目录表
在肿瘤学方面,我们正在推进基于SNAPvax癌症免疫治疗平台或SNAPvax CI的资产,该平台共同交付肿瘤抗原和免疫刺激剂(TLR7/8a),用于治疗晚期癌症的异种Prime-Boost方案。与其他癌症免疫治疗候选药物相比,SNAPvax CI旨在提供独特的优势,因为它可以重复使用,参与癌症免疫周期的每个阶段(T细胞启动和扩张;肿瘤炎症;T细胞招募;以及T细胞许可),从而潜在地实现治疗效益的最大化。
在肿瘤学之外,我们正在开发基于SNAPvax耐受免疫治疗平台或SNAPvax TI的资产,以使用已知的自身抗原治疗自身免疫性疾病。SNAPvax TI含有自身抗原和免疫调节剂(例如:,mTOR抑制剂)特异性地诱导调节性T细胞,这对维持对自身抗原的耐受性至关重要。其独特的设计允许转运到淋巴器官和局部组织,在这些组织中,Treg诱导发生,其活性需要最大化治疗效益。
我们正在筹备几个治疗项目,重点是传染病、肿瘤学和免疫耐受。
我们的管道
下面的图表提供了有关我们计划的关键信息。
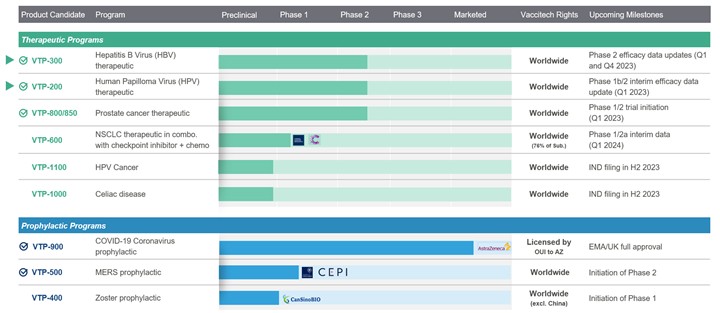
VTP-300:一种针对慢性乙肝病毒感染的免疫疗法
2022年5月,我们在英国完成了HBV001第一阶段临床试验的最后一次患者访问。纳入了两类参与者:健康参与者和感染已被口服抗病毒治疗抑制的慢性乙肝患者。HBV001试验的主要目标是评估单次接种不同剂量ChAdOx1-乙肝疫苗的安全性和耐受性。此外,次要目标是确定ChAdOx1-乙肝病毒的免疫原性,并确定ChAdOx1-乙肝病毒对慢性乙肝感染参与者的乙肝表面抗原水平的影响。所有健康志愿者和慢性乙肝患者队列均已完成治疗和随访。截至本报告日期,尚未报告任何严重不良事件。
我们已经在我们的VTP-300载体中使用了C型乙肝病毒抗原序列来针对最流行的CHB基因型。然而,我们认为VTP-300可以诱导与其他流行的基因型别的交叉反应T细胞反应。因此,我们还旨在确定本试验中使用的ChAdOx1-HBV病毒载体诱导的T细胞反应是否可能与其他常见的HBV型发生交叉反应。CHB患者入选本试验的标准是(I)口服抗病毒药物(HBVDNA)抑制的感染
11