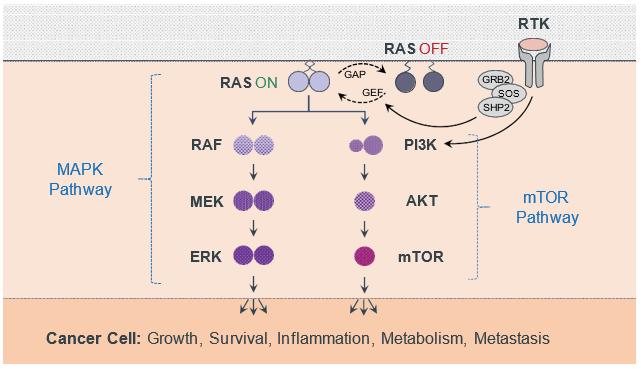
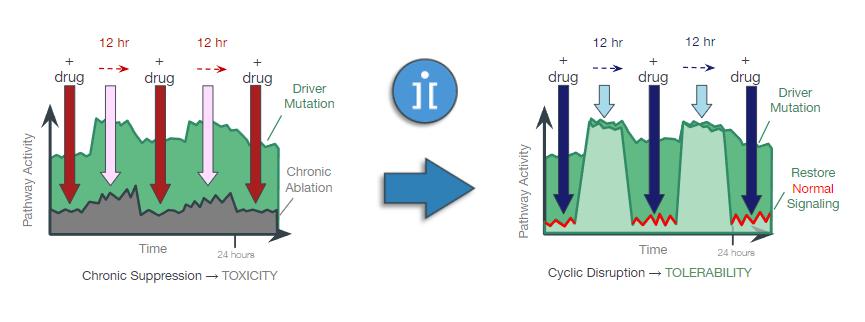
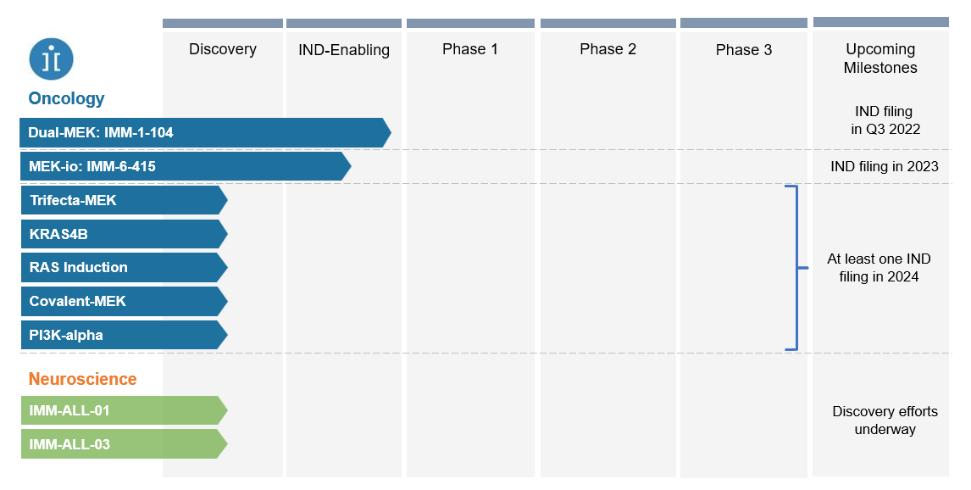
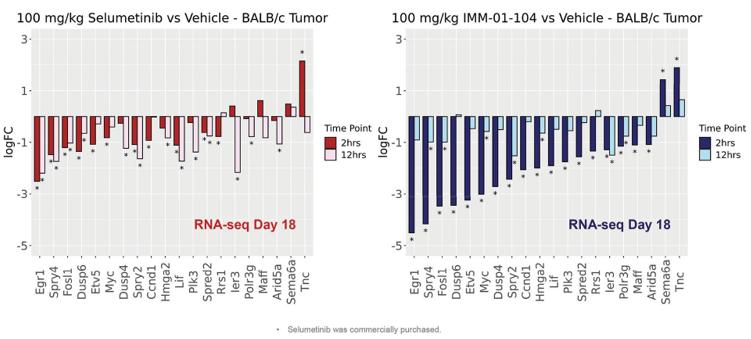
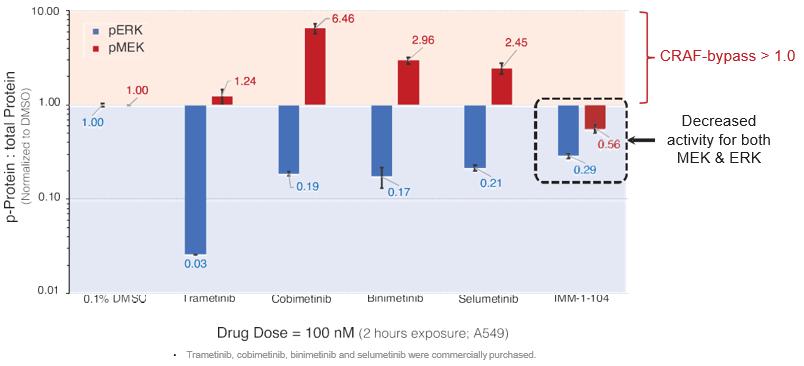
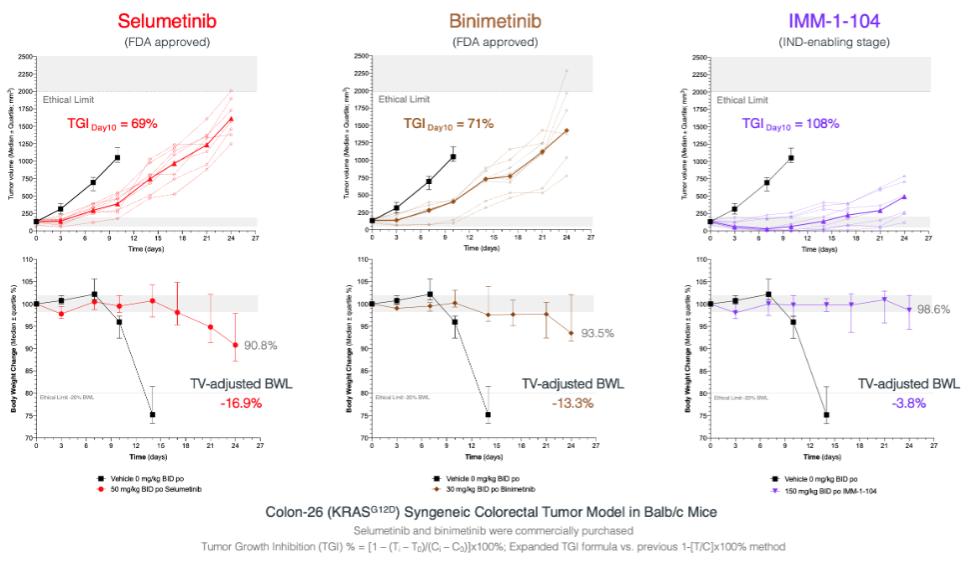
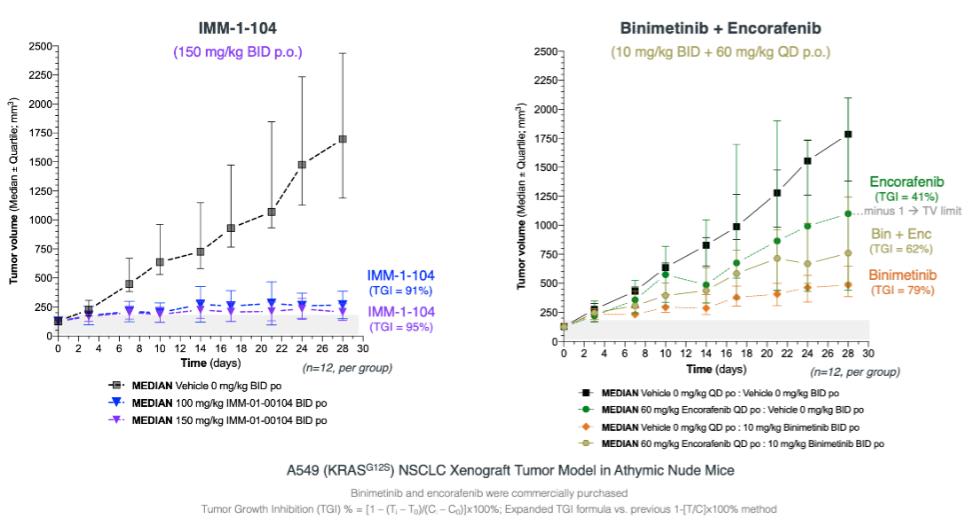
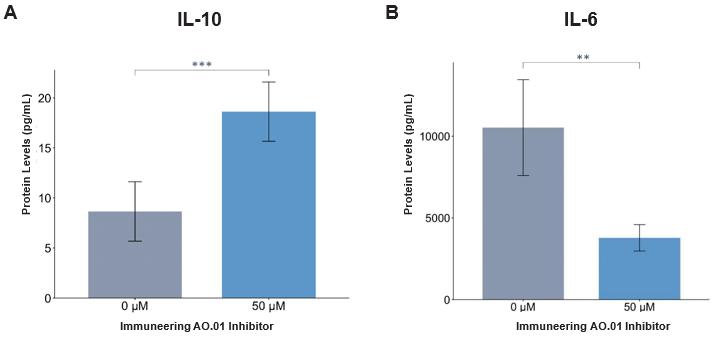
注8-可转换优先股
A系列优先股
2019年9月,本公司授权出售和发行最多1,987,979A系列优先股的股票,$0.001每股面值,原始发行价为$8.5514每股。2020年1月,A系列优先股的授权股票数量增加到2,495,933股份。A系列优先股融资的结构是在2019年至2020年期间滚动结束发行。
2019年9月20日,本公司额外发布了一份1,122,458A系列优先股,现金收益总额为$9,598,847并发布了785,706A系列优先股,连同转换已发行的可转换票据金额。于2019年,本公司产生的发行成本为$200,587与这份供品有关的信息。
该公司收到了一笔资金,用于发行一份额外的468,315A系列优先股,现金收益总额为$4,004,975到2019年12月31日。在这些股份中,410,436A系列优先股,现金收益总额为$3,509,802超过公司章程允许的核定金额,导致负债#美元。3,509,802总共有1,966,0432019年12月31日发行的A系列优先股。2020年1月,之前于2019年12月31日被归类为负债的股份在获准增加至A系列优先股的授权股份后被重新归类为临时股本。
2020年1月,本公司发布119,454增发A系列优先股,现金收益总额为$1,021,413。该公司产生的发行成本为#美元。23,610与2020年1月的融资有关。
B系列优先股
2020年12月,本公司授权出售和发行最多6,032,183B系列优先股的股票,$0.001每股面值,原始发行价为$10.2782每股。B系列优先股融资的结构是在二一批一批。第一期于2020年12月结束,本公司发行了3,619,292B系列优先股,现金收益总额为$37,199,929。该公司产生的发行成本为#美元。216,019与2020年12月的融资有关。
公司确定了投资者的购买权2,412,853第二批B系列优先股的股票不符合独立金融工具的定义,因为它不能与第一批发行的B系列优先股分开。第二批优先股的发行取决于本公司达到某些发展里程碑,或由B系列优先股当时至少多数已发行股票的持有人选举,B系列优先股必须包括一名特定股东(“必要持有人”)。B系列优先股的每个持有者可以在任何时候选择购买他们所需的第二批优先股。截至2021年3月31日,本公司没有达到这些发展里程碑,必要的持有人也没有选择在达到这些里程碑之前购买第二批,因此不是发行了第二批股票。