目录表
对于患者,我们向FDA和我们的合作伙伴美国国防部提供了最新的分析。vt.给出联邦新冠肺炎突发公共卫生事件于2023年5月结束,根据FDA、美国国防部和其他顾问对该计划的监管要求、成本、时间表和成功潜力的反馈,我们决定不提交紧急使用授权(EUA)或sNDA。
新冠肺炎治疗发明和疫苗加速试验2/3期试验(ACTV-4宿主组织试验)由美国国立卫生研究院/国家心肺血液研究所进行并赞助,是一项针对住院患者对新冠肺炎的宿主反应的随机安慰剂对照试验。Activ-4宿主组织试验评估了福斯塔替尼在大约600名新冠肺炎住院患者中的目标人群,300名福斯塔替尼与300名安慰剂。2023年第一季度,数据和安全监测委员会(数据安全监测委员会)完成了对试验的中期分析,并建议继续试验。2023年9月,DSMB建议ACTV-4宿主组织试验平台的福斯塔替尼研究分支停止登记。基于dsmb对条件功率分析的审查,dsmb确定,在住院和新冠肺炎氧疗的患者中,福斯塔替尼提供与主要结果(无氧天数)或其他次要结果相关的益处的可能性极低。没有发现任何安全问题。NIH/NHLBI同意DSMBS的建议,并已要求试验调查人员停止登记,完成对已登记的参与者的随访,并完成研究结束。将按先前计划对全部研究数据进行分析和传播。
新冠肺炎对我们业务的当前和潜在未来影响的最新情况
新冠肺炎疫情对我们的业务和运营产生了不利影响。尽管世界卫生组织在2023年5月宣布新冠肺炎PHE结束,但另一场全球大流行未来可能在多大程度上影响我们的业务和运营以及财务状况,将取决于高度不确定、超出我们知识或控制的事态发展。因此,我们无法确定它对我们业务未来可能产生的影响的全部程度。另请参阅本季度报告10-Q表的“第I部分,第1A项,风险因素”,了解与新冠肺炎大流行相关的风险和不确定性的更多信息。
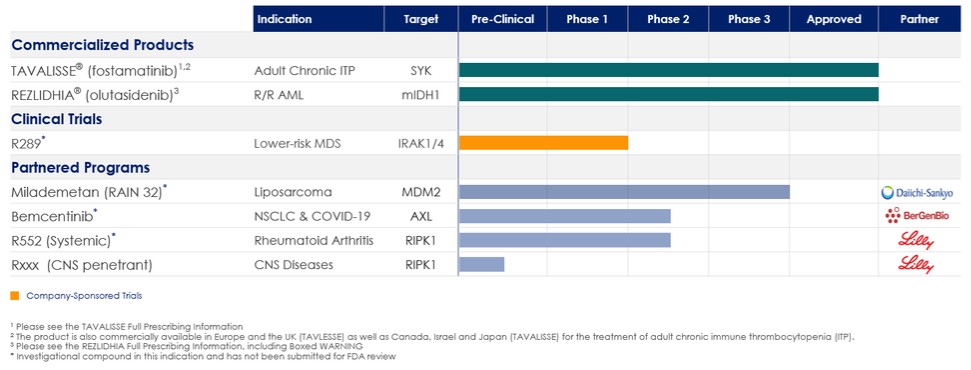
我们的产品组合
下表总结了我们的投资组合:
25