| ☐ |
依据1934年证券交易所ACT第12(B)或(G)条作出的注册声明
|
|
依据1934年证券交易所ACT第13或15(D)条提交的年报
|
| ☐ |
依据1934年证券交易所ACT第13或15(D)条提交的过渡报告
|
| ☐ |
依据证券第13或15(D)条作出的空壳公司报告 1934年交易所
|
|
每一班的职称
|
交易符号
|
注册的每个交易所的名称
|
|
普通股,面值为每股0.12欧元
|
IFRX
|
纳斯达克股票市场有限责任公司
|
|
是的,☐
|
不
|
|
是的,☐
|
不
|
|
是
|
无☐
|
|
是
|
无☐
|
|
大型加速机☐
|
加速机
|
非加速箱☐
|
|
新兴成长型公司
|
| ☐ |
美国公认会计原则
|
|
国际会计准则理事会发布的“国际财务报告准则”
|
| ☐ |
其他
|
|
☐项目17
|
☐项目18
|
|
是的,☐
|
不
|
|
页
|
|||
|
前瞻性陈述
|
1
|
||
|
判决的强制执行
|
2
|
||
|
第一部分
|
3
|
||
|
项目1.董事、高级管理人员和顾问的基本身份
|
3
|
||
|
A.
|
董事和高级管理人员
|
3
|
|
|
B.
|
顾问
|
3
|
|
|
C.
|
审计师
|
3
|
|
|
项目2.提供统计数字和预期时间表
|
3
|
||
|
A.
|
提供统计数据
|
3
|
|
|
B.
|
方法和预期时间表
|
3
|
|
|
项目3.关键信息
|
3
|
||
|
A.
|
选定的财务数据
|
3
|
|
|
B.
|
资本化和负债
|
5
|
|
|
C.
|
提供和使用收益的理由
|
5
|
|
|
D.
|
危险因素
|
5
|
|
|
项目4.关于公司的相关信息
|
55
|
||
|
A.
|
公司的历史和发展
|
55
|
|
|
B.
|
业务概况
|
56
|
|
|
C.
|
组织结构
|
89
|
|
|
D.
|
财产、厂房和设备
|
89 | |
|
项目4A.未得到解决的工作人员意见
|
90
|
||
|
项目5.业务和财务审查及前景
|
90 | ||
|
A.
|
经营成果
|
90 | |
|
B.
|
流动性和资本资源
|
95 | |
|
C.
|
研发、专利和许可证等。
|
96 | |
|
D.
|
趋势信息
|
96 | |
|
E.
|
表外安排
|
97 | |
|
F.
|
合同义务的列表式披露
|
97 | |
|
G.
|
安全港
|
97 | |
|
项目6.高级主管、高级管理人员和雇员
|
98 | ||
|
A.
|
董事和高级管理人员
|
98 | |
|
B.
|
补偿
|
100 | |
|
C.
|
董事会惯例
|
103 | |
|
D.
|
员工
|
104
|
|
|
E.
|
股份所有权
|
105 | |
|
项目7.大股东和关联方交易
|
105 | ||
|
A.
|
大股东
|
105 | |
|
B.
|
关联方交易
|
107 | |
|
C.
|
专家和律师的利益
|
107 | |
|
项目8.财务信息
|
108 | ||
|
A.
|
合并报表和其他财务资料
|
108 | |
|
B.
|
重大变化
|
108 | |
|
第9项.与要约和上市相一致
|
109 | ||
|
A.
|
提供及上市详情
|
109 | |
|
B.
|
分配计划
|
109 | |
|
C.
|
市场
|
109 | |
|
D.
|
出售股东
|
109 | |
|
E.
|
稀释
|
109 | |
|
F.
|
发行费用
|
109 | |
|
项目10.补充资料
|
110 | ||
|
A.
|
股本
|
110 | |
|
B.
|
章程大纲及组织章程细则
|
110 | |
|
C.
|
材料合同
|
110 | |
|
D.
|
外汇管制
|
110 | |
|
E.
|
赋税
|
110 | |
|
F.
|
股息和支付代理人
|
126 | |
|
G.
|
专家发言
|
126
|
|
|
H.
|
展示的文件
|
126 | |
|
I.
|
辅助信息
|
126 | |
|
项目11.市场风险的定量和定性披露
|
127 | ||
|
第12项.股本证券以外的证券类别
|
127 | ||
|
A.
|
债务证券
|
127 | |
|
B.
|
认股权证及权利
|
127 | |
|
C.
|
其他证券
|
127 | |
|
D.
|
美国保存人股份
|
127 | |
|
第二部分
|
128 | ||
|
项目13.基本违约、股利拖欠和拖欠
|
128 | ||
|
A.
|
缺省值
|
128 | |
|
B.
|
拖欠和拖欠款项
|
128 | |
|
项目14.对担保持有人权利的重大修改和收益的使用
|
128 | ||
|
A.
|
对文书的材料修改
|
128 | |
|
B.
|
对权利的实质性修改
|
128 | |
|
C.
|
资产的提取或替换
|
128 | |
|
D.
|
更改受托人或付款代理人
|
128 | |
|
E.
|
收益的使用
|
128 | |
|
项目15.对照控制和程序
|
129 | ||
|
A.
|
披露控制和程序
|
129 | |
|
B.
|
管理层财务报告内部控制年度报告
|
129 | |
|
C.
|
注册会计师事务所认证报告
|
130 | |
|
D.
|
财务报告内部控制的变化
|
130
|
|
|
项目16.保留
|
130
|
||
|
项目16A.审计委员会财务专家
|
130
|
||
|
项目16B.道德守则
|
130 | ||
|
第16C项.主要会计师费用及服务
|
130 | ||
|
A.
|
审计费
|
130 | |
|
B.
|
与审计有关的费用
|
130 | |
|
C.
|
税费
|
130 | |
|
D.
|
所有其他费用
|
131 | |
|
E.
|
审计委员会的审批前政策和程序
|
131 | |
|
F.
|
如审计工作超过50%,则由首席会计师以外的其他人员进行审计工作
|
131 | |
|
项目16D.对审计委员会上市标准的豁免
|
131
|
||
|
第16E条发行人及联营买家以现金方式购买权益证券
|
131
|
||
|
第16F项.注册人核证会计师的变更
|
131 | ||
|
项目16G.公司治理
|
131 | ||
|
项目16H.矿山安全披露
|
131 | ||
|
第III部
|
132 | ||
|
项目17.财务报表
|
132 | ||
|
项目18.财务报表
|
132
|
||
|
项目19.展览品
|
132 | ||
|
综合财务报表索引
|
|||
| • |
IFX-1和任何其他产品候选人临床试验的时间、进展和结果,包括关于研究或试验及有关筹备工作的开始和完成时间的说明、试验结果将公布的期间、这些试验的费用以及我们一般的研究和开发方案;
|
| • |
任何讨论或提交IFX-1或任何其他产品候选产品的监管批准申请的时间和结果,以及我们获得和保持IFX-1的任何指示的监管批准的时间和能力;
|
| • |
我们有能力利用我们专有的抗C5a技术来发现和开发治疗补体介导的自身免疫和炎症性疾病的疗法;
|
| • |
我们保护、维护和执行我们对IFX-1和任何其他产品候选人的知识产权保护的能力,以及这种保护的范围;
|
| • |
FDA、EMA或类似的外国监管机构是否会接受或同意我们临床试验的数量、设计、规模、进行或实施,包括任何建议的此类试验的初级或次要终点;
|
| • |
我们未来对IFX-1和任何其他产品候选产品的临床试验的成功,以及这些临床结果是否会反映在先前进行的临床前研究和临床试验中看到的结果;
|
| • |
我们对IFX-1或任何其他产品候选产品的规模、市场机会和临床效用的期望(如果批准用于商业用途);
|
| • |
我们的制造能力和战略,包括我们的制造方法和工艺的可伸缩性和成本,以及我们的制造方法和工艺的优化,以及我们继续依靠我们现有的第三方制造商进行我们计划中的未来临床试验的能力;
|
| • |
我们对我们的开支、持续亏损、未来收入、资本需求以及我们获得额外融资的需要或能力的估计;
|
| • |
我们对IFX-1任何经批准的指示的范围的期望;
|
| • |
我们的能力,以抵御昂贵和破坏性的责任索赔,由测试我们的产品候选人在诊所或,如果,批准,任何商业销售;
|
| • |
我们将IFX-1或其他产品的候选产品商业化的能力;
|
| • |
如果我们的任何产品候选人获得监管批准,我们的能力,以遵守和满足正在进行的义务和持续的监管概述;
|
| • |
我们的能力,以遵守已颁布和未来的立法,以寻求营销批准和商业化;
|
| • |
我们的未来成长和竞争能力,这取决于我们的关键人才和招聘更多的合格人才;
|
| • |
在发展C5a抑制剂或我们的工业方面,我们的竞争地位以及与我们的竞争对手有关的发展和预测;以及
|
| • |
我们对“就业法案”下的新兴成长型公司或外国私人发行者的期望。
|
|
项目1.
|
董事、高级管理人员和顾问的身份
|
| A. |
董事和高级管理人员
|
| B. |
顾问
|
| C. |
审计师
|
|
项目2.
|
提供统计数据和预期时间表
|
| A. |
提供统计数据
|
| B. |
方法和预期时间表
|
|
项目3.
|
关键信息
|
| A. |
选定的财务数据
|
|
截至12月31日的一年,
|
||||||||||||||||||||
|
2019
|
2018
|
2017
|
2016
|
2015
|
||||||||||||||||
|
(欧元)
|
||||||||||||||||||||
|
营业费用
|
||||||||||||||||||||
|
研发
|
(44,582,136
|
)
|
(25,028,554
|
)
|
(14,414,628
|
)
|
(5,278,252
|
)
|
(3,476,939
|
)
|
||||||||||
|
一般和行政
|
(12,501,048
|
)
|
(12,786,869
|
)
|
(5,138,498
|
)
|
(1,844,248
|
)
|
(438,403
|
)
|
||||||||||
|
业务费用共计
|
(57,083,184
|
)
|
(37,815,422
|
)
|
(19,553,126
|
)
|
(7,122,500
|
)
|
(3,915,342
|
)
|
||||||||||
|
其他收入/(费用)-净额
|
315,011
|
299,058
|
107,881
|
231,601
|
133,998
|
|||||||||||||||
|
运行效果
|
(56,768,173
|
)
|
(37,516,364
|
)
|
(19,445,245
|
)
|
(6,890,899
|
)
|
(3,781,344
|
)
|
||||||||||
|
净财务结果
|
3,513,355
|
7,701,731
|
(4,792,503
|
)
|
(2,048,422
|
)
|
(1,135,640
|
)
|
||||||||||||
|
所得税前损失
|
(53,254,817
|
)
|
(29,814,634
|
)
|
(24,237,748
|
)
|
(8,939,320
|
)
|
(4,916,984
|
)
|
||||||||||
|
所得税
|
—
|
—
|
—
|
—
|
—
|
|||||||||||||||
|
这一期间的损失
|
(53,254,817
|
)
|
(29,814,634
|
)
|
(24,237,748
|
)
|
(8,939,320
|
)
|
(4,916,984
|
)
|
||||||||||
|
基本和稀释普通股净亏损(1)
|
(2.1
|
)
|
(1.2
|
)
|
(2.6
|
)
|
(3.8
|
)
|
(2.1
|
)
|
||||||||||
|
用于计算每股普通股净亏损的加权平均股票数,基本损失和稀释损失
|
26,004,519
|
25,095,027
|
9,410,524
|
2,362,500
|
2,362,500
|
|||||||||||||||
| (1) |
没有未使用的稀释工具
|
|
截至12月31日,
|
||||||||||||||||||||
|
2019
|
2018
|
2017
|
2016
|
2015
|
||||||||||||||||
|
(欧元)
|
||||||||||||||||||||
|
现金和现金等价物
|
33,131,280
|
55,386,240
|
123,281,888
|
29,116,737
|
3,301,715
|
|||||||||||||||
|
总资产
|
121,576,558
|
159,214,161
|
124,213,006
|
29,517,321
|
3,586,105
|
|||||||||||||||
|
负债总额
|
14,429,886
|
7,823,590
|
4,784,189
|
55,206,855
|
21,204,322
|
|||||||||||||||
|
总股本
|
107,146,673
|
151,390,571
|
119,428,816
|
(25,689,533
|
)
|
(17,618,216
|
)
|
|||||||||||||
| B. |
资本化和负债
|
| C. |
提供和使用收益的理由
|
| D. |
危险因素
|
| • |
继续就我们的主要产品候选产品IFX-1开发和进行临床试验,包括评估HS的任何其他临床进展,与AAV和Pyoderma Gangraenosum(简称PG)正在进行的第二阶段临床
试验以及计划中的肿瘤学第二阶段研究有关;
|
| • |
为包括IFX-2在内的任何未来产品候选产品启动并继续开展研究、临床前和临床开发工作;
|
| • |
积极寻找更多的研究项目和更多的产品候选人;
|
| • |
寻求监管和市场批准,我们的产品候选人成功完成临床试验,如果有的话;
|
| • |
在未来建立销售、营销、分销和其他商业基础设施,使各种产品商业化,如果有的话,我们可以获得营销许可;
|
| • |
需要为临床开发和潜在的商业化生产大量的候选产品;
|
| • |
与战略伙伴合作,优化IFX-1和IFX-2的制造工艺;
|
| • |
维护、扩大和保护我们的知识产权组合;
|
| • |
聘用和保留更多人员,如临床、质量管理和科学人员;以及
|
| • |
增加运营,财务和管理信息系统和人员,包括人员,以支持我们的产品开发,并帮助我们履行我们作为一个上市公司的义务。
|
| • |
我们目前和未来的产品候选人,特别是IFX-1的临床试验、研究和临床前开发工作的范围、进展、时间、成本和结果;
|
| • |
我们追求的未来产品候选数量和指标以及它们的开发需求;
|
| • |
寻求监管批准的结果、时间和成本;
|
| • |
任何获得市场营销批准的产品候选人的商业化活动费用不属于任何未来合作者的责任,包括建立
产品销售、营销、分销和商业规模制造能力的成本和时间;
|
| • |
竞争技术和市场发展的影响;
|
| • |
如果收到市场批准,收入,如果有的话,从商业销售我们目前和未来的产品候选人;
|
| • |
我们的能力,以及任何合作、许可或其他安排的条件和时间;
|
| • |
随着研究和开发活动的扩大,我们的员工数量增长和相关成本;
|
| • |
准备、提交和起诉专利申请、维护和保护我们的知识产权,包括执行和维护与知识产权有关的要求的费用;以及
|
| • |
作为一家上市公司经营的成本。
|
| • |
招致额外的计划外费用,包括与所需的额外临床试验或临床前试验有关的费用;
|
| • |
延迟获得IFX-1或我们的任何其他产品的市场批准;
|
| • |
根本没有获得市场许可;
|
| • |
获得批准的适应症或病人群体,但不像预期或期望的那样广泛;
|
| • |
获得包括重大使用或分销限制或重大安全警告,包括装箱警告在内的标签的批准;
|
| • |
须接受额外的售后测试或其他要求;或
|
| • |
获得市场批准后,必须将产品从市场上移除。
|
| • |
监管部门可以撤销对该产品的批准或者扣押该产品;
|
| • |
我们,或任何未来的合作者,可能需要召回该产品,或被要求改变该产品的使用方式或进行额外的临床试验;
|
| • |
可对特定产品的销售或制造工艺施加额外限制;
|
| • |
我们可能会受到罚款、禁制令或民事或刑事处罚;
|
| • |
管理当局可要求添加标签说明,如“黑匣子”警告或禁忌;
|
| • |
我们,或任何未来的合作者,可能需要创建一个药物指南,概述风险的风险,以前不明的副作用分发给病人;
|
| • |
我们,或任何未来的合作者,可能被要求实施限制分配和使用的REMS,或者进行市场后研究或临床试验;
|
| • |
我们或任何未来的合作者可能被起诉,并对对病人造成的伤害承担责任;
|
| • |
产品可能会变得不那么有竞争力;
|
| • |
我们的名声可能会受损。
|
| • |
我们可能无法证明IFX-1是安全和有效的治疗我们的目标适应症,使FDA,EMA或类似的外国监管机构满意;
|
| • |
FDA、EMA或类似的外国监管机构可能需要额外的IFX-1临床试验或非临床研究,而这些试验或非临床研究是在批准之前或作为批准后承诺的,
,这将增加我们的成本,延长IFX-1的开发时间;
|
| • |
我们临床试验的结果可能不符合FDA、EMA或类似的外国监管机构对营销批准所要求的统计或临床意义的水平;
|
| • |
FDA、EMA或类似的外国监管机构可能不同意我们临床试验的数量、设计、规模、实施或实施,包括指定的临床终点,例如在我们计划的IFX-1临床试验中使用HiSCR;
|
| • |
在临床项目中研究的人群可能不够广泛或具有代表性,无法确保我们寻求批准的全部人群的安全;
|
| • |
我们保留进行临床试验的合同研究机构或CRO,可能采取我们无法控制的行动,对我们的临床试验产生重大不利影响;
|
| • |
FDA、EMA或类似的外国监管机构可能不会发现来自临床前研究和临床试验的数据足以证明IFX-1的临床和其他好处大于其安全风险;
|
| • |
FDA、EMA或类似的外国监管机构可能不同意我们对临床前研究和临床试验数据的解释;
|
| • |
FDA、EMA或类似的外国监管机构不得接受在临床试验场所生成的数据;
|
| • |
如果我们的BLA在提交时得到咨询委员会的审查,FDA可能很难及时安排咨询委员会会议,或者咨询委员会可能建议不批准我们的申请,或者建议FDA作为批准的条件,要求额外的临床前研究或临床试验限制经批准的标记或分发和使用限制;
|
| • |
FDA、EMA或类似的外国监管机构可能需要制定风险评估和缓解战略(REMS),作为批准的条件;
|
| • |
fda、epa或类似的外国监管机构可能会发现我们的第三方制造商在制造过程或设施方面的缺陷,包括不遵守现行的良好制造惯例,或cgmp;
或
|
| • |
FDA、EMA或类似的外国监管机构可以改变各自的审批政策或采用新的法规。
|
| • |
被调查疾病的严重程度;
|
| • |
临床试验方案的设计;
|
| • |
病人人数和性质;
|
| • |
有关审判的资格标准;
|
| • |
被试产品候选人的感知风险和利益;
|
| • |
产品的安全性和耐受性;
|
| • |
近距离和可供预期病人使用的临床试验地点;
|
| • |
提供相互竞争的疗法和临床试验;
|
| • |
临床医生和病人对正在研究的药物相对于其他可用疗法的潜在优势的认识,包括护理标准和任何可能被批准用于我们正在调查的适应症的新药;
|
| • |
努力促进临床试验的及时注册;
|
| • |
医生转介病人的做法;及
|
| • |
我们在治疗期间和治疗后对病人进行充分监测的能力。
|
| • |
产品的有效性和安全性;
|
| • |
与竞争疗法相比,该产品具有潜在的优势,尽管在达到或超过临床试验终点方面取得了成功;
|
| • |
任何副作用的发生率和严重程度;
|
| • |
该产品是否根据医生治疗指南指定为第一、第二或第三线治疗;
|
| • |
我们的能力,或任何未来合作者的能力,提供产品的销售,以具有竞争力的价格;
|
| • |
与其他治疗方法相比,该产品的使用方便方便;
|
| • |
目标病人是否愿意尝试,医生是否愿意开处方;
|
| • |
限制或警告,包括产品经批准的标签中所包含的分配或使用限制;
|
| • |
强大的销售、营销和分销支持;
|
| • |
对该产品的有针对性的适应症的护理标准的变化;以及
|
| • |
政府付款人、管理下的照护计划和其他第三方付款人的保险范围和补偿的可得性和金额。
|
| • |
监管机构或机构审查委员会不得授权我们或我们的调查人员在预期的试验地点开始临床试验或进行临床试验;
|
| • |
我们的产品候选产品的临床试验可能产生负面或非决定性的结果,包括未能证明统计意义,我们可能决定,或监管机构可能要求我们进行更多的临床试验或放弃药物开发计划;
|
| • |
我们的产品候选人可能有不良的副作用或其他意想不到的特性,导致我们或我们的调查人员、监管机构或机构审查委员会暂停或终止试验;
|
| • |
我们的第三者承办商可能未能及时或根本不遵守规管规定或履行合约义务;及
|
| • |
监管机构或机构审查委员会可能要求我们或我们的调查人员出于各种原因暂停或终止临床开发,包括不遵守监管要求或发现参与者面临不可接受的健康风险。
|
| • |
外国的不同监管要求;
|
| • |
所谓平行进口的潜力,这是当当地卖方面对高或更高的当地价格,选择从外国市场进口货物(以较低或较低的价格)而不是在当地购买货物时发生的情况;
|
| • |
关税、贸易壁垒、价格和外汇管制及其他监管要求的意外变化;
|
| • |
经济疲软,包括通货膨胀,或政治不稳定,特别是外国经济和市场;
|
| • |
外国偿还、定价和保险制度;
|
| • |
对在国外生活或旅行的雇员遵守税收、就业、移民和劳动法;
|
| • |
外国税收,包括扣缴工资税;
|
| • |
外币波动,可能导致业务费用增加,收入减少,以及在另一国家开展业务的其他义务;
|
| • |
外国业务人员配置和管理方面的困难;
|
| • |
在劳工动乱比美国更普遍的国家,劳动力的不确定性;
|
| • |
根据1977年“外国腐败行为法”或类似的外国条例可能承担的责任;
|
| • |
挑战执行我们的合同和知识产权,特别是在那些与美国一样不尊重和保护知识产权的外国;
|
| • |
任何影响国外原材料供应或制造能力的事件造成的生产短缺;以及
|
| • |
包括战争和恐怖主义在内的地缘政治行动造成的商业中断。
|
| • |
“联邦反Kickback法规”,除其他外,禁止个人和实体故意故意直接或间接地以现金或实物形式索取、提供、接受或提供报酬,以诱使或奖励,或作为交换,要么将个人转介,要么购买、订购或推荐任何商品或服务,这些物品或服务可在联邦和州保健方案下支付,如医疗保险和医疗补助。一个
个人或实体不需要实际了解规约或违反法规的具体意图,就可以实施违法行为。此外,有几家法院对“规约”的意图要求的解释是,如果涉及薪酬的任何一项安排的目的是促使转介联邦医疗保险业务,则违反了“反Kickback规约”。此外,政府可以声称,包括因违反联邦反Kickback法规而产生的物品或服务在内的索赔,就“虚假索赔法”而言,构成虚假或欺诈性索赔;
|
| • |
联邦民事和刑事虚假索赔法,包括但不限于“联邦民事虚假索赔法”(可通过民事举报人或诉讼予以执行),以及民事罚款法,对故意向联邦政府提出或导致向联邦政府提出或导致向联邦政府提出付款的个人或实体处以刑事和民事处罚,包括医疗保险和医疗补助方案、虚假或欺诈性的付款要求,或作出虚假声明,以避免、减少或隐瞒向联邦政府付款的义务;
|
| • |
1996年“联邦健康保险运输和问责法”(HIPAA)规定,除其他外,实施欺骗任何保健福利方案的计划或在医疗事项上作出虚假陈述等行为,应负刑事和民事责任。类似于“联邦反Kickback规约”,个人或实体不需要实际了解法规或违反法规的具体意图即可实施违法行为;
|
| • |
经2009年“经济和临床健康卫生保健信息技术法”(HITECH)或HITECH修订的HIPAA及其各自的实施条例,其中规定受覆盖的保健提供者、保健计划和
保健信息交换所及其业务伙伴有义务为被覆盖实体或代表被覆盖实体创建、接收、维护或传送可单独识别的健康信息,以保障个人可识别的健康信息的隐私、安全和
传输;
|
| • |
“医生付款阳光法”是根据经“保健和教育和解法”修正的“病人保护和平价医疗法案”第6002条制定的,或统称为“平价医疗法案”,及其执行
条例,其中要求指定的药品、设备、生物制品和医疗用品制造商,除具体例外情况外,可根据医疗保险、医疗补助或儿童健康保险方案支付,每年向医疗保险和医疗补助服务中心或合作医疗中心报告与向医生支付或其他“价值转移”有关的信息,其定义包括医生、牙医、视光师、足科医生和脊医,以及教学医院和适用的制造商,每年在每个日历年第90天之前向医师及其直系亲属所持有的合作医疗所有权和投资权益报告。所有这些报告的
信息都是公开的;以及
|
| • |
类似的州和外国法律法规,如州反回扣法和虚假索赔法,可适用于销售或营销安排以及涉及由非政府的第三方付款人(包括私营保险公司)偿还的医疗项目或服务的索赔;要求制药公司遵守制药行业自愿遵守准则和由联邦政府颁布的相关合规指南或以其他方式限制可能支付给保健提供者的付款的州和外国法律;要求药品制造商向医生和其他保健提供者或营销支出报告与付款和其他价值转移有关的信息的州和外国法律;以及在某些情况下关于健康信息的隐私和安全的州和外国法律,其中许多法律在重大方面彼此不同,而且常常不会被HIPAA抢先,从而使遵守工作复杂化。
|
| • |
生产或进口某些品牌处方药和生物制剂的任何实体的年度、不可扣减的费用,由这些实体根据其在某些政府保健项目中的市场份额分摊;
|
| • |
提高法定最低折扣,制造商必须支付医疗补助药品回扣计划,分别为23.1%和13.0%的平均制造商价格品牌和仿制药;
|
| • |
扩大医疗欺诈和滥用法律,包括“虚假索赔法”和“反Kickback法规”,其中除其他外,包括新的政府调查权力和加强对不遵守规定的处罚;
|
| • |
医疗保险D部分覆盖差距折扣方案,其中制造商必须同意在保险差距期间向合格受益人提供75%的可适用品牌药品的销售点折扣,作为制造商门诊药品在医疗保险D部分范围内的一个
条件;
|
| • |
将制造商的医疗补助退税责任扩大到向参加医疗补助管理护理组织的个人发放的涵盖药品;
|
| • |
扩大医疗补助方案的资格标准,除其他外,允许各州向其他个人提供医疗补助保险,从而有可能增加制造商的医疗补助退税责任;
|
| • |
将制造商的医疗补助退税责任扩大到向参加医疗补助管理护理组织的个人发放的涵盖药品;
|
| • |
扩大医疗补助方案的资格标准,除其他外,允许各州向其他个人提供医疗补助保险,从而有可能增加制造商的医疗补助退税责任;
|
| • |
扩大公共卫生服务药品定价方案下有资格享受折扣的实体;
|
| • |
联邦公开支付计划及其实施条例的新要求;
|
| • |
一项新的要求,即每年向医生报告制造商和分销商提供的药品样本;以及
|
| • |
一个新的以病人为中心的结果研究所,负责监督、确定优先事项,并进行比较临床效果研究,并为此类研究提供资金。
|
| • |
限制这类产品的生产;
|
| • |
限制这类产品的标识或销售;
|
| • |
限制产品分配或使用;
|
| • |
进行营销后研究或临床试验的要求;
|
| • |
警告信或无名称信件;
|
| • |
产品退出市场;
|
| • |
拒绝批准我们提交的待批准的申请或对已批准的申请的补充;
|
| • |
召回产品;
|
| • |
对第三方支付人承保范围的限制;
|
| • |
罚款、归还或支配利润或收入;
|
| • |
暂停或撤销销售许可;
|
| • |
拒绝允许进口或出口产品;
|
| • |
扣押产品;或
|
| • |
禁止或判处民事或刑事处罚。
|
| • |
依赖第三方进行生产过程开发、法规遵守和质量保证;
|
| • |
额外扩大或优化工艺所需的新设备和设施的费用和验证;
|
| • |
不符合cGMP和类似的国外标准;
|
| • |
由于第三方的能力和时间限制,对供应供应的限制;
|
| • |
目前从单一或单一来源供应商购买的零部件缺乏合格的备用供应商;
|
| • |
公共卫生危机造成的关闭和对关键设施的限制;
|
| • |
由于我们无法控制的因素,第三方可能违反制造协议;以及
|
| • |
第三方可能终止或不续订制造协议,在对我们来说昂贵或不方便的情况下,以及我们获得替代供应的能力。
|
| • |
生产活动中关键人员的损失可能导致对我们的药品候选人的制造和释放测试出现重大延误,更换这些人员可能会耗费时间,并与我们的额外费用有关;
|
| • |
释放测试中的错误或不当行为可能导致错误的结果,可能导致错误地拒绝生产药物产品的释放,或错误地接受功能失调的药物
产品,从而导致数据和试验结果的错误和可能被错误解释;以及
|
| • |
由于监管机构的检查,GMP法规的不充分可能导致制造或进口许可证的暂时或永久丧失。
|
| • |
协作者在确定他们将适用于这些合作的努力和资源方面有很大的酌处权;
|
| • |
合作者不得按预期履行其义务;
|
| • |
合作者不得追求我们产品候选人的开发和商业化,也不得根据临床试验结果、合作者战略重点的变化或现有资金或外部因素,例如收购,选择不继续或更新开发或商业化方案,以转移资源或创造相互竞争的优先事项;
|
| • |
合作者可以推迟临床试验,为临床试验项目提供资金不足,停止临床试验或者放弃产品候选品,重复或者进行新的临床试验,或者要求新的产品
候选制剂进行临床试验;
|
| • |
合作者可以独立开发或开发与我们的产品候选人直接或间接竞争的第三方产品;
|
| • |
拥有一个或多个产品的营销和分销权的合作者不得为这类产品的营销和分销投入足够的资源;
|
| • |
与合作者的分歧,包括在包括商业机密和其他知识产权、合同解释或优先研究与开发过程在内的所有权方面的分歧,可能造成产品候选者研究、开发或商业化的拖延或终止,可能导致我们对产品候选方承担更多责任,或可能导致诉讼或仲裁,其中任何一种都是费时和昂贵的;
|
| • |
合作者不得适当起诉、维护、捍卫或强制执行我们的知识产权,也不得利用我们的专有信息或其他知识产权,导致可能危及或使我们的知识产权失效或使我们面临潜在诉讼的诉讼;
|
| • |
合作者可能侵犯、挪用或者以其他方式侵犯第三人的知识产权,使我们面临诉讼和潜在责任;
|
| • |
可能终止合作,如果合作终止,则可能需要更多资本,以进一步开发适用的产品候选人或将其商业化;以及
|
| • |
合作协议可能不会以最有效的方式或根本不导致产品候选产品的开发或商业化。如果我们未来的任何合作者参与到一个商业组合中,它可以决定推迟、减少或终止我们授权给它的任何产品的开发或商业化。
|
| • |
美国专利贸易组织和各外国政府专利机构要求在专利过程中遵守若干程序、单据、费用支付和其他规定。在某些情况下,不遵守可能导致专利或专利申请的
放弃或失效,从而导致相关法域的专利权部分或完全丧失。在这种情况下,竞争者可能比其他情况下更早进入市场;
|
| • |
专利申请的范围可以在专利发布前大幅度缩小,其范围可以在发布后重新解释;
|
| • |
专利申请不得导致颁发专利;
|
| • |
可颁发或许可的专利可被质疑、无效、修改、撤销、规避、缩小、认定不可执行或以其他方式不能提供任何竞争优势;
|
| • |
我们的竞争对手,其中许多拥有大得多的资源,其中许多在竞争技术上作了重大投资,可能会寻求或已经获得专利,从而限制、干扰或消除我们制造、使用和销售我们的潜在产品的能力;
|
| • |
美国政府和国际政府机构可能面临很大压力,要求限制美国境内外的专利保护范围,以治疗证明是成功的疾病,这是关于全世界卫生问题的公共政策问题;
|
| • |
美国以外的国家的专利法对专利权人的优惠可能不如美国法院支持的专利法,这使外国竞争对手有更好的机会创造、开发和市场竞争的产品候选人。
|
| • |
我们可能无法产生足够的数据来支持专利申请,以保护我们的一个或多个项目的全部发展,包括我们的Hidradentis Suppurativa(HS)计划;
|
| • |
有可能我们的一项或多项待决专利申请不会成为已颁发的专利,或者该专利将不足以保护我们的技术或产品,为我们提供商业上可行的
产品的基础,或为我们提供任何竞争优势;
|
| • |
如果我们的待决专利申请作为专利颁发,则第三方可能会对其提出质疑,认为根据美国或外国法律,这些申请不受侵犯、无效或无法强制执行;或
|
| • |
如获批出,我们所持有的专利可能无效或不能强制执行。
|
| • |
许可协议规定的权利范围和其他与解释有关的问题;
|
| • |
我们的技术和程序在多大程度上侵犯了不受许可协议约束的许可人的知识产权;
|
| • |
当前和未来任何合作开发关系下的专利和其他权利的再许可;
|
| • |
我们在任何许可协议下的勤勉义务,以及哪些活动符合这些义务;
|
| • |
由于我们的许可对手和我们的合作伙伴共同创造或使用知识产权而产生的发明、技术和其他知识产权的发明权和所有权;以及
|
| • |
专利技术发明的优先地位。
|
| • |
IFX-1和任何其他产品候选产品临床试验的时间、登记和结果;
|
| • |
对IFX-1、我们的其他产品候选者或我们竞争对手的产品和产品候选人采取的管制行动;
|
| • |
现有或新的有竞争力的产品或技术的成功;
|
| • |
我们对IFX-1或任何未来产品候选产品的开发或监管申请的任何延迟,以及在适用的监管当局审查此类文件方面的任何不利发展或可能出现的不利发展,包括不限于林业发展局发出的“拒绝提交”信函或要求提供补充信息的
;
|
| • |
我们或我们的竞争对手宣布重大收购、战略伙伴关系、合资企业、合作或资本承诺;
|
| • |
开始或终止与我们的发展项目的合作;
|
| • |
任何开发项目的失败或中止;
|
| • |
竞争对手产品候选产品临床试验结果;
|
| • |
美国和其他国家的监管或法律发展;
|
| • |
与专利申请、专利或者其他专有权利有关的发展或者争议;
|
| • |
关键人员的征聘或离职;
|
| • |
与我们的任何产品候选人或临床开发项目相关的费用水平;
|
| • |
我们努力开发更多的产品候选人或产品的结果;
|
| • |
财务结果或发展时间表估计数的实际或预期变化;
|
| • |
宣布或期望作出更多的筹资努力;
|
| • |
由我们、我们的内部人士或其他股东出售我们的普通股;
|
| • |
我们的财务业绩或那些被认为与我们相似的公司的财务业绩的变化;
|
| • |
证券分析师(如果有的话)对我们股票的估计或建议的变化;
|
| • |
医疗保健支付系统结构的变化;
|
| • |
制药和生物技术部门的市场条件;
|
| • |
一般经济、工业及市场情况;及
|
|
•
|
在本“项目3.关键信息:-D.风险因素”一节中描述的其他因素。
|
| • |
获准在本年度报告中只提供三年的审定财务报表,并相应减少“第5项.业务和财务审查及前景”披露;
|
| • |
在评估我们对财务报告的内部控制时,不需要遵守审计师的认证要求;
|
| • |
在我们的定期报告、委托书和登记声明中减少了有关行政报酬的披露义务;以及
|
| • |
不被要求举行不具约束力的咨询表决的高管薪酬和股东批准任何黄金降落伞付款之前未获批准。
|
| 项目4. |
有关该公司的资料
|
| A. |
公司的历史和发展
|
| B. |
业务概况
|
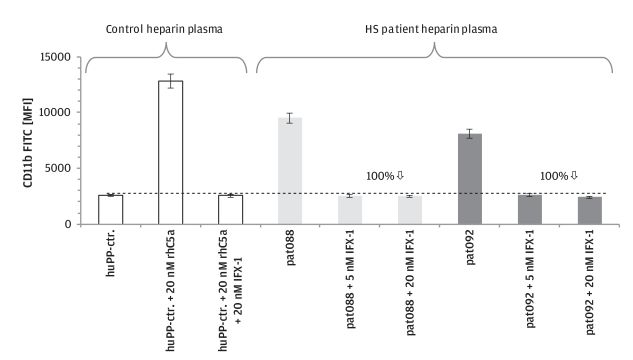
|
IFX-1
|
安慰剂
|
|||
|
最小剂量
|
低剂量
|
中剂量
|
高剂量
|
安慰剂Q2W
|
|
每4次400毫克
周(Q4W)
|
800毫克/4
周(Q4W)
|
800毫克/2
周(Q2W)
|
每2次1200毫克
周(Q2W)
|
|
|
40.0%
|
51.5%
|
38.7%
|
45.5%
|
47.1%
|
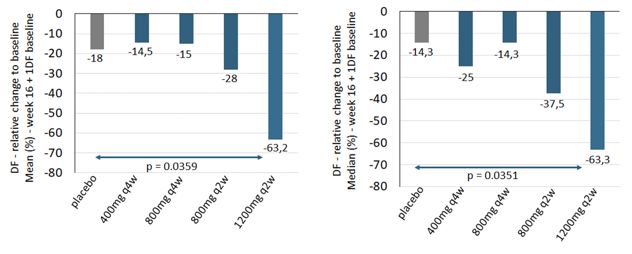
|
图1:
|
所有在基线至少有1个引流瘘管的患者在第16周(左:平均,右:中)减少瘘管引流(DF)。为了进行均值比较和
高剂量与安慰剂的p值,计算了在基线时调整DF和Hurley阶段的ANCOVA模型。高剂量与安慰剂中位数比较的p值是基于Wilcoxon秩和检验的。完成案例分析,不对缺失值进行
计算。
|

|
图2:
|
安慰剂组和高剂量组(IFX-1200mgQ2W)的每次访问(左:平均,右:中)每次访问的Fistula(DF)减少(左:平均,右:中位数),所有基线至少有一个DF
的患者。对于高剂量和安慰剂的平均比较,在基线时计算了一个经过DF和Hurley阶段调整的ANCOVA模型。完整的案例分析,不计算缺失值。
|
|
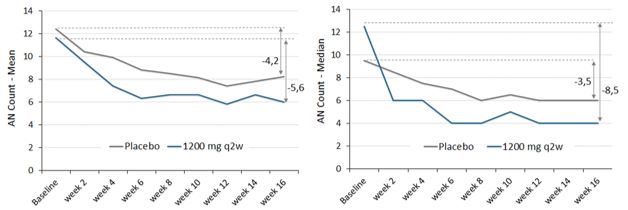
图3:
|
安慰剂组和高剂量组(IFX-1200mgQ2W)每例患者每次访视(左:中,右:中)至第16周的计数(IFX-1200 mg Q2W)。完成案例分析,不对缺失值进行 计算。 |
| • |
70.6%的应答器集团在OLE期间维持其对HiSCR的回应,以及
|
| • |
在第40周,41.8%的非应答者成为应答者.
|
| • |
脓肿和炎性结节(计数)分别为-66.9%(平均)和-75.0%(中位数),以及
|
| • |
排尿瘘-46.0%(平均)和-51.5%(中位数)
|
| • |
快速起效:IFX-1起效快,静脉注射后可完全抑制C5a诱导的信号传导,对C5a诱导的中性粒细胞的启动和活化提供即时保护。与现有的治疗方案相比,这可能导致更快的应答率和更快的缓解诱导。
|
| • |
潜在的潜能优势(超过受体抑制):IFX-1阻断上游配体C5a,C5aR和C5L2,C5a通过C5aR
和C5L2共同抑制信号传导;C5a通过C5aR
和C5L2都被证明对ANCA启动和C5a诱导的中性粒细胞脱颗粒是AAV的主要疾病驱动机制(由Hao和Wang等人2013年发表,PLOS one)。
|
| • |
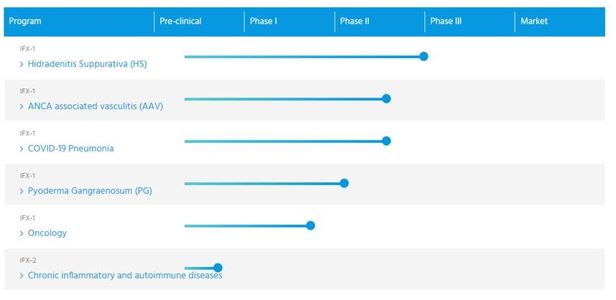
推进我们的领先项目IFX-1为HS.在宣读第IIb阶段的试验之后,我们计划与FDA和EMA一起设计和讨论第三阶段计划,以支持IFX-1治疗HS的监管应用程序
。
|
| • |
IFX-1在AAV、PG、肿瘤性疾病和冠状病毒引起的重症肺炎和其他补体介导的自身免疫性和炎症性疾病中的完整Ⅱ期临床进展。我们正在开发治疗AAV的IFX-1,我们已经启动了一个由两个临床试验组成的二期计划。我们计划在美国和欧洲为AAV寻找孤儿药物。对于PG,我们计划完成一项公开标签的
概念阶段IIa研究,其中我们已经公布了第一批给药队列的初步结果。此外,我们还计划研究IFX-1治疗在肿瘤学指征中的潜在益处,并在2020年下半年开始一项概念研究的临床
第二阶段证明。我们正在开发用于治疗冠状病毒所致重症肺炎的IFX-1,并已启动一项适应性随机临床试验,目的是初步评估IFX-1的安全性和有效性,并在第二步提供有效性证明。我们计划最终开发IFX-1用于其他补体介导的自身免疫性和炎症性疾病。
|
| • |
追求IFX-2的临床发展,并继续扩大我们的抗C5a技术的广度。我们正在开发IFX-2作为一种比IFX-1更长半衰期的注射剂,使其适合于慢性炎症指征和较轻的耀斑或更接近疾病的发病。IFX-2在作用机制、结合表位和选择性等方面与IFX-1具有相同的特征.IFX-2的临床前发展得到了德国政府的资助.我们相信IFX-2具有治疗各种慢性炎症疾病的潜力,这些疾病可以从一种更适合于慢性治疗的剂量方案中获益。
|
| • |
将IFX-1商业化,如果获得批准,可以独立地或与合作伙伴合作.我们打算独立地在美国和欧洲寻求IFX-1的批准和商业化,用于HS和其他潜在的
标志。我们计划采用有针对性的商业基础设施,通过在这些核心市场治疗HS的卓越中心来促进IFX-1的进入。在美国和欧洲之外,我们可以独立地或与他人合作,寻求IFX-1的批准和商业化,用于HS和其他可能的其他迹象。对于其他迹象,我们打算通过与其他各方的合作,独立开发和商业化IFX-1。
|
| • |
巩固我们在反C5a领域的领导地位,充分利用我们专有的抗C5a技术和专业技术在补充和炎症方面的潜力。我们打算继续发现和开发治疗方法,有潜力解决广泛的补体介导或免疫反应介导的迹象与重大的未满足的需要,无论是内部或与合作伙伴。为了实现
这一点,我们继续在密歇根州AnnArbor的发现部门补充我们的研究和开发活动,我们正在进一步建立我们的业务开发能力。
|
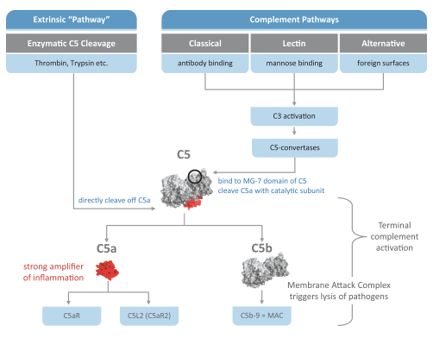
| • |
迅速创造一个煽动性的环境。促炎症分子(如C5a)的产生优化了酶和其他过程对微生物起作用的条件。这些炎症条件包括发烧或中性粒细胞释放侵略性酶和氧自由基。
|
| • |
通过形成膜攻击复合物来溶解微生物。一种快速的第一线防御机制,在入侵微生物的细胞膜上形成孔隙,
导致其解体。
|
| • |
通向适应性免疫系统的桥梁。这种功能是由C3的激活产物C3b促进的,它标记粒子,使它们可见,并且更容易被免疫刺激的
细胞处理。然后这些细胞将这些粒子呈现给B细胞,而B细胞又会产生针对这些粒子的抗体,从而导致靶向清除。这一机制需要几个星期才能充分发挥作用。
|
| • |
清除死细胞颗粒。补体系统还可用于其他各种用途,包括清除身体上的死细胞颗粒。这种功能特别重要,因为未清除的细胞颗粒被认为有可能诱导产生针对正常细胞和组织的抗体,从而导致自身免疫性炎症反应和疾病。
|
| • |
C5a促进多种细胞因子的产生,如IL-8、IL-6、IL 17、TNF-α等。
|
| • |
c5a诱导免疫能力细胞的细胞信号级联发生复杂的变化,导致其他已知信号刺激(如Toll样受体信号)的改变和信号转导的强化。
|
| • |
C5a影响T细胞反应,引起促炎反应,进而产生进一步的促炎细胞因子。
|
| • |
C5a能诱导血管表面粘附分子的表达,导致中性粒细胞粘附于血管内壁,并通过血管向感染部位迁移。
|
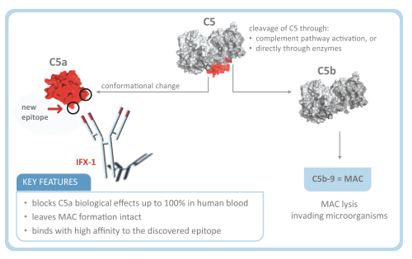
| • |
完全阻断和抑制C5a诱导的效应:人体具有产生C5a的丰富能力,并通过其两种受体C5aR和C5L2诱导炎症作用。
因此,我们的抗C5a抗体被设计为:
|
| • |
产生完全的免疫阻断C5a分子,以实现有效的治疗。缺乏这种质量的抗体或抑制剂可能会为C5a留下一个“信号间隙”,在疾病背景下,这种间隙很可能足以产生强烈的促炎作用。这种信号间隙将限制C5a/C5aR和C5a/C5L2信号轴的沉默能力,以达到预期的治疗效果;
|
| • |
与C5a结合以抵消分子与其两种受体C5aR和C5L2的快速相互作用,C5aR和C5L2在人体绝大多数细胞类型中都有大量存在,在各种
病环境下可被上调。
|
| • |
对MAC形成的影响有限:C5阻断了血液中MAC形成的分子,增加了由脑膜炎球菌等包被细菌引起的危及生命的感染风险。
因此,保持MAC形成完整可能在C5a驱动的疾病中提供显著的优势。
|
| • |
在不直接靶向C5a的情况下,无法完全阻断C5a:在完全没有补体通路的情况下,C5a可以通过各种酶的激活而产生。例如,用C5抑制剂没能阻断C5转化酶驱动的
补体C5酶驱动的切割,在实验条件下不能阻止直接酶C5的激活和C5a的产生。这可能解释了为什么C5a的升高在有效地给药的病人中仍然是可测量的。因此,不直接结合和抑制C5a的非特定方法可能无法完全阻断其效果;以及
|
| • |
缺乏对C5a信号传导能力的控制:C5a受体在人类大多数细胞上大量存在,在某些疾病状态下可被强烈和迅速地上调。因此,
即使在C5a水平较低的情况下,这些受体也会产生一个大的“信号汇”,为即使少量的C5a发送信号提供丰富的能力。因此,完全阻断靶向C5a方法是必要的,以实现对C5a诱导的信号事件的完全
控制,这在高度急性炎症环境中可能特别重要。
|
| • |
完全抑制C5a诱导的信号传递和衍生生物功能,其完全阻止C5a诱导的人全血中性粒细胞活化的能力证明了这一点;
|
| • |
完好无损的MAC形成,如检测完整的补体通路驱动的MAC在红细胞上形成,导致这些细胞的溶解。
|
| • |
美国一项开放的单中心试验,在原计划的21名中、重度HS患者中纳入18名,最近与Secukinumab(一种阻止白细胞介素-17A的单克隆抗体)和初步会议
报告结束,表明HiSCR有了改善,最后观察继续进行。
|
| • |
另一项由Secukinumab在法国一个中心注册17名HS患者的公开试验最近已经进行,并在2019年2月的欧洲HS基金会会议上报告了第一次结果,表明13名患者在4个月的治疗中表现出了
HiSCR的反应。在这项研究中,两名患者在治疗的第四个月就患上了克罗恩氏病,在14个月的试验期内立即停止治疗后仍保持活跃状态。引起Crohn‘s
病是已知的一种副作用,而Crohn’s病已被报道与HS病有关。
|
| • |
Janssen-Cilag‘s ustekinumab的开放标签试验最近在12名HS患者中完成.ustekinumab是针对IL 12和IL 23的单克隆抗体。
|
| • |
一项针对瑞典孤儿biovitrum AB‘s anakinra的小规模安慰剂控制II期研究,以及6名患者的开放标签单中心试验,在HS患者中完成,表明在改良的治疗意向中有潜在的疗效。anakinra是一种IL-1受体拮抗剂.
|
| • |
在佛罗里达皮肤科学术中心进行的一项开放式单一中心20名患者研究,由矫形皮肤科(BauschHealth)赞助,评估使用
hiscr治疗中度HS的siliq™(溴氰菊酯)的疗效,为期24周,随后进行为期4周的观察治疗。
|
| • |
慕尼黑技术大学正在对5名患者进行第二阶段的单臂研究(使用PGA第16周的五点评分,而第0周为主要终点)。
|
| • |
俄亥俄州立大学于2018年与ixekizumab完成了5例患者II期开放标签研究。
|
| • |
苏黎世大学(UniversityofZurich)于2015年完成了一项开放标签研究,评估卡纳基努马(Ilaris)对PG患者的治疗效果。
|
| • |
临床前实验室试验、动物研究和配方研究均按照适用的欧盟良好实验室惯例条例进行;
|
| • |
向有关国家当局提交一份临床试验申请或CTA,该申请或CTA必须在试验开始前在计划登记病人的每个国家获得批准;
|
| • |
充分和良好控制的临床试验的性能,以确定产品的安全性和有效性,为每一个建议的指征;
|
| • |
向有关主管当局提交营销授权申请或MAA,其中包括支持安全性和有效性的数据,以及关于
临床开发和拟议标签中产品的制造和成分的详细信息;
|
| • |
有关国家当局对生产该产品的生产设施,包括第三方设施的检查令人满意地完成,以评估严格执行的现行良好制造做法的遵守情况;
|
| • |
对产生数据以支持MAA的非临床和临床试验地点的潜在审计;以及
|
| • |
在产品的任何商业销售、销售或装运之前,由MAA的有关主管部门进行审查和批准。
|
| • |
用于诊断、预防或治疗一种威胁生命或长期衰弱的状况,在提出申请时影响到欧洲联盟每10 000人中不超过5人,或;
|
| • |
旨在诊断、预防或治疗欧洲联盟的危及生命、严重衰弱或严重和长期的疾病,如果不采取奖励措施,欧洲联盟在
销售这种药物就不太可能产生足够的回报,从而有理由进行必要的投资;
|
| • |
在诊断、预防或治疗欧洲联盟批准的有关疾病方面没有令人满意的方法,或者,如果存在这种方法,这种药物将对受这种情况影响的人有很大的好处。
|
| C. |
组织结构
|
| D. |
财产、厂房和设备
|
| 项目4A。 |
未解决的工作人员意见
|
| 项目5. |
业务和财务审查及前景
|
| A. |
经营成果
|
| • |
评估IFX-1在HS中的临床进展;
|
| • |
继续推进ifx-1的临床开发,以获得更多的适应症,包括aAV、pg和肿瘤学适应症;。
|
| • |
启动并继续开展研究方案和开发活动,包括开发IFX-2;
|
| • |
积极寻找更多的研究项目和更多的产品候选人;
|
| • |
维护、扩大和保护我们的知识产权组合;
|
| • |
聘用及挽留人员,例如业务发展及其他方面的人员;及
|
| • |
增加作为上市公司的运营成本,包括扩大我们的运营、财务和管理团队。
|
| • |
根据与合同研究组织或CRO、合同制造组织或CMOs、顾问和独立承包商签订的协议,代表我们进行研究和开发、临床前和临床
活动的费用;
|
| • |
与雇员有关的开支,包括根据雇员在组织内的角色而支付的薪金、福利及以股票为基础的补偿开支;及
|
| • |
与保护和维护我们的知识产权有关的律师的专业费用。
|
| • |
IFX-1在2019年,我们完成了对HS患者IFX-1的IIb期临床试验的登记和剂量。我们预计,如果我们继续在HS患者中发展IFX-1,我们与IFX-1相关的费用将在2020年减少,2021年将增加,在AAV患者中实施我们的第二期IFX-1临床计划,并继续我们对PG患者的第二阶段临床试验计划,以及我们目前在冠状病毒方面的
运行计划。我们预计,随着这些和任何额外临床试验的开始,我们的研究和开发费用将大幅度增加。此外,我们还承担与临床试验材料的制造和商业规模生产选择有关的费用。
|
| • |
如果是2号。我们正在继续进行IFX-2的临床前开发,其费用主要包括工资、CRO进行临床前测试的费用和临床前材料的
生产费用。
|
| • |
其他发展计划。我们的其他研究和开发费用涉及我们对其他产品候选人和发现活动的临床前研究,费用
主要包括工资、生产临床前化合物的费用和支付给CRO的费用。
|
| • |
临床试验或我们的产品候选产品产生否定或不确定的结果,包括未能证明统计意义;
|
| • |
临床试验、非临床试验和其他相关活动的范围、进度、结果和费用;
|
| • |
拖延或未能就可接受的临床试验合同或临床试验协议与可能的试验地点或潜在的CRO达成协议,这些协议的条件可以进行广泛的谈判,而且在不同的CRO和试验场之间可能会有很大的差异;
|
| • |
生产临床用品和建立我们的产品候选和任何可能开发的产品的商业供应的成本;
|
| • |
第三方承包商未能及时或根本不遵守监管要求或履行对我们的合同义务;
|
| • |
我们所追求的产品候选人的数量和特点;
|
| • |
不良副作用或其他意想不到的特点,导致我们或我们的调查人员、监管机构或机构审查委员会暂停或终止审判;
|
| • |
潜在的额外安全监测或监管机构要求的其他研究;
|
| • |
监管审批的成本、时间和结果;
|
| • |
需要批准的审判数量;
|
| • |
患者随访时间;
|
| • |
建立销售、营销和分销能力的成本和时间;以及
|
| • |
我们可能建立的任何合作、许可和其他安排的条款和时间,包括根据这些安排支付的任何里程碑和特许权使用费。
|
| • |
与员工有关的费用,包括工资、福利和基于员工在组织中的角色的股票补偿费用;
|
| • |
与研究和开发活动无关的审计员和咨询费用的专业费用;
|
| • |
与知识产权的提交、检控、保护及保养无关的律师的专业费用;及
|
| • |
设施、通讯和办公室费用。
|
|
2019
|
2018
|
变化
|
||||||||||
|
(欧元)
|
||||||||||||
|
其他收入和支出(净额)
|
315,011
|
299,058
|
15,953
|
|||||||||
|
研发费用
|
(44,582,136
|
)
|
(25,028,554
|
)
|
(19,553,582
|
)
|
||||||
|
一般和行政费用
|
(12,501,048
|
)
|
(12,786,869
|
)
|
285,821
|
|||||||
|
利息和所得税前的损失
|
(56,768,173
|
)
|
(37,516,364
|
)
|
(19,251,809
|
)
|
||||||
|
净财务结果
|
3,513,355
|
7,701,731
|
(4,188,376
|
)
|
||||||||
|
税前损失
|
(53,254,817
|
)
|
(29,814,634
|
)
|
(23,440,183
|
)
|
||||||
|
所得税费用
|
—
|
—
|
—
|
|||||||||
|
这一期间的损失
|
(53,254,817
|
)
|
(29,814,634
|
)
|
(23,440,183
|
)
|
||||||
|
外汇兑换业务的差异
|
2,177,033
|
50,196
|
2,126,837
|
|||||||||
|
总综合损失
|
(51,077,785
|
)
|
(29,764,438
|
)
|
(21,313,347
|
)
|
||||||
|
2019
|
2018
|
变化
|
||||||||||
|
(欧元)
|
||||||||||||
|
第三方费用
|
36,783,223
|
15,909,366
|
20,873,857
|
|||||||||
|
人事费用
|
6,231,812
|
8,037,082
|
(1,805,270
|
)
|
||||||||
|
其他费用
|
1,567,101
|
1,082,106
|
484,995
|
|||||||||
|
共计
|
44,582,136
|
25,028,554
|
19,553,582
|
|||||||||
|
2019
|
2018
|
变化
|
||||||||||
|
(欧元)
|
||||||||||||
|
人事费用
|
7,534,073
|
9,146,955
|
(1,612,882
|
)
|
||||||||
|
法律、咨询和审计费用
|
2,199,640
|
2,020,447
|
179,193
|
|||||||||
|
其他费用
|
2,767,335
|
1,619,467
|
1,147,868
|
|||||||||
|
共计
|
12,501,048
|
12,786,869
|
(285,821
|
)
|
||||||||
|
2019
|
2018
|
变化
|
||||||||||
|
(欧元)
|
||||||||||||
|
外汇收益
|
3,379,643
|
8,249,853
|
(4,870,210
|
)
|
||||||||
|
利息和其他收入
|
2,840,676
|
2,182,842
|
657,834
|
|||||||||
|
财务费用总额
|
6,220,320
|
10,432,695
|
(4,212,375
|
)
|
||||||||
|
外汇损失
|
2,684,699
|
2,623,782
|
60,917
|
|||||||||
|
其他财务费用
|
22,265
|
107,182
|
(84,917
|
)
|
||||||||
|
财务费用总额
|
2,706,964
|
2,730,964
|
(24,000
|
)
|
||||||||
|
净财务结果
|
3,513,355
|
7,701,731
|
(4,188,376
|
)
|
||||||||
| B. |
流动性和资本资源
|
|
十二月三十一日,
2019
|
十二月三十一日,
2018
|
|||||||
|
(欧元)
|
||||||||
|
用于业务活动的现金净额
|
(43,204,492
|
)
|
(21,549,248
|
)
|
||||
|
投资活动现金净额
|
20,341,554
|
(99,451,341
|
)
|
|||||
|
融资活动现金净额
|
(294,344
|
)
|
49,641,542
|
|||||
|
本期间开始时的现金和现金等价物
|
55,386,240
|
123,281,888
|
||||||
|
现金和现金等价物汇兑收益
|
902,321
|
3,461,399
|
||||||
|
本期间终了时的现金和现金等价物
|
33,131,280
|
55,386,240
|
||||||
| C. |
研发、专利和许可证等。
|
| D. |
趋势信息
|
| E. |
表外安排
|
| F. |
合同义务的列表式披露
|
|
按期间支付的款项
|
||||||||||||||||||||
|
共计
|
不足1年
|
在1至3之间
年数
|
在3至5之间
年数
|
超过5
年数
|
||||||||||||||||
|
(欧元)
|
||||||||||||||||||||
|
不可撤销的经营合同或服务下的合同承诺和其他合同义务:
|
24,447,508
|
10,602,651
|
12,647,879
|
1,196,978
|
—
|
|||||||||||||||
|
合同租赁义务
|
903,951
|
371,105
|
532,845
|
—
|
—
|
|||||||||||||||
|
共计
|
25,351,459
|
10,973,756
|
13,180,724
|
1,196,978
|
—
|
|||||||||||||||
| G. |
安全港
|
| 项目6. |
董事、高级管理人员和雇员
|
| A. |
董事和高级管理人员
|
|
名字
|
位置
|
年龄
|
初年
委任于
英华Rx有限公司或In亚麻Rx N.V.
(视情况而定)*
|
|||
|
阿恩德基督
|
首席财务官
|
53
|
2015
|
|||
|
马克斯
|
首席法律干事
|
44
|
2019
|
|||
|
延斯·霍斯坦
|
非执行主任
|
56
|
2018
|
|||
|
卡特林·乌希曼
|
非执行主任
|
55
|
2007
|
|||
|
马丽娜
|
非执行主任
|
42
|
2016
|
|||
|
马克·库布勒
|
非执行主任
|
44
|
2015
|
|||
|
尼古拉斯·富皮乌斯
|
非执行主任兼董事会主席
|
46
|
2007
|
|||
|
尼尔斯·里德曼
|
执行主任兼首席执行干事
|
48
|
2007
|
|||
|
郭仁峰
|
执行主任兼首席科学干事
|
50
|
2007
|
|||
|
理查德·布鲁尼克
|
非执行主任
|
64
|
2019
|
| B. |
补偿
|
| C. |
董事会惯例
|
| • |
建议股东大会任命独立审计师;
|
| • |
为编制或发布审计报告或提供其他审计服务而聘用的会计师事务所的任命、补偿、保留和监督;
|
| • |
在聘请独立核数师提供该等服务前,预先批准由我们的独立核数师提供的审计服务及非审计服务;
|
| • |
评估独立审计师的资格、业绩和独立性,并至少每年向全面监事会提出其结论;
|
| • |
在分别提交年度和季度报告之前,与董事会和独立审计师审查和讨论审计计划以及我们的年度审定财务报表和季度财务报表;
|
| • |
审查我们遵守法律和条例的情况,包括主要的法律和规章举措,并审查对我们的财务报表可能产生重大影响的任何重大诉讼或调查;
|
| • |
审查内部审计结果,包括设计和运行我们的内部控制的有效性;
|
| • |
检讨道德守则的运作及遵守情况;及
|
| • |
根据我们的相关人员交易政策批准或批准任何相关人交易(如我们的相关人员交易政策中所定义的),并审查涉及我们董事的潜在利益冲突。
|
| • |
确定、审查和批准公司与高管和董事薪酬相关的目标和目标;
|
| • |
分析可变薪酬构成部分的可能结果,以及它们如何影响我们执行官员的薪酬;
|
| • |
根据薪酬政策确定每名行政人员薪酬的任何长期激励成分,并全面检讨我们的行政人员薪酬及福利政策;
|
| • |
为董事会编制定期薪酬报告;
|
| • |
检讨及评估雇员补偿政策及做法所引致的风险,以及该等风险是否合理地可能对我们造成重大不良影响;及
|
| • |
在赔偿委员会认为必要或适当的情况下,保留或征求赔偿顾问、法律顾问或其他顾问的意见,以履行其职责。
|
| • |
制定和审查董事会的遴选标准和任命程序;
|
| • |
审查董事会的规模和组成,并就董事会的组成情况提出建议;
|
| • |
领导董事会进行自我评价,以确定董事会及其委员会是否有效运作;
|
| • |
制订和检讨继任董事的计划;及
|
| • |
就董事的委任或再委任提交建议书。
|
| D. |
员工
|
| E. |
股份所有权
|
|
项目7.
|
大股东与关联方交易
|
| A. |
大股东
|
| • |
据我们所知,每一个人或一组关联人都有权拥有我们5%或更多的已发行普通股(截至该股东向证交会提交的13G附表13G文件);
|
| • |
我们的每一位董事和高级管理人员;以及
|
| • |
所有董事和高级管理人员作为一个整体。
|
|
普通股
|
||||||||
|
数
|
百分比
|
|||||||
|
5%股东
|
||||||||
|
与Staidson香港投资有限公司有关联的实体(1)
|
2,316,644
|
8.9
|
%
|
|||||
|
与贝克兄弟顾问公司有关联的实体LP(2)
|
1,778,415
|
6.8
|
%
|
|||||
|
董事和高级管理人员
|
||||||||
|
Niels Riedemann(3)
|
2,178,740
|
8.3
|
%
|
|||||
|
郭仁峰(4)
|
2,606,747
|
10.0
|
%
|
|||||
|
阿恩德基督(5)
|
346,965
|
1.3
|
%
|
|||||
|
杰森·马克斯(6岁)
|
31,250
|
*
|
||||||
|
马克·库布勒(7岁)
|
996,043
|
3.8
|
%
|
|||||
|
Nicolas Fulpius(8岁)
|
541,977
|
2.1
|
%
|
|||||
|
Katrin Uschmann(9)
|
43,840
|
*
|
||||||
|
Lina Ma(10岁)
|
28,720
|
*
|
||||||
|
Jens Holstein(11岁)
|
10,674
|
*
|
||||||
|
Richard Brudnick(12岁)
|
68,450
|
*
|
||||||
|
全体董事和高级管理人员(11人)
|
6,853,496
|
26.3
|
%
|
|||||
| * |
指未超过已发行普通股总额1%的实益所有权。
|
| (1) |
Staidson Hong Kong Investment Company Limited(“STS”)由Staidson(北京)生物制药有限公司全资拥有,该公司是一家上市公司,其普通股在深圳证券交易所上市。STS地址为香港西贡马耀棠1楼。
|
| (2) |
普通股由Baker Bros.Advisors LP(“顾问”)和Baker Bros.Advisors(GP)LLC(“顾问GP”)持有,如Felix J.Baker和Julian C.Baker(统称为“报告人”)于2020年2月14日向证券交易委员会提交
文件的报告。贝克兄弟生命科学公司每一家公司直接持有的普通股总数为1,631,132和667,L.P.为147283,合为1,778,415。贝克兄弟顾问有限公司的地址是纽约华盛顿街860号,纽约,纽约,10014。
|
| (3) |
包括(A)1,068,908普通股,(B)404,040股普通股,这些普通股可根据根据“2016年计划”发行的期权获得,行使价格为每股3.35美元,于2031年11月
18到期;(C)根据行使根据B系列融资发行的期权可能获得的126,005股普通股,行使价格为每股0.0012欧元,(D)根据
行使根据“2017年计划”发行的期权可能获得的574 378股普通股,行使价格为每股3.35美元,有效期至2025年12月13日,根据根据“2017年计划”发行的期权可获得的5 409股普通股,行使价格为每股3.35美元,将于2026年11月20日到期。
|
| (4) |
(A)1,744,991股普通股,(B)336,672股根据“2016年计划”发行的期权,行使价格为每股3.35美元,于2031年11月
18到期,(C)根据根据2017年计划发行的期权可能获得的519,675股普通股,行使价格为每股3.35美元,于12月13日到期,2025年和5,409股普通股
,它们可能根据根据2017年计划发行的期权获得,行使价格为每股3.35美元,将于2026年11月20日到期。
|
| (5) |
包括(A)6,384股普通股,(B)根据行使根据“2016年计划”发行的期权可能获得的202,020股普通股,行使价格为每股3.35美元,于11月18日到期,
2031,(C)根据根据2017年计划发行的期权可能获得的136,758股普通股,行使价格为每股3.35美元,于12月13日到期,2025年和1,803股普通股,这些股份
可根据根据2017年计划发行的期权获得,行使价格为每股3.35美元,将于2026年11月20日到期。
|
| (6) |
由31,250股普通股组成,这些股份可能根据根据2017年计划发行的期权获得,行使价格为每股3.35美元,将于2026年11月20日到期。
|
| (7) |
包括:(A)960,015股普通股;(B)根据B系列融资发行的期权可获得的7,308股普通股,行使价格为每股0.0012欧元;(C)根据根据2017年计划发行的期权可获得的28,720股普通股,行权价格为每股3.35美元,有效期至2025年12月13日。
|
| (8) |
包括(A)513,257股普通股和(B)28,720股普通股,这些股份可根据根据2017年计划发行的期权获得,行使价格为每股3.35美元,有效期至2025年12月13日
。
|
| (9) |
包括:(A)根据根据B系列融资发行的期权可能获得的15 120股普通股,行使价格为每股0.0012欧元;(B)根据2017年计划发行的期权,可获得28.720股普通股,其行使价格为每股3.35美元,将于2025年12月13日到期。
|
| (10) |
包括根据“2017年计划”发行的期权可能获得的28,720股普通股,行使价格为每股3.35美元,将于2025年12月13日到期。
|
| (11) |
由10,764股普通股组成,这些普通股可能根据根据2017年计划发行的期权获得,行使价格为每股3.35美元,将于2026年9月21日到期。
|
|
(12)
|
(A)50,000股普通股和(B)18,450股普通股,可根据根据2017年计划发行的期权获得,行使价格为每股3.35美元,
将于2027年2月4日到期。
|
| B. |
关联方交易
|
| C. |
专家和律师的利益
|
| 项目8. |
财务信息
|
| A. |
合并报表和其他财务资料
|
| B. |
重大变化
|
| 项目9. |
要约与上市
|
| A. |
提供及上市详情
|
| B. |
分配计划
|
| C. |
市场
|
| D. |
出售股东
|
| E. |
稀释
|
| F. |
发行费用
|
| 项目10. |
补充资料
|
| A. |
股本
|
| B. |
章程大纲及组织章程细则
|
| C. |
材料合同
|
| D. |
外汇管制
|
| E. |
赋税
|
| • |
某些金融机构;
|
| • |
(一)证券交易商、证券交易商采用按市价计价的税务会计方法的;
|
| • |
作为套期保值交易的一部分持有普通股的人、跨行、清洗销售、转换交易或其他综合交易的人或就该普通股进行建设性出售的人;
|
| • |
用于美国联邦所得税目的的功能货币不是美元的人;
|
| • |
被列为美国联邦所得税合作伙伴关系的实体;
|
| • |
免税实体,包括“个人退休账户”或“罗斯爱尔兰共和军”;
|
| • |
持有或被视为持有我们10%或10%以上股份的人(通过投票或价值);
|
| • |
受“守则”第451(B)条规限的人;或
|
| • |
与在美国境外进行的贸易或业务有关而持有普通股的人。
|
| • |
美国公民或居民个人;
|
| • |
在美国、其任何州或哥伦比亚特区内或根据美国法律设立或组织的公司或应作为公司征税的其他实体;
|
| • |
一项财产或信托,其收入不论其来源如何,均须缴纳美国联邦所得税:或
|
| • |
一项信托,如果美国法院可以对信托的管理行使主要监督,并且授权一名或多名美国人控制信托的所有实质性决定。
|
| (i) |
如果持有我们普通股的人,以及个人、其伴侣或其某些亲属通过血亲或直系亲属(包括寄养子女),根据2001年“荷兰所得税法”(Wet Inkompresbeling 2001),在公司中享有相当大的权益(Aanmerkelijk Belang)或被认为是实质性权益(虚构的aanmerkelijk Belang)。一般说来,一家公司的证券持有人被视为持有该公司的重大权益,如果只有该持有人或个人与其
合伙人(2001年“荷兰所得税法”所界定的)直接或间接持有(I)该公司发行和未偿资本总额的5%或以上的权益,或该公司某一类别股份的已发行和未清偿资本的5%或以上;或(2)直接或间接获得该公司股份的权利;或(2)直接或间接获得这种权益的权利;(2)直接或间接获得这种权益的权利;或(2)直接或间接获得这种权益的权利;或(Iii)该公司与该公司5%或以上的年度利润及(或)该公司清盘收益的5%或以上有关的某些利润分享权。如一间公司的一项重大权益(或其部分)已在不获承认的基础上处置或当作已处置,则可产生当作重大权益;
|
| (2) |
如果这些股东持有的股份符合或有资格参加1969年“荷兰企业所得税法”(1969年“荷兰企业所得税法”),则我们的普通股持有人。一般来说,纳税人在一家公司的名义缴足股本中持有5%或以上的股份(或在某些情况下,在投票权方面)符合参与资格。如果持股人没有5%或以上的股份,但相关实体(法定界定的期限)有参与,或者持有股份的公司是相关实体(法律规定的期限),则持有人也可以参与;
|
| (3) |
股份持有人-其股份或从中获得的任何利益是一种报酬,或被视为这类持有者所从事的(就业)活动或服务的报酬,或某些与这些持有人有关的个人
,不论是在雇用关系内还是在雇用关系之外,在经济上向持有人提供与有关工作活动或服务有关的某些福利(如2001年“荷兰所得税法”所界定);以及
|
| (四) |
养恤基金、投资机构(财政方面)、豁免投资机构(Vrijestelde Belegingsinstellingen)(如1969年“荷兰企业所得税法”所界定)和其他实体,这些实体在荷兰全部或部分不受公司所得税或免缴公司所得税,以及在其居住国免征公司所得税的实体,该居住国是欧洲联盟的另一个国家,挪威、列支敦士登、冰岛或荷兰已同意按照国际标准与其交换信息的任何其他国家。
|
| • |
现金或实物的分配、被视为和建设性的分配以及未确认为荷兰股息扣缴税款目的的已付资本的偿还;
|
| • |
清算收益、赎回股份的收益,或我们或我们的一家子公司或其他附属实体回购股份的收益,只要这些收益超过为荷兰股息扣缴税而确认的这些股份的平均已付资本,除非在回购时适用特定的法定豁免;
|
| • |
相当于已发行股票的票面价值或股份票面价值增加的数额,但须符合为荷兰股息预扣缴税的目的而确认的供款,已作出或将会作出;
及
|
| • |
部分偿还已缴入资本,为荷兰股息预扣缴税的目的而确认,但如我们有净利润(Zuivere Winst),则属例外,除非股份持有人已事先在大会上决议作出上述偿还,而有关股份的票面价值已因修订“公司章程”而减少等额。
|
| (i) |
普通股可归因于一家企业,这种股份的持有人从该企业获得利润,无论是作为企业家(Onderneier),还是作为一名共同有权享有该企业净值(Medeerechtigd Tot Het Vermogen)的人,而不是“2001年荷兰所得税法”所界定的股东);或
|
| (2) |
普通股持有人被认为从事的活动超出了普通资产管理范围(Normaal,Actief Vermogensbeheer),或从可作为其他活动的收益征税的
股份中获得收益(Restaat Uit Overige Werkzamheden)。
|
| (i) |
这类持有人在一家企业或被视为企业(如“荷兰所得税法”和“荷兰企业所得税法”中所界定的)中没有利害关系,该企业或企业的全部或部分在荷兰有效管理,或通过在荷兰的常设机构、被视为常设机构或常驻代表进行,企业或其一部分的普通股可归因于该企业或企业的一部分;
|
| (2) |
如果此种持有人是个人,则该持有人在荷兰不就普通股开展超出普通资产管理范围的任何活动(Normaal,Actief
Vermogensbeheer),也不从可作为荷兰其他活动的收益征税的普通股中获得利益(Restaat Uit Overige Werkzaamheden)。
|
| (i) |
被继承人、捐赠人、继承人、受赠人或任何其他受益人在转让时在德国有其住所、住所、注册办事处或管理地点,或者是连续五年以上未在德国居住的德国公民;或
|
| (2) |
(不论个人情况如何)该等股份由死者或捐赠人持有,作为在德国设有常设机构或在德国委任常驻代表的商业资产:或
|
| (3) |
(不论个人情况如何)根据“外国税法”第1条第2款,至少10%的股份直接或间接由遗赠人或赠与人、本人或与关联方共同持有。
|
| F. |
股息和支付代理人
|
| G. |
专家发言
|
| H. |
展示的文件
|
| I. |
辅助信息
|
| 项目11. |
市场风险的定量和定性披露
|
| 项目12. |
证券的描述(股本证券除外)
|
| A. |
债务证券
|
| B. |
认股权证及权利
|
| C. |
其他证券
|
| D. |
美国保存人股份
|
|
项目13.
|
违约、股利拖欠和拖欠
|
| A. |
缺省值
|
| B. |
拖欠和拖欠款项
|
|
项目14.
|
对担保持有人权利和收益使用的实质性修改
|
| A. |
对文书的材料修改
|
| B. |
对权利的实质性修改
|
| C. |
资产的提取或替换
|
| D. |
更改受托人或付款代理人
|
| E. |
收益的使用
|
|
费用
|
数额(欧元)
|
|||
|
公开发行证券保险
|
583,100
|
|||
|
法律费用和开支
|
299,927
|
|||
|
审计费用和费用
|
252,500
|
|||
|
FINRA报名费
|
18,889
|
|||
|
证券交易委员会登记费
|
15,351
|
|||
|
共计
|
1,169,767
|
|||
|
费用
|
数额(美元)
|
|||
|
法律费用和开支
|
1,608,937
|
|||
|
IPO保险
|
583,100
|
|||
|
会计和审计费用和费用
|
393,263
|
|||
|
纳斯达克上市费
|
107,576
|
|||
|
证券交易委员会登记费
|
17,958
|
|||
|
FINRA报名费
|
16,146
|
|||
|
杂项费用
|
2,092
|
|||
|
共计
|
2,729,072
|
|||
|
项目15.
|
管制和程序
|
| A. |
披露控制和程序
|
| B. |
管理层财务报告内部控制年度报告
|
| C. |
注册会计师事务所认证报告
|
| D. |
财务报告内部控制的变化
|
| 项目16. |
预留
|
| 项目16A. |
审计委员会财务专家
|
| 项目16B. |
道德守则
|
| 项目16C. |
首席会计师费用及服务
|
| A. |
审计费
|
| B. |
与审计有关的费用
|
| C. |
税费
|
| D. |
所有其他费用
|
| E. |
审计委员会的审批前政策和程序
|
| F. |
如审计工作超过50%,则由首席会计师以外的其他人员进行审计工作
|
| 项目16D. |
豁免审计委员会的上市标准
|
| 项目16E. |
发行人和关联购买者购买股票证券
|
| 项目16F. |
注册会计师的变更
|
| 项目16G. |
公司治理
|
| 项目16H. |
矿山安全披露
|
| 项目17. |
财务报表
|
| 项目18. |
财务报表
|
| 项目19. |
展品
|
|
陈列品
没有。
|
描述
|
|
|
1.1
|
InaffRx N.V.公司章程(参见本公司于2017年11月9日向
SEC提交的表格F-1(档案号333-220962)上公司注册声明的后生效修正案的附录3.2)。
|
|
|
2.1
|
注册权利协议(参见表4.2中的注册权利协议,表F-1(档案编号333-220962)的公司注册声明于2017年11月9日提交给证交会)。
|
|
|
2.2
|
高级义齿表格(参见本公司于2019年3月28日向证交会提交的F-3ASR表格(档案号333-230560)的注册声明表4.2)。
|
|
|
2.3
|
辅助性义齿表格(参见本公司于2019年3月28日向证交会提交的F-3ASR表格(档案号333-230560)的注册声明表4.3)。
|
|
|
2.4*
|
根据1934年“证券交易法”第12条登记的每一适用类别证券的权利说明。
|
|
|
4.1
|
InaffRx GmbH公司和Ernst-Abbe-Stifong公司2008年1月15日签订的租赁协议英文摘要,并不时加以修订和补充(参见表10.1至
2017年10月13日提交证券交易委员会的公司关于表格F-1的登记声明(档案号333-220962)。
|
|
|
4.2
|
2017年4月10日InaffRx GmbH与Immoprojekt Grundstücksver钨sgesellschaft MBH签订的租赁协议英文摘要(参见2017年10月13日提交给SEC的公司表格F-1的
登记声明(档案号333-220962)图10.2)。
|
|
|
4.3†
|
2015年12月28日InaffRx GmbH公司与北京Defengrei生物技术有限公司之间的共同发展协议,并由2015年12月28日第1号增编补充(此处参考2017年11月7日提交给SEC的公司表格F-1登记声明(档案编号333-220962)的表10.3)。
|
|
|
4.4
|
董事及高级执行人员补偿协议表格(参阅本公司于2017年10月13日向证交会提交的表格F-1(档案编号333-220962)的注册声明图10.4)。
|
|
|
4.5
|
长期激励计划(参见2017年11月17日提交证交会的S-8表格(档案号333-221656)的公司注册声明表99)。
|
|
|
8.1+
|
附属公司名单。
|
|
|
12.1*
|
2002年“萨班斯-奥克斯利法”第302条规定的认证。
|
|
|
12.2*
|
2002年“萨班斯-奥克斯利法”第302条规定的认证。
|
|
|
13.1*
|
根据2002年“萨班斯-奥克斯利法”第906条通过的18 U.S.C.第1350条所规定的认证。
|
|
|
13.2*
|
根据2002年“萨班斯-奥克斯利法”第906条通过的18 U.S.C.第1350条所规定的认证。
|
|
|
15.1*
|
KPMG AG Wirtschaftsprüfunsgesellschaft的同意。
|
|
|
101
|
以XBRL(可扩展的业务报告语言)格式的2019年12月31日终了年度的年度报告中的下列材料:(一)合并财务报表和(二)“综合财务报表说明”,标记为文本和详细内容。
|
| * |
随函提交。
|
| + |
以前的档案。
|
| † |
对展品的部分给予保密待遇。遗漏并单独提交给证券交易委员会的机密资料。
|
|
InaffRx N.V.
|
|||
|
通过:
|
尼尔斯·里德曼
|
||
|
姓名:
|
尼尔斯·里德曼
|
||
|
标题:
|
首席执行官兼主任
|
||
|
日期:2020年4月28日
|
|||
|
通过:
|
/阿恩德基督
|
||
|
姓名:
|
阿恩德基督
|
||
|
标题:
|
首席财务官
|
||
|
日期:2020年4月28日
|
|||
|
经审计的年度合并财务报表
|
页
|
|
独立注册会计师事务所报告
|
F-1
|
|
2019、2018和2017年12月31日终了年度综合损失综合报表
|
F-2
|
|
截至2019年12月31日和2018年12月31日的财务状况综合报表
|
F-3
|
|
2019、2018和2017年12月31日终了年度股东权益变动综合报表
|
F-4
|
|
2019、2018和2017年12月31日终了年度现金流动合并报表
|
F-6
|
|
截至2019和2018年12月31日终了年度合并财务报表附注
|
F-7
|
|
注
|
2019
|
2018
|
2017
|
|||||||||||||
|
(欧元)
|
||||||||||||||||
|
营业费用
|
||||||||||||||||
|
研发费用
|
(44,582,136
|
)
|
(25,028,554
|
)
|
(14,414,628
|
)
|
||||||||||
|
一般和行政费用
|
(12,501,048
|
)
|
(12,786,869
|
)
|
(5,138,498
|
)
|
||||||||||
|
业务费用共计
|
1.
|
(a)
|
(57,083,184
|
)
|
(37,815,422
|
)
|
(19,553,126
|
)
|
||||||||
|
其他收入
|
400,253
|
303,860
|
115,525
|
|||||||||||||
|
其他费用
|
(85,242
|
)
|
(4,802
|
)
|
(7,644
|
)
|
||||||||||
|
运行效果
|
(56,768,173
|
)
|
(37,516,364
|
)
|
(19,445,245
|
)
|
||||||||||
|
财政收入
|
6,220,320
|
10,432,695
|
130,032
|
|||||||||||||
|
财务费用
|
(2,706,964
|
)
|
(2,730,964
|
)
|
(4,922,535
|
)
|
||||||||||
|
净财务结果
|
1.
|
(a)
|
3,513,355
|
7,701,731
|
(4,792,503
|
)
|
||||||||||
|
这一期间的损失
|
(53,254,817
|
)
|
(29,814,634
|
)
|
(24,237,748
|
)
|
||||||||||
|
共享信息
|
4.
|
(d)
|
||||||||||||||
|
加权平均流通股数
|
26,004,519
|
25,095,027
|
9,410,524
|
|||||||||||||
|
欧元每股亏损(基本/稀释)
|
(2.05
|
)
|
(1.19
|
)
|
(2.58
|
)
|
||||||||||
|
这一期间的损失
|
(53,254,817
|
)
|
(29,814,634
|
)
|
(24,237,748
|
)
|
||||||||||
|
其他可在以后各期重新归类为损益的综合收入:
|
||||||||||||||||
|
外汇业务折算中的汇率差异
|
2,177,033
|
50,196
|
—
|
|||||||||||||
|
总综合损失
|
(51,077,785
|
)
|
(29,764,438
|
)
|
(24,237,748
|
)
|
||||||||||
|
注
|
2019
|
2018
|
||||||||||
|
(欧元)
|
||||||||||||
|
资产
|
||||||||||||
|
非流动资产
|
||||||||||||
|
财产、厂房和设备
|
1.
|
(c)
|
1,413,297
|
624,668
|
||||||||
|
无形资产
|
1.
|
(d)
|
452,400
|
222,866
|
||||||||
|
非流动其他非金融资产
|
1.
|
(f)
|
452,217
|
—
|
||||||||
|
非流动金融资产
|
1.
|
(g)
|
272,614
|
207,444
|
||||||||
|
非流动资产共计
|
2,590,528
|
1,054,979
|
||||||||||
|
流动资产
|
||||||||||||
|
流动其他非金融资产
|
1.
|
(f)
|
3,500,884
|
1,588,702
|
||||||||
|
流动金融资产
|
1.
|
(g)
|
82,353,867
|
101,184,240
|
||||||||
|
现金和现金等价物
|
1.
|
(i)
|
33,131,280
|
55,386,240
|
||||||||
|
流动资产总额
|
118,986,031
|
158,159,183
|
||||||||||
|
总资产
|
121,576,558
|
159,214,161
|
||||||||||
|
权益与负债
|
||||||||||||
|
衡平法
|
1.
|
(h)
|
||||||||||
|
已发行资本
|
3,132,631
|
3,115,725
|
||||||||||
|
股票溢价
|
211,006,606
|
211,021,835
|
||||||||||
|
其他资本准备金
|
25,142,213
|
18,310,003
|
||||||||||
|
累积赤字
|
(134,362,006
|
)
|
(81,107,188
|
)
|
||||||||
|
股本的其他组成部分
|
2,227,228
|
50,196
|
||||||||||
|
总股本
|
107,146,673
|
151,390,571
|
||||||||||
|
非流动负债
|
||||||||||||
|
租赁负债
|
1.
|
(e)
|
330,745
|
—
|
||||||||
|
规定和政府赠款
|
39,013
|
67,945
|
||||||||||
|
非流动负债共计
|
369,758
|
67,945
|
||||||||||
|
流动负债
|
||||||||||||
|
租赁负债
|
1.
|
(e)
|
515,203
|
—
|
||||||||
|
雇员福利
|
975,629
|
789,800
|
||||||||||
|
社会证券、其他税收和非金融负债
|
105,634
|
308,533
|
||||||||||
|
贸易和其他应付款
|
1.
|
(g)
|
12,413,662
|
6,657,312
|
||||||||
|
规定
|
50,000
|
—
|
||||||||||
|
流动负债总额
|
14,060,128
|
7,755,645
|
||||||||||
|
负债总额
|
14,429,886
|
7,823,590
|
||||||||||
|
股本和负债共计
|
121,576,558
|
159,214,161
|
||||||||||
|
注
|
股份
突出
|
已发行资本
|
股票溢价
|
|||||||||||||
|
(欧元)
|
||||||||||||||||
|
2017年1月1日结余
|
2,362,500
|
31,428
|
—
|
|||||||||||||
|
这一期间的损失
|
—
|
—
|
—
|
|||||||||||||
|
外汇业务折算中的汇率差异
|
—
|
—
|
—
|
|||||||||||||
|
总综合损失
|
—
|
—
|
—
|
|||||||||||||
|
与公司业主的交易
|
||||||||||||||||
|
捐款
|
||||||||||||||||
|
发行普通股
|
1.
|
(e)
|
7,068,129
|
848,175
|
90,055,312
|
|||||||||||
|
交易成本
|
—
|
—
|
(9,114,770
|
)
|
||||||||||||
|
股权结算股票支付
|
4.
|
(e)
|
—
|
—
|
—
|
|||||||||||
|
捐款总额
|
7,068,129
|
848,175
|
80,940,542
|
|||||||||||||
|
所有权权益的变化
|
||||||||||||||||
|
重组
|
16,482,071
|
1,977,849
|
80,698,025
|
|||||||||||||
|
附属公司的清盘
|
—
|
—
|
—
|
|||||||||||||
|
所有权权益变动共计
|
16,482,071
|
1,977,849
|
80,698,025
|
|||||||||||||
|
与公司业主的交易总额
|
23,550,200
|
2,826,024
|
161,638,566
|
|||||||||||||
|
2017年12月31日结余
|
23,812,100
|
2,857,452
|
161,638,566
|
|||||||||||||
|
这一期间的损失
|
—
|
—
|
—
|
|||||||||||||
|
外汇业务折算中的汇率差异
|
—
|
—
|
—
|
|||||||||||||
|
总综合损失
|
—
|
—
|
—
|
|||||||||||||
|
与公司业主的交易
|
||||||||||||||||
|
捐款
|
||||||||||||||||
|
发行普通股
|
1.
|
(e)
|
1,850,000
|
222,000
|
52,768,733
|
|||||||||||
|
交易成本
|
—
|
—
|
(3,801,265
|
)
|
||||||||||||
|
股权结算股票支付
|
4.
|
(e)
|
—
|
—
|
—
|
|||||||||||
|
行使股票期权
|
302,279
|
36,273
|
415,801
|
|||||||||||||
|
捐款总额
|
2,152,279
|
258,273
|
49,383,269
|
|||||||||||||
|
与公司业主的交易总额
|
2,152,279
|
258,273
|
49,383,269
|
|||||||||||||
|
2018年12月31日结余
|
25,964,379
|
3,115,725
|
211,021,835
|
|||||||||||||
|
这一期间的损失
|
—
|
—
|
—
|
|||||||||||||
|
外汇业务折算中的汇率差异
|
—
|
—
|
—
|
|||||||||||||
|
总综合损失
|
—
|
—
|
—
|
|||||||||||||
|
与公司业主的交易
|
||||||||||||||||
|
捐款
|
||||||||||||||||
|
股权结算股票支付
|
—
|
—
|
—
|
|||||||||||||
|
行使股票期权
|
140,876
|
16,905
|
(15,229
|
)
|
||||||||||||
|
捐款总额
|
140,876
|
16,905
|
(15,229
|
)
|
||||||||||||
|
与公司业主的交易总额
|
140,876
|
16,905
|
(15,229
|
)
|
||||||||||||
|
2019年12月31日结余
|
26,105,255
|
3,132,631
|
211,006,606
|
|||||||||||||
|
注
|
其他资本
储备
|
累积
赤字
|
其他组成部分
衡平法
|
总股本
|
||||||||||||||||
|
(欧元)
|
||||||||||||||||||||
|
2017年1月1日结余
|
1,325,006
|
(27,054,806
|
)
|
8,839
|
(25,689,533
|
)
|
||||||||||||||
|
这一期间的损失
|
—
|
(24,237,748
|
)
|
—
|
(24,237,748
|
)
|
||||||||||||||
|
外汇业务折算中的汇率差异
|
—
|
—
|
—
|
—
|
||||||||||||||||
|
总综合损失
|
—
|
(24,237,748
|
)
|
—
|
(24,237,748
|
)
|
||||||||||||||
|
与公司业主的交易
|
||||||||||||||||||||
|
捐款
|
||||||||||||||||||||
|
发行普通股
|
1.(e
|
)
|
—
|
—
|
—
|
90,903,488
|
||||||||||||||
|
交易成本
|
—
|
—
|
—
|
(9,114,770
|
)
|
|||||||||||||||
|
股权结算股票支付
|
4.(e
|
)
|
4,550,105
|
—
|
—
|
4,550,105
|
||||||||||||||
|
捐款总额
|
4,550,105
|
—
|
—
|
86,338,823
|
||||||||||||||||
|
所有权权益的变化
|
||||||||||||||||||||
|
重组
|
350,242
|
—
|
—
|
83,026,115
|
||||||||||||||||
|
附属公司的清盘
|
—
|
—
|
(8,839
|
)
|
(8,839
|
)
|
||||||||||||||
|
所有权权益变动共计
|
350,242
|
—
|
(8,839
|
)
|
83,017,276
|
|||||||||||||||
|
与公司业主的交易总额
|
4,900,347
|
—
|
(8,839
|
)
|
169,356,099
|
|||||||||||||||
|
2017年12月31日结余
|
6,225,353
|
(51,292,555
|
)
|
0
|
119,428,816
|
|||||||||||||||
|
这一期间的损失
|
—
|
(29,814,634
|
)
|
—
|
(29,814,634
|
)
|
||||||||||||||
|
外汇业务折算中的汇率差异
|
—
|
—
|
50,196
|
50,196
|
||||||||||||||||
|
总综合损失
|
—
|
(29,814,634
|
)
|
50,196
|
(29,764,438
|
)
|
||||||||||||||
|
与公司业主的交易
|
||||||||||||||||||||
|
捐款
|
||||||||||||||||||||
|
发行普通股
|
1.(e
|
)
|
—
|
—
|
—
|
52,990,733
|
||||||||||||||
|
交易成本
|
—
|
—
|
—
|
(3,801,265
|
)
|
|||||||||||||||
|
股权结算股票支付
|
4.(e
|
)
|
12,084,651
|
—
|
—
|
12,084,651
|
||||||||||||||
|
行使股票期权
|
—
|
—
|
—
|
452,075
|
||||||||||||||||
|
捐款总额
|
12,084,651
|
—
|
—
|
61,726,194
|
||||||||||||||||
|
与公司业主的交易总额
|
12,084,651
|
—
|
—
|
61,726,194
|
||||||||||||||||
|
2018年12月31日结余
|
18,310,003
|
(81,107,188
|
)
|
50,196
|
151,390,571
|
|||||||||||||||
|
这一期间的损失
|
—
|
(53,254,817
|
)
|
—
|
(53,254,817
|
)
|
||||||||||||||
|
外汇业务折算中的汇率差异
|
—
|
—
|
2,177,033
|
2,177,033
|
||||||||||||||||
|
总综合损失
|
—
|
(53,254,817
|
)
|
2,177,033
|
(51,077,784
|
)
|
||||||||||||||
|
与公司业主的交易
|
||||||||||||||||||||
|
捐款
|
||||||||||||||||||||
|
股权结算股票支付
|
4.(e
|
)
|
6,832,210
|
—
|
—
|
6,832,210
|
||||||||||||||
|
行使股票期权
|
—
|
—
|
—
|
1,676
|
||||||||||||||||
|
捐款总额
|
6,832,210
|
—
|
—
|
6,833,886
|
||||||||||||||||
|
与公司业主的交易总额
|
6,832,210
|
—
|
—
|
6,833,886
|
||||||||||||||||
|
2019年12月31日结余
|
25,142,213
|
(134,362,006
|
)
|
2,227,228
|
107,146,673
|
|||||||||||||||
|
注
|
2019
|
2018
|
2017
|
|||||||||||||
|
(欧元)
|
||||||||||||||||
|
经营活动
|
||||||||||||||||
|
这一期间的损失
|
(53,254,817
|
)
|
(29,814,634
|
)
|
(24,237,748
|
)
|
||||||||||
|
调整:
|
||||||||||||||||
|
折旧和摊销
|
663,166
|
173,630
|
70,910
|
|||||||||||||
|
净财务结果
|
1.(a)
|
|
(3,513,355
|
)
|
(7,701,731
|
)
|
4,792,503
|
|||||||||
|
股票支付费用
|
4.(e)
|
|
6,832,210
|
12,084,651
|
4,550,105
|
|||||||||||
|
其他非现金调整数
|
(307,849
|
)
|
196,699
|
24,076
|
||||||||||||
|
变动:
|
||||||||||||||||
|
其他非金融资产
|
(2,364,399
|
)
|
(893,602
|
)
|
(522,818
|
)
|
||||||||||
|
流动金融资产
|
—
|
(316,112
|
)
|
89,599
|
||||||||||||
|
雇员福利
|
235,500
|
494,837
|
132,305
|
|||||||||||||
|
社会证券、其他税收和非金融负债
|
(209,948
|
)
|
304,627
|
(30,024
|
)
|
|||||||||||
|
贸易和其他应付款
|
5,734,795
|
2,243,137
|
2,912,740
|
|||||||||||||
|
收到的利息
|
3,001,109
|
1,679,250
|
66,391
|
|||||||||||||
|
已付利息
|
(20,903
|
)
|
—
|
—
|
||||||||||||
|
业务活动现金流量净额
|
(43,204,492
|
)
|
(21,549,248
|
)
|
(12,151,962
|
)
|
||||||||||
|
投资活动
|
||||||||||||||||
|
购置无形资产、实验室和办公设备的现金流出
|
(594,889
|
)
|
(806,531
|
)
|
(148,542
|
)
|
||||||||||
|
非流动其他金融资产投资的现金流出
|
1.(G),2.(B)
|
|
(75,543
|
)
|
(209,705
|
)
|
(18,481
|
)
|
||||||||
|
处置非流动其他金融资产所得收益
|
1.(G),2.(B)
|
|
—
|
21,811
|
—
|
|||||||||||
|
处置流动金融资产或偿还到期证券的收益
|
1.(G),2.(B)
|
|
103,559,395
|
7,990,204
|
—
|
|||||||||||
|
购买流动及非流动金融资产
|
1.(G),2.(B)
|
|
(82,547,409
|
)
|
(106,445,120
|
)
|
—
|
|||||||||
|
投资活动现金流量净额
|
20,341,554
|
(99,449,341
|
)
|
(167,023
|
)
|
|||||||||||
|
筹资活动
|
||||||||||||||||
|
发行股本所得收益
|
—
|
52,990,733
|
90,903,488
|
|||||||||||||
|
发行股本的交易成本
|
—
|
(3,801,265
|
)
|
(9,114,770
|
)
|
|||||||||||
|
行使股票期权所得收益
|
1.(g)
|
|
1,676
|
452,075
|
—
|
|||||||||||
|
发行优先股所得收益
|
—
|
—
|
27,012,050
|
|||||||||||||
|
偿还租赁债务
|
(296,020
|
)
|
—
|
—
|
||||||||||||
|
来自筹资活动的现金流量净额
|
(294,344
|
)
|
49,641,542
|
108,800,767
|
||||||||||||
|
汇率变动的影响
|
902,321
|
3,461,399
|
(2,316,631
|
)
|
||||||||||||
|
现金和现金等价物变动净额
|
(22,254,960
|
)
|
(71,357,047
|
)
|
94,165,152
|
|||||||||||
|
期初现金及现金等价物
|
55,386,240
|
123,281,888
|
29,116,737
|
|||||||||||||
|
期末现金及现金等价物
|
1.(f)
|
33,131,280
|
55,386,240
|
123,281,888
|
||||||||||||
|
1.
|
研发费用
|
|
研发费用
|
2019
|
2018
|
2017
|
|||||||||
|
(欧元)
|
||||||||||||
|
第三方服务
|
36,783,223
|
15,909,366
|
8,856,431
|
|||||||||
|
临床材料的制造
|
13,479,235
|
4,828,534
|
5,558,719
|
|||||||||
|
临床,临床前
|
23,303,988
|
11,080,832
|
3,297,712
|
|||||||||
|
人事费用
|
6,231,812
|
8,037,082
|
4,680,877
|
|||||||||
|
股份补偿费用
|
2,580,983
|
5,256,194
|
3,070,707
|
|||||||||
|
法律和咨询费
|
668,676
|
421,041
|
643,074
|
|||||||||
|
其他费用
|
898,425
|
661,065
|
234,129
|
|||||||||
|
共计
|
44,582,136
|
25,028,554
|
14,414,511
|
|||||||||
|
2.
|
一般和行政费用
|
|
一般和行政费用
|
2019
|
2018
|
2017
|
|||||||||
|
(欧元)
|
||||||||||||
|
人事费用
|
7,534,073
|
9,146,955
|
2,948,229
|
|||||||||
|
股份补偿费用
|
4,251,227
|
6,828,457
|
1,479,398
|
|||||||||
|
法律和咨询费
|
2,199,640
|
2,020,447
|
1,478,210
|
|||||||||
|
其他费用
|
2,767,335
|
1,619,467
|
712,059
|
|||||||||
|
共计
|
12,501,048
|
12,786,869
|
5,138,498
|
|||||||||
|
3.
|
雇员福利
|
|
雇员福利
|
2019
|
2018
|
2017
|
|||||||||
|
(欧元)
|
||||||||||||
|
工资和薪金
|
5,974,807
|
4,501,840
|
2,896,929
|
|||||||||
|
社会保障缴款(雇主份额)
|
562,255
|
350,024
|
182,189
|
|||||||||
|
股权结算股票支付
|
6,832,210
|
12,084,651
|
4,550,105
|
|||||||||
|
其他
|
396,613
|
247,522
|
—
|
|||||||||
|
共计
|
13,765,885
|
17,184,037
|
7,629,223
|
|||||||||
|
4.
|
净财务结果
|
|
财政收入
|
2019
|
2018
|
2017
|
|||||||||
|
(欧元)
|
||||||||||||
|
外汇收入
|
3,379,644
|
8,249,853
|
—
|
|||||||||
|
利息收入
|
2,840,676
|
2,182,842
|
130,032
|
|||||||||
|
共计
|
6,220,320
|
10,432,695
|
130,032
|
|||||||||
|
财务成本
|
||||||||||||
|
外汇费用
|
2,684,699
|
2,623,782
|
2,358,631
|
|||||||||
|
其他
|
22,265
|
107,182
|
2,563,904
|
|||||||||
|
共计
|
2,706,964
|
2,730,964
|
4,922,535
|
|||||||||
|
净财务结果
|
3,513,356
|
7,701,731
|
(4,792,503
|
)
|
||||||||
|
(一九二零九年十二月三十一日)
|
(2018年12月31日)
|
|||||||
|
(欧元)
|
||||||||
|
InaffRx N.V.
|
75,767,524
|
33,571,438
|
||||||
|
英华Rx有限公司
|
34,786,686
|
34,787,686
|
||||||
|
InaffRx制药公司
|
3,816,023
|
1,651,579
|
||||||
|
在德国的InaffRx
|
2019
|
2018
|
2017
|
|||||||||
|
(欧元)
|
||||||||||||
|
税前损失
|
(53,254,817
|
)
|
(29,814,634
|
)
|
(24,237,748
|
)
|
||||||
|
税率
|
29.7%
|
|
29.2%
|
|
31.2%
|
|
||||||
|
按税率计算的税收优惠
|
15,815,083
|
8,715,116
|
7,559,754
|
|||||||||
|
未确认递延税款资产的税务损失
|
(15,815,083
|
)
|
(8,715,116
|
)
|
(7,559,754
|
)
|
||||||
|
所得税
|
—
|
—
|
—
|
|||||||||
|
在美国。
|
2019
|
2018
|
2017
|
|||||||||
|
(以美元计)
|
||||||||||||
|
税前损失
|
(2,177,602
|
)
|
(1,891,058
|
)
|
—
|
|||||||
|
税率
|
27%
|
|
27%
|
|
—
|
|||||||
|
按税率计算的税收优惠
|
587,953
|
510,586
|
—
|
|||||||||
|
未确认递延税款资产的税务损失
|
(587,953
|
)
|
(510,586
|
)
|
—
|
|||||||
|
所得税
|
—
|
—
|
—
|
|||||||||
|
建筑,
办公物业
|
实验室,
办公室和其他
设备
|
前进
付款
|
共计
|
|||||||||||||
|
成本
|
(欧元)
|
|||||||||||||||
|
2018年1月1日
|
—
|
394,609
|
—
|
394,609
|
||||||||||||
|
加法
|
—
|
504,423
|
86,068
|
590,491
|
||||||||||||
|
处置
|
—
|
1,544
|
—
|
1,544
|
||||||||||||
|
重新分类
|
—
|
86,068
|
(86,068
|
)
|
—
|
|||||||||||
|
交换差异
|
—
|
8,534
|
—
|
8,534
|
||||||||||||
|
2018年12月31日
|
—
|
995,179
|
—
|
995,179
|
||||||||||||
|
2019年1月1日的资产使用权,见附注1。
|
695,614
|
35,058
|
—
|
730,672
|
||||||||||||
|
加法
|
636,754
|
259,647
|
54,338
|
950,740
|
||||||||||||
|
处置
|
(266,057
|
)
|
(142,400
|
)
|
—
|
(408,457
|
)
|
|||||||||
|
重新分类
|
—
|
54,408
|
(54,408
|
)
|
—
|
|||||||||||
|
交换差异
|
1,512
|
6,639
|
70
|
8,221
|
||||||||||||
|
2019年12月31日
|
1,067,823
|
1,208,531
|
—
|
2,276,355
|
||||||||||||
|
折旧
|
||||||||||||||||
|
2018年1月1日
|
—
|
(221,970
|
)
|
—
|
(221,970
|
)
|
||||||||||
|
本年度折旧费用
|
—
|
(148,375
|
)
|
—
|
(148,375
|
)
|
||||||||||
|
交换差异
|
—
|
(166
|
)
|
—
|
(166
|
)
|
||||||||||
|
2018年12月31日
|
—
|
(370,510
|
)
|
—
|
(370,510
|
)
|
||||||||||
|
本年度折旧费用
|
(283,350
|
)
|
(273,458
|
)
|
—
|
(556,808
|
)
|
|||||||||
|
处置
|
38,008
|
26,235
|
—
|
64,243
|
||||||||||||
|
交换差异
|
216
|
(198
|
)
|
—
|
18
|
|||||||||||
|
2019年12月31日
|
(245,126
|
)
|
(617,932
|
)
|
—
|
(863,058
|
)
|
|||||||||
|
净账面价值
|
—
|
—
|
—
|
—
|
||||||||||||
|
2019年12月31日
|
822,697
|
590,600
|
—
|
1,413,297
|
||||||||||||
|
2018年12月31日
|
—
|
624,668
|
—
|
624,668
|
||||||||||||
|
专利和
许可证
确定使用寿命
|
建设
正在进行中
|
共计
|
||||||||||
|
成本
|
(欧元)
|
|||||||||||
|
2018年1月1日
|
148,749
|
—
|
148,749
|
|||||||||
|
加法
|
97,620
|
—
|
97,620
|
|||||||||
|
处置
|
(17
|
)
|
—
|
(17
|
)
|
|||||||
|
重新分类
|
—
|
109,852
|
109,852
|
|||||||||
|
交换差异
|
—
|
—
|
—
|
|||||||||
|
2018年12月31日
|
246,351
|
109,852
|
356,204
|
|||||||||
|
加法
|
84,449
|
251,493
|
335,942
|
|||||||||
|
处置
|
—
|
—
|
—
|
|||||||||
|
重新分类
|
353,155
|
(353,155
|
)
|
—
|
||||||||
|
交换差异
|
(64
|
)
|
—
|
(64
|
)
|
|||||||
|
2019年12月31日
|
683,891
|
8,190
|
692,081
|
|||||||||
|
摊销
|
||||||||||||
|
2018年1月1日
|
(108,083
|
)
|
—
|
(108,083
|
)
|
|||||||
|
本年度折旧费用
|
(25,255
|
)
|
—
|
(25,255
|
)
|
|||||||
|
交换差异
|
—
|
—
|
—
|
|||||||||
|
2018年12月31日
|
(133,337
|
)
|
—
|
(133,337
|
)
|
|||||||
|
本年度折旧费用
|
(106,358
|
)
|
—
|
(106,358
|
)
|
|||||||
|
处置
|
—
|
—
|
—
|
|||||||||
|
交换差异
|
14
|
—
|
14
|
|||||||||
|
2019年12月31日
|
(239,681
|
)
|
—
|
(239,681
|
)
|
|||||||
|
净账面价值
|
—
|
—
|
—
|
|||||||||
|
2019年12月31日
|
444,210
|
8,190
|
452,400
|
|||||||||
|
2018年12月31日
|
113,014
|
109,852
|
222,866
|
|||||||||
|
使用权资产
|
外汇
|
|||||||||||||||||||
|
财产
|
汽车
|
差异
|
共计
|
租赁负债
|
||||||||||||||||
|
(欧元)
|
||||||||||||||||||||
|
截至2019年1月1日
|
695,614
|
35,058
|
—
|
730,672
|
730,672
|
|||||||||||||||
|
加法
|
636,754
|
—
|
1,512
|
638,266
|
636,754
|
|||||||||||||||
|
本年度折旧费用
|
(245,342
|
)
|
(20,831
|
)
|
216
|
(265,957
|
)
|
—
|
||||||||||||
|
去认
|
(266,057
|
)
|
—
|
—
|
(266,057
|
)
|
(228,547
|
)
|
||||||||||||
|
利息费用
|
—
|
—
|
—
|
—
|
(12,765
|
)
|
||||||||||||||
|
付款(包括。利息和外汇差额)
|
—
|
—
|
—
|
—
|
(281,535
|
)
|
||||||||||||||
|
截至2019年12月31日
|
820,969
|
14,227
|
1,728
|
836,924
|
844,579
|
|||||||||||||||
|
资本化租赁期限分析
|
契约最小值
租赁义务
|
贴现效果
|
租赁负债
|
|||||||||
|
(欧元)
|
||||||||||||
|
一年内
|
354,878
|
7,175
|
347,703
|
|||||||||
|
一年但不超过五年
|
500,062
|
3,185
|
496,877
|
|||||||||
|
五年以上
|
—
|
—
|
—
|
|||||||||
|
共计
|
854,940
|
10,361
|
844,579
|
|||||||||
|
所有租赁债务的到期日分析
|
共计
|
低值
租赁
|
短期内
租赁
|
资本化
租赁
|
||||||||||||
|
(欧元)
|
||||||||||||||||
|
一年内
|
371,105
|
5,387
|
10,841
|
354,878
|
||||||||||||
|
一年但不超过五年
|
532,845
|
12,779
|
20,005
|
500,062
|
||||||||||||
|
五年以上
|
—
|
—
|
—
|
—
|
||||||||||||
|
共计
|
903,951
|
18,166
|
30,845
|
854,940
|
||||||||||||
|
十二月三十一日,
2019*
|
十二月三十一日,
2018*
|
|||||||
|
(欧元)
|
||||||||
|
使用权资产折旧费用
|
265,957
|
—
|
||||||
|
租赁负债利息费用
|
12,765
|
—
|
||||||
|
租赁费用
|
70,451
|
213,200
|
||||||
|
短期租约(包括行政费用)
|
65,348
|
—
|
||||||
|
低值资产租赁(包括行政费用)
|
5,103
|
—
|
||||||
|
确认的损益总额
|
349,173
|
213,200
|
||||||
|
其他非金融资产
|
十二月三十一日,
2019
|
十二月三十一日,
2018
|
||||||
|
(欧元)
|
||||||||
|
预付费用
|
1,920,153
|
1,032,676
|
||||||
|
预付
|
698,891
|
14,607
|
||||||
|
其他
|
1,334,056
|
541,419
|
||||||
|
共计
|
3,953,100
|
1,588,702
|
||||||
|
金融资产和金融负债
|
十二月三十一日,
2019
|
十二月三十一日,
2018
|
||||||
|
(欧元)
|
||||||||
|
按摊销成本计算的金融资产
|
||||||||
|
非流动金融资产
|
272,614
|
207,444
|
||||||
|
流动金融资产
|
82,353,867
|
101,184,240
|
||||||
|
按摊销成本计算的金融负债
|
||||||||
|
贸易和其他应付款
|
12,413,662
|
6,657,312
|
||||||
|
有息贷款及借款
|
||||||||
|
非流动租赁负债
|
330,745
|
—
|
||||||
|
流动租赁负债
|
513,834
|
—
|
||||||
|
现金和现金等价物
|
十二月三十一日,
2019
|
十二月三十一日,
2018
|
||||||
|
(欧元)
|
||||||||
|
短期存款
|
||||||||
|
以美元持有的存款(3个月原始到期日及以下)
|
27,803,153
|
32,918,604
|
||||||
|
共计
|
27,803,153
|
32,918,604
|
||||||
|
银行现金
|
||||||||
|
以欧元持有的现金
|
1,211,478
|
21,719,699
|
||||||
|
美元现金
|
4,116,649
|
747,937
|
||||||
|
共计
|
5,328,127
|
22,467,636
|
||||||
|
现金和现金等价物共计
|
33,131,280
|
55,386,240
|
||||||
|
1.
|
财务风险管理目标和政策
|
|
曝光
|
量测
|
风险管理
|
|
|
市场风险
|
未来发展成本;确认的非欧元金融资产和负债
|
预测现金流敏感性分析
|
自然树篱的实现
未来
|
|
信用风险
|
现金和现金等价物,
债务投资
|
信用
额定值
|
银行存款多样化,投资指南
债务投资
|
|
流动资金
|
R&D与G&A成本
和贸易应付款项
|
滚压
现金流量预测
|
通过融资轮或公开发行获得资金
|
|
2.
|
市场风险
|
|
属于InaffRx GmbH公司的现金、现金等价物和金融资产
|
十二月三十一日,
2019
|
|||
|
(欧元)
|
||||
|
流动金融资产(证券和应计利息)
|
32,947,491
|
|||
|
现金和现金等价物
|
4,123,532
|
|||
|
面临风险的资产总额
|
37,071,023
|
|||
|
报告日期1/1.1234欧元兑美元的换算率
|
||||
|
敏感性分析:
|
转换
率
|
利润/(亏损)
|
载运
金额
|
|||||||||
|
(欧元)
|
||||||||||||
|
欧元兑美元贬值1%
|
1.1346
|
(367,040
|
)
|
36,703,983
|
||||||||
|
欧元兑美元升值1%
|
1.1122
|
374,455
|
37,445,478
|
|||||||||
|
欧元兑美元贬值5%
|
1.1796
|
(1,765,287
|
)
|
35,305,736
|
||||||||
|
欧元兑美元升值5%
|
1.0672
|
1,951,106
|
39,022,129
|
|||||||||
|
欧元兑美元贬值10%
|
1.2357
|
(3,370,093
|
)
|
33,700,930
|
||||||||
|
欧元兑美元升值10%
|
1.0111
|
4,119,003
|
41,190,026
|
|||||||||
|
3.
|
信用风险
|
|
4.
|
流动性风险
|
|
流动资金
|
十二月三十一日,
2019
|
十二月三十一日,
2018
|
||||||
|
(欧元)
|
||||||||
|
短期存款
|
27,803,153
|
32,918,604
|
||||||
|
银行现金
|
5,328,127
|
22,467,636
|
||||||
|
有价证券(现行)
|
81,895,377
|
100,868,129
|
||||||
|
其他(非流动部分)
|
272,614
|
207,444
|
||||||
|
其他(目前)
|
458,491
|
316,112
|
||||||
|
可动用资金总额
|
115,757,762
|
156,777,925
|
||||||
|
十二月三十一日,
2019
|
十二月三十一日,
2018
|
|||||||
|
(欧元)
|
||||||||
|
与不可取消租赁有关的最低租赁付款承诺
|
||||||||
|
一年内
|
371,105
|
282,711
|
||||||
|
一年但不超过五年
|
532,845
|
292,300
|
||||||
|
五年以上
|
—
|
—
|
||||||
|
共计
|
903,951
|
575,011
|
||||||
|
十二月三十一日,
2019
|
十二月三十一日,
2018
|
|||||||
|
(欧元)
|
||||||||
|
与不可取消的经营合同或服务有关的最低付款承诺:
|
||||||||
|
一年内
|
10,602,651
|
19,623,790
|
||||||
|
一年但不超过五年
|
13,844,857
|
9,683,700
|
||||||
|
五年以上
|
—
|
—
|
||||||
|
共计
|
24,447,508
|
29,307,490
|
||||||
|
名字
|
营业地点/营业地点
成立国
|
功能货币
|
持有的所有权权益
专家组
2019 2018
|
主要活动
|
|
|
英华Rx有限公司
|
德国
|
欧元
|
100%
|
100%
|
生物制药公司主要经营子公司
|
|
InaffRx制药公司
|
美国
|
美元
|
100%
|
100%
|
基础研究附属机构
|
| • |
2019年12月31日:德国2 217 000欧元,美国374 000欧元,
|
| • |
2018年12月31日:德国704 000欧元,美国351 000欧元。
|
| • |
Niels C.Riedemann教授,首席执行官
|
| • |
郭仁峰教授,首席科学官(CSO)
|
| • |
阿恩德·基督,首席财务官(首席财务官)
|
| • |
Jason Marks,首席法律干事,总法律顾问(CLO),自2019年1月1日起
|
| • |
Niels C.Riedemann教授,首席执行官
|
| • |
郭仁峰教授,CSO
|
| • |
Nicolas Fulpius,审计委员会主席
|
| • |
Jens Holstein,审计委员会成员
|
| • |
Richard Brudnick,自2019年5月以来担任审计委员会成员
|
| • |
卡特林·乌希曼
|
| • |
马丽娜
|
| • |
马克·库布勒
|
|
板补偿
|
2019
|
2018
|
2017
|
|||||||||
|
(欧元)
|
||||||||||||
|
行政管理
|
||||||||||||
|
短期雇员福利
|
2,793,529
|
2,524,202
|
1,986,973
|
|||||||||
|
股票支付
|
5,218,324
|
9,801,454
|
3,187,438
|
|||||||||
|
共计
|
8,011,853
|
12,325,656
|
5,174,411
|
|||||||||
|
非执行董事会
|
||||||||||||
|
短期雇员福利
|
269,031
|
238,180
|
80,735
|
|||||||||
|
股票支付
|
710,611
|
1,085,917
|
42,860
|
|||||||||
|
共计
|
979,642
|
1,324,098
|
123,596
|
|||||||||
|
总补偿
|
8,991,495
|
13,649,754
|
5,298,007
|
|||||||||
|
1.
|
股权结算股票支付安排
|
|
2019
数
|
2019
WAEP
|
2018
数
|
2018
WAEP
|
|||||||||||||
|
1月1日未付
|
289,309
|
€
|
0.01
|
533,820
|
€
|
0.01
|
||||||||||
|
年内行使(1)
|
140,876
|
€
|
0.01
|
244,511
|
€
|
0.01
|
||||||||||
|
截至12月31日未缴
|
148,433
|
€
|
0.01
|
289,309
|
€
|
0.01
|
||||||||||
|
12月31日可锻炼
|
148,433
|
€
|
0.01
|
289,309
|
€
|
0.01
|
||||||||||
|
2019
数
|
2019
WAEP
|
2018
数
|
2018
WAEP
|
|||||||||||||
|
1月1日未付
|
1,181,484
|
|
€7,81
|
1,239,252
|
€
|
7,81
|
||||||||||
|
年内行使(1)
|
—
|
—
|
57,768
|
€
|
7,81
|
|||||||||||
|
截至12月31日未缴
|
1,181,484
|
|
$3,35/€2.98
|
*
|
1,181,484
|
€
|
7,81
|
|||||||||
|
12月31日可锻炼
|
1,181,484
|
|
$3,35/€2.98
|
*
|
1,181,484
|
€
|
7,81
|
|||||||||
|
2019
数
|
2019
WAEP
|
2018
数
|
2018
WAEP
|
|||||||||||||
|
1月1日未付
|
2,051,009
|
|
$3.61/€3.16
|
*
|
1,869,192
|
|
$3.35/€2.79
|
*
|
||||||||
|
年内批出
|
242,450
|
|
$3.25/€2.91
|
*
|
208,073
|
|
$5.96/€5.05
|
*
|
||||||||
|
在该年内被没收
|
112,354
|
|
$6.17/€5.51
|
*
|
26,256
|
|
$3.35/€2.84
|
*
|
||||||||
|
截至12月31日未缴
|
2,181,105
|
|
$3.44/€3.06
|
*
|
2,051,009
|
|
$3.61/€3.16
|
*
|
||||||||
|
12月31日可锻炼
|
1,319,548
|
|
$3.52/€3.13
|
*
|
626,933
|
|
$3.35/€2.93
|
*
|
||||||||
|
2.
|
股票期权公允价值的计量
|
|
股票期权
获批
|
数
|
每
期权
|
外汇汇率
截至
格兰特
日期
|
每
期权
|
股价
批给日期/
行使价格
|
预期
波动率
|
预期寿命
(中点)
基础)
|
无风险率
(内插,
美国主权
条曲线)
|
||||||||||||||||||||||||
|
2018
|
||||||||||||||||||||||||||||||||
|
2月7日*
|
28,002
|
$
|
13.79
|
0.82
|
€
|
11.24
|
$
|
22.75
|
0.73
|
4.9
|
2.60
|
%
|
||||||||||||||||||||
|
五月三十日
|
20,000
|
$
|
22.37
|
0.86
|
€
|
19.23
|
$
|
37.85
|
0.73
|
4.6
|
2.70
|
%
|
||||||||||||||||||||
|
七月二十日
|
54,000
|
$
|
19.80
|
0.86
|
€
|
16.96
|
$
|
32.40
|
0.73
|
4.9
|
2.80
|
%
|
||||||||||||||||||||
|
9月9日21*
|
18,450
|
$
|
20.17
|
0.85
|
€
|
17.15
|
$
|
33.06
|
0.73
|
4.9
|
3.00
|
%
|
||||||||||||||||||||
|
11月20日*
|
12,621
|
$
|
13.39
|
0.88
|
€
|
11.75
|
$
|
26.02
|
0.65
|
4.0
|
2.93
|
%
|
||||||||||||||||||||
|
2019年11月20日/1月1日*
|
75,000
|
$
|
14.45
|
0.88
|
€
|
12.69
|
$
|
26.02
|
0.65
|
4.8
|
3,00
|
%
|
||||||||||||||||||||
|
208,073
|
||||||||||||||||||||||||||||||||
|
2019
|
||||||||||||||||||||||||||||||||
|
一月一日
|
—
|
$
|
14.45
|
0.88
|
€
|
12.69
|
$
|
26.02
|
0.65
|
4.8
|
3,00
|
%
|
||||||||||||||||||||
|
二月四日
|
18,450
|
$
|
18.17
|
0.87
|
€
|
15.87
|
$
|
32.63
|
0.65
|
4.9
|
2,60
|
%
|
||||||||||||||||||||
|
5月14日
|
36,000
|
$
|
22.54
|
0.89
|
€
|
20.08
|
$
|
41.39
|
0.65
|
4.7
|
2.30
|
%
|
||||||||||||||||||||
|
重订价格,七月三日
|
—
|
$
|
0.46-$1.08
|
0.89
|
€
|
0.40-€0.96
|
$
|
3.35
|
1.35
|
2.3-4.6
|
2.30
|
%
|
||||||||||||||||||||
|
十月二十四日
|
50,000
|
$
|
1.96
|
0.90
|
€
|
1.76
|
$
|
2.28
|
1.35
|
4.7
|
1,65
|
%
|
||||||||||||||||||||
|
十二月十六日
|
38,000
|
$
|
3.07
|
0.90
|
€
|
2.75
|
$
|
3.57
|
1.35
|
4.7
|
1,79
|
%
|
||||||||||||||||||||
|
12月16日*
|
100,000
|
$
|
3.07
|
0.90
|
€
|
2.75
|
$
|
3.57
|
1.35
|
4.7
|
1,79
|
%
|
||||||||||||||||||||
|
242,450
|
||||||||||||||||||||||||||||||||
| • |
批内提供的期权越多,支付的费用就越高,而且
|
| • |
一批的转归期越短,其费用就越高。
|
|
2022
|
2021
|
2020
|
2019
|
2018
|
2017
|
|||||||||||||||||||
|
(百万欧元)
|
||||||||||||||||||||||||
|
2016年计划
|
—
|
—
|
—
|
—
|
—
|
4.0
|
||||||||||||||||||
|
2017年长期激励计划
|
0.0
|
0.3
|
2.1
|
5.2
|
12.1
|
0.6
|
||||||||||||||||||
|
重新定价于2019年7月3日完成
|
||||||||||||||||||||||||
|
2016年计划
|
—
|
—
|
—
|
0.5
|
—
|
—
|
||||||||||||||||||
|
2017年长期激励计划
|
0.0
|
0.0
|
0.3
|
1.1
|
—
|
—
|
||||||||||||||||||
|
补偿费用总额
|
0.0
|
0.3
|
2.4
|
6.8
|
12.1
|
4.6
|
||||||||||||||||||
|
1.
|
专家组通过的新的和经修正的标准
|
| • |
“国际财务报告准则”第16条租约
|
| • |
IFRIC 23税收待遇的不确定性。
|
| • |
附带负报酬的预付特征(修订“国际财务报告准则”第9条)。
|
| • |
联营和合资企业的长期利益(对国际会计准则第28号的修正)。
|
| • |
计划修订、缩减或结算(对“国际会计准则”第19条的修正)。
|
| • |
对“国际财务报告准则”2015-2017年周期的年度改进-各种标准。
|
| • |
将短期租约豁免适用于在首次申请之日起12个月内届满的租约。
|
| • |
将初始直接费用排除在初次申请之日的资产使用权计量之外。
|
|
2019
|
||||
|
截至2018年12月31日公布的业务租赁承付款
|
575,000
|
|||
|
短期租约在直线基础上确认为费用
|
(17,765
|
)
|
||
|
以直线确认为费用的低值租约
|
(5,993
|
)
|
||
|
由于对延期和终止备选办法的不同处理而作出的调整
|
201,127
|
|||
|
共计
|
752,369
|
|||
|
在首次申请之日使用承租人增量借款利率的折扣
|
(21,697
|
)
|
||
|
截至2019年1月1日已确认的租赁负债
|
730,672
|
|||
|
其现行租赁责任
|
215,312
|
|||
|
非流动租赁负债
|
515,360
|
|||
|
2.
|
新的标准和解释尚未通过
|
| • |
对“国际财务报告准则”中提及概念框架的修正,自2020年1月1日起生效
|
| • |
业务定义(修订“国际财务报告准则”第3条),自2020年1月1日起生效
|
| • |
材料定义(对国际会计准则1和国际会计准则8的修正),自2020年1月1日起生效
|
| • |
“国际财务报告准则”第17号保险合同,2021年1月1日生效
|
|
3.
|
电流和非电流区分
|
|
4.
|
外币交易
|
|
5.
|
现金流量表附注
|
|
6.
|
研发
|
|
7.
|
雇员福利
|
|
8.
|
政府赠款
|
|
9.
|
租赁安排
|
|
10.
|
利息收入
|
|
11.
|
无形资产
|
|
12.
|
实验室和办公设备
|
| • |
实验室设备:3至13年
|
| • |
办公设备:一至五年
|
|
13.
|
金融资产和负债(金融工具)
|
|
14.
|
所得税
|
|
15.
|
公允价值计量
|
| • |
一级,活跃市场相同资产或负债的报价。
|
| • |
第2级,除报价外,还包括在第1级中可观察到的、直接(作为价格)或间接(从价格中得出)的投入。
|
| • |
第3级,非基于可观测的市场数据的仪器的输入(不可观测的输入)。
|