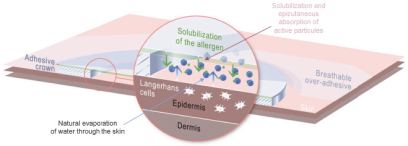
| • | 水的积累溶解了过敏原。由于这个冷凝室,表皮变得更具渗透性,允许过敏原进入表皮。 |
| • | 一旦进入表皮,过敏原就会被一群高度专业化的细胞捕获:朗格汉斯细胞。 这些细胞可以在皮肤表面提取蛋白质,对其进行加工,并将其表位呈现给淋巴结中的淋巴细胞。 |
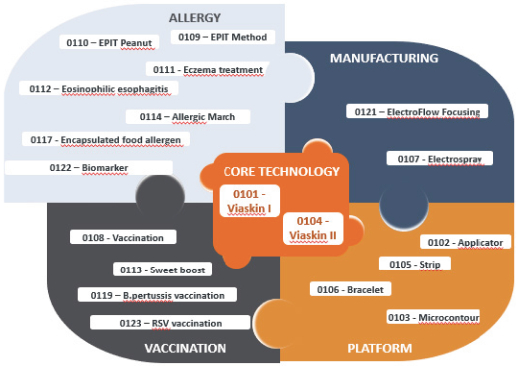
我们的候选产品
我们的产品开发战略是基于利用Viaskin的科学形象。我们选择目标候选产品的目的是解决具有高度未满足医疗需求的 过敏问题。下表总结了我们候选产品的当前开发状况:
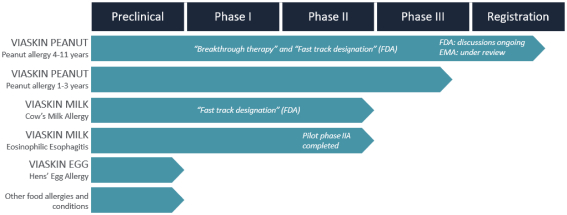
| * | 美国FDA突破性治疗和儿童快速通道指定。 |
| ** | 美国FDA在两岁及以上儿童患者中的快速通道指定。 |
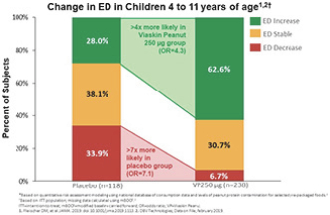
为4-11岁儿童提供维亚斯金花生
我们的主要候选产品Viaskin花生已经完成了全球第三阶段开发计划,用于治疗4至11岁的花生过敏患者 。该计划包括以下研究:
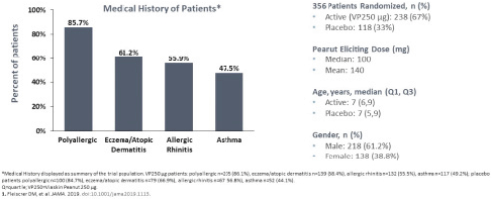
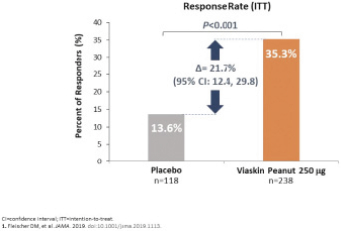
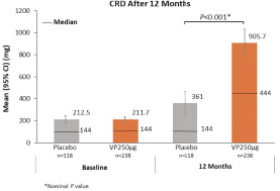
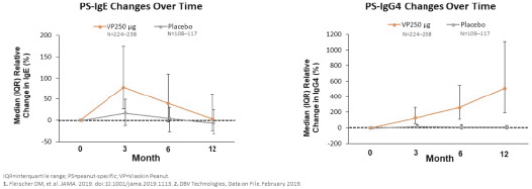
| • | PEPITES(P坚果EPIT E效率和 S安全研究),一项随机、安慰剂对照的关键III期试验,调查治疗12个月后维亚斯金花生250微克在356名患者中的安全性和有效性。 |
| • | 实现(EPIT的实际使用和安全),这是一项随机、安慰剂对照的第三阶段试验,旨在 在6个月的盲目治疗后产生安全数据,并评估Viaskin花生250µg在常规临床实践中的使用情况。 |
5