目录
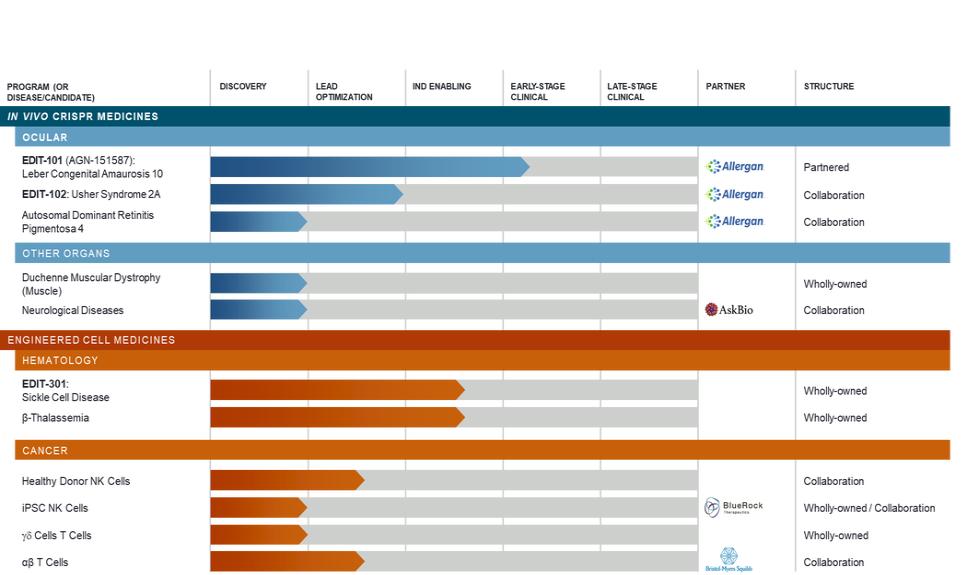
在现场CRISPR药物-眼科
我们已经授予Allergan一个独家的选择,只从我们那里获得五个用于治疗眼部疾病的合作开发项目,包括编辑-101。正如下面所讨论的,Allergan已经在编辑-101方面行使了它的选择,并与我们在美国为这样的项目达成了利润分享安排。见“我们的合作及发牌策略“下面是更多信息。
先天性黑斑病10例
Leber先天性黑质坏死(LCA)是由至少18个不同基因突变引起的一组异质性遗传性视网膜营养不良,是遗传性儿童致盲最常见的原因,全世界每10万活产有2~3例。LCA的症状出现在生命的第一年,严重的视力丧失,眼睛的快速不自主移动,眼睛对强光的痛苦反应,以及由于缺乏功能的光感受器细胞而缺乏可测量的视网膜电图记录。这种疾病最常见的形式是LCA 10,这是一种单基因疾病,约占LCA所有亚型的20-30%。LCA 10是由CEP290基因的常染色体隐性突变引起的,该基因编码光感受器细胞生存和正常功能所需的一种蛋白质。CEP 2 90基因中最常见的突变发生在大约85%的患有LCA 10的北欧和西欧患者中,即A到G核苷酸的改变,破坏了基因信息的正常剪接或处理,最终导致功能上的CEP 2 90蛋白缺乏。随着时间的推移,CEP 2 90蛋白的减少会导致光感受器细胞外段的丢失和功能的发挥,从而导致失明。我们相信在美国和欧洲有2,000到5,000名LCA 10患者。
Edt-101使用AAV 5载体将编码SaCas9的DNA和两个引导RNA传递到眼睛中的感光细胞。这种实验性的CRISPR药物是为了消除CEP 2 90基因非编码区或内含子中引起疾病的A到G核苷酸的变化,方法是切断该核苷酸和周围的DNA。我们相信,这种基因组编辑方法有可能恢复剩余的光感受器细胞的正常蛋白表达和功能,从而改善视力或阻止长期合作行动患者视力的进一步丧失。
8