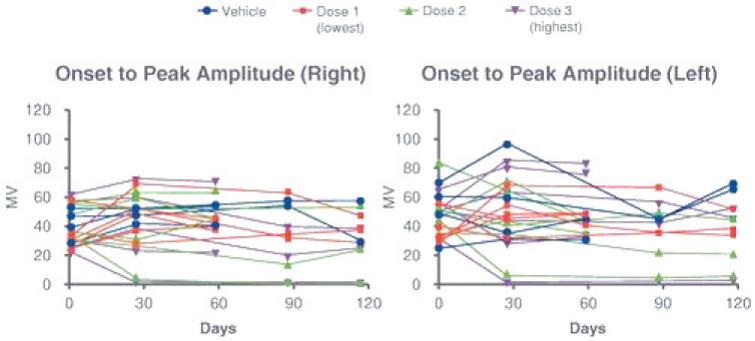
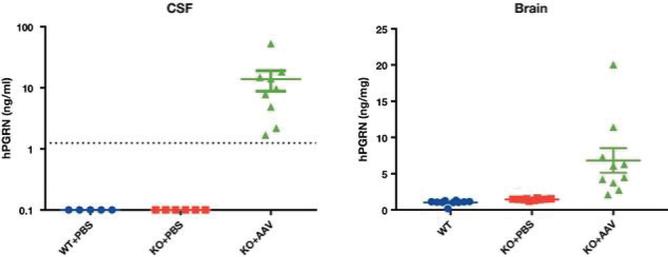
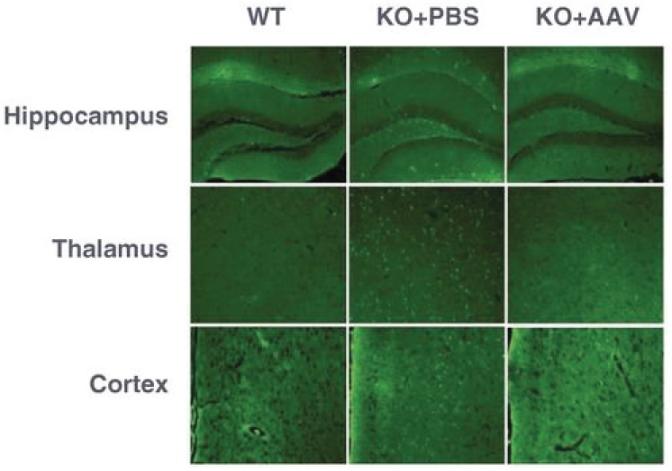
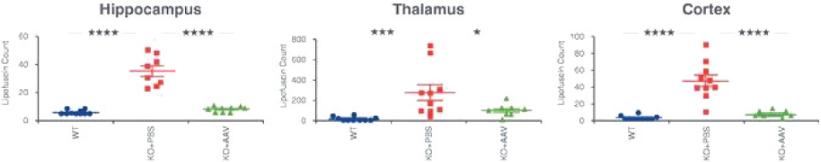
目录
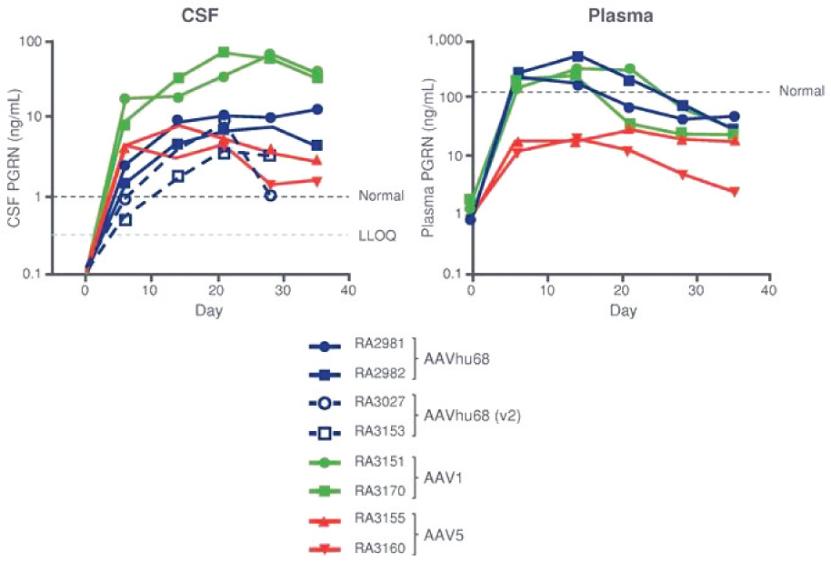
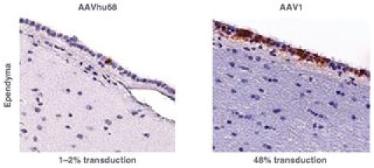
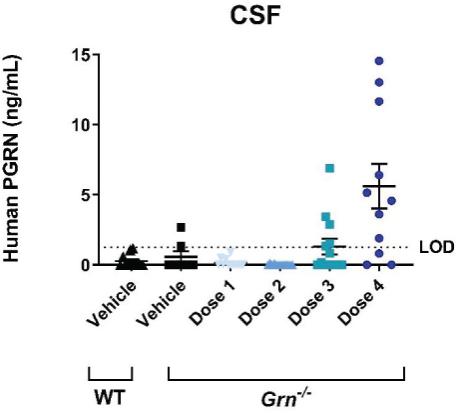
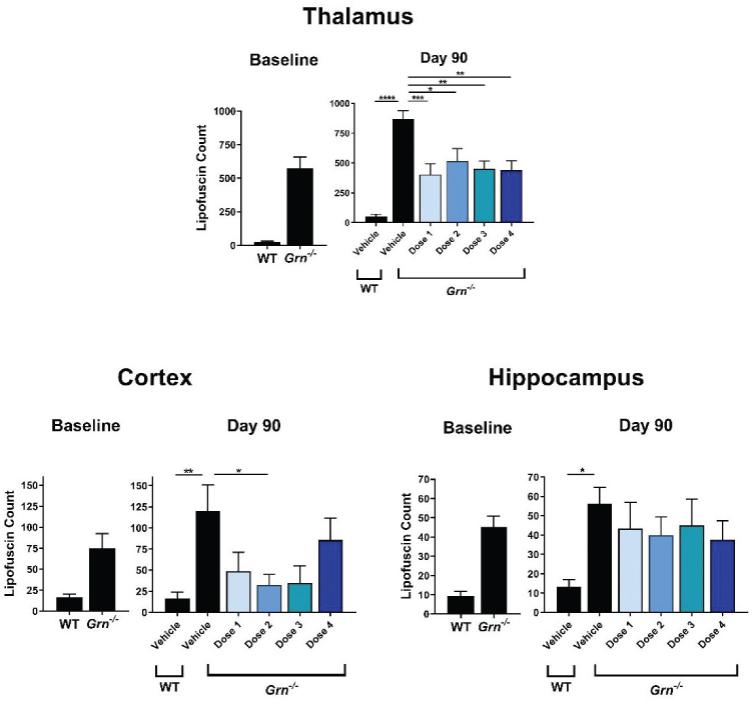
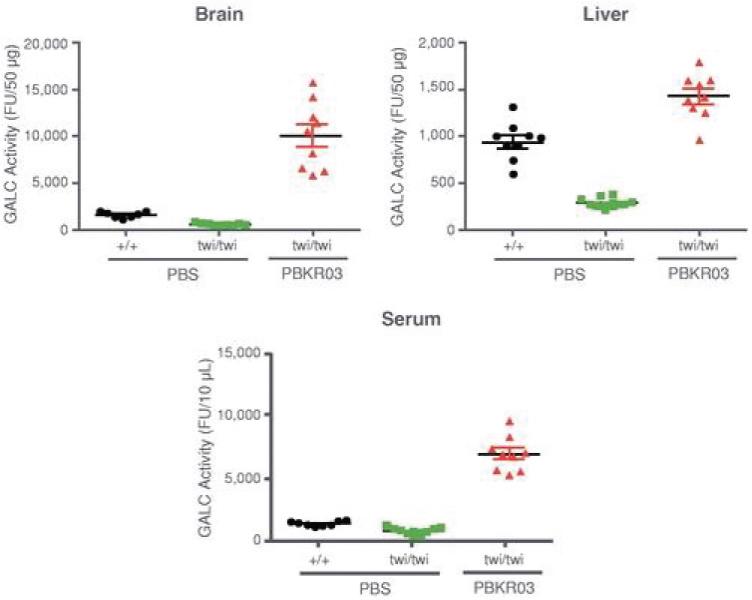
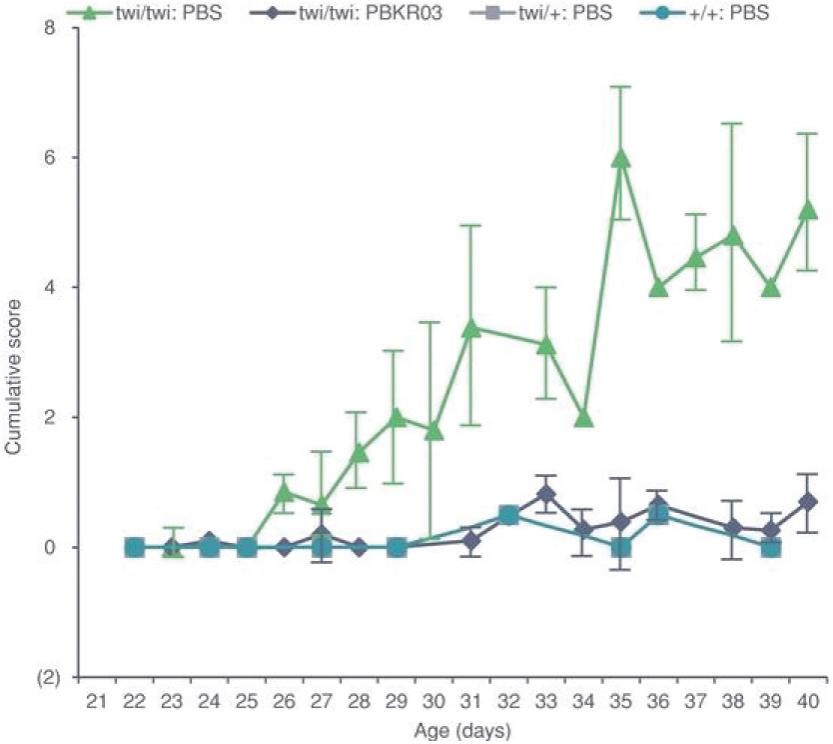
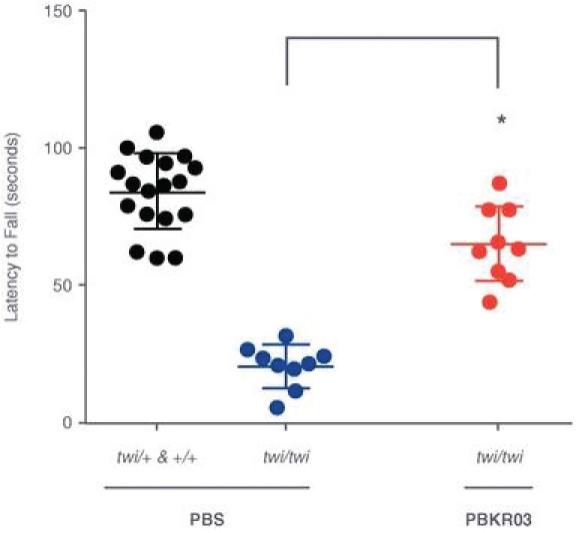
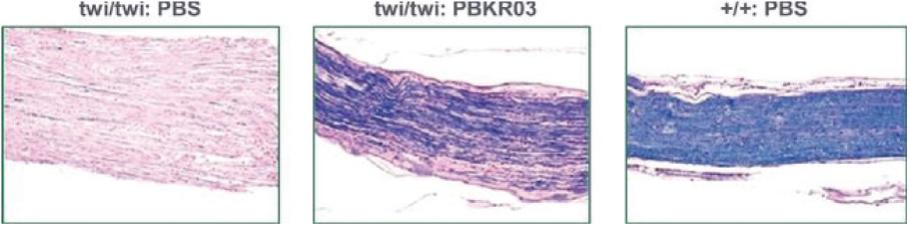
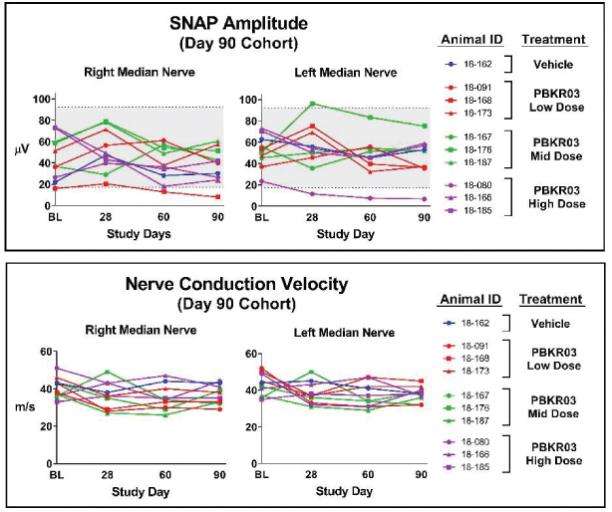
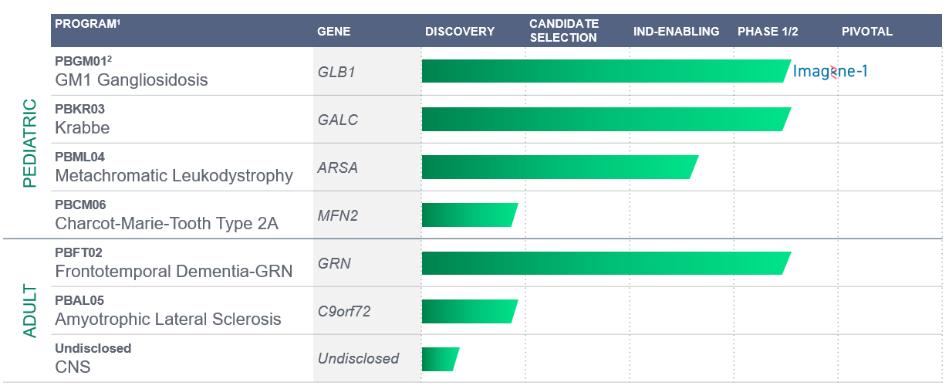
罕见的单基因中枢神经系统疾病的候选患者。我们的第一个候选产品PBGM01利用下一代AAVhu68衣壳向大脑和外周组织输送功能性的GLB1编码gm1溶酶体β-半乳糖苷酶或β-GAL的基因。我们的第二个候选产品PBFT02利用AAV1衣壳向大脑传递功能GRN编码原颗粒(或PGRN)的基因,用于治疗由原颗粒缺乏(FTD-GRN)引起的FTD。我们的第三个候选产品PBKR03利用下一代AAVhu68衣壳向大脑和外周组织输送编码水解酶半乳糖神经酰胺酶的功能基因,即GALC,用于克雷伯病。目前还没有针对这些疾病批准的疾病修正疗法。我们相信我们的主要候选产品有潜力为患者提供显著改善的结果,因为我们选择的枕大池内给药(ICM)给药路线是在颅颈交界处注射,我们的目标选择是利用交叉矫正机制的分泌蛋白,从而减少转导需求以及我们的衣壳和转基因选择过程,使我们能够选择适合特定适应症的载体。
我们还有四个处于研究阶段的计划:PBML04用于异色性脑白质营养不良,或MLD,PBAL05用于肌萎缩侧索硬化症,或ALS,PBCM06用于Charcot-Marie-Tooth 2A型,或CMT2A,以及一个用于未披露靶点的计划。PBML04针对的是MLD患者,这些患者在ARSAPBAL05针对的是在基因中有功能获得突变的肌萎缩侧索硬化症患者C9ORF72基因,PBCM06针对的是CMT2A患者,这些患者在Mfn2基因和我们未公开的计划针对的是成人中枢神经系统的适应症。我们还可以选择在2025年前以罕见的单基因中枢神经系统适应症授权宾夕法尼亚大学的另外10个项目。
我们由在基因药物、罕见疾病药物开发、制造和商业化方面拥有数十年集体经验的先驱和专家领导。我们的科学创始人斯蒂芬·斯奎托博士、詹姆斯·威尔逊博士和山田忠孝博士都是罕见病和基因医学领域的研发领域的世界领先者。威尔逊博士和山田博士与我们公司的持续关系以及对学术研究和临床药物开发的参与使我们能够更早地洞察为我们的商业战略提供指导的新兴技术。我们组建了一支团队,其成员在阿洛斯治疗公司、Biogen公司、葛兰素史克公司、杨森制药公司、Lycera公司、默克公司、Momenta制药公司、NPS制药公司、PharmAsset、Ultragenyx制药公司和ViroPharma等公司成功开发、制造和商业化罕见疾病和遗传药物产品方面拥有丰富的经验。
我们的管道
我们已经为罕见的单基因中枢神经系统疾病汇集了一系列基因药物候选产品,这些疾病的特点是高度未得到满足的医疗需求。随着该领域技术的进步,我们打算进一步扩大我们的产品组合,推出其他罕见的单基因中枢神经系统疾病的候选遗传药物产品,以及其他治疗方法。我们的发展计划包括:
110个额外的新管道许可证选项
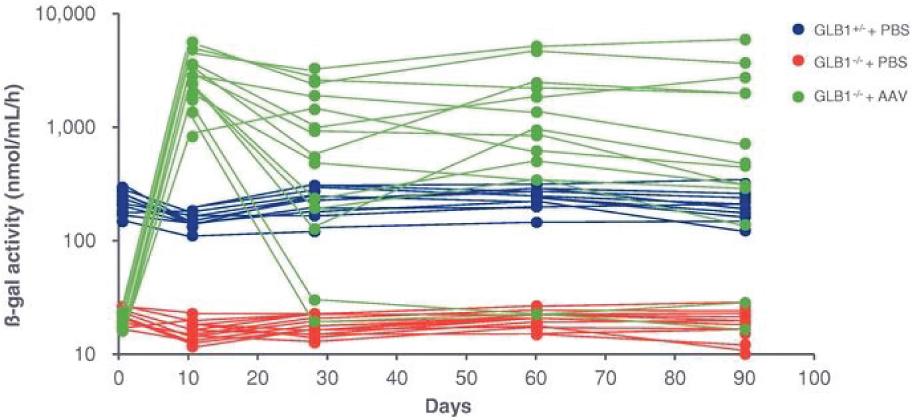
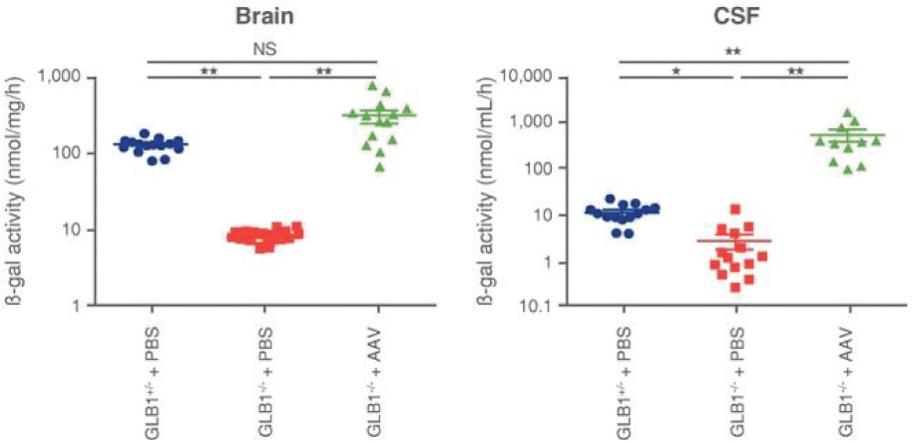
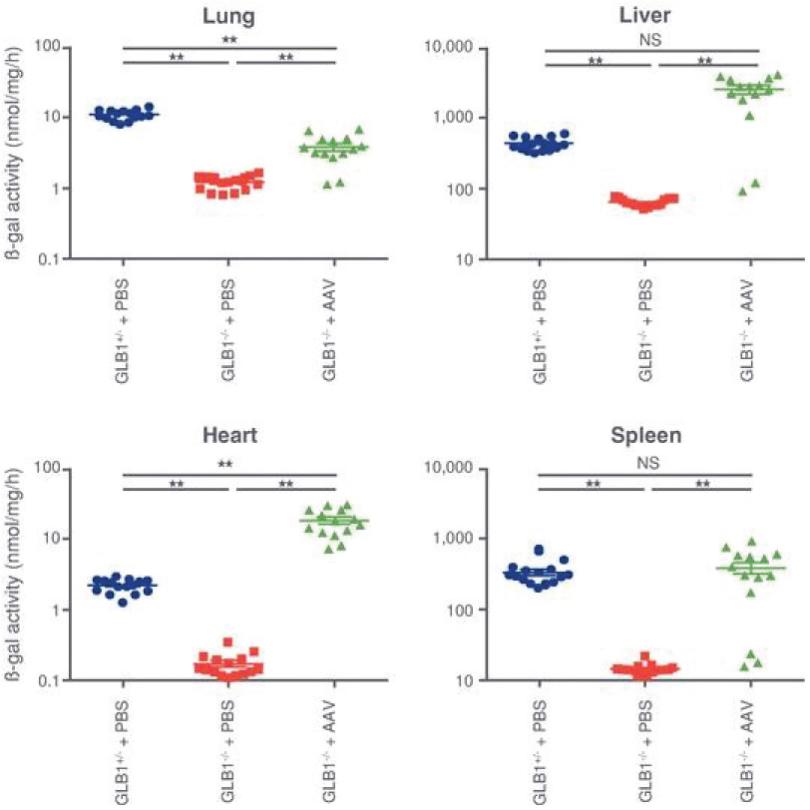
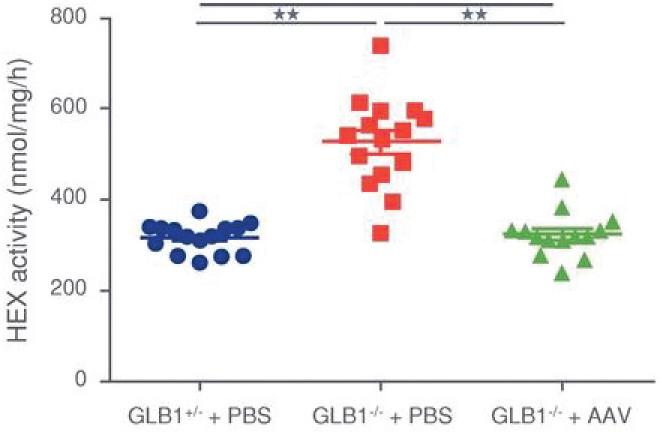
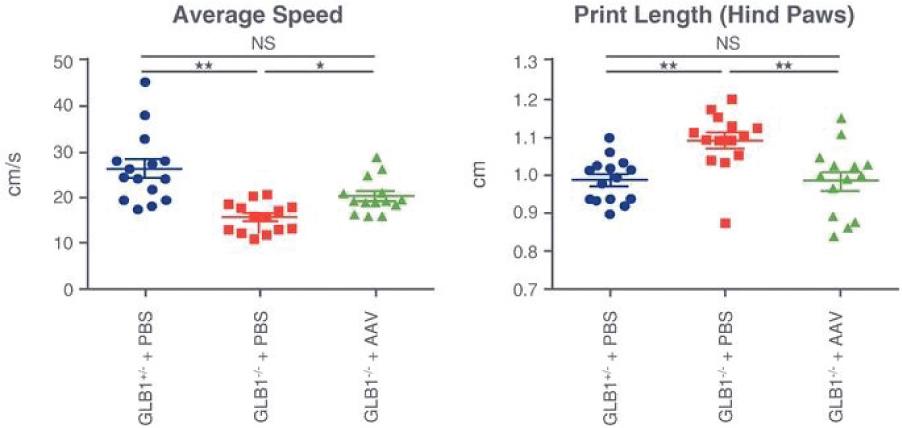
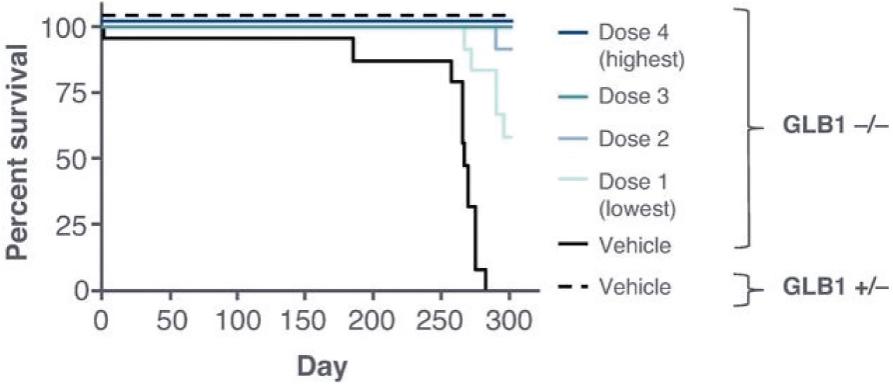
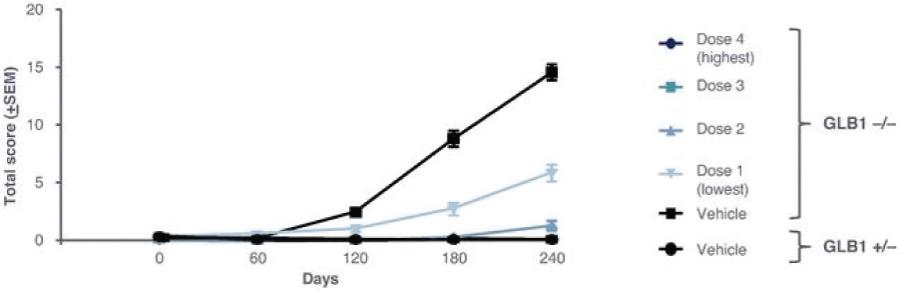
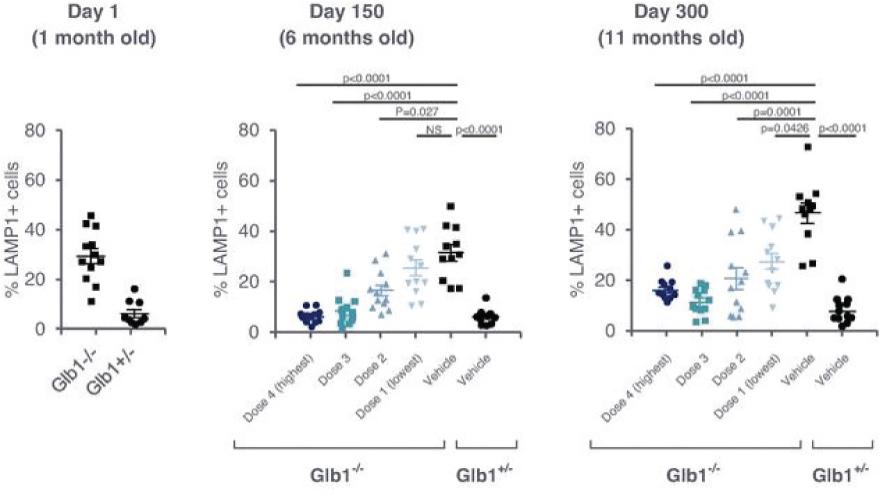
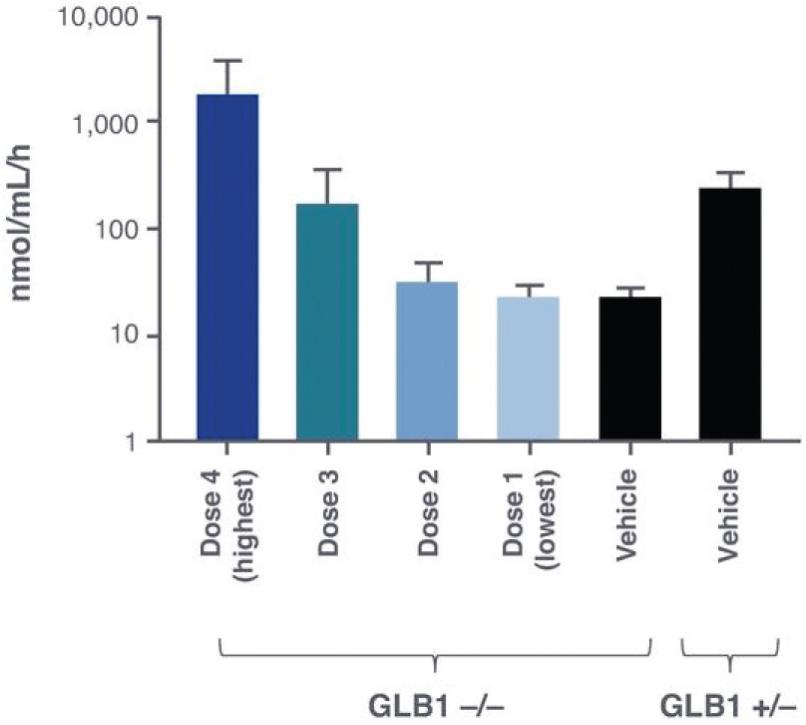
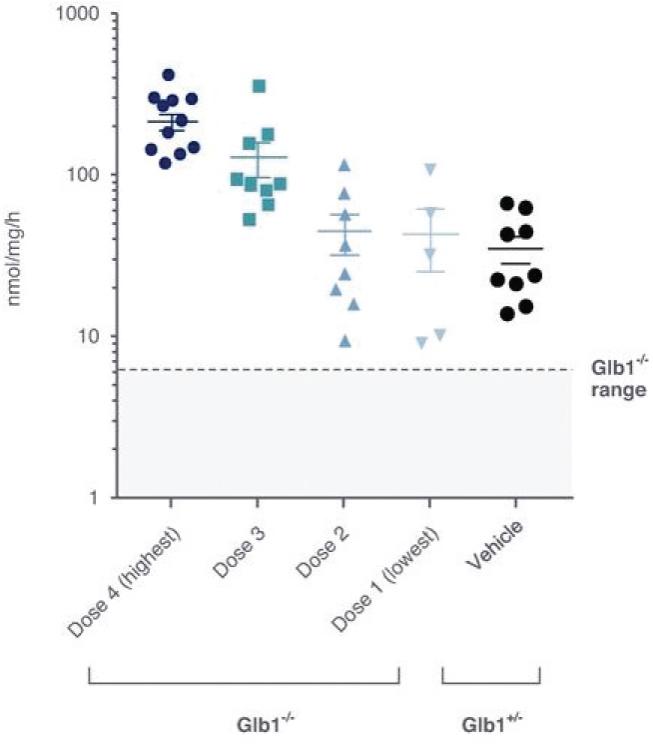
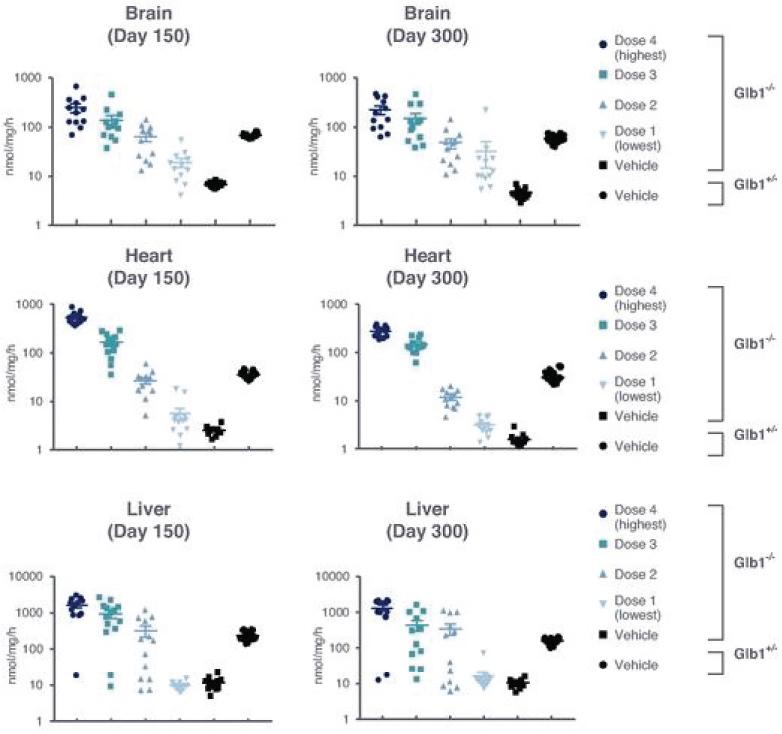
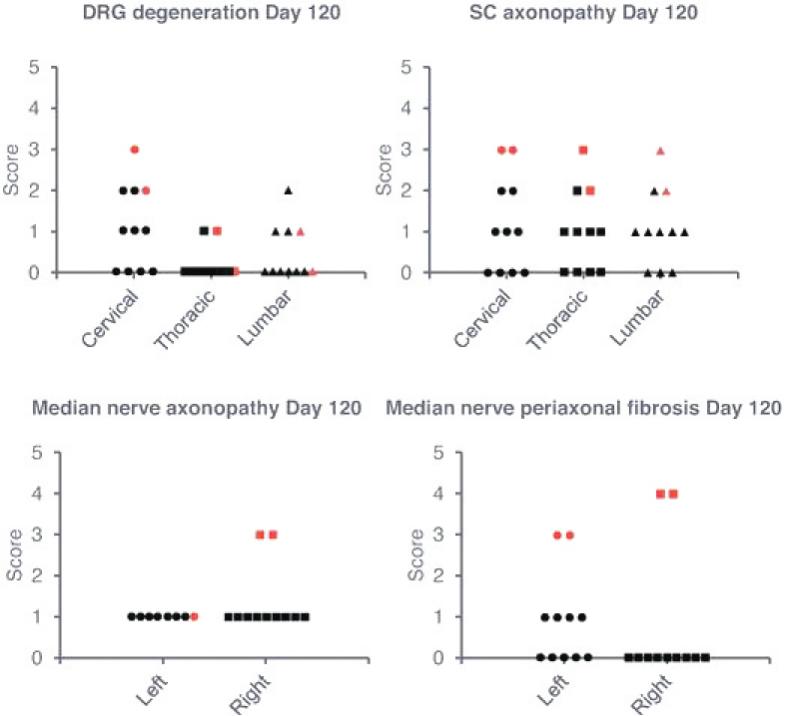
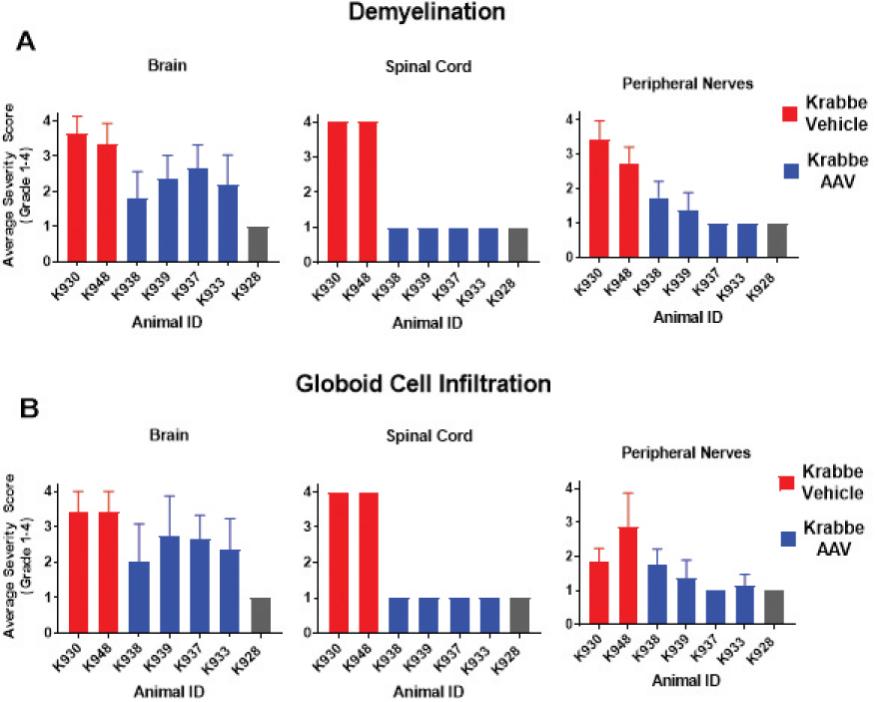
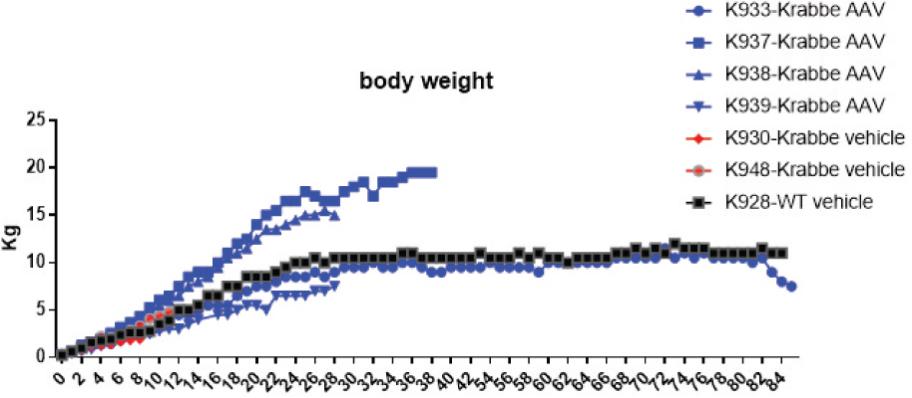
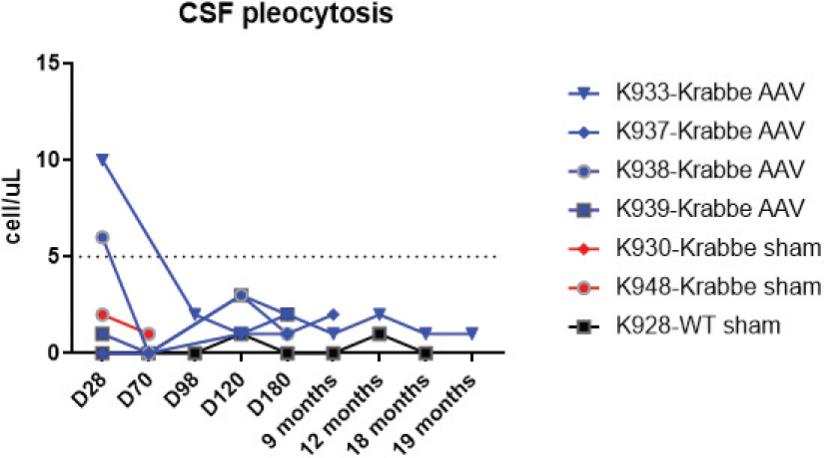
2 该计划包括正在进行的对婴儿和青少年GM1神经节苷脂沉着症患者的自然史研究。
6