目录
通过我们的合作,包括与赛诺菲Genzyme公司或赛诺菲Genzyme的合作,艾伯维生物技术有限公司和AbbVie爱尔兰无限公司,或联合AbbVie和Neurocrine Biosciences,Inc.或Neurocrine。自成立以来,我们的运营一直专注于组织和配备我们的公司、业务规划、筹集资金、建立我们的知识产权组合、确定要研究的神经疾病、推进我们的候选产品(包括交付和制造),以及进行临床前研究和临床试验。我们没有任何候选产品被批准销售,也没有从产品销售中获得任何收入。我们主要通过私募可赎回的可转换优先股、公开发行我们的普通股以及我们的战略合作来为我们的运营提供资金,包括我们与赛诺菲基因酶,或赛诺菲Genzyme合作(始于2015年2月,于2019年6月终止),我们与AbbVie的合作,专注于tau相关疾病的合作,或AbbVie Tau合作,始于2018年2月,我们与AbbVie的合作,专注于α-突触核蛋白的病理物种,或AbbVie Alpha-synuclein合作,始于2019年2月,以及我们与Neurocrine Biosciences,或Neurocrine Collaboration,始于2019年3月
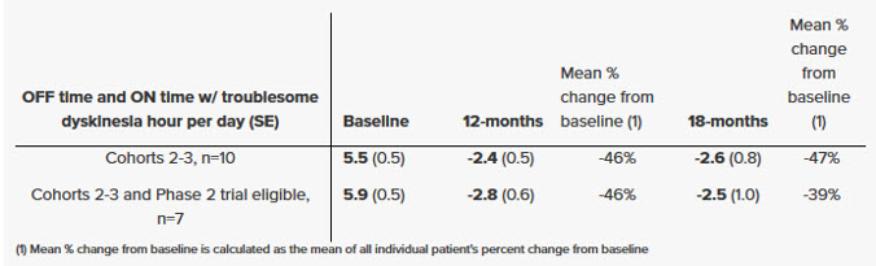
下表汇总了我们的基因治疗项目流水线:
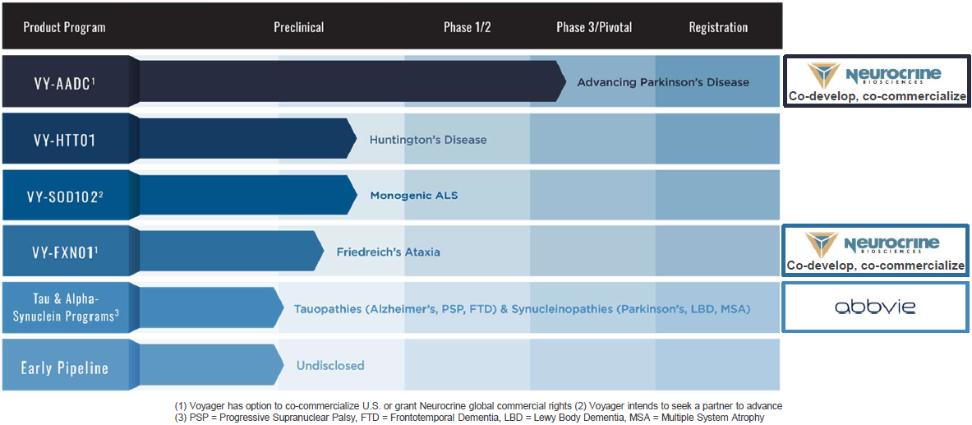
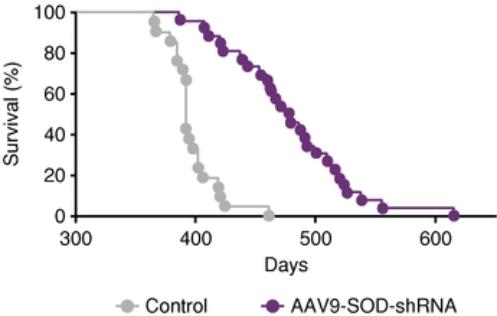
我们的计划包括针对严重神经系统适应症的项目,包括帕金森病、亨廷顿病、单基因形式的肌萎缩侧索硬化症(ALS)、Friedreich共济失调、与tau相关的疾病(包括阿尔茨海默病、额颞叶痴呆或FTD)和进行性核上性瘫痪(PSP),以及与α-突触核蛋白相关的疾病,包括帕金森氏病和其他同核病。我们可能会在美国、欧洲和日本为我们的某些候选产品寻求孤儿药物指定、突破性治疗指定或其他快速审查程序。
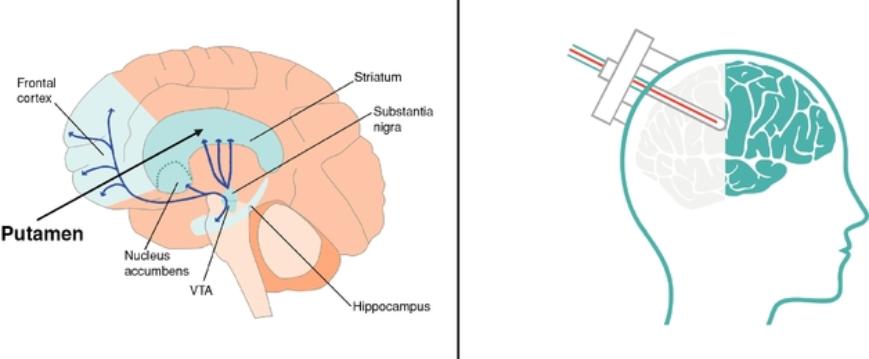
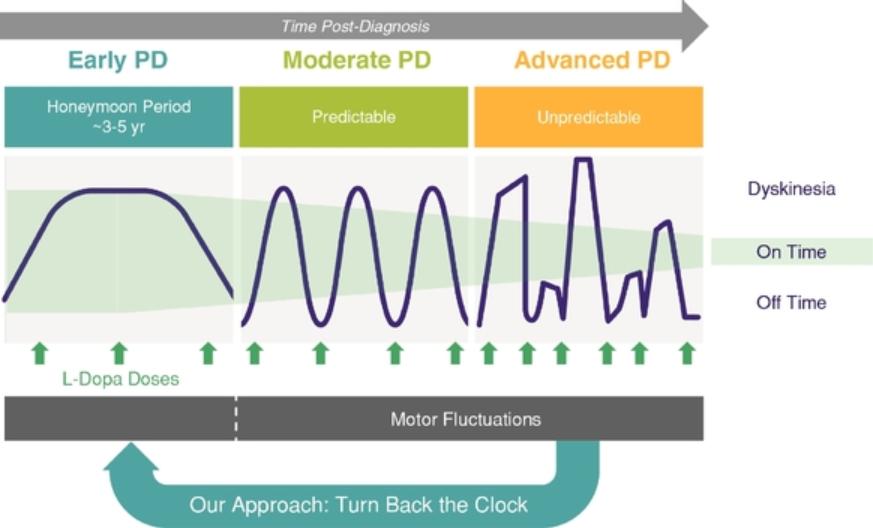
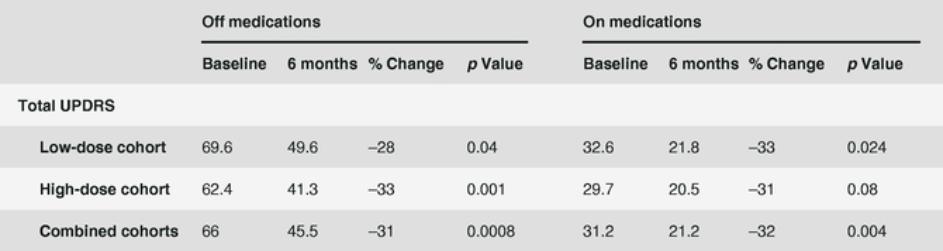
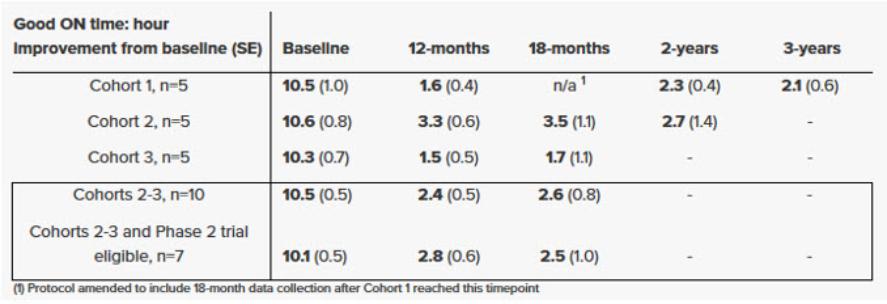
我们正在评估我们最先进的临床候选人,VY-AADC(其Neurocrine称为NBIB-1817)或VY-AADC(NBIB-1817),通过Neurocrine协作治疗帕金森氏症在Restore-1阶段2随机、双盲、安慰剂手术对照试验中,评估了VY-AADC(NBIB-1817)治疗帕金森氏症的安全性和有效性,这些患者患有难以治疗的运动波动。Restore-1阶段2试验计划招募大约85名患者,这些患者被诊断患有帕金森氏症至少四年,对口服药物没有足够的反应,根据一份经过验证的自我报告的患者日记,他们白天至少有三个小时或更多的休息时间。符合资格标准的患者将被随机(2:1)一次性给予VY-AADC(NBIB-1817)(总剂量最高2.5×1012载体基因组或VG)或安慰剂手术。
5