在价值 | ||||||||
| 选项O/S | ||||||||
| 2023年12月31日的内在价值 | $ | $ | ||||||
11. 公允价值计量
下表汇总了公允价值层次中的公允价值和水平,其中公允价值计量属于按经常性基础计量的资产和负债,截至以下日期:
2023年12月31日
| 描述 | 1级 | 2级 | 3级 | 总计 | ||||||||||||
| 负债 | ||||||||||||||||
| 普通股认股权证 | $ | $ | $ | $ | ||||||||||||
| 总计 | $ | $ | $ | $ | ||||||||||||
2022年12月31日
| 描述 | 1级 | 2级 | 3级 | 总计 | ||||||||||||
| 负债 | ||||||||||||||||
| 普通股认股权证 | $ | $ | $ | $ | ||||||||||||
| 2022年可转换票据 | ||||||||||||||||
| 总计 | $ | $ | $ | $ | ||||||||||||
下表呈列截至2023年12月31日止期间按经常性基准计量的第三级工具变动:
| 2021年12月31日的余额 | $ | |||
| 认股权证公允价值变动 | ( | ) | ||
| 发行2022年可转换票据 | ||||
| 2022年可转换票据公允价值变动 | ||||
| 2022年12月31日的余额 | ||||
| 认股权证公允价值变动 | ( | ) | ||
| 2022年可换股票据公允价值变动 | ||||
| 2022年可换股票据的转换 | ( | ) | ||
| 2023年12月31日的余额 |
| F-20 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
11.公平值计量(续)
| 2023年12月31日 | 2022年12月31日 | |||||||
| 基础普通股价值 | $ | $ | ||||||
| 预期期限(年) | ||||||||
| 预期波动率 | % | % | ||||||
| 无风险利率 | % | % | ||||||
| 股息率 | % | % | ||||||
公司普通股公允价值的波动 是每年普通股认股权证负债估值变化的主要驱动因素 。随着普通股公允价值的增加,票据持有人的价值通常也会增加。
赎回前,各种输入数据的波动(包括企业价值、变现时间、波动性及贴现率 )为二零二二年可换股票据各报告期间估值变动的主要驱动因素。随着 企业价值的公允价值、估计变现时间、波动性及贴现率增加,二零二二年可换股 票据持有人的价值普遍增加。
基本 和稀释后每股净亏损计算如下:
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 净亏损 | $ | ( | ) | $ | ( | ) | ||
| 优先股股息 | ( | ) | ( | ) | ||||
| 普通股股东应占净亏损 | $ | ( | ) | $ | ( | ) | ||
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 加权平均已发行普通股、基本普通股和稀释普通股 | ||||||||
| $0.01认股权证 | ||||||||
| 总计 | ||||||||
| F-21 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
12.每股净亏损(续)
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 每股基本和稀释后净亏损 | $ | ) | $ | ) | ||||
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 购买普通股股份的期权 | $ | $ | ||||||
| 未清偿认股权证 | ||||||||
| 总计 | $ | $ | ||||||
13. 所得税
截至2023年12月31日和2022年12月31日止年度的 所得税(福利)准备金包括:
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 当前 | ||||||||
| 联邦制 | $ | $ | ||||||
| 外国 | ||||||||
| 状态 | ||||||||
| 总电流 | ||||||||
| 延期 | ||||||||
| 联邦制 | ||||||||
| 外国 | ||||||||
| 状态 | ||||||||
| 延期合计 | ||||||||
| 所得税拨备总额 | $ | |||||||
使用美国联邦法定税率计算的所得税与截至2023年12月31日和2022年12月31日止年度的营运所得税的 对账包括:
| 2023 | 2022 | |||||||||||||||
| 金额 | 百分比 | 金额 | 百分比 | |||||||||||||
| 所得税使用美国法定税率 | $ | ( | ) | % | $ | ( | ) | % | ||||||||
| 债务公允价值调整 | ( | )% | ( | )% | ||||||||||||
| 扣除联邦福利后的州税 | ( | ) | % | ( | ) | % | ||||||||||
| 研发税收抵免 | ( | ) | % | ( | ) | % | ||||||||||
| 更改估值免税额 | ( | )% | ( | )% | ||||||||||||
| 其他,净额 | ( | )% | ( | )% | ||||||||||||
| $ | $ | ( | )% | $ | % | |||||||||||
| F-22 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
13.所得税(续)
递延 所得税乃根据财务报告目的资产和负债账面值与所得税报告目的资产和负债账面值之间的暂时差异计提。于二零二三年及二零二二年十二月三十一日的递延所得税资产╱(负债)如下:
| 十二月三十一日, | ||||||||
| 2023 | 2022 | |||||||
| 递延税项资产: | ||||||||
| 净营业亏损结转 | $ | $ | ||||||
| 税收抵免 | ||||||||
| 折旧及摊销 | ||||||||
| 应计费用 | ||||||||
| 其他 | ||||||||
| 递延税项资产总额 | $ | |||||||
| 递延税项负债: | ||||||||
| 折旧及摊销 | ||||||||
| 递延税项负债总额 | ||||||||
| 估值免税额 | ( | ) | ( | ) | ||||
| 递延税项净资产 | $ | $ | ||||||
公司的联邦净经营亏损结转额约为美元
公司拥有约为美元的联邦研究税收抵免
美国国税法第382节对所有权超过50%的公司使用其净营业亏损结转来减少纳税义务的能力进行了年度限制。 如果未来本公司的所有权变更超过第382条规定的50%限制门槛,则本公司结转的净营业亏损在特定年度的使用量方面可能受到显著限制。此外,本公司在2018年前产生的全部或部分净营业亏损结转 可能到期而未使用。
| F-23 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
13.所得税(续)
递延税项资产的变现取决于公司在临时差额可扣除期间产生未来应纳税所得额的能力。根据公司最近的盈利历史和预计的美国未来收益,管理层认为,在可预见的未来,其联邦和州递延税项资产更有可能无法完全变现 。作为这项评估的结果,管理层认为需要对其联邦和州递延税项净资产进行全额估值扣除。
公司适用ASC 740-10的规定来处理不确定的税务状况。ASC 740-10解决了是否应在财务报表中记录在纳税申报单上申报或预期申报的税收优惠的确定问题。根据美国会计准则第740-10条,公司只有在税务机关根据税务机关的技术价值审查后更有可能不会维持该税务头寸的情况下,才可确认来自不确定税务头寸的税收优惠。本公司已确定其 并无重大不确定税务状况需要根据美国会计准则740-10予以确认和计量。
该公司需缴纳美国联邦所得税、康涅狄格州所得税和加拿大分行税。本公司尚未接受美国国税局、州或外国税务机关与所得税有关的审计。本公司的纳税年度将继续接受所有联邦和州税务事项的审查 ,直到其净营业亏损结转使用且适用的诉讼时效已过期 。
如果适用,公司将确认与未确认的税收优惠相关的利息和罚款,作为所得税费用的组成部分。
14. 承付款和或有事项
法律
该公司在正常业务过程中受到法律程序或索赔的约束。虽然偶尔会出现不利的决定或和解,但本公司相信该等事宜的最终处置不应对其财务状况、经营业绩或流动资金造成重大不利影响。
许可证 协议
自2022年1月26日起,本公司与无关第三方订立独家许可协议(许可协议)。根据许可协议,本公司被授予某些专利的独家许可和某些专有技术的非独家许可。 许可协议持续到最后一个许可专利到期或第一个许可治疗或非治疗产品商业销售 后十年。公司可通过向第三方提供至少30天的书面通知,随时终止许可协议。如果违反协议规定的重大义务或破产,许可协议也将终止。许可协议终止后,任何一方均不得解除终止前产生的义务 。
在截至2023年12月31日和2022年12月31日的年度内,公司将根据本许可协议支付的款项资本化为$
15. 经营租约
该公司在康涅狄格州布兰福德、康涅狄格州格罗顿和魁北克省拉瓦尔租用办公和实验室空间。本公司的租约将于 至2027年5月31日的不同日期到期。大多数租约都是固定期限和固定金额的。
于2019年,本公司于加拿大拉瓦尔签订了一份新的办公及实验室空间租赁协议。Laval租赁需要每月
支付$
| F-24 |
AZITRA,INC.
合并财务报表附注
截至2023年12月31日及2022年12月31日止年度
15.经营租约(续)
在
2020年期间,公司就公司位于康涅狄格州布兰福德的主要办公和实验室空间签订了新的租赁协议。Branford租约需要每月支付$
2021年5月,公司与康涅狄格州格罗顿的办公室和实验室签订了新租约。格罗顿租约需要每月支付$
未来 在接下来的 五年中,每年初始或剩余期限超过一年的不可取消经营租赁的最低付款如下:
| 2024 | $ | |||
| 2025 | ||||
| 2026 | ||||
| 2027 | ||||
| 2028 | ||||
| 此后 | ||||
| 未来未贴现的租赁付款总额 | ||||
| 更少的兴趣 | ( | ) | ||
| 最低租赁付款现值 | $ |
租金
所有经营租赁的费用为$
融资 租赁
2023年,公司与惠普签订了租赁设备的协议。租赁要求每月支付美元
未来 在接下来的 五年中,每年初始或剩余期限超过一年的不可取消经营租赁的最低付款如下:
| 2024 | $ | |||
| 2025 | ||||
| 2026 | ||||
| 2027 | ||||
| 此后 | ||||
| 未来未贴现的租赁付款总额 | ||||
| 更少的兴趣 | ( | ) | ||
| 最低租赁付款现值 | $ |
融资租赁的租赁
费用为美元
| F-25 |
AZITRA,INC.
合并财务报表附注
截至二零二三年及二零二二年十二月三十一日止年度
16. 退休计划
2019年1月1日起,
17. 关联方
关联方总收入为$
于
二零二二年九月,本公司订立总额为美元的可换股承兑票据
18. 后续事件
2024年2月15日,公司完成了公开募股, 其普通股的发行价为美元每
股。本公司收到的所得款项总额约为美元。
| F-26 |
第 项9.会计和财务披露方面的变更和与会计师的分歧
不适用 。
第 9A项。控制和程序
(A) 对披露控制和程序的评价。
截至2023年12月31日,我们的 管理层在首席执行官和首席财务官的参与下,评估了我们根据《交易法》第13a—15(e)条的披露控制措施和程序的有效性。在评估过程中, 我们发现了一个重大缺陷,因为它与会计职能缺乏适当的分离有关。我们打算在会计基础设施内增加人员 ,以便于适当分离会计职能,并对内部编制的财务报表进行适当审查 。基于上述,我们的首席执行官和首席财务官得出结论, 我们的披露控制和程序截至2023年12月31日尚未生效。
(b) 财务报告内部控制的变化。
在截至2023年12月31日的季度, 我们对财务报告的内部控制(定义见《交易法》第13a—15(f)条)没有发生 的变化,这些变化对我们对财务报告的内部控制产生了重大影响,或合理可能对我们对财务报告的内部控制产生重大影响 。
(c) 管理层关于财务报告内部控制的报告。
本 年度报告不包括管理层对财务报告内部控制的评估报告或我们注册会计师事务所的证明报告 ,因为美国证券交易委员会规则为新上市公司设定了过渡期。
第 9B项。其他信息
不适用 。
第 9C项。关于妨碍检查的外国司法管辖区的披露
不适用 。
| 77 |
第 第三部分
项目 10.董事、高管和公司治理
本项目下所需的 信息将包含在2024年委托书中,并在此以引用方式并入。
第 项11.高管薪酬
本项目下所需的 信息将包含在2024年委托书中,并在此以引用方式并入。
项目 12.某些实益所有人和管理层的担保所有权及相关股东事项
本项目下所需的 信息将包含在2024年委托书中,并在此以引用方式并入。
第 项13.某些关系和关联交易,以及董事独立性
本项目下所需的 信息将包含在2024年委托书中,并在此以引用方式并入。
第 项14.总会计师费用和服务
本项目所需的 信息将包含在2024年委托书中,并在此通过引用并入。
第四部分
第 项15.证物和财务报表附表
(a)财务报表
参照本文件第二部分第8项下的指数和财务报表,其中列出了这些文件。
(B) 财务报表附表
财务 报表明细表不是必需的,或者所要求的信息包含在本文件第二部分第8项下提交的合并财务报表或附注中。
| 78 |
(C) 个展品
本年度报告的表格10-K的展品如下。展品索引表示需要作为展品备案的每个管理合同或补偿性计划或安排。
| 数 | 附件 说明 | 备案方式 | ||
| 3.1 | 二次修订和重新注册的注册人注册证书 | 已注册 本公司于2023年6月21日提交的8—K表格的当前报告。 | ||
| 3.2 | 第二次修订和重新修订注册人章程 | 已注册 本公司于2023年6月21日提交的8—K表格的当前报告。 | ||
| 4.1 | 代表普通股股份的证书样本 | 已注册 本公司于2023年6月13日提交的S—1表格。 | ||
| 4.2 | 向私募投资者发出的认股权证格式 | 已注册 本公司于2023年6月13日提交的S—1表格。 | ||
| 4.3 | 2023年6月20日发布给ThinkEquity LLC的代表权证表格 | 已注册 本公司于2023年6月13日提交的S—1表格。 | ||
| 4.4 | 2024年2月16日向ThinkEquity LLC颁发的代表人认股权证表格(附于2024年2月13日注册人与ThinkEquity,LLC签订的承销协议) | 已注册 本公司于2024年2月14日提交的表格8—K当前报告。 | ||
| 4.5 | 股本说明 | 在此以电子方式提交。 | ||
| 10.1+ | 阿兹特拉公司2016年股票激励计划 | 已注册 本公司于2023年6月13日提交的S—1表格。 | ||
| 10.2+ | 阿兹特拉公司2023年股票激励计划 | 已注册 本公司于2023年6月13日提交的S—1表格。 | ||
| 10.3+ | 2021年4月22日注册人与Francisco D签订的行政雇佣协议。萨尔瓦 | 已注册 本公司于2023年6月13日提交的S—1表格。 | ||
| 10.4+ | 注册人和Travis Whitfill之间的行政雇佣协议2023年7月5日 | 已注册 本公司于2024年1月19日提交的S—1表格。 | ||
| 10.5+ | 注册人与其每一位董事和执行官之间的赔偿协议的形式 | 已注册 本公司于2023年6月13日提交的S—1表格。 | ||
| 10.6 | 第二次修订和重申的投资者权利协议,日期为2020年9月10日,由注册人和其中所列的每个投资者 | 已注册 本公司于2023年6月13日提交的S—1表格。 | ||
| 21.1 | 注册人的子公司名单 | 本文件通过参考公司于2023年3月20日提交的S—1表而并入本文件。 | ||
| 23.2 | Grassi & Co.的同意,注册会计师,独立注册会计师事务所 | 在此以电子方式提交。 | ||
| 31.1 | 2002年《萨班斯—奥克斯利法案》第302条规定的认证 | 在此以电子方式提交。 | ||
| 31.2 | 2002年《萨班斯—奥克斯利法案》第302条规定的认证 | 在此以电子方式提交。 | ||
| 32.1 | 根据2002年《萨班斯—奥克斯利法案》第906条的认证,18 U.S.C.部1350 | 在此以电子方式提交。 | ||
| 97.1 | Azitra公司执行官回扣政策 | 在此以电子方式提交。 |
| + | 指管理层补偿计划、合同或安排。 |
第 项16.表格10-K总结
未提供 。
| 79 |
签名
根据 1934年《证券交易法》第13或15(d)节的要求,注册人已正式促使下列签署人代表注册人签署10—K表格的年度报告 ,并经正式授权。
| AZITRA, Inc. | ||
| 日期: 2024年3月15日 | 发信人: | /S/ 弗朗西斯科·D·萨尔瓦 |
| 弗朗西斯科·D·萨尔瓦, | ||
| 首席执行官兼董事 | ||
根据1934年《证券交易法》的要求,本报告已由以下人员以注册人的身份在指定日期签署。
| 签名 | 标题 | 日期 | ||
| /S/ 弗朗西斯科·D·萨尔瓦 | 总裁, 首席执行官兼董事 |
三月 2024年15月15日 | ||
| 弗朗西斯科·D·萨尔瓦 | (首席执行官 ) | |||
| /S/ 诺曼·斯塔斯基 | 首席财务官、财务主管兼秘书 | 三月 2024年15月15日 | ||
| 诺曼·斯塔斯基 | (首席财务会计官 ) | |||
| /S/ 特拉维斯·惠特菲尔 | 首席运营官兼董事 | 三月 2024年15月15日 | ||
| 特拉维斯 惠特菲尔 | ||||
| /S/ 安德鲁·麦克拉利 | 董事 | 三月 2024年15月15日 | ||
| 安德鲁·麦克拉利 | ||||
| /S/ 芭芭拉·瑞安 | 董事 | 三月 2024年15月15日 | ||
| 芭芭拉 瑞安 | ||||
| S/ 约翰·施罗尔 | 董事 | 三月 2024年15月15日 | ||
| 约翰·施罗尔 |
| 80 |