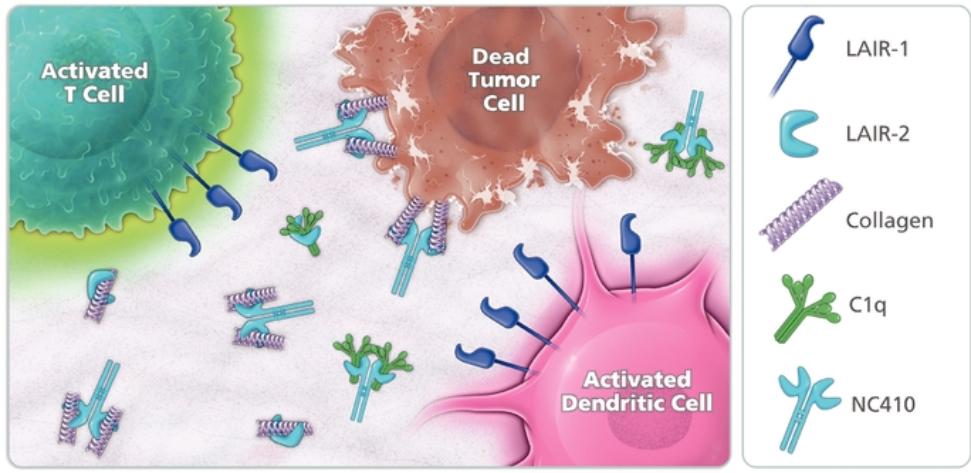
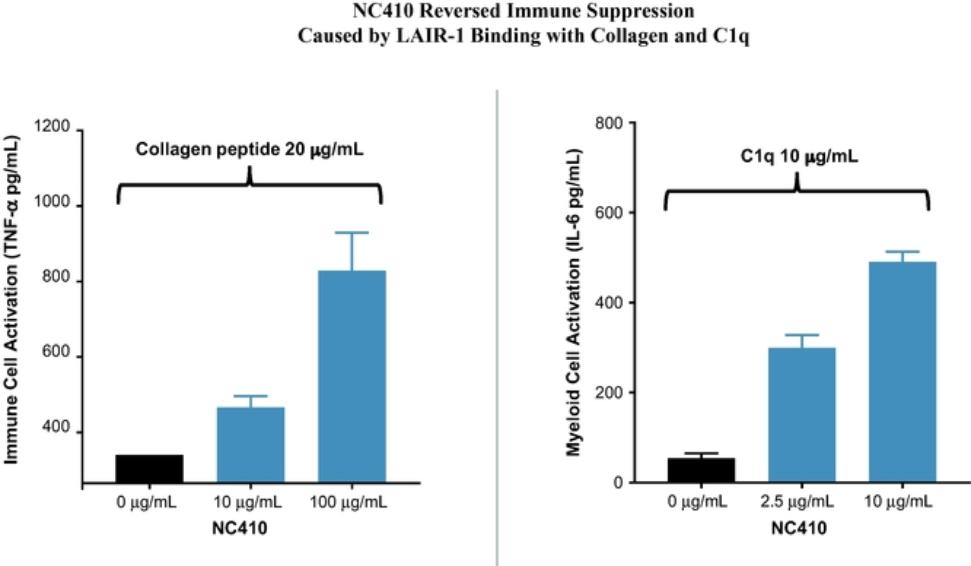
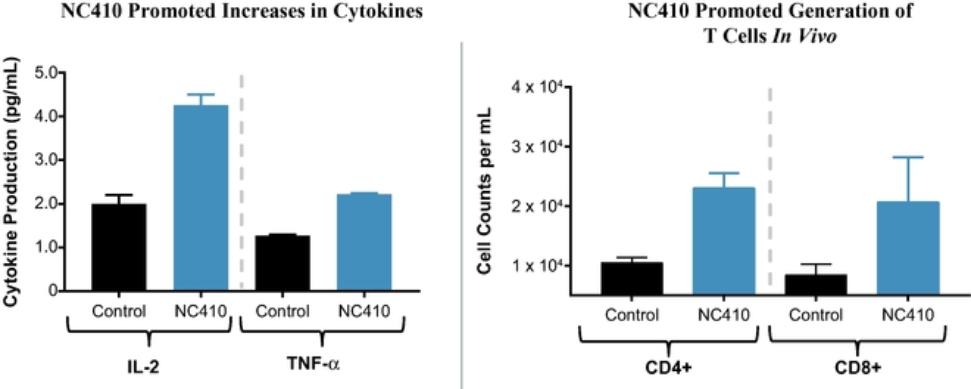
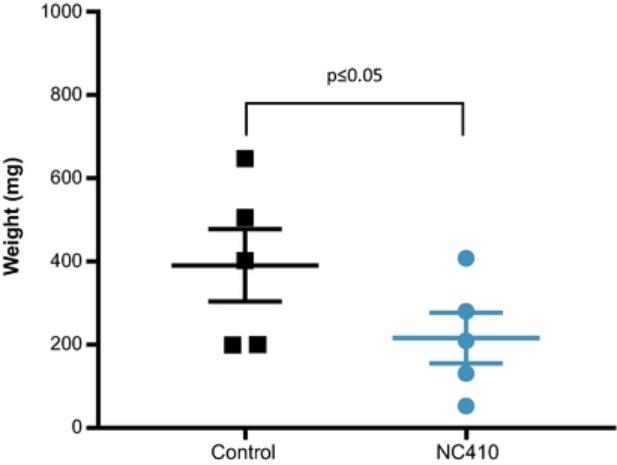
目录
实现更有效的抗癌免疫治疗。我们继续通过与耶鲁大学的独家赞助研究协议,与陈博士合作发现新的免疫药物。
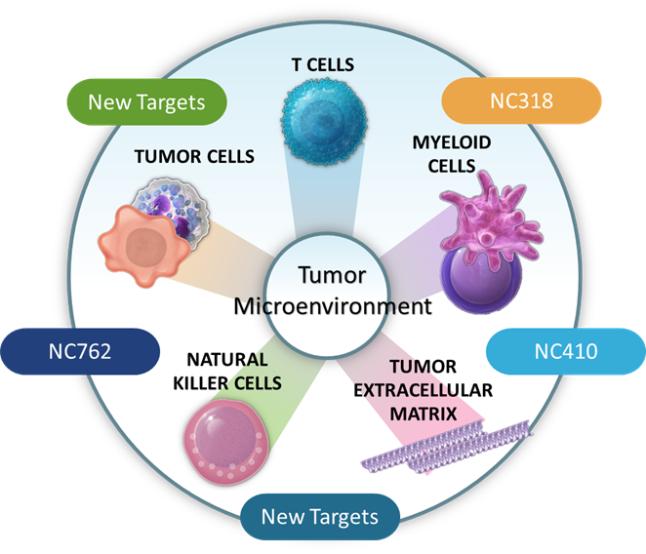
我们的管道
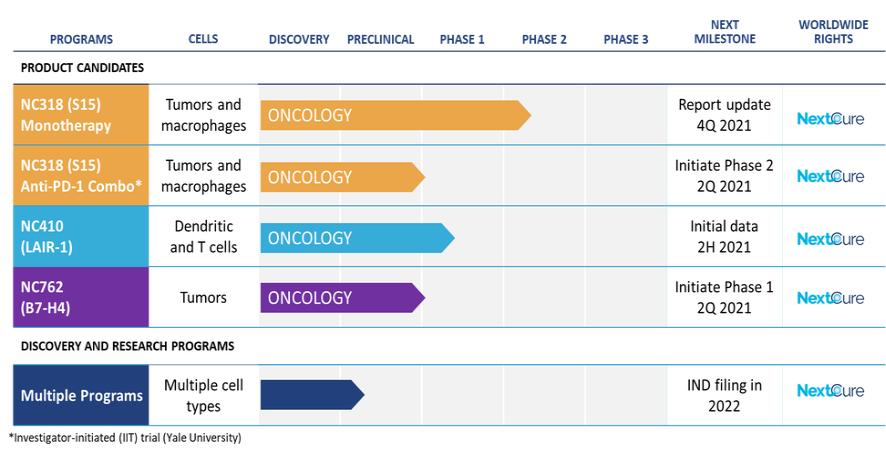
我们正在利用我们对生物途径的理解和我们的Find-IO平台来发现、验证和建立免疫医学候选药物的专有管道。下图详细介绍了我们的候选产品以及主要的发现和研究计划。他说:
我们的战略
我们的战略是利用我们完全集成的发现和产品开发基础设施,建立一条可持续的候选产品管道,以治疗那些目前可用的疗法没有得到充分服务的癌症患者。我们战略的主要内容包括:
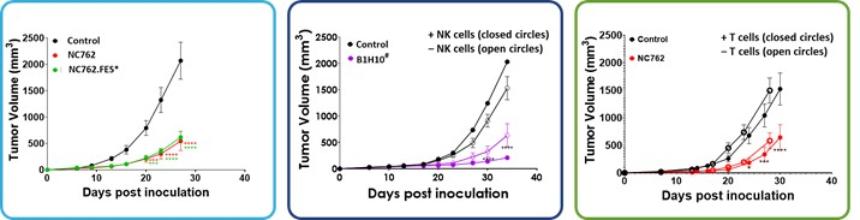
| ● | 推动我们的主要候选产品NC318、NC410和NC762的临床开发。2019年10月,我们开始招募患者参加1/2期临床试验的第二阶段,评估NC318在晚期或转移性肿瘤患者中的应用。此外,我们认为科学证据支持NC318与抗PD-1治疗相结合,耶鲁大学计划在2021年第二季度在NSCLC患者中启动该组合的第二阶段IIT。对于NC410,FDA在2020年第一季度接受了我们的IND,我们在2020年6月启动了1/2期临床试验的1期部分。对于NC762,我们在2021年第一季度提交了我们的IND,我们打算在2021年第二季度启动1/2期临床试验。 |
| ● | 建立针对无反应者的新免疫药物的新靶点的肿瘤学管道。我们将继续利用我们的免疫学专业知识和我们的Find-IO平台来确定与克服免疫抑制相关的新靶点。除了我们的内部发现工作外,我们还寻求利用我们与陈博士在耶鲁的实验室的关系,发现更多的免疫药物靶点。 |
| ● | 充分利用我们完全集成的开发、质量体系和cGMP制造能力。我们的方法是将产品开发的关键方面整合到我们的组织中。我们组建了一支在识别、表征和开发新型免疫药物方面拥有丰富经验的团队。我们寻求将发现重要目标与快速简化目标验证和进行关键的IND使能研究的能力结合起来,从而导致主要候选药物的临床开发。我们专门建造的、专用的、最先进的cGMP制造设施利用一次性使用技术来支持 |
8