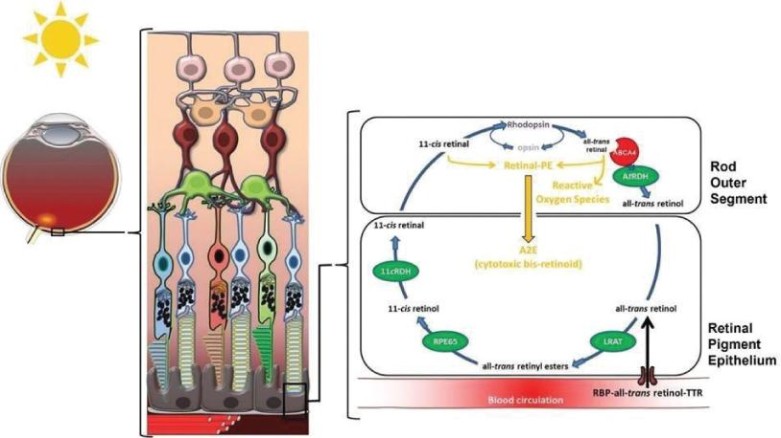
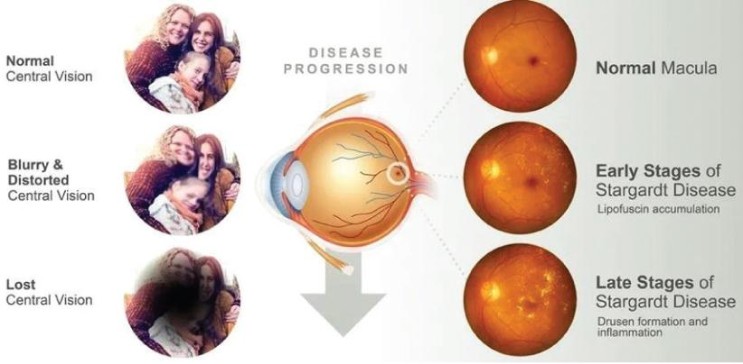
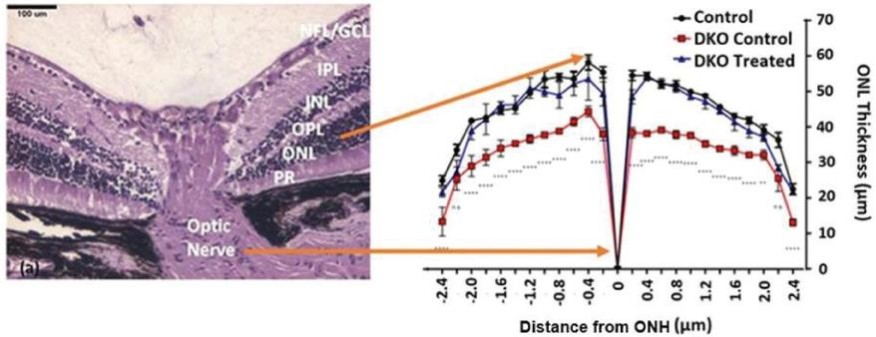
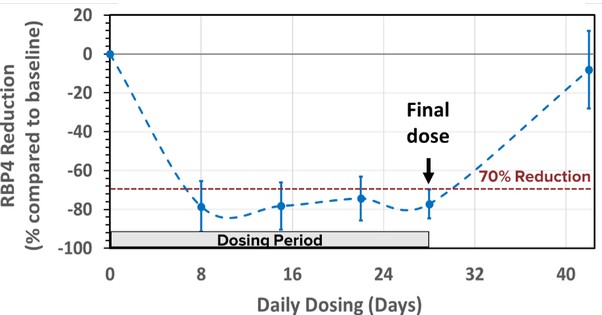
美国
美国证券交易委员会
华盛顿特区,20549
表格:20-F
(标记一)
| ☐ | 根据1934年《证券交易法》第12(B)款或第(G)款作出的注册声明 |
或
| ☒ | 根据1934年《证券交易法》第13或15(D)款提交的年度报告 |
截至本财年的12月31日, 2023
或
| ☐ | 根据1934年《证券交易法》第13或15(D)款提交的过渡报告 |
或
| ☐ | 根据1934年《证券交易法》第13或15(D)款提交的空壳公司报告 |
需要这份空壳公司报告的事件日期:
过渡时期, 到
委托文件编号:001-41359
贝利特生物科技公司
(注册人的确切姓名载于其章程)
开曼群岛
(成立为法团的司法管辖权)
12750高崖驱动套房475,
圣地亚哥,钙92130
(主要执行办公室地址)
林雨欣
电话:+1-858-246-6240
电子邮件:邮箱:Tomlin@beliteBio.com
寄往上述公司的地址
(公司联系人姓名、电话、电子邮件和/或传真号码及地址)
根据该法第12(B)款登记或将登记的证券:
每个班级的标题 | | 交易符号 | | 注册的每个交易所的名称 |
美国存托股份,每股代表一普通股 | | BLTE | | 纳斯达克股市有限责任公司(纳斯达克资本市场) |
普通股,每股票面价值0.0001美元* | | 不适用 | | 纳斯达克股市有限责任公司
(纳斯达克资本市场) |
*不用于交易,仅与美国存托股份在纳斯达克资本市场上市有关。
根据该法第12(G)款登记或将登记的证券:
无
(班级名称)
根据该法第15(D)款负有报告义务的证券:
无
(班级名称)
注明截至年度报告所涵盖的营业时间结束时,发行人所属各类股本或普通股的流通股数量。
29,149,444普通股,每股票面价值0.0001美元,截至2023年12月31日。
用复选标记表示注册人是否为证券法规则第405条所定义的知名经验丰富的发行人。
☐ 是☒ 不是
如果此报告是年度报告或过渡报告,请用复选标记表示注册人是否不需要根据1934年《证券交易法》第13或15(D)节提交报告。
☐ 是☒ 不是
用复选标记表示注册人(1)是否在过去12个月内(或注册人被要求提交此类报告的较短期限内)提交了1934年《证券交易法》第13节或15(D)节要求提交的所有报告,以及(2)在过去90天内是否符合此类提交要求。
☒ 是 ☐ 不是
用复选标记表示注册人是否在过去12个月内(或注册人需要提交和张贴此类文件的较短时间内)以电子方式提交并张贴在其公司网站上(如果有),根据S-T法规第405条规定必须提交和张贴的每个互动数据文件。
☒ 是 ☐ 不是
用复选标记表示注册者是大型加速文件服务器、加速文件服务器、非加速文件服务器还是新兴成长型公司。请参阅《交易法》第12b-2条规则中对“大型加速申报公司”、“加速申报公司”和“新兴成长型公司”的定义。
| | | |
大型加速文件服务器☐ | 加速文件管理器☐ | 非加速文件服务器 ☒ | 新兴成长型公司☒ |
如果一家新兴成长型公司按照美国公认会计原则编制其财务报表,用复选标记表示注册人是否已选择不使用延长的过渡期来遵守†根据交易法第(13)(A)节提供的任何新的或修订的财务会计准则。☐
†“新的或修订的fi财务会计准则”是指财务会计准则委员会在2012年4月5日之后发布的对其会计准则法典fi出版物的任何更新。
用复选标记表示注册人是否提交了一份报告,证明其管理层根据《萨班斯-奥克斯利法案》(《美国联邦法典》第15编,第7262(B)节)第404(B)节对其财务报告内部控制的有效性进行了评估,该评估是由编制或发布其审计报告的注册会计师事务所进行的。☐
如果证券是根据该法第12(B)条登记的,应用复选标记表示登记人的财务报表是否反映了对以前发布的财务报表的错误更正。☐
用复选标记表示其中是否有任何错误更正是需要对任何注册人收到的基于激励的补偿进行恢复分析的重述’在有关的恢复期内,根据§240.10D-1(B)。☐
用复选标记表示注册人在编制本文件所包括的财务报表时使用了哪种会计基础:
美国公认会计原则 ☒ | 发布的国际财务报告准则 | 其他☐ |
| 国际会计准则委员会☐ | |
如果在回答前一个问题时勾选了“其他”,则用复选标记表示登记人选择遵循哪个财务报表项目。
☐第1项17 ☐项目18
如果这是一份年度报告,请用复选标记标明注册人是否为空壳公司(如《交易法》第12B-2条所定义)。
☐ 是☒ 不是