
美国
美国证券交易委员会
华盛顿特区,20549
表格
(标记一)
根据1934年《证券交易法》第13或15(D)条提交的年度报告 |
截至本财政年度止
或
根据1934年《证券交易法》第13或15(D)条提交的过渡报告的过渡期 至 |
委托文件编号:
(注册人的确切姓名载于其章程)
(述明或其他司法管辖权 公司或组织) |
(税务局雇主 识别号码) |
(主要执行办公室地址) |
(邮政编码) |
(
(注册人的电话号码,包括区号)
根据该法第12(B)条登记的证券:
每个班级的标题 |
|
交易 符号 |
|
注册的每个交易所的名称 |
|
|
根据该法第12(G)条登记的证券:无
如果注册人是证券法第405条规定的知名经验丰富的发行人,请用复选标记表示。是 ☐
用复选标记表示注册人是否不需要根据该法第13或15(D)条提交报告。是 ☐
用复选标记表示注册人是否:(1)在过去12个月内(或注册人需要提交此类报告的较短时间内)提交了1934年《证券交易法》第13或15(D)条要求提交的所有报告,以及(2)在过去90天内一直遵守此类提交要求。
用复选标记表示注册人是否在过去12个月内(或在注册人被要求提交此类文件的较短时间内)以电子方式提交了根据S-T规则第405条(本章232.405节)要求提交的每个交互数据文件。
用复选标记表示注册人是大型加速申报公司、加速申报公司、非加速申报公司、较小的报告公司或新兴成长型公司。请参阅《交易法》第12b-2条规则中“大型加速申报公司”、“加速申报公司”、“较小申报公司”和“新兴成长型公司”的定义。
大型加速文件服务器 |
|
☐ |
|
加速文件管理器 |
|
☐ |
|
|
|
|
|
||
|
☒ |
|
规模较小的报告公司 |
|
||
|
|
|
|
|
|
|
|
|
|
|
新兴成长型公司 |
|
|
如果是一家新兴的成长型公司,用复选标记表示注册人是否已选择不使用延长的过渡期来遵守根据《交易所法》第13(A)节提供的任何新的或修订的财务会计准则。
用复选标记表示注册人是否提交了一份报告,证明其管理层根据《萨班斯-奥克斯利法案》(《美国联邦法典》第15编,第7262(B)节)第404(B)条对其财务报告的内部控制的有效性进行了评估,该评估是由编制或发布其审计报告的注册会计师事务所进行的。
如果证券是根据该法第12(B)条登记的,应用复选标记表示登记人的财务报表是否反映了对以前发布的财务报表的错误更正。☐
用复选标记表示这些错误更正中是否有任何重述需要对注册人的任何执行人员在相关恢复期间根据第240.10D-1(B)条收到的基于激励的薪酬进行恢复分析。☐
用复选标记表示注册人是否是空壳公司(如《交易法》第12b-2条所定义)。是 ☐ 不是
截至2022年6月30日,注册人的非关联公司持有的有投票权和无投票权股票的总市值为$
截至2023年3月1日,注册人的普通股流通股数量为
截至2023年3月1日,注册人的已发行优先股数量为647,500股,不包括在上述总市值之外。优先股的每股股份可在任何时间由持有人选择转换为10股普通股,但须受某些限制,包括持有人不得将优先股转换为普通股,条件是持有人连同其联属公司将实益拥有若干普通股,超过当时已发行及紧随该等优先股转换后已发行及已发行的普通股总数的9.99%。优先股的股份通常没有投票权,除非法律要求,而且修改优先股的条款需要得到已发行优先股的多数持有人的同意。在公司清算、解散或清盘的情况下,优先股持有者将与普通股持有者分享任何收益分配。优先股的排名如下:(I)按其条款特定设立的本公司任何类别或系列股本的优先级别低于优先股;(Ii)与普通股及本公司任何类别或系列股本的平价,按其条款与优先股的平价排名;及(Iii)就本公司清算、解散或清盘时的资产分配(不论自愿或非自愿)而言,优先于按其条款特定高于任何优先股的本公司任何类别或系列股本。
目录表
|
|
页面 |
第一部分 |
|
|
第1项。 |
业务 |
1 |
第1A项。 |
风险因素 |
53 |
项目1B。 |
未解决的员工意见 |
106 |
第二项。 |
属性 |
106 |
第三项。 |
法律诉讼 |
106 |
第四项。 |
煤矿安全信息披露 |
106 |
|
|
|
第II部 |
|
|
第五项。 |
注册人普通股市场、相关股东事项与发行人购买股权证券 |
107 |
第六项。 |
已保留 |
107 |
第7项。 |
管理层对财务状况和经营成果的探讨与分析 |
108 |
第7A项。 |
关于市场风险的定量和定性披露 |
119 |
第八项。 |
财务报表和补充数据 |
119 |
第九项。 |
会计与财务信息披露的变更与分歧 |
119 |
第9A项。 |
控制和程序 |
119 |
项目9B。 |
其他信息 |
120 |
项目9C。 |
关于妨碍检查的外国司法管辖区的披露 |
120 |
|
|
|
第三部分 |
|
|
第10项。 |
董事、高管与公司治理 |
121 |
第11项。 |
高管薪酬 |
124 |
第12项。 |
某些实益拥有人的担保所有权以及管理层和相关股东的事项 |
131 |
第13项。 |
某些关系和相关交易,以及董事的独立性 |
134 |
第14项。 |
首席会计师费用及服务 |
137 |
|
|
|
第四部分 |
|
|
第15项。 |
展品和财务报表附表 |
138 |
第16项。 |
表格10-K摘要 |
138 |
关于前瞻性陈述的特别说明
本10-K表格年度报告包含根据1933年证券法(修订)第27A节和1934年证券交易法(“交易法”)第21E节的安全港条款作出的前瞻性陈述,这些陈述涉及风险、不确定因素和其他因素,可能导致实际结果、活动水平、业绩或成就与这些前瞻性陈述明示或暗示的信息大不相同。除有关历史事实的陈述外,本Form 10-K年度报告中包含的所有陈述,包括有关我们的战略、未来运营、未来财务状况、未来收入、预计成本、前景、管理计划和目标以及预期市场增长的陈述,均为前瞻性陈述。“预期”、“相信”、“继续”、“可能”、“估计”、“预期”、“打算”、“可能”、“计划”、“潜在”、“预测”、“项目”、“应该”、“目标”、“将”等类似表述旨在识别前瞻性表述,尽管并不是所有前瞻性表述都包含这些识别词语。
本年度报告中的10-K表格中的任何前瞻性陈述反映了我们对未来事件或我们未来财务表现的当前看法,涉及已知和未知的风险、不确定性和其他因素,可能会导致我们的实际结果、业绩或成就与这些前瞻性陈述明示或暗示的任何未来结果、业绩或成就大不相同。可能导致实际结果与当前预期大不相同的因素包括但不限于本Form 10-K年度报告和提交给证券交易委员会(“美国证券交易委员会”)的其他文件中列出的那些因素,包括:
我们可能无法实际实现我们的前瞻性陈述中披露的计划、意图或预期,投资者不应过度依赖我们的前瞻性陈述。实际结果或事件可能与我们在前瞻性陈述中披露的计划、意图和预期大不相同。我们的前瞻性陈述不反映我们可能进行的任何未来收购、合并、处置、合资或投资或我们可能达成的合作或战略伙伴关系的潜在影响。
您应阅读本年度报告中的Form 10-K以及我们在此引用的文件,这些文件已作为附件完整地存档或合并,并应了解我们未来的实际结果可能与我们预期的大不相同。我们不承担任何义务更新任何前瞻性陈述,无论是由于新信息、未来事件或其他原因,除非法律要求。
这份Form 10-K年度报告还包含关于我们的行业、我们的业务和我们候选产品的市场的估计、预测和其他信息。基于估计、预测、预测、市场研究或类似方法的信息本身就会受到不确定因素的影响,实际事件或情况可能与本信息中假设的事件和情况大不相同。除非另有明确说明,否则我们从我们自己的内部估计和研究以及从市场研究公司和其他第三方准备的报告、研究调查、研究和类似数据、行业、医疗和一般出版物、政府数据和类似来源获得这些行业、商业、市场和其他数据。虽然我们不知道本10-K表格年度报告中关于第三方信息的任何错误陈述,但它们的估计,尤其是与预测有关的估计,涉及许多假设,受风险和不确定性的影响,并可能根据各种因素而变化,包括在题为“风险因素”一节和本10-K表格年度报告其他部分讨论的那些因素。
与我们的业务相关的重大风险和其他风险摘要
以下是使我们的普通股投资具有投机性或风险的主要因素的摘要。这一总结并没有解决我们面临的所有风险。对本风险因素摘要中总结的风险以及我们面临的其他风险的其他讨论在“风险因素”一节中进行了总结,在就我们的普通股做出投资决定之前,应仔细考虑本年度报告Form 10-K和我们提交给美国证券交易委员会的其他文件中的其他信息。
部分 I
项目1.B有用处。
概述
我们是一家临床阶段的生物制药公司,专注于不可知的靶向肿瘤学。我们的战略是识别高影响的癌症靶点,然后选择我们认为对这些靶点最优的治疗方式。我们从内部和外部寻求创新,专注于拥有新技术平台或差异化机制的候选产品。在我们将候选产品推进到临床开发之前,我们评估其作为单一制剂的抗肿瘤活性的潜力,以及其产生免疫系统反应或抑制肿瘤过程的能力。使用这一战略,我们已经建立了广泛而深入的有针对性的肿瘤学计划管道,其中包括六个不同的候选产品,其中五个是临床阶段的,以及多个研究和发现计划。
齐帕替尼(CLN-081/TAS6417)是我们与泰豪制药有限公司(“泰豪”)的一家附属公司共同开发的,是一种口服生物可用小分子不可逆表皮生长因子受体(EGFR)抑制剂,旨在选择性地靶向表达EGFR外显子20插入(“EGFRex20ins”)突变的细胞,相对保留表达野生型EGFR的细胞。美国食品和药物管理局(“FDA”)已授予齐帕替尼突破性治疗称号。在2022年第四季度,我们与我们在泰豪的合作伙伴合作,启动了一项关键的2b期研究,研究对象是EGFR外显子20的非小细胞肺癌(NSCLC)患者,这些患者在之前的系统治疗后进展。2022年6月,大鹏以2.75亿美元的预付款收购了我们部分拥有的子公司库利南珍珠公司(“库利南珍珠”)的股权,该公司为大和提供了日本以外的Zipertinib和大中国的全球权利。作为出售的一部分,我们还有资格获得与EGFR外显子20 NSCLC监管里程碑相关的高达1.3亿美元的额外资金。在完成出售我们在库利南珍珠的股权的同时,我们与泰豪的一家关联公司签订了齐帕尔替尼的共同开发和共同商业化协议,根据协议,我们将合作开发齐帕尔替尼,并将保留在美国(“美国”)共同商业化齐帕尔替尼的选择权。吉帕尔替尼在美国潜在销售的开发成本和任何未来的税前利润将由我们和泰豪平分。
除了齐帕替尼外,我们的产品组合还包括其他四种临床阶段候选产品和一种正在等待FDA批准其研究新药申请(IND)的候选产品:
除了上述候选产品外,我们还在积极开发几个临床前肿瘤学项目,这些项目都处于发现阶段,包括我们与西奈山伊坎医学院合作开发新型造血祖细胞激酶1(HPK1)降解物。
我们的使命是为癌症患者创造新的护理标准。我们寻求通过我们对药物开发的差异化方法来实现我们的使命,该方法以以下核心要素为指导:
1
我们的管道
我们的流水线包括肿瘤学候选产品和计划,这些产品和计划因目标、机制、技术平台和方式而多样化。我们严格评估我们的每个项目,以证明继续投资的合理性,并确定适当的资本分配。当某些计划不符合我们的提升风险标准时,我们将终止这些计划,并保留我们的资本和资源,用于投资于潜力更大的计划。因此,我们的渠道将继续保持活力。我们相信,每个候选项目都有不同的设计特征或行动机制,以及一流和/或一流的潜力。我们目前的渠道如下图所示:
我们的业务战略和目标
我们的战略重点是我们所说的“模式不可知的靶向肿瘤学”:我们首先确定高影响的癌症靶点,然后选择我们认为对这些靶点最优的治疗模式。因此,我们不依赖于单一的技术平台或产品。我们将“高影响”的生物靶标定义为那些发挥关键作用的生物靶标,它们要么是致癌的驱动因素,要么是免疫系统的激活剂。从那时起,我们只发展我们认为具有一流和/或最佳潜力的分子,这些分子可以作为单一制剂产生强大的抗肿瘤反应。体内。通过这种有纪律的方法,我们相信我们将带来具有潜力的差异化分子,为癌症患者创造新的护理标准。我们的策略旨在实现的主要目标如下:
2
我们的结构
我们正在通过库利南肿瘤公司(“库利南”)和我们创建的现有开发子公司开发候选产品。库利南拥有齐帕尔替尼在美国的共同开发权和共同商业化权利,以及美国对CLN-418的权利,以及CLN-978的全球权利。以前,当我们授权或获得我们的几种候选药物的全球独家知识产权时,我们建立了开发子公司,包括CLN-049的库利南佛罗伦萨公司(“库利南佛罗伦萨”),CLN-619的库利南MICA公司(“库利南MICA”),以及CLN-617的库利南琥珀公司(“库利南琥珀”)。库利南目前拥有库利南佛罗伦萨96%的股份、库利南云母95%的股份和库利南琥珀94%的股份。我们与每家子公司的融资安排的结构使我们能够在提供额外资本时增加我们的经济所有权。
在历史上,我们作为一家私人公司创建了开发子公司,以有效地增长和推进我们的投资组合。作为一家上市公司,我们不打算在未来创建新的开发子公司。有关我们子公司的更多信息,包括所有权和治理,请参阅本年度报告的Form 10-K中的“管理层讨论和分析”部分。
3
我们的节目
齐帕替尼
概述
齐帕替尼是一种口服生物利用型小分子药物,旨在与泰豪公司的一家附属公司合作开发下一代不可逆转的EGFR抑制剂,用于治疗基因定义的非小细胞肺癌患者亚群。2022年1月,FDA授予齐帕尔替尼突破性治疗称号,2022年11月,我们与我们在TAIHO的合作伙伴合作,启动了一项关键的2b期试验,评估携带EGFRex20ins突变的成年NSCLC患者在之前的系统治疗后进展的齐帕尔替尼。我们与泰豪的一家附属公司签订了齐帕尔替尼的共同开发和共同商业化协议,根据协议,我们将合作开发齐帕尔替尼,并将保留在美国共同商业化齐帕尔替尼的选择权。我们和泰豪将平均分摊齐帕尔替尼的开发成本,双方将从未来可能在美国销售齐帕尔替尼的任何税前利润中获得50%。
2022年6月,我们在美国临床肿瘤学会会议上提供了临床最新情况,其中包括73名携带EGFRex20ins突变的可评估非小细胞肺癌患者的安全性和有效性数据,这些患者参加了1/2a期试验,剂量水平从每天两次30到150毫克不等。在每天两次100毫克的剂量下,我们观察到以下疗效和安全性亮点,我们认为这反映了齐帕替尼的不同临床特征:
非小细胞肺癌和表皮生长因子受体突变的背景
到目前为止,肺癌是导致男性和女性癌症死亡的主要原因,占所有癌症死亡人数的近25%。美国癌症协会估计,到2023年,美国将有大约238,340例新肺癌病例和大约127,070人死于肺癌。最常见的肺癌亚型是非小细胞肺癌,约占所有肺癌的80%到85%。
EGFR是一种受体酪氨酸激酶(RTK),当生长因子与受体结合时,它通常会触发细胞分裂。酪氨酸激酶结构域的致癌突变可以诱导生长因子非依赖性的EGFR激活,导致细胞生长和增殖失控。最终,这些异常信号可能有助于非小细胞肺癌的发展。大约15%到25%的美国和西欧非小细胞肺癌患者和大约30%到50%的亚洲非小细胞肺癌患者存在EGFR突变。鉴于其在癌症中的重要作用和流行程度,突变的EGFR是肺癌治疗的关键靶点。外显子19缺失和外显子21 L858R突变,统称为经典的EGFR突变,是最常见的突变,占非小细胞肺癌EGFR突变的75%以上。多种EGFR抑制剂,包括gefitinib、erlotinib、afatinib和osimertinib,针对这些常见的突变,已被批准为一线疗法,从而验证了突变的EGFR作为治疗NSCLC的靶点。
外显子20插入占非小细胞肺癌患者所有EGFR突变的7%至13%,是仅次于经典EGFR突变的最常见的突变。我们估计在美国大约有2,000到5,000名非小细胞肺癌患者,在法国、德国、意大利、西班牙和英国(“英国”)大约有1,000到3,000名患者携带EGFRex20ins突变。临床前研究表明,外显子20的插入以及经典的EGFR突变都具有致癌驱动突变的特征,这些突变既与肿瘤的发生有关,也与癌症的进展有关。然而,与经典的EGFR突变不同的是,外显子20的插入不会使激酶结构域对批准的EGFR抑制剂的治疗敏感。
目前,有两种靶向疗法被加速批准,用于携带EGFRex20ins突变的非小细胞肺癌患者,他们的疾病在基于铂的化疗中或之后进展:amivantamab-vmjw(Rybrevant)和mobocertinib(Exkirate)。尽管加速了批准,但我们认为,携带EGFRex20ins突变的非小细胞肺癌患者仍有大量未得到满足的需求。具体地说,我们认为有机会对突变型与野生型EGFR进行高度选择性的口服治疗,这可能会改善安全性和耐受性,特别是在与治疗相关的不良事件方面,如皮疹、腹泻、输液部位反应以及心血管事件。
4
齐帕替尼
齐帕替尼是一种小分子,被设计为一种不可逆转的EGFR抑制剂,带有新型的吡咯嘧啶支架,这在针对EGFRex20ins突变的开发中的治疗方法中是独一无二的。齐帕替尼被设计成适合于EGFR的三磷酸腺苷结合部位,在那里它共价修饰C797,从而形成一种持久的药物-蛋白质连接,不可逆转地抑制突变受体。在临床前研究中,齐帕替尼在表达突变的EGFR蛋白的细胞中表现出高选择性和对EGFR的抑制,而在表达野生型EGFR的细胞中抑制作用明显较小。
评价齐帕替尼与竞争的EGFR抑制剂的选择性指数。体外培养通过表达野生型EGFR的细胞与表达外显子20插入突变的EGFR的细胞的半数最大生长抑制值(IC50)之比来衡量。如下所示,齐帕替尼在一组EGFR靶向治疗中显示出最高的选择性指数,这表明齐帕替尼可能能够在相对较少的野生型EGFR的情况下实现临床相关的抑制EGFRex20ins突变。
齐帕替尼在多种EGFRex20ins突变中显示出卓越的选择性

临床发展
第1/2a期研究
我们在2019年第四季度启动了正在进行的齐帕替尼1/2a阶段试验。这项首例人类开放标签多中心试验旨在评估齐帕替尼对携带EGFRex20ins突变的成年非小细胞肺癌患者的安全性和耐受性、药代动力学、药效学和初步疗效,这些患者在先前的系统治疗后进展。该试验包括两个主要组成部分:剂量递增和队列扩大。剂量递增始于单患者加速滴定设计,并在首次出现2级或更高的Trae时过渡到滚动6个决策规则,发生在每天两次100 mg的剂量水平。这项试验有一个灵活的适应性设计,包括每天两次的30、45、65、100和150毫克剂量队列,并允许扩大任何给定的队列,由可接受的安全性和预先指定的疗效标准控制。根据最初的方案,根据这些标准,队列可以扩大到6名,然后是13名,然后是36名患者。我们扩大了65毫克、100毫克和150毫克的每日两次剂量水平的队列,尽管后来我们在11名患者之后停止了150毫克水平的登记,这是基于对该剂量水平下的总体临床情况的评估。我们总共招募了39名患者,每天两次100毫克的剂量水平,其中包括最初的36名患者,以及另外3名患者,这些患者在我们决定停止登记每天两次150毫克的剂量水平后被重新分配。我们在美国、荷兰、新加坡、香港和台湾招募了患者。
这项研究共招募了73名患者,他们至少接受了一剂齐帕替尼。患者群体接受了大量的预治疗,中位数为两次先前的系统治疗,66%的患者在研究开始时接受了两次或两次以上的先前治疗(即第三线或更高的治疗)。此外,36%的患者之前接受了EGFR抑制剂的治疗,包括4%的患者之前接受了poziotinib或Exkitivity的治疗,这是一种针对第20外显子插入突变的TKIs。超过一半的患者(55%)以前接受过免疫治疗。
5
第1/2a期安全性和药代动力学数据
下表总结了与治疗相关的安全性和耐受性事件,包括皮疹和腹泻,这是与抑制野生型EGFR有关的毒性,以及实验室异常情况,包括贫血和转氨酶升高,用于比较所有两次每日剂量水平,以及我们的1/2a期试验中的总体安全人群。我们认为,这种安全性和耐受性比其他EGFR外显子20抑制剂更有利,特别是在腹泻的发生率和严重性方面。
齐帕替尼每日两次100 mg的分化安全性和耐受性

1不良事件通用术语标准V5.0
在1/2a期试验中接受每日两次100毫克剂量治疗的39名可安全评估的患者中,没有人出现与治疗相关的3级或更严重的皮疹或腹泻。在这个剂量水平下,82%和36%的患者分别经历了治疗相关的皮疹和腹泻,严重程度为1级或2级;然而,经历1级和2级事件的患者的皮疹和腹泻的比例约为3:1。这两个事件都可以通过传统的支持性护理得到控制,而且腹泻治疗不需要实施系统的胃肠道预防措施。与其他EGFR TKI一样,在这个剂量水平上观察到一例3级治疗相关性肺炎,尽管患者最近接受了检查点抑制剂治疗,并同时存在与治疗无关的明显的对侧积液气胸。
在每天两次150毫克剂量水平的11名安全性评估患者中,关键观察包括两名患者出现与治疗相关的3级腹泻,1名患者出现3级皮疹,2名患者出现3级和4级转氨炎。此外,一名因进展性疾病而停用齐帕替尼超过三周的患者被报告患有与三级治疗相关的肺炎;该患者并发甲型肺孢子虫感染。此外,与每天两次100毫克的剂量水平相比,每天两次150毫克治疗的患者的剂量减少率和剂量停减率都有所增加。这些观察结果使我们决定在11名患者之后,停止进一步招募每天两次150 mg的患者。
初步的药代动力学数据显示了暴露剂量的近乎依赖的趋势,由曲线下的非结合面积(AUC)和C最大值价值观。此外,在临床前研究中实现肿瘤消退所需的目标AUC从每天两次30毫克的初始剂量开始达到。齐帕替尼的显著药代动力学特征包括:给药后8小时持续的药物动力学暴露超过EGFRex20ins突变生长抑制50%(“GI50”)所需的浓度,患者间的异质性有限,野生型EGFR每天两次暴露在GI50以上的剂量为100毫克或以下。与每天两次100毫克剂量与150毫克每日两次剂量的临床安全性情况一致,在每天两次150毫克剂量时,我们观察到齐帕替尼的浓度在大约4小时内高于野生型EGFR GI50比率。
6
第1/2a期疗效数据
下表总结了每日两次剂量水平为65 mg或以下(N=23)、100 mg(N=39)和150 mg(N=11)的可反应患者的最佳反应特征,以及截至2022年5月9日数据截止点的总剂量水平的总体人口(N=73)。在每天两次接受100毫克治疗的患者中,16名患者获得了确认的部分缓解,即41%的确认总缓解率(ORR)。在每天两次150毫克剂量队列的11名患者中,这一确认的ORR高于36%的确认ORR。在每天两次服用100毫克的队列中,中位PFS为12个月,而每天服用150毫克的PFS仅为8个月。如上所述,3级或更高级别的皮疹和腹泻以及剂量减少和剂量中断都有利于每天两次100 mg的剂量队列。
齐帕替尼:每日两次100毫克的卓越安全性和有效性

以下是73名患者的额外疗效分析,包括游泳者图表(A)和与基线最好变化百分比的瀑布图(B)。我们还包括了显示PFS(C)中值的Kaplan-Meier曲线。试验中的患者在大约六周的治疗后进行初步的肿瘤成像,然后每九周进行一次。基于这些分析,我们认为齐帕替尼显示出了实质性的抗肿瘤活性,具有广泛的EGFR外显子20变异覆盖;作用起效迅速;PFS测量的令人鼓舞的反应质量。
7
(A)正在进行的齐帕替尼1/2a期试验的初步疗效结果

(B)从基线到最佳反应百分比的变化(目标病变)

8
(C)估计中位数--无进展生存期

第1/2a期食物效应研究
除上述第1/2a期试验外,我们还在2022年完成了16名患者的初步食物效应模块,评估了单剂量150 mg齐帕替尼在全角实体瘤人群中的单剂量药代动力学(PK)。采用随机交叉设计,每个患者在进食和禁食状态下都服用150 mg齐帕替尼,以评估与食物一起服用该药物对暴露的影响。结果表明,与食物一起服用该药物并没有显著改变齐帕替尼的暴露情况。根据FDA审查的决策规则,我们将探索在联邦状态下每天两次150毫克剂量的安全性和有效性,作为研究的关键阶段2b部分的一部分,如下所述。
关键阶段2b研究
2022年11月,我们与我们在泰豪的合作伙伴合作,启动了一项正在进行的关键2b期研究,对携带EGFRex20ins突变的成年NSCLC患者进行齐帕替尼治疗,这些患者在之前的系统治疗后进展,这一患者群体与1/2a期试验一致。2b期试验的关键部分包括两个平行的非随机剂量组:一组患者在禁食状态下每天两次服用100毫克的齐帕替尼,另一组患者在进食状态下每天两次服用150毫克的齐帕替尼。严格的停药规则适用于150毫克的队列,其中该剂量必须显示出相对于100毫克禁食剂量的1/2a期结果更好的疗效和安全性。如果150毫克的队列在安全性和有效性标准上都不能显示出优势,我们将停止招募,继续只招募每天禁食两次的100毫克的患者。试验方案还包括一个单独的队列,旨在评估100毫克禁食剂量水平的齐帕替尼在携带EGFRex20ins突变的成年NSCLC患者中的作用,这些患者之前接受过批准的外显子20疗法的治疗。
9
CLN-049
概述
CLN-049是我们正在开发的用于治疗AML和MDS的人源化双特异性抗体。我们目前正在对复发或难治性急性髓细胞白血病(“r/r AML”)和MDS的成人患者进行多剂量递增试验,以评估CLN-049。CLN-049被设计为同时与靶白血病细胞胞外区的Flt3和T细胞上的CD3结合,触发T细胞杀伤靶癌细胞。Flt3是一种原癌基因,也是一个有效的靶点,几种针对突变flt3的激酶抑制剂已被批准用于治疗r/r AML,但它们的使用仅限于约25%的具有flt3突变的AML人群。通过靶向Flt3的胞外结构域,CLN-049有可能解决高达80%的表达突变或野生型flt3的AML患者。临床前,我们观察到CLN-049导致高度依赖于Flt3的白血病细胞杀伤。体外培养在AML细胞上有广泛的Flt3表达水平,而与Flt3突变状态无关。在临床前研究中,CLN-049的治疗,即使是低剂量,也能在AML异种移植模型中带来存活益处,并在植入AML细胞系或原发患者白血病细胞的小鼠模型中消除白血病母细胞。
急性髓系白血病和Flt3的研究背景
美国癌症协会估计,到2023年,美国将有大约20,380名新诊断的AML患者和大约11,310名死于AML的患者。AML是一种复杂的血液系统恶性肿瘤,其特征是恶性未成熟髓系原始细胞群无法控制地增殖。这些原始细胞可能会完全渗透并取代骨髓,导致正常造血和全血细胞减少的严重破坏,外周血中循环中大量的原始细胞,以及内脏器官和皮肤的渗透。此外,急性髓细胞白血病患者可能容易因血小板减少而出现出血并发症,并经历细胞毒性化疗治疗的并发症。这些患者也可能是继发于疾病的严重免疫受损,并经历了较长时间的中性粒细胞减少和淋巴细胞减少。因此,这些患者往往容易感染危及生命的感染,这些感染也会导致严重的发病率和死亡率。
尽管急性髓细胞白血病的治疗取得了进展,但这些患者的需求仍然很高,尚未得到满足。符合条件的新诊断患者通常接受强化诱导化疗(IC),其中可能包括持续输注阿糖胞苷和蒽环类药物,以试图实现完全缓解。大多数经历完全缓解的患者接受了造血干细胞移植(HSCT)。这种疗法是目前唯一可用的治疗急性髓系白血病的疗法。然而,85%的60岁以上的患者不符合IC和HSCT的条件。鉴于确诊的平均年龄为68岁,大多数AML患者无法获得根治性治疗。尽管积极的一线联合化疗,最近针对分子定义的AML患者亚群的多靶向小分子的批准,以及在匹配合适供者的患者中使用HSCT,AML患者的预后仍然非常差。虽然60%至85%的年轻成人患者获得完全缓解,但60岁以上的患者完全缓解率较低,为40%至60%。此外,大约40%的患者在接受HSCT后复发,大约10%的复发患者存活至少五年。因此,一个重要的未得到满足的需求仍然是一种广泛适用、耐受性良好的治疗方法,它可以产生高比率的持久反应。
Flt3是一种III型RTK,在造血过程中具有重要的作用。在健康人中,Flt3的表达仅限于造血干细胞和祖细胞亚群,诱导其增殖和分化为单核细胞、树突状细胞、B细胞和T细胞。Flt3是一种原癌基因,在促进白血病细胞增殖和存活方面发挥着关键作用。几种针对Flt3突变的小分子激酶抑制剂正在开发中或已被批准用于治疗AML。然而,这些候选产品和批准的疗法仅适用于大约25%具有细胞内Flt3基因突变的AML患者,而不适用于更大规模的具有Flt3细胞外表达的患者,无论癌细胞表面的突变状态如何。
使用流式细胞术的研究表明,在大约80%的AML患者中,无论突变状态如何,Flt3都在AML原始细胞上表达。在一项研究中,对318名新诊断或复发的AML患者的白血病大细胞进行了细胞表面Flt3蛋白表达的评估,发现78%的人Flt3呈阳性,如下图所示。Flt3在AML患者中的广泛表达表明,用一种生物制剂靶向Flt3,即一种结合T细胞的双特异性抗体,招募T细胞杀死细胞表面表达Flt3的肿瘤细胞,与靶向已批准或正在开发的针对Flt3细胞内信号域突变版本的靶向小分子抑制剂相比,可以解决更大的AML患者群体。与AML中发现的其他肿瘤表面抗原,如CD33和CD123相比,Flt3的表达通常局限于骨髓HSPC和循环树突状细胞的一小部分。Flt3在促进白血病的发生和AML的恶性进展、促进白血病细胞的增殖和存活方面发挥着关键作用。我们认为,在大多数AML患者中,Flt3在白血病细胞表面的表达及其作为已知致癌驱动因素的作用使其成为T细胞激活剂方法的一个有吸引力的治疗靶点。
10
约80%的AML患者细胞表面Flt3蛋白表达阳性

CLN-049
如下所示,CLN-049是一种人源化的双特异性抗体,由两个Flt3结合域组成,一个是Fc沉默的人源性免疫球蛋白G1(“IgG1”)主干,另一个是CD3结合的单链可变区(“scFv”),融合在抗体重链的C末端。体外培养和强大的抗肿瘤活性体内。通过靶向细胞外的Flt3,而不考虑突变或野生型状态,我们相信CLN-049有可能解决高达80%的AML患者,这比作用于细胞内区域的现有小分子Flt3激酶抑制剂有更广泛的患者群体,后者仅限于约25%具有Flt3突变的AML患者的子集。
CLN-049机制可能允许广泛依赖于Flt3的AML爆炸杀伤

11
临床前数据
鉴于在患者中观察到的Flt3表达水平的差异性,我们表征了CLN-049在细胞表面表达不同水平的Flt3的多个细胞系中的杀伤潜力。如下图所示,观察到CLN-049可介导强大的靶向依赖的细胞杀伤体外培养在所有测试的AML细胞系中。重要的是,我们观察到杀死50%靶细胞的药物浓度(EC50值)在亚纳摩尔范围内,似乎与AML靶细胞上发现的Flt3受体分子的数量无关。特别是,我们观察到即使在每个细胞表达不到100个拷贝的Flt3受体的情况下,也能观察到强大的靶细胞杀伤作用。我们还观察到具有野生型或突变型Flt3表达的AML细胞株的有效重定向裂解。基于这些结果,我们相信CLN-049可以有效地杀死即使是低水平Flt3表达的AML靶细胞,无论是野生型还是突变状态,这可能会在临床上转化为更深入和更持久的反应,并可能允许我们治疗更大的AML患者亚群。
CLN-049显示了对表达一系列Flt3的靶细胞的杀伤作用,体外培养

CLN-049对表达野生型和突变型Flt3的靶细胞有杀伤作用体外培养

Flt3在正常免疫细胞上不广泛表达,但仅限于骨髓中的某些造血干细胞前体细胞和外周血树突状细胞亚群。如下图所示,最近的一项研究发现,Flt3转录本在AML细胞上的表达水平明显高于正常组织。
12
急性髓系白血病细胞Flt3转录水平高于正常人体实体组织

CLN-049有两个CD3结合臂,可以潜在地使T细胞上的CD3交联,这可能导致靶细胞非依赖性T细胞激活和全身细胞因子相关的毒性。在临床前研究中,我们检查了CLN-049在没有靶细胞的情况下是否会导致虚假的T细胞激活。如下所示,在没有靶表达细胞的情况下,将纯化的人T细胞与CLN-049孵育,并没有在两者的CD4上诱导T细胞激活标记CD25和CD69+或CD8+T细胞,而不是阳性对照抗CD3抗体OKT3和UCHT1(CLN-049亲本抗CD3抗体),它们诱导T细胞激活。
CLN-049在没有靶细胞的情况下对T细胞的激活作用

CLN-049的潜在疗效是在人源化的小鼠模型中评估的,其中人AML细胞系被系统植入。如下图所示,CLN-049控制了植入人外周血单个核细胞(PBMC)的小鼠的AML白血病负担,并以剂量依赖的方式提高了小鼠的存活率,从非常低的剂量开始。我们相信CLN-049通过改变人PBMC群体中的T细胞来杀伤目标AML细胞来影响这一结果。
13
从极低剂量CLN-049开始提高移植小鼠的存活率

CLN-049的抗白血病活性也用患者来源的AML或急性淋巴细胞白血病(“ALL”)母细胞和PBMC在播散性人源化小鼠模型中进行了评估。如下图所示,在原发AML模型和原发ALL模型中,CLN-049治疗都显著降低了骨髓中的总体白血病负担。相反,与未经治疗的对照相比,对照T细胞结合双特异性抗体的形式与CLN-049相同,但包含非特异性靶结合区域,并不影响白血病负担。
CLN-049对小鼠急性髓系白血病细胞的有效清除作用

正在进行的临床开发
2021年12月,我们启动了一项第一阶段临床试验,评估CLN-049在r/r AML患者中的应用。这项研究旨在初步评估CLN-049的药代动力学和安全性。我们完成了第一阶段试验的单一递增剂量部分,测试了静脉注射CLN-049,并于2022年12月利用皮下给药启动了研究的多个递增剂量部分。我们预计在2023年年中提交初步临床数据。
CLN-619
概述
CLN-619是一种针对MICA/B的人源化IgG1单抗,我们打算初步开发用于治疗实体瘤。CLN-619旨在通过多种作用机制促进抗肿瘤反应,包括抑制MICA/B的脱落,介导抗体依赖的细胞介导的细胞毒作用(ADCC),抗体依赖的细胞吞噬作用(ADCP),以及增强NKG2D受体与MICA/B的结合。MICA/B受体NKG2D表达于天然免疫细胞和获得性免疫细胞。尽管几家公司已经披露了临床前MICA/B靶向计划,但我们不知道有任何临床阶段,基于抗体的计划涉及到这一靶点,这意味着CLN-619具有一流的潜力。在多个体内在临床前肿瘤模型中,CLN-619的应用与抗肿瘤活性和降低血清MICA/B水平有关。
我们相信,鉴于MICA/B在肿瘤类型中的广泛表达,以及CLN-619与其他药物联合的生物学原理,CLN-619有可能成为免疫肿瘤学治疗的新型主干药物。我们目前正在进行一项针对晚期实体瘤患者的临床试验,评估CLN-619。试验设计包括将CLN-619作为单一疗法以及与检查点抑制剂疗法相结合的平行评估。
14
NKG2D和MICA/B的背景
NKG2D是NK细胞上的一种关键的激活受体,在与靶细胞上表达的配体结合时负责细胞溶解。NKG2D也表达在其他类型的免疫细胞上,包括CD8+ ?T细胞、自然杀伤T细胞和 T细胞,并可激活这些细胞并增强其作为共激活受体的抗肿瘤活性。健康细胞通常不表达NKG2D的配体,但会在细胞应激,如氧气或营养剥夺、辐射、病毒感染或致癌转化时表达。
Mica/B蛋白被NK细胞广泛识别,T细胞和CD8+ 通过NKG2D受体激活T细胞。NKG2D受体与MICA/B蛋白的结合可触发NK细胞的效应细胞杀伤反应 T细胞对表达MICA/B的肿瘤细胞的杀伤作用+ 通过NKG2D-MICA/B相互作用,T细胞受体介导的效应反应得到增强。NKG2D介导的刺激还可诱导细胞因子,进一步促进免疫细胞的募集和增殖,增强免疫反应。
为了逃避NK细胞和T细胞潜在的细胞毒作用,表达MICA/B的肿瘤细胞将MICA/B从细胞表面脱落作为主要的逃避机制。MICA/Bα-3结构域包含一段氨基酸,允许MICA/B的胞外部分被蛋白酶切割并随后从细胞表面释放,从而降低MICA/B与NKG2D相互作用的能力,导致NKG2D介导的肿瘤细胞杀伤减少。这一机制还伴随着增加循环血清MICA/B(“SmICA/B”)的数量。这种生物学背后的机制如下所示。下面,上面的途径显示了NKG2D的肿瘤相关配体,如MICA/B,可以诱导应激细胞杀伤的正常机制。下面的途径展示了肿瘤细胞如何通过MICA/B的蛋白水解性裂解,逃避免疫监视和免疫细胞介导的杀伤。
Mica/B是免疫系统清除潜在危险细胞的警告信号

对癌症基因组图谱中NKG2D配体表达的分析表明,MICA和MICB是NKG2D的两种最常见的配体,在各种肿瘤类型中都有表达。在下面显示的TCGA分析结果中,红色阴影表示NKG2D配体的高表达水平,蓝色阴影表示低表达水平。我们相信,MICA/B在许多肿瘤类型中的阳性表达情况在各种适应症中提供了有吸引力的发展机会。
15
MICA/B是在多种肿瘤类型中表达最高的NKG2D配体
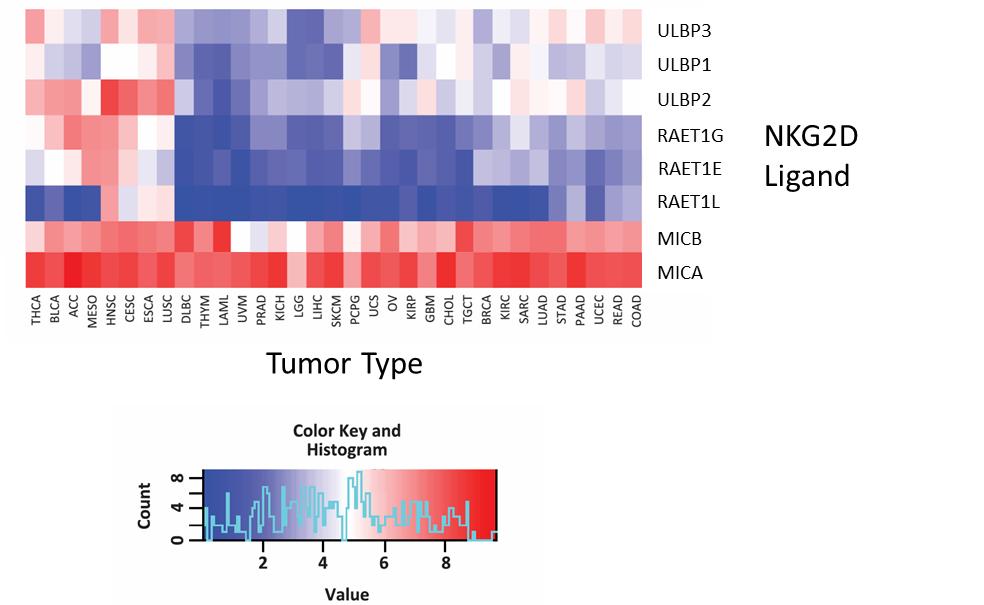
16

通过分析Monoceros BiosSystems的TCGA数据库生成的数据。
CLN-619
CLN-619是一种针对MICA/B的人源化IgG1抗体,具有活性的Fcγ1结构域,能够通过与天然免疫细胞上的Fcγ受体结合来介导效应细胞的功能。
17
我们认为CLN-619可能通过多管齐下的作用机制影响抗肿瘤活性。首先,我们认为CLN-619可以阻止癌细胞上MICA和MICB被肿瘤微环境中常见的蛋白质酶(下图中的“1”)所切割。这一机制将使MICA/B在癌细胞表面积聚。在临床前研究中,用亲本CLN-619克隆治疗导致不同肿瘤细胞系细胞表面表达增加和血清MICA/B水平降低,而CLN-619治疗体内导致血清MICA/B水平下降。癌细胞表面MICA/B的高表达有望通过其NKG2D受体与MICA/B的结合而增强NK细胞对肿瘤细胞的杀伤作用。全球司T细胞和自然杀伤T细胞,其中NKG2D可以起到共激活受体的作用,降低T细胞介导的癌细胞溶解的门槛。其次,CLN-619有一个带有野生型Fc伽玛域的人IgG1骨架,这使得它能够通过结合NK细胞的Fc来与其结合gR(三)A,通向ADCC(在下图中标为“2”)。在临床前研究中,使用CLN-619治疗可诱发ADCC体外培养。第三,CLN-619的野生型Fc伽马结构域使其能够通过巨噬细胞的参与(在下图中标为“3”)来介导靶肿瘤细胞的ADCP。最后,我们的初步临床前数据表明,CLN-619可能具有增强MICA/B与NK细胞或其他免疫细胞上的NKG2D受体的结合的潜力,以改善癌细胞的溶解(下图中标为“4”)。我们认为,所有这些机制可能以协同的方式作用于NK细胞和其他免疫细胞,这可能导致下文描述的临床前研究中观察到的癌细胞溶解。
CLN-619的三种操作模式

临床前数据
CLN-619抗肿瘤活性的关键机制是其稳定和阻止癌细胞表面表达的MICA/B脱落的能力。在临床前研究中,CLN-619防止了多种癌细胞系的脱落。在一个具有代表性的肝癌PLC/PRF/5细胞系中,与对照抗体相比,CLN-619处理后上清液中的可溶性MICA减少,如下图所示。
CLN-619抑制肿瘤细胞MICA/B的脱落

18
CLN-619还证实了增强NK细胞介导的对表达MICA/B的癌细胞的杀伤能力体外培养。在ADCC报告生物测定中,CLN-619的亲本克隆具有来自CLN-619的小鼠杂交瘤的抗体可变区序列,以剂量依赖和MICA/B结合的方式诱导ADCC,如下图所示,其中杀伤活性通过相对发光单位(RLU)来测量。当Fc区的突变被引入h3F9-Dana中时,这种ADCC活性被取消,从而消除了与Fc的结合NK细胞上的RIIIa是介导ADCC的关键。同型对照也未能触发ADCC,证明了MICA/B目标交战的要求。
CLN-619亲本克隆在低浓度下对细胞的杀伤作用

在多种小鼠肿瘤模型上进一步评价了CLN-619的抗肿瘤活性。在一个典型的肺癌异种移植模型中,CLN-619作为单一药物治疗在所有测试剂量下都能抑制肿瘤生长,如下图所示。我们还观察到,根据可溶性MICA血清水平的测量,在测试的所有剂量下,都能抑制MICA的脱落。
19
CLN-619对肿瘤生长有抑制作用
HCC1534肺癌异种移植模型的建立

正在进行的临床发展计划
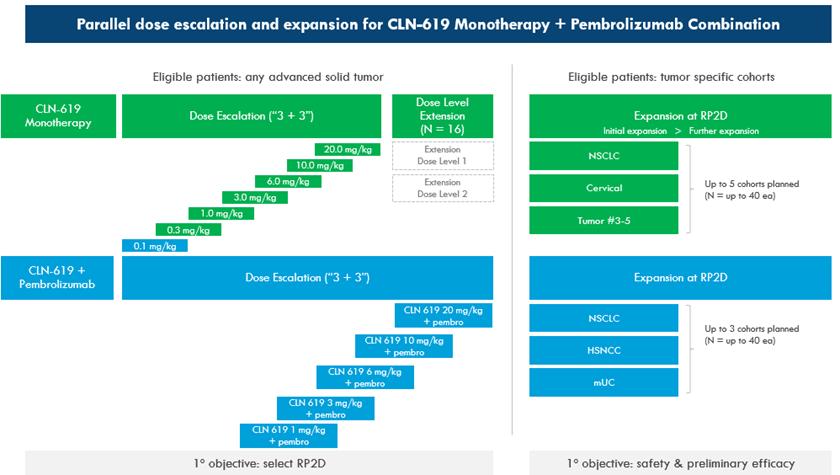
我们目前正在对CLN-619进行评估,这是一项针对晚期实体瘤患者的全球第一阶段临床试验。试验设计包括将CLN-619作为单一疗法的初步评估,以及在平行剂量递增队列中与检查点抑制剂疗法相结合。在确定推荐的第二阶段剂量后,试验设计包括几个扩展队列,以评估CLN-619作为单一疗法以及与检查点抑制药联合治疗多种实体肿瘤类型患者的初步疗效。计划中的单一疗法扩展队列包括一个NSCLC,一个宫颈癌,以及多达三个额外的队列,这些队列将根据研究剂量递增部分的观察确定。计划的联合扩展队列包括非小细胞肺癌、头颈部鳞状细胞癌和转移性尿路上皮癌。此外,我们还将收集和分析包括Smica在内的生物标志物,以了解CLN-619的未来发展。单一治疗剂量升级于2021年12月启动,在3 mg/kg单一治疗剂量水平通过剂量限制毒性评估后,按计划于2022年启动了使用检查点抑制剂治疗的剂量升级。我们预计在2023年年中提交初步临床数据。
20
CLN-619的全球一期临床试验设计
CLN-418
概述
CLN-418是一种针对B7H4和4-1BB的全人双特异性抗体。2023年2月,我们从Harbour BioMed US Inc.(“Harbour”)获得了在美国开发和商业化CLN-418的独家权利,以换取2500万美元的预付许可费,外加高达1.48亿美元的开发和监管里程碑付款,高达4.15亿美元的基于销售的里程碑付款,以及针对潜在的美国商业销售最高可达十几岁的分级特许权使用费。我们目前正在评估CLN-418在美国和澳大利亚进行的一项第一阶段临床研究,用于晚期实体肿瘤患者。
B7H4和4-1BB的背景
我们认为B7H4是癌症免疫治疗的一个有吸引力的靶点。B7H4是B7免疫调节蛋白家族的成员,包括重要的免疫抑制配体,如PD-L1。B7H4表达于肿瘤细胞表面,并已被证明通过抑制T细胞的增殖和扩增而发挥免疫抑制作用。B7H4在患者肿瘤样本中的表达与疾病晚期、预后不良和患者总生存期较低相关。虽然B7H4在一系列实体肿瘤中的表达水平具有潜在的临床意义,包括三阴性乳腺癌、非小细胞肺癌和卵巢癌,但它在正常组织中的表达有限,这表明它有可能成为一个广泛的治疗窗口。最后,B7H4通常与PD-L1的表达有很小的重叠,我们认为这为治疗所谓的“冷肿瘤”创造了可能性,目前PD-1途径的免疫治疗方法对这些肿瘤的疗效有限。下面的小组包括来自Harbour进行的各种研究的B7H4表达数据。
21
B7H4在实体瘤类型中的表达 B7H4与PD-L1在实体瘤细胞中的表达

4-1BB是一种表达在T细胞和NK细胞上的关键共刺激分子,已成为免疫治疗的靶点。从机制上讲,4-1BB活化可诱导T细胞增殖和存活,刺激炎性细胞因子的分泌,促进抗肿瘤免疫。针对4-1BB的单抗已被证明具有刺激免疫细胞并在患者中产生抗肿瘤反应的能力,但并不是没有挑战性的毒性问题,这些问题通常与非特异性4-1BB免疫细胞激活有关,例如肝脏毒性被认为是由肝脏中存在的Fcγ受体表达细胞实现的FC介导的交联性引起的。
CLN-418
CLN-418是一种全人双特异性抗体,含有人B7H4和4-1BB结合域,这些结合域被选择为与B7H4具有高结合亲和力,并旨在使B7H4-交联型4-1BB激活。CLN-418包括一个沉默的Fc结构域,旨在避免由Fcγ受体表达细胞介导的交联。通过靶向肿瘤细胞上的B7H4,我们相信CLN-418代表了一种独特的方法,能够在肿瘤微环境中实现4-1BB的有条件激活。由于这一机制以及由于CLN-418缺乏活性的Fc结构域,我们认为CLN-418有可能避免与Fc功能的单克隆4-1BB激动剂抗体所见的非特异性4-1BB激活相关的毒性。此外,CLN-418可能具有与其他癌症治疗方法,特别是免疫治疗方法协同作用的潜力,使其成为联合治疗的有力候选者。
CLN-418结构

临床前数据
CLN-418在多种肿瘤细胞系中的临床前研究,总结如下,证明了4-1BB T细胞的激活,通过诱导IL-2来测量,取决于B7H4的存在,不同于单克隆4-1BB激动型抗体,如urelumab和utomilumab。
22
CLN-418只能在B7H4存在的情况下诱导T细胞活化

在临床前,CLN-418显示出作为单一药物根除肿瘤的能力体内并驱动记忆反应,防止小鼠再次挑战时肿瘤生长。这些结果的一个例子如下所示,在同基因小鼠肿瘤模型中,CLN-418显示出剂量依赖的抑制肿瘤生长的作用,而使用CLN-418治疗后初始完全反应的小鼠随后在肿瘤再次攻击时获得免疫。
CLN-418清除小鼠体内的肿瘤,并在再次挑战时驱动防止肿瘤生长的记忆反应


正在进行的临床发展计划
我们目前正在评估CLN-418在美国和澳大利亚进行的一项第一阶段临床研究,用于晚期实体肿瘤患者。第一部分剂量升级将测试最多七个剂量水平的CLN-418,从每公斤0.03毫克到20毫克,每三周剂量一次。一旦确定了建议的第二阶段剂量,第二部分剂量扩展将评估三个肿瘤特定扩展队列中的每个队列中的多达30名患者:一个在非小细胞肺癌中,一个在乳腺癌中,第三个将根据在临床研究第一部分中观察到的疗效信号来确定。与此同时,我们正在探索扩大试验范围的机会,以包括一种或多种组合药物,其中可能包括研究研究中批准的免疫疗法药物或其他新出现的治疗候选药物。正在进行的试验第一部分的初步数据预计将于2024年公布。
CLN-978
概述
CLN-978是一种半衰期延长的人源化单链双特异性抗体,旨在同时结合癌细胞上的CD19和T细胞上的CD3,触发重定向的T细胞裂解目标癌细胞。此外,CLN-978具有一个HSA结合域,旨在延长其血清半衰期。CLN-978介导的CD19依赖的细胞裂解体外培养具有一系列CD19目标表达水平的靶细胞系。在临床前阶段体内研究表明,在人源化的淋巴瘤异种移植小鼠模型中,CLN-978在极低和罕见的剂量下治疗,导致了肿瘤生长和肿瘤消退的抑制。我们打算初步评估CLN-978作为治疗B-NHL的新疗法,目前正准备在2023年进入第一阶段临床研究。
我们设计了CLN-978,以CD19和CD3的单链抗体为基础,类似于blinatumomab。此外,我们还以单域抗体(VHH)的形式加入了第三个结构域,用于与人血清白蛋白结合。我们认为CLN-978与白蛋白的结合有可能延长其血清半衰期。下图显示了CLN-978结构的图示。
23
延长血清半衰期的CD19/CD3双特异性T细胞结合蛋白CLN-978的设计

我们已经与ADIMAb,LLC(“ADIMAb”)合作,生成针对CD19、CD3和HSA的抗体衍生结合域,具有优化的生物物理和生化特性、量身定制的结合亲和力以及对候选药物的可开发性、可制造性和临床前测试至关重要的其他参数。在临床前研究中,CLN-978在重定向T细胞裂解低CD19表达细胞方面显示出强大的抗肿瘤活性体外培养和显著的肿瘤生长抑制体内。我们相信这些临床前结果支持对CLN-978的进一步评估,因为它有潜力改善现有疗法的临床疗效。此外,我们相信靶向CD19低表达细胞的能力将潜在地使我们能够解决患者,如那些在CAR-T治疗后进展的患者。
临床前数据
CLN-978结合了一个CD19结合域,该结合域被设计成实现与CD19的微克分子结合亲和力,通过等离子共振测量,我们认为这可能有助于对CD19低表达的细胞株的细胞溶解能力体外培养研究。如下图所示,CLN-978针对工程A20细胞系中的一系列CD19表达水平进行评估,通过重定向细胞裂解的皮摩尔EC50值和最大裂解百分比来衡量。我们相信这一观察结果支持我们的假设,即CLN-978可能有可能更好地解决CD19表达水平较低的患者群体和/或CD19表达下调作为CD19靶向治疗耐药机制的患者。研究还表明,靶细胞的强劲裂解依赖于CD19的表达,因为A20亲本细胞系缺乏CD19的表达,在所测试的任何药物浓度下都不对裂解敏感。
CLN-978重定向CD19低表达细胞系的细胞裂解体外培养细胞毒性检测

CLN-978也显示出抗肿瘤活性。体内在人源化模型中,将人Raji-荧光素酶淋巴瘤细胞系(Raji B.luc)和人PBMC植入小鼠体内。如下图所示,CLN-978在测试的每个剂量水平上都显示出显著的肿瘤生长抑制作用,无论是通过静脉(“IV”)还是皮下(“SC”)给药。此外,在每公斤3微克或更高的剂量水平下,CLN-978治疗导致100%的受试小鼠完全反应,无论给药途径如何。
24
CLN-978在Raji-荧光素酶诱导的人PBMC小鼠模型中的抗肿瘤活性

与肿瘤生长抑制一致,我们观察到Raji B.luc细胞和正常人CD19的数量在统计上显著减少+所有CLN-978处理组的B细胞,如下图所示。
CLN-978对HuPBMC Raji B.luc小鼠模型外周血中正常B细胞和肿瘤细胞的影响

正在进行的临床发展计划
我们在2022年第四季度提交了CLN-978的IND。FDA在2023年1月批准了我们的IND。我们仍将在2023年开始一项第一阶段临床试验,评估CLN-978在复发/难治性B-NHL患者中的应用。
25
CLN-617
概述
CLN-617是一种融合蛋白,在单一试剂中独特地将两种有效的抗肿瘤细胞因子IL-2和IL-12与用于治疗实体瘤的胶原结合结构域结合在一起。IL-2和IL-12的联合应用先前已被证明可以协同增强T和NK细胞的功能体外培养并在临床前肿瘤模型中介导显著的治疗活性,即使在具有原发和/或转移肿瘤的已建立的小鼠模型中也是如此。近50年来,临床研究人员一直在研究细胞因子在刺激癌症免疫反应中发挥的强大作用。然而,与全身应用细胞因子相关的严重毒性和较短的血清半衰期阻碍了它们的临床发展和更广泛的商业应用。尽管在蛋白质工程、递送和靶向机制方面取得了许多进展,但目前只有两种FDA批准的基于细胞因子的癌症治疗方法,最近一次批准是在20多年前。
我们在CLN-617的设计中包括了多种不同的功能,以解决细胞因子治疗的历史局限性。首先,肿瘤内注射后的肿瘤滞留是由胶原蛋白结合域和HSA实现的,由于所有实体肿瘤中都存在胶原蛋白,HSA增加了分子量,进一步降低了肿瘤组织向外扩散的速度。保留可能有助于将IL-2和IL-12的全身扩散和相关毒性降至最低,并延长它们在肿瘤中的免疫刺激抗肿瘤活性。其次,CLN-617是我们所知的唯一可共同传递IL-2和IL-12蛋白的候选产品,在功能上能够协同T细胞和NK细胞激活。第三,CLN-617的结构使用完全野生型结构域,这潜在地降低了与工程细胞因子相关的免疫原性风险。最后,与其他基于细胞因子的肿瘤内治疗不同,CLN-617不依赖于病毒感染或核酸转染就地表情。
CLN-617设计

在临床前研究中,CLN-617的小鼠替代品在治疗和未治疗的肿瘤中都显示出强大的单药抗肿瘤活性,而不会引起全身毒性,以体重减少为衡量标准。鉴于胶原蛋白在多种肿瘤类型中的广泛表达,以及基于细胞因子的治疗的良好验证的抗肿瘤活性,我们相信CLN-617可能在广泛的实体肿瘤中具有实用价值。我们认为CLN-617是一个一流的机会,因为它是我们所知的唯一抗癌候选产品,旨在共同传递IL-2和IL-12细胞因子,并将它们保留在肿瘤微环境中。
CLN-617中使用的胶原蛋白结合保留技术是基于麻省理工学院Karl Dane Wittrup教授的实验室开发的技术。我们进一步开发和改进了这项技术,以创建我们的琥珀平台,我们相信这是一个新的平台,具有拓宽细胞因子和其他免疫刺激剂的治疗窗口的潜力,并大幅降低全身毒性。
临床前数据
产生了一个含有CLN-617组成结构域的小鼠同源物的小鼠替代品,并命名为mCLN-617。在携带B16F10肿瘤的小鼠身上发现了mCLN-617的滞留,无论是瘤内还是静脉注射mCLN-617。治疗2小时后,检测mcln-617在血清中的浓度,我们发现瘤内注射后的药物浓度平均为
26
瘤内给药与静脉给药相比降低全身mCLN-617水平和改善疗效

我们利用MC38同基因肿瘤模型来评估mCLN-617治疗是否可以在肿瘤初次清除后产生长期免疫记忆。如下面的左图所示,mCLN-617在治疗的动物中导致100%的完全反应,而在对照组或抗PD1治疗的组中没有观察到完全反应。随后,我们将肿瘤重新注射到动物体内,取得了完全反应。如下面的右图所示,之前用mCLN-617治疗的小鼠中有五个排斥新注射的肿瘤,并且所有生长出来的肿瘤的生长速度都慢于年龄匹配的幼稚对照组的肿瘤。在CT26同基因肿瘤模型中也获得了类似的结果。
MCLN-617治疗后高完全缓解率和记忆反应

我们证明,除了介导长期记忆外,mCLN-617还能够对已建立的未治疗的远端肿瘤产生反应,这是由于诱导系统免疫,也称为非内窥镜效应。我们使用了两个MC38肿瘤的C57BL/6小鼠,其中只有一个用mCLN-617单独或与系统传递的抗PD1抗体联合治疗。MCLN-617单独介导了治疗和未治疗肿瘤的显著消退,包括完全反应。在未经治疗的肿瘤中,联合抗PD1治疗的效果更加一致和持久,尽管单独使用抗PD1治疗没有显著的生存益处。这些结果显示了mCLN-617与全身注射的抗PD1抗体的异地效应和协同作用。
27
瘤内注射mCLN-617抑制已治疗和未治疗肿瘤的生长并与系统注射的抗PD1治疗协同作用

通过我们正在进行的对mCLN-617的研究,我们正在寻求表征局部保留的细胞因子融合可以产生强大的全身抗肿瘤免疫的作用机制。到目前为止,在携带两种MC38肿瘤的小鼠身上使用mCLN-617的研究表明,在治疗和未治疗的肿瘤中,效应性T细胞和免疫抑制调节性T细胞的比率都有所改善。此外,肿瘤特异性T细胞在外周血中扩增,提供了疫苗样作用机制的证据。
已经产生了完全人类CLN-617临床候选基因,并证实了细胞因子和胶原结合域的生物活性体外培养。非GLP药代动力学研究和使用CLN-617的GLP毒理学研究都已在非人类灵长类动物中进行,以支持IND申请。
根据我们对mCLN-617的临床前研究结果,我们相信,我们的琥珀平台包含一个胶原结合结构域,有可能允许在肿瘤微环境中安全地保留高水平的细胞因子。虽然随着检查点抑制剂的采用,癌症的治疗已经取得了显著的进展,包括pembrolizumab、ipilimumab和nivolumab,但只有少数实体瘤患者对这些治疗有反应。我们相信,一种耐受性良好的药物可以提供IL-2和IL-12的功能协同作用,有可能治疗广泛的实体肿瘤,包括那些对检查点抑制剂无效的肿瘤。
2023年2月,我们提交了CLN-617的IND申请,并打算在2023年底之前启动第一阶段研究,等待IND批准。
我们的其他临床前项目
除了上述候选产品外,我们还在积极开发几个临床前肿瘤学项目,这些项目都处于发现阶段,包括我们与西奈山伊坎医学院合作开发新型造血祖细胞激酶1(HPK1)降解物。
竞争
生物技术和制药行业的特点是技术的快速发展和对疾病病因的了解、激烈的竞争和对知识产权的高度重视。我们相信,我们差异化的商业模式、方法、科学能力、技术诀窍和经验为我们提供了竞争优势。然而,我们面临并将继续面临来自专注于更传统治疗方式的公司的竞争,例如小分子抑制剂。我们预计来自多种来源的激烈竞争,包括世界各地的主要制药、专业制药和现有或新兴的生物技术公司、学术研究机构、政府机构以及公共和私人研究机构。我们的许多竞争对手,无论是单独或通过合作,在研发、制造、临床前测试、进行临床试验、获得监管批准和营销批准的产品方面,都比我们拥有更多的财务资源和专业知识。规模较小或处于初创阶段的公司也可能成为重要的竞争对手,特别是通过与大型和成熟公司的合作安排。这些公司还在招聘和留住合格的科学和管理人员、建立临床试验场地和在临床试验中招募患者以及在获取与我们的计划相辅相成或必要的技术方面与我们竞争。因此,我们的竞争对手可能比我们更早或更成功地发现、开发、许可或商业化产品。
28
我们还面临着更广泛的肿瘤学市场上对具有成本效益和可报销的癌症治疗的竞争。治疗癌症患者最常见的方法是手术、放射和药物治疗,包括化疗、激素治疗、生物治疗(如单抗和双特异性抗体)、免疫治疗、基于细胞的治疗和靶向治疗,或任何这些方法的组合。市场上销售的癌症药物疗法多种多样。在许多情况下,这些药物联合使用以提高疗效。虽然我们的候选产品(如果获得批准)可能会与这些现有的药物和其他疗法竞争,但如果它们最终与这些疗法结合使用或作为这些疗法的附件使用,我们的候选产品可能无法与它们竞争。其中一些药物是品牌药物,受专利保护,其他药物是仿制药。保险公司和其他第三方付款人也可能鼓励使用非专利产品或特定品牌产品。因此,我们可能会面临挑战,以获得市场接受,并获得相当大的市场份额,为我们的任何产品,我们成功地推向市场。此外,许多公司正在开发新的肿瘤学疗法,我们无法预测随着我们的候选产品在临床开发中取得进展,护理标准将是什么。
至于我们正在与泰豪公司的一家附属公司共同开发的齐帕替尼,我们知道还有其他EGFR抑制剂已经加速批准或正在临床开发中,用于治疗携带EGFRex20ins突变的非小细胞肺癌患者。2021年5月,强生开发并上市的EGFR/cMET双特异性抗体Rybrevant(Amivantimab)获得了美国食品和药物管理局的加速批准,用于治疗在铂类化疗期间或之后病情进展的局部晚期或转移性非小细胞肺癌患者。此外,2021年9月,由武田制药公司开发和销售的Exkitivity(Mobocertinib)获得了FDA的加速批准,用于患有局部晚期或转移性非小细胞肺癌(带有EGFRex20ins突变)的成年患者,这些患者的疾病在以铂为基础的化疗中或之后有所进展。我们认为,针对EGFRex20ins患者的最先进的临床阶段计划是Dizal制药有限公司的sunvozertinib,以及Arrient BioPharma,Inc.和Allist制药公司的Furmonertinib。其他临床阶段的EGFR ex20ins TKI计划包括奥瑞克制药公司的ORIC-114(沃罗诺伊公司,中国大陆、香港、澳门和台湾)和Blueprint制药公司的液化天然气-451。
关于CLN-049,我们知道有几家公司正在开发治疗AML的双特异性药物,包括针对CD3和CD33的公司(安进(“安进”)和Amphivena治疗公司)、CD123(MacroGenics公司和Xencor,Inc.)以及CCL1/Clec12a(Merus N.V.和Genentech,Inc.)。安进公司正在开发一种针对AML的Flt3的双特异性T细胞订户。也有几种靶向小分子疗法被批准用于治疗r/r或一线AML,包括用于具有Flt3突变的AML,例如Astellas Pharma Inc.的XOSPATA(吉特替尼)和诺华国际股份公司(Novartis International AG)的RYDAPT(米多妥林)。我们还知道有其他小分子被批准或正在开发中,用于治疗Flt3突变的AML患者,包括IDH抑制剂,如Servier PharmPharmticals LLC的TIBSOVO(Ivosidenib)和Agios PharmPharmticals,Inc.的IDHIFA(Enasidenib),BCL2抑制剂,如艾伯维公司的VENCLEXTA(Ventoclax),以及hedgehog通路抑制剂,如辉瑞公司(Pfizer)的DAURISMO(Glasdegib)。
关于CLN-619,我们知道有几家公司正在开发将MICA/B作为单一疗法和/或与其他药物联合使用的癌症疗法,包括:CanCure LLC、Genentech Inc.、诺华和百时美施贵宝公司(Bristol-Myers Squibb Company)。据我们所知,这些药物都没有进入临床开发阶段。
关于CLN-418,我们不知道有任何其他候选者在开发中将4-1BB和B7H4结合在一个单一的双特异性分子中。然而,有几个候选分子的目标是这些分子中的一个或另一个。例如,Bristol Meyers Squibb公司的urelumab是靶向4-1BB的单抗。几家公司正在开发针对B7H4的单抗、抗体-药物结合物或(CD3)T细胞结合体。这些公司包括安进公司、Genmab A/S公司(“Genmab”)、辉瑞公司、阿斯利康公司(“阿斯利康”)、赛根公司、默萨纳治疗公司和江苏翰森制药有限公司。几家公司还在开发针对其他肿瘤相关抗原而不是B7H4的4-1BB激动剂(如PD-L1、FAP、CD40等)。包括Genmab,Inhibrx,Inc.,Eli Lilly and Company,Boehringer Inglheim Pharma GmbH和Roche AG(以下简称罗氏)。
关于我们的CLN-978计划,我们知道有一些公司正在开发针对CD19或其他与B细胞ALL和B-NHL相关的肿瘤抗原的候选产品,使用免疫细胞或其他细胞毒方式。这些主要包括免疫细胞重定向疗法(例如,T细胞激活剂)、过继细胞疗法(例如,CAR-T)和抗体药物结合物。开发针对CD19的细胞疗法或抗体的公司包括但不限于阿斯利康、Morphosys AG、诺华、吉利德科学、百时美施贵宝、异种基因治疗公司、世纪治疗公司、Nkarta公司、Autolus治疗公司、ADC治疗公司和安进公司。
关于CLN-617,我们不知道目前有任何其他候选药物正在开发中,可以将IL-2和IL-12整合到一个单一的多功能结构中,并以肿瘤特异性的方式刺激免疫系统。我们知道有几家公司正在积极开发单独的IL-2或IL-12疗法的临床阶段计划,包括:Alkermes plc、赛诺菲公司、Philogen S.p.A.、罗氏公司、ApeIron Biologics AG、Werewolf Treateutics,Inc.、Xilio Treateutics,Inc.和蜻蜓Treateutics Inc.。
29
如果我们的竞争对手开发和商业化比我们的候选产品更安全、更有效、副作用更少或更不严重、更方便管理、更便宜或标签更有利的药物,我们可能会看到商业机会减少或消失。我们的竞争对手也可能比我们更快地获得FDA或其他监管机构对其药品的批准,这可能会导致我们的竞争对手在我们能够进入市场之前建立强大的市场地位。影响我们所有候选产品成功的关键竞争因素,如果获得批准,可能是它们的疗效、安全性、便利性、价格、仿制药竞争水平以及政府和其他第三方付款人是否可以报销。
许可协议
DKFZ/Tübingen
我们的部分持股子公司库利南佛罗伦萨是与以下机构签订的独家许可协议(“DKFZ/Tübingen许可协议”):Deutsches Krebsforschungszentum(“DKFZ”)、Eberhard Karls University of Tübingen医学院(“University of Tübingen”)和UFE(“UFE”)。根据DKFZ/Tübingen许可协议,DKFZ和Tübingen大学,统称为许可方,向Cullinan Florentine授予了一项独家的(即使对于许可方、UFE及其附属公司)全球范围内具有里程碑意义和版税的许可,在某些许可专利权、应用、技术信息和专有技术下,有权通过多个层次授予再许可,以研究、开发、商业化或以其他方式开发该领域内的许可产品。库利南佛罗伦萨有起诉和维护全球所有许可专利权的唯一权利,但没有义务,前提是如果库利南佛罗伦萨选择停止起诉或维护许可专利权,许可方可以接管或继续此类起诉和维护。
根据DKFZ/Tübingen许可协议,库利南佛罗伦萨有义务在特定日期(统称为“绩效日期”)之前达到某些监管和研发绩效基准(统称为“绩效基准”)。如果在适用的履行日期前未能达到绩效基准,则库利南佛罗伦萨可通过向许可方发出书面通知并每次延期支付不可退还、不可计入的延期费用,将任何单一绩效基准的履行日期延长中位数至个位数的月数。库利南佛罗伦萨可将任何单一绩效基准的绩效日期延长至较低的个位数次数,前提是库利南佛罗伦萨只能请求延长中位数至个位数的次数。如果库利南佛罗伦萨无法根据前一句话寻求进一步延期,则库利南佛罗伦萨可通过向许可方提供书面通知来寻求进一步延期,任何此类延期应事先获得许可方的书面批准,此类批准不得无理扣留或拖延。截至2022年12月31日,库利南佛罗伦萨已达到创建主细胞库的首个性能基准。
此外,在许可产品发生某些临床和监管事件时,库利南佛罗伦萨公司应向许可方支付某些不可退还、不可贷记的里程碑付款,无论是由库利南佛罗伦萨公司、其附属公司或分被许可人触发的。每笔里程碑式的付款只需支付一次,总金额最高可达2800万美元。根据DKFZ/Tübingen许可协议,到目前为止还没有实现任何里程碑。
此外,库利南佛罗伦萨还需要在特许权使用费期限内,按每个国家和产品的每种许可产品的净销售额支付低至中个位数的特许权使用费百分比,但须进行某些补偿或减少。许可方在一个日历年度内因任何许可产品的净销售额而应向许可方支付的全球版税总额不得低于低至中个位数的百分比。这种专利使用费义务将在下列两者中较晚的一种情况下到期:(A)涵盖该国产品的专利的最后一项有效权利要求到期时,以及(B)产品在该国首次商业销售后两位数的低周年纪念日。在某些情况下,在首次控制权变更时,库利南佛罗伦萨应支付销售收益的不可退还、不可贷记的个位数百分比,但在库利南佛罗伦萨完成首次公开募股后不需要支付此类款项。
任何一方在另一方实质性违约或另一方资不抵债时,均可终止协议。库利南佛罗伦萨可在首次提交IND或临床试验协议后,通过提供事先书面通知,以任何或无理由终止DKFZ/Tübingen许可协议。如果佛罗伦萨库利南公司或其任何附属公司对某些专利权的有效性提出质疑,许可方可以通过事先书面通知的方式终止协议。除非提前终止,否则DKFZ/Tübingen许可协议将永久继续。请参阅注7,许可和协作协议有关DKFZ/Tübingen许可协议的更多详细信息,请参阅本年度报告Form 10-K中包含的综合财务报表。
30
麻省理工学院
我们的部分股权子公司库利南琥珀是与麻省理工学院签订的独家专利许可协议(“麻省理工学院许可协议”)的一方。根据麻省理工学院的许可协议,麻省理工学院根据某些许可的专利权利和申请,向库利南琥珀授予了独家的、全球性的、里程碑式的、具有股权和专利权使用费的许可,有权通过三个层次授予再许可(只要库利南琥珀仍然是该领域全球专利权的独家许可人),以开发、制造、制造、使用、销售、销售、要约销售、租赁和进口在诊断、预后、预防或治疗人类或其他动物癌症领域含有特定融合蛋白的许可产品。麻省理工学院应准备、归档、起诉和维护所有专利权,库利南琥珀将配合检方,就专利起诉文件提供意见,并支付与此类起诉和维护有关的所有费用和费用。
库利南琥珀公司应向麻省理工学院报销麻省理工学院与专利权的准备、备案、起诉、维护和辩护相关的某些有据可查的自付费用。麻省理工学院许可协议还规定了反稀释调整,要求库利南琥珀公司增发麻省理工学院股票,以确保向麻省理工学院发行的股份不低于个位数中位数百分比金额,直到达到代表库利南琥珀公司总投资的融资门槛。麻省理工学院还获得了在库利南琥珀的任何拟议融资中的参与权,最高可达发行证券的两位数百分比。库利南琥珀还负责支付不可退还的、可计入贷方的年度许可证维护费,在许可证的一定年限内递增支付,并在这段时间后支付固定金额。此外,麻省理工学院授予库利南琥珀独家选择权,以修改字段的定义以包括扩展字段,每一项此类修订都将触发向麻省理工学院支付修正费,并导致对库利南琥珀关于特许产品的财务义务的修正案,在行使选择权时进行谈判,以反映所增加的额外权利和价值。
此外,库利南琥珀公司应在其自身或其分被许可人实现某些临床和监管里程碑时,向麻省理工学院支付某些不可退还、不可贷记的里程碑付款,每种不同的许可产品的总金额最高可达700万美元。每笔里程碑付款仅支付一次,最高不超过一定的付款金额,除非授权产品的第二次和第三次指示需要单独支付里程碑付款,每种产品的总金额最高可达550万美元。库利南琥珀还应向麻省理工学院支付某些一次性里程碑付款,以实现某些商业里程碑,该付款是根据所有适应症中所有许可产品的净销售额计算得出的,总金额最高可达1250万美元。截至2022年12月31日,麻省理工学院许可协议下尚未实现任何里程碑。
在某些情况下,在控制权变更时,库利南琥珀需要支付指定的控制权变更费用,库利南琥珀的临床和监管里程碑付款将增加某个较低的三位数百分比。
此外,库利南琥珀公司需要在每个报告期内按所有许可产品的净销售额支付中位数至个位数的版税百分比,但须进行某些补偿或减少。麻省理工学院对所有特许产品的净销售额的版税不得减少超过50%。库利南琥珀公司还被要求分享从许可产品的再许可中获得的任何收入,百分比将由许可产品的临床阶段确定,不超过中低两位数的百分比。此类专利使用费义务将在所有已颁发专利和在专利权范围内提交的专利申请到期或被放弃时,在逐个国家和逐个产品的基础上到期。
根据麻省理工学院许可协议,麻省理工学院必须向库利南·安珀通报在一段时间内构思并付诸实施的某些可专利发明(“改进”),库利南·安珀有权在麻省理工学院批准商业和发展计划时获得这些改进的权利,但不得无理扣留,并支付一定的费用。除了这一指定费用外,库利南琥珀与改进有关的财务义务可能会被修改,以反映所增加的价值,例如通过增加预付款、维护费和里程碑付款。
在提前提供书面通知后,库利南·琥珀可以任何理由自愿终止MIT许可协议,前提是应付给MIT的所有金额都已支付。如果库利南·安珀停止开展与麻省理工学院许可协议相关的业务,麻省理工学院有权在书面通知库利南·安珀后终止麻省理工学院许可协议。任何一方如发生重大违约行为,均可终止麻省理工学院许可协议。除非提前终止,否则麻省理工学院许可协议将保持有效,直到所有已发布的专利到期或被放弃,以及在专利权范围内提交的专利申请。请参阅注7,许可和协作协议有关麻省理工学院许可协议的更多详细信息,请参阅本年度报告Form 10-K中包含的综合财务报表。
31
阿迪玛布
我们与Adimab有合作协议(“Adimab合作协议”)。根据Adimab合作协议,我们选择了个位数的生物靶点,Adimab使用其专有平台技术根据双方商定的研究计划发现和/或优化抗体。根据ADIMAb合作协议,我们有能力选择指定的较低个位数的额外生物靶标,ADIMAb将针对这些靶标提供额外的抗体发现和优化服务。
在Adimab的特定研究计划的研究期限和评估期内,我们拥有Adimab技术下的非独家全球许可,可以执行某些研究活动并评估计划抗体,以确定我们是否希望行使其选择权,根据Adimab的背景专利权获得免版税、全额支付的非独家许可,以利用此类抗体可通过多个层级亚致敏(“Adimab选项”)。如果我们行使Adimab期权,我们将根据一定的调整为每个目标支付期权费用。
根据Adimab合作协议,我们一次性支付了不可计入且不可退还的技术访问费。我们还需要支付与Adimab全职员工履行Adimab合作协议义务相关的年度访问费和研究资金费用。我们还有义务向Adimab支付某些研究交付、临床和销售里程碑付款,每种产品的总金额最高可达1580万美元,按产品分类,受某些折扣和折扣的限制。
此外,我们有义务按产品按年全球净销售额的较低个位数百分比支付某些特许权使用费。此类专利使用费义务将在以下两者中较晚的时间按国家/地区和产品终止:(A)此类产品在该国家/地区首次商业销售后的某个较低的两位数年数,以及(B)涵盖协议中定义的此类产品的计划专利中最后发出且未过期、永久撤销或无效的权利要求到期时。
我们可以在指定期限内提前书面通知,以任何理由随时终止Adimab合作协议。如果没有行使ADIMAB期权,或者如果行使了ADIMAB期权,则ADIMAB合作协议的有效期在最后一个研究项目的评估期届满时到期,或者在ADIMAB期权被行使的情况下,在其特许权使用费期限在指定期限的较晚者或相关产品的无效专利覆盖范围届满后到期。请参阅注7,许可和协作协议,我们的合并财务报表包含在本年度报告的Form 10-K中,以了解有关Adimab合作协议的更多细节。
海港
2023年2月,我们与Harbour BioMed US Inc.(“Harbour”)签订了许可和合作协议(“Harbour许可协议”),根据该协议,Harbour授予我们在美国开发、制造和商业化HBM7008的独家许可,我们现在将其称为CLN-418。
根据海港牌照协议的条款,我们在签订协议时向Harbour预付了2,500万美元的许可费。此外,根据协议,港湾将有资格获得(I)基于预先指定的开发和监管里程碑的实现而获得的高达1.48亿美元的里程碑付款,以及(Ii)高达4.15亿美元的额外基于销售的里程碑,以及基于许可产品的分级特许权使用费,按许可产品占美国商业销售额的百分比,最高可达十几岁。海港将授予我们某些知识产权,使我们能够履行我们在港口许可协议下的义务和行使我们的权利。
除非提前终止,否则海港许可协议将继续有效,直到我们的特许权使用费义务到期。港口许可协议可由任何一方因另一方的重大违约而终止,但须遵守通知和补救条款,或在另一方破产的情况下终止。为方便起见,我们可以提前90天书面通知海港公司终止《海港许可证协议》。
知识产权
我们的知识产权对我们的业务至关重要,我们努力保护它,包括在美国和国际上为我们的专利治疗分子、技术、改进、平台、候选产品及其组件、新的生物发现、新的治疗方法和潜在的适应症以及其他对我们的业务重要的发明获取、维护、捍卫和执行专利和其他知识产权。对于我们的候选产品,通常我们最初寻求的专利保护包括物质的成分、使用方法和生产方法。在我们候选产品的整个开发过程中,我们将寻求确定获得专利保护的其他方法,这些方法可能会提高商业成功,包括改进药物配方、使用方法和生产。
32
截至2023年2月16日,我们的专利组合包括9个专利系列,包括我们拥有的专利申请,以及各自领域的外部技术发起者独家授权的专利申请。具体地说,我们在全球独家授权了至少31项待审的专利申请。我们的待决专利申请可能颁发的专利预计将在2039年至2043年之间到期,如果适用,不包括任何可能的专利期限调整或延长。至于延长专利期限以恢复在授予专利后实际失去的专利期限,但在FDA监管审查过程中,恢复期限不能超过五年,包括恢复期限在内的总专利期限不得超过FDA批准后的14年。
通过我们的子公司库利南MICA,我们拥有与我们的CLN-619候选产品相关的两个专利系列,包括针对组合物的专利系列,以及用于治疗的组合物的方法。如果发布了针对CLN-619组合物的权利要求的专利申请系列,预计将于2039年到期,如果适用,不包括任何专利期限调整或延长。到目前为止,这些专利家族中的第一个已经在澳大利亚、巴西、加拿大、大陆、中国、欧洲专利局、印度、印度尼西亚、以色列、日本、韩国、马来西亚、墨西哥、新西兰、菲律宾、俄罗斯、新加坡、南非、泰国、美国和越南提交了专利申请。如果发布了针对其他抗MICA抗体组合物和使用方法的权利要求,预计专利家族将于2043年到期,如果适用,不包括任何专利期调整或延长。根据这个家庭,美国已经提交了临时申请。
我们与CLN-049候选产品相关的产品组合包括图宾根大学授权的一个针对组合物的专利系列,以及用于治疗的组合物的使用方法。这一系列专利申请包含针对CLN-049组合物的权利要求,如果发布,预计将于2039年到期,如果适用,不包括任何专利期限调整或延长。到目前为止,该家族已在澳大利亚、巴西、加拿大、大陆中国、欧洲专利局、香港、印度、印度尼西亚、以色列、日本、韩国、马来西亚、墨西哥、新西兰、菲律宾、新加坡、南非、泰国、美国和越南提交了专利申请。
我们与CLN-617候选产品和库利南琥珀计划相关的产品组合包括两个专利系列。第一个家庭是从麻省理工学院获得许可的,方向是组合物,以及使用这种组合物进行治疗的方法。这一系列专利申请包含涉及库利南琥珀相关组合物的权利要求,如果发布,预计将于2039年到期,如果适用,不包括任何专利期限调整或延长。到目前为止,这个家族已经在澳大利亚、巴西、加拿大、大陆中国、欧洲专利局、以色列、日本、韩国、新加坡和美国提交了专利申请。第二个家族是库利南·安珀拥有的PCT申请,它针对某些组合物,以及这些组合物的治疗用法。该系列包含其他库利南琥珀相关成分的索赔,如果发布,预计将于2041年到期,如果适用,不包括任何专利期限调整或延长。
我们与CLN-978候选产品相关的产品组合包括三个专利系列,分别针对组合物和用于治疗的组合物的使用方法。前两个专利申请系列包含针对CLN-978组合物的权利要求,如果发布,预计将于2040年到期,如果适用,不包括任何专利期限调整或延长。这两个系列的专利申请都已在澳大利亚、加拿大、大陆中国、欧洲专利局、以色列、日本和美国提交。第三系列专利申请包含针对与CLN-978相关的组合物的替代格式的权利要求,如果发布,预计将于2039年到期。这个家族已经在澳大利亚、加拿大、大陆中国、欧洲专利局、香港、以色列、日本、南非和美国提交了专利申请。
我们的产品组合涉及含有降解HPK1的化合物的组合物和用于治疗的此类化合物的使用方法,包括与西奈山伊坎医学院共同拥有的一个专利系列。该专利家族包含针对含有HPK1降解剂的组合物和使用这种化合物的方法的权利要求。PCT已经为这一专利家族提交了申请。声称优先于PCT申请的申请,如果发出,预计将于2042年到期,不包括任何专利期限调整或延长。
个别专利的展期根据专利申请的提交日期或专利颁发日期以及获得专利的国家的专利法律期限而定。一般来说,在美国为定期提交的申请颁发的专利有效期为20年,自最早有效的非临时申请之日起算。此外,在某些情况下,专利期限可以延长,以重新获得美国专利商标局(“USPTO”)在颁发专利时的部分审查期,以及由于FDA监管审查期而实际上失去的部分期限。
33
制造业
我们不拥有或经营,目前也没有计划建立任何良好的制造规范(“GMP”)、制造设施。我们依赖,并预计将继续依赖第三方生产我们的候选产品,用于临床前和临床测试,以及用于商业生产(如果我们的任何候选产品获得市场批准)。我们还依赖,并预计将继续依赖第三方来包装、标记、存储和分发我们的研究候选产品,如果获得市场批准,还可以包装、标记、存储和分发我们的商业产品。我们相信,这一战略使我们不再需要投资于我们自己的制造设施、设备和人员,从而使我们能够保持更高效的基础设施,同时也使我们能够将我们的专业知识和资源集中在新产品候选产品的开发上。
我们从我们的合同制造组织(“CMO”)获得用于临床前测试的材料。我们收到符合当前GMP要求(“cGMP”)的临床供应材料,并在试验前和试验期间与CMO合作进行审计,以确保符合双方商定的过程描述和cGMP法规。
我们正在与泰豪的一家附属公司共同开发Zipertinib,它是一种小分子,是由可用的原料在合成过程中制造出来的。这种化学物质似乎可以放大,目前在制造过程中不需要特殊的设备。我们通常希望依赖第三方来制造配套诊断,这是一种确定合适的齐帕替尼患者群体的分析或测试。根据我们选择的技术解决方案,我们可能会依赖多个第三方来制造和销售单个测试。
到目前为止,我们已经从单一来源的第三方合同制造商那里获得了药物物质和药物产品,这在现阶段的发展阶段是典型的。我们利用全球制造商网络来开发我们的项目,供应商位于美国、欧洲和亚洲。这些合同制造商的药品或药品供应的任何减少或停止,都可能限制我们开发候选产品的能力,直到我们找到合格的替代合同制造商。然而,我们已经或计划获得足够的药物物质,以供应我们计划的初步临床研究。在开发的适当时候,我们打算签订协议,根据这些协议,我们的第三方合同制造商一般将根据我们预计的开发和商业供应需求,逐个项目地向我们提供必要数量的药物物质和药物产品。
CLN-049、CLN-619、CLN-418、CLN-978和CLN-617是从各自的生产单元线的主细胞库(MCB)的小瓶中制造的。对于根据cGMP和适用法规生产和测试的每个程序,我们都有一个MCB。我们要么已经建立了工作细胞库,要么打算在以后的产品开发中为每个候选产品生产工作细胞库。我们可能会失去来自多个地点的多个细胞库,并因需要更换细胞库而严重影响我们的制造。然而,我们相信,如果任何特定的细胞库在灾难性事件中丢失,我们有足够的后备。
政府监管
美国食品和药物管理局条例
FDA和其他美国联邦、州和地方以及外国的监管机构,除其他外,对我们正在开发的药品和生物制品的研究、开发、测试、制造、质量控制、安全性、有效性、进出口、标签、包装、储存、分销、记录保存、批准、广告、促销、营销、批准后监测和批准后报告等方面进行广泛监管。我们与我们的供应商、合作伙伴、临床研究组织(“CRO”)、临床试验研究人员和CMO将被要求满足我们希望进行研究或寻求批准我们候选产品的国家/地区监管机构的各种临床前、临床、制造和商业批准要求。获得药品监管部门的批准并确保随后遵守适当的美国联邦、州、地方和外国法律法规的过程需要花费大量的时间和财力。在产品开发过程或审批后的任何时候,如果不遵守适用的法规要求,申请人可能会在开发或审批方面受到延误,以及行政和司法制裁。
在美国,FDA根据联邦食品、药物和化妆品法案(修订后的FDCA)监管药品,根据FDCA和公共卫生服务法(修订后的PHSA)及其实施条例监管生物制品。药品和生物制品也受到其他联邦、州和地方法规的约束。我们的候选产品处于早期阶段,尚未获得FDA批准在美国上市。
34
我们的候选产品必须获得FDA批准用于治疗适应症,然后才能在美国上市。对于受FDCA监管的候选药物,如齐帕替尼,FDA必须批准新药申请(“NDA”)。对于受FDCA和PHSA监管的生物制品候选产品,如CLN-049、CLN-619、CLN-418和CLN-978,FDA必须批准生物制品许可证申请(“BLA”)。这一过程类似,通常涉及以下内容:
临床前和临床试验
在人体上测试任何药物或生物之前,候选产品必须经过严格的临床前测试。临床前研究包括化学、配方和稳定性的实验室评估,以及体外培养和动物研究,以评估安全性,并在某些情况下建立治疗使用的理由。临床前研究的进行要遵守联邦和州的法规和要求,包括GLP对安全性和毒理学研究的要求。在美国,临床前研究的结果以及生产信息和分析数据必须作为IND的一部分提交给FDA。
IND是FDA授权给人类使用研究产品的请求,必须在临床试验开始之前生效。IND提交的中心焦点是临床研究的总体研究计划和方案。IND还包括动物和体外培养评估产品的毒理学、药动学、药理学和药效学特征的研究;化学、制造和控制信息;以及任何可用人类数据或文献来支持研究产品的使用。在美国,IND在FDA收到IND后30天自动生效,除非FDA在30天内对临床试验的进行提出担忧或问题,包括担心人类研究对象将面临不合理的健康风险并强制临床搁置。在这种情况下,IND赞助商和FDA必须在临床试验开始之前解决任何悬而未决的问题。在IND提交后,一些长期的临床前试验可能会继续进行。因此,提交IND可能会导致FDA授权开始试验,也可能不会。
35
临床开发阶段涉及到在合格研究人员的监督下,根据GCP要求给健康志愿者或患者服用候选产品,这些调查人员通常是不受试验赞助商雇用或控制的医生,其中包括要求所有研究对象对他们参与任何临床试验提供知情同意。临床试验是在详细说明临床试验的目标、给药程序、受试者选择和排除标准以及用于监测安全性和评估有效性的参数和标准的方案下进行的,其中包括确保在发生某些不良事件时将停止临床研究的停止规则。作为IND的一部分,每项议定书以及随后对议定书的任何修改都必须提交给FDA。此外,每项临床试验都必须由IRB进行中央或每个进行临床试验的机构进行审查和批准,以确保参与临床试验的个人面临的风险降至最低,并与预期效益合理相关。IRB还批准必须提供给每个临床试验受试者或其法律代表的知情同意书,并必须监督临床试验直到完成。
FDA可以在最初30天的IND审查期内或在IND下的临床试验进行期间的任何时候,基于对患者安全和/或不符合监管要求的考虑,实施部分或全部临床暂停。FDA发布的这一命令将推迟拟议的临床研究或导致正在进行的研究暂停,直到所有悬而未决的问题得到充分解决,并且FDA已通知我们,调查可能会继续进行。实施临床搁置可能会导致及时完成计划中的临床研究的重大延误或困难。此外,IRB或赞助商可随时以各种理由暂停或中止临床试验,包括发现受试者面临不可接受的健康风险。一些研究还包括由临床研究赞助商组织的一个独立的合格专家小组的监督,该小组被称为数据安全监测委员会,该委员会根据对研究的某些数据的访问,授权研究是否可以在指定的检查点进行,如果确定对受试者存在不可接受的安全风险或其他理由,如没有显示疗效,可能会停止临床试验。还有关于向公共注册机构报告正在进行的临床试验和已完成的临床试验的要求。在美国,有关适用的临床试验的信息,包括临床试验结果,必须在特定的时间框架内提交,才能在www.Clinicaltrials.gov网站上公布。
希望在美国境外进行临床试验的赞助商可以(但不需要)获得FDA的授权,根据IND进行临床试验。如果研究是根据GCP要求进行的,FDA将接受不是在IND下进行的设计良好和进行良好的外国临床研究,如果认为有必要,FDA能够通过现场检查来验证数据。
评估治疗适应症以支持NDA和BLAS以获得上市批准的临床试验通常分三个连续阶段进行,这三个阶段可能会重叠,也可能会合并。
批准后试验,有时被称为4期临床试验,可能在最初的上市批准后进行。这些试验用于在预期的治疗适应症下从患者的治疗中获得额外的经验,通常是为了产生关于在临床环境中使用该产品的额外安全数据。在某些情况下,FDA可以强制执行4期临床试验,作为批准NDA或BLA的条件。未能在进行所需的4期临床试验方面进行尽职调查可能会导致撤回对产品的批准。
36
在临床开发的所有阶段,监管机构要求对所有临床活动、临床数据和临床研究调查人员进行广泛的监测和审计。除其他信息外,详细说明临床试验结果的进展报告必须至少每年提交给FDA,书面IND安全报告必须在试验赞助商确定信息有资格报告严重和意外的可疑不良事件、来自其他研究或动物或动物的发现后15天提交给FDA和调查人员。体外培养与方案或研究人员手册中列出的测试相比,表明暴露于药物或生物的人类参与者的重大风险,以及任何临床上重要的严重疑似不良反应发生率的增加。赞助商还必须尽快通知FDA任何意外的、致命的或危及生命的疑似不良反应,但在任何情况下,不得晚于赞助商首次收到信息后的七个历日。
在临床试验的同时,公司通常还必须完成额外的动物研究,还必须开发关于候选产品的药物或生物学特性的额外信息,并根据cGMP要求最终确定商业批量生产药物产品的工艺。制造过程必须能够始终如一地生产高质量的候选产品批次,制造商必须开发测试最终药物产品的特性、强度、质量和纯度的方法等。此外,必须选择和测试适当的包装,必须进行稳定性研究,以证明候选产品在保质期内不会发生不可接受的变质,并确定候选产品的适当储存条件。
扩展的访问
扩大使用,有时称为同情使用,是在临床试验之外使用研究产品,在没有可比或令人满意的替代治疗方案的情况下,治疗患有严重或立即危及生命的疾病或条件的患者。FDA的法规允许我们或治疗医生在个案的基础上获得IND项下的研究产品,用于以下群体的治疗:个别患者(在紧急情况下和非紧急情况下治疗的单个患者IND);中等规模的患者群体;以及根据治疗方案或治疗IND使用研究产品的更大人群。
没有要求公司提供更多访问其研究产品的机会。然而,如果一家公司决定将其研究产品用于扩大准入,FDA将审查每一项扩大准入的请求,并确定是否可以继续治疗。当以下所有标准都适用时,扩大准入可能是合适的:患者患有严重或立即危及生命的疾病或状况,并且没有类似或令人满意的替代疗法来诊断、监测或治疗该疾病或状况;潜在的益处证明治疗的潜在风险是合理的,并且潜在风险在要治疗的疾病或状况的背景下并不是不合理的;为所请求的用途提供研究产品不会干扰可能支持扩大准入用途的市场批准的临床研究的启动、进行或完成,或以其他方式损害扩大准入用途的潜在发展。
在美国,除其他事项外,《试用权法案》为患有危及生命的疾病的患者提供了额外的机制,这些患者已经用尽了批准的治疗方法,无法参与临床试验,以获得某些研究产品,这些产品已经完成了第一阶段临床试验,是激活的IND的对象,正在接受调查,等待FDA的批准。与上述扩大的准入框架不同,《试用权法案》不要求FDA审查或批准使用研究产品的请求。
根据FDCA,用于治疗严重疾病或状况的一种或多种研究产品的赞助商必须公开其评估和回应扩大个别患者获得机会的请求的政策。赞助商必须在2期或3期研究启动的较早时间,或在研究药物或生物被指定为突破疗法、快速通道产品或再生医学高级疗法后15天内公开提供此类政策。根据《试用权利法案》,赞助商没有义务向符合条件的患者提供其研究产品,但赞助商必须制定内部政策,并根据该政策回应患者的请求。
37
FDA营销申请审查和审批流程
假设成功完成所需的临床测试,临床前研究和临床试验的结果,以及与产品的化学、制造、控制和拟议的标签等相关的详细信息,将作为NDA或BLA的一部分提交给FDA,请求批准将该产品用于一个或多个适应症的市场。NDA是为一个或多个指定适应症上市新药的批准请求,BLA是为一个或多个指定适应症上市新生物的批准请求。NDA或BLA必须包括从相关的临床前研究和临床研究中获得的所有相关数据,包括否定或模糊的结果以及积极的发现,以及与产品的化学、制造、控制和建议的标签等相关的详细信息。数据可能来自公司赞助的临床试验,旨在测试产品使用的安全性和有效性,或者来自许多替代来源,包括研究人员发起的研究。为了支持上市批准,提交的数据必须在质量和数量上足以确定研究药物的安全性和有效性,或研究生物的安全性、纯度和有效性,以使FDA满意。在药物或生物制剂在美国上市之前,必须获得FDA对NDA或BLA的批准。
此外,根据《儿科研究公平法》(“PREA”),某些NDA和BLA以及某些NDA或BLA的补充剂必须包含数据,以评估候选药物或生物制品在所有相关儿科亚群中声称的适应症的安全性和有效性,并支持该产品对安全有效的每个儿科亚群的剂量和给药。《食品和药物管理局安全与创新法》要求,计划提交营销申请或补充剂申请的药物或生物制品,包括新的活性成分或临床活性成分、新的适应症、新的剂型、新的给药方案或新的给药途径,赞助商必须在第二阶段会议结束后60天内或赞助商与FDA达成一致的情况下,提交初步儿科研究计划。除非法规另有要求,否则PREA不适用于已被授予孤儿称号的药物或生物制品的适应症。
在美国,FDA审查所有提交的NDA和BLA,以确保它们足够完整,以便在接受它们提交之前进行实质性审查,并可能要求提供更多信息,而不是接受NDA或BLA提交。FDA在收到NDA或BLA后60天内做出决定,接受NDA或BLA备案,这一决定可能包括FDA拒绝备案。FDA可以拒绝提交其认为不完整或在提交时不能适当审查的任何BLA,并可能要求提供更多信息。在这种情况下,必须重新提交BLA以及附加信息。重新提交的申请在FDA接受备案之前也要进行审查。一旦提交的申请被接受,FDA就开始对申请进行深入的实质性审查。FDA审查NDA或BLA以确定产品是否安全有效,以及其制造、加工、包装或持有的设施是否符合标准,包括cGMP要求,旨在确保和保持产品的特性、强度、质量和纯度。根据FDA根据《处方药使用费法案》(PDUFA)达成的目标和政策,FDA的目标是自提交日期起十个月内完成对原始NDA或BLA的初步审查并回应申请人,以及自提交优先审查的原始NDA或BLA提交之日起六个月。FDA并不总是满足其PDUFA标准或优先NDA或BLA的目标日期,审查过程经常因FDA要求提供更多信息或澄清而延长。
此外,根据经修订的PDUFA,每个NDA或BLA必须附有使用费,而经批准的NDA或BLA的赞助商也需缴纳年费。FDA每年调整PDUFA用户费用。在某些情况下,可以免除或减免费用,包括免除小企业首次提出申请的申请费。此外,对于被指定为孤儿药物的产品,不对NDA或BLA评估使用费,除非该产品还包括非孤儿适应症。
FDA可以将药物或生物的申请提交给咨询委员会。咨询委员会是由包括临床医生和其他科学专家在内的独立专家组成的小组,负责审查、评估并就是否应批准申请以及在何种条件下提出建议。FDA不受咨询委员会建议的约束,但它在做出决定时会仔细考虑这些建议。
在批准NDA或BLA之前,FDA通常会检查生产该产品的一个或多个设施。FDA将不会批准申请,除非它确定制造工艺和设施符合cGMP要求,并足以确保产品在所要求的规格下一致生产。此外,在批准NDA或BLA之前,FDA可以检查一个或多个临床试验地点,以确保符合GCP和其他要求,并确保提交给FDA的临床数据的完整性。为了确保符合GMP和GCP,申请者必须在培训、记录保存、生产和质量控制方面花费大量的时间、金钱和精力。
38
在评估了申请和所有相关信息后,包括咨询委员会的建议(如果有)以及关于制造设施和临床试验地点的检查报告,FDA可能会发出批准信,或者在某些情况下,发出完整的回复信。一封完整的回复信表明申请的审查周期已经完成,申请尚未准备好批准。一封完整的回复信通常将描述FDA在NDA或BLA中发现的所有缺陷,但如果FDA确定支持申请的数据不足以支持批准,FDA可能会出具完整的回复信,而不首先进行所需的检查、测试提交的产品批次和/或审查拟议的标签。在发布完整的回复信时,FDA可以建议申请人可能采取的行动,以使NDA或BLA处于批准的条件下,包括要求提供更多信息或澄清。即使提交了这一补充信息,FDA最终也可能决定该申请不符合批准的监管标准。如果这些条件得到了FDA的满意,FDA通常会签发一封批准信。批准函授权该产品的商业营销,并提供特定适应症的具体处方信息。
即使FDA根据要解决的具体风险批准产品,FDA也可以限制批准的产品使用适应症,要求在产品标签中包括禁忌症、警告或预防措施,要求进行批准后的研究,包括第四阶段临床试验,以在批准后进一步评估产品的安全性或有效性,要求测试和监督计划在产品商业化后对产品进行监控,或施加其他条件,包括分销和使用限制或风险评估和缓解策略(“REMS”)下的其他风险管理机制,这可能对产品的潜在市场和盈利能力产生重大影响。REMS可以包括药物指南、医疗保健专业人员的沟通计划和确保安全使用的要素(“ETASU”)。ETASU可包括但不限于处方或配药的特殊培训或认证、仅在某些情况下的配药、特殊监测和专利登记处的使用。FDA可以根据上市后研究或监测计划的结果,阻止或限制产品的进一步营销。在批准后,对批准的产品的某些类型的更改,如增加新的适应症、制造更改和额外的标签声明,将受到进一步的测试要求和FDA的审查和批准。
孤儿药物的指定和排他性
根据《孤儿药品法》,FDA可以对用于治疗罕见疾病或疾病的药物或生物授予孤儿药物称号(ODD),该疾病或疾病的定义是患者人数在美国少于20万人,或患者人数超过20万人,但无法合理预期在美国开发和提供药物或生物药物或生物药物的成本将从该药物或生物药物或生物药物在美国的销售中收回。在提交保密协议或BLA之前,必须申请ODD。在FDA批准ODD后,FDA公开披露了治疗剂的仿制药身份及其潜在的孤儿用途。授予ODD并不意味着监管审查和批准过程中的任何好处,也不会缩短监管审查和批准过程的持续时间。
如果一种产品获得了奇数,随后获得了FDA对该药物的首次批准,用于治疗其具有此类名称的疾病,该产品有权获得孤立的产品排他性,这意味着FDA不得批准任何其他申请,包括完整的NDA或BLA,在自NDA或BLA批准后的七年内以相同的适应症销售相同的药物或生物药物,除非在有限的情况下,例如,显示出对具有孤儿药物排他性的产品的临床优势,或者FDA发现孤儿药物排他性持有人没有证明它可以确保获得足够数量的孤儿药物,以满足患有指定药物的疾病或状况的患者的需求。孤儿药物的排他性并不阻止FDA批准不同的药物或生物药物用于相同的疾病或条件,或相同的药物或生物药物用于不同的疾病或条件。ODD的其他好处包括对某些研究的税收抵免,以及免除NDA或BLA申请用户费用。
指定的孤儿药物如果被批准用于比其获得奇数适应症更广泛的用途,则不得获得孤儿药物排他性。此外,如果FDA后来确定指定请求存在重大缺陷,或者如果制造商无法保证足够数量的产品来满足这种罕见疾病或疾病患者的需求,则可能会失去孤儿药物在美国的独家营销权。FDA历来采取的立场是,孤儿排他性的范围与产品的批准适应症或用途一致,而不是产品被指定为孤儿的疾病或状况。然而,2021年9月30日,美国第11巡回上诉法院在Catalyst Pharms,Inc.诉Becera案认为孤儿药物专有的范围必须与该产品被指定为孤儿的疾病或状况相一致,即使该产品的批准用于较窄的用途或适应症。FDA于2023年1月24日宣布,尽管触媒在做出决定时,它将继续适用其长期存在的法规,这些法规将孤儿排他性的范围与药物被批准用于的用途或适应症捆绑在一起,而不是与名称挂钩。FDA对其孤儿药物法规的应用后-触媒可能成为未来立法或在法庭上进一步挑战的主题,这一决定将如何影响未来的孤儿药物排他性仍有待观察。
39
加快发展和审查计划
FDA维持着几个旨在促进和加快新药和生物制品的开发和审查的计划,以满足在治疗严重或危及生命的疾病或条件方面未得到满足的医疗需求。这些计划包括快速通道指定、突破性治疗指定、优先审查和加速批准。快速通道指定、突破性治疗指定、优先审查和加速审批不会改变审批标准,但可能会加快开发或审批过程。
如果一种新药或生物制剂打算用于治疗严重或危及生命的疾病或状况,并显示出满足此类疾病或状况未得到满足的医疗需求的潜力,则该新药或生物制剂有资格获得快速通道指定。快速通道指定适用于产品和正在研究的特定适应症的组合。快速通道指定为赞助商在临床前和临床开发期间与FDA互动提供了更多机会,此外,一旦提交营销申请,FDA还有可能进行滚动审查,这意味着FDA可以在满足某些条件后,在申请完成之前启动对Fast Track产品申请部分的审查。
此外,如果一种新药或生物制品打算用于治疗严重或危及生命的疾病或状况,并且初步临床证据表明,该药物或生物制品单独或与或多个其他药物或生物制品联合使用,可能在一个或多个临床重要终点显示出比现有疗法有显著改善的效果,例如在临床开发早期观察到显著的治疗效果,则该新药或生物制品可能有资格获得突破性疗法指定。突破性治疗指定提供了快速通道指定的所有功能,此外,早在第一阶段就开始了对高效开发计划的密集指导,以及FDA对加快开发的组织承诺,包括在适当的情况下让高级经理和经验丰富的审查人员参与跨学科审查。
任何提交FDA批准的产品,包括具有快速通道或突破疗法称号的产品,也可能有资格接受优先审查。如果产品旨在治疗严重或危及生命的疾病或状况,并且如果获得批准,将在安全性或有效性方面提供显著改善,则该产品有资格接受优先审查。对于原始的NDA和BLAS,优先审查指定意味着FDA的目标是在60天的申请日期后6个月内对营销申请采取行动(而标准审查下为10个月)。
FDA可以加速批准旨在治疗严重或危及生命的疾病或状况的产品,该产品通常为患者提供比现有治疗更有意义的治疗优势,并展示对替代终点的影响,该替代终点合理地可能预测临床益处,或者对临床终点的影响可以比不可逆转的发病率或死亡率(“IMM”)更早地测量,考虑到疾病的严重性、稀有性或流行率以及替代治疗的可获得性或缺乏。
对于加速批准的药物,FDA通常要求赞助商以勤奋的方式进行充分和良好控制的批准后验证性研究,以验证和描述产品的临床益处。没有进行尽职调查的必要的批准后研究,在批准后研究期间没有确认临床益处,或者传播虚假或误导性的宣传材料,将允许FDA迅速撤回产品批准。作为2023年综合拨款法案的一部分,2022年12月29日颁布的2022年食品和药物综合改革法案(FDORA)包括了对加快药品和生物制品审批程序的多项改革,并使FDA能够酌情要求在批准加速批准之前进行批准后研究。FDORA还扩大了FDA可用的快速退出程序,允许该机构在赞助商未能对产品进行尽职调查的任何必要的批准后研究时使用快速程序。FDORA还补充说,在加速批准下获得批准的产品的赞助商没有对该产品进行任何必要的批准后研究,或没有及时提交关于该产品的报告,这是FDCA的禁止行为清单中的一项规定。所有经过加速审批的候选产品的促销材料都必须经过FDA的事先审查,除非FDA另行通知申请人。
FDA批准或批准了Companion诊断
2014年8月,FDA发布了最终指南,澄清了将适用于开发和批准用于治疗的产品的要求体外培养伴随诊断。根据该指南,对于新药和生物制品,治疗产品标签中指示的用途,其配套诊断设备及其相应的治疗设备应同时获得FDA的批准或批准。配套诊断设备的批准或许可将确保该设备已经过充分评估,并在目标人群中具有足够的性能特征。2016年7月,FDA发布了一份指南草案,旨在帮助治疗产品的赞助商和体外培养关于共同开发产品的相关问题的配套诊断设备。
40
在FDCA的领导下,体外培养诊断,包括伴随诊断,被作为医疗设备进行监管。在美国,医疗器械设计和开发、临床前和临床试验、上市前批准或批准、注册和上市、制造、标签、储存、广告和促销、销售和分销、出口和进口以及上市后监督等方面受FDCA及其实施条例和其他联邦和州法规和条例的管辖。除非适用豁免,否则诊断测试在商业分销之前需要获得市场许可或FDA的批准。
FDA此前曾要求体外培养伴随诊断旨在选择将对候选产品做出反应以获得上市前批准(“PMA”)的患者,同时批准治疗产品候选。PMA过程,包括临床和临床前数据的收集,以及提交给FDA和FDA的审查,可能需要几年或更长时间。它包括严格的上市前审查,在此期间,申请人必须准备并向FDA提供有关该设备的安全性和有效性的合理保证,以及有关该设备及其组件的信息,其中包括设备设计、制造和标签。PMA申请须缴交申请费。
PMA申请通常需要临床试验,在一小部分病例中,FDA可能要求进行临床研究,以支持FDCA第510(K)节下的上市前通知。希望进行涉及该设备的临床研究的制造商必须遵守FDA的研究设备豁免(“IDE”)规定。IDE法规区分了重大风险装置研究和非重大风险装置研究,相应地,获得批准开始研究的程序也有所不同。此外,某些类型的研究不受IDE法规的约束。重大风险装置可能会对受试者的健康、安全或福利造成严重风险。重大危险设备是指在诊断、治愈、减轻或治疗疾病或防止损害人类健康方面非常重要的设备。在启动临床研究之前,对具有重大风险的设备的研究需要FDA和IRB的批准。许多伴随诊断被认为是重大风险工具,因为它们在诊断疾病或状况方面发挥了作用。非重大危险装置是指不会对人体构成重大危险的装置。非重大危险装置研究只需在临床研究开始前获得IRB批准。
在设备投放市场后,它仍然受到严格的监管要求。医疗器械的销售只能用于其许可或批准的用途和适应症。设备制造商还必须向FDA建立注册和设备清单。
在美国,设备制造商还必须遵守FDA的医疗设备报告条例,该条例要求制造商向FDA报告其销售的设备可能已导致或促成死亡或重伤,或已发生故障,并且如果故障再次发生,该设备或其销售的类似设备很可能导致或促成死亡或重伤,以及FDA的更正和拆卸报告条例,该条例要求制造商在进行更正或拆卸时向FDA报告,以降低设备对健康构成的风险,或补救可能对健康构成风险的违反FDCA的行为。医疗器械制造商及其供应商的制造流程必须遵守FDA质量体系法规的适用部分,该法规涵盖医疗器械的设计、测试、生产、工艺、控制、质量保证、标签、包装和运输的方法和文档。美国食品和药物管理局会定期对国内工厂的记录和制造流程进行不定期检查。FDA还可能检查向美国出口产品的外国设施。
美国对药品和生物制品的审批后要求
在美国,根据FDA批准生产或分销的药品和生物制品受到FDA普遍和持续的监管,其中包括与记录保存、定期报告、产品抽样和分销、报告产品不良反应、遵守宣传和广告要求有关的要求,其中包括限制为未经批准的用途或患者群体推广产品(称为“标签外使用”),以及对行业赞助的科学和教育活动的限制。尽管医生可能会开出批准的产品用于标签外的用途,但制造商不得销售或推广此类用途。FDA和其他机构积极执行禁止推广标签外用途的法律和法规,不仅包括公司员工,还包括公司代理人或代表公司发言的人,被发现不当推广标签外用途的公司可能会承担重大责任。不遵守这些要求可能会导致不良宣传、警告信、纠正性广告以及可能的民事和刑事处罚,包括根据虚假索赔法案的责任,其中产品根据联邦医疗保健计划获得补偿。经批准的药品和生物制品的宣传材料必须在首次使用或首次发表时提交给FDA。此外,如果产品有任何修改,包括对适应症、标签或制造工艺或设施的拟议更改,申请人可能被要求提交并获得FDA对新的或补充的NDA或BLA的批准,这可能需要开发额外的数据或临床前研究和临床试验。
FDA可以施加一些批准后的要求,作为批准NDA或BLA的条件。例如,FDA可能要求进行上市后测试,包括第四阶段临床试验和监测,以进一步评估和监测产品在商业化后的安全性和有效性。
41
此外,参与生产和分销批准产品的药品和生物制品制造商及其分包商必须向FDA和某些州机构注册其机构,并接受FDA和某些州机构的定期突击检查,以确保其遵守正在进行的法规要求,包括cGMP,这些法规对我们和我们的CMO提出了某些程序和文件要求。对制造工艺的更改受到严格的监管,根据更改的重要性,可能需要FDA事先批准才能实施。FDA的规定还要求调查和纠正任何偏离cGMP的情况,并对我们和我们可能决定使用的任何第三方制造商提出报告要求。因此,制造商必须继续在生产和质量控制方面花费时间、金钱和精力,以保持遵守cGMP和其他方面的法规遵从性。不遵守法定和法规要求的制造商可能会受到可能的法律或法规行动,例如警告信、暂停生产、产品扣押、禁令、民事处罚或刑事起诉。对于任何市场产品,还需要支付持续的年度计划费。
如果没有遵守监管要求和标准,或者如果产品上市后出现问题,FDA可能会撤回批准。后来发现产品存在以前未知的问题,包括预料不到的严重程度或频率的不良事件,或生产工艺,或未能遵守监管要求,可能导致修订批准的标签,以添加新的安全信息,要求上市后研究或临床试验,以评估新的安全风险,或根据REMS实施分销或其他限制。除其他外,其他潜在后果包括:
美国专利期限恢复与市场排他性
根据FDA批准我们未来候选产品的时间、期限和细节,我们的一些美国专利可能有资格根据1984年的《药品价格竞争和专利期限恢复法》(通常称为Hatch-Waxman修正案)获得有限的专利期延长。Hatch-Waxman修正案允许恢复最长五年的专利期,作为对FDA监管审查过程中丢失的专利期的补偿。然而,专利期限恢复不能将专利的剩余期限延长到自产品批准之日起总共14年,只有那些涉及该批准的药物产品、其使用方法或制造方法的权利要求可以延长。专利期恢复期一般为IND的生效日期与NDA或BLA的提交日期之间的时间的一半,加上NDA或BLA的提交日期与该申请获得批准之间的时间的一半,但在申请人未进行尽职调查期间,审查期限将被缩短。只有一项适用于经批准的药物的专利有资格延期,延期申请必须在专利到期之前提交。美国专利商标局与FDA协商,审查和批准任何专利期延长或恢复的申请。未来,我们可能会申请恢复我们目前拥有或许可的专利的专利期,以延长其当前到期日之后的专利寿命,这取决于临床试验的预期长度以及相关保密协议或BLA的提交所涉及的其他因素。
42
FDCA下的监管排他性条款也可能推迟某些申请的提交或批准。FDCA为第一个获得新化学实体保密协议批准的申请者提供了五年的美国境内非专利营销排他性。如果FDA以前没有批准过任何其他含有相同活性部分的新药,药物是一种新的化学实体,活性部分是负责药物物质作用的分子或离子。在专营期内,FDA不得接受另一家公司为该药物的另一版本提交的简化新药申请(ANDA)或505(B)(2)保密协议,但申请人不拥有或拥有合法的参考批准所需的所有数据的权利。但是,如果申请包含专利无效或非侵权的证明,则可以在四年后提交申请。
如果FDA认为申请人进行或赞助的新的临床研究(生物利用度研究除外)对于批准申请是必不可少的,例如,现有药物的新适应症、剂量或强度,FDCA还为NDA、505(B)(2)NDA或现有NDA的补充提供三年的排他性。这项为期三年的排他性只涵盖与新的临床研究相关的使用条件,并不禁止FDA批准含有原始活性物质的药物用于其他使用条件的ANDA。五年和三年的排他性不会推迟提交或批准完整的保密协议。然而,提交完整的保密协议的申请人将被要求进行或获得参考所有必要的临床前研究和充分和良好控制的临床试验的权利,以证明安全和有效。
此外,药物和生物制品也可以在美国获得儿科专有权。如果获得儿科专有权,现有的专有期和专利条款将增加6个月。这一为期六个月的排他性从其他排他性保护或专利期结束开始,可以根据FDA发布的此类研究的书面请求,在自愿完成儿科研究的基础上授予。
美国的生物仿制药和排他性
经2010年《医疗保健和教育协调法》(统称为《ACA》)修订的《患者保护和平价医疗法案》包括一个副标题,称为《生物制品价格竞争和创新法》(以下简称《BPCIA法》),该法案为与FDA许可的参考生物制品生物相似或可互换的生物制品创建了一个简短的审批途径。FDA发布了几份指导文件,概述了美国对生物仿制药的审查和批准方法。生物相似性要求生物制品和参考产品在安全性、纯度和效力方面没有临床上有意义的差异,可以通过分析研究、动物研究和临床研究来证明。互换性要求产品与参考产品生物相似,并且该产品必须证明在任何给定的患者中,它可以预期产生与参考产品相同的临床结果,对于多次给药的产品,在先前给药后,生物和参考生物可以交替或交换,而不会增加安全风险或相对于单独使用参考生物而降低疗效的风险。
根据BPCIA,参考生物制品被授予12年的数据独占权,从该产品首次获得许可之时起,生物相似产品的申请不得提交给FDA,直到该参考产品首次获得FDA许可之日起四年。此外,FDA对生物相似产品的批准可能要到参考产品首次获得许可之日起12年后才能生效。在这12年的独占期内,如果FDA批准竞争产品的完整BLA,包含申请人自己的临床前数据和来自充分和良好控制的临床试验的数据,以证明其产品的安全性、纯度和有效性,另一家公司仍可能销售该参考产品的竞争版本。BPCIA还为被批准为可互换产品的生物仿制药设立了某些排他性期限。在这个节骨眼上,还不清楚FDA认为“可互换”的产品是否真的会被受州药剂法管辖的药房所取代。
BPCIA是复杂的,并继续由FDA解释和实施。此外,政府的建议还试图缩短12年的参考产品专营期。ACA的其他方面,其中一些可能会影响BPCIA的排他性条款,也是最近诉讼的主题。因此,BPCIA的最终影响、实施和监管解释仍然存在重大不确定性。
其他美国监管事项
候选产品的制造、销售、推广和其他在产品批准或商业化后的活动,在适用的情况下,也要受到美国除FDA以外的许多监管机构的监管,这些监管机构可能包括医疗保险和医疗补助服务中心(CMS)、卫生与公众服务部(HHS)的其他部门、司法部、药品监督管理局、消费品安全委员会、联邦贸易委员会、职业安全与健康管理局、环境保护局,以及州和地方政府和政府机构。
43
美国的其他医疗保健法
在美国,医疗保健提供者、医生和第三方付款人将在我们获得营销批准的任何产品的推荐和处方中发挥主要作用。我们的业务运营以及目前或未来与第三方付款人、医疗保健提供者和医生的任何安排可能会使我们面临广泛适用的欺诈和滥用以及其他医疗法律和法规,这些法律和法规可能会限制我们开发、营销、销售和分销我们获得上市批准的任何药物的业务或财务安排和关系。在美国,这些法律包括但不限于州和联邦反回扣、虚假声明、医生透明度以及患者数据隐私和安全法律法规,包括但不限于下述法律法规。
44
此外,制药商还可能受到美国联邦和州消费者保护以及不正当竞争法律和法规的约束,这些法律和法规对市场活动进行了广泛的监管,可能会损害消费者。
药品和生物制品的分销须遵守额外的要求和条例,包括广泛的记录保存、许可、储存和安全要求,以防止未经授权销售药品。
这些法律的全部范围和执行都是不确定的,并受到当前医疗改革环境的快速变化的影响。联邦和州执法机构继续加强对医疗保健公司与医疗保健提供者之间互动的审查,这导致了医疗保健行业的一系列调查、起诉、定罪和和解。政府当局可能会得出结论,认为我们的业务做法不符合当前或未来涉及适用欺诈和滥用或其他医疗保健法律和法规的现行或未来法规、法规或判例法。如果我们的运营被发现违反了任何这些法律或任何其他可能适用于我们的相关政府法规,我们可能会面临重大的民事、刑事和行政处罚、损害赔偿、罚款、监禁、交还、被排除在政府资助的医疗保健计划之外(如Medicare和Medicaid)、声誉损害、额外的监督和报告义务,如果我们受到公司诚信协议或类似和解的约束,以解决有关违反这些法律的指控,并削减或重组我们的业务。如果我们预期与之有业务往来的任何医生或其他医疗保健提供者或实体被发现不遵守适用法律,他们可能会受到类似的行动、处罚和制裁。确保商业安排符合适用的医保法,以及对政府当局可能进行的调查做出回应,可能会耗费时间和资源,并可能分散公司对其业务的注意力。
美国保险和报销
在美国和其他国家的市场上,根据自己的病情接受处方治疗的患者和提供处方服务的提供者通常依赖第三方付款人来报销全部或部分相关的医疗费用。因此,即使候选产品获得批准,该产品的销售在一定程度上也将取决于第三方付款人,包括美国的政府医疗计划,如Medicare和Medicaid,商业健康保险公司和管理型医疗保健组织,为该产品提供保险和建立足够的报销水平的程度。在美国,第三方付款人之间没有统一的药品保险和报销政策。因此,药品的承保范围和报销范围因付款人而异。确定第三方付款人是否将为产品提供保险的过程可以与设置价格或报销率的过程分开,一旦保险获得批准,付款人将为产品支付费用。第三方付款人越来越多地挑战所收取的价格,审查医疗必要性,审查医疗产品和服务的成本效益,并实施控制以管理成本。第三方付款人可以将承保范围限制在批准清单上的特定产品,也称为配方表,该清单可能不包括特定适应症的所有批准产品。
45
为了确保任何可能被批准销售的产品的保险和报销,公司可能需要进行昂贵的药物经济学研究,以证明该产品的医疗必要性和成本效益,以及获得FDA或其他类似监管批准所需的成本。付款人在确定报销时考虑的因素是:产品是否为其健康计划下的承保福利;安全、有效和医学上必要的;适合特定患者的;成本效益;以及既不是试验性的也不是研究性的。此外,公司可能还需要向购买者、私人健康计划或政府医疗计划提供折扣。尽管如此,候选产品可能不被认为是医学上必要的或具有成本效益的。一旦产品获得批准,第三方付款人不承保产品的决定可能会减少医生的使用率,并对销售、我们的运营和财务状况产生实质性的不利影响。此外,第三方付款人决定为产品提供保险并不意味着将批准足够的报销率。此外,一个付款人决定为一种产品提供保险并不能保证其他付款人也会为该产品提供保险和报销,而且不同的付款人的保险和报销水平可能有很大不同。
控制医疗成本已成为联邦、州和外国政府的优先事项,产品价格一直是这一努力的重点。各国政府对实施成本控制计划表现出了极大的兴趣,包括价格控制、限制报销和要求替代仿制药。采取价格控制和成本控制措施,以及在具有现有控制和措施的司法管辖区采取更具限制性的政策,可能会进一步限制公司从销售任何经批准的产品中获得的收入。承保政策和第三方付款人报销率可能随时发生变化。即使一家公司或其协作者获得监管批准的一个或多个产品获得了有利的承保和报销状态,未来也可能实施不太有利的承保政策和报销费率。
医疗改革
在美国和一些外国司法管辖区,关于医疗保健系统的多项立法和监管改革以及拟议的改革已经并可能继续存在,旨在扩大医疗保健的可获得性,提高医疗保健的质量,并遏制或降低医疗保健成本。例如,2010年,美国颁布了ACA,其中包括使生物产品受到低成本生物仿制药的潜在竞争;解决了一种新的方法,即对吸入、输液、滴注、植入或注射的药物计算制造商在医疗补助药物退税计划下所欠的退税;提高了大多数制造商在医疗补助药物退税计划下欠下的最低医疗补助退税;将医疗补助药物退税计划扩大到使用在医疗补助管理的护理组织中登记的个人的处方;要求制造商对某些品牌的处方药征收新的年费和税;创建了一项新的联邦医疗保险D部分承保缺口折扣计划,制造商必须同意在承保间隔期内向符合条件的受益人提供适用品牌药品谈判价格的70%的销售点折扣,作为制造商的门诊药物纳入联邦医疗保险D部分承保的条件;并为增加联邦政府比较有效性研究的计划提供激励。
自颁布以来,ACA的某些方面受到了许多司法、行政、行政和立法方面的挑战,我们预计未来还会对ACA提出更多的挑战和修正案。ACA的各个部分目前正在美国最高法院接受法律和宪法挑战,国会议员提出了几项旨在大幅修改或废除ACA的立法。这起案件挑战了ACA的合宪性,加利福尼亚州诉德克萨斯州,于2020年11月在美国最高法院进行了辩论。2021年6月,最高法院裁定,原告没有资格挑战ACA。尽管最高法院做出了裁决,但我们不能确定未来是否会对ACA提出挑战,或者这些挑战可能会对我们的业务产生什么影响(如果有的话)。ACA的实施仍在进行中,该法律似乎可能会继续对药品定价施加下行压力,特别是在联邦医疗保险药品回扣计划下,还可能增加我们的监管负担和运营成本。与ACA相关的诉讼和立法可能会继续,结果不可预测和不确定。
自ACA颁布以来,美国还提出并通过了其他立法修改。2011年8月,除其他事项外,2011年《预算控制法》包括将向医疗保险提供者支付的医疗保险付款每财年总计减少2%,该法案于2013年4月生效,由于随后对该法规的立法修订,除非国会采取额外行动,否则该法案将一直有效到2030年。2013年1月,《2012年美国纳税人救济法》签署成为法律,其中包括进一步减少了向包括医院、成像中心和癌症治疗中心在内的几家医疗服务提供者支付的医疗保险,并将政府追回向医疗服务提供者多付款项的诉讼时效从三年延长到五年。
46
此外,支付方法可能会受到医疗保健立法和监管举措的影响。例如,在美国,CMS可能会开发新的支付和交付模式,如捆绑支付模式。此外,最近政府对制造商为其商业产品定价的方式进行了更严格的审查,导致国会进行了几次调查,并提出并颁布了州和联邦立法,旨在提高产品定价的透明度,审查定价与制造商患者计划之间的关系,以及改革政府对药品的计划补偿方法。2020年3月10日,特朗普政府向国会提交了药品定价的《原则》,呼吁立法,其中包括限制联邦医疗保险D部分受益人的自付药房费用,提供限制联邦医疗保险D部分受益人每月自付费用的选项,并限制药品价格上涨。此外,特朗普政府此前发布了一份降低药品价格和降低药品自付成本的《蓝图》,其中包含增加药品制造商竞争、增加某些联邦医疗保健计划的谈判力、激励制造商降低产品标价以及降低消费者支付的药品自付成本的建议。卫生和公众服务部已经实施了一些措施。例如,CMS在2019年5月发布了一项最终规则,允许Medicare Advantage计划从2020年1月1日开始选择对B部分药物使用阶梯疗法,这是一种事先授权。这一最终规则编纂了CMS于2019年1月1日生效的政策变化。然而,目前尚不清楚拜登政府是否会挑战、逆转, 撤销或以其他方式修改这些执行和行政操作。此外,美国各州也越来越多地通过立法并实施旨在控制药品定价的法规,包括价格或患者报销限制、折扣、对某些产品准入和营销成本披露的限制以及透明度措施,在某些情况下,旨在鼓励从其他国家进口和批量购买。此外,政府有可能采取更多行动来应对新冠肺炎疫情。
在美国以外,确保产品的覆盖范围和足够的付款也面临挑战。在许多国家,处方药的定价受到政府的管制。与政府当局的定价谈判可能远远超出产品获得监管批准的范围,可能需要进行临床试验,将产品的成本效益与其他可用的疗法进行比较。进行这样的临床试验可能代价高昂,并导致商业化延迟。
在欧洲联盟(“欧盟”),不同国家的定价和补偿方案差别很大。一些国家规定,只有在商定了补偿价格之后,才能销售产品。一些国家可能要求完成额外的研究,将特定候选产品的成本效益与目前可用的疗法或所谓的卫生技术评估进行比较,以获得报销或定价批准。例如,欧盟为其成员国提供了选项,以限制其国家健康保险制度提供补偿的产品范围,并控制供人使用的医疗产品的价格。欧盟成员国可以批准产品的具体价格,也可以采用直接或间接控制公司将产品投放市场的盈利能力的制度。其他成员国允许公司固定自己的产品价格,但监控处方数量,并向医生发布关于处方标准的指导意见,从而对限制处方或使用产生影响。最近,欧盟许多国家提高了药品折扣要求,随着各国试图管理医疗支出,这些努力可能会继续下去,特别是在欧盟许多国家经历严重财政危机和债务危机的情况下。总体上,医疗成本,特别是处方药的下行压力变得很大。因此,对新产品的进入设置了越来越高的壁垒。政治、经济和监管方面的事态发展可能会使定价谈判进一步复杂化,在获得补偿后,定价谈判可能会继续进行。欧盟各成员国使用的参考定价,以及平行贸易,即, 低价和高价成员国之间的套利,可以进一步降低价格。不能保证任何对药品实行价格管制或报销限制的国家,如果在这些国家获得批准,将允许对任何产品作出有利的报销和定价安排。
遵守其他联邦和州法律或要求;改变法律要求
如果我们可能开发的任何产品在美国向美国总务署联邦供应时间表的授权用户提供,则适用其他法律和要求。产品必须符合美国《防止毒物包装法》中适用的儿童保护包装要求。制造、标签、包装、分销、销售、促销和其他活动也可能受到联邦和州消费者保护和不正当竞争法律的约束,以及我们可能受到的其他要求。
医药产品的分销须遵守额外的规定和条例,包括广泛的记录保存、许可、储存和安全要求,以防止未经授权销售医药产品。
47
如果不遵守这些法律或监管要求中的任何一项,公司将面临可能的法律或监管行动。根据情况,未能满足适用的监管要求可能会导致刑事起诉、罚款或其他处罚、禁令、被排除在联邦医疗保健计划之外、请求召回、扣押产品、完全或部分暂停生产、拒绝或撤回产品批准、重新标记或重新包装,或拒绝允许公司签订供应合同,包括政府合同。任何针对我们违反这些法律的索赔或行动,即使我们成功地进行了辩护,也可能导致我们招致巨额法律费用,并转移我们管理层对业务运营的注意力。禁止或限制营销、销售或撤回我们销售的未来产品可能会以不利的方式对我们的业务产生重大影响。
法规、法规或现有法规的解释的变化可能会影响我们未来的业务,例如,要求:(I)改变我们的制造安排;(Ii)增加或修改产品标签或包装;(Iii)召回或停产我们的产品;或(Iv)额外的记录保存要求。如果强制实施任何此类变化,可能会对我们的业务运营产生不利影响。
欧洲药物开发
在欧盟,我们未来的产品也可能受到广泛的监管要求。与美国一样,医药产品只有在获得主管监管机构的营销授权后才能上市。
与美国类似,欧盟的临床前和临床研究的各个阶段都受到重要的监管控制。尽管欧盟临床试验指令2001/20/EC(“临床试验指令”)试图协调欧盟临床试验监管框架,为欧盟临床试验的控制和授权制定了共同规则,但欧盟成员国对临床试验指令的条款的解释和应用有所不同。这导致欧盟成员国的制度发生了重大变化。根据《临床试验指令》规定的制度,在启动临床试验之前,临床试验必须在每个欧盟成员国获得批准,在这些国家,试验将由两个不同的机构进行:国家主管机构(NCA)和一个或多个道德委员会(ECs)。在临床试验期间发生的所有可疑的意外严重不良反应都必须向发生这些反应的欧盟成员国的NCA和ECS报告。
欧盟临床试验立法目前正在进行过渡,主要目的是协调和简化临床试验授权(CTA),简化不良事件报告程序,改善临床试验的监督并增加其透明度。2022年1月,欧盟第536/2014号《临床试验条例》(以下简称《临床试验条例》)开始实施,并废除了《临床试验指令》。临床试验法规直接适用于所有欧盟成员国(因此不需要每个欧盟成员国的国家实施立法),并简化和简化了欧盟临床研究的审批,例如通过称为临床试验信息系统(CTIS)的单一点提供简化的CTA申请程序。《临床试验条例》预计有三年的过渡期。自2023年1月31日起,所有新的CTA申请必须使用CTI,并根据临床试验法规进行。从2025年1月31日起,根据临床试验指令批准的任何仍在进行的试验将需要遵守临床试验条例并记录在CTIS中。
欧洲药品营销
与美国的反回扣法案禁令非常相似,向医生提供福利或优势以诱导或鼓励医生开处方、推荐、背书、购买、供应、订购或使用医疗产品在欧盟也是被禁止的。提供利益或利益以诱导或奖励不当行为一般受欧盟成员国的国家反贿赂法律管辖,英国2010年的《反贿赂法》违反这些法律可能会导致巨额罚款和监禁。管理人用药品的欧盟指令2001/83/EC进一步规定,如果向有资格开处方或供应药品的人推销药品,则不得向这些人提供、提供或承诺任何礼物、金钱利益或实物利益,除非这些礼物、金钱利益或实物利益不贵且与医药或药房业务有关。这一条款已被转移到英国2012年人类药物法规中,因此尽管脱离欧盟,该条款仍适用于英国。
在欧盟和英国,适用于医药产品广告的法定制度辅之以由行业组织制定的行业行为守则所管辖的自我监管制度。这类业务守则只对属有关行业组织成员的公司具约束力。然而,由于它们代表了最佳做法,许多非成员国也选择遵守这些做法守则。这些业务守则要求向医生支付的款项必须公开披露。此外,根据法律规定,在某些欧盟成员国,如法国,支付给医生的费用必须公开披露。此外,与医生达成的协议通常必须事先通知医生的雇主、医生的主管专业组织和/或欧盟成员国的监管机构,并予以批准。不遵守这些要求可能会导致声誉风险、公开谴责、行政处罚、罚款或监禁。
48
欧洲药品审查和审批
在由欧盟成员国以及挪威、冰岛和列支敦士登组成的欧洲经济区(EEA),医药产品只有在获得营销授权(MA)后才能商业化。有两种类型的营销授权。
根据上述程序,在授予MA之前,欧洲经济区成员国的EMA或NCA根据有关产品质量、安全性和有效性的科学标准对产品的风险-效益平衡进行评估。
既然英国(包括大不列颠和北爱尔兰)已经离开欧盟,英国将不再被集中的MA覆盖(根据北爱尔兰议定书,集中的MA将继续在北爱尔兰得到承认)。所有具有当前集中MA的医疗产品于2021年1月1日自动转换为英国MA。自2021年1月1日起的三年内,英国药品和医疗器械监管机构药品和保健产品监管机构(MHRA)可能会依赖欧盟委员会在集中程序中批准新MA的决定,以便更快地批准新的英国MA。然而,仍将需要单独的申请。
49
欧洲数据和营销排他性
在欧洲经济区,创新的医药产品(包括小分子和生物医药产品)受益于八年的MA数据独家授权和另外两年的市场独家授权。数据排他性防止仿制药或生物相似申请者在申请仿制药或生物相似MA时交叉引用创新者的临床前和临床试验数据,这些数据包含在参考药品的档案中,直到数据排他期到期。在额外的两年市场排他期内,可以提交仿制药或生物相似的MA,创新者的临床前和临床数据可以交叉参考,但在市场排他期到期之前,任何仿制药或生物相似产品都不能上市。如果在这十年的头八年中,MA持有者获得了一个或多个新的治疗适应症的授权,而在授权之前的科学评估中,这些适应症被确定为与目前批准的疗法相比具有显著的临床益处,那么整个十年的期限将延长到最多11年。英国国内法遵循同样的监管数据和市场排他性公式。
欧洲的孤儿称号和排他性
在EEA,EMA的孤儿药物产品委员会授予孤儿药物指定,以促进产品的开发,这些产品旨在诊断、预防或治疗危及生命或慢性衰弱的疾病,这些疾病影响到欧盟每10,000人中不超过5人,或者药物的开发不太可能产生足够的回报,以证明对其开发的必要投资是合理的。在每一种情况下,必须没有得到批准的令人满意的诊断、预防或治疗方法(或者,如果存在这样的方法,有关产品将对受这种情况影响的人有重大好处)。
在欧洲药品管理局中,孤儿药物指定使一方有权获得经济激励,如降低费用或免除费用,并在MA批准孤儿产品后授予10年的市场独家经营权。如果在第五年结束时确定不再符合孤儿药物指定标准,包括证明产品的利润足够高,不足以证明维持市场排他性是合理的,则这一期限可缩短至六年。在市场独占期内,只有在下列情况下,才可就同一治疗适应症向“类似医药产品”授予MA:(I)第二申请人能证明其产品虽然与授权产品相似,但更安全、更有效或在临床上更好;(Ii)授权产品的MA持有人同意第二次申请孤儿医药产品;或(Iii)授权产品的MA持有人不能供应足够的孤儿医药产品。“类似医药产品”的定义是含有与批准的孤儿医药产品中所含的一种或多种类似活性物质的医药产品,其目的是用于相同的治疗适应症。在提交上市批准申请之前,必须申请指定孤儿药物。孤儿药物的指定不会在监管审查和批准过程中传递任何优势,也不会缩短监管审查和批准过程的持续时间。
英国国内法也采用了相同的制度来授予孤儿排他权。然而,在英国没有上市前授权的孤儿称号。
欧洲儿科调查计划
在欧洲药品管理局,开发新医药产品的公司必须与欧洲药品管理局的儿科委员会(“PDCO”)就儿科调查计划(“PIP”)达成一致,并必须根据该计划进行儿科临床试验,除非适用豁免。PIP规定了产生数据的时间和建议的措施,以支持正在寻求MA的药物的儿科适应症。PDCO可以批准推迟实施PIP的部分或全部措施的义务,直到有足够的数据证明该产品在成人中的有效性和安全性。此外,当不需要或不适当地提供儿科临床试验数据时,PDCO可以免除提供这些数据的义务,因为该产品可能对儿童无效或不安全,该产品预期用于治疗的疾病或状况仅发生在成人人群中,或者当该产品对儿科患者的现有治疗没有显著的治疗益处时。被授予MA的产品具有根据PIP进行的儿科临床试验的结果(即使结果为阴性),有资格获得6个月的补充保护证书延期(如果在批准时有效)。在孤儿药品的情况下,可以将孤儿市场的专有权延长两年。这一儿科奖励受到特定条件的制约,在开发和提交符合PIP的数据时不会自动获得。
50
英国脱欧与英国的监管框架
2016年6月,英国选民投票赞成脱欧(俗称脱欧)。此后,2017年3月,该国根据《里斯本条约》第50条正式通知欧盟有意退出,英国于2020年1月31日正式退出欧盟。过渡期从2020年2月1日开始,在此期间,欧盟药品立法仍适用于英国。当过渡期于2020年12月31日到期时,英国成为欧盟法律意义上的“第三国”。由于英国已经通过《2012年人类药品条例》(经修订)将欧盟药品立法落实到其国内立法中,因此立即对该立法进行了修改,以反映英国的新地位。《关于大不列颠及北爱尔兰联合王国退出欧洲联盟和欧洲原子能共同体的协定》所载《北爱尔兰议定书》附件2第5条第(4)款规定,在联合王国退出欧盟后,欧盟制药法的某些方面应适用于北爱尔兰。
2021年5月1日,欧盟与英国贸易合作协定(TCA)正式生效。TCA包括关于药品的具体规定,其中包括相互承认GMP检查的结果。申请者和MA持有者可以提交MHRA为位于欧盟/欧洲经济区以外的地点颁发的GMP证书,作为欧盟法规提交的支持信息。然而,TCA预计英国和欧盟的药品法规不会大规模相互承认。目前,英国的监管制度与欧盟的监管制度大体一致。然而,这些制度在未来可能会出现分歧。
欧洲数据收集
欧洲经济区个人健康数据的收集和使用受2018年5月25日生效的《一般数据保护条例》(GDPR)的监管。GDPR适用于在欧洲经济区成立的任何公司,以及在欧洲经济区以外设立的处理个人数据的公司,这些公司与向欧盟内的数据主体提供商品或服务或监测欧盟内数据主体的行为有关。《个人资料保密法》加强了个人资料控权人的资料保护责任,包括有关资料当事人同意的严格规定、有关如何使用个人资料的更广泛披露、就“高风险”处理进行私隐影响评估的规定、对保留个人资料的限制、针对“敏感资料”(包括资料当事人的健康和基因资料)的特别条文、强制性资料泄露通知和“设计隐私权”的规定,以及作为资料处理者的服务提供者的直接责任。GDPR还对将个人数据转移到欧洲经济区以外的国家实施了严格的规则,这些国家没有确保足够的保护水平,比如美国。如果不遵守GDPR的要求和欧洲经济区成员国相关的国家数据保护法,可能会导致高达2000万欧元的罚款或上一财年公司全球年收入的4%,以金额较高者为准。此外,GDPR赋予数据当事人在某些情况下要求删除个人信息的权利,并要求因侵犯GDPR而造成的物质和非物质损害赔偿。鉴于数据保护义务变化的广度和深度,维持遵守GDPR将需要大量的时间、资源和费用, 我们可能需要建立额外的机制,确保遵守新的数据保护规则。这可能是繁重的,并对我们的业务、财务状况、运营结果和前景产生不利影响。
企业信息
我们于2016年9月根据特拉华州的法律注册成立。我们的主要执行办公室位于One Main Street,Suite1350,Cambridge,MA 02142,我们的电话号码是(617)410-4650。
我们在业务中使用各种商标和商品名称,包括但不限于我们的公司名称和徽标。本年度报告中以Form 10-K格式提及的所有其他商标或商号均为其各自所有者的财产。仅为方便起见,本年度报告中的10-K表格中的商标和商号可不使用®和符号,但此类引用不应被解释为其各自所有者不会根据适用法律最大限度地主张其权利的任何指示。我们无意使用或展示其他公司的商标和商号,以暗示与任何其他公司的关系,或由任何其他公司背书或赞助我们。
我们是一家“新兴成长型公司”,在2012年的创业法案中被定义为“新兴成长型公司”。我们将一直是一家新兴的成长型公司,直到:(I)财政年度的最后一天(A)IPO完成五周年后,(B)我们的年度总收入至少为12.35亿美元,或(C)我们被视为大型加速申报公司,这意味着截至前一年6月30日,我们由非关联公司持有的普通股的市值超过7亿美元,以及(Ii)我们在前三年期间发行了超过10亿美元的不可转换债券。
51
人力资本资源
截至2022年12月31日,我们有62名全职员工和两名顾问。我们有19名员工拥有医学或博士学位。在我们的员工队伍中,有35名员工从事研发工作,27名员工从事业务开发、财务、法律以及一般管理和行政工作。我们管理执行团队的十名成员中有四名是女性。在我们更广泛的人口中,大约60%的全职员工是女性。我们的管理执行团队非常关注与我们的人力资本资产有关的问题--特别是我们劳动力的多样性及其能力发展。
我们的办公空间位于大波士顿,我们相信这提供了进入充满活力的生物技术和制药人才库的途径。我们认为,雇员薪酬应该既具有竞争性,又具有公平性。我们有吸引和留住人才的计划,包括基于股票的薪酬和现金绩效奖励。我们也有一个绩效管理和人才培养流程,在这个流程中,经理们定期提供反馈和指导,以发展员工。我们相信,团队成员、经理和领导层之间的公开和诚实的沟通可以营造一个人人都能参与、发展和茁壮成长的工作环境。我们的员工中没有一个是工会代表,也没有集体谈判协议的覆盖范围。我们认为我们与员工的关系很好。
可用信息
我们的公司网站地址是https://www.cullinanoncology.com.本公司网站包含或可通过本公司网站获取的信息不属于本10-K表格年度报告的一部分,本10-K表格年度报告中包含本公司网站地址仅作为非活动文本参考。
我们的10-K表格年度报告、10-Q表格季度报告、8-K表格当前报告,包括证物、委托书和信息声明,以及根据1934年证券交易法(经修订,“交易法”)第13(A)、14和15(D)条提交或提交的报告的修正案,在我们以电子方式向美国证券交易委员会存档或向美国证券交易委员会提供此类材料后,可在合理可行的范围内尽快通过我们网站的“投资者”部分免费获取。我们网站上的信息不是本Form 10-K年度报告或我们任何其他证券备案文件的一部分,除非通过引用特别将其并入本文。此外,我们向美国证券交易委员会提交的文件可通过美国证券交易委员会的电子数据收集、分析和检索系统获取,网址为Https://www.sec.gov。我们在任何证券备案文件中作出的所有陈述,包括所有前瞻性陈述或信息,都是在包含该陈述的文件日期作出的,除非法律要求我们这样做,否则我们不承担或承担任何更新这些陈述或文件的义务。
52
第1A项。RISK因子。
以下资料应与本表格10-K年度报告内的其他资料一并阅读,包括本年度报告中其他表格10-K所载的综合财务报表及相关附注。发生以下任何事件或事态发展都可能损害我们的业务、财务状况或未来业绩,而这些风险因素可能并不是我们面临的唯一风险。我们目前不知道或我们目前认为不重要的其他风险和不确定因素也可能对我们的业务、财务状况或未来业绩产生不利影响。请参阅本年度报告中的Form 10-K“有关前瞻性陈述的特别说明”。
与我们的候选产品开发相关的风险
我们的临床前研究和临床试验可能无法充分证明我们的任何候选产品的安全性和有效性,这将阻碍或推迟开发、监管批准和商业化。
在我们的候选产品,包括齐帕替尼(CLN-081/TAS6417)、CLN-049、CLN-619和CLN-418的商业化销售获得监管部门的批准之前,我们必须通过漫长、复杂和昂贵的临床前研究和临床试验,证明我们用于每个目标适应症的研究产品的安全性和有效性。如果我们正在进行的或未来的临床前研究和临床试验的结果对于我们候选产品的安全性和有效性没有定论,如果我们没有达到具有统计和临床意义的临床终点,或者如果我们的候选产品存在相关的安全问题,我们可能会阻止或推迟获得此类候选产品的上市批准。在某些情况下,由于许多因素,同一候选产品的不同临床前研究和临床试验的安全性或有效性结果可能存在显著差异,包括方案中规定的试验程序的变化、患者群体的大小和类型的差异、临床试验方案的变化和遵守以及临床试验参与者的退学率。
除了我们正在进行的齐帕尔替尼的临床试验外,患者已经并可能继续在扩大使用范围或“同情使用”计划下接受齐帕尔替尼的治疗。如果在该计划中接受治疗的患者的体验与我们正在进行或计划中的公司赞助的齐帕尔替尼试验的结果不一致或不那么有利,它可能会对人们对齐帕尔替尼、我们的其他候选产品或我们业务的看法产生负面影响。此外,美国(“U.S.”)由于这些不一致或不利的结果,美国食品和药物管理局(“FDA”)或外国监管机构可能会要求我们获得并提交额外的临床数据,这可能会推迟齐帕替尼或我们的其他候选产品的临床开发或上市批准。
我们识别、发现和开发目标肿瘤候选产品的方法可能永远不会产生适销对路的产品。
根据我们迄今的发现,支持开发候选产品的可行性的科学证据既是初步的,也是有限的。我们某些候选产品的患者群体仅限于那些具有特定目标突变的患者,我们将需要筛选和识别这些具有目标突变的患者。成功识别患者取决于几个因素,包括确定特定的基因改变和更大类别的突变(如表皮生长因子受体(“EGFR”)外显子20插入突变)如何对我们的候选产品做出反应,以及开发配套诊断方法来识别此类基因改变。此外,即使我们成功地识别了患者,我们也不能确定每个突变或突变类别所产生的患者群体是否足够大,以使我们能够成功地获得每种突变类型的适应症,并将我们的产品商业化并实现盈利。FDA和其他监管机构可能不同意我们为相关突变组而不是单个突变寻求标签的方法,并可能要求我们进行额外的试验并获得每个单个突变的单独批准,这可能会进一步影响我们的产品成功商业化的能力,如果获得批准。此外,即使我们的方法成功地显示了对携带某些靶向突变的肿瘤的临床益处,我们可能永远也无法成功识别更多的致癌突变。因此,我们不知道我们用靶向肿瘤疗法治疗患者的方法是否会成功,如果我们的方法不成功,我们的业务将受到影响。
53
临床前和临床开发涉及一个漫长而昂贵的过程,结果不确定,早期研究和试验的结果可能不能预测未来的临床前研究或临床试验结果。如果我们的临床前研究和临床试验不足以支持监管部门批准我们的任何候选产品,我们可能会在完成或最终无法完成此类候选产品的开发过程中产生额外成本或遇到延迟。
我们的临床前研究和未来的临床试验可能不会成功。我们无法预测我们的候选产品何时或是否会在人体上证明有效和安全,或者是否会获得监管部门的批准。在获得监管部门批准销售任何候选产品之前,我们必须完成临床前研究,然后进行广泛的临床试验,以证明我们的候选产品在人体上的安全性和有效性。一个或多个临床试验的失败可能发生在测试的任何阶段。临床前开发试验和早期临床试验的结果可能不能预测后来的临床试验的成功,临床试验的中期结果也不一定能预测最终结果。制药和生物技术行业的一些公司在后期临床试验中遭遇重大挫折,即使在早期临床试验中取得了令人振奋的结果。此外,临床前和临床数据往往容易受到不同解释和分析的影响,许多公司认为他们的候选产品在临床前研究和临床试验中表现令人满意,但仍未能获得其候选产品的营销批准。
此外,我们进行的一些临床试验在研究设计上可能是开放标签的,并可能在有限数量的临床地点对有限数量的患者进行。“开放标签”临床试验是指患者和研究人员都知道患者是否正在接受研究产品候选,或者是现有的批准药物或安慰剂。最典型的是,开放标签临床试验只测试候选的研究产品,有时可能会在不同的剂量水平上进行测试。开放标签临床试验受到各种限制,可能会夸大任何治疗效果,因为开放标签临床试验的患者在接受治疗时是知道的。开放标签临床试验可能会受到“患者偏见”的影响,即患者认为他们的症状已经改善,仅仅是因为他们意识到接受了实验性治疗。此外,被选中进行早期临床研究的患者通常包括最严重的患者,尽管采用了新的治疗方法,但他们的症状可能肯定会改善。此外,开放标签临床试验可能会受到“调查者偏见”的影响,即那些评估和审查临床试验的生理结果的人知道哪些患者接受了治疗,并可能在了解这一知识的情况下更有利地解释治疗组的信息。鉴于我们与泰豪制药有限公司(“泰豪”)的合作伙伴合作进行的齐帕尔替尼的1/2a期和关键2b期临床试验包括开放标签剂量设计,这些临床试验的结果可能不能预测该产品或其他候选产品的未来临床试验结果,当我们在含有安慰剂或主动对照的受控环境中进行研究时,我们对这些候选产品进行开放标签临床试验。
此外,我们当前和未来临床试验的首席研究员可能会不时担任我们的科学顾问或顾问,并获得与此类服务相关的报酬。在某些情况下,我们可能被要求向FDA或类似的外国监管机构报告其中一些关系。FDA或类似的外国监管机构可能会得出结论,我们与主要研究人员之间的财务关系造成了利益冲突或以其他方式影响了对该研究的解释。因此,FDA或类似的外国监管机构可能会质疑在适用的临床试验地点产生的数据的完整性,临床试验本身的效用可能会受到威胁。这可能会导致FDA或类似的外国监管机构延迟批准或拒绝我们的上市申请,并可能最终导致我们的一个或多个候选产品被拒绝上市批准。
如果我们有足够数量的候选产品被证明是无效的、不安全的或在商业上不可行的,我们的整个流水线可能几乎没有价值,这将对我们的业务、财务状况、运营结果和前景产生实质性的不利影响。
我们可能会在临床前和临床试验方面遇到重大延误,或者可能无法在预期的时间表内进行或完成临床前或临床试验,如果根本没有的话。
我们可能会在启动或完成临床前研究或临床试验方面遇到延误,包括在获得或未能获得FDA批准以启动未来研究性新药申请(IND)的临床试验方面的延误。此外,我们不能确定我们的候选产品的临床前研究或临床试验不需要重新设计,不需要按时招收足够数量的受试者,或者不需要按时完成。在临床前研究和临床试验期间,或由于临床前研究和临床试验的结果,我们可能会遇到许多不利或不可预见的事件,这些事件可能会推迟或终止我们的试验,或者推迟或阻止我们获得上市批准或将我们的候选产品商业化,包括:
54
如果我们被要求对我们目前预期之外的候选产品进行额外的临床前研究、临床试验或其他测试,如果我们无法成功完成候选产品的临床试验或其他测试,如果这些研究、试验或测试的结果不是阳性或仅为中等阳性,或者如果存在安全问题,我们的业务和运营结果可能会受到不利影响,我们可能会产生重大的额外成本。
如果临床试验被我们、IRBs或进行此类临床试验的机构的伦理委员会、用于此类临床试验的数据安全监测委员会(如果有)或FDA或其他监管机构暂停或终止,我们也可能遇到延迟。这些机构可能会因多种因素而暂停、暂停或终止临床试验,这些因素包括未能按照法规要求或我们的临床试验规程进行临床试验、FDA或其他法规机构对临床试验操作或试验地点的检查、不可预见的安全问题或不良副作用、未能证明候选产品带来的益处、政府法规或行政措施的变化或缺乏足够的资金来继续临床试验。
55
我们的开发工作还处于早期阶段,在很大程度上依赖于我们的主要候选产品zipertinib、CLN-049、CLN-619和CLN-418。如果我们无法通过临床开发推进这些或我们的任何其他候选产品,或者无法获得监管部门的批准并最终将任何此类候选产品商业化,无论是我们自己还是与第三方或由第三方合作,或者如果我们在这样做方面遇到重大延误,我们的业务将受到实质性损害。
我们的开发工作还处于早期阶段。我们的主导项目zipertinib正在与我们在大鹏的合作伙伴合作进行关键的2b期临床试验。此外,我们的候选产品CLN-049、CLN-619和CLN-418都处于第一阶段临床试验中。我们创造产品收入的能力将在很大程度上取决于齐帕替尼、CLN-049、CLN-619、CLN-418和我们的其他候选产品的成功临床开发和最终商业化,如果获得批准,我们预计这将在很多年内不会发生。我们候选产品的成功将取决于以下几个因素:
如果我们不能及时或根本地在这些因素中的一个或多个方面取得成功,我们可能会在成功将候选产品商业化的能力方面遇到重大延误,或者根本无法将候选产品商业化。如果我们不能将我们的临床前阶段候选产品推进到临床开发,成功完成我们候选产品的临床试验,获得监管部门的批准,并最终将我们的候选产品商业化,我们的业务将受到实质性的损害。
56
目前的临床前研究或我们正在进行的临床试验中获得的齐帕替尼用于EGFR外显子20插入突变非小细胞肺癌(“NSCLC”)患者、复发或难治性急性髓细胞白血病(“r/r AML”)患者的CLN-049、实体肿瘤患者的CLN-619或晚期实体肿瘤患者的CLN-418将足以获得监管部门的批准或营销授权。例如,FDA可能要求我们在获得监管批准之前,与泰豪的合作伙伴合作,完成正在进行的齐帕替尼1/2a期临床试验和关键2b期试验之外的试验。尽管我们认为我们的候选产品和计划是不相关的,但一个候选产品的开发过程中的负面结果可能会影响其他候选产品或计划。对于我们的每一种候选产品,我们计划在临床试验中评估的每种不同肿瘤类型的抗肿瘤活性可能不同。即使在我们与我们的候选产品建立临床经验的同时,我们也可能需要与FDA进一步讨论或会面,就每项试验的最佳患者群体、研究设计和规模达成一致,以获得监管部门的批准,其中任何一项都可能需要大量额外资源,并推迟我们临床试验的时间,并最终批准我们的任何候选产品(如果有的话)。
招募患者的困难可能会推迟或阻止我们候选产品的临床试验,并最终推迟或阻止监管部门的批准。
确定并使患者有资格参与我们候选产品的临床试验对我们的成功至关重要。我们临床试验的完成时间在一定程度上取决于我们招募患者参与测试我们候选产品的速度,如果我们在登记时遇到困难,我们可能会遇到临床试验的延迟。如果我们无法根据FDA或美国以外的类似监管机构的要求,或为特定试验提供适当的统计数据所需,找到并招募足够数量的合格患者参加这些试验,我们可能无法为我们的候选产品启动或继续临床试验。特别是,由于我们专注于具有特定基因突变的患者来开发齐帕替尼,我们招募符合条件的患者的能力可能会受到限制,或者由于符合条件的患者人数较少,登记可能比我们预期的要慢。对于我们在急性髓细胞白血病患者中评估CLN-049的第一阶段试验,由于登记标准和单一递增剂量设计,我们登记合格患者的能力可能有限,或者登记速度可能比我们预期的要慢。
除了潜在的小群体外,我们计划的临床试验的资格标准将进一步限制可用研究参与者的池,因为我们要求患者具有特定的特征,如疾病进展的特定严重程度或阶段,才能将他们纳入研究。此外,寻找符合条件的患者的过程可能被证明是昂贵的。我们也可能无法识别、招募和招募足够数量的患者来完成我们的临床研究,原因包括接受研究的候选产品的已知风险和益处、竞争疗法和临床试验的可用性和有效性、潜在患者的临床研究地点的邻近和可用性、患者肿瘤的基因测序信息的可用性以便我们能够识别具有目标基因突变的患者以及医生的患者转介做法。如果患者出于任何原因不愿参与我们的研究,招募患者、进行研究和获得监管部门对潜在产品的批准的时间可能会推迟。
患者的登记还取决于许多因素,包括:
57
此外,我们的临床试验将与其他临床试验争夺与我们的候选产品在相同治疗领域的产品,而这一竞争将减少我们可用的患者数量和类型,因为一些可能选择参加我们的临床试验的患者可能会选择参加由我们的竞争对手之一进行的临床试验。由于合格临床研究人员的数量有限,我们预计将在一些竞争对手使用的相同临床试验地点进行我们的一些临床试验,这将减少可供我们在这些临床试验地点进行临床试验的患者数量。此外,由于我们的某些候选产品与更常用的癌症治疗方法不同,而且我们的某些候选产品以前从未在人体上进行过测试,潜在的患者和他们的医生可能倾向于使用传统疗法,如化疗,而不是让患者参加我们候选产品的任何未来临床试验。
如果我们延迟完成或终止任何候选产品的临床试验,我们候选产品的商业前景将受到损害,我们从这些候选产品中获得产品收入的能力可能会被推迟或阻止。
我们宣布或公布的临床试验的临时、“背线”和初步数据可能会随着更多患者数据的出现而发生变化,并受到可能导致最终数据发生重大变化的确认、审计和验证程序的影响。
我们可能会不时公开披露我们的临床前研究和临床试验的初步或主要数据,这些数据基于对当时可用数据的初步分析,在对与特定研究或试验相关的数据进行更全面的审查后,结果及相关发现和结论可能会发生变化。作为数据分析的一部分,我们也会做出假设、估计、计算和结论,而我们可能没有收到或没有机会全面而仔细地评估所有数据。因此,我们报告的背线或初步结果可能与相同研究的未来结果不同,或者一旦收到更多数据并进行充分评估,不同的结论或考虑因素可能会使这些结果合格。背线数据仍需接受审计和核实程序,这可能会导致最终数据与我们之前公布的初步数据大不相同。我们还可能不时地披露临床试验的中期数据。临床试验的中期数据面临这样的风险,即随着患者登记和治疗的继续以及更多患者数据的获得,或者当我们的临床试验中的患者继续进行其他治疗时,一个或多个临床结果可能会发生实质性变化。初步或中期数据与最终数据之间的不利差异可能会严重损害我们的业务前景,以及我们获得批准并将我们的候选产品商业化的能力可能会受到损害。此外,我们或我们的竞争对手披露中期数据可能会导致我们普通股的价格波动。
我们可能无法在我们预期的时间内提交IND或IND修正案以开始额外的临床试验,即使我们能够,FDA或外国监管机构也可能不允许我们继续进行。
我们已经提交了齐帕替尼、CLN-049、CLN-619和CLN-978的IND,这些都是目前有效的。此外,我们在2023年2月提交了CLN-617的IND。但是,我们可能无法按照预期的时间表提交其他候选产品的未来IND。此外,我们可能会遇到生产延迟或IND支持研究的其他延迟,或者FDA或其他监管机构可能需要额外的临床前研究,这是我们没有预料到的。此外,我们不能确定提交IND将导致FDA允许临床试验开始,或者一旦开始,不会出现导致我们、IRBs或独立伦理委员会、FDA或其他监管机构决定暂停或终止临床试验的问题,包括临床暂停。此外,即使FDA或其他监管机构同意IND中规定的临床试验的设计和实施,我们也不能保证他们未来不会改变他们的要求或期望。这些考虑也适用于我们可能提交的新的临床试验,作为现有IND的修正案或新的IND。任何未能在我们预期的时间期限内提交IND或获得监管部门对我们试验的批准可能会阻止我们及时完成临床试验或将我们的产品商业化。
我们打算开发CLN-619和潜在的其他候选产品,与其他疗法相结合,这将使我们面临额外的风险。
我们打算开发CLN-619和潜在的其他候选产品,结合一种或多种已批准或未批准的疗法来治疗癌症或其他疾病。即使我们开发的任何候选产品获得了与其他批准的疗法结合使用的上市批准,FDA、欧洲制造局(“EMA”)或美国以外的类似外国监管机构仍可以撤销与我们产品结合使用的疗法的批准。如果与我们的候选产品结合使用的疗法被取代为适应症的标准护理,我们选择我们的任何候选产品,FDA、EMA或类似的外国监管机构可能会要求我们进行额外的临床试验。任何这些风险的发生都可能导致我们自己的产品,如果获得批准,将被从市场上撤下,或者在商业上不太成功。
58
此外,如果未经批准的癌症疗法最终不能单独或与我们的产品组合获得营销批准,我们将无法营销和销售我们与未经批准的癌症疗法联合开发的任何候选产品。此外,未经批准的癌症疗法面临与我们目前正在开发和临床试验的候选产品相同的风险,包括可能出现严重的不良反应、临床试验延迟以及缺乏FDA批准。
如果FDA、EMA或类似的外国监管机构不批准这些其他产品或撤销其批准,或者如果我们选择结合我们开发的候选产品进行评估的产品出现安全性、有效性、质量、制造或供应问题,我们可能无法获得此类联合疗法的批准或将其推向市场。
如果我们无法成功验证、开发和获得监管机构对我们的候选产品所需的任何配套诊断测试的批准,或者在执行此操作时遇到重大延误,我们可能无法获得批准或可能无法实现这些候选产品的全部商业潜力。
对于我们的某些适应症候选产品的临床开发,我们需要开发或获得体外培养配套的诊断测试,以确定疾病类别中可能从我们的候选产品中受益的患者亚组,因为我们针对的是某些基因定义的人群进行治疗。此类伴随诊断可用于我们的临床试验,并可能在FDA批准我们的候选产品时使用。为了取得成功,我们或我们的合作者需要解决一些科学、技术、监管和后勤方面的挑战。配套诊断作为医疗设备受到FDA、EMA和其他监管机构的监管,在商业化之前需要单独的监管批准。
鉴于我们在开发和商业化诊断方面的经验有限,我们可能会依赖第三方为我们的候选产品设计、开发和制造可能需要此类测试的配套诊断测试。如果我们达成这样的合作协议,我们将依赖于我们未来的合作者在开发和获得对这些伴随诊断的批准方面的持续合作和努力。我们和我们未来的合作者在开发和获得伴随诊断的批准方面可能会遇到困难,包括与伴随诊断的选择性/特异性、分析验证、重复性或临床验证相关的问题。我们和我们未来的合作伙伴在开发、获得监管批准、制造和商业化配套诊断方法方面也可能会遇到困难,这些困难类似于我们针对我们的候选产品本身所面临的问题,包括获得监管许可或批准、以商业规模和适当的质量标准生产足够数量的产品以及获得市场认可的问题。如果我们无法为这些候选产品成功开发配套诊断,或在开发过程中遇到延迟,这些候选治疗产品的开发可能会受到不利影响,或者这些候选产品可能无法获得上市批准或延迟,并且我们可能无法实现这些获得营销批准的候选产品的全部商业潜力。因此,我们的业务、运营结果和财务状况可能会受到实质性的损害。此外,与我们签约的一家诊断公司可能决定停止开发, 销售或制造我们预期用于开发和商业化我们的候选产品或我们与该诊断公司的关系的配套诊断测试可能会终止。我们可能无法与另一家诊断公司达成安排,以获得替代诊断测试的供应,以用于我们候选产品的开发和商业化,或者以商业合理的条款这样做,这可能会对我们候选产品的开发或商业化产生不利影响和/或延迟。
我们的候选产品可能会导致不良副作用或具有其他特性,从而延迟或阻碍其监管审批,限制其商业潜力,或在任何潜在的上市审批后导致重大负面后果。
我们的候选产品可能会引起不良的副作用。此外,给药过程或相关程序也可能导致不良副作用。在我们的试验中发生的不良事件可能会导致我们,或导致FDA、EMA或其他监管机构或IRBs命令我们停止、推迟或修改我们的候选产品的临床前开发或临床开发,并可能导致更严格的标签限制或拒绝监管部门批准任何或所有目标适应症的候选产品。即使严重的不良事件与研究治疗无关,此类事件也可能影响患者登记或登记患者完成试验的能力。此外,如果我们的任何候选产品被测试或与其他药物联合使用,例如我们可能将CLN-619与其他药物联合使用的计划,这些组合可能会产生额外的副作用,可能比单独使用任何一种疗法造成的副作用更严重。
此外,临床试验的本质是利用潜在患者群体的样本。由于患者数量和接触时间有限,我们的候选产品或竞争对手的罕见和严重副作用可能只有在更多的患者接触该药物时才会被发现。例如,虽然我们认为齐帕替尼到目前为止表现出了可控的耐受性,但不能保证它或我们的任何其他候选产品不会在更大比例的患者中引起更严重的副作用。
59
此外,如果我们选择或被要求推迟、暂停或终止我们任何候选产品的临床试验,该等候选产品或我们其他候选产品的商业前景可能会受到损害,我们从任何这些候选产品产生产品收入的能力可能会被推迟或取消。这些情况中的任何一种都可能损害我们开发其他候选产品的能力,并可能严重损害我们的业务、财务状况、运营结果和前景。
如果我们的候选产品获得上市批准,而我们或其他人在获得批准后发现此类候选产品(或任何其他类似药物或生物制品)引起的不良副作用,可能会导致许多潜在的重大负面后果,包括:
我们认为,任何这些事件都可能阻止我们实现或保持市场对受影响的候选产品的接受程度,如果获得批准,可能会大幅增加我们候选产品的商业化成本,并显著影响我们成功将候选产品商业化并创造收入的能力。
由于我们正在进行的临床试验中已经和将接受剂量的患者数量很少,因此一旦完成,此类临床试验的结果可能不如大型临床试验的结果可靠,这可能会阻碍我们为我们的候选产品获得监管部门批准的努力。
样本量较小的临床试验的初步结果可能会受到与进行小型临床试验相关的各种偏差的不成比例的影响,例如,较小的样本量可能无法准确描述更广泛的患者群体的特征,这限制了在更广泛的社区推广结果的能力,从而使临床试验结果的可靠性低于患者数量较大的临床试验。因此,这些候选产品在未来的临床试验中是否会取得统计上的显著效果可能不那么确定。此外,FDA或其他监管机构可能会要求我们进行额外的、规模更大的试验,而不是我们计划支持的上市授权申请。如果我们进行任何未来的临床试验齐帕替尼、CLN-049、CLN-619或CLN-418或我们的其他候选产品,我们可能得不到积极的或有统计学意义的结果或相同水平的统计意义,如果有的话,我们可能会根据先前的结果预期。
60
我们目前正在进行并可能在未来对美国以外的候选产品进行临床试验,FDA和类似的外国监管机构可能不会接受此类试验的数据。
我们正在与我们在TAIHO的合作伙伴合作,评估齐帕替尼的1/2a期试验和关键的2b期试验,CLN-619进行一期临床试验,CLN-418进行一期临床试验,包括设在美国国内外的中心。我们未来还可能选择在美国以外的地区进行一项或多项额外的临床试验,包括在欧洲和澳大利亚。FDA或类似的外国监管机构接受在美国或其他司法管辖区以外进行的临床试验的研究数据可能会受到某些条件的限制,也可能根本不会被接受。如果外国临床试验的数据打算作为在美国上市批准的基础,FDA通常不会仅根据外国数据批准申请,除非(I)数据适用于美国人口和美国医疗实践,以及(Ii)试验由具有公认能力的临床研究人员进行,并符合良好临床实践(GCP)法规。此外,必须满足FDA的临床试验要求,包括足够大的患者群体和统计能力。许多外国监管机构也有类似的审批要求。此外,外国审判受进行审判的外国司法管辖区适用的当地法律管辖。如果FDA或任何类似的外国监管机构不接受在美国或适用的外国司法管辖区以外进行的试验的数据,我们将需要进行额外的试验,这可能是昂贵和耗时的,并可能导致我们可能开发的候选产品在美国或任何此类外国司法管辖区得不到商业化批准。
与我们的财务状况和资本要求有关的风险
我们有限的经营历史可能会使您难以评估我们业务迄今的成功程度,也难以评估我们未来的生存能力。
我们于2017年开始实质性运营。到目前为止,我们的业务仅限于组织和配备我们的公司、业务规划、为我们和我们的子公司筹集资金、提交专利申请、确定并收购和投资潜在的候选产品、进行临床试验、建立我们的知识产权组合,以及为识别、发现和研究活动、临床前研究、临床试验以及制造我们的候选产品和组件材料的第一批产品建立安排并与第三方合作。我们尚未证明我们有能力成功进行后期临床试验、完成任何临床试验、获得市场批准、制造商业规模的产品或安排第三方代表我们这样做,或进行成功的产品商业化所需的销售、营销和分销活动。
因此,你对我们未来成功或生存能力的任何预测都可能不像我们有更长的运营历史时那样准确。
自成立以来,我们遭受了重大亏损,我们预计未来几年将出现亏损,未来可能无法实现或维持收入或盈利能力。
对生物制药产品开发的投资是一项高度投机性的工作,需要大量的前期资本支出,并存在重大风险,即任何潜在的候选产品将无法证明足够的疗效或可接受的安全性,无法获得监管部门的批准,无法在商业上可行。我们仍处于候选产品开发的早期阶段。到目前为止,我们还没有任何产品被批准用于商业销售,也没有从产品销售中获得任何收入,我们继续产生与我们持续运营相关的巨额研发和其他费用。我们主要通过出售股权证券来为我们的业务融资。
自我们开始实质性业务以来,我们在每个时期都发生了重大的净亏损。2022年和2021年,我们分别报告了1.092亿美元的净收益和6750万美元的净亏损。截至2022年12月31日,我们的累计赤字为4770万美元。我们预计在可预见的未来将继续蒙受重大损失,如果我们:
61
由于与开发候选药品相关的众多风险和不确定性,我们无法预测未来亏损的程度或何时实现盈利(如果有的话)。即使我们成功地将我们的一个或多个候选产品商业化,我们也将继续产生大量的研发和其他支出,以开发和寻求监管部门对更多候选产品或更多适应症的批准。我们可能会遇到不可预见的费用、困难、并发症、延误和其他可能对我们的业务产生不利影响的未知因素。我们未来净亏损的规模将在一定程度上取决于我们未来支出的增长率和我们创造收入的能力。我们之前的亏损和预期的未来亏损已经并将继续对我们的股东权益和营运资本产生不利影响。
我们没有从销售我们的候选产品中获得任何收入,而且可能永远不会盈利。
我们盈利的能力取决于我们创造收入的能力。除了之前与再鼎医药签订的许可协议外,我们没有从我们的任何候选产品中获得任何其他许可或协作收入或任何销售收入。我们不希望通过销售或许可我们的一个或多个临床前计划或候选产品获得显著的销售收入或商业收入,除非或直到我们成功完成临床开发,并获得监管部门的批准,然后成功将我们的一个候选产品商业化,或者与第三方就购买、合作或许可我们的一个候选产品达成协议。我们目前正在推进临床开发中的齐帕替尼(根据与大鹏的共同开发协议)、CLN-049、CLN-619和CLN-418,以及我们的其他候选产品,这些产品处于临床前开发阶段,需要进行额外的临床前研究。我们所有的候选产品都需要额外的临床开发、监管审查和批准、大量投资、获得足够的商业制造能力和重大的营销努力,才能从产品销售中产生任何收入。我们的创收能力取决于多个因素,包括但不限于:
62
上面列出的许多因素都是我们无法控制的,可能会导致我们遭遇重大延误,或者阻止我们获得监管部门的批准或将我们的候选产品商业化。即使我们能够将我们的候选产品商业化,我们也可能不会在产生产品销售后不久实现盈利,如果有的话。如果我们无法通过商业销售我们的候选产品或任何未来的候选产品,或通过与第三方就购买、协作或许可我们的一个或多个候选产品达成协议来产生足够的收入,我们可能无法在没有持续资金的情况下继续运营。
我们将需要大量的额外资金来开发和商业化我们的候选产品,并确定和投资于新的候选产品。如果我们无法在需要的时候筹集资金,我们将被迫推迟、减少或取消我们的产品开发计划或其他业务。
医药产品的发展是资本密集型的。我们目前正在推进临床开发中的齐帕替尼(根据与大鹏的共同开发协议)、CLN-049、CLN-619和CLN-418,并对我们的临床前计划进行进一步投资。我们预计我们的费用将与我们正在进行的活动同步增加,正如上文题为“我们自成立以来遭受了重大损失,我们预计在未来几年内将遭受损失,未来可能无法实现或维持收入或盈利”的风险因素所述。因此,我们将需要获得与我们的持续运营相关的大量额外资金,这可能包括通过我们的一个或多个子公司筹集资金,这可能会稀释我们在子公司的股权。我们已经根据可能被证明是错误的假设估计了我们目前的额外资金需求。不断变化的环境可能会导致我们消耗资本的速度比我们目前预期的要快得多,而且由于我们无法控制的情况,我们可能需要花费比目前预期更多的钱。我们不能确定是否会在可接受的条件下提供额外资金,或者根本不能。在此之前,如果我们能够产生可观的产品收入,我们预计将通过公开或私募股权发行、债务融资、政府融资、合作、战略合作伙伴关系和联盟,或者通过我们或我们的一个或多个子公司与第三方的营销、分销或许可安排,为我们的运营提供资金。如果我们或我们的子公司无法在需要时或在有吸引力的条件下筹集资金,我们或适用的子公司将被迫推迟、减少或取消我们的识别、发现和临床前或临床开发计划。, 或任何未来的商业化努力。
截至2022年12月31日,我们的现金及现金等价物和短期投资为4.673亿美元,长期投资和应收利息为8280万美元。我们相信,根据我们目前的运营计划,我们现有的资本资源将足以为我们预期的2026年前的运营提供资金。我们未来的资本需求将取决于许多因素,包括:
63
如果我们或我们的子公司进行收购或战略合作,这可能会增加我们或他们的资本金要求,稀释我们或他们的股东,导致我们或他们产生债务或承担或有负债,并使我们或他们面临其他风险。
于2022年6月,吾等将本公司前部分拥有附属公司库利南珍珠股份有限公司(“库利南珍珠”)的股权出售给泰豪,并与泰豪的一间联营公司订立共同开发协议,以在美国共同开发及按我们的选择共同商业化齐帕尔替尼。根据与泰豪的共同开发协议的条款,出售库利南珍珠股权后产生的齐帕尔替尼的开发成本将由我们及泰豪平分,双方均可从未来潜在的美国销售zipert tinib中获得50%的税前利润。此外,在2022年10月和11月,我们从库利南MICA公司(“库利南MICA”)股东手中购买了股权,并以私募方式获得了额外的股份。我们目前持有库利南MICA 95%的流通股。库利南MICA拥有与CLN-619相关的知识产权。
我们打算在未来从事各种收购和战略合作伙伴关系,包括由我们或我们的一个或多个全资或部分拥有的子公司进行的许可或获取产品、知识产权、技术或业务,包括为进行此类交易而成立的新成立的子公司。任何收购或战略合作伙伴关系,包括与大鹏的共同开发协议,都可能给我们或适用的子公司带来许多风险,包括:
如果我们未能正确评估与创建新的研发计划或维护现有研发计划相关的潜在收购、许可、投资或其他交易,我们可能无法实现任何此类交易的预期收益,我们可能会产生超出预期的成本,管理资源和注意力可能会从其他必要或有价值的活动上转移。
与我们的公司结构相关的风险
我们可能不会成功地建立一条具有商业价值的候选产品渠道。
64
我们战略的一个关键要素是形成或寻求战略联盟,创建合资企业或合作,或与第三方就我们认为新颖、采用差异化行动机制、在开发方面比竞争对手更先进或具有这些属性的项目、候选产品、技术或知识产权达成许可安排。我们在寻找合适的战略合作伙伴以及许可和收购机会方面面临着激烈的竞争,谈判过程既耗时又复杂。我们可能不会成功地通过收购、许可或内部开发来建立用于治疗各种癌症的候选产品的流水线,或者通过临床开发来推进这些候选产品。尽管我们分析是否可以复制在收购或投资候选产品之前观察到的科学结果,但在投资之后,我们可能无法成功做到这一点。例如,我们可能无法成功识别更多的致癌基因突变,这些突变可以被“打包”到一个足够大的群体中,提供足够的商业机会,或者可以用一种化合物下药。
此外,我们正在寻求额外的许可证内或收购开发阶段的资产或计划,这给我们带来了额外的风险。确定、选择和获得有前景的候选产品需要大量的技术、财务和人力资源专业知识。这样做的努力可能不会导致实际获得或许可成功的候选产品,可能会导致我们管理层的时间和资源支出的分流,而不会产生任何好处。例如,如果我们无法确定最终导致批准产品的计划,我们可能会花费大量资本和其他资源来评估、收购和开发最终无法提供投资回报的产品。我们已经终止了项目,如果这些项目不符合我们的晋升标准,我们将在未来终止这些项目。
我们的子公司是某些协议的一方,这些协议为我们子公司的许可人、协作者或其他股东提供的权利可能会推迟或影响我们子公司的潜在出售,或可能影响我们子公司出售资产的能力,或与其他第三方达成战略联盟、合作或许可安排。
我们的每一家子公司都从第三方获得知识产权许可,有几家已经从第三方投资者那里筹集了资金。这些第三方拥有某些权利,可能会推迟与另一第三方的合作、许可或其他安排,这些权利的存在可能会对吸引收购方或合作伙伴的能力产生不利影响。这些权利包括许可协议中包含的出售子公司资产或变更控制权时的谈判权和应付费用,以及与子公司股东达成的协议中的拖拖权等权利。
此外,我们的部分控股子公司库利南佛罗伦萨公司(“库利南佛罗伦萨”)和库利南琥珀公司(“库利南琥珀”)也将在出售或以其他方式处置其大部分资产的情况下向许可人支付成功费用。这些费用将减少我们从任何此类资产出售或处置中获得的净收益。
我们还与第三方投资者签订了投资者权利和投票权协议,这可能会推迟或影响我们出售我们部分拥有的子公司的股权或资产的能力。例如,我们将需要遵守某些通知和其他条款,例如在出售子公司时的拖累条款,这可能会推迟或阻止特定交易,或降低与我们的子公司和我们进行交易对第三方的吸引力。
我们可能会成立更多的子公司,并与未来的合作伙伴或投资者签订类似的协议,或者我们的子公司可能会签订进一步的协议,在每种情况下,这些协议都可能包含对我们不利的类似条款或其他条款。
如果我们将所有权减少到少数股权,或者通过合同协议或其他方式将控制权让给其他投资者,我们从子公司实现价值的能力可能会受到影响。
如果我们的任何子公司需要额外资本,并且其各自的董事会批准了交易,我们在我们子公司的股权可能会进一步减少,前提是这些额外资本是从第三方投资者而不是我们那里获得的。然而,此类交易仍需得到我们保持完全控制的各自子公司董事会的批准。
然而,如果我们不希望或无法向我们的任何子公司提供额外资本,我们可能会批准子公司发行股权,从而稀释我们的所有权并可能失去对子公司的控制。
于2022年6月,吾等完成将本公司前部分拥有附属公司库利南珍珠的股权出售予泰豪,预付款为2.75亿美元。在达到与齐帕替尼相关的某些监管里程碑后,我们可能会额外获得高达1.3亿美元的资金。不能保证实现这些里程碑,也不能保证我们会收到额外的1.3亿美元中的任何一笔。在出售我们在库利南珍珠的股权时,我们与大鹏签订了一项共同开发协议,根据协议,我们和大鹏将共同开发齐帕尔替尼,并根据我们的选择,在美国共同商业化齐帕尔替尼。我们和泰豪将平分未来齐帕尔替尼的临床开发成本,双方将从未来可能在美国销售齐帕尔替尼的任何税前利润中获得50%。不能保证共同开发和共同商业化会成功,也不能保证我们会获得任何净利润,我们可能会赔钱。
65
一家或有限数量的子公司可能构成我们价值的很大比例。
我们价值的很大一部分随时可能存在于我们的一个或两个子公司,包括知识产权和归因于该公司正在开发的候选产品或计划的价值。如果子公司的候选产品或计划的临床开发或潜在商业化前景或特定子公司持有的一个或多个知识产权受到损害,我们的综合财务状况和前景可能会受到严重影响。此外,我们合并收入的很大一部分在任何时候都可能来自一项或少数授权技术,这些技术的许可终止或到期可能会对我们的合并收入产生重大不利影响。对特定子公司价值的任何重大不利影响,包括其知识产权或其候选产品或计划的临床开发,都可能对我们的综合业务、财务状况、运营结果或前景产生重大不利影响。
我们可能会花费有限的资源来追求特定的候选产品或适应症,而无法利用可能更有利可图或成功可能性更大的候选产品或适应症。
由于我们的财务和管理资源有限,我们必须专注于有限数量的研究项目和候选产品以及特定的适应症。因此,我们可能会放弃或推迟寻找其他候选产品或后来被证明具有更大商业潜力的其他迹象的机会,或者无法确认或收购可能比我们收购的资产更有前景的资产。我们的资源分配决策可能会导致我们无法利用可行的商业产品或有利可图的市场机会。我们在当前和未来的识别、发现和临床前开发计划以及特定适应症的候选产品上的支出可能不会产生任何商业上可行的产品。
我们对由有限员工组成的中央团队的依赖带来了运营挑战,可能会对我们的业务产生不利影响。
截至2022年12月31日,我们拥有62名全职员工,我们依赖这些员工在我们的其他运营子公司之间共享各种行政、研发和其他支持服务。我们还有两名顾问,我们依靠他们进行研发、业务发展和其他服务。虽然我们相信这种结构使我们能够降低某些基础设施成本,但我们集中的团队规模较小,可能会限制我们投入足够的人员、时间和资源来支持我们所有子公司的运营,包括它们的研发活动,以及财务、会计和报告事项的管理。鉴于我们的员工和管理层主要是在母公司层面受到激励,这些员工和管理团队成员可能得不到足够的激励来最大化我们整个组织的整体价值。如果我们的集中团队不能在整个组织中提供足够的管理、研发或其他服务,我们的业务、财务状况和运营结果可能会受到损害。
66
我们的一些高级职员和董事目前或将来可能担任我们子公司的董事或高级职员,因此,他们已经并可能继续对我们的子公司负有受托责任和其他职责,这会导致他们对我们的职责和他们对我们子公司的职责产生利益冲突,并决定如何致力于我们的事务和我们子公司的事务。我们子公司的合作伙伴也可能不同意我们向每个子公司提供的资源是否足够。
我们的某些高管同时也是我们的一个或多个子公司的董事和/或高管,因此,他们对我们和我们的子公司都负有受托责任或其他责任。此类责任产生的利益冲突可能会干扰我们子公司及其计划和候选产品的管理,或导致与子公司合作伙伴的分歧。举例来说,既是本公司其中一家子公司的董事,又是库利南的董事的个人对子公司和整个公司负有受托责任,在这种情况下,他或她的决定或行动可能有利于子公司而对公司产生不利影响,反之亦然,或对另一家子公司(包括他或她同时担任董事的子公司)产生不利影响。此外,我们的高管和董事同时也是我们子公司的高管和董事,他们需要将他或她的时间分配给库利南和他或她担任高管或董事的每个子公司的责任,并将代表一个实体做出可能对其他实体产生负面影响的决定。此外,虽然我们的大多数子公司已经放弃向任何董事或同时是董事高管的库利南高管提供或收购、创建或开发的或以其他方式归其所有的任何权益或对公司机会的预期,但我们与子公司的合作伙伴之间可能会因利益冲突而产生纠纷。这些合作伙伴也可能不同意我们的管理人员和员工投入到他们所投资的子公司的资源的数量和质量。任何这样的纠纷或分歧都可能分散我们的管理层的注意力,干扰我们与合作伙伴的关系,并花费大量时间来解决,这可能会扰乱我们候选产品的开发, 推迟我们潜在的商业化努力,导致成本增加,或使其他第三方在未来选择与我们合作的可能性降低。
与潜在商业化相关的风险
即使我们的候选产品获得监管部门的批准,这些产品也可能无法获得医生、患者、医院、癌症治疗中心和医学界其他人的市场接受。
使用靶向肿瘤药物作为潜在的癌症治疗是最近的发展,可能不会被医生、患者、医院、癌症治疗中心和医学界的其他人广泛接受。影响我们的候选产品是否被市场接受的因素有很多,包括:
如果我们的候选产品获得许可,但无法获得医生、患者、医院、癌症治疗中心或医学界其他人的市场认可,我们将无法产生可观的收入。
67
即使我们的候选产品获得了市场认可,但如果推出了比我们的产品更受欢迎、更具成本效益或使我们的产品过时的新产品或技术,我们可能无法随着时间的推移保持市场接受度。
我们面临着激烈的竞争,这可能会导致其他人比我们更早或更成功地发现、开发或商业化产品。
生物技术和制药行业的特点是技术的快速发展和对疾病病因的了解,激烈的竞争,以及对知识产权的高度重视。我们面临并将继续面临来自专注于更传统治疗方式的公司的竞争,例如小分子抑制剂。我们相信,我们差异化的商业模式、方法、科学能力、技术诀窍和经验为我们提供了竞争优势。然而,我们预计来自多种来源的激烈竞争,包括世界各地的主要制药、专业制药和现有或新兴的生物技术公司、学术研究机构、政府机构以及公共和私人研究机构。我们的许多竞争对手,无论是单独或通过合作,在研发、制造、临床前测试、进行临床试验、获得监管批准和营销批准的产品方面,都比我们拥有更多的财务资源和专业知识。规模较小或处于初创阶段的公司也可能成为重要的竞争对手,特别是通过与大型和成熟公司的合作安排。这些公司还在招聘和留住合格的科学和管理人员、建立临床试验场地和在临床试验中招募患者以及在获取与我们的计划相辅相成或必要的技术方面与我们竞争。因此,我们的竞争对手可能比我们更早或更成功地发现、开发、许可或商业化产品。
我们成功开发和商业化的候选产品将与现有疗法和未来可能出现的新疗法展开竞争。我们预计齐帕替尼将与武田制药公司的小分子表皮生长因子受体抑制剂Exkitivity(TAK-788)和强生的EGFR/cMET双特异性抗体Rybrevant竞争。齐帕替尼还可能与来自Dizal制药有限公司的表皮生长因子受体抑制剂sunvozertinib、来自到达生物制药公司和Allist制药公司的Furmonertinib、来自奥利克制药公司(中国大陆、香港、澳门和台湾地区的沃罗诺伊公司)的ORIC114和来自Blueprint制药公司的液化天然气-451LNG-451以及临床前开发中的其他分子竞争。我们预计CLN-049将在治疗AML方面与双特异性药物竞争,包括针对CD3和CD33(Amgen Inc.和Amphivena Treateutics,Inc.)、CD123(MacroGenics Inc.和Xencor,Inc.)、Flt3和CCL1/Clec12a(Merus N.V.和Genentech,Inc.)的药物。我们预计,CLN-619将与将MICA/B作为单一疗法和/或与其他药物结合使用的癌症疗法展开竞争,这些药物包括:CanCure LLC、基因技术公司、诺华国际公司和百时美施贵宝公司。
如果我们的候选产品,包括齐帕替尼、CLN-049和CLN-619,被批准作为其目前建议的目标适应症,它们很可能会与上述竞争对手的产品以及目前正在开发的其他产品竞争。影响我们所有候选产品成功的关键竞争因素,如果获得批准,很可能是它们的疗效、安全性、便利性、价格、仿制药竞争水平,以及政府和其他第三方付款人是否可以报销。我们的竞争对手可能获得专利保护或其他知识产权,这限制了我们开发或商业化我们的候选产品的能力。政府和其他第三方付款人的报销也将对我们产品的定价和竞争力产生重大影响。我们的竞争对手也可能比我们更快地获得FDA或其他监管机构对其产品的批准,这可能会导致我们的竞争对手在我们能够进入市场之前建立强大的市场地位。如果我们的竞争对手开发和商业化比我们的候选产品更安全、更有效、副作用更少或更不严重、更方便管理、更便宜或标签更有利的药物,我们可能会看到商业机会减少或消失。有关我们竞争的更多信息,请参阅本年度报告中题为“商业竞争”的Form 10-K部分。
68
新批产品的保险覆盖范围和报销状况尚不确定。我们的候选产品可能会受到不利的定价法规、第三方保险和报销做法或医疗改革举措的影响,这将损害我们的业务。如果不能为新产品或现有产品获得或保持足够的保险和报销,可能会限制我们营销这些产品的能力,并降低我们创造收入的能力。
管理新药上市审批、定价、覆盖和报销的规定因国家而异。在美国,最近颁布的立法可能会大幅改变审批要求,这可能会涉及额外的成本,并导致获得批准的延迟。一些国家要求药品的销售价格获得批准后才能上市。在许多国家,定价审查期从批准营销或产品许可后开始。在一些外国市场,处方药定价即使在获得初步批准后,仍然受到政府的持续控制。因此,我们可能会获得产品在特定国家/地区的营销批准,但随后会受到价格法规的约束,这些法规会推迟我们的产品的商业发布,可能会推迟很长时间,并对我们在该国家/地区销售该产品所能产生的收入产生负面影响。不利的定价限制可能会阻碍我们收回在一个或多个候选产品上的投资的能力,即使我们可能开发的任何候选产品获得了市场批准。
在美国和其他国家的市场,患者通常依靠第三方付款人来报销与治疗相关的全部或部分费用。政府医疗保健计划(如联邦医疗保险和医疗补助)和商业支付者的足够覆盖和报销对于新产品的接受度至关重要。我们能否成功地将我们的候选产品商业化,在一定程度上将取决于政府卫生行政部门、私人健康保险公司和其他组织为这些产品和相关治疗提供的保险范围和足够的补偿。政府当局和其他第三方付款人,如私人健康保险公司和健康维护组织,决定他们将支付哪些药物并建立报销水平。政府和私人付款人提供的保险范围和报销范围对于大多数患者能够负担得起基因治疗产品等治疗是至关重要的。我们可能确定的这些或其他候选产品的销售将在很大程度上取决于我们产品候选的费用将在多大程度上由健康维护、管理医疗、药房福利和类似的医疗管理组织支付,或由政府卫生行政当局、私人健康保险保险公司和其他第三方付款人报销。如果无法获得保险和足够的报销,或仅限于有限的级别,我们可能无法成功地将我们的候选产品商业化。即使提供了保险,批准的报销金额也可能不够高,不足以让我们建立或保持足够的定价,以实现足够的投资回报。
第三方付款人的报销可能取决于许多因素,包括但不限于第三方付款人对产品使用情况的确定:
美国医疗保健行业和其他地方的一个主要趋势是成本控制。政府当局和其他第三方付款人试图通过限制特定药物的覆盖范围和报销金额来控制成本。在许多国家,作为国家卫生系统的一部分,医疗产品的价格受到不同的价格控制机制的制约。一般来说,这种制度下的药品价格比美国低得多。其他国家允许公司固定自己的药品价格,但监控公司的利润。额外的外国价格管制或定价法规的其他变化可能会限制我们能够向我们的候选产品收取的费用。因此,在美国以外的市场,与美国相比,产品的报销可能会减少,可能不足以产生商业上合理的收入和利润。
69
与新批准的产品的保险覆盖范围和报销有关的不确定性也很大,而且覆盖范围可能比FDA或类似的外国监管机构批准药物的目的更有限。在美国,有关新药报销的主要决定通常由医疗保险和医疗补助服务中心(CMS)做出,该中心是美国卫生与公众服务部(HHS)的一个机构。CMS决定新药是否以及在多大程度上将在联邦医疗保险下覆盖和报销,而私人付款人往往在很大程度上遵循CMS。第三方付款人之间没有统一的产品承保和报销政策,产品的承保和报销水平因付款人而异。因此,承保范围的确定过程通常是一个耗时且昂贵的过程,可能需要我们为每个付款人分别提供使用我们的产品的科学和临床支持,而不能保证承保范围和足够的补偿将得到一致或首先获得。很难预测CMS将对像我们这样的根本性新产品的报销做出什么决定。欧洲的报销机构可能比CMS更保守。例如,一些抗癌药物在美国已获准报销,但在某些欧洲国家尚未获批报销。此外,有资格获得报销并不意味着任何药物在所有情况下都会得到支付,或者支付的费率将覆盖我们的成本,包括研究、开发、制造、销售和分销。新药的临时报销水平(如果适用), 也可能不足以支付我们的成本,也可能不会成为永久性的。报销率可能会因药物的使用和临床环境的不同而有所不同,可能基于已经为低成本药物设定的报销水平,也可能纳入其他服务的现有付款中。我们无法迅速从政府资助和私人付款人那里获得我们可能开发的任何经批准的产品的承保范围和有利可图的支付率,这可能会对我们的经营业绩、我们筹集将候选产品商业化所需的资金的能力以及我们的整体财务状况产生实质性的不利影响。
药品净价可能会因政府医疗保健计划或私人付款人要求的强制性折扣或回扣,以及未来限制从可能以低于美国价格销售的国家进口药品的法律放松而降低。我们无法迅速为我们开发的任何经批准的产品获得保险和有利可图的报销率,第三方付款人可能会对我们的经营业绩、我们筹集产品商业化所需资金的能力以及我们的整体财务状况产生实质性的不利影响。
越来越多的第三方付款人要求制药公司在标价的基础上向他们提供预定的折扣,并对医疗产品的定价提出挑战。我们不能确保我们商业化的任何候选产品都可以得到报销,如果可以报销,报销的级别也是如此。报销可能会影响我们获得市场批准的任何候选产品的需求或价格。为了获得报销,医生可能需要证明,与标准护理药物相比,患者使用我们的产品具有更好的治疗结果,包括价格较低的标准护理药物的仿制药。我们预计,由于管理型医疗保健的趋势、医疗保健组织日益增长的影响力以及额外的立法变化,我们将面临与销售我们的任何候选产品相关的定价压力。总体上,医疗成本的下行压力变得非常大,特别是处方药、外科手术和其他治疗。因此,对新产品的进入设置了越来越高的壁垒。
此外,我们可能会开发与我们的候选产品一起使用的配套诊断测试。我们或我们的合作者可能被要求为这些测试单独获得保险和报销,一旦获得批准,除了我们为我们的候选产品寻求的保险和报销之外。即使我们获得监管机构对此类配套诊断的批准或许可,由于适用于我们的候选产品的相同原因,我们获得保险和足够补偿的能力仍存在重大不确定性。联邦医疗保险报销方法,无论是根据A部分、B部分或临床实验室费用表,可能会不时修改,我们无法预测这些方法的任何更改将对我们获得批准的任何候选产品或配套诊断产生什么影响。对于我们开发并获得监管批准的配套诊断测试,我们无法迅速从第三方付款人那里获得保险和足够的补偿,这可能会对我们的业务、财务状况、运营结果和前景产生实质性的不利影响。
旨在降低医疗成本的医疗立法措施可能会对我们的业务和运营结果产生实质性的不利影响。
美国和许多外国司法管辖区已经制定或提议了影响医疗保健系统的立法和监管改革,这些改革可能会阻止或推迟我们的候选产品或任何未来候选产品的上市审批,限制或监管审批后活动,并影响我们以盈利方式销售获得营销审批的产品的能力。法规、法规或现有法规的解释的变化可能会影响我们未来的业务,例如,要求:(I)改变我们的制造安排;(Ii)增加或修改产品标签;(Iii)召回或停产我们的产品;或(Iv)额外的记录保存要求。如果强制实施任何此类变化,可能会对我们的业务运营产生不利影响。
70
在美国,已经并将继续有许多立法举措来控制医疗成本。例如,2010年3月,《平价医疗法案》(“ACA”)获得通过,它极大地改变了政府和私营保险公司为医疗保健融资的方式,并对美国制药业产生了重大影响。除其他事项外,ACA使生物制品受到低成本生物仿制药的潜在竞争,解决了一种新的方法,通过该方法,对于吸入、输注、滴注、植入或注射的药物,计算制造商在Medicaid药品回扣计划下的回扣,增加Medicaid药品回扣计划下制造商的最低Medicaid回扣,并将回扣计划扩大到在Medicaid管理的护理组织中注册的个人,建立对某些品牌处方药制造商的年费和税收,并创建新的Medicare Part D覆盖缺口折扣计划,其中制造商必须同意提供50%(根据2018年两党预算法增加到70%,自2019年起生效)适用品牌药品在承保间隔期内向符合条件的受益人提供议价的销售点折扣,作为制造商的门诊药品纳入联邦医疗保险D部分承保的条件。
自颁布以来,ACA的某些方面受到了许多司法、行政、行政和立法方面的挑战,我们预计未来还会对ACA提出更多的挑战和修正案。ACA的各个部分目前正在美国最高法院面临法律和宪法挑战,国会议员已经提出了几项旨在大幅修改或废除ACA的立法。ACA的实施仍在进行中,该法律似乎可能会继续对药品定价施加下行压力,特别是在联邦医疗保险计划下,还可能增加我们的监管负担和运营成本。与ACA相关的诉讼和立法可能会继续,结果不可预测和不确定。
美国联邦政府机构目前面临潜在的大幅支出削减。2011年预算控制法案(BCA)设立了一个赤字削减联合特别委员会,其任务是实现至少1.2万亿美元的联邦债务水平削减。该委员会没有在BCA的最后期限前起草一份提案。因此,各种联邦计划中的自动减支,也就是所谓的自动减支,计划从2013年1月开始实施,尽管2012年的《美国纳税人救济法》将BCA的自动减支推迟到了2013年3月1日。虽然联邦医疗保险计划的资格和福利范围通常不受这些削减的影响,但向提供者支付的联邦医疗保险付款和D部分健康计划也不例外。然而,BCA确实规定,除非采取额外的国会行动,否则医疗保险对提供者和D部分医疗计划的削减不会超过2%。总裁奥巴马于2013年3月1日发布了自动减支令,减支自2013年4月1日起生效。根据冠状病毒援助、救济和经济安全法,也被称为CARE法案,以及随后的立法,由于新冠肺炎大流行,这些削减在2020年5月1日至2022年3月31日期间暂停,从2022年4月1日至2022年6月30日,削减幅度将为1%。
在美国,关于特殊药物定价做法的立法和执法兴趣一直在增加。具体地说,美国国会最近进行了几次调查,并提出了联邦和州立法,旨在提高药品定价的透明度,降低联邦医疗保险下处方药的成本,审查定价与制造商患者计划之间的关系,以及改革政府计划的药品报销方法。前特朗普政府的2021财年预算提案包括一项1350亿美元的津贴,用于支持寻求降低药品价格、增加竞争、降低患者自付药品成本、以及增加患者获得成本较低的仿制药和生物相似药物的立法提案。2020年3月10日,前特朗普政府向国会提交了药品定价的《原则》,呼吁立法,其中包括限制联邦医疗保险D部分受益人的自付药房费用,提供限制联邦医疗保险D部分受益人每月自付费用的选项,并限制药品价格上涨。此外,前特朗普政府发布了一份降低药品价格和降低药品自付成本的《蓝图》,其中包含了增加制造商竞争、增加某些联邦医疗保健计划的谈判力、激励制造商降低产品标价以及降低消费者支付的药品自付成本的额外建议。卫生和公众服务部已经实施了一些措施。例如,CMS在2019年5月发布了一项最终规则,允许Medicare Advantage计划从2020年1月1日开始选择对B部分药物使用阶梯疗法,这是一种事先授权。这一最终规则将1月1日生效的CMS政策变化编成了法典, 2019年。然而,目前尚不清楚拜登政府是否会挑战、推翻、撤销或以其他方式修改这些行政和行政行动。
71
2020年,前总裁·特朗普签署了四项旨在降低药品价格的行政命令。作为回应,FDA于2020年9月24日发布了一项最终规则,该规则于2020年11月30日生效,为各州制定和提交从加拿大进口某些处方药的计划提供指导。此外,2020年11月20日,CMS发布了一项实施最惠国模式的暂行最终规则,根据该模式,某些药物和生物制品的联邦医疗保险B部分报销率将根据人均国内生产总值类似的经济合作与发展组织国家的药品制造商收到的最低价格计算。最惠国待遇示范条例要求确定的B部分提供者参与,并将适用于所有美国和地区,从2021年1月1日起至2027年12月31日止的七年内。然而,针对几个行业团体提起的诉讼,12月28日,美国加利福尼亚州北区地区法院发布了一项全国性的初步禁令,要求政府被告在根据《行政程序法》完成通知和评论程序之前,执行最惠国规则。2021年1月13日,在美国马里兰州地区行业团体提起的另一起诉讼中,政府被告提出联合动议,要求搁置诉讼,条件是政府不会对美国加州北区地区法院授予的初步禁令提出上诉,并且最惠国临时最终规则所产生的任何最终规则的执行不得早于该规则在联邦登记册上公布后60天开始。此外,加拿大当局已经通过了旨在保护加拿大药品供应免受短缺的规定。如果实施的话, 从加拿大进口药品和最惠国模式可能会对我们的任何候选产品的价格产生重大和不利的影响。此外,2020年12月2日,HHS发布了一项规定,取消了药品制造商对D部分下计划赞助商降价的安全港保护,直接或通过药房福利经理,除非法律要求降价。该规定还为反映在销售点的降价创造了一个新的安全港,以及为药房福利经理和制造商之间的某些固定费用安排创造了一个安全港。根据美国哥伦比亚特区地区法院的一项命令,根据联邦医疗保险D部分,规则中取消与从制造商向计划发起人销售或购买药品相关的某些回扣的安全港保护的部分已被推迟到2023年1月1日。此外,拜登政府目前正在审查这一变化的实施情况以及处方药产品销售点降价和药房福利经理服务费的新安全港,可能会进行修改或废除。
此外,2018年5月30日,《审判权法案》签署成为法律。除其他事项外,该法律还为某些患者提供了一个联邦框架,允许他们获得某些已完成第一阶段临床试验并正在进行调查以获得FDA批准的研究用新药产品。在某些情况下,符合条件的患者可以在不参加临床试验和根据FDA扩大准入计划获得FDA许可的情况下寻求治疗。根据《试用权法案》,制药商没有义务将其药品提供给符合条件的患者。
在州一级,各州越来越积极地通过立法和实施旨在控制药品和生物制品定价的法规,包括价格或患者报销限制、折扣、对某些产品准入的限制和营销成本披露和透明度措施,在某些情况下,旨在鼓励从其他国家进口和批量购买。此外,地区卫生保健当局和个别医院越来越多地使用招标程序来确定哪些药品和供应商将包括在他们的处方药和其他医疗保健计划中。这些措施一旦获得批准,可能会降低对我们产品的最终需求,或者给我们的产品定价带来压力。
我们预计未来将采取更多的州和联邦医疗改革措施,其中任何一项都可能限制联邦和州政府为医疗产品和服务支付的金额,这可能会导致对我们候选产品的需求减少或额外的定价压力。
我们的收入前景可能会受到美国和海外医疗支出和政策变化的影响。我们在一个高度受监管的行业运营,与医疗保健可用性、医疗保健产品和服务的交付或支付方式相关的新法律、法规或司法裁决,或对现有法律、法规或决定的新解释,可能会对我们的业务、运营和财务状况产生负面影响。
外国、联邦和州各级已经并可能继续提出立法和监管建议,旨在扩大医疗保健的可获得性,控制或降低医疗保健成本。我们无法预测未来可能采取的举措,包括废除、取代或对ACA进行重大修订。政府、保险公司、管理医疗组织和其他医疗服务付款人继续努力控制或降低医疗成本和/或实施价格管制,可能会产生不利影响:
72
联邦医疗保险或其他政府计划报销的任何减少都可能导致私人支付者支付的类似减少,这可能会对我们未来的盈利能力产生不利影响。
如果对我们提起产品责任诉讼,我们可能会承担大量责任,并可能被要求限制我们候选产品的商业化。
由于我们的候选产品的临床测试,我们面临着固有的产品责任风险,如果我们将任何产品商业化,我们将面临更大的风险。例如,如果我们的候选产品在临床测试、制造、营销或销售期间导致或被认为造成伤害,或被发现在其他方面不适合,我们可能会被起诉。任何此类产品责任索赔可能包括对制造缺陷、设计缺陷、未能就产品固有危险发出警告、疏忽、严格责任或违反保修的指控。根据州消费者保护法,索赔也可以主张。如果我们不能成功地在产品责任索赔中为自己辩护,我们可能会招致重大责任或被要求限制我们候选产品的商业化。即使是成功的防御也需要大量的财政和管理资源。无论案情如何或最终结果如何,赔偿责任可能会导致:
未能以可接受的成本获得或保留足够的产品责任保险,以防范潜在的产品责任索赔,可能会阻止或阻碍我们单独或与公司合作伙伴开发的候选产品的商业化。虽然我们有临床试验保险,但我们的保单也有各种排除,我们可能会受到产品责任索赔的影响,而我们没有承保范围。我们可能需要支付任何超出我们的承保范围限制或不在我们的保险覆盖范围内的法院裁决或和解协议中达成的任何金额,并且我们可能没有或能够获得足够的资本来支付这些金额。即使我们与任何未来的公司合作伙伴达成的协议使我们有权获得损失赔偿,如果出现任何索赔,这种赔偿可能是不可用的或足够的。
如果我们不遵守环境、健康和安全法律法规,我们可能会被罚款或罚款,或者产生成本,这可能会对我们的业务成功产生实质性的不利影响。
我们受到许多环境、健康和安全法律和法规的约束,包括那些管理实验室程序以及危险材料和废物的处理、使用、储存、处理和处置的法律和法规。我们的行动涉及使用危险和易燃材料,包括化学品、生物和放射性材料。我们的业务还会产生危险废物产品。我们通常与第三方签订合同,处理这些材料和废物。我们不能消除这些材料造成污染或伤害的风险。如果我们使用危险材料造成污染或伤害,我们可能要对由此造成的任何损害负责,任何责任都可能超出我们的资源范围。我们还可能产生与民事或刑事罚款和处罚相关的巨额费用。
虽然我们维持工人补偿保险以支付我们的成本和开支,但我们可能会因使用危险材料而导致员工受伤,但该保险可能不足以支付潜在的责任。我们不为可能因我们储存或处置生物、危险或放射性材料而对我们提出的环境责任或有毒侵权索赔维持保险。
73
我们候选产品的市场机会可能相对较小,因为可能接受我们产品候选治疗的患者是那些不符合或未能通过先前治疗的患者,而且我们对目标患者群体流行率的估计可能不准确。
癌症疗法有时以治疗路线(一线、二线、三线、四线等)为特征,FDA通常最初只批准针对一条或多条特定路线的新疗法。当癌症被发现得足够早时,一线治疗有时足以治愈癌症或延长生命,而不需要治愈。当一线治疗证明不成功时,通常是化疗、抗体药物、肿瘤靶向小分子、激素治疗、放射治疗、手术或这些疗法的组合,二线治疗可能会被实施。二线治疗通常包括更多的化疗、放射、抗体药物、肿瘤靶向小分子或这些药物的组合。三线治疗可以包括化疗、抗体药物和小分子肿瘤靶向治疗、更具侵入性的手术形式和新技术。不能保证我们的候选产品,即使被批准为第二、第三或随后的治疗路线,也会被批准用于更早的路线治疗,在获得任何此类批准之前,我们可能必须进行额外的临床试验。
我们对患有我们目标癌症的人数、可能对他们的肿瘤进行基因测序的人数,以及能够接受特定治疗路线并有可能从我们的候选产品治疗中受益的这些癌症患者的子集进行的预测,都是基于我们的信念和估计。这些估计来自各种来源,包括科学文献、诊所调查、患者基金会或市场研究,可能被证明是不正确的。此外,新的治疗方法可能会改变我们正在瞄准的癌症的估计发病率或流行率。因此,即使我们的候选产品被批准用于第二或第三线治疗,有资格使用我们的候选产品进行治疗的患者数量可能会比预期的要少得多。此外,我们还没有进行市场研究,以确定如果针对每种肿瘤类型有不同的批准疗法,治疗医生将如何预期开出一种被批准用于多种肿瘤类型的产品。
我们目前没有营销和销售组织,也没有营销产品的经验。如果我们无法建立营销和销售能力,或无法与第三方达成协议来营销和销售我们的候选产品,如果获得批准,我们可能无法产生产品收入。
我们目前没有销售、营销或分销能力,也没有营销产品的经验。如果我们将任何可能获得批准的候选产品商业化,我们将需要建立一个内部营销组织和销售队伍,这将需要大量的资本支出、管理资源和时间。我们将不得不与其他制药和生物技术公司竞争,以招聘、聘用、培训和留住营销和销售人员。
如果我们无法或决定不建立内部销售、营销和分销能力,如果获得批准,我们将寻求与第三方销售、营销和分销合作伙伴就我们产品的销售和营销达成安排。然而,我们不能保证我们能够以有利的条件或根本不能保证我们能够建立或维持这样的安排,或者如果我们能够做到这一点,我们不能保证这些第三方安排将提供有效的销售队伍或营销和分销能力。我们获得的任何收入都将取决于这些第三方的努力,而这可能不会成功。我们可能很少或根本无法控制这些第三方的营销和销售活动,我们的产品销售收入可能会低于我们自己将候选产品商业化的收入。我们在寻找第三方来帮助我们销售和营销我们的候选产品时也面临着竞争。
不能保证我们将能够发展内部销售和分销能力,或者与第三方合作伙伴建立或保持关系,以便在美国或海外将任何产品商业化。
74
与政府监管相关的风险
如果我们不能为我们的候选产品获得或延迟获得所需的监管批准,我们将无法将我们的候选产品商业化,或将推迟将其商业化,我们创造收入的能力将受到严重损害。
我们的候选产品以及与其开发和商业化相关的活动,包括它们的设计、测试、制造、安全、功效、记录保存、标签、储存、审批、广告、促销、销售、分销、进出口,都受到美国FDA和其他监管机构以及其他国家类似机构的全面监管。在我们可以将任何候选产品商业化之前,我们必须获得市场批准。我们目前正在进行的临床试验和其他试验的结果是否足以获得批准将是一个审查问题,FDA可能不会批准,并可能要求我们进行一项或多项受控临床试验才能获得批准。此外,即使FDA确实批准了我们的一个或多个候选产品,它可能是为了一个比我们寻求的更狭窄的适应症。例如,我们打算开发我们的候选产品,并寻求基于生物标记物的肿瘤不可知适应症的批准。FDA只批准了一小部分具有肿瘤不可知适应症的肿瘤学产品,而且FDA可能会不同意或不同意战略或数据,只批准更狭窄的适应症。监管机构,包括FDA,也可能以狭窄的适应症、警告或REMS的形式施加重大限制。这些监管机构可能要求标签包括与使用条件有关的预防或禁忌症,或者他们可能会根据昂贵的上市后临床试验的表现给予批准。此外,监管部门可能不会批准对我们可能开发的任何候选产品成功商业化所必需或可取的标签声明。
到目前为止,我们已经与西班牙、意大利、荷兰、波兰、中国大陆中国、香港、新加坡、韩国和台湾的监管机构就我们的齐帕替尼、CLN-049、CLN-619、CLN-978和CLN-617项目进行了互动。美国以外的监管机构在批准与肿瘤无关的精确癌症药物方面经验有限。
要获得监管批准,还需要向相关监管机构提交有关产品制造过程的信息,并由相关监管机构检查制造设施。此外,我们的候选产品可能没有效果,可能只是中等效果,或者可能被证明具有不良或意外的副作用、毒性或其他特征,这些可能会阻止我们获得上市批准,或者阻止或限制商业使用。
在美国和国外,获得监管批准的过程都是昂贵的,如果需要额外的临床试验,可能需要数年时间,如果真的获得了批准,而且可能会根据各种因素而有很大不同,包括所涉及的候选产品的类型、复杂性和新颖性。开发期间上市审批政策的变化、附加法规或法规的变化,或对每个提交的IND、生物制品许可证申请(“BLA”)、新药申请(“NDA”)或同等申请类型的监管审查的变化,可能会导致申请的批准或拒绝的延迟。FDA和其他国家的类似机构在审批过程中拥有相当大的自由裁量权,可以拒绝接受任何申请,也可以决定我们的数据不足以获得批准,需要进行额外的临床前、临床或其他研究。我们的候选产品可能会因为许多原因而延迟获得或无法获得监管部门的批准,包括以下原因:
75
在大量正在开发的候选产品中,只有一小部分成功完成了FDA或外国监管机构的审批程序并已商业化。漫长的审批过程以及未来临床试验结果的不可预测性可能会导致我们无法获得监管部门的批准,无法将我们的候选产品推向市场,这将严重损害我们的业务、运营结果和前景。
我们未来可能会为Zipertinib、CLN-049和我们未来的一些其他候选产品寻求孤儿药物状态,但我们可能无法获得此类称号或无法保持与孤儿药物状态相关的好处,包括市场排他性,这可能会导致我们的收入减少(如果有的话)。
根据《孤儿药品法》,FDA可以将用于治疗罕见疾病或疾病的药物或生物指定为孤儿,这种疾病或疾病的定义是,在美国,患者人数低于20万人,或在美国患者人数超过20万人,但无法合理预期在美国开发和提供药物或生物药物的成本将从该药物或生物药物或生物药物在美国的销售中收回。在提交BLA或NDA之前,必须申请指定孤儿药物。在美国,孤儿药物指定使一方有权获得财政激励,如为临床试验费用提供赠款资金的机会、税收优惠和用户费用减免。在FDA批准孤儿药物指定后,FDA公开披露药物或生物的仿制药身份及其潜在的孤儿用途。孤儿药物的指定不会在监管审查和批准过程中传递任何优势,也不会缩短监管审查和批准过程的持续时间。
如果具有孤儿药物名称的产品随后获得FDA对其具有这种名称的疾病的特定药物的第一次批准,则该产品有权获得孤儿产品排他性,这意味着FDA可能不会批准任何其他申请,包括BLA或NDA,在七年内销售相同适应症的同一药物。除非在有限的情况下,如显示出对具有孤儿药物排他性的产品的临床优势,或者FDA发现孤儿药物排他性持有者没有证明它可以确保获得足够数量的孤儿药物,以满足患有指定药物的疾病或状况的患者的需求。因此,即使我们的一个候选产品获得了孤立的排他性,这种排他性也可能无法有效地保护该候选产品免受竞争,因为不同的候选产品可以在相同的条件下获得批准。此外,即使在孤儿药物获得批准后,如果FDA得出结论认为,后一种候选产品在临床上更优越,因为与具有孤儿独占性的产品相比,它被证明更安全、更有效,或者对患者护理做出了重大贡献,则FDA随后可以批准与具有孤儿排他性的药物相同的后续候选产品。如果FDA确定指定请求存在重大缺陷,或者如果制造商无法保证足够数量的产品来满足患有罕见疾病或疾病的患者的需求,则也可能失去孤儿药物的排他性。FDA历来采取的立场是,孤儿排他性的范围与产品的批准适应症或用途一致,而不是产品被指定为孤儿的疾病或状况。然而,, 2021年9月30日,美国第11巡回上诉法院在Catalyst Pharms,Inc.诉Becera案认为孤儿药物专有的范围必须与该产品被指定为孤儿的疾病或状况相一致,即使该产品的批准用于较窄的用途或适应症。这一决定将如何影响未来的孤儿药物排他性仍有待观察。
我们可能会在其他孤儿适应症中为齐帕替尼、CLN-049和我们未来的一些或所有其他候选产品寻求孤儿药物名称,在这些适应症中,使用这些产品有医学上可信的基础。即使当我们获得孤儿药物指定时,如果我们寻求批准比孤儿指定适应症更广泛的适应症,在美国的独家营销权可能会受到限制,并且如果FDA后来确定指定请求存在重大缺陷,或者如果制造商无法保证足够数量的产品来满足患有罕见疾病或疾病的患者的需求,则可能会失去独家营销权。此外,尽管我们打算为其他候选产品寻求孤儿药物称号,但我们可能永远不会收到这样的称号。例如,FDA表达了对适用于肿瘤不可知疗法的孤儿药物指定的监管考虑的担忧,FDA可能会解释修订后的联邦食品、药物和化妆品法案(FDCA)及其颁布的法规,以限制或阻止我们获得孤儿药物指定或孤儿药物排他性的能力,如果我们的候选产品获得批准,作为我们的靶向适应症。
76
2017年8月18日,FDA 2017年重新授权法(简称FDARA)颁布。除其他事项外,FDARA还编纂了FDA先前存在的监管解释,要求药品赞助商证明一种孤儿药物的临床优越性,该药物在其他方面与之前批准的治疗同一罕见疾病的药物相同,才能获得孤儿药物排他性。这项立法是对一项法院裁决的回应,该裁决认为,《孤儿药物法》明确要求FDA承认一家公司的孤儿排他期,并获得指定为孤儿药物的批准,无论其临床优势如何。此外,在2021年《综合拨款法》中,国会没有进一步改变这一解释,因为它澄清了FDARA中编纂的解释将适用于FDA在FDARA颁布之前发布孤儿指定但产品批准在FDARA颁布之后的情况。此外,触媒关于解释《孤儿药品法》的排他性条款适用于被批准为孤儿适应症的药物和生物制品的决定,比产品的孤儿指定更窄,可能会极大地扩大此类产品的孤儿排他性范围。FDA于2023年1月24日宣布,尽管触媒在做出决定时,它将继续适用其长期存在的法规,这些法规将孤儿排他性的范围与药物被批准用于的用途或适应症捆绑在一起,而不是与名称挂钩。FDA对其孤儿药物法规的应用后-触媒可能成为未来立法或在法庭上进一步挑战的主题,这可能会影响我们获得或寻求解决孤儿排他性的能力,并可能影响我们保留FDA之前为我们的候选产品承认的孤儿排他性的能力。FDA和立法者可能会进一步重新评估《孤儿药物法》及其法规和政策。我们不知道FDA是否、何时或如何在未来改变孤儿药物的法规和政策,也不确定任何变化可能如何影响我们的业务。根据FDA可能对其孤儿药物法规和政策做出的变化,我们的业务可能会受到不利影响。
FDA的快速通道认证,即使被批准用于Zipertinib、CLN-049和CLN-619或任何其他未来的候选产品,也可能不会导致更快的开发或监管审查或批准过程,也不会增加我们的候选产品获得上市批准的可能性。
如果一种药物或生物制剂用于治疗严重或危及生命的疾病,而该产品显示出解决这种疾病未得到满足的医疗需求的潜力,则产品赞助商可以为特定的适应症申请FDA快速通道认证。我们可能会为Zipertinib、CLN-049和CLN-619以及我们未来的某些候选产品寻求快速通道认证,但不能保证FDA会将这一地位授予我们建议的任何候选产品。根据FDA提供的政策和程序,Fast Track开发产品的赞助商提交的营销申请可能有资格获得优先审查,但Fast Track的指定并不保证任何此类资格或FDA的最终上市批准。FDA拥有广泛的自由裁量权,是否授予快速通道认证,因此,即使我们认为特定的候选产品有资格获得这种认证,也不能保证FDA会决定授予它。即使我们确实获得了Fast Track认证,与传统的FDA程序相比,我们可能不会经历更快的开发过程、审查或批准,而且获得Fast Track认证并不能保证FDA的最终批准。此外,如果FDA认为我们的临床开发项目的数据不再支持快速通道的指定,它可能会撤回该指定。此外,FDA可以随时撤销任何Fast Track的指定。
FDA的突破性疗法指定,如果授予我们的任何候选产品,可能不会导致更快的开发或监管审查或批准过程,也不会增加我们的任何候选产品获得上市批准的可能性。
2022年1月,FDA批准突破性疗法指定齐帕替尼用于治疗局部晚期或转移性非小细胞肺癌患者,这些患者含有表皮生长因子外显子20插入突变,这些患者以前曾接受过基于铂的系统化疗。我们还可能寻求CLN-049和CLN-619的突破性治疗指定,以及我们未来的一些或所有候选产品。突破性疗法被定义为一种药物或生物制剂,旨在单独或与一种或多种其他药物或生物制品联合治疗严重或危及生命的疾病或状况,初步临床证据表明,该药物或生物制剂可能在一个或多个临床重要终点显示出比现有疗法显著改善的效果。被指定为突破性疗法的候选产品的赞助商有资格获得FDA关于制定高效药物开发计划的更深入的指导,有高级管理人员参与的组织承诺,以及滚动审查和优先审查的资格。被FDA指定为突破疗法的药物和生物制品也可能有资格获得其他快速批准计划,包括加速批准。
FDA有权将其指定为突破性疗法。因此,即使我们认为我们的候选产品之一符合被指定为突破性疗法的标准,FDA也可能不同意,而是决定不做出这样的指定。在任何情况下,与开发和考虑批准的未获得突破疗法指定的候选产品相比,收到齐帕替尼或任何其他候选产品的突破疗法指定可能不会导致更快的开发过程、审查或批准,也不能确保FDA的最终批准。即使我们可能寻求CLN-049和CLN-619的突破疗法认证,以及我们未来用于治疗各种癌症的部分或全部候选产品,也不能保证我们将获得此类候选产品的突破疗法认证。
77
FDA的加速批准,即使批准用于zipertinib或任何其他未来的候选产品,也可能不会导致更快的开发或监管审查或批准过程,也不会增加我们的候选产品获得上市批准的可能性。
我们可能会寻求批准齐帕替尼,以及我们目前和未来的某些其他候选产品,使用FDA的加速批准程序。如果一种产品治疗严重或危及生命的疾病,通常比现有的治疗方法提供有意义的优势,并对合理地可能预测临床益处的替代终点产生影响,则该产品可能有资格获得加速批准。作为批准的一项条件,FDA要求获得加速批准的产品的赞助商进行上市后的验证性临床试验。这些验证性试验必须以尽职调查的方式完成。此外,FDA目前要求作为加速批准的条件预先批准宣传材料,这可能会对产品商业推出的时间产生不利影响。作为2023年综合拨款法案的一部分,2022年12月29日颁布的2022年食品和药物综合改革法案(FDORA)包括了对加快药品和生物制品审批程序的多项改革,并使FDA能够酌情要求在批准加速批准之前进行批准后研究。FDORA还扩大了FDA现有的快速退出程序,允许该机构在赞助商未能对产品进行尽职调查的任何必要的批准后研究时使用快速程序。FDORA还补充说,在加速批准下获得批准的产品的赞助商没有对该产品进行任何必要的批准后研究,或没有及时提交关于该产品的报告,这是FDCA的禁止行为清单中的一项规定。即使我们确实获得了加速审批,我们也可能不会经历更快的开发或监管审查或审批过程, 获得加速批准并不能保证最终获得FDA的完全批准。除其他事项外,如果验证该产品的预期临床益处所需的验证性试验未能验证此类益处,或者如果此类试验没有进行尽职调查,则加速批准也可能被撤回。
如果获得批准,我们作为生物制品监管的研究产品可能面临通过简化监管途径获得批准的生物仿制药的竞争。
我们的大多数流水线产品,除了齐帕替尼,都将作为生物制品受到FDA的监管,这些生物制品必须在根据BLA上市之前获得FDA的许可。ACA包括一个副标题,称为2009年生物制品价格竞争和创新法案(BPCIA),它为与FDA许可的参考生物制品生物相似或可互换的生物制品创建了一个简短的批准途径。根据BPCIA,参考生物制品被授予12年的数据独占权,自该产品首次获得许可之时起,FDA将不会接受基于该参考生物制品的生物相似或可互换产品的申请,直到该参考产品首次获得许可之日起四年。此外,FDA对生物相似产品的批准可能要到参考产品首次获得许可之日起12年后才能生效。在这12年的独占期内,如果FDA批准竞争产品的BLA,该竞争产品包含赞助商自己的临床前数据和来自充分且受控良好的临床试验的数据,以证明另一家公司的产品的安全性、纯度和效力,则另一家公司仍可能销售该参考产品的竞争版本。这项法律很复杂,FDA仍在解释和实施。因此,其最终影响、实施和意义都存在不确定性。
我们认为,我们的任何根据BLA被批准为生物制品的候选产品都应该有资格获得12年的专营期。然而,由于国会的行动或其他原因,这种排他性可能会缩短,或者FDA不会将我们的研究药物视为竞争产品的参考产品,这可能会比预期更早地创造仿制药竞争的机会。此外,一旦获得许可,生物相似物将在多大程度上取代我们的任何一种参考产品,其方式类似于非生物产品的传统仿制药替代,目前尚不清楚,这将取决于一些仍在发展中的市场和监管因素。
如果竞争对手能够获得参照我们产品的生物仿制药的营销批准,我们的产品可能会受到此类生物仿制药的竞争,随之而来的是竞争压力和后果。
78
如果FDA或类似的外国监管机构批准了我们任何获得上市批准的小分子研究产品的仿制药版本,或者这些机构在批准这些产品的仿制药版本之前没有给予我们的产品适当的独家专利期,如果批准,我们产品的销售可能会受到不利影响。
一旦NDA获得批准,其所涵盖的产品就成为FDA出版物“已批准的具有治疗等效性评估的药物产品”中的“参考清单药物”,通常被称为橙皮书。在美国,制造商可以通过提交简化的新药申请(ANDA)来寻求参考上市药物的仿制药版本的批准。为了支持ANDA,仿制药制造商不需要进行临床试验来评估安全性和有效性。相反,申请人一般必须证明其产品具有与参考上市药物相同的有效成分、剂型、强度、给药途径和使用条件或标签,并且仿制药与参考上市药物具有生物等效性,这意味着它在体内的吸收速度和程度相同。仿制药上市的成本可能比参考上市药物低得多,生产仿制药的公司通常能够以较低的价格提供这些产品。因此,随着仿制药的推出,任何品牌产品或参考上市药物的销售额中,有相当大一部分通常会流失到仿制药。
在参考上市药物的任何适用的非专利专有期到期之前,FDA不得批准仿制药的ANDA。FDCA为含有新化学实体的新药提供了五年的非专利专有期。具体地说,在已经授予这种排他性的情况下,ANDA在五年期满之前不得向FDA提交ANDA,除非提交的文件附有第四段证明,证明涵盖参考上市药物的专利要么无效,要么不会受到仿制药的侵犯,在这种情况下,申请人可以在参考上市药物获得批准四年后提交申请。
如果我们的产品获得批准,仿制药制造商可能会在我们获得的任何适用的排他期到期后寻求推出仿制药产品,即使我们仍然拥有此类产品的专利保护。我们的产品可能面临来自我们产品的仿制版本的竞争,这可能会对我们未来的收入、盈利能力和现金流产生实质性的不利影响,并极大地限制我们从这些候选产品中获得投资回报的能力。
在一个司法管辖区获得并保持对我们候选产品的监管批准,并不意味着我们将成功地在其他司法管辖区获得我们候选产品的监管批准。
在一个司法管辖区获得并保持我们候选产品的监管批准并不保证我们将能够在任何其他司法管辖区获得或维持监管批准,而在一个司法管辖区未能或延迟获得监管批准可能会对其他司法管辖区的监管审批过程产生负面影响。例如,即使FDA批准了候选产品的上市,外国司法管辖区的可比监管机构也必须批准候选产品在这些国家的制造、营销和推广。审批程序因司法管辖区而异,可能涉及与美国不同或大于美国的要求和行政审查期限,包括额外的临床前研究或临床试验,因为在一个司法管辖区进行的临床试验可能不会被其他司法管辖区的监管机构接受。在美国以外的许多司法管辖区,候选产品必须先获得报销批准,然后才能在该司法管辖区批准销售。在某些情况下,我们打算为我们的产品收取的价格也需要得到批准。
我们也可以在其他国家提交营销申请。美国以外司法管辖区的监管机构对候选产品的批准有要求,我们在这些司法管辖区上市前必须遵守这些要求。获得外国监管批准和遵守外国监管要求可能会给我们带来重大延误、困难和成本,并可能推迟或阻止我们的产品在某些国家/地区推出。如果我们未能遵守国际市场的监管要求和/或获得适用的营销批准,我们的目标市场将会减少,我们充分发挥我们候选产品的市场潜力的能力将受到损害。
即使我们的候选产品获得了监管机构的批准,我们也将受到持续的监管义务和持续的监管审查的约束,这可能会导致大量的额外费用,如果我们没有遵守监管要求或我们的候选产品出现了意想不到的问题,我们可能会受到惩罚。
我们获得上市批准的任何候选产品都将受到广泛和持续的法规要求的约束,这些法规要求管辖我们产品的研究、开发、测试、制造、标签、包装、分销、存储、广告、促销、进出口、记录、监控和报告等。这些要求包括提交安全和其他上市后信息和报告、设施注册和药品上市要求,以及对于我们在批准后进行的任何临床试验,继续遵守cGMP、良好实验室实践、法规和GCP。即使批准了候选产品的上市,批准也可能受到对该产品可能上市的指定用途的限制或批准条件的限制,或者包含对昂贵的上市后测试和监督的要求,以监测产品的安全性或有效性。
79
FDA可能需要REMS,以批准我们的候选产品,这可能需要药物指南、医生沟通计划或确保安全使用的其他要素,如限制分配方法、患者登记和其他风险最小化工具。后来发现我们的候选产品存在以前未知的问题,包括预料不到的严重程度或频率的不良事件,或我们的第三方制造商或制造工艺,或未能遵守监管要求,可能会导致除其他事项外:
FDA和其他监管机构的政策可能会改变,可能会颁布额外的政府法规,以阻止、限制或推迟对我们候选产品的监管批准。我们无法预测美国或国外未来的立法或行政行动可能产生的政府监管的可能性、性质或程度。如果我们缓慢或无法适应现有要求的变化或采用新的要求或政策,或者如果我们无法保持监管合规性,我们可能会失去我们可能获得的任何营销批准,我们可能无法实现或维持盈利。
FDA和其他监管机构积极执行禁止推广非标签使用的法律法规。
FDA和其他监管机构密切监管产品的批准后营销和促销,以确保它们只针对批准的适应症并根据批准的标签的规定进行销售。FDA和其他监管机构对制造商关于非标签使用的沟通施加了严格的限制。如果我们的任何候选产品获得批准,而我们被发现推广了这种标签外的用途,我们可能会承担重大责任。联邦政府已对涉嫌不当推广标签外使用的公司处以巨额民事和刑事罚款,并禁止几家公司从事标签外促销。违反FDCA和其他法规,包括《虚假申报法》(“FCA”)和其他国家与处方产品促销和广告有关的同等法律,也可能导致对违反联邦和州及其他国家的医疗保健欺诈和滥用法律以及州消费者保护法的调查或指控。即使后来确定我们没有违反这些法律,我们也可能面临负面宣传,为我们的行为付出巨额费用,并不得不将大量管理资源从其他事务上转移出去。
美国食品和药物管理局、美国证券交易委员会和其他政府机构的资金不足,包括政府关门或这些机构运营的其他中断,可能会阻碍它们雇用和保留关键领导层和其他人员的能力,阻止新产品和服务的及时开发或商业化,或者以其他方式阻止这些机构履行我们业务运营可能依赖的正常业务职能,这可能会对我们的业务产生负面影响。
FDA审查和批准新产品的能力可能受到各种因素的影响,包括政府预算和资金水平、雇用和留住关键人员以及接受用户费用支付的能力,以及法定、监管和政策变化。FDA和其他机构的中断也可能会减缓新产品候选产品被必要的政府机构审查和/或批准所需的时间,这将对我们的业务产生不利影响。例如,在过去的几年里,美国政府已经关闭了几次,某些监管机构,如FDA,不得不让关键员工休假,并停止关键活动。如果政府长期停摆或发生其他中断,可能会严重影响FDA及时审查和处理我们提交的监管文件的能力,这可能会对我们的业务产生实质性的不利影响。此外,未来政府停摆或其他中断可能会影响我们进入公开市场并获得必要资本的能力,以便适当地为我们的业务提供资本并继续运营。
80
另外,美国食品和药物管理局总体上继续确保在正在进行的新冠肺炎大流行期间,根据其用户收费绩效目标,及时审查医疗产品的申请。然而,FDA可能无法保持这一速度,审查时间表可能会延长。此外,如果需要进行审批前检查或临床站点检查,并且由于持续的新冠肺炎大流行和旅行限制,食品和药物管理局无法在审查期内完成此类必要检查,则可能会推迟或阻止对此类申请采取行动。此外,美国以外的监管机构可能会采取类似的限制或其他政策措施来应对新冠肺炎疫情,并可能在监管活动中遇到延误。
我们的员工、独立承包商、顾问、商业合作伙伴和供应商可能从事不当行为或其他不当活动,包括不遵守监管标准和要求。
我们面临着员工、独立承包商、顾问、商业合作伙伴和供应商进行员工欺诈或其他非法活动的风险。这些当事人的不当行为可能包括故意、鲁莽和/或疏忽的行为,包括未能遵守FDA和其他类似外国监管机构的规定,向FDA和其他类似外国监管机构提供真实、完整和准确的信息,遵守我们制定的制造标准,遵守美国和类似外国欺诈性不当行为法律的医疗欺诈和滥用法律,或准确报告财务信息或数据,或向我们披露未经授权的活动。如果我们的任何候选产品获得FDA的批准,并开始在美国将这些产品商业化,我们在此类法律和法规下的潜在风险将显著增加,我们与遵守此类法律和法规相关的成本也可能增加。这些法律可能会影响我们目前与主要研究人员和研究患者的活动,以及拟议和未来的销售、营销和教育计划。特别是,医疗保健产品和服务的推广、销售和营销,以及医疗保健行业的某些商业安排,都受到旨在防止欺诈、回扣、自我交易和其他滥用行为的广泛法律的约束。这些法律和法规可能限制或禁止广泛的定价、折扣、营销和促销、结构和佣金、某些客户激励计划和其他业务安排。受这些法律约束的活动还涉及不正当使用在招募病人进行临床试验的过程中获得的信息。可能影响我们运作能力的法律包括但不限于:
81
我们采用了商业行为和道德准则,但并不总是能够识别和阻止员工的不当行为,我们为发现和防止不当行为而采取的预防措施可能无法有效控制未知或未管理的风险或损失,或保护我们免受政府调查或其他行动或诉讼,这些调查或诉讼是由于未能遵守此类法律或法规而引起的。
确保我们与第三方的业务安排符合适用的医疗法律和法规的努力将涉及大量成本。由於这些法律的范围广泛,而法定例外情况和可供选择的避风港有限,我们的一些商业活动可能会受到一项或多项这类法律的挑战。政府当局可能会得出结论,我们的业务实践可能不符合当前或未来涉及适用欺诈和滥用或其他医疗保健法律和法规的现行或未来法规、法规或判例法。如果我们的运营被发现违反了这些法律或任何其他可能适用于我们的政府法规,我们可能会受到重大的刑事、民事和行政制裁,包括罚款、损害赔偿、罚款、交还、个人监禁,以及被排除在政府资助的医疗保健计划之外,如联邦医疗保险和医疗补助、合同损害、声誉损害、利润减少和未来收益,我们可能被要求缩减或重组我们的业务,任何这些都可能对我们的业务运营能力和我们的运营结果产生不利影响。
任何因违反这些法律而对我们采取的行动,即使我们成功地进行了辩护,也可能导致我们招致巨额法律费用,并转移我们管理层对业务运营的注意力。不断变化的合规环境,以及建立和维护强大且可扩展的系统以符合具有不同合规和/或报告要求的多个司法管辖区的需要,增加了医疗保健公司可能与一项或多项要求发生冲突的可能性。
82
欧盟也禁止向医生提供福利或优势,以诱导或鼓励医生开处方、推荐、背书、购买、供应、订购或使用医疗产品。提供利益或利益以诱导或奖励不当行为通常受欧盟成员国的国家反贿赂法律和英国2010年《反贿赂法》的管辖。违反这些法律可能会导致巨额罚款和监禁。欧盟指令2001/83/EC是欧盟关于人用药品的指令,它进一步规定,如果向有资格开处方或供应药品的人推销药品,则不得向这些人提供、提供或承诺任何礼物、金钱利益或实物利益,除非这些礼物、金钱利益或实物利益不贵且与医药或药房实践有关。这一条款已被转移到2012年人类药物法规中,因此尽管它脱离了欧盟,但仍适用于英国。在某些欧盟成员国向医生支付的费用必须公开披露。
此外,与医生达成的协议通常必须事先通知医生的雇主、医生的主管专业组织和/或欧盟成员国的监管当局,并予以批准。
这些要求在适用于欧盟成员国的国家法律、行业守则或专业行为守则中作出规定。
如果我们不遵守这些要求,可能会导致行政、民事和刑事处罚、损害赔偿、罚款、交还、被排除在联邦和州医疗保健计划之外、个人监禁、声誉损害以及我们业务的缩减或重组,以及如果我们受到公司诚信协议或其他协议的约束,以解决对不遵守这些法律的指控,以及额外的报告义务和监督。
数据收集受到有关个人信息的使用、处理和跨境转移的限制性规定的约束。
如果我们决定进行临床试验或继续在我们正在进行的或未来的临床试验中招募受试者,我们可能会受到额外的隐私限制。收集、使用、存储、披露、传输或以其他方式处理与欧洲经济区个人有关的个人数据,包括个人健康数据,受2018年5月25日生效的欧盟一般数据保护条例(GDPR)的约束。GDPR的范围很广,对在欧洲经济区设立的公司或不在欧洲经济区设立但收集和使用个人数据的公司提出了许多要求,这些公司涉及(1)向欧洲经济区内的个人提供商品或服务,或(2)监测位于欧洲经济区的个人的行为。GDPR对个人资料的控制人和处理者施加了严格的运作要求,例如,有关处理健康和其他敏感数据、征得与个人资料有关的个人的同意、向个人提供有关数据处理活动的信息、实施和维持处理数据主体权利的程序、实施保障措施以保护个人资料的安全和机密性、向资料当事人和政府当局发出有关违反资料规定的通知,以及在聘用第三者处理者时采取某些措施。GDPR还对向欧洲经济区以外的大多数国家(包括美国)转移个人数据实施了严格的规定,并允许数据保护当局对违反GDPR的行为处以巨额罚款,包括可能高达2,000万欧元或全球年收入4%的罚款,以金额较大者为准。GDPR还授予对数据当事人和消费者协会提起私人诉讼的权利,以向监管当局提出投诉,寻求司法补救, 并获得因违反GDPR而造成的损害赔偿。GDPR增加了我们在处理受GDPR约束的个人数据方面的责任和潜在责任,我们可能需要建立额外的机制来确保遵守GDPR,包括欧洲经济区内个别国家实施的机制。遵守GDPR将继续是一个严格和耗时的过程,可能会增加我们的业务成本或要求我们改变业务做法,尽管我们做出了这些努力,但我们可能面临与我们的欧洲数据处理活动相关的罚款和处罚、诉讼和声誉损害的风险。在英国退出欧盟后,我们对位于英国的人员的个人数据的处理使我们受制于2018年英国数据保护法和经数据保护、隐私和电子通信(修正案等)修订的2018年数据保护法定义的“英国GDPR”。《2019年(欧盟退出)条例》(SI 2019/419)(“英国GDPR”)。英国GDPR对数据控制器和处理器施加的义务与GDPR中的义务类似,并附带与GDPR类似的罚款。
欧盟和英国最近的法律发展也造成了个人数据从欧洲经济区和英国转移到美国和其他国家的复杂性和不确定性。2022年12月13日,欧盟委员会公布了一份欧盟-美国数据充分性决定草案,该草案为跨大西洋数据流动设定了一个新框架,但拟议的框架何时开始运作仍存在不确定性。拟议的框架也可能受到法律挑战。这些事态发展对将个人信息从欧洲经济区和英国合法转移到美国和其他国家/地区的能力产生了影响,导致对从欧洲经济区和英国转移出去的数据进行更严格的审查,并可能增加我们遵守数据隐私立法的成本。
83
此外,美国几个州最近已经制定或正在考虑制定全面的隐私立法。最值得注意的是,加州最近颁布了《加州消费者隐私法》(CCPA),为加州消费者创造了新的类似GDPR的个人隐私权(如法律所定义),并对处理加州消费者或家庭个人数据的承保企业施加了更多隐私和安全义务。CCPA要求覆盖的企业向消费者提供有关此类企业数据收集、使用和共享做法的新披露,为这些消费者提供选择退出某些销售或共享个人信息的新方式,并在数据安全被破坏的情况下为消费者提供额外的诉讼理由。CCPA于2020年1月1日生效,加利福尼亚州总检察长于2020年7月1日开始对违规行为采取执法行动。此外,加州选民于2020年11月3日通过了加州隐私权法案(CPRA)。CPRA修订了CCPA,在处理和存储个人信息方面增加了额外的义务,并于2023年1月1日生效(某些条款具有追溯效力至2022年1月1日)。虽然CCPA目前对受HIPAA约束的受保护健康信息以及根据某些法规进行的研究(包括临床试验)中收集的信息有例外情况,但我们仍在继续监测CCPA可能对我们的业务活动产生的影响。美国超过一半的州和美国国会提出了新的数据隐私法,反映了美国隐私立法更加严格的趋势。, 在现任美国总统任期内,这一趋势可能会加速。这项立法的影响可能是深远的,可能需要我们修改我们的数据处理做法和政策,并为遵守这项立法而招致大量费用和开支。
遵守美国和国际数据保护法律法规可能要求我们在合同中承担更繁重的义务,限制我们收集、使用和披露数据的能力,或者在某些情况下影响我们在某些司法管辖区运营的能力。不遵守美国和国际数据保护法律和法规可能会导致政府执法行动(可能包括民事或刑事处罚)、集体诉讼和/或负面宣传,并可能对我们的经营业绩和业务产生负面影响。此外,我们或我们的潜在合作者获得信息的临床试验对象,以及与我们共享这些信息的提供者,可能会在合同上限制我们使用和披露信息的能力。声称我们侵犯了个人隐私权,未能遵守数据保护法,或违反了我们的合同义务,即使我们不承担责任,辩护也可能代价高昂且耗时,并可能导致负面宣传,可能损害我们的业务。
与我们的知识产权有关的风险
如果我们无法为我们当前和未来的候选产品和技术获得并维护专利和其他知识产权保护,或者如果获得的知识产权保护范围不够广泛,我们的竞争对手可能会开发和商业化与我们类似或相同的产品和技术,我们将Zipertinib、CLN-049、CLN-619和CLN-418或任何其他候选产品或技术商业化的能力可能会受到不利影响。
我们的成功在很大程度上取决于我们是否有能力在美国和其他国家/地区获得和维护我们候选产品的专利和其他知识产权保护,包括齐帕替尼、CLN-049、CLN-619和CLN-418、它们各自的成分、配方、联合疗法、用于制造它们的方法以及对我们的业务至关重要的治疗和开发方法,以及成功地保护这些专利免受第三方挑战。如果我们不充分保护我们的知识产权,竞争对手可能会侵蚀或否定我们可能拥有的任何竞争优势,这可能会损害我们的业务和实现盈利的能力。
我们打算依靠专利申请、保密协议、商业秘密保护和许可协议的组合来保护与我们的候选产品和技术相关的知识产权。任何向第三方披露或盗用我们的机密专有信息都可能使竞争对手迅速复制或超过我们的技术成就,从而侵蚀我们在市场上的竞争地位。我们或任何未来的合作伙伴、合作者或被许可人可能无法识别在开发和商业化活动过程中作出的发明的可专利方面,否则就太晚了,无法获得专利保护。因此,我们可能会错过建立我们专利地位的预期潜在机会。
为了保护我们的专有地位,我们已经在美国和海外提交了专利和专利申请,这些专利和专利与我们的候选产品有关,这些专利和专利申请对我们的业务非常重要,我们已经提交或正在许可,并计划提交或在许可中提交。我们阻止未经授权的第三方制造、使用、销售、提供销售或进口我们的候选产品的能力取决于我们在涵盖这些活动的有效和可强制执行的专利或商业秘密下拥有的权利的程度。如果我们不能确保或保持对Zipertinib、CLN-049、CLN-619和CLN-418或我们开发的任何其他专有产品和技术的专利保护,我们的业务、财务状况、运营结果和前景将受到严重损害。
84
我们在市场上成功竞争所需的专利保护程度在某些情况下可能无法获得或受到严重限制,可能无法充分保护我们的权利或允许我们获得或保持任何竞争优势。我们不能保证我们未来可能拥有或许可的任何专利将拥有,或我们的任何成熟为已发行专利的专利申请将包括范围足以保护我们当前和未来的候选产品或以其他方式提供任何竞争优势的权利要求。此外,就我们目前或未来对知识产权的许可而言,我们不能向您保证这些许可将继续有效。此外,外国法律可能不会像美国法律那样保护我们的权利。此外,专利的寿命有限,我们可能拥有的任何专利或许可中的任何专利的期限可能不足以在足够长的时间内保护我们的候选产品或技术的竞争地位。在美国,专利的自然失效时间通常是申请后20年。可能会有各种延期;然而,专利的有效期及其提供的保护是有限的。考虑到新产品候选产品的开发、测试和监管审查所需的时间,保护这些候选产品的专利可能会在这些候选产品商业化之前或之后不久到期。
即使我们的专利申请不受质疑,我们的专利申请以及我们可能拥有或许可的任何专利可能不会为我们提供任何有意义的保护,或阻止竞争对手围绕我们的专利声明进行设计,通过以非侵权方式开发类似或替代技术或治疗方法来规避我们可能拥有或许可的任何专利。例如,第三方可能开发一种竞争性疗法,该疗法提供与我们的一个或多个候选产品类似的益处,但使用的配方和/或设备不在我们未来可能拥有的任何专利保护的范围内。如果我们的专利申请或任何专利对我们的候选产品提供的专利保护不够广泛,不足以阻碍此类竞争,我们成功将我们的候选产品商业化的能力可能会受到负面影响,这将损害我们的业务。
生命科学公司的专利地位可能是不确定的,涉及复杂的事实和法律问题。在我们寻求专利保护的任何司法管辖区,专利法或其解释的变化可能会削弱我们保护我们的发明、维护和执行我们的知识产权的能力;更广泛地说,可能会影响我们的知识产权的价值,包括缩小我们的专利申请范围或我们可能拥有的或许可中的任何专利。
专利起诉过程复杂、昂贵、耗时,而且在不同司法管辖区之间不一致。专利许可谈判也可能是复杂和旷日持久的,结果不确定。我们可能无法以商业上合理的成本或及时地提交、起诉、维护、强制执行或许可所有必要或可取的专利权。此外,我们可能不会在所有相关市场寻求或获得专利保护。我们可能无法及时确定我们的研究和开发工作中重要的可申请专利的方面,以获得适当的或任何专利保护。虽然我们与能够访问我们研发工作的机密或可专利方面的各方签订了保密和保密协议,例如,包括我们的员工、公司合作者、外部学术科学合作者、CRO、合同制造商、顾问、顾问和其他第三方,但这些各方中的任何一方都可能违反协议并在提交专利申请之前披露结果,从而危及我们寻求专利保护的能力。此外,科学和学术文献中发现的出版物往往落后于实际发现,美国和其他司法管辖区的专利申请通常在申请后18个月才发表,有时甚至根本不发表。因此,我们不能确定我们是第一个就我们的专利申请中要求的发明申请专利保护的公司。
我们的专利申请的准备或提交过程中的形式缺陷,或我们可能拥有的或许可中的任何专利,可能存在或可能在未来出现,例如在适当的优先权权利要求、库存、权利要求范围或专利期限调整请求方面。如果我们或我们的合作伙伴、合作者、被许可人或许可人未能建立、维护或保护此类专利和其他知识产权,此类权利可能会减少或取消。如果我们的合作伙伴、合作者、被许可人或许可人在起诉、维护或执行任何专利权方面与我们不完全合作或不同意,则此类专利权可能会受到损害。如果我们的专利申请或我们可能拥有或许可的专利在形式、准备、起诉或执行方面存在重大缺陷,则此类专利可能无效和/或不可强制执行,并且此类申请可能永远不会产生有效的、可强制执行的专利。这些结果中的任何一个都可能削弱我们阻止第三方竞争的能力,第三方竞争可能会对我们的业务产生不利影响。
此外,我们不能确定我们的专利申请中涉及我们的候选产品或技术的组成的权利要求将被美国专利商标局(USPTO)或外国专利局视为可申请专利,或者我们可能拥有的或许可中的任何已颁发专利中的权利要求将被美国或外国法院视为可申请专利。
使用方法专利保护使用特定方法的产品。这些类型的专利并不阻止竞争对手为超出专利方法范围的指示而制造和销售与我们的产品相同的产品。此外,即使竞争对手没有针对我们的目标适应症积极推广他们的产品,医生也可能会在标签外开出这些产品的处方。尽管标签外的处方可能会导致或助长对使用方法专利的侵犯,但这种做法很常见,这种侵权行为很难预防或起诉。
85
制药和生物技术公司的专利地位通常高度不确定,涉及复杂的法律和事实问题,许多法律原则仍未解决。近年来,专利权一直是重大诉讼的主题。因此,我们可能从专利申请中获得的任何权利的颁发、范围、有效性、可执行性和商业价值都非常不确定。我们的专利申请可能不会导致在美国或其他司法管辖区颁发专利,这些专利保护我们的技术或产品,或有效地阻止其他公司将竞争对手的技术和产品商业化。此外,我们获得和保持有效和可执行的专利的能力取决于我们的发明与现有技术之间的差异,包括我们自己以前提交的专利申请和科学出版物,是否允许我们的发明比现有技术获得专利。即使我们的专利申请以专利的形式发布,第三方也可以基于这样的科学出版物质疑这些专利的有效性,我们可能会失去宝贵的专利权。此外,专利申请中要求保护的发明的范围在专利颁发之前可以显著缩小,该范围在专利颁发后可以重新解释。即使我们的专利申请,无论是拥有的还是许可的,以专利的形式发布,它们的发布形式可能不会为我们提供任何有意义的保护,阻止竞争对手或其他第三方与我们竞争,或以其他方式为我们提供竞争优势。最终颁发的任何专利都可能受到第三方的挑战、缩小范围或使其无效。因此, 我们不知道我们的任何候选产品是否会受到有效和可执行的专利权的保护或继续受到保护。我们的竞争对手或其他第三方可能通过以非侵权方式开发新化合物或替代技术或产品来规避我们可能拥有的任何权利。
就发明性、范围、有效性或可执行性而言,专利的颁发或授予并不是无可辩驳的,我们当前或未来的任何专利,无论是拥有的还是未授权的,都可能在美国和国外的法院或专利局受到挑战。此类挑战可能导致任何此类专利的专利主张缩小、无效或无法执行,这可能会限制我们阻止或阻止他人使用类似或相同的技术和产品或将其商业化的能力,或者限制我们的技术和产品的专利保护期限。可能有一些我们不知道的现有技术可能会影响专利权利要求的有效性或可执行性。也可能存在我们知道但我们不认为会影响索赔的有效性或可执行性的现有技术,但最终可能会被发现影响索赔的有效性或可执行性。在未来,我们可能会受到第三方对现有技术的发行前提交或反对、派生、撤销、重新审查、授予后和各方间审查、干扰程序和其他类似程序,挑战我们可能从我们的专利申请中获得的任何权利,或在USPTO或其他外国专利局或在宣告性判决诉讼或反诉中挑战他人专利权的任何权利。任何此类提交、诉讼或诉讼中的不利裁决可能会缩小我们从我们的专利或专利申请中获得的任何权利的范围或使其无效,允许第三方将我们的技术或产品商业化并直接与我们竞争,而无需向我们付款,或者使我们无法在不侵犯第三方专利权的情况下制造或商业化产品。
此外,我们的一些知识产权可能与第三方共同拥有。如果我们无法获得任何此类第三方共同所有人在此类知识产权中的权益(包括专利或专利申请)的独家许可,这些共同所有人可能能够将其权利许可给其他第三方,包括我们的竞争对手,而我们的竞争对手可以销售竞争对手的产品和技术。此外,我们或我们的许可人可能需要我们拥有和许可的知识产权(包括专利和专利申请)的任何此类共同所有人的合作,以便对第三方强制执行此类知识产权,而此类合作可能不会提供给我们或我们的许可人。上述任何一项都可能对我们的竞争地位、业务、财务状况、经营结果和前景产生重大不利影响。
如果我们未能履行我们在任何协议中的义务,根据这些协议,我们可能会从第三方获得知识产权许可,或者我们与许可方的业务关系受到干扰,我们可能会失去对我们的业务非常重要的许可权。
我们目前是,未来也可能是与第三方签订许可或合作协议,以推进我们的研究或允许候选产品商业化。我们目前的协议对我们施加了许多义务,我们预计未来的协议可能会对我们施加许多义务,如开发、勤奋、付款、商业化、资金、里程碑、特许权使用费、再许可、保险、专利诉讼、执法和其他义务,并可能要求我们遵守开发时间表,或做出商业上合理的努力来开发和商业化授权的产品,以维护许可。如果我们的许可方认定我们严重违反了许可协议,他们可能会寻求终止许可协议,从而取消或限制我们开发和商业化这些许可协议所涵盖的产品和技术的能力。
这些许可的任何终止,或基础专利未能提供预期的排他性,都可能导致重大权利的丧失,并可能损害我们将候选产品商业化的能力,竞争对手或其他第三方将有权寻求监管部门批准并销售与我们相同的产品,我们可能被要求停止对某些候选产品的开发和商业化。上述任何一项都可能对我们的竞争地位、业务、财务状况、经营结果和前景产生重大不利影响。
86
根据许可协议,我们与许可人之间可能还会发生知识产权纠纷,包括:
此外,许可协议很复杂,这类协议中的某些条款可能会受到多种解释的影响。任何可能出现的合同解释分歧的解决可能会缩小我们认为是我们对相关知识产权或技术的权利的范围,或者增加我们认为是相关协议下我们的财务或其他义务,这两种情况中的任何一种都可能对我们的业务、财务状况、运营结果和前景产生实质性的不利影响。例如,如果出售或以其他方式处置其大部分资产,库利南佛罗伦萨和库利南琥珀将各自向许可人支付成功费用。这些费用将减少我们从任何此类资产出售或处置中获得的净收益。
此外,如果知识产权纠纷阻碍或削弱我们以可接受的条款维持许可安排的能力,我们可能无法成功开发受影响的候选产品并将其商业化,这可能对我们的业务、财务状况、运营结果和前景产生重大不利影响。
如果我们不能保护我们的商业秘密的机密性,我们的商业和竞争地位将受到损害。
除了我们拥有和许可的专利提供的保护外,我们还寻求依靠商业秘密保护、保密协议和许可协议来保护不可申请专利的专有技术、难以实施专利的过程以及我们产品识别、发现和开发过程中的任何其他元素,包括我们差异化的中心辐射式业务模式,涉及专利不包括的专有技术、信息或技术。虽然我们要求我们的所有员工、顾问、顾问和能够访问我们专有技术、信息或技术的任何第三方签订保密协议,但商业秘密可能很难保护,而且我们对我们的合作者和供应商使用的商业秘密的保护控制有限。我们不能确定我们已经或将在所有情况下获得这些协议,我们也不能保证我们已经与可能或曾经接触到我们的商业秘密或专有信息的每一方签订了此类协议。与我们签署此类协议的任何一方都可能违反该协议,泄露我们的专有信息,包括我们的商业秘密,我们可能无法就此类违规行为获得足够的补救措施。强制执行一方非法披露或挪用商业秘密的主张是困难、昂贵和耗时的,结果是不可预测的。此外,如果为维护我们的商业秘密而采取的步骤被认为是不充分的,我们可能没有足够的追索权来对抗第三方挪用商业秘密。此外,如果我们的任何商业秘密是由竞争对手合法获取或独立开发的,我们将无权阻止该第三方或他们向其传递此类技术或信息的人。, 利用这些技术或信息与我们竞争。如果我们的任何商业秘密被泄露给竞争对手或由竞争对手独立开发,我们的业务和竞争地位可能会受到损害。
87
第三方对知识产权侵权、挪用或其他侵权行为的索赔可能代价高昂且耗时,并可能阻碍或推迟我们的产品识别、发现和开发工作。
围绕精准医疗的知识产权格局是拥挤的,第三方可能会提起法律诉讼,指控我们侵犯、挪用或以其他方式侵犯他们的知识产权,其结果将是不确定的,并可能对我们的业务成功产生实质性的不利影响。我们的商业成功取决于我们或我们第三方的能力,以开发、制造、营销和销售我们当前和未来的候选产品,并使用我们的专有技术,而不侵犯、挪用或以其他方式侵犯第三方的知识产权。在生物技术和制药行业,有大量涉及专利和其他知识产权的诉讼,以及挑战专利的行政诉讼,包括派生、干扰、复审、各方间审查和批准美国专利商标局的审查程序或外国司法管辖区的异议和其他类似程序。我们或我们的任何许可人或战略合作伙伴可能参与、面临或威胁拥有专利或其他知识产权的第三方未来的对抗诉讼或诉讼,这些诉讼声称我们当前或未来的候选产品和/或专有技术侵犯、挪用或以其他方式侵犯他们的知识产权。我们不能向您保证,我们已经开发、正在开发或未来可能开发的候选产品和其他技术不会或不会侵犯、挪用或以其他方式侵犯第三方拥有的现有或未来专利或其他知识产权。在我们正在开发我们的候选产品的领域中,存在着大量由第三方拥有的美国和外国颁发的专利和未决的专利申请。随着生物技术和制药行业的扩张和专利的颁发,我们的候选产品可能会引发侵犯他人专利权的索赔的风险增加。此外,包括我们在内的行业参与者并不总是清楚哪些专利涵盖各种类型的药物、产品或它们的使用或制造方法。因此,由于在我们的领域中颁发的专利和提交的专利申请数量众多,第三方可能会声称他们拥有包含我们候选产品、技术或方法的专利权。
如果第三方声称我们侵犯、挪用或以其他方式侵犯其知识产权,我们可能面临许多问题,包括但不限于:
我们的一些竞争对手可能比我们更有效地承受复杂专利诉讼的费用,因为他们拥有更多的资源。此外,任何诉讼的发起和继续产生的任何不确定性都可能对我们筹集继续运营所需资金的能力产生重大不利影响,或者可能对我们的业务、运营结果、财务状况和前景产生重大不利影响。上述任何情况的发生都可能对我们的业务、财务状况、经营结果或前景产生重大不利影响。
88
我们可以选择挑战第三方美国专利中权利要求的专利性,方法是请求美国专利商标局审查专利主张中的单方面重考,各方间审查或拨款后审查程序。这些程序是昂贵的,可能会消耗我们的时间或其他资源。我们可以选择在欧洲专利局(“EPO”)或其他外国专利局的专利反对程序中挑战第三方的专利。这些反对诉讼的费用可能是巨大的,并可能消耗我们的时间或其他资源。如果我们未能在美国专利商标局、欧洲专利局或其他专利局获得有利的结果,我们可能会面临第三方的诉讼,指控我们的候选产品或专有技术可能侵犯了我们的专利。
第三方可能会声称我们在未经授权的情况下使用他们的专有技术。在美国依法颁发的专利享有有效性推定,只有在证据“清晰和令人信服”的情况下,才能推翻这一推定,这是一种更高的证明标准。可能已授予第三方专利,但我们目前并不知道这些专利对与使用或制造我们的候选产品相关的成分、配方、制造方法或治疗方法的权利要求。专利申请可能需要很多年的时间才能发布。此外,由于美国的一些专利申请可能在专利颁发之前保密,美国和许多外国司法管辖区的专利申请通常在申请后18个月才公布,而且科学文献中的出版物往往落后于实际发现,因此我们不能确定其他公司没有提交涵盖我们的候选产品或技术的专利申请。如果任何此类专利申请作为专利颁发,并且如果此类专利优先于我们的专利申请或我们可能拥有或许可的专利,我们可能被要求获得第三方拥有的、可能无法以商业合理的条款获得或根本无法获得的权利,或可能仅以非独家基础获得的权利。可能存在当前正在处理的专利申请,这些申请可能会导致我们的候选产品可能会侵犯已颁发的专利。我们知道由第三方拥有的专利,但我们认为这些专利与我们的候选产品或其他技术无关,也有可能被我们的候选产品或其他技术侵犯。此外,第三方可能会在未来获得专利,并声称使用我们的技术侵犯了这些专利。更有甚者, 我们可能无法识别相关专利,或错误地得出专利无效、不可强制执行、用尽或未被我们的活动侵犯的结论。如果有管辖权的法院持有任何第三方专利,以涵盖我们的候选产品的制造过程、在制造过程中使用或形成的分子或任何最终产品本身,则任何此类专利的持有者可能能够阻止我们将候选产品商业化的能力,除非我们根据适用专利获得了许可,或者直到该等专利到期或最终被确定为无效或不可执行。同样,如果有管辖权的法院持有任何第三方专利,涵盖我们的配方、制造工艺或使用方法的各个方面,包括联合疗法或患者选择方法,则任何此类专利的持有者可能能够阻止我们开发候选产品并将其商业化的能力,除非我们获得了许可证,或者直到该专利到期或最终被确定为无效或不可执行。在任何一种情况下,这样的许可可能都不会以商业上合理的条款或根本不存在。如果我们不能以商业上合理的条款获得第三方专利的必要许可,或者根本不能,我们将候选产品商业化的能力可能会受到损害或推迟,这反过来可能会严重损害我们的业务。
对我们提出索赔的各方可能会寻求并获得禁令或其他公平救济,这可能会有效地阻止我们进一步开发和商业化我们的候选产品。对这些索赔的辩护,无论其是非曲直,都可能涉及巨额诉讼费用,并将大量转移我们业务中的员工资源。如果针对我们的侵权、挪用或其他侵权行为索赔成功,我们可能需要支付大量损害赔偿金,包括故意侵权的三倍损害赔偿金和律师费,从第三方获得一个或多个许可证,支付版税或重新设计我们的侵权产品,这可能是不可能的,或者需要大量的时间和金钱支出。我们无法预测是否会有这样的许可证,或者是否会以商业合理的条款提供。此外,即使在没有诉讼的情况下,我们也可能需要或可能选择从第三方获得许可证,以推进我们的研究或允许我们的候选产品商业化。我们可能无法以合理的成本或合理的条款获得这些许可证中的任何一项,如果有的话。在这种情况下,我们将无法进一步开发和商业化我们的候选产品,这可能会严重损害我们的业务。
我们可能无法成功地通过收购和许可证内许可来获得或维护我们开发流程中的产品组件和流程的必要权利。
我们已经授权了一个与CLN-049相关的专利系列。我们拥有与CLN-619相关的三个专利系列。我们拥有两个与CLN-617相关的专利家族。我们拥有与CLN-978相关的三个专利系列。由于其他候选产品可能需要使用第三方持有的专有权,因此我们业务的增长可能在一定程度上取决于我们获取、授权或使用这些专有权的能力。
89
我们的候选产品可能还需要特定的配方才能有效和高效地工作,这些权利可能由其他人持有。同样,高效生产或交付我们的候选产品也可能需要特定的成分或方法,这些权利可能由第三方拥有。我们可能无法从第三方获得我们认为对我们的业务运营必要或重要的任何成分、使用方法、工艺或其他第三方知识产权,或无法对其进行许可。我们可能无法以合理的成本或合理的条款获得这些许可证中的任何一项,如果有的话,这将损害我们的业务。我们可能需要停止使用这种第三方知识产权所涵盖的成分或方法,并可能需要寻求开发不侵犯此类知识产权的替代方法,这可能会导致额外的成本和开发延迟,即使我们能够开发这种可能不可行的替代方法。即使我们能够获得许可,它也可能是非排他性的,从而使我们的竞争对手能够访问向我们许可的相同技术。在这种情况下,我们可能需要花费大量时间和资源来开发或许可替代技术。此外,将与我们的候选产品一起使用的分子可能会受到其他公司的知识产权保护。
此外,我们有时与学术机构合作,根据与这些机构达成的书面协议,加快我们的临床前研究或开发。在某些情况下,这些机构为我们提供了一个选项,可以就合作所产生的机构在技术上的任何权利进行谈判。无论此类选项如何,我们都可能无法在指定的时间范围内或在我们可接受的条款下谈判许可证。如果我们无法做到这一点,该机构可能会将知识产权提供给其他人,可能会阻止我们实施我们的计划,并允许第三方与我们竞争。如果我们不能成功地获得所需的第三方知识产权或维护我们现有的知识产权,我们可能不得不放弃开发此类项目,我们的业务和财务状况可能会受到影响。
第三方知识产权的许可和收购是一个竞争领域,可能比我们更成熟或拥有更多资源的公司也可能采取我们认为必要或有吸引力的第三方知识产权许可或收购战略,以便将我们的候选产品商业化。更成熟的公司可能比我们更具竞争优势,因为它们的规模、现金资源以及更强的临床开发和商业化能力。此外,将我们视为竞争对手的公司可能不愿将权利转让或许可给我们。我们也可能无法以使我们的投资获得适当回报的条款许可或获取第三方知识产权,或者根本无法。不能保证我们能够成功完成此类谈判,并最终获得围绕我们可能寻求收购的其他候选产品的知识产权。如果我们不能成功地获得所需的第三方知识产权或维护我们现有的知识产权,我们可能不得不放弃此类计划的发展,我们的业务、运营结果、财务状况和前景可能会受到影响。
我们可能会卷入保护或执行我们拥有或授权的知识产权的诉讼,这可能是昂贵、耗时和不成功的。
竞争对手可能会侵犯我们可能拥有的或许可中的任何专利。此外,我们可能拥有的或许可中的任何专利都可能涉及发明权、优先权、有效性或不可执行性纠纷。为了对抗侵权或未经授权的使用,我们可能会被要求提出侵权索赔,这可能是昂贵和耗时的。我们可能不会在我们发起的任何诉讼中获胜,而且所判给的损害赔偿或其他补救措施(如果有的话)可能没有商业意义。此外,在侵权诉讼中,法院可以根据《美国法典》第35篇第271(E)(1)节的规定,裁定我们可能拥有或许可的任何专利中的一项或多项无效或不可强制执行,或者另一方使用我们可能获得专利的技术属于专利侵权的安全港。还有一种风险是,即使这些专利的有效性得到支持,法院也可能以我们拥有的或授权内的任何专利不涵盖相关技术或该第三方的活动没有侵犯我们的专利为理由,拒绝阻止另一方使用所涉技术。任何诉讼或辩护程序中的不利结果可能会使我们拥有或授权的一项或多项专利面临被宣布无效、持有不可强制执行或狭义解释的风险,并可能使我们的专利申请面临无法发布的风险。对这些索赔的辩护,无论其是非曲直,都将涉及巨额诉讼费用,并将大量转移我们业务中的员工资源。如果针对我们的侵权索赔成功,我们可能需要支付大量损害赔偿金,包括故意侵权的三倍损害赔偿金和律师费、从第三方获得一个或多个许可证、支付版税或重新设计我们的侵权产品。, 这可能是不可能的,或者需要大量的时间和金钱支出。此类诉讼或诉讼可能大幅增加我们的运营亏损,并减少可用于开发活动或任何未来销售、营销或分销活动的资源。我们可能没有足够的财政或其他资源来适当地进行此类诉讼或诉讼程序。我们的一些竞争对手可能比我们更有效地承担这类诉讼或法律程序的费用,因为他们有更多的财政资源和更成熟和发展的知识产权组合。专利诉讼或其他诉讼的发起和继续所产生的不确定性可能会对我们在市场上的竞争能力产生重大不利影响。
90
由第三方发起或由美国专利商标局提起的授权后程序可能是必要的,以确定关于我们拥有的或许可内的专利或专利申请的发明的有效性或优先权。这些诉讼费用高昂,不利的结果可能导致我们现有专利权的损失,并可能要求我们停止使用相关技术或试图从胜利方那里获得许可权。如果胜利方不以商业上合理的条件向我们提供许可证,我们的业务可能会受到损害。除了可能的USPTO审查程序外,我们还可能成为欧洲专利局的专利反对程序或其他外国专利局的类似程序的一方,在这些程序中,我们的外国专利受到挑战。这些反对或类似诉讼的费用可能是巨大的,并可能导致某些权利要求的范围丧失或整个专利的损失。美国专利商标局、欧洲专利局或其他专利局的不利结果可能会导致我们失去排除他人在相关国家或司法管辖区实施我们的一项或多项发明的权利,这可能会对我们的业务产生实质性的不利影响。
诉讼或赠款后诉讼程序可能导致不利于我们利益的决定,即使我们成功,也可能导致巨额成本,并分散我们的管理层和其他员工的注意力。我们可能无法防止我们的商业秘密或机密信息被盗用,特别是在法律可能没有像美国那样充分保护这些权利的国家。
此外,由于知识产权诉讼需要大量的披露,在这类诉讼期间,我们的一些机密信息可能会因披露而被泄露。此外,可能会公布听证会、动议或其他临时程序或事态发展的结果。如果证券分析师或投资者认为这些结果是负面的,可能会对我们普通股的价格产生重大不利影响。
我们可能无法检测到对我们拥有或授权的任何专利的侵权行为。即使我们发现第三方侵权,我们也可能选择不对第三方提起诉讼或与其达成和解。如果我们后来以专利侵权为由起诉该第三方,该第三方可能有某些法律辩护可用,否则,除了最初发现侵权行为和提起诉讼之间的延迟外,这些辩护是不可用的。这样的法律辩护可能会使我们无法针对该第三方强制执行我们可能拥有的或许可内的任何专利。
获得和维持专利保护取决于遵守政府专利机构提出的各种程序、文件提交、费用支付和其他要求,如果不符合这些要求,专利保护可能会减少或取消。
任何已颁发的专利的定期维护费、续期费、年金费和各种其他政府费用应在专利有效期内分几个阶段支付给美国专利商标局和外国专利代理机构。美国专利商标局和各种外国政府专利机构要求在专利申请过程中和专利颁发后遵守一些程序、文件、费用支付和其他类似条款。虽然在某些情况下,可以根据适用规则通过支付滞纳金或通过其他方式纠正疏忽失效,但在某些情况下,不遵守规定可能导致专利或专利申请被放弃或失效,从而导致相关法域的专利权部分或全部丧失。可能导致专利被放弃或失效的不合规事件包括但不限于未能在规定的时限内对官方行动做出回应、未支付费用以及未能适当地使专利合法化并提交正式文件。在这种情况下,我们的竞争对手可能会以类似或相同的产品或平台进入市场,这可能会对我们的业务前景和财务状况产生重大不利影响。
美国和外国司法管辖区专利法的变化可能会降低专利的整体价值,从而削弱我们保护产品的能力。
与其他生物制药公司一样,我们的成功在很大程度上依赖于知识产权,特别是专利。在生物制药行业获得和实施专利既涉及技术上的复杂性,也涉及法律上的复杂性,因此成本高昂、耗时长,而且具有内在的不确定性。美国最高法院最近的裁决缩小了某些情况下可获得的专利保护范围,并在某些情况下削弱了专利所有者的权利。除了关于我们未来获得专利的能力的不确定性增加之外,这种事件的结合也造成了关于一旦获得专利的价值的不确定性。根据美国国会、联邦法院和美国专利商标局的决定,管理专利的法律和法规可能会以不可预测的方式发生变化,从而削弱我们获得新专利或执行我们未来可能获得的专利的能力。例如,在这种情况下阿索克。分子病理学诉Myriad Genetics,Inc.美国最高法院裁定,对DNA分子的某些主张不能申请专利。其他司法管辖区专利法的任何不利变化都可能对我们的业务和财务状况产生重大不利影响。管理其他司法管辖区专利的法律和法规的变化同样可能对我们获取和有效执行我们在专利申请中拥有的任何权利或我们可能拥有的或许可中的任何专利的能力产生不利影响。
91
最近或未来的专利改革立法也可能增加围绕我们专利申请的起诉以及我们可能拥有或许可的任何专利的执行或保护的不确定性和成本。美国已经颁布并实施了范围广泛的专利改革立法。2011年9月16日,《莱希-史密斯美国发明法》签署成为法律,其中包括对美国专利法的多项重大修改。这些条款包括影响专利申请起诉方式的条款,重新定义现有技术,可能影响专利诉讼,建立新的授权后审查制度,以及将美国专利制度从“先发明”制度转变为“先申请”制度。在“先提交文件”制度下,假设其他可专利性要求得到满足,第一个提交专利申请的发明人通常将有权获得该发明的专利,而无论是否有另一位发明人较早地作出了该发明。
我们的外国知识产权有限,可能无法在全球范围内保护我们的知识产权。
我们可能无法在美国以外的国家对我们的候选产品进行通用覆盖。在世界所有国家/地区对候选产品进行专利申请、起诉和辩护的费用将高得令人望而却步,而且美国以外的一些国家/地区的知识产权可能没有美国那么广泛。此外,一些外国国家的法律对知识产权的保护程度不如美国的联邦和州法律。因此,我们可能无法阻止第三方在美国以外的所有国家/地区实施我们的发明。或在美国或其他司法管辖区销售或进口使用我们的发明制造的产品。竞争对手可以在我们没有获得专利保护的司法管辖区使用我们的技术来开发他们自己的产品,并进一步可能向我们拥有专利保护但执法力度不如美国的地区出口其他侵权产品。这些产品可能与我们的候选产品竞争,在我们没有任何已颁发专利或专利申请或其他知识产权的司法管辖区,这些产品可能不能有效或不足以阻止他们竞争。我们的专利组合还处于非常早期的阶段。我们将需要在适用的最后期限之前决定是否以及在哪些司法管辖区为我们投资组合中的各种发明寻求保护。
许多公司在外国司法管辖区保护和捍卫知识产权方面遇到了重大问题。某些国家的法律制度,特别是某些发展中国家的法律制度,不支持强制执行专利、商业秘密和其他知识产权保护,特别是与生物制药产品有关的专利保护,这可能使我们难以阻止侵犯我们可能拥有的任何专利,或在许可或营销竞争产品时普遍侵犯我们的专有权。在外国司法管辖区强制执行我们在我们的专利和专利申请中可能拥有的任何权利的诉讼程序可能会导致巨额成本,并将我们的努力和注意力从我们业务的其他方面转移出去,可能会使我们拥有的或授权内的任何专利面临被无效或狭义解释的风险,并可能导致我们的专利申请面临无法发布的风险,并可能引发第三方对我们提出索赔。我们可能不会在我们发起的任何诉讼中获胜,所判给的损害赔偿或其他补救措施(如果有的话)可能没有商业意义。因此,我们在世界各地执行我们的知识产权的努力可能不足以从我们开发或许可的知识产权中获得显著的商业优势。
许多国家都有强制许可法,根据这些法律,专利权人可能会被强制向第三方授予许可。此外,许多国家限制专利对政府机构或政府承包商的可执行性。在这些国家,专利权人的补救措施可能是有限的,这可能会大大降低这种专利的价值。如果我们被迫就与我们的业务相关的任何我们拥有的或许可中的专利向第三方授予许可,我们的竞争地位可能会受到损害,我们的业务、财务状况、运营结果和前景可能会受到不利影响。
92
我们可能会受到质疑任何知识产权的发明权或所有权的索赔,包括我们可能拥有的或许可中的任何专利。
我们可能会受到前雇员、合作者或其他第三方作为发明人或共同发明人在我们拥有的或许可内的专利、商业秘密或其他知识产权中的权益的索赔。例如,我们可能会因参与开发我们的候选产品或其他技术的员工、顾问或其他人的义务冲突而产生库存纠纷。我们通常与员工、顾问和承包商签订保密和知识产权转让协议。这些协议一般规定,当事人在向我们提供服务的过程中构思的发明将是我们的专有财产。然而,这些协议可能不会得到遵守,也可能不会有效地将知识产权转让给我们。此外,在某些情况下,我们可能无法就这种所有权进行谈判。关于知识产权所有权或发明权的争端也可能在其他情况下发生,例如合作和赞助研究。如果我们面临一场挑战我们在专利或其他知识产权上或对其权利的纠纷,这样的纠纷可能代价高昂且耗时。可能有必要提起诉讼,以抗辩这些和其他挑战我们拥有或授权的任何专利、商业秘密或其他知识产权的库存的索赔。如果我们失败了,除了支付金钱损害赔偿外,我们还可能失去我们认为是自己的知识产权的宝贵权利,例如对我们的产品候选产品和其他技术非常重要的知识产权的独家所有权或使用权。即使我们成功地对这种说法进行了辩护, 诉讼可能会导致巨额成本,并分散管理层和其他员工的注意力。上述任何一项都可能对我们的业务、财务状况、经营结果和前景产生重大不利影响。
我们可能会受到以下指控的影响:我们的员工、顾问或独立承包商错误地使用或披露了第三方或竞争对手的机密信息或据称的商业秘密,或者违反了与竞争对手的竞业禁止或竞标协议。
我们已从第三方收到机密和专有信息。此外,就像在生物技术和制药行业中常见的那样,我们雇用的个人以前曾受雇于大学或其他生物技术或制药公司,包括我们的竞争对手或潜在竞争对手,在某些情况下,直到最近。我们可能会被指控我们或我们的员工、顾问或独立承包商无意中或以其他方式使用或泄露了这些第三方、我们员工的前雇主、我们的顾问或承包商的现任或前任客户或客户的机密信息或商业秘密。此外,我们可能会受到索赔,即我们导致员工违反了他或她的竞业禁止或竞标协议的条款。可能需要诉讼或仲裁来抗辩这些索赔。如果我们不能为任何此类索赔辩护,除了支付金钱损害赔偿外,我们还可能失去宝贵的知识产权或人员。即使我们成功地对此类索赔进行了辩护,与知识产权索赔相关的诉讼或其他法律程序以及可能的后果也可能导致巨额成本,并分散我们管理层和员工的注意力。任何诉讼或其威胁都可能对我们雇用员工的能力产生不利影响。关键人员或他们的工作产品的流失可能会阻碍或阻止我们将候选产品商业化的能力,这可能会对我们的业务、运营结果和财务状况产生不利影响。此外,可能会公布听证会、动议或其他临时程序或事态发展的结果,如果证券分析师或投资者认为这些结果是负面的,, 这可能会对我们普通股的价格产生实质性的不利影响。这种类型的诉讼或诉讼可能会大幅增加我们的运营损失,并减少我们可用于开发活动的资源。我们可能没有足够的财政或其他资源来适当地进行此类诉讼或法律程序。我们的一些竞争对手可能比我们更有效地承担这类诉讼或诉讼的费用,因为他们的财政资源要大得多。专利诉讼或其他知识产权相关诉讼的发起和继续所产生的不确定性可能会对我们在市场上的竞争能力产生不利影响。
93
如果我们没有为我们当前或未来可能开发的任何候选产品获得专利期延长和数据独占权,我们的业务可能会受到实质性损害。
根据FDA对我们当前或未来可能开发的任何候选产品的上市批准的时间、期限和细节,我们可能拥有或许可的一项或多项美国专利可能有资格根据1984年的《药品价格竞争和专利期限恢复法》(“Hatch-Waxman修正案”)获得有限的专利期延长。哈奇-瓦克斯曼修正案允许专利期限延长最多五年,作为对FDA监管审查过程中失去的专利期限的补偿。一项专利期限的延长不得超过自产品批准之日起共计14年的时间,只能延长一项专利,并且只能延长涉及经批准的药物、其使用方法或者其制造方法的权利要求。但是,我们可能因为在测试阶段或监管审查过程中未能进行尽职调查、未能在适用的最后期限内提出申请、未能在相关专利到期前提出申请或未能满足适用的要求等原因而无法获得延期。此外,专利保护的适用期限或范围可能比我们要求的要短。如果我们无法获得专利期限的延长,或者任何此类延长的期限比我们要求的短,我们的竞争对手可能会在我们专利申请的任何专利到期后获得竞争产品的批准,我们的业务、财务状况、运营结果和前景可能会受到实质性损害。
如果我们的商标和商号没有得到充分的保护,那么我们可能无法在我们感兴趣的商标中建立起知名度,我们的业务可能会受到不利影响。
我们的商标或商号可能会受到挑战、侵犯、稀释、规避或宣布为通用商标,或被认定为侵犯了其他商标。我们打算同时依靠注册和普通法保护我们的商标。我们可能无法保护我们对这些商标和商品名称的权利,或者可能被迫停止使用这些名称,我们需要这些名称才能在我们感兴趣的市场中获得潜在合作伙伴或客户的名称认可。在商标注册过程中,我们可能会收到美国专利商标局对我们商标注册的反对行动。虽然我们将有机会对这些反对意见作出回应,但我们可能无法克服这些反对意见。此外,在美国专利商标局和许多外国司法管辖区的类似机构中,第三方有机会反对未决的商标申请和/或寻求注销注册商标。可能会对我们的商标提起反对或取消诉讼,而我们的商标可能无法继续存在。如果我们无法获得注册商标或根据我们的商标和商号建立名称认可,我们可能无法有效竞争,我们的业务可能会受到不利影响。
与我们对第三方的依赖有关的风险
我们目前依赖并预计将继续依赖将我们的大部分开发职能外包给第三方进行临床前研究和临床试验。如果这些第三方不能正确和成功地履行其合同职责或在预期的最后期限前完成,我们可能无法获得监管部门对我们的候选产品的批准或将其商业化。
我们利用并依赖独立的研究人员和合作者,如医疗机构、CRO、CMO和战略合作伙伴,根据与我们达成的协议进行和支持我们的临床前研究和临床试验,并预计未来将依赖这些各方。
94
我们与CRO、试验点和CMO谈判预算和合同,但我们可能无法以有利的条件这样做,这可能会导致我们的开发时间表延迟和成本增加。在我们的临床前研究和临床试验过程中,我们严重依赖这些第三方,我们只控制他们活动的某些方面。因此,与完全依靠我们自己的员工相比,我们对临床前研究和临床试验的进行、时间和完成以及通过临床前研究和临床试验开发的数据的管理缺乏直接控制力。然而,我们有责任确保我们的每一项研究和试验都按照适用的议定书、法律和法规要求以及科学标准进行,并且我们对第三方的依赖不会免除我们的监管责任。我们和这些第三方必须遵守良好的制造、临床和实验室操作规范(“GXP”),这是FDA和类似的外国监管机构对临床开发中的候选产品执行的法规和指导方针。监管机构通过定期检查试验赞助商、主要调查人员和试验地点来执行这些GXP。如果我们或这些第三方中的任何一方未能遵守适用的GxP法规,在我们的临床试验中产生的临床数据可能被认为是不可靠的,FDA或类似的外国监管机构可能会要求我们在批准我们的上市申请之前进行额外的临床试验。我们不能向您保证,在检查后,这些监管机构将确定我们的任何临床试验是否符合GxP规定。此外, 我们的临床试验必须使用根据GxP规定生产的药品进行,并需要大量的测试患者。我们或这些第三方未能遵守这些规定或未能招募足够数量的患者,可能会推迟正在进行的或计划中的临床试验,或要求我们重复临床试验,这将延误监管批准过程。如果我们或我们承诺的第三方未能遵守监管要求,也可能导致罚款、负面宣传以及民事和刑事制裁。此外,如果其中任何第三方违反联邦或州欺诈和滥用或虚假索赔法律法规或医疗保健隐私和安全法,我们的业务可能会受到牵连。
进行我们临床试验的任何第三方不是我们的员工,也不会是我们的员工,并且,除了我们与这些第三方达成的协议提供的补救措施外,我们不能控制他们是否将足够的时间和资源投入到我们正在进行的临床和临床前候选产品上。这些第三方也可能与其他商业实体有关系,包括我们的竞争对手,他们可能还在为这些实体进行临床试验或其他产品开发活动,这可能会影响他们代表我们的表现。如果这些第三方未能成功履行其合同职责或义务或在预期期限内完成,如果他们需要更换,或者如果他们获得的临床数据的质量或准确性因未能遵守我们的临床方案或监管要求或其他原因而受到影响,我们的临床试验可能会被延长、推迟或终止,我们可能无法完成我们候选产品的开发、获得监管部门的批准或成功地将其商业化。因此,我们的财务业绩和候选产品的商业前景将受到损害,我们的成本可能会增加,我们创造收入的能力可能会被推迟。
更换或增加第三方来进行我们的临床前研究和临床试验涉及大量成本,需要我们的管理人员投入大量时间和精力。此外,当新的第三方开始工作时,有一个自然的过渡期。因此,会出现延迟,这可能会对我们满足期望的临床开发时间表的能力产生实质性影响。
此外,我们不直接控制生产我们候选产品的制造设施,我们必须依赖CMO根据质量和可靠性标准生产我们的候选产品。我们不拥有任何制造设施或设备,也不雇用任何制造人员。我们不能向您保证我们将能够以合理的条件获得合格的合同制造服务。如果与我们签订合同的任何CMO未能履行其义务或更改了原材料采购,我们可能会被迫与其他CMO达成协议,而我们可能无法以合理的条款做到这一点。在这种情况下,随着我们建立替代供应来源,我们的临床试验供应可能会显著延迟。在某些情况下,制造我们的产品或候选产品所需的技术技能可能是原始CMO独有的或专有的,我们可能会遇到困难,或者可能存在合同限制,禁止我们将此类技能转让给备用或替代供应商,或者我们可能根本无法转让此类技能。此外,如果我们因任何原因被要求更换CMO,我们将被要求核实新的CMO保持符合质量标准和所有适用法规的设施和程序。我们还需要验证,例如通过制造可比性或衔接研究,任何新的制造工艺都将根据之前提交给FDA或其他监管机构的规格生产我们的候选产品。与新CMO验证相关的延迟可能会对我们及时或在预算内推进临床试验或开发候选产品或将我们的产品商业化的能力产生负面影响。更有甚者, 一名CMO可能拥有该CMO独立拥有的与我们的候选产品制造相关的技术,这可能会增加我们对该CMO的依赖,或者要求我们获得该CMO的许可,以便让另一名CMO生产我们的候选产品。此外,制造商的变化通常涉及制造程序和工艺的变化,这可能要求我们在临床试验中使用的先前临床供应与任何新制造商的供应之间进行过渡性研究。我们可能不能成功地证明临床用品的可比性,这可能需要进行额外的临床试验。
95
除了我们现有的合作之外,我们可能会在未来形成或寻求更多的合作或战略联盟,或达成更多的许可安排,而我们可能没有意识到此类合作、联盟或许可安排的好处。
如上所述,于2022年6月,我们完成了将我们在库利南珍珠的股权出售给泰豪,我们与泰豪的一家子公司达成了共同开发协议,共同开发并在我们选择的情况下在美国共同商业化齐帕尔替尼。根据与泰豪共同开发协议的条款,我们各自将为齐帕替尼在美国的未来临床开发做出同等贡献,并将各自获得未来可能在美国销售齐帕尔替尼的任何税前利润的50%。
我们可能会结成或寻求其他战略联盟,建立合资企业或合作关系,或与第三方达成额外的许可安排,我们相信这些安排将补充或加强我们关于我们的候选产品和我们可能开发的任何未来候选产品的开发和商业化努力。这些关系中的任何一项都可能要求我们产生非经常性费用和其他费用,增加我们的短期和长期支出,发行稀释我们现有股东或扰乱我们的管理和业务的证券。
此外,涉及我们的候选产品的协作可能会面临许多风险,其中可能包括:
因此,如果我们签订额外的协作协议和战略合作伙伴关系或许可我们的候选产品,如果我们不能成功地将这些交易与我们现有的运营和公司文化相结合,我们可能无法实现此类交易的好处,这可能会推迟我们的时间表或以其他方式对我们的业务产生不利影响。我们也不能确定,在战略交易或许可证之后,我们是否会实现证明此类交易合理的收入或特定净收入。与我们的候选产品相关的新合作或战略合作伙伴协议的任何延迟都可能推迟我们的候选产品在某些地区的开发和商业化,这将损害我们的业务前景、财务状况和运营结果。
96
我们目前依赖并预计未来将依赖于在第三方工厂或第三方中使用制造套件来生产我们的候选产品。如果我们无法使用第三方制造套件,或者第三方制造商无法向我们提供足够数量的候选产品或无法以可接受的质量或价格提供产品,我们的业务可能会受到损害。
我们目前没有任何可用作临床规模制造和加工设施的设施,目前必须依赖外部供应商来生产我们的候选产品。我们将需要与这些外部供应商谈判和维护供应我们候选产品的合同安排,而我们可能无法以有利的条件做到这一点。我们还没有使我们的候选产品进行商业规模的生产,而且可能无法对我们的任何候选产品进行这样的生产。
我们预期对数量有限的第三方制造商的依赖使我们面临许多风险,包括:
这些风险中的每一个都可能延迟或阻止我们的临床试验完成或FDA批准我们的任何候选产品,导致更高的成本或对我们候选产品的商业化产生不利影响。此外,在交付给患者之前,我们将依赖第三方对我们的候选产品进行某些规格测试。如果这些测试做得不恰当,测试数据不可靠,患者可能会面临严重伤害的风险,FDA可能会对我们公司施加重大限制,直到缺陷得到补救。
严重的不遵守也可能导致施加制裁,包括警告或无标题信件、罚款、禁令、民事处罚、监管机构未能为我们的候选产品授予上市批准、延迟、暂停或撤回批准、吊销许可证、扣押或召回产品、运营限制和刑事起诉,任何这些都可能损害我们的声誉和业务。
97
药品,特别是生物制品的生产是复杂的,我们的第三方制造商可能会在生产中遇到困难。如果我们的任何第三方制造商遇到这样的困难,我们为临床试验或为患者提供我们当前候选产品或任何未来候选产品的能力,如果获得批准,可能会被推迟或阻止。
制造药物,特别是生物制剂,尤其是大量生产,往往很复杂,可能需要使用创新技术来处理活细胞。每批经批准的生物必须经过身份、强度、质量、纯度、效力和稳定性的彻底测试。生产生物制品需要专门为此目的而设计和验证的设施,并需要复杂的质量保证和质量控制程序。制造过程中任何地方的微小偏差,包括灌装、标签、包装、储存和运输以及质量控制和测试,都可能导致批次故障、产品召回或变质。当生产过程发生变化时,我们可能被要求提供临床前和临床数据,显示产品在这种变化前后的可比性、强度、质量、纯度或效力。如果在我们制造商的设施中发现微生物、病毒或其他污染,这些设施可能需要关闭很长一段时间以调查和补救污染,这可能会推迟临床试验并对我们的业务造成不利影响。
此外,临床试验或商业规模的大规模生产还存在相关风险,其中包括成本超支、工艺放大的潜在问题、工艺重复性、稳定性问题、符合良好的生产实践、批次一致性和原材料的及时可获得性等。即使我们目前的任何候选产品或任何未来的候选产品获得了市场批准,也不能保证我们的制造商能够按照FDA或其他类似的外国监管机构可以接受的规格生产获得批准的产品,以生产足够数量的产品,以满足该产品潜在的商业投放或未来的潜在需求。如果我们的制造商不能生产足够的数量用于临床试验或商业化,我们的开发和商业化努力将受到损害,这将对我们的业务、财务状况、运营结果和增长前景产生不利影响。
如果我们的第三方制造商以造成伤害或违反适用法律的方式使用危险和生物材料,我们可能要承担损害赔偿责任。
我们的研究和开发活动涉及我们的第三方制造商对潜在危险物质的控制使用,包括化学和生物材料。我们的制造商受美国联邦、州和地方法律法规的约束,管理医疗和危险材料的使用、制造、储存、搬运和处置。尽管我们相信制造商使用、处理、储存和处置这些材料的程序符合法律规定的标准,但我们不能完全消除医疗或危险材料造成的污染或伤害的风险。由于任何此类污染或伤害,我们可能会承担责任,或者地方、城市、州或联邦当局可能会限制这些材料的使用并中断我们的业务运营。一旦发生事故,我们可能被要求承担损害赔偿责任或罚款,责任可能超出我们的资源范围。我们不为医疗或危险材料引起的责任投保。遵守适用的环境法律法规代价高昂,当前或未来的环境法规可能会损害我们的研究、开发和生产努力,从而可能损害我们的业务、前景、财务状况或运营结果。
与管理增长和员工事务相关的风险
新冠肺炎已经并可能继续对我们的业务产生不利影响。
新冠肺炎疫情和政府采取的应对措施对企业和商业产生了直接和间接的重大影响,包括持续的劳动力短缺和供应链中断。由于新冠肺炎大流行,我们的临床试验和临床前开发活动也出现了延误,包括我们在正在进行的临床试验中招募和留住患者的能力。未来大流行对我们的业务、临床前研究和临床试验的影响程度将取决于高度不确定和无法自信预测的事态发展,例如疾病的最终地理传播、大流行的持续时间、美国和其他国家的旅行限制和社会距离、企业关闭或商业中断,以及美国和其他国家采取的控制和治疗疾病的行动的有效性。
98
我们高度依赖我们的关键人员。如果我们不能成功地留住高素质的人才,我们就可能无法成功地实施我们的商业战略。
我们能否在竞争激烈的生物技术和制药行业中竞争,取决于我们能否留住高素质的管理、科学和医疗人员。我们高度依赖我们的高级管理人员,包括科学和医疗人员和其他关键员工。虽然我们希望在整合新任命的干事和管理人员时参与有序的过渡进程,但我们面临与管理过渡有关的各种风险和不确定因素,包括将管理人员的注意力从业务方面转移、未能留住其他关键人员或失去机构知识。此外,失去我们的任何高管、其他关键员工和其他科学和医疗顾问的服务,以及无法找到合适的替代者,都可能导致产品开发的延迟,并损害我们的业务。特别是,由于我们的员工数量较少,与我们同行的损失相比,失去一名员工对我们业务的影响可能更大。
我们在马萨诸塞州剑桥市的设施中开展业务。马萨诸塞州地区是许多其他生物制药公司以及许多学术和研究机构的总部。我们市场对技术人员的竞争非常激烈,可能会限制我们以可接受的条件聘用和留住高素质人员的能力,甚至根本不能。美国移民和工作授权法律法规的变化,包括那些限制科学和专业人才流动的法律法规,可能会受到政治力量和经济活动水平的重大影响。如果移民或签证法律法规的立法或行政变更损害了我们的招聘流程和目标或涉及非美国公民的项目,我们的业务可能会受到实质性的不利影响。
为了鼓励有价值的员工留在我们公司,除了工资和现金激励外,我们还提供了随着时间的推移而授予的股权。随着时间的推移,股票期权对员工的价值可能会受到我们股价波动的重大影响,这些波动超出了我们的控制范围,而且在任何时候都可能不足以抵消其他公司提供的更有利可图的报价。尽管我们努力留住有价值的员工,但我们的管理、科学和开发团队的成员可能会在短时间内终止与我们的雇佣关系。虽然我们与我们的关键员工有雇佣协议,但这些雇佣协议规定可以随意雇用,这意味着我们的任何员工都可以在任何时候离开我们的工作,无论是否通知。我们的成功还取决于我们能否继续吸引、留住和激励高技能的初级、中级和高级管理人员以及初级、中级和高级科学和医疗人员。
我们将需要扩大我们组织的规模,我们在管理这种增长时可能会遇到困难。
截至2022年12月31日,我们有62名全职员工和两名顾问。随着我们的开发和商业化计划和战略的发展,我们预计需要更多的管理、运营、销售、营销、财务和其他人员。未来的增长将使管理层成员承担更多的重大责任,包括:
我们未来的财务业绩以及我们将任何被批准用于营销的候选产品商业化的能力在一定程度上将取决于我们有效管理未来任何增长的能力,我们的管理层可能还必须将不成比例的注意力从日常活动中转移出来,以便将大量时间用于管理这些增长活动。
如果我们不能通过雇佣新员工和扩大我们的顾问和承包商团队来有效地扩大我们的组织,我们可能无法成功地执行进一步开发我们的候选产品并可能将其商业化所需的任务,因此可能无法实现我们的研究、开发和商业化目标。
99
我们的内部计算机系统,或我们的第三方CRO或其他承包商或顾问使用的系统,可能会出现故障或遭遇安全漏洞,这可能会导致我们候选产品的开发计划受到实质性破坏。
尽管实施了安全措施,但我们的内部计算机系统以及我们当前和未来的CRO以及其他承包商和顾问的计算机系统很容易受到计算机病毒、未经授权的访问、自然灾害以及电信和电气故障的破坏。虽然到目前为止,我们还没有经历过任何此类重大系统故障或安全漏洞,但如果发生此类事件并导致我们的运营中断,可能会导致我们的开发计划和业务运营发生重大中断。例如,已完成或未来的临床前研究和临床试验的数据丢失可能会导致我们的监管审批工作延迟,并显著增加我们恢复或复制数据的成本。同样,我们依赖第三方生产我们的候选产品并进行临床试验,与他们的计算机系统相关的类似事件也可能对我们的业务产生重大不利影响。如果任何中断或安全漏洞导致我们的数据或应用程序丢失或损坏,或不适当地披露机密或专有信息,我们可能会招致责任,我们候选产品的进一步开发和商业化可能会被推迟。
我们可能无法充分保护我们的信息系统免受网络攻击,网络攻击可能导致包括个人数据在内的机密或专有信息被泄露,损害我们的声誉,并使我们面临重大的财务和法律风险。
在我们的日常运营中,我们依赖我们或我们的第三方提供商运营的信息技术系统来处理、传输和存储电子信息。在我们的产品发现工作中,我们可能会收集和使用各种个人数据,如姓名、邮寄地址、电子邮件地址、电话号码和临床试验信息。成功的网络攻击可能导致知识产权、数据或其他挪用资产的盗窃或破坏,或以其他方式危及我们的机密或专有信息并扰乱我们的运营。网络攻击的频率、复杂性和强度都在增加,而且越来越难被发现。网络攻击可能包括敌对的外国政府的不法行为、工业间谍活动、电信欺诈和其他形式的网络欺诈、部署有害的恶意软件、拒绝服务、社会工程欺诈或其他威胁数据安全、机密性、完整性和可用性的手段。成功的网络攻击可能会给我们带来严重的负面后果,包括但不限于运营中断,机密商业信息被挪用,包括金融信息、商业秘密、财务损失和公司战略计划的披露。尽管我们投入了资源来保护我们的信息系统,但我们意识到网络攻击是一种威胁,不能保证我们的努力会防止信息安全漏洞,这些漏洞会导致我们的业务、法律、财务或声誉受损,或者会对我们的运营结果和财务状况产生实质性的不利影响。任何未能防止或减轻安全漏洞,或不当访问、使用或披露我们的临床数据或患者个人数据的行为,都可能导致重大责任,根据国家(例如:,州违规通知法),联邦(例如:,HIPAA,经HITECH修订)和国际法(例如:GDPR),并可能对我们的声誉造成实质性的不利影响,影响我们进行新研究的能力,并可能扰乱我们的业务。
此外,我们所依赖的各种第三方的计算机系统,包括我们的CRO和其他承包商、顾问以及法律和会计公司,可能会因计算机病毒、未经授权的访问、数据泄露、网络钓鱼攻击、网络罪犯、自然灾害(包括飓风和地震)、恐怖主义、战争以及电信和电气故障而遭受损害。我们依赖我们的第三方提供商实施有效的安全措施,并识别并纠正任何此类故障、缺陷或违规行为。如果我们或我们的第三方提供商未能有效地维护或保护我们的信息技术系统和数据完整性,或未能预见、计划或管理我们的信息技术系统的重大中断,我们或我们的第三方提供商可能难以预防、检测和控制此类网络攻击,任何此类攻击都可能导致上述损失以及与医生、患者和我们的合作伙伴的纠纷,监管制裁或处罚,运营费用、支出或收入损失或其他不利后果的增加,任何此类攻击都可能对我们的业务、运营结果、财务状况、前景和现金流产生实质性的不利影响。此类第三方未能防止或减轻安全漏洞,或不当获取或披露此类信息,都可能对我们造成类似的不利后果。如果我们无法防止或减轻此类安全或数据隐私泄露的影响,我们可能会面临诉讼和政府调查,这可能会导致我们的业务潜在中断。
100
业务中断可能会严重损害我们未来的收入和财务状况,并增加我们的成本和支出。
我们的业务,以及我们的CRO、CMO和其他承包商和顾问的业务,可能会受到地震、电力短缺、电信故障、缺水、洪水、飓风、台风、火灾、极端天气条件、医疗流行病和其他自然或人为灾难或业务中断的影响,我们主要是自我保险。任何这些业务中断的发生都可能严重损害我们的运营和财务状况,并增加我们的成本和支出。我们依赖第三方制造商来生产我们的候选产品。如果这些供应商的运营受到人为或自然灾害或其他业务中断的影响,我们获得候选产品的临床供应的能力可能会受到干扰。
与我们普通股所有权相关的风险
我们的股票价格可能会波动,你可能会损失你的全部或部分投资。
我们普通股的交易价格可能波动很大,可能会受到各种因素的影响而大幅波动,其中一些因素是我们无法控制的,包括交易量有限。除了“风险因素”一节和本10-K表格年度报告其他部分所讨论的因素外,这些因素还包括:
101
此外,整个股市,特别是纳斯达克全球精选市场和生物制药公司市场,经历了极端的价格和成交量波动,这些波动往往与这些公司的经营业绩无关或不成比例。无论我们的实际经营业绩如何,广泛的市场和行业因素都可能对我们普通股的市场价格产生负面影响。在过去,证券集体诉讼经常是在公司证券的市场价格出现波动后对公司提起的。如果提起这类诉讼,可能会导致巨额费用和分散管理层的注意力和资源,这将损害我们的业务、财务状况、经营结果和未来前景。
我们预计,由于各种因素,我们的财务状况和运营结果将继续在每个季度和每年大幅波动,其中许多因素是我们无法控制的。因此,您不应依赖任何季度或年度的业绩作为未来经营业绩的指标。
我们不打算为我们的普通股支付股息,所以任何回报都将限制在我们股票的价值上。
我们目前预计,我们将保留未来的收益,用于我们业务的发展、运营和扩张,在可预见的未来,我们预计不会宣布或支付任何现金股息。此外,我们可能会签订协议,禁止我们在未经缔约双方事先书面同意的情况下支付现金股息,或订立其他条款,禁止或限制我们的普通股可能宣布或支付的股息数额。因此,对股东的任何回报都将仅限于其股票的升值,而这可能永远不会发生。
我们的主要股东和管理层拥有我们相当大比例的股票,并将能够对有待股东批准的事项施加重大影响。
根据截至2022年12月31日已发行的45,796,449股普通股,我们的高管、董事和5%的股东实益拥有我们约63%的有表决权股票。这些股东有能力通过他们的所有权地位来影响我们。因此,这些股东可能能够决定所有需要股东批准的事项。例如,这些股东可能能够控制董事选举、修改我们的组织文件、批准任何合并、出售资产或其他重大公司交易。这可能会阻止或阻止对我们普通股的主动收购建议或要约,因为您可能认为作为我们的股东之一,这符合您的最佳利益。
我们是一家新兴成长型公司和一家较小的报告公司,我们不能确定适用于新兴成长型公司和较小报告公司的报告要求降低是否会降低我们的普通股对投资者的吸引力。
我们是一家新兴的成长型公司,正如2012年4月颁布的JumpStart Our Business Startups Act(“JOBS Act”)所界定的那样。只要我们继续是一家新兴成长型公司,我们就可以利用适用于其他非新兴成长型公司的上市公司的各种报告要求的豁免,包括不被要求遵守2002年《萨班斯-奥克斯利法案》(修订后的《萨班斯-奥克斯利法案》)第404节的审计师认证要求,减少我们定期报告和委托书中关于高管薪酬的披露义务,以及免除就高管薪酬举行非约束性咨询投票的要求,以及免除股东批准任何先前未获批准的金降落伞付款的要求。我们将一直是一家新兴的成长型公司,直到(1)财政年度的最后一天(A)在我们首次公开募股(IPO)结束五周年后,(B)我们的年总收入至少为12.35亿美元,或(C)我们被视为大型加速申报公司,这要求我们的非关联公司持有的普通股在最近完成的第二财季的最后一个工作日的市值超过7亿美元,以及(2)我们在前三年期间发行了超过10亿美元的不可转换债券的日期。
102
根据《就业法案》,新兴成长型公司还可以推迟采用新的或修订后的会计准则,直到这些准则适用于私营公司。我们已选择不“选择退出”遵守新的或修订的会计准则,因此,我们将在私人公司采用新的或修订的会计准则时采用新的或修订的会计准则,并将这样做,直到我们(I)不可撤销地选择“选择退出”延长的过渡期或(Ii)不再符合新兴成长型公司的资格。
即使我们不再有资格成为一家新兴成长型公司,我们仍有资格成为一家“较小的报告公司”,这将允许我们继续利用许多相同的披露要求豁免,包括不被要求遵守萨班斯-奥克斯利法案第404条的审计师认证要求,以及在我们的定期报告和委托书中减少关于高管薪酬的披露义务。在确定(I)非关联公司持有的有投票权和无投票权普通股的市值超过7亿美元(以最近完成的第二财季的最后一个营业日衡量)或(Ii)非关联方持有的有投票权和无投票权普通股的市值(以我们最近完成的第二财季的最后一个营业日衡量)不到7亿美元但大于2.5亿美元,且我们在最近完成的会计年度的年收入超过1亿美元后,我们可以利用向较小报告公司提供的大规模披露,直到下一财年。我们无法预测投资者是否会因为我们可能依赖这些豁免而发现我们的普通股吸引力下降。如果一些投资者因此发现我们的普通股吸引力下降,我们的普通股可能会有一个不那么活跃的交易市场,我们的股价可能会更加波动。
我们利用我们的净营业亏损结转和某些其他税务属性的能力可能是有限的。
根据修订后的1986年《国税法》第382和383条,如果一家公司经历了“所有权变更”(一般定义为5%的股东在三年期间的股权所有权变动超过50%(按价值计算)),该公司利用变动前净营业亏损(“NOL”)结转和其他变动前税收属性抵销变动后应纳税所得额的能力可能是有限的。由于我们最近的私募和过去三年发生的其他交易,我们可能已经经历了,也可能经历了一次“所有权变更”。我们还可能在未来经历所有权的变化,因为我们的股票所有权随后发生了变化。截至2022年12月31日,我们有美国联邦和州NOL结转的分别为4260万美元和2710万美元。该公司在2018年前产生了580万美元的联邦NOL,这些NOL将于2036年开始到期。州政府的亏损也将于2036年到期。该公司产生了3680万美元的联邦NOL,可以无限期结转。截至2022年12月31日,该公司的联邦和州研发税收抵免结转金额分别为90万美元和30万美元,每项抵免将分别在2037年和2033年的不同日期开始到期,如果我们经历“所有权变更”,这一抵免可能会受到限制。根据2017年减税和就业法案(“TCJA”)降低公司税率,可能会导致我们的NOL结转和其他可供我们使用的递延纳税资产的经济效益减少。根据TCJA,2017年12月31日之后生成的联邦NOL将不会过期,但不会被允许带回。此外,在TCJA下,, 在任何课税年度,我们获准扣除的2017年后净额限制为该年度应纳税所得额的80%,该年度的应纳税所得额是在不考虑净额扣除本身的情况下确定的。截至2021年12月31日,我们有美国联邦和州的NOL结转分别为1.174亿美元和1.19亿美元,这些结转将在2036年之前的不同日期开始到期,如果我们经历“所有权变更”,这一结转可能会受到限制。
我们的章程文件和特拉华州法律中的反收购条款可能会推迟或阻止控制权的变更,这可能会限制我们普通股的市场价格,并可能阻止或挫败我们的股东更换或罢免我们目前管理层的尝试。
我们的第二次修订和重述的公司注册证书以及修订和重述的章程中包含的条款可能会推迟或阻止我们公司的控制权变更或我们的股东可能认为有利的董事会变更。其中一些规定包括:
103
此外,由于我们是在特拉华州注册成立的,我们受特拉华州公司法第203条的规定管辖,该条款可能禁止股东拥有我们已发行有表决权股票的15%或更多的某些业务合并。这些反收购条款和我们第二次修订和重述的公司注册证书以及修订和重述的章程中的其他条款可能会使股东或潜在收购者更难获得我们董事会的控制权,或者发起当时的董事会反对的行动,还可能推迟或阻碍涉及我们公司的合并、收购要约或代理权之争。这些规定还可能阻止代理权竞争,并使您和其他股东更难选举您选择的董事或导致我们采取您希望的其他公司行动。任何延迟或阻止控制权变更、交易或董事会变动都可能导致我们普通股的市场价格下跌。
我们修订和重述的章程指定特定的法院作为可能由我们的股东发起的某些诉讼的独家论坛,这可能限制我们的股东获得有利的司法论坛处理与我们的纠纷的能力。
根据我们修订和重述的章程,除非我们以书面形式同意选择替代法院,否则特拉华州衡平法院将是州法律索赔的唯一和独家论坛,涉及(1)代表我们提起的任何派生诉讼或诉讼;(2)任何声称我们的任何董事、高级管理人员或其他员工违反对我们或我们股东的受托责任的诉讼;(3)根据特拉华州公司法或我们的公司注册证书或章程的任何规定提出索赔的任何诉讼;(4)解释、应用、强制执行或确定我们的公司注册证书或章程的有效性的任何诉讼;或(5)任何主张受内部事务原则或特拉华论坛条款管辖的索赔的诉讼。特拉华州论坛条款将不适用于根据证券法或交易法产生的任何诉讼因由。我们修订和重述的章程将进一步规定,除非我们书面同意选择替代法院,否则美国特拉华州地区法院将是解决根据证券法或联邦论坛条款提出的任何申诉的唯一和独家论坛,因为公司是在特拉华州注册成立的。此外,我们修订和重述的章程将规定,任何购买或以其他方式获得我们股本股份权益的个人或实体将被视为已通知并同意特拉华论坛条款和联邦论坛条款;但前提是,股东不能也不会被视为放弃遵守美国联邦证券法及其下的规则和法规。
我们修订和重述的章程中的特拉华论坛条款和联邦论坛条款可能会给股东在寻求任何此类索赔时带来额外的诉讼费用。此外,这些论坛选择条款可能会限制我们的股东在司法论坛上提出他们认为有利于与我们或我们的董事、高管或员工发生纠纷的索赔的能力,这可能会阻止对我们和我们的董事、高管和员工提起诉讼,即使诉讼如果成功,可能会使我们的股东受益。此外,虽然特拉华州最高法院于2020年3月裁定,根据特拉华州法律,旨在要求根据证券法提出索赔的联邦法院选择条款在表面上是有效的,但其他法院是否会执行我们的联邦论坛条款仍存在不确定性。如果联邦论坛的条款被发现不可执行,我们可能会产生与解决此类问题相关的额外费用。联邦论坛的条款还可能对声称该条款不可执行或无效的股东施加额外的诉讼费用。特拉华州衡平法院和美国特拉华州地区法院也可能做出与其他法院不同的判决或结果,包括考虑诉讼的股东可能位于或将选择提起诉讼的法院,这些判决可能或多或少对我们的股东有利。
如果我们不能建立和保持对财务报告的适当和有效的内部控制,我们的经营业绩和我们经营业务的能力可能会受到损害。
确保我们有足够的内部财务和会计控制和程序,以便我们能够及时编制准确的财务报表,这是一项既昂贵又耗时的工作,需要经常重新评估。我们对财务报告的内部控制是一个旨在为财务报告的可靠性提供合理保证的程序,并根据公认的会计原则编制财务报表。
104
对我们的内部控制实施任何适当的改变可能会分散我们的官员和员工的注意力,需要花费大量成本来修改我们现有的流程,并需要大量时间才能完成。然而,这些变化可能不能有效地维持我们内部控制的充分性,任何未能保持这种充分性,或因此而无法及时编制准确财务报表的情况,都可能增加我们的运营成本,损害我们的业务。此外,投资者认为我们的内部控制不足或我们无法及时编制准确的财务报表,这可能会损害我们的股价,并使我们更难向新老客户有效地营销和销售我们的服务。
如果证券或行业分析师不发表关于我们业务的研究报告,或发表不准确或不利的研究报告,我们的股价和交易量可能会下降。
我们普通股的交易市场在一定程度上取决于证券或行业分析师发布的关于我们或我们业务的研究和报告。如果报道我们的一位或多位分析师下调了我们的股票评级,或者发表了关于我们业务的不准确或不利的研究报告,我们的股价可能会下跌。如果这些分析师中的一位或多位停止对我们公司的报道或未能定期发布有关我们的报告,对我们股票的需求可能会减少,这可能会导致我们的股价和交易量下降。
105
项目1B。取消解析D工作人员评论。
没有。
项目2.新闻歌剧。
我们的公司总部位于马萨诸塞州剑桥市,我们在那里租赁并占用了一栋多租户建筑中约14,000平方英尺的办公空间。我们目前在剑桥的租约将于2026年7月到期。我们还在马萨诸塞州剑桥市的一栋多租户建筑中租赁了约8,000平方英尺的办公空间,我们已将该办公空间的几乎所有剩余租期转租给另一租户。我们相信,我们现有的设施足以满足我们在可预见的未来的需求。
项目3.法律法律程序。
有时,我们可能会卷入诉讼或其他法律程序。我们目前没有参与任何我们管理层认为可能对我们的业务产生重大不利影响的诉讼或法律程序。无论结果如何,由于辩护和和解成本、管理资源转移和其他因素,诉讼可能会对我们的业务、财务状况、运营结果和前景产生不利影响。
第四项:地雷安全TY披露。
不适用。
106
部分第二部分:
项目5.注册人普通股、关联股票的市场持有者很重要,发行者购买股票证券。
我们的普通股在纳斯达克全球精选市场公开交易,代码为“CGEM”。
股东
截至2022年12月31日,我们的普通股共有17个登记在册的股东。我们普通股的实际持有者人数超过了这一记录持有者的数量,包括作为实益所有者的股东,但其股票由经纪商以街头名义持有或由其他被提名者持有。这一数量的登记持有人也不包括其股份可能由其他实体以信托形式持有的股东。
股利政策
我们从未对我们的普通股支付或宣布任何现金股息,我们预计在可预见的未来不会对我们的普通股支付任何现金股息。我们打算保留所有可用资金和未来的任何收益,为我们业务的发展和扩张提供资金。未来是否派发股息将由我们的董事会酌情决定,并将取决于许多因素,包括我们的运营结果、财务状况、未来前景、合同限制、适用法律施加的限制以及我们董事会认为相关的其他因素。
股权补偿计划
表格10-K第5项所要求的有关股权补偿计划的信息,通过引用本年度报告表格10-K第三部分第12项的方式并入本报告。
发行人购买股票证券
在本年度报告(Form 10-K)所涵盖的期间内,吾等并无购买任何注册股本证券。
我们公开发行普通股所得款项的使用
2021年1月7日,我们的S-1表格注册表(注册号:333-251512)被美国证券交易委员会宣布对我们的首次公开募股生效。在2021年1月12日发行结束时,我们出售了13,685,000股普通股,包括承销商全面行使他们的选择权,以每股21.00美元的公开发行价购买最多1,785,000股额外普通股。本次公开发售的净收益总额约为2.645亿美元,包括超额配售以及扣除承销折扣和发售费用后的净收益。
截至2022年12月31日,我们尚未使用首次公开募股的任何净收益。我们已将IPO的净收益投资于货币市场基金和有价证券。根据证券法第424(B)(4)条的规定,我们于2021年1月7日向美国证券交易委员会提交的最终招股说明书中描述了首次公开募股的“收益使用”部分,与注册证券收益的使用有关的信息包含在本文中。我们的最终招股说明书中所描述的募集资金的计划用途没有实质性变化。
项目6.决议草案竖起了。
107
项目7.管理层对以下问题的讨论和分析财务状况及经营业绩。
你应该阅读以下对我们的财务状况和经营结果的讨论和分析,以及我们的综合财务报表和本年度报告中以Form 10-K形式出现的相关附注。本讨论以及本年度报告Form 10-K的其他部分包含涉及风险和不确定性的前瞻性陈述,例如有关我们的计划、目标、预期、意图和预测的陈述。我们的实际结果可能与这些前瞻性陈述中讨论的结果大不相同。可能导致或促成这种差异的因素包括但不限于在本年度报告10-K表格的“风险因素”部分讨论的那些因素。
概述
我们是一家临床阶段的生物制药公司,专注于不可知的靶向肿瘤学。我们的战略是识别高影响的癌症靶点,然后选择我们认为对这些靶点最优的治疗方式。我们从内部和外部寻求创新,专注于拥有新技术平台或差异化机制的候选产品。在我们将候选产品推进到临床开发之前,我们评估其作为单一制剂的抗肿瘤活性的潜力,以及其产生免疫反应或抑制肿瘤过程的能力。使用这一战略,我们已经建立了广泛而深入的有针对性的肿瘤学计划管道,其中包括六个不同的候选产品,其中五个是临床阶段的,以及多个研究和发现计划。
齐帕替尼(CLN-081/TAS6417)是我们与泰豪制药有限公司(“泰豪”)的一家附属公司共同开发的一种口服小分子不可逆表皮生长因子受体(EGFR)抑制剂,旨在选择性地靶向表达EGFR外显子20插入(“EGFRex20ins”)突变的细胞,相对保留表达野生型EGFR的细胞。美国食品和药物管理局(FDA)已授予齐帕替尼突破性疗法称号。在2022年第四季度,我们与我们在泰豪的合作伙伴合作,启动了一项关键的2b期研究,研究对象是EGFR外显子20的非小细胞肺癌(NSCLC)患者,这些患者在之前的系统治疗后进展。2022年6月,大鹏以2.75亿美元的预付款收购了我们部分拥有的子公司库利南珍珠公司(“库利南珍珠”)的股权,该公司为大和提供了日本以外的Zipertinib和大中国的全球权利。作为出售的一部分,我们还有资格获得与EGFR外显子20 NSCLC监管里程碑相关的高达1.3亿美元的额外资金。在完成出售我们在库利南珍珠的股权的同时,我们与泰豪的一家关联公司签订了齐帕尔替尼的共同开发和共同商业化协议,根据协议,我们将合作开发齐帕尔替尼,并将保留在美国(“美国”)共同商业化齐帕尔替尼的选择权。开发成本以及未来可能在美国销售zipertinib的任何税前利润将由我们和泰豪平分。
除了齐帕替尼外,我们的产品组合还包括其他四种临床阶段候选产品和一种正在等待FDA批准其研究新药申请(IND)的候选产品:
除了上述候选产品外,我们还在积极开发几个临床前肿瘤学项目,这些项目都处于发现阶段,包括我们与西奈山伊坎医学院合作开发新型造血祖细胞激酶1降解物。
108
我们拥有CLN-049、CLN-619、CLN-617和CLN-978的全球开发和商业化权利,我们拥有CLN-418的美国开发和商业化权利。我们拥有知识产权和我们早期项目的全球知识产权独家选择权。
自2016年成立以来,我们将所有努力和财务资源集中在筹集资金、组织和配备公司人员、确定、收购或授权和开发产品和技术权利、建立和保护我们的知识产权组合以及开发和推进我们的计划上。我们没有任何产品被批准销售,也没有从产品销售中获得任何收入。
我们主要通过出售股权证券以及向我们的候选产品授权或出售权利来为我们的运营提供资金。截至2022年12月31日,我们从股权融资中获得的净收益为5.412亿美元,其中包括我们首次公开募股(IPO)的净收益2.645亿美元。吾等已从先前与再鼎医药上海有限公司(“再鼎医药”)的许可协议(“ZAI许可协议”)获得1,890,000美元的收入,以及出售吾等于库利南珍珠的股权所得的现金收益27,500,000美元。
截至2022年12月31日,我们拥有现金、现金等价物和短期投资4.673亿美元,长期投资和应收利息8280万美元。应收利息计入综合资产负债表中的预付费用和其他流动资产,代表我们有价证券的应计利息和未付利息。除2022年外,我们自成立以来一直出现运营亏损,运营现金流为负。截至2022年12月31日,我们的累计赤字为4770万美元。除了出售我们在库利南珍珠的股权的一次性收益外,我们预计在可预见的未来将继续产生运营亏损。我们未来的生存能力取决于我们研究和开发的成功,以及我们获得额外资本为我们的运营提供资金的能力。我们不能保证我们目前的运营计划将会实现,也不能保证我们将以我们可以接受的条件提供额外的资金,或者根本不能。
我们受到生物技术行业早期公司常见的风险和不确定因素的影响,包括但不限于新技术创新、对专有技术的保护、对关键人员的依赖、对政府法规的遵守以及获得额外资本以资助运营的能力。我们的治疗计划将需要大量额外的研究和开发努力,包括临床前和临床测试以及商业化之前的监管批准。这些努力需要额外的资本、足够的人员和广泛的合规报告能力。我们不能保证我们的研究和开发将成功完成,我们的知识产权将获得足够的保护,所开发的任何产品将获得必要的政府监管批准,或任何经批准的产品将具有商业可行性。
新冠肺炎大流行的影响
新冠肺炎大流行的持续时间和范围仍不确定。在世界许多地区,不同毒株的毒力和传播仍然很高。新冠肺炎对我们的运营和财务业绩的影响程度和持续时间目前尚不清楚,将取决于未来的发展,这些发展是不确定和不可预测的。我们实施了远程工作和其他保护措施,但到目前为止,我们的运营尚未经历重大中断或延迟,因为这与我们候选产品的临床开发或药物生产有关。然而,新冠肺炎有时会影响我们参加临床试验的速度和临床前研究的进行。未来,与新冠肺炎相关的限制可能会对我们的运营产生不利影响。此类事件可能导致一段时间的业务、供应和药品制造中断,以及业务减少,其中任何一项都可能对我们的业务、财务状况和运营结果产生重大影响。
到目前为止,新冠肺炎还没有对我们产生财务影响。新冠肺炎的蔓延已经在全球范围内造成了广泛的影响,可能会对我们的经济产生实质性的影响。如果近期出现的市场混乱和波动水平持续或恶化,可能会对我们获得资本的能力产生不利影响,这可能在未来对我们的流动性产生负面影响。此外,新冠肺炎传播引发的经济衰退或市场回调可能会对我们的业务造成实质性影响。
列报和合并的基础
自成立以来,我们已创建了全资子公司或对某些受控实体进行了投资。归属于非控股权益的损失在我们的综合经营报表和全面收益(亏损)中单独报告。
当我们还是一家私人公司时,当我们许可或获得我们的几种候选药物的全球独家知识产权时,我们建立了开发子公司,包括CLN-049的库利南佛罗伦萨公司(“库利南佛罗伦萨”),CLN-619的库利南MICA公司(“库利南MICA”),以及CLN-617的库利南琥珀公司(“库利南琥珀”)。我们在我们以前的开发子公司库利南珍珠的股权,该子公司拥有全球权利
109
对日本以外的齐帕替尼和大中华区中国,于2022年第二季度剥离。作为一家上市公司,我们不打算在未来创建新的开发子公司。
以下部分持股子公司合并到我们2022年和2021年的财务报表中:
合并实体 |
|
当前关系 |
|
日期控件 |
|
截至的所有权 |
库利南珍珠公司 |
|
剥离 |
|
2018年11月 |
|
— |
库利南琥珀公司 |
|
部分持股子公司 |
|
2019年12月 |
|
94% |
库利南佛罗伦萨公司 |
|
部分持股子公司 |
|
2019年12月 |
|
96% |
库利南云母公司 |
|
部分持股子公司 |
|
May 2020 |
|
95% |
库利南明珠
2022年6月,我们将部分持股的子公司库利南珍珠的股权出售给了泰豪。有关交易的其他详情,请参阅本年度报告中的综合财务报表附注3(Form 10-K)。
库利南琥珀
Cullinan Amber是我们的部分股权运营子公司,与麻省理工学院(MIT)签订了一项许可协议,提供与Karl Dane Wittrup博士的实验室发明的用于输送免疫刺激药物(如保留在肿瘤微环境中的细胞因子)的新型多功能构建物相关的专利的全球独家权利(“MIT许可协议”)。
2021年6月,我们从库利南琥珀购买了300万股A系列优先股,根据麻省理工学院许可协议,麻省理工学院从库利南琥珀获得了20万股普通股。
2022年6月,我们从库利南琥珀购买了600万股A系列优先股,根据麻省理工学院许可协议,麻省理工学院从库利南琥珀获得了30万股普通股。
2022年11月,我们从库利南琥珀购买了1,000万股A系列优先股,根据麻省理工学院许可协议,麻省理工学院从库利南琥珀获得了50万股普通股,而Wittrup博士根据股权补偿协议从库利南琥珀获得了20万股普通股。
截至2022年12月31日,我们持有普通股和A系列优先股,占库利南琥珀已发行股本的94%。截至2022年12月31日,非控股权益合计持有普通股,占库利南琥珀已发行股本的6%。
佛罗伦萨库利南
根据与德意志银行(DKFZ)、德国图宾根大学医学院的埃伯哈德·卡尔斯大学(Eberhard Karls University of Tübingen)和德国图宾根大学(UFE)的独家许可协议,库利南佛罗伦萨是我们的部分股权运营子公司,拥有CLN-049的独家全球权利,CLN-049是我们针对Flt3和CD3的双特异性抗体。
2021年7月,我们从库利南佛罗伦萨购买了750万股B系列优先股。
2022年7月,我们从库利南佛罗伦萨购买了375万股B系列优先股。
截至2022年12月31日,我们持有普通股、A系列优先股和B系列优先股,占库利南佛罗伦萨已发行股本的96%。截至2022年12月31日,非控股权益合计持有普通股,占库利南佛罗伦萨流通股的4%。
110
库利南云母
库利南MICA,前身为PDI治疗公司,是我们部分拥有的运营子公司,拥有与CLN-619相关的知识产权,CLN-619是我们针对MICA/B的人源化IgG1单抗。
2021年6月,我们从库利南MICA购买了540万股A系列优先股,其他某些现有投资者以90万美元从库利南MICA购买了70万股A系列优先股。
2022年3月,我们从库利南MICA购买了670万股A系列优先股,其他某些现有投资者以120万美元从库利南MICA购买了90万股A系列优先股。
2022年10月,我们从库利南MICA购买了A系列优先股的可转换票据,其他某些现有投资者以20万美元从库利南MICA购买了A系列高级优先股的可转换票据。
2022年10月,我们以3,070万美元从库利南·米卡的两名股东手中购买了150万股库利南·米卡A系列优先股、200万股库利南·米卡A系列初级优先股和1,150万股库利南·米卡A系列A-2初级优先股。
2022年11月,我们以260万美元从库利南米卡的其他五个股东手中购买了40万股库利南米卡普通股和90万股库利南米卡普通股期权。我们还行使了从库利南米卡购买90万股普通股的选择权。
截至2022年12月31日,我们持有库利南米卡95%的完全稀释后流通股,包括96%的其A系列优先股。截至2022年12月31日,非控股权益合计拥有库利南MICA全部稀释后流通股的5%,包括其A系列优先股的4%。
我们运营结果的组成部分
许可证收入
我们自成立以来没有从产品销售中获得任何收入,也不希望在不久的将来从产品销售中获得任何收入,如果有的话。2021年,我们确认了1890万美元的收入,与ZAI许可协议赚取的预付费用有关。
研究和开发费用
研究和开发费用主要包括与我们全资和联合开发的候选产品和计划的研究和开发相关的成本。这些费用包括:
一般和行政费用
一般和行政费用主要包括行政管理、财务、公司和业务发展以及其他行政职能人员的薪酬费用。一般和行政费用还包括与专利和公司事务有关的法律费用;会计、审计、税务和行政咨询服务的专业费用;保险费;行政差旅费用;营销费用和其他运营成本。
我们的一般和行政费用将继续增加,因为我们继续增加我们的员工人数,以支持我们的候选产品和计划的开发以及我们持续的研究活动。
111
库利南珍珠的销售收益
出售库利南珍珠的收益是指所收到的对价超过出售的非金融资产的账面价值。有关交易的其他详情,请参阅本年度报告中的综合财务报表附注3(Form 10-K)。
其他收入
其他收入主要包括从现金、现金等价物、短期投资和长期投资中赚取的利息收入。
所得税
所得税主要由联邦所得税和州所得税组成。
经营成果
2022年和2021年的对比
下表列出了我们2022年和2021年的业务成果:
|
|
截至十二月三十一日止的年度: |
|
|||||
(单位:千) |
|
2022 |
|
|
2021 |
|
||
许可证收入 |
|
$ |
— |
|
|
$ |
18,943 |
|
运营费用: |
|
|
|
|
|
|
||
研发 |
|
|
91,948 |
|
|
|
57,751 |
|
一般和行政 |
|
|
40,189 |
|
|
|
29,146 |
|
总运营费用 |
|
|
132,137 |
|
|
|
86,897 |
|
库利南珍珠的销售收益 |
|
|
276,785 |
|
|
|
— |
|
营业收入(亏损) |
|
|
144,648 |
|
|
|
(67,954 |
) |
其他收入(支出): |
|
|
|
|
|
|
||
利息收入 |
|
|
6,611 |
|
|
|
477 |
|
其他收入(费用),净额 |
|
|
57 |
|
|
|
(8 |
) |
所得税前净收益(亏损) |
|
|
151,316 |
|
|
|
(67,485 |
) |
所得税费用 |
|
|
42,121 |
|
|
|
— |
|
净收益(亏损) |
|
|
109,195 |
|
|
|
(67,485 |
) |
非控股权益应占净亏损 |
|
|
(2,019 |
) |
|
|
(1,915 |
) |
库利南公司普通股股东应占净收益(亏损) |
|
$ |
111,214 |
|
|
$ |
(65,570 |
) |
许可证收入
2021年,我们确认了与从ZAI许可协议赚取的预付费用相关的1890万美元收入。
研究和开发费用
|
|
截至十二月三十一日止的年度: |
|
|||||
(单位:千) |
|
2022 |
|
|
2021 |
|
||
齐帕替尼 |
|
$ |
16,889 |
|
|
$ |
22,723 |
|
CLN-049 |
|
|
6,605 |
|
|
|
6,442 |
|
CLN-619 |
|
|
16,815 |
|
|
|
8,797 |
|
CLN-978 |
|
|
10,822 |
|
|
|
2,805 |
|
CLN-617 |
|
|
12,110 |
|
|
|
2,231 |
|
早期研究 |
|
|
6,894 |
|
|
|
2,073 |
|
其他人员和未分配 |
|
|
10,795 |
|
|
|
3,802 |
|
基于股权的薪酬 |
|
|
11,018 |
|
|
|
8,878 |
|
研发费用总额 |
|
$ |
91,948 |
|
|
$ |
57,751 |
|
在2022年第二季度出售我们在库利南珍珠的股权后,开发成本和未来在美国销售zipertinib的任何潜在税前利润都由我们和泰豪平分。与2021年相比,2022年齐帕替尼的研发费用减少了580万美元,这主要是由于2021年没有在2022年发生的一次性再许可费(300万美元),化学、制造和控制(CMC)成本的下降(270万美元),以及我们与大鹏的共同开发协议带来的好处(90万美元),但被临床前成本的增加(70万美元)部分抵消。
112
与2021年相比,2022年CLN-619的研发费用增加了800万美元,这主要是由于我们进一步参加了我们正在进行的第一阶段剂量递增研究后CRO成本增加(520万美元),以及为获得足够的CLN-619供应以支持当前和未来的临床试验活动而增加的CMC成本(260万美元)。
与2021年相比,2022年CLN-978的研发费用增加了800万美元,这主要是因为CMC为获得足够的CLN-978供应以支持未来的临床试验活动而支付的成本增加(350万美元),支持IND使能活动的临床前活动增加(330万美元),以及由于合同研究组织实现某些监管里程碑而产生的一次性付款(80万美元)。
与2021年相比,2022年CLN-617的研究和开发费用增加了990万美元,这主要是因为CMC获得足够的CLN-617供应以支持当前和未来的临床试验活动的成本增加(550万美元),支持IND使能活动的临床前活动增加(320万美元),以及支持这些增加的活动的与人员相关的成本增加(100万美元)。
与2021年相比,2022年研发费用增加了1,400万美元,这主要是由于早期研究活动的增加(480万美元),增加了员工人数并扩大了运营以支持我们的研发活动(700万美元),以及由于我们增加了员工人数而提高了基于股权的薪酬(210万美元)。
一般和行政费用
与2021年相比,2022年的一般和行政费用增加了1,100万美元,这主要是由于与员工人数增加相关的人员成本增加(350万美元),支持我们扩大业务的专业服务费增加(300万美元),与库利南珍珠销售相关的非经常性成本(200万美元),由于我们增加员工而增加的基于股权的薪酬支出(150万美元),以及设施成本(80万美元)。
库利南珍珠的销售收益
出售库利南珍珠的2.768亿美元收益是所收到的对价超过出售的非金融资产账面价值的部分。有关交易的其他详情,请参阅本年度报告中的综合财务报表附注3(Form 10-K)。
其他收入
与2021年相比,2022年其他收入增加了620万美元,主要是因为利息收入增加。
所得税费用
2022年的所得税支出为4210万美元。2022年确认的所得税支出净额代表库利南珍珠销售收益的税收,包括本年度的利用和某些历史税收属性。
我们在2021年没有记录所得税拨备。
非控股权益应占净亏损
2022年和2021年,可归因于非控股权益的净亏损分别为200万美元和190万美元。非控股权益应占净亏损是在计入部分拥有的附属公司与第三方之间的任何资本交易后,于综合资产负债表中的非控股权益于每个报告期期初与期末的差额厘定。有关部分拥有的附属公司与第三方之间的资本交易的额外详情,请参阅本年度报告(Form 10-K)内的综合财务报表附注8。
流动性与资本资源
概述
除了2022年出售库利南珍珠股权的一次性收益以及自公司成立以来的运营现金流为负外,我们已发生重大运营亏损,预计在可预见的未来将继续产生运营亏损。我们还没有将任何产品商业化,我们预计在几年内不会从产品销售中获得收入,如果有的话。到目前为止,我们的运营资金主要来自出售股权证券以及向我们的候选产品发放许可证或出售权利的收益。截至2022年12月31日,我们拥有现金、现金等价物和短期投资4.673亿美元,长期投资和应收利息8280万美元。
113
根据我们目前的运营计划和假设,我们预计我们目前的现金、现金等价物、短期投资和长期投资将足以为2026年的运营提供资金。我们基于可能被证明是错误的假设做出了这些估计,我们可以比预期更快地利用可用的资本资源。我们不能保证我们将能够以合理的条件筹集额外的资本,或者根本不能。
2022年6月,我们将部分持股的子公司库利南珍珠的股权出售给泰豪,预付款为2.75亿美元。
2022年10月和11月,我们以3330万美元从库利南MICA的其他股东手中收购了库利南MICA的股权,使我们对库利南MICA的持股比例增加到95%。
2023年2月,我们与Harbour BioMed US Inc.(“Harbour”)签订了许可和合作协议(“Harbour许可协议”),根据该协议,Harbour授予我们在美国开发、制造和商业化CLN-418的独家许可。根据Harbour许可协议的条款,我们在签署时向Harbour支付了2500万美元的预付许可费。
现金流
2022年和2021年的对比
下表汇总了我们在所列每个期间的现金来源和用途:
|
|
截至十二月三十一日止的年度: |
|
|||||
(单位:千) |
|
2022 |
|
|
2021 |
|
||
用于经营活动的现金净额 |
|
$ |
(126,664 |
) |
|
$ |
(43,433 |
) |
投资活动提供(用于)的现金净额 |
|
|
248,975 |
|
|
|
(333,775 |
) |
融资活动提供(用于)的现金净额 |
|
|
(25,933 |
) |
|
|
268,784 |
|
现金及现金等价物净增(减) |
|
$ |
96,378 |
|
|
$ |
(108,424 |
) |
经营活动现金流
在2022年,我们将1.267亿美元的现金用于经营活动,主要包括1.321亿美元的运营费用和3780万美元的库利南珍珠销售收益的估计税负付款,部分被2980万美元的非现金费用和我们运营资产和负债净变化带来的112万美元的收益所抵消。非现金费用主要包括基于股权的薪酬支出以及我们有价证券的摊销和增值。
于2021年,我们使用4,340万美元现金进行经营活动,主要包括8,690万美元的营运开支及我们的营运资产及负债净变动350万美元,但由2,760万美元的非现金费用及根据我们与再鼎医药的许可协议收到的1,890万美元预付款部分抵销。非现金费用主要包括基于股权的薪酬支出以及我们有价证券的摊销和增值。
投资活动产生的现金流
在2022年,我们的投资活动提供了2.49亿美元的现金,其中主要包括出售和到期有价证券的3.529亿美元的收益,以及出售库利南珍珠股权的2.75亿美元的收益,但部分抵消了用于购买有价证券的3.779亿美元和用于购买物业和设备的110万美元,以改善我们租赁的办公空间并提供家具。
2021年,我们的投资活动使用了3.338亿美元的现金,其中主要包括用于购买有价证券的5.258亿美元,部分被有价证券的销售和到期日收益1.92亿美元所抵消。
融资活动产生的现金流
2022年,我们的融资活动使用了2,590万美元的现金,其中主要包括收购由非控股权益持有的Cullinan MICA股份所支付的3,330万美元,部分被行使股票期权的收益600万美元和发行非控股权益的收益120万美元所抵消。
在2021年,我们的融资活动提供了2.688亿美元的现金,其中主要包括我们首次公开募股的净收益2.673亿美元和行使股票期权的收益330万美元,但部分被270万美元的递延发行成本的支付所抵消。
未来的资金需求
我们预计,与我们正在进行的活动相关的费用将继续增加,特别是随着我们推进候选产品的临床前活动、制造和临床试验。此外,我们已经并将继续招致
114
与上市公司运营相关的额外成本,包括重大的法律、会计、投资者关系和其他我们作为私人公司没有产生的费用。我们的支出也将增加,因为我们:
根据我们目前的运营计划和假设,我们预计我们目前的现金、现金等价物以及短期和长期投资将足以为2026年前的运营提供资金。我们基于可能被证明是错误的假设做出了这些估计,我们可以比预期更快地利用可用的资本资源。随着我们开发计划和监管审查过程的进展,我们预计将产生与产品制造、商业化前活动和商业化相关的巨额商业化费用。我们还可能需要额外的资金来寻求许可证内或收购其他项目,以进一步扩大我们的渠道。
由于与我们的候选产品和计划的研究、开发和商业化相关的众多风险和不确定性,我们无法估计我们营运资金需求的确切金额。我们未来的资金需求将取决于许多因素,并可能因此而大幅增加,这些因素包括:
115
在我们能够产生足以实现盈利的产品收入之前,我们预计将通过股权发行、债务融资、政府或其他第三方资金、营销和分销安排以及其他合作、战略联盟和许可安排来满足我们的现金需求。在一定程度上,我们通过出售股权来筹集额外资本,目前的所有权权益将被稀释。如果我们通过政府或第三方资金、合作协议、战略联盟、许可安排或营销和分销安排筹集额外资金,我们可能不得不放弃对我们的技术、未来收入来源、研究计划或候选产品的宝贵权利,或者以可能对我们不利的条款授予许可。债务融资如果可行,可能涉及一些协议,其中包括限制或限制我们采取特定行动的能力的契约,如招致额外债务、进行资本支出或宣布股息。如果我们无法在需要时筹集更多资金,我们可能会被要求推迟、限制、减少或终止我们的产品开发或未来的商业化努力,或者授予我们开发和营销我们本来更愿意自己开发和营销的产品或候选产品的权利。
合同义务和其他承诺
根据各种许可和协作协议,我们有一定的付款义务。根据这些协议,我们必须在成功完成和实现某些知识产权、临床、监管和销售里程碑时支付里程碑式的付款。许可和合作协议下的付款义务取决于未来的事件,例如我们是否实现了特定的开发、临床、监管和商业里程碑,我们将被要求就根据这些协议开发的产品的销售支付里程碑和特许权使用费。由于这些未来里程碑付款的实现和时间不可能或不可估量,截至2022年12月31日和2021年12月31日,这些金额尚未包括在我们的综合资产负债表中。
截至2022年12月31日,未来最低租赁付款总额为600万美元,其中190万美元应在12个月内支付。关于我们的租赁义务和预期未来付款的时间,请参阅本年度报告Form 10-K中包含的综合财务报表的附注13。
此外,我们在正常业务过程中与CRO签订临床试验协议,并与其他供应商签订临床前研究、制造服务和用于运营目的的其他服务和产品的协议,这些协议通常可在书面通知下取消。
关键会计政策和估算
我们的综合财务报表是根据美国公认会计原则(“GAAP”)编制的。在编制我们的合并财务报表和相关披露时,我们需要做出影响综合财务报表中资产、负债、成本和费用的报告金额以及或有资产和负债的披露的估计和判断。我们基于历史经验、已知趋势和事件以及我们认为在当时情况下合理的各种其他因素进行估计,这些因素的结果构成了对资产和负债账面价值的判断的基础,而这些资产和负债的账面价值并不是从其他来源很容易看出的。我们在持续的基础上评估我们的估计和假设。在不同的假设或条件下,我们的实际结果可能与这些估计不同。
虽然我们的主要会计政策在本年度报告10-K表格所载的综合财务报表附注2中有更详细的描述,但我们相信以下会计政策对编制综合财务报表所使用的判断和估计最为关键。
收入确认
当客户获得对承诺的商品或服务的控制权时,我们确认收入。确认的收入数额反映了我们预期有权换取这些商品和服务的对价。为了实现这一核心原则,我们采用了以下五个步骤:1)确定客户合同;2)确定合同的履约义务;3)确定交易价格;4)将交易价格分配给履约义务;以及5)在履行履约义务时确认收入。
交易价格是根据我们有权获得的对价来确定的。交易价格可以包括固定金额、可变金额或两者兼有。我们根据相关履约义务的估计独立销售价格来分配交易价格。我们利用关键假设来确定独立销售价格,这可能包括其他可比交易、交易谈判中考虑的定价以及完成各自履约义务的估计成本。我们还利用判断来评估可变对价是否受到限制,或者是否可以专门分配给安排中的一项或多项履约义务。
116
当履行责任已履行时,收入按按相对独立销售价格基准分配给该履行责任的交易价格确认,这不包括受约束的可变对价估计。对于由许可证和其他承诺组成的履约义务,我们利用判断来评估合并的履约义务是否随着时间或在某个时间点得到履行,以及分配给履行义务的交易价格部分的确认模式。于2021年第一季度履行对再鼎医药的履行义务后,我们从与再鼎医药的许可协议中确认了1,890万美元的收入。确认的金额是预付费用减去外国预扣税款,因为这些金额预计不会收回。
研究与开发合同成本和应计项目
研究和开发成本在发生时计入费用。我们记录第三方服务提供商进行的研究和开发活动的估计成本的应计负债,其中包括进行临床前研究、临床试验和合同制造活动。我们根据提供但尚未开具发票的估计服务金额来记录研发活动的估计成本,并将这些成本计入我们合并资产负债表中的应计研发负债中,并将这些成本计入我们的综合经营报表和全面收益(亏损)中的研发费用中。这些成本是我们研发费用的重要组成部分。
我们根据与内部人员和外部服务提供商就服务的进度或完成阶段进行讨论而完成的工作量估计等因素,以及根据与我们的第三方服务提供商就此类服务达成的协议等因素,应计这些费用。我们在确定每个报告期的应计研究和开发负债余额时作出重大判断和估计。随着实际成本的了解,我们调整了应计估计数。尽管我们预计我们的估计不会与实际发生的金额有实质性差异,但所提供服务的状态和时间、参加临床试验的患者数量和患者参保率可能与我们的估计不同,并可能导致我们报告的任何特定时期的金额过高或过低。我们的应计费用在一定程度上取决于从临床研究组织和其他第三方服务提供商收到的及时和准确的报告。我们将对服务提供商的预付款记录为预付资产,在履行合同服务时计入费用。
基于股权的薪酬费用
我们使用蒙特卡罗模拟模型来衡量授予日基于市场的RSU的公允价值。我们使用Black-Scholes期权定价模型来估计股票期权的公允价值。蒙特卡洛模拟模型和Black-Scholes期权定价模型都需要输入客观和主观假设。所使用的某些假设,包括首次公开募股前我们普通股的公允价值和股价波动,代表管理层的估计,涉及内在不确定性以及管理层对可比公司的判断和选择的应用。我们没有足够的普通股历史或隐含波动率数据来估计与我们股票期权奖励的预期期限相称的一段时间内的预期波动率。在此类报告期内,我们根据选定的同类上市公司的普通股估计预期波动率,这些公司拥有足够的历史波动率数据。因此,如果因素发生变化,管理层使用不同的假设,基于股权的薪酬支出对于未来的奖励可能会有实质性的不同。
所得税
所得税按资产负债法核算。递延税项资产及负债因现有资产及负债的账面值及其各自的课税基础与营业亏损及税项抵免结转之间的差额而产生的未来税项影响予以确认。当递延税项资产无法变现的可能性不大时,就需要减少递延税项资产的账面价值。当判断发生变化时,需要判断某些所得税状况是否更有可能持续,并可能在不同时期发生变化。我们记录了2022年的所得税支出,这是由于库利南珍珠销售收益的预期税项,但因释放了本年度预期使用的估值准备和某些历史税项属性而部分抵消了销售收益。由于我们在本财年之前没有盈利历史,管理层决定需要全额估值准备金来抵消截至2022年12月31日的递延税项净资产。
新兴成长型公司和较小的报告状态
2012年4月,《就业法案》颁布。JOBS法案第107条规定,“新兴成长型公司”或EGC可以利用证券法第7(A)(2)(B)条规定的延长过渡期,以遵守新的或修订的会计准则。因此,企业会计准则委员会可以推迟采用某些会计准则,直到这些准则适用于私营公司。在我们仍是一家新兴成长型公司期间,我们已选择将延长的过渡期用于新的或修订的会计准则;然而,我们可能会提前采用某些新的或修订的会计准则。
117
我们将一直是一家新兴的成长型公司,直到(1)财政年度的最后一天(A)在我们首次公开募股(IPO)结束五周年之后,(B)我们的年总收入至少为12.35亿美元,或(C)我们被视为大型加速申报公司,这要求我们非关联公司持有的普通股的市值在前一年6月30日超过7亿美元,以及(2)我们在之前三年期间发行了超过10亿美元的不可转换债券。
我们也是一家“较小的报告公司”,即非关联公司持有的我们股票的市值不到7亿美元,在最近结束的财年中,我们的年收入不到1亿美元,或者非关联公司持有的我们股票的市值不到2.5亿美元。如果(I)非关联公司持有的我们股票的市值低于2.5亿美元,或(Ii)在最近结束的财年中,我们的年收入低于1亿美元,并且非关联公司持有的我们股票的市值低于7亿美元,我们可能会继续成为一家规模较小的报告公司。如果我们当时是一家较小的报告公司,我们不再是一家新兴的成长型公司,我们可能会继续依赖较小的报告公司可以获得的某些披露要求的豁免。具体地说,作为一家较小的报告公司,我们可能会选择在我们的Form 10-K年度报告中只公布最近两个财政年度的经审计财务报表,与新兴成长型公司类似,较小的报告公司减少了有关高管薪酬的披露义务。
近期发布和采纳的会计公告
对最近发布的可能影响我们财务状况和经营结果的会计声明的描述,在本年度报告Form 10-K中包括的综合财务报表的附注2中披露。
118
第7A项。量化与高质关于市场风险的披露。
我们是S-K法规第10项所界定的较小的报告公司,不需要提供这一项所要求的其他信息。
项目8.财务报表S和补充数据。
根据本项目8要求提交的财务报表附在本年度报告的表格10-K之后。这些财务报表的索引载于本年度报告表格10-K的第15项“物证和财务报表附表”。
项目9.与Accou的变更和分歧会计与财务信息披露专业。
在本项目下要求报告的财务披露的任何会计原则或做法问题上,没有更换会计师,也没有与会计师有任何分歧。
第9A项。控制和程序。
信息披露控制和程序的评估
本公司已根据1934年《证券交易法》(修订后的《交易法》)第13a-15(E)和15d-15(E)条规定的规则建立了披露控制程序和程序,旨在确保本公司根据交易法提交或提交的报告中要求披露的信息在美国证券交易委员会规则和表格指定的时间段内被记录、处理、汇总和报告,并被积累并传达给管理层,包括首席执行官(首席执行官)和首席财务官(首席财务官),以便及时做出关于所需披露的决定。此外,披露控制和程序的设计必须反映这样一个事实,即存在资源限制,要求管理层在评估可能的控制和程序相对于其成本的益处时作出判断。
截至2022年12月31日,我们的管理层在首席执行官和首席财务官的参与下,评估了我们的披露控制和程序(如《交易法》第13a-15(E)和15d-15(E)条所定义)的有效性。管理层认识到,任何控制和程序,无论设计和操作多么良好,都只能为实现预期的控制目标提供合理的保证,并且管理层必须在评估可能的控制和程序的成本-效益关系时运用其判断。我们的披露控制和程序旨在为实现其目标提供合理的保证。基于这一评估,我们的首席执行官和首席财务官得出结论,截至2022年12月31日,我们的披露控制和程序(如《交易法》规则13a-15(E)和15d-15(E)中定义的)在合理保证水平下有效。
管理层财务报告内部控制年度报告
我们的管理层负责建立和维护对我们财务报告的充分内部控制。对财务报告的内部控制在《交易法》下的规则13a-15(F)和15d-15(F)中定义为由我们的首席执行官和我们的首席财务官设计或监督,并由我们的董事会、管理层和其他人员实施的程序,以提供关于我们财务报告的可靠性和根据GAAP为外部目的编制我们的财务报表的合理保证,并包括以下政策和程序:
由于其固有的局限性,财务报告的内部控制可能无法防止或发现错误陈述。此外,对未来期间的有效性进行任何评估的预测都有这样的风险,即由于条件的变化,控制可能会变得不充分,或者遵守政策或程序的程度可能会恶化。
我们的管理层根据特雷德韦委员会赞助组织委员会发布的《内部控制-综合框架(2013)》中提供的框架,评估了我们对财务报告的内部控制的有效性。根据这项评估,我们的管理层得出结论,我们对财务报告的内部控制自2022年12月31日起有效。
119
作为JOBS法案中定义的新兴成长型公司,我们的独立注册会计师事务所不需要出具财务报告内部控制的认证报告。
财务报告内部控制的变化
在截至2022年12月31日的财政季度内,我们对财务报告的内部控制(根据《交易法》第13a-15(F)或15(D)-15(F)条的定义)没有发生重大影响或合理地可能对我们的财务报告内部控制产生重大影响的变化。
项目9B。其他信息。
没有。
项目9C。披露妨碍检查的外国司法管辖区。
没有。
120
第三部分
项目10.董事、高管职务ICERS与公司治理。
关于我们的执行官员的信息
下表列出了我们每一位高管的姓名、截至2023年2月15日的年龄和职位。
名字 |
|
库利南肿瘤科的职位 |
|
警员自 |
|
年龄 |
纳迪姆·艾哈迈德 |
|
董事首席执行官总裁 |
|
2021 |
|
55 |
帕特里克·鲍尔,博士。 |
|
首席科学官 |
|
2016 |
|
65 |
杰弗里·琼斯医学博士 |
|
首席医疗官 |
|
2022 |
|
52 |
詹妮弗·迈克尔森博士。 |
|
首席发展官 |
|
2018 |
|
56 |
科琳·萨维尔,博士。 |
|
首席商务官 |
|
2017 |
|
63 |
Jacquelyn Sumer,Esq. |
|
首席法律官、首席合规官兼秘书 |
|
2022 |
|
45 |
杰弗里·特里格里奥 |
|
首席财务官兼财务主管 |
|
2020 |
|
39 |
纳迪姆·艾哈迈德自2021年10月以来,艾哈迈德先生一直担任我们的总裁和首席执行官兼董事会成员。在加入本公司之前,Ahmed先生于2019年11月至2021年1月在百时美施贵宝(“百时美施贵宝”)担任血液科主任总裁。在此之前,他从2010年3月至2019年11月在Celgene Corporation(“Celgene”)担任越来越多的职责,最近的职务是全球血液与肿瘤学总裁。在Celgene任职之前,Ahmed先生从1998年6月到2010年3月在葛兰素史克(“葛兰素史克”)担任越来越多的职责,最近担任的职务是董事血液肿瘤学特许经营部高级营销经理。艾哈迈德先生拥有拉夫堡大学的理学硕士学位和伦敦大学学院的理学学士学位。我们相信,艾哈迈德先生有资格担任我们的董事会成员,因为他在生物制药行业拥有丰富的领导经验。
帕特里克·鲍尔,博士。Baeuerle博士是该公司的联合创始人,自2022年4月以来一直担任我们的首席科学官。此前,他在2016年9月至2022年4月期间担任公司生物制品代理首席科学官。Baeuerle博士目前还担任医疗保健投资公司MPM Capital的执行合伙人、Cross弓治疗公司的代理首席科学官和Aktis Oncology,Inc.的科学顾问。在加入该公司之前,Baeuerle博士在2012年3月至2015年3月期间担任安进慕尼黑有限公司研究副总裁兼总经理总裁。在此之前,他于1998年10月至2012年3月担任Micromet,Inc.的首席科学官,并在被安进收购的上市生物技术公司Tularik Inc.领导小分子药物发现。Baeuerle博士于1994年至1996年担任弗莱堡大学医学院教授兼生物化学主席,在那里他对转录因子NF-kappaB进行了开创性的研究。他与他人共同创立了MPM的肿瘤学公司Harpoon Treateutics,Inc.、TCR2 Treateutics,Inc.(“TCR2”)、iOmx Treateutics AG(“iOmx”)、Maverick Treateutics,Inc.和Werewolf Treateutics,Inc.,目前在TCR2和iOmx的董事会任职。Baeuerle博士拥有理学学士学位。他在慕尼黑大学获得生物学博士和博士学位,并在麻省理工学院怀特黑德研究所与巴尔的摩的David博士一起进行博士后研究。
杰弗里·琼斯医学博士琼斯博士自2022年2月以来一直担任我们的首席医疗官。在加入本公司之前,Jones博士曾于2020年4月至2022年2月在拜耳医疗服务公司担任全球药物开发、淋巴瘤和骨髓疾病部副主任总裁,并于2017年8月至2020年4月在拜耳医疗服务公司担任全球临床研究与开发部医学董事主管。在此之前,琼斯博士曾担任俄亥俄州立大学医学院临床内科副教授。在此之前,琼斯博士于2006年至2014年在俄亥俄州立大学担任临床讲师。琼斯博士在密歇根州安娜堡的密歇根大学医学院获得医学博士学位,并在蒙特利尔的麦吉尔大学医学院完成内科住院医师资格,并在德克萨斯州休斯敦的MD Anderson癌症中心完成血液学和肿瘤学研究员学位。琼斯博士还拥有俄亥俄州立大学费舍尔商学院的工商管理硕士学位和德克萨斯大学公共卫生学院的公共卫生硕士学位。
詹妮弗·迈克尔森博士。迈克尔森博士自2022年2月以来一直担任我们的首席开发官。此前,她于2020年1月至2022年2月担任公司生物制品首席开发官,并于2018年1月至2019年12月担任公司临床前研究和早期开发副总裁。在加入公司之前,迈克尔森博士在2017年7月至2017年12月期间担任生物科技公司Celsius Treateutics,Inc.的生物制品部门负责人。在此之前,她从2012年9月到2017年7月在乔斯治疗公司担任越来越多的责任职位,最近担任董事高级和执行项目负责人,之前担任肿瘤免疫学董事和顾问。在此之前,在Biogen IDEC Inc.任职的10年间,Michaelson博士曾担任肿瘤学和免疫学治疗领域的几个单抗和双特异性抗体项目的项目负责人。迈克尔森博士拥有普林斯顿大学生物学学士学位和阿尔伯特·爱因斯坦医学院细胞生物学博士学位,并在哈佛医学院遗传学系菲利普·莱德的实验室完成了博士后研究。
121
科琳·萨维尔,博士。萨维尔博士自2017年2月以来一直担任我们的首席商务官。在加入公司之前,Savill博士在诺华制药公司(“Novartis”)担任越来越多的职责,包括于2013年6月至2017年2月担任全球业务开发及许可部主管,于2010年9月至2013年6月担任定价及市场准入全球主管,于2005年1月至2010年8月担任全球搜索及评估部主管、业务发展及许可部主管,并于2002年8月至2005年1月担任移植及免疫业务部欧洲区域经理。在诺华任职之前,萨维尔博士是英国生物技术公司Imutran的首席执行官,该公司被诺华收购。她之前还曾在阿斯利康从事研究工作。萨维尔博士拥有曼彻斯特大学生物化学学士学位,并在大学学院、米德尔塞克斯医学院和伦敦查令十字森利研究中心获得博士学位。
Jacquelyn Sumer,Esq.苏默尔女士自2022年8月以来一直担任我们的首席法律干事。在加入本公司之前,Sumer女士于2021年2月至2022年6月在Genocea Biosciences,Inc.(“Genocea”)担任首席法律和合规官。在加入Genocea之前,Sumer女士于2019年11月至2021年2月担任BMS助理总法律顾问总裁副总经理。在此之前,她曾于2018年7月至2019年11月担任Celgene的CAR T法律团队负责人。她之前曾在Kaye Scholer,LLP工作,并在华盛顿特区的美国地区法院为Gladys Kessler阁下担任书记员。Sumer女士拥有杜克大学法学院的法学博士和国际法和比较法硕士学位,以及巴克内尔大学的学士学位。
杰弗里·特里格里奥Trigilio先生自2020年9月以来一直担任我们的首席财务官。在加入公司之前,Trigilio先生在2020年1月至2020年7月期间担任制药公司Amylyx PharmPharmticals,Inc.的首席财务官。在此之前,他于2018年8月至2020年1月在蓝石治疗公司(以下简称蓝石公司)担任企业财务副总裁总裁。在加入Bluerock之前,Trigilio先生于2017年11月至2018年8月在加拿大皇家银行资本市场有限责任公司担任董事医疗保健投资银行业务负责人。2013年4月至2017年11月,他曾在Alexion PharmPharmticals,Inc.担任越来越多的职责,2008年7月至2013年3月,他在瑞士信贷证券(Credit Suisse Securities)任职。特里格里奥先生拥有康奈尔大学的工业和劳动关系学士学位和哥伦比亚大学的工商管理硕士学位。
在过去五年中,我们每一位高管的主要职业和工作都是在一个不是我们的母公司、子公司或其他附属公司的公司或组织进行的,除非本年度报告中明确指出了这一点。我们的任何执行官员与任何其他人之间没有任何安排或谅解,根据这些安排或谅解,他或她将被选为执行官员。
本公司并无任何行政人员为不利本公司或本公司附属公司的一方的重大法律程序,或任何该等人士在该等诉讼中拥有对本公司或本公司附属公司不利的重大利益。
董事
下表列出了截至2023年2月15日我们每位非雇员董事的姓名和年龄。
名字 |
|
董事自 |
|
年龄 |
托马斯·埃贝林 |
|
2017 |
|
64 |
安妮-玛丽·马丁,博士。 |
|
2022 |
|
51 |
安东尼·罗森伯格 |
|
2017 |
|
69 |
David·P·瑞安医学博士。 |
|
2022 |
|
56 |
斯蒂芬·韦伯斯特 |
|
2020 |
|
61 |
托马斯·埃贝林Ebeling先生自2017年8月以来一直担任我们的董事会成员。他还担任MPM的顾问。自2018年以来,他一直担任咨询和高管培训公司TE Convest AG的首席执行官。在此之前,Ebeling先生于2009年3月至2017年2月期间担任大众媒体公司ProSieben Media SE的首席执行官。Ebeling先生曾于2007年9月至2008年9月担任诺华消费者健康公司的首席执行官,并于2000年7月至2007年9月担任诺华制药公司的首席执行官。从1991年到1996年,他在百事可乐德国公司担任过多个领导职务。他是启根公司和奥纳治疗公司的董事会成员。此外,他目前在几家私人公司的董事会任职。此前,Ebeling先生于2012年4月至2019年9月担任拜耳股份公司董事会成员,并于2013年3月至2017年3月担任龙沙集团董事会成员。Ebeling先生拥有汉堡大学心理学学士学位。我们相信,Ebeling先生有资格担任我们的董事会成员,因为他在生命科学行业拥有丰富的领导经验。
122
安妮-玛丽·马丁,博士。马丁博士自2022年3月以来一直担任我们的董事会成员。马丁博士目前担任葛兰素史克实验医学部全球负责人高级副总裁,自2020年8月以来一直担任这一职位。2016年2月至2020年7月,她曾担任诺华精密医学全球主管高级副总裁。在加入诺华之前,她于2015年5月至2016年2月担任Adaptimmune治疗公司生物标记物研究和诊断开发副总裁总裁。在加入Adaptimmune之前,马丁博士在2005年3月至2015年4月期间在葛兰素史克担任过越来越多的职责。此外,她在2019年8月至2020年7月期间担任私营生物技术公司Freenome Holdings,Inc.的董事会观察员。她在谢菲尔德哈勒姆大学获得生物医学学士学位,并在MCP-Hahnemann大学获得免疫遗传学博士学位。我们相信,马丁博士有资格在我们的董事会任职,因为她在生物技术和制药领域拥有丰富的经验。
安东尼·罗森伯格罗森博格先生自2017年8月以来一直担任我们的董事会成员,并自2020年4月以来担任我们的董事会主席。目前,罗森博格先生担任TRConsulting Services GmbH的首席执行官,该公司是一家咨询公司,为业务发展、许可和并购提供咨询。2015年4月至2020年4月,罗森博格先生担任MPM董事董事总经理。2012年1月至2015年2月,罗森博格先生担任诺华公司并购和许可业务主管。罗森博格先生目前在Argenx SE的董事会任职。在此之前,罗森博格先生是Radius Health,Inc.的董事会成员。他还在两家私人公司的董事会任职。罗森博格先生拥有理学学士学位。从莱斯特大学获得理科硕士学位。伦敦大学生理学专业。我们相信,罗森博格先生有资格担任我们的董事会成员,因为他在生物技术运营和战略管理方面拥有丰富的经验。
David·P·瑞安医学博士。瑞安博士自2022年11月以来一直担任我们的董事会成员。自2012年以来,Ryan博士一直担任临床董事和马萨诸塞州总医院癌症中心主任。自1998年以来,他在MGH担任的职责越来越多,专门从事癌症患者的研究和治疗。此外,瑞安博士目前是哈佛医学院癌症治疗领域的谢尔比纪念医学教授。瑞安博士也是美国临床肿瘤学会的成员,并担任MPM和生物影响资本的顾问,BioImpact Capital是MPM的附属管理公司。瑞安博士拥有哥伦比亚内科和外科医学院的医学博士学位和圣十字学院的学士学位。我们相信,瑞安博士有资格担任我们的董事会成员,因为他在肿瘤学临床研究方面拥有丰富的经验。
斯蒂芬·韦伯斯特韦伯斯特先生自2020年9月以来一直担任我们的董事会成员。韦伯斯特从2014年7月开始担任基因治疗公司Spark Treateutics,Inc.的首席财务官,直到2019年12月被罗氏控股公司以48亿美元收购。从2012年7月到2013年10月被立体主义制药公司收购之前,他曾是Optimer PharmPharmticals Inc.的高级副总裁和首席财务官。韦伯斯特先生目前在NextCure公司、Nabriva治疗公司(前身为Nabriva治疗公司)和TCR2公司的董事会任职。韦伯斯特在达特茅斯学院获得经济学学士学位,在宾夕法尼亚大学沃顿商学院获得金融MBA学位。我们相信韦伯斯特先生有资格担任我们的董事会成员,因为他在生物制药行业有丰富的经验,包括他以前担任首席财务官和其他管理职位的经验。
我们的任何董事或高管之间都没有家族关系。在过去五年中,我们每一位董事的主要职业和工作都是在一个不是我们的母公司、子公司或其他关联公司的公司或组织进行的,除非在本年度报告10-K表格中明确指出的情况除外。我们的任何董事与任何其他人士之间并无任何安排或谅解,据此他或她将被选为董事。
吾等并无任何董事为不利吾等或吾等任何附属公司的一方的重大法律程序,或任何该等人士在该等诉讼中拥有不利吾等或吾等附属公司的重大权益。
商业行为和道德准则
我们已经通过了适用于我们的董事、高级管理人员和员工的书面商业行为和道德准则,包括我们的首席执行官、首席财务官、首席会计官、财务总监或执行类似职能的人员。代码的最新副本发布在我们网站的公司治理部分,该网站位于https://investors.cullinanoncology.com/documents-charters.如果我们对任何高级管理人员的商业行为和道德准则进行任何实质性修订或给予任何豁免,我们将在我们的网站或当前的Form 8-K报告中披露此类修订或豁免的性质。
股东提名程序
我们之前披露的股东可以推荐董事会候选人的程序没有实质性的变化。
123
审计委员会
我们的董事会成立了一个审计委员会。托马斯·埃贝林、安东尼·罗森伯格和斯蒂芬·韦伯斯特是审计委员会的成员,该委员会由斯蒂芬·韦伯斯特担任主席。我们的董事会已确定,就审计委员会而言,审计委员会的每位成员都是“独立的”,因为该词由美国证券交易委员会(“美国证券交易委员会”)和纳斯达克证券市场有限责任公司(“纳斯达克”)的规则定义,并且每位成员都拥有足够的金融和审计知识来担任审计委员会成员。我们的董事会已经指定斯蒂芬·韦伯斯特为“审计委员会财务专家”,这是根据美国证券交易委员会的适用规则定义的。
拖欠款项第16(A)条报告
交易法第16(A)条要求我们的董事、高管和持有我们普通股超过10%的实益所有者向美国证券交易委员会提交公司持有和交易证券的报告。美国证券交易委员会已经为这些报告指定了具体的截止日期,我们必须在我们的Form 10-K年度报告的这一部分中指出哪些人没有在到期时提交这些报告。
仅根据对就最近一个财政年度以电子方式向美国证券交易委员会提交的表格3、4和5及其任何修正案以及报告人的书面陈述的审查,我们相信2022年所有第16(A)条的备案要求都已满足,但Corinne Savill于2022年7月21日提交的表格4除外,该表格报告了2022年7月18日出售5,042股普通股的情况。
项目11.行政人员E补偿。
2022年和2021年向我们指定的执行干事提供的薪酬详细载于薪酬汇总表以及紧随其后的脚注和说明。我们提名的2022年高管包括在2022年担任我们首席执行官的个人,以及在上一个财政年度结束时担任高管的接下来两名薪酬最高的高管,他们是:
薪酬汇总表
下表列出了在以下列出的年度内,我们的指定执行官员因以各种身份向我们提供服务而获得的或支付给我们的总薪酬。
名称和主要职位 |
|
年 |
|
薪金 |
|
|
奖金(1) |
|
|
股票大奖(2) |
|
|
期权大奖(2) |
|
|
所有其他补偿 |
|
|
总计 |
|
||||||
纳迪姆·艾哈迈德(3) |
|
2022 |
|
$ |
618,000 |
|
|
$ |
370,800 |
|
|
$ |
2,762,750 |
|
(4) |
$ |
— |
|
|
$ |
148,977 |
|
(6) |
$ |
3,900,527 |
|
总裁与首席执行官 |
|
2021 |
|
$ |
125,000 |
|
|
$ |
699,000 |
|
|
$ |
— |
|
|
$ |
37,723,200 |
|
|
$ |
— |
|
|
$ |
38,547,200 |
|
帕特里克·鲍尔,博士。 |
|
2022 |
|
$ |
463,500 |
|
|
$ |
266,234 |
|
|
$ |
510,000 |
|
(5) |
$ |
686,250 |
|
|
$ |
34,000 |
|
(7) |
$ |
1,959,984 |
|
首席科学官 |
|
2021 |
|
$ |
450,000 |
|
|
$ |
218,000 |
|
|
$ |
— |
|
|
$ |
3,973,177 |
|
|
$ |
34,800 |
|
(7) |
$ |
4,675,977 |
|
杰弗里·特里格里奥 |
|
2022 |
|
$ |
452,000 |
|
|
$ |
260,352 |
|
|
$ |
510,000 |
|
(5) |
$ |
686,250 |
|
|
$ |
15,634 |
|
(8) |
$ |
1,924,236 |
|
首席财务官 |
|
2021 |
|
$ |
400,000 |
|
|
$ |
220,000 |
|
|
$ |
— |
|
|
$ |
2,032,777 |
|
|
$ |
14,858 |
|
(8) |
$ |
2,667,635 |
|
124
薪酬汇总表说明
概述
我们的高管薪酬计划旨在吸引、留住和奖励关键员工,并使他们的利益与我们股东的利益保持一致。我们的首席执行官向我们的薪酬委员会建议他的直接下属的薪酬(除了他自己的薪酬),我们的薪酬委员会负责确定我们高管的薪酬。
直到2022年11月,我们的薪酬委员会一直聘请Radford作为其独立的薪酬顾问,随后聘请Compensia担任这一职务。雷德福和康彭西亚都是独立的薪酬咨询公司,视情况而定,协助或协助薪酬委员会评估公司高管和董事的薪酬实践,包括方案设计,为薪酬比较确定合适的同行群体,以及提供具有竞争力的市场薪酬数据。在与雷德福和康彭西亚各自聘用之前,我们的薪酬委员会评估了雷德福和康彭西亚各自独立于管理层的情况,并在评估的基础上,考虑了适用的证券交易所上市标准和美国证券交易委员会规则所要求考虑的独立性因素后,认定雷德福或康彭西亚所做的任何工作都不会产生利益冲突,也不会损害雷德福或康彭西亚的独立性。
基本工资
2022年和2021年期间,艾哈迈德先生的年基本工资分别为61.8万美元和60万美元。2022年和2021年,鲍耶勒的年化基础咨询费分别为46.4万美元和45万美元。在2022年和2021年期间,特里吉里奥的年基本工资分别为45.2万美元和40万美元。
奖金安排
根据艾哈迈德的雇佣协议条款,他在2022年和2021年的目标奖励奖金都是基本工资的50%。根据薪酬委员会和董事会对他在2022年至2021年期间表现的评估,艾哈迈德分别获得了相当于基本工资60%和59%的可自由支配奖金。2021年,他的奖金按比例计算,以反映他受雇于本公司的那段时间。此外,根据雇用协议的条款,2021年,艾哈迈德先生获得了625 000美元的一次性签到奖金。
根据鲍埃勒博士的咨询协议条款,他的目标激励奖金是2022年和2021年年化基本咨询费的40%。根据薪酬委员会对他在2022年至2021年期间表现的评估,鲍耶博士获得了相当于其年化基本咨询费的47%和48%的可自由支配奖金。
根据特里吉里奥的雇佣协议条款,他在2022年和2021年的目标奖金都是基本工资的40%。根据薪酬委员会对他在2022年和2021年期间表现的评估,特里吉里奥获得了相当于基本工资的48%和55%的可自由支配奖金。
125
此外,作为一项在全体员工中广泛实施的计划的一部分,鲍耶尔博士和特里格里奥先生还获得了留任奖金。留任计划的目的是确保过渡期内劳动力的稳定和连续性,特别是关键职位,因为截至2021年12月31日,该公司有31名全职员工。根据2022年2月签订的补充奖金协议,Baeuerle博士和Trigilio先生各自有权获得与继续为本公司服务至2023年8月相关的现金留任奖金。奖金总额按照以下支付时间表分期支付:2022年8月20%,2023年2月40%,2023年8月剩余40%。2022年,鲍尔博士和特里吉利奥分别获得了46350美元和4.52万美元的奖金,以表彰他们继续服务到2022年8月的支付日期。
股权补偿
根据我们的2021年股票期权和激励计划(“2021年股票计划”),我们的员工和高管有资格获得股票期权和其他基于股票的奖励。
虽然我们没有向高管授予股权激励奖励的正式政策,但我们相信,股权奖励为我们的高管提供了与我们的长期业绩密切相关的联系,创造了一种所有权文化,并有助于协调我们高管和股东的利益。此外,我们认为,具有多年归属要求的股权奖励有助于留住高管,因为它们激励我们的高管在归属期间继续留任。我们的薪酬委员会定期审查我们的高管(包括我们任命的高管)的股权激励薪酬,并可能不定期向他们颁发股权激励奖励。我们通常在高管开始受雇时以股票期权的形式授予股权奖励,此后每年以股票期权和RSU的组合形式授予股权奖励。我们将期权行权价格设定为授予日普通股股票的收盘价(如果授予日没有收盘价,则为紧接报告收盘价的前一天)。
2022年3月5日,根据雇用协议的条款,艾哈迈德先生获得了以市场为基础的RSU,即有权在结算时获得215,000股我们的普通股。受制于基于市场的RSU的股票数量代表归属时可发行的股票数量,假设公司在目标业绩水平上实现了公司股票价格指标。奖励授予时可发行的股份数量(如果有的话)将取决于公司股价指标在归属日期的实现程度,范围在目标股份数量的0%至200%之间。基于市场的RSU将在授予之日起三年内授予和结算,金额将根据当时我们普通股的每股价格确定。如本公司控制权于归属日期前发生变更,则以市场为基础的RSU须加速归属。2021年10月18日,在艾哈迈德先生开始受雇于我们时,他获得了以每股21.12美元的行使价购买2,710,000股我们普通股的选择权,行权开始日期为2021年10月18日。认购权相关股份的四十八分之一按月归属,但须持续服务至每个归属日期。此购股权的未授出部分须因本公司控制权变更(亦称为“出售事件”)或吾等无故终止Ahmed先生的雇用而加速归属(每项事项均见2021年股票计划的定义)。
2022年2月11日,Baeuerle博士被授予(I)以每股13.60美元的行使价购买75,000股我们普通股的选择权和(Ii)RSU代表在结算时获得37,500股我们普通股的权利(“2022 Bauerle Equity Awards”)。2022年Baeuerle Equity Awards相关股票的四十八分之一每月归属,但在每个归属日期继续服务。2021年1月7日,就我们首次公开招股前与重组相关的股权交换,Baeuerle博士获得(I)以每股4.30美元的行使价购买262,114股我们普通股的期权,归属开始日期为2018年9月1日;以及(Ii)以每股4.30美元的行权价购买我们普通股407,993股的期权,归属开始日期为2019年5月15日,(统称为“Baeuerle股权交换期权”)。2021年1月7日,Baeuerle博士还获得了以每股21.00美元的行使价购买286,665股我们普通股的期权,反映了我们普通股在该日期的首次公开募股价格,归属开始日期为2021年1月7日(“Baeuerle IPO期权”,与Baeuerle Equity Exchange期权一起,“2021 Baeuerle期权”)。就每项2021 Baeuerle购股权而言,25%的购股权相关股份于归属开始日期一周年归属,其余股份于其后按月按月分36期等额归属,直至每个归属日期持续提供服务。2022年Baeuerle股权奖和2021年Baeuerle期权的未归属部分因公司控制权的变更而加速归属,也称为“出售事件”, 或由我们无故终止Baeuerle博士的咨询关系(每一项均在2021年股票计划中定义)。
126
2022年2月11日,Trigilio先生被授予(I)以每股13.60美元的行使价购买75,000股我们普通股的选择权,以及(Ii)代表有权在结算时获得37,500股我们普通股的RSU(“2022 Trigilio Equity Awards”)。2022年Trigilio Equity Awards相关股份的四十八分之一每月归属,但在每个归属日期继续服务。于2021年1月7日,就本公司首次公开招股前与重组有关的股权交换事宜,Trigilio先生获授予一项选择权,可按行使价每股4.30美元购买本公司普通股252,891股,归属开始日期为2020年9月8日(“Trigilio股权交换期权”)。2021年1月7日,Trigilio先生还获得了以每股21.00美元的行使价购买146,665股我们普通股的期权,反映了我们普通股在该日期的首次公开募股价格,归属开始日期为2021年1月7日(“Trigilio IPO期权”,以及Trigilio股权交换期权,“2021 Trigilio期权”)。就2021年Trigilio购股权而言,25%的购股权相关股份于归属开始日期一周年归属,其余股份于其后按月分36期等额归属,但须持续服务至每个归属日期。2022年Trigilio股权奖和2021年Trigilio期权的未归属部分因本公司控制权的变更(也称为“出售事件”)或吾等无故终止Trigilio先生的雇用(定义见2021年股票计划)而加速归属。
与我们指定的高级管理人员签订雇佣协议和服务协议
我们已经与我们任命的每一位高管签订了雇佣协议或服务协议。每份协议都规定了这些高管的基本工资或基本咨询费(视情况而定)、目标奖金百分比以及参加我们福利计划的一般资格。艾哈迈德先生和特里吉利奥先生还须遵守保密、委派和禁止邀约协议,其中规定了一项永久的离职后保密协定,以及在雇用期间和离职后一年内适用的雇员和咨询人不得邀约的协定。2020年12月,我们还与Baeuerle博士签订了一项新的服务协议,该协议在我们首次公开募股时生效。下文提及的“原因”、“充分理由”和“控制权变更”等术语在每个指定的执行干事的雇用协议或服务协议中作了适当的定义。
纳迪姆·艾哈迈德
2021年10月,我们与艾哈迈德先生签订了聘用协议,艾哈迈德先生目前担任我们的总裁兼首席执行官。雇佣协议规定了年度基本工资,由我们的董事会或薪酬委员会定期审查,以及相当于Ahmed先生年度基本工资的50%的年度目标奖金,任何奖金的实际金额由我们的董事会或薪酬委员会根据里程碑决定。根据雇用协议,Ahmed先生收到了625 000美元的签约奖金,如果他非正当理由辞职或被我们以正当理由解雇,在他开始受雇后18个月内(任何还款义务在一周年后按比例分摊),这笔奖金由他偿还。Ahmed先生还有权获得每月6,000美元的住房津贴,以获得临时住房,直至他开始工作四周年或他不再需要临时住房之日的较早者。雇佣协议还规定,只要艾哈迈德先生仍然是我们的首席执行官,他将被提名参加我们的董事会选举,并向我们的股东推荐选举进入我们的董事会。
根据艾哈迈德先生的雇用协议,如果我们无故终止艾哈迈德先生的雇用,或艾哈迈德先生因正当理由辞职,但须遵守离职协议的执行和效力,包括全面解除对我们有利的索赔,并由公司自行决定签订为期一年的离职后竞业禁止协议,他将有权获得:(1)相当于当时12个月基本工资的数额,外加按比例计算的年度奖金部分,以当年的实际业绩为基础;和(Ii)如果Ahmed先生在紧接他被解雇之前参加了我们的集团健康计划,并选择了COBRA继续健康保险,则继续这种集团健康保险的费率与他是在职雇员的费率相同,直到(A)他被解雇12个月周年,(B)他根据任何其他雇主的集团医疗计划有资格获得集团医疗计划福利,或(C)他根据COBRA继续享有权利。雇佣协议还规定,作为上述付款和福利的替代,如果艾哈迈德先生的雇佣被我们无故终止,或艾哈迈德先生在公司控制权变更后12个月内辞职,取决于对我们有利的全面索赔的执行和有效性,他将有权获得:(1)相当于其当时的基本工资(或紧接控制权变更之前有效的基本工资,如果更高)的数额加上按比例计算的年度奖金,根据本年度的实际业绩,及(Ii)相当于本公司于Ahmed先生额外雇用24个月时应支付予其集团健康计划提供者或眼镜蛇提供者作为每月雇主供款的保费。此外, 如果我们或他在公司控制权变更后12个月内无故或有充分理由终止雇用Ahmed先生,所有仅基于时间授予的未偿还股权奖励将被完全授予,任何基于业绩的未偿还RSU将根据Ahmed先生在适用的履约期间的受雇期间按比例授予。
127
帕特里克·鲍尔,博士。
关于我们2021年的首次公开募股,我们与目前担任我们代理首席科学官的Baeuerle博士签订了一项服务协议。本服务协议取代Baeuerle博士与本公司于2019年1月1日签订的先前咨询协议。服务协议规定固定的年化咨询费,由我们的董事会或薪酬委员会定期审查,以及相当于Baeuerle博士年化咨询费的40%的年度目标奖金,应支付的任何奖金的实际金额由我们的董事会或薪酬委员会全权决定,符合公司任何适用的激励性薪酬计划的条款。
根据Baeuerle博士的服务协议,如果Baeuerle博士的聘用被我们无故终止,或者Baeuerle博士有正当理由辞职,取决于分居协议的执行和有效性,包括对我们有利的全面索赔释放,他将有权获得相当于当时年化咨询费的9个月的金额,外加按比例计算的年度目标奖金部分。服务协议还规定,作为上述付款的替代,如果我们无故终止Baeuerle博士的聘用,或Baeuerle博士有充分理由辞职,在公司控制权变更后12个月内,取决于对我们有利的全面索赔的执行和有效性,他将有权获得相当于(I)当时年化咨询费(或其在紧接控制权变更前生效的年化咨询费,如果更高)的12个月加上(Ii)当时本年度的年度目标奖金(或紧接控制权变更前生效的年度目标奖金,如果更高)的一笔现金付款。
服务协议还规定,如果Baeuerle博士的聘用因其死亡或残疾而终止,除非奖励协议中另有规定,证明以业绩为基础归属的股权奖励,每一笔未归属股权奖励的25%,加上为本公司提供的每一整年服务的额外5%,将立即加速并于终止日变得完全归属、可行使或不可没收。服务协议还规定,如果Baeuerle博士的聘用被我们无故终止或Baeuerle博士有充分理由辞职,在公司控制权变更后12个月内,每笔未偿还的股权奖励将立即加速,并在终止之日变得完全归属、可行使或不可没收。
杰弗里·特里格里奥
关于我们2021年的首次公开募股,我们与目前担任我们首席财务官的Triglio先生签订了一份新的雇佣协议。他的雇佣协议规定,Trigilio先生的年度基本工资由我们的董事会或薪酬委员会定期审查,年度目标奖金相当于Trigilio先生年度基本工资的40%,应支付的任何奖金的实际金额由我们的董事会或薪酬委员会全权决定,符合本公司任何适用的激励性薪酬计划的条款。
根据Trigilio先生的雇佣协议,如果Trigilio先生的雇佣被我们无故终止,或Trigilio先生有正当理由辞职,取决于离职协议的执行和效力,包括对我们有利的全面索赔的释放,以及公司唯一酌情决定的一年离职后竞业禁止协议,他将有权获得:(1)相当于他当时当前基本工资的9个月的金额加上他年度目标奖金的按比例部分,以及(Ii)如果Trigilio先生在紧接他被解雇之前参加了我们的集团健康计划,并且选择了COBRA继续健康保险,则继续该集团健康保险的费率与他是在职雇员的相同,直到(A)他被解雇九个月周年,(B)他根据任何其他雇主的集团医疗计划有资格获得集团医疗计划福利,或(C)他根据COBRA继续享有权利。Trigilio先生的雇佣协议还规定,作为上述付款和福利的替代,如果我们无故终止他的雇用或他有充分理由辞职,在公司控制权变更后12个月内,根据以我们为受益人的全面解除索赔的执行和效力,他将有权获得(I)相当于其当时当前基本工资(或紧接控制权变更前有效的基本工资)12个月的一次性现金付款。如果更高)加上他当时本年度的年度目标奖金(或紧接控制权变更之前生效的年度目标奖金,如果更高),以及(Ii)如果Trigilio先生在紧接他被解雇之前参加了我们的集团健康计划并选择眼镜蛇健康继续, (A)其被解雇12个月周年纪念日,(B)其根据任何其他雇主的团体医疗计划有资格获得团体医疗计划福利,或(C)其根据《眼镜蛇》规定的延续权利终止。
128
Trigilio先生的雇佣协议还规定,如果他的雇佣因他的死亡或残疾而终止,除非奖励协议中另有规定,证明以业绩为基础归属的股权奖励,当时未归属的股权奖励的25%,再加上为本公司服务的每一整年额外的5%,将立即加速,并于终止日变得完全归属且可行使或不可没收。雇佣协议进一步规定,倘若吾等无故终止聘用Trigilio先生或Trigilio先生于本公司控制权变更后12个月内因正当理由辞职,除非奖励协议另有规定,证明须按表现归属的股权奖励,每笔尚未支付的股权奖励将立即加速,并于终止日期变得完全归属及可行使或不可没收。
财政年度年终评选中的未偿还股票奖励
下表列出了截至2022年12月31日,我们每位被任命的高管持有的未偿还股权奖励。
|
|
期权大奖 |
|
|
股票大奖 |
|
||||||||||||||||||||||||||
名字 |
|
可行使的未行使期权标的证券数量 |
|
|
未行使期权未行使的证券标的数量 |
|
|
期权行权价 |
|
|
期权到期日 |
|
|
未归属的股份或单位的数目 |
|
|
未归属的股份或单位的市值(1) |
|
|
股权激励计划奖励:未归属的未赚取的股份、单位或其他权利的数量 |
|
|
股权激励计划奖励:尚未归属的未赚取的股份、单位或其他权利的市场或派息价值(1) |
|
||||||||
纳迪姆·艾哈迈德 |
|
|
790,416 |
|
|
|
1,919,584 |
|
(2) |
$ |
21.12 |
|
|
10/17/2031 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
53,750 |
|
(3) |
$ |
567,063 |
|
帕特里克·鲍尔,博士。 |
|
|
240,114 |
|
|
|
— |
|
|
$ |
4.30 |
|
|
10/28/2030 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
365,493 |
|
|
|
42,500 |
|
(4) |
$ |
4.30 |
|
|
10/28/2030 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
137,360 |
|
|
|
149,305 |
|
(4) |
$ |
21.00 |
|
|
12/30/2030 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
15,625 |
|
|
|
59,375 |
|
(4) |
$ |
13.60 |
|
|
2/11/2032 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
3,903 |
|
(5) |
$ |
41,177 |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2,774 |
|
(5) |
$ |
29,266 |
|
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
29,688 |
|
(6) |
$ |
313,208 |
|
|
|
— |
|
|
|
— |
|
杰弗里·特里格里奥 |
|
|
119,250 |
|
|
|
110,641 |
|
(4) |
$ |
4.30 |
|
|
10/28/2030 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
70,276 |
|
|
|
76,389 |
|
(4) |
$ |
21.00 |
|
|
12/30/2030 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
15,625 |
|
|
|
59,375 |
|
(4) |
$ |
13.60 |
|
|
2/11/2032 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
29,688 |
|
(6) |
$ |
313,208 |
|
|
|
— |
|
|
|
— |
|
薪酬风险评估
我们不认为我们的高管薪酬计划鼓励过度或不必要的冒险。我们的薪酬计划,包括任何基于绩效的薪酬,旨在鼓励我们指定的高管和其他员工继续专注于短期和长期战略目标。因此,我们不认为我们的补偿计划有可能对我们产生实质性的不利影响。
129
员工福利
我们的合格美国员工参与了一项符合税务条件的退休计划,该计划提供了在税收优惠的基础上为退休储蓄的机会。计划参与者可以根据修订后的《1986年国税法》(下称《国税法》),根据适用的年度限额,延期支付符合条件的补偿。我们提供相当于员工延期的100%的安全港匹配贡献,最高可达员工工资的5%,但须遵守适用的法规限制。
我们的所有全职员工,包括我们的高管,都有资格参加我们提供的某些医疗、残疾和人寿保险福利计划。我们为所有员工支付定期人寿保险和短期和长期残疾的保费,包括我们的高管。我们还为所有员工,包括我们的高管,提供带薪休假福利,包括假期、病假和节假日。我们不为我们的任何员工或高管提供任何合格或不合格的固定福利计划。
根据我们的内幕交易政策,我们的董事和高管可以采用书面计划,称为规则10b5-1计划,在该计划中,他们将与经纪商签订合同,定期买卖我们普通股的股票。根据规则10b5-1计划,经纪商根据董事或官员在进入计划时建立的参数执行交易,而不需要董事或官员的进一步指示。此外,当我们的董事和高管不掌握重要的非公开信息时,他们可以在规则10b5-1计划之外购买或出售额外的股票。
董事薪酬
董事薪酬表
下表显示了我们的非雇员董事在2022年内赚取或支付的所有薪酬。我们的首席执行官纳迪姆·艾哈迈德没有为董事服务获得任何报酬,因此不在此表中。艾哈迈德先生在2022年期间获得的薪酬列于《高管薪酬--薪酬汇总表》。
名字 |
|
以现金支付或赚取的费用 |
|
|
期权大奖 |
|
|
总计 |
|
|||
托马斯·埃贝林 |
|
$ |
50,500 |
|
|
$ |
149,923 |
|
|
$ |
200,423 |
|
安斯伯特·加迪克医学博士(3) |
|
$ |
36,667 |
|
|
$ |
149,923 |
|
|
$ |
186,590 |
|
安妮-玛丽·马丁,博士。(4) |
|
$ |
— |
|
(5) |
$ |
349,991 |
|
|
$ |
349,991 |
|
安东尼·罗森伯格 |
|
$ |
86,500 |
|
|
$ |
149,923 |
|
|
$ |
236,423 |
|
David·P·瑞安医学博士。(6) |
|
$ |
6,667 |
|
|
$ |
249,994 |
|
|
$ |
256,661 |
|
斯蒂芬·韦伯斯特 |
|
$ |
59,000 |
|
|
$ |
149,923 |
|
|
$ |
208,923 |
|
130
非员工董事薪酬政策
根据我们的非雇员董事薪酬政策,我们向非雇员董事支付一笔现金预付金,用于支付在董事会和董事所在的每个委员会的服务。我们的董事会主席每年会因此而获得额外的聘用费。这些费用旨在使我们能够长期吸引和留住高素质的非雇员董事。非员工董事现金托管人的时间表如下所示。
|
|
年度定额 |
|
|
董事会: |
|
|
|
|
成员 |
|
$ |
35,000 |
|
非执行主席的额外聘任 |
|
$ |
30,000 |
|
审计委员会: |
|
|
|
|
委员(主席除外) |
|
$ |
7,500 |
|
主席 |
|
$ |
15,000 |
|
薪酬委员会: |
|
|
|
|
委员(主席除外) |
|
$ |
5,000 |
|
主席 |
|
$ |
10,000 |
|
提名和公司治理委员会: |
|
|
|
|
委员(主席除外) |
|
$ |
4,000 |
|
主席 |
|
$ |
8,000 |
|
此外,我们的非雇员董事薪酬政策规定,在首次当选为我们的董事会成员后,每位非雇员董事将被授予一项选择权,以购买授予日期公允价值为250,000美元的普通股(“初始授予”)。初始授出于授出日期的第一、二及三周年纪念日以等额分期付款方式授予,但须在适用的归属日期前继续作为董事服务。此外,在每次股东周年大会当日,每位在股东周年大会后继续为非雇员董事的非雇员董事将获授购入本公司普通股的选择权,于授出日期公平值为150,000美元(“年度授出”)。年度授出将于(I)授出日期一周年或(Ii)本公司下一年度股东大会日期(以较早者为准)全数授予,但须继续作为董事服务至适用的归属日期为止。董事在进入董事会满一年后才有资格获得年度补助金。初始奖助金及年度奖助金于公司出售时全数归属。
最初授予马丁博士的是一项选择权,即购买我们普通股的股票,授予日期的公允价值为350,000美元,而不是我们董事薪酬政策中规定的250,000美元,原因是马丁博士没有收到标准的董事会和委员会费用。该奖项在授予日的第一、二和三周年纪念日分成等额的分期付款,但在适用的归属日期之前继续作为董事服务。
我们报销非雇员董事出席董事会和委员会会议所产生的所有合理的自付费用。
根据非雇员董事薪酬政策,在任何年度向非雇员董事支付作为董事服务的报酬总额,包括股权补偿和现金补偿,不得超过500,000美元(非雇员董事最初当选或被任命为我们董事会成员的当年为750,000美元)。
项目12.某些受益者的担保所有权员工和管理层及相关股东事宜。
下表列出了关于截至2023年2月15日我们普通股的实益所有权的信息,在我们已知的范围内或从公开备案文件中可以确定:
题为“实益拥有的股份百分比”的栏目基于截至2023年2月15日的已发行普通股总数39,327,298股。
131
实益所有权是根据美国证券交易委员会的规则和规定确定的,包括对我们普通股的投票权或投资权。受当前可行使或可在2023年2月15日起60天内行使的期权约束的普通股股票被视为已发行股票,并由持有该期权的人实益拥有,目的是为了计算该人的所有权百分比,而不是为了计算任何其他人的所有权百分比。除非另有说明,本表中的个人和实体对其实益拥有的我们普通股的所有股份拥有唯一投票权和投资权,但须遵守适用的社区财产法。除非下表另有说明,否则指定受益所有人的地址由库利南肿瘤公司保管,地址为One Main Street,Suite1350,Cambridge,MA 02142。
实益拥有人姓名或名称 |
|
实益拥有的股份 |
|
|
实益拥有的股份百分比 |
|
||
5%或更大股东 |
|
|
|
|
|
|
||
瑞银肿瘤学影响基金L.P.(1) |
|
|
7,648,268 |
|
|
|
19.45 |
% |
隶属于BVF,Inc.的实体。(2) |
|
|
7,509,059 |
|
|
|
16.39 |
% |
附属于F2风险投资公司的实体(3) |
|
|
3,783,971 |
|
|
|
9.62 |
% |
CHI顾问有限责任公司(4) |
|
|
3,317,544 |
|
|
|
8.44 |
% |
先锋集团(5) |
|
|
2,812,965 |
|
|
|
7.15 |
% |
贝莱德股份有限公司(6) |
|
|
2,506,215 |
|
|
|
6.37 |
% |
获任命的行政人员及董事 |
|
|
|
|
|
|
||
纳迪姆·艾哈迈德(7) |
|
|
961,411 |
|
|
|
2.39 |
% |
帕特里克·鲍尔,博士。(8) |
|
|
1,175,101 |
|
|
|
2.93 |
% |
杰弗里·特里格里奥(9) |
|
|
254,627 |
|
|
* |
|
|
托马斯·埃贝林(10) |
|
|
159,528 |
|
|
* |
|
|
安妮-玛丽·马丁,博士。(11) |
|
|
12,294 |
|
|
* |
|
|
安东尼·罗森伯格(12) |
|
|
260,686 |
|
|
* |
|
|
David·P·瑞安医学博士。 |
|
|
— |
|
|
* |
|
|
斯蒂芬·韦伯斯特(13) |
|
|
23,605 |
|
|
* |
|
|
全体执行干事和董事(12人)(14) |
|
|
3,517,920 |
|
|
|
8.34 |
% |
*不到1%
132
133
根据股权补偿计划获授权发行的证券
下表提供了截至2022年12月31日根据我们现有的股权补偿计划可能发行的普通股的信息。
|
截至2022年12月31日的股权薪酬计划 |
|
|||||||||
计划类别 |
在行使尚未行使的期权、认股权证及权利时须发行的证券数目 |
|
|
未偿还期权、权证和权利的加权平均行权价 |
|
|
证券数量 |
|
|||
证券持有人批准的股权补偿计划(1)(2)(3) |
|
7,695,780 |
|
|
$ |
18.03 |
|
|
|
2,980,918 |
|
未经证券持有人批准的股权补偿计划(4) |
|
1,638,500 |
|
|
$ |
12.38 |
|
|
|
— |
|
总计 |
|
9,334,280 |
|
|
$ |
17.04 |
|
|
|
2,980,918 |
|
除在Form 10-K年报本部第III部分“高管薪酬”及“董事薪酬”项下所述的薪酬协议及其他安排,以及下文所述的交易外,自2021年1月1日以来,并没有亦目前并无建议进行任何交易或一系列类似的交易,而吾等曾经或将会参与的任何交易或一系列类似交易所涉及的金额超过或将会超过120,000美元,而董事的任何人士、高管、持有任何类别股本百分之五或以上的任何人士、或任何前述人士的直系亲属或关联实体的任何成员、或将会有直接或间接的物质利益。
134
参与我们的IPO
我们的某些高管和现有股东在2021年1月的首次公开募股(IPO)中以首次公开募股的价格购买了我们的普通股。下表列出了我们的高管和现有股东以每股21.00美元的首次公开募股价格购买的普通股数量。
股东 |
|
普通股股份 |
|
|
购进总价 |
|
||
欧文·休斯(1) |
|
|
2,500 |
|
|
$ |
52,500 |
|
杰弗里·特里格里奥(2) |
|
|
2,250 |
|
|
$ |
47,250 |
|
附属于F2风险投资公司的实体(3) |
|
|
200,000 |
|
|
$ |
4,200,000 |
|
瑞银肿瘤学影响基金L.P.(4) |
|
|
300,000 |
|
|
$ |
6,300,000 |
|
________________________________________
与我们的股东达成协议
优先股融资协议
关于首次公开募股前的优先股融资,我们分别与优先股的购买者和普通股的某些持有人签订了投资者权利协议和股东协议。该等协议的所有重大条款于紧接吾等首次公开招股完成前终止,但与注册权有关的条款除外,该等条款在吾等首次公开招股完成后继续有效,并授权该等权利的持有人要求吾等提交一份注册声明,但须受某些限制所规限,并有权要求吾等以其他方式提交的一份注册声明涵盖其股份。
专利权使用费转让协议
库利南琥珀公司(“库利南琥珀”)、库利南佛罗伦萨公司(“库利南佛罗伦萨”)和库利南MICA公司(“库利南MICA”)是与MPM肿瘤学慈善基金会公司和瑞银擎天柱基金会(统称为“基金会”)签订的特许权使用费转移协议(“特许权使用费转移协议”)的每一方。根据这些各自的协议,每个基金会有权获得适用子公司开发的任何产品全球净销售额的较低个位数版税百分比,但受专利到期后的限制和控制权变更后开发的知识产权的限制。
除非较早终止,否则每项特许权使用费转让协议应在(I)该子公司的产品在该国首次商业销售的12周年之日和(Ii)涉及该子公司的产品在该国的组成或使用的任何收购前知识产权的最后一项到期之日终止,以下列日期为准。这些基金会隶属于OIF和Gadicke博士,OIF实益拥有我们5%以上的已发行普通股,Gadicke博士是我们的前董事会成员。
在签署每项特许权使用费转让协议的同时,库利南琥珀、库利南佛罗伦萨和库利南米卡也分别与基金会和公司签订了一份书面协议(“特许权使用费函”)。根据特许权使用费函件,双方同意,本公司为购买证券而向适用附属公司支付的现金代价的一部分,将被视为有权获得适用附属公司收到的任何公司产品全球净销售额中较低的个位数特许权使用费百分比(“特许权使用费流”)。此外,在本公司购买版税流后立即生效,本公司通过指示适用的子公司签署、交付和履行版税转让协议,将其在版税流下的权利转让给基金会。于2022年6月,就将吾等于库利南珍珠股份有限公司(“库利南珍珠”)的股权出售予泰豪药业有限公司(“泰豪”)一事,吾等修订了适用于库利南珍珠的特许权使用费转让协议及特许权使用费函件,以规定适用的特许权使用费流不包括由泰豪拥有或控制并由泰豪或其代表在日本商业化的产品的净销售额。特许权使用费转让协议表格作为我们于2020年12月18日提交的S-1表格注册声明的附件10.21提交。
就业和咨询安排
我们已经与我们的高管签订了雇佣或咨询协议。有关与我们指定的高管签订的协议的更多信息,请参阅“高管薪酬-雇佣协议”。
135
与科琳·萨维尔博士签订的咨询协议。
自2021年1月12日起,我们与Savill博士签订了一项服务协议,根据该协议,我们同意向Savill博士支付每年380,000美元的咨询费,Savill博士有资格获得40%的年度绩效奖金,但须经我们的董事会批准。2022年,萨维尔博士的基本咨询费涨到了391,400美元。服务协议的有效期将在萨维尔博士向公司提供服务的最后一天到期。
赔偿协议
我们已经达成协议,对我们的董事和高管进行赔偿。除其他事项外,这些协议将要求我们在特拉华州法律允许的最大范围内,赔偿这些个人在任何诉讼或诉讼中合理地招致的某些费用(包括律师费)、判决、罚款和和解金额,包括由我们或根据我们的权利提起的任何诉讼,因为该人代表我们公司提供的任何服务或该人作为我们董事会成员的身份。
关联人交易政策
我们的董事会审查和批准与我们5%或更多有投票权证券的董事、高级管理人员和持有者及其附属公司的交易,每个附属公司都是关联方。我们的关联方交易政策规定,此类交易必须得到我们的审计委员会的批准。根据这项政策,审核委员会主要负责审核和批准或不批准“关联方交易”,即吾等与关联人之间的交易,涉及的总金额超过或可能超过120,000美元,而关联人在其中拥有或将拥有直接或间接重大利益。就本政策而言,关连人士定义为董事、行政总裁、董事的被提名人或自最近完成年度开始以来持有董事普通股超过5%的实益拥有者,及其直系亲属。
董事独立自主
适用的《纳斯达克》规则要求上市公司董事会的多数成员必须是独立董事。此外,除指定的例外情况外,纳斯达克规则要求上市公司的审计、薪酬和提名委员会及公司管治委员会的每名成员均为独立成员,而审计委员会成员亦须符合交易法第10A-3条所载的独立性标准,而薪酬委员会成员亦须符合交易法第10C-1条所载的独立性标准。根据适用的纳斯达克规则,董事只有在上市公司董事会认为该人与董事之间的关系不会干扰其在履行董事责任时行使独立判断的情况下,才有资格成为“独立董事”。上市公司审计委员会成员除以审计委员会、董事会或任何其他董事会委员会成员的身份外,不得直接或间接从上市公司或其任何附属公司接受任何咨询、咨询或其他补偿费,或以其他方式成为该上市公司或其任何附属公司的关联人。此外,在肯定地确定任何将在公司薪酬委员会任职的董事的独立性时,根据交易法,规则10C-1要求公司董事会必须考虑与确定董事是否与该公司有关系的所有具体相关因素,这些因素对于董事在薪酬委员会成员的职责方面独立于管理层的能力是至关重要的,包括:董事的薪酬来源,包括任何咨询, 该公司向董事支付的咨询费或其他补偿费,以及董事是否隶属于该公司或其任何子公司或关联公司。
我们的董事会已经决定,除了纳迪姆·艾哈迈德之外,现任董事会的所有成员都是独立董事,包括就纳斯达克和美国证券交易委员会的规则而言。在作出这种独立性决定时,我们的董事会考虑了每一位非雇员董事与我们的关系以及董事会认为与确定他们的独立性相关的所有其他事实和情况,包括每一位非雇员董事对我们的股本的实益所有权。在考虑上述董事的独立性时,我们的董事会考虑了我们的董事与持有我们超过5%的普通股的人的关联。我们的任何董事或高管之间都没有家族关系。根据这些规则,艾哈迈德先生不是独立的董事,因为他是我们的总裁和首席执行官。
136
第14项.本金账户律师费和服务费。
我们向毕马威有限责任公司支付了以下费用,用于审计综合财务报表和2022年至2021年期间提供的其他服务。
费用类别 |
|
2022 |
|
|
2021 |
|
||
审计费(1) |
|
$ |
702,500 |
|
|
$ |
470,000 |
|
审计相关费用(2) |
|
|
— |
|
|
|
— |
|
税费(3) |
|
|
307,420 |
|
|
|
104,210 |
|
所有其他费用(4) |
|
|
— |
|
|
|
— |
|
总费用 |
|
$ |
1,009,920 |
|
|
$ |
574,210 |
|
________________________________________
审计委员会预审政策和程序
我们的审计委员会通过了关于批准所有审计和非审计服务的政策和程序,这些服务将由我们的独立注册会计师事务所进行。这项政策规定,我们不会聘请我们的独立注册会计师事务所提供审计或非审计服务,除非该服务事先得到了我们的审计委员会的特别批准,或者是根据下文所述的预先批准程序进行的。
我们的审计委员会可能会不时预先批准我们的独立注册会计师事务所在未来12个月内预期向我们提供的特定类型的服务。任何此类预先批准都将详细说明将提供的特定服务或服务类型,并且通常也受最高金额的限制。
在2022年至2021年期间,毕马威有限责任公司除按照上述审批前政策和程序向我们提供任何服务外,并无向我们提供任何服务。
137
部分IV
项目15.展品和资金ALI对帐表。
项目16.表格10-K摘要
没有。
138
库利南肿瘤公司。
财务报表索引
|
页面 |
|
|
独立注册会计师事务所报告(马萨诸塞州波士顿毕马威会计师事务所ID: |
F-2 |
合并资产负债表 |
F-3 |
合并经营表和全面损益表(亏损) |
F-4 |
股东权益合并报表 |
F-5 |
合并现金流量表 |
F-6 |
合并财务报表附注 |
F-7 |
F-1
《独立注册会计师报告》艾瑞德会计师事务所
致股东和董事会
库利南肿瘤公司:
对合并财务报表的几点看法
我们已经审计了库利南肿瘤公司及其子公司的合并资产负债表
(本公司)截至2022年12月31日及2021年12月31日止年度的相关综合经营报表及综合收益(亏损)、股东权益及现金流量,以及相关附注(统称为综合财务报表)。我们认为,综合财务报表在所有重要方面都公平地反映了公司截至2022年12月31日和2021年12月31日的财务状况,以及截至2022年12月31日和2021年12月31日止年度的经营结果和现金流量,符合美国公认会计原则。
意见基础
这些合并财务报表由公司管理层负责。我们的责任是根据我们的审计对这些合并财务报表发表意见。我们是一家在美国上市公司会计监督委员会(PCAOB)注册的公共会计师事务所,根据美国联邦证券法以及美国证券交易委员会和PCAOB的适用规则和法规,我们必须与公司保持独立。
我们是按照PCAOB的标准进行审计的。这些准则要求我们计划和执行审计,以获得关于合并财务报表是否没有重大错报的合理保证,无论是由于错误还是舞弊。本公司并无被要求对其财务报告的内部控制进行审计,我们也没有受聘进行审计。作为我们审计的一部分,我们被要求了解财务报告的内部控制,但不是为了对公司财务报告内部控制的有效性发表意见。因此,我们不表达这样的意见。
我们的审计包括执行评估合并财务报表重大错报风险的程序,无论是由于错误还是欺诈,以及执行应对这些风险的程序。这些程序包括在测试的基础上审查关于合并财务报表中的金额和披露的证据。我们的审计还包括评价管理层使用的会计原则和作出的重大估计,以及评价合并财务报表的整体列报。我们相信,我们的审计为我们的观点提供了合理的基础。
/s/
自2018年以来,我们一直担任本公司的审计师。
March 9, 2023
F-2
库利南肿瘤公司。
合并B配额单
(单位为千,不包括份额)
|
|
十二月三十一日, |
|
|
十二月三十一日, |
|
||
资产 |
|
|
|
|
|
|
||
流动资产: |
|
|
|
|
|
|
||
现金和现金等价物 |
|
$ |
|
|
$ |
|
||
短期投资 |
|
|
|
|
|
|
||
预付费用和其他流动资产 |
|
|
|
|
|
|
||
流动资产总额 |
|
|
|
|
|
|
||
财产和设备,净额 |
|
|
|
|
|
|
||
经营性租赁使用权资产 |
|
|
|
|
|
|
||
其他资产 |
|
|
|
|
|
|
||
长期投资 |
|
|
|
|
|
|
||
总资产 |
|
$ |
|
|
$ |
|
||
负债与股东权益 |
|
|
|
|
|
|
||
流动负债: |
|
|
|
|
|
|
||
应付帐款 |
|
$ |
|
|
$ |
|
||
应计费用和其他流动负债 |
|
|
|
|
|
|
||
应付所得税 |
|
|
|
|
|
|
||
经营租赁负债,流动 |
|
|
|
|
|
|
||
流动负债总额 |
|
|
|
|
|
|
||
长期负债: |
|
|
|
|
|
|
||
经营租赁负债,扣除当期部分 |
|
|
|
|
|
|
||
递延租金 |
|
|
|
|
|
|
||
总负债 |
|
|
|
|
|
|
||
(注12) |
|
|
|
|
|
|
||
股东权益: |
|
|
|
|
|
|
||
优先股,$ |
|
|
|
|
|
|
||
普通股,$ |
|
|
|
|
|
|
||
额外实收资本 |
|
|
|
|
|
|
||
累计其他综合损失 |
|
|
( |
) |
|
|
( |
) |
累计赤字 |
|
|
( |
) |
|
|
( |
) |
库利南股东权益总额 |
|
|
|
|
|
|
||
非控制性权益 |
|
|
|
|
|
|
||
股东权益总额 |
|
|
|
|
|
|
||
总负债和股东权益 |
|
$ |
|
|
$ |
|
||
见合并财务报表附注。
F-3
库利南肿瘤公司。
业务处合并报表折旧及综合收益(亏损)
(以千为单位,每股除外)
|
|
截至十二月三十一日止的年度: |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
$ |
|
|
$ |
|
|||
运营费用: |
|
|
|
|
|
|
||
研发 |
|
|
|
|
|
|
||
一般和行政 |
|
|
|
|
|
|
||
总运营费用 |
|
|
|
|
|
|
||
库利南珍珠的销售收益 |
|
|
|
|
|
|
||
营业收入(亏损) |
|
|
|
|
|
( |
) |
|
其他收入(支出): |
|
|
|
|
|
|
||
利息收入 |
|
|
|
|
|
|
||
其他收入(费用),净额 |
|
|
|
|
|
( |
) |
|
所得税前净收益(亏损) |
|
|
|
|
|
( |
) |
|
所得税费用 |
|
|
|
|
|
|
||
净收益(亏损) |
|
|
|
|
|
( |
) |
|
非控股权益应占净亏损 |
|
|
( |
) |
|
|
( |
) |
库利南公司普通股股东应占净收益(亏损) |
|
$ |
|
|
$ |
( |
) |
|
|
|
|
|
|
|
|
||
综合收益(亏损): |
|
|
|
|
|
|
||
净收益(亏损) |
|
$ |
|
|
$ |
( |
) |
|
投资未实现亏损 |
|
|
( |
) |
|
|
( |
) |
综合收益(亏损) |
|
$ |
|
|
$ |
( |
) |
|
可归属于非控股权益的综合损失 |
|
|
( |
) |
|
|
( |
) |
可归因于库利南的全面收益(亏损) |
|
$ |
|
|
$ |
( |
) |
|
|
|
|
|
|
|
|
||
每股收益(净亏损): |
|
|
|
|
|
|
||
基本信息 |
|
$ |
|
|
$ |
( |
) |
|
稀释 |
|
$ |
|
|
$ |
( |
) |
|
|
|
|
|
|
|
|
||
用于计算每股收益(净亏损)的加权平均股份数: |
|
|
|
|
|
|
||
基本信息 |
|
|
|
|
|
|
||
稀释 |
|
|
|
|
|
|
||
见合并财务报表附注。
F-4
库利南肿瘤公司。
ST合并报表OCKHOLDERS的股权
(单位为千,不包括份额)
|
|
普通股 |
|
|
其他内容 |
|
|
累计 |
|
|
留存收益(累计 |
|
|
非控制性 |
|
|
总计 |
|
||||||||||
|
|
股票 |
|
|
金额 |
|
|
资本 |
|
|
损失 |
|
|
赤字) |
|
|
附属公司 |
|
|
权益 |
|
|||||||
2020年12月31日的余额 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|||||
首次公开募股,扣除发行成本$ |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
员工购股计划下普通股的发行 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
非控制性权益的贡献 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
按股权薪酬计划净发行普通股 |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
基于股权的薪酬 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|||
投资未实现亏损 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
净亏损 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
|
|
( |
) |
2021年12月31日的余额 |
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|||||
非控制性权益的贡献 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
||
收购非控制性权益 |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
按股权薪酬计划净发行普通股 |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
基于股权的薪酬 |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|||
投资未实现亏损 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
|
|
|
( |
) |
|
净收益(亏损) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
( |
) |
|
|
|
||
2022年12月31日的余额 |
|
|
|
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
( |
) |
|
$ |
|
|
$ |
|
|||||
见合并财务报表附注。
F-5
库利南肿瘤公司。
合并状态现金流NTS
(单位:千)
|
|
截至十二月三十一日止的年度: |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
经营活动: |
|
|
|
|
|
|
||
净收益(亏损) |
|
$ |
|
|
$ |
( |
) |
|
对净收益(亏损)与业务活动中使用的现金净额进行调整: |
|
|
|
|
|
|
||
库利南珍珠的销售收益 |
|
|
( |
) |
|
|
|
|
基于股权的薪酬费用 |
|
|
|
|
|
|
||
有价证券的摊销或增值 |
|
|
|
|
|
|
||
非控制性权益的非现金贡献 |
|
|
|
|
|
|
||
有价证券已实现亏损 |
|
|
|
|
|
|
||
折旧及摊销 |
|
|
|
|
|
|
||
处置固定资产收益 |
|
|
( |
) |
|
|
|
|
经营性资产和负债变动情况: |
|
|
|
|
|
|
||
预付费用和其他流动资产 |
|
|
( |
) |
|
|
( |
) |
应付帐款 |
|
|
( |
) |
|
|
( |
) |
应计费用和其他负债 |
|
|
|
|
|
|
||
应付所得税 |
|
|
|
|
|
|
||
用于经营活动的现金净额 |
|
|
( |
) |
|
|
( |
) |
投资活动: |
|
|
|
|
|
|
||
购买有价证券 |
|
|
( |
) |
|
|
( |
) |
有价证券的出售和到期日 |
|
|
|
|
|
|
||
出售库利南珍珠,扣除出售现金后的净额为$ |
|
|
|
|
|
|
||
购置财产和设备 |
|
|
( |
) |
|
|
|
|
出售财产和设备所得收益 |
|
|
|
|
|
|
||
投资活动提供(用于)的现金净额 |
|
|
|
|
|
( |
) |
|
融资活动: |
|
|
|
|
|
|
||
收购非控制性权益 |
|
|
( |
) |
|
|
|
|
按股权薪酬计划净发行普通股 |
|
|
|
|
|
|
||
发行可转换票据 |
|
|
|
|
|
|
||
可转换票据的偿还 |
|
|
( |
) |
|
|
|
|
非控制性权益的贡献 |
|
|
|
|
|
|
||
首次公开招股所得收益 |
|
|
|
|
|
|
||
支付递延发售费用 |
|
|
|
|
|
( |
) |
|
融资活动提供(用于)的现金净额 |
|
|
( |
) |
|
|
|
|
现金及现金等价物净增(减) |
|
|
|
|
|
( |
) |
|
期初现金及现金等价物 |
|
|
|
|
|
|
||
期末现金及现金等价物 |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
||
补充非现金披露 |
|
|
|
|
|
|
||
非现金投融资活动和补充现金流量信息 |
|
|
|
|
|
|
||
应付账款和应计费用及其他负债中所列财产和设备的购置 |
|
$ |
|
|
$ |
|
||
缴纳所得税的现金 |
|
$ |
|
|
$ |
|
||
支付利息的现金 |
|
$ |
|
|
$ |
|
||
上一年支付的递延发行成本 |
|
$ |
|
|
$ |
|
||
见合并财务报表附注。
F-6
库利南肿瘤公司。
合并后的注释财务报表
组织
库利南肿瘤公司及其合并的子公司(“库利南”或“本公司”)是一家临床阶段的生物制药公司,专注于模式不可知的靶向肿瘤学。库利南的前身公司库利南制药有限责任公司成立于2016年9月,随后于2017年11月更名为库利南肿瘤有限责任公司(“有限责任公司”)。有限责任公司的全资子公司库利南管理公司(“管理”)成立于2016年9月,并于2021年1月在与有限责任公司的反向合并中成为幸存实体。2021年2月,Management更名为Cullinan Oncology,Inc.
本公司于2022年6月完成将其部分拥有的附属公司库利南珍珠公司(“库利南珍珠”)的全部股权出售给泰豪药业有限公司(“泰豪”)。有关交易的其他详情,请参阅附注3。出售该公司在库利南珍珠公司的股权不符合根据美国公认会计原则(“美国公认会计原则”)报告为非持续经营的标准。因此,上期合并财务报表和披露没有追溯重述,以反映公司出售库利南珍珠股权的影响。
重组、反向股权分置与首次公开发行
2021年1月,公司完成了首次公开募股(IPO)并进行了发行和出售
紧接于本公司的注册声明生效前,本公司完成重组,据此,合并后的有限责任公司与Management and Management合并为尚存实体(“重组”)。管理层是此次IPO的注册人。
流动性
除2022年出售库利南珍珠的一次性收益和自成立以来的运营现金流为负外,该公司已发生运营亏损,预计在可预见的未来将继续产生运营亏损。该公司的最终成功取决于其研究和开发活动的结果以及将该公司的候选产品商业化的能力。该公司面临许多风险,包括但不限于,需要为其候选产品的正在进行和计划中的临床开发获得足够的额外资金。由于与医药产品和开发相关的许多风险和不确定性,公司无法准确预测完成其候选产品开发所需的时间或资金数额,成本可能会因多种原因超过公司的预期,包括公司无法控制的原因。
于2022年6月,本公司完成将本公司在其部分拥有的附属公司库利南珍珠的股权出售给泰豪,预付款为#美元
自成立以来,该公司主要通过出售股本证券以及向其候选产品发放许可证或出售权利来为其运营提供资金。该公司预计其现金、现金等价物和短期投资为#美元。
陈述的基础
随附的本公司综合财务报表乃根据美国公认会计原则及美国证券交易委员会(“美国证券交易委员会”)适用的财务报告规则及规定编制,包括本公司及其综合附属公司的账目。公司间余额和交易已在合并中冲销。
F-7
合并原则
本公司合并其拥有控股权的实体。本公司评估其每一间附属公司,以确定该实体是否代表应根据VIE模型评估其合并的可变权益实体(“VIE”),或如该实体为具表决权的权益实体,则应使用具表决权的权益模型(“VOE”)评估其合并。该公司的结论是,其子公司都不是VIE,并已将每个子公司合并到VOE之下。根据VOE,本公司在以下情况下合并实体:1)直接或间接拥有超过50%的有表决权股份,或其他股权持有人没有实质性的投票权、参与权或清算权,或2)当公司通过控制董事会拥有控股权,且实体的重大决策在董事会层面做出。
2022年至2021年期间,该公司在以下部分持股的子公司进行了投资:
合并实体 |
|
截至2022年12月31日的关系 |
|
日期控件优先 |
库利南珍珠公司 |
|
剥离 |
|
2018年11月 |
库利南琥珀公司 |
|
部分持股子公司 |
|
2019年12月 |
库利南佛罗伦萨公司 |
|
部分持股子公司 |
|
2019年12月 |
库利南云母公司 |
|
部分持股子公司 |
|
May 2020 |
预算的使用
根据美国公认会计原则编制公司的综合财务报表和附注时,公司管理层需要作出影响财务报表中报告的金额的估计和判断。公司管理层持续评估其估计,包括但不限于与预付和应计研发费用以及专利费转移协议的公允价值相关的估计。管理层的估计可能会根据事实和情况的变化而一期又一期地改变。本公司管理层根据过往经验及其他被认为合理的相关假设作出估计。实际结果可能与这些估计大不相同。
细分市场
本公司已确定其首席执行官为首席运营决策者(“CODM”)。公司以下列方式经营和管理业务
风险集中
截至2022年12月31日,该公司没有明显的信用风险集中。截至2022年12月31日,现金和现金等价物主要与美国的两家金融机构保持。银行存款可能会超过为此类存款提供的保险。这些存款可以按需赎回,因此风险最小。根据我们的投资政策,本公司按投资类型、信用评级、到期日、行业集团和发行人限制投资于此类证券的金额。我们的投资政策的目标是(I)安全、保全本金和分散风险,以及(Ii)投资的流动性足以满足现金流要求。
公司会受到某些风险和不确定因素的影响,并认为以下任何方面的变化都可能对公司未来的财务状况或经营结果产生重大不利影响:获得未来融资的能力;监管部门对候选产品的批准和市场接受程度,以及对产品候选产品的补偿;公司所依赖的第三方临床研究机构和制造商的表现;对公司知识产权的保护;基于知识产权、专利、产品、监管或其他因素对公司的诉讼或索赔;以及公司吸引和留住支持其增长所需的员工的能力。
F-8
现金和现金等价物
本公司将购买时原始到期日为三个月或以下的所有高流动性投资视为现金等价物。截至2022年12月31日和2021年12月31日,现金等价物包括政府支持的货币市场基金。
投资
该公司一般持有有价证券投资。未被归类为现金等价物且到期日少于12个月的投资在综合资产负债表中被归类为短期投资。本公司有意及有能力持有超过12个月的投资,而其到期日超过12个月的投资在综合资产负债表中列为长期投资。
有价证券的摊销成本根据溢价摊销和到期折价增加进行调整。这样的摊销包括在利息收入中。利息和股息也包括在利息收入中。应收利息计入综合资产负债表中的预付费用和其他流动资产,代表公司有价证券的应计利息和未付利息。本公司定期审查其有价证券的减值情况,并在市场价值下降被视为非暂时性时将这些投资调整为其公允价值。公允价值的下降被判断为非暂时性的有价证券,如果有的话,计入其他收入(费用)净额。
金融工具的公允价值
本公司有若干按公允价值记录的金融资产及负债,在公允价值计量会计准则所述的公允价值层级中被分类为1、2或3级。公允价值层次的三个层次如下所述:
第1级-公司有能力进入的活跃市场上相同资产或负债的未调整报价;
第2级--活跃市场中类似资产和负债的报价,或其他市场可观察到的信息,如利率、收益率曲线和外币现货汇率;以及
第3级-需要对公允价值计量具有重大意义且不可观察的输入的定价或估值。
第一级和第二级之间没有按公允价值计量的金融资产或负债转移,2022年或2021年期间也没有第三级投资。
本公司按公允价值计入的金融资产包括短期投资和长期投资。本公司短期和长期投资的公允价值主要使用市场报价或从独立定价来源获得的价格来确定。
财产和设备,净额
财产和设备按成本减去累计折旧列报。折旧费用在每项资产的估计使用年限内使用直线法确认,具体如下:
资产类别 |
|
预计使用寿命 |
办公家具和设备 |
|
|
租赁权改进 |
|
没有改善或延长资产寿命的维护和维修在发生时计入费用。在处置或报废资产时,成本及累计折旧和摊销将从综合资产负债表中剔除,由此产生的任何收益或亏损将反映在综合经营表和全面收益(亏损)中。
F-9
租契
公司在一开始就确定一项安排是否为租约。如果合同转让了在一段时间内控制已确定资产的使用权以换取对价,则该合同是租赁或包含租赁。本公司于租赁开始日将租赁分类为经营性租赁或融资租赁,并将初始租期超过12个月的所有租赁的使用权资产(“ROU”)和租赁负债记录在综合资产负债表上。租约的初始期限为
本公司订立既包含租赁内容又包含非租赁内容的合同。非租赁部分可能包括维护、水电费和其他运营成本。本公司在其租赁安排中将固定成本的租赁和非租赁部分合并为单一租赁部分。变动成本,如水电费或维护费,不计入净资产和租赁负债的计量,而是在决定支付变动对价金额的事件发生时计入费用。
经营租赁资产及负债于租赁开始日按租赁期间租赁付款的现值按租赁隐含的贴现率确认。如果贴现率不能轻易确定,本公司将根据租赁开始日的现有信息对其递增借款利率进行估计。经营租赁资产根据预付或应计租赁付款进行进一步调整。经营租赁付款采用直线法作为租赁期间的经营费用计提。本公司的租赁条款可包括在合理确定本公司将行使该选择权时延长或终止租约的选择权。
长期资产减值准备
只要发生事件或环境变化表明某项资产的账面价值可能无法收回,就会对长期资产进行减值审查。将持有和使用的资产的可回收性是通过将资产的账面价值与预期产生的未来未贴现现金流量进行比较来衡量的。减值费用按一项资产的账面价值超过该资产公允价值的金额确认。待处置资产按账面值或公允价值减去出售成本中较低者列报。
递延发售成本
本公司对与正在进行的股权融资直接相关的成本进行资本化,直至完成该等融资为止,届时该等成本将计入发售的总收益中。如果放弃正在进行的股权融资,递延发售成本将立即在综合经营报表和全面收益(亏损)表中计入营业费用。该公司产生了$
非控制性权益
非控股权益代表本公司部分拥有的子公司中的第三方权益。本公司于每个资产负债表日以账面价值(“)以假设清算方式厘定本公司部分拥有附属公司的非控股权益在净资产中的金额。”HLBV“)方法。根据HLBV方法,在合并资产负债表中报告为非控股权益的金额代表根据部分拥有的子公司的清算条款,第三方在每个资产负债表日假设将收到的金额,假设部分拥有的子公司的净资产按照根据美国公认会计准则确定的记录金额清算,并分配给部分拥有的子公司的所有者。可归因于综合经营报表非控制性权益的净收益(亏损)及综合收益(亏损),在计入部分拥有的附属公司与第三方之间的任何资本交易后,按综合资产负债表中的非控制性权益在每个报告期开始及结束时的差额厘定。
收入确认
当客户获得对承诺的商品或服务的控制权时,收入即被确认。确认的收入金额反映了公司预期有权换取这些商品和服务的对价。为了实现这一核心原则,公司采用了以下五个步骤:1)确定客户合同;2)确定合同的履约义务;3)确定交易价格;4)将交易价格分配给履约义务;5)在履行履约义务时确认收入。
对许可安排进行分析,以确定所承诺的货物或服务--可能包括许可证以及研发材料和服务--是不同的,还是必须作为合并履行义务的一部分加以核算。
F-10
交易价格是根据公司将有权获得的对价确定的。交易价格可以包括固定金额、可变金额或两者兼有。本公司在每个报告期重新评估实现该可变对价的可能性和任何相关限制。本公司在交易价格中包括可变对价,只要确认的累计收入很可能不会发生重大逆转。
本公司根据相关履约责任的估计独立售价分配交易价格。公司必须建立假设,需要判断来确定合同中确定的每项履约义务的独立销售价格。该公司利用关键假设来确定独立销售价格,其中可能包括其他可比交易、交易谈判中考虑的定价以及完成各自履约义务的估计成本。本公司还利用判断来评估可变对价是否受到限制,或者是否可以专门分配给安排中的一项或多项履约义务。
当履行责任已履行时,收入按按相对独立销售价格基准分配给该履行责任的交易价格确认,这不包括受约束的可变对价估计。在决定一项安排所需的努力程度及预期本公司在一项安排下完成其履约责任的期间时,需要有重大的管理层判断。
对于由许可证和其他承诺组成的履约义务,公司利用判断来评估合并的履约义务是否在一段时间或某个时间点得到履行,以及分配给履约义务的交易价格部分的确认模式。
研究和开发费用
研究和开发成本在发生时计入费用。研发费用主要包括员工薪酬成本及因提供产品候选开发、临床及临床前开发及相关供应及制造服务而与第三方产生的金额,以及合规成本。在报告期末,该公司将向第三方服务提供商支付的款项与在完成研究或开发目标方面的估计进展进行比较。随着获得更多信息,这些估计可能会发生变化。根据向服务提供商付款的时间和公司估计所提供服务取得的进展,公司可能会记录与这些成本相关的预付或应计费用净额。
如果被许可的技术在未来没有其他用途,则获得许可所产生的成本被确认为研究和开发费用。将来收到的用于研究和发展活动的货物或服务的预付款被记为预付费用。预付金额在收到相关货物或提供服务时计入费用。
该公司已与美国境内外的各方签订了各种与研发相关的合同。与这些协议相关的付款在发生时被记录为研发费用。本公司记录估计的持续研究和开发成本的应计负债。在评估应计负债的充分性时,公司分析研究或临床试验的进展情况,包括事件的阶段或完成情况、收到的发票和合同成本。在确定任何报告期结束时的应计结余时,会作出重大判断和估计。实际结果可能与公司的估计不同。
专利费用
基于股权的薪酬
以权益为基础的薪酬于授予日按授予雇员及非雇员的所有基于权益的奖励以奖励的公允价值计量,并于必需的服务期(通常为归属期间)内确认为开支。没收行为在发生时予以确认。
F-11
该公司向员工和非员工授予基于服务的RSU、基于市场的RSU和股票期权。基于服务的限制性股票单位(“RSU”)的公允价值是公司普通股在授予日的收盘价。基于市场的RSU的公允价值在授予日使用蒙特卡洛模拟模型进行计量。公司采用布莱克-斯科尔斯期权定价模型估算股票期权的公允价值,该模型需要输入客观和主观假设。使用的某些假设,包括首次公开募股前公司普通股的公允价值和股价波动,代表管理层的估计,涉及内在不确定性和管理层判断的应用。因此,如果因素发生变化,管理层使用不同的假设,基于股权的薪酬支出对于未来的奖励可能会有实质性的不同。
公司在其综合经营报表和全面收益(亏损)表中对基于权益的薪酬进行分类的方式与对获奖者的工资成本进行分类或对获奖者的服务付款进行分类的方式相同。
所得税
只有在所得税头寸更有可能持续的情况下,公司才会确认这些头寸的影响。确认的所得税头寸是按结算时可能实现的最大利益金额计算的。计量的变化反映在判断发生变化的期间。
综合收益(亏损)
综合收益(亏损)被定义为企业在一段时期内因非所有者来源的交易和其他事件和情况而发生的权益变化。该公司其他全面亏损的唯一要素是投资的未实现损益。
每股收益(净亏损)
每股基本收益(净亏损)是通过普通股股东应占收益(净亏损)除以当期已发行普通股的加权平均数来确定的。每股摊薄收益(净亏损)是通过普通股股东应占收益(净亏损)除以期内已发行普通股的加权平均数,并根据使用库存股方法确定的普通股等价物股份的稀释影响进行调整而确定的。
新兴成长型公司的地位
本公司是一家新兴的成长型公司,如2012年的JumpStart Our Business Startups Act(“JOBS法案”)所界定。根据《就业法案》,新兴成长型公司可以推迟采用在《就业法案》颁布后发布的新的或修订后的会计准则,直到这些准则适用于私营公司。本公司已选择使用此延长过渡期以符合新的或经修订的会计准则,该等准则对上市公司及私人公司具有不同的生效日期,直至(I)不再是新兴成长型公司或(Ii)明确及不可撤销地选择退出《就业法案》所规定的延长过渡期,两者以较早者为准。因此,这些合并财务报表可能无法与截至上市公司生效日期遵守新的或修订的会计声明的公司进行比较。
《就业法案》并不排除新兴成长型公司在新的或修订后的会计准则适用于私营公司之前采用该准则。该公司预计在其仍是一家新兴成长型公司期间,将把延长的过渡期用于任何其他新的或修订的会计准则。
最近采用的会计公告
2022年1月1日,公司通过了新的租赁准则(修订后的ASC 842),要求承租人在资产负债表上确认除某些短期租赁外的所有租赁的租赁负债和ROU资产。在实施ASC 842的过程中,本公司采用了一套由三项实际措施组成的权宜之计,使其能够延续其先前的租赁分类和嵌入的租赁评估,而不重新评估截至采用之日的初始直接成本。该公司还采取了一项实际的权宜之计,允许其在其房地产租赁中合并租赁和非租赁部分。
公司与单一公司地点相关的现有租赁义务受新标准的约束,导致经营租赁负债和净资产在实施日记入公司的综合资产负债表。现有租赁义务被归类为经营性租赁。
F-12
下表详细说明了2022年1月1日记录的与公司采用ASC 842相关的资产负债表调整(单位:千):
|
|
2021年12月31日 |
|
|
|
|
|
2022年1月1日 |
|
|||
|
|
根据ASC 840报告 |
|
|
ASC 842调整 |
|
|
根据ASC 842报告 |
|
|||
资产 |
|
|
|
|
|
|
|
|
|
|||
经营性租赁使用权资产 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
负债 |
|
|
|
|
|
|
|
|
|
|||
经营租赁负债的当期部分 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
递延租金 |
|
$ |
|
|
$ |
( |
) |
|
$ |
|
||
经营租赁负债的非流动部分 |
|
$ |
|
|
$ |
|
|
$ |
|
|||
2019年12月,财务会计准则委员会发布了ASU第2019-12号,旨在简化所得税的会计处理。本公司于2022年1月1日采用本标准。本标准的采用对本公司的综合财务状况及综合经营业绩并无重大影响。
2022年6月,本公司出售其部分拥有的子公司库利南珍珠的股权,库利南珍珠拥有齐帕替尼(CLN-081/TAS6417)的全球权利,不包括日本和中国内地中国以及香港、澳门和台湾(“大中国”)。,向泰豪支付一笔预付款$
本公司得出的结论是,该交易是对主要由知识产权和相关无形资产组成的非金融资产的出售,并在出售结束时转移了对非金融资产的控制权。该公司确认出售库利南珍珠的收益为#美元。
|
|
(单位:千) |
|
|
出售资产的账面价值 |
|
|
|
|
现金 |
|
$ |
|
|
预付费用和其他流动资产 |
|
|
|
|
可归因于出售资产的金额 |
|
|
|
|
已出售负债的账面价值 |
|
|
|
|
应计费用和其他流动负债 |
|
|
|
|
可归因于已出售负债的金额 |
|
|
|
|
出售的可识别净资产总额 |
|
|
|
|
预付对价,包括转账现金#美元 |
|
|
|
|
库利南珍珠的销售收益 |
|
$ |
|
|
在2022年期间,库利南珍珠发行了$
与大鹏签订共同开发协议
2022年6月,在完成出售公司在库利南珍珠的股权的同时,公司与泰豪的一家关联公司签订了一项共同开发协议,根据协议,公司将合作开发齐帕尔替尼,并将保留在美国共同商业化齐帕替尼的选择权。出售公司在库利南珍珠的股权后产生的齐帕尔替尼的开发成本将由泰豪和本公司平分,每一方都将收到
F-13
该公司的结论是,与大鹏公司的共同开发协议是一项合作安排,因为该公司是齐帕替尼开发的积极参与者。出售后,向泰豪支付或从泰豪收到的齐帕替尼开发活动的付款记录在研究和开发费用中。2022年,可由泰豪偿还并反映为研究和开发费用减少的费用为#美元
投资
本公司于2022年12月31日按证券类别确认其短期和长期投资如下:
|
|
摊销 |
|
|
毛收入 |
|
|
毛收入 |
|
|
估计数 |
|
||||
|
|
(单位:千) |
|
|||||||||||||
短期投资 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
公司票据 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
资产支持证券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
商业票据 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
美国政府注意到 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
短期投资总额 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
长期投资 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
公司票据 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
长期投资总额 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
总投资 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
本公司于2021年12月31日按证券类别确认其短期和长期投资如下:
|
|
摊销 |
|
|
毛收入 |
|
|
毛收入 |
|
|
估计数 |
|
||||
|
|
(单位:千) |
|
|||||||||||||
短期投资 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
公司票据 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
商业票据 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
美国政府注意到 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
短期投资总额 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
长期投资 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
公司票据 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
资产支持证券 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
美国政府注意到 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
长期投资总额 |
|
|
|
|
|
|
|
|
( |
) |
|
|
|
|||
总投资 |
|
$ |
|
|
$ |
|
|
$ |
( |
) |
|
$ |
|
|||
金融工具的公允价值
下表列出了截至2022年12月31日按公允价值经常性计量的公司金融资产的公允价值:
|
|
1级 |
|
|
2级 |
|
|
3级 |
|
|
总计 |
|
||||
|
|
(单位:千) |
|
|||||||||||||
短期投资 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
公司票据 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
资产支持证券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
商业票据 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
美国政府注意到 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
短期投资总额 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
长期投资 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
公司票据 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
长期投资总额 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
总投资 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
F-14
下表列出了截至2021年12月31日按公允价值经常性计量的公司金融资产的公允价值:
|
|
1级 |
|
|
2级 |
|
|
3级 |
|
|
总计 |
|
||||
|
|
(单位:千) |
|
|||||||||||||
短期投资 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
公司票据 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
商业票据 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
美国政府注意到 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
短期投资总额 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
长期投资 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
公司票据 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
资产支持证券 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
美国政府注意到 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
长期投资总额 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
总投资 |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
||||
截至2022年12月31日和2021年12月31日,财产和设备净额如下:
|
|
十二月三十一日, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(单位:千) |
|
|||||
电脑 |
|
$ |
|
|
$ |
|
||
办公家具和设备 |
|
|
|
|
|
|
||
租赁权改进 |
|
|
|
|
|
|
||
财产和设备总额(毛额) |
|
|
|
|
|
|
||
减去:累计折旧 |
|
|
( |
) |
|
|
( |
) |
财产和设备合计(净额) |
|
$ |
|
|
$ |
|
||
折旧费用为$
截至2022年12月31日和2021年12月31日,应计费用和其他流动负债包括:
|
|
十二月三十一日, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(单位:千) |
|
|||||
应计研究和开发成本 |
|
$ |
|
|
$ |
|
||
应计奖金 |
|
|
|
|
|
|
||
其他流动负债 |
|
|
|
|
|
|
||
可转换票据和应计利息 |
|
|
|
|
|
|
||
|
|
$ |
|
|
$ |
|
||
阿迪玛布
本公司与Adimab,LLC有合作协议(“Adimab”)(“Adimab合作协议”)。根据Adimab合作协议,该公司选择了一些生物靶点,Adimab使用其专有平台技术根据双方商定的研究计划发现和/或优化抗体。根据Adimab合作协议,该公司有能力选择特定数量的额外生物靶标,Adimab将针对这些靶标提供额外的抗体发现和优化服务。
在与Adimab的特定研究计划的研究期和评估期内,该公司在Adimab的技术下拥有非独家的全球许可,可以执行某些研究活动并评估该计划的抗体,以确定该公司是否想要行使其选择权,以获得利用此类抗体的免版税、全额、非独家许可,并通过多个级别进行再许可(“Adimab选项”)。
F-15
根据Adimab合作协议,该公司一次性支付了不可贷记且不可退还的技术访问费。该公司还需要支付与Adimab全职员工履行Adimab合作协议义务相关的年度访问费和研究资金费用。该公司还有义务按计划向Adimab支付某些研究交付、临床和销售里程碑付款,但有一定的折扣和折扣。
该公司有义务按年全球净销售额的较低个位数百分比按产品逐一支付某些特许权使用费。此类专利使用费义务将在以下两者中较晚的时间按国家/地区和产品终止:(A)此类产品在该国家/地区首次商业销售后的某一较低的两位数年数和(B)涵盖该产品的计划专利中最后一次发布且未过期、永久撤销或无效的权利要求到期时。
本公司可随时以任何理由在规定期限内提前书面通知终止Adimab合作协议。如果没有行使ADIMAB选择权或行使了ADIMAB选择权,则ADIMAB合作协议的有效期在最后一项研究计划的评估期结束时到期,在特定期限较晚到期或相关产品的无效专利覆盖范围之后。
在2022年至2021年期间,该公司记录了
库利南·琥珀-麻省理工学院
库利南琥珀公司(“库利南琥珀”)与麻省理工学院(“麻省理工学院”)签署了一项独家专利许可协议,将在全球范围内开发一种癌症免疫治疗产品(“麻省理工学院许可协议”)。库利南琥珀还负责支付不可退还的、可计入贷方的年度许可证维护费,在一定年限内递增支付,并在此期间之后支付固定金额。此外,麻省理工学院授予库利南·琥珀修改最初确定的字段以包括扩展字段的独家选择权,这种修改将触发向麻省理工学院支付修改费。
此外,库利南琥珀有义务支付某些不可退还、不可贷记的里程碑付款,最高可达$
在某些情况下,当库利南琥珀的控制权发生变更时,库利南琥珀需要支付指定的控制权变更费用和库利南Amber的临床和监管里程碑付款将增加
此外,库利南琥珀公司需要在每个报告期内按所有特许产品的净销售额支付较低的个位数特许权使用费百分比,但须进行某些抵消或减少。麻省理工学院对特许产品净销售额的版税减幅不得超过两位数的中位数百分比。库利南琥珀公司还被要求分享从许可产品的再许可中获得的任何收入,百分比将由许可产品的临床阶段确定,不超过低至中两位数的百分比。此类专利使用费义务将在所有已颁发专利和在专利权范围内提交的专利申请到期或被放弃时,在逐个国家和逐个产品的基础上到期。
在2022年至2021年期间,该公司记录了
库利南佛罗伦萨-图宾根许可协议
库利南佛罗伦萨公司(“库利南佛罗伦萨”)与德意志银行(DKFZ)、图宾根大学埃伯哈德·卡尔斯大学医学院(“图宾根大学”)和德国图宾根州立大学(UFE)签订了独家许可协议(Tübingen许可协议)。根据Tübingen许可协议,DKFZ和Tübingen大学,统称为许可方,根据某些许可的专利权、申请、技术信息和专有技术,向佛罗伦萨授予了具有里程碑和特许权使用费的全球独家许可,并有权通过多个层次授予再许可,以在该领域内研究、开发、商业化或以其他方式开发许可产品。
当与许可产品相关的某些临床和监管事件发生时,佛罗伦萨应向许可方支付某些不可退还、不可计入的里程碑付款。每笔里程碑付款只支付一次,最高可达一定的付款金额。
F-16
此外,库利南佛罗伦萨还需要在特许权使用费期限内,按每个国家和产品的每种许可产品的净销售额支付低至中个位数的特许权使用费百分比,但须进行某些补偿或减少。许可方在一个日历年度内因任何许可产品的净销售额而应向许可方支付的全球版税总额不得低于低至中个位数的百分比。这种专利使用费义务将在下列两者中较晚的一种情况下失效:(A)涵盖该国产品的专利的最后有效权利要求到期时,以及(B)产品在该国首次商业销售后的两位数低周年纪念日。在某些情况下,在首次控制权变更时,库利南佛罗伦萨应支付销售收益的不可退还、不可贷记的个位数百分比,但在库利南佛罗伦萨完成首次公开募股后不需要支付此类款项。
任何一方在另一方实质性违约或另一方资不抵债时,均可终止协议。在首次提交研究新药申请或临床试验协议或CTA后,库利南佛罗伦萨可通过提供事先书面通知,以任何或无理由终止DKFZ/Tübingen许可协议。如果佛罗伦萨库利南公司或其任何附属公司对某些专利权的有效性提出质疑,许可方可以通过事先书面通知的方式终止协议。除非提早终止,否则Tübingen许可协议将永久继续。根据Tübingen许可协议,到目前为止还没有实现任何里程碑。本公司在2022年至2021年期间未发生与本协议相关的许可费支出。
库利南MICA-Steinle协议
库利南MICA与Alexander Steinle博士达成了一项协议(“Steinle协议”),后者为库利南MICA提供了发现、设计和开发单抗的服务,这种单抗可以防止MICA从癌细胞表面蛋白质分解,并增强表达NKG2D受体的免疫细胞(“MICA抗体”)对这些癌细胞的杀伤作用。
根据这项协议,库利南MICA将在与MICA抗体有关的某些临床和管理事件发生时向斯坦勒博士支付某些不可退还、不可贷记的里程碑付款。每笔里程碑付款只支付一次,最高可达一定的付款金额。库利南MICA还被要求在特许权使用期内,根据每个国家和产品的每一种MICA抗体的净销售额,向Steinle博士支付较低的个位数特许权使用费百分比,但受某些补偿或减少的限制。在2022年期间,公司记录了$
库利南珍珠-泰豪和再鼎医药许可协议
于二零二零年,库力南明珠与再鼎医药上海有限公司(“再鼎医药”)订立许可协议(“载许可协议”),授予再鼎医药于大区中国研究、开发、商业化及制造CLN-081及含有CLN-081的产品的独家许可使用费。作为授予再鼎医药的许可和权利的部分对价,再鼎医药向库利南·珀尔一次性支付了不可撤销、不可退还的许可费#美元。
库利南·珀尔收到的预付款需缴纳外国预扣税款。2021年,当许可的知识产权和技术诀窍转让给再鼎医药时,公司确认收入为1美元。
根据与大鹏的收入分成协议,该公司还记录了$
本公司于2022年6月完成将其于库利南珍珠的全部股权出售给泰豪。有关交易的其他详情,请参阅附注3。
普通股
附属公司的非控股权益
某些子公司根据许可协议发行普通股,并根据子公司股权激励计划向员工、董事和顾问发行普通股。子公司普通股的持有者每股享有一票投票权。附属公司普通股持有人有权在子公司董事会宣布时获得股息,而在任何一种情况下,只有在向各自子公司的优先股持有人支付了所有需要支付的优先金额后,才有权获得股息。
F-17
库利南琥珀
2021年6月,公司购买了
2022年6月,公司购买了
2022年11月,公司购买了
截至2022年12月31日,公司持有普通股和A系列优先股
In 2022 and 2021, $
佛罗伦萨库利南
2021年7月,公司购买了
2022年7月,公司购买了
截至2022年12月31日,公司持有普通股、A系列优先股和B系列优先股
在2022年和2021年,
库利南云母
2021年6月,公司购买了
2022年3月,公司购买了
2022年10月,公司购买了
2022年11月,公司购买了
截至2022年12月31日,公司持有
In 2022 and 2021, $
在被公司收购之前,库利南MICA董事会授权向该实体的员工、董事、顾问和其他关键人员授予股票期权。2022年10月,库利南MICA董事会授权加速归属约
库利南明珠
F-18
2022年6月,公司将其部分持股的子公司库利南珍珠的股权出售给泰豪。有关交易的其他详情,请参阅附注3。
在出售前,本公司采用HLBV法对非控股权益进行会计处理。2022年和2021年,
在2022年和2021年的综合经营报表和全面收益(亏损)中,公司在以下费用类别中记录了基于股权的补偿:
|
|
截至十二月三十一日止的年度: |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(单位:千) |
|
|||||
研发 |
|
$ |
|
|
$ |
|
||
一般和行政 |
|
|
|
|
|
|
||
基于股权的薪酬总额 |
|
$ |
|
|
$ |
|
||
2021年股票期权和激励计划
库利南通过《2021年股票期权和激励计划》(《2021年股票计划》),以股票期权、限制性股票奖励(RSA)和RSU的形式向其员工、董事、顾问和其他关键人员授予股权奖励。公司还在2021年股票计划之外以股票期权的形式授予股权奖励,作为个人进入公司就业的激励材料。截至2022年12月31日,大约有
2021年股票计划规定,根据2021年股票计划保留和可供发行的股票数量将在每年1月1日自动增加
授予的期权具有
确定期权的公允价值
期权的公允价值是使用Black-Scholes期权定价模型估计的,该模型考虑了诸如行权价格、授予日相关普通股的价值、预期期限、预期波动率、无风险利率和股息收益率等因素。2022年和2021年期间每次授予期权的公允价值是使用以下讨论的方法和假设确定的:
F-19
2022年和2021年,授予期权的加权平均授予日公允价值为#美元。
|
|
截至的年度 |
|
截至的年度 |
无风险利率 |
|
|
||
预期期限(以年为单位) |
|
|
||
预期波动率 |
|
|
||
预期股息收益率 |
|
|
确定以市场为基础的RSU的公允价值
该公司使用蒙特卡洛模拟模型在授予之日计量基于市场的RSU的公允价值。蒙特卡洛模拟需要输入假设,包括公司的股票价格、股票价格的波动性、剩余期限(年数)、预期股息收益率和无风险比率。该公司使用自己的交易历史来计算所授予的基于市场的RSU的预期波动率。无风险利率是通过参考美国国债的隐含收益来确定的,剩余期限等于授予日假定的预期期限。
下表详细说明了蒙特卡洛模拟模型中用来估计2022年期间授予的基于市场的RSU的公允价值的假设:
|
|
截至的年度 |
|
|
股票价格 |
|
$ |
|
|
波动率 |
|
|
% |
|
剩余期限(年) |
|
|
|
|
无风险利率 |
|
|
% |
|
预期股息收益率 |
|
|
|
|
股票期权
下表汇总了2022年和2021年的股票期权活动(股票和总内在价值,以千为单位):
|
|
数量 |
|
|
加权平均行权价 |
|
|
加权平均剩余合同期限(年) |
|
|
聚合内在价值 |
|
||||
截至2020年12月31日的未偿还债务 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
授与 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
已锻炼 |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
被没收 |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
截至2021年12月31日的未偿还债务 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
授与 |
|
|
|
|
$ |
|
|
|
|
|
|
|
||||
已锻炼 |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
被没收 |
|
|
( |
) |
|
$ |
|
|
|
|
|
|
|
|||
截至2022年12月31日的未偿还债务 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
自2022年12月31日起可行使 |
|
|
|
|
$ |
|
|
|
|
|
$ |
|
||||
截至2022年12月31日和2021年12月31日,
F-20
期权的总内在价值计算为行使价格低于公司普通股公允价值的期权的行权价格与公司普通股公允价值之间的差额。2022年和2021年行使的期权的内在价值总额为#美元。
RSU
2022年2月,公司向员工和非员工发放了基于服务的RSU。2022年6月,该公司签订了一项协议,向其首席执行官授予基于市场的RSU。
|
|
数量 |
|
|
加权平均授予日期公允价值 |
|
||
截至2021年12月31日的未清偿未归属资产 |
|
|
|
|
$ |
|
||
授与(1) |
|
|
|
|
$ |
|
||
既得 |
|
|
( |
) |
|
$ |
|
|
被没收 |
|
|
( |
) |
|
$ |
|
|
截至2022年12月31日的未偿还未归属资产 |
|
|
|
|
$ |
|
||
截至2022年12月31日,
RSA
作为2021年1月重组的一部分,公司用重组前公司及其部分拥有的子公司颁发的股权奖励交换了公司普通股的RSA。
|
|
数量 |
|
|
加权平均授予日期公允价值 |
|
||
截至2020年12月31日的未归属余额 |
|
|
|
|
$ |
|
||
既得 |
|
|
( |
) |
|
$ |
|
|
截至2021年12月31日的未清偿未归属资产 |
|
|
|
|
$ |
|
||
既得 |
|
|
( |
) |
|
$ |
|
|
被没收 |
|
|
( |
) |
|
$ |
|
|
截至2022年12月31日的未偿还未归属资产 |
|
|
|
|
$ |
|
||
截至2022年12月31日和2021年12月31日,
2021年员工购股计划
2021年员工股票购买计划(“ESPP”)授权向参与计划的合格员工发行普通股,并规定每年两个为期六个月的发售期限。截至2022年12月31日,大约有
ESPP规定,根据ESPP保留和可供发行的股份数量将在每年1月1日自动增加较小的
于2022年及2021年期间,本公司每年发行少于
F-21
库利南·安珀、库利南·佛罗伦萨和库利南·米卡分别是与MPM肿瘤学慈善基金会和瑞银擎天柱基金会(统称为基金会)签订的特许权使用费转移协议的一方。根据这些各自的协议,每个基金会有权获得适用子公司开发的任何产品全球净销售额的较低个位数版税百分比,但受专利到期后的限制和控制权变更后开发的知识产权的限制。本公司认为该等特许权使用费转让协议为独立的金融工具,应按公允价值入账。本公司的结论是,这些票据在协议开始时没有价值。
鉴于基础技术的早期性质,以及与获得批准和商业化相关的固有技术、监管和竞争风险,截至2022年12月31日和2021年12月31日,该公司没有将专利费转让协议归因于任何价值。该公司目前没有任何适用的产品净销售额,因此,
于2022年期间,本公司录得当期所得税开支为#美元
公司2022年和2021年法定所得税率与公司有效所得税率的对账如下:
|
|
截至十二月三十一日止的年度: |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
联邦法定利率 |
|
|
% |
|
|
% |
||
扣除联邦福利后的州税 |
|
|
% |
|
|
% |
||
估值免税额 |
|
|
% |
|
|
( |
)% |
|
其他 |
|
|
% |
|
|
% |
||
|
|
|
% |
|
|
% |
||
截至2022年12月31日和2021年12月31日,递延所得税净资产余额与以下相关:
|
|
十二月三十一日, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(单位:千) |
|
|||||
递延税项资产: |
|
|
|
|
|
|
||
净营业亏损 |
|
$ |
|
|
$ |
|
||
资本化研究与开发 |
|
|
|
|
|
|
||
资本化的组织和启动费用 |
|
|
|
|
|
|
||
许可证 |
|
|
|
|
|
|
||
应计费用 |
|
|
|
|
|
|
||
研发信贷 |
|
|
|
|
|
|
||
基于股权的薪酬 |
|
|
|
|
|
|
||
库利南珍珠销售收益基差 |
|
|
|
|
|
|
||
租赁责任 |
|
|
|
|
|
|
||
递延税项总资产 |
|
|
|
|
|
|
||
估值免税额 |
|
|
( |
) |
|
|
( |
) |
递延税项净资产 |
|
|
|
|
|
( |
) |
|
递延税项负债 |
|
|
|
|
|
|
||
ROU资产 |
|
|
|
|
|
|
||
折旧及摊销 |
|
|
|
|
|
( |
) |
|
递延税项净资产 |
|
$ |
|
|
$ |
|
||
F-22
公司的净营业亏损(“NOL”)和税收抵免结转受到美国国税局(IRS)的审查和可能的调整。和州税务机关。
截至2022年12月31日和2021年12月31日,公司拥有联邦NOL结转,金额为
截至2022年12月31日和2021年12月31日,公司拥有联邦研发税收抵免结转$
本公司已评估影响其实现递延税项资产能力的正面和负面证据,主要包括资本化的研发成本、股权薪酬的暂时性差异和NOL结转。除2022年出售库利南珍珠的一次性收益外,本公司已考虑其累积净亏损历史、估计未来应课税收入及审慎可行的税务筹划策略,并得出结论认为本公司更有可能无法实现其递延税项资产的收益。因此,截至2022年12月31日,该公司对其剩余的递延税项净资产保持了全额估值准备金。
|
|
截至十二月三十一日止的年度: |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(单位:千) |
|
|||||
年初的估值免税额 |
|
$ |
|
|
$ |
|
||
计入所得税拨备的增加 |
|
|
|
|
|
|
||
计入权益的增加 |
|
|
|
|
|
|
||
年终估值免税额 |
|
$ |
|
|
$ |
|
||
在计算公司的纳税义务时,涉及到对联邦税收和公司经营或经营所在州的复杂税收法律和法规的应用中的不确定性。
本公司确认一项来自不确定税务状况的税务利益,前提是该状况极有可能会在审核后得以维持,包括任何相关上诉或诉讼程序的解决方案,以技术价值为基础。本公司将不确定的税务状况记录为负债,并在其判断因评估以前无法获得的新信息而发生变化时对这些负债进行调整。由于其中一些不确定因素的复杂性,最终解决方案可能导致的支付与公司目前对未确认税收优惠负债的估计大不相同。这些差异将在可获得新信息的期间反映为所得税费用的增加或减少。截至2022年12月31日和2021年12月31日,公司已
该公司在随附的综合经营报表和全面收益(亏损)中确认与所得税支出项目中未确认的税收优惠相关的利息和罚款。截至2022年和2021年12月31日,
公司按照其经营所在司法管辖区的税法的规定提交纳税申报单。在正常业务过程中,本公司须接受美国联邦和州司法管辖区的审查。目前没有悬而未决的税务审查。该公司的联邦和州所得税申报单通常在2016及以后的纳税年度接受税务审查。在本公司具有税收结转属性的情况下,产生该属性的纳税年度仍可经美国国税局和国家税务机关审核后在未来一段时间内进行调整。
F-23
该公司在正常业务过程中与合同研究机构、合同制造机构和其他第三方就临床前研究、临床试验以及测试和制造服务签订合同。这些协议通常包括取消条款。
赔偿协议
在正常业务过程中,公司可以就某些事项向供应商、出租人、业务合作伙伴和其他各方提供不同范围和条款的赔偿,包括但不限于因违反此类协议或第三方提出的知识产权侵权索赔而产生的损失。此外,本公司已与其董事会成员及高级管理人员订立弥偿协议,要求本公司(其中包括)就他们作为董事或高级管理人员的身份或服务而可能产生的若干责任作出弥偿。根据这些赔偿协议,公司未来可能需要支付的最大潜在金额在某些情况下是无限制的。到目前为止,本公司尚未因该等赔偿而产生任何重大成本。本公司并不知悉任何可能对其财务状况、经营业绩或现金流产生重大影响的赔偿安排,而本公司已
法律程序
本公司目前并未参与或知悉任何重大法律程序。于每个报告日期,本公司评估潜在亏损金额或潜在亏损范围是否根据权威性指引处理或有事项的规定而可能及合理地评估。本公司在发生与该等法律诉讼有关的费用时支出.
该公司的经营租约约为
下表汇总了2022年的补充现金流信息(单位:千):
|
|
截至的年度 |
|
|
为计量租赁负债所包括的金额支付的现金: |
|
|
|
|
来自经营租赁的经营现金流(1) |
|
$ |
|
|
为换取经营租赁负债而获得的净资产 |
|
$ |
|
|
下表汇总了截至2022年12月31日的加权平均租赁期限和折扣率:
|
|
2022年12月31日 |
|
|
加权平均剩余租赁年限(年) |
|
|
||
加权平均贴现率 |
|
|
% |
|
由于本公司的经营租赁没有提供隐含利率,本公司根据现有信息使用递增借款利率来确定租赁付款的现值。本公司的递增借款利率基于租赁期限、经济环境,并反映了本公司在有担保的基础上必须支付的借款利率。
F-24
下表汇总了截至2022年12月31日,公司在新租赁会计准则下的未来最低租赁支付金额(单位:千):
|
|
2022年12月31日 |
|
|
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
2026 |
|
|
|
|
未来最低租赁付款总额 |
|
|
|
|
减去:推定利息 |
|
|
( |
) |
按现值计算的租赁负债总额 |
|
$ |
|
|
下表汇总了公司截至2021年12月31日根据先前租赁会计准则支付的未来最低租赁金额(单位:千):
截至12月31日止的年度, |
|
(单位:千) |
|
|
2023 |
|
$ |
|
|
2024 |
|
|
|
|
2025 |
|
|
|
|
|
|
$ |
|
|
转租协议
于2022年9月,本公司就Suite 520租赁订立了一份至2024年5月的分租协议。于2022年,本公司录得分租收入#美元。
下表列出了2022年和2021年基本和稀释后每股收益(净亏损)的计算方法:
|
|
截至十二月三十一日止的年度: |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
|
|
(单位为千,每股数据除外) |
|
|||||
分子: |
|
|
|
|
|
|
||
库利南公司普通股股东应占净收益(亏损) |
|
$ |
|
|
$ |
( |
) |
|
分母: |
|
|
|
|
|
|
||
加权平均已发行普通股-基本 |
|
|
|
|
|
|
||
假定行使股权奖励对可发行普通股的稀释效应 |
|
|
|
|
|
|
||
加权平均已发行普通股-摊薄 |
|
|
|
|
|
|
||
每股收益(净亏损): |
|
|
|
|
|
|
||
基本信息 |
|
$ |
|
|
$ |
( |
) |
|
稀释 |
|
$ |
|
|
$ |
( |
) |
|
公司采用库存股方法确定稀释股数。下表列出了不包括在2022年和2021年稀释后每股收益(净亏损)计算中的潜在普通股,因为它们的影响将是反稀释的:
|
|
截至十二月三十一日止的年度: |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
股票期权 |
|
|
|
|
|
|
||
RSA |
|
|
|
|
|
|
||
RSU |
|
|
|
|
|
|
||
ESPP |
|
|
|
|
|
|
||
总计 |
|
|
|
|
|
|
||
F-25
BVF普通股交易所
于二零二三年一月,本公司与Biotech Value Fund,L.P.、Biotech Value Fund II,L.P.、Biotech Value Trading Fund OS LP及MSI BVF SPV,LLC(“股东”)订立交换协议,据此,股东交换
CLN-418的入网许可
2023年2月,公司与港湾生物医药美国公司(“港湾”)签订了一项许可和合作协议(“港湾许可协议”),根据该协议,港湾授予公司在美国开发、制造和商业化HBM7008(CLN-418)的独家许可。
根据海港特许协议的条款,该公司向海港支付了一笔预付许可费#美元。
除非较早前终止,否则海港许可协议将继续有效,直至本公司的专利权使用费义务届满。港口许可协议可由任何一方因另一方的重大违约而终止,但须遵守通知和补救条款,或在另一方破产的情况下终止。为方便起见,本公司可于90天前以书面通知海港公司,终止《海港许可证协议》。在《海港许可证协议》中,每一方都作出了习惯陈述和保证,并同意了习惯契约,包括但不限于对这类交易的赔偿。
展品索引
展品 数 |
|
描述 |
|
|
|
3.1 |
|
经修订证书修订的第二次修订和重新注册的注册人注册证书,自2021年2月25日起生效(通过引用2021年3月30日提交给美国证券交易委员会的注册人截至2020年12月31日的10-K表格年度报告的附件3.1并入)。 |
|
|
|
3.2 |
|
第二次修订和重新修订的注册人章程,自2021年2月25日起生效(合并时参考了注册人于2021年3月30日向美国证券交易委员会提交的截至2020年12月31日的10-K表格年度报告的附件3.2)。 |
|
|
|
3.3 |
|
A系列可转换优先股的优先股、权利和限制指定证书(合并内容参考注册人于2023年1月19日提交给美国证券交易委员会的8-K表格当前报告的附件3.1)。 |
|
|
|
4.1 |
|
普通股证书样本(参考2020年12月28日提交给美国证券交易委员会的注册人S-1表格登记说明书(文件编号333-251512)附件4.1并入)。 |
|
|
|
4.2 |
|
注册人及其若干股东之间于2021年1月7日签订的登记权协议(合并内容参考了注册人于2021年3月30日向美国证券交易委员会提交的截至2020年12月31日的10-K表格年度报告附件4.2)。 |
|
|
|
4.3 |
|
证券说明(参考注册人于2021年3月30日向美国证券交易委员会提交的截至2020年12月31日的10-K表格年度报告的附件4.3)。 |
|
|
|
10.1# |
|
2021年股票期权和奖励计划及其奖励协议的格式(通过引用注册人于2021年1月4日提交给美国证券交易委员会的S-1表格登记声明(第333-251512号文件)附件10.1而并入)。 |
|
|
|
10.2# |
|
2021年员工购股计划(参考2021年1月4日提交给美国证券交易委员会的注册人S-1表格登记说明书(文件编号333-251512)附件10.2并入)。 |
|
|
|
10.3# |
|
高级管理人员现金激励奖金计划(参考2020年12月18日提交给美国证券交易委员会的注册人S-1表注册说明书(文件编号333-251512)附件10.3并入)。 |
|
|
|
10.4# |
|
赔偿协议表,由注册人及其每名董事(通过参考注册人于2020年12月18日提交给美国证券交易委员会的注册人S-1表格S-1(文件编号333-251512)附件10.4合并而成)。 |
|
|
|
10.5# |
|
赔偿协议表,由登记人与其每一名执行人员(通过参考登记人于2020年12月18日提交给美国证券交易委员会的S-1表格登记说明书(第333-251512号文件)附件10.5合并而成)。 |
|
|
|
10.6 |
|
独家专利许可协议,日期为2019年12月12日,由麻省理工学院和库利南琥珀公司之间签署,于2020年4月3日修订(通过参考2020年12月18日提交给美国证券交易委员会的注册人S-1表格注册声明(文件编号333-251512)的附件10.6而并入)。 |
|
|
|
10.7 |
|
Adimab,LLC与注册人之间于2018年11月28日签署的合作协议(通过引用注册人于2020年12月18日提交给美国证券交易委员会的S-1表格注册声明(文件编号333-251512)附件10.7而并入)。 |
|
|
|
10.8 |
|
股份购买协议,日期为2022年5月11日,由注册人、泰豪药业有限公司和库利南珍珠公司签订并相互之间(通过参考注册人于2022年8月10日提交给美国证券交易委员会的10-Q表格季度报告的附件10.1而合并)。 |
|
|
|
10.9 |
|
注册人与泰豪肿瘤学公司之间的共同开发协议,日期为2022年6月21日(合并内容参考注册人于2022年8月10日提交给美国证券交易委员会的10-Q表格季度报告的附件10.2)。 |
|
|
|
10.10 |
|
独家许可协议,日期为2020年8月31日,由德国Krebsforschungszentum公司、图宾根埃伯哈德·卡尔大学、图宾根大学医学系、Medizin Gesellschaft皮草公司、Tubingen公司和库利南佛罗伦萨公司签订(合并通过引用2020年12月18日提交给美国证券交易委员会的注册人S-1表格注册声明(第333-251512号文件)第10.9号附件)。 |
|
|
|
10.11# |
|
行政人员聘用协议表格(参考注册人于2020年12月18日向美国证券交易委员会提交的S-1表格注册说明书(文件编号333-251512)附件10.13)。 |
|
|
|
10.12# |
|
注册人和科琳·萨维尔之间于2019年1月1日签订的咨询协议(通过引用注册人于2020年12月18日提交给美国证券交易委员会的S-1表格注册声明(第333-251512号文件)附件10.14合并而成)。 |
|
|
|
10.13 |
|
转租,于2017年12月14日生效,由Teva PharmPharmticals USA,Inc.和注册人之间进行(通过引用2020年12月18日提交给美国证券交易委员会的注册人S-1表格注册声明(文件编号333-251512)第10.17号附件合并)。 |
|
|
|
10.14 |
|
表决协议表格(通过引用登记人于2020年12月18日提交给美国证券交易委员会的S-1表格登记声明(第333-251512号文件)附件10.18而并入)。 |
|
|
|
10.15 |
|
投资者权益协议书表格(参考注册人于2020年12月18日向美国证券交易委员会提交的S-1表格登记说明书(第333-251512号文件)附件10.19而合并)。 |
|
|
|
10.16 |
|
服务格式协议(通过引用注册人于2020年12月18日提交给美国证券交易委员会的S-1表格注册声明(文件编号333-251512)的附件10.20而并入)。 |
|
|
|
10.17 |
|
特许权使用费转让协议表(通过引用注册人于2020年12月18日提交给美国证券交易委员会的S-1表格登记声明(第333-251512号文件)附件10.21而并入)。 |
|
|
|
10.18 |
|
出资协议格式(通过参考登记人于2021年1月4日提交给美国证券交易委员会的S-1表格注册说明书(文件编号333-251512)附件10.22并入)。 |
|
|
|
10.19# |
|
董事非雇员薪酬政策(参考2020年12月18日提交给美国证券交易委员会的注册人S-1表格登记说明书(文件编号333-251512)第10.23条纳入)。 |
|
|
|
10.20# |
|
服务协议,由注册人和Patrick Baeuerle(通过参考注册人于2021年1月4日提交给美国证券交易委员会的注册人S-1表格注册声明(文件编号333-251512)的第10.25号附件合并而成)。 |
|
|
|
10.21# |
|
登记人与Jeffrey Jones之间于2022年2月28日生效的雇佣协议(合并内容参考登记人于2022年3月3日提交给美国证券交易委员会的当前8-K表格报告的附件10.1)。 |
|
|
|
10.22# |
|
登记人与纳迪姆·艾哈迈德之间于2022年6月9日签订的业绩单位奖励协议(合并内容参考了登记人于2022年8月10日提交给美国证券交易委员会的当前10-Q表格报告的附件10.3)。 |
|
|
|
10.23* |
|
登记人和Jeffrey Trigilio之间的雇佣协议,2021年1月7日生效 |
|
|
|
10.24* |
|
机构转让方股份购买转让协议格式 |
|
|
|
10.25* |
|
个人转让人股份购买和转让协议格式 |
|
|
|
10.26* |
|
麻省理工学院和库利南琥珀公司之间的独家专利许可协议第二修正案,日期为2022年12月20日。 |
|
|
|
10.27* |
|
注册人、MPM肿瘤学慈善基金会和瑞银擎天柱基金会之间于2022年6月6日签署的特许权使用费转移协议修正案1 |
|
|
|
21.1* |
|
注册人的子公司名单。 |
|
|
|
23.1* |
|
经本公司独立注册会计师事务所毕马威有限责任公司同意。 |
|
|
|
31.1* |
|
根据根据2002年《萨班斯-奥克斯利法案》第302节通过的1934年《证券交易法》第13a-14(A)和15d-14(A)条颁发的首席执行干事证书。 |
|
|
|
31.2* |
|
根据依照2002年萨班斯-奥克斯利法案第302节通过的1934年《证券交易法》第13a-14(A)和15d-14(A)条认证首席财务干事。 |
|
|
|
32.1** |
|
根据2002年《萨班斯-奥克斯利法案》第906节通过的《美国法典》第18编第1350条对首席执行官和首席财务官的认证。 |
|
|
|
101.INS |
|
内联XBRL实例文档(该实例文档不会出现在交互数据文件中,因为其XBRL标记嵌入在内联XBRL文档中) |
|
|
|
101.SCH |
|
内联XBRL分类扩展架构文档 |
|
|
|
101.CAL |
|
内联XBRL分类扩展计算链接库文档 |
|
|
|
101.DEF |
|
内联XBRL分类扩展定义Linkbase文档 |
|
|
|
101.LAB |
|
内联XBRL分类扩展标签Linkbase文档 |
|
|
|
101.PRE |
|
内联XBRL分类扩展演示文稿Linkbase文档 |
|
|
|
104 |
|
封面交互数据文件(格式为内联XBRL,包含在附件101中) |
* |
现提交本局。 |
** |
随信提供。 |
# |
指管理合同或补偿计划、合同或安排。 |
|
本展品的某些部分(用星号表示)已被省略,因为注册人已确定它们不是实质性的,如果公开披露可能会对注册人造成竞争损害。 |
签名
根据1934年《证券交易法》第13或15(D)节的要求,登记人已正式安排由正式授权的下列签署人代表其签署本报告.
|
|
库利南肿瘤公司。 |
|
|
|
|
|
日期:2023年3月9日 |
|
发信人: |
/s/纳迪姆·艾哈迈德 |
|
|
|
姓名:纳迪姆·艾哈迈德 |
|
|
|
总裁与首席执行官 |
授权委托书
每名个人签名如下的人士现授权并委任纳迪姆·艾哈迈德和杰弗里·特里格里奥为其真正合法的事实受权人和代理人,他们各自拥有完全的替代和再替代的权力,并有完全的权力在没有对方的情况下行事,以其名义、地点和代理行事,并以每一个人的名义和代表,以个人和下文所述的每一身份行事,并以表格10-K的形式提交对本年度报告的任何和所有修订,并将其连同所有证物和与此相关的其他文件提交证券交易委员会。授予上述事实代理人和代理人及其每一人作出和执行每一项作为和事情的全部权力和权力,批准和确认上述事实代理人和代理人或他们中的任何一人或他们或他们的替代者可以合法地作出或导致作出的所有行为和事情。
根据修订后的1934年《证券交易法》的要求,本报告已由以下注册人以登记人的身份在指定日期签署。
名字 |
|
标题 |
|
日期 |
|
|
|
|
|
|
|
|
|
|
/s/纳迪姆·艾哈迈德 |
|
董事首席执行官总裁 |
|
March 9, 2023 |
纳迪姆·艾哈迈德 |
|
(首席行政主任) |
|
|
|
|
|
|
|
|
|
|
|
|
/s/Jeffrey Trigilio |
|
首席财务官 |
|
March 9, 2023 |
杰弗里·特里格里奥 |
|
(首席财务会计官) |
|
|
|
|
|
|
|
|
|
|
|
|
/s/托马斯·埃贝林 |
|
|
|
March 9, 2023 |
托马斯·埃贝林 |
|
董事 |
|
|
|
|
|
|
|
安妮-玛丽·马丁 |
|
|
|
March 9, 2023 |
安妮-玛丽·马丁 |
|
董事 |
|
|
|
|
|
|
|
/s/安东尼·罗森伯格 |
|
|
|
March 9, 2023 |
安东尼·罗森伯格 |
|
董事 |
|
|
|
|
|
|
|
/David P.瑞安,医学博士 |
|
|
|
March 9, 2023 |
David·P·瑞安医学博士。 |
|
董事 |
|
|
|
|
|
|
|
/s/斯蒂芬·韦伯斯特 |
|
|
|
March 9, 2023 |
斯蒂芬·韦伯斯特 |
|
董事 |
|
|