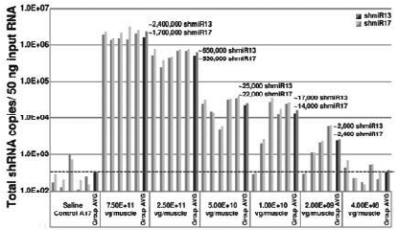
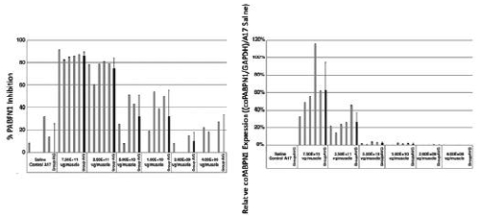
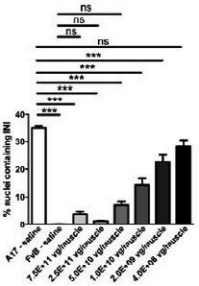
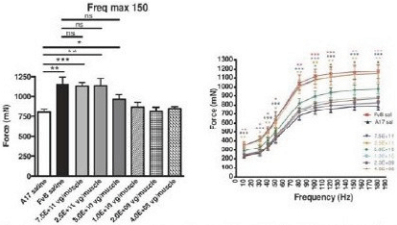
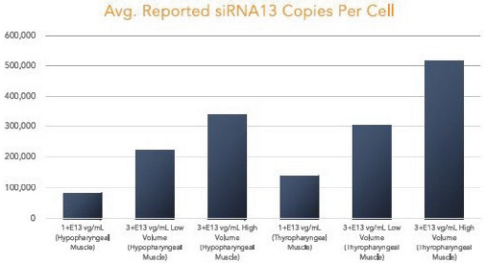
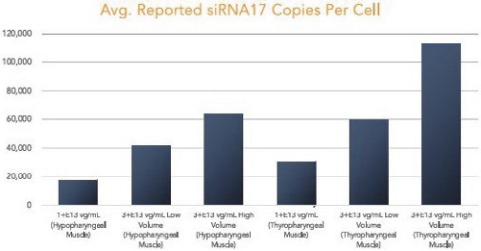
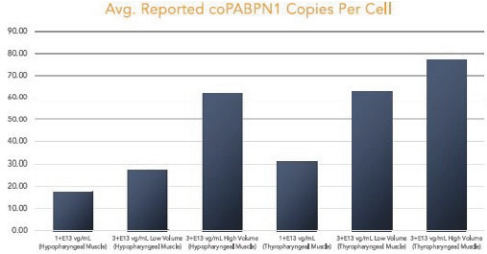
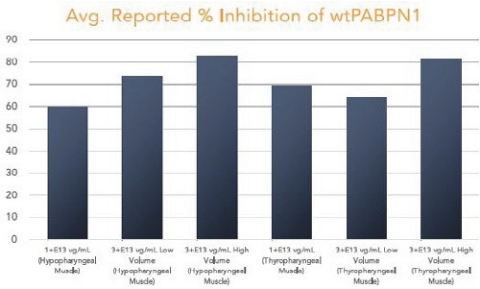
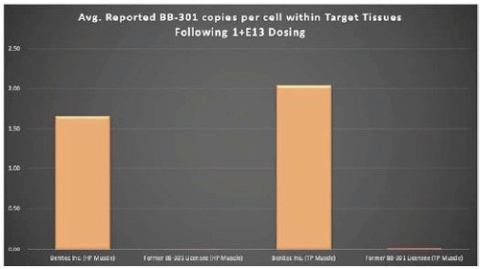
图1
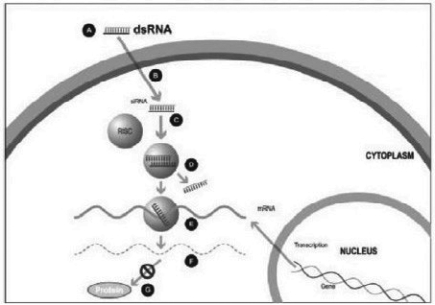
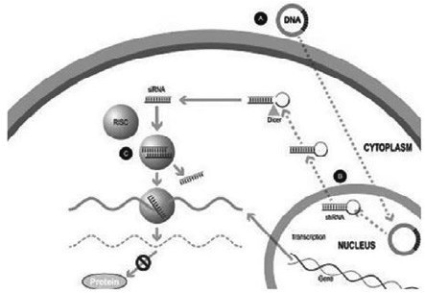
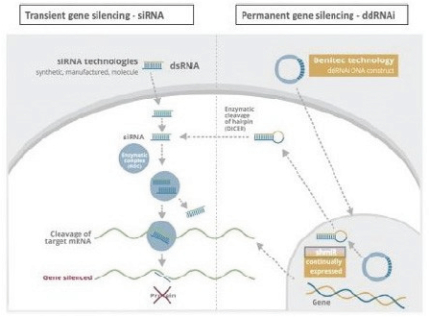
在实验室合成了一种小的双链RNA或dsRNA分子(A,图1),由一条称为正义链的链和另一条称为反义链的链组成,这两条链相互补充。这些小的dsRNA被称为小干扰RNA,或siRNA。正义链的序列对应于靶基因mRNA的一小段区域。SiRNA被传递到靶细胞(B,图1),在那里有一组酶,称为RNA诱导沉默复合体或RISC处理siRNA(C,图1),其中一条链(通常是正义链)被释放(D,图1)。RISC使用反义链来寻找具有互补序列(E,图1)的mRNA,从而导致目标mRNA(F,图1)的切割。因此,不会产生信使核糖核酸(蛋白质生产)的输出(G,图1)。包括Alnylam制药公司(“Alnylam”)在内的几家公司在他们的RNAi产品候选中使用了这种方法。
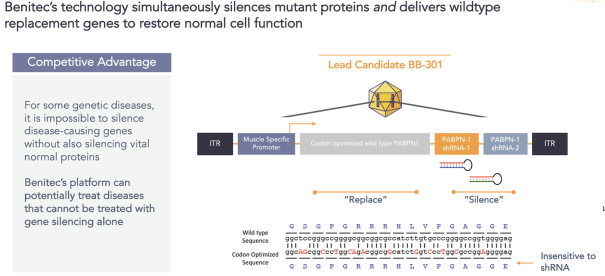
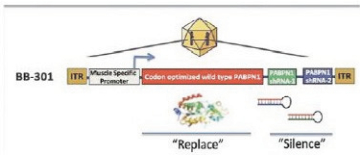
重要的是,许多遗传疾病不符合图1中概述的传统基因沉默方法,因为患病细胞可能产生感兴趣的野生型蛋白质和致病蛋白质的突变变体的混合物,潜在的基因突变可能太小,无法通过专门使用基于siRNA的方法选择性地靶向致病蛋白质变体。在这些情况下,在不同时沉默对正常细胞功能至关重要的野生型细胞内蛋白的情况下,选择性地沉默致病蛋白是非常困难的。
我们专有的沉默和替换技术利用了RNAi独特的特异性和强大的基因沉默能力,同时克服了基于siRNA的疾病管理方法的许多关键限制。
在标准的RNAi方法中,合成双链siRNA,然后通过对RNA的化学修饰或替代递送方法将其引入靶细胞。虽然通过使用这种方法已经在几个临床适应症中证明了有效性,但基于siRNA的方法仍然存在一些局限性,包括:
| • | 临床治疗需要重复给予以siRNA为基础的治疗剂多个周期以维持疗效; |
5