目录表
我们的候选产品包括交付和制造,以及进行临床前研究和早期临床试验。我们没有任何候选产品被批准销售,也没有从产品销售中产生任何收入,我们目前所有的候选产品都处于发现和临床前开发阶段。
我们主要通过私募可赎回的可转换优先股、公开发行我们的普通股以及与我们的战略合作相关的费用、里程碑付款和成本补偿来为我们的运营提供资金,其中包括我们之前与赛诺菲Genzyme Corporation的合作,或赛诺菲Genzyme合作(始于2015年2月并于2019年6月终止),我们先前与AbbVie Biotech Ltd.的合作(专注于tau相关疾病),或AbbVie Tau合作(始于2018年2月并于2020年8月终止),我们先前与AbbVie爱尔兰无限公司的合作,专注于α-突触核蛋白的病理物种,或AbbVie Alpha-突触核蛋白合作,我们与Neurocrine的合作始于2019年2月,终止于2020年8月,我们与Neurocrine的持续合作始于2019年3月,我们与辉瑞的期权和许可协议始于2021年10月,我们与诺华的期权和许可协议始于2022年3月。我们将我们与Neurocrine的合作协议称为Neurocrine合作协议。我们将我们与辉瑞的选择权和许可协议称为辉瑞协议,将我们与诺华的选择权和许可协议称为诺华协议。
我们仍有兴趣推进我们的其他计划,并在发现和临床前开发阶段寻求其他候选产品,包括我们治疗亨廷顿病的第二代计划、治疗脊髓性肌萎缩症(SMA)的计划以及治疗人类表皮生长因子受体阳性(HER2+)脑转移的计划。我们正在评估通过合作伙伴关系这样做的机会,并将继续为我们的第二代亨廷顿病治疗计划和我们的HER2+脑转移治疗计划进行临床前研究。我们预计将继续为我们治疗SMA的第二代计划进行早期临床前研究,同时我们正在为该计划寻找合作伙伴。
我们继续与Neurocrine合作治疗包括Friedreich共济失调在内的疾病。我们目前所有的候选产品都处于开发的早期阶段。我们还在继续评估可以使用AAV基因疗法或我们专有抗体治疗的其他疾病,并积极探索其他可能的治疗方法,这些方法可以利用我们专有的示踪剂衣壳。
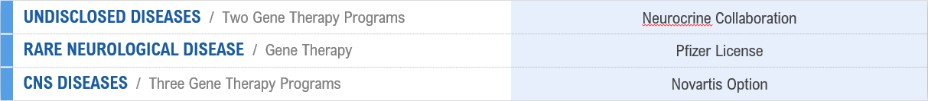
我们的优先基因治疗和其他生物治疗计划都处于临床前开发阶段,总结如下:

*早期研究计划包括针对转移性乳腺癌脑转移的vHER2矢量化抗体和亨廷顿病的HTT基因治疗。
*第一阶段结束后,Voyager可以选择在美国共同开发/共同商业化FA计划,将40%的成本/利润分配给Voyager,60%分配给Neurocrine,或者授予Neurocrine全球商业权。
31