依据第424(B)(4)条提交
注册号码333-249020
招股说明书

8,563,300股美国存托股份
购买936,700股美国存托股份的预先出资认股权证
相当于7600万股普通股
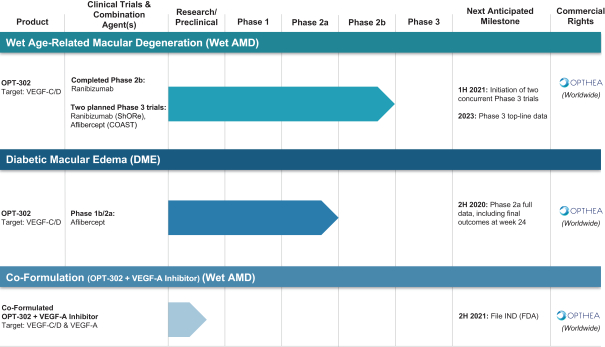
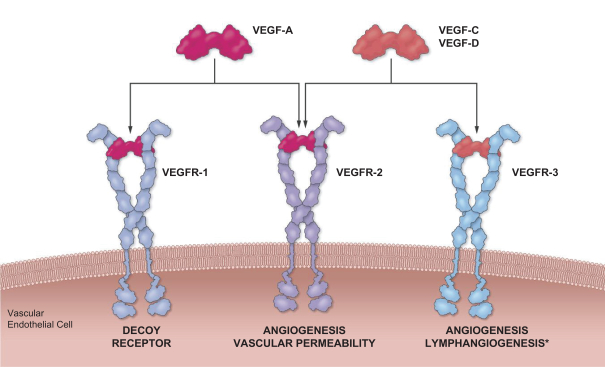
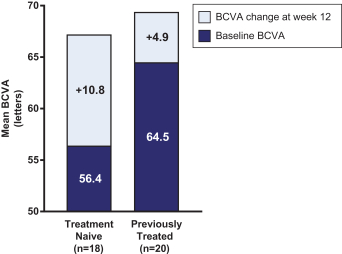
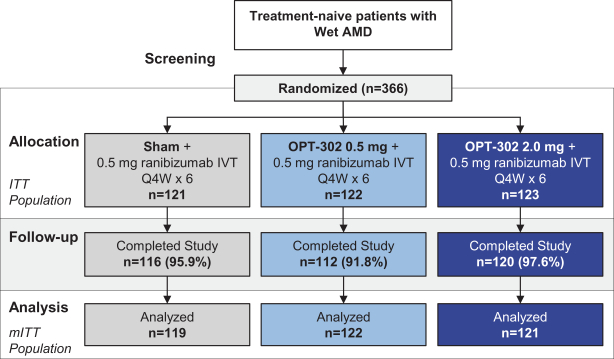
这是8,563,300股美国存托股份(ADS)在美国的首次公开发行,相当于Opthea Limited的68,506,400股普通股。每个美国存托股份代表8股没有面值的普通股,存放在纽约梅隆银行作为托管机构。
我们还向某一买家提出购买936,700股美国存托凭证,相当于Opthea Limited的7,493,600股普通股,以代替美国存托凭证。每一份预先出资的权证将可行使一份美国存托股份。每份预资金权证的购买价将等于 美国存托股份在此次发行中向公众出售的价格减去0.00001美元,每份预资金权证的行使价为每美国存托股份0.00001美元。预资资权证将可立即行使,并可随时行使,直至所有预资资权证全部行使为止,但须受澳大利亚法律在预资资权证的描述中所载的某些条件所规限。本次发售亦涉及在行使本次发售的预资资权证后可发行的美国存托凭证(及美国存托凭证相关普通股) 。
我们已授予承销商购买至多1,425,000份美国存托凭证的选择权,以弥补超额配售。
在此次发行之前,美国存托凭证尚未公开上市。这两只美国存托凭证已获准在纳斯达克全球精选市场上市,交易代码为?OPT。这些预先融资的权证目前还没有成熟的公开交易市场,我们预计市场也不会发展起来。此外, 我们不打算申请在任何国家证券交易所或其他国家认可的交易系统上市预融资权证。
我们的普通股在澳大利亚证券交易所挂牌上市,代码为JOPT。2020年10月16日,我们普通股在澳大利亚证券交易所的最后一次公布销售价格为每股普通股2.78澳元,相当于每股美国存托股份15.73美元,此前澳元兑美元汇率于2020年10月16日生效,澳元兑美元汇率为1.4134澳元至1美元。美国存托股份到普通股份比例为1比8。
投资我们的证券涉及高度风险 。见本招股说明书第12页开始的风险因素。
我们是一家新兴的成长型公司,因为该术语在2012年的JumpStart Our Business Startups Act中使用,因此, 已选择遵守某些降低的上市公司报告要求。
美国证券交易委员会和美国任何州证券委员会都没有批准或不批准这些证券,也没有确定本招股说明书是否真实或完整。 任何相反的陈述都是刑事犯罪。
| 每个美国存托股份 | 每个Pre- 资金支持 搜查令 |
总计 | ||||||||||
| 公开发行价 |
美元 | 13.50000 | 美元 | 13.49999 | 美元 | 128,249,991 | ||||||
| 承保折扣和佣金 |
美元 | 0.94500 | 美元 | 0.94500 | 美元 | 8,977,499 | ||||||
| 向Opthea Limited出售扣除费用前的收益(1) |
美元 | 12.55500 | 美元 | 12.55499 | 美元 | 119,272,491 | ||||||
| (1) | 我们已同意向保险商赔偿某些费用。见承销。 |
承销商预计在2020年10月20日左右将美国存托凭证和预筹资权证交付给买家。
联合簿记管理经理
| 花旗集团 | SVB Leerink |
主管经理
| 奥本海默公司 | Truist证券 |
2020年10月16日。