
根据424(B)(4)提交
注册 第333-275361号声明
4,806,226股
FibroBiologics, 公司
普通股 股票
本招股说明书涉及 与我们在纳斯达克全球市场或纳斯达克直接上市相关的登记,由本招股说明书中确定的股东或登记股东转售最多4,806,226股我们的普通股。 与首次公开募股不同,登记股东的转售不会由 任何投资银行以确定承诺的基础上承销。登记股东可以选择,也可以不选择出售本招股说明书涵盖的普通股, 取决于他们决定的范围。登记股东可按现行市价或协议价格,公开或以私下交易方式发售、出售或分配本公司登记的全部或部分普通股。如果登记股东选择出售其普通股股份,我们将不会从登记股东出售普通股股份中获得任何收益。
我们的董事会和我们的股东已于2023年10月6日各自批准了我们所有类别的已发行 和已发行股本的1/4反向股票拆分,或反向股票拆分。2023年10月31日,我们向特拉华州提交了修订和重述的公司注册证书,以 立即实施反向股票拆分。 本招股说明书中的所有股票和每股信息已进行调整,以反映反向股票拆分,除非另有说明。
我们的普通股目前不存在公开市场,我们的普通股在私下交易中的交易历史有限。 2022年12月,我们以私募方式向投资者发行了总计相当于381,658股B系列优先股,价格相当于每股6.76美元,相当于318,049股,其余相当于63,609股 为红股。从2023年2月至2023年4月,我们以监管众筹方式向投资者发行了总计相当于890,310股B系列优先股 ,价格相当于每股6.76美元,相当于724,937股,其余相当于143,225股和相当于22,148股分别为红股和佣金支付股, 。2023年3月和4月,我们以非公开配售的方式向投资者发行了相当于1,680,084股B系列优先股,价格相当于每股6.76美元,相当于1,527,349股,其余相当于152,735股为红股。2023年4月至2023年9月,我们以私募方式向投资者发行了相当于74,922股B-1系列优先股 ,价格从相当于每股18.00美元到相当于64,070股 相当于20.00美元,其余相当于10,852股为红股。对于此类B-1系列优先股的非公开配售的一部分,我们还同意发行认股权证,从我们的 直接上市起可行使三年,以相当于每股20.00美元的行使价 购买总计相当于8,890股我们的普通股。2023年11月,本公司向认购B-1系列优先股的投资者增发了14,859股B-1优先股和1,431股额外认股权证,认购普通股的每股价格超过直接上市预期的每股参考价。
直接上市后,我们当时发行的所有B系列优先股和B-1系列优先股将自动转换为我们的普通股,而不需要由其持有人或向其持有人支付额外的代价 。
我们普通股最近在私下交易中的购买价格可能与我们普通股在纳斯达克上的开盘公开价格或我们普通股在纳斯达克上的后续交易价格几乎没有 关系。有关详细信息,请参阅“我们股本的销售价格历史。此外,我们的普通股在纳斯达克上市,没有确定承销发行,是开始公开交易我们普通股股票的一种新方法 ,因此,我们普通股的股票交易量和价格可能比我们普通股的 股票最初上市时在确定承诺承销的首次公开募股 基础上上市时更不稳定。
自我们的普通股在纳斯达克首次上市之日起,纳斯达克将开始接受但不执行开盘前的买入和卖出订单,并将开始在此类 接受的订单的基础上持续生成当前指示性参考价(定义如下)。当前参考价格每秒计算一次,在10分钟的“仅供展示”期间,纳斯达克通过其noii和图书查看器工具将其与其他指示性失衡信息一起传播给市场参与者。 在“仅供展示”期限之后,将开始一个“上市前”期限,在此期间,Maxim Group LLC或顾问必须以我们财务顾问的身份通知纳斯达克,我们的股票已“可以交易”。一旦顾问通知纳斯达克我们的普通股可以交易,纳斯达克将根据纳斯达克规则确认我们普通股的当前参考价格。如果顾问随后批准以当前参考价格继续进行,则已输入的适用订单将以该价格执行,我们的普通股将开始在纳斯达克上的常规交易,但纳斯达克 将根据纳斯达克规则进行验证检查。根据纳斯达克规则,“当前参考价”是指:(I) 最大买入或卖出指令可匹配的单一价格;(Ii)如果有多个价格可匹配 最大买入或卖出指令数,则是将买卖指令之间的不平衡降至最低的价格 (即将在该价格下无法匹配的股票数量降至最低);(Iii)如果(Ii)项下存在多个价格,则 它是输入的价格(即客户在买入或卖出订单中输入的指定价格),在此价格下,我们的普通股将保持不匹配(即不会被买卖);及(Iv)如果(Iii)项下存在多个价格,则是纳斯达克 以我们财务顾问的身份与顾问协商后确定的价格。如果第(Iii)项下存在多个价格,则顾问将仅在符合联邦证券法的反操纵条款 (包括法规M)或根据其授予的适用救济的范围内行使任何咨询权。注册股东不会 参与纳斯达克的定价机制,包括决定推迟或继续交易,也不会控制或影响顾问履行其财务顾问的角色。顾问将主要根据成交量、时机和价格的考虑,确定我们的普通股股票何时准备好以当前参考价进行交易和审批。 具体而言,顾问将主要根据开盘前的买入和卖出订单,确定何时将有合理数量的成交量 在开盘交易中 ,以充分的价格发现以当前参考价开盘交易。有关更多 信息,请参阅“配送计划“从本招股说明书第126页开始。
我们 已申请将我们的普通股在纳斯达克全球市场上市,代码为“FBLG.我们预计我们的普通股 将于2024年1月31日左右在纳斯达克开始交易。
如果我们的纳斯达克申请未获批准,或者我们以其他方式确定我们将无法确保我们的普通股在纳斯达克上市,我们将不会完成此次直接上市 。此次上市是此次上市的一个条件。不能保证我们的纳斯达克申请会获得批准,也不能保证我们的普通股永远会在纳斯达克上市。如果我们的上市申请没有得到纳斯达克的批准,我们将无法完成此次发行,我们将终止此次直接上市。
本次发行完成后,我们的创始人兼首席执行官皮特·奥希隆将共同实益拥有我们已发行的有投票权证券约59%的投票权,我们将成为纳斯达克上市规则所指的“控股公司”。我们不打算依赖受控公司可以获得的任何公司治理要求的豁免。
我们 是联邦证券法所定义的“新兴成长型公司”和“较小的报告公司” ,因此,我们已选择遵守本招股说明书中某些降低的上市公司报告要求,并可能选择在未来的备案文件中这样做。请参阅“招股说明书摘要-成为新兴成长型公司和小型报告公司的意义 .”
投资 我们的普通股涉及高度风险。见“风险因素“ 本招股说明书第9页开始的部分,了解您在投资我们的普通股之前应考虑的风险和不确定性。
美国证券交易委员会或任何州证券委员会都没有批准或不批准这些证券,也没有确定本招股说明书是否真实或完整。任何相反的陈述都是刑事犯罪。
招股说明书 日期为2024年1月23日
目录表
| 关于本招股说明书 | 1 |
| 招股说明书摘要 | 2 |
| 财务和其他数据摘要 | 8 |
| 风险因素 | 9 |
| 有关前瞻性陈述的警示说明 | 62 |
| 市场和行业数据 | 63 |
| 商标、服务标记和商标名 | 64 |
| 收益的使用 | 65 |
| 股利政策 | 65 |
| 大写 | 65 |
| 管理层对财务状况和经营成果的讨论与分析 | 66 |
| 生意场 | 77 |
| 管理 | 101 |
| 高管和董事薪酬 | 108 |
| 某些关系和关联人交易 | 113 |
| 主要股东和注册股东 | 114 |
| 股本说明 | 116 |
| 有资格在未来出售的股份 | 120 |
| 我国股本销售价格历史 | 121 |
| 美国联邦所得税对非美国持有者的重大影响 | 121 |
| 配送计划 | 126 |
| 法律事务 | 129 |
| 专家 | 129 |
| 在那里您可以找到更多信息 | 129 |
| 财务报表索引 | F-1 |
您 应仅依赖本招股说明书中包含的信息或提交给证券和交易委员会的任何免费撰写的招股说明书中包含的信息。吾等或任何注册股东均未授权任何人提供与本招股章程及本招股章程及任何免费撰写招股章程所载资料不同的 任何资料,或提供不同于该等资料的任何资料 。我们或任何注册股东均不对 负责,也不能保证其他人可能向您提供的任何其他信息的可靠性。登记股东仅在其 合法出售或购买其普通股的情况下,才提出出售或寻求购买其普通股的要约。本招股说明书中包含的信息仅为截止日期的最新信息,无论本招股说明书的交付时间或我们普通股的任何出售时间。自该日期以来,我们的业务、财务状况、运营结果和前景可能已发生变化 。
对于美国以外的投资者:我们或任何注册股东都没有做任何事情,允许在美国以外的任何司法管辖区使用或拥有或分发本招股说明书或任何相关的免费写作招股说明书 。持有本招股说明书的美国境外人士必须告知 注册股东发行本公司普通股的情况,并遵守与此相关的任何限制。 本招股说明书在美国境外的分销。
| i |
关于 本招股说明书
本招股说明书是我们向美国证券交易委员会或美国证券交易委员会提交的S-1表格注册声明的一部分,该注册声明使用 “搁置”注册或持续发售流程。在这一过程中,登记股东可不时地以标题为“配送计划。 此外,我们还可能提供招股说明书补充资料,以添加、更新或更改本招股说明书中包含的信息, 包括标题为配送计划“。”您可以免费获取此信息,方法是按照“在那里您可以找到更多信息”部分。在决定投资我们的普通股之前,您应该阅读本 招股说明书和任何招股说明书补充文件。
本 招股说明书包含本文所述部分文件中包含的某些条款的摘要,但完整信息请参考 实际文件。所有的摘要都是由实际文件完整地限定的。本招股章程所述的部分 文件的副本已作为或将作为本招股章程 所属的注册声明的附件存档,阁下可按“在那里您可以找到更多信息.”
| 1 |
招股说明书 摘要
本 摘要重点介绍了本招股说明书其他地方包含的部分信息,但不包含您在做出投资决定之前 应考虑的所有信息。在 做出投资决定之前,您应仔细阅读整份招股说明书,包括标题为“风险因素"、 “关于前瞻性陈述的警示性说明”、 “管理层对财务状况和经营业绩的讨论与分析”的章节以及本招股说明书其他地方包含的我们的财务报表和随附注释。除非另有说明或文义另有所指,否则本招股章程中所有提述“我们"、 “我们”、“我们的”、”本公司”、“FibroBiologics”及类似词语均指FibroBiologics, Inc.
概述
我们 是一家临床阶段的细胞治疗公司,专注于开发和商业化基于成纤维细胞的治疗方法,用于 患有慢性疾病且医疗需求严重未得到满足的患者,包括退行性椎间盘疾病、多发性硬化症、伤口愈合 和某些癌症,并用于潜在的生命延长应用,包括胸腺和脾脏退化逆转。
我们 于2021年4月以Fibrobiologics,LLC的名称成立为德克萨斯州有限责任公司,并于2021年12月以Fibrobiologics,Inc的名称转换为特拉华州公司 。2023年4月12日,我们更名为FibroBiologics,Inc.。在 我们成立时,我们向当时的母公司SpinalCyte LLC (以FibroGenesis的名义开展业务)或FibroGenesis发行了A系列优先股,以换取通过专利转让协议和知识产权交叉许可协议获得的某些知识产权的权利。开发从FibroGenesis获得的知识产权是我们成立的基础。在我们成立之前,与转让的知识产权相关的临床前研究和开发 在FibroGenesis下进行。
成纤维细胞
技术平台
成纤维细胞 和干细胞是人体中仅有的两种可以再生组织和器官的细胞类型。研究表明,间充质干细胞和成纤维细胞具有许多共同的表面标志物,并且可以分化成许多细胞,包括脂肪细胞、软骨细胞、成骨细胞、肝细胞和心肌细胞,并且可以调节免疫系统。然而,转录组学和表观遗传学研究表明两种细胞类型之间存在明显差异。
成纤维细胞 包括结缔组织的主要细胞类型,具有纺锤形形态,其经典功能在历史上被 认为是产生负责维持组织结构完整性的细胞外基质。成纤维细胞还在维持器官中的干细胞小生境中发挥 重要作用,并且参与伤口愈合的每个阶段。
成纤维细胞 有利于干细胞作为细胞疗法治疗平台,因为成纤维细胞:
| ● | 可以 可以从来自外科手术的各种皮肤供体非侵入性地获取, 腹部折叠皮瓣或简单的活检穿孔; |
| ● | 有 培养中的倍增时间比干细胞快; |
| ● | 拥有 与干细胞相比,具有更好的免疫调节活性; |
| ● | 展品 与干细胞相比,产生再生细胞因子和生长因子的能力增强 细胞;以及 |
| ● | 是 与干细胞相比,分离、培养和扩增更经济,因为成纤维细胞 不需要使用昂贵的组织培养基和添加剂。 |
研究 表明,同种异体成纤维细胞与间充质干细胞非常相似,具有免疫豁免性,不会引发免疫应答 体外培养和体内.如果需要自体成纤维细胞,这意味着必须从每个患者身上收集细胞,进行处理和培养,然后给予同一患者,这将更加昂贵和低效。由于 同种异体成纤维细胞不会引起免疫应答,因此我们计划建立我们自己的现行药品生产质量管理规范(cGMP) 生产设施,以获取同种异体成纤维细胞,用于我们候选产品的临床试验和商业销售 (如果我们的候选产品获得上市批准)。
然而,迄今为止,还没有成纤维细胞治疗产品 获得批准,并且只有少数涉及成纤维细胞的临床试验。利用我们的成纤维细胞技术平台开发、制造和商业化 候选产品的成本可能超出我们的估计。此外,生物技术和制药 行业的特点是技术发展迅速、竞争激烈,并高度重视专有和新型产品 和候选产品,因此我们成功开发和商业化的任何候选产品都将与现有疗法 和未来可能出现的新疗法竞争。有关与我们的候选产品 技术和业务相关的风险和不确定性的其他信息,请参见标题为“-风险因素摘要“和”风险因素 “在这份招股说明书中。
| 2 |
我们的 管理团队和监督
我们 组建了一个执行领导团队,由我们的创始人、首席执行官和董事会主席、 我们的首席科学官和首席财务官组成,他们在初创企业和 生命科学行业拥有成功的业绩记录。我们的执行领导团队在董事会的监督下工作,董事会成员都是具有实际行业经验的公认领导者 。我们的科学顾问 委员会还拥有一支由世界知名科学家组成的团队,他们拥有相关的专业知识,可帮助指导我们的研发工作。
我们的 当前渠道
我们 拥有一系列处于不同开发阶段的候选产品,包括:
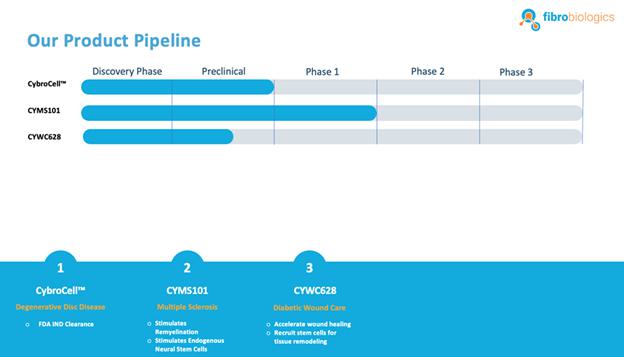
CybroCell™治疗椎间盘退行性疾病: CybroCell™是一种基于同种异体成纤维细胞的治疗退行性椎间盘疾病的方法。这项新技术被设计 作为修复椎间盘软骨(或任何其他关节软骨)的替代方法。该方法是基于 使用人皮肤成纤维细胞或HDF,其被迫分化为软骨细胞样细胞 体内使用脊柱中发现的机械力和间歇静水压力,用于成纤维细胞的软骨形成分化。我们相信我们的解决方案 将优于现有的治疗方法,因为我们预计它的侵入性更小,并且可以再生椎间盘,恢复功能 并减轻疼痛,而不会造成长期影响。我们已经完成了两轮动物实验。这些研究的结果 是积极的,并导致我们的研究性新药(IND)在向 美国食品药品监督管理局(FDA)提交的申请中获得了“首次人体”试验批准。我们已收到FDA的IND批准,条件是我们的主细胞库获得批准,对患有退行性椎间盘疾病的患者进行1/2期临床试验。我们将在 美国境内进行此试验。将通过与FDA讨论确定时间轴。
| 3 |
CYMS 101治疗多发性硬化症:We are developing CYMS101 as an allogeneic fibroblast cell-based therapy to treat multiple sclerosis, or MS. After completing animal studies using CYMS101 (allogeneic fibroblast cells), we received approval from Mexico to conduct clinical investigations using the fibroblast cell composition for patients with MS and have completed a Phase 1 clinical trial called “Feasibility Study of Tolerogenic Fibroblasts in Patients with Refractory Multiple Sclerosis.” The study was conducted in five participants. The primary objective of the study was to assess safety, and the secondary objective was to assess efficacy. The results of the study for safety were no adverse effects during intravenous injection of the tolerogenic fibroblasts, no short or long-impact in complete blood count test during the 16-week monitoring period, and no short or long impact in electrocardiogram results during the 16-week monitoring period. In addition, the results of the study for efficacy included general improvement of Paced Auditory Serial Addition Test, or PASAT, score for all patients during the 16-week monitoring period, general improvement of 9-hole Peg test completion time for all patients during the 16-week testing period, no general improvement or deterioration noted with the Timed 25-Foot walk test, no general improvement or deterioration noted with Expanded Disability Status Scale, or EDSS, test, and no patient exhibited further deterioration during the trial. We are currently conducting further research to determine the mode of action of fibroblasts in oligodendrocyte expansion and expect to file an IND application for a Phase 2 clinical trial in MS. We will likely seek a strategic partner to collaborate with us on the development of CYMS101 either before initiating the Phase 2 clinical trial, or after its completion, if successful, and prior to commencing with a Phase 3 clinical trial.
CYWC 628用于伤口愈合:我们正处于开发CYWC628作为一种基于同种异体成纤维细胞的伤口愈合疗法的临床前后期阶段。我们的研究目前集中在利用成纤维细胞和成纤维细胞来源的细胞治疗糖尿病小鼠的伤口。到目前为止,我们的数据是从四个独立的动物模型研究(手稿正在出版中)汇编而成的。每项研究都使用了16只野生型以及瘦素突变的NONcNZO10LTJ 小鼠,这些小鼠在喂食高脂肪食物时会患上2型糖尿病。我们所有实验的伤口大小和面积都是使用eKare Insight™设备测量的,该设备是FDA批准的,用于测量和监测伤口大小、面积和深度。我们的临床前研究的第一阶段 研究了皮下和局部给药的单细胞小鼠真皮成纤维细胞(两种处理每两天给药一次), 以及小鼠真皮成纤维细胞来源的外体。这项研究的结果表明在伤口愈合方面有显著的改善 (p
我们的竞争优势
我们的优势在于我们以成纤维细胞的力量为核心的技术平台,以及我们经验丰富的领导团队。成纤维细胞是人体中发现的最常见的细胞,我们相信它们比干细胞更健壮和强大。我们的知识产权组合包括48项已颁发专利 和108项在不同治疗领域使用成纤维细胞的未决专利。我们还拥有一支经验丰富的领导团队,在创业型初创公司和生命科学行业拥有成功的记录,拥有具有生命科学运营领导经验的董事会,以及具有相关专业知识的世界知名科学顾问委员会。
我们的 战略
我们 正在利用成纤维细胞作为技术平台,研究和开发针对治疗需求未得到满足的慢性疾病的创新疗法。我们的愿景是通过严格的科学流程和承诺满足患者的需求,成为再生医学领域的世界领先者。为实现我们的愿景,我们将重点抓好以下战略:
| ● | 将我们最初的临床开发工作与重要的未满足的治疗需求、较低的风险和较高的市场潜力相结合,优先考虑候选产品。 |
| 4 |
| ● | 与具有相关专业知识和经验的合同研究组织或CRO合作,成功并及时地执行临床试验,以生成可靠的关键数据, 可用于寻求批准。 | |
| ● | 吸引并留住具备开展临床前研究和确定临床试验最佳途径所需技能的科学家。 |
| ● | 投资 生产和供应成纤维细胞用于临床试验和初步商业化所需的关键能力。 |
| ● | 保护、扩大和捍卫我们围绕成纤维细胞的知识产权组合。 |
| ● | 在资金允许的情况下,在开发时间更长、风险更大且有重大未满足治疗需求的候选产品中,扩大 开发力度。 |
风险因素摘要
我们的业务受到许多风险和不确定性的影响,您在做出投资决定之前应该意识到这些风险和不确定性,包括标题为“风险因素“在这份招股说明书中。这些风险包括但不限于以下 :
| ● | 生物制药产品的成功开发具有很高的不确定性。 |
| ● | 我们 的运营历史有限,目前的候选产品均未获得商业销售的批准 。 |
| ● | 自成立以来,我们 已发生重大净亏损,预计在可预见的未来将继续出现重大净亏损,并且可能永远无法实现或保持盈利。 |
| ● | 我们 将需要大量额外资本来为我们的运营提供资金。如果我们无法在需要时或在可接受的条件下筹集此类资金,我们可能会被迫推迟、减少 和/或取消我们的一个或多个研究和药物开发计划或未来的商业化工作 。 |
| ● | FDA、欧洲药品管理局或欧洲药品管理局以及其他类似的外国监管机构的监管审批流程 既冗长又耗时,而且本质上是不可预测的。 |
| ● | 我们 在完成我们的候选产品的开发和商业化方面可能会遇到很大的延迟,或者最终无法完成。 |
| ● | 临床前研究或早期临床试验的结果可能不能预测后续临床试验的成功,我们的临床试验结果可能不符合FDA、EMA或其他类似外国监管机构的要求 。 |
| ● | 随着更多的患者数据可用,我们不时公布或公布的临床试验的临时、 基本和初步数据可能会发生变化,并受到审计和验证程序的影响,这些程序可能会导致最终数据发生重大变化。 |
| ● | 我们当前或未来的候选产品在单独使用或与其他批准的产品或研究中的新药联合使用时,可能会导致不良事件、毒性或其他不良副作用 可能会导致可能会阻碍监管部门批准的安全状况,妨碍 市场接受,限制其商业潜力或造成重大负面后果。 |
| ● | 即使获得批准,我们的候选产品也可能无法获得足够的市场接受度。 |
| ● | 我们的 候选冷藏产品需要在 临床站点进行特定的存储、处理和管理。 |
| ● | 我们 打算识别和开发新的候选细胞疗法产品,这使得 很难预测候选产品开发的时间、成本和潜在成功。 |
| 5 |
| ● | 由于 细胞疗法是新颖的,而且我们可能开发的任何候选细胞疗法产品的监管环境都是严格、复杂、不确定和可能发生变化的,因此我们无法预测获得监管批准的时间和成本,如果我们真的获得了批准,对于 我们可能开发的任何候选产品。 |
| ● | 我们 可能无法获得美国或外国监管部门的批准,因此可能无法 将我们的候选产品商业化。 |
| ● | 我们打算寻求批准作为生物制品的任何 候选产品可能比预期更早面临竞争 。 |
| ● | 我们在设计临床试验方面的经验有限。 |
| ● | 我们的长期前景在一定程度上取决于发现、开发和商业化其他 候选产品,这些候选产品可能会在开发中失败或出现延迟,从而对其商业可行性产生不利影响。 |
| ● | 我们 以前从未将基于成纤维细胞的候选治疗产品商业化,可能 缺乏必要的专业知识、人员和资源,无法单独或与合适的合作伙伴一起成功将任何候选产品商业化。 |
| ● | 我们面临着激烈的竞争。 |
| ● | 如果 我们无法建立销售或营销能力,或无法与 第三方达成协议来销售或营销我们的候选产品,则我们可能无法成功 销售或营销获得监管部门批准的候选产品。 |
| ● | 为了成功实施我们的计划和战略,我们需要扩大我们组织的规模,我们在管理这种增长时可能会遇到困难。 |
| ● | 我们 依赖第三方(I)进行临床前研究和临床试验的某些方面,以及(Ii)制造过程的某些部分受到风险的影响。 |
| ● | 我们 高度依赖我们在德克萨斯州休斯敦的设施,任何未能维持该设施的使用都将对我们的业务产生重大和不利的影响。 |
| ● | 我们 受到广泛的政府法规的约束。 |
| ● | 我们的业务存在重大的产品责任风险。 |
| ● | FDA、EMA和其他类似的外国监管机构可能不接受在其管辖范围外进行的 试验数据。 |
| ● | 即使我们的候选产品获得监管部门的批准,他们也将受到重大的上市后监管要求和监督。 |
| ● | 我们的成功取决于我们保护我们的知识产权和专有技术的能力, 我们面临与我们的知识产权相关的各种风险。 |
| ● | 我们的上市与公司承销的首次公开募股(IPO)有很大不同。 | |
| ● | 作为上市公司的 要求 可能会使我们的资源紧张,分散管理层的注意力,并 影响我们吸引和留住执行管理层和合格董事会成员的能力。 | |
| ● | 我们 将成为纳斯达克直接上市规则所指的“受控公司”,因为我们的内部人士将实益拥有我们已发行有投票权证券的50%以上的投票权。 | |
| ● | 直接上市后,我们将拥有2,500股具有超级投票权的C系列优先股 。 | |
| ● | 我们 发现,由于缺乏职责分工,我们对财务报告的内部控制存在重大缺陷。 |
| ● | 我们的 普通股目前没有公开市场。活跃的交易市场可能不会发展或继续保持流动性,我们普通股的市场价格可能会 波动。 |
反向 股票拆分
2023年10月6日,我们的董事会和我们的股东各自批准了4取1的反向股票拆分,2023年10月31日,我们向特拉华州提交了修订和重述的公司注册证书,以立即生效反向股票拆分。除非另有说明,本招股说明书中的所有股票和每股信息均已调整,以反映反向股票拆分。
| 6 |
对法定股本的调整
关于反向股票拆分,我们的董事会和股东还批准减少股本数量 ,以及我们授权发行的各自构成我们股本的证券。
紧接在反向股票拆分之前,我们被授权发行的所有类别股本的总股数为600,000,000股,包括(I)400,000,000股有表决权的普通股(在本招股说明书中我们有时将其称为“普通股”),(Ii)120,000,000股无表决权的普通股和(Iii)80,000,000股优先股, 其中35,000,000股被指定为A系列优先股,20,000,000股被指定为B系列优先股, 20,000,000,000被指定为B-1系列优先股,10,000,000被指定为C系列优先股。
根据我们对法定股本的调整,紧接反向股票拆分后,我们被授权发行的所有类别股本的股份总数为150,000,000股,包括(I)100,000,000股有表决权的普通股(在本招股说明书中有时称为我们的“普通股”),(Ii)30,000,000股无表决权的普通股和(Iii)20,000,000股优先股,其中8,750,000股被指定为A系列优先股, 5,000,000股被指定为B系列优先股,5,000,000股被指定为B-1系列优先股 ,2,500股被指定为C系列优先股。在本招股说明书中,我们有时将我们股本的上述调整称为“授权股本调整”。
于完成直接上市后,我们将获授权发行110,000,000股普通股,其中包括:(I)100,000,000股普通股,每股面值0.00001美元及(Ii)10,000,000股优先股,每股面值0.00001美元,其中2,500股被指定为C系列优先股。有关更多细节,请参阅“股本说明”。
作为受控公司的含义
直接上市完成后,我们的创始人兼首席执行官皮特·奥希隆将共同实益拥有我们已发行有表决权证券约59%的投票权,我们将成为 纳斯达克股票市场有限责任公司上市规则所指的“受控公司”。
只要我们的主要股东拥有我们公司至少50%的投票权,我们就是纳斯达克上市规则所定义的“控股公司” 。作为一家受控公司,我们被允许依赖纳斯达克公司治理规则的某些豁免,包括:
| ● | 免除我们董事会的大多数成员必须是独立董事的规定; |
| ● | 豁免我们首席执行官的薪酬必须完全由独立董事确定或推荐的规则;以及 |
| ● | 我们的董事提名人选必须完全由独立董事挑选或推荐,这一规定获得豁免。 |
虽然我们目前不打算依靠纳斯达克上市规则下的“受控公司”豁免,但我们可以选择在未来 依靠这一豁免。因此,您未来可能无法获得受这些公司治理要求约束的公司的股东所享有的同等保护 。
作为一家新兴成长型公司和较小的报告公司的影响
我们 是根据1933年《证券法》或《证券法》的定义,经《2012年创业法案》或《JOBS法案》修订的《新兴成长型公司》。因此,只要我们继续作为一家新兴成长型公司,我们就有资格并打算利用适用于其他非新兴成长型公司的上市公司的各种报告要求的某些豁免,包括:(I)根据2002年萨班斯-奥克斯利法案或萨班斯-奥克斯利法案第404(B)条关于财务报告的内部控制 豁免审计师认证要求,(Ii)豁免 说明性薪酬,在我们的定期报告和委托书中减少了关于高管薪酬的披露义务。
我们 将一直是一家新兴成长型公司,直到(I)2028年12月31日,(Ii)本财年的最后一天,我们的年总收入至少达到12.35亿美元,(Iii)本财年的最后一天,我们被视为《1934年证券交易法》(经修订)或《交易法》下的第12b-2条规则所定义的“大型加速申请者”,如果截至本年度第二个财政季度的最后一个营业日,非关联公司持有的我们普通股的市值为7.00亿美元或更多,或者(Iv)我们在前三年期间发行了超过10亿美元的不可转换债务证券,就会发生这种情况。
此外,《就业法案》还规定,新兴成长型公司可以利用延长的过渡期来遵守新的或修订后的会计准则。这允许新兴成长型公司推迟采用某些会计准则,直到 这些准则适用于私营公司。我们选择利用这一延长的过渡期,因此,我们可能会在非上市公司需要采用新的或修订的会计准则的相关日期采用此类准则,而不是其他上市公司要求采用的日期。
我们 也是《交易法》中定义的“小型报告公司”。即使我们不再是一家新兴成长型公司,我们也可能继续成为一家规模较小的报告公司。我们可能会利用某些适用于较小 报告公司的按比例披露,直到确定非关联公司持有的有表决权和无表决权普通股 在我们第二财政季度的最后一个工作日达到2.5亿美元或更多之后的财政年度,或者在最近完成的财政年度中,我们的年收入低于1亿美元 ,并且非关联公司持有的有表决权和无表决权普通股为7亿美元或更多 在我们第二财政季度的最后一个工作日测量。
企业信息
我们 成立于2021年4月,是一家得克萨斯州有限责任公司,名称为FibroBiologics,LLC,并于2021年12月以Fibrobiologics,Inc.的名称转换为特拉华州公司 。2023年4月12日,我们更名为FibroBiologics,Inc.。我们的主要高管 办公室位于德克萨斯州休斯敦,Suite300,455 E,Suite 300,Texas 77598。我们的电话号码是(281)671-5150,我们的网站地址是www.fifibiologics.com。我们网站上包含的或可以通过我们网站访问的信息既不是本招股说明书的一部分,也不是通过引用并入本招股说明书的,您不应将我们网站上的信息视为本招股说明书的一部分。我们的网站地址 仅作为非活动文本参考包含在本招股说明书中。
| 7 |
摘要 财务和其他数据
下面列出的汇总财务和其他数据应与我们的财务报表和这些报表的相关附注以及“管理层对财务状况和经营结果的讨论和分析“本招股说明书的一节。截至2022年12月31日和2021年12月31日的年度经营报表数据,以及截至2022年12月31日和2021年12月31日的年度现金流量表数据,均来自本招股说明书其他部分包括的经审计财务报表。截至2023年9月30日和2022年9月30日的9个月的运营报表数据 、截至2023年9月30日和2022年9月30日的9个月的现金流量表数据以及截至2023年9月30日的资产负债表数据均来自本招股说明书中其他部分包含的未经审计的中期财务报表。未经审核的中期财务报表是在与我们经审核的财务报表一致的基础上编制的,并在管理层的意见中包括我们认为为公平列报该等报表所载财务信息所必需的所有调整,包括正常经常性调整 。我们的历史业绩 不一定代表未来任何时期的预期结果,我们的中期业绩也不一定代表我们对截至2023年12月31日的年度的预期业绩。
下表中的所有股票编号和每股金额 均已调整,以反映反向股票拆分。
对于 止九个月 9月30日 |
截至 年度 12月31日, |
|||||||||||||||
| 2023 | 2022 | 2022 | 2021 | |||||||||||||
| (未经审计,以千为单位,不包括股票和每股数据) | (单位为 千,不包括股份和每股数据) | |||||||||||||||
| 运营数据报表 : | ||||||||||||||||
| 运营费用 : | ||||||||||||||||
| 研发 | $ | 1,595 | $ | 802 | $ | 1,147 | $ | 521 | ||||||||
| 常规、 管理和其他 | 4,814 | 2,361 | 3,320 | 1,057 | ||||||||||||
| 运营费用总额 | 6,409 | 3,163 | 4,467 | 1,578 | ||||||||||||
| 运营亏损 | (6,409 | ) | (3,163 | ) | (4,467 | ) | (1,578 | ) | ||||||||
| 其他 收入/(亏损) | (213 | ) | — | — | — | |||||||||||
| 利息 费用 | (146 | ) | (434 | ) | (654 | ) | (4 | ) | ||||||||
| 净亏损 | $ | (6,768 | ) | $ | (3,597 | ) | $ | (5,121 | ) | $ | (1,582 | ) | ||||
| 视为 股息 | (2,573 | ) | — | — | — | |||||||||||
| 普通股股东应占净亏损 | $ | (9,341 | ) | $ | (3,597 | ) | $ | (5,121 | ) | $ | (1,582 | ) | ||||
| 每股基本和稀释后净亏损 | $ | (.33 | ) | $ | (.13 | ) | $ | (.18 | ) | $ | 不适用 | |||||
| 加权平均 已发行、基本和稀释后的股票 | 28,230,842 | 28,230,842 | 28,230,842 | 不适用 | ||||||||||||
| 现金流量数据报表 : | ||||||||||||||||
| 净额 经营活动中使用的现金 | $ | (4,800 | ) | $ | (2,893 | ) | $ | (4,066 | ) | $ | (1,410 | ) | ||||
| 用于投资活动的现金净额 | $ | (493 | ) | $ | — | $ | — | $ | — | |||||||
| 净额 融资活动提供的现金 | $ | 13,793 | $ | 3,775 | $ | 5,925 | $ | 1,817 | ||||||||
自.起 9月30日, 2023 |
||||
| (未经审计,以千为单位) | ||||
| 资产负债表数据: | ||||
| 现金 和现金等价物 | $ | 10,766 | ||
| 流动资金 资金? | $ | 9,600 | ||
| 总资产 | $ | 13,299 | ||
| 总负债 | $ | 2,761 | ||
| 股东权益总额 | $ | 10,538 | ||
?我们将营运资本定义为流动资产减去流动负债。
| 8 |
风险因素
投资我们的普通股涉及很高的风险。在决定是否投资我们的普通股之前,您应仔细考虑以下风险和不确定性,以及本招股说明书中包含的所有其他信息,包括本招股说明书中其他部分的财务报表和相关附注 。发生这些风险因素中描述的一个或多个事件或情况 单独或与其他事件或情况一起发生,可能会对我们的业务、声誉、收入、财务状况、运营结果和未来前景产生重大不利影响,在这种情况下,您可能会损失全部或 部分投资。以下描述的风险和不确定因素并非详尽无遗,也不是我们面临的唯一风险和不确定因素。 我们目前不知道或认为不重要的其他风险和不确定因素也可能影响我们的业务运营。 本招股说明书还包含涉及风险和不确定因素的前瞻性陈述。请参阅“有关前瞻性 声明的警告说明”。由于某些因素,包括下文描述的因素,我们的实际结果可能与这些前瞻性陈述中预期的结果大不相同。
与我们的财务状况和资本要求有关的风险
生物制药产品的成功开发具有很高的不确定性。
生物制药产品的成功开发具有很高的不确定性,并取决于许多因素,其中许多因素是我们无法控制的。 在开发早期阶段看起来很有希望的候选产品可能无法投放市场,原因包括:
| ● | 临床 试验结果显示候选产品的有效性低于预期(例如,临床试验可能无法满足其主要或关键的次要终点(S))或具有 不可接受的安全性或耐受性概况; |
| ● | 未能获得必要的监管批准或延迟获得此类批准,这可能是由于患者未能通过试验筛查过程、临床试验登记缓慢、患者退出试验、患者失去后续行动、 达到试验终点的时间长度,数据分析或NDA准备的额外时间要求,与FDA的讨论,FDA要求额外的临床前或临床数据,或意外的安全或制造问题; |
| ● | 临床前 研究结果显示,候选产品不如预期效果或具有有害副作用; |
| ● | 上市后 审批要求;或 |
| ● | 其他人及其竞争产品和技术的专有权利,可能会阻止我们的候选产品 商业化。 |
完成临床试验并提交上市批准申请以供监管机构做出最终决定所需的时间长度 因候选产品的不同以及不同国家或司法管辖区的不同而有很大差异,可能很难预测。
即使我们成功地获得了市场批准,已批准产品的商业成功也可能在很大程度上取决于第三方付款人的承保范围和足够的报销。第三方付款人包括政府付款人,如Medicare和Medicaid计划 和美国的管理式医疗保健组织或外国特定国家的政府组织,这些组织可能会 受到旨在降低医疗成本的现有和未来医疗改革措施的影响。第三方付款人可能要求 我们进行其他研究,包括与批准产品的成本效益相关的上市后研究,以使 有资格获得报销,这可能代价高昂,并会转移我们的资源。如果政府和其他医疗保健付款人在获得批准后不为我们的产品提供保险和足够的补偿,市场认可度和商业成功可能会降低。
| 9 |
此外,如果我们的任何候选产品获得上市批准,我们将受到有关提交安全和其他上市后信息、报告和注册的重大监管义务的约束,并且对于我们在批准后进行的任何临床试验,我们将需要继续遵守(或确保任何第三方提供商遵守)cGMP和良好临床实践或GCP。 此外,我们、监管机构或第三方始终存在这样的风险,即我们、监管机构或第三方可能会发现 产品审批后存在以前未知的问题,例如意外严重或频率的不良事件。遵守这些要求的成本很高, 如果我们的候选产品在审批后未能遵守或出现其他问题,可能会对我们的业务、财务状况和运营结果产生不利影响。
我们 的运营历史有限,我们当前的候选产品都没有获得商业销售的批准,这可能会使您很难评估我们当前的业务并预测我们未来的成功和生存能力。
生物制药 产品开发是一项投机性很强的工作,涉及很大程度的风险。我们是一家临床阶段的细胞治疗公司,运营历史有限,您可以据此评估我们的业务和前景。我们目前的候选产品 均未被批准用于商业销售,我们也未从此类候选产品中获得任何收入。到目前为止,我们已经投入了大量的 所有资源和努力来组织和配备我们的公司、业务规划、执行合作伙伴关系、筹集资金、发现、确定和开发潜在的候选产品、保护相关的知识产权以及对我们的候选产品进行和 规划临床前研究和临床试验。对于我们目前的候选产品,我们 尚未证明我们有能力成功完成任何第三阶段临床试验、获得市场批准、制造商业规模的产品或安排第三方代表我们这样做,或进行成功的产品商业化所需的销售和营销活动。因此,与我们拥有更长的运营历史或成功开发和商业化生物制药产品的历史相比,您可能更难准确预测我们未来的成功或生存能力 。
此外,我们还可能遇到临床阶段的生物制药公司在快速发展的领域中经常遇到的不可预见的费用、困难、并发症、延误和其他已知和未知的因素和风险。我们还可能需要从一家专注于研究的公司转型为一家能够支持商业活动的公司。如果我们不能充分应对这些风险和困难 或成功实现这样的过渡,我们的业务将受到影响。
我们 自成立以来发生了重大净亏损,预计在可预见的未来将继续出现重大净亏损, 可能永远不会实现或保持盈利。
自我们 成立以来,我们发生了巨大的净亏损,到目前为止,我们没有从产品销售中产生任何收入,我们的运营主要通过私人融资。 截至2022年12月31日和2021年12月31日的年度以及截至2023年9月30日的9个月,我们分别发生了510万美元、160万美元和680万美元的净亏损。截至2022年12月31日和2023年9月30日,我们的累计赤字分别为790万美元和1460万美元。我们的亏损主要源于研究和开发我们的候选产品所产生的费用,以及我们在构建业务基础设施时发生的管理和行政成本以及其他费用。我们预计需要数年时间(如果有的话)才能拥有商业化的产品并从产品销售中获得收入。即使我们成功获得了一个或多个候选产品的营销批准并将其商业化, 我们预计,随着我们发现、开发和营销其他潜在的候选产品,我们将继续产生大量的研发和其他费用。
我们 预计在可预见的未来将继续蒙受重大损失,我们预计这些损失将大幅增加,如果和 我们:
| ● | 通过临床开发推进我们的主要候选产品的开发,如果FDA批准,则将其商业化; |
| ● | 将我们的临床前开发计划推进到临床开发; |
| ● | 为供应我们的候选产品而产生的电池生产制造成本; |
| 10 |
| ● | 为我们成功完成临床试验的任何候选产品寻求监管批准 ; |
| ● | 增加我们的研究和开发活动,以确定和开发新的候选产品; |
| ● | 雇用 名额外人员; |
| ● | 扩展我们的运营、财务和管理系统; |
| ● | 满足上市公司的要求和要求; |
| ● | 投资 进一步发展,保护和扩大我们的知识产权; |
| ● | 建立 销售、营销、医疗事务和分销基础设施,将我们可能获得营销批准并打算商业化的任何候选产品 商业化; |
| ● | 扩大我们的制造并发展我们的商业化努力。 |
我们产生的净亏损可能会在不同时期之间波动很大,因此我们运营结果的期间间比较 可能不是我们未来业绩的良好指示。我们未来净亏损的规模将在一定程度上取决于我们未来支出的增长率和我们创造收入的能力。我们之前的亏损和预期的未来亏损已经并将继续 对我们的营运资本以及我们实现和维持盈利的能力产生不利影响。
我们实现盈利并保持盈利的能力取决于我们创造收入或执行其他业务发展安排的能力。 除非我们能够获得监管部门的批准,并成功地将我们正在开发或可能开发的一个或多个候选产品 商业化,否则我们预计不会产生可观的收入。成功的商业化将需要实现许多关键里程碑,包括在临床试验中证明安全性和有效性,获得这些候选产品的监管批准,制造、营销和销售我们可能获得监管批准的产品,满足任何上市后要求,以及从私人保险或政府付款人那里获得我们产品的报销。由于与这些活动相关的不确定性和风险,我们无法准确和准确地预测收入的时间和金额、任何进一步亏损的程度或我们是否或何时可能实现盈利。
我们 在这些活动中可能永远不会成功,即使我们成功了,我们也可能永远不会产生足以让我们实现 盈利的收入。即使我们确实实现了盈利,我们也可能无法维持或提高季度或年度盈利能力 。我们未能实现并保持盈利,可能会削弱我们筹集资金、扩大业务、使产品多样化或继续运营的能力。如果我们继续像我们成立以来那样蒙受损失,投资者可能无法从他们的投资中获得任何回报 ,并可能失去他们的全部投资。
我们 将需要大量额外资本来为我们的运营提供资金。如果我们无法在需要时或在可接受的条件下筹集此类资金,我们可能会被迫推迟、减少和/或取消我们的一个或多个研究和药物开发计划或未来的商业化努力 。
开发生物制药产品,包括进行临床前研究和临床试验,是一个非常耗时、昂贵和不确定的过程,需要数年时间才能完成。自成立以来,我们的运营消耗了大量现金,我们预计我们与持续活动相关的费用 将会增加,特别是当我们启动和进行临床试验,并为我们当前的候选产品和任何未来的候选产品寻求营销 批准。即使我们 开发的一个或多个候选产品获准用于商业销售,我们预计也会产生与任何批准的候选产品 商业化相关的巨额成本。如果FDA、EMA或其他类似的监管机构要求我们在目前预期的基础上进行临床试验或临床前研究,我们的费用可能会超出预期。还可能产生其他意想不到的成本。此外,如果我们的任何候选产品获得市场批准,我们预计将产生与药品销售、营销、制造和分销相关的巨额商业化费用。由于我们预期的临床试验的设计和结果高度不确定,我们无法合理估计成功完成我们开发的任何候选产品的开发和商业化所需的实际金额。因此,我们将需要获得大量额外资金以维持我们的持续运营。
| 11 |
截至2023年9月30日,我们拥有约1,080万美元的现金和现金等价物。根据我们目前的业务计划, 我们相信,我们的现有资本将使我们能够为我们的运营提供资金,至少持续到2024年9月30日。我们预计我们的现有资本能够在多长时间内继续为我们的运营提供资金,这是基于可能被证明是错误的假设, 我们可以比目前预期的更快地使用可用的资本资源。不断变化的情况,其中一些可能超出我们的控制,可能会导致我们消耗资本的速度大大快于我们目前的预期,我们可能需要比计划更早地寻求额外的 资金。
我们未来的资金需求将取决于许多因素,包括但不限于:
| ● | 我们候选产品的临床试验的启动、进度、时间表、成本和结果; |
| ● | 其他研究和临床前研究的启动、进度、时间表、成本和结果 与流水线开发和我们未来启动的其他研究计划相关的 ; |
| ● | 制造活动的 成本和时间安排,包括我们计划的与我们的候选产品和其他计划相关的生产扩展活动,以及我们通过商业化推进它们的临床前和临床开发 ; |
| ● | 我们当前发展计划的潜在扩展,以寻求新的适应症; |
| ● | 满足FDA和其他可比外国监管机构制定的监管要求的结果、时间和成本; |
| ● | 提交、起诉、辩护和执行专利权利要求和其他知识产权的费用,无论是许可内的还是其他方面的; |
| ● | 竞争的技术和市场发展的影响; |
| ● | 支付许可费、可能的特许权使用费和可能的里程碑付款; |
| ● | 一般业务费用的成本; |
| ● | 为我们可能获得监管批准的任何候选产品建立销售、营销和分销能力的成本 在我们选择自行将产品商业化的地区 ;以及 |
| ● | 作为一家上市公司的运营成本。 |
推进我们候选产品的开发将需要大量资金。为了为完成我们的候选产品开发所必需的所有活动提供资金,我们将被要求通过股权发行、债务融资、合作和许可安排或其他来源获得更多资金,这可能会稀释我们的股东或限制我们的运营活动 。在可接受的条件下,我们可能无法获得足够的额外资金,或者根本没有。
我们 未能在需要时或在可接受的条件下筹集资金,将对我们的财务状况和我们执行业务战略的能力产生负面影响,我们可能不得不推迟、缩小、暂停或取消我们的一个或多个研究阶段计划、临床试验或未来的商业化努力,授予开发和营销我们原本 更愿意开发和营销的候选产品的权利,通过与合作者的安排以对我们不利的条款获得资金,或者寻求合并 或收购战略,所有这些都可能对我们的持股或股东的权利产生不利影响。
| 12 |
筹集额外资本可能会稀释我们的现有股东,限制我们的运营,或者要求我们以对我们不利的条款放弃对我们的 产品候选产品的权利。
我们 可以通过各种方式寻求更多资金,包括股权、债务融资或其他来源,包括预付款和战略合作的里程碑付款。由于有利的市场条件或战略考虑,我们可能会寻求额外的资本,即使我们认为我们目前或未来的运营计划有足够的资金。如果我们通过出售股权或可转换债务证券来筹集额外的 资本,您的所有权权益将被稀释,条款可能包括清算或其他优惠和反稀释保护,对您作为股东的权利产生不利影响。
此类 融资还可能导致强制实施债务契约、增加固定付款义务或其他限制,从而可能对我们开展业务的能力产生不利影响。如果我们通过合作、战略联盟或营销、分销或与第三方的许可安排来筹集更多资金,我们可能不得不放弃对我们的技术、未来收入流、研究计划或候选产品的宝贵权利,或者以对我们不利的条款授予许可证。
我们 与某些投资者签订了日期为2021年11月12日的股份购买协议或股份购买协议,根据该协议,我们可以选择向该等投资者发行和出售股票,如果我们被选中,该等投资者将有义务购买 自我们的普通股在美国主要证券交易所交易的第一天起至自该日起60个月止的时间, 价值不超过100,000,000美元的普通股,或总限额。股份购买协议取决于我们实现我们普通股的公开上市。根据协议,我们需要向投资者支付相当于总限额的2%的承诺费,以现金或普通股的形式支付。即使我们不使用任何提款,承诺费也是要支付的。
此外,协议还要求我们在公开上市之日向投资者发行认股权证,购买最多为我们普通股的 股,相当于我们公开上市后紧随其后已发行的总股权的4%。每股价格相等于(I)每股公开招股价(如属首次公开招股) 或每股于公开上市日的收市价(如属首次公开招股以外的公开上市) 或(Ii)700,000,000元除以股权总数所得的商数。
我们选择根据股份购买协议向投资者发行和出售我们普通股的股份,或行使我们将有义务在本次直接上市完成时发行的认股权证,将导致我们的现有股东 和在此次发行中购买我们普通股股份的投资者进一步稀释。
涉及开发、监管审批和商业化的风险
FDA、EMA和其他类似的外国监管机构的监管审批过程冗长、耗时,而且 本质上是不可预测的。如果我们最终无法获得监管机构对我们的候选产品的批准,我们将无法 产生产品收入,我们的业务将受到严重损害。
未经FDA批准,我们 不得在美国商业化、营销、推广或销售任何候选产品。外国监管机构,如EMA,也实施了类似的要求。获得FDA、EMA和其他类似外国监管机构的批准所需的时间是不可预测的,通常在临床试验开始 后需要数年时间,并取决于许多因素,包括所涉及的候选产品的类型、复杂性和新颖性。 此外,批准政策、法规或获得批准所需的临床数据的类型和数量可能会在候选产品的临床开发过程中发生变化,并可能因司法管辖区而异,这可能会导致批准延迟或 决定不批准申请。监管机构在审批过程中拥有相当大的自由裁量权,可以拒绝接受任何申请,也可以决定我们的数据不足以获得批准,需要额外的临床前、临床或其他 数据。即使我们最终完成了临床测试,并获得了对我们候选产品的任何监管申请的批准,FDA、EMA和其他类似的外国监管机构可能会批准我们的候选产品,因为这些候选产品的适应症或患者人数可能比我们最初要求的更有限。我们尚未为任何候选产品提交或获得监管批准, 我们现有的候选产品或我们未来可能寻求开发的任何产品都可能永远不会 获得监管批准。
此外, 我们候选产品的开发和/或监管审批可能会因我们无法控制的原因而延迟。
我们候选产品的申请 可能会因多种原因而无法获得监管部门的批准,包括以下原因:
| ● | FDA、EMA或其他类似的外国监管机构可能不同意我们临床试验的设计、实施或结果; |
| ● | 该 FDA、EMA或其他类似的外国监管机构可能会确定我们的产品 候选人不安全和有效,只有中等有效性,或具有不受欢迎或非预期的 副作用、毒性或妨碍我们获得上市许可的其他特征 防止或限制商业用途; |
| 13 |
| ● | 该 临床试验中研究的人群可能不够广泛或具有代表性 确保我们寻求批准的全部人群的有效性和安全性; |
| ● | 该 FDA、EMA或其他类似的外国监管机构可能不同意我们的解释 来自临床前研究或临床试验的数据; |
| ● | 该 从我们候选产品的临床试验中收集的数据可能不足以支持 提交生物制品许可申请或BLA或其他提交材料,或获得 美国或其他地方的监管批准; |
| ● | 我们 可能无法向FDA、EMA或其他类似的外国监管机构证明 候选产品针对其拟定适应症的风险受益比是可接受的; |
| ● | 该 FDA、EMA或其他类似的外国监管机构可能无法批准我们的生产 流程、测试程序和规范或设施或第三方制造商的流程、测试程序和规范或设施 我们与之签订临床和商业供应合同;以及 |
| ● | 该 FDA、EMA或其他类似外国监管机构的批准政策或法规 当局可能会以使我们的临床数据不足的方式发生重大变化 等待批准 |
这种 漫长且不确定的审批过程,以及临床试验结果的不可预测性,可能会导致我们无法 获得监管机构批准以销售我们的任何候选产品,这将严重损害我们的业务、经营业绩 和前景。此外,FDA、EMA或类似的外国监管机构可能会改变其政策、采用额外的 法规或修订现有法规或采取其他行动,这可能会阻止或延迟我们未来正在开发的候选产品的及时批准。此类政策或法规变更可能会对我们提出额外要求,从而可能会延迟 我们获得批准的能力、增加合规成本或限制我们 维持可能已获得的任何营销授权的能力。
我们 可能会在完成或最终无法完成候选产品 的开发和商业化过程中遇到重大延误。
在 从FDA、EMA或其他类似的外国监管机构获得销售我们的候选产品的上市批准之前,我们必须完成临床前开发和广泛的临床试验,以证明我们候选产品的安全性和有效性。 临床测试昂贵、难以设计和实施,可能需要多年才能完成,其最终结果也不确定。 在该过程的任何阶段都可能发生一项或多项临床试验失败。临床前研究和早期临床试验的结果可能不能预测后续临床试验的成功。
我们 不知道我们未来的临床试验是否会按时开始或按时招募患者,也不知道我们正在进行的和/或未来的临床试验是否会如期完成,或者根本不知道。临床试验可能会因各种原因而延迟,包括与以下方面有关的延迟:
| ● | FDA、EMA或其他类似的外国监管机构对我们临床试验的设计或实施持不同意见; |
| ● | 获得开始试验的监管授权或与监管机构就试验设计达成共识的 ; |
| ● | 与CRO和临床试验站点达成协议的任何 失败或延迟, 的条款可以进行广泛的谈判,并且可能在不同的CRO和试验站点之间存在显著差异; |
| ● | 获得一个或多个独立机构审查委员会或IRBs的批准; |
| ● | IRBs 拒绝批准、暂停或终止调查地点的试验,禁止 招募更多受试者,或撤回对试验的批准; |
| 14 |
| ● | 因旅行或检疫政策、或其他与流行病有关的因素或 其他我们无法控制的事件而延迟注册; |
| ● | 将 更改为临床试验方案; |
| ● | 临床 站点偏离试验方案或退出试验; |
| ● | 生产足够数量的候选产品或获得足够数量的组合疗法以用于临床试验; |
| ● | 受试者 未能以我们预期的速度登记或留在我们的试验中,或未能返回进行 治疗后跟进; |
| ● | 受试者 为我们正在开发产品的适应症选择替代疗法 候选者,或参与竞争性临床试验; |
| ● | 缺乏足够的资金来继续临床试验; |
| ● | 出现严重或意想不到的药物不良反应的受试者; |
| ● | 在其他公司进行的同类药物试验中发生严重不良事件的; |
| ● | 选择需要长时间临床观察或对结果数据进行分析的临床终点。 |
| ● | 因违反cGMP法规或其他适用要求,FDA、EMA或类似的外国监管机构责令生产我们的候选产品或其任何组件的工厂暂时或永久关闭。或制造过程中候选产品的感染或交叉污染; |
| ● | 可能需要或希望对我们的制造流程进行的任何 更改; |
| ● | 第三方临床研究人员 失去进行临床试验所需的许可证或许可, 未按预期时间表或与临床试验方案、GCP或其他法规要求一致的情况下进行临床试验; |
| ● | 第三方承包商未及时或准确地进行数据收集或分析;或 |
| ● | 第三方承包商因违反监管要求而被FDA、EMA或其他政府或监管机构禁止或暂停或以其他方式处罚,在这种情况下,我们可能需要寻找替代承包商,我们可能无法使用此类承包商提供的部分或全部数据来支持我们的营销应用程序。 |
在国外进行临床试验,就像我们可能为我们的候选产品所做的那样,会带来额外的风险,可能会推迟我们的临床试验的完成 。这些风险包括在外国登记的患者由于医疗服务或文化习俗的差异而未能遵守临床方案,管理与外国监管计划相关的额外行政负担,以及与此类外国相关的政治和经济风险。
| 15 |
此外, 如果我们的临床试验结果不确定,或者如果我们的候选产品存在安全问题或严重不良事件,我们可以:
| ● | 在获得上市批准方面被推迟(如果有的话); |
| ● | 获得 批准的适应症或患者群体不像预期或期望的那样广泛; |
| ● | 获得包括重大使用或分发限制或安全警告的标签的批准 ; |
| ● | 被要求进行额外的临床试验以支持批准或接受额外的上市后测试要求; |
| ● | 添加诸如警告或禁忌症等标签说明; |
| ● | 被起诉;或 |
| ● | 经历 对我们声誉的损害。 |
如果我们在测试或获得市场批准方面遇到延误,我们的开发成本也会增加。我们不知道我们的任何临床前研究或临床试验是否将按计划开始、需要重组或按计划完成(如果有的话)。 我们临床试验的任何延迟或终止都将推迟向FDA或具有类似外国监管机构的类似申请提交BLA,并最终影响我们将候选产品商业化的能力(如果获得批准),并产生产品 收入。即使我们的临床试验按计划完成,我们也不能确定他们的结果是否支持我们的差异化声明或我们候选产品的有效性或安全性。FDA在审查和批准过程中拥有很大的自由裁量权,可能会 不同意我们的数据支持我们提出的主张。
此外,我们临床试验的首席研究员可能会不时担任我们的科学顾问或顾问,并获得与此类服务相关的报酬 。在某些情况下,我们可能被要求向FDA、EMA或其他类似的外国监管机构报告其中一些关系。FDA、EMA或其他类似的外国监管机构可能会得出结论 我们与主要调查员之间的财务关系造成了利益冲突或以其他方式影响了对研究的解释 。因此,FDA、EMA或其他类似的外国监管机构可能会质疑在适用的临床试验地点产生的数据的完整性 ,临床试验本身的效用可能会受到威胁。这可能会导致FDA、EMA或其他类似的外国监管机构延迟批准或拒绝我们的营销申请,具体视情况而定,并可能最终导致我们的一个或多个候选产品被拒绝上市批准。
如果 我们的候选产品临床试验延迟完成或终止,我们候选产品的商业前景将受到损害,我们从这些候选产品中获得产品收入的能力将被推迟。 此外,完成临床试验的任何延迟都将增加我们的成本,减慢我们候选产品的开发和审批流程 ,并危及我们开始产品销售和创造收入的能力。
此外,许多导致或导致临床试验终止或暂停、或延迟开始或完成的因素也可能最终导致候选产品的监管审批被拒绝。我们的临床试验因此出现的任何延迟都可能缩短我们可能拥有独家商业化候选产品权利的任何期限 ,而我们的竞争对手可能会在我们之前将产品推向市场,并且我们候选产品的商业可行性可能会显著降低。任何这些情况都可能对我们的业务、财务状况和前景造成重大损害。
| 16 |
临床前研究或早期临床试验的结果可能不能预测后续临床试验的成功,我们临床试验的结果可能不符合FDA、EMA或其他类似外国监管机构的要求。
临床前研究和早期临床试验的阳性结果并不意味着未来的临床试验将会成功。在临床试验过程中,任何时候都可能出现失败。我们不知道我们的候选产品是否会像在临床前研究和早期临床试验中那样在当前或未来的临床试验中表现。后期临床试验的候选产品 可能无法证明足够的安全性和有效性,使FDA、EMA和其他类似的外国监管机构满意,尽管已通过临床前研究和早期临床试验取得进展。
在 某些情况下,由于多种因素,同一候选产品的不同临床试验的安全性和有效性结果可能存在显著差异,这些因素包括试验方案的变化、患者群体的大小和类型的差异、给药方案和其他试验方案的差异和遵守情况以及临床试验参与者之间的退学率。使用我们的候选产品治疗的患者 还可能正在接受手术、放射和化疗,并且可能正在使用其他经批准的 产品或研究新药,这些药物可能会导致与我们的候选产品无关的副作用或不良事件。因此,对特定患者以及临床试验中不同患者和不同地点的疗效评估可能会有很大差异。 这种主观性会增加我们临床试验结果的不确定性,并对其产生不利影响。我们不知道我们可能进行的任何临床试验是否会证明一致或足够的有效性和安全性,足以获得市场批准以销售我们的候选产品。大多数开始临床试验的候选产品从未获得监管机构的商业化批准。
此外, 我们计划的一些临床试验可能会使用“开放标签”试验设计。“开放标签”临床试验是指患者和研究人员都知道患者是否正在接受研究候选产品或现有批准的药物或安慰剂。最典型的情况是,开放标签临床试验只测试候选研究产品,有时可能会以不同的剂量水平进行测试。开放标签临床试验受到各种限制,这些限制可能会夸大任何治疗效果,因为开放标签临床试验中的患者在接受治疗时是知道的。开放标签临床试验 可能会受到“患者偏见”的影响,即患者认为他们的症状改善仅仅是因为他们知道接受实验性治疗 。此外,开放标签临床试验可能会受到“调查者偏见”的影响 在这种情况下,评估和审查临床试验的生理结果的人知道哪些患者接受了治疗 ,并可能在了解这一知识的情况下更有利地解释治疗组的信息。开放标签试验的结果可能无法 预测我们的任何候选产品的未来临床试验结果,当在受控环境中使用安慰剂或主动对照进行研究时,我们包括开放标签临床试验 。
此外,临床前和临床数据往往容易受到不同解释和分析的影响,许多公司认为他们的产品 候选产品在临床前研究或临床试验中表现令人满意,但仍未能获得FDA、EMA或类似的外国监管机构的批准。我们不能保证FDA、EMA或类似的外国监管机构会像我们一样解释试验结果,在我们能够提交寻求批准我们的候选产品的申请之前,可能需要进行更多的试验。 罕见疾病的临床试验尤其如此,患者人数非常少,因此很难进行 两项传统、充分和受控的研究,因此FDA、EMA或类似的外国监管机构经常被要求在批准此类疾病的治疗方案时具有灵活性。如果试验结果不能满足FDA、EMA或类似的外国监管机构对营销申请的支持,我们可能需要 花费我们可能无法获得的大量资源来进行额外的试验,以支持我们的 候选产品的潜在批准。即使我们的任何候选产品获得了监管部门的批准,此类批准的条款也可能会限制我们候选产品的范围和用途,这也可能限制其商业潜力。此外,FDA、EMA或类似的外国监管机构的审批政策或法规可能会发生重大变化,导致我们的临床数据不足以获得批准 ,这可能会导致FDA、EMA或类似的外国监管机构推迟、限制或拒绝批准我们的候选产品 。
| 17 |
随着更多的患者数据可用,我们不时宣布或公布的临床试验的临时、 基本和初步数据可能会发生变化,并受到审计和验证程序的影响,这可能会导致最终数据发生重大变化。
我们可能会不时地公开披露我们的临床前研究或临床试验的中期、初步或主要数据,这些数据是基于对当时可用数据的初步分析而得出的,在对与特定研究或试验相关的数据进行更全面的审查后,结果及相关发现和结论可能会发生变化。作为数据分析的一部分,我们还会进行假设、估计、计算和结论,我们可能没有收到或没有机会全面仔细地评估 所有数据。因此,一旦收到更多数据并进行充分评估,我们报告的中期、最新或初步结果可能与相同研究的未来结果不同, 或不同的结论或考虑因素可能会使这些结果合格。临时数据、初步数据和背线数据也将继续接受审核和验证程序,这可能会导致最终数据与我们之前发布的初步数据大不相同。因此,在最终数据可用之前,应谨慎查看中期数据、背线数据和初步数据。初步、背线或中期数据与最终数据之间的不利差异可能会显著 损害我们的业务前景。
此外, 其他人,包括监管机构,可能不接受或同意我们的假设、估计、计算、结论或分析 ,或者可能以不同的方式解释或权衡数据的重要性,这可能会影响特定计划的价值、特定候选产品和我们公司的总体商业化。此外,我们选择公开披露的有关特定研究或临床试验的信息是基于通常广泛的信息,您或其他人可能不会 同意我们确定的重要信息或其他适当信息包含在我们的披露中。如果我们报告的中期、背线或 初步数据与实际结果不同,或者如果包括监管机构在内的其他人不同意得出的结论,我们获得我们候选产品的批准并将其商业化的能力可能会受到损害,这可能会损害我们的业务, 经营结果、前景或财务状况。
我们当前或未来的候选产品在单独使用或与其他批准的产品或正在研究的新药一起使用时,可能会导致不良事件、毒性或其他不良副作用 ,这可能会导致安全状况,可能会阻碍监管部门的批准, 阻止市场接受,限制其商业潜力或导致严重的负面后果。
由于生物制药一般都是这种情况,因此很可能会出现与我们的候选产品 使用相关的副作用和不良事件。我们的临床试验结果可能会揭示出严重且不可接受的副作用或意外特征的严重程度和流行程度。我们的候选产品引起的不良副作用可能会导致我们或监管机构中断、推迟或暂停临床试验,并可能导致更严格的标签,或者FDA、EMA或类似的外国监管机构推迟或拒绝监管批准。与药物相关的副作用可能会影响患者招募或 注册患者完成试验的能力或导致潜在的产品责任索赔。这些情况中的任何一种都可能严重损害我们的业务、财务状况和前景。
如果我们的候选产品单独使用或与其他批准的产品或正在研究的新药联合使用时,在临床前研究或临床试验中出现不良副作用或意外特征,我们可能需要中断、推迟或 放弃它们的开发,或将开发限制在更狭窄的用途或子群中,在这些用途或子群中,不良副作用或其他特征 不太普遍、不太严重或从风险效益的角度来看更容易接受。与治疗相关的副作用也可能影响患者招募或受试者完成试验的能力,或导致潜在的产品责任索赔。 任何此类事件都可能阻止我们实现或保持市场对受影响候选产品的接受程度,并可能严重损害我们的业务、财务状况和前景。
我们正在进行和计划在未来进行的临床试验中的患者 可能会出现严重的不良事件或其他副作用,这在我们的临床前研究或之前的临床试验中没有观察到。我们的一些候选产品可能用于慢性疗法或用于儿科人群,监管机构可能会特别审查这些人群的安全性问题。此外,如果我们的候选产品 与其他疗法联合使用,我们的候选产品可能会加剧与该疗法相关的不良事件。接受我们候选产品治疗的患者 也可能正在接受手术、放疗或化疗,这可能会导致副作用或不良事件,这些副作用与我们的候选产品无关,但仍可能影响我们临床试验的成功。将危重患者纳入我们的临床试验可能会由于此类患者可能正在使用的其他疗法或药物,或由于此类患者病情的严重性而导致死亡或其他不良医疗事件 。
如果在我们当前或未来的任何临床试验中观察到重大不良事件或其他副作用,我们可能难以招募患者参加临床试验,患者可能会退出我们的试验,或者我们可能被要求完全放弃试验或我们对该候选产品的开发 努力。我们、FDA、EMA、其他类似的监管机构或IRB可随时出于各种原因暂停候选产品的临床试验,包括认为此类试验的受试者面临不可接受的健康风险或不良副作用。
| 18 |
此外, 如果我们的任何候选产品获得监管部门的批准并成为产品,而我们或其他人后来发现该产品造成的不良副作用 ,可能会导致许多潜在的重大负面后果。例如,FDA可以要求我们采用风险评估和缓解策略,或REMS,以确保使用此类产品治疗的好处大于每个潜在患者的风险,其中可能包括与医疗从业者的沟通计划、患者教育、 高度受控、严格限制且成本高于行业典型水平的广泛患者监测或分配系统和流程。如果我们或我们的合作者后来发现我们或我们的合作者开发的任何产品造成的不良副作用 ,我们或我们的合作者也可能被要求采用REMS或参与类似的操作,例如患者 教育、医疗保健专业人员认证或特定监测。其他潜在的重大负面后果包括:
| ● | 我们 可能被迫暂停该产品的营销,或决定将该产品从市场中移除 ; |
| ● | 监管机构可以撤回或更改对该产品的批准; |
| ● | 监管当局可能要求在标签上附加警告,或限制该产品进入有附加安全报告的选择性专科中心,并要求 患者在地理上靠近这些中心进行全部或部分治疗; |
| ● | 我们 可能需要创建一份药物指南,概述该产品对患者的风险,或者进行上市后研究; |
| ● | 我们 可能被要求更改产品的管理方式; |
| ● | 我们 可能受到罚款、禁令或施加刑事或民事处罚, 或被起诉并对对受试者或患者造成的伤害承担责任;以及 |
| ● | 产品的竞争力可能会降低,我们的声誉可能会受到影响。 |
这些事件中的任何一项都可能减少我们候选产品的使用或以其他方式限制其商业成功,并阻止我们实现 或保持市场对受影响候选产品的接受程度(如果获得适用监管机构的批准)。
即使 获得批准,我们的候选产品也可能无法在医生、患者、医疗保健付款人和医学界取得商业成功所必需的其他 人中获得足够的市场接受度。
即使我们的候选产品获得监管部门的批准并成为一种产品,它们也可能无法在医生、患者、医疗保健付款人和医疗界其他人中获得足够的市场接受度。市场对我们批准的任何候选产品的接受度 将取决于许多因素,包括:
| ● | 临床试验中证明的与替代疗法相比的有效性和安全性; |
| ● | 产品和竞争产品投放市场的时间; |
| ● | 该产品获批的临床适应症; |
| ● | 对我们产品的使用限制,如标签中的方框警告或禁忌症,或替代疗法和竞争产品可能不要求的REMS(如果有); |
| ● | 产品相对于替代疗法的潜在和公认的优势; |
| ● | 与替代治疗相关的治疗费用; |
| 19 |
| ● | 包括政府当局在内的第三方付款人提供保险和适当的补偿,以及定价。 |
| ● | 相对 管理的便利性和易用性; |
| ● | 目标患者群体尝试新疗法的意愿和医生开出这些疗法的意愿; |
| ● | 销售和营销工作的有效性; |
| ● | 与我们的产品或类似的经批准的产品或第三方开发的候选产品有关的不利宣传 ;以及 |
| ● | 批准用于相同适应症的其他新疗法。 |
如果 我们的任何候选产品获得批准,但没有获得医生、医院、医疗保健付款人和患者的足够程度的接受,我们可能无法从该候选产品产生或获得足够的收入,我们的财务业绩可能会受到负面影响 。
我们的候选冷藏产品需要在临床现场进行特定的存储、处理和管理。
我们的 候选冷藏药品必须低温储存在专门的冷藏容器中,直到立即使用 。在给药时,药品容器必须小心地从存储中取出,加热到室温,倒置细胞悬浮液,然后将产品拉入注射器。细胞疗法产品的处理、升温和给药必须按照特定说明进行。未能正确处理产品、遵循升温说明,以及在升温后未在指定时间内给药和/或未能给药,可能会对产品的功效和/或安全性产生负面影响。
由于 细胞疗法是新颖的,管理我们可能开发的任何候选细胞疗法产品的监管环境是严格、复杂、不确定和可能发生变化的,因此我们无法预测我们可能开发的任何候选产品获得监管批准的时间和成本(如果我们获得批准的话)。目前,只有一小部分细胞治疗产品在美国和欧盟获得批准。
我们开发的任何新的细胞疗法候选产品的监管要求并不完全明确,可能会发生变化。在更广泛的遗传医学领域,很少有治疗产品获得FDA或EMA的营销授权。即使是符合基因疗法或细胞疗法类别的更成熟的产品,监管格局仍在发展中。细胞治疗产品的监管要求经常发生变化,未来可能会继续变化。此外,负责监管现有细胞治疗产品的人员有很大的重叠。 例如,在美国,FDA在其生物评估和研究中心内设立了组织和高级治疗办公室 以整合对细胞治疗和相关产品的审查。尽管FDA已经批准了其他基于细胞的疗法,但不能保证这些先前的批准会影响FDA对我们候选产品的审查。
我们的候选细胞治疗产品将需要满足FDA管理的监管框架下适用于任何新生物的安全性和有效性标准。除了FDA的监督和IRBs的监督外,根据美国国立卫生研究院涉及重组或合成核酸分子的研究指南或NIH指南,细胞治疗临床试验也要接受机构生物安全委员会(IBC)的审查和监督,IBC是一个地方机构委员会,负责审查和监督该机构使用重组或合成核酸分子的研究。IBC评估研究的安全性,并确定对公共卫生或环境的任何潜在风险。虽然NIH指南不是强制性的,除非相关研究是在接受美国国立卫生研究院(NIH)重组或合成核酸分子研究资金的机构进行的或由其赞助的,但许多公司和其他不受NIH指南约束的机构自愿遵循这些指南。尽管FDA决定是否可以继续进行个别细胞治疗方案,但其他审查机构的审查过程和决定可能会阻碍或推迟临床试验的启动,即使FDA已经审查了试验并批准了其启动。
| 20 |
欧盟的情况也是如此。EMA的高级治疗委员会(CAT)负责评估高级治疗药物产品的质量、安全性和有效性。先进治疗药物包括细胞治疗药物、体细胞治疗药物和组织工程药物。CAT的作用是为提交给EMA的细胞疗法候选药物的营销申请准备一份意见草案。在欧盟,细胞治疗产品的开发和评估必须在相关欧盟指南的背景下进行考虑。EMA可能会发布有关细胞治疗产品的开发和营销授权的新指南,并要求我们遵守这些新指南。因此,应用于细胞治疗产品的程序和标准可能适用于我们可能开发的任何候选细胞治疗产品,但 目前仍不确定。
细胞治疗和细胞调节产品领域中其他人进行的临床前研究或临床试验的不利发展 可能会导致FDA、EMA和其他监管机构修改对我们可能开发的任何候选产品的审批要求 或限制使用细胞治疗技术的产品,这两种情况中的任何一种都可能损害我们的业务。此外,FDA、EMA和其他监管机构的临床试验要求以及这些监管机构用来确定候选产品安全性和有效性的标准因潜在产品的类型、复杂性、新颖性以及预期用途和市场而有很大不同。像我们这样的候选产品的监管审批流程可能比 其他更知名或更广泛研究的候选药品或其他产品更昂贵,花费的时间也更长。此外,由于我们正在为疾病开发潜在的新疗法,在某些情况下,几乎没有潜在的新终点和方法的临床经验, FDA、EMA或其他监管机构可能不会认为临床试验终点提供临床有意义的结果的风险增加,由此产生的临床数据和结果可能更难分析。此外,我们可能无法 识别或开发适当的动物疾病模型来支持或支持计划中的临床开发。我们在临床开发中可能进行或依赖的任何自然历史研究 可能不会被FDA、EMA或其他监管机构接受。 管理现有或未来法规或立法的监管机构可能不允许及时或在技术或商业可行的条件下利用 细胞治疗技术生产和销售产品。此外,监管行动或 私人诉讼可能会对我们的研究计划或最终产品的商业化造成费用、延迟或其他障碍 。此外,一个监管机构的批准可能不代表其他监管机构可能需要批准什么。
上述监管审查委员会和顾问组及其发布的新指南可能会延长监管审查流程,要求我们进行额外的临床前研究或临床试验,增加我们的开发成本,导致 监管立场和解释的变化,推迟或阻止这些候选产品的批准和商业化,或者导致重大的批准后限制或限制。随着我们推进研究计划和开发未来的候选产品,我们将被要求 与这些监管和咨询小组进行磋商,并遵守适用的指导方针。如果我们未能做到这一点,我们可能会被要求 推迟或停止我们确定和开发的任何候选产品的开发。这些额外的流程可能会导致审核 和审批流程比我们预期的更长。由于监管审批流程增加或延长或对我们候选产品开发的进一步限制而导致的延迟可能代价高昂,并可能对我们及时完成临床试验和将我们当前和未来的候选产品商业化的能力 产生负面影响。
我们 可能无法获得美国或外国监管部门的批准,因此可能无法将我们的候选产品商业化。
我们的候选产品必须遵守广泛的政府法规,其中包括研究、测试、开发、 制造、安全性、有效性、审批、记录保存、报告、标签、储存、包装、广告和促销、定价、 药品营销和分销。严格的临床前研究和临床试验以及广泛的监管审批程序必须在美国和许多外国司法管辖区成功完成,然后新药才能上市。满足这些 和其他法规要求是昂贵、耗时、不确定的,并且会受到意外延迟的影响。我们不能保证 我们可能开发的任何候选产品都将通过所需的临床测试,并获得我们开始销售它们所需的监管批准 。
| 21 |
我们 没有进行、管理或完成大规模或关键的临床试验,也没有管理FDA、EMA或任何其他监管机构对我们当前候选产品的监管审批过程。从FDA和其他监管机构获得批准所需的时间是不可预测的,需要成功完成广泛的临床试验,这通常需要数年时间,具体取决于候选产品的类型、复杂性和新颖性。FDA及其外国同行在评估临床试验数据时使用的标准在药物开发过程中可能且经常发生变化,这使得 很难确定地预测这些标准将如何应用。由于新的政府法规,包括未来的立法或行政行动,或在药物开发、临床试验和FDA监管审查期间FDA政策的变化,我们还可能遇到意想不到的延迟或成本增加。
在寻求或获得所需批准方面的任何延误或失败都将对我们从我们正在开发和寻求批准的特定候选产品获得收入的能力产生重大和不利的影响 。此外,任何监管部门对候选产品的上市批准都可能受到我们销售候选产品的批准用途或适应症的重大限制 或标签或其他限制。此外,FDA有权要求REMS作为批准NDA或BLA的一部分,或在批准后要求REMS,这可能会对批准的候选产品的分发或使用施加进一步的要求或限制。 这些要求或限制可能包括限制某些医生或经过专门培训的医疗中心开出处方,将治疗限制为满足某些安全使用标准的患者,并要求接受治疗的患者登记注册。这些 限制和限制可能会显著限制候选产品的市场规模,并影响第三方付款方的报销。
我们 还受制于许多外国监管要求,其中包括临床试验的进行、生产和营销授权、定价和第三方报销。外国监管审批流程因国家/地区而异, 通常包括与上述FDA审批相关的所有风险,以及可归因于满足外国司法管辖区当地法规的风险。此外,获得批准所需的时间可能与获得FDA批准所需的时间不同。
我们 可能会结合其他疗法开发我们当前和未来的候选产品,这会使我们面临额外的风险,并且我们的某些候选产品被监管为组合产品。
我们 可能会结合一种或多种其他已批准或未批准的疗法来开发我们当前和未来的候选产品,以治疗 皮肤和结缔组织疾病或其他疾病。我们还可能开发某些候选产品作为生物/药物组合产品。 我们的候选产品可能需要额外的时间才能获得监管部门的批准,因为它们是组合产品。我们的候选产品 是生物/药物组合产品,需要在FDA和类似的外国监管机构内进行协调,以 审查其生物和药物成分。尽管FDA和类似的外国监管机构已经建立了对像我们这样的组合产品进行 审查和批准的系统,但由于监管时间限制以及产品开发和批准过程中的不确定性,我们可能会在候选产品的开发和商业化方面遇到延误。
此外,即使我们开发的任何候选产品获得上市批准或商业化,以便与其他现有疗法结合使用,我们也将继续面临FDA、EMA或类似的外国监管机构 可能撤销与我们产品结合使用的疗法的批准,或者这些现有疗法中的任何一种可能出现安全性、有效性、制造或供应问题的风险。如果我们与候选产品结合使用的疗法被替换为我们为任何候选产品选择的适应症的标准护理 ,FDA、EMA或类似的外国监管机构 可能会要求我们进行额外的临床试验。任何这些风险的发生都可能导致我们自己的候选产品,如果获得批准,将被从市场上移除或在商业上不太成功。
我们 还可以选择结合FDA、EMA或类似的外国监管机构批准上市的一种或多种疗法来评估我们当前的候选产品或任何未来的候选产品 。如果未经批准的 疗法最终不能单独或与我们的候选产品组合获得市场批准,我们将无法 营销和销售我们与未经批准的疗法组合开发的候选产品。此外,未经批准的 疗法面临与我们目前正在开发和临床试验的候选产品相同的风险,包括可能出现严重不良反应、临床试验延迟以及未获得FDA批准。
如果FDA、EMA或类似的外国监管机构不批准这些其他产品或撤销其批准,或者如果我们选择结合我们开发的候选产品进行评估的产品出现安全、疗效、质量、制造或供应问题 ,我们可能无法获得批准或无法销售此类联合疗法。
| 22 |
任何我们打算寻求批准作为生物制品的候选产品都可能比预期的更早面临竞争。
《患者保护和平价医疗法案》经《医疗保健和教育协调法案》修订,或统称为《ACA》,其中包括一个副标题《2009年生物制品价格竞争与创新法案》或《BPCIA》,该法案为与FDA许可的参考生物制品生物相似或可互换的生物制品创建了一条简化的批准途径。 根据《BPCIA》,生物相似产品的申请必须在参考产品首次获得FDA许可之日起四年后才能提交给FDA。此外,自参考产品首次获得许可之日起12年后,FDA才能批准生物相似产品 。在这12年的独占期内,如果FDA批准竞争产品的完整BLA,则另一家公司 仍可销售该参考产品的竞争版本,该竞争产品包含发起人自己的临床前数据和充分且受控良好的临床试验数据,以证明其产品的安全性、纯度和 效力。这项法律很复杂,FDA仍在解释和实施。因此,其最终影响、实施和意义都存在不确定性。虽然目前还不确定FDA何时可以完全采用这些旨在实施BPCIA的工艺,但任何此类工艺都可能对我们生物制品未来的商业前景产生不利影响。
存在这样的风险:根据BLA被批准为生物制品的我们的任何候选产品都没有资格获得12年的独占期,或者由于国会的行动或其他原因,这种独占性可能会缩短,或者FDA不会将我们的 候选产品视为竞争产品的参考产品,这可能会比预期更早地创造仿制药竞争的机会 。BPCIA的其他方面,其中一些可能会影响BPCIA的排他性条款,也一直是诉讼的主题。此外,一旦获得批准,生物相似物将在多大程度上以类似于非生物制品的传统仿制药替代的方式取代我们的任何一种参考产品,目前尚不清楚,这将取决于许多仍在发展中的市场和监管因素。如果竞争对手能够参照我们的候选产品获得营销批准,如果获得批准,我们的产品可能会受到此类生物仿制药的竞争,随之而来的是竞争压力 和潜在的不利后果。
我们 可能会花费有限的资源来追求特定的候选产品或指示,而无法利用可能更有利可图或成功可能性更大的候选产品或指示 。
由于 我们的财务和管理资源有限,我们专注于我们为特定适应症确定的研究计划、治疗平台和候选产品。因此,我们可能会放弃或推迟寻求其他治疗平台或候选产品,或后来被证明具有更大商业潜力或更大成功可能性的其他适应症。 我们的资源分配决策可能会导致我们无法利用可行的商业产品或有利可图的市场机会。 我们在当前和未来研发计划、治疗平台和特定适应症候选产品上的支出可能无法产生任何商业上可行的产品。如果我们没有准确评估特定候选产品的商业潜力或目标市场,我们可能会通过协作、许可或其他版税安排向该候选产品放弃宝贵的权利 如果我们保留独家开发权和商业化权利会更有利。
与我们的业务相关的风险
我们的 公司在设计临床试验方面经验有限,在为我们当前和未来的候选产品获得监管 批准时可能会遇到延迟或意想不到的困难。
我们 在设计临床试验方面经验有限,可能无法设计和执行临床试验以支持市场审批。 我们无法确定我们计划的临床试验或任何未来的临床试验是否会成功。FDA可能会 拒绝接受我们计划的任何或全部BLA进行实质性审查,或者可能在审查我们的数据后得出结论,认为我们的申请 不足以获得监管部门对任何候选产品的批准。如果FDA不批准我们计划中的任何BLAS,它可能会 要求我们进行额外昂贵的临床试验、临床前研究或生产验证研究,然后才会重新考虑我们的申请 。根据这些或任何其他FDA要求的研究的范围,我们 提交的任何BLA或其他申请的批准可能会大幅推迟,可能会推迟几年,或者可能需要我们花费比可用的资源更多的资源。任何未能或延迟获得监管部门批准的情况都将妨碍我们将候选产品商业化、产生收入 以及实现和维持盈利。如果进行并完成其他研究,FDA也可能认为 不足以批准我们提交的任何BLA或其他申请。如果出现上述任何结果,我们可能会被迫放弃开发我们的候选产品,这将对我们的业务产生重大不利影响,并可能导致我们停止运营 。我们在外国司法管辖区的申请也面临类似的风险。
| 23 |
我们 打算确定和开发新的候选细胞治疗产品,这使得我们很难预测候选产品开发的时间、成本和潜在的成功。
我们的 战略是使用我们专有的成纤维细胞技术来识别、开发和商业化候选细胞治疗产品, 该技术包括从捐赠者患者身上收集皮肤活检组织,分离细胞并在培养中扩大它们。我们未来的成功取决于这些新的治疗方法的成功开发。到目前为止,还没有成纤维细胞治疗产品获得批准。此外,与其他更传统的治疗方式相比,已经有一些涉及成纤维细胞的临床试验。
我们候选产品的市场规模是预估的,这些市场可能比预估的要小。
本招股说明书中对我们候选产品的年度潜在市场的估计是基于多个第三方估计。 虽然我们认为这些估计所依据的假设和数据是合理的,但这些假设和估计可能不正确 并且支持这些假设或估计的条件可能随时发生变化,从而降低这些 潜在因素的预测准确性。因此,对我们候选产品的年度潜在市场的估计可能被证明是不正确的。
我们的长期前景在一定程度上取决于发现、开发和商业化其他候选产品,这些候选产品可能会在 开发中失败或出现延迟,从而对其商业可行性产生不利影响。
我们未来的经营业绩取决于我们能否成功发现、开发、获得监管部门对候选产品的批准并将其商业化 我们目前正在进行临床开发的产品。候选产品在临床前或临床开发的任何阶段都可能意外失败。由于与安全性、有效性、临床执行、不断变化的医疗护理标准和其他不可预测的变量有关的风险,候选产品的历史失败率很高。候选产品的临床前研究或早期临床试验的结果可能无法预测候选产品的后期临床试验将获得的结果。
我们可能开发的其他候选产品的成功取决于许多因素,包括:
| ● | 生成足够的数据来支持临床试验的启动或继续; |
| ● | 获得启动临床试验的监管许可; |
| ● | 与必要的各方签订进行临床试验的合同; |
| ● | 成功地 招募患者并及时完成临床试验; |
| ● | 及时生产临床试验所需的足够数量的候选产品和其他关键材料;以及 |
| ● | 临床试验中的不良事件。 |
即使我们成功地将任何其他候选产品推进到临床开发中,它们的成功也将受制于本文中其他部分描述的所有临床、监管和商业风险。风险因素“部分。因此,我们无法向您保证 我们将能够发现、开发、获得监管部门的批准、将我们的候选产品商业化或产生可观的收入。
| 24 |
我们 之前从未将基于成纤维细胞的候选治疗产品商业化,并且可能缺乏必要的专业知识、人员和 资源来成功将任何候选产品商业化,如果获得批准,我们将独自或与合适的合作伙伴合作。
我们 从未将基于成纤维细胞的治疗产品候选产品商业化,目前我们没有销售队伍、营销或分销能力 。为了使我们当前的候选产品取得商业成功,我们可能会授权给其他人,我们将依靠这些合作者的帮助和指导。对于我们保留商业化权利的任何已批准的候选产品,我们必须 建立自己的销售、营销和供应组织,或将这些活动外包给第三方。
可能影响我们将候选产品自行商业化的因素 包括招聘和保留足够数量的有效销售和营销人员、接触或说服足够数量的医生为我们的候选产品开处方,以及与创建独立的销售和营销组织相关的其他不可预见的成本。开发销售和营销组织将是昂贵和耗时的,如果获得批准,可能会推迟我们候选产品的发布。我们可能无法建立一个 有效的销售和营销组织。如果我们无法建立自己的分销和营销能力,或无法找到合适的合作伙伴将我们的候选产品商业化,我们可能无法从他们那里获得收入,也可能无法达到或保持盈利。
我们 面临着激烈的竞争,如果我们的竞争对手比我们更快地开发和营销技术或产品,或者比我们开发的候选产品更有效、更安全或更便宜,我们的商业机会将受到负面影响。
生物技术和制药行业的特点是技术快速进步、竞争激烈,并且非常重视专利和创新产品以及候选产品。我们的竞争对手已经开发、正在开发或可能开发与我们的候选产品竞争的产品、候选产品和工艺。我们成功开发和商业化的任何候选产品都将与现有疗法和未来可能推出的新疗法展开竞争。我们认为,有相当数量的 产品目前正在开发中,并可能在未来投入商业使用,用于治疗我们可能尝试开发的候选产品的条件。请参阅“商业-竞争“以了解更多详情。此外, 我们的产品可能需要与医生用于治疗我们寻求批准的适应症的标签外药物竞争。这可能 使我们难以用我们的产品替代现有疗法。
Many current and potential competitors have significantly greater financial, manufacturing, marketing, drug development, technical and human resources and commercial expertise than we do. Large pharmaceutical and biotechnology companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and manufacturing biotechnology products. These companies also have significantly greater research and marketing capabilities than we do and may also have products that have been approved or are in late stages of development, and collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical and biotechnology companies may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the product candidates that we develop obsolete. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies, as well as in acquiring technologies complementary to, or necessary for, our programs. As a result, our competitors may succeed in obtaining approval from the FDA, the EMA or other comparable foreign regulatory authorities or in discovering, developing and commercializing products in our field before we do.
如果我们的竞争对手开发和商业化的产品更安全、更有效、 影响更少或更不严重、更方便、标签更广泛、营销更有效、可报销或比我们可能开发的任何候选产品 更便宜,我们的 商业机会可能会减少或消除。我们的竞争对手也可能比我们更快地从FDA、EMA或 其他类似的外国监管机构获得其产品的上市批准,这可能导致 我们的竞争对手在我们进入市场之前就建立了强大的市场地位。即使我们开发的候选产品 获得了市场批准,如果届时已经有任何产品获得批准,它们的定价可能会比竞争产品高出很多, 从而降低竞争力。我们的竞争对手开发的技术进步或产品可能会使我们的技术或 候选产品过时、竞争力下降或不经济。如果我们无法有效竞争,我们通过销售我们可能开发的任何产品(如果获得批准)产生收入的机会 可能会受到不利影响。
| 25 |
在本次发行后,我们将成为纳斯达克股票市场规则意义上的“受控 公司”,因为我们的内部人士将实益拥有我们已发行投票证券的投票权的 50%以上。
本次发行完成后, 我们的创始人兼首席执行官Pete O 'Heeron将共同实益拥有我们已发行有投票权证券的约59%的投票权 ,我们将成为纳斯达克股票市场有限责任公司上市规则 意义上的“受控公司”。我们可以依赖公司治理规则的某些豁免,包括豁免 董事会多数成员必须是独立董事的规则。虽然我们目前不打算依赖纳斯达克上市规则下的 “受控公司”豁免,但我们可以选择在未来依赖此豁免。 如果我们选择依赖“受控公司”豁免, 董事会的大多数成员可能不是独立董事,我们的提名和公司治理及薪酬委员会可能 不完全由独立董事组成。我们作为受控公司的地位可能会导致我们的普通股对某些投资者的吸引力降低 ,或以其他方式损害我们的交易价格。因此,您将无法获得向受这些公司治理要求约束的公司的 股东提供的相同保护。
我们 将因作为上市公司运营而增加成本,我们的管理层将需要投入大量时间 执行新的合规计划。我们将遵守财务报告和其他要求,我们的会计和其他管理 系统和资源可能无法充分准备。
作为 一家上市公司,特别是在我们不再是一家新兴成长型公司之后,我们将产生大量的法律、会计和 其他费用,而这些费用是我们作为一家私人公司不会产生的。此外,联邦证券法,包括《萨班斯-奥克斯利法案》、 2010年《多德-弗兰克华尔街改革和消费者保护法案》以及随后由SEC 和纳斯达克实施的规则和法规,对上市公司施加了各种要求,包括要求提交关于其业务和财务状况的年度、季度和事件驱动 报告,并建立和保持有效的披露和财务控制 以及公司治理实践。这些规则和法规将增加我们的法律和财务合规成本,使某些 活动更加耗时且成本更高,并要求我们的管理层和其他人员将大量时间投入到合规 计划中。
尽管我们尽了最大努力,但我们可能无法编制可靠的财务报表,或将此类财务报表作为定期报告的一部分及时提交给美国证券交易委员会或遵守纳斯达克的上市要求。我们还预计,这些规章制度可能会 使我们获得董事和高级管理人员责任保险变得更加困难和昂贵。
根据《萨班斯-奥克斯利法案》第404条,我们将被要求提交一份管理层关于我们财务报告的内部控制的报告,包括由我们的独立注册会计师事务所出具的关于财务报告的内部控制的证明报告,从我们成为上市公司后的第一个全年开始。然而,尽管我们仍是一家新兴的成长型公司, 我们将不会被要求包括由我们的独立注册会计师事务所出具的财务报告内部控制的证明报告。为了符合《萨班斯-奥克斯利法案》第404条的规定,我们将参与记录和评估我们对财务报告的内部控制的过程,这既成本高昂,又具有挑战性。我们将需要继续投入 内部资源,可能聘请外部顾问,采用详细的工作计划来评估和记录财务报告内部控制的充分性,继续适当地改进控制流程,通过测试验证控制 是否按照文档所述发挥作用,并实施财务报告内部控制的持续报告和改进过程。 尽管我们做出了努力,但我们和我们的独立注册会计师事务所都有可能无法在规定的时间框架内得出结论,即我们对财务报告的内部控制是有效的,如萨班斯-奥克斯利法案第404条所要求的那样。这可能会导致金融市场因对我们的财务报表的可靠性失去信心而产生不良反应。我们还可能成为美国证券交易委员会或其他监管机构的调查对象,这可能需要额外的 财务和管理资源。
作为一家上市公司,我们还将被要求保持披露控制和程序。披露控制和程序是指我们的 控制和其他程序,旨在确保我们提交或根据交易法提交的报告中要求我们披露的信息在美国证券交易委员会规则和 表格中指定的时间段内得到记录、处理、汇总和报告。我们不期望我们的披露控制和程序或我们对财务报告的内部控制将 防止或检测所有错误和所有欺诈。我们相信,无论控制系统的设计和操作有多好,都只能提供合理的、而不是绝对的保证,确保控制系统的目标能够实现。由于所有控制系统的固有限制, 任何控制评估都不能绝对保证不会发生因错误或舞弊而导致的错误陈述,或已检测到所有控制问题和舞弊实例(如果有)。任何控制系统的设计在一定程度上都是基于对未来事件可能性的某些假设 ,任何设计都可能在所有潜在的未来条件下都不能成功实现其规定的目标。 随着时间的推移,控制可能会因为条件的变化或遵守政策或程序的程度的恶化而变得不充分。因此,由于我们控制系统的固有限制,可能会发生因错误或舞弊而导致的错误陈述 并且无法检测到。
我们 发现,由于缺乏职责分工,我们对财务报告的内部控制存在重大弱点。未能 对财务报告保持有效的内部控制可能会导致我们的投资者对我们失去信心,并对我们普通股的市场价格产生不利影响。如果我们对财务报告的内部控制不有效,我们可能无法准确地 报告我们的财务结果或防止欺诈。
在编制截至2022年12月31日的财年的财务报表期间,我们的管理层发现,由于缺乏职责分工,财务报告的内部控制存在重大缺陷。重大缺陷被定义为财务报告内部控制的缺陷或缺陷的组合,使得我们的年度或中期财务报表的重大错报有合理的可能性无法及时防止或发现。
具体地说, 我们的管理层发现财务报告职能内的内部控制存在缺陷,这是由于在我们的首席财务官于2022年6月加入我们之前,我们在财务报表所涵盖的时间段缺乏职责分工,当时所有财务职能都由一个人处理。
随着我们首席财务官的加入,以及我们在2022财年下半年对会计和财务报告流程以及内部控制做出的改变,我们加强了内部控制,并将继续评估职责分离 ,并随着我们的发展主动改善我们对财务报告的内部控制。然而,这些 计划的实施可能无法完全解决我们在财务报告方面的内部控制的重大弱点,我们不能向您保证 我们不会发现其他重大弱点或缺陷,这些弱点或缺陷可能会对我们未来的运营业绩产生负面影响。
| 26 |
与我们依赖第三方有关的风险
我们依赖,并预计将继续依赖第三方,包括独立的临床研究人员和CRO,进行我们临床前研究和临床试验的某些方面 。如果这些第三方不能成功履行其合同职责、遵守适用的法规要求或在预期的最后期限前完成,我们可能无法获得监管部门对我们的候选产品的批准或将其商业化 我们的业务可能会受到不利影响。
我们 一直依赖并计划继续依赖第三方,包括独立的临床研究人员和第三方CRO, 进行我们临床前研究和临床试验的某些方面,并为我们正在进行的临床前研究和临床计划监测和管理数据。我们依赖这些方来执行我们的临床前研究和临床试验,并且只控制他们活动的某些方面。然而,我们有责任确保我们的每一项研究和试验都是按照适用的协议、法律、法规和科学标准进行的,我们对这些第三方的依赖不会免除我们的监管责任。我们、我们的第三方承包商和CRO必须遵守GCP要求,这是FDA和类似的外国监管机构对我们临床开发中的所有候选产品执行的 法规和指导方针。监管机构通过定期检查试验赞助商、主要调查人员和试验场地来执行这些GCP。如果我们或我们的任何第三方或我们的CRO未能遵守适用的GCP,在我们的临床试验中产生的临床数据可能被认为是不可靠的,FDA、EMA或类似的外国监管机构可能会要求我们在批准我们的营销申请之前进行额外的 临床试验。我们不能向您保证,在特定监管机构进行检查后,该监管机构将确定我们的任何临床试验是否符合GCP规定。此外,我们的临床试验必须使用根据cGMP规定生产的产品进行。如果我们不遵守这些规定,我们可能需要重复 临床试验,这将推迟监管审批过程。此外,如果这些 第三方中的任何一方违反联邦或州欺诈和滥用或虚假索赔法律法规或医疗保健隐私和安全法律,我们的业务可能会受到不利影响。
此外, 不能保证我们所依赖的任何此类CRO、调查人员或其他第三方将投入足够的时间和资源 用于我们的开发活动或按照合同要求执行任务。这些第三方还可能与包括我们的竞争对手在内的其他商业实体有关系,他们可能还在为这些实体进行临床试验或其他产品开发活动,这 可能会影响我们的表现。如果独立研究人员或CRO未能投入足够的资源来开发我们的候选产品 ,或者CRO未能成功履行其合同职责或义务或在预期的截止日期前完成, 如果他们需要更换,或者如果他们获得的临床数据的质量或准确性因未能遵守我们的临床方案、法规要求或其他原因而受到影响,我们的临床试验可能会被延长、推迟或终止,并且我们可能无法获得监管部门的批准,也无法成功地将我们的候选产品商业化。因此,我们的运营结果和候选产品的商业前景将受到损害,我们的成本可能会增加,我们创造 收入的能力可能会被推迟或完全停止。
如果发生未治愈的重大违规行为,我们的CRO有权终止与我们的协议。此外,如果能够合理地证明参与我们临床试验的受试者的安全性,如果我们为了债权人的利益进行一般转让,或者如果我们被清算,我们的一些CRO 有能力终止与我们各自的协议。
如果我们与这些第三方CRO的任何关系终止,我们可能无法与替代CRO或 以商业合理的条款达成协议。更换或添加额外的CRO涉及额外成本,并且需要管理时间 和重点。此外,当新的CRO开始工作时,有一个自然的过渡期。因此,会出现延迟,这可能会对我们满足所需临床开发时间表的能力产生重大影响。此外,CRO可能缺乏能力来吸收更高的工作负载 或承担额外的容量来支持我们的需求。尽管我们谨慎地管理与CRO的关系,但不能保证我们在未来不会遇到类似的挑战或延误,也不能保证这些延误或挑战不会对我们的业务、财务状况和前景产生实质性的不利影响。
| 27 |
如果 我们决定建立更多合作,但无法以合理的商业条款建立这些合作, 我们可能不得不更改我们的开发和商业化计划。
我们的候选产品开发计划和候选产品的潜在商业化将需要大量额外的 现金来支付费用。我们可能会继续寻求有选择地形成合作,以扩展我们的能力,潜在地加速 研发活动,并为第三方的商业化活动提供支持。这些关系中的任何一项都可能要求我们产生非经常性费用和其他费用,增加我们的近期和长期支出,发行稀释我们现有股东的证券,或扰乱我们的管理和业务。
我们 在寻找合适的合作伙伴方面将面临激烈的竞争,谈判过程既耗时又复杂。 我们是否就协作达成最终协议将取决于我们对协作者的资源和专业知识的评估、拟议协作的条款和条件以及提议的协作者对 多个因素的评估。这些因素可能包括临床试验的设计或结果,FDA、EMA或类似的外国监管机构批准的可能性,候选研究产品的潜在市场,制造和向患者交付候选产品的成本和复杂性,竞争产品的潜力,与我们知识产权所有权和行业及市场状况有关的不确定性的存在。潜在的协作者还可以考虑替代 候选产品或技术,以获得类似的可供协作的指示,以及此类协作是否会 比我们与我们的候选产品更具吸引力。此外,我们为未来的候选产品建立协作或其他替代安排的努力可能不会成功,因为它们可能被认为处于协作努力的开发阶段 太早,第三方可能认为它们没有展示安全性和有效性所需的潜力。
此外,大型生物制药公司之间的合并可能会导致未来潜在合作伙伴的数量减少。即使我们 成功达成协作,该协作的条款和条件也可能会限制我们未来与潜在协作者签订特定条款的 协议。
如果 并且当我们寻求进行协作时,我们可能无法以可接受的条款及时协商协作, 或根本无法进行协商。如果我们无法做到这一点,我们可能不得不减少候选产品的开发,减少或推迟其开发计划或我们的一个或多个其他开发计划,推迟其潜在的商业化或缩小任何销售或营销活动的范围,或者增加我们的支出,并自费进行开发或商业化活动。如果我们选择增加支出以资助我们自己的开发或商业化活动,我们可能需要获得额外的资金,而这些资金可能无法以可接受的条款提供给我们或根本无法获得。如果我们没有足够的资金,我们可能无法进一步开发我们的 候选产品或将它们推向市场并产生产品收入。
在 未来,我们可能会与第三方合作开发候选产品并将其商业化。如果这些 协作不成功,我们可能无法利用这些候选产品的市场潜力。
我们 未来可能会为我们的一个或多个候选产品的开发和商业化寻找第三方合作伙伴。 我们未来任何合作安排的可能合作伙伴包括大中型生物制药公司、地区性和 全国性生物制药公司和生物技术公司。我们已经并可能有限地控制我们的合作者用于我们候选产品的开发或商业化的资源数量和时间 。我们从这些安排中获得收入的能力将取决于我们的合作者成功履行这些安排中分配给他们的职能的能力和努力。涉及我们候选产品的协作可能会给我们带来许多风险,包括 :
| ● | 协作者 在确定他们将应用于这些协作的工作和资源方面有很大的自由裁量权,并且可能无法按预期履行其义务; |
| 28 |
| ● | 合作者可以根据临床试验结果、合作者战略重点的变化、包括 出售或处置业务部门或开发职能的结果,或可用的 资金或外部因素,如转移资源或产生相互竞争的 优先事项的收购; |
| ● | 合作者 可以推迟临床试验,为临床试验计划提供资金不足,停止临床试验或放弃候选产品,重复或进行新的临床试验,或者 要求临床试验候选产品的新配方; |
| ● | 合作者 可以独立开发,也可以与第三方开发,直接或间接与我们的候选产品竞争的产品,如果合作者认为有竞争力的产品 更有可能被成功开发或能够以比我们的产品更具经济吸引力的条款进行商业化; |
| ● | 拥有多个产品营销和分销权限的 协作者可能不会投入足够的 资源来相对于其他产品营销和分销我们的候选产品; |
| ● | 协作者 可能无法正确获取、维护捍卫或强制执行我们的知识产权,或者 可能以某种方式使用我们的专有信息和知识产权,导致 诉讼或其他与知识产权相关的诉讼,从而危及或使我们的专有信息和知识产权无效或暴露我们可能的诉讼或其他与知识产权有关的诉讼; |
| ● | 合作者和我们之间可能发生纠纷,导致我们候选产品的研究、开发或商业化的延迟或终止,或者导致成本高昂的诉讼或仲裁,从而分散管理层的注意力和资源; |
| ● | 协作 可能会终止,如果终止,可能需要额外的资金来进一步开发适用的候选产品或将其商业化。 |
| ● | 合作协议可能不会以最有效的方式或根本不会导致候选产品的开发或商业化;以及 |
| ● | 如果 我们的合作伙伴参与业务合并,我们对候选产品开发或商业化计划的持续追求和重视可能会被推迟、减少或终止。 |
我们的 员工、独立承包商、顾问、商业合作者、主要调查人员、CRO、供应商和供应商可能从事不当行为或其他不当活动,包括不遵守监管标准和要求,这可能会对我们的运营结果产生不利的 影响。
我们 面临员工、独立承包商、顾问、商业合作者、主要调查人员、CRO、供应商和供应商可能从事不当行为或其他不当活动的风险。这些当事人的不当行为可能包括未能 遵守FDA法规,向FDA提供准确信息,遵守联邦和州医疗欺诈和滥用法律和法规 ,准确报告财务信息或数据,或向我们披露未经授权的活动。特别是,医疗保健行业的销售、营销和商业安排受到旨在防止欺诈、不当行为、回扣、自我交易和其他滥用行为的广泛法律法规的约束。这些法律法规可能会限制或禁止各种定价、折扣、营销和促销、销售佣金、客户激励计划和其他业务安排。这些各方的不当行为还可能涉及对临床试验过程中获得的信息的不当使用,这可能会导致监管制裁和 严重损害我们的声誉。并非总是能够识别和阻止这些各方的不当行为,我们为检测和防止此类活动而采取的预防措施可能无法有效控制未知或未管理的风险或损失,或保护我们免受因未能遵守这些法律或法规而引起的政府调查或其他行动或诉讼。如果对我们提起任何此类 诉讼,而我们未能成功为自己辩护或维护自己的权利,这些诉讼可能会对我们的业务产生重大影响,包括施加重大处罚,包括民事、刑事和行政处罚、损害赔偿、罚款、交还、个人监禁、被排除在政府资助的医疗保健计划之外、 联邦医疗保险和医疗补助、诚信监督和报告义务、合同损害、声誉损害、利润减少和未来收益以及我们业务的削减或重组。
| 29 |
与制造相关的风险
细胞治疗产品的制造是复杂的,并受到人体和系统风险的影响。我们的第三方制造商或我们可能在生产和采购方面遇到困难,并可能受到关键组件的变化和供应限制的影响。如果我们或我们的任何第三方制造商遇到此类困难,我们为临床试验提供候选产品或为患者提供产品的能力可能会被推迟或阻止。 如果获得批准。
生物细胞疗法候选产品和产品的制造(如果获得批准)是复杂的,需要大量的专业知识和资本投资,包括开发先进的制造技术和工艺控制。生物产品制造商经常在生产和采购方面遇到困难,特别是在扩大或缩小规模、验证生产过程以及确保制造过程的高可靠性(包括没有污染)方面,考虑到关键组件的变化和供应限制 。这些问题包括物流和运输、生产成本和产量方面的困难、质量控制、 产品的一致性、稳定性、纯度和功效、产品测试、操作员错误和合格人员的可用性,以及是否遵守严格执行的联邦、州和外国法规。此外,如果在我们的候选产品供应或制造设施中发现污染物 ,此类制造设施可能需要关闭 一段较长的时间以调查和补救污染。我们不能向您保证未来不会发生任何稳定性、纯度和功效故障、 缺陷或其他与生产我们的候选产品相关的问题。
此外, 我们的候选产品来自从人类收集的细胞。由于捐赠者的年龄、病史和许多其他因素不同,这种细胞的类型和质量可能会有所不同。我们对供体细胞材料和我们的候选产品有严格的规格。供体细胞材料的可变性可能超出我们的制造工艺能力或偏离指定的范围,导致细胞治疗产品的生产失败、批量质量下降,甚至需要调整权威机构批准的规格。 供体细胞材料的因素也可能是可变的,我们目前使用的分析方法可能无法检测到这些因素,或者可能不知道如何测量。我们也可能在生产后发现材料有故障。因此,我们可能无法提供我们需要或可能需要重新收集的细胞材料所需的细胞治疗产品的质量和一致性,这可能会增加 成本和/或导致延迟,对患者结果产生不利影响,并以其他方式损害我们的临床试验、声誉、业务和前景。
我们 可能无法管理收集患者材料并将其运送到制造现场、将候选产品运回相关方的物流,以及遇到某些临床或商业级供应和组件的延迟或短缺。物流 和发货延迟以及由我们、我们的供应商或我们无法控制的其他因素引起的问题,包括业务中断、全球供应链问题和天气,可能会阻止或延迟向患者交付候选产品。此外,在捐献材料运往制造设施、通过制造流程并最终到达患者的过程中,我们必须维护与捐献材料相关的复杂身份链和保管链。未能维护身份链和监护链可能导致患者 死亡、产品丢失或监管行动。
将我们的细胞库 转移或生产到合同开发制造组织可能会失败,并导致延误、额外成本或技术故障。
我们目前从合同开发和制造组织或CDMO购买我们的细胞疗法候选产品。我们正在与CDMO签订合同,以转让我们的实验细胞库,以生产我们的主细胞库、工作细胞库和基于成纤维细胞的产品 候选产品,以便进行临床试验。如果将我们的实验细胞库转移到CDMO不成功,我们可能会遇到延迟、 额外成本或我们的一个或多个候选产品的技术故障。同样,如果CDMO无法从实验细胞库中生产我们的主细胞库、工作细胞库和我们的基于成纤维细胞的候选产品以进行临床试验, 我们可能会遇到一个或多个候选产品的延迟、额外成本或技术故障。
更改候选产品制造或配方的方法 可能会导致额外成本或延迟。
随着候选产品通过临床前研究到后期临床试验,以获得潜在的上市批准和商业化,开发计划的各个方面,如制造方法、配方、材料和工艺,在此过程中进行更改是很常见的 ,以努力优化工艺和产品特性。此类更改也可能因制造商的更改而发生。这样的变化存在无法实现预期目标的风险。任何此类更改都可能导致我们的 候选产品表现不同,并影响使用改进的制造方法、材料和流程生产的候选产品进行的计划临床试验或其他未来临床试验的结果。此类变更还可能需要额外的 测试、FDA通知或FDA批准。这可能会推迟临床试验的完成,需要进行过渡临床试验或重复一项或多项超出我们目前预期的临床试验,增加临床试验成本,推迟我们候选产品的批准 ,并危及我们将候选产品商业化的能力。此外,我们可能需要 对我们整个管道的上下游流程进行重大更改,这可能会推迟未来候选产品的开发 。
| 30 |
如果我们或我们的第三方制造商以造成伤害或违反适用法律的方式使用危险和生物材料,我们可能要承担损害赔偿责任。
研究和开发活动涉及我们和我们的第三方制造商对潜在危险物质的控制使用,包括化学和生物材料。我们目前将所有制造业务外包给第三方。尽管如此,我们和我们的制造商仍受美国联邦、州和地方法律法规的约束,管理医疗和危险材料的使用、制造、储存、搬运和处置 。尽管我们相信制造商使用、处理、储存和处置这些材料的程序符合法律规定的标准,但我们不能完全消除医疗或危险材料造成污染或伤害的风险。由于任何此类污染或伤害,我们可能会承担责任,或者地方、城市、州或联邦当局可能会限制这些材料的使用并中断我们的业务运营。如果发生事故,我们可能 被追究损害赔偿责任或被罚款,责任可能超出我们的资源范围。我们目前没有为医疗或危险材料引起的责任 投保。遵守适用的环境法律法规代价高昂, 当前或未来的环境法规可能会损害我们的研发和生产努力,这可能会损害我们的业务、前景、财务状况或运营结果。
我们的制造流程依赖第三方,未来我们的候选产品可能依赖第三方制造商,这增加了与我们的候选产品及时和充足生产相关的风险。
我们 不能完全控制我们的合同制造合作伙伴的制造过程的所有方面,并且依赖我们的合同制造合作伙伴 遵守cGMP法规来生产我们的候选细胞治疗产品。第三方制造商可能无法 遵守cGMP法规或美国以外的类似法规要求。如果我们的合同制造商不能成功地 生产出符合我们的规格和FDA、EMA或其他机构严格监管要求的材料,他们将无法 确保和/或保持其制造设施的上市批准。此外,我们无法控制我们的合同制造商保持足够的质量控制、质量保证和合格人员的能力。如果FDA、EMA或类似的外国监管机构不批准这些设施来生产我们的候选产品 或如果它在未来撤回任何此类批准,我们可能需要寻找替代制造设施,这将显著 影响我们开发、获得营销批准或营销我们的候选产品的能力。我们或我们的第三方制造商未能遵守适用的法规可能会导致对我们实施制裁,包括罚款、禁令、民事处罚、延迟、暂停或撤回审批、吊销许可证、扣押或召回候选产品、 运营限制和刑事起诉,其中任何一项都可能对我们候选产品的供应造成重大不利影响 并损害我们的业务和运营结果。此外,我们候选产品的原材料可能来自单一供应商,在某些情况下, 。如果我们的任何候选产品或任何未来候选产品因任何原因(无论是制造、供应或存储问题或其他原因)意外失去供应,我们可能会遇到 任何未决或正在进行的临床试验的延迟、中断、暂停或终止,或者需要重新启动或重复。
我们 目前依赖第三方 制造商在我们组织成员的指导下生产我们的候选产品,用于开发和商业化。 如果我们或我们的任何第三方制造商未能遵守此类要求或执行与质量、时间或其他方面有关的 某些要求,或者如果我们的组件或其他材料的供应受到限制 或由于其他原因中断,我们可能被迫与另一方达成协议,而我们可能无法以商业上合理的条款 执行 。特别是,我们第三方制造商的任何更换都可能需要大量的工作和专业知识,因为合格的更换数量可能是有限的。在某些情况下,制造我们的候选产品所需的技术技能或技术可能是我们或第三方制造商独有的或专有的。 我们可能难以将此类技能或技术转让给其他第三方,并且可能不存在可行的替代方案。此外,我们的某些候选产品和我们自己的专有方法从未在我们公司以外生产或实施过。因此, 如果我们尝试为这些候选产品或方法建立新的第三方制造安排,我们的开发计划可能会出现延迟。这些因素将增加我们对这些制造商的依赖,或者要求我们获得 这些制造商的许可证,以便让其他第三方生产我们的候选产品。如果我们因任何原因被要求或自愿停止生产我们的候选产品,我们将被要求验证新制造商是否拥有符合质量标准和所有适用法规和指南的设施和程序,以及所生产的产品是否与我们工厂生产的产品等同。与新制造商和同等产品的验证相关的延迟可能会对我们及时或在预算范围内开发候选产品的能力产生负面影响。
| 31 |
如果我们的 或第三方未能执行我们的制造要求,按照商业上合理的条款和时间表执行,并且 遵守cGMP要求,可能会在多个方面对我们的业务产生不利影响,包括:
| ● | 无法始终如一地满足我们的产品规格和质量要求; |
| ● | 无法启动或继续我们正在开发的候选产品的临床试验; |
| ● | 延迟提交法规申请或收到我们候选产品的上市批准,如果有的话, ; |
| ● | 无法将任何及时获得上市批准的候选产品商业化; |
| ● | 失去未来合作者的合作; |
| ● | 让第三方制造设施或我们的制造设施接受监管机构的额外检查 ; |
| ● | 要求 停止开发或召回我们的候选产品批次;以及 |
| ● | 在 如果我们的候选产品获准上市和商业化,无法 满足我们候选产品或任何未来候选产品的商业需求。 |
我们的制造过程中的任何 污染或中断、原材料短缺或供应商未能交付必要的 组件都可能导致我们的临床开发或营销计划延迟。
鉴于 细胞疗法生产的性质,存在污染风险。任何污染都可能对我们按时生产 候选产品的能力产生不利影响,因此可能损害我们的运营结果并造成声誉损害。此外,虽然 我们的细胞疗法在放行前进行了污染测试,但如果将受污染的候选产品给予患者,则 可能会对患者造成伤害。我们生产过程中所需的一些原材料来自生物来源。 这些原材料很难采购,可能会受到污染或被召回。材料短缺、污染、召回、 或在候选产品生产中限制使用生物衍生物质可能会对 商业生产或临床材料生产产生不利影响或中断,从而可能对我们的开发时间表和业务、 财务状况、经营业绩和前景产生不利影响。
与法律和监管合规事项相关的风险
我们与医疗保健专业人士、临床研究者、CRO和第三方付款人之间的 关系与我们当前和未来的 业务活动有关,可能会受到联邦和州医疗保健欺诈和滥用法律、虚假索赔法律、透明度法律、政府 价格报告以及健康信息隐私和安全法律的约束,这可能会使我们面临刑事制裁、 民事处罚、合同损害、被排除在政府医疗保健计划之外、名誉损害、行政负担以及 利润和未来收入减少。
医疗保健 提供者和第三方付款人在我们获得 营销批准的任何候选产品的推荐和处方中发挥主要作用。我们目前和未来与医疗保健专业人士、临床研究者、CRO、第三方付款人 和客户的安排可能会使我们面临广泛适用的欺诈和滥用以及其他医疗保健法律法规,这些法律法规可能会限制我们营销、销售和分销我们获得营销 批准的候选产品的业务 或财务安排和关系。适用的联邦和州医疗保健法律和法规的限制包括以下内容:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, offering, receiving or paying any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce, or in return for the purchase, lease, order, arrangement, or recommendation of any good, facility, item or service for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. A person or entity does not need to have actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it to have committed a violation. Violations are subject to civil fines and criminal penalties for each violation, plus up to three times the remuneration involved, imprisonment, and exclusion from government healthcare programs. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act or federal civil money penalties; |
| 32 |
| ● | 联邦民事和刑事虚假申报法和民事金钱惩罚法,如联邦虚假申报法,对个人或实体施加刑事和民事处罚,并授权对其提起民事举报人或刑事诉讼,除其他事项外: 或导致向联邦政府提交虚假或欺诈性的付款要求;故意作出、使用或导致作出或使用记录材料的虚假陈述,以虚假或欺诈性的索赔或义务向联邦政府支付或传输金钱或财产,或故意隐瞒或故意不正当地逃避或减少向联邦政府支付金钱的义务。根据联邦虚假索赔法案,制造商可能被追究责任,即使他们没有直接 向政府付款人提交索赔,如果他们被认为“导致”提交虚假或欺诈性索赔 。联邦虚假申报法还允许作为“告密者”行事的个人代表联邦政府提起诉讼,指控 违反联邦虚假申报法,并参与任何金钱追回; |
| ● | 1996年的《健康保险可转移性和责任法案》,或称HIPAA,除其他事项外,禁止任何人在知情的情况下故意执行或试图执行诈骗任何医疗福利计划或获得的计划,通过虚假或欺诈性的 借口、陈述或承诺,任何医疗福利计划拥有或托管或控制的任何金钱或财产,而无论付款人(例如公共或私人),并故意伪造,以任何诡计或手段隐瞒或掩盖重大事实,或作出任何重大虚假、虚构或欺诈性陈述或陈述,与提供或支付医疗福利、与医疗事宜有关的物品或服务有关。与联邦反回扣法规类似, 个人或实体不需要实际了解法规或违反该法规的具体意图 即可实施违规; |
| ● | HIPAA,经2009年《卫生信息技术促进经济和临床卫生法案》(HITECH)及其各自实施条例的进一步修订,包括2013年1月发布的最终综合规则,它们对某些承保医疗服务提供者、健康计划和医疗信息交换中心及其各自的商业伙伴、独立承包商或承保实体的代理施加了某些要求,这些服务涉及使用、创建、维护、接收或披露、 个人可识别的健康信息,与个人可识别的健康信息的隐私、安全和传输有关。HITECH还创建了新的民事罚款等级,修改了HIPAA,使民事和刑事处罚直接适用于企业 合伙人,并赋予州总检察长新的权力,可以向联邦法院提起损害赔偿民事诉讼或禁制令,以执行联邦HIPAA法律,并寻求律师与提起联邦民事诉讼相关的费用和费用。此外,还有其他联邦、州和非美国法律对健康和其他个人信息在某些情况下的隐私和安全进行管理,我们可能会受到这些情况的影响,其中许多法律在重大方面彼此不同,可能不会产生相同的效果。从而使合规工作复杂化; |
| ● | 联邦政府价格报告法,要求制造商准确、及时地计算和报告复杂的定价指标给政府项目; |
| 33 |
| ● | 联邦消费者保护法和不正当竞争法,广泛监管市场活动和可能损害消费者的活动; |
| ● | ACA,包括通常被称为医生支付阳光法案及其实施条例的条款,该条款要求适用的药品、器械、生物制品和医疗用品制造商可以在联邦医疗保险下获得付款,Medicaid或儿童健康保险计划每年向美国卫生与人类服务部内的美国医疗保险和医疗补助服务中心(CMS)报告与向医生支付款项或其他价值转移有关的信息, 执业护士、注册护士麻醉师、医师助理、临床护士专科医生、注册助产士以及教学医院,并披露医生及其直系亲属持有的所有权和投资权益;和 |
| ● | 许多州的法律在特定情况下管理个人信息的隐私。例如,在加利福尼亚州,2020年1月1日生效的《加州消费者隐私法》(California Consumer Privacy Act,简称CCPA)通过 创建个人信息的扩展定义,为覆盖的企业建立了一个新的隐私框架,为加利福尼亚州的消费者建立新的数据隐私权,对个人信息的销售实施特殊规则 ,并为违反《反海外腐败法》以及未能实施合理安全程序和 做法以防止数据泄露的企业创建一个新的、可能严重的法定损害赔偿框架。虽然受HIPAA管辖的临床试验数据和信息目前不受CCPA的约束,但其他个人信息收集做法 可能会受到CCPA的约束,可能对CCPA进行的更改可能会扩大其范围。 |
此外, 我们受制于上述每个医疗保健法律法规的州和国外等价物,其中包括一些可能范围更广且可能适用于任何付款人的 。美国许多州已经通过了类似于联邦《反回扣法令和虚假索赔法案》的法律,可能适用于我们的商业实践,包括但不限于研究、分销、销售或营销安排以及涉及由非政府付款人(包括私人保险公司)报销的医疗项目或服务的索赔。 此外,一些州还通过了法律,要求生物制药公司遵守2003年4月的《制药制造商检查员办公室一般合规计划指南》和/或《美国制药研究和制造商与医疗保健专业人员互动准则》。几个州还实施了其他营销限制或要求生物制药公司向该州进行营销或价格披露,并要求生物制药销售代表注册。 美国以外的隐私和数据保护法,包括欧盟一般数据保护 法规和英国2018年数据保护法,或统称为GDPR,也管理个人信息的隐私和安全, 在某些情况下包括健康信息,并且这些法律中的许多在重大方面相互不同,从而使合规工作复杂化。此外,在美国,有许多州已经制定了管理个人信息隐私和安全的法律,其中许多法律在很大程度上彼此不同,而且往往不会被HIPAA抢先,因此 使合规工作复杂化。对于遵守这些州的要求需要什么是模棱两可的,如果我们未能遵守适用的州法律要求,我们可能会受到惩罚。
这些法律的范围和执行都是不确定的,在当前的医疗改革环境中受到快速变化的影响。 特别是考虑到缺乏适用的先例和法规。联邦和州执法机构最近加强了对医疗保健公司和医疗保健提供者之间互动的审查,这导致了医疗保健行业的一系列调查、起诉、定罪和和解。确保业务安排符合适用的医疗保健和隐私法律,以及对政府当局可能进行的调查做出回应,可能会耗费时间和资源,并可能分散 公司对业务的注意力。
| 34 |
确保 我们的内部运营和未来与第三方的业务安排符合适用的医疗法律法规 将涉及大量成本。政府当局可能会得出结论,我们的业务实践不符合当前或未来涉及适用欺诈和滥用或其他医疗保健法律和法规的现行或未来法律、法规、机构指导或判例法 。如果我们的业务被发现违反了上述任何法律或可能适用于我们的任何其他政府法律和法规,我们可能会受到重大处罚,包括行政、民事和刑事处罚、损害赔偿、罚款、交还、被排除在联邦和州医疗保健计划之外、个人监禁、声誉损害和削减或重组我们的业务,以及如果我们受到公司诚信协议或其他协议的约束,以解决有关不遵守这些法律的指控,则我们可能会受到额外的报告义务和监督。此外,防御 任何此类操作都可能既昂贵又耗时,并可能需要大量的财力和人力资源。因此,即使我们成功抵御了可能对我们提起的任何此类诉讼,我们的业务也可能会受到损害。如果发现与我们有业务往来或预期与我们有业务往来的任何医生或其他提供者或实体违反适用法律,他们 可能会受到刑事、民事或行政制裁,包括被排除在政府资助的医疗保健计划之外和监禁。 如果发生上述任何情况,我们的业务运营能力和运营结果可能会受到不利影响。
我们 可能或将受到不断变化的全球数据保护法律和法规的约束,这可能要求我们产生大量合规成本 ,如果我们未能或被认为未能遵守此类法律和法规,可能会损害我们的业务和运营。
全球数据保护格局正在迅速发展,我们可能会受到众多联邦、州和外国 法律法规以及监管指南的制约或影响,这些法律法规和法规对个人数据的收集、使用、披露、传输、安全和处理进行管理,例如我们收集的与临床试验相关的参与者和医疗保健提供者的信息。实施 标准和执法实践在可预见的未来可能仍然不确定,这可能会给我们的业务带来不确定性, 影响我们在某些司法管辖区运营或收集、存储、转移、使用和共享个人数据的能力,导致责任 或将额外的合规或其他成本强加给我们。如果我们未能或被认为未能遵守联邦、州或国外的法律或自律标准,可能会导致负面宣传、转移管理时间和精力,以及政府实体或其他人对我们提起诉讼。例如,加利福尼亚州、弗吉尼亚州、科罗拉多州、犹他州和康涅狄格州等州最近颁布了消费者隐私法,授予数据主体权利,并将隐私和安全义务强加给处理消费者或家庭个人数据的实体。一些观察人士指出,CCPA和类似立法可能标志着美国 更严格隐私立法趋势的开始,这可能会增加我们的潜在责任并对我们的业务产生不利影响。
除了我们在美国的业务可能受到医疗保健和其他与健康信息和其他个人信息的隐私和安全相关的法律的约束 之外,我们还可能寻求在英国或欧洲经济区或欧洲经济区进行临床试验,并可能受到其他欧洲数据隐私法律、法规和指南的约束。对于我们从这些地区收集的个人数据,我们将受 欧盟和英国的数据保护法约束。这些 法律对我们的业务施加了额外的义务和风险,包括需要 遵守这些法律的巨额费用和业务运营变更。英国退出欧盟或英国退欧,以及随后这些地区的数据保护制度分离 意味着我们必须遵守欧洲联盟和英国的单独数据保护法律,这可能会导致额外的合规成本,并可能增加我们的总体风险。
GDPR涉及个人数据的处理和此类数据的自由流动,规定了广泛的严格要求,包括有关拥有处理个人数据和将此类信息转移到欧洲经济区/英国以外的合法基础的要求,包括向美国提供有关个人数据处理的细节,确保个人数据安全,与处理个人数据的第三方有数据处理协议,回应个人对其个人数据行使权利的请求 ,向主管的国家数据保护机构和受影响的个人报告涉及个人数据的安全漏洞,任命数据保护官员,进行数据保护影响评估 并保存记录。
| 35 |
GDPR对将个人数据从欧洲经济区/英国转移到欧盟委员会和英国政府认为没有提供足够保护的国家或第三国(包括美国)实施了严格的规则。除非实施了数据保护法规定的适当保障措施,如欧盟委员会批准的标准合同条款或SCC,否则禁止此类转让,或适用减损。英国已经发布了自己的转移机制,即国际数据转移协议和国际数据转移附录,允许从英国转移,并实施了类似的转移 等价性测试。欧盟和英国数据保护制度下的国际转移义务需要付出努力和成本,并可能 导致我们需要就EEA/英国个人数据的位置以及我们利用哪些服务提供商处理EEA/英国个人数据做出战略考虑,特别是在围绕GDPR国际转移合规义务的执行情况 目前尚不清楚的情况下。英国政府现已将《数据保护和数字信息法案》或《英国法案》引入英国立法程序,意在改革英国的数据保护制度。如果获得通过,英国法案的最终版本可能会进一步改变英国和欧盟数据保护制度之间的相似之处。这可能导致 额外的合规成本,并可能增加我们的总体风险。
我们 不能向您保证,任何遵守欧洲隐私法规定的义务的努力都是足够的。如果我们受到欧洲数据保护机构的调查,我们可能会面临罚款和其他处罚。欧洲数据保护机构的任何此类调查或指控都可能对我们的声誉产生负面影响,并对我们的业务造成实质性损害。
我们的 业务存在重大的产品责任风险,如果我们无法获得足够的保险范围,这种无法承保的情况 可能会对我们的业务和财务状况产生不利影响。
我们的 业务使我们面临治疗性 治疗的开发、测试、制造和营销过程中固有的重大产品责任风险。产品责任索赔可能会推迟或阻止我们开发计划的完成。如果我们成功营销候选产品 ,此类声明可能导致FDA、EMA或其他监管机构对我们的 候选产品、我们的制造工艺和设施或营销计划的安全性和有效性进行调查。FDA、EMA或其他监管机构的调查 可能会导致召回我们的候选产品或采取更严重的执法行动,限制其可用于的批准适应症 或暂停或撤回批准。无论是非曲直或最终结果如何,责任索赔可能 还会导致对我们候选产品的需求减少(如果获得批准)、对我们声誉的损害、相关诉讼的辩护成本、 管理层的时间和资源的分流以及对试验参与者或患者的巨额金钱奖励。我们目前 有产品责任保险,我们认为该保险适合我们的开发阶段,如果获得批准,可能需要在 营销我们的任何候选产品之前获得更高的级别。我们拥有或可能获得的任何保险可能无法为潜在责任提供足够的承保范围。此外,临床试验和产品责任保险正变得越来越昂贵。因此,我们 可能无法以合理的成本获得足够的保险,以保护我们免受产品责任索赔造成的损失,而产品责任索赔 可能对我们的业务和财务状况产生不利影响。
我们开发的任何候选产品都可能会受到不利的第三方覆盖和报销做法以及定价规定的约束。
第三方付款人(包括政府卫生行政部门、私人健康保险公司、管理式医疗组织和其他第三方付款人)的可获得性和承保范围以及足够的报销对大多数患者能够 支付昂贵的治疗费用至关重要。我们的任何候选产品获得营销批准的销售将在很大程度上取决于我们候选产品的成本将在多大程度上由第三方付款方支付和报销,无论是在美国还是在国际上都是如此。如果无法获得报销或仅限于有限级别,我们可能无法成功将我们的 候选产品商业化。即使提供了保险,批准的报销金额也可能不足以让我们建立或 保持足够的定价来实现足够的投资回报。承保范围和报销可能会影响我们获得市场批准的任何候选产品的需求或价格。如果无法获得承保和报销,或者报销仅限于有限的水平,我们可能无法成功地将我们获得营销批准的任何候选产品商业化。 对于在医生监督下管理的产品,获得承保和足够的报销可能特别困难 ,因为此类药物通常价格较高。
| 36 |
与新批准产品的第三方付款人覆盖范围和报销相关的重大不确定性。在美国, 第三方付款人之间没有统一的产品承保范围和报销政策, 产品的承保范围和报销级别可能因付款人而异。联邦医疗保险和医疗补助计划越来越多地被用作私人付款人和其他政府付款人如何为药品和生物制剂制定保险和报销政策的典范。但是,第三方付款人决定为候选产品提供保险并不能保证其他付款人也会为该候选产品提供保险 。因此,确定覆盖范围的过程往往既耗时又昂贵。此流程将要求 我们单独向每个第三方付款人提供有关使用我们的候选产品的科学和临床支持,但不能保证将始终如一地应用承保范围和适当的报销范围或首先获得足够的报销。付款人在确定报销金额时考虑的因素基于产品是否:(I)其健康计划下的承保福利;(Ii)安全、有效且具有医疗必要性; (Iii)适用于特定患者;(Iv)成本效益;以及(V)既不是试验性的也不是研究性的。
越来越多的第三方付款人要求制药公司在标价的基础上向他们提供预定的折扣,并对医疗产品的定价提出挑战。此外,这些付款人越来越多地挑战价格,审查医疗必要性,并审查候选医疗产品的成本效益。对于新批准的药品,在获得保险和报销方面可能会出现特别严重的延误。第三方付款人可以将承保范围限制到已批准清单(称为处方集)上的特定候选产品,该清单可能不包括FDA批准的特定适应症的所有药物。我们可能需要进行昂贵的药物经济学研究,以证明我们的候选产品的医疗必要性和成本效益。尽管如此,我们的候选产品可能 不被认为是医学上必要的或具有成本效益的。我们不能确保我们商业化的任何候选产品都可以获得保险和报销,如果可以报销,报销级别是多少。
在美国以外,国际运营通常受到广泛的政府价格控制和其他市场监管,我们认为,欧洲、加拿大和其他国家对成本控制举措的日益重视已经并将继续 给我们的候选产品等治疗药物的定价和使用带来压力。在许多国家,特别是欧盟国家,作为国家卫生系统的一部分,医疗产品价格受到不同的价格控制机制的制约。在这些国家/地区,在产品获得上市批准后,与政府当局进行定价谈判可能需要相当长的时间。 要在某些国家/地区获得报销或定价批准,我们可能需要进行临床试验,将我们候选产品的成本效益 与其他可用的疗法进行比较。一般来说,这种制度下的产品价格比美国的价格低很多。其他国家允许公司为产品定价,但监控公司利润。额外的 外国价格控制或定价法规的其他变化可能会限制我们能够向候选产品收取的费用。 因此,在美国以外的市场,我们候选产品的报销可能会比美国减少 ,可能不足以产生商业上合理的收入和利润。
如果 我们无法为第三方付款人提供的任何未来候选产品建立或维持承保范围并获得足够的报销, 这些候选产品的采用和销售收入将受到不利影响,这反过来又可能对 营销或销售这些候选产品的能力产生不利影响(如果获得批准)。承保政策和第三方付款人报销费率可能随时更改 。即使我们获得监管部门 批准的一个或多个候选产品获得了有利的承保范围和报销状态,未来也可能会实施不太有利的承保政策和报销费率。
我们 面临与我们从我们赞助的临床试验中获得的健康信息的隐私相关的潜在责任。
大多数医疗保健提供者,包括我们从其获取患者健康信息的研究机构,都受HIPAA颁布并经HITECH修订的隐私和安全 法规的约束。我们目前未被归类为HIPAA下的承保实体或业务伙伴 ,因此不会直接受到其要求或处罚。然而,任何人都可以根据HIPAA的刑事条款直接或根据协助教唆或共谋原则被起诉。因此,根据事实和情况, 如果我们在知情的情况下从HIPAA覆盖的医疗保健提供者或研究机构收到个人可识别的健康信息,而该医疗保健提供者或研究机构未满足HIPAA关于披露个人可识别的健康信息的要求,则我们可能面临重大刑事处罚。此外,我们可能会保留敏感的个人身份信息,包括健康信息,这些信息是我们在整个临床试验过程中、在我们的研究协作过程中以及直接从注册了我们的患者援助计划的个人(或他们的医疗保健提供者)那里获得的。因此,我们可能需要遵守州法律,要求在个人信息被泄露的情况下通知受影响的个人和州监管机构,这是比HIPAA保护的健康信息更广泛的信息类别 。
| 37 |
此外, 某些健康隐私法、数据泄露通知法、消费者保护法和基因测试法可能直接适用于我们和/或我们的合作者的运营,并可能对我们收集、使用和传播个人健康信息 施加限制。我们或我们的合作者获取健康信息的患者以及与我们共享此信息的提供者可能拥有法定或合同权利,从而限制我们使用和披露信息的能力。我们可能需要花费大量资本和其他资源,以确保持续遵守适用的隐私和数据安全法律。声称我们侵犯了个人隐私权或违反了我们的合同义务,即使我们没有被发现负有责任,辩护也可能是昂贵的 和耗时的,并可能导致负面宣传,可能会损害我们的业务。
如果我们或第三方合同制造组织、CRO或其他承包商或顾问未能遵守适用的联邦、州或地方监管要求,我们可能会受到一系列监管行动的影响,这些监管行动可能会影响我们或我们的承包商开发和商业化我们的候选产品的能力,并可能损害或阻止我们 能够商业化的任何受影响的候选产品的销售,或者可能大幅增加开发、商业化和营销我们的候选产品的成本和费用。任何威胁或实际的政府执法行动也可能产生负面宣传,并要求我们投入大量资源,否则这些资源可能会用于我们业务的其他方面。越来越多地使用社交媒体可能会导致责任、 违反数据安全或声誉受损。
如果 我们未能遵守环境、健康和安全法律法规,我们可能会受到罚款或处罚,或产生可能对我们的业务产生重大不利影响的费用。
我们 受众多环境、健康和安全法律法规的约束,包括管理实验室程序以及危险材料和废物的处理、使用、储存、处理和处置的法律法规。我们的业务涉及使用危险和易燃材料,包括化学品和生物材料。我们的业务还会产生危险废物产品。我们通常与第三方签订处理这些材料和废物的合同。我们无法消除这些材料造成污染或伤害的风险。如果我们使用危险材料造成污染或伤害,我们可能要对由此产生的任何损害承担责任,任何责任都可能超出我们的资源范围。我们还可能产生与民事或刑事罚款相关的巨额成本 和处罚。
虽然 我们维持工人补偿保险,以支付因使用危险材料而对员工造成的伤害而产生的成本和开支,但该保险可能不足以承担潜在的责任。我们不为因我们储存或处置危险和易燃材料(包括化学品和生物材料)而对我们提出的环境责任或有毒侵权索赔维护 保险。
此外,为了遵守当前或未来的环境、健康和安全法律法规,我们可能会产生巨额成本。 这些当前或未来的法律法规可能会损害我们的研究、开发或商业化努力。不遵守这些法律法规还可能导致巨额罚款、处罚或其他制裁。
FDA、EMA和其他类似的外国监管机构可能不接受在其管辖范围以外的地点进行的试验数据。
我们 未来可能会选择进行国际临床试验。FDA、EMA或其他类似的外国监管机构接受在其各自管辖范围外进行的临床试验的研究数据可能会受到某些条件的限制。 如果外国临床试验的数据打算作为在美国上市批准的基础,FDA通常不会仅根据外国数据批准申请,除非(I)该数据适用于美国 人群和美国的医疗实践;(Ii)试验由具有公认能力并符合当前GCP要求的临床研究人员进行;以及(Iii)FDA能够通过现场检查或其他适当手段验证数据。此外,还必须满足FDA的临床试验要求,包括研究的患者群体的充分性和统计能力。 此外,此类外国审判将受进行审判的外国司法管辖区适用的当地法律管辖。不能保证FDA、EMA或任何适用的外国监管机构会接受在其适用管辖权范围外进行的试验数据。如果FDA、EMA或任何适用的外国监管机构不接受此类数据,将导致需要额外的试验,这将是昂贵和耗时的,并延误我们业务计划的各个方面,这可能导致我们的候选产品无法在适用司法管辖区获得商业化批准。
| 38 |
在一个司法管辖区获得并保持对我们候选产品的监管批准并不意味着我们将在其他司法管辖区成功获得 我们候选产品的监管批准。
在一个司法管辖区获得并保持对我们的候选产品的监管批准并不保证我们能够在任何其他司法管辖区获得或 保持监管批准。例如,即使FDA或EMA批准了候选产品的上市 ,外国司法管辖区的可比监管机构也必须批准候选产品在这些国家/地区的制造、营销和推广以及 报销。但是,在一个司法管辖区未能或延迟获得监管审批 可能会对其他司法管辖区的监管审批流程产生负面影响。审批程序因司法管辖区而异,可能涉及与美国不同的 要求和行政审查期限,包括额外的临床前研究或临床试验,因为在一个司法管辖区进行的临床试验可能不会被其他司法管辖区的监管机构接受。 在美国以外的许多司法管辖区,候选产品必须先获得报销批准,然后才能在该司法管辖区 销售。在某些情况下,我们打算对我们的候选产品收取的价格也需要批准。
获得外国监管批准以及建立和维护对外国监管要求的合规可能会导致我们的重大延误、困难和成本,并可能推迟或阻止我们的候选产品在某些国家/地区推出。如果我们或任何未来的合作伙伴未能遵守国际市场的法规要求或未能获得适用的 营销批准,我们的目标市场将会减少,我们充分发挥我们候选产品的市场潜力的能力将受到损害。
即使我们的候选产品获得监管部门的批准并成为产品,它们也将受到重大的上市后监管要求和监督。
我们可能获得的任何候选产品的监管批准都将要求向监管机构提交报告 并进行监督以监控产品的安全性和有效性,可能包含与特定年龄段的使用限制、警告、预防措施或禁忌症相关的重大限制,并可能包括繁重的批准后研究或风险管理 要求。例如,FDA可能需要REMS才能批准我们的候选产品,这可能需要 药物指南、医生培训和沟通计划或确保安全使用的其他要素,例如限制分发 方法、患者登记和其他风险最小化工具。此外,如果FDA、EMA或外国监管机构批准了我们的候选产品,我们候选产品的制造流程、标签、包装、分销、不良事件报告、储存、广告、促销、进口、出口和记录保存将受到广泛和持续的监管要求。 这些要求包括提交安全性和其他上市后信息和报告、注册,以及我们在批准后进行的任何临床试验的持续 遵守cGMP和GCP。此外,药品制造商及其设施将接受FDA和其他监管机构的持续审查和定期突击检查,以确保 遵守cGMP法规和标准。如果我们或监管机构发现某一产品存在以前未知的问题,如意外严重性或频率的不良事件,或该产品的制造设施存在问题,监管机构可对该产品、该制造设施或我们施加限制,包括要求从市场上召回或撤回该产品 或暂停生产。此外,如果不遵守FDA、EMA或其他类似的外国监管要求,我们公司可能会受到行政或司法制裁,包括:
| ● | 推迟或拒绝产品审批; |
| ● | 对我们进行临床试验的能力的限制,包括正在进行的或计划中的试验的全部或部分临床搁置 ; |
| 39 |
| ● | 对产品、制造商或制造工艺的限制; |
| ● | 警告 或无标题信件; |
| ● | 民事和刑事处罚; |
| ● | 禁制令; |
| ● | 暂停或撤回监管审批; |
| ● | 产品:扣押、拘留或禁止进口; |
| ● | 自愿或强制性的产品召回和宣传要求; |
| ● | 全部或部分停产;以及 |
| ● | 对运营实施限制,包括成本高昂的新制造要求。 |
发生上述任何事件或处罚可能会抑制我们将候选产品商业化并创造收入的能力 ,可能需要我们花费大量时间和资源来应对,并可能产生负面宣传。
FDA和其他监管机构的政策可能会发生变化,可能会颁布额外的政府法规,以阻止、限制或推迟监管部门对我们候选产品的审批。如果我们缓慢或无法适应现有要求的变化或新要求或政策的采用,或者如果我们无法保持合规性,我们可能会失去我们可能获得的任何营销批准 ,并且我们可能无法实现或保持盈利。
我们 也无法预测美国或国外未来的立法或行政 或行政行动可能产生的政府监管的可能性、性质或程度。例如,本届美国政府的某些政策可能会影响我们的商业和行业。也就是说,上届美国政府采取了几项行政行动,包括发布多项行政命令,这些行政命令可能会对FDA参与常规监管和监督活动的能力造成重大负担,或以其他方式严重拖延,例如通过制定规则、发布指导意见以及审查和批准营销申请来实施法规。很难预测这些行政行动,包括行政命令,是否或如何执行, 或者它们是否会在美国新政府的领导下被撤销或取代。如果这些行政行动限制了FDA在正常过程中从事监督和执行活动的能力,我们的业务可能会受到负面影响。
FDA和其他监管机构积极执行禁止推广非标签用途的法律法规。
如果 我们的任何候选产品获得批准,并且我们被发现不正当地推广这些产品的标签外用途,我们可能会承担重大责任。FDA和其他监管机构严格监管有关处方产品的促销声明。特别是,产品不得用于未经FDA、EMA或其他监管机构批准的用途,如该产品批准的标签所反映的那样。如果我们获得了候选产品的营销批准, 医生可能会以与批准的标签不一致的方式给他们的患者开这种药。如果我们被发现推广此类标签外使用,我们可能会承担重大责任。美国联邦政府已对涉嫌不当推广标签外使用的公司处以巨额民事和刑事罚款,并禁止几家公司从事标签外推广。政府还要求公司签订同意法令或实施永久禁令,以改变或限制特定的促销行为。如果我们不能成功管理我们候选产品的促销, 如果获得批准,我们可能会承担重大责任,这将对我们的业务和财务状况产生重大不利影响。
| 40 |
我们 可能会面临当前法规和未来立法的变化带来的困难。
现有的 监管政策可能会发生变化,可能会颁布额外的政府法规,以阻止、限制或推迟对我们候选产品的监管审批 。我们无法预测美国或国外未来立法或行政行动可能产生的政府监管的可能性、性质或程度。如果我们缓慢或无法适应现有要求的变化或新要求或政策的采用,或者如果我们无法保持合规性,我们可能会失去我们可能获得的任何营销批准 ,我们可能无法实现或保持盈利。
例如,ACA极大地改变了政府和私营保险公司为医疗保健融资的方式,并对美国生物制药行业产生了重大影响。自ACA颁布以来,美国还提出并通过了其他立法修改。这些变化包括联邦医疗保险支付总额的减少,并可能导致联邦医疗保险和其他医疗保健资金的进一步减少,如果获得批准,所有这些都可能对我们候选产品的客户以及我们的财务运营产生实质性的不利影响。
340B药品定价计划也有几个变化和挑战,该计划对药品制造商 可以向某些医疗保健机构销售的药品设定最高价格。目前尚不清楚这些发展如何影响承保医院 谁可能购买我们未来的候选产品,以及影响我们未来可能向我们批准的候选产品收取此类设施的费率 。
此外, 近年来,政府对药品制造商为其市场上销售的产品定价的方式进行了更严格的审查,导致美国国会进行了几次调查,并提出并颁布了联邦和州立法,旨在提高产品定价的透明度,审查定价与制造商患者 计划之间的关系,以及改革政府计划对药品的补偿方法。美国国会表示,将继续寻求新的立法措施来控制药品成本。
此外,2018年5月30日,《审判权法案》签署成为法律。除其他事项外,该法律还为某些患者 提供了一个联邦框架,以访问已完成第一阶段临床试验并正在接受调查 以获得FDA批准的某些研究用新产品。在某些情况下,符合条件的患者可以在不参加临床试验和根据FDA扩大准入计划获得FDA许可的情况下寻求治疗。根据《试用权利法案》,药品制造商没有义务 向符合条件的患者提供其产品。
我们 预计ACA以及未来可能采取的其他医疗改革措施可能会导致更严格的覆盖范围 标准,并对我们收到的任何经批准的候选产品的价格造成额外的下行压力。联邦医疗保险或其他政府计划的任何报销减少 都可能导致私人支付者支付的类似减少。实施成本控制措施或其他医疗改革可能会阻止我们创造收入、实现盈利或将我们的候选产品商业化 。
已经提出了立法和监管建议,以扩大批准后的要求,并限制生物技术产品的销售和促销活动 。我们无法确定是否会颁布额外的法律变更,或FDA的法规、指南或解释是否会更改 ,或者此类更改可能会对我们的候选产品的上市审批产生什么影响(如果有)。此外, 国会对FDA审批过程的更严格审查可能会显著推迟或阻止上市审批,以及 使我们受到更严格的产品标签和上市后测试和其他要求的影响。
与我们知识产权相关的风险
我们的成功取决于我们保护知识产权和专有技术的能力。
我们的商业成功在一定程度上取决于我们是否有能力为我们的候选产品、专有技术及其用途获得并维护专利保护和商业秘密保护,从而在不侵犯他人专有权利的情况下运营。如果我们或我们的许可方 无法保护我们的知识产权,或者如果我们的知识产权不适用于我们的技术或我们的 候选产品,我们的竞争地位可能会受到损害。我们和我们的许可方通常通过在美国和海外提交与我们的候选产品、专有技术及其用途相关的专利申请来保护我们的专有地位 这些对我们的业务非常重要。我们授权内的专利申请不能针对应用此类申请中所声称的技术的第三方强制执行,除非且直到此类申请颁发专利,且仅限于所发布的权利要求涵盖该技术的范围内。不能保证我们的许可内专利申请将导致专利被颁发,或已颁发的专利将针对具有类似技术的竞争对手提供足够的保护,也不能保证专利 在颁发后不会被第三方侵犯、设计、失效或无法强制执行。即使已颁发的专利也可能在以后被认定为无效或不可强制执行,或者可能在第三方向各专利局或法院提起的诉讼中被修改或撤销。未来对我们和我们的许可人的所有权的保护程度是不确定的。可能只提供有限的保护 ,可能无法充分保护我们或我们的许可人的权利,或不允许我们或我们的许可人获得或保持任何 竞争优势。我们和我们的许可方在适当保护与我们的候选产品相关的知识产权方面的这些不确定性和/或限制 可能会对我们的财务状况和运营结果产生重大不利影响。
| 41 |
虽然 我们可以在美国和其他国家/地区授权已颁发的专利,但我们不能确定我们在其他未授权的 美国未决专利申请、相应的国际专利申请和在某些国家/地区的专利申请中的权利要求将被美国专利商标局或美国专利商标局或外国的专利机构和法院视为可申请专利,也不能确定我们在授权内已颁发的专利中的权利要求不会在受到挑战时被认定为无效或不可强制执行。
专利申请过程存在许多风险和不确定性,不能保证我们或我们的许可方或我们未来的任何潜在合作伙伴都能成功地通过获得和保护专利来保护我们的候选产品。 这些风险和不确定性包括:
| ● | 美国专利商标局和各种外国政府专利机构要求在专利过程中遵守一些程序、文件、费用支付和其他规定,不遵守这些规定可能导致专利或专利申请被放弃或失效,在有关司法管辖区内部分或全部丧失专利权; |
| ● | 专利申请不得导致任何专利被授予; |
| ● | 专利 可能被质疑、宣布无效、修改、撤销、规避、被发现不可执行 或以其他方式可能不提供任何竞争优势; |
| ● | 任何颁发的专利将为我们提供针对竞争对手的保护程度和范围,包括 第三方是否会找到方法使我们的专利无效或以其他方式规避我们的专利; |
| ● | 其他人是否会申请或获得与我们的专利和专利申请所涵盖的方面类似的专利; |
| ● | 我们拥有的或许可中的专利申请是否会导致已颁发的专利,其权利要求 涵盖我们的候选产品或其在美国或其他外国的使用 ; |
| ● | 我们的 竞争对手,其中许多拥有比我们或我们的许可方更多的资源,并且许多在竞争技术上进行了重大投资,他们可能寻求或 可能已经获得了将限制、干扰或阻止我们制作、使用和销售我们的候选产品的能力; |
| ● | 美国政府和国际政府机构可能面临巨大压力,要求限制被证明成功的疾病治疗方法在美国国内外的专利保护范围。作为涉及全球卫生问题的公共政策问题 ;和 |
| ● | 美国以外的其他国家的专利法对专利权人的优惠程度可能不如美国法院所支持的专利法,从而使外国竞争者有更好的机会创造、开发和营销竞争产品。 |
| 42 |
专利起诉过程也既昂贵又耗时,我们或我们的许可人可能无法以合理的成本或及时提交并起诉所有必要的或理想的专利申请,或者在保护可能在商业上有利的所有司法管辖区 。我们或我们的许可方也有可能在获得专利保护为时已晚之前,无法识别我们研发成果的可专利方面 。此外,在某些情况下,我们无权控制针对我们许可的技术的专利申请的准备、提交和起诉,或维护专利,包括来自我们许可方和第三方的专利。我们还可能需要许可方的合作以强制执行许可的专利权,但可能不会提供此类合作。因此,这些专利和申请可能不会以符合我们业务最佳利益的方式进行起诉和强制执行。我们不能确定许可人的专利起诉和维护活动已经或将会遵守适用的法律法规,这可能会影响此类专利或此类申请可能颁发的任何专利的有效性和可执行性。如果他们不这样做,这可能会导致我们失去我们许可的任何适用知识产权的权利,因此我们开发和商业化产品或候选产品的能力可能会受到不利影响,我们可能无法阻止竞争对手制造、使用和销售竞争产品。
生物和医药产品(如细胞疗法候选产品)的物质专利构成通常为这些类型的产品提供强有力的知识产权保护,因为这种专利提供的保护与任何使用方法无关。然而,我们不能 确定我们未决的专利申请中涉及我们候选产品的物质组成的权利要求将被美国专利商标局或外国专利局 视为可申请专利,或者我们已颁发的任何专利中的权利要求将被美国或外国法院视为有效和可强制执行。使用方法专利保护指定方法的产品的使用。此类专利不会阻止竞争对手制造和销售与我们的产品完全相同的产品,以获得超出专利方法范围的指示。此外,即使竞争对手没有针对我们的目标适应症积极推广他们的产品,医生也可能会为我们的使用方法专利 涵盖的那些用途开出这些产品的标签外处方。尽管标签外的处方可能会侵犯或助长对使用方法专利的侵犯,但这种做法很常见,这种侵权行为很难防止或起诉。
此外,尽管我们与可以访问我们研发成果的可专利方面的各方签订了保密和保密协议,例如我们的员工、外部科学合作者、CRO、第三方制造商、顾问、 顾问、许可人和其他第三方,但这些各方中的任何一方都可能违反此类协议,并在提交专利 申请之前披露此类成果,从而危及我们寻求专利保护的能力。
如果我们的许可方获得的任何专利保护的范围不够广泛,或者如果我们的许可方失去了我们许可的任何专利保护,我们阻止竞争对手将类似或相同的候选产品商业化的能力将受到不利影响。
生物制药公司的专利地位通常高度不确定,涉及复杂的法律和事实问题,近年来一直是许多诉讼的主题。因此,我们授权的专利权的存在、颁发、范围、有效性、可执行性和商业价值都具有高度的不确定性。我们的未决专利申请和未来的授权专利申请可能不会导致 保护我们的候选产品或有效阻止他人将竞争产品商业化的专利 候选产品。
此外,专利申请中权利要求的范围可以在专利权利要求发布之前大幅缩小,权利要求范围在专利发布后可以 重新解释。即使我们当前或未来许可的专利申请作为专利发布,它们也不会以能够为我们提供任何有意义的保护、阻止竞争对手或其他第三方与我们竞争或以其他方式为我们提供任何竞争优势的形式发布 。我们许可的任何专利可能会受到第三方的挑战或规避,或者 可能会因第三方的挑战而缩小范围或使其失效。因此,我们不知道我们的候选产品 是否会受到有效和可强制执行的专利的保护。我们的竞争对手或其他第三方可能能够通过以非侵权方式开发类似或替代技术或产品来规避我们的专利,这可能会对我们的业务、财务状况、运营结果和前景产生重大不利影响 。
| 43 |
专利的颁发对于其发明性、范围、有效性或可执行性并不是决定性的,我们的许可专利可能不包括我们的候选产品,或者可能会在美国和国外的法院或专利局受到挑战。我们可能会 接受第三方向美国专利商标局提交的现有技术的发行前提交,或参与反对、派生、撤销、复审、授予后审查或PGR,以及各方间在USPTO或外国专利局挑战我们专利权的审查、知识产权或其他类似程序 。在法律上断言无效和不可执行之后的结果是不可预测的。例如,关于我们授权的专利的有效性,我们不能确定没有无效的先前技术,我们或我们的许可人和专利审查员在起诉期间并不知道这些技术。不能保证已找到与我们的许可内专利和专利申请或我们许可方的专利相关的所有潜在相关的先前技术。也不能保证存在我们或许可方知晓但我们不认为会影响我们的专利和专利申请或许可方的专利和专利申请中的权利要求的有效性或可执行性的 现有技术,但最终可能会被发现影响权利要求的有效性或可执行性 。任何此类提交、诉讼或诉讼中的不利裁决可能会缩小我们许可内的专利权的范围,或使其无效或使其不可执行,允许第三方将我们的候选产品商业化并直接与我们竞争,而无需向我们付款。此类许可专利权的丧失、排他性的丧失或专利主张的缩小、无效或无法强制执行,可能会限制我们阻止他人使用类似或相同的技术和产品或将其商业化的能力,或者限制我们候选产品的专利保护期限。即使最终结果对我们有利,这样的程序也可能导致大量成本,并需要我们的科学家和管理层花费大量时间。此外,如果我们的专利和专利申请提供的保护的广度或强度受到威胁,无论结果如何,都可能阻止公司 与我们合作,授权、开发或商业化当前或未来的候选产品。
我们 可能无法识别相关的第三方专利,或可能错误地解释第三方专利的相关性、范围或到期时间,这可能会对我们开发和营销我们的候选产品的能力产生不利影响。
我们 不能保证我们的任何专利搜索或分析,包括相关专利的识别、专利权利要求的范围或相关专利的到期,都是完整或彻底的,我们也不能确保我们已经识别了与我们产品商业化相关或对我们的产品在任何司法管辖区的商业化所必需的每一个第三方 专利和在美国和国外的待处理申请。专利权利要求的范围由法律解释、专利中的书面披露和专利的起诉历史确定。我们对专利或待处理申请的相关性或范围的解释可能不正确 ,这可能会对我们的产品营销能力产生负面影响。我们可能会错误地确定我们的产品不受第三方专利的保护,或者可能会错误地预测第三方的待决申请是否会提出相关范围的权利要求 。我们对美国或国外任何我们认为相关的专利的到期日期的确定可能是不正确的 ,这可能会对我们开发和营销我们的候选产品的能力产生负面影响。我们未能识别并正确解释相关专利,可能会对我们开发和营销我们的候选产品的能力产生负面影响。
确定我们的发明的可专利性的一个方面取决于“现有技术”的范围和内容,即在要求保护的发明的优先权日期之前,相关领域的技术人员可以获得或被认为可获得的信息。可能存在我们不知道的现有技术,这些技术可能会影响我们的专利权利要求的可专利性,或者如果发布,可能会影响专利权利要求的有效性或可执行性 。此外,我们可能不知道与我们的候选产品或其预期用途有关的所有第三方知识产权,因此,这些第三方知识产权对我们自己的专利和专利申请的可专利性的影响,以及这些第三方知识产权对我们运营自由的影响,是非常不确定的 。由于美国和大多数其他国家/地区的专利申请通常在申请后18个月内保密,或者可能根本不会公布,因此我们不能确定我们是第一个提交与我们的候选产品相关的专利申请的公司。因此,我们专利权的颁发、范围、有效性、可执行性和商业价值都非常不确定。 此外,对于所有权利要求都享有2013年3月16日之前的优先日期的美国申请,第三方可以提起干扰诉讼程序,或者由美国专利商标局提起诉讼,以确定谁最先发明了我们申请的专利权利要求所涵盖的任何标的 。对于在2013年3月16日之前包含无权享有优先权的权利要求的美国申请, 由于《美国发明法》的通过,专利法中存在更大的不确定性,该法案使美国专利法发生了重大的 变化,包括挑战未决专利申请和已颁发专利的新程序。
| 44 |
我们的专利或悬而未决的专利申请可能会在美国和国外的法院或专利局受到挑战。例如, 我们可能在美国或其他地方被第三方向USPTO提交先前技术的发行前提交,或参与PGR程序、异议、派生、重新审查或知识产权诉讼,挑战我们的专利权或其他人的专利权 。在任何此类挑战中做出不利裁决可能会导致排他性丧失或专利主张全部或部分缩小、无效、 或无法执行,从而限制我们阻止他人使用类似或相同的技术和产品或将其商业化的能力,或者限制我们的技术和产品的专利保护期限。此外,考虑到新产品候选产品的开发、测试和监管审查所需的时间,保护这些候选产品的专利可能会在这些候选产品商业化之前或之后不久 到期。任何未能获得或保持与我们的候选产品相关的专利保护的情况都可能对我们的业务、财务状况、运营结果和前景产生重大不利影响。
在 未来,我们的一些知识产权可能会通过政府资助的计划被发现,因此可能会受到联邦 法规的约束,例如“游行”权利、某些报告要求以及对美国公司的偏好。遵守此类法规可能会限制我们的独家权利,并限制我们与非美国制造商签订合同的能力。
我们未来可能获得或许可的一些知识产权可能是通过使用美国政府资金而产生的 ,因此可能受到某些联邦法规的约束。美国政府的这些权利可能包括知识产权中保留的权利,包括将发明用于任何政府目的的非独家、不可转让、不可撤销的全球许可。 此外,在某些有限的情况下,如果美国政府确定:(I)没有采取足够的步骤将发明商业化;(Ii)为满足公共卫生或安全需要,政府有权要求我们向第三方授予这些发明的独家、部分独家或非独家许可;或(Iii)政府 必须采取行动以满足联邦法规对公众使用的要求(也称为“进行权”)。 如果授权者未能向政府披露发明或未能在规定的时限内提交知识产权登记申请,美国政府也有权取得这些发明的所有权。根据政府资助的计划产生的知识产权 也受某些报告要求的约束,遵守这些要求可能需要我们 花费大量资源。此外,美国政府要求任何包含任何这些发明的产品或通过使用任何这些发明而生产的产品必须基本上在美国制造。如果知识产权所有者或受让人能够证明以类似条款向可能在美国大量生产的潜在被许可人授予许可,或者在这种情况下,国内制造在商业上是不可行的,则提供资金的联邦机构可以 放弃对美国行业的这种偏爱。这种对美国行业的偏爱 可能会限制我们与非美国产品制造商签订此类知识产权所涵盖产品的合同的能力。 如果我们未来的任何知识产权也是通过使用美国政府资金产生的,那么《贝赫-多尔法案》的条款也可能同样适用。
知识产权不一定能解决我们竞争优势面临的所有潜在威胁。
我们的知识产权所提供的未来保护 程度不确定,因为知识产权具有局限性, 可能无法充分保护我们的业务或使我们保持竞争优势。例如:
| ● | 其他 可能能够开发与我们的候选产品相似但不在我们拥有或许可的专利权利要求范围内的产品; |
| ● | 我们 或我们的许可人或未来的合作者可能不是第一个使我们拥有或许可的已发布专利或未决专利申请涵盖的发明 ; |
| ● | 我们 或我们的许可人或未来的合作者可能不是第一个提交专利申请的人 ,涉及我们的某些发明; |
| 45 |
| ● | 其他 可以独立开发类似或替代技术或复制我们的任何技术 而不侵犯我们的知识产权; |
| ● | 我们许可方的未决专利申请有可能不会导致已颁发的专利 ; |
| ● | 由于竞争对手的法律挑战,我们拥有或许可的已颁发的 专利可能被认定为无效或不可执行; |
| ● | 我们的竞争对手可能会在我们没有专利权的国家开展研发活动,然后利用从这些活动中学到的信息来开发有竞争力的 产品,在我们的主要商业市场销售; |
| ● | 我们 不得开发其他可申请专利的专有技术; |
| ● | 我们 无法根据我们的专利申请来预测任何专利发布的保护范围, 包括我们拥有或许可的专利申请是否将导致已颁发的 项专利,其权利要求涵盖我们的候选产品或其在美国或其他国家/地区的使用 ; |
| ● | 基于我们的专利申请而进行的任何专利申请的权利要求可能无法提供针对竞争对手或任何竞争优势的保护,或可能受到第三方的挑战; |
| ● | 如果强制执行,法院可能不会认为我们的专利是有效的、可执行的和被侵犯的; |
| ● | 我们 可能需要启动诉讼或行政诉讼来执行和/或捍卫我们的 专利权,这将是代价高昂的,无论我们是赢是输; |
| ● | 我们 可以选择不提交专利申请以保护某些商业秘密或专有技术,第三方随后可以提交专利申请并获得涵盖此类知识产权的已颁发 专利; |
| ● | 我们 可能无法充分保护和监管我们的商标和商业机密;以及 |
| ● | 其他人的专利可能会对我们的业务产生不利影响,包括如果其他人获得了 项要求与我们的专利和专利申请所涵盖的主题相似或更好的专利。 |
如果发生其中任何事件,可能会严重损害我们的业务、运营结果和前景。
我们的商业成功在很大程度上取决于我们在不侵犯第三方专利和其他专有权利的情况下运营的能力 。第三方声称我们侵犯了他们的专有权,可能会导致损害赔偿责任,或者阻碍或推迟我们的开发和商业化努力。
我们的商业成功在一定程度上取决于避免侵犯第三方的专利和所有权。但是,我们的研究、开发和商业化活动可能会受到侵犯或以其他方式侵犯第三方拥有或控制的专利或其他知识产权的指控。其他实体可能拥有或获得专利或专有权利,这可能会限制我们制造、使用、销售、提供销售或进口我们的候选产品和将来可能获得批准的产品的能力,或者损害我们的竞争地位。在美国国内外,生物制药行业有大量涉及专利和其他知识产权的诉讼,包括专利侵权诉讼、异议、复审、知识产权诉讼和向美国专利商标局和/或外国专利局提起的PGR诉讼。在我们正在开发候选产品的领域中,存在着大量的第三方美国和外国颁发的专利 和未决的专利申请。可能存在与使用或制造我们的候选产品相关的第三方专利或 专利申请,这些专利要求材料、配方、制造方法或处理方法。
| 46 |
As the biopharmaceutical industry expands and more patents are issued, the risk increases that our product candidates may be subject to claims of infringement of the patent rights of third parties. We cannot provide any assurances that third-party patents do not exist which might be enforced against our current product candidates or future products, resulting in either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation to third parties. Because patent applications are maintained as confidential for a certain period of time, until the relevant application is published, we may be unaware of third-party patents that may be infringed by commercialization of any of our product candidates, and we cannot be certain that we were the first to file a patent application related to a product candidate or technology. Moreover, because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our product candidates may infringe. In addition, identification of third-party patent rights that may be relevant to our technology is difficult because patent searching is imperfect due to differences in terminology among patents, incomplete databases and the difficulty in assessing the meaning of patent claims. It is also possible that patents owned by third parties of which we are aware, but which we do not believe are relevant to our product candidates and other proprietary technologies we may develop, could be found to be infringed by our product candidate. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Any claims of patent infringement asserted by third parties would be time consuming and could:
| ● | 结果 诉讼费用高昂,可能造成负面宣传; |
| ● | 转移我们技术人员和管理人员的时间和注意力; |
| ● | 造成 开发延迟; |
| ● | 防止 我们不得将我们的任何候选产品商业化,直到所主张的专利到期或 在法庭上被最终认定为无效或不可强制执行或未被侵犯; |
| ● | 要求我们开发非侵权技术,这在成本效益的基础上可能是不可能的; |
| ● | 主题 我们对第三方承担重大责任;或 |
| ● | 要求 我们签订版税或许可协议,这些协议可能无法按商业上的 合理条款获得,或者根本无法获得,或者可能是非独家的,这可能会导致我们的竞争对手 获得相同的技术。 |
Although, to our knowledge, no third party has asserted a claim of patent infringement against us as of the date of this prospectus, others may hold proprietary rights that could prevent our product candidates from being marketed. Any patent-related legal action against us claiming damages and seeking to enjoin activities relating to our product candidates or processes could subject us to potential liability for damages, including treble damages if we were determined to willfully infringe, and require us to obtain a license to manufacture or develop our product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of management and employee resources from our business. We cannot predict whether we would prevail in any such actions or that any license required under any of these patents would be made available on commercially acceptable terms, if at all. Moreover, even if we or our future strategic partners were able to obtain a license, the rights may be nonexclusive, which could result in our competitors gaining access to the same intellectual property. In addition, we cannot be certain that we could redesign our product candidates or processes to avoid infringement, if necessary. Accordingly, an adverse determination in a judicial or administrative proceeding, or the failure to obtain necessary licenses, could prevent us from developing and commercializing our product candidates, which could harm our business, financial condition and operating results.
对我们提出索赔的当事人 可能比我们更有效地承受复杂专利诉讼的成本,因为他们拥有 更多的资源或更成熟和发达的知识产权组合,或两者兼而有之。此外,由于在知识产权诉讼或行政诉讼中需要大量 发现,因此存在 我们的某些机密信息可能因披露而受到损害的风险。此外,任何诉讼的发起和继续导致的任何不确定性可能对我们筹集额外资金的能力产生重大不利影响,或对我们的业务、我们在市场上的竞争能力、经营业绩、财务状况和前景产生重大不利影响。
| 47 |
我们 可能会卷入诉讼以保护或执行我们的专利或我们的许可方的专利,这可能是昂贵、耗时 且不成功的。此外,倘在法庭上受到质疑,我们的授权专利可能会被视为无效或不可执行。
竞争对手 可能会侵犯我们的专利或其他知识产权或我们许可方的知识产权。要停止此类侵权或未经授权的使用,我们和/或我们的许可方可能被要求提出侵权索赔,这可能既昂贵又耗时。 此外,我们的许可方可能需要提交侵权索赔,而我们的许可方可能选择不提交此类索赔。我们的未决专利 申请不能针对实践此类申请中所要求的技术的第三方强制执行,除非且直到专利 从此类申请中颁发。此外,在专利侵权诉讼中,法院可以裁定我们拥有或许可的专利 无效、不可强制执行和/或未被侵犯。如果我们或我们的任何许可人或未来的潜在合作者对第三方提起法律诉讼,以强制执行针对我们的候选产品之一的专利,被告可以反诉 我们的专利全部或部分无效和/或不可强制执行。在专利诉讼中,被告声称无效和/或不可执行的反诉很常见。质疑有效性的理由包括据称未能满足几项法定要求中的任何一项,包括缺乏新颖性或书面描述、明显、书面描述或未实施。不可强制执行的理由 可能包括与专利诉讼有关的人故意向美国专利商标局隐瞒重要信息或在起诉期间做出误导性陈述的指控。
如果被告在无效和/或不可强制执行的法律主张上获胜,我们将失去对该候选产品的至少部分甚至全部专利保护。还有一种风险是,即使此类专利的有效性得到维持,法院也会狭隘地解释专利权利要求,或以我们的专利权利要求不包括发明为理由,裁定我们无权阻止另一方使用所涉发明,或根据《美国法典》第35篇第271(E)(1)条,裁定对方使用我们的专利技术属于专利侵权的安全港。此外,如果我们的专利和专利申请或我们的许可方提供的保护的广度或强度 受到威胁,可能会阻止公司 与我们合作许可、开发或商业化当前或未来的候选产品。这种专利保护的丧失将对我们的业务产生实质性的不利影响。同样,如果我们主张商标侵权索赔,法院可能会裁定我们主张的商标无效或不可强制执行,或者我们主张商标侵权的一方对相关商标拥有更高的 权利。在这种情况下,我们最终可能会被迫停止使用此类商标。
即使 如果解决方案对我们有利,与我们知识产权相关的诉讼或其他法律程序也可能导致我们产生巨额 费用,并可能分散我们的技术和管理人员的正常责任。此类诉讼或诉讼 可能大幅增加我们的运营亏损,并减少可用于开发活动或任何未来销售、营销或分销活动的资源。我们可能没有足够的财政或其他资源来充分开展此类诉讼或诉讼程序 。我们的一些竞争对手可能比我们更有效地承担此类诉讼或诉讼的费用,因为他们拥有更大的财力。专利诉讼或其他诉讼的发起和继续所产生的不确定性 可能会影响我们在市场上的竞争能力。
即使我们认定侵权行为成立,法院也可能决定不对进一步的侵权活动颁发禁令,而只判给 金钱损害赔偿,这可能是也可能不是足够的补救措施。此外,由于知识产权诉讼或与我们的知识产权相关的其他法律程序需要进行大量的披露,因此存在在此类诉讼或其他程序中披露我们的某些机密信息的风险。
知识产权诉讼可能会导致负面宣传,损害我们的声誉,并导致我们普通股的市场价格下跌。
在任何知识产权诉讼的过程中,可能会有关于诉讼启动的公告以及 诉讼中的听证结果、动议裁决和其他临时程序。如果证券分析师或投资者认为 这些公告是负面的,我们现有候选产品、计划或知识产权的感知价值可能会降低。 此类公告还可能损害我们未来候选产品的声誉或市场,这可能会对我们的业务产生实质性的不利影响 。
| 48 |
可能需要派生 或干预程序来确定发明的优先权,而不利的结果可能要求我们停止使用相关技术或尝试许可胜利方的权利。
派生 或由第三方引发或由我们或我们的许可人提起的干扰程序,或由美国专利商标局或外国专利局的类似程序 宣布的程序,可能对于确定与我们的 或我们的许可人的专利或专利申请有关的发明的优先权或更正发明是必要的。不利的结果可能会导致我们失去当前的专利权,并且 要求我们停止使用相关技术或尝试从胜利方那里获得许可权。如果胜利方不以商业上合理的条款向我们提供许可,我们的业务可能会受到损害。我们或我们的许可人对此类诉讼的辩护可能会失败,即使成功,也可能导致巨额成本,并分散我们的管理层和其他员工的注意力。此外,与此类程序相关的不确定性可能会对我们筹集必要资金的能力产生重大不利影响,以继续我们的临床试验、继续我们的研究计划、从第三方获得必要的技术许可或建立开发或制造合作伙伴关系,以帮助我们将候选产品推向市场。
专利改革立法可能会增加围绕我们专利申请的起诉以及执行或保护我们已颁发的专利的不确定性和成本。
2011年9月,《莱希-史密斯美国发明法》或《莱希-史密斯法案》签署成为法律。Leahy-Smith法案可能会增加围绕我们专利申请的起诉以及我们已发布专利的执行或保护的不确定性和成本。Leahy-Smith 法案包括对美国专利法的多项重大修改。这些条款影响了专利申请的起诉方式,重新定义了现有技术,并为竞争对手提供了更有效、更具成本效益的途径来挑战专利的有效性。 具体而言,根据《莱希-史密斯法案》,美国于2013年3月过渡到“第一发明人提交申请”制度 ,在该制度下,假设满足其他可专利性要求,第一个提交专利申请的发明人将有权 获得专利,而无论所要求的发明是否是第三方首先发明的。因此,在2013年3月之后但在我们提交涉及同一发明的申请之前向美国专利商标局提交专利申请的第三方可以获得涵盖我们的发明的专利,即使我们在发明由该第三方做出之前就已经做出了该发明。这将要求我们认识到从发明到专利申请的提交时间。此外,我们获得和维护有效且可强制执行的专利的能力取决于我们的技术与现有技术之间的差异是否允许我们的技术比现有技术获得专利。由于在美国和大多数其他国家/地区的专利申请在提交后或在 颁发之前的一段时间内是保密的,我们不能确定我们或我们的许可人是第一个(I)提交与我们的产品 候选和我们可能开发的其他专有技术有关的任何专利申请,或(Ii)发明我们或我们的许可人的 专利或专利申请中声称的任何发明。即使我们拥有有效且可强制执行的专利,我们也不能排除其他人实施所要求保护的发明,如果对方能够证明他们在我们的申请日期之前在商业上使用了该发明,或者另一方 受益于强制许可。
《莱希-史密斯法案》还包括一些重大变化,这些变化将影响专利申请的起诉方式,也可能影响专利诉讼。这些措施包括在专利诉讼期间允许第三方向USPTO提交现有技术,以及通过USPTO管理的授予后诉讼程序(包括PGR、IPR和派生程序)来攻击专利有效性的额外程序。 在任何此类提交或程序中做出不利裁决可能会缩小我们专利权利的范围或可执行性,或使其无效,这可能会对我们的竞争地位产生不利影响。
由于USPTO诉讼中的证据标准低于美国联邦法院宣布专利权利要求无效所需的证据标准,因此第三方可能会在USPTO程序中提供足以让USPTO裁定权利要求无效的证据,即使如果在地区法院诉讼中首次提交相同的证据将不足以使权利要求无效。 因此,第三方可能会尝试使用USPTO程序来使我们的专利主张无效 如果第三方首先作为被告在地区法院诉讼中提出质疑。因此,Leahy-Smith法案及其实施可能会 增加围绕起诉我们或许可人的专利申请以及执行或保护我们已发布的专利的不确定性和成本,所有这些都可能对我们的业务、财务状况、运营结果和前景产生重大不利影响 。
| 49 |
美国专利法或其他国家/地区的法律变更可能会降低专利的整体价值,从而削弱我们保护候选产品的能力。
与其他生物制药公司的情况一样,我们的成功在很大程度上依赖于知识产权,特别是专利。 在生物制药行业获得和实施专利涉及高度的技术和法律复杂性。因此,获得和执行生物制药专利既昂贵又耗时,而且本身就不确定。专利法 或美国和其他国家/地区专利法解释的变化可能会削弱我们保护我们发明的能力, 获取、维护和执行我们的知识产权,更广泛地说,可能会影响我们知识产权的价值 ,并可能增加围绕专利申请的起诉和已颁发专利的执行或保护的不确定性和成本 。我们无法预测在我们的专利或第三方专利中可能允许或执行的权利要求的广度。此外,国会或其他外国立法机构可能会通过对我们不利的专利改革立法,或缩小我们拥有和许可的专利的范围。
例如,美国最高法院近年来对几起专利案件做出了裁决,要么缩小了某些情况下可用的专利保护范围 ,要么在某些情况下削弱了专利所有者的权利。除了关于我们或我们的许可人未来获得专利的能力的不确定性增加之外,这种事件的组合还造成了关于获得专利后的价值的不确定性。根据美国国会、美国联邦法院、USPTO或外国司法管辖区类似机构的决定,管理专利的法律和法规可能会以不可预测的方式发生变化,从而削弱我们或我们的许可人获得新专利或强制执行我们现有专利和我们未来可能获得的专利的能力。我们无法预测国会、联邦法院或美国专利商标局未来的裁决会如何影响我们的专利价值。
我们 或我们的许可人可能会受到质疑我们或我们的许可内专利和其他知识产权的发明权或所有权的索赔。
作为发明人或共同发明人,我们 还可能会受到前员工、合作者或其他第三方对我们授权的专利或其他知识产权的所有权权益的影响。未能在专利申请中指定适当的发明人 可能导致在其上颁发的专利无法强制执行。发明权纠纷可能是由于以下原因引起的:关于被指定为发明人的不同个人的贡献的相互冲突的意见 、外国公民参与专利标的开发的外国法律的影响、参与开发我们候选产品的第三方的义务冲突,或者由于关于潜在联合发明的共同所有权的问题。例如,我们可能会因参与开发我们的候选产品的顾问或其他人的义务冲突而产生库存纠纷。作为替代或补充,我们可以签订协议以澄清我们在此类知识产权中的权利范围。可能需要通过诉讼来对抗这些 和其他挑战库存或所有权的索赔。如果我们或我们的许可人未能为任何此类索赔辩护,除了支付 金钱损害赔偿外,我们还可能失去宝贵的知识产权,如独家所有权或使用权。这样的结果可能会对我们的业务产生实质性的不利影响。即使我们或我们的许可方成功地对此类索赔进行了辩护,诉讼 也可能导致巨额成本,并分散管理层和其他员工的注意力。
此外,尽管我们的政策是要求可能参与知识产权构思或开发的员工和承包商执行将此类知识产权转让给我们的协议,但我们可能无法成功地与 实际上构思或开发我们视为自己的知识产权的每一方执行此类协议。知识产权转让可能不是自动执行的,或者转让协议可能被违反,我们可能被迫向第三方提出索赔,或者他们可能对我们提出的索赔进行抗辩,以确定我们所认为的知识产权的所有权。此类索赔 可能会对我们的业务、财务状况、运营结果和前景产生实质性的不利影响。
| 50 |
专利 条款可能不足以在足够长的时间内保护我们在候选产品上的竞争地位。
专利 的寿命有限。在美国,如果及时支付所有维护费,专利的自然失效时间通常是其首次生效申请日期后20年。可能有各种延期,但专利的期限及其提供的保护是有限的。即使获得了针对我们候选产品的专利,一旦专利期到期,我们可能会 接受来自竞争产品的竞争。考虑到候选产品的开发、测试和监管审查所需的时间,针对我们候选产品的专利可能会在这些候选产品商业化之前或之后不久到期。因此,我们的专利组合可能无法为我们提供足够的权利来排除其他公司将与我们相似或相同的产品商业化 。此外,虽然在美国颁发专利时,可以根据美国专利商标局造成的某些延迟 延长专利的寿命,但由于专利申请人在专利诉讼期间造成的某些延迟,这种增加可以减少或消除。
如果我们或我们的许可方没有为我们的候选产品获得专利期限延长,我们的业务可能会受到严重损害。
根据我们候选产品的FDA上市批准的时间、持续时间和细节(如果有),我们的一个或多个美国专利 可能有资格根据1984年的《药品价格竞争和专利期限恢复法》或《哈奇-瓦克斯曼修正案》获得有限的专利期恢复。Hatch-Waxman修正案允许最长五年的专利恢复期限,作为对产品开发和FDA监管审查过程中失去的专利期限的补偿。每个FDA批准的产品最多可以延长一项专利,作为FDA监管审查过程中失去的专利期的补偿。专利期限延长不得超过自产品批准之日起共计14年的剩余专利期,只有涉及经批准的 药品、其使用方法或制造方法的权利要求方可延长。在监管机构批准我们的候选产品后,专利期限延长也可能在 某些外国国家/地区可用。但是,我们或我们的许可人可能无法获得延期 ,例如,未能在适用的截止日期内申请、未能在相关专利到期前申请或未能满足适用的要求。此外,适用的时间段或提供的专利保护范围可能比我们要求的少。如果我们或我们的许可方无法获得专利期限的延长或恢复,或者任何此类延长的期限 低于我们的要求,我们的竞争对手可能会在我们的专利到期后获得竞争产品的批准,我们的收入可能会 大幅减少。此外,如果发生这种情况,我们的竞争对手可能会参考我们的临床和临床前数据,利用我们在开发和临床试验方面的投资,比其他情况下更早推出他们的产品。如果我们没有足够的专利期来保护我们的产品,我们的业务和经营结果将受到不利影响。
我们 可能无法在全球范围内保护我们的知识产权。
尽管我们在美国和某些其他国家/地区有未获许可的待决专利申请,但在全球所有国家/地区提交、起诉和保护专利的费用将高得令人望而却步,而且我们在美国以外的一些国家/地区的知识产权可能没有美国的知识产权广泛。此外,一些外国的法律,特别是某些发展中国家的法律,对知识产权的保护程度不如美国的联邦法律和州法律。因此,我们可能无法阻止第三方在美国以外的所有国家/地区实施我们的许可内发明,也无法阻止第三方在美国或其他司法管辖区销售或进口使用我们许可内的发明制造的产品。竞争对手可以在我们未获得专利保护的司法管辖区使用我们的许可内技术来开发自己的产品,此外,还可以将其他侵权产品出口到我们或我们的许可方拥有专利保护的地区,但执法力度不如美国。这些产品可能会与我们的候选产品竞争,而我们或我们的许可方专利或其他知识产权可能不能有效或不足以阻止他们竞争。
许多公司在保护和捍卫外国司法管辖区的知识产权方面遇到了严重的问题。 许多国家/地区的法律制度不支持强制执行专利、商业秘密和其他知识产权保护, 这可能会使我们在这些司法管辖区很难阻止侵犯或挪用我们或我们许可人的专利或其他知识产权,或以侵犯我们的专有权利的方式营销竞争产品。在外国司法管辖区强制执行我们或我们许可人的专利权的诉讼程序 可能会导致巨大的成本,并将我们的努力和注意力从我们业务的其他方面转移出去,可能会使我们或我们的许可人的专利面临无效、无法强制执行、 或被狭义解释的风险,我们或我们的许可人的专利申请有可能无法发放,并可能引发第三方对我们提出索赔。我们或我们的许可人可能不会在我们或我们的许可人发起的任何诉讼中获胜,所判给的损害赔偿或其他 补救措施(如果有)可能没有商业意义。同样,如果我们的商业秘密在外国司法管辖区泄露, 世界各地的竞争对手可能会访问我们的专有信息,而我们可能没有令人满意的追索权。这样的披露可能会对我们的业务产生实质性的不利影响。因此,我们或我们的许可人在全球范围内强制执行我们的知识产权的努力可能不足以从我们开发或许可的知识产权中获得显著的商业优势。
| 51 |
此外,包括中国和印度在内的某些国家有强制许可法,根据这些法律,专利权人可能会被强制 向第三方授予许可。在这些国家/地区,如果专利被侵犯,或者如果我们或我们的许可人被迫向第三方授予许可,我们和我们的许可人可能会获得有限的补救措施,这可能会大幅降低这些专利的价值。此外, 许多国家限制专利对政府机构或政府承包商的可执行性。在这些国家,专利所有者的补救措施可能有限,这可能会大幅降低此类专利的价值。如果我们或我们的许可人被迫向第三方授予与我们业务相关的任何专利的许可,我们的竞争地位可能会受到损害,我们的业务、财务状况、运营结果和前景可能会受到不利影响。
由于在某些外国司法管辖区进行诉讼的费用和不确定性,我们可能会得出结论,即使第三方侵犯了我们已发布的专利、由于我们未决的或未来的专利申请或其他知识产权而可能发布的任何专利、提起和执行此类索赔或诉讼的风险调整成本(通常持续数年)、 可能太高或不符合我们公司或我们股东的最佳利益,或者 针对某些第三方强制执行我们的知识产权是不切实际或不可取的。我们的竞争对手或其他第三方可能比我们更有效地承担复杂的专利诉讼或诉讼的费用,因为他们有更多的财力和更成熟的 和发达的知识产权组合。在这种情况下,我们可能会决定,此类诉讼的金钱成本以及我们管理层和科学人员注意力的转移可能会超过我们从诉讼中获得的任何好处,并且 更谨慎的做法是简单地监控情况或发起或寻求其他非诉讼诉讼或解决方案。 此外,与诉讼相关的不确定性可能会损害我们筹集必要资金的能力,以继续我们的临床试验、继续我们的内部研究计划、获得许可所需的技术或其他候选产品,或建立合作伙伴关系,以帮助我们将候选产品推向市场。
获得和维护我们的专利保护取决于遵守法规和政府专利机构提出的各种程序、文件、费用支付和其他要求 ,如果不符合这些要求,我们的专利保护可能会减少或取消 。
定期 专利和/或申请的维护费、续期费、年金费和各种其他政府费用将在我们的专利和/或申请的有效期内的不同时间点支付给美国专利商标局和各个外国专利局。我们有系统提醒我们支付这些费用,我们依赖第三方在到期时支付这些费用。此外,美国专利商标局和各种外国专利局要求在专利申请过程中遵守一些程序、文件、费用支付和其他类似规定 。我们聘请信誉良好的律师事务所和其他专业人员来帮助我们遵守,在许多情况下,可以通过支付滞纳金或根据适用于特定司法管辖区的规则通过其他方式纠正疏忽。但是,在某些情况下,不遵守规定可能会导致专利或专利申请被放弃或失效,从而导致相关司法管辖区的专利权部分或全部丧失。如果发生这样的事件,可能会对我们的业务产生实质性的不利影响。
如果我们无法保护我们的商业秘密的机密性,我们的业务和竞争地位将受到损害。
除了其他类型的知识产权提供的保护外,我们还依靠保护我们的商业秘密,包括 非专利技术诀窍、技术和其他专有信息来维持我们的竞争地位。尽管我们已采取步骤 保护我们的商业秘密和非专利专有技术,包括与第三方签订保密协议(包括但不限于承包商、合作者和外部科学顾问),以及与员工、顾问、许可人和顾问签订保密信息和发明协议,但我们不能保证所有这些协议都已正式执行, 并且这些各方中的任何一方都可能违反协议并泄露我们的专有信息,包括我们的商业秘密,我们可能 无法针对此类违规行为获得足够的补救措施。我们要求我们的员工签订书面保密协议 ,向我们分配他们在受雇过程中构思、简化为实践或发展的任何类型的发明、开发、创造性作品和有用的想法。此外,我们要求我们的第三方承包商签订书面的 保密协议,要求第三方在规定的时间内不得以任何方式或出于与其义务相关的必要和/或适当以外的任何 目的披露我们的某些机密信息,但受某些 排除的限制。执行一方非法披露或挪用商业秘密的主张是困难、昂贵和耗时的, 结果不可预测。此外,美国国内外的一些法院不太愿意或不愿意保护商业秘密。我们可能需要与我们当前和未来的业务合作伙伴、合作者、承包商和位于商业机密被窃取风险较高的国家/地区的其他人共享我们的专有信息,包括商业秘密,包括通过私人或外国行为者的直接入侵,以及与国家行为者有关联或由国家行为者控制的行为。
| 52 |
此外, 第三方仍可能获取此信息或可能独立获取此信息或类似信息,我们无权阻止他们使用该技术或信息与我们竞争。如果发生任何此类事件,或者如果我们失去了对我们商业秘密的保护 ,这些信息的价值可能会大大降低,我们的竞争地位将受到损害。如果我们或我们的 许可人在专利发布之前没有申请专利保护,或者如果我们不能以其他方式对我们的专有技术和其他机密信息保密,那么我们获得专利保护或保护我们的商业秘密的能力可能会受到威胁。
我们 可能会受到以下指控:我们错误地从竞争对手那里雇佣了一名员工,或者我们或我们的员工错误地使用了 ,或者泄露了他们前雇主的所谓机密信息或商业秘密。
我们 可能会受到员工、顾问或独立承包商不当使用或泄露第三方机密信息的索赔 。
由于 在生物制药行业很常见,除了我们的员工外,我们还聘请顾问来帮助我们开发我们的候选产品。这些顾问中的许多人以及我们的许多员工以前受雇于或可能曾为其他生物制药公司提供或目前可能正在为其提供咨询服务,包括我们的竞争对手 或潜在竞争对手。我们可能会受到以下索赔的影响:我们、我们的员工、独立承包商或顾问无意中 或以其他方式使用或泄露了其前雇主或其前客户或现任客户的商业秘密或其他专有信息。 可能需要提起诉讼来抗辩这些索赔。如果我们未能为任何此类索赔辩护,除了支付金钱损失外, 我们可能会失去宝贵的知识产权或人员,这可能会对我们的业务造成不利影响。即使我们成功地对这些索赔进行了辩护,诉讼也可能导致巨额成本,并分散我们的管理团队和其他 员工的注意力。
如果我们的商标和商号得不到充分保护,我们可能无法在我们感兴趣的市场建立知名度 ,我们的业务可能会受到不利影响。
我们当前或未来的商标或商号可能会受到质疑、侵犯、规避,或被宣布为通用商标或描述性商标,或被确定为侵犯了其他商标。我们可能无法保护我们对这些商标和商品名称的权利,或者可能被迫停止 使用这些名称,我们需要这些名称才能在我们感兴趣的市场中获得潜在合作伙伴或客户的名称认可。在商标注册过程中,我们可能会收到美国专利商标局或其他外国司法管辖区对我们的申请的拒绝。尽管我们 将有机会回应这些拒绝,但我们可能无法克服这些拒绝。此外,在美国专利商标局和许多外国司法管辖区的类似机构中,第三方有机会反对未决的商标申请,并 寻求注销注册商标。可能会对我们的商标提起反对或取消诉讼,而我们的商标可能无法继续存在。如果我们不能根据我们的商标和商品名称建立名称识别,我们可能无法 有效竞争,我们的业务可能会受到不利影响。我们可能会将我们的商标和商品名称授权给第三方,例如分销商。尽管这些许可协议可能会为如何使用我们的商标和商号提供指导原则,但被许可人违反这些协议或滥用我们的商标和商号可能会危及我们的权利或削弱与我们的商标和商号相关的商誉。
| 53 |
此外, 我们建议在美国的候选产品中使用的任何名称都可能需要FDA批准,无论我们是否已将其注册或申请注册为商标。欧洲也有类似的要求。FDA通常对建议的产品名称进行审查,包括评估可能与其他产品名称混淆的可能性。如果FDA(或外国司法管辖区内的同等管理机构)反对我们建议的任何专有产品名称,我们可能需要花费大量额外的 资源,以努力确定符合适用商标法资格、不侵犯第三方现有权利并为FDA接受的合适替代名称。此外,在许多国家,拥有和维护商标注册 可能不能针对高级商标所有人随后提出的侵权索赔提供充分的辩护。有时,竞争对手 或其他第三方可能采用与我们类似的商号或商标,从而阻碍我们建立品牌标识的能力,并可能导致市场混乱。此外,其他注册商标或商标的所有者 可能会提出商号或商标侵权索赔,这些商标或商标包含我们的注册或未注册商标或商号的变体。 如果我们主张商标侵权索赔,法院可能会裁定我们主张的商标无效或不可强制执行,或者 我们主张商标侵权的一方对相关商标拥有更高的权利。在这种情况下,我们最终可能会 被迫停止使用此类商标。
与员工事务和管理我们的增长相关的风险
如果 我们无法建立销售或营销能力,或无法与第三方达成协议来销售或营销我们的候选产品 ,我们可能无法成功销售或营销获得监管部门批准的候选产品。
我们 目前没有,也从未拥有过营销或销售团队。为了将任何候选产品商业化,如果获得批准,我们 必须建立营销、销售、分销、管理和其他非技术能力,或与第三方达成安排,以便 在我们可能获得批准销售或营销我们候选产品的每个地区执行这些服务。我们可能无法 成功完成这些必需的任务。
建立一个具有技术专业知识和支持分销能力的内部销售或营销团队来将我们的候选产品商业化将是一项昂贵且耗时的工作,并且需要我们的管理人员投入大量精力进行管理。如果我们没有与第三方达成协议,代表我们提供此类 服务,则在开发我们的内部销售、营销和分销能力方面的任何失败或延迟 都可能对我们获得市场批准的任何候选产品的商业化产生不利影响。或者,如果我们选择在全球范围内或在逐个地区的基础上与拥有直接销售队伍和建立分销系统的第三方 合作,以增强我们自己的销售队伍和分销系统 或代替我们自己的销售队伍和分销系统,我们将被要求与这些第三方就拟议的合作进行谈判并达成协议。如果我们无法在需要时以可接受的条款或根本无法达成此类安排,我们可能无法成功地将任何获得监管部门批准的候选产品商业化,或者任何此类 商业化可能会遇到延迟或限制。如果我们无法单独或通过与一个或多个第三方合作成功地将我们批准的候选产品成功商业化,我们未来的产品收入将受到影响,我们可能会遭受重大的额外损失。
我们的成功在很大程度上取决于我们吸引和留住高技能高管和员工的能力。
要取得成功,我们必须招聘、留住、管理和激励合格的临床、科学、技术和管理人员,我们面临着对经验丰富的人员的激烈竞争。我们高度依赖管理层、科研人员和医务人员的主要成员。如果我们不能成功地吸引和留住合格的人才,特别是在管理层,这可能会对我们执行业务计划的能力产生不利影响,并损害我们的经营业绩。特别是,如果我们不能及时招聘合适的接班人,失去一名或多名高管可能对我们不利。生物技术领域对合格人才的竞争非常激烈,因此,我们可能无法继续吸引和留住我们业务未来成功所需的合格人员 。我们未来可能难以吸引有经验的人员到我们的公司 ,并且可能需要花费大量的财务资源来招聘和留住员工。
与我们竞争人才的许多其他生物技术公司 拥有比我们更多的财务和其他资源、不同的 风险状况和更长的行业历史。它们还可能为职业发展提供更多样化的机会和更好的前景 。这些特点中的一些可能比我们所提供的更吸引高素质的应聘者。如果我们不能继续吸引和留住高素质的人才,我们发现、开发和商业化候选产品的速度和成功率将受到限制,我们成功发展业务的潜力将受到损害。
| 54 |
为了成功实施我们的计划和战略,我们需要扩大组织的规模,我们在管理这种增长时可能会遇到困难。
截至2023年12月31日,我们有10名全职员工。为了成功实施我们的开发和商业化计划和战略,我们预计需要更多的管理、运营、销售、营销、财务和其他人员。未来的增长 将使管理层成员承担更多的重大责任,包括:
| ● | 确定、招聘、整合、维护和激励更多员工; |
| ● | 有效管理我们的内部开发工作,包括临床、FDA、EMA和其他类似的外国监管机构对我们的候选产品和我们开发的任何其他候选产品的审查流程,在履行我们可能对承包商和其他第三方承担的任何合同义务的同时,和 |
| ● | 改进我们的运营、财务和管理控制、报告系统和程序。 |
我们未来的财务业绩以及我们成功开发当前候选产品 并将其商业化的能力以及我们可能开发的任何其他候选产品将在一定程度上取决于我们有效管理任何未来增长的能力,我们的 管理层可能还必须将不成比例的注意力从日常活动中转移出来,以便投入大量的 时间来管理这些增长活动。
我们 目前和在可预见的未来将继续主要依靠某些独立组织、顾问和顾问来提供某些服务,包括临床开发和制造的关键方面。我们不能向您保证, 在需要时,独立组织、顾问和顾问的服务将继续及时提供给我们,或者我们可以找到合格的替代者。此外,如果我们无法有效管理我们的外包活动,或者如果第三方服务提供商提供的服务的质量或准确性因任何原因而受到影响,我们的临床试验可能会延长、 推迟或终止,并且我们可能无法获得任何当前或未来候选产品的营销批准或以其他方式推进我们的业务 。我们不能向您保证,我们将能够管理我们现有的第三方服务提供商,或以经济合理的条款找到其他称职的 外部承包商和顾问,或者根本不能。
如果
我们无法通过招聘新员工和/或聘用更多第三方服务提供商来有效地扩展我们的组织,
我们可能无法成功执行进一步开发和商业化我们的候选产品和任何未来候选产品所需的任务,因此可能无法实现我们的研究、开发和商业化目标。
与本次发行和我们普通股所有权相关的风险
直接上市流程不同于在公司承诺基础上承销的首次公开募股。
此 不是承销的首次公开发行普通股。我们普通股在纳斯达克的上市与承销的首次公开募股有几个重要方面的不同,包括但不限于以下几个方面:
| ● | 没有承销商在固定承诺的基础上受聘。因此,在纳斯达克开盘交易 之前,将不会有传统的询价流程,也不会有承销商最初向公众出售股票的 价格,以帮助提供关于纳斯达克开盘交易的有效和充分的 价格发现。因此,在我们的普通股在纳斯达克开盘前和开盘时提交的买入和卖出订单 将不会 获得已公布的价格区间或承销商最初向公众出售股票的价格的通知,就像首次公开募股(IPO)中的情况一样, 是以坚定承诺为基础承销的。此外,将不会有承销商承担与首次转售我们普通股 股票相关的风险。在以确定承诺为基础承销的首次公开募股中,承销商 可以从事相当于承销商 购买额外股份的选择权的股票的“备兑”卖空。要平仓备兑空头头寸,承销商应在公开市场购买股票,或行使承销商购买额外股票的选择权。在确定平仓的股票来源时, 承销商通常会特别考虑公开市场上可购买的股票价格与他们通过 承销商购买额外股票的选择权购买股票的价格进行比较。公开市场买入以回补空头头寸,以及承销商可能为其 自己的账户进行的其他买入,可能起到防止股价下跌的作用。 鉴于不会有承销商认购额外股份的选择权,也没有承销商从事稳定交易的行为,在紧随上市之后的一段时间内,我们普通股的公开价格可能会有更大的波动。 另见“-我们的普通股没有以前的公开市场。活跃的交易市场可能不会发展或继续保持流动性,我们普通股的市场价格可能会波动。.” |
| 55 |
| ● | 没有固定数量的普通股可供出售。因此, 不能保证任何注册股东或其他现有股东会出售其任何或所有普通股,而且我们在纳斯达克上的普通股最初可能出现供应不足或需求 。或者,我们可能有大量登记的 股东或其他现有股东选择在短期内出售其普通股,导致我们的普通股供过于求,这可能会对我们的普通股在纳斯达克上市后的 公开价格产生不利影响。 |
| ● | 我们的注册股东或其他现有股东均未 签订合同锁定协议或其他合同转让限制。在承销公司承诺的首次公开募股(IPO)中,发行人的高管、董事、以及其大多数其他股东与承销商签订为期180天的合同锁定安排,以帮助在首次公开募股后立即促进有序交易。 因此,我们的任何股东,包括拥有我们普通股的董事和高级管理人员以及其他重要股东,可以在任何时间出售其任何或全部普通股(受适用法律的任何限制),包括上市后立即出售。 如果在我们 上市后的短时间内大量出售我们的普通股,可能会导致我们的普通股在市场上供过于求,这可能会对我们的普通股的公开价格产生不利的 影响。 |
| ● | 我们 不会在纳斯达克开盘 之前与承销商进行传统的路演。取而代之的是,我们打算举办投资者日,并参与其他一些投资者教育会议。在投资者日之前,我们将通过财经新闻媒体宣布这一天的日期 ,方式与典型的企业向投资者宣传的方式一致 。我们将为此次投资者日准备一个电子演示文稿,它将 包含类似于传统路演演示文稿的内容,并在网站上公开提供一个版本的 演示文稿,不受限制。不能保证 投资者日和其他投资者教育会议对投资者教育的影响将与与公司承诺承销的首次公开募股(IPO)相关的传统“路演”一样。因此,我们的普通股在上市后可能没有有效的 价格发现或投资者的足够需求 ,这可能导致我们普通股的公开价格波动更大。 |
这种与公司承诺承销的首次公开募股的差异可能会导致我们普通股的交易价格波动 和交易量不确定,这可能会对您出售您可能购买的任何普通股的能力产生不利影响。
我们的 普通股目前没有公开市场。活跃的交易市场可能不会发展或继续保持流动性,我们普通股的市场价格可能会波动。
我们 希望我们的普通股在纳斯达克上市交易。在纳斯达克上市之前,我们的任何证券都没有公开市场 ,上市后我们普通股的活跃市场可能无法发展或持续,这可能会压低我们普通股的市场价格,并可能影响我们股东出售我们普通股的能力。在没有活跃的公开交易市场的情况下,投资者可能无法变现他们对我们普通股的投资。不活跃的市场 还可能削弱我们通过出售普通股筹集资金的能力、我们通过股权奖励激励员工的能力 以及我们以普通股作为对价收购其他公司、产品或技术的能力。
| 56 |
In addition, we cannot predict the prices at which our common stock may trade on Nasdaq following the listing of our common stock, and the market price of our common stock may fluctuate significantly in response to various factors, some of which are beyond our control. In particular, as this listing is taking place through a novel process that is not a firm-commitment underwritten initial public offering, there will be no traditional book building process and no price at which traditional underwriters initially sold shares to the public to help inform efficient price discovery with respect to the opening trades on Nasdaq. On the day that our shares of common stock are initially listed on Nasdaq, Nasdaq will begin accepting, but not executing, pre-opening buy and sell orders and will begin to continuously generate the indicative Current Reference Price on the basis of such accepted orders. The Current Reference Price is calculated each second and, during a 10-minute “Display Only” period, is disseminated, along with other indicative imbalance information, to market participants by Nasdaq on its NOII and BookViewer tools. Following the “Display Only” period, a “Pre-Launch” period begins, during which the Advisor, in its capacity as our financial advisor, must notify Nasdaq that our shares are “ready to trade.” Once the Advisor has notified Nasdaq that our shares of common stock are ready to trade, Nasdaq will confirm the Current Reference Price for our shares of common stock, in accordance with Nasdaq rules. If the Advisor then approves proceeding at the Current Reference Price, the applicable orders that have been entered will be executed at such price and regular trading of shares of our common stock on Nasdaq will commence, subject to Nasdaq conducting validation checks in accordance with Nasdaq rules. The Advisor will determine when our shares of common stock are ready to trade and approve proceeding at the Current Reference Price primarily based on considerations of volume, timing and price. In particular, the Advisor will determine, based primarily on pre-opening buy and sell orders, when a reasonable amount of volume will cross on the opening trade such that sufficient price discovery has been made to open trading at the Current Reference Price. If the Advisor does not approve proceeding at the Current Reference Price (for example, due to the absence of adequate preopening buy and sell interest), the Advisor will request that Nasdaq delay the open until such a time that sufficient price discovery has been made to ensure a reasonable amount of volume crosses on the opening trade. For more information, see “配送计划.”
此外, 在开盘交易之前,承销商最初向公众出售普通股的价格将不会像 在公司承诺承销的首次公开发行中那样。没有预先确定的首次公开发行价格可能会影响纳斯达克从各经纪自营商收集的买卖订单范围。因此,在纳斯达克上市后,我们普通股的公开 价格可能比公司承诺承销的首次公开发行更不稳定,并可能大幅 迅速下跌。
此外, 由于我们在纳斯达克的上市流程新颖,纳斯达克确保遵守其初始上市标准的规则(例如, 要求估值或其他令人信服的价值证据的规则)未经测试。在我们的普通股没有事先活跃的公开交易市场 的情况下,如果我们的普通股价格或我们的市值低于纳斯达克资格 标准所要求的价格或市值,我们可能无法满足正在进行的上市标准,并可能被要求摘牌。
此外,由于我们的上市流程新颖,个人投资者(零售或其他)可能对设定我们的普通股在纳斯达克的 开盘公开价格和随后的公开价格具有更大的影响力,并且可能比 公司承诺承销的首次公开发行的典型情况更多地参与我们的首次交易。这些因素可能导致我们的普通股 公开价格高于其他投资者(如机构投资者)愿意支付,这可能会导致我们的普通股交易价格波动,如果我们的普通股价格在上市后大幅上涨,并且机构投资者认为我们的普通股价值低于散户投资者,在这种情况下,我们的普通股价格可能会随着时间的推移而下降。此外,如果我们普通股的公开价格高于投资者认为对我们普通股合理的水平, 一些投资者可能会在交易开始后试图做空我们的普通股,这将对我们普通股的公开价格 造成额外的下行压力。在零售投资者中缺乏消费者意识的情况下,这种消费者意识的缺乏 可能会降低我们普通股的价值,并导致我们普通股交易价格的波动。
| 57 |
上市后我们普通股的 公开价格也可能会因本招股说明书中所述的风险因素和我们无法控制的其他因素而大幅波动,这些因素包括:
| ● | 更改 在我们经营的行业中; |
| ● | 我们的经营业绩和竞争对手的总体业绩存在差异 ; |
| ● | 本公司季度或年度经营业绩的实际或预期波动; |
| ● | 出版物 证券分析师关于我们或我们的竞争对手或我们的行业的研究报告; |
| ● | 该 公众对我们的新闻稿、其他公告和文件的反应 与SEC; |
| ● | 我们的 我们的竞争对手未能满足分析师的预测或指导 我们或我们的竞争对手可能向市场提供的信息; |
| ● | 关键人员增减离任; |
| ● | 影响我们业务的法律法规变化 ; |
| ● | 启动或参与涉及我们的诉讼; |
| ● | 我们资本结构的变化,如未来发行证券或产生额外的 债务; |
| ● | 该 可公开出售的普通股数量;以及 |
| ● | 一般 经济和政治条件,如经济衰退、利率、燃料价格、外国 货币波动、国际关税、社会、政治和经济风险和行为 战争或恐怖主义。 |
此外,证券交易所经历了价格和交易量波动,这些波动已经并将继续影响许多公司的股票证券的市场价格 。许多公司的股票价格波动的方式往往与这些公司的经营业绩无关 。由于上述供求力量,在我们的普通股在纳斯达克上市 后不久,这些波动在我们的普通股交易市场上可能会更加明显。过去,在市场波动时期之后,股东提起了 证券集体诉讼。如果我们卷入证券诉讼, 可能会使我们承担巨额成本,转移资源和管理层对我们业务的注意力,并损害我们的业务、经营业绩和财务状况。
我们的注册股东和其他现有股东未来 出售普通股可能会导致我们的股价下跌。
We currently expect our common stock to be listed and traded on Nasdaq. Prior to listing on Nasdaq, there has been no public market for our common stock and there has not been a sustained history of trading in our common stock in “over-the-counter” markets. While our common stock may be sold after our listing on Nasdaq by the Registered Stockholders pursuant to this prospectus or by our other existing stockholders in accordance with Rule 144 under the Securities Act, unlike a firm-commitment underwritten initial public offering, there can be no assurance that any Registered Stockholders or other existing stockholders will sell any of their shares of common stock and there may initially be a lack of supply of, or demand for, common stock on Nasdaq. As described herein, certain shares of our common stock outstanding as of the date hereof will be registered under this registration statement. There can be no assurance that the Registered Stockholders and other existing stockholders will not sell all of their shares of common stock, resulting in an oversupply of our common stock on Nasdaq. In the case of a lack of supply of our common stock, the trading price of our common stock may rise to an unsustainable level. Further, institutional investors may be discouraged from purchasing our common stock if they are unable to purchase a block of our common stock in the open market due to a potential unwillingness of our existing stockholders to sell a sufficient amount of common stock at the price offered by such institutional investors and the greater influence individual investors have in setting the trading price. If institutional investors are unable to purchase our common stock, the market for our common stock may be more volatile without the influence of long-term institutional investors holding significant amounts of our common stock. In the case of a lack of market demand for our common stock, the trading price of our common stock could decline significantly and rapidly after our listing. Therefore, an active, liquid and orderly trading market for our common stock may not initially develop or be sustained, which could significantly depress the public price of our common stock and/or result in significant volatility, which could affect your ability to sell your shares of common stock.
| 58 |
直接上市后,我们将拥有2,500股具有超级投票权的C系列优先股。
截至本协议日期,我们的 股本包括有表决权的普通股(我们有时称之为“普通股”)、 无表决权的普通股、A系列优先股、B系列优先股和B-1系列优先股。我们的董事会和 股东已分别批准创建和发行总计2,500(反向股票分割生效后) C系列优先股,所有C系列优先股将发行给我们的创始人兼首席执行官Pete O 'Heeron, 因此,在直接上市之前,我们的股本将由普通股组成,无投票权普通股、B系列优先股、 B-1系列优先股和C系列优先股。
在直接上市方面,(I)我们当时所有未偿还的A系列优先股,全部由FibroGenesis持有, 将自动注销,而不向其持有人或向其持有人支付额外代价,(Ii)我们当时尚未偿还的 无投票权普通股将自动转换为 普通股,而无需向其持有人或向其持有人支付额外代价,一对一的基础上,(Iii)我们所有当时尚未发行的B系列优先股和我们当时尚未发行的所有B系列B-1优先股将按一对一的基础自动转换为普通股,而无需向其持有人或向其持有人支付额外代价;及(Iv)我们所有当时尚未发行的C系列优先股将保持C系列优先股,这样,在直接上市后,我们的已发行和已发行股本将立即包括普通股和C系列优先股 。
C系列优先股(I)没有股息权,(Ii)在从初始持有人转让时转换为普通股,(Iii) 在我们的清算、解散或清盘时,C系列优先股的清算优先权为每股18.00美元(在任何股票拆分、合并或其他类似资本重组的情况下需进行适当调整),以及(Iv)直接上市时,C系列优先股的每股股份将有权获得 13,000投票权(直接上市前,除特拉华州法律要求外,C系列优先股将无权在任何事项上投票 ,在这种必要情况下,C系列优先股每股将有一票投票权)。
C系列优先股将受制于Pete O 'Heeron(所有C系列优先股 的持有人)以我们董事会为受益人签发的不可撤销的委托书,只要 C系列优先股尚未发行,以董事会全权酌情决定的任何方式,就C系列优先股有权投票的所有事项对所有C系列优先股进行投票;但是, 未经Pete O 'Heeron的书面同意,该不可撤销的代理人不得允许我们的董事会就任何修订提案对 系列优先股进行投票,删除或放弃Pete O 'Heeron在我们修订和重述的公司注册证书中规定的关于C系列优先股的任何权利。鉴于 与C系列优先股相关的优先投票权,不可撤销的委托书旨在确保此类优先投票权的使用符合我们的最佳利益,并避免或减轻Pete O 'Heeron作为个人股东员工未来可能出现的冲突。
参见 “股本说明-C系列优先股”有关我们C系列优先 股票的更多信息。
除了对我们普通股的投票权和价值的稀释作用外,我们股本的上述结构可能使 我们的普通股不符合纳入某些证券市场指数的条件,从而对我们普通股或其他证券的价格和流动性 以及公众情绪产生不利影响。我们的 C系列优先股的存在及其相关的投票权,无论是单独还是与我们经修订和重述的公司注册证书的某些其他规定( 如交错董事会的要求)一起,也可能会延迟、阻止或防止我们的控制权发生变化, 或使我们的管理层更难被免职。
直接上市后,我们将成为纳斯达克股票市场规则意义上的“受控公司”,因为我们的内部人士将实益拥有我们已发行投票证券的50%以上投票权。
本次发行完成后,我们的创始人 兼首席执行官Pete O 'Heeron将共同实益拥有我们已发行 有投票权证券的约59%投票权,我们将成为纳斯达克股票市场 有限责任公司上市规则意义上的“受控公司”。我们可以依赖公司治理规则的某些豁免,包括豁免我们的 董事会多数成员必须是独立董事的规则。虽然我们目前不打算依赖纳斯达克上市规则下的“受控公司” 豁免,但我们可以选择在未来依赖此豁免。如果我们选择依赖 “受控公司”豁免,我们董事会的大多数成员可能不是独立董事, 我们的提名委员会、公司治理委员会和薪酬委员会可能不完全由独立董事组成。我们作为受控公司的地位 可能会导致我们的普通股对某些投资者的吸引力降低,或以其他方式损害我们的交易价格 。因此,您将无法获得向受这些公司 治理要求约束的公司的股东提供的相同保护。
您 可能会被未来发行的优先股或与我们的激励计划、收购 或其他有关的额外普通股稀释;未来在公开市场出售此类股票或预期此类出售可能会发生,可能会降低我们的股票 价格。
在注册声明(本招股说明书构成其一部分)生效之前,我们将采用经修订和 重述的公司注册证书,该证书将授权我们发行普通股股份以及与我们的普通股有关的期权、权利、认股权证和增值权,其代价和条款及条件由我们的董事会 全权决定。我们可以在未来发行大量普通股,用于投资或收购。 这些发行中的任何一项都可能稀释我们现有的股东,而且这种稀释可能是显著的。此外,这种稀释可能 对我们普通股的市场价格产生重大不利影响。
未来发行具有投票权的优先股可能会对 普通股股东的投票权产生不利影响,如果优先股与普通股作为 单一类别一起投票,则会稀释普通股的投票权,或者赋予任何此类优先股的持有人阻止其拥有单独类别投票的行动的权利 ,即使我们的普通股持有人同意了这一行动。
未来发行的优先股(具有股息或转换权、清算优先权或其他有利于优先股持有人的经济条款)可能会降低普通股的投资吸引力,从而对我们普通股的市场价格产生不利影响。 例如,普通股投资者可能不希望以高于一系列可转换优先股转换价格的价格购买普通股,因为优先股持有人实际上有权以较低的转换价格购买普通股,从而导致普通股持有人的经济稀释。
由于
我们目前没有为普通股支付现金股息的计划,除非您以高于您购买价格的价格出售您的普通股,否则您可能无法获得任何投资回报。
我们 目前打算保留所有可用资金和任何未来收益,为我们业务的发展、商业化和增长提供资金,因此,我们预计在可预见的未来不会宣布或支付任何现金股息给我们的普通股。未来宣布派息的任何决定将由我们的董事会酌情决定,并将取决于我们的财务状况、经营业绩、 资本要求、一般业务状况以及我们董事会可能认为相关的其他因素。我们未来为普通股支付现金股息的能力也可能受到任何未来债务证券或信贷安排条款的限制. 因此,您在本次发行中购买的普通股的资本增值(如果有的话)将是您在可预见的未来的唯一收益来源 。
我们 是一家新兴成长型公司和一家较小的报告公司,适用于新兴成长型公司和较小报告公司的披露要求降低可能会降低我们的普通股对投资者的吸引力。
根据《就业法案》的定义,我们 是一家新兴成长型公司。只要我们继续是一家新兴成长型公司, 我们就可以利用适用于其他非新兴成长型公司的上市公司的各种报告要求的某些豁免和减免,包括(I)不需要遵守萨班斯-奥克斯利法案 404节的审计师认证要求,(Ii)可以选择推迟采用某些新的或修订的财务会计准则, (Iii)减少了本招股说明书以及我们的定期报告和委托书中有关高管薪酬的披露义务 和(Iv)免除了就高管薪酬和股东批准之前未获批准的任何金降落伞付款进行不具约束力的咨询投票的要求。我们可能会利用这些豁免,直到我们不再是一家新兴成长型公司。因此,此处包含的信息可能与您从您持有股票的其他 上市公司收到的信息不同。此外,根据《就业法案》第107条,我们选择利用延长的过渡期来遵守新的或修订的会计准则,直到这些准则适用于私营 公司。因此,我们的经营业绩和财务报表可能无法与采用新会计准则或修订后的会计准则的其他公司的经营业绩和财务报表 相比较。
| 59 |
我们 将一直是一家新兴的成长型公司,直到(I)2028年12月31日,(Ii)我们 的年度总收入至少为12.35亿美元的财年的最后一天,(Iii)我们被视为《交易法》第12b-2条规则所定义的“大型加速申请者”的财年的最后一天,如果非关联公司持有的普通股市值在该年度第二财季的最后一个营业日为7.00亿美元或以上,或(Iv)我们在前三年期间发行了超过10亿美元的不可转换债务证券的日期 ,就会发生这种情况。
我们 也是《交易法》中定义的“小型报告公司”。即使我们不再是一家新兴成长型公司,我们也可能继续成为一家规模较小的报告公司。我们可能会利用某些适用于较小 报告公司的按比例披露,直到确定非关联公司持有的有表决权和无表决权普通股 在我们第二财政季度的最后一个工作日达到2.5亿美元或更多之后的财政年度,或者在最近完成的财政年度中,我们的年收入低于1亿美元 ,并且非关联公司持有的有表决权和无表决权普通股为7亿美元或更多 在我们第二财政季度的最后一个工作日测量。
由于上述原因, 一些投资者可能会发现我们的普通股吸引力下降,这可能会导致我们普通股的交易市场不那么活跃,我们的股价波动更大。
我们的 管理层和主要股东拥有我们相当大比例的股票,并将能够对 经股东批准的事项施加重大控制。
截至2023年12月31日,我们的高管、董事和5%或以上的股东以及他们各自的关联公司, 在转换后的基础上总共实益拥有我们约19%的已发行普通股。如果同一集团在此次发行后继续拥有我们相当大比例的普通股,这些股东如果 齐心协力,将能够控制我们公司的管理和事务,以及大多数需要股东批准的事项,包括 董事选举、修改我们的组织文件以及批准任何合并、出售我们几乎所有资产或其他重大公司交易。这种所有权集中可能会阻止或阻止您或其他股东可能认为符合您或其他股东作为我们股东之一的最佳利益的主动收购提议或对我们普通股的要约。
在每种情况下,我们修订和重述的公司注册证书和章程的条款 将与注册说明书(招股说明书是其中的一部分)的有效性相关而生效,这些条款可能会推迟或阻止收购,而收购可能 不符合我们股东的最佳利益。
在每种情况下,我们修订和重述的公司注册证书和章程的条款将与注册说明书(招股说明书是其中的一部分)的有效性相关而生效,这些条款可能被视为具有反收购效果,其中包括:(I)我们的C系列优先股的存在,有权获得每股13,000票的C系列优先股,如本招股说明书中其他地方更详细地描述的那样,(Ii)一个交错的三年任期的分类董事会,(Iii)谁可以填补我们的董事会空缺,(Iv)罢免本公司董事会成员的绝对多数投票门槛,以及(V)何时及由谁召开股东特别会议,并可能延迟、推迟或阻止 收购尝试。
此外,我们修订和重述的公司注册证书将授权发行优先股股票,这些优先股将拥有由我们的董事会不时确定的权利和优惠。在通过修订和重述的公司注册证书后,我们的董事会可以不经股东批准(纳斯达克规则可能要求的除外), 发行额外的优先股,包括股息、清算、转换、投票权或其他可能对我们普通股持有人的投票权或其他权利产生不利影响的权利。 此外,我们修改和重述的公司证书将授权 发行“空白支票”优先股,我们的董事会可以使用这些优先股来实施股东权利计划 (也称为“毒丸”)。
| 60 |
在每种情况下,我们的 修改和重述的公司注册证书将在注册说明书(招股说明书是其中的一部分)生效时生效,它将在特拉华州衡平法院为我们与我们的股东之间的某些纠纷提供专属论坛,这可能会限制我们的股东获得 与我们或我们的董事、高级管理人员或员工的纠纷的有利司法论坛的能力。
在每种情况下,我们修改和重述的公司注册证书将与注册说明书(招股说明书是其中的一部分)的有效性相关而生效,该证书将规定,除非我们书面同意选择替代论坛,(I)特拉华州衡平法院(或,如果该法院没有管辖权,则为特拉华州联邦地区法院)应在法律允许的最大范围内,成为(A)代表我们提起的任何衍生诉讼或诉讼的唯一和排他性论坛,(B)任何声称违反本公司任何董事、高级职员或其他雇员对本公司或本公司股东所负受托责任的诉讼,(C)根据特拉华州《一般公司法》或DGCL、我们的公司注册证书或本公司章程的任何规定而引起的任何诉讼,或(D)任何主张受内部事务原则管辖的索赔的诉讼,及(Ii)在法律允许的最大范围内,美国联邦地区法院应是解决根据《证券法》提出的一项或多项诉因的任何申诉的独家论坛,包括针对该申诉的任何被告提出的所有诉因。根据我们计划修订和重述的公司注册证书 ,购买或以其他方式收购或持有我们普通股的任何股份的任何个人或实体将被视为已知悉并同意本段所述我们计划修订和重述的公司注册证书 中的论坛选择条款。
上述条款不会阻止根据《交易所法案》提出索赔的股东向联邦法院提出此类索赔,前提是《交易所法案》授予联邦政府对此类索赔的独家管辖权,但须遵守适用法律。
我们 相信,我们选择的法院条款可能会使我们受益,因为它提高了院长和法官在解决公司纠纷方面对特拉华州法律的应用的一致性,相对于其他论坛 更快地高效地管理案件,并保护我们免受多个法院诉讼的负担。然而,我们选择的法院条款可能会给股东在索赔过程中增加额外的 诉讼成本,并可能限制股东在司法法院提出其认为有利于与我们或我们的任何董事、高级管理人员或其他员工发生纠纷的索赔的能力,这可能会阻碍与此类索赔有关的诉讼 。此外,虽然特拉华州法院认定这种法院条款的选择在表面上是有效的,但股东仍然可以寻求在法院条款选择中指定的地点以外的地点提出索赔,而且不能保证这些其他司法管辖区的法院将强制执行此类规定。如果法院发现我们公司注册证书中选择的法院条款在诉讼中不适用或不可执行,我们可能会因在其他司法管辖区解决此类诉讼而产生额外的 费用,这可能会对我们的业务和财务状况产生不利影响。
一般风险
分析师发布的报告 ,包括与我们实际结果不同的报告中的预测,可能会对我们普通股的价格和交易量 产生不利影响。
证券 研究分析师可以建立并发布他们自己对我们公司的定期预测。这些预测可能差异很大,可能无法准确预测我们实际实现的结果。如果我们的实际结果与这些证券研究分析师的预测不符,我们普通股的价格可能会下跌。同样,如果为我们撰写报告的一位或多位分析师下调了我们的股票评级,或者发表了关于我们业务的不准确或不利的研究报告,我们的股价可能会下跌。如果其中一位或多位分析师 停止对我们的报道或未能定期发布有关我们的报告,我们的股价或交易量可能会下降。
我们的 内部计算机系统或我们的任何CRO、制造商、其他承包商、顾问、合作者或未来的潜在合作者的计算机系统可能会出现故障或遭受安全或数据隐私泄露,或以其他未经授权或不正当的方式访问、使用或销毁我们的专有或机密数据、员工数据或个人数据,这可能会导致额外的成本、收入损失、重大 负债、对我们的品牌造成损害,并对我们的运营造成实质性中断。
尽管实施了安全措施,但我们的内部计算机系统以及我们当前和未来的任何CRO和其他承包商、顾问、合作者和第三方服务提供商的计算机系统仍容易受到计算机病毒、网络安全威胁、 未经授权访问、自然灾害、恐怖主义、战争、电信和电气故障的破坏。由于用于获得对系统的未经授权访问或破坏的技术经常变化,并且通常在针对目标启动之前无法识别,因此我们 可能无法预测这些技术或实施足够的预防措施。我们还可能遇到安全漏洞,这些漏洞可能会在较长时间内无法检测到。如果发生此类事件并导致我们的运营中断,或导致 未经授权获取或访问个人身份信息或个人身份的健康信息(违反了某些隐私法律,如HIPAA和GDPR),则可能会导致我们的药物发现和开发计划以及我们的业务 业务严重中断,无论是由于我们的商业机密丢失还是其他类似的中断。联邦、州和外国政府的一些要求包括公司有义务通知个人涉及特定个人身份信息的安全漏洞,这些漏洞可能是我们或我们的供应商、承包商或与我们建立了战略关系的组织所经历的漏洞造成的。与安全漏洞相关的通知和后续行动可能会影响我们的声誉,导致我们产生大量成本,包括法律费用和补救费用。例如,已完成或未来临床试验中的临床试验数据丢失可能会导致我们的监管审批工作延迟,并显著增加我们恢复或复制丢失数据的成本 。我们的制造流程的某些部分也依赖于第三方,与他们的计算机系统相关的类似事件也可能对我们的业务产生实质性的不利影响。如果任何中断或安全漏洞 导致我们的数据丢失或损坏,或不适当地披露机密或专有信息,我们可能会面临诉讼和政府调查,我们候选产品的进一步开发和商业化可能会被推迟, 我们可能会因违反某些州、联邦和/或国际隐私和安全法律而受到巨额罚款或处罚。
我们的 保险单可能不足以补偿任何此类中断、故障或安全漏洞造成的潜在损失 。此外,我们未来可能不会以经济上合理的条款获得这种保险,或者根本不能。此外,我们的保险可能无法覆盖针对我们提出的所有索赔,并且在任何情况下都可能有很高的免赔额,而为诉讼辩护,无论其是非曲直,都可能代价高昂,并分散管理层的注意力。
我们的运营很容易受到火灾、恶劣天气条件、停电、电信故障、恐怖活动 和其他我们无法控制的事件的影响,这可能会损害我们的业务。
我们的工厂位于一个时不时会经历恶劣天气的地区。我们没有对重大龙卷风、洪水、火灾、地震、断电、恐怖活动或其他灾难对我们的业务和财务业绩的潜在后果进行系统分析,也没有针对此类灾难的恢复计划。此外,我们没有提供足够的保险来赔偿我们可能发生的业务中断造成的实际损失,我们造成的任何损失或损害都可能损害我们的业务。 任何这些业务中断的发生都可能严重损害我们的运营和财务状况,并增加我们的成本 和费用。
| 61 |
有关前瞻性陈述的警示性说明
本招股说明书包含可能涉及重大风险和不确定性的前瞻性陈述。除本招股说明书中包含的有关历史事实的陈述外,其他所有陈述,包括有关我们未来的经营结果和财务状况、 业务战略、预期产品、产品批准、研发成本、未来收入、成功的时机和可能性、未来经营的计划和目标、预期产品和前景的未来结果、管理计划和目标的陈述,均为前瞻性陈述。这些表述涉及已知和未知的风险、不确定因素和其他重要因素,可能导致我们的实际结果、业绩或成就与前瞻性表述中明示或暗示的任何未来结果、业绩或成就大不相同。
在某些情况下,您可以通过“预期”、“相信”、“考虑”、“继续”、“可能”、“估计”、“预期”、“打算”、“可能”、“计划”、“ ”、“潜在”、“预测”、“项目”、“应该”、“目标”、“意志”等术语来识别前瞻性陈述,“ 或”将“或这些术语的否定或其他类似表述,尽管并非所有前瞻性表述都包含这些词语。本招股说明书中包含的前瞻性陈述包括但不限于以下陈述:
| ● | 我们当前和未来候选产品的临床前研究和临床试验的时间、进度和结果,包括关于研究或试验和相关准备工作的开始和完成时间的声明。试验结果将在多长时间内公布,以及我们的研发计划; |
| ● | 监管提交、备案和批准的时间、范围或可能性,包括我们产品候选产品的最终监管批准; |
| ● | 我们开发和推进候选产品并成功完成临床试验的能力 ; |
| ● | 如果 被批准用于商业用途,我们对候选产品的患者群体大小的预期; |
| ● | 实施我们的业务模式和我们对业务、候选产品和技术的战略计划; |
| ● | 我们的商业化、营销和制造能力和战略; |
| ● | 如果获得批准,我们的候选产品的定价和报销; |
| ● | 我们的候选产品的市场接受度和临床效用的比率和程度,尤其是一般的细胞疗法; |
| ● | 我们 能够建立或维护协作或战略关系或获得额外的 资金; |
| ● | 我们的竞争地位; |
| ● | 我们和/或我们的许可方能够为我们的候选产品建立和维护知识产权的 保护范围; |
| ● | 与我们的竞争对手和我们的行业相关的发展情况和预测; |
| ● | 我们对费用、未来收入、资本需求和额外融资需求的估计 ; |
| ● | 我们估计我们现有的现金和现金等价物将足以为我们未来的运营费用和资本支出需求提供资金的 期间;以及 |
| ● | 法律法规的影响。 |
我们 这些前瞻性陈述主要基于我们目前对我们的业务、我们经营的行业以及我们认为可能会影响我们的业务、财务状况、运营结果和前景的财务趋势的预期和预测, 这些前瞻性陈述不是对未来业绩或发展的保证。截至本招股说明书发布之日,这些前瞻性陈述仅代表 ,可能会受到标题为 的章节中所述的一系列风险、不确定性和假设的影响。风险因素“以及在本招股说明书的其他地方。由于前瞻性陈述固有地受到风险和不确定性的影响,其中一些风险无法预测或量化,因此您不应依赖这些前瞻性陈述作为对未来事件的预测 。我们的前瞻性陈述中反映的事件和情况可能无法实现或发生,实际结果可能与前瞻性陈述中预测的结果大不相同。除适用法律另有要求外,在我们分发本招股说明书之前,我们不打算 公开更新或修改本文中包含的任何前瞻性陈述,无论是由于任何新信息、未来事件或其他原因。
此外,“我们相信”的陈述和类似的陈述反映了我们对相关主题的信念和意见。这些 陈述基于截至本招股说明书发布之日向我们提供的信息,虽然我们认为这些信息构成了此类陈述的合理基础,但此类信息可能是有限或不完整的,我们的陈述不应被解读为表明我们已对所有可能获得的相关信息进行了详尽的调查或审查。这些陈述本质上是不确定的,提醒您不要过度依赖这些陈述。
| 62 |
市场 和行业数据
本招股说明书包括有关市场和行业数据的估计。除非另有说明,否则有关我们所在行业和我们所在市场的信息,包括我们的总体预期、市场地位、市场机会和市场规模,均基于我们管理层对我们所在市场的知识和经验,以及从各种来源获得的当前可用信息,包括公开信息、行业报告和出版物、调查、我们的客户、行业和商业组织,以及我们所在市场的其他联系人。某些信息基于管理层估计, 来自第三方来源,以及我们内部研究的数据。
在提供此信息时,我们根据此类数据和其他类似来源,以及我们对我们所在市场的了解和迄今的经验,做出了我们认为合理的某些假设。虽然我们认为本招股说明书中包含的估计市场和行业数据总体上是可靠的,但此类信息本质上是不确定和不准确的。市场和行业数据 可能会发生变化,可能会受到原始数据的可用性、数据收集过程的自愿性以及此类数据的任何统计调查所固有的其他 限制。此外,由于各种因素的影响,对我们经营的市场未来表现的预测、假设和估计必然会受到不确定性和风险的影响,包括《风险因素“和”有关前瞻性陈述的注意事项“这些因素和其他因素 可能会导致结果与第三方和我们的估计中所表达的结果大不相同。因此,我们告诫您不要过度依赖此类市场和行业数据或任何其他此类估计。
本招股说明书中包含的某些统计数据、估计和预测的 来源为以下独立行业出版物或报告:
| ● | 《2022-2029年退行性腰椎病治疗药物全球市场分析、洞察与预测》 《财富商业洞察》; | |
| ● | 《全球再生药物市场2022-2029》《财富商业洞察》; | |
| ● | 《全球多发性硬化症药物市场2022-2029》《财富商业洞察》;以及 | |
| ● | 《全球伤口护理市场2022-2029》《财富商业洞察》。 |
除本招股说明书明确规定的范围外,上述来源的内容不构成本招股说明书的一部分 ,也不包含在本说明书中。
| 63 |
商标、服务标记和商标名
我们 拥有或以其他方式拥有商标的权利,包括本招股说明书中提及的商标,与我们业务的运营 一起使用。本招股说明书包括受适用知识产权法保护的我们自己的商标,以及其他实体的商标、服务标志和商标名,这些商标、服务标志和商标名是其各自所有者的财产。仅为方便起见, 本招股说明书中提及的商标、商号和服务标志可在不使用®, TM或SM 符号,但此类引用并不意味着适用的许可方不会根据适用法律在最大程度上主张其对这些商标、服务标记和商号的权利。我们不打算使用或展示其他 实体的商标、服务标记或商标名,以暗示与任何其他 实体之间的关系,或对我们的背书或赞助。
| 64 |
使用收益的
登记股东可以选择也可以不选择出售本招股说明书所涵盖的普通股。如果任何登记的 股东选择出售本招股说明书涵盖的我们普通股的股份,我们将不会从出售我们的普通股 中获得任何收益。请参阅“主要股东和注册股东。”
分红政策
我们 从未宣布或支付过普通股的现金股息。我们目前打算保留所有可用资金和任何未来收益,为我们业务的发展、商业化和增长提供资金,因此我们预计在可预见的未来不会宣布或支付任何现金 普通股股息。未来关于宣布和支付股息的任何决定(如果有)将由我们的董事会酌情决定。任何此类决定也将取决于我们的业务前景、经营业绩、财务状况、资本要求、一般业务状况以及我们董事会可能认为相关的其他因素。我们未来为普通股支付现金股息的能力也可能受到任何未来债务证券或信贷安排条款的限制。.
大写
下表列出了我们截至2023年9月30日的现金和现金等价物及资本化情况如下。
| ● | 按实际基础计算(在实施反向股票拆分和股本调整后); | |
| ● | 在 形式基础上,以实现(I)自动取消我们的A系列优先股 ,无需考虑直接上市,(Ii)自动转换, 无需任何考虑,对于我们的可转换优先股的所有流通股(即我们的B系列优先股和B-1系列优先股)和我们的无投票权普通股的所有流通股 ,在每种情况下,一对一的基础上,总计32,477,209股我们的普通股(在实施反向股票拆分后), 于2023年10月31日生效,(Iii)设立和发行2,500股我们的C系列优先股 (在实施反向股票拆分后),这将在本招股说明书构成其组成部分的登记声明生效之前 发生,以及(Iv)本公司经修订及重述的注册证书的存档及效力,以及本公司经修订及重述的章程的采纳情况,以上各项均与本招股说明书 所包含的登记声明的效力有关。 |
下表中的 股票编号已进行调整,以反映反向股票拆分和股本调整。
本 表应结合本招股说明书中其他地方的我们的财务报表和相关附注 阅读,并对其全文进行限定。
| 截至2023年9月30日 | ||||||||
| 实际 | 形式上 | |||||||
(未经审计) (以千为单位,但份额和 | ||||||||
| 现金和现金等价物 | $ | 10,766 | $ | 10,766 | ||||
| 股东权益/(亏损): | ||||||||
| 优先股,每股面值0.00001美元;共计20,000,000股授权、实际和预计股份;8,750,000股A系列优先股 已授权、已发行和已发行的实际优先股;没有A系列优先股已授权、已发行和已发行的, 预计 | — | — | ||||||
| 优先股,每股面值0.00001美元;共计20,000,000股授权、实际和预计股份;5,000,000股B系列优先股,实际;4,171,445股B系列已发行和已发行优先股,实际;没有B系列优先股,已授权、已发行和已发行,预计 | — | — | ||||||
| 优先股,每股面值0.00001美元;授权、实际和预计总股份2,000,000股;5,000,000股B-1系列优先股,实际;74,922股B-1系列已发行和已发行优先股,实际;没有B-1系列优先股,已授权、已发行和已发行,预计 | — | — | ||||||
| 优先股,每股面值0.00001美元;总授权、实际和预计总股份2,000,000股;未授权、已发行和已发行的C系列优先股,实际;2,500股C系列优先股,预计已发行和已发行 | — | — | ||||||
| 无投票权普通股,每股面值0.00001美元; 3,000,000股已授权股票和28,230,842股已发行和已发行股票,实际;没有股份已授权、已发行和已发行股票, 形式上 | 1 | — | ||||||
| 有投票权的普通股,每股面值0.00001美元;授权、实际和预计100,000,000股;没有已发行或已发行的实际股份;已发行和已发行的32,477,209股,预计 | — | 1 | ||||||
| 额外实收资本 | 25,177 | 25,177 | ||||||
| 累计赤字 | (14,640 | ) | (14,640 | ) | ||||
| 股东权益总额 | $ | 10,538 | $ | 10,538 | ||||
| 总市值 | $ | 10,538 | $ | 10,538 | ||||
上表所列实际和形式信息中反映的我们有投票权普通股的股份数量 不包括:
| ● | 截至2023年9月30日,根据我们的2022年股票计划(本文定义),可根据我们的2022年股票计划(定义)行使已发行股票期权的普通股3,788,500股,加权平均 行权价为每股2.36美元; | |
| ● | 根据我们的2022年股票计划,预留8,711,500股普通股供发行 ; | |
| ● | 2023年9月30日后发行的14,859股B-1系列优先股 ,直接上市时将自动一对一地转换为普通股 ; | |
| ● | 发行10,321股普通股标的认股权证,与发行B-1系列优先股的某些股份有关;以及 | |
| ● | 普通股相关认股权证的股份 购买将根据股份认购协议于直接上市时向某些投资者发行的普通股,普通股相关股份应相等于紧接直接上市完成后本公司已发行权益总额的4% ,详见“风险因素-筹集额外的资本可能会稀释我们现有的股东,限制我们的运营,或者要求我们以对我们不利的条款放弃对我们产品的权利 候选产品。 |
| 65 |
管理层的讨论和分析
的财务状况和经营结果
您 应阅读以下对我们的财务状况和运营结果的讨论和分析,以及我们的财务报表 和本招股说明书中其他部分的相关说明和其他财务信息。本讨论和分析中包含的一些信息以及本招股说明书中其他地方陈述的信息,包括与我们的业务计划和战略有关的信息, 包括涉及风险和不确定性的前瞻性陈述。由于许多因素的影响,包括本招股说明书“风险因素”部分所列的因素,我们的实际结果可能与 中描述的结果或以下讨论和分析中包含的前瞻性陈述所暗示的结果大不相同。
概述
我们是一家临床阶段的细胞治疗公司,专注于为患有慢性疾病的患者开发基于成纤维细胞的治疗方法并将其商业化,这些患者的重大医疗需求尚未得到满足,包括退行性腰椎间盘疾病、多发性硬化症、伤口愈合和某些癌症,以及包括胸腺和脾退化逆转在内的潜在延长生命的应用。目前,我们的新制造工艺需要从外科手术中收集多余的组织,并使用同种异体成纤维细胞培养细胞库,用于我们的手术。 我们最先进的候选产品是Cybrocell™和CYMS101。
Cybrocell™ 是一种基于同种异体成纤维细胞的治疗退行性间盘疾病的方法,正在被设计为修复间盘软骨(或任何其他关节软骨)的替代方法。我们已经完成了两项动物研究。这些研究的结果是积极的,并导致了“人类首例”试验批准。我们已经从FDA获得了IND许可,条件是我们的主细胞库获得批准,可以为退行性间盘疾病患者进行1/2期研究 ,并将在美国境内进行这项研究。时间表将通过与FDA的讨论确定。
我们正在开发CYMS101作为一种基于同种异体成纤维细胞的疗法来治疗多发性硬化症。在使用同种异体成纤维细胞完成动物研究后,我们获准在墨西哥使用成纤维细胞成分对多发性硬化症患者进行临床研究,并已完成第一阶段研究。这项研究是在五名参与者中进行的。这项研究的主要目标是评估安全性,次要目标是评估疗效。我们目前正在进行进一步的研究,以确定成纤维细胞在少突胶质细胞扩增中的作用模式,并预计在MS提交IND第二阶段临床试验申请 我们可能会在启动第二阶段研究之前寻找战略合作伙伴与我们合作开发CYMS101,如果成功,也可能在其完成后,开始第三阶段临床试验之前与我们合作开发CYMS101。
| 66 |
我们正处于开发CyWC628作为一种基于同种异体成纤维细胞的伤口愈合疗法的早期阶段。我们的研究目前集中在利用成纤维细胞和成纤维细胞来源的细胞治疗糖尿病小鼠和大鼠的伤口。根据我们迄今取得的成果,我们计划最早在2024年向FDA提交伤口愈合方面的IND申请。
我们 也有处于开发早期阶段的癌症和延长生命计划 ,我们计划在资金允许的情况下加快这类计划的实施。
我们目前从CDMO购买我们的候选细胞治疗产品。我们正在与CDMO签订合同,将我们的实验细胞库转移 ,以生产我们的主细胞库、工作细胞库和基于成纤维细胞的产品 候选产品,以便进行临床试验。如果我们的候选产品获得市场批准,我们将评估建立自己的cGMP制造设施或继续将生产外包给CDMO进行临床测试和商业销售的长期可行性。
自我们于2021年4月从FibroGenesis剥离以来,我们的运营包括业务规划、招聘人员、筹集资金、 建立我们的知识产权组合以及对我们的候选产品和我们的成纤维细胞技术进行研究和开发, 利用成纤维细胞的临床优势作为我们细胞治疗平台的基础。
我们 自成立以来已出现净亏损,并预计在未来继续我们的研发活动时会出现亏损。 到目前为止,我们主要通过来自FibroGenesis的投资、发行560万美元的可转换 期票(从2021年12月至2022年4月发行)以及发行1860万美元的优先股来为我们的运营提供资金。
截至2023年9月30日,我们拥有约1,080万美元的现金和现金等价物。自成立以来,我们遭受了重大的运营亏损。截至2023年9月30日及2022年9月30日止九个月,本公司分别录得约680万美元及360万美元净亏损,截至2022年12月31日及2021年12月31日止年度分别录得510万美元及160万美元净亏损。截至2023年9月30日,我们的累计赤字约为1,460万美元。我们预计未来几年将继续产生巨额费用和运营亏损。见“-资金需求“下面。
我们 预计在可预见的未来将继续蒙受重大损失,我们预计这些损失将大幅增加,如果和 我们:
| ● | 通过临床开发推进我们的主要候选产品的开发,如果FDA批准,则将其商业化; |
| ● | 将我们的临床前开发计划推进到临床开发; |
| ● | 为供应我们的候选产品而产生的电池生产制造成本; |
| ● | 为我们成功完成临床试验的任何候选产品寻求监管批准 ; |
| ● | 增加我们的研究和开发活动,以确定和开发新的候选产品; |
| ● | 雇用 名额外人员; |
| ● | 扩展我们的运营、财务和管理系统; |
| ● | 满足上市公司的要求和要求; |
| ● | 投资 进一步发展,保护和扩大我们的知识产权; |
| ● | 建立 销售、营销、医疗事务和分销基础设施,将我们可能获得营销批准并打算商业化的任何候选产品 商业化; |
| ● | 扩大我们的制造并发展我们的商业化努力。 |
| 67 |
由于与生物制药产品开发相关的众多风险和不确定性以及经济和发展的不确定性,我们可能无法准确预测所有费用的时间或金额。我们最终产生收入以实现盈利的能力将在很大程度上取决于我们候选产品的开发、审批和随后的商业化。如果我们未能 实现盈利或无法持续盈利,则我们可能无法在 计划水平上继续运营,并被迫减少或终止运营。
因此,我们将需要大量的额外资金来支持我们的长期持续运营和实施我们的增长战略。 在我们能够从产品销售中获得可观的收入之前,我们预计将通过出售股权、债务融资或其他资本来源来为我们的运营提供资金,其中可能包括与其他公司的合作或其他战略交易。 我们可能无法在需要时以有利的条款筹集额外资金或达成此类协议或安排。如果我们不能在需要时筹集资金或达成此类协议,我们将不得不大幅推迟、减少或取消我们一个或多个候选产品的开发和商业化,或者推迟我们潜在的许可证内或收购。
运营结果的组成部分
收入
截至 日期,我们尚未从产品销售中获得任何收入,并且预计在可预见的未来也不会从产品销售中获得任何收入。如果我们对任何候选产品的开发工作取得成功并获得监管部门的批准,我们 未来可能会从产品销售中获得收入。我们无法预测我们是否、何时或在多大程度上会从我们的任何候选产品的商业化和销售中获得收入。我们的任何候选产品都可能永远无法获得监管部门的批准。
研究和开发费用
我们的研发费用包括与开发我们的候选产品相关的费用,包括:
| ● | 与员工相关的费用,包括我们研发人员的工资、福利、差旅和股票薪酬 ; |
| ● | 实验室设备和用品; |
| ● | 直接 第三方费用,如根据与CRO和CMO达成的协议发生的费用; |
| ● | 代表我们进行研究和开发活动的顾问 ; |
| ● | 与进行临床前研究和临床试验相关的费用; |
| ● | 与技术相关的成本;以及 |
| ● | 设施 和其他分摊费用,包括租金和其他设施相关成本和其他用品的费用。 |
我们 按所发生的费用来支付研发费用。我们为将来收到的产品或服务支付的不可退还的预付款 用于研发活动的预付款被记录为预付费用。预付金额在 相关商品交付或服务执行时计入费用。
| 68 |
我们 预计我们的研究和开发费用在可预见的未来将大幅增加,因为我们将继续投资于与开发我们的候选产品相关的研究和开发活动,因为它们进入临床开发的后期阶段,以及我们的其他候选产品进入临床前开发阶段。进行必要的临床研究以获得监管批准的过程既昂贵又耗时,我们候选产品的成功开发也非常不确定 。因此,我们无法确定我们研发项目的持续时间和完成成本 ,也无法确定我们将在何时以及在多大程度上从我们的任何候选产品的商业化和销售中获得收入。这是由于与开发候选产品相关的许多风险和不确定性,包括与以下方面相关的不确定性:
| ● | 我们当前开发计划的临床试验的持续时间、成本和时间,以及与新产品候选相关的任何进一步的临床试验; |
| ● | 我们的财政和其他资源是否足以完成必要的临床前研究和临床试验; |
| ● | 接受用于未来临床试验的IND申请; |
| ● | 成功和及时地登记和完成临床试验; |
| ● | 该 成功完成临床前研究和临床试验; |
| ● | 成功 来自我们临床项目的数据,支持我们产品的可接受风险-受益概况 目标人群中的候选人; |
| ● | 该 从适用的监管机构接收并维护监管和营销批准 当局; |
| ● | 建立 与第三方制造商就我们的临床试验的临床供应达成协议,以及 商业制造,如果我们的任何候选产品获得批准; |
| ● | 该 进行合作,以进一步开发我们的候选产品; |
| ● | 获取 并维护我们产品的专利和商业秘密保护以及监管排他性 候选人;及 |
| ● | 成功 如果获得批准,推出我们的候选产品并实现商业销售。 |
如果 与我们的任何项目或我们开发的任何候选产品的开发相关的任何这些变量的结果发生变化, 将显著改变与此类项目或候选产品的开发和/或监管批准相关的成本、时间安排和可行性。
一般、 行政和其他费用
我们的 一般、行政和其他费用主要包括人员成本、分配的设施成本以及 外部专业服务的其他费用,包括法律、营销、投资者关系、人力资源服务和会计服务。人员 成本包括我们的一般和行政人员的工资、福利和基于股票的薪酬。作为上市公司运营,我们预计会产生额外的 费用,包括与遵守 SEC、纳斯达克的规则和法规相关的费用、额外的保险费用、投资者关系活动以及其他行政和专业服务。我们还希望 增加行政职能部门的规模,以支持我们的业务增长。
利息 费用
我们的 利息支出主要包括应计利息支出和可换股票据折价摊销。
| 69 |
营运说明书
运营结果
2022年12月31日和2021年12月31日财政年度比较
下表载列我们截至二零二二年及二零二一年十二月三十一日止年度的经营业绩。
| 截至12月31日的财年, | 变化量 | |||||||||||
| 2022 | 2021 | |||||||||||
| (单位:千) | ||||||||||||
| 运营费用: | ||||||||||||
| 研发 | $ | 1,147 | $ | 521 | $ | 626 | ||||||
| 一般、行政和其他 | 3,320 | 1,057 | 2,263 | |||||||||
| 总运营费用 | 4,467 | 1,578 | 2,889 | |||||||||
| 运营亏损 | (4,467 | ) | (1,578 | ) | (2,889 | ) | ||||||
| 利息支出 | (654 | ) | (4 | ) | (650 | ) | ||||||
| 净亏损 | $ | (5,121 | ) | $ | (1,582 | ) | $ | (3,539 | ) | |||
研究和开发费用
截至2022年和2021年12月31日止年度,研究和开发费用分别为110万美元和50万美元。增加 60万美元的主要原因是:
| ● | 增加 由于在 雇用我们的首席科学官, 2021年7月,2022年12月增加两名研究科学家; |
| ● | 增加 由于许可和扩大临时实验室,研究设施成本为10万美元 办公空间;以及 |
| ● | 增加 由于实验室人员和临床前人员增加,研究用品增加了10万美元 问题研究 |
研究和开发费用不按候选产品跟踪。
一般、 行政和其他费用
截至2022年及2021年12月31日止年度的一般、行政及其他开支分别为330万元及110万元。 增加230万美元的主要原因是:
| ● | 增加 由于聘用我们的首席执行官的时间安排,人事相关费用为90万美元 2021年8月,我们的首席执行官和2022年6月,我们的首席财务官; |
| ● | 增加 会计、法律、营销和咨询相关费用为70万美元 准备成为一家上市公司; |
| ● | 增加 40万美元的设施费用,用于我们租赁办公空间的成本,以及 |
| ● | 增加 由于建立独立董事会,董事会费用为20万美元 2021年成立后。 |
| 70 |
利息 费用
截至2022年和2021年12月31日止年度的利息支出分别为70万美元和00万美元。增加70万美元是由于2021年12月、2022年1月和2022年4月发行了可转换票据。利息开支于二零二二年就 名义利率6. 0%加上二零二二年可换股票据折让摊销入账。
所得税 税
所有期间的有效所得税率均为0.0%。目前,我们已经记录了对我们的净递延税项资产的全部估值备抵。
截至二零二三年九月三十日止九个月与二零二二年九月三十日止九个月的比较
下表载列我们截至二零二三年及二零二二年九月三十日止九个月的经营业绩。
九个月结束 9月30日, | 变化量 | |||||||||||
| 2023 | 2022 | |||||||||||
| (未经审计,以千计) | ||||||||||||
| 运营费用: | ||||||||||||
| 研发 | $ | 1,595 | $ | 802 | $ | 793 | ||||||
| 一般、行政和其他 | 4,814 | 2,361 | 2,453 | |||||||||
| 总运营费用 | 6,409 | 3,163 | 3,246 | |||||||||
| 运营亏损 | (6,409 | ) | (3,163 | ) | (3,246 | ) | ||||||
| 其他损失 | (213 | ) | — | (213 | ) | |||||||
| 利息支出 | (146 | ) | (434 | ) | 288 | |||||||
| 净亏损 | $ | (6,768 | ) | $ | (3,597 | ) | $ | (5,744 | ) | |||
研究和开发费用
截至2023年及2022年9月30日止九个月,研究及开发开支分别为160万元及80万元。增加80万美元的主要原因是:
| ● | 增加 由于在 年额外雇用三名研究科学家并授予股票期权, 2023年; |
| ● | 增加 研究顾问费用10万元,以提供技术写作支援;以及 |
| ● | 由于增加了实验室人员和临床前研究,研究设施和实验室用品增加了 20万美元。 |
研究和开发费用不按候选产品进行跟踪。
| 71 |
一般、 行政和其他费用
总体而言,截至2023年、2023年和2022年9月30日的9个月,行政和其他费用分别为480万美元和240万美元。增加240万美元的主要原因是:
| ● | 由于2022年6月聘用首席财务官的时间安排、2023年授予的股票期权和2023年的高管奖金,与人事相关的费用增加了 140万美元; |
| ● | 与准备上市相关的成本增加了 会计、法律、营销和咨询费用50万美元。 |
| ● | 我们租用办公空间的成本增加了 30万美元的设施费用,以及 |
| ● | 由于2023年授予的股票期权,董事会支出增加了 20万美元。 |
其他 损失
截至2023年9月30日和2022年9月30日的9个月,其他亏损分别为20万美元和零。其他亏损 包括向FibroGenesis支付的ROFN款项超出于2023年1月ROFN协议开始时确立的衍生负债。
利息 费用
截至2023年和2022年9月30日的9个月,利息支出分别为10万美元和40万美元。减少30万美元是由于截至2023年9月30日的九个月内票据的到期日和转换。2022年和2023年录得利息支出,名义利率为6.0%,外加2022年可转换票据的折价摊销 。
所得税 税
所有期间的有效所得税率均为0.0%。目前,我们已经记录了对我们的净递延税项资产的全部估值备抵。
流动性 与资本资源
概述
到目前为止,我们的运营资金主要来自FibroGenesis的投资、根据我们的可转换贷款协议 借款的收益以及发行优先股的收益。自成立以来至2023年9月30日,我们已从出售可转换票据和发行优先股中获得总计约560万美元的收益。 截至2023年9月30日,我们的现金和现金等价物约为1080万美元,累计赤字约为1460万美元。截至2023年9月30日,我们没有未偿债务。
现金流
下表概述了截至2022年12月31日和2021年12月31日的年度现金流。
| 截至十二月三十一日止的年度: | ||||||||
| 2022 | 2021 | |||||||
| (单位:千) | ||||||||
| 用于经营活动的现金净额 | $ | (4,066 | ) | $ | (1,410 | ) | ||
| 融资活动提供的现金净额 | 5,925 | 1,817 | ||||||
| 现金及现金等价物净增加情况 | $ | 1,859 | $ | 407 | ||||
操作 活动
截至2022年和2021年12月31日止年度,经营活动中使用的现金净额分别为410万美元和140万美元,主要包括净亏损510万美元和160万美元。应付帐款和应计费用增加50万美元,加上基于股票的薪酬支出30万美元和可转换票据债务贴现摊销40万美元的非现金支出,部分抵消了截至2022年12月31日的年度净亏损。应付账款和应计费用增加了20万美元,部分抵消了截至2021年12月31日的年度的净亏损。
| 72 |
为 活动提供资金
融资活动提供的现金净额在截至2022年12月31日和2021年12月31日的年度分别约为590万美元和180万美元。 2021年,我们通过贷款协议从FibroGenesis获得近100万美元,并在同一年 偿还了约80万美元,FibroGenesis在2021年第一季度投资了30万美元,作为我们从FibroGenesis创业的一部分。我们还从2021年12月发行的可转换票据中获得了130万美元。2022年,我们从2022年1月和4月发行的可转换票据中获得了430万美元,在2022年12月通过发行优先股获得了220万美元。我们在2022年偿还了20万美元 ,并借给了FibroGenesis 40万美元,FibroGenesis在2022年偿还了10万美元。
下表概述了截至2023年9月30日和2022年9月30日的9个月的现金流。
| 截至9月30日的9个月 | ||||||||
| 2023 | 2022 | |||||||
| (单位:千) | ||||||||
| 用于经营活动的现金净额 | $ | (4,800 | ) | $ | (2,893 | ) | ||
| 用于投资活动的现金净额 | (493 | ) | — | |||||
| 融资活动提供的现金净额 | 13,793 | 3,775 | ||||||
| 现金及现金等价物净增加情况 | $ | 8,500 | $ | 882 | ||||
操作 活动
截至2023年和2022年9月30日止九个月,经营活动所用现金净额分别为480万美元和290万美元,主要由净亏损680万美元和360万美元组成。应付账款和应计费用增加了40万美元,加上基于股票的薪酬费用增加了130万美元,衍生债务损失增加了20万美元,可转换票据债务折价摊销增加了10万美元,这部分抵消了截至2023年9月30日的9个月的净亏损。应付账款和应计费用增加30万美元,加上基于股票的薪酬支出20万美元和可转换票据债务折价摊销20万美元,部分抵消了截至2022年9月30日的9个月的净亏损。
投资 活动
在截至2023年9月30日、2023年和2022年的9个月中,用于投资活动的现金净额约为50万美元,没有现金。增加的原因是购买了实验室设备。
为 活动提供资金
在截至2023年9月30日和2022年9月30日的9个月中,融资活动提供的现金净额分别约为1380万美元和380万美元。我们从FibroGenesis收到了一笔30万美元的贷款,根据ROFN协议(在此定义)向FibroGenesis支付了260万美元,在截至2023年9月30日的9个月内,我们从发行B系列优先股和B-1系列优先股中总共获得了1610万美元。在截至2022年9月30日的9个月里,我们向FibroGenesis偿还了20万美元的贷款,借给了FibroGenesis 30万美元,并从发行可转换票据中获得了430万美元。
资金需求
我们 没有任何获准销售的产品,我们从未从与客户的合同中获得任何收入。我们不希望 产生任何有意义的收入,除非我们获得监管机构对我们当前或未来的任何候选产品的批准并将其商业化 ,我们不知道这将在何时或是否发生。我们预计在可预见的未来将继续遭受重大损失, 随着我们继续开发当前和未来的候选产品并寻求监管部门的批准,并开始将任何批准的产品商业化,损失将会增加。我们面临所有通常与开发新产品候选产品相关的风险,我们可能会遇到不可预见的费用、困难、复杂情况、延误和其他未知因素, 可能会对我们的业务产生不利影响。此外,在此次发行完成后,我们预计将产生与上市公司运营相关的额外成本。
财务报表的编制就像我们将继续作为一家持续经营的企业一样,考虑在正常业务过程中实现资产和偿还负债。自成立以来,我们因运营而产生了运营亏损和负现金流。截至2023年9月30日,我们的累计赤字约为1,460万美元。管理层预计 将继续出现运营亏损和负现金流。
我们 将需要筹集额外资本以继续为我们的运营提供资金。我们相信,我们将能够通过 股权融资或其他安排为运营提供资金;但不能保证此类额外融资(如果可用)能够以可接受的条款获得。如果我们无法获得此类额外融资,未来的业务将需要缩减或停止。
我们 相信,我们的现有资本将使我们能够为我们的运营提供资金,至少持续到2024年12月31日。我们可能需要筹集 额外资本,以满足2024年12月31日之后资本支出和营运资金的现金需求。我们 基于可能被证明是不正确的假设进行上述估计,我们可能会比我们 预期的更早使用我们的资本资源。
我们未来的资金需求将取决于许多因素,包括但不限于:
| ● | 我们候选产品的临床试验的启动、进度、时间表、成本和结果; |
| ● | 其他研究和临床前研究的启动、进度、时间表、成本和结果 与流水线开发和我们未来启动的其他研究计划相关的 ; |
| ● | 制造活动的 成本和时间安排,包括我们计划的与我们的候选产品和其他计划相关的生产扩展活动,以及我们通过商业化推进它们的临床前和临床开发 ; |
| ● | 我们当前发展计划的潜在扩展,以寻求新的适应症; |
| ● | 满足FDA和其他可比外国监管机构制定的监管要求的结果、时间和成本; |
| 73 |
| ● | 提交、起诉、辩护和执行专利权利要求和其他知识产权的费用,无论是许可内的还是其他方面的; |
| ● | 竞争的技术和市场发展的影响; |
| ● | 支付许可费、可能的特许权使用费和可能的里程碑付款; |
| ● | 一般业务费用的成本; |
| ● | 为我们可能获得监管批准的任何候选产品建立销售、营销和分销能力的成本 在我们选择自行将产品商业化的地区 ;以及 |
| ● | 作为一家上市公司的运营成本。 |
此外, 我们的运营计划可能会改变,我们可能需要额外的资金来满足临床试验的运营需求和资本要求 以及其他研发支出。
如果 我们需要筹集额外资本来支持我们的运营,我们可能无法以可接受的条款获得资金,或者根本无法获得资金。如果我们 无法在需要时获得足够的资金,我们可能不得不推迟、缩小或暂停我们的一个或多个临床前 研究、临床试验、研发计划或商业化努力。我们可能寻求通过公开或私募股权发行、债务融资、合作和其他许可安排来筹集任何必要的额外资本。 如果我们通过债务融资筹集额外资本,我们可能会受到契约的限制或限制我们采取特定行动的能力,例如招致额外债务、进行资本支出或宣布股息。如果我们通过 营销和分销安排或与第三方的其他合作、战略联盟或许可安排来筹集额外资本,我们 可能不得不放弃对我们的候选产品、技术、未来收入流或研究计划的某些有价值的权利,或者 以可能对我们不利的条款授予许可。
合同义务和承诺
截至2022年12月31日,我们的承诺包括可转换应付票据和德克萨斯州休斯敦的两个实验室和办公空间租赁 。截至2023年9月30日,我们的承诺包括租用德克萨斯州休斯敦的两个实验室和办公空间。
下表汇总了截至2022年12月31日我们的合同义务。
| 总计 | 不到一年 | 1–3 年份 | 3–5 年份 | 多过 5年 | ||||||||||||||||
| (单位:千) | ||||||||||||||||||||
| 可转换票据应付债务 | $ | 5,600 | $ | 5,600 | $ | — | $ | — | $ | — | ||||||||||
| 经营租赁义务 | 2,484 | 466 | 1,509 | 509 | — | |||||||||||||||
| 总计 | $ | 8,084 | $ | 6,066 | $ | 1,509 | $ | 509 | $ | — | ||||||||||
在截至2023年9月30日的9个月内,应付可转换票据已转换为B系列优先股。
下表汇总了截至2023年9月30日我们的合同义务。
| 按期间到期付款 | ||||||||||||||||||||
| 总计 | 减
比 一年 | 1–3 年 | 3–5 年 | 更多
比 5年 | ||||||||||||||||
| (单位:千) | ||||||||||||||||||||
| 运营 租赁义务 | $ | 2,104 | $ | 475 | $ | 982 | $ | 647 | $ | — | ||||||||||
| 总计 | $ | 2,104 | $ | 475 | $ | 982 | $ | 647 | $ | — | ||||||||||
表外安排 表内安排
在本报告所述期间,我们没有,目前也没有任何表外安排或持有任何可变利息 实体。
| 74 |
关键会计估算
我们的管理层对我们的财务状况和经营结果的讨论和分析是基于我们的财务报表, 这些报表是根据公认会计准则编制的。在编制这些财务报表时,我们需要做出估计、判断和假设,以影响资产和负债的报告金额、截至资产负债表日期的或有资产和负债的披露以及报告期内报告的费用金额。根据公认会计原则,我们会持续评估我们的估计和判断。我们根据历史经验及我们认为 在当时情况下属合理的其他各种因素作出估计,而这些因素的结果构成对资产及负债的账面价值作出判断的基础,而该等资产及负债的账面价值并非从其他来源轻易可见。在不同的假设或条件下,实际结果可能与这些估计值不同。
我们 将我们的关键会计估计定义为根据GAAP要求我们对 不确定并可能对我们的财务状况和运营结果产生重大影响的事项做出主观估计和判断,以及我们应用这些原则的具体方式。虽然我们的重要会计政策在本招股说明书其他部分的财务报表附注2中有更全面的描述,但我们认为以下是编制财务报表时使用的关键会计估计 ,需要进行重大估计和判断。
净额
母公司投资
由于我们在2021年4月8日成立之前的财务报表来自FibroGenesis的历史记录,因此母公司的净投资在资产负债表上的股东赤字中列示。作为FibroGenesis当时的子公司, 在签订可转换票据协议之前,我们所有的营运资金和融资要求都依赖于FibroGenesis。与FibroBiologics有关但发生在母公司一级的财务交易通过 净母公司投资账户入账。因此,FibroGenesis的现金、现金等价物或债务均未在财务报表中分配给我们。母公司净投资代表FibroGenesis对我们记录的净资产的兴趣。
研究和开发
研究和开发成本在发生时计入费用。研发成本包括进行研究和开发活动所产生的成本,包括工资和奖金、科学家招聘成本、员工福利、设施成本、实验室用品、制造费用、临床前费用、研究材料、咨询和其他合同服务。某些研发活动的成本根据个别安排的条款确认,这些条款可能不同于已发生成本的模式,并在财务报表中反映为预付或应计研发。
可转换票据 应付票据
我们在2021年12月签订了多个可转换本票协议,或统称为2021年票据。根据2021年债券,我们收到了130万美元, 这笔利息按6.0%的年利率累算,并将在我们首次公开发行时到期。 2021年债券到期时,持有人可以选择全额现金支付未偿还本金和利息 或选择延期一年。
如果我们发行了 并出售了超过1,000万美元的股本,2021年票据的未偿还余额和应计利息可以 转换为固定数量的普通股,仅受可能发生的任何资本重组的惯例反稀释条款的约束 。
根据2021年债券的条款,我们根据ASC 815衍生工具和套期保值评估了转换期权功能。它提供了三个标准,如果满足,要求公司从其宿主工具中分离出转换选项,并将其作为独立的衍生品金融工具进行核算。该三项准则包括以下情况:(I)嵌入衍生工具的经济特征及风险与宿主合约的经济特征及风险并不明显及密切相关;(Ii) 同时包含嵌入衍生工具及宿主合约的混合工具并未根据其他适用的公认会计原则按公允价值重新计量,而公允价值的变动在发生时于收益中报告;及(Iii) 与嵌入衍生工具条款相同的独立工具将被视为衍生工具。
| 75 |
在发行2021年票据时,以及在2021年、2021年和2022年12月31日,我们确定转换特征的嵌入衍生工具 不符合标准,因为它符合“与实体自己的股票挂钩”的例外,因此不需要 从宿主工具中分离出来。如下所述,在截至2023年9月30日的九个月内,所有2021年债券均转换为我们的B系列优先股。
我们 在2022年1月至4月期间额外发行了本金总额为430万美元、期限为一年的可转换本票,或统称为2022年票据。2022年债券可按以下两者中较低者转换:(I)在首次公开发售时我们的普通股发行价有15%的折让,或(Ii)商数2.0亿美元除以发行前的总股本 权益。2022年票据中的转换期权功能根据ASC 815进行了评估,并在票据发行时和截至2022年12月31日记录了转换期权估计公允价值50万美元的衍生负债。票据发行已录得抵销折扣,并正摊销至2022年票据预期年期的利息支出。
2023年2月,我们将2022年债券本金价值370万美元的本金和利息转换为相当于我们B系列优先股的799,603股。2023年4月,我们将2021年债券本金价值160万美元和2022年债券本金价值30万美元的本金和利息转换为相当于我们B系列优先股的353,713股 。2023年6月,我们将2022年债券剩余的30万美元本金价值转换为相当于我们B系列优先股的66,077股 。截至2022年9月30日,2022年债券没有未偿还本金余额。
截至2023年9月30日和2022年12月31日,可转换债务余额分别为零和560万美元。
基于股票的薪酬
我们 根据员工、董事和非员工在授予之日的公允价值来衡量所有授予员工、董事和非员工的股票期权,并使用直线法在必要的服务期内确认这些奖励的相应薪酬支出,这通常是相应奖励的获得期。没收是按发生的情况计算的。
我们在运营报表中对基于股票的薪酬费用进行分类的方式与对获奖者的工资成本进行分类或对获奖者的服务付款进行分类的方式相同。
我们使用Black-Scholes期权定价模型估计每个股票期权授予的公允价值,该模型使用普通股的公允价值和我们对普通股波动性、股票期权的预期期限、接近我们股票期权预期期限的无风险利率和我们的预期股息收益率所做的假设作为输入。
基于我们的股票奖励的普通股的估计公允价值 是由我们的董事会根据管理层的意见在每个期权授予日确定的,考虑到我们对普通股的最新第三方估值,以及我们董事会对其他客观和主观因素的评估,它认为这些因素是相关的,并且从最近的估值日期到授予日期可能发生了变化。这些第三方估值是根据《美国注册会计师协会会计和估值指南,私人持股公司的估值》 作为补偿发行的股权证券(《执业援助》)中概述的指南进行的。
一旦完成直接上市,我们普通股的公开交易市场已经建立,我们的董事会将不再需要评估我们普通股的公允价值,因为我们普通股的公允价值将基于我们普通股的市场报价 价格来确定。
有关我们在确定基于股票的薪酬时使用的假设的更多信息,请参阅本招股说明书中其他部分包括的经审计的财务报表的附注11。
ROFN协议
2023年1月,我们与FibroGenesis达成了一项关于第一谈判权的协议,即ROFN协议。为了换取FibroGenesis同意修改我们的公司注册证书,以(I)在我们承销的首次公开募股或我们的普通股在证券交易所直接上市(我们统称为IPO)或出售我们的公司时消除A系列优先股的清算优先权,(Ii)使B系列优先股 清算优先权等于A系列优先股,以及(Iii)规定在我们的 公司首次公开募股或出售时,A系列优先股将被免费取消,我们同意在首次公开募股或出售公司之前,向FibroGenesis支付对我们进行任何股权投资的总收益的15%。此外,如果FibroGenesis决定向外部许可其任何技术,我们 将获得为期五年的第一谈判权。根据我们的 管理层在执行ROFN协议时对上市前将筹集的资本的估计,我们 为预期未来向FibroGenesis支付的款项记录了260万美元的衍生品负债。作为被视为股息,衍生债务 首先计入母公司净投资,然后在母公司净投资取消后计入额外实收资本 。支付给FibroGenesis的金额超过衍生品负债的金额在经营报表中记为其他亏损。 在计算普通股股东在确定每股收益时可用金额时,视为股息作为净亏损的减少额计入。
就业法案
根据《就业法案》的定义,我们 是一家“新兴成长型公司”。根据《就业法案》,新兴成长型公司可以推迟采用在《就业法案》颁布后发布的新的或修订后的会计准则,直到这些准则适用于私营 公司。
我们 已选择使用这一延长的过渡期来遵守新的或修订的会计准则,这些新的或修订的会计准则对上市公司和非上市公司具有不同的生效日期 ,直到我们(I)不再是新兴成长型公司或(Ii)肯定地 并不可撤销地选择退出《就业法案》中规定的延长过渡期。因此,我们的财务报表可能无法 与截至上市公司生效日期遵守新的或修订的会计声明的公司进行比较。如果我们随后 选择遵守这些上市公司生效日期,则根据JOBS 法案,此类选择将不可撤销。
我们 将一直是一家新兴的成长型公司,直到(I)2028年12月31日,(Ii)我们 的年度总收入至少为12.35亿美元的财年的最后一天,(Iii)我们被视为《交易法》第12b-2条规则所定义的“大型加速申请者”的财年的最后一天,如果非关联公司持有的普通股市值在该年度第二财季的最后一个营业日为7.00亿美元或以上,或(Iv)我们在前三年期间发行了超过10亿美元的不可转换债务证券的日期 ,就会发生这种情况。
关于市场风险的定量和定性披露
我们投资活动的主要目标是确保流动性和保存资本。我们在正常的业务过程中面临着市场风险。这些风险主要包括利率敏感性。截至2023年9月30日和2022年12月31日,我们的现金和现金等价物分别约为1080万美元和230万美元,其中包括银行存款。利率的历史波动对我们来说并不重要。由于我们的现金等价物的到期日较短,立即调整利率100个基点不会对我们的现金等价物的公平市场价值产生实质性影响。我们不认为 通胀、利率变化或外币汇率波动对我们在本报告所述任何时期的经营业绩产生重大影响 。
| 76 |
生意场
概述
我们是一家临床阶段的细胞治疗公司,专注于为患有慢性疾病的患者开发基于成纤维细胞的疗法并将其商业化,这些慢性疾病具有重大的医疗需求,包括退行性腰椎间盘疾病、多发性硬化症、伤口愈合、 和某些癌症,以及包括胸腺和脾退化逆转在内的潜在延长寿命的应用。
我们 成立于2021年4月,是一家得克萨斯州有限责任公司,名称为FibroBiologics,LLC,并于2021年12月以FibroBiologics,Inc.的名称转换为特拉华州公司 。2023年4月14日,我们更名为FibroBiologics,Inc.。与我们的成立相关,我们向当时的母公司FibroGenesis发行了A系列优先股或A系列优先股,以通过专利转让协议和知识产权交叉许可协议 换取某些知识产权的权利 。开发从FibroGenesis获得的知识产权是我们成立的基础。在我们成立之前,与上述疾病途径相关的临床前研究和开发是在FibroGenesis下进行的。
成纤维细胞 技术平台
成纤维细胞 和干细胞是人体中仅有的两种可以再生组织和器官的细胞类型。研究表明,间充质干细胞和成纤维细胞具有许多共同的表面标志物,并且可以分化成许多细胞,包括脂肪细胞、软骨细胞、成骨细胞、肝细胞和心肌细胞,并且可以调节免疫系统。然而,转录组学和表观遗传学研究表明两种细胞类型之间存在明显差异。
成纤维细胞是结缔组织的主要细胞类型,具有纺锤形的形态,其经典功能一直被认为是产生负责维持组织结构完整性的细胞外基质。成纤维细胞也在伤口愈合的增殖期发挥重要作用,导致细胞外基质的沉积。
成纤维细胞 有利于干细胞作为细胞疗法治疗平台,因为成纤维细胞:
| ● | 是否可以从各种外科手术的供皮者身上非侵入性地获取皮肤,例如 腹部褶皱皮瓣; |
| ● | 有 培养中的倍增时间比干细胞快; |
| ● | 拥有 与干细胞相比,具有更好的免疫调节活性; |
| ● | 展品 与干细胞相比,产生再生细胞因子和生长因子的能力增强 细胞;以及 |
| ● | 与干细胞相比,分离、培养和扩增更经济,因为成纤维细胞不需要使用昂贵的组织培养介质。 |
| 77 |
研究表明,同种异体成纤维细胞很像间充质干细胞,具有免疫特免性,不会引起免疫反应。体外培养 和体内。这些研究包括瓦伦特和他的同事(PMID 7646145),他们观察了心脏移植后的主动脉瓣,注意到即使是急性心肌排斥的病例似乎也不会影响瓣膜的长期活性和耐久性,组织活性得到了组织学上的确认,显示成纤维细胞保存完好。1在O‘Brien及其同事的另一项研究(PMID 3682851)中,研究人员使用染色体分析说明了男性捐赠者的成纤维细胞长期存活,该细胞来自植入女性受体9年后取出的瓣膜叶。这说明供体成纤维细胞能够在免疫系统破坏的情况下在宿主体内存活和增殖。2 如果需要自体成纤维细胞,这将意味着必须从每个患者身上获取细胞,进行处理和培养,然后再应用于同一患者,这将更加昂贵和效率低下。由于同种异体成纤维细胞不会引起免疫反应,我们正计划建立自己的cGMP制造工厂,以采购同种异体成纤维细胞,以供我们的候选产品进行临床测试,并在候选产品获得上市批准的情况下用于商业销售。
我们的 战略
我们 正在利用成纤维细胞作为技术平台,研究和开发针对治疗需求未得到满足的慢性疾病的创新疗法。我们的愿景是通过严格的科学流程和承诺满足患者的需求,成为再生医学领域的世界领先者。为实现我们的愿景,我们将重点抓好以下战略:
| ● | 吸引并留住具备开展临床前研究和确定临床试验最佳途径所需技能的科学家。 | |
| ● | 将我们最初的临床开发工作与重要的未满足的治疗需求、较低的风险和市场潜力相结合,将我们最初的临床开发工作放在候选产品上。 | |
| ● | 与具有相关专业知识和经验的CRO合作,成功、及时地执行临床 试验,以生成可用于寻求批准的可靠关键数据。 | |
| ● | 投资 生产和供应成纤维细胞用于临床试验和初步商业化所需的关键能力。 | |
| ● | 保护、扩大和捍卫我们围绕成纤维细胞的知识产权组合。 | |
| ● | 在资金允许的情况下,在开发时间更长、风险更大且有重大未满足治疗需求的候选产品中,扩大 开发力度。 |
截至2023年9月30日,我们拥有约1,080万美元的现金和现金等价物。为了在未来12个月推进我们的上述战略 ,除了人员和基础设施方面的持续支出外,以下研发计划预计将由可用资金提供资金:
| ● | 实验室设备的资本支出约为50万至70万美元,以支持继续研究和扩大能力; | |
| ● | 约30万至40万美元用于获取临床皮肤样本; | |
| ● | 约20万至30万美元用于组织和成纤维细胞的特性和优化; | |
| ● | 大约20万至40万美元用于开发主细胞库; | |
| ● | 大约10万至30万美元,用于开发一个可用的细胞库; | |
| ● | 大约10万至30万美元,用于开发转移到CDMO的流程; | |
| ● | 大约10万至30万美元,用于完成另一项关于CYWC628伤口愈合的动物研究 ; | |
| ● | 为伤口愈合的1/2期临床试验提交CYWC628的IND申请,金额约为10万美元。 | |
| ● | 大约20万至30万美元,用于完成临床前动物研究,以确定CYMS101在MS中的作用模式,然后提交IND申请,在MS中进行1/2期临床试验;以及 | |
| ● | 大约10万美元,用于在CyTER915中启动胸腺退化逆转的临床前动物研究。 |
我们 相信我们的可用资金将允许我们开发主细胞库和工作细胞库,以转移到CDMO,最终将 生产我们的药物产品,这些产品是启动CybroCell、CYMS101和CYWC628的1/2期临床试验所需的。
以上估计是初步估计,可能会更改。我们无法在未来12个月内确定我们的可用资金的所有特定用途。由于开发过程中固有的不确定性,很难估计我们将用于任何特定目的的可用资金的确切金额。此外,我们实际支出的金额、分配和时间将取决于许多因素,包括我们的研发工作的结果。
我们的 人员
我们 组建了一个执行领导团队,由我们的创始人、首席执行官和董事会主席、 我们的首席科学官和首席财务官组成,他们在初创企业和 生命科学行业拥有成功的业绩记录。我们的执行领导团队在董事会的监督下工作,董事会成员都是具有实际行业经验的公认领导者 。我们的科学顾问 委员会还拥有一支由世界知名科学家组成的团队,他们拥有相关的专业知识,可帮助指导我们的研发工作。
1 资料来源:瓦伦特·M、费吉恩·G、比林厄姆·ME、塔伦蒂·E、卡拉布雷斯·F、卡苏拉·R、沙姆韦·NE、蒂恩·G,心脏移植后的主动脉瓣。胸外科,1995年8月;60(2补充):S135-40。电话:00251-f,电话:10.1016。7646145。
2 来源:O‘Brien MF,Stafford EG,Gardner MA,Pohlner PG,McGiffin DC冷冻保存和新鲜同种异体瓣膜置换术的比较,并附染色体研究报告。胸心外科杂志1987年12月;94(6):812-23.PMID: 3682851。
| 78 |
我们的 当前渠道
我们 拥有一系列处于不同开发阶段的候选产品,包括:
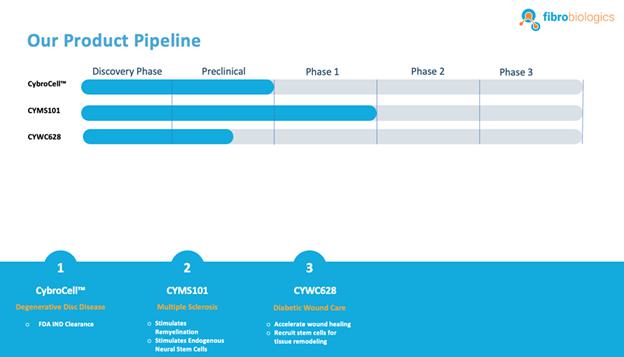
CybrocellTM 治疗退行性腰椎间盘疾病
退行性腰椎病
背痛与椎间盘退变有很强的相关性。椎间盘退变,虽然在许多情况下没有症状,但也与坐骨神经痛和椎间盘突出、疼痛或脱垂有关。它会改变椎间高度和脊柱其余部分的力学性能,对肌肉和韧带等其他脊柱结构的行为产生不利影响。从长远来看,它可能导致脊柱狭窄,这是老年人疼痛和残疾的主要原因。随着当前人口结构的变化和老年人口的增加,其发病率正在上升。
椎间盘充当两个椎骨之间的关节,并执行以下关键功能:
| ● | 吸收 冲击; | |
| ● | 保持 运动;以及 | |
| ● | 保持 稳定性。 |
椎间盘退化的时间比其他肌肉骨骼组织要早得多。腰椎间盘退变的第一个明确发现是在11-16岁年龄组。大约20%的青少年患有轻度退变的椎间盘。这一比例随着年龄的增长而急剧增加,特别是在男性中,因此大约10%的50年年龄段和60%的70年年龄段椎间盘严重退化(现代腰背痛流行病学研究进展“Mattiuzzi等人,2020年)。
| 79 |
在生长和骨骼成熟过程中,环与核之间的边界变得不那么明显,随着年龄的增长,核一般变得更纤维,凝胶状更少。随着年龄的增长和退变,椎间盘的形态发生变化,变得越来越无序。通常,环形板层变得不规则、分叉和交错,胶原和弹性蛋白网络也变得更加杂乱无章。
有裂隙的 形成常在椎间盘内形成,特别是在细胞核。神经和血管越来越多地出现变性。细胞发生增殖,导致在细胞核内形成簇状。细胞死亡也会发生,出现坏死和凋亡的细胞。据报道,在成人椎间盘中,超过50%的细胞是坏死性的。随着年龄的增长,退行性改变的发生率增加,包括细胞死亡、细胞增殖、粘膜变性、颗粒状改变和同心性撕裂。很难区分完全由于衰老而发生的变化和那些可能被认为是“病态”的变化。
根据发表在《全球脊柱杂志》上的一项研究,题为退行性腰椎疾病:估计全球发病率和全球范围Ravindra等人于2018年发表,每年约有2.66亿人患有退行性脊椎疾病和下腰痛。此外,每年有4.03亿人患有症状性腰椎间盘退变,1.03亿人患有椎管狭窄症,3900万人患有腰椎滑脱症。此外,下背部疼痛被认为是主要主诉之一,可能表明潜在的脊柱相关疾病。根据发表在《医院管理与健康政策》杂志上的这项研究,《医院管理与健康政策》现代腰背痛流行病学研究进展Mattiuzzi等人指出,2020年,下腰痛的发病率、患病率和伤残调整寿命年分别为2.459亿例(全球第15个原因)、5.77亿例 (全球第15个原因)和6490万例(全球第6个原因)。这篇论文进一步指出,女性患下腰痛的风险比男性略高。慢性下腰痛是常见的主诉之一,可能预示着潜在的严重脊椎疾病。
这些 统计数据表明退行性脊柱疾病可能对患者的生活产生重大影响。这些适应症与一系列不同的临床症状有关,如虚弱、下肢疼痛和背部疼痛,并可能导致生活质量的显著下降 。目前使用的治疗主要是保守和姑息治疗,旨在让患者重返工作岗位。 从卧床休息(不再推荐)到止痛,使用肌肉松弛药或注射皮质类固醇,或局部麻醉和手法治疗。也使用了各种干预措施(例如,椎间盘内电疗),但尽管有传闻称成功,但到目前为止,试验发现它们的使用几乎没有直接好处。椎间盘退变相关的疼痛也可以通过手术治疗 通过人工椎间盘置换或固定受影响的椎骨。
| 80 |
退行性腰椎间盘疾病的可用治疗方法
大多数患有退行性腰椎间盘疾病的患者,至少在一开始,通过物理治疗、髓核强化和拉伸等非手术干预措施显示出改善。当这些干预措施不再起到缓解作用时,患者通常使用治疗药物,其中包括用于缓解疼痛的常规药物,如阿片类药物、非类固醇消炎药和皮质类固醇。当这些非手术疗法不再有效时,患者可以接受手术治疗,包括使用医疗设备或植入物, 以提供缓解。
矫正退变椎间盘的原始外科治疗方法是进行椎间盘切除术或脊柱融合术。椎间盘切除术是一种适当的手术,常规的做法是通过开窗在环内摘除变性的髓核。它允许同时移除突出的髓核(疝气切除术)和变性的剩余椎核碎片。虽然此手术是减压和缓解神经系统(根或马尾)的理想方法,但对于脊柱来说,这是一个糟糕的手术,因为它会导致致残,导致下跌退行性变,并可能需要额外的侵入性外科手术,如融合或关节成形术。腰椎间盘摘除在缓解神经根性疼痛方面有较好的短期效果,但会导致椎间隙高度降低,神经孔狭窄,治疗节段不稳,腰背痛,和/或并发症,如椎管狭窄或小关节痛。
接受这些手术的患者通常会服用几周的止痛药,并至少有三到六个月的恢复时间。因此,需要一种痛苦更小、侵入性更小、更有效的方法。原始治疗程序的缺陷导致了对非融合技术发展的探索,例如椎间盘或髓核假体。人工椎间盘关节成形术是治疗腰椎间盘退变患者的一种新兴疗法。它的优点是保持运动,减少相邻节段退变的发生率,避免融合相关的并发症,使功能早日恢复。目前,市场上有两种设备: 全盘替换和核替换。然而,这两种设备都有重大缺陷。
全球市场对脊柱人工椎间盘的需求一直在增长。这些设备越来越受欢迎,因为它们的设计目的是在提供稳定和消除疼痛的同时保持功能脊柱单位的运动。2021年7月,Aesculap植入系统有限责任公司宣布了其Actil L®人工椎间盘关键试验的长期报告。

Centinel Spine的prodisL是一种全椎间盘置换(TDR)技术平台,为患有颈椎和腰椎腰椎退变的合格患者提供了一种手术替代融合的方法。ProdisL植入物旨在缓解疼痛,同时允许患病脊柱节段的运动潜力。

用于单节段腰椎的Actil人工椎间盘是一种承重模块植入物,由两个终板和一个聚乙烯嵌体组成,旨在作为融合术的替代方案。它的设计允许在手术水平上进行受控运动。
| 81 |
全盘置换术是一种笨重的金属假体,旨在替换整个椎间盘:环隙、髓核和终板。这些假体使用侵入性前入路(经腹膜或后腹膜),需要血管外科医生在场。人工髓核替代物保留了剩余的椎间盘组织及其功能,已有文献报道此类假体移位、磨损、邻近椎间盘退变、关节突关节及下沉。它的设计允许通过后路植入髓核假体,但这种髓核假体的主要限制是它只能用于椎间盘退变处于早期或中期的患者,因为它需要有合适的自然环存在。作为一种基于水凝胶的设备,它是脆弱的,因此无法抵抗腰椎突出的生物力学约束(剪切力)。作为惰性材料, 随着时间的推移,它们可能会失去其机械性能,并且已有撕裂和断裂的报告。仅更换髓核并保留受损的环状物会产生植入物挤压或腰椎间盘突出复发的条件。
除椎间盘置换外,目前还有组织工程和再生医学的治疗选择,这代表了治疗退行性间盘疾病的新选择。再生组织的方法多种多样。这些方法 可分为以下三类:
| (i) | 生物材料,不含额外细胞,用于发送信号以吸引细胞并促进再生; | |
| (Ii) | 可单独使用细胞 来形成组织;以及 | |
| (Iii) | 细胞 可以与生物材料支架一起使用,该支架充当组织发育的框架。 |
自体软骨细胞移植或ACT用于修复关节软骨已经有几年了,但组织工程学用于关节盘修复仍处于初级阶段。目前正在进行密集的研究,动物实验已经证明了组织工程化椎间盘的可行性。通常情况下,关节软骨是一种一旦受损就不能自然再生的组织。最近,人们努力通过在实验室中再生部分受损组织来重建受损的生物组织。这种被定义为“组织工程学”的方法受到了极大的关注。
组织 工程涉及开发能够与生物组织特异性相互作用以产生 功能组织等同物的生物相容性材料。组织工程的基本概念是从患者收集所需的组织,从组织样本中分离细胞 ,增殖细胞,将增殖的细胞接种到可生物降解的聚合物支架上,体外培养 细胞预定的时间,并将细胞/聚合物构建体移植回患者体内。更有趣的是,最近的 试点临床试验表明,ACT是椎间盘突出的有效治疗方法。ACT用于椎间盘修复的主要缺点 是需要椎间盘活检。因此,需要一种改进的方法来恢复椎间盘解剖结构并改善其功能, 并且仍然需要一种改进的软骨修复方法。
我们的 解决方案
CybroCell™ 是一种基于同种异体成纤维细胞的治疗退行性椎间盘疾病的方法。这项新技术被设计为修复椎间盘软骨(或任何其他关节软骨)的替代 方法。该方法是基于使用人 真皮成纤维细胞,或HDF,这是被迫分化成软骨细胞样细胞 体内using the mechanical force and intermittent hydrostatic pressure found in the spine, for chondrogenic differentiation of fibroblasts. We believe our solution will prove superior to existing treatments because it is less invasive, regenerates the disc, restores function and reduces pain, without debilitating long-term effects. We received IND clearance from the FDA on November 7, 2018, with an IND number of 18151. The trial is designed to assess the safety and efficacy of CybroCell™ administered through injection directly into a damaged intervertebral disc. The trial will enroll up to 15 participants with a primary outcome of safety and a secondary outcome of efficacy. Safety will be assessed as the occurrence/frequency of adverse events during the study procedures and for up to 12 months afterwards and will be recorded regardless of severity or relatedness to treatment. Serious adverse events will be documented throughout the 12-month follow-up period. The incidence and nature of adverse events will be tabulated and analyzed at baseline, pre, and post procedure including the three, six, and 12-months evaluations. These include complete physical exam (including vital signs of blood pressure, temperature, and heart rate), laboratory determinations (including urinalysis, hematology, and biochemistry), review of medical history, review of medication history, Dallas pain questionnaire, and patient questionnaire (Oswestry Disability Index). Secondary efficacy outcome will assess subjective and objective parameters at three months, six months, and 12 months post CybroCell™ implantation and compare to those measured at baseline pre-implantation. These parameters will include visual analog scale, patient questionnaire (Oswestry Disability Index), Beck Depression Inventory, Dallas pain questionnaire, range of motion test, and radiological assessment using MRI to evaluate morphological changes to the treated discs.
我们目前正在完成实验细胞库 的生产,该生产将转移至合同开发和生产组织(CDMO),以按照FDA要求生产 主细胞库和工作细胞库,并将向FDA提交必要的文件。我们已从 CDMO处获得了开展此项工作的报价。
我们已经完成了两项动物实验。第一项初步研究(PMID 27853661)中使用了16只动物3 目的是通过测量椎间盘高度、磁共振成像或MRI、信号强度、基因表达和胶原蛋白免疫染色来确定新生人皮肤成纤维细胞或nHDF 椎间盘内移植对椎间盘或IVD退变的影响。结果表明,nHDF组治疗8周后椎间盘高度指数增加10%,p值为 <.05 while there was no significant difference in the saline treated group. when compared with group discs nhdfs showed reduced expression of inflammatory markers a higher ratio collagen type ii over i gene and more intense immunohistochemical staining for both types ii. second study>4使用了38只动物,目的是 在比较兔真皮成纤维细胞或RDF与nHDF时,确定供体来源对真皮成纤维细胞治疗对椎间盘退变和炎症的治疗效果的影响 。治疗8周后,nHDF治疗的椎间盘高度指数显著增加7.8%(p<.01 whereas those treated with saline or rdf increased by and respectively. gene expression analysis showed that discs transplanted nhdfs rdfs displayed similar inflammatory responses to .8 compared intact of both collagen types i ii significantly in nhdf-treated trending significant rdf-treated not discs. the ratio type was higher ivds than last proteoglycan contents nhdf were toward significance rdf- saline. results from studies positive resulted human trial approval. technology allowed for differentiation hdfs into chondrocytes cells thrived spinal disc environment. remained did migrate. further created a biologic condition which appeared increase height.>
3资料来源: Chee A,Shi P,Cha T,Kao TH,Yang SH,Zhu J,Chen D,Zhang Y,An HS.人真皮成纤维细胞的细胞疗法在兔模型中增强椎间盘修复并减少炎症。Global Spine J. 2016年12月;6(8):771-779。doi:10.1055/s-0036-1582391。电子版 2016年4月13日。PMID:27853661; PMCID:PMC5110358。
4资料来源: Shi P,Chee A,Liu W,Chou PH,Zhu J,An HS.新生人真皮成纤维细胞和兔真皮成纤维细胞对椎间盘退变和炎症的细胞治疗作用。脊柱杂志2019年1月;19(1):171-181。doi:10.1016/j.spinee.2018.08.005。Epub 2018 Aug 22. PMID:30142460。
| 82 |
以下 是Howard An,M.D.的动物研究结果总结,拉什大学医学院脊柱奖学金项目主任:
我们的研究表明,这种使用人真皮成纤维细胞的生物治疗方法,在治疗椎间盘退变方面具有巨大的潜力。当这些细胞被注射到退变的兔椎间盘中时,它们在椎间盘中保留了长达8周。作为椎间盘修复和再生标志的II型胶原基因在经人真皮成纤维细胞处理的椎间盘中的表达高于对照组。细胞处理组的椎间盘高度和细胞数也较高。总而言之,这些数据 表明,人类真皮成纤维细胞是一种很有希望的细胞治疗选择,可以恢复生物功能,减轻中间或进行性退变的症状 。
我们 已获得FDA的IND批准,条件是我们的主细胞库获得批准,可以对患有退行性腰椎间盘疾病的患者进行1/2期研究,并将在美国境内进行这项研究。时间表将通过与FDA的讨论确定。
市场机会
退行性腰椎间盘疾病疗法每年的市场价值约为260亿美元。除治疗方法外,退行性腰椎间盘疾病仅在美国每年就导致约120万例整形外科手术,每个手术的费用约为60,000至100,000美元。如果证明Cybrocell™在再生椎间盘软骨和减轻疼痛方面取得成功,它可能会取代治疗方法或推迟许多此类手术。
治疗多发性硬化症的CYMS101
多发性硬化症
多发性硬化症(MS)被分为四种不同的临床亚型,在发病年龄、疾病的侵袭性和进展以及复发频率上有所不同。大多数MS病例(85%)遵循复发-缓解模式,或RRMS,在未经治疗的人群中平均每12至18个月复发 ,神经功能缺失的短期发作完全或几乎完全消失。 MS复发通常被定义为持续24小时的新的或恶化的症状,在没有发烧或感染的情况下发生。 其他患者可能过渡到更具侵袭性的疾病形式,称为继发性进展性MS,或将经历稳步进行性神经恶化,称为原发进展性MS。
多发性硬化症尚无主要指征检查,但疑似多发性硬化症的常见检查包括核磁共振检查、诱发电位检查、腰椎穿刺术/脊椎抽液及其他客观功能检查。
一旦确定多发性硬化症的诊断,将作为标准的临床实践进行持续的定期残疾测量测试。第一个残疾状态量表是由Kurtzke在1955年提出的,后来在1983年被加强为扩展的残疾状态量表,或EDSS。随着时间的推移,EDSS已成为比较大多数MS临床结果指标的标准。EDSS测量了8个神经功能系统,包括视觉、脑干、锥体、小脑、感觉、肠道/膀胱、精神/大脑和行走 (500米步行)。
其他残疾测量测试包括斯克里普斯神经评定量表,这是一种全面的神经评估;九孔针测试,测量手臂功能;以及计时25英尺步行测试,测量行走功能。也可以使用兰德36问题健康调查 ,这是管理型医疗组织和Medicare用来对成年患者的护理结果进行常规监测和评估的一般生活质量调查。
适用于多发性硬化症的 治疗方法
目前尚无已知的治疗多发性硬化症的方法,可用于治疗多发性硬化症的药物包括用于暂时发作的类固醇、疾病修正药物和针对特定症状的药物,如平衡、视力、痉挛、性功能障碍和膀胱或肠道控制。目前治疗多发性硬化症的药物的作用机制是阻断宿主免疫介导的神经攻击,以抑制或最大限度地减少髓鞘的进行性破坏。虽然这些药物可能会减少恶化的频率,并减缓疾病的进展,导致进一步的神经损伤,但它们中的任何一种都没有髓鞘或神经再生能力来恢复已经存在的累积损伤 。此外,随着疾病的进一步发展,这些药物中的任何一种有效地阻断免疫介导的髓鞘或神经破坏的能力变得更加迟钝。大多数多发性硬化症药物都有已确定的风险和副作用,包括“黑匣子”警告。
| 83 |
提供现有多发性硬化症治疗的主要 公司包括:
| ● | Biogen, Inc.: 在全球市场上的强势地位,加上多发性硬化症药物的多样化组合; | |
| ● | 霍夫曼-拉罗氏有限公司(俗称罗氏):专注于研究和开发活动,以开发治疗多发性硬化症的新药;以及 | |
| ● | 诺华公司:加大对创新分子研发的投资力度。 |
正在开发新的 疗法
神经治疗领域的研究和发展一直很活跃。正在研究治疗多发性硬化症的各种新分子 一些主要的制药公司正在强调改善与多发性硬化症相关的残疾。不同公司的流水线投资组合 包括具有不同作用机制的药物,预计将促进医生对它们的需求, 旨在未来几年改变治疗算法。
十年来,赛诺菲、强生、诺华制药等多家公司一直在投资治疗多发性硬化症,为患者带来高效、高效的新疗法。这些公司最近推出了针对最流行的MS的疗法。2020年8月,FDA批准了诺华制药的Kesimpta,这是唯一一种针对复发性MS患者的自我给药、靶向B细胞疗法,2021年3月,强生获得FDA批准,推出Ponvory作为治疗MS的每日口服药物。
批准的药物的成本、结果和质量是医生和患者的优先事项。医生在开发跨学科方法管理多发性硬化症方面发挥着重要作用,这是制造商专注于具有不同作用机制的新分子的关键原因。例如,TG治疗公司的S ublituximab最近被食品和药物管理局批准用于治疗复发形式的多发性硬化症,包括临床孤立综合征、复发缓解性疾病和活动性继发性进展性疾病。
这些最近批准的治疗多发性硬化症相关症状的药物的批准和商业化预计将在不久的将来促进全球多发性硬化症药物市场的增长。
我们的 解决方案
我们正在开发CYMS101作为一种基于同种异体成纤维细胞的疗法来治疗多发性硬化症。在完成了使用同种异体成纤维细胞的动物研究后,我们获得了墨西哥的批准,可以使用成纤维细胞成分进行针对多发性硬化症患者的临床研究,并已完成了名为“难治性多发性硬化患者耐受性成纤维细胞的可行性研究“这项研究是在五名参与者中进行的。这项研究的主要目标是评估安全性,次要目标是评估疗效。安全性研究的结果是,静脉注射耐受性成纤维细胞期间没有不良反应,16周监测期内全血计数无短期或长期影响 ,16周监测期内心电图结果无短期或长期影响。此外, 疗效研究结果包括:
| ● | 听觉序列加法测试(PASAT)。该测试是由Gronwell于1977年开发的,后来由Rao于1989年修订用于MS。该测试是一种认知功能的衡量标准,用于评估听觉信息处理速度和计算能力。在测试中,每隔3秒显示一位数字,患者必须将每个新数字添加到紧接其前的一个数字上。评分是60个可能答案中正确答案的总数。在16周的监测期间,所有患者的PASAT评分都有普遍的改善。 |
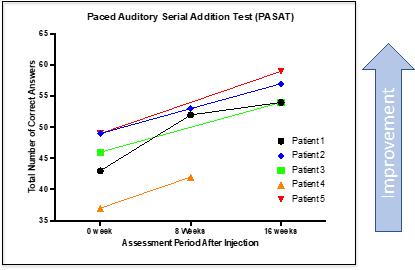
| 84 |
| ● | 九孔 桩试验。这是一项标准化的上肢功能定量测试。测试 是MS最常用的上肢功能指标。在测试中,对优势手和非优势手进行了两次测试。在测试中,一次拿起一个钉子,然后将 放入九个洞中的一个。一旦所有九个钉子都放好了,病人就会一次取下一个钉子,然后把它们放回容器里。打分是在孔中放置钉子,然后从孔中取出钉子所需的时间。在16周的测试期内,所有患者的九孔PEG测试完成时间都有了普遍的改善。 |
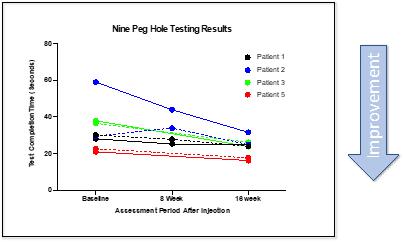
| ● | 计时 25英尺步行测试。这项测试是一项定量的活动能力和腿部功能测试。 在测试中,患者被指导快速而安全地走一条25英尺长的路径。通过让患者向后走相同的距离,再次进行测试。 测试的分数是两次完成步行的平均时间。计时的25英尺步行测试没有发现总体上的改善或恶化。 |
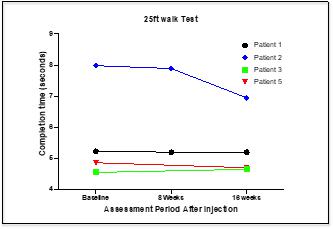
| ● | 扩展了 残疾状态量表(EDSS)。该测试用于量化MS中的残疾,并监控残疾级别随时间的变化。EDSS被广泛用于临床试验中,用于对MS参与者进行 评估,该量表基于神经学检查和对代表大脑神经元网络的功能系统的影响。由于复杂的评分规则和神经学测试的主观性质,EDSS评分可能变化很大。 EDSS测试没有发现总体改善或恶化,在研究期间也没有患者表现出 进一步恶化。 |
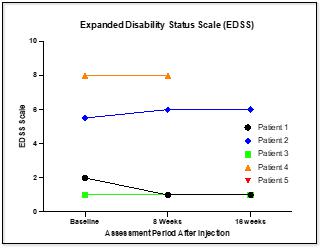
尽管确定作用模式不是申请FDA批准所必需的(Brown和Wobst,2020),而且到目前为止,已批准的药物中有10%-20%的药物尚未确定已知的作用模式(Moffat等人,2017),但我们目前正在进行进一步的研究,以确定成纤维细胞在少突胶质细胞扩增中的作用模式。我们将在美国提交多发性硬化症第二阶段临床试验的IND申请。虽然确定行动模式将是最佳的,但对可能的行动模式有一个大致的感觉将在开发、优化和缓解可能的副作用方面有切实的好处。我们可能会寻找战略合作伙伴,在启动第二阶段研究之前,或在研究完成后(如果成功),在开始第三阶段临床试验之前, 与我们合作开发CYMS101。
市场机会
多发性硬化症药物市场目前在全球的年收入约为240亿美元,其中48%的收入来自美国。这个市场上的主要公司包括生物遗传公司、F·霍夫曼-拉罗氏有限公司、赛诺菲和诺华制药。虽然批准的治疗方法有20多种,但大多数都有严重的不良反应,目前还没有治愈方法。私营和公共组织都在增加投资,以寻求这种复杂疾病的更好治疗方法,包括恢复丧失功能的治疗方法,以及政府改善发展中经济体获得多发性硬化症药物的举措,这是多发性硬化症市场未来增长的另一个驱动力。
| 85 |
CYWC 628用于伤口愈合
伤口护理/愈合
慢性伤口通常停留在炎症阶段,不能进展到愈合的增殖和重塑阶段。由坏死组织、异物和细菌产生的促炎细胞因子允许炎症阶段继续。 此外,细胞脱氧核糖核酸或DNA合成的变化会导致金属蛋白酶的形成增加,从而通过压倒正常伤口愈合所需的构建块(化学活性因子、生长因子和有丝分裂原)来阻碍身体的愈合尝试。成纤维细胞是伤口愈合过程中的重要细胞,在慢性伤口的形成过程中会发生表观遗传变化,因此其复制和产生肉芽组织所需的构建块的能力也会发生变化。此外,伤口周围的角质形成细胞表型不同,因此在能够增殖的同时,它们不能完全分化为迁移性角质形成细胞。这解释了伤口边缘经常可见的上皮堆积。
糖尿病足溃疡是最常见的慢性伤口类型。全球慢性病患病率上升,导致慢性伤口发病率增加,包括糖尿病足溃疡、压疮和静脉性腿部溃疡。这些慢性伤口,特别是晚期的“难以治愈的溃疡”,给全球医疗机构带来了巨大的经济成本负担。此外,超过50%的糖尿病足部溃疡被感染,这增加了住院、截肢和死亡的风险。
可用的 伤口护理治疗
目前有几种疗法可用于治疗慢性创面,包括Apligraf、Grafix、DermACELL和TheraSkin。Apligraf由新生角质形成细胞和牛胶原基质中的新生成纤维细胞组成,可用于治疗静脉性小腿溃疡和糖尿病足溃疡。Grafix是一种冷冻保存的人类胎盘膜,可用作伤口覆盖、包裹和/或屏障,用于治疗急慢性伤口、糖尿病溃疡、压力性损伤、手术伤口、烧伤和静脉溃疡。DermACELL是一种技术先进的真皮基质,由完整的细胞基质组成,可去除至少97%的DNA,可用于治疗糖尿病足部溃疡等慢性伤口。TheraSkin是一种冷冻保存的人类同种异体皮肤,具有表皮和真皮两层,可用于促进伤口愈合。
正在开发新的 疗法
皮肤移植和生长因子等生物活性疗法在糖尿病足部溃疡等伤口的紧急治疗中的应用日益增多,导致市场参与者在这些疗法的研发上投入了大量资金。因此,伤口快速愈合的先进生物活性疗法采用率的上升预计将推动伤口护理市场的增长。例如,2021年2月,Axio生物解决方案私人有限公司的MaxioCel高级伤口护理产品获得了欧洲的CE标志。生物活性超细纤维 凝胶技术可帮助伤口快速愈合。
负压伤口治疗(NPWT)提供了显著的 临床益处,再加上新的先进功能的引入,如单次使用和便携性等,以实现有效的伤口护理,推动了全球医疗专业人员对NPWT设备的需求。 2021年1月,Smith&Nephew plc发布了其Pico一次性负压伤口治疗系统,将手术部位感染和裂开显著减少了63.0%和30.0%。2019年1月,应用组织技术有限责任公司的平台伤口敷料-NPWT设备获得了FDA的批准 ,该设备消除了泡沫或纱布敷料的使用。2019年4月,PolarityTE,Inc. 启动了针对慢性伤口的SkinTE再生组织产品的临床试验。这些试验将评估Skinte在治疗糖尿病足溃疡和静脉性腿部溃疡方面的有效性。
此外,新的参与者正在进入伤口护理市场,专注于同种异体移植、异种移植、纳米纤维、真皮替代品和基于细胞的疗法 ,以满足未得到满足的需求和患者对紧急和有效治疗日益增长的需求。
| 86 |
我们的 解决方案
我们正处于开发CyWC628作为一种基于同种异体成纤维细胞的伤口愈合疗法的早期阶段。我们的研究目前集中在利用成纤维细胞和成纤维细胞来源的细胞治疗糖尿病小鼠和大鼠的伤口。根据我们迄今取得的成果,我们计划最早在2024年向FDA提交伤口愈合方面的IND申请。
市场机会
2021年,全球伤口护理市场规模约为170亿美元,其中一半以上的收入来自美国和欧洲,根据2022年3月发布的《财富商业洞察》,到2029年,预计将增长至约280亿美元。全球慢性病患病率上升导致慢性伤口发病率增加,包括糖尿病足部溃疡、压疮和静脉性腿部溃疡。慢性和急性伤口造成的巨大经济成本负担导致全球各国政府采取更多主动行动,在普通民众中提高对伤口早期诊断的认识 。这些举措,加上这些国家不断完善的伤口护理报销政策,预计将推动伤口护理产品的采用,并导致这一市场的持续增长。
我们的早期研究
延长寿命的CyTER915
延长寿命
成纤维细胞不再仅仅被认为是器官的结构成分,而是免疫过程中的动态参与者。成纤维细胞产生一种环境,影响调节性T细胞的迁移、增殖和活性,以确保免疫耐受。
免疫系统的关键器官之一是胸腺。它在T细胞的成熟和选择、消除自身反应性细胞、建立中枢耐受和T细胞迁移以识别广泛的病原体方面起着至关重要的作用。已经在胸腺内发现了各种细胞。这些细胞包括上皮细胞、胸腺细胞、树突状细胞或DC、巨噬细胞、B淋巴细胞、肌样细胞、内皮细胞和成纤维细胞。随着年龄的增长,胸腺的功能会下降,这一过程被称为胸腺或胸腺退化。文献 表明,退化过程增强了调节性T细胞的生成,从而增加了对病原体感染、肿瘤和自身免疫性疾病的易感性。
胸腺对免疫系统至关重要,免疫系统是人体的防御机制,提供对各种病原体、肿瘤、抗原和组织损伤介质的监视和保护。免疫系统由细胞和分子组成的复杂网络组成,分子分为胸腺非依赖性(先天)和胸腺依赖性(适应性)臂,它们在所有免疫反应中协同发挥作用。先天免疫是机体的第一道防线,由组织中的巨噬细胞、树突状细胞和粒细胞等先天免疫细胞在暴露抗原后数分钟至数小时内发挥效应。天然细胞通过胚系编码的模式识别受体激活,包括Toll样受体和核苷酸寡聚结构域样受体 ,它们识别病原体的不变特征(病原体相关的分子模式)和组织损伤。
一旦被激活,巨噬细胞和中性粒细胞等天然细胞就可以通过吞噬有效地清除抗原。其他类型的固有细胞,如DC,吸收和处理抗原,导致抗原表位与其主要组织相容性复合体或MHC或人类白细胞抗原分子一起表达。然后,这些DC可以作为抗原提呈细胞,启动适应性免疫系统。通过这种方式,早期的先天反应与获得性免疫相连,并促进了获得性免疫。
获得性免疫系统由T和B淋巴细胞组成,它们表达特异性抗原识别受体,并具有高度专门化的效应器功能,具有形成长期免疫记忆的能力。B细胞和T细胞都来自骨髓来源的祖细胞;而成熟的B细胞直接从骨髓输出到外周,T细胞的发育、成熟和输出 需要在胸腺发生关键的分化步骤。胸腺依赖的T细胞分化过程包括通过生殖系编码基因片段的重组表达抗原特异性细胞表面T细胞受体,以及胸腺“教育” 包括负选择潜在的自我反应性T细胞和正选择有能力识别外周遇到的抗原的T细胞。这些重要的胸腺过程确保T细胞可以在自身MHC的背景下识别抗原,但不会引发自我反应。
| 87 |
脾是关键的次级淋巴器官之一,负责免疫系统对血液中病原体的快速反应,并维持对这些病原体的长期适应性反应。脾也是铁代谢和红细胞动态平衡的关键器官。该器官也是血小板和白细胞的关键储存部位。在脾中已鉴定出多种细胞,包括内皮细胞、间皮细胞、网状细胞、红细胞、粒细胞、单核细胞、造血细胞、巨噬细胞、树突状细胞、浆细胞、CD4+和CD8+T细胞以及移行B细胞。随着年龄的增长,脾的结构和功能会发生变化,导致对疫苗接种的积极反应能力降低,对病毒和细菌病原体感染的易感性增加,自身免疫性疾病的发病率增加。因此,有必要通过细胞疗法改善和延长胸腺和脾的生产性寿命,这可能会通过击败在这些重要腺体衰退过程中被允许扩散的疾病来延长人类的寿命。
我们的 解决方案
我们的研究计划处于早期阶段,旨在再生或重振胸腺和/或脾的生产。 再生包括器官发生和/或T细胞发育,其中组织分化和/或上皮细胞的扩张 使用激活或灭活的成纤维细胞。除了成纤维细胞,我们预计还可以使用其他药物,如核酸、细胞因子、趋化因子、转录因子、表观遗传因子、生长因子、激素或它们的组合。可以激活细胞群 体外培养或离体。开发用于胸腺或脾退化逆转的成纤维细胞的下一步将是设计和进行临床前研究,以证明能否在动物模型中实现胸腺或脾退化逆转 。
市场机会
全球抗衰老药物市场预计在2021年超过5.0亿美元,随着全球老龄化人口和生活水平的提高,预计未来十年将经历两位数的年增长。老龄化人口对有效的再生药物解决方案的需求比以往任何时候都要高。因此,几家公司正在开发抗衰老疗法,使用干细胞和再生医学来识别、预防、治愈和逆转与年龄相关的功能障碍、疾病和疾病。
TCB190用于某些癌症的治疗
我们对某些癌症的研究才刚刚开始,有关这一机会的进一步信息将在可用时发布。
制造和供应
我们目前在德克萨斯州休斯敦的实验室生产我们的候选细胞治疗产品。我们正在与CDMO签订合同,转让我们的实验细胞库,以生产我们的主细胞库、工作细胞库和我们基于成纤维细胞的 候选产品,以便进行临床试验。如果我们的候选产品获得市场批准,我们将评估建立我们自己的cGMP制造设施或继续将生产外包给CDMO进行临床测试和商业供应的长期可行性 我们目前依赖第三方提供细胞疗法制造过程的某些部分,未来可能会继续这样做 。
知识产权
WE 成立于2021年4月,是FibroGenesis的衍生产品。在我们的组建过程中,我们向FibroGenesis发行了相当于8,750,000股A系列优先股,以换取专利转让协议或专利转让协议 和知识产权交叉许可协议。专利转让协议 将某些专利/应用程序的所有权利、所有权和利益从FibroGenesis转让给我们,而知识产权交叉许可协议 在FibroGenesis和我们之间分配,为已转让和保留的已颁发/待批专利分配独家使用领域。
| 88 |
通过专利转让协议和知识产权交叉许可协议,FibroGenesis有效地授予了我们在以下用途领域开发成纤维细胞的独家 权利:
| ● | 脊椎疾病、障碍或状况的诊断、治疗、预防和缓解; | |
| ● | 某些癌症; | |
| ● | 整形外科疾病、障碍或状况;以及 | |
| ● | 多发性硬化症。 |
对于转让给我们或由FibroGenesis保留的已颁发专利和专利申请,FibroGenesis 保留所有其他使用领域的独家权利。
已授予我们的专利和转让给我们的专利申请,以及我们在成立后独立提交的其他专利申请, 截至本文件之日,共有48项专利和109项专利申请正在申请中。我们发布的专利的专利保护期一般在2027到2043年之间到期。
我们所有的已授权专利均受《专利转让协议》的保护,包括在美国的10项已授权专利、在澳大利亚的八项已授权专利、在日本的四项已授权专利、在英国的四项已授权专利、在法国的三项已授权专利、在德国的三项已授权专利、在意大利的三项已授权专利、在西班牙的三项已授权专利、在香港的三项已授权专利、在加拿大的两项已授权专利、在中国的两项已授权专利、在瑞士的两项已授权专利,以及在欧洲的剩余已授权专利。我们的一项已颁发专利也包含在《知识产权交叉许可协议》中,该专利在美国颁发。
鉴于目前关于天然产品的专利不合格法律,目前没有涉及Cybrocell™的物质组成专利,尽管 有与生产Cybrocell™相关的专利。我们目前有针对CYMS101和CYWC628的物质成分 的专利申请正在申请中。
除了专利,我们还依靠商业秘密和技术诀窍来发展和保持我们的竞争地位。我们通常依靠商业秘密来保护我们业务中不受专利保护或我们认为不适合专利保护的方面。我们通过与员工、顾问、科学顾问、承包商和合作伙伴建立保密协议和发明转让协议来保护商业秘密和专有技术。这些协议一般规定,在个人或实体与我们的关系过程中开发或公布的所有机密信息必须在关系期间和之后保密。这些 协议还一般规定,为我们完成的工作或与我们的业务相关的工作产生的所有发明、构思或在受雇或转让期间完成的所有发明(如果适用)应为我们的专有财产。此外,我们还采取了其他适当的预防措施,如物理和技术安全措施,以防止我们的财产被挪用。
竞争
生物技术和制药行业的特点是技术快速进步、竞争激烈,并且非常重视专利和创新产品以及候选产品。我们的竞争对手已经开发、正在开发或可能开发与我们的候选产品竞争的产品、候选产品和工艺。我们成功开发和商业化的任何候选产品都将与现有疗法和未来可能推出的新疗法展开竞争。我们认为,有相当数量的 产品目前正在开发中,并可能在未来投入商业使用,用于治疗我们可能尝试开发的候选产品的条件。此外,我们的产品可能需要与医生使用的标签外药物竞争 以治疗我们寻求批准的适应症。这可能会使我们很难用我们的产品取代现有的疗法。
我们 正在开发CybroCellTM用于治疗退行性间盘疾病。我们在退行性腰椎间盘疾病市场上的竞争对手包括Aesculap植入物系统公司、诺华制药、辉瑞、礼来公司、DiscGenics公司、脊柱生物制药公司和Ferring B.V.。2021年7月,Aesculap植入物系统有限责任公司宣布了其Actil®人工椎间盘关键试验的长期报告。
我们 正在开发CYMS101作为一种基于同种异体成纤维细胞的疗法来治疗多发性硬化症。目前提供多发性硬化症治疗的主要公司包括生物遗传公司、F.霍夫曼-拉罗氏有限公司和诺华制药。赛诺菲和诺华制药等多家公司一直在投资治疗多发性硬化症,为患者带来高效、高效的新疗法。这些公司最近推出了针对最流行的MS的疗法。2020年8月,FDA批准了诺华制药的Kesimpta,这是唯一一种针对复发MS患者的自我给药、靶向B细胞疗法。2021年3月,强生获得FDA批准,推出Ponvory作为治疗TG女士的日常口服药物。用于治疗MS的适应症S的ublituximab最近也被FDA批准用于治疗MS。
我们正处于开发CyWC628作为一种基于同种异体成纤维细胞的伤口愈合疗法的早期阶段。我们面临着来自目前可用于治疗慢性伤口的几种疗法的竞争,包括Apligraf、Grafix、DermACELL和TheraSkin。此外,皮肤移植和生长因子等生物活性疗法在糖尿病足溃疡等伤口的紧急治疗中的应用日益增多,导致企业在这些疗法的研发上投入了大量资金。2019年1月,应用组织技术有限公司因其平台伤口敷料NPWT设备而获得FDA批准,该设备消除了泡沫或纱布敷料的使用。2019年4月,PolarityTE,Inc.启动了针对慢性伤口的SkinTE再生组织产品的临床试验。2021年1月,Smith &Nephew plc发布了其PICO一次性负压伤口治疗系统,将手术部位感染和裂开显著减少了63.0%和30.0%,2021年2月,Axio Biosolutions Private Limited因其MaxioCel高级伤口护理产品获得了欧洲CE认证,MaxioCel是一种生物活性超细纤维凝胶技术,有助于伤口快速愈合。
| 89 |
我们当前和潜在的许多竞争对手可能比我们拥有更多的财务、制造、营销、药品开发、技术以及人力资源和商业专业知识。尤其是大型制药和生物技术公司,在临床测试、获得监管批准、招募患者和制造生物技术产品方面拥有丰富的经验。这些公司 的研究和营销能力也比我们强得多,可能也有已获批准的产品或 处于开发后期阶段的产品,以及在我们的目标市场与领先公司和研究机构的合作安排。 老牌制药和生物技术公司还可能投入巨资加快新化合物的发现和开发 或授权使用可能使我们开发的候选产品过时的新化合物。生物技术和制药行业的合并和收购可能会导致更多的资源集中在数量较少的竞争对手中。规模较小或处于初创阶段的公司也可能成为重要的竞争对手,特别是通过与大型且成熟的公司的协作安排,以及在获取补充我们计划或为其提供必要技术方面。因此,我们的竞争对手可能会在我们之前获得FDA、EMA或其他类似外国监管机构的批准,或在我们之前发现、开发和商业化我们领域的产品。如果我们的竞争对手开发和商业化比我们可能开发的任何产品更安全、更有效、影响更少或更不严重、更方便、标签更宽、营销更有效、得到报销或更便宜的产品,我们的商业机会可能会减少或消失。我们的竞争对手开发的技术进步或产品 可能会使我们的技术或候选产品过时、缺乏竞争力或不经济。如果我们无法 有效竞争,我们通过销售我们可能开发的任何产品获得收入的机会,如果获得批准,可能会受到不利的 影响。
监管环境
政府 法规和产品审批
FDA和州和地方司法管辖区以及其他国家和司法管辖区的类似监管机构对参与我们正在开发的产品的临床开发、制造、营销和分销的公司提出了大量 和繁重的要求 。这些实体对我们候选产品的研究、开发、测试、制造、质量控制、包装、安全、有效性、标签、储存、记录保存、审批、广告、促销、分销、审批后监控和报告、抽样、出口和进口等进行监管。我们开发的任何候选产品必须获得FDA的批准,然后才能在美国合法销售,并必须得到相应的外国监管机构的批准,才能 在这些国家合法销售。通常,我们在其他国家的活动将受到与美国实施的类似 性质和范围的监管,尽管可能存在重要差异。
美国 产品开发流程
在美国,FDA根据美国联邦食品、药物和化妆品法(FDCA)监管药品,根据FDCA和公共卫生服务法(Public Health Service Act)及其实施条例监管生物制品。在美国以及其他国家和司法管辖区获得监管批准的流程,以及随后对适用的法规和法规的遵守,需要花费大量的时间和财力。在产品开发过程、批准过程或批准后的任何时间未能遵守适用的美国要求可能会使申请人和/或赞助商受到各种行政或司法制裁,包括FDA拒绝批准未决申请、撤回批准、实施临床搁置、发布警告信和其他类型的信、产品扣押、完全或部分暂停生产或分销、禁令、罚款、拒绝政府合同、恢复原状、返还利润、或FDA和美国司法部或其他政府实体提起的民事或刑事调查和处罚 。此外,申请者可能需要召回 产品。此外,我们的某些候选产品作为组合产品在美国受到监管。如果单独销售,每个组件将受到不同的监管途径,并需要FDA批准独立销售 申请。然而,组合产品被分配给FDA内的一个中心,该中心将根据对组合产品的主要作用模式的确定,对其监管拥有主要管辖权,这是提供最重要治疗作用的单一作用模式 。就我们的CybroCell™候选产品而言,我们认为主要的作用模式应归因于产品的生物成分。我们预计将通过BLA寻求该组合候选产品的批准 ,我们预计FDA不会要求该产品的每个药物和生物成分 单独获得营销授权。
| 90 |
FDA要求的新产品在美国上市前的流程通常涉及以下内容:
| ● | 根据FDA的良好实验室实践或GLP要求和其他适用法规,完成非临床或临床前实验室测试、动物研究和配方研究。 |
| ● | 向FDA提交IND,该IND必须在人体临床试验开始之前生效; |
| ● | 在每个临床试验开始之前,每个临床站点的IRB或伦理委员会的批准; |
| ● | 根据GCP要求进行充分且受控良好的人体临床试验,以确定拟用药物的安全性和有效性,或就生物制品而言,确定候选产品对每个拟用适应症的安全性、纯度和效力; |
| ● | 在所有关键试验完成后,向FDA提交NDA或BLA; |
| ● | FDA在收到NDA或BLA后60天内决定将申请提交审查 ; |
| ● | 令人满意的 完成FDA咨询委员会的审查,如果适用; |
| ● | 令人满意的 完成FDA对生产产品或其组件的一个或多个制造设施的一次或多次检查,以评估符合cGMP要求的情况 以确保设施,方法和控制足以保持药物的特性、强度、质量和纯度,以及选定的临床调查地点,以评估 对GCP的依从性; |
| ● | FDA可能对产生支持NDA或BLA的数据的临床前和/或临床试验地点进行审计;以及 |
| ● | FDA对NDA或BLA的审查和批准,以允许在美国使用的特定适应症的产品 进行商业营销。 |
临床前研究包括产品化学、毒性和配方的实验室评估,以及评估潜在安全性和有效性的体外和动物研究。临床前研究的实施受联邦法规和要求的约束,包括针对安全性/毒理学研究的GLP法规。
在美国开始对候选产品进行首次临床试验之前,我们必须向FDA提交IND。IND是FDA授权对人类进行研究的新药产品的请求。IND提交的中心焦点是临床试验的总体研究计划和方案(S)。即使在提交IND之后,一些临床前研究也可能继续进行。IND还包括动物和体外培养评估产品的毒理学、药动学、药理学和药效学特征的研究;化学、制造和控制信息;以及任何可用来支持研究产品使用的人类数据或文献。IND必须在人体临床试验开始前生效 。IND在FDA收到后30天自动生效,除非FDA在30天内对拟议的临床试验提出安全顾虑或问题。在这种情况下,IND可能被临床搁置,IND赞助商和FDA必须在临床试验开始之前解决任何悬而未决的问题或问题。因此,提交IND可能会导致FDA授权开始临床试验,也可能不会。
| 91 |
除了在美国启动临床试验前向FDA提交IND之外,涉及重组或合成核酸分子的某些人体临床试验 还必须接受NIH指南规定的地方层面的监督。 具体而言,根据NIH指南,对人类基因转移试验的监督包括由机构生物安全委员会(IBC)进行评估和评估,IBC是当地机构委员会,负责审查和监督该机构利用重组或合成核酸分子进行的研究。IBC评估研究的安全性,并确定对公共卫生或环境的任何潜在风险,这种审查可能会导致临床试验启动之前的一些延迟。虽然NIH指南不是强制性的 ,除非相关研究是在接受NIH重组或合成核酸分子研究资助的机构进行的或由其赞助的,但许多公司和其他不受NIH指南约束的机构自愿遵循这些指南 。
临床试验涉及在符合GCP要求的 合格研究人员的监督下给人类受试者服用研究产品,其中包括要求所有研究受试者或其法定代表人就其参与任何临床研究提供其 知情同意。临床试验是在详细说明纳入和排除标准、研究目标、用于监测安全性的参数和要评估的有效性标准的方案下进行的。在产品开发期间进行的每个后续临床试验以及后续的任何方案修改都必须单独提交给现有的IND。此外,提议进行临床试验的每个地点的独立IRB必须在该地点开始临床试验之前审查和批准任何临床试验的计划及其知情同意书,并且必须监督研究直到完成。IRB负责保护试验参与者的福利和权利 ,并考虑参与临床试验的个人的风险是否降至最低,以及相对于预期利益是否合理。监管机构、IRB或赞助商可随时以各种理由暂停临床试验,包括发现受试者面临不可接受的健康风险或试验不太可能达到其声明的目标 。一些临床试验还包括由临床试验赞助商组织的独立的合格专家小组的监督,该小组称为数据安全监测委员会或委员会,该委员会根据对试验的某些数据的访问授权 试验是否可以在指定的检查点进行,并可能建议 如果确定对受试者存在不可接受的安全风险或其他原因,如没有疗效证明 ,则可能建议停止临床试验。还有关于向公共注册机构报告正在进行的临床试验和临床试验结果的要求。
人类 临床试验通常分三个连续阶段进行,这些阶段可能重叠或合并:
| ● | 阶段 1:候选产品最初被引入健康的人体受试者,或者在某些情况下,如某些癌症、目标疾病或疾病的患者。 这些试验旨在测试研究产品的安全性、剂量耐受性、吸收、代谢 和在人体中的分布,与增加剂量相关的副作用,并在可能的情况下,获得有效性的早期证据。如果某些产品用于治疗严重或危及生命的疾病,如某些癌症,尤其是当产品本身毒性太大而无法合乎道德规范地给健康志愿者使用时, 最初的人体试验通常在患者身上进行。 |
| ● | 阶段 2:该候选产品适用于患有特定疾病或状况的有限患者群体,以评估初步疗效、最佳剂量、剂量耐受性和剂量计划,并确定可能的不良副作用和安全风险。在开始更大的 和更昂贵的3期临床试验之前,可能会进行多个2期临床试验以获取信息。 |
| ● | 阶段 3:将该候选产品应用于扩大的患者群体,以进一步评估剂量,提供临床疗效的统计显著证据,并进行安全性的进一步测试,通常在多个地理上分散的临床试验地点进行。 这些临床试验旨在确定研究产品的总体风险/收益比,并为医生标签提供充分的基础。通常,FDA需要两个充足且控制良好的3期临床试验才能批准NDA 或BLA。 |
上市后 研究,有时称为4期临床试验,可在初步上市批准后进行。这些临床试验用于从预期治疗适应症的患者的治疗中获得更多经验。在某些情况下,例如加速批准的药物,FDA可能会强制要求进行第四阶段临床试验作为批准的条件。
| 92 |
在新药开发期间,赞助商有机会在特定时间与FDA会面。这些要点可能在提交IND之前、在第二阶段临床试验结束时或在提交NDA或BLA之前。可以要求在其他时间召开会议。 这些会议可以为赞助商提供机会,让赞助商共享有关迄今收集的数据的信息,为FDA提供建议,并为赞助商和FDA就下一阶段的开发达成一致。赞助商通常利用第二阶段临床试验结束时的会议来讨论临床试验的结果,并提交他们认为将支持其新药批准的关键第三阶段临床试验计划。
在临床试验的同时,公司通常会完成额外的动物研究,还必须开发有关药物化学和物理特性的更多信息,并根据cGMP要求确定商业批量生产产品的工艺。制造过程必须能够始终如一地生产高质量的候选产品批次 ,此外,制造商还必须开发测试最终产品的特性、强度、质量和纯度的方法。 此外,必须选择和测试适当的包装,并且必须进行稳定性研究,以证明候选产品在保质期内不会发生不可接受的变质。
在IND处于活动状态且在产品批准之前,必须至少每年向FDA提交总结自上次进度报告以来进行的临床试验和非临床研究结果的进度报告,并且必须向FDA和调查人员提交书面IND安全报告,包括严重和意外的可疑不良事件、暴露于相同或类似药物的其他研究表明对人类有重大风险的其他研究结果、动物或体外试验的结果表明对人类有重大风险。 与方案或研究人员手册中列出的相比,任何临床上重要的可疑不良反应的发生率都会增加 。赞助商必须在确定该信息符合报告条件后15个日历日内提交IND安全报告。赞助商还必须在首次收到信息后的七个日历日内通知FDA任何意外的、致命的或危及生命的疑似不良反应。
美国 审核和审批流程
假设 根据所有适用的法规要求成功完成所有必需的测试,产品开发、临床前和其他非临床研究和临床试验的结果,以及对制造过程的描述、对药物化学进行的分析测试、建议的标签和其他相关信息将作为NDA或 BLA的一部分提交给FDA,以请求批准该产品上市。数据可能来自公司赞助的临床试验,旨在测试使用产品的安全性和有效性,或者来自许多替代来源,包括研究人员发起的研究。为支持上市批准,提交的数据必须在质量和数量上足够,以确定研究药物产品的安全性和有效性,使FDA满意。提交保密协议或BLA需要支付可观的使用费;在某些有限的情况下,可获得免除此类费用。此外,对于被指定为孤儿药物的产品,不会在NDA或BLA上评估使用费,除非该产品还包括非孤儿药物。
FDA审查NDA或BLA,以确定产品对于其预期用途是否安全有效,以及其制造是否符合cGMP,以确保和保持产品的特性、强度、质量和纯度。根据目前生效的处方 药物使用者费用法案(PDUFA)指南,FDA的目标是自提交标准NDA之日起十个月内对新的分子实体或BLA进行审查并采取行动。该审查通常需要12个月的时间,从NDA或BLA提交给FDA之日起算,因为FDA在提交申请后有大约两个月的时间做出“备案”决定 。FDA在接受备案之前,在提交后的前60天内对所有NDA或BLA进行初步审查,以确定它们是否足够完整,允许进行实质性审查。FDA可能会要求提供更多信息,而不是接受NDA或BLA申请。在这种情况下,必须重新提交保密协议或BLA以及附加的 信息。重新提交的申请在FDA接受备案之前也要进行审查。
| 93 |
FDA可以将新药的申请提交给咨询委员会。咨询委员会是由包括临床医生和其他科学专家在内的独立专家组成的小组,负责审查、评估申请,并就申请是否应获得批准以及在何种条件下批准提出建议。FDA不受咨询委员会建议的约束,但它在做出决定时会仔细考虑这些建议。
在批准保密协议或BLA之前,FDA通常会检查生产该产品的工厂。FDA将不会批准申请,除非它确定制造工艺和设施符合cGMP,并有足够的 确保产品在要求的规格下持续生产。此外,在批准NDA或BLA之前,FDA 通常会检查一个或多个临床地点,以确保符合GCP。如果FDA确定申请、制造工艺或制造设施不可接受,它将在提交中列出不足之处,并经常要求进行额外的 测试或提供信息。尽管提交了任何要求的附加信息,FDA最终仍可能判定该申请不符合审批的监管标准。
FDA评估保密协议或BLA后,可能会签发批准信或完整的回复信。批准函授权该药物的商业销售,并提供特定适应症的处方信息。完整的回复信表明申请的审查周期 已经结束,目前的申请将不会获得批准。完整的回复信通常描述FDA确定的NDA或BLA中的具体缺陷,可能需要额外的临床数据,例如额外的关键的3期临床试验或与临床试验、非临床研究或制造相关的其他重要且耗时的要求。 如果发出完整的回复信,赞助商必须重新提交NDA或BLA,以解决 信中确定的所有缺陷,或撤回申请。即使提交了这样的数据和信息,FDA也可能决定NDA或BLA不符合批准标准。
如果产品获得监管批准,则此类批准将针对特定适应症授予,并可能包含对此类产品可能上市的指定用途的 限制。例如,FDA可能会批准带有REMS的NDA或BLA,以确保产品的好处大于其风险。RMS是一种安全策略,用于管理与药物相关的已知或潜在严重风险,并通过管理药物的安全使用使患者能够继续获得此类药物,可能包括药物指南、医生沟通计划或确保安全使用的要素,如受限分配方法、患者登记和其他 风险最小化工具。FDA还可能以更改拟议的标签或制定适当的控制和规范为条件进行批准。一旦获得批准,如果没有遵守上市前和上市后的要求,或者产品上市后出现问题,FDA可能会撤回产品批准。FDA还可能要求进行一项或多项上市后研究和监测,以进一步评估和监控产品在商业化后的安全性和有效性,并可能根据这些上市后研究的结果 限制产品的进一步销售。此外,可能会建立新的政府要求, 包括新立法产生的要求,或者FDA的政策可能会改变,这可能会影响监管审批的时间表 或以其他方式影响正在进行的开发计划。
孤儿 药品名称
根据《孤儿药品法》,FDA可以将用于治疗罕见疾病或疾病的药物授予孤儿称号,这种疾病或疾病在美国影响不到200,000人,如果在美国影响超过200,000人,则无法合理预期在美国开发和生产针对此类疾病或疾病的药物产品的成本将从产品的销售中收回。在提交保密协议或BLA之前,必须申请孤立指定。在FDA授予孤儿称号后,FDA将公开披露治疗剂的身份及其潜在的孤儿用途。孤立指定不会在监管审批流程中传达任何优势,也不会缩短流程持续时间 。孤儿指定使一方有权获得财政激励,例如为临床试验费用提供赠款资金的机会、 税收优惠和用户费用减免。
| 94 |
如果具有孤儿指定的产品随后获得FDA对其具有此类指定的疾病或病症的第一次批准,则该产品有权获得孤儿产品独家经营权,这意味着FDA在七年内不得批准任何其他申请,以销售相同适应症的相同药物或生物制品,除非在有限情况下,例如显示 临床优于具有孤儿独占权的产品。但是,竞争对手可能会获得不同产品的批准,以获得 孤立产品具有排他性的指示,或者获得相同产品的批准,但获得不同指示的 孤立产品具有排他性。如果 竞争对手获得FDA定义的相同药物的批准,或者如果我们的候选产品被确定包含在 竞争对手的产品中用于相同的适应症或疾病,则孤立独占也可能在七年内阻止我们的产品获得批准。如果指定的孤立产品获得的营销批准范围大于指定范围,则可能无权获得独家销售。此外,如果FDA后来确定指定请求存在重大缺陷,或者制造商 无法保证足够数量的产品来满足患有这种罕见疾病或疾病的患者的需求,则可能会失去在美国的独家营销权。
加快开发和审查计划
FDA有许多计划旨在加快符合特定标准的产品的开发或审查。例如,如果新药用于治疗严重或危及生命的疾病或病症,并显示出 满足该疾病或病症未得到满足的医疗需求的潜力,则有资格获得快速通道指定。快速通道指定适用于产品 和正在研究的特定适应症的组合。快速通道产品的赞助商在产品开发期间有机会与审查团队进行更频繁的互动 ,FDA可以在提交完整申请之前考虑滚动审查NDA或BLA的部分,如果赞助商提供了提交NDA或BLA部分的时间表,FDA 同意接受NDA或BLA的部分,并确定时间表是可接受的,赞助商在提交NDA或BLA的第一部分时支付任何必要的用户费用 。
产品,包括具有Fast Track称号的产品,也可能符合FDA旨在加快 开发和审查的其他类型计划的资格,例如优先审查和加速审批。如果产品有可能在没有令人满意的替代疗法的情况下提供安全有效的治疗,或者与市场上销售的产品相比,在治疗、诊断或预防疾病方面有显著改善,则有资格优先审查该产品。FDA将尝试将额外资源用于评估指定优先审查的新药申请,以努力促进审查。FDA努力在提交日期后6个月内审查具有优先审查指定的申请 ,而根据其当前的PDUFA审查目标,标准NDA或BLAS的审查时间为10个月 。
此外,产品可能有资格获得加速审批。用于治疗严重或危及生命的疾病或病症的产品在确定该产品对可合理地 预测临床益处的替代终点有效,或对可在不可逆转发病率或死亡率之前测量的临床终点有效时, 可合理预测对不可逆转发病率或死亡率或其他临床益处的影响的产品有资格获得加速批准,同时考虑疾病的严重性、罕见性或流行率以及可用或缺乏替代治疗。作为批准的条件,FDA可以要求获得加速批准的药物的赞助商进行充分和良好控制的上市后临床试验 。此外,FDA目前要求将促销材料的预先审批作为加速审批的条件,这可能会对产品的商业发布时间产生不利影响。
用于治疗严重或危及生命的疾病或状况的候选产品也有资格获得突破性治疗 指定,以加快其开发和审查。如果初步临床证据 表明,产品单独或与一种或多种其他药物或生物制品联合使用,可能在一个或多个临床重要终点显示比现有疗法有显著改善,例如在临床开发早期观察到显著的治疗效果,则该产品可获得突破性的治疗指定。该指定包括Fast Track计划的所有功能,以及早在第一阶段就开始的更密集的FDA互动和指导,以及加快候选产品开发和审查的组织承诺,包括 高级经理的参与。
| 95 |
此外,FDA可能会将一种产品指定为再生医学高级疗法或RMAT。指定RMAT旨在促进对符合以下标准的任何候选产品的有效开发计划和快速审查:(I)候选产品 符合RMAT的定义,其定义为细胞疗法、治疗性组织工程产品、人体细胞和组织产品或使用此类疗法或产品的任何组合产品;(Ii)候选产品旨在治疗、修改、逆转或治愈严重或危及生命的疾病或状况;以及(Iii)初步临床证据表明,候选产品有可能解决此类疾病或状况未得到满足的医疗需求。RMAT指定提供了潜在的 好处,包括更频繁地与FDA开会讨论候选产品的开发计划,以及滚动审查和优先审查BLAS的资格。获得RMAT资格的细胞治疗候选者也有资格根据合理地可能预测长期临床益处的替代物或中间终点,或依赖于从大量地点获得的数据,包括适当地扩展到其他地点,获得加速批准。获得加速批准的RMAT指定单元候选疗法可视情况通过完成临床试验和患者登记,或通过提交其他真实证据来源(如电子健康记录),通过收集更大的验证性数据集,或通过在批准治疗之前对所有接受此类治疗的患者进行批准后监测,来满足批准后的要求。
快速跟踪指定、突破性治疗指定、优先审查、RMAT指定和加速审批不会改变审批标准 ,但可能会加快开发或审批过程。即使候选产品符合其中一个或多个计划的资格, FDA也可以在以后决定该候选产品不再符合资格条件,或决定不会缩短FDA审查或批准的时间段。
审批后要求
根据FDA批准生产或分销的任何产品均受FDA普遍和持续的监管,其中包括与记录保存、不良经历报告、定期报告、产品抽样和分销以及产品广告和促销相关的要求。批准后,对已批准产品的大多数更改,如增加新的 适应症或其他标签声明,均需事先接受FDA的审查和批准。还有持续的使用费要求,根据这一要求,FDA将评估批准的保密协议或BLA中确定的每种产品的年度计划费用。药品和生物制造商 及其分包商必须向FDA和某些州机构注册其机构,并接受FDA和某些州机构的定期 突击检查是否符合cGMP,这对我们和我们的第三方制造商施加了某些程序和文件要求 。对制造流程的更改受到严格监管,根据更改的重要性,可能需要FDA事先批准才能实施。FDA法规还要求调查和纠正与cGMP的任何偏差,并对我们和我们可能决定使用的任何第三方制造商提出报告要求。因此,制造商必须继续在生产和质量控制领域花费时间、金钱和精力,以保持符合cGMP和其他方面的法规遵从性。
如果没有遵守监管要求和标准,或者产品上市后出现问题,FDA可能会撤回批准。如果后来发现产品存在以前未知的问题,包括未预料到的严重程度或频率的不良事件,或与制造工艺有关的不良事件,或未能遵守法规要求,可能会导致修订已批准的标签以添加新的安全信息;实施上市后研究或临床试验以评估新的安全风险;或根据REMS计划实施分销限制或其他限制。
除其他事项外,其他 潜在后果包括:
| ● | 限制产品的销售或制造,将产品从市场上完全撤回或召回; |
| ● | 罚款、 警告信或无标题信; |
| ● | 临床 坚持临床试验; |
| ● | FDA拒绝批准待决申请或已批准申请的补充申请,或暂停或撤销产品许可证批准; |
| ● | 产品 扣押或扣押,或者拒绝允许产品进出口的; |
| ● | 同意 法令、企业诚信协议、取消联邦医疗保健计划的资格或将其排除在外; |
| ● | 强制 修改宣传材料和标签,并发布更正信息; |
| ● | 发布安全警报、亲爱的医疗保健提供者信函、新闻稿和包含有关产品的警告或其他安全信息的其他通信 ;或 |
| ● | 禁令或施加民事或刑事处罚。 |
| 96 |
FDA还可能要求进行上市后测试,即所谓的第四阶段测试,并进行监控以监控已批准产品的效果。新发现或开发的安全性或有效性数据可能需要更改产品已批准的标签,包括添加新的警告和禁忌症,还可能需要实施其他风险管理措施。
FDA严格监管药品的营销、标签、广告和促销。一家公司只能提出那些与安全性和有效性、纯度和效力有关的声明,这些声明是由FDA批准的,并符合批准的标签的规定。 FDA和其他机构积极执行禁止推广非标签用途的法律和法规。不遵守这些要求可能会导致负面宣传、警告信、改正广告以及潜在的民事和刑事处罚。医生可根据其独立的专业医学判断,为产品标签中未说明且与我们测试并经FDA批准的产品不同的用途开出合法可用产品。医生 可能认为,在不同的情况下,这种非标签使用是许多患者的最佳治疗方法。FDA不规范医生在选择治疗时的行为。然而,FDA确实限制了制造商在产品标签外使用问题上的沟通。联邦政府已对涉嫌不正当使用标签外促销的公司处以巨额民事和刑事罚款,并禁止公司从事标签外促销活动。FDA和其他监管机构还要求公司签订同意法令或永久禁令,根据这些法令或永久禁令,改变或限制特定的促销行为。然而,公司可能会分享与FDA批准的产品标签一致的真实且不具误导性的信息。
此外,处方药生物医药产品的分销受《处方药营销法》(PDMA)的约束,该法案 监管联邦一级的药品和药品样品的分销,并为各州对药品分销商的注册和监管设定最低标准。PDMA和州法律都限制了处方药生物制药产品样品的分发 ,并要求确保分发中的责任。
生物仿制药和参考产品排他性
《2009年生物制品价格竞争与创新法》(简称BPCIA)为那些与FDA批准的参考生物制品高度相似、或与其高度相似或可互换的生物制品开辟了一条简化的审批途径。FDA已经发布了几份指导文件,概述了审查和批准生物仿制药的方法。生物相似性,即要求生物制品和参考制品在安全性、纯度和效力方面不存在临床上有意义的差异, 通常通过分析研究、动物研究和一项或多项临床试验证明。互换性要求 产品与参考产品生物相似,并且该产品必须证明其在任何给定患者中都能产生与参考产品相同的临床结果,对于多次给药的产品,生物制剂和参考生物制剂可以在先前给药后交替或交换,而不会增加安全风险或与独家使用参考生物制剂相比疗效降低的风险。被证明与FDA批准的参考生物制品生物相似或可互换的产品可能在一定程度上依赖于FDA先前对待批准的参考产品的安全性和有效性的确定 ,这可能会减少获得批准将该产品推向市场所需的成本和时间。
根据BPCIA,生物相似产品的申请必须在参考产品首次获得FDA许可的四年后才能提交给FDA。此外,FDA对生物相似产品的批准可能要到参考产品首次获得许可之日起12年后才能生效。在这12年的独占期内,如果FDA批准竞争产品的完整BLA,且该竞争产品包含申请人自己的临床前数据和来自充分且控制良好的临床试验的数据,以证明其产品的安全性、纯度和效力,则另一家公司可能仍在销售该参考产品的竞争版本。BPCIA还为被批准为可互换产品的生物仿制药设立了某些排他性期限。在这一关头, 尚不清楚被FDA视为“可互换”的产品是否真的会被受国家药剂法管辖的药房所取代。
生物制品也可以在美国获得儿科市场的排他性。如果授予儿科专营权,将在现有专营期和专利条款上增加六个月 。这项为期六个月的专营权从其他专有性保护或专利期结束时开始,可根据FDA发布的此类研究的书面请求,在自愿完成儿科研究的基础上授予。
| 97 |
数据 隐私和安全
其他联邦立法可能会影响我们在研究活动中获取某些健康信息的能力。我们可能 受到联邦政府和我们开展业务的州的数据隐私和安全监管。经2009年健康信息技术促进经济和临床健康法案及其实施条例修订的1996年《健康保险可携带性和责任法案》,统称为HIPAA,规定了保护个人可识别健康信息的隐私、安全和传输方面的义务,包括强制性合同条款。HIPAA还禁止在知情的情况下故意伪造、隐瞒或掩盖重大事实,或作出任何重大虚假、虚构或欺诈性的陈述或陈述,或作出或使用明知其包含任何重大虚假、虚构或欺诈性陈述或条目的任何虚假书写或文件 与医疗福利、项目或服务的交付或付款相关的任何虚假、虚构或欺诈性陈述或条目。 我们可能会从受HIPAA隐私和安全要求约束的第三方(如研究机构)获取健康信息。虽然除提供某些员工福利外,我们不直接受到HIPAA的约束,但如果我们、我们的附属公司或我们的代理故意获取或披露由HIPAA覆盖的实体以未经HIPAA授权或允许的方式维护的可单独识别的健康信息,我们可能会受到刑事处罚。
此外,许多涉及隐私和数据安全的联邦和州法律法规,包括州数据泄露通知 法律、州健康信息隐私法以及联邦和州消费者保护法(例如,联邦贸易委员会法案第5条)管理与健康相关的个人信息和其他个人信息的收集、使用、披露和保护。不遵守数据保护法律和法规可能会导致政府采取执法行动,并为我们带来责任,这可能包括 民事和/或刑事处罚、私人诉讼和/或可能对我们的业务产生负面影响的负面宣传。
如果 未能达到并持续遵守适用的联邦和州隐私、安全和欺诈法,可能会导致政府采取执法行动并为我们承担责任,这可能包括民事和/或刑事处罚、私人诉讼和/或负面宣传 ,这可能会对我们的运营结果和业务产生负面影响。
其他 美国法规要求
生物制药 公司受到联邦政府以及它们开展业务的州和外国司法管辖区当局的额外医疗法规和执法的约束,这可能会限制我们通过 进行研究以及销售、营销和分销我们获得营销授权的任何产品的财务安排和关系。此类法律包括但不限于州和联邦反回扣、欺诈和滥用、虚假声明以及与药品定价和付款以及向医生和其他医疗保健提供者进行的其他价值转移有关的透明度法律和法规。如果他们的运营被发现违反了任何此类法律或任何其他适用的政府法规,他们可能会受到惩罚,包括但不限于行政、民事和刑事处罚、损害赔偿、罚款、返还、削减或重组运营、 诚信监督和报告义务、被排除在联邦和州医疗保健计划之外以及负责任的 个人可能被监禁。
| 98 |
承保 和报销
任何产品的销售 在一定程度上取决于第三方付款人对该产品的承保范围,例如联邦、州和外国政府医疗保健计划、商业保险和托管医疗机构,以及第三方付款人对该产品的报销水平。在美国,第三方付款人之间没有统一的产品承保和报销政策,产品的承保和报销级别因付款人而异。联邦医疗保险和医疗补助计划 越来越多地被用作私人支付者和其他政府支付者如何制定药品和生物制剂的保险和报销政策的典范 。付款人在确定报销时考虑的因素基于产品是否(I)其健康计划下的承保福利 ,(Ii)安全、有效和医学上必要的,(Iii)适合特定患者,(Iv)具有成本效益的 和(V)既不是试验性的也不是研究性的。关于将提供的补偿范围和金额的决定 是在逐个计划的基础上作出的。这些第三方付款人越来越多地减少对医疗产品、药品和服务的报销。 对于在医生监督下管理的产品,获得保险和足够的报销可能特别困难 ,因为此类药品通常价格较高。
此外,美国政府、州立法机构和外国政府继续实施成本控制计划,包括 价格控制、覆盖范围和报销限制以及非专利产品替代要求。采取价格控制和成本控制措施,以及在拥有现有控制和措施的司法管辖区采取更严格的政策,可能会 进一步限制任何产品的销售。减少任何产品的第三方报销或第三方付款人决定不承保产品可能会减少医生使用量和患者对产品的需求,还会对销售产生实质性的不利影响。
医疗保健 改革
2010年3月,ACA颁布,极大地改变了政府和私营保险公司为医疗保健提供资金的方式。 并显著影响了生物制药行业。ACA包含许多条款,包括管理联邦医疗保健计划的登记、报销调整以及对欺诈和滥用法律的修改。此外,ACA:
| ● | 增加 品牌药品制造商应支付的最低医疗补助回扣水平从15.1% 平均制造商价格的23.1%; |
| ● | 必需 对医疗补助管理的医疗机构支付的药品收取回扣; |
| ● | 必需 制造商必须同意参加覆盖范围差距折扣计划 提供50%(根据2018年两党预算法案增加到70%,自 2019年1月1日)适用品牌药品协议价格的销售点折扣 作为制造商 Medicare Part D承保的门诊药物;以及 |
| ● | 对向指定的联邦政府计划销售“品牌处方药”的药品制造商或进口商征收不可扣除的年费。 |
自ACA颁布以来,已提出并通过了其他 立法变更,包括每个财政年度将向 提供者支付的Medicare付款总额减少2%。此外,最近政府加强了对制造商 为其销售产品定价的方式的审查,这导致了几次国会调查,提出并颁布了立法和前特朗普政府发布的行政命令,旨在提高产品定价的透明度,审查 定价与制造商患者计划之间的关系,改革药品政府项目报销办法。美国各州也越来越积极地实施旨在控制 药品定价的法规,包括价格或患者报销限制、折扣、对某些产品获取的限制 以及营销成本披露和透明度措施,在某些情况下,这些法规旨在鼓励从其他国家进口和 批量购买。
国际 法规
为了 在美国境外销售任何产品,公司还必须遵守其他国家和司法管辖区关于质量、安全性和有效性的众多不同的监管要求 ,并管理(除其他事项外)临床试验、营销 授权、商业销售和产品分销。无论是否获得FDA对产品的批准,申请人都需要 获得类似外国监管机构的必要批准,然后才能在这些国家或司法管辖区开始临床试验或销售 产品。
| 99 |
对美国以外的候选产品的监管因国家/地区而异。某些国家将人体组织产品 作为药品进行监管,这将要求我们在销售我们的候选产品 之前进行大量备案并获得监管批准。某些其他国家将我们的候选产品归类为用于移植的人体组织,但可能会限制其进口或 销售。其他国家/地区可能没有关于进口或销售与我们的候选产品类似的产品的应用法规, 这对我们可能需要满足的标准造成了不确定性。
员工
截至 2023年12月31日,我们有10名全职员工,其中6名员工拥有医学或博士学位, 8名员工直接从事研发工作,其余员工提供行政、业务和运营支持。 我们的员工没有工会代表,也没有集体谈判协议。我们认为与 员工的关系良好。
我们的 设施
我们的 主要行政办公室位于455 E。医疗中心大道,套房300,休斯敦,得克萨斯州,在那里我们租用约23,000平方英尺的办公空间。该空间是我们公司总部的所在地。租约将于二零二七年四月到期。此外, 我们还在德克萨斯州休斯顿租赁了研究实验室和办公室,用于我们的研究和细胞制造业务。
我们 相信,我们的设施足以满足我们当前和预期的近期需求,如果需要,我们将提供适当的额外或替代空间 。
法律诉讼
我们可能不时成为日常业务过程中产生的诉讼的一方。我们目前并非任何重大 法律程序的当事方,且据我们所知,目前并无任何重大法律程序待决或受到威胁。无论 结果如何,由于辩护和和解成本、管理资源转移和其他 因素,诉讼都可能对我们产生不利影响。
| 100 |
管理
执行官员
下表载列截至本招股说明书日期有关本公司行政人员的若干资料:
| 名字 | 年龄 | 职位 | ||
| 皮特·奥希隆,MSHA | 60 | 创始人、 董事长兼首席执行官 | ||
| 马克·安德森,注册会计师CFA | 52 | 首席财务官 | ||
| 哈米德·霍贾,博士。 | 54 | 首席科学官 |
以下是我们高管经验的传记总结。
皮特·奥希隆, MSHA。皮特·奥希隆创立了我们的公司,自2021年4月公司成立以来,一直担任我们的首席执行官、董事长和董事会成员。奥赫隆先生也是我们的附属公司FibroGenesis的创始人,自2006年1月以来一直担任FibroGenesis的首席执行官。奥赫隆先生是一位杰出的生物制药发明家,在生物制药、细胞治疗和医疗设备领域拥有300多项已颁发和正在申请的专利。 奥赫隆先生是该领域经验丰富的领导者,拥有超过25年的医疗技术和生物技术开发经验。作为首席执行官,他的目标是使我们成为基于成纤维细胞的细胞疗法的全球领导者,开发可以治愈和治疗慢性病患者的疗法并将其商业化。奥赫隆先生汇集了将独特技术商业化所需的多学科团队和资源。在创建我们的公司和FibroGenesis之前, 他创建了运营投资集团Advanced Medical Technologies,LLC,该集团在2006年发现了医疗领域具有强大知识产权潜力的早期机会。他还于1998年创立了NeoSurg Technologies,该公司开发了T2000微创接入系统。NeoSurg Technologies于2006年被出售给库珀外科公司。奥赫隆先生还曾在1988至1995年间在Christus Health Care Corporation担任过多个高管级别的职位,并在生物制剂、先进手术器械和远程医疗领域为医疗保健公司提供了战略性的咨询服务。奥希隆先生拥有德克萨斯州立大学的医疗保健管理学士学位,休斯敦大学克利尔湖大学的医疗保健管理硕士学位,以及芝加哥大学的并购管理高级管理证书。根据我们对奥赫隆先生的经验、资历、属性和技能(包括共同创立我们的公司以及他在生物技术行业的行政领导经验)的审查,我们相信他有资格担任我们的董事会成员。
马克·安徒生,注册会计师CFA。马克·安德森自2022年6月以来一直担任我们的首席财务官。在加入我们之前,Andersen先生在2016年5月至2022年5月期间在印第安纳州印第安纳波利斯的印第安纳生物科学研究所担任首席财务官兼行政副总裁总裁。在该职位上,他负责该研究所的财务、人力资源、法律和信息技术。安徒生帮助该研究所创建了运营基础设施,协助筹款,并为捐赠投资组合提供监督,该投资组合已增长至近1.5亿美元。在此之前, 2015年8月至2016年2月,安徒生先生担任MiMedx副财务兼企业总监总裁,负责美国证券交易委员会的报告和财务职能。此前,从2004年1月至2015年8月,安徒生先生在礼来公司担任过多个财务领导职务,包括负责公司养老金计划的投资董事、负责合并和收购的财务董事以及礼来美国公司的财务总监。Andersen先生拥有南犹他大学会计学学士学位和会计学硕士学位,以及密歇根大学罗斯商学院工商管理硕士学位。
| 101 |
哈米德·霍贾博士 哈米德·霍贾自2021年8月以来一直担任我们的首席科学官。Khoja博士在科学团队、基于细胞的基因组、蛋白质组、表观遗传学分析以及用于药物发现和开发以及临床诊断的工具、方案和技术方面拥有超过25年的领导经验。在加入我们之前,Khoja博士在2009年3月至2021年8月期间担任Covaris,LLC首席科学家,Covaris,LLC是一家私营科学工具公司,重点研究基因组学、表观遗传学和蛋白质组学,他向首席执行官提供长期战略应用建议,管理产品和应用开发的外部合作,评估用于收购和OEM机会的新技术,并在许多科学会议上展示海报和演讲。Khoja博士领导了将Covaris技术成功地整合到Illumina下一代测序技术协议中的努力,导致超过15,000篇引用。Khoja博士还开发了Covaris染色质免疫沉淀方法,在同行评议的出版物中被引用了3,000多篇,并领导了使用Covaris技术简化表观遗传学分析工作流程的努力,用于药物开发和发现以及临床应用。 Khoja博士还领导了与美国国家癌症研究所的合作,成功地开发了使用Covaris技术的微生物组DNA提取,并完成了FDA EUA Sara-CoC-2桥梁研究设计,以批准新的样本采集和使用Covaris技术的病毒核糖核酸提取。Khoja博士还开发了一种制造合成无细胞DNA的专利工作流程,用作基于测序的液体活组织临床肿瘤学分析的参考标准。在加入Covaris之前,Khoja博士于2022年3月至2009年3月领导开发用于制药 行业药物开发的高通量蛋白质结晶平台,管理科学应用小组,在科学会议上介绍公司资源,并评估用于收购和OEM机会的新技术。在Sequenom公司2000年1月至2003年3月的创业阶段,Khoja博士建立了高度多重聚合酶链式反应(PCR)的方法学,用于开发Sequenom的基于MALDI-TOF MS的单核苷酸多态和遗传病分析技术。Khoja博士利用Sequenom的MassExTEND技术开发了基于MS的血色素沉着症、囊性纤维化和十种以犹太为主的遗传病的诊断分析方法,然后将其转移到一家大型临床诊断公司。Khoja博士还曾于1998年11月至1999年9月在礼来公司工作,并于1995年10月至1998年10月在奇龙公司工作。在礼来公司的职业生涯中,Khoja博士利用1999年提供的各种测序策略和生物信息学工具建立了高通量的PCR和测序策略,以获得高覆盖率的基因组测序,从而最终完成了有史以来第一个肺炎链球菌基因组的完整序列 。在后来被诺华公司收购的CHIRON公司,Khoja博士帮助设计、开发和优化了FGFR、VEGF、PDGF和EPO受体的HTP结合分析,鉴定了新的g蛋白偶联的七个跨膜受体,鉴定了参与肿瘤坏死因子信号通路的新蛋白质,并开发了基于分支DNA的HTP筛选 用于配体诱导的癌基因定量。
Khoja博士在南加州大学获得分子生物学学士学位,在波士顿大学获得分子生物学博士学位。
非雇员董事
下表列出了截至本招股说明书发布之日有关我们董事会中非雇员成员的某些信息 :
| 名字 | 年龄 | 职位 | ||
| Robert Hoffman,CPA(非在职) | 58 | 董事 | ||
| 维多利亚 Niklas,医学博士 | 65 | 董事 | ||
| 理查德 小奇伦托MBA | 61 | 董事 | ||
| Stacy 科恩,MBA | 53 | 董事 | ||
| Matthew 链路 | 49 | 董事 |
以下为非雇员董事的履历摘要。
Robert Hoffman,CPA(inactive).Robert Hoffman has served on our board of directors since April 2021. Mr. Hoffman currently serves as President, Chief Executive Officer and Chairperson of the board of directors of Kintara Therapeutics, Inc. (Nasdaq: KTRA), a clinical stage, biopharmaceutical company focused on the development and commercialization of new cancer therapies, a member of the board of directors of ASLAN Pharmaceuticals Limited (Nasdaq: ASLN), an oncology-focused biotechnology company developing a portfolio of immuno-oncology agents and targeted therapies, and Chairperson, and a member, of the board of directors of Antibe Therapeutics Inc., a Toronto, Canada-based pharmaceutical company listed on the Toronto Stock Exchange. Mr. Hoffman previously served as Senior Vice President and Chief Financial Officer of Heron Therapeutics, Inc., (Nasdaq: HRTX), a commercial-stage biotechnology company, from April 2017 to October 2020, and as Chief Financial Officer of AnaptysBio, Inc. (Nasdaq: ANAB), a specialty pharmaceutical company, from July 2015 to September 2016. From June 2012 to July 2015, Mr. Hoffman served as the Senior Vice President, Finance and Chief Financial Officer of Arena Pharmaceuticals, Inc., or Arena, a biopharmaceutical company, prior to its acquisition by Pfizer Inc. in March 2022. From August 2011 to June 2012 and previously from December 2005 to March 2011, Mr. Hoffman served as Arena’s Vice President, Finance and Chief Financial Officer and in a number of various roles of increasing responsibility from 1997 to December 2005. Mr. Hoffman formerly served as a member of the board of directors of Saniona AB, a biopharmaceutical company, from September 2021 to May 2022, and as a member of the board of directors of Kura Oncology, Inc. (Nasdaq: KURA), a cancer research company, from March 2015 to August 2021. He also previously served as a member of the board of directors of CombiMatrix Corporation, a molecular diagnostics company, MabVax Therapeutics Holdings, Inc., a biopharmaceutical company, and Aravive, Inc. (Nasdaq: ARAV), a clinical stage biotechnology company. Mr. Hoffman serves as a member of the steering committee of the Association of Bioscience Financial Officers. Mr. Hoffman formerly served as a director and President of the San Diego Chapter of Financial Executives International and was an advisor to the Financial Accounting Standard Board, or FASB, from 2010 to 2020, advising the U.S. accounting rulemaking organization on emerging issues and new financial guidance. Mr. Hoffman holds a B.B.A. from St. Bonaventure University. We believe Mr. Hoffman’s financial and executive business experience qualifies him to serve on our board of directors.
| 102 |
Victoria Niklas,医学博士. Victoria Niklas has served on our board of directors since April 2021. Dr. Niklas has a distinguished career spanning more than two decades in translational research, clinical care and teaching at academic health centers, and is currently the Chief Medical Officer of Oak Hill Bio, a clinical-stage neonatology and rare disease therapeutics company, a position she has held since 2022. Prior to joining Oak Hill Bio, Dr. Niklas served in Global Medical Affairs and as Global Program Leader of the OHB-607 program in Rare Disease and Hematology at Takeda Pharmaceuticals. Before Takeda, she was Chief Medical and Scientific Officer at Prolacta Bioscience, a neonatal nutritional product development company based on human donor milk. Dr. Niklas has over 20 years of experience as an academic neonatologist with expertise in developmental and acquired inflammatory disorders of the gut, the lung and the mucosal immune system with relevance to diseases across the lifespan. She has held positions as Chief, Division of Newborn Medicine at Nemours Children’s Hospital, Chief of Neonatology at UCLA Olive View Medical Center, and Visiting Professor of Clinical Pediatrics at the David Geffen School of Medicine at UCLA. Dr. Niklas is board certified in Perinatal and Neonatal Medicine and holds a California medical license. In addition to being a co-author on numerous scientific and clinical publications, she has helped lead the development of patented products and has served as a board member for multiple biotech and early-stage companies in functional foods. Dr. Niklas received her MD from Harvard Medical School, her MA in Biochemistry and Molecular Biology from Harvard University, and her bachelor’s in Biological Sciences from Goucher College. We believe Dr. Niklas’ extensive experience and knowledge in the biotechnology sector qualifies her to serve on our board of directors.
Richard Cilento,Jr.,工商管理硕士理查德·西伦托自2021年4月以来一直在我们的董事会任职。Cilento先生是生命科学研发公司GlyCosBio Inc.的创始人、董事会主席兼首席执行官。 Cilento先生是FuelQuest,Inc.的创始人、总裁和首席执行官,FuelQuest,Inc.是一家信息技术、供应链管理和税务自动化技术提供商,于2007年5月被Saracen Energy Advisors LP收购。齐伦托曾在多家科技公司担任过高级管理职位,其中包括施乐公司,他在施乐公司担任战略服务副总裁总裁。在此之前,他是XLConnect解决方案公司服务副总裁总裁,在那里他担任了先进系统的首席技术专家 ,并通过该组织的首次公开募股以及最终与施乐的合并为该组织提供了支持。作为一名航空和天文工程师,Cilento先生在美国国家航空航天局(NASA)开始了他的职业生涯,在那里,他和他的团队为美国国防部的星球大战计划以及一系列政府资助的技术和生命科学实验制定了航天飞机飞行计划。Cilento先生是为建造国际空间站而设计和规划空间站组装顺序的首席工程师。Cilento先生拥有伊利诺伊大学航空和天文工程学士学位和休斯顿大学MBA学位。我们相信,Cilento先生在广泛的技术行业中的商业经验和对资本市场(包括风险资本、私募股权和公开市场)的高管级别的知识, 使他有资格在我们的董事会任职。
史黛西·科恩,MBA。Stacy Coen自2021年7月以来一直担任我们的董事会成员。Coen女士在领先的肿瘤学和罕见疾病公司拥有超过25年的商业和企业发展经验。她目前是免疫基因公司的首席商务官,该公司正在开发下一代抗体-药物结合物,以改善癌症患者的预后。 在加入免疫基因之前,科恩女士曾在Editas Medicine,Inc.工作,这是一家开发罕见疾病疗法的生物技术公司,在那里她 担任业务开发副总裁总裁,负责业务发展、战略、交易和联盟管理。 在加入Editas之前,Coen女士在Genzyme Corporation(现为赛诺菲Genzyme)担任过多个不断增加的职位, 包括副总裁总裁、担任罕见疾病业务发展和许可主管,以及多发性硬化症战略和业务发展全球主管总裁副主任等人。科恩女士目前是美国亨廷顿病协会中心项目和教育咨询委员会的成员,也是MassBio和再生医学联盟的成员。科恩女士拥有马萨诸塞大学金融与经济学学士学位和弗吉尼亚大学达顿商学院工商管理硕士学位。我们相信,科恩女士在生物技术行业的丰富高管经验使她有资格在我们的董事会任职。
| 103 |
马修 链接。马修·林克自2021年4月以来一直在我们的董事会任职。林克先生在医疗保健和医疗技术行业拥有20多年的经验,目前担任视觉科学(SGHT)首席商务官。从2021年到2023年,他担任咨询服务提供商Orion Healthcare Advisors,LLC的管理合伙人。2006年至2021年,林克先生在NuVasive Inc.担任区域和执行领导职位,NuVasive Inc.是外科植入物和脊柱外科和矫形外科技术的全球领导者。作为NuVasive,Inc.的总裁,他的职责包括监督脊柱、神经生理学和整形外科的全球业务部门。在加入NuVasive,Inc.之前,林克先生在Depuy Orthopedics和Depuy Spine担任过商业领导职务。他目前还担任Galen Robotics的董事会主席,以及Springbok Analytics和Dinamicor以及弗吉尼亚大学库尔特翻译研究基金会的董事会成员。林克先生获得了弗吉尼亚大学体育教育和运动医学学士学位。我们相信,林克先生丰富的医疗技术行业和管理经验使他有资格在我们的董事会中任职。
家庭关系
我们的任何董事或高管之间没有家族关系。
科学顾问委员会
我们 有一个科学顾问委员会,由以下具有相关专业知识的世界知名科学家组成,帮助 指导我们的研发工作。
| ● | 克劳迪娅·卢奇内蒂,医学博士,博士 | |
| ● | 托马斯·卡迈克尔,医学博士,博士。 | |
| ● | 凯特·鲁宾斯,博士。 | |
| ● | 伊丽莎白·施帕尔,医学博士。 | |
| ● | 尼尔·布拉米克,博士。 |
董事会
我们的董事会目前由六名董事组成。本公司的公司注册证书规定,在符合本公司任何系列优先股持有人选举董事的权利的情况下,本公司董事会的董事人数应不时通过授权董事总数的多数决议确定为 次,无论以前的授权董事职位是否有任何空缺。我们每一位董事的任期在该董事当选或任命后的下一次股东年会结束,但受该董事提前去世、被取消资格、辞职或免职的限制。
根据我们的公司注册证书,在符合我们任何系列优先股持有人的优先权利的情况下,因董事人数增加或董事会任何空缺而设立的任何新设立的董事职位只能由在任董事总数的多数票(即使不足法定人数)或唯一剩余的
董事投票填补,并且不能由股东填补。此外,我们董事会的任何成员或整个董事会只能
只有在有理由的情况下才能被免职,而且必须经过至少66名股东的赞成票2/3我们的
股票的投票权为%。
根据我们修订和重述的公司注册证书,该证书将与本招股说明书构成的注册说明书的效力相关 在本招股说明书构成的注册说明书生效时生效。我们的董事会将分为三类,董事交错任职三年。
当 考虑董事是否具备根据我们的业务和结构有效履行其监督职责所需的经验、资质、属性或技能时,董事会主要关注每个董事的背景和经验,这些信息反映在每个董事的个人传记中 。我们相信,我们的董事提供与我们业务的规模和性质相关的经验和技能的适当组合。
| 104 |
董事 独立
我们的董事会已经决定,除了皮特·奥希隆之外,我们的所有董事会成员都是独立董事, 就纳斯达克和美国证券交易委员会的规则而言。在做出这一决定时,我们的董事会考虑了每个非员工董事与我们的关系以及董事会认为相关的所有其他事实和情况,包括每个非员工董事对我们普通股的实益所有权。
在 本招股说明书所包含的注册生效后,我们期望 我们董事会和每个委员会的组成和运作将符合纳斯达克的所有适用要求和美国证券交易委员会的规章制度 ,并受委员会的适用逐步引入期限的限制。
交错 板
根据我们修订和重述的公司注册证书的条款,该条款将与本招股说明书所包含的注册说明书的效力同时生效,我们的董事会将分为 三个交错的董事级别,每个董事会将被分配到三个级别中的一个。在我们的每一次股东年会上,将选出一类董事,任期三年,以接替同级董事的任期到期满。 董事任期将在2024年举行的股东年会上选出继任董事并获得资格后终止。 2024年为I类董事,2025年为II类董事,2026年为III类董事。
| ● | 我们的一级董事将是罗伯特·霍夫曼和小理查德·西伦托; |
| ● | 我们的第二类董事将是马修·林克和维多利亚·尼克拉斯;以及 |
| ● | 我们的三级导演将是史黛西·科恩和皮特·奥希隆。 |
我们的董事会分为三个级别,交错三年任期,这可能会推迟或阻止股东实施 我们管理层或控制权变更的努力。
董事会 领导结构
我们的董事会目前由我们的创始人皮特·奥希隆担任主席。我们的公司治理准则进一步为我们的董事会提供了灵活性,使其可以在未来根据其认为合适的情况修改我们的领导结构。
董事会委员会
我们的董事会成立了一个审计委员会、一个薪酬委员会和一个提名和公司治理委员会,每个委员会都按照我们董事会通过的章程运作。我们的董事会也可能不定期成立其他委员会来协助董事会。我们所有委员会的组成和运作均符合《萨班斯-奥克斯利法案》、《纳斯达克》和《美国证券交易委员会》规则和规定的所有适用要求。当我们在纳斯达克上上市后,每个委员会的章程 将在我们的网站www.fifibiologics.com上查阅。
审计委员会
我们审计委员会的成员是霍夫曼先生、尼克拉斯博士和西伦托先生。霍夫曼先生担任审计委员会主席。 我们的董事会已经确定,审计委员会的每一名成员都是“独立的”,这一术语在 纳斯达克规则中有定义,并且在财务和审计事务方面拥有足够的知识,可以在审计委员会任职。此外,我们的 董事会已经确定,审计委员会的每位成员都符合交易所法案第10A节以及美国证券交易委员会和纳斯达克相关规则对审计委员会的更高独立性要求 。我们的董事会已经确定,霍夫曼先生 是美国证券交易委员会适用规则所定义的“审计委员会财务专家”。审计委员会的职责 包括:
| ● | 任命、批准我们独立注册会计师事务所的薪酬和评估其独立性。 |
| 105 |
| ● | 预先批准由我们的独立注册会计师事务所提供的审计和允许的非审计服务,以及此类服务的条款 ; |
| ● | 与我们的独立注册会计师事务所和负责编制财务报表的管理层成员一起审查整体审计计划; |
| ● | 审查 并与管理层和我们的独立注册会计师事务所讨论我们的 年度和季度财务报表和相关披露,以及我们使用的关键会计政策和做法。 |
| ● | 协调监督并审查我们对财务报告的内部控制是否充分; |
| ● | 制定政策和程序,以接收和保留与会计有关的投诉和关切; |
| ● | 根据审计委员会与管理层和我们的独立注册会计师事务所的审查和讨论, 建议我们的经审计的财务报表是否应包括在我们的10-K年度报告中; |
| ● | 监督 我们财务报表的完整性以及我们遵守与财务报表和会计事项相关的法律和法规要求 ; |
| ● | 准备 美国证券交易委员会规则要求的审计委员会报告,该报告应包括在我们的年度委托书中; |
| ● | 审查 所有相关人员交易是否存在潜在的利益冲突情况,并批准所有此类交易;以及 |
| ● | 审查 季度收益发布。 |
薪酬委员会
我们薪酬委员会的成员是霍夫曼先生、科恩女士和林克先生。霍夫曼先生担任该委员会的主席。 我们的董事会已经确定,薪酬委员会的每位成员都是纳斯达克规则中定义的“独立的” ,并且是根据交易所法案第16b-3条规则的“非员工董事”。此外,我们的董事会 已确定薪酬委员会的每位成员都符合交易所法案第10C节以及美国证券交易委员会和纳斯达克相关规则对薪酬委员会宗旨的更高独立性要求。薪酬委员会的职责 包括:
| ● | 审查并批准我们关于首席执行官薪酬的理念、政策和计划; |
| ● | 就首席执行官和其他高管的薪酬向董事会提出建议; |
| ● | 审查和评估薪酬顾问的独立性; |
| ● | 监督和管理我们的股权激励计划; |
| ● | 审查 并就董事薪酬向我们的董事会提出建议; 和 |
| ● | 准备美国证券交易委员会要求的薪酬委员会报告,包括我们的《薪酬 讨论与分析》披露。 |
| 106 |
提名 和公司治理委员会
我们提名和公司治理委员会的成员是科恩女士、尼克拉斯博士和林克先生。科恩女士是该委员会的主席。我们的董事会已经确定,提名和公司治理委员会的每一名成员都是纳斯达克规则中定义的“独立的” 。提名和公司治理委员会的职责包括:
| ● | 制定并向董事会推荐董事会和委员会成员的标准; |
| ● | 建立识别和评估董事候选人董事会的程序,包括股东推荐的被提名者 ; |
| ● | 审查董事会的组成,以确保董事会由具备适当技能和专业知识的成员组成,以向我们提供建议; |
| ● | 确定和筛选有资格成为董事会成员的个人; |
| ● | 向董事会推荐董事候选人和董事会各委员会的提名人选; |
| ● | 制定并向董事会推荐一套商业行为和道德准则以及一套公司治理准则;以及 |
| ● | 监督我们董事会和管理层的评估。 |
行为准则
我们 已通过书面商业行为和道德准则,适用于我们的董事、高级管理人员和员工,包括我们的首席执行官、首席财务官、首席会计官或主计长,或执行类似职能的人员。 为确保招股说明书的有效性,我们将在我们的网站www.fifibiologics.com上发布 准则的最新副本。如果我们对任何高级职员或董事的商业行为和道德守则进行任何实质性修改或批准豁免,我们将在我们的网站 或当前的Form 8-K报告中披露此类修改或豁免的性质。
| 107 |
高管 和董事薪酬
高管薪酬
这一节讨论了高管薪酬计划的主要组成部分,这些薪酬计划适用于我们的高管,这些高管的名字在“-2023 薪酬汇总表“下面。在截至2023年12月31日的财政年度中,我们的“指定高管 官员”及其职位如下:
| ● | 董事长兼首席执行官皮特·奥希隆; | |
| ● | 哈米德·霍贾博士,首席科学官;以及 | |
| ● | 首席财务官马克·安徒生。 |
此 讨论可能包含基于我们当前的计划、考虑事项、预期和对未来薪酬计划的确定的前瞻性陈述 。我们在此产品完成后采用的实际薪酬计划可能与本讨论中总结的当前计划计划存在实质性差异。作为美国证券交易委员会规则定义的“新兴成长型公司”和“较小的报告公司”,我们不需要包括薪酬讨论和分析部分,并已选择遵守适用于新兴成长型公司和/或较小的报告公司的分级披露要求。
2023薪酬汇总表
下表显示了在截至2023年12月31日的财年中,我们指定的高管所获得的薪酬总额。
| 名称和主要职位 | 年 | 薪金(元) | 奖金 ($)(2) | 选项 奖励($)(3) | 所有 其他薪酬(美元)(4) | 总计(美元) | ||||||||||||||||||
| 皮特·奥希隆 | 2022 | 600,000 | — | — | 22,485 | 622,485 | ||||||||||||||||||
| 董事长兼首席执行官 | 2023 | 600,000 | 300,000 | 3,335,400 | 50,723 | 3,452,273 | ||||||||||||||||||
| 哈米德·霍贾博士 | 2022 | 300,208 | 101,500 | 23,900 | 20,505 | 446,113 | ||||||||||||||||||
| 首席科学官 | 2023 | 325,000 | 47,396 | 818,100 | 50,723 | 1,241,219 | ||||||||||||||||||
| 马克·安徒生(1) | 2022 | 189,583 | 15,000 | 20,100 | 88,401 | 313,084 | ||||||||||||||||||
| 首席财务官 | 2023 | 325,000 | 66,354 | 820,350 | 70,523 | 1,282,227 | ||||||||||||||||||
| (1) | 马克·安徒生于2022年6月加入我们。 |
| (2) | 奖金金额反映 日历年度的实际奖金支出。 |
| (3) | 根据美国证券交易委员会规则,此栏中的金额反映根据ASC718计算的授予日股票期权的公允价值合计,而不是被点名个人支付或变现的金额。我们提供有关本招股说明书其他部分所载经审核财务报表附注11中授予的股票期权价值计算所用的假设 。 |
| (4) | “所有其他补偿”栏中的金额 由下表所列金额组成: |
| 被任命为高管 官员(2022) | 401(K)计划匹配 捐款(美元) | 医疗福利(美元) | 搬迁(美元) | |||||||||
| 皮特·奥希隆 | — | 22,485 | — | |||||||||
| 哈米德·霍贾博士 | — | 20,505 | — | |||||||||
| 马克·安徒生 | 6,500 | 20,512 | 61,389 | |||||||||
| 被任命为首席执行官(2023) | 401(K)计划匹配 捐款(美元) | 医疗福利(美元) | 搬迁(美元) | |||||||||
| 皮特·奥希隆 | — | 50,723 | — | |||||||||
| 哈米德·霍贾博士 | — | 50,723 | — | |||||||||
| 马克·安徒生 | 19,800 | 50,723 | — | |||||||||
2022年工资
在 2022年,我们任命的高管获得了年度基本工资,以补偿他们为我们提供的服务。支付给每位指定高管的基本工资 旨在提供反映高管技能、经验、角色和责任的固定薪酬组成部分。
在2022财年,奥希隆先生的年基本工资为60万美元,安徒生先生的年基本工资为325,000美元。 Khoja博士的年基本工资在2022财年从29万美元增加到325,000美元。
2023年工资
在 2023年,我们任命的高管获得了年度基本工资,以补偿他们为我们提供的服务。支付给每位指定高管的基本工资 旨在提供反映高管技能、 经验、角色和责任的固定薪酬部分。
在2023财年,奥希隆先生的年基本工资为60万美元,安徒生先生的年基本工资为325,000美元,霍贾博士的年基本工资为325,000美元。
| 108 |
2022年奖金
在2022财年,每一位被任命的高管都有资格获得奖金,这是根据我们董事会自行决定的预先设定的公司年度业绩目标和个人业绩目标来确定的。奥赫隆先生有资格获得年度现金奖金,奖金目标为基本工资的50%,并根据2022年的业绩在2023年获得30万美元的奖金。 Khoja博士有资格获得年度现金奖金,目标为基本工资的35%,并在2022年一周年后于2022年获得奖金101,500美元,2023年根据他与我们的一周年纪念日起的2022年业绩按比例获得47,396美元的奖金。未来的奖金将以历年为基础。Andersen先生有资格获得以基本工资35%为目标的年度现金奖金 ,并根据2022年的业绩在2023年获得66,354美元的奖金,并根据他于2022年6月开始受雇于我们的 按比例分配到2022年。
2023年奖金
在2023财年,每一位被任命的高管都有资格获得奖金,这是根据我们董事会自行决定的预先设立的年度公司业绩和个人业绩目标来确定的。奥赫隆有资格获得相当于基本工资50%的年度现金奖金,Khoja博士有资格获得相当于基本工资35%的年度现金奖金,安徒生有资格获得相当于基本工资35%的年度现金奖金。
年度奖金 根据公司业绩和个人在本财年的贡献确定,通常确定 并在下一年1月发放。
公平薪酬
Khoja博士和Andersen先生在他们的雇佣协议中分别获得了相当于7,500份股票期权的承诺。这些 期权是在《2022年股票计划》(本文定义)获得批准和授权后于2022年授予的。在制定2022年股票计划之前,Khoja博士还在2022年获得了相当于1,250股无投票权普通股的奖励。2022年授予指定高管的股票期权在受聘一周年日授予1/3,在36周年日授予1/36这是此后每个月,直至完全 归属,但须继续服务,并将在公司发生“控制权变更”时全面加速 (定义见2022年股票计划)。
2023年,奥希隆先生、霍贾博士和安徒生先生分别获得了2022年股票计划下的股票期权。奥希隆先生获得等值1,853,000股,霍贾博士获得等值454,500股,安徒生先生获得等值455,750股。2023年授予的股票期权在归属开始日期的一周年时授予四分之一,即 2023年1月1日,其余的按月授予超过36个月。
有关2022年股票计划的更多信息,请参阅下面标题为“-股权补偿计划”的章节。
其他 薪酬要素
退休 计划
我们 为符合特定资格要求的员工(包括我们指定的高级管理人员)参加Inperity的401(K)退休储蓄计划。我们指定的高管有资格以与其他全职员工相同的条款 参加Inperity 401(K)计划。在2022年和2023年,401(K)计划参与者的缴款与指定执行干事的雇员缴款的特定百分比相匹配。这些匹配的缴费自缴费之日起 完全归属。我们预计,在完成直接上市后,我们指定的高管 将继续以与其他全职员工相同的条件参与这项Inperity 401(K)计划。
员工 福利和津贴
健康/福利计划 。我们的所有全职员工,包括我们指定的高管,都有资格参加Inperity的健康和福利计划,包括:
| ● | 医疗、牙科和视力福利; | |
| ● | 医疗和受抚养人护理灵活支出账户; | |
| ● | 短期和长期伤残保险;以及 | |
| ● | 人寿保险。 |
我们 认为,上述员工福利是必要和适当的,可以为我们 任命的高管提供具有竞争力的薪酬方案。
| 109 |
雇佣 与我们指定的高管签订的协议
皮特·奥希隆雇佣协议
2023年12月1日,我们与彼得·奥希隆先生签订了聘用协议,根据协议,奥希隆先生同意 担任我们的总裁兼首席执行官。根据协议,奥赫隆先生的雇佣是“随意的” ,可由任何一方以任何理由终止,通知或不通知。
根据雇佣协议,奥赫隆先生有权获得600,000美元的初始基本工资,该基本工资将由董事会或薪酬委员会进行年度审查,但未经奥赫隆先生同意不得下调。此外, 协议规定,奥赫隆先生有资格获得由董事会 合理厘定的年度绩效花红,或在董事会授权的范围内由薪酬委员会根据董事会或委员会每年厘定的一个或多个绩效目标 厘定,但前提是在达到所有绩效目标的情况下,奖金须不少于其基本工资的50%。百分比奖金目标将由董事会或薪酬委员会定期审查。
协议还规定,奥赫隆先生有资格参加由我们为员工的利益而维护的健康和福利福利计划和计划。
根据该协议,如本公司在无任何理由(定义见协议)或 奥赫隆先生有充分理由(定义见协议)的情况下终止聘用O‘Heeller先生,则他将有资格领取相当于12个月基本工资的遣散费,犹如他在该12个月期间仍受雇一样,以及在该12个月期间应支付的目标奖金金额 (须于终止通知后60天支付)。此外,奥希隆先生 将在该12个月期间继续授予期权。如果协议因任何原因终止,奥赫隆先生应 支付终止之日获得的所有补偿,包括未使用的和应计的假期、他应支付的任何未付奖金,以及终止年度应按比例计算的奖金部分(该等奖金金额与支付给其他公司高管的奖金同时支付)。
如果在控制权变更后的12个月内(根据协议的定义),或在控制权变更前的两个月内,或在控制权变更发生后的六个月内,发生非自愿终止奥赫隆先生的雇用的情况 ,并且受奥赫隆先生与公司签订的一份新闻稿、奥赫隆先生持有的所有股票期权和基于股票的奖励的限制,自通知终止之日起, 将被授予并可行使或不可没收。
协议 包含奥赫隆先生惯常的发明转让和保密义务,以及终止雇佣关系后12个月的竞业禁止/竞业禁止。
雇佣协议下的薪酬(包括奖金目标)可由薪酬委员会、 或董事会(根据薪酬委员会的建议)不时增加,增加的薪酬不需要签订修订的雇佣协议。
薪酬委员会或董事会还可根据薪酬委员会的建议,随时酌情支付或发放可自由支配的现金奖金或股权奖金。股权红利可以是普通股、股票期权或其他股权对价的形式,其金额和条款由薪酬委员会或董事会根据薪酬委员会的建议而不时决定。
哈米德·霍贾博士就业协议
我们 已与Khoja博士签订雇佣协议,日期为2021年7月20日,根据协议,Khoja博士将担任我们的首席科学官。根据协议,Khoja博士的雇用是“随意的”,可由任何一方以任何理由终止,并可事先通知或不通知。
根据他的协议,Khoja博士有权获得29万美元的初始基本工资,并在2022年增加到325,000美元。 此外,协议还规定,Khoja博士有资格获得高达基本工资的35%的年度绩效奖金, 根据公司和个人绩效目标的完成情况支付。在加入聘书时,Khoja博士获得了相当于7,500股普通股的股票期权奖励,这相当于在受雇一周年和1/36周年时授予股票期权相关股份的三分之一。这是此后每月支付,直至完全归属为止,但须在适用归属日期之前继续受雇。根据协议,Khoja博士还获得了与其开始就业有关的相当于15,000美元的一次性现金奖金,并有权获得最高45,000美元的搬迁费用。该协议还规定,Khoja博士有资格参加我们为员工的利益而维护的健康和福利福利计划和计划。
根据协议,如果公司无故终止雇用Khoja博士,他将有资格获得相当于九个月基本工资的遣散费。
马克 安徒生雇佣协议
我们 已于2022年5月20日与Andersen先生签订雇佣协议,根据该协议,Andersen先生担任我们的首席财务官。根据协议,Andersen先生的雇佣是“随意的”,任何一方均可在通知或不通知的情况下以任何 原因终止。
根据他的协议,Andersen先生有权获得325,000美元的初始基本工资。此外,协议规定,Andersen先生有资格获得高达基本工资35%的年度绩效奖金,奖金将根据公司 的业绩和个人绩效目标支付。与签订协议相关的是,Andersen先生获得了相当于7,500股普通股的股票期权奖励,这相当于在雇佣日期1周年和36月1日授予股票期权1/3的股份。这是此后每月支付,但须持续受雇至适用的归属日期。根据该协议,Andersen先生还获得了与其开始工作相关的相当于15,000美元的一次性现金红利,并有权获得最高45,000美元的搬迁费用。该协议还规定,Andersen先生有资格参加我们为员工的利益而维护的健康和福利计划和计划。
根据 协议,如果Andersen先生的雇佣关系被公司无故终止,那么他将有资格获得 相当于九个月基本工资的遣散费。
| 110 |
股权 薪酬计划
以下总结了FibroBiologics,Inc.的材料术语。2022年股票计划或2022年股票计划。
2022年库存计划
我们的董事会于2022年8月10日通过,我们的股东于2022年8月18日批准了我们的2022年股票计划。《2022年股票计划》规定授予激励性股票期权、非法定股票期权、股票增值权、限制性股票奖励、限制性股票单位奖励和其他股票奖励。2022年股票计划通过授予股票奖励,旨在帮助我们 确保和保留合格获奖者的服务,激励这些人为我们的成功尽最大努力 并提供一种手段,使符合资格的获奖者可以从我们普通股的增值中受益。截至2023年12月31日,我们已根据2022年股票计划向员工、董事、 和科学顾问董事会成员发行了相当于101,250份期权,执行价为每股3.28美元,向员工和董事发行了相当于3,689,750份执行价格为每股2.28美元的期权 。2023年8月,共有2,500个期权被没收,执行价为每股3.28美元 。一般来说,我们授予的奖励期限为四年,行使价等于我们董事会确定的我们普通股的估计公允价值,并考虑了由独立第三方估值公司对我们普通股进行的同期估值 。
截至2023年12月31日,根据2022年股票计划,可供未来发行的股票相当于8,711,500股。
2023年12月31日的未偿还股权奖
下表提供了截至2023年12月31日我们指定的高管持有的未偿还股权奖励的信息。 除了在2022年股票计划制定之前于2022年授予Khoja博士的等值1,250股无投票权普通股外,所有奖励都是根据我们的2022年股票计划授予的。
| 名字 | 未行使期权标的证券数量 (#) 可行使 | 证券数量 潜在的 未锻炼身体 选项 (#) 不可执行 | 选择权 行权 价格 ($) | 选择权 过期日期 | ||||||||||
| 皮特·奥希隆 | — | 1,853,000 | 2.28 | 2033年2月16日 | ||||||||||
| 哈米德·霍贾博士 | 4,861 | 2,639 | 3.28 | 2032年9月25日 | ||||||||||
| 哈米德·霍贾博士 | — | 454,500 | 2.28 | 2033年2月16日 | ||||||||||
| 马克·安徒生 | 3,472 | 4,028 | 3.28 | 2032年9月25日 | ||||||||||
| 马克·安徒生 | — | 455,750 | 2.28 | 2033年2月16日 | ||||||||||
董事 薪酬
非在职董事 薪酬表
下表列出了截至2022年12月31日的财政年度内担任我们董事会非雇员成员的每位人员的薪酬总额。除下表所载及更详细描述者外,我们于二零二二年并无 就董事会任何非雇员成员作为董事会成员提供的服务向其支付任何报酬、作出任何股权奖励或非股权奖励或支付任何其他报酬。我们的董事长 兼首席执行官Pete O 'Heeron作为董事没有获得额外的报酬。有关截至2022年12月31日止年度O 'Heeron作为雇员支付或赚取的薪酬的更多信息,请参阅标题为“高管薪酬”的章节。
| 名字 | 以现金形式赚取或支付的费用(美元) | 股票 奖励(美元)(1)(3) | 选项 奖励($)(2)(3) | 总计(美元) | ||||||||||||
| 罗伯特·霍夫曼 | 50,000 | 24,600 | 12,800 | 87,400 | ||||||||||||
| 维多利亚·尼克拉斯 | 43,000 | 24,600 | 12,800 | 80,400 | ||||||||||||
| 小理查德·奇伦托 | 43,000 | 24,600 | 12,800 | 80,400 | ||||||||||||
| 斯泰西·科恩 | 41,000 | 24,600 | 12,800 | 78,400 | ||||||||||||
| 马修·林克 | 41,000 | 24,600 | 12,800 | 78,400 | ||||||||||||
| (1) | 在 于2022年1月,我们的每名非雇员董事均获授相当于7,500股股票的奖励。 | |
| (2) | 2022年9月,我们的每位非雇员董事获得了相当于5,000份股票期权的期权,行权价相当于每股3.28美元。 | |
| (3) | 报告的 金额代表根据ASC主题718计算的2022财年授予非雇员董事的股票和股票期权的公允价值合计。该授予日期公允价值不考虑任何估计的没收。 在计算授予日期公允价值时使用的假设在本 招股说明书其他部分包括的我们财务报表的附注中阐述。本栏中报告的金额反映了股票和股票期权的会计成本,与行使股票期权或出售普通股的任何标的股份时可能收到的实际经济价值并不相符。 |
下表显示了在截至2023年12月31日的财年中担任董事会非雇员成员的每位员工的总薪酬。 除下表所列及下文更全面描述外,于2023年,本公司并无向本公司董事会任何非雇员成员支付任何薪酬、任何股权奖励或非股权奖励,或向他们支付任何其他薪酬。我们的董事长兼首席执行官皮特·奥希隆不会因为他作为董事的服务而获得额外的薪酬。有关截至2023年12月31日的年度作为员工支付给或赚取的薪酬的详细信息,请参阅“高管薪酬” 一节。
| 名字 | 以现金形式赚取或支付的费用(美元) | 股票奖励(美元) | 选项 奖励($)(1)(2) | 总计(美元) | ||||||||||||
| 罗伯特·霍夫曼 | 55,000 | — | 333,540 | 388,540 | ||||||||||||
| 维多利亚·尼克拉斯 | 48,000 | — | 333,540 | 381,540 | ||||||||||||
| 小理查德·奇伦托 | 43,000 | — | 333,540 | 376,540 | ||||||||||||
| 斯泰西·科恩 | 51,000 | — | 333,540 | 384,540 | ||||||||||||
| 马修·林克 | 46,000 | — | 333,540 | 379,540 | ||||||||||||
| (1) | 2023年2月,我们的每位非雇员董事被授予相当于185,300份股票期权,行权价为每股2.28美元。 | |
| (2) | 报告的金额代表根据ASC主题718计算的2023财年授予非雇员董事的股票期权的授予日期公允价值合计。此类授予日期的公允价值不考虑任何预计的 没收。计算本专栏所报告奖励的授予日期公允价值时使用的假设载于本招股说明书其他部分的财务报表附注 。本栏报告的金额反映了股票期权的会计成本,与行使股票期权或出售任何普通股标的股份时可能收到的实际经济价值并不相符。 |
| 111 |
截至2022年12月31日,我们董事会的非雇员成员持有以下未行使期权的总数:
| 名字 | 未行使期权的证券标的数量 | |||
| 罗伯特·霍夫曼 | 5,000 | |||
| 维多利亚·尼克拉斯 | 5,000 | |||
| 理查德·西伦托 | 5,000 | |||
| 斯泰西·科恩 | 5,000 | |||
| 马修·林克 | 5,000 | |||
除上文所述的 外,截至2022年12月31日,我们的董事会没有非雇员成员持有我们普通股的未行使期权或未归属股份。
截至2023年12月31日,我们董事会的非雇员成员持有以下未行使的期权总数 :
| 名字 | 证券数量 潜在的未行使期权 | |||
| 罗伯特·霍夫曼 | 190,300 | |||
| 维多利亚·尼克拉斯 | 190,300 | |||
| 理查德·西伦托 | 190,300 | |||
| 斯泰西·科恩 | 190,300 | |||
| 马修·林克 | 190,300 | |||
除上文所述的 外,截至2023年12月31日,我们的董事会没有非雇员成员持有我们普通股的未行使期权或未归属股份。
非员工董事薪酬政策
我们的 董事会采用了非员工董事薪酬政策,该政策将在本招股说明书 生效时继续实施。该政策旨在使我们能够长期吸引和留住高素质的非雇员董事。根据该政策,每位非员工董事将在本次发售完成后获得现金补偿 ,具体如下:
| 职位 | 年度定额 | |||
| 董事会: | ||||
| 委员(主席除外) | $ | 35,000 | ||
| 审计委员会: | ||||
| 委员(主席除外) | $ | 8,000 | ||
| 座椅固位器 | $ | 10,000 | ||
| 薪酬委员会: | ||||
| 委员(主席除外) | $ | 6,000 | ||
| 座椅固位器 | $ | 10,000 | ||
| 提名和公司治理委员会: | ||||
| 委员(主席除外) | $ | 5,000 | ||
| 座椅固位器 | $ | 10,000 | ||
此外,非员工董事薪酬政策规定,在首次选举进入董事会时,每位非员工董事 将获得相当于7,500股普通股的股权奖励,或初始授予。此外,在我们每次年度股东大会的 日,每位非员工董事在会议后继续作为非员工董事 将被授予年度股票期权奖励,以购买相当于5,000股的股票,或年度授予。年度授予将于(I)授予日期一周年或(Ii)下一届年度会议日期(以较早者为准)全数归属;但若董事辞去董事会职务或以其他方式不再担任董事的职务,则所有归属均应停止,除非董事会认为情况需要继续 归属。此外,如果董事退出董事会或因其他原因停止担任董事,所有已授予的期权仍可在12个月内行使。尽管有上述规定,如果外部董事在股东周年大会前12个月内首次当选为董事会成员,则该外部董事将获得按月按比例分配的年度补助金,作为外部董事的一段时间内。
| 112 |
某些 关系和相关人员交易
以下是自成立以来的交易或一系列交易的摘要,或当前提议的交易或一系列交易的摘要,其中我们是或将成为当事人,涉及的金额超过或将超过120,000美元,并且我们的任何董事、高管或据我们所知,拥有5%或更多股本的实益所有者,或5%以上的证券持有人,或任何前述人士的直系亲属或关联实体的任何成员,直接或间接利益 。
系列 A优先股
2021年5月,作为我们组建的一部分,我们向FibroGenesis发行了相当于8,750,000股A系列优先股 ,以换取专利转让协议和知识产权交叉许可协议,其中专利转让协议将某些专利/应用转让给我们,知识产权交叉许可协议向我们提供定义使用领域内FibroGenesis保留的专利/申请的独家许可,并且 向FibroGenesis提供转让给FibroBiologics的专利/申请用于所有其他使用领域的独家许可。
对于直接上市,我们当时尚未发行的所有A系列优先股将自动注销,持有人不需要支付额外的对价 。
纤维起源 贷款
2022年7月,我们向FibroGenesis提供了300,000美元的贷款,利率为0%,到期日为一年。2022年10月,我们额外向FibroGenesis提供了60,000美元的贷款,利率为0%,期限为一年。6万美元已于2022年12月全额偿还,30万美元已于2023年4月全额偿还 。
ROFN 协议
2023年1月,我们与FibroGenesis签订了关于第一谈判权的协议,或ROFN协议。作为交换,FibroGenesis同意修改我们的公司注册证书,以(I)在我们承销的首次公开募股或我们的普通股在证券交易所(我们统称为IPO)或出售时取消 A系列优先股的清算优先权,(Ii)使B系列优先股清算优先权等于A系列优先股,以及(Iii)规定 在我们公司首次公开募股或出售时,A系列优先股将被免费取消,我们同意在首次公开募股或出售公司之前,向FibroGenesis支付对我们进行任何股权投资所得毛收入的15%。此外,如果FibroGenesis决定向外部许可其任何技术,我们还获得了为期五年的第一谈判权。截至2023年9月30日,根据ROFN协议,根据截至2023年9月30日收到的股权投资总收益,我们 已向FibroGenesis支付了总计260万美元。
2021年和2022年可转换票据
2021年12月,我们以私募方式向投资者发行并出售了130万美元的可转换本票,即2021年债券,其中一些投资者持有超过5%的股份。2021年债券的初始年利率为6.0%,在合格融资的情况下将自动转换为我们普通股的 股票。2021年债券的转换价格等于2亿美元 除以从发行中稀释之前的股权总数。2021年票据为无抵押票据,在向本公司所有商业金融贷款人、保险公司、租赁融资机构或经本公司董事会批准并定期从事贷款业务的其他贷款机构全额付款之前,有权优先付款。2023年4月,这些票据中的130万美元被转换为我们B系列优先股的股票,2021年发行的票据中没有一种是流通股。
2022年1月和2022年4月,我们分别以私募方式向投资者发行和出售了我们的可转换本票,即2022年票据,分别为35万美元和395万美元,其中一些投资者持有超过5%的股份。2022年债券的初始年利率为6.0%,期限为一年,如果获得合格融资,可应持有人的要求转换为我们的普通股。2022年票据的换股价格为(I)于首次公开招股时本公司普通股的发行价折让15%或(I)商数200,000,000美元除以发售前的总股本权益 。2022年票据为无抵押票据,优先于向本公司所有商业融资贷款人、保险公司、租赁融资机构或经本公司董事会批准并定期从事贷款业务的其他贷款机构全额支付。从2023年2月到2023年6月,这些票据中的430万美元 被转换为我们B系列优先股的股票,2022年的票据都没有流通股。
股权 和薪酬安排
我们于2022年8月10日通过,我们的股东于2022年8月18日批准了我们的2022年股票计划或2022年计划。2022年计划规定 授予激励性股票期权、非法定股票期权、股票增值权、限制性股票奖励、限制性股票单位奖励和其他股票奖励。2022年,我们根据2022年计划向员工、董事和科学顾问委员会成员发行了相当于101,250份期权的行权价 相当于每股3.28美元。2023年2月,我们向员工和董事额外发行了等值3,689,750份期权,行权价相当于每股2.28美元。一般来说,本公司授予的奖励为期三年,行使价等于本公司董事会在考虑独立第三方评估公司对本公司普通股的同期估值后确定的普通股的估计公允价值。
| 113 |
委托人和注册股东
安全 某些受益所有者和管理层的所有权
下表列出了截至2024年1月23日的情况:
| ● | 截至1月23日,关于我们有投票权的证券(即我们有投票权的普通股和我们的C系列优先股)受益所有权的某些 信息,2024通过(I)我们所知的持有我们5%以上有投票权证券的每个人或关联人集团,(Ii)我们的每一位高管,(Iii)我们的每一位董事 和(Iv)我们的所有董事和高管作为一个团体。除非另有说明, 下列所有人对其普通股拥有(I)唯一投票权和投资权,但根据适用法律由配偶分享的除外,以及(Ii)关于其普通股的记录和受益所有权;以及 |
| ● | 通过本招股说明书为登记股东持有并登记转售的普通股数量 。 |
注册股东包括(I)我们的联属公司和某些持有“受限制证券”(如证券法第144条所界定的 )的其他股东,他们因根据第144条成为联属公司或在之前12个月内从联属公司或吾等购入其普通股而不能根据第144条出售其证券,直至 吾等已遵守证券交易法第13条或第15(D)条的申报要求至少90天 及(Ii)吾等的员工。登记股东可以选择,也可以不选择出售本招股说明书所涵盖的普通股,出售范围由他们决定。登记股东可以公开或私下以现行市价或协议价格发售、出售或分配在此登记的普通股的全部或部分股份。登记股东可选择出售与本次直接上市相关的股份以及在本次直接上市后的市场交易中出售其股份。 因此,如果以及何时任何登记股东可以或不可以选择出售其普通股或任何此类出售的价格,我们将不会有任何意见。请参阅“配送计划.”
有关注册股东的信息 可能会不时更改,如有必要,任何更改的信息将在本招股说明书的附录中列出。由于注册股东可以出售本招股说明书所涵盖的全部、部分或全部普通股,因此我们无法确定注册股东将出售的普通股数量,或在完成任何特定出售后注册股东将持有的普通股的金额或百分比。此外,在提供下表所列信息的日期之后,下表中所列的注册股东可能已经出售、转让或以其他方式处置,或可能出售、转让或以其他方式处置我们在交易中的普通股,而不受证券法的登记要求的约束。
登记股东无权享有与普通股相关的任何登记权利。但是,我们目前打算 尽我们的合理努力,使注册声明在注册声明生效后的90天内有效。 注册声明。我们不与任何注册股东或任何经纪交易商就注册股东出售普通股 达成任何安排。但是,我们将就与我们上市相关的某些其他事宜聘请财务顾问。 请参阅“配送计划.”
| 114 |
根据美国证券交易委员会的规则,受益所有权包括证券的投票权或投资权,并包括根据可在2024年1月23日起60天内行使或交收的期权和认股权证可发行的普通股。 根据期权和认股权证可发行的普通股在计算持有此类证券的人实益持有的类别的百分比时视为未偿还普通股 但在计算任何其他人实益拥有的类别的百分比时并不视为未偿还的普通股。
在下表中,在构成本招股说明书一部分的登记声明生效之前的受益所有权百分比是基于适用的: (I)在实施反向股票拆分和 (A)与直接上市相关的自动注销我们所有已发行的A系列优先股之后, (I)截至2024年1月23日已发行的32,492,068股我们的已发行普通股,(B)与直接上市相关的我们所有已发行的无投票权普通股一对一的自动转换,(C)与直接上市相关的所有已发行的B系列优先股一对一地自动转换为总计4,171,445股我们的普通股,以及(D)与直接上市相关的所有已发行的B-1系列优先股自动转换为总计89,781股我们的普通股;以及(Ii)在反向股票拆分生效后,截至2024年1月23日我们的C系列优先股流通股2,500股。
在直接上市时,我们C系列优先股的每股股票将有权 获得每股13,000个投票权。下表中总投票权的百分比是根据(br}前一段第(I)和(Ii)款所述交易生效和C系列优先股直接上市时每股13,000票)的总和,即(I)32,492,068票,即与本公司普通股32,492,068股(每股普通股有一票)和(Ii)32,500,000票的总投票数计算的。为与2,500股C系列优先股相关的总投票数(C系列优先股每股拥有13,000票)。
FibroGenesis是本公司所有已发行A系列优先股的实益拥有人(在实施反向股票分拆后,总数为8,750,000股),所有已发行的A系列优先股将自动注销,而不加考虑,与直接上市有关。除上述将全部注销的A系列优先股外,FibroGenesis并不拥有我们股本中的任何其他股份。
除本招股说明书所披露的外,注册股东在过去三年内与本公司并无任何职位、职务或其他重大关系。请参阅“管理层对财务业绩和财务状况的讨论与分析“和”某些关系和关联方交易“了解有关注册股东的更多信息。除非另有说明,否则下列个人和实体的营业地址为:c/o FibroBiologics,Inc.,455 E.Medical Center,Blvd.,Suite300,Houston,Texas 77598。
| 登记声明生效前的实益所有权 | ||||||||||||||||||||||||
| 普通股 | C系列优先股 | 总数的百分比 | 根据以下条件登记的普通股 | |||||||||||||||||||||
| 实益拥有人姓名或名称及地址 | 股票 | % | 股票 | % | 投票权(1) | 本招股说明书 | ||||||||||||||||||
| 5%的股东: | ||||||||||||||||||||||||
| 黄金骑士股份有限公司,L.P.(2) | 2,125,001 | 6.5 | % | — | — | 3.3 | % | 218,351 | ||||||||||||||||
| 丹和帕姆·林斯库姆(3) | 1,627,219 | 5.0 | % | — | — | 2.5 | % | 1,627,219 | ||||||||||||||||
| 行政人员及董事 | ||||||||||||||||||||||||
| 皮特·奥希隆,MSHA(4) | 6,048,147 | 18.6 | % | 2,500 | 100 | % | 59.3 | % | — | |||||||||||||||
| 马克·安德森,注册会计师CFA | — | — | — | — | — | — | ||||||||||||||||||
| 哈米德·霍贾博士(5) | 1,250 | * | — | — | * | — | ||||||||||||||||||
| 罗伯特·霍夫曼,注册会计师(非活跃)(6) | 7,500 | * | — | — | * | — | ||||||||||||||||||
| 维多利亚·尼克拉斯,医学博士。(7) | 7,500 | * | — | — | * | — | ||||||||||||||||||
| 小理查德·西伦托,MBA(8) | 93,225 | * | — | — | * | — | ||||||||||||||||||
| 斯泰西·科恩,MBA(9) | 7,500 | * | — | — | * | — | ||||||||||||||||||
| 马修·林克(10) | 7,500 | * | — | — | * | — | ||||||||||||||||||
| 全体董事及行政人员(8人)(11) | 6,172,622 | 19.0 | % | 2,500 | 100 | % | 59.5 | % | — | |||||||||||||||
| 其他登记股东: | ||||||||||||||||||||||||
| 非执行干事雇员、顾问和服务提供商 | — | — | — | — | — | — | ||||||||||||||||||
| 基思·丹纳牧师信托基金(12) | 677,367 | 2.1 | % | — | — | 1.0 | % | 507,245 | ||||||||||||||||
| 本尼·布朗(13) | 565,155 | 1.7 | % | — | — | * | 455,856 | |||||||||||||||||
| 柯林斯合伙有限公司(14) | 509,489 | 1.6 | % | — | — | * | 40,065 | |||||||||||||||||
| 迈克尔·M·拉沃(15) | 497,754 | 1.5 | % | — | — | * | 5,873 | |||||||||||||||||
| 全球能源公司。(16) | 465,744 | 1.4 | % | — | — | * | 272,500 | |||||||||||||||||
| 摩登资本有限责任公司(17) | 403,799 | 1.2 | % | — | — | * | 4,438 | |||||||||||||||||
| 穆斯塔西姆·鲁米(18) | 350,280 | 1.1 | % | — | — | * | 38,462 | |||||||||||||||||
| 标记Monical(19) | 313,223 | 1.0 | % | — | — | * | 183,956 | |||||||||||||||||
| 菲利普·P·洛维尔和伊丽莎白·G·洛维尔(20) | 212,803 | * | — | — | * | 154,890 | ||||||||||||||||||
| 詹姆斯·P·多尔蒂,III Trust,日期:2000年11月14日(21) | 189,868 | * | — | — | * | 91,250 | ||||||||||||||||||
| Barchil Ltd合作伙伴关系(22) | 66,077 | * | — | — | * | 66,077 | ||||||||||||||||||
| David·M·卡瓦纳(23) | 251,574 | * | — | — | * | 92,557 | ||||||||||||||||||
| 梅根·多尔蒂(24) | 45,000 | * | — | — | * | 45,000 | ||||||||||||||||||
| 肖恩·瑞安信托基金2007年3月15日(25) | 106,579 | * | — | — | * | 44,822 | ||||||||||||||||||
| 斯莱德·刘易斯(26) | 99,359 | * | — | — | * | 44,436 | ||||||||||||||||||
| 史蒂文·卡伯顿(27) | 98,600 | * | — | — | * | 62,167 | ||||||||||||||||||
| 安东尼·J·莱登(28) | 105,044 | * | — | — | * | 44,379 | ||||||||||||||||||
| 达雷尔·琼斯(29) | 135,749 | * | — | — | * | 38,462 | ||||||||||||||||||
| 林恩·戴维斯DDS(30) | 62,735 | * | — | — | * | 33,332 | ||||||||||||||||||
| 阿凡达投资有限责任公司(31) | 132,164 | * | — | — | * | 24,424 | ||||||||||||||||||
| 沙娜拉·杰普投资服务有限责任公司(32) | 25,934 | * | — | — | * | 25,934 | ||||||||||||||||||
| 理查德·基思利(33) | 29,576 | * | — | — | * | 22,225 | ||||||||||||||||||
| 桑德拉·刘易斯(34) | 22,218 | * | — | — | * | 22,218 | ||||||||||||||||||
| 约翰·希利斯(35) | 79,081 | * | — | — | * | 22,190 | ||||||||||||||||||
| StartEngine Capital LLC(36) | 22,148 | * | — | — | * | 22,148 | ||||||||||||||||||
| 杰伊·邓普西(37) | 21,779 | * | — | — | * | 21,779 | ||||||||||||||||||
| 乔治·内塞梅尔(38) | 75,486 | * | — | — | * | 19,231 | ||||||||||||||||||
| J·David陶尔米诺(39) | 44,686 | * | — | — | * | 19,231 | ||||||||||||||||||
| 瑞安·塔布洛夫(40) | 18,677 | * | — | — | * | 18,677 | ||||||||||||||||||
| 迈克尔·琼斯(41) | 46,618 | * | — | — | * | 17,752 | ||||||||||||||||||
| 威廉·奥斯汀(42) | 17,752 | * | — | — | * | 17,752 | ||||||||||||||||||
| 迈克尔·摩西(43) | 17,752 | * | — | — | * | 17,752 | ||||||||||||||||||
| 斯科特·德卢森(44) | 16,346 | * | — | — | * | 16,346 | ||||||||||||||||||
| 琳达·路德维希(45) | 19,949 | * | — | — | * | 11,199 | ||||||||||||||||||
| GNGI投资服务 LLLP(46) | 12,774 | * | — | — | * | 12,774 | ||||||||||||||||||
| 维克托·埃斯特拉达(47) | 143,543 | * | — | — | * | 9,764 | ||||||||||||||||||
| 杰里·柯林斯(48) | 8,876 | * | — | — | * | 8,876 | ||||||||||||||||||
| 伊凡一世(49) | 25,412 | * | — | — | * | 8,506 | ||||||||||||||||||
| 詹姆斯·塞奇(50) | 8,506 | * | — | — | * | 8,506 | ||||||||||||||||||
| 克里斯·麦考尔(51) | 8,387 | * | — | — | * | 8,387 | ||||||||||||||||||
| 格拉迪斯·E皮尔森(52) | 10,644 | * | — | — | * | 10,644 | ||||||||||||||||||
| Schnoy,LLC(53) | 38,825 | * | — | — | * | 8,188 | ||||||||||||||||||
| 马歇尔A.刘易斯(54) | 8,136 | * | — | — | * | 8,136 | ||||||||||||||||||
| 约翰·梅(55) | 7,692 | * | — | — | * | 7,692 | ||||||||||||||||||
| 芭芭拉·佩拉斯凯瓦斯(56) | 7,316 | * | — | — | * | 7,316 | ||||||||||||||||||
| 萨比奥·维洛里亚(57) | 12,982 | * | — | — | * | 7,101 | ||||||||||||||||||
| 查德·伯恩斯(58) | 7,101 | * | — | — | * | 7,101 | ||||||||||||||||||
| 格雷格·马赫(59) | 21,174 | * | — | — | * | 6,472 | ||||||||||||||||||
| 罗伯特·安东尼(60) | 7,096 | * | — | — | * | 7,096 | ||||||||||||||||||
| 史蒂夫·赖斯(61) | 5,104 | * | — | — | * | 5,104 | ||||||||||||||||||
| 里奇·桑伯格(62) | 4,863 | * | — | — | * | 4,863 | ||||||||||||||||||
| 查尔斯·卡登黑德(63) | 4,808 | * | — | — | * | 4,808 | ||||||||||||||||||
| RoboSpine LLC(64) | 4,623 | * | — | — | * | 4,623 | ||||||||||||||||||
| 泰德·福勒(65) | 52,421 | * | — | — | * | 4,582 | ||||||||||||||||||
| J·迪克森·马普斯(66) | 29,893 | * | — | — | * | 4,438 | ||||||||||||||||||
| 马克·德里克沃特(67) | 17,166 | * | — | — | * | 4,438 | ||||||||||||||||||
| 达林·尚德利(68) | 4,438 | * | — | — | * | 4,438 | ||||||||||||||||||
| 玛丽·陶尔米诺(69) | 4,438 | * | — | — | * | 4,438 | ||||||||||||||||||
| Wichuta Prochnow(70) | 4,068 | * | — | — | * | 4,068 | ||||||||||||||||||
| 巴特·帕斯捷尔纳克(71) | 3,938 | * | — | — | * | 3,938 | ||||||||||||||||||
| 查尔斯·里德(72) | 3,883 | * | — | — | * | 3,883 | ||||||||||||||||||
| 谢丽尔·摩西(73) | 3,872 | * | — | — | * | 3,872 | ||||||||||||||||||
| 罗伯特·迈尔斯退休计划-罗斯(74) | 3,750 | * | — | — | * | 3,750 | ||||||||||||||||||
| 威廉姆·福特(75) | 41,865 | * | — | — | * | 3,551 | ||||||||||||||||||
| 格洛丽亚·洛佩兹(76) | 1,480 | * | — | — | * | 1,480 | ||||||||||||||||||
| 唐娜·L·布朗(77) | 740 | * | — | — | * | 740 | ||||||||||||||||||
| 杰森·D·塞勒斯(78) | 5,925 | * | — | — | * | 5,925 | ||||||||||||||||||
| 所有其他登记股东(79) | 612,747 | 1.9 | % | — | — | % | * | % | 236,533 | |||||||||||||||
| 正在登记的股份总数 | 4,806,226 | |||||||||||||||||||||||
* 不到1%。
| (1) | 在 C系列优先股的权利生效后,直接上市时,每股有13,000个投票权。 |
| (2) | 持有的普通股数量包括:(I)1,906,650股无投票权普通股,与直接上市相关,将一对一地自动转换为1,906,650股普通股;(Ii)218,351股B系列优先股,与直接上市相关,将一对一地自动转换为218,351股普通股。Michael F.Newlin和Cindy L.Newlin作为Golden Knight Inc.,L.P.的普通合伙人, 拥有投票和处置Golden Knight Inc.,L.P.直接持有的股份的酌情决定权,可被视为该等股份的实益拥有人。黄金骑士股份有限公司的地址是3773Howard Hughes Pkwy,Suite 500S,拉斯维加斯,邮编:89189-6014. |
| (3) | 持有的普通股数量包括1,627,219股B系列优先股,与直接上市 相关,将按一对一原则自动转换为总计1,627,219股普通股。丹和帕姆·林斯库姆的地址是德克萨斯州休斯敦圣菲利佩大街5110号,邮编:77056。 |
| (4) | 持有的普通股数量包括6,048,147股无投票权普通股,与直接上市有关,将按一对一原则自动转换为6,048,147股普通股。持有的2,500股C系列优先股 构成我们被授权发行的C系列优先股的最大数量。直接上市后,C系列优先股每股将有权获得13,000票。只要C系列优先股仍未发行,C系列优先股将受到皮特·奥希隆签发的不可撤销的委托书的约束,并有利于我们的董事会, 如本招股说明书中更详细地描述的那样。 |
| (5) | 持有的普通股数量包括1,250股无投票权普通股,与直接上市有关, 将按一对一原则自动转换为总计1,250股普通股。 |
| (6) | 持有的普通股数量包括7,500股无投票权普通股,与直接上市有关, 将按一对一的原则自动转换为总计7,500股普通股。 |
| (7) | 持有的普通股数量包括7,500股无投票权普通股,与直接上市有关, 将按一对一的原则自动转换为总计7,500股普通股。 |
| (8) | 持有的普通股数量包括93,225股无投票权普通股,与直接上市有关, 将按一对一原则自动转换为总计93,225股普通股。 |
| (9) | 持有的普通股数量包括7,500股无投票权普通股,与直接上市有关, 将按一对一的基础自动转换为总计7,500股普通股 |
| (10) | 持有的普通股数量包括7,500股无投票权普通股,与直接上市有关, 将按一对一的原则自动转换为总计7,500股普通股。 |
| (11) | 持有的普通股数量包括6,172,622股无投票权普通股,与直接上市有关,将按一对一原则自动转换为总计6,172,622股普通股。持有的2,500股C系列优先股 构成我们被授权发行的C系列优先股的最大数量。直接 上市后,C系列优先股每股将有权获得13,000票。只要C系列优先股仍未发行,C系列优先股将受制于皮特·奥希隆签发的不可撤销的委托书,该委托书将受惠于我们的董事会 ,这在本招股说明书中有更详细的描述。 |
| (12) | 持有的普通股数量包括:(I)170,122股无投票权普通股,与直接上市相关,将按一对一原则自动转换为总计170,122股普通股;(Ii) 507,245股B系列优先股,与直接上市相关,按1对1原则自动转换为507,245股普通股。Keith Denner Rev Trust的地址是C/o FibroBiologics,地址是德克萨斯州休斯敦,Ste300,E Medical Center Blvd,455E,TX 77598。 |
| (13) | 持有的普通股数量包括:(I)109,299股无投票权普通股,与直接上市相关,将一对一地自动转换为109,299股普通股;(Ii)455,856股B系列优先股,与直接上市相关,将一对一自动转换为455,856股普通股。本尼·布朗的地址是田纳西州富兰克林教堂北路4338号,邮编37067。 |
| (14) | 持有的普通股数量包括:(1)469,424股无投票权普通股,与直接上市相关,将一对一自动转换为469,424股普通股;(2)20,711股B系列优先股,与直接上市相关,将一对一自动转换为20,711股普通股;(3)19,354股B-1系列优先股,关于直接上市,将按一对一的方式自动转换为19,354股普通股。柯林斯合伙有限公司的地址是德克萨斯州奥斯汀伯内特路5000号,邮编78756。 |
| (15) | 持有的普通股总数 迈克尔A。拉沃尔,野猫海军有限责任公司,任务管理和信托公司FBO迈克尔M。Lavor SEP IRA with Michael A. Lavor 作为受益人,包括(i)491,881股无投票权普通股,就直接上市而言, 将在一对一的基础上自动转换为491,881股普通股和(ii)5,873股B系列优先股 与直接上市相关的股票将在一对一的基础上自动转换为5,873股普通股 车辆.迈克尔·A的地址地址:3650 N Camino Ojo DeAgua,Tucson,AZ 85749 |
| (16) | 该 持有的普通股数量包括465,744股无投票权普通股,与直接 上市,将自动转换,在一对一的基础上,为465,744股普通股。Global Energy Source的地址 Inc是6233 N。75这是St,Scottsdale, AZ 85250. |
| (17) | 持有的普通股数量包括 总计(i)399,361股无投票权普通股,与直接上市相关,将自动转换, 以一对一的方式,分为399,361股普通股和(ii)4,438股B系列优先股, 直接上市后,将自动转换为4,438股普通股。Mod的地址 资本有限责任公司是14840 Jolenta Ln,榆树林,威斯康星州53122。 |
| (18) | 持有的普通股总数 作者:Mustasim Rumi、MDR Strategic Investments,LLC、Quest IRA FBO Mustasim Rumi、Sarah V Investments,LLC和Vendal-Rumi Irrigable 以Mustasim Rumi为受益人的信托包括(i)311,818股无投票权普通股, 与直接上市,将自动转换,在一对一的基础上,为311,818股普通股和(ii)38,462 B系列优先股的股份,与直接上市有关,将自动转换,一对一的基础上, 转换成38,462股普通股地址:5919 Bold Ruler Way,Austin,TX 78746 |
| (19) | 该 持有的普通股股份总数包括(i)129,267股无投票权普通股, 与直接上市,将自动转换,在一对一的基础上,为129,267股普通股和(ii)183,956 B系列优先股的股份,与直接上市有关,将自动转换,一对一的基础上, 183,956股普通股地址:78 Rosewood St,Lake Jackson,TX 77566 |
| (20) | 持有的普通股总数 由洛弗尔家族信托基金于2002年3月12日和Vantage Roth IRA FBO菲利普洛弗尔与菲利普P洛弗尔和伊丽莎白G。洛弗尔(Lovell) 受益人包括(i)194,163股无投票权普通股,与直接上市相关, 将自动转换为194,163股普通股和(ii)18,640股B系列优先股 与直接上市相关的股票,将在一对一的基础上自动转换为18,640股普通股 车辆.菲利普P洛弗尔和伊丽莎白G。洛弗尔是4601 E。Foothill Drive,Paradise Valley,AZ 85253. |
| (21) | 持有的普通股数量包括189,868股 与直接上市相关的无投票权普通股,将以一对一的方式自动转换为 189,868股普通股2000年11月14日詹姆斯·P·多尔蒂三世信托基金的地址是107 Whisper Dunes Dr., 密歇根州,邮编46360。 |
| (22) | 该 持有的普通股数量包括66,077股B系列优先股,与直接 上市后,将自动转换为66,077股普通股。Barchil Ltd Partnership的地址 是邮政信箱11739,沃思堡,TX 76110。 |
| (23) | 该 大卫·卡瓦纳、大卫·卡瓦纳信托、迪尔伯恩资本管理有限公司、 及David Kavanagh 2010信托(以David M Kavanagh及其家人为受益人)包括(i)159,017股无投票权 普通股,与直接上市相关,将在一对一的基础上自动转换为159,017股 普通股和(ii)92,557股B系列优先股,与直接上市有关,将自动 一对一转换为92,557股普通股。David M Kavanagh的地址是2001 Knollwood Rd,Lake Forest,IL 60045. |
| (24) | 该 持有的普通股数量包括45,000股无投票权普通股,就直接上市而言, 将在一对一的基础上自动转换为45,000股普通股。Megan Doherty的地址是145 Montgomery St,Glencoe IL 60022. |
| (25) | 该 持有的普通股股份总数包括(i)61,757股无投票权普通股, 直接上市后,将按一对一的方式自动转换为61,757股普通股和(ii)44,822股 B系列优先股,与直接上市相关,将在一对一的基础上自动转换为 44,822股普通股。地址为肖恩瑞安信托dtd 3/15/07是1425 E平托Ct,吉尔伯特,亚利桑那州85233。 |
| (26) | 该 持有的普通股数量包括:(i)54,923股无投票权普通股, 直接上市后,将按一对一的方式自动转换为54,923股普通股和(ii)44,436股 B系列优先股,与直接上市相关,将在一对一的基础上自动转换为 44,436股普通股。斯莱德·刘易斯的地址是501 Hunters Lane,Pastswood,TX 77546。 |
| (27) | 该 Steven Caperton和Painted River Royalties LLC(Steven Caperton为受益人)持有的普通股总数 包括(i)36,433股无投票权普通股,就直接上市而言,该等股份将自动 在一对一的基础上转换为36,433股普通股和(ii)62,167股B系列优先股, 与直接上市相关的股票将自动转换为62,167股普通股。地址 史蒂文·卡珀顿的地址是1855 Painted River Rd,Valley Mills,TX 76689。 |
| (28) | 该 Anthony J. Lydon和Anthony J. Lydon Living Trust以Anthony J. Lydon为 受益人包括(i)60,665股无投票权普通股,就直接上市而言, 将在一对一的基础上自动转换为60,665股普通股和(ii)44,379股B系列优先股 与直接上市相关的股票,将在一对一的基础上自动转换为44,379股普通股 车辆.安东尼·J·莱登的地址是4300 E地址:Camelback Rd,Ste 100,Phoenix,AZ 85018 |
| (29) | 该 Darrell Jones和Darrell Matthew Jones Trust(以Darrell Jones为受益人)持有的普通股总数 包括(i)97,287股无投票权普通股,就直接上市而言,该等股份将自动 在一对一的基础上转换为97,287股普通股和(ii)38,462股B系列优先股, 与直接上市相关,将自动转换为38,462股普通股。地址 达雷尔·琼斯的地址是堪萨斯州塞当市68号邮政信箱67361 |
| (30) | 该 持有的普通股股份总数包括(i)29,403股无投票权普通股, 直接上市后,将按一对一的方式自动转换为29,403股普通股和(ii)33,332股 B系列优先股,与直接上市相关,将在一对一的基础上自动转换为 33,332股普通股。Lynn Davis DDS的地址是19 Ellis Rd,League City,TX 77573。 |
| (31) | 该 Avatar Investments,LLC以Galen Miler为受益人持有的普通股总数为 (i)107,740股无投票权普通股,与直接上市有关,将以一对一的方式自动转换 107,740股普通股和(ii)24,424股B系列优先股,与直接 上市,将自动转换,在一对一的基础上,为24,424股普通股。盖伦米勒的地址是20539 N.熊峡谷CT,惊喜吧,亚利桑那州85387。 |
| (32) | 持有的普通股数量包括25,934股B-1系列优先股,与直接上市有关,将按一对一的原则自动转换为25,934股普通股。沙娜拉 Jap Investment Services LLLP的地址是AZ 85712,Tucson,Suite200 Grant Rd5995E。 |
| (33) | 持有的普通股数量包括:(I)7,351股无投票权普通股,与直接上市相关,将一对一地自动转换为7,351股普通股;(Ii)22,225股B系列优先股,与直接上市相关,将一对一自动转换为22,225股普通股。理查德·基斯利的地址是德克萨斯州弗里德斯伍德安德森牧场104号,邮编:77546。 |
| (34) | 持有的普通股数量包括22,218股B系列优先股,与直接上市 相关,将按一对一的原则自动转换为22,218股普通股。桑德拉·刘易斯的地址是德克萨斯州联盟城隐藏松树州204号,邮编:77573。 |
| (35) | 持有的普通股数量包括:(I)56,891股无投票权普通股,与直接上市相关,将一对一地自动转换为56,891股普通股;(Ii)22,190股B系列优先股,与直接上市相关,将一对一自动转换为22,190股普通股。约翰·希利斯的地址是加州戴维斯拉塞尔公园400号,邮编:95616。 |
| (36) | 持有的普通股数量包括22,148股B系列优先股,与直接上市 相关,将按一对一的原则自动转换为22,148股普通股。StartEngine Capital的地址是C/o FibroBiologics,C/o FibroBiologics,455E Medical Center Blvd,Ste300,Houston,TX 77598。 |
| (37) | 持有的普通股数量包括21,779股B系列优先股,与直接上市 相关,将按一对一的原则自动转换为21,779股普通股。杰伊·邓普西的地址是C/o FibroBiologics,455 E Medical Center Blvd,Ste300,Houston,TX 77598。 |
| (38) | 持有的普通股数量包括:(I)56,255股无投票权普通股,与直接上市相关,将一对一地自动转换为56,255股普通股;(Ii)19,231股B系列优先股,与直接上市相关,将一对一自动转换为19,231股普通股。George Nesemeier的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,455E,邮编:77598。 |
| (39) | 持有的普通股数量包括:(I)25,455股无投票权普通股,与直接上市相关,将一对一地自动转换为25,455股普通股;(Ii)19,231股B系列优先股,与直接上市相关,将一对一自动转换为19,231股普通股。David的地址是加州戴维斯5号F街429F号,邮编:95616。 |
| (40) | 持有的普通股数量包括18,677股B系列优先股,与直接上市 相关,将按一对一的原则自动转换为18,677股普通股。瑞安·塔布洛夫的地址是亚利桑那州斯科茨代尔市克莱蒙特7343E,邮编:85250。 |
| (41) | 持有的普通股数量包括:(I)28,866股无投票权普通股,与直接上市相关,将一对一地自动转换为28,866股普通股;(Ii)17,752股B系列优先股,与直接上市相关,将一对一自动转换为17,752股普通股。迈克尔·琼斯的地址是KS 67361,Sedan 20路234号。 |
| (42) | 持有的普通股数量包括17,752股B系列优先股,与直接上市 相关,这些优先股将按一对一的原则自动转换为17,752股普通股。威廉·奥斯汀的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,455E,邮编:77598。 |
| (43) | 持有的普通股数量包括17,752股B系列优先股,与直接上市 相关,这些优先股将按一对一的原则自动转换为17,752股普通股。迈克尔·摩西的地址是德克萨斯州休斯敦,Ste300,医疗中心大道455E号,C/o FibroBiologics,邮编:77598。 |
| (44) | 持有的普通股数量包括16,346股B系列优先股,与直接上市 相关,这些优先股将按一对一的原则自动转换为16,346股普通股。斯科特·德鲁岑的地址是德克萨斯州休斯敦,Ste300,E Medical Center Blvd,455E,FibroBiologics,C/o,邮编:77598。 |
| (45) | 持有的普通股数量包括:(I)8,750股无投票权普通股,与直接上市相关,将一对一地自动转换为8,750股普通股;(Ii)11,199股B系列优先股,与直接上市相关,将一对一自动转换为11,199股普通股。琳达·路德维希的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,455E, 77598。 |
| (46) | 持有的普通股数量包括12,774股B-1系列优先股,与直接上市有关,将按一对一的原则自动转换为12,774股普通股。GNGI投资服务有限责任公司的地址是亚利桑那州图森市格兰特大道5995E Suite200,邮编:85712。 |
| (47) | 麦迪逊信托公司、托管FBO Victor Estrada M1604015和Victor A Estrada以Victor A Estrada为受益人持有的普通股总数包括:(I)133,779股无投票权普通股,与直接上市 将按一对一的基础自动转换为133,779股普通股,以及(Ii)9,764股B系列优先股,与直接上市将一对一地自动转换,转换为9,764股普通股。维克多·A·埃斯特拉达的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,455E,邮编:77598。 |
| (48) | 持有的普通股数量包括8,876股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为8,876股普通股。Jerry·柯林斯的地址是15604 E。 菊苣博士,喷泉山庄,邮编85268。 |
| (49) | 持有的普通股数量包括:(I)16,906股无投票权普通股,与直接上市相关,将一对一自动转换为16,906股普通股;(Ii)8,506股B系列优先股,与直接上市相关,将一对一自动转换为 8,506股普通股。Ivan I Smith III的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,455 E,邮编:77598。 |
| (50) | 持有的普通股数量包括8,506股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为8,506股普通股。James Sage的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,邮编:77598。 |
| (51) | 持有的普通股数量包括8,387股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为8,387股普通股。克里斯·麦考尔的地址是C/o FibroBiologics,德克萨斯州休斯顿,Ste300,E Medical Center Blvd,邮编:77598。 |
| (52) | 持有的普通股数量包括10,644股B-1系列优先股,与直接上市有关,将按一对一的原则自动转换为10,644股普通股。格拉迪斯·E·皮尔森的地址是德克萨斯州康罗市,Ste110,West Davis Street 122号,邮编:77301。 |
| (53) | 持有的普通股数量包括:(I)30,637股无投票权普通股,与直接上市相关,将一对一地自动转换为30,637股普通股;(Ii)8,188股B系列优先股,与直接上市相关,将一对一自动转换为8,188股普通股。Schnoy,LLC的地址是7411E.Calle Cabo,Tucson,AZ 85750。 |
| (54) | 该 持有的普通股数量包括8,136股B系列优先股,就直接上市而言, 将在一对一的基础上自动转换为8,136股普通股。马歇尔A的地址路易斯是c/o FibroBiologics, 455 E Medical Center Blvd,Ste 300,Houston,TX 77598. |
| (55) | 该 持有的普通股数量包括7,692股B系列优先股,就直接上市而言, 将在一对一的基础上自动转换为7,692股普通股。John Mai的地址是c/o FibroBiologics, 455 E Medical Center Blvd,Ste 300,Houston,TX 77598. |
| (56) | 该 持有的普通股数量包括7,316股B系列优先股,就直接上市而言, 将在一对一的基础上自动转换为7,316股普通股。Barbara Peraskevas的地址为c/o FibroBiologics,455 E Medical Center Blvd,Ste 300,Houston,TX 77598。 |
| (57) | 该 持有的普通股数量包括:(i)5,881股无投票权普通股, 直接上市后,将按一对一的方式自动转换为5,881股普通股和(ii)7,101股 B系列优先股,与直接上市相关,将在一对一的基础上自动转换为 7,101股普通股。Sabio Viloria的地址为c/o FibroBiologics,455 E Medical Center Blvd,Ste 300,Houston,TX 77598. |
| (58) | 该 持有的普通股数量包括7,101股B系列优先股,就直接上市而言, 将在一对一的基础上自动转换为7,101股普通股。Chad Burns的地址是215 Magnolia Ridge Pl,Apt 206,Dothan,AL 36303. |
| (59) | 该 Greg Maher和Vantage FBO Gregory Maher IRA以Gregory Maher为受益人持有的普通股总数 包括(i)14,702股无投票权普通股,就直接上市而言,该等股份将自动 在一对一的基础上转换为14,702股普通股和(ii)6,472股B系列优先股,其中, 直接上市后,将自动转换为6,472股普通股。Greg的地址 马赫是13020 N 82发送斯科茨代尔街,亚利桑那州85260。 |
| (60) | 持有的普通股数量包括7,096股B-1系列优先股,与直接上市有关,将按一对一的原则自动转换为7,096股普通股。罗伯特·J·安东尼的地址是德克萨斯州休斯敦602单元白令博士661号,邮编:77057。 |
| (61) | 持有的普通股数量包括5,104股B系列优先股,与直接上市相关, 将按一对一的基础自动转换为5,104股普通股。史蒂夫·赖斯的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,邮编:77598。 |
| (62) | 持有的普通股数量包括4,863股B系列优先股,与直接上市有关, 将按一对一的原则自动转换为4,863股普通股。Rich Sangberg的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,邮编:77598。 |
| (63) | 持有的普通股数量包括4,808股B系列优先股,与直接上市有关, 将按一对一的基础自动转换为4,808股普通股。查尔斯·卡登黑德的地址是德克萨斯州布拉索里亚582a县路2303号,邮编77422。 |
| (64) | 持有的普通股数量包括4,623股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为4,623股普通股。RoboSpine LLC的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,邮编:77598。 |
| (65) | Ted Fowler、2XD Productions 401K FBO Ted Fowler和以Ted Fowler为受益人的Vantage Retiering Ted Fowler合计持有的普通股数量包括(I)47,839股无投票权普通股,与直接上市 将按一对一的基础自动转换为47,839股普通股,以及(Ii)B系列优先股的4,582股 将与直接上市一对一自动转换,换成4,582股普通股。泰德·福勒的地址是德克萨斯州帕克城铁峡谷博士2422号,邮编:84060。 |
| (66) | 持有的普通股数量包括:(I)25,455股无投票权普通股,与直接上市相关,将一对一地自动转换为25,455股普通股;(Ii)4,438股B系列优先股,与直接上市相关,将一对一自动转换为4,438股普通股。J.Dickson Mappus的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,455E,邮编:77598。 |
| (67) | 持有的普通股数量包括:(I)12,728股无投票权普通股,与直接上市相关,将一对一地自动转换为12,728股普通股;(Ii)4,438股B系列优先股,与直接上市相关,将一对一自动转换为4,438股普通股。马克·德里克沃特的地址是澳大利亚斯科茨代尔蒙特贝洛大道7715E号,邮编:85250。 |
| (68) | 持有的普通股数量包括4,438股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为4,438股普通股。达林·尚德利的地址是德克萨斯州波特兰Wildcat大道1540号,邮编:78374。 |
| (69) | 持有的普通股数量包括4,438股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为4,438股普通股。玛丽·陶尔米诺的地址是加州戴维斯布坎南大街615号,邮编:95616。 |
| (70) | 持有的普通股数量包括4,068股B系列优先股,与直接上市有关, 将按一对一的原则自动转换为4,068股普通股。Wichuta Prochnow的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,邮编:77598。 |
| (71) | 持有的普通股数量包括3,938股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为3,938股普通股。巴特·帕斯捷尔纳克的地址是康涅狄格州诺沃克市坎菲尔德路1号,邮编:06855。 |
| (72) | 持有的普通股数量包括3,883股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为3,883股普通股。Charles Read的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,邮编:77598。 |
| (73) | 持有的普通股数量包括3,872股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为3,872股普通股。谢丽尔·摩西的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,邮编:77598。 |
| (74) | 持有的普通股数量包括3,750股B系列优先股,与直接上市相关, 将按一对一的基础自动转换为3,750股普通股。罗伯特·迈尔斯退休计划的地址是8390 E Via de Ventura Ste F110#169,Scott dale,AZ 85258。 |
| (75) | 持有的普通股数量包括:(I)38,314股无投票权普通股,与直接上市相关,将一对一地自动转换为38,314股普通股;(Ii)3,551股B系列优先股,与直接上市相关,将一对一自动转换为3,551股普通股。威廉·福特的地址是C/o FibroBiologics,德克萨斯州休斯敦,Ste300,E Medical Center Blvd,455E, 77598。 |
| (76) | 持有的普通股数量包括1,480股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为1,480股普通股。格洛丽亚·E·洛佩兹的地址是田纳西州富兰克林教堂路4330 N,邮编37067。 |
| (77) | 持有的普通股数量包括740股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为740股普通股。唐娜·L·布朗的地址是加利福尼亚州西萨克拉门托格林布里尔路1292号,邮编95691。 |
| (78) | 持有的普通股数量包括5,925股B系列优先股,与直接上市相关, 将按一对一的原则自动转换为5,925股普通股。Jason D Sellers的地址是邮政信箱 41504,田纳西州纳什维尔,邮编37204。 |
| (79) | 675名股东持有的普通股数量包括:(1)376,214股无投票权普通股 ,将在一对一的基础上自动转换为376,214股普通股, (Ii)222,554股B系列优先股,将在1:1的基础上自动转换为222,554股普通股 ,和(2)11,172股B系列优先股,与Direct 上市相关,将以一对一的方式自动转换为11,172股普通股。这些将与直接上市相关登记的 股票的总投票权不到总投票权的1%。 |
| 115 |
股本说明
一般信息
以下说明概述了本公司股本的某些重要条款,因为这些条款预计将与本招股说明书所包含的注册说明书的有效性有关。我们预计将通过修订和重述的公司注册证书和修订和重述的章程,该修订和重述的章程将与本招股说明书构成的注册声明的有效性相关而生效,本说明概述了预计将包括在此类 文件中的条款。因为它只是一个摘要,所以它不包含可能对您重要的所有信息。有关本部分所述事项的完整说明,请参阅标题为“股本说明,“您应该参考我们修订和重述的公司证书和我们修订和重述的章程,它们作为本招股说明书的一部分包括在注册声明中 ,并参考特拉华州法律的适用条款。
在直接上市方面,(I)我们当时所有未偿还的A系列优先股,全部由FibroGenesis持有, 将自动注销,而无需向其持有人或向其持有人支付额外代价,(Ii)我们当时尚未偿还的 无投票权普通股将自动转换为 普通股,而无需向其持有人或其持有人支付额外代价,一对一的基础上,(Iii)我们所有当时尚未发行的B系列优先股和我们当时尚未发行的所有B系列B-1优先股将按一对一的基础自动转换为普通股,而不需要由其持有人或向其持有人支付额外代价,以及(Iv)我们所有当时未偿还的C系列优先股将保持C系列优先股,这样,在直接上市后,我们的已发行和已发行股本将立即包括普通股和C系列优先股 。
于 完成直接上市后,于吾等经修订及重述的公司注册证书的存档及生效及吾等经修订及重述的附例通过后,吾等将获授权发行110,000,000股股本,包括:(I)100,000,000股普通股,每股面值0.00001美元及(Ii)10,000,000股优先股,每股面值0.00001美元,其中2,500股被指定为C系列优先股。
在实施反向股票拆分和与直接上市相关的自动 转换后,截至2023年9月30日,我们所有当时已发行的无投票权普通股和可转换优先股(即我们的B系列优先股和B-1系列优先股)共有32,477,209股已发行普通股,由1,178名登记股东持有,我们的C系列优先股有2,500股,全部为C系列已发行优先股,由一名股东持有。根据我们的 修订和重述的公司注册证书,我们的董事会将有权增发我们的股本,而不需要股东批准,但纳斯达克规则要求的除外。
普通股 股票
我们修订和重述的公司注册证书将规定:
| ● | 普通股的持有者 将有投票权选举我们的董事以及需要股东采取行动的所有其他事项,但如果受影响的优先股系列的持有者有权就该修正案进行表决,则我们的公司注册证书修正案将 变更或改变任何已发行优先股的权力、优先股、权利或其他条款。 | |
| ● | 普通股持有者 将有权对股东表决的事项享有每股一票的投票权,还将有权获得我们董事会可能不时宣布的股息(如果有),从合法的可用资金中提取 ; | |
| ● | 支付普通股股息(如果有的话)将以优先股股利优先股股息为条件; | |
| ● | 在我们清算或解散后,普通股持有人将有权获得按比例在偿还所有债务和准备清算当时已发行的任何优先股后,剩余可供分配给股东的所有资产 ;以及 | |
| ● | 我们的 股东没有转换、优先认购权或其他认购权,也没有适用于普通股的偿债基金或赎回条款。 |
优先股 股票
我们的 修订和重述的公司注册证书将规定,优先股股份可能会不时以一个 或多个系列发行。我们的董事会将被授权确定适用于 每个系列股票的投票权(如有)、指定、权力、优先权、相对的、 参与的、可选的或其他特殊权利(如有),以及任何资格、限制和约束。我们的董事会将能够在未经股东批准的情况下,发行具有投票权和 其他权利的优先股,这些优先股可能对普通股持有人的投票权和其他权利产生不利影响,并可能产生反收购 效果。我们的董事会在未经股东批准的情况下发行优先股的能力可能会延迟、 推迟或阻止我们的控制权发生变化或我们现有管理层的离职。
| 116 |
C系列优先股
完成直接上市后,将有一个系列的指定优先股,即C系列优先股,其中2,500股获得授权 ,所有2,500股C系列优先股将由我们的创始人、首席执行官兼董事会主席皮特·奥希隆发行、发行和持有。C系列优先股的流通股均已缴足股款且不可评估。
在我们清算、解散、清盘或其他情况下,C系列优先股应 优先于我们的普通股。
完成直接上市后, C系列优先股有权就待我们股东表决的任何事项投票,在每种情况下,与我们普通股的持有人作为一个单一类别一起投票,C系列优先股每股将有权获得13,000票。 C系列优先股应有权收到与我们普通股股东相同的任何股东会议的事先通知 。
C系列优先股无权获得任何股息,无论是以现金、股票还是财产支付。
在受 其他类别或系列优先股的优先权利的约束下,如果我公司发生任何清算、解散或清盘,则C系列优先股有权优先于此类清算、解散或清盘我们的任何资产给普通股持有人,获得每股18.00美元的清算优先权(如果发生任何股票拆分、合并或其他类似的资本重组,则受适当的 调整)。
C系列优先股可按如下方式随时转换:
| ● | 根据持有人的选择权,C系列优先股的一股可转换为我们普通股的一股。 | |
| ● | 在选出C系列优先股当时的大多数流通股的持有人后,C系列优先股的所有流通股可以一对一的方式转换为同等数量的普通股。 |
此外,在转让C系列优先股时,C系列优先股 必须进行强制转换。在转让C系列优先股的任何股份时,C系列优先股的每股股票将自动转换为我们普通股的一股,而不需要由其持有人或向其持有人支付额外的代价。任何按上述方式转换的C系列优先股股票必须注销并注销,不得作为该系列股票重新发行。
只要C系列优先股仍未发行,则当时已发行的C系列优先股的总股数应按比例调整 因我们普通股的细分或组合或其他类似资本重组而导致的普通股已发行股数的增加或减少,在每种情况下均不收取我们的对价。
C系列优先股将 受制于所有C系列优先股的持有者皮特·奥希隆签发的不可撤销的委托书,授予我们的董事会不可撤销的代理权,只要C系列优先股仍未发行,就C系列优先股有权 投票的所有事项,投票方式由我们的董事会以其唯一和绝对的酌情决定权决定;但是,如果该不可撤销的委托书未经皮特·奥赫隆的书面同意,不得允许我们的董事会就修改、删除或放弃皮特·奥赫隆关于C系列优先股的任何权利的任何提议进行表决 ,如我们修订和重述的公司注册证书所述。鉴于与C系列优先股相关的优越投票权,不可撤销的委托书旨在确保该等优越投票权的使用符合我们的最佳利益,并 避免或缓解作为个人股东员工的皮特·奥希隆未来可能出现的冲突。
| 117 |
反收购 公司注册证书、附则和特拉华州法律的影响
我们的 修订和重述的公司注册证书以及修订和重述的章程将包括许多条款,这些条款可能会延迟、推迟或阻止另一方获得对我们的控制权,并鼓励考虑主动要约或其他单方面收购提议的人与我们的董事会进行谈判,而不是进行非谈判收购 尝试。这些规定包括以下所述的项目。
分类 板
我们修改和重述的公司注册证书将要求我们的董事会分为三个级别,交错 三年任期,每年选举一个级别。董事分类的效果是使股东更难改变我们董事会的组成。
股东 书面同意的诉讼
我们的 修订和重述的公司注册证书将要求,我们的股东 必须采取或允许采取的任何行动都必须在正式召开的股东年度或特别会议上生效,并且不得通过书面同意代替 会议来生效。
提前 通知要求
我们的 修订和重述的章程将建立有关股东提案的预先通知程序,这些提案涉及提名 董事候选人或提交给股东会议的新业务。这些程序将规定 股东提案的通知必须在采取行动 的会议召开之前及时以书面形式提交给公司秘书,并定义什么是及时的。我们修订和重述的章程还将规定所有股东通知的格式和 内容要求。这些要求可能会阻止股东在 年度或特别会议上向股东提出问题。
董事 撤换和空缺
我们修订和重述的公司注册证书将要求,我们的董事会成员或整个董事会成员只能 因原因被免职,然后必须得到至少66名持股人的赞成票2/3本公司股票有权投票除名的投票权比例为%。此外,我们修改和重述的公司注册证书将要求,任何因增加董事人数或董事会任何空缺而新设立的董事职位,必须由在任董事总数的多数票(即使不足法定人数)或唯一剩余的 董事完全 投票填补,不得由股东填补。
绝对多数投票要求
我们修改和重述的公司注册证书将需要至少66名持有人的赞成票。2/3%投票权 有权表决的本公司股票有权(I)修订、更改或废除本公司的章程并采纳新的章程,或(Ii)修订、更改、更改 或废除或采纳任何与本公司注册证书的某些条款不一致的条款,包括有关设立分类董事会的条款、与罢免董事有关的条款、禁止 股东以书面同意方式采取行动的条款,以及我们经修订及重述的公司注册证书中的选择法院条款(如下所述)。
未指定的 优先股
我们修订和重述的公司注册证书将规定优先股的授权股份。授权但未发行的优先股的存在可能使我们的董事会能够阻止通过合并、要约收购、代理竞争或其他方式获得对我们的控制权的企图。例如,如果我们的董事会在适当行使其受托责任时确定收购提议不符合我们股东的最佳利益,我们的董事会可以在一个或多个非公开发行或其他可能稀释拟议收购者或叛逆股东或股东团体的投票权或其他权利的交易中,在未经股东批准的情况下发行优先股股票 。在这方面,我们修订和重述的公司注册证书将赋予我们的董事会广泛的权力,以确立优先股的授权和 未发行股份的权利和优先权。发行优先股可能会减少可供分配给普通股持有者的收益和资产。发行还可能对这些持有人的权利和权力产生不利影响,包括投票权,并可能具有延迟、威慑或阻止我们控制权变更的效果。
| 118 |
独家 论坛
我们修订和重述的公司注册证书将规定,除非我们书面同意选择替代的 法院,(I)特拉华州衡平法院(或,如果该法院没有管辖权,则为特拉华州地区的联邦地区法院)应在法律允许的最大范围内,作为(A) 代表我们提起的任何派生诉讼或诉讼的唯一和独家论坛,(B)任何声称违反我们任何董事所欠受托责任的诉讼,(C)根据DGCL、我们的公司注册证书或我们的章程的任何规定而引起的任何诉讼,或(D)任何主张受内部事务 原则管辖的索赔的诉讼,以及(Ii)在法律允许的最大范围内,美利坚合众国联邦地区法院应成为解决根据证券法提出的一项或多项诉因的投诉的 独家论坛,包括 针对该等投诉的任何被告所主张的所有诉讼理由。上述条款不会阻止根据《交易所法案》提出索赔的股东向联邦法院提出此类索赔,前提是《交易所法案》授予联邦政府对此类索赔的独家管辖权,但须遵守适用法律。我们对法院条款的选择可能会使 股东在提出索赔时承担额外的诉讼成本,并可能限制股东在司法法院提出其认为有利于与我们或我们的任何董事、高级管理人员或其他员工发生纠纷的索赔的能力,这可能会阻碍与此类索赔有关的诉讼。
董事和高级职员的责任限制和赔偿
我们修订和重申的章程将规定,我们的董事和高级管理人员将在 特拉华州法律授权的最大程度上得到我们的赔偿。
这些条款可能会阻止股东以违反受托责任为由对我们的董事提起诉讼。这些条款 还可能降低针对董事和高级管理人员提起衍生品诉讼的可能性,即使此类诉讼如果成功, 可能会使我们和我们的股东受益。此外,如果我们根据这些赔偿条款向董事和高级管理人员支付和解和损害赔偿金的费用,股东的投资可能会受到不利影响 我们认为这些条款和保险对于吸引和留住有才华和经验的董事和高级管理人员是必要的。此外,就本招股说明书所包含的注册声明的有效性而言,我们打算与我们的每一位董事和高管签订单独的赔偿协议。
DGCL第(Br)203节
作为特拉华州的一家公司,我们将遵守DGCL第203节的规定。该法规禁止特拉华州的某些公司在某些情况下与“有利害关系的股东”进行“商业合并”。一般而言,第203条将“有利害关系的股东”定义为一个实体或个人,该实体或个人连同个人的关联公司和关联公司一起,实益拥有公司15%或更多的已发行有表决权股票。
“业务合并”包括合并或出售我们10%以上的资产。但是,在下列情况下,DGCL第 203节的上述规定不适用:
| ● | 业务合并发生在感兴趣的股东成为“感兴趣的股东”三年多之后; | |
| ● | 我们的 董事会批准了在交易日期 之前使股东成为“利益股东”的交易; | |
| ● | 在导致股东成为利益股东的交易完成后,该股东至少拥有我们已发行有表决权股票的85%,但法定排除在外的普通股除外;或 | |
| ● | 在交易发生之日或之后,企业合并由我们的董事会批准,并在我们的股东会议上授权,而不是通过书面同意,至少三分之二的已发行有表决权的 股票不为感兴趣的股东所拥有。 |
上市
我们 已申请将我们的普通股在纳斯达克全球市场上市,代码为“FBLG”。
转接 代理和注册表
我们普通股的转让代理和登记机构是VStock Transfer LLC。转让代理和登记员的地址是18Lafayette Place, 伍德米尔,NY 11598。转会代理和登记员可以通过电话联系:(212)828-8436。
| 119 |
有资格在未来出售的股票
在我们的普通股上市之前在 纳斯达克上,我们的普通股没有公开市场。在我们在纳斯达克上市后,在公开市场上出售大量我们的普通股,或者 认为可能发生这种出售,可能会对我们普通股的公开价格产生不利影响, 可能会使您更难在您认为合适的时间和价格出售您的股票。如果以及何时任何登记股东可以或不可以选择出售他们的股票或任何此类出售的价格,我们将不会有任何投入。
直接上市后,我们的普通股将共有32,492,068股 流通股,包括4,806,226股我们的普通股,根据注册说明书登记转售 本招股说明书是其中的一部分。任何未在本协议下登记的股票都将被称为“受限证券”,这一术语在证券法第144条中有定义。这些受限制证券只有在根据《证券法》登记(包括但不限于根据《证券法》登记的股份),或有资格获得豁免登记,包括根据《证券法》第144或701条登记的情况下,才有资格公开出售,概述如下。根据S规则第904条,受限证券也可以在美国境外出售给非美国人,但我们的董事、高级管理人员和某些股东拥有的股票 除外,这几乎是我们所有的普通股可能在本公司首次在纳斯达克上市后由登记股东根据本招股说明书 或由我们的其他现有股东根据证券法第144条出售。
规则 144
总体而言,根据目前有效的规则144,一旦我们遵守并遵守上市公司报告要求 第13条或交易所法案第15(D)条至少90天,符合条件的股东有权在不遵守规则144的销售方式、成交量限制或通知条款的情况下出售此类股票,但须遵守规则144的公开信息 要求。要成为第144条规定的合格股东,该股东不得被视为在出售前90天内的任何时间就证券法而言是我们的关联公司之一,并且实益拥有普通股 股票建议出售至少六个月,包括我们附属公司以外的任何先前所有者的持有期 。如果该人实益拥有拟出售的普通股股份至少一年,包括我们关联公司以外的任何先前所有人的持有期,则该人有权在不遵守规则144的任何要求的情况下出售该等股份。
一般而言,根据目前有效的第144条,我们的关联公司或销售普通股的人员代表我们的附属公司有权在我们成为报告公司后90天内出售股票 。在任何三个月内,该等股东可出售数量不超过下列较大者的股份:
| ● | 当时已发行普通股数量的1%,这将相当于紧随我们注册后的大约1股; 或 | |
| ● | 在提交表格144中有关出售的通知之前的四个日历周内,我们普通股的平均每周交易量。 |
我们的关联公司或出售普通股的人根据规则144进行的销售 代表我们的关联公司也受某些销售条款和通知 要求以及关于我们的最新公开信息的可用性的约束。
规则 701
规则 701一般允许根据书面补偿计划或合同发行股票且在紧接之前90天内未被视为我们的关联公司的股东依据规则144出售这些股票,但不需要 遵守规则144的公开信息、持有期、成交量限制或通知条款。规则701还允许我们的 关联公司根据规则144出售其规则701股票,而无需遵守规则144的持有期要求。然而,根据该规则,所有规则701股票的持有者必须等到我们成为报告公司后90天 才能根据规则701出售这些股票。
注册 S-8表格声明
我们打算根据证券法提交一份或多份S-8表格的登记声明,以便在证券法允许的情况下尽快登记受已发行股票 期权约束的普通股或根据我们的2022年股票计划保留供发行的普通股。此类注册声明 将自向美国证券交易委员会备案后自动生效。然而,以S-8表格登记的股票可能受规则第144条的数量限制 以及销售方式、通知和公开信息要求的限制。
| 120 |
销售我们股本的价格历史记录
我们 已申请将我们的普通股在纳斯达克上市。在我们的普通股在纳斯达克上市之前,我们的普通股还没有公开上市 。我们的普通股在私人交易方面的历史有限。2022年12月,我们以私募方式向投资者发行了总计相当于381,658股B系列优先股的 ,价格相当于每股6.76美元,相当于318,049股,其余相当于63,609股为红股。红股 发行给在签署认购协议一周内付款的投资者。从2023年2月至2023年4月,我们在监管众筹中向投资者发行了总计相当于890,310股B系列优先股,价格相当于每股6.76美元,相当于724,937股,其余相当于143,225股 和相当于22,148股分别为红股和佣金支付股。红股是根据向满足忠诚度、早起时机、基于金额和/或StartEngine所有者红利要求之一或组合的投资者发行的 。FibroBiologics的现有股东投资于法规众筹发行,获得了忠诚度10%的红股。在法规众筹发行的前两周内投资至少1,000美元的投资者将获得基于时机的5%红股。在法规众筹发行的前72小时内投资至少25,000美元的投资者 将获得基于时机的10%红股。在监管众筹中投资至少25,000美元的投资者将获得按金额计算的5%红股。在法规众筹产品中投资至少10万美元的投资者将获得基于金额的10%的红股。 符合StartEngine众筹公司所有者红利的投资者将获得10%的红股。法规 众筹发行的投资者获得他们有资格获得的最高金额或基于时机的奖金,以及他们有资格获得的忠诚奖金和 所有者奖金。2023年3月和4月,我们以私募方式向投资者发行了相当于1,680,084股B系列优先股 ,价格相当于每股6.76美元,相当于1,527,349股,其余相当于152,735股为红股。红股是向签署认购协议后 周内付款的投资者发行的。自2023年4月至2023年9月,我们以私募方式向投资者发行了相当于B-1系列优先股的74,922股,价格从相当于每股18.00美元到相当于64,070股的20.00美元不等,其余相当于10,852股为红股。红股 发行给在签署认购协议一周内付款的投资者。对于此类B-1系列优先股的非公开配售的一部分,我们还同意发行认股权证,从我们的 直接上市起可行使三年,以相当于每股20.00美元的行使价 购买总计相当于8,890股我们的普通股。2023年11月,本公司向认购B-1系列优先股的投资者增发了14,859股B-1优先股和1,431股额外认股权证,认购普通股的每股价格超过直接上市预期的每股参考价。虽然预计顾问将在制定我们普通股的开盘公开价格时考虑此信息,但此信息可能与市场对我们普通股的更广泛需求以及我们在纳斯达克上的普通股的开盘公开价格和后续公开价格几乎没有 或没有任何关系。因此,您不应过度依赖这些历史私下销售价格,因为它可能与我们在纳斯达克上的普通股的开盘价和随后的公开价存在实质性差异。
材料:美国联邦所得税对非美国持有者的影响
以下是关于美国联邦所得税的重要考虑事项和某些美国联邦遗产税考虑事项的一般性讨论 适用于购买我们普通股的非美国持有者 在本次发行中购买我们的普通股并将其作为“资本资产”持有的非美国持有者 修订后的《1986年美国国内收入法》第1221节或该守则(通常是为投资而持有的财产)的含义。在本讨论中, “非美国持有人”是指我们普通股的实益所有人(不包括在美国联邦所得税中被视为合伙企业的实体或安排),而在美国联邦所得税中,该实体或安排不属于下列任何一项:
| ● | 是美国公民或居民的个人; | |
| ● | 在美国、其任何一个州或哥伦比亚特区内或根据美国法律设立或组织的公司(或被视为美国联邦所得税目的公司的实体)。 | |
| ● | 遗产,指 的收入,不论其来源如何,均可计入美国联邦所得税总收入;或 | |
| ● | 如果(I)美国境内的法院能够对信托的管理进行主要监督,并且 一个或多个《守则》所定义的美国人或美国人有权控制信托的所有 重大决定,或(Ii)出于美国联邦收入 纳税的目的,该信托已作出有效选择,被视为美国人,则为信托。 |
如果合伙企业(或在美国联邦所得税方面被视为合伙企业的其他实体或安排)持有我们的普通股,则该合伙企业中合伙人的纳税待遇通常取决于合伙人的身份和合伙企业的活动。持有我们普通股的合伙企业的合伙人 应咨询其税务顾问,了解适用于他们的特定美国联邦所得税后果 。
本讨论基于《准则》的当前条款、根据其颁布的最终、临时和拟议的财政部条例、 或美国国税局(IRS)的《财政部条例》、司法裁决、已公布的裁决和行政声明,所有这些都在本招股说明书的日期生效,所有这些都可能会发生变化或有不同的解释, 可能具有追溯力。任何变化都可能改变本文所述的非美国持有者的税收后果。无法保证 国税局不会对本文所述的一个或多个税收后果提出质疑。
| 121 |
本讨论不涉及可能与特定非美国持有人的个人情况相关的美国联邦所得税和遗产税的所有方面 ,也不涉及美国州税、地方税或非美国税、其他美国联邦税、替代最低税或对净投资收入征收的非劳动所得联邦医疗保险缴费税的任何方面。此 讨论也不考虑可能适用于非美国持有人的任何特定事实或情况,也不涉及适用于特定非美国持有人的特殊 税收规则,例如:
| ● | 银行、保险公司和其他金融机构; | |
| ● | 证券经纪商或交易商或交易商 ; | |
| ● | 免税组织; | |
| ● | 养老金计划; | |
| ● | 持有我们的普通股作为跨境、套期保值、转换交易、合成证券或其他综合投资的一部分,或选择将证券按市价计价的人; | |
| ● | 受控外国公司、被动外国投资公司和为逃避美国联邦所得税而积累收益的公司; | |
| ● | 非美国政府;以及 | |
| ● | 美国侨民和前美国公民或在美国的长期居民。 |
| 122 |
本摘要并非针对非美国持有者与我们普通股的所有权和处置有关的所有税收后果的完整描述。我们普通股的潜在持有者应就收购、拥有和处置我们普通股对他们的税收后果(包括任何州、当地、非美国所得税和其他税法的适用和影响) 咨询他们的税务顾问。
分配
正如 在“股利政策“如上所述,我们预计在可预见的未来不会对我们的普通股进行分配。然而,如果我们在普通股上进行现金或财产分配,这种分配将构成美国联邦所得税目的的股息,根据美国联邦 所得税原则,从我们当前或累计的收入和利润中支付。就美国联邦所得税而言,未被视为股息的分派金额将首先构成非美国持有者投资的免税资本回报,并适用于非美国持有者在其普通股中调整后的 税基,但不低于零。任何剩余的超额部分将被视为资本利得,并将按照以下条款 处理:普通股出售收益或其他处置收益“由于我们可能不知道在作出分配时 在多大程度上是美国联邦所得税的股息,为了下面讨论的扣缴规则的目的,我们或适用的扣缴义务人可能会将整个分配视为股息。任何此类分发也将受制于下面标题下的 讨论“FATCA“和”备份扣留、信息报告和其他报告要求 .”
根据接下来的两段讨论,支付给非美国持有者的股息通常将被扣缴美国联邦 所得税,税率为30%,或美国与该持有者居住国家之间适用的所得税条约规定的较低税率。
我们向非美国持有者支付的股息,如果与该非美国持有者在美国境内开展贸易或业务有效相关(如果适用的税收条约要求,可归因于由该非美国持有者维持的美国常设机构或固定基地),通常将免除上述美国联邦预扣税,如果非美国持有者符合适用的认证和披露要求(通常包括提供有效的IRS表W-8ECI(或适用的继承者表),证明股息与非美国持有者在美国境内进行的贸易或业务有效相关)。相反,此类股息一般将在净收入的基础上缴纳美国联邦所得税,税率与持股人为美国个人(如守则所定义)时适用的税率相同。非美国持有者收到的任何美国有效关联收入 在美国联邦所得税中被归类为公司,也可以按30%的税率(或适用所得税条约规定的较低税率)缴纳额外的 “分支机构利得税”。
| 123 |
我们普通股的非美国持有者如果声称受益于美国和其居住国之间适用的所得税条约,通常将被要求提供一份正确签署的IRS表格W-8BEN或W-8BEN-E(或后续表格),并满足适用的认证和其他要求。敦促非美国持有者咨询他们的税务顾问,了解他们根据相关所得税条约享有的福利 以及他们可以使用的满足这些要求的具体方法。
出售或以其他方式处置普通股的收益
主题 以下标题下的讨论“FATCA“和”备份扣留、信息报告和其他 报告要求,非美国持有者出售或以其他方式处置我们普通股的任何收益,一般不需缴纳美国联邦所得税,除非:
| ● | 收益实际上与非美国持有者在美国境内进行的贸易或业务有关(如果适用的所得税条约要求,应归因于该非美国持有者在美国设立的常设机构或固定基地); | |
| ● | 非美国持有人是个人,在纳税年度内在美国停留183天或以上,并满足其他某些条件 ;或 | |
| ● | 就美国联邦所得税而言,我们是或曾经是“美国不动产控股公司”,在该处置之前的五年期间或该非美国持有人持有我们普通股的较短期间内的任何时间,如果我们的普通股 定期在适用的财政部法规所指的成熟证券市场交易,则非美国持有人 在上述期间内的任何时间直接、间接或建设性地持有:超过我们普通股的5%。 |
如果收益 与在美国的贸易或业务活动有效相关,一般将在净所得税的基础上缴纳美国联邦 所得税,适用于美国个人的常规美国联邦所得税税率。如果非美国持有者是 非美国公司,上述分支机构利得税也可能适用于此类有效关联收益。因非美国持有人在销售或以其他方式处置我们的普通股的年度内在美国停留183天或以上而缴纳美国联邦所得税的个人 将对此类出售或其他处置所获得的收益缴纳30%的统一税(或适用的所得税条约可能指定的较低税率),这可能会被某些美国来源 资本损失抵消。我们认为,我们不是,也不打算成为美国联邦所得税目的的美国房地产控股公司。非美国持有者应就可能适用的可能规定不同规则的所得税条约 咨询他们的税务顾问。
FATCA
根据《外国账户税收合规法》(FATCA),可以对向非美国金融机构和某些其他非美国实体支付的某些类型的款项征收预扣税 。具体而言,可对支付给“外国金融机构”或“非金融外国实体”的普通股股息(包括视为股息) 征收30%的预扣税,除非(I)该外国金融机构承担一定的尽职调查和报告义务,(Ii) 非金融外国实体证明其没有任何“主要美国所有者”(定义见 准则)或提供有关每个主要美国所有者的身份信息,或者(三)境外金融机构或者非金融境外实体在其他方面有资格豁免本规则。位于司法管辖区的外国金融机构如果与美国管辖的FATCA有政府间协议,则可能受该政府间协议的报告规则的约束。由于我们在作出分配时可能不知道分配在多大程度上是美国联邦所得税的股息 ,因此,为了这些扣缴规则的目的,我们或适用的扣缴义务人可能会将整个分配视为股息。 尽管FATCA下的扣缴也适用于在2019年1月1日或之后出售或以其他方式处置股票的毛收入的支付,但拟议的财政部法规将完全取消FATCA对支付毛利的扣缴。纳税人通常可以依赖这些拟议的财政部条例,直到最终的财政部条例发布。在某些情况下,非美国持有人将有资格通过及时提交美国联邦收入 纳税申报单,获得根据FATCA征收的预扣税款的退款或抵免。潜在投资者应咨询他们的税务顾问,了解这些预提条款的潜在适用情况。
| 124 |
备份 扣缴、信息报告和其他报告要求
我们 必须每年向美国国税局和每个非美国持有人报告支付给每个非美国持有人的任何分配金额和扣缴税款 。无论适用的所得税条约是否减少或取消了预提,这些报告要求都适用。根据特定所得税条约的规定或与非美国持有人居住或设立的国家税务机关达成的协议,也可以提供此信息报告的副本。
非美国持有人一般会因支付给该持有人的普通股股息而被备用扣留,除非该持有人 在伪证处罚下证明其为非美国持有人(前提是付款人并不实际知道或没有理由知道该持有人是美国人)或以其他方式确立豁免。
信息 报告和备份扣缴一般适用于非美国持有人处置我们普通股的收益,该收益由或通过任何经纪人(美国或非美国)的美国办事处实现,除非持有人证明其非美国持有人身份并满足 某些其他要求,或以其他方式确立豁免。通常,如果交易是通过经纪商在美国以外的办事处完成的,则信息报告和备份扣缴不适用于向非美国持有人支付处置收益。但是,出于信息报告的目的,通过拥有大量美国所有权或业务的经纪人的非美国办事处进行的处置通常将以类似于通过经纪人的美国办事处进行的处置的方式处理。 非美国持有人应就信息报告和备份扣缴规则对其适用的问题咨询其税务顾问 。
备份 预扣不是额外的所得税。根据备份预扣规则从向非美国持有人的付款中扣留的任何金额 通常可以从非美国持有人的美国联邦所得税义务(如果有)中扣除或退还,前提是及时向美国国税局提供所需的信息。非美国持有者应就信息申报和备份扣缴规则的应用 咨询其税务顾问。
美国联邦遗产税
我们的普通股股票 在去世时被视为非美国公民或居民的个人所有(根据为美国联邦遗产税目的而特别定义的),被视为美国所在地资产,并将计入个人的 美国联邦遗产税目的总遗产。因此,此类股份可能需要缴纳美国联邦遗产税,除非适用的遗产税或其他条约另有规定。
前面讨论的重要美国联邦所得税考虑事项和某些美国联邦遗产税考虑事项仅供参考 。这不是法律或税务建议。潜在投资者应就收购、拥有和处置我们的普通股的特定美国联邦、州、地方和非美国税收后果咨询他们的税务顾问,包括任何拟议的适用法律变更的后果。
| 125 |
分销计划
登记股东及其质权人、受让人或其他利益继承人可以在普通股上市交易后,根据经纪交易在纳斯达克、其他公开交易所或登记的另类交易场所,随时按现行市场价格出售所持 普通股。我们不参与与任何登记股东或任何经纪交易商就登记股东出售普通股作出的任何安排,除非我们已就与登记普通股登记及上市有关的某些其他事宜聘请财务顾问,详情如下。因此,我们预计不会收到关于任何登记股东是否以及何时可以或不可以选择出售其普通股或任何此类出售的价格的通知,并且不能保证任何登记股东将出售本招股说明书所涵盖的任何或全部普通股。
我们 不会收到登记股东出售普通股的任何收益。我们将确认与此次直接上市和我们向上市公司转型相关的成本,包括专业费用和其他费用。我们将在发生的期间支出 这些金额,不会像在首次公开募股时那样从发行人的净收益中扣除这些成本 。
在我们的普通股首次在纳斯达克上市的那一天,纳斯达克将开始接受但不执行开盘前的买入和卖出 订单,并将开始在此类接受的订单的基础上连续生成当前参考价格。 当前参考价格每秒计算一次,在10分钟的“仅展示”期间,纳斯达克将通过其noii和图书查看器工具向市场参与者发布 和其他指示性失衡信息。在“仅供展示 ”之后,将开始一个“发行前”阶段,在此期间,顾问必须以我们的财务顾问的身份, 通知纳斯达克我们的股票已经“可以交易”。一旦顾问通知纳斯达克我们的普通股已经可以交易 ,纳斯达克将根据纳斯达克规则确认我们普通股的当前参考价格。 如果顾问随后批准以当前参考价格进行操作,则已经输入的适用订单将按该价格执行,我们的普通股将开始在纳斯达克上的常规交易,但纳斯达克将根据纳斯达克规则进行验证检查 。
根据纳斯达克规则,当前参考价意味着:(1)买入或卖出的最大指令数可以匹配的单一价格; (2)如果有多个价格可以匹配最大买入或卖出指令数,则是将买入或卖出指令之间的失衡降至最低的价格(即将在该价格下无法匹配的股票数量降至最低);(Iii)如果(Ii)项下存在多个价格,则为输入的价格(即客户在买入或卖出订单中输入的指定价格),在该价格下,我们的普通股将保持不匹配(即不会被买卖);及(Iv)如果(Iii)项下存在多个价格 ,则为纳斯达克以吾等财务顾问的身份与顾问磋商后确定的价格。如果 第(Iii)项下存在多个价格,顾问将仅在符合联邦证券法(包括法规M)的反操纵条款或根据其授予的适用救济的范围内行使任何咨询权。
在确定当前参考价格时,纳斯达克的交叉算法将匹配已输入并被纳斯达克系统接受的订单。当以大于或等于潜在当前参考价的输入买入价买入普通股的订单与以小于或等于该潜在当前参考价的输入要价出售相同数量的 普通股的订单相匹配时,潜在当前参考价会发生这种情况。为了举例说明, 作为当前参考价计算的假设示例,如果纳斯达克的交叉算法如上所述匹配所有接受的订单,并且仍然存在两个限制单--以每股10.01美元的输入出价买入500股普通股的限价指令和以每股10.00美元的输入要价出售200股普通股的限价指令--当前的 参考价将被选择如下:
| ● | 根据第(I)条,如果当前参考价为10.00美元,则可匹配的额外股票的最大数量为200股。如果 当前参考价为10.01美元,则可以匹配的最大额外股票数量也是200股,这意味着 将以10.00美元或10.01美元的价格匹配相同的最大额外股票数量。 |
| 126 |
| ● | 由于第(I)款下存在多个价格,因此第(Ii)款中,当前参考价将是将买卖订单之间的不平衡降至最低的价格(即,将在该价格下保持不匹配的股票数量降至最低)。 选择10.00美元或10.01美元作为当前参考价将在限价订单中产生相同的不平衡,无法 匹配,因为在这两个价格中,都不会匹配300股。 | |
| ● | 由于第(Ii)款下存在多个价格,因此根据第(Iii)款,当前参考价将是输入价格,在该输入价格下,普通股股票的订单将保持不匹配。在这种情况下,选择10.01美元将导致输入价格为10.01美元的500股限价指令中的300股 保持不匹配,而选择10.00美元将匹配所有 200股输入价格为10.00美元的限价指令,并且没有输入价格为10.01美元的股票保持不匹配。 因此,纳斯达克将选择10.01美元作为当前参考价,因为以此输入价格的股票订单将保持不匹配。 以上示例(包括价格)仅供说明之用。 |
顾问将主要根据成交量、时机和价格的考虑,确定我们的普通股何时准备好进行交易,并批准以当前参考价进行交易。特别是,顾问将主要根据开盘前买入和 卖出订单,确定开盘交易何时会有合理数量的成交量,从而充分发现以当前参考价进行的开盘交易 。如果顾问不批准按当前参考价继续进行交易(例如,由于缺乏足够的开盘前买卖兴趣),顾问将请求纳斯达克将开盘时间推迟至 充分的价格发现以确保开盘交易中有合理数量的交易量交叉。此外, 如果纳斯达克按照当前参考价定义第(Iv)款的规定与顾问磋商的可能性极小,顾问将要求纳斯达克推迟开盘,以确保在当前参考价定义第(I)、(Ii)或(Iii)款内实现单一开盘价。根据纳斯达克的规定,如果出现此类延迟,在终止此类延迟之前, 将有一个10分钟的“仅供展示”时间段,在此期间市场参与者可以在纳斯达克系统中输入我们普通股的报价和订单。另外,从凌晨4点开始,市场参与者可以在纳斯达克上输入我们普通股的订单。 这样的订单将被接受并进入系统。在10分钟的“仅供展示”期间结束后,我们的 普通股将进入不确定持续时间的“发行前”期间。预售期将结束,当满足某些条件时,纳斯达克将释放我们的普通股进行交易,包括纳斯达克收到顾问的 通知我们的普通股已经可以交易,之后纳斯达克系统将计算当时的当前参考价格并将其显示给顾问。如果顾问随后批准继续进行,纳斯达克系统将执行某些 验证检查。顾问在征得纳斯达克的同意后,可在“上市前”期限结束前的任何时间决定推迟及重新安排直接上市。登记股东不会参与纳斯达克的定价机制,也不会与顾问进行协调或沟通,包括与顾问就延迟或继续交易的任何决定进行协调。
类似于在纳斯达克上市的公司承诺承销的首次公开募股(IPO),在我们的普通股上市方面, 已认购的买家和卖家在提交买入或卖出订单之前,将可以访问纳斯达克的订单失衡指标或净订单失衡指标, 一个广泛可用的基于认购的数据馈送。纳斯达克的电子交易平台 每秒模拟拍卖,计算当前参考价,计算 当前参考价可以配对的普通股数量,按当前参考价剩余未执行的普通股数量,以及 是否存在买方或卖方失衡,或者是否存在失衡,通过净订单失衡指标数据馈送将这些信息持续发布给买家和 卖家。
但是, 由于这不是在确定承销的基础上进行的首次公开募股,因此不会有传统的 询价流程(即,一个有组织的流程,根据该流程,买卖兴趣提前协调到某一规定的 水平--“预订”)。此外,在开盘交易之前,承销商最初向公众出售普通股的价格将不会像公司承诺承销的首次公开募股(IPO)那样。缺乏首次公开募股价格可能会影响纳斯达克从多家经纪自营商那里获得的买入和卖出订单范围。因此,我们普通股的公开价格可能比在坚定承诺基础上承销的首次公开募股(IPO)的波动性更大 ,在纳斯达克上市后可能会大幅快速下跌。
此外,要在纳斯达克上市,我们还需要至少有四名注册的活跃做市商。我们预计, 顾问将作为注册的、活跃的做市商,并将与其他做市商接洽。
除根据本招股说明书进行的销售外,本招股说明书涵盖的普通股还可由登记的股东以非公开交易方式出售,不受证券法的登记要求。根据一些州的证券法,普通股只能通过注册或持牌的经纪人或交易商在这些州出售。
| 127 |
登记股东可以不时转让、分发(包括登记股东作为投资基金的实物分配)、质押、转让或授予其拥有的部分或全部普通股的担保权益,如果登记股东在履行其担保债务时违约,受让人、分配人、质权人、受让人或担保当事人可以根据本招股说明书,或根据证券法适用条款对招股说明书的修订,将登记股东名单修改为包括受让人、分配人、质权人、受让人或作为本招股说明书登记股东的其他利益继承人。登记股东还可以在其他情况下转让股份 ,在这种情况下,受让人、分派人、质权人或其他利益继承人将是本招股说明书中的登记实益拥有人。
作为实体的登记股东可以选择将普通股实物分配给其成员、合伙人或股东 根据本招股说明书组成的登记说明书,提交招股说明书。
如果任何登记股东利用经纪交易商出售本招股说明书所提供的普通股股份,则该经纪交易商可从该登记股东那里获得折扣、优惠或佣金形式的佣金,或从普通股购买者那里收取佣金 他们可以作为代理或作为委托人向其出售普通股。
我们 已聘请顾问Maxim Group LLC作为我们的财务顾问,就与直接上市有关的某些事宜向我们提供建议和协助。顾问预计将执行的服务将包括提供有关定义目标、分析、构建和规划直接上市的建议和协助,以及制定和协助我们的投资者与直接上市有关的沟通策略。关于聘请顾问作为我们的财务顾问,顾问将有权在成功完成直接上市后获得200,000美元的费用。顾问还将有权 获得与其聘用相关的所有合理、有记录的费用报销,但条件是:(I)未经我们事先授权,此类费用(律师费除外)不得超过5,000美元;以及(Ii)未经我们事先授权,构成法律费用的此类费用不得超过10,000美元。
此外,根据吾等与顾问的协议,自完成直接上市之日起九个月内,如吾等建议(I)在美国主要交易所公开发售吾等证券,(Ii)私募吾等证券,(Iii)与顾问介绍给吾等的第三方进行某些融资交易,或(Iv)建议与顾问介绍给吾等的第三方进行其他交易,包括但不限于合并、收购或出售股票或资产,或其他类似交易,我们有义务根据顾问与我们共同同意的合理和惯例条款,在与该融资或交易有关的情况下, 有义务提出保留顾问作为我们的主承销商和账簿管理经理、我们的潜在配售或销售代理,或我们的 首席顾问(主要但非排他性顾问,至少有70%的经济价值,最多向其他人提供30%的经济价值)。
顾问不会与我们协商,以其他方式协助或协调价格发现活动或招揽和/或出售我们普通股的股票,并且不会被允许,也不会受我们的指示,计划或积极参与任何投资者教育活动, ,除非本文所述。
在顾问向我们提供与证券上市相关的财务咨询服务之前,顾问及其任何关联公司均未向我们提供任何类型的服务。
| 128 |
法律事务
特此提供的普通股的有效性将由德克萨斯州休斯敦的诺顿·罗斯·富布赖特美国有限责任公司为我们传递。
专家
FibroBiologics,Inc.截至2022年和2021年12月31日及截至2021年12月31日的经审计财务报表 已由独立注册会计师事务所Withum Smith+Brown,PC审计,如本报告中所述 。此类经审计的财务报表是根据该公司作为会计和审计专家提供的报告 列入的。
此处 您可以找到其他信息
我们 已根据证券法以S-1表格向美国证券交易委员会提交了关于本招股说明书 提供的普通股股份的登记说明书。本招股说明书是注册说明书的一部分,并不包含注册说明书或随附的证物和附表所载的所有信息。本招股说明书中包含的关于作为登记说明书附件的任何合同或任何其他文件的内容的陈述不一定完整, 并且每个此类陈述通过参考作为登记说明书附件存档的该合同或其他文件的全文在所有方面都是合格的。
本招股说明书所包含的注册声明生效后,我们将立即遵守交易所法案的信息和报告要求,并将根据此类法律向美国证券交易委员会提交年度、季度和当前报告、代理声明和其他信息。美国证券交易委员会维护一个网站,其中包含以电子方式向美国证券交易委员会提交的报告、委托书和信息声明以及 其他有关发行人的信息。您可以获得我们向美国证券交易委员会提交的文件,网址为www.sec.gov。 我们的网站地址是www.fifibiologics.com。我们不会将我们网站上的信息或通过我们网站访问的信息合并到本招股说明书中,您也不应将我们网站上的任何信息或可以通过我们网站访问的任何信息作为本招股说明书的一部分。我们的网站地址 仅作为非活动文本参考包含在本招股说明书中。
| 129 |
财务报表索引
| 截至2022年和2021年12月31日的年度经审计财务报表 | |
| 独立注册会计师事务所报告 | F-2 |
| 截至2022年12月31日和2021年12月31日的资产负债表 | F-3 |
| 截至2022年和2021年12月31日止年度的业务报表 | F-4 |
| 截至2022年12月31日和2021年12月31日止年度股东赤字变动表 | F-5 |
| 截至2022年12月31日和2021年12月31日的年度现金流量表 | F-6 |
| 财务报表附注 | F-7 |
| 截至2023年9月30日和2022年9月30日的9个月未经审计的 简明财务报表 | |
| 截至2023年9月30日(未经审计)和2022年12月31日的简明资产负债表 | F-17 |
| 截至2023年9月30日和2022年9月30日的9个月未经审计的 简明营业报表 | F-18 |
| 未经审计的 截至2023年和2022年9月30日的9个月股东权益/(亏损)简明变动表 | F-19 |
| 截至2023年9月30日和2022年9月30日的9个月未经审计的现金流量表简明报表 | F-20 |
| 截至2023年9月30日和2022年9月30日的9个月未经审计简明财务报表附注{br | F-21 |
| F-1 |
独立注册会计师事务所报告{br
致 公司股东和董事会
FibroBiologics, 公司:
对财务报表的意见
我们审计了FibroBiologics,Inc. (“本公司”)截至2022年12月31日和2021年12月31日的资产负债表,以及截至2022年12月31日期间各年度的相关经营报表、股东赤字变化和现金流量,以及相关附注(统称为“财务报表”)。我们认为,该等财务报表在各重大方面均公平地反映了本公司于2022年、2022年及2021年12月31日的财务状况,以及截至2022年12月31日期间各年度的经营业绩及现金流量,符合美国公认的会计原则。
征求意见的依据
这些财务报表由公司管理层负责。我们的责任是根据我们的审计对这些财务报表发表意见 。我们是一家在美国上市公司会计监督委员会(PCAOB)注册的公共会计师事务所,根据美国联邦证券法以及美国证券交易委员会和PCAOB的适用规则和法规,我们必须与公司保持独立。
我们 按照PCAOB的标准和美国公认的审计标准进行审计。这些标准要求我们计划和执行审计,以获得关于财务报表是否没有重大错报的合理保证,无论是由于错误还是舞弊。本公司不需要对其财务报告的内部控制进行审计, 也没有聘请我们进行审计。作为我们审计的一部分,我们需要 了解财务报告的内部控制,但不是为了对公司财务报告内部控制的有效性发表意见 。因此,我们不表达这样的意见。
我们的 审计包括执行程序,以评估财务报表重大错报的风险,无论是由于错误还是欺诈,以及执行应对这些风险的程序。这些程序包括在测试的基础上审查与财务报表中的数额和披露有关的证据。我们的审计还包括评估使用的会计原则和管理层做出的重大估计,以及评估财务报表的整体列报。 我们认为我们的审计为我们的意见提供了合理的基础。
/S/ 史密斯+布朗电脑
我们 自2022年以来一直担任本公司的审计师。
新泽西州布伦瑞克东部
2023年4月28日,除附注13中描述的反向股票拆分的影响外,日期为2023年11月7日
| F-2 |
FibroBiologics, 公司
资产负债表
(单位为 千,不包括股份和每股数据)
| 十二月三十一日, | ||||||||
| 2022 | 2021 | |||||||
| 资产 | ||||||||
| 流动资产 | ||||||||
| 现金和现金等价物 | $ | 2,266 | $ | 407 | ||||
| 预付费用 | 29 | 37 | ||||||
| 母公司应收账款 | 300 | — | ||||||
| 其他流动资产 | 30 | 24 | ||||||
| 流动资产总额 | 2,625 | 468 | ||||||
| 经营性租赁使用权资产净额 | 2,199 | — | ||||||
| 总资产 | $ | 4,824 | $ | 468 | ||||
| 负债和股东赤字 | ||||||||
| 流动负债 | ||||||||
| 应付账款和应计费用 | $ | 758 | $ | 233 | ||||
| 母公司应付 | — | 225 | ||||||
| 短期经营租赁负债 | 326 | — | ||||||
| 衍生负债 | 538 | — | ||||||
| 可转换票据,扣除债务贴现后的净额 | 5,451 | 1,300 | ||||||
| 流动负债总额 | 7,073 | 1,758 | ||||||
| 长期经营租赁负债 | 1,747 | — | ||||||
| 总负债 | 8,820 | 1,758 | ||||||
| 股东亏损额 | ||||||||
| 母公司净投资 | 1,461 | 1,461 | ||||||
| 优先股,面值0.00001美元;授权总股份12,500,000股;截至2022年和2021年12月31日,授权、发行和发行的8,750,000系列“A”优先股 | — | — | ||||||
| 优先股,面值0.00001美元;授权总股本12,500,000股;授权发行2,500,000股“B”系列优先股;截至2022年12月31日,已发行381,658股,流通股 ;截至2021年12月31日,未发行和流通股 | — | — | ||||||
| 无投票权普通股,面值0.00001美元;授权发行62,500,000股;截至2022年12月31日,已发行和已发行28,230,842股;截至2021年12月31日,未发行股票和已发行股票 | 1 | — | ||||||
| 额外实收资本 | 2,414 | — | ||||||
| 累计赤字 | (7,872 | ) | (2,751 | ) | ||||
| 股东总亏损额 | (3,996 | ) | (1,290 | ) | ||||
| 总负债和股东赤字 | $ | 4,824 | $ | 468 | ||||
附注是这些财务报表的组成部分。
| F-3 |
FibroBiologics, 公司
营运说明书
(单位为 千,不包括股份和每股数据)
| 截至12月31日止年度, | ||||||||
| 2022 | 2021 | |||||||
| 运营费用: | ||||||||
| 研发 | $ | 1,147 | $ | 521 | ||||
| 一般、行政和其他 | 3,320 | 1,057 | ||||||
| 总运营费用 | 4,467 | 1,578 | ||||||
| 运营亏损 | (4,467 | ) | (1,578 | ) | ||||
| 利息支出 | (654 | ) | (4 | ) | ||||
| 净亏损 | $ | (5,121 | ) | $ | (1,582 | ) | ||
| 每股基本和稀释后净亏损 | $ | (.18 | ) | $ | 不适用 | |||
| 加权平均流通股、基本股和稀释股 | 28,230,842 | 不适用 | ||||||
附注是这些财务报表的组成部分。
| F-4 |
FibroBiologics, 公司
股东亏损变动表
截至2022年12月31日和2021年12月31日的年度
(单位:千,不包括股票)
| 净额 父级 | 系列 “A” 优先股 股票 |
系列 “B” 优先股 股票 |
无表决权 普通股 股票 |
额外的 个实收 | 累计 | 合计 股东 |
||||||||||||||||||||||||||||||||||
| 投资 | 股票 | 金额 | 股票 | 金额 | 股票 | 金额 | 资本 | 赤字 | 赤字 | |||||||||||||||||||||||||||||||
| 余额 -2020年12月31日 | $ | 1,169 | — | $ | — | — | $ | — | — | $ | — | $ | — | $ | (1,169 | ) | $ | — | ||||||||||||||||||||||
| 公司成立时发行股本 | — | 8,750,000 | — | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||||
| 资本 缴款 | 292 | — | — | — | — | — | — | — | — | 292 | ||||||||||||||||||||||||||||||
| 净亏损 | — | — | — | — | — | — | — | — | (1,582 | ) | (1,582 | ) | ||||||||||||||||||||||||||||
| 余额 -2021年12月31日 | 1,461 | 8,750,000 | — | — | — | — | — | — | (2,751 | ) | (1,290 | ) | ||||||||||||||||||||||||||||
| 向母公司成员发行 无投票权普通股 | — | — | — | — | — | 28,179,592 | 1 | (1 | ) | — | — | |||||||||||||||||||||||||||||
| 发行“B”系列优先股 | — | — | — | 381,658 | — | — | — | 2,150 | — | 2,150 | ||||||||||||||||||||||||||||||
| 基于股票的 薪酬费用 | — | — | — | — | — | 51,250 | — | 265 | — | 265 | ||||||||||||||||||||||||||||||
| 净亏损 | — | — | — | — | — | — | — | — | (5,121 | ) | (5,121 | ) | ||||||||||||||||||||||||||||
| 余额 -2022年12月31日 | $ | 1,461 | 8,750,000 | $ | — | 381,658 | $ | — | 28,230,842 | $ | 1 | $ | 2,414 | $ | (7,872 | ) | $ | (3,996 | ) | |||||||||||||||||||||
附注是这些财务报表的组成部分。
| F-5 |
FibroBiologics, 公司
现金流量表
(单位:千)
| 截至12月31日止年度, | ||||||||
| 2022 | 2021 | |||||||
| 经营活动的现金流 | ||||||||
| 净亏损 | $ | (5,121 | ) | $ | (1,582 | ) | ||
| 对净亏损与经营活动中使用的现金净额进行的调整: | ||||||||
| 基于股票的薪酬费用 | 265 | — | ||||||
| 可换股票据债务折价摊销 | 389 | — | ||||||
| 经营性租赁使用权资产摊销 | 94 | — | ||||||
| 经营性资产和负债变动情况: | ||||||||
| 预付费用的变动 | 8 | (37 | ) | |||||
| 应付账款和应计费用变动 | 525 | 233 | ||||||
| 其他流动资产变动 | (6 | ) | (24 | ) | ||||
| 经营租赁负债变动 | (220 | ) | — | |||||
| 用于经营活动的现金净额 | (4,066 | ) | (1,410 | ) | ||||
| 融资活动产生的现金流 | ||||||||
| 向母公司借款所得款项 | — | 975 | ||||||
| 向父母还款 | (225 | ) | (750 | ) | ||||
| 贷款给父母 | (360 | ) | — | |||||
| 母公司还款 | 60 | |||||||
| 母公司净投资收益 | — | 292 | ||||||
| 发行可转换票据所得款项 | 4,300 | 1,300 | ||||||
| 发行“B”系列优先股的收益 | 2,150 | — | ||||||
| 融资活动提供的现金净额 | 5,925 | 1,817 | ||||||
| 现金及现金等价物净增加情况 | 1,859 | 407 | ||||||
| 现金和现金等价物,年初 | 407 | — | ||||||
| 现金和现金等价物,年终 | $ | 2,266 | $ | 407 | ||||
| 补充披露现金流量信息: | ||||||||
| 缴纳所得税的现金 | $ | — | $ | — | ||||
| 支付利息的现金 | $ | — | $ | — | ||||
| 补充披露非现金投资和融资活动: | ||||||||
| 债务发行贴现的衍生负债附加 | $ | 538 | $ | — | ||||
| 取得经营性租赁使用权资产和负债 | $ | 2,293 | $ | — | ||||
附注是这些财务报表的组成部分。
| F-6 |
FibroBiologics, 公司
财务报表附注
2022年12月31日
| 1. | 组织机构、业务描述和流动资金 |
组织 和业务
FibroBiologics, Inc.(“公司”,“FibroBiologics”)最初是根据德克萨斯州法律于2021年4月8日(“初始”)成立的有限责任公司,然后于2021年12月14日转变为特拉华州公司。FibroBiologics是一家处于早期阶段的细胞治疗公司,总部设在得克萨斯州休斯敦,利用成纤维细胞开发慢性疾病的创新治疗方法。 该公司的主要重点是启动和进展与成纤维细胞治疗退行性腰椎间盘疾病、多发性硬化症、癌症、伤口愈合和其他疾病有关的临床前研究和FDA临床阶段试验。在此之前,与这些疾病途径相关的临床前研究和开发是在我们的母公司SpinalCyte,LLC(“母公司”, “FibroGenesis”)的领导下进行的。
持续关注和管理的计划
公司自创始以来已出现运营亏损,预计随着基础设施建设、知识产权开发和研发活动的开展,此类亏损将继续存在。该公司主要依靠天使投资者和私人债务配售的组合为其运营提供资金。截至2022年12月31日,公司累计亏损7,872,000美元,现金及现金等价物为2,266,000美元。向盈利的过渡将取决于候选产品的成功开发、审批和商业化,以及能否获得足够的收入来支持公司的 成本结构。该公司目前没有产生收入,可能永远不会实现盈利。除非并直至产生收入和净收入,否则公司将需要继续筹集额外资本。如附注7所述, 管理层已于2021年11月12日签订购股协议。如果在国家认可的美国证券交易所直接上市或首次公开募股,本协议将为公司提供获得额外流动资金的途径。 如附注12中进一步描述的,在2023年前三个月,公司通过众筹筹集了500万美元 ,通过私募募集了1000多万美元。因此,本公司相信,自该等财务报表发布之日起,至少在未来12个月内,本公司有足够的资本为其目前的营运计划提供资金。
细分市场
运营部门被确定为企业的组成部分,其独立的离散财务信息可供首席运营决策者在做出有关资源分配的决策和评估业绩时进行评估 。首席执行官是首席运营决策者,他在汇总的基础上审查财务信息,以分配资源和评估财务业绩。本公司将其业务作为一个单一的运营部门进行运营和管理,因此是一个可报告的部门。
| 2. | 摘要 主要会计政策 |
演示基础
在2021年4月8日成立之前的2021年的最初阶段,本公司作为FibroGenesis的一个业务线运营,而不是作为一个独立的实体运营。因此,历史上没有为FibroBiologics编制独立的财务报表。 这些财务报表是与FibroBiologics的成立和计划公开上市相关编制的 在公司于2021年4月8日成立之前,这些财务报表来自母公司的历史会计记录。与公司业务活动直接相关的所有费用、资产和负债以及母公司的某些分配都包括在财务报表中。此类分配包括公司应承担的一般和行政费用,以及母公司在2021年4月8日成立之前为FibroBiologics目前研究的疾病途径而产生的研发费用。
费用分配由管理层确定,并根据FibroBiologics和FibroGenesis之间的专利转让和转让协议转让给公司的专利数量得出。专利被确定为最合理的分配基础,因为专利开发是每个实体在临床前阶段业务活动的主要驱动力,并且它们是母公司代表公司产生的费用的最强代理。管理层认为财务报表所依据的假设,包括有关母公司费用分配的假设是合理的。然而,公司确认的金额不一定代表本公司在列报期间作为一个独立实体独立运营的情况下应反映在财务报表中的金额。
所附财务报表是根据美国公认的会计原则(“公认会计原则”)编制的。
| F-7 |
净额 母公司投资
由于财务报表来自母公司的历史记录,母公司的净投资在资产负债表上的股东赤字内列示。作为母公司的附属公司,在订立可换股票据协议前,本公司所有营运资金及融资需求均依赖母公司 。与FibroBiologics有关但发生在母公司级别的财务交易 通过净母公司投资账户入账。因此,母公司的现金、现金等价物或债务均未在财务报表中分配给本公司。母公司净投资 代表母公司在记录的公司净资产中的权益。
使用预估的
根据公认会计原则编制财务报表要求管理层作出估计和假设,使其影响财务报表日期的已报告资产和负债额、或有资产和负债的披露以及报告期内已报告的费用金额。这些估计是基于截至财务报表日期可获得的信息;因此,实际结果可能与这些估计和假设不同。
信用风险集中度
可能使公司面临集中信用风险的金融工具主要包括现金和现金等价物。 公司在金融机构有大量现金余额,全年经常超过联邦保险限额250,000美元。发生的任何损失或无法获得此类资金可能会对公司的财务状况、经营业绩和现金流产生重大不利影响。
风险 和不确定性
公司受某些风险和不确定性的影响,包括但不限于公司认为可能对未来财务状况或经营结果产生重大不利影响的以下任何领域的变化:将当前和未来的候选产品提前到临床开发并通过临床开发的时间和公司的能力;与生产公司候选产品的临床用品相关的成本和时间表;监管部门对其候选产品的批准和市场接受度;第三方合同研究组织(“CRO”)和合同制造组织(“CMO”)的业绩;来自财力或专业知识更强的制药公司的竞争;知识产权保护、基于知识产权的诉讼或针对公司的索赔或其他因素;需要获得额外资金; 以及吸引和留住支持公司发展所需的员工的能力。CRO、CMO或供应商的运营中断可能会对公司的业务、财务状况和运营结果产生负面影响。
现金 和现金等价物
现金 和现金等价物包括无限制的现金余额和购买时原始到期日为三个月或更短的短期流动性投资。
公允价值计量
会计准则编纂(“ASC”)主题820,公允价值计量(“ASC 820”),为按公允价值计量的工具建立了公允价值等级,区分了基于市场数据的假设(可观察到的投入)和公司自己的假设(不可观察到的投入)。可观察到的投入是指市场参与者将根据从本公司以外的来源获得的市场数据为资产或负债定价时使用的投入。不可观察的投入是反映公司对市场参与者将用于为资产或负债定价的投入的 假设,并根据当时可获得的最佳 信息制定。ASC 820将公允价值确定为在计量日期在市场参与者之间有序交易中出售资产或支付 转移负债的价格。作为在公允价值计量中考虑市场参与者假设的基础,ASC 820建立了一个三级价值层次结构,该层次结构区分了 以下各项:
第1级-相同资产或负债的活跃市场报价。
第2级-直接或间接可观察到的第1级输入以外的其他输入,如报价市场价格、利率和收益率曲线。
第 3级--资产或负债的不可观察的投入(即很少或没有市场活动的支持)。3级输入包括管理层自己对市场参与者在为资产或负债定价时将使用的假设的假设(包括对风险的假设)。
| F-8 |
研究和开发
研究和开发成本在发生时计入费用。研发成本包括进行研究和开发活动所产生的成本,包括工资和奖金、科学家招聘成本、员工福利、设施成本、实验室用品、制造费用、临床前费用、研究材料、咨询和其他合同服务。某些研发活动的成本根据个别安排的条款确认,这可能与已发生成本的模式 不同,并在财务报表中反映为预付或应计研发。
专利成本
由于公司继续为专利技术获得市场批准而产生成本,专利成本在发生时计入费用。成本包括已确认的无形资产续期或延长期限的费用、专利保护费和专利申请费。管理层将继续支出此类成本,直到通过适当的管理机构的监管批准获得市场批准。
所得税 税
2021年12月8日,该公司从合伙有限责任公司转变为C公司。自该日起,本公司开始按资产负债法核算所得税。根据此方法,递延税项资产及负债乃根据财务报表与资产及负债的计税基准之间的差额而厘定,按预期差额将拨回的 年度的现行税率厘定。当有必要将递延税项资产减少至更有可能变现的数额时,将设立估值免税额。
根据ASC 740-10《所得税》的规定,本公司通过对照适用的税法审查本公司就诉讼时效仍未生效的纳税年度所持的所有立场来评估不确定的税务状况。ASC 740-10规定, 如果基于技术上的是非曲直,通过审查,包括任何相关上诉或诉讼程序的解决,更有可能维持来自不确定税收状况的税收优惠,则可确认该税收优惠。公司确认 与未确认税收优惠责任相关的利息和罚款(如果有的话),作为所附营业报表中所得税支出项目的组成部分 。
最近 采用了会计公告
2020年8月,财务会计准则委员会发布了ASU第2020-06号《债务与转换及其他期权(次级主题470-20)》和《衍生工具和实体自有权益中的套期保值合约(次级主题815-40):实体自有权益中可转换工具和合同的会计处理(ASU 2020-06)》,通过减少可转换债务工具可用会计模型的数量,简化了可转换工具的会计核算。本指引还取消了计算可转换工具稀释每股收益的库存股方法,并要求使用IF-转换方法。自2021年1月1日起,该公司已提前采用该标准,目前已反映在其财务报表中。
2019年12月,FASB发布了ASU第2019-12号,所得税(主题740)。ASU第2019-12号修正案通过删除主题740中一般原则的某些例外,简化了所得税的会计处理。修正案还通过澄清和修改现有指南,改进和简化了美国公认会计原则或主题740的其他领域的一致应用。新标准自2022年1月1日起对本公司生效,并自2023年1月1日起对过渡期生效。本公司采用了这一准则,该准则目前已反映在其财务报表中。
| 3. | 普通股股东每股净亏损 |
下表汇总了公司普通股股东应占每股基本净亏损和摊薄净亏损的计算方法:
| 截至十二月三十一日止的年度: | ||||||||
| (单位为千,不包括每股和每股金额) | 2022 | 2021 | ||||||
| 分子: | ||||||||
| 普通股股东应占净亏损: | $ | (5,121 | ) | $ | (1,582 | ) | ||
| 分母: | ||||||||
| 加权-已发行普通股、基本普通股和稀释后普通股的平均数 | 28,230,842 | 不适用 | ||||||
| 普通股股东应占每股基本亏损和摊薄后每股净亏损(1) | $ | (0.18 | ) | $ | 不适用 | |||
(1) 截至2021年12月31日止年度,本公司并无已发行及已发行普通股。
| F-9 |
如附注6所述,公司于2022年8月18日发行了28,230,842股无投票权普通股,并于2022年12月发行了381,658股“B”系列优先股。截至2022年12月31日的年度的加权平均流通股数量是根据2022年8月18日发行的无投票权普通股计算的。
截至2022年12月31日,公司有5,600,000美元的已发行可转换票据,如附注5所述,如果公司发行和出售的股本超过10,000,000美元,这些票据可能会转换为普通股。截至2022年12月31日,转换后将发行的普通股估计数量为801,145股。于截至2022年及2021年12月31日止年度,本公司录得净亏损,因此,潜在普通股并未计入 ,因为此类计入会产生反摊薄作用。因此,该公司的每股基本和摊薄净亏损是相同的 ,因为它在所有呈报的期间都产生了净亏损。
| 4. | 金融工具的公允价值 |
截至2022年12月31日,本公司按公允价值计量其与2022年票据中转换期权功能相关的衍生负债,如附注5所述。该衍生负债被归类在价值层次结构的第三级,因为该负债基于使用投入和假设(包括潜在结果、利率、概率和时机)的估值模型。截至2021年12月31日,本公司并无任何按公允价值经常性计量的金融工具。
由于到期日较短,现金及现金等价物、预付费用、其他流动资产、应付帐款、应计费用、可转换票据以及母公司应付和应收账款的账面金额与其公允价值相近。
截至2022年12月31日及2021年12月31日止年度,本集团并无将1级、2级或3级资产及负债转进或转出。
| 5. | 可转换票据 应付票据 |
本公司于2021年12月订立多项可转换本票协议(统称为“2021年票据”)。根据2021年债券,公司收到1,300,000美元,按6.0%的年利率计算单利,并在公司首次公开募股 时到期。2021年债券到期时,持有人可选择全数收取未偿还本金及利息的现金付款 ,或由2021年债券持有人酌情选择延期一年。
如果本公司发行和出售的股本超过10,000,000美元,2021年票据的未偿还余额和应计利息可转换为固定数量的普通股,仅受可能发生的任何资本重组的典型反摊薄拨备的限制。
根据2021年票据的条款,本公司根据ASC 815衍生工具和对冲评估转换选择权功能。 它提供了三个标准,如果满足,要求公司从其宿主工具中分离转换选择权,并将其作为独立的衍生金融工具进行核算。这三项准则包括以下情况:(A)嵌入衍生工具的经济特征 及风险与嵌入衍生工具的经济特征及风险并无明确而密切的关系,(Br)同时包含嵌入衍生工具及宿主合约的混合工具并未根据其他适用的公认会计原则按公允价值重新计量 ,而公允价值的变动在盈利中报告为 其发生及(C)条款与嵌入衍生工具相同的独立工具将被视为衍生工具。
在2021年票据开始时,以及在2021年、2021年和2022年12月31日,公司确定用于转换的嵌入式衍生工具 不符合标准,因为它符合“与实体自己的股票挂钩”的例外,因此不需要从主机工具中分离出来。
公司于2022年1月至4月期间增发本金总额为4,300,000美元、期限为一年的可转换本票(统称为“2022年票据”)。2022年债券可按a)在公司首次公开发行时普通股发行价的15%折扣或b)20万美元的商数除以发行稀释前的总股本权益中的较小者进行转换。2022年债券中的转换期权功能根据ASC 815进行了评估,并在债券发行时和截至2022年12月31日记录了转换期权估计公允价值538,000美元的衍生负债。票据发行有抵销折扣 ,现正摊销至2022年票据的预期年期的利息开支。
截至2021年12月31日、2022年和2021年的年度,不包括2022年和2022年债券的折价摊销的利息支出分别为265,000美元和4,000美元,未偿还并计入应付账款和应计支出 。
| F-10 |
截至2022年12月31日和2021年12月31日,可转换债务余额如下:
| 十二月三十一日, | ||||||||
| (单位:千) | 2022 | 2021 | ||||||
| 可转换票据本金 | $ | 5,600 | $ | 1,300 | ||||
| 可转换票据折扣 | (149 | ) | — | |||||
| 应付可转换票据,扣除折扣后的净额 | $ | 5,451 | $ | 1,300 | ||||
| 6. | 股东赤字/母公司净投资 |
授权资本-截至2022年和2021年12月31日,公司已授权12,500,000股优先股,并已向FibroGenesis发行8,750,000股“A”系列优先股,这些优先股是根据公司的成立进行投标的,以换取通过专利转让和知识产权交叉许可协议贡献某些正在进行的研发和专利资产 。在支付股息和分配以及在清算、解散、清盘或其他方面,“A”系列优先股享有投票权和排名高于无投票权的普通股和普通股。 此外,“A”系列优先股具有相当于35,000,000美元的清算优先权,在公司发生清算、解散或清盘的情况下分配给“A”系列优先股持有人。且“A”系列优先股的每股股份可在“A”系列优先股的持有人选择 时,随时转换为一股普通股。除非持有人(S)另有选择,否则本公司并非多数尚存实体或出售本公司全部或实质所有资产的合并或合并 将被视为清盘事件。本公司还授权发行62,500,000股无投票权普通股,并在截至2022年12月31日的年度内发行了28,230,842股。2022年8月,公司向母公司发行了28,179,592股无投票权普通股,母公司又将股份分配给其成员。本次发行的无投票权普通股 计入股票拆分,公司未收到任何收益。公司还在2022年向董事会、一名顾问和一名员工额外发行了51,250股,并记录了16.8万美元的发行费用,这是根据发行时的第三方股票估值计算的。截至2021年12月31日,无投票权普通股均未发行和发行。
2022年12月,公司修改了公司注册证书,授权发行2,500,000股“B”系列优先股,发行381,658股,换取2,150,000美元。“B”系列优先股优先于“A”系列优先股,优先于普通股和非投票权普通股。“B”系列优先股具有投票权,并将在IPO交易完成时自动转换为普通股,这一点见公司修订和重新签署的公司注册证书。
| 7. | 共享 认购协议 |
于2021年11月12日,本公司与若干投资者订立股份购买协议,出售最多100,000,000股普通股(“合计限额”)。该协议取决于公司能否实现其普通股的公开上市。协议的主要条款包括不迟于公开上市后一年 到期的总限额2%的承诺费,以及在公开上市时向投资者发行的五年期认股权证,以购买相当于本公司总股权的4%的普通股,以a)上市时的每股价格或b)700,000,000美元的商数除以股权总数(完全稀释的普通股 股份)中的较小者为准。除非提前终止,否则本公司可要求在本协议上市后的五年内提取普通股或向投资者出售普通股。所要求的减持金额受减持前30天内公司普通股交易量的限制,每股价格相当于同期每股平均价格的90%。如果公司是以私下销售交易方式出售的,而不是完成其股票的公开上市,则必须向投资者支付1%的费用。
| F-11 |
| 8. | 所得税 税 |
A 使用联邦法定所得税率计算的所得税优惠与公司实际所得税率的对账如下:
| 截至十二月三十一日止的年度: | ||||||||
| 2022 | 2021 | |||||||
| 联邦法定利率 | (21.0 | )% | (21.0 | )% | ||||
| 永久性物品 | 0.8 | — | ||||||
| 真实上年度NOL递延税项资产 | (6.1 | ) | — | |||||
| 其他变化 | 1.9 | — | ||||||
| 更改估值免税额 | 24.4 | 21.0 | % | |||||
| 总计 | 0.0 | % | 0.0 | % | ||||
公司递延税项净资产的 组成部分如下:
| (单位:千美元) | 2022年12月31日 | 2021年12月31日 | ||||||
| 递延税项资产: | ||||||||
| 净营业亏损结转 | $ | 896 | $ | — | ||||
| 租赁责任 | 435 | |||||||
| 资本化研究与开发 | 299 | |||||||
| 衍生负债 | 31 | |||||||
| 应计负债 | 81 | 17 | ||||||
| 股票薪酬 | 17 | |||||||
| 递延税项资产 | 1,759 | 17 | ||||||
| 递延税项负债: | ||||||||
| 租赁使用权资产 | (462 | ) | — | |||||
| 未摊销债务贴现 | (31 | ) | — | |||||
| 递延税项负债 | (493 | ) | — | |||||
| 减去:估值免税额 | (1,266 | ) | (17 | ) | ||||
| 递延税项净资产 | $ | — | $ | — | ||||
该公司最初是一家有限责任公司,2021年12月转变为特拉华州的一家公司。由于在截至2022年12月31日和2021年12月31日的年度内产生净营业亏损,公司在截至2022年12月31日和2021年12月31日的年度内没有所得税支出。截至2022年12月31日,该公司的美国联邦净营业亏损(NOL)结转为4,265,000美元。联邦NOL 无限期结转,在所有权 权益发生某些累积变化的情况下,可能会受到年度限制。这可能会限制每年可用于抵销未来应税收入或纳税义务的税收属性的数量。 后续所有权变更可能会进一步影响未来几年的限制。
自2021年12月31日之后的纳税年度起生效,纳税人必须将根据IRC第174条规定与研究和实验(R&E)活动相关的任何支出资本化。虽然纳税人历来可以根据IRC第174条选择扣除这些费用,但2017年12月的减税和就业法案要求在2021年12月31日之后的纳税年度对R&E费用进行资本化和摊销 。与在美国的研发活动相关的费用必须在5年内摊销 ,在美国以外发生的研发费用必须在15年内摊销。R&E活动 的范围比IRC第41条(与研究税收抵免有关)下考虑的合格研究活动的范围更广。对于截至2022年12月31日的年度,本公司根据现有指引进行了分析,并确定即使在对其R&E费用进行了必要的资本化和摊销后,它仍将继续处于亏损状态。该公司将继续关注这一问题,以了解未来的发展,但预计R&E资本化和摊销不会要求其现在或在不久的将来缴纳现金税 。我们已经包括了这一准备金的影响,这导致截至2022年12月31日的递延税项资产约为29.9万美元 。
| F-12 |
管理层已评估与本公司递延税项净资产变现有关的正面及负面证据,并已确定本公司更有可能不会确认递延税项净资产的利益。因此,本公司已于2022年12月31日及2021年12月31日录得全额估值津贴。本公司将继续评估其递延税项资产未来的变现能力,并将根据需要调整估值拨备。
截至2022年、2022年和2021年12月31日,公司没有不确定的税务状况。本公司确认与未确认的税收优惠相关的利息和罚款均为所得税支出的组成部分。自成立以来,本公司没有记录任何未确认的税收优惠的利息或罚款。
| 9. | 租赁、承诺和或有事项 |
自2020年1月1日起,本公司采用ASU 2016-02租赁(主题842)来说明本公司的租赁。本公司选择在采用时适用短期租赁的实际做法。由于租赁的短期性质,本公司选择了一项会计政策,不在资产负债表上记录短期租赁。ASC 842-20-25-2允许承租人选择不在资产负债表上记录短期租赁(定义为12个月或以下的短期租赁)的会计政策 。根据公认会计原则,用于财务报表的租金 费用是根据可获得的最新合同 条款在租赁期内以直线方式确认的。
截至2021年12月31日,公司已签订了两份实验室和办公空间的短期租赁协议。该公司于2022年3月30日选择延长其其中一项实验室和办公空间租约的租期,租约续期的开始日期为2022年5月1日。延长的租赁期为126个月,并根据ASC 842租赁会计准则 入账为经营租赁。于延期时,应收使用权租赁资产及租户改善津贴合共2,799,000美元及租赁负债2,799,000美元。本租赁于2022年7月31日终止,剩余的使用权资产、应收租户改善津贴和租赁负债余额 已注销。
公司于2022年7月1日扩大了临时实验室和办公用房的剩余租赁范围,并将租期延长了6个月,随后于2022年8月1日和2022年10月7日进一步扩大了范围。每月许可费增加到每月15,000美元。这笔临时实验室和办公空间的租约将继续作为短期租约入账。
本公司于2022年10月订立为期62个月的写字楼租赁协议,于2027年11月30日届满。 本租约将按ASC 842租赁会计指引入账为营运租赁。使用权租赁资产和 租赁负债各为2,293,000美元,于租赁期开始时以7.5%的贴现率入账。
截至2022年12月31日和2021年12月31日止年度的租金 分别为39.2万美元和2.6万美元。截至2022年12月31日,不可取消的租赁付款为2,484,000美元。
截至2022年12月31日的经营租赁负债到期日 如下:
| (单位:千美元) | ||||
| 2023 | $ | 466 | ||
| 2024 | 477 | |||
| 2025 | 488 | |||
| 2026 | 544 | |||
| 2027 | 509 | |||
| 此后 | — | |||
| 租赁付款总额 | 2,484 | |||
| 减去:推定利息 | (411 | ) | ||
| 租赁总负债 | 2,073 | |||
| 减去:流动租赁负债 | (326 | ) | ||
| 非流动租赁负债总额 | $ | 1,747 | ||
| 10. | 相关的 方交易 |
截至2021年12月31日,本公司有一家尚未清偿的关联方母公司,应付金额为225,000美元。这笔债务由FibroGenesis持有,应于2022年4月1日到期,并可由FibroBiologics酌情选择延期六个月。 公司于2022年4月偿还了剩余母公司应付的225,000美元。2022年7月,该公司向母公司 提供了30万美元的一年期无息票据贷款。2022年10月,公司向母公司借出6万美元一年期无息票据,该票据已于2022年12月31日前偿还。
如附注6所述,公司通过专利转让和知识产权交叉许可协议从FibroGenesis收购了某些正在进行的研发和专利资产。专利转让协议将FibroGenesis的某些专利的权利、所有权和权益 转让给本公司进行进一步开发。《知识产权交叉许可协议》授予本公司对FibroGenesis在有限使用领域拥有的专利的独家权利,这些专利包括a)脊柱疾病、障碍或状况、b)癌症、c)骨科疾病、障碍或状况、 和d)多发性硬化症的诊断、治疗、预防和缓解。
| F-13 |
| 11. | 基于股份的薪酬 |
公司于2022年8月10日通过,股东于2022年8月18日批准《2022年股票计划》(以下简称《计划》)。该计划规定授予激励性股票期权、非法定股票期权、股票增值权、限制性股票奖励、限制性股票单位奖励和其他股票奖励。本计划通过授予股票奖励,旨在帮助公司获得并保留合格获奖者的服务,激励该等人士为公司的成功尽最大努力,并提供一种手段,使符合资格的获奖者可以从普通股的增值中受益。公司 在2022年根据本计划向员工、董事和科学顾问委员会成员共发行了101,250份期权,执行价为每股3.28美元。一般而言,本公司授予的奖励为期三年,行使价等于董事会在考虑独立第三方估值公司编制的本公司普通股当时估值后确定的普通股估计公允价值。
截至2022年12月31日,根据该计划,可供未来发行的股票为12,398,750股。
基于库存的 薪酬费用在运营报表中确认如下:
| 截至12月31日止年度, | ||||||||
| (单位:千美元) | 2022 | 2021 | ||||||
| 研发 | $ | 115 | $ | — | ||||
| 一般和行政 | 150 | — | ||||||
| 基于股票的薪酬总支出 | $ | 265 | $ | — | ||||
截至2022年12月31日的年度基于股票的薪酬支出,包括向董事会和顾问发行的无投票权普通股支出16.8万美元。
截至2022年12月31日,与未归属奖励相关的未确认的基于股票的薪酬成本以及预计确认这些成本的加权平均期间 如下:
| 股票期权 | ||||
| 未确认的基于股票的薪酬支出(千) | $ | 169 | ||
| 待确认的预期加权平均期间薪酬成本(年) | 1.7 | |||
本公司股票期权活动摘要如下:
| 股票期权 | 加权平均每股行权价 | 加权-平均剩余合同寿命(年) | 聚合内在价值(以千为单位) | |||||||||||||
| 截至2021年12月31日的未偿还债务 | — | — | — | — | ||||||||||||
| 授与 | 101,250 | $ | 3.28 | 10.0 | — | |||||||||||
| 已锻炼 | — | — | — | — | ||||||||||||
| 被没收/取消 | — | — | — | — | ||||||||||||
| 截至2022年12月31日的未偿还债务 | 101,250 | $ | 3.28 | 9.7 | — | |||||||||||
| 自2022年12月31日起可行使 | 19,445 | $ | 3.28 | 9.7 | — | |||||||||||
授予员工、董事和顾问的股票期权的公允价值在授予之日使用Black-Scholes期权定价模型进行估算,该模型采用以下假设:
| 假设: | 截至的年度 2022年12月31日 | |||
| 无风险利率 | 4.1 | % | ||
| 预期波动率 | 100 | % | ||
| 预期期限(年) | 5.4至6.4 | |||
| 预期股息 | 0 | % | ||
于截至2022年12月31日止年度内,授出购股权之加权平均授出日期公允价值为每股2.64美元。
| F-14 |
| 12. | 后续 事件 |
公司评估了截至2023年4月28日(财务报表发布之日)的后续事件,并确定除以下披露的事件外,没有其他事件需要在财务报表中披露或进行调整 。
于2023年1月,本公司与其母公司FibroGenesis订立关于第一次谈判权的协议(“ROFN协议”)。作为交换,FibroGenesis同意修改公司注册证书,以a)在IPO、直接上市或出售公司时取消“A”系列优先股$35,000,000清算优先权,b)使“B”系列优先股清算优先权等于“A”系列优先股,以及c)规定在IPO时,直接上市或出售公司“A”系列优先股将被免费取消,FibroBiologics将 同意在IPO前向FibroGenesis支付对FibroBioics的任何股权投资所得毛收入的15%,直接上市 或出售公司。此外,如果FibroGenesis决定向外部许可其任何技术,FibroBiologics将获得为期五年的第一谈判权。2023年1月,公司修改了公司注册证书以反映这些变化,并向FibroGenesis支付了23.2万美元,以获得公司2022年12月发行的股权所得毛收入的15%。
2023年1月,该公司发起了一项活动,通过 超额认购的监管CF发售“B”系列优先股,筹集至多5,000,000美元。本次发行已募集4,990,000美元,本公司迄今已收到净收益4,230,000美元。该公司正在收到此次发行的剩余收益,预计在收到并核对最终分配后,将发行867,913股。根据ROFN协议,本公司已于2023年3月向FibroGenesis支付该等收益中的63.4万美元(15%),并预计在收到最终分派后于2023年4月向FibroGenesis额外支付11.4万美元。
2023年1月,该公司将其临时实验室和办公空间的剩余租约再延长六个月。
于2023年2月,本公司将2022年债券本金价值3,700,000元的本金及利息转换为799,603股“B”系列优先股。
2023年3月,公司以10,325,000美元的私募方式额外出售了1,680,084股“B”系列优先股。根据ROFN协议,该公司已向FibroGenesis支付了1,549,000美元(15%)的收益。
于2023年4月,本公司修订公司注册证书,授权发行10,000,000股普通股,将“B”系列优先股数量增加至5,000,000股,并批准5,000,000股“B-1”系列优先股,清算优先权相当于“A”和“B”系列优先股 。该公司还将2021年债券本金价值160万美元和2022年债券本金价值30万美元的本金和利息转换为353,713股“B”系列优先股,并以15.3万美元的非公开配售方式出售了8,388股“B-1”系列优先股。根据ROFN协议,公司将于2023年4月向FibroGenesis支付2.3万美元(15%)的收益。
2023年4月,FibroGenesis全额偿还母公司应收账款30万美元。
| 13. | 反向 股票拆分 |
2023年10月,该公司向特拉华州提交了修订和重述的公司注册证书,以立即 实施4股1股的反向股票拆分。所有股票和每股金额 均已追溯调整,以反映反向股票拆分的影响。
| F-15 |
FibroBiologics, 公司
未经审计的 简明财务报表
和
未经审计简明财务报表附注
截至2023年和2022年9月30日的9个月
| F-16 |
FibroBiologics, 公司
精简的资产负债表
(单位为 千,不包括股份和每股数据)
9月30日, 2023 | 十二月三十一日, 2022 | |||||||
| (未经审计) | ||||||||
| 资产 | ||||||||
| 流动资产 | ||||||||
| 现金和现金等价物 | $ | 10,766 | $ | 2,266 | ||||
| 预付费用 | 27 | 29 | ||||||
| 母公司应收账款 | — | 300 | ||||||
| 其他流动资产 | 121 | 30 | ||||||
| 流动资产总额 | 10,914 | 2,625 | ||||||
| 财产和设备,净额 | 477 | — | ||||||
| 经营性租赁使用权资产净额 | 1,908 | 2,199 | ||||||
| 总资产 | $ | 13,299 | $ | 4,824 | ||||
| 负债和股东权益/(赤字) | ||||||||
| 流动负债 | ||||||||
| 应付账款和应计费用 | $ | 820 | $ | 758 | ||||
| 应付给父母 | 141 | — | ||||||
| 短期经营租赁负债 | 353 | 326 | ||||||
| 衍生负债 | — | 538 | ||||||
| 可转换票据,扣除债务贴现后的净额 | — | 5,451 | ||||||
| 流动负债总额 | 1,314 | 7,073 | ||||||
| 长期经营租赁负债 | 1,447 | 1,747 | ||||||
| 总负债 | 2,761 | 8,820 | ||||||
| 股东权益/(亏损) | ||||||||
| 母公司净投资 | — | 1,461 | ||||||
| 优先股,面值0.00001美元;授权总股本2000万股;截至2023年9月30日、2023年12月31日和2022年12月31日,授权、发行和发行的A系列优先股8,750,000股 | — | — | ||||||
| 优先股,面值0.00001美元;授权总股本2000万股;授权B系列优先股500万股;截至2023年9月30日已发行和已发行4,171,445股;截至2022年12月31日已发行和已发行381,658股 | — | — | ||||||
优先股,面值0.00001美元;授权总股本2000万股;授权B-1系列优先股500万股 ;截至2023年9月30日已发行和已发行股票74,922股;截至2022年12月31日未发行和已发行股票 | — | — | ||||||
| 优先股,面值0.00001美元;授权总股本2000万股;授权C系列优先股2,500股;截至2023年9月30日和2022年12月31日,没有发行和发行的股票 | — | — | ||||||
| 无投票权普通股,面值0.00001美元;授权发行30,000,000股;截至2023年9月30日和2022年12月31日,已发行和已发行28,230,842股 | 1 | 1 | ||||||
| 普通股,面值0.00001美元;授权发行100,000,000股;截至2023年9月30日和2022年12月31日,没有发行和发行的股票 | — | — | ||||||
| 额外实收资本 | 25,177 | 2,414 | ||||||
| 累计赤字 | (14,640 | ) | (7,872 | ) | ||||
| 股东权益合计/(亏损) | 10,538 | (3,996 | ) | |||||
| 总负债和股东权益/(赤字) | $ | 13,299 | $ | 4,824 | ||||
附注是这些未经审计的简明财务报表的组成部分。
| F-17 |
FibroBiologics, 公司
简明的操作报表
(未经审计,以千为单位,不包括股票和每股数据)
在截至的9个月中 9月30日, | ||||||||
| 2023 | 2022 | |||||||
| 运营费用: | ||||||||
| 研发 | $ | 1,595 | $ | 802 | ||||
| 一般、行政和其他 | 4,814 | 2,361 | ||||||
| 总运营费用 | 6,409 | 3,163 | ||||||
| 运营亏损 | (6,409 | ) | (3,163 | ) | ||||
| 其他收入/(亏损) | (213 | ) | — | |||||
| 利息支出 | (146 | ) | (434 | ) | ||||
| 净亏损 | $ | (6,768 | ) | $ | (3,597 | ) | ||
| 当作股息 | (2,573 | ) | — | |||||
| 普通股股东应占净亏损 | (9,341 | ) | $ | (3,597 | ) | |||
| 每股基本和稀释后净亏损 | $ | (.33 | ) | $ | (.13 | ) | ||
| 加权平均流通股、基本股和稀释股 | 28,230,842 | 28,230,842 | ||||||
附注是这些未经审计的简明财务报表的组成部分。
| F-18 |
FibroBiologics, 公司
简明 股东权益变动表/(亏损)
截至2023年和2022年9月30日的9个月
(未经审计,以千为单位,股票除外)
| 净父节点 | 系列 A类优先股 | 系列 “B”优先股 | 系列 “B-1”优先股 | 无投票权 普通股 | 额外实收 | 累计 | 股东总股本 股权/ | |||||||||||||||||||||||||||||||||||||||||
| 投资 | 股票 | 金额 | 股票 | 金额 | 股票 | 金额 | 股票 | 金额 | 资本 | 赤字 | (赤字) | |||||||||||||||||||||||||||||||||||||
| 余额-2022年12月31日 | $ | 1,461 | 8,750,000 | $ | — | 381,658 | $ | — | — | $ | — | 28,230,842 | $ | 1 | $ | 2,414 | $ | (7,872 | ) | $ | (3,996 | ) | ||||||||||||||||||||||||||
| 出售“B”系列优先股股份, 直接费用净额 | — | — | — | 2,570,394 | — | — | — | — | — | 14,945 | — | 14,945 | ||||||||||||||||||||||||||||||||||||
| 出售“B-1”系列优先股 扣除直接费用后的份额 | — | — | — | — | — | 74,922 | — | — | — | 1,193 | — | 1,193 | ||||||||||||||||||||||||||||||||||||
| 发行“B”系列优先股 转换票据的股份及应计利息 | — | — | — | 1,219,393 | — | — | — | — | — | 6,404 | — | 6,404 | ||||||||||||||||||||||||||||||||||||
| 与ROFN协议衍生产品相关的视同股息 责任 | (1,461 | ) | — | — | — | — | — | — | — | — | (1,112 | ) | — | (2,573 | ) | |||||||||||||||||||||||||||||||||
| 基于股票的薪酬费用 | — | — | — | — | — | — | — | — | — | 1,333 | — | 1,333 | ||||||||||||||||||||||||||||||||||||
| 净亏损 | — | — | — | — | — | — | — | — | — | — | (6,768 | ) | (6,768 | ) | ||||||||||||||||||||||||||||||||||
| 余额(未审计) -2023年9月30日 | $ | — | 8,750,000 | $ | — | 4,171,445 | $ | — | 74,922 | $ | — | 28,230,842 | $ | 1 | $ | 25,177 | $ | (14,640 | ) | $ | 10,538 | |||||||||||||||||||||||||||
| 净父节点 | 系列 A类优先股 | 系列 “B”优先股 | 系列 “B-1”优先股 | 无投票权 普通股 | 额外实收 | 累计 | 总计 股东权益/ | |||||||||||||||||||||||||||||||||||||||||
| 投资 | 股票 | 金额 | 股票 | 金额 | 股票 | 金额 | 股票 | 金额 | 资本 | 赤字 | (赤字) | |||||||||||||||||||||||||||||||||||||
| 余额-2021年12月31日 | $ | 1,461 | 8,750,000 | $ | — | — | $ | — | — | $ | — | — | $ | — | $ | — | $ | (2,751 | ) | $ | (1,290 | ) | ||||||||||||||||||||||||||
| 向母公司成员发行无投票权普通股 | — | — | — | — | — | — | — | 28,179,592 | 1 | (1 | ) | — | — | |||||||||||||||||||||||||||||||||||
| 基于股票的薪酬费用 | — | — | — | — | — | — | — | 51,250 | — | 233 | — | 233 | ||||||||||||||||||||||||||||||||||||
| 净亏损 | — | — | — | — | — | — | — | — | — | — | (3,597 | ) | (3,597 | ) | ||||||||||||||||||||||||||||||||||
天平 (未经审计)-2022年9月30日 | $ | 1,461 | 8,750,000 | $ | — | — | $ | — | — | $ | — | 28,230,842 | $ | 1 | $ | 232 | $ | (6,348 | ) | $ | (4,654 | ) | ||||||||||||||||||||||||||
附注是这些未经审计的简明财务报表的组成部分。
| F-19 |
FibroBiologics, 公司
简明现金流量表
(未经审计,以千为单位)
| 截至9月30日的9个月, | ||||||||
| 2023 | 2022 | |||||||
| 经营活动的现金流 | ||||||||
| 净亏损 | $ | (6,768 | ) | $ | (3,597 | ) | ||
| 对净亏损与经营活动中使用的现金净额进行的调整: | ||||||||
| 基于股票的薪酬费用 | 1,333 | 233 | ||||||
| 衍生产品负债的其他损失 | 72 | — | ||||||
| 可换股票据债务折价摊销 | 81 | 254 | ||||||
| 经营性租赁使用权资产摊销 | 291 | — | ||||||
| 折旧费用 | 16 | — | ||||||
| 经营性资产和负债变动情况: | ||||||||
| 预付费用的变动 | 2 | (21 | ) | |||||
| 其他流动资产变动 | (91 | ) | (22 | ) | ||||
| 应付账款和应计费用变动 | 396 | 260 | ||||||
| 应支付给父母的金额发生变化 | 141 | — | ||||||
| 经营租赁负债变动 | (273 | ) | — | |||||
| 用于经营活动的现金净额 | (4,800 | ) | (2,893 | ) | ||||
| 投资活动产生的现金流 | ||||||||
| 购置财产和设备 | (493 | ) | — | |||||
| 用于投资活动的现金净额 | (493 | ) | — | |||||
| 融资活动产生的现金流 | ||||||||
| 向父母偿还贷款 | — | (225 | ) | |||||
| ROFN协议向母公司付款 | (2,645 | ) | — | |||||
| 母公司应收票据的还款和收益 | 300 | (300 | ) | |||||
| 发行可转换票据所得款项 | — | 4,300 | ||||||
| 发行B系列优先股的收益,扣除直接成本 | 14,945 | — | ||||||
| 发行B-1系列优先股的收益,扣除直接成本 | 1,193 | — | ||||||
| 融资活动提供的现金净额 | 13,793 | 3,775 | ||||||
| 现金及现金等价物净增加情况 | 8,500 | 882 | ||||||
| 期初现金及现金等价物 | 2,266 | 407 | ||||||
| 期末现金和现金等价物 | $ | 10,766 | $ | 1,289 | ||||
| 补充披露现金流量信息: | ||||||||
| 缴纳所得税的现金 | $ | — | $ | — | ||||
| 支付利息的现金 | $ | 1 | $ | — | ||||
| 补充披露非现金投资和融资活动 : | ||||||||
| 债务发行贴现的衍生负债附加 | $ | — | $ | 538 | ||||
| 取得经营性租赁使用权资产和负债 | $ | — | $ | 2,799 | ||||
| 发行B系列优先股以转换票据和应计利息 | $ | 5,866 | $ | — | ||||
| 将票据转换为B系列优先股的衍生负债重新分类 | $ | 538 | $ | — | ||||
附注是这些未经审计的简明财务报表的组成部分。
| F-20 |
FibroBiologics, 公司
未经审计简明财务报表附注
2023年9月30日
| 1. | 组织机构、业务描述和流动资金 |
组织 和业务
FibroBiologics, Inc.(“公司”或“FibroBiologics”)最初是根据德克萨斯州法律于2021年4月8日(“初始”)成立的有限责任公司(“LLC”),然后于2021年12月14日转变为特拉华州的 公司。FibroBiologics是一家处于早期阶段的细胞治疗公司,总部设在德克萨斯州休斯顿,利用成纤维细胞开发慢性疾病的创新治疗方法。该公司的主要关注点是启动和推进美国食品和药物管理局关于成纤维细胞治疗退行性腰椎间盘疾病、多发性硬化症、癌症、伤口愈合和其他疾病的临床前研究和临床阶段试验。在此之前,与这些疾病途径相关的临床前研究和开发是在母公司SpinalCyte,LLC(“母公司”或 “FibroGenesis”)的领导下进行的。
持续关注和管理的计划
该公司自创始以来已出现运营亏损,预计未来随着基础设施建设、知识产权开发和研发活动的开展,此类亏损还将继续。该公司主要依靠天使投资者和私人债务配售的组合来为其运营提供资金。截至2023年9月30日,公司累计亏损14,640,000美元,现金及现金等价物为10,766,000美元。向盈利的过渡将取决于候选产品的成功开发、审批和商业化,以及能否获得足够的收入来支持公司的成本结构。 公司目前不会产生收入,可能永远不会实现盈利。除非并直至产生收入和净收入,否则公司将需要继续筹集额外资本。如附注7所述,管理层已于2021年11月12日签订股份购买协议。如果在国家认可的美国证券交易所直接上市或首次公开募股,本协议将为公司提供获得额外流动资金的途径。如附注6所述,于2023年首九个月内,本公司透过众筹及私募分别筹得4,620,000元及11,518,000元。因此,本公司相信自该等未经审核简明财务报表发布之日起至少未来12个月内,本公司有足够资本为其目前的营运计划提供资金。
细分市场
运营部门被确定为企业的组成部分,其独立的离散财务信息可供首席运营决策者在做出有关资源分配的决策和评估业绩时进行评估 。首席执行官是首席运营决策者,他在汇总的基础上审查财务信息,以分配资源和评估财务业绩。本公司将其业务作为一个单一的运营部门进行运营和管理,因此是一个可报告的部门。
| 2. | 摘要 主要会计政策 |
演示基础
随附的未经审计简明财务报表已根据美国公认会计原则(“GAAP”)编制。 随附的截至2023年9月30日的简明资产负债表、截至2023年和2022年9月30日的九个月的简明经营报表、截至 2023年和2022年9月30日的九个月的股东权益/(赤字)简明报表以及截至2023年、2023年和2022年9月30日的九个月的简明现金流量表均未经审计。这些未经审计的简明财务报表是根据美国证券交易委员会关于中期财务信息的规则和条例 编制的。因此,它们不包括《公认会计准则》要求的完整财务报表所需的所有信息和脚注。这些未经审计的简明财务报表应与截至2022年12月31日的年度的已审计财务报表及附注一并阅读。未经审计的简明财务报表已按照与年度合并财务报表相同的基础编制,管理层认为反映了为公平陈述公司截至2023年9月30日的财务状况、截至2023年和2022年9月30日的9个月的经营业绩、截至2023年9月30日、2023年和2022年9月30日的9个月的未经审计的股东权益/(亏损)简明报表以及截至9月30日的9个月的未经审计的简明现金流量表所必需的所有调整(包括正常经常性调整)。2023年和2022年。 本文所包括的2022年12月31日简明资产负债表源自经审计的财务报表,但不包括《公认会计准则》要求的完整财务报表所需的所有披露或附注。
| F-21 |
这些简明财务报表附注所披露的与截至2023年9月30日和2022年9月30日的九个月相关的财务数据和其他信息未经审计。中期业绩不一定代表全年或未来任何时期的业绩 。
从2021年1月1日至2021年4月8日成立期间,本公司作为FibroGenesis的一个业务线运营,而不是作为一个独立的实体运营。因此,在公司于2021年4月8日成立之前,财务报表 来自母公司的历史会计记录。母公司自2021年1月1日至本公司于2021年4月8日成立时最初发生的与本公司业务活动直接相关的所有一般及行政费用及研发费用均已分配并计入本公司的财务报表。由此产生的母公司投资净额计入股东权益/(赤字),代表母公司在 本公司记录的净资产中的权益。
随附的未经审计的 简明财务报表不包括母公司的任何分配,但母公司的净投资包括在开始的 股东权益/(亏损),并已根据公认会计准则编制。
于 2023年10月,本公司修订及重列其于特拉华州的注册证书,以立即实施1比4股份反向股份拆细。所有股份和每股金额均已追溯调整,以反映反向 股票分割的影响。
使用预估的
编制符合GAAP的未经审计的 简明财务报表要求管理层做出估计和假设,这些估计和假设会影响未经审计的简明财务报表日期的资产和负债的报告 金额以及或有资产和负债的披露,以及报告期间的费用报告金额。该等估计乃基于截至未经审核简明财务报表日期可得的资料;因此,实际结果可能与该等估计及假设有所不同。
信用风险集中度
可能使公司面临集中信用风险的金融工具主要包括现金和现金等价物。 公司在金融机构有大量现金余额,全年经常超过联邦保险限额250,000美元。发生的任何损失或无法获得此类资金可能会对公司的财务状况、经营业绩和现金流产生重大不利影响。
风险 和不确定性
公司面临一定的风险和不确定性,包括但不限于公司 认为可能对未来财务状况或经营业绩产生重大不利影响的以下任何领域的变化:公司 将其当前和未来候选产品推进临床开发的时机和能力;与 公司候选产品的临床供应品生产相关的成本和时间表;其候选产品的监管批准和市场接受度;第三方合同研究组织(“CRO”)和合同生产组织 (“CMO”)的绩效;来自拥有更多财务资源或专业知识的制药公司的竞争;知识产权保护、基于知识产权或其他因素针对公司的诉讼或索赔;获得额外资金的需要; 以及吸引和留住支持公司发展所需员工的能力。CRO、CMO或供应商的运营中断 可能会对公司的业务、财务状况和运营业绩产生负面影响。
现金 和现金等价物
现金 和现金等价物包括无限制的现金余额和购买时原始到期日为三个月或更短的短期流动性投资。
财产 和设备
财产和设备包括 实验室设备,这些设备按成本记录,并在五年的估计使用寿命内使用直线法折旧。截至2023年9月30日止九个月确认折旧开支16,000元。截至2022年9月30日止九个月并无确认折旧 开支。折旧费用在随附的简明经营报表中分类为研究和 开发费用。
当有事件或情况变化显示资产账面值可能无法收回时,会 检讨物业及设备的减值。 需要进行减值评估的条件包括:资产的可观察市场价值显著下降, 资产使用的程度或方式发生重大变化,或表明资产或资产组的 账面金额无法收回的重大不利变化。对于长期持有并使用的资产,本公司仅在其账面价值无法通过未折现现金流量收回的情况下确认 减值损失,并根据其账面价值与预计公允价值之间的差额 计量减值损失。截至二零二三年及二零二二年九月三十日止九个月并无该等亏损。
| F-22 |
公允价值计量
会计 标准编码(“ASC”)820, 公允价值计量,为以公允价值计量的工具建立公允价值等级,以区分基于市场数据的假设(可观察输入)和公司自己的假设(不可观察输入)。可观察输入数据是市场参与者根据从独立于本公司的来源获得的市场 数据为资产或负债定价时使用的输入数据。不可观察输入值是指反映公司对市场参与者在资产或负债定价中使用的 输入值的假设的输入值,并且是根据当时情况下可获得的最佳信息 制定的。ASC 820将公允价值确定为市场参与者之间在计量日进行的有序交易中出售资产所收到的价格或转让负债所支付的价格。作为在公允价值计量中考虑市场参与者假设 的基础,ASC 820建立了一个三层价值层级,区分以下内容:
第1级-相同资产或负债的活跃市场报价。
第2级-直接或间接可观察到的第1级输入以外的其他输入,如报价市场价格、利率和收益率曲线。
第 3级--资产或负债的不可观察的投入(即很少或没有市场活动的支持)。3级输入包括管理层自己对市场参与者在为资产或负债定价时将使用的假设的假设(包括对风险的假设)。
估值层级中的分类 基于对公允价值计量具有重大意义的最低输入级别。
研究和开发
研究和开发成本在发生时计入费用。研发成本包括进行研究和开发活动所产生的成本,包括工资和奖金、科学家招聘成本、员工福利、设施成本、实验室用品、制造费用、临床前费用、研究材料、咨询和其他合同服务。某些研发活动的成本根据个别安排的条款确认,可能与已发生成本的模式 不同,并在未经审核的简明财务报表中反映为预付或应计研发。
专利成本
由于公司继续为专利技术获得市场批准而产生成本,专利成本在未经审计的简明经营报表中计入一般费用、行政费用和其他费用。成本包括确认的无形资产的续期或延期费用、专利保护费用和专利申请费用。管理层将继续支出此类成本,直到通过适当的管理机构的监管批准获得市场批准为止。
所得税 税
2021年12月14日,该公司从合伙有限责任公司转变为C公司。此后,本公司开始按资产负债法对所得税进行会计处理。根据此方法,递延税项资产及负债乃根据资产及负债的财务报表与税基之间的差额而厘定,并采用预期差额将拨回的年度的现行税率 。当有必要将递延税项资产减少至更有可能变现的金额时,将设立估值免税额。
根据ASC 740-10的规定,所得税,本公司通过对照适用的税法评估不确定的税务状况 本公司就诉讼时效仍未生效的纳税年度持有的所有仓位。ASC 740-10规定,如果基于技术上的是非曲直,通过审查(包括任何相关上诉或诉讼程序的解决方案)更有可能维持不确定的税收状况,则可确认来自该不确定税收状况的税收优惠。公司确认 与未确认税收优惠责任相关的利息和罚款(如果有的话),作为附带的简明经营报表中所得税支出项目的组成部分。
| F-23 |
| 3. | 普通股股东每股净亏损 |
下表汇总了公司普通股股东应占每股基本净亏损和摊薄净亏损的计算方法:
九个月结束 9月30日, | ||||||||
| (单位为千,不包括每股和每股金额) | 2023 | 2022 | ||||||
| 分子: | ||||||||
| 净亏损 | $ | (6,768 | ) | $ | (3,597 | ) | ||
| 对每股收益分子的调整: | ||||||||
| 当作股息 | (2,573 | ) | — | |||||
| 普通股股东应占净亏损 | $ | (9,341 | ) | $ | (3,597 | ) | ||
| 分母: | ||||||||
| 加权-已发行普通股、基本普通股和稀释后普通股的平均数 | 28,230,842 | 28,230,842 | ||||||
| 普通股股东应占普通股每股净亏损,基本亏损和稀释亏损 | $ | (0.33 | ) | $ | (0.13 | ) | ||
如附注 6所述,本公司于2022年8月18日发行了28,230,842股无投票权普通股。截至2023年9月30日和2022年9月30日的9个月的加权平均流通股数量 以2022年8月18日发行的无投票权普通股为基础。由于无投票权普通股的发行在会计上被视为股票拆分,这些股票被视为于2022年1月1日发行,并在截至2023年9月30日和 2022年的整个9个月内流通无阻。
如附注 10所述,本公司同意在首次公开招股、直接上市或出售本公司之前,向FibroGenesis支付FibroBiologics任何股权投资所得毛收入的15%,以在该等事件发生时消除A系列优先股及其3,500万美元的清算优先权。优先股的赎回产生的衍生负债比母公司净投资1,461,000美元高出1,112,000美元,在这里反映为普通股股东在计算每股收益时可用金额的减少。
截至2022年9月30日,公司有5,600,000美元的已发行可转换票据,如果公司出售和发行的股本超过10,000,000美元,则这些可转换票据可以转换为普通股,并在2023年9月30日之前全部转换为B系列优先股,如附注5中进一步描述。截至2022年9月30日,转换后发行的普通股估计数量为790,459股。于截至2023年、2023年及2022年9月30日止九个月,本公司报告净亏损,因此,潜在普通股不包括在内,因为此类纳入将 反摊薄。因此,该公司的每股基本和摊薄净亏损是相同的,因为它在所有呈报的期间都产生了 净亏损。
| 4. | 金融工具的公允价值 |
截至2022年12月31日, 公司按公允价值计量其与2022年票据中转换期权功能相关的衍生负债,如附注5所述。这一衍生负债被归类在价值层次结构的第三级,因为该负债基于一个估值模型,该模型使用了投入和假设,包括潜在结果、利率、概率和时机。截至2023年9月30日,2022年票据已转换,消除了衍生工具负债,本公司 并无任何按公允价值经常性计量的重大金融工具。
现金及现金等价物、预付费用、其他流动资产、应付帐款、应计费用、应付可转换票据及母公司应付及应收账款的账面值因到期日较短而接近其公允价值。
截至2023年9月30日和2022年9月30日的9个月内,1级、2级或3级资产和负债没有转移或 流出。
| F-24 |
| 5. | 可转换票据 应付票据 |
本公司于2021年12月订立多项可转换本票协议(统称为“2021年票据”)。根据2021年债券,公司收到1,300,000美元,按6.0%的年利率计算单利,并在公司首次公开募股 时到期。2021年债券到期时,持有人可选择全数收取未偿还本金及利息的现金付款 ,或由2021年债券持有人酌情选择延期一年。
如果本公司发行和出售的股本超过10,000,000美元,2021年票据的未偿还余额和应计利息可转换为固定数量的普通股,仅受可能发生的任何资本重组的典型反摊薄拨备的限制。
根据2021年票据的条款,本公司根据ASC 815评估转换期权功能,衍生工具和套期保值。 它提供了三个标准,如果得到满足,要求公司将转换期权从其宿主工具中分离出来,并将其作为独立的衍生金融工具进行核算。这三项准则包括以下情况:(A)嵌入衍生工具的经济特征 及风险与嵌入衍生工具的经济特征及风险并无明确而密切的关系,(Br)同时包含嵌入衍生工具及宿主合约的混合工具并未根据其他适用的公认会计原则按公允价值重新计量 ,而公允价值的变动在盈利中报告为 其发生及(C)条款与嵌入衍生工具相同的独立工具将被视为衍生工具。
在2021年票据开始时和2022年12月31日,公司确定转换功能的嵌入衍生工具 不符合标准,因为它符合“与实体自己的股票挂钩”的例外,因此不需要 从主机工具中分离出来。
公司于2022年1月至4月期间增发本金总额为4,300,000美元、期限为一年的可转换本票(统称为“2022年票据”)。2022年债券可按a)在公司首次公开发行时普通股发行价的15%折扣或b)20万美元的商数除以发行稀释前的总股本权益中的较小者进行转换。2022年债券中的转换期权功能根据ASC 815进行了评估,并在债券发行时和截至2022年12月31日记录了转换期权估计公允价值538,000美元的衍生负债。票据发行有抵销折扣 ,现正摊销至2022年票据的预期年期的利息开支。
截至2023年9月30日和2022年9月30日的9个月,2021年和2022年债券的利息支出分别为6.5万美元和18万美元,其中不包括2022年债券上记录的折价摊销。截至2022年12月31日,应计利息尚未支付, 已计入应付帐款和应计费用。
于2023年2月,本公司将2022年债券本金价值3,700,000元的本金及利息转换为799,603股“B”系列优先股。2023年4月,本公司还将2021年债券本金价值130万美元 和2022年债券本金价值30万美元的本金和利息转换为353,713股“B”系列优先股。2023年9月,本公司将2022年发行的债券本金价值30万美元的本金及利息转换为66,077股“B”系列优先股。截至2023年9月30日,没有未付利息,也没有计入应付账款和应计费用。
截至2023年9月30日和2022年12月31日的可转换债务余额 包括:
| (单位:千) | 2023年9月30日 | 2022年12月31日 | ||||||
| 可转换票据本金 | $ | — | $ | 5,600 | ||||
| 可转换票据折扣 | — | (149 | ) | |||||
| 应付可转换票据,扣除折扣后的净额 | $ | — | $ | 5,451 | ||||
| F-25 |
| 6. | 股东权益/(赤字)/母公司净投资 |
授权资本-截至2023年和2022年9月30日,公司分别授权了20,000,000股和12,500,000股优先股 ,并已向FibroGenesis发行了8,750,000股“A”系列优先股,这些优先股是根据公司的成立 进行投标的,以换取某些正在进行的研发和专利资产的贡献,通过专利转让和知识产权交叉许可协议。在支付股息和分派以及清算、解散、清盘或其他方面,“A”系列优先股有权投票并排在无投票权普通股和普通股之前。此外,“A”系列优先股拥有相当于35,000,000美元的清算优先权,可在公司发生清算、解散、 或清盘时分配给“A”系列优先股持有人,该优先股随后作为如下进一步描述的ROFN协议的一部分被注销,且“A”系列优先股的每股股份可于“A”系列优先股的 持有人的选择下随时转换为一股普通股。除非持有人(S)另有选择,否则本公司并非多数尚存实体或出售本公司全部或实质所有资产的合并或合并 将被视为清盘事件。本公司还授权发行62,500,000股无投票权普通股,并在截至2022年12月31日的年度内发行了28,230,842股。2022年8月,公司向母公司发行了28,179,592股无投票权普通股,母公司又将股份分配给其成员。本次发行无投票权普通股计入 股票拆分,公司未收到任何收益。公司还在2022年向董事会、一名顾问和一名员工额外发行了51,250股,并记录了16.8万美元的发行费用,这是 根据发行时的第三方股票估值计算的。
2022年12月,公司修改了公司注册证书,授权发行2,500,000股“B”系列优先股,发行381,658股,换取2,150,000美元。“B”系列优先股优先于“A”系列优先股,优先于普通股和非投票权普通股。“B”系列优先股具有投票权,并将在IPO交易完成时自动转换为普通股,这一点见公司修订和重新签署的公司注册证书。
于2023年1月,为反映与母公司订立的ROFN协议,如附注10所述,本公司修订其注册证书,以a)于本公司首次公开发售、直接上市或出售时取消“A”系列优先股$35,000,000 清算优先权,b)使“B”系列优先股清算优先权等于“A”系列优先股 股份,及c)规定于首次公开发售、直接上市或出售本公司“A”系列优先股时,“A”系列优先股将作废 。
于2023年4月,本公司修订公司注册证书,授权发行10,000,000股普通股,将“B”系列优先股数量增加至5,000,000股,并批准5,000,000股“B-1”系列优先股,清算优先权相当于“A”和“B”系列优先股 。B-1系列优先股拥有投票权,并将在完成IPO交易时自动转换为普通股,这一点在公司修订和重新发布的公司注册证书中有定义。
于2023年1至9个月期间,本公司透过发行890,310股“B”系列优先股、私募发行1,680,084股“B”系列优先股及于2023年9月30日后发行74,922股“B-1”优先股及发行8,890份认股权证,共筹集费用净额4,620,000元。
2023年10月,公司修改并重述了其与特拉华州的公司注册证书,将其有表决权普通股的授权股份 增加到100,000,000股,每股面值0.00001美元,将其无投票权普通股的授权股份减少到30,000,000股,每股面值0.00001美元,并授权2,500股C系列优先股,每股面值0.00001美元。C系列优先股 在清算、解散、清盘或其他情况下,优先于普通股和无投票权普通股,低于A系列优先股、B系列优先股和B-1系列优先股。C系列优先股在IPO前没有投票权 ,IPO结束时每股拥有13,000个投票权。C系列优先股无权获得股息, 具有每股18.00美元的清算优先权,可根据持有人的选择随时将1:1转换为普通股,并且在IPO结束时,如果转让,将自动将1:1转换为普通股。
| 7. | 共享 认购协议 |
于2021年11月12日,本公司与若干投资者订立股份购买协议,出售最多100,000,000股普通股(“合计限额”)。该协议取决于公司能否实现其普通股的公开上市。协议的主要条款包括不迟于公开上市后一年 到期的总限额2%的承诺费,以及在公开上市时向投资者发行的五年期认股权证,以购买相当于本公司总股权的4%的普通股,以a)上市时的每股价格或b)700,000,000美元的商数除以股权总数(完全稀释的普通股 股份)中的较小者为准。除非提前终止,否则本公司可要求在本协议上市后的五年内提取普通股或向投资者出售普通股。所要求的减持金额受减持前30天内公司普通股交易量的限制,每股价格相当于同期每股平均价格的90%。如果公司是以私下销售交易方式出售的,而不是完成其股票的公开上市,则必须向投资者支付1%的费用。
| F-26 |
| 8. | 所得税 税 |
A 使用联邦法定所得税率计算的所得税优惠与公司实际所得税率的对账如下:
| 截至2023年9月30日的9个月 | 截至9个月
个月 2022年9月30日 | |||||||
| 联邦法定利率 | (21.0 | )% | (21.0 | )% | ||||
| 永久性物品 | — | — | ||||||
| 真实上年度净营业亏损递延税项资产 | — | (8.7 | ) | |||||
| 其他变化 | 0.2 | (1.2 | ) | |||||
| 更改估值免税额 | 20.8 | 30.9 | ||||||
| 总计 | 0.0 | % | 0.0 | % | ||||
公司递延税项净资产的 组成部分如下:
| (单位:千美元) | 2023年9月30日 | 2022年12月31日 | ||||||
| 递延税项资产: | ||||||||
| 净营业亏损结转 | $ | 1,862 | $ | 896 | ||||
| 租赁责任 | 378 | 435 | ||||||
| 资本化研究与开发 | 614 | 299 | ||||||
| 衍生负债 | — | 31 | ||||||
| 应计负债 | 124 | 81 | ||||||
| 股票薪酬 | 99 | 17 | ||||||
| 递延税项资产 | 3,077 | 1,759 | ||||||
| 递延税项负债: | ||||||||
| 租赁使用权资产 | (400 | ) | (462 | ) | ||||
| 未摊销债务贴现 | — | (31 | ) | |||||
| 递延税项负债 | (400 | ) | (493 | ) | ||||
| 减去:估值免税额 | (2,677 | ) | (1,266 | ) | ||||
| 递延税项净资产 | $ | — | $ | — | ||||
该公司最初是一家有限责任公司,2021年12月转变为特拉华州的一家公司。由于在截至2022年12月31日和2021年12月31日的年度内产生净营业亏损,公司在截至2022年12月31日和2021年12月31日的年度内没有所得税支出。截至2023年9月30日,该公司的美国联邦净营业亏损(“NOL”)为8,867,000美元。联邦NOL无限期结转,在所有权权益发生某些累积 变化的情况下,可能会受到年度限制。这可能会限制每年可用于抵销未来应纳税所得额或纳税义务的纳税属性数量。 随后的所有权变更可能会进一步影响未来几年的限制。
对于2021年12月31日之后开始的纳税年度,纳税人必须资本化根据国内税法(IRC)第174条被认为是研究和试验(R&E)活动附带 的任何费用。 尽管纳税人历来可以根据IRC第174条选择扣除这些费用,但2017年12月的减税和就业法案 要求对2021年12月31日之后的纳税年度的R&E费用进行资本化和摊销。在美国与R&E活动有关的费用必须在5年内摊销,而在美国以外发生的R&E费用必须在15年内摊销。R&E活动的范围比IRC第41条(与研究税收抵免有关)下考虑的合格研究活动 更广泛。在截至2022年12月31日的年度,本公司根据现有指引进行了 分析,并确定即使在对其R&E费用进行了必要的资本化 和摊销后,该公司仍将继续处于亏损状态。该公司将继续关注这一问题的未来发展,但它预计 R&E资本化和摊销不会要求它现在或在不久的将来支付现金税款。本公司已计入这一拨备的影响,截至2023年9月30日,该拨备产生的递延税项资产约为61.4万美元。
| F-27 |
管理层已评估对本公司递延税项净资产变现有影响的正面及负面证据,并确定本公司更有可能不会确认递延税项净资产的利益。因此,公司 已在2023年9月30日和2022年12月31日记录了全额估值津贴。本公司将继续评估未来其递延税项资产的可变现能力,并将根据需要调整估值拨备。
截至2023年9月30日和2022年12月31日,本公司没有不确定的税务头寸。本公司确认与 未确认税收优惠相关的利息和罚款均为所得税支出的组成部分。自成立以来,本公司没有记录任何未确认的税收优惠的利息或罚款。
| 9. | 租赁、承付款和或有事项 |
本公司已根据ASC 842-20-25-2选择了一项会计政策,不在简明资产负债表中记录短期租赁,其定义为12个月或以下的租赁。出于财务报表的目的,租赁项下记录的租金费用以直线基础确认 基于可获得的最新合同条款的租赁期限。
截至2021年12月31日,公司已签订了两份实验室和办公空间的短期租赁协议。该公司于2022年3月30日选择延长其其中一项实验室和办公空间租约的租期,租约续期的开始日期为2022年5月1日。延长的租赁期为126个月,并根据ASC 842租赁会计准则 入账为经营租赁。于延期时,应收使用权租赁资产及租户改善津贴合共2,799,000美元及租赁负债2,799,000美元。本租赁于2022年7月31日终止,剩余的使用权资产、应收租户改善津贴和租赁负债余额 已注销。
公司于2022年7月1日扩大了范围,将临时实验室和办公空间的剩余租赁期限延长了6个月,并于2022年8月1日和2022年10月7日进一步扩大范围,将每月许可费提高到每月1.5万美元 。2023年1月,本公司将本租约再延长六个月,并于2023年6月底到期。这一临时实验室和办公空间的租约被计入短期租约。
本公司于2022年10月订立为期62个月的写字楼租赁协议,于2027年11月30日届满。 本租约将按ASC 842租赁会计指引入账为营运租赁。使用权租赁资产和 租赁负债各为2,293,000美元,于租赁期开始时以7.5%的贴现率入账。
2023年6月,该公司签订了一份新的租约,为其研究业务提供临时实验室和办公空间。本租赁期限为12个月,月租金为6,000美元,将作为短期租赁入账。本租约于2023年8月开始。2023年9月,公司 签订了一份关于增加空间的租约修正案,每月租金增加到7000美元。
截至2023年9月30日和2022年9月30日的9个月的租金支出分别为504,000美元和198,000美元。截至2023年9月30日,经营租赁项下的不可取消租赁付款为2,104,000美元,短期租赁项下的不可取消租赁付款为65,000美元。
截至2023年9月30日, 剩余期间和未来五年的未来最低付款如下(以千为单位):
| (单位:千美元) | ||||
| 2023 | $ | 86 | ||
| 2024 | 477 | |||
| 2025 | 488 | |||
| 2026 | 544 | |||
| 2027 | 509 | |||
| 此后 | — | |||
| 租赁付款总额 | 2,104 | |||
| 减去:推定利息 | (304 | ) | ||
| 租赁总负债 | 1,800 | |||
| 减去:流动租赁负债 | (353 | ) | ||
| 非流动租赁负债总额 | $ | 1,447 | ||
| F-28 |
| 10. | 相关的 方交易 |
截至2021年12月31日,本公司有一家尚未清偿的关联方母公司,应付金额为225,000美元。这笔债务由FibroGenesis持有,应于2022年4月1日到期,并可由FibroBiologics酌情选择延期六个月。 公司于2022年4月偿还了剩余母公司应付的225,000美元。2022年7月,该公司向母公司 提供了30万美元的一年期无息票据贷款。2022年10月,公司向母公司借出6万美元一年期无息票据,该票据已于2022年12月31日前偿还。2023年4月,FibroGenesis全额偿还母公司应收账款30万美元。
如附注6所述,公司通过专利转让和知识产权交叉许可协议从FibroGenesis收购了某些正在进行的研发和专利资产。专利转让协议将FibroGenesis的某些专利的权利、所有权和权益 转让给本公司进行进一步开发。《知识产权交叉许可协议》授予本公司对FibroGenesis在有限使用领域拥有的专利的独家权利,这些专利包括a)脊柱疾病、障碍或状况、b)癌症、c)骨科疾病、障碍或状况、 和d)多发性硬化症的诊断、治疗、预防和缓解。
2023年1月,本公司 与其母公司FibroGenesis签订了关于第一谈判权的协议(“ROFN协议”)。作为交换,FibroGenesis同意修改公司注册证书,以a)在公司首次公开募股、直接上市或出售时消除A系列优先股$35,000,000清算优先权,b)使B系列优先股清算优先权等于 与“A”系列优先股,以及c)规定在首次公开募股、直接上市或出售公司时,A系列优先股将被免费取消 FibroBiologics同意在公司首次公开募股、直接上市或出售之前向FibroGenesis支付对FibroBiologics的任何股权投资所得毛收入的15%。此外,如果FibroGenesis决定向外部许可其任何技术,FibroBiologics将获得为期五年的第一次谈判权。2023年1月,公司修订了公司注册证书以反映这些变化,记录了预期未来向FibroGenesis支付的衍生品负债2,573,000美元,并向FibroGenesis支付了323,000美元,以支付公司于2022年12月发行的股权所得毛收入的15%。根据如上所述其与A系列优先股的关系,衍生负债首先计入母公司净投资,然后在母公司净投资取消后计入额外实收资本。在2023年的前九个月,该公司通过股票发行筹集了16,418,000美元的总收益,并向 FibroGenesis支付了2,322,000美元。超出衍生产品负债的金额在简明经营报表中记为其他亏损。 截至2023年9月30日,没有衍生产品负债,应支付给FibroGenesis的金额为14.1万美元。
| 11. | 基于股份的薪酬 |
公司于2022年8月10日通过,股东于2022年8月18日批准的《2022年股票计划》(以下简称《计划》)。该计划规定授予激励性股票期权、非法定股票期权、股票增值权、限制性股票奖励、限制性股票单位奖励和其他股票奖励。本计划通过授予股票奖励,旨在帮助公司获得并保留合格获奖者的服务,激励该等人士为公司的成功尽最大努力,并提供一种手段,使符合资格的获奖者可以从普通股的增值中受益。2022年9月,公司根据本计划向员工、董事和科学顾问 董事会成员共发行了101,250份期权,执行价为每股3.28美元。2023年2月,公司根据本计划向员工和董事额外发行了总计3,689,750份执行价格为每股2.28美元的期权。2023年8月,共有2,500个期权被没收,执行价为每股3.28美元 。一般而言,本公司授予的奖励为期四年,其行使价格等于董事会在考虑由独立第三方估值公司编制的 公司普通股同期估值后确定的普通股的估计公允价值。
截至2023年9月30日和2022年12月31日,根据该计划,可供未来发行的股票分别为8,711,500股和12,398,750股。
| F-29 |
以库存为基础的 薪酬费用在简明经营报表中确认如下:
截至 前九个月 9月30日, | ||||||||
| (单位:千美元) | 2023 | 2022 | ||||||
| 研发 | $ | 196 | $ | 100 | ||||
| 一般、行政和其他 | 1,137 | 133 | ||||||
| 基于股票的薪酬总支出 | $ | 1,333 | $ | 233 | ||||
截至2022年9月30日的9个月的基于股票的薪酬支出,包括向董事会和顾问发行的无投票权普通股的16.8万美元支出。
截至2023年9月30日,与未归属奖励相关的未确认的基于股票的薪酬成本以及预计确认这些成本的加权平均期间 如下:
| 股票 期权 | ||||
| 未确认的基于股票的薪酬支出(千) | $ | 5,470 | ||
| 待确认的预期加权平均期间薪酬成本(年) | 3.2 |
本公司股票期权活动摘要如下:
| 股票期权 | 加权平均行权价 每股 | 加权-平均剩余合同寿命(年) | 聚合内在价值(以 千为单位) | |||||||||||||
| 截至2022年12月31日的未偿还债务 | 101,250 | $ | 3.28 | 9.7 | — | |||||||||||
| 授与 | 3,689,750 | $ | 2.28 | 10.0 | — | |||||||||||
| 已锻炼 | — | $ | — | — | — | |||||||||||
| 被没收/取消 | 2,500 | $ | 3.28 | 9.0 | — | |||||||||||
| 截至2023年9月30日未偿还 | 3,788,500 | $ | 2.36 | 9.4 | — | |||||||||||
| 自2023年9月30日起可行使 | 56,298 | $ | 3.28 | 9.0 | — | |||||||||||
授予员工、董事和顾问的股票期权的公允价值在授予之日使用Black-Scholes期权定价模型进行估算,该模型采用以下假设:
| 假设: | 九个 个月 告一段落 2023年9月30日 | |||
| 无风险利率 | 3.9 | % | ||
| 预期波动率 | 90 | % | ||
| 预期期限(年) | 7.0 | |||
| 预期股息 | 0 | % | ||
于截至2023年9月30日止九个月内,授出购股权之加权平均授出日期公允价值为每股1.80美元。
| 12. | 后续 事件 |
本公司已评估截至2023年11月27日(即未经审核简明财务报表可供发出的日期)的后续事件,并已确定除附注2及6及以下所披露的事项外,并无其他事项需要披露未经审核简明财务报表或对其作出调整。
于2023年11月,本公司向认购B-1系列优先股股份的投资者共发行14,859股额外股份及10,321份认股权证。
| F-30 |
2024年1月23日
到2024年2月25日(包括我们普通股上市后的第25天),所有交易这些证券的交易商,无论是否参与此次发行,都可能被要求提交招股说明书。