目录
第 2 项。管理层对财务状况和经营业绩的讨论和分析。
您应阅读以下关于我们财务状况和经营业绩的讨论和分析,以及我们的财务报表以及本10-Q季度报告中其他地方包含的相关附注和其他财务信息。除历史财务信息外,本讨论和分析还包含基于当前预期的前瞻性陈述,涉及风险和不确定性,例如我们的计划、目标、预期、意图和信念的陈述。由于各种因素,包括下文第二部分第1A项下标题为 “风险因素” 的部分中列出的因素,我们的实际业绩可能与这些前瞻性陈述中的预期存在重大差异。
概述和管道
我们是一家临床阶段的遗传药物公司,专注于为中枢神经系统或中枢神经系统(CNS)的疾病开发变革性疗法,这些疾病的治疗选择有限或没有获得批准的治疗选择。我们的愿景是通过开发改变中枢神经系统疾病患者生活的开创性疗法来兑现基因疗法的承诺。遗传医学领域正在迅速扩大,我们相信我们在开发中枢神经系统疾病的治疗方法时采用了差异化的方法,这使我们能够选择和推进更有可能在技术和监管方面取得成功的候选产品。我们已经与宾夕法尼亚大学(Penn)的基因疗法项目(GTP)的受托人建立了战略研究合作,该项目由遗传药物领域的领导者詹姆斯·威尔逊博士领导。我们还利用与宾夕法尼亚大学孤儿病中心(ODC)的密切合作关系,制定历史和前瞻性可比的自然史患者档案,以便与介入试验的参与者进行比较。通过此次合作,我们组建了强大的遗传医学候选产品组合,我们保留了这些产品的全球版权,其详细信息如下表所示:
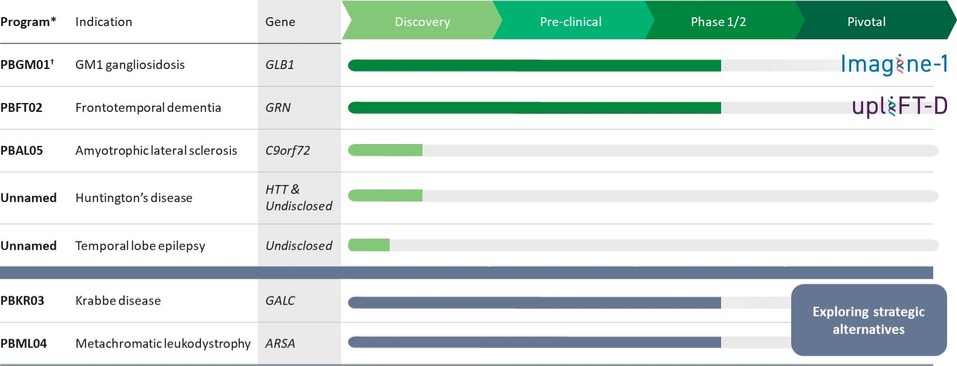
*还剩下8个额外的CNS管道许可选项;此前已行使了3种许可选项,随后权利归还给了宾夕法尼亚大学。
† 项目包括正在进行的对婴幼儿和青少年 GM1 神经节苷脂症患者的自然史研究
22