| • | 一种精确的肿瘤学方法:在任何有益和可行的情况下,我们都追求精确 方法,最初选择我们认为最有可能在涉及严重或危及生命的情况下做出反应的患者,在这些情况下,存在重大的未得到满足的医疗需求。在这些适应症中,我们认为,根据我们临床前研究的 结果,我们的候选产品可能会被考虑用于FDA的加速审批过程,因为它们可能证明对合理地可能预测 临床益处的替代终点有影响。 |
下表总结了我们的候选产品渠道:

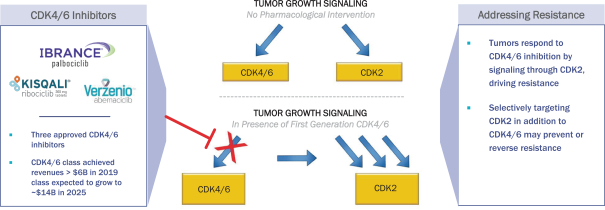
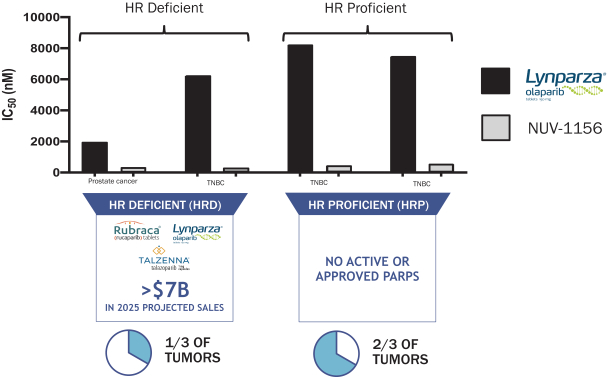
我们的主要候选产品NUV-422被设计为第二代脑穿透性CDK4/6抑制剂,具有抑制CDK2的额外能力,CDK2是一种细胞周期检查点,在包括胶质母细胞瘤(GBM)在内的许多高级别胶质瘤患者中被发现改变,也被认为是乳腺癌对目前上市的CDK4/6抑制剂产生耐药性的 机制。NUV-422还被设计用来限制CDK1抑制,这是目前正在开发的第二代抑制剂的一个潜在毒性原因。目前上市的CDK4/6抑制剂在2019年产生了超过60亿美元的销售额,预计2025年将增长到140亿美元,这反映了这一治疗类别药物对患者的显著好处。 我们相信,避免CDK1抑制的CDK2/4/6抑制剂可以为更广泛的患者群体带来更大的好处。我们还在推进NUV-422,用于治疗乳腺癌患者和可能存在脑转移的其他癌症患者 ,FDA批准的CDK4/6抑制剂在这些患者中显示的疗效有限。此外,我们正在推进NUV-422,用于治疗激素受体阳性的转移性乳腺癌(ER+MBC)患者,CDK4/6抑制剂已经成功,但CDK2驱动的耐药性可能会产生。此外,我们还打算开发NUV-422治疗转移性去势耐受前列腺癌(mCRPCα)。FDA已经批准NUV-422作为孤儿药物,用于治疗恶性胶质瘤患者。我们于2020年12月开始对高级别胶质瘤患者进行NUV-422的1/2期临床试验。我们计划扩大到CDKN2A缺失的基因定义的高级别胶质瘤患者队列中, CDK2活性的遗传标记。CDKN2A基因缺失存在于近70%的高级别胶质瘤患者中。我们预计将于2022年报告该试验第一阶段的数据。
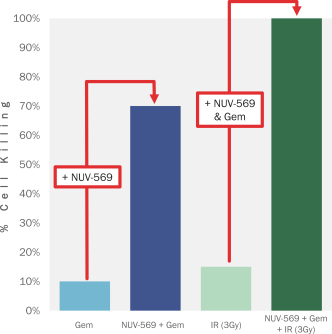
我们的第二个候选产品NUV-868是表观遗传 转录调节因子BET家族的选择性抑制剂。NUV-868特异性地抑制BRD4蛋白,BRD4是BET家族中的一个关键成员,它从表观上调节控制肿瘤生长和分化的蛋白。值得注意的是,BET 蛋白被认为是癌基因c-myc的重要调节因子。由于c-myc在高达70%的癌症中被认为是肿瘤生长的驱动因素,BET 抑制剂被认为是一种潜在的重要方法来抑制这种癌基因,而这种癌基因一直很难产生治疗药物。我们设计的NUV-868具有 避免目前正在开发的BRD4抑制剂的治疗限制毒性的特性,专注于优化BD2与BD1的选择性。一些第一代BET抑制剂对BD2的抑制作用仅为BD1的1.4-1.5倍 。这些第一代BET抑制剂已经被观察到有显著的毒性,特别是在胃肠道和骨髓。NUV-868 对BRD4的BD2亚域的抑制作用几乎是对BD1亚域的1500倍,我们相信这将提高其耐受性。我们计划在2021年下半年提交NUV-868的IND,并在2022年上半年启动急性髓系白血病(AML)和/或潜在的c-myc驱动实体瘤患者的第一阶段临床试验。
3