目录表
我们正在筹备的所有项目都处于临床前开发阶段,总结如下:
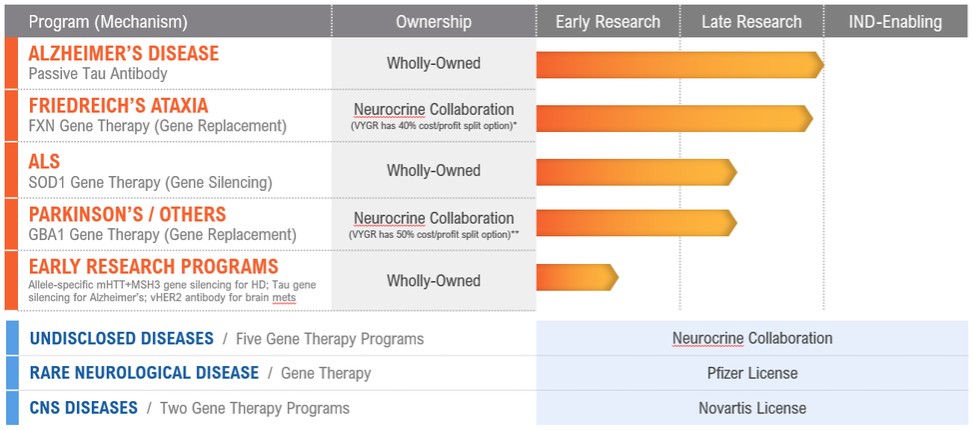
*在第一阶段读出后,旅行者公司可以选择:(1)根据60/40的成本和利润分享安排(Neurocrine/Voyager)与Neurocrine Biosciences在美国共同开发和共同商业化,或(2)授予Neurocrine Biosciences在美国的全部商业权,以换取基于美国销售的里程碑式付款和特许权使用费。
*第一阶段读出后,旅行者公司可以选择:(1)根据50/50的成本和利润分享安排,在美国与Neurocrine Biosciences共同开发和共同商业化,或(2)授予Neurocrine Biosciences在美国的全部美国商业权,以换取基于美国销售的里程碑式付款和特许权使用费。
抗Tau抗体方案治疗阿尔茨海默病
疾病概述
我们正在开发专有抗体,选择性地靶向并减少病理性tau的传播,用于治疗tauopathy,我们的主要适应症是阿尔茨海默病(AD)。Tau病理的传播与AD的疾病进展和认知能力下降密切相关,在美国约有600万人受到影响,并给社会带来越来越大的医疗负担。最近,抗淀粉样蛋白抗体已被批准用于治疗阿尔茨海默病,但仍有大量药物需求未得到满足。
我们的治疗方法
长期以来,我们一直致力于开发专有的和互补的方法来干扰tau病理的进展,据信tau病理是AD和其他tauopathy的核心。减少有毒的tau聚集体可能会减缓这些疾病的疾病进展和认知能力下降。我们正在探索被动使用我们的抗tau抗体。我们的抗tau抗体具有不同的特性,包括改进了对tau蛋白特定区域的靶向,与第一代方法相比,可以提供更好的轮廓。我们相信,我们针对C末端的抗体与其他方法有很大的不同。此外,我们相信,在IND申请获得批准后,利用正电子发射断层扫描(PET)对人类tau进行成像的临床评估,加上测量血浆和脑脊液生物标记物,有可能实现有效和加速的人类生物学证据演示。
临床前研究
在2022年8月举行的阿尔茨海默病协会国际会议上,我们展示了我们专有的抗tau抗体的数据,该抗体针对的是具有高亲和力的中间结构域和C末端,并在小鼠模型的临床前研究中显示出良好的生物物理特性和强大的活性。在P301S播种繁殖互作中
10