目录表
直接改变肿瘤的免疫表型。肿瘤免疫表型的改变是独一无二的,并通过一种我们称为抗原播种的机制利用ETB固有的细胞内路由特性。
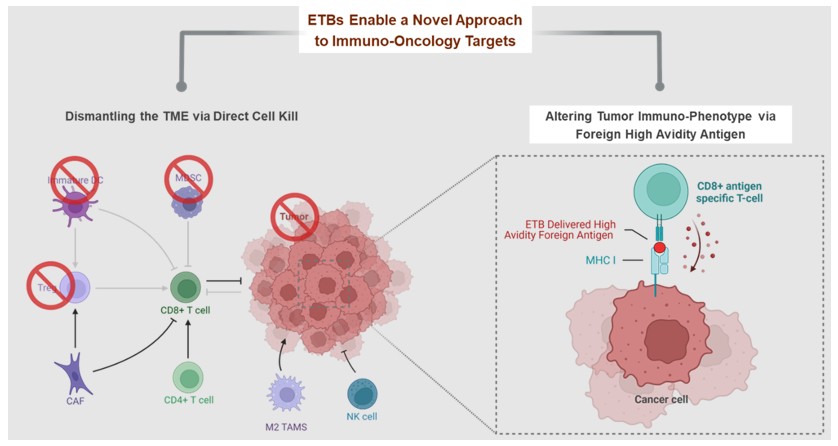
免疫肿瘤学ETBS
MT-6402-ETB针对PD-L1
我们于2020年12月提交了针对我们针对PD-L1的ETB MT-6402的研究新药(IND)申请,并于2021年1月接受了IND申请。MT-6402在PD-L1表达肿瘤的复发/难治性患者中的I期研究于2021年7月开始,起始剂量为16微克/公斤。MT-6402的第一阶段研究是一项多中心、开放标签、剂量递增和剂量扩大试验。确诊为PD-L1表达肿瘤或在TME中证实PD-L1表达的患者有资格参加登记,无论是HLA型还是CMV状态。2021年11月,MT-6402获得快速通道指定,用于治疗表达PD-L1的晚期非小细胞肺癌(NSCLC)患者。
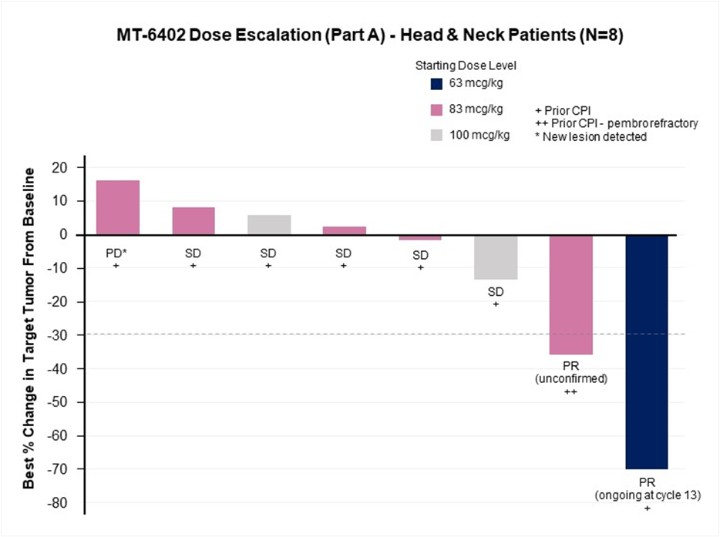
在MT-6402对复发/难治性肿瘤患者的研究中,48名患者接受了16、24、32、42、63、83和100微克/公斤的7个剂量递增队列的治疗。我们继续观察药效学(PD)效应,包括耗尽PD-L1+单核细胞、MDSCs、PD-L1+树突状细胞,以及伴随的T细胞激活。到目前为止,还没有观察到与药物有关的4级或5级不良事件。MT-6402在复发/难治性患者中的单一治疗活性也被观察到。
MT-6402第一阶段研究的A部分剂量升级已经完成。100微克/公斤的剂量被认为是不可耐受的,因为3级皮疹和1级高敏感性肌钙蛋白升高的两种剂量限制毒性(DLT)均未出现临床后遗症,导致药物中断超过两周。皮疹和高敏感性肌钙蛋白升高是免疫相关的不良事件,已被批准的检查点疗法记录在案。63和83微克/公斤剂量将在B部分剂量扩展研究中进一步探讨。
在A部分剂量递增中,10例头颈部癌症患者按63、83或100微克/公斤的剂量进行治疗。头颈部肿瘤的TME通常富含免疫抑制细胞。其中两名患者由于早期进展而无法评估第1周期DLT期,分别在仅接受一剂或两剂MT-6402后停止研究。其余8名头颈部癌症病人的最佳反应如下:
28