目录表
我们的产品组合
下表总结了我们的投资组合:
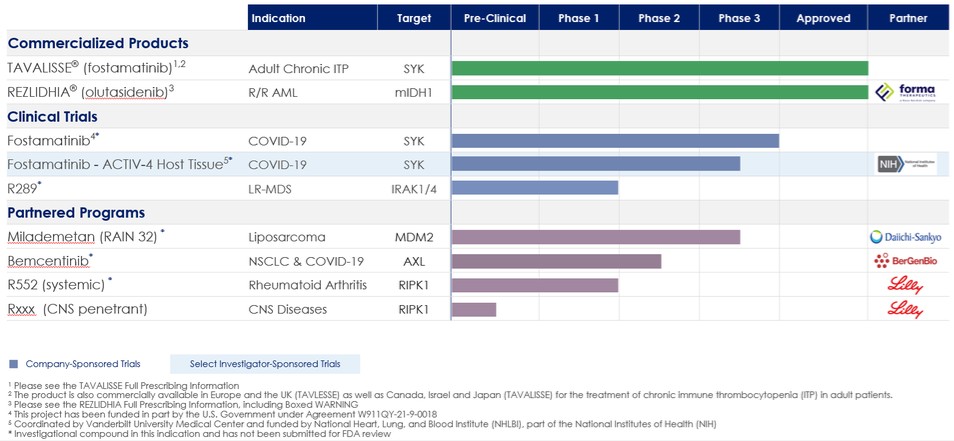
商业产品
他伐他利/福司他替尼在ITP中的应用
疾病背景。据估计,美国有81,300名成人患者患有慢性ITP。在ITP患者中,免疫系统攻击并破坏人体自身的血小板,后者在血液凝结和愈合中发挥积极作用。由于血小板计数低,ITP患者可能会遭受严重的瘀伤、出血和疲劳。目前治疗ITP的方法包括类固醇、模拟血小板生成素(TPO)的血小板生成增强剂和脾切除术。
口服福司他替尼程序。福斯塔替尼以片剂形式服用,可阻止免疫细胞内SYK的激活。ITP的典型特征是身体产生抗体,附着在血流中的健康血小板上。免疫细胞识别这些抗体,并将其附着在它们上,从而激活免疫细胞内的SYK酶,并触发抗体和附着的血小板的破坏。当SYK被福斯塔替尼抑制时,它会中断这种免疫细胞功能,使血小板逃脱破坏。在我们的第二阶段临床试验中,16名成人慢性ITP患者口服福斯塔替尼,结果发表在血样研究表明,福斯塔替尼显著增加了某些ITP患者的血小板计数,包括那些目前可用的其他药物失败的患者。
我们的福斯塔替尼治疗免疫性血小板减少症(FIT)第三阶段临床计划共有150名ITP患者,他们被随机分成两个相同的多中心、双盲、安慰剂对照临床试验。这些患者被诊断为持续性或慢性ITP,血小板计数始终低于每微升血液30,000。三分之二的受试者口服福斯塔替尼,每天两次,每次100毫克,另三分之一的受试者接受相同时间表的安慰剂。受试者预计将继续接受长达24周的治疗。在治疗的第四周,未能达到某些血小板计数和达到某些耐受性阈值的受试者,可以将他们的福斯塔替尼(或相应的安慰剂)剂量增加到150毫克,每日2次。该计划的主要疗效终点是在24周前出现稳定的血小板反应,在最后6次合格抽血中,至少有4次的血小板计数达到或超过每微升血液50,000。2016年8月,我们宣布了第一项FIT研究的结果,报告称福斯塔替尼达到了研究的主要疗效终点。研究表明,接受福斯塔替尼治疗的患者中有18%的患者实现了稳定的血小板反应,而接受安慰剂对照组的患者中没有患者。2016年10月,我们公布了第二项FIT研究的结果,报告了应答率(治疗组为16%,安慰剂组为4%)与第一项研究一致,尽管差异不具有统计学意义。在ITP中
26