重新定居
2020年4月15日,即实施日期,根据西澳大利亚州法律注册成立的上市公司Benitec Biopharma Limited或Benitec Limited,根据Benitec Limited和我们于2020年1月30日修订和重述的计划实施协议,完成了对Benitec Biopharma Limited的重新注册或重新注册。由于重新注册,我们的注册管辖区从澳大利亚变更为特拉华州,Benitec Limited成为我们的全资子公司。
重新注册是根据澳大利亚法律规定的法定安排计划或该计划进行的,根据该计划,在实施日期,Benitec Limited的所有已发行和流通普通股均按Benitec Limited每发行和流通的300股普通股兑换成我们新发行的普通股,每股面值0.0001美元,基准是Benitec Limited每发行和流通300股普通股。Benitec Limited的美国存托股(ADS)(每股代表200股普通股)的持有人每持有三个ADS就会获得两股普通股。
我们的普通股于实施日开始在纳斯达克资本市场(纳斯达克)上市,股票代码为 “BNTC”。
新冠肺炎
COVID-19 已被世界卫生组织宣布为大流行,并已蔓延到几乎所有国家,包括澳大利亚和美国。这种流行病已经并将继续对社会的许多方面产生广泛影响,这已经导致并可能继续对全球企业和资本市场造成重大干扰。冠状病毒继续影响我们的程度将取决于未来的发展,这些发展高度不确定且无法预测,包括可能出现的有关冠状病毒及其变种严重程度的新信息,以及遏制冠状病毒或治疗其影响的行动,包括该病毒疫苗的有效性和采用情况等。
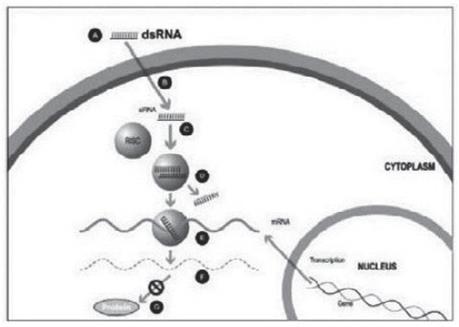
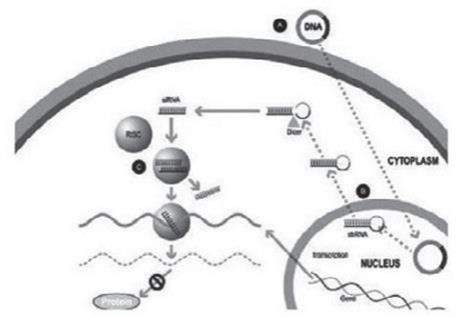
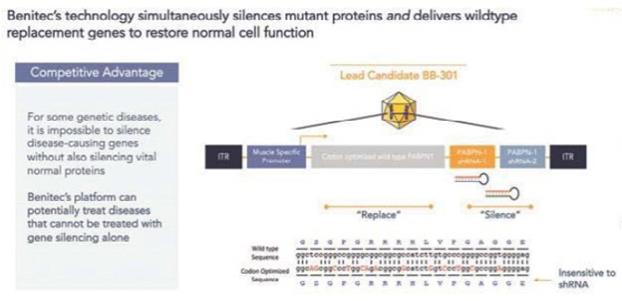
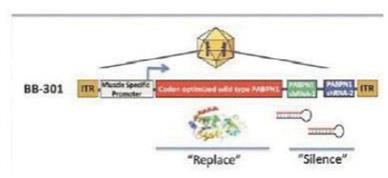
我们的某些研发工作是在全球范围内进行的,包括正在进行的静默治疗和替代治疗眼咽肌肉萎缩症(OPMD)的开发,这将取决于我们在新冠肺炎(COVID-19)疫情和任何类似事件的情况下继续进行临床前和临床研究及相关工作的能力。
反向股票分割
2023年7月26日,公司对其普通股进行了1比17的反向股票拆分(“反向股票拆分”)。根据反向股票拆分,公司拆分前的17股普通股被自动转换为一股已发行和拆分后流通的股票。还根据各自的条款,对所有未偿还的股票期权、预先融资的认股权证和普通认股权证进行了按比例调整。反向股票拆分没有改变公司普通股的面值或授权的股票数量。没有发行与反向股票拆分相关的部分股票。与普通股相比,所有零股四舍五入至最接近的整股。本表格10-Q中列出的所有股票和每股收益金额都反映了这种反向拆分的影响。
可用信息
我们的电话号码是 (510) 780-0819,我们的互联网网站是 www.benitec.com。我们网站上的信息或可通过我们网站访问的信息不属于本10-Q表季度报告,也未以引用方式纳入此处。
特许权使用费、里程碑付款和其他许可费
我们需要支付与向第三方许可知识产权相关的特许权使用费、里程碑付款和其他许可费,包括下文讨论的内容。
外币折算和其他综合收益(亏损)
公司的功能货币和报告货币是美元。BBL 的功能货币是澳元 (AUD)。资产和负债按资产负债表日的有效汇率折算。收入和支出按报告所述期间的平均汇率折算。股票交易按每个历史交易日期即期汇率进行折算。因不同时期使用不同汇率而产生的折算调整作为股东权益的组成部分列为 “累计其他综合亏损”。外币折算产生的损益列入合并经营报表,综合亏损作为其他综合收益(亏损)列入。
2021 年 4 月筹集资金
2021年4月30日,公司宣布完成普通股和普通股等价物的承销公开发行。该公司从本次发行中获得的总收益约为1,430万美元,净收益约为1,270万美元。
2022 年 9 月筹集资金
2022年9月15日,公司宣布完成普通股和普通股等价物的承销公开发行。公司从本次发行中获得了约1,790万美元的总收益和约1,600万美元的净收益。
2023 年 8 月筹集资金
2023年8月11日,我们完成了承销公开发行,出售了875,949股普通股,15,126,226份预先筹资的认股权证以购买15,126,226股普通股,以及16,002,175份普通认股权证,购买最多16,002,175股普通股。预先注资的认股权证可立即行使,直至以每股普通股0.0001美元的行使价全额行使。普通认股权证可立即行使,普通股每股价格为3.86美元,并在首次行使日期五周年时到期。每股普通股和随附普通认股权证的合并购买价格为1.93美元,分配为每股普通股1.9299美元,普通认股权证每股0.0001美元。每份预先注资的认股权证与一份普通认股权证一起出售,总价格为1.9299美元,每份预筹认股权证分配为1.9298美元,每份普通认股权证分配为0.0001美元。此外,公司授予承销商30天的期权,可以额外购买最多2,331,606股普通股和/或最多2,331,606份额外的普通认股权证。截至2023年12月31日,承销商已部分行使该期权,并额外购买了458,134股普通股和458,134份普通认股权证。此次发行的净收益,包括承销商部分行使期权的影响以及扣除承保折扣、佣金和其他发行费用,共计2790万美元。
公司拥有未偿还的第二系列认股权证(“系列2认股权证”),在反向股票拆分生效并在截至2023年12月31日的三个月内行使后,该认股权证目前可行使1,733,503股普通股。系列2认股权证包含行使价调整机制,规定某些普通股(或普通股等价物)的发行价格如果低于该系列2认股权证的现有行使价11.22美元,则会将行使价重置为较低的价格。由于2023年8月11日的公开募股,自该公开募股截止之日起,系列2认股权证的行使价已自动重置为1.9299美元。
运营结果
收入
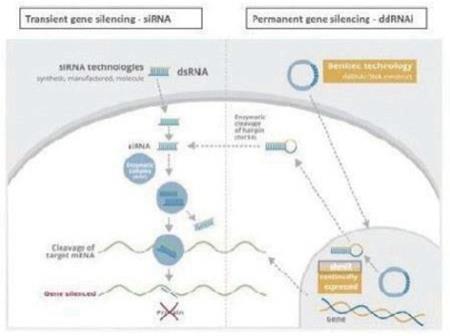
该公司没有从产品销售中产生任何收入。许可费收入包含在我们合并运营报表和综合亏损报表中的客户收入项中。我们的许可费是通过向生物制药公司许可我们的ddrNAI技术产生的。在截至2023年12月31日的六个月中,该公司没有确认任何收入,在截至2022年12月31日的六个月中确认了14,000美元。
48